modeling snp mediated differential targeting of homologous 3'utr by microrna
TRANSCRIPT

Modeling SNP mediated differential targetingof homologous 3'UTR by MicroRNA
Jasmine Kaur Ahluwalia,1,2 Kartik Soni,1 Sridhar Sivasubbu1 and Vani Brahmachari2,*
1CSIR Institute of Genomics and Integrative Biology; Mall Road; Delhi, India; 2Dr. B.R. Ambedkar Center for Biomedical Research; University of Delhi; Delhi, India
Keywords: Differential regulation of homologous alleles, microRNA, Incomplete penetrance, Variable expressivity, JAG1, TGFBR2
Abbreviations: IP-VE, Incomplete penetrance-variable expressivity; miRNA, Micro RNA; 3'UTR, 3'Untranslated region;Fluc, Firefly luciferase; Rluc, Renilla luciferase
We have previously proposed the post-transcriptional regulation through microRNA as a mechanism for incompletepenetrance and variable expressivity leading to lack of correlation between genotype and phenotype. Here we report thevalidation of miRNA-target interactions we predicted earlier and demonstrate the regulation of endogenous JAG1 by hsa-miR-214 and hsa-miR-124, and TGFBR2 by hsa-miR-34b*, through luciferase activity of reporter constructs and also theexpression levels of the endogenous genes. Using these targets, we have modeled the diploid state for miRNA target sitewith heterozygosity for the SNP and demonstrate the differential targeting of an otherwise identical 39UTR. We show thatSNP rs8708 (A . G) at the target site of hsa-miR-214 can relieve the repression while an SNP rs11466532 (C . T)enhances the repression of reporter expression by hsa-miR-34b*. We discuss the results in the light of its implications inthe context of penetrance of dominant mutations in miRNA targeted genes, using JAG1 as an example. Theseobservations imply that disease causing mutations in JAG1 linked to the SNP rs8708G will be poorly targeted by hsa-miR-214 when present against a normal allele of JAG1 with rs8708A and will show penetrance of JAG1 mutations as Alagillesyndrome, while mutant JAG1 linked to rs8708A against rs8708G on the normal allele will show either no disease ormuch attenuated symptoms and hence exhibit incomplete penetrance.
Introduction
Most single gene disorders are expected to follow Mendelianinheritance; dominant mutations showing the phenotype in allindividuals who carry the mutations and recessive mutationsexpressing phenotype only when they are in homozygouscondition. However, in several dominant disorders heterozygousindividuals carrying identical mutation exhibit variability inclinical features ranging from the absence of phenotype to severesymptoms.1 Such mutations/diseases are said to exhibit incom-plete penetrance and variable expressivity (IP-VE). The molecularmechanisms leading to IP-VE are known to be diverse andinclude variation in epigenetic marking of the alleles.2 Morerecently, deep sequencing strategies have led to the identificationof SNPs resulting in splice site variation and low expression of thenormal allele as a basis of IP-VE. In erythropoietic protoporphyria(EPP), an autosomal dominant disorder of heme biosynthesis,the clinical phenotype results from coinheritance of a mutantallele and a low-expressed wild-type allele of FECH.3,4 Brugadasyndrome, a cardiac disorder, is inherited as an autosomaldominant mutation but shows incomplete penetrance. Poelzinget al. reported that individuals carrying a H558R polymorphismin trans, on the normal allele of the gene, in addition to the
disease causing R282H mutation in SCN5A are asymptomatic,suggesting that the H558R polymorphism over rides the effect ofthe mutant allele.5 Similarly, incomplete penetrance is attributedto the differential expression of the normal allele in retinitispigmentosa 11 caused due to mutations in PRPF31gene.6,7 Inthese cases, allelic variations at the sequence level, leading toclinically variable phenotype adequately explain the observeddeviation in penetrance and expressivity. However, the molecularbasis of incomplete penetrance remains unexplained in a numberof cases such as Camurati-Engelmann disease, vitelliform maculardystrophy, senile systemic amyloidosis, autosomal dominanthypophosphatemic rickets, familial thoracic aortic aneurysm 3,autosomal dominant spastic paraplegia 4, among many others.Variation in allelic expression is not limited to IP-VE, and isobserved in epigenetic phenomena like genomic imprinting andinactivation of whole chromosome as in the X chromosome infemale mammals.8,9
We have earlier proposed a model for the mechanism of IP-VEinvoking microRNA (miRNA) in differential regulation ofhomologous alleles independent of the primary disease causingmutation.10 MiRNAs have emerged as one of the major post-transcriptional regulators of gene expression. The interaction ofmiRNA with target site even with partial complementarity, offers
*Correspondence to: Vani Brahmachari; Email: [email protected]: 08/12/11; Revised: 01/10/12; Accepted: 01/11/12http://dx.doi.org/10.4161/rna.9.3.19318
RESEARCH PAPER
RNA Biology 9:3, 1–10; March 2012; G 2012 Landes Bioscience
www.landesbioscience.com RNA Biology 1

immense scope for variation in kinetics of miRNA and targetinteraction and the resultant dynamics in gene expressionproviding specificity and variability at the same time. Thereforethe presence of SNP in the 3'UTR at the target site for miRNAcan modulate the level of expression leading to individualdifferences in susceptibility to diseases.11-14 The differentialfrequency of SNP in let-7 target site in 3'UTR of KRAS inpatients compared with world population is implicated in risk ofnon-small cell lung cancer. Similarly in small-cell lung cancer SNPin 3'UTR of MYCL1 targeted by hsa-miR-1827 is also associatedwith disease risk.11,12 Significant effect of SNP in miRNA-mediated regulation is demonstrated for BRCA1, XCCR1,TGFB1, BMPR1B in luciferase reporter assays on one hand andcorrelation with population susceptibility to breast cancer on theother.13,14 The role of SNP in miRNA mediated regulation isbeing widely acknowledged and allelic imbalance sequencing ininter-strain crosses suggest that we cannot ignore the effect ofpolymorphic miRNA regulatory sites on gene expression variationbetween individuals.15
These studies have indicated that a SNP in a miRNA targetsite can either stabilize or destabilize the target site-miRNAinteraction and thus impact the differential expression ofhomologous alleles. We have examined this paradigm for itsrelevance to incomplete-penetrance and variable expressivity.
Based on the rationale that variable expressivity is an inherentproperty of alleles resulting in subtle changes in the concentrationof gene product, we proposed the involvement of miRNAs thatcan fine-tune gene expression.10 We showed that a significantlyhigh number of IP-VE genes are targeted by multiple miRNAsand are co-expressed with the cognate miRNA in the same tissue.Meta-analysis of data from the public domain indicates a negativecorrelation between the miRNA and target protein levels.10 Ouranalysis indicated that SNP in the miRNA target site canpotentially affect its interaction with miRNA based on thepredictions using dbSMR database, which utilizes structuralparameters associated with variation.16
For our current study, we selected 2 IP-VE genes namely,JAG1 and TGFBR2 from a list of 88 genes previously predicted astargets of miRNA mediated regulation.10 Apart from diseaseimplication, the selection of genes was based on the co-expressionof the miRNA and the genes in the same tissue as reported inliterature, as well as presence of SNP in the miRNA target sites.
JAG1, a ligand for NOTCH receptor is fundamental in variousdevelopmental processes. Deletions and frame shift mutations inJAG1 is implicated in Alagille syndrome (AGS), a multisystemautosomal dominant disorder that shows IP-VE.17-19 Wepredicted binding sites for two miRNAs namely, hsa-miR-124and hsa-miR-214 on a 1.8Kb long JAG13'UTR.10 Additionally,the binding site of hsa-miR-214 on JAG1 contains a SNP(rs8708, A . G). Mutations in TGFBR2 cause Aortic aneurysmfamilial thoracic type 3 (OMIM 610380) an autosomal dominantcardiovascular disorder showing reduced penetrance and markedintra-familial variability in the age of disease onset.20 According toour in silico analysis, a 2.5Kb 3'UTR of TGFBR2 mRNA containsthe binding sites for hsa-miR-34b* and -642, each containing avalidated SNP in the target region namely, rs11466532 (C . T)
in the target site of hsa-miR-34b* and rs11466534 (G. A) in thesequence complementary to the seed region of hsa-miR-642.
In the present report we have modeled homologous miRNAtarget site to analyze differential gene expression in addition tovalidating miRNA-target predictions of IP-VE genes.
Results
Selection and validation of miRNA targets. The miRNA targetpredictions were based on the consensus of three differentsoftwares, namely miRanda, TargetScan and RNAhybrid.10 Weprioritized 4 IP-VE genes in our current study from a list of 88genes previously predicted as targets of miRNA.10 Apart fromdisease implication, the selection of genes was based on co-expression of miRNA and the gene in the same tissue as reportedin literature. The effect of miRNA was assessed by cloning theputative target site of around 90 base pairs including the miRNAtarget sequence, in pMIR-REPORTTM Luciferase vector and co-transfection of the target construct with miRNA mimics. Co-transfection with hRLucpGL4.75 with Renilla luciferase reporter(Rluc) was used as the control. In each construct, target site for asingle miRNA was cloned to unambiguously delineate theirindependent effect. In certain cases, longer 3'UTR was cloned toinclude multiple miRNA target sites and is indicated appro-priately. The effect of miRNA was evaluated as the ratio of Fireflyluciferase (Fluc) expression normalized to that of Renilla luciferase(Rluc), as detailed in the methods section. BEST1 and FGF23contain overlapping binding sites for two miRNAs each (hsa-miR-370 and -552 for BEST1; hsa-miR-200b and -200c for FGF23).However, upon transfection of the target luciferase constructswith cognate miRNA mimic, no miRNA-mediated repressionwas evident. The other two genes, JAG1 and TGFBR2 containnon-overlapping target sites for two miRNAs each (hsa-miR-124and -214 for JAG1; hsa-miR-34b* and -642 for TGFBR2). Co-transfection of their constructs with the respective miRNA mimicsshowed significant reduction in luciferase activity and is discussedin the following sections. Since, we want to model differentialeffect of target site SNP on miRNA-mediated regulation, thepresence of SNP in the miRNA target sites of JAG1 and TGFBR2was the major consideration in analyzing these two targets ingreater detail. The choice of SNP for analysis was based on thepredictions made using dbSMR database, which utilizes structuralparameters associated with variation.16 As mentioned in dbSMR,the effect of SNP is scored on the basis of the change it creates inthe intramolecular base-pairing of the 3'UTR at the binding siteof miRNA. The authors considered the number of bases changingtheir conformation relative to the total number of bases binding tothe miRNA to calculate the degree of change in the overallstructural variation. Increase in the number of structured bases inthe target site due to a SNP implies a loss of a legitimate miRNAbinding site, while a decrease in the number of structured bases atthe target site implies a gain.16 Initial selection of both the SNPsrs8708 (A.G; JAG1) and rs11466532 (C.T; TGFBR2) wasbased on their occurrence within the miRNA binding site. TheSNP at the target site on the 3'UTR of JAG1 maps at the positionpairing with 6th nucleotide from the 3' end of the miRNA.
2 RNA Biology Volume 9 Issue 3
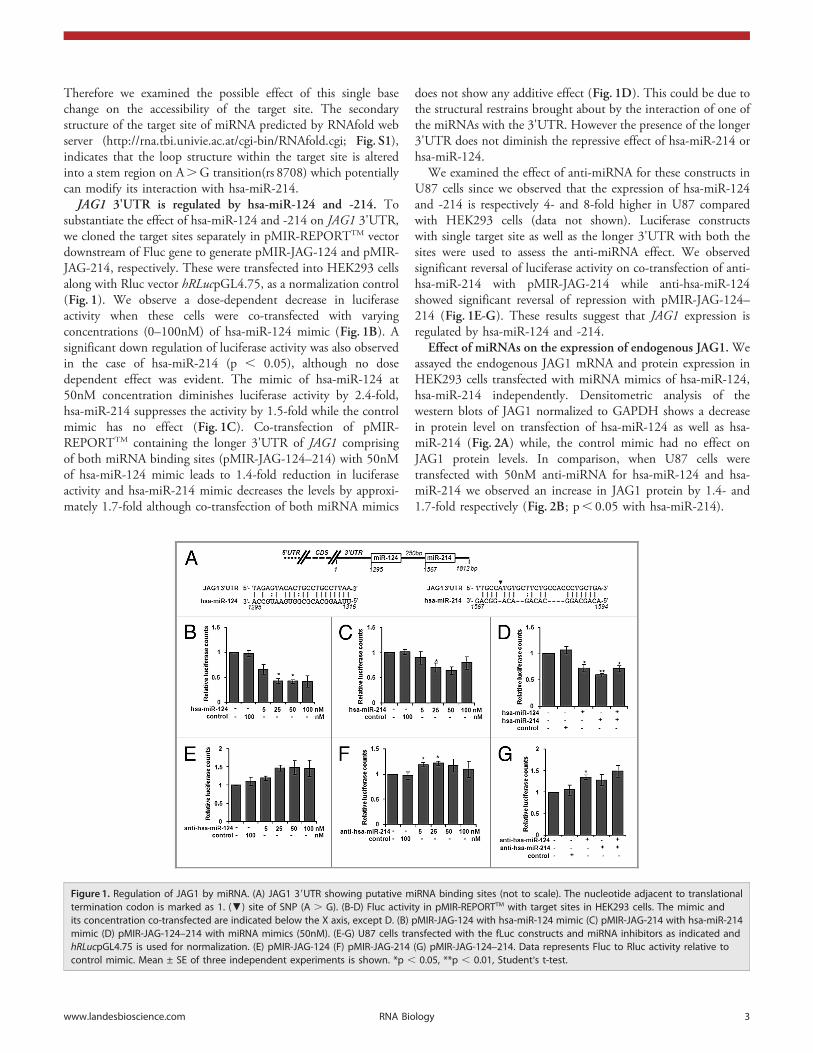
Therefore we examined the possible effect of this single basechange on the accessibility of the target site. The secondarystructure of the target site of miRNA predicted by RNAfold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi; Fig. S1),indicates that the loop structure within the target site is alteredinto a stem region on A.G transition(rs 8708) which potentiallycan modify its interaction with hsa-miR-214.
JAG1 3'UTR is regulated by hsa-miR-124 and -214. Tosubstantiate the effect of hsa-miR-124 and -214 on JAG1 3'UTR,we cloned the target sites separately in pMIR-REPORTTM vectordownstream of Fluc gene to generate pMIR-JAG-124 and pMIR-JAG-214, respectively. These were transfected into HEK293 cellsalong with Rluc vector hRLucpGL4.75, as a normalization control(Fig. 1). We observe a dose-dependent decrease in luciferaseactivity when these cells were co-transfected with varyingconcentrations (0–100nM) of hsa-miR-124 mimic (Fig. 1B). Asignificant down regulation of luciferase activity was also observedin the case of hsa-miR-214 (p , 0.05), although no dosedependent effect was evident. The mimic of hsa-miR-124 at50nM concentration diminishes luciferase activity by 2.4-fold,hsa-miR-214 suppresses the activity by 1.5-fold while the controlmimic has no effect (Fig. 1C). Co-transfection of pMIR-REPORTTM containing the longer 3'UTR of JAG1 comprisingof both miRNA binding sites (pMIR-JAG-124–214) with 50nMof hsa-miR-124 mimic leads to 1.4-fold reduction in luciferaseactivity and hsa-miR-214 mimic decreases the levels by approxi-mately 1.7-fold although co-transfection of both miRNA mimics
does not show any additive effect (Fig. 1D). This could be due tothe structural restrains brought about by the interaction of one ofthe miRNAs with the 3'UTR. However the presence of the longer3'UTR does not diminish the repressive effect of hsa-miR-214 orhsa-miR-124.
We examined the effect of anti-miRNA for these constructs inU87 cells since we observed that the expression of hsa-miR-124and -214 is respectively 4- and 8-fold higher in U87 comparedwith HEK293 cells (data not shown). Luciferase constructswith single target site as well as the longer 3'UTR with both thesites were used to assess the anti-miRNA effect. We observedsignificant reversal of luciferase activity on co-transfection of anti-hsa-miR-214 with pMIR-JAG-214 while anti-hsa-miR-124showed significant reversal of repression with pMIR-JAG-124–214 (Fig. 1E-G). These results suggest that JAG1 expression isregulated by hsa-miR-124 and -214.
Effect of miRNAs on the expression of endogenous JAG1.Weassayed the endogenous JAG1 mRNA and protein expression inHEK293 cells transfected with miRNA mimics of hsa-miR-124,hsa-miR-214 independently. Densitometric analysis of thewestern blots of JAG1 normalized to GAPDH shows a decreasein protein level on transfection of hsa-miR-124 as well as hsa-miR-214 (Fig. 2A) while, the control mimic had no effect onJAG1 protein levels. In comparison, when U87 cells weretransfected with 50nM anti-miRNA for hsa-miR-124 and hsa-miR-214 we observed an increase in JAG1 protein by 1.4- and1.7-fold respectively (Fig. 2B; p, 0.05 with hsa-miR-214).
Figure 1. Regulation of JAG1 by miRNA. (A) JAG1 39UTR showing putative miRNA binding sites (not to scale). The nucleotide adjacent to translationaltermination codon is marked as 1. (▼) site of SNP (A . G). (B-D) Fluc activity in pMIR-REPORTTM with target sites in HEK293 cells. The mimic andits concentration co-transfected are indicated below the X axis, except D. (B) pMIR-JAG-124 with hsa-miR-124 mimic (C) pMIR-JAG-214 with hsa-miR-214mimic (D) pMIR-JAG-124–214 with miRNA mimics (50nM). (E-G) U87 cells transfected with the fLuc constructs and miRNA inhibitors as indicated andhRLucpGL4.75 is used for normalization. (E) pMIR-JAG-124 (F) pMIR-JAG-214 (G) pMIR-JAG-124–214. Data represents Fluc to Rluc activity relative tocontrol mimic. Mean ± SE of three independent experiments is shown. *p , 0.05, **p , 0.01, Student’s t-test.
www.landesbioscience.com RNA Biology 3
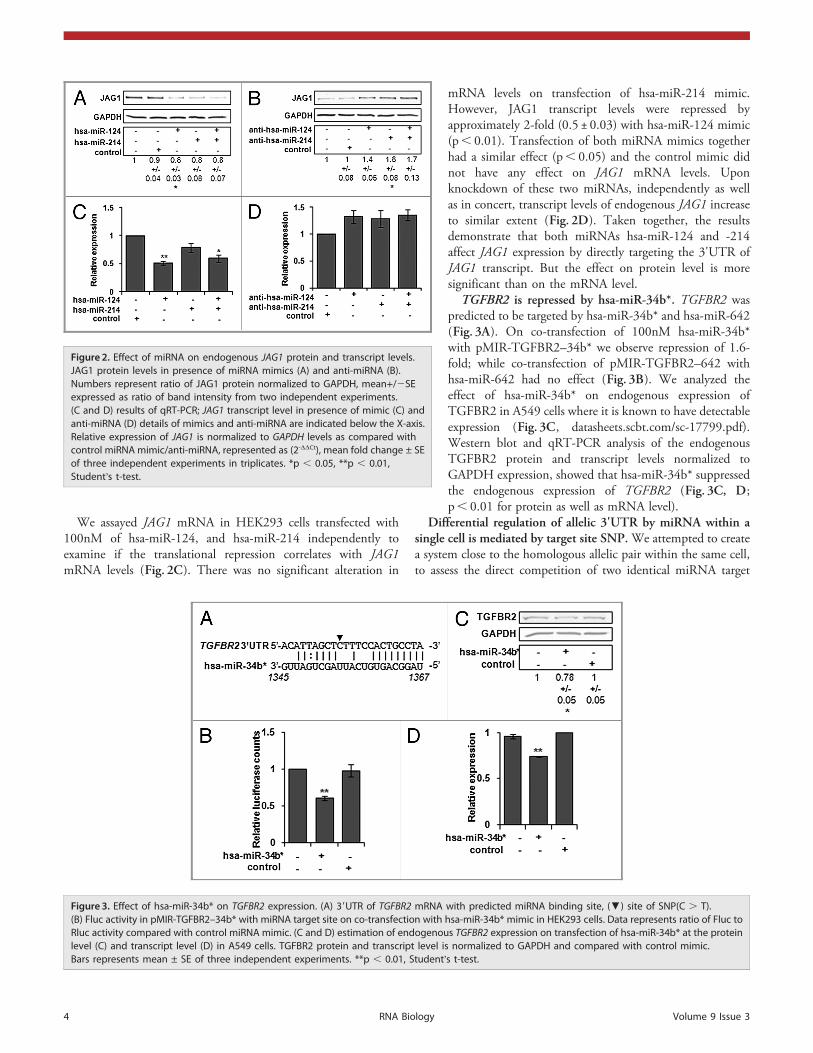
We assayed JAG1 mRNA in HEK293 cells transfected with100nM of hsa-miR-124, and hsa-miR-214 independently toexamine if the translational repression correlates with JAG1mRNA levels (Fig. 2C). There was no significant alteration in
mRNA levels on transfection of hsa-miR-214 mimic.However, JAG1 transcript levels were repressed byapproximately 2-fold (0.5 ± 0.03) with hsa-miR-124 mimic(p, 0.01). Transfection of both miRNA mimics togetherhad a similar effect (p, 0.05) and the control mimic didnot have any effect on JAG1 mRNA levels. Uponknockdown of these two miRNAs, independently as wellas in concert, transcript levels of endogenous JAG1 increaseto similar extent (Fig. 2D). Taken together, the resultsdemonstrate that both miRNAs hsa-miR-124 and -214affect JAG1 expression by directly targeting the 3'UTR ofJAG1 transcript. But the effect on protein level is moresignificant than on the mRNA level.
TGFBR2 is repressed by hsa-miR-34b*. TGFBR2 waspredicted to be targeted by hsa-miR-34b* and hsa-miR-642(Fig. 3A). On co-transfection of 100nM hsa-miR-34b*with pMIR-TGFBR2–34b* we observe repression of 1.6-fold; while co-transfection of pMIR-TGFBR2–642 withhsa-miR-642 had no effect (Fig. 3B). We analyzed theeffect of hsa-miR-34b* on endogenous expression ofTGFBR2 in A549 cells where it is known to have detectableexpression (Fig. 3C, datasheets.scbt.com/sc-17799.pdf).Western blot and qRT-PCR analysis of the endogenousTGFBR2 protein and transcript levels normalized toGAPDH expression, showed that hsa-miR-34b* suppressedthe endogenous expression of TGFBR2 (Fig. 3C, D;p, 0.01 for protein as well as mRNA level).
Differential regulation of allelic 3'UTR by miRNA within asingle cell is mediated by target site SNP.We attempted to createa system close to the homologous allelic pair within the same cell,to assess the direct competition of two identical miRNA target
Figure 2. Effect of miRNA on endogenous JAG1 protein and transcript levels.JAG1 protein levels in presence of miRNA mimics (A) and anti-miRNA (B).Numbers represent ratio of JAG1 protein normalized to GAPDH, mean+/2SEexpressed as ratio of band intensity from two independent experiments.(C and D) results of qRT-PCR; JAG1 transcript level in presence of mimic (C) andanti-miRNA (D) details of mimics and anti-miRNA are indicated below the X-axis.Relative expression of JAG1 is normalized to GAPDH levels as compared withcontrol miRNA mimic/anti-miRNA, represented as (2-DDCt), mean fold change ± SEof three independent experiments in triplicates. *p , 0.05, **p , 0.01,Student’s t-test.
Figure 3. Effect of hsa-miR-34b* on TGFBR2 expression. (A) 39UTR of TGFBR2 mRNA with predicted miRNA binding site, (▼) site of SNP(C . T).(B) Fluc activity in pMIR-TGFBR2–34b* with miRNA target site on co-transfection with hsa-miR-34b* mimic in HEK293 cells. Data represents ratio of Fluc toRluc activity compared with control miRNA mimic. (C and D) estimation of endogenous TGFBR2 expression on transfection of hsa-miR-34b* at the proteinlevel (C) and transcript level (D) in A549 cells. TGFBR2 protein and transcript level is normalized to GAPDH and compared with control mimic.Bars represents mean ± SE of three independent experiments. **p , 0.01, Student’s t-test.
4 RNA Biology Volume 9 Issue 3
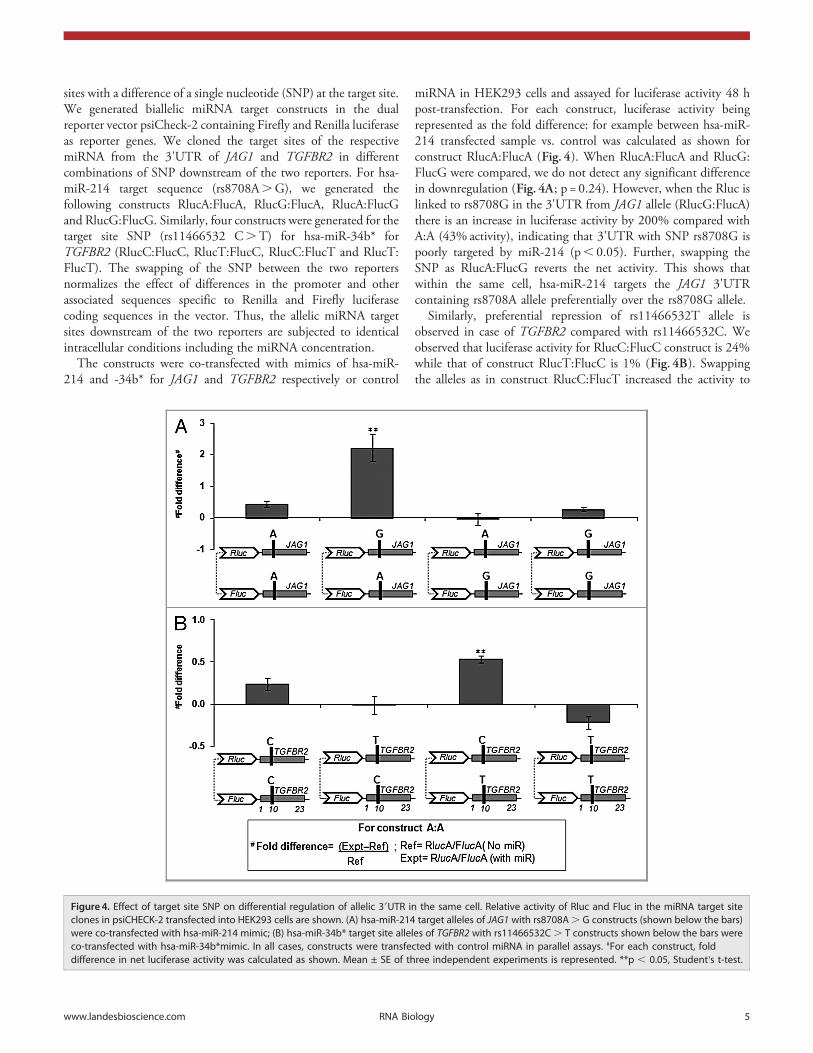
sites with a difference of a single nucleotide (SNP) at the target site.We generated biallelic miRNA target constructs in the dualreporter vector psiCheck-2 containing Firefly and Renilla luciferaseas reporter genes. We cloned the target sites of the respectivemiRNA from the 3'UTR of JAG1 and TGFBR2 in differentcombinations of SNP downstream of the two reporters. For hsa-miR-214 target sequence (rs8708A.G), we generated thefollowing constructs RlucA:FlucA, RlucG:FlucA, RlucA:FlucGand RlucG:FlucG. Similarly, four constructs were generated for thetarget site SNP (rs11466532 C.T) for hsa-miR-34b* forTGFBR2 (RlucC:FlucC, RlucT:FlucC, RlucC:FlucT and RlucT:FlucT). The swapping of the SNP between the two reportersnormalizes the effect of differences in the promoter and otherassociated sequences specific to Renilla and Firefly luciferasecoding sequences in the vector. Thus, the allelic miRNA targetsites downstream of the two reporters are subjected to identicalintracellular conditions including the miRNA concentration.
The constructs were co-transfected with mimics of hsa-miR-214 and -34b* for JAG1 and TGFBR2 respectively or control
miRNA in HEK293 cells and assayed for luciferase activity 48 hpost-transfection. For each construct, luciferase activity beingrepresented as the fold difference: for example between hsa-miR-214 transfected sample vs. control was calculated as shown forconstruct RlucA:FlucA (Fig. 4). When RlucA:FlucA and RlucG:FlucG were compared, we do not detect any significant differencein downregulation (Fig. 4A; p = 0.24). However, when the Rluc islinked to rs8708G in the 3'UTR from JAG1 allele (RlucG:FlucA)there is an increase in luciferase activity by 200% compared withA:A (43% activity), indicating that 3'UTR with SNP rs8708G ispoorly targeted by miR-214 (p, 0.05). Further, swapping theSNP as RlucA:FlucG reverts the net activity. This shows thatwithin the same cell, hsa-miR-214 targets the JAG1 3'UTRcontaining rs8708A allele preferentially over the rs8708G allele.
Similarly, preferential repression of rs11466532T allele isobserved in case of TGFBR2 compared with rs11466532C. Weobserved that luciferase activity for RlucC:FlucC construct is 24%while that of construct RlucT:FlucC is 1% (Fig. 4B). Swappingthe alleles as in construct RlucC:FlucT increased the activity to
Figure 4. Effect of target site SNP on differential regulation of allelic 39UTR in the same cell. Relative activity of Rluc and Fluc in the miRNA target siteclones in psiCHECK-2 transfected into HEK293 cells are shown. (A) hsa-miR-214 target alleles of JAG1 with rs8708A. G constructs (shown below the bars)were co-transfected with hsa-miR-214 mimic; (B) hsa-miR-34b* target site alleles of TGFBR2 with rs11466532C . T constructs shown below the bars wereco-transfected with hsa-miR-34b*mimic. In all cases, constructs were transfected with control miRNA in parallel assays. #For each construct, folddifference in net luciferase activity was calculated as shown. Mean ± SE of three independent experiments is represented. **p , 0.05, Student’s t-test.
www.landesbioscience.com RNA Biology 5
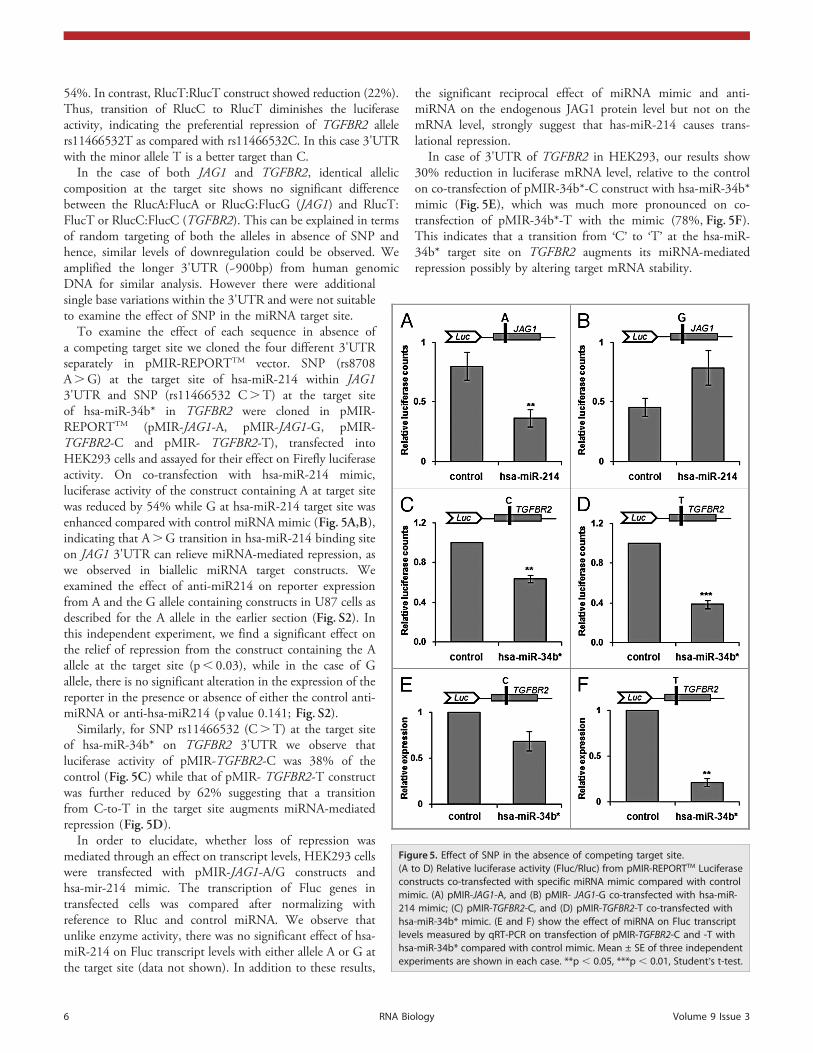
54%. In contrast, RlucT:RlucT construct showed reduction (22%).Thus, transition of RlucC to RlucT diminishes the luciferaseactivity, indicating the preferential repression of TGFBR2 allelers11466532T as compared with rs11466532C. In this case 3'UTRwith the minor allele T is a better target than C.
In the case of both JAG1 and TGFBR2, identical alleliccomposition at the target site shows no significant differencebetween the RlucA:FlucA or RlucG:FlucG (JAG1) and RlucT:FlucT or RlucC:FlucC (TGFBR2). This can be explained in termsof random targeting of both the alleles in absence of SNP andhence, similar levels of downregulation could be observed. Weamplified the longer 3'UTR (~900bp) from human genomicDNA for similar analysis. However there were additionalsingle base variations within the 3'UTR and were not suitableto examine the effect of SNP in the miRNA target site.
To examine the effect of each sequence in absence ofa competing target site we cloned the four different 3'UTRseparately in pMIR-REPORTTM vector. SNP (rs8708A.G) at the target site of hsa-miR-214 within JAG13'UTR and SNP (rs11466532 C.T) at the target siteof hsa-miR-34b* in TGFBR2 were cloned in pMIR-REPORTTM (pMIR-JAG1-A, pMIR-JAG1-G, pMIR-TGFBR2-C and pMIR- TGFBR2-T), transfected intoHEK293 cells and assayed for their effect on Firefly luciferaseactivity. On co-transfection with hsa-miR-214 mimic,luciferase activity of the construct containing A at target sitewas reduced by 54% while G at hsa-miR-214 target site wasenhanced compared with control miRNA mimic (Fig. 5A,B),indicating that A.G transition in hsa-miR-214 binding siteon JAG1 3'UTR can relieve miRNA-mediated repression, aswe observed in biallelic miRNA target constructs. Weexamined the effect of anti-miR214 on reporter expressionfrom A and the G allele containing constructs in U87 cells asdescribed for the A allele in the earlier section (Fig. S2). Inthis independent experiment, we find a significant effect onthe relief of repression from the construct containing the Aallele at the target site (p, 0.03), while in the case of Gallele, there is no significant alteration in the expression of thereporter in the presence or absence of either the control anti-miRNA or anti-hsa-miR214 (p value 0.141; Fig. S2).
Similarly, for SNP rs11466532 (C.T) at the target siteof hsa-miR-34b* on TGFBR2 3'UTR we observe thatluciferase activity of pMIR-TGFBR2-C was 38% of thecontrol (Fig. 5C) while that of pMIR- TGFBR2-T constructwas further reduced by 62% suggesting that a transitionfrom C-to-T in the target site augments miRNA-mediatedrepression (Fig. 5D).
In order to elucidate, whether loss of repression wasmediated through an effect on transcript levels, HEK293 cellswere transfected with pMIR-JAG1-A/G constructs andhsa-mir-214 mimic. The transcription of Fluc genes intransfected cells was compared after normalizing withreference to Rluc and control miRNA. We observe thatunlike enzyme activity, there was no significant effect of hsa-miR-214 on Fluc transcript levels with either allele A or G atthe target site (data not shown). In addition to these results,
the significant reciprocal effect of miRNA mimic and anti-miRNA on the endogenous JAG1 protein level but not on themRNA level, strongly suggest that has-miR-214 causes trans-lational repression.
In case of 3'UTR of TGFBR2 in HEK293, our results show30% reduction in luciferase mRNA level, relative to the controlon co-transfection of pMIR-34b*-C construct with hsa-miR-34b*mimic (Fig. 5E), which was much more pronounced on co-transfection of pMIR-34b*-T with the mimic (78%, Fig. 5F).This indicates that a transition from ‘C’ to ‘T’ at the hsa-miR-34b* target site on TGFBR2 augments its miRNA-mediatedrepression possibly by altering target mRNA stability.
Figure 5. Effect of SNP in the absence of competing target site.(A to D) Relative luciferase activity (Fluc/Rluc) from pMIR-REPORTTM Luciferaseconstructs co-transfected with specific miRNA mimic compared with controlmimic. (A) pMIR-JAG1-A, and (B) pMIR- JAG1-G co-transfected with hsa-miR-214 mimic; (C) pMIR-TGFBR2-C, and (D) pMIR-TGFBR2-T co-transfected withhsa-miR-34b* mimic. (E and F) show the effect of miRNA on Fluc transcriptlevels measured by qRT-PCR on transfection of pMIR-TGFBR2-C and -T withhsa-miR-34b* compared with control mimic. Mean ± SE of three independentexperiments are shown in each case. **p , 0.05, ***p , 0.01, Student’s t-test.
6 RNA Biology Volume 9 Issue 3

Discussion
The experiments described here were undertaken to generate amodel to test the hypothesis we proposed based on in silicoanalysis, wherein we invoked a role for miRNA in regulatingpenetrance and expressivity for dominantly inherited diseases.Despite the differences in mechanisms, variations in penetranceand expressivity of mutations resulting in the lack of correlationbetween the genotype and the phenotype can be viewed as amanifestation of differential expression of homologous alleles.Therefore, mechanisms resulting in fine tuning of expression ofthe homologs can bias the expression of one of the alleles. Weproposed that miRNA-mediated regulation brings about non-equivalence in expression of homologous alleles and thus, can be amechanism of IP-VE.10 We hypothesized that under normalcircumstances the homologous alleles are expected to have nearlyidentical sequence and therefore, any one of them may be targetedfor post-transcriptional regulation by the cognate miRNA. Inabsence of mutation in the target gene, cellular level of proteincoded by the target gene is adequate for normal phenotype.However, when there is a mutation in a heterozygous state, thephenotype is dictated by which of the two products of trans-cription from the homologous alleles is targeted by miRNA, thusresulting in IP-VE. Furthermore, we envisaged the occurrence ofSNP in a heterozygous state in the target sequence as a basis ofenhancing the differential effect of miRNA on the mRNA fromthe homologs. The differential effect of miRNA on genes leadingto variation in risk for cancer is reported in many cases.11-14 Herewe created a paradigm of homologous 3'UTR with a single basechange in the miRNA target site, challenged with an identicalconcentration of miRNA using the 3'UTR of JAG1 andTGFBR2. The differential targeting, measured in terms of activityof luciferase clearly indicates preferential targeting of one of thealleles against the other. In case of JAG1, in the SNP (rs8708A.G) at 6th nucleotide from the +1 position of the miRNAbinding site on the target 3'UTR, G which is the minor allele issignificantly poorly targeted as compared with the major allele A.
Deletions and frame shift mutations in JAG1, coding for aligand for the NOTCH receptor is implicated in Alagillesyndrome (AGS) which shows IP-VE.17-19 Genetic modifiers havebeen proposed to influence the severity of AGS phenotypeindicating that the ligand-receptor interaction is sensitive to theconcentration of both the receptor and the ligand.21 Our resultsindicate that miRNA can bring about variation in JAG1 proteinlevel in the cells. Taking this into account, the possibleimplication of our observations on variable penetrance ofAlagille syndrome is shown in Figure 6. A heterozygous individualhaving the disease-causing JAG1 mutation (exonic mutation)linked to SNP rs8708A in the 3' UTR and the normal JAG1 allelelinked to rs8708G will show an absence or attenuation of thedisease phenotype (Fig. 6, Case I). Because, the mutant JAG1mRNA will be preferentially targeted by miRNA and repressed,the level of the normal protein will predominate. Thus JAG1mutation will not be penetrant in the background of the SNP onthe normal allele. However, if the normal allele has rs8708A andthe mutant allele has rs8708G, then mutant protein levels will behigher than that of the normal functional protein and Alagillesyndrome will manifest (Fig. 6, Case II). Similarly, the manifesta-tion of AAT3 (Aortic aneurysm familial thoracic type 3) will beinfluenced by the preferentially targeted rs11466532T in thenormal TGFBR2 homolog and rs11466532C in the mutantTGFBR2 in a heterozygous individual. Our results suggest thatboth translational inhibition as observed in JAG1 3'UTR andtranscript instability as in TGFBR2 can contribute to the endeffect of differential expression of the homologous alleles.
The comparison of constructs with identical 3'UTR (RlucA:FlucA and RlucG:FlucG) show that the phenotype of individualswith heterozygosity for disease-causing JAG1 mutation butidentical sequence at the miRNA target site at the 3'UTR ofthe homologous genes, would depend on the amount of wild typeJAG1 protein. Nevertheless it may be predicted that theseindividuals would have comparable phenotypes. It is significantthat the heterozygosity for rs8708A . G is 0.45 (www.ncbi.nlm.nih.gov/snp). Further, under HGDP (Human Genome Diversity
Figure 6. A model for the impact of SNP on penetrance of Alagille syndrome. Heterozygosity for disease causing mutation (shown as star) in JAG1 andour observation that rs8708A is targeted better than rs8708G by hsa-miR-214 is considered in proposing this model. JAG1 alleles, normal and mutant,their relevant mRNA and protein are shown respectively as green and pink colored symbols. The levels of protein products are shown as boxes withgraded colors. SNP in the miRNA target site in the 39UTR region is shown as a yellow bar. In Case I, mRNA from mutant allele linked to rs8708A will bepreferentially targeted and repressed, normal JAG1 protein will be higher in concentration and hence, no Alagille syndrome; in Case II, normal JAG1 allelelinked to rs8708A will be preferentially targeted and repressed, therefore Alagille syndrome will manifest.
www.landesbioscience.com RNA Biology 7

Project) a significantly different allele frequency across worldpopulations is observed for rs8708 of JAG1 (Fig. S3). Therefore,differential targeting of the normal and mutant allele and hence,penetrance can vary between populations. Presently no epide-miological data are available.
In the miRNA paradigm elucidated here the SNP at the targetsite on the normal allele will be critical in determining theexpressivity and the penetrance of the disease. Similarly, thecontrasting phenotype in a family segregating Erythropoieticprotoporphyria is known to be associated with inheritance of amutant FECH along with a normal allele containing an SNPleading to aberrant transcript. In absence of the SNP an individualwith identical mutation in FECH is unaffected.3,4
The effect of SNP at miRNA target site modulating post-transcriptional regulation is increasingly been reported.22,23 Suchcases could be examined for variations in penetrance of theassociated mutations. It is interesting to note that Tourette’ssyndrome is estimated to have variable penetrance in the range of28% to 50% in heterozygotes.24,25 Recently a rare variantmapping in the target site for hsa-miR-189 in the 3'UTR ofSLITRK1 has been shown in two independent cases of Tourette'ssyndrome along with modest but significant effect of the varianton regulation of luciferase activity by hsa-miR-189.22 It remains tobe investigated if the observed variable penetrance in Tourette’ssyndrome is related to the nature of the mutation. The differencein expression attributable to a SNP is modest in several cases.However, in an analysis of the effect of has-miR-10a, it isobserved that variation in the range of 0.5- to 1.7-fold inexpression of the direct target of has-miR-10a and its downstreamgenes is correlated with pathological changes in atherosclerosis.26
The occurrence of SNP in the miRNA target site within thegenome and in different populations has been analyzed.27,28
Saunders et al. observed higher variation in miRNA target sitethan in the miRNA coding regions and predicted that SNP inmiRNA target sites can confer phenotypic differences betweenindividuals.27 The lower frequency of polymorphism at themiRNA target site relative to rest of the genome argues forthe importance of sequence conservation of the target sites.28 Theallelic imbalance sequencing data from mice heterozygous forparental alleles from five inter-strain crosses led to the estimationthat hundreds of genes in the mammalian genome could differ intheir level of expression between individuals due to polymorphicmiRNA sites and significantly contribute to gene expressionvariation between individuals within the species.15 Theseobservations strongly support our paradigm of SNP at themiRNA target site as a contributor to variable penetrance ofdisease causing mutations. In other words allelic differences inexpression are accentuated by the occurrence of disease causingmutations in the background of miRNA target site SNP. Theestimates of allelic imbalance based on mRNA could be an under-estimate as miRNA can also bring about translational repression.15
In summary, we have shown that IP-VE genes JAG1andTGFBR2 are directly regulated by hsa-miR-214 and hsa-miR-34b*, respectively. The SNP in the miRNA binding site have thepotential to result in differential targeting of the 3'UTR of JAG1and TGFBR2, such that the alleles having A and T of JAG1 and
TGFBR2 respectively, are the preferred targets for hsa-miR-214and hsa-miR-34b*. This provides a novel paradigm formechanism of differential expression of homologous alleles whichresults in incomplete penetrance and variable expressivity.
Materials and Methods
Cell culture and transfection. HEK293 and A549 cells werepropagated in MEM, U87 in DMEM high glucose media (Gibco,Invitrogen USA), supplemented with 10% FBS, 1mM sodiumpyruvate, 2mM L-glutamine and 1X Antibiotic solutionPenicillin-Streptomycin Amphotericin B (Biological Industries(Kibbutz Beit Haemek, Israel) at 37°C in presence of 5% CO2.For reporter assays, 2.5x105 cells were transfected with 150ngpMIR-REPORTTM Luciferase or psiCHECKTM-2 construct and5–100nM specific miRIDIAN miRNA mimics/ miRIDIANmiRNA mimic negative Control #1 and specific miRIDIAN anti-miRNA / miRIDIAN miRNA Hairpin Inhibitor Negative Control#1 (Dharmacon, Thermo Scientific) using 2ul siPORTTM
NeoFXTM transfection agent (Ambion) per well of a 24-well plate.For western blot and qRT-PCR analysis, 5�105 cells weretransfected with 100nM miRNA mimics or inhibitors using 4ulsiPORTTM NeoFXTM per well of a 12-well plate.
All the primers used in the study are given in Table S1, S2,S3and S4.
Reporter constructs and assays. 3'UTR sequence(s) containingmiRNA binding sites were cloned downstream of Firefly luciferasegene in pMIR-REPORTTM Luciferase vector (Ambion/AppliedBiosystems) using HindIII and SpeI restriction sites.Computation used to evaluate the effect of miRNA mimic ontarget construct is represented by fold change calculated as a ratioof normalized Fluc luminescence with miRNA mimic andnormalized Fluc with control miRNA mimic, where normalizedFluc is Fluc of target 3'UTR /Rluc. Vector psiCHECKTM-2(Promega Corporation USA), which contains two different reporters(Renilla and Firefly) was used for cloning both, rs8708A.G ofJAG1 3' UTR as well as and rs11466532C.T ofTGFBR2 3'UTR.For cloning downstream of Rluc, XhoI and Not I restriction siteswere used while XbaI restriction site was used for cloningdownstream of Fluc gene. JAG1 rs8708-A and -G were cloned inthe following combinations, RlucA:FlucA, RlucG:FlucA, RlucA:FlucG and RlucG:FlucG. Similarly, TGFBR2 psiCHECKTM-2constructs were generated, RlucC:FlucC, RlucT:FlucC, RlucC:FlucT and RlucT:FlucT.
For cloning the 3'UTR fragments, overlapping oligonucleotidescorresponding to the miRNA target site along with 35–40 bpupstream and downstream of the target site and a suitablerestriction site were used. The oligonucleotides were annealed andextended to form double stranded fragments in reactionscontaining 50pmol each of the overlapping oligonucleotidesalong with 0.2mM dNTPs, 2.5U Taq DNA Polymerase and 1XTaq Polymerase buffer (New England Biolabs), and subjected to asingle cycle of denaturation (95°C, 5 mins), annealing (65°C,5 mins) and extension (72°C, 2 mins). The products of thisextension reaction were restriction digested and ligated into therestriction digested-CiP treated vector. The longer 3'UTR region
8 RNA Biology Volume 9 Issue 3

of target genes were PCR amplified from human genomic DNAusing appropriate primers and Taq DNA Polymerase followingcycling conditions of, initial denaturation at 95°C for 10minfollowed by 35 cycles of, denaturation at 95°C for 40 sec,annealing at 60°C for 40 sec, extension at 72°C for 40 sec, andfinal extension at 72°C for 5min. The PCR products werepurified, digested and cloned into pMIR-REPORTTM vector. Allthe clones were confirmed by DNA sequencing.
Cells were transfected in 24-well plates and lysed 48 h post-transfection using 100ul, 1X Passive Lysis Buffer (PromegaCorporation) according to manufacturer’s protocol. The Fireflyand Renilla luminescence was measured using Dual-LuciferaseReporter (DLRTM) Assay System (Promega Corporation) accord-ing to manufacturer’s protocol and read on the Orion IImicroplate luminometer (Berthold detection systems).
RNA isolation and quantitative RT-PCR. Cells transfectedwith reporter constructs and/or miRNA mimics or inhibitors werelysed with TRIzol1 Reagent (Invitrogen) and total RNA preparedaccording to manufacturer’s instructions. 1–2ug total RNAwastreated with RNase free-DNaseand reverse transcribed tosynthesize cDNA using SuperScriptTM II Reverse Transcriptase(Invitrogen) according to manufacturer’s protocol. QuantitativePCR for detection of JAG1 (Jagged1), TGFBR2 (Transforminggrowth factor-β receptor type II) and GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) was performed using 2.5 pmol each offorward and reverse primer along with 1ul cDNA and 1X SYBRGreen PCR master mix (Roche Applied Science) with the cyclingconditions: 95°C for 10 min, followed by 40 cycles of, 95°C for10 sec, 60°C for 10 sec, 72°C for 10 sec; and 72°C for 5 min, onLightCycler 480 machine (Roche Applied Science). Relative gene
expression of JAG1 or TGFBR2 with and without miRNA mimicor inhibitor was calculated using 2-DDCt method.
Western Blotting. Cells were harvested, washed with 1X PBSand lysed in RIPA buffer (150 mMNaCl, 1% NP40, 0.5%sodium deoxycholate, 0.1% SDS, 50 mMTRIS-HCl at pH 8,2 mM EDTA, 1 mM DTT) and 1X, Protease Inhibitor cocktail(Roche Applied Science). Protein concentration was measuredusing BCA (bicinchoninic acid) method. Western blot wasperformed using 30 or 60ug total protein per lane. Anti-Jagged1(#2620, 1:500 dilution), anti-GAPDH (#2118, 1:2000), anti-rabbit (#7054, 1:1000) (Cell Signaling technologies, USA) andanti-TGFBRII (ab53168, 1:500; Abcam) were used and signaldeveloped using BCIP (5-Bromo-4-chloro-3-indolyl phosphate,Sigma Chemical Co. St.Louis) and NBT (Nitro BlueTetrazolium, Sigma) in Alkaline Phosphatase Buffer (100mMTRIS-HCl, 100mM NaCl, 5mM MgCl2, 0.05% Tween 20,pH 9.5).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Acknowledgments
We thank Council of Scientific and Industrial Research (CSIR)India, NWP0036 grant for funding the project, Indian Council ofMedical Research (ICMR) for fellowship to JA and Beena Pillaifor critical reading of the manuscript and help with experiments.
Supplemental Materials
Supplemental materials can be found at:www.landesbioscience.com/journals/rnabiology/article/19318
References1. Van Heyningen V, Yeyati PL. Mechanisms of non-
Mendelian inheritance in genetic disease. Hum MolGenet 2004; 13(Spec No 2):R225-33; PMID:15358729;http://dx.doi.org/10.1093/hmg/ddh254
2. Harder A, Titze S, Herbst L, Harder T, Guse K,Tinschert S, et al. Monozygotic twins with neurofibro-matosis type 1 (NF1) display differences in methylationof NF1 gene promoter elements, 5' untranslated region,exon and intron 1. Twin Res Hum Genet 2010; 13:582-94; PMID:21142935; http://dx.doi.org/10.1375/twin.13.6.582
3. Di Pierro E, Brancaleoni V, Moriondo V, Besana V,Cappellini MD. Co-existence of two functional muta-tions on the same allele of the human ferrochelatasegene in erythropoietic protoporphyria. Clin Genet 2007;71:84-8; PMID:17204051; http://dx.doi.org/10.1111/j.1399-0004.2007.00733.x
4. Gouya L, Puy H, Robreau AM, Bourgeois M, LamorilJ, Da Silva V, et al. The penetrance of dominanterythropoietic protoporphyria is modulated by expres-sion of wildtype FECH. Nat Genet 2002; 30:27-8;PMID:11753383; http://dx.doi.org/10.1038/ng809
5. Poelzing S, Forleo C, Samodell M, Dudash L,Sorrentino S, Anaclerio M, et al. SCN5A poly-morphism restores trafficking of a Brugada syndromemutation on a separate gene. Circulation 2006; 114:368-76; PMID:16864729; http://dx.doi.org/10.1161/CIRCULATIONAHA.105.601294
6. Rivolta C, McGee TL, Rio Frio T, Jensen RV, BersonEL, Dryja TP. Variation in retinitis pigmentosa-11(PRPF31 or RP11) gene expression between sympto-matic and asymptomatic patients with dominant RP11mutations. Hum Mutat 2006; 27:644-53; PMID:16708387; http://dx.doi.org/10.1002/humu.20325
7. Vithana EN, Abu-Safieh L, Pelosini L, Winchester E,Hornan D, Bird AC, et al. Expression of PRPF31 mRNAin patients with autosomal dominant retinitis pigmen-tosa: a molecular clue for incomplete penetrance? InvestOphthalmol Vis Sci 2003; 44:4204-9; PMID:14507862;http://dx.doi.org/10.1167/iovs.03-0253
8. Zakharova IS, Shevchenko AI, Zakian SM. Monoallelicgene expression in mammals. Chromosoma 2009; 118:279-90; PMID:19242715; http://dx.doi.org/10.1007/s00412-009-0206-8
9. Serre D, Gurd S, Ge B, Sladek R, Sinnett D, Harmsen E,et al. Differential allelic expression in the human genome:a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoSGenet 2008; 4:e1000006; PMID:18454203; http://dx.doi.org/10.1371/journal.pgen.1000006
10. Ahluwalia JK, Hariharan M, Bargaje R, Pillai B,Brahmachari V. Incomplete penetrance and variableexpressivity: is there a microRNA connection? Bioessays2009; 31:981-92; PMID:19642110; http://dx.doi.org/10.1002/bies.200900066
11. Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I,et al. A SNP in a let-7 microRNA complementary sitein the KRAS 3' untranslated region increases non-smallcell lung cancer risk. Cancer Res 2008; 68:8535-40;PMID:18922928; http://dx.doi.org/10.1158/0008-5472.CAN-08-2129
12. Xiong F, Wu C, Chang J, Yu D, Xu B, Yuan P, et al.Genetic variation in an miRNA-1827 binding site inMYCL1 alters susceptibility to small-cell lung cancer.Cancer Res 2011; 71:5175-81; PMID:21676885;http://dx.doi.org/10.1158/0008-5472.CAN-10-4407
13. NicolosoMS, Sun H, Spizzo R, KimH,WickramasingheP, Shimizu M, et al. Single-nucleotide polymorphismsinside microRNA target sites influence tumor suscepti-bility. Cancer Res 2010; 70:2789-98; PMID:20332227;http://dx.doi.org/10.1158/0008-5472.CAN-09-3541
14. Saetrom P, Biesinger J, Li SM, Smith D, Thomas LF,Majzoub K, et al. A risk variant in an miR-125bbinding site in BMPR1B is associated with breastcancer pathogenesis. Cancer Res 2009; 69:7459-65;PMID:19738052; http://dx.doi.org/10.1158/0008-5472.CAN-09-1201
15. Kim J, Bartel DP. Allelic imbalance sequencing revealsthat single-nucleotide polymorphisms frequently altermicroRNA-directed repression. Nat Biotechnol 2009;27:472-7; PMID:19396161; http://dx.doi.org/10.1038/nbt.1540
www.landesbioscience.com RNA Biology 9

16. Hariharan M, Scaria V, Brahmachari SK. dbSMR: anovel resource of genome-wide SNPs affectingmicroRNA mediated regulation. BMC Bioinformatics2009; 10:108; PMID:19371411; http://dx.doi.org/10.1186/1471-2105-10-108
17. Spinner NB, Colliton RP, Crosnier C, Krantz ID,Hadchouel M,Meunier-Rotival M. Jagged1 mutations inalagille syndrome. Hum Mutat 2001; 17:18-33; PMID:11139239; http://dx.doi.org/10.1002/1098-1004(2001)17:1,18::AID-HUMU3.3.0.CO;2-T
18. Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome.J Med Genet 1997; 34:152-7; PMID:9039994; http://dx.doi.org/10.1136/jmg.34.2.152
19. Elmslie FV, Vivian AJ, Gardiner H, Hall C, Mowat AP,Winter RM. Alagille syndrome: family studies. J MedGenet 1995; 32:264-8; PMID:7643353; http://dx.doi.org/10.1136/jmg.32.4.264
20. Milewicz DM, Chen H, Park ES, Petty EM, Zaghi H,Shashidhar G, et al. Reduced penetrance and variableexpressivity of familial thoracic aortic aneurysms/dissec-tions. Am J Cardiol 1998; 82:474-9; PMID:9723636;http://dx.doi.org/10.1016/S0002-9149(98)00364-6
21. McCright B, Lozier J, Gridley T. A mouse model ofAlagille syndrome: Notch2 as a genetic modifier of Jag1haploinsufficiency. Development 2002; 129:1075-82;PMID:11861489
22. Abelson JF, Kwan KY, O’Roak BJ, Baek DY, StillmanAA, Morgan TM, et al. Sequence variants in SLITRK1are associated with Tourette’s syndrome. Science 2005;310:317-20; PMID:16224024; http://dx.doi.org/10.1126/science.1116502
23. Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, BibéB, et al. A mutation creating a potential illegitimatemicroRNA target site in the myostatin gene affectsmuscularity in sheep. Nat Genet 2006; 38:813-8; PMID:16751773; http://dx.doi.org/10.1038/ng1810
24. Hasstedt SJ, Leppert M, Filloux F, van de Wetering BJ,McMahon WM. Intermediate inheritance of Tourettesyndrome, assuming assortative mating. Am J HumGenet 1995; 57:682-9; PMID:7668298
25. Comings DE, Comings BG, Devor EJ, Cloninger CR.Detection of major gene for Gilles de la Tourettesyndrome. Am J Hum Genet 1984; 36:586-600; PMID:6587774
26. Fang Y, Shi C, Manduchi E, Civelek M, DaviesPF. MicroRNA-10a regulation of proinflammatoryphenotype in athero-susceptible endothelium in vivoand in vitro. Proc Natl Acad Sci U S A 2010; 107:13450-5; PMID:20624982; http://dx.doi.org/10.1073/pnas.1002120107
27. Saunders MA, Liang H, Li WH. Human polymorphismat microRNAs and microRNA target sites. Proc NatlAcad Sci U S A 2007; 104:3300-5; PMID:17360642;http://dx.doi.org/10.1073/pnas.0611347104
28. Chen K, Rajewsky N. Natural selection on humanmicroRNA binding sites inferred from SNP data. NatGenet 2006; 38:1452-6; PMID:17072316; http://dx.doi.org/10.1038/ng1910
10 RNA Biology Volume 9 Issue 3