micromolar l-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of...
TRANSCRIPT

Neuropharmacology 37 (1998) 1419–1429
Micromolar L-glutamate induces extensive apoptosis in anapoptotic-necrotic continuum of insult-dependent, excitotoxic
injury in cultured cortical neurones
Nam S. Cheung, Catherine J. Pascoe, Sarah F. Giardina, Christopher A. John,Philip M. Beart *
Department of Pharmacology, Monash Uni6ersity, Clayton, 3168, Victoria, Australia
Accepted 24 July 1998
Abstract
Excitotoxicity induced by L-glutamate (Glu), when examined in a pure neuronal cortical culture, involved widespread apoptosisat concentrations of 1–10 mM as part of a continuum of injury, which at its most servere was purely necrotic. Cells, maintainedin chemically defined neurobasal/B27 medium, were exposed at d7 for 2 h to Glu (1–500 mM), and cellular injury was analysed2 and 24 h after insult using morphology (phase-contrast microscopy), a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) viability assay, nuclear staining with 4,6-diamidino-2-phenylindole (DAPI), terminal transferase-mediated dUTPnick end-labelling (TUNEL) and DNA fragmentation by gel electrophoresis. Glu-mediated neurotoxicity was prevented byMK-801 (5 mM), whilst CNQX (20 mM) attenuated injury by 20%. Exposure to intensive insults (100 and 500 mM Glu) inducednecrosis characterized by rapid cell swelling (B2 h) and lack of chromatin condensation, confirmed by DAPI nuclear staining.In contrast, mild insults (B20 mM Glu) failed to produce acute neuronal swelling at B2 h, but 24 h after injury resulted in alarge number of apoptotic nuclei as confirmed by TUNEL and electrophoretic evidence of DNA fragmentation, which wasattenuated by cycloheximide (0.1 mg/ml). Our findings indicate for the first time that physiological concentrations of Glu produceneuronal injury across a continuum involving apoptosis (B20 mM) and increasingly necrosis(\20 mM), dependent on the severityof the initial insult. © 1998 Elsevier Science Ltd. All rights reserved.
Keywords: Apoptosis; Necrosis; Glutamate; Cortical neurones; DNA fragmentation; TUNEL
1. Introduction
L-glutamate (Glu) is a major excitatory transmitter ofthe mammalian central nervous system and under nor-mal circumstances plays a key role in neurologicalprocesses including cognition, learning and memory(Collingridge and Lester, 1989). The family of gluta-mate receptors is categorized into two subgroups:ionotropic and metabotropic. Ionotropic glutamate re-ceptors (iGluRs) are ligand-gated ion channels and aresubdivided into three subtypes by their selective ago-nists: N-methyl-D-aspartate (NMDA), kainate and a-amino-3-hydroxy-5-methylisoxazole-4-propionate(AMPA) (Hollmann and Heinemann, 1994). Over stim-
ulation of iGluRs produces excitotoxic neuronal death,which has been suggested to be involved in severalneurological disorders including ischemic-hypoxic in-jury (Choi and Rothman, 1990; Meldrum and Garth-waite, 1990), epileptic seizures (Olney et al., 1983) andchronic neurodegenerative disorders including amy-otrophic lateral sclerosis, Huntington’s disease andAlzheimer’s disease (Lipton and Rosenberg, 1994; Si-monian et al., 1996).
Overactivation of iGluRs also causes toxicity in neu-ronal cell culture (Choi, 1992), however, the mode ofcell death after injury induced by excitatory aminoacids is still controversial (Bonfoco et al., 1995; Finielset al., 1995; Choi, 1996; Nicotera et al., 1997). Severallines of evidence suggest that cell death occurs by oneof two general pathways, necrosis or apoptosis. Somegroups have reported features of necrosis (Choi et al.,
* Corresponding author. Tel.: +61 3 99053817; fax: +61 399055851; e-mail: [email protected].
0028-3908/98/$ - see front matter © 1998 Elsevier Science Ltd. All rights reserved.
PII: S0028-3908(98)00123-3
N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–14291420
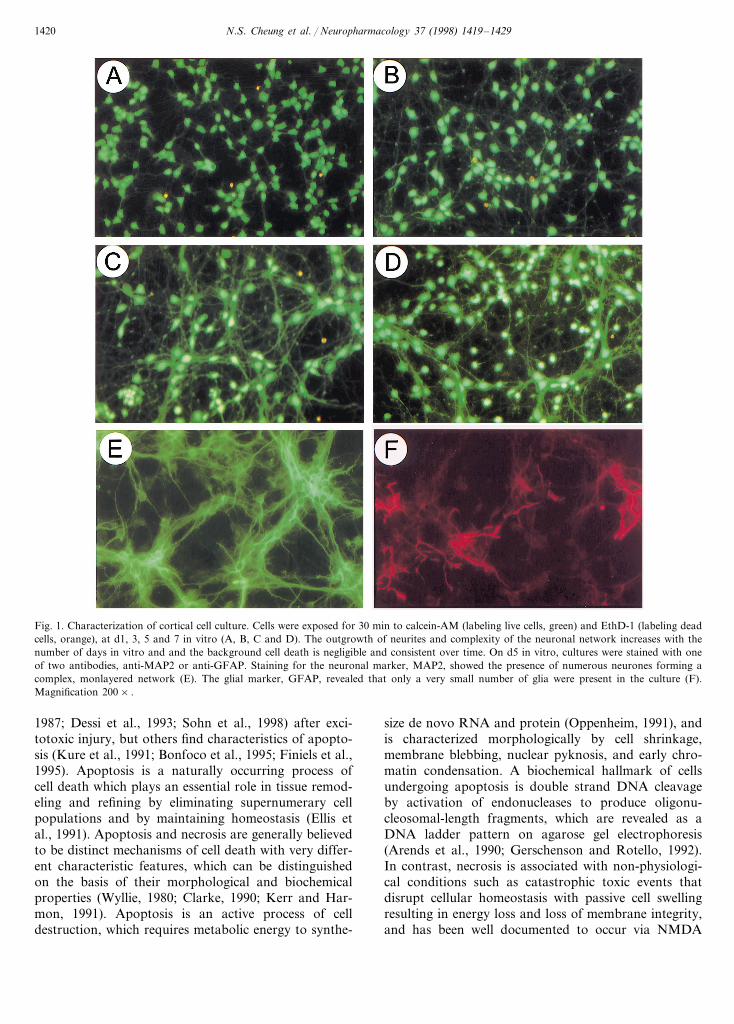
Fig. 1. Characterization of cortical cell culture. Cells were exposed for 30 min to calcein-AM (labeling live cells, green) and EthD-1 (labeling deadcells, orange), at d1, 3, 5 and 7 in vitro (A, B, C and D). The outgrowth of neurites and complexity of the neuronal network increases with thenumber of days in vitro and and the background cell death is negligible and consistent over time. On d5 in vitro, cultures were stained with oneof two antibodies, anti-MAP2 or anti-GFAP. Staining for the neuronal marker, MAP2, showed the presence of numerous neurones forming acomplex, monlayered network (E). The glial marker, GFAP, revealed that only a very small number of glia were present in the culture (F).Magnification 200× .
1987; Dessi et al., 1993; Sohn et al., 1998) after exci-totoxic injury, but others find characteristics of apopto-sis (Kure et al., 1991; Bonfoco et al., 1995; Finiels et al.,1995). Apoptosis is a naturally occurring process ofcell death which plays an essential role in tissue remod-eling and refining by eliminating supernumerary cellpopulations and by maintaining homeostasis (Ellis etal., 1991). Apoptosis and necrosis are generally believedto be distinct mechanisms of cell death with very differ-ent characteristic features, which can be distinguishedon the basis of their morphological and biochemicalproperties (Wyllie, 1980; Clarke, 1990; Kerr and Har-mon, 1991). Apoptosis is an active process of celldestruction, which requires metabolic energy to synthe-
size de novo RNA and protein (Oppenheim, 1991), andis characterized morphologically by cell shrinkage,membrane blebbing, nuclear pyknosis, and early chro-matin condensation. A biochemical hallmark of cellsundergoing apoptosis is double strand DNA cleavageby activation of endonucleases to produce oligonu-cleosomal-length fragments, which are revealed as aDNA ladder pattern on agarose gel electrophoresis(Arends et al., 1990; Gerschenson and Rotello, 1992).In contrast, necrosis is associated with non-physiologi-cal conditions such as catastrophic toxic events thatdisrupt cellular homeostasis with passive cell swellingresulting in energy loss and loss of membrane integrity,and has been well documented to occur via NMDA

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–1429 1421
Fig. 2. Morphological analysis of Glu-mediated neurotoxicity in cultured cortical neurones. On d7 in vitro, cultured cells were exposed to Glu:Control (A and B), 10 mM (C and D), 20 mM (E and F), 100 mM (G and H), 500 mM (I and J) for 2 h. Phase-contrast microscopy showed intenseinsults (100 and 500 mM Glu; G and I) induced rapid cell swelling after 2 h exposure compared with control cultures (A). Almost all neuroneshad been lysed and were detached from the plate 24 h after stimulation (H and J). In contrast, mild insults (10 and 20 mM Glu; C and E) producedlittle neuronal swelling and 24 h after exposure revealed shrinkage of neuronal cell bodies and membrane blebbing (D and F). Magnification200× .

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–14291422
Fig. 3. Fluorescent microscopy images of DAPI-stained necrotic nuclei. At 2 h after Glu exposure, (A) Control; (B) 20 mM; (C) 100 mM; (D) 500mM, cells were stained with DAPI (1 mg/ml) for 10 min and apoptotic nuclei were not observed in neurones exposed to Glu (B, C, D). Instead,exposure to 100 and 500 mM Glu induced necrosis and revealed normal labeling of nuclei due to the lysed membrane. Magnification 200× .
receptors (Choi and Rothman, 1990). These changesare accompanied by random nuclear breakdown andthe fragmented DNA appears as a diffuse smear pat-tern on agarose gel electrophoresis (Wyllie, 1980; Kerrand Harmon, 1991).
How Glu itself produces neuronal injury is relevantto physiological and pathological cell death in vivo.Historically, there is evidence that Glu appears to pro-duce rapid neuronal death, involving cellular swellingand lysis, and that is presumed to be via necrosis.However, even in older reports there are suggestions ofexcitotoxic death not involving necrosis (Schwob et al.,1980; Hajos et al., 1986), whilst one more recent studyfound only apoptosis (Kure et al., 1991). Recently,based on studies employing the iGluR agonistsNMDA, AMPA and KA (Bonfoco et al., 1995; Simo-nian et al., 1996; Larm et al., 1997), excitotoxic injuryhas been suggested to occur across a complex contin-uum, involving apoptosis and necrosis, and which isdependent on the intensity of the insult. However, sucha pattern of injury has not been demonstrated for Gluitself at concentrations of physiological relevance, al-though earlier data are suggestive of Glu-mediatedneurotoxicity being very dependent on the experimentalconditions (Rosenberg and Aizenman, 1989; Nicoteraet al., 1997). Precise details of the death mechanismsare difficult to dissect in vivo because of the inherentcomplexity of the synaptic milieu and thus studies withprimary neuronal cultures have considerable experi-mental advantages, especially in terms of drug deliveryand their convenience for pathological assessment. Inthe present study, we have combined morphological
and biochemical techniques to examine whether toxicityinduced by Glu involves apoptosis or necrosis in pureneuronal cortical cultures, and to better understand thenature of excitotoxic cell death. Our data demonstratefor the first time that Glu produces a physiologicallyrelevant pattern of excitotoxicity that occurs across aninsult-dependent continuum, i.e. necrosis predominatesafter intense insults with Glu, whereas mild insultsresult in widespread apoptotic neuronal death.
2. Materials and methods
2.1. Cell culture
Primary cultures of cerebrocortical neurones wereobtained from embryonic d15 or 16 Swiss-white miceusing previously described procedures (Larm et al.,1996) with modifications. In brief, cortices were dis-sected from the brains under sterile condition and weredigested in 0.2% trypsin and 0.02% deoxyribonucleasein Hank’s balanced salt solution (HBSS) for 5 min at37°C. The digestion was terminated by the addition ofHBSS containing 0.1% soybean trypsin inhibitor andcentrifugation for 5 min at 1000×g. The resultanttissue pellet was resupended in HBSS and mechanicallydissociated by trituration (20 strokes with a 24-gaugeneedle). The cells were spun down for 5 min at 1000×gand resuspended in Neurobasal medium™ containingB27 (Brewer et al., 1993), 100 U/ml penicillin, 100mg/ml streptomycin, 0.5 mM L-glutamine and 10% dia-lyzed foetal calf serum (FCS). Cells were seeded in

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–1429 1423
Fig. 4. Glu receptor-mediated neurotoxicity in cultured cortical neurones was determined using the MTT cell viability assay. At d7 cells wereexposed for 2 h to Glu (3–500 mM) and cell injury was found to be concentration-dependent whether assessed 2 h () or 24 h (�) after insult(A). Glu-mediated neurotoxicity was fully prevented by MK-801 (5 mM; ), whilst CNQX (20 mM; �) only attenuated injury by 20% (B). Datawere standardized relative to untreated (100% cell viability) and 500 mM H2O2 treated cells (100% cell death), and are plotted as mean9S.E.M.,and were routinely from three to six replicate cultures. Further details are give in the text.
24-well plates previously coated with poly-D-lysine (50mg/ml) to a density of 1.6×105 cells/cm2. After 24 h invitro, the culture medium was replaced with serum-freegrowth medium. The cultures were maintained in ahumidified CO2 incubator (5% CO2, 8.5% O2; 37°C).
2.2. Cell 6iability assay
On d1, 3, 5 and 7 in vitro, live and dead cells weredetermined by live/dead viability/cytotoxicity proce-dure. In brief, culture medium was replaced by Dulbec-co’s phosphate buffered saline (2.7 mM KCl, 1.5 mMKH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, pH 7.4)and incubated at 37°C for 30 min with calcein acetoxymethyl ester (calcein-AM, 2 mM) and ethidium homod-imer-1 (EthD-1, 2 mM). Calcein-AM is cleaved by theintracellular esterases of living cells to a fluorescentproduct (green), while EthD-1 only penetrates themembranes of dead cells and binds to nucleic acid toproduce a bright red fluorescense. Photomicrographswere then taken on Kodak Ektachrome P1600 filmusing fluorescent microscopy (Olympus, IMT-2).
2.3. Immunocytochemistry
On d5 in vitro, cells were fixed for 20 min in 4%paraformaldehyde in 0.1 M phosphate buffer (pH 7.4)at room temperature (RT). Cells were then washedthree times with Tris buffered saline (TBS; 50 mMTris–HCl, 0.9% NaCl, pH 7.6) and quenched in 3%w/w H2O2 for 5 min at RT. After three washes in TBSthe cells were then blocked for 1 h at 4°C with 10%normal goat serum (NGS) and 0.1% Triton X-100(TX-100) in TBS. After washing twice with TBS, the
cells were incubated overnight at 4°C with the primaryantibody, either mouse anti-microtubule associatedprotein-2 (MAP2; 1:250 dilution) or rabbit anti-glialfibrillary acidic protein (GFAP; 1:3 dilution), each insolution with 2% NGS and 0.1% TX-100 in TBS. Thecells were then washed three times with TBS and incu-bated for 3 h at 25°C with the secondary antibody(either sheep antimouse IgG conjugated fluorescein-isothiocyanate (against MAP2; 1:100 dilution) or sheepantirabbit IgG conjugated tetramethylrhodamine(against GFAP; 1:100 dilution)), in solution with 2%NGS and 0.1% TX-100 in TBS. Cells were then washedthree times with TBS, the buffer was thoroughly aspi-rated and the cells were sealed with antifade solution(1,4-diazabicyclo[2.2.2]octane) under coverslips and ex-amined under fluorescent microscopy.
2.4. Glutamate-mediated excitotoxicity
At d7 cultures were exposed for 2 h to Glu (3–500mM), and then cells were returned to an antioxidantfree N-2 medium (Neurobasal medium™, 100 U/mlpenicillin, 100 mg/ml streptomycin, 0.25% BSA, 83 mMD(+ )galactose, 16 mM ethanolamine, 6 mM L-carnitine,0.4 mM biotin, and N-2 supplement (Bottenstein andSato, 1979)). Neuronal cell injury was assessed afterinsult (2 and 24 h) using morphology (phase-contrastand fluorescence microscopy) and a 3-(4,5-dimethylthi-azol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as-say (Larm et al., 1996). All drugs and controltreatments were prepared in N-2 medium. MK-801 (5mM), 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX; 20mM) or cycloheximide (CHX; 0.1 mg/ml) were added 1h before Glu exposure.

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–14291424
Fig. 5. Glu-mediated DNA fragmentation as shown by agarose gel electrophoresis. DNA was isolated from control (lane 2) and 24 h Glu-treatedcultured neurones in the presence or absence of CHX (0.1 mg/ml), 3 mM (lane 3), 10 mM (lane 4), 20 mM (lane 5), 30 mM (lane 6), 100 mM (lane7), 500 mM (lane 8), 3 mM+CHX (lane 9), 10 mM+CHX (lane 10), 20 mM+CHX (lane 11), 30 mM+CHX (lane 12), 100 mM+CHX (lane 13),(lane 1) (EcoRI/HindIII) standard DNA and run on 1.5% agarose gel for 4 h at 80 V. Glu induced concentration-dependent, internucleosomalgenomic DNA fragmentation (‘laddering’) not seen in the control lane, whereas, CHX attenuated the fragmentation in a concentration-dependentmanner. Note higher concentration of Glu (30 and 100 mM) produced little laddering because injury occurred by necrosis.
2.5. Fluorescent DNA staining of necrotic nuclei
After 2 h Glu exposure cells, were stained with4,6-diamidino-2-phenylindole (DAPI, 1 mg/ml) for 10min and visualized by fluorescence microscopy.
2.6. Analysis of DNA fragmentation by electrophoresis
Genomic DNA was isolated from control and 24 hGlu (3–500 mM) treated cultured neurones in the pres-ence or absence of CHX (0.1 mg/ml) as described previ-ously (Larm et al., 1997). Equal amounts of purifiedDNA samples (2 mg) were subjected to electrophoresison a 1.5% agarose gel and then stained with ethidiumbromide. DNA was visualized by ultraviolet transillu-mination and photographed.
2.7. In situ labeling of nick-end DNA
Cortical cell cultures were stained by the TUNELtechnique (TdT-mediated dUTP-digoxigenin nick end-labeling) by a modification of a previously describedprocedure (Larm et al., 1997). Briefly, 24 h after expo-sure to Glu (1–10 mM), cultures were fixed in 4%paraformaldehyde/phosphate buffer saline (pH 7.4) for1 h. Cells were then permeabilized with 0.1% TX-100 inphosphate-buffered saline (30 min) and processed asdescribed (Larm et al., 1997). Cells were visualizedunder bright field microscopy.
2.8. Data analysis
All data were expressed as mean9S.E.M. Data fromMTT assays were standardized relative to control (notreatment, 0% cell death) and 100% cell death (500 mMH2O2). Concentration–response curves were generatedby non-linear regression (variable slope) using theGraphPad PRISM™ computer program. Cellular in-jury data were normalized by log transformation priorto two-way analysis of variance (ANOVA) with a post-hoc Newman–Keul’s test.
2.9. Materials
Cell culture plates were obtained from Nunc™ (Den-mark). Dialyzed FCS, 1,4-diazabicyclo[2.2.2]octane andcycloheximide were purchased from Sigma (Sydney,Australia). L-glutamine, B27, Neurobasal medium™,N-2 supplement, penicillin/streptomycin and HBSSwere bought from GibcoBRL (Melbourne, Australia).Calcein-AM and EthD-1 were obtained from Molecu-lar Probes (Eugene, Oregon). Antibodies for MAP2and anti-digoxigenin-alkaline phosphatase were pur-chased from Boehringer Mannheim (NSW, Australia),and GFAP was obtained from INCSTAR Corporation(Stillwater, USA). MK-801 was a gift from MerckSharp and Dohme (Harlow, UK). CNQX was obtainedfrom Tocris Cookson (Bristol, UK).
N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–1429 1425
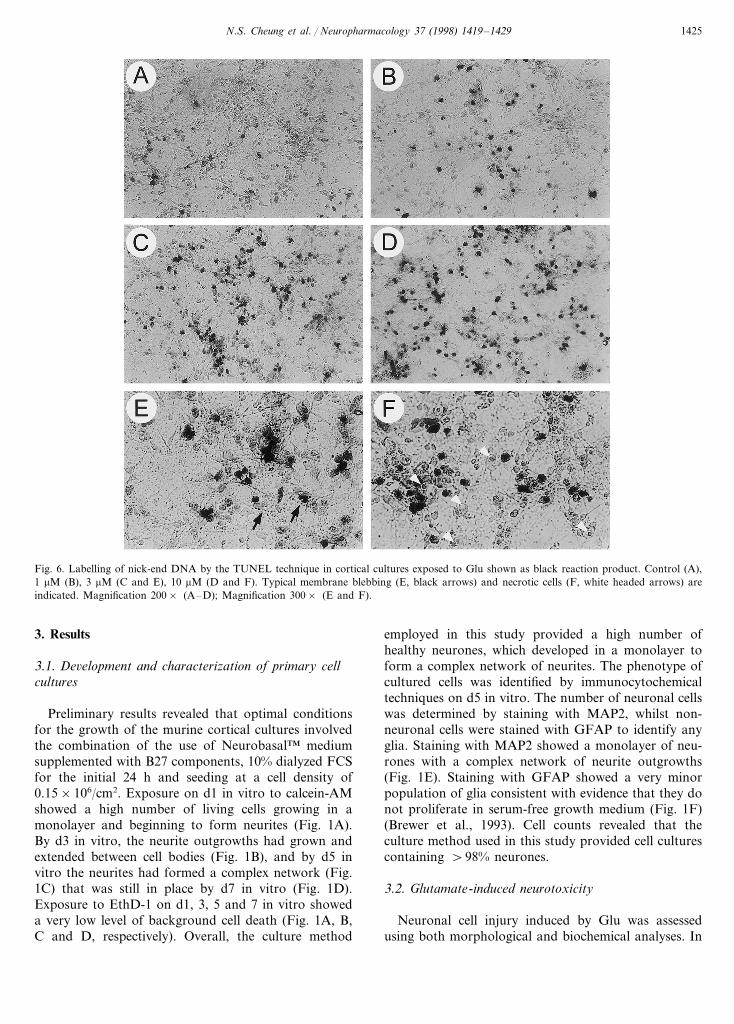
Fig. 6. Labelling of nick-end DNA by the TUNEL technique in cortical cultures exposed to Glu shown as black reaction product. Control (A),1 mM (B), 3 mM (C and E), 10 mM (D and F). Typical membrane blebbing (E, black arrows) and necrotic cells (F, white headed arrows) areindicated. Magnification 200× (A–D); Magnification 300× (E and F).
3. Results
3.1. De6elopment and characterization of primary cellcultures
Preliminary results revealed that optimal conditionsfor the growth of the murine cortical cultures involvedthe combination of the use of Neurobasal™ mediumsupplemented with B27 components, 10% dialyzed FCSfor the initial 24 h and seeding at a cell density of0.15×106/cm2. Exposure on d1 in vitro to calcein-AMshowed a high number of living cells growing in amonolayer and beginning to form neurites (Fig. 1A).By d3 in vitro, the neurite outgrowths had grown andextended between cell bodies (Fig. 1B), and by d5 invitro the neurites had formed a complex network (Fig.1C) that was still in place by d7 in vitro (Fig. 1D).Exposure to EthD-1 on d1, 3, 5 and 7 in vitro showeda very low level of background cell death (Fig. 1A, B,C and D, respectively). Overall, the culture method
employed in this study provided a high number ofhealthy neurones, which developed in a monolayer toform a complex network of neurites. The phenotype ofcultured cells was identified by immunocytochemicaltechniques on d5 in vitro. The number of neuronal cellswas determined by staining with MAP2, whilst non-neuronal cells were stained with GFAP to identify anyglia. Staining with MAP2 showed a monolayer of neu-rones with a complex network of neurite outgrowths(Fig. 1E). Staining with GFAP showed a very minorpopulation of glia consistent with evidence that they donot proliferate in serum-free growth medium (Fig. 1F)(Brewer et al., 1993). Cell counts revealed that theculture method used in this study provided cell culturescontaining \98% neurones.
3.2. Glutamate-induced neurotoxicity
Neuronal cell injury induced by Glu was assessedusing both morphological and biochemical analyses. In

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–14291426
preliminary experiments we evaluated different Glu ex-posure times (5–120 min) and the results showed thatshorter times of exposure (B120 min) induced necroticcell death only at high concentrations of Glu (100–500mM). So in subsequent experiments, 2 h was chosen asthe initial insult. Cortical cultures exposed to intenseexcitotoxic insult (100 and 500 mM Glu) in antioxidant-free N-2 medium exhibited substantial signs of neuro-toxicity after a 2 h initial exposure. Within 2 h ofintense insults, almost all neurones were swollen (Fig.2G, I) relative to control (Fig. 2A), and none of theneurones appeared to possess apoptotic characteristics(Fig. 3C, D). As shown in Fig. 3, DAPI nuclear stain-ing, observed by fluorescent microscopy 2 h after initialinsults (100 and 500 mM) showed normal nuclear label-ing as very diffuse large intact structures due to thelysed membrane (i.e. necrosis), rather than bright clus-tered bodies which is a typical feature of chromatincondensation (i.e. apoptosis). The number of necroticcells labeled by DAPI appeared to increase in a concen-tration-dependent manner after Glu exposure (Fig. 3C,D) relative to control (Fig. 3A). At 24 h after stimula-tion (100 and 500 mM Glu) the neurite network wasextensively degenerated and almost all cells had beenlysed and were detached from the plate (Fig. 2H, J). Incontrast, mild insults (10 and 20 mM Glu) did notinduce rapid cell swelling (Fig. 2C, E) and 24 h afterexposure revealed typical signs of neuronal apoptosis,such as shrinkage of neuronal cell body, membraneblebbing (Fig. 2D, F) and condensation of nuclearchromatin as indicated by staining with propidiumiodide (data not shown).
Neurotoxicity was assessed using a MTT assay, abiochemical index of cellular viability (Mosmann,1983), which showed that injury was concentration-de-pendent whether assessed 2 or 24 h after insult. Intenseinsults (50–500 mM) reduced neuronal viability to 60%within 2 h compared to control cultures, however, mildinsults (B20 mM) did not affect \30% of the neuronsuntil 24 h after stimulation (Fig. 4A). The EC50 valuesfor Glu at 2 and 24 h exposures were 2491.1 (n=18cultures) and 2193.6 mM (n=18 cultures), respec-tively. Analyses of neuronal viability (Fig. 4A) revealeda significant effects of Glu concentration (F [5, 35]=226, PB0.0001), time (F [1, 35]=85.5, PB0.0001) anda significant time x concentration interaction(F [5, 35]=5.3, PB0.002). The pharmacological profileof total injury was assessed 24 h after insult by evaluat-ing the ability of iGluR antagonists to block Glu-medi-ated cell injury. Glu-mediated neurotoxicity was fullyprevented by MK-801 (5 mM), a non-competitive antag-onist of the NMDA receptor (Wong et al., 1986), whilstthe non-NMDA antagonist CNQX (20 mM) only atten-uated injury by approximately 20%. Both antagonistsexerted significant effects (F [2, 53]=519, PB0.0001),with the neuroprotection provided by MK-801 being
different from that observed with CNQX (PB0.05;Fig. 4B).
3.3. Glutamate-induced DNA fragmentation
To characterize the mode of cell death induced byGlu, total DNA was extracted from the Glu-treatedcortical cultures and examined by gel electrophoresis.DNA fragmentation, a specific biochemical marker inapoptosis (Gerschenson and Rotello, 1992), was ob-served. Exposure of cortical cultures to 3–100 mM Gluinduced DNA laddering (Fig. 5, lanes 3–7) after 24 hstimulation compared to control cultures (Fig. 5, lane2). In contrast, this pattern of DNA fragmentation wasnot seen with 500 mM Glu alone because death was bynecrosis (Fig. 5, lane 8). CHX (0.1 mg/ml) was used todetermine whether Glu-mediated apoptosis was depen-dent on de novo protein synthesis for its mode of actionand attenuated the pattern of Glu-mediated DNA frag-mentation. These results indicate that the inhibition ofprotein synthesis also prevented Glu-mediated internu-cleosomal DNA cleavage in cultured cells.
The involvement of DNA fragmentation after mildGlu exposure was further investigated by the TUNELtechnique to provide additional evidence for the occur-rence of apoptosis. Cortical cultures exposed to 1–10mM Glu exhibited a concentration-dependent increasein the number of positively stained apoptotic cellscompared with control cultures (Fig. 6). Compared tocontrol cultures where TUNEL-positive neurones wererarely observed (Fig. 6A), the exposure to 3 and 10 mMGlu resulted in numerous positively stained cells (Fig.6C, D). At 10 mM Glu the number of TUNEL-positiveneurones was estimated to be \50% of the totalnumber of cells. As shown in Fig. 6E, TUNEL-positivestaining was observed only in neurons with shrunken,pyknotic morphology and staining was not seen in theswollen necrotic cells (Fig. 6F), which appeared moreabundant at increasing concentrations of Glu (Fig. 6E,F).
4. Discussion
In the present investigation we have combined state-of-the-art morphological and biochemical techniques(Poirier, 1997) to demonstrate for the first time thatphysiologically relevant concentrations of Glu producewidespread apoptotic death of cultured cortical neu-rones. At concentrations of Glu B20 mM acute rapidswelling of neurones, indicative of necrosis, was notobserved and death was exclusively by apoptosis asshown by extensive TUNEL-positive labelling of neu-rones and by electrophoretic evidence of cycloheximide-sensitive DNA fragmentation; two indices widelyaccepted to be indicative of apoptosis, and reflecting

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–1429 1427
chromatin condensation and internucleosomal cleav-age of DNA, respectively. Other evidence consistentwith low micromolar concentrations of Glu producingapoptosis were our morphological observations ofneurite blebbing and cellular shrinkage. Another ma-jor finding was the concentration-dependent, Glu-in-duced increase of TUNEL-positive apoptotic cellsover the range 1–10 mM—these data are indicativeof an injury process dependent on the severity of theinsult as has been found previously using selectiveiGluR agonists (Bonfoco et al., 1995; Simonian et al.,1996; Larm et al., 1997). Under our experimentalconditions, necrosis began to occur in the presence ofapoptosis at intermediate concentrations (20–50 mM)of Glu, and eventually became the sole death processas the insult became extreme (\50 mM). Taken to-gether with evidence that Glu-mediated injury wascompletely attenuated by MK-801 and CNQX, ourunique findings are indicative of iGluR receptor-medi-ated neurotoxicity, subsequent to exposure to Glu,occurring across a continuum of insult-dependent in-jury involving apoptosis and necrosis. Given that therole of iGluRs in neuronal death has implications forneurodegenerative diseases (Lipton and Rosenberg,1994), it is interesting to consider our evidence inrelation to the patterns of injury found in stroke,which is considered to involve excitotoxicity, where inthe inner core adjacent to the epicentre of traumaneuronal death is by necrosis, whilst in the surround-ing penumbra apoptosis is certainly involved (Choi,1996; Kuroda and Siesjo, 1997). Whilst the relation-ship between Glu and apoptotic cell death in vivo isdifficult to study because the elimination of apoptoticcells by phagocytes engulfs dead and dying cells cellsso rapidly and ultrastructural analyses are indicated(Schwartzman and Cidlowski, 1993), the advantage ofcultured neurones as a model where phagocytes areabsent is that cell death mechanisms can be carefullydissected as described herein. We combined morpho-logical and biochemical techniques to investigate thecontribution of Glu to apoptosis or necrosis in a pureneuronal cortical culture to help increase understand-ing about the nature of excitotoxic cell death in vivo.Thus under our experimental conditions, mild excito-toxic insult induced cell death by apoptosis ratherthan necrosis, which nevertheless begins to occur withapoptosis at intermediate concentrations of Glu, andeventually becomes the sole death process as the in-sult becomes extreme.
In experiments not described here, we examinedGlu-mediated cell death using a previously estab-lished, totally serum-free cell culture system (Larm etal., 1996). Surprisingly, Glu induced necrotic celldeath even at very mild insult (B1 mM Glu for 5min) and hence under these conditions the death pro-
cesses were difficult to analyse because of the extremesensitivity (unpublished data). The fact that this corti-cal cell culture appears to be so sensitive to Glu maydue to the different receptor expression on their cellsurface, and for example Cox et al. (1990) showedthat high potassium alters the expression of NMDAand AMPA receptors in cortical cultures. We there-fore combined the use of 10% dialyzed FCS and B27components in the growth medium for the initial 24h. Staining with calcein-AM revealed that the opti-mized culture method produced viable cell culturesthat developed neurite outgrowths with increasingcomplexity over time. Staining with EthD-1 at thesame time points showed consistent and minimalbackground cell death (B1%), indicating a viable cellculture. Immunocytochemistry using the neuronalmarker, MAP2, and the glial marker, GFAP, demon-strated that the culture contained \98% neurones.Therefore, this modified culture method is able toproduce a viable and essentially pure neuronal popu-lation, in which based on our findings, it is possibleto dissect temporally different mechanisms of iGluR-mediated cell death.
Many previous reports have suggested that over-stimulation of the NMDA receptor triggers only ne-crosis characterized as rapid cellular swelling followedby membrane rupture (Choi et al., 1987; Dessi et al.,1993). However, some groups recently reported thatonly apoptosis is involved in Glu receptor-mediatedcell death (Kure et al., 1991; Finiels et al., 1995). Incontrast, under our culture conditions we found thatcortical neurones can be activated by Glu to undergoapoptosis and necrosis under appropriate culture andexperimental conditions. The reasons for our differentresults are likely to be the: (1) the use of chemicallydefined, antioxidant-rich supplements and of 10% dia-lyzed FCS only in the initial 24 h growth medium;and (2) the model being a pure neuronal culturerather than a mixed neuronal-glial cell culture. It iswell established that glia possess a high affinity Gluuptake system which is an extremely rapid process forremoving extracellular Glu to maintain a concentra-tion below 1–10 mM, a concentration that damagesneurones under normal conditions (Robinson et al.,1993; Schousboe and Westergaard, 1995). It is possi-ble that glia, because of their high Glu transport ca-pacity, protect against the mild insult which inducesapoptosis, and only necrosis can be observed at ex-tensive insult i.e. high concentration of Glu, such as1–2 mM. (3) We stimulated our cultures with Glu ond7–d8 (mature) instead of d3–d5 (immature) or 2–3weeks (aged) (Kure et al., 1991; Choi, 1992; Dessi etal., 1993; Finiels et al., 1995). In preliminary experi-ments, we challenged our cultures with Glu from d2–d12 and the results indicated that immature neurones

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–14291428
(Bd4) were not affected by Glu (500 mM, 24 h), whilstaged neurones seemed to be extremely sensitive toinsults including rapid wash (\50% necrotic cell deathin control). Nevertheless, our observations are in agree-ment with the observations of Rosenberg and Aizen-man (1989), who reported that low concentrations ofGlu were neurotoxic (EC50 2 mM) in 3–5 week oldastrocyte-poor cortical cultures, although they did notaddress the mechanism involved. We found substantialapoptosis in this concentration range and the pattern ofinjury was insult-dependent, as indicated by Nicotera etal. (1997), although they found a rather different con-centration-dependence of neurotoxicity with millimolarGlu killing B50% of neurones.
The non-competitive NMDA antagonist, MK-801,was effective at 5 mM in preventing Glu-mediated celldeath, a concentration at which this antagonist hasbeen shown to block NMDA receptors specifically.However, CNQX only attenuated injury by 20%. Takentogether these results indicate that Glu is acting pre-dominantly under the present experimental conditonsvia NMDA-selective receptors to produce concentra-tion-dependent neurotoxicity. This evidence agrees withextensive observations that NMDA receptor-mediatedcell death is generally the prime mechanism operative inneuronal cultures (Choi and Rothman, 1990) and sup-ports the contention that injury via NMDA-receptors isinsult-dependent (Bonfoco et al., 1995). This is not tosay that non-NMDA receptors are not involved inexcitotoxicity because certainly situations could be en-visaged where in a particular synaptic milieu, for exam-ple during injury, programmed cell death or in agingbrain in a neurodegenerative condition, AMPA andKA receptors may be responsible for specific injurypatterns. Indeed, we have found that over stimulationof AMPA receptors produces a very slow form ofneurotoxicity, which is apoptotic, and where the timecourse of injury takes place over days not hours (Larmet al., 1997; Cheung et al., 1998).
Our findings suggest that the intensity of the initialGlu insult determines the fate of neurones to be eithernecrotic or apoptotic death. Moreover, Glu-inducedneuronal death does meet the currently accepted mor-phological and biochemical criteria of apoptosis underour culture conditions. In summary, injury occurredacross a continuum with low concentrations of Gluinducing only apoptosis, slighter higher concentrationscausing a mix of injury patterns with apoptosis pre-dominating considerably over necrosis, and only necro-sis observed at moderate-high concentrations of Glu.To our knowledge, this is the first demonstration thatat physiologically relevant concentrations, Glu per secan indeed trigger neuronal necrosis and apoptosis,providing new insights into the mechanisms underlyinghuman neurological disorders related to the excito-toxicity.
Acknowledgements
This study was supported by the National Heath andMedical Research Council (Australia), of which P.M.Beart is a Senior Principal Research Fellow, and bygrants from the Ramaciotti, Rebecca L. Cooper andWilliam Buckland Foundations, and PerpetualTrustees.
References
Arends, M.J., Morris, R.G., Wyllie, A.H., 1990. Apoptosis. The roleof the endonuclease. American Journal of Pathology 136, 593–608.
Bonfoco, E., Krainc, D., Ankarcrona, M., Nicotera, P., Lipton, S.A.,1995. Apoptosis and necrosis: two distinct events induced, respec-tively, by mild and intense insults with N–methyl-D-aspartate ornitric oxide/superoxide in cortical cell cultures. Proceedings of theNational Academy of Sciences of the United States of America92, 7162–7166.
Bottenstein, J.E., Sato, G.H., 1979. Growth of a rat neuroblastomacell line in serum-free supplemented medium. Proceedings of theNational Academy of Sciences of the United States of America76, 514–517.
Brewer, G.J., Torricelli, J.R., Evege, E.K., Price, P.J., 1993. Opti-mized survival of hippocampal neurons in B27-supplementedNeurobasal, a new serum-free medium combination. Journal ofNeuroscience Research 35, 567–576.
Cheung, N.S., John, C.A., Giardina, S.F., Pascoe, C.J., Beart, P.M.,1998. AMPA receptor-mediated neurotoxicity is very slow, exten-sive and apoptotic in nature—a unique mechanism contributingto neuronal loss in degenerative diseases? (submitted).
Choi, D.W., 1992. Excitotoxic cell death. Journal of Neurobiology23, 1261–1276.
Choi, D.W., 1996. Ischemia-induced neuronal apoptosis. CurrentOpinion in Neurobiology 6, 667–672.
Choi, D.W., Maulucci-Gedde, M., Kriegstein, A.R., 1987. Glutamateneurotoxicity in cortical cell culture. Journal of Neuroscience 7,357–368.
Choi, D.W., Rothman, S.M., 1990. The role of glutamate neurotoxi-city in hypoxic-ischemic neuronal death. Annual Review of Neu-roscience 13, 171–182.
Clarke, P.G., 1990. Developmental cell death: morphological diver-sity and multiple mechanisms. Anatomy and Embryology 181,195–213.
Collingridge, G.L., Lester, R.A., 1989. Excitatory amino acid recep-tors in the vertebrate central nervous system. PharmacologicalReviews 41, 143–210.
Cox, J.A., Felder, C.C., Henneberry, R.C., 1990. Differential expres-sion of excitatory amino acid receptor subtypes in cultured cere-bellar neurons. Neuron 4, 941–947.
Dessi, F., Charriaut-Marlangue, C., Khrestchatisky, M., Ben-Ari, Y.,1993. Glutamate-induced neuronal death is not a programmedcell death in cerebellar culture. Journal of Neurochemistry 60,1953–1955.
Ellis, R.E., Yuan, J.Y., Horvitz, H.R., 1991. Mechanisms and func-tions of cell death. Annual Review of Cell Biology 7, 663–698.
Finiels, F., Robert, J.J., Samolyk, M.L., Privat, A., Mallet, J., Revah,F., 1995. Induction of neuronal apoptosis by excitotoxins associ-ated with long-lasting increase of 12-O-tetradecanoylphorbol 13-acetate-responsive element-binding activity. Journal ofNeurochemistry 65, 1027–1034.

N.S. Cheung et al. / Neuropharmacology 37 (1998) 1419–1429 1429
Gerschenson, L.E., Rotello, R.J., 1992. Apoptosis: a different type ofcell death. FASEB Journal 6, 2450–2455.
Hajos, F., Garthwaite, G., Garthwaite, J., 1986. Reversible andirreversible neuronal damage caused by excitatory amino acidanalogues in rat cerebellar slices. Neuroscience 18, 417–436.
Hollmann, M., Heinemann, S., 1994. Cloned glutamate receptors.Annual Review of Neuroscience 17, 31–108.
Kerr, J.F.R., Harmon, B.V., 1991. Definition and incidence of apop-tosis: an historical perspective. In: Tomei, L.D., Cope, F.O.(Eds.), Apoptosis: the molecular basis of cell death. Cold SpringHarbor Laboratory, New York, pp. 5–29.
Kure, S., Tominaga, T., Yoshimoto, T., Tada, K., Narisawa, K.,1991. Glutamate triggers internucleosomal DNA cleavage in neu-ronal cells. Biochemical and Biophysical Research Communica-tions 179, 39–45.
Kuroda, S., Siesjo, B.K., 1997. Reperfusion damage following focalischemia: pathophysiology and therapeutic windows. ClinicalNeuroscience 4, 199–212.
Larm, J.A., Cheung, N.S., Beart, P.M., 1997. Apoptosis induced viaAMPA-selective glutamate receptors in cultured murine corticalneurons. Journal of Neurochemistry 69, 617–622.
Larm, J.A., Cheung, N.S., Beart, P.M., 1996. (S)-5-Fluorowillardi-ine-mediated neurotoxicity in cultured murine cortical neuronesoccurs via AMPA and kainate receptors. European Journal ofPharmacology 314, 249–254.
Lipton, S.A., Rosenberg, P.A., 1994. Excitatory amino acids as afinal common pathway for neurologic disorders. New EnglandJournal of Medicine 330, 613–622.
Meldrum, B., Garthwaite, J., 1990. Excitatory amino acid neurotoxi-city and neurodegenerative disease. Trends in PharmacologicalSciences 11, 379–387.
Mosmann, T., 1983. Rapid colorimetric assay for cellular growth andsurvival: application to proliferation and cytotoxicity assays.Journal of Immunological Methods 65, 55–63.
Nicotera, P., Ankarcrona, M., Bonfoco, E., Orrenius, S., Lipton,S.A., 1997. Neuronal necrosis and apoptosis: two distinct eventsinduced by exposure to glutamate or oxidative stress. Advances inNeurology 72, 95–101.
Olney, J.W., deGubareff, T., Sloviter, R.S., 1983. ‘Epileptic’ braindamage in rats induced by sustained electrical stimulation of the
perforant path. II. Ultrastructural analysis of acute hippocampalpathology. Brain Research Bulletin 10, 699–712.
Oppenheim, R.W., 1991. Cell death during development of the ner-vous system. Annual Review of Neuroscience 14, 453–501.
Poirier, J., 1997. In: Boulton, A.A., Baker, G.B. Jr. (Eds.), ApoptosisTechniques and Protocols. Neuromethods 29. Human Press, NewJersey.
Robinson, M.B., Sinor, J.D., Dowd, L.A., Kerwin, J.F. Jr., 1993.Subtypes of sodium-dependent high-affinity L-[3H]glutamatetransport activity: pharmacologic specificity and regulation bysodium and potassium. Journal of Neurochemistry 60, 167–179.
Rosenberg, P.A., Aizenman, E., 1989. Hundred-fold increase in neu-ronal vulnerability to glutamate toxicity in astrocyte-poor culturesof rat cerebral cortex. Neuroscience Letters 103, 162–168.
Schousboe, A., Westergaard, N., 1995. Transport of neuroactiveamino acids in astrocytes. In: Kettenmann, H., Ransom, B.R.(Eds.), Neuroglial cells. Oxford University Press, New York, pp.246–258.
Schwartzman, R.A., Cidlowski, J.A., 1993. Apoptosis: the biochem-istry and molecular biology of programmed cell death. EndocrineReviews 14, 133–151.
Schwob, J.E., Fuller, T., Price, J.L., Olney, J.W., 1980. Widespreadpatterns of neuronal damage following systemic or intracerebralinjections of kainic acid: a histological study. Neuroscience 5,991–1014.
Simonian, N.A., Getz, R.L., Leveque, J.C., Konradi, C., Coyle, J.T.,1996. Kainic acid induces apoptosis in neurons. Neuroscience 75,1047–1055.
Sohn, S., Kim, E.Y., Gwag, B.J., 1998. Glutamate neurotoxicity inmouse cortical neurons: atypical necrosis with DNA ladders andchromatin condensation. Neuroscience Letters 240, 147–150.
Wong, E.H., Kemp, J.A., Priestley, T., Knight, A.R., Woodruff,G.N., Iversen, L.L., 1986. The anticonvulsant MK-801 is a potentN-methyl-D-aspartate antagonist. Proceedings of the NationalAcademy of Sciences of the United States of America 83, 7104–7108.
Wyllie, A.H., 1980. Glucocorticoid-induced thymocyte apoptosis isassociated with endogenous endonuclease activation. Nature 284,555–556.
..