mechanisms of antiapoptotic effects of estrogens in nigral dopaminergic neurons
TRANSCRIPT

Mechanisms of antiapoptotic effects of estrogens innigral dopaminergic neurons
HIDEYUKI SAWADA, MASAKAZU IBI,* TAKESHI KIHARA, MAKOTO URUSHITANI,KAZUHIRO HONDA, MIKI NAKANISHI,* AKINORI AKAIKE,* AND SHUN SHIMOHAMA1
Department of Neurology, Graduate School of Medicine, Kyoto University, Sakyoku, Kyoto 606-8507,Japan; and *Department of Pharmacology, Graduate School of Pharmaceutical Sciences, KyotoUniversity, Sakyoku, Kyoto 606-8501, Japan
ABSTRACT Parkinson’s disease is characterized bythe mesencephalic dopaminergic neuronal loss, pos-sibly by apoptosis, and the prevalence is higher inmales than in females. The estrogen receptor (ER)subtype in the mesencephalon is exclusively ER b, arecently cloned novel subtype. Bound with estradiol,it enhances gene transcription through the estrogenresponse element (ERE) or inhibits it through theactivator protein-1 (AP-1) site. We demonstrated that17b-estradiol provided protection against nigral neu-ronal apoptosis caused by exposure to either bleo-mycin sulfate (BLM) or buthionine sulfoximine(BSO). BLM and BSO-induced nigral apoptosis wasblocked by inhibitors for caspase-3 or c-Jun/AP-1.The antiapoptotic effect by estradiol was blocked byICI 182,780, an antagonist for ER, but not by asynthesized peptide that inhibits binding of the ER tothe ERE. Estradiol had no effects on caspase-3activation and c-Jun NH2-terminal kinase (JNK),which were activated by BLM. It also suppressedapoptosis by serum deprivation, which was indepen-dent of caspase-3 activation. Therefore, the anti-apoptotic neuroprotection by estradiol is mediatedby transcription through AP-1 site downstream fromJNK and caspase-3 activation. Furthermore, 17a-estradiol, a stereoisomer without female hormoneactivity, also provided an antiapoptotic effect. There-fore, the antiapoptotic effect is independent offemale hormone activity.—Sawada, H., Ibi, M.,Kihara, T., Urushitani, M., Honda, K., Nakanishi, M.,Akaike, A., Shimohama, S. Mechanisms of antiapo-ptotic effects of estrogens in nigral dopaminergicneurons. FASEB J. 14, 1202–1214 (2000)
Key Words: estrogen receptor b z AP-1 element z Parkinson’sdisease z neuroprotection
Parkinson’s disease is a chronic, progressive de-generative disorder characterized by selective loss ofmesencephalic dopaminergic neurons. In the devel-opment of the disease, exaggerated metabolic dopa-mine turnover, deposition of free iron, and decreasein glutathione content (1) cause oxidative stress
against mesencephalic dopaminergic neurons. Oxi-dative stress can cause neuronal apoptosis (2, 3), andit has been revealed that radical induced-apoptosis isinvolved in the degenerative process of dopaminer-gic neurons (4, 5).
Epidemiological studies have indicated that theincidence and prevalence of Parkinson’s disease ishigher in men than in women (6). The ratio of theprevalence in females vs. males is 1:1.36 (7) ;1:3.7(8). Estrogens, which are female sex steroid hor-mones, are modulators of apoptosis in the uterineepithelium (9) and in breast carcinoma cells (10). Ithas been discovered recently that estrogens provideneuroprotection against neuronal degeneration.There have been several reports that the decline ofestrogen is related to the risk of Alzheimer’s diseasein postmenopausal women and that replacement ofestrogen reduces the morbidity of the disease (11–14). Also, estradiol provides neuroprotection againstglutamate- or radical-induced neurotoxicity in vitro(15–19), including in nigral dopaminergic neurons(20). Recent investigations reported that the neuro-protection by estradiol is at least partially mediatedby conjugation of estradiol with glutathione (19) andnot mediated by the estrogen receptor (21). Further-more, others have reported that activation of extra-cellular signal-regulated protein kinase (ERK), anisoform of mitogen-activated kinase, by estradiolmediates the neuroprotection (22, 23); however, it iscontroversial whether or not the ERK activation byestradiol is mediated by estrogen receptor (22–25).
Recently, a novel subtype (named ER b) of theestrogen receptor (ER) was cloned and the classicalsubtype was renamed ER a (26). Estradiol regulatesgene transcription in two ways: through binding tothe estrogen response element (ERE) and throughthe activator protein-1 (AP-1) enhance element ofDNA. Estradiol enhances gene transcription throughthe ERE when bound to either ER a or b. In
1 Correspondence: Department of Neurology, GraduateSchool of Medicine, Kyoto University, 54 Shogoin-Kawahara-cho, Sakyoku, Kyoto 606-8507, Japan. E-mail: [email protected]
1202 0892-6638/00/0014-1202/$02.25 © FASEB

contrast, it enhances transcription through the AP-1site when bound to ER a, but suppresses it whenbound to ER b (27). In the central nervous system,the dominant ER subtype is ER b, especially in thecerebral cortex, cerebellum, and brainstem. In thesubstantia nigra of the mesencephalon, the subtypeis exclusively ER b (28, 29). In the process ofapoptosis, the activator proteins c-Fos and c-Jun playa pivotal role and may regulate gene transcriptionthrough heteromer formation with the ER (30, 31).Therefore, estradiol may inhibit gene transcriptionthrough the AP-1 site, inhibiting apoptosis in nigraldopaminergic neurons.
L-buthionine-[S,R]-sulfoximine (BSO) is an irre-versible specific inhibitor of g-glutamylcysteine syn-thetase (32) and causes deprivation of glutathione(33). BSO-induced neurotoxicity can be used as amodel of toxicity induced by oxidative stress inParkinson’s disease because glutathione is depletedin the nigral dopaminergic neurons in the disease(34–36). Bleomycin sulfate (BLM) is an anti-cancerdrug with two molecular domains: one binds tonuclear DNA and the other acts as a radical genera-tor (37). BSO and BLM have been reported toinduce apoptosis (38–41) and can be used as modelsof oxidative stress. In the present study, we examinedthe hypothesis that estrogens provide neuroprotec-tion against apoptosis induced by experimental oxi-dative stress, achieved by exposure to BSO or BLM,in nigral dopaminergic neurons, and that the anti-apoptotic effects are independent of female hor-mone activity and mediated by gene transcriptionalregulation through the AP-1 site.
MATERIALS AND METHODS
Materials
Eagle’s minimum essential medium (EMEM) was pur-chased from Nissui Pharmaceutical Co. (Tokyo, Japan).BLM was obtained from Calbiochem-Novabiochem AG(Laufelfingen, Switzerland). 1,7-bis(4-hydroxy-3-methoxy-phenyl)-1,6-heptadiene-3,5-dione (curcumin), BSO, andtamoxifen citrate were purchased from Sigma ChemicalCo. (St. Louis, Mo.). ICI 182,780 was obtained from TocrisCookson Incorporation (Ballwin, Mo.). Antiestrogen pep-tide (Yp537; H-Cys-Asn-Val-Val-Pro-Leu-Tyr (PO3H2)-Asp-Leu-Leu-Leu-Glu-OH) was purchased from Bachem Bio-science Inc. (Bubendorf, Switzerland). Specific inhibitorsfor caspase-1 (Ac-Trp-Glu-His-Asp-H; Ac-WEHD-CHO) (42)and caspase-3 (Ac-Asp-Met-Gln-Asp-H; Ac-DMQD-CHO)(43) were obtained from Peptide Institute Inc. (Osaka,Japan). Monoclonal anti-tyrosine hydroxylase (TH) anti-body was obtained from Eugene Tech (Ridgefield Park,N.J..), and monoclonal anti-microtubule associated protein2 (MAP 2) antibody for immunocytochemical studies wasobtained from Sigma. Anti-c-Jun NH2-terminal kinase 1(JNK1), anti-c-Jun-NH2-terminal kinase 2/3 (JNK 2/3), andanti-c-Jun antibodies were purchased from Santa CruzBiotechnology (Santa Cruz, Calif.).
Cell culture
Cultures of the rat mesencephalon were performed accord-ing to methods described previously (20, 44–46). The ventraltwo-thirds of the mesencephalon was dissected from ratembryos on the 16th day of gestation. The dissected regionsincluded dopaminergic neurons in the substantia nigra andthe ventral tegmental area, but not noradrenergic neurons inthe locus ceruleus. Neurons were incubated with 0.1% trypsinat 37°C for 20 min and incubated with EMEM containing40% fetal calf serum (FCS). They were gently triturated andplated out onto 0.1% polyethyleneimine-coated plastic cover-slips at a density of 1.3 3 105 cells/cm2. The culture mediumconsisted of EMEM containing 10% FCS for the first 1 to 4days in culture and horse serum (HS) from the 5th dayonward. The animals were treated in accordance with guide-lines published in the NIH Guide for the Care and Use ofLaboratory Animals (47).
Treatment of the cultures
In a pilot study, 24 h incubation was required to induceapoptotic morphological changes which were detected byTUNEL and the nuclear dye Hoechst 33258. Therefore, toinvestigate BSO- or BLM-induced neurotoxicity, culturedneurons were exposed to BSO or BLM for 24 h on the 8th dayof culture, incubated in EMEM containing 10% HS for anadditional 72 h, and fixed on the 12th day. To determine theeffects of preincubation with estrogens on BSO- or BLM-induced neurotoxicity, the cultures were preincubated with10 nM 17a- or 17b-estradiol on the 7th day in culture for 24 hprior to BSO or BLM exposure. On the 8th day in culture,cells were exposed to EMEM containing BSO or BLM with17a- or 17b-estradiol for 24 h. Cells were incubated inmedium without drugs for an additional 72 h and fixed onthe 12th day. Control experiments were similar to treatment,using EMEM containing no drugs. Incubation with less than0.1% dimethyl sulfoxide (DMSO) or 0.1% ethanol for 24 hwas found to have no effect on neuronal survival rates, and so17a- and 17b-estradiol, ICI 182,780, and curcumin weredissolved in DMSO. Tamoxifen, Ac-WEHD-CHO, Ac-DMQD-CHO, and Yp537 were dissolved in ethanol. BSO and BLMwere dissolved in water.
In a pilot study, concentrations of tamoxifen greater than1 mM, Yp537 greater than 10 mM, Ac-WEHD-CHO greaterthan 100 mM and Ac-DMQD-CHO greater than 100 mM weretoxic to cultured neurons (data not shown). Therefore, thedrug concentrations used were 10–100 nM for tamoxifen, 1mM for Yp537, and 1–10 mM for Ac-WEHD-CHO or Ac-DMQD-CHO.
Evaluation of neurotoxicity
The number of surviving neurons was determined usingimmunostaining as described in our previous studies (20,44–46). Briefly, cultured cells were fixed and incubated withanti-TH (diluted 1:1000) or anti-MAP2 (diluted 1:400) anti-bodies for 24 h, with the secondary biotinylated antibody for1 h, and with avidin–biotin complex solution (Vectastain) for1 h. Finally, the cultures were reacted with diaminobenzidinesolution for 6 min. The number of cells stained with anti-THantibody in 10 to 20 randomly selected fields (3200, totalmagnification) was taken as the numbers of surviving dopa-minergic neurons and those stained with anti-MAP2 antibodyin 15 randomly selected fields (3400) as that of total neurons.Counts were made blind to the experimental treatment. Incontrol cultures, 100 to 200 TH-positive neurons or 100 to200 MAP2-positive neurons were counted for dopaminergic
1203ANTIAPOPTOTIC MECHANISM OF ESTRADIOL
and total neurons, respectively. Neurotoxicity was evaluatedby the reduction in neuronal survival rate in each experi-ment.
Statistical analysis
Statistical analysis was performed by one-way ANOVA andpost hoc multiple comparison by Newman-Keuls’ methodwhen variance in the data was statistically equivalent. Unifor-mity of variance was analyzed statistically by Bartlett’s test.Statistical significance was defined as P,0.05.
LD50 of the neurotoxicity induced by BSO and BLMexposure
The dose response curve for BSO toxicity was fitted to thecurve using the following Michaelis Menten’s equation.
@Survival~%!# 5 100 3 S1 2@BSO#
@BSO# 1 LD50D
where [Survival (%)] is survival rate of the cultured neurons,[BSO] is the BSO concentration (mM), LD50 is the 50% lethaldose of BSO toxicity. The value of LD50 and the 95%confidential intervals were determined by the computer soft-ware (GraphPad Prism Version 3.0, GraphPad Software Inc.)and convergence was reached when three consecutive itera-tions changed the sum-of-squares by less than 0.001%. Curvefitting for BLM toxicity was performed similarly.
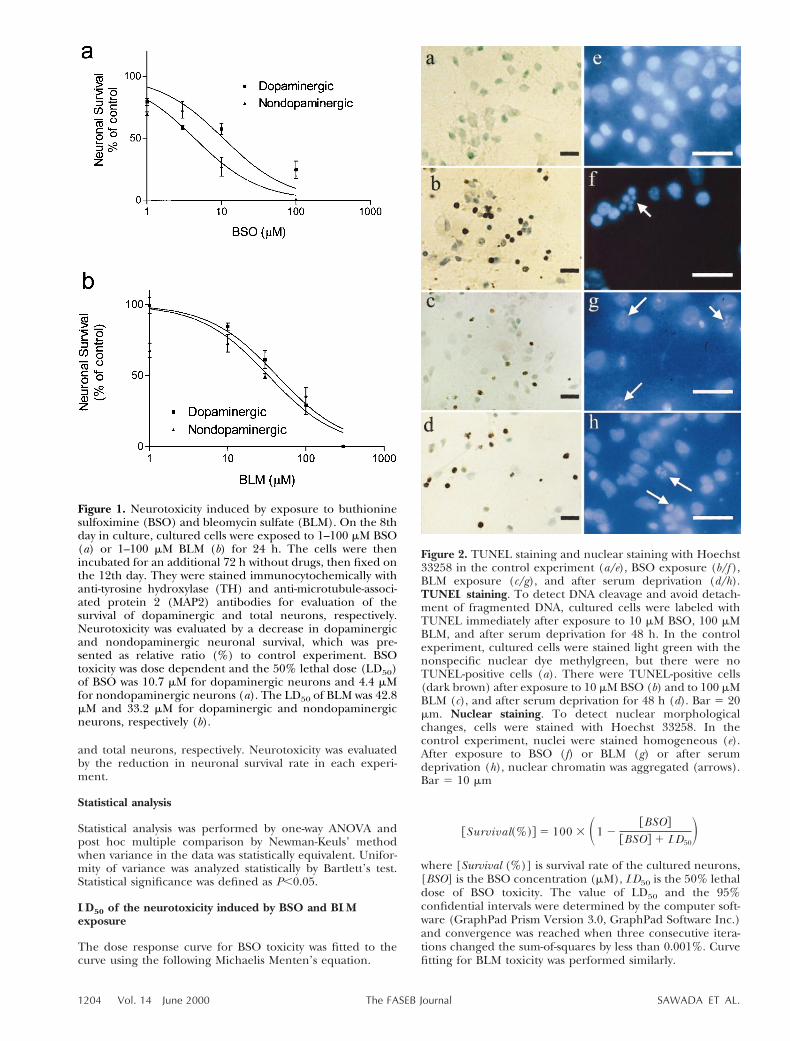
Figure 1. Neurotoxicity induced by exposure to buthioninesulfoximine (BSO) and bleomycin sulfate (BLM). On the 8thday in culture, cultured cells were exposed to 1–100 mM BSO(a) or 1–100 mM BLM (b) for 24 h. The cells were thenincubated for an additional 72 h without drugs, then fixed onthe 12th day. They were stained immunocytochemically withanti-tyrosine hydroxylase (TH) and anti-microtubule-associ-ated protein 2 (MAP2) antibodies for evaluation of thesurvival of dopaminergic and total neurons, respectively.Neurotoxicity was evaluated by a decrease in dopaminergicand nondopaminergic neuronal survival, which was pre-sented as relative ratio (%) to control experiment. BSOtoxicity was dose dependent and the 50% lethal dose (LD50)of BSO was 10.7 mM for dopaminergic neurons and 4.4 mMfor nondopaminergic neurons (a). The LD50 of BLM was 42.8mM and 33.2 mM for dopaminergic and nondopaminergicneurons, respectively (b).
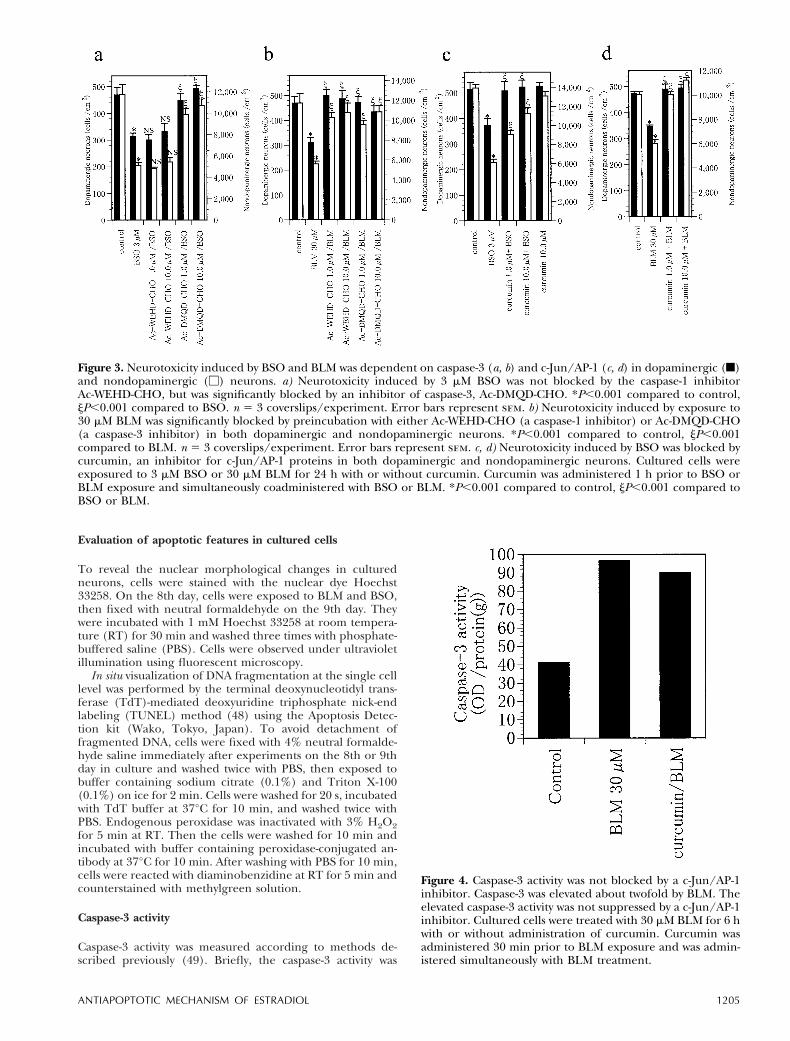
Figure 2. TUNEL staining and nuclear staining with Hoechst33258 in the control experiment (a/e), BSO exposure (b/f ),BLM exposure (c/g), and after serum deprivation (d/h).TUNEL staining. To detect DNA cleavage and avoid detach-ment of fragmented DNA, cultured cells were labeled withTUNEL immediately after exposure to 10 mM BSO, 100 mMBLM, and after serum deprivation for 48 h. In the controlexperiment, cultured cells were stained light green with thenonspecific nuclear dye methylgreen, but there were noTUNEL-positive cells (a). There were TUNEL-positive cells(dark brown) after exposure to 10 mM BSO (b) and to 100 mMBLM (c), and after serum deprivation for 48 h (d). Bar 5 20mm. Nuclear staining. To detect nuclear morphologicalchanges, cells were stained with Hoechst 33258. In thecontrol experiment, nuclei were stained homogeneous (e).After exposure to BSO (f) or BLM (g) or after serumdeprivation (h), nuclear chromatin was aggregated (arrows).Bar 5 10 mm
1204 Vol. 14 June 2000 SAWADA ET AL.The FASEB Journal
Evaluation of apoptotic features in cultured cells
To reveal the nuclear morphological changes in culturedneurons, cells were stained with the nuclear dye Hoechst33258. On the 8th day, cells were exposed to BLM and BSO,then fixed with neutral formaldehyde on the 9th day. Theywere incubated with 1 mM Hoechst 33258 at room tempera-ture (RT) for 30 min and washed three times with phosphate-buffered saline (PBS). Cells were observed under ultravioletillumination using fluorescent microscopy.
In situ visualization of DNA fragmentation at the single celllevel was performed by the terminal deoxynucleotidyl trans-ferase (TdT)-mediated deoxyuridine triphosphate nick-endlabeling (TUNEL) method (48) using the Apoptosis Detec-tion kit (Wako, Tokyo, Japan). To avoid detachment offragmented DNA, cells were fixed with 4% neutral formalde-hyde saline immediately after experiments on the 8th or 9thday in culture and washed twice with PBS, then exposed tobuffer containing sodium citrate (0.1%) and Triton X-100(0.1%) on ice for 2 min. Cells were washed for 20 s, incubatedwith TdT buffer at 37°C for 10 min, and washed twice withPBS. Endogenous peroxidase was inactivated with 3% H2O2for 5 min at RT. Then the cells were washed for 10 min andincubated with buffer containing peroxidase-conjugated an-tibody at 37°C for 10 min. After washing with PBS for 10 min,cells were reacted with diaminobenzidine at RT for 5 min andcounterstained with methylgreen solution.
Caspase-3 activity
Caspase-3 activity was measured according to methods de-scribed previously (49). Briefly, the caspase-3 activity was
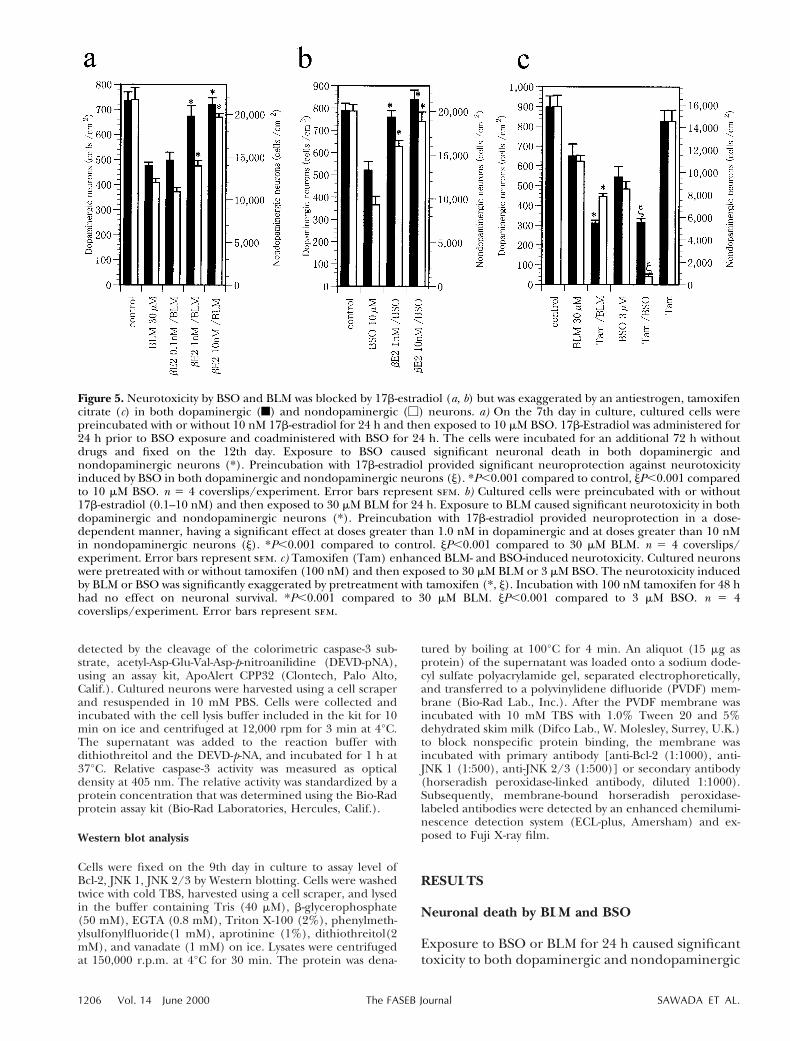
Figure 3. Neurotoxicity induced by BSO and BLM was dependent on caspase-3 (a, b) and c-Jun/AP-1 (c, d) in dopaminergic (f)and nondopaminergic (M) neurons. a) Neurotoxicity induced by 3 mM BSO was not blocked by the caspase-1 inhibitorAc-WEHD-CHO, but was significantly blocked by an inhibitor of caspase-3, Ac-DMQD-CHO. *P,0.001 compared to control,jP,0.001 compared to BSO. n 5 3 coverslips/experiment. Error bars represent sem. b) Neurotoxicity induced by exposure to30 mM BLM was significantly blocked by preincubation with either Ac-WEHD-CHO (a caspase-1 inhibitor) or Ac-DMQD-CHO(a caspase-3 inhibitor) in both dopaminergic and nondopaminergic neurons. *P,0.001 compared to control, jP,0.001compared to BLM. n 5 3 coverslips/experiment. Error bars represent sem. c, d) Neurotoxicity induced by BSO was blocked bycurcumin, an inhibitor for c-Jun/AP-1 proteins in both dopaminergic and nondopaminergic neurons. Cultured cells wereexposured to 3 mM BSO or 30 mM BLM for 24 h with or without curcumin. Curcumin was administered 1 h prior to BSO orBLM exposure and simultaneously coadministered with BSO or BLM. *P,0.001 compared to control, jP,0.001 compared toBSO or BLM.
Figure 4. Caspase-3 activity was not blocked by a c-Jun/AP-1inhibitor. Caspase-3 was elevated about twofold by BLM. Theelevated caspase-3 activity was not suppressed by a c-Jun/AP-1inhibitor. Cultured cells were treated with 30 mM BLM for 6 hwith or without administration of curcumin. Curcumin wasadministered 30 min prior to BLM exposure and was admin-istered simultaneously with BLM treatment.
1205ANTIAPOPTOTIC MECHANISM OF ESTRADIOL
detected by the cleavage of the colorimetric caspase-3 sub-strate, acetyl-Asp-Glu-Val-Asp-p-nitroanilidine (DEVD-pNA),using an assay kit, ApoAlert CPP32 (Clontech, Palo Alto,Calif.). Cultured neurons were harvested using a cell scraperand resuspended in 10 mM PBS. Cells were collected andincubated with the cell lysis buffer included in the kit for 10min on ice and centrifuged at 12,000 rpm for 3 min at 4°C.The supernatant was added to the reaction buffer withdithiothreitol and the DEVD-p-NA, and incubated for 1 h at37°C. Relative caspase-3 activity was measured as opticaldensity at 405 nm. The relative activity was standardized by aprotein concentration that was determined using the Bio-Radprotein assay kit (Bio-Rad Laboratories, Hercules, Calif.).
Western blot analysis
Cells were fixed on the 9th day in culture to assay level ofBcl-2, JNK 1, JNK 2/3 by Western blotting. Cells were washedtwice with cold TBS, harvested using a cell scraper, and lysedin the buffer containing Tris (40 mM), b-glycerophosphate(50 mM), EGTA (0.8 mM), Triton X-100 (2%), phenylmeth-ylsulfonylfluoride(1 mM), aprotinine (1%), dithiothreitol(2mM), and vanadate (1 mM) on ice. Lysates were centrifugedat 150,000 r.p.m. at 4°C for 30 min. The protein was dena-
tured by boiling at 100°C for 4 min. An aliquot (15 mg asprotein) of the supernatant was loaded onto a sodium dode-cyl sulfate polyacrylamide gel, separated electrophoretically,and transferred to a polyvinylidene difluoride (PVDF) mem-brane (Bio-Rad Lab., Inc.). After the PVDF membrane wasincubated with 10 mM TBS with 1.0% Tween 20 and 5%dehydrated skim milk (Difco Lab., W. Molesley, Surrey, U.K.)to block nonspecific protein binding, the membrane wasincubated with primary antibody [anti-Bcl-2 (1:1000), anti-JNK 1 (1:500), anti-JNK 2/3 (1:500)] or secondary antibody(horseradish peroxidase-linked antibody, diluted 1:1000).Subsequently, membrane-bound horseradish peroxidase-labeled antibodies were detected by an enhanced chemilumi-nescence detection system (ECL-plus, Amersham) and ex-posed to Fuji X-ray film.
RESULTS
Neuronal death by BLM and BSO
Exposure to BSO or BLM for 24 h caused significanttoxicity to both dopaminergic and nondopaminergic
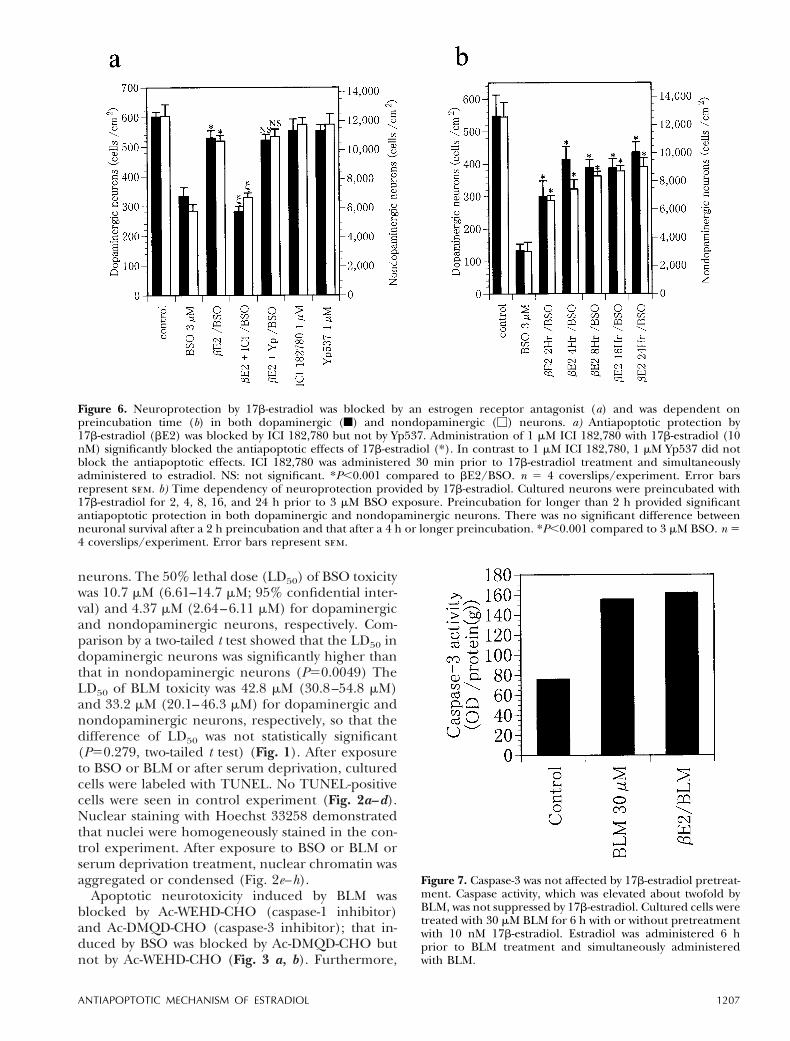
Figure 5. Neurotoxicity by BSO and BLM was blocked by 17b-estradiol (a, b) but was exaggerated by an antiestrogen, tamoxifencitrate (c) in both dopaminergic (f) and nondopaminergic (M) neurons. a) On the 7th day in culture, cultured cells werepreincubated with or without 10 nM 17b-estradiol for 24 h and then exposed to 10 mM BSO. 17b-Estradiol was administered for24 h prior to BSO exposure and coadministered with BSO for 24 h. The cells were incubated for an additional 72 h withoutdrugs and fixed on the 12th day. Exposure to BSO caused significant neuronal death in both dopaminergic andnondopaminergic neurons (*). Preincubation with 17b-estradiol provided significant neuroprotection against neurotoxicityinduced by BSO in both dopaminergic and nondopaminergic neurons (j). *P,0.001 compared to control, jP,0.001 comparedto 10 mM BSO. n 5 4 coverslips/experiment. Error bars represent sem. b) Cultured cells were preincubated with or without17b-estradiol (0.1–10 nM) and then exposed to 30 mM BLM for 24 h. Exposure to BLM caused significant neurotoxicity in bothdopaminergic and nondopaminergic neurons (*). Preincubation with 17b-estradiol provided neuroprotection in a dose-dependent manner, having a significant effect at doses greater than 1.0 nM in dopaminergic and at doses greater than 10 nMin nondopaminergic neurons (j). *P,0.001 compared to control. jP,0.001 compared to 30 mM BLM. n 5 4 coverslips/experiment. Error bars represent sem. c) Tamoxifen (Tam) enhanced BLM- and BSO-induced neurotoxicity. Cultured neuronswere pretreated with or without tamoxifen (100 nM) and then exposed to 30 mM BLM or 3 mM BSO. The neurotoxicity inducedby BLM or BSO was significantly exaggerated by pretreatment with tamoxifen (*, j). Incubation with 100 nM tamoxifen for 48 hhad no effect on neuronal survival. *P,0.001 compared to 30 mM BLM. jP,0.001 compared to 3 mM BSO. n 5 4coverslips/experiment. Error bars represent sem.
1206 Vol. 14 June 2000 SAWADA ET AL.The FASEB Journal
neurons. The 50% lethal dose (LD50) of BSO toxicitywas 10.7 mM (6.61–14.7 mM; 95% confidential inter-val) and 4.37 mM (2.64–6.11 mM) for dopaminergicand nondopaminergic neurons, respectively. Com-parison by a two-tailed t test showed that the LD50 indopaminergic neurons was significantly higher thanthat in nondopaminergic neurons (P50.0049) TheLD50 of BLM toxicity was 42.8 mM (30.8–54.8 mM)and 33.2 mM (20.1–46.3 mM) for dopaminergic andnondopaminergic neurons, respectively, so that thedifference of LD50 was not statistically significant(P50.279, two-tailed t test) (Fig. 1). After exposureto BSO or BLM or after serum deprivation, culturedcells were labeled with TUNEL. No TUNEL-positivecells were seen in control experiment (Fig. 2a–d).Nuclear staining with Hoechst 33258 demonstratedthat nuclei were homogeneously stained in the con-trol experiment. After exposure to BSO or BLM orserum deprivation treatment, nuclear chromatin wasaggregated or condensed (Fig. 2e–h).
Apoptotic neurotoxicity induced by BLM wasblocked by Ac-WEHD-CHO (caspase-1 inhibitor)and Ac-DMQD-CHO (caspase-3 inhibitor); that in-duced by BSO was blocked by Ac-DMQD-CHO butnot by Ac-WEHD-CHO (Fig. 3 a, b). Furthermore,
Figure 6. Neuroprotection by 17b-estradiol was blocked by an estrogen receptor antagonist (a) and was dependent onpreincubation time (b) in both dopaminergic (f) and nondopaminergic (M) neurons. a) Antiapoptotic protection by17b-estradiol (bE2) was blocked by ICI 182,780 but not by Yp537. Administration of 1 mM ICI 182,780 with 17b-estradiol (10nM) significantly blocked the antiapoptotic effects of 17b-estradiol (*). In contrast to 1 mM ICI 182,780, 1 mM Yp537 did notblock the antiapoptotic effects. ICI 182,780 was administered 30 min prior to 17b-estradiol treatment and simultaneouslyadministered to estradiol. NS: not significant. *P,0.001 compared to bE2/BSO. n 5 4 coverslips/experiment. Error barsrepresent sem. b) Time dependency of neuroprotection provided by 17b-estradiol. Cultured neurons were preincubated with17b-estradiol for 2, 4, 8, 16, and 24 h prior to 3 mM BSO exposure. Preincubation for longer than 2 h provided significantantiapoptotic protection in both dopaminergic and nondopaminergic neurons. There was no significant difference betweenneuronal survival after a 2 h preincubation and that after a 4 h or longer preincubation. *P,0.001 compared to 3 mM BSO. n 54 coverslips/experiment. Error bars represent sem.
Figure 7. Caspase-3 was not affected by 17b-estradiol pretreat-ment. Caspase activity, which was elevated about twofold byBLM, was not suppressed by 17b-estradiol. Cultured cells weretreated with 30 mM BLM for 6 h with or without pretreatmentwith 10 nM 17b-estradiol. Estradiol was administered 6 hprior to BLM treatment and simultaneously administeredwith BLM.
1207ANTIAPOPTOTIC MECHANISM OF ESTRADIOL

BSO- and BLM-induced neurotoxicity was blockedby coadministration of curcumin, an inhibitor forc-Jun/AP-1 (Fig. 3c, d). Caspase-3 activity was ele-vated by BLM treatment. The elevated caspase activ-ity by BLM was not affected by curcumin (Fig. 4).
Protection by estradiol against BLM- and BSO-induced neuronal death
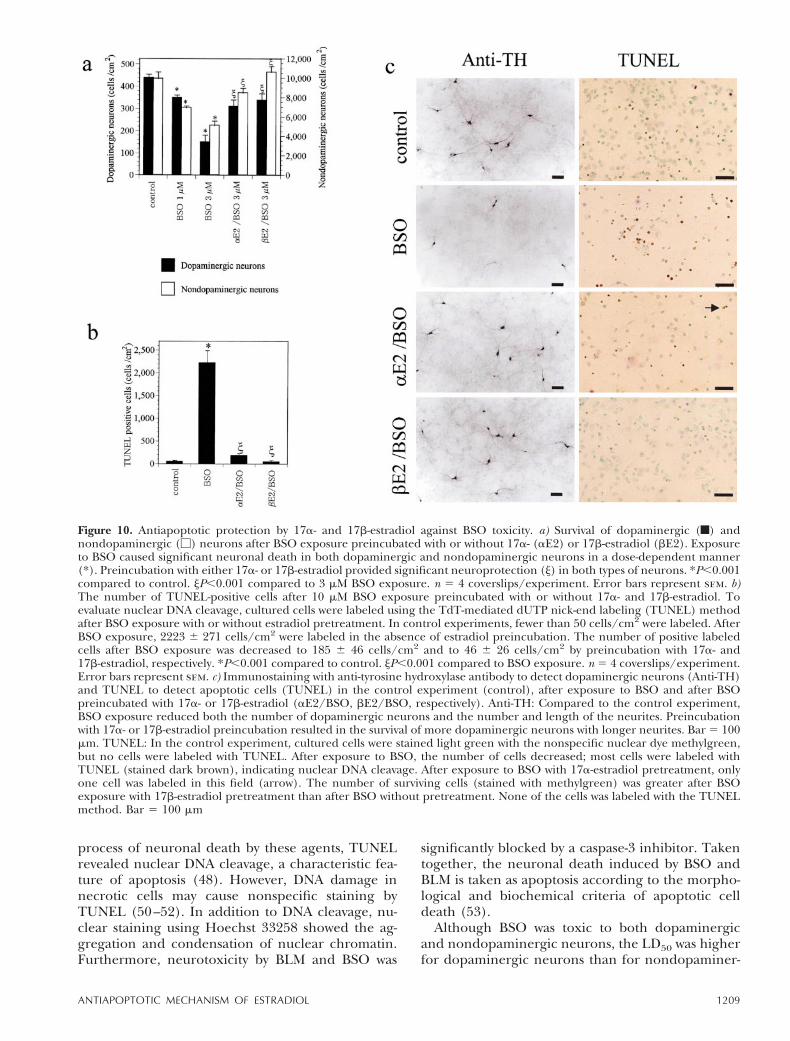
Preincubation with 1 or 10 nM 17b-estradiol signifi-cantly protected both types of neurons against tox-icity induced by BSO. 17b-Estradiol preincubationexerted significant neuroprotection against BLMneurotoxicity in both dopaminergic and nondopam-inergic neurons dose dependently (Fig. 5a, b). Incontrast, 100 nM tamoxifen, an antiestrogen agent,exaggerated BLM neurotoxicity, and the survivingneuronal number was decreased from 650 6 60 to311 6 15 cells/cm2 for dopaminergic neurons andfrom 10,966 6 537 to 7,841 6 256 cells/cm2 fornondopaminergic neurons. It also significantly exag-gerated the BSO neurotoxicity to dopaminergic neu-rons (from 544654 to 314621 cells/cm2) and non-dopaminergic neurons (from 8,5076652 to7236224 cells/cm2) (Fig. 5c). Coadministration with1 mM ICI 182,780, a pure antagonist for ERb,antagonized the neuroprotection provided by 17b-estradiol, but an inhibitor for ERb homodimeriza-tion, Yp537 (1 mM), did not block the neuroprotec-tion. ICI 182,780 (1 mM) or Yp537 (1 mM) alone didnot exert any detectable effects on neuronal survival(Fig. 6a). The neuroprotection provided by 17b-estradiol was significant when administered forlonger than 2 h; preincubation for 4 h or longer didnot provide additional neuroprotection (Fig. 6b).
The activity of caspas-3, which was elevated byBLM, was not suppressed by preincubation with 10nM 17b-estradiol (Fig. 7). Immunoblotting assay ofBcl-2 revealed that 17b-estradiol increased theamount of Bcl-2, but the up-regulation of Bcl-2required preincubation for 8 h or longer (Fig. 8).JNK 1 and JNK 2/3 were elevated by BSO or BLMtreatment. The elevated activity of JNK 1 was sup-
pressed neither by 17b-estradiol nor by Ac-DMQD-CHO. In contrast to JNK 1, JNK 2/3 activity wasslightly suppressed by estradiol but was almost com-pletely suppressed by Ac-DMQD-CHO (Fig. 9).
17a-Estradiol, a stereoisomer with little biologicalactivity as a female sex hormone, also significantlyprotected dopaminergic and nondopaminergic neu-rons against apoptosis induced by BSO (Fig. 10).Preincubation with either 17a- or 17b-estradiol sig-nificantly reduced the number of TUNEL-labeledcells after BSO exposure (the number of TUNEL-positive cells induced by BSO was decreased from2,2236271 cells/cm2 to 185646 cells/cm2 and46626 cells/cm2 by 17a- and 17b-estradiol, respec-tively) (Fig. 10). Table 1 presents the neuroprotec-tion of 17a- and 17b-estradiol against apoptosisinduced by various agents. Preincubation with either17a- or 17b-estradiol provided significant neuropro-tection against apoptosis induced by exposure toBLM and by serum deprivation, as well as thatinduced by BSO. In contrast to preincubation withestradiol, preincubation with other steroids, such ascorticosterone, testosterone, and cholesterol, had noeffect on neuronal apoptosis induced by BLM expo-sure (Table 2).
As shown in Fig. 3a, b, apoptotic neurotoxicityinduced by BLM was blocked by Ac-WEHD-CHO(caspase-1 inhibitor) and Ac-DMQD-CHO (caspase-3inhibitor), and that induced by BSO was blocked byAc-DMQD-CHO but not by Ac-WEHD-CHO. In con-trast, neuronal death induced by serum deprivationwas not blocked by either inhibitor (Fig. 11).
DISCUSSION
BSO- and BLM-induced neurotoxicity
Exposure to BSO or BLM caused significant neuro-nal death in cultured mesencephalic neurons. In the
Figure 8. Western blotting analysis for Bcl-2. Cultured cellswere incubated with 10 nM 17b-estradiol for 2, 4, 8, 24 h onthe 8th day in culture and analyzed for Bcl-2 by Westernblotting. Bcl-2 was elevated by estradiol incubation, but theelevation required 4 h or longer incubation.
Figure 9. Western blotting analysis for JNK. Cultured cellswere exposed to 30 mM BLM or 3 mM BSO for 24 h, with orwithout preincubation with 10 nM 17b-estradiol or 10 mMAc-DMQD-CHO, and analyzed for JNK 1 and JNK 2/3. BLMcaused the elevation of JNK1 or JNK2/3. Estradiol providedno or little effect on the elevation of JNK1 and JNK2/3. Theelevation of JNK2/3 was suppressed by Ac-DMQD-CHO.
1208 Vol. 14 June 2000 SAWADA ET AL.The FASEB Journal
process of neuronal death by these agents, TUNELrevealed nuclear DNA cleavage, a characteristic fea-ture of apoptosis (48). However, DNA damage innecrotic cells may cause nonspecific staining byTUNEL (50–52). In addition to DNA cleavage, nu-clear staining using Hoechst 33258 showed the ag-gregation and condensation of nuclear chromatin.Furthermore, neurotoxicity by BLM and BSO was
significantly blocked by a caspase-3 inhibitor. Takentogether, the neuronal death induced by BSO andBLM is taken as apoptosis according to the morpho-logical and biochemical criteria of apoptotic celldeath (53).
Although BSO was toxic to both dopaminergicand nondopaminergic neurons, the LD50 was higherfor dopaminergic neurons than for nondopaminer-
Figure 10. Antiapoptotic protection by 17a- and 17b-estradiol against BSO toxicity. a) Survival of dopaminergic (f) andnondopaminergic (M) neurons after BSO exposure preincubated with or without 17a- (aE2) or 17b-estradiol (bE2). Exposureto BSO caused significant neuronal death in both dopaminergic and nondopaminergic neurons in a dose-dependent manner(*). Preincubation with either 17a- or 17b-estradiol provided significant neuroprotection (j) in both types of neurons. *P,0.001compared to control. jP,0.001 compared to 3 mM BSO exposure. n 5 4 coverslips/experiment. Error bars represent sem. b)The number of TUNEL-positive cells after 10 mM BSO exposure preincubated with or without 17a- and 17b-estradiol. Toevaluate nuclear DNA cleavage, cultured cells were labeled using the TdT-mediated dUTP nick-end labeling (TUNEL) methodafter BSO exposure with or without estradiol pretreatment. In control experiments, fewer than 50 cells/cm2 were labeled. AfterBSO exposure, 2223 6 271 cells/cm2 were labeled in the absence of estradiol preincubation. The number of positive labeledcells after BSO exposure was decreased to 185 6 46 cells/cm2 and to 46 6 26 cells/cm2 by preincubation with 17a- and17b-estradiol, respectively. *P,0.001 compared to control. jP,0.001 compared to BSO exposure. n 5 4 coverslips/experiment.Error bars represent sem. c) Immunostaining with anti-tyrosine hydroxylase antibody to detect dopaminergic neurons (Anti-TH)and TUNEL to detect apoptotic cells (TUNEL) in the control experiment (control), after exposure to BSO and after BSOpreincubated with 17a- or 17b-estradiol (aE2/BSO, bE2/BSO, respectively). Anti-TH: Compared to the control experiment,BSO exposure reduced both the number of dopaminergic neurons and the number and length of the neurites. Preincubationwith 17a- or 17b-estradiol preincubation resulted in the survival of more dopaminergic neurons with longer neurites. Bar 5 100mm. TUNEL: In the control experiment, cultured cells were stained light green with the nonspecific nuclear dye methylgreen,but no cells were labeled with TUNEL. After exposure to BSO, the number of cells decreased; most cells were labeled withTUNEL (stained dark brown), indicating nuclear DNA cleavage. After exposure to BSO with 17a-estradiol pretreatment, onlyone cell was labeled in this field (arrow). The number of surviving cells (stained with methylgreen) was greater after BSOexposure with 17b-estradiol pretreatment than after BSO without pretreatment. None of the cells was labeled with the TUNELmethod. Bar 5 100 mm
1209ANTIAPOPTOTIC MECHANISM OF ESTRADIOL
gic neurons, showing that dopaminergic neuronswere relatively resistant to the toxicity. The reasonwhy dopaminergic neurons were less vulnerable toBSO is unknown. However, dopaminergic neuronsare resistant to another oxygen radical, nitric oxide(44, 45), and they may have a great ability toscavenge radicals (34). Therefore, they may be rela-tively resistant in general to toxicity induced byreactive oxygen species.
BSO-induced apoptosis is thought to be mediatedby an increase in intracellular H2O2 because itdeprives the cells of glutathione and suppressesglutathione peroxidase. Apoptosis induced by reac-tive oxygen species such as H2O2 or O2
- is mediatedby caspase-1 (54, 55) or caspase-3 (56). In thepresent study, BSO-induced apoptosis was notblocked by Ac-WEHD-CHO, a caspase-1 inhibitor,but by Ac-DMQD-CHO, a caspase-3 inhibitor. There-fore, BSO-induced neurotoxicity was mediated by
caspase-3 activation. BLM-induced apoptosis is medi-ated by the activation of caspases-1 and -3, because itwas blocked by caspase-1 and caspase-3 inhibitors.Caspase-3 is thought to be activated downstream ofcaspase-1 (57). Therefore, the pathway of BLM-induced neuronal apoptosis involves activation ofcaspase-1, followed by activation of caspase-3.
Apoptosis induced by anti-cancer drugs and oxy-gen radicals such as hydrogen peroxide requires theactivation of AP-1 and transcriptional activationthrough the AP-1 element (30, 31, 58–62). Also, inthe present study apoptosis by BSO or BLM wasblocked by curcumin, a pharmacological inhibitorfor c-Jun/AP-1 proteins. c-Jun was activated by JNK, afamily of mitogen activator protein kinases, and JNK2/3 was suppressed by a caspase-3 inhibitor, as shownin Fig. 9. Furthermore, activation of caspase-3 byBLM was not blocked by curcumin, as shown in Fig.4. Therefore, the c-Jun/AP-1 proteins were activated
TABLE 1. Neuroprotection provided by estrogens against apoptosisa
Ligand for ER Induction of apoptosis NNeuroprotectionfor dopaminergic
Neurons(%) N
Neuroprotection fornondopaminergic
Neurons(%)
None BLM (30 mM) 4 0 6 5 4 0 6 5bE2 1.0 nM BLM (30 mM) 4 76 6 16 P , 0.001 4 20 6 6 P , 0.05bE2 10 nM BLM (30 mM) 4 94 6 10 P , 0.001 4 79 6 4 P , 0.001None BLM (100 mM) 4 0 6 25 4 0 6 14bE2 10 nM BLM (100 mM) 4 104 6 19 P , 0.001 4 83 6 17 P , 0.001aE2 10 nM BLM (100 mM) 4 105 6 22 P , 0.001 4 84 6 19 P , 0.001None BSO (3 mM) 4 0 6 10.6 4 0 6 9bE2 10 nM BSO (3 mM) 4 65 6 10 P , 0.01 4 114 6 12 P , 0.001aE2 10 nM BSO (3 mM) 4 55 6 10 P , 0.01 4 69 6 10 P , 0.01None BSO (10 mM) 4 0 6 7 4 0 6 3bE2 10 nM BSO (10 mM) 4 41 6 10 P , 0.01 4 70 6 3 P , 0.001None 48 hr Serum Deprivation 4 0 6 12 4 0 6 3bE2 10 nM 48 hr Serum Deprivation 4 44 6 14 P , 0.05 4 72 6 33 P , 0.01aE2 10 nM 48 hr Serum Deprivation 4 63 6 8 P , 0.05 4 35 6 0 P , 0.05
a Neuroprotection is defined by the following equation in the case where apoptosis was induced by BLM and the ligand was bE2:Neuroprotection 5 {([bE2 1 BLM] 2 [BLM])/([Control] 2 [BLM])} 3 100 (%). [bE2 1 BLM]: neuronal survival number after treatmentwith BLM and bE2 preincubation. [BLM]: that after BLM exposure. [Control]: that in the control experiment. ER: estrogen receptor; aE2:17a-estradiol; bE2: 17b-estradiol; BLM: bleomycin sulfate; BSO: buthionine sulfoximine; Statistical analysis was performed by one-way ANOVA[compared to the experiment without preincubation (neuroprotection 0%)]; NS: not significant.
TABLE 2. Anti-apoptotic effects provided by steroidsa
Treatment
Dopaminergic neurons (cells/cm2) Nondopaminergic neurons (cells/cm2)
N Mean sem N Mean sem
Control 4 787 35 4 20014 761BLM (100 mM) 4 328 19 4 8102 625bE2 (1 nM)/BLM 4 553 31 P , 0.001 4 11140 1556 P , 0.05bE2 (10 nM)/BLM 4 594 31 P , 0.001 4 15963 427 P , 0.001Cholesterol (1 nM)/BLM 4 373 46 NS 4 9018 749 NSCholesterol (10 nM)/BLM 4 332 45 NS 4 6559 361 NSTestosterone (1 nM)/BLM 4 356 28 NS 4 7427 320 NSTestosterone (10 nM)/BLM 4 374 6 NS 4 8825 475 NSCorticosterone (1 nM)/BLM 4 365 28 NS 4 8391 940 NSCorticosterone (10 nM)/BLM 4 245 37 NS 4 8825 988 NS
a BLM: bleomycin sulfate; bE2: 17b-estradiol; N: number of data set; sem: standard error of means; P , 0.001, P , 0.05: compared toBLM (100 mM) by ANOVA and Newmans-Keuls’ test. NS: not significant.
1210 Vol. 14 June 2000 SAWADA ET AL.The FASEB Journal
downstream of caspase-3. These data are consistentwith a recent report by Srivastava (63).Therefore,apoptosis by BSO or BLM is possibly mediated by theAP-1 activation downstream of caspase-3.
Neuroprotection by estrogens
17b-Estradiol provides neuroprotection against theapoptosis induced by these agents. The antiapoptoticneuroprotection was not attributable to a generalsteroid structure because other steroids provided noprotection against apoptosis. A possible mechanismis the antioxidant property of estradiol. Steroids withhydroxyl group in C3 position of the A ring providedan antioxidant property (17). In our previous report,17b-estradiol suppressed intracellular oxygen radi-cals and provided neuroprotection against oxidativestress; however, antioxidant property requires amuch higher concentration of estradiol (10–100mM) (20). Furthermore, antiapoptotic neuroprotec-tion was blocked by ICI 182,780, which has hydroxyl
group in C3. Therefore, it is unlikely that the anti-apoptotic effect of estradiol is due to the antioxidantproperty of estradiol.
Recently, it has been reported that estradiol causesup-regulation of Bcl-2 (64–66), which suppressescytochrome c release and the ensuing caspase-3activation. In our mesencephalic culture, estradiolcaused up-regulation of Bcl-2, but it required estra-diol treatment for longer than 8 h (Fig. 8). As shownin Fig. 6, preincubation for only 2 h providedsignificant antiapoptotic neuroprotection. It is un-likely that up-regulation of Bcl-2 plays a pivotal rolein antiapoptotic protection because of different timedependency. Furthermore, we recently reported thatglial cell line-derived neurotrophic factor (GDNF)causes up-regulation of Bcl-2 in the mesencephalicneurons and provides neuroprotection against BSO-and BLM-induced apoptosis, but GDNF does notprotect neurons from apoptosis by serum depriva-tion (49). In contrast to GDNF, estradiol providedneuroprotection from apoptosis induced by serumdeprivation treatment. Therefore, antiapoptotic pro-tection by estradiol in the present study includesother mechanisms besides Bcl-2 up-regulation.
The neuroprotection provided by 17b-estradiolwas thought to be mediated by the ER because it wasantagonized by ICI 182,780, a pure antagonist forER b. 17b-Estradiol has affinity for both subtypes ofER (29), ER a and ER b, but the neuroprotection ofnigral dopaminergic neurons is probably mediatedby ER b, because it has been revealed that the ERsubtype in the substantia nigra of the mesencepha-lon is exclusively ER b (28, 29). Gene expression asa result of ligand-bound ER includes both primaryand secondary responses, with the former directlyregulated by the ER and the latter regulated byproducts of the primary response. The primary re-sponse to ligand-bound ER is mediated in two ways:through the ERE and through the AP-1 element. ERb activated by 17b-estradiol binds to DNA as ahomodimer and enhances gene transcriptionthrough the ERE. However, it also suppresses genetranscription from the AP-1 element by couplingwith he AP-1 proteins c-Fos and c-Jun (27). Tamox-ifen acts as a pure antagonist for ER b (67) and,when bound to ER b, regulates transcriptionthrough either the ERE or the AP-1 element in areverse manner: it suppresses transcription throughERE and enhances it through AP-1 site (27). In thisstudy, it exaggerated the apoptotic toxicity inducedby BSO and BLM. Therefore, the antiapoptoticeffect of 17b-estradiol could be mediated by theactivation of antiapoptotic genes through the ERE orby the suppression of proapoptotic genes throughthe AP-1 enhancer element.
The neuroprotection provided by 17b-estradiol inthe present study was not antagonized by Yp537.
Figure 11. Neurotoxicity induced by serum deprivation treat-ment was not blocked by inhibitors for caspases-1 and -3.Serum deprivation (SD) for 48 h caused significant neuronaldeath in both dopaminergic (f) and nondopaminergic (M)neurons. Neurotoxicity induced by sd was not affected byinhibitors for caspases-1 (Ac-WEHD-CHO) and -3 (Ac-DMQD-CHO). N.S., not significant. n 5 3 coverslips/experi-ment. Error bars represent sem.
1211ANTIAPOPTOTIC MECHANISM OF ESTRADIOL

Yp537 is a synthesized 12-amino acid phosphotyrosylpeptide containing a selected sequence surroundingtyrosine-537, a constitutively phosphorylated site, onthe estrogen receptor (68). It blocks the homodimerformation of the ER and effects of the pathwayregulated by the ERE, but not the effects of theactivation of the AP-1 site, because ligand-bound ERattaches to the AP-1 consensus element by making aheteromer with he AP-1 proteins Fos and Jun. Thesefindings suggest that the 17b-estradiol-ER b complexinhibits the transcription of certain proapoptoticgenes from the AP-1 element that suppress apopto-sis. Therefore, the antiapoptotic effect by estradiol isthought to be mediated by suppression of proapop-totic genes through the AP-1, but not ERE.
Serum deprivation for 48 h caused significantneurotoxicity resulting in a large number of TUNEL-positive cells. Nuclear staining using Hoechst 33258showed that there was nuclear aggregation afterserum deprivation treatment. Therefore, neuronaldeath caused by serum deprivation was accompaniedby the nuclear morphological features characteristicof apoptosis and by DNA cleavage, and was thoughtto be apoptosis. In contrast to apoptosis induced byBSO or BLM, that induced by serum deprivation wasnot blocked by either Ac-WEHD-CHO or Ac-DMQD-CHO. These data are consistent with a recent reportthat apoptosis induced by potassium deprivationdoes not require activation of caspase-3 (69) andindicate that serum deprivation caused apoptosis atleast partially by a direct pathway to cascades down-stream from the activation of caspase-3, such as
activation of the AP-1 proteins or endonuclease inthe apoptotic process (70). Estradiol provided anti-apoptotic protection against serum deprivation treat-ment. Furthermore, estradiol had no effect oncaspase-3 or JNK activation. Therefore, the key pro-apoptotic gene products inhibited by estradiol aremost likely downstream from the activation ofcaspase-3 and JNK (Fig. 12). In addition to 17b-estradiol, 17a-estradiol also provided significant an-tiapoptotic neuroprotection, consistent with a recentstudy by Green et al. (71) using a neuroblastoma cellline. However, the estradiol-induced neuroprotec-tion reported by Green et al. might be mediated bya different mechanism from that in nigral neuronsbecause the neuroprotection in their report was notantagonized by tamoxifen, in contrast to our results.Although it is not known which subtype of ERmediates neuroprotection in the neuroblastoma cellline, the different response to tamoxifen may berelated to the subtypes of ER mediating neuropro-tection, since tamoxifen does not antagonize tran-scription regulation through AP-1 when mediated byER a (27, 67).
Compared to 17b-estradiol, 17a-estradiol has fewof the biological effects of female sex steroid hor-mones. A ligand binding study showed that 17a-estradiol has affinity for ER b (relative bindingaffinity of 17a- and 17b-estradiol is 0.11:1) (29).Therefore, 17a-estradiol can activate ER b and causethe same primary response as that of 17b-estradiol, atleast when an appropriate dose is applied. Theantiapoptotic protection by estradiol is probably the
Figure 12. Proposed model forthe antiapoptotic protectionprovided by estradiol to mesen-cephalic neurons. Buthioninesulfoximine (BSO), a g-glu-tamylcysteine synthetase inhibi-tor, causes depletion of glutathi-one (GSH), results in elevationof hydrogen peroxide, andcauses apoptosis mediated bycaspase-3 (Casp-3) activation.Bleomycin sulfate (BLM) causesapoptosis mediated by activa-tion of caspases-1 (Casp-1) and-3. Neurotoxicity by BSO and byBLM is blocked by curcumin, aninhibitor for c-Jun/AP-1. BLMactivates caspase-3 and JNK 2/3.Neuroprotection by estradiolwas blocked by ICI 182,780 butnot by Yp537. Furthermore, ac-tivation of caspase-3 or JNK 2/3was not blocked by estradiol.Therefore, estradiol providedneuroprotection by suppressionof proapoptotic gene transcrip-tion through the AP-1 site,which was downstream ofcaspase-3 activation and the en-suing JNK activation.
1212 Vol. 14 June 2000 SAWADA ET AL.The FASEB Journal

results of a primary response initiated by estradiolbecause preincubation for only 2 h provided signif-icant antiapoptotic protection in the present study.The negative biological estrogenic effects of 17a-estradiol may be a result not of the difference inaffinity, but rather of the difference in secondaryresponse induced by 17a- and 17b-estradiol, sincethe 17-a isomer is bound to ER a and ER b for ashorter duration than the 17b-isomer (72). 17a-Estradiol, or its analog, may be a candidate forantiapoptotic therapy in neurodegenerative diseasesaffecting neurons expressing ER b, such as Parkin-son’s disease, because it has few of the biologicaleffects associated with female sex hormones.
This work was supported by Grants-in-Aid for ScientificResearch on Priority Areas from the Ministry of Education,Science, Sports and Culture, and by grants from the Ministryof Welfare of Japan, the Smoking Research Foundation, theInamori Foundation, and the Yamanouchi Foundation forResearch on Metabolic Disorders.
REFERENCES
1. Fahn, S., and Cohen, G. (1992) The oxidant stress hypothesis inParkinson’s disease: evidence supporting it. Ann. Neurol. 32,804–812
2. Tan, S., Wood, M., and Maher, P. (1998) Oxidative stressinduces a form of programmed cell death with characteristics ofboth apoptosis and necrosis in neuronal cells. J. Neurochem. 71,95–105
3. Ratan, R. R., Murphy, T. H., and Baraban, J. M. (1994)Oxidative stress induces apoptosis in embryonic cortical neu-rons. J. Neurochem. 62, 376–379
4. Mochizuki, H., Goto, K., Mori, H., and Mizuno, Y. (1996)Histochemical detection of apoptosis in Parkinson’s disease.J. Neurol. Sci. 137, 120–123
5. Hunot, S., Brugg, B., Ricard, D., Michel, P. P., Muriel, M. P.,Ruberg, M., Faucheux, B. A., Agid, Y., and Hirsch, E. C. (1997)Nuclear translocation of NF-kB is increased in dopaminergicneurons of patients with Parkinson disease. Proc. Natl. Acad. Sci.USA 94, 7531–7536
6. Mayeux, R., Marder, K., Cote, L. J., Denaro, J., Hemenegildo, N.,Mejia, H., Tang, M. X., Lantigua, R., Wilder, D., and Gurland, B.(1995) The frequency of idiopathic Parkinson’s disease by age,ethnic group, and sex in northern Manhattan, 1988–1993.Am. J. Epidemiol. 142, 820–827
7. Bauer, R. B., Stevens, C., Reveno, W. S., and Rosenbaum, H.(1982) L-dopa treatment of Parkinson’s disease: a ten-yearfollow up study. J. Am. Geriatr. Soc. 30, 322–325
8. Li, S. C., Schoenberg, B. S., Wang, C. C., Cheng, X. M., Rui,D. Y., Bolis, C. L., and Schoenberg, D. G. (1985) A prevalencesurvey of Parkinson’s disease and other movement disorders inthe People’s Republic of China. Arch. Neurol. 42, 655–657
9. Pollard, J. W., Pacey, J., Cheng, S. V., and Jordan, E. G. (1987)Estrogens and cell death in murine uterine luminal epithelium.Cell Tissue Res. 249, 533–540
10. Kyprianou, N., English, H. F., Davidson, N. E., and Isaacs, J. T.(1991) Programmed cell death during regression of the MCF-7human breast cancer after estrogen ablation. Cancer Res. 51,162–166
11. Tang, M. X., Jacobs, D., Stern, Y., Marder, K., Schofield, P.,Gurland, B., Andrews, H., and Mayeux, R. (1996) Effect ofoestrogen during menopause on risk and age at onset ofAlzheimer’s disease. Lancet 348, 429–432
12. Henderson, V. W. (1997) Estrogen, cognition, and a woman’srisk of Alzheimer’s disease. Am. J. Med. 103, 11S–18S
13. Birge, S. J. (1997) The role of estrogen in the treatment ofAlzheimer’s disease. Neurology 48, S36–S41
14. Birge, S. J. (1996) Is there a role for estrogen replacementtherapy in the prevention and treatment of dementia? J. Am.Geriatr. Soc. 44, 865–870
15. Singer, C. A., Rogers, K. L., Strickland, T. M., and Dorsa, D. M.(1996) Estrogen protects primary cortical neurons from gluta-mate toxicity. Neurosci. Lett. 212, 13–16
16. Weaver, C. E., Jr., Park-Chung, M., Gibbs, T. T., and Farb, D. H.(1997) 17b-Estradiol protects against NMDA-induced excitotox-icity by direct inhibition of NMDA receptors. Brain Res. 761,338–341
17. Behl, C., Skutella, T., Lezoualc’h, F., Post, A., Widmann, M.,Newton, C. J., and Holsboer, F. (1997) Neuroprotection againstoxidative stress by estrogens: structure–activity relationship. Mol.Pharmacol. 51, 535–541
18. Green, P. S., Gridley, K. E., and Simpkins, J. W. (1996) Estradiolprotects against b-amyloid (25–35)-induced toxicity in SK-N-SHhuman neuroblastoma cells. Neurosci. Lett. 218, 165–168
19. Green, P. S., Gridley, K. E., and Simpkins, J. W. (1998) Nuclearestrogen receptor-independent neuroprotection by estra-trienes: a novel interaction with glutathione. Neuroscience 84,7–10
20. Sawada, H., Ibi, M., Kihara, T., Urushitani, M., Akaike, A., andShimohama, S. (1998) Estradiol protects mesencephalic dopa-minergic neurons from oxidative stress-induced neuronaldeath. J. Neurosci. Res. 54, 707–719
21. Gridley, K. E., Green, P. S., and Simpkins, J. W. (1998) A novel,synergistic interaction between 17 b-estradiol and glutathione inthe protection of neurons against b-amyloid 25–35-inducedtoxicity in vitro. Mol. Pharmacol. 54, 874–880
22. Singer, C. A., Figueroa-Masot, X. A., Batchelor, R. H., andDorsa, D. M. (1999) The mitogen-activated protein kinasepathway mediates estrogen neuroprotection after glutamatetoxicity in primary cortical neurons. J. Neurosci. 19, 2455–2463
23. Singh, M., Setalo, G., Jr., Guan, X., Warren, M., and Toran-Allerand, C. D. (1999) Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: conver-gence of estrogen and neurotrophin signaling pathways.J. Neurosci. 19, 1179–1188
24. Watters, J. J., Campbell, J. S., Cunningham, M. J., Krebs, E. G.,and Dorsa, D. M. (1997) Rapid membrane effects of steroids inneuroblastoma cells: effects of estrogen on mitogen activatedprotein kinase signalling cascade and c-fos immediate earlygene transcription. Endocrinology 138, 4030–4033
25. Sawada, H., and Shimohama, S. (2000). Neuroprotective effecsof estradiol in mesencephalic dopaminergic neurons. Neurosci.Biobehav. Rev. 24, 143–147
26. Kuiper, G. G., Enmark, E., Pelto-Huikko, M., Nilsson, S., andGustafsson, J. A. (1996) Cloning of a novel receptor expressedin rat prostate and ovary. Proc. Natl. Acad. Sci. USA 93, 5925–5930
27. Paech, K., Webb, P., Kuiper, G. G., Nilsson, S., Gustafsson, J.,Kushner, P. J., and Scanlan, T. S. (1997) Differential ligandactivation of estrogen receptors ERa and ERb at AP1 sites.Science 277, 1508–1510
28. Shughrue, P. J., Lane, M. V., and Merchenthaler, I. (1997)Comparative distribution of estrogen receptor-a and -b mRNAin the rat central nervous system. J. Comp. Neurol. 388, 507–525
29. Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad,J., Nilsson, S., and Gustafsson, J. A. (1997) Comparison of theligand binding specificity and transcript tissue distribution ofestrogen receptors a and b. Endocrinology 138, 863–870
30. Sawai, H., Okazaki, T., Yamamoto, H., Okano, H., Takeda, Y.,Tashima, M., Sawada, H., Okuma, M., Ishikura, H., and Ume-hara, H. (1995) Requirement of AP-1 for ceramide-inducedapoptosis in human leukemia HL-60 cells. J. Biol. Chem. 270,27326–27331
31. Kasibhatla, S., Brunner, T., Genestier, L., Echeverri, F., Mah-boubi, A., and Green, D. R. (1998) DNA damaging agentsinduce expression of Fas ligand and subsequent apoptosis in Tlymphocytes via the activation of NF-k B and AP-1. Mol Cell 1,543–551
32. Griffith, O. W., and Meister, A. (1979) Potent and specificinhibition of glutathione synthesis by buthionine sulfoximine(S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254, 7558–7560
33. Griffith, O. W., and Meister, A. (1979) Glutathione: interorgantranslocation, turnover, and metabolism. Proc. Natl. Acad. Sci.USA 76, 5606–5610
1213ANTIAPOPTOTIC MECHANISM OF ESTRADIOL

34. Ambani, L. M., Van Woert, M. H., and Murphy, S. (1975) Brainperoxidase and catalase in Parkinson disease. Arch. Neurol. 32,114–118
35. Perry, T. L., Godin, D. V., and Hansen, S. (1982) Parkinson’sdisease: a disorder due to nigral glutathione deficiency? Neuro-sci. Lett. 33, 305–310
36. Kish, S. J., Morito, C., and Hornykiewicz, O. (1985) Glutathioneperoxidase activity in Parkinson’s disease brain. Neurosci. Lett.58, 343–346
37. Caspary, W. J., Lanzo, D. A., and Niziak, C. (1982) Effect ofdeoxyribonucleic acid on the production of reduced oxygen bybleomycin and iron. Biochemistry 21, 334–338
38. Li, Y., Maher, P., and Schubert, D. (1997) Requirement forcGMP in nerve cell death caused by glutathione depletion. J. CellBiol. 139, 1317–1324
39. Higuchi, Y., and Matsukawa, S. (1997) Appearance of 1–2 Mbpgiant DNA fragments as an early common response leading tocell death induced by various substances that cause oxidativestress. Free Rad. Biol. Med. 23, 90–99
40. Marini, M., Musiani, D., Sestili, P., and Cantoni, O. (1996)Apoptosis of human lymphocytes in the absence or presence ofinternucleosomal DNA cleavage. Biochem. Biophys. Res. Commun.229, 910–915
41. Hamilton, R. F., Jr., Li, L., Felder, T. B., and Holian, A. (1995)Bleomycin induces apoptosis in human alveolar macrophages.Am. J. Physiol. 269, L318–L325
42. Rano, T. A., Timkey, T., Peterson, E. P., Rotonda, J., Nicholson,D. W., Becker, J. W., Chapman, K. T., and Thornberry, N. A.(1997) A combinatorial approach for determining proteasespecificities: application to interleukin-1b converting enzyme(ICE). Chem. Biol. 4, 149–155
43. Hirata, H., Takahashi, A., Kobayashi, S., Yonehara, S., Sawai, H.,Okazaki, T., Yamamoto, K., and Sasada, M. (1998) Caspases areactivated in a branched protease cascade and control distinctdownstream processes in Fas-induced apoptosis. J. Exp. Med.187, 587–600
44. Sawada, H., Kawamura, T., Shimohama, S., Akaike, A., and Kimura,J. (1996) Different mechanisms of glutamate-induced neuronaldeath between dopaminergic and non-dopaminergic neurons inrat mesencephalic culture. J. Neurosci. Res. 43, 503–510
45. Sawada, H., Shimohama, S., Kawamura, T., Akaike, A., Kita-mura, Y., Taniguchi, T., and Kimura, J. (1996) Mechanism ofresistance to NO-induced neurotoxicity in cultured rat dopami-nergic neurons. J. Neurosci. Res. 46, 509–518
46. Sawada, H., Ibi, M., Kihara, T., Urushitani, M., Akaike, A.,Kimura, J., and Shimohama, S. (1998) Dopamine D2-typeagonists protect mesencephalic neurons from glutamate neuro-toxicity: mechanisms of neuroprotective treatment against oxi-dative stress. Ann. Neurol. 44, 110–119
47. Bayne, K. (1996) Revised Guide for the Care and Use ofLaboratory Animals available. Physiologist 39, 199 208–199
48. Gavrieli, Y., Sherman, Y., and Ben-Sasson, S. A. (1992) Identifi-cation of programmed cell death in situ via specific labeling ofnuclear DNA fragmentation. J. Cell Biol. 119, 493–501
49. Sawada, H., Ibi, M., Kihara, T., Urushitani, M., Nakanishi, M.,Akaike, A., and Shimohama, S. (2000) Neuroprotective mecha-nism of GDNF in mesencephalic neurons. J. Neurochem. 74,1175–1184
50. Charriaut-Marlangue, C., and Ben-Ari, Y. (1995) A cautionarynote on the use of the TUNEL stain to determine apoptosis.NeuroReport 7, 61–64
51. Rink, A., Fung, K. M., Trojanowski, J. Q., Lee, V. M., Neuge-bauer, E., and McIntosh, T. K. (1995) Evidence of apoptotic celldeath after experimental traumatic brain injury in the rat. Am. J.Pathol. 147, 1575–1583
52. Grasl-Kraupp, B., Ruttkay-Nedecky, B., Koudelka, H., Bukowska,K., Bursch, W., and Schulte-Hermann, R. (1995) In situ detec-tion of fragmented DNA (TUNEL assay) fails to discriminateamong apoptosis, necrosis, and autolytic cell death: a cautionarynote. Hepatology 21, 1465–1468
53. Saraste, A. (1999) Morphologic criteria and detection of apo-ptosis. Herz 24, 189–195
54. Sengpiel, B., Preis, E., Krieglstein, J., and Prehn, J. H. (1998)NMDA-induced superoxide production and neurotoxicity in
cultured rat hippocampal neurons: role of mitochondria. Eur.J. Neurosci. 10, 1903–1910
55. Tan, S., Sagara, Y., Liu, Y., Maher, P., and Schubert, D. (1998)The regulation of reactive oxygen species production duringprogrammed cell death. J. Cell Biol. 141, 1423–1432
56. Matsura, T., Kai, M., Fujii, Y., Ito, H., and Yamada, K. (1999)Hydrogen peroxide-induced apoptosis in HL-60 cells requirescaspase-3 activation. Free Rad. Res. 30, 73–83
57. Enari, M., Talanian, R. V., Wong, W. W., and Nagata, S. (1996)Sequential activation of ICE-like and CPP32-like proteases dur-ing Fas-mediated apoptosis. Nature (London) 380, 723–726
58. Ishikawa, Y., Yokoo, T., and Kitamura, M. (1997) c-Jun/AP-1,but not NF-k B, is a mediator for oxidant-initiated apoptosis inglomerular mesangial cells. Biochem. Biophys. Res. Commun. 240,496–501
59. Amato, S. F., Swart, J. M., Berg, M., Wanebo, H. J., Mehta, S. R.,and Chiles, T. C. (1998) Transient stimulation of the c-Jun-NH2-terminal kinase/activator protein 1 pathway and inhibition ofextracellular signal-regulated kinase are early effects in paclitax-el-mediated apoptosis in human B lymphoblasts. Cancer Res. 58,241–247
60. Liebermann, D. A., Gregory, B., and Hoffman, B. (1998) AP-1(Fos/Jun) transcription factors in hematopoietic differentiationand apoptosis. Int. J. Oncol. 12, 685–700
61. Jacobs-Helber, S. M., Wickrema, A., Birrer, M. J., and Sawyer,S. T. (1998) AP1 regulation of proliferation and initiation ofapoptosis in erythropoietin-dependent erythroid cells. Mol. Cell.Biol. 18, 3699–3707
62. Watabe, M., Ito, K., Masuda, Y., Nakajo, S., and Nakaya, K.(1998) Activation of AP-1 is required for bufalin-induced apo-ptosis in human leukemia U937 cells. Oncogene 16, 779–787
63. Srivastava, R. K., Sollott, S. J., Khan, L., Hansford, R., Lakatta,E. G., and Longo, D. L. (1999) Bcl-2 and Bcl-X(L) blockthapsigargin-induced nitric oxide generation, c-Jun NH(2)-terminal kinase activity, and apoptosis. Mol. Cell. Biol. 19,5659–5674
64. Singer, C. A., Rogers, K. L., and Dorsa, D. M. (1998) Modulationof Bcl-2 expression: a potential component of estrogen protec-tion in NT2 neurons. NeuroReport 9, 2565–2568
65. Garcia-Segura, L. M., Cardona-Gomez, P., Naftolin, F., andChowen, J. A. (1998) Estradiol upregulates Bcl-2 expression inadult brain neurons. NeuroReport 9, 593–597
66. Dubal, D. B., Shughrue, P. J., Wilson, M. E., Merchenthaler, I.,and Wise, P. M. (1999) Estradiol modulates bcl-2 in cerebralischemia: a potential role for estrogen receptors. J. Neurosci. 19,6385–6393
67. Barkhem, T., Carlsson, B., Nilsson, Y., Enmark, E., Gustafsson,J., and Nilsson, S. (1998) Differential response of estrogenreceptor a and estrogen receptor b to partial estrogen agonists/antagonists. Mol. Pharmacol. 54, 105–112
68. Arnold, S. F., and Notides, A. C. (1995) An antiestrogen: aphophotyrosyl peptide that blocks dimerization of the humanestrogen receptor. Proc. Natl. Acad. Sci. USA 92, 7475–7479
69. D’Mello, S. R., Kuan, C.-Y., Flavell, R. A., and Rakic, P. Caspase-3is required for apoptosis-associated DNA fragmentation but notfor cell death in neurons deprived of potassium. J. Neurosci. Res.In press
70. Pike, B. R., Zhao, X., Newcomb, J. K., Wang, K. K., Posmantur,R. M., and Hayes, R. L. (1998) Temporal relationships betweende novo protein synthesis, calpain and caspase 3-like proteaseactivation, and DNA fragmentation during apoptosis in septo-hippocampal cultures. J. Neurosci. Res. 52, 505–520
71. Green, P. S., Bishop, J., and Simpkins, J. W. (1997) 17 a-Estra-diol exerts neuroprotective effects on SK-N-SH cells. J. Neurosci.17, 511–515
72. Clark, J. H., Schrader, W. T., and O’Malley, B. W. (1992)Mechanisms of action of steroid hormones. In Textbook ofEndocrinology (Wilson, J., and Foster, D. W., eds) pp 35–90, W.B.Saunders Company, Philadelphia
Received for publication April 7, 1999.Revised for publication January 10, 2000.
1214 Vol. 14 June 2000 SAWADA ET AL.The FASEB Journal