mechanism and kinetics of the spontaneous thermal copolymerization of styrene/maleic anhydride....
TRANSCRIPT
Full Paper
222
Mechanism and Kinetics of the SpontaneousThermal Copolymerization of Styrene/MaleicAnhydride. Experimental and SimulationStudies in the Presence of 4-oxo-TEMPOa
Josue D. Mota-Morales, Iraıs Quintero-Ortega, Enrique Saldıvar-Guerra,*Gabriel Luna-Barcenas, Martha Albores-Velasco, Judith Percino,Vıctor Chapela, Miguel A. Ocampo
J. D. Mota-Morales, I. Quintero-Ortega, G. Luna-BarcenasCinvestav Queretaro, Libramiento Norponiente No. 2000, Fracc,Real de Juriquilla, Queretaro, Qro. 76230, MexicoE. Saldıvar-GuerraCentro de Investigacion en Quımica Aplicada (CIQA), Blvd. EnriqueReyna No. 140, Saltillo Coah. 25253, MexicoE-mail: [email protected]. Albores-VelascoFacultad de Quımica, Universidad Nacional Autonoma de Mexico,CU Coyoacan 04510, Mexico DF, MexicoJ. Percino, V. ChapelaLaboratorio de Polımeros, Benemerita Universidad Autonoma dePuebla, Puebla, MexicoM. A. OcampoCentro de Fısica Aplicada y Tecnologıa Avanzada, UniversidadNacional Autonoma de Mexico, Queretaro, Qro 76230, MexicoaMost of this work was performed during a stay of J. Mota-Morales at CIQA.
The mechanism and kinetics of the spontaneous copolymerization of styrene (S) and maleicanhydride (MA) in the presence of 4-oxo-TEMPO nitroxide (N) were studied. Experiments wereperformed at 125 8C varying the S/MA and the N/S ratios and the evolution of conversion wasmeasured by dilatometry up to 20% conversion.Clean induction periods or severe retardation inthe initial stage of the reaction were observed.From a proposed kinetic mechanism amathemat-ical model was built, which was used for fittingthe relevant kinetic constants for the self-initiation steps, and the ratio kp=k
1=2
t . The modelperforms well in certain concentration regimes,but it remains a challenge to completely under-stand this complex system in other concentrationregimes.
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Introduction
The commercial production of styrene (S)/maleic anhydride
(MA) copolymers is based in a mature technology; however
and despite its commercial importance, it is remarkable
that the corresponding reaction mechanism and kinetics,
especially those of spontaneous initiation, are poorly
understood and have not been studied in detail yet.
Copolymers of S and MA are produced by bulk
polymerization in a process similar to that used for the
production of crystal and impact grade polystyrene. The MA
content of these polymers typically ranges from 7 to 15%.
From public information made available by commercial
companies, we estimate that the annual world consumption
DOI: 10.1002/mren.200900061

Mechanism and Kinetics of the Spontaneous Thermal . . .
of S/MA in the 2 000–2 010 decade is in the order of 50–70 kt
per year. On the other hand, several groups have recently
reported the commercial use of the nitroxide mediated
radical polymerization (NMRP) in industry, in applications
oriented to high added value specialties, such as dispersants
and compatibilizers, some of them involving the copoly-
merization of S/MA.[1–3] In the last cases the mechanism of
autoinitiation becomes even more complicated due to the
influence that the nitroxide radical has on the spontaneous
generation of radicals that can initiate reaction.
Various mechanisms have been proposed to explain the
initiation process of the spontaneous copolymerization of S
with electron acceptor monomers such as MA and
acrylonitrile. The copolymerization of S with electron
acceptor monomers has been the subject of extensive
mechanistic discussions.[4,5]
With regard to the autoinitiation process, these mechan-
isms are based on those proposed for the self-initiated S
homopolymerization. In the case of S itself, two competing
initiation mechanisms have been proposed: i.e., the Mayo
and Flory mechanisms. In the Mayo mechanism,[6] initial
Diels Alder reaction [p4sþ p2s] between two molecules of S
leads the semibenzene dimer to the so-called Mayo dimer
form. This dimerization is followed by a hydrogen atom
abstraction by another S molecule to form two initiating
monoradicals. Detection of an accompanying cycload-
duct,[7] and some mechanistic behavior when acid cata-
lysts[8] are present, supports the Mayo mechanism for S. In
an earlier mechanism proposed by Flory for S spontaneous
polymerization,[9] the two monomer molecules generate a
tetramethylene diradical intermediate by bond formation
between two b-carbons. This diradical may actually ring-
close to form either a Mayo dimer, which can then transfer
hydrogen and initiate free radical polymerization, or the
1,2-diphenylcyclobutane derivative, a species that is
inactive to polymerization.[9] Isolation of a small
amount of 1,2-diphenylcyclobutane-1-phenyltetralin and
1-phenyl-1,2-dihydronaphthalene supports the Flory
mechanism for S to a minor competing extent.[10–12]
Hall and Padias[4] propose that in a copolymerization
between electron-rich monomers and electron-poor mono-
mers, the tetramethylene diradical formed may initiate
spontaneous polymerizations, either by an anionic (via
zwitterion tetramethylene) or free radical mechanism (via
tetramethylene diradical). In addition, from radical-
trapping experiments, several authors have concluded that
the tetramethylene diradical plays and intermediating role
in alternating copolymerizations, including the S and MA
case, and is the key element to understand their experi-
mental results.[13–16]
Part of the problem in studying the mechanism and
kinetics of S/MA copolymerization lies in the difficulty of
separating the initiation and propagation steps. Kothe and
Fischer[17] first used induction period experiments in the
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
self-initiated S polymerization in the presence of TEMPO, in
order to estimate kinetic coefficients associated with the
self-initiation reactions. In this way they first estimated the
value of the kinetic coefficient for S dimerization (see
Scheme 1) which is the first step of the radical self-
generation mechanism. This concept was further extended
by Bonilla-Cruz et al.[18] who used nitroxide radicals in order
to separate the initiation and propagation steps in the self
initiated S/MA copolymerization, and managed to provide
a preliminary order of magnitude estimate of the rate
constant for the dimerization of S and MA, assuming the
existence in this system of a reaction homologous to the S
dimerization for the formation of the Mayo dimer. An
interesting feature of the autoinitiation mechanism in the
case of the S/MA copolymerization in the presence of a
nitroxide radical is that, even for small amounts of MA in
the system, the induction period is dramatically reduced by
about one order of magnitude in comparison with the
nitroxide mediated S autopolymerization at 120–125 8C.[18]
From the scientific and technological viewpoint the
understanding and confirmation of mechanisms present in
the traditional and controlled radical (e.g., nitroxide-
mediated) copolymerization of S/MA, and the estimation
of the associated kinetic constants, are important.
In this work we undertake an experimental and modeling
study of the initial stages of the S/MA copolymerization in
order to provide a better understanding of the mechanism
and kinetics of this system of industrial and scientific
relevance. To this end, we synthesized copolymers of S and
MA by thermal autoinitiation in the presence and absence of
a nitroxide radical in an industrially relevant wide range of
S/MA and S/nitroxide ratios at 125 8C. Conversion rates in the
initial reaction stages were measured up to 20% conversion.
From these experiments, clean induction periods or severe
retardation behavior of the initial polymerization stages
wereobtained and then analyzed and discussed with the aim
of extracting mechanistic and kinetic information. A
mathematical model was developed to facilitate a better
understanding of the kinetic mechanism for this polymer-
ization process. The analysis and simulation of the experi-
mental data indicate the convenience of separating the
study of this system in different regimes that depend upon
the nitroxide concentration. In this study, with the help of
the mathematical model and simulations, we evaluate the
rate constant values for some of the proposed reactions in an
intermediate concentration regime. The extension of the
model to other concentration regimes, which may require
the inclusion of additional reactions not fully understood yet,
remains a challenge.
Experimental Part
In order to obtain kinetic data we performed thermal polymeriza-
tion reactions for the system S/MA in the presence or absence of
www.mre-journal.de 223

J. D. Mota-Morales et al.
Scheme 1. Mechanism proposed for the copolymerization of S/MA in the presence of 4-oxo-TEMPO.
224
4-oxo-2,2,6,6-tetramethylpiperidine-1-oxyl (4-oxo-TEMPO) in a
capillary dilatometer. The volume of the capillary bulb used is
5 mL; the capillary tube has a 2.2 mm diameter and a length of
10 cm. Fresh distilled S, recently sublimated MA and N,N’-
dimethylformamide (DMF) were put in a capillary in the presence
of 4-oxo-TEMPO (N) or without it and heated at 125 8C, after oxygen
evacuation from the capillary by bubbling the solutions with
high purity argon gas and sealing. Measurements of the induction
period and the conversion versus time curve after the induction
period were done according to literature.[19] DMF was used in
order to achieve complete dissolution of the MA in the reaction
media; without the presence of DMF the solubility of MA in S is
limited to about 3–5 wt.-%. Since the amount of DMF used in
the experiments is very small, the experiments are in practical
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
terms very similar to bulk polymeriza-
tions and therefore in the following we
refer to the reactions as bulk experi-
ments. Different compositions of the
pair S/MA and of the nitroxide mixture
were tested. In each experiment
the mixture initially increased its
volume by thermal expansion until
thermal equilibrium was established,
and then the zero-time point was
marked and the volume contraction
of the reaction mixture with time was
recorded and correlated with conver-
sion via standard calculations that use
the density of the monomer mixture
and the polymer.
In the capillary, the conversion in a
time increment can be calculated as
follows:
1
Dt
D M½ �M½ � ¼
1
Dt
DV=V0
d2 � d1ð Þ=d2(1)
whereD[M] is the monomer concentra-
tion change in a time increment Dt, DV
is the corresponding change of volume,
and V0 is the initial volume; d1 and d2
are the monomer and polymer density,
respectively. The fractional volume
change at total conversion is (d2� d1)/
d2, and DV/V0 is the fractional volume
change at time t. The ratio of these two
quantities is equivalent to the mono-
mer conversion at time t. Most of the
dilatometric experiments were run in
triplicate.
The induction period is estimated by
linear extrapolation of the non-zero
conversion-time data until they cross
the time axis. In those cases where
some data points exist with very small
conversions just before a well-defined
change of slope marking the end of the
induction period, this is estimated as
the crossing point of the two lines with different slopes.
Modeling
Kinetic Mechanism
Scheme 1 includes the main reactions in a plausible
mechanism of nitroxide-mediated radical thermal poly-
merization of S/MA.[18] In the mechanism proposed, the
homopolymerization of S may in principle compete with
the alternating polymerization of S with MA. Also, the
autoinitiation may arise either from the dimerization
reaction between the S and MA, either in a concerted way or
DOI: 10.1002/mren.200900061

Mechanism and Kinetics of the Spontaneous Thermal . . .
not, or between two S molecules (based on the Mayo
mechanism). The dimer 8 can further react with more
monomer to form initiating radicals in the absence of a
nitroxide radical (Scheme 1 path A), or undergo a faster
hydrogen abstraction by a 4-oxo-TEMPO molecule
(Scheme 1 path B), in the same way as dimer 3 (paths C
and D).
Paths A and B have been proposed by Bonilla et al.[18] by
analogy with the spontaneous initiation mechanism that
seems to be occurring in S thermal homopolymerization in
presence or absence of a nitroxide radical,[20] and provided
spectroscopic evidence of the formation of the S–MA dimer
8. Connolly and Scaiano[21] (see Scheme 2) proposed
the direct addition of nitroxide to an S molecule
generating another propagating radical in autothermal
homopolymerization at very large concentrations of
TEMPO (50:50 mol/mol S/TEMPO). The rest of the reactions:
propagation, reversible capping of growing radicals by 4-
oxo-TEMPO, and irreversible termination, are well estab-
lished for this system.
Reaction Kinetics and Mathematical Model
The mechanism described in detail in Scheme 1 and 2 is
written in a simpler way in Table 1. Based on this
representation and doing the material balances for a batch
reactor one ends up with a set of ordinary differential
equations (see Appendix) that describe the evolution of the
species in the system. In order to simplify the mathematical
model we lumped together the primary radicals and the
growing polymeric radicals of Scheme 1, independently of
their chain length. Although the kinetic scheme can in
principle describe the molecular weight distribution, the
study of this feature is out of the scope of the present work
and therefore we decided to omit the description of the
chain length of the growing radicals. However, the
mathematical model contains enough detail to simulate
the autoinitiation mechanisms in Scheme 1 and the
Scheme 2. Radical generation mechanism by the direct addition of4-oxo-TEMPO to the monomers in the system S/MA.
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
occurrence of an induction period in the presence of
the nitroxide radical. On the other hand, due to the
incomplete understanding of the propagation step, we
modeled this as a pseudo-homopolymerization using an
effective propagation rate constant kp and the total
concentration of monomer and radicals, independently of
their detailed chemical nature as follows:
Rp ¼ kp M½ � R½ � (2)
where Rp is the overall propagation rate (or polymerization
rate), [M] the total monomer concentration, and [P] is the
total polymeric radical concentration. Based on abundant
empirical evidence,[22] we assumed that in the presence of
MA in the system, the propagation occurs in an alternate
fashion consuming one molecule of each one of the
monomers in two consecutive propagation steps:
R�n þ 2M �!Kp
R�nþ2
We have also confirmed experimentally that the S/MA
system produces an alternate copolymer[18] in the presence
of TEMPO derivatives; in addition, we have previously
modeled this copolymerization by using the terminal
model with reactivity ratios (close to zero) available in
the literature;[23] therefore, we do not consider necessary to
model the system as a copolymerization. This would have
unnecessarily complicated the modeling and would
have introduced additional parameters which would make
the parameter estimation task more difficult.
The reaction of Scheme 2 was assumed to occur only for
nitroxide concentrations higher than 0.1%, by analogy with
the findings of Connolly and Scaiano. No quasi-steady state
(QSS) assumption was made for the radical species and the
set of differential equations was solved by the DDASSL
algorithm which can handle systems of stiff differential
equations as well as differential/algebraic systems.[24]
Results and Discussion
Table 2 shows the experimental design that was performed;
this is essentially a 32 factorial in the variables nitroxide and
MA concentration, with additional points to explore
specific effects. In order to verify the reproducibility of
the dilatometric technique we ran two sets of experiments
using this method and then compared the results with
another experiment in which the conversion was measured
by gravimetry at the same conditions. The comparison of
these techniques can be observed in Figure 1, which shows
excellent reproducibility for the dilatometric technique and
very good agreement with the gravimetric determination.
From the triplicate runs of the dilatometric technique, an
estimation of the standard deviation was made for every
www.mre-journal.de 225
J. D. Mota-Morales et al.
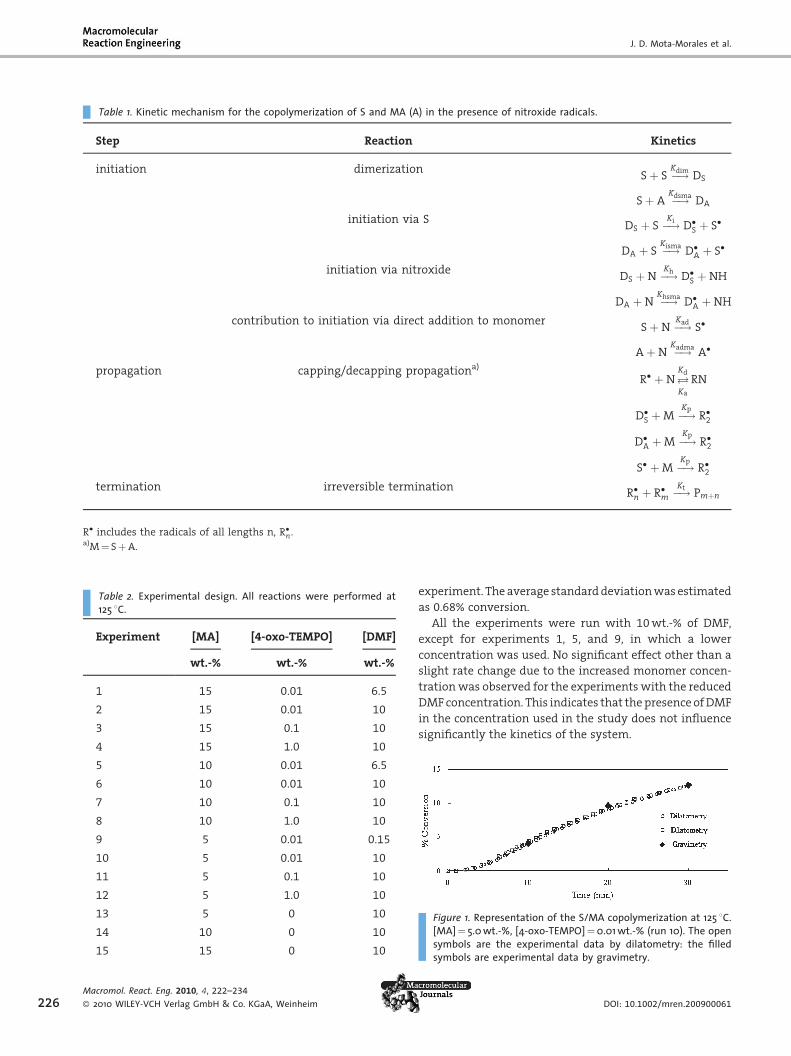
Table 1. Kinetic mechanism for the copolymerization of S and MA (A) in the presence of nitroxide radicals.
Step Reaction Kinetics
initiation dimerizationSþ S �!
KdimDS
Sþ A �!Kdsma
DA
initiation via SDS þ S �!Ki
D�S þ S�
DA þ S �!Kisma
D�A þ S�
initiation via nitroxideDS þN �!
KhD�S þNH
DA þN �!Khsma
D�A þNH
contribution to initiation via direct addition to monomerSþN �!
KadS�
AþN �!Kadma
A�
propagation capping/decapping propagationa)
R� þ N !Kd
Ka
RN
D�S þM �!Kp
R�2
D�A þM �!Kp
R�2
S� þM �!Kp
R�2
termination irreversible terminationR�n þ R�m �!
KtPmþn
R� includes the radicals of all lengths n, R�n.a)M¼ SþA.
Table 2. Experimental design. All reactions were performed at125 8C.
Experiment [MA] [4-oxo-TEMPO] [DMF]
wt.-% wt.-% wt.-%
1 15 0.01 6.5
2 15 0.01 10
3 15 0.1 10
4 15 1.0 10
5 10 0.01 6.5
6 10 0.01 10
7 10 0.1 10
8 10 1.0 10
9 5 0.01 0.15
10 5 0.01 10
11 5 0.1 10
12 5 1.0 10
13 5 0 10
14 10 0 10
15 15 0 10
226Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
experiment. The average standard deviation was estimated
as 0.68% conversion.
All the experiments were run with 10 wt.-% of DMF,
except for experiments 1, 5, and 9, in which a lower
concentration was used. No significant effect other than a
slight rate change due to the increased monomer concen-
tration was observed for the experiments with the reduced
DMF concentration. This indicates that the presence of DMF
in the concentration used in the study does not influence
significantly the kinetics of the system.
Figure 1. Representation of the S/MA copolymerization at 125 8C.[MA]¼ 5.0 wt.-%, [4-oxo-TEMPO]¼0.01 wt.-% (run 10). The opensymbols are the experimental data by dilatometry: the filledsymbols are experimental data by gravimetry.
DOI: 10.1002/mren.200900061
Mechanism and Kinetics of the Spontaneous Thermal . . .
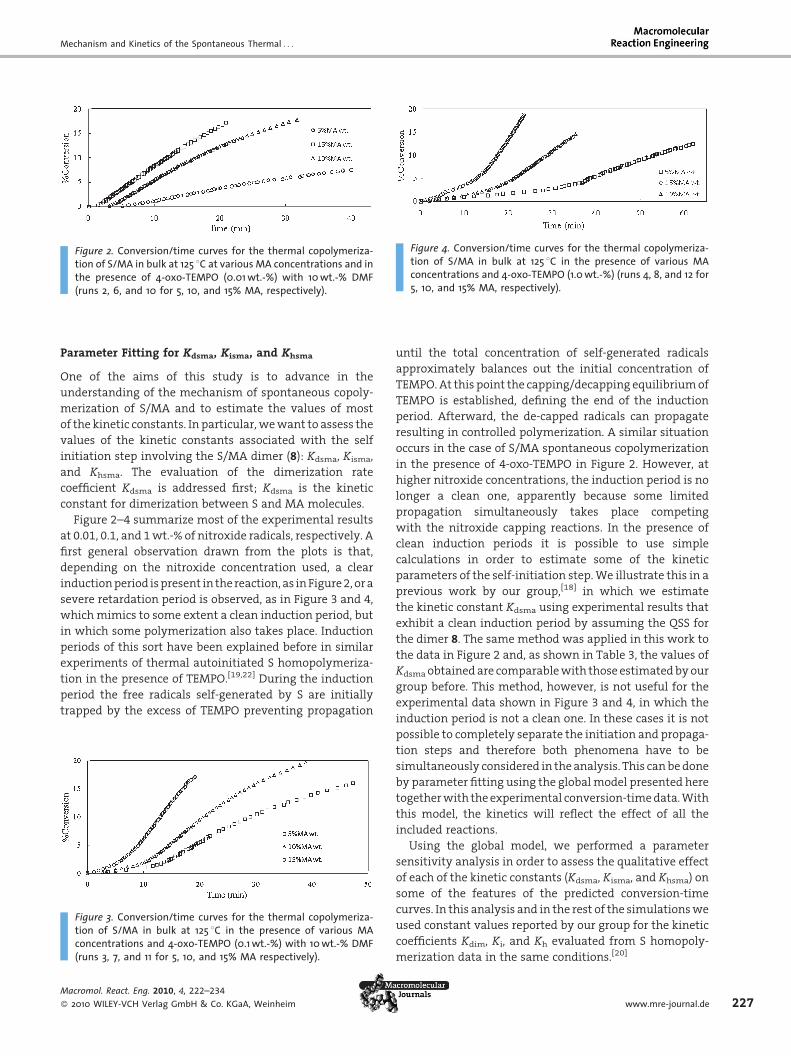
Figure 2. Conversion/time curves for the thermal copolymeriza-tion of S/MA in bulk at 125 8C at various MA concentrations and inthe presence of 4-oxo-TEMPO (0.01 wt.-%) with 10 wt.-% DMF(runs 2, 6, and 10 for 5, 10, and 15% MA, respectively).
Figure 4. Conversion/time curves for the thermal copolymeriza-tion of S/MA in bulk at 125 8C in the presence of various MAconcentrations and 4-oxo-TEMPO (1.0 wt.-%) (runs 4, 8, and 12 for5, 10, and 15% MA, respectively).
Parameter Fitting for Kdsma, Kisma, and Khsma
One of the aims of this study is to advance in the
understanding of the mechanism of spontaneous copoly-
merization of S/MA and to estimate the values of most
of the kinetic constants. In particular, we want to assess the
values of the kinetic constants associated with the self
initiation step involving the S/MA dimer (8): Kdsma, Kisma,
and Khsma. The evaluation of the dimerization rate
coefficient Kdsma is addressed first; Kdsma is the kinetic
constant for dimerization between S and MA molecules.
Figure 2–4 summarize most of the experimental results
at 0.01, 0.1, and 1 wt.-% of nitroxide radicals, respectively. A
first general observation drawn from the plots is that,
depending on the nitroxide concentration used, a clear
induction period is present in the reaction, as in Figure 2, or a
severe retardation period is observed, as in Figure 3 and 4,
which mimics to some extent a clean induction period, but
in which some polymerization also takes place. Induction
periods of this sort have been explained before in similar
experiments of thermal autoinitiated S homopolymeriza-
tion in the presence of TEMPO.[19,22] During the induction
period the free radicals self-generated by S are initially
trapped by the excess of TEMPO preventing propagation
Figure 3. Conversion/time curves for the thermal copolymeriza-tion of S/MA in bulk at 125 8C in the presence of various MAconcentrations and 4-oxo-TEMPO (0.1 wt.-%) with 10 wt.-% DMF(runs 3, 7, and 11 for 5, 10, and 15% MA respectively).
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
until the total concentration of self-generated radicals
approximately balances out the initial concentration of
TEMPO. At this point the capping/decapping equilibrium of
TEMPO is established, defining the end of the induction
period. Afterward, the de-capped radicals can propagate
resulting in controlled polymerization. A similar situation
occurs in the case of S/MA spontaneous copolymerization
in the presence of 4-oxo-TEMPO in Figure 2. However, at
higher nitroxide concentrations, the induction period is no
longer a clean one, apparently because some limited
propagation simultaneously takes place competing
with the nitroxide capping reactions. In the presence of
clean induction periods it is possible to use simple
calculations in order to estimate some of the kinetic
parameters of the self-initiation step. We illustrate this in a
previous work by our group,[18] in which we estimate
the kinetic constant Kdsma using experimental results that
exhibit a clean induction period by assuming the QSS for
the dimer 8. The same method was applied in this work to
the data in Figure 2 and, as shown in Table 3, the values of
Kdsma obtained are comparable with those estimated by our
group before. This method, however, is not useful for the
experimental data shown in Figure 3 and 4, in which the
induction period is not a clean one. In these cases it is not
possible to completely separate the initiation and propaga-
tion steps and therefore both phenomena have to be
simultaneously considered in the analysis. This can be done
by parameter fitting using the global model presented here
together with the experimental conversion-time data. With
this model, the kinetics will reflect the effect of all the
included reactions.
Using the global model, we performed a parameter
sensitivity analysis in order to assess the qualitative effect
of each of the kinetic constants (Kdsma, Kisma, and Khsma) on
some of the features of the predicted conversion-time
curves. In this analysis and in the rest of the simulations we
used constant values reported by our group for the kinetic
coefficients Kdim, Ki, and Kh evaluated from S homopoly-
merization data in the same conditions.[20]
www.mre-journal.de 227
J. D. Mota-Morales et al.
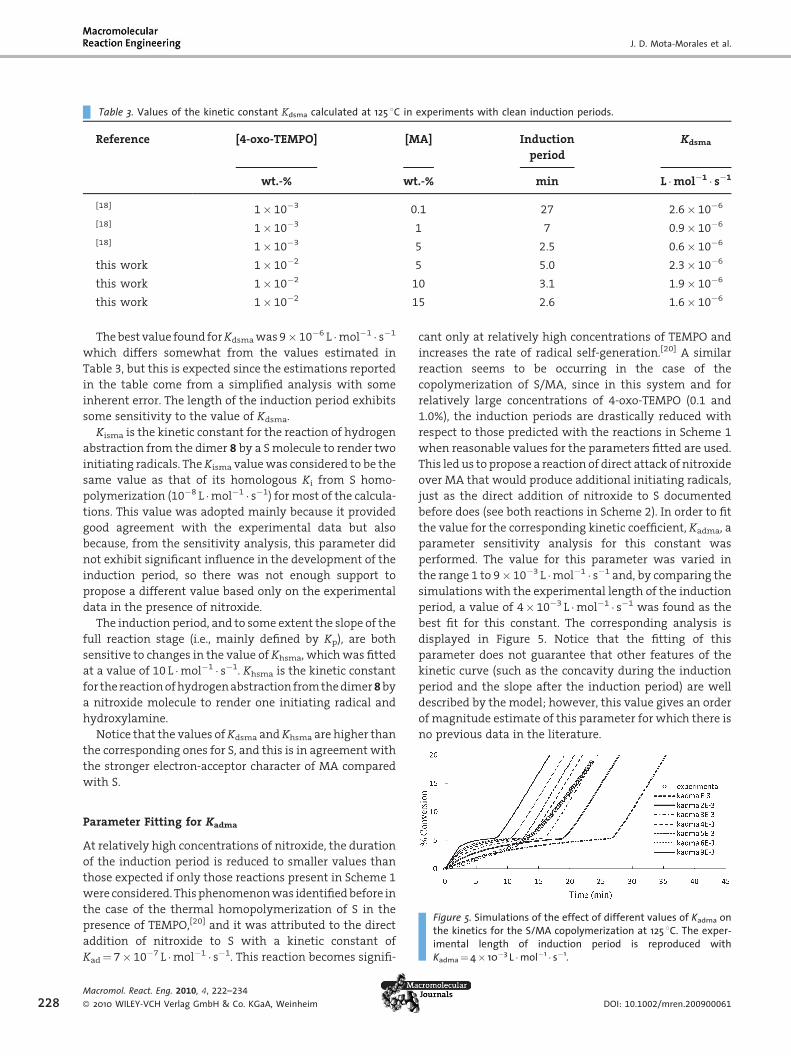
Table 3. Values of the kinetic constant Kdsma calculated at 125 8C in experiments with clean induction periods.
Reference [4-oxo-TEMPO] [MA] Induction
period
Kdsma
wt.-% wt.-% min L �mol�1 � s�1
[18] 1� 10�3 0.1 27 2.6� 10�6
[18] 1� 10�3 1 7 0.9� 10�6
[18] 1� 10�3 5 2.5 0.6� 10�6
this work 1� 10�2 5 5.0 2.3� 10�6
this work 1� 10�2 10 3.1 1.9� 10�6
this work 1� 10�2 15 2.6 1.6� 10�6
228
The best value found forKdsma was 9� 10�6 L �mol�1 � s�1
which differs somewhat from the values estimated in
Table 3, but this is expected since the estimations reported
in the table come from a simplified analysis with some
inherent error. The length of the induction period exhibits
some sensitivity to the value of Kdsma.
Kisma is the kinetic constant for the reaction of hydrogen
abstraction from the dimer 8 by a S molecule to render two
initiating radicals. The Kisma value was considered to be the
same value as that of its homologous Ki from S homo-
polymerization (10�8 L �mol�1 � s�1) for most of the calcula-
tions. This value was adopted mainly because it provided
good agreement with the experimental data but also
because, from the sensitivity analysis, this parameter did
not exhibit significant influence in the development of the
induction period, so there was not enough support to
propose a different value based only on the experimental
data in the presence of nitroxide.
The induction period, and to some extent the slope of the
full reaction stage (i.e., mainly defined by Kp), are both
sensitive to changes in the value of Khsma, which was fitted
at a value of 10 L �mol�1 � s�1. Khsma is the kinetic constant
for the reaction of hydrogen abstraction from the dimer8by
a nitroxide molecule to render one initiating radical and
hydroxylamine.
Notice that the values of Kdsma and Khsma are higher than
the corresponding ones for S, and this is in agreement with
the stronger electron-acceptor character of MA compared
with S.
Figure 5. Simulations of the effect of different values of Kadma onthe kinetics for the S/MA copolymerization at 125 8C. The exper-imental length of induction period is reproduced withKadma¼4� 10�3 L �mol�1 � s�1.
Parameter Fitting for Kadma
At relatively high concentrations of nitroxide, the duration
of the induction period is reduced to smaller values than
those expected if only those reactions present in Scheme 1
were considered. This phenomenon was identified before in
the case of the thermal homopolymerization of S in the
presence of TEMPO,[20] and it was attributed to the direct
addition of nitroxide to S with a kinetic constant of
Kad¼ 7� 10�7 L �mol�1 � s�1. This reaction becomes signifi-
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
cant only at relatively high concentrations of TEMPO and
increases the rate of radical self-generation.[20] A similar
reaction seems to be occurring in the case of the
copolymerization of S/MA, since in this system and for
relatively large concentrations of 4-oxo-TEMPO (0.1 and
1.0%), the induction periods are drastically reduced with
respect to those predicted with the reactions in Scheme 1
when reasonable values for the parameters fitted are used.
This led us to propose a reaction of direct attack of nitroxide
over MA that would produce additional initiating radicals,
just as the direct addition of nitroxide to S documented
before does (see both reactions in Scheme 2). In order to fit
the value for the corresponding kinetic coefficient, Kadma, a
parameter sensitivity analysis for this constant was
performed. The value for this parameter was varied in
the range 1 to 9� 10�3 L �mol�1 � s�1 and, by comparing the
simulations with the experimental length of the induction
period, a value of 4� 10�3 L �mol�1 � s�1 was found as the
best fit for this constant. The corresponding analysis is
displayed in Figure 5. Notice that the fitting of this
parameter does not guarantee that other features of the
kinetic curve (such as the concavity during the induction
period and the slope after the induction period) are well
described by the model; however, this value gives an order
of magnitude estimate of this parameter for which there is
no previous data in the literature.
DOI: 10.1002/mren.200900061
Mechanism and Kinetics of the Spontaneous Thermal . . .
0
5
10
15
20
25
4035302520151050
Time (min)
% C
onve
rsio
n
5%AM
Model 5% MA
10%Am
Model 10% MA
15%AM
Model 15% MA
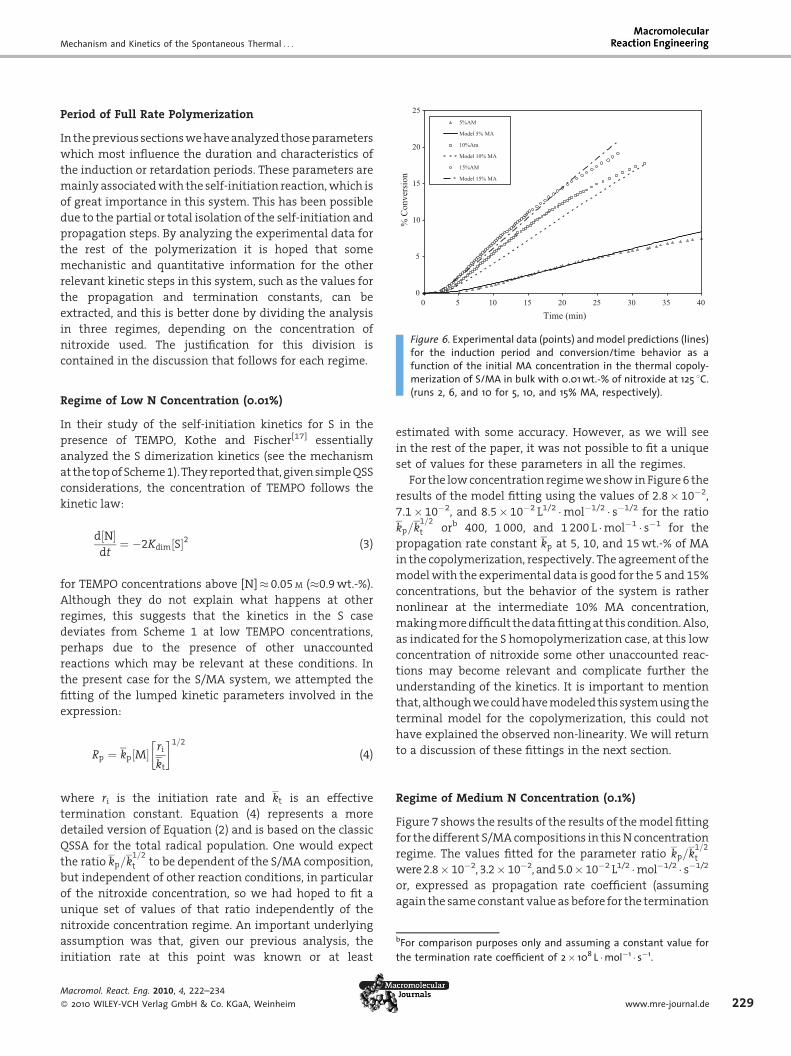
Figure 6. Experimental data (points) and model predictions (lines)for the induction period and conversion/time behavior as afunction of the initial MA concentration in the thermal copoly-
Period of Full Rate Polymerization
In the previous sections we have analyzed those parameters
which most influence the duration and characteristics of
the induction or retardation periods. These parameters are
mainly associated with the self-initiation reaction, which is
of great importance in this system. This has been possible
due to the partial or total isolation of the self-initiation and
propagation steps. By analyzing the experimental data for
the rest of the polymerization it is hoped that some
mechanistic and quantitative information for the other
relevant kinetic steps in this system, such as the values for
the propagation and termination constants, can be
extracted, and this is better done by dividing the analysis
in three regimes, depending on the concentration of
nitroxide used. The justification for this division is
contained in the discussion that follows for each regime.
merization of S/MA in bulk with 0.01 wt.-% of nitroxide at 125 8C.(runs 2, 6, and 10 for 5, 10, and 15% MA, respectively).
Regime of Low N Concentration (0.01%)
In their study of the self-initiation kinetics for S in the
presence of TEMPO, Kothe and Fischer[17] essentially
analyzed the S dimerization kinetics (see the mechanism
at the top of Scheme 1). They reported that, given simple QSS
considerations, the concentration of TEMPO follows the
kinetic law:
Macrom
� 2010
d N½ �dt¼ �2Kdim S½ �2 (3)
for TEMPO concentrations above [N]� 0.05 M (�0.9 wt.-%).
Although they do not explain what happens at other
regimes, this suggests that the kinetics in the S case
deviates from Scheme 1 at low TEMPO concentrations,
perhaps due to the presence of other unaccounted
reactions which may be relevant at these conditions. In
the present case for the S/MA system, we attempted the
fitting of the lumped kinetic parameters involved in the
expression:
Rp ¼ kp M½ � ri
kt
� �1=2
(4)
bFor comparison purposes only and assuming a constant value forthe termination rate coefficient of 2� 108 L �mol�1 � s�1.
where ri is the initiation rate and kt is an effective
termination constant. Equation (4) represents a more
detailed version of Equation (2) and is based on the classic
QSSA for the total radical population. One would expect
the ratio kp=k1=2
t to be dependent of the S/MA composition,
but independent of other reaction conditions, in particular
of the nitroxide concentration, so we had hoped to fit a
unique set of values of that ratio independently of the
nitroxide concentration regime. An important underlying
assumption was that, given our previous analysis, the
initiation rate at this point was known or at least
ol. React. Eng. 2010, 4, 222–234
WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
estimated with some accuracy. However, as we will see
in the rest of the paper, it was not possible to fit a unique
set of values for these parameters in all the regimes.
For the low concentration regime we show in Figure 6 the
results of the model fitting using the values of 2.8� 10�2,
7.1� 10�2, and 8.5� 10�2 L1/2 �mol�1/2 � s�1/2 for the ratio
kp=k1=2
t orb 400, 1 000, and 1 200 L �mol�1 � s�1 for the
propagation rate constant kp at 5, 10, and 15 wt.-% of MA
in the copolymerization, respectively. The agreement of the
model with the experimental data is good for the 5 and 15%
concentrations, but the behavior of the system is rather
nonlinear at the intermediate 10% MA concentration,
making more difficult the data fitting at this condition. Also,
as indicated for the S homopolymerization case, at this low
concentration of nitroxide some other unaccounted reac-
tions may become relevant and complicate further the
understanding of the kinetics. It is important to mention
that, although we could have modeled this system using the
terminal model for the copolymerization, this could not
have explained the observed non-linearity. We will return
to a discussion of these fittings in the next section.
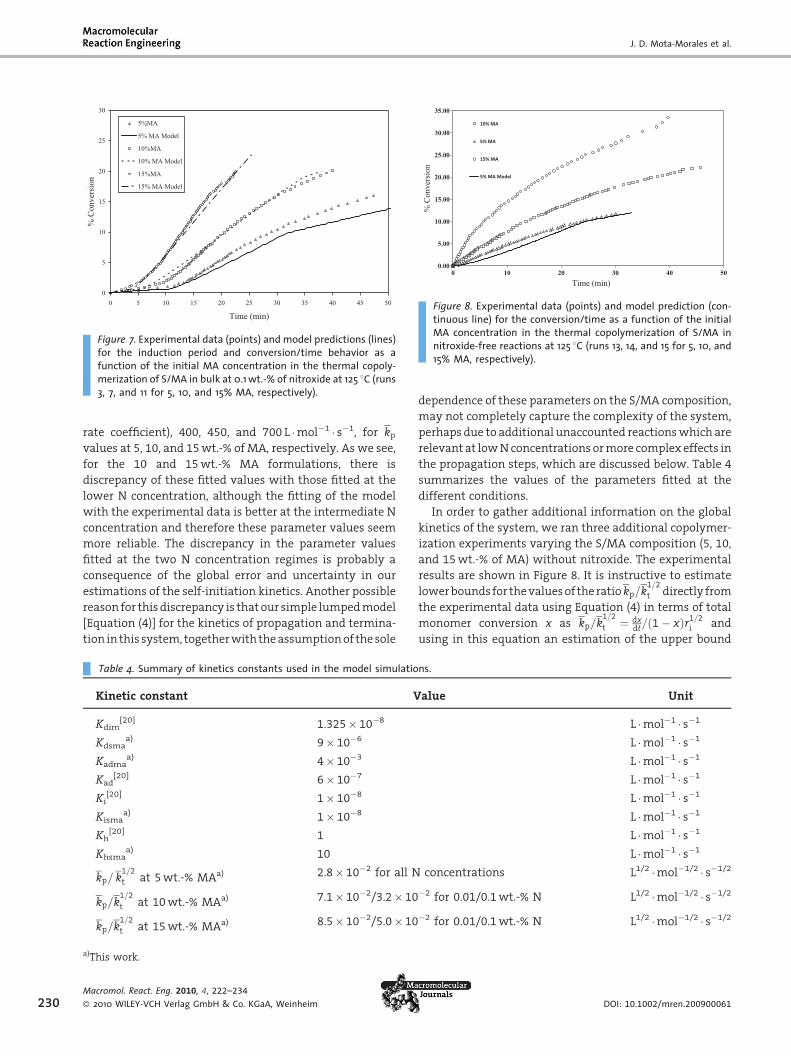
Regime of Medium N Concentration (0.1%)
Figure 7 shows the results of the results of the model fitting
for the different S/MA compositions in this N concentration
regime. The values fitted for the parameter ratio kp=k1=2
t
were 2.8� 10�2, 3.2� 10�2, and 5.0� 10�2 L1/2 �mol�1/2 � s�1/2
or, expressed as propagation rate coefficient (assuming
again the same constant value as before for the termination
www.mre-journal.de 229
J. D. Mota-Morales et al.
0
5
10
15
20
25
30
50454035302520151050
Time (min)
% C
onve
rsio
n
5%MA
5% MA Model
10%MA
10% MA Model
15%MA
15% MA Model
Figure 7. Experimental data (points) and model predictions (lines)for the induction period and conversion/time behavior as afunction of the initial MA concentration in the thermal copoly-merization of S/MA in bulk at 0.1 wt.-% of nitroxide at 125 8C (runs3, 7, and 11 for 5, 10, and 15% MA, respectively).
0.00
5.00
10.00
15.00
20.00
25.00
30.00
35.00
50403020100Time (min)
% C
onve
rsio
n
10% MA
5% MA
15% MA
5% MA Model
Figure 8. Experimental data (points) and model prediction (con-tinuous line) for the conversion/time as a function of the initialMA concentration in the thermal copolymerization of S/MA innitroxide-free reactions at 125 8C (runs 13, 14, and 15 for 5, 10, and15% MA, respectively).
230
rate coefficient), 400, 450, and 700 L �mol�1 � s�1, for kp
values at 5, 10, and 15 wt.-% of MA, respectively. As we see,
for the 10 and 15 wt.-% MA formulations, there is
discrepancy of these fitted values with those fitted at the
lower N concentration, although the fitting of the model
with the experimental data is better at the intermediate N
concentration and therefore these parameter values seem
more reliable. The discrepancy in the parameter values
fitted at the two N concentration regimes is probably a
consequence of the global error and uncertainty in our
estimations of the self-initiation kinetics. Another possible
reason for this discrepancy is that our simple lumped model
[Equation (4)] for the kinetics of propagation and termina-
tion in this system, together with the assumption of the sole
Table 4. Summary of kinetics constants used in the model simulatio
Kinetic constant V
Kdim[20] 1.325� 10�8
Kdsmaa) 9� 10�6
Kadmaa) 4� 10�3
Kad[20] 6� 10�7
Ki[20] 1� 10�8
Kismaa) 1� 10�8
Kh[20] 1
Khsmaa) 10
kp= k1=2
t at 5 wt.-% MAa) 2.8� 10�2 for all N
kp=k1=2
t at 10 wt.-% MAa) 7.1� 10�2/3.2� 10
kp=k1=2
t at 15 wt.-% MAa) 8.5� 10�2/5.0� 10
a)This work.
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
dependence of these parameters on the S/MA composition,
may not completely capture the complexity of the system,
perhaps due to additional unaccounted reactions which are
relevant at low N concentrations or more complex effects in
the propagation steps, which are discussed below. Table 4
summarizes the values of the parameters fitted at the
different conditions.
In order to gather additional information on the global
kinetics of the system, we ran three additional copolymer-
ization experiments varying the S/MA composition (5, 10,
and 15 wt.-% of MA) without nitroxide. The experimental
results are shown in Figure 8. It is instructive to estimate
lower bounds for the values of the ratiokp=k1=2
t directly from
the experimental data using Equation (4) in terms of total
monomer conversion x as kp=k1=2
t ¼ dxdt= 1� xð Þr1=2
i and
using in this equation an estimation of the upper bound
ns.
alue Unit
L �mol�1 � s�1
L �mol�1 � s�1
L �mol�1 � s�1
L �mol�1 � s�1
L �mol�1 � s�1
L �mol�1 � s�1
L �mol�1 � s�1
L �mol�1 � s�1
concentrations L1/2 �mol�1/2 � s�1/2
�2 for 0.01/0.1 wt.-% N L1/2 �mol�1/2 � s�1/2
�2 for 0.01/0.1 wt.-% N L1/2 �mol�1/2 � s�1/2
DOI: 10.1002/mren.200900061
Mechanism and Kinetics of the Spontaneous Thermal . . .
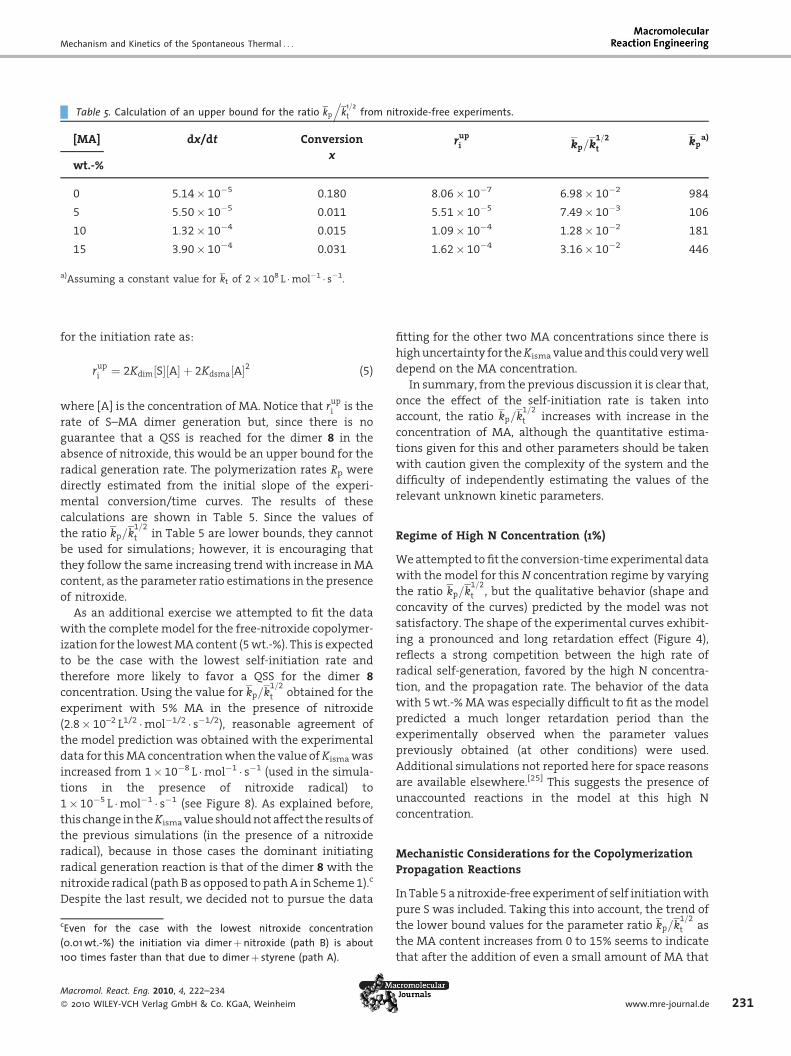
Table 5. Calculation of an upper bound for the ratio kp
.k1=2
t from nitroxide-free experiments.
[MA] dx/dt Conversion
xrupi kp=k
1=2
tkp
a)
wt.-%
0 5.14� 10�5 0.180 8.06� 10�7 6.98� 10�2 984
5 5.50� 10�5 0.011 5.51� 10�5 7.49� 10�3 106
10 1.32� 10�4 0.015 1.09� 10�4 1.28� 10�2 181
15 3.90� 10�4 0.031 1.62� 10�4 3.16� 10�2 446
a)Assuming a constant value for kt of 2� 108 L �mol�1 � s�1.
for the initiation rate as:
cEven(0.01 w100 tim
Macrom
� 2010
rupi ¼ 2Kdim S½ � A½ � þ 2Kdsma A½ �2 (5)
where [A] is the concentration of MA. Notice that rupi is the
rate of S–MA dimer generation but, since there is no
guarantee that a QSS is reached for the dimer 8 in the
absence of nitroxide, this would be an upper bound for the
radical generation rate. The polymerization rates Rp were
directly estimated from the initial slope of the experi-
mental conversion/time curves. The results of these
calculations are shown in Table 5. Since the values of
the ratio kp=k1=2
t in Table 5 are lower bounds, they cannot
be used for simulations; however, it is encouraging that
they follow the same increasing trend with increase in MA
content, as the parameter ratio estimations in the presence
of nitroxide.
As an additional exercise we attempted to fit the data
with the complete model for the free-nitroxide copolymer-
ization for the lowest MA content (5 wt.-%). This is expected
to be the case with the lowest self-initiation rate and
therefore more likely to favor a QSS for the dimer 8
concentration. Using the value for kp=k1=2
t obtained for the
experiment with 5% MA in the presence of nitroxide
(2.8� 10–2 L1/2 �mol�1/2 � s�1/2), reasonable agreement of
the model prediction was obtained with the experimental
data for this MA concentration when the value of Kisma was
increased from 1� 10�8 L �mol�1 � s�1 (used in the simula-
tions in the presence of nitroxide radical) to
1� 10�5 L �mol�1 � s�1 (see Figure 8). As explained before,
this change in theKisma value should not affect the results of
the previous simulations (in the presence of a nitroxide
radical), because in those cases the dominant initiating
radical generation reaction is that of the dimer 8 with the
nitroxide radical (path B as opposed to path A in Scheme 1).c
Despite the last result, we decided not to pursue the data
for the case with the lowest nitroxide concentrationt.-%) the initiation via dimerþnitroxide (path B) is aboutes faster than that due to dimerþ styrene (path A).
ol. React. Eng. 2010, 4, 222–234
WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
fitting for the other two MA concentrations since there is
high uncertainty for theKisma value and this could very well
depend on the MA concentration.
In summary, from the previous discussion it is clear that,
once the effect of the self-initiation rate is taken into
account, the ratio kp=k1=2
t increases with increase in the
concentration of MA, although the quantitative estima-
tions given for this and other parameters should be taken
with caution given the complexity of the system and the
difficulty of independently estimating the values of the
relevant unknown kinetic parameters.
Regime of High N Concentration (1%)
We attempted to fit the conversion-time experimental data
with the model for this N concentration regime by varying
the ratio kp=k1=2
t , but the qualitative behavior (shape and
concavity of the curves) predicted by the model was not
satisfactory. The shape of the experimental curves exhibit-
ing a pronounced and long retardation effect (Figure 4),
reflects a strong competition between the high rate of
radical self-generation, favored by the high N concentra-
tion, and the propagation rate. The behavior of the data
with 5 wt.-% MA was especially difficult to fit as the model
predicted a much longer retardation period than the
experimentally observed when the parameter values
previously obtained (at other conditions) were used.
Additional simulations not reported here for space reasons
are available elsewhere.[25] This suggests the presence of
unaccounted reactions in the model at this high N
concentration.
Mechanistic Considerations for the CopolymerizationPropagation Reactions
In Table 5 a nitroxide-free experiment of self initiation with
pure S was included. Taking this into account, the trend of
the lower bound values for the parameter ratio kp=k1=2
t as
the MA content increases from 0 to 15% seems to indicate
that after the addition of even a small amount of MA that
www.mre-journal.de 231

J. D. Mota-Morales et al.
Scheme 4. Mechanism for the cross-propagation steps in the S/MA copolymerization.
232
parameter ratio decreases with respect to the value
corresponding to the S homopolymerization case, and then
increases again as the MA concentration increases. This is
confirmed by comparing the values of the parameter ratio
kp=k1=2
t fitted for the experiments with 0.1 wt.-% N (see
Table 4) with the value known for S homopolymerization.
For this last system the ratio kp=k1=2
t at 125 8C can be
calculated as 0.125 L1/2 �mol�1/2 � s–1/2 using kinetic con-
stants taken from the literature and measured via pulsed
laser polymerization (PLP) experiments. When 5 wt.-% of
MA is used in the reaction, the value of the ratio kp=k1=2
t
drops to 2.8� 10�2 and then increases up to
5.0� 10�2 L1/2 �mol�1/2 � s�1/2 as the MA proportion is
increased up to 15%. These calculations seem to indicate
that after the addition of even a small amount of MA the
propagation mechanism drastically changes. The old
literature ascribed these mechanistic changes to the
intermediation of a charge transfer complex; more recent
studies[4,5] discard this hypothesis and explain related
experimental observations through the influence of a
tetramethylene diradical which is formed by the reaction of
S and MA molecules (see Scheme 3). According to these
authors this diradical acts as an intermediate in the
propagation reaction and is responsible for the alternating
character of the copolymer formed. Their theory has been
generalized for pairs or monomers having electron donor-
acceptor characteristics of which the S–MA system is an
example.
Radical stability considerations may also play a role in
the propagation reaction. The cross-propagation in the
S/MA system can be represented by the two reactions in
Scheme 4. Reaction 1 in Scheme 4 generates a more stable
radical than that produced in reaction 2 (the ionization
energy for obtaining the MA radical is around 414 kJ �mol�1
while the corresponding energy of the benzylic radical is
around 330 kJ �mol�1). Since the benzylic radical is more
stable it is also less reactive, therefore reaction 2 is slower,
the energy necessary to reach the transition state is larger
and as a consequence this reaction is the rate controlling
step in the propagation process. The former discussion
assumes that the propagation has alternating character and
Scheme 3. Mechanism for the formation of a diradical that mayinfluence the self-initiation and propagation steps in the S/MAcopolymerization.
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
that homo-propagation is negligible, which turns out to be
an experimental fact in the case of the S/MA system. Finally,
since the rate of a reaction depends on the concentration of
reactants, as the MA concentration increases the rate of
reaction 2 increases too, leading to an overall faster
polymerization.
With respect to the preference of this system for
alternating propagation over homopolymerization (at least
S homopolymerization), Fischer and Radom[26] conclude for
model systems that in general this behavior is due to the
difference in the energy barrier for the two reaction paths
with the path having the lower energy barrier being the
most favored one. This is due to a complex combination of
factors including the enthalpy of the reaction, charge
transfer during the reaction, steric effect contributions and,
finally, the electrophilic or nucleophilic character of the
substituents attached to the reacting group.
Conclusion
A detailed kinetic study of the NMR autopolymerization of
S/MA at 125 8C was performed in order to make an initial
assessment of the values of the kinetic parameters for the
self-initiation and propagation steps in this system. Care-
fully executed dilatometric experiments provided kinetic
data in the range of 5–15 wt.-% of MA at various
concentrations of the nitroxide radical (4-oxo-TEMPO).
Depending on the nitroxide concentration, two types of
induction period were observed: one that is a true induction
period and another one that is rather a severe retardation
rate period in which propagation and radical-capping
reactions compete.
A kinetic mechanism previously proposed by our group
was used as the basis for a kinetic model aimed at
describing the kinetics of NMR copolymerization of S/MA.
From the experimental data and simulations with the
model, estimates for the values of the relevant self-
initiation and propagation parameters, for which there is
very little information available in the literature, were
DOI: 10.1002/mren.200900061

Mechanism and Kinetics of the Spontaneous Thermal . . .
provided. However, given the complexity of the system and
the uncertainty associated with the mechanism, the
numerical values should be taken with caution and as a
first approximation to the rate constant values for the
proposed reactions. The model based in the mechanism
proposed reproduces well the experimental data at
different MA contents at an intermediate nitroxide
concentration (0.1 wt.-%), but for other concentration
regimes, in general, some deviations of the model with
the experiment are present, which suggests that the
proposed kinetic mechanism may be overlooking some
reactions that become relevant in those concentration
regimes. Remarkably, however, the experimental data for
all the reactions with an MA concentration of 5 wt.-% are
reasonably well reproduced by the model at all the
nitroxide concentrations below or equal to 0.1 wt.-%,
including the nitroxide-free experiment, using a single
set of kinetic parameter values. The separation of initiation
and propagation steps by the use of radical trapping
experiments is promissory for mechanistic studies of this
system, but it is recommendable to try other families of
radical trapping species as a way to improve the separation
of the kinetic steps. A similar method can be followed to
study the mechanism and kinetics of other relevant self-
initiating copolymerizations involving an electron-donor
and an electron-acceptor monomer, such as the system S/
acrylonitrile.
To our knowledge this is the first attempt at measuring
the relevant self-initiation and propagation/termination
kinetic coefficients for the S/MA system. More experi-
mental work is required to get a better understanding of the
mechanisms and kinetics of the copolymerization of S/MA.
Appendix: Mathematical Model
d S½ �dt¼ Kdim S½ �2�Kdsma S½ � A½ � � Ki DS½ � S½ � � Kisma DA½ � S½ �
� Kp G�½ � S½ � þ A½ �ð Þ
d A½ �dt¼ Kdsma S½ � A½ � � Kp G�½ � S½ � þ A½ �ð Þ
d DS½ �dt¼ Kdim S½ �2�Ki DS½ � S½ � � Kh DS½ � N½ �
d DA½ �dt¼ Kdsma S½ � A½ � � Kisma DA½ � S½ � � Khsma DA½ � N½ �
d N½ �dt¼ Kh DS½ � N½ � � Khsma DA½ � N½ � � Kd R�½ � N½ � þ Ka RN½ �
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
d RN½ � �
dt¼ Ka RN½ � þ Kd R½ � N½ �d R�½ �þ G�½ �ð Þdt
¼2Ki DS½ � S½ �þ 2Kisma DA½ � S½ �þ Khsma DA½ � N½ �
þKh DS½ � N½ � þ Ka RN½ � � Kd½ � R�½ � N½ � � Kt R�½ �2
For 1% N:
d S½ �dt¼ Kad S½ � N½ �
� Kdim S½ �2�Kdsma S½ � A½ � � Ki DS½ � S½ � � Kisma DA½ � S½ �
� Kp G�½ � S½ � þ A½ �ð Þ
d A½ �dt¼ �Kadma A½ � N½ � � Kdsma S½ � A½ � � Kp G�½ � S½ � þ A½ �ð Þ
d G�½ �dt¼ �Kad S½ � N½ � � Kadma A½ � N½ � þ 2Ki DS½ � S½ � þ 2Kisma DA½ �
� S½ � þ Khsma DA½ � N½ � þ Kh DS½ � N½ � � Kp G�½ �M½ �
d R½ � þ G½ �ð Þdt
¼ �Kad S½ � N½ � � Kadma A½ � N½ � þ 2Ki DS½ � S½ �
þ 2Kisma DA½ � S½ � þ Khsma DA½ � N½ � þ Kh DS½ � N½ �
þ Ka RN½ � � Kd R�½ � N½ � � Kt R�½ �2
Acknowledgements: Financial support from the National Councilfor Science and Technology of Mexico (CONACyT) through grant2004-46048 is gratefully acknowledged. Josue Mota acknowledgesthe M. Sci. scholarship and his present scholarship for PhD studiesfrom CONACyT. One of us (GLB) acknowledges support fromCONACyT grants 58239 & 78798.
Received: October 2, 2009; Revised: December 22, 2009;DOI: 10.1002/mren.200900061
Keywords: copolymerization; kinetics; 4-oxo-TEMPO; sponta-neous polymerization; styrene/maleic anhydride
[1] A. Gonzalez, E. Saldıvar, E. Fernandez, G. Zacahua, ‘‘A NovelStyrene-Maleic Anhydride Block-Copolymer Compatibilizer’’,in: Additives 2004 Conference, Tampa, Florida 2004.
[2] US 7 323 528 B2 (2008), invs.: E. Saldıvar-Guerra, A. Gonzalez-Montiel.
[3] F. O. H. Pirrung, C. Aushra, Polym. Prepr. 2005, 46, 316.[4] H. K. Hall, Jr., A. B. Padias, J. Polym. Sci., Part A: Polym. Chem.
2001, 39, 2069.[5] H. K. Hall, Jr., A. B. Padias, Helv. Chim. Acta 2005, 88, 1553.[6] F. R. Mayo, J. Am. Chem. Soc. 1953, 75, 6133.[7] W. Choi, X. Han, H. K. Hall, Jr., Polym. Bull. 2005, 54, 179.
www.mre-journal.de 233

J. D. Mota-Morales et al.
234
[8] W. C. Buzanowsky, J. D. Graham, D. B. Priddy, E. Shero,Polymer 1992, 33, 3055.
[9] P. J. Flory, J. Am. Chem. Soc. 1937, 59, 241.[10] V. J. Kurze, D. J. Stein, K. P. Syima, R. Kaiser, Angew. Makromol.
Chem. 1970, 12, 25.[11] W. G. Brown, Makromol. Chem. 1969, 128, 130.[12] G. Moad, E. Rizzardo, D. H. Solomon, Polym. Bull. 1982, 6, 589.[13] L. Eberson, O. Persson, Acta Chem. Scand. 1999, 53, 680.[14] L. Eberson, O. Persson, Macromolecules 2000, 33, 2021.[15] R. Huisgen, Acc. Chem. Res. 1977, 10, 199.[16] T. Sato, M. Abe, T. Otsu, J. Macromol. Sci. Chem. 1981, A15, 367.[17] T. Kothe, H. Fischer, J. Polym. Sci., Part A: Polym. Chem. 2001,
39, 4009.[18] J. Bonilla-Cruz, L. Caballero, M. Albores-Velasco, E. Saldıvar-
Guerra, J. Percino, V. Chapela, Macromol. Symp. 2007, 248,132.
[19] M. J. Percino, V. M. Chapela, A. Jimenez, J. Appl. Polym. Sci.2004, 94, 1662.
Macromol. React. Eng. 2010, 4, 222–234
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
[20] E. Saldıvar-Guerra, J. Bonilla, G. Zacahua, M. Albores-Velasco,J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 6962.
[21] T. J. Connolly, J. C. Scaiano, Tetrahedron Lett. 1997, 38, 1133.[22] [22a] E. Tsuchida, T. Tomono, Die Makromol. Chem. 1971, 141,
265; [22b] W. Zeng, Y. Shirota, Macromolecules 1989, 22, 11;[22c] M.-Q. Zhu, L.-H. Wei, F.-S. Du, Z.-C. Li, F.-M. Li, M. Li, L.Jiang, Chem. Commun. 2001, 4, 365; [22d] J.-F. Lutz, B. Kirci, K.Matyjaszewski, Macromolecules 2003, 36, 3136; [22e] D. Brau,F. Hu, Prog. Polym. Sci. 2006, 31, 239.
[23] C. Guerrero-Sanchez, E. Saldıvar, M. Hernandez, A. Jimenez,Polym. React. Eng. 2003, 11, 457.
[24] K. E. Brennan, S. L. Campbell, L. R. Petzold, ‘‘Numerical Solutionof Initial-Value Problems in Differential-Algebraic Equations’’,Elsevier Science Publishing Company, New York 1989.
[25] J. Mota-Morales, M. Sc. Thesis, Centro de Investigacion yEstudios Avanzados (CINVESTAV) Queretaro, Mexico 2009.
[26] H. Fischer, L. Radom, Angew. Chem., Int. Ed. 2001, 40,1340.
DOI: 10.1002/mren.200900061