late transition metal complexes bearing functionalized n
TRANSCRIPT

Late Transition Metal Complexes Bearing Functionalized N-Heterocyclic Carbenes
and the Catalytic Hydrogenation of Polar Double Bonds
by
Wylie Wing Nien O
A thesis submitted in conformity with the requirements
for the degree of Doctor of Philosophy
Graduate Department of Chemistry
University of Toronto
© Copyright by Wylie Wing Nien O 2012

iiLATE TRANSITION METALS COMPLEXES BEARING FUNCTIONALIZED
N-HETETROCYCLIC CARBENES AND THE CATALYTIC HYDROGENATION OF POLAR
DOUBLE BONDS
Wylie Wing Nien O
Doctor of Philosophy
Department of Chemistry
University of Toronto
2012
Abstract
Late transition metal complexes of silver(I), rhodium(I), ruthenium(II), palladium(II) and
platinum(II) containing a nitrile-functionalized N-heterocyclic carbene ligand (C–CN) were
prepared. The nitrile group on the C–CN ligand was shown to undergo hydrolysis under basic
conditions, leading to a silver(I) carbene complex with a primary-amido functional group, and a
trimetallic complex of palladium(II) with a partially hydrolyzed C–N–N–C donor ligand.
The reduction of a nitrile-functionalized imidazolium salt in the presence of nickel(II)
chloride under mild conditions yielded an axially chiral square-planar nickel(II) complex
containing a unique primary-amino functionalized N-heterocyclic carbene ligand (C–NH2). A
transmetalation reaction moved this chelating C–NH2 ligand from nickel(II) to ruthenium(II),
osmium(II), and iridium(III), yielding important catalysts for the hydrogenation of polar double
bonds.
The ruthenium(II) complex, [Ru(p-cymene)(C–NH2)Cl]PF6 catalyzed the transfer and H2-
hydrogenation of ketones. The bifunctional hydride complex, [Ru(p-cymene)(C–NH2)H]PF6,
which contains a Ru–H/N–H couple showed no activity under catalytic conditions unless when
activated by a base. The outer-sphere mechanism involving bifunctional catalysis of ketone
reduction is disfavored according to experimental and theoretical studies and an inner-sphere

iiimechanism is proposed involving the decoordination of the amine donor from the C–NH2 ligand.
The ruthenium(II) complex [RuCp*(C–NH2)py]PF6 showed higher activity than the
iridium(III) complex [IrCp*(C–NH2)Cl]PF6 in the hydrogenation of ketones. This ruthenium(II)
complex also catalyzes the hydrogenation of an aromatic ester, a ketimine, and the
hydrogenolysis of styrene oxide. We proposed an alcohol-assisted outer sphere bifunctional
mechanism for both systems based on experimental findings and theoretical calculations. The
cationic iridium(III) hydride complex, [IrCp*(C–NH2)H]PF6 , was prepared and this failed to react
with a ketone in the absence of base. The crucial role of the alkoxide base was demonstrated in
the activation of this hydride complex in catalysis. Calculations support the proposal that the
base deprotonates the amine group of this hydride complex and triggers the migration of the
hydride to the η5-Cp* ring producing a neutral iridium(I) amido complex. This system contains an
active Ir–H/N–H couple required for the outer sphere hydrogenation of ketones in the
bifunctional mechanism.

ivAcknowledgements
I would like to thank first my supervisor, Professor Bob Morris. He is the kindest and the
most helpful person that I have ever met. At times he was busy with other duties in the
department, he could still come up with brilliant ideas to aid research, and spent quality times
with me for the betterment of my professional development. His encouragement can never be
forgotten.
Many thanks to all of the Morris group members, past and present. The joys and talks we
shared and the fruitful discussions we had are memorable and treasurable. They have made my
life in the lab “more interesting”, and a very enjoyable experience. Special thanks to the
undergraduate students Ali Rizvi and Hisashi Ohara for their dedicated efforts to this research
project.
More thanks go to the Department of Chemistry at the University of Toronto and all of the
supporting staff for their help for the past four years. In particular, I would like to thank Dr. Alan
Lough for his kind help and expertise in X-ray crystallography. He is a great person to work
with.
Last but not least, I would like to thank my family and all of my friends for the support, care
and love for my years at graduate studies, especially for the ups and downs that we went through
together.
I shall remember a quote from my chemistry teacher at high school, “ Chemistry = Chem Is
Try”. Indeed, this is very true!

vTable of Contents
Abstract
Acknowledgements
Table of Contents
List of Figures
List of Schemes
List of Tables
List of Abbreviations
Chapter 1: Introduction
1.1 Homogeneous Catalysis Involving Metal-Dihydrogen and Metal-Hydrides
Complexes
1.1.1 The Metal-Dihydrogen Bond.
1.1.2 The Acid-base Reactivity of Metal Hydride and Metal Dihydrogen
Complexes.
1.1.3 The Heterolytic Splitting of H2 at a Transition Metal Center and Implications
for Catalysis.
1.1.4 Metal Hydrides in Homogeneous Hydrogenation Reactions.
1.2 Mechanisms of the Hydrogenation of Polar Double Bonds
1.2.1 Ketone Hydrogenation, the Outer-sphere Mechanism and the “NH Effect”.
1.2.2 Effect of Alcohols on the Outer-sphere Bifunctional Mechanism using the
“NH Effect”.
1.2.3 Concerted or Stepwise Transfer of a Proton/Hydride Couple from the Metal
Center to the Polar Double bond.
ii
iv
v
xvi
xxviii
xxxi
xxxiii
1
1
1
2
3
5
7
7
9
10

vi1.2.4 The Inner Sphere Mechanism.
1.3 Applications of Phosphines and N-Heterocyclic Carbenes in Catalysis
1.3.1 Applications of Phosphines in Catalysis.
1.3.2 N-heterocyclic carbenes and their Late Transition Metal Complexes.
1.3.3 Donor-functionalized N-heterocyclic carbenes and applications in catalysis.
1.4 Thesis Goals
Chapter 2: Synthesis and Characterization of Nitrile-Functionalized N-Heterocyclic
Carbenes and Their Complexes of Silver(I), Rhodium(I) and Ruthenium(II)
2.1 Abstract
2.2 Introduction
2.3 Results and Discussion
2.3.1 Synthesis of Imidazolium Salt Precursors.
2.3.2 Nitrile Functionalized N-Heterocyclic Carbene Complexes of Silver(I).
2.3.4. Hydrolysis of Nitrile groups on the Silver(I) Complex 2b.
2.3.5. Nitrile Functionalized N-Heterocyclic Carbene Complex of Rhodium(I) and
Ruthenium(II).
2.4 Conclusion
2.5 Experimental Section
2.5.1 General Considerations.
2.5.2. Synthesis of 1-(2-Cyanophenyl)-3-methylimidazolium Tetrafluoroborate
(1a).
2.5.3. Synthesis of 3-(Cyanomethyl)-1-(2-cyanophenyl)imidazolium
Hexafluorophosphate (1b).
11
13
13
14
18
21
24
24
24
28
28
30
34
37
40
41
41
41
42

vii2.5.4. Synthesis of 1-(2-Cyanophenyl)-3-(2-pyridinylmethyl)imidazolium
Hexafluoro-phosphate (1c).
2.5.5. Synthesis of 1-(2-Cyanophenyl)-3-(2-pyridinyl)imidazolium
Hexafluorophosphate (1d).
2.5.6. Synthesis of 1-(2-Cyanophenyl)-3-(1-phenylethyl)imidazolium
Tetrafluoroborate (1e).
2.5.7. Synthesis of 1-(2-(Inden-3-yl)ethyl)-3-(2-cyanophenyl)imidazolium
Tetrafluoro-borate (1f).
2.5.8. Synthesis of Bis[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene]silver(I)
Tetrafluoroborate ([Ag(C–CN)2]BF4, 2a) .
2.5.9. Observation of Silver(I) Complex of (3-(Cyanomethyl)-1-(2-cyanophenyl)-
imidazol-2-ylidene (2b).
2.5.10. Observation of Silver(I) Complex of (3-(Carbomoylmethyl)-1-(2-
cyanophenyl)imidazol-2-ylidene (2e).
2.5.11. Synthesis of Bis{[1-(2-cyanophenyl)-3-(2-pyridinylmethyl)imidazol-2-
ylidene]silver(I)} Hexafluorophosphate (2c).
2.5.12. Synthesis of Bis{[1-(2-cyanophenyl)-3-(2-pyridinyl)imidazol-2-
ylidene]silver(I)} Hexafluorophosphate (2d).
2.5.13. Synthesis of Bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)-(η4-1,5-
cycloctadiene)rhodium(I)] Tetrafluoroborate ([Rh(C–CN)(cod)]2(BF4)2, 3).
2.5.14. Synthesis of Bis[chloro-(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)-
(η6-p-cymene)ruthenium(II)] Tetrafluoroborate ([Ru(p-cymene)(C–CN)Cl]2(BF4)2,
4).
Chapter 3: Palladium(II) and Platinum(II) Complexes Featuring a Nitrile-Functionalized
N-Heterocyclic Carbene Ligand
3.1 Abstract
42
43
43
44
45
45
46
46
46
47
48
49
49

viii3.2 Introduction
3.3 Results and Discussion
3.3.1 Transmetalation of a Nitrile-Functionalized N-heterocyclic Carbene Ligand
from Silver(I) to Palladium(II).
3.3.2 Hydrolysis of Nitrile-Functionalized N-Heterocyclic Carbene Ligands on
Palladium(II) Centers.
3.3.3 Palladium(II) Complex Bearing Nitrile-Functionalized N-Heterocyclic
Carbene (C–CN) and Methoxycyclooctenyl Ligands.
3.3.4 Nitrile-Functionalized N-Heterocyclic Carbene Complex of Platinum(II).
3.3.5 Nucleophilic Attack of Methoxide on the Coordinated 1,5-Cyclo-octadiene
Ligand of 7.
3.3.6 Proposed Reaction Pathways Leading to the Formation of Rotamers A and B
of Complexes 6, 8 and 9.
3.3.7 Platinum(II) Complex with Coordinated 1,3-bis(2,4,6-trimethylphenyl)-
imidazol-2-ylidene ligand (IMes).
3.4 Conclusion
3.5 Experimental Section
3.5.1 General Considerations.
3.5.2 Synthesis of Bis(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)-
palladium(II)(μ-dichloro)bis(acetonitrile)palladium(II) Tetrafluoroborate ([(C–
CN)2Pd(μ-Cl)2Pd(CH3CN)2](BF4)2, 5a).
3.5.3 Synthesis of (Acetonitrile)(1-(2-cyanophenyl)-3-methylimidzol-2-ylidene)
(η1:η2-2-methoxycyclooct-5-enyl)palladium(II) Tetrafluoroborate ([Pd(C–CN)
(η1:η2-coe-OMe)(CH3CN)]BF4, 6a).
3.5.4 Synthesis of Bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-2-
methoxy-cyclooct-5-enyl)palladium(II)] Tetrafluoroborate ([Pd(C–CN)(η1:η2-coe-
50
51
51
53
55
59
61
65
66
68
69
69
70
71

ixOMe)]2(BF4)2, 6b).
3.5.5 Synthesis of Chloro[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene](η4-1,5-
cyclooctadiene)platinum(II) Tetrafluoroborate ([Pt(C–CN)(cod)Cl]BF4, 7).
Hexafluorophosphate (1d).
3.5.6 Synthesis of Chloro[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-
2-methoxy-cyclooct-5-enyl)platinum(II) (Pt(C–CN)(η1:η2-coe-OMe)Cl, 8).
3.5.7 Synthesis of (Acetonitrile)(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)
(η1:η2-2-methoxycyclooct-5-enyl)platinum(II) Tetrafluoroborate ([Pt(C–CN)
(η1:η2-coe-OMe)(CH3CN)]BF4, 9a).
3.5.8 Synthesis of Bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-2-
methoxy-cyclooct-5-enyl)platinum(II)] Tetrafluoroborate ([Pt(C–CN)(η1:η2-coe-
OMe)]2(BF4)2, 9b).
3.5.9 Synthesis of Chloro[1,3-bis-(2,4,6-trimethylphenyl)imidazol-2-ylidene)](η4-
1,5-cyclo-octadiene)platinum(II) Tetrafluoroborate ([Pt(IMes)(cod)Cl]BF4, 11).
Chapter 4: Transmetalation of a Primary Amino-Functionalized N-Heterocyclic Carbene
Ligand from an Axially Chiral Square-Planar Nickel(II) Complex to Ruthenium(II) and
Osmium(II) Catalysts for the Hydrogenation of Polar Bonds
4.1 Abstract
4.2 Introduction
4.3 Results and Discussion
4.3.1 Reduction of a Nitrile-Functionalized Imidazolium Salt to the First
Homoleptic Primary Amino-Functionalized N-Heterocyclic Carbene Complex of
Nickel(II).
4.3.2 Solution NMR Studies of the Axial Chirality of the Homoleptic Primary
Amino-Functionalized N-Heterocyclic Carbene Complex of Nickel(II).
4.3.3 Transmetalation Reaction of a Primary Amino-Functionalized N-
Heterocyclic Carbene from Nickel(II) to Ruthenium(II) and Osmium(II)
71
72
73
74
74
75
77
77
78
79
79
82

xComplexes with an Arene ligand.
4.3.4 Synthesis of a Ruthenium(II) Complex with a Primary Amino-
Functionalized N-Heterocyclic Carbene and Pentamethylcyclopentadienyl ligands.
4.3.5 The Transfer Hydrogenation of Acetophenone Catalyzed by Complexes 12,
13 and 14.
4.3.6 The H2-Hydrogenation of Ketones Catalyzed by Complexes 15 and 16.
4.3.7 The H2-Hydrogenation of Other Polar Bonds Catalyzed by Complex 15.
4.4 Conclusion
4.5 Experimental Section
4.5.1 Synthesis.
4.5.2 Synthesis of Bis[1-(2-aminomethylphenyl)-3-methylimidazol-2-ylidene]-
nickel(II) Hexafluorophosphate ([Ni(C–NH2)2](PF6)2, 12).
4.5.3 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-
chloro(η6-p-cymene)ruthenium(II) Hexafluorophosphate ([Ru(p-cymene)(C–
NH2)Cl]PF6, 13).
4.5.4 Synthesis of [1-(2-(Aminomethyl)phenyl)-3-methylimidazol-2-ylidene]-
chloro(η6-p-cymene)osmium(II) Hexafluorophosphate ([Os(p-cymene)(C–
NH2)Cl]PF6, 14).
4.5.5 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene](η5-
pentamethyl-cyclopentadienyl)(pyridine)ruthenium(II) Hexafluorophosphate
([RuCp*(C–NH2)(py)]PF6, 15).
4.5.6 Synthesis of [2-(Diphenylphosphino)benzylamine](η5-pentamethyl-
cyclopentadienyl)(pyridine)ruthenium(II) Hexafluorophosphate ([RuCp*(P–NH2)
(py)]PF6, 16a).
4.5.7 Catalysis.
84
88
91
94
96
98
99
99
100
101
102
102
103
104

xi4.5.8 General Procedure for Transfer Hydrogenation Studies.
4.5.9 General Procedure for H2-Hydrogenation Studies.
Chapter 5: Mechanistic Investigation of the Hydrogenation of Ketones Catalyzed by a
Ruthenium(II) Complex Featuring an N-Heterocyclic Carbene with a Tethered Primary
Amine Donor: Evidence for an Inner Sphere Mechanism
5.1 Abstract
5.2 Introduction
5.3 Results and Discussion
5.3.1 H2-Hydrogenation of Acetophenone Catalyzed by Complex 13.
5.3.2 H2-Hydrogenation of Other Ketones Catalyzed by Complex 13.
5.3.3 Kinetic Studies.
5.3.4 Effect of the Base on Catalysis.
5.3.5 Effect of Alcohols on Catalysis.
5.3.6 Isotope Effects and Deuterium Labelling Studies.
5.3.7 The Disfavored Outer-Sphere Bifunctional Mechanism.
5.3.8 The Favored Inner-Sphere Mechanism.
5.3.9 Theoretical Considerations: The Outer-sphere Bifunctional Mechanism.
5.3.10 Theoretical Considerations: The Inner-sphere Mechanism.
5.3.11 Possible Mechanism for Transfer Hydrogenation.
5.3.12 Role of Complex 17 in Catalysis.
5.4 Conclusion
5.5 Experimental Section
105
105
106
106
107
110
110
111
113
119
120
120
125
130
133
137
141
142
142
144

xii5.5.1 Synthesis.
5.5.2 Synthesis of [1-(2-(Aminomethyl)phenyl)-3-methylimidazol-2-ylidene]-
hydrido(η6-p-cymene)ruthenium(II) Hexafluorophosphate ([Ru(p-cymene)(C–
NH2)H]PF6, 17).
5.5.3 Synthesis of [1-(N,N-Dimethylaminopropyl)-3-methylimidazol-2-
ylidene]chloro(η6-p-cymene)ruthenium(II) Hexafluorophosphate Dimethyl
Sulfoxide Solvate ([Ru(p-cymene)(C–NMe2)Cl]PF6·1.5 DMSO, 18).
5.5.4 Catalysis.
5.5.5 Kinetics.
5.5.6 Kinetic Isotope Effect Studies.
5.5.7 Computational Details.
Chapter 6: Conventional Bifunctional Mechanism for Ketone Hydrogenation Catalyzed
by Structurally Similar Ruthenium and Iridium Complexes but with Unconventional
Intermediates for Iridium
6.1 Abstract
6.2 Introduction
6.3 Results and Discussion
6.3.1 Synthesis of Ruthenium(II) and Iridium(III) Complexes Containing a C–NH2
Ligand.
6.3.2 Synthesis, Observation, and Reactivity of Hydride Complexes of
Ruthenium(II) and Iridium(III).
6.3.3 Synthesis of an Iridium(III) Complex Containing a C–NMe2 Ligand.
6.3.4 General Features of the H2-Hydrogenation of Ketones Catalyzed by
Complexes 15, 19, 20 and 21.
144
145
146
147
148
148
149
150
150
151
157
157
159
162
163

xiii6.3.5 Effect of Alkoxide Base on the H2-Hydrogenation of Acetophenone
Catalyzed by Complex 19.
6.3.6 The Importance of the NH2 Group in the Iridium(III) System on its Activity
in the Catalytic Hydrogenation of Ketones.
6.3.7 Selectivity in the Hydrogenation of an α,β-unsaturated ketone Catalyzed by
Complexes 15 and 19.
6.3.8 Effect of Alcohol and Other Additives on the H2-Hydrogenation of
Acetophenone Catalyzed by the Ruthenium(II) Complex 15.
6.3.9 Deuterium Labelling Studies Using the Ruthenium(II) Complex 15.
6.3.10 Effect of Alcohol on the H2-Hydrogenation of Acetophenone Catalyzed by
Iridium(III) Complexes 19 and 21.
6.3.11 Deuterium Labelling Studies Using the Iridium(III) Complex 19.
6.3.12 Stoichiometric Reactions Using Complex 19.
6.3.13 The Conventional Non-Alcohol Assisted Outer Sphere Bifunctional
Mechanism.
6.3.14 The Alcohol-Assisted Outer Sphere Bifunctional Mechanism for the
Ruthenium(II) System.
6.3.15 The Alcohol-Assisted Outer Sphere Bifunctional Mechanism Involving
Iridium(I) Intermediates.
6.3.16 Disfavored Inner Sphere Mechanisms Involving Cationic Iridium(III)
Intermediates.
6.3.17 Electronic Properties of Ruthenium(II) Complexes that Relate to their
Reactivity in Catalytic Hydrogenation.
6.4 Conclusion
6.5 Experimental Section
167
168
168
169
171
171
173
174
176
180
182
185
188
191
193

xiv6.5.1 Synthesis.
6.5.2 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-
chloro-(η5-pentamethylcyclopentadienyl)iridium(III) Hexafluorophosphate
([IrCp*(C–NH2)Cl]PF6, 19).
6.5.3 Synthesis of [2-(Diphenylphosphino)benzylamine]-chloro-(η5-pentamethyl-
cyclopentadienyl)iridium(III) Hexafluorophosphate ([IrCp*(P–NH2)Cl]PF6, 20).
6.5.4 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-
hydrido-(η5-pentamethylcyclopentadienyl)iridium(III) Hexafluorophosphate
([IrCp*(C–NH2)H]PF6, 21).
6.5.5 Synthesis of [1-(N,N-Dimethylaminopropyl)-3-methylimidazol-2-ylidene]-
chloro-(η5-pentamethylcyclopentadienyl)iridium(III) Hexafluorophosphate
([IrCp*(C–NMe2)Cl]PF6, 22).
6.5.6 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-
carbonyl-(η5-pentamethylcyclopentadienyl)ruthenium(II) Hexafluorophosphate
([RuCp*(C–NH2) (CO)]PF6, 23).
6.5.7 Synthesis of [2-(Diphenylphosphino)benzylamine]-carbonyl-(η5-
pentamethylcyclopentadienyl)ruthenium(II) Hexafluorophosphate ([RuCp*(P–
NH2)(CO)]PF6, 24).
6.5.8 Representative Stoichiometric Reaction Using High Pressure of H2.
6.5.9 Stoichiometric Reaction Using 19 and 1-Phenylethoxide.
6.5.10 Catalysis.
6.5.11 Computational Details.
Chapter 7: Conclusions and Future Work
7.1 Conclusions
7.2 Future Work
193
194
194
195
196
197
197
198
199
199
201
202
202
206

xvAppendix
Derivation of the Rate Law (eq 5.1, Chapter 5)
Complete Citation for Gaussian 03 and 09 Packages
210
211
214

xviList of Figures
Figure 1.1. Bonding scheme of the η2-H2 ligand on a transition metal center.
Figure 1.2. Reversible proton exchange of a η2-H2 ligand with a pendant amine arm in a
ruthenium(II) complex containing a diphosphine ligand.
Figure 1.3. The catalytic hydrogenation of iminium cations (above) and quinolines
(below) catalyzed by ruthenium(II) and iridium(III) catalysts involving an [M](η2-H2)
intermediate (M = Ru or Ir).
Figure 1.4. Examples of ruthenium(II) complexes with hydride-amine or an amido group
that catalyze the hydrogenation of ketones under base free conditions.
Figure 1.5. The transition state of the alcohol-assisted heterolytic splitting of H2 calculated
by Andersson and co-workers using computational methods.
Figure 1.6. Concerted transfer of a proton/hydride couple from the Shvo's type catalyst to
benzaldehyde.
Figure 1.7. Orbital interaction diagram showing the hydride attack on a σ-bonded ketone.
Figure 1.8. The production of an (S)-N-alkylated aniline (NAA) using an iridium complex
containing a JOSIPHOS ligand as a catalyst.
Figure 1.9. Schematics showing the mesomeric effect to the electronic configuration and
the interactions of π-orbitals in the acyclic, bent singlet carbene N–C–N.
Figure 1.10. The structures of Bertrand's and Arduengo's carbenes.
Figure 1.11. Examples of late transition metal complexes and their use in (a) the oxidation
of alcohols, (b) the hydrosilylation of a terminal alkyne, and in (c) the carboxylation of
aromatic and heteroaromatic C–H bonds (from top to bottom).
Figure 1.12. Structures of some metal complexes containing an amino (NH)-
functionalized NHC ligand.
Figure 2.1. Nitrile-functionalized imidazolium salts 1a-1f.
2
4
5
9
10
11
12
13
15
16
17
21
26

xviiFigure 2.2. Nitrile-functionalized N-heterocyclic carbene (C–CN) complexes of
palladium(II) (left) and nickel(II) (right).
Figure 2.3. ORTEP diagram of 1a depicted with thermal ellipsoids at 30% probability. The
counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and
bond angles (deg): C(1)-N(1), 1.340(3); C(1)-N(2), 1.324(3); C(2)-C(3), 1.342(3); C(11)-
N(3), 1.139(3); N(1)-C(1)-N(2), 108.2(2).
Figure 2.4. ORTEP diagram of 1b depicted with thermal ellipsoids at 30% probability. The
counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and
bond angles (deg): C(1)-N(1), 1.342(5); C(1)-N(2), 1.328(5); C(2)-C(3), 1.349(6); C(12)-
N(4), 1.134(6); C(5)-N(3), 1.142(6); N(1)-C(1)-N(2), 107.8(4).
Figure 2.5. ORTEP diagram of 2a depicted with thermal ellipsoids at 30% probability. The
counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and
bond angles (deg): Ag(1)-C(1), 2.094(5); Ag(1)-C(4), 2.095(5); C(1)-N(1), 1.359(6); C(1)-
N(2), 1.362(6); C(13)-N(5), 1.140(7); C(1)-Ag(1)-C(4), 171.1(2); N(1)-C(1)-N(2),
104.4(4).
Figure 2.6. Proposed structures of Ag(I) complexes 2c and 2d forming 12- and 10
membered rings, respectively.
Figure 2.7. 1H NMR spectra (400 MHz) in the amide-NH region of (a) acetamide, (b)
acetamide prepared from hydrolysis of acetonitrile with silver oxide and water, and (c) 2e
in acetonitrile-d3.
Figure 2.8. ORTEP diagram of 3 depicted with thermal ellipsoids at 30% probability. The
counteranions and hydrogens have been omitted for clarity. Selected bond distances (Å)
and bond angles (deg): Rh(1)-C(16), 2.040(5); Rh(1)-N(1), 2.064(4); Rh(1)-cod(trans to
N)cent, 2.016; Rh(1)-cod(trans to C)cent, 2.100; C(9)-N(1), 1.139(6); C(9)-N(1)-Rh(1),
172.0(5); C(16)-Rh(1)-N(1), 89.9(2); N(1)-Rh(1)-cod(trans to N)avg, 160.6; N(1)-Rh(1)-
cod(cis to N)avg, 91.15; C(16)-Rh(1)-cod(trans to C)avg, 162.1; C(16)-Rh(1)-cod(cis to
C)avg, 92.37.
Figure 2.9. ORTEP diagram of 4 depicted with thermal ellipsoids at 30% probability. The
counteranions and hydrogens have been omitted for clarity. Only one asymmetric unit is
27
30
30
32
33
35
38

xviiishown. Selected bond distances (Å) and bond angles (deg): Ru(1a)-C(1a), 2.078(7);
Ru(1a)-N(3a), 2.028(8); Ru(1a)-Cl(1a), 2.408(2); Ru(1a)-C(12a), 2.238(8); C(11a)-N(3a),
1.13(1); C(11a)-N(3a)-Ru(1a), 173.3(6); C(1a)-Ru(1a)-N(3a), 84.9(3); C(1a)-Ru(1a)-
Cl(1a), 90.9(2); N(3a)-Ru(1a)-Cl(1a), 83.7(2).
Figure 3.1. Nitrile-functionalized N-heterocyclic carbene complexes of Ag(I) and Rh(I)
and the general structures of the complexes reported in this chapter (M = Pd(II), Pt(II); D =
olefin or alkyl ligand; L = nitrile donor or a chloro group).
Figure 3.2. ORTEP diagram of 5b⋅1.5 CH3CN depicted with thermal ellipsoids at the 30%
probability level. The counteranions, hydrogens, and solvent molecules have been omitted
for clarity. Selected bond distances (Å) and bond angles (deg): Pd(1)-C(1), 1.969(6);
Pd(3)-C(20), 1.957(6); Pd(1)-N(3), 2.015(5); Pd(3)-N(4), 2.018(5); Pd(2)-N(3), 1.976(5);
Pd(2)-N(4), 1.972(5); Pd(1)-N(8), 2.094(6); Pd(1)-N(7), 2.020(5); C(10)-N(3), 1.272(8);
C(11)-N(4), 1.265(8); C(10)-O(1), 1.361(7); C(11)-O(1), 1.382(7); N(3)-Pd(2)-N(4),
91.9(2); C(1)-Pd(1)-N(3), 85.4(2); C(10)-O(1)-C(11), 126.7(5); C(10)-N(3)-Pd(2),
125.3(4).
Figure 3.3. Diastereomers and rotamers (A and B) of Pd(II) (6a) and Pt(II) (9a) complexes
bearing 2-methoxycyclooct-5-enyl and the C–CN ligands.
Figure 3.4. ORTEP diagram of 6a depicted with thermal ellipsoids at the 30% probability
level. The counteranion and hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Pd(1)-C(1), 2.012(4); Pd(1)-C(19), 2.040(4); Pd(1)-
N(4), 2.164(3); Pd(1)-coe(trans to C)cent, 2.161; C(11)-N(3), 1.148(6); C(18)-O(1),
1.441(5); C(1)-Pd(1)-coe(trans to C)avg, 161.2; C(19)-Pd(1)-N(4), 177.2(9); C(1)-Pd(1)-
C(19), 87.7(1).
Figure 3.5. ORTEP diagram of 7 depicted with thermal ellipsoids at the 30% probability
level. The counteranion and hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Pt(1)-C(1), 2.011(5); Pt(1)-Cl(1), 2.317(6); Pt(1)-
cod(trans to C)cent, 2.152; Pt(1)-cod(cis to C)cent, 2.048; C(11)-N(3), 1.134(7); C(1)-Pt-(1)-
cod(trans to C)avg, 162.3; Cl(1)-Pt(1)-cod(cis to C)avg, 161.0; C(1)-Pt(1)-cod(cis to C)avg,
94.0.
Figure 3.6. ORTEP diagrams of 8 (left) and 9a (right) depicted with thermal ellipsoids at
39
51
54
56
57
61

xixthe 30% probability level. The counteranion and hydrogens have been omitted for clarity.
Selected bond distances (Å) and bond angles (deg): 8 (left): Pt(1)-C(1), 1.969(1); Pt(1)-
C(12), 2.095(1); Pt(1)-Cl(1), 2.430(4); Pt(1)-coe(trans to C)cent, 2.102; C(11)-N(3),
1.135(7); C(19)-O(1), 1.418(2); C-(1)-Pt(1)-coe(trans to C)avg, 160.7; C(12)-Pt(1)-Cl(1),
178.4(4); C(1)-Pt(1)-C(12), 88.9(5). 9a (right): Pt(1)-C(1), 2.001(5); Pt(1)-C(17), 2.044(5);
Pt(1)-N(4), 2.122(5); Pt(1)-coe(trans to C)cent, 2.100; C(10)-N(3), 1.145(8); C(16)-O(1),
1.449(6); C(1)-Pt(1)-coe(trans to C)avg, 161.0; C(17)-Pt(1)-N(4), 178.3(0); C-(1)-Pt(1)-
C(17), 88.9(7).
Figure 3.7. ORTEP diagram of 11⋅0.5 CH2Cl2 depicted with thermal ellipsoids at the 30%
probability level. The counteranion, hydrogens, and solvent molecules have been omitted
for clarity. Only one asymmetric unit is shown. Selected bond distances (Å) and bond
angles (deg): Pt(1A)-C(1A), 2.037(1); Pt(1A)-Cl(1A), 2.315(3); Pt(1A)-cod(trans to C)cent,
2.173; Pt(1A)-cod(cis to C)cent, 2.084; C(1A)-Pt(1A)-cod(trans to C)avg, 162.3; Cl(1)-Pt(1)-
cod(cis to C)avg, 160.9; C(1)-Pt(1)-cod(cis to C)avg, 95.1.
Figure 4.1. Examples of transition metal complexes bearing amino-functionalized N-
heterocyclic carbene ligands reported by Douthwaite and Oro. See references 9a, 9b and
9e.
Figure 4.2. ORTEP diagram of 12 depicted with thermal ellipsoids at 30% probability. The
counteranions and most of the hydrogens have been omitted for clarity. Selected bond
distances (A° ) and bond angles (deg): Ni(1)-C(1), 1.870(6); Ni(1)-C(12), 1.883(6); Ni(1)-
N(5), 1.963(5); Ni(1)-N(6), 1.937(5); C(1)-Ni(1)-N(5), 91.8(3); C(1)-Ni(1)-C(12), 88.1(3).
Figure 4.3. Selected sections of the 1H NMR spectra of complex 12 in acetonitrile-d3 (400
MHz, 298 K) and the assignments of the imidazolylidene ring (left) and methyl protons
(right) in the presence of (a) 0 equiv, (b) 1 equiv, (c) 2 equiv, and (d) 3 equiv of [Bu4N][Δ-
TRISPHAT].
Figure 4.4. ORTEP diagram of 13⋅THF depicted with thermal ellipsoids at 30%
probability. The counteranion, solvent molecule, and most of the hydrogens have been
omitted for clarity. Selected bond distances (Å) and bond angles (deg): Ru(1)-C(1),
2.092(5); Ru(1)-N(3), 2.146(4); Ru(1)-Cl(1), 2.4180(13); Ru(1)-C(15), 2.248(5); C(1)-
Ru(1)-N(3), 91.98(17); C(1)-Ru(1)-Cl(1), 88.24(13); Cl(1)-Ru(1)-N(3), 81.81(11).
63
68
79
81
83
85

xxFigure 4.5. ORTEP diagram of 14 depicted with thermal ellipsoids at the 30% probability
level. The counteranion and most of the hydrogens have been omitted for clarity. Selected
bond distances (Å) and bond angles (deg): Os(1)-C(1), 2.07(1); Os(1)-N(3), 2.137(9);
Os(1)-Cl(1), 2.422(3); Os(1)-C(15), 2.201(6); C(1)-Os(1)-N(3), 91.1(4); C(1)-Os(1)-Cl(1),
87.5(3); Cl(1)-Os(1)-N(3), 80.0(3).
Figure 4.6. Selected sections of the 1H NMR spectra of complex 13 in dichloromethane-d2
(400 MHz, 298 K) and the assignments of the imidazolylidene ring (left), p-cymene ring
(middle) and the isopropyl-methyl protons (right) in the presence of (a) 0 equiv, (b) 1
equiv, (c) 2 equiv, and (d) 3 equiv of [Bu4N][Δ-TRISPHAT].
Figure 4.7. ORTEP diagram of 15 depicted with thermal ellipsoids at 30% probability. The
counteranions and most of the hydrogens have been omitted for clarity. Only one
asymmetric unit is shown. Selected bond distances (Å) and bond angles (deg): Ru(1a)-
C(1a), 2.03(1); Ru(1a)-N(3a), 2.195(7); Ru(1a)-N(4a), 2.156(8); Ru(1a)-C(14a), 2.215(9);
C(1a)-Ru(1a)-N(3a), 91.9(3); N(3a)-Ru(1a)-N(4a), 90.9(4); C(1a)-Ru(1a)-N(3a), 81.3(3).
Figure 4.8. ORTEP diagram of 16a depicted with thermal ellipsoids at 30% probability.
The counteranions and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Ru(1)-P(1), 2.314(1); Ru(1)-N(1), 2.188(2); Ru(1)-
N(2), 2.171(2); Ru(1)-C(8), 2.207(3); P(1)-Ru(1)-N(1), 84.89(7); N(1)-Ru(1)-N(2),
87.35(9); P(1)-Ru(1)-N(2), 95.72(7).
Figure 4.9. Catalytic transfer hydrogenation of acetophenone to 1-phenylethanol in the
presence of 13, potassium tert-butoxide, and 2-propanol (6 mL) at 75°C (C/B/S = 1/8/200).
The conversions from two runs and an average of these are shown.
Figure 4.10. Addition of acetophenone (200 mg, C/S = 1/200) after 180 min of transfer
hydrogenation of acetophenone in the presence of 13, potassium tert-butoxide, and 2-
propanol (6 mL) at 75°C (C/B/S = 1/8/200).
Figure 4.11. Catalytic H2-hydrogenation of 4'-bromoacetophenone to 1-(4'-
bromophenyl)ethanol (Table 4.3, entry 4) in the presence of catalyst 15, KOtBu, and THF
(6 mL) in 8 bar of H2 pressure at 25°C (C/B/S = 1/8/1500).The concentrations of the
product alcohol from two runs and an average of these are shown.
86
88
90
90
91
92
96

xxiFigure 4.12. Catalytic H2-hydrogenation of 4'-chloroacetophenone to 1-(4'-chlorophenyl)-
ethanol in the presence of catalyst 15, KOtBu, and THF (6 mL) in 8 bar of H2 at 25°C
(C/B/S = 1/14/2250). Mercury poisoning test was conducted by adding a drop of mercury
to the reaction mixture against a flow of hydrogen at 15 min.
Figure 5.1. Transfer hydrogenation of acetophenone catalyzed by complex 13 in basic 2-
propanol.
Figure 5.2. Catalytic H2-hydrogenation of benzophenone to benzhydrol (Table 5.2, entries
2 and 3) in the presence of catalyst 13, KOtBu, and THF (6 mL) in 25 bar of H2 pressure at
50°C with a C/B/S ratio of (a) 1/8/200 (triangles) and (b) 1/8/400 (squares).
Figure 5.3. Kinetic data showing the production of 1-phenylethanol from acetophenone
catalyzed by complex 13 in basic THF: (a) dependence on the catalyst concentration (13);
(b) dependence on the hydrogen concentration; (c) dependence on the acetophenone
concentration; (d) dependence on the base concentration (KOtBu); (e) dependence on the
1-phenylethanol concentration. The inset shows the dependence of initial rates (v0, 10-5 M
s-1) and the concentration of the analyte of interest.
Figure 5.4. Linear plot showing the relationship between the reciprocal of the initial rate
(1/v0 in 103 M-1 s) and acetophenone concentration (M). The rate (kH) and equilibrium (Keq)
constants were derived from the slope and the y intercept according to eq 5.2, respectively.
Figure 5.5. Kinetic data showing the production of 1-phenylethanol from acetophenone
catalyzed by complex 13 in basic THF: (a) under 8 bar of D2 gas with different
concentrations of acetophenone; (b) under 25 bar of H2 gas with different concentrations of
acetophenone-d3.
Figure 5.6. Linear plots showing the relationship between the reciprocal of the initial rate
(1/v0 in 103 M-1 s) and acetophenone concentration (M), using (a) D2 gas (8 bar) and
acetophenone, (b) H2 gas (25 bar) and acetophenone, and (c) H2 gas (25 bar) and
acetophenone-d3 (0.16 – 0.49 M), in the production of 1-phenylethanol from acetophenone
catalyzed by complex 13 in basic THF.
Figure 5.7. ORTEP diagram of 17⋅THF depicted with thermal ellipsoids at the 30%
probability level. The counteranion, solvent molecule, and most of the hydrogens have
97
109
113
115
119
122
123

xxiibeen omitted for clarity. Selected bond distances (Å) and bond angles (deg): Ru(1)-C(1),
2.029(7); Ru-(1)-N(3), 2.145(5); Ru(1)-H(1ru), 1.79(7); Ru(1)-C(15), 2.232(6); C(1)-
Ru(1)-N(3), 91.9(2); C(1)-Ru(1)-H(1ru), 81(2); H(1ru)-Ru(1)-N(3), 84(2).
Figure 5.8. Reaction profiles showing the hydrogenation of acetophenone catalyzed by
complex 13 (blue squares) and complex 17 (red circles) in (a) basic THF at 25 bar of H2
pressure and 50°C and (b) 2-propanol at 75°C. The C/B/S ratio was 1/8/200. The transfer
hydrogenation of acetophenone catalyzed by complex 13 in basic 2-propanol was
described in Chapter 4.
Figure 5.9. Reaction profile showing the hydrogenation of trans-4-phenyl-but-3-en-2-one
catalyzed by complex 13 in basic THF at 25 bar of H2 pressure and 50°C: (blue circles)
trans-4-phenylbut-3-en-2-ol; (red squares) 4-phenylbutan-2-ol; (green triangles) 4-
phenylbutan-2-one. The C/B/S ratio was 1/8/200.
Figure 5.10. Possible reaction intermediates for the inner-sphere mechanism involving
hydride migration to the coordinated ketone substrate: (left) decoordination of the
chelating amine group from the NHC ligand; (right) ring slippage of the arene ring.
Figure 5.11. Catalytic H2-hydrogenation of acetophenone to 1-phenylethanol in the
presence of catalyst, KOtBu, and THF (6 mL) in 25 bar of H2 pressure at 50°C with a
C/B/S ratio of (a) 1/8/200 (red squares), catalyst 13, [13] = 0.83 mM and (b) 1/10/240,
catalyst 18, [18] = 0.71 mM (blue triangles).
Figure 5.12. The free energy profile for an outer-sphere mechanism for the H2-
hydrogenation of acetone starting from A1 and moving to the right and the enolate
formation starting from A1 and moving to the left. The gas phase free energies (1 atm, 298
K) are reported relative to G in kcal/mol.
Figure 5.13. Selected computed structures A-C and G for the outer-sphere mechanism in
the H2-hydrogenation of acetone and the transition state structures for the heterolytic
splitting of H2 (TSB,C) and for the concerted transfer of a hydride/proton pair to the ketone
(TSD,E). The bond lengths (Å) are given in the structures.
Figure 5.14. Computed structures and energies (at 298 K and 1 atm relative to G + H2 in
kcal/mol) for the ring slippage mechanism.
127
128
130
131
133
135
136
137

xxiiiFigure 5.15. The free energy profile for the inner-sphere mechanism in the H2-
hydrogenation of acetone starting from H and moving to the right. The amino group is
decoordinated throughout the catalytic cycle. Moving to the left from H leads to the
unstable enolate complex M. The gas phase free energies (1 atm, 298 K) are reported
relative to G, hydrogen and acetone in kcal/mol.
Figure 5.16. Selected computed structures H-K for the inner-sphere mechanism involving
decoordination of the amine group of the NHC ligand in the H2-hydrogenation of ketone
and the transition state structures for the heterolytic splitting for H2 (TSI,J) and for the
hydride attack on the coordinated ketone (TSK,H). The bond lengths (Å) are given in the
structures.
Figure 6.1. Late transition metal complexes containing a chelating N-heterocyclic carbene
(NHC)-primary amine (C–NH2) or a chelating phosphine-primary amine (P–NH2) ligand.
Figure 6.2. An inner sphere mechanism involving the decoordination of the primary amine
group proposed for the H2-hydrogenation of ketones catalyzed by complex 13 in the
presence of an alkoxide base.
Figure 6.3. ORTEP diagram of 19 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Ir(1)–C(1), 2.067(7); Ir(1)–N(3), 2.127(5); Ir(1)–
Cl(1), 2.424(2); Ir(1)–C(13), 2.199(6); C(1)–Ir(1)–N(3), 90.9(2); C(1)–Ir(1)–Cl(1),
92.2(2); Cl(1)–Ir(1)–N(3), 80.7(1).
Figure 6.4. ORTEP diagram of 20 depicted with thermal ellipsoids at 30% probability.
The counteranion and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Ir(1)–P(1), 2.307(2); Ir(1)–N(1), 2.146(8); Ir(1)–
Cl(1), 2.407(2); Ir(1)–C(24), 2.208(9); P(1)–Ir(1)–N(1), 88.9(2); P(1)–Ir(1)–Cl(1),
89.02(9); Cl(1)–Ir(1)–N(1), 82.3(2).
Figure 6.5. ORTEP diagram of 21 depicted with thermal ellipsoids at 30% probability.
The counteranion and most of the hydrogens have been omitted for clarity. The position of
the hydride ligand is not refined and thus not shown. Selected bond distances (Å) and bond
angles (deg): Ir(1)–C(1), 2.015(5); Ir(1)–N(3), 2.151(4); Ir(1)–C(14), 2.233(3); C(1)–Ir(1)–
N(3), 89.6(2).
138
140
154
155
158
159
161

xxivFigure 6.6. ORTEP diagram of 22 depicted with thermal ellipsoids at 30% probability.
The counteranion and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Ir(1)–C(1), 2.036(4); Ir(1)–N(3), 2.225(4); Ir(1)–
Cl(1), 2.426(1); Ir(1)–C(11), 2.264(4); C(1)–Ir(1)–N(3), 87.5(2); C(1)–Ir(1)–Cl(1),
95.0(1); Cl(1)–Ir(1)–N(3), 84.4(1).
Figure 6.7. Reaction profiles showing the effect of the concentrations of catalyst,
hydrogen and substrate to the H2-hydrogenation of benzophenone catalyzed by complex
19. (a) [19] = 0.72 mM, [benzophenone] = 0.14 M, P(H2) = 25 bar, red circles; (b) [19] =
0.72 mM, [benzophenone] = 0.14 M, P(H2) = 15 bar, blue diamonds; (c) [19] = 0.72 mM,
[benzophenone] = 0.29 M, P(H2) = 25 bar, green squares; (d) [19] = 1.2 mM,
[benzophenone] = 0.14 M, P(H2) = 25 bar, purple crosses. All of the reactions were
conducted in 25 bar H2 at 50°C in THF and KOtBu was used as a base (5.9 mM).
Figure 6.8. Reaction profiles showing the effect of alcohols and pyridine on the
hydrogenation of acetophenone catalyzed by complex 15 in (a) basic THF, red circles; (b)
basic 2-propanol, blue diamonds; (c) basic THF, [1-phenylethanol] = 0.2 M, green squares;
(d) basic THF, [1-phenylethanol] = 0.4 M, orange triangles; (e) basic THF, [pyridine] =
0.030 M, purple crosses. All of the reactions were conducted with 8 bar H2 at 25°C and
KOtBu was used as the base. The C/B/S ratio was 1/8/2515.
Figure 6.9. Reaction profiles showing the effect of the concentrations of catalyst,
hydrogen and substrate on the hydrogenation of acetophenone catalyzed by complex 15,
(a) [15] = 0.77 mM, [acetophenone] = 1.9 M, P(H2) = 8 bar, red circles; (b) [15] = 1.3 mM,
[acetophenone] = 1.9 M, P(H2) = 8 bar, blue squares; (c) [15] = 0.77 mM, [acetophenone]
= 0.97 M, P(H2) = 8 bar, green triangles; (d) [15] = 0.77 mM, [acetophenone] = 1.9 M,
P(H2) = 2 bar, orange crosses. All of the reactions were conducted with 25oC in THF and
KOtBu was used as the base (5.9 mM).
Figure 6.10. Reaction profiles showing the effect of alcohols on the H2-hydrogenation of
acetophenone catalyzed by the chloride complex 19 in the presence of KOtBu, in (a) basic
THF, red circles, C/B = 1/8; (b) basic 2-propanol, blue diamonds, C/B = 1/8; (c) basic
THF, [1-phenylethanol] = 0.015 M, C/B = 1/8, green squares; (d) basic 2-propanol, C/B =
1/16, purple triangles. All of the reactions were conducted using 25 bar H2 at 50°C. The
C/S ratio was 1/200.
163
164
170
170
172

xxvFigure 6.11. Reaction profiles showing the effect of alcohols on the H2-hydrogenation of
acetophenone catalyzed by the hydride-amine complex 21 in (a) basic 2-propanol, blue
diamonds; (b) basic THF, red squares; (c) basic 2-propanol, [1-phenylethanol] = 0.030 M,
green triangles. All of the reactions were conducted using 25 bar H2 at 50°C and KOtBu
was used as the base. The C/B/S ratio was 1/8/200.
Figure 6.12. The free energy profile for the outer sphere bifunctional mechanism in the H2-
hydrogenation of acetone starting from A and moving to the right. Pathway colored in blue
represents the ruthenium(II) system and the one in red represents the iridium(III) system.
The gas phase free energies (1 atm, 298 K) are reported relative to A, hydrogen and
acetone in kcal/mol.
Figure 6.13. Computed transition state structures for the heterolytic splitting of H2 (TSC,D)
of the ruthenium(II) (left) and the iridium(III) system (right). The bond lengths (Å) are
given in the structures. The color code for the atoms are: ruthenium (orange), iridium
(yellow), nitrogen (blue), carbon (grey) and hydrogen (white).
Figure 6.14. Computed transition state structures for the transfer of a proton/hydride pair
to acetone (TSE,F) from the ruthenium(II) (above) and iridium(III) hydrides (below). The
bond lengths (Å) are given in the structures. The color code for the atoms are: ruthenium
(orange), iridium (yellow), nitrogen (blue), oxygen (blue), carbon (grey) and hydrogen
(white).
Figure 6.15. The free energy profile for the outer sphere bifunctional mechanism in the
activation of H2 starting from A and moving to the right (blue pathway), and in the
presence of 2-propanol staring from Aalc and moving to the right (red pathway). The gas
phase free energies (1 atm, 298 K) are reported relative to A, hydrogen and acetone in
kcal/mol.
Figure 6.16. Computed transition state structure for the heterolytic splitting of H2 (TSC,Dalc)
by the ruthenium(II) system in the presence of 2-propanol. The bond lengths (Å) are given
in the structure. The color code for the atoms are: ruthenium (orange), nitrogen (blue),
oxygen (red), carbon (grey) and hydrogen (white).
Figure 6.17. Computed transition state structures for the non-alcohol assisted (TS(HI)3,4,
left) and the alcohol-assisted heterolytic splitting of H2 (TS(HI)3,4alc, right) by the
173
178
179
179
181
182

xxviiridium(I) system. The bond lengths (Å) are given in the structures. The color code for the
atoms are: iridium (yellow), nitrogen (blue), oxygen (red), carbon (grey) and hydrogen
(white).
Figure 6.18. The free energy profile for the (a) alcohol-assisted outer sphere bifunctional
mechanism (blue pathway) and (b) the non alcohol-assisted outer sphere bifunctional
mechanism (red pathway) in the H2-hydrogenation of acetone starting from the amido
complex HI1 and moving to the right. The gas phase free energies are reported relative to
HI1, hydrogen, acetone and 2-propanol for the blue pathway, and to HI1, hydrogen and
acetone for the red pathway, in kcal/mol.
Figure 6.19. Computed transition state structure for the attack of hydride on the
coordinated acetone (TSM,J) of the iridium(III) system for the inner sphere mechanism
involving the decoordination of the amine group. The bond lengths (Å) are given in the
structure. The colour code for the atoms are: iridium (yellow), nitrogen (blue), oxygen
(red), carbon (grey) and hydrogen (white).
Figure 6.20. ORTEP diagram of 23 depicted with thermal ellipsoids at 30% probability.
The counteranion and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Ru(1)–C(1), 2.065(4); Ru(1)–N(3), 2.178(3); Ru(1)–
C(22), 1.859(4); Ru(1)–C(15), 2.223(4); C(22)–O(1), 1.149(5); C(1)–Ru(1)–N(3), 90.8(2);
C(1)–Ru(1)–C(22), 95.1(2); C(22)–Ru(1)–N(3), 91.7(2).
Figure 6.21. ORTEP diagram of 24 depicted with thermal ellipsoids at 30% probability.
The counteranion and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Ru(1)–P(1), 2.320(1); Ru(1)–N(1), 2.166(4); Ru(1)–
C(30), 1.877(6); Ru(1)–C(24), 2.218(5); C(30)–O(1), 1.140(6); P(1)–Ru(1)–N(1), 87.3(1);
P(1)–Ru(1)–C(30), 88.6(2); C(30)–Ru(1)–N(1), 90.8(2).
Figure 6.22. The carbonyl stretching wavenumbers (cm-1 in KBr) of a series of
ruthenium(II) complexes, [RuCp*(D–NH2)CO]+, where D is a donor group.
Figure 7.1. The structures of ruthenium(II) complexes containing the C–NH2 ligand
forming a six-membered ring metal-chelate (-M–C–NH2-).
185
186
188
189
190
191
208

xxviiFigure 7.2. Proposed structure of an iron(II) complex containing a C–N–N–C type donor
ligand. 209

xxviiiList of Schemes
Scheme 1.1. The Proposed Mechanism for the Hydrogenation of Terminal Alkenes
Catalyzed by the Rhodium(I)-based Wilkinson's Catalyst.
Scheme 1.2. The Outer Sphere Bifunctional Mechanism Using the “NH effect” for the
Hydrogenation of Aromatic Ketones.
Scheme 1.3. The Inner Sphere Mechanism for the H2-Hydrogenation of Aromatic Ketones.
Scheme 2.1. Synthesis of the Imidazolium Salt 1a and its Ag(I), Rh(I) and Ru(II)
Complexes 2a, 3 and 4.
Scheme 2.2. Synthesis of Imidazolium Salt 1b and the Ag(I) complexes 2b and 2e.
Scheme 2.3. Synthesis of Imidazolium Salts 1c to 1f and the Ag(I) Complexes 2c and 2d.
Scheme 2.4. Proposed Reaction Pathways to Hydrolysis of Ag(I) Complex 2b to 2e.
Scheme 3.1. Synthesis of Pd(II) and Pt(II) Complexes (5, 6a, and 7) Starting from a
Nitrile- Functionalized N-Heterocyclic Carbene Complex of Ag(I) (2a).
Scheme 3.2. Interconversion of 6a to 6b and 9a to 9b in Different Recrystallization
Solvents.
Scheme 3.3. Nucleophilic Attack of Methoxide at the Coordinated Cyclooctadiene Ligand
of 7 to 8 and Chloride Abstraction of 8 with AgBF4 in CH3CN to 9a.
Scheme 3.4. Proposed Reaction Pathways Leading to the Formation of Rotamers A and B
of Complexes 6, 8, and 9.
Scheme 3.5. Synthesis of Pt(II) Complex 11 from an N-Heterocyclic Carbene Complex of
Ag(I) (10).
Scheme 4.1. Synthesis of a Homoleptic Primary Amino-Functionalized N-Heterocyclic
Carbene Complex of Nickel(II) (12).
Scheme 4.2. Synthesis of Complexes 13 and 14 Bearing the C–NH2 Ligand by the
Transmetalation Reaction Involving Complex 12 and [M(p-cymene)Cl2]2 (M = Ru, Os).
6
8
12
28
28
29
37
52
58
62
66
67
80
84

xxixScheme 4.3. Synthesis of the Ruthenium(II) Complexes Containing the C–NH2 (15) and
P–NH2 (l6a) Ligands.
Scheme 5.1. Reaction Scheme Showing the Definitions of the Rate and Equilibrium
Constants.
Scheme 5.2. Possible Outer-Sphere Mechanism That Involves Bifunctional Catalysis in the
Hydrogenation of Acetophenone.
Scheme 5.3. Synthesis of Complex 17 from Reaction of Complex 13 and Basic 2-
Propanol.
Scheme 5.4. Hydrogenation of trans-4-Phenyl-but-3-en-2-one Catalyzed by Complex 13.
Scheme 5.5. Synthesis of Complex 18 by in Situ Generation of the Silver(I) Carbene
Complex and Subsequent Transmetalation of the NHC Ligand to Ruthenium(II).
Scheme 5.6. Proposed Mechanism for the H2-Hydrogenation of Ketones Catalyzed by
Complex 13 and an Alkoxide Base.
Scheme 6.1. An Alcohol-assisted Outer Sphere Bifunctional Mechanism of H2-
Hydrogenation of Ketones Catalyzed by Ruthenium(II) and Iridium(I) Systems Containing
a C–NH2 Ligand.
Scheme 6.2. Synthesis of Iridium(III) and Ruthenium(II) Complexes Containing a C–NH2
Ligand.
Scheme 6.3. Synthesis of Iridium(III) and Ruthenium(II) Complexes Containing a P–NH2
Ligand.
Scheme 6.4. Synthesis of a Iridium(III) Complex Containing a C–NMe2 ligand.
Scheme 6.5. The Hydrogenation of trans-4-Phenyl-but-3-ene-2-one Catalyzed by
Complexes 15 and 19.a
Scheme 6.6. Computed Outer Sphere Bifunctional Mechanism in the Hydrogenation of
Acetone Catalyzed by Complexes of Ruthenium(II) and Iridium(III).
Scheme 6.7. Computed Reaction Pathways of the Activation of H2 by Complex A in the
89
117
125
126
129
132
144
156
157
158
162
169
177

xxxPresence of 2-Propanol.
Scheme 6.8. Computed Hydride Migration Pathway from the Ir–H bond to the
Coordinated Cp Ligand Starting from R.
Scheme 6.9. Computed Alcohol-assisted Outer Sphere Bifunctional Mechanism in the H2-
Hydrogenation of Acetone Catalyzed by Complexes of Iridium(I).
Scheme 6.10. The Disfavoured Pathways Involving Either an η5 to η3 Ring Slip or a
Decoordination of the Amine Group Leading to the Activation of H2 by the Iridium(III)
System.
180
183
184
187

xxxiList of Tables
Table 4.1. The transfer hydrogenation of acetophenone to 1-phenylethanol in basic 2-
propanol catalyzed by complexes 12, 13 and 14.
Table 4.2. The hydrogenation of acetophenone catalyzed by complex 15 in the presence of
KOtBu.a
Table 4.3. The hydrogenation of substituted acetophenones catalyzed by complex 15 in the
presence of KOtBu.a
Table 4.4. The hydrogenation of organic molecules with polar bonds catalyzed by complex
15 in the presence of KOtBu.a
Table 5.1. H2-Hydrogenation of Acetophenone to 1-Phenylethanol Catalyzed by Complex
13.
Table 5.2. H2-Hydrogenation of Ketones Catalyzed by Complex 13.
Table 5.3. Kinetic Data for the Hydrogenation of Acetophenone Catalyzed by Complex 13.
Table 5.4. Kinetic Data for Kinetic Isotope Effect Studies of the Hydrogenation of
Acetophenone Catalyzed by Complex 13.
Table 5.5. Isotope Effect for the Hydrogenation of Acetophenone Catalyzed by Complex
13.
Table 5.6. Atomic Polar Tensor (APT) Charges (in ESU) on Selected Atoms for the
Computed Structures A-C, G, TSB,C, and TSD,E.
Table 5.7. Atomic Polar Tensor (APT) Charges (in ESU) on Selected Atoms for the
Computed Structures H-K, TSI,J and TSK,H.
Table 6.1. The H2-Hydrogenation of Acetophenone Catalyzed by Iridium(III) Complexes.
Table 6.2. The H2-Hydrogenation of Benzophenone Catalyzed by Iridium(III) Complexes.
Table 6.3. Transfer Hydrogenation of Acetophenone Catalyzed by Iridium(III)
Complexes.
93
94
95
98
111
112
118
124
124
137
141
165
166
166

xxxiiTable 6.4. Deuteration of Acetophenone and 2-Propanol Catalyzed by Complex 15.
Table 6.5. Deuteration of Acetophenone and 1-phenylethanol Catalyzed by Complex 19.
Table A.1. The oven temperatures, retention times (tR, tp, /min) for all of the substrates and
alcohol products reported from GC analyses.
171
174
210

xxxiiiList of Abbreviations
1a
1b
1c
1d
1e
1f
2a
2b
2c
2d
2e
3
4
5a
1-(2-cyanophenyl)-3-methylimidazolium tetrafluoroborate
3-(cyanomethyl)-1-(2-cyanophenyl)imidazolium
hexafluorophosphate
1-(2-cyanophenyl)-3-(2-pyridinylmethyl)imidazolium
hexafluorophosphate
1-(2-cyanophenyl)-3-(2-pyridinyl)imidazolium
hexafluorophosphate
1-(2-cyanophenyl)-3-(1-phenylethyl)imidazolium
tetrafluoroborate
1-(2-(inden-3-yl)ethyl)-3-(2-cyanophenyl)imidazolium
tetrafluoroborate
[Ag(C–CN)2]BF4
silver(I) complex of (3-(cyanomethyl)-1-(2-cyanophenyl)-
imidazol-2-ylidene
bis{[1-(2-cyanophenyl)-3-(2-pyridinylmethyl)imidazol-2-
ylidene]silver(I)} hexafluorophosphate
bis{[1-(2-cyanophenyl)-3-(2-pyridinyl)imidazol-2-
ylidene]silver(I)} hexafluorophosphate
silver(I) complex of (3-(carbomoylmethyl)-1-(2-cyanophenyl)-
imidazol-2-ylidene
[Rh(C–CN)(cod)]2(BF4)2
[Ru(p-cymene)(C–CN)Cl]2(BF4)2
[(C–CN)2Pd(μ-Cl)2Pd(CH3CN)2](BF4)2

xxxiv5b
6a
6b
7
8
9a
9b
10
11
12
13
14
15
16a
16b
17
18
19
20
21
22
23
{Pd(CH3CN)2}3(C–N–N–C)](BF4)4
[Pd(C–CN)(η1:η2-coe-OMe)(CH3CN)]BF4
[Pd(C–CN)(η1:η2-coe-OMe)]2(BF4)2
[Pt(C–CN)(cod)Cl]BF4
(Pt(C–CN)(η1:η2-coe-OMe)Cl
[Pt(C–CN)(η1:η2-coe-OMe)(CH3CN)]BF4
[Pt(C–CN)(η1:η2-coe-OMe)]2(BF4)2
[Ag(IMes)2]BF4
[Pt(IMes)(cod)Cl]BF4
[Ni(C–NH2)2](PF6)2
[Ru(p-cymene)(C–NH2)Cl]PF6
[Os(p-cymene)(C–NH2)Cl]PF6
[RuCp*(C–NH2)(py)]PF6
[RuCp*(P–NH2)(py)]PF6
RuCp*(κ2(P,N)-PPh2CH2CH2NH2)Cl
[Ru(p-cymene)(C–NH2)H]PF6
[Ru(p-cymene)(C–NMe2)Cl]PF6·1.5 DMSO
[IrCp*(C–NH2)Cl]PF6
[IrCp*(P–NH2)Cl]PF6
[IrCp*(C–NH2)H]PF6
[IrCp*(C–NMe2)Cl]PF6
[RuCp*(C–NH2)(CO)]PF6

xxxv24
app
APT
atm
BINAP
BINOL
Bu
Bu4N
C/B/S
C–CN
CD
C–NH2
C–NMe2
cod
coe-OMe
Cp
Cp*
CpH
DFT
DMSO
[RuCp*(P–NH2)(CO)]PF6
2-amido-2-(2-pyridyl)-propane
atomic polar tensor
atmosphere
2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
1,1'-bi-2-naphthol
butyl
tetrabutylammonium
catalyst to base to substrate
nitrile functionalized N-heterocyclic carbene/
1-(2-cyanophenyl)-3-methylimidazol-2-ylidene
circular dichroism
primary amino-functionalized N-heterocyclic carbene/
1-(2-aminomethylphenyl)-3-methylimidazol-2-ylidene
1-(N,N-dimethylaminopropyl)-3-methylimidazol-2-ylidene
1,5-cyclooctadiene
2-methoxycyclooct-5-enyl
cyclopentadienyl
pentamethylcyclopentadienyl
cyclopentadiene
Density Functional Theory
dimethylsulfoxide

xxxvi(L)-DOPA
dpen
dpim
dpmp
dppe
dppm
en
eq
equiv
ESI-MS
ESU
Et
FID
GC
HC–NMe2
HMBC
HRMS
HSQC
ICy
IMes
IMesH
imid
(L)-3,4-dihydroxyphenylalanine
1,2-diphenylethylenediamine
1,3-diphenyl-2-imidazolidinylidenato-2-C,2'-C
bis((diphenylphosphino)methyl)phenylphosphine
1,2-bis(diphenylphosphino)ethane
1,1-bis(diphenylphosphino)methane
ethylenediamine
equation/equilibrium
equivalent
Electrospray Ionization Mass Spectrometry
electrostatic unit
ethyl
flame ionization detector
gas chromatography
1-(N,N-dimethylamino)propyl-3-methylimidazolium
Heteronuclear Mutiple-bond Correlation
High Resolution Mass Spectrometry
Heteronuclear Single-quantum Correlation
1,3-dicyclohexylimidazol-2-ylidene
1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene
1,3-bis(2,4,6-trimethylphenyl)imidazolium
imidazol-2-ylidene

xxxviiiPr
IPr
IR
KIE
MEA
MS
NAA
NHC
NMR
N–N
NOESY
ORTEP
papH
PCy3
PiPr3
P–NH2
P–N–N–P
P2(NH)2
PPh3
ppm
py
pyCH2
isopropyl
bis(2,6-diisopropylphenyl)imidazol-2-ylidene
infrared
kinetic isotope effect
2-methyl-5-ethyl-aniline
mass spectrometry
N-alkylated aniline
N-heterocyclic carbene
Nuclear Magnetic Resonance
2-hydroxylphenylbis(pyrazol-1-yl)methane and other derivatives
Nuclear Overhauser Effect Spectroscopy
Oak Ridge Thermal-Ellipsoid Plot
2-phenyl-6-(2 aminoisopropyl)pyridine
tricyclohexylphosphine
triisopropylphosphine
phosphine-amine/2-(diphenylphosphino)benzylamine
tetradentate diphosphinediimine ligand
tetradentate diphosphinediamine ligand
triphenylphosphine
parts per million
pyridine
2'-pyridylmethyl

xxxviiiQST
quinCH2
rac
ROMP
tBu
tert
THF
tmen
TMS
TOF
tot
TRISPHAT
Ts
UV-vis
Quadratic Synchronous Transit
2'-quinolylmethyl
racemic
ring opening metathesis polymerization
tert-butyl
tertiary
tetrahydrofuran
2,3-dimethylbutane-2,3-diamine
tetramethylsilane
turnover frequency
total
tris[tetrachlorobenzene-1,2-bis(olato)]phosphate
p-toluenesulfonyl
ultraviolet-visible

1Chapter 1: Introduction
The catalytic homogeneous hydrogenation of polar double bonds using molecular hydrogen is
an important industrial process to give valuable organic building blocks such as alcohols and
amines for use as fragrance and pharmaceutical precursors. Late transition metal catalysts using
certain phosphine ligands and phosphine-amine ligands have exceptional activity in the
hydrogenation of polar double bonds. The studies of these, including the proposed mechanisms
of action, have been extensively reviewed in the literature.1 The emergence of N-heterocyclic
carbenes (NHC) as ligands for use in homogeneous catalysis in the past two decades has exciting
implications as these ligands in general form strong metal-carbon bonds, and they are greener
and less toxic compared to their phosphine counterparts.2 This introduction reviews the
importance of metal-dihydrogen and metal hydride complexes in homogeneous catalysis, the
mechanisms of polar double bond reduction by molecular hydrogen, and the recent applications
of phosphines and donor-functionalized NHC in homogeneous catalysis.
1.1 Homogeneous Catalysis Involving Metal-Dihydrogen and Metal-Hydrides Complexes
1.1.1 The Metal-Dihydrogen Bond. A dihydrogen complex is often an intermediate in the
oxidative addition of dihydrogen to a transition metal center leading to the formation of a metal
dihydride complex. Such a complex is considered to be an important intermediate in the
hydrogenation of alkenes catalyzed by rhodium(I)- and ruthenium(II)-based Wilkinson's
catalysts.3 Kubas isolated the first octahedral side-on dihydrogen metal complex, W(η2-H2)
(CO)3(PCy3)2 (PCy3 = tricyclohexylphosphine) in 1984. He described such a interaction between
an η2-H2 ligand and the transition metal as a three-center two electron bond.4 Crabtree called this 1. (a) Rosales, M., Coord. Chem. Rev. 2000, 196, 249-280; (b) Noyori, R.; Yamakawa, M.; Hashiguchi, S.,
J. Org. Chem. 2001, 66, 7931-7944; (c) Clapham, S. E.; Hadzovic, A.; Morris, R. H., Coord. Chem. Rev. 2004, 248, 2201-2237; (d) Samec, J. S. M.; Bäckvall, J. E.; Andersson, P. G.; Brandt, P., Chem. Soc. Rev. 2006, 35, 237-248; (e) Ikariya, T.; Murata, K.; Noyori, R., Org. Biomol. Chem. 2006, 4, 393-406; (f) Ito, M.; Ikariya, T., Chem. Commun. 2007, 5134-5142; (g) Conley, B. L.; Pennington-Boggio, M. K.; Boz, E.; Williams, T. J., Chem. Rev. 2010, 110, 2294-2312; (h) Kuwata, S.; Ikariya, T., Dalton Trans. 2010, 39, 2984-2992; (i) Ikariya, T., Bull. Chem. Soc. Jpn. 2011, 84, 1-16.2. (a) Herrmann, W. A., Angew. Chem. Int. Ed. 2002, 41, 1290-1309; (b) Lee, H. M.; Lee, C. C.; Cheng, P. Y., Curr. Org. Chem. 2007, 11, 1491-1524; (c) Hahn, F. E.; Jahnke, M. C., Angew. Chem. Int. Ed. 2008, 47, 3122-3172; (d) Normand, A. T.; Cavell, K. J., Eur. J. Inorg. Chem. 2008, 2781-2800; (e) Diez-Gonzalez, S.; Marion, N.; Nolan, S. P., Chem. Rev. 2009, 109, 3612-3676; (f) Poyatos, M.; Mata, J. A.; Peris, E., Chem. Rev. 2009, 109, 3677-3707.3. Kubas, G. J., Acc. Chem. Res. 1988, 21, 120-128.4. Kubas, G. J.; Ryan, R. R.; Swanson, B. I.; Vergamini, P. J.; Wasserman, H. J., J. Am. Chem. Soc. 1984, 106, 451-452.

2class of η2-H2 metal complexes as σ-complexes to cover a variety of η2-σ-bonded ligands (C–H
and Si–H) in order to describe accurately the synergistic interaction between the σ-orbital of H2
and the dσ orbital on the metal, and between the σ*-orbital of H2 and the dπ orbital of the metal
(Figure 1.1).5
Figure 1.1. Bonding scheme of the η2-H2 ligand on a transition metal center.
Now hundreds of dihydrogen complexes have been isolated and structurally characterized by
diffraction methods (X-ray and neutron) and various NMR techniques. These non-classical
dihydride complexes contain a range of H–H bond lengths (0.8 – 1.4 Å) as a continuum
depending on the magnitude of the σ and σ* interactions.6 Metal dihydrides are at one end of the
continuum with H–H distances (dHH) of more than 1.6 Å, while dihydrogen complexes have dHH
between 0.8 – 1.2 Å on the other extreme.6 Although the H–H distances can rarely be directly
measured, especially the ones in between the two regimes, they can be accurately determined
using mathematical models based on T1 measurements and JHD coupling constants that are
obtained spectroscopically.6 Morris proposed the first equation to relate dHH with the JHD values
that were measured for several late transition metal dihydrogen complexes.7 This has been
extended recently to cover more precisely those of iron, ruthenium, and osmium.6f
1.1.2 The Acid-base Reactivity of Metal Hydrides and Metal Dihydrogen Complexes. The
acidities of those complexes have been measured and placed on pKa scales. Following the work
that was conducted by Heinekey, Norton and Angelici in the determination of the pKa values and
the bond dissociation energies of a collection of hydride complexes,8 the Morris group
constructed pKa ladders in tetrahydrofuran to cover the acidities of metal hydride and phosphine-
containing compounds over a 35 pKa unit range.9 In one case, a hydride ligand on trans- 5. (a) Crabtree, R. H.; Hamilton, D. G., Adv. Organomet. Chem. 1988, 28, 299-338; (b) Crabtree, R. H.,
Angew. Chem. Int. Ed. 1993, 32, 789-805.6. (a) Jessop, P. G.; Morris, R. H., Coord. Chem. Rev. 1992, 121, 155-284; (b) Heinekey, D. M.; Oldham, W. J., Chem. Rev. 1993, 93, 913-926; (c) Morris, R. H., Can. J. Chem. 1996, 74, 1907-1915; (d) Peruzzini, M.; Poli, R., Eds. Recent Advances in Hydride Chemistry; Elsevier: Amsterdam, 2001; (e) Kubas, G. J., Chem. Rev. 2007, 107, 4152-4205; (f) Morris, R. H., Coord. Chem. Rev. 2008, 252, 2381-2394.7. Maltby, P. A.; Schlaf, M.; Steinbeck, M.; Lough, A. J.; Morris, R. H.; Klooster, W. T.; Koetzle, T. F.; Srivastava, R. C., J. Am. Chem. Soc. 1996, 118, 5396-5407.
HH
M
dπ
σ *H
HM
dσ
σ

3Fe(H)2(diphosphine)2 was protonated by alcohols (HOR) giving the dihydrogen complex trans-
[Fe(η2-H2)(H)(diphosphine)2]OR10 (pKaTHF of trans-Fe(η2-H2)(H)(dppe)2]BPh4 = 13,9a dppe =
1,2-bis(diphenylphosphino)ethane). The polyhydride complex IrH5(PiPr3)2 (pKaTHF ≥ 43, PiPr3 =
triisopropylphosphine),9a when reacted with KH and a diaza-crown ether, forms the anionic
polyhydride complex [IrH4(PiPr3)2]-, in which one of the hydride ligands forms a hydridic-
protonic bond with the N–H group of the diaza-crown ether.11 In this case, the hydride ligand
acts as a hydrogen bond acceptor. The dihydrogen ligand, on the other hand, can act as a
hydrogen bond donor in the complex trans-Ru(dppe)2(η2-H2... OSO2CF3)CN, when the parent
complex Ru(dppe)2(OSO2CF3)CN is reacted with an atmosphere of hydrogen in methylene
chloride at -10°C.12 All these reactions are relevant to the intramolecular heterolytic splitting of
H2 at a transition metal center.
1.1.3 The Heterolytic Splitting of H2 at a Transition Metal Center and Implications for
Catalysis. One could view the heterolytic splitting of H2 when reacted with a base (B) in the
presence of a metal center (M) as a type of Brønsted acid chemistry giving an H+ and H-
equivalent.6d The extent of this reaction depends on the relative acidities of the coordinated
dihydrogen ligand and the Brønsted-base partner as discussed above (eq 1.1):
M(H2) + B M(H-) + B(H+) (1.1)
When the ligand (L) of a transition metal complex is suitably functionalized and is in close
proximity to the coordinated η2-H2 ligand, the intramolecular heterolytic splitting of the η2-H2
may occur to give a M–H/L–H couple. Chaudret, Lau and others have shown that a
ruthenium(II) diphosphine complex can undergo reversible proton exchange from the amine arm
of the functionalized cyclopentadienyl ring with the η2-H2 ligand, forming a quaternary amine
8. (a) Chinn, M. S.; Heinekey, D. M., J. Am. Chem. Soc. 1987, 109, 5865-5867; (b) Chinn, M. S.; Heinekey, D. M., J. Am. Chem. Soc. 1990, 112, 5166-5175; (c) Kristjansdottir, S. S.; Moody, A. E.; Weberg, R. T.; Norton, J. R., Organometallics 1988, 7, 1983-1987; (d) Weberg, R. T.; Norton, J. R., J. Am. Chem. Soc. 1990, 112, 1105-1108; (e) Kristjansdottir, S. S.; Loendorf, A. J.; Norton, J. R., Inorg. Chem. 1991, 30, 4470-4471; (f) Angelici, R. J., Acc. Chem. Res. 1995, 28, 51-60; (g) Wang, D. M.; Angelici, R. J., J. Am. Chem. Soc. 1996, 118, 935-942.9. (a) Jia, G.; Lough, A. J.; Morris, R. H., Organometallics 1992, 11, 161-171; (b) Abdur-Rashid, K.; Fong, T. P.; Greaves, B.; Gusev, D. G.; Hinman, J. G.; Landau, S. E.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2000, 122, 9155-9171; (c) Li, T.; Lough, A. J.; Zuccaccia, C.; Macchioni, A.; Morris, R. H., Can. J. Chem. 2006, 84, 164-175; (d) Li, T.; Lough, A. J.; Morris, R. H., Chem. Eur. J. 2007, 13, 3796-3803.10. Baker, M. V.; Field, L. D.; Young, D. J., Chem. Commun. 1988, 546-548.11. (a) Abdur-Rashid, K.; Gusev, D. G.; Landau, S. E.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 1998, 120, 11826-11827; (b) Landau, S. E.; Groh, K. E.; Lough, A. J.; Morris, R. H., Inorg. Chem. 2002, 41, 2995-3007.12. Fong, T. P.; Forde, C. E.; Lough, A. J.; Morris, R. H.; Rigo, P.; Rocchini, E.; Stephan, T., Dalton Trans. 1999, 4475-4486.

4salt on the ligand and a metal hydride (Figure 1.2).13 Our group has also observed such an
exchange process from an iridium hydride with a proton on a nitrogen of a sulfur-bonded
thiopyridine ligand.14 In fact, the “NH effect” in polar double bond hydrogenation is based on
this concept (see Section 1.2).
Figure 1.2. Reversible proton exchange of a η2-H2 ligand with a pendant amine arm in a
ruthenium(II) complex containing a diphosphine ligand.
The aforementioned reaction involving the intramolecular heterolytic splitting of H2 can be
extended to the intermolecular regime using external bases. This has important implications for
catalysis. Norton and coworkers have shown that iminium ions can be hydrogenated at 50 psi at
room temperature giving primary amine salts using CpRu(diphosphine)H as the catalyst. In the
catalytic cycle, they have proposed a stepwise mechanism involving first the hydride transfer to
the iminium ion forming a primary amine. The coordination of H2 to the acidic metal center then
follows. Protonation of the product amine by the η2-H2 ligand leads to a primary amine salt. This
is an example of an ionic hydrogenation as the dihydrogen ligand on the transition metal must be
acidic enough to protonate the substrate during the catalytic cycle and the resulting hydride is
hydridic enough to attack the iminium ion.15 A more recent example was reported by Crabtree
and workers in the hydrogenation of quinolines catalyzed by an iridium(I) complex giving
tetrahydroquinolines. A polyhydride iridium(III) complex, [Ir(η2-H2)(H)2(NHC)(PPh3)2]+, was
proposed to be an intermediate in the catalytic cycle. This again involves stepwise protonation of
the substrate by the η2-H2 ligand, and a hydride attack by the Ir–H to the double bond of the
substrate, all occurring in the outer coordination sphere (Figure 1.3).16 These examples show that
molecular hydrogen, when coordinated to a transition metal center, can be acidic enough to
13. (a) Chu, H. S.; Lau, C. P.; Wong, K. Y.; Wong, W. T., Organometallics 1998, 17, 2768-2777; (b) Ayllon, J. A.; Sayers, S. F.; Sabo-Etienne, S.; Donnadieu, B.; Chaudret, B.; Clot, E., Organometallics 1999, 18, 3981-3990.14. Lough, A. J.; Park, S.; Ramachandran, R.; Morris, R. H., J. Am. Chem. Soc. 1994, 116, 8356-8357.15. (a) Magee, M. P.; Norton, J. R., J. Am. Chem. Soc. 2001, 123, 1778-1779; (b) Guan, H. R.; Iimura, M.; Magee, M. P.; Norton, J. R.; Zhu, G., J. Am. Chem. Soc. 2005, 127, 7805-7814.16. Dobereiner, G. E.; Nova, A.; Schley, N. D.; Hazari, N.; Miller, S. J.; Eisenstein, O.; Crabtree, R. H., J. Am. Chem. Soc. 2011, 133, 7547-7562.
Ru
PPh2
Ph2P+ HH
NMe2n
Ru
PPh2
Ph2P
+
H
H
NMe2n
n = 2, 3

5undergo heterolytic splitting with an aid of an external base (B), for example, an amine, giving a
metal hydride and the protonated base (HB).
Figure 1.3. The catalytic hydrogenation of iminium cations (above) and quinolines (below)
catalyzed by ruthenium(II) and iridium(III) catalysts involving an [M](η2-H2) intermediate (M =
Ru or Ir).
1.1.4 Metal Hydrides in Homogeneous Hydrogenation Reactions. Metal hydrides play a
central role in homogeneous hydrogenation reactions. Many important reactions, including the
hydrogenation and isomerization of alkenes, involve the addition of a hydride to a coordinated
double bond (C=C).17 The hydricities of transition metal hydrides can be quantified to compare
the propensity of hydride loss from the metal center, and therefore, the reactivity of these
hydrides. In most cases, however, this has proven to be difficult. There have been some reports
of their thermodynamic properties.18
One of the important reactions is the hydrogenation of terminal alkenes catalyzed by
Wilkinson's catalyst.19 The catalytic hydrogenation reaction using the rhodium(I) catalyst,
RhCl(PPh3)3 (PPh3 = triphenylphosphine), proceeds by first the dissociation of PPh3. Hydrogen
oxidatively adds on rhodium(I) to form a rhodium(III) dihydride complex Rh(H)2Cl(PPh3)2. The
alkene coordinates to the metal center, and hydride migration to the alkene then occurs.
Reductive elimination of the hydride ligand and the metal alkyl affords the product alkane and
17. de Vries, J. G.; Elsevier, C. J., Eds. The Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim, Germany, 2004; Vol 1-3.18. (a) Nietlispach, D.; Bakhmutov, V. I.; Berke, H., J. Am. Chem. Soc. 1993, 115, 9191-9195; (b) Ciancanelli, R.; Noll, B. C.; DuBois, D. L.; DuBois, M. R., J. Am. Chem. Soc. 2002, 124, 2984-2992.19. (a) Evans, D.; Osborn, J. A.; Jardine, F. H.; Wilkinson, G., Nature 1965, 208, 1203-1204; (b) Osborn, J. A.; Jardine, F. H.; Young, J. F.; Wilkinson, G., J. Chem. Soc. A 1966, 1711-1732.
Ru
PPh2
Ph2P H
N+BF4
NH+
BF4
cat.
H2 (50 psi), rt
IrN
N
HPh3P
PPh3
H
H H
cat.
H2 (1 atm), 35°CN N
HToluene
CH2Cl2
+

6regenerates the rhodium(I) form of the catalyst. The cycle then turns over (Scheme 1.1).20 The
ruthenium(II) analogue, RuHCl(PPh3)3, is known to catalyze the hydrogenation of alkenes and
the mechanism of action has been well studied by Halpern.19a, 21 Other catalysts such as
Crabtree's catalyst, [Ir(cod)(py)(PCy3)]+ (cod = 1,5-cyclooctadiene; py = pyridine)22 and the
cationic rhodium complex, [Rh(diphosphine)(CH3OH)2]+, are used for the hydrogenation of
substituted alkenes with functional group tolerance.23 The use of a chiral diphosphine with the
latter catalyst system allows the mass production of the enantiopure product, (L)-DOPA, in one
of Monsanto's processes.24 This is an important drug in the treatment of Parkinson's disease.
These catalysts are shown to have the oxidative addition of dihydrogen to the metal center as one
of the key step in the catalytic cycle.
Scheme 1.1. The Proposed Mechanism for the Hydrogenation of Terminal Alkenes Catalyzed by
the Rhodium(I)-based Wilkinson's Catalyst.
20. (a) Meakin, P.; Jesson, J. P.; Tolman, C. A., J. Am. Chem. Soc. 1972, 94, 3240-3242; (b) Halpern, J.; Okamoto, T.; Zakhariev, A., J. Mol. Cat. 1977, 2, 65-68.21. Fordyce, W. A.; Wilczynski, R.; Halpern, J., J. Organomet. Chem. 1985, 296, 115-125.22. (a) Crabtree, R. H.; Felkin, H.; Morris, G. E., J. Organomet. Chem. 1977, 141, 205-215; (b) Crabtree, R. H.; Felkin, H.; Fillebeenkhan, T.; Morris, G. E., J. Organomet. Chem. 1979, 168, 183-195; (c) Cui, X. H.; Burgess, K., Chem. Rev. 2005, 105, 3272-3296; (d) Church, T. L.; Andersson, P. G., Coord. Chem. Rev. 2008, 252, 513-531.23. (a) Halpern, J.; Riley, D. P.; Chan, A. S. C.; Pluth, J. J., J. Am. Chem. Soc. 1977, 99, 8055-8057; (b) Chan, A. S. C.; Pluth, J. J.; Halpern, J., J. Am. Chem.. Soc. 1980, 102, 5952-5954; (c) Landis, C. R.; Halpern, J., J. Am. Chem. Soc. 1987, 109, 1746-1754.24. Knowles, W. S., Angew. Chem. Int. Ed. 2002, 41, 1998-2007.
RhPh3P
PPh3ClH2
RhH
PPh3
HCl
PPh3
RhH
HClPPh3 R
Rh
PPh3
HPPh3
R
H
R
HH
- PPh3
RhPPh3Ph3P
PPh3Cl
RPPh3
Cl

71.2 Mechanisms of the Hydrogenation of Polar Double Bonds
1.2.1 Ketone Hydrogenation, the Outer-sphere Mechanism and the “NH Effect”. Early
attempts to hydrogenate aldehydes and ketones used ruthenium catalysts Ru(η2-H2)(H)2(PPh3)325
and cis-RuH2(PPh3)426 with hydrogen or alcohol as the reductant, respectively. Although these
reactions proceed smoothly at room temperature, the hydride affinity to the polar double bond is
low and the substrate scope is limited. For the design of highly active catalysts for polar double
bond reduction, the use of strong donor ligands is needed to stabilize the conjugate Lewis acid
from the metal hydride complex in order to favor the attack of the polar double bond. In addition,
the use of a strong trans-influence ligand across the metal from the hydride can labilize the metal
hydride bond and makes it more reactive.1c
Early work conducted by Noyori and co-workers used a diphosphine ligand on ruthenium for
ketone hydrogenation. The asymmetric hydrogenation of ketones using H2 was achieved with an
enantiomeric excess of up to 99% using an enantiopure BINAP ligand (BINAP = 2,2'-
bis(diphenylphosphino)-1,1'-binaphthyl) in the system [Ru(η6-arene)((S)-BINAP)(halide)]+.27
This process uses a relatively high pressure of H2 (up to 100 atm) at 30°C. Later the same
research group discovered that the addition of a diamine ligand in the reaction pot containing
RuCl2(diphosphine)(solvent)2 and potassium hydroxide catalyzed the H2-hydrogenation of
ketones under a low pressure of H2 (4 atm) at room temperature with a substrate to catalyst
loading of 500/1.28 When a chiral diphosphine was used, an enantiomeric excess up to 99% was
obtained.28-29 Noyori called the positive influence of the diamine on catalysis the “NH effect”,1b,
30 as it was shown that the absence of an NH group in these catalytic systems resulted in lower
activity. Much effort has been devoted to study the importance of the “NH effect” in bifunctional
catalysis, including the work of Baratta,31 Bergens,32 Clarke,33 Ikariya,34 Wills35 and many
others.36
25. (a) Cole-Hamilton, D. J.; Wilkinson, G., New J. Chem. 1977, 1, 141-155; (b) Linn, D. E.; Halpern, J., J. Am. Chem. Soc. 1987, 109, 2969-2974.26. (a) Imai, H.; Nishiguchi, T.; Fukuzumi, K., J. Org. Chem. 1976, 41, 665-671; (b) Hayashi, Y.; Komiya, S.; Yamamoto, T.; Yamamoto, A., Chem. Lett. 1984, 1363-1366.27. Mashima, K.; Kusano, K. H.; Ohta, T.; Noyori, R.; Takaya, H., Chem. Commun. 1989, 1208-1210.28. Ohkuma, T.; Ooka, H.; Hashiguchi, S.; Ikariya, T.; Noyori, R., J. Am. Chem. Soc. 1995, 117, 2675-2676.29. Doucet, H.; Ohkuma, T.; Murata, K.; Yokozawa, T.; Kozawa, M.; Katayama, E.; England, A. F.; Ikariya, T.; Noyori, R., Angew. Chem. Int. Ed. 1998, 37, 1703-1707.30. (a) Noyori, R.; Ohkuma, T., Angew. Chem. Int. Ed. 2001, 40, 40-73; (b) Noyori, R., Angew. Chem. Int. Ed. 2002, 41, 2008-2022.31. Baratta, W.; Ballico, M.; Esposito, G.; Rigo, P., Chem. Eur. J. 2008, 14, 5588-5595.32. Leong, C. G.; Akotsi, O. M.; Ferguson, M. J.; Bergens, S. H., Chem. Commun. 2003, 750-751.33. Phillips, S. D.; Fuentes, J. A.; Clarke, M. L., Chem. Eur. J. 2010, 16, 8002-8005.

8Noyori and co-workers proposed that the bifunctional catalysis of ketone hydrogenation
involves the dual action of the metal hydride and the protic amine group. It was proposed this
proceeds via a six-membered pericyclic transition state involving hydrogen bonding of the N-H
group with the oxygen of the ketone and attack of the carbonyl group by the metal hydride. The
concerted transfer of the H+/H- pair to the ketone affords the product alcohol, and a metal-amido
bond is formed. A hydrogen molecule then coordinates to the metal center in a side-on fashion.
The amido nitrogen, which acts as a strong Lewis base, is responsible for the heterolytic splitting
of the η2-H2 ligand, forming an N–H and an M–H bond (Scheme 1.2).1b-f, 1i Significantly, Noyori
and co-workers have isolated all the intermediates in this bifunctional mechanism using the
precatalyst RuCl(η6-p-cymene){(S,S)-TsNCHPhCHPhNH2} (TsNCHPhCHPhNH2 = N-(p-
toluenesulfonyl)-1,2-diphenylethylene-diamine) and KOH in the transfer hydrogenation of
ketones in 2-propanol, which serves as the hydrogen source. These include the hydride-amine
complex RuH(η6-p-cymene){(S,S)-TsNCHPhCHPhNH2} and the amido complex Ru(η6-p-
cymene){(S,S)-TsNCHPhCHPhNH}. They showed identical activities compared to the activated
precatalyst in the transfer hydrogenation of ketones in 2-propanol. No base was required for
catalysis (Figure 1.4).37
Scheme 1.2. The Outer Sphere Bifunctional Mechanism Using the “NH effect” for the
Hydrogenation of Aromatic Ketones.
34. Ito, M.; Hirakawa, M.; Murata, K.; Ikariya, T., Organometallics 2001, 20, 379-381.35. Soni, R.; Cheung, F. K.; Clarkson, G. C.; Martins, J. E. D.; Graham, M. A.; Wills, M., Org. Biomol. Chem. 2011, 9, 3290-3294.36. (a) Standfest-Hauser, C.; Slugovc, C.; Mereiter, K.; Schmid, R.; Kirchner, K.; Xiao, L.; Weissensteiner, W., Dalton Trans. 2001, 2989-2995; (b) Lundgren, R. J.; Rankin, M. A.; McDonald, R.; Schatte, G.; Stradiotto, M., Angew. Chem. Int. Ed. 2007, 46, 4732-4735; (c) Ma, G. B.; McDonald, R.; Ferguson, M.; Cavell, R. G.; Patrick, B. O.; James, B. R.; Hu, T. Q., Organometallics 2007, 26, 846-854.37. Haack, K. J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R., Angew. Chem. Int. Ed. 1997, 36, 285-288.
[M]
N
D
H
H2
OR'
OHH
R'
R
R
[M]
N
D
H
H
H
[M]
N
D
H
HH
[M]H
N
D
HH
O
R'
R
D = Donor ligand

9Our recent research program involved a study of the bifunctional action of the catalyst
systems RuHX(diamine)(phosphine)2 (X = H, BH4, H, OR) in the asymmetric H2-hydrogenation
of ketones and imines.38 The key intermediates of the catalytic cycle involving a trans-dihydride-
amine group and a monohydrido-amido group attached to the metal center have been identified
and structurally characterized.38a, 38b, 38g, 38h Among all of these catalysts, the complexes trans-
Ru(H)2((R)-BINAP)(tmen) and (OC-6-22)-Ru(H)2(PPh3)2(tmen) (tmen = 2,3-dimethylbutane-
2,3-diamine) were found to have the heterolytic splitting of a coordinated η2-H2 ligand on the
active species as the rate-determining step according to various experimental and theoretical
studies.38a, 38b, 38g, 38i These are active catalysts, without prior activation by base, and catalyze
efficiently the reduction of ketones by H2 under very mild conditions in non-polar solvents. The
corresponding coordinatively unsaturated complexes containing a ruthenium-amido bond were
isolated, and these were also found to activate dihydrogen to give the trans-dihydride complexes
(Figure 1.4).38a, 38b, 38g
Figure 1.4. Examples of ruthenium(II) complexes with hydride-amine or an amido group that
catalyze the hydrogenation of ketones under base free conditions.
1.2.2 Effect of Alcohols on the Outer-sphere Bifunctional Mechanism using the “NH
Effect”. Many research groups have found that the presence of alcohols in bifunctional catalysis
accelerates the catalytic hydrogenation of ketones. For example, Ikariya34 and Andersson39 have
found that their ruthenium(II) catalyst systems, RuCp*(diamine)Cl, catalyze efficiently the
hydrogenation of ketones using molecular hydrogen. Deuterium labelling studies conducted by
38. (a) Abdur-Rashid, K.; Faatz, M.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2001, 123, 7473-7474; (b) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2002, 124, 15104-15118; (c) Rautenstrauch, V.; Hoang-Cong, X.; Churlaud, R.; Abdur-Rashid, K.; Morris, R. H., Chem. Eur. J. 2003, 9, 4954-4967; (d) Guo, R.; Lough, A. J.; Morris, R. H.; Song, D., Organometallics 2004, 23, 5524-5529; (e) Abdur-Rashid, K.; Guo, R. W.; Lough, A. J.; Morris, R. H.; Song, D. T., Adv. Synth. Catal. 2005, 347, 571-579; (f) Guo, R. W.; Chen, X. H.; Elpelt, C.; Song, D. T.; Morris, R. H., Org. Lett. 2005, 7, 1757-1759; (g) Abbel, R.; Abdur-Rashid, K.; Faatz, M.; Hadzovic, A.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2005, 127, 1870-1882; (h) Hadzovic, A.; Song, D.; MacLaughlin, C. M.; Morris, R. H., Organometallics 2007, 26, 5987-5999; (i) Zimmer-De Iuliis, M.; Morris, R. H., J. Am. Chem. Soc. 2009, 131, 11263-11269.39. Hedberg, C.; Kallstrom, K.; Arvidsson, P. I.; Brandt, P.; Andersson, P. G., J. Am. Chem. Soc. 2005, 127, 15083-15090.
RuH
HNN
H H
PPh2
Ph2PRu
N NH SO
ORu
H
NN
HPh3P
Ph3PH H
Ru
H
NN
HPh3P
Ph3PH H
H H
H H

10Ikariya and co-workers revealed that there was significant deuterium scrambling between the
hydrogen/deuterium gas and the hydroxyl group of the alcohol solvent. 34 Andersson have
computed a smaller energy barrier in the heterolytic splitting of the η2-H2 ligand with an alcohol
serving as a proton shuttle.39 The six-membered ring transition state of the heterolytic splitting
has the hydroxyl group of the alcohol in close proximity to the dihydrogen ligand and the N–H
group, forming two hydrogen-bonds (Figure 1.5). It was concluded that the presence of alcohol
can act as proton shuttle to assist the heterolytic splitting of the η2-H2 ligand.1f, 39-40 We have also
observed a similar effect of alcohol recently where the transition state has been computed.38h
Figure 1.5. The transition state of the alcohol-assisted heterolytic splitting of H2 calculated by
Andersson and co-workers using computational methods.
1.2.3 Concerted or Stepwise Transfer of a Proton/Hydride Couple from the Metal Center
to the Polar Double bond. There has been a debate whether the transfer of the M–H/N–H group
to the polar double bond is a concerted or a stepwise process. Bergens and co-workers have
observed a ruthenium(II) alkoxide complex trans-Ru((R)-BINAP)(H)(PhMeCHO)((R,R)-dpen))
(dpen = 1,2-diphenylethylenediamine) upon the reaction of the trans-dihydride complex, trans-
Ru((R)-BINAP)(H)2((R,R)-dpen)), and acetophenone at -78°C.41 They have also studied a similar
intermediate by using intermolecular trapping agents42 and in the hydrogenation of lactones.43
Gusev and co-workers have recently computed the catalytic cycle for the dehydrogenation of
alcohols catalyzed by the trans-dihydride osmium(IV) complex trans-Os(H)2(CO)(κ3(P,N,P)-
HN(C2H4PiPr2)2), and found there is a significant interaction of the N–H group with the 2-
propoxide anion in the transition state structure.44 An alkoxide complex of ruthenium(II), trans-
Ru(H)(OiPr)(CO)(κ3(P,N,P)-HN(C2H4PiPr2)2) was structurally characterized, and this was in fast
40. (a) Sandoval, C. A.; Ohkuma, T.; Muniz, K.; Noyori, R., J. Am. Chem. Soc. 2003, 125, 13490-13503; (b) Casey, C. P.; Johnson, J. B.; Singer, S. W.; Cui, Q., J. Am. Chem. Soc. 2005, 127, 3100-3109; (c) Casey, C. P.; Johnson, J. B.; Jiao, X. D.; Beetner, S. E.; Singer, S. W., Chem. Commun. 2010, 46, 7915-7917.41. Hamilton, R. J.; Bergens, S. H., J. Am. Chem. Soc. 2008, 130, 11979-11987.42. Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H., J. Am. Chem. Soc. 2011, 133, 9666-9669.43. Takebayashi, S.; Bergens, S. H., Organometallics 2009, 28, 2349-2351.44. Bertoli, M.; Choualeb, A.; Gusev, D. G.; Lough, A. J.; Major, Q.; Moore, B., Dalton Trans. 2011, 40, 8941-8949.
Ru
NN
H
HH
OH H

11equilibrium with the trans-dihydride complex, trans-Ru(H)2(CO)(κ3(P,N,P)-HN(C2H4PiPr2)2) in
solution.45
Casey and co-workers provided evidence to support a concerted transfer process for the
hydrogenation of aldehydes using the Shvo's type catalyst 2,5-Ph2-3,4-Tol2(η5-C4COH)
Ru(CO)2H (Figure 1.6).46 The products of the kinetic isotope effects measured upon deuteration
only at the metal hydride (kRuH/kRuD = 1.5 ± 0.2), and only at the hydroxyl group of the
pentadienyl ligand (kOH/kOD = 2.2 ± 0.1) is in good agreement with that measured for deuteration
at both sites (kRuH/OH/kRuD/OD = 3.6 ± 0.3). They further demonstrated that both the hydroxyl
group and the metal hydride were required for catalysis as no alkoxide complex was observed
when benzaldehyde was reacted with the complex, 2,5-Ph2-3,4-Tol2(η5-C4COSiEt3)Ru(CO)2H,
which lacked a hydroxyl group.46 Recent studies has been devoted to the analogous iron(II)
system, and a concerted transfer step was supported by experimental studies.47
Figure 1.6. Concerted transfer of a proton/hydride couple from the Shvo's type catalyst to
benzaldehyde.
1.2.4 The Inner Sphere Mechanism. The coordination of a ketone to a metal center during its
catalytic hydrogenation is a key step in the inner sphere mechanism (Scheme 1.3). In the case of
H2-hydrogenation, an alkoxide base is required to activate the precatalyst. This forms a strong
metal-alkoxide bond. A dihydrogen molecule then coordinates to one of the vacant coordination
sites in a side-on fashion. The alkoxide then acts as an internal base to heterolytically split the η2-
H2 ligand, giving a metal hydride and alcohol. The ketone then coordinates to the metal center
and is attacked by the metal-hydride in a four-membered ring transition state, leading back to the
metal alkoxide complex. The protonation of this alkoxide by H2 releases the product alcohol.1c
45. Bertoli, M.; Choualeb, A.; Lough, A. J.; Moore, B.; Spasyuk, D.; Gusev, D. G., Organometallics 2011, 30, 3479-3482. 46. Casey, C. P.; Singer, S. W.; Powell, D. R.; Hayashi, R. K.; Kavana, M., J. Am. Chem. Soc. 2001, 123, 1090-1100.47. Casey, C. P.; Guan, H., J. Am. Chem. Soc. 2009, 131, 2499-2507.
RuH
OC
O
OC
H
PhTolTol
PhO
H
RuH
OC
O
OC
H
PhTolTol
PhRu
OC
O
OC
PhTolTol
Ph
O
H OH
H H+

12Scheme 1.3. The Inner Sphere Mechanism for the H2-Hydrogenation of Aromatic Ketones.
The hydrogenation of β-keto-esters was a well known process using enantiomerically pure
catalysts of the types RuCl2(BINAP)(solvent)2 or [Ru(BINAP)(bisolefin)(solvent)2]2+ prior to the
discovery of the “NH effect”.48 It was proposed that the active catalyst was generated by
dehydrochlorination using hydrogen. The β-keto-ester then chelates to the metal center during
the catalytic cycle. Protonation of the ketone oxygen by HCl in the alcoholic solvent makes the
carbonyl carbon more electrophilic to facilitate the attack of the metal-hydride on the carbonyl
carbon. This releases the product alcohol. The acidic hydrogen on the η2-H2 ligand was
deprotonated by the alcoholic solvent to regenerate the active species for catalysis.17 Noyori and
co-workers have described the intramolecular attack of the metal hydride on the ketone σ-bonded
through the oxygen atom as a [2 + 2] addition. The required change in the geometry of the metal
complex to align the orbitals makes this process energetically demanding (Figure 1.7). The
presence of the chelating group, in the case of the hydrogenation of a β-keto-ester, favours a π-
interaction of the metal-hydride and the carbonyl group of the ketone.1b, 30a
Figure 1.7. Orbital interaction diagram showing the hydride attack on a σ-bonded ketone.
48. Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, S., J. Am. Chem. Soc. 1987, 109, 5856-5858.
[M]H2
OR'
OHH
R'
R
R
OHR'
R
[M]
OHR'
R
H
H
[M]
O
R' R
H
[M]
OR'
R
H
MH
OH
CR
R'π *
MH
OH
CR
R'π *

131.3 Applications of Phosphines and N-Heterocyclic Carbenes in Catalysis
1.3.1 Applications of Phosphines in Catalysis. A phosphine ligand (PR3) is known to be a
strong σ-donor and a moderate π-acceptor using its σ* orbital to interact with the dπ orbital at the
metal center.49 The tuneable electronic and steric properties of the R substituent at phosphorus
makes a phosphine a very attractive and useful ligand in catalysis.50 Phosphines are used in many
of the important catalytic reactions including cross-coupling reactions, hydroformylation
reactions, metathesis reactions and more. Many of these have been studied and reviewed
extensively in the literature.51
Functionalized phosphine ligands containing either a phosphine donor (diphosphine) or a
nitrogen-based donor (P,N-type ligand) are important in the design of highly active catalysts
systems.52 The use of a chiral diphosphine ligand, JOSIPHOS (Figure 1.8), for example, in an
iridium catalyst provides high activity and high enantiomeric excess in the asymmetric
hydrogenation of imines.53 This was applied in the large scale production of an enantiomerically
pure N-alkylated aniline (NAA) from a MEA imine (MEA = 2-methyl-5-ethyl-aniline, Figure
1.8), an important intermediate for the synthesis of metolachlor.53b, 53d, 53e The chiral BINAP
ligand also has rich applications in the hydrogenation of alkenes and ketones (see Section 1.2).
Figure 1.8. The production of an (S)-N-alkylated aniline (NAA) using an iridium complex
containing a JOSIPHOS ligand as a catalyst.
49. Orpen, A. G.; Connelly, N. G., Chem. Commun. 1985, 1310-1311.50. (a) Tolman, C. A., Chem. Rev. 1977, 77, 313-348; (b) Mason, R.; Meek, D. W., Angew. Chem. Int. Ed. 1978, 17, 183-194; (c) Dias, P. B.; Depiedade, M. E. M.; Simoes, J. A. M., Coord. Chem. Rev. 1994, 135, 737-807.51. (a) Puddephatt, R. J., Chem. Soc. Rev. 1983, 12, 99-127; (b) Farina, V.; Krishnan, B., J. Am. Chem. Soc. 1991, 113, 9585-9595; (c) Nguyen, S. T.; Grubbs, R. H.; Ziller, J. W., J. Am. Chem. Soc. 1993, 115, 9858-9859; (d) Kranenburg, M.; Vanderburgt, Y. E. M.; Kamer, P. C. J.; Vanleeuwen, P.; Goubitz, K.; Fraanje, J., Organometallics 1995, 14, 3081-3089; (e) Hartwig, J. F., Acc. Chem. Res. 1998, 31, 852-860; (f) Tang, W. J.; Zhang, X. M., Chem. Rev. 2003, 103, 3029-3069; (g) Grubbs, R. H., Tetrahedron 2004, 60, 7117-7140; (h) Trnka, T. M.; Grubbs, R. H., Acc. Chem. Res. 2001, 34, 18-29; (i) Surry, D. S.; Buchwald, S. L., Chem. Sci. 2011, 2, 27-50.52. Guiry, P. J.; Saunders, C. P., Adv. Synth. Catal. 2004, 346, 497-537.
N
OCH3
NH
OCH3cat.
H2
FePR2
PR'2
0.5 equiv [Ir(cod)Cl]2

14P,N-type ligands (P–N) are used in the asymmetric hydrogenation of hindered alkenes.22c, 54
These have a phosphine donor and a functionalized chiral oxazoline donor. Pfaltz and Burgess
showed that iridium(I) catalysts of the type [Ir(P–N)(cod)]+ have excellent activity in catalyzing
the asymmetric hydrogenation of tetrasubstituted alkenes to chiral alkanes in good enantiomeric
excess.22c, 54d On the other hand, certain phosphine ligands with a primary amine donor
(phosphine-amine ligand) form an important class of ligands that are used in the asymmetric
hydrogenation of polar double bonds. With well established synthetic procedures of such ligands
reported in the literature,55 the ruthenium(II) complexes containing such ligands have found
wide applications in the hydrogenation of ketones,33, 56 imines,38e imides,57 esters58 and in the
dehydrogenation of amine boranes.59 Many of these catalytic systems were found to utilize the
bifunctional mechanism using the “NH effect”.33, 57, 59
1.3.2 N-heterocyclic carbenes and their Late Transition Metal Complexes. Singlet carbenes
are excellent Lewis bases by virtue of their highest energy occupied molecular orbital on carbon.
Although free carbenes are highly reactive in nature, many organometallic complexes with
coordinated carbene ligands are known in the literature.60 When heteroatoms are attached to the
carbene carbon of an acyclic, bent singlet carbene, σ-withdrawing substituents tends to lower the
energy level of the non-bonding σ orbital (inductive effect); while π-donating substituents, for
example nitrogen atoms, raise the energy level of the empty π-orbital (mesomeric effect, Figure
1.9). This results in a large σ–π energy gap, providing an extra stabilization of the singlet ground
state on the sp2-hybridized carbene carbon.2c, 60e, 61 N-heterocyclic carbenes are the most
important cyclic, bent singlet carbene in which stabilization of the singlet state is achieved by
presence of two nitrogen π-donors adjacent to the carbene carbon. 53. (a) Spindler, F.; Pugin, B.; Blaser, H. U., Angew. Chem. Int. Ed. 1990, 29, 558-559; (b) Blaser, H. U.;
Buser, H. P.; Coers, K.; Hanreich, R.; Jalett, H. P.; Jelsch, E.; Pugin, B.; Schneider, H. D.; Spindler, F.; Wegmann, A., Chimia 1999, 53, 275-280; (c) Dai, L. X.; Tu, T.; You, S. L.; Deng, W. P.; Hou, X. L., Acc. Chem. Res. 2003, 36, 659-667; (d) Blaser, H. U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M., Adv. Synth. Catal. 2003, 345, 103-151; (e) Blaser, H. U.; Pugin, B.; Spindler, F.; Thommen, M., Acc. Chem. Res. 2007, 40, 1240-1250.54. (a) Helmchen, G.; Pfaltz, A., Acc. Chem. Res. 2000, 33, 336-345; (b) Braunstein, P.; Naud, F., Angew. Chem. Int. Ed. 2001, 40, 680-699; (c) McManus, H. A.; Guiry, P. J., Chem. Rev. 2004, 104, 4151-4202; (d) Roseblade, S. J.; Pfaltz, A., Acc. Chem. Res. 2007, 40, 1402-1411.55. Ito, M.; Osaku, A.; Kobayashi, C.; Shiibashi, A.; Ikariya, T., Organometallics 2009, 28, 390-393.56. (a) Guo, R.; Morris, R. H.; Song, D., J. Am. Chem. Soc. 2005, 127, 516-517; (b) Jia, W. L.; Chen, X. H.; Guo, R. W.; Sui-Seng, C.; Amoroso, D.; Lough, A. J.; Abdur-Rashid, K., Dalton Trans. 2009, 8301-8307.57. (a) Ito, M.; Sakaguchi, A.; Kobayashi, C.; Ikariya, T., J. Am. Chem. Soc. 2007, 129, 290-291; (b) Ito, M.; Kobayashi, C.; Himizu, A.; Ikariya, T., J. Am. Chem. Soc. 2010, 132, 11414-11415.58. (a) Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P., Angew. Chem. Int. Ed. 2007, 46, 7473-7476; (b) Clarke, M. L.; Diaz-Valenzuela, M. B.; Slawin, A. M. Z., Organometallics 2007, 26, 16-19; (c) Kuriyama, W.; Ino, Y.; Ogata, O.; Sayo, N.; Saito, T., Adv. Synth. Catal. 2010, 352, 92-96.59. Blaquiere, N.; Diallo-Garcia, S.; Gorelsky, S. I.; Black, D. A.; Fagnou, K., J. Am. Chem. Soc. 2008, 130, 14034-14035.

15
Figure 1.9. Schematics showing the mesomeric effect to the electronic configuration and the
interactions of π-orbitals in the acyclic, bent singlet carbene N–C–N.
Since the initial discovery of Bertrand's carbene62 and Arduengo's stable cyclic
diaminocarbene, or N-heterocyclic carbene (NHC)63 (Figure 1.10), much work has been devoted
to study the properties of carbene donors. 2c, 60e, 61, 64 A study of the thermodynamic properties of
the NHC suggests its σ-donor property is superior to that of phosphines, as a result of the
presence of a high energy filled σ-orbital.65 Similar to phosphines, the tuneable electronics and
steric properties of an N-heterocyclic carbene makes it very attractive to be used as a ligand in
catalysis. Pioneering work conducted by Herrmann and co-workers on the use of the NHC-
containing complex, Pd(NHC)2I2, in Heck-coupling reactions revealed that such complexes are
more reactive, and are moisture, heat and oxygen tolerant compared to their phosphine
60. (a) Fischer, E. O.; Maasbol, A., Angew. Chem. Int. Ed. 1964, 3, 580-581; (b) Cardin, D. J.; Cetinkay.B; Lappert, M. F., Chem. Rev. 1972, 72, 545-574; (c) Schrock, R. R., J. Am. Chem. Soc. 1974, 96, 6796-6797; (d) Schrock, R. R., Acc. Chem. Res. 1979, 12, 98-104; (e) Bourissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G., Chem. Rev. 2000, 100, 39-91.61. de Frémont, P.; Marion, N.; Nolan, S. P., Coord. Chem. Rev. 2009, 253, 862-892.62. Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G., J. Am. Chem. Soc. 1988, 110, 6463-6466.63. Arduengo, A. J.; Harlow, R. L.; Kline, M., J. Am. Chem. Soc. 1991, 113, 361-363.64. (a) Leuthausser, S.; Schwarz, D.; Plenio, H., Chem. Eur. J. 2007, 13, 7195-7203; (b) Fantasia, S.; Petersen, J. L.; Jacobsen, H.; Cavallo, L.; Nolan, S. P., Organometallics 2007, 26, 5880-5889; (c) Fey, N.; Haddow, M. F.; Harvey, J. N.; McMullin, C. L.; Orpen, A. G., Dalton Trans. 2009, 8183-8196; (d) Gusev, D. G., Organometallics 2009, 28, 763-770; (e) Gusev, D. G., Organometallics 2009, 28, 6458-6461; (f) Droge, T.; Glorius, F., Angew. Chem. Int. Ed. Engl. 2010, 49, 6940-6952.65. Huang, J.; Jafarpour, L.; Hillier, A. C.; Stevens, E. D.; Nolan, S. P., Organometallics 2001, 20, 2878-2882.66. Herrmann, W. A.; Elison, M.; Fischer, J.; Kocher, C.; Artus, G. R. J., Angew. Chem. Int. Ed. 1995, 34, 2371-2374.
pπ
pπ
π
π ∗
n.b.
n.b. = non bonding
NC
N
NC
N
NC
N
π
π ∗
n.b.
pπ
pπ
π
π ∗
n.b.
O OC
N NC

16counterparts.66 Many of the palladium(II) based systems with NHC ligands are now used in C–C
coupling reactions.2a, 2b, 2d, 2e, 67 Grubbs and co-workers have successfully replaced one of the
tricyclohexylphosphines of their first generation catalyst, Ru(PCy3)2Cl2(CHPh) with an N-
heterocyclic carbene to give highly active catalysts, Ru(NHC)(PCy3)Cl2(CHPh), for diverse
applications in alkene metathesis reactions.68 The application of ruthenium complexes containing
N-heterocyclic carbene ligands in alkene metathesis reactions has recently been reviewed.69
Figure 1.10. The structures of Bertrand's and Arduengo's carbenes.
Many ruthenium(II) complexes containing N-heterocyclic carbenes were found to be useful in
the activation of heteroatom bonds of small molecules. Our group observed and isolated the first
ruthenium(II) hydride complex containing 1,3-bis(2,4,6-trimethylphenyl)-imidazol-2-ylidene
(IMes) as a ligand, with C–H bond activation on the mesityl group of the NHC ligand.70
Whittlesey and co-workers have studied this further and extended this to the activation of C–F
bonds.71 Of note, the highly active four-coordinate ruthenium(II) complex RuH(CO)(IMes)2+
induces C–H bond activation on the IMes ligand as indicated by labelling studies using D2 gas.72
On the other hand, the five coordinate ruthenium(II) complex RuH(NHC)4+ allows coordination
of H2, CO, N2, and O2. The hydrogen and oxygen molecules were found to coordinate in a side-
on fashion to the coordinatively unsaturated metal center.73 A coordinatively unsaturated
67. Kantchev, E. A. B.; O'Brien, C. J.; Organ, M. G., Angew. Chem. Int. Ed. 2007, 46, 2768-2813.68. Trnka, T. M.; Morgan, J. P.; Sanford, M. S.; Wilhelm, T. E.; Scholl, M.; Choi, T. L.; Ding, S.; Day, M. W.; Grubbs, R. H., J. Am. Chem.. Soc. 2003, 125, 2546-2558.69. Vougioukalakis, G. C.; Grubbs, R. H., Chem. Rev. 2010, 110, 1746-1787.70. Abdur-Rashid, K.; Fedorkiw, T.; Lough, A. J.; Morris, R. H., Organometallics 2004, 23, 86-94.71. (a) Chilvers, M. J.; Jazzar, R. F. R.; Mahon, M. F.; Whittlesey, M. K., Adv. Synth. Catal. 2003, 345, 1111-1114; (b) Burling, S.; Paine, B. M.; Nama, D.; Brown, V. S.; Mahon, M. F.; Prior, T. J.; Pregosin, P. S.; Whittlesey, M. K.; Williams, J. M. J., J. Am. Chem. Soc. 2007, 129, 1987-1995; (c) Diggle, R. A.; Kennedy, A. A.; Macgregor, S. A.; Whittlesey, M. K., Organometallics 2008, 27, 938-944; (d) Reade, S. P.; Acton, A. L.; Mahon, M. F.; Martin, T. A.; Whittlesey, M. K., Eur. J. Inorg. Chem. 2009, 1774-1785; (e) Reade, S. P.; Mahon, M. F.; Whittlesey, M. K., J. Am. Chem. Soc. 2009, 131, 1847-1861; (f) Panetier, J. A.; Macgregor, S. A.; Whittlesey, M. K., Angew. Chem. Int. Ed. 2011, 50, 2783-2786.72. Lee, J. P.; Ke, Z. F.; Ramirez, M. A.; Gunnoe, T. B.; Cundari, T. R.; Boyle, P. D.; Petersen, J. L., Organometallics 2009, 28, 1758-1775.73. (a) Burling, S.; Haller, L. J. L.; Mas-Marza, E.; Moreno, A.; Macgregor, S. A.; Mahon, M. F.; Pregosin, P. S.; Whittlesey, M. K., Chem. Eur. J. 2009, 15, 10912-10923; (b) Haller, L. J. L.; Mas-Marza, E.; Moreno, A.; Lowe, J. P.; Macgregor, S. A.; Mahon, M. F.; Pregosin, P. S.; Whittlesey, M. K., J. Am. Chem. Soc. 2009, 131, 9618-9619.
NP SiMe3N N N

17rhodium(I) complex containing NHC ligands (RhCl(NHC)2) reacts with oxygen forming an η2-
O2 complex and retains the same oxidation state on rhodium.74
Other late transition metal NHC complexes such as those of iridium and platinum have also
found applications in catalysis. Iridium(III) complexes containing [IrCp*(NHC)]2+ are useful
catalysts in the Oppenauer-type oxidation reaction, providing useful carbonyl compounds from
the corresponding alcohols.75 Peris and co-workers have found that these complexes are useful in
the transfer hydrogenation of carbon dioxide to a formate anion.76 Platinum(0) complexes
containing an NHC are useful for the hydrosilylation of terminal alkynes.77 Nolan and co-
workers have recently reported the use of stable gold(I) NHC complexes in the hydration of
alkenes, alkynes and nitriles under silver free conditions, and in the carboxylation of aromatic
and heteroaromatic C–H bonds (Figure 1.11).78
Figure 1.11. Examples of late transition metal complexes and their use in (a) the oxidation of
alcohols, (b) the hydrosilylation of a terminal alkyne, and in (c) the carboxylation of aromatic
and heteroaromatic C–H bonds (from top to bottom).
Some ruthenium(II) complexes containing NHC ligand were found to be useful catalysts for
the hydrogenation of alkenes.72, 79 Iridium(I) complexes containing a functionalized chiral
oxazoline group on the NHC ligand was found to be useful in the asymmetric hydrogenation of 74. Praetorius, J. M.; Allen, D. P.; Wang, R. Y.; Webb, J. D.; Grein, F.; Kennepohl, P.; Crudden, C. M., J.
Am. Chem. Soc. 2008, 130, 3724-3725.
cat.
Ir
NN
NNH3C
H3C
2+
O
K2CO3 , acetone40°C
HO H
N N
Pt
SiOSi
o-xylene80°C
cat.C6H13 + Me3Si O
SiO
SiMe3H3C H C6H13
SiMe
OSiMe3
OSiMe3
N N
Au
OHcat.
KOH, THF20°C
R
NHR'CO2 + HCl
R
NR'O
OH

18hindered alkenes (see Section 1.3.3).22c, 80 In addition, many ruthenium(II) and iridium(III)
complexes containing NHC ligands are highly active catalysts for the transfer hydrogenation of
ketones in basic 2-propanol solution.81 Recently,Whittlesey and co-workers have investigated
their ruthenium(II) complexes in the formation of C–C and C–N bonds from alcohols and amines
by the hydrogen borrowing methodology.82
1.3.3 Donor-functionalized N-heterocyclic carbenes and applications in catalysis. The
installation of a donor group adjacent to the nitrogen atom of the imidazolylidene ring provides a
donor-functionalized NHC ligand. There are many reports that the presence of such a donor
group provides extra stability to the catalyst, and sometimes unique activity to catalysis.2c, 2d, 2f, 83
The synthesis of the donor-functionalized NHC ligand requires the synthesis of an
unsymmetrical imidazolium salt.84 The most common way to access this class of imidazolium
salt is by direct alkylation of a substituted imidazole,85 or via a copper-catalyzed Ullman-
coupling reaction of imidazole and a functionalized aryl group, and then alkylation of the
resulting aryl-imidazole.86 Douthwaite and co-workers prepared chiral bis-imidazoles by the
reaction of a chiral diimine and tosylmethyl isocyanide in a 1,3-cycloaddition reaction.87 A
change in reaction conditions leads to the reaction of one imine group leading to the formation of
a chiral imidazole with an imine functional group.88
75. (a) Hanasaka, F.; Fujita, K.; Yamaguchi, R., Organometallics 2005, 24, 3422-3433; (b) Hanasaka, F.; Fujita, K.-i.; Yamaguchi, R., Organometallics 2006, 25, 4643-4647.76. Sanz, S.; Benitez, M.; Peris, E., Organometallics 2010, 29, 275-277.77. (a) Marko, I. E.; Sterin, S.; Buisine, O.; Mignani, G.; Branlard, P.; Tinant, B.; Declercq, J. P., Science 2002, 298, 204-206; (b) Buisine, O.; Berthon-Gelloz, G.; Briere, J. F.; Sterin, S.; Mignani, G.; Branlard, P.; Tinant, B.; Declercq, J. P.; Marko, I. E., Chem. Commun. 2005, 3856-3858; (c) De Bo, G.; Berthon-Gelloz, G.; Tinant, B.; Marko, I. E., Organometallics 2006, 25, 1881-1890.78. Nolan, S. P., Acc. Chem. Res. 2011, 44, 91-100.79. (a) Lee, H. M.; Smith, D. C.; He, Z. J.; Stevens, E. D.; Yi, C. S.; Nolan, S. P., Organometallics 2001, 20, 794-797; (b) Dharmasena, U. L.; Foucault, H. M.; dos Santos, E. N.; Fogg, D. E.; Nolan, S. P., Organometallics 2005, 24, 1056-1058; (c) Gandolfi, C.; Heckenroth, M.; Neels, A.; Laurenczy, G.; Albrecht, M., Organometallics 2009, 28, 5112-5121; (d) Horn, S.; Albrecht, M., Chem. Commun. 2011, 47, 8802-8804; (e) Horn, S.; Gandolfi, C.; Albrecht, M., Eur. J. Inorg. Chem. 2011, 2863-2868.80. (a) Hou, D. R.; Reibenspies, J.; Colacot, T. J.; Burgess, K., Chem. Eur. J. 2001, 7, 5391-5400; (b) Powell, M. T.; Hou, D.-R.; Perry, M. C.; Cui, X.; Burgess, K., J. Am. Chem. Soc. 2001, 123, 8878-8879.81. (a) Albrecht, M.; Miecznikowski, J. R.; Samuel, A.; Faller, J. W.; Crabtree, R. H., Organometallics 2002, 21, 3596-3604; (b) Burling, S.; Whittlesey, M. K.; Williams, J. M. J., Adv. Synth. Catal. 2005, 347, 591-594; (c) Corberan, R.; Peris, E., Organometallics 2008, 27, 1954-1958; (d) Zinner, S. C.; Rentzsch, C. F.; Herdtweck, E.; Herrmann, W. A.; Kuhn, F. E., Dalton Trans. 2009, 7055-7062; (e) Dyson, G.; Frison, J. C.; Whitwood, A. C.; Douthwaite, R. E., Dalton Trans. 2009, 7141-7151; (f) Gnanamgari, D.; Sauer, E. L. O.; Schley, N. D.; Butler, C.; Incarvito, C. D.; Crabtree, R. H., Organometallics 2009, 28, 321-325; (g) Cheng, Y.; Sun, J.-F.; Yang, H.-L.; Xu, H.-J.; Li, Y.-Z.; Chen, X.-T.; Xue, Z.-L., Organometallics 2009, 28, 819-823.82. Nixon, T. D.; Whittlesey, M. K.; Williams, J. M. J., Dalton Trans. 2009, 753-762.83. Kuhl, O., Chem. Soc. Rev. 2007, 36, 592-607.84. Benhamou, L.; Chardon, E.; Lavigne, G.; Bellemin-Laponnaz, S.; César, V., Chem. Rev. 2011, 111, 2705-2733.85. Chan, B.; Chang, N.; Grimmett, M., Aust. J. Chem. 1977, 30, 2005-2013.

19The general strategies in the complexation of these donor-functionalized NHC ligand starting
from their corresponding imidazolium salts requires (a) the generation of free carbene in situ, or
(b) the use of a transmetalation reagent. The former was achieved by Arnold and co-workers89,
for example, but free carbenes are known to react with functional groups that have an acidic
proton.90 The more desirable method is to generate a stable carbene complex such as those of
silver(I) in situ, and transmetalate the NHC ligand to another metal in a separate reaction or in
the same pot.91 Many late transition metal complexes have been successfully synthesized by this
method, although there are reports that the choice of the silver base (silver(I) oxide or silver(I)
carbonate) is crucial.92 Some of these silver(I) complexes containing donor-functionalized NHC
ligands such as those of pyridyl or amido are isolated and structurally characterized.92-93
The Grubbs-Hoveyda metathesis catalyst containing a tethered oxygen donor from the NHC
ligand to the ruthenium(II) center is an active catalyst for the asymmetric cross-metathesis and
ring-opening metathesis reactions of prochiral alkenes.94 This catalyst has a bidentate NHC
ligand functionalized with a chiral BINOL (1,1'-bi-2-naphthol) group attached to the nitrogen
atom of the NHC ring. Burgess and co-workers have found that the iridium(I) catalysts of the
type [Ir(C–N)(cod)]+, where the C–N ligand represents an NHC donor containing a chiral
oxazoline group, gave superior activity in the asymmetric hydrogenation of tetrasubstituted
alkenes with good enantiomeric excess compared to phosphine-based catalysts. Double bond
migration of the substrate molecule was not observed.22c, 80 Elsevier and co-worker have recently
developed a highly active catalyst for the transfer hydrogenation of alkynes to Z-alkenes using a
palladium(0) catalysts containing an NHC ligand with a tertiary amine donor. It was found that
amine group acts as an internal base during catalysis and no external base is required.95
86. Cristau, H. J.; Cellier, P. P.; Spindler, J. F.; Taillefer, M., Chem. Eur. J. 2004, 10, 5607-5622.87. Bonnet, L. G.; Douthwaite, R. E.; Hodgson, R., Organometallics 2003, 22, 4384-4386.88. Bonnet, L. G.; Douthwaite, R. E.; Kariuki, B. M., Organometallics 2003, 22, 4187-4189.89. Arnold, P. L.; Mungur, S. A.; Blake, A. J.; Wilson, C., Angew. Chem. Int. Ed. 2003, 42, 5981-5984.90. (a) Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J. H.; Melder, J. P.; Ebel, K.; Brode, S., Angew. Chem. Int. Ed. 1995, 34, 1021-1023; (b) Frey, G. D.; Lavallo, V.; Donnadieu, B.; Schoeller, W. W.; Bertrand, G., Science 2007, 316, 439-441.91. (a) Garrison, J. C.; Youngs, W. J., Chem. Rev. 2005, 105, 3978-4008; (b) Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B., Chem. Rev. 2009, 109, 3561-3598.92. Tulloch, A. A. D.; Danopoulos, A. A.; Winston, S.; Kleinhenz, S.; Eastham, G., Dalton Trans. 2000, 4499-4506.93. (a) Catalano, V. J.; Malwitz, M. A., Inorg. Chem. 2003, 42, 5483-5485; (b) Catalano, V. J.; Moore, A. L., Inorg. Chem. 2005, 44, 6558-6566; (c) Samantaray, M. K.; Pang, K.; Shaikh, M. M.; Ghosh, P., Inorg. Chem. 2008, 47, 4153-4165.94. (a) Van Veldhuizen, J. J.; Garber, S. B.; Kingsbury, J. S.; Hoveyda, A. H., J. Am. Chem. Soc. 2002, 124, 4954-4955; (b) VanVeldhuizen, J. J.; Campbell, J. E.; Giudici, R. E.; Hoveyda, A. H., J. Am. Chem. Soc. 2005, 127, 6877-6882; (c) Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H., Nature 2008, 456, 933-937.

20Palladium(II) catalysts containing pyridyl-functionalized NHC donor ligands have found
important applications in cross-coupling reactions in the formation of C–C and C–N bonds.2d
Among all of the donor-functionalized NHC ligand, the amino (NH)-functionalized NHC
ligand is an important class of ligand system as it has a potential amphoteric character of the
basic carbene ligand and the acidic NH group (Figure 1.12). Arnold and co-workers reported the
coordination chemistry of such an NHC ligand with a secondary amine and an anionic amido
donor to lanthanide metals.96 Douthwaite reported the first primary amine-functionalized NHC
ligand on palladium(II) and silver(I),97 and a pincer-type C–NH–C ligand on palladium(II) as
well.98 Busetto reported the synthesis of silver(I) complexes with a protected NH functional
group tethered to the NHC ligand.99 Fryzuk reported rhodium(I) complexes100 and a Grubbs-like
ruthenium(II) complex containing an aniline-type nitrogen donor functionalized on the NHC
ligand.101 He showed that the hemilabile amine arm of the NHC donor did not improve catalytic
activity of ring opening metathesis polymerization reactions (ROMP) using their ruthenium(II)
complexes. Luo and co-workers reported the use of the anilido-type C–N–C pincer ligand on
palladium(II).102 More recently, Cross and co-workers reported palladium(II),103 platinum(II),103
ruthenium(II),104 rhodium(III)104 and iridium(III)104 complexes of bidentate or tridentate C–N or
C–N–C donor ligand with aniline-type NH and anilido-type donor. There are still no important
applications of these complexes to date in catalysis, however.
95. Hauwert, P.; Boerleider, R.; Warsink, S.; Weigand, J. J.; Elsevier, C. J., J. Am. Chem. Soc. 2010, 132, 16900-16910.96. Liddle, S. T.; Edworthy, I. S.; Arnold, P. L., Chem. Soc. Rev. 2007, 36, 1732-1744.97. Bonnet, L. G.; Douthwaite, R. E.; Hodgson, R.; Houghton, J.; Kariuki, B. M.; Simonovic, S., Dalton Trans. 2004, 3528-3535.98. (a) Douthwaite, R. E.; Houghton, J.; Kariuki, B. M., Chem. Commun. 2004, 698-699; (b) Houghton, J.; Dyson, G.; Douthwaite, R. E.; Whitwood, A. C.; Kariuki, B. M., Dalton Trans. 2007, 3065-3073.99. Busetto, L.; Cassani, M. C.; Femoni, C.; Macchioni, A.; Mazzoni, R.; Zuccaccia, D., J. Organomet. Chem. 2008, 693, 2579-2591.100. Jong, H.; Patrick, B. O.; Fryzuk, M. D., Can. J. Chem. 2008, 86, 803-810.101. Jong, H.; Patrick, B. O.; Fryzuk, M. D., Organometallics 2011, 30, 2333-2341.102. Wei, W.; Qin, Y.; Luo, M.; Xia, P.; Wong, M. S., Organometallics 2008, 27, 2268-2272.103. Cross, W. B.; Daly, C. G.; Ackerman, R. L.; George, I. R.; Singh, K., Dalton Trans. 2011, 40, 495-505.104. Cross, W. B.; Daly, C. G.; Boutadla, Y.; Singh, K., Dalton Trans. 2011, 40, 9722-9730.

21
Figure 1.12. Structures of some metal complexes containing an amino (NH)-functionalized
NHC ligand.
1.4 Thesis Goals
The goal of the present research is to develop new catalyst systems that mimic the highly
active ruthenium(II) based catalysts containing phosphine-amine ligands (P–NH2). The new
catalyst systems will have the features required for bifunctional catalysis using the “NH effect”;
notably a highly active metal-hydride and a protic amine pair. Replacing the phosphine donor on
the P–NH2 ligand with an N-heterocyclic carbene donor, leading to a primary-amino
functionalized N-heterocyclic carbene ligand (C–NH2), will allow a comparison of their
activities in the H2-hydrogenation of polar bonds. The mechanisms of action for ketone
hydrogenation will be studied in detail. Throughout all of the chapters, Dr. Alan Lough collected
all of the X-ray crystallographic data and solved all of the structures for publication purposes.
Our first research direction is to investigate the synthesis of amino-functionalized
imidazolium salts as precursor for the C–NH2 ligand. Results obtained from past research in our
group and in the recent literature99 suggested that such a synthesis and subsequent complexation
to late transition metals is indeed difficult. We turned to the synthesis of nitrile-functionalized
imidazolium salts, in hope that the nitrile-functionalized group can be reduced to a primary
amine when the N-heterocyclic carbene donor is coordinated to the metal. The synthesis of a
N
NPh
nPr
NPd
ClClHH
N
NN
N
NnBu nBu
Pd
Cl
NN
Rh+ N
H N N
N Ru
PMe3
Ph
H
Ir
NN
NI
HH
NN
NSm
N(SiMe3)2
N(SiMe3)2

22library of nitrile-functionalized imidazolium salts (Chapter 2) and the coordination chemistry of
the nitrile-functionalized N-heterocyclic carbene (C–CN) on Ag, Rh, Ru (Chapter 2), Pd and Pt
(Chapter 3) were achieved. These materials have been described in articles in Organometallics
in the years 2009105 (Chapter 2) and 2010106 (Chapter 3).
The reduction of the nitrile-functionalized salt was achieved in the presence of nickel(II) and
reducing agents, leading to the first nickel(II) complex containing the primary-amino
functionalized (C–NH2) ligand. The C–NH2 ligand on this nickel(II) complex can be
transmetalated to late transition metals such as ruthenium(II) and osmium(II). All of these are
described in Chapter 4. This reaction generates active ruthenium(II) catalysts for the transfer
hydrogenation of acetophenone in basic 2-propanol, and the H2-hydrogenation of a variety of
polar double bonds under very mild conditions. The catalytic results pertaining to these catalysts
are also described in the same chapter. The materials including the synthesis of the first nickel(II)
complex containing such a C–NH2 ligand, and the catalytic results of the transfer hydrogenation
reactions have been published in Organometallics in 2009 as a full article.107 The results for H2-
hydrogenation of polar double bonds have been published in Chemical Communications in
2010.108
We are interested in studying the mechanism of action in the H2-hydrogenation of ketones
using the ruthenium(II) catalyst described in Chapter 4 that contains the C–NH2 ligand and an η6-
arene ligand. Chapter 5 describes our detailed experimental studies of this system in ketone
hydrogenation. The cationic hydride-amine complex of this ruthenium(II) complex was found to
be inactive during catalysis, and we showed that the N–H group in this system is not required for
catalysis. We compared the energies by computational studies of the catalytic cycles involving
the outer-sphere bifunctional and the inner-sphere mechanisms, and proposed an inner-sphere
mechanism based on our findings. These studies have been published in Organometallics in
2011 as a full article.109
We are also interested to compare the diagonal relationship between ruthenium(II) and
iridium(III) in the H2-hydrogenation of ketones, and to study the donor properties of phosphines
versus N-heterocyclic carbene ligands in catalysis. Chapter 6 describes our detailed studies of the
105. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 853-862.106. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2010, 29, 570-581.107. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 6755-6761.108. O, W. W. N.; Lough, A. J.; Morris, R. H., Chem. Commun. 2010, 46, 8240 – 8242.109. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2011, 30, 1236-1252.

23mechanism of action of ketone hydrogenation catalyzed by a ruthenium(II) catalyst described in
Chapter 4 containing a C–NH2 ligand and a pentamethylcyclopentadienyl ligand. The
structurally similar iridium(III) complex and its catalytic activity were also described. A similar
cationic hydride-amine complex of iridium(III) complex was isolated, but we found that this is
not responsible for the catalytic activity that was observed. We were surprised to find that both
systems undergo an alcohol-assisted outer-sphere bifunctional mechanism, and our computation
studies suggested the operation of an unprecedented iridium(I) intermediate, which is formed
from hydride migration to the pentamethylcyclopentadienyl ligand, is the active species during
catalysis. We also studied the catalytic activity in ketone hydrogenation of the ruthenium(II) and
the iridium(III) catalysts containing the P–NH2 ligand and compared to its C–NH2 counterpart.
These results have been submitted for publication as a full article.110
110. O, W. W. N.; Lough, A. J.; Morris, R. H., Submitted 2011.

24Chapter 2: Synthesis and Characterization of Nitrile-Functionalized
N-Heterocyclic Carbenes and Their Complexes of Silver(I),
Rhodium(I) and Ruthenium(II)
2.1 Abstract
A series of unsymmetrical nitrile-functionalized imidazolium salts were synthesized by N-
alkylation and N-arylation of 2-cyanophenylimidazole. These imidazolium salts were used to
synthesize nitrile-functionalized N-heterocyclic carbene (C–CN) complexes of silver(I), all of
which were fully characterized by spectroscopic means. The compound bis[1-(2-cyanophenyl)-3-
methylimidazol-2-ylidene]silver(I) tetrafluoroborate (2a) was structurally characterized by a X-
ray diffraction study. Interestingly, the preparation of the silver(I) carbene complex using 3-
(cyanomethyl)-1-(2-cyanophenyl)-imidazolium hexafluorophosphate (1b) and excess silver(I)
oxide leads to the formation of the desired bis-carbene complex, and the bis-carbene complex
with hydrolyzed cyanomethyl groups on the ligands. The selectivity in hydrolysis of the
cyanomethyl group over the cyanophenyl group on the ligand, as evident from the NMR data,
suggests that the process must be mediated by silver(I) centers. The use of an N-heterocyclic
carbene complex of silver(I) as a carbene transfer reagent was demonstrated by the reaction of 2a
with [Rh(cod)Cl]2 and [Ru(p-cymene)Cl2]2, respectively. The crystal structures of the dimeric
complexes, bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)-(η4-1,5-cyclooctadiene)-
rhodium(I)] tetrafluoroborate (3) and bis[chloro(1-(2-cyanophenyl)-3-methylimidazol-2-
ylidene)-(η6-p-cymene)ruthenium(II)] tetrafluoroborate (4) showed that the nitrile nitrogen and
the carbene carbon of the C–CN ligand were bridged to two different metal centers, with a slight
distortion of the M–N≡C (M = Rh, Ru) bond angles.
2.2 Introduction
The use of N-heterocyclic carbenes (NHC) as ligands in homogeneous catalysis has been
extensively explored in the past decades because of the higher stability and reactivity they impart
*Reproduced in part with permission from O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 853-862. Copyright 2009 American Chemical Society.1. (a) Herrmann, W. A., Angew. Chem. Int. Ed. 2002, 41, 1290-1309; (b) Cui, X. H.; Burgess, K., Chem. Rev. 2005, 105, 3272-3296; (c) Hahn, F. E.; Jahnke, M. C., Angew. Chem. Int. Ed. 2008, 47, 3122-3172; (d) Diez-Gonzalez, S.; Marion, N.; Nolan, S. P., Chem. Rev. 2009, 109, 3612-3676; (e) Vougioukalakis, G. C.; Grubbs, R. H., Chem. Rev. 2010, 110, 1746-1787.

25to homogeneous catalysts and their lower toxicity relative to phosphine ligands.1 The carbene
ligands, in particular, imidazol-2-ylidene-type ligands isolated by Arduengo and co-workers,2
have comparable or even stronger donor capacity compared to phosphine ligands, and are
capable of stabilizing metal centers with different oxidation states.3 With different synthetic
routes reported for these imidazolium salt precursors,4 the steric bulk of the ligand can be tuned
either on the imidazole backbone, or on the N,N-substituted groups. This is beneficial in order to
tune the activity and selectivity of a catalyst.
N-heterocyclic carbenes are viewed not only as spectator monodentate ligands in
homogeneous catalysis, but also, when suitably functionalized at nitrogen of the imidazolylidene
ring, as useful building blocks in the design of highly active homogeneous catalysts for various
important chemical transformations.5 These donor-functionalized NHC ligands show
coordination versatility when complexed to metal centers, and more importantly, provide ligand-
metal bifunctionality in small molecule activation.6
Our group has been interested in the hydrogenation of polar multiple bonds including ketones,
imines and nitriles catalyzed by ruthenium(II)7 and iron(II)8 based systems. In the continuous
pursuit of active catalysts for polar bond hydrogenation, we are interested in the use of N-
heterocyclic carbene ligands to enhance catalytic activities, so as to replace traditional phosphine
donors in the construction of highly active catalysis. From our initial report on the synthesis of
coordinatively unsaturated hydridoruthenium(II) NHC systems,9 we are interested in
constructing new donor-functionalized NHC ligands that would assist in the ligand-metal
bifunctional catalysis involved the action of M–H/H–X (X = N, O) in the hydrogenation of polar
2. (a) Kliegman, J. M.; Barnes, R. K., J. Org. Chem. 1970, 35, 3140-3143; (b) Arduengo, A. J.; Harlow, R. L.; Kline, M., J. Am. Chem. Soc. 1991, 113, 361-363; (c) Arduengo, A. J.; Krafczyk, R.; Schmutzler, R.; Craig, H. A.; Goerlich, J. R.; Marshall, W. J.; Unverzagt, M., Tetrahedron 1999, 55, 14523-14534; 3. (a) Crudden, C. M.; Allen, D. P., Coord. Chem. Rev. 2004, 248, 2247-2273; (b) Scott, N. M.; Nolan, S. P., Eur. J. Inorg. Chem. 2005, 1815-1828; (c) Leuthausser, S.; Schwarz, D.; Plenio, H., Chem. Eur. J. 2007, 13, 7195-7203; (d) Fantasia, S.; Petersen, J. L.; Jacobsen, H.; Cavallo, L.; Nolan, S. P., Organometallics 2007, 26, 5880-5889; (e) Fey, N.; Haddow, M. F.; Harvey, J. N.; McMullin, C. L.; Orpen, A. G., Dalton Trans. 2009, 8183-8196; (f) Gusev, D. G., Organometallics 2009, 28, 763-770; (g) Gusev, D. G., Organometallics 2009, 28, 6458-6461; (h) Droge, T.; Glorius, F., Angew. Chem. Int. Ed. 2010, 49, 6940-6952.4. (a) Bon, R. S.; deKanter, F. J. J.; Lutz, M.; Spek, A. L.; Jahnke, M. C.; Hahn, F. E.; Groen, M. B.; Orru, R. V. A., Organometallics 2007, 26, 3639-3650; (b) Benhamou, L.; Chardon, E.; Lavigne, G.; Bellemin-Laponnaz, S.; Cesar, V., Chem. Rev. 2011, 111, 2705-2733 and references therein.5. (a) Lee, H. M.; Lee, C. C.; Cheng, P. Y., Curr. Org. Chem. 2007, 11, 1491-1524; (b) Kuhl, O., Chem. Soc. Rev. 2007, 36, 592-607; (c) Normand, A. T.; Cavell, K. J., Eur. J. Inorg. Chem. 2008, 2781-2800; (d) Poyatos, M.; Mata, J. A.; Peris, E., Chem. Rev. 2009, 109, 3677-3707; (e) Corberan, R.; Mas-Marza, E.; Peris, E., Eur. J. Inorg. Chem. 2009, 1700-1716.6. Liddle, S. T.; Edworthy, I. S.; Arnold, P. L., Chem. Soc. Rev. 2007, 36, 1732-1744.

26bonds.10 The nitrile group, when hydrogenated at a late transition metal center, could produce a
H–M–NH2CH2– (M = Ru, Os, Rh or Ir) group known to be very effective at the hydrogenation
of ketones and imines.10c, 11
Figure 2.1. Nitrile-functionalized imidazolium salts 1a-1f.
In this chapter, we report the synthesis and characterization of a series of nitrile-
functionalized imidazolium salts (Figure 2.1), and their corresponding metal complexes of
silver(I), rhodium(I) and ruthenium(II) containing such a NHC ligand.12 Although an analogous
nitrile-functionalized NHC (C–CN) complex of palladium(II) was reported by Dyson and co-
workers, the nitrile moiety on the ligand (Figure 2.2) was remote from the metal center.13 More
recently, an intramolecular C–H bond activation of the CH2CN group of a nitrile-functionalized
NHC ligand on nickel(II) was reported by Chetcuti and Veiros forming new nickelacycles
(Figure 2.2).14 The nitrile group on these new ligands, on the other hand, shows coordination
versatility and reactivity with metal centers. The hydrolysis of the nitrile-functionalized carbene
ligand mediated by silver(I) centers is reported here.
7. (a) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2002, 124, 15104-15118; (b) Guo, R.; Lough, A. J.; Morris, R. H.; Song, D., Organometallics 2004, 23, 5524-5529; (c) Abdur-Rashid, K.; Guo, R. W.; Lough, A. J.; Morris, R. H.; Song, D. T., Adv. Synth. Catal. 2005, 347, 571-579; (d) Guo, R. W.; Elpelt, C.; Chen, X. H.; Song, D. T.; Morris, R. H., Chem. Commun. 2005, 3050-3052; (e) Li, T.; Bergner, I.; Haque, F. N.; Iuliis, M. Z.-D.; Song, D.; Morris, R. H., Organometallics 2007, 26, 5940-5949.8. Morris, R. H., Chem. Soc. Rev. 2009, 38, 2282-2291 and references therein.9. Abdur-Rashid, K.; Fedorkiw, T.; Lough, A. J.; Morris, R. H., Organometallics 2004, 23, 86-94.10. (a) Noyori, R.; Yamakawa, M.; Hashiguchi, S., J. Org. Chem. 2001, 66, 7931-7944; (b) Clapham, S. E.; Hadzovic, A.; Morris, R. H., Coord. Chem. Rev. 2004, 248, 2201-2237; (c) Ikariya, T.; Murata, K.; Noyori, R., Org. Biomol. Chem. 2006, 4, 393-406.11. (a) Ito, M.; Ikariya, T., Chem. Commun. 2007, 5134-5142; (b) Kuwata, S.; Ikariya, T., Dalton Trans. 2010, 39, 2984-2992; (c) Ikariya, T., Bull. Chem. Soc. Jpn. 2011, 84, 1-16.12. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 853-862.13. Fei, Z.; Zhao, D.; Pieraccini, D.; Ang, W. H.; Geldbach, T. J.; Scopelliti, R.; Chiappe, C.; Dyson, P. J., Organometallics 2007, 26, 1588-1598.
N
N N
N
N N RX
2'-PyCH2-2'-Py
-+
CH3CH2CN
R X
CH(CH3)Ph
BF4 (1a)
CH2CH2(3'-C9H7)
PF6 (1b)PF6 (1c)PF6 (1d)BF4 (1e)BF4 (1f)

27
Figure 2.2. Nitrile-functionalized N-heterocyclic carbene (C–CN) complexes of palladium(II)
(left) and nickel(II) (right).
2.3 Results and Discussion
2.3.1 Synthesis of Imidazolium Salt Precursors. Initial attempts utilized the standard protocol
for imidazolium salt synthesis starting from glyoxal and 2-aminobenzonitrile to synthesize 1,3-
bis(2-cyanophenyl)imidazolium halide. These failed because the nucleophilic character of the
amine-nitrogen of 2-aminobenzonitrile is diminished due to the presence of an ortho-cyano
group and so did not condense with glyoxal to yield the desired glyoxal diimine, even under
templating conditions in the presence of nickel(II) or zinc(II) cations.2a,2c We, therefore,
developed a synthesis of unsymmetrical imidazolium salts using the N-alkylation and N-arylation
of a substituted imidazole. The starting substituted imidazole, 2-cyanophenylimidazole, is
conveniently prepared in a large scale with very good yields using literature methods.15 The
syntheses of the new imidazolium salts were conducted by means of electrophilic alkylation with
trimethyloxonium tetrafluoroborate (Meerwein’s salt) to give 1a and SN2 reactions with
bromoacetonitrile, picolyl chloride and 3-(2-bromoethyl)indene to yield 1b, 1c and 1f,
respectively, in good yields (Schemes 2.1-2.3). N-alkylation using a secondary alkyl bromide is
slow giving 1e in low yield (Scheme 2.3). N-Arylation of 2-cyanophenylimidazole with phenyl
halides, on the other hand, did not proceed, even at elevated temperatures.16
14. Oertel, A. M.; Freudenreich, J.; Gein, J.; Ritleng, V.; Veiros, L. F.; Chetcuti, M. J., Organometallics 2011, 30, 3400-3411.15. (a) Johnson, A. L.; Kauer, J. C.; Sharma, D. C.; Dorfman, R. I., J. Med. Chem. 1969, 12, 1024-1028; (b) Ren, L.; Chen, A. C.; Decken, A.; Crudden, C. M., Can. J. Chem. 2004, 82, 1781-1787. 16. (a) Chan, B.; Chang, N.; Grimmett, M., Aust. J. Chem. 1977, 30, 2005-2013; (b) Harlow, K. J.; Hill, A. F.; Welton, T., Synthesis 1996, 697.
N NNN
NNN N
PdCl ClNi
N N RH
N
R = CH3, 2,4,6-(CH3)3C6H2 (Mes)n = 2-5
n-1
Chetcuti and VeirosDyson
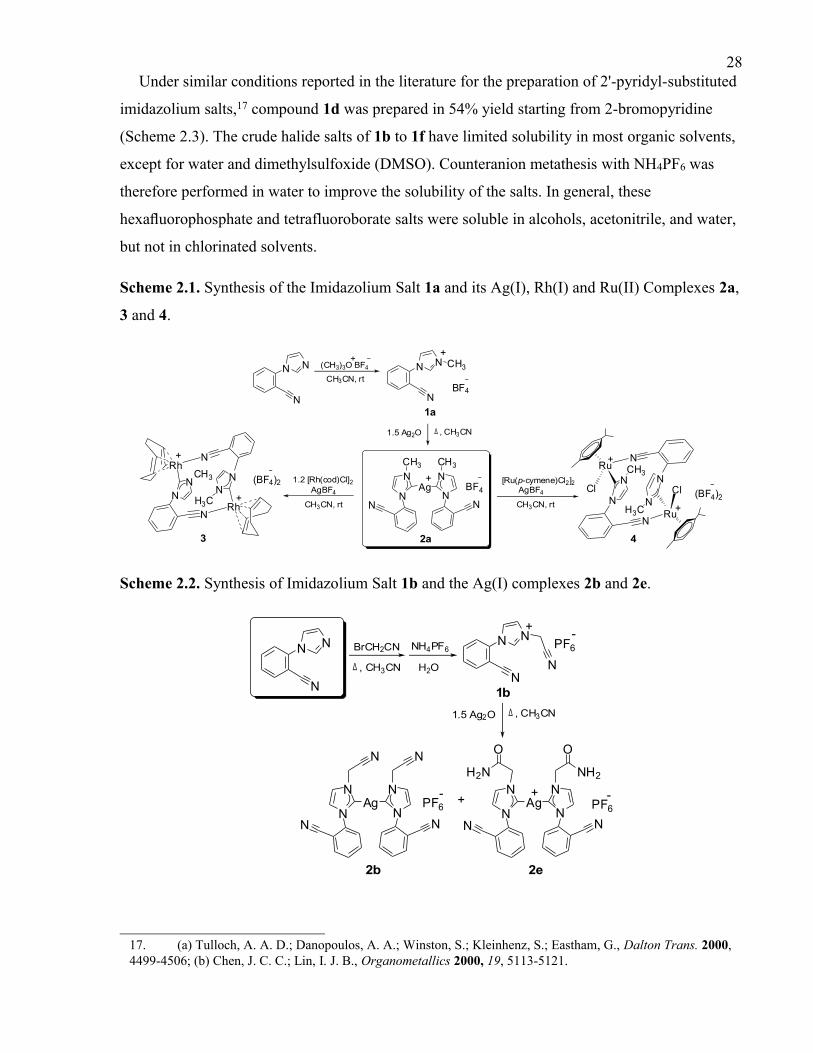
28Under similar conditions reported in the literature for the preparation of 2'-pyridyl-substituted
imidazolium salts,17 compound 1d was prepared in 54% yield starting from 2-bromopyridine
(Scheme 2.3). The crude halide salts of 1b to 1f have limited solubility in most organic solvents,
except for water and dimethylsulfoxide (DMSO). Counteranion metathesis with NH4PF6 was
therefore performed in water to improve the solubility of the salts. In general, these
hexafluorophosphate and tetrafluoroborate salts were soluble in alcohols, acetonitrile, and water,
but not in chlorinated solvents.
Scheme 2.1. Synthesis of the Imidazolium Salt 1a and its Ag(I), Rh(I) and Ru(II) Complexes 2a,
3 and 4.
Scheme 2.2. Synthesis of Imidazolium Salt 1b and the Ag(I) complexes 2b and 2e.
17. (a) Tulloch, A. A. D.; Danopoulos, A. A.; Winston, S.; Kleinhenz, S.; Eastham, G., Dalton Trans. 2000, 4499-4506; (b) Chen, J. C. C.; Lin, I. J. B., Organometallics 2000, 19, 5113-5121.
N
N N CH3
BF4
NN
NCH3
BF4Ag
NN
NCH3
(BF4)2
1.5 Ag2O
(CH3)3O BF4CH3CN, rt
1.2 [Rh(cod)Cl]2
CH3CN, rt
N
N N
AgBF4
RhN
NN
RhN
NN
H3C
CH3
+
+
+ +
+
1a
2a3
Ru
ClN
N
N
H3C Ru
ClN
N
N
CH3
+
+(BF4)2
[Ru(p-cymene)Cl2]2
CH3CN, rt
AgBF4
∆ , CH3CN
4
N
N N
1bN
N N
N
PF6
NN
NPF6Ag
NN
NPF6
N N
NN
NAg
NN
NH2N NH2
+
O O
2e2b
BrCH2CN
H2O
NH4PF6
1.5 Ag2O
∆ , CH3CN
∆ , CH3CN
+ -
+- -
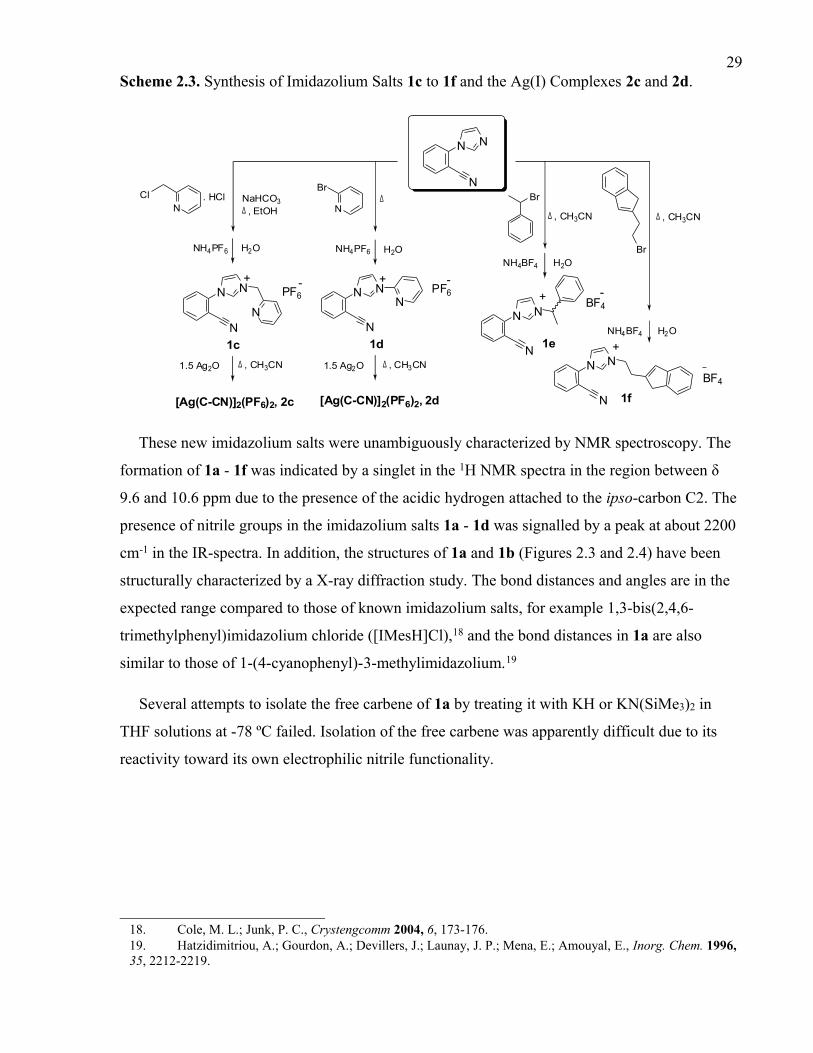
29Scheme 2.3. Synthesis of Imidazolium Salts 1c to 1f and the Ag(I) Complexes 2c and 2d.
These new imidazolium salts were unambiguously characterized by NMR spectroscopy. The
formation of 1a - 1f was indicated by a singlet in the 1H NMR spectra in the region between δ
9.6 and 10.6 ppm due to the presence of the acidic hydrogen attached to the ipso-carbon C2. The
presence of nitrile groups in the imidazolium salts 1a - 1d was signalled by a peak at about 2200
cm-1 in the IR-spectra. In addition, the structures of 1a and 1b (Figures 2.3 and 2.4) have been
structurally characterized by a X-ray diffraction study. The bond distances and angles are in the
expected range compared to those of known imidazolium salts, for example 1,3-bis(2,4,6-
trimethylphenyl)imidazolium chloride ([IMesH]Cl),18 and the bond distances in 1a are also
similar to those of 1-(4-cyanophenyl)-3-methylimidazolium.19
Several attempts to isolate the free carbene of 1a by treating it with KH or KN(SiMe3)2 in
THF solutions at -78 ºC failed. Isolation of the free carbene was apparently difficult due to its
reactivity toward its own electrophilic nitrile functionality.
18. Cole, M. L.; Junk, P. C., Crystengcomm 2004, 6, 173-176.19. Hatzidimitriou, A.; Gourdon, A.; Devillers, J.; Launay, J. P.; Mena, E.; Amouyal, E., Inorg. Chem. 1996, 35, 2212-2219.
N
N N
N
N NN
N
N N
N
PF6PF6
1c 1d
N
Br
NCl
∆ , EtOHNaHCO3. HCl
NH4BF4 H2ONH4PF6 H2O
1.5 Ag2O ∆ , CH3CN 1.5 Ag2O ∆ , CH3CN
[Ag(C-CN)]2(PF6)2, 2c [Ag(C-CN)]2(PF6)2, 2d
+ - +
NH4PF6 H2O
N
N NBF4
+
N
N NBF4
+
--
Br
∆ , CH3CN
∆
NH4BF4 H2O
Br
∆ , CH3CN
1e
1f
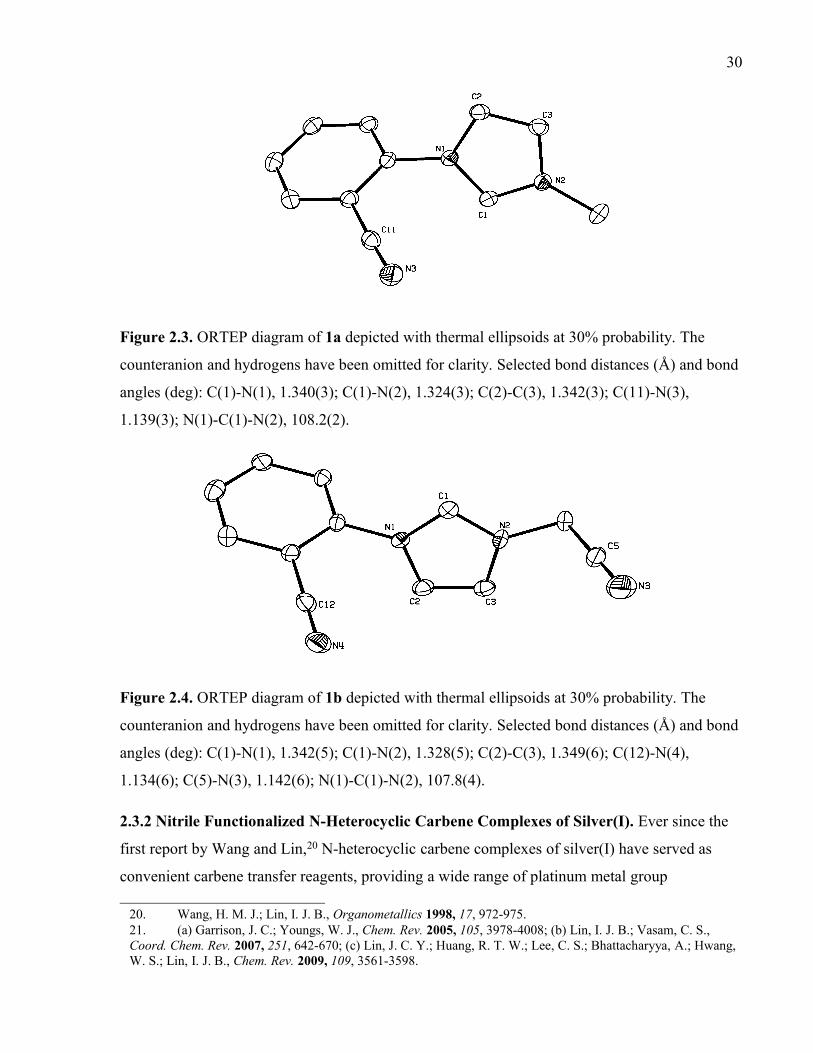
30
Figure 2.3. ORTEP diagram of 1a depicted with thermal ellipsoids at 30% probability. The
counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and bond
angles (deg): C(1)-N(1), 1.340(3); C(1)-N(2), 1.324(3); C(2)-C(3), 1.342(3); C(11)-N(3),
1.139(3); N(1)-C(1)-N(2), 108.2(2).
Figure 2.4. ORTEP diagram of 1b depicted with thermal ellipsoids at 30% probability. The
counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and bond
angles (deg): C(1)-N(1), 1.342(5); C(1)-N(2), 1.328(5); C(2)-C(3), 1.349(6); C(12)-N(4),
1.134(6); C(5)-N(3), 1.142(6); N(1)-C(1)-N(2), 107.8(4).
2.3.2 Nitrile Functionalized N-Heterocyclic Carbene Complexes of Silver(I). Ever since the
first report by Wang and Lin,20 N-heterocyclic carbene complexes of silver(I) have served as
convenient carbene transfer reagents, providing a wide range of platinum metal group
20. Wang, H. M. J.; Lin, I. J. B., Organometallics 1998, 17, 972-975.21. (a) Garrison, J. C.; Youngs, W. J., Chem. Rev. 2005, 105, 3978-4008; (b) Lin, I. J. B.; Vasam, C. S., Coord. Chem. Rev. 2007, 251, 642-670; (c) Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B., Chem. Rev. 2009, 109, 3561-3598.

31complexes.21 These silver(I)-NHC complexes were prepared by the treatment of the imidazolium
salt precursors with mildly basic silver(I) oxide or silver(I) carbonate,17a normally under mild
conditions. Given the ease of their preparation, their relative stability toward air and moisture,
and their structural diversity, we were prompted to prepare analogous silver(I) complexes
bearing nitrile-functionalized N-heterocyclic carbene ligands.
The silver(I) complexes, 2a-2d, were prepared by the reaction of excess silver(I) oxide with
the imidazolium salts 1a-1d, respectively, under reflux for overnight with protection from light
(Schemes 2.1-2.3). Stirring the aforementioned solutions at room temperature did not work
efficiently. In addition, an excess of silver(I) oxide, preferably 1.5 equiv with respect to the
imidazolium salt, is required for the successful generation of these bis-silver(I)-NHC complexes.
A full conversion under the same reaction conditions could not be achieved if a stoichiometric
amount of base was used. The use of molecular sieves was crucial for obtaining a pure product
by preventing the hydrolysis of the nitrile group in the presence of silver hydroxide and water
(vide infra).
The formation of these silver(I) complexes was established by observing a sharp singlet above
182 ppm in the 13C{1H} NMR spectra which was assigned to the carbene carbon attached to the
silver center (Ag–C). No one-bond coupling 1J(107Ag-13C) or 1J(109Ag-13C) was observed, even at
-40 ºC for 2a in acetonitrile-d3 solution.21a, 21c This may be the result of a rapid exchange of
carbene ligands between silver ions. On the other hand, the absence of a peak in the region of δ
9.6-10.6 ppm in the 1H NMR spectra further suggested full conversion to silver(I) NHC
complexes.
The silver(I) complex, [Ag(C-CN)2]BF4 (2a) has been structurally characterized by use of X-
ray diffraction (Figure 2.5). The complex crystallizes in the monoclinic space group P21/n with
four units residing in the unit cell. The structure shows a slightly distorted geometry from
linearity about the silver(I) center, and the C–Ag–C bond angle of 171.1(2)º is smaller than those
of typical [Ag(NHC)2]+ complexes, at around 174-177º. The average Ag–C distance is 2.095 Å,
which is in the expected range of typical [Ag(NHC)2]+ complexes. The decrease in the C–N bond
lengths C(1)-N(1) and C(1)-N(2) and the bond angle N(1)-C(1)-N(2) in the imidazol-2-ylidene
22. (a) Paas;, M.; Wibbeling;, B.; Fröhlich;, R.; Hahn, F. E., Eur. J. Inorg. Chem. 2006, 2006, 158-162; (b) Samantaray, M. K.; Pang, K.; Shaikh, M. M.; Ghosh, P., Inorg. Chem. 2008, 47, 4153-4165; (c) Liao, C. Y.; Chan, K. T.; Chiu, P. L.; Chen, C. Y.; Lee, H. M., Inorg. Chim. Acta 2008, 361, 2973-2978.

32ring compared to the imidazolium salt suggests an increase in p-character at the carbene
carbon.17a, 20-22
In addition, the phenyl rings are twisted with respect to the imidazole rings in opposite
enantiomeric configurations with dihedral angles of 43.18º and 51.05º, in order to facilitate
stacking of phenyl and imidazole rings in the crystal lattice, with the contact distance between
them being 3.40 Å, which is comparable to the sum of van der Waals radius of carbon atoms
(Figure 2.5).
Figure 2.5. ORTEP diagram of 2a depicted with thermal ellipsoids at 30% probability. The
counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and bond
angles (deg): Ag(1)-C(1), 2.094(5); Ag(1)-C(4), 2.095(5); C(1)-N(1), 1.359(6); C(1)-N(2),
1.362(6); C(13)-N(5), 1.140(7); C(1)-Ag(1)-C(4), 171.1(2); N(1)-C(1)-N(2), 104.4(4).
The complexes 2c and 2d showed different structural motifs compared to 2a. We would
expect the structures of these compounds would be similar to those of other pyridyl-
functionalized N-heterocyclic carbene complexes of silver(I).17a, 23 However, the analytical data
(NMR, Fourier transform infrared (FT-IR), and elemental analysis) suggest the presence of a
[Ag(NHC)]PF6 unit, which could exist in a monomeric, dimeric, or trimeric structure. Although
crystals suitable for X-ray diffraction were not obtained successfully, the structures can be
partially assigned on the basis of NMR data. Only one set of signals are present in the 1H NMR 23. (a) Catalano, V. J.; Malwitz, M. A., Inorg. Chem. 2003, 42, 5483-5485; (b) Catalano, V. J.; Malwitz, M.
A.; Etogo, A. O., Inorg. Chem. 2004, 43, 5714-5724; (c) Catalano, V. J.; Moore, A. L., Inorg. Chem. 2005, 44, 6558-6566; (d) Catalano, V. J.; Etogo, A. O., J. Organomet. Chem. 2005, 690, 6041-6050.

33spectra, indicative of a symmetric single component complex. A monomeric structure is highly
unlikely, as the C–Ag–NPy angle will be bent away from linearity, if the pyridyl group were to
bond to the metal center. A trimeric structure is not possible, as metal-metal interactions of the
silver(I) clusters will give rise to extensive Ag-C couplings at the carbene carbon in the 13C{1H}
NMR spectrum. Only a sharp singlet was observed in our system at δ 183 ppm. This signal is
more downfield than those of silver(I) cluster systems, such as [Ag3((pyCH2)2imid)3](BF4)3 and
[Ag3((quinCH2)2imid]3](BF4)3 (py = 2'-pyridyl; quin = 2'-quinolyl; imid = imidazol-2-
ylidene).23a, 23b Dimeric structures are most probable, with either the nitrile or the pyridyl group
bound to the silver(I) center to produce a dication with a 10- or 12-membered
macrometallocyclic ring. Silver(I)-NHC complexes forming a 12-membered ring
macrometallocycle are known, at least with donor-functionalized NHC ligands of alkylated
amides22b, 24 and pyrazoles.25 Bridging via the nitrile group on the carbene is not likely because
the wavenumbers of the nitrile stretch of these compounds and their imidazolium precursors
observed in the IR spectra are comparable (see the Section 2.5). The only additional binding site
is through the pyridyl group of the ligand. Possible structures are given in Figure 2.6, with each
silver(I) center bonding to the carbene carbon and nitrogen (2c-1, 2d-1), or with one silver(I)
center bonding to two carbene carbons, and the others bonding to two nitrogens (2c-2, 2d-2),
similar to those reported with pyrazole-functionalized NHC ligands.25
Figure 2.6. Proposed structures of Ag(I) complexes 2c and 2d forming 12- and 10 membered
rings, respectively.
24. Legault, C. Y.; Kendall, C.; Charette, A. B., Chem. Commun. 2005, 3826-3828.25. (a) Chiu, P. L.; Chen, C. Y.; Lee, C.-C.; Hsieh, M.-H.; Chuang, C.-H.; Lee, H. M., Inorg. Chem. 2006, 45, 2520-2530; (b) Zhou, Y. B.; Zhang, X. M.; Chen, W. Z.; Qiu, H. Y., J. Organomet. Chem. 2008, 693, 205-215.
N NN
N
N NN
N
Ag (PF6)2
2c-1
(PF6)2
N NN
N
N N N
N
Ag Ag
Ag
(PF6)2
N NN
N
N N N
N
NNN
N
Ag Ag
NNN
N
AgAg
(PF6)2
2c-2
2d-1 2d-2
+ + + +
++
+ +
- -
--

342.3.3. Hydrolysis of Nitrile groups on the Silver(I) Complex 2b. In the preparation of the
silver(I) complex using the imidazolium salt 1b, a mixture of products 2b and 2e was always
observed, even when molecular sieves were used in the syntheses. The appearance of two broad
peaks at δ 6.12 and 6.49 ppm in the 1H NMR spectrum of the compounds in acetonitrile-d3
suggested the presence of an amide group (Figure 2.7c), and this is further confirmed by their
disappearance upon the addition of D2O to the NMR sample.
We initially believed that these peaks were due to the presence of acetamide formed by the
hydrolysis of acetonitrile during the course of reaction, as similar peaks were observed in the
preparation of 2a without using molecular sieves. In order to identify these products that resulted
from hydrolysis, we attempted to synthesize acetamide under the same conditions that were used
in the preparation of 2a-2d. The 1H NMR (400 MHz) of the reaction mixture resulting from
refluxing a solution of acetonitrile, water, and silver(I) oxide in acetonitrile-d3 showed two broad
peaks at δ 6.03 and 6.64 ppm (Figure 2.7b). This was compared to the 1H NMR (400 MHz) of
pure acetamide in acetonitrile-d3, which gave two broad peaks at δ 6.18 and 6.51 ppm (Figure
2.7a). The peak separations of these amide-NH peaks are quite different for these compounds. In
particular, it is smaller in pure acetamide (0.33 ppm) than in the ones prepared from silver(I)
oxide (0.61 ppm). Even though the peak separation between the amido-protons of 2e are small
(0.37 ppm) like that of acetamide, the peak width of these protons are the smallest among all.
These observations suggest the NH2 group in 2e has a higher barrier to rotation about the C–N
bond compared to free acetamide. We postulate that a weak silver-oxygen electrostatic
interaction may contribute to the observed NMR behavior, and this could also explain the
smaller peak separation for acetamides prepared from silver(I) oxide when compared to pure
acetamide.
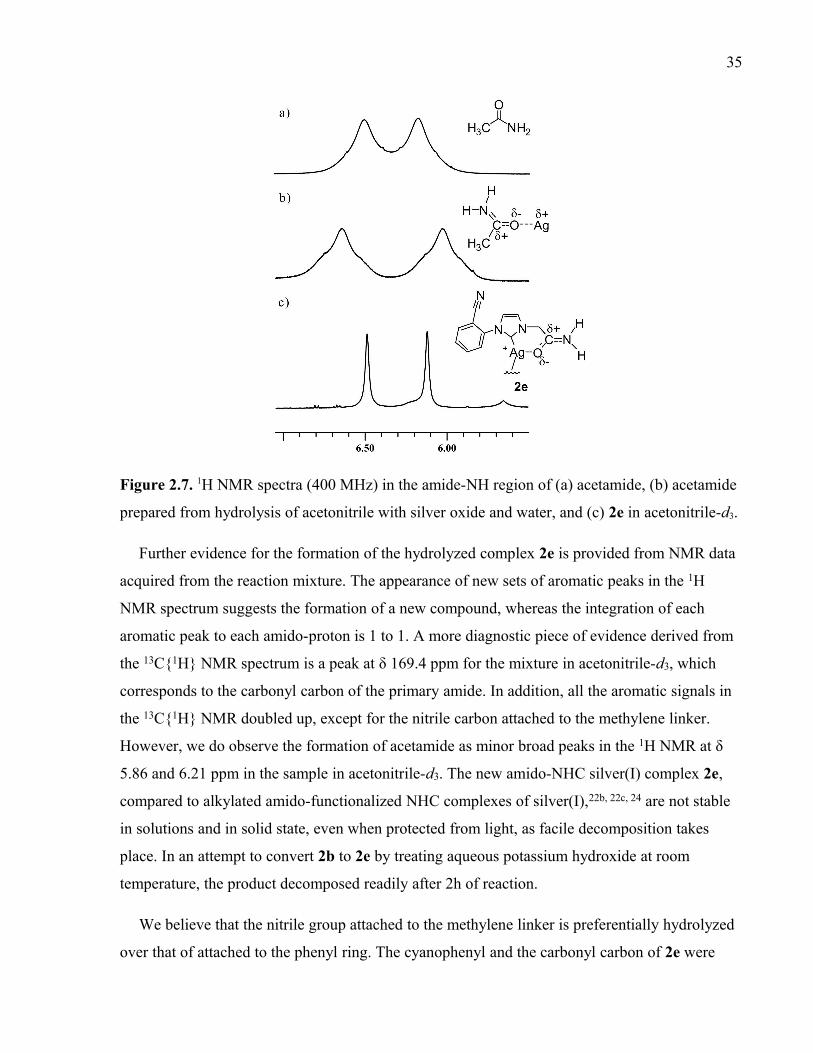
35
Figure 2.7. 1H NMR spectra (400 MHz) in the amide-NH region of (a) acetamide, (b) acetamide
prepared from hydrolysis of acetonitrile with silver oxide and water, and (c) 2e in acetonitrile-d3.
Further evidence for the formation of the hydrolyzed complex 2e is provided from NMR data
acquired from the reaction mixture. The appearance of new sets of aromatic peaks in the 1H
NMR spectrum suggests the formation of a new compound, whereas the integration of each
aromatic peak to each amido-proton is 1 to 1. A more diagnostic piece of evidence derived from
the 13C{1H} NMR spectrum is a peak at δ 169.4 ppm for the mixture in acetonitrile-d3, which
corresponds to the carbonyl carbon of the primary amide. In addition, all the aromatic signals in
the 13C{1H} NMR doubled up, except for the nitrile carbon attached to the methylene linker.
However, we do observe the formation of acetamide as minor broad peaks in the 1H NMR at δ
5.86 and 6.21 ppm in the sample in acetonitrile-d3. The new amido-NHC silver(I) complex 2e,
compared to alkylated amido-functionalized NHC complexes of silver(I),22b, 22c, 24 are not stable
in solutions and in solid state, even when protected from light, as facile decomposition takes
place. In an attempt to convert 2b to 2e by treating aqueous potassium hydroxide at room
temperature, the product decomposed readily after 2h of reaction.
We believe that the nitrile group attached to the methylene linker is preferentially hydrolyzed
over that of attached to the phenyl ring. The cyanophenyl and the carbonyl carbon of 2e were

36identified in the 13C{1H} NMR spectrum, and the assignment was further established by two-
dimensional 1H-13C HSQC and HMBC NMR experiments. In addition, under identical reaction
conditions in the preparation of 2a and 2b when molecular sieves were used, the amido-protons
were absent in 2a but not in 2b, even though hydrolysis can be promoted without the use of
molecular sieves in the preparation of 2a.
This selectivity is reversed compared to conventional base hydrolysis where the benzonitrile
is hydrolyzed more readily. This suggests the hydrolysis is mediated by the metal center. In fact,
as there are more degrees of freedom of the cyanomethyl group compared to the cyanophenyl
group, the nitrogen on the nitrile group can be activated by two silver(I) cations to facilitate
attack by the hydroxide ion on the electrophilic carbon, which leads to the formation of a six-
membered ring intermediate 2e-i (Scheme 2.4). A similar behavior was observed by Mao and co-
workers, when the nitrogen on the acetonitrile was activated by two silver(I) centers of a silver(I)
cryptate system, and thus underwent catalytic hydrolysis,26 although the nitrile was hydrolyzed
stoichiometrically by silver hydroxide in our system.
We propose that the hydrolysis occurs during the preparation of 2b according to Scheme 2.4.
The first equivalent of the imidazolium salt reacts with silver(I) oxide, leading to the formation
of the monocarbene complex 2b-i and silver hydroxide. Silver hydroxide and a second
equivalent of the imidazolium salt react to generate the more thermodynamically stable complex
2b.27 However, as water is generated and in the presence of silver(I) oxide, more silver
hydroxide is generated and this starts to hydrolyze the cyanomethyl group on the ligand, forming
the six-member ring intermediate 2e-i. Further hydrolysis lead to the final product 2e.
26. Luo, R.-S.; Mao, X.-A.; Pan, Z.-Q.; Luo, Q.-H., Spectrochim. Acta, Part A 2000, 56, 1675-1680.27. Hayes, J. M.; Viciano, M.; Peris, E.; Ujaque, G.; Lledos, A., Organometallics 2007, 26, 6170-6183.

37Scheme 2.4. Proposed Reaction Pathways to Hydrolysis of Ag(I) Complex 2b to 2e.
2.3.4. Nitrile Functionalized N-Heterocyclic Carbene Complex of Rhodium(I) and
Ruthenium(II). To demonstrate the use of a silver(I)-NHC complex as a carbene transfer
reagent, we reacted [Ag(C–CN)2]BF4 (2a) with [Rh(cod)Cl]2 and 1 equiv of AgBF4 in
acetonitrile, and [Ru(p-cymene)Cl2]2 and 1 equiv of AgBF4 in acetonitrile to afford 3 and 4 in
71% and 79% yields, respectively. These yellow complexes are very soluble in acetonitrile,
where they tend to react, and soluble in nitromethane and nitroethane, but only slightly soluble in
acetone, alcohols, and chlorinated solvents. Interestingly, both complexes crystallized as dimers
and were structurally characterized by X-ray diffraction (Figures 2.8 and 2.9).
In the solid state structure of 3 ([Rh(C–CN)(cod)]2(BF4)2, Figure 2.8), the structure shows a
slightly distorted square planar geometry about the rhodium(I) center. The Rh–C22d, 28 distance is
in the expected range for analogous compounds. The Rh–cod bond distances measured from the
centroid of the olefin are different with respect to its coordination sphere, the one trans to the
carbene (2.100 Å) being longer than the one trans to nitrile (2.016 Å). The carbene ligand should
impart a stronger trans influence compared to the nitrile group, and this is also seen in the
28. (a) Dastgir, S.; Coleman, K. S.; Cowley, A. R.; Green, M. L. H., Organometallics 2006, 25, 300-306; (b) Messerle, B. A.; Page, M. J.; Turner, P., Dalton Trans. 2006, 3927-3933; (c) Jong, H.; Patrick, B. O.; Fryzuk, M. D., Can. J. Chem. 2008, 86, 803-810; (d) Jimenez, M. V.; Perez-Torrente, J. J.; Bartolome, M. I.; Gierz, V.; Lahoz, F. J.; Oro, L. A., Organometallics 2008, 27, 224-234; (e) Dyson, G.; Frison, J. C.; Simonovic, S.; Whitwood, A. C.; Douthwaite, R. E., Organometallics 2008, 27, 281-288; (f) Dyson, G.; Frison, J. C.; Whitwood, A. C.; Douthwaite, R. E., Dalton Trans. 2009, 7141-7151; (g) Praetorius, J. M.; Wang, R. Y.; Crudden, C. M., Eur. J. Inorg. Chem. 2009, 1746-1751; (h) Yu, X. Y.; Sun, H. S.; Patrick, B. O.; James, B. R., Eur. J. Inorg. Chem. 2009, 1752-1758.
Ag2O
-AgOH
xs AgOHH2O
NN
NAg
NN
NC N N
NN
N
N
xs AgOH
H2O
AgOH
- H2O, AgPF6CH3CN
1b
δ+
δ -AgO
HH2O
δ +δ -
NN
NAg
NH2
O δ -
2b-i
2e-i
1bN
N N
N
PF6+ -
NN
NAg
N
2b
NN
NPF6Ag
NN
N
N N
-
N CH3+
+
PF6
NN
NAg
NN
NH2N NH2
O O
2e
+ -+ +
+-
PF6-
PF6-
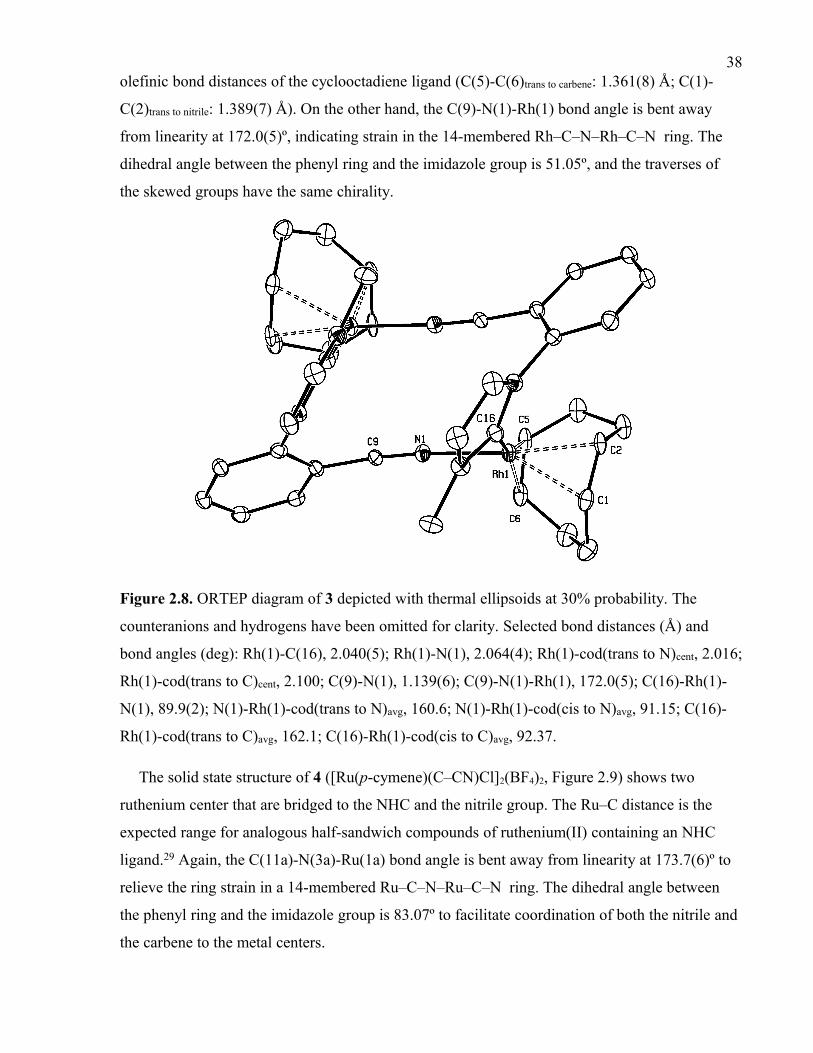
38olefinic bond distances of the cyclooctadiene ligand (C(5)-C(6)trans to carbene: 1.361(8) Å; C(1)-
C(2)trans to nitrile: 1.389(7) Å). On the other hand, the C(9)-N(1)-Rh(1) bond angle is bent away
from linearity at 172.0(5)º, indicating strain in the 14-membered Rh–C–N–Rh–C–N ring. The
dihedral angle between the phenyl ring and the imidazole group is 51.05º, and the traverses of
the skewed groups have the same chirality.
Figure 2.8. ORTEP diagram of 3 depicted with thermal ellipsoids at 30% probability. The
counteranions and hydrogens have been omitted for clarity. Selected bond distances (Å) and
bond angles (deg): Rh(1)-C(16), 2.040(5); Rh(1)-N(1), 2.064(4); Rh(1)-cod(trans to N)cent, 2.016;
Rh(1)-cod(trans to C)cent, 2.100; C(9)-N(1), 1.139(6); C(9)-N(1)-Rh(1), 172.0(5); C(16)-Rh(1)-
N(1), 89.9(2); N(1)-Rh(1)-cod(trans to N)avg, 160.6; N(1)-Rh(1)-cod(cis to N)avg, 91.15; C(16)-
Rh(1)-cod(trans to C)avg, 162.1; C(16)-Rh(1)-cod(cis to C)avg, 92.37.
The solid state structure of 4 ([Ru(p-cymene)(C–CN)Cl]2(BF4)2, Figure 2.9) shows two
ruthenium center that are bridged to the NHC and the nitrile group. The Ru–C distance is the
expected range for analogous half-sandwich compounds of ruthenium(II) containing an NHC
ligand.29 Again, the C(11a)-N(3a)-Ru(1a) bond angle is bent away from linearity at 173.7(6)º to
relieve the ring strain in a 14-membered Ru–C–N–Ru–C–N ring. The dihedral angle between
the phenyl ring and the imidazole group is 83.07º to facilitate coordination of both the nitrile and
the carbene to the metal centers.

39
Figure 2.9. ORTEP diagram of 4 depicted with thermal ellipsoids at 30% probability. The
counteranions and hydrogens have been omitted for clarity. Only one asymmetric unit is shown.
Selected bond distances (Å) and bond angles (deg): Ru(1a)-C(1a), 2.078(7); Ru(1a)-N(3a),
2.028(8); Ru(1a)-Cl(1a), 2.408(2); Ru(1a)-C(12a), 2.238(8); C(11a)-N(3a), 1.13(1); C(11a)-
N(3a)-Ru(1a), 173.3(6); C(1a)-Ru(1a)-N(3a), 84.9(3); C(1a)-Ru(1a)-Cl(1a), 90.9(2); N(3a)-
Ru(1a)-Cl(1a), 83.7(2).
Additional spectroscopic evidence supports the coordination of nitrile as well as the carbene
carbon. The 13C{1H} NMR of 3 in dichloromethane-d2 shows a doublet at δ 178.4 ppm (1JRh,C =
50.47 Hz), which is typical for most Rh(NHC) systems.22a, 28 A doublet is also located at δ 108.0
ppm (2JRh,C = 7.60 Hz) for the carbon on the nitrile group coordinated to rhodium(I). This
29. (a) Herrmann, W. A.; Kocher, C.; Goossen, L. J.; Artus, G. R. J., Chem. Eur. J. 1996, 2, 1627-1636; (b) Geldbach, T. J.; Laurenczy, G.; Scopelliti, R.; Dyson, P. J., Organometallics 2006, 25, 733-742; (c) Ozdemir, I.; Demir, S.; Cetinkaya, B.; Toupet, L.; Castarlenas, R.; Fischmeister, C.; Dixneuf, P. H., Eur. J. Inorg. Chem. 2007, 2862-2869; (d) Ozdemir, I.; Demir, S.; Gurbuz, N.; Cetinkaya, B.; Toupet, L.; Bruneau, C.; Dixneuf, P. H., Eur. J. Inorg. Chem. 2009, 1942-1949.29. (e) Gandolfi, C.; Heckenroth, M.; Neels, A.; Laurenczy, G. b.; Albrecht, M., Organometallics 2009, 28, 5112-5121; (f) Horn, S.; Gandolfi, C.; Albrecht, M., Eur. J. Inorg. Chem. 2011, 2863-2868.

40coupling is small compared to ones in systems such as the pincer-type compound Rh(PCN)
(PhCN) (PCN = PiPr2CH2-2-(3,5-(CH3)2-C6H)-6-CH2NEt2, 2JRh,C = 19.1 Hz).30 The carbene
resonance was observed for 4 at 170.4 ppm in its 13C{1H} NMR in nitromethane-d3. Further, the
nitrile stretching frequency observed in the IR spectrum (2183 cm-1) is significantly lower than
that of the imidazolium salt (2283 cm-1). The nitrile stretch for 4, however, was not observed in
its IR spectrum.
2.4 Conclusion
We have reported the synthesis of unsymmetrical nitrile-functionalized imidazolium salts
from 2-cyanophenylimidazole. The reactions of these imidazolium salts with silver(I) oxide
yielded silver(I) carbene complexes 2a-2d. The preparations of these complexes were sensitive
to the presence of water. In particular, the reaction of 1b with excess silver(I) oxide in the
presence of molecular sieves leads to the formation of the desired bis-carbene complex, but also
leads to the hydrolysis of the cyanomethyl group of the carbene ligand. This is the first report of
the observation of a NHC complex of silver(I) with a primary-amido donor. The selectivity in
hydrolysis of cyanomethyl group over the cyanophenyl group suggests the process must be
mediated by silver centers. We have observed hindered rotation about the C–N bond of the
amide by means of NMR, and suggested that an interaction between the silver(I) center and the
oxygen of the amido-group was present.
The use of these N-heterocyclic carbene complexes of silver(I) as a carbene transfer reagent
was further demonstrated by the reaction of [Ag(C–CN)2]BF4 (2a) with [Rh(cod)Cl]2 and [Ru(p-
cymene)Cl2]2. This C–CN ligand has shown versatility in its coordination in 3 and 4. These
complexes with bridging carbene ligands crystallized as dimers where the nitrile on the ligand
and the carbene ligand were coordinated to two different metal centers.
30. Cohen, R.; Rybtchinski, B.; Gandelman, M.; Shimon, L. J. W.; Martin, J. M. L.; Milstein, D., Angew.Chem. Int. Ed. 2003, 42, 1949-1952.

412.5 Experimental Section
2.5.1 General Considerations. All of the preparations and manipulations, except where
otherwise stated, were carried out under an argon or nitrogen atmosphere using standard
Schlenk-line and glovebox techniques. Dry and oxygen-free solvents were always used. The
syntheses of (2-cyanophenyl)imidazole15b, 3-(2-bromoethyl)indene31 and [Rh(cod)Cl]232 were
reported in the literature. All other reagents were purchased from commercial sources and were
used as received. Deuterated solvents were purchased from Cambridge Isotope Laboratories and
degassed and dried over activated molecular sieves prior to use. NMR spectra were recorded on a
Varian 400 spectrometer operating at 400 MHz for 1H, 100 MHz for 13C and 376 MHz for 19F.
The 1H and 13C NMR were measured relative to partially deuterated solvent peaks but are
reported relative to tetramethylsilane (TMS). All 19F chemical shifts were measured relative to
trichlorofluoromethane as an external reference. All infrared spectra were recorded on a Nicolet
550 Magna-IR spectrometer. The elemental analysis was performed at the Department of
Chemistry, University of Toronto, on a Perkin-Elmer 2400 CHN elemental analyzer. Samples
were handled under argon where it was appropriate. Single-crystal X-ray diffraction data were
collected using a Nonius Kappa-CCD diffractometer with Mo Kα radiation (λ = 0.71073 Å). The
CCD data were integrated and scaled using the Denzo-SMN package. The structures were solved
and refined using SHELXTL V6.1. Refinement was by full-matrix least-squares on F2 using all
data.
2.5.2. Synthesis of 1-(2-Cyanophenyl)-3-methylimidazolium Tetrafluoroborate (1a). A
Schlenk flask was charged with (2-cyanophenyl)imidazole (1.69 g, 10 mmol) and
trimethyloxonium tetrafluoroborate (Meerwein’s salt; 2.22 g, 15 mmol). Dry acetonitrile (20 mL)
was then added to the reaction mixture, for which a yellow color was observed. The solution was
stirred at ambient temperature for 2 h under argon, and the solvent was removed in vacuo. The
crude product was recrystallized with hot methanol to give a white crystalline solid. Yield: 2.45
g, 90%. 1H NMR (DMSO-d6, δ): 9.66 (s, 2-CH of imid., 1H), 8.27 (t, JHH= 1.84 Hz, 5-CH of
imid., 1H), 8.20 (dd, JHH = 1.03, 7.63 Hz, 3-CH of Ph, 1H), 8.03 (dt, JHH= 1.03, 8.83 Hz, 5-CH
of Ph, 1H), 8.00 (t, JHH = 1.84 Hz, 4-CH of imid., 1H), 7.88 (dd, JHH = 1.45, 8.83 Hz, 6-CH of
Ph, 1H), 7.86 (dt, JHH = 1.45, 7.63 Hz, 4-CH of Ph, 1H), 4.01 (s, CH3, 3H). 13C{1H} NMR
(DMSO-d6, δ): 137.7 (NCN), 136.1 (CPh), 134.9 (CPh), 134.3 (CPh), 131.1(CPh), 127.1(CPh), 124.1
31. Deppner, M.; Burger, R.; Alt, H. G., J. Organomet. Chem. 2004, 689, 1194-1211.32. Giordano, G.; Crabtree, R. H., Inorg. Synth. 1990, 28, 88-90.

42(Cimid.), 123.2 (Cimid.), 114.9 (CN), 108.4 (CPh), 36.1 (CH3). IR (KBr, cm-1): 2238 (v(CN)). MS
(ESI, methanol/water; m/z): 184.1 (M+). Anal. Calcd for C11H10BF4N3: C, 48.75; H, 3.72; N,
15.50. Found: C, 48.45; H, 3.74; N, 15.43.
2.5.3. Synthesis of 3-(Cyanomethyl)-1-(2-cyanophenyl)imidazolium Hexafluorophosphate
(1b). A two-necked Schlenk flask was charged with (2-cyanophenyl)imidazole (1.69 g, 10
mmol) in dry acetonitrile (20 mL). Bromoacetonitrile (1.80 g, 15 mmol) was added through a
syringe to the refluxing solution during the course of 0.5 h under argon. The reflux was
continued for 3.5 h, whereupon the bromide salt of the imidazolium precipitated out from the
reaction mixture. After the reaction mixture was cooled to ambient temperature, the white solids
were filtered and rinsed with diethyl ether. The crude bromide salt of the imidazolium was
dissolved in water (10 mL), and was added to a saturated aqueous solution (10 mL) of
ammonium hexafluorophosphate (2.45 g, 15 mmol). The white precipitate was then filtered,
rinsed with cold water, and recrystallized with hot ethanol to give a white crystalline solid.
Yield: 2.96 g, 84%. 1H NMR (DMSO-d6, δ): 9.87 (s, 2-CH of imid., 1H), 8.39 (t, JHH = 1.83 Hz,
4-CH of imid., 1H), 8.23 (t, JHH = 1.83 Hz, 5-CH of imid., 1H), 8.22 (dd, JHH = 1.37, 7.67 Hz, 3-
CH of Ph, 1H), 8.04 (dt, JHH = 1.37, 8.09 Hz, 5-CH of Ph, 1H), 7.92 (dd, JHH = 1.22, 8.09 Hz, 6-
CH of Ph, 1H), 7.87 (dt, JHH = 1.22, 7.67 Hz, 4-CH of Ph, 1H), 5.74 (s, CH2, 2H). 13C{1H} NMR
(DMSO-d6, δ): 138.9 (NCN), 136.0 (CPh), 134.9 (CPh), 134.4 (CPh), 131.4 (CPh), 127.3 (CPh),
124.2 (Cimid), 123.2 (Cimid), 115.0 (PhCN), 114.3 (CH2CN), 108.7 (CPh), 37.3 (CH2). IR (KBr,
cm-1): 2236 (v(CN)). MS (ESI, methanol/water; m/z): 209.1 (M+). Anal. Calcd for C12H9F6N4P:
C, 40.69; H, 2.56; N, 15.82. Found: C, 40.68; H, 2.51; N, 15.63.
2.5.4. Synthesis of 1-(2-Cyanophenyl)-3-(2-pyridinylmethyl)imidazolium Hexafluoro-
phosphate (1c). A solution of 2-picolyl chloride hydrochloride (1.23 g, 7.5 mmol) and
potassium carbonate (0.95 g, 11.3 mmol) in degassed 95% ethanol (10 mL) was stirred at
ambient temperature for 15 min under argon. The reddish solution was added through a syringe,
to a refluxing solution of (2-cyanophenyl)imidazole (0.846 g, 5 mmol) in degassed 95% ethanol
(10 mL). The solution was then refluxed for overnight under argon until a deep red color solution
was obtained. The solvent was removed in vacuo, and the residue was dissolved in water (10
mL) and filtered to remove any unreacted (2-cyanophenyl)imidazole. The aqueous solution
containing the crude chloride salt of the imidazolium salt was then added to a saturated aqueous
solution (10 mL) of ammonium hexafluorophosphate (1.22 g, 7.5 mmol). The red-brown
precipitate was then filtered, rinsed with cold water, recrystallized with hot ethanol and chilled at

43-78ºC to give a tan solid. Yield: 1.01 g, 50%. 1H NMR (DMSO-d6, δ): 9.92 (s, 2-CH of imid.,
1H), 8.60 (m, 6-CH of Py, 1H), 8.32 (t, JHH = 1.84 Hz, 5-CH of imid., 1H), 8.21 (dd, JHH = 1.53,
7.65 Hz, 3-CH of Ph, 1H), 8.12 (t, JHH = 1.84 Hz, 4-CH of imid., 1H), 8.03 (dt, JHH = 1.53, 7.80
Hz, 5-CH of Ph, 1H), 7.94 (dd, JHH = 1.11, 7.65 Hz, 6-CH of Ph, 1H), 7.92 (m, 4-CH of Py, 1H),
7.86 (dt, JHH = 1.11, 7.80 Hz, 4-CH of Ph, 1H), 7.57 (m, 3-CH of Py, 1H), 7.44 (m, 5-CH of Py,
1H), 5.73 (s, CH2, 2H). 13C{1H} NMR (DMSO-d6, δ): 152.9 (CPy), 149.6 (CPy), 138.1 (NCN),
137.5 (CPy), 136.2 (CPh), 134.9 (CPh), 134.4 (CPh), 131.2 (CPh), 127.3 (CPh), 123.9 (Cimid.), 123.7
(Cimid), 123.6 (CPy), 122.6 (CPy), 115.0 (CN), 108.7 (CPh), 53.6 (CH2). IR (KBr, cm-1): 2236
(v(CN)). MS (ESI, methanol/water; m/z): 261.1 (M+). Anal. Calcd for C16H13F6N4P: C, 47.30; H,
3.23; N, 13.79. Found: C, 47.03; H, 3.29; N, 13.22.
2.5.5. Synthesis of 1-(2-Cyanophenyl)-3-(2-pyridinyl)imidazolium Hexafluorophosphate
(1d). A 10 mL round-bottom flask was charged with (2-cyanophenyl)imidazole (0.846 g, 5
mmol) and 2-bromopyridine (1.19 g, 7.5 mmol), and was heated neat at 140ºC under argon for 1
day with vigorous stirring. The liquid was cooled down to give a deep red solid, which was
extracted with water and filtered through activated charcoal to give a bright red solution. The red
solution containing the crude bromide salt of the imidazolium salt was then added to a saturated
aqueous solution (10 mL) of ammonium hexafluorophosphate (1.22 g, 7.5 mmol). The red
precipitate was filtered, rinsed with cold water, and was recrystallized with hot ethanol and
chilled at -78ºC to give a red solid. Yield: 1.05 g, 54%. 1H NMR (DMSO-d6, δ): 10.63 (s, 2-CH
of imid., 1H), 8.83 (dd, JHH = 1.72, 2.12 Hz, 5-CH of imid., 1H), 8.72 (m, 3-CH of Py, 1H), 8.58
(dd, JHH = 1.72, 2.12 Hz, 4-CH of imid., 1H), 8.30 (m, 5-CH of Py, 1H), 8.26 (dd, JHH = 1.55,
8.06 Hz, 3-CH of Ph, 1H), 8.14 (m, 6-CH of Py, 1H), 8.08 (dd, JHH = 1.40, 7.52 Hz, 6-CH of Ph,
1H), 8.04 (dt, JHH = 1.40, 8.06 Hz, 5-CH of Ph, 1H), 7.91 (dt, JHH = 1.55, 7.52 Hz, 4-CH of Ph,
1H), 7.73 (m, 4-CH of Py, 1H). 13C{1H} NMR (DMSO-d6, δ): 149.3 (CPy), 146.0 (CPy) , 140.7
(CPy), 136.6 (NCN), 136.0 (CPh), 134.9 (CPh), 134.4 (CPh), 131.5 (CPh), 127.4 (CPh), 125.7 (CPy),
124.5 (Cimid.), 119.7 (Cimid.), 115.0 (CN), 114.7 (CPy), 108.7 (CPh). IR (KBr, cm-1): 2242 (v(CN)).
MS (ESI, methanol/water; m/z): 247.1 (M+). Anal. Calcd for C15H11F6N4P: C, 45.93; H, 2.83; N,
14.28. Found: C, 46.03; H, 3.05; N, 14.51.
2.5.6. Synthesis of 1-(2-Cyanophenyl)-3-(1-phenylethyl)imidazolium Tetrafluoroborate (1e).
A Schlenk flask was charged with (2-cyanophenyl)imidazole (300 mg, 1.8 mmol) and excess (1-
bromoethyl)benzene (1.77g, 8.9 mmol) in dry acetonitrile (10 mL). The reaction mixture was
refluxed for 3 d. The solvent was then removed, and the residue was rinsed with diethyl ether (10

44mL) and dried in vacuo. This residue was extracted further with water (8 mL) and filtered to
remove unreacted (2-cyanophenyl)imidazole. The clear aqueous solution was added to a
saturated aqueous solution (10 mL) of ammonium tetrafluoroborate (223 mg, 2.0 mmol). The
white precipitate was then filtered, rinsed with cold water, and dried in vacuo to give the
analytically pure product. Yield: 119 mg, 19%. 1H NMR (DMSO-d6, δ): 10.01 (s, 2-CH of imid.,
1H), 8.33 (t, JHH = 1.71 Hz, 5-CH of imid., 1H), 8.22 (dd, JHH = 1.17, 7.76 Hz, 3-CH of Ph, 1H),
8.16 (t, JHH = 1.71 Hz, 4-CH of imid., 1H), 8.06 (dd, JHH = 1.08, 7.89 Hz, 6-CH of Ph, 1H), 8.00
(t, JHH = 7.89, 5-CH of Ph, 1H), 7.87 (dt, JHH = 1.21, 7.76 Hz, 4-CH of Ph, 1H), 7.52 – 7.42 (m,
Ph, 5H), 5.95 (q, JHH = 6.96 Hz, CH, 1H), 1.98 (d, JHH = 6.96 Hz, CH3, 3H). 13C{1H} NMR
(DMSO-d6, δ): 138.9 (NCN), 136.7 (CPhCH), 136.3 (CPh), 134.9 (CPh), 134.3 (CPh), 131.3 (CPh),
129.0 (CPhCH), 128.8 (CPh), 127.4 (CPhCH), 126.6 (CPhCH), 124.0 (Cimid), 122.0 (Cimid), 115.1 (CN),
108.9 (CPh), 59.5 (CH), 20.5 (CH3). MS (ESI, methanol/water; m/z): 274.1 (M+). Anal. Calcd for
C18H16F4 BN3: C, 59.86; H, 4.47; N, 11.64. Found: C, 59.77; H, 4.69; N, 11.99.
2.5.7. Synthesis of 1-(2-(Inden-3-yl)ethyl)-3-(2-cyanophenyl)imidazolium Tetrafluoro-
borate (1f). A Schlenk flask was charged with (2-cyanophenyl)imidazole (500 mg, 2.96 mmol)
and 3-(2-bromoethyl)indene (659 mg, 2.96 mmol) in dry acetonitrile (12 mL). The reaction
mixture was refluxed for 4.5 d. After the reaction mixture was cooled to ambient temperature,
the beige solid that was precipitated from the pale-purple solution was filtered and rinsed with
diethyl ether (10 mL). The crude bromide salt of the imidazolium was dissolved in water (5 mL),
and was added to a saturated aqueous solution (10 mL) of ammonium tetrafluoroborate (464 mg,
4.43 mmol). The pale-brown precipitate was then filtered, rinsed with cold water, and dried in
vacuo to give the analytically pure product. Yield: 562 mg, 48%. 1H NMR (DMSO-d6, δ): 9.78
(s, 2-CH of imid., 1H), 8.30 (t, JHH = 1.60 Hz, 4-CH of imid., 1H), 8.21 (dd, JHH = 1.23, 7.75 Hz,
3-CH of Ph, 1H), 8.15 (t, JHH = 1.60 Hz, 5-CH of imid., 1H), 8.03 (dt, JHH = 1.20, 7.90 Hz, 5-CH
of Ph, 1H), 7.85 (m, 4-CH and 6-CH of Ph, 2H), 7.50 (d, JHH = 7.50 Hz, 5-CH of Ar-Ind, 1H),
7.49 (d, JHH = 7.50 Hz, 8-CH of Ar-Ind, 1H), 7.33 (t, JHH = 7.48 Hz, 6-CH of Ar-Ind, 1H), 7.23
(t, JHH = 7.48 Hz, 7-CH of Ar-Ind, 1H), 6.37 (s, CH of Ind, 1H), 4.66 (t, JHH = 7.13 Hz, CH2,
2H), 3.37 (s, CH2 of Ind, 2H), 3.24 (t, JHH = 6.83 Hz, CH2, 2H). 13C{1H} NMR (DMSO-d6, δ):
144.1 (CAr-Ind), 144.0 (CAr-Ind), 138.8 (NCN), 137.6 (CInd), 136.3 (CPh), 135.1 (CPh), 134.5 (CPh),
131.4 (CPh), 130.9 (CInd), 127.4 (CPh), 126.1 (CAr-Ind), 125.0 (CAr-Ind), 123.9 (Cimid), 123.7(CAr-Ind),
123.3 (Cimid), 118.9 (CAr-Ind), 115.1 (CN), 108.8 (CPh), 48.4 (CH2), 37.7 (CH2 of Ind), 27.6 (CH2).

45MS (ESI, methanol/water; m/z): 312.1 (M+). Anal. Calcd for C21H18F4 BN3: C, 63.18; H, 4.54; N,
10.53. Found: C, 63.07; H, 4.75; N, 10.50.
2.5.8. Synthesis of Bis[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene]silver(I)
Tetrafluoroborate ([Ag(C–CN)2]BF4, 2a) . A Schlenk flask was charged with 1a (542 mg, 2
mmol) and silver(I) oxide (695 mg, 3 mmol) in dry acetonitrile (15 mL). The reaction mixture
was refluxed over 3 Å molecular sieves under argon overnight. It was then filtered through a pad
of Celite to give a gold-yellow solution. The volume of solvent was reduced to 3 mL ca., and
diethyl ether (10 mL) was added, which caused fine tan crystals to precipitate from the reaction
mixture. The crystals were then collected on a glass frit and dried in vacuo. Yield: 456 mg, 81%.
Suitable crystals for an X-ray diffraction study were obtained by slow diffusion of diethyl ether
into a saturated solution of 2a in acetonitrile. 1H NMR (CD3CN, δ): 7.88 (dd, JHH = 1.63, 7.68
Hz, 3-CH of Ph, 1H), 7.77 (dt, JHH = 1.63, 7.84 Hz, 5-CH of Ph, 1H), 7.68 (dt, JHH = 1.26, 7.68
Hz, 4-CH of Ph, 1H), 7.55 (dd, JHH = 1.26, 7.84 Hz, 6-CH of Ph, 1H), 7.47 (d, JHH = 1.86 Hz, 5-
CH of imid., 1H), 7.39 (dt, JHH = 1.86 Hz, 4-CH of imid., 1H), 3.85 (s, CH3, 3H). 19F NMR
(CD3CN, δ): -152.2 (s), -152.3 (s). 13C{1H} NMR (CD3CN, δ): 182.6 (Ag–C), 142.1 (CPh) ,
135.0 (CPh), 134.6 (CPh), 130.6 (CPh), 128.5 (CPh), 124.1 (Cimid.), 123.5 (Cimid.), 116.1 (CN), 110.7
(CPh), 39.0 (CH3). IR (KBr, cm-1): 2242, 2227 (v(CN)). Anal. Calcd for C22H18AgBF4N6: C,
47.09; H, 3.23; N, 14.98. Found: C, 46.95; H, 3.39; N, 14.75.
2.5.9. Observation of Silver(I) Complex of (3-(Cyanomethyl)-1-(2-cyanophenyl)-imidazol-2-
ylidene (2b). A Schlenk flask was charged with 1b (354 mg, 1 mmol) and silver(I) oxide (348
mg, 1.5 mmol) in dry acetonitrile (10 mL). The reaction mixture was refluxed over 3Å molecular
sieves under argon overnight. It was then filtered through a pad of Celite to give a gold-yellow
solution. The volume of solvent was removed, and 3 mL of acetonitrile-d3 was added to prepare
samples for NMR analysis. The compound was too unstable to be isolated in its solid state, and
prone to decomposition under light in both solution and solid state. 1H NMR (CD3CN, δ): 7.88
(dd, JHH = 1.57, 7.70 Hz, 3-CH of Ph, 1H), 7.77 (dt, JHH = 1.57, 7.76 Hz, 5-CH of Ph, 1H), 7.70
(dt, JHH = 0.96, 7.70 Hz, 4-CH of Ph, 1H), 7.56 (m, CH of imid., 2H), 7.55 (dd, JHH = 0.96, 7.76
Hz, 6-CH of Ph., 1H), 5.23 (s, CH2, 2H). 19F NMR (CD3CN, δ): -73.3 (d, JPF =707 Hz). 13C{1H}
NMR (CD3CN, δ): 184.4 (Ag–C), 142.0 (CPh) , 135.6 (CPh), 135.1 (CPh), 131.5 (CPh), 128.8
(CPh), 125.0 (Cimid.), 123.8 (Cimid.), 116.4 (PhCN), 116.1 (CH2CN), 110.9 (CPh), 40.4 (CH2).

462.5.10. Observation of Silver(I) Complex of (3-(Carbomoylmethyl)-1-(2-
cyanophenyl)imidazol-2-ylidene (2e). The amide complex of 2b was generated in situ in small
quantities during the preparation of 2b, and was observed by NMR as with 2b. 1H NMR
(CD3CN, δ): 7.86 (m, 3-CH of Ph, 1H), 7.76 (m, 5-CH of Ph, 1H), 7.70 (m, 4-CH of Ph, 1H),
7.55 (m, 6-CH of Ph, 1H), 7.48 (d, JHH = 1.86 Hz, 5-CH of imid., 1H), 7.48 (d, JHH = 1.86 Hz, 4-
CH of imid., 1H), 6.49 (s, br, NH2, 1H), 6.12 (s, br, NH2, 1H), 4.89 (s, CH2, 2H). 19F NMR
(CD3CN, δ): -73.3 (d, JPF = 707 Hz). 13C{1H} NMR (CD3CN, δ): 183.9 (Ag–C), 169.4 (CONH2),
142.3 (CPh) , 135.6 (CPh), 135.1 (CPh), 131.2 (CPh), 128.7 (CPh), 124.6 (Cimid.), 123.9 (Cimid.),
116.6 (PhCN), 110.8 (CPh), 54.2 (CH2).
2.5.11. Synthesis of Bis{[1-(2-cyanophenyl)-3-(2-pyridinylmethyl)imidazol-2-
ylidene]silver(I)} Hexafluorophosphate (2c). A Schlenk flask was charged with 1c (203 mg,
0.5 mmol) and silver(I) oxide (174 mg, 0.75 mmol) in dry acetonitrile (10 mL). The reaction
mixture was refluxed over 3 Å molecular sieves under argon overnight. It was then filtered
through a pad of Celite to give a gold-yellow solution. The volume of solvent was reduced to 1
mL ca., and diethyl ether (8 mL) was added for which tan solids were precipitated from the
reaction mixture. The precipitate was filtered and dried under vacuo to give a tan powder. Yield:
174 mg, 68%. 1H NMR (CD3CN, δ): 8.44 (m, 3-CH of Py, 1H), 7.79 (dd, JHH = 1.16, 7.76 Hz, 3-
CH of Ph, 1H), 7.75 (m, 5-CH of Py, 1H), 7.69 (dt, JHH = 1.16, 8.07 Hz, 5-CH of Ph, 1H), 7.61
(dt, JHH = 1.23, 7.76 Hz, 4-CH of Ph, 1H), 7.52 (dd, JHH = 1.23, 8.07 Hz, 6-CH Ph, 1H), 7.49 (d,
JHH = 1.86 Hz, 4-CH of imid., 1H), 7.48 (d, JHH = 1.86 Hz, 5-CH of imid., 1H), 7.31 (m, 4-CH of
Py, 1H), 7.27 (m, 6-CH of Py, 1H), 5.41 (s, CH2, 2H). 19F NMR (CD3CN, δ): -73.3 (d, JPF = 707
Hz). 13C{1H} NMR (CD3CN, δ): 183.5 (Ag–C), 156.1 (CPy), 151.0 (CPy), 142.5 (CPh), 138.8
(CPy), 135.5 (CPh), 135.1 (CPh), 131.1 (CPh), 128.9 (CPh), 124.7 (CPy), 124.4 (Cimid.), 124.1 (Cimid.),
123.6 (CPy), 116.6 (CN), 111.1 (CPh), 57.8 (CH2). IR (KBr, cm-1): 2236 (v(CN)). Anal. Calcd for
C32H24Ag2F12N8P2: C, 37.45; H, 2.36; N, 10.91. Found: C, 38.04; H, 2.83; N, 10.42.
2.5.12. Synthesis of Bis{[1-(2-cyanophenyl)-3-(2-pyridinyl)imidazol-2-ylidene]silver(I)}
Hexafluorophosphate (2d). A Schlenk flask was charged with 1d (189 mg, 0.48 mmol) and
silver(I) oxide (168 mg, 0.72 mmol) in dry acetonitrile (10 mL). The reaction mixture was
refluxed over 3 Å molecular sieves under argon for overnight. It was then filtered through a pad
of Celite to give a gold-yellow solution. The volume of solvent was reduced to ca. 1 mL, and
diethyl ether (8 mL) was added was added for which beige solids were precipitated from the
reaction mixture. The precipitate was filtered and dried under vacuo to give a beige powder.

47Yield: 150 mg, 63%. 1H NMR (CD3CN, δ): 8.19 (m, 3-CH of Py, 1H), 8.01 (d, JHH = 1.44 Hz, 4-
CH of imid., 1H), 7.88 (m, 5-CH of Py, 1H), 7.86 (d, JHH = 7.40 Hz, 3-CH of Ph, 1H), 7.80 (m,
6-CH of Py, 1H), 7.72 (t, JHH = 7.79 Hz, 5-CH of Ph, 1H), 7.68 (d, JHH = 1.44 Hz, 5-CH of imid.,
1H), 7.67 (t, JHH = 7.40 Hz, 4-CH of Ph, 1H), 7.62 (d, JHH = 7.79 Hz, 6-CH of Ph, 1H), 7.41 (m,
4-CH of Py, 1H). 19F NMR (CD3CN, δ): -73.3 (d, JPF = 707 Hz). 13C{1H} NMR (CD3CN, δ):
183.8 (Ag–C), 151.2 (CPy), 149.9 (CPy), 142.7 (CPh), 140.9 (CPy), 135.5 (CPh), 135.1 (CPh), 131.4
(CPh), 128.9 (CPh), 125.5 (Cimid.), 125.2 (CPy), 121.6 (Cimid.), 116.5 (CPy.), 116.4 (CN), 111.1
(CPh). IR (KBr, cm-1): 2232 (v(CN)). Anal. Calcd for C30H20Ag2F12N8P2: C, 36.10; H, 2.02; N,
11.23. Found: C, 37.17; H, 2.26; N, 11.30.
2.5.13. Synthesis of Bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)-(η4-1,5-
cycloctadiene)rhodium(I)] Tetrafluoroborate ([Rh(C–CN)(cod)]2(BF4)2, 3). A solution of 2a
(56 mg, 0.1 mmol) and silver tetrafluoroborate (10 mg, 0.05 mmol) in acetonitrile (3 mL) was
added to a solution of [RhCl(cod)2]2 (57 mg, 0.115 mmol) in acetonitrile (2 mL). A pale-brown
precipitate was formed instantaneously. The reaction mixture continued to be stirred for 2 h. It
was then filtered through a pad of Celite to give a bright-yellow solution. The solvent was then
removed in vacuo, and the residue was dissolved in dichloromethane (1 mL). Addition of diethyl
ether (8 mL) caused precipitation of a bright-yellow precipitate. The precipitate was then filtered,
rinsed with diethyl ether (2 mL), and dried in vacuo. Yield: 75 mg, 71%. Suitable crystals for an
X-ray diffraction study were obtained by slow diffusion of diethyl ether into a saturated solution
of 3 in dichloromethane. 1H NMR (CD2Cl2, δ): 8.21 (dd, JHH = 1.40, 7.80 Hz, 3-CH of Ph, 1H),
8.00 (dt, JHH = 1.40, 7.97 Hz, 5-CH of Ph, 1H), 7.90 (dt, JHH = 1.13, 7.80 Hz, 4-CH of Ph, 1H),
7.61 (d, JHH = 2.00 Hz, 5-CH of imid., 1H), 7.55 (dd, JHH = 1.13, 7.97 Hz, 6-CH of Ph, 1H), 7.41
(d, JHH = 2.00 Hz, 4-CH of imid., 1H), 5.16 (m, olefinic-CH of cod, 1H), 4.76 (m, olefinic-CH of
cod, 1H), 4.07 (m, olefinic-CH of cod, 1H), 3.90 (s, CH3, 3H), 2.88 (m, olefinic-CH of cod, 1H),
2.51 (m, CH2 of cod, 1H), 2.24 (m, CH2 of cod, 1H), 2.18 (m, CH2 of cod, 1H), 2.04 (m, CH2 of
cod, 1H), 1.94 (m, CH2 of cod, 1H), 1.88 (m, CH2 of cod, 1H), 1.65 (m, CH2 of cod, 1H), 1.40
(m, CH2 of cod, 1H). 19F NMR (CD2Cl2, δ): -152.3 (s), -152.4 (s). 13C{1H} NMR (CD2Cl2, δ):
178.4 (Rh–C, 1JRh,C = 50.47 Hz), 142.56 (CPh), 136.7 (CPh) , 136.3 (CPh), 131.3 (CPh), 128.6
(CPh), 125.9 (Cimid.), 123.9 (Cimid.), 110.5 (CPh), 108.0 (CN, 2JRh,C = 7.60 Hz), 100.1 (olefinic-C of
cod, 1JRh,C = 7.20 Hz), 99.2 (olefinic-C of cod, 1JRh,C = 6.71 Hz), 83.3 (olefinic-C of cod, 1JRh,C =
13.30 Hz), 79.1 (olefinic-C of cod, 1JRh,C = 12.69 Hz), 38.3 (CH3), 34.2 (CH2 of cod), 30.4 (CH2
of cod), 29.9 (CH2 of cod), 27.8 (CH2 of cod). IR (KBr, cm-1): 2183 (v(CN)). MS (ESI,

48methanol/water; m/z): 394.1 ([0.5 M]+). Several attempts at elemental analyses failed to give an
acceptable carbon content, while hydrogen and nitrogen content are in the acceptable range.
Typical results: Anal. Calcd for C38H42B2F8N6Rh2: C, 47.43; H, 4.40; N, 8.73. Found: C, 45.49;
H, 4.39; N, 8.51.
2.5.14. Synthesis of Bis[chloro-(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)-(η6-p-
cymene)ruthenium(II)] Tetrafluoroborate ([Ru(p-cymene)(C–CN)Cl]2(BF4)2, 4). A solution
of 2a (50 mg, 0.089 mmol) and silver tetrafluoroborate (18 mg, 0.092 mmol) in acetonitrile (4
mL) was added to a solution of [Ru(p-cymene)Cl2]2 (55 mg, 0.090 mmol) in acetonitrile (6 mL).
A pale-brown precipitate was formed instantaneously. The reaction mixture continued to be
stirred for 2 h. It was then filtered through a pad of Celite to give a red-orange solution. The
solvent was then removed in vacuo, and the residue was dissolved in methanol (3 mL). A yellow
precipitate was formed gradually from the homogeneous solution. It was then filtered, rinsed
with diethyl ether (4 mL), and dried in vacuo. Yield: 152 mg, 79%. Suitable crystals for an X-ray
diffraction study were obtained by slow diffusion of diethyl ether into a saturated solution of 4 in
nitroethane. 1H NMR (CD3NO2, δ): 7.99 (m, 4-CH and 6-CH of Ph, 2H), 7.73 (t, JHH = 7.73 Hz,
5-CH of Ph, 1H), 7.66 (d, JHH = 7.46 Hz, 3-CH of Ph, 1H), 7.34 (d, JHH = 2.00 Hz, 4-CH of
imid., 1H), 7.16 (d, JHH = 2.00 Hz, 5-CH of imid., 1H), 6.01 (d, JHH = 5.87 Hz, 2-Ar-CH of p-
cymene, 1H), 5.45 (m, 3-Ar-CH and 6-Ar-CH of p-cymene, 2H), 5.29 (d, JHH = 6.14 Hz, 5-Ar-
CH of p-cymene, 1H), 4.01 (s, CH3, 3H), 2.71 (sept, JHH = 6.91 Hz, CH of (CH3)2CH of p-
cymene, 1H), 2,01 (s, CH3 of p-cymene, 3H), 1.26 (d, JHH = 6.91 Hz, CH3 of (CH3)2CH of p-
cymene, 1H), 1.21 (d, JHH = 6.91 Hz, CH3 of (CH3)2CH of p-cymene, 1H).19F NMR (CD3NO2,
δ): -153.0 (s), -153.1 (s). 13C{1H} NMR (CD3NO2, δ): 170.4 (Ru–C), 144.7 (CPh), 135.8 (CPh) ,
134.0 (CPh), 131.3 (CPh), 130.0 (CPh), 127.4 (Cimid.), 127.1 (Cimid.), 124.8 (CPh), 116.6 (CN), 113.7
(CAr-p-cymene), 105.4 (CAr-p-cymene), 95.2 (CAr-p-cymene), 90.1 (CAr-p-cymene), 84.5 (CAr-p-cymene), 83.6
(CAr-p-cymene), 40.5(CH3), 32.6 (CH of (CH3)2CH of p-cymene), 23.5 (CH3 of p-cymene), 21.9,
(CH3 of (CH3)2CH of p-cymene), 19.0 (CH3 of (CH3)2CH of p-cymene). MS (ESI, methanol/
water; m/z): 454.1 (0.5 M+), 418.1 ([0.5 (M – Cl)]+). Anal. Calcd for C42H46B2Cl2F8N6Ru2: C,
46.64; H, 4.29; N, 7.77. Found: C, 46.26; H, 3.96; N, 7.86

49Chapter 3: Palladium(II) and Platinum(II) Complexes Featuring a
Nitrile-Functionalized N-Heterocyclic Carbene Ligand
3.1 Abstract
The transmetalation reaction of 2 equiv of trans-PdCl2(CH3CN)2 or Pd(cod)Cl2 with a nitrile-
functionalized N-heterocyclic carbene (C–CN) complex of silver(I), bis[1-(2-cyanophenyl)-3-
methylimidazol-2-ylidene]silver(I) tetrafluoroborate ([Ag(C–CN)2]BF4 2a), and 1 equiv of
AgBF4 afforded a bimetallic complex formulated as [(C–CN)2Pd(μ-Cl)2Pd(CH3CN)2](BF4)2 (5a).
The interesting trimetallic complex 5b was obtained during an attempt to crystallize 5a from wet
diethyl ether and acetonitrile. The solid-state structure of 5b reveals the presence of a novel C–
N–N–C donor ligand providing two bridging imido nitrogens to adjacent palladium(II) centers in
the trimetallic complex [{Pd(CH3CN)2}3(C–N–N–C)](BF4)4 (5b). The C–N–N–C ligand, which
has a central –N=C–O–C=N– linkage, was formed from the hydrolysis and condensation of the
nitrile groups of two carbene ligands. Palladium(II) and platinum(II) complexes bearing
chelating olefin and N-heterocyclic carbene (NHC) ligands were synthesized by transmetalation
reactions of N-heterocyclic carbene complexes of silver(I), complex 2a and bis[1,3-bis(2,4,6-
trimethyl-phenyl)imidazol-2-ylidene]silver(I) tetrafluoroborate ([Ag(IMes)2]BF4, 10), with
[Pd(η1:η2-coe-OMe)(μ-Cl)]2 and Pt(cod)Cl2 (coe-OMe = 2-methoxycyclooct-5-enyl, cod = 1,5-
cyclooctadiene) or by addition of a methoxide anion to the coordinated diolefin ligand of the
platinum(II) complex, chloro[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene](η4-1,5-
cyclooctadiene)-platinum(II) tetrafluoroborate (7). The complexes bearing the C–CN and 2-
methoxycyclooct-5-enyl ligands ([M(C–CN)(η1:η2-coe-OMe)]+: 6, M = Pd; 9, M = Pt) exists in
both monomeric and dimeric forms depending on the choice of recrystallization solvents. All of
these complexes were isolated and studied by NMR and infrared spectroscopy. Different ratios
of rotamers were observed for the complexes bearing a 2-methoxycyclooct-5-enyl ligand owing
to its orientation relative to the carbene ligand. Rotamers are believed to form because of steric
restrictions to rotation about the M–Ccarbene bond (M = Pd, Pt).
*Reproduced in part with permission from O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2010, 29, 570-581. Copyright 2010 American Chemical Society.

503.2 Introduction
Donor functionalized N-heterocyclic carbenes (NHC) have exceptional utility in the synthesis
of novel and highly active homogeneous catalysts in the fields of organometallic and synthetic
organic chemistry.1 The versatility in the mode of coordination of these ligands and, more
importantly, their ability to promote oxidative addition, reductive elimination, and
dehydrohalogenation reactions in catalytic reactions have been extensively reviewed in the
literature in recent years.2
Our group has been interested in the development of catalysts for the hydrogenation of polar
bonds, including those of ketones, imines, and nitriles. High activities were reported for
ruthenium(II)3 and iron(II)4 based catalysts bearing phosphino-amino ligands. We have reported
the syntheses of coordinatively unsaturated hydridoruthenium(II) complexes bearing N-
heterocyclic carbene ligands,5 and nitrile-functionalized N-heterocyclic carbene ligands and their
metal complexes of silver(I), rhodium(I) and ruthenium(II) (Chapter 2 and Figure 3.1).6 The
nitrile functionality on these carbene ligands can bridge to metal centers, and can be hydrolyzed
with the assistance of silver ions, giving primary amido-functionalized N-heterocyclic carbene
ligands.6
In our continuing investigation of the coordinating ability and reactivity of these nitrile-
functionalized N-heterocyclic carbene ligands, and N-heterocyclic carbene ligands in general and
their late transition metal complexes, we report in this chapter the synthesis and studies of
palladium(II) and platinum(II) complexes bearing N-heterocyclic carbene and olefin ligands,
which predominantly have the general structures shown in Figure 3.1. Of particular interest is the
reactivity of the coordinated olefin and nitriles in these metal complexes.7
1. (a) Kuhl, O., Chem. Soc. Rev. 2007, 36, 592-607; (b) Liddle, S. T.; Edworthy, I. S.; Arnold, P. L., Chem. Soc. Rev. 2007, 36, 1732-1744; (c) Hahn, F. E.; Jahnke, M. C., Angew. Chem. Int. Ed. 2008, 47, 3122-3172; (d) Diez-Gonzalez, S.; Marion, N.; Nolan, S. P., Chem. Rev. 2009, 109, 3612-3676.2. (a) Crudden, C. M.; Allen, D. P., Coord. Chem. Rev. 2004, 248, 2247-2273; (b) Lee, H. M.; Lee, C. C.; Cheng, P. Y., Curr. Org. Chem. 2007, 11, 1491-1524; (c) Normand, A. T.; Cavell, K. J., Eur. J. Inorg. Chem. 2008, 2781-2800; (d) Poyatos, M.; Mata, J. A.; Peris, E., Chem. Rev. 2009, 109, 3677-3707; (e) Corberan, R.; Mas-Marza, E.; Peris, E., Eur. J. Inorg. Chem. 2009, 1700-1716.3. (a) Guo, R.; Lough, A. J.; Morris, R. H.; Song, D., Organometallics 2004, 23, 5524-5529; (b) Abdur-Rashid, K.; Guo, R. W.; Lough, A. J.; Morris, R. H.; Song, D. T., Adv. Synth. Catal. 2005, 347, 571-579; (c) Guo, R.; Morris, R. H.; Song, D., J. Am. Chem. Soc. 2005, 127, 516-517.4. Morris, R. H., Chem. Soc. Rev. 2009, 38, 2282-2291 and references therein.5. Abdur-Rashid, K.; Fedorkiw, T.; Lough, A. J.; Morris, R. H., Organometallics 2004, 23, 86-94.6. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 853-862.7. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2010, 29, 570-581.

51
Figure 3.1. Nitrile-functionalized N-heterocyclic carbene complexes of Ag(I) and Rh(I) and the
general structures of the complexes reported in this chapter (M = Pd(II), Pt(II); D = olefin or
alkyl ligand; L = nitrile donor or a chloro group).
3.3 Results and Discussion
3.3.1 Transmetalation of a Nitrile-Functionalized N-heterocyclic Carbene Ligand from
Silver(I) to Palladium(II). The transmetalation reaction8 of [Ag(C–CN)2]BF4 (2a) with 2 equiv
of trans-PdCl2(CH3CN)2 and 1 equiv of AgBF4 afforded the bimetallic complex 5a as a yellow
powder in 62% yield. Alternatively, the complex could also be prepared using Pd(cod)Cl2 in
higher yields (79%) (Scheme 3.1). The reaction did not proceed cleanly when only 1 equiv of the
palladium(II) precursor was used. Although crystals suitable for X-ray diffraction were not
obtained successfully, the structure of 5a can be elucidated by spectroscopic data and elemental
analysis. The 13C{1H} NMR spectrum in acetonitrile-d3 solution shows a singlet at 141.4 ppm,
which was assigned to the carbene carbon (Pd–Ccarbene) according to a 1H-13C HMBC
experiment. Although it is shifted upfield compared to most Pd(NHC)2X2 (X = halide) systems,9
it is in the expected range when compared to cationic cis-[Pd(NHC)2(CH3CN)X]+ or cis-
[Pd(NHC)2(CH3CN)2]2+ systems.10 The nitrile stretching frequencies in the infrared spectrum
8. Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B., Chem. Rev. 2009, 109, 3561-3598.9. (a) Herrmann, W. A.; Elison, M.; Fischer, J.; Kocher, C.; Artus, G. R. J., Angew. Chem. Int. Ed. 1995, 34, 2371-2374; (b) Ku, R. Z.; Huang, J. C.; Cho, J. Y.; Kiang, F. M.; Reddy, K. R.; Chen, Y. C.; Lee, K. J.; Lee, J. H.; Lee, G. H.; Peng, S. M.; Liu, S. T., Organometallics 1999, 18, 2145-2154; (c) Magill, A. M.; McGuinness, D. S.; Cavell, K. J.; Britovsek, G. J. P.; Gibson, V. C.; White, A. J. P.; Williams, D. J.; White, A. H.; Skelton, B. W., J. Organomet. Chem. 2001, 617-618, 546-560; (d) Grundemann, S.; Albrecht, M.; Kovacevic, A.; Faller, J. W.; Crabtree, R. H., Dalton Trans. 2002, 2163-2167; (e) Fu, C. F.; Lee, C. C.; Liu, Y. H.; Peng, S. M.; Warsink, S.; Elsevier, C. J.; Chen, J. T.; Liu, S. T., Inorg. Chem. 2010, 49, 3011-3018.10. Scherg, T.; Schneider, S. K.; Frey, G. D.; Schwarz, J.; Herdtweck, E.; Herrmann, W. A., Synlett 2006, 2894-2907.11. (a) Andrews, M. A.; Chang, T. C. T.; Cheng, C. W. F.; Emge, T. J.; Kelly, K. P.; Koetzle, T. F., J. Am. Chem. Soc. 1984, 106, 5913-5920; (b) Mitsudo, K.; Kaide, T.; Nakamoto, E.; Yoshida, K.; Tanaka, H., J. Am. Chem. Soc. 2007, 129, 2246-2247.
NN
NCH3
MD
L
M = Pd(II), Pt(II)
This chapter
n+
NN
NCH3
BF4Ag
NN
NCH3
(BF4)2
RhN
NN
RhN
NN
H3C
CH3
+
+
+
2a 3
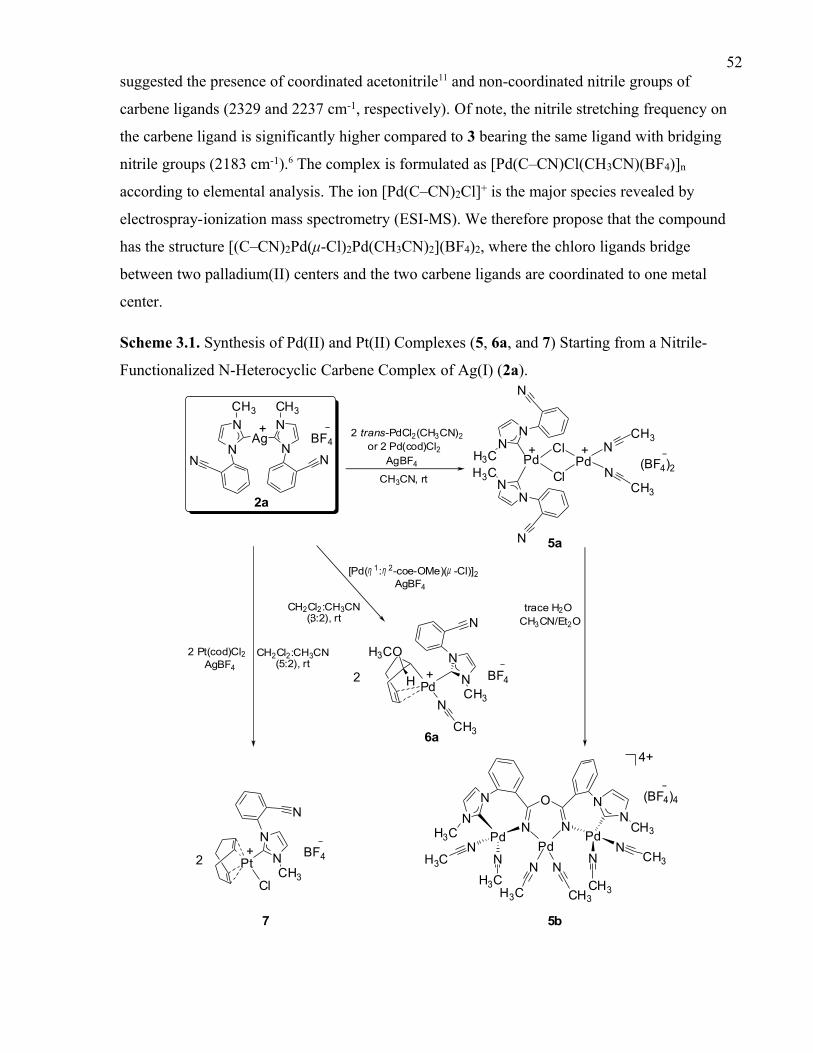
52suggested the presence of coordinated acetonitrile11 and non-coordinated nitrile groups of
carbene ligands (2329 and 2237 cm-1, respectively). Of note, the nitrile stretching frequency on
the carbene ligand is significantly higher compared to 3 bearing the same ligand with bridging
nitrile groups (2183 cm-1).6 The complex is formulated as [Pd(C–CN)Cl(CH3CN)(BF4)]n
according to elemental analysis. The ion [Pd(C–CN)2Cl]+ is the major species revealed by
electrospray-ionization mass spectrometry (ESI-MS). We therefore propose that the compound
has the structure [(C–CN)2Pd(μ-Cl)2Pd(CH3CN)2](BF4)2, where the chloro ligands bridge
between two palladium(II) centers and the two carbene ligands are coordinated to one metal
center.
Scheme 3.1. Synthesis of Pd(II) and Pt(II) Complexes (5, 6a, and 7) Starting from a Nitrile-
Functionalized N-Heterocyclic Carbene Complex of Ag(I) (2a).
AgBF4
2
2 Pt(cod)Cl2AgBF4
CH2Cl2:CH3CN(5:2), rt
N
NCH3
Pt
Cl
N
BF4
[Pd(η 1:η 2-coe-OMe)(µ -Cl)]2
2
Pd (BF4)2Cl
ClPd
NN
H3C
N
NCH3
NCH3N
NH3C
N
2 trans-PdCl2(CH3CN)2
AgBF4CH3CN, rt
5a
or 2 Pd(cod)Cl2
Pd
H3CO N
N
N
NCH3
CH3
H BF4
4+
PdPd
Pd
ON
N
NN
NN
NN
NNNN
CH3
CH3CH3H3CH3C
H3C
CH3H3C
trace H2OCH3CN/Et2O
5b
(BF4)4
7
CH2Cl2:CH3CN(3:2), rt:
6a
NN
NCH3
BF4Ag
NN
NCH3
2a
+
+
+
+ +

533.3.2 Hydrolysis of Nitrile-Functionalized N-Heterocyclic Carbene Ligands on
Palladium(II) Centers. In an attempt to obtain a crystal structure of 5a using wet diethyl ether
layered on top a saturated solution of the complex in acetonitrile, the C–CN ligands were
hydrolyzed, leading to formation of very small amounts of crystals, which in turn were
characterized by X-ray diffraction as the trimetallic complex 5b (Figure 3.2). Despite the
presence of disordered diethyl ether molecules in the lattice, this contribution to the electron
density was removed from the observed data during refinement and resulted in a significant
increase in the precision of the geometric parameters.7 Complex 5b contains a C–N–N–C donor
ligand providing two bridging imido nitrogens in the trimetallic complex [{Pd(CH3CN)2}3(C–N–
N–C)](BF4)4; the C–N–N–C ligand, which has a central –N=C–O–C=N– linkage, is formed from
the partial hydrolysis of the nitrile moiety to a primary amido group6 followed by a condensation
reaction with elimination of water. Each square-planar palladium(II) center has two cis
acetonitrile ligands. The coordination geometry around the central palladium(II) center
comprises of a six-membered ring with a –Pd–N=C–O–C=N– linkage and resembles that of a β-
diketiminato complex.12 To our knowledge, there are only few examples of organic compounds
with the general formula R–N=CR'–O–R'C=N–R reported in the literature (R = H, alkyl).13 The
outer palladium(II) cations are coordinated to the bridging imido nitrogen and the carbene carbon
of the N-heterocyclic carbene, thus forming a seven-membered ring (Figure 3.2). Bridging
groups with this type of nitrogen are rare. Shriver and co-workers have reported the structural
characterization of metal carbonyl cluster complexes with an acetamido ligand bridging between
three heterometal centers (Ru, Mn, Re) via nitrogen and coordination to ruthenium via its
oxygen.14 Mehrotra and Verkade have reported structures with trialkylstannates bridged by
acetamido ligands.15 More recently, Besenyei have reported the synthesis of some bridging
phenylacetamido complexes of palladium(II) from reactions of benzoyl azides with
[PdCl(dppm)]2 (dppm = 1,1-bis(diphenylphosphino)methane).16
12. Hadzovic, A.; Song, D., Organometallics 2008, 27, 1290-1298.13. (a) Gramstad, T.; Husebye, S.; Saebo, J., Tet. Lett. 1983, 24, 3919-3920; (b) Hitzler, M. G.; Lutz, M.; Shrestha-Dawadi, P. B.; Jochims, J. C., Liebigs Ann. 1996, 247-257.14. (a) Voss, E. J.; Sabat, M.; Shriver, D. F., Inorg. Chem. 1991, 30, 2705-2707; (b) Voss, E. J.; Sabat, M.; Shriver, D. F., Inorg. Chim. Acta 1995, 240, 49-61.15. (a) Sharma, K. K.; Mehrotra, S. K.; Mehrotra, R. C., J. Organomet. Chem. 1977, 142, 165-169; (b) Geetha, S.; Ye, M. C.; Verkade, J. G., Inorg. Chem. 1995, 34, 6158-6162.16. Besenyei, G.; Parkanyi, L.; Szalontai, G.; Holly, S.; Papai, I.; Keresztury, G.; Nagy, A., Dalton Trans. 2004, 2041-2050.

54
Figure 3.2. ORTEP diagram of 5b⋅1.5 CH3CN depicted with thermal ellipsoids at the 30%
probability level. The counteranions, hydrogens, and solvent molecules have been omitted for
clarity. Selected bond distances (Å) and bond angles (deg): Pd(1)-C(1), 1.969(6); Pd(3)-C(20),
1.957(6); Pd(1)-N(3), 2.015(5); Pd(3)-N(4), 2.018(5); Pd(2)-N(3), 1.976(5); Pd(2)-N(4),
1.972(5); Pd(1)-N(8), 2.094(6); Pd(1)-N(7), 2.020(5); C(10)-N(3), 1.272(8); C(11)-N(4),
1.265(8); C(10)-O(1), 1.361(7); C(11)-O(1), 1.382(7); N(3)-Pd(2)-N(4), 91.9(2); C(1)-Pd(1)-
N(3), 85.4(2); C(10)-O(1)-C(11), 126.7(5); C(10)-N(3)-Pd(2), 125.3(4).
The solid state structure of 5b shows square-planar geometries about the metal centers, and
the phenyl rings are twisted with respect to the imidazolylidene rings at dihedral angles 41.00
and 41.13° to facilitate chelation. The C(10)-N(3) and C(11)-N(4) bond distances (1.272(8) and
1.265(8) Å) reveal a double bond character between carbon and nitrogen atoms, forming an
imido linkage (cf. CPh–C(sp2)=N–Cavg = 1.279 Å; C(sp3)–Navg = 1.46 - 1.48 Å).17 On the other
hand, the C–O distances (1.361(7) and 1.382(7) Å) are shorter than a C(sp3)–O bond (C(sp3)–
Oavg = 1.42 - 1.45 Å for ethers) and slightly longer than the C(sp2)–O bonds of organic amides
(C(sp2)–Oavg = 1.23 Å).17 These all suggest delocalization of electrons through the π-orbitals of
the –N=C–O–C=N– bonds. In addition, the Pd(2)-N(3) and Pd(2)-N(4) bond distances (1.976(5)
and 1.972(5) Å) are significantly shorter than a Pd–RN=CR2 motif (Pd–Navg = 2.037Å),18
17. Allen, F. H.; Kennard, O.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R., J. Chem. Soc., Perkin Trans. 2 1987, S1-S19.18. Orpen, A. G.; Brammer, L.; Allen, F. H.; Kennard, O.; Watson, D. G.; Taylor, R., Dalton Trans. 1989, S1-S83.

55reflecting the presence of highly charged bridging imido nitrogen atoms. The Pd–Ccarbene bond
distances lie within the typical range for Pd(NHC) complexes.9a, 9d, 9e, 10, 19 The carbene ligand has
a stronger trans influence than the bridging-imido groups, as reflected by the Pd–Nacetonitrile bond
lengths (Pd(1)-N(8)trans to C = 2.094(6), Pd(1)-N(7)trans to N = 2.020(5) Å).
The mechanism in the formation of complex 5b from a solution of 5a is not well understood.
It is believed that the carbene ligands on the palladium(II) center of 5a serve as a base in the
deprotonation of water in the cooperative hydrolysis of the nitrile moieties. Cooperative
hydrolysis of nitrile was observed in cis-dialkylcyanamide complexes of platinum(IV)
(cis-PtCl4(NCNR2)2, R = CH3, C2H5, C5H10) to a diimino complex of platinum(IV)
(cis-PtCl4(NC–R2N–μ-O–NR2–CN)) under base-free conditions.20 We attempted to synthesize 5b
independently to obtain spectroscopic information, by stoichiometric reaction of 3 equiv of
trans-PdCl2(CH3CN)2, 2 equiv of 2a, 4 equiv of silver tetrafluoroborate and water in acetonitrile.
This gave a mixture of a palladium complex and free imidazolium salt (1a, see chapter 2). The 13C{1H} NMR spectrum of this mixture in acetonitrile-d3 revealed a peak at 141.4 ppm of the
carbene carbon (Pd–Ccarbene), and a peak at 169.1 ppm which is suggestive of an amido-carbon.
The nitrile carbons on the ligands were not observed. No primary or secondary amido groups
were observed in the 1H NMR spectrum. However, these spectroscopic data are insufficient to
identify the presence of 5b in this stoichiometric reaction.
3.3.3 Palladium(II) Complex Bearing Nitrile-Functionalized N-Heterocyclic Carbene
(C–CN) and Methoxycyclooctenyl Ligands. In order to obtain more well-defined palladium(II)
complexes with the C–CN ligand, the reactions of other palladium(II) precursors with the
silver(I) complex [Ag(C–CN)2]BF4 (2a) were explored. Attempts using PdCl2(en),21
[Pd(CH3CN)4](BF4)2,22 and [Pd(cod)(μ-Cl)]2(BF4)223 (en = ethylenediamine, cod = 1,5-
cyclooctadiene) as starting materials failed. The palladium(II) precursor [Pd(η1:η2-coe-OMe)(μ-
Cl)]2 (coe-OMe = 2-methoxycyclooct-5-enyl)24 can be conveniently prepared as the exo isomer
19. (a) Meyer, A.; Taige, M. A.; Strassner, T., J. Organomet. Chem. 2009, 694, 1861-1868; (b) Kantchev, E. A. B.; Ying, J. Y., Organometallics 2009, 28, 289-299; (c) Tang, Y. Q.; Lu, J. M.; Wang, X. R.; Shao, L. X., Tetrahedron 2010, 66, 7970-7974; (d) Warsink, S.; Drost, R. M.; Lutz, M.; Spek, A. L.; Elsevier, C. J., Organometallics 2010, 29, 3109-3116; (e) Hashmi, A. S. K.; Lothschutz, C.; Bohling, C.; Rominger, F., Organometallics 2011, 30, 2411-2417.19.20. Bokach, N. A.; Pakhomova, T. B.; Kukushkin, V. Y.; Haukka, M.; Pombeiro, A. J. L., Inorg. Chem. 2003, 42, 7560-7568.21. Hohmann, H.; Vaneldik, R., Inorg. Chim. Acta 1990, 174, 87-92.22. Thomas, R. R.; Sen, A.; Beck, W.; Leidl, R., Inorg. Synth. 1989, 26, 128-134.23. Eaborn, C.; Farrell, N.; Pidcock, A., Dalton Trans. 1976, 289-292.24. Bailey, C. T.; Lisensky, G. C., J. Chem. Educ. 1985, 62, 896-897.

56by the reaction of Pd(cod)Cl2 and sodium methoxide in methanol. When this was reacted with 1
equiv of 2a and AgBF4, a white complex 6a, [Pd(η1:η2-coe-OMe)(C–CN)(CH3CN)]BF4, was
isolated in 67% yield. The product exists as mixture of diastereomers, which includes rotamers A
and B and each of their enantiomers, with chiral centers at the C(1) and C(2) carbons (see Figure
3.3 for the numbering schemes). The rotamers are believed to form via restricted rotation about
the M–Ccarbene bond (M = Pd, Pt; vide infra) owing to the steric crowd of the neighboring 2-
methoxycyclooct-5-enyl ligand. Grubbs and co-workers showed that a ruthenium alkylidene
complex with a saturated N-heterocyclic carbene with o-tolyl groups on the nitrogens has a
restricted rotation around the Ru–Ccarbene bond. The o-tolyl groups have a smaller barrier to
rotation than the Ru–Ccarbene bond but do cause the formation of rotamers about the CPh–Nimid
bonds.25 A second explanation is that there are geometric isomers with the carbene ligand trans to
the chelating olefin and the carbene ligand trans to the σ-alkyl group. These, however, are
unlikely, as the 13C{1H} NMR resonances of these carbons fortuitously have the same chemical
shifts (with a difference less than 10 Hz), whereas a chemical shift difference of up to 150 Hz
was observed if these geometric isomers do exist.26
Figure 3.3. Diastereomers and rotamers (A and B) of Pd(II) (6a) and Pt(II) (9a) complexes
bearing 2-methoxycyclooct-5-enyl and the C–CN ligands.
25. Stewart, I. C.; Benitez, D.; O'Leary, D. J.; Tkatchouk, E.; Day, M. W.; Goddard, W. A.; Grubbs, R. H., J. Am. Chem. Soc. 2009, 131, 1931-1938.26. Cooper, D. G.; Powell, J., Inorg. Chem. 1977, 16, 142-148.
18BF4
6a: M = Pd9a: M = Pt
A-(1S,2S) A-(1R,2R)
B-(1S,2S) B-(1R,2R)
M
H3CON
N
N
NCH3
CH3
6
54
3
2H
7
BF4
NN N
H3C
NN
N
CH3
BF4M
OCH3NN
N
NCH3
CH3
H
18
M
H3CO
N
CH3
6
54
3
2H7
BF4M
OCH3
N
CH3
H
+ +
+ +

57
Figure 3.4. ORTEP diagram of 6a depicted with thermal ellipsoids at the 30% probability level.
The counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and
bond angles (deg): Pd(1)-C(1), 2.012(4); Pd(1)-C(19), 2.040(4); Pd(1)-N(4), 2.164(3); Pd(1)-
coe(trans to C)cent, 2.161; C(11)-N(3), 1.148(6); C(18)-O(1), 1.441(5); C(1)-Pd(1)-coe(trans to
C)avg, 161.2; C(19)-Pd(1)-N(4), 177.2(9); C(1)-Pd(1)-C(19), 87.7(1).
Slow diffusion of diethyl ether into a saturated solution of 6a afforded white needles which
were then characterized by X-ray diffraction (Figure 3.4). The square-planar complex
crystallizes in the chiral triclinic space group P-1 with the exo-S,S rotamer A being observed
(Figures 3.3 and 3.4). The carbene ligand is oriented trans to the cyclooctene ligand. The Pd–C
bond distances of the carbene9c-e, 19d, 27 (Pd–Ccarbene) and methoxycyclooctenyl (Pd–C(1))28 ligands
are within expected ranges. On the other hand, the acetonitrile ligand is weakly coordinated,
owing to the strong trans influence of the σ-alkyl group (2.164(3) Å; cf. 2.06 Å in
PdCl2(CH3CN)(μ-dpmp)PdCl229 and 2.01 - 2.09 Å in 5b; dpmp = bis((diphenylphosphino)-
methyl)phenylphosphine). The dihedral angle between the 2-cyanophenyl and the
27. Li, D. C.; Liu, D. J., J. Chem. Crystallogr. 2003, 33, 989-991.28. (a) Hoel, G. R.; Stockland, R. A.; Anderson, G. K.; Ladipo, F. T.; Braddock-Wilking, J.; Rath, N. P.; Mareque-Rivas, J. C., Organometallics 1998, 17, 1155-1165; (b) Macchioni, A.; Bellachioma, G.; Cardaci, G.; Travaglia, M.; Zuccaccia, C.; Milani, B.; Corso, G.; Zangrando, E.; Mestroni, G.; Carfagna, C.; Formica, M., Organometallics 1999, 18, 3061-3069; (c) Binotti, B.; Bellachioma, G.; Cardaci, G.; Macchioni, A.; Zuccaccia, C.; Foresti, E.; Sabatino, P., Organometallics 2002, 21, 346-354; (d) Binotti, B.; Bellachioma, G.; Cardaci, G.; Carfagna, C.; Zuccaccia, C.; Macchioni, A., Chem. Eur. J. 2007, 13, 1570-1582.29. Olmstead, M. M.; Guimerans, R. R.; Farr, J. P.; Balch, A. L., Inorg. Chim. Acta 1983, 75, 199-208.

58imidazolylidene rings of the carbene ligand is 59.38°, large enough to relieve steric repulsion
between the coordinated ligands. The infrared spectrum of 6a displays two nitrile stretches at
2277 cm-1 for the carbene ligand and 2229 cm-1 for the coordinated acetonitrile.
In nitromethane-d3 solution, the carbene atom (Pd–Ccarbene) of the NHC ligand was observed
as a singlet at 174.2 ppm in the 13C{1H} NMR spectrum. All of the proton and carbon resonances
of the methoxycyclooctenyl ligand were assigned by 1H-1H COSY, 1H-13C HSQC, and 1H-13C
HMBC experiments based on the distinctive resonance28 of the C(2) carbon in the 13C{1H} NMR
spectrum. The two enantiomers of a single rotamer are not distinguishable by one-dimensional 1H NMR spectroscopy. However, the rotamers A and B of 6a in nitromethane-d3 solutions are
distinguishable in the 13C{1H} NMR spectrum, where the resonance of each carbon appears in a
1:4 ratio, and in the 1H NMR spectrum, where there are two resonances of the H(1) proton of the
methoxycyclooctenyl ligand. The other peaks of the methoxycyclooctenyl ligand were
overlapping with each other so that the exact ratio could not be determined. As no single crystal
was isolated as the rotamer B, it is therefore expected, on the basis of the NMR data, that there
exists approximately 20 mol% of the isolated product containing the rotamer B. The 1H and 13C{1H} NMR spectra of the platinum(II) analogues also provide evidence for the existence of
these rotamer (vide infra).
Scheme 3.2. Interconversion of 6a to 6b and 9a to 9b in Different Recrystallization Solvents.
When 6a was recrystallized in dichloromethane and diethyl ether mixtures, the acetonitrile
ligand was lost and the corresponding dimer 6b was isolated (Scheme 3.2). The rhodium(I)
complex 3 bearing the same bridging C–CN ligand was isolated as a dimer (See Chapter 2 and
Figure 3.1).6 The identity of 6b was established for its nitromethane-d3 solution by the absence of
the resonance at 2.01 ppm for coordinated acetonitrile ligand in the 1H NMR spectrum. The
nitrile stretch of the acetonitrile ligand was absent in the infrared spectrum. There is a slight
NNH3C
N(BF4)2
M
M
H3CO
OCH3
6b:M = Pd9b:M = Pt
BF4M
H3CO N
N
N
NCH3
CH3
6a: M = Pd9a: M = Pt
H
H
H
NN CH3
N
CH3CN/Et2O
CH2Cl2/Et2O
+
++

59decrease in the stretching frequency of the nitrile moiety of the carbene ligand on going from
complex 6a (2277 cm-1) to 6b (2260 cm-1), signaling the effect of coordination to palladium(II)
center. Complex 6a can be crystallized from a solution of 6b in acetonitrile and diethyl ether,
suggesting the lability of the nitrile groups of both the acetonitrile and carbene ligand. Of note,
the 2-cyanophenyl group of the carbene ligand has to rotate about the C–N bond by 180° to
allow this interconversion of the structures.
It should be noted that solutions of 6a and 6b are extremely sensitive to light and are prone to
β-hydride elimination to release cyclooctadienyl methyl ethers and then reductive elimination of
the palladium hydride to release the imidazolium salt with the formation of black palladium(0)
metal. Reductive elimination of N-heterocyclic carbene ligands can occur when they are not
tethered to the metal by a second donor group from the carbene ligand.1c The organic products
were identified by 1H NMR.30 In addition, complex 6a is also light sensitive in its solid state to
form palladium metal. Pure 6a can be recovered by filtration of the palladium metal from a
solution of the compound exposed to light and then recrystallization in acetonitrile and diethyl
ether solution. Complex 6b in its solid state is less sensitive to light and can be stored without
further decomposition.
In fact, attempts at catalysis using complex 6b (2 mol% of Pd) for the Buchwald-Hartwig
amination reactions31 was not successful, starting from 4-chlorotoluene or 4-bromotoluene,
morpholine and base (K2CO3, Cs2CO3, NaOtBu, KOtBu) in various temperatures (up to 100°C)
and different solvents (dimethoxyethane, dimethylacetamide and toluene). The decomposition of
the complex and the formation of palladium(0) metal were observed in all cases.
3.3.4 Nitrile-Functionalized N-Heterocyclic Carbene Complex of Platinum(II). Initial
attempts using both isomers of PtCl2(CH3CN)232 as starting materials in the transmetalation
reaction with [Ag(C–CN)2]BF4 (2a) and AgBF4 failed. Only starting materials were recovered,
even if the reactions were carried out at elevated temperatures. On the other hand, the reaction of
2a with 2 equiv of Pt(cod)Cl2 and 1 equiv of AgBF4 in acetonitrile and dichloromethane
mixtures afforded complex 7, [Pt(C–CN)(cod)Cl]BF4, as a white solid in 59% yield. The slightly
distorted square-planar complex (Figure 3.5) crystallizes in the monoclinic space group P21/n
30. Anderson, C. B.; Burreson, B. J.; Michalowski, J. T., J. Org. Chem. 1976, 41, 1990-1994.31. (a) Hartwig, J. F., Angew. Chem.-Int. Edit. 1998, 37, 2047-2067; (b) Hartwig, J. F., Acc. Chem. Res. 1998, 31, 852-860; (c) Surry, D. S.; Buchwald, S. L., Chem. Sci. 2011, 2, 27-50.32. Fraccarollo, D.; Bertani, R.; Mozzon, M.; Belluco, U.; Michelin, R. A., Inorg. Chim. Acta 1992, 201, 15-22.

60with four units residing in the unit cell. The cyclooctadiene ligand is coordinated to the metal
center with bond distances from the centroid of each olefin being 2.152 and 2.048 Å. The longer
distance (2.152 Å) corresponds to that for the olefin trans to the carbene ligand, and the shorter
C=C bond distance of this olefin compared to that trans to the chloro is consistent with a higher
trans influence of the carbene ligand (C(16)-C(17)trans to C, 1.372(8) Å; C(12)-C(13)trans to Cl,
1.408(7) Å). The carbene ligand that is bonded to the metal center is oriented with a dihedral
angle between the phenyl and imidazolylidene ring of 51.70°, and the nitrile moiety is directed
away from the metal center. The Pt–Ccarbene bond distance is in the expected range for
platinum(II) compounds bearing NHC ligands.33 A similar cationic platinum(II) complex with a
diolefin and an N-heterocyclic carbene ligand with a cyclometalated phenyl ring of the carbene
ligand, [Pt(dpim)(cod)]ClO4 (dpim = 1,3-diphenyl-2-imidazolidinylidenato-2-C,2'-C), was
reported by Hiraki and co-workers.34
The 13C{1H} NMR spectrum in acetonitrile-d3 solution of 7 shows a singlet for the carbene
carbon (Pt–Ccarbene) at 151.4 ppm; this is shifted upfield compared to the case for most
Pt(NHC)2X2 systems.9b, 19a, 33a, 33c-e, 34-35 Platinum satellites were observed at the four olefinic
carbons bonded to the metal center (1JPt-C = 31.54, 31.92, 75.12, and 80.20 Hz) and for two
olefinic protons (2JPt-H = 30.72 and 32.61 Hz). The resonances with smaller 1JPt-C coupling
constants are assigned to the olefinic C–H trans to the carbene ligand. Similar observations were
reported and discussed by Clark and Manzer,36 Appleton,37 Anderson38 and Klein.39 Attempts to
remove the cyclooctadiene and the chloro ligands by reduction of the olefin and reaction with
silver(I) salts, respectively, were not successful.
33. (a) Bacciu, D.; Cavell, K. J.; Fallis, I. A.; Ooi, L. L., Angew. Chem. Int. Ed. 2005, 44, 5282-5284; (b) Baker, M. V.; Brown, D. H.; Simpson, P. V.; Skelton, B. W.; White, A. H.; Williams, C. C., J. Organomet. Chem. 2006, 691, 5845-5855; (c) Brissy, D.; Skander, M.; Retailleau, P.; Marinetti, A., Organometallics 2007, 26, 5782-5785; (d) Fantasia, S.; Petersen, J. L.; Jacobsen, H.; Cavallo, L.; Nolan, S. P., Organometallics 2007, 26, 5880-5889; (e) Zhang, Y. Z.; Garg, J. A.; Michelin, C.; Fox, T.; Blacque, O.; Venkatesan, K., Inorg. Chem. 2011, 50, 1220-1228.34. Hiraki, K.; Onishi, M.; Ohnuma, K.; Sugino, K., J. Organomet. Chem. 1981, 216, 413-419.35. (a) Riederer, S. K. U.; Bechlars, B.; Herrmann, W. A.; Kuhn, F. E., Eur. J. Inorg. Chem. 2011, 249-254; (b) Taige, M. A.; Ahrens, S.; Strassner, T., J. Organomet. Chem. 2011, 696, 2918-2927.36. Clark, H. C.; Manzer, L. E., J. Organomet. Chem. 1973, 59, 411-428.37. Appleton, T. G.; Clark, H. C.; Manzer, L. E., Coord. Chem. Rev. 1973, 10, 335-422.38. Anderson, G. K.; Clark, H. C.; Davies, J. A., Inorg. Chem. 1981, 20, 1636-1639.39. Klein, A.; Klinkhammer, K. W.; Scheiring, T., J. Organomet. Chem. 1999, 592, 128-135.
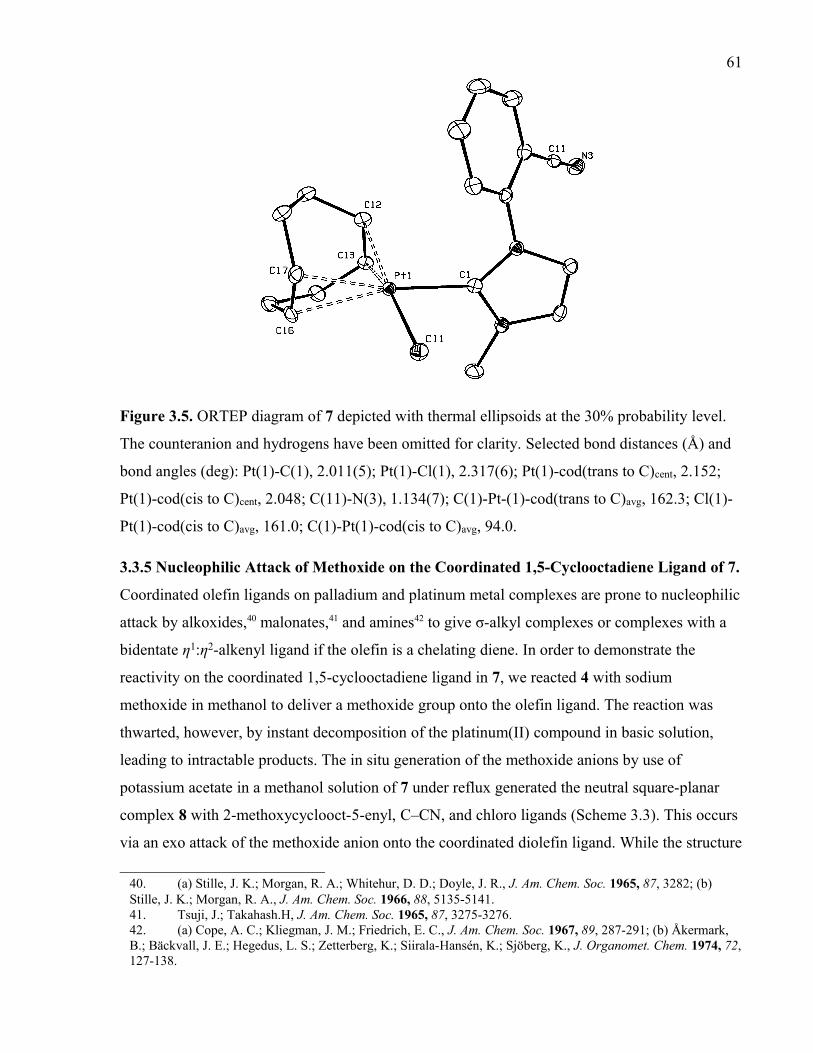
61
Figure 3.5. ORTEP diagram of 7 depicted with thermal ellipsoids at the 30% probability level.
The counteranion and hydrogens have been omitted for clarity. Selected bond distances (Å) and
bond angles (deg): Pt(1)-C(1), 2.011(5); Pt(1)-Cl(1), 2.317(6); Pt(1)-cod(trans to C)cent, 2.152;
Pt(1)-cod(cis to C)cent, 2.048; C(11)-N(3), 1.134(7); C(1)-Pt-(1)-cod(trans to C)avg, 162.3; Cl(1)-
Pt(1)-cod(cis to C)avg, 161.0; C(1)-Pt(1)-cod(cis to C)avg, 94.0.
3.3.5 Nucleophilic Attack of Methoxide on the Coordinated 1,5-Cyclooctadiene Ligand of 7.
Coordinated olefin ligands on palladium and platinum metal complexes are prone to nucleophilic
attack by alkoxides,40 malonates,41 and amines42 to give σ-alkyl complexes or complexes with a
bidentate η1:η2-alkenyl ligand if the olefin is a chelating diene. In order to demonstrate the
reactivity on the coordinated 1,5-cyclooctadiene ligand in 7, we reacted 4 with sodium
methoxide in methanol to deliver a methoxide group onto the olefin ligand. The reaction was
thwarted, however, by instant decomposition of the platinum(II) compound in basic solution,
leading to intractable products. The in situ generation of the methoxide anions by use of
potassium acetate in a methanol solution of 7 under reflux generated the neutral square-planar
complex 8 with 2-methoxycyclooct-5-enyl, C–CN, and chloro ligands (Scheme 3.3). This occurs
via an exo attack of the methoxide anion onto the coordinated diolefin ligand. While the structure
40. (a) Stille, J. K.; Morgan, R. A.; Whitehur, D. D.; Doyle, J. R., J. Am. Chem. Soc. 1965, 87, 3282; (b) Stille, J. K.; Morgan, R. A., J. Am. Chem. Soc. 1966, 88, 5135-5141.41. Tsuji, J.; Takahash.H, J. Am. Chem. Soc. 1965, 87, 3275-3276.42. (a) Cope, A. C.; Kliegman, J. M.; Friedrich, E. C., J. Am. Chem. Soc. 1967, 89, 287-291; (b) Åkermark, B.; Bäckvall, J. E.; Hegedus, L. S.; Zetterberg, K.; Siirala-Hansén, K.; Sjöberg, K., J. Organomet. Chem. 1974, 72, 127-138.

62of the isolated crystals of 8 was characterized as rotamer A, solutions of 8 reveal the presence of
rotamers A and B in equal amounts, as evidenced by NMR spectroscopy (see below). The
retention of the chloro ligand and the formation of both rotamers A and B are unusual
observations for such reactions of palladium(II) and platinum(II) diolefin complexes.28a, 40b
Scheme 3.3. Nucleophilic Attack of Methoxide at the Coordinated Cyclooctadiene Ligand of 7
to 8 and Chloride Abstraction of 8 with AgBF4 in CH3CN to 9a.
The square-planar complex 8 as the exo-R,R rotamer A crystallizes in the chiral triclinic space
group P1 (Figure 3.6, left). The relative orientation of carbene and methoxycyclooctenyl ligands
is similar to that of its palladium analogue (6a). The dihedral angle between the phenyl and the
imidazolylidene rings of the carbene ligand is 58.77°, to relieve sterics between the coordinated
ligands. The Pt–C bond distances of the carbene9b, 33d, 43 (Pt–Ccarbene) and methoxycyclooctenyl28c,
44 (Pt–C(1)) ligands are within expected ranges, while the Pt–Ccarbene bond distance (1.970(1)Å) is
shorter than that of 7 (2.011(5) Å).
43. Ahrens, S.; Herdtweck, E.; Goutal, S.; Strassner, T., Eur. J. Inorg. Chem. 2006, 2006, 1268-1274.44. (a) Goel, A. B.; Goel, S.; Vanderveer, D. G., Inorg. Chim. Acta 1981, 54, L169-L170; (b) Aucott, S. M.; Slawin, A. M. Z.; Woollins, J. D., Dalton Trans. 2000, 2559-2575; (c) Angurell, I.; Martínez-Ruiz, I.; Rossell, O.; Seco, M.; Gómez-Sal, P.; Martín, A.; Font-Bardia, M.; Solans, X., J. Organomet. Chem. 2007, 692, 3882-3891.
NNCH3
Pt
Cl
N
BF4KOAc
CH3CN, rtAgBF4
7 8 9a
PtCl
OCH3NN
N
H3CH
BF4PtN
OCH3NN
N
H3C
H3C
H+ +∆ , CH3OH

63
Figure 3.6. ORTEP diagrams of 8 (left) and 9a (right) depicted with thermal ellipsoids at the
30% probability level. The counteranion and hydrogens have been omitted for clarity. Selected
bond distances (Å) and bond angles (deg): 8 (left): Pt(1)-C(1), 1.969(1); Pt(1)-C(12), 2.095(1);
Pt(1)-Cl(1), 2.430(4); Pt(1)-coe(trans to C)cent, 2.102; C(11)-N(3), 1.135(7); C(19)-O(1),
1.418(2); C-(1)-Pt(1)-coe(trans to C)avg, 160.7; C(12)-Pt(1)-Cl(1), 178.4(4); C(1)-Pt(1)-C(12),
88.9(5). 9a (right): Pt(1)-C(1), 2.001(5); Pt(1)-C(17), 2.044(5); Pt(1)-N(4), 2.122(5); Pt(1)-
coe(trans to C)cent, 2.100; C(10)-N(3), 1.145(8); C(16)-O(1), 1.449(6); C(1)-Pt(1)-coe(trans to
C)avg, 161.0; C(17)-Pt(1)-N(4), 178.3(0); C-(1)-Pt(1)-C(17), 88.9(7).
Complex 8 is soluble in polar solvents such as methanol, acetonitrile, dichloromethane, and
tetrahydrofuran, as well as in aromatic solvents such as benzene and toluene. The absence of the
tetrafluoroborate counteranion is established by the lack of requisite signals in the 19F NMR and
infrared spectra.
A dichloromethane-d2 solution of 8 at 298 K shows a significant downfield shift of the
carbene resonance (Pt–Ccarbene) to 175.8 ppm in the 13C{1H} NMR spectrum compared to the
signal in 7. In addition, the 1H NMR spectrum shows a 1:1 ratio of the methyl resonances for the
carbene (4.05 and 4.02 ppm) and methoxycyclooctenyl ligands (3.05 and 2.95 ppm). In addition,
the H(3) proton of the phenyl ring of the carbene ligand appears as a pair of doublets (3JHH = 7.86
Hz) at 8.66 and 8.39 ppm. Therefore, the complex exists as a 1:1 mixture of rotamers A and B
because of the restricted rotation about the Pt–Ccarbene bond similar to complex 6a. The barrier to

64rotation must be high, because heating a solution of 8 in nitromethane-d3 at 323 K afforded no
change in the 1H NMR spectrum in comparison to that acquired at 298 K. A close examination of
the 13C{1H} NMR spectrum of 8 showed a doubling of all of the resonances in a 1:1 ratio.
Rotamer B is more abundant than that of the palladium(II) complex 6a. Attempts to establish by 1H-1H NOESY NMR spectroscopy the proximity of groups of each rotamer of 8 was not
successful, as no cross-peaks of the relevant protons were observed.
In order to synthesize the platinum(II) analogues of complexes 6a and 6b, we first attempted
to react the silver(I) complex 2a with 1 equiv of [Pt(η1:η2-coe-OMe)(μ-Cl)]2 and AgBF4 in
acetonitrile solutions. These, however, led to intractable products along with decomposition. On
the other hand, adding 1 equiv of AgBF4 to an acetonitrile-d3 solution of 8 afforded the
immediate precipitation of silver chloride. Subsequent filtration gave the white acetonitrile
complex 9a (Scheme 3.3), [Pt(η1:η2-coe-OMe)(C–CN)(CH3CN)]BF4. The carbene carbon
(Pt–Ccarbene) of the NHC ligand was observed as two singlets at 168.9 and 168.7 ppm in a 1:4
ratio in its 13C{1H}NMR spectrum in dichloromethane-d2. All the proton and carbon resonances
of the methoxycyclooctenyl ligand were assigned similarly as with complex 6a on the basis of
the resonance of the C(2) carbon.28c, 43b, 43c In general, the resonances of the platinum(II)
compound appeared at lower frequencies compared to those of its palladium(II) analogue,
consistent with the observation reported by Macchioni and co-workers.28c Again, the presence of
both rotamers A and B in solution are detectable by NMR techniques. In accordance with the
previous assignments made to complexes 6a and 8, we believe there exists at least 20 mol% of
rotamer B in the solution sample of 9a. This is much smaller than the amounts of rotamer B in a
solution for a sample of 8.
Evaporation of the deuterated solvent and slow diffusion of diethyl ether into a solution of 9a
in acetonitrile provided crystals suitable for X-ray diffraction (Figure 3.6, right). The square-
planar complex crystallizes as the exo-R,R rotamer A. The phenyl ring is twisted at a dihedral
angle of 61.36° to the plane of the imidazolylidene ring of the carbene ligand. The acetonitrile
ligand is weakly coordinated (Pt(1)-N(4), 2.122(5)Å) owing to the strong trans influence of the
σ-alkyl group (cf. the average Pt–N distances of other acetonitrile complexes, 2.003Å18). A
comparison of the M–C and M–N distances (M = Pd, Pt) of 6a and 9a reveal similarities in their
magnitudes.

65When 9a was resynthesized in a larger scale and recrystallized in dichloromethane and diethyl
ether mixtures, the corresponding dimer 9b was obtained, analogous to complex 6b (Scheme
3.2). The loss of the acetonitrile ligand was identified by the absence of the resonance at 1.94
ppm in the 1H NMR spectrum in dichloromethane-d2 and the nitrile stretch of acetonitrile in the
infrared spectrum (2227 cm-1). As for the palladium(II) complexes 6a and 6b, there is a decrease
in the nitrile stretching frequency on going from 9a to 9b (2282 to 2262 cm-1). The acetonitrile
complex 9a and the dimeric complex 9b, like 6a and 6b, are interconvertible by using different
recrystallization solvents (Scheme 3.2). Unlike the palladium(II) complexes, the platinum(II)
complexes are stable enough for storage without any precautions taken.
3.3.6 Proposed Reaction Pathways Leading to the Formation of Rotamers A and B of
Complexes 6, 8 and 9. In order to account for the presence of both rotamers A and B in
complexes 6, 8, and 9, and the difference in the distributions of rotamers of complexes 8 and 9, it
is proposed that the carbene ligand can freely rotate in a pentacoordinate complex produced
before the solvent molecule or a chloride ligand that occupies the fifth coordination site of the
square pyramidal complex dissociates. The transmetalation reaction of complex 2a with
[Pd(η1:η2-coe-OMe)(μ-Cl)]2 first forms the transient complex [Pd(η1:η2-coe-OMe)(C–CN)Cl].
The solvent/nitrile ligand then occupies the fifth coordination site of the square-planar complex,
forming a pentacoordinate, possibly a square-pyramidal complex with the carbene ligand
occupying the axial position. The carbene ligand of this intermediate might freely rotate, as the
steric congestion brought about by the 2-methoxycyclooct-5-enyl ligand is relieved. Silver
chloride precipitates from acetonitrile/dichloromethane solutions, forming the square planar
complex 6a. The presence of 80 mol% of rotamer A in solution and the fact that only the single
rotamer A was found in the crystal suggest that this is the thermodynamically preferred product
(Scheme 3.4).
On the other hand, an exo attack by methoxide onto the diolefin of complex 7 occurs at the
double bond that is trans to the chloride ligand. This displaces the chloride ligand and forms a
similar transient complex as the [Pt(η1:η2-coe-OMe)(C–CN)(CH3OH)]+ cation, or it dimerizes
with a bridging group such as the carbene ligand. In this case the coordination of solvated
chloride gives the same pentacoordinate intermediate. Upon dissociation of the coordinated
methanol, which is a poor ligand compared to chloride, this gave the same amounts of rotamers
A and B of 8 in solution. In fact, removal of the chloride ligand from 8 by precipitation using

66silver tetrafluoroborate in acetonitrile gives the same distribution of rotamers (favoring A over
B) of complex 9a as for complex 6a (Scheme 3.4).
Scheme 3.4. Proposed Reaction Pathways Leading to the Formation of Rotamers A and B of
Complexes 6, 8, and 9.
3.3.7 Platinum(II) Complex with Coordinated 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-
ylidene ligand (IMes). For comparison, the analogous coordination chemistry of platinum(II)
was investigated with 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes). The silver(I)
precursor complex [Ag(IMes)2]BF4 (10) was prepared by an established method6 and was reacted
with 2.5 equiv of Pt(cod)Cl2 and 1 equiv of AgBF4 under reflux overnight (Scheme 3.5). The
corresponding complex 11, with coordinated chloro, 1,5-cyclooctadiene, and IMes ligands, was
isolated in 85% yield. Reactions carried out at room temperature did not proceed and recovered
exclusively the starting materials. The carbene carbon (Pt–Ccarbene) was observed as a singlet at
151.4 ppm in the 13C{1H} NMR spectrum in acetonitrile-d3 solution, consistent with that of
complex 7 with a carbene bearing an electron-withdrawing group. Platinum satellites were also
observed at the four olefinic protons and carbons bonded to the metal center (2JPt-H = 22.03 and
31.55 Hz; 1JPt-C = 33.11 and 76.88 Hz), whereas the protons and carbons with smaller coupling
constants are bonded trans to the carbene ligand.36-39 A small coupling of the imidazolylidene
carbon with platinum satellites was also observed (3JPt-C = 16.14 Hz).
M
N
N
N
ClCH3
BF4 CH3OH- KCl- HOAc
+ KCl- CH3OH
M
OCH3NN
N
Cl
CH3
HM
OCH3N
N
N
ClCH3
H
M
H3CON
N
N
CH3H
Cl
M
OCH3NN
N
N
CH3
HM
OCH3N
N
N
NCH3
H
H3C H3CBF4
8: M = PtRotamer A, 50 mol%
8: M = PtRotamer B, 50 mol%
6a: M = Pd; 9a: M = PtRotamer A, Major
6a: M = Pd; 9a: M = PtRotamer B, Minor
OCH3
2
KOAc
AgBF4- AgClCH3CN
BF4M
H3CO
N N
N
CH3
HCl
L
L = CH3OH/CH3CN
CH3OH
+ L AgBF4
-AgClCH3CN
" "
CH2Cl2:CH3CN
+
+ +
BF4
+
+

67Scheme 3.5. Synthesis of Pt(II) Complex 11 from an N-Heterocyclic Carbene Complex of Ag(I)
(10).
When complex 11 was subjected to similar reaction conditions for the synthesis of 8, namely
nucleophilic attack of the coordinated diolefin by methoxide anion, only a trace amount of the
desired product was observed along with the generation of imidazolium salt, 1,3-bis(2,4,6-
trimethylphenyl)imidazolium. On the other hand, reaction of the silver(I) complex 10 with 1
equiv of [Pt(η1:η2-coe-OMe)(μ-Cl)]2 and AgBF4 in refluxing acetonitrile overnight afforded
complex 11, along with trace amounts of the desired product ([Pt(η1:η2-coe-OMe)(IMes)
(CH3CN)]BF4), imidazolium salt, and other decomposition products (Scheme 3.5). All these
results can be attributed to the steric bulk of the IMes ligand directed toward the metal center and
to the methoxycyclooctenyl ligand, therefore destabilizing the metal complex and inducing β-
elimination of H(1) and methoxide of the olefin ligand, generating imidazolium salts and
complex 11, respectively (Scheme 3.5). Attempts to prepare palladium(II) analogues of complex
6 with IMes from the reaction of 10, 1 equiv of [Pd(η1:η2-coe-OMe)(μ-Cl)]2, and AgBF4 was
thwarted by instant decomposition under light and in solutions, in particular, chlorinated
solvents.
Slow diffusion of diethyl ether solutions into the aforementioned reaction mixture in
dichloromethane afforded crystals of complex 11, which were thereby characterized by X-ray
diffraction (Figure 3.7). The slightly distorted square-planar complex crystallizes in the
monoclinic space group Cc with four pairs of asymmetric units residing in the unit cell. The
AgBF4
NN
BF4Ag
NN
[Pt(η 1:η 2-coe-OMe)(µ -Cl)]2
2.5 Pt(cod)Cl2AgBF4
NN
Cl
BF4Pt∆ , CH2Cl2:CH3CN (1:1)
∆ , CH3CN
10
11
NN
Cl
BF4Pt
11Major Minor Minor
NN
BF4BF4
PtN
NN
H3C
OCH3
H
+ +
+++ + +

68carbene ligand that is bonded to the metal center is oriented with the dihedral angles between the
mesityl and imidazolylidene rings of 80.78 and 97.40°. The Pt–Ccarbene bond distance is in the
expected range for most platinum(II) compounds bearing IMes ligands,33a, 33d but is slightly longer
than that of 7 because of its bulkiness. Overall, the metal-carbon bonds of 11 are slightly longer
compared to those of 7.
Figure 3.7. ORTEP diagram of 11⋅0.5 CH2Cl2 depicted with thermal ellipsoids at the 30%
probability level. The counteranion, hydrogens, and solvent molecules have been omitted for
clarity. Only one asymmetric unit is shown. Selected bond distances (Å) and bond angles (deg):
Pt(1A)-C(1A), 2.037(1); Pt(1A)-Cl(1A), 2.315(3); Pt(1A)-cod(trans to C)cent, 2.173; Pt(1A)-
cod(cis to C)cent, 2.084; C(1A)-Pt(1A)-cod(trans to C)avg, 162.3; Cl(1)-Pt(1)-cod(cis to C)avg,
160.9; C(1)-Pt(1)-cod(cis to C)avg, 95.1.
3.4 Conclusion
In summary, we have synthesized and fully characterized new palladium(II) and platinum(II)
complexes bearing chelating olefin and N-heterocyclic carbene ligands. Initial attempts using
silver(I) carbene complex 2a and trans-PdCl2(CH3CN)2 led to the dimeric complex 5a, along
with the partially hydrolyzed trimetallic complex 5b as a side product when wet diethyl ether and
acetonitrile were used as recrystallization solvents. The structure of 5b reveals the presence of a

69novel C–N–N–C donor ligand, comprised of a central –N=C–O–C=N– linkage and two bridging
imido nitrogens.
Platinum(II) complexes with 1,5-cyclooctadiene ligands and N-heterocyclic carbene ligands
(7 and 11) were prepared by transmetalation of the corresponding silver(I) carbene complexes
(2a and 10). The reaction of methoxide with the coordinated diolefin ligand of 7 afforded the
neutral complex 8 and complex 9 upon removal of the chloro ligand. The palladium(II) analogue,
complex 6, was independently prepared by direct transmetalation of 2a and [Pd(η1:η2-coe-OMe
(μ-Cl)]2. Both the monomer and dimer of complexes 6 and 9 were isolated and studied by NMR
and infrared spectroscopy. In addition, reactions leading to complexes 6, 8, and 9 produced
different amounts of rotamers A and B (Figure 3.3), owing to the orientation of C–CN ligand
relative to the 2-methoxycyclooct-5-enyl ligand, and restricted rotation about the M–CCarbene
bond originated from the steric crowding of the 2-methoxycyclooct-5-enyl ligand by the C–CN
ligand. These are the first examples of structurally characterized N-heterocyclic carbene
complexes of palladium(II) and platinum(II) bearing 1,5-cyclooctadiene and 2-methoxy-
cyclooct-5-enyl ligands.
3.5 Experimental Section
3.5.1 General Considerations. All of the preparations and manipulations, except where
otherwise stated, were carried out under an argon or nitrogen atmosphere using standard
Schlenk-line and glovebox techniques. Dry and oxygen-free solvents were always used.
Deuterated solvents were purchased from Cambridge Isotope Laboratories and Sigma Aldrich
and degassed and dried over activated molecular sieves prior to use. NMR spectra were recorded
on a Varian 400 spectrometer operating at 400 MHz for 1H, 100 MHz for 13C, and 376 MHz for 19F. The 1H and 13C{1H} NMR spectra were measured relative to partially deuterated solvent
peaks but are reported relative to tetramethylsilane (TMS). All 19F chemical shifts were
measured relative to trichlorofluoromethane as an external reference. The 1H-1H NOESY NMR
spectrum of complex 8 was recorded on a Varian 600 spectrometer operating at 600MHz for 1H.
All infrared spectra were recorded on a Nicolet 550 Magna-IR spectrometer. The elemental
analysis was performed at the Department of Chemistry, University of Toronto, on a Perkin-
Elmer 2400 CHN elemental analyzer. Samples were handled under argon where it was
appropriate. Single-crystal X-ray diffraction data were collected using a Nonius Kappa-CCD

70diffractometer with Mo Kα radiation (λ = 0.71073 Å). The CCD data were integrated and scaled
using the Denzo-SMN package. The structures were solved and refined using SHELXTL V6.1.
Refinement was by full-matrix least squares on F2 using all data.
The synthesis of the silver(I) complex, [Ag(C–CN)2]BF4 (2a) was described in Chapter 2.6
The complex [Ag(IMes)2]BF4 (10) was prepared using procedures analogous to those for 2a
from (IMesH)BF4, which was prepared by counteranion metathesis of (IMesH)Cl45 and NH4BF4
in water. The characterization data are similar to those reported in the literature.46 The syntheses
of trans-PdCl2(CH3CN)2,11a Pd(cod)Cl2, Pt(cod)Cl247 and [Pd(η1:η2-coe-OMe)(μ-Cl)]2
24 were
reported in the literature. All other reagents were purchased from commercial sources and were
used as received.
3.5.2 Synthesis of Bis(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)palladium(II)(μ-
dichloro)bis(acetonitrile)palladium(II) Tetrafluoroborate ([(C–CN)2Pd(μ-Cl)2Pd(CH3CN)2]
(BF4)2, 5a). A solution of 2a (39 mg, 0.07 mmol) and silver tetrafluoroborate (14 mg, 0.07
mmol) in acetonitrile (2 mL) was added to a solution of trans-PdCl2(CH3CN)2 (36 mg, 0.14
mmol) in acetonitrile (4 mL). A pale brown precipitate was formed instantaneously. The reaction
mixture continued to be stirred for 2 h. It was then filtered through a pad of Celite to give a
yellow solution. The volume of solvent was reduced (ca. 1 mL), and diethyl ether (8 mL) was
added. The solution was kept at -10°C to give a yellow oil. The supernatant liquid was decanted,
and the oil was triturated with diethyl ether (3 × 3 mL) to give a yellow powder, which was
collected and dried in vacuo. Yield: 39 mg, 62%. Alternatively, the title compound can be
prepared from 2a (75 mg, 0.13 mmol), silver tetrafluoroborate (26 mg, 0.13 mmol), and
Pd(cod)Cl2 (77 mg, 0.27 mmol) using the same procedure. Yield: 96 mg, 79%. 1H
NMR(CD3CN, δ): 8.03 (dd, JHH = 1.58, 7.70 Hz, 3-CH of Ph, 1H), 8.00 (dt, JHH = 1.58, 7.85 Hz,
5-CH of Ph, 1H), 7.89 (dd, JHH = 1.22, 7.85 Hz, 6-CH of Ph, 1H), 7.84 (dt, JHH = 1.22, 7.70 Hz,
4-CH of Ph, 1H), 7.56 (d, JHH = 2.10 Hz, 4-CH of imid, 1H), 7.48 (d, JHH = 2.10 Hz, 5-CH of
imid, 1H), 4.15 (s, CH3 of C–CN, 3H), 1.96 (s, integration cannot be determined because of
exchange with CD3CN,CH3 of CH3CN). 19F NMR (CD3CN, δ): -152.2 (s), -152.3 (s). 13C{1H}
NMR (CD3CN, δ): 141.4 (Pd–C), 140.7 (CPh), 135.4 (CPh), 135.1 (CPh), 131.9 (CPh), 130.7 (CPh),
126.5 (Cimid), 126.2 (Cimid), 118.4 (CN of CH3CN), 116.2 (CN of C–CN), 112.1 (CPh), 38.6 (CH3
45. Arduengo, A. J.; Krafczyk, R.; Schmutzler, R.; Craig, H. A.; Goerlich, J. R.; Marshall, W. J.; Unverzagt, M., Tetrahedron 1999, 55, 14523-14534.46. Yu, X.-Y.; Patrick, B. O.; James, B. R., Organometallics 2006, 25, 2359-2363.47 Drew, D.; Doyle, J. R., Inorg. Synth. 1990, 28, 346-349.

71of C–CN), 1.69 (CH3 of CH3CN). IR (KBr, cm-1): 2329 (v(CN) of CH3CN), 2237 (v(CN) of C–
CN). MS(ESI, methanol/water; m/z): 507.0 ([Pd(C–CN)2Cl]+), 471.1 ([Pd(C–CN)2]•+). Anal.
Calcd for C26H24B2Cl2F8N8Pd2: C, 34.47; H, 2.67; N, 12.37. Found: C, 34.74; H, 2.58; N, 12.00.
3.5.3 Synthesis of (Acetonitrile)(1-(2-cyanophenyl)-3-methylimidzol-2-ylidene)(η1:η2-2-
methoxycyclooct-5-enyl)palladium(II) Tetrafluoroborate ([Pd(C–CN)(η1:η2-coe-OMe)
(CH3CN)]BF4, 6a). A solution of 2a (69 mg, 0.12 mmol) and silver tetrafluoroborate (24 mg,
0.12 mmol) in acetonitrile (4 mL) was added to a solution of [Pd(η1:η2-coe-OMe)(μ-Cl)]2 (70
mg, 0.12 mmol) in dichloromethane (6 mL). A white precipitate was formed instantaneously.
The reaction mixture continued to be stirred for 2 h, protected from light. It was then filtered
through a pad of Celite to give a golden yellow solution. The solvent was then removed in vacuo,
and the crude product was recrystallized with acetonitrile (2 mL) and diethyl ether (10 mL) at
-25°C to give white needles. The needles were then collected on a glass frit and dried in vacuo.
Yield: 92 mg, 67%. Alternatively, the title compound can be obtained by recrystallization of 6b
in acetonitrile and diethyl ether. Suitable crystals for an X-ray diffraction study were obtained by
slow diffusion of diethyl ether into a saturation solution of 6a in acetonitrile. In addition to a
peak at 2.01 ppm in the 1H NMR spectrum (CD3NO2), all other spectroscopic information are
identical with those of compound 6b. IR (KBr, cm-1): 2277 (v(CN) of C–CN), 2229 (v(CN) of
CH3CN). MS (ESI, methanol/water; m/z): 428.1 ([M – CH3CN]+). Anal. Calcd for
C22H27BF4N4OPd: C,47.46; H, 4.89; N, 10.06. Found: C, 47.18; H, 4.04; N, 10.26.
3.5.4 Synthesis of Bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-2-methoxy-
cyclooct-5-enyl)palladium(II)] Tetrafluoroborate ([Pd(C–CN)(η1:η2-coe-OMe)]2(BF4)2, 6b).
A solution of 2a (148 mg, 0.26 mmol) and silver tetrafluoroborate (52 mg, 0.27 mmol) in
acetonitrile (4 mL) was added to a solution of [Pd(η1:η2-coe-OMe)(μ-Cl)]2 (149 mg, 0.27 mmol)
in dichloromethane (10 mL). A white precipitate was formed instantaneously. The reaction
mixture continued to be stirred for 2 h, protected from light. The solvent was then removed in
vacuo. The residue was extracted with dichloromethane (10 mL) and filtered through a pad of
Celite to give a golden yellow solution. The solvent was removed, and the crude product was
recrystallized with dichloromethane (2 mL) and diethyl ether (10 mL) to give a white precipitate.
The precipitate was filtered and dried in vacuo to give a white powder. Yield: 210 mg, 77%.
Alternatively, the title compound can be obtained by recrystallization of 6a in dichloromethane
and diethyl ether. 1H NMR (CD3NO2, δ): 8.24 (d, JHH = 7.90 Hz, 3-CH of Ph, 1H), 8.06 (t, JHH =
7.88 Hz, 5-CH of Ph, 1H), 7.88 (t, JHH = 7.90 Hz, 4-CH of Ph, 1H), 7.78 (d, JHH = 7.80 Hz, 6-CH

72of Ph, 1H), 7.66 (d, JHH = 1.86Hz, 4-CH of imid, 1H), 7.54 (d, JHH = 1.86 Hz, 5-CH of imid, 1H),
6.39 (m, 6-CH of olefinic CH, 1H), 6.10 (m, 5-CH of olefinic CH, 1H), 3.91 (d, CH3 of C–CN,
3H), 3.29 (m, 2-CH of coe-OMe, 1H), 2.95 (d, CH3 of coe-OMe, 3H), 2.65 (m, 1A-CH or 1B-
CH and 4-CH of coe-OMe, 1.5H), 2.36 (m, 1A-CH or 1B-CH of coe-OMe, 0.5H), 2.15 (m, 4-CH
and 7-CH of coe-OMe, 2H), 1.91 (m, 3-CH2 of coe-OMe, 2H), 1.77 (m, 7-CH of coe-OMe, 1H),
1.42 (m, 8-CH of coe-OMe, 1H), 0.42 (m, 8-CH of coe-OMe, 1H). 19F NMR (CD3NO2, δ):
-152.9 (s), -153.0 (s). 13C{1H} NMR (CD3NO2, δ): 174.6, 174.2 (Pd–CB/A), 144.5, 144.4
(CPh,B/A), 136.4, 136.0 (CPh,B/A), 134.8, 134.3 (CPh,A/B), 130.5, 130.2 (CPh,B/A), 128.3, 127.9
(CPh,B/A), 124.4, 124.2 (Cimid,B/A), 123.4, 123.2 (Cimid,A/B), 119.5, 119.4 (6-CA/B of coe-OMe),
117.7, 117.4 (CNB/A), 115.4, 115.2 (5-CA/B of coe-OMe), 109.4, 109.1 (CPh,A/B), 81.9, 80.8 (2-
CA/B of coe-OMe), 54.9, 54.8 (CB/A-CH3 of coe-OMe), 41.4, 41.2 (1-CA/B of coe-OMe), 37.5,
37.4 (CA/B-CH3 of C–CN), 33.5, 33.5 (8-CB/A of coe-OMe), 30.1, 29.4 (3-CA/B of coe-OMe),
28.0, 27.6 (4-CA/B of coe-OMe), 24.8, 24.4 (7-CB/A of coe-OMe). IR (KBr, cm-1): 2260 (v(CN) of
C–CN). MS (ESI, methanol/ water; m/z): 428.1 ([0.5 M]+). Anal. Calcd for C40H48B2F8N6O2Pd2:
C, 46.58; H, 4.69; N, 8.15. Found: C, 45.90; H, 5.18; N, 7.82.
3.5.5 Synthesis of Chloro[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene](η4-1,5-cyclo-
octadiene)platinum(II) Tetrafluoroborate ([Pt(C–CN)(cod)Cl]BF4, 7). A solution of 2a (109
mg, 0.19 mmol) and silver tetrafluoroborate (38 mg, 0.19 mmol) in acetonitrile (4 mL) was
added to a solution of Pt(cod)Cl2 (145 mg, 0.39 mmol) in dichloromethane (10 mL). A white
precipitate was formed instantaneously. The reaction mixture continued to be stirred for 2 h. It
was then filtered through a pad of Celite to give a pale yellow solution. The solvent was then
removed in vacuo, and the crude product was recrystallized with dichloromethane (3 mL) at
-25°C to give white crystalline solids. The product was then collected on a glass frit, rinsed with
dichloromethane (2 × 3 mL) and diethyl ether (3 × 3 mL), and dried in vacuo. Yield: 140 mg,
59%. Suitable crystals for an X-ray diffraction study were obtained by slow diffusion of diethyl
ether into a saturation solution of 7 in acetonitrile. 1H NMR (CD3CN, δ): 8.08 (dd, JHH = 1.23,
8.03 Hz, 3-CH of Ph, 1H), 8.03 (dd, JHH = 1.53, 7.70 Hz, 6-CH of Ph, 1H), 8.01 (dt, JHH = 1.53,
8.03 Hz, 4-CH of Ph, 1H), 7.84 (dt, JHH = 1.23, 7.70 Hz, 5-CH of Ph, 1H), 7.68 (d, JHH = 2.10
Hz, 4-CH of imid, 1H), 7.54 (d, JHH = 2.10 Hz, 5-CH of imid, 1H), 5.94 (m, olefinic CH of
codtrans to C, 2H), 5.32 (m, JPt-H = 32.61 Hz, olefinic CH of codtrans to Cl, 1H), 4.35 (m, JPt-H = 30.72
Hz, olefinic CH of codtrans to Cl, 1H), 4.00 (s, CH3, 3H), 2.69 (m, CH2 of cod, 1H), 2.56 (m, CH2
of cod, 1H), 2.38 (m, CH2 of cod, 1H), 2.24 (m, CH2 of cod, 1H), 2.07 (m, CH2 of cod, 1H), 1.72

73(m, CH2 of cod, 1H). 19F NMR (CD3CN, δ): -152.2 (s), -152.3 (s). 13C{1H} NMR (CD3CN, δ):
151.4 (Pt–C), 140.1 (CPh), 135.8 (CPh), 135.6 (CPh), 132.0 (CPh), 130.1 (CPh), 126.5 (Cimid), 125.8
(Cimid), 119.5 (JPt-C = 31.54 Hz, olefinic C of codtrans to C), 119.4 (JPt-C = 31.92 Hz, olefinic C of
codtrans to C), 115.9 (CN), 111.3 (CPh), 97.7 (JPt-C = 75.12 Hz, olefinic C of codtrans to Cl), 94.9 (JPt-C
= 80.20 Hz, olefinic C of codtrans to Cl), 38.6 (CH3), 33.1 (CH2 of cod), 31.7 (CH2 of cod), 29.1
(CH2 of cod), 28.5 (CH2 of cod). IR (KBr, cm-1): 2231 (v(CN)). MS (ESI, methanol/water; m/z):
521.1 ([M]+), 517.2 ([M – Cl + MeOH)]•+), 503.1 ([M – Cl + H2O)]•+). Anal. Calcd for
C19H21BF4ClN3Pt: C, 37.49; H, 3.48; N, 6.90. Found: C, 37.68; H, 3.55; N, 7.14.
3.5.6 Synthesis of Chloro[1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-2-methoxy-
cyclooct-5-enyl)platinum(II) (Pt(C–CN)(η1:η2-coe-OMe)Cl, 8). A mixture of 7 (100 mg, 0.16
mmol) and potassium acetate (19 mg, 0.19 mmol) were suspended in methanol (12 mL). The
reaction mixture was then refluxed under an argon atmosphere for 3 h to a golden yellow
solution, and all solids were completely dissolved. The solvent was then removed in vacuo. The
residue was extracted with dichloromethane (3 mL) and filtered through a pad of Celite. The
crude product was then recrystallized overnight at -25°C with the addition of n-pentane (15 mL)
to the dichloromethane solution to give white crystalline solids. The product was then collected
on a glass frit and dried in vacuo. Yield: 63 mg, 69%. Suitable crystals for an X-ray diffraction
study were obtained by slow diffusion of diethyl ether into a saturation solution of 8 in
dichloromethane. 1H NMR (CD2Cl2, δ): 8.66 (d, JHH = 7.86 Hz, 3A-CH or 3B-CH of Ph, 0.5H),
8.39 (d, JHH = 7.86 Hz, 3A-CH or 3B-CH of Ph, 0.5H), 7.85 (t, JHH = 7.86 Hz, 4-CH of Ph, 1H),
7.80 (d, JHH = 7.75 Hz, 6-CH of Ph, 1H), 7.61 (dd, JHH = 7.75, 14.37 Hz, 5-CH of Ph, 1H), 7.42
(d, JHH = 1.94 Hz, 4-CH of imid, 1H), 7.23 (d, JHH = 1.94 Hz, 5-CH of imid, 1H), 5.39 (m, JPt-H =
24.46 Hz, 5-CH of olefinic CH, 1H), 5.26 (m, JPt-H = 31.14 Hz, 6-CH of olefinic CH, 1H), 4.05
(s, CH3-A or CH3-B of C–CN, 1.5H), 4.02 (s, CH3-A or CH3-B of C–CN, 1.5H), 3.20 (m, 2A-
CH or 2B-CH of coe-OMe, 0.5H), 3.05 (s, CH3-A or CH3-B of coe-OMe, 1.5H), 2.95 (s, CH3-A
or CH3-B of coe-OMe, 1.5H), 2.71 (m, 4A-CH or 4B-CH of coe-OMe, 1H), 2.57 (m, 4A-CH or
4B-CH of coe-OMe, 1H), 2.28 (m, 1A-CH or 1B-CH of coe-OMe, 0.5H), 2.11 (m, 7A-CH or
7B-CH of coe-OMe, 1H), 1.97 (m, 1A-CH or 1B-CH, 2A-CH or 2B-CH, and 8A-CH or 8B-CH
of coe-OMe, 2H), 1.74 (m, 7A-CH or 7B-CH, and 8A-CH or 8B-CH of coe-OMe, 2H), 1.59 (m,
3A-CH or 3B-CH of coe-OMe, 2H). 13C{1H} NMR (CD2Cl2, δ): 176.2, 175.8 (Pt–CA/B), 142.0,
141.7 (CPh,A/B), 133.9, 133.8 (CPh,A/B), 133.6, 133.5 (CPh,A/B), 130.9, 130.6 (CPh,A/B), 129.8, 129.6
(CPh,A/B), 123.5, 123.3 (Cimid,A/B), 122.2, 122.1 (Cimid,A/B), 116.1, 116.0 (CNA/B), 109.6, 109.5

74(CPh,A/B), 102.9, 101.4 (6-CA/B of coe-OMe), 101.9, 101.2 (5-CA/B of coe-OMe), 83.9, 83.1 (2-
CA/B of coe-OMe), 55.7, 55.6 (CA/B-CH3 of coe-OMe), 38.2, 38.1 (CA/B-CH3 of C–CN), 35.4,
34.1 (3-CA/B of coe-OMe), 29.9, 29.7 (4-CA/B of coe-OMe), 29.6, 29.4 (7-CA/B of coe-OMe),
27.4, 26.9 (8-CA/B of coe-OMe), 22.2, 20.1 (1-CA/B of coe-OMe). IR (KBr, cm-1): 2232 (v(CN)).
MS (ESI, methanol/water; m/z): 517.2 ([M – Cl]+). Anal. Calcd for C20H24ClN3OPt: C, 43.44; H,
4.37; N, 7.60. Found: C, 43.14; H, 4.95; N, 7.92.
3.5.7 Synthesis of (Acetonitrile)(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-2-
methoxycyclooct-5-enyl)platinum(II) Tetrafluoroborate ([Pt(C–CN)(η1:η2-coe-OMe)
(CH3CN)]BF4, 9a). Complex 8 (11 mg, 0.08 mmol) was dissolved in acetonitrile-d3 (3 mL).
Silver tetrafluoroborate (15 mg, 0.08 mmol) was then added to the reaction mixture, whereupon
a pale brown precipitate was formed instantaneously. The solution was then stirred for 1 h. The
reaction mixture was filtered through a pad of Celite to give a golden yellow solution. 1H NMR
showed quantitative conversion of complex 8 to the title compound. The solution was then
evaporated to dryness to give a yellow oil as the crude product. Suitable crystals for an X-ray
diffraction study were obtained by slow diffusion of diethyl ether into a saturated solution of the
crude product in acetonitrile. In addition to a peak at δ 1.99 ppm in the 1H NMR spectrum
(CD2Cl2), all other spectroscopic data are identical with those for compound 9b. IR (KBr, cm-1):
2282 (v(CN) of C–CN), 2227 (v(CN) of CH3CN). MS (ESI, methanol/water; m/z): 517.2 ([M –
CH3CN]+). HRMS (ESI, methanol/water; m/z): calcd for C20H24N3OPt+ ([M – BF4 – CH3CN]+)
517.1561, found 517.1537.
3.5.8 Synthesis of Bis[(1-(2-cyanophenyl)-3-methylimidazol-2-ylidene)(η1:η2-2-methoxy-
cyclooct-5-enyl)platinum(II)] Tetrafluoroborate ([Pt(C–CN)(η1:η2-coe-OMe)]2(BF4)2, 9b).
Complex 8 (42 mg, 0.08 mmol) was dissolved in acetonitrile (5 mL). Silver tetrafluoroborate (15
mg, 0.08 mmol) was then added to the reaction mixture, whereupon a pale brown precipitate was
formed instantaneously. The solution was then stirred for 1 h. The reaction mixture was filtered
through a pad of Celite to give a golden yellow solution, and the solvent was evaporated to
dryness. The crude product was then recrystallized with dichloromethane (1 mL) and diethyl
ether (8 mL) to give a white precipitate. The precipitate was then collected and dried in vacuo to
give a white powder. Yield: 40 mg, 87%. 1H NMR (CD2Cl2, δ): 8.24 (dd, JHH = 1.56, 7.98 Hz, 3-
CH of Ph, 1H), 7.96 (dt, JHH = 1.56, 7.80 Hz, 5-CH of Ph, 1H), 7.85 (dt, JHH = 1.18, 7.98 Hz, 4-
CH of Ph, 1H), 7.83 (d, JHH = 1.88 Hz, 4-CH of imid, 1H), 7.60 (dt, JHH = 1.18, 7.80 Hz, 6-CH
of Ph, 1H), 7.55 (d, JHH = 1.88Hz, 5-CH of imid, 1H), 5.95 (m, JPt-H = 29.49 Hz, 6-CH of olefinic

75CH, 1H), 5.57 (m, JPt-H = 27.08 Hz, 5-CH of olefinic CH, 1H), 3.80 (d, CH3 of C–CN, 3H), 3.01
(d, CH3 of coe-OMe, 3H), 2.90 (m, 2-CH of coe-OMe, 1H), 2.81 (m, 4A-CH or 4B-CH of coe-
OMe, 1H), 2.52 (m, 4A-CH or 4B-CH of coe-OMe, 1H), 2.16 (m, 8A-CH or 8B-CH of coe-
OMe, 1H), 1.87 (m, 1-CH, 7A-CH or 7B-CH, and 8A-CH or 8B-CH of coe-OMe, 3H), 1.62 (m,
3-CH2 and 7A-CH or 7B-CH of coe-OMe, 3H). 19F NMR (CD3CN, δ): -152.2 (s), -152.3 (s). 13C{1H} NMR (CD2Cl2, δ): 168.9, 168.7 (Pt–CB/A), 141.5, 141.3 (CPh,B/A), 136.7, 136.6 (CPh,A/B),
136.1, 136.0 (CPh,A/B), 131.6, 131.2 (CPh,B/A), 128.8, 128.5 (CPh,B/A), 126.1, 125.7 (Cimid,B/A),
123.8, 123.6 (Cimid,A/B), 118.3, 117.9 (CNB/A), 110.5, 110.4 (CPh,A/B), 109.1, 108.9 (JPt-C = 48.51,
28.31 Hz, 6-CA/B of coe-OMe), 105.7, 104.0 (5-CA/B of coe-OMe), 83.1, 81.5 (2-CA/B of coe-
OMe), 56.0, 54.8 (CA/B-CH3 of coe-OMe), 38.3, 38.1 (CA/B-CH3 of C–CN), 34.4, 33.9 (3-CB/A of
coe-OMe), 29.5 (4-CA/B of coe-OMe), 28.9, 28.2 (7-CA/B of coe-OMe), 26.6, 26.1 (8-CB/A of coe-
OMe), 24.1, 23.9 (1-CA/B of coe-OMe). IR (KBr, cm-1): 2262 (v(CN) of C–CN). MS (ESI,
methanol/water; m/z): 517.2 ([0.5 M]+). Anal. Calcd for C40H48B2F8N6O2Pt2: C, 39.75; H, 4.00;
N, 6.95. Found: C, 39.36; H, 3.91; N, 6.92.
3.5.9 Synthesis of Chloro[1,3-bis-(2,4,6-trimethylphenyl)imidazol-2-ylidene)](η4-1,5-cyclo-
octadiene)platinum(II) Tetrafluoroborate ([Pt(IMes)(cod)Cl]BF4, 11). A solution of 10 (106
mg, 0.13 mmol) and silver tetrafluoroborate (26 mg, 0.13 mmol) in acetonitrile (6 mL) was
added to a solution of Pt(cod)Cl2 (123 mg, 0.33mmol) in dichloromethane (6 mL). A white
precipitate was formed instantaneously. The reaction mixture was then refluxed under an argon
atmosphere overnight. The solvent was removed in vacuo after the reaction has gone to
completion. The residue was extracted with acetonitrile (6 mL) and filtered through a pad of
Celite to give a golden yellow solution. The solvent was then removed from vacuum, extracted
with dichloromethane (3 mL), and filtered through a pad of Celite. The crude product was
recrystallized at -25°C with the addition of diethyl ether (15 mL) to the dichloromethane solution
to give tan crystalline solids. The product was then collected on a glass frit and dried in vacuo.
Yield: 163 mg, 85%. 1H NMR (CD3CN, δ): 7.52 (s, CH of imid, 2H), 7.16 (d, 2-CH of Ph, 4H),
5.61 (m, JPt-H = 22.03 Hz, olefinic CH of codtrans to C, 2H), 4.87 (m, JPt-H = 31.55 Hz, olefinic CH
of codtrans to Cl, 2H), 2.39 (s, o-CH3 of Ph,12H), 2.25 (s, p-CH3 of Ph, 6H), 2.25-2.20 (m, CH2 of
cod, 8H). 19F NMR (CD3CN, δ): -152.2 (s), -152.3 (s). 13C{1H} NMR (CD3CN, δ): 151.4 (Pt–C),
142.5 (CPh), 137.3 (CPh), 135.5 (CPh), 134.6 (CPh), 130.8 (CPh), 130.1 (CPh), 127.3 (JPt-C = 16.14
Hz, Cimid), 119.1 (JPt-C = 33.11 Hz, olefinic C of codtrans to C), 96.6 (JPt-C = 76.88 Hz, olefinic C of
codtrans to Cl), 32.6 (CH2 of cod), 28.4 (CH2 of cod), 21.1 (o-C-CH3 of Ph), 19.4 (p-C-CH3 of Ph).

76MS(ESI, methanol/water; m/z): 642.2 ([M]+), 638.3 ([M – Cl + MeOH]•+), 624.3 ([M – Cl +
H2O]•+). Anal. Calcd for C29H36BClF4N2Pt⋅CH2Cl2: C, 44.22; H, 4.70; N, 3.44. Found: C, 44.44;
H, 4.61; N, 3.70.

77Chapter 4: Transmetalation of a Primary Amino-Functionalized N-
Heterocyclic Carbene Ligand from an Axially Chiral Square-Planar
Nickel(II) Complex to Ruthenium(II) and Osmium(II) Catalysts for
the Hydrogenation of Polar Bonds
4.1 Abstract
The first homoleptic nickel(II) complex with primary amino-functionalized N-heterocyclic
carbene (C–NH2) ligands ([Ni(C–NH2)2](PF6)2, 12) was prepared under mild conditions by the
reduction of a nitrile-functionalized imidazolium salt 1a. This axially chiral, square-planar
nickel(II)complex was characterized by NMR spectroscopy and an X-ray diffraction study.
Enantiopure Δ-TRISPHAT was used as an NMR chiral shift reagent to observe the
diastereotopic ion pairs by 1H NMR in acetonitrile-d3. A novel transmetalation reaction moved
the C–NH2 ligand from the nickel(II) complex 12 to the [M(p-cymene)Cl2]2 dimers (M = Ru,
Os), yielding the ruthenium(II) and osmium(II) complexes [M(p-cymene)(C–NH2)Cl]PF6, (13,
M = Ru; 14, M = Os), the first ruthenium(II) and osmium(II) complexes with such a chelating C–
NH2 ligand. Complex 13 is a catalyst for the transfer hydrogenation of acetophenone to 1-
phenylethanol in basic 2-propanol at 75°C with a turnover frequency (TOF) of up to 880 h-1 and
a conversion of 96%. The complex [RuCp*(C–NH2)(py)]PF6 (15), on the other hand, was
prepared from the transmetalation reaction of complex 12 and RuCp*(cod)Cl, and subsequent
addition of pyridine (cod = η4-1,5-cyclooctadiene, py = pyridine). This complex is an active
catalyst for the H2-hydrogenation of ketones, an epoxide, ester and ketimine in basic solution. A
maximum TOF of 17 600 h-1 is achieved under mild reaction conditions (25°C) and economical
use of hydrogen (8 bar) while the TOF of a related complex with a 2-(diphenylphosphino)-
benzylamine ligand (P–NH2), [RuCp*(P–NH2)(py)]PF6 (16a), is much smaller.
*Reproduced in part with permission from O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 6755-6761. Copyright 2009 American Chemical Society.*O, W. W. N.; Lough, A. J.; Morris, R. H., Chem. Commun. 2010, 46, 8240 – 8242. Reproduced in part by the permission of The Royal Society of Chemistry (RSC). http://pubs.rsc.org/en/content/articlelanding/2010/cc/ c0cc02664f

784.2 Introduction
A powerful concept in the design of homogeneous hydrogenation catalysts is the heterolytic
splitting of dihydrogen1 across a transition metal–amido bond.2 This provides a bifunctional
metal–hydride and protic amine grouping for the efficient, selective reduction of polar bonds to
produce valuable alcohols and amines.2 Ruthenium(II) complexes bearing a phosphine–amine
ligand (P–NH2) that catalyze efficiently the hydrogenation of ketones,3 imines,4 esters5 and other
polar bonds6 may utilize this mechanism.
The notion of replacing a phosphine with an N-heterocyclic carbene (NHC) donor is attractive
with the promise of a reduction in the toxicity of catalyst precursors and contaminants in the
hydrogenated products.7 The use of donor-functionalized N-heterocyclic carbenes as ligands in
the design of homogeneous catalysts is of particular interest as they provide coordination
versatility and metal-ligand bifunctionality that will enhance catalytic activity.8 Among those,
both secondary and tertiary amino-functionalized NHC ligands are found to be important
building blocks of active transition metal catalysts that are used for cross-coupling and
hydrosilylation reactions (Figure 4.1).9
1. (a) Morris, R. H., Can. J. Chem. 1996, 74, 1907-1915; (b) Ito, M.; Ikariya, T., Chem. Commun. 2007, 5134-5142; (c) Kubas, G. J., Chem. Rev. 2007, 107, 4152-4205.2. (a) Clapham, S. E.; Hadzovic, A.; Morris, R. H., Coord. Chem. Rev. 2004, 248, 2201-2237; (b) Ikariya, T.; Murata, K.; Noyori, R., Org. Biomol. Chem. 2006, 4, 393-406; (c) Ikariya, T., Bull. Chem. Soc. Jpn. 2011, 84, 1-16.3. (a) Guo, R.; Lough, A. J.; Morris, R. H.; Song, D., Organometallics 2004, 23, 5524-5529; (b) Jia, W. L.; Chen, X. H.; Guo, R. W.; Sui-Seng, C.; Amoroso, D.; Lough, A. J.; Abdur-Rashid, K., Dalton Trans. 2009, 8301-8307; (c) Phillips, S. D.; Fuentes, J. A.; Clarke, M. L., Chem.-Eur. J. 2010, 16, 8002-8005.4. Abdur-Rashid, K.; Guo, R. W.; Lough, A. J.; Morris, R. H.; Song, D. T., Adv. Synth. Catal. 2005, 347, 571-579.5. (a) Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P., Angew. Chem. Int. Ed. 2007, 46, 7473-7476; (b) Clarke, M. L.; Diaz-Valenzuela, M. B.; Slawin, A. M. Z., Organometallics 2007, 26, 16-19; (c) Kuriyama, W.; Ino, Y.; Ogata, O.; Sayo, N.; Saito, T., Adv. Synth. Catal. 2010, 352, 92-96.6. (a) Ito, M.; Hirakawa, M.; Osaku, A.; Ikariya, T., Organometallics 2003, 22, 4190-4192; (b) Ito, M.; Sakaguchi, A.; Kobayashi, C.; Ikariya, T., J. Am. Chem. Soc. 2007, 129, 290-291; (c) Ito, M.; Kobayashi, C.; Himizu, A.; Ikariya, T., J. Am Chem Soc. 2010, 132, 11414-11415.7. (a) Hahn, F. E.; Jahnke, M. C., Angew. Chem. Int. Ed. 2008, 47, 3122-3172; (b) Diez-Gonzalez, S.; Marion, N.; Nolan, S. P., Chem. Rev. 2009, 109, 3612-3676.8. (a) Kuhl, O., Chem. Soc. Rev. 2007, 36, 592-607; (b) Liddle, S. T.; Edworthy, I. S.; Arnold, P. L., Chem. Soc. Rev. 2007, 36, 1732-1744; (c) Lee, H. M.; Lee, C. C.; Cheng, P. Y., Curr. Org. Chem. 2007, 11, 1491-1524; (d) Normand, A. T.; Cavell, K. J., Eur. J. Inorg. Chem. 2008, 2781-2800; (e) Poyatos, M.; Mata, J. A.; Peris, E., Chem. Rev. 2009, 109, 3677-3707; (f) Corberan, R.; Mas-Marza, E.; Peris, E., Eur. J. Inorg. Chem. 2009, 1700-1716.9. (a) Douthwaite, R. E.; Houghton, J.; Kariuki, B. M., Chem. Commun. 2004, 698-699; (b) Bonnet, L. G.; Douthwaite, R. E.; Hodgson, R.; Houghton, J.; Kariuki, B. M.; Simonovic, S., Dalton Trans. 2004, 3528-3535; (c) Houghton, J.; Dyson, G.; Douthwaite, R. E.; Whitwood, A. C.; Kariuki, B. M., Dalton Trans. 2007, 3065-3073; (d) Jong, H.; Patrick, B. O.; Fryzuk, M. D., Can. J. Chem. 2008, 86, 803-810; (e) Jimenez, M. V.; Perez-Torrente, J. J.; Bartolome, M. I.; Gierz, V.; Lahoz, F. J.; Oro, L. A., Organometallics 2008, 27, 224-234; (f) Wei, W.; Qin, Y.; Luo, M.; Xia, P.; Wong, M. S., Organometallics 2008, 27, 2268-2272; (g) Jong, H.; Patrick, B. O.; Fryzuk, M. D., Organometallics 2011, 30, 2333-2341.

79We are interested in the design of late transition metal catalysts for polar bond hydrogenation
with chelating primary amino-functionalized NHC (C–NH2) ligands that resemble those of the
highly active phosphine-amine analogues to achieve greener catalysis. This is synthetically
challenging since free NHC are known to react with amines,10 and the syntheses of these
imidazolium salt precursors were thwarted by their low yields.11 We show in this chapter that a
nitrile-functionalized N-heterocyclic carbene ligand12 provides direct access to such a C–NH2
ligand by the reduction of a nitrile-functionalized imidazolium salt under mild conditions. Once
attached to nickel(II), this new type of C–NH2 ligand can be moved to ruthenium(II)13 and
osmium(II)14 in an efficient transmetalation reaction to yield catalysts for the transfer
hydrogenation of ketone, and H2-hydrogenation of a variety of molecules with polar bonds.15 The
ruthenium(II) complex containing a phosphine-amine (P–NH2 = 2-(diphenylphosphino)-
benzylamine herein) and pentamethylcyclopentadienyl ligand (Cp*) ligands was also prepared
for comparison.15 The related complex RuCp*(κ2(P,N)-PPh2CH2CH2NH2)Cl (16b), was prepared
in situ by Ikariya and co-workers.6a, 16
Figure 4.1. Examples of transition metal complexes bearing amino-functionalized N-
heterocyclic carbene ligands reported by Douthwaite and Oro. See references 9a, 9b and 9e.
4.3. Results and Discussion
4.3.1 Reduction of a Nitrile-Functionalized Imidazolium Salt to the First Homoleptic
Primary Amino-Functionalized N-Heterocyclic Carbene Complex of Nickel(II). Caddick et
10. (a) Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J. H.; Melder, J. P.; Ebel, K.; Brode, S., Angew. Chem. Int. Ed. 1995, 34, 1021-1023; (b) Frey, G. D.; Lavallo, V.; Donnadieu, B.; Schoeller, W. W.; Bertrand, G., Science 2007, 316, 439-441.11. Busetto, L.; Cassani, M. C.; Femoni, C.; Macchioni, A.; Mazzoni, R.; Zuccaccia, D., J. Organomet. Chem. 2008, 693, 2579-2591.12. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 853-862.13. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 6755-6761.14. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2011, 30, 1236-1252.15. O, W. W. N.; Lough, A. J.; Morris, R. H., Chem. Commun. 2010, 46, 8240 - 8242.16. Ito, M.; Hirakawa, M.; Murata, K.; Ikariya, T., Organometallics 2001, 20, 379-381.
NN
N
RRh
N
N N N
NR R
Pd
ClN
NPh
nPr
NPd
ClClHH
H+ +

80al. and Khurana and Kukreja have reported the use of nickel(II) chloride and sodium borohydride
in alcoholic solvents to reduce both aromatic and aliphatic nitriles to primary and secondary
amines or to protected primary amines in high yields under mild conditions.17 We attempted to
synthesize amino-functionalized imidazolium salts by the reduction of the corresponding nitriles
by use of such an established procedure.17a The reaction of a stoichiometric amount of hydrated
nickel(II) chloride and imidazolium salt 1a (Scheme 4.1) with an excess of sodium borohydride
at low temperature produced an intractable mixture of products; nevertheless all of the
imidazolium salt 1a was consumed. An aqueous workup of the reaction mixture in air, followed
by counteranion metathesis with NH4PF6 in water, afforded a yellow solid, which was
characterized as the first homoleptic primary amino-functionalized N-heterocyclic carbene
complex of nickel(II) ([Ni(C–NH2)2](PF6)2, 12) in about 20% yield based on 1a. The reaction
conditions were further optimized by using anhydrous nickel(II) chloride to improve the purity
of the product. The yield was increased to 30% by carrying out the reduction under a hydrogen
atmosphere and using large volumes of methanol to dissolve the imidazolium salt, which
otherwise has limited solubility (Scheme 4.1). A hydrogen atmosphere is not a prerequisite for
the reduction to occur although a drop of the isolated yield of up to 5% was observed. To our
knowledge, there are only two primary amino-functionalized N-heterocyclic carbene complexes,
those of silver bromide and palladium dichloride reported by Douthwaite and co-workers.9b Only
a few crystals of the palladium(II) complexes containing a chelating C–NH2 ligand were
synthesized by slow hydrolysis of an imine linkage. Williams and co-workers reported the use of
primary amino-functionalized imidazolium salts in enantioselective copper-catalyzed conjugate
addition reactions.18 None of these copper complexes, however, were fully characterized or
isolated.
Scheme 4.1. Synthesis of a Homoleptic Primary Amino-Functionalized N-Heterocyclic Carbene
Complex of Nickel(II) (12).
17. (a) Caddick, S.; Haynes, A. K. D.; Judd, D. B.; Williams, M. R. V., Tetrahedron Lett. 2000, 41, 3513-3516; (b) Khurana, J. M.; Kukreja, G., Synth. Commun. 2002, 32, 1265-1269; (c) Caddick, S.; Judd, D. B.; Lewis, A. K. D.; Reich, M. T.; Williams, M. R. V., Tetrahedron 2003, 59, 5417-5423.18. Moore, T.; Merzouk, M.; Williams, N., Synlett 2008, 21-24.
1) NiCl2, CH3OH2) 7 NaBH4, -78
°C, Ar or H23) NH4PF6, H2O, rt
0.5
N
N N CH3
BF4
+
Ni
NN
N N
NN
H3CCH3
2+
(PF6)2
H H H H1a 12

81Complex 12 has been structurally characterized by the use of X-ray diffraction (Figure 4.2).
The complex crystallizes in the orthorhombic chiral space group P212121 with four units residing
in the unit cell. The structure shows a square-planar geometry about the metal center, with C(1)-
Ni(1)-N(5) and C(1)-Ni(1)-C(12) bond angles of 91.8 and 88.1°, respectively. The Ni–Ccarbene
bond distances are typical of analogous cationic nickel(II) NHC systems.19 The Ni–Namine bond
distances are slightly longer than those of a nickel(II)-(2-methyl-1,2-propanediamine) complex
(1.913 Å)20 because of the higher trans influence of the carbene ligand. The amino protons on the
carbene ligands were hydrogen bonded to the fluorine atoms of the hexafluorophosphate anions
at distances between 2.18 and 2.60 Å.13 The phenyl rings are twisted with respect to the
imidazolylidene ring at dihedral angles of 52.76 and 55.01° to facilitate chelation.
Figure 4.2. ORTEP diagram of 12 depicted with thermal ellipsoids at 30% probability. The
counteranions and most of the hydrogens have been omitted for clarity. Selected bond distances
(A° ) and bond angles (deg): Ni(1)-C(1), 1.870(6); Ni(1)-C(12), 1.883(6); Ni(1)-N(5), 1.963(5);
Ni(1)-N(6), 1.937(5); C(1)-Ni(1)-N(5), 91.8(3); C(1)-Ni(1)-C(12), 88.1(3).
Furthermore, the identity of the title compound was established by the observation of a sharp
singlet in the 13C{1H} NMR spectrum in acetonitrile-d3 solution at 159.0 ppm assigned to the
carbene carbon (Ni–Ccarbene), which is in the expected range for analogous cationic nickel(II)
NHC systems.19 The identity of the complex was further established by the disappearance of a 19. (a) Wang, X.; Liu, S.; Jin, G. X., Organometallics 2004, 23, 6002-6007; (b) Winston, S.; Stylianides, N.;
Tulloch, A. A. D.; Wright, J. A.; Danopoulos, A. A., Polyhedron 2004, 23, 2813-2820; (c) Xi, Z.; Zhang, X.; Chen, W.; Fu, S.; Wang, D., Organometallics 2007, 26, 6636-6642; (d) Shibata, T.; Ito, S.; Doe, M.; Tanaka, R.; Hashimoto, H.; Kinoshita, I.; Yano, S.; Nishioka, T., Dalton Trans. 2011, 40, 6778-6784.20. (a) García-Granda, S.; Beurskens, P. T.; Behm, H. J. J.; Gómez-Beltrán, F., Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 1987, 43, 236-238; (b) García-Granda, S.; Díaz, M. R.; Gómez-Beltrán, F., Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 1990, 46, 598-600.

82broad resonance at 2.80 ppm upon addition of D2O to a solution of 12, as a result of complete
deuteration of the coordinated amino groups of the carbene ligands.
4.3.2 Solution NMR Studies of the Axial Chirality of the Homoleptic Primary Amino-
Functionalized N-Heterocyclic Carbene Complex of Nickel(II). The solid state structure of
complex 12 reveals that the metal complex is axially chiral about the two chelating ligands,
analogous to a 1,1ꞌ-binaphthyl linkage, but where the chiral and C2 rotational axes pass through
the metal center, lying in the same plane as the carbene carbon and amine nitrogen atoms. In
solution, the racemic mixture of complex 12 shows two diastereotopic protons on the methylene
linker at 2.53 and 3.56 ppm, as a result of the chirality established on the 3-methyl-
imidazolylidene rings. Chiral square-planar complexes with no stereogenic centers on the
coordinating ligands are rare. Mills and Quibell have reported the first optically resolved square-
planar complex of platinum(II) with meso-1,2-diphenylethylenediamine (dpen) and 1,1-
dimethylethylenediamine ligands, but these have carbon stereocenters.21 Thereafter, there were
reports of chiral square-planar complexes of pyridines without stereogenic centers, some of
which have been optically resolved.22 Attempts to resolve the two enantiomers of 12, for
example, by sodium L-tartrate failed, as indicated by circular dichroism (CD) spectra of the
isolated solids.
The use of enantiopure Δ-TRISPHAT as a NMR chiral shift reagent allows the observation of
diastereotopic ion pairs if the nickel(II) cation is configurationally stable on the time scale of the
NMR experiment.23 In this case the lifetime of the diastereomers must be greater than the inverse
of the chemical shift difference (in Hz) between resonances of the diastereomers. The addition of
2 equiv of [Bu4N][Δ-TRISPHAT] to an acetonitrile-d3 solution of complex 12 caused
diamagnetic shifting and doubling of the resonances of the methyl and imidazolylidene ring
protons in the 1H NMR spectrum. The integration of signals remained in a ratio of 1:1 as
expected starting with a configurationally stable racemic mixture (Figure 4.3). The spectra are
consistent with an ion pair structure with the Δ-TRISPHAT anion located near the 3-methyl-
21. Mills, W. H.; Quibell, T. H. H., J. Chem. Soc. 1935, 839-846.22. (a) Gianini, M.; Forster, A.; Haag, P.; von Zelewsky, A.; Stoeckli-Evans, H., Inorg. Chem. 1996, 35, 4889-4895; (b) Biagini, M. C.; Ferrari, M.; Lanfranchi, M.; Marchio, L.; Pellinghelli, M. A., Dalton Trans. 1999, 1575-1580.23. (a) Lacour, J.; Ginglinger, C.; Favarger, F.; Torche-Haldimann, S., Chem. Commun. 1997, 2285-2286; (b) Lacour, J.; Frantz, R., Org. Biomol. Chem. 2005, 3, 15-19 and references therein; (c) Lacour, J., C. R. Chim. 2010, 13, 985-997 and references therein.24. (a) Macchioni, A., Chem. Rev. 2005, 105, 2039-2073 and references therein; (b) Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D., Chem. Soc. Rev. 2008, 37, 479-489.
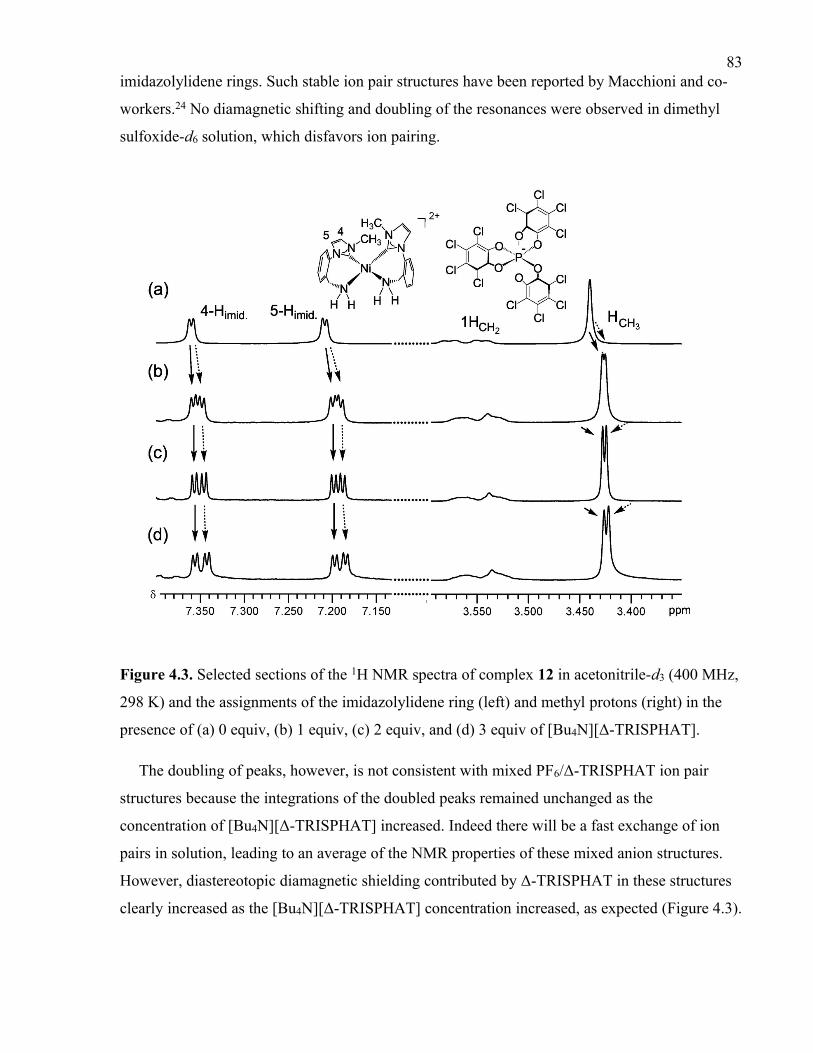
83imidazolylidene rings. Such stable ion pair structures have been reported by Macchioni and co-
workers.24 No diamagnetic shifting and doubling of the resonances were observed in dimethyl
sulfoxide-d6 solution, which disfavors ion pairing.
Figure 4.3. Selected sections of the 1H NMR spectra of complex 12 in acetonitrile-d3 (400 MHz,
298 K) and the assignments of the imidazolylidene ring (left) and methyl protons (right) in the
presence of (a) 0 equiv, (b) 1 equiv, (c) 2 equiv, and (d) 3 equiv of [Bu4N][Δ-TRISPHAT].
The doubling of peaks, however, is not consistent with mixed PF6/Δ-TRISPHAT ion pair
structures because the integrations of the doubled peaks remained unchanged as the
concentration of [Bu4N][Δ-TRISPHAT] increased. Indeed there will be a fast exchange of ion
pairs in solution, leading to an average of the NMR properties of these mixed anion structures.
However, diastereotopic diamagnetic shielding contributed by Δ-TRISPHAT in these structures
clearly increased as the [Bu4N][Δ-TRISPHAT] concentration increased, as expected (Figure 4.3).

844.3.3 Transmetalation Reaction of a Primary Amino-Functionalized N-Heterocyclic
Carbene from Nickel(II) to Ruthenium(II) and Osmium(II) Complexes with an Arene
ligand. The transmetalation reaction to move the chelating C–NH2 ligand from complex 12 to
the ruthenium(II) or osmium(II) dimers [M(p-cymene)Cl2]2 (M = Ru, Os) in refluxing
acetonitrile solution afforded a deep green solution after 2.5 h (Scheme 4.2). Subsequent
extraction with tetrahydrofuran (THF) or dichloromethane and recrystallization afforded yellow-
colored solids, which were characterized as [M(p-cymene)(C–NH2)Cl]PF6 (13, M = Ru; 14, M =
Os) by NMR spectroscopy and X-ray diffraction study (Figures 4.4 and 4.5). Complex 13 is an
air-stable solid, but it slowly decomposes in solution under prolonged exposure to air. Complex
14 is infinitely stable in both solid state and in solution. To our knowledge, these are the first
group 8 metal complexes bearing a primary amino-functionalized N-heterocyclic carbene ligand.
Scheme 4.2. Synthesis of Complexes 13 and 14 Bearing the C–NH2 Ligand by the
Transmetalation Reaction Involving Complex 12 and [M(p-cymene)Cl2]2 (M = Ru, Os).
Complex 13 crystallizes in the orthorhombic chiral space group Pbca, with four pairs of
enantiomers residing in the unit cell, as the metal complex is chiral about the ruthenium(II)
center (Figure 4.4). The structure has a piano-stool geometry about the metal center with a planar
arene ring coordinated to the metal center in an η6 fashion. The Ru–Ccarbene bond distance is
typical of analogous ruthenium(II) complexes of the form [Ru(η6-arene)(NHC)D2]n+ (D = halides
or mixed neutral/halide ligands, n = 0 or +1).25 The Ru–Namine bond distance is comparable to
known bifunctional catalysts for polar bond hydrogenation with the general formula [Ru(η6
25. (a) Geldbach, T. J.; Laurenczy, G.; Scopelliti, R.; Dyson, P. J., Organometallics 2006, 25, 733-742; (b) Ozdemir, I.; Demir, S.; Cetinkaya, B.; Toupet, L.; Castarlenas, R.; Fischmeister, C.; Dixneuf, P. H., Eur. J. Inorg. Chem. 2007, 2862-2869; (c) Ozdemir, I.; Demir, S.; Gurbuz, N.; Cetinkaya, B.; Toupet, L.; Bruneau, C.; Dixneuf, P. H., Eur. J. Inorg. Chem. 2009, 1942-1949; (d) Gandolfi, C.; Heckenroth, M.; Neels, A.; Laurenczy, G.; Albrecht, M., Organometallics 2009, 28, 5112-5121; (e) Horn, S.; Gandolfi, C.; Albrecht, M., Eur. J. Inorg. Chem. 2011, 2863-2868.26. (a) Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R., J. Am. Chem. Soc. 1995, 117, 7562-7563; (b) Haack, K. J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R., Angew. Chem. Int. Ed. 1997, 36, 285-288; (c) Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R., J. Am. Chem. Soc. 1996, 118, 4916-4917; (d) Hannedouche, J.; Clarkson, G. J.; Wills, M., J. Am. Chem. Soc. 2004, 126, 986-987; (e) Hayes, A. M.; Morris, D. J.; Clarkson, G. J.; Wills, M., J. Am. Chem. Soc. 2005, 127, 7318-7319.
[M(p-cymene)Cl2]2∆ , CH3CN
Ni
NN
N N
NN
H3CCH3
2+
(PF6)2
H H H H
MCl NNN
CH3
HH
PF6
13: M = Ru;14: M = Os
+
12
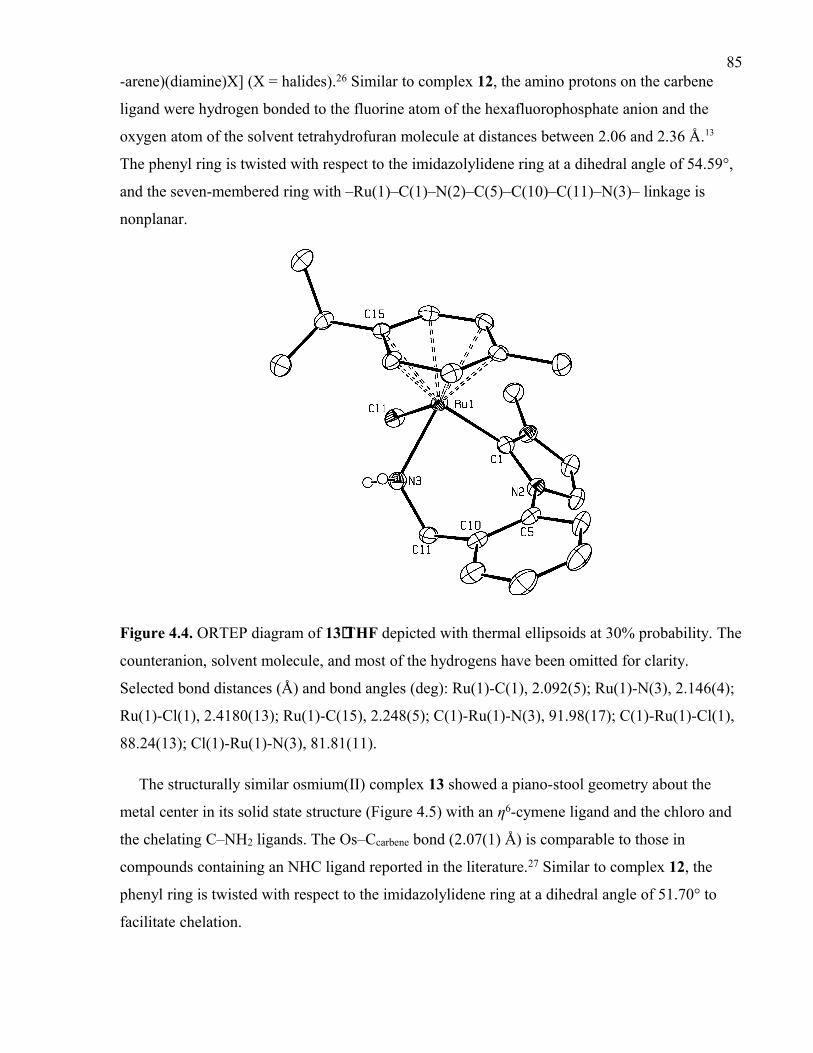
85-arene)(diamine)X] (X = halides).26 Similar to complex 12, the amino protons on the carbene
ligand were hydrogen bonded to the fluorine atom of the hexafluorophosphate anion and the
oxygen atom of the solvent tetrahydrofuran molecule at distances between 2.06 and 2.36 Å.13
The phenyl ring is twisted with respect to the imidazolylidene ring at a dihedral angle of 54.59°,
and the seven-membered ring with –Ru(1)–C(1)–N(2)–C(5)–C(10)–C(11)–N(3)– linkage is
nonplanar.
Figure 4.4. ORTEP diagram of 13⋅THF depicted with thermal ellipsoids at 30% probability. The
counteranion, solvent molecule, and most of the hydrogens have been omitted for clarity.
Selected bond distances (Å) and bond angles (deg): Ru(1)-C(1), 2.092(5); Ru(1)-N(3), 2.146(4);
Ru(1)-Cl(1), 2.4180(13); Ru(1)-C(15), 2.248(5); C(1)-Ru(1)-N(3), 91.98(17); C(1)-Ru(1)-Cl(1),
88.24(13); Cl(1)-Ru(1)-N(3), 81.81(11).
The structurally similar osmium(II) complex 13 showed a piano-stool geometry about the
metal center in its solid state structure (Figure 4.5) with an η6-cymene ligand and the chloro and
the chelating C–NH2 ligands. The Os–Ccarbene bond (2.07(1) Å) is comparable to those in
compounds containing an NHC ligand reported in the literature.27 Similar to complex 12, the
phenyl ring is twisted with respect to the imidazolylidene ring at a dihedral angle of 51.70° to
facilitate chelation.

86
Figure 4.5. ORTEP diagram of 14 depicted with thermal ellipsoids at the 30% probability level.
The counteranion and most of the hydrogens have been omitted for clarity. Selected bond
distances (Å) and bond angles (deg): Os(1)-C(1), 2.07(1); Os(1)-N(3), 2.137(9); Os(1)-Cl(1),
2.422(3); Os(1)-C(15), 2.201(6); C(1)-Os(1)-N(3), 91.1(4); C(1)-Os(1)-Cl(1), 87.5(3); Cl(1)-
Os(1)-N(3), 80.0(3).
In dichloromethane-d2 solution, the carbene carbon Ru–Ccarbene and Os–Ccarbene were observed
as a singlet at 175.1 and 159.4 ppm in the 13C{1H} NMR spectrum, respectively. They are in the
expected range for analogous ruthenium(II)25d, 25e, 28 and osmium(II)27 complexes. Complete
deuteration of the amino protons of the chelating carbene ligand in complex 13 by D2O was
observed by the disappearance of two broad multiplets at 5.12 and 3.68 ppm in the 1H NMR
spectrum.
The use of enantiopure Δ-TRISPHAT is again useful to observe the diastereotopic ion pairs
originating from the presence of two enantiomers in solution. Thus the addition of 1 equiv of
[Bu4N][Δ-TRISPHAT] to a dichloromethane-d2 solution of complex 13 caused diamagnetic
27. (a) Castarlenas, R.; Esteruelas, M. A.; Onate, E., Organometallics 2005, 24, 4343-4346; (b) Eguillor, B.; Esteruelas, M. A.; Olivan, M.; Puerta, M., Organometallics 2008, 27, 445-450; (c) Castarlenas, R.; Esteruelas, M. A.; Lalrempuia, R.; Olivan, M.; Onate, E., Organometallics 2008, 27, 795-798; (d) Castarlenas, R.; Esteruelas, M. A.; Onate, E., Organometallics 2008, 27, 3240-3247. 28. (a) Arnold, P. L.; Scarisbrick, A. C., Organometallics 2004, 23, 2519-2521; (b) Csabai, P.; Joo, F., Organometallics 2004, 23, 5640-5643.

87shifting and doubling of the resonances of the imidazolylidene ring protons and complete
splitting of the C(2) proton of the η6-arene ring in the 1H NMR spectrum, while the relative
integration of the split peaks remained in a ratio of 1:1 even when the concentration of [Bu4N]
[Δ-TRISPHAT] increased (Figure 4.6). Of note, an increase in the concentration of [Bu4N][Δ-
TRISPHAT] caused a diamagnetic shifting of the NH proton, which is not observed in the same
experiment with complex 12. A loss of the hydrogen bonding between the amino protons and the
hexafluorophosphate anion and an increase in the shielding effect of the amino protons by the
ring-current induced by the arene rings of the Δ-TRISPHAT anion might be responsible for such
an observation. However, the observed diamagnetic shifting and doubling of the resonances of
the protons of the 3-methylimidazolylidene ring, the amino group, and parts of the arene ligand
indicate that the Δ-TRISPHAT anion takes up various positions of ion pairing and rapidly moves
between these locations to produce averaged NMR properties. The methyl protons of the
isopropyl group of the η6-arene ligand are least affected by the addition of up to 3 equiv of
[Bu4N][Δ-TRISPHAT] (Figure 4.6). Similar to the case of complex 12, the extent of the
diastereotopic diamagnetic shielding of the protons in 13 caused by Δ-TRISPHAT increased as
the anion concentration increased, and this dominated over effects caused by anion exchange
between the various ion pairs and solvated anions.

88
Figure 4.6. Selected sections of the 1H NMR spectra of complex 13 in dichloromethane-d2 (400
MHz, 298 K) and the assignments of the imidazolylidene ring (left), p-cymene ring (middle) and
the isopropyl-methyl protons (right) in the presence of (a) 0 equiv, (b) 1 equiv, (c) 2 equiv, and
(d) 3 equiv of [Bu4N][Δ-TRISPHAT].
4.3.4 Synthesis of a Ruthenium(II) Complex with a Primary Amino-Functionalized N-
Heterocyclic Carbene and Pentamethylcyclopentadienyl ligands. The transmetalation
reaction between 12 and 1.5 equiv. of RuCp*(cod)Cl (cod = 1,5-cyclooctadiene) in acetonitrile,
and subsequent workup in THF and excess pyridine afforded complex 15 in 64% yield as
oxygen-sensitive orange-red needles (Scheme 4.3). Of note, the use of 2 equiv. of RuCp*(cod)Cl
and subsequent workup in THF and toluene mixtures afforded crystallization of small amounts
of [RuCp*(η6-toluene)]PF6 as a side product.29 The phosphine-amine complex 16a was
synthesized from the reaction of 2-(diphenylphosphino)benzylamine30 and RuCp*(cod)Cl in
dichloromethane. Subsequent halide abstraction with AgPF6 and addition of excess pyridine at

89ambient temperature (25°C) afforded a moderately oxygen-sensitive yellow solid in 67% yield
(Scheme 4.3).
Scheme 4.3. Synthesis of the Ruthenium(II) Complexes Containing the C–NH2 (15) and P–NH2
(l6a) Ligands.
Complexes 15 and 16a were unambiguously characterized by X-ray diffraction studies
(Figures 4.7 and 4.8). Both complexes adopt a piano-stool geometry about the ruthenium center,
with the corresponding chelating and pyridine ligands. The Ru–Ccarbene bond (2.03(1) Å) in 15 is
shorter than that of complex 13 (2.092(5)Å),13 but in the expected range of analogous
RuCp*(NHC)L2 complexes.25d, 31 The Ru–PPh2 bond length is similar to those reported by
Ikariya and co-workers.32
Diagnostic NMR features include the Ru–Ccarbene resonance at 198.0 ppm in the 13C{1H}
NMR spectrum of complex 15 in acetone-d6 and the PPh2 peak at 44.7 ppm as a singlet in the 31P{1H} NMR spectrum of 16a in dichloromethane-d2. These complexes demonstrate fluxional
behaviour at ambient temperature (25°C) due to restricted rotation of the Ru–Npyridine bond.
Cooling these samples to -40°C allows the observation of the aromatic protons, as well as the
diastereotopic protons for the CH2 linker between the primary amine group and the phenylene
spacer.15
29. O, W. W. N.; Lough, A. J.; Morris, R. H., Acta Crystallogr. Sect. E: Struct Rep. Online 2010, 66, m1264.30. Cahill, J. P.; Bohnen, F. M.; Goddard, R.; Kruger, C.; Guiry, P. J., Tetrahedron: Asymmetry 1998, 9, 3831-3839.31. (a) Baratta, W.; Herdtweck, E.; Herrmann, W. A.; Rigo, P.; Schwarz, J. D., Organometallics 2002, 21, 2101-2106; (b) Miranda-Soto, V.; Grotjahn, D. B.; Cooksy, A. L.; Golen, J. A.; Moore, C. E.; Rheingold, A. L., Angew. Chem. Int. Ed. 2011, 50, 631-635.32. Ito, M.; Osaku, A.; Kobayashi, C.; Shiibashi, A.; Ikariya, T., Organometallics 2009, 28, 390-393.
1.5 RuCp*Cl(cod)∆ , CH3CN
xs pyridine Ru
NN
N
CH3 PF6N1.5HH
NiN
NN N
NN
H3CCH3
2+
(PF6)2
H H H H
NH2 PPh2RuCp*Cl(cod) Ru
PPh2NPF6N
CH2Cl2, rt
1) AgPF6, CH3CN, rt2) xs pyridine, THF, rt
HH
15
16a
12
THF, rt
+
+
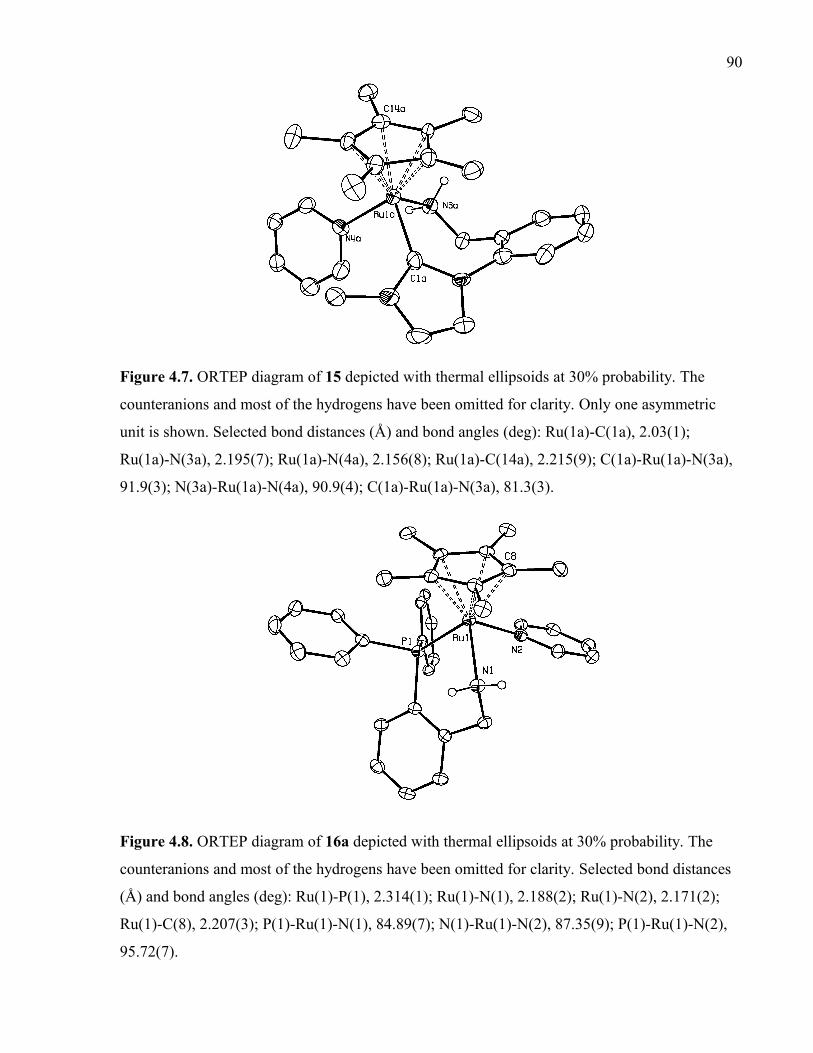
90
Figure 4.7. ORTEP diagram of 15 depicted with thermal ellipsoids at 30% probability. The
counteranions and most of the hydrogens have been omitted for clarity. Only one asymmetric
unit is shown. Selected bond distances (Å) and bond angles (deg): Ru(1a)-C(1a), 2.03(1);
Ru(1a)-N(3a), 2.195(7); Ru(1a)-N(4a), 2.156(8); Ru(1a)-C(14a), 2.215(9); C(1a)-Ru(1a)-N(3a),
91.9(3); N(3a)-Ru(1a)-N(4a), 90.9(4); C(1a)-Ru(1a)-N(3a), 81.3(3).
Figure 4.8. ORTEP diagram of 16a depicted with thermal ellipsoids at 30% probability. The
counteranions and most of the hydrogens have been omitted for clarity. Selected bond distances
(Å) and bond angles (deg): Ru(1)-P(1), 2.314(1); Ru(1)-N(1), 2.188(2); Ru(1)-N(2), 2.171(2);
Ru(1)-C(8), 2.207(3); P(1)-Ru(1)-N(1), 84.89(7); N(1)-Ru(1)-N(2), 87.35(9); P(1)-Ru(1)-N(2),
95.72(7).
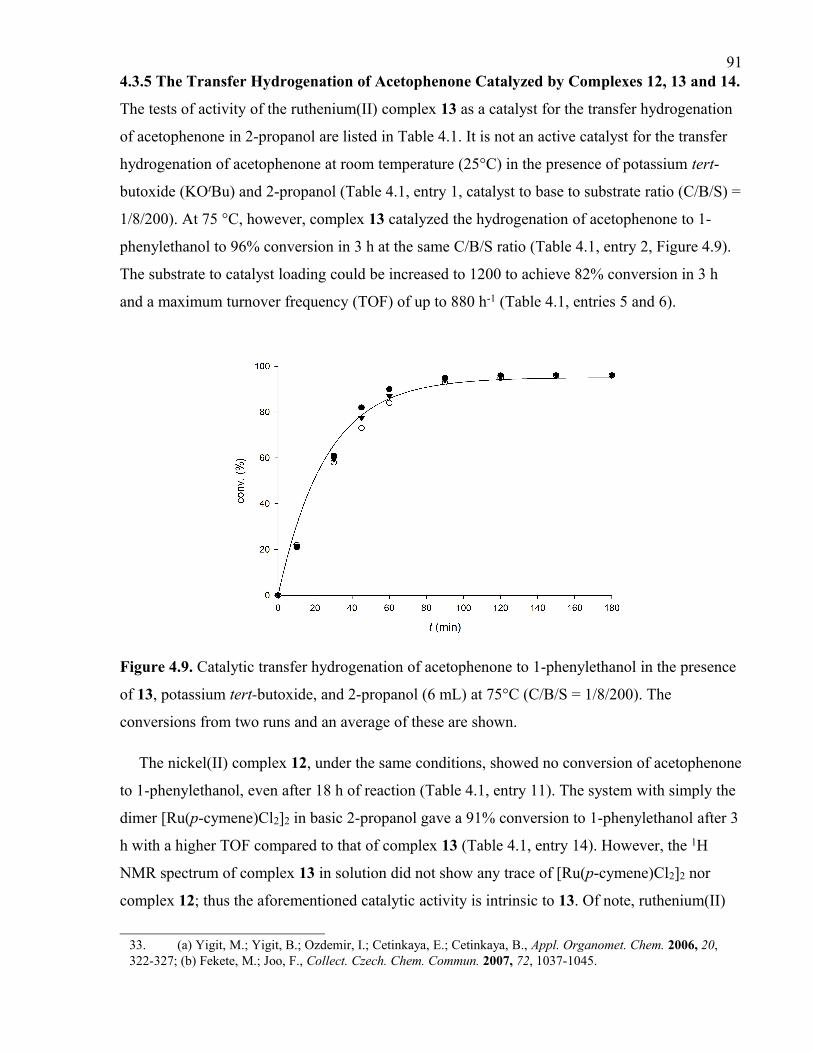
914.3.5 The Transfer Hydrogenation of Acetophenone Catalyzed by Complexes 12, 13 and 14.
The tests of activity of the ruthenium(II) complex 13 as a catalyst for the transfer hydrogenation
of acetophenone in 2-propanol are listed in Table 4.1. It is not an active catalyst for the transfer
hydrogenation of acetophenone at room temperature (25°C) in the presence of potassium tert-
butoxide (KOtBu) and 2-propanol (Table 4.1, entry 1, catalyst to base to substrate ratio (C/B/S) =
1/8/200). At 75 °C, however, complex 13 catalyzed the hydrogenation of acetophenone to 1-
phenylethanol to 96% conversion in 3 h at the same C/B/S ratio (Table 4.1, entry 2, Figure 4.9).
The substrate to catalyst loading could be increased to 1200 to achieve 82% conversion in 3 h
and a maximum turnover frequency (TOF) of up to 880 h-1 (Table 4.1, entries 5 and 6).
Figure 4.9. Catalytic transfer hydrogenation of acetophenone to 1-phenylethanol in the presence
of 13, potassium tert-butoxide, and 2-propanol (6 mL) at 75°C (C/B/S = 1/8/200). The
conversions from two runs and an average of these are shown.
The nickel(II) complex 12, under the same conditions, showed no conversion of acetophenone
to 1-phenylethanol, even after 18 h of reaction (Table 4.1, entry 11). The system with simply the
dimer [Ru(p-cymene)Cl2]2 in basic 2-propanol gave a 91% conversion to 1-phenylethanol after 3
h with a higher TOF compared to that of complex 13 (Table 4.1, entry 14). However, the 1H
NMR spectrum of complex 13 in solution did not show any trace of [Ru(p-cymene)Cl2]2 nor
complex 12; thus the aforementioned catalytic activity is intrinsic to 13. Of note, ruthenium(II)
33. (a) Yigit, M.; Yigit, B.; Ozdemir, I.; Cetinkaya, E.; Cetinkaya, B., Appl. Organomet. Chem. 2006, 20, 322-327; (b) Fekete, M.; Joo, F., Collect. Czech. Chem. Commun. 2007, 72, 1037-1045.
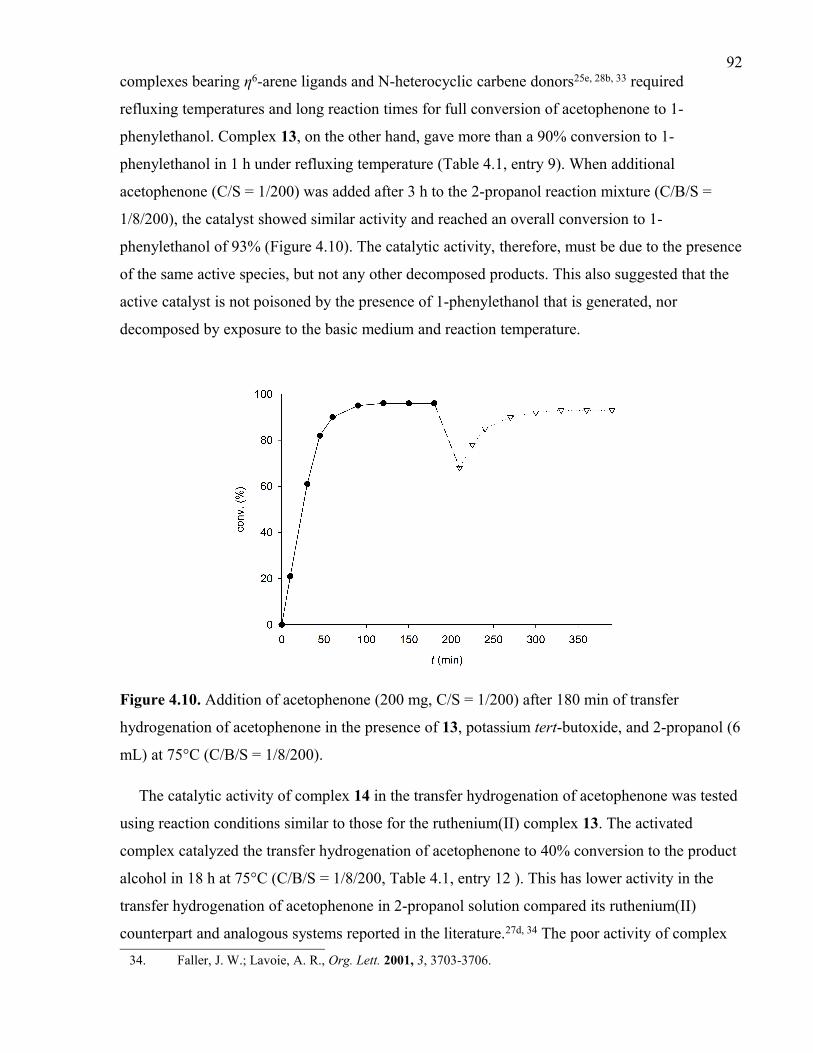
92complexes bearing η6-arene ligands and N-heterocyclic carbene donors25e, 28b, 33 required
refluxing temperatures and long reaction times for full conversion of acetophenone to 1-
phenylethanol. Complex 13, on the other hand, gave more than a 90% conversion to 1-
phenylethanol in 1 h under refluxing temperature (Table 4.1, entry 9). When additional
acetophenone (C/S = 1/200) was added after 3 h to the 2-propanol reaction mixture (C/B/S =
1/8/200), the catalyst showed similar activity and reached an overall conversion to 1-
phenylethanol of 93% (Figure 4.10). The catalytic activity, therefore, must be due to the presence
of the same active species, but not any other decomposed products. This also suggested that the
active catalyst is not poisoned by the presence of 1-phenylethanol that is generated, nor
decomposed by exposure to the basic medium and reaction temperature.
Figure 4.10. Addition of acetophenone (200 mg, C/S = 1/200) after 180 min of transfer
hydrogenation of acetophenone in the presence of 13, potassium tert-butoxide, and 2-propanol (6
mL) at 75°C (C/B/S = 1/8/200).
The catalytic activity of complex 14 in the transfer hydrogenation of acetophenone was tested
using reaction conditions similar to those for the ruthenium(II) complex 13. The activated
complex catalyzed the transfer hydrogenation of acetophenone to 40% conversion to the product
alcohol in 18 h at 75°C (C/B/S = 1/8/200, Table 4.1, entry 12 ). This has lower activity in the
transfer hydrogenation of acetophenone in 2-propanol solution compared its ruthenium(II)
counterpart and analogous systems reported in the literature.27d, 34 The poor activity of complex 34. Faller, J. W.; Lavoie, A. R., Org. Lett. 2001, 3, 3703-3706.
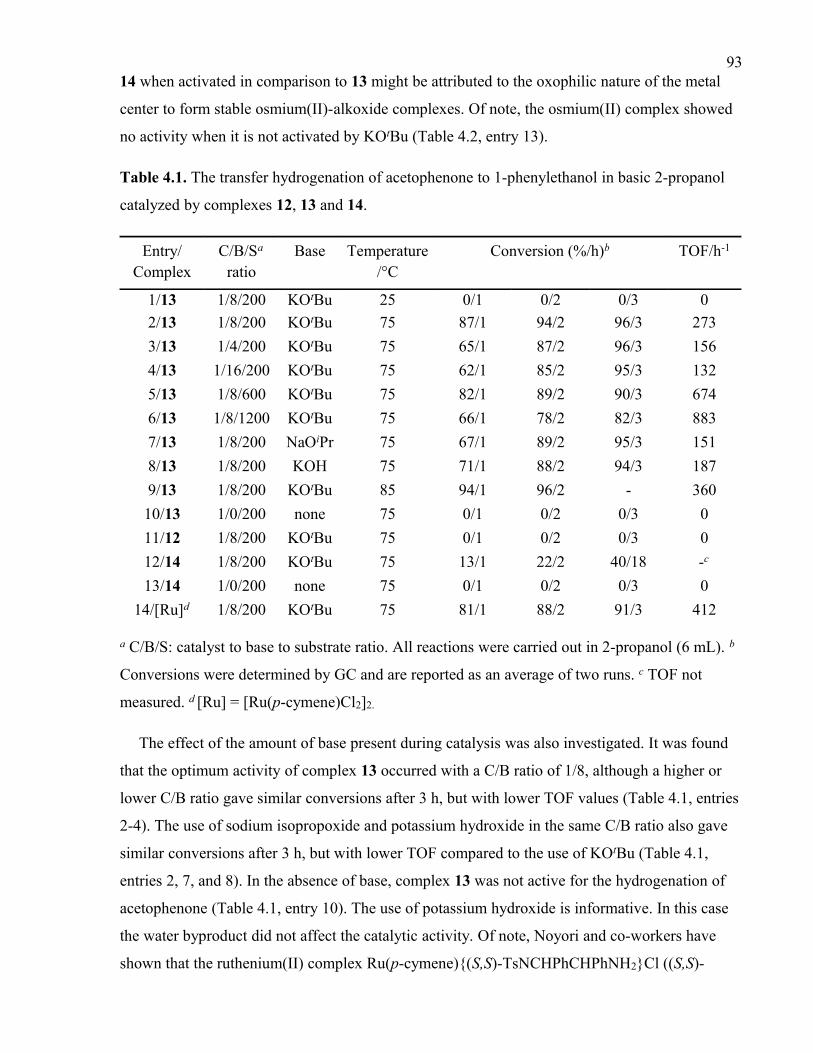
9314 when activated in comparison to 13 might be attributed to the oxophilic nature of the metal
center to form stable osmium(II)-alkoxide complexes. Of note, the osmium(II) complex showed
no activity when it is not activated by KOtBu (Table 4.2, entry 13).
Table 4.1. The transfer hydrogenation of acetophenone to 1-phenylethanol in basic 2-propanol
catalyzed by complexes 12, 13 and 14.
Entry/Complex
C/B/Sa
ratioBase Temperature
/°CConversion (%/h)b TOF/h-1
1/13 1/8/200 KOtBu 25 0/1 0/2 0/3 02/13 1/8/200 KOtBu 75 87/1 94/2 96/3 2733/13 1/4/200 KOtBu 75 65/1 87/2 96/3 1564/13 1/16/200 KOtBu 75 62/1 85/2 95/3 1325/13 1/8/600 KOtBu 75 82/1 89/2 90/3 6746/13 1/8/1200 KOtBu 75 66/1 78/2 82/3 8837/13 1/8/200 NaOiPr 75 67/1 89/2 95/3 1518/13 1/8/200 KOH 75 71/1 88/2 94/3 1879/13 1/8/200 KOtBu 85 94/1 96/2 - 36010/13 1/0/200 none 75 0/1 0/2 0/3 011/12 1/8/200 KOtBu 75 0/1 0/2 0/3 012/14 1/8/200 KOtBu 75 13/1 22/2 40/18 -c
13/14 1/0/200 none 75 0/1 0/2 0/3 014/[Ru]d 1/8/200 KOtBu 75 81/1 88/2 91/3 412
a C/B/S: catalyst to base to substrate ratio. All reactions were carried out in 2-propanol (6 mL). b
Conversions were determined by GC and are reported as an average of two runs. c TOF not
measured. d [Ru] = [Ru(p-cymene)Cl2]2.
The effect of the amount of base present during catalysis was also investigated. It was found
that the optimum activity of complex 13 occurred with a C/B ratio of 1/8, although a higher or
lower C/B ratio gave similar conversions after 3 h, but with lower TOF values (Table 4.1, entries
2-4). The use of sodium isopropoxide and potassium hydroxide in the same C/B ratio also gave
similar conversions after 3 h, but with lower TOF compared to the use of KOtBu (Table 4.1,
entries 2, 7, and 8). In the absence of base, complex 13 was not active for the hydrogenation of
acetophenone (Table 4.1, entry 10). The use of potassium hydroxide is informative. In this case
the water byproduct did not affect the catalytic activity. Of note, Noyori and co-workers have
shown that the ruthenium(II) complex Ru(p-cymene){(S,S)-TsNCHPhCHPhNH2}Cl ((S,S)-

94TsNCHPhCHPhNH2 = (1S,2S)-N-p-toluenesulfonyl-1,2-diphenylethylenediamine) in the
presence of potassium hydroxide and 2-propanol catalyzed the transfer hydrogenation of
acetophenone to 1-phenylethanol in 97% ee and 98% yield at 28°C after 10 h (C/B/S =
1/2/200).26a, 26b The related active catalysts, the amido complex Ru(p-cymene){(S,S)-
TsNCHPhCHPhNH} and the hydride-amine complex RuH(p-cymene){(S,S)-
TsNCHPhCHPhNH2}, showed the same catalytic activity without the use of base.2b, 26b, 35 These
are referred to as bifunctional catalysts since both the metal and the amido ligand work together
in the formation of the active hydride-amine species from 2-propanol and the transfer of the a
Ru-H/N-H pair from the hydride-amine complex to the carbonyl group of the ketone.2b, 35
Table 4.2. The hydrogenation of acetophenone catalyzed by complex 15 in the presence of
KOtBu.a
O 15 cat.H2, KOtBu
25°C
HO H
Entry P(H2)/bar C/B/S ratio Conversion (%/min)b TOF/h-1 c
1 2 1/8/2515 16/60 99/120 31102 8 1/8/2515 30/20 98/30 103003d 8 1/8/11500 29/30 97/90 72404e 8 1/8/2515 46/4 98/10 176005f -/Ar 1/8/1200 53/30 82/120 1270
a Reactions were carried out in a 50 mL Parr hydrogenation reactor in THF (6 mL) at 25°C
unless otherwise stated; KOtBu was used as the base. b Conversions were determined by GC and
are reported as an average of two runs. c Determined from the slope of the linear portion of
[alcohol] vs. time plot. d No solvent was used. e 2-Propanol was used as solvent. f Conducted
under an argon environment and 2-propanol was used as solvent.
4.3.6 The H2-Hydrogenation of Ketones Catalyzed by Complexes 15 and 16. Complex 15,
when reacted with KOtBu in THF, is an efficient catalyst for the H2-hydrogenation of
acetophenone (Table 4.2). Full conversion to 1-phenylethanol is achieved within 30 min under 8
bar of H2 in THF at 25°C with a C/B/S ratio of 1/8/2515. On the other hand, full conversion is
achieved in 10 min under similar catalytic conditions when 2-propanol was used with a TOF of
17 600 h-1. Complex 15 also catalyzed the transfer hydrogenation of acetophenone in 2-propanol 35. (a) Ikariya, T.; Blacker, A. J., Acc. Chem. Res. 2007, 40, 1300-1308; (b) Kuwata, S.; Ikariya, T., Dalton
Trans. 2010, 39, 2984-2992.

95at 25°C, but with a smaller TOF value (Table 4.2, entry 5). By comparison, complex 16a is a
poor catalyst which when activated (C/B/S=1/8/200) produces a maximum conversion of 8% in
4 h under 25 bar of H2 in THF at 50°C. The related complex 16b catalyzed the hydrogenation of
acetophenone under 1 atm of H2 in 2-propanol at 50°C (Ru/KOH/substrate = 1/1/100) with 16%
conversion in 1 h.16 In terms of selectivity and broad substrate scope (vide infra), complex 15 is
superior to these and a few other ruthenium(II) complexes with NHC ligands that have been
reported to catalyze the H2-hydrogenation of ketones and aldehydes,25d, 36 and it is comparable to
or better than many P–NH2 systems of ruthenium(II).3b, 3c, 4, 5b, 37
Some results of the H2-hydrogenation of substituted acetophenones are listed in Table 4.3. In
general, TOF values decrease with increasing donor ability at the 4' position of the aryl group on
the ketone. On the other hand, an increase in substituent bulk on the acyl group gave variable
TOF values. Pinacolone is effectively hydrogenated but not 1R-(–)-fenchone (Table 4.4, entries
1–5).
Table 4.3. The hydrogenation of substituted acetophenones catalyzed by complex 15 in the
presence of KOtBu.a
O 15 cat.H2, KOtBu
25°C
RHO
RH
Entry R Conversion (%/min) TOF/h-1
1 2'-Chloro- 46/40 99/60 24002 3'-Chloro- 41/15 99/20 80503 4'-Chloro- 45/15 99/25 78004 4'-Bromo- 18/15 99/30 69705 4'-Methoxy- 42/15 99/25 5180
a Reactions were carried out in a 50 mL Parr hydrogenation reactor in THF (6 mL) at 8 bar of H2
pressure at 25°C. KOtBu was used as the base. The C/B/S ratio was 1/8/1500.
36. (a) Chantler, V. L.; Chatwin, S. L.; Jazzar, R. F. R.; Mahon, M. F.; Saker, O.; Whittlesey, M. K., Dalton Trans. 2008, 2603-2614; (b) Lee, J. P.; Ke, Z. F.; Ramirez, M. A.; Gunnoe, T. B.; Cundari, T. R.; Boyle, P. D.; Petersen, J. L., Organometallics 2009, 28, 1758-1775.37. Diaz-Valenzuela, M. B.; Phillips, S. D.; France, M. B.; Gunn, M. E.; Clarke, M. L., Chem. Eur. J. 2009, 15, 1227-1232.
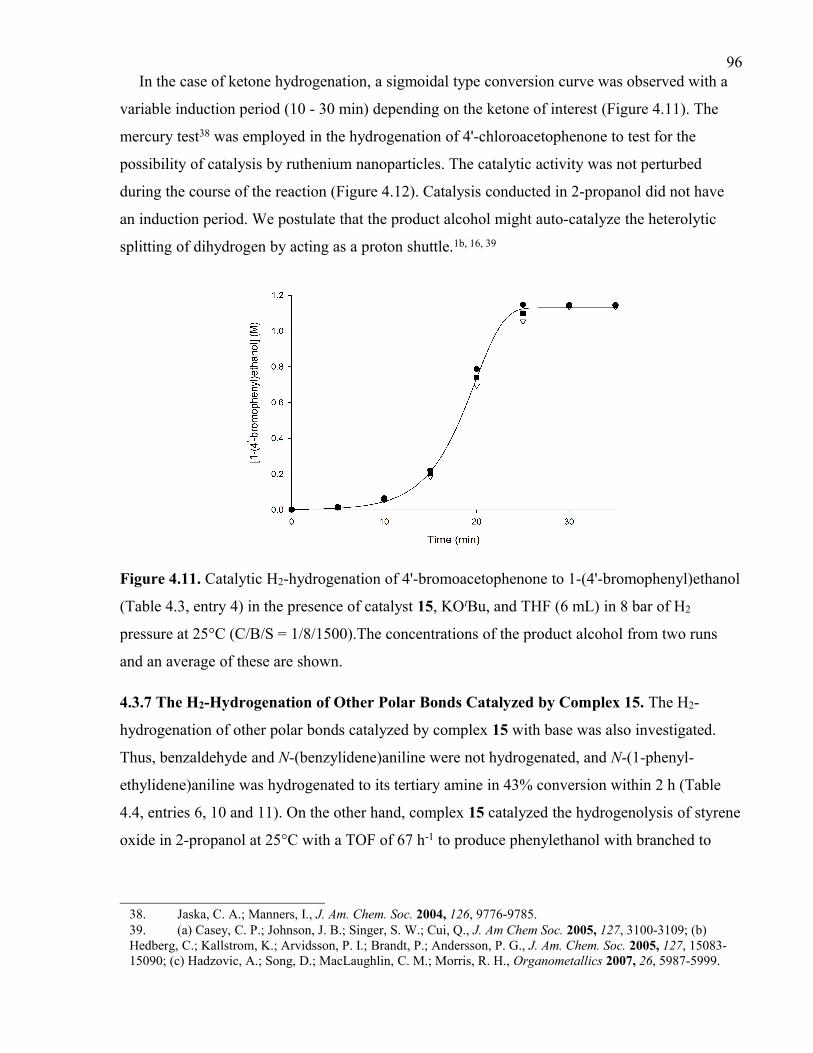
96In the case of ketone hydrogenation, a sigmoidal type conversion curve was observed with a
variable induction period (10 - 30 min) depending on the ketone of interest (Figure 4.11). The
mercury test38 was employed in the hydrogenation of 4'-chloroacetophenone to test for the
possibility of catalysis by ruthenium nanoparticles. The catalytic activity was not perturbed
during the course of the reaction (Figure 4.12). Catalysis conducted in 2-propanol did not have
an induction period. We postulate that the product alcohol might auto-catalyze the heterolytic
splitting of dihydrogen by acting as a proton shuttle.1b, 16, 39
Figure 4.11. Catalytic H2-hydrogenation of 4'-bromoacetophenone to 1-(4'-bromophenyl)ethanol
(Table 4.3, entry 4) in the presence of catalyst 15, KOtBu, and THF (6 mL) in 8 bar of H2
pressure at 25°C (C/B/S = 1/8/1500).The concentrations of the product alcohol from two runs
and an average of these are shown.
4.3.7 The H2-Hydrogenation of Other Polar Bonds Catalyzed by Complex 15. The H2-
hydrogenation of other polar bonds catalyzed by complex 15 with base was also investigated.
Thus, benzaldehyde and N-(benzylidene)aniline were not hydrogenated, and N-(1-phenyl-
ethylidene)aniline was hydrogenated to its tertiary amine in 43% conversion within 2 h (Table
4.4, entries 6, 10 and 11). On the other hand, complex 15 catalyzed the hydrogenolysis of styrene
oxide in 2-propanol at 25°C with a TOF of 67 h-1 to produce phenylethanol with branched to
38. Jaska, C. A.; Manners, I., J. Am. Chem. Soc. 2004, 126, 9776-9785.39. (a) Casey, C. P.; Johnson, J. B.; Singer, S. W.; Cui, Q., J. Am Chem Soc. 2005, 127, 3100-3109; (b) Hedberg, C.; Kallstrom, K.; Arvidsson, P. I.; Brandt, P.; Andersson, P. G., J. Am. Chem. Soc. 2005, 127, 15083-15090; (c) Hadzovic, A.; Song, D.; MacLaughlin, C. M.; Morris, R. H., Organometallics 2007, 26, 5987-5999.
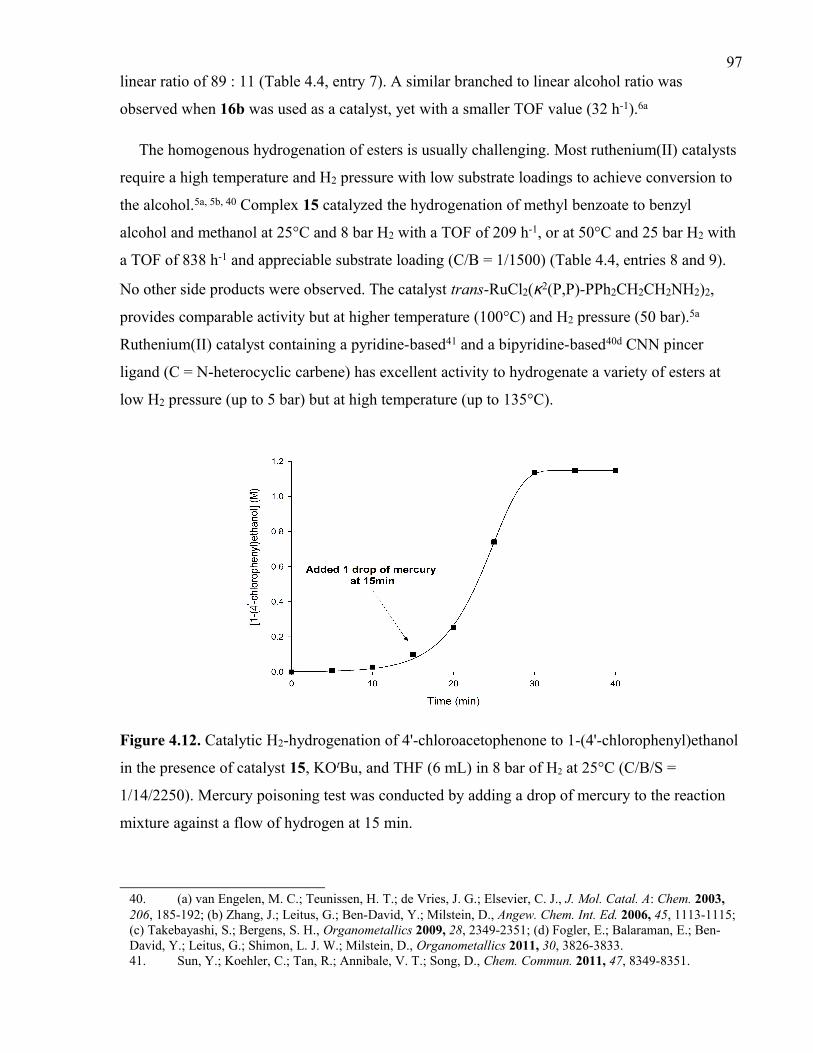
97linear ratio of 89 : 11 (Table 4.4, entry 7). A similar branched to linear alcohol ratio was
observed when 16b was used as a catalyst, yet with a smaller TOF value (32 h-1).6a
The homogenous hydrogenation of esters is usually challenging. Most ruthenium(II) catalysts
require a high temperature and H2 pressure with low substrate loadings to achieve conversion to
the alcohol.5a, 5b, 40 Complex 15 catalyzed the hydrogenation of methyl benzoate to benzyl
alcohol and methanol at 25°C and 8 bar H2 with a TOF of 209 h-1, or at 50°C and 25 bar H2 with
a TOF of 838 h-1 and appreciable substrate loading (C/B = 1/1500) (Table 4.4, entries 8 and 9).
No other side products were observed. The catalyst trans-RuCl2(κ2(P,P)-PPh2CH2CH2NH2)2,
provides comparable activity but at higher temperature (100°C) and H2 pressure (50 bar).5a
Ruthenium(II) catalyst containing a pyridine-based41 and a bipyridine-based40d CNN pincer
ligand (C = N-heterocyclic carbene) has excellent activity to hydrogenate a variety of esters at
low H2 pressure (up to 5 bar) but at high temperature (up to 135°C).
Figure 4.12. Catalytic H2-hydrogenation of 4'-chloroacetophenone to 1-(4'-chlorophenyl)ethanol
in the presence of catalyst 15, KOtBu, and THF (6 mL) in 8 bar of H2 at 25°C (C/B/S =
1/14/2250). Mercury poisoning test was conducted by adding a drop of mercury to the reaction
mixture against a flow of hydrogen at 15 min.
40. (a) van Engelen, M. C.; Teunissen, H. T.; de Vries, J. G.; Elsevier, C. J., J. Mol. Catal. A: Chem. 2003, 206, 185-192; (b) Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D., Angew. Chem. Int. Ed. 2006, 45, 1113-1115; (c) Takebayashi, S.; Bergens, S. H., Organometallics 2009, 28, 2349-2351; (d) Fogler, E.; Balaraman, E.; Ben-David, Y.; Leitus, G.; Shimon, L. J. W.; Milstein, D., Organometallics 2011, 30, 3826-3833.41. Sun, Y.; Koehler, C.; Tan, R.; Annibale, V. T.; Song, D., Chem. Commun. 2011, 47, 8349-8351.

98Table 4.4. The hydrogenation of organic molecules with polar bonds catalyzed by complex 15 in
the presence of KOtBu.a
OO O
O
H
O O
O
MeO N
h i j k
l m n
NO
g
o p
Entry Substrate Conversion (%/min) TOF/h-1
1 g 75/5 99/15 134002 h 49/60 99/120 11103 i 42/5 99/15 81004 j 58/5 99/15 104005 k 0/60 1/120 06 l 1/60 1/120 07b m 9/120 13/180e 678c n 10/120 23/180 2099c,d n 48/60 78/120 83810e o 0/60 0/120 011e p 28/60 43/120 247
a Unless otherwise stated, all reactions were carried out in a 50 mL Parr hydrogenation reactor in
THF (6 mL) at 8 bar of H2 pressure and 25°C. KOtBu was used as base. The C/B/S ratio was
1/8/1500. b 2-Propanol was used as solvent. Branched to linear alcohol ratio: 89 : 11. c Tridecane
was used as an internal standard for GC analysis. d Reaction was carried out at 25 bar of H2
pressure and 50°C. e Conversion determined by 1H NMR spectroscopy.
4.4 Conclusion
In summary, we have reported the facile synthesis of a primary amino-functionalized N-
heterocyclic carbene (C–NH2) complex by reduction under mild conditions of a nitrile-
functionalized imidazolium salt. The resulting nickel(II) complex 12 is an axially chiral square-
planar complex, and the use of Δ-TRISPHAT as a NMR chiral shift reagent is particularly useful

99to observe the diastereotopic ion pairs. A transmetalation reaction that moved the chelating C–
NH2 ligand from complex 12 to the [M(p-cymene)Cl]2 dimer (M = Ru, Os) and RuCp*(cod)Cl
afforded the first ruthenium(II) and osmium complexes 13 – 15 with such a C–NH2 ligand.
Complex 13 catalyzed the transfer hydrogenation of acetophenone in basic 2-propanol at 75°C in
3 h to give 1-phenylethanol with a TOF of up to 880 h-1. The osmium(II) analogue, complex 14,
was less active in the transfer hydrogenation of acetophenone in basic 2-propanol compared to its
ruthenium(II) counterpart. Complex 15, on the other hand, provides an active catalyst for the
hydrogenation of a variety of polar bonds under mild conditions using hydrogen gas in basic
medium.
4.5 Experimental Section
4.5.1 Synthesis. All of the preparations and manipulations, except where otherwise stated, were
carried out under a nitrogen, argon, or hydrogen atmosphere using standard Schlenk-line and
glovebox techniques. Dry and oxygen-free solvents were always used unless otherwise stated.
Methanol was stirred over magnesium turnings and iodine chips overnight under an argon
atmosphere, refluxed for 2-3 h, and distilled prior to use. The synthesis of the imidazolium salt
1a was described in Chapter 2.12 The syntheses of [Ru(p-cymene)Cl2]2,42 [Os(p-cymene)Cl2]2,27a,
43 RuCp*(cod)Cl44 and 2-(diphenylphosphino)benzylamine (P–NH2)30 were reported in the
literature. All other reagents and solvents were purchased from commercial sources and were
used as received. Deuterated solvents were purchased from Cambridge Isotope Laboratories and
Sigma Aldrich and degassed and dried over activated molecular sieves prior to use. NMR spectra
were recorded on a Varian 400 spectrometer operating at 400 MHz for 1H, 100 MHz for 13C, 161
MHz for 31P and 376 MHz for 19F. The 1H and 13C{1H} NMR were measured relative to partially
deuterated solvent peaks but are reported relative to tetramethylsilane (TMS). All 19F chemical
shifts were measured relative to trichlorofluoromethane as an external reference. All 31P
chemical shifts were measured relative to 85% phosphoric acid as an external reference. All
infrared spectra were recorded on a Nicolet 550 Magna-IR spectrometer. All UV-vis spectra
were recorded on a Hewlett-Packard Agilent 8453 UV-vis spectrophotometer. The elemental
analysis was performed at the Department of Chemistry, University of Toronto, on a Perkin- 42. Bennett, M. A.; Smith, A. K., Dalton Trans. 1974, 233-241.
43. Cabeza, J. A.; Maitlis, P. M., Dalton Trans. 1985, 573-578.44. (a) Fagan, P. J.; Ward, M. D.; Calabrese, J. C., J. Am. Chem. Soc. 1989, 111, 1698-1719; (b) Fagan, P. J.; Mahoney, W. S.; Calabrese, J. C.; Williams, I. D., Organometallics 1990, 9, 1843-1852.

100Elmer 2400 CHN elemental analyzer. Samples were handled under argon where it was
appropriate. Single-crystal X-ray diffraction data were collected using a Nonius Kappa-CCD
diffractometer with Mo Kα radiation (λ = 0.71073 Å). The CCD data were integrated and scaled
using the Denzo-SMN package. The structures were solved and refined using SHELXTL V6.1.
Refinement was by full-matrix least-squares on F2 using all data.
4.5.2 Synthesis of Bis[1-(2-aminomethylphenyl)-3-methylimidazol-2-ylidene]nickel(II)
Hexafluorophosphate ([Ni(C–NH2)2](PF6)2, 12). A Schlenk flask was charged with anhydrous
nickel(II) chloride (239 mg, 1.8 mmol) and the imidazolium salt 1a (500 mg, 1.8 mmol). A warm
methanol solution (48 mL) was added to the solid mixture under an argon or a hydrogen
atmosphere, and the solution was stirred until all the imidazolium salt dissolved. A fresh, cold
methanolic solution (12 mL) of sodium borohydride (489 mg, 12.9 mmol, previously prepared
by dissolving sodium borohydride in methanol at 0°C) was added via a syringe and a needle
slowly to the orange slurry containing anhydrous nickel(II) chloride and the imidazolium salt at
-78°C, and vigorous effervescence occurred. The dark black slurry was stirred for 1 h at -78°C,
slowly warmed to room temperature, and stirred overnight. After the reaction had gone to
completion, the solvent was removed in vacuo. The residue was extracted with commercial grade
dichloromethane (20 mL) in air and filtered through a pad of Celite. If the filtrate was not clear,
water (2 mL) was added to the dichloromethane solution and filtered through a pad of Celite
again to remove all the black residue. The organic layer was then extracted with water (5 × 8
mL). The orange aqueous solution was filtered through a plug of cotton wool, and the clear
solution was added to a saturated aqueous solution (1 mL) of ammonium hexafluorophosphate
(363 mg, 2.2 mmol). The yellow-orange precipitate was then collected, rinsed with water (5 mL),
and dried in vacuo. Yield: 200 mg, 30%. Suitable crystals for X-ray diffraction studies were
obtained by slow diffusion of diethyl ether solution into a saturated solution of 12 in acetonitrile. 1H NMR (CD3CN, δ): 7.78 (dt, JHH = 1.19, 7.77 Hz, 5-CH of Ph, 1H), 7.75 (dd, JHH = 1.73, 7.77
Hz, 6-CH of Ph, 1H), 7.69 (dt, JHH = 1.73, 7.40 Hz, 4-CH of Ph, 1H), 7.62 (dd, JHH = 1.19, 7.40
Hz, 3-CH of Ph, 1H), 7.36 (d, JHH = 1.79 Hz, 5-CH of imid., 1H), 7.21 (d, JHH = 1.79, 4-CH of
imid., 1H), 3.56 (dd, JHH = 3.89, 12.13 Hz, CH2, 1H), 3.44 (s, CH3, 3H), 2.80 (m, br, NH2, 2H),
2.53 (dt, JHH = 4.37, 12.22 Hz, CH2, 1H). 19F NMR (CD3CN, δ): -73.3 (d, JPF = 706 Hz). 13C{1H}
NMR (CD3CN, δ): 159.0 (Ni–C), 139.1 (CPh), 133.9 (CPh), 133.0 (CPh), 131.6 (CPh), 130.5(CPh),
126.5 (Cimid.), 126.2 (CPh), 125.2 (Cimid.), 44.1 (CH2), 37.8 (CH3). IR (KBr, cm-1): 3347, 3275
(ν(NH) stretch). UV-vis (acetonitrile; λmax (nm), ε (M-1cm-1)): 380, 364. MS (ESI,

101methanol/water; m/z): 431.1 ([M – H]+). Anal. Calcd for C22H26F12N6NiP2: C, 36.54; H, 3.62; N,
11.62. Found: C, 36.78; H, 3.71; N, 12.69.
4.5.3 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]chloro(η6-p-
cymene)ruthenium(II) Hexafluorophosphate ([Ru(p-cymene)(C–NH2)Cl]PF6, 13). A
Schlenk flask was charged with 12 (135 mg, 0.19 mmol) and [Ru(p-cymene)Cl2]2 dimer (114
mg, 0.19 mmol). Dry acetonitrile (16 mL) was added to the reaction mixture, and it was refluxed
under an argon atmosphere for 2.5 h to give a green solution. The solvent was then evacuated.
The residue was extracted with tetrahydrofuran or dichloromethane (8 mL) and filtered through a
pad of Celite. The volume of solvent was reduced (2 mL), and addition of diethyl ether (10 mL)
to the tetrahydrofuran or dichloromethane solution yielded a yellow precipitate, which was
collected and dried in vacuo. Yield: 135 mg, 60%. Suitable crystals for an X-ray diffraction
studies were obtained by slow evaporation of the filtrate solution in tetrahydrofuran and diethyl
ether under a nitrogen atmosphere. 1H NMR(CD2Cl2, δ): 7.69 (m, 3-CH of Ph, 1H), 7.60 (m, 4-
CH and 5-CH of Ph, 2H), 7.48 (m, 6-CH of Ph, 1H), 7.37 (d, JHH = 1.92 Hz, 5-CH of imid., 1H),
7.35 (d, JHH = 1.92 Hz, 4-CH of imid., 1H), 5.50 (d, JHH = 5.80 Hz, 2-Ar-CH of p-cymene, 1H),
5.29 (d, JHH = 5.74 Hz, 6-Ar-CH of p-cymene, 1H), 5.15 (d, JHH = 5.74 Hz, 5-Ar-CH of p-
cymene, 1H), 5.12 (m, br, NH2, 1H), 4.74 (d, JHH = 5.80 Hz, 3-Ar-CH of p-cymene, 1H), 4.07 (s,
CH3, 3H), 3.96 (m, CH2, 1H), 3.68 (m, br, NH2, 1H), 2.86 (t, JHH = 11.19 Hz, CH2, 1H), 2.55
(sept, JHH = 7.01 Hz, CH of (CH3)2CH of p-cymene, 1H), 1.67 (s, CH3 of p-cymene, 3H), 1.13
(dd, JHH = 1.29, 7.01 Hz, CH3 of (CH3)2CH of p-cymene, 1H). 19F NMR (CD2Cl2, δ): -72.3 (d,
JPF = 712 Hz). 13C{1H} NMR (CD2Cl2, δ): 175.1 (Ru–C), 138.8 (CPh), 132.8 (CPh), 131.6 (CPh),
130.4 (CPh), 130.0 (CPh), 125.9 (Cimid.), 125.6 (Cimid.), 124.5 (CPh), 111.6 (CAr-p-cymene), 101.1 (CAr-
p-cymene), 86.6 (CAr-p-cymene), 85.4 (CAr-p-cymene), 84.3 (CAr-p-cymene), 82.2 (CAr-p-cymene), 46.6 (CH2),
39.8 (CH3), 31.1 (CH of (CH3)2CH of p-cymene), 23.6 (CH3 of p-cymene), 21.0, (CH3 of
(CH3)2CH of p-cymene), 18.7 (CH3 of (CH3)2CH of p-cymene). IR(KBr, cm-1): 3326, 3279
(ν(NH) stretch). UV-vis (acetonitrile; λmax (nm), ε (M-1 cm-1)): 391, 360. MS (ESI,
methanol/water; m/z): 458.1 ([M]+). HRMS (ESI, methanol/water; m/z): calcd for
C21H27N3ClRu+ ([M]+) 458.0931, found 458.0915. Several attempts at elemental analyses failed
to give an acceptable carbon content, while hydrogen and nitrogen content are in the acceptable
range. Typical results: Anal. Calcd for C21H27F6N3ClPRu: C, 41.83; H, 4.51; N, 6.97. Found: C,
38.22; H, 4.41; N, 6.47.

1024.5.4 Synthesis of [1-(2-(Aminomethyl)phenyl)-3-methylimidazol-2-ylidene]chloro(η6-p-
cymene)osmium(II) Hexafluorophosphate ([Os(p-cymene)(C–NH2)Cl]PF6, 14). A Schlenk
flask was charged with 12 (46 mg, 0.064 mmol) and the [Os(p-cymene)Cl2]2 dimer (50 mg,
0.063 mmol). Dry acetonitrile (8 mL) was added to the reaction mixture, and it was refluxed
under an argon atmosphere for 2.5 h to give a green solution. The solvent was then evacuated.
The residue was extracted with THF (4 mL) and filtered through a pad of Celite. The volume of
solvent was reduced (2 mL), and addition of diethyl ether (12 mL) to the THF solution yielded a
pale yellow precipitate which was collected and dried in vacuo. Yield: 81 mg, 93%. Crystals
suitable for an X-ray diffraction study were obtained by slow diffusion of diethyl ether into a
saturated solution of 14 in a 1/1 acetonitrile/methanol mixture under a nitrogen atmosphere. 1H
NMR(CD2Cl2, δ): 7.70 (m, 3-CH of Ph, 1H), 7.59 (m, 4-CH and 5-CH of Ph, 2H), 7.41 (m, 6-
CH of Ph, 1H), 7.24 (d, JHH = 1.95 Hz, 5-CH of imid, 1H), 7.22 (d, JHH = 1.95, 4-CH of imid,
1H), 5.88 (m, br, NH2, 2H), 5.58 (d, JHH = 5.44 Hz, 2-Ar-CH of p-cymene, 1H), 5.51 (d, JHH =
5.51 Hz, 6-Ar-CH of p-cymene, 1H), 5.30 (d, JHH = 5.51 Hz, 5-Ar-CH of p-cymene, 1H), 4.98
(d, JHH = 5.44 Hz, 3-Ar-CH of p-cymene, 1H), 4.21 (m, CH2, 1H), 4.03 (s, CH3, 3H), 3.15 (dt,
JHH = 2.54, 12.28 Hz, CH2, 1H), 2.48 (sept, JHH = 6.88 Hz, CH of (CH3)2CH of p-cymene, 1H),
1.77 (s, CH3 of p-cymene, 3H), 1.14 (d, JHH = 6.91 Hz, CH3 of (CH3)2CH of p-cymene, 3H), 1.07
(d, JHH = 6.91 Hz, CH3 of (CH3)2CH of p-cymene, 3H). 19F NMR (CD2Cl2, δ): -72.0 (d, JPF = 712
Hz). 13C{1H} NMR (CD2Cl2, δ): 159.4 (Os–C), 138.9 (CPh), 132.8 (CPh), 131.0 (CPh), 130.5
(CPh), 130.0 (CPh), 125.6 (Cimid.), 124.9 (CPh), 123.5 (Cimid.), 102.8 (CAr-p-cymene), 92.5 (CAr-p-
cymene), 78.2 (CAr-p-cymene),76.9 (CAr-p-cymene), 75.6 (CAr-p-cymene), 73.1 (CAr-p-cymene), 47.7 (CH2), 39.8
(CH3), 31.0 (CH of (CH3)2CH of p-cymene), 23.9 (CH3 of p-cymene), 21.2, (CH3 of (CH3)2CH
of p-cymene), 18.7 (CH3 of (CH3)2CH of p-cymene). MS (ESI, methanol/water; m/z): 548.2 ([M]+). HRMS (ESI, methanol/water; m/z): calcd for C21H27N3ClOs+ ([M]+) 548.1502, found
548.1463. Anal. Calcd for C21H27ClF6N3OsP: C, 36.44; H, 3.93; N, 6.07. Found: C, 37.04; H,
3.44; N, 5.51.
4.5.5 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene](η5-pentamethyl-
cyclopentadienyl)(pyridine)ruthenium(II) Hexafluorophosphate ([RuCp*(C–NH2)(py)]PF6,
15). A Schlenk flask was charged with 12 (73 mg, 0.10 mmol) and RuCp*(cod)Cl (58 mg, 0.15
mmol). Dry acetonitrile (10 mL) was added to the reaction mixture, and it was refluxed under an
argon atmosphere for 2.5 h. The colour of the solution turned from yellow to cloudy yellow and
then to deep green. The solvent was evaporated under reduced pressure, and the residue was

103extracted with oxygen-free tetrahydrofuran (4 mL) and filtered through a pad of Celite under a
nitrogen atmosphere. To the yellow-brown solution was added pyridine (1 mL), whereupon the
colour of the solution turned into deep orange-red. The solution was evaporated under reduced
pressure, and the solid residue was extracted with ice-cold acetone (2 mL) and filtered through a
pad of Celite. Addition of diethyl ether (15 mL) to the acetone solution and slow cooling of the
solution at -25°C afforded orange-red needles, which were collected on a glass frit, washed with
diethyl ether (1 mL) and dried in vacuo. Yield: 63 mg, 64%. Suitable crystals for an X-ray
diffraction study were obtained by slow diffusion of diethyl ether into a saturated solution of 15
in acetone under a nitrogen atmosphere. 1H NMR (acetone-d6, 233K, δ): 8.49 (m, 4-CH of py,
1H), 7.88 (m, 2-CH of py, 2H), 7.77 (m, 3-CH of Ph, 1H), 7.67 (d, JHH = 1.99 Hz, 5-CH of
imid.,1H), 7.63 (m, 4-CH and 5-CH of Ph, 2H), 7.60 (d, JHH = 1.99 Hz, 4-CH of imid., 1H), 7.49
(m, 6-CH of Ph, 1H), 7.42 (m, 3-CH of py, 2H), 4.23 (t, JHH = 13.37 Hz, CH2, 1H), 3.48 (s, CH3,
3H), 3.30 (t, JHH = 9.22 Hz, CH2, 1H), 2.83 (m, br, NH2, 1H), 2.59 (m, br, NH2, 1H), 1.23 (s,
CH3 of Cp*, 15H). 19F NMR (acetone-d6, 233K, δ): -72.0 (d, JPF = 708 Hz). 13C{1H} NMR
(acetone-d6, 233K, δ): 198.0 (Ru–C), 156.0 (Cpy), 141.8 (Cpy), 136.3 (CPh), 132.9 (CPh), 132.5
(CPh), 130.0 (CPh), 128.3 (CPh), 127.1 (CPh), 126.6 (Cpy), 124.8 (Cimid.), 124.3 (Cimid.), 80.7 (CAr-
Cp*), 48.7 (CH2), 37.6 (CH3), 9.6 (CH3 of Cp*). MS (ESI, methanol/water; m/z): 456.1 ([M – py +
CH3OH]+), 424.1 ([M – py]+). Anal. Calcd for C26H33F6N4PRu: C, 48.22; H, 5.14; N, 8.65.
Found: C, 47.73; H, 4.20; N, 9.25.
4.5.6 Synthesis of [2-(Diphenylphosphino)benzylamine](η5-pentamethylcyclopentadienyl)-
(pyridine)ruthenium(II) Hexafluorophosphate ([RuCp*(P–NH2)(py)]PF6, 16a). A
scintillation vial with a threaded screw cap was charged with RuCp*(cod)Cl (40 mg, 0.11 mmol)
in dry dichloromethane (3 mL) under a nitrogen atmosphere. A solution of 2-(diphenyl-
phosphino)benzylamine (32 mg, 0.10 mmol) in dry dichloromethane (3 mL) was added to the
aforementioned yellow solution and stirred for 1 h at room temperature (25°C), whereupon the
reaction mixture turned into orange in colour. Silver hexafluorophosphate (27 mg, 0.11 mmol) in
dry acetonitrile (1 mL) was added to the reaction mixture, and a yellow-brown suspension was
obtained. After stirring the reaction mixture for 0.5 h, it was filtered through a pad of Celite
under a nitrogen atmosphere. To the yellow solution was added pyridine (1 mL), and the colour
of the solution turned into deep orange-yellow. Addition of pentane (15 mL) yielded an orange-
yellow precipitate, which was washed with pentane (3 mL) and dried in vacuo. Alternatively, the
crude product can be recrystallized with acetone and pentane or acetone and diethyl ether

104mixtures at -25°C to afford an orange-yellow solid. Yield: 53 mg, 67%. Suitable crystals for an
X-ray diffraction study were obtained by slow diffusion of diethyl ether into a saturated of 16a in
acetone under a nitrogen atmosphere. 1H NMR (CD2Cl2, 233K, δ): 7.76 (d, JHH = 5.20 Hz, 2-CH
of py, 2H), 7.56 (m, 6-CH of Ph, 1H), 7.47 (m, 5-CH of Ph, 1H), 7.45 (m, 4-CH of py, 1H), 7.42
(m, Ar-CH of PPh2, 10H), 7.19 (t, JHH = 8.32 Hz, 4-CH of Ph, 1H), 7.07 (dd, JHH = 7.44, 7.37
Hz, 3-CH of Ph, 1H), 6.93 (t, JHH = 7.40 Hz, 3-CH of py, 1H), 3.91 (m, CH2, 1H), 3.67 (m, CH2,
1H), 3.54 (m, br, NH2, 1H), 3.32 (m, br, NH2, 1H), 1.21 (s, CH3 of Cp*, 15H). 19F NMR
(CD2Cl2, 233K, δ): -72.4 (d, JPF = 712 Hz). 31P{1H} NMR (CD2Cl2, 233K, δ): 44.7 (s), -144.6
(sept, JPF = 709 Hz). 13C{1H} NMR (CD2Cl2, 233K, δ): 154.7 (Cpy), 136.0 (Cpy), 134.9 (CPh),
132.9 (t, JCP = 11.61 Hz, CPh), 130.4 (d, JCP = 8.64 Hz, CPh), 130.0 (CPh), 129.6 (d, JCP = 17.86
Hz, CPh), 128.7 (d, JCP = 9.05 Hz, CPh), 128.3 (m, CPPh), 124.8 (Cpy), 84.0 (CAr-Cp*), 46.6 (CH2),
9.0 (CH3 of Cp*). MS (ESI, methanol/water; m/z): 528.1 ([M – py]+). Anal. Calcd for
C34H38F6N2P2Ru: C, 54.33; H, 5.10; N, 3.73. Found: C, 54.00; H, 5.30; N, 3.75.
4.5.7 Catalysis. 2-Propanol that was used for all of the transfer hydrogenation runs was stirred
over magnesium turnings and iodine chips overnight under an argon atmosphere, refluxed for 2-3
h, and distilled prior to use. Oxygen-free tetrahydrofuran that was used for all of the catalytic
runs for H2-hydrogenation was stirred over sodium for 2-3 days under argon, and freshly distilled
from sodium benzophenone ketyl prior to use. Acetophenone was vacuum distilled over
phosphorus pentoxide (P2O5) and stored under nitrogen prior to use. All of the other substrates
were vacuum distilled, dried over activated molecular sieves, and stored under nitrogen prior to
use. All of the H2-hydrogenation reactions were performed at constant pressures using a stainless
steel 50 mL Parr hydrogenation reactor. The temperature was maintained at 25°C using a
constant-temperature water bath. The reactor was flushed several times with hydrogen gas at 2-4
bar prior to the addition of catalyst/substrate and base solutions.
A Perkin-Elmer Clarus 400 chromatograph equipped with a chiral column (CP chirasil-Dex
CB 25 m × 2.5 mm) with an auto-sampling capability was used for gas chromatography (GC)
analyses. Hydrogen was used as a mobile phase at a column pressure of 5 psi with a split flow
rate of 50 mL/min. The injector temperature was 250°C and the FID temperature was 275°C.
The oven temperatures and the retention times (tR, tp, /min) for all of the substrates and alcohol
products are given in the Appendix. All of the conversions are reported as an average of two GC
runs. The reported conversions were reproducible.

1054.5.8 General Procedure for Transfer Hydrogenation Studies. A solution of acetophenone
(200 mg, 1.7 mmol) in 2-propanol (6 mL) was added via a syringe and needle to a Schlenk flask
charged with a mixture of 13 (5 mg, 8.3 µmol) and potassium tert-butoxide (7 mg, 0.062 mmol)
at 75°C under an argon atmosphere. The solution became homogeneous upon stirring. Samples
were taken from the reaction mixture periodically by a syringe and needle and were quenched by
exposure to air. All samples for GC analyses were diluted to a total volume of approximately
0.75 mL using oxygenated 2-propanol.
4.5.9 General Procedure for H2-Hydrogenation Studies. In a typical run (Table 4.3, Entry 4),
the catalyst 15 (3 mg, 4.6 µmol) and 4'-bromoacetophenone (1.383 g, 6.9 mmol), and potassium
tert-butoxide (4 mg, 0.036 mmol) were dissolved in tetrahydrofuran (4 mL and 2 mL,
respectively) under a nitrogen atmosphere. The catalyst/substrate and base solutions were taken
up by means of two separate syringes and needles in a glovebox. The needles were stoppered and
the syringes were taken to the reactor. The solutions were then injected into the reactor against a
flow of hydrogen gas. The hydrogen gas was adjusted to the desired pressure. Small aliquots of
the reaction mixture were quickly withdrawn with a syringe and needle under a flow of hydrogen
at timed intervals by venting the Parr reactor at reduced pressure. Alternatively, small aliquots of
the reaction mixture were sampled from a stainless steel sampling dip tube attached to a
modified Parr reactor. The dip tube was 30 cm in length with an inner diameter of 0.01 in., and a
swing valve was attached to the end of the sampling tube. Other technical details were reported
elsewhere.45 Two small aliquots of samples were thereby withdrawn quickly at timed intervals
by opening the swing valve, and the first two aliquots were discarded. All samples for GC
analyses were diluted to a total volume of approximately 0.75 mL using oxygenated THF.
45. Zimmer-De Iuliis, M.; Morris, R. H., J. Am. Chem. Soc. 2009, 131, 11263-1126

106Chapter 5: Mechanistic Investigation of the Hydrogenation of
Ketones Catalyzed by a Ruthenium(II) Complex Featuring an N-
Heterocyclic Carbene with a Tethered Primary Amine Donor:
Evidence for an Inner Sphere Mechanism
5.1 Abstract
The complex [Ru(p-cymene)(C–NH2)Cl]PF6 (13) catalyzes the H2-hydrogenation of ketones
in basic THF under 25 bar of H2 at 50°C with a turnover frequency (TOF) of up to 461 h-1 and a
maximum conversion of 99%. When the substrate is acetophenone, the TOF decreases
significantly as the catalyst to substrate ratio is increased. The rate law is then determined to be
rate = kH[Ru]tot[H2]/(1 + Keq[ketone]), and [13] is equal to [Ru]tot if catalyst decomposition does
not occur. This is consistent with the heterolytic splitting of dihydrogen at the active ruthenium
species as the rate-determining step. In competition with this reaction is the reversible addition of
acetophenone to the active species to give an enolate complex. The transfer to the ketone of a
hydride and proton equivalent that are produced in the heterolytic splitting reaction yields the
product in a fast, low activation barrier step. The kinetic isotope effect was measured using D2
gas and acetophenone-d3, and this gave values (kH/kD) of 1.33 ± 0.15 and 1.29 ± 0.15,
respectively. The ruthenium hydride complex [Ru(p-cymene)(C–NH2)H]PF6 (17) was prepared,
as this was postulated to be a crucial intermediate in the outer-sphere bifunctional mechanism.
This is inactive under catalytic conditions unless it is activated by a base. DFT computations
suggest that the energy barriers for the addition of dihydrogen, heterolytic splitting of
dihydrogen, and concerted transfer of H+/H- to the ketone for the outer-sphere mechanism would
be respectively 18.0, 0.2, and 33.5 kcal/mol uphill at 298 K and 1 atm. On the other hand, the
energy barriers for an inner-sphere mechanism involving the decoordination of the amine group
of the NHC ligand, the heterolytic splitting of dihydrogen across a Ru–O(alkoxide) bond, and
hydride migration to the coordinated ketone, are respectively 15.5, 17.5, and 15.6 kcal/mol uphill
at 298 K and 1 atm. This is more consistent with the experimental observation that the
heterolytic splitting of dihydrogen is the turnover-limiting step. This was confirmed by showing
*Reproduced in part with permission from O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2011, 30, 1236-1252. Copyright 2011 American Chemical Society.

107that an analogous complex with a tethered tertiary amine group, [Ru(p-cymene)(C–NMe2)Cl]
PF6⋅1.5 DMSO (18), has comparable activity for the H2-hydrogenation of acetophenone.
5.2 Introduction
The hydrogenation of polar bonds using molecular hydrogen has remained by far the most
efficient process in terms of atom economy.1 However, dihydrogen is inert to reaction with most
organic substrates of interest, and so it must be activated by a transition metal complex,2 forming
metal hydride complexes, or more recently by main-group compounds such as frustrated Lewis
pairs.3 Bifunctional catalysis,4 which was originally proposed by Noyori and co-workers in the
pioneering work in the hydrogenation of polar bonds using the catalyst RuH(η6-p-cymene)((S,S)-
Tsdpen) (Tsdpen = N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine)5 and, more generally,
late transition metal catalysts containing M–H and N–H groups6 have superior activities in the
selective reduction of polar bonds to produce valuable alcohols and amines. Several studies, both
theoretical7 and experimental,8 including the study of kinetic isotope effects,9 have been
conducted to understand the mechanism of action using the “NH effect” and the bifunctional
nature of the true form of catalytically active species. The presence of the N–H group and its
relationship to the outer-sphere bifunctional10 and inner-sphere mechanisms11 have also been
studied by many research groups.
1. (a) de Vries, J. G.; Elsevier, C. J., Eds. The Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim, Germany, 2004; Vols 1-3; (b) Ito, M.; Ikariya, T., Chem. Commun. 2007, 5134-5142; (c) Kubas, G. J., Chem. Rev. 2007, 107, 4152-4205.2. (a) Jessop, P. G.; Morris, R. H., Coord. Chem. Rev. 1992, 121, 155-284; (b) Morris, R. H., Can. J. Chem. 1996, 74, 1907-1915; (c) Rosales, M., Coord. Chem. Rev. 2000, 196, 249-280; (d) DuBois, M. R.; DuBois, D. L., Chem. Soc. Rev. 2009, 38, 62-72; (e) Gloaguen, F.; Rauchfuss, T. B., Chem. Soc. Rev. 2009, 38, 100-108.3. Stephan, D. W.; Erker, G., Angew. Chem. Int. Ed. 2010, 49, 46-76.4. (a) Muniz, K., Angew. Chem. Int. Ed. 2005, 44, 6622-6627; (b) Samec, J. S. M.; Bäckvall, J. E.; Andersson, P. G.; Brandt, P., Chem. Soc. Rev. 2006, 35, 237-248; (c) Ikariya, T.; Murata, K.; Noyori, R., Org. Biomol. Chem. 2006, 4, 393-406; (d) Grützmacher, H., Angew. Chem. Int. Ed. 2008, 47, 1814-1818; (e) Ito, M.; Ikariya, T., J. Synth. Org. Chem. Jpn. 2008, 66, 1042-1048; (f) Conley, B. L.; Pennington-Boggio, M. K.; Boz, E.; Williams, T. J., Chem. Rev. 2010, 110, 2294-2312; (g) Kuwata, S.; Ikariya, T., Dalton Trans. 2010, 39, 2984-2992; (h) Grotjahn, D. B., Top. Catal. 2010, 53, 1009-1014.5. Haack, K. J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R., Angew. Chem. Int. Ed. 1997, 36, 285-288.6. (a) Noyori, R.; Yamakawa, M.; Hashiguchi, S., J. Org. Chem. 2001, 66, 7931-7944; (b) Clapham, S. E.; Hadzovic, A.; Morris, R. H., Coord. Chem. Rev. 2004, 248, 2201-2237.7. (a) Alonso, D. A.; Brandt, P.; Nordin, S. J. M.; Andersson, P. G., J. Am Chem Soc. 1999, 121, 9580-9588; (b) Yamakawa, M.; Ito, H.; Noyori, R., J. Am. Chem. Soc. 2000, 122, 1466-1478; (c) Handgraaf, J. W.; Reek, J. N. H.; Meijer, E. J., Organometallics 2003, 22, 3150-3157; (d) Hedberg, C.; Kallstrom, K.; Arvidsson, P. I.; Brandt, P.; Andersson, P. G., J. Am. Chem. Soc. 2005, 127, 15083-15090; (e) Handgraaf, J. W.; Meijer, E. J., J. Am. Chem. Soc. 2007, 129, 3099-3103; (f) Di Tommaso, D.; French, S. A.; Catlow, C. R. A., J. Mol. Struct. (THEOCHEM) 2007, 812, 39-49; (g) Puchta, R.; Dahlenburg, L.; Clark, T., Chem. Eur. J. 2008, 14, 8898-8903; (h) Zhang, H. H.; Chen, D. Z.; Zhang, Y. H.; Zhang, G. Q.; Liu, J. B., Dalton Trans. 2010, 39, 1972-1978; (i) Chen, Z.; Chen, Y.; Tang, Y. H.; Lei, M., Dalton Trans. 2010, 39, 2036-2043; (j) Lei, M.; Zhang, W. C.; Chen, Y.; Tang, Y. H., Organometallics 2010, 29, 543-548.

108The many catalyst systems that were studied by our research group12 which undergo efficient
H2-hydrogenation of ketones and imines, including those of trans-Ru(H)2((R)-BINAP)(tmen),
(OC-6-22)-Ru(H)2(PPh3)2(tmen) (tmen = 2,3-dimethylbutane-2,3-diamine), and trans-Ru-
(H)2(κ4(P,N,N,P)-P2(NH)2)12d, 13 (P2(NH)2 = tetradentate diphosphinediamine ligand), were found
to have the heterolytic splitting of the coordinated η2-H2 ligand on the active species as the rate-
determining step from various mechanistic and computational studies.12c-g These are active
catalysts, without prior activation with base, and catalyze efficiently the reduction of ketones by
H2 under mild conditions.12b-f The energy barrier calculated for the model complex (OC-6-22)-
Ru(H)2(PH3)2(en) (en = ethylenediamine) was found to be higher in the heterolytic splitting of H2
compared to the concerted transfer of H+/H- to the ketone in a six-membered-ring transition state. 6, 7a-g, 7j The corresponding coordinatively unsaturated complexes containing a ruthenium-amido
bond were isolated, and these were also found to activate dihydrogen to give the trans-dihydride
complexes.12a, 12c-f For the system trans-Ru(H)2(diamine)((R)-BINAP), Bergens and co-workers
suggested a ruthenium(II) alkoxide complex was indeed formed prior to the formation of such
amido complexes.8b, 14
Not much work has been devoted to study the mechanism of action of catalysts containing
phosphine-amine ligands (P–NH2).15 The ruthenium catalysts containing these ligands effect not
only the reduction of ketones15c, 15f but also the hydrogenation of a broad range of substrates,
including imines,15d esters,16 epoxides,15a and other polar bonds.17 All these may utilize the same
bifunctional mechanism involving the action of the M–H and N–H groups. The notion of
replacing the phosphine with an N-heterocyclic carbene (NHC) donor, in particular, a donor-
functionalized NHC, is therefore attractive to achieve the goal of greener chemistry.18 We have
previously reported that the transfer hydrogenation catalyst [Ru(p-cymene)(C–NH2)Cl]PF6 (13,
8. (a) Maire, P.; Buttner, T.; Breher, F.; Le Floch, P.; Grützmacher, H., Angew. Chem. Int. Ed. 2005, 44, 6318-6323; (b) Hamilton, R. J.; Leong, C. G.; Bigam, G.; Miskolzie, M.; Bergens, S. H., J. Am. Chem. Soc. 2005, 127, 4152-4153; (c) Sandoval, C. A.; Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Noyori, R., Chem.-Asian J. 2006, 1, 102-110; (d) Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Sandoval, C.; Noyori, R., J. Am. Chem. Soc. 2006, 128, 8724-8725; (e) Friedrich, A.; Drees, M.; auf der Gunne, J. S.; Schneider, S., J. Am. Chem. Soc. 2009, 131, 17552-17553; (f) Cheung, F. K.; Clarke, A. J.; Clarkson, G. J.; Fox, D. J.; Graham, M. A.; Lin, C. X.; Criville, A. L.; Wills, M., Dalton Trans. 2010, 39, 1395-1402.9. (a) Sandoval, C. A.; Ohkuma, T.; Muniz, K.; Noyori, R., J. Am. Chem. Soc. 2003, 125, 13490-13503; (b) Kass, M.; Friedrich, A.; Drees, M.; Schneider, S., Angew. Chem. Int. Ed. 2009, 48, 905-907; (c) Zimmer-De Iuliis, M.; Morris, R. H., J. Am. Chem. Soc. 2009, 131, 11263-11269.10. (a) Noyori, R.; Ohkuma, T., Angew. Chem. Int. Ed. 2001, 40, 40-73; (b) Ito, M.; Hirakawa, M.; Murata, K.; Ikariya, T., Organometallics 2001, 20, 379-381; (c) Ma, G. B.; McDonald, R.; Ferguson, M.; Cavell, R. G.; Patrick, B. O.; James, B. R.; Hu, T. Q., Organometallics 2007, 26, 846-854; (d) Baratta, W.; Ballico, M.; Esposito, G.; Rigo, P., Chem. Eur. J. 2008, 14, 5588-5595; (e) Sandoval, C. A.; Shi, Q. X.; Liu, S. S.; Noyori, R., Chem. Asian J. 2009, 4, 1221-1224; (f) Soni, R.; Cheung, F. K.; Clarkson, G. C.; Martins, J. E. D.; Graham, M. A.; Wills, M., Org. Biomol. Chem. 2011, 9, 3290-3294.

109see Chapter 4) is effective for the hydrogenation of acetophenone to 1-phenylethanol in basic 2-
propanol at 75°C. This system reached a maximum conversion of 96% and a turnover frequency
(TOF) of 880 h-1 (Figure 5.1).19 Here we present our study toward its H2-hydrogenation activity
in the reduction of ketones and a detailed mechanistic investigation, including kinetic studies and
theoretical computations that were performed to study the possibility of the cooperative nature of
the ligand and the metal center in this new class of ligand system.20
Figure 5.1. Transfer hydrogenation of acetophenone catalyzed by complex 13 in basic 2-
propanol.
11. (a) Standfest-Hauser, C.; Slugovc, C.; Mereiter, K.; Schmid, R.; Kirchner, K.; Xiao, L.; Weissensteiner, W., Dalton Trans. 2001, 2989-2995; (b) Leong, C. G.; Akotsi, O. M.; Ferguson, M. J.; Bergens, S. H., Chem. Commun. 2003, 750-751; (c) Lundgren, R. J.; Rankin, M. A.; McDonald, R.; Schatte, G.; Stradiotto, M., Angew. Chem. Int. Ed. 2007, 46, 4732-4735; (d) Phillips, S. D.; Fuentes, J. A.; Clarke, M. L., Chem. Eur. J. 2010, 16, 8002-8005.12. (a) Abdur-Rashid, K.; Faatz, M.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2001, 123, 7473-7474; (b) Abdur-Rashid, K.; Lough, A. J.; Morris, R. H., Organometallics 2001, 20, 1047-1049; (c) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2002, 124, 15104-15118; (d) Rautenstrauch, V.; Hoang-Cong, X.; Churlaud, R.; Abdur-Rashid, K.; Morris, R. H., Chem. Eur. J. 2003, 9, 4954-4967; (e) Abbel, R.; Abdur-Rashid, K.; Faatz, M.; Hadzovic, A.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2005, 127, 1870-1882; (f) Clapham, S. E.; Morris, R. H., Organometallics 2005, 24, 479-481; (g) Hadzovic, A.; Song, D.; MacLaughlin, C. M.; Morris, R. H., Organometallics 2007, 26, 5987-5999.13. (a) Li, T.; Churlaud, R.; Lough, A. J.; Abdur-Rashid, K.; Morris, R. H., Organometallics 2004, 23, 6239-6247; (b) Li, T.; Bergner, I.; Haque, F. N.; Iuliis, M. Z.-D.; Song, D.; Morris, R. H., Organometallics 2007, 26, 5940-5949.14. (a) Hamilton, R. J.; Bergens, S. H., J. Am. Chem. Soc. 2008, 130, 11979-11987; (b) Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H., J. Am Chem Soc. 2011, 133, 9666-9669.15. (a) Ito, M.; Hirakawa, M.; Osaku, A.; Ikariya, T., Organometallics 2003, 22, 4190-4192; (b) Dahlenburg, L.; Gotz, R., Eur. J. Inorg. Chem. 2004, 888-905; (c) Guo, R.; Lough, A. J.; Morris, R. H.; Song, D., Organometallics 2004, 23, 5524-5529; (d) Abdur-Rashid, K.; Guo, R. W.; Lough, A. J.; Morris, R. H.; Song, D. T., Adv. Synth. Catal. 2005, 347, 571-579; (e) Blaquiere, N.; Diallo-Garcia, S.; Gorelsky, S. I.; Black, D. A.; Fagnou, K., J. Am. Chem. Soc. 2008, 130, 14034-14035; (f) Jia, W. L.; Chen, X. H.; Guo, R. W.; Sui-Seng, C.; Amoroso, D.; Lough, A. J.; Abdur-Rashid, K., Dalton Trans. 2009, 8301-8307.16. (a) Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P., Angew. Chem. Int. Ed. 2007, 46, 7473-7476; (b) Kuriyama, W.; Ino, Y.; Ogata, O.; Sayo, N.; Saito, T., Adv. Synth. Catal. 2010, 352, 92-96.17. (a) Ito, M.; Sakaguchi, A.; Kobayashi, C.; Ikariya, T., J. Am. Chem. Soc. 2007, 129, 290-291; (b) Ito, M.; Koo, L. W.; Himizu, A.; Kobayashi, C.; Sakaguchi, A.; Ikariya, T., Angew. Chem. Int. Ed. 2009, 48, 1324-1327; (c) Ito, M.; Kobayashi, C.; Himizu, A.; Ikariya, T., J. Am Chem Soc. 2010, 132, 11414-11415.18. (a) Kuhl, O., Chem. Soc. Rev. 2007, 36, 592-607; (b) Lee, H. M.; Lee, C. C.; Cheng, P. Y., Curr. Org. Chem. 2007, 11, 1491-1524; (c) Normand, A. T.; Cavell, K. J., Eur. J. Inorg. Chem. 2008, 2781-2800; (d) Diez-Gonzalez, S.; Marion, N.; Nolan, S. P., Chem. Rev. 2009, 109, 3612-3676; (e) Corberan, R.; Mas-Marza, E.; Peris, E., Eur. J. Inorg. Chem. 2009, 1700-1716.
TOF up to 880 h-1
RuCl NNN
CH3
HH
HOBase, iPrOH
O
75°C
13 cat.
PF6
H
+

1105.3 Results and Discussion
5.3.1 H2-Hydrogenation of Acetophenone Catalyzed by Complex 13. The results pertaining to
the catalytic activity of complex 13 in the H2-hydrogenation of acetophenone are given in Table
5.1. Full conversion to 1-phenylethanol is achieved in 1.5 h under 25 bar of H2 in 2-propanol at
50°C with a catalyst to base to substrate (C/B/S) ratio of 1/8/200 on activation in the presence of
potassium tert-butoxide (KOtBu) as the base. This took somewhat longer (2 h) when an aprotic
solvent, tetrahydrofuran (THF), was used instead (Table 5.1, entries 1 and 2). The ruthenium(II)
complex is not a catalyst when not activated by base. It exhibited low activity when sodium tert-
butoxide, which has a low solubility in THF, was used. In addition, the ruthenium(II) complex is
not a catalyst when activated by base in the absence of H2 in THF (Table 5.1, entries 3, 4 and 8).
More importantly, an increase in substrate loading to a catalyst to substrate (C/S) ratio of 1/1200
decreased the TOF value by 3-fold, giving a value of 73 h-1 (Table 5.1, entries 6 and 7). The
catalyst [RuCp*(C–NH2)(py)]PF6 (15, Cp* = pentamethylcyclopentadienyl; py = pyridine, see
Chapter 4), which contains the same chelating primary amine-NHC ligand, tolerates high
substrate loading (C/S ratio up to 1/11500) when activated by base.21
On the other hand, the osmium(II) complex, [Os(p-cymene)(C–NH2)Cl]PF6 (14, see Chapter
4),20 when activated by base (KOtBu) in THF, gave 23% conversion to 1-phenylethanol using 25
bar of H2 as the hydrogen source (C/B/S=1/8/200) in 3 h at 50°C. It gave 99% conversion in 20 h
at 50°C if 2-propanol was used as solvent. This has lower activity in the H2-hydrogenation of
acetophenone compared to its ruthenium(II) counterpart.
19. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 6755-6761.20. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2011, 30, 1236-1252.21. O, W. W. N.; Lough, A. J.; Morris, R. H., Chem. Commun. 2010, 46, 8240-8242.
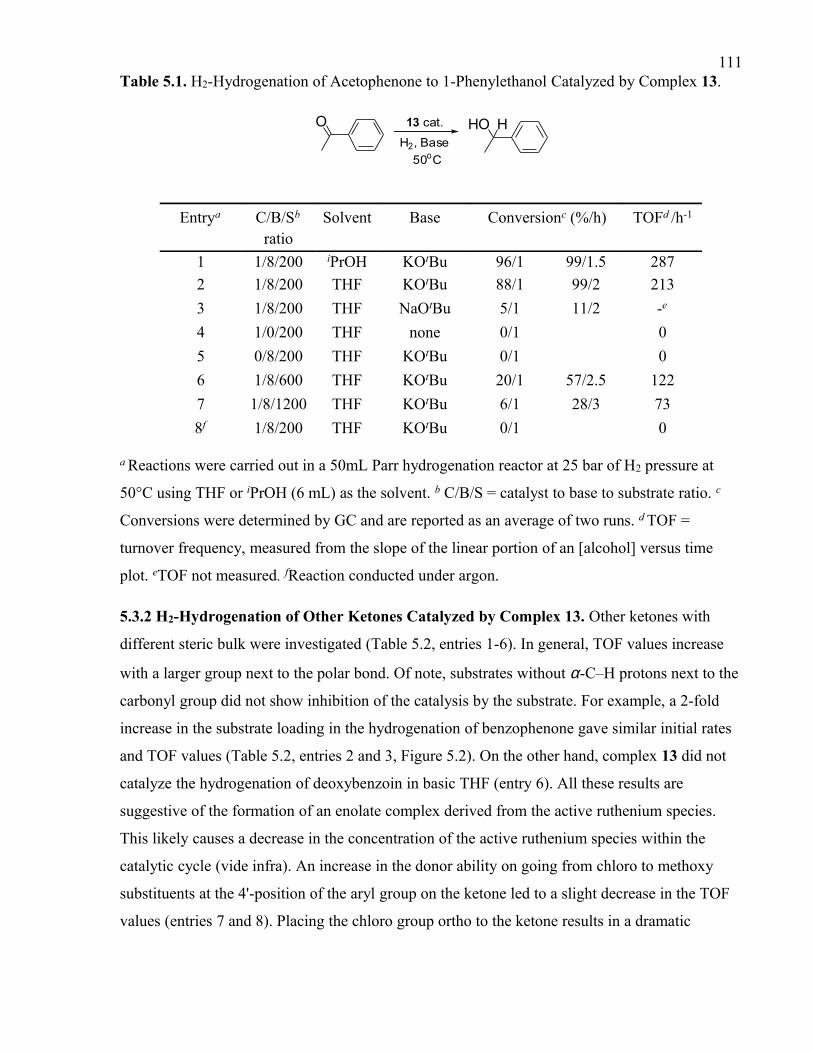
111Table 5.1. H2-Hydrogenation of Acetophenone to 1-Phenylethanol Catalyzed by Complex 13.
O 13 cat.
H2, Base50oC
HO H
Entrya C/B/Sb
ratioSolvent Base Conversionc (%/h) TOFd /h-1
1 1/8/200 iPrOH KOtBu 96/1 99/1.5 2872 1/8/200 THF KOtBu 88/1 99/2 2133 1/8/200 THF NaOtBu 5/1 11/2 -e
4 1/0/200 THF none 0/1 05 0/8/200 THF KOtBu 0/1 06 1/8/600 THF KOtBu 20/1 57/2.5 1227 1/8/1200 THF KOtBu 6/1 28/3 738f 1/8/200 THF KOtBu 0/1 0
a Reactions were carried out in a 50mL Parr hydrogenation reactor at 25 bar of H2 pressure at
50°C using THF or iPrOH (6 mL) as the solvent. b C/B/S = catalyst to base to substrate ratio. c
Conversions were determined by GC and are reported as an average of two runs. d TOF =
turnover frequency, measured from the slope of the linear portion of an [alcohol] versus time
plot. eTOF not measured. fReaction conducted under argon.
5.3.2 H2-Hydrogenation of Other Ketones Catalyzed by Complex 13. Other ketones with
different steric bulk were investigated (Table 5.2, entries 1-6). In general, TOF values increase
with a larger group next to the polar bond. Of note, substrates without α-C–H protons next to the
carbonyl group did not show inhibition of the catalysis by the substrate. For example, a 2-fold
increase in the substrate loading in the hydrogenation of benzophenone gave similar initial rates
and TOF values (Table 5.2, entries 2 and 3, Figure 5.2). On the other hand, complex 13 did not
catalyze the hydrogenation of deoxybenzoin in basic THF (entry 6). All these results are
suggestive of the formation of an enolate complex derived from the active ruthenium species.
This likely causes a decrease in the concentration of the active ruthenium species within the
catalytic cycle (vide infra). An increase in the donor ability on going from chloro to methoxy
substituents at the 4'-position of the aryl group on the ketone led to a slight decrease in the TOF
values (entries 7 and 8). Placing the chloro group ortho to the ketone results in a dramatic

112decrease in rate (entry 9). Pinacolone and benzaldehyde were also effectively hydrogenated
under similar reaction conditions (entries 10 and 11).
Table 5.2. H2-Hydrogenation of Ketones Catalyzed by Complex 13.
O O O O
OCl
OOCH3
OCl
O O
H
O
a b c d e
f g h i j
Entrya Substrate Conversionc (%/h) TOF/h-1
1 a 88/1 99/2 2132 b 78/1 98/2 2083b b 40/1 77/3 1794 c 94/1 99/1.5 4615 d 94/1 99/1.5 3776 e 0/1 0/2 07 f 94/1 99/1.5 2608 g 79/1 96/2.5 2159 h 3/1 4/1.5 510 i 74/1 99/2.5 16411 j 70/0.16 99/0.5 838
a Reactions were carried out in a 50 mL Parr hydrogenation reactor at 25 bar of H2 pressure at
50°C using THF (6 mL). KOtBu was used as base. The C/B/S ratio was 1/8/200. b The C/B/S
ratio was 1/8/400. c Conversions were determined by GC and are reported as an average of two
runs.
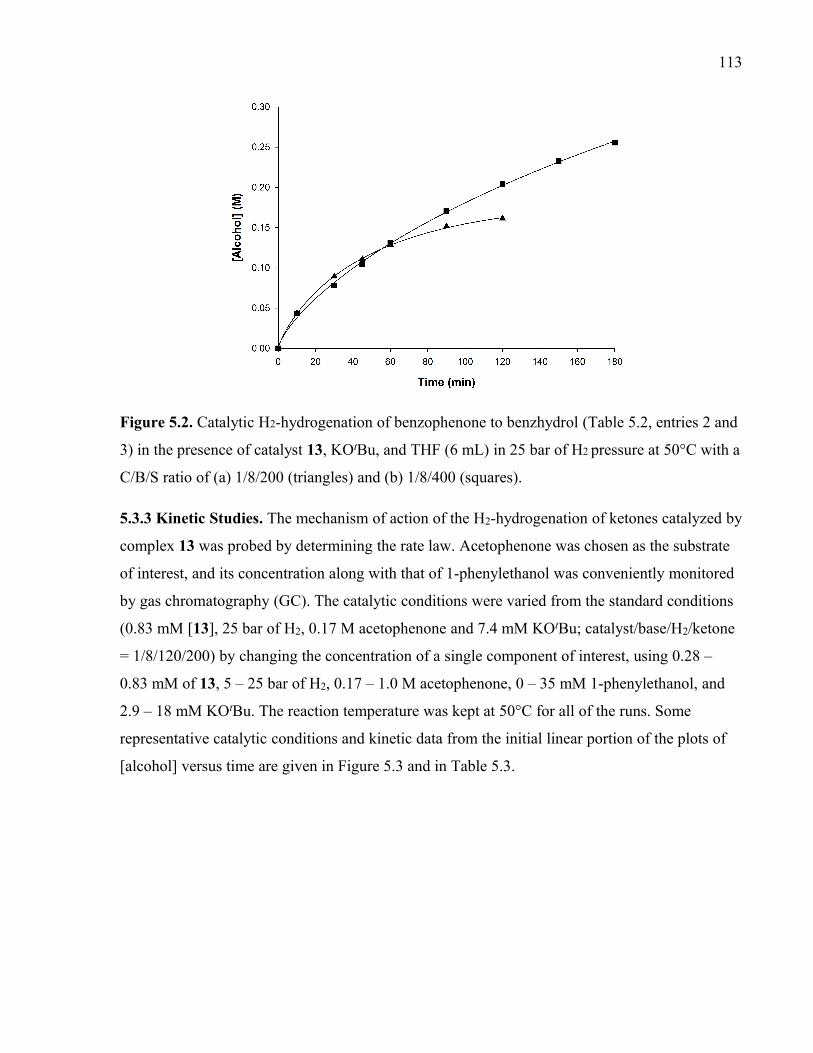
113
Figure 5.2. Catalytic H2-hydrogenation of benzophenone to benzhydrol (Table 5.2, entries 2 and
3) in the presence of catalyst 13, KOtBu, and THF (6 mL) in 25 bar of H2 pressure at 50°C with a
C/B/S ratio of (a) 1/8/200 (triangles) and (b) 1/8/400 (squares).
5.3.3 Kinetic Studies. The mechanism of action of the H2-hydrogenation of ketones catalyzed by
complex 13 was probed by determining the rate law. Acetophenone was chosen as the substrate
of interest, and its concentration along with that of 1-phenylethanol was conveniently monitored
by gas chromatography (GC). The catalytic conditions were varied from the standard conditions
(0.83 mM [13], 25 bar of H2, 0.17 M acetophenone and 7.4 mM KOtBu; catalyst/base/H2/ketone
= 1/8/120/200) by changing the concentration of a single component of interest, using 0.28 –
0.83 mM of 13, 5 – 25 bar of H2, 0.17 – 1.0 M acetophenone, 0 – 35 mM 1-phenylethanol, and
2.9 – 18 mM KOtBu. The reaction temperature was kept at 50°C for all of the runs. Some
representative catalytic conditions and kinetic data from the initial linear portion of the plots of
[alcohol] versus time are given in Figure 5.3 and in Table 5.3.
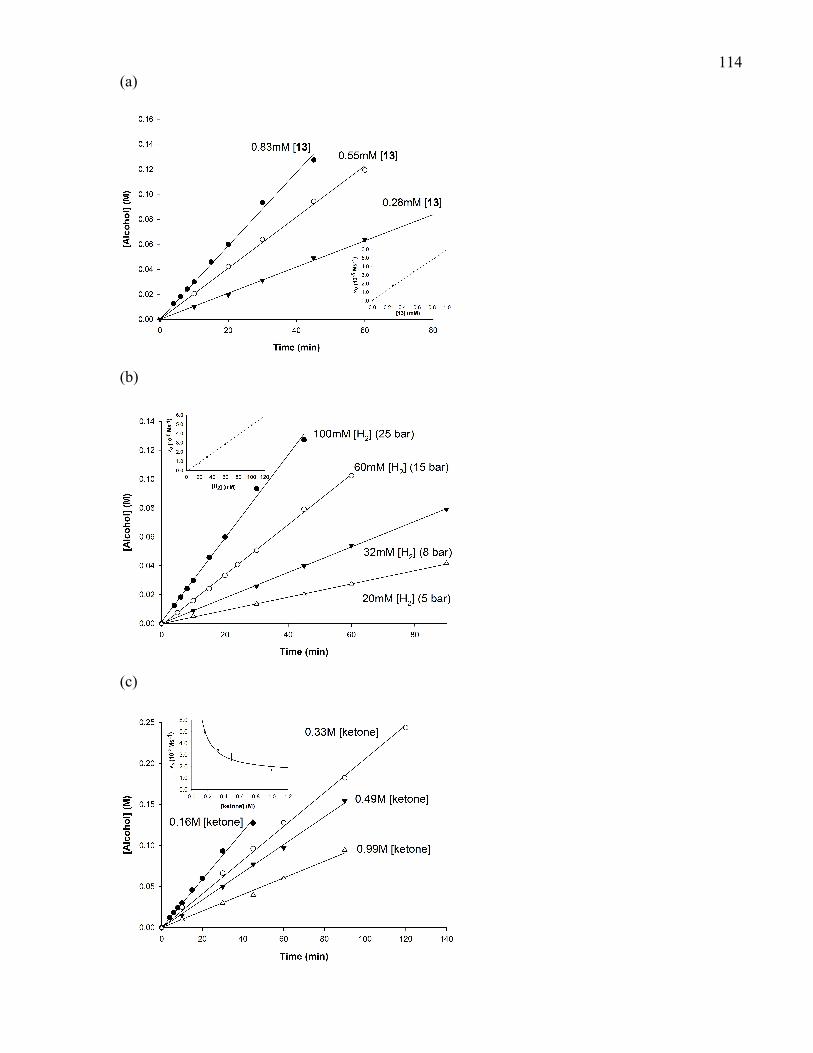
114(a)
(b)
(c)
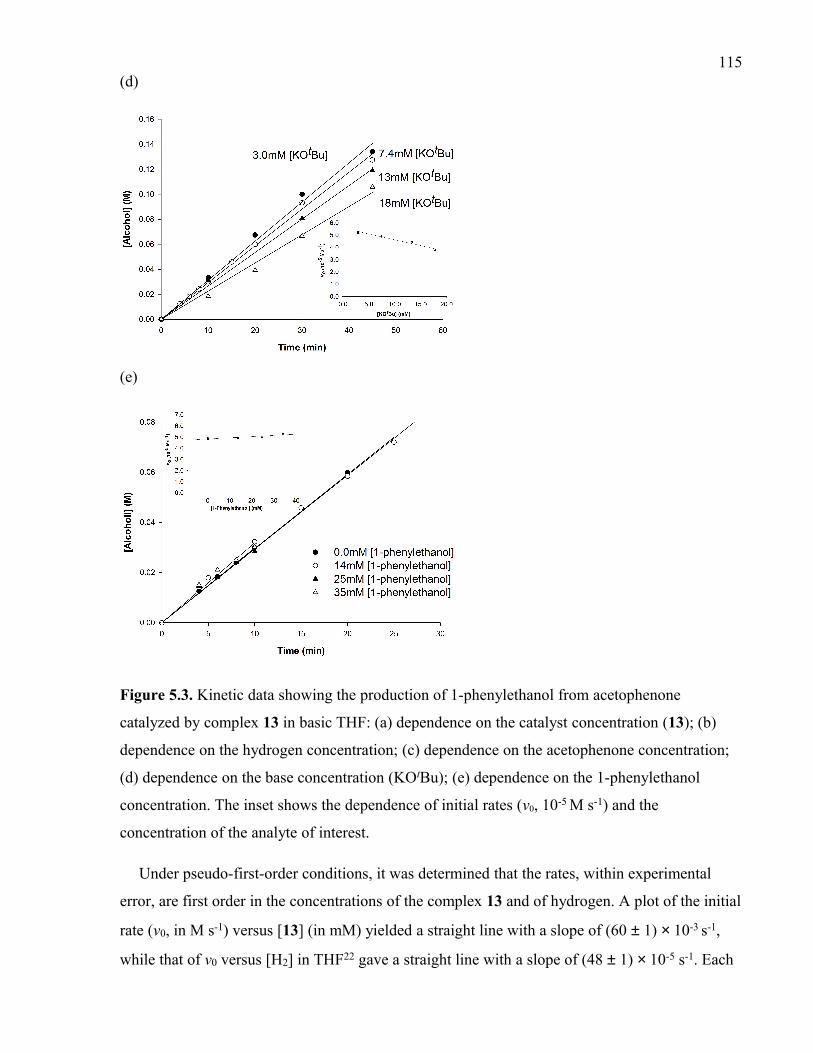
115(d)
(e)
Figure 5.3. Kinetic data showing the production of 1-phenylethanol from acetophenone
catalyzed by complex 13 in basic THF: (a) dependence on the catalyst concentration (13); (b)
dependence on the hydrogen concentration; (c) dependence on the acetophenone concentration;
(d) dependence on the base concentration (KOtBu); (e) dependence on the 1-phenylethanol
concentration. The inset shows the dependence of initial rates (v0, 10-5 M s-1) and the
concentration of the analyte of interest.
Under pseudo-first-order conditions, it was determined that the rates, within experimental
error, are first order in the concentrations of the complex 13 and of hydrogen. A plot of the initial
rate (v0, in M s-1) versus [13] (in mM) yielded a straight line with a slope of (60 ± 1) × 10-3 s-1,
while that of v0 versus [H2] in THF22 gave a straight line with a slope of (48 ± 1) × 10-5 s-1. Each

116plot passed through the origin. A plot of initial rate (in M s-1) versus [ketone] (in M) yielded a
hyperbola which does not pass through the origin (Figure 5.3) and gave a reaction order of -0.6
on the ketone. The rate law, on the other hand, is zero order in the concentrations of 1-
phenylethanol and KOtBu within the range of concentrations of interest (vide infra).
A possible general form of the course of the reaction is proposed as shown in Scheme 5.1 to
explain the kinetic data and the experimental findings. First, an active species is quickly formed
by the reaction of complex 13 with a base. This species can then either reversibly react with
acetophenone (Keq(H/D)) to produce an enolate complex or, in the rate-determining step (kH/D),
irreversibly react with hydrogen (or deuterium) gas to give a second reactive ruthenium complex
responsible for the reduction of the ketone to the product alcohol. This second complex is likely
to be a hydride (or deuteride) formed by the heterolytic splitting of dihydrogen (or dideuterium).
Similar observations were reported in the H2-hydrogenation of acetophenone using the
precatalyst trans-RuHCl-(κ4(P,N,N,P)-(S,S)-cyP2(NH)2) when activated by base under 12 bar of
H2 in 2-propanol at 20°C: a 3-fold increase in the C/S ratio to 1/12500 led to a decrease in the
initial rate by 25%.12d For the system which might undergo the outer-sphere bifunctional
mechanism, it was postulated that a reversible reaction between acetophenone and the
ruthenium-amido complex RuH(κ4(P,N,N,P)-(S,S)-cyP2(NH)-(NH2)) forms the ruthenium-ketone
adduct, and this equilibrium outcompetes the coordination of dihydrogen to ruthenium. This had
the effect of slowing catalysis, since it was proposed that the heterolytic splitting of η2-H2 by the
amido complex to afford the bifunctional dihydride catalyst trans-Ru(H)2(κ4(P,N,N,P)-(S,S)-
cyP2(NH)2) was rate-limiting. The binding of the ketone to the amido complex might also lead to
the formation of a stable enolate complex: this could occur by the deprotonation of an α-C–H
proton from the coordinated acetophenone by the ruthenium-amido complex (Scheme 5.1).
22. This was modeled using the data obtained for 1,2-dimethoxyethane at 323K. The Henry's Law constant for the solubility of H2 in THF at 50°C was therefore modeled to 3.98 × 10-2 M/bar, see: Solubility Data Series, Hydrogen and Deuterium, 5/6; Young, C. L., Ed.; Pergamon Press: New York, 1981.

117Scheme 5.1. Reaction Scheme Showing the Definitions of the Rate and Equilibrium Constants.
The rate law (eq 5.1) can be derived from this general reaction scheme (see the Appendix for
derivation):
rate =−d [ ketone]
dt=
k H[Ru ]tot [H2]1+K eq[ ketone]
(5.1)
in which [Ru]tot is the total concentration of ruthenium species, after complex 13 is activated
with base. This suggests that [13] is equal to [Ru]tot if catalyst decomposition does not occur. By
taking the reciprocal of eq 5.1, the reciprocal of the rate and the ketone concentration is related
by eq 5.2.
1rate
= 1k H[Ru]tot[H2]
+K eq[ ketone]
k H [Ru]tot [H2](5.2)
This will allow the calculation of kH and Keq values, respectively, using the slope and the y
intercept of such a linear plot. Indeed, a plot of the reciprocal of initial rate (in M-1 s) versus
[acetophenone] (in M) using the kinetic data is linear with a y intercept of (12.9 ± 0.7) × 103 M-1
s-1 and a slope of (46.6 ± 1.2) × 103 M-2 s-1 (Figure 5.4). Thus the rate and equilibrium constants
obtained using the rate law given in eq 5.1 for the range of acetophenone, complex 13, hydrogen,
1-phenylethanol and base concentrations studied are
kH = 0.94 ± 0.05 M-1 s-1
[Ru](H+/D+)
O
H H
O
O
-
[Ru]
+Keq(H/D) kH/D
+ H2/D2 [Ru](H+/D+)
H/D
RuCl NNN
CH3
HH
13
PF6 KO tBu++
Keq(H/D)=[Ru]
[Ru-enolate] [acetophenone][Ru-enolate]
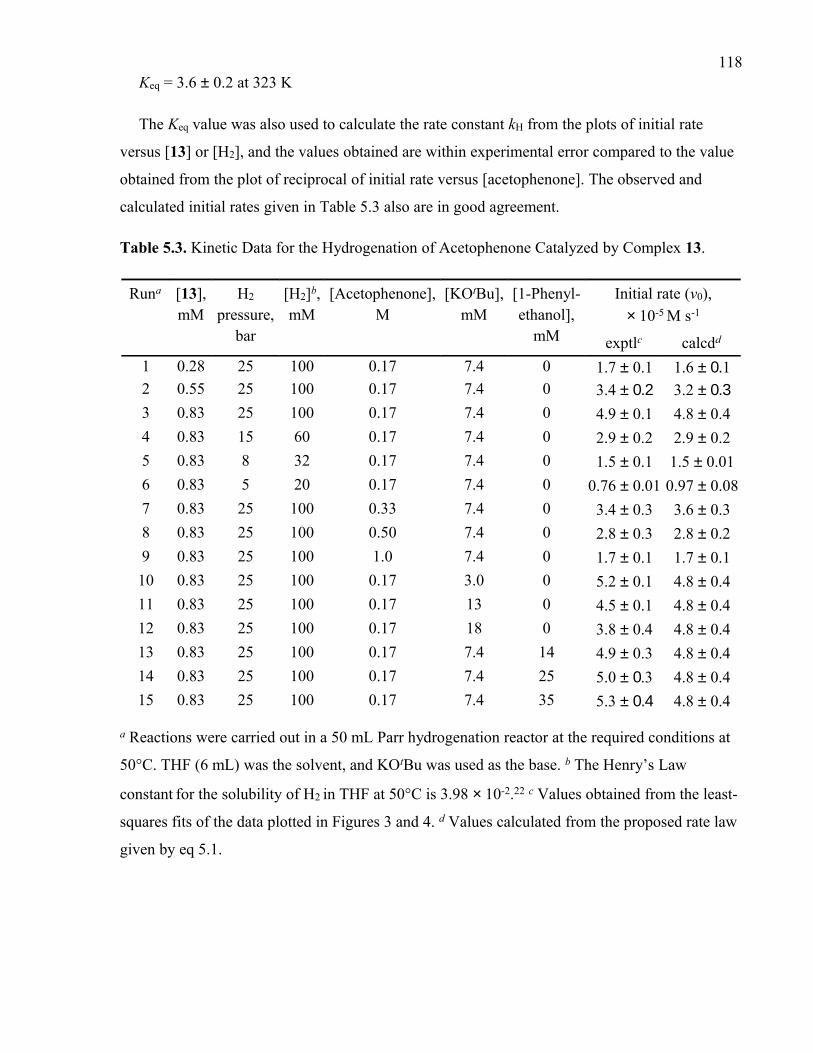
118Keq = 3.6 ± 0.2 at 323 K
The Keq value was also used to calculate the rate constant kH from the plots of initial rate
versus [13] or [H2], and the values obtained are within experimental error compared to the value
obtained from the plot of reciprocal of initial rate versus [acetophenone]. The observed and
calculated initial rates given in Table 5.3 also are in good agreement.
Table 5.3. Kinetic Data for the Hydrogenation of Acetophenone Catalyzed by Complex 13.
Runa [13], mM
H2
pressure, bar
[H2]b, mM
[Acetophenone], M
[KOtBu], mM
[1-Phenyl-ethanol],
mM
Initial rate (v0),× 10-5 M s-1
exptlc calcdd
1 0.28 25 100 0.17 7.4 0 1.7 ± 0.1 1.6 ± 0.12 0.55 25 100 0.17 7.4 0 3.4 ± 0.2 3.2 ± 0.33 0.83 25 100 0.17 7.4 0 4.9 ± 0.1 4.8 ± 0.44 0.83 15 60 0.17 7.4 0 2.9 ± 0.2 2.9 ± 0.25 0.83 8 32 0.17 7.4 0 1.5 ± 0.1 1.5 ± 0.016 0.83 5 20 0.17 7.4 0 0.76 ± 0.01 0.97 ± 0.087 0.83 25 100 0.33 7.4 0 3.4 ± 0.3 3.6 ± 0.38 0.83 25 100 0.50 7.4 0 2.8 ± 0.3 2.8 ± 0.29 0.83 25 100 1.0 7.4 0 1.7 ± 0.1 1.7 ± 0.110 0.83 25 100 0.17 3.0 0 5.2 ± 0.1 4.8 ± 0.411 0.83 25 100 0.17 13 0 4.5 ± 0.1 4.8 ± 0.412 0.83 25 100 0.17 18 0 3.8 ± 0.4 4.8 ± 0.413 0.83 25 100 0.17 7.4 14 4.9 ± 0.3 4.8 ± 0.414 0.83 25 100 0.17 7.4 25 5.0 ± 0.3 4.8 ± 0.415 0.83 25 100 0.17 7.4 35 5.3 ± 0.4 4.8 ± 0.4
a Reactions were carried out in a 50 mL Parr hydrogenation reactor at the required conditions at
50°C. THF (6 mL) was the solvent, and KOtBu was used as the base. b The Henry’s Law
constant for the solubility of H2 in THF at 50°C is 3.98 × 10-2.22 c Values obtained from the least-
squares fits of the data plotted in Figures 3 and 4. d Values calculated from the proposed rate law
given by eq 5.1.
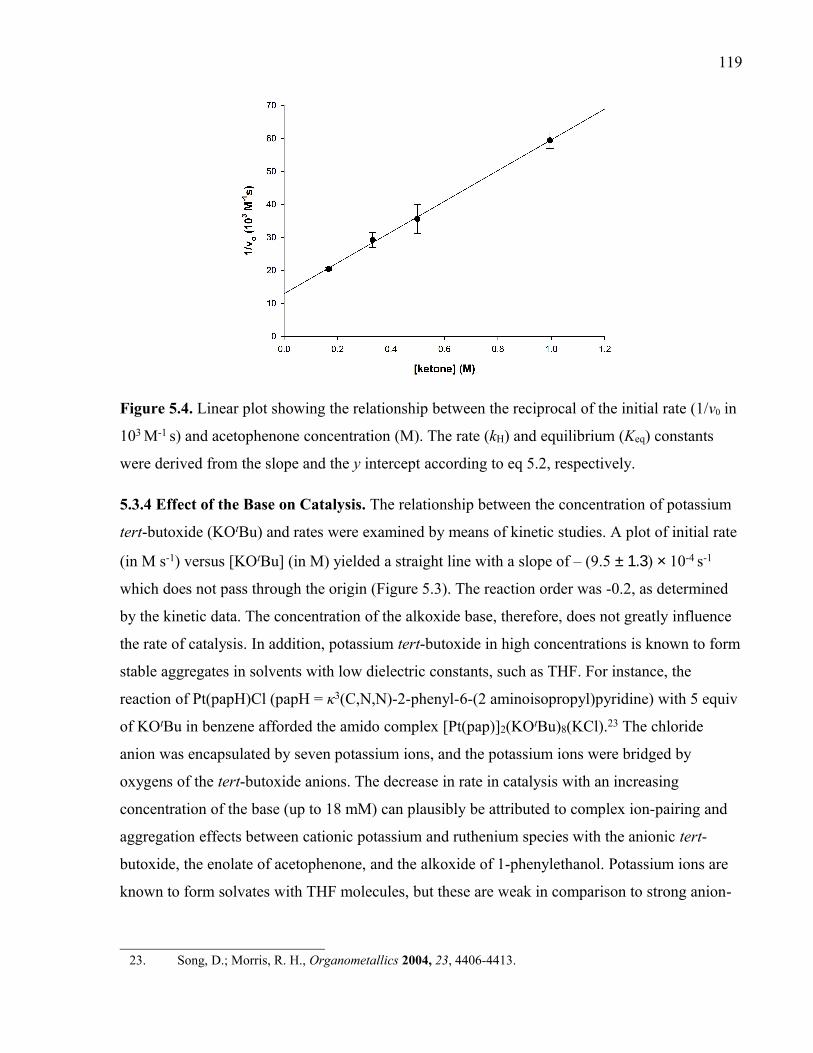
119
Figure 5.4. Linear plot showing the relationship between the reciprocal of the initial rate (1/v0 in
103 M-1 s) and acetophenone concentration (M). The rate (kH) and equilibrium (Keq) constants
were derived from the slope and the y intercept according to eq 5.2, respectively.
5.3.4 Effect of the Base on Catalysis. The relationship between the concentration of potassium
tert-butoxide (KOtBu) and rates were examined by means of kinetic studies. A plot of initial rate
(in M s-1) versus [KOtBu] (in M) yielded a straight line with a slope of – (9.5 ± 1.3) × 10-4 s-1
which does not pass through the origin (Figure 5.3). The reaction order was -0.2, as determined
by the kinetic data. The concentration of the alkoxide base, therefore, does not greatly influence
the rate of catalysis. In addition, potassium tert-butoxide in high concentrations is known to form
stable aggregates in solvents with low dielectric constants, such as THF. For instance, the
reaction of Pt(papH)Cl (papH = κ3(C,N,N)-2-phenyl-6-(2 aminoisopropyl)pyridine) with 5 equiv
of KOtBu in benzene afforded the amido complex [Pt(pap)]2(KOtBu)8(KCl).23 The chloride
anion was encapsulated by seven potassium ions, and the potassium ions were bridged by
oxygens of the tert-butoxide anions. The decrease in rate in catalysis with an increasing
concentration of the base (up to 18 mM) can plausibly be attributed to complex ion-pairing and
aggregation effects between cationic potassium and ruthenium species with the anionic tert-
butoxide, the enolate of acetophenone, and the alkoxide of 1-phenylethanol. Potassium ions are
known to form solvates with THF molecules, but these are weak in comparison to strong anion-
23. Song, D.; Morris, R. H., Organometallics 2004, 23, 4406-4413.

120cation ion pairing interactions. On the other hand, a decrease in rate was also observed when a
low concentration of base (1.2 mM) was used in catalysis (C/B/S = 1/1.4/200).
It has been shown that for certain transfer24 and H2-hydrogenation25 systems, particular alkali-
metal cations can accelerate the catalysis by acting as a Lewis acid. This possibility was
examined by our system. The addition of [2.2.2]cryptand in equimolar amount to potassium ions
(C/B/S = 1/1.4/200) led to comparable reaction rates with respect to the standard condition for
catalysis (C/B/S = 1/8/200). An equimolar amount of 18-crown-6 that was added with respect to
base (C/B/S=1/8/200, [KOtBu] = 7.4 mM) had no effect on the reaction rate. The use of a base
with sodium ions (NaOtBu, Table 5.1, entry 3) gave low activity in catalysis. This, however, was
attributed to the limited solubility of the base in THF solution. Therefore, the cations of the
alkoxide base do not significantly affect the rate of catalysis.12d
5.3.5 Effect of Alcohols on Catalysis. A plot of initial rate (in M s-1) versus [1-phenylethanol]
(in M) added in excess obtained in kinetic studies yielded a straight line with a slope of (1.1
± 0.5) × 10-4 s-1 which does not pass through the origin (Figure 5.3). The reaction order was -0.1,
as determined by the kinetic data. The product alcohol and 2-propanol, therefore, have minimal
effect on catalysis and do not contribute to the rate law. Of note, the rate of conversion was
somewhat higher using 2-propanol (Table 5.1), due to the combined activities of both H2 and
transfer hydrogenation.19
5.3.6 Isotope Effects and Deuterium Labelling Studies. The measurement of kinetic isotope
effects (KIE)26 is a powerful technique to gain important mechanistic insight in catalysis. The
use of both hydrogen and deuterium gas in the measurement of the kinetic isotope effect for
hydrogenation catalysts is common,9b, 27 but it is less well known for homogeneous bifunctional
catalysts for the H2-hydrogenation of polar bonds.9a, 9c Noyori and co-workers have reported a
KIE value (kH/kD) of 2 for the hydrogenation of acetophenone catalyzed by trans-RuH(η1-BH4)
((S)-tolBINAP)((S,S)-dpen) in the presence of base in H2/2-propanol versus D2/2-propanol-d8.9a
We have recently reported a KIE value of 2.0 ± 0.1 for the hydrogenation of acetophenone
24. (a) Vastila, P.; Zaitsev, A. B.; Wettergren, J.; Privalov, T.; Adolfsson, H., Chem. Eur. J. 2006, 12, 3218-3225; (b) Wettergren, J.; Buitrago, E.; Ryberg, P.; Adolfsson, H., Chem. Eur. J. 2009, 15, 5709-5718.25. Hartmann, R.; Chen, P., Angew. Chem. Int. Ed. 2001, 40, 3581-3585.26. Westheimer, F. H., Chem. Rev. 1961, 61, 265-273.27. (a) Chock, P. B.; Halpern, J., J. Am. Chem. Soc. 1966, 88, 3511- 3514; (b) Brown, J. M.; Parker, D., Organometallics 1982, 1, 950-956; (c) Kitamura, M.; Tsukamoto, M.; Bessho, Y.; Yoshimura, M.; Kobs, U.; Widhalm, M.; Noyori, R., J. Am. Chem. Soc. 2002, 124, 6649-6667; (d) Imamoto, T.; Itoh, T.; Yoshida, K.; Gridnev, I. D., Chem. Asian J. 2008, 3, 1636-1641.

121catalyzed by trans-Ru(H)2((R)-BINAP)(tmen). This was interpreted as an early transition state
involving the heterolytic splitting of dihydrogen (dideuterium) by the ruthenium catalyst where
there is little weakening of the H–H/D–D bond.9c
The rates of the reaction using D2 gas were examined within a given range of acetophenone
concentrations (0.083 – 0.16 M) at low D2 gas pressure (8 bar)28 and 50°C in basic THF to obtain
meaningful kinetic data. The kD and Keq(D) values were calculated from the slope and y intercept
of a plot of the reciprocal of the initial rate (in M-1 s) and [acetophenone] (in M) as in eq 5.2.
This gave a linear plot with a y intercept of (51 ± 5) × 103 M-1 s-1 and a slope of (15 ± 4) × 104
M-2 s-1 The catalysis conditions along with the results of the kinetic data and calculated values of
kD and Keq(D) are given in Figures 5.5 and 5.6, and in Tables 5.4 and 5.5. The experimentally
determined kinetic isotope effect value (kH/kD), within experimental error, is 1.33 ± 0.15 for
using H2 versus D2 gas.
Since the presence of α-C–H groups adjacent to the carbonyl group of a ketone was shown to
inhibit catalysis by allowing enolate formation, it was important to determine the effect of
deuteration of these positions in acetophenone. Kinetic studies were therefore performed using
acetophenone-d3 (0.16 – 0.49 M) at 25 bar of H2 and 50°C in basic THF. The kD and Keq(D)
values were computed in a fashion similar to that described above by first plotting the reciprocal
of initial rate (in M-1 s) versus [acetophenone-d3] (in M, Figure 5.6) to yield a y intercept of (17
± 1) × 103 M-1 s-1 and a slope of (22 ± 3) × 103 M-2 s-1. The catalysis conditions, results of the
kinetic data, and calculated values of kD and Keq(D) are given in Figures 5.5 and 5.6, and in Tables
5.4 and 5.5.
28. Deuterium gas is 1.046 times more soluble than hydrogen gas in water at 50°C; see Muccitelli, J.; Wen, W. Y., J. Solut. Chem. 1978, 7, 257-267.
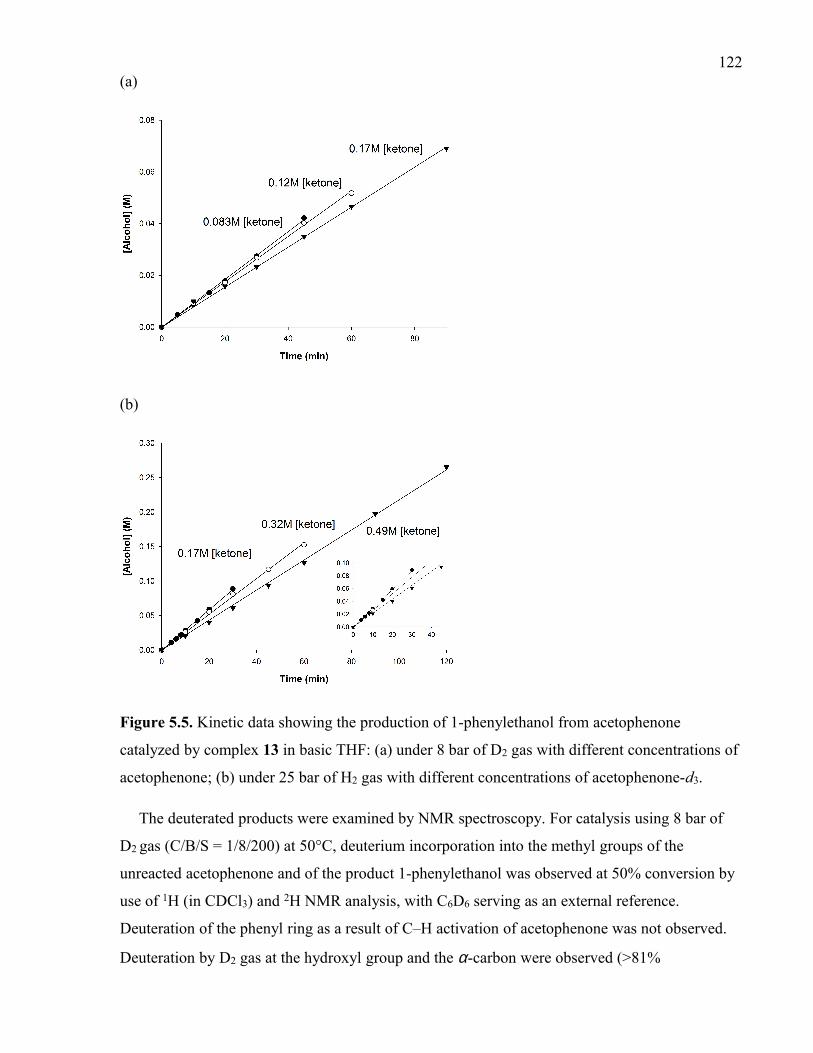
122(a)
(b)
Figure 5.5. Kinetic data showing the production of 1-phenylethanol from acetophenone
catalyzed by complex 13 in basic THF: (a) under 8 bar of D2 gas with different concentrations of
acetophenone; (b) under 25 bar of H2 gas with different concentrations of acetophenone-d3.
The deuterated products were examined by NMR spectroscopy. For catalysis using 8 bar of
D2 gas (C/B/S = 1/8/200) at 50°C, deuterium incorporation into the methyl groups of the
unreacted acetophenone and of the product 1-phenylethanol was observed at 50% conversion by
use of 1H (in CDCl3) and 2H NMR analysis, with C6D6 serving as an external reference.
Deuteration of the phenyl ring as a result of C–H activation of acetophenone was not observed.
Deuteration by D2 gas at the hydroxyl group and the α-carbon were observed (>81%
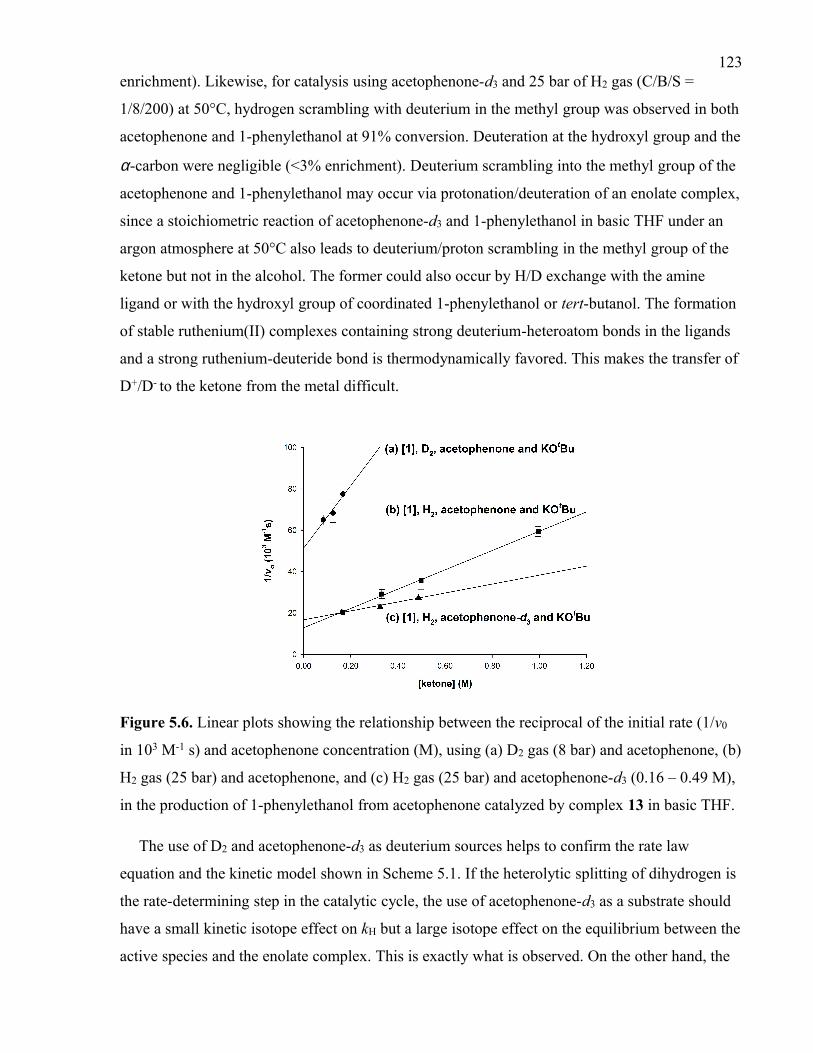
123enrichment). Likewise, for catalysis using acetophenone-d3 and 25 bar of H2 gas (C/B/S =
1/8/200) at 50°C, hydrogen scrambling with deuterium in the methyl group was observed in both
acetophenone and 1-phenylethanol at 91% conversion. Deuteration at the hydroxyl group and the
α-carbon were negligible (<3% enrichment). Deuterium scrambling into the methyl group of the
acetophenone and 1-phenylethanol may occur via protonation/deuteration of an enolate complex,
since a stoichiometric reaction of acetophenone-d3 and 1-phenylethanol in basic THF under an
argon atmosphere at 50°C also leads to deuterium/proton scrambling in the methyl group of the
ketone but not in the alcohol. The former could also occur by H/D exchange with the amine
ligand or with the hydroxyl group of coordinated 1-phenylethanol or tert-butanol. The formation
of stable ruthenium(II) complexes containing strong deuterium-heteroatom bonds in the ligands
and a strong ruthenium-deuteride bond is thermodynamically favored. This makes the transfer of
D+/D- to the ketone from the metal difficult.
Figure 5.6. Linear plots showing the relationship between the reciprocal of the initial rate (1/v0
in 103 M-1 s) and acetophenone concentration (M), using (a) D2 gas (8 bar) and acetophenone, (b)
H2 gas (25 bar) and acetophenone, and (c) H2 gas (25 bar) and acetophenone-d3 (0.16 – 0.49 M),
in the production of 1-phenylethanol from acetophenone catalyzed by complex 13 in basic THF.
The use of D2 and acetophenone-d3 as deuterium sources helps to confirm the rate law
equation and the kinetic model shown in Scheme 5.1. If the heterolytic splitting of dihydrogen is
the rate-determining step in the catalytic cycle, the use of acetophenone-d3 as a substrate should
have a small kinetic isotope effect on kH but a large isotope effect on the equilibrium between the
active species and the enolate complex. This is exactly what is observed. On the other hand, the
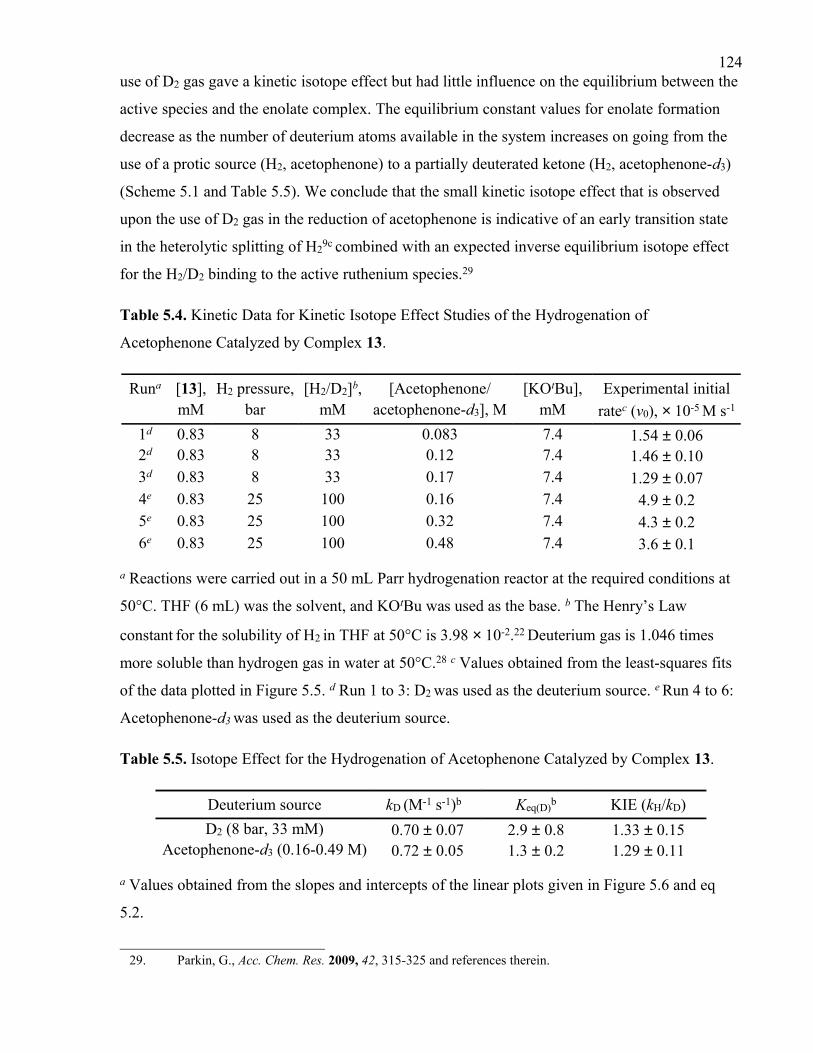
124use of D2 gas gave a kinetic isotope effect but had little influence on the equilibrium between the
active species and the enolate complex. The equilibrium constant values for enolate formation
decrease as the number of deuterium atoms available in the system increases on going from the
use of a protic source (H2, acetophenone) to a partially deuterated ketone (H2, acetophenone-d3)
(Scheme 5.1 and Table 5.5). We conclude that the small kinetic isotope effect that is observed
upon the use of D2 gas in the reduction of acetophenone is indicative of an early transition state
in the heterolytic splitting of H29c combined with an expected inverse equilibrium isotope effect
for the H2/D2 binding to the active ruthenium species.29
Table 5.4. Kinetic Data for Kinetic Isotope Effect Studies of the Hydrogenation of
Acetophenone Catalyzed by Complex 13.
Runa [13], mM
H2 pressure, bar
[H2/D2]b, mM
[Acetophenone/ acetophenone-d3], M
[KOtBu], mM
Experimental initial ratec (v0), × 10-5 M s-1
1d 0.83 8 33 0.083 7.4 1.54 ± 0.062d 0.83 8 33 0.12 7.4 1.46 ± 0.103d 0.83 8 33 0.17 7.4 1.29 ± 0.074e 0.83 25 100 0.16 7.4 4.9 ± 0.25e 0.83 25 100 0.32 7.4 4.3 ± 0.26e 0.83 25 100 0.48 7.4 3.6 ± 0.1
a Reactions were carried out in a 50 mL Parr hydrogenation reactor at the required conditions at
50°C. THF (6 mL) was the solvent, and KOtBu was used as the base. b The Henry’s Law
constant for the solubility of H2 in THF at 50°C is 3.98 × 10-2.22 Deuterium gas is 1.046 times
more soluble than hydrogen gas in water at 50°C.28 c Values obtained from the least-squares fits
of the data plotted in Figure 5.5. d Run 1 to 3: D2 was used as the deuterium source. e Run 4 to 6:
Acetophenone-d3 was used as the deuterium source.
Table 5.5. Isotope Effect for the Hydrogenation of Acetophenone Catalyzed by Complex 13.
Deuterium source kD (M-1 s-1)b Keq(D)b KIE (kH/kD)D2 (8 bar, 33 mM) 0.70 ± 0.07 2.9 ± 0.8 1.33 ± 0.15
Acetophenone-d3 (0.16-0.49 M) 0.72 ± 0.05 1.3 ± 0.2 1.29 ± 0.11
a Values obtained from the slopes and intercepts of the linear plots given in Figure 5.6 and eq
5.2.
29. Parkin, G., Acc. Chem. Res. 2009, 42, 315-325 and references therein.

1255.3.7 The Disfavored Outer-Sphere Bifunctional Mechanism. Two mechanistic proposals
should be considered in light of the experimental findings described above. While the outer-
sphere bifunctional mechanism6b is expected, there is stronger evidence for an inner-sphere
mechanism as described below. In an outer-sphere mechanism which would involve the
bifunctional nature of a primary amine group (NH2) and the metal-hydride bond (Ru–H), the
activation of complex 13 with base should give a metal-amido complex containing a trigonal-
planar nitrogen donor atom (Scheme 5.2). Coordination of dihydrogen to such a complex and
subsequent heterolytic splitting of the molecule should give the bifunctional metal-hydride and
protic amine grouping. The hydride and the proton equivalent would then transfer to the ketone
in the outer coordination sphere, giving the product alcohol, without coordination to the metal
center. In this mechanism the observed inhibition by enolizable ketones would be explained by
the reaction of the ketone with the metal-amido complex.12d Deprotonation of the methyl group
should afford the enolate complex with the primary amine group restored.
Scheme 5.2. Possible Outer-Sphere Mechanism That Involves Bifunctional Catalysis in the
Hydrogenation of Acetophenone.
Ru NNN
CH3
H
RuH NNN
CH3
HH
O
RuH NNN
CH3
HH
17
Ru NNN
CH3
H
HH
Ru NNN
CH3
H
OH
HH
O
O
-
KOtBu - KCl- HOtBu
RuCl NNN
CH3
HH
13
PF6+
H2
- H2
OOH
H
+
+
+
+
+

126In order to identify reaction intermediates in the outer-sphere mechanism, reactions of excess
or stoichiometric amounts of base (KOtBu, KH, NaBH4, or K-Selectride) with complex 13 in
THF were tried, but these gave multiple unidentifiable products. Attempts to isolate such
products failed and instead led to instant decomposition. Attempts to prepare the ruthenium-
hydride complex 17 ([Ru(p-cymene)(C–NH2)H]PF6, Scheme 5.2) by using dihydrogen (up to 5
bar) in excess KOtBu in stirring THF at 25 or 50°C also failed. However, complex 17 could be
prepared by warming complex 13 and 3 equiv of sodium 2-propoxide in 2-propanol solution at
50°C for 3 h, giving a brown solid upon isolation in 57% yield (Scheme 5.3). The use of KOtBu
or a reaction that was conducted in refluxing 2-propanol gave impure products. Complex 17 is
oxygen sensitive both in solution and in the solid state. A solution of 17 in dichloromethane
readily converts to the cation of complex 13 at room temperature in hours and slowly reacts with
solvents such as acetonitrile after prolonged standing for days.
Scheme 5.3. Synthesis of Complex 17 from Reaction of Complex 13 and Basic 2-Propanol.
Complex 17 was unambiguously characterized by an X-ray diffraction study (Figure 5.7). The
compound adopts a piano-stool geometry about the metal center. The Ru–Ccarbene bond (2.029(7)
Å) was shorter than that of complex 13 (2.092(5)Å).19 The large bite angle between the carbene
and primary amine donor containing the metal center (C(1)-Ru(1)-N(3) = 91.9(2)°) is typical of
L–Ru–L angles for ruthenium arene complexes [Ru(η6-arene)HL2]+ containing a hydride
ligand.5, 11c, 30 Note that the contact distance of 2.45 Å between the ruthenium hydride and the
primary amine protons (Ru–H⋅⋅⋅H–N–) is comparable to that for the complex RuH(η6-p-cymene)
((S,S)-Tsdpen) (contact distance 2.29 Å) reported by Noyori and co-workers.5 In solution, the
Ru–Ccarbene resonance was observed at 185.3 ppm in the 13C{1H} NMR spectrum. The Ru–H
resonance was observed at -7.82 ppm in the 1H NMR spectrum in CD3CN. This lies in the region
for analogous ruthenium-hydride complexes of the type [Ru(η6-arene)HL2]+.5, 11c, 30
30. Chaplin, A. B.; Dyson, P. J., Organometallics 2007, 26, 4357-4360.
RuCl NNN
CH3
HH
3 NaOiPr RuH NNN
CH3
HH
PF6 PF6
13 17
+ +iPrOH, 50°C
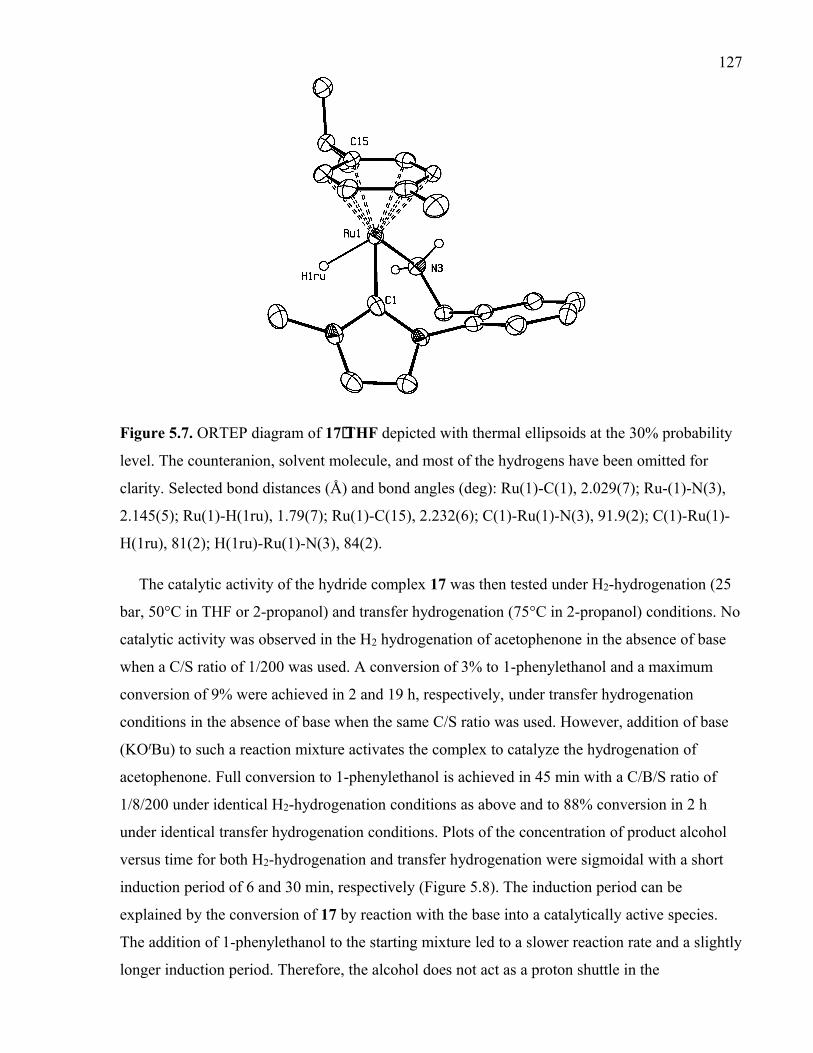
127
Figure 5.7. ORTEP diagram of 17⋅THF depicted with thermal ellipsoids at the 30% probability
level. The counteranion, solvent molecule, and most of the hydrogens have been omitted for
clarity. Selected bond distances (Å) and bond angles (deg): Ru(1)-C(1), 2.029(7); Ru-(1)-N(3),
2.145(5); Ru(1)-H(1ru), 1.79(7); Ru(1)-C(15), 2.232(6); C(1)-Ru(1)-N(3), 91.9(2); C(1)-Ru(1)-
H(1ru), 81(2); H(1ru)-Ru(1)-N(3), 84(2).
The catalytic activity of the hydride complex 17 was then tested under H2-hydrogenation (25
bar, 50°C in THF or 2-propanol) and transfer hydrogenation (75°C in 2-propanol) conditions. No
catalytic activity was observed in the H2 hydrogenation of acetophenone in the absence of base
when a C/S ratio of 1/200 was used. A conversion of 3% to 1-phenylethanol and a maximum
conversion of 9% were achieved in 2 and 19 h, respectively, under transfer hydrogenation
conditions in the absence of base when the same C/S ratio was used. However, addition of base
(KOtBu) to such a reaction mixture activates the complex to catalyze the hydrogenation of
acetophenone. Full conversion to 1-phenylethanol is achieved in 45 min with a C/B/S ratio of
1/8/200 under identical H2-hydrogenation conditions as above and to 88% conversion in 2 h
under identical transfer hydrogenation conditions. Plots of the concentration of product alcohol
versus time for both H2-hydrogenation and transfer hydrogenation were sigmoidal with a short
induction period of 6 and 30 min, respectively (Figure 5.8). The induction period can be
explained by the conversion of 17 by reaction with the base into a catalytically active species.
The addition of 1-phenylethanol to the starting mixture led to a slower reaction rate and a slightly
longer induction period. Therefore, the alcohol does not act as a proton shuttle in the
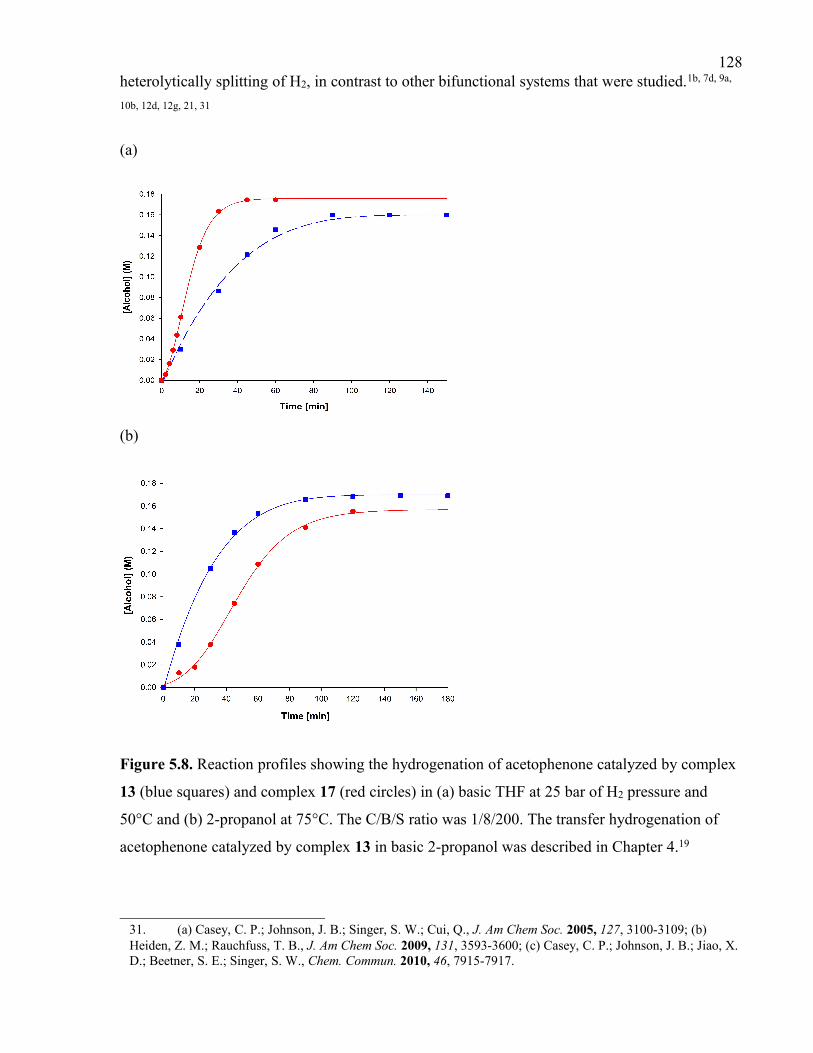
128heterolytically splitting of H2, in contrast to other bifunctional systems that were studied.1b, 7d, 9a,
10b, 12d, 12g, 21, 31
(a)
(b)
Figure 5.8. Reaction profiles showing the hydrogenation of acetophenone catalyzed by complex
13 (blue squares) and complex 17 (red circles) in (a) basic THF at 25 bar of H2 pressure and
50°C and (b) 2-propanol at 75°C. The C/B/S ratio was 1/8/200. The transfer hydrogenation of
acetophenone catalyzed by complex 13 in basic 2-propanol was described in Chapter 4.19
31. (a) Casey, C. P.; Johnson, J. B.; Singer, S. W.; Cui, Q., J. Am Chem Soc. 2005, 127, 3100-3109; (b) Heiden, Z. M.; Rauchfuss, T. B., J. Am Chem Soc. 2009, 131, 3593-3600; (c) Casey, C. P.; Johnson, J. B.; Jiao, X. D.; Beetner, S. E.; Singer, S. W., Chem. Commun. 2010, 46, 7915-7917.

129Interestingly, the cationic complex 17 containing a ruthenium hydride and primary amine
grouping does not transfer its hydride and proton equivalent to a ketone. This was confirmed by
the lack of reaction between complex 17 and a stoichiometric amount of acetophenone in THF-d8
at 25 or 50°C. It was expected that the metal hydride and a protic amine grouping would be
suitable to effect bifunctional catalysis of polar bonds.1b, 4c, 6 The use of an N-heterocyclic
carbene as the ligand in this cationic complex clearly shows that there is no activity in this case.
Stradiotto and co-workers have recently reported a highly active zwitterionic ruthenium catalyst
containing a phosphine-amine ligand that catalyzes the transfer hydrogenation of ketones in
refluxing 2-propanol. On the other hand, the hydride complex of the ruthenium catalyst showed
no activity toward ketone reduction, regardless of whether an excess amount of base was added.
Such a hydride complex, however, has no protic N–H functionality on its ligand.11c The cationic
charge seems to reduce the nucleophilicity of the hydride ligand so that there is diminished
catalytic activity.
If the outer sphere mechanism operates in the H2-hydrogenation of ketone catalyzed by
complex 13 in basic solvents, the hydrogenation of an α,β-unsaturated ketone would most likely
afford the reduction of the carbonyl group,6b although hydrogenation of the conjugated olefin
could also occur by a 1,4-addition reaction.32 A ligand in the active ruthenium species might also
rearrange or dissociate to allow the coordination of the olefin and its reduction to occur. This was
tested using trans-4-phenyl-but-3-en-2-one as the substrate (Scheme 5.4). Under similar catalytic
conditions (C/B/S = 1/8/200, 25 bar of H2 at 50°C), complex 13 catalyzed the reduction of the
polar bond and the olefin, giving the products trans-4-phenyl-but-3-en-2-ol (33% conversion), 4-
phenylbutan-2-one (10% conversion), and the fully hydrogenated product 4-phenylbutan-2-ol
(24% conversion) in 3 h (Scheme 5.4 and Figure 5.9).
Scheme 5.4. Hydrogenation of trans-4-Phenyl-but-3-en-2-one Catalyzed by Complex 13.
32. (a) Ito, M.; Kitahara, S.; Ikariya, T., J. Am. Chem. Soc. 2005, 127, 6172-6173; (b) Ikariya, T.; Gridnev, I. D., Chem. Rec. 2009, 9, 106-123.
13 cat.
25 bar H2, KO tBuTHF, 50°C
O
OH
O
OH
33%
10%
24% in 3 hC/B/S = 1/8/200
130
Figure 5.9. Reaction profile showing the hydrogenation of trans-4-phenyl-but-3-en-2-one
catalyzed by complex 13 in basic THF at 25 bar of H2 pressure and 50°C: (blue circles) trans-4-
phenylbut-3-en-2-ol; (red squares) 4-phenylbutan-2-ol; (green triangles) 4-phenylbutan-2-one.
The C/B/S ratio was 1/8/200.
5.3.8 The Favored Inner-Sphere Mechanism. Given the experimental data that are not
supportive of the outer-sphere mechanism, an inner-sphere mechanism is proposed.4b, 6b, 33 The
ketone must coordinate to the metal center to allow the hydride migration from the metal to the
carbonyl group. To effect coordination of the ketone, the primary amine group can decoordinate
from the metal center, or the arene ligand on the ruthenium center can slip to η4 coordination
(Figure 5.10). Of note, a seven-membered ring is formed with the chelating primary amine-NHC
ligand, including the nitrogen and carbene donor and the metal center. Facile coordination and
recoordination of the tethered group can occur under catalytic conditions. This has been
proposed for the transfer hydrogenation of ketones using a complex of type Ru(η6-arene)
(κ2(N,N)-N–N)Cl (N–N = 2-hydroxylphenylbis(pyrazol-1-yl)methane and other derivatives)
under base free conditions. The authors proposed that a nitrogen donor of a pyrazolyl group
decoordinates from the metal center during catalysis and acts as a base in the deprotonation of
the coordinated alcohol.34 Elsevier and co-workers recently reported the use of N-heterocyclic
carbene complexes with a tethered tertiary-amine group of palladium(0), [Pd(NHC-amine)
(alkene)], in the transfer hydrogenation of alkynes to (Z)-alkenes under base-free conditions.
Experimental findings suggest the tethered amine group dissociates from the metal center and
acts as a base in catalysis.35 Chu and co-workers reported the use of a half-sandwich complex of

131ruthenium containing a tethered tertiary amine group on the cyclopentadienyl ligand, which acts
as a base to assist the heterolytic cleavage of dihydrogen.36 Nolan and co-workers have recently
reported the mechanism of racemization of chiral alcohols catalyzed by ruthenium(II) systems
containing N-heterocyclic carbene ligands. This was suggested to undergo an inner-sphere
mechanism which involved the dissociation of a coordinated carbonyl ligand. An Ru–
O(alkoxide) bond was formed in the presence of alkoxide, and this is responsible for the
formation of a ruthenium hydride intermediate in the racemization of chiral alcohol. This is
further supported by NMR studies and DFT computations.37 The feasibility of the amino group
dissociation has been explored using computational methods (vide infra). Of note, it is less likely
for the primary amine group to act as a base in catalysis, as complex 13 must be activated by an
alkoxide base to become active.
Figure 5.10. Possible reaction intermediates for the inner-sphere mechanism involving hydride
migration to the coordinated ketone substrate: (left) decoordination of the chelating amine group
from the NHC ligand; (right) ring slippage of the arene ring.
In order to probe the importance of the primary amine group tethered to the N-heterocyclic
carbene ligand in complex 13, we attempted to make and test the N,N-dimethylamine analogue.
Methylation of the primary amine group of complex 13 was not successful. We did succeed in
preparing a closely related compound by reaction of the imidazolium salt, 1-(N,N-dimethyl-
amino)propyl-3-methylimidazolium chloride hydrochloride (HC–NMe2⋅HCl, 1g),38 with silver(I)
oxide and [Ru(p-cymene)Cl2]2 in one pot (Scheme 5.5). Note that the carbene ligand derived
from 1g, if it chelates to the metal, will form a seven-membered ring as observed in complex 13.
The in situ generation of the silver(I) carbene complex derived from 1g and subsequent
33. Daley, C. J. A.; Bergens, S. H., J. Am. Chem. Soc. 2002, 124, 3680-3691.34. Carrion, M. C.; Sepulveda, F.; Jalon, F. A.; Manzano, B. R.; Rodriguez, A. M., Organometallics 2009, 28, 3822-3833.35. Hauwert, P.; Boerleider, R.; Warsink, S.; Weigand, J. J.; Elsevier, C. J., J. Am. Chem. Soc. 2010, 132, 16900-16910.36. Chu, H. S.; Lau, C. P.; Wong, K. Y.; Wong, W. T., Organometallics 1998, 17, 2768-2777.37. Bosson, J.; Poater, A.; Cavallo, L.; Nolan, S. P., J. Am. Chem. Soc. 2010, 132, 13146-13149.
CH3
NNRu
NH
H
O
H
+CH3
NN
RuNH
H OH +

132transmetalation of the NHC ligand to ruthenium(II),39 followed by counteranion metathesis, gave
a compound formulated on the basis of elemental analysis as [Ru(p-cymene)(C–NMe2)Cl]PF6⋅
1.5 DMSO (18, DMSO = dimethyl sulfoxide). The use of DMSO as cosolvent for the reaction is
unavoidable, as ligand 1g has limited solubility in most organic solvents except alcohols.
Attempts to isolate the intermediate silver(I) carbene complex containing the corresponding
imidazolylidene ligand failed, as this quickly decomposed.
Scheme 5.5. Synthesis of Complex 18 by in Situ Generation of the Silver(I) Carbene Complex
and Subsequent Transmetalation of the NHC Ligand to Ruthenium(II).
The NMR spectra of 18 in CD2Cl2 at 253 K indicate that compound is a mixture of two
complexes, one with a bidentate NHC ligand and another with a monodentate NHC and
coordinated DMSO ligand. Infrared spectroscopy usually provides a means of distinguishing
between O- and S-bound forms of the dimethyl sulfoxide ligand40 but did not prove helpful
because of the complex spectrum in the fingerprint region.
Complex 18 is an effective catalyst for the H2-hydrogenation of acetophenone with an activity
comparable to that of complex 13 under similar reaction conditions (C/B/S=1/10/240, 25 bar of
H2 at 50°C) in basic THF with KOtBu as a base. A conversion of 76% to 1-phenylethanol was
achieved within 2 h of reaction. Like complex 13, no induction period was observed (Figure
5.11). Therefore, the protons of the amino group in complex 13 are not needed for catalysis. This
is another example showing that the “NH effect” is not always operational in catalysts for polar
bond hydrogenation.11
38. Jimenez, M. V.; Perez-Torrente, J. J.; Bartolome, M. I.; Gierz, V.; Lahoz, F. J.; Oro, L. A., Organometallics 2008, 27, 224-234.39. Warsink, S.; de Boer, S. Y.; Jongens, L. M.; Fu, C. F.; Liu, S. T.; Chen, J. T.; Lutz, M.; Spek, A. L.; Elsevier, C. J., Dalton Trans. 2009, 7080-7086.40. James, B. R.; Morris, R. H., Can. J. Chem. 1980, 58, 399-408.
0.5 [Ru(p-cymene)Cl2]2DMSO:CH3CN (1:4), rt
181g
N NCH3 N HCl1.5 Ag2O
AgPF6CH2Cl2, rt
[Ru(p-cymene)(C NMe2)Cl]PF6 •1.5 DMSO
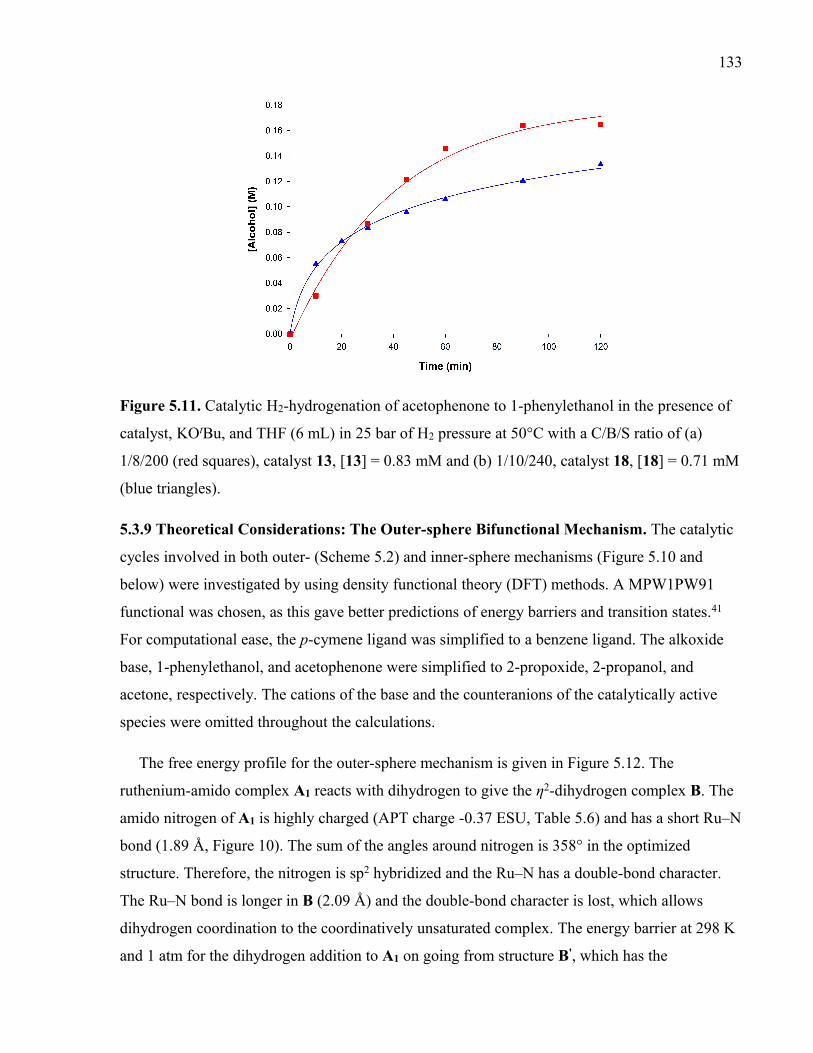
133
Figure 5.11. Catalytic H2-hydrogenation of acetophenone to 1-phenylethanol in the presence of
catalyst, KOtBu, and THF (6 mL) in 25 bar of H2 pressure at 50°C with a C/B/S ratio of (a)
1/8/200 (red squares), catalyst 13, [13] = 0.83 mM and (b) 1/10/240, catalyst 18, [18] = 0.71 mM
(blue triangles).
5.3.9 Theoretical Considerations: The Outer-sphere Bifunctional Mechanism. The catalytic
cycles involved in both outer- (Scheme 5.2) and inner-sphere mechanisms (Figure 5.10 and
below) were investigated by using density functional theory (DFT) methods. A MPW1PW91
functional was chosen, as this gave better predictions of energy barriers and transition states.41
For computational ease, the p-cymene ligand was simplified to a benzene ligand. The alkoxide
base, 1-phenylethanol, and acetophenone were simplified to 2-propoxide, 2-propanol, and
acetone, respectively. The cations of the base and the counteranions of the catalytically active
species were omitted throughout the calculations.
The free energy profile for the outer-sphere mechanism is given in Figure 5.12. The
ruthenium-amido complex A1 reacts with dihydrogen to give the η2-dihydrogen complex B. The
amido nitrogen of A1 is highly charged (APT charge -0.37 ESU, Table 5.6) and has a short Ru–N
bond (1.89 Å, Figure 10). The sum of the angles around nitrogen is 358° in the optimized
structure. Therefore, the nitrogen is sp2 hybridized and the Ru–N has a double-bond character.
The Ru–N bond is longer in B (2.09 Å) and the double-bond character is lost, which allows
dihydrogen coordination to the coordinatively unsaturated complex. The energy barrier at 298 K
and 1 atm for the dihydrogen addition to A1 on going from structure B', which has the

134dihydrogen molecule outside the coordination sphere, to the transition state TSB',B is 18.0
kcal/mol uphill. The entropy change (ΔS‡) for dihydrogen coordination is -25.9 cal/(mol K). The
dihydrogen is weakly activated in the transition state and also in structure B, as the H–H
distances are short (0.76 and 0.83 Å, respectively).42 Structure B leads to the transition state
TSB,C, in which the H–H distance increases to 0.88 Å and the negative charge on nitrogen
increases from -0.37 to -0.40 ESU. Note that the energy difference between B and TSB,C is only
0.2 kcal/mol.
The heterolytic splitting of H2 by A1 leads to C, which is -39.5 kcal/mol downhill from TSB,C.
The Ru–H distance is 1.58 Å , which is similar within the ESD to the value of the Ru–H distance
(1.79(7) Å) in the crystal structure of complex 17. The single-bond character of Ru–N is retained
(2.16 Å), and the charge on nitrogen is further reduced to -0.24 ESU. The charges on ruthenium
and hydride are -0.72 and -0.042 ESU, respectively. The smaller charge on the hydride may
explain the poor activity of complex 17 toward reaction with acetophenone. The hydrides of the
model complex of a ketone hydrogenation catalyst, (OC-6-22)- Ru(H)2(PH3)2(en), have charges
of -0.20 and -0.15 ESU.9c From structure C, acetone is hydrogen-bonded to the complex,
forming D, and this goes +33.5 kcal/mol uphill from C plus acetone at 298 K and 1 atm, leading
to the transition state TSD,E, where the concerted transfer of dihydrogen to the ketone from Ru–H
and Ru–NH2 occurs via a six-membered ring transition state.4b, 6b, 7a-g, 7j The Ru–H bond is
elongated (to 1.84 Å), as is the N–H bond (from 1.02 to 1.30 Å). In addition, the C–H and C–O
bonds of the alcohol are formed simultaneously, with bond distances of 1.27 and 1.33 Å ,
respectively. The charge of ruthenium is reduced (-0.32 ESU) and the hydride is more negatively
charged (-0.43 ESU). This leads to the ruthenium-amido-isopropyl alcohol adduct E, held by
weak electrostatic interactions. The elimination of the alcohol product results in the regeneration
of the amido complex A1, which is -15.6 kcal/mol downhill from TSD,E. The alcohol could also
coordinate to the amido complex, forming F. Subsequent deprotonation of the alcohol by the
amide ligand causes the formation of the ruthenium-alkoxide complex G. This has a basic
alkoxide ligand (APT charge on O -0.82 ESU) and is more thermodynamically stable than A1
plus 2-propanol by 2.4 kcal/mol. Of note, the Ru–Ccarbene bond distances vary from 2.03 to 2.08
Å for all the computed structures, and they are comparable to those of complexes 13 and 17 in
the crystal structures (Figure 5.13).19
41. Lynch, B. J.; Truhlar, D. G., J. Phys. Chem. A 2001, 105, 2936-2941.42. Morris, R. H., Coord. Chem. Rev. 2008, 252, 2381-2394.

135
Figure 5.12. The free energy profile for an outer-sphere mechanism for the H2-hydrogenation of
acetone starting from A1 and moving to the right and the enolate formation starting from A1 and
moving to the left. The gas phase free energies (1 atm, 298 K) are reported relative to G in
kcal/mol.
The enolate complex A3 is formed from A1 via coordination of acetone, giving A2, and this
leads to the transition state TSA2,A3, where deprotonation of the methyl group occurs. This is
+17.9 kcal/mol uphill from A1 plus acetone. The product A3 is thermodynamically less stable
than A1 plus acetone by 7.3 kcal/mol (Figure 5.12). The computations thus accurately reflect
comparable energy barriers for the H2 addition and enolate formation from the amido complex
A1 (18.0 versus 17.9 kcal/mol at 298 K and 1 atm). The concerted transfer of a H+/H- pair to
acetone has a much higher barrier of +33.5 kcal/mol in comparison to the aforementioned steps,
which contradicts the experimental findings. The calculation also predicts that the ruthenium-
hydride complex C (complex 17) is the resting state of the catalytic cycle. This is more stable
than the ruthenium-alkoxide and ruthenium-amido complexes by 15.5 and 22.4 kcal/mol.
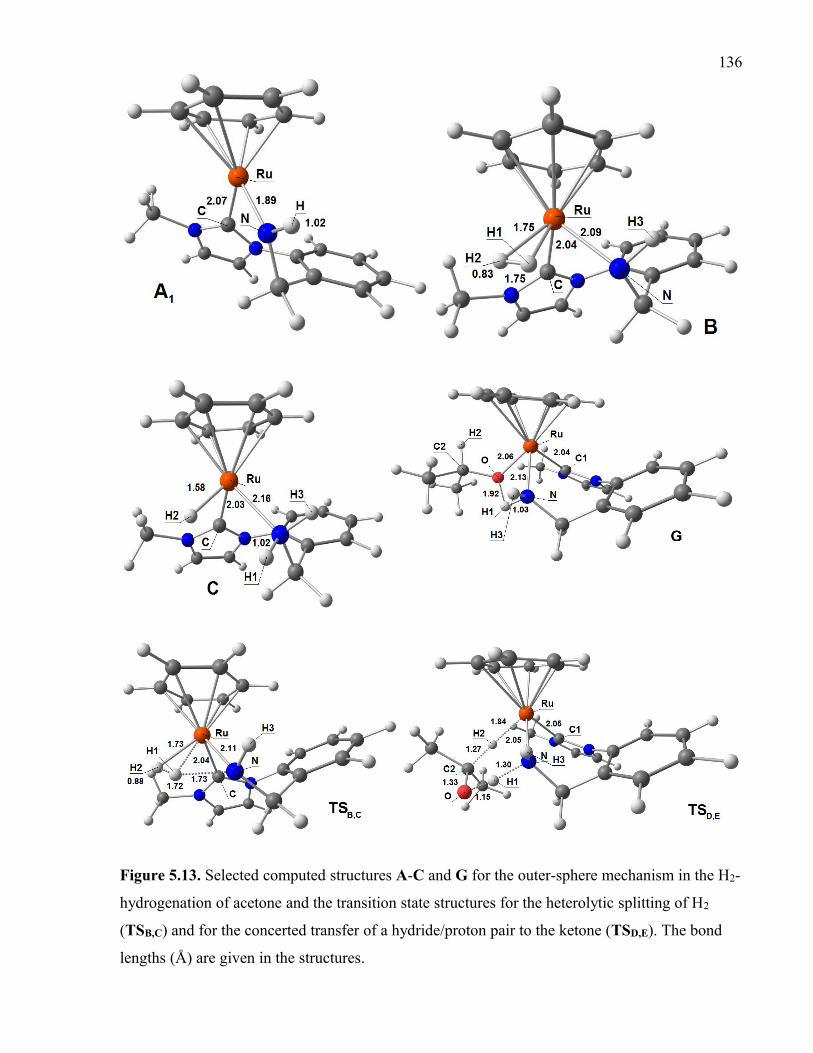
136
Figure 5.13. Selected computed structures A-C and G for the outer-sphere mechanism in the H2-
hydrogenation of acetone and the transition state structures for the heterolytic splitting of H2
(TSB,C) and for the concerted transfer of a hydride/proton pair to the ketone (TSD,E). The bond
lengths (Å) are given in the structures.

137Table 5.6. Atomic Polar Tensor (APT) Charges (in ESU) on Selected Atoms for the Computed
Structures A-C, G, TSB,C, and TSD,E.
Computed structures
APT charges (ESU)
Ru NHC-C C–O C–O N H1/N–H H2/Ru–H H3/N–H
A -0.25 0.51 - - -0.37 - - 0.088B -0.63 0.60 - - -0.37 0.18 0.31 0.063C -0.72 0.71 - - -0.24 0.15 -0.042 0.13G -0.21 0.60 0.57 -0.82 -0.29 0.23 -0.11 0.12
TSB,C -0.68 0.61 - - -0.40 0.097 0.020 0.064TSD,E -0.32 0.60 1.11 -0.88 -0.33 0.40 -0.43 0.084
5.3.10 Theoretical Considerations: The Inner-sphere Mechanism. An alternative proposal is
the inner-sphere mechanism, which involves the decoordination of the amine group from the
NHC ligand or ring slippage of the coordinated arene ring.7b The computed ring slipped
structures were higher in energy than those structures computed for the mechanism involving the
decoordination of the amine group (see below). Thus, they are not considered as the possible
intermediates (Figure 5.14).
Figure 5.14. Computed structures and energies (at 298 K and 1 atm relative to G + H2 in
kcal/mol) for the ring slippage mechanism.
Starting from the alkoxide complex G, the decoordination of the amine group is an uphill
process by +15.5 kcal/mol at 298 K and 1 atm (Figure 5.15). This process is entropically favored
(ΔS = 16.6 cal/(mol K)). This afterward provides the coordinatively unsaturated complex H,
which has a short Ru–O distance compared to G (1.90 and 2.06 Å , respectively, Figures 5.13
and 5.16). The charges on the oxygens of the alkoxide are comparable in H and G (-0.82 and
-0.77, respectively; see Tables 5.6 and 5.7). The variation in the Ru–Ccarbene bond distance is
similar to that of the outer-sphere mechanism for all of the computed structures (Figure 5.16). In
CH3
NNRu
NH
H
O
H
CH3
NNRu
NH
H
O
H
HCH3NN
Ru
NH
H
O
HH
HCH3NN
Ru
NH
H
OH
+H
+ + +
+ H2 + H2
*Free energy (kcal/mol) given in parentheses.
(25.1) (33.9) (28.7) (26.7)

138addition, the Ru–N distances are more than 5.0 Å for all of the computed structures. For all, the
dihedral angle of the phenyl and the imidazolylidene rings range from 83.3° for H and 95.0° for
K. For comparison, the dihedral angles for the crystal structures of complexes 13 and 17 are
54.6° and 54.3°.19
Figure 5.15. The free energy profile for the inner-sphere mechanism in the H2-hydrogenation of
acetone starting from H and moving to the right. The amino group is decoordinated throughout
the catalytic cycle. Moving to the left from H leads to the unstable enolate complex M. The gas
phase free energies (1 atm, 298 K) are reported relative to G, hydrogen and acetone in kcal/mol.
Coordination of molecular hydrogen in a side-on fashion to H then occurs, giving I. Both
QST3 and QST2 searches were performed, but these failed to locate a transition state for
dihydrogen addition to H. The dihydrogen is again weakly activated in I with a short H–H
distance of 0.85 Å.42 This process works against entropy (ΔS = -16.0 cal/(mol K)). Heterolytic
splitting of η2-H2 takes place through transition state TSI,J. The H–H distance increases to 0.92
Å, and the charge on oxygen increases from -0.68 to -0.73 ESU. The energy barrier for the
heterolytic splitting of H2 is +33.0 kcal/mol uphill from G plus dihydrogen or +17.5 kcal/mol

139from H plus dihydrogen at 298 K and 1 atm. The energy difference between I and TSI,J is 3.7
kcal/mol, indicative of an early transition state. Again, the heterolytic splitting of H2 is
entropically demanding (ΔS‡ = -19.9 cal/(mol K)). Similar DFT calculations were also performed
with the osmium(II) complex ([Os(p-cymene)(C–NH2)Cl]PF6, complex 14, see Chapter 4) using
the inner-sphere mechanism. The osmium(II) dihydrogen complex with a decoordinated amine
group similar to I is 30.7 kcal/mol uphill from the alkoxide complex analogous to G plus
dihydrogen. The related complex [Os(p-cymene)(NHC)(OH)]OTf (NHC = bis(2,6-diisopropyl-
phenyl)imidazol-2-ylidene, IPr), was believed to undergo an inner-sphere mechanism for the
transfer hydrogenation of aldehydes in 2-propanol solution, whereas the hydroxyl ligand acts as
an internal base in the formation of an Os–O(alkoxide) bond.43
The ruthenium-hydride complex containing a coordinated alcohol is then formed from TSI,J,
leading to J, which is -27.7 kcal/mol downhill. The structure has a short Ru–H bond (1.57 Å).
The charges on the ruthenium and the hydride are -0.52 and -0.039 ESU. Decoordination of 2-
propanol and recoordination of acetone forms K, and a further energy of +15.6 kcal/mol uphill is
required, leading to the transition state TSK,H. The hydride ligand on the ruthenium complex
attacks the coordinated acetone via a four-membered-ring transition state.4b, 6b, 37 The Ru–H bond
elongates to 1.65 Å and the Ru–O bond shortens (from 2.14 to 2.09 Å), due to an increased
charge on oxygen (from -0.57 to -0.60 ESU; see Table 5.7). Likewise, the C–H (1.58 Å) and C–
O (1.30 Å) bonds of the alcohol are formed simultaneously. The charge of ruthenium is reduced
(-0.70 ESU), and the hydride is more highly charged (-0.10 ESU). This leads to structure H and
completes the cycle.
43. Castarlenas, R.; Esteruelas, M. A.; Onate, E., Organometallics 2008, 27, 3240-3247.
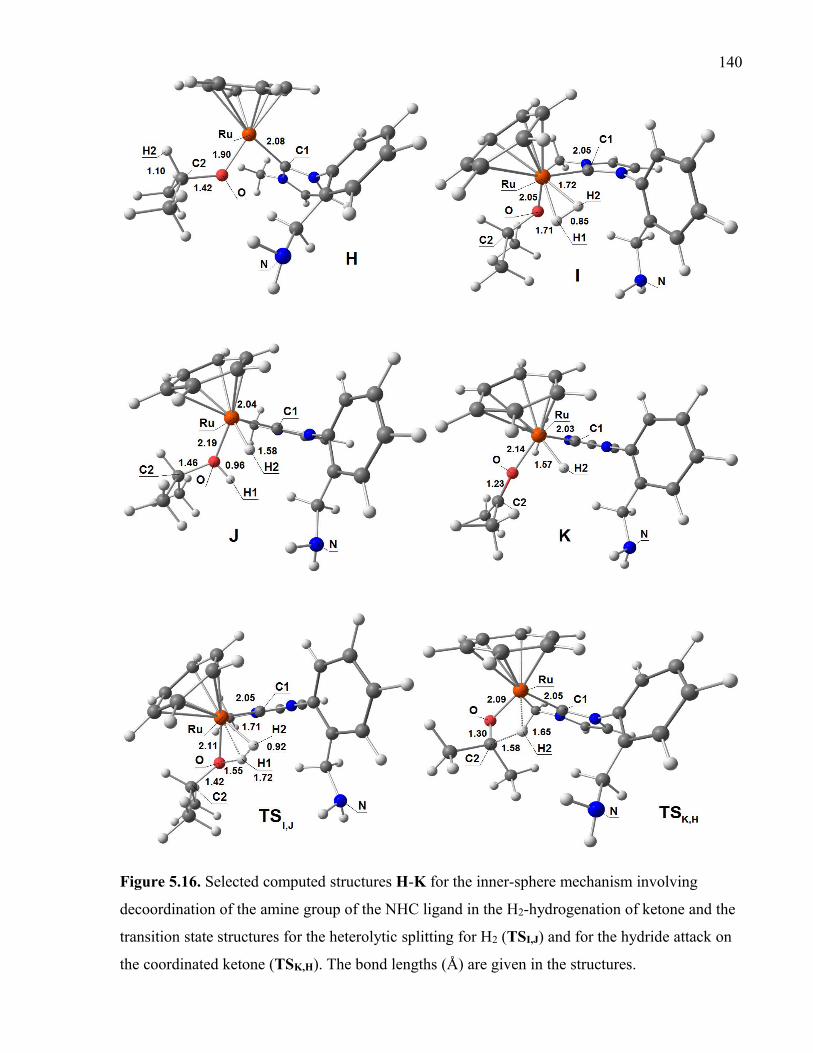
140
Figure 5.16. Selected computed structures H-K for the inner-sphere mechanism involving
decoordination of the amine group of the NHC ligand in the H2-hydrogenation of ketone and the
transition state structures for the heterolytic splitting for H2 (TSI,J) and for the hydride attack on
the coordinated ketone (TSK,H). The bond lengths (Å) are given in the structures.
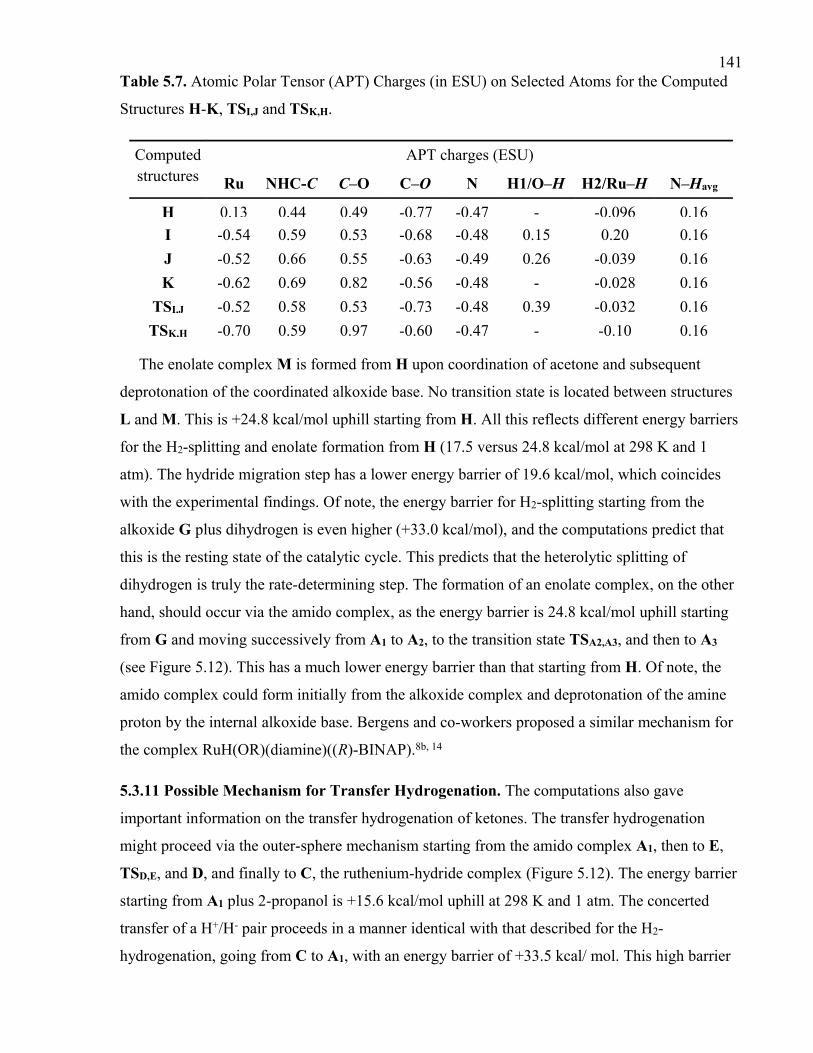
141Table 5.7. Atomic Polar Tensor (APT) Charges (in ESU) on Selected Atoms for the Computed
Structures H-K, TSI,J and TSK,H.
Computed structures
APT charges (ESU)
Ru NHC-C C–O C–O N H1/O–H H2/Ru–H N–Havg
H 0.13 0.44 0.49 -0.77 -0.47 - -0.096 0.16I -0.54 0.59 0.53 -0.68 -0.48 0.15 0.20 0.16J -0.52 0.66 0.55 -0.63 -0.49 0.26 -0.039 0.16K -0.62 0.69 0.82 -0.56 -0.48 - -0.028 0.16
TSI,J -0.52 0.58 0.53 -0.73 -0.48 0.39 -0.032 0.16TSK,H -0.70 0.59 0.97 -0.60 -0.47 - -0.10 0.16
The enolate complex M is formed from H upon coordination of acetone and subsequent
deprotonation of the coordinated alkoxide base. No transition state is located between structures
L and M. This is +24.8 kcal/mol uphill starting from H. All this reflects different energy barriers
for the H2-splitting and enolate formation from H (17.5 versus 24.8 kcal/mol at 298 K and 1
atm). The hydride migration step has a lower energy barrier of 19.6 kcal/mol, which coincides
with the experimental findings. Of note, the energy barrier for H2-splitting starting from the
alkoxide G plus dihydrogen is even higher (+33.0 kcal/mol), and the computations predict that
this is the resting state of the catalytic cycle. This predicts that the heterolytic splitting of
dihydrogen is truly the rate-determining step. The formation of an enolate complex, on the other
hand, should occur via the amido complex, as the energy barrier is 24.8 kcal/mol uphill starting
from G and moving successively from A1 to A2, to the transition state TSA2,A3, and then to A3
(see Figure 5.12). This has a much lower energy barrier than that starting from H. Of note, the
amido complex could form initially from the alkoxide complex and deprotonation of the amine
proton by the internal alkoxide base. Bergens and co-workers proposed a similar mechanism for
the complex RuH(OR)(diamine)((R)-BINAP).8b, 14
5.3.11 Possible Mechanism for Transfer Hydrogenation. The computations also gave
important information on the transfer hydrogenation of ketones. The transfer hydrogenation
might proceed via the outer-sphere mechanism starting from the amido complex A1, then to E,
TSD,E, and D, and finally to C, the ruthenium-hydride complex (Figure 5.12). The energy barrier
starting from A1 plus 2-propanol is +15.6 kcal/mol uphill at 298 K and 1 atm. The concerted
transfer of a H+/H- pair proceeds in a manner identical with that described for the H2-
hydrogenation, going from C to A1, with an energy barrier of +33.5 kcal/ mol. This high barrier

142argues against this mechanism, and in fact, the ruthenium-hydride complex 17 has very low
activity under transfer hydrogenation conditions in the absence of base.
For the inner-sphere mechanism, decoordination of the amine group of the NHC ligand takes
place from the 2-propoxide complex G to allow the β-hydride elimination to take place, forming
H. This proceeds through the transition state TSK,H, leading to the hydride complex K (Figure
5.15). The β-hydride elimination of the alkoxide is uphill by 6.6 kcal/mol from H. The hydride
ligand from K can then attack the carbonyl group of the substrate; this is the same step as
described in the inner-sphere H2 hydrogenation mechanism, with the same energy barrier of
+15.6 kcal/mol uphill (Figure 5.15). Overall, this has lower energy barrier compared with the
outer-sphere mechanism and seems more likely to occur during catalysis.
5.3.12 Role of Complex 17 in Catalysis. Complex 17 is not likely to be an intermediate in the
H2-hydrogenation reaction, as attempts to prepare this from complex 13 in basic THF under H2
failed. In addition, it does not react with acetophenone (see below). This, however, may be the
resting state of the catalyst in the transfer hydrogenation, as it can be prepared under similar
catalytic conditions. According to the calculations, the amine group of structure K can
recoordinate, which displaces the coordinated acetone to give structure C, which is analogous to
complex 17. This is -33.1 kcal/mol downhill from the transition state TSH,K. The fact that a base
is needed to activate complex 17 in both H2-hydrogenation and transfer hydrogenation reactions
is difficult to explain. In fact, a stoichiometric reaction of complex 17 and KOtBu in THF-d8 at
50°C under argon produces a reaction mixture that lacks a hydride signal in the 1H NMR
spectrum. Subsequent addition of a stoichiometric amount of acetophenone to this reaction
mixture did not lead to the formation of 1-phenylethanol or 1-phenylethoxide. The species that is
formed from the reaction of complex 17 and an alkoxide base may be responsible for the
activation of H2.
5.4 Conclusion
In summary, the H2-hydrogenation of ketones catalyzed by complex 13 was studied and the
mechanism of action was investigated by both experimental and theoretical means. The
ruthenium-hydride complex 17, which was thought to be the crucial intermediate for bifunctional
catalysis, was isolated. This, however, showed no activity under catalytic conditions unless when

143activated by a base. This hydride complex is believed to be the resting state in the transfer
hydrogenation mechanism. The cationic charge on the metal center is likely to decrease the
nucleophilicity of the hydride ligand. In fact, this is a rare example of a catalyst with an M–H
and N–H grouping that fails to undergo bifunctional catalysis using the “NH effect”, which was
originally proposed by Noyori and co-workers.6a Kinetic studies including the studies of isotope
effects using D2 gas and acetophenone-d3 support the theoretical predictions that an early
transition state in the heterolytic splitting of H2 is the rate-determining step of the catalytic cycle.
The outer-sphere mechanism involving bifunctional catalysis of ketone reduction is disfavored
according to experimental studies. Computational studies also suggest a high energy barrier for
the concerted transfer of H+/H- to the ketone compared to dihydrogen addition and subsequent
heterolytic splitting.
An alternative to the outer-sphere bifunctional mechanism is therefore proposed on the basis
of experimental and theoretical findings. First, complex 13, when activated, leads to a
ruthenium-alkoxide complex. The alkoxide ligand labilizes the cis-amine ligand.44
Decoordination of the amine group of the NHC ligand provides a vacant site for the coordination
of dihydrogen (Scheme 5.6). Ring-opening reactions are common for chelating ligands that form
a seven-membered ring with the metal. Catalysis then proceeds with the heterolytic splitting of
dihydrogen by the internal alkoxide base, and then hydride attacks the coordinated ketone. In
addition, the alkoxide complex can convert to the ruthenium-amido complex which is
responsible for the formation of an enolate complex. The current study should assist in the
rational design of more robust and active hydrogenation catalysts using N-heterocyclic carbenes
as ligands.
44. (a) Atwood, J. D.; Brown, T. L., J. Am. Chem. Soc. 1976, 98, 3160-3166; (b) Bryndza, H. E.; Domaille, P. J.; Paciello, R. A.; Bercaw, J. E., Organometallics 1989, 8, 379-385; (c) Caulton, K. G., New J. Chem. 1994, 18, 25-41; (d) Flood, T. C.; Lim, J. K.; Deming, M. A.; Keung, W., Organometallics 2000, 19, 1166-1174.
144Scheme 5.6. Proposed Mechanism for the H2-Hydrogenation of Ketones Catalyzed by Complex
13 and an Alkoxide Base.
5.5 Experimental Section
5.5.1 Synthesis. All of the preparations and manipulations, except where otherwise stated, were
carried out under a nitrogen or argon atmosphere using standard Schlenk-line and glovebox
techniques. Dry and oxygen-free solvents were always used. The synthesis of [1-(2-(amino-
methyl)phenyl)-3-methylimidazol-2-ylidene]chloro(η6-p-cymene)ruthenium(II) hexafluoro-
phosphate (13) has been described in Chapter 4.19 The synthesis of 1-(N,N-
dimethylamino)propyl-3-methylimidazolium chloride hydrochloride (1g)38 and [Ru(p-
cymene)Cl2]245 were reported in the literature. All other reagents and solvents were purchased
from commercial sources and were used as received. Deuterated solvents were purchased from
Cambridge Isotope Laboratories and Sigma Aldrich and degassed and dried over activated 45. Bennett, M. A.; Smith, A. K., Dalton Trans. 1974, 233-241.
CH3NN
RuNH
H HR'
O
CH3NN
RuNH
H O
R'
H
Ru NNN
CH3
H
OR'
H
H
H2
OR'
CH3NN
RuNH
H R'O HHH
CH3NN
RuNH
H HO
R'
R
R
R
R
R
R
Ru NNN
CH3
HRu N
NN
CH3
H
OR''
H H-
++O
R'
R
OR'
R
R HOH R'
R
HOH R'
R-
HOH R'
R
+
+
+
+
+

145molecular sieves prior to use. NMR spectra were recorded on a Varian 400 spectrometer
operating at 400 MHz for 1H, 100 MHz for 13C, and 376 MHz for 19F. The 1H and 13C{1H} NMR
spectra were measured relative to partially deuterated solvent peaks but are reported relative to
tetramethylsilane (TMS). All 19F chemical shifts were measured relative to
trichlorofluoromethane as an external reference. Elemental analyses were performed at the
Department of Chemistry, University of Toronto, on a Perkin-Elmer 2400 CHN elemental
analyzer. Samples were handled under argon where it was appropriate. Single-crystal X-ray
diffraction data were collected using a Nonius Kappa-CCD diffractometer with Mo Kα radiation
(λ = 0.71073 Å). The CCD data were integrated and scaled using the Denzo-SMN package. The
structures were solved and refined using SHELXTL V6.1. Refinement was by full-matrix least
squares on F2 using all data.
5.5.2 Synthesis of [1-(2-(Aminomethyl)phenyl)-3-methylimidazol-2-ylidene]hydrido(η6-p-
cymene)ruthenium(II) Hexafluorophosphate ([Ru(p-cymene)(C–NH2)H]PF6, 17). A Schlenk
flask was charged with 13 (50 mg, 0.083 mmol) in 2-propanol solution (14 mL). The solution
was warmed to 50°C under an argon atmosphere. A solution of sodium 2-propoxide (20 mg, 0.24
mmol) in 2-propanol (6 mL) was added to this stirred solution over the course of 0.5 h,
whereupon the color of the reaction mixture turned from yellow to deep red and then to brown.
The solution was stirred for a further 3 h. After the reaction had gone to completion, the solvent
was removed under vacuum. The solid residue was extracted with tetrahydrofuran (THF, 4 mL)
and filtered through a pad of Celite. The addition of pentane (16 mL) to the THF solution yielded
a brown precipitate, which was collected and dried in vacuo. Yield: 27 mg, 57%. Crystals
suitable for an X-ray diffraction study were obtained as a THF solvate by slow diffusion of
pentane into a saturated solution of 17 in tetrahydrofuran under a nitrogen atmosphere. 1H NMR
(CD3CN, δ): 7.56 (m, 3-CH and 4-CH of Ph, 2H), 7.46 (m, 5-CH of Ph, 1H), 7.39 (d, JHH = 7.34
Hz, 6-CH of Ph, 1H), 7.33 (d, JHH = 1.72 Hz, 5-CH of imid, 1H), 7.21 (d, JHH = 1.72 Hz, 4-CH
of imid, 1H), 5.06 (d, JHH = 5.65 Hz, 2-Ar-CH of p-cymene, 1H), 5.02 (d, JHH = 5.92 Hz, 6-Ar-
CH of p-cymene, 1H), 4.82 (d, JHH = 5.92 Hz, 5-Ar-CH of p-cymene, 1H), 4.40 (d, JHH = 5.65
Hz, 3-Ar-CH of p-cymene, 1H), 3.68 (m, CH2, 1H), 3.61 (s, CH3, 3H), 3.40 (m, br, NH2, 2H),
2.64 (dt, JHH = 3.66, 12.17 Hz, CH2, 1H), 2.40 (sept, JHH = 6.80 Hz, CH of (CH3)2CH of p-
cymene, 1H), 1.52 (s, CH3 of p-cymene, 3H), 1.21 (d, JHH = 6.80 Hz, CH3 of (CH3)2CH of p-
cymene, 3H), 1.14 (d, JHH = 6.80 Hz, CH3 of (CH3)2CH of p-cymene, 3H), -7.82 (s, Ru–H). 19F
NMR (CD3CN, δ): -72.9 (d, JPF = 706 Hz). 13C{1H} NMR (CD3CN, δ): 185.3 (Ru–C), 141.4

146(CPh), 134.0 (CPh), 132.2 (CPh), 130.5 (CPh), 129.3 (CPh), 126.6 (CPh), 124.3 (Cimid.), 123.5 (Cimid.),
108.7 (CAr-p-cymene), 105.1 (CAr-p-cymene), 88.9 (CAr-p-cymene), 85.6 (CAr-p-cymene), 81.3 (CAr-p-cymene),
80.9 (CAr-p-cymene), 47.4 (CH2), 39.8 (CH3), 32.4 (CH of (CH3)2CH of p-cymene), 24.1 (CH3 of
(CH3)2CH of p-cymene), 23.9 (CH3 of (CH3)2CH of p-cymene), 19.1 (CH3 of p-cymene). MS
(ESI, methanol/water; m/z): 424.1 ([M]+). Attempts at elemental analyses failed to give an
acceptable carbon content, while hydrogen and nitrogen contents are in the acceptable range.
Typical results are as follows. Anal. Calcd for C21H28F6N3PRu: C, 44.37; H, 4.96; N, 7.39.
Found: C, 42.91; H, 4.80; N, 7.34.
5.5.3 Synthesis of [1-(N,N-Dimethylaminopropyl)-3-methylimidazol-2-ylidene]chloro(η6-p-
cymene)ruthenium(II) Hexafluorophosphate Dimethyl Sulfoxide Solvate ([Ru(p-cymene)
(C–NMe2)Cl]PF6·1.5 DMSO, 18). A Schlenk flask was charged with silver(I) oxide (145 mg,
0.63 mmol) and the [Ru(p-cymene)Cl2]2 dimer (127 mg, 0.21 mmol) in dry acetonitrile (15 mL)
under molecular sieves (3 Å). In a separate Schlenk flask was charged with 1g (100 mg, 0.42
mmol) and anhydrous dimethyl sulfoxide (DMSO, 5 mL) under an argon atmosphere. The
DMSO solution containing the dissolved imidazolium salt was then added to the stirring solution
of silver(I) oxide and the [Ru(p-cymene)Cl2]2 dimer in acetonitrile. Acetonitrile washing (5 mL)
was applied to the residual DMSO solution containing 1g, and this was also added to the reaction
mixture. This was stirred under an argon atmosphere at room temperature (25°C) for overnight.
After the reaction has gone to completion, the reaction mixture was filtered through a pad of
Celite under an argon atmosphere. The solvent was evaporated under reduced pressure. The
residue that obtained was dissolved in dichloromethane (10 mL) and this was added silver
hexafluorophosphate (105 mg, 0.42 mmol) and stirred further for 1 h. The suspension that
formed was filtered through a pad of Celite. The volume of solvent was reduced (2 mL), and
addition of diethyl ether (12 mL) to the dichloromethane solution yielded an orange-yellow oil.
The supernatant liquid was decanted and the residue was washed with diethyl ether (3 mL).
Trituration of the oil with diethyl ether (3 × 3 mL) afforded an orange powder which was
collected and dried in vacuo. Yield: 120 mg, 41%. 1H NMR (CD2Cl2, 253K, δ): 7.19 (d, JHH =
1.94 Hz, CH of imid., 1H), 7.16 (d, JHH = 1.96 Hz, CH of imid., 1H), 7.12 (d, JHH = 1.96 Hz, CH
of imid., 1H), 7.10 (d, JHH = 1.94 Hz, CH of imid., 1H), 5.63 (d, JHH = 6.03 Hz, Ar-CH of p-
cymene, 1H), 5.53-5.50 (m, Ar-CH of p-cymene, 2H), 5.44-5.41 (m, Ar-CH of p-cymene, 3H),
5.21 (m, Ar-CH of p-cymene, 2H), 4.43 (m, CH2, 2H), 4.26 (m, CH2, 4H), 3.91 (s, CH3, 3H),
3.82 (s, CH3, 3H), 3.10 (s, CH3 of DMSO, 3H), 3.00 (s, CH3 of N(CH3)2, 3H), 2.92 (s, CH3 of

147DMSO, 3H), 2.78 (m, CH of (CH3)2CH of p-cymene and CH2, 4H), 2.30 (m, CH2, 2H), 2.10 (s,
CH3 of N(CH3)2, 6H), 2.05 (m, CH2, 2H), 1.50 (s, CH3 of p-cymene, 3H), 1.25 (m, CH3 of
(CH3)2CH of p-cymene, CH3 of p-cymene and CH3 of N(CH3)2, 12H), 0.97 (d, JHH = 6.81 Hz,
CH3 of (CH3)2CH of p-cymene, 6H). 13C{1H} NMR (CD2Cl2, 253K, δ): 173.9, 170.8 (Ru–C),
125.4, 124.8, 124.6, 122.5 (Cimid.), 112.9, 112.2, 102.4, 99.5, 87.8, 87.0, 85.8, 85.2, 85.1, 83.7,
81.6, 80.3 (CAr-p-cymene), 60.9, 60.6 (CH3 of DMSO), 58.4, 50.0, 45.2, 43.3 (CH2), 39.4, 38.8
(CH3), 31.0 (CH of (CH3)2CH of p-cymene), 30.9 (CH3 of N(CH3)2), 30.5 (CH of (CH3)2CH of
p-cymene), 26.7, 26.4 (CH2), 24.2 (CH3 of p-cymene), 23.5 (CH3 of (CH3)2CH of p-cymene),
21.9 (CH3 of p-cymene), 21.6, 20.9, 19.2 (CH3 of (CH3)2CH of p-cymene), 18.0 (CH3 of
N(CH3)2). MS (ESI, methanol/water; m/z) 438.1 ([M]+), 402.1 ([M – Cl]+). Anal. Calcd for
C19H31ClF6N3PRu·1.5 C2H6SO: C, 37.74; H, 5.76; N, 6.00. Found: C, 37.11; H, 5.26; N, 6.62.
5.5.4 Catalysis. Oxygen-free tetrahydrofuran (THF) used for all of the catalytic runs was stirred
over sodium for 2-3 days under argon, and freshly distilled from sodium benzophenone ketyl
prior to use. Acetophenone was vacuum-distilled over phosphorus pentoxide (P2O5) and stored
under nitrogen prior to use. All of the other substrates were vacuum-distilled, dried over
activated molecular sieves, and stored under nitrogen prior to use. All of the H2-hydrogenation
reactions were performed at constant pressure using a stainless steel 50 mL Parr hydrogenation
reactor. The temperature was maintained at 50°C using a constant-temperature water bath. The
reactor was flushed several times with hydrogen gas at 2-4 bar prior to the addition of
catalyst/substrate and base solutions.
In a typical run (Table 5.2, entry 2), the catalyst 13 (3 mg, 5.0 μmol), benzophenone (181 mg,
0.99 mmol), and potassium tert-butoxide (5 mg, 0.044 mmol) were dissolved in THF (4 and
2mL, respectively) under a nitrogen atmosphere. The catalyst/substrate and base solutions were
taken up by means of two separate syringes and needles in a glovebox. The needles were
stoppered, and the syringes were taken to the reactor. The solutions were then injected into the
reactor against a flow of hydrogen gas. The hydrogen gas was adjusted to the desired pressure.
Small aliquots of the reaction mixture were quickly withdrawn with a syringe and needle under a
flow of hydrogen at timed intervals by venting the Parr reactor at reduced pressure.
Alternatively, small aliquots of the reaction mixture were sampled from a stainless steel
sampling dip tube attached to a modified Parr reactor. The dip tube was 30 cm in length with an
inner diameter of 0.01 in., and a swing valve was attached to the end of the sampling tube. Other
technical details were previously reported.9c Two small aliquots of samples were thereby

148withdrawn quickly at timed intervals by opening the swing valve, and the first two aliquots were
discarded. All samples for gas chromatography (GC) analyses were diluted to a total volume of
approximately 0.75 mL using oxygenated THF.
A Perkin-Elmer Clarus 400 chromatograph equipped with a chiral column (CP chirasil-Dex
CB 25 m × 2.5 mm) with an auto-sampling capability was used for GC analyses. Hydrogen was
used as a mobile phase at a column pressure of 5 psi with a split flow rate of 50 mL/min. The
injector temperature was 250°C and the FID temperature was 275°C. The oven temperatures and
the retention times (tR, tp, /min) for all of the substrates and alcohol products are given in the
Appendix. All of the conversions are reported as an average of two GC runs. The reported
conversions were reproducible.
5.5.5 Kinetics. A standard solution of the catalyst 13 (1.66 mM) was prepared by dissolving the
complex (25 mg) in THF (25 mL). Reaction mixtures were then prepared in THF by dispensing
the required amount of the catalyst and weighed amounts of acetophenone, phenylethanol, and
potassium tert-butoxide (or a standard solution of potassium tert-butoxide (17.8 mM) in THF for
base-dependent studies) and diluted to a total volume of 6 mL. The hydrogenation reaction and
sampling techniques were described as above in order to obtain at least five data points including
the origin (time = 0 min) at below 40% conversion to the product alcohol. The tabulated results
of initial rates with varying concentrations of catalyst, acetophenone, hydrogen, and potassium
tert-butoxide are given in Table 5.3. All of the conversions are reported as an average of at least
two GC runs. The reported conversions were reproducible.
5.5.6 Kinetic Isotope Effect Studies. D2 (99.8% D) gas and acetophenone-β,β,β-d3 (99% D)
were purchased from Cambridge Isotope Laboratories. Acetophenone-d3 was vacuum-distilled,
dried over activated molecular sieves, and stored under nitrogen prior to use. D2 gas was used as
received without further purification. For experiments using deuterium gas, the modified Parr
reactor with a stainless steel sampling dip tube was used. The reactor was degassed by
evacuation for 10 min, followed by refilling with 2-4 bar of deuterium gas. This was repeated
three times. The reaction mixtures were then injected into the reactor against a flow of deuterium
gas, and the gas pressure was adjusted to 8 bar. The tabulated results of the initial rates with
varying concentrations of acetophenone are given in the Table 5.4. Experiments using
acetophenone-d3 were conducted similarly to the kinetic studies described above.

1495.5.7 Computational Details. All density functional theory (DFT) calculations were performed
using the Gaussian0346 package with the restricted hybrid mPW1PW91functional.47 Ruthenium
was treated with the SDD48 relativistic effective core potential and an associated basis set. All
other atoms were treated with the double-ζ basis set 6-31++G**, which includes diffuse
functionals49 and additional p orbitals on hydrogen as well as additional d orbitals on carbon,
nitrogen, and oxygen.50 All geometry optimizations were conducted in the gas phase, and the
stationary points were characterized by normal mode analysis. Reported free energies were
obtained at 1 atm and 298 K using unscaled vibrational frequencies. The QST3 method was used
to locate transition states. All transition states reported were found to have a single imaginary
frequency. The calculated atomic polar tensor (APT) charges were used to reflect more
accurately the charge distribution on each atom under the derivative of dipole moments with
respect to an applied external electric field.51
46. Frisch, M. J., et al., Gaussian 03, Revision C.02 ed; Gaussian Inc.: Wallingford, CT 2004. Full citation is given in the Appendix.47. (a) Burke, K.; Perdew, J. P.; Wang, Y., Electronic Density Functional Theory: Recent Progress and New Directions; Plenum: New York, 1997; (b) Adamo, C.; Barone, V., J. Chem. Phys. 1998, 108, 664-675.48. (a) Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H., Theor. Chim. Acta 1990, 77, 123-141; (b) Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P., J. Chem. Phys. 1996, 105, 1052-1059.49. (a) Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V., J. Comput. Chem. 1983, 4, 294-301; (b) Lynch, B. J.; Zhao, Y.; Truhlar, D. G., J. Phys. Chem. A 2003, 107, 1384-1388.50. Frisch, M. J.; Pople, J. A.; Binkley, J. S., J. Chem. Phys. 1984, 80, 3265-3269.51. Cioslowski, J., J. Am. Chem. Soc. 1989, 111, 8333-8336.

150Chapter 6: Conventional Bifunctional Mechanism for Ketone
Hydrogenation Catalyzed by Structurally Similar Ruthenium and
Iridium Complexes but with Unconventional Intermediates for
Iridium
6.1 Abstract
An alcohol-assisted outer sphere bifunctional mechanism for the H2-hydrogenation of ketones
catalyzed by similar ruthenium and iridium systems is presented based on experimental and
theoretical evidence. The ruthenium(II) complex containing an N-heterocyclic carbene (NHC)
with a tethered primary amine donor (C–NH2), [RuCp*(C–NH2)py]PF6 (15, Cp* = pentamethyl-
cyclopentadienyl ligand, py = pyridine), when activated by an alkoxide base, has a higher
activity but with similar selectivity for the H2-hydrogenation of ketones in comparison to the
structurally similar iridium(III) complex, [IrCp*(C–NH2)Cl]PF6 (19). The presence of alcohol
accelerates catalysis in the ruthenium(II) system, and in the iridium(III) system but only when
the alkoxide base is used in large excess with respect to iridium. Computational studies using
density functional theory (DFT) methods reveal a higher free energy barrier for the cationic
iridium(III) system to transfer a proton/hydride couple from [MCp(C–NH2)H]n+ (M = Ir, n = 1;
Cp = cyclopentadienyl) to the ketone in an outer sphere bifunctional mechanism compared to the
neutral ruthenium(II) counterpart (M = Ru, n = 0). This supports an outer sphere bifunctional
mechanism for the ruthenium(II) system, but this is energetically unfavourable for the
iridium(III) counterpart. Consistent with this the iridium(III) hydride-amine complex, [IrCp*(C–
NH2)H]PF6 (21), was isolated and found to be inactive as a catalyst for ketone hydrogenation.
The cationic charge is thought to contribute to a diminished hydricity and reactivity of this metal
hydride. Nevertheless we present evidence that this iridium system, in the presence of excess
alkoxide base, does efficiently hydrogenate ketones via an outer sphere bifunctional mechanism
involving a novel, neutral hydride intermediate Ir(η4-Cp*H)(C–NH2)H. The formation of this
intermediate relies on the uncommon migration of a hydride ligand to the η5-Cp* ligand which
appears to be promoted by the unique NHC ligand of the present system. The important role of
the N-H group is illustrated by the poor catalytic activity of the structurally similar iridium(III)
complex, [IrCp*(C–NMe2) Cl]PF6 (22), which does not contain an N–H group. The carbonyl

151stretching wavenumber of the complexes [RuCp*(D–NH2)CO]X decreases as D is changed in the
order phosphine, NHC, 2'-pyridine, amine. This electronic effect results in the poorer
performance in the catalytic H2-hydrogenation of ketones of the phosphine complexes [RuCp*(P–
NH2)py]PF6 (16a) and [IrCp*(P–NH2)Cl]PF6 (20) compared to the more electron rich C–NH2
analogues.
6.2 Introduction
Catalytic hydrogenation of unsaturated bonds using dihydrogen is an attractive, atom
economical, chemical process.1 The activation of dihydrogen by homogenous catalysts is a
process of continuing interest.2 The use of an ancillary ligand such as an amido3 or an amine
donor,4 to heterolytically split dihydrogen has emerged as an effective strategy in the design of
catalysts for the hydrogenation of polar double bonds.5 This provides a bifunctional metal–
hydride and protic amine grouping for the efficient and selective reduction of organic molecules,
including ketones and imines, to produce valuable alcohols and amines.6 This idea has been
extended to the development of catalysts for C–C,6b, 7 C–O8 and C–N9 bond formation to provide
access to new synthetic building blocks which are otherwise difficult to synthesize using
conventional methods.10
1. (a) de Vries, J. G.; Elsevier, C. J., Eds, The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, Germany, 2004; Vol 1-3 ; (b) Kubas, G. J., Chem. Rev. 2007, 107, 4152-4205.2. (a) Samec, J. S. M.; Bäckvall, J. E.; Andersson, P. G.; Brandt, P., Chem. Soc. Rev. 2006, 35, 237-248; (b) Gloaguen, F.; Rauchfuss, T. B., Chem. Soc. Rev. 2009, 38, 100-108; (c) Nixon, T. D.; Whittlesey, M. K.; Williams, J. M. J., Dalton Trans. 2009, 753-762; (d) Conley, B. L.; Pennington-Boggio, M. K.; Boz, E.; Williams, T. J., Chem. Rev. 2010, 110, 2294-2312.3. (a) Noyori, R.; Yamakawa, M.; Hashiguchi, S., J. Org. Chem. 2001, 66, 7931-7944; (b) Ito, M.; Endo, Y.; Ikariya, T., Organometallics 2008, 27, 6053-6055; (c) Kuwata, S.; Ikariya, T., Dalton Trans. 2010, 39, 2984-2992.4. (a) Chu, H. S.; Lau, C. P.; Wong, K. Y.; Wong, W. T., Organometallics 1998, 17, 2768-2777; (b) Lee, D. H.; Patel, B. P.; Clot, E.; Eisenstein, O.; Crabtree, R. H., Chem. Commun. 1999, 297-298; (c) Ayllon, J. A.; Sayers, S. F.; Sabo-Etienne, S.; Donnadieu, B.; Chaudret, B.; Clot, E., Organometallics 1999, 18, 3981-3990; (d) Henry, R. M.; Shoemaker, R. K.; DuBois, D. L.; DuBois, M. R., J. Am Chem Soc. 2006, 128, 3002-3010; (e) Grützmacher, H., Angew. Chem. Int. Ed. 2008, 47, 1814-1818; (f) Kayaki, Y.; Ikeda, H.; Tsurumaki, J. I.; Shimizu, I.; Yamamoto, A., Bull. Chem. Soc. Jpn. 2008, 81, 1053-1061.5. (a) Jessop, P. G.; Morris, R. H., Coord. Chem. Rev. 1992, 121, 155-284; (b) Morris, R. H., Can. J. Chem. 1996, 74, 1907-1915; (c) Clapham, S. E.; Hadzovic, A.; Morris, R. H., Coord. Chem. Rev. 2004, 248, 2201-2237; (d) DuBois, M. R.; DuBois, D. L., Chem. Soc. Rev. 2009, 38, 62-72.6. (a) Muniz, K., Angew. Chem. Int. Ed. 2005, 44, 6622-6627; (b) Ikariya, T.; Murata, K.; Noyori, R., Org. Biomol. Chem. 2006, 4, 393-406; (c) Ito, M.; Ikariya, T., Chem. Commun. 2007, 5134-5142.7. (a) Guo, R.; Morris, R. H.; Song, D., J. Am. Chem. Soc. 2005, 127, 516-517; (b) Gridnev, I. D.; Watanabe, M.; Wang, H.; Ikariya, T., J. Am. Chem. Soc. 2010, 132, 16637-16650; (c) Ikariya, T.; Gridnev, I. D., Top. Catal. 2010, 53, 894-901.8. (a) Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D., J. Am. Chem. Soc. 2005, 127, 10840-10841; (b) Zweifel, T.; Naubron, J. V.; Grützmacher, H., Angew. Chem. Int. Ed. 2009, 48, 559-563.

152The “NH effect”, which involves the bifunctional action of M–H/N–H groups to attack a polar
bond in the outer coordination sphere, has been studied the most both experimentally11 and
theoretically.12 The ketone hydrogenation is proposed to proceed via a six-membered pericyclic
transition state involving hydrogen bonding of the N-H group with the oxygen of the ketone and
attack of the carbonyl group by the metal hydride.3a, 5c Many research groups,12e, 13 including
ours,12l, 14 have shown that the heterolytic splitting of a coordinated η2-H2 ligand is rate
determining.12d, 12e, 12h, 12i, 12k-m, 13-15 The key intermediates of the catalytic cycle involving the trans-
dihydride-amine and the monohydrido-amido catalysts of RuXY(diamine)(phosphine)2 type
systems (X = H, Cl; Y = H, BH4, H, OR) have been identified and isolated.14, 16 Ruthenium(II)
alkoxide complexes have been observed at low temperature prior to the formation of an amido
species17 and one of these has been recently isolated.18 Under optimum conditions, such catalysts
allow hydrogenation to occur under mild conditions: room temperature, low hydrogen pressures
with no added base.14, 16a, 19 The importance of the N-H group was demonstrated by the fact that
systems containing only a metal hydride were found to be less active and selective and required
forcing conditions in the hydrogenation of ketones.20 However, the N–H group is not needed for
the hydrogenation of β-keto-esters which chelate to the metal center.21
9. (a) Gunanathan, C.; Ben-David, Y.; Milstein, D., Science 2007, 317, 790-792; (b) Gnanaprakasam, B.; Milstein, D., J. Am. Chem. Soc. 2011, 133, 1682-1685.10. Milstein, D., Top. Catal. 2010, 53, 915-923 and references therein.11. (a) Rosales, M., Coord. Chem. Rev. 2000, 196, 249-280; (b) Maire, P.; Buttner, T.; Breher, F.; Le Floch, P.; Grützmacher, H., Angew. Chem. Int. Ed. 2005, 44, 6318-6323; (c) Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Sandoval, C.; Noyori, R., J. Am. Chem. Soc. 2006, 128, 8724-8725; (d) Kass, M.; Friedrich, A.; Drees, M.; Schneider, S., Angew. Chem. Int. Ed. 2009, 48, 905-907; (e) Friedrich, A.; Drees, M.; auf der Gunne, J. S.; Schneider, S., J. Am. Chem. Soc. 2009, 131, 17552-17553; (f) Cheung, F. K.; Clarke, A. J.; Clarkson, G. J.; Fox, D. J.; Graham, M. A.; Lin, C. X.; Criville, A. L.; Wills, M., Dalton Trans. 2010, 39, 1395-1402; (g) Picot, A.; Dyer, H.; Buchard, A.; Auffrant, A.; Vendier, L.; Le Floch, P.; Sabo-Etienne, S., Inorg. Chem. 2010, 49, 1310-1312; (h) Bertoli, M.; Choualeb, A.; Gusev, D. G.; Lough, A. J.; Major, Q.; Moore, B., Dalton Trans. 2011, 40, 8941-8949.12. (a) Alonso, D. A.; Brandt, P.; Nordin, S. J. M.; Andersson, P. G., J. Am. Chem. Soc. 1999, 121, 9580-9588; (b) Petra, D. G. I.; Reek, J. N. H.; Handgraaf, J. W.; Meijer, E. J.; Dierkes, P.; Kamer, P. C. J.; Brussee, J.; Schoemaker, H. E.; van Leeuwen, P., Chem. Eur. J. 2000, 6, 2818-2829; (c) Yamakawa, M.; Ito, H.; Noyori, R., J. Am. Chem. Soc. 2000, 122, 1466-1478; (d) Handgraaf, J. W.; Reek, J. N. H.; Meijer, E. J., Organometallics 2003, 22, 3150-3157; (e) Hedberg, C.; Kallstrom, K.; Arvidsson, P. I.; Brandt, P.; Andersson, P. G., J. Am. Chem. Soc. 2005, 127, 15083-15090; (f) Handgraaf, J. W.; Meijer, E. J., J. Am. Chem. Soc. 2007, 129, 3099-3103; (g) Comas-Vives, A.; Ujaque, G.; Lledos, A., Organometallics 2007, 26, 4135-4144; (h) Di Tommaso, D.; French, S. A.; Catlow, C. R. A., J. Mol. Struct. (THEOCHEM) 2007, 812, 39-49; (i) Puchta, R.; Dahlenburg, L.; Clark, T., Chem. Eur. J. 2008, 14, 8898-8903; (j) Chen, Y.; Liu, S. B.; Lei, M., J. Phys. Chem. C 2008, 112, 13524-13527; (k) Chen, Y.; Tang, Y. H.; Lei, M., Dalton Trans. 2009, 2359-2364; (l) Zimmer-De Iuliis, M.; Morris, R. H., J. Am. Chem. Soc. 2009, 131, 11263-11269; (m) Lei, M.; Zhang, W. C.; Chen, Y.; Tang, Y. H., Organometallics 2010, 29, 543-548; (n) Li, H. X.; Lu, G.; Jiang, J. L.; Huang, F.; Wang, Z. X., Organometallics 2011, 30, 2349-2363.13. (a) Sandoval, C. A.; Ohkuma, T.; Muniz, K.; Noyori, R., J. Am. Chem. Soc. 2003, 125, 13490-13503; (b) Hamilton, R. J.; Leong, C. G.; Bigam, G.; Miskolzie, M.; Bergens, S. H., J. Am. Chem. Soc. 2005, 127, 4152-4153; (c) Sandoval, C. A.; Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Noyori, R., Chem. Asian J. 2006, 1, 102-110.

153The effect of alcohol in bifunctional catalysis using the “NH effect” has also been well
studied. Combined computational and experimental studies on many systems have shown that
the presence of alcohol in catalysis plays an active role to serve as a proton shuttle that assists in
the heterolytic splitting of the η2-H2 ligand. 6c, 12e, 13a, 14b, 19b, 20b, 22 This decreases the energy barrier
for the activation of H2 but does not significantly change the energy barrier for the transfer of a
proton/hydride couple to the ketone in a hydrogen-bonded network.12e, 19b
Certain ruthenium(II) complexes bearing a phosphine–amine (P–NH2) ligand, including the
complexes trans-Ru(H)2((S)-BINAP)(P–NH2)23 and RuCp*(κ2(P,N)-PPh2CH2CH2NH2)H24
(Figure 6.1) catalyze efficiently the hydrogenation of a variety of polar bonds including those of
ketones,20h, 23, 25 imines,16b esters and lactones,26 epoxides24 and cyclic imides.27 The notion of
replacing a phosphine with a more electron-donating N-heterocyclic carbene (NHC) donor is
attractive with the promise of a reduction in the toxicity of catalyst precursors and contaminants
in the hydrogenated products leading to greener catalysis.28 Thus, the replacement of phosphine
by an NHC donor would give a donor-functionalized NHC29 containing an NHC ligand and a
14. (a) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A.; Harvey, J. N.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2002, 124, 15104-15118; (b) Rautenstrauch, V.; Hoang-Cong, X.; Churlaud, R.; Abdur-Rashid, K.; Morris, R. H., Chem. Eur. J. 2003, 9, 4954-4967.15. Abbel, R.; Abdur-Rashid, K.; Faatz, M.; Hadzovic, A.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2005, 127, 1870-1882.16. (a) Abdur-Rashid, K.; Faatz, M.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2001, 123, 7473-7474; (b) Abdur-Rashid, K.; Guo, R. W.; Lough, A. J.; Morris, R. H.; Song, D. T., Adv. Synth. Catal. 2005, 347, 571-579.17. (a) Hamilton, R. J.; Bergens, S. H., J. Am. Chem. Soc. 2008, 130, 11979-11987; (b) Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H., J. Am. Chem. Soc. 2011, 133, 9666-9669.18. Bertoli, M.; Choualeb, A.; Lough, A. J.; Moore, B.; Spasyuk, D.; Gusev, D. G., Organometallics 2011, 30, 3479-3482 19. (a) Clapham, S. E.; Morris, R. H., Organometallics 2005, 24, 479-481; (b) Hadzovic, A.; Song, D.; MacLaughlin, C. M.; Morris, R. H., Organometallics 2007, 26, 5987-5999.20. (a) Standfest-Hauser, C.; Slugovc, C.; Mereiter, K.; Schmid, R.; Kirchner, K.; Xiao, L.; Weissensteiner, W., Dalton Trans. 2001, 2989-2995; (b) Ito, M.; Hirakawa, M.; Murata, K.; Ikariya, T., Organometallics 2001, 20, 379-381; (c) Leong, C. G.; Akotsi, O. M.; Ferguson, M. J.; Bergens, S. H., Chem. Commun. 2003, 750-751; (d) Lundgren, R. J.; Rankin, M. A.; McDonald, R.; Schatte, G.; Stradiotto, M., Angew. Chem. Int. Ed. 2007, 46, 4732-4735; (e) Ma, G. B.; McDonald, R.; Ferguson, M.; Cavell, R. G.; Patrick, B. O.; James, B. R.; Hu, T. Q., Organometallics 2007, 26, 846-854; (f) Baratta, W.; Ballico, M.; Esposito, G.; Rigo, P., Chem. Eur. J. 2008, 14, 5588-5595; (g) Sandoval, C. A.; Shi, Q. X.; Liu, S. S.; Noyori, R., Chem. Asian J. 2009, 4, 1221-1224; (h) Phillips, S. D.; Fuentes, J. A.; Clarke, M. L., Chem. Eur. J. 2010, 16, 8002-8005; (i) Soni, R.; Cheung, F. K.; Clarkson, G. C.; Martins, J. E. D.; Graham, M. A.; Wills, M., Org. Biomol. Chem. 2011, 9, 3290-3294.21. (a) Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, S., J. Am. Chem. Soc. 1987, 109, 5856-5858; (b) Daley, C. J. A.; Wiles, J. A.; Bergens, S. H., Can. J. Chem. 1998, 76, 1447-1456; (c) Noyori, R.; Ohkuma, T., Angew. Chem. Int. Ed. 2001, 40, 40-73; (d) Daley, C. J. A.; Bergens, S. H., J. Am. Chem. Soc. 2002, 124, 3680-3691.22. (a) Casey, C. P.; Johnson, J. B.; Singer, S. W.; Cui, Q., J. Am. Chem. Soc. 2005, 127, 3100-3109; (b) Heiden, Z. M.; Rauchfuss, T. B., J. Am. Chem. Soc. 2009, 131, 3593-3600; (c) Casey, C. P.; Johnson, J. B.; Jiao, X. D.; Beetner, S. E.; Singer, S. W., Chem. Commun. 2010, 46, 7915-7917.

154primary amine donor (C–NH2). Douthwaite and co-workers reported the synthesis of the first
palladium complex containing such a C–NH2 ligand (Figure 6.1).30
Figure 6.1. Late transition metal complexes containing a chelating N-heterocyclic carbene
(NHC)-primary amine (C–NH2) or a chelating phosphine-primary amine (P–NH2) ligand.
We then began to explore the use of a more conveniently prepared C–NH2 ligand in metal
complexes for the catalytic hydrogenation of ketones using H2 or 2-propanol as the hydrogen
source. The complexes [M(p-cymene)(C–NH2)Cl]PF6 (13, M = Ru;31 14, M = Os,32 Figure 6.1),
were prepared by the transmetalation reaction31 of a nickel(II) complex, [Ni(C–NH2)2](PF6)2 (12,
Figure 6.1 and see Chapter 4), with the appropriate precursor complex. The hydride-amine
complex, [Ru(p-cymene)(C–NH2)H]PF6 (17, see Chapter 5), was prepared, but this failed to
transfer its proton/hydride couple to acetophenone in either stoichiometric or catalytic reactions.
Experimental and computational studies suggested that an outer sphere bifunctional mechanism
23. Guo, R.; Lough, A. J.; Morris, R. H.; Song, D., Organometallics 2004, 23, 5524-5529.24. Ito, M.; Hirakawa, M.; Osaku, A.; Ikariya, T., Organometallics 2003, 22, 4190-4192.25. Jia, W. L.; Chen, X. H.; Guo, R. W.; Sui-Seng, C.; Amoroso, D.; Lough, A. J.; Abdur-Rashid, K., Dalton Trans. 2009, 8301-8307.26. (a) Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P., Angew. Chem. Int. Ed. 2007, 46, 7473-7476; (b) Clarke, M. L.; Diaz-Valenzuela, M. B.; Slawin, A. M. Z., Organometallics 2007, 26, 16-19; (c) Kuriyama, W.; Ino, Y.; Ogata, O.; Sayo, N.; Saito, T., Adv. Synth. Catal. 2010, 352, 92-96.27. (a) Ito, M.; Sakaguchi, A.; Kobayashi, C.; Ikariya, T., J. Am. Chem. Soc. 2007, 129, 290-291; (b) Ito, M.; Kobayashi, C.; Himizu, A.; Ikariya, T., J. Am. Chem. Soc. 2010, 132, 11414-11415.28. (a) Lee, H. M.; Lee, C. C.; Cheng, P. Y., Curr. Org. Chem. 2007, 11, 1491-1524; (b) Hahn, F. E.; Jahnke, M. C., Angew. Chem. Int. Ed. 2008, 47, 3122-3172; (c) Diez-Gonzalez, S.; Marion, N.; Nolan, S. P., Chem. Rev. 2009, 109, 3612-3676.29. (a) Normand, A. T.; Cavell, K. J., Eur. J. Inorg. Chem. 2008, 2781-2800; (b) Corberan, R.; Mas-Marza, E.; Peris, E., Eur. J. Inorg. Chem. 2009, 1700-1716.30. Bonnet, L. G.; Douthwaite, R. E.; Hodgson, R.; Houghton, J.; Kariuki, B. M.; Simonovic, S., Dalton Trans. 2004, 3528-3535.31. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2009, 28, 6755-6761.32. O, W. W. N.; Lough, A. J.; Morris, R. H., Organometallics 2011, 30, 1236-1252.
RuCl NNN
CH3
HH
NiN
NN N
NN
H3CCH3
2+
(PF6)2
H H H H
PF6
Ru
N PPh2HHH
Ru
NN
N
CH3 PF6NHH
Ikariya
12
15
N
NPh
nPr
NPd
ClClHH
Douthwaite 13
Ru
H
HN
Ph2P
CH3
Ph
H H
PPh2
Ph2P
t rans-Ru(H)2((S)-BINAP)(P-NH2)
+
+

155was not viable for this cationic hydride-amine complex, and that an inner sphere mechanism
involving the decoordination of the primary amine group was more likely to occur (Figure 6.2).32
Figure 6.2. An inner sphere mechanism involving the decoordination of the primary amine
group proposed for the H2-hydrogenation of ketones catalyzed by complex 13 in the presence of
an alkoxide base.
Although there are many metal complexes containing an amine-functionalized NHC ligand,30,
33 none were reported to be useful for ketone hydrogenation33c, 33f apart from our work.34 We have
prepared the ruthenium(II) complex, [RuCp*(C–NH2)py]PF6 (15, Cp* = pentamethyl-
cyclopentadienyl ligand; py = pyridine, Figure 6.1) and found it to be very active for the
hydrogenation of a variety of ketones, styrene oxide, an aromatic ester and a ketimine with high
substrate to catalyst loadings under very mild reaction conditions (8 bar of H2 pressure, 25°C): a
maximum turnover frequency (TOF) of 17300 h-1 is achieved with this catalyst in the H2-
hydrogenation of acetophenone in 2-propanol.34 This system has activity that is superior to the
related catalysts, RuCp*(κ2(P,N)-PPh2CH2CH2NH2)Cl (16b), and RuCp*(diamine)Cl.12e, 20b, 24
In the current study35 we show that when 15 is activated by an alkoxide base, it follows an
alcohol-assisted outer sphere bifunctional mechanism in the H2-hydrogenation of ketones
involving the action of a metal hydride and a protic amine group. To our surprise, a structurally
similar iridium(III) complex when it is activated with an excess of alkoxide base in 2-propanol,
33. (a) Arnold, P. L.; Mungur, S. A.; Blake, A. J.; Wilson, C., Angew. Chem. Int. Ed. 2003, 42, 5981-5984; (b) Douthwaite, R. E.; Houghton, J.; Kariuki, B. M., Chem. Commun. 2004, 698-699; (c) Jong, H.; Patrick, B. O.; Fryzuk, M. D., Can. J. Chem. 2008, 86, 803-810; (d) Wei, W.; Qin, Y.; Luo, M.; Xia, P.; Wong, M. S., Organometallics 2008, 27, 2268-2272; (e) Arnold, P. L.; McMaster, J.; Liddle, S. T., Chem. Commun. 2009, 818-820; (f) Dyson, G.; Frison, J. C.; Whitwood, A. C.; Douthwaite, R. E., Dalton Trans. 2009, 7141-7151; (g) Cross, W. B.; Daly, C. G.; Ackerman, R. L.; George, I. R.; Singh, K., Dalton Trans. 2011, 40, 495-505; (h) Jong, H.; Patrick, B. O.; Fryzuk, M. D., Organometallics 2011, 30, 2333-2341.34. O, W. W. N.; Lough, A. J.; Morris, R. H., Chem. Commun. 2010, 46, 8240 - 8242.35. O, W. W. N.; Lough, A. J.; Morris, R. H., Submitted 2011.
OR
HOR
H
Inner Sphere
H2
CH3NN
RuNH
H HO
R'
R
+

156likely undergoes the same mechanism involving iridium(I) intermediates (Scheme 6.1). Herein,
we will present both experimental and theoretical evidences to support the two elementary steps
in the alcohol-assisted outer sphere bifunctional mechanism: a) the heterolytic splitting of
dihydrogen at the active metal center, aided by a 2-propanol molecule acting as an proton shuttle
(Step A in Scheme 6.1); b) the transfer of an M–H/N–H couple to the ketone in the outer sphere
(Step B in Scheme 6.1). Other possible mechanisms including a hydride attack on a coordinated
ketone in the inner coordination sphere will be described briefly. Of note, diagonal relationships
between late transition metals were studied for the dihydrogen complexes [M(η2-
H2)2(H)2(PCy3)]n+ (M = Ru, n = 0; M = Ir, n = 1),36 and more recently their use in the activation
of B–H bonds.37 Also relevant to the current work is a computational study of the transfer
hydrogenation of ketones catalyzed by structurally similar ruthenium(II) and iridium(I)
systems.12d In addition, a comparison between the C–NH2 and analogous P–NH2 ligand in
ruthenium(II) and iridium(III) catalysts allows us to evaluate the relative merits of these ligands
in the homogenous hydrogenation of ketones.
Scheme 6.1. An Alcohol-assisted Outer Sphere Bifunctional Mechanism of H2-Hydrogenation of
Ketones Catalyzed by Ruthenium(II) and Iridium(I) Systems Containing a C–NH2 Ligand.
36. (a) Crabtree, R. H.; Lavin, M.; Bonneviot, L., J. Am. Chem. Soc. 1986, 108, 4032-4037; (b) Arliguie, T.; Chaudret, B.; Morris, R. H.; Sella, A., Inorg. Chem. 1988, 27, 598-599.37. Alcaraz, G.; Chaplin, A. B.; Stevens, C. J.; Clot, E.; Vendier, L.; Weller, A. S.; Sabo-Etienne, S., Organometallics 2010, 29, 5591-5595.
M NNN
CH3
H
MH NNN
CH3
HH
H2
OR'
OHH
R'
M NNN
CH3
H
HH
MH NNN
CH3
HH
O
R'
RR
R
O HH
OHH
OHH
OH
HAlcohol-Assisted Outer Sphere
Bifunctional Mechanism
M = Ru,
H
= η 5-
= η 4-M = Ir,
Step A
Step B

1576.3 Results and Discussion
6.3.1 Synthesis of Ruthenium(II) and Iridium(III) Complexes Containing a C–NH2 Ligand.
The ruthenium(II) complex, [RuCp*(C–NH2)py]PF6 (15), was prepared by a transmetalation
reaction31, 34 of 12 and RuCp*(cod)Cl in refluxing acetonitrile followed by the addition of pyridine
in tetrahydrofuran (THF, Scheme 6.2). The characterization of this complex has been described
in Chapter 4.34 The structurally similar iridium(III) complex was thereby synthesized similarly
using 1231, 34 and [IrCp*Cl2]2 in refluxing acetonitrile. This afforded the complex, [IrCp*(C–
NH2)Cl]PF6 (19), as an air stable yellow powder upon isolation (Scheme 6.2). The solid state
structure of 19 has a piano-stool geometry at the iridium center with coordinated Cp*, C–NH2
and chloride ligands (Figure 6.3). The Ir–Ccarbene distance and the Ir–Ccarbene resonance in its 13C{1H} NMR spectrum are typical of most iridium(III) complexes containing an NHC ligand.38
To compare the donor strength properties of a phosphine with an N-heterocyclic carbene, the
diphenylphosphino-containing complex [IrCp*(P–NH2)Cl]PF6 (20, (P–NH2 = 2-(diphenyl-
phosphino)benzylamine herein, Scheme 6.3 and Figure 6.4) was prepared and has been
structurally characterized. The ruthenium(II) analogue (complex 16a) was previously described
in Chapter 4.34
Scheme 6.2. Synthesis of Iridium(III) and Ruthenium(II) Complexes Containing a C–NH2
Ligand.
NiN
NN N
NN
H3CCH3
2+
(PF6)2
H H H H
12
THF, rtCO (1 atm)
Ru
NN
N
CH3 PF6OCHH
23
1) 1.5 RuCp*(cod)Cl∆ , CH3CN
Ru
NN
N
CH3 PF6N
HH
15
2) pyridine, THF, rt
[IrCp*Cl2]2 ∆ , CH3CN
Ir
NN
N
CH3 PF6Cl2HH
19
1.5
1.5
Ir
NN
N
CH3 PF6H2HH
iPrOH, 50°C
6 NaOiPr
+
+
+
+
21
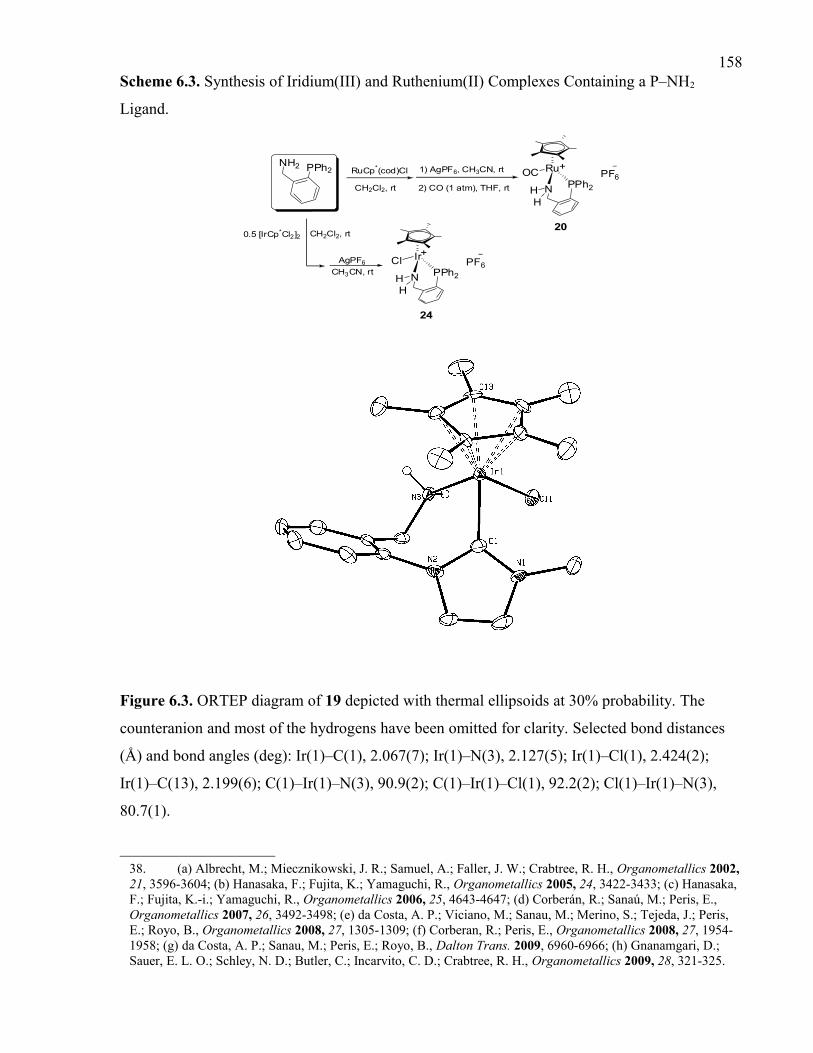
158Scheme 6.3. Synthesis of Iridium(III) and Ruthenium(II) Complexes Containing a P–NH2
Ligand.
Figure 6.3. ORTEP diagram of 19 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. Selected bond distances
(Å) and bond angles (deg): Ir(1)–C(1), 2.067(7); Ir(1)–N(3), 2.127(5); Ir(1)–Cl(1), 2.424(2);
Ir(1)–C(13), 2.199(6); C(1)–Ir(1)–N(3), 90.9(2); C(1)–Ir(1)–Cl(1), 92.2(2); Cl(1)–Ir(1)–N(3),
80.7(1).
38. (a) Albrecht, M.; Miecznikowski, J. R.; Samuel, A.; Faller, J. W.; Crabtree, R. H., Organometallics 2002, 21, 3596-3604; (b) Hanasaka, F.; Fujita, K.; Yamaguchi, R., Organometallics 2005, 24, 3422-3433; (c) Hanasaka, F.; Fujita, K.-i.; Yamaguchi, R., Organometallics 2006, 25, 4643-4647; (d) Corberán, R.; Sanaú, M.; Peris, E., Organometallics 2007, 26, 3492-3498; (e) da Costa, A. P.; Viciano, M.; Sanau, M.; Merino, S.; Tejeda, J.; Peris, E.; Royo, B., Organometallics 2008, 27, 1305-1309; (f) Corberan, R.; Peris, E., Organometallics 2008, 27, 1954-1958; (g) da Costa, A. P.; Sanau, M.; Peris, E.; Royo, B., Dalton Trans. 2009, 6960-6966; (h) Gnanamgari, D.; Sauer, E. L. O.; Schley, N. D.; Butler, C.; Incarvito, C. D.; Crabtree, R. H., Organometallics 2009, 28, 321-325.
NH2 PPh2 RuPPh2N
PF6OCCH2Cl2, rt H
H
20
RuCp*(cod)Cl
2) CO (1 atm), THF, rt
1) AgPF6, CH3CN, rt
IrPPh2N
PF6Cl
CH2Cl2, rt
HH
24
0.5 [IrCp*Cl2]2
AgPF6CH3CN, rt
+
+

159
Figure 6.4. ORTEP diagram of 20 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. Selected bond distances
(Å) and bond angles (deg): Ir(1)–P(1), 2.307(2); Ir(1)–N(1), 2.146(8); Ir(1)–Cl(1), 2.407(2);
Ir(1)–C(24), 2.208(9); P(1)–Ir(1)–N(1), 88.9(2); P(1)–Ir(1)–Cl(1), 89.02(9); Cl(1)–Ir(1)–N(1),
82.3(2).
6.3.2 Synthesis, Observation, and Reactivity of Hydride Complexes of Ruthenium(II) and
Iridium(III). Hydride intermediates have been proposed in the catalytic cycles for the
hydrogenation of polar bonds and the racemization of chiral alcohols. The complex,
RuCp*(κ2(P,N)-PPh2CH2CH2NH2)H, which was suggested to be present during the catalytic
hydrogenolysis of epoxides, has been observed but not isolated.24 Hydride intermediates have
been observed spectroscopically in the racemization catalyst system containing Ru(η5-Cp*)
(ICy)Cl, NaOtBu and (S)-1-phenylethanol (ICy = 1,3-dicyclohexylimidazol-2-ylidene).39 The
complex, Ru(η5-C5Ph5)(CO)2H, has been isolated and this catalyzes the racemization of (S)-1-
phenylethanol40 via an inner sphere mechanism upon the dissociation of the carbonyl ligand.41
In order to provide evidence to support an alcohol-assisted outer sphere bifunctional
mechanism, we sought to observe or isolate the hydride-amine complex, [MCp*(C–NH2)H]n+ (M
= Ru, n = 0; M = Ir, n = 1), which can potentially transfer a proton/hydride couple from its M–
H/N–H group to the ketone (Step B in Scheme 6.1).3a, 6b, 6c Attempts to isolate such a complex for
the ruthenium(II) system were not successful. Reactions of excess or stoichiometric amounts of 39. Bosson, J.; Poater, A.; Cavallo, L.; Nolan, S. P., J. Am. Chem. Soc. 2010, 132, 13146-13149.
40. Martín-Matute, B.; Edin, M.; Bogár, K.; Kaynak, F. B.; Bäckvall, J. E., J. Am. Chem. Soc. 2005, 127, 8817-8825.41. Warner, M. C.; Verho, O.; Backvall, J. E., J. Am. Chem. Soc. 2011, 133, 2820-2823.

160base (potassium tert-butoxide (KOtBu), NaOiPr, NaOMe, NaBH4, KOH, KH or K-Selectride)
with complex 15 in THF under a hydrogen atmosphere (up to 8 bar) or in 2-propanol solution
gave intractable products. Nevertheless, a hydride peak was observed at -9.23 ppm in THF-d8
when the reaction was carried out under a hydrogen atmosphere in the presence of KOtBu.
Attempts to utilize such reaction mixture upon removal of excess KOtBu in the catalytic
hydrogenation of acetophenone (0.15 or 1.9 M) using 8 bar of H2 pressure at 25°C in the absence
of base gave no conversion to the product alcohol.
However, reactions of complex 15 with sodium 2-propoxide (2.6 equiv.) or potassium 1-
phenylethoxide (1.3 equiv.) in THF-d8 at 25°C under argon gave a species containing a new
hydride peak at -9.47 ppm. The relative integration of this peak against the alkoxide suggested
that 6% of the starting material was converted to this hydride. A hydride peak at -9.23 ppm was
also observed in a smaller amount in these two experiments. Acetone and acetophenone that
were produced from β-hydride elimination of the alkoxides were observed as well with 2% and
8% conversion, respectively. After leaving the solutions for 1 day in sealed NMR tubes at 25°C,
the hydride peaks disappeared and both samples contained less than 2% of the ketone. These
results suggest that the hydride-containing species at -9.47 ppm in THF-d8 may represent the true
catalytically active species, instead of the one that was observed at -9.23 ppm. These hydride
complexes, however, are difficult to identify due to the complexity of the reaction mixture and
their high reactivity.
The iridium(III) hydride-amine complex [IrCp*(C–NH2)H]PF6 (21), on the other hand, was
successfully prepared from a warm 2-propanol solution of 19 containing three equiv. of sodium
2-propoxide. The analytically pure compound can be isolated in moderate yields (Scheme 6.2).
In determining its solid state structure (Figure 6.5), the hydride position in the piano-stool
complex was not refined but instead was located approximately by use of an electron density
difference map.35 The Ir–H distance was thus determined to be 1.54 Å, which is typical of
iridium hydride complexes containing a Cp* ligand.42 The Ir–Ccarbene distance for 21 (2.015(5) Å)
is shorter than that of 19 (2.067(7) Å). The Ir–Ccarbene and Ir–H resonances of 21 in acetonitrile-d3
were observed at 157.0 and -13.59 ppm in the 13C{1H} and 1H NMR spectra, respectively. The
characteristic Ir–H stretch was observed at 2068 cm-1 in the infrared spectrum.
42. (a) Buchanan, J. M.; Stryker, J. M.; Bergman, R. G., J. Am. Chem. Soc. 1986, 108, 1537-1550; (b) Glueck, D. S.; Winslow, L. J. N.; Bergman, R. G., Organometallics 1991, 10, 1462-1479; (c) Ohki, Y.; Sakamoto, M.; Tatsumi, K., J. Am. Chem. Soc. 2008, 130, 11610-11611.

161
Figure 6.5. ORTEP diagram of 21 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. The position of the
hydride ligand is not refined and thus not shown. Selected bond distances (Å) and bond angles
(deg): Ir(1)–C(1), 2.015(5); Ir(1)–N(3), 2.151(4); Ir(1)–C(14), 2.233(3); C(1)–Ir(1)–N(3),
89.6(2).
The bifunctional Ir–H and N–H groups of the hydride-amine complex 21 should react with a
polar double bond if it is truly an intermediate of the outer sphere mechanism that utilizes the
“NH effect”. This was tested in a stoichiometric reaction. The reaction mixture of 21 and
acetophenone in THF-d8 when heated at 50°C did not result in any product alcohol. The addition
of 1.5 to 2 equiv. of KOtBu prior to or after the addition of acetophenone to 21 in THF-d8 did not
give the reduced product. It appears that metal hydride is not hydridic enough and therefore not
reactive towards the polar double bond, in line with the properties of the cationic complex [Ru(p-
cymene)(C–NH2)H]PF6 (17) that we have isolated.32 Crabtree and co-workers have isolated an
iridium(III) complex, [Ir(η5-HOC5H4)(PPh3)2H]BF4¸which failed to react with aldehydes.43
Attempts to observe an iridium(III) dihydrogen complex [IrCp*(C–NH2)(η2-H2)]2+ similar to the
bis-NHC complex [IrCp*(C–C)(η2-H2)]2+ 44 and measure its pKa value by equilibrating known
amounts of 21, the phosphonium salts [HPCy3]BPh4 (pKαTHF
= 9.7)45 or [HPtBu3]BPh4 (pKαTHF
=
10.6)45 in THF-d8 were not successful due to the facile displacement of the coordinated
43. Li, X. W.; Chen, P.; Faller, J. W.; Crabtree, R. H., Organometallics 2005, 24, 4810-4815.44. Vogt, M.; Pons, V.; Heinekey, D. M., Organometallics 2005, 24, 1832-1836.45. Abdur-Rashid, K.; Fong, T. P.; Greaves, B.; Gusev, D. G.; Hinman, J. G.; Landau, S. E.; Lough, A. J.; Morris, R. H., J. Am. Chem. Soc. 2000, 122, 9155-9171.

162dihydrogen by the phosphine that forms. We conclude that complex 21 is unlikely to be an active
species involved in step B of the alcohol-assisted outer sphere bifunctional mechanism (Scheme
6.1).
6.3.3 Synthesis of an Iridium(III) Complex Containing a C–NMe2 Ligand. The importance of
the N–H group in the outer sphere bifunctional catalysis involving the action of M–H/N–H (Step
B in Scheme 6.1) has been demonstrated for several catalysts.20b, 20f-i, 32 However, there are active
systems for the hydrogenation of ketones that lack the N–H group; these, in most cases, operate
by an inner sphere mechanism which involves the coordination of the ketone to the metal
center.20a, 20c-e We attempted to address this issue by the preparation of an analogous iridium(III)
complex with an N-heterocyclic carbene containing a tethered tertiary amine donor (C–NMe2).
When chelated, this ligand forms a seven-membered ring including the metal center. The desired
complex was synthesized in one pot in two steps, by first the in situ generation of a silver(I)
complex containing the C–NMe2 ligand from the reaction of silver(I) oxide and 1-(N,N-
dimethylamino)propyl-3-methylimidazolium chloride hydrochloride (HC–NMe2·HCl, 1g),46 and
the C–NMe2 ligand was transmetalated from silver(I) to [IrCp*Cl2]2 in the same pot in the second
step (Scheme 6.4).32, 47 A salt metathesis reaction with AgPF6 afforded a pale yellow powder
upon isolation, which was identified as [IrCp*(C–NMe2)Cl]PF6 (22) by NMR spectroscopy and
an X-ray diffraction study (Figure 6.6). In comparison to 19, the piano-stool complex has a
shorter Ir–Ccarbene distance (19: 2.067(7)Å, 22: 2.036(4)Å), a longer Ir–N distance (19: 2.127(5)Å,
22: 2.225(4)Å) and a smaller C(1)–Ir(1)–N(3) bite angle of the chelate (19: 90.9(2)o, 22:
87.5(2)o), otherwise it has similar NMR spectroscopic properties to those of other iridium(III)
complexes containing an NHC ligand.38c
Scheme 6.4. Synthesis of an Iridium(III) Complex Containing a C–NMe2 ligand.
46. Jimenez, M. V.; Perez-Torrente, J. J.; Bartolome, M. I.; Gierz, V.; Lahoz, F. J.; Oro, L. A., Organometallics 2008, 27, 224-234.47. Warsink, S.; de Boer, S. Y.; Jongens, L. M.; Fu, C. F.; Liu, S. T.; Chen, J. T.; Lutz, M.; Spek, A. L.; Elsevier, C. J., Dalton Trans. 2009, 7080-7086.
0.5 [IrCp*Cl2]2DMSO:CH3CN (1:4), rt
N NCH3 N HCl1.5 Ag2O
AgPF6
CH3CN:CH2Cl2 (1:1), rt
1g
Ir
NN
N
CH3 PF6Cl
22
+
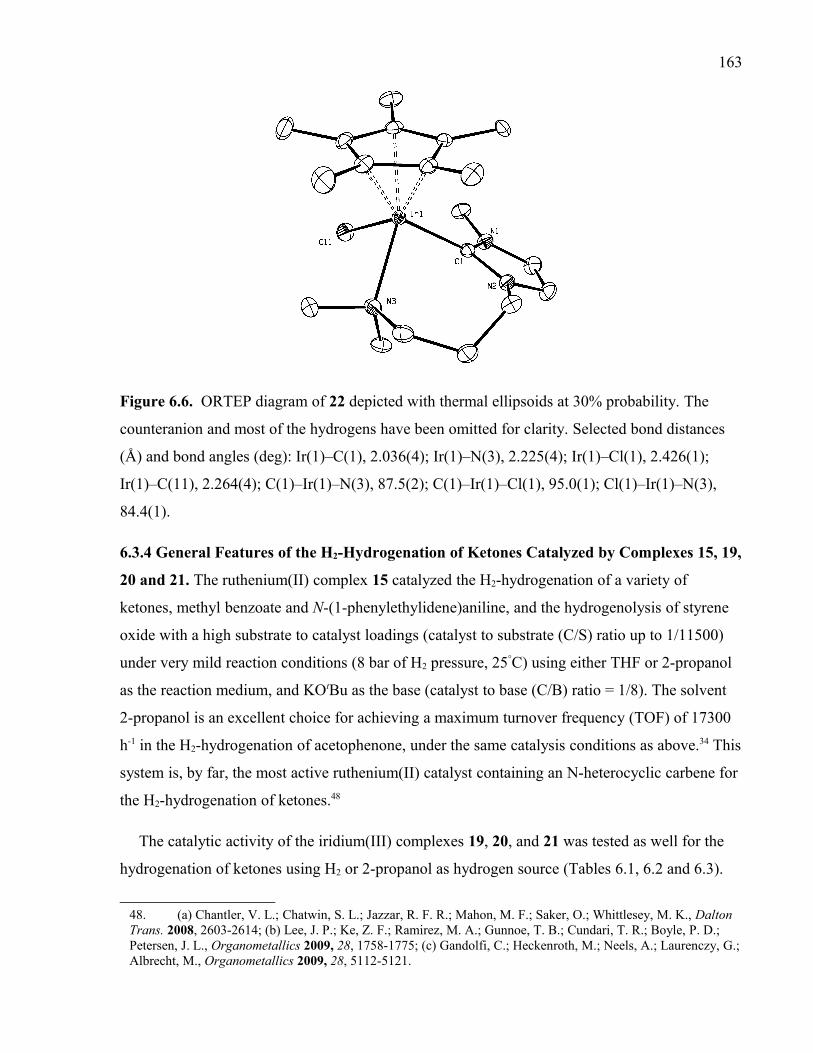
163
Figure 6.6. ORTEP diagram of 22 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. Selected bond distances
(Å) and bond angles (deg): Ir(1)–C(1), 2.036(4); Ir(1)–N(3), 2.225(4); Ir(1)–Cl(1), 2.426(1);
Ir(1)–C(11), 2.264(4); C(1)–Ir(1)–N(3), 87.5(2); C(1)–Ir(1)–Cl(1), 95.0(1); Cl(1)–Ir(1)–N(3),
84.4(1).
6.3.4 General Features of the H2-Hydrogenation of Ketones Catalyzed by Complexes 15, 19,
20 and 21. The ruthenium(II) complex 15 catalyzed the H2-hydrogenation of a variety of
ketones, methyl benzoate and N-(1-phenylethylidene)aniline, and the hydrogenolysis of styrene
oxide with a high substrate to catalyst loadings (catalyst to substrate (C/S) ratio up to 1/11500)
under very mild reaction conditions (8 bar of H2 pressure, 25°C) using either THF or 2-propanol
as the reaction medium, and KOtBu as the base (catalyst to base (C/B) ratio = 1/8). The solvent
2-propanol is an excellent choice for achieving a maximum turnover frequency (TOF) of 17300
h-1 in the H2-hydrogenation of acetophenone, under the same catalysis conditions as above.34 This
system is, by far, the most active ruthenium(II) catalyst containing an N-heterocyclic carbene for
the H2-hydrogenation of ketones.48
The catalytic activity of the iridium(III) complexes 19, 20, and 21 was tested as well for the
hydrogenation of ketones using H2 or 2-propanol as hydrogen source (Tables 6.1, 6.2 and 6.3).
48. (a) Chantler, V. L.; Chatwin, S. L.; Jazzar, R. F. R.; Mahon, M. F.; Saker, O.; Whittlesey, M. K., Dalton Trans. 2008, 2603-2614; (b) Lee, J. P.; Ke, Z. F.; Ramirez, M. A.; Gunnoe, T. B.; Cundari, T. R.; Boyle, P. D.; Petersen, J. L., Organometallics 2009, 28, 1758-1775; (c) Gandolfi, C.; Heckenroth, M.; Neels, A.; Laurenczy, G.; Albrecht, M., Organometallics 2009, 28, 5112-5121.
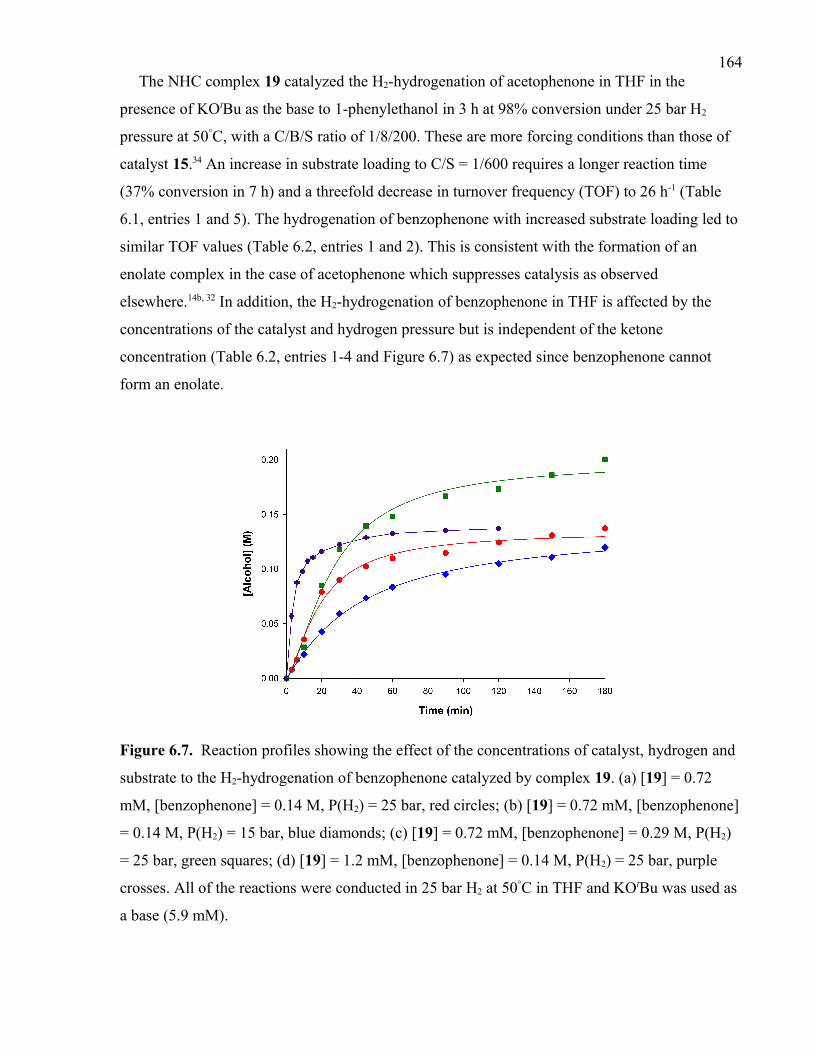
164The NHC complex 19 catalyzed the H2-hydrogenation of acetophenone in THF in the
presence of KOtBu as the base to 1-phenylethanol in 3 h at 98% conversion under 25 bar H2
pressure at 50°C, with a C/B/S ratio of 1/8/200. These are more forcing conditions than those of
catalyst 15.34 An increase in substrate loading to C/S = 1/600 requires a longer reaction time
(37% conversion in 7 h) and a threefold decrease in turnover frequency (TOF) to 26 h-1 (Table
6.1, entries 1 and 5). The hydrogenation of benzophenone with increased substrate loading led to
similar TOF values (Table 6.2, entries 1 and 2). This is consistent with the formation of an
enolate complex in the case of acetophenone which suppresses catalysis as observed
elsewhere.14b, 32 In addition, the H2-hydrogenation of benzophenone in THF is affected by the
concentrations of the catalyst and hydrogen pressure but is independent of the ketone
concentration (Table 6.2, entries 1-4 and Figure 6.7) as expected since benzophenone cannot
form an enolate.
Figure 6.7. Reaction profiles showing the effect of the concentrations of catalyst, hydrogen and
substrate to the H2-hydrogenation of benzophenone catalyzed by complex 19. (a) [19] = 0.72
mM, [benzophenone] = 0.14 M, P(H2) = 25 bar, red circles; (b) [19] = 0.72 mM, [benzophenone]
= 0.14 M, P(H2) = 15 bar, blue diamonds; (c) [19] = 0.72 mM, [benzophenone] = 0.29 M, P(H2)
= 25 bar, green squares; (d) [19] = 1.2 mM, [benzophenone] = 0.14 M, P(H2) = 25 bar, purple
crosses. All of the reactions were conducted in 25 bar H2 at 50°C in THF and KOtBu was used as
a base (5.9 mM).

165Interestingly, catalysis conducted in 2-propanol under H2 with low substrate and base loadings
(C/B/S = 1/8/200) using 19 was slow, and its activity was nearly identical to that of transfer
hydrogenation (Table 6.1, entry 6 and Table 6.3, Entry 1).
The phosphine complex 20, on the other hand, has low activity in the H2-hydrogenation and
similar activity in the transfer hydrogenation of acetophenone under identical reaction conditions
as those used for 3 (Table 6.1, entry 2 and Table 6.3, Entry 2). The structurally similar
diphenylphosphino-containing complex [RuCp*(P–NH2)py]PF6 (16a) also showed poor activity
in the catalytic H2-hydrogenation of acetophenone compared to 15.34
Table 6.1. The H2-Hydrogenation of Acetophenone Catalyzed by Iridium(III) Complexes.
IrPPh2N
PF6Cl
HH
20
Ir
NN
N
CH3 PF6Cl
22
Ir
NN
N
CH3 PF6HHH
21
Ir
NN
N
CH3 PF6Cl
HH
19
+ + + +
Entrya Complex C/B/Sb ratio Solvent Conversionc (%/hr) TOF (h-1)d
1 19 1/8/200 THF 62/1 98/3 1542 20 1/8/200 THF 6/1 15/4 -f
3 21 1/8/200 THF 9/2 - -4 22 1/8/200 THF 7/2 - -5 19 1/8/600 THF 19/3 37/7 266 19 1/8/200 iPrOH 28/3 99/20 177 19 1/16/200 iPrOH 52/0.3 99/1 4168e 19 1/16/200 iPrOH 20/0.3 98/1 1529 21 1/8/200 iPrOH 41/0.5 99/2 21310 22 1/8/200 iPrOH 3/2 - -f
aReactions were carried out in a 50 mL Parr hydrogenation reactor at 25 bar of H2 pressure at
50°C using THF or iPrOH (6 mL) as the solvent. KOtBu was used as the base. bC/B/S = catalyst
to base to substrate ratio . cConversions were determined by GC and are reported as an average
of two runs. dTOF = turnover frequency, measured from the slope of the linear portion of an
[alcohol] versus time plot. eAn equimolar amount of [2.2.2]cryptand to potassium ions was
added. fTOF not measured.
166Table 6.2. The H2-Hydrogenation of Benzophenone Catalyzed by Iridium(III) Complexes.
Entrya Complex C/B/S ratio
Solvent H2 (bar) Conversionb (%/hr) TOF (h-1)
1 19 1/8/200 THF 25 63/0.5 96/3 3642 19 1/8/400 THF 25 41/0.5 70/3 4723 19 1/8/200 THF 15 41/0.5 83/3 1724c 19 1/5/120 THF 25 61/0.15 95/2 12205 22 1/8/200 THF 25 15/1 25/3 166 22 1/16/200 iPrOH 25 4/2 - -d
aReactions were carried out in a 50 mL Parr hydrogenation reactor at the required H2 pressure at
50°C using THF or iPrOH (6 mL) as the solvent. KOtBu was used as base. bConversions were
determined by GC and are reported as an average of two runs. c[19] = 1.2 mM, the
concentrations of the ketone and base are the same as of entries 1 and 3. dTOF not measured.
Table 6.3. Transfer Hydrogenation of Acetophenone Catalyzed by Iridium(III) Complexes.
Entrya Complex Conversionb (%/hr)
1 19 28/3 69/172 20 27/3 65/203 21 23/3 44/19
aReactions were carried out in iPrOH (6 mL) at 75°C under an argon atmosphere, the C/B/S ratio
was 1/8/200. KOtBu was used as the base. bConversions were determined by GC and are reported
as an average of two runs.
Attempts at catalysis using the hydride-amine complex 21 in the absence of an alkoxide,
whether conducted as direct hydrogenation under H2 in THF or 2-propanol (25 bar H2 pressure,
50°C), or transfer hydrogenation under argon at 75°C in 2-propanol resulted in no conversion of
the starting ketone. Significantly, with an alkoxide base, 21 becomes active in 2-propanol (TOF
= 213 h-1) but less active in THF for the hydrogenation of acetophenone using H2 gas (Table 6.1,
entries 3 and 9). Under argon, 21 is somewhat active for the transfer hydrogenation of
acetophenone in basic 2-propanol solution (Table 6.3, entry 3). Such observations are similar to
those reported for the cationic ruthenium hydride-amine complex 17.32 The neutral complexes
containing a cyclometalated C–NH2 ligand, IrCp*(κ2(C,N)-2-C6H4CR2NH2)H49 and Ru(η6-C6H6)
49. Arita, S.; Koike, T.; Kayaki, Y.; Ikariya, T., Organometallics 2008, 27, 2795-2802.

167(κ2(C,N)-2-C6H4CR2NH2)H,50 are active in the transfer hydrogenation of ketones in the absence
of base at room temperature.
6.3.5 Effect of Alkoxide Base on the H2-Hydrogenation of Acetophenone Catalyzed by
Complex 19. When the ratio of the alkoxide base (KOtBu) to catalyst is increased from 8/1 to
16/1, the chloride complex 19 achieves a similar activity (99% conversion to 1-phenylethanol,
TOF = 416 h-1) to the hydride-amine complex 21 in the presence of 8 equiv. of base (Table 6.1,
entries 7 and 9). The addition of excess metal alkoxides are reported to accelerate the H2- and
transfer hydrogenation of ketones catalyzed by certain ruthenium(II) systems, where the cations
of the added base act as a Lewis acid to stabilize the transition states for the H+/H- transfer from
the catalyst to the ketone.14b, 51 Addition of [2.2.2]cryptand in an equimolar amount to the
potassium ions (C/B/S = 1/16/200) gave full conversion to 1-phenylethanol in 1 h, but with a
smaller TOF value (152 h-1, Table 6.1, entry 8). This suggests that the cations do play a minor
role.14b, 32 In fact, stoichiometric reactions of 19 with different amounts of KOtBu in 2-propanol
gave drastically different products as determined by 1H NMR (vide infra).
Some conclusions can be drawn from the catalytic results obtained by these experiments.
1. Reactions conducted in basic 2-propanol solution starting from the chloride complex 19
lead to the formation of the hydride-amine complex 21 (Scheme 6.2), which has a poor catalytic
activity if the concentration of the alkoxide base present in the beginning of the reaction is small
(for example, a C/B ratio of 1/8). This explains the similar catalytic behaviour when 19 was used
in either the H2- or transfer hydrogenation of acetophenone.
2. A critical ratio of catalyst to base (C/B = 1/8 for the hydride-amine complex 21 and C/B
= 1/16 for the chloride complex 19) is needed for comparable H2-hydrogenation activity. It
appears that 21 is the resting state in catalysis and that the alkoxide base must play a crucial role
in the activation of this hydride.
50. (a) Sortais, J. B.; Ritleng, V.; Voelklin, A.; Holuigue, A.; Smail, H.; Barloy, L.; Sirlin, C.; Verzijl, G. K. M.; Boogers, J. A. F.; de Vries, A. H. M.; de Vries, J. G.; Pfeffer, M., Org. Lett. 2005, 7, 1247-1250; (b) Sortais, J. B.; Barloy, L.; Sirlin, C.; de Vries, A. H. M.; de Vries, J. G.; Pfeffer, M., Pure Appl. Chem. 2006, 78, 457-462; (c) Pannetier, N.; Sortais, J. B.; Dieng, P. S.; Barloy, L.; Sirlin, C.; Pfeffer, M., Organometallics 2008, 27, 5852-5859.51. (a) Hartmann, R.; Chen, P., Angew. Chem. Int. Ed. 2001, 40, 3581-3585; (b) Vastila, P.; Zaitsev, A. B.; Wettergren, J.; Privalov, T.; Adolfsson, H., Chem. Eur. J. 2006, 12, 3218-3225; (c) Wettergren, J.; Buitrago, E.; Ryberg, P.; Adolfsson, H., Chem. Eur. J. 2009, 15, 5709-5718.

1683. Catalysis by 21 conducted in basic 2-propanol was faster than in a basic aprotic solution
using the same concentration of the alkoxide base. This is reversed when 19 was used. The
alcohol solvent must participate in catalysis which acts as a reactant and/or as a proton shuttle.
6.3.6 The Importance of the NH2 Group in the Iridium(III) System on its Activity in the
Catalytic Hydrogenation of Ketones. Complex 22 which does not contain an N–H
functionality catalyzed the hydrogenation of acetophenone and benzophenone in THF and
KOtBu very slowly to 1-phenylethanol in 2 h to 7% conversion, and to diphenylmethanol in 3h
to 25% conversion. respectively, under 25 bar H2 and 50°C (C/B/S = 1/8/200, Table 6.1, entry 4
and Table 6.3, entry 5). Catalysis was even slower when 2-propanol was used as the solvent (C/B
= 1/16, Table 6.1, entry 10 and Table 6.3, entry 6). This is slow compared to 19 (96% conversion
in 3 h), which contains an N–H group, under identical catalysis conditions. This is in contrast to
the previously reported systems, [Ru(η6-p-cymene)(C–NH2)Cl]PF6 (13) and the N,N-dimethyl
analogue ([Ru(η6-p-cymene)(C–NMe2)Cl]PF6·1.5 DMSO (18), see Chapter 5), both of which
showed similar activities in catalyzing the hydrogenation of acetophenone.32 The N–H group of
our iridium system, therefore, must be important in catalysis, consistent with an outer sphere
bifunctional mechanism for ketone hydrogenation (Step B in Scheme 6.1).
6.3.7 Selectivity in the Hydrogenation of an α,β-unsaturated ketone Catalyzed by
Complexes 15 and 19. Under similar reaction conditions to the hydrogenation of acetophenone
(Scheme 6.5), the ruthenium(II) complex 15 catalyzed the selective reduction of the polar bond
to give trans-4-phenyl-but-3-en-2-ol (89% conversion) and 4-phenylbutan-2-ol (4% conversion)
and none of the saturated ketone, 4-phenylbutan-2-one within 1 h. The iridium(III) complex 19
also catalyzed the selective reduction of the ketone in the same substrate giving trans-4-phenyl-
but-3-en-2-ol (88% conversion) and 4-phenylbutan-2-ol (8% conversion), and the saturated
ketone, 4-phenylbutan-2-one (3% conversion) in 5 h, under 25 bar of H2 pressure at 50°C in 2-
propanol, and in the presence of an excess of alkoxide base (16 equiv. with respect to catalyst,
Scheme 6.5). The selectivity that complexes 15 and 19 exhibit is characteristic of an outer sphere
bifunctional mechanism. It is known that the reduction of the olefin can also proceed via a 1,4-
addition reaction utilizing the same mechanism.52

169Scheme 6.5. The Hydrogenation of trans-4-Phenyl-but-3-ene-2-one Catalyzed by Complexes 15
and 19.a
aReactions were carried out in a 50 mL Parr hydrogenation reactor at the required H2 pressure
and temperature in THF or iPrOH (6 mL) and KOtBu was used as base. Conversions were
determined by GC and are reported as an average of two runs.
6.3.8 Effect of Alcohol and Other Additives on the H2-Hydrogenation of Acetophenone
Catalyzed by the Ruthenium(II) Complex 15. Reactions carried out in THF gave sigmoidal-
type reaction profiles with variable induction periods (10 – 60 min). On the other hand, a
reaction that was conducted in 2-propanol did not show any sigmoidal behavior in the reaction
profile (Figure 6.8).
The effect of different additives on this induction period was then investigated. The addition
of the product alcohol, 1-phenylethanol, at the beginning of the reaction (up to 0.4 M in THF)
decreased the induction period. In addition, the induction period increased by roughly six fold if
the pressure of H2 is decreased from 8 to 2 bar. There was little effect on changing the
concentrations of acetophenone or the catalyst by two fold (Figure 6.9). The addition of pyridine
to the reaction mixture retarded catalysis (Figure 6.8). As well, the possibility of the formation of
ruthenium nanoparticles was examined and this was deemed unlikely by conducting the mercury
test in the hydrogenation of 4'-chloroacetophenone in basic THF.34
It appears that there exists a competition between H2 and pyridine to coordinate to the
catalytically active species. The presence of alcohol selectively favors the binding and
heterolytic splitting of η2-H2 by acting as a proton shuttle.6c, 12e, 19b, 20b, 22a This system will have a
different rate law reported for the amido complex RuH((S)-BINAP)(app) (app = 2-amido-2-(2-
pyridyl)-propane) which displayed autocatalysis in the presence of alcohols.19b
52. (a) Ito, M.; Kitahara, S.; Ikariya, T., J. Am. Chem. Soc. 2005, 127, 6172-6173; (b) Ikariya, T.; Gridnev, I. D., Chem. Rec. 2009, 9, 106-123.
[15] or [19] cat.
[15]: 8 bar H2, 25°C, THF
O
OH
O
OH
89% in 1 h
0%
4%
KOtBu
[15] [19]
88 % in 5 h
3%
8 %
C/B/S = 1/8/600 C/B/S = 1/16/200
[19]: 25 bar H2, 50°C, iPrOH
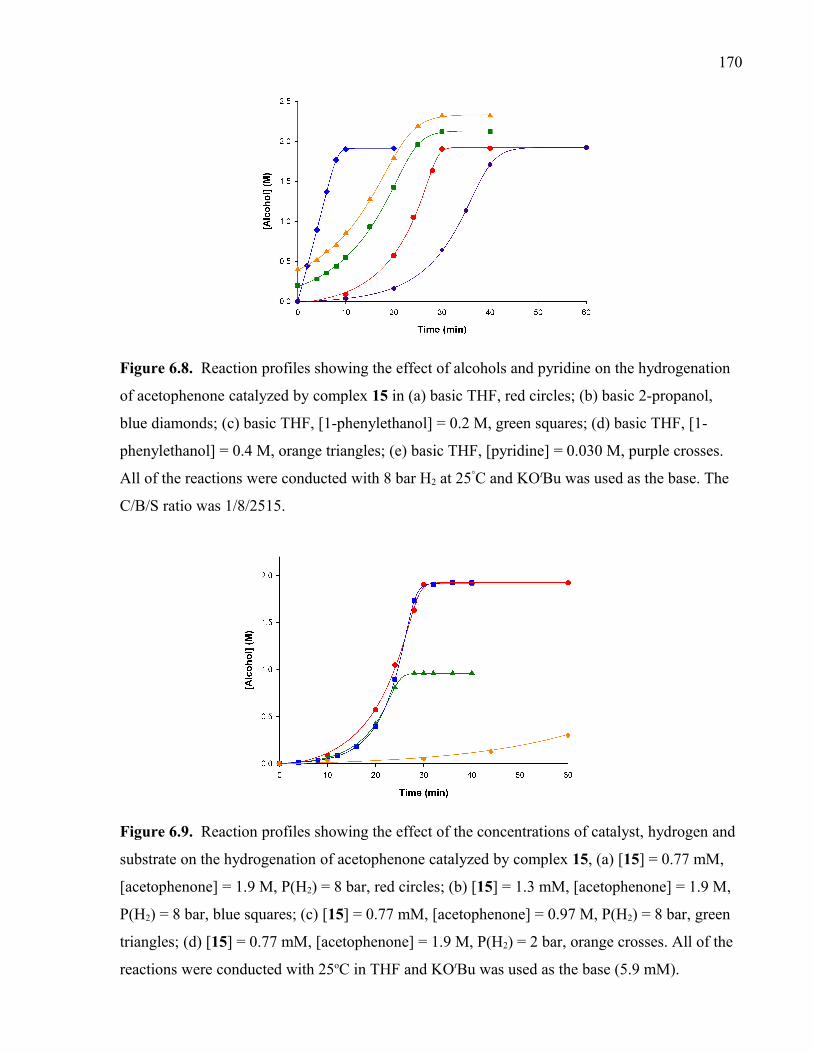
170
Figure 6.8. Reaction profiles showing the effect of alcohols and pyridine on the hydrogenation
of acetophenone catalyzed by complex 15 in (a) basic THF, red circles; (b) basic 2-propanol,
blue diamonds; (c) basic THF, [1-phenylethanol] = 0.2 M, green squares; (d) basic THF, [1-
phenylethanol] = 0.4 M, orange triangles; (e) basic THF, [pyridine] = 0.030 M, purple crosses.
All of the reactions were conducted with 8 bar H2 at 25°C and KOtBu was used as the base. The
C/B/S ratio was 1/8/2515.
Figure 6.9. Reaction profiles showing the effect of the concentrations of catalyst, hydrogen and
substrate on the hydrogenation of acetophenone catalyzed by complex 15, (a) [15] = 0.77 mM,
[acetophenone] = 1.9 M, P(H2) = 8 bar, red circles; (b) [15] = 1.3 mM, [acetophenone] = 1.9 M,
P(H2) = 8 bar, blue squares; (c) [15] = 0.77 mM, [acetophenone] = 0.97 M, P(H2) = 8 bar, green
triangles; (d) [15] = 0.77 mM, [acetophenone] = 1.9 M, P(H2) = 2 bar, orange crosses. All of the
reactions were conducted with 25oC in THF and KOtBu was used as the base (5.9 mM).

1716.3.9 Deuterium Labelling Studies Using the Ruthenium(II) Complex 15. In order to find
evidence for an alcohol-assisted mechanism in the hydrogenation of ketone by complex 15,
acetophenone was deuterated using D2 gas in 2-propanol (OH) and 2-propanol-d1 (OD, Table
6.4). Full conversion to the deuterated product was achieved in 30 min. Analysis of the 1H NMR
spectra of the products revealed significant deuteration at the α-carbon and the hydroxyl group of
1-phenylethanol when both D2 and 2-propanol-d1 were used, but less when D2 and 2-propanol
were used (Table 6.4). These results suggest significant H/D scrambling took place at the active
ruthenium species with participation of the alcohol solvent. Further analysis of the 2H NMR
spectra showed deuteration of the hydroxyl group of 2-propanol but not at its α-carbon.
Table 6.4. Deuteration of Acetophenone and 2-Propanol Catalyzed by Complex 15.
Deuteriuma Source Deuterium Content in 1-Phenylethanol (%)b In 2-Propanol (%)b
α-CD OD group CDnH3-n group (n = 1-3)
OD group
D2/iPrOH 42 11 - 27D2/iPrOD 80 76 71 84
aReactions were carried out in a 50 mL Parr hydrogenation reactor at 8 bar of D2 pressure at 25°C
using the appropriate solvent (6 mL). KOtBu was used as the base. The catalyst/base/
substrate/solvent ratio was 1/8/898/17060. Complete conversion to the product alcohol was
achieved in 30 min. bThe deuterium content of the 1-phenylethanol and 2-propanol were
determined by 1H and 2H NMR.
On the other hand, the hydroxyl deuterium of 2-propano1-d1 was not retained after catalysis
(lowered from 100% to 84% deuteration, Table 6.4) further supporting the idea that H/D
scrambling has taken placed. This is evidence that 2-propanol is acting as a proton shuttle.20b Of
note, deuteration of the methyl group of 1-phenylethanol was observed when D2 and 2-propanol-
d1 were used in catalysis. This is expected due to the base-catalyzed deuteration of enolizable
ketones as reported elsewhere.14b, 32 These experiments support the alcohol-assisted heterolytic
splitting of η2-H2 on the active ruthenium species starting from complex 15 when activated (Step
A in Scheme 6.1).
6.3.10 Effect of Alcohol on the H2-Hydrogenation of Acetophenone Catalyzed by
Iridium(III) Complexes 19 and 21. The effect of alcohols on the hydrogenation of
acetophenone catalyzed by the chloride complex 19 and hydride-amine complex 21 was further
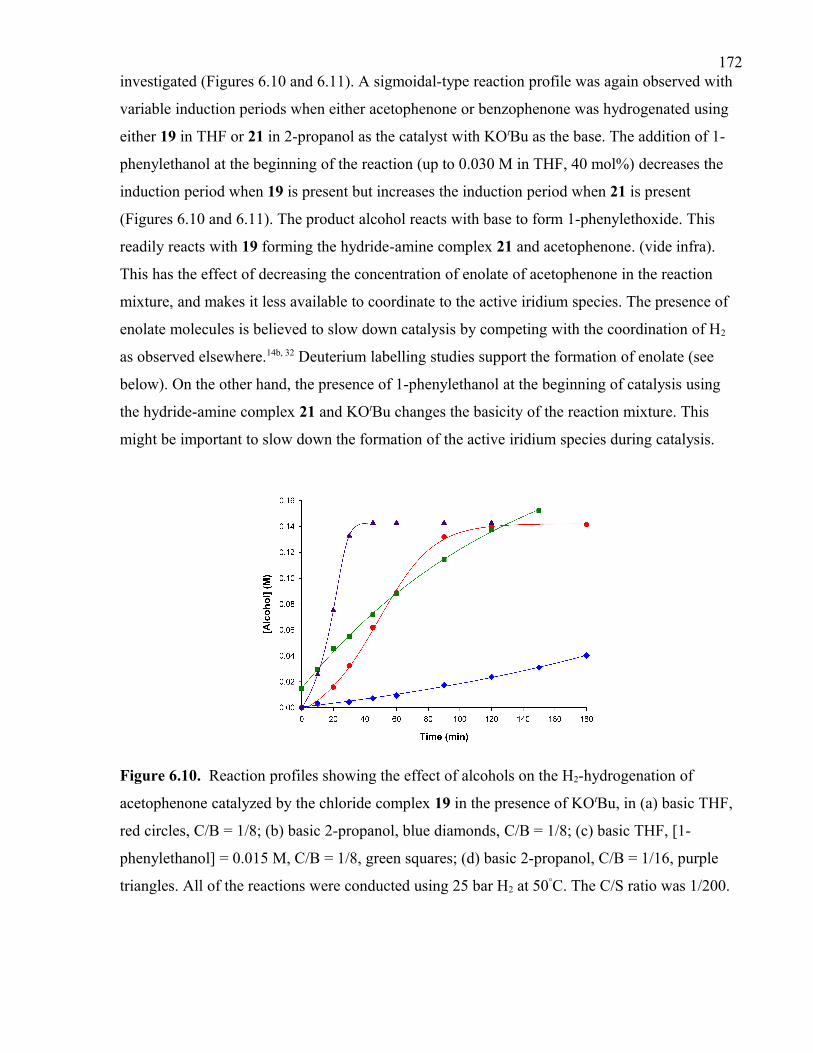
172investigated (Figures 6.10 and 6.11). A sigmoidal-type reaction profile was again observed with
variable induction periods when either acetophenone or benzophenone was hydrogenated using
either 19 in THF or 21 in 2-propanol as the catalyst with KOtBu as the base. The addition of 1-
phenylethanol at the beginning of the reaction (up to 0.030 M in THF, 40 mol%) decreases the
induction period when 19 is present but increases the induction period when 21 is present
(Figures 6.10 and 6.11). The product alcohol reacts with base to form 1-phenylethoxide. This
readily reacts with 19 forming the hydride-amine complex 21 and acetophenone. (vide infra).
This has the effect of decreasing the concentration of enolate of acetophenone in the reaction
mixture, and makes it less available to coordinate to the active iridium species. The presence of
enolate molecules is believed to slow down catalysis by competing with the coordination of H2
as observed elsewhere.14b, 32 Deuterium labelling studies support the formation of enolate (see
below). On the other hand, the presence of 1-phenylethanol at the beginning of catalysis using
the hydride-amine complex 21 and KOtBu changes the basicity of the reaction mixture. This
might be important to slow down the formation of the active iridium species during catalysis.
Figure 6.10. Reaction profiles showing the effect of alcohols on the H2-hydrogenation of
acetophenone catalyzed by the chloride complex 19 in the presence of KOtBu, in (a) basic THF,
red circles, C/B = 1/8; (b) basic 2-propanol, blue diamonds, C/B = 1/8; (c) basic THF, [1-
phenylethanol] = 0.015 M, C/B = 1/8, green squares; (d) basic 2-propanol, C/B = 1/16, purple
triangles. All of the reactions were conducted using 25 bar H2 at 50°C. The C/S ratio was 1/200.
173
Figure 6.11. Reaction profiles showing the effect of alcohols on the H2-hydrogenation of
acetophenone catalyzed by the hydride-amine complex 21 in (a) basic 2-propanol, blue
diamonds; (b) basic THF, red squares; (c) basic 2-propanol, [1-phenylethanol] = 0.030 M, green
triangles. All of the reactions were conducted using 25 bar H2 at 50°C and KOtBu was used as the
base. The C/B/S ratio was 1/8/200.
The fact that the solvent alcohol and an excess of alkoxide base is required for catalysis
involving either catalyst 19 or 21 in order to achieve high and comparable activity, suggests that
they have a role to play in catalysis. The alcohol may play a similar role as in the ruthenium(II)
system to accelerate catalysis by acting as a proton shuttle in the heterolytic splitting of η2-H2 in
step A of Scheme 6.1.
6.3.11 Deuterium Labelling Studies Using the Iridium(III) Complex 19. To gain further
insight into the effect of 2-propanol and 1-phenylethanol on the hydrogenation of acetophenone
catalyzed by complex 19, acetophenone was first deuterated using D2 gas in THF in the presence
of 20 mol% 1-phenylethanol and KOtBu (Table 6.5, entry 1). Full conversion to the product
alcohol was achieved in 18 h. Analysis of the 1H NMR and 2H NMR spectra of the deuterated
product suggested 80% deuteration at the α-carbon, 14% at the hydroxyl group, and 36% at the
methyl group of 1-phenylethanol. No deuteration was observed at the phenyl ring. Base-
catalyzed deuteration of the enolizable acetophenone is responsible for such a deuterium
distribution on 1-phenylethanol.14b, 32 In contrast, when 1-phenylethanol was deuterated for 6 h
under similar reaction conditions as above, deuteration was also observed at the α-carbon,
hydroxyl and the methyl group, but to a minor extent (Table 6.5, entry 2). This supports the

174formation of acetophenone and the hydride-amine complex 21 via the formation of an alkoxide
intermediate, [IrCp*(C–NH2)(O-CHPhCH3)]+, in which the β-hydrogen of 1-phenylethoxide
ligand can eliminate.2a, 5c, 14a, 18, 39-40, 53 Acetophenone that formed was then deuterated in the
presence of an active iridium species.
Table 6.5. Deuteration of Acetophenone and 1-phenylethanol Catalyzed by Complex 19.
Entry Reaction Conditionsa Substrate Deuterium Content in 1-Phenylethanol (%)b
α-CD OD group CDnH3-n group (n = 1-3)
1 10 bar D2, THF, 18 hc Acetophenone,1-phenylethanol
80 14 36
2 10 bar D2, THF, 6 hc 1-Phenylethanold 9 42 153 8 bar D2, iPrOH, 6 he Acetophenone 30 -f 14 10 bar D2, iPrOH, 6 he 1-Phenylethanol - Negligiblef -
aReactions were carried out in a 50 mL Parr hydrogenation reactor at the required D2 pressure at
50°C using the appropriate solvent (6 mL). KOtBu was used as the base. bThe deuterium content
of the 1-phenylethanol and 2-propanol were determined by 1H and 2H NMR, no deuteration was
observed on the phenyl ring of the product alcohol. cThe C/B/S ratio was 1/8/200. d20 mol% of
1-phenylethanol was used with respect to [19]. eThe C/B/S ratio was 1/16/200. fDeuteration at the
OD group of 2-propanol was observed.
When acetophenone was deuterated using D2 gas in 2-propanol in the presence complex 19
and excess base (Table 6.5, entry 3), deuteration at the α-carbon was significantly smaller (30%
deuteration) and deuteration at the hydroxyl and the methyl group was negligible. However, 2H
NMR of the reaction mixture showed that the extent of deuteration of the hydroxyl group of 2-
propanol was much greater than that at the α-carbon of 1-phenylethanol. In fact, the effect of 2-
propanol by acting as a proton shuttle in the heterolytic splitting of D2 is more important than the
product alcohol, as evident by a reaction of 1-phenylethanol with 0.5 mol% of complex 19 in
basic 2-propanol (Table 6.5, entry 4). In this case, the hydroxyl group of 2-propanol was
deuterated but not that of 1-phenylethanol.
6.3.12 Stoichiometric Reactions Using Complex 19. In order to identify possible intermediates
in the catalytic cycle involving 19, KOtBu, acetophenone and H2, stoichiometric reactions were
53. Laxmi, Y. R. S.; Backvall, J. E., Chem. Commun. 2000, 611-612.

175monitored by 1H NMR spectroscopy. The reaction of 19 and 2 equiv. of KOtBu under 1 bar of H2
at 50°C in THF only led to 5% conversion to the hydride-amine complex 21. A reaction
conducted at higher H2 pressure (25 bar) and with more base (5 or 12 equiv.) in THF followed by
evaporation of the solvent and dissolution in acetonitrile-d3 afforded at least three species as
identified by three resonances at about 3.6 ppm in the 1H NMR spectrum for the CH3 group on
the C–NH2 ligand. These were the monohydride complex 21, a cis-dihydride complex containing
two peaks at -17.92 and -18.34 ppm with JHH = 9.83 Hz,54 and another species that does not
contain any hydride peaks. The ratio between the aforementioned hydride containing complexes
varies with the initial concentrations of 19 and base.
When 19 was reacted with KOtBu in 25 bar of H2 at 50°C in 2-propanol, the products that
were formed depended on the amount of base that was added. The addition of 8 equiv. of KOtBu
at the beginning of the reaction gave, upon evaporation of the solvent and dissolution in
acetonitrile-d3, at least three species that were observed by three resonances at around 3.6 ppm in
the 1H NMR spectrum for the CH3 group on the C–NH2 ligand associated with 21 (63%) and two
other species that did not contain any hydride peaks (37%, CH3 peak resonates at 3.69 and 3.72
ppm). When more base was added (16 equiv.), the species that has the CH3 peak at 3.69 was
observed in 67% abundance. There were two other species that were observed in equal amounts
as well, one of which gave a hydride peak at -19.72 ppm. The signals pertaining to the hydride-
amine complex 21 were lost. Although the species in this complicated mixture were not
identified further, this shows that complex 21 reacts further with excess alkoxide base.
Further, a reaction of complex 19 with potassium 1-phenylethoxide in warm THF (50°C)
under argon afforded 21 and acetophenone in 21% conversion as a result of β-hydrogen
elimination of 1-phenylethoxide. This also left behind the starting complex as identified by 1H
NMR in acetonitrile-d3. Heating such sample overnight in a sealed NMR tube afforded
conversion to 21 in 63% and acetophenone in 70%. All these results establish the formation of
complex 21 at the initial stage of catalysis, especially when the concentration of the active
catalyst is low and there exists a similar amount of alkoxide anions containing β-hydrogen in the
reaction mixture. The activation of such a complex by an alkoxide base, therefore, becomes
important at a later stage of catalysis.
54. (a) Grundemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J. W.; Crabtree, R. H., J. Am. Chem. Soc. 2002, 124, 10473-10481; (b) Piras, E.; Lang, F.; Ruegger, H.; Stein, D.; Worle, M.; Grützmacher, H., Chem. Eur. J. 2006, 12, 5849-5858; (c) Dzik, W. I.; Smits, J. M. M.; Reek, J. N. H.; de Bruin, B., Organometallics 2009, 28, 1631-1643.

1766.3.13 The Conventional Non-Alcohol Assisted Outer Sphere Bifunctional Mechanism. The
catalytic cycles that involve the outer sphere bifunctional mechanism in the absence of alcohol
for the ruthenium(II) (complex 15) and iridium(III) (complexes 19 and 21) systems were
investigated by Density Functional Theory (DFT). The MPW1PW91 functional was used as this
gives better prediction of energy barriers and transition states for 4d and 5d metals.55
Simplifications were made to ease computation by replacing the Cp* ligand with Cp; acetone and
2-propanol were used to model the ketone and the product alcohol.
In the conventional outer sphere mechanism that involves the action of M–H and N–H groups
(Scheme 6.6), the addition of base to the precatalyst afforded the amido complex A, which
results from the deprotonation of the N–H group. Such a complex is responsible for activating
dihydrogen to yield a metal hydride complex D via an H2 addition step to the metal center
(TSB,C) and subsequent heterolytic splitting at the metal center (TSC,D). The activation of H2 at
the amido complex is rate-determining and this should have the largest energy barrier of the
overall catalytic cycle. The fast, low barrier step, then follows for the transfer of H+/H- from D to
the ketone in the outer sphere fashion to afford the product alcohol and A. 6c, 12e, 12k, 12m, 13a, 14a, 14b, 19b
According to calculations (Figure 6.12), the free energy barriers for the activation of H2 (the
coordination and heterolytic splitting of H2) by the neutral ruthenium(II) and cationic iridium(III)
species are 14.1 and 22.6 kcal/mol, respectively. The heterolytic splitting of H2 leads to a more
reactive ruthenium(II) hydride DRu (-8.8 kcal/mol relative to ARu and H2) but a more
thermodynamically stable iridium(III) hydride DIr (-23.5 kcal/mol relative to AIr and H2). The
free energy barriers for the transfer of H+/H- from DRu to acetone and to acetophenone are 16.9
and 12.3 kcal/mol, respectively. By contrast it is unfavourable to transfer H+/H- from DIr to
acetone (35.9 kcal/mol). In addition to a stronger Ir–H bond that is formed due to relativistic
effect on going from a 4d (Ru) to a 5d (Ir) metal, the cationic charge of the iridium may
deactivate the metal hydride as was observed for the cationic ruthenium hydride-amine complex
17.32
55. Lynch, B. J.; Truhlar, D. G., J. Phys. Chem. A 2001, 105, 2936-2941.

177Scheme 6.6. Computed Outer Sphere Bifunctional Mechanism in the Hydrogenation of Acetone
Catalyzed by Complexes of Ruthenium(II) and Iridium(III).
The computed results suggest well-balanced energy barriers for the ruthenium(II) system. The
hydride complex (DRu) which is formed upon the activation of H2 by ARu is readily consumed
when it transfers its H+/H- to the ketone in the outer sphere. On the other hand, the calculated
results show that the iridium(III) hydride complex (DIr) is the resting state of the catalytic cycle,
and the transfer of H+/H- to the ketone in the outer sphere is very difficult. This is in line with the
observation that the hydride-amine complex 21 does not react with acetophenone in either
stoichiometric or catalytic reactions in the absence of base.
Of interest, the geometric parameters for the optimized transition state structures for the
heterolytic splitting of H2 (TSC,D, Figure 6.13) and the transfer of H+/H- to acetone (TSE,F, Figure
6.14) of both systems are surprisingly similar, except for a longer O–H interaction (O–H…N
distance: 1.61 Å for ruthenium and 1.38 Å for iridium) for the ruthenium(II) system. An Intrinsic
Reaction coordinate (IRC) calculation that was performed on TS(Ru)E,F located a local minima,
M NNN
CH3
H
MH NNN
CH3
HH
H2
OOH
H
n+ n+
M NNN
CH3
H
n+
HH
MH NNN
CH3
HH
n+
OM = Ru, n = 0M = Ir, n = 1
MH NNN
CH3
HH
n+
M NNN
CH3
H
n+
HH M N
NN
CH3
H
n+
H
H
H M NNN
CH3
H
n+
H
O
M NNN
CH3
H
n+
O
H
H
A
B
TSB,C C
TSC,D
D
EF
TSE,F
Outer SphereBifunctional Mechanism

178which revealed a 2-propoxide anion that is hydrogen-bonded via oxygen to the N–H group on
the C–NH2 ligand, along with an agostic C–H interaction of the 2-propoxide anion with the
ruthenium(II) center. Subsequent geometry optimization and normal mode analysis provided that
this structure (FRu') is 0.9 kcal/mol more stable than TS(Ru)E,F (∆G = 7.2 kcal/mol relative to
ARu, H2 and acetone). This has a O–H distance of 1.39 Å, a C–H distance of 1.26 Å, and a Ru…H
distance of 1.85 Å. These geometric parameters are similar to an analogous structure reported by
Gusev and co-workers.11h A QST3 or a QST2 calculation that were performed for the protonation
of the 2-propoxide anion by the N–H group on the C–NH2 ligand giving FRu failed to locate a
transition state structure. All these suggest that the transfer of a H+/H- couple from the hydride-
amine complex to the ketone in the outer-sphere for the ruthenium(II) system might occur in a
stepwise fashion.11h, 17
Figure 6.12. The free energy profile for the outer sphere bifunctional mechanism in the H2-
hydrogenation of acetone starting from A and moving to the right. Pathway colored in blue
represents the ruthenium(II) system and the one in red represents the iridium(III) system. The gas
phase free energies (1 atm, 298 K) are reported relative to A, hydrogen and acetone in kcal/mol.
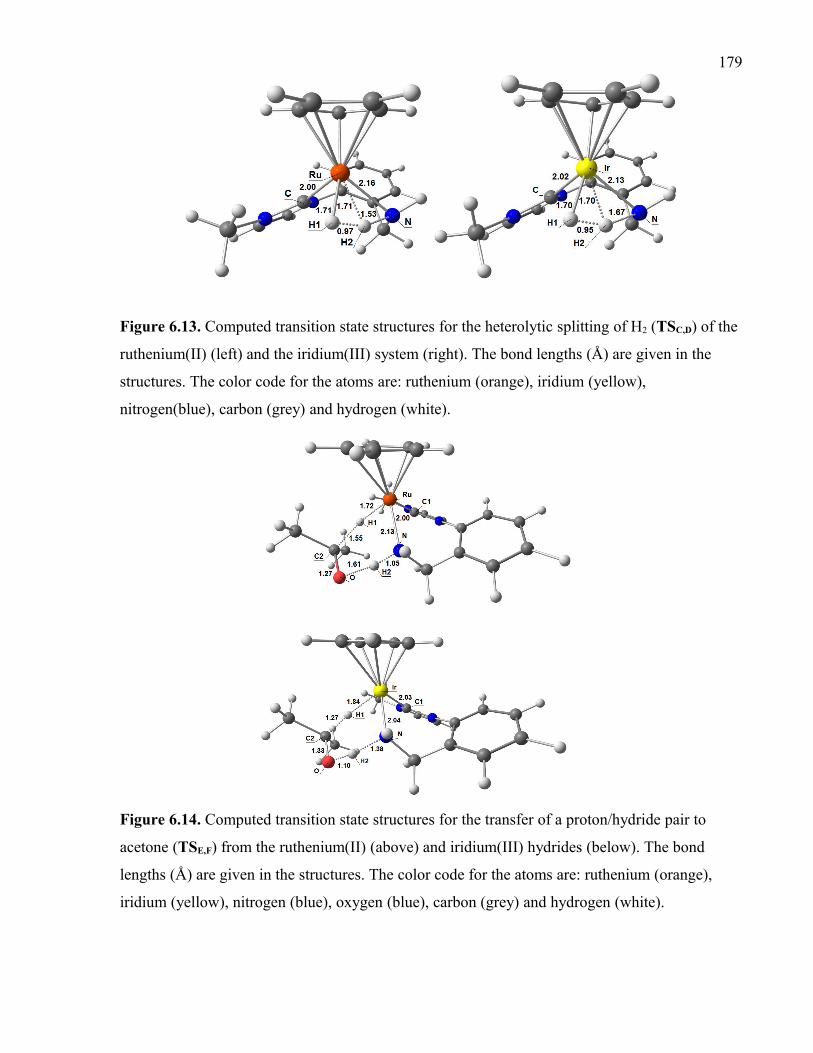
179
Figure 6.13. Computed transition state structures for the heterolytic splitting of H2 (TSC,D) of the
ruthenium(II) (left) and the iridium(III) system (right). The bond lengths (Å) are given in the
structures. The color code for the atoms are: ruthenium (orange), iridium (yellow),
nitrogen(blue), carbon (grey) and hydrogen (white).
Figure 6.14. Computed transition state structures for the transfer of a proton/hydride pair to
acetone (TSE,F) from the ruthenium(II) (above) and iridium(III) hydrides (below). The bond
lengths (Å) are given in the structures. The color code for the atoms are: ruthenium (orange),
iridium (yellow), nitrogen (blue), oxygen (blue), carbon (grey) and hydrogen (white).

1806.3.14 The Alcohol-Assisted Outer Sphere Bifunctional Mechanism for the Ruthenium(II)
System. In the presence of 2-propanol (Scheme 6.7), or when the concentration of the product
alcohol builds up, the free energy barriers for the coordination and the heterolytic splitting of H2
become lower (ΔGǂ = 11.1 and 8.5 kcal/mol respectively) starting from Aalc (Figure 6.15, Aalc
corresponds to F in Scheme 6.6).
The transition state structure of the alcohol-assisted heterolytic splitting of H2 shows that the
protons neighbouring to the alcoholic oxygen are held in close proximity by the formation of two
hydrogen-bonds via O...H interactions (H2–O: 1.74; H3–O: 1.36 Å, Figure 6.16). These results
are supportive of an alcohol-assisted mechanism where the alcoholic proton is shuttled between
the coordinated dihydrogen and the amido nitrogen via a six-membered ring transition state 6c, 12e,
12m, 13a, 14b, 19b, 20b, 22 Andersson and co-workers have specifically calculated a similar transition state
structure for the alcohol-assisted heterolytic splitting of H2 of a structurally similar
RuCp*(diamine)X system, and the effect of methanol on the free energy barriers of H2
coordination and its activation are pronounced given the level of theories of calculations.12e
Scheme 6.7. Computed Reaction Pathways of the Activation of H2 by Complex A in the
Presence of 2-Propanol.
In addition, the free energy barrier for the transfer of a proton/hydride couple to acetone is
16.2 kcal/mol if a six-membered ring transition state is assumed.19b The alcohol, therefore, has a
Ru NNN
CH3
H
RuH NNN
CH3
HH
H2
O
OH
H
Ru NNN
CH3
H
HH
Ru NNN
CH3
H
HH
H Ru NNN
CH3
H
H
Aalc
Balc TSB,C
Calc
TSC,DDalc
Alcohol-Assisted Outer SphereBifunctional Mechanism
O
H
H
O
H
H
O
H
H
Ru NNN
CH3
H
HH
O
H
H
OH HO
HH
alc
alc
Ealc → TSE,F → Falcalc

181minor effect on such a transition state as the energy barriers with and without alcohol were
similar (ΔGǂ = 16.9 kcal/mol).19b On the other hand, the induction period that was observed when
an aprotic solvent was used during catalysis can be explained by a competition between pyridine
and dihydrogen to coordinate to the amido complex RuCp*(C–NH). The energy barriers for the
coordination of pyridine and hydrogen to A are very similar (ΔGǂ = 15.7 and 14.1 kcal/mol),35
and the presence of alcohol provides a lower energy pathway to effectively convert Aalc and H2 to
the hydride complex Dalc, which will be readily consumed by transferring its H+/H- to acetone in
the outer sphere. This has the effect of driving the equilibrium between A plus pyridine and its
adduct, RuCp*(C–NH)(py), to the left.
Figure 6.15. The free energy profile for the outer sphere bifunctional mechanism in the
activation of H2 starting from A and moving to the right (blue pathway), and in the presence of 2-
propanol staring from Aalc and moving to the right (red pathway). The gas phase free energies (1
atm, 298 K) are reported relative to A, hydrogen and acetone in kcal/mol.

182
Figure 6.16. Computed transition state structure for the heterolytic splitting of H2 (TSC,Dalc) by
the ruthenium(II) system in the presence of 2-propanol. The bond lengths (Å) are given in the
structure. The color code for the atoms are: ruthenium (orange), nitrogen (blue), oxygen (red),
carbon (grey) and hydrogen (white).
6.3.15 The Alcohol-Assisted Outer Sphere Bifunctional Mechanism Involving Iridium(I)
Intermediates. As the cationic hydride-amine complex 21 must be activated during catalysis by
an alkoxide base, H2 and 2-propanol, we proposed, on the basis of computational results, the
formation of a neutral amido-hydride complex R that results from the deprotonation of the N-H
group of 21 (modeled as DIr of Scheme 6.6 in calculations) by 2-propoxide. This has a 2-
propanol molecule hydrogen-bonded to the amido nitrogen (Scheme 6.8). An alternative
structure Q can also form by the reaction of the cationic hydride complex DIr and 2-propoxide.
This has a coordinated η5-Cp, hydride and 2-propoxide and a decoordinated amine group of the
C–NH2 ligand. This structure is thermodynamically less stable than R (ΔG = 11.4 kcal/mol,
Scheme 6.8). As a coordination site is required for the activation of H2 starting from structures Q
or R, we have evaluated three feasible pathways, which included reductive elimination of 2-
propanol from Q, ring slippage of the Cp* ligand of R, and hydride migration to the Cp* ligand of
R. Bergman and co-workers proposed a similar pathways in the reductive elimination of ethanol
from IrCp*(PPh3)(H)(OEt), and subsequent coordination of a neutral ligand L forming the
iridium(I) species IrCp*(PPh3)L.42b To our surprise, the alcohol-assisted outer-sphere bifunctional
mechanism involving a hydride migration to the Cp* ligand was an energetically favourable
pathway for ketone hydrogenation: this will be presented in detail. The migration of a
coordinated ligand (hydrides,56 alkyl or aryl groups57) to the η5-Cp* ligand56b, 56c, in particular in
an endo fashion,56a, 56b, 57b, 58 is not common, but there are examples of stable iron(II) and

183ruthenium(II) complexes containing such a η4-Cp ligand as a result of an exo59 addition of the
hydride ligand to the η5-Cp ring. The two other pathways, reductive elimination of alcohol and
the ring slippage of the η5-Cp* ring, are characterized by higher free energy barriers to transfer a
hydride to acetone in the outer sphere (71.5 kcal/mol for the reductive elimination mechanism
and 34.1 kcal/mol for the ring slippage mechanism).35 The reductive elimination pathway,
however, is not feasible as this involves the decoordination of the amine throughout the catalytic
cycle, while experimental evidence supports the role of the N-H group.
Scheme 6.8. Computed Hydride Migration Pathway from the Ir–H bond to the Coordinated Cp
Ligand Starting from R.
According to the mechanism shown in Scheme 6.8, the amido-hydride complex S was formed
by the loss of a 2-propanol molecule from R. The hydride ligand from S can then add to the Cp
ligand, which is an uphill process and slightly entropically disfavoured (ΔGǂ = 35.1 kcal/mol
from S, ΔSǂ = -3.0 cal/mol·K, Scheme 6.8). Interestingly, replacing the Cp with a Cp* ligand
gave a very similar barrier to this process (ΔGǂ = 35.2 kcal/mol from S*, ΔSǂ = -1.2 cal/mol·K).
This contributes to the induction period that was observed in the H2-hydrogenation of
acetophenone in 2-propanol catalyzed by 21 in the presence of KOtBu. The analogous hydride
migration product of ruthenium(0), Ru(η4-CpH)(C–NH2) is thermodynamically less stable than D
(ΔG = 23.8 kcal/mol), although the barrier for hydride migration is similar in energy (ΔGǂ = 35.3
kcal/mol from D).
56. (a) Hirsekorn, F. J.; Rakowski, M. C.; Muetterties, E. L., J. Am. Chem. Soc. 1975, 97, 237-238; (b) McAlister, D. R.; Erwin, D. K.; Bercaw, J. E., J. Am. Chem. Soc. 1978, 100, 5966-5968; (c) Gusev, O. V.; Morozova, L. N.; Peganova, T. y. A.; Petrovskii, P. V.; Ustynyuk, N. A.; Maitlis, P. M., J. Organomet. Chem. 1994, 472, 359-363; (d) Lin, S. B.; Day, M. W.; Agapie, T., J. Am. Chem. Soc. 2011, 133, 3828-3831.57. (a) Meanwell, N. J.; Smith, A. J.; Maitlis, P. M., Dalton Trans. 1986, 1419-1424; (b) Wu, F.; Dash, A. K.; Jordan, R. F., J. Am. Chem. Soc. 2004, 126, 15360-15361.58. Cadenbach, T.; Gemel, C.; Schmid, R.; Fischer, R. A., J. Am. Chem. Soc. 2005, 127, 17068-17078.59. (a) Devies, S. G.; Hibberd, J.; Simpson, S. J.; Thomas, S. E.; Watts, O., Dalton Trans. 1984, 701-709; (b) DiBiase Cavanaugh, M.; Gregg, B. T.; Chiulli, R. J.; Cutler, A. R., J. Organomet. Chem. 1997, 547, 173-182; (c) Brown, D. A.; Deignan, J. P.; Fitzpatrick, N. J.; Fitzpatrick, G. M.; Glass, W. K., Organometallics 2001, 20, 1636-1645.
S (-0.2)
Ir NNN
CH3
HO
H
HH
R (0.0)
Ir NNN
CH3
H
H
TSS,HI (34.9)
Ir NNN
CH3
H
H
1
OHH
HI1 (3.3)
Ir NNN
CH3
H
HH
*Free energy (kcal/mol) given in parentheses.
CH3N
N
Ir
NHH
OH
H
Q (11.4)

184Scheme 6.9. Computed Alcohol-assisted Outer Sphere Bifunctional Mechanism in the H2-
Hydrogenation of Acetone Catalyzed by Complexes of Iridium(I).
Significantly, this neutral iridium(I) system, containing a η4-cyclopentadiene ligand, allows
efficient bifunctional catalysis via the outer sphere mechanism (Scheme 6.9). The square planar
amido complex HI1, can heterolytically split a coordinated H2 molecule (ΔGǂ = 28.1 kcal/mol) in
the transition state TS(HI)3,4 (Figure 6.17). The distorted square pyramidal hydride-amine
complex HI4, which has an apical hydride ligand (Ir–H distance = 1.64 Å), then completes the
cycle via the transition state TS(HI)5,6 by transferring a proton/hydride couple to acetone in the
outer sphere forming the amido complex HI1 and 2-propanol with a free energy barrier of 16.5
kcal/mol (Figure 6.18). It appears the neutral charge of the iridium(I) system is an important
factor in determining the reactivity of the bifunctional M–H/N–H pair during catalysis.
The presence of 2-propanol in the system (Scheme 6.9, Figure 6.18) effectively lowers the
free energy barrier for the heterolytic splitting of H2 by acting as a proton shuttle (ΔGǂ = 20.7
HH
HI2 TS(HI)3,4
TS(HI)5,6
O
H
H
H2
O
O
HI5
HI4
HI3Ir N
NN
CH3
H
Ir NNN
CH3
H
HH
Ir NNN
CH3
H
HH
Ir NNN
CH3
H
H
Ir NNN
CH3
H
HH
O Ir NNN
CH3
H
H
H
H
H
H
H
H
HH
H
H
H
H
H
Ir NNN
CH3
HO
H
H
HI6
HH
Ir NNN
CH3
HO
H
H
HI1
HH
O
H
H
O
H
H
HOH H
OH H
O
H
H
O
H
H
OH H
Alcohol-Assisted Outer SphereBif unctional Mechanism Involving
Iridium( I) intermediatesalc
alc
alc
alc
alc
alc
alc
alc
TS(HI)2,3
Ir NNN
CH3
H
HH
HH
O
H
H
alc

185kcal/mol). The transition state structure TS(HI)3,4alc shows that the protons neighbouring the
hydroxyl oxygen are even in closer proximity by the formation of two hydrogen-bonds via O...H
interactions (H2–O: 1.67; H3–O: 1.24 Å, Figure 6.17). On the other hand, the free energy
barriers for the coordination of H2 to HI1alc and the transfer of proton/hydride couple to acetone
in the outer sphere are less affected by the presence of a hydrogen-bonded 2-propanol molecule
in the system (ΔGǂ = 20.5 and 17.0 kcal/mol, respectively). All these provide evidence to support
an alcohol-assisted outer sphere bifunctional mechanism (Scheme 6.1), and serve to explain the
importance of 2-propanol as a solvent during catalysis.
Figure 6.17. Computed transition state structures for the non-alcohol assisted (TS(HI)3,4, left)
and the alcohol-assisted heterolytic splitting of H2 (TS(HI)3,4alc, right) by the iridium(I) system.
The bond lengths (Å) are given in the structures. The color code for the atoms are: iridium
(yellow), nitrogen (blue), oxygen (red), carbon (grey) and hydrogen (white).
6.3.16 Disfavored Inner Sphere Mechanisms Involving Cationic Iridium(III) Intermediates.
As an alternative to the outer sphere bifunctional mechanism, we investigated the possibility that
a cationic iridium(III) alkoxide complex G, [Ir(η5-Cp)(C–NH2)(OiPr)]+, is first formed instead of
the amido complex A upon activation of the precatalyst (Complex 19) by 2-propoxide. The
energy difference between A and G is 1.7 kcal/mol.12d In order to provide a vacant coordination
site for the activation of H2, ring slippage of the Cp ligand from η5 to η3 or the decoordination of
the amine group might occur.12d, 20a, 32 The energy barriers that were computed for the activation
of H2 are 55.0 kcal/mol and 35.6 kcal/mol, respectively (Scheme 6.10). Bäckvall and co-workers
have calculated a higher energy barrier for a η5 to η3 ring slip compared to the dissociation of CO
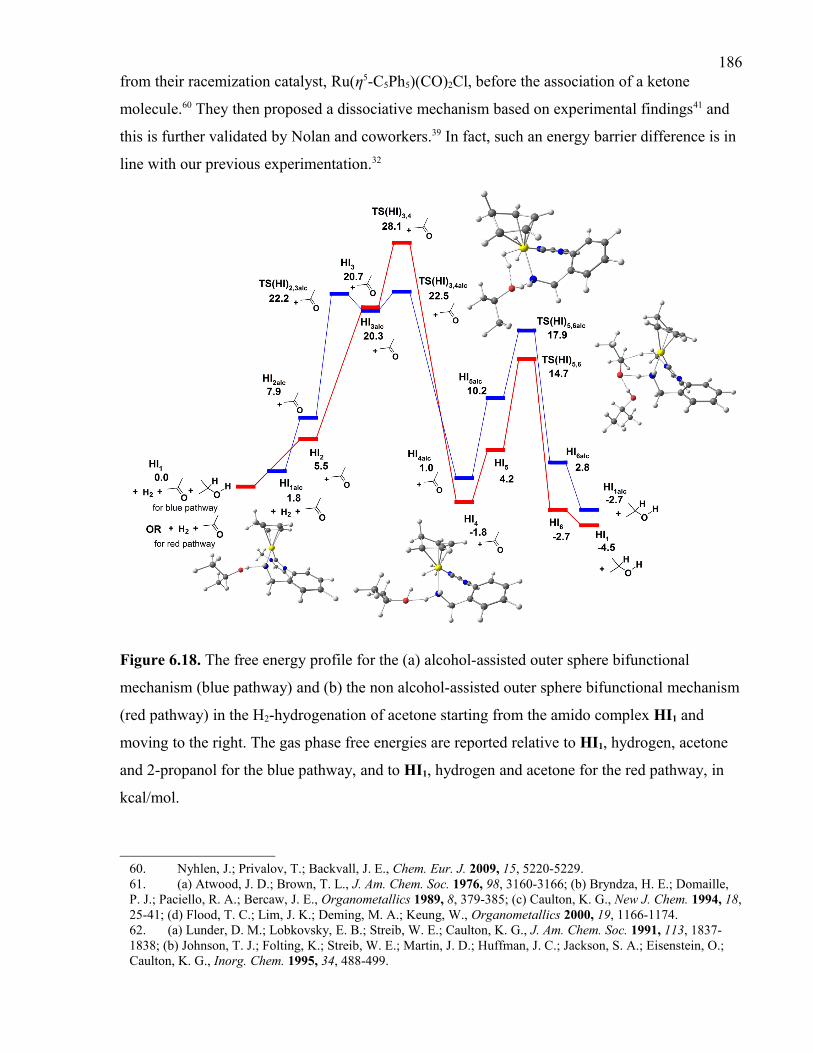
186from their racemization catalyst, Ru(η5-C5Ph5)(CO)2Cl, before the association of a ketone
molecule.60 They then proposed a dissociative mechanism based on experimental findings41 and
this is further validated by Nolan and coworkers.39 In fact, such an energy barrier difference is in
line with our previous experimentation.32
Figure 6.18. The free energy profile for the (a) alcohol-assisted outer sphere bifunctional
mechanism (blue pathway) and (b) the non alcohol-assisted outer sphere bifunctional mechanism
(red pathway) in the H2-hydrogenation of acetone starting from the amido complex HI1 and
moving to the right. The gas phase free energies are reported relative to HI1, hydrogen, acetone
and 2-propanol for the blue pathway, and to HI1, hydrogen and acetone for the red pathway, in
kcal/mol.
60. Nyhlen, J.; Privalov, T.; Backvall, J. E., Chem. Eur. J. 2009, 15, 5220-5229.61. (a) Atwood, J. D.; Brown, T. L., J. Am. Chem. Soc. 1976, 98, 3160-3166; (b) Bryndza, H. E.; Domaille, P. J.; Paciello, R. A.; Bercaw, J. E., Organometallics 1989, 8, 379-385; (c) Caulton, K. G., New J. Chem. 1994, 18, 25-41; (d) Flood, T. C.; Lim, J. K.; Deming, M. A.; Keung, W., Organometallics 2000, 19, 1166-1174.62. (a) Lunder, D. M.; Lobkovsky, E. B.; Streib, W. E.; Caulton, K. G., J. Am. Chem. Soc. 1991, 113, 1837-1838; (b) Johnson, T. J.; Folting, K.; Streib, W. E.; Martin, J. D.; Huffman, J. C.; Jackson, S. A.; Eisenstein, O.; Caulton, K. G., Inorg. Chem. 1995, 34, 488-499.

187In an inner sphere mechanism involving dissociation of the tethered-amine donor, the
alkoxide ligand in G labilizes the cis-amine ligand61 giving the 16 electron iridium(III) alkoxide
complex J with a decoordinated amine group (Scheme 6.10) The Ir–O bond length (1.91 Å) is
shorter in J in comparison to G (2.07 Å) to account for partial π-stabilization as observed
elsewhere.61-62 The free energy barrier for the labilization of the tethered amine donor by the
alkoxide ligand is 14.5 kcal/mol and it is entropically favoured (ΔS = 11.6 cal/mol·K). Complex
J then activates dihydrogen via oxidative addition to produce a dihydride63 complex (dH–H = 1.64
Å). Reductive elimination of the hydride and alkoxide releases the product alcohol. The substrate
ketone then coordinates and is attacked by the metal hydride in a four-membered ring transition
state 2a, 3a, 5c to give J again, with a free energy barrier of 20.5 kcal/mol comprised of an
unfavourable entropy change (ΔSǂ = 16.4 cal/mol·K). The transition state structure (TSM,J, Figure
6.19), was located on a very flat potential energy surface64 after numerous computational
attempts. It shows an electronegative carbon (APT charge = -0.68 ESU) of the coordinated
acetone and a charged hydride ligand (APT charge = -0.05 ESU).
Scheme 6.10. The Disfavoured Pathways Involving Either an η5 to η3 Ring Slip or a
Decoordination of the Amine Group Leading to the Activation of H2 by the Iridium(III) System.
In this mechanism, the N–H group does not play an active role in catalysis. However, a
comparison of the observed catalytic activities of 19 and 22 does not support this. The formation
of the hydride-amine complex 21 (or D) using H2 when either basic 2-propanol or THF is used as
the reaction medium could well proceed by only the outer sphere mechanism according to
63. Morris, R. H., Coord. Chem. Rev. 2008, 252, 2381-2394.64. (a) Bosch, E.; Moreno, M.; Lluch, J. M.; Bertran, J., Chem. Phys. Lett. 1989, 160, 543-548; (b) Koseki, S.; Gordon, M. S., J. Phys. Chem. 1989, 93, 118-125; (c) Espinosagarcia, J.; Corchado, J. C., J. Phys. Chem. 1995, 99, 8613-8616.
Ir NNN
CH3
H
OH
H
Decoordination ofNH2 group
+ H2
+ H2
Ring slippageof the Cp ring
∆ G‡ = 14.6 kcal/mol
∆ G‡ = 21.1 kcal/mol
NNN
CH3
H
Ir
OHH
∆ G = -1.7 kcal/mol GA
Ir
NN
N
CH3
HO
H
H
CH3NN
IrNH
H H O
J
CH3NN
IrN
OH HH
L
Ir
NN
N
CH3
HH O H
HH
N P
∆ G= 14.5 kcal/mol
∆ G= 40.4 kcal/mol
HH
+ +
+ +
++

188computations (ΔGǂ = 22.6 versus 38.3 kcal/mol relative to A, H2 and acetone for the activation
of H2 in the inner sphere mechanism).
Figure 6.19. Computed transition state structure for the attack of hydride on the coordinated
acetone (TSM,J) of the iridium(III) system for the inner sphere mechanism involving the
decoordination of the amine group. The bond lengths (Å) are given in the structure. The colour
code for the atoms are: iridium (yellow), nitrogen (blue), oxygen (red), carbon (grey) and
hydrogen (white).
6.3.17 Electronic Properties of Ruthenium(II) Complexes that Relate to their Reactivity in
Catalytic Hydrogenation. As the presence of an N-heterocyclic carbene provides enhanced
catalytic activity of the ruthenium(II) and iridium(I/III) system, we set out to prepare the
carbonyl complexes [RuCp*(C–NH2)(CO)]PF6 (23) and [RuCp*(P–NH2)(CO)]PF6 (24) in order to
compare their electronic properties. Both complexes 23 and 24 were characterized by NMR and
an X-ray diffraction study (Schemes 6.1 and 6.2, Figures 6.20 and 6.21) The carbonyl stretching
wavenumbers of 23 and 24 were observed at 1940 and 1952 cm-1, respectively.

189
Figure 6.20. ORTEP diagram of 23 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. Selected bond distances
(Å) and bond angles (deg): Ru(1)–C(1), 2.065(4); Ru(1)–N(3), 2.178(3); Ru(1)–C(22), 1.859(4);
Ru(1)–C(15), 2.223(4); C(22)–O(1), 1.149(5); C(1)–Ru(1)–N(3), 90.8(2); C(1)–Ru(1)–C(22),
95.1(2); C(22)–Ru(1)–N(3), 91.7(2).
A series of ruthenium(II) complexes formulated as RuCp*(D–NH2)Cl, where D is a donor
group, have been evaluated by Ikariya and co-workers by preparing the corresponding cationic
carbonyl complexes.6c, 65 The carbonyl stretching wavenumber of 23 lies in between those of the
complex [RuCp*(κ2(P,N)-PPh2CH2CH2NH2)CO]OTf, in which the D–NH2 ligand is less
donating, and complexes containing nitrogen donors, which are more electron donating (Figure
6.22). Of note, the complexes RuCp*(κ2(N,N)-N(CH3)2CH2CH2NH2)Cl and RuCp*(κ2(N,N)-(2'-
C5H4N) CH2NH2)Cl are ketones and aldehydes hydrogenation catalysts upon activation by KOH,
yet are poor catalysts for the hydrogenolysis of epoxides; the complex, RuCp*(κ2(P,N)-
PPh2CH2CH2NH2)Cl (16b), on the other hand, shows the reverse trend.20b, 24 Ikariya and co-
workers have attributed the stronger Brønsted acidity of the coordinated NH2 group of the latter
to the facile proton and hydride transfer to the polar bond.6c A counter-example to this is the
rhodium(I) amine complex, [Rh(η4-diene)(C–NHR)]BF4 (C–NHR = 1-mesityl-3-(2-
(mesitylamino)ethyl)imidazolylidene), which contains an aniline-type N–H group, is not active
towards the transfer hydrogenation of benzophenone.33c The phosphine-amine complex,
65. Ito, M., Pure Appl. Chem. 2008, 80, 1047-1053.
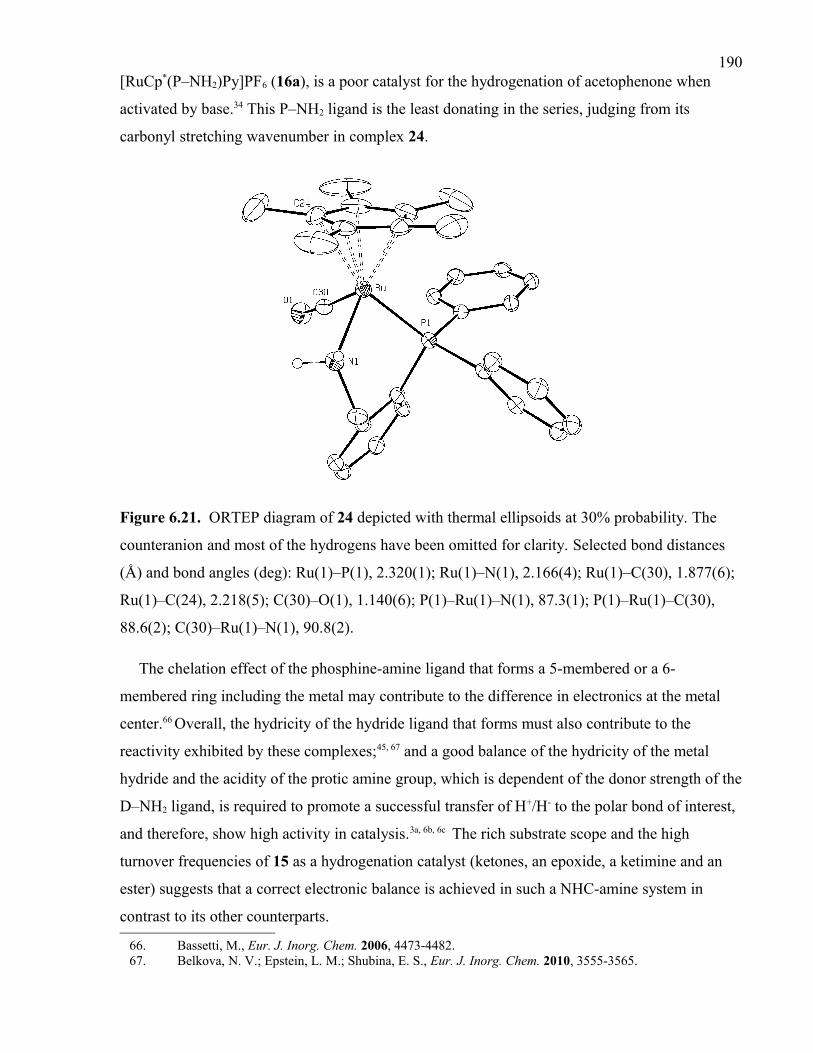
190[RuCp*(P–NH2)Py]PF6 (16a), is a poor catalyst for the hydrogenation of acetophenone when
activated by base.34 This P–NH2 ligand is the least donating in the series, judging from its
carbonyl stretching wavenumber in complex 24.
Figure 6.21. ORTEP diagram of 24 depicted with thermal ellipsoids at 30% probability. The
counteranion and most of the hydrogens have been omitted for clarity. Selected bond distances
(Å) and bond angles (deg): Ru(1)–P(1), 2.320(1); Ru(1)–N(1), 2.166(4); Ru(1)–C(30), 1.877(6);
Ru(1)–C(24), 2.218(5); C(30)–O(1), 1.140(6); P(1)–Ru(1)–N(1), 87.3(1); P(1)–Ru(1)–C(30),
88.6(2); C(30)–Ru(1)–N(1), 90.8(2).
The chelation effect of the phosphine-amine ligand that forms a 5-membered or a 6-
membered ring including the metal may contribute to the difference in electronics at the metal
center.66 Overall, the hydricity of the hydride ligand that forms must also contribute to the
reactivity exhibited by these complexes;45, 67 and a good balance of the hydricity of the metal
hydride and the acidity of the protic amine group, which is dependent of the donor strength of the
D–NH2 ligand, is required to promote a successful transfer of H+/H- to the polar bond of interest,
and therefore, show high activity in catalysis.3a, 6b, 6c The rich substrate scope and the high
turnover frequencies of 15 as a hydrogenation catalyst (ketones, an epoxide, a ketimine and an
ester) suggests that a correct electronic balance is achieved in such a NHC-amine system in
contrast to its other counterparts.
66. Bassetti, M., Eur. J. Inorg. Chem. 2006, 4473-4482.67. Belkova, N. V.; Epstein, L. M.; Shubina, E. S., Eur. J. Inorg. Chem. 2010, 3555-3565.

191
Figure 6.22. The carbonyl stretching wavenumbers (cm-1 in KBr) of a series of ruthenium(II)
complexes, [RuCp*(D–NH2)CO]+, where D is a donor group.
6.4 Conclusion
A comparison of the activity and selectivity of two structurally similar ruthenium(II) (15) and
iridium(III) (19 and 21) catalyst systems in the H2-hydrogenation of ketones reveals that the
ruthenium system has the higher activity but with similar selectivity in comparison to the iridium
system in the hydrogenation of ketones. An alcohol-assisted outer sphere bifunctional
mechanism is proposed for both systems. There are several pieces of evidence to support this:
(a) the pronounced effect of alcohols in catalysis, and H/D scrambling that is observed when
deuterated sources are used;
(b) an N–H group is required by the chloride complex 19 for catalysis since the structurally
similar iridium(III) complex 22 with no N–H group is much less active;
(c) a low free energy barrier is calculated for the transfer of a proton/hydride couple from the
neutral ruthenium(II) hydride Ru(η5-Cp)(C–NH2)H to the ketone in the outer sphere; however,
the cationic hydride complex [Ir(η5-Cp)(C–NH2)H]+ is calculated to have a high barrier;
(d) an accessible energy barrier is calculated for the transfer a proton/hydride couple from the
neutral hydride complex Ir(η4-CpH)(C–NH2)H to acetone in the outer sphere;
(f) a significant decrease in the free energy barriers to the heterolytic splitting of the η2-H2
ligand on Ru(η5-Cp)(C–NH) and Ir(η4-CpH)(C–NH) is calculated when a 2-propanol molecule
acts a proton shuttle by participating in a six-membered ring transition state.6c, 12e, 13a, 14b, 19b, 20b, 22
Ru
NN
N
CH3PF6OC
HH
RuPPh2N
PF6OC
HH
2423
Ru
N NOTfOC
HH
RuPPh2N
OTfOC
HH
RuN
N
OTfOC
HH
CO Stretch(cm-1, KBr)
1931 1938 1940 1948 1952
+ + + + +

192 The cationic charge of the intermediates of the iridium(III) system lead to a diminished
ability of the metal hydride-amine complex 21 to react with a ketone in the absence of base. The
neutral, well-defined complexes containing a cyclometalated C–NH2 ligand and an anionic
carbon donor, IrCp*(κ2(C,N)-2-C6H4CR2NH2)H49 and Ru(η6-C6H6)(κ2(C,N)-2-C6H4CR2NH2)H,50
react with acetophenone in the absence of base. The decreased hydricity of such piano-stool
cationic metal hydrides in catalysis appears to be a general phenomenon.45, 67
The activation of the iridium(III) hydride-amine complex 21 by alkoxide base seems to be
crucial for catalysis. Our computational studies support the proposal that the base deprotonates
the amine group of the cationic hydride-amine complex 21 and this then triggers the migration of
the hydride to the η5-Cp ring producing a neutral iridium(I) amido complex. This amido complex
with an η4-cyclopentadiene (CpH) group splits η2-H2 with the assistance of alcohol to produce the
Ir–H/N–H couple required for the outer sphere hydrogenation of ketones in the bifunctional
mechanism. These steps involve novel iridium(I) intermediates and proceed with reasonable free
energy barriers. The two other possibilities, which are the reductive elimination of 2-propanol
and the ring slippage pathway, are characterized by a high energy barrier to transfer the metal
hydride to the ketone in the outer sphere within the catalytic cycle in comparison to our proposed
mechanism. As experimental evidence supports the role of the N–H group, the reductive
elimination pathway is not feasible as this involves the decoordination of the amine throughout
the catalytic cycle. Although the migration of the hydride ligand to the η5-Cp* ligand is not
common but is precedented,42b, 56b, 56c this might be favoured by the presence of the unique NHC
ligand in our catalytic system.28b, 28c It appears that the activation of the cationic hydride-amine
complex 21 is responsible for the poor activity of the iridium(III) system for catalytic ketone
hydrogenation in comparison to its ruthenium(II) counterpart.
This role of the alkoxide base in the catalysis should be considered for other systems
involving catalytic ketone hydrogenation. Although it has been suggested that the alkali metal
cations of these bases may play a crucial role to stabilize the transition state during hydride
transfer to the ketone,51 we have found that the alkoxide base reacts with the metal hydride
complex by changing the geometry and the electronics of the piano-stool complex in order to
make it reactive. It was known that certain ruthenium(II)-based catalysts required a high base
loading (C/B > 1/1000) in the hydrogenation of ketones using H2.14b, 68 The role of the alkoxide
68. Doucet, H.; Ohkuma, T.; Murata, K.; Yokozawa, T.; Kozawa, M.; Katayama, E.; England, A. F.; Ikariya, T.; Noyori, R., Angew. Chem. Int. Ed. 1998, 37, 1703-1707.

193base remains unclear but it may play a similar role to transform the metal hydride complex to a
more reactive species which activates hydrogen and promotes H+/H- transfer to the ketone.
Finally, the donor ability of the D–NH2 type ligand (D = NHC, phosphine, amine, 2'-pyridine)
was also investigated by use of the carbonyl stretching wavenumbers of the complexes
[RuCp*(D–NH2)CO]X to help explain the poorer performance in the catalytic H2-hydrogenation
of ketones of the phosphine-amine complexes [RuCp*(P–NH2)py]PF6 (16a) and [IrCp*(P–
NH2)Cl]PF6 (20) compared to C–NH2 analogues. The C–NH2 ligand was found to be more
donating than the P–NH2 ligand.
We conclude that a fine-tuning of the Brønsted acidities of the N–H and the M–H groups by
choosing the right donor ligand, the right metal center, as well as the correct overall charge of the
catalyst is crucial to maximize the activity for the hydrogenation of polar bonds. The present
study will provide guidelines in catalyst architecture and in the rational use of alkoxide base to
maximize the potential of bifunctional catalysts in the direct hydrogenation of polar bonds.
6.5 Experimental Section
6.5.1 Synthesis. All of the preparations and manipulations, except where otherwise stated, were
carried out under a nitrogen or argon atmosphere using standard Schlenk-line and glovebox
techniques. Dry and oxygen-free solvents were always used. The syntheses of bis[1-(2-
aminomethylphenyl)-3-methylimidazol-2-ylidene]nickel(II) hexafluorophosphate (12) and [1-(2-
aminomethylphenyl)-3-methylimidazol-2-ylidene](η5-pentamethylcyclopentadienyl)-
(pyridine)ruthenium(II) hexafluorophosphate (15) have been reported previously.31, 34 The
syntheses of RuCp*(cod)Cl,69 [IrCp*Cl2]2,70 2-(diphenylphosphino)benzylamine (P–NH2),71 and 1-
(N,N-dimethylamino)propyl-3-methylimidazolium chloride hydrochloride (1g)46 were reported in
the literature. All other reagents and solvents were purchased from commercial sources and
were used as received. Deuterated solvents were purchased from Cambridge Isotope
Laboratories and Sigma Aldrich and degassed and dried over activated molecular sieves prior to
use. NMR spectra were recorded on a Varian 400 spectrometer operating at 400 MHz for 1H, 100
MHz for 13C, 161 MHz for 31P and 376 MHz for 19F. The 1H and 13C{1H} NMR were measured
69. Boren, B. C.; Narayan, S.; Rasmussen, L. K.; Zhang, L.; Zhao, H. T.; Lin, Z. Y.; Jia, G. C.; Fokin, V. V., J. Am. Chem. Soc. 2008, 130, 8923-8930.70. White, C.; Yates, A.; Maitlis, P. M., Inorg. Synth. 1992, 29, 228-234.71. Cahill, J. P.; Bohnen, F. M.; Goddard, R.; Kruger, C.; Guiry, P. J., Tetrahedron: Asymmetry 1998, 9, 3831-3839.

194relative to partially deuterated solvent peaks but are reported relative to tetramethylsilane (TMS).
All 19F chemical shifts were measured relative to trichlorofluoromethane as an external reference.
All 31P chemical shifts were measured relative to 85% phosphoric acid as an external reference.
All infrared spectra were recorded on a Nicolet 550 Magna-IR spectrometer. The elemental
analysis was performed at the Department of Chemistry, University of Toronto, on a Perkin-
Elmer 2400 CHN elemental analyzer. Samples were handled under argon where it was
appropriate. Single-crystal X-ray diffraction data were collected using a Nonius Kappa-CCD
diffractometer with Mo Kα radiation (λ = 0.71073 Å). The CCD data were integrated and scaled
using the Denzo-SMN package. The structures were solved and refined using SHELXTL V6.1.
Refinement was by full-matrix least-squares on F2 using all data.
6.5.2 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-chloro-(η5-penta-
methylcyclopentadienyl)iridium(III) Hexafluorophosphate ([IrCp*(C–NH2)Cl]PF6, 19). A
Schlenk flask was charged with 12 (71 mg, 0.098 mmol) and [IrCp*Cl2]2 (78 mg, 0.098 mmol).
Dry acetonitrile (12 mL) was added to the reaction mixture, and it was refluxed under an argon
atmosphere for 2.5 h until a deep green solution was obtained. The solvent was evaporated under
reduced pressure, and the residue was extracted with tetrahydrofuran (4 mL) and filtered through
a pad of Celite under a nitrogen atmosphere. Addition of diethyl ether (15 mL) to this and slow
cooling of the solution at -25°C afforded a yellow precipitate. This was collected on a glass frit,
washed with diethyl ether (1 mL) and dried in vacuo. Yield: 127 mg, 93%. Suitable crystals for
an X-ray diffraction study were obtained by slow diffusion of diethyl ether into a saturated solu-
tion of 19 in acetonitrile under a nitrogen atmosphere. 1H NMR (CD2Cl2, δ): 7.75 (dd, JHH = 1.70,
7.32 Hz, 3-CH of Ph, 1H), 7.59 (m, 4-CH and 5-CH of Ph, 2H), 7.48 (dd, JHH = 1.51, 7.55 Hz, 6-
CH of Ph, 1H), 7.34 (d, JHH = 2.06 Hz, 5-CH of imid.,1H), 7.32 (d, JHH = 2.06 Hz, 4-CH of imid.,
1H), 4.38 (br, CH2 and NH2, 3H), 4.04 (s, CH3, 3H), 3.29 (m, CH2, 1H), 1.33 (s, CH3 of Cp*,
15H). 19F NMR (CD2Cl2, δ): -72.2 (d, JPF = 712 Hz). 13C{1H} NMR (CD2Cl2, δ): 155.8 (Ir–C),
139.1 (CPh), 132.7 (CPh), 131.5 (CPh), 130.9 (CPh), 130.1 (CPh), 125.8 (CPh), 125.4 (Cimid.), 124.2
(Cimid.), 90.5 (CAr-Cp*), 47.4 (CH2), 38.9 (CH3), 8.6 (CH3 of Cp*). MS (ESI, methanol/water; m/z):
550.2 ([M]+), 514.2 ([M – Cl]+). HRMS (ESI, methanol/water; m/z): calcd for C21H28N3ClIr+ ([M]
+): 550.1595, found: 550.1566. Anal. Calcd for C21H28N3ClF6PIr: C, 36.29; H, 4.06; N, 6.05.
Found: C, 36.72; H, 3.97; N, 5.23.
6.5.3 Synthesis of [2-(Diphenylphosphino)benzylamine]-chloro-(η5-pentamethyl-cyclo-
pentadienyl)iridium(III) Hexafluorophosphate ([IrCp*(P–NH2)Cl]PF6, 20). A scintillation

195vial with a threaded screw cap was charged with [IrCp*Cl2]2 (47 mg, 0.059 mmol) in dry di-
chloromethane (6 mL) under a nitrogen atmosphere. A solution of 2-(diphenylphosphino)-ben-
zylamine (36 mg, 0.12 mmol) in dry dichloromethane (6 mL) was added to the aforementioned
yellow solution and stirred for 1 h at room temperature (25oC). Silver hexafluorophosphate (30
mg, 0.12 mmol) in dry acetonitrile (1 mL) was added to the reaction mixture, and a pale-yellow
suspension was obtained. After stirring the reaction mixture further for 0.5 h, it was filtered
through a pad of Celite under a nitrogen atmosphere. The solvent was removed at reduced pres-
sure. The yellow crude product was recrystallized with dichloromethane (2 mL) and a diethyl
ether and pentane mixture (1:8, 12 mL) to yield a yellow solid, which was filtered and dried in
vacuo. Yield: 54 mg, 57%. Suitable crystals for an X-ray diffraction study were obtained by slow
diffusion of diethyl ether into a saturated solution of 20 in dichloromethane under a nitrogen at-
mosphere. 1H NMR (CD2Cl2, δ): 7.76 (m, Ar-CH of PPh2, 2H), 7.59 (m, Ar-CH of PPh2, 8H),
7.44 (m, 3-CH of Ph, 1H), 7.38 (m, 4-CH and 5-CH of Ph, 2H), 7.22 (m, 6-CH of Ph, 1H), 5.52
(br, NH2, 1H), 4.39 (m, CH2, 1H), 3.73 (m, br, CH2 and NH2, 2H), 1.55 (s, CH3 of Cp*, 15H). 19F
NMR (CD2Cl2, δ): -72.3 (d, JPF = 712 Hz). 31P{1H} NMR (CD2Cl2, δ): 4.5 (s), -144.4 (sept, JPF =
712 Hz). 13C{1H} NMR (CD2Cl2, δ): 138.5 (d, JCP = 13.62 Hz, CPPh), 135.4 (d, JCP = 12.07 Hz,
CPPh), 134.3 (d, JCP = 9.54 Hz, CPPh), 132.8 (d, JCP = 2.54 Hz, CPPh), 132.3 (d, JCP = 2.69 Hz, CPPh),
131.8 (CPh), 131.7 (CPh), 131.6 (d, JCP = 2.42 Hz, CPh), 131.0 (d, JCP = 2.36 Hz, CPh), 130.8 (d, JCP
= 55.83 Hz, CPPh), 129.6 (d, JCP = 10.96 Hz, CPh), 129.0 (d, JCP = 11.08 Hz, CPh), 127.5 (d, JCP =
60.80 Hz, CPPh), 124.8 (d, JCP = 56.53 Hz, CPPh), 94.5 (CAr-Cp*), 48.8, (d, JCP = 11.00 Hz, CH2), 8.8
(CH3 of Cp*). MS (ESI, methanol/water; m/z): 654.2 ([M]+), 618.2 ([M – Cl]+). HRMS (ESI,
methanol/water; m/z): calcd for C29H33NPClIr+ ([M]+): 654.1663, found: 654.1638. Anal. Calcd
for C29H33NClF6P2Ir: C, 43.58; H, 4.16; N, 1.75. Found: C, 43.11; H, 4.11; N, 1.79.
6.5.4 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-hydrido-(η5-pen-
tamethylcyclopentadienyl)iridium(III) Hexafluorophosphate ([IrCp*(C–NH2)H]PF6, 21). A
Schlenk flask was charged with 19 (50 mg, 0.072 mmol) in 2-propanol solution (14 mL). The
solution was warmed to 50°C under an argon atmosphere. A solution of sodium 2-propoxide (18
mg, 0.22 mmol) in 2-propanol (6 mL) was added to this stirring solution during a course of 0.5 h,
whereupon the color of the reaction mixture turned from yellow to red, then to deep brown. The
solution was stirred for a further 3 h. After the reaction had gone to completion, the solvent was
removed under vacuum. The solid residue was extracted with THF (4 mL) and filtered through a
pad of Celite. The addition of pentane (16 mL) to the THF solution yielded a beige-coloured pre-

196cipitate, which was collected and dried in vacuo. Yield: 36 mg, 76%. Suitable crystals for an X-
ray diffraction study were obtained by slow diffusion of diethyl ether into a saturated solution of
21 in THF under a nitrogen atmosphere at -30°C. 1H NMR (CD3CN, δ): 7.60 (m, 3-CH and 4-CH
of Ph, 2H), 7.53 (m, 5-CH of Ph, 1H), 7.49 (m, 6-CH of Ph, 1H), 7.37 (d, JHH = 2.13 Hz, 5-CH of
imid.,1H), 7.34 (d, JHH = 2.13 Hz, 4-CH of imid., 1H), 4.80 (br, NH2, 1H), 4.01 (m, CH2, 1H),
3.73 (s, CH3, 3H), 3.55 (br, NH2, 1H), 2.85 (dt, JHH = 3.50, 12.40 Hz, CH2, 1H), 1.51 (s, CH3 of
Cp*, 15H), -13.59 (s, Ir–H, 1H). 19F NMR (CD3CN, δ): -73.0 (d, JPF = 706 Hz). 13C{1H} NMR
(CD3CN, δ): 157.0 (Ir–C), 140.8 (CPh), 133.0 (CPh), 132.5 (CPh), 130.6 (CPh), 129.4 (CPh), 126.4
(CPh), 123.8 (Cimid.), 123.5 (Cimid.), 90.6 (CAr-Cp*), 49.2 (CH2), 39.6 (CH3), 9.8 (CH3 of Cp*). IR
(KBr, cm-1): 2068 (v(Ir–H)). MS (ESI, methanol/water; m/z): 516.2 ([M]+). HRMS (ESI, meth-
anol/water; m/z): calcd for C21H29N3ClIr+ ([M]+): 516.1985, found: 516.1961. Anal. Calcd for
C21H29N3F6PIr: C, 38.18; H, 4.42; N, 6.36. Found: C, 38.02; H, 4.35; N, 6.01.
6.5.5 Synthesis of [1-(N,N-Dimethylaminopropyl)-3-methylimidazol-2-ylidene]-chloro-(η5-
pentamethylcyclopentadienyl)iridium(III) Hexafluorophosphate ([IrCp*(C–NMe2)Cl]PF6,
22). A Schlenk flask was charged with silver(I) oxide (65 mg, 0.28 mmol) and [IrCp*Cl2]2 (75
mg, 0.094 mmol) in dry acetonitrile (8 mL) under molecular sieves (3 Å). In a separate Schlenk
flask was charged with 1g (45 mg, 0.19 mmol) and anhydrous dimethyl sulfoxide (DMSO, 3
mL) under an argon atmosphere. The DMSO solution containing the dissolved imidazolium salt
was then added to the stirring solution of silver(I) oxide and [IrCp*Cl2]2 in acetonitrile. Acetoni-
trile washing (4 mL) was applied to the residual DMSO containing the imidazolium salt, and this
was also added to the reaction mixture. This was stirred under an argon atmosphere at room tem-
perature (25°C) for overnight. After the reaction has gone to completion, the reaction mixture
was filtered through a pad of Celite under an argon atmosphere. The solvent was evaporated un-
der reduced pressure. The residue that obtained was washed with toluene (6 mL) and then diethyl
ether (8 mL). It was dissolved in a dichloromethane and acetonitrile mixture (1:1, 10 mL) and
this was added silver hexafluorophosphate (47 mg, 0.19 mmol) and stirred further for 1 h. The
suspension that formed was filtered through a pad of Celite. The solvent was evaporated under
reduced pressure, and the residue that obtained was precipitated from tetrahydrofuran (3 mL) and
pentane (15 mL). This was filtered and dried in vacuo to give a bright yellow solid. Yield: 77
mg, 61%. Suitable crystals for an X-ray diffraction study were obtained by slow diffusion of di-
ethyl ether into a saturated of 22 in dichloromethane under a nitrogen atmosphere 1H NMR
(CD2Cl2, δ): 7.17 (d, JHH = 2.00 Hz, 5-CH of imid., 1H), 7.06 (d, JHH = 2.00 Hz, 4-CH of imid.,

1971H), 4.28 (m, CH2, 1H), 4.18 (m, CH2, 1H), 3.92 (s, CH3, 3H), 3.49 (t, JHH = 13.12 Hz, CH2, 1H),
3.24 (m, CH2, 1H), 2.21 (t, JHH = 13.41 Hz, CH2, 2H), 1.70 (s, CH3 of N(CH3)2, 6H), 1.56 (s, CH3
of Cp*, 15H). 19F NMR (CD2Cl2, δ): -73.1 (d, JPF = 711 Hz). 13C{1H} NMR (CD2Cl2, δ): 155.4
(Ir–C), 124.6 (Cimid.), 124.3 (Cimid.), 91.6 (CAr-Cp*), 69.8 (CH2), 49.3 (CH2), 38.7 (CH3), 26.8 (CH2),
9.9 (CH3 of Cp*), 8.8 (CH3 of N(CH3)2). MS (ESI, methanol/water; m/z): 496.2 ([M – Cl]·+),
451.2 ([M – Cl – NMe2]·+). Anal. Calcd for C19H32N3ClF6 PIr: C, 33.80; H, 4.78; N, 6.22. Found:
C, 33.96; H, 4.74; N, 6.34.
6.5.6 Synthesis of [1-(2-Aminomethylphenyl)-3-methylimidazol-2-ylidene]-carbonyl-(η5-
pentamethylcyclopentadienyl)ruthenium(II) Hexafluorophosphate ([RuCp*(C–NH2)
(CO)]PF6, 23). A Schlenk flask was charged with 15 (32 mg, 0.049 mmol). It was evacuated and
backfilled with a CO atmosphere (1 atm) for two times. A solution of tetrahydrofuran (10 mL)
saturated with Ar was injected into the Schlenk flask against a flow of CO by means of a syringe
and a needle. The colour of the solution turned immediately from orange-yellow to pale yellow
upon dissolution. The solution was stirred at room temperature (25°C) for 3 h. The volume of the
solvent was reduced (2 mL). Addition of diethyl ether or hexanes (12 mL) to this and slow cool-
ing of the solution at -25°C afforded a pale yellow crystalline solid, which was filtered on a glass
frit and dried in vacuo. Yield: 22 mg, 75%. Suitable crystals for an X-ray diffraction study were
obtained by slow diffusion of hexanes into a saturated solution of 23 in THF under a nitrogen at-
mosphere. 1H NMR (CD2Cl2, δ): 7.62 (m, 3-CH and 5-CH of Ph, 2H), 7.51 (m, 4-CH and 6-CH
of Ph, 2H), 7.35 (d, JHH = 2.07 Hz, 5-CH of imid.,1H), 7.29 (d, JHH = 2.07 Hz, 4-CH of imid.,
1H), 4.21 (td, JHH = 2.69, 11.53 Hz, CH2, 1H), 3.89 (s, CH3, 3H), 3.84 (br, NH2, 1H), 2.98 (m,
CH2, 1H), 2.17 (m, br, NH2, 1H), 1.40 (s, CH3 of Cp*, 15H). 19F NMR (CD2Cl2, δ): -72.5 (d, JPF =
712 Hz). 13C{1H} NMR (CD2Cl2, δ): 207.1 (Ru–CCO), 181.8 (Ru–C), 139.5 (CPh), 132.6 (CPh),
131.8 (CPh), 130.5 (CPh), 129.7 (CPh), 126.2 (CPh), 125.0 (Cimid.), 124.7 (Cimid.), 94.6 (CAr-Cp*), 48.9
(CH2), 39.4 (CH3), 9.5 (CH3 of Cp*). IR (KBr, cm-1): 1940 (v(CO)). MS (ESI, methanol/water;
m/z): 452.1 ([M]+). HRMS (ESI, methanol/water; m/z): calcd for C22H28N3ORu+ ([M]+): 452.1270,
found: 452.1269. Attempts at elemental analyses failed to give an acceptable carbon content,
while hydrogen and nitrogen content are in the acceptable range. Typical results: Anal. Calcd for
C22H38F6N3OPRu: C, 44.30; H, 4.73; N, 7.04. Found: C, 42.99; H, 4.64; N, 7.35.
6.5.7 Synthesis of [2-(Diphenylphosphino)benzylamine]-carbonyl-(η5-pentamethyl-cyclo-
pentadienyl)ruthenium(II) Hexafluorophosphate ([RuCp*(P–NH2)(CO)]PF6, 24). A scintilla-
tion vial with a threaded screw cap was charged with RuCp*(cod)Cl (50 mg, 0.13 mmol) in dry

198dichloromethane (5 mL) under a nitrogen atmosphere. A solution of 2-(diphenyl-phosphino)ben-
zylamine (40 mg, 0.13 mmol) in dry dichloromethane (5 mL) was added to the aforementioned
yellow solution and stirred for 1 h at room temperature (25°C), whereupon the reaction mixture
turned into orange in colour. Silver hexafluorophosphate (33 mg, 0.13 mmol) in dry acetonitrile
(1 mL) was added to the reaction mixture, and a yellow-brown suspension was obtained. After
stirring the reaction mixture for 0.5 h, it was filtered through a pad of Celite, and the solvent was
collected into a Schlenk flask under a nitrogen atmosphere. The solvent was then removed under
reduced pressure. The Schlenk flask containing the residue was evacuated and backfilled with a
CO atmosphere (1 atm) for two times. A solution of tetrahydrofuran (10 mL) saturated with Ar
was injected into the Schlenk flask against a flow of CO by means of a syringe and needle. The
colour of the solution turned immediately from orange-yellow to pale yellow upon dissolution.
The solution was stirred at room temperature for 3 h. The volume of the solvent was reduced (2
mL). Addition of diethyl ether (15 mL) afforded a yellow precipitate, which was filtered and
dried in vacuo to give a pale-yellow solid. Yield: 72 mg, 78%. Suitable crystals for an X-ray dif-
fraction study were obtained by slow diffusion of diethyl ether into a saturated solution of 24 in
dichloromethane under a nitrogen atmosphere. 1H NMR (CD2Cl2, δ): 7.56-7.49 (m, Ar-CH of Ph-
PPh2, 6H), 7.47 (m, Ar-CH of Ph-PPh2, 1H), 7.45-7.36 (m, Ar-CH of Ph-PPh2, 5H), 7.32-7.25 (m,
Ar-CH of Ph-PPh2, 2H), 4.00 (m, CH2, 1H), 3.71 (br, NH2, 1H), 3.58 (m, CH2, 1H), 3.05 (br,
NH2, 1H), 1.67 (s, CH3 of Cp*, 15H). 19F NMR (CD2Cl2, δ): -72.6 (d, JPF = 712 Hz). 31P{1H}
NMR (CD2Cl2, δ): 42.1 (s), -144.5 (sept, JPF = 712 Hz). 13C{1H} NMR (CD2Cl2, δ): 203.9 (d, JCP
= 16.83 Hz, Ru–Cco), 140.7 (d, JCP = 15.08 Hz, CPh-PPh), 134.1 (d, JCP = 12.48 Hz, CPh-PPh), 133.4 (d,
JCP = 10.93 Hz, CPh-PPh), 132.5 (d, JCP = 22.03 Hz, CPh-PPh), 132.3 (d, JCP = 31.09 Hz, CPh-PPh), 131.8
(d, JCP = 2.20 Hz, CPh-PPh), 131.6 (d, JCP = 2.11 Hz, CPh-PPh), 131.4 (d, JCP = 2.51 Hz, CPh-PPh), 131.3
(CPh-PPh), 130.9 (d, JCP = 1.95 Hz, CPh-PPh), 129.7 (CPh-PPh), 129.4 (d, JCP = 10.38 Hz, CPh-PPh), 129.3
(d, JCP = 10.51 Hz, CPh-PPh), 129.1 (d, JCP = 7.11 Hz, CPh-PPh), 96.8 (d, JCP = 1.91 Hz, CAr-Cp*), 50.4,
(d, JCP = 9.41 Hz, CH2), 9.7 (CH3 of Cp*). IR (KBr, cm-1): 1952 (v(CO)). MS (ESI, methanol/wa-
ter; m/z): 556.1 ([M]+). HRMS (ESI, methanol/water; m/z): calcd for C30H33NOPRu+ ([M]+):
556.1337, found: 556.1355. Anal. Calcd for C30H33NF6OP2Ru: C, 51.43; H, 4.75; N, 2.00. Found:
C, 51.10; H, 4.70; N, 1.91.
6.5.8 Representative Stoichiometric Reaction Using High Pressure of H2. Complex 19 (5 mg,
7.2 µmol) and potassium tert-butoxide (10 mg, 0.089 mmol) were dissolved separately in THF (4
mL and 2 mL, respectively) under a nitrogen atmosphere. These solutions were taken up by

199means of two separate syringes and needles in a glovebox. The needles were stoppered and the
syringes were taken to the reactor. The solutions were then injected into the reactor against a
flow of hydrogen gas. The hydrogen gas was adjusted to 25 bar and the reaction mixture was
stirred at 50°C. After 3 h of reaction, the reactor was detached from the hydrogen source and a H2
pressure of 2-4 bar was maintained. The reactor was attached to a Schlenk-line and backfilled
with argon gas using standard Schlenk-line techniques. The reaction mixture was then
transferred to an empty Schlenk flask filled with argon by syringe and a needle and the solvent
was removed in vacuum. The residue was taken up in acetonitrile-d3 and filtered through a pad of
Celite, and a 1H NMR spectrum was measured.
6.5.9 Stoichiometric Reaction Using 19 and 1-Phenylethoxide. A solution of complex 19 (15
mg, 0.022 mmol) in THF (6 mL) was added potassium 1-phenylethoxide (5 mg, 0.031 mmol) in
THF (4 mL) under an argon atmosphere at 50°C. The reaction was stirred for 3 h and the solvent
was removed in vacuum. The residue was taken up in acetonitrile-d3 and filtered through a pad of
Celite, and a 1H NMR spectrum was measured. The 1H NMR spectrum was compared to
authentic samples of 19, 21 and acetophenone in acetonitrile-d3 and the conversion was
measured by comparing the integration of the methyl groups of each.
6.5.10 Catalysis. Oxygen-free tetrahydrofuran (THF) used for all of the catalytic runs was stirred
over sodium for 2-3 days under argon, and freshly distilled from sodium benzophenone ketyl
prior to use. Oxygen-free 2-propanol used for all catalytic runs was stirred over magnesium
turnings and a single chip of iodine for several hours under argon, and freshly distilled prior to
use. Acetophenone was vacuum distilled over phosphorus pentoxide (P2O5) and stored under
nitrogen prior to use. 2-Propanol-d1 (purchased from Cambridge Isotope Laboratories) and 1-
phenylethanol were vacuum-distilled, dried over activated molecular sieves, and stored under
nitrogen prior to use. D2 gas was purchased from Cambridge Isotope Laboratories. All of the
hydrogenation reactions were performed at constant pressures using a stainless steel 50 mL Parr
hydrogenation reactor. The temperature was maintained at 25 or 50°C using a constant
temperature water bath. The reactor was flushed several times with hydrogen gas at 2-4 bar prior
to the addition of catalyst and substrate, and base solutions.
In a typical run (Table 6.1, entry 1), catalyst 19 (3 mg, 4.3 µmol) and acetophenone (104 mg,
0.87 mmol), and potassium tert-butoxide (4 mg, 0.036 mmol) were dissolved in THF (4 mL and
2 mL, respectively) under a nitrogen atmosphere. The catalyst/substrate and base solutions were

200taken up by means of two separate syringes and needles in a glovebox. The needles were
stoppered and the syringes were taken to the reactor. The solutions were then injected into the
reactor against a flow of hydrogen gas. The hydrogen gas was adjusted to 25 bar. Small aliquots
of the reaction mixture were quickly withdrawn with a syringe and a needle under a flow of
hydrogen at timed intervals by venting the Parr reactor at reduced pressure. Alternatively, small
aliquots of the reaction mixture were sampled from a stainless steel sampling dip tube attached to
a modified Parr reactor. The dip tube was 30 cm in length with an inner diameter of 0.01 inches,
and a swing valve was attached to the end of the sampling tube. Other technical details were
previously reported.12l Two small aliquots of samples were thereby withdrawn quickly at timed
intervals by opening the swing valve, and the first two aliquots were discarded. All samples for
gas chromatography (GC) analyses were diluted to a total volume of approximately 0.75 mL
using oxygenated THF.
A Perkin–Elmer Clarus 400 chromatograph equipped with a chiral column (CP chirasil-Dex
CB 25 m x 2.5 mm) with an auto-sampling capability was used for GC analyses. Hydrogen was
used as the mobile phase at a column pressure of 5 psi with a split flow rate of 50 mL/min. The
injector temperature was 250°C, the FID temperature was 275°C and the oven temperature was
130°C. Retention times (tR/min) for acetophenone: 4.56; (R)-1-phenylethanol: 7.58; (S)-1-
phenylethanol: 8.03; benzophenone: 7.94; diphenylmethanol: 12.57. All of the conversions were
reported as an average of two GC runs. The reported conversions were reproducible.

2016.5.11 Computational Details. All density functional theory (DFT) calculations were performed
using the Gaussian 0372 and 0973 packages with the restricted hybrid mPW1PW91 functional.74
Ruthenium and iridium were treated with the SDD75 basis relativistic effective core potential and
an associated basis set. All other atoms were treated with the double-ζ basis set 6-31++G**
which includes diffuse functionals76 and additional p-orbitals on hydrogen as well as additional
d-orbitals on carbon, nitrogen and oxygen.77 All geometry optimization were conducted in the
gas phase, and the stationary points were characterized by normal mode analysis. Reported free
energies were obtained at 1 atm and 298 K using unscaled vibrational frequencies. All transition
states reported were found to have a single imaginary frequency. The calculated Atomic Polar
Tensor (APT) charges were used to reflect more accurately the charge distribution on each atom
under the derivative of dipole moments with respect to an applied external electric field.78
72. Frisch, M. J., et al., Gaussian 03, Revision C.02 ed; Gaussian Inc.: Wallingford, CT 2004. Full citation is given in the Appendix.73. Frisch, M. J., et al., Gaussian 09, Revision A.1; Gaussian Inc.: Wallingford, CT 2009. Full citation is given in the Appendix.74. (a) Burke, K.; Perdew, J. P.; Wang, Y., Electronic Density Functional Theory: Recent Progress and New Directions; Plenum: New York, 1997; (b) Adamo, C.; Barone, V., J. Chem. Phys. 1998, 108, 664-675.75. (a) Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H., Theor. Chim. Acta 1990, 77, 123-141; (b) Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P., J. Chem. Phys. 1996, 105, 1052-1059.76. (a) Clark, T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. V., J. Comput. Chem. 1983, 4, 294-301; (b) Lynch, B. J.; Zhao, Y.; Truhlar, D. G., J. Phys. Chem. A 2003, 107, 1384-1388.77. Frisch, M. J.; Pople, J. A.; Binkley, J. S., J. Chem. Phys. 1984, 80, 3265-3269.78. Cioslowski, J., J. Am. Chem. Soc. 1989, 111, 8333-8336.

202Chapter 7: Conclusions and Future Work
7.1 Conclusions
A library of unsymmetrical nitrile-functionalized imidazolium salts (1a-1f) were synthesized
starting from 2-cyanophenylimidazole. The reactions of these imidazolium salts with silver(I)
oxide yielded silver(I) carbene complexes containing nitrile-functionalized N-heterocyclic
carbenes (C–CN). The use of these N-heterocyclic carbene (NHC) complexes of silver(I) as a
carbene transfer reagent was demonstrated by the reaction of the fully characterized complex,
[Ag(C–CN)2]BF4 (2a), with [Rh(cod)Cl]2 and [Ru(p-cymene)Cl2]2 leading to the rhodium(I)
complex ([Rh(C–CN)(cod)]2(BF4)2 (3) and the ruthenium(II) complex [Ru(p-cymene)
(C–CN)Cl]2(BF4)2 (4). An X-ray diffraction study and the infrared spectra of these complexes
established that the nitrile nitrogen and the carbene carbon of such a C–CN ligand are bridged to
two different metal centers.
The nitrile functional group on these C–CN ligands is prone to hydrolysis under basic
conditions. For instance, we showed that hydrolysis of the cyanomethyl group of a C–CN ligand
occurred during the preparation of a silver(I) carbene complex starting from the imidazolium salt
1b, which has cyanomethyl and cyanophenyl groups. This leads to the first N-heterocyclic
carbene complex of silver(I) with a primary-amido donor. The selectivity in hydrolysis of
cyanomethyl group over the cyanophenyl group suggests the process must be mediated by silver
centers. In addition, we have observed hindered rotation about the C–N bond of the amide by
means of 1H NMR. This suggested that an interaction between the silver(I) center and the oxygen
of the amido-group was indeed present.
When the silver(I) carbene complex 2a was reacted with trans-PdCl2(CH3CN)2, this led to the
dimeric complex [(C–CN)2Pd(μ-Cl)2Pd(CH3CN)2](BF4)2 (5a), along with the trimetallic complex
5b as a side product when wet diethyl ether and acetonitrile were used as recrystallization
solvents. Interestingly, the solid state structure of 5b ([{Pd(CH3CN)2}3(C–N–N–C)](BF4)4)
reveals the presence of a novel C–N–N–C donor ligand providing two bridging imido nitrogens
to adjacent palladium(II) centers. This C–N–N–C ligand comprised of a central –N=C–O–C=N–
linkage, which was formed from the partial hydrolysis and condensation of the two nitrile groups
of two adjacent C–CN ligands.

203To further explore the coordination chemistry of the C–CN ligand on group 10 metals, the
Platinum(II) complex with 1,5-cyclooctadiene, chloro and the C–CN ligands ([Pt(C–CN)
(cod)Cl]BF4, 7) was prepared in a transmetalation reaction of the silver(I) complex 2a with
Pt(cod)Cl2. The reaction of methoxide with the coordinated diolefin ligand on 7 afforded the
neutral complex 8 (Pt(C–CN)(η1:η2-coe-OMe)Cl) and complex 9 ([Pt(C–CN)(η1:η2-coe-
OMe)CH3CN]BF4) upon removal of the chloro ligand. For comparison, the palladium(II)
analogue, complex 6, was independently prepared by direct transmetalation of 2a and [Pd(η1:η2-
coe-OMe(μ-Cl)]2. These complexes bearing the C–CN and 2-methoxycyclooct-5-enyl ligands
([M(C–CN)(η1:η2-coe-OMe)]+: 6, M = Pd; 9, M = Pt) exists in both monomeric and dimeric
forms depending the choice of recrystallization solvents. Bridging C–CN ligands were observed
in the dimeric form by 1H NMR and infrared spectroscopy. On the other hand, different amounts
of rotamers were observed, owing to the orientation of C–CN ligand relative to the 2-
methoxycyclooct-5-enyl ligand, and restricted rotation about the M–CCarbene bond caused by the
steric crowding of the 2-methoxycyclooct-5-enyl ligand by the C–CN ligand,.
We have developed a facile synthetic route to access a primary amino-functionalized N-
heterocyclic carbene (C–NH2). This was achieved by the reduction of a nitrile-functionalized
imidazolium salt in the presence of nickel(II) chloride under mild conditions. The resulting
nickel(II) complex, [Ni(C–NH2)2](PF6)2 (12), is an axially chiral square-planar complex, and the
use of enantiopure Δ-TRISPHAT anions as an NMR chiral shift reagent is particularly useful to
observe the diastereotopic ion pairs by 1H NMR in acetonitrile-d3. A novel transmetalation
reaction moved the chelating C–NH2 ligand from complex 12 to a variety of platinum group
metal precursor complexes, including those of ruthenium(II), osmium(II), and iridium(III),
yielding important catalysts for the hydrogenation of polar double bonds. The ruthenium(II) and
osmium(II) complexes, [M(p-cymene)(C–NH2)Cl]PF6 (13, M = Ru; 14, M = Os), upon activation
by potassium tert-butoxide (KOtBu) as the base are transfer hydrogenation catalysts for the
hydrogenation of acetophenone in basic 2-propanol at 75°C to give 1-phenylethanol. A maximum
turnover frequency (TOF) of up to 880 h-1 was achieved with the ruthenium(II) complex 13,
giving a conversion of 96% to 1-phenylethanol in 3 h.
In addition, this ruthenium(II) complex (13), upon activation by an excess KOtBu (up to 8
equiv with respect to 13), catalyzes the H2-hydrogenation of ketones in tetrahydrofuran (THF)
under 25 bar of H2 pressure at 50°C with a TOF of up to 461 h-1 and a maximum conversion of
99%. The mechanism of action was thereby investigated by kinetic studies using acetophenone

204as the model substrate, including the studies of kinetic isotope effects using D2 gas and
acetophenone-d3. The rate law is determined to be rate = kH[Ru]tot[H2]/(1 + Keq[ketone]). Our
kinetic studies results are consistent with the heterolytic splitting of dihydrogen at the active
ruthenium species as the rate-determining step. In competition with this reaction is the reversible
addition of acetophenone to the active species to give an enolate complex. The transfer to the
ketone of a hydride and proton equivalent that are produced in the heterolytic splitting reaction
yields the product in a fast, low activation barrier step.
The bifunctional ruthenium hydride-amine complex [Ru(p-cymene)(C–NH2)H]PF6 (17)
containing the Ru–H/N–H couple was prepared. This complex, however, showed no activity
under catalytic conditions unless when activated by a base. This hydride-amine complex is
believed to be the resting state in the transfer hydrogenation mechanism. In fact, this is a rare
example of a catalyst with an M–H and N–H grouping that fails to undergo bifunctional catalysis
using the “NH effect”. The outer-sphere mechanism involving bifunctional catalysis of ketone
reduction is thus disfavored according to experimental studies. This was further confirmed by
showing that an analogous complex with a tethered tertiary amine group (C–CMe2), [Ru(p-
cymene)(C–NMe2)Cl]PF6⋅1.5 DMSO (18, DMSO = dimethyl sulfoxide), has comparable activity
for the H2-hydrogenation of acetophenone. Computational studies also suggest a high energy
barrier for the concerted transfer of a H+/H- pair to the ketone compared to dihydrogen addition
and subsequent heterolytic splitting if an outer-sphere bifunctional mechanism is operative.
An alternative to the outer-sphere bifunctional mechanism is therefore proposed on the basis
of experimental and theoretical findings. First, complex 13, when activated, leads to a
ruthenium-alkoxide complex. The alkoxide ligand labilizes the cis-amine ligand which leads to
the decoordination of the amine group of the C–NH2 ligand. This provides a vacant site for the
coordination of dihydrogen. Catalysis then proceeds with the heterolytic splitting of dihydrogen
by the internal alkoxide base, and then hydride attacks the coordinated ketone in the inner
sphere. The computed energy barriers for the heterolytic splitting of H2 and the attack of a ketone
by a metal hydride in the inner coordination sphere are consistent with our experimental
findings.
The ruthenium(II) ([RuCp*(C–NH2)py]PF6, 15, Cp* = pentamethylcyclopentadienyl ligand, py
= pyridine) and iridium(III) ([IrCp*(C–NH2)Cl]PF6, 19) complexes were prepared from the
transmetalation reaction of the nickel(II) complex 12 with RuCp*(cod)Cl and [IrCp*Cl2]2,

205respectively. A comparison to the activity in H2-hydrogenation of ketones reveals that the
ruthenium system has the higher activity and selectivity than the iridium system. The
ruthenium(II) complex 15 has a rich substrate scope in the H2-hydrogenation of ketones, and this
also catalyzes the hydrogenation of an aromatic ester, a ketimine, and the hydrogenolysis of
styrene oxide. A high substrate to catalyst loadings (catalyst to substrate ratio up to 1/11500) can
be used in ketone hydrogenation, under very mild reaction conditions (8 bar of H2 pressure at
25°C) in basic THF or 2-propanol. A maximum TOF of 17300 h-1 was achieved in the
hydrogenation of acetophenone in 2-propanol. On the other hand, the iridium(III) complex 19
catalyzes the H2-hydrogenation of acetophenone and benzophenone at a higher temperature
(50°C) and H2 pressure (25 bar) in basic THF with a lower substrate to catalyst loading. The use
of an excess alkoxide base (KOtBu, up to 16 equiv with respect to complex 19) is required to
achieve complete reduction of acetophenone to 1-phenylethanol by H2 in 2-propanol at a higher
rate. The phosphine-amine (P–NH2) complexes, [RuCp*(P–NH2)py]PF6 (16a) and [IrCp*(P–
NH2)Cl]PF6 (20), are poorer catalysts in the H2-hydrogenation of ketones compared to their C–
NH2 analogues.
On the basis of experimental findings and theoretical calculations, we propose an alcohol-
assisted outer sphere bifunctional mechanism for both the ruthenium(II) and iridium(III) systems.
First, H/D scrambling in acetophenone and the product alcohol was observed in both systems
when deuterated sources (D2 and 2-propanol-d1) were used. Significantly, we found that an N-H
group is required by complex 19 for catalysis since the structurally similar iridium(III) complex
22 containing a C–NMe2 ligand with no N–H group is much less active. We have computed the
outer-sphere bifunctional mechanism using DFT methods for both ruthenium(II) and iridium(III)
systems. A significant decrease in the free energy barrier to the heterolytic splitting of the η2-H2
ligand on the model complex, Ru(η5-Cp)(C–NH), is calculated when a 2-propanol molecule acts
a proton shuttle by participating in a six-membered ring transition state. On the other hand, a low
free energy barrier is found for the transfer of a proton/hydride couple from the neural hydride-
amine complex, Ru(η5-Cp)(C–NH2)H to the ketone in the outer sphere; however, the cationic
hydride-amine complex [Ir(η5-Cp)(C–NH2)H]+ is calculated to have a high barrier. In fact, we
have prepared the iridium(III) hydride-amine complex, [IrCp*(C–NH2)H]PF6 (21), and this failed
to react with a ketone in the absence of base. The decreased hydricity of such piano-stool
cationic metal hydrides in catalysis appears to be a general phenomenon based on our findings.

206We also showed that the activation of the iridium(III) hydride-amine complex 21 by alkoxide
base is crucial for catalysis. Our computational studies support the proposal that the base
deprotonates the amine group of the cationic hydride-amine complex 21. This triggers the
migration of the hydride to the η5-Cp ring producing a neutral iridium(I) amido complex. This
amido complex with an η4-cyclopentadiene (CpH) group splits η2-H2 with the assistance of
alcohol to produce the model complex, Ir(η4-CpH)(C–NH2)H, which contains a Ir–H/N–H couple
required for the outer sphere hydrogenation of ketones in the bifunctional mechanism. These
steps involve novel iridium(I) intermediates and proceed with accessible free energy barriers.
We also studied the donor ability of the D–NH2 type ligand (D = NHC, phosphine, amine, 2'-
pyridine) by use of the carbonyl stretches of the complexes [RuCp*(D–NH2)CO]X to help
explain the poorer performance in the catalytic H2-hydrogenation of ketones of the phosphine-
amine (P–NH2) complexes of ruthenium(II) (16a) and iridium(III) (20) compared to their C–NH2
analogues. The C–NH2 ligand was found to be more donating than the P–NH2 ligand.
We conclude, based on our present work using a primary-amino functionalized N-
heterocyclic carbene ligand, a right balance of the Brønsted acidities of the N–H and the M–H
groups is crucial to maximize the activity for the hydrogenation of polar bonds. This is achieved
by choosing the right donor ligand, the right metal center, as well as the correct overall charge of
the catalyst system.
7.2 Future Work
The discovery of late transition metal catalyst systems containing primary-amino
functionalized N-heterocyclic carbene ligand (C–NH2) for ketone hydrogenation is proven to be
important in order to understand the “NH effect” in bifunctional catalysis. We showed that the
correct choice of ligand sets and the metal center can provide highly active catalysts. There are
several future research directions that can stem from this work. The effect of the steric properties
of the ligand on catalysis has not been studied in this work. A series of C–NH2 ligands that
contains different substituents on the imidazolylidene ring is worth exploring, and analogous
ruthenium(II) and iridium(III) complexes can be synthesized based on a similar transmetalation
reaction of the corresponding nickel(II) complex with the appropriate precursor complex. This
1. Satoh, T.; Suzuki, S.; Suzuki, Y.; Miyaji, Y.; Imai, Z., Tetrahedron Lett. 1969, 4555-4558.

207will require the expansion of the family of nitrile-functionalized imidazolium salts, including the
search for new synthetic routes leading to these imidazolium salts, and to extend the scope of the
nitrile-reduction reaction based on the use of nickel(II) chloride. In fact, the use of other first row
metals such as iron(II) and cobalt(III) halides should be of target as their utility as reducing
agents have been demonstrated in the reduction of aromatic nitriles to amines.1
The emergence of pincer-type ligands, in particular their use to introduce metal-ligand
cooperativity upon activation, has become a new direction of research. Milstein and co-workers
have showed that ruthenium(II)2 and iron(II)3 based systems containing P–N–P pincer type
ligands are useful in the hydrogenation of esters2 and organic carbonates,4 the synthesis of amides
from esters and amines,5 and the photochemical splitting of water.6 Some of our group members
have began to explore these areas. The reduction of the imidazolium salt 1b, which contains
cyanomethyl and cyanophenyl groups, and 1c, which contains a methylpyridine and cyanophenyl
groups, could lead to interesting pincer type ligands. This will provide a novel NHC based N–C–
N ligand containing primary amine donors when coordinated to late transition metals. The metal
complexes of these could be important for the aforementioned processes by demonstrating metal-
ligand cooperativity.
During the course of our study to the ketone hydrogenation mechanisms, we found that the
decoordination of the amine group of the C–NH2 ligand could be facile under catalytic
conditions. Further research could be directed to study the effect on catalysis of changing the
metal-chelate (-M–C–NH2-) ring size from a seven-membered ring to a six-membered ring.
Results using the ruthenium(II) based complex, [Ru(p-cymene)(C–6–NH2)Cl]PF6, show that this
is a more active transfer hydrogenation catalyst of ketones upon activation by an alkoxide base in
comparison to its seven-membered ring analogue (Figure 7.1, left).7 The ruthenium(II) complex
containing the pentamethylcyclopentadienyl ligand could be important and useful for the
hydrogenation of polar double bonds such as those of esters and amides (Figure 7.1, right). This
system can be used for dehydrogenative coupling reactions8 as well as in the activation of other
2. Milstein, D., Top. Catal. 2010, 53, 915-923.3. Langer, R.; Leitus, G.; Ben-David, Y.; Milstein, D., Angew. Chem. Int. Ed. 2011, 50, 2120-2124.4. Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L. J. W.; Milstein, D., Nat. Chem. 2011, 3, 609-614.5. Gnanaprakasam, B.; Milstein, D., J. Am Chem Soc. 2011, 133, 1682-1685.6. Kohl, S. W.; Weiner, L.; Schwartsburd, L.; Konstantinovski, L.; Shimon, L. J. W.; Ben-David, Y.; Iron, M. A.; Milstein, D., Science 2009, 324, 74-77.7. Ohara, H.; O, W. W. N.; Morris, R. H.; Lough, A. J., Mansucript in preparation.8. Nixon, T. D.; Whittlesey, M. K.; Williams, J. M. J., Dalton Trans. 2009, 753-762.9. Ikariya, T., Bull. Chem. Soc. Jpn. 2011, 84, 1-16.

208small molecules.9 As a well-defined synthetic route that is developed for this new class of amino-
functionalized imidazolium salts based on our study,7 chiral catalyst systems can be designed and
this will be important for the asymmetric hydrogenation of ketones and imines, which can
potentially utilize the bifunctional mechanism. Williams and Roland have reported the synthesis
of some chiral imidazolium salts with a primary10 or a secondary11 amine group, respectively.
The silver(I) complexes of secondary-amine functionalized NHC were isolated.11
Figure 7.1. The structures of ruthenium(II) complexes containing the C–NH2 ligand forming a
six-membered ring metal-chelate (-M–C–NH2-).
Our research results have showed that the ruthenium(II)12 and iron(II)13 based complexes
containing an amine- or imine-based tetradentate P–N–N–P ligands provide highly active
catalysts for the hydrogenation of ketones and nitriles. The N-heterocyclic cyclic carbene based
ligand containing such a C–N–N–C type donor ligand should be of target and this might even
exhibit higher activity in these hydrogenation reactions based on our findings (Figure 7.2). One
approach to access this class of ligand is a reaction of a 1,2-alkyl-dibromide with the nickel(II)
complex, [Ni(C–NH2)2](PF6)2, which could yield the amine-based tetradentate ligand of
nickel(II), [Ni(C–N–N–C)](PF6)2. The transmetalation reaction of this complex with the
appropriate ruthenium(II) and iron(II) sources should yield the desired complex. This class of
tetradentate ligands containing a chiral diamine backbone should be pursued as well as this will
lead to highly active asymmetric hydrogenation catalysts.
10. Moore, T.; Merzouk, M.; Williams, N., Synlett 2008, 21-24.11. (a) Alexandre, F.; Baltaze, J. P.; Roland, S.; Mangeney, P., J. Organomet. Chem. 2006, 691, 3498-3508; (b) Flahaut, A.; Roland, S.; Mangeney, P., Tetrahedron: Asymmetry 2007, 18, 229-236.12. (a) Li, T.; Bergner, I.; Haque, F. N.; Iuliis, M. Z.-D.; Song, D.; Morris, R. H., Organometallics 2007, 26, 5940-5949; (b) Li, T.; Churlaud, R.; Lough, A. J.; Abdur-Rashid, K.; Morris, R. H., Organometallics 2004, 23, 6239-6247.13. Morris, R. H., Chem. Soc. Rev. 2009, 38, 2282-2291.
PF6Ru
NN
N
CH3XHH
+
X = halides
Ru
NN
N
CH3ClHH

209
Figure 7.2. Proposed structure of an iron(II) complex containing a C–N–N–C type donor ligand.
We succeeded to replace a phosphine-amine ligand (P–NH2) with a C–NH2 ligand in second
and third row transition metals-based ketone hydrogenation catalysts to achieve green catalysis.
New research directions for an even greener catalyst system should be sought by using first row
transition metal ions including those of iron(II), cobalt(II) and cobalt(III). These closely
resembles those of their ruthenium(II) and iridium(III) analogues, and could be beneficial for
catalysis. These systems will open new reaction opportunities other than the hydrogenation of
polar double bonds, and studies to the mechanism of actions of these systems could lead to
unexplored metal-ligand cooperativity.
FeNN
N
R
NN
N
R
n n
Ph PhBr
CO
2+
n = 0, 1

210Appendix
Table A.1. The oven temperatures, retention times (tR, tp, /min) for all of the substrates and
alcohol products reported from GC analyses.
Substrates/Alcohol Products Oven Temperature
(°C)
tR(min) tP(min)
130 4.56 7.58, 8.03
145 4.63 10.34, 12.16
140 6.46 14.81, 16.05
145 5.96 11.03, 12.09
155 7.02 12.79, 13.72
140 10.61 13.50, 14.28
140 5.19 10.06 (rac)
140 5.67 13.15, 13.78
170 9.67 14.43, 14.76
180 7.94 12.57
60 5.62 17.64, 18.79
130 3.53 7.31
130 4.81, 4.95 8.15, 8.65; 9.47
140 3.83 5.70, 5.93(tridecane)
O
O
O
O
O
Cl
Cl
Cl
Br
OOCH3
HO
HOCl
HOCl
HOCl
HOBr
HOOCH3
O
O
HO
HO
HO
HO
O
H
O
O
HO
O
O
O HO
O
HOHO
HO
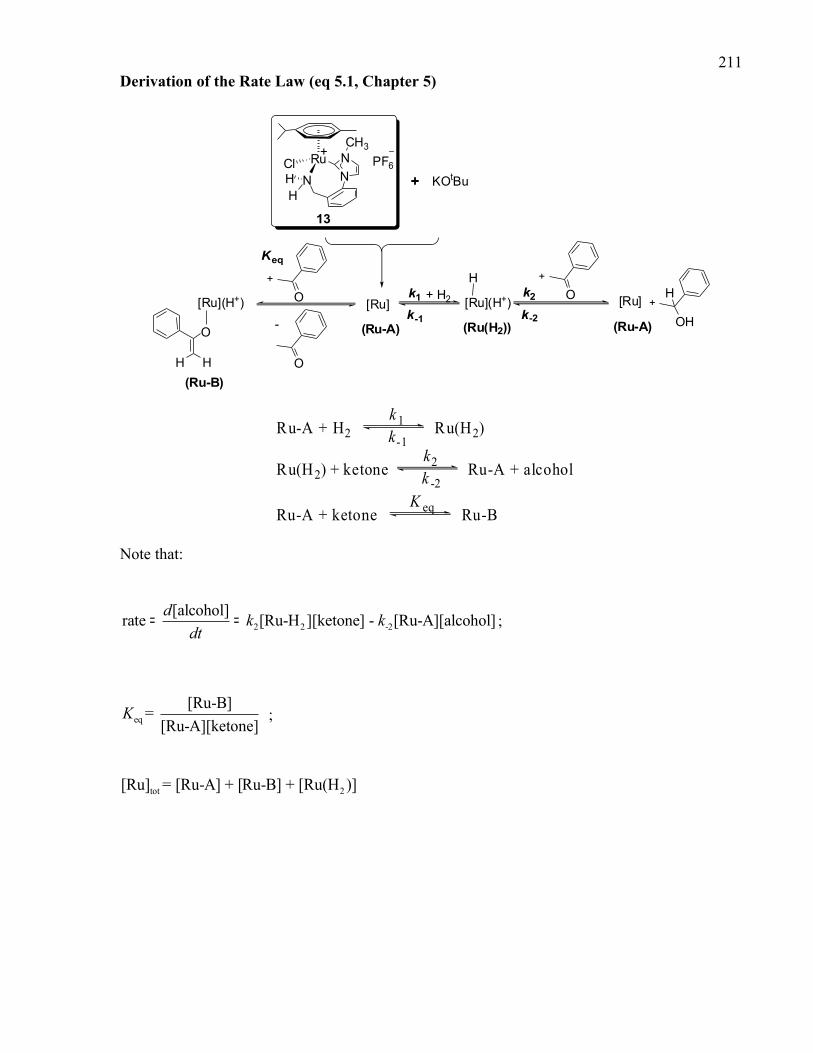
211Derivation of the Rate Law (eq 5.1, Chapter 5)
Note that:
2 2 -2[alcohol]rate [Ru-H ][ketone] - [Ru-A][alcohol]d k k
dt= = ;
eq[Ru-B]=
[Ru-A][ketone]K ;
tot 2[Ru] = [Ru-A] + [Ru-B] + [Ru(H )]
RuCl NNN
CH3
HH
PF6
13
+
[Ru](H+)
O
H H
O
O
-[Ru]
+
Keq
k1 + H2 [Ru](H+)
H
KOtBu+
(Ru-B)
(Ru-A)
O+
k-1
[Ru]
(Ru-A)
k2
k-2 OH+
H
(Ru(H2))
k1k-1
Ru-A + H2 Ru(H2)
k2k -2
Ru(H2) + ketone Ru-A + alcohol
K eqRu-A + ketone Ru-B

212By steady state approximation:
21 2 -1 2 -2 2 2
1 2 -2 2 -1 2
-1 22
1 2 -2
[Ru(H )] [Ru-A][H ] - [Ru(H )] + [Ru-A][alcohol] - [Ru(H )][ketone] 0
[Ru-A]( [H ]+ [alcohol]) - [Ru(H )]( + [ketone]) 0
+ [ketone][Ru-A] = [Ru(H )][H ]+ [alcohol]
d k k k kdt
k k k k
k kk k
= ≈
=
As well,
tot eq 2
eq 2
[Ru] = [Ru-A] + [Ru-A][ketone] + [Ru(H )]
= [Ru-A](1 + [ketone]) + [Ru(H )]
KK
Hence,
tot 1 2 -22
-1 2 eq 1 2 -2
[Ru] ( [H ]+ [alcohol])[Ru(H )] = ( + [ketone])(1 + [ketone]) + [H ]+ [alcohol]
k kk k K k k
tot -1 2
-1 2 eq 1 2 -2
[Ru] ( + [ketone])[Ru-A] = ( + [ketone])(1 + [ketone]) + [H ]+ [alcohol]
k kk k K k k
The rate law, expressed in terms of [Ru]tot, will be:
2 tot 1 2 -2 -2 tot -1 2
-1 2 eq 1 2 -2
tot 1 2 2 -1 -2
-1
[Ru] [ketone]( [H ]+ [alcohol])- [Ru] [alcohol]( + [ketone])[alcohol]rate( + [ketone])(1 + [ketone]) + [H ]+ [alcohol]
[Ru] ( [H ][ketone]- [alcohol])+
k k k k k kddt k k K k k
k k k kk
= =
=1 2 -2 2 -1 eq 2 eq
[H ] + [alcohol] + [ketone]( + [ketone]) k k k k K k K+
For initial rates measurement, [alcohol] ~ 0, then:
1 2 tot 2
-1 1 2 2 -1 eq 2 eq
[Ru] [H ][ketone]rate + [H ] + [ketone]( + [ketone])
k kk k k k K k K
=+

213
If -1 1 2 2 -1 eq 2 eq 2+ [H ] << [ketone]( + [ketone]), as [H ] << [ketone]k k k k K k K+ , then:
1 2 tot 2
2 -1 eq 2 eq
1 tot 2
-1eq
2
[Ru] [H ][ketone] rate [ketone]( + [ketone])
[Ru] [H ]
1+ [ketone] eq
k kk k K k K
kk K
Kk
=+
=+
If -1 eq
2
0k K
k: when -1 eq 2k K k= , in which k2 is the rate constant that represents the fastest step
during catalysis, i.e. not rate determining, this gives:
H tot 2
eq
[Ru] [H ] rate (5.1) 1 [ketone]
kK
=+

214Complete Citation for Gaussian 03 and 09 Packages
Gaussian 03:
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.;
Zakrzewski, V. G.; Montgomery, J. A. J.; E., S. R.; Burant, J.; C.; Dapprich, S.; Millam, J. M.;
Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.;
Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.;
Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.;
Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.;
Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.;
Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.;
Wong, M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 03,
Revision C.02 ed; Gaussian Inc.: Wallingford, CT, 2004.
Gaussian 09:
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.;
Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.;
Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.;
Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.;
Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.;
Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.;
Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene,
M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.
E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.;
Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.;
Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J., Gaussian 09,
Revision A.1; Gaussian Inc.: Wallingford, CT, 2009.