krüppel-type zinc finger proteins ifn-regulatory factor 4 function by stage-specific modulation of
TRANSCRIPT

of June 23, 2015.This information is current as
Finger ProteinsFactor 4 Function by Krüppel-Type Zinc Stage-Specific Modulation of IFN-Regulatory
Sanjay Gupta, Alissa Anthony and Alessandra B. Pernis
http://www.jimmunol.org/content/166/10/6104doi: 10.4049/jimmunol.166.10.6104
2001; 166:6104-6111; ;J Immunol
Referenceshttp://www.jimmunol.org/content/166/10/6104.full#ref-list-1
, 23 of which you can access for free at: cites 43 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2001 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 23, 2015
http://ww
w.jim
munol.org/
Dow
nloaded from

Stage-Specific Modulation of IFN-Regulatory Factor 4Function by Kruppel-Type Zinc Finger Proteins1
Sanjay Gupta, Alissa Anthony, and Alessandra B. Pernis2
Optimal humoral responses depend on the activation of Ag-specific B cells, followed by their progression toward a fully differ-entiated phenotype. Acquisition of stage-appropriate patterns of gene expression is crucial to this differentiation program. How-ever, the molecular mechanisms used by B cells to modulate gene expression as they complete their maturation program are poorlyunderstood. IFN-regulatory factor 4 (IRF-4) plays a critical role in mature B cell function. Using the transcriptional regulationof the human B cell activation marker CD23 as a model system, we have previously demonstrated that IRF-4 is induced in responseto B cell-activating stimuli and that it acts as a transactivator of CD23 gene expression. We have furthermore found that IRF-4function can be blocked by B cell lymphomas 6 (BCL-6) protein, a Kruppel-type zinc finger repressor normally expressed ingerminal center B cells. However, CD23 expression is known to be down-regulated in plasma cells despite high level expressionof IRF-4 and the lack of BCL-6, suggesting that in plasma cells the IRF-4-mediated induction of CD23 is prevented by itsinteraction with a distinct repressor. In this set of studies, we demonstrate that IRF-4 interacts with B lymphocyte-inducedmaturation protein/positive regulatory domain I-binding factor 1 (Blimp1/PRD1-BF1), a Kruppel-type zinc finger protein whoseexpression correlates with terminal B cell differentiation. Functional studies indicate that Blimp1, like BCL-6, can block IRF-4-transactivating ability. These findings thus support a model whereby IRF-4 function is modulated in a stage-specific manner byits interaction with developmentally restricted sets of Kruppel-type zinc finger proteins. The Journal of Immunology,2001, 166:6104–6111.
Effective humoral responses involve the activation of Ag-specific B cells, followed by their differentiation towardmemory or plasma cells. Progression of an activated B
cell along its terminal differentiation pathway requires the acqui-sition of stage-appropriate patterns of gene expression. Therefore,once the molecular machinery responsible for the activation pro-gram of a B cell has been implemented, it must be selectivelymodulated to ensure successful completion of terminal differenti-ation. The molecular mechanisms used by B cells to selectivelyregulate gene expression as they proceed along their maturationprogram are largely unknown.
IRF-4 is a novel member of the IFN-regulatory factor (IRF)3
family of transcriptional regulators, whose expression is primarilyrestricted to the lymphoid compartment (1–3). IRF-4 expression inB cells is induced in response to stimuli known to drive B cellactivation (2, 4–6). Genetic evidence indicates that IRF-4 plays acrucial role in controlling the activation and homeostasis of im-mune responses (7). Despite exhibiting a normal B cell develop-ment, IRF-4-deficient mice display profound defects in the func-
tion of mature B cells and are unable to mount Ab responses toeither T-dependent or T-independent Ags. Functional studies haveshown that IRF-4 can subserve a dual role in lymphoid cells. Byitself, IRF-4 can bind to IFN-stimulated response elements andrepress the expression of IFN-inducible genes (3). In the presenceof a cofactor, the protooncogene PU.1, IRF-4 can act as a trans-activator of the Igk- and l-light chain enhancers and the CD20promoter (1, 8, 9).
Our studies have used the regulation of the B cell activationmarker CD23 as a model system. CD23 is a type II integral mem-brane protein that belongs to the C-type lectin superfamily (10–12). This family includes a subset of killer-inhibitory receptors thatexert inhibitory functions on NK cells (10–14). Two differentiallyspliced forms of CD23 (termed CD23a and CD23b) exist in hu-mans, whereas only one form, which more closely resemblesCD23a, has been consistently found in mice. The two humanCD23 isoforms are the result of use of alternative transcriptionalstart sites and are regulated by two distinct promoters (15). CD23aand CD23b only differ by a 6/7-aa substitution, leading to theremoval of an immunoreceptor tyrosine-based inhibition motif(ITIM) present in CD23a, but not in CD23b. ITIMs have beenshown to play a critical role in the ability of the killer-inhibitoryreceptors to inhibit NK cell activation (16, 17). Although the exactfunctional role of the CD23a ITIM has not been elucidated, thedifferential expression of an ITIM motif in the two CD23 isoformssuggests that CD23a and CD23b may mediate distinct signalingpathways.
We have focused our attention on the regulation of CD23b, theITIM-less isoform of CD23. We have previously found that IRF-4participates in a multiprotein or “enhanceosome-like” complexthat targets the CD23b IFN-g activation site (GAS), a critical reg-ulatory element in the promoter of CD23b (4). The IRF-4-medi-ated transactivation of CD23b is blocked by B cell, lymphomas 6protein (BCL-6), a Kruppel-type zinc finger transcriptional repres-sor present in germinal center B cells (18, 19). Because BCL-6
Department of Medicine, Columbia University, New York, NY 10032
Received for publication September 20, 2000. Accepted for publication February16, 2001.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the New York City Council Speaker’s Fund for Bio-medical Research: Toward the Science of Patient Care, and by an Arthritis FoundationScience Grant.2 Address correspondence and reprint requests to Dr. Alessandra Pernis, Departmentof Medicine, Columbia University, 630 West 168th Street, New York, NY 10032.E-mail address: [email protected] Abbreviations used in this paper: IRF, IFN-regulatory factor; Blimp1, B lympho-cyte-induced maturation protein; CIITA, MHC class II transactivator; GAS, IFN-gactivation site; HA, hemagglutinin; ITIM, immunoreceptor tyrosine-based inhibitionmotif; PRDI-BF1, positive regulatory domain I-binding factor 1; wt, wild type; MYC-PRF site, myc-plasmacytoma repressor factor site.
Copyright © 2001 by The American Association of Immunologists 0022-1767/01/$02.00
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

expression is down-regulated upon B cell activation (4, 18, 20), wehave proposed that induction of high levels of IRF-4 coupled withthe loss of BCL-6 plays a key role in the ability of activated B cellsto up-regulate CD23b expression. Interestingly, upon terminal dif-ferentiation of a B cell into a plasma cell, CD23 expression isnormally down-regulated (21), although plasma cells continue toexpress high levels of IRF-4 (1) and lack BCL-6 (19). This obser-vation led us to hypothesize that, in plasma cells, the IRF-4-me-diated induction of CD23b might be repressed by a differentmechanism.
Previous work has demonstrated that another Kruppel-type zincfinger protein, Blimp-1 (B lymphocyte-induced maturation pro-tein), is mainly detected in late B and plasma cells and is a criticalregulator of terminal B cell differentiation (22). Although the fullextent of B lymphocyte-induced maturation protein (Blimp1) func-tions is not known, Blimp1 is a critical transcriptional repressor ofthe c-myconcogene (23) and the MHC class II transactivator (CI-ITA) (24), both of which are down-regulated upon terminal B celldifferentiation. Although Blimp1 is believed to play a key role interminal B cell differentiation, its expression is not confined tolymphoid cells. Indeed, the human homologue of Blimp1, PRDI-BF1 (positive regulatory domain I-binding factor 1), is inducedupon viral infection of fibroblasts and was cloned because of itsability to target the IRF binding site within theb-IFN enhanceo-some, and to repressb-IFN gene expression (25). Interestingly,recent immunohistochemical analysis has revealed that plasmacells as well as a subset of germinal center B cells with a partialplasma cell phenotype strongly express both Blimp1 and IRF-4 (26).
In this study, we report that PRDI-BF1/Blimp1 can bind to thesame functional element in the human CD23b promoter to whichBCL-6 and IRF-4 had previously been shown to bind, and that,like BCL-6, Blimp1 can repress IRF-4-transactivating ability.Thus, IRF-4 function can be modulated in a stage-specific mannerby its interaction with developmentally restricted sets of Kruppel-type zinc finger proteins.
Materials and MethodsCell lines and cultures
The human B cell line Ramos (obtained from Dr. S. Lederman, ColumbiaUniversity, New York, NY) is an EBV-negative Burkitt’s lymphoma.U266 (American Type Culture Collection (ATCC), Manassas, VA) is de-rived from a multiple myeloma cell line. U937 (obtained from Dr. K.Calame, Columbia University) is a monocytic cell line. Human embryokidney 293T cells (obtained from Dr. S. Lederman) were cultured inDMEM supplemented with 10% FCS (Atlanta Biologicals, Norcross, GA).All other cells were grown in IMDM supplemented with 10% FCS (AtlantaBiologicals), as previously described (4). U266 cells (10–203 106) werestimulated with 1mg/ml of either anti-CD40 or an isotype-matched controlAb in a final volume of 10 ml at 37°C for 24 h.
Antibodies
The rabbit polyclonal antiserum against IRF-4 used in EMSA experimentswas a generous gift of Hisamaru Hirai (University of Tokyo, Tokyo, Japan)(3). We subsequently generated our own rabbit polyclonal anti-IRF-4 an-tiserum using a similar GST-IRF-4 (nucleotides 441–924) fusion protein asthe immunogen (Berkeley Antibody, Richmond, CA). This antiserum wasused for immunoprecipitations and immunoblot analyses (4). Blots werealso reprobed with a commercially available anti-IRF-4 antiserum (SantaCruz Biotechnology, Santa Cruz, CA), which gave identical results. Rabbitpolyclonal antisera against human Stat6, or BCL-6, were purchased fromSanta Cruz Biotechnology. The mAb against hemagglutinin (HA) epitope(clone 12CA5) was from Boehringer Mannheim (Indianapolis, IN). Thehybridomas secreting the anti-CD40 mAb G28-5 (IgG1) or an isotype-matched control mAb were obtained from ATCC.
DNA constructs
The human IRF-4 expression plasmid (pCEP4-IRF-4) and the GST-IRF-4expression plasmid were previously described (4). Various deletion mu-
tants of IRF-4 were prepared by Pfu PCR from pBSKS-IRF-4 plasmidtemplate using appropriate primers. The cDNA segments encoding thesedeletion mutants were then subcloned, in frame, into the filledEcoRI siteof pGEX-3X vector to generate GST-IRF-4 deletion mutants. The in framejunction in the GST-IRF-4 fusion constructs was confirmed by DNA se-quencing in an automated cycle sequencer (Perkin-Elmer, Norwalk, CT).The full-length human BCL-6 expression vector was a kind gift of R.Dalla-Favera (Columbia University, New York, NY) (27). HA epitope-tagged murine Blimp1 cDNA cloned into pBluescript vector (pBSKS-HA-Blimp1) was a kind gift of K. Calame (23). The Blimp1 expression con-struct (pCEP4-HA-Blimp1) was generated by cloning the coding region ofthe HA-Blimp1 cDNA into pCEP4 expression vector. The full-length hu-man Blimp1/PRDI-BF1 cDNA cloned into pXM mammalian expressionvector was a kind gift of T. Maniatis (Harvard University, Cambridge, MA)(25). The CD23b promoter firefly luciferase reporter construct in the pGL3-enhancer vector was previously described (4).
DNA-binding assays and cell extracts
The preparation and employment of DNA oligonucleotide probes for EM-SAs have been described previously (4). Thedouble-stranded oligonucleo-tides used as probes or cold competitors in these studies were as follows:CD23b GAS (wild-type, wt), 59-gatcGGGTGAATTTCTAAGAAAGGGAC-39; CD23b GAS M1, 59-gatcGGGTGAATTTCTAAGGTCGGGAC-39;CD23b GAS M2, 59-gatcGGGTGGTCTTCTAAGAAAGGGAC-39; CD23bGAS M3, 59-gatcGGGTGAATGCTGAAGAAAGGGAC-39; CD20, 59-gatcGGGGTCTTTTTCAAGAAGTGAAACCT-39 (9); k39 enhancer, 59-gatcCCTTTGAGGAACTGAAAACAGAACCT-39 (28); CIITA, 59-gatcACAGTAAGGAAGTGAAATTAATTTCAG-39; MYC-PRF site, 59-gatcCGCGTACAGAAAGGGAAAGGACTAG-39 (23); Ie GAS, 59-gatcAACTTCCCAAGAACA-39 (29). Oligonucleotide competition and Ab interference assayswere performed as previously described (4). Nuclear extracts were prepared aspreviously described (4).
Immunoprecipitations and Western blot analysis
Cell extracts were immunoprecipitated with an anti-IRF-4, or anti-HA Ab,as previously described (4). The immunoprecipitates were resolved by 7%SDS-PAGE. The gel was transferred to a nitrocellulose membrane, andthen immunoblotted with an IRF-4 or HA epitope Ab. The bands werevisualized by ECL (Amersham Pharmacia Biotech, Piscataway, NJ).
GST pull-down assays and transient transfections
GST pull-down assays were conducted as previously described (4). Thebound proteins were eluted from the beads by boiling them in SDS-PAGEsample buffer, fractionated on a 7% SDS-polyacrylamide gel, and thenblotted onto a nitrocellulose membrane. The blot was probed with either aBCL-6, HA epitope, or Stat6 Ab.
For expression of recombinant proteins, 293T cells were transfectedwith expression plasmids by calcium phosphate precipitation method. After24 h of incubation, the transfected cells were harvested for nuclear extractpreparation.
Transient transfection assays for reporter experiments were performedas previously described (4).
ResultsBlimp1 binds to the CD23b GAS
The human homologue of Blimp1, PRDI-BF1, has previously beenshown to target the IRF binding site in the IFN-b enhancer (25).Furthermore, the known DNA-binding elements for murine IRF-4(1, 8) and Blimp1 (23, 24) are strikingly similar (Table I). We thusdecided to investigate whether Blimp1/PRDI-BF1 can target theIRF-4 binding site in the human CD23b promoter (4). Therefore,we performed EMSAs with a CD23b GAS probe on extracts fromU266. This is a myeloma cell line, representative of a terminallydifferentiated plasma cell (30), which lacks BCL-6 and expresseshigh levels of Blimp1/PRDI-BF1 (data not shown). As shown inFig. 1A, the pattern of CD23b GAS-binding complexes in U266extracts was markedly different from that detected in extracts fromRamos cells, which phenotypically resemble germinal center Bcells and contain BCL-6, but not Blimp1/PRDI-BF1 (data notshown) (31). As we previously demonstrated (4), in Ramos cells,IRF-4 participates in the formation of a slow mobility complex,whose visualization is normally obscured by the presence of a
6105The Journal of Immunology
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

strong BCL-6-containing complex. In contrast, extracts from U266cells contained a clearly visible IRF-4 complex and lacked theBCL-6 complex. Interestingly, U266 contained a distinct CD23bGAS-binding complex (complex X), which was absent in Ramoscells. The mobility of this complex was faster than that of theBCL-6-containing complex. Furthermore, BCL-6 expression isnormally down-regulated upon CD40 stimulation (4, 18, 20). Incontrast, stimulation of U266 cells with an anti-CD40 Ab did notaffect the appearance of complex X, despite appropriately decreas-ing the intensity of the BCL-6 complex in Ramos cells (Fig. 1A).Supershifting experiments confirmed the presence of IRF-4 inU266 cells and demonstrated that complex X did not contain eitherIRF-4 or BCL-6 (Fig. 1Band data not shown). EMSA analysis ofthree additional myeloma cell lines revealed a similar pattern ofCD23b GAS-binding complexes (data not shown).
To determine whether the faster CD23b GAS-binding complexpresent in U266, but not in Ramos cells contained Blimp1/PRDI-BF1, we subjected U266 extracts to oligonucleotide competitionexperiments using the known Blimp1 binding site from the mycpromoter (MYC-PRF site) (23) as a cold competitor of the radio-labeled CD23b GAS wt probe. These oligonucleotide competitionexperiments revealed that the MYC-PRF element could efficientlycompete both the IRF-4-containing complex as well as the fastermobility complex, suggesting that the latter complex might containBlimp1/PRDI-BF1 (Fig. 2A,left panel). Competition of IRF-4 wasnot surprising because MYC-PRF has been described as an IFN-stimulated response-like element (23). To confirm this observa-tion, we then proceeded to directly test whether rBlimp1 couldtarget the CD23b GAS. 293T cells (which lack BCL-6, Blimp1,and IRF-4) were transiently transfected with an expression plasmidencoding either Blimp1 or BCL-6. Cells transfected with an emptyvector were included as control. Extracts from the various 293Ttransfectants were then subjected to EMSA experiments using theCD23b GAS as a probe (Fig. 2A, right panel). These experimentsclearly revealed that 293T transfectants expressing Blimp1 orBCL-6 contained CD23b GAS-binding complexes, which were ab-sent in 293T cells tranfected with an empty vector. The mobilityexhibited by rBlimp1 was similar to that of the CD23b GAS-bind-ing complex, which we had detected in U266 cells, but not inRamos, and was clearly distinct from that displayed by rBCL-6.
Oligonucleotide competition experiments with a panel of CD23bGAS mutant elements (Fig. 2B) revealed that the CD23b GAS M2mutant, which contains mutations in the IRF-4 binding site (4), isunable to efficiently compete the Blimp1 complex. The competi-tion pattern for the Blimp1 complex is furthermore distinct fromthat we have previously shown for BCL-6, which is competed bythe M2, but not by the M1 or M3 mutants (4). These results thussuggest that at different stages of B cell differentiation, distinctKruppel-type zinc finger proteins target the CD23b GAS.
To assess whether the ability of BCL-6, Blimp1, and IRF-4 totarget a common DNA element was confined to the CD23b GAS,we then subjected extracts from 293T transfectants expressingeither BCL-6 or Blimp1 to an oligonucleotide competition exper-iment with a panel of DNA elements from different promoters/enhancers. The elements used in this experiment included: 1) sitesknown to bind Blimp1 (MYC-PRF, CIITA) (23, 24), 2) sites knownto bind IRF-4 (k39 enhancer, CD20) (8, 9), and 3) a site known tobind BCL-6 (Ie GAS) (32). Self-competition with the CD23b GASelement was included as control. As shown inFig. 2C, this experi-ment indicated that all three known IRF-4 binding sites (k39 en-hancer, CD20, and CD23b GAS) could efficiently block the bind-ing of both the BCL-6 as well as the Blimp1 complexes to theCD23b GAS. In contrast, the known Blimp1 binding sites (MYC-PRF, CIITA) only competed the Blimp1, but not the BCL-6 com-plex, whereas the addition of an oligonucleotide containing theBCL-6 binding site (Ie GAS) only blocked binding of the BCL-6complex, but not that of the Blimp1 complex. These data thussuggest that both BCL-6 and Blimp1 can also target other IRF-4binding sites in addition to the CD23b GAS. However, the targetsof the two Kruppel-type zinc finger proteins do not completelyoverlap. Of particular interest is the finding that the Blimp1 com-plex was not competed by the Ie GAS, which is known to bindStat6 in addition to BCL-6 (32). In contrast to the CD23b GAS, theIe GAS does not appear to be targeted by IRF-4, because it fails tocompete IRF-4 complexes in oligonucleotide competition assays(data not shown). This result thus suggests that the known abilityof BCL-6 to modulate Stat6 activity may not be shared by Blimp1.
IRF-4 physically interacts with Blimp1
In our previous studies, we had detected a very strong interactionbetween BCL-6 and IRF-4 (4). To determine whether Blimp1could also physically associate with IRF-4, we first performedpull-down assays with a GST-IRF-4 fusion protein. As shown inFig. 3A, incubation of a GST-IRF-4 fusion protein with extractsfrom 293T cells transfected with an HA epitope-tagged Blimp1expression vector revealed that IRF-4 can indeed associate withBlimp1. No interaction was observed with the GST moiety aloneor upon incubation of the GST-IRF-4 with a control extract(Ramos) or with 293T cells transfected with an empty vector. We
FIGURE 1. Different patterns of CD23b GAS-bind-ing complexes are displayed by human cell lines cor-responding to distinct stages of B cell differentiation.A,Nuclear extracts from U266 (a myeloma cell line) werecompared with those from Ramos (a Burkitt’s lym-phoma) by EMSA using a32P-labeled CD23b GAS wtprobe. Ramos and U266 cells were either unstimulatedor stimulated with an anti-CD40 Ab (1mg/ml) or acontrol Ab (1mg/ml) for 24 h at 37°C.B, Ab interfer-ence mobility shift assays were conducted by additionof antisera against IRF-4 or control. All antisera wereadded at a final dilution of 1/20 for 30 min at 4°C be-fore incubation with the probe for 20 min at 25°C.U266 cells were unstimulated.
Table I. Sequence comparison of IRF-4 and Blimp1 binding sites
Binding site Sequence (59to 39)
IRF-4 k39 Enhancer CTTTGA GGAACTGAAA ACAGAlB Enhancer ATAAAA GGAAGTGAAA CCAAG
Blimp 1 MYC-PRF GGTACA GAAAGGGAAA GGACTCIITA CAGTAA GGAAGTGAAA TTAAT
6106 MODULATION OF IRF-4 FUNCTION BY BLIMP1
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

also cotransfected 293T cells with both IRF-4 as well as HAepitope-tagged Blimp1 expression vectors and then subjected theextracts to immunoprecipitation assays with either an anti-IRF-4 oranti-HA Ab (Fig. 3B). Consistent with our GST pull-down assays,presence of the IRF-4 protein could be detected in anti-HA im-munoprecipitates of 293T cells cotransfected with both IRF-4 andHA epitope-tagged Blimp1 (Fig. 3B, upper panel), but not of con-trol transfectants. Stripping and reprobing of the filter with an anti-HA Ab demonstrated the presence of HA-Blimp1 in the IRF-4immunoprecipitates of 293T transfectants expressing both IRF-4and HA epitope-tagged Blimp1, but not of 293T control transfec-tants (Fig. 3B,lower panel). Taken together, these data indicatethat IRF-4 can physically interact not only with BCL-6, but alsowith Blimp1.
To further dissect the interaction of IRF-4 with the two Kru¨p-pel-type zinc finger proteins, we proceeded to map the IRF-4domains involved in the association of IRF-4 with BCL-6 andwith Blimp1. We thus generated a panel of different GST-IRF-4mutant proteins, and compared them with wt IRF-4 in GSTpull-down assays (Fig. 4A). Incubation of the different GST-IRF-4 deletion mutants with extracts from 293T transfectantsexpressing HA epitope-tagged Blimp1 revealed that interactionof IRF-4 with Blimp1 requires the aa 1–150 region of IRF-4,which is known to contain its DNA binding domain (Fig. 4A,upper panel) (1). Contribution of the proline-rich region ofIRF-4 to its interaction with Blimp1 is also likely because de-
letion of this region (D151–199) resulted in a diminished inter-action of the mutant protein with Blimp1. When the GST-IRF-4deletion mutants were incubated with extracts from Ramos cellsand the IRF-4/BCL-6 interaction visualized by Western blottingwith an anti-BCL-6 antiserum, the aa 1–150 region of IRF-4was again found to be essential for its interaction with BCL-6(Fig. 4A, middle panel). Similarly to the Blimp1/IRF-4 inter-action, the IRF-4 mutant lacking the proline-rich region (D151–199) also displayed a decreased ability to associate with BCL-6.However, in striking contrast to the results obtained withBlimp1, association of IRF-4 with BCL-6 also required the aa200 –360 region of IRF-4. We have previously shown that, inRamos cells, IRF-4 can interact not only with BCL-6, but alsowith Stat6 (4). Interestingly, stripping and reprobing of thisWestern blot with a Stat6 Ab (Fig. 4A, lower panel) demon-strated that the IRF-4D1–150 interacted normally with Stat6.Association of Stat6 with IRF-4 was instead found to requirethe aa 200 –360 region. As summarized in Fig. 4B, this exper-iment thus revealed that the aa 1–150 region of IRF-4, whichcontains the DNA binding domain of IRF-4, is required for itsinteraction with both BCL-6 and Blimp1, but not with Stat6. Incontrast, the aa 200 –360 region of IRF-4, which contains res-idues previously shown to be critical for its interaction withPU.1 (33), is necessary for the association of IRF-4 with BCL-6and Stat6, but not with Blimp1.
FIGURE 2. Blimp1 can target the CD23bGAS. A, Nuclear extracts from unstimulatedU266 cells were obtained and assayed as de-scribed in Fig. 1. Oligonucleotide competi-tion assays were performed either in the ab-sence or in the presence of a 100-fold molarexcess of a cold MYC-PRF or CD23b GASoligonucleotide added to the shift reaction asindicated (left panel). 293T cells were tran-siently transfected with an empty vector orwith an expression vector encoding eitherBCL-6 or Blimp1. Nuclear extracts werethen prepared and analyzed by EMSA usinga 32P-labeled CD23b GAS wt probe (rightpanel).B, 293T cells were transiently trans-fected with an empty vector or with an ex-pression vector encoding Blimp1 and thenassayed by EMSA, as described above. Oli-gonucleotide competition assays were per-formed either in the absence or in the pres-ence of a 100-fold molar excess of cold wt ormutant CD23b GAS oligonucleotides addedto the shift reaction as indicated.C, 293Tcells were transiently transfected with anempty vector or with an expression vectorencoding either BCL-6 or Blimp1 and thenassayed as described above. Oligonucleotidecompetition assays were performed either inthe absence or in the presence of a 100-foldmolar excess of cold oligonucleotides corre-sponding to previously described Blimp1,IRF-4, or BCL-6 binding sites. Cold compet-itors were added to the shift reaction asindicated.
6107The Journal of Immunology
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

Blimp1 selectively blocks the ability of IRF-4 to transactivateCD23b
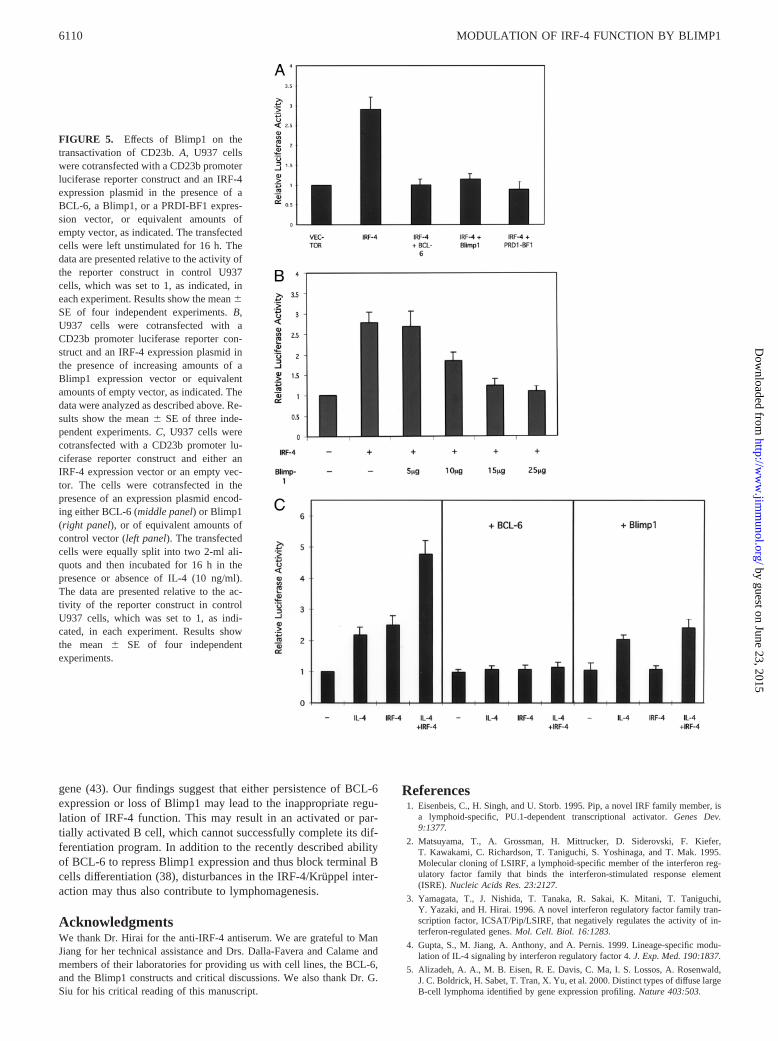
To determine whether Blimp1/PRDI-BF1 could repress the abilityof IRF-4 to transactivate the CD23b promoter, we performed tran-sient transfection assays in U937 cells, a monocytic cell line thatis capable of activating Stat6 in response to IL-4, but lacks IRF-4,BCL-6, and Blimp1 (Fig. 5A). Consistent with our previous results(4), cotransfection of an IRF-4 expression vector with a luciferasereporter construct driven by the CD23b promoter resulted in a3-fold induction in luciferase activity. As in the case of BCL-6,coexpression of Blimp1 with IRF-4 repressed the ability of IRF-4to transactivate the CD23b promoter reporter construct. A similarinhibitory effect was also detected when the human homologue ofBlimp1, PRDI-BF1, was cotransfected with IRF-4. The effect ofBlimp1 on IRF-4-mediated transactivation was dose dependent be-cause addition of increasing amounts of the Blimp1 expressionvector led to a progressive decrease in reporter activity (Fig. 5B).
These studies thus indicate that Blimp1/PRDI-BF1 can blockIRF-4 function.
To further examine the functional role of Blimp1 on the regu-lation of CD23b, we then proceeded to determine the effects ofBlimp1 cotransfection on the IL-4 inducibility of CD23b (Fig. 5C).As we previously demonstrated, similar inducibility of the CD23bpromoter construct can be achieved upon IL-4 stimulation of U937or upon cotransfection of IRF-4. However, when cotransfection ofIRF-4 is conducted in the presence of IL-4 stimulation, higherlevels of luciferase activity are detected, suggesting that optimalCD23b inducibility requires cooperation between Stat6 and IRF-4(4). Consistent with previous results, addition of BCL-6 blockedthe IRF-4-mediated transactivation of this reporter construct aswell as the IL-4 inducibility of CD23b (4, 34). Cotransfection ofBlimp1 blocked the ability of IRF-4 to drive CD23b promoteractivity and to augment the IL-4 inducibility of this reporter con-struct. However, in contrast to BCL-6, presence of Blimp1 did notaffect the IL-4 inducibility of CD23b, regardless of the presence ofIRF-4. These findings thus suggest that, in contrast to BCL-6,Blimp1 selectively targets IRF-4 function and does not interferewith Stat6 activity.
DiscussionIRF-4, an IRF family member whose expression is mostly re-stricted to lymphocytes (1–3), has been shown by genetic studiesto play a crucial role in mature B cell function (7). Consistent withthe phenotype displayed by the IRF-4-deficient mice, IRF-4 ex-pression is up-regulated in response to B cell activation stimuli (2,4–6). Our previous studies have indicated that IRF-4 function canbe regulated by BCL-6, a Kruppel-type zinc finger transcriptionalrepressor (27, 35, 36). In this study, we report that IRF-4 functioncan also be modulated by a distinct Kruppel-type zinc finger pro-tein, Blimp1/PRDI-BF1, previously shown to be a critical regula-tor of terminal B cell differentiation (22). Whereas expression ofBCL-6 is mostly restricted to germinal center B cells (19), highlevels of Blimp-1 are primarily found in terminally differentiatedcells (22). Thus, the ability of IRF-4 to pair with developmentallyrestricted sets of Kruppel-type zinc finger proteins may regulateIRF-4 activity in a stage-appropriate manner. Furthermore, be-cause Blimp1 expression can be detected in nonhemopoietic cells(25), interaction of Blimp1 with IRF-4 may confer lineage speci-ficity to the actions of Blimp1.
The interaction of IRF-4 with Blimp1 may have broad biolog-ical implications. Indeed, our competition experiments suggest thatthis interaction is not restricted solely to the regulation of CD23gene expression, but may extend to other IRF-4 target genes. In-terestingly, one of these genes is CD20, a B cell surface marker,which, in humans, is differentially expressed in memory cells(CD201) vs plasma cells (CD202) (37). Because IRF-4 has beenshown to participate in the control of CD20 (9), this finding sug-gests that modulation of IRF-4 function by Blimp1 may constituteone of the molecular events driving activated B cells toward aspecific cell fate.
Our studies reveal not only similarities between the two Kru¨p-pel-type Zinc finger proteins, but also profound differences be-tween them. In both cases, association with IRF-4 maps to a regionof IRF-4 that contains its DNA binding domain (1). However,physical interaction of IRF-4 with BCL-6, but not with Blimp1,also requires a region of IRF-4 that mediates its association withcofactors such as PU.1 (33) or Stat6 (Fig. 4A). This finding sug-gests that BCL-6 may modulate not only the DNA-binding abilityof IRF-4, but also its ability to interact with cofactors and possiblyto assemble into multiprotein complexes. In contrast, Blimp1 mayprimarily target the IRF-4 DNA binding domain. Because plasma
FIGURE 3. Blimp1 interacts with IRF-4.A, GST-IRF-4 fusion proteinbinds Blimp1. Nuclear extracts from 293T cells transiently transfected ei-ther with an empty vector or with an HA epitope-tagged Blimp1 expressionvector were prepared and incubated with immobilized GST-IRF-4 fusionprotein. Bound proteins were eluted, resolved by 7% SDS-PAGE, blottedonto a nitrocellulose membrane, and then probed with an anti-HA Ab.Pull-downs with GST-alone or from Ramos extracts are shown as controls.B, Coimmunoprecipitation of Blimp1 with IRF-4. The 293T cells werecotransfected with an HA epitope-tagged Blimp1 expression vector as wellas an IRF-4 expression vector. 293T cells cotransfected with empty vectorswere simultaneously set up as control. Nuclear extracts were then preparedand immunoprecipitated with either an IRF-4 antiserum or an anti-HA Ab.The immunoprecipitated proteins were resolved by 7% SDS-PAGE andthen analyzed by Western blotting using an anti-IRF-4 Ab (upper panel).The blot was later stripped and reprobed with an anti-HA Ab (lower panel).
6108 MODULATION OF IRF-4 FUNCTION BY BLIMP1
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

cells express high levels of both IRF-4 and Blimp1, we suspect thatthe interaction of IRF-4 with Blimp1 does not necessarily lead torepression of IRF-4 function, but it may rather direct IRF-4 towardstage-appropriate targets by modulating its DNA-binding proper-ties. Thus, one may predict that, in a different promoter context,cooperative interactions between IRF-4 and Blimp1 may also beobserved. Another important distinction uncovered by our studiesis that, in contrast to BCL-6 (34), Blimp1 selectively repressesIRF-4 function and neither targets Stat6 binding sites nor blocksStat6-mediated transactivation. Thus, it will be interesting to de-termine whether terminally differentiated B cells possess distinctmechanisms to modulate Stat6 activity.
Although our studies suggest that all of the IRF-4 binding sitestested could serve as targets for both BCL-6 and Blimp1 (Fig. 2C),the regulation of IRF-4-mediated gene expression is likely to dis-play additional complexities. In particular, different subsets ofIRF-4 target genes may exist, which can be selectively targeted bydifferent combinations of Kruppel-type zinc finger proteins. Con-sistent with this notion, although the IRF-4 binding sites in both
CD23a and CD23b promoters can efficiently compete Blimp1, wehave so far been unable to detect any targeting of the CD23a iso-form by BCL-6 (S.G., unpublished observations). A selective ef-fect of BCL-6 only on the CD23b (ITIM-less) isoform may thusexplain the failure of microarray analysis to detect CD23 as aBCL-6 target gene (38). It is intriguing to speculate that employ-ment of different mechanisms for regulating the two CD23 iso-forms may allow a B cell to modulate the ratio, and thus the sig-naling outcome, of this pair of potentially inhibitory/activatingreceptors.
Consistent with the central role exerted by IRF-4 in B cell ac-tivation, deregulation of IRF-4 expression has been postulated toplay a role in a variety of lymphoid malignancies (5, 39). Trans-locations of the BCL-6 gene leading to inappropriate expression ofBCL-6 are a common event in non-Hodgkin’s lymphomas (35,40–42). Interestingly, deletion of chromosome 6q21-q22.1, whichcontains the PRDI-BF1/Blimp1 gene, has been consistently de-tected in high-grade non-Hodgkin’s lymphoma and PRDI-BF1/Blimp1 has been suggested to be a candidate B-NHL suppressor
FIGURE 4. Identification of IRF-4 do-main sequences important for interactionwith Blimp1 and BCL-6. A, Binding ofvarious GST-IRF-4 mutant proteins withBlimp1 or BCL-6. To examine the IRF-4domains mediating the IRF-4/Blimp1 in-teraction, nuclear extracts from 293T cellstransfected with an HA epitope-taggedBlimp1 expression vector were incubatedwith various GST-IRF-4 mutant proteinsimmobilized onto reduced glutathione-aga-rose beads, as indicated. Bound proteinswere eluted, fractionated by 7% SDS-PAGE, transferred to a nitrocellulosemembrane, and then probed with an anti-HA Ab (upper panel). Binding to immobi-lized GST alone is shown as a control. Toassess the IRF-4 regions involved in theIRF-4/BCL-6 interaction, nuclear extractsfrom unstimulated Ramos cells were incu-bated with the same panel of GST-IRF-4mutant proteins as above. Bound proteinswere eluted, fractionated by 7% SDS-PAGE, transferred to a nitrocellulosemembrane, and then probed with an anti-BCL-6 Ab (middle panel). The blot wasthen stripped and reprobed with a Stat6 Ab(lower panel).B, Summary of the deletionmutation analysis. The IRF-4 mutant pro-teins used in the GST pull-down assays arediagrammed with the deleted sequences in-dicated on theleft of each mutant. The po-sitions of the DNA binding domain (DBD),the proline-rich region (PRO-RICH), andthe glutamine-rich region (GLN-RICH) areindicated. The relative ability of each mu-tant to interact with Blimp1, BCL-6, orStat6 is shown on theright of each mutant.11 indicates binding comparable withthat demonstrated by wt IRF-4;1 indi-cates a diminished interaction;2 indicatesno interaction.
6109The Journal of Immunology
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
gene (43). Our findings suggest that either persistence of BCL-6expression or loss of Blimp1 may lead to the inappropriate regu-lation of IRF-4 function. This may result in an activated or par-tially activated B cell, which cannot successfully complete its dif-ferentiation program. In addition to the recently described abilityof BCL-6 to repress Blimp1 expression and thus block terminal Bcells differentiation (38), disturbances in the IRF-4/Kruppel inter-action may thus also contribute to lymphomagenesis.
AcknowledgmentsWe thank Dr. Hirai for the anti-IRF-4 antiserum. We are grateful to ManJiang for her technical assistance and Drs. Dalla-Favera and Calame andmembers of their laboratories for providing us with cell lines, the BCL-6,and the Blimp1 constructs and critical discussions. We also thank Dr. G.Siu for his critical reading of this manuscript.
References1. Eisenbeis, C., H. Singh, and U. Storb. 1995. Pip, a novel IRF family member, is
a lymphoid-specific, PU.1-dependent transcriptional activator.Genes Dev.9:1377.
2. Matsuyama, T., A. Grossman, H. Mittrucker, D. Siderovski, F. Kiefer,T. Kawakami, C. Richardson, T. Taniguchi, S. Yoshinaga, and T. Mak. 1995.Molecular cloning of LSIRF, a lymphoid-specific member of the interferon reg-ulatory factor family that binds the interferon-stimulated response element(ISRE).Nucleic Acids Res. 23:2127.
3. Yamagata, T., J. Nishida, T. Tanaka, R. Sakai, K. Mitani, T. Taniguchi,Y. Yazaki, and H. Hirai. 1996. A novel interferon regulatory factor family tran-scription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of in-terferon-regulated genes.Mol. Cell. Biol. 16:1283.
4. Gupta, S., M. Jiang, A. Anthony, and A. Pernis. 1999. Lineage-specific modu-lation of IL-4 signaling by interferon regulatory factor 4.J. Exp. Med. 190:1837.
5. Alizadeh, A. A., M. B. Eisen, R. E. Davis, C. Ma, I. S. Lossos, A. Rosenwald,J. C. Boldrick, H. Sabet, T. Tran, X. Yu, et al. 2000. Distinct types of diffuse largeB-cell lymphoma identified by gene expression profiling.Nature 403:503.
FIGURE 5. Effects of Blimp1 on thetransactivation of CD23b.A, U937 cellswere cotransfected with a CD23b promoterluciferase reporter construct and an IRF-4expression plasmid in the presence of aBCL-6, a Blimp1, or a PRDI-BF1 expres-sion vector, or equivalent amounts ofempty vector, as indicated. The transfectedcells were left unstimulated for 16 h. Thedata are presented relative to the activity ofthe reporter construct in control U937cells, which was set to 1, as indicated, ineach experiment. Results show the mean6SE of four independent experiments.B,U937 cells were cotransfected with aCD23b promoter luciferase reporter con-struct and an IRF-4 expression plasmid inthe presence of increasing amounts of aBlimp1 expression vector or equivalentamounts of empty vector, as indicated. Thedata were analyzed as described above. Re-sults show the mean6 SE of three inde-pendent experiments.C, U937 cells werecotransfected with a CD23b promoter lu-ciferase reporter construct and either anIRF-4 expression vector or an empty vec-tor. The cells were cotransfected in thepresence of an expression plasmid encod-ing either BCL-6 (middle panel) or Blimp1(right panel), or of equivalent amounts ofcontrol vector (left panel). The transfectedcells were equally split into two 2-ml ali-quots and then incubated for 16 h in thepresence or absence of IL-4 (10 ng/ml).The data are presented relative to the ac-tivity of the reporter construct in controlU937 cells, which was set to 1, as indi-cated, in each experiment. Results showthe mean 6 SE of four independentexperiments.
6110 MODULATION OF IRF-4 FUNCTION BY BLIMP1
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

6. Glynne, R., S. Akkaraju, J. I. Healy, J. Rayner, C. C. Goodnow, and D. H. Mack.2000. How self-tolerance and the immunosuppressive drug FK506 prevent B-cellmitogenesis.Nature 403:672.
7. Mittrucker, H., T. Matsuyama, A. Grossman, T. Kundig, J. Potter, A. Shahinian,A. Wakeham, B. Patterson, P. Ohashi, and T. Mak. 1997. Requirement for thetranscription factor LSIRF/IRF4 for mature B and T lymphocyte function.Sci-ence 275:540.
8. Pongubala, J. M. R., S. Nagulapalli, M. J. Klemsz, S. R. McKercher, R. A. Maki,and M. L. Atchinson. 1992. PU.1 recruits a second nuclear factor to a site im-portant for immunoglobulink 39 enhancer activity.Mol. Cell. Biol. 12:368.
9. Himmelmann, A., A. Riva, G. L. Wilson, B. P. L. C. Thevenin, and J. H. Kehrl.1997. PU.1/Pip and basic helix loop helix zipper transcription factors interactwith binding sites in the CD20 promoter to help confer lineage- and stage-specificexpression of CD20 in B lymphocytes.Blood 90:3984.
10. Bonnefoy, J.-Y., S. Lecoanet-Henchoz, J.-P. Aubry, J.-F. Gauchat, and P. Graber.1995. CD23 and B-cell activation.Curr. Opin. Immunol. 7:355.
11. Conrad, D. 1990. FceRII/CD23: the low affinity receptor for IgE.Annu. Rev.Immunol. 8:623.
12. Richards, M., and D. Katz. 1991. Biology and chemistry of the low affinity IgEreceptor (FceRII/CD23). Crit. Rev. Immunol. 11:65.
13. Raulet, D., and W. Held. 1995. Natural killer receptors: the offs and ons of NKcell recognition.Cell 82:697.
14. Yokoyama, W. 1997. What goes up must come down: the emerging spectrum ofinhibitory receptors.J. Exp. Med. 186:1803.
15. Kikutani, H., and T. Kishimoto. 1990. Molecular genetics and biology of twodifferent species of FceRII. Res. Immunol. 141:249.
16. Vivier, E., and M. Daeron. 1997. Immunoreceptor tyrosine-based inhibition mo-tifs. Immunol. Today 18:286.
17. Unkeless, J., and J. Jin. 1997. Inhibitory receptors, ITIM sequences and phos-phatases.Curr. Opin. Immunol. 9:338.
18. Allman, D., A. Jain, A. Dent, R. Maile, T. Selvaggi, M. Kehry, and L. Staudt.1996. BCL-6 expression during B-cell activation.Blood 87:5257.
19. Catoretti, G., C.-C. Chang, K. Cechova, J. Zhang, B. H. Ye, B. Falini,D. C. Louie, K. Offit, R. S. K. Chaganti, and R. Dalla-Favera. 1995. BCL-6protein is expressed in germinal center B cells.Blood 1:45.
20. Catoretti, G., C. Chang, K. Cechova, J. Zhang, B. Ye, B. Falini, D. Louie,K. Offit, R. Chaganti, and R. Dalla-Favera. 1997. Down-regulation of BCL-6gene expression by CD40 and EBV latent membrane protein-1 (LMP1) and itsblock in lymphoma carrying BCL-6 rearrangements.Blood 90(Suppl. 1):175a.
21. Banchereau, J., and F. Rousset. 1992. Human B lymphocytes: phenotype, pro-liferation, and differentiation.Adv. Immunol. 52:125.
22. Turner, C. A., D. Mack, and M. M. Davis. 1994. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immu-noglobulin-secreting cells.Cell 77:297.
23. Lin, Y., K. Wong, and K. Calame. 1997. Repression of c-myctranscription byBlimp1, an inducer of terminal B cell differentiation.Science 276:596.
24. Piskurich, J. F., K. Lin, Y. Lin, Y. Wang, J. P.-Y. Ting, and K. Calame. 2000.Blimp1/PRDI-BF1 mediates extinction of major histocompatibility class II trans-activator (CIITA) expression in plasma cells.Nat. Immun. 1:526.
25. Keller, A. D., and T. Maniatis. 1991. Identification and characterization of anovel repressor ofb-interferon gene expression.Genes Dev. 5:868.
26. Angelin-Duclos, C., G. Catoretti, K.-I. Lin, and K. Calame. 2000. Commitmentof B lymphocytes to a plasma cell fate is associated with Blimp-1 expression invivo. J. Immunol. 165:5462.
27. Chang, C., B. Ye, R. Chaganti, and R. Dalla-Favera. 1996. BCL-6, aPOZ/zinc-finger protein, is a sequence-specific transcriptional repressor.Proc. Natl. Acad.Sci. USA 93:6947.
28. Perkel, J. M., and M. L. Atchison. 1998. A two-step mechanism for recruitmentof Pip by PU.1.J. Immunol. 160:241.
29. Kotanides, H., and N. C. Reich. 1993. Requirement of tyrosine phosphorylationfor rapid activation of a DNA binding factor by IL-4.Science 262:1265.
30. Goldstein, M., J. Hoxie, D. Zembryki, D. Matthews, and A. I. Levinson. 1985.Phenotypic and functional analysis of B cell lines from patients with multiplemyeloma.Blood 66:444.
31. Tuscano, J. M., K. M. Druey, A. Riva, J. Pena, C. B. Thompson, and J. H. Kehrl.1996. Bcl-x rather than Bcl-2 mediates CD40-dependent centrocyte survival inthe germinal center.Blood 88:1359.
32. Harris, M. B., C.-C. Chang, M. T. Berton, N. N. Danial, J. Zhang, D. Kuehner,B. H. Ye, M. Kvatyuk, P. P. Pandolfi, G. Catoretti, et al. 1999. Transcriptionalrepression of Stat6-dependent interleukin-4-induced genes by BCL-6: specificregualtion of Ie transcription and immunoglobulin E switching.Mol. Cell. Biol.19:7264.
33. Ortiz, M. A., J. Light, R. A. Maki, and N. Assa-Munt. 1999. Mutation analysisof the Pip interaction domain reveals critical residues for protein-protein inter-actions.Proc. Natl. Acad. Sci. USA 96:2740.
34. Dent, A. L., A. L. Shaffer, X. Yu, D. Allman, and L. M. Staudt. 1997. Control ofinflammation, cytokine expression, and germinal center formation by BCL-6.Science 276:589.
35. Ye, B., F. Lista, F. L. Coco, D. Knowles, K. Offit, R. Chaganti, andR. Dalla-Favera. 1993. Alterations of a zinc finger-encoding gene, BCL-6, indiffuse large-cell lymphoma.Science 262:747.
36. Seyfert, V., D. Allman, Y. He, and L. Staudt. 1996. Transcriptional repression bythe proto-oncogene BCL-6.Oncogene 12:2331.
37. Liu, Y.-J. 1997. Tracing antigen-driven B cell development in humans.Chem.Immunol. 67:14.
38. Shaffer, A. L., X. Yu, Y. He, J. Boldrick, E. P. Chan, and L. M. Staudt. 2000.BCL-6 represses genes that function in lymphocyte differentiation, inflammation,and cell cycle control.Immunity 13:199.
39. Iida, S., P. Rao, M. Butler, P. Corradini, M. Boccadoro, B. Klein, R. Chaganti,and R. Dalla-Favera. 1997. Deregulation of MUM1/IRF4 by chromosomal trans-location in multiple myeloma.Nat. Genet. 17:226.
40. Baron, B. W., G. Nucifora, N. McCabe, R. Espinosa, M. M. L. Beau, andT. W. McKeithan. 1993. Identification of the gene associated with the recurringchromosomal translocations t(3;14)(q27;q32) and t(3;22)(q27;q11) in B-cell lym-phomas.Proc. Natl. Acad. Sci. USA 90:5262.
41. Kerckaert, J. P., C. Deweindt, H. Tilly, S. Quief, G. Lecocq, and C. Bastard.1993. LAZ3, a novel zinc-finger encoding gene, is disrupted by recurring chro-mosome 3q27 translocations in human lymphomas.Nat. Genet. 5:66.
42. Miki, T., N. Kawamata, S. Hirosawa, and N. Aoki. 1994. Gene involved in the3q27 translocation associated with B-cell lymphoma, BCL5, encodes a Kruppel-like zinc-finger protein.Blood 83:26.
43. Mock, B. A., L. Liu, D. L. Paslier, and S. Huang. 1996. The B-lymphocytematuration promoting transcription factor BLIMP1/PRDI-BF1 maps to D6S447on human chromosome 6q21–q22.1 and the syntenic region of mouse chromo-some 10.Genomics 37:24.
6111The Journal of Immunology
by guest on June 23, 2015http://w
ww
.jimm
unol.org/D
ownloaded from