ji1100285 2202..2212
TRANSCRIPT

of August 21, 2011This information is current as
http://www.jimmunol.org/content/187/5/2202doi:10.4049/jimmunol.1100285August 2011;
2011;187;2202-2212; Prepublished online 1J Immunol Hanzhong WangMao, Bingke Bai, Baojing Lu, Panyong Mao, Qinxue Hu and Zhenhua Zheng, Hongxia Li, Zhenfeng Zhang, Jin Meng, Da
PhosphorylationβB Kinase κB by Suppressing IκMediated Activation of NF-
−αEnterovirus 71 2C Protein Inhibits TNF-
DataSupplementary
85.DC1.htmlhttp://www.jimmunol.org/content/suppl/2011/08/02/jimmunol.11002
References http://www.jimmunol.org/content/187/5/2202.full.html#ref-list-1
, 28 of which can be accessed free at:cites 67 articlesThis article
Subscriptions http://www.jimmunol.org/subscriptions
is online atThe Journal of ImmunologyInformation about subscribing to
Permissions http://www.aai.org/ji/copyright.html
Submit copyright permission requests at
Email Alerts http://www.jimmunol.org/etoc/subscriptions.shtml/
Receive free email-alerts when new articles cite this article. Sign up at
Print ISSN: 0022-1767 Online ISSN: 1550-6606.Immunologists, Inc. All rights reserved.
by The American Association ofCopyright ©2011 9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from

The Journal of Immunology
Enterovirus 71 2C Protein Inhibits TNF-a–MediatedActivation of NF-kB by Suppressing IkB Kinase bPhosphorylation
Zhenhua Zheng,* Hongxia Li,* Zhenfeng Zhang,* Jin Meng,* Da Mao,* Bingke Bai,†
Baojing Lu,‡ Panyong Mao,† Qinxue Hu,* and Hanzhong Wang*
Enterovirus 71 (EV71), a single, positive-stranded RNA virus, has been regarded as the most important neurotropic enterovirus
after the eradication of the poliovirus. EV71 infection can cause hand, foot, and mouth disease or herpangina. Cytokine storm with
elevated levels of proinflammatory and inflammatory cytokines, including TNF-a, has been proposed to explain the pathogenesis of
EV71-induced disease. TNF-a–mediated NF-kB signaling pathway plays a key role in inflammatory response. We hypothesized
that EV71 might also moderate host inflammation by interfering with this pathway. In this study, we tested this hypothesis and
identified EV71 2C protein as an antagonist of TNF-a–mediated activation of NF-kB signaling pathway. Expression of 2C protein
significantly reduced TNF-a–mediated NF-kB activation in 293T cells as measured by gene reporter and gel mobility shift assays.
Furthermore, overexpression of TNFR-associated factor 2-, MEK kinase 1-, IkB kinase (IKK)a-, or IKKb-induced NF-kB
activation, but not constitutively active mutant of IKKb (IKKb SS/EE)-induced NF-kB activation, was inhibited by 2C protein.
These data together suggested that the activation of IKKb is most likely targeted by 2C; this notion was further strengthened by
immunoblot detection of IKKb phosphorylation and IkBa phosphorylation and degradation. Coimmunoprecipitation and coloc-
alization of 2C and IKKb expressed in mammalian cells provided compelling evidence that 2C interacts with IKKb. Collectively,
our data indicate that EV71 2C protein inhibits IKKb activation and thus blocks NF-kB activation. The Journal of Immunology,
2011, 187: 2202–2212.
Enterovirus 71 (EV71) is known as one of the most im-portant pathogens causing emerging infectious disease inhumans. EV71 has been reported to be associated with
sporadic cases and outbreaks of a wide spectrum of diseases, in-cluding hand, foot, and mouth disease, herpangina, aseptic men-ingitis, encephalitis, poliomyelitis-like syndrome, or even fataldisease (1). EV71 is a single, positive-stranded RNA virus thatbelongs to Picornaviridae family (2). The ∼7.4-kb genome ofEV71 encodes a single polyprotein that is proteolytically cleavedto various structural and nonstructural viral proteins. The non-structural protein 2C in the P2 region of the polyprotein isa multifunctional and highly conserved viral protein among allpicornaviruses. In human enterovirus family, for instance, thepoliovirus 2C protein is composed of 329-aa–containing mem-
brane, RNA, and NTP binding regions (3–7). The 2C is believed tohave two functions during viral RNA synthesis, as follows: as anNTPase and directing replication complexes to cell membranes(8). Recently, EV71 2C protein has been reported to interact withhost protein reticulon 3, and this interaction is required for viralreplication (9).TNF-a and cell products induced by viral and bacterial in-
fection (e.g., IL-1, dsRNA, LPS) or cellular stresses (e.g., phorbolesters, UV) activate the NF-kB signaling pathway (10, 11). NF-kBtarget genes encode numerous mediators important for malignanttransformation, cell adhesion, cell growth control, apoptosis con-trol, immune functions, and embryonic development (for reviews,see Refs. 12 and 13). In addition, NF-kB may also play acrucial role in host antivirus responses as NF-kB activation caninduce the expression of b IFN, MHC class I, and several in-flammatory cytokines (for review, see Ref. 14). A wide varietyof viruses from various viral families are capable of modulatingNF-kB activation in infected cells, such as hepatitis C virus, para-myxoviruses, influenza virus, African swine fever virus, poxvi-ruses, and so on (15, 16). In most cases, these viruses encodeproteins that disrupt or modulate immune responses by targetingspecific aspects of the NF-kB signaling pathway.TNF-a is a pleiotropic proinflammatory cytokine involved in
protecting the host from pathogen infections (17). On TNF-a li-gand binding, its cognate receptor TNFRI recruits three signalingmolecules, TNFR-associated death domain, receptor-interactingprotein, and TNFR-associated factor 2 (TRAF2), to form a re-ceptor-based signalsome (18). Afterward, IkB kinase (IKK)complex, composed of at least two catalytic subunits (IKKa,IKKb) and an essential regulatory subunit (IKKg) (19), migratesto the signalsome. IKKa and IKKb are subsequently phosphory-lated and converted to activated kinase forms, and they in turn lead
*State Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Academyof Sciences, Wuhan, 430071, China; †Beijing Institute of Infectious Diseases, Beijing302 Hospital, Beijing, 100045, China; and ‡Department of Microbiology, AnhuiMedical University, Hefei, 230032, China
Received for publication January 28, 2011. Accepted for publication June 10, 2011.
This work was supported in part by the National 973 Program of China(2010CB530100 and 2011CB504902), the National Natural Science Foundation ofChina (81071351), and the Chinese Academy of Sciences (KSCX2-YW-R-144).
Address correspondence and reprint requests to Dr. Hanzhong Wang and Dr. QinxueHu, State Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Acad-emy of Sciences, Xiaohongshan NO. 44, Wuhan, 430071, China. E-mail addresses:[email protected] and [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: EV71, enterovirus 71; HA, hemagglutinin; HLH,helix–loop–helix; IKK, IkB kinase; KD, kinase domain; LZ, leucine zipper; MEKK,MEK kinase; MOI, multiplicity of infection; ORF, open reading frame; PVDF, poly-vinylidene difluoride; TRAF2, TNFR-associated factor 2.
Copyright� 2011 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100285
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from

to phosphorylation-induced degradation of IkBs (the inhibitors ofNF-kB) and the consequent translocation of NF-kB to nucleus(20–22). Current data indicate that the critical IKK complex is thebottleneck for most NF-kB activation pathways (19, 23), includingthose initiated through TLRs and TNFRs (24).Poliovirus has been reported to suppress TNF-mediated NF-kB
activation through its 3A protein, eliminating TNFR from the cellsurface. Other picornaviruses such as coxsackievirus B3 (25) andfoot-mouth-disease virus (26) have been reported to inhibit NF-kBactivation through viral protease proteins. EV71 infection cancause cytokine storm with elevated levels of proinflammatory andinflammatory cytokines, including TNF-a (27, 28), one of thestimuli in NF-kB activation pathway. However, the role of EV71in the TNF-a–mediated activation of NF-kB has yet to be ad-dressed.We hypothesized that EV71 might evolve strategies to interfere
with TNF-a–induced NF-kB activation to evade the immune re-sponses of the host. To test this hypothesis, we examined whe-ther TNF-a–induced NF-kB activation could be modulated byEV71. In the current study, we demonstrate the ability of EV712C protein to inhibit TNF-a–mediated NF-kB activation. Furtherstudy indicates that IKKb highly likely serves as the target for 2Cprotein.
Materials and MethodsVirus, cells, plasmids, Abs, and cytokines
Human EV71 BrCr strain was provided by the Institute of Medical Biology,Chinese Academy of Medical Science. The human embryonic kidney293T cell line and HeLa cell line were cultured in DMEM supplementedwith 10% FBS (Life Technologies).
The open reading frames (ORFs) of EV71 2B, 2BC, 2C, 3A, 3AB, 3C,and 3D were amplified by RT-PCR from EV71-BrCr genomic RNA usingprimers, as described in Table I, and subsequently cloned into thepCAGGS eukaryotic expression vector, respectively, with hemagglutinin(HA) tag at the carbon or N terminus (29). The deletion mutants of IKKband 2C were generated by PCR and subcloned into pCAGGS with Flag orHA tag at the carbon or N terminus. Plasmid pHA-TRAF2 was a gift ofJ. Ashwell (National Institutes of Health) (30). Plasmids pFLAG-IKKband pFLAG-IKKb SS/EE were provided by R. Anjana (Immune DiseaseInstitute, Harvard Medical School) (21). Plasmids pHA-IKKa and pHA-IKKb were gifts of G. Hur (Chungnam National University, South Korea)(31). pFLAG-2C and pMyc-2C were constructed by inserting a codingsequence of FLAG or Myc tag to the N terminus of 2C in pCAGGSeukaryotic expression vector. p2C-GFP was constructed by inserting theORF encoding the 2C protein into the pEGFP-C1 vector (Clontech). pFC-MEKK and pNF-kB-luc were purchased from Stratagene, and pRL-TKwas purchased from Promega (Madison, WI).
Polyclonal Abs against EV71 2C protein were produced by immunizingrabbits with His tag-2C fusion protein. Mouse anti-HA and anti-FLAGmAbs were obtained from Sigma-Aldrich. Rabbit polyclonal anti-mycAb was purchased from Genscript (Nanjing, China). Rabbit polyclonalanti-IKKb and anti-IkBa Abs were acquired from Proteintech (Wuhan,China). Mouse anti–phospho-IkBa mAb and rabbit phospho-IKKb(Ser177/181) mAb were purchased from Cell Signaling Technology. HumanrTNF-a was purchased from Biovision (San Francisco, CA).
Luciferase reporter assays
In assays involving TNF-a–mediated NF-kB activation, 293T cells wereseeded into 6-well plates and cotransfected with 0.5 mg reporter plasmidpNF-kB-luc, 0.1 mg Renilla luciferase expression plasmid pRL-TK, andthe indicated expression plasmids using ProFection Mammalian Trans-fection System (Promega). At 24 h posttransfection, cells were mocktreated or treated with TNF-a (10 ng/ml) for 6 h, lysed in passive lysisbuffer (Promega), and assayed for firefly and Renilla luciferase activitiesby using the dual luciferase reporter system (Promega). NF-kB can beactivated by overexpression of TRAF2, MEK kinase (MEKK) 1, IKKa,IKKb, or IKKb SS/EE signaling proteins. Briefly, 293T cells were co-transfected with plasmids expressing TRAF2, MEKK1, IKKa, IKKb, orIKKb SS/EE along with pNF-kB-luc (0.5 mg), pRL-TK (0.1 mg), andempty vector pCAGGS or pHA-2C. In some cases, large amounts ofpCAGGS were included in the transfection mixture to equalize thequantity of DNA. At 24 h posttransfection, cells were lysed and the ac-tivities of firefly and Renilla luciferases were measured.
For all assays, experiments were performed in triplicate. For each ex-periment, firefly luciferase activity was divided by Renilla luciferase ac-tivity to correct for differences in transfection efficiencies. The resultantratios were normalized to fold change value obtained from TNF-a–untreated cells cotransfected with pCAGGS, pNF-kB-luc, and pRL-TK.
EMSA
In transfection assays, subconfluent monolayers of 293T cells weretransfected with 2 mg of either pCAGGS or pHA-2C. At 24 h post-transfection, cells were mock treated or treated with TNF-a (10 ng/ml), asindicated. In infection assays, HeLa cells were infected with EV71 ata multiplicity of infection (MOI) of 5. At 10 h postinfection, cells weremock treated or treated with TNF-a (10 ng/ml), as indicated. Sub-sequently, the transfected or infected cells were collected at various times,and nuclear proteins were extracted by cytoplasmic/nuclear protein ex-traction kit (Beyotime Institute of Biotechnology). Single-stranded oligo-nucleotides containing binding sites of NF-kB transcription factor (59-AGTTGAGGGGACTTTCCCAGGC-39) were synthesized and end labeledwith biotin (Invitrogen). Single-stranded oligonucleotides were annealedto double-strand probe for EMSA. Detection of NF-kB–oligonucleotidecomplex was performed using a LightShift chemiluminescent EMSA kit(Pierce). Briefly, nuclear protein (5 mg) was incubated with 20 fmol biotin-labeled oligonucleotides for 20 min at room temperature in binding bufferconsisting of 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM DTT, 2.5%glycerol, 5 mM MgCl2, 50 ng poly(dG·dC), and 0.05% Nonidet P-40. Thespecificity of the NF-kB–DNA binding was determined in competitionreactions in which a 200-fold molar excess (4 pmol) of unlabeled oligo-nucleotide was added. Products of binding reactions were resolved by6% PAGE using 0.53 Tris-borate EDTA buffer. Protein–oligonucleotidecomplex was electroblotted to a nylon membrane. After incubation inblocking buffer for 15 min at room temperature, the membrane was in-cubated with streptavidin–HRP conjugate for 30 min at room temperature.The membrane was then incubated with chemiluminescent substrate for 5min before analyzed in Fluochem HD2 Imaging System (Alpha Innotech).
Immunoblot and coimmunoprecipitation assays
Transfected or infected cells were harvested at the indicated time points andlysed with cell lysis buffer for Western and immunoprecipitation (BeyotimeInstitute of Biotechnology) containing complete protease inhibitor tablets(Roche). Cell lysates were resolved by SDS-PAGE and transferred ontopolyvinylidene difluoride (PVDF) membranes (Millipore). Blots wereblocked in 20 mM Tris-HCl buffer (pH 7.4) containing 37 mM NaCl
Table I. Primers used for cloning of cDNA
Primer Name Sequence (59–39) Purpose
EV-2BHA-F GGTACCACCATGTACCCATACGATGTTCCAGATTACGCTGGGGTATCTGATTACATC Cloning of EV71 2B ORFEV-2B-R CCCGGGTTACTGTTTTTGTGCCATGGGAATGEV-2BC-F GCGGGTACCACCATGGGGGTATCTGATTACATCAAAGGTC Cloning of EV71 2BC ORFEV-2C-F GCGCTCGAGCCACCATGAGTGCCTCATGGCTAAAGAAGTTCEV-2CHA-R CCCGGGTTAAGCGTAATCTGGAACATCGTATGGGTATTGAAAGAGTGCTTCTATAG Cloning of EV71 2C ORFEV-3A-R CCCGGGTTACTGGAACCCGGCAAACAGCEV-3AHA-F CTCGAGCCACCATGTACCCATACGATGTTCCAGATTACGCTGGACCCCCTAAATTCAG Cloning of EV71 3A ORFEV3C-F GCGCTCGAGCCACCATGGGGCCCAGCTTAGACTTCGC Cloning of EV71 3C ORFEV3CHA-R CCCGGGTTAAGCGTAATCTGGAACATCGTATGGGTATTGCTCGCTGGCAAAATAACEV3D-F GCGGGTACCACCATGGGAGAGATCCAGTGGATGAAGCEV3DHA-R CCCGGGTTAAGCGTAATCTGGAACATCGTATGGGTAAAATAACTCGAGCCAATTTC Cloning of EV71 3D ORF
The Journal of Immunology 2203
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from

(TBS), with 2% BSA (Sigma-Aldrich) and reacted overnight at 4˚C withprimary Abs diluted 1:500 to 1:1000 in TBS containing 0.1% Tween 20and 2% BSA. After being washed with TBS containing 0.1% Tween 20,membranes were incubated with HRP-conjugated secondary Ab (Pierce).Following 1-h incubation at 37˚C, the membranes were washed, as de-scribed above, and then incubated with Super Signal West Pico Chemi-luminescent Substrate (Pierce) for signal detection. The membranes wereanalyzed in Fluochem HD2 Imaging System (Alpha Innotech).
For the fractionation of lysates, HeLa cells were mock infected orinfected with EV71 for 10 h and left untreated or treated with 10 ng/mlTNF-a for 30 min. Cells were washed, and cytoplasmic and nuclearfractions were isolated using cytoplasmic/nuclear protein extraction kit(Beyotime Institute of Biotechnology). The fractions were processed, asdescribed above, for immunoblotting. Tubulin Ab was used as a loadingcontrol for cytoplasmic fractions, and HDAC1 was used as a control fornuclear fractions.
For coimmunoprecipitation experiment, 293T cells were cotransfectedwith equal amounts of plasmids, as indicated. In some assays, at 18 hposttransfection, cells were infected with EV71 at a MOI of 5 for 12 h.Coimmunoprecipitation assay was performed using the protein G immu-noprecipitation kit (Roche), according to the manufacturer’s instructions.Briefly, cells were washed with PBS and homogenized in 1 ml lysis buffercontaining 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40,and 0.5% sodium deoxycholate with protease inhibitors. A total of 1 mllysates was precleared by incubation with 50 ml 50% slurry of protein G-agarose beads for 3 h at 4˚C. The extracts were then incubated with mouseanti-FLAG mAb (M2) or nonspecific mouse IgG for 1 h at 4˚C. Immu-nocomplexes were precipitated with 50 ml protein G-agarose overnight at4˚C. After washing with high-salt wash buffer (50 mM Tris-HCl [pH 7.5],500 mM NaCl, 0.1% Nonidet P-40, and 0.05% sodium deoxycholate) andlow-salt wash buffer (50 mM Tris-HCl [pH 7.5], 0.1% Nonidet P-40, and0.05% sodium deoxycholate), immunoprecipitates were boiled with 30 mlelectrophoresis sample buffer, and analyzed by immunoblotting.
Immunofluorescence microscopy
For colocalization assay, 293T cells grown on glass slides were cotrans-fected with plasmids pEGFP-2C and pFLAG-IKKb, as described above. At30 h posttransfection, slides were washed in PBS, fixed in 4% para-formaldehyde, and permeabilized with 0.2% Triton X-100 for 15 min.Cells were incubated in 5% goat serum in PBS at 4˚C for 12 h and thenincubated with mouse anti-FLAG Ab overnight. After being washed inPBS three times, cells were further incubated with tetramethylrhodamineisothiocyanate-conjugated goat anti-mouse IgG (Pierce). After two washesin PBST (0.1% Triton X-100 in PBS) and three washes in PBS, cells wereanalyzed using Revolution XD confocal microscope (Andor).
ResultsEV71 inhibits TNF-a–induced NF-kB activation
TNF-a plays an important role in host defense against viral in-fection. In EV71 infection, the elevated level of TNF-a is detectedin serum (32). To investigate the role of EV71 in the TNF-a–mediated activation of NF-kB, the nuclear translocalization andDNA-binding activity were determined by immunoblotting andelectromobility shift assay.To confirm the inhibition of NF-kB translocation, cytoplasmic
and nuclear fractions were prepared and detected by immuno-blotting analysis. Mock-infected or EV71-infected lysates wereseparated into cytoplasmic and nuclear fractions following nostimulation or stimulation with TNF-a. Immunoblotting analysisof cytoplasmic p65 indicated that similar levels of p65 werepresent in mock- and EV71-infected lysates of unstimulated cells(Fig. 1A). No p65 was detected in nuclear fractions of untreatedmock- and EV71-infected cell lysates (Fig. 1A). After the treat-ment of cells with TNF-a, we observed less p65 in nuclear lysatesof EV71-infected cells than in those of mock-infected cells (Fig.1A). Similar levels of HDAC1 and tubulin proteins were observedin cytoplasmic and nuclear fractions from infected and mock-infected cells.NF-kB activation by TNF-a was also examined by using EMSA
to measure the binding of nucleus-located NF-kB to a biotin-labeled oligonucleotide containing a NF-kB consensus sequence.
As shown in Fig. 1B, nuclear extracts from unstimulated cellpopulations failed to alter oligonucleotide mobility, indicating thatNF-kB was inactive. Compared with the samples from mock-in-fected cells, the amount of NF-kB–containing band was greatlyreduced in samples from EV71-infected cells when exposed toTNF-a for 30 min.
The 2C protein of EV71 suppresses TNF-a–induced NF-kBactivation
To illustrate whether EV71 nonstructural proteins account for theinhibition of TNF-a–induced NF-kB activation, we cotransfected293T cells with NF-kB–luciferase reporter vector together with2.5 mg 2B-, 2C-, 2BC-, 3A-, 3AB-, 3C-, or 3D-expressing plas-mid. At 24 h posttransfection, cells were either mock treated ortreated with human TNF-a (10 ng/ml) for 6 h. In 2C- or 3C-expressing cells, NF-kB–regulated luciferase activity was 4-foldlower than empty vector-transfected cells, respectively. However,in other nonstructural protein-expressing cells, NF-kB–regulatedluciferase activity was not significantly affected (Fig. 2A). We
FIGURE 1. The effect of EV71 on TNF-a–induced NF-kB activation.
A, EV71 inhibits TNF-a–induced p65 nuclear translocation. HeLa cells
were mock infected or infected with EV71 at a MOI of 5. At 10 h post-
infection, mock- and EV71-infected cells were left untreated (TNF-a2) or
treated (TNF-a+) with 10 ng/ml TNF-a for 30 min. Cell lysates were
separated into cytoplasmic and nuclear fractions. The results shown are
representative of three independent experiments. Both cytoplasmic and
nuclear proteins were analyzed by SDS-PAGE and immunoblotting with
anti-p65 Ab to reveal the localization of NF-kB subunits. Nucleus-specific
anti-HDAC1 Ab and cytoplasmic-specific anti-tubulin Ab were used as
controls. B, EV71 inhibits TNF-a–induced NF-kB DNA-binding activity.
The 293T cells were incubated with TNF-a (10 ng/ml) for indicated times.
A total of 5 mg nuclear protein from each sample was incubated with
biotin-labeled oligonucleotides containing consensus NF-kB binding sites.
Products of binding reactions were resolved by 6% PAGE under non-
denaturing conditions. A mobility-retarded complex containing NF-kB and
the bands composed of unbound oligonucleotides are indicated as * and –,
respectively. The lane labeled N indicates that no nucleus-extracted protein
was present in the reaction mixture, and C indicates reaction that nuclear
extracts from pCAGGS-transfected cells treated with TNF-a for 30 min
were incubated with labeled probes and unlabeled oligonucleotides com-
petitor.
2204 ENTEROVIRUS 71 2C PROTEIN INHIBITS NF-kB ACTIVATION
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from
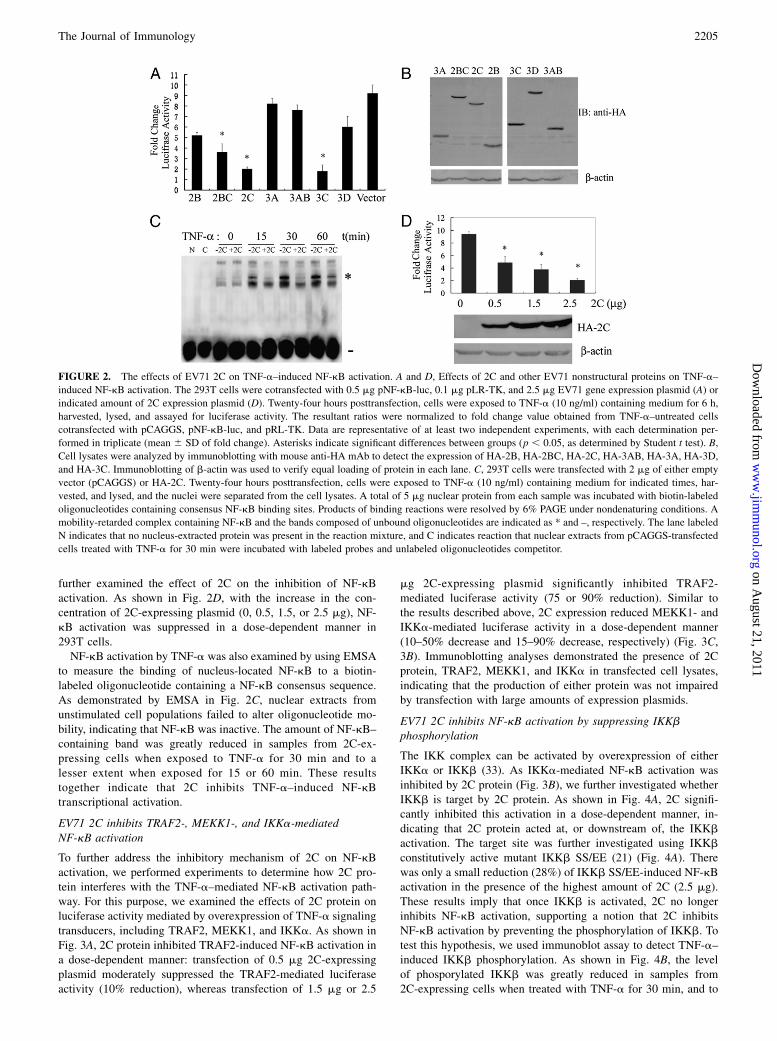
further examined the effect of 2C on the inhibition of NF-kBactivation. As shown in Fig. 2D, with the increase in the con-centration of 2C-expressing plasmid (0, 0.5, 1.5, or 2.5 mg), NF-kB activation was suppressed in a dose-dependent manner in293T cells.NF-kB activation by TNF-a was also examined by using EMSA
to measure the binding of nucleus-located NF-kB to a biotin-labeled oligonucleotide containing a NF-kB consensus sequence.As demonstrated by EMSA in Fig. 2C, nuclear extracts fromunstimulated cell populations failed to alter oligonucleotide mo-bility, indicating that NF-kB was inactive. The amount of NF-kB–containing band was greatly reduced in samples from 2C-ex-pressing cells when exposed to TNF-a for 30 min and to alesser extent when exposed for 15 or 60 min. These resultstogether indicate that 2C inhibits TNF-a–induced NF-kBtranscriptional activation.
EV71 2C inhibits TRAF2-, MEKK1-, and IKKa-mediatedNF-kB activation
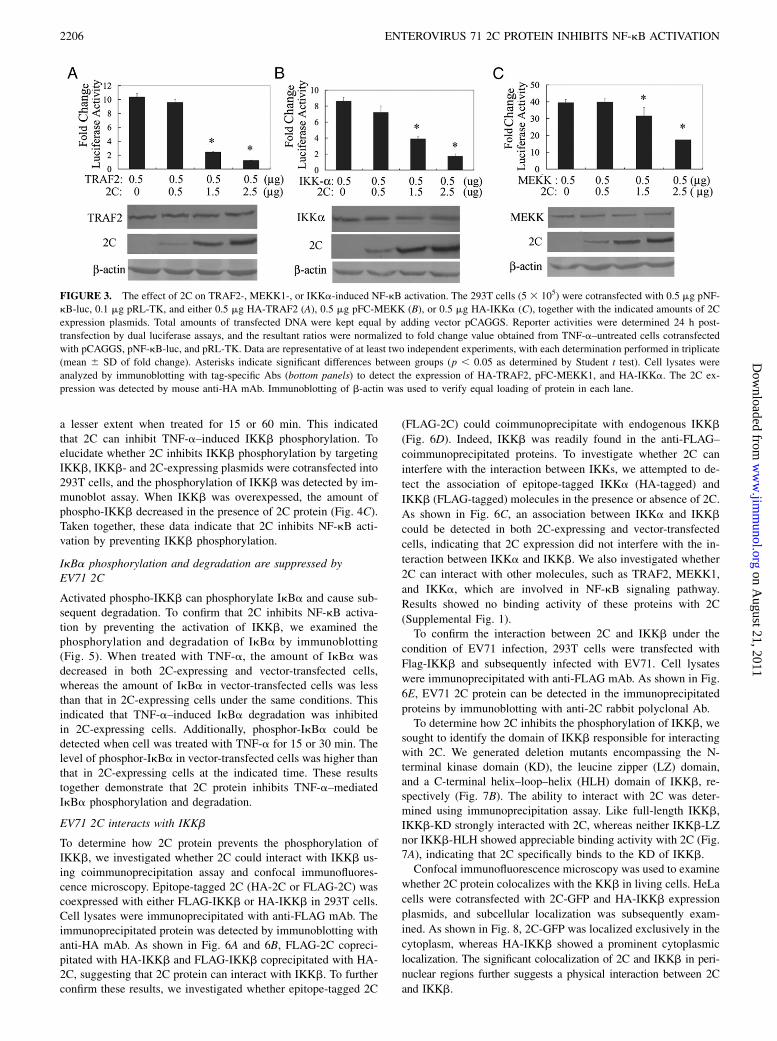
To further address the inhibitory mechanism of 2C on NF-kBactivation, we performed experiments to determine how 2C pro-tein interferes with the TNF-a–mediated NF-kB activation path-way. For this purpose, we examined the effects of 2C protein onluciferase activity mediated by overexpression of TNF-a signalingtransducers, including TRAF2, MEKK1, and IKKa. As shown inFig. 3A, 2C protein inhibited TRAF2-induced NF-kB activation ina dose-dependent manner: transfection of 0.5 mg 2C-expressingplasmid moderately suppressed the TRAF2-mediated luciferaseactivity (10% reduction), whereas transfection of 1.5 mg or 2.5
mg 2C-expressing plasmid significantly inhibited TRAF2-mediated luciferase activity (75 or 90% reduction). Similar tothe results described above, 2C expression reduced MEKK1- andIKKa-mediated luciferase activity in a dose-dependent manner(10–50% decrease and 15–90% decrease, respectively) (Fig. 3C,3B). Immunoblotting analyses demonstrated the presence of 2Cprotein, TRAF2, MEKK1, and IKKa in transfected cell lysates,indicating that the production of either protein was not impairedby transfection with large amounts of expression plasmids.
EV71 2C inhibits NF-kB activation by suppressing IKKbphosphorylation
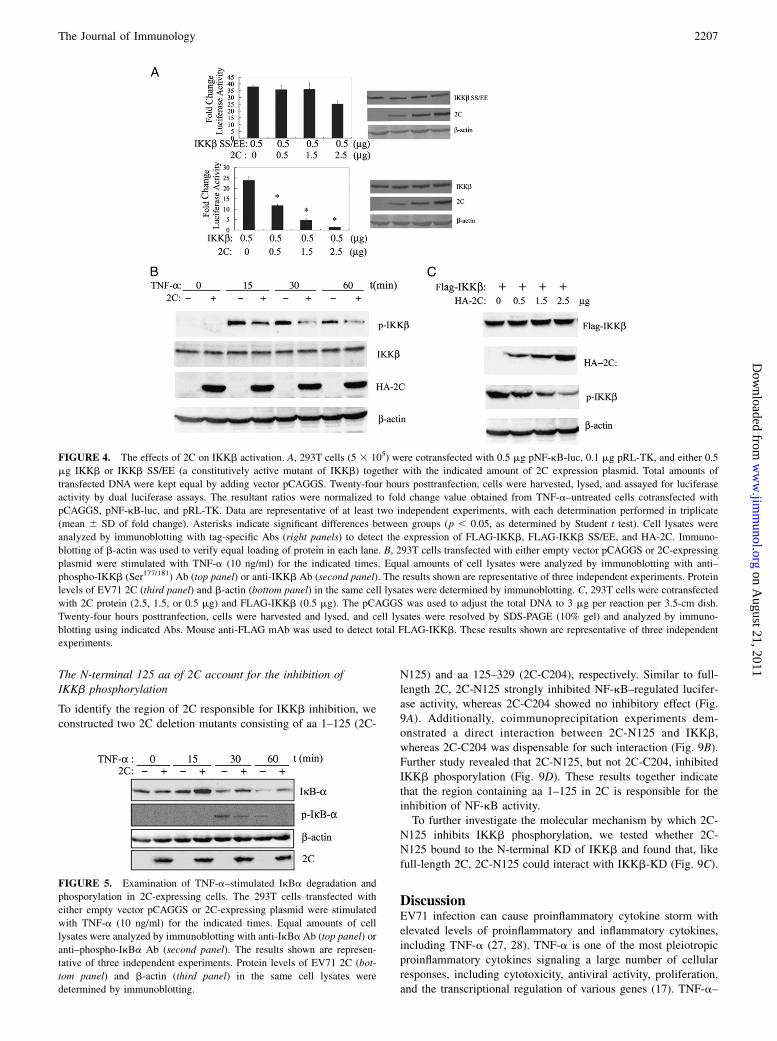
The IKK complex can be activated by overexpression of eitherIKKa or IKKb (33). As IKKa-mediated NF-kB activation wasinhibited by 2C protein (Fig. 3B), we further investigated whetherIKKb is target by 2C protein. As shown in Fig. 4A, 2C signifi-cantly inhibited this activation in a dose-dependent manner, in-dicating that 2C protein acted at, or downstream of, the IKKbactivation. The target site was further investigated using IKKbconstitutively active mutant IKKb SS/EE (21) (Fig. 4A). Therewas only a small reduction (28%) of IKKb SS/EE-induced NF-kBactivation in the presence of the highest amount of 2C (2.5 mg).These results imply that once IKKb is activated, 2C no longerinhibits NF-kB activation, supporting a notion that 2C inhibitsNF-kB activation by preventing the phosphorylation of IKKb. Totest this hypothesis, we used immunoblot assay to detect TNF-a–induced IKKb phosphorylation. As shown in Fig. 4B, the levelof phosporylated IKKb was greatly reduced in samples from2C-expressing cells when treated with TNF-a for 30 min, and to
FIGURE 2. The effects of EV71 2C on TNF-a–induced NF-kB activation. A and D, Effects of 2C and other EV71 nonstructural proteins on TNF-a–
induced NF-kB activation. The 293T cells were cotransfected with 0.5 mg pNF-kB-luc, 0.1 mg pLR-TK, and 2.5 mg EV71 gene expression plasmid (A) or
indicated amount of 2C expression plasmid (D). Twenty-four hours posttransfection, cells were exposed to TNF-a (10 ng/ml) containing medium for 6 h,
harvested, lysed, and assayed for luciferase activity. The resultant ratios were normalized to fold change value obtained from TNF-a–untreated cells
cotransfected with pCAGGS, pNF-kB-luc, and pRL-TK. Data are representative of at least two independent experiments, with each determination per-
formed in triplicate (mean 6 SD of fold change). Asterisks indicate significant differences between groups (p , 0.05, as determined by Student t test). B,
Cell lysates were analyzed by immunoblotting with mouse anti-HA mAb to detect the expression of HA-2B, HA-2BC, HA-2C, HA-3AB, HA-3A, HA-3D,
and HA-3C. Immunoblotting of b-actin was used to verify equal loading of protein in each lane. C, 293T cells were transfected with 2 mg of either empty
vector (pCAGGS) or HA-2C. Twenty-four hours posttransfection, cells were exposed to TNF-a (10 ng/ml) containing medium for indicated times, har-
vested, and lysed, and the nuclei were separated from the cell lysates. A total of 5 mg nuclear protein from each sample was incubated with biotin-labeled
oligonucleotides containing consensus NF-kB binding sites. Products of binding reactions were resolved by 6% PAGE under nondenaturing conditions. A
mobility-retarded complex containing NF-kB and the bands composed of unbound oligonucleotides are indicated as * and –, respectively. The lane labeled
N indicates that no nucleus-extracted protein was present in the reaction mixture, and C indicates reaction that nuclear extracts from pCAGGS-transfected
cells treated with TNF-a for 30 min were incubated with labeled probes and unlabeled oligonucleotides competitor.
The Journal of Immunology 2205
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from
a lesser extent when treated for 15 or 60 min. This indicatedthat 2C can inhibit TNF-a–induced IKKb phosphorylation. Toelucidate whether 2C inhibits IKKb phosphorylation by targetingIKKb, IKKb- and 2C-expressing plasmids were cotransfected into293T cells, and the phosphorylation of IKKb was detected by im-munoblot assay. When IKKb was overexpessed, the amount ofphospho-IKKb decreased in the presence of 2C protein (Fig. 4C).Taken together, these data indicate that 2C inhibits NF-kB acti-vation by preventing IKKb phosphorylation.
IkBa phosphorylation and degradation are suppressed byEV71 2C
Activated phospho-IKKb can phosphorylate IkBa and cause sub-sequent degradation. To confirm that 2C inhibits NF-kB activa-tion by preventing the activation of IKKb, we examined thephosphorylation and degradation of IkBa by immunoblotting(Fig. 5). When treated with TNF-a, the amount of IkBa wasdecreased in both 2C-expressing and vector-transfected cells,whereas the amount of IkBa in vector-transfected cells was lessthan that in 2C-expressing cells under the same conditions. Thisindicated that TNF-a–induced IkBa degradation was inhibitedin 2C-expressing cells. Additionally, phosphor-IkBa could bedetected when cell was treated with TNF-a for 15 or 30 min. Thelevel of phosphor-IkBa in vector-transfected cells was higher thanthat in 2C-expressing cells at the indicated time. These resultstogether demonstrate that 2C protein inhibits TNF-a–mediatedIkBa phosphorylation and degradation.
EV71 2C interacts with IKKb
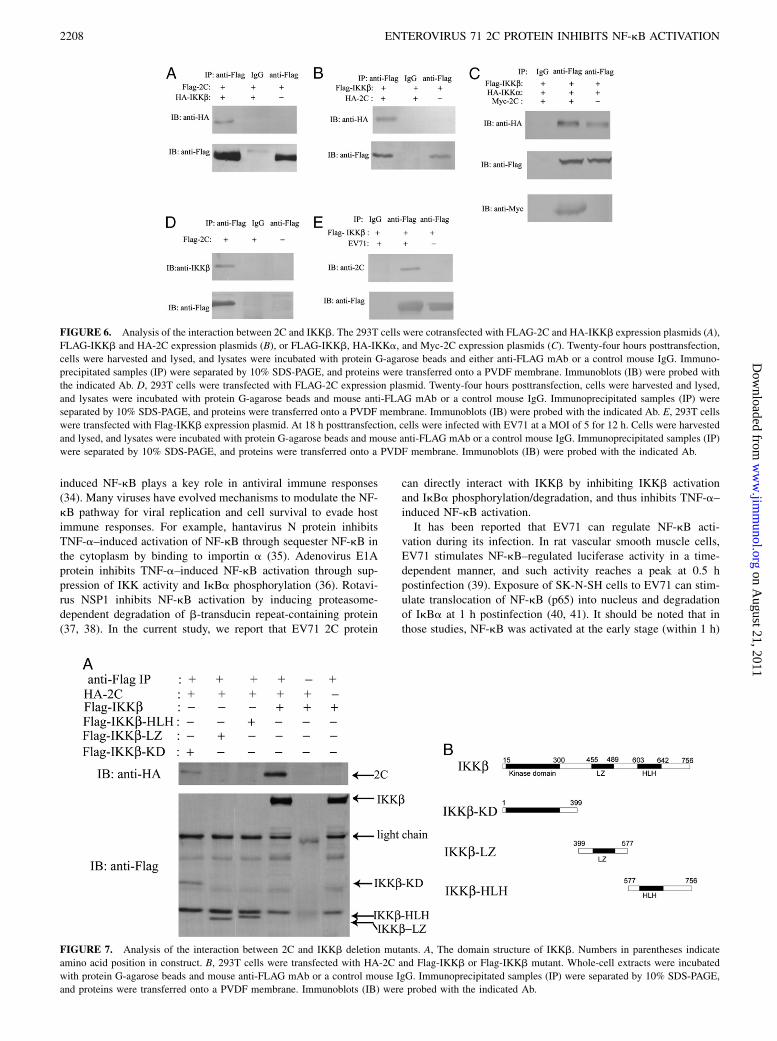
To determine how 2C protein prevents the phosphorylation ofIKKb, we investigated whether 2C could interact with IKKb us-ing coimmunoprecipitation assay and confocal immunofluores-cence microscopy. Epitope-tagged 2C (HA-2C or FLAG-2C) wascoexpressed with either FLAG-IKKb or HA-IKKb in 293T cells.Cell lysates were immunoprecipitated with anti-FLAG mAb. Theimmunoprecipitated protein was detected by immunoblotting withanti-HA mAb. As shown in Fig. 6A and 6B, FLAG-2C copreci-pitated with HA-IKKb and FLAG-IKKb coprecipitated with HA-2C, suggesting that 2C protein can interact with IKKb. To furtherconfirm these results, we investigated whether epitope-tagged 2C
(FLAG-2C) could coimmunoprecipitate with endogenous IKKb
(Fig. 6D). Indeed, IKKb was readily found in the anti-FLAG–
coimmunoprecipitated proteins. To investigate whether 2C can
interfere with the interaction between IKKs, we attempted to de-
tect the association of epitope-tagged IKKa (HA-tagged) and
IKKb (FLAG-tagged) molecules in the presence or absence of 2C.
As shown in Fig. 6C, an association between IKKa and IKKb
could be detected in both 2C-expressing and vector-transfected
cells, indicating that 2C expression did not interfere with the in-
teraction between IKKa and IKKb. We also investigated whether
2C can interact with other molecules, such as TRAF2, MEKK1,
and IKKa, which are involved in NF-kB signaling pathway.
Results showed no binding activity of these proteins with 2C
(Supplemental Fig. 1).To confirm the interaction between 2C and IKKb under the
condition of EV71 infection, 293T cells were transfected with
Flag-IKKb and subsequently infected with EV71. Cell lysates
were immunoprecipitated with anti-FLAG mAb. As shown in Fig.
6E, EV71 2C protein can be detected in the immunoprecipitated
proteins by immunoblotting with anti-2C rabbit polyclonal Ab.To determine how 2C inhibits the phosphorylation of IKKb, we
sought to identify the domain of IKKb responsible for interactingwith 2C. We generated deletion mutants encompassing the N-terminal kinase domain (KD), the leucine zipper (LZ) domain,and a C-terminal helix–loop–helix (HLH) domain of IKKb, re-spectively (Fig. 7B). The ability to interact with 2C was deter-mined using immunoprecipitation assay. Like full-length IKKb,IKKb-KD strongly interacted with 2C, whereas neither IKKb-LZnor IKKb-HLH showed appreciable binding activity with 2C (Fig.7A), indicating that 2C specifically binds to the KD of IKKb.Confocal immunofluorescence microscopy was used to examine
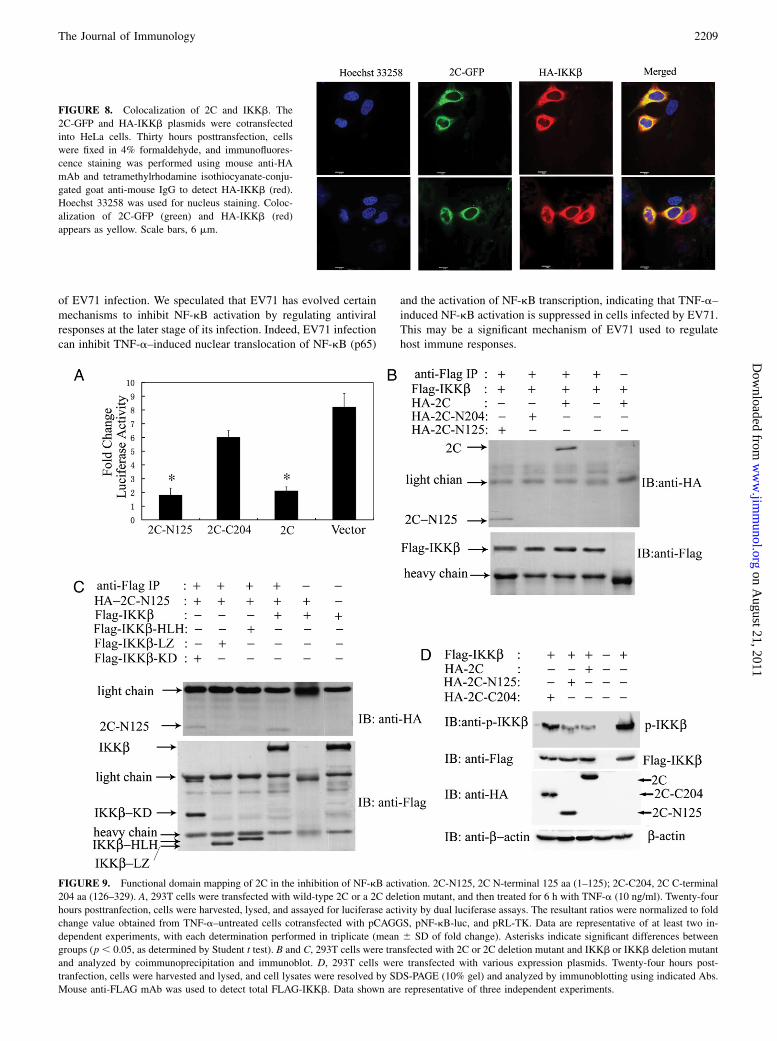
whether 2C protein colocalizes with the KKb in living cells. HeLa
cells were cotransfected with 2C-GFP and HA-IKKb expression
plasmids, and subcellular localization was subsequently exam-
ined. As shown in Fig. 8, 2C-GFP was localized exclusively in the
cytoplasm, whereas HA-IKKb showed a prominent cytoplasmic
localization. The significant colocalization of 2C and IKKb in peri-
nuclear regions further suggests a physical interaction between 2C
and IKKb.
FIGURE 3. The effect of 2C on TRAF2-, MEKK1-, or IKKa-induced NF-kB activation. The 293T cells (5 3 105) were cotransfected with 0.5 mg pNF-
kB-luc, 0.1 mg pRL-TK, and either 0.5 mg HA-TRAF2 (A), 0.5 mg pFC-MEKK (B), or 0.5 mg HA-IKKa (C), together with the indicated amounts of 2C
expression plasmids. Total amounts of transfected DNA were kept equal by adding vector pCAGGS. Reporter activities were determined 24 h post-
transfection by dual luciferase assays, and the resultant ratios were normalized to fold change value obtained from TNF-a–untreated cells cotransfected
with pCAGGS, pNF-kB-luc, and pRL-TK. Data are representative of at least two independent experiments, with each determination performed in triplicate
(mean 6 SD of fold change). Asterisks indicate significant differences between groups (p , 0.05 as determined by Student t test). Cell lysates were
analyzed by immunoblotting with tag-specific Abs (bottom panels) to detect the expression of HA-TRAF2, pFC-MEKK1, and HA-IKKa. The 2C ex-
pression was detected by mouse anti-HA mAb. Immunoblotting of b-actin was used to verify equal loading of protein in each lane.
2206 ENTEROVIRUS 71 2C PROTEIN INHIBITS NF-kB ACTIVATION
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from
The N-terminal 125 aa of 2C account for the inhibition ofIKKb phosphorylation
To identify the region of 2C responsible for IKKb inhibition, weconstructed two 2C deletion mutants consisting of aa 1–125 (2C-
N125) and aa 125–329 (2C-C204), respectively. Similar to full-length 2C, 2C-N125 strongly inhibited NF-kB–regulated lucifer-ase activity, whereas 2C-C204 showed no inhibitory effect (Fig.9A). Additionally, coimmunoprecipitation experiments dem-onstrated a direct interaction between 2C-N125 and IKKb,whereas 2C-C204 was dispensable for such interaction (Fig. 9B).Further study revealed that 2C-N125, but not 2C-C204, inhibitedIKKb phosporylation (Fig. 9D). These results together indicatethat the region containing aa 1–125 in 2C is responsible for theinhibition of NF-kB activity.To further investigate the molecular mechanism by which 2C-
N125 inhibits IKKb phosphorylation, we tested whether 2C-N125 bound to the N-terminal KD of IKKb and found that, likefull-length 2C, 2C-N125 could interact with IKKb-KD (Fig. 9C).
DiscussionEV71 infection can cause proinflammatory cytokine storm withelevated levels of proinflammatory and inflammatory cytokines,including TNF-a (27, 28). TNF-a is one of the most pleiotropicproinflammatory cytokines signaling a large number of cellularresponses, including cytotoxicity, antiviral activity, proliferation,and the transcriptional regulation of various genes (17). TNF-a–
FIGURE 4. The effects of 2C on IKKb activation. A, 293T cells (5 3 105) were cotransfected with 0.5 mg pNF-kB-luc, 0.1 mg pRL-TK, and either 0.5
mg IKKb or IKKb SS/EE (a constitutively active mutant of IKKb) together with the indicated amount of 2C expression plasmid. Total amounts of
transfected DNA were kept equal by adding vector pCAGGS. Twenty-four hours posttranfection, cells were harvested, lysed, and assayed for luciferase
activity by dual luciferase assays. The resultant ratios were normalized to fold change value obtained from TNF-a–untreated cells cotransfected with
pCAGGS, pNF-kB-luc, and pRL-TK. Data are representative of at least two independent experiments, with each determination performed in triplicate
(mean 6 SD of fold change). Asterisks indicate significant differences between groups (p , 0.05, as determined by Student t test). Cell lysates were
analyzed by immunoblotting with tag-specific Abs (right panels) to detect the expression of FLAG-IKKb, FLAG-IKKb SS/EE, and HA-2C. Immuno-
blotting of b-actin was used to verify equal loading of protein in each lane. B, 293T cells transfected with either empty vector pCAGGS or 2C-expressing
plasmid were stimulated with TNF-a (10 ng/ml) for the indicated times. Equal amounts of cell lysates were analyzed by immunoblotting with anti–
phospho-IKKb (Ser177/181) Ab (top panel) or anti-IKKb Ab (second panel). The results shown are representative of three independent experiments. Protein
levels of EV71 2C (third panel) and b-actin (bottom panel) in the same cell lysates were determined by immunoblotting. C, 293T cells were cotransfected
with 2C protein (2.5, 1.5, or 0.5 mg) and FLAG-IKKb (0.5 mg). The pCAGGS was used to adjust the total DNA to 3 mg per reaction per 3.5-cm dish.
Twenty-four hours posttranfection, cells were harvested and lysed, and cell lysates were resolved by SDS-PAGE (10% gel) and analyzed by immuno-
blotting using indicated Abs. Mouse anti-FLAG mAb was used to detect total FLAG-IKKb. These results shown are representative of three independent
experiments.
FIGURE 5. Examination of TNF-a–stimulated IkBa degradation and
phosporylation in 2C-expressing cells. The 293T cells transfected with
either empty vector pCAGGS or 2C-expressing plasmid were stimulated
with TNF-a (10 ng/ml) for the indicated times. Equal amounts of cell
lysates were analyzed by immunoblotting with anti-IkBaAb (top panel) or
anti–phospho-IkBa Ab (second panel). The results shown are represen-
tative of three independent experiments. Protein levels of EV71 2C (bot-
tom panel) and b-actin (third panel) in the same cell lysates were
determined by immunoblotting.
The Journal of Immunology 2207
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from
induced NF-kB plays a key role in antiviral immune responses(34). Many viruses have evolved mechanisms to modulate the NF-kB pathway for viral replication and cell survival to evade hostimmune responses. For example, hantavirus N protein inhibitsTNF-a–induced activation of NF-kB through sequester NF-kB inthe cytoplasm by binding to importin a (35). Adenovirus E1Aprotein inhibits TNF-a–induced NF-kB activation through sup-pression of IKK activity and IkBa phosphorylation (36). Rotavi-rus NSP1 inhibits NF-kB activation by inducing proteasome-dependent degradation of b-transducin repeat-containing protein(37, 38). In the current study, we report that EV71 2C protein
can directly interact with IKKb by inhibiting IKKb activationand IkBa phosphorylation/degradation, and thus inhibits TNF-a–induced NF-kB activation.It has been reported that EV71 can regulate NF-kB acti-
vation during its infection. In rat vascular smooth muscle cells,EV71 stimulates NF-kB–regulated luciferase activity in a time-dependent manner, and such activity reaches a peak at 0.5 hpostinfection (39). Exposure of SK-N-SH cells to EV71 can stim-ulate translocation of NF-kB (p65) into nucleus and degradationof IkBa at 1 h postinfection (40, 41). It should be noted that inthose studies, NF-kB was activated at the early stage (within 1 h)
FIGURE 6. Analysis of the interaction between 2C and IKKb. The 293T cells were cotransfected with FLAG-2C and HA-IKKb expression plasmids (A),
FLAG-IKKb and HA-2C expression plasmids (B), or FLAG-IKKb, HA-IKKa, and Myc-2C expression plasmids (C). Twenty-four hours posttransfection,
cells were harvested and lysed, and lysates were incubated with protein G-agarose beads and either anti-FLAG mAb or a control mouse IgG. Immuno-
precipitated samples (IP) were separated by 10% SDS-PAGE, and proteins were transferred onto a PVDF membrane. Immunoblots (IB) were probed with
the indicated Ab. D, 293T cells were transfected with FLAG-2C expression plasmid. Twenty-four hours posttransfection, cells were harvested and lysed,
and lysates were incubated with protein G-agarose beads and mouse anti-FLAG mAb or a control mouse IgG. Immunoprecipitated samples (IP) were
separated by 10% SDS-PAGE, and proteins were transferred onto a PVDF membrane. Immunoblots (IB) were probed with the indicated Ab. E, 293T cells
were transfected with Flag-IKKb expression plasmid. At 18 h posttransfection, cells were infected with EV71 at a MOI of 5 for 12 h. Cells were harvested
and lysed, and lysates were incubated with protein G-agarose beads and mouse anti-FLAG mAb or a control mouse IgG. Immunoprecipitated samples (IP)
were separated by 10% SDS-PAGE, and proteins were transferred onto a PVDF membrane. Immunoblots (IB) were probed with the indicated Ab.
FIGURE 7. Analysis of the interaction between 2C and IKKb deletion mutants. A, The domain structure of IKKb. Numbers in parentheses indicate
amino acid position in construct. B, 293T cells were transfected with HA-2C and Flag-IKKb or Flag-IKKb mutant. Whole-cell extracts were incubated
with protein G-agarose beads and mouse anti-FLAG mAb or a control mouse IgG. Immunoprecipitated samples (IP) were separated by 10% SDS-PAGE,
and proteins were transferred onto a PVDF membrane. Immunoblots (IB) were probed with the indicated Ab.
2208 ENTEROVIRUS 71 2C PROTEIN INHIBITS NF-kB ACTIVATION
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from
of EV71 infection. We speculated that EV71 has evolved certainmechanisms to inhibit NF-kB activation by regulating antiviralresponses at the later stage of its infection. Indeed, EV71 infectioncan inhibit TNF-a–induced nuclear translocation of NF-kB (p65)
and the activation of NF-kB transcription, indicating that TNF-a–induced NF-kB activation is suppressed in cells infected by EV71.This may be a significant mechanism of EV71 used to regulatehost immune responses.
FIGURE 8. Colocalization of 2C and IKKb. The
2C-GFP and HA-IKKb plasmids were cotransfected
into HeLa cells. Thirty hours posttransfection, cells
were fixed in 4% formaldehyde, and immunofluores-
cence staining was performed using mouse anti-HA
mAb and tetramethylrhodamine isothiocyanate-conju-
gated goat anti-mouse IgG to detect HA-IKKb (red).
Hoechst 33258 was used for nucleus staining. Coloc-
alization of 2C-GFP (green) and HA-IKKb (red)
appears as yellow. Scale bars, 6 mm.
FIGURE 9. Functional domain mapping of 2C in the inhibition of NF-kB activation. 2C-N125, 2C N-terminal 125 aa (1–125); 2C-C204, 2C C-terminal
204 aa (126–329). A, 293T cells were transfected with wild-type 2C or a 2C deletion mutant, and then treated for 6 h with TNF-a (10 ng/ml). Twenty-four
hours posttranfection, cells were harvested, lysed, and assayed for luciferase activity by dual luciferase assays. The resultant ratios were normalized to fold
change value obtained from TNF-a–untreated cells cotransfected with pCAGGS, pNF-kB-luc, and pRL-TK. Data are representative of at least two in-
dependent experiments, with each determination performed in triplicate (mean 6 SD of fold change). Asterisks indicate significant differences between
groups (p, 0.05, as determined by Student t test). B and C, 293T cells were transfected with 2C or 2C deletion mutant and IKKb or IKKb deletion mutant
and analyzed by coimmunoprecipitation and immunoblot. D, 293T cells were transfected with various expression plasmids. Twenty-four hours post-
tranfection, cells were harvested and lysed, and cell lysates were resolved by SDS-PAGE (10% gel) and analyzed by immunoblotting using indicated Abs.
Mouse anti-FLAG mAb was used to detect total FLAG-IKKb. Data shown are representative of three independent experiments.
The Journal of Immunology 2209
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from

Other picornaviruses, such as foot-and-mouth disease virus andcoxsackievirus B3, are reported to inhibit NF-kB activation viaviral proteases. Foot-and-mouth disease virus Lpro protein inhib-its NF-kB activation through degrading p65/RelA, whereas cox-sackievirus B3 3Cpro cleaves IkBa and subsequently inhibits NF-kB transactivation (25). Beyond the scope of this study, we alsofound that EV71 3Cpro can inhibit NF-kB activation. We speculate3C and 2C protein affect different targets in NF-kB signaling infavor of EV71. In addition to viral protease proteins, poliovirus3A has been reported to suppress TNF signaling, including NF-kB activation, by eliminating TNFR from the cell surface (42).The nonstructural protein 2C, which is highly conserved amongpicornaviruses, has multiple functions during viral replicationcycle, including uncoating (43), host cell membrane rearrange-ment (44), RNA replication (for review, see Ref. 45), encapsida-tion (46), and morphogenesis (47). EV71 2C has been reported tointeract with host protein reticulon 3, and thus modulate viralreplication (9). In this study, we demonstrate a novel function of2C protein in modulating TNF-a–mediated NF-kB activation. Inour study, luciferase reporter assays showed that 2C suppressedTNF-a–stimulated NF-kB activity in a dose-dependent manner.Further electromobility shift assays indicated that 2C proteincould specifically inhibit TNF-a–stimulated transcriptional acti-vity of NF-kB, suggesting that EV71 2C may be a novel viru-lence factor. Given that 2C protein of another picornavirus, avianencephalomyelitis virus, can induce apoptosis by activating cy-tochrome c/caspase-9 pathway (48), our data and others togethersuggest that 2C proteins in picornavirus family can regulate hostantiviral responses.We found that EV71 2C protein inhibited NF-kB activation by
targeting IKKb. In our observation, IKKb-induced NF-kB acti-vation was suppressed by 2C protein, whereas IKKb constitutivelyactive mutant-IKKb SS/EE-induced NF-kB activation was notsuppressed. Further study indicated that 2C inhibited TNF-a andIKKb induced phosphorylation of IKKb. IKKb is a crucial kinaseof the IKK-signalosome for NF-kB activation by most inflamma-tory stimuli. The phosphorylation in the activation loop of IKKbleads to the activation of IKKb and the subsequent activationof NF-kB (33, 49). Usually, in NF-kB activation pathway, ubi-quitylation of IKKg allows the recruitment of kinases, whichphosphorylates IKKb in its activation loop at Ser177 and Ser181
(50–52). Some viral proteins can inhibit phosporylation of IKKbby targeting IKKb itself, regulatory subunit IKKg, the upstreamkinase such as TGF-b–activated kinase-1, or other IKK complexcomponent. For example, hepatitis C virus nonstructural 5B pro-tein interacts with IKKa (53); vaccinia virus virulence factor B14binds to IKKbg (54); poxvirus MC160 interacts with heat shockprotein 90 (a component of IKK complex) (55, 56); and thepoxvirus protein N1L associates with TRAF-associated NF-kBactivator-binding kinase 1 (57). Our finding that EV71 2C inter-acts with IKKb provides a mechanistic explanation for the in-hibition of IKKb phosphorylation by 2C.In the current study, 2C region containing the N-terminal 125 aa
was found to inhibit IKKb phosphorylation and the binding of 2Cto IKKb, whereas aa 125–329 of 2C did not have such effects.These results indicate that the binding of 2C to IKKb is in-dispensable for its inhibitory effects on IKKb phosphorylation.A number of biologically active IKKb inhibitors, such as aspi-rin, salicylate, sulindac, vitamin C, and thiol-reactive metal com-pounds, have been reported to inhibit the catalytic activity ofIKKb (58–61). Parthenolide, a compound from the medicinal herbfeverfew, has been found to bind directly to IKKa and IKKb (62).More importantly, arsenite can block IKKb activation by inter-acting with Cys179, which lies between the activating phosphor-
ylation sites Ser177 and Ser181 (63). The 2C may be an EV71-encoded IKKb inhibitor sharing mechanism similar to those de-scribed above.IKKb consists of an N-terminal KD, a predicted LZ domain,
and a HLH domain at its C terminus (64). Our data revealed that2C specifically bound to the KD of IKKb containing the twophosphorylation sites Ser177 and Ser181. We also found that 2C didnot affect NF-kB–regulated luciferase activity induced by con-stitutively active IKKb mutant-IKKb SS/EE (a IKKb mutantcontains substitutions of Ser177 and Ser181 with glutamic acidresidues). In addition, TNF-a– and IKKb-induced phosphoryla-tion level of IKKb in Ser177 or Ser181 was also decreased in thepresence of 2C. The 2C may inhibit the two phosphorylation sitesof IKKb by binding to the KD of IKKb, interrupting the upstreamkinase to interact with Ser177 or Ser181. Therefore, IKKb is unableto be phosphorylated and remains in the unactivated state.Taken together, it is reasonable to conclude that 2C binds to
the KD of IKKb through its N-terminal region and inhibits IKKbphosphorylation. The binding between 2C and IKKb is mostlikely reversible. In resting cells without TNF-a stimulation, 2Cmay interact competitively with IKKb, and 2C-unbound IKKbcan still be phosphated by upstream kinase. This may be thereason that 2C has a partial inhibitory effect on IKKb phosphor-ylation and NF-kB activation. Recent studies indicate that hostproteins NLRC5 and CUEDC2 can bind to IKK and negativelyregulate NF-kB activation (65, 66). NLRC5 competes with IKKgfor IKKb and inhibits phosphorylation and kinase activity (66).CUEDC2 acts as an adaptor protein to target IKK for de-phosphorylation and inactivation by recruiting protein phospha-tase 1 (65). EV71 2C is likely to function as a negative regulatorof IKKb. A partial inhibition of NF-kB may be beneficial to viralreplication, as a complete inhibition of TNF-mediated NF-kB willlead to apoptosis of host cells, which is harmful to EV71 repli-cation (67).Due to the difficulty in manipulating EV71 by using conven-
tional reverse genetic system, we are currently unable to determinethe significance of 2C in viral pathogenesis. Nevertheless, wedemonstrate in this study that 2C protein prevents TNF-a–inducedNF-kB activation, revealing a potential mechanism by whichEV71 inhibits the production of antiviral immune responses. Inconclusion, our observation identifies a novel role of EV71 2C asa viral protein with anti-inflammatory and immunomodulatoryproperties. More recently, it has been reported that another EV71protein 3C inhibits RIG-I–mediated type I IFN response by inter-acting with RIG-I (68). A more detailed understanding of EV71viral proteins involved in counteracting the host immune responsewill provide ideas for developing antiviral strategies.
AcknowledgmentsWe thank Dr. Jonathan Ashwell (National Institutes of Health), Dr. Rao
Anjana (Immune Disease Institute, HarvardMedical School), and Dr. Gang-
min Hur (Chungnam National University) for providing plasmids.
DisclosuresThe authors have no financial conflicts of interest.
References1. Bible, J. M., P. Pantelidis, P. K. Chan, and C. Y. Tong. 2007. Genetic evolution of
enterovirus 71: epidemiological and pathological implications. Rev. Med. Virol.17: 371–379.
2. Palacios, G., and M. S. Oberste. 2005. Enteroviruses as agents of emerging in-fectious diseases. J. Neurovirol. 11: 424–433.
3. Rodrıguez, P. L., and L. Carrasco. 1995. Poliovirus protein 2C contains tworegions involved in RNA binding activity. J. Biol. Chem. 270: 10105–10112.
2210 ENTEROVIRUS 71 2C PROTEIN INHIBITS NF-kB ACTIVATION
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from

4. Rodrıguez, P. L., and L. Carrasco. 1993. Poliovirus protein 2C has ATPase andGTPase activities. J. Biol. Chem. 268: 8105–8110.
5. Echeverri, A., R. Banerjee, and A. Dasgupta. 1998. Amino-terminal region ofpoliovirus 2C protein is sufficient for membrane binding. Virus Res. 54: 217–223.
6. Echeverri, A. C., and A. Dasgupta. 1995. Amino terminal regions of poliovirus2C protein mediate membrane binding. Virology 208: 540–553.
7. Banerjee, R., A. Echeverri, and A. Dasgupta. 1997. Poliovirus-encoded 2Cpolypeptide specifically binds to the 39-terminal sequences of viral negative-strand RNA. J. Virol. 71: 9570–9578.
8. Racaniello, V. R. 2007. Picornaviridae: the viruses and their replication. In FieldsVirology, 5th Ed. D. M. Knipe, and P. M. Howley, eds., eds. Lippincott Williams& Wilkins, Philadelphia, p. 796–839.
9. Tang, W. F., S. Y. Yang, B. W. Wu, J. R. Jheng, Y. L. Chen, C. H. Shih, K. H. Lin,H. C. Lai, P. Tang, and J. T. Horng. 2007. Reticulon 3 binds the 2C protein ofenterovirus 71 and is required for viral replication. J. Biol. Chem. 282: 5888–5898.
10. Ghosh, S., M. J. May, and E. B. Kopp. 1998. NF-kappa B and Rel proteins:evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol.16: 225–260.
11. Campbell, K. J., and N. D. Perkins. 2006. Regulation of NF-kappaB function.Biochem. Soc. Symp. 73: 165–180.
12. Baeuerle, P. A., and D. Baltimore. 1996. NF-kappa B: ten years after. Cell 87:13–20.
13. Barkett, M., and T. D. Gilmore. 1999. Control of apoptosis by Rel/NF-kappaBtranscription factors. Oncogene 18: 6910–6924.
14. Pahl, H. L. 1999. Activators and target genes of Rel/NF-kappaB transcriptionfactors. Oncogene 18: 6853–6866.
15. Hiscott, J., T. L. Nguyen, M. Arguello, P. Nakhaei, and S. Paz. 2006. Manipu-lation of the nuclear factor-kappaB pathway and the innate immune response byviruses. Oncogene 25: 6844–6867.
16. Unterholzner, L., and A. G. Bowie. 2008. The interplay between viruses andinnate immune signaling: recent insights and therapeutic opportunities. Biochem.Pharmacol. 75: 589–602.
17. Herbein, G., and W. A. O’Brien. 2000. Tumor necrosis factor (TNF)-alpha andTNF receptors in viral pathogenesis. Proc. Soc. Exp. Biol. Med. 223: 241–257.
18. Chen, G., and D. V. Goeddel. 2002. TNF-R1 signaling: a beautiful pathway.Science 296: 1634–1635.
19. Scheidereit, C. 2006. IkappaB kinase complexes: gateways to NF-kappaB acti-vation and transcription. Oncogene 25: 6685–6705.
20. DiDonato, J. A., M. Hayakawa, D. M. Rothwarf, E. Zandi, and M. Karin. 1997.A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388: 548–554.
21. Mercurio, F., H. Y. Zhu, B. W. Murray, A. Shevchenko, B. L. Bennett, J. W. Li,D. B. Young, M. Barbosa, M. Mann, A. Manning, and A. Rao. 1997. IKK-1 andIKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation.Science 278: 860–866.
22. Hayden, M. S., and S. Ghosh. 2004. Signaling to NF-kappaB. Genes Dev. 18:2195–2224.
23. Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: thecontrol of NF-[kappa]B activity. Annu. Rev. Immunol. 18: 621–663.
24. Chen, F., L. M. Demers, and X. Shi. 2002. Upstream signal transduction of NF-kappaB activation. Curr. Drug Targets Inflamm. Allergy 1: 137–149.
25. Zaragoza, C., M. Saura, E. Y. Padalko, E. Lopez-Rivera, T. R. Lizarbe, S. Lamas,and C. J. Lowenstein. 2006. Viral protease cleavage of inhibitor of kappaBalphatriggers host cell apoptosis. Proc. Natl. Acad. Sci. USA 103: 19051–19056.
26. de Los Santos, T., F. Diaz-San Segundo, and M. J. Grubman. 2007. Degradationof nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol.81: 12803–12815.
27. Lin, T. Y., S. H. Hsia, Y. C. Huang, C. T. Wu, and L. Y. Chang. 2003. Proin-flammatory cytokine reactions in enterovirus 71 infections of the central nervoussystem. Clin. Infect. Dis. 36: 269–274.
28. Lin, T. Y., L. Y. Chang, Y. C. Huang, K. H. Hsu, C. H. Chiu, and K. D. Yang.2002. Different proinflammatory reactions in fatal and non-fatal enterovirus 71infections: implications for early recognition and therapy. Acta Paediatr. 91:632–635.
29. Chen, Z., Y. Sahashi, K. Matsuo, H. Asanuma, H. Takahashi, T. Iwasaki,Y. Suzuki, C. Aizawa, T. Kurata, and S.-i. Tamura. 1998. Comparison of theability of viral protein-expressing plasmid DNAs to protect against influenza.Vaccine 16: 1544–1549.
30. Li, X., Y. Yang, and J. D. Ashwell. 2002. TNF-RII and c-IAP1 mediate ubiq-uitination and degradation of TRAF2. Nature 416: 345–347.
31. Park, K. A., H. S. Byun, M. Won, K.-J. Yang, S. Shin, L. Piao, J. M. Kim,W.-H. Yoon, E. Junn, J. Park, et al. 2007. Sustained activation of protein kinaseC downregulates nuclear factor-kappaB signaling by dissociation of IKK-gamma and Hsp90 complex in human colonic epithelial cells. Carcinogenesis28: 71–80.
32. Chen, C. S., Y. C. Yao, S. C. Lin, Y. P. Lee, Y. F. Wang, J. R. Wang, C. C. Liu,H. Y. Lei, and C. K. Yu. 2007. Retrograde axonal transport: a major transmissionroute of enterovirus 71 in mice. J. Virol. 81: 8996–9003.
33. Schmid, J. A., and A. Birbach. 2008. IkappaB kinase beta (IKKbeta/IKK2/IKBKB): a key molecule in signaling to the transcription factor NF-kappaB.Cytokine Growth Factor Rev. 19: 157–165.
34. Santoro, M. G., A. Rossi, and C. Amici. 2003. NF-kappaB and virus infection:who controls whom. EMBO J. 22: 2552–2560.
35. Taylor, S. L., N. Frias-Staheli, A. Garcıa-Sastre, and C. S. Schmaljohn. 2009.Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits
tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J.Virol. 83: 1271–1279.
36. Shao, R. P., M. C. T. Hu, B. P. Zhou, S. Y. Lin, P. J. Chiao, R. H. von Lindern,B. Spohn, and M. C. Hung. 1999. E1A sensitizes cells to tumor necrosis factor-induced apoptosis through inhibition of IkappaB kinases and nuclear factorkappaB activities. J. Biol. Chem. 274: 21495–21498.
37. Holloway, G., T. T. Truong, and B. S. Coulson. 2009. Rotavirus antagonizescellular antiviral responses by inhibiting the nuclear accumulation of STAT1,STAT2, and NF-kappaB. J. Virol. 83: 4942–4951.
38. Graff, J. W., K. Ettayebi, and M. E. Hardy. 2009. Rotavirus NSP1 inhibitsNFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5: e1000280.
39. Tung, W. H., C. C. Sun, H. L. Hsieh, S. W. Wang, J. T. Horng, and C. M. Yang.2007. EV71 induces VCAM-1 expression via PDGF receptor, PI3-K/Akt, p38MAPK, JNK and NF-kappaB in vascular smooth muscle cells. Cell. Signal. 19:2127–2137.
40. Tung, W. H., H. L. Hsieh, and C. M. Yang. 2010. Enterovirus 71 induces COX-2expression via MAPKs, NF-kappaB, and AP-1 in SK-N-SH cells: role of PGE(2)in viral replication. Cell. Signal. 22: 234–246.
41. Tung, W. H., I. T. Lee, H. L. Hsieh, and C. M. Yang. 2010. EV71 induces COX-2expression via c-Src/PDGFR/PI3K/Akt/p42/p44 MAPK/AP-1 and NF-kappaB inrat brain astrocytes. J. Cell. Physiol. 224: 376–386.
42. Neznanov, N., A. Kondratova, K. M. Chumakov, B. Angres, B. Zhumabayeva,V. I. Agol, and A. V. Gudkov. 2001. Poliovirus protein 3A inhibits tumor ne-crosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from thecell surface. J. Virol. 75: 10409–10420.
43. Li, J. P., and D. Baltimore. 1990. An intragenic revertant of a poliovirus 2Cmutant has an uncoating defect. J. Virol. 64: 1102–1107.
44. Cho, M. W., N. Teterina, D. Egger, K. Bienz, and E. Ehrenfeld. 1994. Membranerearrangement and vesicle induction by recombinant poliovirus 2C and 2BC inhuman cells. Virology 202: 129–145.
45. Wimmer, E., C. U. Hellen, and X. Cao. 1993. Genetics of poliovirus. Annu. Rev.Genet. 27: 353–436.
46. Vance, L. M., N. Moscufo, M. Chow, and B. A. Heinz. 1997. Poliovirus 2Cregion functions during encapsidation of viral RNA. J. Virol. 71: 8759–8765.
47. Liu, Y., C. L. Wang, S. Mueller, A. V. Paul, E. Wimmer, and P. Jiang. 2010.Direct interaction between two viral proteins, the nonstructural protein 2C andthe capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog.6: e1001066.
48. Liu, J., T. Wei, and J. Kwang. 2004. Avian encephalomyelitis virus nonstructuralprotein 2C induces apoptosis by activating cytochrome c/caspase-9 pathway.Virology 318: 169–182.
49. Karin, M. 2005. The IkappaB kinase (IKK) complex as a critical regulator ofimmune responses. Int. Congr. Ser. 1285: 97–103.
50. Perkins, N. D. 2006. Post-translational modifications regulating the activity andfunction of the nuclear factor kappa B pathway. Oncogene 25: 6717–6730.
51. Chen, Z. J. 2005. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol.7: 758–765.
52. Krappmann, D., and C. Scheidereit. 2005. A pervasive role of ubiquitin conju-gation in activation and termination of IkappaB kinase pathways. EMBO Rep. 6:321–326.
53. Choi, S. H., K. J. Park, B. Y. Ahn, G. Jung, M. M. Lai, and S. B. Hwang. 2006.Hepatitis C virus nonstructural 5B protein regulates tumor necrosis factor alphasignaling through effects on cellular IkappaB kinase. Mol. Cell. Biol. 26: 3048–3059.
54. Chen, R. A. J., G. Ryzhakov, S. Cooray, F. Randow, and G. L. Smith. 2008.Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoSPathog. 4: e22.
55. Nichols, D. B., and J. L. Shisler. 2006. The MC160 protein expressed by thedermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosisfactor alpha-induced NF-kappaB activation via inhibition of I kappa kinasecomplex formation. J. Virol. 80: 578–586.
56. Nichols, D. B., and J. L. Shisler. 2009. Poxvirus MC160 protein utilizes multiplemechanisms to inhibit NF-kappaB activation mediated via components of thetumor necrosis factor receptor 1 signal transduction pathway. J. Virol. 83: 3162–3174.
57. DiPerna, G., J. Stack, A. G. Bowie, A. Boyd, G. Kotwal, Z. Zhang, S. Arvikar,E. Latz, K. A. Fitzgerald, and W. L. Marshall. 2004. Poxvirus protein N1Ltargets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by thetumor necrosis factor superfamily of receptors, and inhibits NF-kappaB andIRF3 signaling by Toll-like receptors. J. Biol. Chem. 279: 36570–36578.
58. Yin, M. J., Y. Yamamoto, and R. B. Gaynor. 1998. The anti-inflammatory agentsaspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396:77–80.
59. Yamamoto, Y., M. J. Yin, K. M. Lin, and R. B. Gaynor. 1999. Sulindac inhibitsactivation of the NF-kappaB pathway. J. Biol. Chem. 274: 27307–27314.
60. Carcamo, J. M., A. Pedraza, O. Borquez-Ojeda, B. Zhang, R. Sanchez, andD. W. Golde. 2004. Vitamin C is a kinase inhibitor: dehydroascorbic acidinhibits IkappaBalpha kinase beta. Mol. Cell. Biol. 24: 6645–6652.
61. Jeon, K. I., J.-Y. Jeong, and D.-M. Jue. 2000. Thiol-reactive metal compoundsinhibit NF-kappa B activation by blocking I kappa B kinase. J. Immunol. 164:5981–5989.
62. Kwok, B. H., B. Koh, M. I. Ndubuisi, M. Elofsson, and C. M. Crews. 2001. Theanti-inflammatory natural product parthenolide from the medicinal herb Fever-few directly binds to and inhibits IkappaB kinase. Chem. Biol. 8: 759–766.
63. Reynaert, N. L., A. van der Vliet, A. S. Guala, T. McGovern, M. Hristova,C. Pantano, N. H. Heintz, J. Heim, Y. S. Ho, D. E. Matthews, et al. 2006.
The Journal of Immunology 2211
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from

Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA103: 13086–13091.
64. Xu, G., Y.-C. Lo, Q. Li, G. Napolitano, X. Wu, X. Jiang, M. Dreano, M. Karin,and H. Wu. 2011. Crystal structure of inhibitor of kB kinase b. Nature 472: 325–330.
65. Li, H.-Y., H. Liu, C.-H. Wang, J.-Y. Zhang, J.-H. Man, Y.-F. Gao, P.-J. Zhang,W.-H. Li, J. Zhao, X. Pan, et al. 2008. Deactivation of the kinase IKK byCUEDC2 through recruitment of the phosphatase PP1. Nat. Immunol. 9: 533–541.
66. Cui, J., L. Zhu, X. Xia, H. Y. Wang, X. Legras, J. Hong, J. Ji, P. Shen, S. Zheng,Z. J. Chen, and R. F. Wang. 2010. NLRC5 negatively regulates the NF-kappaBand type I interferon signaling pathways. Cell 141: 483–496.
67. Liu, J., and A. Lin. 2007. Wiring the cell signaling circuitry by the NF-kappa Band JNK1 crosstalk and its applications in human diseases. Oncogene 26: 3267–3278.
68. Lei, X., X. Liu, Y. Ma, Z. Sun, Y. Yang, Q. Jin, B. He, and J. Wang. 2010. The 3Cprotein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated in-terferon regulatory factor 3 activation and type I interferon responses. J. Virol.84: 8051–8061.
2212 ENTEROVIRUS 71 2C PROTEIN INHIBITS NF-kB ACTIVATION
on August 21, 2011
ww
w.jim
munol.org
Dow
nloaded from