influence of the molecular stacking pattern on the excited
TRANSCRIPT

CHINESE JOURNAL OF CHEMICAL PHYSICS AUGUST 3, 2021
ARTICLE
Influence of the Molecular Stacking Pattern on the Excited State
Dynamics of Copper Phthalocyanine Films
Meng Lia, Wen-hui Lia, Yu-jie Hua, Jing Lengb, Wen-ming Tianb, Chun-yi Zhaob, Jun-xue Liub,
Rong-rong Cuib, Sheng-ye Jinb, Chuan-hui Chenga∗, Shu-lin Conga
a. School of Physics, Dalian University of Technology, Dalian 116024, Chinab. State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, ChineseAcademy of Sciences, Dalian 116023, China
(Dated: Received on March 22, 2021; Accepted on July 5, 2021)
A better understanding of the photophysical pro-
cesses occurring within organic semiconductors is
important for designing and fabricating organic so-
lar cells (OSCs). Copper phthalocyanine (CuPc)
is a typical electron acceptor. In this work, the
triplet exciton lifetime is prolonged by altering
the molecular stacking pattern of the CuPc film.
For CuPc thin films, the excited state decays are
mainly determined by the triplet-triplet annihila-
tion process. The ultrafast transient absorption
measurements indicate that the primary annihila-
tion mechanism is one-dimensional exciton diffu-
sion collision destruction. The decay kinetics show
a clearly time-dependent annihilation rate constant
with γ∝t−1/2. Annihilation rate constants are deter-
mined to be γ0=(2.87±0.02)×10−20 cm3·s−1/2 and
(1.42±0.02)×10−20 cm3·s−1/2 for upright and lying-
down configurations, respectively. Compared to the CuPc thin film with an upright config-
uration, the thin film with a lying-down configuration shows a longer exciton lifetime and a
higher absorbance, which are beneficial for OSCs. The results in this work have important
implications on the design and mechanistic understanding of organic optoelectronic devices.
Key words: CuPc, Photophysics, Excited state, Triplet-triplet annihilation, Organic solar
cell
I. INTRODUCTION
The energy problem is a serious modern concern. In
recent years, organic solar cells (OSCs) have attracted
considerable attention owing to their potential to pro-
vide a low-cost, light-weight, and flexible strategy for
solar energy conversion [1, 2]. A better understanding of
∗Author to whom correspondence should be addressed. E-mail:
the photophysical processes within such cells is impor-
tant for designing new materials and devices. Ultrafast
pump-probe techniques are powerful tools for studying
excited-state dynamics in OSC systems [3–6]. Phthalo-
cyanines are archetypical electron donors which are de-
posited by vacuum evaporation to form films for use
in such cells [7–10]. The molecular stacking configura-
tion and crystal structure can markedly affect material
characteristics, such as light absorption, charge trans-
port, exciton diffusion, and molecular energy levels [11–
14]. In our previous work, we considerably improved the
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society

2 Chin. J. Chem. Phys. Meng Li et al.
device performance by altering the molecular stacking
configuration of copper phthalocyanine (CuPc) from an
upright to a lying-down configuration in OSCs based on
the heterojunction of CuPc/C60 [15]. Herein, we report
a study of the excited-state dynamics of the CuPc films
with lying-down and upright molecular stacking con-
figuration using an ultrafast transient absorption (TA)
spectroscopy. Our results suggest that compared with
the upright configuration, the excited state lifetime is
longer, and the absorbance is higher for the lying-down
configuration, which are of benefit for OSCs. We also
elucidate the excited state decay mechanism.
II. EXPERIMENTS
The quartz substrates were ultrasonicated in ace-
tone, alcohol, and deionized water in sequence, and
then blown dry with pure N2 gas. CuPc (sublimed
grade, 99.0%) and CuI (99.9%) were purchased from
Nichem and Aldrich, respectively. All the films were
prepared by the vacuum evaporation under a base pres-
sure of ≤5×10−4 Pa. The film thicknesses were mon-
itored online by a quartz-crystal microbalance. Atom
force microscopy (AFM) morphologies were measured
on a Bruker Dimension Icon. Steady state absorp-
tion spectra were measured on a Shimadzu UV3600
spectrophotometer. TA spectra (Time-Tech Spec-
tra, Femto-TA100) were measured using previously de-
scribed methods [16]. The full-width at half-maximum
of the pump pulse was ∼80 fs, and the time resolution
was ∼150 fs. All the TA spectra were measured with a
700-nm pump pulse. The pump beam was focused to a
diameter of∼300 µm. The sample was moved with a ve-
locity of 0.3 mm/s during the TA measurements. There
is no evident difference in the dynamics of the samples
with and without an encapsule, and all the measure-
ments were performed in ambient atmosphere at room
temperature without encapsulation.
III. RESULTS AND DISCUSSION
We prepared two kinds of CuPc films, one with a
lying-down and the other with an upright molecular
stacking configuration, which were determined by X-ray
diffraction in our previous study [15]. The schematic
diagrams of the molecular stacking configuration in the
film and energy levels are shown in FIG. 1. The stacking
configuration was converted from upright to lying-down
by introducing a 3.0-nm thick CuI buffer layer [15].
FIG. 1 Molecular structure of CuPc. Schematic diagramsof energy levels and molecular stacking configurations. Theπ-stacking direction is given.
FIG. 2 AFM surface morphologies of the films with (a) anupright and (b) a lying-down configuration.
AFM surface morphologies of the films are given in
FIG. 2. It can be seen that the surface morphologies
are very different from the films with lying-down and
upright configurations. The crystal grains can be seen
clearly for the lying-down configuration. The absorp-
tion spectra of CuI and CuPc films with different stack-
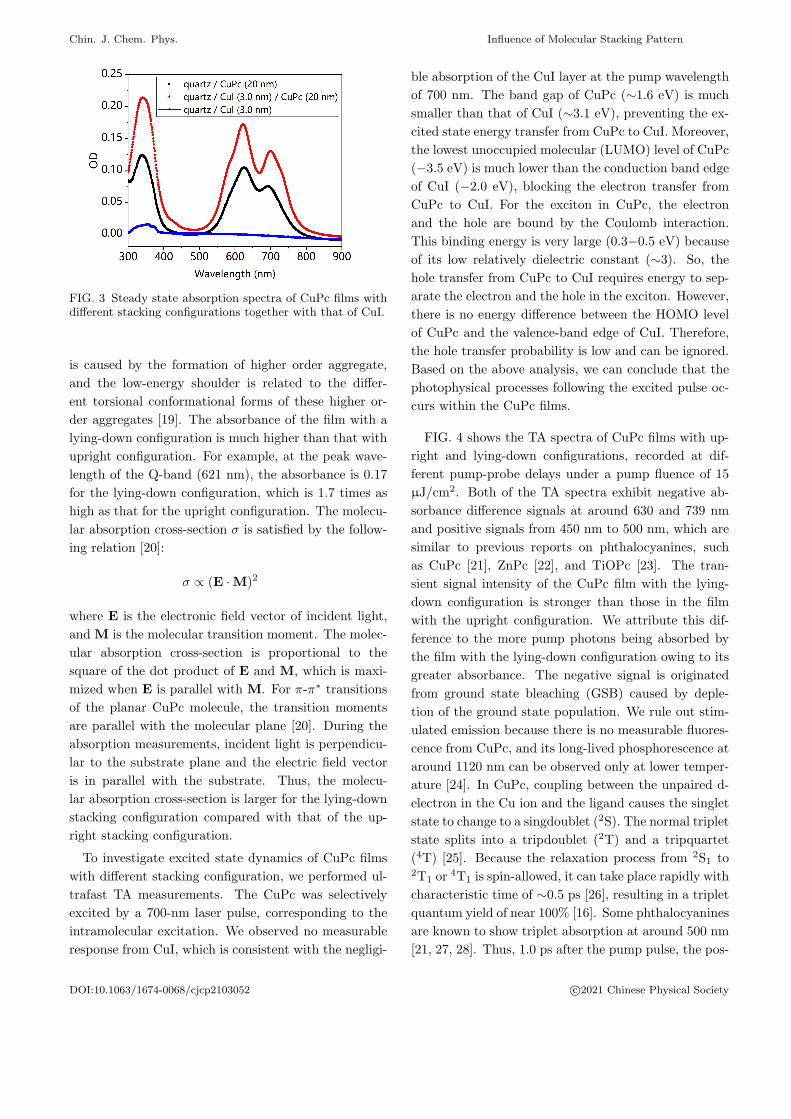
ing configurations are shown in FIG. 3. Both spectra
of the CuPc films showed a Soret-band at around 350
nm and a Q-band from 550 nm to 800 nm. The Soret-
band has been assigned to the transition of S0→S2, and
the Q-band corresponds to the transition of S0→S1 [17].
Here, S0, S1, and S2 are the ground, the first, and the
second excited singlet states. There are two peaks in
the Q-band. The longer wavelength feature arises from
intramolecular excitations, and the shorter wavelength
peak is from intermolecular charge transfer excitations
in molecular aggregates [18]. Furthermore, there are
two shoulders located at the low- and high-energy ab-
sorption edge of the Q-band. The high-energy shoulder
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society
Chin. J. Chem. Phys. Influence of Molecular Stacking Pattern
FIG. 3 Steady state absorption spectra of CuPc films withdifferent stacking configurations together with that of CuI.
is caused by the formation of higher order aggregate,
and the low-energy shoulder is related to the differ-
ent torsional conformational forms of these higher or-
der aggregates [19]. The absorbance of the film with a
lying-down configuration is much higher than that with
upright configuration. For example, at the peak wave-
length of the Q-band (621 nm), the absorbance is 0.17
for the lying-down configuration, which is 1.7 times as
high as that for the upright configuration. The molecu-
lar absorption cross-section σ is satisfied by the follow-
ing relation [20]:
σ ∝ (E ·M)2
where E is the electronic field vector of incident light,
and M is the molecular transition moment. The molec-
ular absorption cross-section is proportional to the
square of the dot product of E and M, which is maxi-
mized when E is parallel with M. For π-π∗ transitions
of the planar CuPc molecule, the transition moments
are parallel with the molecular plane [20]. During the
absorption measurements, incident light is perpendicu-
lar to the substrate plane and the electric field vector
is in parallel with the substrate. Thus, the molecu-
lar absorption cross-section is larger for the lying-down
stacking configuration compared with that of the up-
right stacking configuration.
To investigate excited state dynamics of CuPc films
with different stacking configuration, we performed ul-
trafast TA measurements. The CuPc was selectively
excited by a 700-nm laser pulse, corresponding to the
intramolecular excitation. We observed no measurable
response from CuI, which is consistent with the negligi-
ble absorption of the CuI layer at the pump wavelength
of 700 nm. The band gap of CuPc (∼1.6 eV) is much
smaller than that of CuI (∼3.1 eV), preventing the ex-
cited state energy transfer from CuPc to CuI. Moreover,
the lowest unoccupied molecular (LUMO) level of CuPc
(−3.5 eV) is much lower than the conduction band edge
of CuI (−2.0 eV), blocking the electron transfer from
CuPc to CuI. For the exciton in CuPc, the electron
and the hole are bound by the Coulomb interaction.
This binding energy is very large (0.3−0.5 eV) because
of its low relatively dielectric constant (∼3). So, the
hole transfer from CuPc to CuI requires energy to sep-
arate the electron and the hole in the exciton. However,
there is no energy difference between the HOMO level
of CuPc and the valence-band edge of CuI. Therefore,
the hole transfer probability is low and can be ignored.
Based on the above analysis, we can conclude that the
photophysical processes following the excited pulse oc-
curs within the CuPc films.
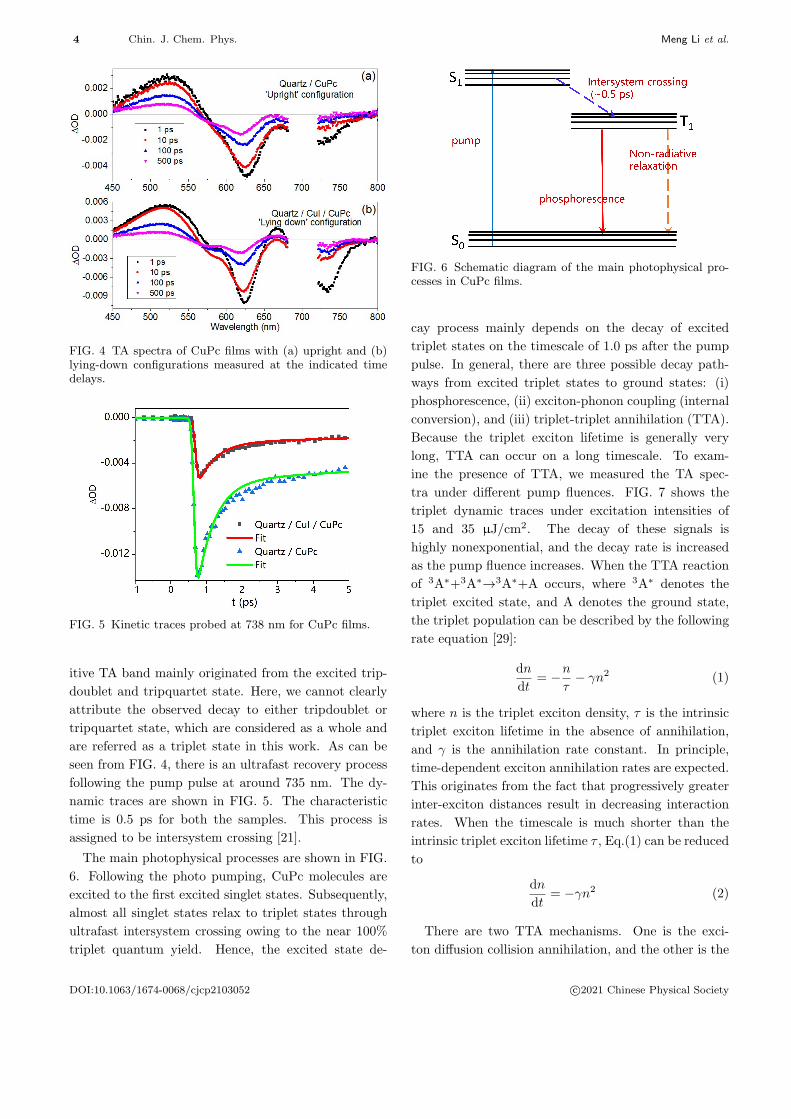
FIG. 4 shows the TA spectra of CuPc films with up-
right and lying-down configurations, recorded at dif-
ferent pump-probe delays under a pump fluence of 15
µJ/cm2. Both of the TA spectra exhibit negative ab-
sorbance difference signals at around 630 and 739 nm
and positive signals from 450 nm to 500 nm, which are
similar to previous reports on phthalocyanines, such
as CuPc [21], ZnPc [22], and TiOPc [23]. The tran-
sient signal intensity of the CuPc film with the lying-
down configuration is stronger than those in the film
with the upright configuration. We attribute this dif-
ference to the more pump photons being absorbed by
the film with the lying-down configuration owing to its
greater absorbance. The negative signal is originated
from ground state bleaching (GSB) caused by deple-
tion of the ground state population. We rule out stim-
ulated emission because there is no measurable fluores-
cence from CuPc, and its long-lived phosphorescence at
around 1120 nm can be observed only at lower temper-
ature [24]. In CuPc, coupling between the unpaired d-
electron in the Cu ion and the ligand causes the singlet
state to change to a singdoublet (2S). The normal triplet
state splits into a tripdoublet (2T) and a tripquartet
(4T) [25]. Because the relaxation process from 2S1 to2T1 or
4T1 is spin-allowed, it can take place rapidly with
characteristic time of ∼0.5 ps [26], resulting in a triplet
quantum yield of near 100% [16]. Some phthalocyanines
are known to show triplet absorption at around 500 nm
[21, 27, 28]. Thus, 1.0 ps after the pump pulse, the pos-
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society
4 Chin. J. Chem. Phys. Meng Li et al.
FIG. 4 TA spectra of CuPc films with (a) upright and (b)lying-down configurations measured at the indicated timedelays.
FIG. 5 Kinetic traces probed at 738 nm for CuPc films.
itive TA band mainly originated from the excited trip-
doublet and tripquartet state. Here, we cannot clearly
attribute the observed decay to either tripdoublet or
tripquartet state, which are considered as a whole and
are referred as a triplet state in this work. As can be
seen from FIG. 4, there is an ultrafast recovery process
following the pump pulse at around 735 nm. The dy-
namic traces are shown in FIG. 5. The characteristic
time is 0.5 ps for both the samples. This process is
assigned to be intersystem crossing [21].
The main photophysical processes are shown in FIG.
6. Following the photo pumping, CuPc molecules are
excited to the first excited singlet states. Subsequently,
almost all singlet states relax to triplet states through
ultrafast intersystem crossing owing to the near 100%
triplet quantum yield. Hence, the excited state de-
FIG. 6 Schematic diagram of the main photophysical pro-cesses in CuPc films.
cay process mainly depends on the decay of excited
triplet states on the timescale of 1.0 ps after the pump
pulse. In general, there are three possible decay path-
ways from excited triplet states to ground states: (i)
phosphorescence, (ii) exciton-phonon coupling (internal
conversion), and (iii) triplet-triplet annihilation (TTA).
Because the triplet exciton lifetime is generally very
long, TTA can occur on a long timescale. To exam-
ine the presence of TTA, we measured the TA spec-
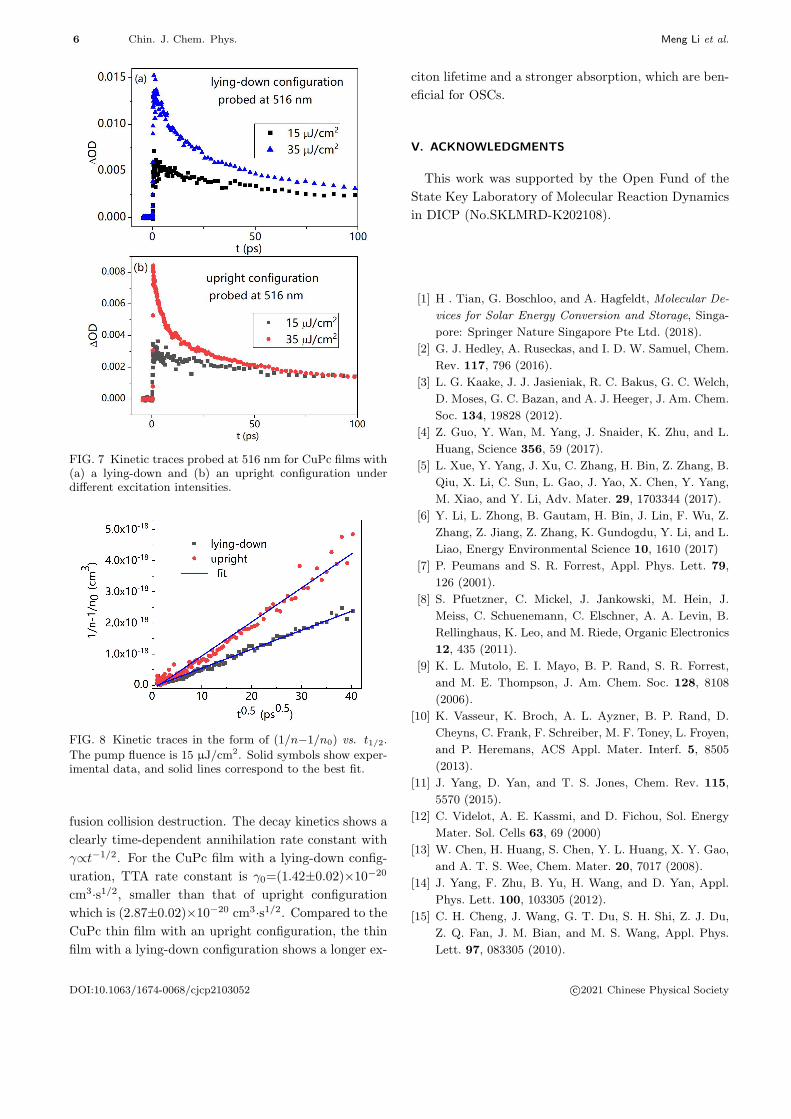
tra under different pump fluences. FIG. 7 shows the
triplet dynamic traces under excitation intensities of
15 and 35 µJ/cm2. The decay of these signals is
highly nonexponential, and the decay rate is increased
as the pump fluence increases. When the TTA reaction
of 3A∗+3A∗→3A∗+A occurs, where 3A∗ denotes the
triplet excited state, and A denotes the ground state,
the triplet population can be described by the following
rate equation [29]:
dn
dt= −n
τ− γn2 (1)
where n is the triplet exciton density, τ is the intrinsic
triplet exciton lifetime in the absence of annihilation,
and γ is the annihilation rate constant. In principle,
time-dependent exciton annihilation rates are expected.
This originates from the fact that progressively greater
inter-exciton distances result in decreasing interaction
rates. When the timescale is much shorter than the
intrinsic triplet exciton lifetime τ , Eq.(1) can be reduced
to
dn
dt= −γn2 (2)
There are two TTA mechanisms. One is the exci-
ton diffusion collision annihilation, and the other is the
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society

Chin. J. Chem. Phys. Influence of Molecular Stacking Pattern
static annihilation via Forster long-range dipole-dipole
interaction. Forster energy transfer requires that the
transition of the energy donor is allowed. However, the
transition of 3A∗→A is spin-forbidden for CuPc. There-
fore, we can conclude that the static annihilation mech-
anism is not important. Our conclusion consists with
that the energy transport in CuPc film is dominated
by short-range Dexter mechanism [21]. In contrast to
the triplet exciton in CuPc, the singlet exciton annihi-
lation in polycrystalline film of H2Pc is dominated by
the static annihilation via Forster energy transfer mech-
anism [38]. Since the static annihilation mechanism is
ruled out, the diffusion collision annihilation mechanism
is dominant in the TTA process in CuPc films. In this
case, annihilation rate constant can be described as,
for one-dimensional diffusion:
γ1D(t) =
√2D
πt(3)
for three-dimensional diffusion [30, 31]:
γ3D(t) = 4πRaD
(1 +
Ra√2πDt
)(4)
where D is the diffusion coefficient, Ra is the critical
distance at which exciton annihilation reaction takes
place, and t is the time. In general Ra is assumed to be
the separation of adjacent molecules. When t is much
larger than Ra2/2πD, the Eq.(4) can be reduced to
γ3D = 4πRaD (5)
In the case of one-dimensional diffusion, annihilation
rate Eq.(2) can be rewritten as
dn
dt= −γ0t
−1/2n2 (6)
where γ0 is a constant. Integration of Eq.(6) yields an
expression of the time-dependent exciton density:
n(t) =
(2γ0
√t+
1
n0
)−1
(7)
where n0 is the initial exciton concentration at t=0. If
we plot the kinetic traces in the form of (1/n−1/n0) vs.
t1/2, kinetics obeying rate equation (Eq.(6)) will yield a
straight line with a slope of 2γ0. The initial triplet den-
sities n0 for the lying-down and upright configurations
were estimated to be 6.85×1018 cm−3 and 3.95×1018
cm−3 based on their absorbance values of 0.13 and
0.07, respectively. FIG. 8 shows the kinetic traces in
the form of (1/n−1/n0) vs. t1/2, which yield straight
lines for both the lying-down and the upright configu-
rations. The results suggest that one-dimensional dif-
fusion collision annihilation is the dominant mechanism
in CuPc films. It is similar to exciton annihilation in
polycrystalline film of H2Pc, which shows a clearly time-
dependent annihilation rate constant with γ∝t−1/2 [38].
The kinetic traces were fitted to Eq.(7), and solid lines
are the fitting results as shown in FIG. 8. For the
CuPc film with a lying-down configuration, TTA rate
constant is γ0=(1.42±0.02)×10−20 cm3·s−1/2, which is
smaller than that for upright configuration of (2.87±0.02)×10−20 cm3·s−1/2. The results indicate that the
triplet exciton lifetime is longer for the CuPc film with
a lying-down configuration, which is of benefit for OSCs.
In our experiment the film thickness is 20 nm, which
is the optimized thickness of our solar cell based on
CuPc/C60 heterojunction [15]. This thin thickness
should thus limit the exciton diffuse along the direction
perpendicular to the substrate. Hence, collision anni-
hilation mainly depends on exciton hopping in parallel
with the substrate. In the anisotropic CuPc film, ex-
citon hopping along the π-π stacking direction is more
favorable owing to the stronger electron coupling, in
agreement with the above conclusion of one-dimensional
diffusion model. In order to elucidate this issue more
clearly, the π-π stacking direction is given in FIG. 1. As
shown in FIG. 1, for the CuPc film with a lying-down
configuration, exciton hopping is more difficult in the
direction parallel with the substrate, leading to a lower
collision annihilation probability and a longer exciton
lifetime, which are desirable for OSCs. Furthermore, π-
π stacking in direction perpendicular to the substrate is
in favor of the carrier collection and the exciton diffu-
sion to the interface of heterojunction. Therefore, the
CuPc film with a lying-down configuration is more suit-
able for OSCs.
IV. CONCLUSION
In summary, we investigate the photophysical pro-
cesses in CuPc films with lying-down and upright molec-
ular stacking configurations. The absorbance of the film
with a lying-down configuration is much higher than
that of the film with an upright configuration. The
ultrafast TA measurements indicate that the primary
annihilation mechanism is one-dimensional exciton dif-
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society
6 Chin. J. Chem. Phys. Meng Li et al.
FIG. 7 Kinetic traces probed at 516 nm for CuPc films with(a) a lying-down and (b) an upright configuration underdifferent excitation intensities.
FIG. 8 Kinetic traces in the form of (1/n−1/n0) vs. t1/2.
The pump fluence is 15 µJ/cm2. Solid symbols show exper-imental data, and solid lines correspond to the best fit.
fusion collision destruction. The decay kinetics shows a
clearly time-dependent annihilation rate constant with
γ∝t−1/2. For the CuPc film with a lying-down config-
uration, TTA rate constant is γ0=(1.42±0.02)×10−20
cm3·s1/2, smaller than that of upright configuration
which is (2.87±0.02)×10−20 cm3·s1/2. Compared to the
CuPc thin film with an upright configuration, the thin
film with a lying-down configuration shows a longer ex-
citon lifetime and a stronger absorption, which are ben-
eficial for OSCs.
V. ACKNOWLEDGMENTS
This work was supported by the Open Fund of the
State Key Laboratory of Molecular Reaction Dynamics
in DICP (No.SKLMRD-K202108).
[1] H . Tian, G. Boschloo, and A. Hagfeldt, Molecular De-
vices for Solar Energy Conversion and Storage, Singa-
pore: Springer Nature Singapore Pte Ltd. (2018).
[2] G. J. Hedley, A. Ruseckas, and I. D. W. Samuel, Chem.
Rev. 117, 796 (2016).
[3] L. G. Kaake, J. J. Jasieniak, R. C. Bakus, G. C. Welch,
D. Moses, G. C. Bazan, and A. J. Heeger, J. Am. Chem.
Soc. 134, 19828 (2012).
[4] Z. Guo, Y. Wan, M. Yang, J. Snaider, K. Zhu, and L.
Huang, Science 356, 59 (2017).
[5] L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z. Zhang, B.
Qiu, X. Li, C. Sun, L. Gao, J. Yao, X. Chen, Y. Yang,
M. Xiao, and Y. Li, Adv. Mater. 29, 1703344 (2017).
[6] Y. Li, L. Zhong, B. Gautam, H. Bin, J. Lin, F. Wu, Z.
Zhang, Z. Jiang, Z. Zhang, K. Gundogdu, Y. Li, and L.
Liao, Energy Environmental Science 10, 1610 (2017)
[7] P. Peumans and S. R. Forrest, Appl. Phys. Lett. 79,
126 (2001).
[8] S. Pfuetzner, C. Mickel, J. Jankowski, M. Hein, J.
Meiss, C. Schuenemann, C. Elschner, A. A. Levin, B.
Rellinghaus, K. Leo, and M. Riede, Organic Electronics
12, 435 (2011).
[9] K. L. Mutolo, E. I. Mayo, B. P. Rand, S. R. Forrest,
and M. E. Thompson, J. Am. Chem. Soc. 128, 8108
(2006).
[10] K. Vasseur, K. Broch, A. L. Ayzner, B. P. Rand, D.
Cheyns, C. Frank, F. Schreiber, M. F. Toney, L. Froyen,
and P. Heremans, ACS Appl. Mater. Interf. 5, 8505
(2013).
[11] J. Yang, D. Yan, and T. S. Jones, Chem. Rev. 115,
5570 (2015).
[12] C. Videlot, A. E. Kassmi, and D. Fichou, Sol. Energy
Mater. Sol. Cells 63, 69 (2000)
[13] W. Chen, H. Huang, S. Chen, Y. L. Huang, X. Y. Gao,
and A. T. S. Wee, Chem. Mater. 20, 7017 (2008).
[14] J. Yang, F. Zhu, B. Yu, H. Wang, and D. Yan, Appl.
Phys. Lett. 100, 103305 (2012).
[15] C. H. Cheng, J. Wang, G. T. Du, S. H. Shi, Z. J. Du,
Z. Q. Fan, J. M. Bian, and M. S. Wang, Appl. Phys.
Lett. 97, 083305 (2010).
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society

Chin. J. Chem. Phys. Influence of Molecular Stacking Pattern
[16] J. Liu, J. Leng, K. Wu, J. Zhang, and S. Jin, J. Am.
Chem. Soc. 139, 1432 (2017).
[17] A. W. Snow and W. R. Barger, Phthalocyanine Proper-
ties and Applications, C. C. Leznoff and A. B. P. Lever
Ed., New York: Wiley (1989).
[18] J. H. Sharp and M. Abkowitz, J. Phys. Chem. 77, 481
(1973).
[19] T. Gunaratne, V. O. Kennedy, M. E. Kenney, and M.
A. J. Rodgers, J. Physical Chem. A 108, 2576 (2004).
[20] B . Valeur, Molecular Fluorescence: Principles and Ap-
plications, New York: Wiley (2001).
[21] B. W. Caplins, T. K. Mullenbach, R. J. Holmes, and D.
A. Blank, Phys. Chem. Chem. Phys. 18, 11454 (2016).
[22] X. He, G. Zhu, J. Yang, H. Chang, Q. Meng, H. Zhao,
X. Zhou, S. Yue, Z. Wang, J. Shi, L. Gu, D. Yan, and
Y. Weng, Sci. Rep. 5, (2015).
[23] J. Park, O. G. Reid, and G. Rumbles, J. Phys. Chem.
B 119, 7729 (2014).
[24] K. Yoshino, M. Hikida, K. Tatsuno, K. Kaneto, and Y.
Inuishi, J. Phys. Soc. Jpn. 34, 441 (1973).
[25] M. Liao and S. Scheiner, J. Chem. Phys. 114, 9780
(2001).
[26] G. J. Dutton and S. W. Robey, J. Phys. Chem. C 116,
19173 (2012).
[27] B. W. Caplins, T. K. Mullenbach, R. J. Holmes, and D.
A. Blank, J. Phys. Chem. C 119, 27340 (2015).
[28] A. V. Nikolaitchik and M. A. J. Rodgers, J. Phys.
Chem. A 103, 7597 (1999).
[29] A. Suna, Phys. Rev. B 1, 1716 (1970).
[30] S. Chandrasekhar, Rev. Modern Phys. 15, 1 (1943)
[31] H. Marciniak, X. Li, F. Wurthner, and S. Lochbrunner,
J. Phys. Chem. A 115, 648 (2011).
[32] J. Baumann and M. D. Fayer, J. Chem. Phys. 85, 4087
(1986).
[33] P. Peumans, A. Yakimov, and S. R. Forrest, J. Appl.
Phys. 93, 3693 (2003).
[34] Y. Terao, H. Sasabe, and C. Adachi, Appl. Phys. Lett.
90, 103515 (2007).
[35] M. Ichikawa, Thin Solid Films 527, 239 (2013).
[36] T. Stbinger and W. Brtting, J. Appl. Phys. 90, 3632
(2001).
[37] F. Piersimoni, D. Cheyns, K. Vandewal, J. V. Manca,
and B. P. Rand, J. Phys. Chem. Lett. 3, 2064 (2012).
[38] B. I. Greene and R. R. Millard, Phys. Rev. Lett. 55,
1331 (1985).
DOI:10.1063/1674-0068/cjcp2103052 c⃝2021 Chinese Physical Society