improved scfv anti-hiv-1 p17 binding affinity guided from the theoretical calculation of pairwise...
TRANSCRIPT

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013 Article ID 713585 12 pageshttpdxdoiorg1011552013713585
Research ArticleImproved scFv Anti-HIV-1 p17 Binding Affinity Guided fromthe Theoretical Calculation of Pairwise Decomposition Energiesand Computational Alanine Scanning
Panthip Tue-ngeun1 Kanchanok Kodchakorn1 Piyarat Nimmanpipug12
Narin Lawan1 Sawitree Nangola3 Chatchai Tayapiwatana45 Noorsaadah Abdul Rahman6
Sharifuddin Md Zain6 and Vannajan Sanghiran Lee126
1 Computational Simulation Modelling Laboratory (CSML) Department of Chemistry and Center of Excellence for Innovation inChemistry and Materials Science Research Center Faculty of Science Chiang Mai University Chiang Mai 50200 Thailand
2Thailand Center of Excellence in Physics Commission on Higher Education 328 Sri Ayutthaya Road Bangkok 10400 Thailand3Department of Medical Technology School of Allied Health Sciences University of Phayao Phayao 56000 Thailand4Division of Clinical Immunology Department of Medical Technology Faculty of Associated Medical Sciences Chiang Mai UniversityChiang Mai 50200 Thailand
5 Biomedical Technology Research Unit National Center for Genetic Engineering and Biotechnology National Science andTechnology Development Agency Faculty of Associated Medical Sciences Chiang Mai University Chiang Mai 50200 Thailand
6Department of Chemistry Faculty of Science University of Malaya 50603 Kuala Lumpur Malaysia
Correspondence should be addressed to Chatchai Tayapiwatana asimi002chiangmaiacth andVannajan Sanghiran Lee vannajangmailcom
Received 30 April 2013 Revised 3 September 2013 Accepted 10 September 2013
Academic Editor Carmen Domene
Copyright copy 2013 Panthip Tue-ngeun et al This is an open access article distributed under the Creative Commons AttributionLicense which permits unrestricted use distribution and reproduction in any medium provided the original work is properlycited
Computational approaches have been used to evaluate and define important residues for protein-protein interactions especiallyantigen-antibody complexes In our previous study pairwise decomposition of residue interaction energies of single chain Fv withHIV-1 p17 epitope variants has indicated the key specific residues in the complementary determining regions (CDRs) of scFv anti-p17 In this present investigation in order to determinewhether a specific side chain group of residue inCDRs plays an important rolein bioactivity computational alanine scanning has been appliedMolecular dynamics simulations were donewith several complexesof original scFv anti-p17 and scFv anti-p17mutants with HIV-1 p17 epitope variants with a production run up to 10 ns With thecombination of pairwise decomposition residue interaction and alanine scanning calculations the pointmutation has been initiallyselected at the positionMET100 to improve the residue binding affinityThe calculated docking interaction energy between a singlemutation frommethionine to either arginine or glycine has shown the improved binding affinity contributed from the electrostaticinteraction with the negative favorably interaction energy compared to the wild type Theoretical calculations agreed well with theresults from the peptide ELISA results
1 IntroductionOne of the challenges inmolecular biology consists in impro-ving the structural functional properties or binding activitiesof proteins The antibodies constitute an excellent model totest the potential approaches to this problem because theyconstitute a homogeneous family of proteins and a largeamount of structural and functional data is available The
antigen-binding sites of immunoglobulins are embedded intothe variable heavy and light chain domains (V
119867 V119871) and
are specially separated from the effector function-mediatingregions located in the Fc fragment One type of geneti-cally engineered antibody is the single chain Fv fragment(scFv) Single chain Fv fragments are genetically engineeredpolypeptides that contain a heavy chain variable region (VH)
2 BioMed Research International
linked to a light chain variable region (VL) via a flexiblepeptide linker Each VH and VL domain contains threecomplementarity determining regions (CDRs) CDRs areshort amino acid sequences that vary greatly among antibodymolecules and thus are responsible for generating the greatdiversity of antibody binding specificity The combinationof the CDRs of the VH plus the CDRs of the VL deter-mines the binding specificity of any given antibody Singlechain Fv fragments display the binding specificity mono-valent binding affinity of full-size antibodies and providethe added benefit of relative ease of genetic manipulationand expression Given their advantages of small size andantigen specificity encompassed within a single polypeptidechain scFvs are the most common type of recombinantantibody fragment used for intracellular antibody expression[1]
Several computation approaches have been applied tostudy the protein-ligand interaction by the combination ofcomputational alanine scanning and free energy decomposi-tion methods The molecular mechanics poisson-Boltzmannsurface area (MMPBSA) [2] and molecular mechanics gen-eralized born surface area (MMGBSA) [3] approaches areused to evaluate binding free energy in the computationalalanine scanning Both PB and GB implicit solvent modelswere used to calculate the energy contribution of each residuein the binding free energy on a decomposition basis Theresults from computational alanine scanning and free energydecomposition methods indicated the important residues forbinding 4 of lipopeptide inhibitor to E coli SPase [4] andrecognize the key residues in the ATP binding site of GyrBsubunit fromEscherichia coli boundwith the inhibitors cloro-biocin novobiocin and 51015840-adenylyl-120573-120574-imidodiphosphate[5]
In our previous work scFv anti-p17 was simulated basedon molecular modeling of its homologue structure Theantibody-antigen complex models were generated using themolecular docking that predicted the most favorable bindinginteraction Then the interactions between nine peptideepitopes and the scFv anti-p17 in water were analyzedusing molecular dynamics (MDs) simulation to evaluate thebinding free energy and pairwise decomposition or residue-based energy calculation of complexes in solution using themolecularmechanicspoisson-Boltzmann surface area (MM-PBSA) and molecular mechanicsgeneralized born surfacearea (MM-GBSA) methods The latter analysis can provideinteresting information in terms of electrostatic and van derWaals energies solvation energies and entropic contribu-tions at the binding interface Pairwise decomposition ofresidue interaction energies of the complexes between scFvanti-p17 and its variants have indicated that the specificresidues located in the complementary determining regions(CDRs) of scFv anti-p17 MET100 LYS101 ASN169 HIS228and LEU229 play a crucial role in the effective bindinginteraction [6]
To determine whether the side chain of the specific resi-due in CDRs of scFv anti-p17 plays an important rolein bioactivity computational alanine scanning was carriedout in this study Computational alanine scanning uses asimple free energy function to calculate the effects of alanine
mutations on the binding free energy of a protein-proteincomplex It involves the free energy decomposition involvingMM-PBSA method [2 3 7ndash11] and MM-GBSA method[3 12] to investigate the binding modes in detail at theatomic level and also to estimate protein stabilities [13] Theinput for the computational alanine scanning consisted ofa three-dimensional modeled structure of the scFv anti-p17Potential amino acids that involved changing nonalanineamino acids into alanines will be listed up The relevantchange of more than 1 kcal molminus1 in decomposition energycalculation indicated the important residues involving in thebinding [14]
In this study theoreticalmodeling andmolecular dynam-ics simulations investigation of scFv against HIV-1 epitopeat C-terminal on p17 (scFv anti-p17) has been performedto specify the key residues in the binding From the searchin National Center for Biotechnology Information (NCBI)database 9 different variants of HIV-1 epitope at C-terminalwhich show different binding activities upon binding withscFv were found Computational alanine scanning was uti-lized to investigate the effect of side chain atoms of theresidues in CDR loops of scFv anti-p17With the combinationof the two techniques the selected point mutation to improvethe binding affinity between the scFv anti-p17 and its variantswas identified These residues were mutated to improve thebinding activities guided from statistically preferred substi-tutions observed in buried residues and exposed residuesdue to their solvent exposed area and side chain chargewhich generally correlate with side chain physicochemicalproperties [15] Finally as validated by the experimentalresults we have designed new scFv anti-p17 which bindsbetter with HIV-1 epitope at C-terminal on p17
2 Materials and Methods
21 Molecular Docking The built model reported in theprevious work [6] of primary sequence of the scFv anti-p17protein obtained by Tewari [16] was used in this study Thegeneral docking protocol and potential functions employedin CDOCKER have been described in prior articles [17]In this work docking of the peptides to scFv anti-p17 wasconducted using CDOCKER CDOCKER is a grid-basedmolecular docking method that employs CHARMm Thereceptor is held rigid while the ligands are allowed to flexduring the refinement Ligands are assumed to have alreadybeen roughly docked into the receptor binding site Theactive site pocket of the receptor was found on the CDRs ofscFv anti-p17 by the Discovery Studio 20 (Accelrys SoftwareInc) A site sphere radius of 25 A was set to assign thebinding pocket and the ligand partial charge method forassigning partial charges to the ligands during force fieldassignment was CHARMm Other parameters were set asdefault The lowest docking interaction structure where thepeptide lied in the CDRs similar to the X-ray crystal structureof HIV-1 p24 bound with antibody (PDB ID 1AFV) was thenselected
22 Molecular Dynamics Simulation and Binding Free EnergyCalculations Energy minimization and MD simulations
BioMed Research International 3
were performed using PMEMDCUDA from AMBER12 [18]on GPUs Quadro 2000D produced by NVIDIA which speedup the simulation wall time required to obtain the trajectoryfiles from each simulation MD simulations were carriedout at the molecular mechanics level using the AMBER03force field Antibody-peptide structures were solvated ina cubic box of TIP3P water extending at least 10 A ineach direction from the solute and the cut-off distancewas kept to 12 A to compute the nonbonded interactionsAll simulations were performed under periodic boundaryconditions [19] and long-range electrostatics were treatedby using the particle-mesh-Ewald method [20 21] The timestep was set to 1 fs and the trajectory was recorded every01 ps Prior to MD simulations the systems were relaxedby a series of steepest descent (SD) and conjugated gradient(CG) minimizations MD simulations were performed basedon each of the minimized systems by gradually heating over60 ps from 0 to 310K with the protein atoms fixed usinga force constant of 5 kcalmolA2 Then a 200 ps pressure-constant period (NPT) was applied to obtain an equilibrateddensity of the constrained protein atoms The followingstep was a 40 ps-volume-constant period (NVT) at a forceconstant of 25 kcalmolA2 followed by 100 ps dynamics ata force constant of 125 kcalmolA2 Finally an unrestrainedMD simulation (no force applied on any protein atoms) wasperformed for each fully flexible system in theNVT ensembleat a constant temperature of 310 K for a total simulation timeof 10 ns 500 snapshots were collected from the last 5 ns ofMDsimulations for binding free energy analysis Equilibrationwas monitored by convergence in terms of the temperatureenergy and density of the system and the root-mean-squareddeviations (RMSD) of the backbone atoms compared to thedocking structure
The binding free energies (Δ119866bind)were evaluated accord-ing to the strategy described by Massova and Kollman [2]Below it is summarized the Δ119866bind was determined from thefree energies of the complex protein and peptide accordingto the following equation
Δ119866bind = Δ119866water (complex)
minus [Δ119866water (protein) + Δ119866water (peptide)] (1)
Based on the selected MD snapshots the binding freeenergy for each antibody-peptide system could be estimatedusing MM-PBSA [22] The binding free energy Δ119866bind iswritten as the sum of the gas phase contribution Δ119866gasthe desolvation free energy of the system upon bindingΔ119866desolv and an entropic contribution minus119879Δ119878 as seenin Figure 7 where the term Δ119867gas contains the van derWaals (Δ119864vdW) and electrostatic (Δ119864elec) interaction energiesbetween the two partners in the complex and the internalenergy variation (including bond angle and torsional angleenergies) between the complex and the isolated molecules(Δ119864intra) respectively minus119879Δ119878 is the change of conformationalentropy upon peptide binding which is not considered herebecause of its high computational demand and relativelylow accuracy of prediction [5] All energies are averagedalong the MD trajectories Δ119866desolv is the difference between
the solvation free energy Δ119866solv of the complex and thatof the isolated parts Δ119866solv is divided into the electro-static Δ119866elecsolv and the nonpolar Δ119866npsolv contributionsΔ119866solv = Δ119866elecsolv + Δ119866npsolv
For the MMPBSA calculations Δ119866elecsolv was calculatedwith a built-in module the PBSA program in AMBER12which solves the Poisson-Boltzmann equation The gridsize for the PB calculations was 05 A The values of theinterior and exterior dielectric constants were set to 1 and80 respectively Δ119866npsolv was estimated based on the solventaccessible surface area (SASA) as Δ119866npsolv = 00072 times SASAusing the MolSurf program The scFv anti-p17peptide inter-action energy profiles were generated by decomposing thetotal binding free energies into residue-residue interactionpairs by the MM-GBSA decomposition process in the MM-PBSA program of AMBER12The calculated binding energiesherein were not absolute ones since we do not include theentropic changes of the solute molecule It is a computa-tionally expensive to estimate entropic changes using normalmode analysis and the calculation tends to have a large errorthat introduces significant uncertainty in the result Involvingentropy in the calculation would not make much differencefor the comparison of the binding free energies because ofthe similarity of the short peptides binding to the samescFv
23 Computational Alanine Scanning Alanine scanning [22]a computational method of systematic alanine substitutionhas been particularly useful for the identification of func-tional epitopes Substitution with alanine removes the sidechain atoms of the residues in CDR loops of wild type scFvanti-p17 All the alanine mutant structures were obtainedby deleting atoms and truncating the mutated residue inthe hypervariable portions of the loops on the heavy chain(H1ndashH3) and light chain (L1ndashL3) of wild-type scFv anti-p17 All parameters in the topology files for the mutatedresidues were accordingly replaced by the alanine residueparameters Proline residues were not mutated since theirbackbone conformations differ significantly from the alanineresidue [2] As a result the Pro58 andPro230 which belong tothe CDR loops H2 and L3 respectively are not selectedThenthe modified parameter files were generated again by usingthe LEaP module [5] This was extrapolated to the snapshotscollected from the trajectories at the last 500 ps resultingfrom the MD simulations by using the script mm pbsaplimplemented in the AMBER package From the decomposedenergy and alanine scanning result the point mutation hasbeen selected and investigated
24 Site-Directed Mutagenesis and the Evaluation of theBinding Activity of scFv Anti-p17 To generate scFv anti-p17mutant phagemid was subjected to perform mutation pro-cedure following the instruction of site-directed mutagenesis(Stratagene) Briefly ten nanograms of phagemid templatewere mixed with 125 ng of mutated primers in providedbuffer PfuTurbo DNA polymerase (Stratagene 25U) wasadded to the mixture for cycle amplification The reactionstarted with one round of 95∘C for 30 s followed by 16 roundsconsisting of 95∘C for 30 s 55∘C for 1min and 68∘C for
4 BioMed Research International
Table 1 ndashCDOCKER interaction energy (kcalmol) of mutant (M100G) scFv anti-p17 bound to its variants
AntibodyndashCDOCKER interaction energy
p171 p172 p173 p174 p175 p176 p177 p178 p179Competitive ELISA percentageinhibition values ()Mean plusmn STDEV
(1) 759(2) 901830 plusmn 100
mdash552596574 plusmn 31
mdash mdash mdash446601524 plusmn 110
795876836 plusmn 57
mdash885885 plusmn 00
scFv anti-p17 wild type 6184 7048 6410 5669 6027 5565 4639 6455 4675M100G 6086 4502 6516 6023 6305 5329 5259 6516 5455M100A 4689 5679 4692 5962 7361 5666 5136 5633 5878M100C 5926 6416 4763 6066 4772 5067 6226 7026 6937M100F 4375 5351 6593 5219 5525 5470 7620 6093 5983M100H 6023 5855 5807 6374 7578 7233 6568 5238 7155M100K 6772 6093 6099 5223 5908 7371 5363 6375 7268M100R 6978 7214 7264 8519 7063 7299 6685 5433 6646M100E 5924 6082 5407 5895 4789 5639 5502 6042 6495M100P 5772 5158 4461 6683 4935 5987 4563 5804 7810Note Highlight in bold letter indicates the lower interaction energy than original scFv Sequences of p171ndashp179 are 121DTGHSSQVSQNY132121DTGHSNQVSQNY132 121DTGHSSQISQNY132 121DTGHNSQVSQNY132 121NTGHSSQVSQNY132 121DTGNSSQVSQNY132 121DTGHSSQASQNY132121DTGHSKQVSQNY132 and 121DTGNNSQVSQNY132 respectively
9min 10U of DpnI restriction enzyme (Stratagene) wassubsequently added to eliminate phagemid template andincubated for 1 hour at 37∘C The reaction tube was subse-quently placed on ice for 2minThis synthesized product wasfurther transformed into E coli XL-1 Blue Bacterial contain-ing mutant phagemid was then cultured for production ofphage-displayedmutant scFv anti-p17 as described elsewhere[6] To evaluate the binding activity of wild type and mutantscFv anti-p17 with a series of synthetic peptides (GenScriptPiscataway New Jersey USA) phage ELISA was set up asdescribed in our previous study [6]
3 Results and Discussion
31 Pairwise Decomposition Energies and ComputationalAlanine Scanning The comparison of experimental activi-ties peptide ELISA with the results of CDOCKER intera-ction energy derived from molecular docking (CDOCKER)suggested that the experimental value had a high cor-relation (1199032 = 084) with the CDOCKER interactionenergy (Figure 2) From Table 1 peptide p177 had the lowestscore (less favorable interaction energy) and p178 had thehighest score (more favorable interaction energy) whereasp173 had very similar score to that of the wild-type pep-tide The peptide ELISA was used to describe the bindingactivity of scFv anti-p17 to its target peptide (p171) andfour mutant peptides (p173 p177 p178 and p179) Positivesignals were observed in all peptide coated wells indi-cating that this scFv anti-p17 could bind to all mutantpeptides Peptide p178 gave the highest signal followed byp171 p173 p179 and p177 respectively Although a goodcorrelation between the docking scores and ELISA compet-itive binding activity was found there is some difference
between p178 and p173 It should be remarked here thatonly slightly difference between p178 and p173 was observedwith a limitation of receptor rigidity in molecular dockingin the screening process Furthermore we selected fivepeptide epitopes consisting of one wild-type peptide (p171)and four mutated peptides (p173 p177 p178 and p179)for further investigation by molecular dynamics simulations(MDs) and MM-PBSA To validate the dynamic stabilityof the complexes total potential energy and the RMSDfor the backbone atoms along the 10 ns MD trajectoriesusing the initial minimized docking structure as a ref-erence were monitored in Figure 1 The RMSD values ofmost complexes converged around 25ndash35 A which meansthe MD trajectories of the complexes appear to be wellequilibrated Some systems of scFv anti-p17 mutant com-plexes were not stable and RMSD still did not converge after10 ns In order to investigate protein binding capability theresults derived fromMM-PBSA andMM-GBSA calculations(Figures 3(c) and 3(d)) were compared and MM-PBSAshows higher correlation with experimental valueThereforethe value of PBTOT was used to compare the simulationwith the peptide ELISA results The more negative thevalue the more favorable the binding The binding energiesidentified by the MM-PBSA protocol were ranked as followspeptide p171 gt p178 gt p173simp177 gt p179 with the val-ues of minus2993 minus2570 minus2515 minus2515 and minus1488 kcalmolrespectively The major contributions to the binding freeenergy arise from the electrostatic energy as calculated bythe molecular mechanic (MM) force field (ELE) and fromthe electrostatic contribution to the solvation free energyas calculated by PB (PBCAL) van der Waals contributionarises from MM (VDW) For the five binding peptides withthe wild type scFv Δ119864vdW Δ119866PB and Δ119866GB are quite similar
BioMed Research International 5
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus96
0 1 2 3 4 5 6 7 8 9 10Ep
tot (
kcal
mol
)Time (ns)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
(a)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
Time (ns)
(b)
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus960 1 2 3 4 5 6 7 8 9 10
Epto
t (kc
alm
ol)
M100R-p171M100R-p177M100R-p179
Time (ns)
(c)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
Time (ns)
M100R-Lg1M100R-Lg7M100R-Lg9
(d)
Figure 1 The potential energy (kcalmol) and the root-mean-squared deviations (RMSD) of the backbone atoms compared to the dockingstructure for mutants (M100G M100R) of scFv anti-p17 bound to its variants during the production run were monitored
but the electrostatic energies were quite varied among the lowand high activities peptides indicating that the main factordetermining the binding activitymight arise from the electro-static contribution as peptide p177 had a very low electrostatic
contribution (minus4628 kcalmol) compared to other sequences(Figure 3(a))
In order to elucidate the key residues in the bindingpocket of the scFv anti-p17 and themost favorable interaction
6 BioMed Research International
6184
7048641
5669 60275565
4639
6455
4675
0
10
20
30
40
50
60
70
80
90
p171 p172 p173 p174 p175 p176 p177 p178 p179
Wild typeM100GM100AM100CM100F
M100HM100KM100RM100EM100P
scFv anti-p17
minusCDOCKER interaction energy(kcalmol)
Figure 2 The ndashCDOCKER interaction energy (kcalmol) of orig-inal scFv anti-p17 bound to its variants was compared with thedifferent mutants from point mutation at MET100
modes in this study computational alanine scanning wasperformed This method depends on the assumption thatlocal changes of the protein do not influence the wholeconformation of the complex significantly In the calcula-tion with alanine scanning the positive and negative rela-tive values in Figure 4 indicate the unfavorable and favor-able for alanine substitution respectively All the residuesin the hypervariable portions of the loops on the heavychain (H1ndashH3) and light chain (L1ndashL3) of wild-type scFvanti-p17 have been mutated to alanine except for the prolineresidue whose backbone is remarkably different from that ofalanine In comparison with wild type the calculated bindingfree energy and computational alanine scanning analysis(Figure 4) demonstrated the importance of the residues ofscFv anti-p17 in the binding pocket which are TRP50 ASN52GLU57 MET100 LEU185 and LYS188 with the relativedecomposed energy below minus200 kcalmol Our previousresults demonstrated that the specific residues located inthe complementary determining regions (CDRs) of scFvanti-p17 MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities withthe HIV-1 p17 variants [6] Therefore from both resultsthe importance of the mutationrsquos location has 10 positionsin the CDRs of scFv anti-p17 consist of TRP50 ASN52GLU57 MET100 LYS101 ASN169 LEU185 LYS188 HIS228and LEU229 The methionine at position 100 (MET100)is initially selected for mutation due to the high con-tribution in binding energy of this region from position100ndash104 residues and the substitution of alanine at this
position showed the significant change in the bindinginteraction
32 Structural Analysis of scFv Anti-p17 Point MutationsWith the combination of pairwise decomposition energiesand computational alanine scanning the key amino acidsin binding TRP50 ASN52 GLU57 MET100 LYS101ASN169 LEU185 LYS188 HIS228 and LEU229 were theimportant residues to binding efficiency of natural HIVepitope at the C-terminal on p17 Therefore these residuesare subjected to mutation in order to improve the bindingactivities The residues roughly equivalent were groupedtogether in five subsets according to statistically preferredsubstitutions which generally correlate with side chainphysicochemical properties observed in buried residuesand exposed residues reported by Bordo and Argos [15] asfollows (a) buried in the protein core (solvent-accessiblesurface for both residues le 10 A2) (b) exposed (solvent-accessible surface area ge 30 A2) and (c) all the possibleaccessibility states allowed The mutation of amino acidsin the CDRs of scFv anti-p17 has been initially selectedat position M100 since the pairwise decomposed energyindicated the unfavorable residue interaction and thesubstitution of the residue with alanine can lead to significantchange in binding affinity Mutation of methionine residuesin position 100 of scFv anti-p17 (wild type) was mutatedto another polarity group with electrically charged oruncharged or hydrophobic side chains which have theimpact on the development of protein cores in structuresmaintaining main-chain fold [23] according to Table 1Finally the mutated scFv anti-p17 was docked with ninepeptides by CDOCKER The energies obtained from thedocking of each peptide with the mutated scFv are listedin Table 1 The more negative interaction energies exhibitthe more favorable binding The prediction of interactionenergy with most peptides and mutant forms (M100G) ismore than that of wild type scFv anti-p17 and the improvedelectrostatic contribution was observed as in Figure 3(a)for the mutant type The hydrogen bond interactions ofmutant form of scFv anti-p17 with five peptides at theresidues ASP31 TYR32 ASN52 THR59 SER99 GLY100LYS101 LYS165 TYR184 LYS188 LEU189 LEU229 andGLN231 were shown in Figure 5 Both of the two peptides(p171 and p173) showed interactions with the receptormostly at TYR32 THR59 and LYS165 and other threepeptides (p177 p178 and p179) showed interactions withthe receptor mostly at ASN52 GLY100 and TYR184 Fromthe conformation with the lowest CDOCKER interactionenergy structure of M100G scFv anti-p17 the peptides boundin two orientations where the N-terminal (p171ndashp176)and the C-terminal (p177ndashp179) of peptide sequenceswere directed toward the binding pocket The calculateddocking interaction energy between single mutation frommethionine 100 to glycine (M100G) and peptide sequencesp171 (DTGHSSQVSQNY) p173 (DTGHSSQISQNY) p177(DTGHSSQASQNY) p178 (DTGHSKQVSQNY) p179(DTGNNSQVSQNY) has shown the favorably interactionenergy compared to wild type which correlated well with
BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

2 BioMed Research International
linked to a light chain variable region (VL) via a flexiblepeptide linker Each VH and VL domain contains threecomplementarity determining regions (CDRs) CDRs areshort amino acid sequences that vary greatly among antibodymolecules and thus are responsible for generating the greatdiversity of antibody binding specificity The combinationof the CDRs of the VH plus the CDRs of the VL deter-mines the binding specificity of any given antibody Singlechain Fv fragments display the binding specificity mono-valent binding affinity of full-size antibodies and providethe added benefit of relative ease of genetic manipulationand expression Given their advantages of small size andantigen specificity encompassed within a single polypeptidechain scFvs are the most common type of recombinantantibody fragment used for intracellular antibody expression[1]
Several computation approaches have been applied tostudy the protein-ligand interaction by the combination ofcomputational alanine scanning and free energy decomposi-tion methods The molecular mechanics poisson-Boltzmannsurface area (MMPBSA) [2] and molecular mechanics gen-eralized born surface area (MMGBSA) [3] approaches areused to evaluate binding free energy in the computationalalanine scanning Both PB and GB implicit solvent modelswere used to calculate the energy contribution of each residuein the binding free energy on a decomposition basis Theresults from computational alanine scanning and free energydecomposition methods indicated the important residues forbinding 4 of lipopeptide inhibitor to E coli SPase [4] andrecognize the key residues in the ATP binding site of GyrBsubunit fromEscherichia coli boundwith the inhibitors cloro-biocin novobiocin and 51015840-adenylyl-120573-120574-imidodiphosphate[5]
In our previous work scFv anti-p17 was simulated basedon molecular modeling of its homologue structure Theantibody-antigen complex models were generated using themolecular docking that predicted the most favorable bindinginteraction Then the interactions between nine peptideepitopes and the scFv anti-p17 in water were analyzedusing molecular dynamics (MDs) simulation to evaluate thebinding free energy and pairwise decomposition or residue-based energy calculation of complexes in solution using themolecularmechanicspoisson-Boltzmann surface area (MM-PBSA) and molecular mechanicsgeneralized born surfacearea (MM-GBSA) methods The latter analysis can provideinteresting information in terms of electrostatic and van derWaals energies solvation energies and entropic contribu-tions at the binding interface Pairwise decomposition ofresidue interaction energies of the complexes between scFvanti-p17 and its variants have indicated that the specificresidues located in the complementary determining regions(CDRs) of scFv anti-p17 MET100 LYS101 ASN169 HIS228and LEU229 play a crucial role in the effective bindinginteraction [6]
To determine whether the side chain of the specific resi-due in CDRs of scFv anti-p17 plays an important rolein bioactivity computational alanine scanning was carriedout in this study Computational alanine scanning uses asimple free energy function to calculate the effects of alanine
mutations on the binding free energy of a protein-proteincomplex It involves the free energy decomposition involvingMM-PBSA method [2 3 7ndash11] and MM-GBSA method[3 12] to investigate the binding modes in detail at theatomic level and also to estimate protein stabilities [13] Theinput for the computational alanine scanning consisted ofa three-dimensional modeled structure of the scFv anti-p17Potential amino acids that involved changing nonalanineamino acids into alanines will be listed up The relevantchange of more than 1 kcal molminus1 in decomposition energycalculation indicated the important residues involving in thebinding [14]
In this study theoreticalmodeling andmolecular dynam-ics simulations investigation of scFv against HIV-1 epitopeat C-terminal on p17 (scFv anti-p17) has been performedto specify the key residues in the binding From the searchin National Center for Biotechnology Information (NCBI)database 9 different variants of HIV-1 epitope at C-terminalwhich show different binding activities upon binding withscFv were found Computational alanine scanning was uti-lized to investigate the effect of side chain atoms of theresidues in CDR loops of scFv anti-p17With the combinationof the two techniques the selected point mutation to improvethe binding affinity between the scFv anti-p17 and its variantswas identified These residues were mutated to improve thebinding activities guided from statistically preferred substi-tutions observed in buried residues and exposed residuesdue to their solvent exposed area and side chain chargewhich generally correlate with side chain physicochemicalproperties [15] Finally as validated by the experimentalresults we have designed new scFv anti-p17 which bindsbetter with HIV-1 epitope at C-terminal on p17
2 Materials and Methods
21 Molecular Docking The built model reported in theprevious work [6] of primary sequence of the scFv anti-p17protein obtained by Tewari [16] was used in this study Thegeneral docking protocol and potential functions employedin CDOCKER have been described in prior articles [17]In this work docking of the peptides to scFv anti-p17 wasconducted using CDOCKER CDOCKER is a grid-basedmolecular docking method that employs CHARMm Thereceptor is held rigid while the ligands are allowed to flexduring the refinement Ligands are assumed to have alreadybeen roughly docked into the receptor binding site Theactive site pocket of the receptor was found on the CDRs ofscFv anti-p17 by the Discovery Studio 20 (Accelrys SoftwareInc) A site sphere radius of 25 A was set to assign thebinding pocket and the ligand partial charge method forassigning partial charges to the ligands during force fieldassignment was CHARMm Other parameters were set asdefault The lowest docking interaction structure where thepeptide lied in the CDRs similar to the X-ray crystal structureof HIV-1 p24 bound with antibody (PDB ID 1AFV) was thenselected
22 Molecular Dynamics Simulation and Binding Free EnergyCalculations Energy minimization and MD simulations
BioMed Research International 3
were performed using PMEMDCUDA from AMBER12 [18]on GPUs Quadro 2000D produced by NVIDIA which speedup the simulation wall time required to obtain the trajectoryfiles from each simulation MD simulations were carriedout at the molecular mechanics level using the AMBER03force field Antibody-peptide structures were solvated ina cubic box of TIP3P water extending at least 10 A ineach direction from the solute and the cut-off distancewas kept to 12 A to compute the nonbonded interactionsAll simulations were performed under periodic boundaryconditions [19] and long-range electrostatics were treatedby using the particle-mesh-Ewald method [20 21] The timestep was set to 1 fs and the trajectory was recorded every01 ps Prior to MD simulations the systems were relaxedby a series of steepest descent (SD) and conjugated gradient(CG) minimizations MD simulations were performed basedon each of the minimized systems by gradually heating over60 ps from 0 to 310K with the protein atoms fixed usinga force constant of 5 kcalmolA2 Then a 200 ps pressure-constant period (NPT) was applied to obtain an equilibrateddensity of the constrained protein atoms The followingstep was a 40 ps-volume-constant period (NVT) at a forceconstant of 25 kcalmolA2 followed by 100 ps dynamics ata force constant of 125 kcalmolA2 Finally an unrestrainedMD simulation (no force applied on any protein atoms) wasperformed for each fully flexible system in theNVT ensembleat a constant temperature of 310 K for a total simulation timeof 10 ns 500 snapshots were collected from the last 5 ns ofMDsimulations for binding free energy analysis Equilibrationwas monitored by convergence in terms of the temperatureenergy and density of the system and the root-mean-squareddeviations (RMSD) of the backbone atoms compared to thedocking structure
The binding free energies (Δ119866bind)were evaluated accord-ing to the strategy described by Massova and Kollman [2]Below it is summarized the Δ119866bind was determined from thefree energies of the complex protein and peptide accordingto the following equation
Δ119866bind = Δ119866water (complex)
minus [Δ119866water (protein) + Δ119866water (peptide)] (1)
Based on the selected MD snapshots the binding freeenergy for each antibody-peptide system could be estimatedusing MM-PBSA [22] The binding free energy Δ119866bind iswritten as the sum of the gas phase contribution Δ119866gasthe desolvation free energy of the system upon bindingΔ119866desolv and an entropic contribution minus119879Δ119878 as seenin Figure 7 where the term Δ119867gas contains the van derWaals (Δ119864vdW) and electrostatic (Δ119864elec) interaction energiesbetween the two partners in the complex and the internalenergy variation (including bond angle and torsional angleenergies) between the complex and the isolated molecules(Δ119864intra) respectively minus119879Δ119878 is the change of conformationalentropy upon peptide binding which is not considered herebecause of its high computational demand and relativelylow accuracy of prediction [5] All energies are averagedalong the MD trajectories Δ119866desolv is the difference between
the solvation free energy Δ119866solv of the complex and thatof the isolated parts Δ119866solv is divided into the electro-static Δ119866elecsolv and the nonpolar Δ119866npsolv contributionsΔ119866solv = Δ119866elecsolv + Δ119866npsolv
For the MMPBSA calculations Δ119866elecsolv was calculatedwith a built-in module the PBSA program in AMBER12which solves the Poisson-Boltzmann equation The gridsize for the PB calculations was 05 A The values of theinterior and exterior dielectric constants were set to 1 and80 respectively Δ119866npsolv was estimated based on the solventaccessible surface area (SASA) as Δ119866npsolv = 00072 times SASAusing the MolSurf program The scFv anti-p17peptide inter-action energy profiles were generated by decomposing thetotal binding free energies into residue-residue interactionpairs by the MM-GBSA decomposition process in the MM-PBSA program of AMBER12The calculated binding energiesherein were not absolute ones since we do not include theentropic changes of the solute molecule It is a computa-tionally expensive to estimate entropic changes using normalmode analysis and the calculation tends to have a large errorthat introduces significant uncertainty in the result Involvingentropy in the calculation would not make much differencefor the comparison of the binding free energies because ofthe similarity of the short peptides binding to the samescFv
23 Computational Alanine Scanning Alanine scanning [22]a computational method of systematic alanine substitutionhas been particularly useful for the identification of func-tional epitopes Substitution with alanine removes the sidechain atoms of the residues in CDR loops of wild type scFvanti-p17 All the alanine mutant structures were obtainedby deleting atoms and truncating the mutated residue inthe hypervariable portions of the loops on the heavy chain(H1ndashH3) and light chain (L1ndashL3) of wild-type scFv anti-p17 All parameters in the topology files for the mutatedresidues were accordingly replaced by the alanine residueparameters Proline residues were not mutated since theirbackbone conformations differ significantly from the alanineresidue [2] As a result the Pro58 andPro230 which belong tothe CDR loops H2 and L3 respectively are not selectedThenthe modified parameter files were generated again by usingthe LEaP module [5] This was extrapolated to the snapshotscollected from the trajectories at the last 500 ps resultingfrom the MD simulations by using the script mm pbsaplimplemented in the AMBER package From the decomposedenergy and alanine scanning result the point mutation hasbeen selected and investigated
24 Site-Directed Mutagenesis and the Evaluation of theBinding Activity of scFv Anti-p17 To generate scFv anti-p17mutant phagemid was subjected to perform mutation pro-cedure following the instruction of site-directed mutagenesis(Stratagene) Briefly ten nanograms of phagemid templatewere mixed with 125 ng of mutated primers in providedbuffer PfuTurbo DNA polymerase (Stratagene 25U) wasadded to the mixture for cycle amplification The reactionstarted with one round of 95∘C for 30 s followed by 16 roundsconsisting of 95∘C for 30 s 55∘C for 1min and 68∘C for
4 BioMed Research International
Table 1 ndashCDOCKER interaction energy (kcalmol) of mutant (M100G) scFv anti-p17 bound to its variants
AntibodyndashCDOCKER interaction energy
p171 p172 p173 p174 p175 p176 p177 p178 p179Competitive ELISA percentageinhibition values ()Mean plusmn STDEV
(1) 759(2) 901830 plusmn 100
mdash552596574 plusmn 31
mdash mdash mdash446601524 plusmn 110
795876836 plusmn 57
mdash885885 plusmn 00
scFv anti-p17 wild type 6184 7048 6410 5669 6027 5565 4639 6455 4675M100G 6086 4502 6516 6023 6305 5329 5259 6516 5455M100A 4689 5679 4692 5962 7361 5666 5136 5633 5878M100C 5926 6416 4763 6066 4772 5067 6226 7026 6937M100F 4375 5351 6593 5219 5525 5470 7620 6093 5983M100H 6023 5855 5807 6374 7578 7233 6568 5238 7155M100K 6772 6093 6099 5223 5908 7371 5363 6375 7268M100R 6978 7214 7264 8519 7063 7299 6685 5433 6646M100E 5924 6082 5407 5895 4789 5639 5502 6042 6495M100P 5772 5158 4461 6683 4935 5987 4563 5804 7810Note Highlight in bold letter indicates the lower interaction energy than original scFv Sequences of p171ndashp179 are 121DTGHSSQVSQNY132121DTGHSNQVSQNY132 121DTGHSSQISQNY132 121DTGHNSQVSQNY132 121NTGHSSQVSQNY132 121DTGNSSQVSQNY132 121DTGHSSQASQNY132121DTGHSKQVSQNY132 and 121DTGNNSQVSQNY132 respectively
9min 10U of DpnI restriction enzyme (Stratagene) wassubsequently added to eliminate phagemid template andincubated for 1 hour at 37∘C The reaction tube was subse-quently placed on ice for 2minThis synthesized product wasfurther transformed into E coli XL-1 Blue Bacterial contain-ing mutant phagemid was then cultured for production ofphage-displayedmutant scFv anti-p17 as described elsewhere[6] To evaluate the binding activity of wild type and mutantscFv anti-p17 with a series of synthetic peptides (GenScriptPiscataway New Jersey USA) phage ELISA was set up asdescribed in our previous study [6]
3 Results and Discussion
31 Pairwise Decomposition Energies and ComputationalAlanine Scanning The comparison of experimental activi-ties peptide ELISA with the results of CDOCKER intera-ction energy derived from molecular docking (CDOCKER)suggested that the experimental value had a high cor-relation (1199032 = 084) with the CDOCKER interactionenergy (Figure 2) From Table 1 peptide p177 had the lowestscore (less favorable interaction energy) and p178 had thehighest score (more favorable interaction energy) whereasp173 had very similar score to that of the wild-type pep-tide The peptide ELISA was used to describe the bindingactivity of scFv anti-p17 to its target peptide (p171) andfour mutant peptides (p173 p177 p178 and p179) Positivesignals were observed in all peptide coated wells indi-cating that this scFv anti-p17 could bind to all mutantpeptides Peptide p178 gave the highest signal followed byp171 p173 p179 and p177 respectively Although a goodcorrelation between the docking scores and ELISA compet-itive binding activity was found there is some difference
between p178 and p173 It should be remarked here thatonly slightly difference between p178 and p173 was observedwith a limitation of receptor rigidity in molecular dockingin the screening process Furthermore we selected fivepeptide epitopes consisting of one wild-type peptide (p171)and four mutated peptides (p173 p177 p178 and p179)for further investigation by molecular dynamics simulations(MDs) and MM-PBSA To validate the dynamic stabilityof the complexes total potential energy and the RMSDfor the backbone atoms along the 10 ns MD trajectoriesusing the initial minimized docking structure as a ref-erence were monitored in Figure 1 The RMSD values ofmost complexes converged around 25ndash35 A which meansthe MD trajectories of the complexes appear to be wellequilibrated Some systems of scFv anti-p17 mutant com-plexes were not stable and RMSD still did not converge after10 ns In order to investigate protein binding capability theresults derived fromMM-PBSA andMM-GBSA calculations(Figures 3(c) and 3(d)) were compared and MM-PBSAshows higher correlation with experimental valueThereforethe value of PBTOT was used to compare the simulationwith the peptide ELISA results The more negative thevalue the more favorable the binding The binding energiesidentified by the MM-PBSA protocol were ranked as followspeptide p171 gt p178 gt p173simp177 gt p179 with the val-ues of minus2993 minus2570 minus2515 minus2515 and minus1488 kcalmolrespectively The major contributions to the binding freeenergy arise from the electrostatic energy as calculated bythe molecular mechanic (MM) force field (ELE) and fromthe electrostatic contribution to the solvation free energyas calculated by PB (PBCAL) van der Waals contributionarises from MM (VDW) For the five binding peptides withthe wild type scFv Δ119864vdW Δ119866PB and Δ119866GB are quite similar
BioMed Research International 5
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus96
0 1 2 3 4 5 6 7 8 9 10Ep
tot (
kcal
mol
)Time (ns)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
(a)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
Time (ns)
(b)
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus960 1 2 3 4 5 6 7 8 9 10
Epto
t (kc
alm
ol)
M100R-p171M100R-p177M100R-p179
Time (ns)
(c)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
Time (ns)
M100R-Lg1M100R-Lg7M100R-Lg9
(d)
Figure 1 The potential energy (kcalmol) and the root-mean-squared deviations (RMSD) of the backbone atoms compared to the dockingstructure for mutants (M100G M100R) of scFv anti-p17 bound to its variants during the production run were monitored
but the electrostatic energies were quite varied among the lowand high activities peptides indicating that the main factordetermining the binding activitymight arise from the electro-static contribution as peptide p177 had a very low electrostatic
contribution (minus4628 kcalmol) compared to other sequences(Figure 3(a))
In order to elucidate the key residues in the bindingpocket of the scFv anti-p17 and themost favorable interaction
6 BioMed Research International
6184
7048641
5669 60275565
4639
6455
4675
0
10
20
30
40
50
60
70
80
90
p171 p172 p173 p174 p175 p176 p177 p178 p179
Wild typeM100GM100AM100CM100F
M100HM100KM100RM100EM100P
scFv anti-p17
minusCDOCKER interaction energy(kcalmol)
Figure 2 The ndashCDOCKER interaction energy (kcalmol) of orig-inal scFv anti-p17 bound to its variants was compared with thedifferent mutants from point mutation at MET100
modes in this study computational alanine scanning wasperformed This method depends on the assumption thatlocal changes of the protein do not influence the wholeconformation of the complex significantly In the calcula-tion with alanine scanning the positive and negative rela-tive values in Figure 4 indicate the unfavorable and favor-able for alanine substitution respectively All the residuesin the hypervariable portions of the loops on the heavychain (H1ndashH3) and light chain (L1ndashL3) of wild-type scFvanti-p17 have been mutated to alanine except for the prolineresidue whose backbone is remarkably different from that ofalanine In comparison with wild type the calculated bindingfree energy and computational alanine scanning analysis(Figure 4) demonstrated the importance of the residues ofscFv anti-p17 in the binding pocket which are TRP50 ASN52GLU57 MET100 LEU185 and LYS188 with the relativedecomposed energy below minus200 kcalmol Our previousresults demonstrated that the specific residues located inthe complementary determining regions (CDRs) of scFvanti-p17 MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities withthe HIV-1 p17 variants [6] Therefore from both resultsthe importance of the mutationrsquos location has 10 positionsin the CDRs of scFv anti-p17 consist of TRP50 ASN52GLU57 MET100 LYS101 ASN169 LEU185 LYS188 HIS228and LEU229 The methionine at position 100 (MET100)is initially selected for mutation due to the high con-tribution in binding energy of this region from position100ndash104 residues and the substitution of alanine at this
position showed the significant change in the bindinginteraction
32 Structural Analysis of scFv Anti-p17 Point MutationsWith the combination of pairwise decomposition energiesand computational alanine scanning the key amino acidsin binding TRP50 ASN52 GLU57 MET100 LYS101ASN169 LEU185 LYS188 HIS228 and LEU229 were theimportant residues to binding efficiency of natural HIVepitope at the C-terminal on p17 Therefore these residuesare subjected to mutation in order to improve the bindingactivities The residues roughly equivalent were groupedtogether in five subsets according to statistically preferredsubstitutions which generally correlate with side chainphysicochemical properties observed in buried residuesand exposed residues reported by Bordo and Argos [15] asfollows (a) buried in the protein core (solvent-accessiblesurface for both residues le 10 A2) (b) exposed (solvent-accessible surface area ge 30 A2) and (c) all the possibleaccessibility states allowed The mutation of amino acidsin the CDRs of scFv anti-p17 has been initially selectedat position M100 since the pairwise decomposed energyindicated the unfavorable residue interaction and thesubstitution of the residue with alanine can lead to significantchange in binding affinity Mutation of methionine residuesin position 100 of scFv anti-p17 (wild type) was mutatedto another polarity group with electrically charged oruncharged or hydrophobic side chains which have theimpact on the development of protein cores in structuresmaintaining main-chain fold [23] according to Table 1Finally the mutated scFv anti-p17 was docked with ninepeptides by CDOCKER The energies obtained from thedocking of each peptide with the mutated scFv are listedin Table 1 The more negative interaction energies exhibitthe more favorable binding The prediction of interactionenergy with most peptides and mutant forms (M100G) ismore than that of wild type scFv anti-p17 and the improvedelectrostatic contribution was observed as in Figure 3(a)for the mutant type The hydrogen bond interactions ofmutant form of scFv anti-p17 with five peptides at theresidues ASP31 TYR32 ASN52 THR59 SER99 GLY100LYS101 LYS165 TYR184 LYS188 LEU189 LEU229 andGLN231 were shown in Figure 5 Both of the two peptides(p171 and p173) showed interactions with the receptormostly at TYR32 THR59 and LYS165 and other threepeptides (p177 p178 and p179) showed interactions withthe receptor mostly at ASN52 GLY100 and TYR184 Fromthe conformation with the lowest CDOCKER interactionenergy structure of M100G scFv anti-p17 the peptides boundin two orientations where the N-terminal (p171ndashp176)and the C-terminal (p177ndashp179) of peptide sequenceswere directed toward the binding pocket The calculateddocking interaction energy between single mutation frommethionine 100 to glycine (M100G) and peptide sequencesp171 (DTGHSSQVSQNY) p173 (DTGHSSQISQNY) p177(DTGHSSQASQNY) p178 (DTGHSKQVSQNY) p179(DTGNNSQVSQNY) has shown the favorably interactionenergy compared to wild type which correlated well with
BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

BioMed Research International 3
were performed using PMEMDCUDA from AMBER12 [18]on GPUs Quadro 2000D produced by NVIDIA which speedup the simulation wall time required to obtain the trajectoryfiles from each simulation MD simulations were carriedout at the molecular mechanics level using the AMBER03force field Antibody-peptide structures were solvated ina cubic box of TIP3P water extending at least 10 A ineach direction from the solute and the cut-off distancewas kept to 12 A to compute the nonbonded interactionsAll simulations were performed under periodic boundaryconditions [19] and long-range electrostatics were treatedby using the particle-mesh-Ewald method [20 21] The timestep was set to 1 fs and the trajectory was recorded every01 ps Prior to MD simulations the systems were relaxedby a series of steepest descent (SD) and conjugated gradient(CG) minimizations MD simulations were performed basedon each of the minimized systems by gradually heating over60 ps from 0 to 310K with the protein atoms fixed usinga force constant of 5 kcalmolA2 Then a 200 ps pressure-constant period (NPT) was applied to obtain an equilibrateddensity of the constrained protein atoms The followingstep was a 40 ps-volume-constant period (NVT) at a forceconstant of 25 kcalmolA2 followed by 100 ps dynamics ata force constant of 125 kcalmolA2 Finally an unrestrainedMD simulation (no force applied on any protein atoms) wasperformed for each fully flexible system in theNVT ensembleat a constant temperature of 310 K for a total simulation timeof 10 ns 500 snapshots were collected from the last 5 ns ofMDsimulations for binding free energy analysis Equilibrationwas monitored by convergence in terms of the temperatureenergy and density of the system and the root-mean-squareddeviations (RMSD) of the backbone atoms compared to thedocking structure
The binding free energies (Δ119866bind)were evaluated accord-ing to the strategy described by Massova and Kollman [2]Below it is summarized the Δ119866bind was determined from thefree energies of the complex protein and peptide accordingto the following equation
Δ119866bind = Δ119866water (complex)
minus [Δ119866water (protein) + Δ119866water (peptide)] (1)
Based on the selected MD snapshots the binding freeenergy for each antibody-peptide system could be estimatedusing MM-PBSA [22] The binding free energy Δ119866bind iswritten as the sum of the gas phase contribution Δ119866gasthe desolvation free energy of the system upon bindingΔ119866desolv and an entropic contribution minus119879Δ119878 as seenin Figure 7 where the term Δ119867gas contains the van derWaals (Δ119864vdW) and electrostatic (Δ119864elec) interaction energiesbetween the two partners in the complex and the internalenergy variation (including bond angle and torsional angleenergies) between the complex and the isolated molecules(Δ119864intra) respectively minus119879Δ119878 is the change of conformationalentropy upon peptide binding which is not considered herebecause of its high computational demand and relativelylow accuracy of prediction [5] All energies are averagedalong the MD trajectories Δ119866desolv is the difference between
the solvation free energy Δ119866solv of the complex and thatof the isolated parts Δ119866solv is divided into the electro-static Δ119866elecsolv and the nonpolar Δ119866npsolv contributionsΔ119866solv = Δ119866elecsolv + Δ119866npsolv
For the MMPBSA calculations Δ119866elecsolv was calculatedwith a built-in module the PBSA program in AMBER12which solves the Poisson-Boltzmann equation The gridsize for the PB calculations was 05 A The values of theinterior and exterior dielectric constants were set to 1 and80 respectively Δ119866npsolv was estimated based on the solventaccessible surface area (SASA) as Δ119866npsolv = 00072 times SASAusing the MolSurf program The scFv anti-p17peptide inter-action energy profiles were generated by decomposing thetotal binding free energies into residue-residue interactionpairs by the MM-GBSA decomposition process in the MM-PBSA program of AMBER12The calculated binding energiesherein were not absolute ones since we do not include theentropic changes of the solute molecule It is a computa-tionally expensive to estimate entropic changes using normalmode analysis and the calculation tends to have a large errorthat introduces significant uncertainty in the result Involvingentropy in the calculation would not make much differencefor the comparison of the binding free energies because ofthe similarity of the short peptides binding to the samescFv
23 Computational Alanine Scanning Alanine scanning [22]a computational method of systematic alanine substitutionhas been particularly useful for the identification of func-tional epitopes Substitution with alanine removes the sidechain atoms of the residues in CDR loops of wild type scFvanti-p17 All the alanine mutant structures were obtainedby deleting atoms and truncating the mutated residue inthe hypervariable portions of the loops on the heavy chain(H1ndashH3) and light chain (L1ndashL3) of wild-type scFv anti-p17 All parameters in the topology files for the mutatedresidues were accordingly replaced by the alanine residueparameters Proline residues were not mutated since theirbackbone conformations differ significantly from the alanineresidue [2] As a result the Pro58 andPro230 which belong tothe CDR loops H2 and L3 respectively are not selectedThenthe modified parameter files were generated again by usingthe LEaP module [5] This was extrapolated to the snapshotscollected from the trajectories at the last 500 ps resultingfrom the MD simulations by using the script mm pbsaplimplemented in the AMBER package From the decomposedenergy and alanine scanning result the point mutation hasbeen selected and investigated
24 Site-Directed Mutagenesis and the Evaluation of theBinding Activity of scFv Anti-p17 To generate scFv anti-p17mutant phagemid was subjected to perform mutation pro-cedure following the instruction of site-directed mutagenesis(Stratagene) Briefly ten nanograms of phagemid templatewere mixed with 125 ng of mutated primers in providedbuffer PfuTurbo DNA polymerase (Stratagene 25U) wasadded to the mixture for cycle amplification The reactionstarted with one round of 95∘C for 30 s followed by 16 roundsconsisting of 95∘C for 30 s 55∘C for 1min and 68∘C for
4 BioMed Research International
Table 1 ndashCDOCKER interaction energy (kcalmol) of mutant (M100G) scFv anti-p17 bound to its variants
AntibodyndashCDOCKER interaction energy
p171 p172 p173 p174 p175 p176 p177 p178 p179Competitive ELISA percentageinhibition values ()Mean plusmn STDEV
(1) 759(2) 901830 plusmn 100
mdash552596574 plusmn 31
mdash mdash mdash446601524 plusmn 110
795876836 plusmn 57
mdash885885 plusmn 00
scFv anti-p17 wild type 6184 7048 6410 5669 6027 5565 4639 6455 4675M100G 6086 4502 6516 6023 6305 5329 5259 6516 5455M100A 4689 5679 4692 5962 7361 5666 5136 5633 5878M100C 5926 6416 4763 6066 4772 5067 6226 7026 6937M100F 4375 5351 6593 5219 5525 5470 7620 6093 5983M100H 6023 5855 5807 6374 7578 7233 6568 5238 7155M100K 6772 6093 6099 5223 5908 7371 5363 6375 7268M100R 6978 7214 7264 8519 7063 7299 6685 5433 6646M100E 5924 6082 5407 5895 4789 5639 5502 6042 6495M100P 5772 5158 4461 6683 4935 5987 4563 5804 7810Note Highlight in bold letter indicates the lower interaction energy than original scFv Sequences of p171ndashp179 are 121DTGHSSQVSQNY132121DTGHSNQVSQNY132 121DTGHSSQISQNY132 121DTGHNSQVSQNY132 121NTGHSSQVSQNY132 121DTGNSSQVSQNY132 121DTGHSSQASQNY132121DTGHSKQVSQNY132 and 121DTGNNSQVSQNY132 respectively
9min 10U of DpnI restriction enzyme (Stratagene) wassubsequently added to eliminate phagemid template andincubated for 1 hour at 37∘C The reaction tube was subse-quently placed on ice for 2minThis synthesized product wasfurther transformed into E coli XL-1 Blue Bacterial contain-ing mutant phagemid was then cultured for production ofphage-displayedmutant scFv anti-p17 as described elsewhere[6] To evaluate the binding activity of wild type and mutantscFv anti-p17 with a series of synthetic peptides (GenScriptPiscataway New Jersey USA) phage ELISA was set up asdescribed in our previous study [6]
3 Results and Discussion
31 Pairwise Decomposition Energies and ComputationalAlanine Scanning The comparison of experimental activi-ties peptide ELISA with the results of CDOCKER intera-ction energy derived from molecular docking (CDOCKER)suggested that the experimental value had a high cor-relation (1199032 = 084) with the CDOCKER interactionenergy (Figure 2) From Table 1 peptide p177 had the lowestscore (less favorable interaction energy) and p178 had thehighest score (more favorable interaction energy) whereasp173 had very similar score to that of the wild-type pep-tide The peptide ELISA was used to describe the bindingactivity of scFv anti-p17 to its target peptide (p171) andfour mutant peptides (p173 p177 p178 and p179) Positivesignals were observed in all peptide coated wells indi-cating that this scFv anti-p17 could bind to all mutantpeptides Peptide p178 gave the highest signal followed byp171 p173 p179 and p177 respectively Although a goodcorrelation between the docking scores and ELISA compet-itive binding activity was found there is some difference
between p178 and p173 It should be remarked here thatonly slightly difference between p178 and p173 was observedwith a limitation of receptor rigidity in molecular dockingin the screening process Furthermore we selected fivepeptide epitopes consisting of one wild-type peptide (p171)and four mutated peptides (p173 p177 p178 and p179)for further investigation by molecular dynamics simulations(MDs) and MM-PBSA To validate the dynamic stabilityof the complexes total potential energy and the RMSDfor the backbone atoms along the 10 ns MD trajectoriesusing the initial minimized docking structure as a ref-erence were monitored in Figure 1 The RMSD values ofmost complexes converged around 25ndash35 A which meansthe MD trajectories of the complexes appear to be wellequilibrated Some systems of scFv anti-p17 mutant com-plexes were not stable and RMSD still did not converge after10 ns In order to investigate protein binding capability theresults derived fromMM-PBSA andMM-GBSA calculations(Figures 3(c) and 3(d)) were compared and MM-PBSAshows higher correlation with experimental valueThereforethe value of PBTOT was used to compare the simulationwith the peptide ELISA results The more negative thevalue the more favorable the binding The binding energiesidentified by the MM-PBSA protocol were ranked as followspeptide p171 gt p178 gt p173simp177 gt p179 with the val-ues of minus2993 minus2570 minus2515 minus2515 and minus1488 kcalmolrespectively The major contributions to the binding freeenergy arise from the electrostatic energy as calculated bythe molecular mechanic (MM) force field (ELE) and fromthe electrostatic contribution to the solvation free energyas calculated by PB (PBCAL) van der Waals contributionarises from MM (VDW) For the five binding peptides withthe wild type scFv Δ119864vdW Δ119866PB and Δ119866GB are quite similar
BioMed Research International 5
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus96
0 1 2 3 4 5 6 7 8 9 10Ep
tot (
kcal
mol
)Time (ns)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
(a)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
Time (ns)
(b)
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus960 1 2 3 4 5 6 7 8 9 10
Epto
t (kc
alm
ol)
M100R-p171M100R-p177M100R-p179
Time (ns)
(c)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
Time (ns)
M100R-Lg1M100R-Lg7M100R-Lg9
(d)
Figure 1 The potential energy (kcalmol) and the root-mean-squared deviations (RMSD) of the backbone atoms compared to the dockingstructure for mutants (M100G M100R) of scFv anti-p17 bound to its variants during the production run were monitored
but the electrostatic energies were quite varied among the lowand high activities peptides indicating that the main factordetermining the binding activitymight arise from the electro-static contribution as peptide p177 had a very low electrostatic
contribution (minus4628 kcalmol) compared to other sequences(Figure 3(a))
In order to elucidate the key residues in the bindingpocket of the scFv anti-p17 and themost favorable interaction
6 BioMed Research International
6184
7048641
5669 60275565
4639
6455
4675
0
10
20
30
40
50
60
70
80
90
p171 p172 p173 p174 p175 p176 p177 p178 p179
Wild typeM100GM100AM100CM100F
M100HM100KM100RM100EM100P
scFv anti-p17
minusCDOCKER interaction energy(kcalmol)
Figure 2 The ndashCDOCKER interaction energy (kcalmol) of orig-inal scFv anti-p17 bound to its variants was compared with thedifferent mutants from point mutation at MET100
modes in this study computational alanine scanning wasperformed This method depends on the assumption thatlocal changes of the protein do not influence the wholeconformation of the complex significantly In the calcula-tion with alanine scanning the positive and negative rela-tive values in Figure 4 indicate the unfavorable and favor-able for alanine substitution respectively All the residuesin the hypervariable portions of the loops on the heavychain (H1ndashH3) and light chain (L1ndashL3) of wild-type scFvanti-p17 have been mutated to alanine except for the prolineresidue whose backbone is remarkably different from that ofalanine In comparison with wild type the calculated bindingfree energy and computational alanine scanning analysis(Figure 4) demonstrated the importance of the residues ofscFv anti-p17 in the binding pocket which are TRP50 ASN52GLU57 MET100 LEU185 and LYS188 with the relativedecomposed energy below minus200 kcalmol Our previousresults demonstrated that the specific residues located inthe complementary determining regions (CDRs) of scFvanti-p17 MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities withthe HIV-1 p17 variants [6] Therefore from both resultsthe importance of the mutationrsquos location has 10 positionsin the CDRs of scFv anti-p17 consist of TRP50 ASN52GLU57 MET100 LYS101 ASN169 LEU185 LYS188 HIS228and LEU229 The methionine at position 100 (MET100)is initially selected for mutation due to the high con-tribution in binding energy of this region from position100ndash104 residues and the substitution of alanine at this
position showed the significant change in the bindinginteraction
32 Structural Analysis of scFv Anti-p17 Point MutationsWith the combination of pairwise decomposition energiesand computational alanine scanning the key amino acidsin binding TRP50 ASN52 GLU57 MET100 LYS101ASN169 LEU185 LYS188 HIS228 and LEU229 were theimportant residues to binding efficiency of natural HIVepitope at the C-terminal on p17 Therefore these residuesare subjected to mutation in order to improve the bindingactivities The residues roughly equivalent were groupedtogether in five subsets according to statistically preferredsubstitutions which generally correlate with side chainphysicochemical properties observed in buried residuesand exposed residues reported by Bordo and Argos [15] asfollows (a) buried in the protein core (solvent-accessiblesurface for both residues le 10 A2) (b) exposed (solvent-accessible surface area ge 30 A2) and (c) all the possibleaccessibility states allowed The mutation of amino acidsin the CDRs of scFv anti-p17 has been initially selectedat position M100 since the pairwise decomposed energyindicated the unfavorable residue interaction and thesubstitution of the residue with alanine can lead to significantchange in binding affinity Mutation of methionine residuesin position 100 of scFv anti-p17 (wild type) was mutatedto another polarity group with electrically charged oruncharged or hydrophobic side chains which have theimpact on the development of protein cores in structuresmaintaining main-chain fold [23] according to Table 1Finally the mutated scFv anti-p17 was docked with ninepeptides by CDOCKER The energies obtained from thedocking of each peptide with the mutated scFv are listedin Table 1 The more negative interaction energies exhibitthe more favorable binding The prediction of interactionenergy with most peptides and mutant forms (M100G) ismore than that of wild type scFv anti-p17 and the improvedelectrostatic contribution was observed as in Figure 3(a)for the mutant type The hydrogen bond interactions ofmutant form of scFv anti-p17 with five peptides at theresidues ASP31 TYR32 ASN52 THR59 SER99 GLY100LYS101 LYS165 TYR184 LYS188 LEU189 LEU229 andGLN231 were shown in Figure 5 Both of the two peptides(p171 and p173) showed interactions with the receptormostly at TYR32 THR59 and LYS165 and other threepeptides (p177 p178 and p179) showed interactions withthe receptor mostly at ASN52 GLY100 and TYR184 Fromthe conformation with the lowest CDOCKER interactionenergy structure of M100G scFv anti-p17 the peptides boundin two orientations where the N-terminal (p171ndashp176)and the C-terminal (p177ndashp179) of peptide sequenceswere directed toward the binding pocket The calculateddocking interaction energy between single mutation frommethionine 100 to glycine (M100G) and peptide sequencesp171 (DTGHSSQVSQNY) p173 (DTGHSSQISQNY) p177(DTGHSSQASQNY) p178 (DTGHSKQVSQNY) p179(DTGNNSQVSQNY) has shown the favorably interactionenergy compared to wild type which correlated well with
BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

4 BioMed Research International
Table 1 ndashCDOCKER interaction energy (kcalmol) of mutant (M100G) scFv anti-p17 bound to its variants
AntibodyndashCDOCKER interaction energy
p171 p172 p173 p174 p175 p176 p177 p178 p179Competitive ELISA percentageinhibition values ()Mean plusmn STDEV
(1) 759(2) 901830 plusmn 100
mdash552596574 plusmn 31
mdash mdash mdash446601524 plusmn 110
795876836 plusmn 57
mdash885885 plusmn 00
scFv anti-p17 wild type 6184 7048 6410 5669 6027 5565 4639 6455 4675M100G 6086 4502 6516 6023 6305 5329 5259 6516 5455M100A 4689 5679 4692 5962 7361 5666 5136 5633 5878M100C 5926 6416 4763 6066 4772 5067 6226 7026 6937M100F 4375 5351 6593 5219 5525 5470 7620 6093 5983M100H 6023 5855 5807 6374 7578 7233 6568 5238 7155M100K 6772 6093 6099 5223 5908 7371 5363 6375 7268M100R 6978 7214 7264 8519 7063 7299 6685 5433 6646M100E 5924 6082 5407 5895 4789 5639 5502 6042 6495M100P 5772 5158 4461 6683 4935 5987 4563 5804 7810Note Highlight in bold letter indicates the lower interaction energy than original scFv Sequences of p171ndashp179 are 121DTGHSSQVSQNY132121DTGHSNQVSQNY132 121DTGHSSQISQNY132 121DTGHNSQVSQNY132 121NTGHSSQVSQNY132 121DTGNSSQVSQNY132 121DTGHSSQASQNY132121DTGHSKQVSQNY132 and 121DTGNNSQVSQNY132 respectively
9min 10U of DpnI restriction enzyme (Stratagene) wassubsequently added to eliminate phagemid template andincubated for 1 hour at 37∘C The reaction tube was subse-quently placed on ice for 2minThis synthesized product wasfurther transformed into E coli XL-1 Blue Bacterial contain-ing mutant phagemid was then cultured for production ofphage-displayedmutant scFv anti-p17 as described elsewhere[6] To evaluate the binding activity of wild type and mutantscFv anti-p17 with a series of synthetic peptides (GenScriptPiscataway New Jersey USA) phage ELISA was set up asdescribed in our previous study [6]
3 Results and Discussion
31 Pairwise Decomposition Energies and ComputationalAlanine Scanning The comparison of experimental activi-ties peptide ELISA with the results of CDOCKER intera-ction energy derived from molecular docking (CDOCKER)suggested that the experimental value had a high cor-relation (1199032 = 084) with the CDOCKER interactionenergy (Figure 2) From Table 1 peptide p177 had the lowestscore (less favorable interaction energy) and p178 had thehighest score (more favorable interaction energy) whereasp173 had very similar score to that of the wild-type pep-tide The peptide ELISA was used to describe the bindingactivity of scFv anti-p17 to its target peptide (p171) andfour mutant peptides (p173 p177 p178 and p179) Positivesignals were observed in all peptide coated wells indi-cating that this scFv anti-p17 could bind to all mutantpeptides Peptide p178 gave the highest signal followed byp171 p173 p179 and p177 respectively Although a goodcorrelation between the docking scores and ELISA compet-itive binding activity was found there is some difference
between p178 and p173 It should be remarked here thatonly slightly difference between p178 and p173 was observedwith a limitation of receptor rigidity in molecular dockingin the screening process Furthermore we selected fivepeptide epitopes consisting of one wild-type peptide (p171)and four mutated peptides (p173 p177 p178 and p179)for further investigation by molecular dynamics simulations(MDs) and MM-PBSA To validate the dynamic stabilityof the complexes total potential energy and the RMSDfor the backbone atoms along the 10 ns MD trajectoriesusing the initial minimized docking structure as a ref-erence were monitored in Figure 1 The RMSD values ofmost complexes converged around 25ndash35 A which meansthe MD trajectories of the complexes appear to be wellequilibrated Some systems of scFv anti-p17 mutant com-plexes were not stable and RMSD still did not converge after10 ns In order to investigate protein binding capability theresults derived fromMM-PBSA andMM-GBSA calculations(Figures 3(c) and 3(d)) were compared and MM-PBSAshows higher correlation with experimental valueThereforethe value of PBTOT was used to compare the simulationwith the peptide ELISA results The more negative thevalue the more favorable the binding The binding energiesidentified by the MM-PBSA protocol were ranked as followspeptide p171 gt p178 gt p173simp177 gt p179 with the val-ues of minus2993 minus2570 minus2515 minus2515 and minus1488 kcalmolrespectively The major contributions to the binding freeenergy arise from the electrostatic energy as calculated bythe molecular mechanic (MM) force field (ELE) and fromthe electrostatic contribution to the solvation free energyas calculated by PB (PBCAL) van der Waals contributionarises from MM (VDW) For the five binding peptides withthe wild type scFv Δ119864vdW Δ119866PB and Δ119866GB are quite similar
BioMed Research International 5
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus96
0 1 2 3 4 5 6 7 8 9 10Ep
tot (
kcal
mol
)Time (ns)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
(a)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
Time (ns)
(b)
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus960 1 2 3 4 5 6 7 8 9 10
Epto
t (kc
alm
ol)
M100R-p171M100R-p177M100R-p179
Time (ns)
(c)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
Time (ns)
M100R-Lg1M100R-Lg7M100R-Lg9
(d)
Figure 1 The potential energy (kcalmol) and the root-mean-squared deviations (RMSD) of the backbone atoms compared to the dockingstructure for mutants (M100G M100R) of scFv anti-p17 bound to its variants during the production run were monitored
but the electrostatic energies were quite varied among the lowand high activities peptides indicating that the main factordetermining the binding activitymight arise from the electro-static contribution as peptide p177 had a very low electrostatic
contribution (minus4628 kcalmol) compared to other sequences(Figure 3(a))
In order to elucidate the key residues in the bindingpocket of the scFv anti-p17 and themost favorable interaction
6 BioMed Research International
6184
7048641
5669 60275565
4639
6455
4675
0
10
20
30
40
50
60
70
80
90
p171 p172 p173 p174 p175 p176 p177 p178 p179
Wild typeM100GM100AM100CM100F
M100HM100KM100RM100EM100P
scFv anti-p17
minusCDOCKER interaction energy(kcalmol)
Figure 2 The ndashCDOCKER interaction energy (kcalmol) of orig-inal scFv anti-p17 bound to its variants was compared with thedifferent mutants from point mutation at MET100
modes in this study computational alanine scanning wasperformed This method depends on the assumption thatlocal changes of the protein do not influence the wholeconformation of the complex significantly In the calcula-tion with alanine scanning the positive and negative rela-tive values in Figure 4 indicate the unfavorable and favor-able for alanine substitution respectively All the residuesin the hypervariable portions of the loops on the heavychain (H1ndashH3) and light chain (L1ndashL3) of wild-type scFvanti-p17 have been mutated to alanine except for the prolineresidue whose backbone is remarkably different from that ofalanine In comparison with wild type the calculated bindingfree energy and computational alanine scanning analysis(Figure 4) demonstrated the importance of the residues ofscFv anti-p17 in the binding pocket which are TRP50 ASN52GLU57 MET100 LEU185 and LYS188 with the relativedecomposed energy below minus200 kcalmol Our previousresults demonstrated that the specific residues located inthe complementary determining regions (CDRs) of scFvanti-p17 MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities withthe HIV-1 p17 variants [6] Therefore from both resultsthe importance of the mutationrsquos location has 10 positionsin the CDRs of scFv anti-p17 consist of TRP50 ASN52GLU57 MET100 LYS101 ASN169 LEU185 LYS188 HIS228and LEU229 The methionine at position 100 (MET100)is initially selected for mutation due to the high con-tribution in binding energy of this region from position100ndash104 residues and the substitution of alanine at this
position showed the significant change in the bindinginteraction
32 Structural Analysis of scFv Anti-p17 Point MutationsWith the combination of pairwise decomposition energiesand computational alanine scanning the key amino acidsin binding TRP50 ASN52 GLU57 MET100 LYS101ASN169 LEU185 LYS188 HIS228 and LEU229 were theimportant residues to binding efficiency of natural HIVepitope at the C-terminal on p17 Therefore these residuesare subjected to mutation in order to improve the bindingactivities The residues roughly equivalent were groupedtogether in five subsets according to statistically preferredsubstitutions which generally correlate with side chainphysicochemical properties observed in buried residuesand exposed residues reported by Bordo and Argos [15] asfollows (a) buried in the protein core (solvent-accessiblesurface for both residues le 10 A2) (b) exposed (solvent-accessible surface area ge 30 A2) and (c) all the possibleaccessibility states allowed The mutation of amino acidsin the CDRs of scFv anti-p17 has been initially selectedat position M100 since the pairwise decomposed energyindicated the unfavorable residue interaction and thesubstitution of the residue with alanine can lead to significantchange in binding affinity Mutation of methionine residuesin position 100 of scFv anti-p17 (wild type) was mutatedto another polarity group with electrically charged oruncharged or hydrophobic side chains which have theimpact on the development of protein cores in structuresmaintaining main-chain fold [23] according to Table 1Finally the mutated scFv anti-p17 was docked with ninepeptides by CDOCKER The energies obtained from thedocking of each peptide with the mutated scFv are listedin Table 1 The more negative interaction energies exhibitthe more favorable binding The prediction of interactionenergy with most peptides and mutant forms (M100G) ismore than that of wild type scFv anti-p17 and the improvedelectrostatic contribution was observed as in Figure 3(a)for the mutant type The hydrogen bond interactions ofmutant form of scFv anti-p17 with five peptides at theresidues ASP31 TYR32 ASN52 THR59 SER99 GLY100LYS101 LYS165 TYR184 LYS188 LEU189 LEU229 andGLN231 were shown in Figure 5 Both of the two peptides(p171 and p173) showed interactions with the receptormostly at TYR32 THR59 and LYS165 and other threepeptides (p177 p178 and p179) showed interactions withthe receptor mostly at ASN52 GLY100 and TYR184 Fromthe conformation with the lowest CDOCKER interactionenergy structure of M100G scFv anti-p17 the peptides boundin two orientations where the N-terminal (p171ndashp176)and the C-terminal (p177ndashp179) of peptide sequenceswere directed toward the binding pocket The calculateddocking interaction energy between single mutation frommethionine 100 to glycine (M100G) and peptide sequencesp171 (DTGHSSQVSQNY) p173 (DTGHSSQISQNY) p177(DTGHSSQASQNY) p178 (DTGHSKQVSQNY) p179(DTGNNSQVSQNY) has shown the favorably interactionenergy compared to wild type which correlated well with
BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

BioMed Research International 5
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus96
0 1 2 3 4 5 6 7 8 9 10Ep
tot (
kcal
mol
)Time (ns)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
(a)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
M100G-p171M100G-p173M100G-p177
M100G-p178M100G-p179
Time (ns)
(b)
minus110
minus108
minus106
minus104
minus102
minus100
minus98
minus960 1 2 3 4 5 6 7 8 9 10
Epto
t (kc
alm
ol)
M100R-p171M100R-p177M100R-p179
Time (ns)
(c)
00
10
20
30
40
50
60
0 1 2 3 4 5 6 7 8 9 10
RMSD
(Ang
s)
Time (ns)
M100R-Lg1M100R-Lg7M100R-Lg9
(d)
Figure 1 The potential energy (kcalmol) and the root-mean-squared deviations (RMSD) of the backbone atoms compared to the dockingstructure for mutants (M100G M100R) of scFv anti-p17 bound to its variants during the production run were monitored
but the electrostatic energies were quite varied among the lowand high activities peptides indicating that the main factordetermining the binding activitymight arise from the electro-static contribution as peptide p177 had a very low electrostatic
contribution (minus4628 kcalmol) compared to other sequences(Figure 3(a))
In order to elucidate the key residues in the bindingpocket of the scFv anti-p17 and themost favorable interaction
6 BioMed Research International
6184
7048641
5669 60275565
4639
6455
4675
0
10
20
30
40
50
60
70
80
90
p171 p172 p173 p174 p175 p176 p177 p178 p179
Wild typeM100GM100AM100CM100F
M100HM100KM100RM100EM100P
scFv anti-p17
minusCDOCKER interaction energy(kcalmol)
Figure 2 The ndashCDOCKER interaction energy (kcalmol) of orig-inal scFv anti-p17 bound to its variants was compared with thedifferent mutants from point mutation at MET100
modes in this study computational alanine scanning wasperformed This method depends on the assumption thatlocal changes of the protein do not influence the wholeconformation of the complex significantly In the calcula-tion with alanine scanning the positive and negative rela-tive values in Figure 4 indicate the unfavorable and favor-able for alanine substitution respectively All the residuesin the hypervariable portions of the loops on the heavychain (H1ndashH3) and light chain (L1ndashL3) of wild-type scFvanti-p17 have been mutated to alanine except for the prolineresidue whose backbone is remarkably different from that ofalanine In comparison with wild type the calculated bindingfree energy and computational alanine scanning analysis(Figure 4) demonstrated the importance of the residues ofscFv anti-p17 in the binding pocket which are TRP50 ASN52GLU57 MET100 LEU185 and LYS188 with the relativedecomposed energy below minus200 kcalmol Our previousresults demonstrated that the specific residues located inthe complementary determining regions (CDRs) of scFvanti-p17 MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities withthe HIV-1 p17 variants [6] Therefore from both resultsthe importance of the mutationrsquos location has 10 positionsin the CDRs of scFv anti-p17 consist of TRP50 ASN52GLU57 MET100 LYS101 ASN169 LEU185 LYS188 HIS228and LEU229 The methionine at position 100 (MET100)is initially selected for mutation due to the high con-tribution in binding energy of this region from position100ndash104 residues and the substitution of alanine at this
position showed the significant change in the bindinginteraction
32 Structural Analysis of scFv Anti-p17 Point MutationsWith the combination of pairwise decomposition energiesand computational alanine scanning the key amino acidsin binding TRP50 ASN52 GLU57 MET100 LYS101ASN169 LEU185 LYS188 HIS228 and LEU229 were theimportant residues to binding efficiency of natural HIVepitope at the C-terminal on p17 Therefore these residuesare subjected to mutation in order to improve the bindingactivities The residues roughly equivalent were groupedtogether in five subsets according to statistically preferredsubstitutions which generally correlate with side chainphysicochemical properties observed in buried residuesand exposed residues reported by Bordo and Argos [15] asfollows (a) buried in the protein core (solvent-accessiblesurface for both residues le 10 A2) (b) exposed (solvent-accessible surface area ge 30 A2) and (c) all the possibleaccessibility states allowed The mutation of amino acidsin the CDRs of scFv anti-p17 has been initially selectedat position M100 since the pairwise decomposed energyindicated the unfavorable residue interaction and thesubstitution of the residue with alanine can lead to significantchange in binding affinity Mutation of methionine residuesin position 100 of scFv anti-p17 (wild type) was mutatedto another polarity group with electrically charged oruncharged or hydrophobic side chains which have theimpact on the development of protein cores in structuresmaintaining main-chain fold [23] according to Table 1Finally the mutated scFv anti-p17 was docked with ninepeptides by CDOCKER The energies obtained from thedocking of each peptide with the mutated scFv are listedin Table 1 The more negative interaction energies exhibitthe more favorable binding The prediction of interactionenergy with most peptides and mutant forms (M100G) ismore than that of wild type scFv anti-p17 and the improvedelectrostatic contribution was observed as in Figure 3(a)for the mutant type The hydrogen bond interactions ofmutant form of scFv anti-p17 with five peptides at theresidues ASP31 TYR32 ASN52 THR59 SER99 GLY100LYS101 LYS165 TYR184 LYS188 LEU189 LEU229 andGLN231 were shown in Figure 5 Both of the two peptides(p171 and p173) showed interactions with the receptormostly at TYR32 THR59 and LYS165 and other threepeptides (p177 p178 and p179) showed interactions withthe receptor mostly at ASN52 GLY100 and TYR184 Fromthe conformation with the lowest CDOCKER interactionenergy structure of M100G scFv anti-p17 the peptides boundin two orientations where the N-terminal (p171ndashp176)and the C-terminal (p177ndashp179) of peptide sequenceswere directed toward the binding pocket The calculateddocking interaction energy between single mutation frommethionine 100 to glycine (M100G) and peptide sequencesp171 (DTGHSSQVSQNY) p173 (DTGHSSQISQNY) p177(DTGHSSQASQNY) p178 (DTGHSKQVSQNY) p179(DTGNNSQVSQNY) has shown the favorably interactionenergy compared to wild type which correlated well with
BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990
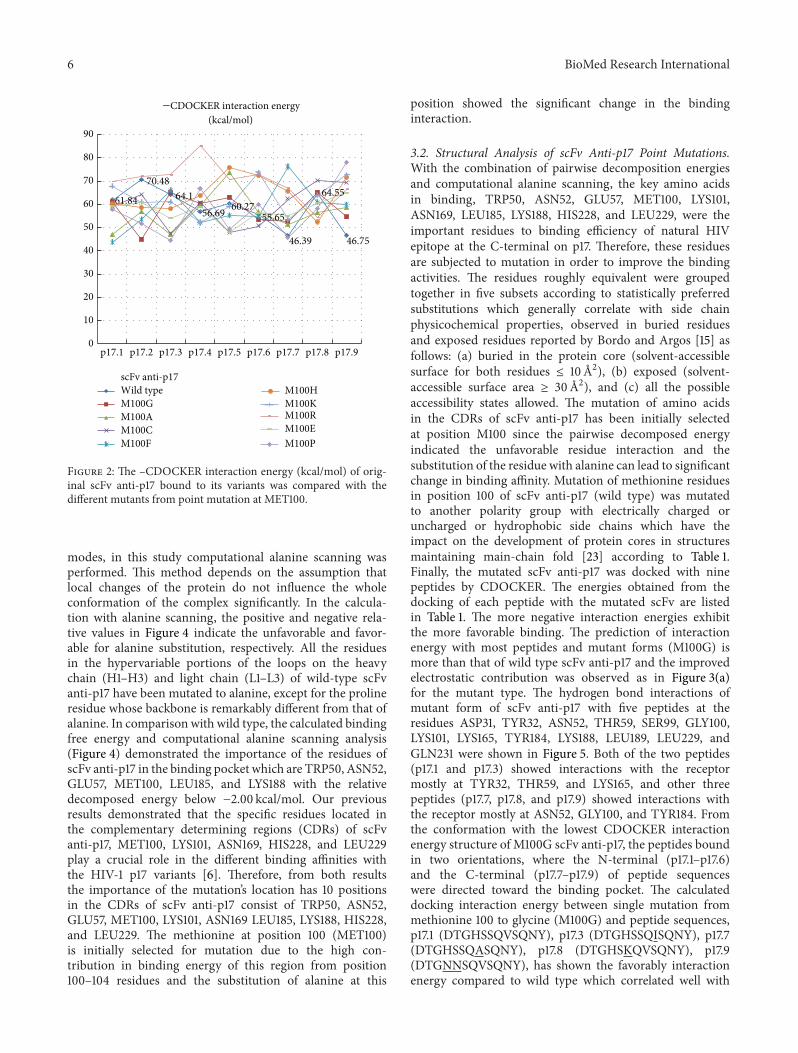
6 BioMed Research International
6184
7048641
5669 60275565
4639
6455
4675
0
10
20
30
40
50
60
70
80
90
p171 p172 p173 p174 p175 p176 p177 p178 p179
Wild typeM100GM100AM100CM100F
M100HM100KM100RM100EM100P
scFv anti-p17
minusCDOCKER interaction energy(kcalmol)
Figure 2 The ndashCDOCKER interaction energy (kcalmol) of orig-inal scFv anti-p17 bound to its variants was compared with thedifferent mutants from point mutation at MET100
modes in this study computational alanine scanning wasperformed This method depends on the assumption thatlocal changes of the protein do not influence the wholeconformation of the complex significantly In the calcula-tion with alanine scanning the positive and negative rela-tive values in Figure 4 indicate the unfavorable and favor-able for alanine substitution respectively All the residuesin the hypervariable portions of the loops on the heavychain (H1ndashH3) and light chain (L1ndashL3) of wild-type scFvanti-p17 have been mutated to alanine except for the prolineresidue whose backbone is remarkably different from that ofalanine In comparison with wild type the calculated bindingfree energy and computational alanine scanning analysis(Figure 4) demonstrated the importance of the residues ofscFv anti-p17 in the binding pocket which are TRP50 ASN52GLU57 MET100 LEU185 and LYS188 with the relativedecomposed energy below minus200 kcalmol Our previousresults demonstrated that the specific residues located inthe complementary determining regions (CDRs) of scFvanti-p17 MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities withthe HIV-1 p17 variants [6] Therefore from both resultsthe importance of the mutationrsquos location has 10 positionsin the CDRs of scFv anti-p17 consist of TRP50 ASN52GLU57 MET100 LYS101 ASN169 LEU185 LYS188 HIS228and LEU229 The methionine at position 100 (MET100)is initially selected for mutation due to the high con-tribution in binding energy of this region from position100ndash104 residues and the substitution of alanine at this
position showed the significant change in the bindinginteraction
32 Structural Analysis of scFv Anti-p17 Point MutationsWith the combination of pairwise decomposition energiesand computational alanine scanning the key amino acidsin binding TRP50 ASN52 GLU57 MET100 LYS101ASN169 LEU185 LYS188 HIS228 and LEU229 were theimportant residues to binding efficiency of natural HIVepitope at the C-terminal on p17 Therefore these residuesare subjected to mutation in order to improve the bindingactivities The residues roughly equivalent were groupedtogether in five subsets according to statistically preferredsubstitutions which generally correlate with side chainphysicochemical properties observed in buried residuesand exposed residues reported by Bordo and Argos [15] asfollows (a) buried in the protein core (solvent-accessiblesurface for both residues le 10 A2) (b) exposed (solvent-accessible surface area ge 30 A2) and (c) all the possibleaccessibility states allowed The mutation of amino acidsin the CDRs of scFv anti-p17 has been initially selectedat position M100 since the pairwise decomposed energyindicated the unfavorable residue interaction and thesubstitution of the residue with alanine can lead to significantchange in binding affinity Mutation of methionine residuesin position 100 of scFv anti-p17 (wild type) was mutatedto another polarity group with electrically charged oruncharged or hydrophobic side chains which have theimpact on the development of protein cores in structuresmaintaining main-chain fold [23] according to Table 1Finally the mutated scFv anti-p17 was docked with ninepeptides by CDOCKER The energies obtained from thedocking of each peptide with the mutated scFv are listedin Table 1 The more negative interaction energies exhibitthe more favorable binding The prediction of interactionenergy with most peptides and mutant forms (M100G) ismore than that of wild type scFv anti-p17 and the improvedelectrostatic contribution was observed as in Figure 3(a)for the mutant type The hydrogen bond interactions ofmutant form of scFv anti-p17 with five peptides at theresidues ASP31 TYR32 ASN52 THR59 SER99 GLY100LYS101 LYS165 TYR184 LYS188 LEU189 LEU229 andGLN231 were shown in Figure 5 Both of the two peptides(p171 and p173) showed interactions with the receptormostly at TYR32 THR59 and LYS165 and other threepeptides (p177 p178 and p179) showed interactions withthe receptor mostly at ASN52 GLY100 and TYR184 Fromthe conformation with the lowest CDOCKER interactionenergy structure of M100G scFv anti-p17 the peptides boundin two orientations where the N-terminal (p171ndashp176)and the C-terminal (p177ndashp179) of peptide sequenceswere directed toward the binding pocket The calculateddocking interaction energy between single mutation frommethionine 100 to glycine (M100G) and peptide sequencesp171 (DTGHSSQVSQNY) p173 (DTGHSSQISQNY) p177(DTGHSSQASQNY) p178 (DTGHSKQVSQNY) p179(DTGNNSQVSQNY) has shown the favorably interactionenergy compared to wild type which correlated well with
BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

BioMed Research International 7
p171 p173 p177 p178 p179ΔE
elect
rosta
tic(k
calm
ol)
minus250
minus200
minus150
minus100
minus50
0
(a)
minus60
minus50
minus40
minus30
minus20
minus10
0
ΔE
vDW
(kca
lmol
)
p171 p173 p177 p178 p179
(b)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
ΔG
PB(k
calm
ol)
Wild typeM100G
(c)
ΔG
GB
(kca
lmol
)
minus60
minus50
minus40
minus30
minus20
minus10
0p171 p173 p177 p178 p179
Wild typeM100G
(d)
Figure 3 Decomposition energy of amino acids in the CDRs from 2 ns simulations with series of peptides 171 173 177 178 and 179 forwild and mutant (M100G) type scFv anti-p17 (a) Δ119864electrostatic (b) Δ119864vdW (c) Δ119866PB (d) Δ119866GB A total of 500 snapshots were collected at 1 psintervals from the last 500 ps of 2 ns MD for binding free energy analysis
the indirect ELISA (Figure 6) Detail on experimental resultswill be discussed in the following section
33 Comparison of CalculatedBinding Free Energywith Exper-imental Data From the indirect ELISA results in Figure 6the different signal from the same number of added phageparticle (1011 and 1010 CFUml) was observed We identifiedp177 and p179 as the low affinity binding peptides whereasthe p171 p173 and p178 were identified as the high affinitybinding peptides with our scFv wild type In the same figurethe signal of phage displaying scFv anti-p17 mutant (M100Rand M100G) was higher than that of scFv wild type reflectedthe higher binding activity suggested that both mutants canimprove the binding affinity with the peptide HoweverM100G in phage 1010 CFUml shows significantly improve-ment in binding affinity a lot more than M100R The binding
efficiencieswere ranked as follows peptide p178gtp171gtp173gt p179 gt p177 for M100G However the energy calculationfrom CDOCKER did not explain very well why M100G isbetter than M100R According to amino acids characteristicsand structures for our residue mutation arginine has thelong side chain with the electrically positive charged aminogroups glycine is considered small nonpolar side chain andweakly hydrophobic where methionine as in the scFv wildtype has larger side chains and is more strongly hydrophobicPossibly the primary effect of arginine at M100R might bedue to the disruption of the assembly between 119881
119867and 119881
119871
as it causes the loss of important hydrogen bonds mediatedby the M100R side chain including a conserved interfacehydrogen bond Comparison of the complex stability wasmonitored fromRMSD in Figure 1We have observed severalinstable complexes of peptides p173 and p178 with M100Rand cannot process for MD simulations From Figure 3 the
8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

8 BioMed Research International
ASP
31 (H
1)TY
R32
(H1)
MET
34 (H
1)A
SN35
(H1)
TRP5
0 (H
2)IL
E51
(H2)
ASN
52 (H
2)TH
R53
(H2)
TYR5
4 (H
2)TH
R55
(H2)
GLU
57 (H
2)TH
R59
(H2)
TYR6
0 (H
2)A
SP62
(H2)
GLU
63 (H
2)PH
E64
(H2)
LYS6
5 (H
2)SE
R99
(H3)
MET
100
(H3)
LYS1
01 (H
3)SE
R103
(H3)
TYR1
04 (H
3)LY
S154
(L1)
SET1
55 (L
1)SE
R156
(L1)
GLN
157
(L1)
SER1
58 (L
1)LE
U15
9 (L
1)LE
U16
0 (L
1)SE
R162
(L1)
ASP
163
(L1)
LYS1
65 (L
1)TH
R166
(L1)
PHE1
67 (L
1)LE
U16
8 (L
1)A
SN16
9 (L
1)LE
U18
5 (L
2)V
AL1
86 (L
2)SE
R187
(L2)
LYS1
88 (L
2)LE
U18
9 (L
2)A
SP19
0 (L
2)TR
P224
(L3)
GLN
225
(L3)
THR2
27 (L
3)H
IE22
8 (L
3)LE
U22
9 (L
3)G
LN23
1 (L
3)TH
R232
(L3)
minus600
minus500
minus400
minus300
minus200
minus100
000
100
200
300
Pairwise decomposition energy of scFv with p171 (2 ns)Pairwise decomposition energy of scFv with p171 (10 ns)Relative decomposition energy from alanine scanningcompared to wild type scFv anti-HIV-1 p17
Dec
ompo
sitio
nre
lativ
e dec
ompo
sitio
nen
ergy
(kca
lmol
)
Figure 4 Histograms reporting the calculated pairwise decomposition energy which negative numbers in Δ119866bind mean highly favorablebinding and the relative decomposition energy (Δ119866bind(ala) minus Δ119866bind(wt)) from the computational alanine scanning mutagenesis experimentscompared to wild type of scFv with p171The total bar height reflects the relative binding free energies of each amino acid in CDRs loops withwild type of scFv anti-p17 whose mutation to alanine by alanine scanning mutagenesis The negative numbers indicated the preference foralanine mutation Pairwise decomposition analysis 2 ns was extracted from 500 snapshots from 15ndash2 ns whereas the analysis 10 ns was from500 snapshots from 5ndash10 ns simulations
electrostatic contributions have been significantly improvedwith series of peptides 171 173 177 178 and 179 for mutant(M100G) compared towild-type scFv-p17while other param-eters such as Δ119864vdW Δ119866PB and Δ119866GB did not show muchvariation
4 Conclusion
The identification of the key residues of scFv in the com-plementarity determining regions (CDRs) from the combi-nation of the computational alanine scanning and pairwisedecomposition energy calculation can be used to design thenew potential scFv anti-p17 From the result the importanceof the residues which highly effect by alanine scanning of
scFv anti-p17 are TRP50 ASN52 GLU57 MET100 LEU185and LYS188 whereas from pairwise decomposition energycalculation MET100 LYS101 ASN169 HIS228 and LEU229play a crucial role in the different binding affinities with theHIV-1 p17 variants The new antibodies were designed bymutating the potential amino acid residues in CDRs of scFvanti-p17 With the guide from both methods the key residueat MET100 was initially selected to a single point mutationThe fast protocol of docking interaction energies can beused to estimate the binding affinity of the new scFvs withthe series of natural peptides The electrostatic contributionshave been a major part in the antibody design while otherparameters such as Δ119864vdW Δ119866PB and Δ119866GB did not showmuch variation Long time scaleMD simulations canmonitorthe stability of the novel scFv anti-p17 complexes Concern
BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

BioMed Research International 9
Asp163
Ser99Lys165
Thr59
C-terminal
N-terminal
Gly226
(a1)
Asp31
Asn52Lys165
Tyr32
Thr59
C-terminal
N-terminal
Asp31
2Ly
Tyr32
Thr59ThTh
C-termin
inal
(a2)
Gln231
Gly100
Lys101
Lys165
C-terminal
N-terminalLeu229
231
Gly1000000000000000000000000
Lys10111111111111111111111111111
Lys165
C
N-terminalLeu229
(b2)
Tyr184
Asp31
Lys165
C-terminal
N-terminal
Met100
Lys188
(b1)
Thr59
Tyr184
Asn52
Lys165
Trp50
C-terminal
N-terminal
Ser99
Lys188
(c1)
Asn52
C-terminal
N-terminal
Ser99
n5222222222
C-terminal
N terminal
Ser99999999
Tyr184
Lys165
Gly100
Leu189
(c2)
Asp31
Gly33
Asn52
Lys188
Tyr184
C-terminal
N-terminal
Ser99
(d1)
Asp31
Gly100
Asn52
Lys101 Lys188
Leu189
Tyr184
C-terminal
N-terminal
Asp31
Gly100
Asn52
LysLLLyyyyy 101 Lys18
Leu18
Tyr184yy
C-terminal
N term(d2)
(a)
Figure 5 Continued
10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

10 BioMed Research International
Asp31
Asn52
Asn52Ser99
Lys165
Lys165
Lys188Lys188
Tyr184
C-terminal C-terminal
N-terminal N-terminal
Asn52
Lys165
LTyr184yy
C-terminal
l
Gly100
(e1) (e2)
Thr30
(b)
Figure 5 Docking structural comparison of wild type (1) and mutant form (M100G 2) of scFv anti-p17 against five peptides (a) p171 (b)p173 (c) p177 (d) p178 and (e) p179 respectively A stick representation of side chains of scFv predicted to interact through hydrogen bondto peptides (scaled ball and stick) Hydrogen bonds are presented as dashed lines
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
p171p173p177
p178p179
M100R1010
(a)
p171p173p177
p178p179
0
02
04
06
08
1
12
14
16
WT 1011 WT 10
10
Bacteriophage (cfumL)
OD
450
(nm
)
M100R1010
(b)
Figure 6 The indirect ELISA results of binding efficiency for scFv anti-p17 wild type and selected mutation M100R (a) and M100G (b) Thevalues shown here are the results from second experiments
on the disruption of the scFv which affects the bindingactivity due to the mutation is subject to further investiga-tion Peptide ELISA results confirmed the improved bindingaffinity of novel scFv anti-p17 mutants from the theoreticalcalculations
Conflict of Interests
There was no conflict of interests nor a financial disclosurefor any of the authors
Acknowledgments
The authors would like to express grateful acknowledgementto the Thailand Research Fund (TRF) the Commission onHigher Education (Thailand) the NSTDA Research ChairGrant National Sciences and Technology Development Age-ncy (Thailand) the Center for Innovation in Chemistry(PERCH-CIC) and the National Research University Projectunder Thailandrsquos Office of the Higher Education Commiss-ion for support for support This research is partly funded
BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

BioMed Research International 11
Solvent
Vacuo
epitope
epitope
complex
complex
ΔGbind
ΔGgas
GP
solv minusΔminusΔ GL
solv ΔGC
solv
ΔGbind = ΔGgas minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
= ΔHgas minus TΔS minus ΔGP
solv minus ΔGL
solv + ΔGC
solv
ΔHgas asymp ΔEgas = ΔEelec + ΔEvdW + ΔEintra
scFv anti-p17 +
scFv anti-p17 +
Figure 7
with the support of University Malaya Research Grantunder UMBIO Research Cluster (UMRG-Project no RP002-2012D) Computation and Informatics (C+i) ResearchClusterHigh Performance Scientific Computing Program(UMRG Project no RP001C-13ICT) and Fundamental Res-earch Grant Scheme (FRGS Project no FP008-2012A andFP013-2013A)
References
[1] A Worn and A Pluckthun ldquoStability engineering of antibodysingle-chain Fv fragmentsrdquo Journal of Molecular Biology vol305 no 5 pp 989ndash1010 2001
[2] IMassova and P A Kollman ldquoComputational alanine scanningto probe protein-protein interactions a novel approach to eval-uate binding free energiesrdquo Journal of the American ChemicalSociety vol 121 no 36 pp 8133ndash8143 1999
[3] H Gohlke C Kiel and D A Case ldquoInsights into protein-protein binding by binding free energy calculation and freeenergy decomposition for the Ras-Raf and Ras-RalGDS com-plexesrdquo Journal of Molecular Biology vol 330 no 4 pp 891ndash9132003
[4] T Li M Froeyen and P Herdewijn ldquoComputational alaninescanning and free energy decomposition for E coli type Isignal peptidase with lipopeptide inhibitor complexrdquo Journalof Molecular Graphics amp Modelling vol 26 no 5 pp 813ndash8232008
[5] L Saız-Urra M A Cabrera and M Froeyen ldquoExploring theconformational changes of the ATP binding site of gyraseB from Escherichia coli complexed with different establishedinhibitors by using molecular dynamics simulation protein-ligand interactions in the light of the alanine scanning and freeenergy decomposition methodsrdquo Journal of Molecular Graphicsamp Modelling vol 29 no 5 pp 726ndash739 2011
[6] V S Lee P Tue-ngeun S Nangola et al ldquoPairwise decomposi-tion of residue interaction energies of single chain Fv with HIV-1 p17 epitope variantsrdquoMolecular Immunology vol 47 no 5 pp982ndash990 2010
[7] H B Thorsteinsdottir T Schwede V Zoete and M MeuwlyldquoHow inaccuracies in protein structure models affect estimatesof protein-ligand interactions computational analysis of HIV-Iprotease inhibitor bindingrdquo Proteins vol 65 no 2 pp 407ndash4232006
[8] S Huo I Massova and P A Kollman ldquoComputational alaninescanning of the 11 human growth hormone-receptor complexrdquoJournal of Computational Chemistry vol 23 no 1 pp 15ndash272002
[9] O Villacanas and J Rubio-Martinez ldquoReducing CDK46-p16INK4a interface computational alanine scanning of a pep-tide bound to CDK6 proteinrdquo Proteins vol 63 no 4 pp 797ndash810 2006
[10] I S Moreira P A Fernandes and M J Ramos ldquoUnravelingthe importance of protein-protein interaction application of acomputational alanine-scanningmutagenesis to the study of theIgG1 streptococcal protein G (C2 Fragment) complexrdquo Journalof Physical Chemistry B vol 110 no 22 pp 10962ndash10969 2006
[11] L T Chong W C Swope J W Pitera and V S Pande ldquoKin-etic computational alanine scanning application to p53 oligo-merizationrdquo Journal of Molecular Biology vol 357 no 3 pp1039ndash1049 2006
[12] V Zoete M Meuwly and M Karplus ldquoStudy of the insulindimerization binding free energy calculations and per-residuefree energy decompositionrdquo Proteins vol 61 no 1 pp 79ndash932005
[13] V Zoete and M Meuwly ldquoImportance of individual sidechains for the stability of a protein fold computational alaninescanning of the insulin monomerrdquo Journal of ComputationalChemistry vol 27 no 15 pp 1843ndash1857 2006
[14] A Malik A Firoz V Jha E Sunderasan and S Ahmad ldquoMod-eling the three-dimensional structures of an unbound single-chain variable fragment (scFv) and its hypothetical complexwith a Corynespora cassiicola toxin cassiicolinrdquo Journal ofMol-ecular Modeling vol 16 no 12 pp 1883ndash1893 2010
[15] D Bordo and P Argos ldquoSuggestions for lsquosafersquo residue substitu-tions in site-directedmutagenesisrdquo Journal ofMolecular Biologyvol 217 no 4 pp 721ndash729 1991
[16] D Tewari S L Goldstein A L Notkins and P Zhou ldquocDNAencoding a single-chain antibody to HIV p17 with cytoplasmicor nuclear retention signals inhibits HIV-1 replicationrdquo Journalof Immunology vol 161 no 5 pp 2642ndash2647 1998
[17] MVieth J DHirst A Kolinski andC L Brooks III ldquoAssessingenergy functions for flexible dockingrdquo Journal of ComputationalChemistry vol 19 no 14 pp 1612ndash1622 1998
[18] D A Case T A Darden T E Cheatham III et al AMBER 12University of California San Francisco Calif USA 2012
12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990

12 BioMed Research International
[19] U Essmann L Perera M L Berkowitz T Darden H Lee andL G Pedersen ldquoA smooth particle mesh Ewald methodrdquo TheJournal of Chemical Physics vol 103 no 19 pp 8577ndash8593 1995
[20] P A Kollman I Massova C Reyes et al ldquoCalculating stru-ctures and free energies of complex molecules combiningmolecular mechanics and continuum modelsrdquo Accounts ofChemical Research vol 33 no 12 pp 889ndash897 2000
[21] T Hou S Guo and X Xu ldquoPredictions of binding of a diverseset of ligands to gelatinase-A by a combination of moleculardynamics and continuum solvent modelsrdquo Journal of PhysicalChemistry B vol 106 no 21 pp 5527ndash5535 2002
[22] T Kortemme D E Kim and D Baker ldquoComputational alaninescanning of protein-protein interfacesrdquo Sciencersquos STKE vol2004 no 219 p pl2 2004
[23] D Burdo and P Argos ldquoEvolution of protein cores Constraintsin point mutations as observed in globin tertiary structuresrdquoJournal of Molecular Biology vol 211 no 4 pp 975ndash988 1990