immobilization of mercury by pyrite (fes2)
TRANSCRIPT

Available online at www.sciencedirect.com
Environmental Pollution 156 (2008) 504e514www.elsevier.com/locate/envpol
Immobilization of mercury by pyrite (FeS2)
Julia Bower a, Kaye S. Savage b, Beth Weinman b, Mark O. Barnett a,*,William P. Hamilton c, Willie F. Harper a
a Department of Civil Engineering, Auburn University, 238 Harbert Engineering Center, Auburn, AL 36849, USAb Department of Earth and Environmental Sciences, Vanderbilt University, 2301 Vanderbilt Place, Nashville, TN 37235, USA
c Department of Civil and Environmental Engineering, Vanderbilt University, 2301 Vanderbilt Place, Nashville, TN 37235, USA
Received 9 August 2007; received in revised form 7 January 2008; accepted 11 January 2008
Pyrite may be an effective subsurface reactive barrier for Hg in groundwater.
Abstract
Elemental mercury (Hg0) is a metal with a number of atypical properties, which has resulted in its use in myriad anthropogenic processes.However, these same properties have also led to severe local subsurface contamination at many places where it has been used. As such, westudied the influence of various parameters on Hg(II) sorption onto pyrite (pH, time, Hg(II) concentration), a potential subsurface reactive bar-rier. Batch sorption studies revealed that total Hg(II) removal increases with both pH and time. X-ray absorption spectroscopy analysis showedthat a transformation in the coordination environment at low pH occurred during aging over 2 weeks, to form an ordered monolayer of mono-dentate HgeCl complexes on pyrite. In column studies packed with pure quartz sand, the transport of Hg(II) was significantly retarded by thepresence of a thin pyrite-sand reactive barrier, although dissolved oxygen inhibited Hg(II) sorption onto pyrite in the column.� 2008 Elsevier Ltd. All rights reserved.
Keywords: Mercury; Pyrite; Remediation; Sulfide
1. Introduction
Mercury is an important yet complex and poorly under-stood pollutant. Fish advisories for Hg have been issuedthroughout the United States and Europe, arguably makingHg the most important persistent bioaccumulative toxin. An-thropogenic combustion processes (e.g., coal-fired powerplants) are typically considered the largest source of Hg inthe environment (EPA, 1997), which has resulted in a substan-tial increase in the global atmospheric Hg burden dating fromthe onset of the industrial revolution (Fitzgerald et al., 1998).In addition to combustion processes, there are also a largenumber of Hg-contaminated industrial and mining sites, whichhas resulted in many instances of gross local subsurface con-tamination. Although the use of Hg is decreasing due to
* Corresponding author. Tel.: þ1 334 844 6291.
E-mail address: [email protected] (M.O. Barnett).
0269-7491/$ - see front matter � 2008 Elsevier Ltd. All rights reserved.
doi:10.1016/j.envpol.2008.01.011
environmental concerns, there is still a tremendous amountof Hg at industrial and government facilities here and abroad.There are eight chlor-alkali plants in the United States (downfrom a high of 35), which together possess almost 2400 metrictons of Hg, with emissions from these facilities possibly ex-ceeding emissions for coal-fired power plants (Johnson,2004; Hogue, 2007). In addition, the U.S. Departments of De-fense and Energy own over 5600 metric tons of stockpiled Hg,and there are over 12,000 metric tons of Hg at chlor-alkaliplants in Europe (Hogue, 2007).
The unique physical properties (very high density, liquid atroom temperature, volatility) that have made Hg attractive toindustry have also made it difficult to contain and recover,and rarely is Hg used without significant environmental re-leases (Turner and Southworth, 1999). In the subsurface,non-aqueous phase Hg0 serves as a long-term source of Hgto groundwater and, eventually, surface water. In an effort toremediate the problem at contaminated sites, Hg-laden soils

Fig. 1. SEM photomicrograph of pyrite showing a distribution of particle sizes.
Average particle size is w75 mm.
505J. Bower et al. / Environmental Pollution 156 (2008) 504e514
have been excavated and incinerated. However, this process isexpensive and can result in Hg air emissions. Furthermore,disturbing the soils often causes the contamination to spread.An alternative approach to immobilizing Hg at contaminatedsites is in situ stabilization.
Mercury (II) is a soft Lewis acid and complexes readilywith soft Lewis bases such as reduced-S ligands. HgS(s) isa natural and perhaps dominant sink for Hg in the global cycle(Stein et al., 1996), including oxic environments, where itsstrong affinity for S may outcompete oxygen for bonding,according to spectroscopic measurements of oxidized mack-inawite (Wolfenden et al., 2005). Addition of H2S(g) to a sus-pension containing dissolved Hg(II) and goethite led to rapidprecipitation of HgS, with Hg in a coordination environmentsimilar to metacinnabar (cubic, black: b-HgS(s)), whether ornot the Hg was initially adsorbed to the goethite; additionalH2S(g) produced polysulfides that promoted subsequent disso-lution of some of the metacinnabar (Slowey and Brown, 2007).HgeS precipitates in nitrate solution initially experience trans-formations among different geometric configurations, includingclusters and chains with varying degrees of order, eventuallyforming crystalline metacinnabar (Charnock et al., 2003). Add-ing reduced S to form HgS(s) has been proposed as a method tostabilize Hg-contaminated soils and materials (Oji, 1998;Fuhrmann et al., 2002; Zhang and Bishop, 2002; Piao andBishop, 2006), including as a stabilizing agent for a long-termHg storage subsurface repository (Svensson et al., 2006).
HgS(s) is relatively insoluble and less volatile than otherforms of Hg and thus potentially less harmful (Willet et al.,1992; Barnett and Turner, 2001). Cinnabar (hexagonal, red,a-HgS(s)) is persistent in exposed ore bodies and mine wastesdue to slow oxidation kinetics, although ore processing activi-ties such as roasting may produce more reactive Hg compounds(Kim et al., 2000). Hence, Hg0 is a higher long-term potentialrisk than HgS(s) in soils and mine spoils (Navarro et al., 2006).
An alternative to in situ stabilization by adding S to soilwould be to construct a subsurface reactive barrier of S-con-taining minerals to intercept and immobilize Hg in groundwa-ter, preventing (or significantly dampening) its effect onnatural water bodies. Sulfide minerals, for which Hg hasa high affinity (Brown et al., 1979), have great thermody-namic potential for Hg immobilization. The most commonsulfide minerals are Fe sulfides, such as pyrite (FeS2), pyrrho-tite (Fe1�xSx), and mackinawite (FeS). Pyrite, the most abun-dant metal sulfide in nature, is a commonly available,inexpensive material that is capable of adsorbing a varietyof toxic elements, including Hg, Pb, Cu, Cd, Cr, and As(Bostick and Fendorf, 2003; Bostick et al., 2003; Doyle etal., 2004; Borah and Senapati, 2006; Ozverdi and Erdem,2006). Pyrite effectively removes Hg from aqueous solutionalong a wide pH range (Brown et al., 1979; Morse andArakaki, 1993; Ehrhardt et al., 2000; Behra et al., 2001).X-ray photoelectron spectroscopy (XPS) studies have beenconducted on Hgepyrite interactions to determine the sorp-tion mechanisms. Both weakly and strongly bound speciesare formed including Hgechloro and Hgesulfhydryl com-plexes, although not HgS(s) itself (Hyland et al., 1990;
Ehrhardt et al., 2000; Behra et al., 2001), perhaps due tothe relatively greater kinetic stability of pyrite than other sul-fide minerals (Hyland et al., 1990).
The objective of this study was to gain an improved under-standing of the potential for using pyrite as a reactive barrier toimmobilize Hg in groundwater. Mercury(II) interactions withpyrite were studied using both batch and column experiments,the latter of which simulate groundwater flow and provide in-sight to behavior under hydrodynamic conditions. ElementalHg often dissolves to form Hg(II), particularly in the presenceof Cl� (Amyot et al., 2005), and studies with dissolved Hg0
are ongoing. Experiments were conducted both in the absenceand presence of O2(aq) to determine both the thermodynamicpotential of pyrite to remove Hg(II) and the potential effectsthat O2(aq) could have on a pyrite-based reactive barrier. Com-plementary X-ray absorption spectroscopy was carried out toassess the molecular bonding environment of Hg that was incontact with pyrite under different solution conditions andaging times.
2. Methods and materials
2.1. Pyrite
The pyrite used in this study was obtained from Wards Natural Science Co.
It was crushed, pulverized with a mortar and pestle, and then sieved to
<250 mm. The pyrite was washed with 0.01 M HCl to remove oxidation prod-
ucts and stored under anaerobic conditions until use. The purity of the pyrite
was confirmed by X-ray diffraction. Scanning electron microscopy (SEM) was
utilized to examine the morphology and size distribution of the pyrite (Fig. 1).
The particle sizes ranged from about 1 to 250 mm with an average particle size
of w75 mm and appeared nonporous. The specific surface area of the powder
was determined to be 0.42 m2 g�1 using Kr BET at 77 K, which is very similar
to the value of 0.40 m2 g�1 found by Behra (2001) for similar particle sizes.
2.2. Batch experiments
Batch experiments were conducted in the absence of oxygen because py-
rite is thermodynamically unstable in the presence of O2. All solutions were

506 J. Bower et al. / Environmental Pollution 156 (2008) 504e514
sparged with N2(g) to drive off any dissolved O2(g) prior to reaction, and ex-
periments were conducted in an anaerobic chamber.
Kinetic, isotherm, and pH edge batch experiments were performed in
which the effect of reaction time, Hg(II) concentration, and pH were exam-
ined, respectively. For all experiments, the final concentrations were kept
below 100 mM (20 mg L�1) Hg(II) to avoid the potential influence of
Hg(II)-containing solids. Adjustments in pH were made using N2(g)-sparged
0.05 M NaOH or HCl. Each vial contained 2 g L�1 pyrite. All suspensions
were prepared in a constant ionic strength background matrix of NaCl
(0.1 M) because Hg(II) forms very strong complexes with chloride ions, which
helped to stabilize the Hg(II) in solution and prevent volatile losses. Although
Hg(II) is non-volatile, Hg0, which can be produced from the interaction of
Hg(II) with pyrite, is volatile (log KH ¼ 1.3 atm M�1), and the presence of
chloride ions promotes the stability of Hg(II) (Amyot et al., 2005). Under
the conditions of our experiments, Hgehydroxo complexes theoretically pre-
dominated at high pH (�8.5) while Hgechloro complexes predominated at
lower pH. In natural systems, Hg speciation is also strongly influenced by nat-
ural organic matter (NOM). However, NOM can also reduce Hg(II) and inter-
act strongly with metal sulfides. Thus we chose to conduct these experiments
with well-understood inorganic ligands only. Experiments examining the ef-
fect of NOM on the performance of metal sulfide reactive barriers are ongoing.
Samples were gently rotated end-over-end. After the desired reaction time, the
samples were centrifuged at 2500 � g for 20 min and filtered through
a 0.45 mm acrodisk syringe filter membrane to separate pyrite solids from
the liquid, with adsorbed Hg concentration calculated by difference. The re-
maining pyrite solids from one in every ten samples were digested using the
aqua regia method (EPA method 1631) for quality control purposes. Full Hg
recovery (>90%) was obtained in over 95% of the samples tested. Subsamples
of the filtrate were taken to measure the pH of each sample.
2.3. Column experiments
Column experiments were conducted to provide insight into the environ-
mental behavior of Hg(II) under hydrodynamic conditions. Studies were car-
ried out under both aerobic and anaerobic conditions, as both of these could
be encountered in the field. Anaerobic conditions were achieved by bubbling
the influent continuously with N2(g) to minimize dissolved oxygen. No other
special precautions were utilized to remove oxygen.
Two 1-cm diameter borosilicate glass columns were packed with 6.30 g of
pure quartz sand. One column (control) contained no pyrite (sand only), while
the other column also contained pyrite at the column exit. The barrier material
consisted of 0.25 g of washed pyrite mixed with 0.75 g of the pure quartz
sand; 0.25 g of this mixture was distributed evenly at the top of the column
producing a w0.1 cm thick permeable barrier. The total column length was
5 cm, resulting in a porosity of 0.37 and a bulk density of 1.67 g cm�3. A non-
reactive (KBr) tracer test was performed to determine the dispersion coeffi-
cient (D) to verify the uniformity of the packed material.
A 0.5 mM (100 mg L�1) Hg(II) solution was prepared in a 1000 ml volu-
metric flask using 100 ml of a 5 mM (1 mg L�1) standard solution of
Hg(NO3)2 and 0.1 M NaCl. This solution concentration was used based on
the approximate effluent Hg concentration found in a study using 1 g Hg0 in
the sand column (not shown here). A four-way valve was set up so that two
influents could be used, a 0.5 mM (100 mg L�1) Hg(II) in 0.1 M NaCl and
a Hg-free 0.1 M NaCl solution at pH 4. At this pH, the Hg(II) solution pH
remained stable without added buffers that could affect the performance of
the barrier and the results of the column experiments could be compared to
batch results at the corresponding pH. Additionally, this pH is similar to
that found in acidic, Fe-oxide-rich Ultisols common to Oak Ridge, Tennessee
(Widner et al., 1996), which served as a model Hg-contaminated site for our
study. The columns were run in up-flow mode at a specific discharge rate of
0.076 cm min�1, which is on the high end of typical groundwater velocities,
and samples were obtained using a fraction collector. For comparison pur-
poses, a subsequent column experiment was conducted at a specific discharge
rate of 0.255 cm min�1. Initially, the Hg-free 0.1 M NaCl solution was used as
the influent until the column was saturated; then, the valve was switched so
that the 0.5 mM (100 mg L�1) Hg(II) solution was the influent. Once the pyrite
was close to saturation with Hg(II), the valve was turned back to the Hg-free
0.1 M NaCl solution influent and run until the effluent concentration fell below
the Hg detection limit (0.5 mg L�1). The pH was measured immediately after
each sample was collected. The laboratory room temperature remained stable
at 21 � 2 �C. One of the column experiments was run in duplicate to quantify
process error, with w2% difference in effluent concentrations at breakthrough.
2.4. Analytical methods
Mercury analysis was conducted by Cold Vapor Atomic Absorption Spec-
trophotometry (CVAAS-USEPA Methods 7470A and 7471A). Mercury sam-
ple preparation and preservation was conducted similar to these two
procedures and those described in EPA Method 1631. Prior to analysis, all
samples were first preserved and oxidized with 1% BrCl, followed by 1% hy-
droxylamine hydrochloride to destroy the unreacted BrCl. Although BrCl is
typically used for preservation of samples containing organic matter, inconsis-
tent results were obtained when BrCl was not utilized. Mercury has been
shown to sorb to container walls and volatilize from reactors, causing signif-
icant mass balance problems. Teflon containers were used to minimize adsorp-
tion to container walls. Spikes, blanks, and duplicates were run to estimate
process error and quantify possible losses to containers.
X-ray absorption spectra were collected at the Advanced Photon Source
(Argonne National Laboratories) bending magnet beam line 20-BM, operated
by the Pacific Northwest Consortium Collaborative Access Team (PNC-CAT).
Current within the electron storage ring was maintained at w100 mA through
continuous electron top-up injections. X-ray energy at the beamline was con-
trolled using an N2-cooled Si(111) double-crystal monochromator. Samples
were loaded into Teflon sample holders. For anaerobically prepared samples,
this was accomplished inside a Plas-Labs glove box with a mix of hydrogen
and helium gases to maintain an anaerobic atmosphere. The sample was
placed inside a sealed cell in the glove box for transfer to the beam line.
The cell was continuously flushed with N2(g) during data collection.
Hg L(III) edge X-ray absorption data were collected in fluorescence mode
using a multi-element Ge solid-state detector. Incident and transmitted inten-
sities were measured with ionization chambers. The beam energy was cali-
brated using an Au foil. Layers of Al foil were placed in front of the
detector to improve the ratio of Hg to Fe counts reaching the detector. Between
6 and 12 spectra were collected from 150 eV below the Hg L(III) absorption
edge (12,284 eV) to k ¼ 12, where k represents the momentum wave-vector.
Data from the detector channels were summed to obtain the total Hg signal.
Data files were imported into the EXAFS processing program Athena
(Ravel and Newville, 2005). Hg L(III)-edge EXAFS data scans for each sam-
ple were grouped. Glitches (single-point excursions away from the data trend)
were removed, and scans were averaged. Spectral backgrounds were sub-
tracted and the fine-structure portions of the spectra were expressed in k-space
(A�1); a Fourier transform was performed to express the data in real space as
a radial distance function. The resulting spectra were imported into the EX-
AFS processing program Artemis (Ravel and Newville, 2005) for analysis.
Least squares fitting algorithms of the EXAFS function c(k) were applied
to determine close-neighbor atomic identities, coordination numbers, and dis-
tances from the target Hg atoms. Models for the backscattering paths were
generated from the crystalline structures of cinnabar (a-HgS, hexagonal),
metacinnabar (b-HgS, cubic), montroydite (HgO) and mercuric chloride
(HgCl2), and for simple HgeFeeHg paths, using the computer code FEFF
8.20 (Ankudinov et al., 1998; Rehr and Albers, 2000) via the Artemis
interface. Peaks in the radial structure function were isolated using the
KaisereBessel window function, back transformed and fit in q-space using
integer-value coordination numbers. The results of these fits provided a set
of parameters that were used together to initiate the fit over the full K range.
Between 13 and 30 free-floating parameters were permitted according to the
Stern (1993) formulation; resolvable bond distances are <0.05 A for all spec-
tra. Coordination numbers were initially held to the integer values from the
q-space fits while backscattering distances and DebyeeWaller disorder factors
were refined. They were adjusted to new integer values in cases where the
refined DebyeeWaller factor fell outside the range 0.0005e0.01, and another
refinement cycle was undertaken to determine the parameters reported in
Table 1. In the case of two atoms of the same element at similar distances,
the DebyeeWaller factors were constrained to be the same value for both.

Table 1
EXAFS fitting and Hg coordination environments
Identity CN r, A s2
A. Results of EXAFS fitting
Fresh, anaerobic (01F) S 1 2.55 0.003
Cl 1 2.43 0.006
R-factor ¼ 0.16
Fresh, aerobic (02F) S 1 2.53 0.003
Cl 1 2.47 0.005
R-factor ¼ 0.22
Aged, anaerobic (01A) S 1 2.36 0.0056
Cl 1 2.38 0.0064
R-factor ¼ 0.43 S 1 3.29 0.0056
Fe 1 3.82 0.0040
Hg 3 4.55 0.0018
Aged, aerobic (02A) S 1 2.42 0.0041
Cl 1 2.33 0.0031
R-factor ¼ 0.46 S 1 3.27 0.0042
Fe 2 3.80 0.0100
Hg 2 4.65 0.0020
B. Hg coordination environments in model compoundsCinnabar (hexagonal HgS) S 2 2.37
S 2 3.09
S 2 3.90
Hg 2 3.75
4 4.10
6 4.15
Sa 6 4.6e4.9
Hg 4 5.59
Metacinnabar (cubic HgS) S 4 2.54
Hg 12 4.14
Sa 12 4.85
Hg 6 5.85
HgCl2 Cl 2 2.28
Cl 6 3.36e3.46
Hg 4 4.33e4.41
Cl 6 4.58e4.84
Hg 4 4.86
Montroydite (HgO) O 2 2.05
O 4 2.83
Hg 12 3.31e3.74
Oa 6 4.08e4.48
Hg 4 4.83
a Backscattering from oxygen or sulfur at these distances is not expected to
contribute significantly to the EXAFS.
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0 50 100 150 200t (hr)
C/C
0
0.00.1
0.20.30.40.5
0.60.70.8
0.91.0
0 5 10 15 20 25t (hr)
C/C
0
pH 10.4 pH 6.4
pH 4.1
Fig. 2. Hg(II) sorption onto pyrite as a function of time at various pH. Initial
pyrite and Hg(II) concentrations were 2 g L�1 and 10 mM (2 mg L�1), respec-
tively. Background electrolyte was 0.1 M NaCl. Experiments were carried out
at pH 4.1, 6.4, and 10.4 � 0.2.
-6.0
-5.0
-4.0
-3.0
-2.0
-1.0
0.0
ln
C
/C
0
pH 4.1
pH 6.4
pH10.4
507J. Bower et al. / Environmental Pollution 156 (2008) 504e514
3. Results and discussion
-7.0
3.1. Batch experiments-9.0
-8.0
0 2 4 6 8 10 12 14
t1/2
Fig. 3. Linearization of Hg(II) sorption onto pyrite as a function of time. Initial
pyrite and Hg(II) concentrations were 2 g L�1 and 10 mM (2 mg L�1), respec-
tively. Background electrolyte was 0.1 M NaCl. Experiments were carried out
at pH 4.1, 6.4, and 10.4 � 0.2.
Fig. 2 illustrates the effect of pH and time on the rate ofHg(II) sorption onto pyrite, with the rate of sorption increasingsharply with pH. Sorption was initially rapid and then slowedwith time, behavior generally attributed to either the initialrapid filling of high-affinity sites followed by a slower fillingof lower-affinity sites (Axe and Anderson, 1995) or intrapar-ticle diffusion (Axe and Anderson, 1998). Under the given
conditions, the reaction time required to reach >95% removalvaried from w7 days at pH 4.1, w2 days at pH 6.4, and w2 hat pH 10.4. The actual time required to reach equilibriumcould not be determined at any pH value because the Hg(II)concentrations either fell below the detection limit or contin-ued to decrease over the duration of the experiment. Maximumsorption under these conditions, i.e. 100% uptake, was equiv-alent to 11.9 mmol Hg m�2 pyrite (1000 mg Hg g�1 pyrite).
Transformations of Hg(II) concentration and time wereperformed, and it was observed that at all pH values thedata could be linearized by plotting the natural log of the di-mensionless Hg(II) concentration versus the square root oftime, resulting in coefficient of determination (r2) valuesgreater than 0.9 for all cases (Fig. 3). In general, when theplot of concentration versus square root of time is linear, intra-particle diffusion is present (Jenne, 1998). Although published

0
10
20
30
40
50
60
70
80
90
100
0 2 4 6 8 10 12 14pH
% H
g R
em
oval
1
2
3
4
5
0 2 4 6 8 10 12 14
lo
g K
D (m
l/g
)
pH
Fig. 4. Hg(II) sorption onto pyrite as a function of pH. Initial pyrite and Hg(II)
concentrations were 2 g L�1 and 10 mM (2 mg L�1), respectively. Background
electrolyte was 0.1 M NaCl.
0
2
4
6
8
10
12
14
16
18
20
0 2 4 6 8 10 12C (µM)
q (µm
ol/g
)
pH 4.1
pH 6.4
pH 10.4
02468
101214161820
0.00 0.02 0.04 0.06 0.08 0.10C (nM)
q (µm
ol/g
)
Fig. 5. Hg(II) sorption isotherm data at pH 4.1, 6.4, and 10.4 � 0.2, respec-
tively. Initial pyrite concentration was 2 g L�1. Initial Hg(II) concentration
ranged from 2 to 40 mM. Background electrolyte was 0.1 M NaCl.
508 J. Bower et al. / Environmental Pollution 156 (2008) 504e514
Hg(II)-pyrite kinetic data is scarce, ease of diffusion has beenreported to be an important property in the reaction of Hg(II)with other metal sulfides (Phillips and Kraus, 1965). However,neither our SEM (Fig. 1) nor EXAFS (below) results wereparticularly suggestive of a diffusion-controlled sorptionmechanism.
Standard kinetic analysis was used to model the adsorptionrate data, which resulted in the following equation:
lnC
C0
¼ exph� 2k
�Hþ�n
t1=2i
ð1Þ
where C is the aqueous Hg concentration as a function of time,C0 is the initial Hg concentration, {Hþ} is the proton activity, tis the time (h), and k and n are constants. The model was fit tothe experiment data via non-linear regression over all datapoints at all pH values yielding k ¼ 2.25 � 10�2 andn ¼ �0.175. The modeled Hg concentrations as a functionof time are also shown in Figs. 2 and 3. The resulting modelwas able to describe the uptake of Hg over a wide rangeof pH (6 units), Hg concentration (�10 mM), and time(�1 week). However, some preliminary data at pH 3.3 indi-cated that the model may not extrapolate quantitatively outsidethe ranges noted above.
The model presented above is based on a diffusion-con-trolled adsorption rate, which was consistent with data shownin Fig. 3. For ideal spherical particles, the specific surfacearea is given by 6/(rd ) where r and d are the density and diam-eter of the particles, respectively. Using a pyrite density of5 g cm�3 and the measured specific surface area of0.42 m g�2 yields an equivalent-spherical diameter of 2.8 mm,in the same range as the smallest particles shown in Fig. 1.The relatively low specific surface area and the absence of vis-ible porosity visible in Fig. 1 suggest that traditional intrapar-ticle diffusion is unlikely. However, this does not considerHg(II) incorporation into the pyrite lattice via diffusion, whichis addressed in the discussion of the XAS results.
Although the potential for longer-term uptake is clearlypresent, particularly at lower pH, for practical reasons and asa basis for comparison we chose two days as a standard reac-tion time for subsequent batch (quasi-)equilibrium experi-ments. Batch sorption studies were conducted at a constantinitial Hg(II) concentration of 10 mM over a pH range of 1.5to 11.5 and a reaction time of 2 days (Fig. 4). Hg(II) removalfrom solution increased sharply with pH over a relatively nar-row pH range, a common characteristic of cationic adsorptionon variably charged surfaces, where surface charge becomesmore negative as pH increases above the isoelectric point,which for pyrite is w1.4 (Bebie et al., 1998). Nearly completeHg(II) removal (>95%) was achieved above pH 4. Log KD
versus pH is also shown, as plots of log KD (as opposed to per-cent removal) versus pH are generally relatively insensitive tothe solids concentration (Jenne, 1998).
Because these pH-dependent differences in sorption behav-ior were observed, adsorption isotherms were measured atacidic, neutral and basic pH values to examine the effect ofHg(II) concentration on sorption (Fig. 5). There was evidence
of sorption maxima of w14.6 mmol m�2 (1230 mg Hg(II) g�1)and w23.7 mmol m�2 (2000 mg Hg(II) g�1) at pH 4.1 and 6.4,respectively. As a comparison, Behra (2001) found the sorp-tion capacity to be at least 23.7 mmol m�2 (2000 mg g�1)Hg(II) for pH >4.
Much greater sorption was observed at pH 10.4 (Fig. 5),and the resulting isotherm was linear (KD ¼ 169 L g�1 pyrite)with no evidence of an approaching adsorption maximum.However, we were limited in what the initial (and thus final)Hg(II) concentration could be at pH 10.4, as we did notwant to supersaturate the initial solution with respect toHgO(s). At pH 10.4, pyrite can sorb Hg(II) at concentrations>17 mM g�1 (3500 mg g�1).
The sorption capacity can be related to the atomic structureat the pyrite surface by considering the dimensions of the unitcell; the lower uptake concentration, at pH 4.1, is equivalent toapproximately 2.6 Hg sorption sites per exposed face of theunit cell (29.2 A2). Pyrite fractures conchoidally rather thanproducing perfect cleavage surfaces, but considering an ideal

509J. Bower et al. / Environmental Pollution 156 (2008) 504e514
surface parallel to (100) provides a basis for considering po-tential sorption geometry. On this surface, there are four Satoms at the unit cell face. Two in a plane projecting abovethe near-surface Fe provide the most plausible sorption sitesfor Hg, consistent with the adsorption density at pH 4.1.Two more are in a plane below the Fe; adsorption of a cationon these S atoms is unlikely because it would require unreal-istically short cation-cation distances between Hg and Fe(2.7 A). Similar geometric constraints apply for other latticeplane-cut surfaces. Hence, sorption greater than w2 sites perunit cell face indicates that a different mechanism operates,such as formation of a solid Hg-bearing phase, developmentof a reaction product with higher specific surface area andsorption capacity, or diffusion into the pyrite. Observationsof Hg behavior in the batch reactors may be interpreted inthe context of EXAFS data that suggest differences in Hg ad-sorption over time.
3.2. EXAFS analysis of Hg on pyrite
Three potential mechanisms may account for association ofHg with pyrite: precipitation of a new phase such as HgS;adsorption of Hg onto the pyrite surface, possibly followingreaction and rearrangement of its atomic configuration; or dif-fusion into the pyrite, perhaps in exchange for Fe (a thermody-namically favorable reaction; Hyland et al., 1990). EXAFSanalysis can help to distinguish among these possibilities.
Best-fit parameters for the local atomic environment aroundHg in four samples at pH 4 (combinations of aerobic andanaerobic preparation conditions, and aged 24 h vs. 2 weeks)are summarized in Table 1, and the corresponding fits areshown in Fig. 6. The major differences occur between freshlyprepared (24 h) and aged (2 weeks) samples, where the agedsamples show a greater degree of long-range order and closerinteratomic distances for atoms bonded to Hg.
Fig. 6. Results of EXAFS fitting. Solid line, data; das
Aerobically and anaerobically prepared samples did notshow strong differences, and none of the samples exposedto air during preparation and/or aging showed evidence fornear-neighbor O. Attempts to include it led to unrealisticvalues for e0 (energy offset), or to negative values for the am-plitude of the function. The presence or absence of oxygenwas assessed using theoretical phase and amplitude pathsbased on the mineral montroydite (orthorhombic HgO;HgeO 2.04 A), in which each Hg atom is bonded to two ox-ygen atoms in a linear arrangement (Aurivillius, 1964), asa model. Schuetteite (Hg3(SO4)O2), in which Hg is bondedto sulfate groups via O atoms arranged in a distorted octahe-dron, has its nearest-neighbor O atoms at a similar distance(Weil, 2001), as does Hg adsorbed onto goethite (Collinset al., 1999). The occurrence of multiple scattering wasalso considered and tested, as this phenomenon has beenfound to contribute to EXAFS spectra collected on HgeScomplexes in solution (Lennie et al., 2003); however, noneof the fits were improved by including a multiple scatteringcomponent.
The freshly prepared samples show evidence of close-neighbor backscatters at two different distances. Two plausi-ble fits were determined for each sample e one using threeS-neighbors, and one using one S- and one Cl-neighbor,which provided a slightly better fit. In the S-only cases, thebest fit was achieved with one S at the w2.37 A distancecharacteristic of hexagonal HgS (cinnabar) and two S at thelonger w2.54 A distance characteristic of cubic HgS (meta-cinnabar; see Table 1). The SeHgeCl geometry is more con-sistent with Hg settings in crystalline phases; here, Hgtypically occurs in either linear coordination (e.g. SeHgeSin cinnabar, OeHgeO in montroydite (HgO), CleHgeCl inmercuric chloride), or in tetrahedral coordination (HgS4 inmetacinnabar). The fits with two backscatterers, with S atw2.54 A and Cl at w2.45 A, are consistent with the linear
hed line, fit. Fit parameters are shown in Table 1.

Fig. 7. Proposed model of HgeCl sorption complexes on pyrite surface, show-
ing one Hg per unit cell face on (001).
510 J. Bower et al. / Environmental Pollution 156 (2008) 504e514
configuration, and are also the best model fits to the EXAFSdata. The HgeCl distances are somewhat longer than those inmercuric chloride but shorter than those in minerals such aseglestonite (Hg6Cl3O2) (Mereiter et al., 1992) or corderoite(Hg3S2Cl2) (Frueh and Gray, 1968). The radial distance func-tions (Fig. 6B) show that there may potentially be backscatter-ers at longer distances, but these could not be confidentlyresolved and their low amplitudes suggest a lack of order be-yond the first shell.
The coordination environment of Hg appears to becomemore ordered over time. The samples aged for two weeksshow evidence for additional backscattering atoms at longerdistances, as observed in the more complex shape of the oscil-lations in the EXAFS and the additional peaks on the radialdistance function plots (Fig. 6). In these samples, like thefreshly prepared samples, the first coordination shell wasbest fitted with contributions from both S and Cl, but withshorter bond distances than the freshly prepared samples(2.36e2.42 A and 2.33e2.38 A, respectively). They could befit nearly as well with only S in the first shell, at the shorterHgeS distance characteristic of cinnabar, but the configura-tion at longer distances is not consistent with crystalline cinna-bar. Rather, best overall fits were achieved by including S atw3.28 A, Fe at 3.8 A, and Hg at w4.6 A (Table 1). Thefirst-shell coordination environment in the aged samples issimilar to that observed by Behra et al. (2001) for Hg(II) ad-sorbed on pyrite in a pH 3 solution with 0.1 M NaCl; thetime between sample preparation and EXAFS analysis wasnot specified.
To summarize, in the freshly prepared samples, the lack ofsecond-shell backscattering contributions to the EXAFS indi-cates disorder beyond the first S shell around Hg; it has neitherbecome incorporated in a second crystalline phase nor in thepyrite itself. In contrast, the aged samples show evidence forFe backscatterers, suggesting that Hg distribution has becomemore ordered with respect to the pyrite surface over time. TheHg backscattering contribution to the EXAFS does not con-form to the cinnabar or metacinnabar structure, indicatingthat HgS was not precipitated.
A plausible explanation for this aging behavior is dissolu-tion and reconfiguration of atoms at the pyrite surface in re-sponse to the introduction of Hg. Ubiquitous reactive defectsare present on pyrite surfaces (Nesbitt et al., 2000). Underideal circumstances at a pristine pyrite (100) surface, danglingbonds from both Fe and S are autocompensated and the sur-face is stable, although surface S atoms expand outward andFe atoms contract inward (Rosso and Vaughan, 2006). How-ever, in the presence of Hg, its affinity for S could draw elec-tron density away from surface S atoms, weakening theirbonds to Fe and resulting in a disordered set of Hg complexesthat are weakly attracted to the pyrite surface. A wide varietyof Hg sulfide complexes have been described (summarizedin Rickard and Luther, 2006). Over time, a more structuredenvironment develops in which Hg atoms are located in con-sistent positions with respect to a (likely modified) pyritesurface. Indeed, to obtain the surface coverage observed inthe batch experiments conducted at higher pH, surface
modification to produce a larger number of active sorptionsites is required.
Many studies have suggested adsorption as a predominantmechanism for Hg association with pyrite and other sulfidemineral surfaces (Jean and Bancroft, 1986; Walcarius et al.,1999; Ehrhardt et al., 2000; Behra et al., 2001; Genin et al.,2001). Other interactions, such as surface rearrangement andprecipitation of HgS, have been proposed for Hg interactionwith the Fe sulfide mackinawite (FeS) (Wolfenden et al., 2005).
An ordered monolayer of HgeCl complexes adsorbed onthe pyrite in the aged samples is also plausible (Fig. 7).Such an arrangement is consistent with XPS analyses of sul-fide mineral surfaces that were exposed to HgCl2 solutions(Jean and Bancroft, 1986; Hyland et al., 1990) where bothHg and Cl were distributed across the mineral surface in pro-portion to surface S, but Cl did not adsorb without Hg present.Genin et al. (2001) also suggested monolayer adsorption of Hgon galena (PbS). The fits to their EXAFS data indicated threecoordinating S atoms for Hg, but they dismissed this value asunrealistic given the typical coordination environments of Hg.Instead they suggested adjacent SeHgeS complexes arrangedin a square planar configuration on the galena surface.
The model proposed in Fig. 7 for HgeCl adsorption on py-rite assumes a simplified representation of the pyrite surface,i.e. an unbroken plane. Pyrite exhibits very poor cleavage anda fractured surface is expected to expose complex nanotopogra-phy with a diverse array of S functional groups (Schaufusset al., 1998). However, small areas of the dominant growthplane could provide the geometric basis for a consistently ori-ented Hg complex. Based on the results of EXAFS fitting, lin-ear SeHgeCl arrangements may extend away from one of theS atoms in 50% of the disulfide pairs at the pyrite {001} surface(one site per unit cell face) with an FeeSeHg bond angle of109�. Depending on the surface coverage, each SeHgeClcomplex could be adjacent to up to two similar complexes,with 4.6 A between Hg atoms. Each Hg would be coordinatedwith two Fe at w3.8 A; two more Fe expected at w4.4 A werenot fit in the EXAFS as they are weak in comparison with thephotoelectron wave-vector interactions of the neighboringHg. The S at w3.28 A may be accounted for by a location equi-distant from three Hg atoms; this position is also equidistantfrom three Cl atoms at approximately 2.9 A, and lies directlyabove an Fe at the pyrite surface at w4.7 A. As discussedabove, this model implies partial pyrite dissolution to producethe S at these sites. Behra et al. (2001) also found both Fe and Sreleased to solution at acidic pH.
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0 100 200 300 400V/V
0
C/C
0
4000 100 200 300 500 600 700infiltration (cm)
a
b
0.0
0.1
0.2
0.3
0.4
0.9
1.0
0 100 200 300 400V/V
0
C/C
0
0 100 200 300 400 500 600 700infiltration (cm)
0.5
0.6
0.7
0.8
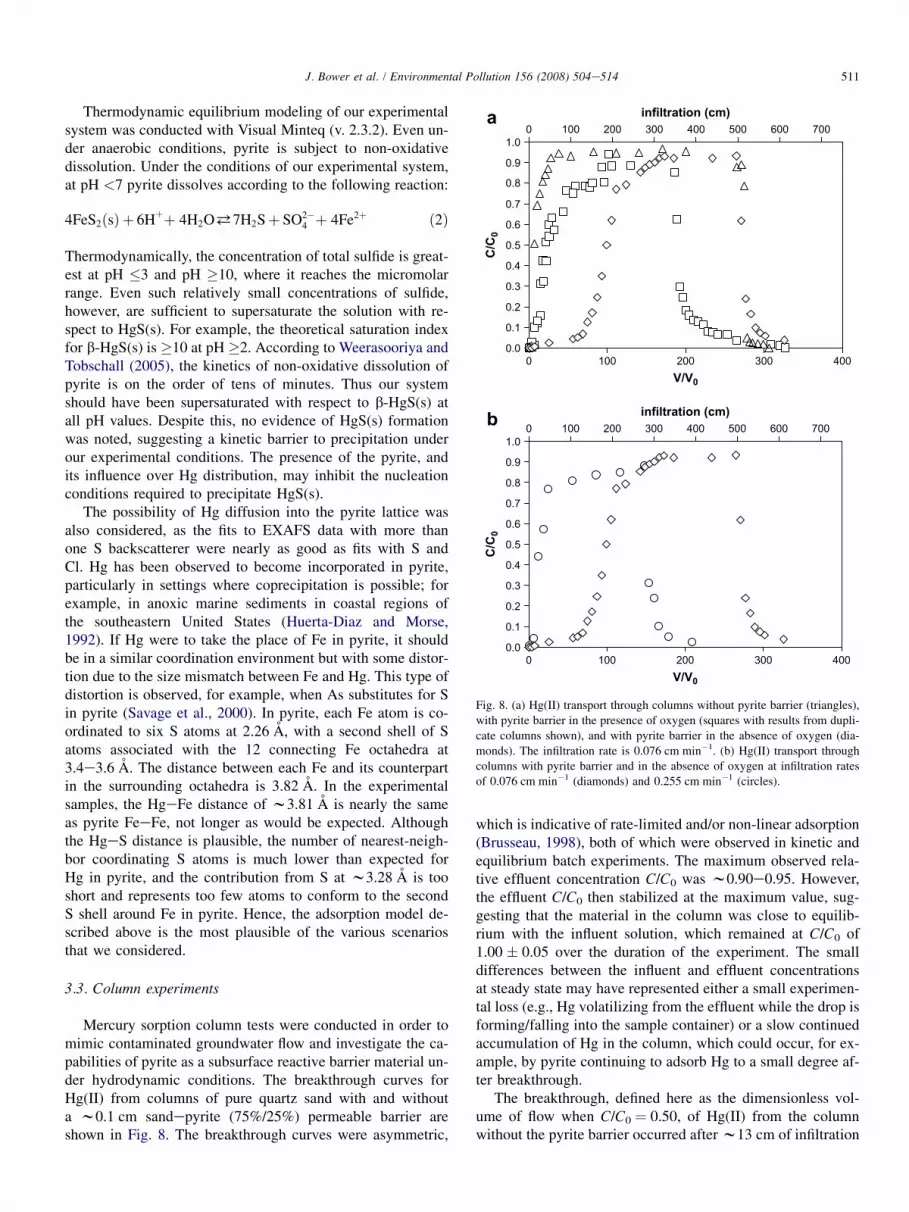
Fig. 8. (a) Hg(II) transport through columns without pyrite barrier (triangles),
with pyrite barrier in the presence of oxygen (squares with results from dupli-
cate columns shown), and with pyrite barrier in the absence of oxygen (dia-
monds). The infiltration rate is 0.076 cm min�1. (b) Hg(II) transport through
columns with pyrite barrier and in the absence of oxygen at infiltration rates
of 0.076 cm min�1 (diamonds) and 0.255 cm min�1 (circles).
511J. Bower et al. / Environmental Pollution 156 (2008) 504e514
Thermodynamic equilibrium modeling of our experimentalsystem was conducted with Visual Minteq (v. 2.3.2). Even un-der anaerobic conditions, pyrite is subject to non-oxidativedissolution. Under the conditions of our experimental system,at pH <7 pyrite dissolves according to the following reaction:
4FeS2ðsÞ þ 6Hþþ 4H2O%7H2Sþ SO2�
4 þ 4Fe2þ ð2Þ
Thermodynamically, the concentration of total sulfide is great-est at pH �3 and pH �10, where it reaches the micromolarrange. Even such relatively small concentrations of sulfide,however, are sufficient to supersaturate the solution with re-spect to HgS(s). For example, the theoretical saturation indexfor b-HgS(s) is �10 at pH �2. According to Weerasooriya andTobschall (2005), the kinetics of non-oxidative dissolution ofpyrite is on the order of tens of minutes. Thus our systemshould have been supersaturated with respect to b-HgS(s) atall pH values. Despite this, no evidence of HgS(s) formationwas noted, suggesting a kinetic barrier to precipitation underour experimental conditions. The presence of the pyrite, andits influence over Hg distribution, may inhibit the nucleationconditions required to precipitate HgS(s).
The possibility of Hg diffusion into the pyrite lattice wasalso considered, as the fits to EXAFS data with more thanone S backscatterer were nearly as good as fits with S andCl. Hg has been observed to become incorporated in pyrite,particularly in settings where coprecipitation is possible; forexample, in anoxic marine sediments in coastal regions ofthe southeastern United States (Huerta-Diaz and Morse,1992). If Hg were to take the place of Fe in pyrite, it shouldbe in a similar coordination environment but with some distor-tion due to the size mismatch between Fe and Hg. This type ofdistortion is observed, for example, when As substitutes for Sin pyrite (Savage et al., 2000). In pyrite, each Fe atom is co-ordinated to six S atoms at 2.26 A, with a second shell of Satoms associated with the 12 connecting Fe octahedra at3.4e3.6 A. The distance between each Fe and its counterpartin the surrounding octahedra is 3.82 A. In the experimentalsamples, the HgeFe distance of w3.81 A is nearly the sameas pyrite FeeFe, not longer as would be expected. Althoughthe HgeS distance is plausible, the number of nearest-neigh-bor coordinating S atoms is much lower than expected forHg in pyrite, and the contribution from S at w3.28 A is tooshort and represents too few atoms to conform to the secondS shell around Fe in pyrite. Hence, the adsorption model de-scribed above is the most plausible of the various scenariosthat we considered.
3.3. Column experiments
Mercury sorption column tests were conducted in order tomimic contaminated groundwater flow and investigate the ca-pabilities of pyrite as a subsurface reactive barrier material un-der hydrodynamic conditions. The breakthrough curves forHg(II) from columns of pure quartz sand with and withouta w0.1 cm sandepyrite (75%/25%) permeable barrier areshown in Fig. 8. The breakthrough curves were asymmetric,
which is indicative of rate-limited and/or non-linear adsorption(Brusseau, 1998), both of which were observed in kinetic andequilibrium batch experiments. The maximum observed rela-tive effluent concentration C/C0 was w0.90e0.95. However,the effluent C/C0 then stabilized at the maximum value, sug-gesting that the material in the column was close to equilib-rium with the influent solution, which remained at C/C0 of1.00 � 0.05 over the duration of the experiment. The smalldifferences between the influent and effluent concentrationsat steady state may have represented either a small experimen-tal loss (e.g., Hg volatilizing from the effluent while the drop isforming/falling into the sample container) or a slow continuedaccumulation of Hg in the column, which could occur, for ex-ample, by pyrite continuing to adsorb Hg to a small degree af-ter breakthrough.
The breakthrough, defined here as the dimensionless vol-ume of flow when C/C0 ¼ 0.50, of Hg(II) from the columnwithout the pyrite barrier occurred after w13 cm of infiltration

512 J. Bower et al. / Environmental Pollution 156 (2008) 504e514
(Fig. 8a), well before the breakthrough from the columns withthe pyrite barrier, which occurred at w190 cm of Hg-contain-ing infiltration. Considering the annual average rainfall infiltra-tion in the southeastern USA is about 63 cm (Viessman andHammer, 1998), this very thin pyrite barrier had not completelyexhausted its capacity even after the equivalent of w3 yearsof infiltration. The surface coverage, assuming all of the re-tained Hg was on pyrite surfaces, is about 4.7 mmol g�1
(950 m g�1)dapproximately two sites per unit cell face.The total amount of Hg(II) adsorbed onto the pyrite, q, at
the end of the input pulse was determined using both graphicaland numerical integration of the breakthrough curves. At theeffluent Hg concentration and pH (4.54) at the end of the inputpulse, the resulting average distribution between adsorbed andaqueous Hg(II) (expressed as log KD) was calculated to be 3.5,a little lower than the corresponding equilibrium log KD ofw4.3e4.4 shown in Fig. 4, suggesting that the column had in-deed not yet reached equilibrium with the influent Hg concen-tration. The slow approach to equilibrium in a column witha residence time of 24 min was consistent with the slow ap-proach to equilibrium observed in batch kinetic experiments(Fig. 2), particularly at lower pH. Increasing the infiltrationrate also shifted the breakthrough curve to the left (Fig. 8b),further evidence of kinetic limitations in the column.
A second noteworthy difference between the columns withand without the reactive pyrite barrier was the reversibility ofthe sorbed Hg. In the column without the pyrite barrier,w100% of the adsorbed Hg(II) was desorbed when the inletwas switched to Hg-free 0.1 M NaCl. In contrast, in the col-umn with the pyrite barrier, only w8% of the immobilizedHg was recovered in the Hg-free solution, revealing that themajority of the Hg(II) was not readily desorbed, and that theadsorptionedesorption process is not reversible and/or it ex-hibits hysteresis. This could be due to the strong binding ofHg(II) to high affinity S sites or the formation of an essentiallyirreversible Hg species (e.g., incorporated into the pyrite struc-ture). Irreversible (or only very slowly reversible) immobiliza-tion would be a very desirable characteristic of a Hg-reactivesubsurface barrier.
Pyrite is thermodynamically unstable in the presence ofO2(aq), ultimately reacting to form Fe(III) (hydr)oxides, whichare generally less reactive to Hg(II) than pyrite (Behra et al.,2001). Therefore, the presence of O2(aq) could compromisethe effectiveness of a pyrite-based barrier, consistent withour experimental data. The presence of O2(aq) significantlydecreased the performance of the pyrite barrier (Fig. 8a),with breakthrough occurring at w45 cm of infiltration, a de-crease in the volume of infiltration before breakthrough bya factor of w4. However, even in the presence of O2(aq),most of the Hg remained immobilized, with only w24% des-orbed by the Hg-free 0.1 M NaCl solution. Thus the presenceof O2(aq) appears to have a stronger effect on Hg(II) adsorp-tion than desorption. Pyrite oxidation results in the generationof acidity (e.g., acid mine drainage). However, theoretical cal-culations suggest that observed pH change should be negligi-ble under our experimental conditions if only surface sites areoxidized. There was not a marked difference in pH between
the column in the presence and absence of O2(aq), withsteady-state effluent pH values of w4.5 in each, and the efflu-ent pH never dropped below the influent pH. Furthermore, ef-fluent Fe concentrations were measured and determined to bebelow the detection limit (<20 mM). The lack of effect on ef-fluent Fe and pH may have been due to the small quantity ofpyrite that was initially in the columns (0.0625 g), althoughthe presence of Hg(II) has also previously been shown to in-hibit pyrite oxidation (Behra et al., 2001).
4. Conclusions
This study reveals that pyrite has a strong effect on thesorption, and thus potentially the subsurface transport, ofHg(II). Both the Hg(II) adsorption rate and capacity increasewith increasing pH, most likely due to increased hydrolysis,pH dependent surface charge, and a greater surface densityof high affinity sorption sites. EXAFS analysis showed thata transformation in the coordination environment at low pHoccurred during aging over 2 weeks, to form an ordered mono-layer of HgeCl complexes on pyrite, while at high pH, Hg isbound to S on the pyrite surface as HgeOH.
The effluent of Hg(II) from column experiments was signif-icantly retarded in the presence of a thin reactive barrier of py-rite, with breakthrough delayed w15-fold. Further, the Hg(II)was not then readily desorbed, indicating the relative irrevers-ibility of the immobilization reaction, a desirable characteris-tic of a subsurface barrier. The ultimate degree of retardationin the column studies agree relatively well with values mea-sured in batch experiments, indicating that relatively simplesite-specific batch treatability experiments may provide a po-tential indicator of in situ barrier performance under flowingconditions.
The effectiveness of the barrier decreased approximately4-fold in the presence of O2(aq). Accordingly, pyrite may notbe the optimal barrier mineral in oxic groundwater. However,once sorbed, the Hg was still largely irreversibly bound, indi-cating that O2(aq) had a greater effect on Hg(II) immobiliza-tion than on mobilization. The ultimate goal of this work isto develop a fundamental understanding of the potential of us-ing pyrite as a subsurface reactive barrier. The fundamentalapproach to construction and implementation of immobiliza-tion strategies such as permeable reactive barriers to treat othercontaminants is well developed, which could lead to acceler-ated implementation. We are currently investigating the useof other metal sulfides to capture Hg in permeable reactivebarriers as well.
Acknowledgments
This research was funded by the Environmental ProtectionAgency (EPA) grant number GR832212. PNC/XOR facilitiesat the Advanced Photon Source, and research at these facili-ties, are supported by the U.S. Department of EnergydBasicEnergy Sciences, a major facilities access grant from NSERC,the University of Washington, Simon Fraser University, thePacific Northwest National Laboratory and the Advanced

513J. Bower et al. / Environmental Pollution 156 (2008) 504e514
Photon Source. Use of the Advanced Photon Source is alsosupported by the U.S. Department of Energy, Office of Sci-ence, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. Beamline technical support from SteveHeald and Dale Brewe of the PNC-CAT is deeply appreciated.The authors would also like to thank Jinling Zhuang and JieLing for their help with analytical equipment.
References
Amyot, M., Morel, F.M.M., Ariya, P.A., 2005. Dark oxidation of dissolved and
liquid elemental mercury in aquatic environments. Environmental Science
& Technology 39, 110e114.
Ankudinov, A.L., Ravel, B., Rehr, J.J., Conradson, S.D., 1998. Real-space
multiple-scattering calculation and interpretation of x-ray-absorption
near-edge structure. Physical Review B 58, 7565e7576.
Aurivillius, K., 1964. Least-squares refinement of the crystal structures of or-
thorhombic HgO and of Hg2O2NaI. Acta Chemica Scandinavica 18,
1305e1306.
Axe, L., Anderson, P.R., 1995. Sr diffusion and reaction within Fe oxides:
Evaluation of the rate-limiting mechanism for sorption. Journal of Colloid
and Interface Science 175, 157e165.
Axe, L., Anderson, P.R., 1998. Intraparticle diffusion of metal contaminants in
amorphous oxide minerals. In: Jenne, E.A. (Ed.), Adsorption of Metals by
Geomedia. Academic Press, pp. 193e205.
Barnett, M.O., Turner, R.R., 2001. Bioaccessibility of mercury in soils. Soil
and Sediment Contamination 10, 301e316.
Bebie, J., Schoonen, M.A.A., Fuhrmann, M., Strongin, D.R., 1998. Surface
charge development on transition metal sulfides: An electrokinetic study.
Geochimica et Cosmochimica Acta 62, 633e642.
Behra, P., Bonnissel-Gissinger, P., Alnot, M., Revel, R., Ehrhardt, J.J., 2001.
XPS and XAS study of the sorption of Hg(II) onto pyrite. Langmuir 17,
3970e3979.
Borah, D., Senapati, K., 2006. Adsorption of Cd(II) from aqueous solution
onto pyrite. Fuel 85, 1929e1934.
Bostick, B.C., Fendorf, S., 2003. Arsenite sorption on troilite (FeS) and pyrite
(FeS2). Geochimica Et Cosmochimica Acta 67, 909e921.
Bostick, B.C., Fendorf, S., Helz, G.R., 2003. Differential adsorption of molyb-
date and tetrathiomolybdate on pyrite (FeS2). Environmental Science &
Technology 37, 285e291.
Brown, J.R., Bancroft, G.M., Fyfe, W.S., 1979. Mercury removal from water
by iron sulfide minerals. An electron spectroscopy for chemical analysis
(ESCA) study. Environmental Science and Technology 13, 1142e1144.
Brusseau, M.L., 1998. Impact of chemical and biochemical reactions on trans-
port of environmental pollutants in porous media. In: Huang, P.M.,
Adriano, D.C., Logan, T.J., Checkai, R.T. (Eds.), Soil Chemistry and Eco-
system Health. SSSA Spec. Publ., 52, pp. 173e189. Madison, WI.
Charnock, J.M., Moyes, L.N., Pattrick, R.A.D., Mosselmans, J.F.W.,
Vaughan, D.J., Livens, F.R., 2003. The structural evolution of mercury sulfide
precipitate: an XAS and XRD study. American Mineralogist 88, 1197e1203.
Collins, C.R., Sherman, D.M., Ragnarsdottir, K.V., 1999. Surface complexation of
Hg2þ on goethite: Mechanism from EXAFS spectroscopy and density func-
tional calculations. Journal of Colloid and Interface Science 219, 345e350.
Doyle, C.S., Kendelewicz, T., Bostick, B.C., Brown, G.E., 2004. Soft X-ray
spectroscopic studies of the reaction of fractured pyrite surfaces with
Cr(Vl)-containing aqueous solutions. Geochimica Et Cosmochimica Acta
68, 4287e4299.
Ehrhardt, J.J., Behra, P., Bonnissel-Gissinger, P., Alnot, M., 2000. XPS study
of the sorption of Hg(II) onto pyrite (FeS2). Surface and Interface Analysis
30, 269e272.
EPA, 1997. Mercury Study Report to Congress, Volume I.U.S. Environmental
Protection Agency, Washington, DC.
Fitzgerald, W.F., Engstrom, D.R., Mason, R.P., Nater, E.A., 1998. The case for
atmospheric mercury contamination in remote areas. Environmental Sci-
ence and Technology 32, 1e7.
Frueh, A.J., Gray, N., 1968. Confirmation and refinement of structure of
Hg3S2Cl2. Acta Crystallographica Section B Structural Crystallography
and Crystal Chemistry B 24, 156e157.
Fuhrmann, M., Melamed, D., Kalb, P.D., Adams, J.W., Milian, L.W., 2002.
Sulfur polymer solidification/stabilization of elemental mercury waste.
Waste Management 22, 327e333.
Genin, F., Alnot, M., Ehrhardt, J.J., 2001. Interaction of vapours of mercury
with PbS(001): a study by X-ray photoelectron spectroscopy, RHEED
and X-ray absorption spectroscopy. Applied Surface Science 173, 44e53.
Hogue, C., 2007. Quicksilver Quandry: Mercury in aging chemical plants
could end up in and on the hands of gold miners. Chemical and Engineer-
ing News 85, 26e29.
Huerta-Diaz, M.A., Morse, J.W., 1992. Pyritization of trace metals in
anoxic marine sediments. Geochimica et Cosmochimica Acta 56,
2681e2702.
Hyland, M.M., Jean, G.E., Bancroft, G.M., 1990. XPS and AES Studies of
Hg(II) Sorption and Desorption Reactions on Sulfide Minerals. Geochi-
mica et Cosmochimica Acta 54, 1957e1967.
Jean, G.E., Bancroft, G.M., 1986. Heavy metal adsorption by sulphide mineral
surfaces. Geochimica et Cosmochimica Acta 50, 1455e1463.
Jenne, E.A., 1998. Adsorption of metals by geomedia: data analysis, modeling,
controlling factors, and related issues. In: Jenne, E.A. (Ed.), Adsorption of
Metals by Geomedia. Academic Press, San Diego, pp. 1e73.
Johnson, J., 2004. Where goes the missing mercury? Chemical and Engineer-
ing News 82, 31e32.
Kim, C.S., Brown, G.E., Rytuba, J.J., 2000. Characterization and speciation of
mercury-bearing mine wastes using X-ray absorption spectroscopy. Sci-
ence of the Total Environment 261, 157e168.
Lennie, A.R., Charnock, J.M., Pattrick, R.A.D., 2003. Structure of mercu-
ry(II)-sulfur complexes by EXAFS spectroscopic measurements. Chemical
Geology 199, 199e207.
Mereiter, K., Zemann, J., Hewat, A.W., 1992. Eglestonite, [Hg2]3Cl3O2H:
Confirmation of the chemical formula by neutron powder diffraction.
American Mineralogist 77, 839e842.
Morse, J.W., Arakaki, T., 1993. Adsorption and coprecipitation of divalent
metals with mackinawite (FeS). Geochimica et Cosmochimica Acta 57,
3635e3640.
Navarro, A., Biester, H., Mendoza, J.L., Cardellach, E., 2006. Mercury speci-
ation and mobilization in contaminated soils of the Valle del Azogue Hg
mine (SE, Spain). Environmental Geology 49, 1089e1101.
Nesbitt, H.W., Scaini, M., Hochst, H., Bancroft, G.M., Schaufuss, A.G.,
Szargan, R., 2000. Synchrotron XPS evidence for Fe2þ-S and Fe3þ-S sur-
face species on pyrite fracture-surfaces, and their 3D electronic states.
American Mineralogist 85, 850e857.
Oji, L.N., 1998. Mercury disposal via sulfur reactions. Journal of Environmen-
tal Engineering-Asce 124, 945e952.
Ozverdi, A., Erdem, M., 2006. Cu2+, Cd2+ and Pb2+ adsorption from aqueous
solutions by pyrite and synthetic iron sulphide. Journal of Hazardous
Materials 137, 626e632.
Phillips, H.O., Kraus, K.A., 1965. Adsorption on inorganic materials. VI.
Reaction of insoluble sulfides with metal ions in aqueous media. Journal
of Chromatography 17, 549e557.
Piao, H.S., Bishop, P.L., 2006. Stabilization of mercury-containing wastes us-
ing sulfide. Environmental Pollution 139, 498e506.
Ravel, B., Newville, M., 2005. ATHENA, ARTEMIS, HEPHAESTUS: data
analysis for X-ray absorption spectroscopy using IFEFFIT. Journal of Syn-
chrotron Radiation 12, 537e541.
Rehr, J.J., Albers, R.C., 2000. Theoretical approaches to X-ray absorption fine
structure. Reviews of Modern Physics 72, 621e654.
Rickard, D., Luther, G.W., 2006. Metal sulfide complexes and clusters. Re-
views in Mineralogy and Geochemistry: Sulfide Mineralogy and Geo-
chemistry 61, 421e504.
Rosso, K.M., Vaughan, D.J., 2006. Reactivity of sulfide mineral surfaces. Sul-
fide Mineralolgy and Geochemistry 61, 557e607.
Savage, K.S., Tingle, T.N., O’Day, P.A., Waychunas, G.A., Bird, D.K., 2000.
Arsenic speciation in pyrite and secondary weathering phases, Mother
Lode Gold District, Tuolumne County, California. Applied Geochemistry
15, 1219e1244.

514 J. Bower et al. / Environmental Pollution 156 (2008) 504e514
Schaufuss, A.G., Nesbitt, H.W., Kartio, I., Laajalehto, K., Bancroft, G.M.,
Szargan, R., 1998. Reactivity of surface chemical states on fractured py-
rite. Surface Science 411, 321e328.
Slowey, A.J., Brown, G.E., 2007. Transformations of mercury, iron, and sulfur
during the reductive dissolution of iron oxyhydroxide by sulfide. Geochi-
mica et Cosmochimica Acta 71, 877e894.
Stein, E.D., Cohen, Y., Winer, A.M., 1996. Environmental distribution and
transformation of mercury compounds. Critical Reviews in Environmental
Science and Technology 26, 1e43.
Stern, E.A., 1993. Number of relevant independent points in x-ray absorption
fine-structure spectra. Physical Review B 48, 9825e9827.
Svensson, M., Allard, B., Duker, A., 2006. Formation of HgS: mixing HgO or
elemental Hg with S, FeS or FeS2. Science of the Total Environment 368,
418e423.
Turner, R.-R., Southworth, G.R., 1999. Mercury-contaminated industrial and
mining sites in North America: An overview with selected case studies.
In: Ebinghaus, R., Turner, R.R., de Lacerda, L.D. (Eds.), Mercury Contam-
inated Sites: Characterization, Risk Assessment, and Remediation.
Springer, Berlin, pp. 89e112.
Viessman, W., Hammer, M., 1998. Water Supply and Pollution Control,
sixth ed. Prentice Hall.
Walcarius, A., Devoy, J., Bessiere, J., 1999. Electrochemical recognition of se-
lective mercury adsorption on minerals. Environmental Science & Tech-
nology 33, 4278e4284.
Weerasooriya, R., Tobschall, H.J., 2005. Pyrite-water interactions: Effects of
pH and pFe on surface charge. Colloids and Surfaces A Physicochemical
and Engineering Aspects 264, 68e74.
Weil, M., 2001. Schuetteite, Hg3(SO4)O2, a re-investigation. Acta Crystallog-
raphica Section E Structure Reports Online 57, I98eI100.
Widner, T.E., Ripple, S.R., Buddenbaum, J.E., 1996. Identification and screen-
ing evaluation of key historical materials and emission sources at the Oak
Ridge Reservation. Health Physics 71, 457e469.
Willet, K.L., Turner, R.R., Beauchamp, J.J., 1992. Effect of chemical form
of mercury on the performance of dosed soils in standard leaching
protocols: EP and TCLP. Hazardous Waste and Hazardous Materials 9,
275e288.
Wolfenden, S., Charnock, J.M., Hilton, J., Livens, F.R., Vaughan, D.J., 2005.
Sulfide species as a sink for mercury in lake sediments. Environmental Sci-
ence & Technology 39, 6644e6648.
Zhang, J., Bishop, P.L., 2002. Stabilization/solidification (S/S) of mercury-
containing wastes using reactivated carbon and Portland cement. Journal
of Hazardous Materials 92, 199e212.