il-6 modulates cd5 expression in b cells from patients with lupus by regulating dna methylation
TRANSCRIPT

IL-6 Modulates CD5 Expression in B Cells from Patients withLupus by Regulating DNA Methylation1
Soizic Garaud,2* Christelle Le Dantec,2* Sandrine Jousse-Joulin,† Catherine Hanrotel-Saliou,†
Alain Saraux,*† Rizgar A. Mageed,‡ Pierre Youinou,3*† and Yves Renaudineau*†
B lymphocytes from patients with systemic lupus erythematosus (SLE) are characterized by reduced expression levels of mem-brane CD5. Recent studies from our laboratory have revealed that the level of membrane CD5 is determined by the relative levelof two alternative CD5 isoforms; CD5-E1A, which is expressed on the membrane, and CD5-E1B, which is retained in the cyto-plasm. Using bisulfite sequencing and methylation-sensitive endonuclease assays we show that the promoter for the alternativeCD5-E1B isoform is demethylated in B cells from patients with SLE but not in healthy controls. We go on to show that differentialmethylation is more pronounced following BCR engagement. As a result of this demethylation, CD5-E1B mRNA is transcribedat the expense of CD5-E1A mRNA transcription. We provide further evidence that production of high IL-6 levels by SLE B cellsabrogates the ability of SLE B cells to induce DNA methyl transferase (DNMT1) and then to methylate DNA, an effect that isreversed in the presence of a blocking Ab to the IL-6 receptor. The pattern of demethylation of CpG islands in the CD5-E1Bpromoter in SLE B cells is similar to those in B cells from healthy controls stimulated in the presence of IL-6, or treated withthe methylation inhibitor PD98059. The study reveals that engagement of the BCR with constitutive IL-6 down-regulates thelevel of membrane CD5, which negatively regulates BCR signaling, in SLE B cells. This altered signaling could, in turn,promote the activation and expansion of autoreactive B cells in SLE patients. The Journal of Immunology, 2009, 182:5623–5632.
S ystemic lupus erythematosus (SLE)4 is associated with di-verse clinical manifestations (1). The main features of au-toimmunity in SLE are B cell hyperactivity, spontaneous
lymphocyte proliferation, and the production of pathogenic Abs toself-Ags. B cell abnormalities in SLE also include excess cytokineproduction, autoantigen presentation to T cells and modulation ofthe function of other immune cells (2). Thus, SLE is generallyconsidered a B cell disease, a theme strengthened by the efficacy oftherapies targeting B cells. For example, therapeutic approachesusing depleting anti-CD20 has proved to be highly beneficial intreatment of SLE (3). Further, neutralization of cytokines that pro-mote B cell responses such as IL-6, or interruption of cognate Tcell and B cell interactions has been successful in early clinicaltrials (4, 5). All this provides the rationale for further investigation
of mechanisms of B cell involvement in driving autoimmunity andto develop more selective therapeutic targets.
The central role played by B cells in immunity necessitates thatits responses are tightly regulated. B cell responses are initiated bysignaling through the BCR. Signaling initiated following BCR en-gagement is regulated by coreceptors and by a network of proteintyrosine kinases and phosphatases. Recent findings suggest thatdefects in BCR-mediated signaling can result in lupus autoimmu-nity. For example, there is an association between SLE autoim-munity and mutations in a number of genes that encode B cell-specific signaling molecules including protein tyrosine kinases,non-receptor phosphatase type 22, B cell scaffold protein withankyrin repeats 1 and the inhibitory IgG Fc�RIIb (6). In additionto direct evidence for the effect of mutations in signaling moleculeson BCR-mediated signaling, the contribution of epigenetic factorshas also been proposed (7). The most commonly observed epige-netic abnormality implicated in SLE pathology is altered DNAmethylation at the 5-carbon position of cytosines of CpG dinucle-otides. DNA methylation is regulated by three DNA methyl trans-ferases (DNMTs), DNMT1, DNMT3a, and DNMT3b. DNMT1preferentially targets hemi-methylated DNA over non-methylatedDNA. DNMT3a and DNMT3b, in contrast, exhibit de novo activ-ities. The methyl-CpG-binding domain (MBD) protein-4 is a DNAglycosylase that acts preferentially on hemi-methylated CpGs andinitiates demethylation by replacing a 5-methylcytosine with anunmethylated cytosine (8). The action of MBD2 on DNA de-meth-ylation has been suggested, but remains controversial (9). DNAde-methylation activates the expression of many genes, such asCD21 in B cells in patients with rheumatoid arthritis (10), CD40ligand in T cells from patients with SLE (11), and the expressionof silenced retroviral genes (12). Increased expression of genesresulting from de-methylation was confirmed using DNA methyl-ation inhibitors in T cells from patients with SLE. Among thoseinhibitors the ras signal blocker PD98059 appears to be the more
*Research Unit EA2216 Immunology and Pathology, IFR148 ScInBioS, Universitede Brest and Universite Europeenne de Bretagne; †CHU Brest, Hopital Morvan andHopital de la Cavale Blanche, Brest, France; ‡William Harvey Research Institute,Barts and the London School of Medicine and Dentistry, Queen Mary University ofLondon, London, United Kingdom
Received for publication July 30, 2008. Accepted for publication February 18, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by grants from the Conseil Regional de Bretagne, theConseil General du Finistere, and the French Ministry for Education and Research.2 S.G. and C.L.D. contributed equally to the work.3 Address correspondence and reprint requests to Prof. Pierre Youinou, Laboratory ofImmunology, Brest University Medical School Hospital, BP824, F-29609, Brest,France. E-mail address: [email protected] Abbreviations used in this paper: SLE, systemic lupus erythematosus; ChIP, chro-matin immunoprecipitation; DNMT, DNA methyl transferase; HERV, human endog-enous retrovirus; HC, healthy control; MBD, methyl-CpG-binding domain protein;LTR, long terminal repeat; SLEDAI, SLE disease activity index.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0802412
relevant inhibitor because it induces cellular defects similar tothose observed in SLE (13).
B cells that express the CD5 protein, also known as B1 cells,primarily express BCRs that are autoreactive, have a reduced ca-pacity to enter the cell cycle, and have a longer lifespan. Onemodel for the role of CD5 in intracellular signaling suggests thatsurface CD5 modulates signaling from the BCR and thereby con-trols autoreactivity (14). According to this model, it is necessarythat the level of membrane CD5 is maintained to control thethreshold of BCR-mediated signaling. In humans, three mecha-nisms have been shown to be involved in regulating the level ofmembrane CD5. These are shedding (15), internalization of theprotein (16), and transcriptional regulation through altered level ofexpression of two alternative CD5 isoforms (17). The first isoform,designated CD5-E1A, corresponds to the full-length CD5 proteinwhich is synthesized and translocated to the cell membrane. Thesecond isoform derived from an alternative promoter, designatedCD5-E1B, encodes a truncated form of the protein that lacks aleader peptide and is retained intracellularly. Indeed, CD5-E1B isa fusion transcript from a new exon (4) that incorporates the 5�long terminal repeat (LTR) of a human endogenous retrovirus(HERV) sequence integrated at the time of old and new worldmonkeys divergence estimated at �25–30 million years ago (18).We recently observed that transcription of mRNA for this isoforminversely correlates with the level of DNMT1 mRNA (19).
Here, we provide evidence that relative to B cells from healthycontrols (HCs), the level of DNA methylation in BCR-mediated Bcell activation in patients with SLE is reduced. A consequence ofthis hypomethylation is increased expression of CD5-E1B. Excessproduction of IL-6 augments CD5-E1B transcription. Based onthis observation, we propose that modulation of B cell autoreac-tivity in SLE could be achieved by targeting IL-6.
Materials and MethodsB lymphocyte isolation
PBMCs were isolated from the blood of 25 patients with SLE and 25 HCsby centrifugation on Ficoll-Hypaque. All patients fulfilled the 1982 Amer-ican College of Rheumatology criteria for SLE (20). SLE activity wasassessed by the SLE disease activity index (SLEDAI), and those withSLEDAI of �5 were considered active. The characteristics of SLE patientsand HCs are shown in Table I. The cells were stained with FITC-anti-CD19and PE-anti-CD5 Abs, and CD5�CD19� B cells sorted on an Epics EliteFACS (Beckman-Coulter). All sorted cells were �98% CD19�. Informedconsent was obtained from the patients before taking blood, and the study
protocol approved by the Institutional Review Board at Brest University.The Daudi human B cell line was purchased from American Type CultureCollection.
FACS analysis
FITC-anti-CD19 (clone J4-119) and PE-anti-CD5 (clone BL1a) were ob-tained from Beckman-Coulter, whereas anti-DNMT1 and anti-p27kip1 wereobtained from Abcam. Intracellular staining was performed after perme-abilization of the cells with 70% methanol. Binding of primary unconju-gated Abs was revealed with FITC-conjugated anti-mouse Abs (JacksonImmunoResearch).
In pilot experiments, the number of CD5 molecules per cell was quan-tified by determining the amount of Ab binding to the cells (ABC) atsaturating concentrations, using the Quantum Simply Cellular kit (FlowCytometry Standards). Arbitrary ABC value was then determined from astandard ABC curve generated from the mean fluorescence intensity ob-tained from the FACS analysis of 50 �l calibrated microspheres stainedwith 20 �l of the same anti-CD5 Ab.
Cell culture
FACS-sorted B cells were suspended in RPMI 1640 supplemented with10% heat-inactivated FCS, 2 mM L-glutamine, 200 U/ml penicillin and 100�g/ml streptomycin. B lymphocytes were seeded at 2 � 105 cells per well,and incubated with 1 �g/ml anti-IgM Ab-coated Sepharose beads (BioRad)and 10 U/ml IL-2, in the presence or absence of 10–40 ng/ml anti-IL-6RAb (R&D Systems), or 100 ng/ml rhIL-6 (Immuno Tools). Repressionof DNMTs was achieved by incubating the cells with 50 �M of the rassignal blocker PD98059. IL-6 and IFN-� were detected in sera and IL-6 inthe supernatant of cultured cells using ELISA kits according to the man-ufacturer’s instructions (Beckman Coulter).
mRNA extraction and quantitative RT-PCR
Total mRNA was extracted using the RNAble method (Eurobio), andcDNA synthesized by reverse transcription in 20 �l volume with Super-script II RNase H-RT (Invitrogen Corporation). Quantitative RT-PCR wasconducted in 20 �l mixtures containing 50 ng template cDNA, 1X SybrGreen PCR Master mix (Applied Biosystems), and 500 nM of each primer(Table II). As described in detail elsewhere (17), CD5 promoter usage wasevaluated using two sets of primers. All assays included a negative controlwhich consisted of the reaction mixture with no template, and a positivecontrol which consisted of the mixture with 18S rRNA. Comparison ofcycle thresholds was completed with the 2���ct method using 18S as aninternal control.
RACE
The 5� transcript ends were amplified from mRNA using SMART-RACEkit (Clontech). As described previously (17), the first strand of cDNA wassynthesized using the sense UPM primer and the gene-specific antisenseprimer CD5 E5 (Table II). The PCR protocol included an initial denatur-ation step at 94°C for 5 min, starting 5 touchdown-PCR cycles of dena-turation at 94°C for 30 s and annealing at 72°C for 3 min. These cycleswere followed by another 5 cycles of 94°C for 30 s, 70°C for 30 s, and72°C for 3 min, then with a decreasing temperature for 35 cycles of 94°Cfor 30 s, 68°C for 30 s, and 72°C for 3 min. A nested PCR was performedusing the sense NUP primer and the gene-specific antisense primer CD5E3. The second PCR round was for 40 cycles of 30 s at 94°C, 1 min at56°C, and 1 min at 72°C with a final extension at 72°C for 10 min.
Methylation-specific PCR
This assay is based on the inability of some restriction enzymes to digesta methylated 5�-CmG-3� site. Genomic DNA was purified using QIAmp 96DNA blood kit (Qiagen) and digested with 20 U of the methylation-sen-sitive restriction enzymes HpaII, HaeII, FauI, HgaI, or the methylation-insensitive restriction enzyme MspI for 3 h at 37°C. Undigested genomicDNA was used as positive control. The PCR primers were positioned up-stream and downstream of the recognition site in the promoters of E1A andE1B of the cd5, cd19, cd70, Pax5, Syk, and HRES-1 genes (11, 19, 21, 22).The PCR protocol included an initial denaturation at 94°C for 5 min, fol-lowed by 35 cycles of denaturation at 94°C for 30 s, annealing at 56°C for1 min, and primer extension at 72°C for 1 min; PCR cycles were followedby final extension at 72°C for 10 min. The PCR products were separated onagarose gel and visualized with 0.5 �g/ml ethidium bromide.
Table I. Demographic and clinical characteristics of SLE patients andHCs included in the study
Patients Controls
n � 25 n � 25
Age (years) 47.6 15.1 22–74�a 43.3 10.7 28–68�Female (%) 19 (76) 17 (68)Ethnicity, Caucasian 25 25SLEDAI score 3.7 5.3 0–19� –SLE manifestations
Diseases duration 11.1 8.1 0–19� –Skin disease 52% –Arthritis 72% –CNS disease 12% –Lupus nephritis 48% –Heart 32% –ANA 100% –Anti-dsDNA 64% –
Medication usage (%)Steroids 60% –Antimalarials 72% –Cytotoxic agents 20% –
a Mean SD min � max�.
5624 IL-6 CONTROLS DNA METHYLATION AND CD5 IN LUPUS B CELLS

Bisulfite sequencing
To determine the methylation status of DNA, non-methylated cytosineswere converted to uridines by bisulfite treatment using the EZ-DNA meth-ylation-Gold kit according to the manufacturer’s instructions (Zymo Re-search). Unmodified DNA (100 ng) was amplified 40 times at 56°C usingspecific primers. The bisulfite-converted DNA was amplified by nestedPCR using two rounds of 40 cycles each at 56°C with primers specific formethylated cytosines (Table II). PCR products were purified using the highpure PCR product purification kit (Roche), and directly sequenced withinternal primers by means of the BigDye Terminator Cycle Sequencing kitusing an automated ABI-310 genetic analyzer (Applied Biosystems). Theelectrophoregram T and C peaks were quantified and methylation statusdetermined as [peak (C)/peak (T) � peak (C)] � 100. At the same time, theunmodified DNA was amplified and sequenced using specific primers.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was conducted using the EpiQuikkit (Epigentek Group) according to the manufacturer’s instructions to eval-uate activation of the CD5-E1B promoter. In brief, sonicated DNA (200–1000 bp) was transferred into strip wells precoated either with mouse anti-RNA polymerase II, or with a nonspecific mouse IgG, used as a negativecontrol. After a 90-min incubation at room temperature and extensivewashes, precipitated DNA-protein complexes were treated with 250 �g/mlproteinase K in the DNA release buffer for 15 min, and left in the samebuffer for 90 min at 65°C. The DNA samples were collected by the P-spin
columns, washed with ethanol, and eluted. Using purified DNA as a tem-plate, PCR was performed using GAPDH and CD5-E1B-specific primers(Table II) and 40 cycles at 56°C. PCR products were separated on agarosegel, and visualized with 0.5 �g/ml ethidium bromide.
Computational promoter analysis
Putative transcription factor binding sites were identified by using Alibaba,version 2.1, the transcription element search system (http://www.cbil.open.edu/tess/index.html) and Genomatix (http://www.genomatix.de).
Statistics
The results were expressed as arithmetic means with SD. Comparisonswere made using the Mann-Whitney U test for unpaired data and the Wil-coxon test for paired data. Correlations were established using Spearman’srank correlation.
ResultsCD5 expression in B cells
The percentage of CD5-expressing B cells was similar in the 25SLE patients and 25 HCs (Fig. 1A). However, membrane densityof CD5 on B1 cells was lower in the patients (Fig. 1B) comparedwith the controls (49,971 15,124 vs 80,703 22,462; p �0.001). A representative example is depicted in Fig. 1C.
Table II. Oligonucleotide primers used in the studya
Oligonucleotide Sequence Method
CD5-E1A sense 5�-ATGCCCATGGGGTCTCTGCAAC-3� Quantitative PCRCD5-E1B sense 5�-GCTGAACACAGGGAAGGAAC-3� Quantitative PCRCD5-E3 antisense 5�-GCCTGGAAATCTGGGTCATA-3� Quantitative PCRDNMT1 sense 5�-CCTGTACCGAGTTGGTGATGGT-3� Quantitative PCRDNMT1 antisense 5�-CCTTCCGTGGGCGTTTC-3� Quantitative PCRDNMT3a sense 5�-CTCCTGTGGGAGCCTCAATGTTACC-3� Quantitative PCRDNMT3a antisense 5�-CAGTTCTTGCAGTTTTGGCACATTCC-3� Quantitative PCRDNMT3b sense 5�-CTCGAAGACGCACAGCTGACGAC-3� Quantitative PCRDNMT3b antisense 5�-CCTATAACAACGGCAAAGACCGAGC-3� Quantitative PCRMBD2 sense 5�-CCATGGAACTACCCAAAGGTCTT-3� Quantitative PCRMBD2 antisense 5�-CAGCAGATAAAAGGGTCTCATCATT-3� Quantitative PCRMBD4 sense 5�-TCTAGTGAGCGCCTAGTCCCAG-3� Quantitative PCRMBD4 antisense 5�-TTCCAATTCCATAGCAACATCTTCT-3� Quantitative PCR18S sense 5�-GGCTACCACATCCAAGGAAGGCAG-3� Quantitative PCR18S antisense 5�-CCAATTACAGGGCCTCGAAAGAGTC-3� Quantitative PCRCD5-E5 antisense 5�-AGTCTCTGGCAGCTGCTTCAAGAA-3� 5�RACECD5-E3 antisense 5�-TGCCATCCGTCCTTGAGGTAGAC-3� 5�RACECD5 promoter-1B sense 5�-GTGAAGGGCTGCTTACTGCACACAGC-3� Methylation-PCRCD5-E1B antisense 5�-CAGCCACTGCGTTGATCCTCCACAG-3� Methylation-PCRCD5 promoter-1A sense 5�-CTGGAAGGGTAAAGCAGGGTTCTC-3� Methylation-PCRCD5-E1A antisense 5�-GGAGTCTGCAACAAGAACTGGCATC-3� Methylation-PCRCD5 promoter-1B sense 5�-TTTTATTTGTGAAATGGAAAGTTGT-3� Methylation-PCRCD5-E1B antisense 5�-AAATTCCCAAAACCAATCCTATC-3� Bisulfite sequencingCD5 promoter-1B sense 5�-TGTGAAATGGAAAGTTGTGTTTATT-3� Bisulfite sequencingCD5-E1B antisense 5�-AACATACCATAAATAATTAAACCAC-3� Bisulfite sequencingCD5 promoter-1A sense 5�-GAATTGGTATTATGTTGTTTATTTTT-3� Bisulfite sequencingCD5-E1A antisense 5�-CTCCCTACCAACCTAAAAACTACTC-3� Bisulfite sequencingCD5 promoter-1A sense 5�-ATTGTTTTAGTTTTGGGTATTTTGG-3� Bisulfite sequencingCD5-E1A antisense 5�-CCCCCTACAATCTCTCTTACACTAA-3� Bisulfite sequencingCD5-E1B sense 5�-TCTGCCAAAGAGGTTCAAGC-3� ChIPCD5-E1B antisense 5�-TGCATGCACCGGTAATTAGA-3� ChIPGAPDH sense 5�-TACTAGCGGTTTTACGGGCG-3� ChIPGAPDH antisense 5�-TCGAACAGGAGGAGCAGAGAGCGA-3� ChIPCD19 promoter sense 5�-AGCGTGGCAGGGAGGAGGCAAGTGTT-3� Methylation PCRCD19 promoter antisense 5�-GCGAGGAGGTGGCATGGTGGTCAGA-3� Methylation PCRCD70 promoter sense 5�-TCACCCAAGGTCAGGAGTTC-3� Methylation PCRCD70 promoter antisense 5�-CCATCTCAACTCACCCCAAG-3� Methylation PCRPax5 promoter sense 5�-GCAATAGTCAGGACCCCAAC-3� Methylation PCRPax5 promoter antisense 5�-TTCTCGCCAACATCACAAGA-3� Methylation PCRSyk promoter sense 5�-GGCAGCCCCACCTTCTCT-3� Methylation PCRSyk promoter antisense 5�-CGCGGCTCTTCCTCATTT-3� Methylation PCRHRES-1 promoter sense 5�-GCATATGCACTGGGAAAGGT-3� Methylation PCRHRES-1 promoter antisense 5�-CCGCCTTTTCAAGTTTCCTC-3� Methylation PCR
a E1A, exon 1A; E1B, exon 1B; DNMT, DNA methyltransferase; MBD, methyl-CpG binding domain protein.
5625The Journal of Immunology

Expression of CD5-E1B, which is retained in the cytoplasm,reduces membrane expression of CD5-E1A (17, 19). This suggeststhat CD5-expressing B cells may, under certain physiological con-ditions (such as activation), preferentially up-regulate CD5-E1Brelative to CD5-E1A. To test this proposition, levels of CD5-E1Aand CD5-E1B mRNA were determined using a 5� specific realtime PCR following 24h anti-IgM stimulation of FACS-sorted
CD5-negative B cells from the blood of 10 SLE patients and 15HCs (Fig. 1D). BCR engagement of the cells raised the level ofCD5-E1B mRNA in B cells from the patients (54.4 68.1-fold)but not in those from the controls (0.5 0.2-fold). In contrast,after BCR engagement expression of CD5-E1A is increased bothin SLE patients and in HCs (7.7 4.9-fold and 5.5 5.6-fold,NS). The 5�-RACE analysis of CD5 cDNA using B cells from HCs
FIGURE 2. Amplification of methylation-sensitive, endonuclease-digested genomic DNA reveals methylation status of the alternative promoters of CD5in resting B cells. A, Schematic representation of the affected promoters. The CD5-E1B promoter arises from a LTR element subdivided into U3, R, andU5 regions. The two splice donors (SD) are indicated, and positions of DNA-specific primers shown. The 1177-bp CD5-E1B amplicon contains sixHpaII/MspI motifs (5�-CCGG-3�) at positions �8736, �8726, �8522, �8268, �8114, and �7795 bp, and one HaeII motif (5�(A/G)GCGC(T/C)3�) atposition �8393bp according to numbering from the established ATG initiation site in exon 1A (22). The 783-bp CD5-E1A amplicon contains one HgaImotif (5�-GACGC(N)5-3�) at position �151, one FauI motif (5�-CCCGC(N)4-3�) at position �96, and two HpaII/MspI motifs at positions �65 and �87.B, Analysis of CD5-E1B promoter methylation by amplification of genomic DNA digested with methylation-sensitive HaeII, HpaII, or methylation-insensitive MspI enzymes. C, Analysis of the CD5-E1A promoter region using HgaI, FauI, HpaII, and MspI enzymes. Undigested genomic DNA isamplified by PCR, separated on agarose gel, and visualized with ethidium bromide.
FIGURE 1. CD5 expression in B cells. A, A scatter-gram depicting percentage of CD5-expressing B cells in25 SLE patients and 25 HCs. B, The number of CD5molecules per cell is given in the scattergram. The num-bers are expressed as the amount of anti-CD5 Ab boundto the cell membrane. C, Representative FACS profilefor cell surface expression of CD5 in blood B cells fromone SLE patient (bold line) and one HC (thin line). D,Quantitative RT-PCR results presenting as histogramsrevealing that a 24-h stimulation of B cells with anti-IgM increased CD5-E1B transcription in B cells fromSLE patients but not HCs. Incubation of anti-IgM-activated B cells from HCs with PD98059 resultedin up-regulation of CD5-E1B. Mean and SD of datafrom 10 SLE patients and 15 HCs are shown. E, CD55�-RACE analysis of cDNA revealing that CD5-E1B(639 bp) and CD5-E1A (259 bp) are induced when Bcells from the HCs were stimulated with anti-IgM in thepresence of PD98059, whereas only CD5-E1A (259 bp)is induced in the presence of anti-IgM. These data in-dicate that MAPK/Erk has a role in regulating CD5-E1B up-regulation.
5626 IL-6 CONTROLS DNA METHYLATION AND CD5 IN LUPUS B CELLS

confirmed that BCR engagement induce CD5-E1A but did not in-duce CD5-E1B transcripts (Fig. 1E).
To test the hypothesis that epigenetic changes in the cd5 genecontribute to the generation of alternative transcripts in activated Bcells from patients with SLE, BCR engagement was re-evaluatedin the presence of PD98059 which decreases DNA methylation. Inthese conditions, an up-regulation of CD5-E1B upon anti-IgM/PD98059 stimulation was observed in B cells from HCs usingreal-time PCR (61.5 11.9-fold) and 5�-RACE analysis (Fig. 1, Dand E).
The CD5-E1B promoter is hypomethylated in resting SLEB cells
To determine whether epigenetic changes result in increased CD5-E1B in SLE B cells, genomic DNA from six randomly selectedpatients and seven HCs were analyzed for the level of DNA meth-ylation restriction enzyme treatment of the DNA followed by PCR(Fig. 2A). The protocol is based on the inability of methylation-sensitive restriction enzymes to cut methylated DNA. The resultsof treatment with HaeII enzyme revealed that the CD5-E1B pro-moter was de-methylated in B cells from all 6 SLE patients but innone of the 7 HCs (Fig. 2B). Treatment with HpaII confirmeddemethylation in two of six SLE patients but in none of the HCs.The positive control was the DNA methylation of Daudi cell line
cells (23). The negative control was the HpaII isoschizomer, MpsI,that cut the CpG sequences so that there were no PCR products.
In addition to this data, the methylation status of the CD5-E1Apromoter was sought using HgaII, FauI and HpaII (Fig. 2C). Theresults showed that the CpG motifs were de-methylated, or hemi-methylated in B cells from both the SLE patients and HCs.
The U3 region in CD5-E1B is hypomethylated in B cells fromSLE patients
The presence of HaeII site (site 4) in the U3 region of the 5�-LTRelement of HERV-CD5, and three HpaII sites upstream U3-LTR(site 1) or in the R region of the 5�-LTR (sites 7 and 13) raises thepossibility that the HERV U3-R-U5 regulatory elements may bede-methylated in B cells from patients with SLE (Fig. 3A). Toaddress this issue, bisulfite-treated genomic DNA were amplifiedand sequenced.
Bisulfite C3T transition in SLE CD5-E1B amplicons were an-alyzed and quantified as shown in Fig. 3B and supplemental Fig.1.5 Among 26 CpG sites studied in the 1439-bp amplicon of CD5-EIB, five CpG sites are hemi- or de-methylated. These CpGs (sites2 to 6) are located in the U3-LTR at positions �8466 from the
5 The online version of this article contains supplemental material.
FIGURE 3. The U3-LTR HERV-CD5 region is de-methylated in B cells from SLE patients. A, Regulatory motifs for transcription factor binding andTATA box location within the U3 region. Circles and boxes identify the CpG and U3-R-U5 regions. The HpaII and HaeII sites are also indicated. B, Thelevel of CpG methylation was determined by bisulfite sequencing using genomic DNA obtained from six SLE patients (white) and seven HCs (black). CpGsites are numbered as in A. C, Correlation between CD5 cell surface expression and methylation status for CpG sites 3 to 6. White circle, SLE patients;black circle, HC subjects. �, p � 0.05; p � 0.001.
5627The Journal of Immunology

known ATG1 initiation site (24), �8396, �8393 (HaeII), �8347,and �8363, respectively. De-methylated CpG motifs contain, orare located near, the binding sites of E2/Rb family proteins (con-sensus TTT(C/G)(C/G)CGC, position �8394 to �8391), E-boxfamily proteins (CA(N)3TG, �8391 to �8388), and STAT familyproteins (inverted consensus TT(N)4AA, �8353 to �8344).
When comparing SLE patients to HCs, the cytosine residuesnear the E2/Rb binding sites 3 and 4, plus the CpG at site 5, weresignificantly less affected by the bisulfite treatment in the SLEpatients (Fig. 3B, p � 0.05). Furthermore, the level of methylationof CpG sites 3 to 5, are inversely correlated with CD5 cell surfaceexpression in CD5� B cells (Fig. 3C, p � 0.01). However, de-methylation of the CD5-E1B promoter identified in B cells fromthe SLE patients could be due to gene polymorphisms. Such aproposition was discounted because only one association betweena methylated CpG motif at position �8001 and a SNP site wasobserved (supplemental Fig. 2).
In contrast to CD5-E1B promoter, the 791bp amplicon for CD5-E1A which contains 12 CpG was equally de-methylated in B cellsfrom the SLE patients and HCs (Fig. 2C and supplemental Fig. 1).De-methylated CpGs were located at positions: �148, �123, �102,�73 and �64. These regions contain, or are located nearby to con-sensus binding sites for AP-1 (�151), Sp-1 (�120, �96), E-box fam-ily proteins (�50) and the INR transcriptional start site (�61).
Activation modifies methylation of the CD5-E1B promoter
To assess the influence of BCR engagement on methylation of theCD5 locus, FACS-sorted CD5-negative B cells from six SLE pa-tients and six HCs were stimulated with anti-IgM for 24 h. Meth-ylation patterns were then compared in these activated cells withthe pattern of methylation in the cells before activation (Fig. 4). Inthe SLE patients, engagement of the BCR did not modify CD5promoter methylation status, whereas in the HCs, it resulted inmethylation of the CD5-E1B promoter at positions 3 and 4 (Fig.4D), but not that of CD5-E1A (not shown). Interestingly, when B
cells from the HCs were stimulated with anti-IgM in the presenceof PD98059, CpG sites 3, 4, and 5 were prone to demethylationsimilar to what was seen in the SLE B cells. All these results areconsistent with previous findings that the U3-LTR regions inHERV elements are prone to demethylation (25).
Analysis of DNA methylation enzymes
To gain insights into the cause(s) of de-methylation of the CD5-E1B promoter in B cells from SLE patients, levels of mRNA forDNMTs and MBDs were determined in B cells from ten SLEpatients and 15 HCs. Levels of DNMT1, DNMT3a, DNMT3b,MBD2, and MBD4 were not different in B cells from the patientsand the controls (Table III). With regard to SLE activity, we failedto find correlations between patients with SLEDAI � 5 (n � 3)and changes in DNMTs or MBDs activity. On the basis of ourobservation that B cell activation through the BCR influences U3-LTR methylation, we measured the level of mRNA for DNMT1and MBDs by real-time PCR in B cells stimulated for 24 h withanti-IgM from ten SLE patients and 15 HCs (Fig. 5A). The ex-pression of DNMT1 was increased by 2.3 0.2-fold in B cellsfrom the patients and by 16.6 13.4-fold in the controls ( p �0.005). Importantly, the expression of MBDs was not affected by
FIGURE 4. Effect of BCR engagement on cd5 genemethylation. A, Methylation of CD5-E1B promoter an-alyzed by restriction enzymes and bisulfite sequencing.Enzymes and symbols used are the same as in the leg-end to Fig. 3. B, Effect of restriction enzymes on CD5-E1A. C, The level of CpG methylation measured bybisulfite sequencing with (white) or without (black)BCR engagement in six SLE patients. D, CpG methyl-ation in B cells from six HCs stimulated with anti-IgMin the presence (gray) or absence (white) of PD98059.�, p � 0.05.
Table III. Level of DNMTs and MBDs mRNA in resting B cells from10 SLE patients and 15 HCsa
Patients Controls
DNMT1 14.8 8.4 15.4 10.4DNMT3a 1.03 0.52 0.61 0.76DNMT3b undetectable undetectableMBD2 44.2 22.9 37.1 19.4MBD4 34.2 21.5 23.5 12.5
a cDNA were subjected to quantitative RT-PCR (primers indicated in Table I).Their relative expression was adjusted to 18S levels (�10�6).
5628 IL-6 CONTROLS DNA METHYLATION AND CD5 IN LUPUS B CELLS

stimulation of the BCR or by the addition of the DNMTs inhibitorPD98059.
FACS analyses verified that BCR engagement modulated DNMT1expression. Results from four of ten independent experiments are de-picted in Fig. 5B. Interestingly, DNMT1 staining showed two peaks,DNMT1dim and DNMT1bright. According to this dichotomy, resting Bcells could be divided into 76.1 7.9% DNMT1dim and 23.8 7.9%DNMT1bright cells in the patients and into 74.7 12.3% DNMT1dim
and 25.3 12.3% DNMT1bright cells in the controls ( p � NS). A24-h culture of B cells with anti-IgM increased the proportion of theDNMT1bright population to 86.1 12.2% in the HCs compared with46.1 7.8% in the SLE patients ( p � 0.05). Thus, these resultsindicate that induction of DNMT1 following BCR engagement is re-duced in patients with SLE.
The role of IL-6 in de-methylation and CD5-E1B expression
Based on our previous observations that IL-6 over-expression con-trols the cell cycle in BCR-activated in B cells from SLE patients,we predicted that IL-6 by arresting the cell cycle at late G1 phasemay control the expression of DNMT1 and its capacity to meth-ylate DNA and subsequently CD5 cell surface expression (26–28).
To test whether IL-6 acts on its own, or requires engagement ofthe BCR, FACS-sorted B cells from six HCs were stimulated for48 h with rhIL-6 in the presence or absence of anti-IgM. Expres-sion of CD5-E1B increased by 4.1 3.13-fold in B cells culturedwith rhIL-6, and by 54.8 11.3-fold in B cells cultured withrhIL-6 and anti-IgM (Fig. 6A, p � 0.05). The induction of CD5-E1B upon IgM/rhIL-6 stimulation was also demonstrated by ChIP
FIGURE 5. Involvement of DNMTs on CD5-E1Bexpression. A, Quantitative PCR measurement ofDNMT1, MBD2, and MBD4 mRNA levels in ten SLEpatients and 15 HCs following BCR engagement. Roleof activation of MAPK/Erk pathway in DNMT1 induc-tion was examined in HCs using the PD98059 inhibitor.B, Cytoplasmic staining of DNMT1 in methanol-per-meabilized non-activated, or B cells activated withanti-IgM.
FIGURE 6. The effect of IL-6 on CD5-E1B expres-sion and promoter methylation. A, FACS-sorted B cellsfrom HCs were incubated with IL-6 in the presence, orabsence of anti-IgM, or anti-IL-6R Abs. QuantitativePCR measurement of CD5-E1B (white boxes) andDNMT1 (black boxes) in six HCs. B, CD5-E1B pro-moter ChIP analysis using a nonspecific mouse IgG asnegative control (C�), or a mouse anti-RNA polymer-ase as positive control (C�). C, Cytoplasmic staining ofDNMT1 and p27kip1 as a marker of cell cycle arrest inthe G1 phase in methanol-permeabilized B cells fromthe HCs incubated with IL-6 in the presence or absenceof anti-IgM.
5629The Journal of Immunology
analysis. This experiment showed that RNA polymerase II wasrecruited to the CD5-E1B promoter upon anti-IgM/rhIL-6 stimu-lation (Fig. 6B). Thus, rhIL-6 induces CD5-E1B expression andthis effect is more pronounced when B cells are activated throughthe BCR.
The induction of DNMT1 mRNA by anti-IgM was negated inthe presence of rhIL-6 (Fig. 6A). The experiments were re-peated in the presence of anti-IL-6R Ab to inhibit the activity ofIL-6. DNMT1 induction was restored (10.0 3.6 vs 0.63 0.25-fold without anti-IL-6R Ab, p � 0.05) and CD5-E1B wasreduced (54.8 11.3 vs 35.6 5.2-fold without anti-IL-6R Ab,p � 0.05). Moreover, FACS analyses showed that the numberof DNMT1bright cells was reduced after anti-IgM stimulation inthe presence of rhIL-6 (86.1 12.2% vs 33.5 3.7%, p �0.05). A representative example of two experiments is shown inFig. 6C. We also studied whether these differences could beattributed to a cell cycle blockade. As suspected, the cyclin-dependent kinases inhibitor p27kip1 was over-expressed in anti-IgM/rhIL-6-stimulated B cells compared with rhIL-6 or anti-IgM stimulation alone (Fig. 6C). Overall, IL-6 appears tocontrol CpG methylation in SLE B cells resulting, probably,from its effect on arresting cells at the late G1 phase of the cellcycle.
Effects of IgM/IL-6 stimulation on promoters of other genes
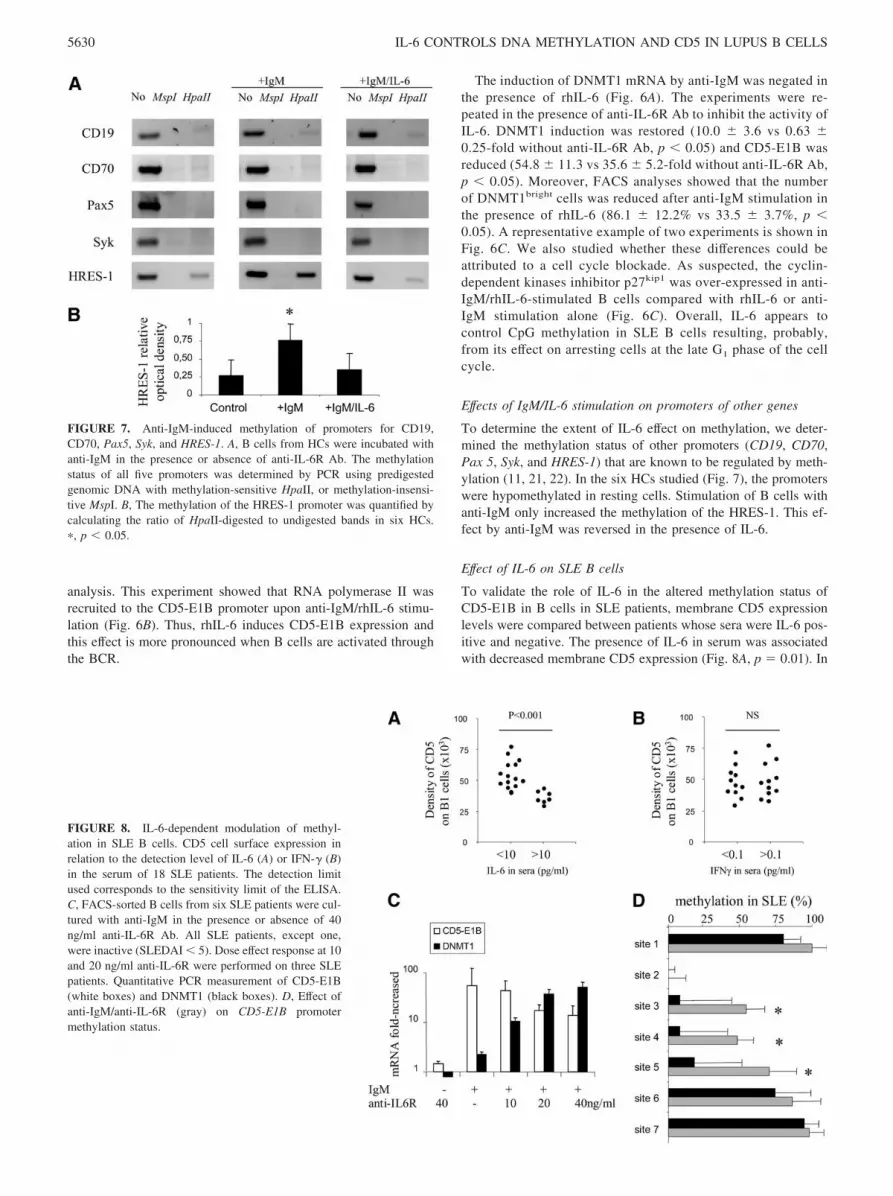
To determine the extent of IL-6 effect on methylation, we deter-mined the methylation status of other promoters (CD19, CD70,Pax 5, Syk, and HRES-1) that are known to be regulated by meth-ylation (11, 21, 22). In the six HCs studied (Fig. 7), the promoterswere hypomethylated in resting cells. Stimulation of B cells withanti-IgM only increased the methylation of the HRES-1. This ef-fect by anti-IgM was reversed in the presence of IL-6.
Effect of IL-6 on SLE B cells
To validate the role of IL-6 in the altered methylation status ofCD5-E1B in B cells in SLE patients, membrane CD5 expressionlevels were compared between patients whose sera were IL-6 pos-itive and negative. The presence of IL-6 in serum was associatedwith decreased membrane CD5 expression (Fig. 8A, p � 0.01). In
FIGURE 7. Anti-IgM-induced methylation of promoters for CD19,CD70, Pax5, Syk, and HRES-1. A, B cells from HCs were incubated withanti-IgM in the presence or absence of anti-IL-6R Ab. The methylationstatus of all five promoters was determined by PCR using predigestedgenomic DNA with methylation-sensitive HpaII, or methylation-insensi-tive MspI. B, The methylation of the HRES-1 promoter was quantified bycalculating the ratio of HpaII-digested to undigested bands in six HCs.�, p � 0.05.
FIGURE 8. IL-6-dependent modulation of methyl-ation in SLE B cells. CD5 cell surface expression inrelation to the detection level of IL-6 (A) or IFN-� (B)in the serum of 18 SLE patients. The detection limitused corresponds to the sensitivity limit of the ELISA.C, FACS-sorted B cells from six SLE patients were cul-tured with anti-IgM in the presence or absence of 40ng/ml anti-IL-6R Ab. All SLE patients, except one,were inactive (SLEDAI � 5). Dose effect response at 10and 20 ng/ml anti-IL-6R were performed on three SLEpatients. Quantitative PCR measurement of CD5-E1B(white boxes) and DNMT1 (black boxes). D, Effect ofanti-IgM/anti-IL-6R (gray) on CD5-E1B promotermethylation status.
5630 IL-6 CONTROLS DNA METHYLATION AND CD5 IN LUPUS B CELLS

contrast, changes in membrane CD5 expression were not associ-ated with serum IFN-� (Fig. 8B). CD5 cell surface expression wasreduced in patients who had SLEDAI � 5 (39,789 11,450 vs51,294 15,215), but this was not significant (data not shown).
Finally, the effect of a blocking anti-IL-6R Ab was studied be-cause we previously observed that anti-IgM stimulation of B cellsfrom SLE patients produced higher levels of IL-6 than matchedHCs (514.1 159.7 pg/ml vs 99.0 116.0 pg/ml, data not shown)(26). Blocking the autocrine loop of IL-6 increased DNMT1 ex-pression (58.9 13.5-fold with 40 ng/ml anti-IL-6R) and contrib-uted to the methylation of the U3-LTR sites 3 to 5 (Fig. 8, C andD). In addition, expression of CD5-E1B was reduced (17.2 3.5-fold with 40 ng/ml anti-IL-6R vs 54.4 68.1-fold without, p �0.05). Further, this effect was dose-dependent.
DiscussionThis study provides evidence for the notion that reduction in thelevel of membrane CD5 on circulating CD5� B cells SLE is dueto increased expression of the cytoplasmic isoform of CD5, CD5-E1B. As a consequence of reduced membrane CD5, increasedCD5-E1B limits the negative regulatory effects of CD5 on BCR-mediated signaling. The available evidence indicates that mem-brane CD5 increases the threshold of BCR-mediated responses(14). The implication of such data is that regulation of CD5-E1B,which in turn regulates expression of CD5, is involved in main-taining the anergy status of autoreactive B cells. Results generatedfrom our previous studies indicated that the modulatory effects ofCD5 protein on BCR signaling are attributed to the CD5 isoformwhich contains E1A, the previously documented exon 1. Introduc-tion of cDNA for the recently identified CD5-E1B isoform intoCD5� T and B cells reduced membrane expression of CD5 protein(17, 19). Coexpression experiments have also shown that in thepresence of CD5-E1B, CD5-E1A was not translocated to the mem-brane but associated with CD5-E1B in cytoplasmic aggregates(19). The exact dynamics of the interaction between CD5-E1B andCD5-E1A remain unknown. However, we have established thatthe stretch of amino acids from positions 286–400 in CD5-E1B iscrucial for reducing membrane CD5-E1A translocation (19). Func-tionally, these data suggest that up-regulated CD5-E1B expressionin B cells could reduce the BCR activation threshold by Ags andthus, promote autoimmunity.
Because CD5-E1B arises from the integration of an HERV el-ement known to be silenced by DNA methylation (19), we pre-dicted that epigenetic modifications could be involved in regulat-ing CD5 isoform expression in B cells. Normally, methylation ofDNA is evident in CpG-poor regions and in regions of repetitivesequences including LTR in HERV sequences. In contrast, CpGislands present within gene promoters resist to methylation. In nor-mal B cells, this paradigm holds true for the cd5 locus, where theU3-LTR element is methylated while the CD5-E1A promoter isnot. However, in SLE B cells, the rate of de-methylation is higherin the U3 region of the HERV-CD5 element. Consistent with thisobservation is the finding that another HERV element, HRES-1,was also integrated at the stage of old world primate divergence(22). Indeed, expression of HRES-1 retroviral proteins, HRES-1/p28 and HRES-1/Rab4 (29), and our observations on HRES-1 pro-moter methylation status (Fig. 7) may hint to a global defect incontrolling repetitive elements in SLE. Our preliminary results areindeed indicative that HRES-1 methylation pattern was not in-creased in SLE patients upon BCR engagement. In this respect,global DNA methylation has been reported to be altered in almostall forms of SLE including drug-induced lupus (30). As a conse-quence, it was postulated that epigenetic transformation modifies Bcell physiology resulting in polyclonal activation, IgG1 class-
switching (31), V(D)J rearrangement by RAG1/RAG2 enzymes(32) and a shift in cytokine profiles (33). Altered methylation alsoinfluences T cells in SLE causing up-regulation of T cell costimu-lating molecules (CD70, CD40 ligand), and a shift in cytokineprofile from Th1 to Th2 (for review, see Ref. 34). We propose thatthese alterations, together with our findings on CD5 down-regula-tion, contribute to promoting autoreactivity.
De-methylation of DNA can be result from the inhibition or lackof DNMTs or be due to specific enzymatic reactions includingMBDs or a combination of both. Thus, DNA methylation patternscan be altered owing to a shift in the balance between MBD de-methylation and DNMT methylation enzymes. Quantitative PCRassays were performed to evaluate the relative levels of MBD andDNMT transcripts in resting and in anti-IgM stimulated B cells. Atbasal level, B cells from SLE patients and HCs had similar levelsof DNMT1, DNMT3a, MBD2, and MBD4. The amount ofDNMT3b was very low or undetectable in both SLE patients andHCs as previously described for T cells (35). A recent study re-ported reductions in DNMT1 in a subgroup of SLE patients (36).However, this study was conducted using a relatively small cohortand was not confirmed either in CD4� T cells (35) or in B cells bythe present study. In addition, the recently reported increase inMBD2 and MBD4 in patients with SLE (37) was also not con-firmed in our study. These differences may be because we haveexamined B cells instead of CD4� T cells, or that the patientpopulation included in the other study was different. Interestingly,analysis of stimulated B cells revealed that expression of DNMT1was reduced following engagement of the BCR in SLE in agree-ment with findings in stimulated CD4� T cells (13).
Our results, suggesting that DNA hypomethylation in activatedSLE B cells, could be attributed to cell cycle arrest in G1 is sur-prising. The surprising aspect of the observation is based on thefact that proliferation of lymphocytes did not differ between thepatients and HCs implying that decreased DNMT1 activity in SLEwas not due to a cell cycle arrest (13). However, inhibition of theMAPK/Erk2 pathway was clearly associated with DNMT1 reduc-tion in those patients (13, 38). Further, inhibition of this pathwayis likely to elicit cell cycle arrest through p27kip1 induction (39,40). However, inhibition of MEK/Erk2 pathway using PD98059 inpro-B induces cell growth arrest with p27kip1 accumulation (39).Therefore, in such a setting, it was not surprising that PD98059decreased methylation in HCs as in stimulated SLE B cells. Hence,these data indicate that decreased activation of the Erk pathwaycould be important for the development of autoimmunity (7). Thiswas previously confirmed in animal models showing that treatingnormal mice with inhibitors of DNA methylation, or with Erk in-hibitors causes a lupus-like disease (41). Further, Tg mice express-ing dominant-negative MEK leads to overexpression of methyla-tion-sensitive genes and the production of anti-dsDNAautoantibodies by B cells (38).
The proposed involvement of IL-6 in promoting autoreactivityis supported by in vitro and in vivo studies. For example, highlevels of serum IL-6 is correlated with lupus disease activity andtargeting IL-6 with blocking Abs is an effective treatment for SLE(4). However, the precise mechanism by which IL-6-mediated Bcell proliferation, differentiation, and autoantibody production inSLE remains to be elucidated. Repression of IL-6 is associatedwith hypermethylation of its promoter in HCs whereas its overex-pression in SLE is associated with promoter hypomethylation (42).Interestingly, by treating B cells from HCs with IL-6 in the pres-ence of anti-IgM, we have shown that CD5-E1B expression can besignificantly increased and that this correlates with a cell cyclearrest in late G1 phase. In addition, this effect is associated with anIg gene rearrangement following RAG re-expression (26). The loss
5631The Journal of Immunology

of, or reduction in, the inhibitory effects of CD5 is associated withRAG re-expression in B cells in patients with SLE and these maysynergize to induce autoantibody production.
In conclusion, the finding that IL-6 activates CD5-E1B tran-scription is relevant for understanding mechanism of action andtherapeutic benefits of anti-IL-6. Thus, treatment of patients withSLE with anti-IL-6R mAb could inhibit autoreactive B cell expan-sion by restoring DNA methylation and cell cycle progression. Inaddition, the data provide further evidence for the pivotal role ofCD5 in maintaining anergy in autoreactive B cells.
AcknowledgmentsWe are grateful to the Conseil Regional de Bretagne, the Conseil Generaldu Finistere, the College Doctoral International, and the Universite Europ-eenne de Bretagne for support. Thanks are also due to Cindy Sene andSimone Forest for excellent secretarial assistance.
DisclosuresThe authors have no financial conflict of interest.
References1. Rahman, A., and D. A. Isenberg. 2008. Systemic lupus erythematosus. N. Engl.
J. Med. 358: 929–939.2. Renaudineau, Y., P. O. Pers, B. Bendaoud, C. Jamin, and P. Youinou. 2004.
Dysfunctional B cells in SLE. Autoimmun. Rev. 3: 516–523.3. Leandro, M. J., J. C. Edwards, G. Cambridge, M. R. Ehrenstein, and
D. A. Isenberg. 2002. An open study of B lymphocyte depletion in systemic lupuserythematosus. Arthritis Rheum. 46: 2673–2677.
4. Illei, G., C. Yarboro, Y. Shirota, E. Tackey, L. Lapteva, and T. Fleisher. 2006.Tocilizumab (humanized anti IL-6 receptor monoclonal antibody) in patients withsystemic lupus erythematosus (SLE): safety, tolerability and preliminary efficacy.Arthritis Rheum. 54 (Suppl): 4043 (Abstract).
5. Finck, B. K., P. S. Linsley, and D. Wofsy. 1994. Treatment of murine lupus withCTLA4Ig. Science 265: 1225–1227.
6. Hom, G., R. R. Graham, B. Modrek, K. E. Taylor, W. Ortmann, S. Garnier,A. T. Lee, S. A. Chung, R. C. Ferreira, P. V. Pant, et al. 2008. Association ofsystemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl.J. Med. 358: 900–909.
7. Ballestar, E., M. Esteller, and B. C. Richardson. 2006. The epigenetic face ofsystemic lupus erythematosus. J. Immunol. 176: 7143–7147.
8. Zhu, B., Y. Zheng, H. Angliker, S. Schwarz, S. Thiry, M. Siegmann, andJ. P. Jost. 2000. 5-Methylcytosine DNA glycosylase activity is also present in thehuman MBD4 (G/T mismatch glycosylase) and in a related avian sequence. Nu-cleic Acids Res. 28: 4157–4165.
9. Hendrich, B., and A. Bird. 1998. Identification and characterization of a familyof mammalian methyl-CpG binding proteins. Mol. Cell Biol. 18: 6538–6547.
10. Schwab, J., and H. Illges. 2001. Regulation of CD21 expression by DNA meth-ylation and histone acetylation. Int. Immunol. 13: 705–710.
11. Lu, Q., A. Wu, L. Tesmer, D. Ray, N. Yousif, and B. Richardson. 2007. De-methylation of CD40LG on the inactive X in T cells from women with lupus.J. Immunol. 179: 6352–6358.
12. Piotrowski, P. C., S. Duriagin, and P. P. Jagodzinski. 2005. Expression of HERVclone 4-1 may correlate with blood plasma concentration of anti-U1 RNP andanti-Sm nuclear antibodies. Clin. Rheumatol. 24: 620–624.
13. Deng, C., M. J. Kaplan, J. Yang, D. Ray, Z. Zhang, W. J. McCune, S. M. Hanash,and B. C. Richardson. 2001. Decreased ras-mitogen-activated protein kinase sig-naling may cause DNA hypomethylation in T lymphocytes from lupus patients.Arthritis Rheum. 44: 397–407.
14. Hippen, K. L., L. E. Tze, and T. W. Behrens. 2000. CD5 maintains tolerance inanergic B cells. J. Exp. Med. 191: 883–890.
15. Jamin, C., G. Magadur, A. Lamour, L. MacKenzie, P. M. Lydyard, P. Katsikis,and P. Youinou. 1992. Cell-free CD5 in patients with rheumatic diseases. Im-munol. Lett. 31: 79–84.
16. Lu, X., R. C. Axtell, J. F. Collawn, A. Gilson, L. B. Justement, and C. Raman.2002. AP2 adaptor complex-dependent internalization of CD5: differential reg-ulation in T and B cells. J. Immunol. 168: 5612–5620.
17. Renaudineau, Y., S. Hillion, A. Saraux, R. A. Mageed, and P. Youinou. 2005. Analternative exon 1 of the CD5 gene regulates CD5 expression in human B lym-phocyte. Blood 106: 2781–2789.
18. Renaudineau, Y., S. Vallet, C. Le Dantec, S. Hillion, A. Saraux, and P. Youinou.2005. Characterization of the human CD5 endogenous retrovirus-E in B lym-phocytes. Gene Immun. 6: 663–671.
19. Garaud, S., C. Le Dantec, C. Berthou, P. M. Lydyard, P. Youinou, andY. Renaudineau. 2008. Selection of the alternative exon 1 from the cd5 genedown-regulates membrane level of the protein in B lymphocytes. J. Immunol.181: 2010–2018.
20. Tan, E. M., A. S. Cohen, J. F. Fries, A. T. Masi, D. J. McShane, N. F. Rothfield,J. G. Schaller, N. Talal, and R. J. Winchester. 1982. The 1982 revised criteria forthe classification of SLE. Arthritis Rheum. 25: 1271–1277.
21. Ushmorov, A., F. Leithauser, O. Sakk, A. Weinhausel, S. W. Popov, P. Moller,and T Wirth. 2006. Epigenetic processes play a major role in B-cell-specific genesilencing in classical Hodgkin lymphoma. Blood 107: 2493–2500.
22. Perl, A., J. D. Rosenblatt, I. S. Chen, J. P. DiVincenzo, R. Bever, B. J. Poiesz, andG. N. Abraham. 1989. Detection and cloning of new HTLV-related endogenoussequences in man. Nucleic Acids Res. 17: 6841–6854.
23. Ruchusatsawat, K., J. Wongpiyabovorn, S. Shuangshoti, N. Hirankarn, andA. Mutirangura. 2006. SHP-1 promoter 2 methylation in normal epithelial tissuesand demethylation in psoriasis. J. Mol. Med. 84: 175–182.
24. Jones, N. H., M. L. Clabby, D. P. Dialynas, H. J. Huang, L. A. Herzenberg, andJ. L. Strominger. 1986. Isolation of complementary DNA clones encoding thehuman lymphocyte glycoprotein T1/Leu-1. Nature 323: 346–349.
25. Reiss, D., Y. Zhang, and D. L. Mager. 2007. Widely variable endogenous ret-roviral methylation levels in human placenta. Nucleic Acids Res. 35: 4743–4754.
26. Hillion, S., S. Garaud, V. Devauchelle, A. Bordron, C. Berthou, P. Youinou, andC. Jamin. 2007. IL-6 is responsible for aberrant BCR-mediated regulation ofRAG expression in SLE. Immunology 122: 371–380.
27. Robertson, K. D., K. Keyomarsi, F. A. Gonzales, M. Velicescu, and P. A. Jones.2000. Differential mRNA expression of the human DNA methyltransferases(DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal andtumor cells. Nucleic Acids Res. 28: 2108–2113.
28. Brown, S. E., M. F. Fraga, I. C. Weaver, M. Berdasco, and M. Szyf. 2007.Variations in DNA methylation patterns during the cell cycle of HeLa cells.Epigenetics 2: 54–65.
29. Pullmann, R., Jr., E. Bonilla, P. E. Phillips, F. A. Middleton, and A. Perl. 2008.Haplotypes of the HRES-1 endogenous retrovirus are associated with develop-ment and disease manifestations of systemic lupus erythematosus. ArthritisRheum. 58: 532–540.
30. Zhou, Y., and Q. Lu. 2008. DNA methylation in T cells from idiopathic lupus anddrug-induced lupus patients. Autoimmun. Rev. 7: 376–383.
31. Vigorito, E., K. L. Perks, C. Abreu-Goodger, S. Bunting, Z. Xiang, S. Kohlhaas,P. P. Das, E. A. Miska, A. Rodriguez, A. Bradley, et al. 2007. microRNA-155regulates the generation of Ig class-switched plasma cells. Immunity 27:847–859.
32. Wang, H., J. Feng, C. F. Qi, Z. Li, H. C. Morse 3rd, and S. H. Clarke. 2007.Transitional B cells lose their ability to receptor edit but retain their potential forpositive and negative selection. J. Immunol. 179: 7544–7552.
33. Pang, Y., Y. Norihisa, D. Benjamin, R. R. Kantor, and H. A. Young. 1992. IFN-�gene expression in human B-cell lines: induction by IL-2, PKC, and possibleeffect of hypomethylation on gene regulation. Blood 80: 724–732.
34. Huber, L. C., J. Stanczyk, A. Jungel, and S. Gay. 2007. Epigenetics in inflam-matory rheumatic diseases. Arthritis Rheum. 56: 3523–3531.
35. Balada, E., J. Ordi-Ros, S. Serrano-Acedo, L. Martinez-Lostao, M. Rosa-Leyva,and M. Vilardell-Tarres. 2008. Transcript levels of DNA methyltransferasesDNMT1, DNMT3A and DNMT3B in CD4� T cells from patients with systemiclupus erythematosus. Immunology 124: 339–347.
36. Ogasawara, H., M. Okada, H. Kaneko, T. Hishikawa, I. Sekigawa, andH. Hashimoto. 2003. Possible role of DNA hypomethylation in the induction ofSLE: relationship to the transcription of HERV. Clin. Exp. Rheumatol. 21:733–738.
37. Balada, E., J. Ordi-Ros, S. Serrano-Acedo, L. Martinez-Lostao, andM. Vilardell-Tarres. 2007. Transcript overexpression of the MBD2 and MBD4genes in CD4� T cells from SLE patients. J. Leukocyte Biol. 81: 1609–1616.
38. Sawalha, A. H., M. Jeffries, R. Webb, Q. Lu, G. Gorelik, D. Ray, J. Osban,N. Knowlton, K. Johnson, and B. Richardson. 2008. Defective T-cell ERK sig-naling induces IFN-regulated gene expression and overexpression of methyla-tion-sensitive genes similar to lupus patients. Genes Immun. 9: 368–378.
39. Qiang, Y. W., M. Kitagawa, M. Higashi, G. Ishii, C. Morimoto, and K. Harigaya.2000. Activation of MAPK through alpha5/�1 integrin is required for cell cycleprogression of B progenitor cell line, Reh, on human marrow stromal cells. Exp.Hematol. 28: 1147–1157.
40. Gysin, S., S. H. Lee, N. M. Dean, and M. McMahon. 2005. Pharmacologic in-hibition of RAF–�MEK–�ERK signaling elicits pancreatic cancer cell cyclearrest through induced expression of p27Kip1. Cancer Res. 65: 4870–4880.
41. Richardson, B., D. Ray, and R. Yung. 2004. Murine models of lupus induced byhypomethylated T cells. Methods Mol. Med. 102: 285–294.
42. Mi, X. B., and F. Q. Zeng. 2008. Hypomethylation of IL-4 and -6 promoters inT cells from SLE patients. Acta Pharmacol. Sin. 29: 105–112.
5632 IL-6 CONTROLS DNA METHYLATION AND CD5 IN LUPUS B CELLS