host interaction for phage therapy against avian pathogenic
TRANSCRIPT

List of Abbreviations
Contribution to the understanding of the bacteriophage-
host interaction for phage therapy against avian pathogenic
Escherichia coli (APEC)
Patricia Espenhain Sørensen
This dissertation has been submitted in the fulfilment of the requirements for the degree of Doctor of
Philosophy (PhD) in Veterinary Sciences, Faculty of Veterinary Medicine, Ghent University and Ross
University School of Veterinary Medicine, 2022.
Promoters:
Prof. Dr. Patrick Butaye
Prof. Dr. An Garmyn
Prof. Dr. Hanne Ingmer
Faculty of Veterinary Medicine
Department of Pathobiology, Pharmacology and Zoological Medicine

List of Abbreviations
Contribution to the understanding of the bacteriophage-host interaction for phage
therapy against avian pathogenic Escherichia coli (APEC)
PhD thesis, Ghent University and Ross University School of Veterinary Medicine, 2022
© Patricia Espenhain Sørensen
This research was funded by the European Union’s Horizon 2020 research and innovation program
under the Marie Skłodowska-Curie grant agreement no. 765147 and the Special Research Fund (BOF)
of Ghent University under the grant no. BOF.ITN.2021.0007.02. Conference attendance was supported
by Ghent University Mobility Fund. Additional funding was received from Augustinus Fonden and
Christian og Ottilia Brorsons Rejselegat.
Printed by: University Press, Belgium
Cover image: Electron microscopy pictures of Caudovirales coliphages by Liesbeth Couck.

Examination board
Prof. dr. Niek Sanders (Chair)
Prof. dr. Gunther Antonissen (Secretary)
Dr. Ilias Chantziaras
Dr. Steven Van Borm
Prof. dr. Rob Lavigne
Prof. dr. Felix Toka

List of Abbreviations
“Impossible means that you haven’t found a solution yet”
Henry Ford (1863-1947)


List of Abbreviations
i
Table of contents
List of Abbreviations ....................................................................................................... iii
Chapter 1: General Introduction...................................................................................... 1
1.1 Antibiotics and resistance ............................................................................................ 1
1.2 Bacteriophages ........................................................................................................... 4
1.2.1 General characteristics .......................................................................................... 4
1.2.2 Phage life cycle .................................................................................................... 6
1.2.3 Phage taxonomy ................................................................................................... 9
1.2.4 Phage diversity and signature genes.......................................................................11
1.2.5 Phage therapy......................................................................................................15
1.3 Phage-host interactions...............................................................................................21
1.3.1 Population growth dynamics.................................................................................22
1.3.2 Bacterial phage resistance.....................................................................................24
1.4 Avian pathogenic Escherichia coli (APEC) ..................................................................30
1.4.1 Diseases, transmission, and reservoirs ...................................................................30
1.4.2 Virulence factors .................................................................................................31
1.4.3 Strain typing and population genetics ....................................................................33
1.4.4 Current strategies to prevent and control APEC ......................................................33
1.4.5 Phage therapy against APEC infections .................................................................35
Chapter 2: Scientific Aims.............................................................................................. 61
Chapter 3: Experimental Studies ................................................................................... 63
3.1 New insights into the biodiversity of coliphages in the intestine of poultry ......................65
3.2 Classification of in vitro phage-host population growth dynamics................................. 115
3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli .............................. 145
3.4 Schematic overview of the experimental studies and main findings .............................. 182
Chapter 4: General Discussion ..................................................................................... 185
4.1 Avian pathogenic E. coli (APEC) - The need for alternative treatment options............... 185
4.2 The remarkable diversity of E. coli-infecting phages ................................................... 186
4.2.1 Phage host spectrum .......................................................................................... 187
4.2.2 Hypothetical proteins of unknown function .......................................................... 188
4.3 Phage-host population growth dynamics .................................................................... 189
4.4 Phage resistance in APEC......................................................................................... 190
4.4.1 The cost of phage resistance ............................................................................... 193

ii
4.5 Acquisition and selection of suitable phages for phage therapy ..................................... 194
4.6 Conclusions and future perspectives .......................................................................... 196
Summary ...................................................................................................................... 207
Samenvatting ................................................................................................................ 211
Curriculum Vitae ......................................................................................................... 215
Bibliography ................................................................................................................. 217
Conference contributions ............................................................................................. 219
Acknowledgements ....................................................................................................... 221

List of Abbreviations
iii
List of Abbreviations
Abi Abortive infection
Acr Anti-CRISPR
AMR Antimicrobial resistance
AP Agar plate
APEC Avian pathogenic E. coli
AUC Area under the curve
BLAST Basic Local Alignment Search Tool
bp Base pairs
BREX Bacteriophage exclusion
BWA Burrows-Wheeler Aligner
C Cytosine
CARD Comprehensive Antibiotic Resistance Database
Cas CRISPR-associated proteins
CDS Coding sequence
CFU Colony forming unit
Coliphage E. coli-infecting phage
ColV Colicin V
CRISPR Clustered regularly interspaced short palindromic repeats
crRNA CRISPR RNA
crRNP crRNA-Cas protein
DLA Double-layer agar
DNA Deoxyribonucleic acid
ds Double-stranded
E. coli Escherichia coli
EPS Exopolysaccharides
EU European Union
ExPEC Extraintestinal E. coli
FAO Food and Agriculture Organization
G Guanine
GI Gastrointestinal
GMO Genetically modified organism

List of Abbreviations
iv
GMP Good Manufacturing Practices
HGT Horizontal gene transfer
IC Intracranial
ICTV International Committee on Taxonomy of Viruses
IM Intramuscular
IT Intratracheal
Kbp Kilobase pairs
LB Lysogeny Broth
LCBs Local collinear blocks
LPS Lipopolysaccharide
MCP Major capsid protein
MDR Multidrug-resistant
MEGA Molecular evolutionary genetics analyses
MLST Multilocus sequence typing
MOI Multiplicity of infection
MTase Methyltransferase
MTP Major tail protein
NASP Northern Arizona SNP Pipeline
NCBI National Center for Biotechnology Information
NMDS Non-metric multidimensional scaling
OD Optical density
OIE World Organization for Animal Health
OMP Outer membrane protein
PCA Principal component analysis
PCR Polymerase chain reaction
PD Pharmacodynamics
Phage Bacteriophage
PHASTER PHAge Search Tool Enhanced Release
PK Pharmacokinetics
PFA Paraformaldehyde
PFU Plaque forming units
R-M Restriction-modification
RAST Rapid Annotation using Subsystem Technology
RBP Receptor-binding protein

List of Abbreviations
v
REase Restriction endonuclease
RNA Ribonucleic acid
rpm Revolutions per minute
RT-qPCR Real-time quantitative PCR
SC Secondary culture
SEA-PHAGES Science Education Alliance-Phage Hunters Advancing Genomics and
Evolutionary Science
SNPs Single nucleotide polymorphisms
ST Sequence type
TA Toxin-antitoxin
TEM Transmission electron microscopy
TLS Terminase large subunit
TMP Tape measure protein
TPR Tetratricopeptide repeat
UPGMA Unweighted pair group method with arithmetic mean
WGS Whole-genome sequencing
WHO World Health Organization
WT Wild type

List of Abbreviations
vi

Chapter 1: General Introduction
1
Chapter 1: General Introduction
Chapter 1
General Introduction
1.1 Antibiotics and resistance
The modern era of antibiotics began with the discovery of penicillin by Sir Alexander Fleming
in 1928 [1, 2]. Since then, antibiotics have transformed modern medicine, veterinary as well as
human, and saved millions of lives. The “golden age” of antibiotics began in the 1940s and
continued for over four decades, with more than 20 classes of antibiotics being discovered and
introduced for clinical use [3–5]. During this period, bacterial resistance emerged, but was met
with minimal concern, as new compounds, often exhibiting better pharmacokinetics (PK) and
pharmacodynamics (PD), were quickly developed and provided alternative treatments [6, 7].
From the 1990s, as the number of novel antibiotics introduced steadily decreased, the
consequences of the link between antibiotic use/overuse and occurrence of resistance became
more apparent. This period has been described as a “dry pipeline” or “discovery void” in
antibiotic research and development, with fewer new drugs introduced, and with the majority
of these being either modified or combined versions of previously known compounds (Figure
1). As a result, many decades after the first patients were treated with antibiotics, more and
more bacterial infections are becoming a threat and difficult to treat once again [3, 8, 9].

Chapter 1: General Introduction
2
Figure 1 | Timeline of the discovery of different antibiotic classes in clinical use. The “golden age” refers to the
period from 1940s to 1960 as one-half of the drugs commonly used today were discovered in this period. The
“discovery void” refers to the period from 1987 until today with only few new drugs introduced, and with majority
of these being semi- or fully synthetic [adapted from reference [8, 10, 11]].
The excessive use of antimicrobials in various areas, including clinical, industrial and
agricultural settings, and release into the environment for over half a century drives the
evolution of resistance and have generated a constant selective pressure for resistant bacterial
strains in all ecological niches [3, 12–14]. Studies have now demonstrated the direct association
between antibiotic consumption and the emergence and dissemination of resistant bacterial
strains [6, 7, 15–17]. The consecutive acquisition of antibiotic resistant traits has resulted in
the emergence of multidrug-resistant (MDR) and even pan-resistant pathogens. The resistance
can be caused by spontaneous mutations, recombination, or the acquisition of genes through
horizontal gene transfer (HGT), which is the major mechanisms involved in dissemination of
antibiotic resistance. In bacteria, HGT occurs mainly by conjugation, transformation and
transduction [18–20]. Conjugation is thought to be the main mode responsible for the spread
of antibiotic resistance [21]. During conjugation, deoxyribonucleic acid (DNA) is transferred
from the donor cell to a recipient through cell-to-cell contact and occurs either through plasmid
transfer or chromosomally integrated conjugation elements [19]. In transformation,
extracellular DNA is taken up by the bacterium from the environment and incorporated into
the genome, while in transduction, gene transfer is mediated by bacteriophages (phages) [18,
22, 23]. Also, resistance can automatically be transferred from one generation to the next

Chapter 1: General Introduction
3
through replication (vertical gene transmission), unless the resistance-conferring element is lost
[24–26].
The global emergence of widespread antimicrobial resistance (AMR) has forced us to consider
the One Health approach to effectively control AMR and reduce the dissemination of resistance
genes between microorganisms [27]. The term “One Health” refers to the collaborative,
multisectoral, and transdisciplinary approach - working at the local, national, and global level
- to achieve optimal health outcomes by recognising the interdependency between people,
animals, plants, and their shared environment [28]. This holistic approach is required since
many of the antimicrobials used in human medicine are also used in veterinary medicine and
livestock production, as well as in plant production and their use drives selection of AMR,
regardless of the specific context in which they are used [27, 29]. Moreover, there is increasing
evidence that clinically relevant resistant bacteria and/or resistance genes are able to transfer
between animals and humans by overcoming both ecological and geographical barriers,
although the impact of this remains unclear [27, 29, 30]. Actions are needed to preserve the
continued effectiveness of existing antimicrobials by for example eliminating their
inappropriate use and by limiting the spread of infections through biosecurity measures [28,
31]. Remaining concerns are mass medication of animals in the animal production sector with
antimicrobials that are critically important for humans and the in-feed use of medically
important antimicrobials for growth promotion of healthy animals in some countries [28].
Numerous countries and several international agencies, such as the World Health Organization
(WHO), the World Organization for Animal Health (OIE) and the Food and Agriculture
Organization (FAO), have included a One Health approach within their action plans to assess,
control and prevent the spread of AMR as well as zoonotic diseases [28, 29]. Necessary actions
include improvement of antimicrobial use regulations, surveillance, infection control, animal
husbandry, and alternatives to antimicrobials [28]. Accordingly, as current antimicrobials
become increasingly inadequate, alternative treatment options are urgently needed. As such,
the use of phages as therapeutics (phage therapy) may help cope with the burden of
antimicrobial resistance [3, 12, 32–35].

Chapter 1: General Introduction
4
1.2 Bacteriophages
1.2.1 General characteristics
Phages are viruses that specifically infect bacteria. They were discovered independently by
William Twort in 1915 and by Felix d’Herelle in 1917 who realised that they had antimicrobial
potential [36, 37]. d’Herelle used the term “bacteriophage” meaning “bacteria eater”, to
describe the organism’s antimicrobial ability. Phages are the most abundant organisms on
Earth, estimated about 4.8 x 1031 entities, and can be found in all known ecosystems, including
soil, wastewater, sewage water, seawater and in and on humans and animals [38–41]. Phages
outnumber their hosts by more than an order of magnitude, and are thought to play essential
roles in shaping the microbial communities, including driving the diversity, ecology and
evolution [38, 42–44]. Like other viruses, phages are unable to replicate independently of a
susceptible cellular host, and both their abundance and distribution are likely to be based on
that of their host. While some phages are able to infect hosts from different genera, families,
or orders, most studied phages are extremely specific and only capable of infecting a narrow
range of bacteria that are closely related [45–48]. The host range is determined by a
combination of various factors, including phage specificity, host attachment factors and
receptors, biochemical interactions during infection, presence of related (pro)phages in the
bacterial cell, and bacterial phage-resistance mechanisms [43, 45, 49–51]. In nature, phage host
range can be broadened or changed through mutation of receptor-binding proteins or exchange
and/or acquisition of new tail fiber genes by recombination, which may allow the phages to
move between related hosts [45, 52, 53].
The majority of phages, known to date, belong to the Caudovirales order (also known as the
tailed phages) (see section 1.2.3 Phage taxonomy) [54–56]. They have a double-stranded
(ds)DNA genome that range from about 18 to 500 Kilobase pairs (Kbp) [54]. The dsDNA is
enclosed in a polyhedral head, often being icosahedral, to which a tubular tailed is attached
(Figure 2). Most often, the head size of these phages range between about 45 nm and 170 nm
and the tail length between 3 nm and 825 nm. [54].

Chapter 1: General Introduction
5
Figure 2 | Tailed phage. A) Negative staining transmission electron microscopy (TEM) images of tailed phage.
x60,000 magnification. Bar indicates 100 nm. Source: Lisbeth Couck, Department of Veterinary medical imaging
and small animal orthopaedics, Faculty of Veterinary Medicine, Ghent University. B) A schematic representation
of a tailed phage.
The tail morphology has traditionally been used for classification of the Caudovirales phages
into families: the Myoviridae family with a complex long contractile tail, the Siphoviridae
family with a long non-contractile tail, and the Podoviridae family with a short non-contractile
tail [56]. However, phage classification is currently undergoing extensive reorganisation,
primarily using genomic-based methods [57, 58]. Recently, it was suggested that these three
morphotypes are kept only as descriptors, and not as basis for establishment of phage families
as these would not be monophyletic [59].
The head-to-tail connecting region, termed connector or neck, ensures the interaction of the
phage capsid with its tail in all Caudovirales phages. It is often made of three different
components organised as consecutive rings: the portal protein and two head completion
proteins. The portal protein is located at the top of the “neck”/collar of the phage and is involved
in DNA packaging during assembly and release at the onset of infection [56, 60].
The composition of the Siphoviridae tail is rather simple and is based on three components: the
central TMP, the tail tube protein or major tail protein (MTP), and the tail terminator protein.
These components are also present and assembled in a similar way in Myoviridae tails, in
combination with the sheath protein that provides the contractile nature to the tail [61, 62]. At
the distal tail end, a special organelle (varying in size, composition, and morphology) dedicated
A) B)

Chapter 1: General Introduction
6
to specific host recognition is found, and which controls phage specificity. The composition of
this organelle can be as simple as a tail tip or consists of a larger macromolecular complex
termed the baseplate. Despite these major conformational differences between tail tip and
baseplate, common scaffolding principles apply to both of these structural elements [60]. Also,
for many long-tail phages (Siphoviridae and Myoviridae phages), a similar consecutive open
reading frame order is often observed, including: the tail terminator, the MTP, the two tail
chaperones, the TMP, the baseplate hub, the tail-associated lysozyme or tail fiber, and varying
numbers of baseplate/tip proteins [60].
1.2.2 Phage life cycle
Phage adsorption to the bacterial host is one of the key aspects in phage life cycles [63]. When
a tailed phage encounters a susceptible host cell, the adsorption is facilitated by specific
recognition of host receptor surface proteins, such as lipopolysaccharide (LPS) (on the outer
membrane of Gram-negative bacteria), or other molecules (fimbria, flagella) on the bacterial
cell wall [64]. Successful recognition of bacterial surface receptor(s) leads to permanent phage
adhesion and allows for penetration of the bacterial cell wall using specialised enzymes,
followed by injection of the phage DNA thorough the cytoplasmic membrane and into the
cytoplasm. Depending on the phage type and host cell physiological condition, the phage will
enter a specific phage life cycle. There are four common phage life cycles, including lytic,
lysogenic, pseudolysogenic and chronic infections [42, 43, 63, 65]. As chronic infection is
typical for only filamentous phages (out of scope of this dissertation), this life cycle will not be
described in more detail but we refer the reader to the papers of [66, 67].
Virulent or obligate lytic phages are strict pathogens of the bacterial host. These phages can
only replicate through the lytic cycle and infection results in the production of new phage
particles and lysis of the host [42]. The lytic cycle includes infection, transcription, phage
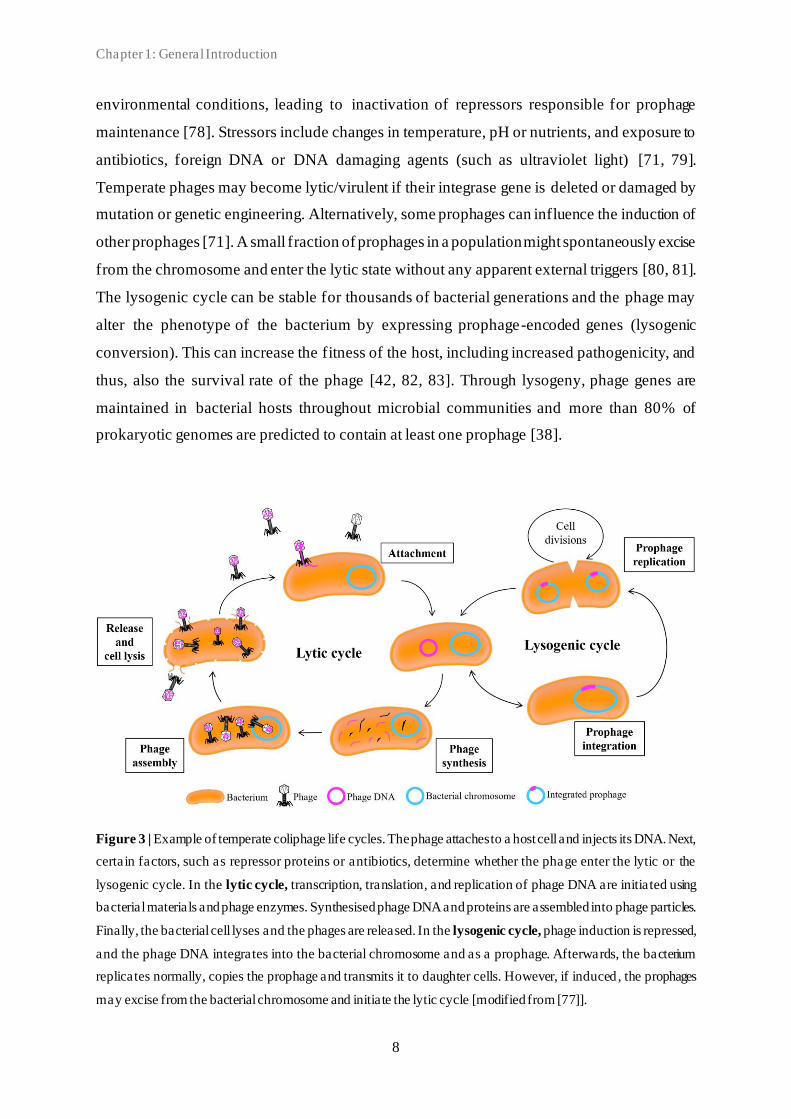
replication, and particle assembly and release (Figure 3). After successful infection the phage
DNA is transcribed. The phage genome encodes early proteins, including endonucleases and
exonucleases to degrade host DNA. The phage takes over host metabolism to replicate,
transcribe and translate phage structural component-encoding genes. Phages have evolved a
variety of transcriptional control strategies that range from full dependence on host
transcription machinery to near-complete independence [65]. Moreover, phages can conserve
energy for infection by shutting off “non-essential” host process, such as host replication and

Chapter 1: General Introduction
7
cell division [68, 69]. The efficiency of host hijacking differs between different phages.
Specialist phages infecting their preferred host seem more efficient compared to generalist
phages that infect multiple hosts. These generalist phages tend to have less efficient infections
and fail to completely suppress host translation and transcription [68]. Once all the structural
components have been translated, they are assembled into new phage particles, and the phage
DNA (or ribonucleic acid (RNA)) is packed into the capsid. Finally, the newly produced phages
are released from the bacterial host to the environment through lysis. The bacterial membrane
and cell wall disruption is facilitated by a combination of specific phage-encoded lysins, such
as holins and endolysins. Holins are small proteins that cause non-specific lesions in the
bacterial plasma membrane, allowing the endolysins to reach the peptidoglycan and attack the
murein layer of the bacterial cell wall. Phages infecting Gram-negative hosts can utilise
additional proteins, called spanins, that aid the lysis through destabilisation of the outer
membrane [70].
Temperate phages have the ability to switch between the lytic and lysogenic life cycle. Whether
the phage will follow the lytic or lysogenic pathway is decided at the start of each infection in
response to phage-, host-, and environmental factors [71]. Among others, host abundance,
defined by multiplicity of infection (MOI), may determine which pathway is followed; low
MOI favours lytic replication, whereas high MOI favours lysogeny [72–74]. Factors such as
host cell activity may play a role in phage replication, as conditions that cause reduced activity,
such as low nutrients or reduced host fitness favour lysogeny [71, 75]. Also, some phages use
a phage-encoded signal peptide to coordinate lysis-lysogeny initiation [76]. During the
lysogenic cycle the phage DNA integrates into the bacterial genome (or stays as a plasmid
inside the host cell), rather than replicates and produces new phages. Following successful
infection, the phage DNA is integrated into the bacterial genome as a prophage by a specific
phage-encoded DNA insertion enzyme called integrase. This integration includes breakage and
re-joining of the phage and bacterial host DNA. Following prophage integration, the bacterial
cell remains alive and continues to grow and replicate together with the prophage. The
prophage genes are replicated as part of the bacterial genome and are transmitted to the
daughter cells, resulting in a large population of bacteria infected with prophages [43, 77]. The
prophage genome is maintained by phage-encoded repressors, which controls expression of
genes required for prophage excision, but can be excised from the genome and enter the lytic
cycle when induced. The induction signals vary among phages but prophages are commonly
induced when the bacterial SOS response is activated due to exposure to stress or adverse
Chapter 1: General Introduction
8
environmental conditions, leading to inactivation of repressors responsible for prophage
maintenance [78]. Stressors include changes in temperature, pH or nutrients, and exposure to
antibiotics, foreign DNA or DNA damaging agents (such as ultraviolet light) [71, 79].
Temperate phages may become lytic/virulent if their integrase gene is deleted or damaged by
mutation or genetic engineering. Alternatively, some prophages can influence the induction of
other prophages [71]. A small fraction of prophages in a population might spontaneously excise
from the chromosome and enter the lytic state without any apparent external triggers [80, 81].
The lysogenic cycle can be stable for thousands of bacterial generations and the phage may
alter the phenotype of the bacterium by expressing prophage-encoded genes (lysogenic
conversion). This can increase the fitness of the host, including increased pathogenicity, and
thus, also the survival rate of the phage [42, 82, 83]. Through lysogeny, phage genes are
maintained in bacterial hosts throughout microbial communities and more than 80% of
prokaryotic genomes are predicted to contain at least one prophage [38].
Figure 3 | Example of temperate coliphage life cycles. The phage attaches to a host cell and injects its DNA. Next,
certain factors, such as repressor proteins or antibiotics, determine whether the phage enter the lytic or the
lysogenic cycle. In the lytic cycle, transcription, translation, and replication of phage DNA are initiated using
bacterial materials and phage enzymes. Synthesised phage DNA and proteins are assembled into phage particles.
Finally, the bacterial cell lyses and the phages are released. In the lysogenic cycle, phage induction is repressed,
and the phage DNA integrates into the bacterial chromosome and as a prophage. Afterwards, the bacterium
replicates normally, copies the prophage and transmits it to daughter cells. However, if induced , the prophages
may excise from the bacterial chromosome and initiate the lytic cycle [modified from [77]].

Chapter 1: General Introduction
9
Pseudolysogeny can be defined as the stage of stalled development of phage in a host cell [84].
It is often caused by unfavourable growth conditions for the bacterial host (such as starvation)
where there is insufficient energy available for the phage to initiate genetic expression and
replication [63]. After entering the host cell, the phage DNA resides inactive within the cell,
and the replication cycle is halted until environmental conditions improve [84].
Pseudolysogeny occurs in both lytic and temperate phages. While the lytic replication cycle is
simply stopped, the lysogenic infection may lead to two subpopulations of bacteria: lysogens
and phage-carrying cells, resulting in infected and non-infected cell lineages [63]. The
pseudolysogenic state may explain the long-term survival of phages in unfavourable
environments in nature [84, 85].
1.2.3 Phage taxonomy
In contrast to bacteria, no single conserved gene is present in all phages. As a consequence, the
taxonomic classification of phages is based on host range, physical characteristics, including
size, structure, and morphology, genetic makeup, and overall genomic similarity [86, 87].
Moreover, defining characteristics can be determined for each phage genus, including average
genome length and number of coding sequences (CDSs), percentage of shared CDSs, and the
presence of specific signature genes in genus member phages [88]. The phage classification
scheme is regularly updated, refined and approved by the International Committee on the
Taxonomy of Viruses (ICTV) [89]. In recent years several genome-based phage taxonomy
schemes have been introduced [87, 90] and taxonomy has changed considerably [57, 91][59].
Currently (October 29, 2021), more than 11.000 complete phage genomes have been included
in the National Center for Biotechnology information (NCBI) Nucleotide database. However,
despite a continuously increasing number of sequenced genomes, most phages remain
unclassified and poorly characterised.
Most E. coli-infecting phages, or named also coliphages, belong to the highly heterogeneous
Caudovirales order, which constitute ~94% of all known isolated phages [54, 58]. To this date,
this order contains 14 families of tailed phages with dsDNA genomes (Figure 4):
Ackermannviridae, Autographiviridae, Chaseviridae, Demerecviridae, Drexlerviridae,
Guelinviridae, Herelleviridae, Myoviridae, Podoviridae, Rountreeviridae, Salasmaviridae,
Schitoviridae, Siphoviridae and Zobellviridae [92]. Following the ICTV taxonomy
(https://talk.ictvonline.org/taxonomy/), these families comprise 73 subfamilies, 927 genera,

Chapter 1: General Introduction
10
and 2814 species. In addition, this order includes a single genus with no designated family. The
number of coliphages in the Caudovirales order constitute ~13% (n=374) of the registered
species. Coliphage species are found in nine of the 14 families (Figure 4).
Figure 4 | The Caudovirales order according to ICTV taxonomy. Numbers in the Subfamily column indicate
number of subfamilies within each family. Numbers in the Genus column indicate number of genera within each
family. Numbers in the Species column indicate number of species within each family. Numbers in brackets
indicate the number of coliphage species. Figure based on numbers from ICTV taxonomy, accessed on October
28, 2021.
For tailed phages, it has been reported that conserved genes such as the terminase large subunit
(TLS), the portal protein and major capsid protein (MCP), can be used as phylogenetic markers
for the diversity as well as their evolutionary relationship [48, 93]. Furthermore, automated
classification of tailed phages can be done according to their neck organisation [94].

Chapter 1: General Introduction
11
1.2.4 Phage diversity and signature genes
Characterisation of phage abundance and diversity traditionally involves phage culture-based
techniques and plaque assays [51, 95–97]. The advantage of these methods includes the viable
counts of phage particles and the potential for phenotypic characterisation of phages, including
host range determination. Challenges associated with these methods, include the requirement
of phages to produce plaques, that plaques can be formed under the plaquing conditions
employed, and the demand of a suitable host bacterium. Consequently, the re are strong
selective biases in determinations of phage environmental diversity using these traditional
methods [98]. Biases may be even stronger when methods of pre-enrichment and propagation
prior to plaquing are included. Also, there exists no guarantee that the host strain used is
susceptible to a fair representation of the phages in the environment of interest. Most phages
have been isolated using a single bacterial host strain [40, 99]. While this procedure is widely
used, it may likely produce narrow rather than broad host range phage. One way to obtain more
broad host range phages is using a sequential multiple host strains-approach during isolation
[47]. For isolation studies, suitable host strains can either be isolated specifically from the
environment of interest, or be a “model” host, such as a well-characterised laboratory strain,
chosen based on specific desirable traits [40]. Evidently, only phages that can infect the specific
host strain(s) will be identified, and accordingly, it is difficult to establish what proportion of
phages present in the environment are being isolated [42, 43]. For phage therapy (see section
1.2.5), testing on clinical bacterial isolates may be more relevant than testing on laboratory
strains [49].
Transmission electron microscopy (TEM) has traditionally been used for direct assessment of
the phenotypic diversity and abundance of phages. It offers powerful magnification and
provides information of surface features, shape, size, and structure. The number of detected
phage morphological types varies significantly between studies and might reflect the diversity
of different microbial communities. However, variations between studies might also reflect
low sensitivity as well as low specificity [40]. Tailed phage morphologies are unique and
different from other viruses. However, tailed phages with highly similar morphology may in
fact have very different genomes. Thus, nucleotide sequence information (preferably whole
genome sequence) is required to fully understand the diversity, relationships, and dynamics
among the members of any set of phages being compared [48, 56].
Recent advances in viral omics and high-throughput sequencing methods have enabled the
rapid discovery of various phages in numerous environments and have broadened our view of

Chapter 1: General Introduction
12
phage abundance and diversity [56, 100]. Although these advances have expanded our
understanding of phage genomic diversity, they also revealed that we have only scratched the
surface of the abundance of phage diversity. It is predicted that more than 99% of viral genetic
diversity remains to be revealed [56, 101].
Whole-genome sequencing (WGS) is a comprehensive method for determination of the DNA
sequence of an organism’s genome. Decreasing sequencing cost and the ability to produce large
volumes of sequence data in a short amount of time make WGS a powerful tool for genomic
research. Different sequencing technologies are available. Short-read sequencing (such as the
Illumina platform), also referred to as second generation sequencing, offers the potential to
rapidly sequence hundreds of phage genomes with high accuracy (~99%). This sequencing
method generates high read counts of short reads (150-300 bp) within a single run producing
high coverage, and the base-by-base sequencing protocol enables the accurate data acquisition
[102, 103]. However, all short-read sequencing technologies have a common limitation – the
inability to assemble long stretches of DNA resulting in relatively fragmented genome contigs.
Long-read sequencing (such as Nanopore, and PacBio singe-molecule real-time sequencing),
also referred to as third generation sequencing, address the shortcomings of short-read
sequencing with read length of >10.000 on average [104]. Longer reads are especially useful
when sequencing complex genomic regions such as repeats and phages. However, these longer
reads are more prone to errors resulting in sequencing accuracy of ~92-97%. Though more
expensive, combining short-read and long-read sequencing has emerged as a promising
approach to overcome pitfalls associated with singe-technology use and generate fully resolved
and accurate genome assemblies [103, 105]. Different sequencing approaches can be applied
depending on the sequencing goal. Metagenomics is the sequencing of all DNA present in a
sample as opposed to sequencing just a single microorganism [106]. This approach has
extraordinary potential to improve our understanding of for example (complex) microbial
populations in their natural environments or primary sample, regardless of whether they belong
to microorganisms that can be cultured in the laboratory. Also, this isolation-free, culture-
independent method does not rely on amplification of specific genomic sequences, which can
otherwise introduce bias [107]. However, this approach does not provide high-resolution
needed for in-depth characterisation of single genomes and may produce biases towards certain
sequences rather than abundance [108].
Yet, despite the remarkable diversity of phages at the nucleotide sequence level and the lack of
a universal conserved marker gene found throughout all phage families, the structural proteins

Chapter 1: General Introduction
13
that form viral particles show strong similarity and conservation, representing important
taxonomic characteristics [56, 109]. Accordingly, diversity and abundance can be assessed
using family-specific signature “marker” genes shared by all members [110, 111]. One of the
most conserved marker gene type of the tailed phages are the terminases. These genes are
phage-coded proteins that bind to and cut DNA. They consist of a large and a small subunit
with molecular weights of 44-73 and 10-45 kDa, respectively. The small subunit is responsible
for DNA recognition and binding, initiating the packaging of the viral genome. The larger
subunit (TLS) ensures DNA cutting, binding of the terminase to the connector, and DNA
translocation into the empty phage head (capsid) to finalise the packaging process [112, 113].
Moreover, the two conserved structural proteins: portal protein and MCP, involved in the phage
head-assembly process, appear to be universally present in the tailed phage genome. The use
of these “conserved” genes has been shown useful for the characterisation of Myoviridae
phages, including when used as signature marker genes either alone or in combination. The
T4-like g20 portal protein gene has been shown to be conserved among phages that inhabit
marine and freshwater environments as well as eukaryotic hosts [60, 110], and the T4-like gp23
MCP has been shown useful in assessing Myoviridae diversity [111, 114]. Also, multi-marker
gene studies have been done to assess phage diversity using a multilocus sequence typing
(MLST) scheme approach [111]. By using (degenerate) polymerase chain reaction (PCR)
primers specific for identified marker genes and sequencing of the amplified environmental
sequences, these marker genes offer new ways of providing estimates of diversity and
quantifying phage abundance without the cultivation-based complications. Based on those
marker gene studies, the global phage diversity far exceeds that represented by cultured isolates
[115–117]. However, despite providing insights into the overall distribution of specific phage
genes, even in the best cases, signature genes fail to capture the full diversity present in natural
communities [38]. While amplicon methods such as PCR are fast and low-cost, they can suffer
from amplification bias [118], which might be more apparent when examining less abundant
genes or genomes [119]. Marker gene abundance only reflects phage abundance if the gene is
present and detected. Scaffolding proteins and procapsid proteases are often but not universally
encoded. Head-tail joining or connecting proteins are likely present in all tailed phage virions
but are too diverse to be recognisable in all of them at present. As such, no universal primer
can be designed, making it cumbersome to design primers that target the whole phage diversity
[97]. Recent studies have shown that the different tail tape measure proteins (TMPs) correlates
well with clusters for Siphoviridae and Myoviridae. However, the short tails of the Podoviridae
have no TMP. Moreover, there is no known tail protein that is common to all types of tails, and

Chapter 1: General Introduction
14
both phage DNA replication/metabolism and lysis mechanism are too diverse so that no
homologue proteins can be used universally for all phage families [48].
The diversification of phage genomes is driven by multiple mechanisms, including the
accumulation of mutations, gene acquisition and loss , and recombination events [48, 56, 120].
For tailed phages, especially HGT contributes to evolution as well as diversification of the
genomes [48]. The horizontal transfer of genetic material occurs via both homologues and non-
homologues recombination events, and both within and between phages as well as bacterial
hosts. Especially homologues recombination between genomes of co-infecting phages is
thought to be the main mechanism of HGT [121]. Temperate phages have been shown to
acquire DNA from defective prophages through homologues recombination [122]. Some
phages are able to exchange up to 79% of their genome [123, 124]. Phage genomes exhibit
genetic mosaicism with conserved genetic modules that encode exchangeable functional units
such as the virion coat-encoding genes, the virion tail-encoding genes, or the genome
integration proteins [123, 125]. The creation of this mosaicism is an ongoing process, driven
by the HGT and recombination events [52, 120, 122, 126, 127]. Such horizontal exchange
among phages complicates whole genome comparisons and might make any strictly
hierarchical classification scheme insufficient and potentially misleading if the exchange is
great enough [125]. Accordingly, single (marker) gene locus use, such as the terminase, is
usually not used independently, but rather in combination with other marker genes and/or other
approaches to minimise the potential impact of HGT events on the taxonomy [59]. However,
horizontal exchange does not appear to be rapid enough to destroy the overall relationships
within or between ‘phage types’ [48, 52, 99, 127]. Also, the rate of HGT is associated with
phage lifestyle: most phages with a high rate of HGT are temperate whereas the majority of
phages with low rates of HGT are strictly virulent [120, 128].
In silico analyses of phage genomes have shown that there is a large number of phage genes
whose function cannot be predicted due to very low or no similarity to already known genes
[129]. However, due to high degree of conservation of function-associated gene orders in
regions encoding morphogenesis modules in tailed phages, it is possible to identify protein
functions in the absence of detectable sequence similarity [60].

Chapter 1: General Introduction
15
1.2.5 Phage therapy
Phage therapy is defined as the application of phages to treat or prevent bacterial infections
[130]. The therapeutic potential of phages was first discovered over a century ago, but the
discovery and widespread use of antibiotics led to a loss of interest in the therapeutic
application of phages [3]. Still, phage therapy research and application did continue in some
countries, as in Georgia (part of the former Soviet Union) and Poland, where phages were, and
continue to be, routinely used to treat a large number of diseases [3, 131]. Nowadays, we are
facing a worldwide increase in the prevalence of antibiotic resistant bacteria, and lack of
discovery of new antimicrobials, urging for alternative treatment options. This, along with
advances in modern molecular biology, biotechnology, and genetic engineering, have led to a
renewed interest in phage therapy [9, 132–134]. Furthermore, advances in bioinformatics and
WGS technologies, including genome sequencing of phages and entire microbiomes, have
broadened our understanding of phage taxonomy, host-specificity, population structure and
genomic evolution [38, 59, 129, 135–137]. Also, with recent advances the phage application
spectrum has been expanded to various medical, biotechnological and agricultural fields,
including the use of phages in phage therapy in humans and animals, surface dis infections,
bacterial detection, gene delivery, food bio-preservation and safety, biocontrol of food and
plant pathogens, and biofilm control [138–142].
Phages, as therapeutic agents, have numerous advantages that make them good alternatives or
supplements to antibiotics (Table 1). 1) New phages are often relatively easily discovered and
isolated due to the great biodiversity of phages in nature [143, 144]. Any environment that
contains the pathogen of interest is likely to contain phages that are able to infect and kill that
organism [51]. 2) Strictly lytic phages are by nature bactericidal [145]. Phages’ ability to
effectively eliminate bacterial pathogens in animals and humans has convincingly been
demonstrated, and doses as low as 102 plaque forming units (PFU) have been shown to be able
to prevent disease in animals by pathogenic E. coli [146, 147]. 3) As phages infect and kill
bacteria using mechanisms that differ from those of antibiotics, phages can be used to target
bacterial states such as biofilms, persistence, and bacteria that are antibiotic resistant [148].
The high number of bacteria present within the biofilm(s) facilitate rapid and efficient phage
infection of the host and consequent phage replication. Also, phages can produce specific
enzymes that degrade the extracellular matrix of the biofilm. Phages are able to infect persister
cells where they remain dormant, but re-activate when the host cells become metabolically
active [149, 150]. 4) Phages hijack multiple essential cellular processes, including DNA

Chapter 1: General Introduction
16
replication, transcription, and translation upon infection [144]. 5) Due to the host specificity of
phages, they tend to only minimally disrupt the normal microflora by selectively targeting only
pathogens [151]. By contrast, many antibiotics, which tend to have broader spectrums of
activity, might cause damage to all bacterial cells independently of whether they are pathogenic
or commensal [152]. Such disturbance of normal microbiota can amongst others result in
diarrhoea as well as increased risk of secondary infections [40, 144]. Also, the relatively narrow
host range exhibited by most phages limits the risk of cross-resistance between different phages
[144, 153]. 6) As phages co-evolve with bacteria over time, the administered phage population
may evolve to (re-)infect the phage-resistant bacteria (an arms race), which is not possible for
antibiotics [132, 154, 155]. 7) If phage resistance should develop, careful choice of phage(s)
that select for resistant bacterial mutant types with lower fitness, such as reduced cell division
rates and pathogenicity traits expression, could be an advantage despite resistance development
[144]. Bacteria resistant to LPS-targeting phages, are typically reduced in both fitness and
virulence [132, 156]. 8) Phages are self-regulating and can increase in number over the course
of treatment, specifically at the site of infection, where most target bacteria are present. This
also allows for less frequent dosing and low-dosage use of phage compared to antibiotics [144].
9) Finally, phages are self-limiting as they will decrease in abundance as soon as susceptible
host cells are eliminated [152].
Despite the numerous advantages of phage therapy, phages still have limitations (Table 1).
Both regulatory and technical hurdles must be overcome before phage therapy can be fully
accepted in modern clinical practice. Not all phages make for good therapeutics. Good
therapeutic phage candidates should have a high potential to reach and then kill target bacteria
without negatively modify the environments to which they are applied. Such high “virulence”
includes good adsorption properties, high potential to evade bacterial defences, good
replication characteristics, and/or high fecundity (short latent period and large burst size) [157,
158]. These characteristics can be reasonably assured using phages that are obligately lytic,
viable, stable under typical storage conditions and temperatures, subject to appropriate efficacy
and safety studies, and, ideally, fully sequenced to, among others, confirm the absen ce of
bacterial virulence factors [132, 144, 159]. To achieve therapeutic efficacy, the phages applied
should be able to replicate (or, at least, infect) at the expense of their bacterial target faster than
they are removed from the site of treatment such as by the host immune system or by
environmental turnover (in vivo persistence) [157, 160].

Chapter 1: Genera l Introduction
17
Table 1 | Advantages and disadvantages of phage therapy
Trait Advantages Disadvantages
Bactericidal
agents
Lytic phages cause host cell lysis.
Active against Gram-positive and
Gram-negative bacteria, including
MDR-variants
Not accessible to intracellular pathogens
Specificity
Highly specific, minimal or no
disruption to normal microbial
community
Narrow host spectrum, host bacterium
needs to be identified.
Resistance
Phage-resistant mutants are often
less virulent, as phage receptors are
commonly associated with
pathogenicity. Able to evolve to
overcome bacterial resistance.
No cross-resistance to antibiotics
Risk of phage resistance development in
bacteria
Dosage
Simplifying dosage. Self-regulating
in proportion with target bacteria,
replicates at the site of infection
Depend on susceptible host for
replication. When target organism is not
present the phages will not replicate.
Perceived by the immune system as
invaders and can be rapidly degraded
Toxicity
Generally considered as safe due to
nucleic acid and protein
composition
Rapid lysis of bacteria may lead to the
release of endotoxins and induce
inflammatory immune response
Discovery Rapid and relatively easy discovered
due to their ubiquitous nature
Depend on susceptible host bacterium
for isolation and replication
Phage cocktails Can broaden host range and reduce
risk of resistance development
Lack of standardised guidelines to
generate phage cocktails
Implementation New regulatory framework for
phage therapy in Belgium.
Regulatory hurdles. Not accepted as
pharmaceutical drugs in most countries.
Difficulties in patenting
Bioengineering Can be genetically modified Requires host bacterium, expertise, and
technology
Other Exhibit anti-biofilm properties and
can target persister cells -
Information adapted from [3, 32, 144, 157, 161].

Chapter 1: General Introduction
18
Temperate phages are usually avoided as their genomes may contain genes which alter the
phenotype of the host after infection (lysogenic conversion). These phages are capable of
generalised transduction, and thus, are able to transfer large amounts of bacterial DNA from
one host to another, including virulence and antibiotic resistance genes [130, 160]. Also,
administration of temperate phages may not result in an immediate bactericidal effect on the
target pathogen if the phages integrate as prophages. Furthermore, when integrated in the
bacterial chromosome, prophages may display superinfection immunity, making bacteria
resistant to further phage infections [162, 163].
Advances in the field of biotechnology, synthetic biology and genetic engineering facilitate
engineering of phages in various ways that potentially improve the antimicrobial properties of
the phages as well as create new strategies for fighting bacterial infections [32, 130, 163, 164].
Genetic engineering potentially improves phage efficiency. Phages engineered to express
biofilm matrix-degrading enzymes penetrates biofilms more readily than the non-engineered
wild type (WT) phage [165]. Phage host range can be altered to serve practical purposes.
Synthetic phage variants with altered host range have been constructed by swapping tail
component-encoding genes [166]. To bypass concerns regarding uncontrolled self -
amplification and sudden release of bacterial endotoxins upon lysis, phages can be engineered
to be non-replicative and used to deliver genes, interfering with important bacterial intracellular
processes, into specific bacterial populations through transduction [167]. As mentioned above,
strictly lytic phages might be preferable for phage therapy. However, some bacterial species
appear to produce only temperate phages [168]. Genetic engineering can be used to obtain
strictly lytic derivatives of temperate phages in which the repressor and/or integrases are
deleted. Also, gene encoding bacterial virulence factors and integrases can similarly be
removed [163]. However, considerable considerations have been given to the use, design, and
associated risks [169]. Also, there are still a lot of difficulties in engineering phages. Many
strategies require the ability to genetically modify the bacterial host(s) or to efficiently deliver
exogenous DNA into these hosts, which is still a challenge for many bacterial species [170].
Most phage genomes are too large (>20 Kbp) for easy in vitro manipulation and are lethal to
their bacterial host [167]. Temperate phages can be stably maintained in the bacterial genome,
enabling modification of the phage genomes using the same methodologies as those used for
engineering of bacterial genomes. However, genomes of virulent phages cannot be cloned
whole into bacteria for subsequent genetic modification. As a result, virulent phage genomes
are commonly edited using homologues recombination (allelic exchange), whereby the gene(s)

Chapter 1: General Introduction
19
to be modified are cloned into a plasmid and modified as needed before being introduced to
the host bacterium. Homologues recombination efficiency might be low and many phage loci
cannot be cloned due to their adverse effect on bacteria [167]. Consequently, there is a strong
pressure for developing new phage genome engineering methods. Intracellular bacterial
pathogens constitute another limitation of phage therapy as phages do not have a mechanism
of entry into eukaryotic cells. Also, as a phage population can undergo rapid exponential
growth, widespread lysis of target bacteria may potentially release endotoxins that could be
harmful to the patient [132, 171]. Limited knowledge on the phage interaction with patient
immune system is another matter of concern [132, 172, 173]. The great specificity of phages
might represent a challenge to phage therapy as it requires the potential need of characterisation
of bacterial susceptibility prior to the phage application. Thus, it is essential to know exactly
which bacterial species is the causative agent of the infection, and success of phage therapy is
associated with careful choice of phage capable of infecting the causative bacterial agent [132].
However, genetic engineering can potentially address the phage specificity shortcomings and
increase the therapeutic potential of natural phages [167]. The second method of overcoming
a too narrow spectrum of activity is combining phages (so-called phage “cocktails”) [133, 152,
157, 160, 164, 174]. Emergence of phage resistant bacteria constitute another limitation to
phage therapy [175, 176], as bacteria are readily capable of evolving resistance to phages
through a variety of different mechanisms (see section 1.3.2).
Using a cocktail of phages might reduce the probability of phage-resistant bacteria emerging
as different types of phages infecting the same species and/or strains are present [177, 178].
Nevertheless, even resistance to phage cocktails have been described and as such cannot be
regarded as the most optimal solution [179, 180]. When designing phage cocktails the
following should be taken into account to overcome the emergence of resistance. First, the
mechanism(s) whereby phage resistance can evolve should be taken into consideration. By
knowing the specific mechanisms, phages that select for resistance associated with reduced
fitness and pathogenicity can be chosen. Also, phages with desired properties to overcome
these resistance mechanisms can be chosen. Second, the potential of bacteria to develop cross-
resistance to multiple phages should be considered. The risk of cross-resistance could be
overcome by combining phages utilising different receptors, as the fitness cost associated with
resistance development to all might be too high for the host bacterium and therefore unlikely.
Finally, it should be confirmed that the cocktail phages do not compete with one another and
as such reduce the overall efficacy [144]. Currently, phage therapy is in its infancy. The current

Chapter 1: General Introduction
20
strategy to have phage therapy readily available is development of phage banks. Such banks
are collections of previously characterised phage isolates, which are available as phage stocks,
for direct matching to a specific recently isolated target bacterium and application in cocktails
[160, 181]. Few places around the world have such bank available and one of the biggest phage
therapy centers is located at the Eliava Institute, Georgia [182, 183].
However, despite thorough phage characterisation, in vitro phage performance does not always
match experimental outcomes observed in vivo [157, 181, 184]. The reasons for this may be
diverse. Phages may not adsorb with the same efficiency in vivo due to differences in the
chemical composition of the adsorption conditions [185]. The phage latent periods might be
extended, or the burst sizes might be smaller in vivo, thereby slowing the magnitude or rates of
phage population growth. This might be of a concern especially for lytic infections, where
phages are produced [186]. Notably, phages in vitro are often cultured with bacteria under
somewhat optimal growth conditions, which could differ substantially from in vivo and/or in
situ circumstances. Finally, the target bacteria can differ considerably from the bacterial hosts
against which the phages may have been characterised in vitro [157, 184]. It is clear that
increased knowledge on the host-pathogen interaction is necessary as well as the PK and PD
behaviour of phages is deciphered for a more certain and reliable in vitro and/or in vivo outcome
and hereby successful phage therapy application [160, 187, 188].
Apart from the clinical hurdles, there are also regulatory problems that represent a significant
barrier for the implementation of phage therapy in modern medicine. Unlike the well-
established path to approval for antibiotics, the path for phage therapeutics is currently under
development. The main challenges are the traditional large-scale clinical trials that should be
in accordance with official guidelines and the Good Manufacturing Practices (GMP) in the
production of phage cocktails. Usually, these procedures are very expensive and take several
years, while for phage therapy, for each infection, there may be the need for using another
cocktail [152]. In Belgium, however, a group of researchers recently worked with regulatory
authorities to successfully set up a new regulatory framework for phage therapy [189]. This
new framework classifies phages not as drugs but as active pharmaceutical ingredients, thus
exempting them from clinical trial requirements and allowing them to be administered by
pharmacists on a per-patient basis upon medical prescription. Even though progress has been
made towards overcoming some of the hurdles associated with phage therapy, in order for
phage therapy to gain widespread acceptance or worldwide application profound interest from
big pharmaceutical companies and funding bodies is still needed [152].

Chapter 1: General Introduction
21
An alternative to some of the problems described above could be the combined therapy of
phages and antibiotics that take advantage of each treatment’s differing strengths constituting
an ideal synergistic approach [132, 152, 190]. The mechanism of this relies on phages that use
an antibiotic efflux pump to infect the bacterium. This may select against the expression of the
pump, rendering the bacteria more sensitive to antibiotics that were previously pumped out.
This interaction selects for phage-resistant variants, however, as they become more sensitive
to antibiotics, the combined therapy is still effective in inhibiting/killing the target bacteria.
This type of combined therapy has been shown to have an increased effect on several bacterial
species, including E. coli in broiler chickens, compared to when used separately [132, 191,
192].
1.3 Phage-host interactions
Phage-bacterium co-evolution is an important driving force for the ecology and evolution of
microbial communities [193]. However, the nature of some interactions within phage
populations or between different phages and bacteria is only now becoming clear and we are
just starting to understand the complexity of these interactions [194–196]. From an
evolutionary point of view, interactions can be classified as parasitic, predatory, cheating,
mutualistic, or altruistic depending on the system [194, 197, 198]. This classification depends
by large on the life cycle of the phage, including determinants that play a role in the phage host
range and bacterial defence mechanisms [43, 50]. Phages are considered to be parasites when
they exploit bacterial cells for their survival and replication. In the lytic life cycle, when phages
infect and kill their infected host cell(s) they are considered predatory, likewise, they shape the
bacterial population dynamics and may assist in their long-term evolution through generalised
transduction [43]. As phages replicate inside a bacterial cell, pools of public goods (enzymes,
capsid, proteins etc.) are produced. Phage cheaters can emerge and do not contribute to the
production of common goods or consume the goods at a higher rate than the ancestral phages
[194]. In response to phage predation, infected bacterial cells can altruistically arrest their
growth trapping immature phages inside the infected bacterial cell to protect the overall
bacterial population [199]. Also, infected bacterial cells may commit altruistic suicide to halt
phage replication. In the lysogenic life cycle, phages can stably integrate into the host cell
genome or stay as a plasmid inside the cell and may confer lysogenic conversion. Some of
these phages may cause an increased host fitness and diversity as well as function as a survival
strategy for both phage and their host and as such interact mutualistically [43, 194, 198].

Chapter 1: General Introduction
22
Interactions between phages and their host(s) have profound effects on biological processes,
prokaryotic metabolism, and diversity and composition of microbial communities [50, 193,
200]. Thus, understanding these interactions not only provides new insights into phage biology
and evolution, the use of phages in genetic engineering and other application(s) but most
importantly may lead to advances in the development of phage therapies [50, 65].
1.3.1 Population growth dynamics
Population dynamics is the study of how and why the population of one or more species
changes in size and structure over time [201]. Accordingly, when studying phage population
growth dynamics, parameters such as phage attachment and adsorption rate, burst size (the
number of phages produced per infected bacterium), latent period (the time period between
adsorption and cell lysis), and life cycle as well as bacterial growth rate and defence
mechanisms affect the dynamics [202, 203]. Phage resistance can occur through various
mechanism in populations of phage and bacteria (see section 1.3.2 ), resulting in partial or
complete resistance, and can differ in the extent of the physiological cost associated with
resistance, and in whether the mutation can be countered by a mutation in the infecting phage.
These important differences determine the effect of the phage infection on the population
dynamics and may have significant consequences for the resulting structure of the microbial
community [204, 205]. Phages can subvert the host’s cellular processes to optimise the
intracellular environment for the phage replication. This is achieved by specific, often toxic,
protein-protein interactions that occur early during phage infection, influencing the
intracellular molecular interactions [206, 207].
Phages may evolve to infect multiple hosts. Such extended host range properties can give rise
to an “arms race” between resistance mutations in the bacterium and the changing host range
[208]. When examining experimental phage communities, however, there seems to be an
asymmetry in the arms race in favour of the bacteria, as some resistance mutations cannot be
countered by host range mutations [204, 209]. However, in natural settings, the arms race
dynamics seems different. The bacteria are more resistant to their contemporary phages than
to ancestor phages, allowing fluctuating selection dynamics and continuous cycles of co -
evolution [205, 210]. This difference in dynamics may be due to the additional biotic and
abiotic selection pressures that are found outside of controlled laboratory conditions [197, 211].

Chapter 1: General Introduction
23
Several mathematical models have been developed to predict and explain the behaviour and
dynamics of phage and bacteria populations based on fundamental phage-bacteria biological
parameters [202, 212–214]. Most often, such models are validated using in vitro data obtained
from phage-interaction studies. While no single model to date has been able to capture all
aspects of the complex in vivo interaction between phage and host, together, suitable models
can be selected to predict and explain basic behaviours of selected population dynamics in a
given environment [202, 212, 214]. Microbial model communities have been shown to be ideal
to provide insights into complex microbial community interactions [204]. One important
advantage of phage–bacteria systems is that the initial complexity of a community is
controllable. The complexity can be reduced to a minimum (one phage and one bacterium) and
then increased gradually to examine its effects on population dynamics, community properties,
and evolutionary change. Moreover, both environmental and genetic parameters can relatively
easily be manipulated [204, 215].
When studying phage population dynamics in an animal-associated microbial environment,
one should not only consider the interaction between the phage and the host bacterium, but also
the interplay with the environment within the animal host. The host environment includes a
direct influence of digestive enzymes as well as the influence of non-enzymatic secreted
compounds, such as bile salts, which have been shown to inhibit phage adsorption and
components of the eukaryotic hosts immunity [40, 194, 216]. On the other hand, phages can
have profound effects on the outcome of bacterial infection by modulating the immune
responses of the animal host, either indirectly via effects on the eukaryotic microbiome or
directly, often in anti-inflammatory ways. Phages can modulate the innate immunity of the
mammalian and avian host via the stimulation of phagocytosis and cytokine response, as well
as impact on the adaptive immunity via stimulating the antibody production [173, 217, 218].
Essential knowledge on phage-host interactions may be obtained using mathematical model
outputs using in vitro data from controlled laboratory conditions, followed by in vivo
verification in more complex environments [187]. Subsequent model refinement could be
applied if the experimental data do not reflect the simulated ones [202]. Gaining an
understanding of the phage-bacterium interactions and population dynamics in natural
environments seem essential for future successful phage therapy application as well as
exploiting the full potential of phages for our benefits [219].

Chapter 1: General Introduction
24
1.3.2 Bacterial phage resistance
Faced with a strong selection pressure, bacteria can evolve resistance to a phage infection,
either complete or partial, through various mechanisms. These include among others
spontaneous mutations, innate immune systems, including restriction-modification (R-M)
systems, abortive infection (Abi) mechanisms, bacteriophage exclusion (BREX) systems, and
adaptive immunity via the Clustered Regularly Interspaced Short Palindromic Repeats
(CRISPR)-Cas (CRISPR-associated proteins) systems [175, 176, 208, 220–222]. These
antiviral mechanisms can be used to target different steps of the phage life cycle, including
inhibition of phage adsorption and blockage of phage DNA injection, replication of the phage,
or lysis of the bacterium.
Both phage resistance and phage-bacterial co-evolution are mainly driven by spontaneous
mutations [193, 204]. The resistance is largely affecting the adhesin-receptor binding, through
mutation of the receptor or loss of the receptor. This adhesin-receptor binding is highly specific.
The main phage adhesin is the phage tail fiber [223, 224]. The host cell receptors (reviewed in
[224, 225]) of tailed coliphages are surface structures, of which the most often involved surface
structures are outer membrane proteins (OMPs): FhuA (involved in iron uptake, previously
called TonA), OmpC (involved in iron transmembrane transport), OmpF (involved in iron
transmembrane transport), FadL (involved in translocation of long-chain fatty acids across the
membrane), BtuB (involved transports vitamin B12 across the membrane), LamB (involved in
the transport of maltose and maltodextrins), NfrA (required for irreversible adsorption of N4-
like phages), TolC (involved in efflux of antibiotics and other toxic compounds from the cell)
and the TonB protein (involved in the transduction of energy from the cytoplasmic membrane
to the Omps). Other surface structures involved are the O-antigen (part of the LPS of Gram-
negative bacteria), the LPS core, and the pilus (colonisation factor) [226, 227]. In some cases
however, resistance through mutations may lead to an extended host range of the phage [228,
229]. This has been shown for both T2 phages that attach to OmpF, LPS and/or FadL and for
T7 phages that attach to the LPS core. While phage T7 and T4 are phylogenetically unrelated,
both bind to the LPS core, and mutations in the LPS that confer resistance to T4 often gives
cross-resistance to T7. However, LPS mutations that confer cross-resistance do not select for
extended T4 host range. Thus, utilising the same receptor does not guarantee same type of
dynamics [204, 226]. The mutations in the host, giving resistance to the T4 phage and confer
cross‐resistance to phage T7, tend to have a larger fitness cost than the mutations giving only
resistance to T4. This is because the mutation(s), giving cross-resistance, occur deeper in the

Chapter 1: General Introduction
25
LPS (the initial binding site for T7 is deeper in the LPS core than is the T4 site) and as a
consequence have a greater effects on the E. coli physiology [204].
Different phage-host dynamics can arise depending on the cost of the mutation. Coliphage T5
has two sets of tail fibers which bind to the O-antigen and/or the FhuA protein [226]. E. coli
resistance to coliphage T5 happens without any fitness costs (under laboratory experimental
settings). The mutations occurring in T5 to counter these resistant bacteria do not confer host‐
range changes, and as a consequence, phage T5 rapidly goes extinct in experimental bacterial
communities when resistance arises [204]. Phage resistance development has also been shown
to influence bacterial virulence. Phage-resistant bacteria may become less virulent in case of
mutations in surface virulence factors, such as LPS, though it depends on which part of the
LPS is affected [156].
In addition to receptor mutations, mechanisms that prevent phage adsorption can be divided
into three categories: blocking of phage receptors, production of extracellular matrix and
presence of competitive inhibitors (Figure 5).
Figure 5 | Defence strategies used by bacteria to prevent phage adsorption. A) Bacteria can produce proteins that
mask the phage receptor. B) Phage adsorption can be prevented by the production of EPS, but some phages
overcome the EPS layer by producing enzymes (lyase or hydrolase) that cleave EPS. C) Bacteria can produce
competitive inhibitors that bind to the phage receptor and reduce or prevent phage adsorption [adapted from
reference [175]].

Chapter 1: General Introduction
26
The first category is related to masking the phage receptor(s) through production of masking
proteins that block the access to the receptor from the phage. The second category also includes
hindering access of the phage, but through the production of extracellular matrix of
exopolysaccharides (EPSs) that provides a physical barrier between phages and their receptors
on the host cell surface. However, some phages have evolved to either utilise these extracellular
polymers as receptors or to degrade them. The third category involves competition between
molecules that bind to the same receptors. By procuring competitive inhibitors that bind to the
phage attachment site, the bacterium renders these receptors unavailable for the phage(s). Also,
when the phage receptors play important roles in bacterial metabolism, such as substrate intake,
molecules are binding to the receptor as part of the normal cell activity and might block the
access of the phage [175, 176, 225].
R-M systems are widespread innate defence systems in bacteria [230, 231]. They prevent entry
of foreign DNA into the cell and comprise two contrasting enzymatic activities: a restriction
endonuclease (REase) and a methyltransferase (MTase). The REase recognizes and cleaves
foreign phage DNA sequences at specific sites, while MTase activity ensures discrimination
between host and foreign DNA. As such, these systems may cause phage resistance by cutting
the phage DNA and likewise block the intercellular phage development [40, 231]. Some
phages, however, have evolved several strategies to evade these R-M systems (reviewed by
[232]). One strategy is to select against specific restriction sites. Phages that possess fewer
restriction sites in their genomes are less prone to DNA cleavage by the host REases. A second
strategy includes modification and change of the orientation of restriction-recognition sites to
avoid cleavage by the host REases. A third strategy is for the phage to synthesise proteins that
prevent cleavage by masking the restriction sites. Also, some coliphages while injecting their
own DNA into the host cell, also co-inject host-genome-binding proteins that mask the
restriction sites. A fourth strategy is to disrupt the structural conformation of the REase-MTase
complex. A fifth strategy is for the phage to code for proteins that directly inhibit REase.
Finally, many phages encode their own MTases, which they have acquired from their bacterial
host(s). These enzymes enable phage self -methylation of the DNA hereby protecting against
host restriction enzymes. In turn, some bacteria can evolve to encode modification-specific
endonucleases to restrict and specifically counteract these adapted phages and their
modifications, resulting in a co-evolutionary arms race [231, 233].
Abi systems constitute another mechanism of the bacterial innate immunity. These systems
function as “suicide” systems and induce an altruistic death (or dormancy) of the bacterial cell

Chapter 1: General Introduction
27
upon phage infection, and thus ensure no multiplication of the phage [175, 232, 234]. Abi
systems are diverse and can act at any stage of phage replication cycle. They often consist of a
single protein or protein complex encoded by mobile genetic elements, including prophages
and plasmids [232]. Abi can be mediated by toxin-antitoxin (TA) systems encoded by the
bacterium. The TA systems are composed of a toxin, which targets essential cellular process,
such as DNA replication and translation, and a neutralising antitoxin that inhibits the toxin
during normal bacterial growth [234, 235]. Compared to their toxins, antitoxins are more labile
and degrade more rapidly. As a consequence, when stress is encountered (e.g. during phage
infection) and the production of both components is inhibited, the antitoxin is preferentially
degraded, allowing the toxin to induce either bacterial dormancy or cell death [236]. Phages
can evade Abi systems effect by a variety of mechanisms. First, phages can produce
spontaneous "escape" mutants, often conferred by mutation in a single gene (differs between
phages) [237, 238]. Such mutation may hinder depolarisation of the bacterial membrane or
prevent the activation of the Abi system [232]. Second, some virulent phages can become
resistant to specific Abi systems by recombining with the genome of a res ident prophage.
However, the exact mode of action of this evasion strategy remains unclear. Third, phages can
hijack and/or produce antitoxins that neutralises the bacterial toxin [124, 232, 239]. Finally,
some phages are able to produce small RNAs that act as a molecular mimic of the antitoxin
RNA (pseudo-antitoxin), leading to toxin inhibition and avoiding cell death leading to
continued phage replication [240, 241].
In addition to the innate immune systems, CRISPR-Cas systems are found among ~36% of
bacteria and confer a sequence specific adaptive immunity against invading foreign DNA,
including phages [242]. The CRISPR loci consist of arrays of short direct repeats of ~30 base
pairs (bp), separated by similar sized highly variable spacer sequences, derived from invading
nucleic acids, and associated cas genes, encoding Cas proteins [220, 243]. CRISPR-Cas system
immunity involves three main stages (Figure 6). First, the adaptation (or acquisition), where
short DNA fragments from newly encountered foreign genomes are incorporated into the
CRISPR loci as a new unique spacer by the Cas proteins. Second, the expression, whe re the
CRISPR loci is transcribed from a leader sequence upstream of the loci and processed into
small guide CRISPR RNAs (crRNAs). Third, the interference, whereby Cas protein(s) form
complexes with the crRNAs. Using the crRNAs as guides, the crRNA-Cas protein (crRNP)
complexes specifically recognise, bind, and degrade complementary foreign nucleic acids, and
as such kill the phage [244–248].
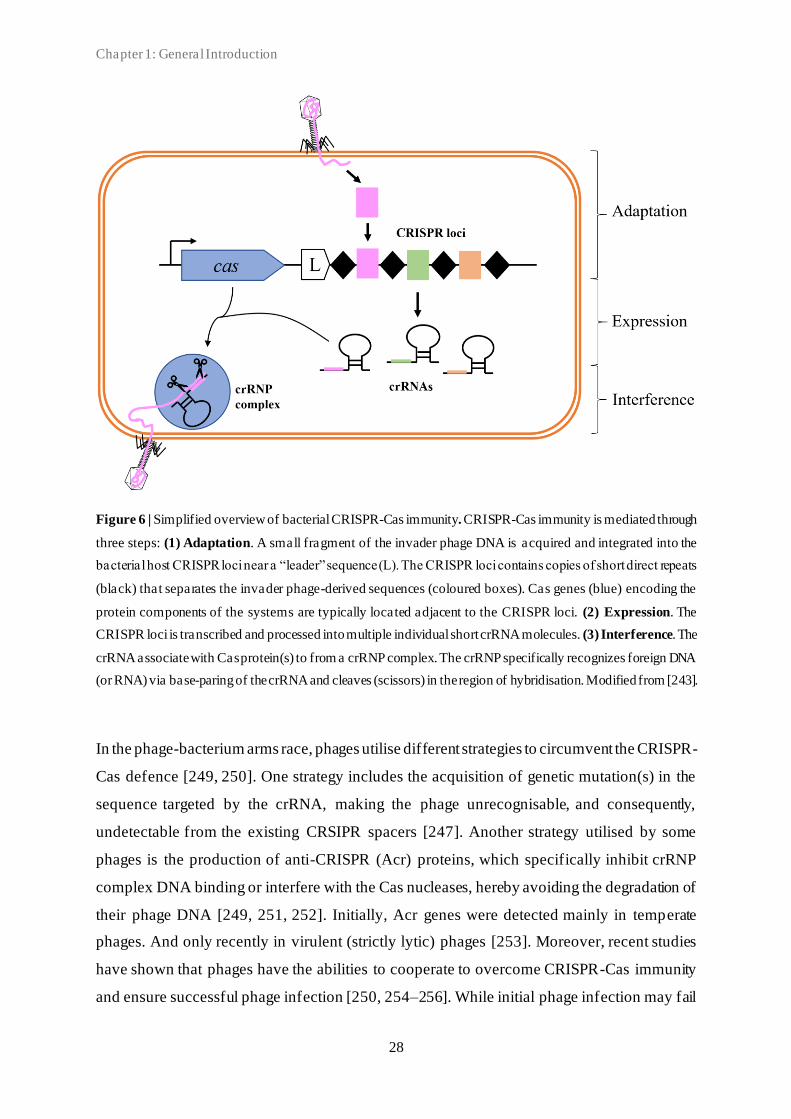
Chapter 1: General Introduction
28
Figure 6 | Simplified overview of bacterial CRISPR-Cas immunity. CRISPR-Cas immunity is mediated through
three steps: (1) Adaptation. A small fragment of the invader phage DNA is acquired and integrated into the
bacterial host CRISPR loci near a “leader” sequence (L). The CRISPR loci contains copies of short direct repeats
(black) that separates the invader phage-derived sequences (coloured boxes). Cas genes (blue) encoding the
protein components of the systems are typically located adjacent to the CRISPR loci. (2) Expression. The
CRISPR loci is transcribed and processed into multiple individual short crRNA molecules. (3) Interference. The
crRNA associate with Cas protein(s) to from a crRNP complex. The crRNP specifically recognizes foreign DNA
(or RNA) via base-paring of the crRNA and cleaves (scissors) in the region of hybridisation. Modified from [243].
In the phage-bacterium arms race, phages utilise different strategies to circumvent the CRISPR-
Cas defence [249, 250]. One strategy includes the acquisition of genetic mutation(s) in the
sequence targeted by the crRNA, making the phage unrecognisable, and consequently,
undetectable from the existing CRSIPR spacers [247]. Another strategy utilised by some
phages is the production of anti-CRISPR (Acr) proteins, which specifically inhibit crRNP
complex DNA binding or interfere with the Cas nucleases, hereby avoiding the degradation of
their phage DNA [249, 251, 252]. Initially, Acr genes were detected mainly in temperate
phages. And only recently in virulent (strictly lytic) phages [253]. Moreover, recent studies
have shown that phages have the abilities to cooperate to overcome CRISPR-Cas immunity
and ensure successful phage infection [250, 254–256]. While initial phage infection may fail

Chapter 1: General Introduction
29
due to degradation by the host CRISPR-Cas system(s), Acr production prior to the phage
degradation leaves the cell immunosuppressed. If the cell is re-infected by other phages, Acr
proteins from the initial phage infection increase the likelihood of subsequent successful phage
infection. Moreover, Borges et al. (2018) demonstrated that complete CRISPR-Cas
inactivation by Acr proteins is challenging, and that the concentration of Acr required for the
inactivation and successful infection is contributed by multiple phages. Accordingly, these
initially failed phage infections represent a form of altruism within phage populations: phages
that initiate failed infections suffer a cost in suppressing CRISPR-Cas immunity while kin
phages benefit by initiation successful infection.
Active defence systems, such as R-M enzymes and CRISPR-Cas adaptive immunity, are
widely distributed among bacteria and confer a specific defence against phage infection.
However, the maintenance of such systems has its own fitness costs [231, 257]. For R-M
systems, bacterial fitness cost associated with the production of enzymes involved in the
restriction of foreign DNA exists, while expression of Cas proteins is particularly costly and
associated with lower competitive abilities [258].
It remains unclear what is the effect of the co-evolutionary arms race between phage and host
and the possible emergence of phage-resistant bacteria variants [176]. This has major
implications for the use of phages in therapy as the speed of the emergence of resistance may
influence the outcome. In vitro, resistance has been shown to develop in the timespan of hours
to days [147]. Whether the evolution of phage resistance in vitro is comparable to in vivo and/or
in situ conditions where bacteria may be replicating more slowly and challenged with a greater
set of environmental conditions needs further investigation and most likely will depend on the
type of phage, pathogen and their interaction(s) [176]. Accordingly, in vitro resistance selection
experiments might not fully account for the complexity of the phage-host interaction and co-
evolution dynamics [133, 147]. Nevertheless, in vitro phage resistance characterisation might
be an important first step in assessing the relative likelihood of emerging phage-resistant
bacterial populations, the most likely phenotype(s) of resistant mutants, as well as the effect of
combinations of phages on the resistance development [214, 259].

Chapter 1: General Introduction
30
1.4 Avian pathogenic Escherichia coli (APEC)
1.4.1 Diseases, transmission, and reservoirs
Escherichia coli (E. coli) belongs to the Enterobacteriaceae family, and is a Gram-negative,
facultative anaerobe, motile rod-shaped bacterium with approximately 0.5 µm in diameter and
1-3 µm in length [260, 261]. E. coli is a ubiquitous bacterium widely distributed in various
environments and the global population size has been estimated to be 10 20 [262]. The
gastrointestinal (GI) tract of humans and animals is considered to be the primary habitat of E.
coli, whereas the external environment serve as a secondary habitat, in which the bacteria are
excreted (e.g. water, soil, and sediments) [263, 264]. Although most E. coli strains are harmless
or beneficial commensals of the GI tract, some strains are highly virulent pathogens that can
cause a variety of infections [265–267]. The majority of pathogenic E. coli strains are
considered opportunistic pathogens as they exist most of the time harmlessly as commensals
of the microflora in a wide spectrum of hosts, and only cause infections under certain
conditions, including weakened immune system of the host and presence of bacterial stress
factors [267, 268].
Pathogenic E. coli can be classified into two main groups according to the infection site and
clinical outcome: intestinal or diarrheagenic E. coli, if they cause infections inside the intestine,
and extraintestinal E. coli (ExPEC), if they cause infections in extra-intestinal sites [265].
ExPEC strains that cause disease in poultry are characterised as avian pa thogenic E. coli
(APEC). APEC is recognised as one of the most important bacterial pathogens of poultry and
other avian species. APEC causes a large range of localised or systemic extra -intestinal
infections, which collectively are referred to as colibacillosis [269]. These infections can result
in morbidity and mortality, and hereby, significant economic losses to the poultry industry
globally [268–270]. Healthy birds with a normal immune system generally do not develop
disease. Thus, the different infection types of colibacillosis and corresponding pathological
manifestations depend on the routes of entry of APEC, the tissue(s) affected, virulence
properties of the strain, host status (e.g. species, age, type of production, and health status), and
the presence of predisposing factors [271]. Such factors include damaged mucosal epithelial
barriers (e.g. skin wounds and mucosal damage from viral, bacterial, and parasitic infections),
impaired or suppressed immune system (e.g. due to viral infections or nutritional deficiencies),
or inappropriate husbandry practices (e.g. contaminated environments and abnormal stress).
Accordingly, colibacillosis in poultry often occur as a secondarily localised or systemic disease

Chapter 1: General Introduction
31
when the host defences have been impaired [268]. In Belgium, APEC infections were identified
as a major factor in poultry disease, and the incidence of APEC infections in layers, breeders,
and broilers has been shown to be 38.6%, 26.9%, and 17.7%, respectively [272]. The primary
reservoir of APEC strains is the intestinal tract of poultry. Poultry may carry up to 106 colony
forming units (CFU) of E. coli per gram of faeces, of which an estimate of 10-15% belong to
potentially APEC serogroups [268, 273]. Excretion to the environment allow for bird-to-bird
transmission of E. coli strains via the faecal-oral route, and the bacteria can survive in dust of
poultry houses reaching concentrations of ~105-106 CFU/g [268, 269]. Poultry litter may reach
levels as high as 108 CFU/g [274].
1.4.2 Virulence factors
APEC possesses and utilises various virulence factors, including adhesins, iron acquisition
systems, protectins, and toxins to cause infection in poultry (reviewed in [253]) (Table 2).
These factors are essential for APEC to adhere, invade, evade the host immune responses,
colonise, and cause infection in extraintestinal sites [267, 269]. Adherence of the bacterium to
epithelial surfaces of the host is an essential step in the APEC infection required for host
colonisation. The adherence is primary mediated by pili/fimbriae located on the outer
membrane of most strains [275]. The P pili are thought to play a role by means of their PapG
adhesin, which is found in three variants: PapGI, PapGII, and PapGIII, encoded by the three
alleles of the corresponding gene, papG. Especially papGII is more likely to be found among
APEC isolates compared to avian faecal E. coli isolates [271, 276, 277]. Besides, the outer
membrane protease OmpT, encoded by the ompT gene, is thought to participate in the adhesion
of APEC as well as antibiotic resistance through peptide cleavage [278, 279]. Pathogenic E.
coli have been shown to possess high prevalence of genes, such as sitA, iutA, fyuA, irp2, ireA
and iroN, coding for iron acquisition systems [275, 280]. Once the bacteria have successfully
colonised the host, these systems may contribute to the APEC virulence by facilitating
acquisition of iron, which is essential for bacterial growth, proliferation, and protection against
environmental stresses. Moreover these systems may promote adaptation to colonise and infect
sites where iron is depleted by antibacterial host defences [269, 275, 276, 281]. Protectins
provide protection of the bacteria against the host immune system as well as unfavourable
conditions, and include, among others, bacterial capsule, OMPs, and LPS components [271,
275]. Multiple toxin types have been reported in APEC and assist in the ability of the bacteria
to invade and cause damage to the tissues [269].

Chapter 1: General Introduction
32
Table 2 | Prevalent virulence-associated traits of avian pathogenic E. coli (APEC)
Category Gene(s) Description Ref
Adhesins pap P fimbriae [271]
ompT Outer membrane protein T (protease) [278]
Toxins hlyF* Hemolysin F [282]
tsh Temperature-sensitive haemagglutinin auto-transporter [283]
Iron acquisition
systems
fyuA Yersiniabactin (siderophore) synthesis and receptor [283]
ireA Iron regulated element (catecholate siderophore receptor) [284]
iroN* Catecholate siderophore receptor [285]
irp2 Yersiniabactin (siderophore) synthesis and receptor [286]
iutA* Aerobactin (siderophore) synthesis and receptor [285]
sitABCD Iron and manganese transport system [281]
Protectins
cvaC* Colicin V (ColV) [276]
iss* Increased serum survival, complement resistance [285]
neu Capsular polysialic acid biosynthesis [287]
wzy O-antigen polymerase [288]
Other fliC Flagellar antigen H7 [276]
* Plasmid-encoded. Modified from [269, 289].
The majority of APEC strains are often characterised by the presence of large conjugative
plasmids, harbouring a number of virulence genes [276, 290, 291]. Several of these plasmid-
encoded genes, such as the ColV-type plasmid-encoded genes cvaC, and iss provide a
competitive advantage for nutrient acquisition through colicin production and increased serum
survival of the bacterium, respectively [292–299]. Also frequently located on a ColV-type
plasmid is the temperature-sensitive hemagglutinin gene, tsh, which encodes the
autotransporter Tsh protein that play a role in the pathogenicity of E. coli in the early stages of
infection [300]. It has been shown that transferring APEC ColV-type plasmids into
avirulent/commensal E. coli strains enhances their ability to colonise and infect hosts in
experimental models, supporting a role of plasmid-encoded genes in the pathogenicity of E.
coli [301]. Compared to avian faecal E. coli isolates, several virulence genes have more
frequently been observed in APEC, including the five plasmid-encoded genes (iutA, hlyF, iss,
iroN, and ompT), which have been identified as predictors of APEC virulence [276, 285, 293,
302, 303].

Chapter 1: General Introduction
33
1.4.3 Strain typing and population genetics
Since the 1940s, bacteria have been serotyped based on their surface antigens: the somatic O-
LPS antigen, the flagellar H-antigen, and the capsular K-antigen, which define serogroup (O
antigen only) or serotype (O, H, and K antigens) [304]. More than 185 O-, 60 H-, and 80 K-
antigens have been recognised, and variations of various different serotypes combinations have
been identified [305, 306]. Multiple APEC serogroups have been associated with colibacillosis
cases, however, the three O serogroups O1, O2, and O78 constitute more than 80% of the cases
[269, 307, 308]. Other APEC infection-associated serogroups include O8, O18, O35, O109,
and O115 [276, 309].
In the late 1990s, MLST emerged as a powerful method for analysis of bacterial population
genetics and phylogenetic relationship [310]. The principle of MLST is to identity nucleotide
sequence variation in 400-500 bp of seven conserved housekeeping genes. The genetic
variation is characterised by sequencing the fragments of housekeeping genes. An identifier
number is given to each unique sequence (allele) recognised for each locus, and a sequence
type (ST) is given to the specific combination of alleles at all loci. Together, this loci
combination is used to generate a DNA fingerprint of the bacterial isolate. The most widely
used MLST scheme for typing of E. coli strains is Achtman’s scheme
(https://pubmlst.org/data/), which uses seven housekeeping genes for strain discrimination
[311]. Recently, APEC (isolates belonging to the ST95 and ST131) has been p resented as a
potential foodborne zoonotic pathogen and as such, is a pathogen of importance to the poultry
industry as well as the public health [269].
In recent years, high-throughput sequencing technologies have made it possible to understand
the population genomics at the single gene level as well as the whole-genome level. WGS data
has allowed for the development of high-resolution typing methods that circumvents previous
typing challenges and enable comparisons of inter- and intraspecies bacterial genomes.
Besides, WGS typing approaches, such as discovery of single nucleotide polymorphisms
(SNPs), offer greater phylogenetic resolution as well as genetic markers to study evolution
[312–314].
1.4.4 Current strategies to prevent and control APEC
The commercial poultry industry depends on cost efficiently raising birds in large quantities
[315]. The current prevention and control of APEC infections in poultry relies on biosecurity

Chapter 1: General Introduction
34
measures to reduce the pathogen load, management of environmental stressors, application of
feed additives, vaccination strategies, and antimicrobial treatment [269, 315, 316].
Prevention and control of APEC infections (colibacillosis) in poultry depends largely on
identifying and eliminating the predisposing cause(s) of the disease outbreak [289]. Keeping
strict biosecurity measures (such as segregation, traffic control, cleaning, and disinfection)
helps to prevent a large proportion of harmful bacteria and viruses from entering and spread
within the poultry facilities [269, 315, 317], however, it is necessary to make these measures
practical, enforceable and cost effective [269, 315]. Environmental factors should be well
monitored, controlled, and adjusted. Proper ventilation as well as maintaining optimum
temperature, humidity, and bird density will keep ammonia and dust in poultry facilities low
and thereby reducing the risk of APEC infections [269, 289].
A variety of different feed additives are commonly used to control E. coli in poultry, including
prebiotics, probiotics, enzymes, acidifiers, vitamins, immune enhances, and other
antimicrobials [269, 289].These may enhance wound healing or promote intestinal health, and
thus, directly or indirectly help preventing infections [315].
Vaccination is commonly used in the control of infections. While some vaccines are given to
protect the individual bird against disease, others are given to increase maternal immunity
[315]. Various E. coli vaccines have been developed to prevent colibacillosis (to varying effect)
[269, 317]. These include inactivated, live-attenuated, recombinant and subunit vaccines [289].
Despite multiple vaccine candidates with proven in vivo efficacy, only two vaccines (live-
attenuated APEC O78 ΔaroA Poulvac® and inactivated Nobilis® comprising F11 fimbrial and
FT flagellar antigens) are currently commercially available for use in chickens [269, 316]. The
major drawback of these vaccines is the lack of protection against heterogenous APEC
infections, comprised of multiple serotypes [289]. In large-scale poultry productions, efficient
and economic application of vaccines is a challenge. The most commonly used application
techniques include in ovo injection, subcutaneous or intramuscular injection, spray, intraocular
or nasal drop, and through the drinking water [315]. Application technique depends on the
vaccine used [269, 315]. The ideal APEC vaccine should be able to confer cross-protection
against multiple APEC serotypes and be deliverable by mass-vaccination methods, such as in
ovo, oral (feed or water), or spray routes [269].
Antimicrobials are commonly used to treat colibacillosis [269]. When thousands of birds are
grouped together in commercial facilities, segregation and treatment of individuals is

Chapter 1: General Introduction
35
impractical and labour-intensive, so metaphylactic use is applied [315]. Accordingly, the
majority of antibiotics used in poultry production are administered orally, mixed into feed or
drinking water, using automated systems [318, 319]. As sick birds often have little or no
appetite and are unable to compete for feed, water medication may be preferred over feed
medication, as sick animals will still frequently drink [289, 315]. While antimicrobial treatment
may not be completely successful in curing sick animals, it may hold the disease in check until
cleared by the immune system of the host [269]. However, the increasing resistance of APEC
strains to multiple antibiotics limits the use of antimicrobials [269]. Moreover, restrictions on
antimicrobial use in poultry have been imposed by regulatory and public concern [289]. The
United States of America and European Union (EU) have employed strategies to restrict the
non-therapeutic use (for growth promotion) of antibiotics in food–animal production and to
limit the therapeutic use (for treatment of sick animals with a diagnosis) of medically important
antibiotics [320].
1.4.5 Phage therapy against APEC infections
Phage therapy has proven to be a potential therapeutic against APEC. However, while several
studies have described efficacies, others have not been able to show efficacy in avian species
[321–330]. Moreover, comparing studies is challenging as different infection models and
treatment protocols were used.
When comparing the infection models used, great variation can be observed between different
variables. 1) The age of the birds (range between 1-day-old and 10-weeks-old). 2) The route of
infection, including direct injection to the air sac, intratracheal (IT) inoculation, oral
inoculation, intramuscular (IM) inoculation, intracranial (IC) injection, or the use of naturally
infected birds. 3) The challenge dose of E. coli used in the infection models (range between
103 and 108 CFU/ml). 4) The bacterial strains used, including APEC and ExPEC isolates with
serogroup O1, O2, O18, O78, O86, O126 or unknown. As for the infection model, several
parameters differ in the treatment protocols used. 1) Prevention and/or therapeutical use has
been reported for respiratory infection/colibacillosis, septicaemia and meningitis. 2) The routes
of administration of phage treatment include air sac inoculation, drinking water, coarse spray
(on chicken or litter), fine spray, orally syringe combined with spray, IT inoculation, IM
inoculation, or IC injection. 3) The dose of administration and formulation. The concentration
of phage(s) applied ranges between 102 and 109 PFU/ml. Most studies include only a single

Chapter 1: General Introduction
36
phage in the formulation. Few studies have investigated the difference in efficacy between a
single phage alone and a cocktail of phages. 4) The treatment models include great differences
when comparing time and type of phage administration. Single-dose administration included
prophylactic administration (between one and five days prior to E. coli challenge),
simultaneous administration of phage and E. coli, and therapeutic administration (eight hours
post E. coli challenge). Multiple-dose administration included once a day for seven days prior
to challenge as well as once a day for seven days both prior and post E. coli challenge.
Several variables have been identified that influence differences in outcome (Table 3 ). 1)
Concentration and timing of administered phage(s) are known to effect the success rate of
phage therapy [331]. Independent of infection and treatment model (aerosol spray, orally,
thoracic air sac inoculation, IM inoculation, or IC inoculation), higher phage dosages have
shown more effective (decreased mortality and/or morbidity) [321, 325, 329, 330]. However,
timing of the phage administration has been shown to be of extremely importance for a
successful outcome, as administration of the same high phage titer on different timepoint
(before and/or after bacterial inoculation) resulted in significant differences in outcomes [328,
331]. 2) Phage therapy success rates also depend on the site and route of administration of the
phages. Oliveira et al. (2010), confirmed that colibacillosis-induced morbidity and mortality
may be significantly reduced by spraying of housing systems with phage cocktails as well as
oral administration of phages. However, Huff et al. (2002) found that the best results were
obtained through direct application of the phages on the site of infection. Likewise, Huff et al.
(2013) did not obtain a reduced mortality when phages were administrated via spray while IT
administration was more successful, which was attributed to the fact that only direct
administration could bring high enough phage titers at the site of infection. Similarly, IM
delivery during septicaemia was very effective. On the contrary, high phage titers present in
drinking water did not prevent infection after severe thoracic air sac challenge, as the birds
were not able to absorb enough phages from the water [325]. Thus, while phages targeting
systemic infections may be delivered IM or intravenous, it might be preferable to deliver
phages targeting respiratory infection by inhalation [332]. Moreover, when phages are
administered by spray, the spray type should be taken into considera tion, as smaller droplet
aerosols may penetrate the respiratory tract to a greater extent compared to coarse spray [325].

Chapter 1: General Introduction
37
Table 3 | Phage therapy study outcomes based on the body weight and mortality of the birds.
Positive outcomes Negative outcomes
Prevention
• High titer (108 and 109 PFU/ml) phage
in aerosol spray applied
simultaneously with E. coli challenge
significantly decreased mortality [329].
• High titer (108 PFU/ml) phage sprayed
on litter applied simultaneously with
high (108 CFU/ml) oral E. coli
challenge reduced mortality and
morbidity [327].
• High titer (108 PFU/ml) phage mixed
with E. coli prior to challenging birds
via thoracic air sac inoculation
provided complete protection of the
birds [325].
• Low titer (104 PFU/ml) phage mixed
with E. coli prior to challenging birds
via thoracic air sac inoculation
significantly reduced mortality from
85% to % [325].
• High titer (108 PFU/ml) phage
administered IT just before IT E. coli
challenge provided complete
protection [323].
High titer (109 PFU/ml) phage coarse
or fine spray did not protect birds
from IT E. coli challenge that cause
significant decrease in body weight
and significant increase in mortality
[323].
• Phage titer (103-106) in drinking
water had little or no efficacy to
prevent infection when birds were
challenged with large inoculum of E.
coli via thoracic air sac inoculation,
resulting in significant decrease in
body weight and increase in mortality
[325].
Treatment
• High titer (107 PFU/ml) phage cocktail
administered in both drinking water
and fine spray decreased mortality in
large commercial flock naturally
infected [321].
• High titer (106 or 108 PFU/ml) phage
administered IC 8 hours post IC E. coli
challenge significantly reduced
morbidity and mortality [328].
• High titer (109 PFU/ml) phage
administered IM 8 hours post E. coli
challenge via thoracic air sac
significantly reduced the mortality.
Phage cocktail showed more efficient
than single phage [324].
• High titer (108 and 109 PFU/ml)
phage in aerosol spray applied
simultaneously with E. coli challenge
was not an effective treatment as only
low or no titers of phages was
delivered to the blood [329].
• Low titer (102 PFU/ml) phage
administrated IM did not result in any
statistically significant protection
against IM E. coli challenge [328].
IT = Intratracheally, IC = Intracranially, IM = Intramuscularly.

Chapter 1: General Introduction
38
3) Phage cocktails have been shown more efficient in treating colibacillosis than a single phage
treatments, probably due to a synergistic effect between the individual phages [324, 333].
However, a cocktail of phages does not necessarily lead to successful application as
demonstrated by Tsonos et al. (2014). Though the cocktail was composed of well-characterised
phages and administered via three different routes (IT, intraoesophageally or via the drinking
water) in high dose, they could not detect a decrease in mortality, lesion scores or weight loss.
Essentially in phage therapy is that the appropriate phage(s) are delivered timely in sufficient
quantities at the site of bacterial infection. Even though this seems to be a relatively easy
criterion to meet, reality show that it is not. As modern poultry production facilities contain
thousands of birds, it would not be feasible to administer an IM treatment of phage during an
outbreak of colibacillosis. If successful, treatment through drinking water would be a practical
method to administer phage in the poultry industry [334].
Despite many promising phage therapy results, phage-based product for treatment of
colibacillosis in poultry are still not available on the market [269, 334]. A great deal of work is
required to select the most efficacious phage(s) against specific pathogenic target bacteria, and
to determine how, when, and how much phage should be administered to provide sufficient
protection to poultry [334, 335]. If we learn practical ways to exploit the phage therapy
potential, phage(s) could provide a valuable alternative or supplement to the use of antibiotics
to prevent and/or control bacterial diseases in poultry [269].

Chapter 1: General Introduction
39
References
[1] Gaynes, R. The Discovery of Penicillin—New Insights After More Than 75 Years of Clinical Use.
Emerg. Infect. Dis., 2017, 23 (5), 849–853. https://doi.org/10.3201/eid2305.161556.
[2] Aminov, R. I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future.
Front. Microbiol., 2010, 1, 134. https://doi.org/10.3389/fmicb.2010.00134.
[3] Gordillo Altamirano, F. L.; Barr, J. J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev.,
2019, 32 (2), 1–25. https://doi.org/10.1128/CMR.00066-18.
[4] Gould, K. Antibiotics: From Prehistory to the Present Day. J. Antimicrob. Chemother., 2016, 71 (3),
572–575. https://doi.org/10.1093/jac/dkv484.
[5] Hutchings, M. I.; Truman, A. W.; Wilkinson, B. Antibiotics: Past, Present and Future. Curr. Opin.
Microbiol., 2019, 51 (October 2019), 72–80. https://doi.org/10.1016/j.mib.2019.10.008.
[6] Ventola, C. L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. P T, 2015, 40 (4), 277–
283. https://doi.org/10.24911/IJMDC.51-1549060699.
[7] Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev.,
2010, 74 (3), 417–433. https://doi.org/10.1128/mmbr.00016-10.
[8] Silver, L. L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev., 2011, 24 (1), 71–109.
https://doi.org/10.1128/CMR.00030-10.
[9] World Health Organization (WHO). Antimicrobial Resistance: Global Report on Surveillance;
Geneva, Switzerland, 2014. https://doi.org/10.1007/s13312-014-0374-3.
[10] Ribeiro da Cunha; Fonseca; Calado. Antibiotic Discovery: Where Have We Come from, Where Do
We Go? Antibiotics, 2019, 8 (2), 45. https://doi.org/10.3390/antibiotics8020045.
[11] ReAct Group. No Title https://www.reactgroup.org/toolbox/understand/how-did-we-end-up-
here/few-antibiotics-under-development/.
[12] Centers for Disease Control and Prevention (CDC). Antibiotic Resistance Threats in the United
States, 2019; Atlanta, Georgia, 2019. https://doi.org/10.15620/cdc:82532.
[13] zur Wiesch, P. A.; Kouyos, R.; Engelstädter, J.; Regoes, R. R.; Bonhoeffer, S. Population Biological
Principles of Drug-Resistance Evolution in Infectious Diseases. Lancet Infect. Dis., 2011, 11 (3),
236–247. https://doi.org/10.1016/S1473-3099(10)70264-4.
[14] Rodríguez-Rojas, A.; Rodríguez-Beltrán, J.; Couce, A.; Blázquez, J. Antibiotics and Antibiotic
Resistance: A Bitter Fight against Evolution. Int. J. Med. Microbiol., 2013, 303 (6–7), 293–297.
https://doi.org/10.1016/j.ijmm.2013.02.004.
[15] Goossens, H. Antibiotic Consumption and Link to Resistance. Clin. Microbiol. Infect., 2009, 15
(SUPPL. 3), 12–15. https://doi.org/10.1111/j.1469-0691.2009.02725.x.
[16] Llor, C.; Bjerrum, L. Antimicrobial Resistance: Risk Associated with Antibiotic Overuse and
Initiatives to Reduce the Problem. Ther. Adv. Drug Saf., 2014, 5 (6), 229–241.
https://doi.org/10.1177/2042098614554919.
[17] Holmes, A. H.; Moore, L. S. P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P. J.;
Piddock, L. J. V. Understanding the Mechanisms and Drivers of Antimicrobial Resistance. Lancet,
2016, 387 (10014), 176–187. https://doi.org/10.1016/S0140-6736(15)00473-0.
[18] von Wintersdorff, C. J. H.; Penders, J.; van Niekerk, J. M.; Mills, N. D.; Majumder, S.; van Alphen,
L. B.; Savelkoul, P. H. M.; Wolffs, P. F. G. Dissemination of Antimicrobial Resistance in Microbial
Ecosystems through Horizontal Gene Transfer. Front. Microbiol., 2016, 7 (Feb), 173.
https://doi.org/10.3389/fmicb.2016.00173.

Chapter 1: General Introduction
40
[19] Wozniak, R. A. F.; Waldor, M. K. Integrative and Conjugative Elements : Mosaic Mobile Genetic
Elements Enabling Dynamic Lateral Gene Flow. Nat. Rev. Microbiol., 2010, 8, 552–563.
https://doi.org/10.1038/nrmicro2382.
[20] Skippington, E.; Ragan, M. A. Lateral Genetic Transfer and the Construction of Genetic Exchange
Communities. FEMS Microbiol. Rev., 2011, 35 (5), 707–735. https://doi.org/10.1111/j.1574-
6976.2010.00261.x.
[21] Lopatkin, A. J.; Sysoeva, T. A.; You, L. Dissecting the Ef fects of Antibiotics on Horizontal Gene
Transfer: Analysis Suggests a Critical Role of Selection Dynamics. BioEssays, 2016, 38 (12), 1283–
1292. https://doi.org/10.1002/bies.201600133.
[22] Chen, I.; Christie, P. J.; Dubnau, D. The Ins and Outs of DNA Transfer in Bacteria. Science (80-. ).,
2005, 310 (5753), 1456–1460. https://doi.org/10.1126/science.1114021.
[23] Balcazar, J. L. Bacteriophages as Vehicles for Antibiotic Resistance Genes in the Environment. PLoS
Pathog., 2014, 10 (7), e1004219. https://doi.org/10.1371/journal.ppat.1004219.
[24] Partridge, S. R.; Kwong, S. M.; Firth, N.; Jensen, S. O. Mobile Genetic Elements Associated with
Antimicrobial Resistance. Clin. Microbiol. Rev., 2018, 31 (4), e00088-17.
https://doi.org/10.1128/CMR.00088-17.
[25] Gómez-Gómez, C.; Blanco-Picazo, P.; Brown-Jaque, M.; Quirós, P.; Rodríguez-Rubio, L.; Cerdà-
Cuellar, M.; Muniesa, M. Infectious Phage Particles Packaging Antibiotic Resistance Genes Found
in Meat Products and Chicken Feces. Sci. Rep., 2019, 9 (13281). https://doi.org/10.1038/s41598-
019-49898-0.
[26] Berglund, B. Environmental Dissemination of Antibiotic Resistance Genes and Correlation to
Anthropogenic Contamination with Antibiotics. Infect. Ecol. Epidemiol., 2015, 5 (1), 28564.
https://doi.org/10.3402/iee.v5.28564.
[27] White, A.; Hughes, J. M. Critical Importance of a One Health Approach to Antimicrobial Resistance.
Ecohealth, 2019, 16, 404–409. https://doi.org/10.1007/s10393-019-01415-5.
[28] McEwen, S. A.; Collignon, P. J. Antimicrobial Resistance: A One Health Perspective. Microbiol.
Spectr., 2018, 6 (2), ARBA-0009-2017. https://doi.org/10.1128/microbiolspec.ARBA-0009-2017.
[29] Guardabassi, L.; Butaye, P.; Dockrell, D. H.; Fitzgerald, J. R.; Kuijper, E. J. One Health: A
Multifaceted Concept Combining Diverse Approaches to Prevent and Control Antimicrobial
Resistance. Clin. Microbiol. Infect., 2020, No. xxxx. https://doi.org/10.1016/j.cmi.2020.07.012.
[30] Robinson, T. P.; Bu, D. P.; Carrique-Mas, J.; Fèvre, E. M.; Gilbert, M.; Grace, D.; Hay, S. I.;
Jiwakanon, J.; Kakkar, M.; Kariuki, S.; et al. Antibiotic Resistance Is the Quintessential One Health
Issue. Trans. R. Soc. Trop. Med. Hyg., 2016, 110 (7), 377–380.
https://doi.org/10.1093/trstmh/trw048.
[31] Collignon, P. J.; McEwen, S. A. One Health-Its Importance in Helping to Better Control
Antimicrobial Resistance. Trop. Med. Infect. Dis., 2019, 4 (1), 22.
https://doi.org/10.3390/tropicalmed4010022.
[32] Pires, D. P.; Costa, A. R.; Pinto, G.; Meneses, L.; Azeredo, J. Current Challenges and Future
Opportunities of Phage Therapy. FEMS Microbiol. Rev., 2020, fuaa017, 1–17.
https://doi.org/10.1093/femsre/fuaa017.
[33] Lin, D. M.; Koskella, B.; Lin, H. C. Phage Therapy: An Alternative to Antibiotics in the Age of
Multi-Drug Resistance. World J. Gastrointest. Pharmacol. Ther., 2017, 8 (3), 162–173.
https://doi.org/10.4292/wjgpt.v8.i3.162.
[34] Interagency Coordination Group on Antimicrobial Resistance (IACG). No Time to Wait: Securing
the Future from Drug-Resistant Infections; 2019.
[35] Brives, C.; Pourraz, J. Phage Therapy as a Potential Solution in the Fight against AMR: Obstacles
and Possible Futures. Palgrave Commun., 2020, 6, 100.

Chapter 1: General Introduction
41
[36] Twort, F. W. An Investigation on the Nature of Ultra -Microscopic Viruses. Lancet, 1915, 186 (4814),
1241–1243. https://doi.org/10.1016/S0140-6736(01)20383-3.
[37] Summers, W. C. Felix D’Herelle and the Origins of Molecular Biology; Yale University Press: New
Haven, Conn, 1999.
[38] Güemes, A. G. C.; Merry Youle; Cantú, V. A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners
in the Game of Life. Annu. Rev. Virol., 2016, 3 (1), 197–214. https://doi.org/10.1146/annurev-
virology-100114-054952.
[39] Hatfull, G. F. Dark Matter of the Biosphere: The Amazing World of Bacteriophage Diversity. J.
Virol., 2015, 89 (16), 8107–8110. https://doi.org/10.1128/jvi.01340-15.
[40] Letarov, A.; Kulikov, E. The Bacteriophages in Human- and Animal Body-Associated Microbial
Communities. J. Appl. Microbiol., 2009, 107 (1), 1–13. https://doi.org/10.1111/j.1365-
2672.2009.04143.x.
[41] Batinovic; Wassef; Knowler; Rice; Stanton; Rose; Tucci; Nittami; Vinh; Drummond; et al.
Bacteriophages in Natural and Artificial Environments. Pathogens, 2019, 8 (3), 100.
https://doi.org/10.3390/pathogens8030100.
[42] Abedon, S. T. Phages, Ecology, Evolution. In Bacteriophage Ecology: Population Growth,
Evolution, and Impact of Bacterial Viruses; Abedon, S. T., Ed.; Cambridge University Press:
Cambridge, 2008; pp 1–28. https://doi.org/10.1017/CBO9780511541483.004.
[43] Clokie, M. R. J.; Millard, A. D.; Letarov, A. V; Heaphy, S. Phages in Nature. Bacteriophage, 2011,
1 (1), 31–45. https://doi.org/10.4161/bact.1.1.14942.
[44] Weinbauer, M. G.; Rassoulzadegan, F. Are Viruses Driving Microbial Diversification and Diversity?
Environ. Microbiol., 2004, 6 (1), 1–11. https://doi.org/10.1046/j.1462-2920.2003.00539.x.
[45] de Jonge, P. A.; Nobrega, F. L.; Brouns, S. J. J.; Dutilh, B. E. Molecular and Evolutionary
Determinants of Bacteriophage Host Range. Trends Microbiol., 2019, 27 (1), 51–63.
https://doi.org/10.1016/j.tim.2018.08.006.
[46] Bielke, L.; Higgins, S.; Donoghue, A.; Donoghue, D.; Hargis, B. M. Salmonella Host Range of
Bacteriophages That Infect Multiple Genera. Poult. Sci., 2007, 86 (12), 2536–2540.
https://doi.org/10.3382/ps.2007-00250.
[47] Yu, P.; Mathieu, J.; Li, M.; Dai, Z.; Alvarez, P. J. J. Isolation of Polyvalent Bacteriophages by
Sequential Multiple-Host Approaches. Appl. Environ. Microbiol., 2016, 82 (3), 808–815.
https://doi.org/10.1128/AEM.02382-15.
[48] Grose, J. H.; Casjens, S. R. Understanding the Enormous Diversity of Bacteriophages: The Tailed
Phages That Infect the Bacterial Family Enterobacteriaceae. Virology, 2014, 468, 421–443.
https://doi.org/10.1016/j.virol.2014.08.024.
[49] Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages.
Front. Microbiol., 2016, 7, 1352. https://doi.org/10.3389/fmicb.2016.01352.
[50] Díaz-Muñoz, S. L.; Koskella, B. Bacteria –Phage Interactions in Natural Environments. In Advances
in Applied Microbiology; Elsevier Inc., 2014; Vol. 89, pp 135–183. https://doi.org/10.1016/B978-0-
12-800259-9.00004-4.
[51] Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth.
Pharmaceuticals, 2019, 12 (1), 35. https://doi.org/10.3390/ph12010035.
[52] Casjens, S. R. Comparative Genomics and Evolution of the Tailed -Bacteriophages. Curr. Opin.
Microbiol., 2005, 8 (4), 451–458. https://doi.org/10.1016/j.mib.2005.06.014.
[53] Scholl, D.; Rogers, S.; Adhya, S.; Merril, C. R. Bacteriophage K1-5 Encodes Two Different Tail
Fiber Proteins, Allowing It To Infect and Replicate on Both K1 and K5 Strains of Escherichia Coli.
J. Virol., 2001, 75 (6), 2509–2515. https://doi.org/10.1128/jvi.75.6.2509-2515.2001.

Chapter 1: General Introduction
42
[54] International Committee on Taxonomy of Viruses (ICTV), A. M. K. Virus Taxonomy: Ninth Report
of the International Committee on Taxonomy of Viruses, 1st ed.; King, A. M. ., Adams, M. J.,
Carstens, E. B., Lefkowitz, E. J., Eds.; Elsevier, 2011.
[55] Ackermann, H.-W. 5500 Phages Examined in the Electron Microscope. Arch. Virol., 2007, 152 (2),
227–243. https://doi.org/10.1007/s00705-006-0849-1.
[56] Dion, M. B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev.
Microbiol., 2020, 12. https://doi.org/10.1038/s41579-019-0311-5.
[57] Krupovic, M.; Turner, D.; Morozova, V.; Dyall, M.; Hanna, S.; Edwards, R.; Dutilh, B. E.; Lehman,
S. M.; Reyes, A.; Baquero, D. P. Bacterial Viruses Subcommittee and Archaeal Viruses
Subcommittee of the ICTV : Update of Taxonomy Changes in 2021. Arch. Virol., 2021, 1–6.
https://doi.org/10.1007/s00705-021-05205-9.
[58] Zrelovs, N.; Dislers, A.; Kazaks, A. Motley Crew: Overview of the Current ly Available Phage
Diversity. Front. Microbiol., 2020, 11. https://doi.org/10.3389/fmicb.2020.579452.
[59] Turner, D.; Kropinski, A. M.; Adriaenssens, E. M. A Roadmap for Genome-Based Phage Taxonomy.
Viruses, 2021, 13 (3), 506. https://doi.org/10.3390/v13030506.
[60] Veesler, D.; Cambillau, C. A Common Evolutionary Origin for Tailed-Bacteriophage Functional
Modules and Bacterial Machineries. Microbiol. Mol. Biol. Rev., 2011, 75 (3), 423–433.
https://doi.org/10.1128/mmbr.00014-11.
[61] Aksyuk, A. A.; Leiman, P. G.; Kurochkina, L. P.; Shneider, M. M.; Kostyuchenko, V. A.;
Mesyanzhinov, V. V.; Rossmann, M. G. The Tail Sheath Structure of Bacteriophage T4: A Molecular
Machine for Infecting Bacteria. EMBO J., 2009, 28 (7), 821–829.
https://doi.org/10.1038/emboj.2009.36.
[62] Leiman, P. G.; Chipman, P. R.; Kostyuchenko, V. A.; Mesyanzhinov, V. V.; Rossmann, M. G. Three-
Dimensional Rearrangement of Proteins in the Tail of Bacteriophage T4 on Infection of Its Host.
Cell, 2004, 118 (4), 419–429. https://doi.org/10.1016/j.cell.2004.07.022.
[63] Olszak, T.; Latka, A.; Roszniowski, B.; Valvano, M. A.; Drulis-Kawa, Z. Phage Life Cycles Behind
Bacterial Biodiversity. Curr. Med. Chem., 2017, 24 (36), 3987–4001.
https://doi.org/10.2174/0929867324666170413100136.
[64] Rakhuba, D. V.; Kolomiets, E. I.; Dey, E. S.; Novik, G. I. Bacteriophage Receptors, Mechanisms of
Phage Adsorption and Penetration into Host Cell. Polish J. Microbiol., 2010, 59 (3), 145–155.
https://doi.org/10.33073/pjm-2010-023.
[65] De Smet, J.; Hendrix, H.; Blasdel, B. G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas
Predators: Understanding and Exploiting Phage-Host Interactions. Nat. Rev. Microbiol., 2017, 15
(9), 517–530. https://doi.org/10.1038/nrmicro.2017.61.
[66] Rakonjac, J.; Bennett, N. J.; Spagnuolo, J.; Gagic, D.; Russel, M. Filamentous Bacteriophage:
Biology, Phage Display and Nanotechnology Applications. Curr. Issues Mol. Biol., 2011, 13 (2), 51–
76. https://doi.org/10.21775/cimb.013.051.
[67] Hay, I. D.; Lithgow, T. Filamentous Phages: Masters of a Microbial Sharing Economy. EMBO Rep.,
2019, 20 (6), e47427. https://doi.org/10.15252/embr.201847427.
[68] Warwick-Dugdale, J.; Buchholz, H. H.; Allen, M. J.; Temperton, B. Host-Hijacking and Planktonic
Piracy: How Phages Command the Microbial High Seas. Virol. J., 2019, 16 (1), 15.
https://doi.org/10.1186/s12985-019-1120-1.
[69] Kutter, E.; Bryan, D.; Ray, G.; Brewster, E.; Blasdel, B.; Guttman, B. From Host to Phage
Metabolism: Hot Tales of Phage T4’s Takeover of E. Coli. Viruses, 2018, 10 (7), 387.
https://doi.org/10.3390/v10070387.
[70] Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol., 2014, 52 (3), 243–
258. https://doi.org/10.1007/s12275-014-4087-z.

Chapter 1: General Introduction
43
[71] Howard-Varona, C.; Hargreaves, K. R.; Abedon, S. T.; Sullivan, M. B. Lysogeny in Nature:
Mechanisms, Impact and Ecology of Temperate Phages. ISME J., 2017, 11 (7), 1511–1520.
https://doi.org/10.1038/ismej.2017.16.
[72] Maurice, C. F.; Bouvier, C.; De Wit, R.; Bouvier, T. Linking the Lytic and Lysogenic Bacteriophage
Cycles to Environmental Conditions, Host Physiology and Their Variability in Coastal Lagoons.
Environ. Microbiol., 2013, 15 (9), 2463–2475. https://doi.org/10.1111/1462-2920.12120.
[73] Coleman, S.; Yao, T.; Nguyen, T. V. P.; Golding, I.; Igoshin, O. Bacteriophage Self -Counting in the
Presence of Viral Replication. bioRxiv, 2021.
[74] Knowles, B.; Silveira, C. B.; Bailey, B. A.; Barott, K.; Cantu, V. A.; Cobia n-Guëmes, A. G.;
Coutinho, F. H.; Dinsdale, E. A.; Felts, B.; Furby, K. A.; et al. Lytic to Temperate Switching of Viral
Communities. Nature, 2016, 531 (7595), 466–470. https://doi.org/10.1038/nature17193.
[75] Payet, J. P.; Suttle, C. A. To Kill or Not to Kill: The Balance between Lytic and Lysogenic Viral
Infection Is Driven by Trophic Status. Limnol. Oceanogr., 2013, 58 (2), 465–474.
https://doi.org/10.4319/lo.2013.58.2.0465.
[76] Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.;
Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between Viruses Guides Lysis–Lysogeny
Decisions. Nature, 2017, 541 (7638), 488–493. https://doi.org/10.1038/nature21049.
[77] Campbell, A. The Future of Bacteriophage Biology. Nat. Rev. Genet., 2003, 4 (6), 471–477.
https://doi.org/10.1038/nrg1089.
[78] Casjens, S. R.; Hendrix, R. W. Bacteriophage Lambda: Early Pioneer and Still Relevant. Virology,
2015, 479–480, 310–330. https://doi.org/10.1016/j.virol.2015.02.010.
[79] Zhang, Y.; Liao, Y. Te; Salvador, A.; Sun, X.; Wu, V. C. H. Prediction, Diversity, and Genomic
Analysis of Temperate Phages Induced From Shiga Toxin-Producing Escherichia Coli Strains.
Front. Microbiol., 2020, 10, 3093. https://doi.org/10.3389/fmicb.2019.03093.
[80] Nanda, A. M.; Thormann, K.; Frunzke, J. Impact of Spontaneous Prophage Induction on the Fitness
of Bacterial Populations and Host-Microbe Interactions. J. Bacteriol., 2015, 197 (3), 410–419.
https://doi.org/10.1128/JB.02230-14.
[81] Czyz, A.; Los, M.; Wrobel, B.; Wegrzyn, G. Inhibition of Spontaneous Induction of Lambdoid
Prophages in Escherichia Coli Cultures: Simple Procedures with Possible Biotechnological
Applications. BMC Biotechnol., 2001, 1, 1. https://doi.org/10.1186/1472-6750-1-1.
[82] Brussow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From
Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev., 2004, 68 (3), 560–
602. https://doi.org/10.1128/MMBR.68.3.560-602.2004.
[83] Fortier, L.; Sekulovic, O. Importance of Prophages to Evolution and Virulence of Bacterial
Pathogens. Virulence, 2013, 4 (5), 354–365. https://doi.org/10.4161/viru.24498.
[84] Łoś, M.; Węgrzyn, G. Pseudolysogeny. In Advances in Virus Research; 2012; Vol. 82, pp 339–349.
https://doi.org/10.1016/B978-0-12-394621-8.00019-4.
[85] Cenens, W.; Makumi, A.; Mebrhatu, M. T.; Lavigne, R.; Aertsen, A. Phage–Host Interactions during
Pseudolysogeny. Bacteriophage, 2013, 3 (1), e25029. https://doi.org/10.4161/bact.25029.
[86] Ackermann, H. W. Phage Classification and Characterization. In Bacteriophages: Methods and
Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M. R. J., Kropinski, A.
M., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2009; Vol. 501, pp 127 –140.
https://doi.org/10.1007/978-1-60327-164-6.
[87] Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A Genome-Based Taxonomy for Phage. J.
Bacteriol., 2002, 184 (16), 4529–4535. https://doi.org/10.1128/JB.184.16.4529-4535.2002.
[88] Adriaenssens, E. M.; Rodney Brister, J. How to Name and Classify Your Phage: An Informal Guide.

Chapter 1: General Introduction
44
Viruses, 2017, 9 (4), 1–9. https://doi.org/10.3390/v9040070.
[89] Lefkowitz, E. J.; Dempsey, D. M.; Hendrickson, R. C.; Orton, R. J.; Siddell, S. G.; Smith, D. B.
Virus Taxonomy: The Database of the International Committee on Taxonomy of Viruses (ICTV).
Nucleic Acids Res., 2018, 46 (D1), D708–D717. https://doi.org/10.1093/nar/gkx932.
[90] Meier-Kolthoff, J. P.; Göker, M. VICTOR: Genome-Based Phylogeny and Classification of
Prokaryotic Viruses. Bioinformatics, 2017, 33 (21), 3396–3404.
https://doi.org/10.1093/bioinformatics/btx440.
[91] Gorbalenya, A. E.; Krupovic, M.; Mushegian, A.; Kropinski, A. M.; Siddell, S. G.; Varsani, A.;
Adams, M. J.; Davison, A. J.; Dutilh, B. E.; Harrach, B.; et al. The New Scope of Virus Taxonomy:
Partitioning the Virosphere into 15 Hierarchical Ranks. Nat. Microbiol., 2020, 5 (5), 668–674.
https://doi.org/10.1038/s41564-020-0709-x.
[92] Barylski, J.; Enault, F.; Dutilh, B. E.; Schuller, M. B.; Edwards, R. A.; Gillis, A.; Klumpp, J.;
Knezevic, P.; Krupovic, M.; Kuhn, J. H.; et al. Analysis of Spounaviruses as a Case Study for the
Overdue Reclassification of Tailed Phages. Syst. Biol., 2019, 69 (1), 110–123.
https://doi.org/10.1093/sysbio/syz036.
[93] Casjens, S. R.; Thuman-Commike, P. A. Evolution of Mosaically Related Tailed Bacteriophage
Genomes Seen through the Lens of Phage P22 Virion Assembly. Virology, 2011, 411 (2), 393–415.
https://doi.org/10.1016/j.virol.2010.12.046.
[94] Lopes, A.; Tavares, P.; Petit, M. A.; Guérois, R.; Zinn-Justin, S. Automated Classification of Tailed
Bacteriophages According to Their Neck Organization. BMC Genomics, 2014, 15, 1027.
https://doi.org/10.1186/1471-2164-15-1027.
[95] Wilhem, S. W.; Poorvin, L. Quantification of Algal Viruses in Marine Samples. In Methods in
Microbiology; Paul, J. H., Ed.; 2001; Vol. 30, pp 53–65. https://doi.org/10.1016/S0580-
9517(01)30039-9.
[96] Anderson, B.; Rashid, M. H.; Carter, C.; Pasternack, G.; Rajanna, C.; Revazishvili, T.; Dean, T.;
Senecal, A.; Sulakvelidze, A. Enumeration of Bacteriophage Particles. Bacteriophage, 2011, 1 (2),
86–93. https://doi.org/10.4161/bact.1.2.15456.
[97] Ács, N.; Gambino, M.; Brøndsted, L. Bacteriophage Enumeration and Detection Methods. Front.
Microbiol., 2020, 11, 594868. https://doi.org/10.3389/fmicb.2020.594868.
[98] Snyder, J. C.; Spuhler, J.; Wiedenheft, B.; Roberto, F. F.; Douglas, T.; Young, M. J. Effects of
Culturing on the Population Structure of a Hyperthermophilic Virus. Microb. Ecol., 2004, 48 (4),
561–566. https://doi.org/10.1007/s00248-004-0246-9.
[99] Hatfull, G. F. Mycobacteriophages: Windows into Tuberculosis. PLoS Pathog., 2014, 10 (3), 12–14.
https://doi.org/10.1371/journal.ppat.1003953.
[100] Olsen, N. S.; Hendriksen, N. B.; Hansen, L. H.; Kot, W. A New High-Throughput Screening Method
for Phages: Enabling Crude Isolation and Fast Identification of Diverse Phages with Therapeutic
Potential. Phage, 2020, 1 (3), 137–148. https://doi.org/10.1089/phage.2020.0016.
[101] Mokili, J. L.; Rohwer, F.; Dutilh, B. E. Metagenomics and Future Perspectives in Virus Discovery.
Curr. Opin. Virol., 2012, 2 (1), 63–77. https://doi.org/10.1016/j.coviro.2011.12.004.
[102] Bharti, R.; Grimm, D. G. Current Challenges and Best-Practice Protocols for Microbiome Analysis.
Brief. Bioinform., 2021, 22 (1), 178–193. https://doi.org/10.1093/bib/bbz155.
[103] De Maio, N.; Shaw, L. P.; Hubbard, A.; George, S.; Sanderson, N. D.; Swann, J.; Wick, R.; Oun, M.
A.; Stubberfield, E.; Hoosdally, S. J.; et al. Comparison of Long-Read Sequencing Technologies in
the Hybrid Assembly of Complex Bacterial Genomes. Microb. Genomics, 2019, 5 (9), e000294.
https://doi.org/10.1099/mgen.0.000294.
[104] Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front.
Genet., 2019, 10, 426. https://doi.org/10.3389/fgene.2019.00426.

Chapter 1: General Introduction
45
[105] Tyler, A. D.; Mataseje, L.; Urfano, C. J.; Schmidt, L.; Antonation, K. S.; Mulvey, M. R.; Corbett, C.
R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome
Sequencing Applications. Sci. Rep., 2018, 8, 10931. https://doi.org/10.1038/s41598-018-29334-5.
[106] Roumpeka, D. D.; Wallace, R. J.; Escalettes, F.; Fotheringham, I.; Watson, M. A Review of
Bioinformatics Tools for Bio-Prospecting from Metagenomic Sequence Data. Front. Genet., 2017,
8, 23. https://doi.org/10.3389/fgene.2017.00023.
[107] Pearman, W. S.; Freed, N. E.; Silander, O. K. Testing the Advantages and Disadvantages of Short-
And Long- Read Eukaryotic Metagenomics Using Simulated Reads. BMC Bioinformatics, 2020, 21
(1), 220. https://doi.org/10.1186/s12859-020-3528-4.
[108] Gawad, C.; Koh, W.; Quake, S. R. Single-Cell Genome Sequencing: Current State of the Science.
Nat. Rev. Genet., 2016, 17 (3), 175–188. https://doi.org/10.1038/nrg.2015.16.
[109] Paul, J. H.; Sullivan, M. B.; Segall, A. M.; Rohwer, F. Marine Phage Genomics. Comp. Biochem.
Physiol. - B Biochem. Mol. Biol., 2002, 133 (4), 463–476. https://doi.org/10.1016/S1096-
4959(02)00168-9.
[110] Sullivan, M. B.; Coleman, M. L.; Quinlivan, V.; Rosenkrantz, J. E.; DeFrancesco, A. S.; Ta n, G.; Fu,
R.; Lee, J. A.; Waterbury, J. B.; Bielawski, J. P.; et al. Portal Protein Diversity and Phage Ecology.
Environ. Microbiol., 2008, 10 (10), 2810–2823. https://doi.org/10.1111/j.1462-2920.2008.01702.x.
[111] Adriaenssens, E. M.; Cowan, D. A. Using Signature Genes as Tools To Assess Environmental Viral
Ecology and Diversity. Appl. Environ. Microbiol., 2014, 80 (15), 4470–4480.
https://doi.org/10.1128/AEM.00878-14.
[112] Ackermann, H. W. Tailed Bacteriophages: The Order Caudovirales. Adv. Virus Res., 1998, 51, 135–
201.
[113] Roy, A.; Bhardwaj, A.; Datta, P.; Lander, G. C.; Cingolani, G. Small Terminase Couples Viral DNA
Binding to Genome-Packaging ATPase Activity. Structure, 2012, 20 (8), 1403–1413.
https://doi.org/10.1016/j.str.2012.05.014.
[114] Zhong, Y.; Chen, F.; Wilhelm, S. W.; Poorvin, L.; Hodson, R. E. Phylogenetic Diversity of Marine
Cyanophage Isolates and Natural Virus Communities as Revealed by Sequences of Viral Capsid
Assembly Protein Gene G20. In Applied and Environmental Microbiology; 2001; Vol. 30, pp 1576–
1584. https://doi.org/10.1128/AEM.68.4.1576-1584.2002.
[115] Comeau, A. M.; Krisch, H. M. The Capsid of the T4 Phage Superfamily: The Evolution, Diversity,
and Structure of Some of the Most Prevalent Proteins in the Biosphere. Mol. Biol. Evol., 2008, 25
(7), 1321–1332. https://doi.org/10.1093/molbev/msn080.
[116] Filée, J.; Tétart, F.; Suttle, C. A.; Krisch, H. M. Marine T4-Type Bacteriophages, a Ubiquitous
Component of the Dark Matter of the Biosphere. Proc. Natl. Acad. Sci. U. S. A., 2005, 102 (35),
12471–12476. https://doi.org/10.1073/pnas.0503404102.
[117] Dwivedi, B.; Schmieder, R.; Goldsmith, D. B.; Edwards, R. A.; Breitbart, M. PhiSiGns: An Online
Tool to Identify Signature Genes in Phages and Design PCR Primers for Examining Phage Diversity.
BMC Bioinformatics, 2012, 13 (1), 37. https://doi.org/10.1186/1471-2105-13-37.
[118] Gołębiewski, M.; Tretyn, A. Generating Amplicon Reads for Microbial Community Assessment
with Next-Generation Sequencing. J. Appl. Microbiol., 2019, 128 (2), 330–354.
https://doi.org/10.1111/jam.14380.
[119] Hsieh, S. Y.; Tariq, M. A.; Telatin, A.; Ansorge, R.; Adriaenssens, E. M.; Savva, G. M.; Booth, C.;
Wileman, T.; Hoyles, L.; Carding, S. R. Comparison of PCR versus PCR-Free DNA Library
Preparation for Characterising the Human Faecal Virome. Viruses, 2021, 13 (10), 2093.
https://doi.org/10.3390/v13102093.
[120] Mavrich, T. N.; Hatfull, G. F. Bacteriophage Evolution Differs by Host, Lifestyle and Genome. Nat.
Microbiol., 2017, 2, 17112. https://doi.org/10.1038/nmicrobiol.2017.112.

Chapter 1: General Introduction
46
[121] Glazko, G.; Makarenkov, V.; Liu, J.; Mushegian, A. Evolutionary History of Bacteriophages with
Double-Stranded DNA Genomes. Biol. Direct, 2007, 2 (36), 1–14. https://doi.org/10.1186/1745-
6150-2-36.
[122] De Paepe, M.; Hutinet, G.; Son, O.; Amarir-Bouhram, J.; Schbath, S.; Petit, M. A. Temperate Phages
Acquire DNA from Defective Prophages by Relaxed Homologous Recombination: The Role of
Rad52-Like Recombinases. PLoS Genet., 2014, 10 (3).
https://doi.org/10.1371/journal.pgen.1004181.
[123] Botstein, D. A Theory of Modular Evolution for Bacteriophages. Ann. N. Y. Acad. Sci., 1980, 354,
484–491. https://doi.org/10.1111/j.1749-6632.1980.tb27987.x.
[124] Labrie, S. J.; Moineau, S. Abortive Infection Mechanisms and Prophage Sequences Significantly
Influence the Genetic Makeup of Emerging Lytic Lactococcal Phages. J. Bacteriol., 2007, 189 (4),
1482–1487. https://doi.org/10.1128/JB.01111-06.
[125] Lawrence, J. G.; Hatfull, G. F.; Hendrix, R. W. Imbroglios of Viral Taxonomy: Genetic Exchange
and Failings of Phenetic Approaches. J. Bacteriol., 2002, 184 (17), 4891–4905.
https://doi.org/10.1128/JB.184.17.4891-4905.2002.
[126] Hendrix, R. W. Bacteriophages: Evolution of the Majority. Theor. Popul. Biol., 2002, 61 (4), 471–
480. https://doi.org/10.1006/tpbi.2002.1590.
[127] Pedulla, M. L.; Ford, M. E.; Houtz, J. M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J. A.; Jacobs-
Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N. R.; et al. Origins of Highly Mosaic Mycobacteriophage
Genomes. Cell, 2003, 113 (2), 171–182. https://doi.org/10.1016/S0092-8674(03)00233-2.
[128] Moura de Sousa, J. A.; Pfeifer, E.; Touchon, M.; Rocha, E. P. C. Causes and Consequences of
Bacteriophage Diversification via Genetic Exchanges across Lifestyles and Bacterial Taxa. Mol.
Biol. Evol., 2021, 38 (6), 2497–2512. https://doi.org/10.1093/molbev/msab044.
[129] Pope, W. H.; Bowman, C. A.; Russell, D. A.; Jacobs-Sera, D.; Asai, D. J.; Cresawn, S. G.; Jacobs,
W. R.; Hendrix, R. W.; Lawrence, J. G.; Hatfull, G. F. Whole Genome Comparison of a Large
Collection of Mycobacteriophages Reveals a Continuum of Phage Genetic Diversity. Elife, 2015, 4,
e06416. https://doi.org/10.7554/elife.06416.
[130] Viertel, T. M.; Ritter, K.; Horz, H. P. Viruses versus Bacteria -Novel Approaches to Phage Therapy
as a Tool against Multidrug-Resistant Pathogens. J. Antimicrob. Chemother., 2014, 69 (9), 2326–
2336. https://doi.org/10.1093/jac/dku173.
[131] Rohde, C.; Wittmann, J.; Kutter, E. Bacteriophages: A Therapy Concept against Multi-Drug-
Resistant Bacteria. Surg. Infect. (Larchmt)., 2018, 19 (8), 737–744.
https://doi.org/10.1089/sur.2018.184.
[132] Kortright, K. E.; Chan, B. K.; Koff, J. L.; Turner, P. E. Phage Therapy: A Renewed Approach to
Combat Antibiotic-Resistant Bacteria. Cell Host Microbe, 2019, 25 (2), 219–232.
https://doi.org/10.1016/j.chom.2019.01.014.
[133] Lu, T. K.; Koeris, M. S. The next Generation of Bacteriophage Therapy. Curr. Opin. Microbiol.,
2011, 14 (5), 524–531. https://doi.org/10.1016/j.mib.2011.07.028.
[134] Gibb, B.; Hyman, P.; Schneider, C. L. The Many Applications of Engineered Bacteriophages—An
Overview. Pharmaceuticals, 2021, 14 (7), 634. https://doi.org/10.3390/ph14070634.
[135] Hatfull, G. F.; Hendrix, R. W. Bacteriophages and Their Genomes. Curr. Opin. Virol., 2011, 1 (4),
298–303. https://doi.org/10.1016/j.coviro.2011.06.009.
[136] Hayes, S.; Mahony, J.; Nauta, A.; Van Sinderen, D. Metagenomic Approaches to Assess
Bacteriophages in Various Environmental Niches. Viruses, 2017, 9 (6), 127.
https://doi.org/10.3390/v9060127.
[137] Ofir, G.; Sorek, R. Contemporary Phage Biology: From Classic Models to New Insights. Cell, 2018,
172 (6), 1260–1270. https://doi.org/10.1016/j.cell.2017.10.045.

Chapter 1: General Introduction
47
[138] Harada, L. K.; Silva, E. C.; Campos, W. F.; Del Fiol, F. S.; Vila, M.; Dąbrowska, K.; Krylov, V. N.;
Balcão, V. M. Biotechnological Applications of Bacteriophages: State of the Art. Microbiol. Res.,
2018, 212–213, 38–58. https://doi.org/10.1016/j.micres.2018.04.007.
[139] Moye, Z. D.; Woolston, J.; Sulakvelidze, A. Bacteriophage Applications for Food Production and
Processing. Viruses, 2018, 10 (4), 205. https://doi.org/10.3390/v10040205.
[140] Nikolich, M. P.; Filippov, A. A. Bacteriophage Therapy: Developments and Directions. Antibiotics,
2020, 9 (3). https://doi.org/10.3390/antibiotics9030135.
[141] Monk, A. B.; Rees, C. D.; Barrow, P.; Hagens, S.; Harper, D. R. Bacteriophage Applications: Where
Are We Now? Lett. Appl. Microbiol., 2010, 51 (4), 363–369. https://doi.org/10.1111/j.1472-
765X.2010.02916.x.
[142] Toribio-Avedillo, D.; Blanch, A. R.; Muniesa, M.; Rodríguez-Rubio, L. Bacteriophages as Fecal
Pollution Indicators. Viruses, 2021, 13 (6), 1089. https://doi.org/10.3390/v13061089.
[143] Fair, R. J.; Tor, Y. Perspectives in Medicinal Chemistry Antibiotics and Bacterial Resistance in the
21st Century. Perspect. Medicin. Chem., 2014, 25–64.
https://doi.org/10.4137/PMC.S14459.Received.
[144] Loc-Carrillo, C.; Abedon, S. T. Pros and Cons of Phage Therapy. Bacteriophage, 2011, 1 (2), 111–
114. https://doi.org/10.4161/bact.1.2.14590.
[145] Principi, N.; Silvestri, E.; Esposito, S. Advantages and Limitations of Bacteriophages for the
Treatment of Bacterial Infections. Front. Pharmacol., 2019, 10, 513.
https://doi.org/10.3389/fphar.2019.00513.
[146] Sulakvelidze, A.; Barrow, P. Phage Therapy in Animals and Agribusiness. In Bacteriophages:
Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, 2005; pp
335–380.
[147] Brüssow, H. Phage Therapy: The Escherichia Coli Experience. Microbiology, 2005, 151 (7), 2133–
2140. https://doi.org/10.1099/mic.0.27849-0.
[148] Ferriol-González, C.; Domingo-Calap, P. Phages for Biofilm Removal. Antibiotics, 2020, 9 (5), 268.
https://doi.org/10.3390/antibiotics9050268.
[149] Harper, D. R.; Parracho, H. M. R. T.; Walker, J.; Sharp, R.; Hughes, G.; Werthén, M.; Lehman, S.;
Morales, S. Bacteriophages and Biofilms. Antibiotics, 2014, 3 (3), 270–284.
https://doi.org/10.3390/antibiotics3030270.
[150] Sharma, U.; Vipra, A.; Channabasappa, S. Phage-Derived Lysins as Potential Agents for Eradicating
Biofilms and Persisters. Drug Discov. Today, 2018, 23 (4), 848–856.
https://doi.org/10.1016/j.drudis.2018.01.026.
[151] Hyman, P.; Abedon, S. T. Bacteriophage Host Range and Bacterial Resistance. In Advances in
Applied Microbiolog; Elsevier Inc., 2010; Vol. 70, pp 217–248. https://doi.org/10.1016/S0065-
2164(10)70007-1.
[152] Kutateladze, M.; Adamia, R. Bacteriophages as Potential New Therapeutics to Replace or
Supplement Antibiotics. Trends Biotechnol., 2010, 28 (12), 591–595.
https://doi.org/10.1016/j.tibtech.2010.08.001.
[153] Wright, R. C. T.; Friman, V. P.; Smith, M. C. M.; Brockhurst, M. A. Resistance Evolution against
Phage Combinations Depends on the Timing and Order of Exposure. MBio, 2019, 10 (5), e01652-
19. https://doi.org/10.1128/mBio.01652-19.
[154] Buckling, A.; Rainey, P. B. Antagonistic Coevolution between a Bacterium and a Bacteriophage.
Proc. R. Soc. B Biol. Sci., 2002, 269 (1494), 931–936. https://doi.org/10.1098/rspb.2001.1945.
[155] Laanto, E.; Hoikkala, V.; Ravantti, J.; Sundberg, L.-R. Long-Term Genomic Coevolution of Host-
Parasite Interaction in the Natural Environment. Nat. Commun., 2017, 8, 111.

Chapter 1: General Introduction
48
https://doi.org/10.1038/s41467-017-00158-7.
[156] León, M.; Bastías, R. Virulence Reduction in Bacteriophage Resistant Bacteria. Front. Microbiol.,
2015, 6 (343), 1–7. https://doi.org/10.3389/fmicb.2015.00343.
[157] Chan, B. K.; Abedon, S. T. Phage Therapy Pharmacology: Phage Cocktails. In Advances in Applied
Microbiology; Elsevier Inc., 2012; Vol. 78, pp 1–23. https://doi.org/10.1016/B978-0-12-394805-
2.00001-4.
[158] Bull, J. J.; Gill, J. J. The Habits of Highly Effective Phages: Population Dynamics as a Framework
for Identifying Therapeutic Phages. Front. Microbiol., 2014, 5 (618).
https://doi.org/10.3389/fmicb.2014.00618.
[159] Skurnik, M.; Pajunen, M.; Kiljunen, S. Biotechnological Challenges of Phage Therapy. Biotechnol.
Lett., 2007, 29 (7), 995–1003. https://doi.org/10.1007/s10529-007-9346-1.
[160] Gill, J.; Hyman, P. Phage Choice, Isolation, and Preparation for Phage Therapy. Curr. Pharm.
Biotechnol., 2010, 11 (1), 2–14. https://doi.org/10.2174/138920110790725311.
[161] Bretaudeau, L.; Tremblais, K.; Aubrit, F.; Meichenin, M.; Arnaud, I. Good Manufacturing Practice
(GMP) Compliance for Phage Therapy Medicinal Products. Front. Microbiol., 2020, 11, 1161.
https://doi.org/10.3389/fmicb.2020.01161.
[162] Dedrick, R. M.; Jacobs-Sera, D.; Bustamante, C. A. G.; Garlena, R. A.; Mavrich, T. N.; Pope, W.
H.; Reyes, J. C. C.; Russell, D. A.; Adair, T.; Alvey, R.; et al. Prophage-Mediated Defence against
Viral Attack and Viral Counter-Defence. Nat. Microbiol., 2017, 2, 16251.
https://doi.org/10.1038/nmicrobiol.2016.251.
[163] Monteiro, R.; Pires, D. P.; Costa, A. R.; Azeredo, J. Phage Therapy: Going Temperate? Trends
Microbiol., 2019, 27 (4), 368–378. https://doi.org/10.1016/j.tim.2018.10.008.
[164] Goodridge, L. Designing Phage Therapeutics. Curr. Pharm. Biotechnol., 2010, 11 (1), 15–27.
https://doi.org/10.2174/138920110790725348.
[165] Lu, T. K.; Collins, J. J. Dispersing Biofilms with Engineered Enzymatic Bacteriophage. Proc. Natl.
Acad. Sci. U. S. A., 2007, 104 (27), 11197–11202. https://doi.org/10.1073/pnas.0704624104.
[166] Ando, H.; Lemire, S.; Pires, D. P.; Lu, T. K. Engineering Modular Viral Scaffolds for Targeted
Bacterial Population Editing. Cell Syst., 2015, 1 (3), 187–196.
https://doi.org/10.1016/j.cels.2015.08.013.
[167] Lemire, S.; Yehl, K. M.; Lu, T. K. Phage-Based Applications in Synthetic Biology. Annu. Rev. Virol.,
2018, 5 (1), 453–476. https://doi.org/10.1146/annurev-virology-092917-043544.
[168] Hargreaves, K. R.; Clokie, M. R. J. Clostridium Difficile Phages: Still Difficult? Front. Microbiol.,
2014, 5, 184. https://doi.org/10.3389/fmicb.2014.00184.
[169] Meile, S.; Du, J.; Dunne, M.; Kilcher, S.; Loessner, M. J. Engineering Therapeutic Phages for
Enhanced Antibacterial Efficacy. Curr. Opin. Virol., 2022, 52, 182–191.
https://doi.org/10.1016/j.coviro.2021.12.003.
[170] Pires, D. P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T. K. Genetically Engineered Phages: A
Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev., 2016, 80 (3), 523–543.
https://doi.org/10.1128/mmbr.00069-15.
[171] Sulakvelidze, A.; Alavidze, Z.; Morris, J. G. Bacteriophage Therapy. Antimicrob. Agents
Chemother., 2001, 45 (3), 649–659. https://doi.org/10.1128/AAC.45.3.649-659.2001.
[172] Roach, D. R.; Leung, C. Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J. P.; Weitz, J. S.;
Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for
Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe, 2017, 22 (1),
38-47.e4. https://doi.org/10.1016/j.chom.2017.06.018.

Chapter 1: General Introduction
49
[173] Van Belleghem, J.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.; Bollyky, P. Interactions between
Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses, 2018, 11 (1), 10.
https://doi.org/10.3390/v11010010.
[174] Abedon, S. T.; Kuhl, S. J.; Blasdel, B. G.; Kutter, E. M. Phage Treatment of Human Infections.
Bacteriophage, 2011, 1 (2), 66–85. https://doi.org/10.4161/bact.1.2.15845.
[175] Labrie, S. J.; Samson, J. E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev.
Microbiol., 2010, 8 (5), 317–327. https://doi.org/10.1038/nrmicro2315.
[176] Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy.
Viruses, 2018, 10 (7). https://doi.org/10.3390/v10070351.
[177] Taati Moghadam, M.; Amirmozafari, N.; Shariati, A.; Hallajzadeh, M.; Mirkalantari, S.;
Khoshbayan, A.; Masjedian Jazi, F. How Phages Overcome the Challenges of Drug Resistant
Bacteria in Clinical Infections. Infect. Drug Resist., 2020, 13, 45–61.
https://doi.org/10.2147/IDR.S234353.
[178] Nobrega, F. L.; Costa, A. R.; Kluskens, L. D.; Azeredo, J. Revisiting Phage Therapy: New
Applications for Old Resources. Trends Microbiol., 2015, 23 (4), 185–191.
https://doi.org/10.1016/j.tim.2015.01.006.
[179] Tanji, Y.; Shimada, T.; Yoichi, M.; Miyanaga, K.; Hori, K.; Unno, H. Toward Rational Control of
Escherichia Coli O157:H7 by a Phage Cocktail. Appl. Microbiol. Biotechnol., 2004, 64 (2), 270–
274. https://doi.org/10.1007/s00253-003-1438-9.
[180] Örmälä, A.-M.; Jalasvuori, M. Phage Therapy: Should Bacterial Resistance to Phages Be a Concern,
Even in the Long Run? Bacteriophage, 2013, 3 (1), e24219. https://doi.org/10.4161/bact.24219.
[181] Chan, B. K.; Abedon, S. T.; Loc-Carrillo, C. Phage Cocktails and the Future of Phage Therapy.
Future Microbiol., 2013, 8 (6), 769–783. https://doi.org/10.2217/fmb.13.47.
[182] McCallin, S.; Sacher, J. C.; Zheng, J.; Chan, B. K. Current State of Compassionate Phage Therapy.
Viruses, 2019, 11 (4), 343. https://doi.org/10.3390/v11040343.
[183] Yerushalmy, O.; Khalifa, L.; Gold, N.; Rakov, C.; Alkalay-Oren, S.; Adler, K.; Ben-Porat, S.;
Kraitman, R.; Gronovich, N.; Ginat, K. S.; et al. The Israeli Phage Bank (IPB). Antibiotics, 2020, 9
(5), 269. https://doi.org/10.3390/antibiotics9050269.
[184] Tsonos, J.; Oosterik, L. H.; Tuntufye, H. N.; Klumpp, J.; Butaye, P.; De Greve, H.; Hernalsteens, J.
P.; Lavigne, R.; Goddeeris, B. M. A Cocktail of in Vitro Efficient Phages Is Not a Guarantee for in
Vivo Therapeutic Results against Avian Colibacillosis. Vet. Microbiol., 2014.
https://doi.org/10.1016/j.vetmic.2013.10.021.
[185] Hyman, P.; Abedon, S. T. Practical Methods for Determining Phage Growth Parameters. In
Bacteriophages. Methods in Molecular Biology; Clokie, M. R. J., Kropinski, A. M., Eds.; Methods
in Molecular Biology; Humana Press: Totowa, NJ, 2009; Vol. 501, pp 175 –202.
https://doi.org/10.1007/978-1-60327-164-6.
[186] T. Abedon, S. Phage “Delay” towards Enhancing Bacterial Escape from Biofilms: A More
Comprehensive Way of Viewing Resistance to Ba cteriophages. AIMS Microbiol., 2017, 3 (2), 186–
226. https://doi.org/10.3934/microbiol.2017.2.186.
[187] Casey, E.; van Sinderen, D.; Mahony, J. In Vitro Characteristics of Phages to Guide ‘Real Life’
Phage Therapy Suitability. Viruses, 2018, 10 (4), 163. https://doi.org/10.3390/v10040163.
[188] Levin, B. R.; Bull, J. J. Population and Evolutionary Dynamics of Phage Therapy. Nat. Rev.
Microbiol., 2004, 2 (2), 166–173. https://doi.org/10.1038/nrmicro822.
[189] Pirnay, J. P.; Verbeken, G.; Ceyssens, P. J.; Huys, I.; de Vos, D.; Ameloot, C.; Fauconnier, A. The
Magistral Phage. Viruses, 2018, 10 (2), 64. https://doi.org/10.3390/v10020064.
[190] Mangalea, M. R.; Duerkop, B. A. Fitness Trade-Offs Resulting from Bacteriophage Resistance

Chapter 1: General Introduction
50
Potentiate Synergistic Antibacterial Strategies. Infect. Immun., 2020, 88 (7), e00926-19.
https://doi.org/10.1128/IAI.00926-19.
[191] Huff, W. E.; Huff, G. R.; Rath, N. C.; Balog, J. M.; Donoghue, A. M. Therapeutic Efficacy of
Bacteriophage and Baytril (Enrofloxacin) Individually and in Combination to Treat Colibacillosis in
Broilers. Poult. Sci., 2004, 83 (12), 1944–1947. https://doi.org/10.1093/ps/83.12.1944.
[192] Lu, T. K.; Collins, J. J. Engineered Bacteriophage Targeting Gene Networks as Adjuvants for
Antibiotic Therapy. Proc. Natl. Acad. Sci., 2009, 106 (12), 4629–4634.
https://doi.org/10.1073/pnas.0800442106.
[193] Koskella, B.; Brockhurst, M. A. Bacteria –Phage Coevolution as a Driver of Ecological and
Evolutionary Processes in Microbial Communities. FEMS Microbiol. Rev., 2014, 38 (5), 916–931.
https://doi.org/10.1111/1574-6976.12072.
[194] Secor, P. R.; Dandekar, A. A. More than Simple Parasites: The Sociobiology of Bacteriophages and
Their Bacterial Hosts. MBio, 2020, 11 (2), 1–13. https://doi.org/10.1128/mBio.00041-20.
[195] Weinbauer, M. G. Ecology of Prokaryotic Viruses. FEMS Microbiol. Rev., 2004, 28 (2), 127–181.
https://doi.org/10.1016/j.femsre.2003.08.001.
[196] Chaturongakul, S.; Ounjai, P. Phage-Host Interplay: Examples from Tailed Phages and Gram -
Negative Bacterial Pathogens. Front. Microbiol., 2014, 5, 442.
https://doi.org/10.3389/fmicb.2014.00442.
[197] Sausset, R.; Petit, M. A.; Gaboriau-Routhiau, V.; De Paepe, M. New Insights into Intestinal Phages.
Mucosal Immunol., 2020, 13 (2), 205–215. https://doi.org/10.1038/s41385-019-0250-5.
[198] Obeng, N.; Pratama, A. A.; Elsas, J. D. van. The Significance of Mutualistic Phages for Bacterial
Ecology and Evolution. Trends Microbiol., 2016, 24 (6), 440–449.
https://doi.org/10.1016/j.tim.2015.12.009.
[199] Labrie, S. J.; Moineau, S. Phosphorylation, an Altruistic Bacterial Trick to Halt Phages. Cell Host
Microbe, 2016, 20 (4), 409–410. https://doi.org/10.1016/j.chom.2016.09.017.
[200] Chibani-chennoufi, S.; Bruttin, A.; Brüssow, H.; Dillmann, M.; Bru, H. Phage-Host Interaction : An
Ecological Perspective. J. Bacteriol., 2004, 186 (12), 3677–3686.
https://doi.org/10.1128/JB.186.12.3677.
[201] Juliano, S. A. Population Dynamics. J. Am. Mosq. Control Assoc., 2007, 23 (2 Suppl), 265–275.
https://doi.org/10.2987/8756-971X(2007)23[265:PD]2.0.CO;2.
[202] Santos, S. B.; Carvalho, C.; Azeredo, J.; Ferreira, E. C. Population Dynamics of a Salmonella Lytic
Phage and Its Host: Implications of the Host Bacterial Growth Rate in Modelling. PLoS One, 2014,
9 (7), e102507. https://doi.org/10.1371/journal.pone.0102507.
[203] Krysiak-Baltyn, K.; Martin, G. J. O.; Stickland, A. D.; Scales, P. J.; Gras, S. L. Computational
Models of Populations of Bacteria and Lytic Phage. Crit. Rev. Microbiol., 2016, 42 (6), 942–968.
https://doi.org/10.3109/1040841X.2015.1114466.
[204] Bohannan, B. J. M.; Lenski, R. D. Linking Genetic Change to Community Evolution: Insights from
Studies of Bacteria and Bacteriophage. Ecol. Lett., 2000, 3 (4), 362–377.
https://doi.org/https://doi.org/10.1046/j.1461-0248.2000.00161.x.
[205] Weissman, J. L.; Holmes, R.; Barrangou, R.; Moineau, S.; Fagan, W. F.; Levin, B.; Johnson, P. L.
F. Immune Loss as a Driver of Coexistence during Host-Phage Coevolution. ISME J., 2018, 12 (2),
585–597. https://doi.org/10.1038/ismej.2017.194.
[206] Roucourt, B.; Lavigne, R. The Role of Interactions between Phage and Bacterial Proteins within the
Infected Cell: A Diverse and Puzzling Interactome. Environ. Microbiol., 2009, 11 (11), 2789–2805.
https://doi.org/10.1111/j.1462-2920.2009.02029.x.
[207] Mahmoudabadi, G.; Milo, R.; Phillips, R. Energetic Cost of Building a Virus. Proc. Natl. Acad. Sci.

Chapter 1: General Introduction
51
U. S. A., 2017, 114 (22), E4324–E4333. https://doi.org/10.1073/pnas.1701670114.
[208] Stern, A.; Sorek, R. The Phage-Host Arms Race: Shaping the Evolution of Microbes. BioEssays,
2011, 33 (1), 43–51. https://doi.org/10.1002/bies.201000071.
[209] Holguín, A.; Cárdenas, P.; Prada -Peñaranda, C.; Rabelo Leite, L.; Buitrago, C.; Clavijo, V.; Oliveira,
G.; Leekitcharoenphon, P.; Møller Aarestrup, F.; Vives, M. Host Resistance, Genomics and
Population Dynamics in a Salmonella Enteritidis and Phage System . Viruses, 2019, 11 (2), 188.
https://doi.org/10.3390/v11020188.
[210] Gómez, P.; Buckling, A. Bacteria -Phage Antagonistic Coevolution in Soil. Science (80-. )., 2011,
332 (6025), 106–109. https://doi.org/10.1126/science.1198767.
[211] Gorter, F. A.; Scanlan, P. D.; Buckling, A. Adaptation to Abiotic Conditions Drives Local Adaptation
in Bacteria and Viruses Coevolving in Heterogeneous Environments. Biol. Lett., 2016, 12, 20150879.
https://doi.org/10.1098/rsbl.2015.0879.
[212] Storms, Z. J.; Sauvageau, D. Modeling Tailed Bacteriophage Adsorption: Insight into Mechanisms.
Virology, 2015, 485, 355–362. https://doi.org/10.1016/j.virol.2015.08.007.
[213] Storms, Z. J.; Teel, M. R.; Mercurio, K.; Sauvageau, D. The Virulence Index: A Metric for
Quantitative Analysis of Phage Virulence. Phage, 2020, 1 (1), 27–36.
https://doi.org/10.1089/phage.2019.0001.
[214] Cairns, B. J.; Timms, A. R.; Jansen, V. A. A.; Connerton, I. F.; Payne, R. J. H. Quantitative Models
of in Vitro Bacteriophage-Host Dynamics and Their Application to Phage Therapy. PLoS Pathog.,
2009, 5 (1), e1000253. https://doi.org/10.1371/journal.ppat.1000253.
[215] Yehl, K.; Lemire, S.; Yang, A. C.; Ando, H.; Mimee, M.; Torres, M. D. T.; de la Fuente-Nunez, C.;
Lu, T. K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail
Fiber Mutagenesis. Cell, 2019, 179 (2), 459-469.e9. https://doi.org/10.1016/j.cell.2019.09.015.
[216] Gabig, M.; Herman-Antosiewicz, A.; Kwiatkowska, M.; Los, M.; Thomas, M. S.; Wȩgrzyn, G. The
Cell Surface Protein Ag43 Facilitates Phage Infection of Escherichia Coli in the Presence of Bile
Salts and Carborhydrates. Microbiology, 2002, 148 (5), 1533–1542.
https://doi.org/10.1099/00221287-148-5-1533.
[217] Park, S. H.; Biswas, D.; Lingbeck, J.; Koo, O. K.; Ricke, S. C. Enhancement of Chicken Macrophage
Cytokine Response to Salmonella Typhimurium When Combined with Bacteriophage P22. FEMS
Microbiol. Lett., 2013, 339 (2), 137–144. https://doi.org/10.1111/1574-6968.12064.
[218] Carroll-Portillo, A.; Lin, H. C. Bacteriophage and the Innate Immune System: Access and Signaling.
Microorganisms, 2019, 7 (12), 625. https://doi.org/10.3390/microorganisms7120625.
[219] Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage–Host
Interactions. Viruses, 2019, 11 (6), 567. https://doi.org/10.3390/v11060567.
[220] Vale, P. F.; Little, T. J. CRISPR-Mediated Phage Resistance and the Ghost of Coevolution Past.
Proc. R. Soc. Biol. Sci., 2010, 277 (1691), 2097–2103. https://doi.org/10.1098/rspb.2010.0055.
[221] Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak‐Amikam, Y.; Afik, S.; Ofir,
G.; Sorek, R. <scp>BREX</Scp> Is a Novel Phage Resistance System Widespread in Microbial
Genomes. EMBO J., 2015, 34 (2), 169–183. https://doi.org/10.15252/embj.201489455.
[222] Ofir, G.; Melamed, S.; Sberro, H.; Mukamel, Z.; Silverman, S.; Yaakov, G.; Doron, S.; Sorek, R.
DISARM Is a Widespread Bacterial Defence System with Broad Anti-Phage Activities. Nat.
Microbiol., 2018, 3 (1), 90–98. https://doi.org/10.1038/s41564-017-0051-0.
[223] Swanson, N. A.; Cingolani, G. A Tail of Phage Adhesins. Structure, 2018, 26 (12), 1565–1567.
https://doi.org/10.1016/j.str.2018.11.008.
[224] Letarov, A. V.; Kulikov, E. E. Adsorption of Bacteriophages on Bacterial Cells. Biochem., 2017, 82
(13), 1632–1658. https://doi.org/10.1134/S0006297917130053.

Chapter 1: General Introduction
52
[225] Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host Receptors for Bacteriophage Adsorption. FEMS
Microbiol. Lett., 2016, 363 (4), 1–11. https://doi.org/10.1093/femsle/fnw002.
[226] Islam, M. Z.; Fokine, A.; Mahalingam, M.; Zhang, Z.; Garcia -Doval, C.; Van Raaij, M. J.; Rossmann,
M. G.; Rao, V. B. Molecular Anatomy of the Receptor Binding Module of a Bacteriophage Long
Tail Fiber. PLoS Pathog., 2019, 15 (12), e1008193. https://doi.org/10.1371/journal.ppat.1008193.
[227] Maffei, E.; Shaidullina, A.; Burkolter, M.; Heyer, Y.; Estermann, F.; Druelle, V.; Sauer, P.; Willi,
L.; Michaelis, S.; Hilbi, H.; et al. Systematic Exploration of Escherichia Coli Phage–Host
Interactions with the BASEL Phage Collection. PLOS Biol., 2021, 19 (11), e3001424.
https://doi.org/10.1371/journal.pbio.3001424.
[228] Wu, L. T.; Chang, S. Y.; Yen, M. R.; Yang, T. C.; Tseng, Y. H. Characterization of Extended -Host-
Range Pseudo-T-Even Bacteriophage Kpp95 Isolated on Klebsiella Pneumoniae. Appl. Environ.
Microbiol., 2007, 73 (8), 2532–2540. https://doi.org/10.1128/AEM.02113-06.
[229] Nobrega, F. L.; Vlot, M.; de Jonge, P. A.; Dreesens, L. L.; Beaumont, H. J. E.; Lavigne, R.; Dutilh,
B. E.; Brouns, S. J. J. Targeting Mechanisms of Tailed Bacteriophages. Nat. Rev. Microbiol., 2018,
16 (12), 760–773. https://doi.org/10.1038/s41579-018-0070-8.
[230] Dimitriu, T.; Szczelkun, M. D.; Westra, E. R. Evolutionary Ecology and Interplay of Prokaryotic
Innate and Adaptive Immune Systems. Curr. Biol., 2020, 30 (19), R1189–R1202.
https://doi.org/10.1016/j.cub.2020.08.028.
[231] Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to
Cellular Defense. Microbiol. Mol. Biol. Rev., 2013, 77 (1), 53–72.
https://doi.org/10.1128/mmbr.00044-12.
[232] Samson, J. E.; Magadán, A. H.; Sabri, M.; Moineau, S. Revenge of the Phages: Defeating Bacterial
Defences. Nat. Rev. Microbiol., 2013, 11 (10), 675–687. https://doi.org/10.1038/nrmicro3096.
[233] Nechaev, S.; Severinov, K. The Elusive Object of Desire—Interactions of Bacteriophages and Their
Hosts. Curr. Opin. Microbiol., 2008, 11 (2), 186–193. https://doi.org/10.1016/j.mib.2008.02.009.
[234] Lopatina, A.; Tal, N.; Sorek, R. Abortive Infection: Bacterial Suicide as an Antiviral Immune
Strategy. Annu. Rev. Virol., 2020, 7 (1), 371–384. https://doi.org/10.1146/annurev-virology-011620-
040628.
[235] Fineran, P. C.; Blower, T. R.; Foulds, I. J.; Humphreys, D. P.; Lilley, K. S.; Salmond, G. P. C. The
Phage Abortive Infection System, ToxIN, Functions as a Protein–RNA Toxin–Antitoxin Pair. Proc.
Natl. Acad. Sci., 2009, 106 (3), 894–899. https://doi.org/10.1073/pnas.0808832106.
[236] Gerdes, K.; Christensen, S. K.; Løbner-Olesen, A. Prokaryotic Toxin-Antitoxin Stress Response
Loci. Nat. Rev. Microbiol., 2005, 3 (5), 371–382. https://doi.org/10.1038/nrmicro1147.
[237] Chen, B.; Akusobi, C.; Fang, X.; Salmond, G. P. C. Environmental T4-Family Bacteriophages
Evolve to Escape Abortive Infection via Multiple Routes in a Bacterial Host Employing “Altruistic
Suicide” through Type III Toxin-Antitoxin Systems. Front. Microbiol., 2017, 8 (MAY), 1–11.
https://doi.org/10.3389/fmicb.2017.01006.
[238] Blower, T. R.; Chai, R.; Przybilski, R.; Chindhy, S.; Fang, X.; Kidman, S. E.; Tan, H.; Luisi, B. F.;
Fineran, P. C.; Salmond, G. P. C. Evolution of Pectobacterium Bacteriophage ΦM1 To Escape Two
Bifunctional Type III Toxin-Antitoxin and Abortive Infection Systems through Mutations in a Single
Viral Gene. Appl. Environ. Microbiol., 2017, 83 (8), 1–17. https://doi.org/10.1128/AEM.03229-16.
[239] Seed, K. D. Battling Phages: How Bacteria Defend against Viral Attack. PLOS Pathog., 2015, 11
(6), e1004847. https://doi.org/10.1371/journal.ppat.1004847.
[240] Blower, T. R.; Evans, T. J.; Przybilski, R.; Fineran, P. C.; Salmond, G. P. C. Viral Evasion of a
Bacterial Suicide System by RNA–Based Molecular Mimicry Enables Infectious Altruism. PLoS
Genet., 2012, 8 (10), e1003023. https://doi.org/10.1371/journal.pgen.1003023.
[241] Blower, T. R.; Short, F. L.; Fineran, P. C.; Salmond, G. P. C. Viral Molecular Mimicry Circumvents

Chapter 1: General Introduction
53
Abortive Infection and Suppresses Bacterial Suicide to Make Hosts Permissive for Replication.
Bacteriophage, 2012, 2 (4), e23830. https://doi.org/10.4161/bact.23830.
[242] Pourcel, C.; Touchon, M.; Villeriot, N.; Vernadet, J.-P.; Couvin, D.; Toffano-Nioche, C.; Vergnaud,
G. CRISPRCasdb a Successor of CRISPRdb Containing CRISPR Arrays and Cas Genes from
Complete Genome Sequences, and Tools to Download and Query Lists of Repeats and Spacers.
Nucleic Acids Res., 2020, 48 (D1), D535–D544. https://doi.org/10.1093/nar/gkz915.
[243] Barrangou, R.; Marraffini, L. A. CRISPR-Cas Systems: Prokaryotes Upgrade to Adaptive Immunity.
Mol. Cell, 2014, 54 (2), 234–244. https://doi.org/10.1016/j.molcel.2014.03.011.
[244] Sorek, R.; Lawrence, C. M.; Wiedenheft, B. CRISPR-Mediated Adaptive Immune Systems in
Bacteria and Archaea. Annu. Rev. Biochem., 2013, 82 (1), 237–266. https://doi.org/10.1146/annurev-
biochem-072911-172315.
[245] Jackson, S. A.; McKenzie, R. E.; Fagerlund, R. D.; Kieper, S. N.; Fineran, P. C.; Brouns, S. J. J.
CRISPR-Cas: Adapting to Change. Science (80-. )., 2017, 356 (6333).
https://doi.org/10.1126/science.aal5056.
[246] van der Oost, J.; Westra, E. R.; Jackson, R. N.; Wiedenheft, B. Unravelling the Struc tural and
Mechanistic Basis of CRISPR–Cas Systems. Nat. Rev. Microbiol., 2014, 12 (7), 479–492.
https://doi.org/10.1038/nrmicro3279.
[247] Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D. a;
Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science (80-. ).,
2007, 315 (5819), 1709–1712. https://doi.org/10.1126/science.1138140.
[248] Fineran, P. C. Resistance Is Not Futile: Bacterial ‘Innate’ and CRISPR -Cas ‘Adaptive’ Immune
Systems. Microbiology, 2019, 165 (8), 834–841. https://doi.org/10.1099/mic.0.000802.
[249] Stanley, S. Y.; Maxwell, K. L. Phage-Encoded Anti-CRISPR Defenses. Annu. Rev. Genet., 2018, 52
(1), 445–464. https://doi.org/10.1146/annurev-genet-120417-031321.
[250] Li, Y.; Bondy-Denomy, J. Anti-CRISPRs Go Viral: The Infection Biology of CRISPR-Cas
Inhibitors. Cell Host Microbe, 2021, 29 (5), 704–714. https://doi.org/10.1016/j.chom.2020.12.007.
[251] Bondy-Denomy, J.; Pawluk, A.; Maxwell, K. L.; Davidson, A. R. Bacteriophage Genes That
Inactivate the CRISPR/Cas Bacterial Immune System. Nature, 2013, 493 (7432), 429–432.
https://doi.org/10.1038/nature11723.
[252] Marino, N. D.; Zhang, J. Y.; Borges, A. L.; Sousa, A. A.; Leon, L. M.; Rauch, B. J.; Walton, R. T.;
Berry, J. D.; Joung, J. K.; Kleinstiver, B. P.; et al. Discovery of Widespread Type I and Type V
CRISPR-Cas Inhibitors. Science (80-. )., 2018, 362 (6411), 240–242.
https://doi.org/10.1126/science.aau5174.
[253] Hynes, A. P.; Rousseau, G. M.; Agudelo, D.; Goulet, A.; Amigues, B.; Loehr, J.; Romero, D. A.;
Fremaux, C.; Horvath, P.; Doyon, Y.; et al. Widespread Anti-CRISPR Proteins in Virulent
Bacteriophages Inhibit a Range of Cas9 Proteins. Nat. Commun., 2018, 9 (1), 1–10.
https://doi.org/10.1038/s41467-018-05092-w.
[254] Borges, A. L.; Zhang, J. Y.; Rollins, M. F.; Osuna, B. A.; Wiedenheft, B.; Bondy-denomy, J.
Bacteriophage Cooperation Suppresses CRISPR- Article Bacteriophage Cooperation Suppresses
CRISPR-Cas3 and Cas9 Immunity. Cell, 2018, 174 (4), 917-925.e10.
https://doi.org/10.1016/j.cell.2018.06.013.
[255] Chevallereau, A.; Meaden, S.; Fradet, O.; Gandon, S.; Houte, S. Van; Westra, E. R.; Chevallereau,
A.; Meaden, S.; Fradet, O.; Landsberger, M.; et al. Exploitation of the Cooperative Behaviors of
Anti- Article Exploitation of the Cooperative Behaviors of Anti-CRISPR Phages. Cell Host Microbe,
2020, 27 (2), 189-198.e6. https://doi.org/10.1016/j.chom.2019.12.004.
[256] Landsberger, M.; Gandon, S.; Meaden, S.; Rollie, C.; Chevallereau, A.; Chabas, H.; Buckling, A.;
Westra, E. R.; van Houte, S. Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity.

Chapter 1: General Introduction
54
Cell, 2018, 174 (4), 908-916.e12. https://doi.org/10.1016/j.cell.2018.05.058.
[257] Bradde, S.; Mora, T.; Walczak, A. M. Cost and Benefits of Clustered Regularly Interspaced Short
Palindromic Repeats Spacer Acquisition. Philos. Trans. R. Soc. B Biol. Sci., 2019, 374 (1772).
https://doi.org/10.1098/rstb.2018.0095.
[258] Vale, P. F.; Lafforgue, G.; Gatchitch, F.; Gardan, R.; Moineau, S.; Gandon, S. Costs of CRISPR -
Cas-Mediated Resistance in Streptococcus Thermophilus. Proc. R. Soc. B Biol. Sci., 2015, 282
(1812), 20151270. https://doi.org/10.1098/rspb.2015.1270.
[259] Hesse, S.; Rajaure, M.; Wall, E.; Johnson, J.; Bliskovsky, V.; Gottesman, S.; Adhya, S. Phage
Resistance in Multidrug-Resistant Klebsiella Pneumoniae ST258 Evolves via Diverse Mutations
That Culminate in Impaired Adsorption. MBio, 2020, 11 (1), e02530-19.
https://doi.org/10.1128/mBio.02530-19.
[260] Welch, R. A. The Genus Escherichia. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E.,
Schleifer, K.-H., Stackebrandt, E., Eds.; Springer, New York, NY, 2006; Vol. 6: Proteob, pp 60–71.
https://doi.org/110.1007/0-387-30746-x_9.
[261] Reshes, G.; Vanounou, S.; Fishov, I.; Feingold, M. Cell Shape Dynamics in Escherichia Coli.
Biophys. J., 2008, 94 (1), 251–264. https://doi.org/10.1529/biophysj.107.104398.
[262] Whitman, W. B.; Coleman, D. C.; Wiebe, W. J. Prokaryotes: The Unseen Majority . Proc. Natl. Acad.
Sci., 1998, 95 (12), 6578–6583. https://doi.org/10.1073/pnas.95.12.6578.
[263] Gordon, D. M.; Bauer, S.; Johnson, J. R. The Genetic Structure of Escherichia Coli Populations in
Primary and Secondary Habitats. Microbiology, 2002, 148 (5), 1513–1522.
https://doi.org/10.1099/00221287-148-5-1513.
[264] Savageau, M. A. Escherichia Coli Habitats, Cell Types, and Molecular Mechanisms of Gene Control.
Am. Nat., 1983, 122 (6), 732–744. https://doi.org/10.1086/284168.
[265] Kaper, J. B.; Nataro, J. P.; Mobley, H. L. T. Pathogenic Escherichia Coli. Nat. Rev. Microbiol., 2004,
2 (2), 123–140. https://doi.org/10.1038/nrmicro818.
[266] Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The Population Genetics of Commensal
Escherichia Coli. Nat. Rev. Microbiol., 2010, 8 (3), 207–217. https://doi.org/10.1038/nrmicro2298.
[267] Denamur, E.; Picard, B.; Tenaillon, O. Population Genetics of Pathogenic Escherichia Coli. In
Bacterial Population Genetics in Infectious Disease; Robinson, D. A., Falush, D., Feil, E. J., Eds.;
John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp 269–286.
https://doi.org/10.1002/9780470600122.ch14.
[268] Nolan, L. K.; Barnes, H. J.; Vaillancourt, J.; Abdul-aziz, T.; Logue, C. M. Colibacillosis. In Diseases
of Poultry; Swayne, D. E., Ed.; John Wiley & Sons, Inc., 2013; pp 751–805.
https://doi.org/10.1002/9781119421481.
[269] Kathayat, D.; Lokesh, D.; Ranjit, S.; Rajashekara, G. Avian Pathogenic Escherichia Coli (APEC):
An Overview of Virulence and Pathogenesis Factors, Zoonotic Potential, and Control Strategies.
Pathogens, 2021, 10 (4), 467. https://doi.org/10.3390/pathogens10040467.
[270] Lutful Kabir, S. M. Avian Colibacillosis and Salmonellosis: A Closer Look at Epidemiology,
Pathogenesis, Diagnosis, Control and Public Health Concerns. Int. J. Environ. Res. Public Health,
2010, 7 (1), 89–114. https://doi.org/10.3390/ijerph7010089.
[271] Dziva, F.; Stevens, M. P. Colibacillosis in Poultry: Unravelling the Molecular Basis of Virulence of
Avian Pathogenic Escherichia Coli in Their Natural Hosts. Avian Pathol., 2008, 37 (4), 355–366.
https://doi.org/10.1080/03079450802216652.
[272] Vandemaele, F.; Vereecken, M.; Derijcke, J.; Goddeeris, B. M. Incidence and Antiobiotic Resistance
of Pathogenic Escherichia Coli among Poultry in Belgium. Vet. Rec., 2002, 151 (12), 355–356.
https://doi.org/10.1136/vr.151.12.355.

Chapter 1: General Introduction
55
[273] Ewers, C.; Antão, E. M.; Diehl, I.; Philipp, H. C.; Wieler, L. H. Intestine and Environment of the
Chicken as Reservoirs for Extraintestinal Pathogenic Escherichia Coli Strains with Zoonotic
Potential. Appl. Environ. Microbiol., 2009, 75 (1), 184–192. https://doi.org/10.1128/AEM.01324-08.
[274] Macklin, K. S.; Hess, J. B.; Bilgili, S. F.; Norton, R. A. Bacterial Levels of Pine Shavings and Sand
Used as Poultry Litter. J. Appl. Poult. Res., 2005, 14 (2), 238–245.
https://doi.org/10.1093/japr/14.2.238.
[275] Sarowska, J.; Futoma-Koloch, B.; Jama-Kmiecik, A.; Frej-Madrzak, M.; Ksiazczyk, M.; Bugla -
Ploskonska, G.; Choroszy-Krol, I. Virulence Factors, Prevalence and Potential Transmission of
Extraintestinal Pathogenic Escherichia Coli Isolated from Different Sources: Recent Reports. Gut
Pathog., 2019, 11, 10. https://doi.org/10.1186/s13099-019-0290-0.
[276] Rodriguez-Siek, K. E.; Giddings, C. W.; Doetkott, C.; Johnson, T. J.; Nolan, L. K. Characterizing
the APEC Pathotype. Vet. Res., 2005, 36 (2), 241–256. https://doi.org/10.1051/vetres:2004057.
[277] Vandemaele, F. J.; Mugasa, J. P.; Vandekerchove, D.; Goddeeris, B. M. Predominance of the PapGII
Allele with High Sequence Homology to That of Human Isolates among Avian Pathogenic
Escherichia Coli (APEC). Vet. Microbiol., 2003, 97 (3–4), 245–257.
https://doi.org/10.1016/j.vetmic.2003.09.017.
[278] McCarter, J. D.; Stephens, D.; Shoemaker, K.; Rosenberg, S.; Kirsch, J. F.; Georgiou, G. Substrate
Specificity of the Escherichia Coli Outer Membrane Protease OmpT. J. Bacteriol., 2004, 186 (17),
5919–5925. https://doi.org/10.1128/JB.186.17.5919-5925.2004.
[279] Hejair, H. M. A.; Ma, J.; Zhu, Y.; Sun, M.; Dong, W.; Zhang, Y.; Pan, Z.; Zhang, W.; Yao, H. Role
of Outer Membrane Protein T in Pathogenicity of Avian Pathogenic Escherichia Coli. Res. Vet. Sci.,
2017, 115, 109–116. https://doi.org/10.1016/j.rvsc.2017.01.026.
[280] Torres, A. G.; Redford, P.; Welch, R. A.; Payne, S. M. TonB-Dependent Systems of Uropathogenic
Escherichia Coli: Aerobactin and Heme Tra nsport and TonB Are Required for Virulence in the
Mouse. Infect. Immun., 2001, 69 (10), 6179–6185. https://doi.org/10.1128/IAI.69.10.6179-
6185.2001.
[281] Sabri, M.; Caza, M.; Proulx, J.; Lymberopoulos, M. H.; Bree, A.; Moulin-Schouleur, M.; Curtiss, R.;
Dozois, C. M. Contribution of the SitABCD, MntH, and FeoB Metal Transporters to the Virulence
of Avian Pathogenic Escherichia Coli O78 Strain Χ7122. Infect. Immun., 2008, 76 (2), 601–611.
https://doi.org/10.1128/IAI.00789-07.
[282] Murase, K.; Martin, P.; Porcheron, G.; Houle, S.; Helloin, E.; Pénary, M.; Nougayrède, J. P.; Dozois,
C. M.; Hayashi, T.; Oswald, E. HlyF Produced by Extraintestinal Pathogenic Escherichia Coli Is a
Virulence Factor That Regulates Outer Membrane Vesicle Biogenesis. J. Infect. Dis., 2015, 212 (11),
856–865. https://doi.org/10.1093/infdis/jiv506.
[283] Janßen, T.; Schwarz, C.; Preikschat, P.; Voss, M.; Philipp, H. C.; Wieler, L. H. Virulence-Associated
Genes in Avian Pathogenic Escherichia Coli (APEC) Isolated from Internal Organs of Poultry
Having Died from Colibacillosis. Int. J. Med. Microbiol., 2001, 291 (5), 371–378.
https://doi.org/10.1078/1438-4221-00143.
[284] Li, Y.; Dai, J.; Zhuge, X.; Wang, H.; Hu, L.; Ren, J.; Chen, L.; Li, D.; Tang, F. Iron-Regulated Gene
IreA in Avian Pathogenic Escherichia Coli Participates in Adhesion and Stress-Resistance. BMC Vet.
Res., 2016, 12 (1), 167. https://doi.org/10.1186/s12917-016-0800-y.
[285] Johnson, T. J.; Wannemuehler, Y.; Doetkott, C.; Johnson, S. J.; Rosenberger, S. C.; Nolan, L. K.
Identification of Minimal Predictors of Avian Pathogenic Escherichia Coli Virulence for Use as a
Rapid Diagnostic Tool. J. Clin. Microbiol., 2008, 46 (12), 3987–3996.
https://doi.org/10.1128/JCM.00816-08.
[286] Schubert, S.; Rakin, A.; Karch, H.; Carniel, E. Prevalence of the “ High-Pathogenicity Island ” of
Yersinia Species among Escherichia Coli Strains That Are Pathogenic to Humans. Infect. Immun.,
1998, 66 (2), 480–485.

Chapter 1: General Introduction
56
[287] Daines, D. A.; Wright, L. F.; Chaffin, D. O.; Rubens, C. E.; Silver, R. P. NeuD Plays a Role in the
Synthesis of Sialic Acid in Escherichia Coli K1. FEMS Microbiol. Lett., 2000, 189 (2), 281–284.
https://doi.org/10.1111/j.1574-6968.2000.tb09244.x.
[288] Zuo, J.; Tu, C.; Wang, Y.; Qi, K.; Hu, J.; Wang, Z.; Mi, R.; Yan huang; Chen, Z.; Han, X. The Role
of the Wzy Gene in Lipopolysaccharide Biosynthesis and Pathogenesis of Avian Pathogenic
Escherichia Coli. Microb. Pathog., 2019, 127, 296–303.
https://doi.org/10.1016/j.micpath.2018.12.021.
[289] Nolan, L. K.; Vaillancourt, J.; Barbieri, N. L.; Logue, C. M. Colibacillosis. In Diseases of Poultry;
Swayne, D. E., Boulianne, M., Logue, C. M., McDougald, L. R., Nair, V., Suarez, D. L., Wit, S.,
Grimes, T., Johnson, D., Kromm, M., et al., Eds.; Wiley, 2020; pp 770–830.
https://doi.org/10.1002/9781119371199.ch18.
[290] Johnson, T. J.; Wannemeuhler, Y. M.; Scaccianoce, J. A.; Johnson, S. J.; Nolan, L. K. Complete
DNA Sequence, Comparative Genomics, and Prevalence of an IncHI2 Plasmid Occurring among
Extraintestinal Pathogenic Escherichia Coli Isolates. Antimicrob. Agents Chemother., 2006, 50 (11),
3929–3933. https://doi.org/10.1128/AAC.00569-06.
[291] Johnson, T. J.; Johnson, S. J.; Nolan, L. K. Complete DNA Sequence of a ColBM Plasmid from
Avian Pathogenic Escherichia Coli Suggests That It Evolved from Closely Related ColV Virulence
Plasmids. J. Bacteriol., 2006, 188 (16), 5975–5983. https://doi.org/10.1128/JB.00204-06.
[292] Johnson, T. J.; Wannemuehler, Y. M.; Johnson, S. J.; Logue, C. M.; White, D. G.; Doetkott, C.;
Nolan, L. K. Plasmid Replicon Typing of Commensal and Pathogenic Escherichia Coli Isolates.
Appl. Environ. Microbiol., 2007, 73 (6), 1976–1983. https://doi.org/10.1128/AEM.02171-06.
[293] Johnson, T. J.; Siek, K. E.; Johnson, S. J.; Nolan, L. K. DNA Sequence of a ColV Plasmid and
Prevalence of Selected Plasmid-Encoded Virulence Genes among Avian Escherichia Coli Strains. J.
Bacteriol., 2006, 188 (2), 745–758. https://doi.org/10.1128/JB.188.2.745-758.2006.
[294] Ginns, C. A.; Benham, M. L.; Adams, L. M.; Whithear, K. G.; Bettelheim, K. A.; Crabb, B. S.;
Browning, G. F. Colonization of the Respiratory Tract by a Virulent Strain of Avian Escherichia Coli
Requires Carriage of a Conjugative Plasmid. Infect. Immun., 2000, 68 (3), 1535–1541.
https://doi.org/10.1128/IAI.68.3.1535-1541.2000.
[295] Tivendale, K. A.; Allen, J. L.; Ginns, C. A.; Crabb, B. S.; Browning, G. F. Association of Iss and
IucA, but Not Tsh, with Plasmid-Mediated Virulence of Avian Pathogenic Escherichia Coli. Infect.
Immun., 2004, 72 (11), 6554–6560. https://doi.org/10.1128/IAI.72.11.6554.
[296] Mellata, M.; Dho-Moulin, M.; Dozois, C. M.; Curtiss, R.; Brown, P. K.; Arné, P.; Brée, A.;
Desautels, C.; Fairbrother, J. M. Role of Virulence Factors in Resistance of Avian Pathogenic
Escherichia Coli to Serum and in Pathogenicity. Infect. Immun., 2003, 71 (1), 536–540.
https://doi.org/10.1128/IAI.71.1.536-540.2003.
[297] Gibbs, P. S.; Maurer, A. J. J.; Nolan, B. L. K.; Wooleyc, R. E. Prediction of Chicken Embryo
Lethality with the Avian Escherichia Coli Traits Complement Resistance, Co licin V Production, and
Presence of the Increased Serum Survival Gene Cluster (Iss). Avian Dis., 2003, 47 (2), 370–379.
[298] Johnson, T. J.; Giddings, C. W.; Horne, S. M.; Gibbs, P. S.; Wooley, R. E.; Skyberg, J.; Olah, P.;
Kercher, R.; Sherwood, J. S.; Foley, S. L.; et al. Location of Increased Serum Survival Gene and
Selected Virulence Traits on a Conjugative R Plasmid in an Avian Escherichia Coli Isolate. Avian
Dis., 2002, 46 (2), 342–352. https://doi.org/10.1637/0005-2086(2002)046[0342:loissg]2.0.co;2.
[299] Mellata, M.; Ameiss, K.; Mo, H.; Curtiss, R. Characterization of the Contribution to Virulence of
Three Large Plasmids of Avian Pathogenic Escherichia Coli Χ7122 (O78:K80:H9). Infect. Immun.,
2010, 78 (4), 1528–1541. https://doi.org/10.1128/IAI.00981-09.
[300] Dozois, C. M.; Dho-Moulin, M.; Brée, A.; Fairbrother, J. M.; Desautels, C.; Curtiss, R. Relationship
between the Tsh Autotransporter and Pathogenicity of Avian Escherichia Coli and Localization and
Analysis of the Tsh Genetic Region. Infect. Immun., 2000, 68 (7), 4145–4154.
https://doi.org/10.1128/IAI.68.7.4145-4154.2000.

Chapter 1: General Introduction
57
[301] Skyberg, J. A.; Johnson, T. J.; Johnson, J. R.; Clabots, C.; Logue, C. M.; Nolan, L. K. Acquisition of
Avian Pathogenic Escherichia Coli Plasmids by a Commensal E. Coli I solate Enhances Its Abilities
to Kill Chicken Embryos, Grow in Human Urine, and Colonize the Murine Kidney. Infect. Immun.,
2006, 74 (11), 6287–6292. https://doi.org/10.1128/IAI.00363-06.
[302] McPeake, S. J. W.; Smyth, J. A.; Ball, H. J. Characterisation of Avian Pathogenic Escherichia Coli
(APEC) Associated with Colisepticaemia Compared to Faecal Isolates from Healthy Birds. Vet.
Microbiol., 2005, 110 (3–4), 245–253. https://doi.org/10.1016/j.vetmic.2005.08.001.
[303] Kawano, M.; Yaguchi, K.; Osawa, R. Genotypic Analyses of Escherichia Coli Isolated from
Chickens with Colibacillosis and Apparently Healthy Chickens in Japan. Microbiol. Immunol., 2006,
50 (12), 961–966. https://doi.org/10.1111/j.1348-0421.2006.tb03872.x.
[304] Kauffmann, F. The Serology of the Coli Group. J Immunol., 1947, 57 (1), 71–100.
[305] DebRoy, C.; Fratamico, P. M.; Yan, X.; Baranzoni, G. M.; Liu, Y.; Needleman, D. S.; Tebbs, R.;
O’Connell, C. D.; Allred, A.; Swimley, M.; et al. Comparison of O-Antigen Gene Clusters of All O-
Serogroups of Escherichia Coli and Proposal for Adopting a New Nomenclature for O-Typing. PLoS
One, 2016, 11 (1), 1–13. https://doi.org/10.1371/journal.pone.0147434.
[306] Stenutz, R.; Weintraub, A.; Widmalm, G. The Structures of Escherichia Coli O-Polysaccharide
Antigens. FEMS Microbiol. Rev., 2006, 30 (3), 382–403. https://doi.org/10.1111/j.1574-
6976.2006.00016.x.
[307] Huja, S.; Oren, Y.; Trost, E.; Brzuszkiewicz, E.; Biran, D.; Blom, J.; Goesmann, A.; Gottschalk, G.;
Hacker, J.; Ron, E. Z.; et al. Genomic Avenue to Avian Colisepticemia. MBio, 2015, 6 (1), 1–13.
https://doi.org/10.1128/mBio.01681-14.
[308] Zhao, S.; Maurer, J. J.; Hubert, S.; De Villena, J. F.; McDermott, P. F.; Meng, J.; Ayers, S.; English,
L.; White, D. G. Antimicrobial Susceptibility and Molecular Characterization of Avian Pathogenic
Escherichia Coli Isolates. Vet. Microbiol., 2005, 107 (3–4), 215–224.
https://doi.org/10.1016/j.vetmic.2005.01.021.
[309] Blanco, J.; Blanco, M.; Mora, A.; WH, J.; García, V.; Vá zquez, M. Serotypes of E Coli Isolated from
Septicaemic Chickens in Galicia (Northwest Spain). Vet. Microbiol., 1998, 61 (3), 229–235.
[310] Maiden, M. C. J.; Bygraves, J. A.; Feil, E.; Morelli, G.; Russell, J. E.; Urwin, R.; Zhang, Q.; Zhou,
J.; Zurth, K.; Caugant, D. A.; et al. Multilocus Sequence Typing: A Portable Approach to the
Identification of Clones within Populations of Pathogenic Microorganisms. Proc. Natl. Acad. Sci.,
1998, 95 (6), 3140–3145. https://doi.org/10.1073/pnas.95.6.3140.
[311] Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L. H.; Karch, H.; Reeves, P. R.; Maiden,
M. C. J.; Ochman, H.; et al. Sex and Virulence in Escherichia Coli: An Evolutionary Perspective.
Mol. Microbiol., 2006, 60 (5), 1136–1151. https://doi.org/10.1111/j.1365-2958.2006.05172.x.
[312] Mardis, E. R. The Impact of Next-Generation Sequencing Technology on Genetics. Trends Genet.,
2008, 24 (3), 133–141. https://doi.org/10.1016/j.tig.2007.12.007.
[313] Fratamico, P. M.; DebRoy, C.; Liu, Y.; Needleman, D. S.; Baranzoni, G. M.; Feng, P. Advances in
Molecular Serotyping and Subtyping of Escherichia Coli†. Front. Microbiol., 2016, 7 (644).
https://doi.org/10.3389/fmicb.2016.00644.
[314] Joensen, K. G.; Tetzschner, A. M. M.; Iguchi, A.; Aarestrup, F. M.; Scheutz, F. Rapid and Easy in
Silico Serotyping of Escherichia Coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin.
Microbiol., 2015, 53 (8), 2410–2426. https://doi.org/10.1128/JCM.00008-15.
[315] Collett, S. R.; Smith, J. A.; Boulianne, M.; Owen, R. L.; Gingerich, E.; Singer, R. S.; Johnson, T. J.;
Hofacre, C. L.; Berghaus, R. D.; Stewart‐Brown, B. Principles of Disease Prevention, Diagnosis, and
Control. In Diseases of Poultry; Swayne, D. E., Boulianne, M., Logue, C. M., McDougald, L. R.,
Nair, V., Suarez, D. L., Wit, S., Grimes, T., Johnson, D., Kromm, M., et al., Eds.; Wiley, 2020; pp
1–78. https://doi.org/10.1002/9781119371199.ch1.

Chapter 1: General Introduction
58
[316] Christensen, H.; Bachmeier, J.; Bisgaard, M. New Strategies to Prevent and Control Avian
Pathogenic Escherichia Coli (APEC). Avian Pathol., 2021, 50 (5), 370–381.
https://doi.org/10.1080/03079457.2020.1845300.
[317] Swelum, A. A.; Elbestawy, A. R.; El-Saadony, M. T.; Hussein, E. O. S.; Alhotan, R.; Suliman, G.
M.; Taha, A. E.; Ba-Awadh, H.; El-Tarabily, K. A.; Abd El-Hack, M. E. Ways to Minimize Bacterial
Infections, with Special Reference to Escherichia Coli, to Cope with the First -Week Mortality in
Chicks: An Updated Overview. Poult. Sci., 2021, 100 (5), 101039.
https://doi.org/10.1016/j.psj.2021.101039.
[318] Caekebeke, N.; Jonquiere, F. J.; Ringenier, M.; Tobias, T. J.; Postma, M.; van den Hoogen, A.;
Houben, M. A. M.; Velkers, F. C.; Sleeckx, N.; Stegeman, J. A.; et al. Comparing Farm Biosecurity
and Antimicrobial Use in High-Antimicrobial-Consuming Broiler and Pig Farms in the Belgian–
Dutch Border Region. Front. Vet. Sci., 2020, 7, 558455. https://doi.org/10.3389/fvets.2020.558455.
[319] U.S. Food and Drug Administration. Summary Report On Antimicrobials Sold or Distributed for Use
in Food-Producing Animals; 2020.
[320] Tang, K. L.; Caffrey, N. P.; Nóbrega, D. B.; Cork, S. C.; Ronksley, P. E.; Barkema, H. W.; Polachek,
A. J.; Ganshorn, H.; Sharma, N.; Kellner, J. D.; et al. Restricting the Use of Antibiotics in Food-
Producing Animals and Its Associations with Antibiotic Resistance in Food-Producing Animals and
Human Beings: A Systematic Review and Meta -Analysis. Lancet Planet. Heal., 2017, 1 (8), e316–
e327. https://doi.org/10.1016/S2542-5196(17)30141-9.
[321] Oliveira, A.; Sereno, R.; Azeredo, J. In Vivo Efficiency Evaluation of a Phage Cocktail in Controlling
Severe Colibacillosis in Confined Conditions and Experimental Poultry Houses. Vet. Microbiol.,
2010, 146 (3–4), 303–308. https://doi.org/10.1016/j.vetmic.2010.05.015.
[322] Li, H.; Ma, M. L.; Xie, H. J.; Kong, J. Biosafety Evaluation of Bacteriophages for Treatment of
Diarrhea Due to Intestinal Pathogen Escherichia Coli 3-2 Infection of Chickens. World J. Microbiol.
Biotechnol., 2012, 28 (1), 1–6. https://doi.org/10.1007/s11274-011-0784-5.
[323] Huff, W. E.; Huff, G. R.; Rath, N. C.; Donoghue, A. M. Method of Administration Affects the Ability
of Bacteriophage to Prevent Colibacillosis in 1-Day-Old Broiler Chickens. Poult. Sci., 2013, 92 (4),
930–934. https://doi.org/10.3382/ps.2012-02916.
[324] Naghizadeh, M.; Torshizi, M. A. K.; Rahimi, S.; Dalgaard, T. S. Synergistic Effect of Phage Therapy
Using a Cocktail Rather than a Single Phage in the Control of Severe Colibacillosis in Quails. Poult.
Sci., 2019, 98 (2), 653–663. https://doi.org/10.3382/ps/pey414.
[325] Huff, W. E.; Huff, G. R.; Rath, N. C.; Balog, J. M.; Xie, H.; Moore, P. A.; Donoghue, A. M.
Prevention of Escherichia Coli Respiratory Infection in Broiler Chickens with Bacteriophage
(SPR02). Poult. Sci., 2002, 81 (4), 437–441. https://doi.org/10.1093/ps/81.4.437.
[326] Wernicki, A.; Nowaczek, A.; Urban-Chmiel, R. Bacteriophage Therapy to Combat Bacterial
Infections in Poultry. Virol. J., 2017, 14 (1), 179. https://doi.org/10.1186/s12985-017-0849-7.
[327] El-Gohary, F. A.; Huff, W. E.; Huff, G. R.; Rath, N. C.; Zhou, Z. Y.; Donoghue, A. M. Environmental
Augmentation with Bacteriophage Prevents Colibacillosis in Broiler Chickens. Poult. Sci., 2014, 93
(11), 2788–2792. https://doi.org/10.3382/ps.2014-04282.
[328] Barrow, P.; Lovell, M.; Berchieri, A. Use of Lytic Bacteriophage for Control of Experimental
Escherichia Coli Septicemia and Meningitis in Chickens and Calves. Clin. Diagn. Lab. Immunol.,
1998, 5 (3), 294–298.
[329] Huff, W. E.; Huff, G. R.; Rath, N. C.; Balog, J. M.; Donoghue, A. M. Prevention of Escherichia Coli
Infection in Broiler Chickens with a Bacteriophage Aerosol Spray. Poult. Sci., 2002, 81 (10), 1486–
1491. https://doi.org/10.1093/ps/81.10.1486.
[330] Huff, W. E.; Huff, G. R.; Rath, N. C.; Balog, J. M.; Donoghue, A. M. Alternatives to Antibiotics:
Utilization of Bacteriophage to Treat Colibacillosis and Prevent Foodborne Pathogens. Poult. Sci.,
2005, 84 (4), 655–659. https://doi.org/10.1093/ps/84.4.655.

Chapter 1: General Introduction
59
[331] Ryan, E. M.; Gorman, S. P.; Donnelly, R. F.; Gilmore, B. F. Recent Advances in Bacteriophage
Therapy: How Delivery Routes, Formulation, Concentration and Timing Influence the Success of
Phage Therapy. J. Pharm. Pharmacol., 2011, 63 (10), 1253–1264. https://doi.org/10.1111/j.2042-
7158.2011.01324.x.
[332] Pirnay, J.-P.; Ferry, T.; Resch, G. Recent Progress toward the Implementation of Phage Therapy in
Western Medicine. FEMS Microbiol. Rev., 2021, fuab040. https://doi.org/10.1093/femsre/fuab040.
[333] Huff, G.; Huff, W.; Rath, N.; Donoghue, A. Critical Evaluation of Bacteriophage to Prevent and
Treat Colibacillosis in Poultry. J. Ark. Acad. Sci., 2009, 63 (1), 93–98.
[334] Żbikowska, K.; Michalczuk, M.; Dolka, B. The Use of Bacteriophages in the Poultry Industry.
Animals, 2020, 10 (5), 872. https://doi.org/10.3390/ani10050872.
[335] Fernández, L.; Gutiérrez, D.; García, P.; Rodríguez, A. The Perfect Bacteriophage for Therapeutic
Applications—A Quick Guide. Antibiotics, 2019, 8 (3), 126.
https://doi.org/10.3390/antibiotics8030126.

60

Chapter 2: Scientific Aims
61
Chapter 2: Scientific Aims
Chapter 2
Scientific Aims
Avian pathogenic E. coli (APEC) is one of the most important bacterial pathogens affecting
poultry. The emergence of multidrug-resistant pathogens has renewed the interest in alternative
treatment options, such as the therapeutic use of phages (phage therapy). Phages are viruses
that specifically infect bacteria and are the most abundant organism on Earth. In recent years,
an increasing amount of sequencing data becoming available has expanded our understanding
of phage diversity, but also revealed that we have only scratched the surface. A key first step
to develop a successful therapy is to build a collection of well-characterised candidate phages
targeting the pathogen of interest, as not all isolated phages meet the criteria for therapeutic
application. Nevertheless, there are still major problems with therapy, indicating our
understanding of the host-pathogen interaction is not well developed. We see that the dynamics
of interaction of the pathogen is variable and that a major concern of phage therapy is the
emergence of phage-resistant bacterial mutants. Therefore, the general aim of this PhD project
was to provide new insight into the diversity of coliphages, and to determine the coliphage-
bacterium interaction and population dynamics.

Chapter 2: Scientific Aims
62
The specific aims of the project included:
1) Establish a well-characterised collection of lytic coliphages, using whole-genome
sequencing (WGS) analysis and bioinformatics tools (chapter 3.1).
2) Determine coliphage-host in vitro interactions and population dynamics in order to
have an in vitro model for better understanding of the pharmacodynamics and
pharmacokinetics (chapter 3.2).
3) Determine factors involved in APEC phage resistance, by generating in vitro resistant
combinations and determine the genetic background of resistance (chapter 3.3).

Chapter 3: Experimental Studies
63
Chapter 3: Experimental Studies
Chapter 3
Experimental Studies
3.1 New insights into the biodiversity of coliphages in the intestine of poultry
3.2 Classification of in vitro phage-host population growth dynamics
3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
3.4 Schematic overview of the experimental studies and main findings

Chapter 3: Experimental Studies
64

Chapter 3: Experimental Studies
65
3.1
New insights into the biodiversity of coliphages in the intestine of
poultry
Patricia E. Sørensen1,2, Wim Van Den Broeck3, Kristoffer Kiil4, Dziuginta Jasinskyte5,
Arshnee Moodley5,6, An Garmyn1, Hanne Ingmer5, and Patrick Butaye1,2
1 Department of Pathology, Bacteriology and Poultry diseases, Ghent University, Belgium
2 Department of Biomedical Sciences, Ross University School of Veterinary Medicine, St. Kitts, West
Indies
3 Department of Morphology, Ghent University, Belgium
4 Department of Microbiology and Infection Control, Statens Serum Institut, Denmark
5 Department of Veterinary and Animal Sciences, University of Copenhagen, Denmark
6 CGIAR Antimicrobial Resistance Hub, International Livestock Research Institute, Nairobi, Kenya
Published in Scientific Reports 2020, 10 (15220)
3.1 New insights into the biodiversity of coliphages in the intestine of poultry

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
66
Abstract
Despite phages’ ubiquitous presence and great importance in shaping microbial communities,
little is known about the diversity of specific phages in different ecological niches. Here, we
isolated, sequenced, and characterised 38 Escherichia coli-infecting phages (coliphages) from
poultry faeces to gain a better understanding of the coliphage diversity in the poultry intestine.
All phages belonged to either the Siphoviridae or Myoviridae family and their genomes ranged
between 44,324-173,384 bp, with a G+C content between 35.5-46.4%. Phylogenetic analysis
was performed based on single “marker” genes; the terminase large subunit, portal protein, and
exonucleases, as well as the full draft genomes. Single gene analysis resulted in six distinct
clusters. Only minor differences were observed between the different phylogenetic analyses,
including branch lengths and additional duplicate or triplicate subclustering. Cluster formation
was according to genome size, G+C content and phage subfamily. Phylogenetic analysis based
on the full genomes supported these clusters. Moreover, several of our Siphoviridae phages
might represent a novel unclassified phage genus. This study allowed for identification of
several novel coliphages and provides new insights to the coliphage diversity in the intestine
of poultry. Great diversity was observed amongst the phages, while they were isolated from an
otherwise similar ecosystem.

Chapter 3: Experimental Studies
67
Introduction
Bacteriophages (phages) are viruses that have the ability to specifically infect bacteria. They
are estimated to be the most abundant form of life on Earth (~1031 organisms) and can be found
in almost every ecosystem, including soil, wastewater, sewage water, seawater and in and on
humans and animals [1–4]. Phages are thought to play essential roles in shaping the microbial
ecology, including driving the diversity of the bacterial communities [5]. As no single gene is
present in all phages, their taxonomic classification is based on host range, physical
characteristics, including size and morphology, genetic structure and composition, and overall
genome similarity [6, 7]. The phage classification scheme is regularly updated, refined and
approved by the International Committee on the Taxonomy of Viruses (ICTV) [8].
Furthermore, in recent years several genome-based phage taxonomy schemes have been
implemented [7, 9]. According to the National Center for Biotechnology information (NCBI),
as of February 2020, 9,238 complete phage genomes have been sequenced. However, despite
a continuously rising number of sequenced phage genomes, most of them remain unclassified
and poorly characterised. According to the ICTV, a phage genus can be defined as a group of
viruses with >50% nucleotide sequence similarity, which is distinct from viruses of other
genera. Moreover, defining characteristics can be determined for each genus, including average
genome length and number of coding sequences (CDSs), percentage of shared CDSs, and the
presence of specific signature genes in genus members [10].
Most phages that infect Escherichia coli, coliphages, belong the highly heterogeneous
Caudovirales order, which constitute ~96% of all known isolated phages [11]. This order
contains five families of tailed phages with dsDNA genomes: Myoviridae, Siphoviridae,
Podoviridae, Ackermannviridae and Herelleviridae [12]. According to ICTV taxonomy (data
of February 2020), these families comprise five, eleven, three, two, and five subfamilies,
respectively, and 87, 210, 48, three, and 15 genera, respectively. The currently analysed
Myoviridae coliphages belong to four subfamilies, including Ounavirinae, Peduovirinae,
Tevenvirinae, and Vequintavirinae, and 17 genera. Siphoviridae coliphages are found in only
two subfamilies: Guernseyvirinae and Tunavirinae, and in 13 genera. Podoviridae coliphages
belong to two subfamilies, the Autographivirinae and the Sepvirinae, and to 10 genera.
Ackermannviridae coliphages belong only to the Cvivirinae subfamily and the Kuttervirus
genus. To date, there have been no Herelleviridae coliphages isolated. To understand the
diversity, relationships, and dynamics among any group of phages, nucleotide sequence
information is needed [1]. For tailed phages, it has been reported that conserved genes such as

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
68
the terminase large subunit, the portal protein and major capsid protein (MCP), can be used as
phylogenetic markers for the diversity as well as their evolutionary relationship [1, 13].
Compared to their bacterial hosts, relatively few phages have been fully characterised
[14]. Besides, despite the phages’ significant role and ubiquitous presence in various areas,
little is known on the nature and extent of phage diversity in different ecosystems [3]. Recently,
there has been an interest in the diversity of coliphages [1, 15, 16]. Here, we performed a
detailed genome-based characterisation and phylogenetic analysis of 38 fully sequenced
coliphages, all isolated from a single, relatively unexplored environmental source: poultry
faecal material.
Methods
Phage isolation and propagation
The phages were isolated from poultry faecal material, collected randomly from 27 poultry
houses in Belgium in 2013. Phages were propagated according to Adams (1959) and Bonilla
et al. (2016) with minor modifications [17, 18]. Briefly, the samples (5g) were emulsified in
Lysogeny broth(LB) broth (Miller)(Sigma-Aldrich, Saint Louis, MO, USA). The decanted
supernatant obtained from each emulsion was enriched by the addition of two early -log phase
host bacteria, E. coli K-12 derived laboratory strains C600 [19] or K514 [20], a non-restricting,
modifying derivative of strain C600. Suspensions were incubated overnight at 37°C, with
shaking (120 rpm) and were then centrifuged at 4,000 rpm for 30 min to pellet the cellular
debris. The supernatant containing the phage(s) was centrifuged and filtered using a 0.45 µm
membrane filter followed by a 0.2 µm Minisart Filter (Fisher Scientific, Waltham, MA, USA).
The enriched phage suspensions were enumerated and tested for lytic activity on the host
bacteria using the double-layer agar (DLA) technique [17, 21, 22]. Briefly, phage suspensions
were serial diluted and spotted on an overlay of the respective host bacteria on solid LB medium
supplemented with 0.8% agar and 0.5 mM CaCl2. A clear zone in the plate, a plaque, resulting
from the lysis of host bacterial cells, indicated the presence of virulent coliphage(s). Samples
with lytic activity against the indicator strain were further processed for single phage plaque
isolation, including three rounds of plaque purification, and propagation. All phage lysates
were stored at 4°C until required.
Phage morphological analysis

Chapter 3: Experimental Studies
69
The morphology of unique coliphages (≤ 95% nucleotide similarity) isolated in this study was
investigated using transmission electron microscopy (TEM). Phage suspension was applied to
the surface of Formvar carbon-coated grids, the phages were fixed using paraformaldehyde
(PFA) (4% w/v), washed, and negatively stained with UrAC (1% w/v). After drying, grids were
examined using a JEM-1400 Plus transmission electron microscope (JEOL, Benelux).
Genomic DNA extraction and sequencing
DNA extraction from phage lysates was performed using DNeasy Blood & Tissue Kit (Qiagen,
Hilden, Germany) as previously described [23]. The DNA concentration and quality was
assessed using the NanoDrop (Thermo Scientific, Roskilde, Denmark) and Qubit fluorometer
(Thermo Scientific, Roskilde, Denmark) according to the manufacturer’s instructions.
Preparation of paired-end 2 x 250 bp sequencing libraries was done using the Nextera XT Kit
(Illumina, San Diego, USA) with adaptations for phage genomes as shown elsewhere [24] and
sequenced on the Illumina MiSeq platform using MiSeq Reagent Kit v2 (500-cycles) and
manufacturer’s instructions, yielding a total of 16,270-237,128 paired end reads for each phage
lysate. Read-pair contigs were generated for each MiSeq cluster prior to assembly.
Phage genome sequence analysis and annotation
FastQC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), v0.11.3, was
used for quality control validation of the raw reads sequence data. Low-quality sequences were
excluded from further analysis. The raw reads were trimmed for quality, adaptor sequences
were removed using default parameters. The sequence reads were de novo assembled using
QIAGEN Bioinformatics CLC Genomic Workbench, v11.0.1, using default settings, with
minimum contig length changed to 250 bp. An overview of assembly statistics is provided in
Supplementary Table S3. The assembled draft phage sequences were compared with phage
homologues from the NCBI nucleotide database (https://blast.ncbi.nlm.nih.gov/Blast.cgi)
using the Basic Local Alignment Search Tool (BLAST) software [25], and from the custom
PHAge Search Tool Enhanced Release (PHASTER) phage database [26]. Newly assembled
phage sequences were compared using both BLAST and PHASTER to identify unique and
identical (>95% nucleotide sequence similarity) phages. Assembled contigs were submitted to
the ResFinder database, v3.2 [27] and the VirulenceFinder database, v2.0 [28] to identify any
acquired antimicrobial resistance and virulence associated genes, respectively. By default,
selected threshold for %ID was 90% and 60% for minimum length. All 15 databases for

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
70
antimicrobial resistance genes were selected. The taxonomic group of E. coli bacteria was
selected for VirulenceFinder. PHASTER and The Rapid Annotation using Subsystem
Technology (RAST) server and the SEED viewer, v2.0, were used for identification of CDSs
and initial annotation of the phage genomes, including identification of the phage terminase
large subunit [29]. Gene function of genes defined as “hypothetical protein” was predicted by
comparison to homologue genes with defined functions in other related phage genomes. The
G+C content of the phages was calculated using the SEED viewer.
Phage phylogeny and taxonomy
Multiple genome alignment of the WGS sequences was performed using Applied Maths
BioNumerics software, v7.6. According to the ICTV taxonomy guidelines, the 38 coliphages
were classified into phage family, subfamily and genera based on nucleotide similarity to
known Siphoviridae and Myoviridae coliphages. Known reference coliphages included were
limited to isolates with complete genomes found in the ICTV database and the NCBI database
(data of November 2019).
Phage diversity
Phylogenetic trees based on the phage whole genome sequences were constructed using R,
v3.5.1 [30], for comparison of the coliphages with published Siphoviridae or Myoviridae
reference phage genomes (accessed f rom the NCBI database). The trees were constructed using
unweighted pair group method with arithmetic mean (UPGMA) from a distance matrix of
binary distances calculated from either, gene presence/absence within the full genomes of the
phages determined using Roary v3.12.0 [31], Prokka, v1.13.7 [32] and prodigal, v2.6.3 [33],
or Kmer presence/absence (using 10 and 21mer) based on de novo assembled contigs, as
calculated using a python script (Supplementary Script kmer.py).
For phylogenetic analysis based on single marker gene, phage gene sequences were aligned
using Clustal X, v2.1 [34]. A maximum likelihood phylogenetic trees (unrooted) was
constructed and supported by bootstrap analysis (inferred from 1000 replicates) with default
substitution model (Tamura-Nei model) to assess the diversity of the coliphages using the
phylogenetic and molecular evolutionary genetics analyses (MEGA) software, vX [35]. The
phylogenetic trees were based on the nucleotide sequences of the CDSs of the following genes:

Chapter 3: Experimental Studies
71
phage terminase large subunit, phage portal protein, or phage exonucleases. The reference
genomes included, represented the best matching published sequences to the phages in this
study (selected based on the BLAST max score) and core reference genomes for comparison.
The degree of topological and branch length agreement between the different phylogenetic
methods and between the three marker genes was investigated using the R packages Analysis
of Phylogenetics and Evolution (ape) [36] and phangorn [37].
Phage comparative genomics
A more detailed analysis of the most closely related coliphage genomes was carried out.
Genomes were re-annotated using Prokka and pan genome analysis was carried out with Roary
using script “roary -e -n -s -p 20 -i 90 *.gff”, including identification of core genome, including
core and softcore genes, and accessory genome, including shell and cloud genes.
To investigate the level of synteny and genomic rearrangement, whole genome alignment and
comparison of coliphages and related reference phages from each cluster or subcluster were
performed with the Mauve software using progressiveMauve [38] with default parameters. No
more than 19 of the most related phages were included in each comparison for simplification.
Relatedness of the phages were based on percentage of nucleotide similarity and number of
shared core genes. Reference genomes were included for annotation references.
Results
Phage isolation
In this study, 38 coliphages were isolated from poultry faecal samples collected from 27
Belgian poultry farms located in five different regions, including West Flanders, East Flanders,
Antwerp, and Limburg. Between one and seven phages were isolated from each farm using E.
coli C600 or K514 as host strain.
Phage morphological analysis
Based on a sequencing cut-off value of ≤95% nucleotide similarity, 18 coliphages were
selected and subjected to TEM to determine phage morphology and confirm phage

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
72
classification. Based on the morphological features, the phages were classified into the
Caudovirales order and either the Siphoviridae family or the Myoviridae family. Analysis of
the isolated Siphoviridae phages showed a long flexible non-contractile tail with a length
varying between ~100 nm and ~200 nm and icosahedral heads with widths ranging from ~52
nm and ~77 nm (Fig. 1a-h).
Figure 1 | Negative staining electron microscopy images of Siphoviridae and Myoviridae coliphages. Siphoviridae
phages: a) Phage 17. b) Phage 53. c) Phage 54. d) Phage 61. e) Phage 70. f) Phage 74. g) Phage 76. h) Phage 77.
Myoviridae phages: i) Phage 10. j) Phage 11. k) Phage 15. l) Phage 18. m) Phage 30. n) Phage 55. o) Phage 60.
p) Phage 62 q) Phage 78. r) Phage 79. The black bars represent 100 nm.

Chapter 3: Experimental Studies
73
Among the isolated Myoviridae phages a long straight contractile tail was observed with a
tailed length varied between ~100 nm and ~120 nm, head widths ranging from ~65 nm to ~84
nm, and head lengths from ~60 nm to ~110 nm (Fig. 1i-r). Taxonomic classification of each of
the coliphages is shown in Table 1.
Phage genome sequence analysis and annotation
All 38 coliphages isolated in this study were characterised based on WGS data. An overview
of the genomic characteristics and properties are listed in Table 1. According to FastQC
parameters, good quality of the raw sequence data for all phages was confirmed. The phage
genomes ranged in size between 44,324 bp to 173,384 bp, with a G+C content between 35.5%
and 46.4%. Genomes smaller than 90,000 bp had a G+C content between 38.9% and 46.4%,
whereas the larger genomes had a G+C content of 35.5-38%. For each coliphage, 72 to 275
putative CDSs were identified using both automatic and manual annotation. CDSs encoding
the phage terminase small subunit, the phage terminase large subunit, the phage portal protein,
and phage capsid and scaffold proteins were identified within all 38 coliphage genome
sequences. They presented the same conserved genome structure with a general gene order: the
terminase small subunit upstream from the terminase large subunit, the phage portal protein
and two genes encoding phage capsid and scaffold proteins. In general, one phage terminase
small subunit, one phage portal protein, and up to four phage capsid and scaffold proteins were
found within each of the phage genomes. Besides, phage exonucleases were identified in all
phage genomes. For each phage, one to three CDSs for exonucleases were found. No gene
encoding for an integrase was found, indicating that these phages are strictly virulent/lytic
phages. No known acquired antimicrobial resistance or virulence genes were detected in any
of the 38 phage genomes.
Phage phylogeny and taxonomy
Taxonomic classification of the 38 coliphages was performed through multiple WGS genome
comparisons. These coliphages included 27 (71%) Siphoviridae coliphages and 11 (29%)
Myoviridae coliphages. The Siphoviridae phages were compared with 146 published phages
from this family. The Myoviridae phages were compared with 171 published Myoviridae
phages. According to ICTV guidelines, phage family, subfamily and genus were predicted
based on genome similarity. Results are shown in Table 1.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
74
Ta
ble
1 | C
hara
cte
rist
ics
of
the 3
8 E
. co
li-i
nfe
cti
ng p
hages
invest
igate
d in
th
is s
tud
y
Ph
age
nam
e
Reg
-
ion
Farm
ID
E. co
li
ho
st
Gen
om
e
size
(b
p)
%
G+
C
#
CD
Ss
Rel
ate
d r
ef.
ph
ag
e
Ph
age
fam
ily
Ph
age
sub
fam
ily
P
hage
gen
us
Ph
age
clu
ster
**
WG
S T
LS
P
P E
xo
Phag
e 47
O
VL
1
5
C6
00
5
10
63
43
.6
83
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 48
W
VL
1
2
C6
00
5
10
31
43
.7
85
Hen
u8
Sip
ho
T
unavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 53
W
VL
1
0
K5
14
5
08
35
44
.2
87
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 54
W
VL
1
4
K5
14
5
26
02
43
.5
88
Hen
u8
Sip
ho
T
unavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 59
L
IM
22
K
51
4
51
70
2
43
.6
85
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 63
O
VL
1
9
K5
14
4
91
32
44
.0
79
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A2
A1
A1
Phag
e 64
O
VL
1
9
K5
14
5
13
52
43
.7
85
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 65
O
VL
1
9
K5
14
5
10
31
43
.6
83
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 68
O
VL
2
1
K5
14
5
12
91
43
.7
84
G2
9-2
S
iph
o
Tunavi
rinae
Han
rive
rvir
us
A1
A1
A1
A1
Phag
e 71
O
VL
19
K
514
51
44
6
43.6
85
G29-2
Sip
ho
T
unavi
rinae
Hanri
verv
irus
A1
A1
A1
A1
Phag
e 72
O
VL
17
K
514
51
28
4
43.7
84
G29-2
Sip
ho
T
unavi
rinae
Hanri
verv
irus
A1
A1
A1
A1
Phag
e 75
O
VL
20
K
514
50
44
5
44.1
88
G29-2
Sip
ho
T
unavi
rinae
Hanri
verv
irus
A1
A1
A1
A1
Phag
e 77
A
NT
6
K514
51
07
3
44.0
85
G29-2
Sip
ho
T
unavi
rinae
Hanri
verv
irus
A1
A1
A1
A1
Phag
e 8
V
BR
27
C
600
51
03
1
43.6
83
pS
f-1
Sip
ho
T
unavi
rinae
Hanri
verv
irus
A1
A1
A1
A1
Phag
e 28
O
VL
19
K
514
52
97
0
44.4
87
SE
Cphi2
7
Sip
ho
T
unavi
rinae
Sw
anvi
rus*
A
2
A2
A2
A2
Phag
e 56_1
A
NT
1
K514
52
71
6
44.5
86
Eco
S-9
5
Sip
ho
T
unavi
rinae
Sw
anvi
rus*
A
2
A2
A2
A2
Phag
e 76
W
VL
11
K
514
51
90
5
45.0
93
SE
Cphi2
7
Sip
ho
T
unavi
rinae
Sw
anvi
rus*
A
2
A4
A2
A2
Phag
e 80
O
VL
18
K
514
52
70
3
44.5
88
Eco
S-9
5
Sip
ho
T
unavi
rinae
Sw
anvi
rus*
A
2
A2
A2
A2
Phag
e 52
W
VL
13
K
514
53
01
8
45.9
90
Jahat
MG
145
Sip
ho
T
unavi
rinae
New
gen
us
A3
A3
A3
A3
Phag
e 56_2
A
NT
1
K514
50
82
9
45.7
87
Jahat
MG
145
Sip
ho
T
unavi
rinae
New
gen
us
A3
A3
A3
A3
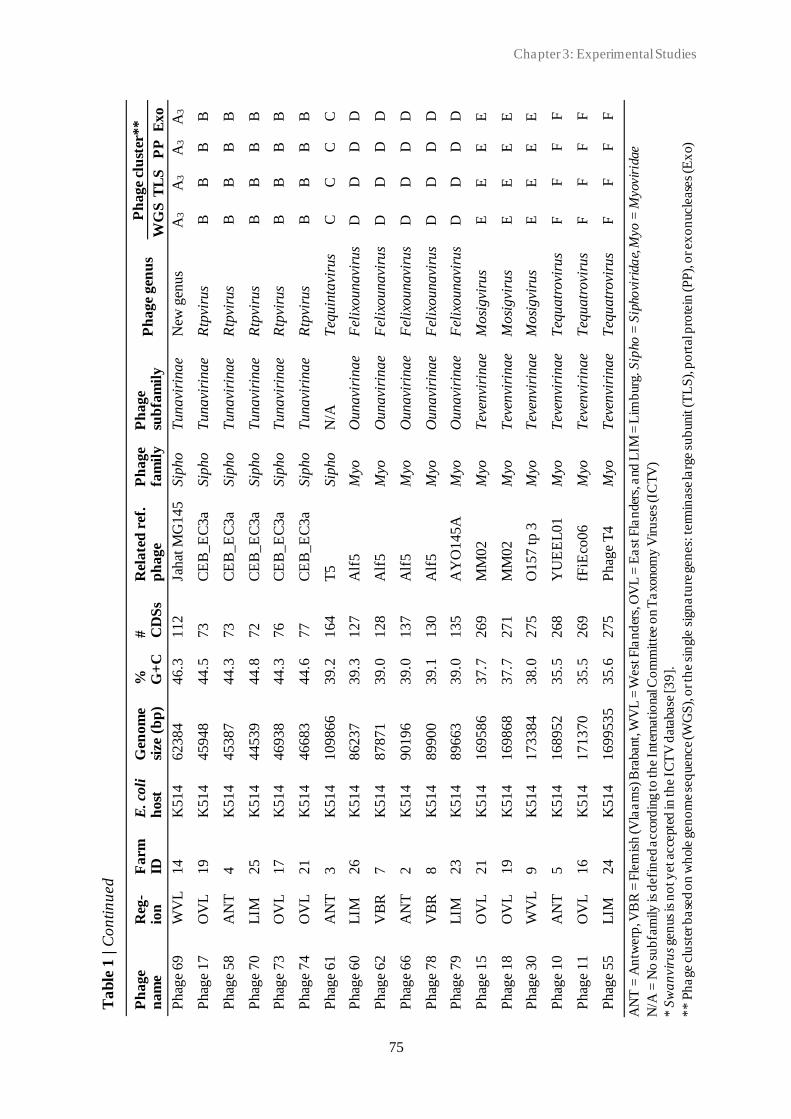
Chapter 3: Experimental Studies
75
Ta
ble
1 | C
on
tin
ued
Ph
age
nam
e R
eg-
ion
F
arm
ID
E
. co
li
host
G
eno
me
size
(b
p)
%
G+
C
#
CD
Ss
Rel
ate
d r
ef.
ph
age
Ph
ag
e fa
mil
y
Ph
age
sub
fam
ily
P
hage
gen
us
Ph
age
clu
ster
**
WG
S T
LS
P
P
Exo
Phag
e 69
W
VL
14
K
514
62
38
4
46.3
112
Jahat
MG
145
Sip
ho
T
unavi
rinae
New
gen
us
A3
A3
A3
A3
Phag
e 17
O
VL
19
K
514
45
94
8
44.5
73
CE
B_E
C3a
Sip
ho
T
unavi
rinae
Rtp
viru
s B
B
B
B
Phag
e 58
A
NT
4
K514
45
38
7
44.3
73
CE
B_E
C3a
Sip
ho
T
unavi
rinae
Rtp
viru
s B
B
B
B
Phag
e 70
L
IM
25
K
514
44
53
9
44.8
72
CE
B_E
C3a
Sip
ho
T
unavi
rinae
Rtp
viru
s B
B
B
B
Phag
e 73
O
VL
17
K
514
46
93
8
44.3
76
CE
B_E
C3a
Sip
ho
T
unavi
rinae
Rtp
viru
s B
B
B
B
Phag
e 74
O
VL
21
K
514
46
68
3
44.6
77
CE
B_E
C3a
Sip
ho
T
unavi
rinae
Rtp
viru
s B
B
B
B
Phag
e 61
A
NT
3
K514
10
98
66
39.2
164
T5
Sip
ho
N
/A
Teq
uin
tavi
rus
C
C
C
C
Phag
e 60
L
IM
26
K
514
86
23
7
39.3
127
Alf
5
Myo
O
unavi
rinae
Fel
ixoun
avi
rus
D
D
D
D
Phag
e 62
V
BR
7
K514
87
87
1
39.0
128
Alf
5
Myo
O
unavi
rinae
Fel
ixoun
avi
rus
D
D
D
D
Phag
e 66
A
NT
2
K514
90
19
6
39.0
137
Alf
5
Myo
O
unavi
rinae
Fel
ixoun
avi
rus
D
D
D
D
Phag
e 78
V
BR
8
K
51
4
89
90
0
39
.1
13
0
Alf
5
Myo
O
unavi
rinae
Fel
ixou
na
viru
s D
D
D
D
Phag
e 79
L
IM
23
K
514
89
66
3
39.0
135
AY
O145
A
Myo
O
unavi
rinae
Fel
ixoun
avi
rus
D
D
D
D
Phag
e 15
O
VL
2
1
K5
14
1
69
58
6
37
.7
26
9
MM
02
M
yo
Tev
envi
rinae
Mosi
gvi
rus
E
E
E
E
Phag
e 18
O
VL
1
9
K5
14
1
69
86
8
37
.7
27
1
MM
02
M
yo
Tev
envi
rinae
Mosi
gvi
rus
E
E
E
E
Phag
e 30
W
VL
9
K
51
4
17
33
84
38
.0
27
5
O1
57
tp
3
Myo
T
even
viri
nae
Mosi
gvi
rus
E
E
E
E
Phag
e 10
A
NT
5
K
51
4
16
89
52
35
.5
26
8
YU
EE
L0
1
Myo
T
even
viri
nae
Teq
uatr
ovi
rus
F
F
F
F
Phag
e 11
O
VL
1
6
K5
14
1
71
37
0
35
.5
26
9
fFiE
co0
6
Myo
T
even
viri
nae
Teq
uatr
ovi
rus
F
F
F
F
Phag
e 55
L
IM
24
K
51
4
16
99
53
5
35
.6
27
5
Ph
age
T4
Myo
T
even
viri
nae
Teq
uatr
ovi
rus
F
F
F
F
AN
T =
An
twerp
, VB
R =
Fle
mis
h (V
laa
ms)
Bra
bant,
WV
L =
West
Fla
nd
ers,
OV
L =
Ea
st F
lan
der
s, a
nd
LIM
= L
imb
urg
. S
ipho =
Sip
ho
viri
da
e, M
yo
= M
yovir
idae
N/A
= N
o s
ub
fam
ily
is d
efi
ned a
cco
rdin
g to
th
e I
nte
rnati
onal
Co
mm
itte
e o
n T
axonom
y V
iru
ses
(IC
TV
)
* S
wa
nvir
us
gen
us
is n
ot y
et acce
pte
d in
th
e I
CT
V d
ata
base
[3
9].
**
Ph
age c
lust
er b
ase
d o
n w
hole
gen
om
e se
quence
(WG
S),
or th
e s
ingle
sig
na
ture
gen
es:
term
inase
larg
e s
ub
unit
(T
LS
), p
ort
al p
rote
in (P
P),
or ex
on
ucle
ase
s (E
xo)

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
76
All Siphoviridae phages belonged to the Tunavirinae subfamily, except for Phage 61. This
phage was predicted to belong to the Tequintavirus genus, which do not have any ICTV
subfamily. The 10 phages, Phage 8, 53, 54, 63, 65, 68, 69, 71, 72, and 75 all belonged to the
Hanrivervirus genus. Three phages belonged to the Rtpvirus genus, including Phage 17, 70,
and 73. No existing ICTV genus could be assigned to the remaining 13 coliphages. Phage 28,
56_1 and 76 could be assigned to the same unknown genus. Phage 58 and Phage 74 were found
to be in the same genus. Phage 47, 48, 59, 64, and 77 were predicted to belong to the same
genus. Phage 52, 56_2, and 80 were predicted to belong to the same genus. The 11 Myoviridae
belonged either to the Tevenvirinae or the Ounavirinae subfamily. Tevenvirinae phages
included the six phages: Phage 10, 11, 15, 18, 30 and 55. Phage 10, 11 and 55 belonged to the
Tequatrovirus genus, and Phage 18 and 30 belonged to the Mosigvirus genus. Ounavirinae
phages included the remaining five phages; Phage 60, 62, 66, 78 and 79. All phages belonged
to the Felixounavirus genus.
Phage diversity
To investigate the diversity of the coliphages, phage genomes were first clustered based on
whole genome sequence. A total of 173 Siphoviridae and 182 Myoviridae coliphage genomes
were included. Characteristics of selected reference genomes are listed in Supplementary Table
S1. Siphoviridae phages isolated in this study were found in five different (sub)clusters, cluster
A1-3, B and C, with a cut-off value of 0.82 (Fig. 2). Cluster A was divided into three
subclusters. Fourteen of our phages, formed subcluster A1 together with the three pSf-1-like
reference phages from the NCBI database. Phage 80, 28, 56_1, and 76 formed subcluster A2
with the three Swan01-like reference phages. Phages 69, 52, and 56_2 formed subcluster A3
with phage Jahat_MG145. Phage 73, 70, 17, 58 and 74 formed cluster B without any known
reference phages. Phage 61 was placed in cluster C with 13 T5-like reference phages. For the
Myoviridae phages, the resulting phylogeny placed phages isolated in this study in three
different clusters with a cut-off height of 0.52 (Fig. 3). Phages 62, 78, 66, 60 and 79 formed
cluster D with Felix01-like reference phage Alf5. Phage 30, 15 and 18 formed cluster E with
19 T4-like reference phages (cut-off height of 0.36). Phages 55, 11 and 10 were placed in
cluster F with 57 reference phages. At the cut-off height of 0.39 Phage 55 was found in a
different subcluster than Phage 10 and 11.

Chapter 3: Experimental Studies
77
Fig
ure 2
| P
hy
logenetic a
naly
sis
of
Sip
hovir
idae c
olip
hages
base
d o
n W
GS
seq
uence.
Ph
ages
iso
late
d in
th
is s
tudy a
re h
igh
ligh
ted. E
ach c
olo
ur
rep
rese
nts
a c
lust
er:
Clu
ster A
(b
lue),
clu
ster B
(gre
en
), a
nd
clu
ster C
(re
d).
Clu
ster A
su
bclu
ster
s in
clu
de A
1 (ligh
t b
lue),
A2
(b
lue),
an
d A
3 (d
ark
blu
e). D
ista
nce
matr
ices a
nd c
lust
ering a
re
ba
sed o
n k
mer le
ngth
= 1
0.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
78
Fig
ure 3
| P
hy
logenetic a
nal
ysi
s o
f M
yo
viri
dae c
olip
hages
base
d o
n W
GS
seq
uence
. P
hages
iso
late
d in
th
is s
tudy a
re h
igh
ligh
ted. E
ach c
olo
ur
rep
rese
nts
a c
lust
er:
Clu
ster D
(o
ran
ge),
clu
ster E
(p
urp
le),
and
clu
ster F
(b
row
n).
Dis
tan
ce m
atr
ices
and c
lust
eri
ng a
re b
ase
d o
n k
mer le
ngth
= 1
0

Chapter 3: Experimental Studies
79
Coliphages were further assessed based on the presence/absence of families of orthologues
genes in their pan genome. Similar clusters were observed with only minor changes. For the
Siphoviridae phage analysis, 5227 gene groups were included (Supplementary Fig. S1). The
resulting phylogenetic analysis placed phages isolated in this study in the same five clusters,
cluster A1-3, B and C, with a cut-off height of 0.81 (Supplementary Fig. S2). One additional
reference phage was found in cluster B and C, including the T1-like reference phage
CEB_EC3a and the T5-like reference phage EPS7, respectively. For the Myoviridae phage
analysis, 9420 gene groups were included (Supplementary Fig. S3). The resulting phylogeny
placed phages isolated in this study in the same three clusters, cluster D, E, and F, with a cut-
off height of 0.58 (Supplementary Fig. S4). For cluster D, additionally 13 Felix01-like
reference phages were found. In contrast to the WGS-based analysis, at a cut-off height of 0.39,
all cluster F phages isolated in this study, were found in one subcluster with 10 T4 -like
reference phages. The degree of topological and branch length agreement between the different
phylogenetic methods were compared (Supplementary Table S2).
The coliphage diversity was further assessed based on three phage marker genes: the terminase
large subunit and phage portal protein, and the phage exonuclease. Selected gene sequences
from known phages were included for reference. Results are summarised in Table 1. For all
three marker genes, cluster formation was in accordance with resulting clusters of the pan
genome- and WGS-based phylogeny, cluster A-F, only with minor differences. Results based
on the terminase large subunit analysis are shown below (Fig. 4).
For cluster A, all coliphages isolated in this study were found within same subclusters as for
the WGS-based phylogeny except for Phage 63, which was found in the A2 subcluster instead
of A1. Analysis based on the phage portal protein resulted in the division of our A2 subcluster
phages into two groups: Phage 56_1, 80 and 28 in one group and Phage 76 in the other group
(Supplementary Fig. S5). Analysis based on the exonuclease resulted in multiple clusters of
cluster C and F, as phages from these clusters encoded two or two-three exonuclease genes,
respectively (Supplementary Fig. S6). Comparison of the cluster construction of the three
single genes analysis showed only minor topological and branch length differences
(Supplementary Table S2). Moreover, cluster construction was in accordance with phage
subfamily defined based on the whole genome. Siphoviridae phages from cluster A and B
belonged to the Tunavirinae subfamily, and Siphoviridae phages form cluster C had no defined
ICTV subfamily. Myoviridae phages from cluster D belonged to the Ounavirinae subfamily,
and Myoviridae phages from cluster E and F belonged to the Tevenvirinae subfamily.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
80
Figure 4 | Maximum likelihood tree based on the nucleotide sequences of the phage terminase large subunit. The
analysis resulted in six clusters: A-F, according to phage family and subfamily. Cluster A and B: Siphoviridae,
Tunavirinae, cluster C: Siphoviridae and Tequintavirus genus, cluster D: Myoviridae, Ounavirinae, and cluster E
and F: Myoviridae, Tevenvirinae. Cluster A was divided into three subclusters: A1, A2 and A3. The tree was
constructed using the MEGA X software [35]. The percent of data coverage for internal nodes is indicated. The
scale bar indicates the number of nucleotide sequence substitutions per site. The analysis included 62 nucleotide
sequences, including 24 reference phages listed in Supplementary Table S1 for comparison.
Phage comparative genomics
Pan genome analysis of Siphoviridae and Myoviridae phages isolated in this study revealed
that neither of the two groups had any core genes. Analysis of coliphage genomes from each
of the six clusters, A-F, identified core genes (core and softcore) and accessory genes (shell
and cloud). As cluster A phages had only five core genes (2% of the total genome), analysis of
subclusters, A1, A2 and A3, were performed additionally. Results are summarised in Table 2.

Chapter 3: Experimental Studies
81
The pan genome included between 81 and 333 genes, and core genes constituted between 22%
and 73% of the pan genome. The level of synteny and genomic rearrangement within each
cluster or subcluster of related phages was assessed by genome comparison (Table 2). Eight
comparisons were performed, corresponding to the eight (sub)clusters, A1, A2, A3, B, C, D,
E, and F resulting from the phage diversity analysis above (Supplementary Fig. S7 -14).
Genome comparison of the phages resulted in identification of local collinear blocks (LCBs),
indicating homologues DNA regions shared by two or more genomes without sequence
rearrangements. The LCBs comprised different modules of genes with different functions,
including modules for DNA packaging, structural proteins, head and tail morphogenesis, and
host cell lysis. Several modules comprised only hypothetical proteins with unknown function.
The average level of conservation varied between the different type of genes.
Genes encoding the terminase large and small subunit, the MCP, DNA primase, portal protein,
recombinase, specific tail protein and holin were the most conserved genes between all phages,
whereas genes with the lowest level of conservation included, tail fiber proteins, tail tape
measure proteins and HNH homing endonucleases. Hypothetical proteins were found with
large variation in level of conservation. Each phage genome comprised between four and 17
LCBs. Genome comparison subcluster A1, A2 and A3 phages identified 16, seven and four
LCBs, respectively. All phages in each cluster comprised all LCBs. All cluster B phages
comprised all six LCBs. For the cluster C phages, between six and 10 LCBs were identified
for each phage. Phage 61 comprised all nine regions. Variation in number of LCBs was due to
a variable repeat region comprising multiple LCBs, which was found only in some of the
cluster C phages. For the cluster D comparison, 14-17 different LCBs were identified for each
phage. Variation in number of LCBs was due to four different small variable regions of which
some of all were missing in the majority of the phages. Phages isolated in this study, including
Phages 79, 78, 60, 66, and 62, comprised 17, 17, 16, 15, and 14 LCBs, respectively.
Comparison of phages belonging to cluster E identified 18 LCBs. All phages lacked one or
both of the same two LCBs. Phages isolated in this study, including Phage 30, Phage 15, and
Phage 18, comprised 16, 17, and 17 LCBs, respectively. All 13 cluster F phages included in
the comparison comprised all five LCBs. The comparison confirmed the presence of
homologue regions between the phages within the clusters but also highlighted that re -
arrangement and/or gain/loss of LCBs must have occurred at some point during the evolution
of the phages. The region encoding the terminase large subunit and portal protein were present
in a conserved region all genomes in all eight comparisons.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
82
T
ab
le 2
| O
verv
iew
of
co
mpara
tiv
e g
en
om
ics
an
aly
sis
Clu
ster
# o
f
ph
ag
es
Ph
age
nam
es
# o
f
LC
Bs
# c
ore
gen
es
Core
gen
om
e
#
acc
esso
ry
gen
es
Acc
esso
ry
gen
om
e
Pan
gen
om
e
gen
es
A1*
1
7
Phag
e 77, 53, 75, 63, 54, 72, 64, 71, 47, 4
8, 59, 6
8, 65, 8,
Hen
u8, G
29
-2, an
d p
Sf-
1
16
2
8
22%
98
78%
1
26
A2*
7
Phag
e 80, 28, 56_1,7
6, S
wan
01, S
EC
phi2
7 a
nd E
coS_95
7
46
40%
69
60%
1
15
A3*
4
Phag
es 6
9, 52, 56_2, an
d J
ahat
_M
G145
4
62
73%
23
27%
8
5
B
6
Phag
e 73, 70, 17, 58, 74, an
d C
EB
_E
C3a
6
27
33%
54
67
8
1
C
10
Phag
e 61, T
5, E
AS
G3, H
AS
G4, A
KF
V33, O
SY
SP
,
phiL
LS
, S
P15, F
FH
1, an
d H
dH
2
6-1
0
86
37%
147
63%
2
33
D
19
Phag
e 60, 78, 62, 66, 79, E
C6, V
paE
1, X
TG
1, K
hF
1,
KhF
2, K
hF
3, H
Y02, R
o111lw
, O
157_1,
O157_12, W
V8,
O157_11, A
lf5, an
d A
YO
145A
14-1
7
72
39%
115
61%
1
87
E
18
Phag
e 30, 15, 18, H
X01, K
AW
3E
185, W
FbE
185, G
53,
AP
EC
c01, M
M02, H
P3, A
TK
47, A
TK
48, O
157_ 3
,
O157_ 6
, S
T0, JS
09, G
2285, an
d G
2469
16-1
7
184
58%
133
42%
3
17
F
13
Phag
e 55, 11, 10,
G2540-3
, G
29, G
4500, D
5505, G
9062,
CF
2, Y
UE
EL
01, fF
iEco
06,
AC
G_C
40, an
d O
E55
05
5
1
97
59%
136
41%
3
33
*S
ub
clu
ster in
stea
d o
f clu
ster
. Lo
cal c
ollin
ear b
locks
(LC
B) =
ind
ica
tin
g h
om
olo
go
us
DN
A regio
ns
sha
red
by tw
o o
r m
ore
gen
om
es
wit
ho
ut se
quence
rea
rrangem
ents
.

Chapter 3: Experimental Studies
83
Discussion
In this study, 38 coliphages were isolated from poultry faecal material, sequenced and
characterised. The high number of coliphage genomes included in the analysis allowed for a
better understanding of both coliphage diversity in poultry and the global coliphage diversity
within the Siphoviridae and Myoviridae families. However, one should be aware of the possible
biases as the coliphages were isolated using two E. coli K12-derived host strains only and, as
such, cannot be seen as the complete coliphage diversity. Both host strains are mutated for the
FhuA (previously called TonA) gene, which is used as receptor for some phages, including
Siphoviridae coliphages T1 and T5 [40]. However, phage adsorption is not always restricted to
one receptor. Accordingly, we were able to isolate a T5-like phage using the selected host
strains. Also, as none of the host strains has the F pili, phages utilising a F pili encoded receptor
for absorption, such as Inoviridae phages, will most likely not be isolated [40]. The reasons
why we could not isolate coliphages from the Podoviridae family remains obscure, however,
this is in accordance with the finding of Korf et al. (2019), who also did not isolate any
Podoviridae coliphages from poultry while they could isolate them from sewage and surface
water. On the other hand, other studies have successfully isolated Podoviridae phages from
poultry faeces [41–44]. Several factors might play a role in the type of phage being isolated,
including culture and isolation method, host strain, and isolation source. In Podoviridae studies
mentioned above, phages were isolated using a single or multiple E. coli host-strains and the
DLA method was similar to this study. However, a notable difference is the type of host-strain
used. In our study, laboratory strains were used whereas the Podoviridae study host-strains
only included E. coli strains isolated from poultry. As our 11 Myoviridae phages were isolated
from samples from 11 different farms, no correlation between phage isolated and geographical
location (poultry farm) was found (data not shown). In accordance with the findings of Olsen
et al. (2020), the most prevalent genus of the Myoviridae and Siphoviridae phages isolated in
our study was the Felixounavirus (45.5%) and the Hanrivervirus (33.3%), respectively. Both
studies used E. coli K-12 derived laboratory strains as host strain for phage isolation. While in
a collection of 50 coliphages isolated from surface water, manure, sewage, or animal faeces,
29 different E. coli host strains were used [15]. No Felixounavirus phages could be isolated
and most Myoviridae phages belonged to the Tequatrovirus genus, followed by the Mosigvirus
genus. Those two genera were also found in our study.
Currently, the polyphasic approach is the most commonly used for bacterial classification [45,
46], and a similar approach combining multiple methods is recommended when working with

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
84
phages [12]. However, studying phage taxonomy has proven challenging since no universal
conserved marker gene, as the 16S rRNA gene used for bacteria, exists throughout all phage
families. Several semi-conserved family-specific marker genes have been proposed as
candidates to support taxonomical classification of tailed phages, including DNA packaging
and head assembly genes [1, 47, 48]. Accordingly, in this study we used the terminase large
subunit, portal protein and exonucleases as markers, and were able to determine the family and
subfamily to all coliphages isolated in this study in accordance with the TEM-based and whole
genome-based classification, respectively. Furthermore, the analysis based on the terminase
large subunit and portal protein resulted in a clear distinction between the two Tevenvirinae
clusters, indicating different genera (Mosigvirus and Tequatrovirus). Thus, single gene-based
analysis provides good initial indication in which taxonomic cluster the phages belong to.
However, a single gene does not provide a global view of the structural organisation of the
phage nor accounts for genomic rearrangements, mutations, and mosaicism. Moreover, in
accordance with findings from previous studies the selected gene was not always detected in
the phage genomes [47], thus excluding these phages from classification. When using single
genes, one should be aware of the possibility of multiple distinct variants of the same gene
within one genome. Several of our phages encoded up to three different exonuclease genes.
Depending on which gene variant used for analysis, the risk of “false” cluster formation and
distance, and hereby a faulty classification at subfamily or genus level was present. Thus, for
more comprehensive phage taxonomy, including genus classification, the single gene analysis
should be accompanied by whole genome-based analysis as well as functional gene studies.
When investigating the evolutionary relationship between phages studies have shown the
advantage of combining different proteomic and comparative genomic approaches, including
WGS data and well-characterised reference dataset, which take into account the effect of
horizontal gene transfer (HGT) and recombination events on the phage genome evolution [7,
49].
In this study, genome-based phylogenetic and taxonomic analysis were performed in
combination with traditional morphological examination of the phage using TEM. Through the
genome-based analysis we identified a potential new Siphoviridae genus. The three
unclassified A3 subcluster from this study clustered together with the Jahat_MG145 reference
phage, which was a singleton [16]. Thus, we propose that this group of phages represents a
new unclassified genus with currently four phages, including Phage 52, Phage 56_2 Phage 69,
and Jahat_MG145.

Chapter 3: Experimental Studies
85
We aimed to expand our knowledge on the coliphage diversity, and observed great diversity
among these phages, while they were isolated from a similar ecosystem. The diversity was
characterised by a great span in genome size (44.3 kb to 173.1 kb) and G+C content range
(35.5-46.4%), as well as cluster-specific characteristics of the six phage clusters, A-F. Cluster
B phages had the smallest genomes and lowest number of CDSs followed by phages from
cluster A, D, C, and E/F. Similar to findings from other studies[16, 50], lower genome size
seemed to be correlated with an increase in G+C content. The largest variation in genome size
and number of CDSs were observed for the group of Myoviridae phages, whereas the largest
variation in G+C content (7.2%) was overserved for the group of Siphoviridae phages. Notably,
the Tequintavirus Phage 61 showed to be more similar to the group of Myoviridae compared
to the other Siphoviridae phages based on the above-mentioned characteristics. When omitting
Phage 61, G+C content variation for the Siphoviridae phages was only 2.9%. Phage G+C
content has been shown to be correlated with the G+C content of the phage host [50, 51].
Accordingly, differences in G+C content observed for the coliphages might reflect phage-host
interactions and co-evolution with past and current host(s). Gene content, including number of
core genes appeared to be associated with the cluster. Moreover, number of exonucleases
encoded by the phage appeared to be cluster associated as well, as only phages from cluster C
and cluster F encoded multiple exonucleases. Encoding multiple exonucleases could be a result
of adaptive evolution conferring fitness advantage over other phages. However, it could just as
well reflect some of the challenges to accurate phage genome annota tion, including false
negatives (undetected genes) and incorrect functional annotation [52, 53]. Gene content
variation has been shown to be related to recombination events resulting in acquisition or loss
of gene(s) [54]. Through our comparative genomics analysis of related phages, LCBs with
modules with varying level of gene conservation were identified and highlighted the different
levels of heterogeneity between different phage clusters. Repeat regions were observed in
several of the phage genomes and resulted in variation of LCBs. The presence of these regions
should be considered when assessing the gene content variation, as these regions are shown to
be prone to genome assembly mistakes, and as such, might represent false level of variation
[55]. LCBs modules are also called for mosaic sections and the two terms have been used
interchangeably throughout history, referring to exchangeable genomic segments between two
or more phages in the population [13]. The genome comparison showed the mosaic nature of
the phage genomes, with modules with high level of conservation interspersed with low-
similarity sections. These sections could be acquired though HGT from other phages, which is
thought to happen when phages are found in the same host. Most often this happens through

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
86
phage co-infection or single-phage infection of a host that carries one or more prophages [56,
57]. Moreover, phages have been shown to acquire genes from their host [58]. The comparative
genomics approach hereby underlines the continuous evolution of phage genomes as well as
the great phage diversity.
In conclusion, this study has identified a potential new coliphage genus and several new
species and provides insight not only to the coliphage diversity of the intestine of poultry but
the global coliphage diversity as well. Moreover, classification of phages isolated in this study
brings us one step closer to a more refined taxonomic understanding of coliphages. Our
comparative genomic analysis showed different levels of heterogeneity between different
phage clusters and highlighted the mosaic nature of the phage genomes as well as the
continuous evolution of phages in a single environment source. However, to fully understand
the complexity and underlying mechanisms of the phage diversity further studies are needed.

Chapter 3: Experimental Studies
87
References
[1] Grose, J. H.; Casjens, S. R. Understanding the Enormous Diversity of Bacteriophages: The Tailed
Phages That Infect the Bacterial Family Enterobacteriaceae. Virology, 2014, 468, 421–443.
https://doi.org/10.1016/j.virol.2014.08.024.
[2] Güemes, A. G. C.; Merry Youle; Cantú, V. A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners
in the Game of Life. Annu. Rev. Virol., 2016, 3 (1), 197–214. https://doi.org/10.1146/annurev-
virology-100114-054952.
[3] Hatfull, G. F. Dark Matter of the Biosphere: The Amazing World of Bacteriophage Diversity. J.
Virol., 2015, 89 (16), 8107–8110. https://doi.org/10.1128/jvi.01340-15.
[4] Pope, W. H.; Bowman, C. A.; Russell, D. A.; Jacobs-Sera, D.; Asai, D. J.; Cresawn, S. G.; Jacobs,
W. R.; Hendrix, R. W.; Lawrence, J. G.; Hatfull, G. F. Whole Genome Comparison of a Large
Collection of Mycobacteriophages Reveals a Continuum of Phage Genetic Diversity. Elife, 2015, 4,
e06416. https://doi.org/10.7554/elife.06416.
[5] Clokie, M. R. J.; Millard, A. D.; Letarov, A. V; Heaphy, S. Phages in Nature. Bacteriophage, 2011,
1 (1), 31–45. https://doi.org/10.4161/bact.1.1.14942.
[6] Ackermann, H. W. Phage Classification and Characterization. In Bacteriophages: Methods and
Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M. R. J., Kropinski, A.
M., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2009; Vol. 501, pp 127 –140.
https://doi.org/10.1007/978-1-60327-164-6.
[7] Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A Genome-Based Taxonomy for Phage. J.
Bacteriol., 2002, 184 (16), 4529–4535. https://doi.org/10.1128/JB.184.16.4529-4535.2002.
[8] Lefkowitz, E. J.; Dempsey, D. M.; Hendrickson, R. C.; Orton, R. J.; Siddell, S. G.; Smith, D. B.
Virus Taxonomy: The Database of the International Committee on Taxonomy of Viruses (ICTV).
Nucleic Acids Res., 2018, 46 (D1), D708–D717. https://doi.org/10.1093/nar/gkx932.
[9] Meier-Kolthoff, J. P.; Göker, M. VICTOR: Genome-Based Phylogeny and Classification of
Prokaryotic Viruses. Bioinformatics, 2017, 33 (21), 3396–3404.
https://doi.org/10.1093/bioinformatics/btx440.
[10] Adriaenssens, E. M.; Rodney Brister, J. How to Name and Classify Your Phage: An Informal Guide.
Viruses, 2017, 9 (4), 1–9. https://doi.org/10.3390/v9040070.
[11] International Committee on Taxonomy of Viruses (ICTV), A. M. K. Virus Taxonomy: Ninth Report
of the International Committee on Taxonomy of Viruses, 1st ed.; King, A. M. ., Adams, M. J.,
Carstens, E. B., Lefkowitz, E. J., Eds.; Elsevier, 2011.
[12] Barylski, J.; Enault, F.; Dutilh, B. E.; Schuller, M. B.; Edwards, R. A.; Gillis, A.; Klumpp, J.;
Knezevic, P.; Krupovic, M.; Kuhn, J. H.; et al. Analysis of Spounaviruses as a Case Study for the
Overdue Reclassification of Tailed Phages. Syst. Biol., 2019, 69 (1), 110–123.
https://doi.org/10.1093/sysbio/syz036.
[13] Casjens, S. R.; Thuman-Commike, P. A. Evolution of Mosaically Related Tailed Bacteriophage
Genomes Seen through the Lens of Phage P22 Virion Assembly. Virology, 2011, 411 (2), 393–415.
https://doi.org/10.1016/j.virol.2010.12.046.
[14] Hatfull, G. F.; Hendrix, R. W. Bacteriophages and Their Genomes. Curr. Opin. Virol., 2011, 1 (4),
298–303. https://doi.org/10.1016/j.coviro.2011.06.009.
[15] Korf, I. H. E.; Meier-Kolthoff, J. P.; Adriaenssens, E. M.; Kropinski, A. M.; Nimtz, M.; Rohde, M.;
van Raaij, M. J.; Wittmann, J. Still Something to Discover: Novel Insights into Escherichia Coli
Phage Diversity and Taxonomy. Viruses, 2019, 11 (5), 1–29. https://doi.org/10.3390/v11050454.
[16] Olsen, N. S.; Kot, W.; Junco, L. M. F.; Hansen, L. H. Exploring the Remarkable Diversity of
Escherichia Coli Phages in the Danish Wastewater Environment, Including 91 Novel Phage Species.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
88
bioRxiv, 2020.
[17] Adams, M. H. Bacteriophages; Interscience Publishers: New York, 1959.
[18] Bonilla, N.; Rojas, M. I.; Netto Flores Cruz, G.; Hung, S.-H.; Rohwer, F.; Barr, J. J. Phage on Tap–
a Quick and Efficient Protocol for the Preparation of Bacteriophage Laboratory Stocks. PeerJ, 2016.
https://doi.org/10.7717/peerj.2261.
[19] Appleyard, R. K. Segregation of New Lysogenic Types during Growth of a Doubly Lysogenic Strain
Derived from Escherichia Coli K12. Genetics, 1954, 39 (4), 440–452.
[20] Colson, C.; Glover, S. W.; Symonds, N.; Stacey, K. A. The Location of the Genes for Host -
Controlled Modification and Restriction in Escherichia Coli K-12. Genetics, 1965, 52 (5), 1043–
1050.
[21] Kropinski, A. M.; Mazzocco, A.; Waddell, T. E.; Lingohr, E.; Johnson, R. P. Enumeration of
Bacteriophages by Double Agar Overlay Plaque Assay. In Bacteriophages. Methods in Molecular
Biology; Clokie, M. R., Kropinski, A. M., Eds.; Humana Press, 2009; Vol. 501, pp 69 –76.
https://doi.org/10.1007/978-1-60327-164-6_7.
[22] Oliveira, A.; Sillankorva, S.; Quinta, R.; Henriques, A.; Sereno, R.; Azeredo, J. Isolation and
Characterization of Bacteriophages for Avian Pathogenic E. Coli Strains. J. Appl. Microbiol., 2009,
106 (6), 1919–1927. https://doi.org/10.1111/j.1365-2672.2009.04145.x.
[23] Jakočiūnė, D.; Moodley, A. A Rapid Bacteriophage DNA Extraction Method. Methods Protoc.,
2018, 1 (3), 27. https://doi.org/10.3390/mps1030027.
[24] Kot, W.; Vogensen, F. K.; Sørensen, S. J.; Hansen, L. H. DPS - A Rapid Method for Genome
Sequencing of DNA-Containing Bacteriophages Directly from a Single Plaque. J. Virol. Methods,
2014, 196, 152–156. https://doi.org/10.1016/j.jviromet.2013.10.040.
[25] Altschul, S. F.; Gish, W.; Miller, W.; Myers, E. W.; Lipman, D. J. Basic Local Alignment Search
Tool. J. Mol. Biol., 1990, 215 (3), 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2.
[26] Arndt, D.; Grant, J. R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D. S. PHASTER: A Better,
Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res., 2016, 44, W16–W21.
https://doi.org/10.1093/nar/gkw387.
[27] Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.
M.; Larsen, M. V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob.
Chemother., 2012, 67 (11), 2640–2644. https://doi.org/10.1093/jac/dks261.
[28] Joensen, K. G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R. S.; Nielsen, E. M.; Aarestrup, F. M.
Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection
of Verotoxigenic Escherichia Coli. J. Clin. Microbiol., 2014, 52 (5), 1501–1510.
https://doi.org/10.1128/JCM.03617-13.
[29] Overbeek, R.; Olson, R.; Pusch, G. D.; Olsen, G. J.; Davis, J. J.; Disz, T.; Edwards, R. A.; Gerdes,
S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using
Subsystems Technology (RAST). Nucleic Acids Res., 2014, 42 (D1), 206–214.
https://doi.org/10.1093/nar/gkt1226.
[30] R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for
Statistical Computing: Vienna, Austria 2020.
[31] Page, A. J.; Cummins, C. A.; Hunt, M.; Wong, V. K.; Reuter, S.; Holden, M. T. G.; Fookes, M.;
Falush, D.; Keane, J. A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis.
Bioinformatics, 2015, 31 (22), 3691–3693. https://doi.org/10.1093/bioinformatics/btv421.
[32] Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics, 2014, 30 (14), 2068–
2069. https://doi.org/10.1093/bioinformatics/btu153.
[33] Hyatt, D.; Chen, G.; Locascio, P. F.; Land, M. L.; Larimer, F. W.; Hauser, L. J. Prodigal: Prokaryotic

Chapter 3: Experimental Studies
89
Gene Recognition and Translation Initiation Site Identification. BMC Bioinformatics, 2010, 11 (119).
https://doi.org/10.1186/1471-2105-11-119.
[34] Larkin, M. A.; Blackshields, G.; Brown, N. P.; Chenna, R.; Mcgettigan, P. A.; McWilliam, H.;
Valentin, F.; Wallace, I. M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0.
Bioinformatics, 2007, 23 (21), 2947–2948. https://doi.org/10.1093/bioinformatics/btm404.
[35] Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics
Analysis across Computing Platforms. Mol. Biol. Evol., 2018, 35 (6), 1547–1549.
https://doi.org/10.1093/molbev/msy096.
[36] Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary
Analyses in R. Bioinformatics, 2019, 35 (3), 526–528.
https://doi.org/10.1093/bioinformatics/bty633.
[37] Schliep, K. P. Phangorn: Phylogenetic Analysis in R. Bioinformatics, 2011, 27 (4), 592–593.
https://doi.org/10.1093/bioinformatics/btq706.
[38] Darling, A. E.; Mau, B.; Perna, N. T. Progressivemauve: Multiple Genome Alignment with Gene
Gain, Loss and Rearrangement. PLoS One, 2010, 5 (6).
https://doi.org/10.1371/journal.pone.0011147.
[39] Besler, I.; Sazinas, P.; Harrison, C.; Gannon, L.; Redgwell, T.; Michniewski, S.; Hooton, S. P.;
Hobman, J. L.; Millard, A. Genome Sequence and Characterisation of Coliphage VB_Eco_SLUR29.
PHAGE Ther. Appl. Res., 2020, 1 (1), 38–44. https://doi.org/10.1089/phage.2019.0009.
[40] Bertozzi Silva, J.; Storms, Z.; Sauva geau, D. Host Receptors for Bacteriophage Adsorption. FEMS
Microbiol. Lett., 2016, 363 (4), 1–11. https://doi.org/10.1093/femsle/fnw002.
[41] Mullally, J.; Stockdale, Stephen R. Smith, M. K.; Draper, L. A.; Ross, P. R.; Hill, C. Complete
Genome Sequence of Escherichia Coli Phage APC_JM3.2 Isolated from a Chicken Cecum. Genome
Announc., 2018, 6 (1), e01292-17. https://doi.org/10.1128/genomeA.01292-17.
[42] Li, Y.; Chen, M.; Tang, F.; Yao, H.; Lu, C.; Zhang, W. Complete Genome Sequence of the Novel
Lytic Avian Pathogenic Coliphage NJ01. J. Virol., 2012, 86 (24), 13874–13875.
https://doi.org/10.1128/jvi.02727-12.
[43] Chen, M.; Xu, J.; Yao, H.; Lu, C.; Zhang, W. Isolation, Genome Sequencing and Functional Analysis
of Two T7-like Coliphages of Avian Pathogenic Escherichia Coli. Gene, 2016, 582 (1), 47–58.
https://doi.org/10.1016/j.gene.2016.01.049.
[44] Lau, G. L.; Sieo, C. C.; Tan, W. S.; Ho, Y. W. Characteristics of a Phage Effective for Colibacillosis
Control in Poultry. J. Sci. Food Agric., 2012, 92 (13), 2657–2663. https://doi.org/10.1002/jsfa.5683.
[45] Vandamme, P.; Pot, B.; Gillis, M.; De Vos, P.; Kersters, K.; Swings, J. Polyphasic Taxonomy, a
Consensus Approach to Bacterial Systematics. Microbiol. Rev., 1996, 60 (2), 407–438.
https://doi.org/10.1128/mmbr.60.2.407-438.1996.
[46] Prakash, O.; Verma, M.; Sharma, P.; Kumar, M.; Kumari, K.; Singh, A.; Kumari, H.; Jit, S.; Gupta,
S. K.; Khanna, M.; et al. Polyphasic Approach of Bacterial Classification - An Overview of Recent
Advances. Indian J. Microbiol., 2007, 47 (2), 98–108. https://doi.org/10.1007/s12088-007-0022-x.
[47] Lopes, A.; Tavares, P.; Petit, M. A.; Guérois, R.; Zinn-Justin, S. Automated Classification of Tailed
Bacteriophages According to Their Neck Organization. BMC Genomics, 2014, 15, 1027.
https://doi.org/10.1186/1471-2164-15-1027.
[48] Sullivan, M. B.; Coleman, M. L.; Quinlivan, V.; Rosenkrantz, J. E.; DeFrancesco, A. S.; Tan, G.; Fu,
R.; Lee, J. A.; Waterbury, J. B.; Bielawski, J. P.; et al. Portal Protein Diversity and Phage Ecology.
Environ. Microbiol., 2008, 10 (10), 2810–2823. https://doi.org/10.1111/j.1462-2920.2008.01702.x.
[49] Glazko, G.; Makarenkov, V.; Liu, J.; Mushegian, A. Evolutionary History of Bacteriophages with
Double-Stranded DNA Genomes. Biol. Direct, 2007, 2 (36), 1–14. https://doi.org/10.1186/1745-
6150-2-36.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
90
[50] Almpanis, A.; Swain, M.; Gatherer, D.; McEwan, N. Correlation between Bacterial G+C Content,
Genome Size and the G+C Content of Associated Plasmids and Bacteriophages. Microb. Genomics,
2018, 4 (4), e000168. https://doi.org/10.1099/mgen.0.000168.
[51] Jacobs-Sera, D.; Marinelli, L. J.; Bowman, C.; Broussard, G. W.; Guerrero Bustamante, C.; Boyle,
M. M.; Petrova, Z. O.; Dedrick, R. M.; Pope, W. H.; Modlin, R. L.; et al. On the Nature of
Mycobacteriophage Diversity and Host Preference. Virology, 2012, 434 (2), 187–201.
https://doi.org/10.1016/j.virol.2012.09.026.
[52] McNair, K.; Aziz, R. K.; Pusch, G. D.; Overbeek, R.; Dutilh, B. E.; Edwards, R. Phage Genome
Annotation Using the RAST Pipeline. In Bacteriophages. Methods in Molecular Biology; Humana
Press, New York, NY, 2018; Vol. 1681, pp 231–238. https://doi.org/10.1007/978-1-4939-7343-
9_17.
[53] Salisbury, A.; Tsourkas, P. K. A Method for Improving the Accuracy and Efficiency of
Bacteriophage Genome Annotation. Int. J. Mol. Sci., 2019, 20 (3391), 1–17.
https://doi.org/10.3390/ijms20143391.
[54] Kupczok, A.; Neve, H.; Huang, K. D.; Hoeppner, M. P.; Heller, K. J.; Franz, C. M. A. P.; Dagan, T.
Rates of Mutation and Recombination in Siphoviridae Phage Genome Evolution over Three
Decades. Mol. Biol. Evol., 2018, 35 (5), 1147–1159. https://doi.org/10.1093/molbev/msy027.
[55] Klumpp, J.; Fouts, D. E.; Sozhamannan, S. Next Generation Sequencing Technologies and the
Changing Landscape of Phage Genomics. Bacteriophage, 2012, 2 (3), 190–199.
https://doi.org/10.4161/bact.22111.
[56] Hendrix, R. W. Bacteriophage Genomics. Curr. Opin. Microbiol., 2003, 6 (5), 506–511.
https://doi.org/10.1016/j.mib.2003.09.004.
[57] Hendrix, R. W.; Smith, M. C. M.; Burns, R. N.; Ford, M. E.; Hatfull, G. F. Evolutionary
Relationships among Diverse Bacteriophages and Prophages: All the World’s a Phage. Proc. Natl.
Acad. Sci., 1999, 96 (5), 2192–2197. https://doi.org/10.1073/pnas.96.5.2192.
[58] Pedulla, M. L.; Ford, M. E.; Houtz, J. M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J. A.; Jacobs-
Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N. R.; et al. Origins of Highly Mosaic Mycobacteriophage
Genomes. Cell, 2003, 113 (2), 171–182. https://doi.org/10.1016/S0092-8674(03)00233-2.

Chapter 3: Experimental Studies
91
Supplementary Script Kmer.py
#!/usr/bin/env python3
import Utils
import argparse
import sys
import math
import collections
#import timeit
parser=argparse.ArgumentParser(description="Creates a kmer profile from a fastq or fasta file")
parser.add_argument('seq_files',type=str,nargs='+')
parser.add_argument('-l','--length',type=int,default=10)
parser.add_argument('-e','--euclidian',action="store_true")
args=parser.parse_args()
kmerlength=args.length
def kmer_fq(fil):
kmers=dict()
fqs=Utils.FqStream(fh=fil)
for fq in fqs:
for i in range(len(fq)-kmerlength):
try:
kmers[fq.sseq()[i:i+kmerlength]]+=1.0
except KeyError:
kmers[fq.sseq()[i:i+kmerlength]]=1.0
unfounded=list()
for (k,v) in kmers.items():
try:
if kmers[k[::-1].translate(Utils.tr)] + v <3:
unfounded.append(k)
except KeyError:
unfounded.append(k)
for k in unfounded:
del kmers[k]
if args.euclidian:
normalize(kmers)
return kmers
def kmer_fasta(fil):
kmers=dict()
try:
fasta="".join([line.decode().strip() if line.decode()[0]!=">" else "" for line in fil])
except AttributeError:
fasta="".join([line.strip() if line[0]!=">" else "" for line in fil])
for i in range(len(fasta)-kmerlength):
try:
kmers[fasta[i:i+kmerlength]]+=1.0
except KeyError:
kmers[fasta[i:i+kmerlength]]=1.0
if args.euclidian:
normalize(kmers)
return kmers

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
92
def normalize(kmers):
s=0.0
for val in kmers.values():
s+=val*val
s=math.sqrt(s)
for k in kmers.keys():
kmers[k]/=s
return kmers
def dist2(kmer1,kmer2):
dist=0.0
(L1,L2)=(sum(kmer1.values()),sum(kmer2.values()))
for (k,v) in kmer1.items():
try:
dist+=min(v,kmer2[k])
except KeyError:
pass
return dist/(min(L1,L2))
def dist(kmer1,kmer2):
dist=0.0
ignore=set()
for (k,v) in kmer1.items():
try:
d=v-kmer2[k]
dist+=d*d
ignore.add(k)
except KeyError:
dist+=v*v
remainder=set(kmer2.keys()).difference(ignore)
for k in remainder:
d=kmer2[k]
dist+=d*d
return math.sqrt(dist)
kmers=list()
print("Reading data...",file=sys.stderr)
for filename in args.seq_files:
fil=Utils.gzopen(filename)
line=next(fil)
try:
line=line.decode()
except AttributeError:
pass
if line.startswith(">"):
filetype="Fasta"
kmers.append(kmer_fasta(fil))
elif line.startswith("@"):
filetype="Fastq"
fil.seek(0)
kmers.append(kmer_fq(fil))
else:
print("Unrecognized file format:\n{}".format(line),file=sys.stdout)
fil.close()
print("Done",file=sys.stderr)
for i in range(len(kmers)):
print(args.seq_files[i],end="\t")

Chapter 3: Experimental Studies
93
for j in range(i):
print("\t",end="")
for j in range(i+1,len(kmers)):
print("\t{}".format(dist(kmers[i],kmers[j]) if args.euclidian else dist2(kmers[i],kmers[j])),end="")
print("")

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
94
Su
pp
lem
en
tary
Ta
ble
S1
| C
hara
cte
rist
ics
of
refe
ren
ce g
en
om
es m
en
tio
ned in
th
is s
tud
y
Ph
ag
e n
am
e P
ha
ge
Gen
om
e
size (b
p)
G+
C%
#
CD
Ss
Ph
ag
e
fam
ily
Ph
ag
e
sub
fam
ily
Ph
ag
e
clu
ster
Ph
ag
e g
en
us
Access
ion
nu
mb
er
pS
f-1
Shig
ella
phag
e pS
f-1
51821
44.0
93
Sip
hovi
ridae
Tunavi
rinae
A1
Hanri
verv
irus
KC
710998.1
YU
EE
L01
E
scher
ichia
phag
e Y
UE
EL
01
169621
35.4
268
Myo
viri
dae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
KY
290975.2
fFiE
co0
6
Esc
her
ich
ia p
hag
e v
B_
Eco
M-f
FiE
co06
167076
35.5
267
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MG
781190.1
CE
B_
EC
3a
Esc
her
ich
ia p
hag
e v
B_
Eco
s_C
EB
_E
C3
a 44234
44.2
72
Sip
hovi
ridae
Tu
navi
rinae
B
Rtp
viru
s K
Y398841.1
ST
0
Esc
her
ichia
phag
e S
T0
170496
37.7
270
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
MF
044457.1
O157_ 3
E
scher
ichia
coli O
157
typin
g p
hag
e 3
168733
37.3
268
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
KP
869101.1
Sw
an0
1
Esc
her
ich
ia p
hag
e v
B_
Eco
_sw
an0
1
50865
44.7
85
Sip
hovi
ridae
Tu
navi
rinae
A2
Uncl
assi
fied
L
T841304.1
RB
14
E
nte
rob
acte
ria
ph
age
RB
14
165429
35.3
264
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
FJ8
39692.1
Alf
5
Esc
her
ichia
phag
e vB
_E
coM
_A
lf5
87662
39.0
130
Myo
viri
dae
Ounavi
rinae
C
Fel
ixounavi
rus
KX
377933.1
OS
YS
P
Esc
her
ichia
phag
e O
SY
SP
110901
39.2
161
Sip
hovi
ridae
N/A
D
T
equin
tavi
rus*
M
F402939.1
VpaE
1
Esc
her
ichia
phag
e vB
_E
coM
-VpaE
1
88403
38.9
126
Myo
viri
dae
Ounavi
rinae
C
Fel
ixounavi
rus
KM
657822.1
AY
O1
45
A
Esc
her
ich
ia p
hag
e v
B_
Eco
M_
AY
O14
5A
87372
39.0
128
M
yovi
ridae
Ou
navi
rinae
C
Fel
ixounavi
rus
KR
014248.1
SU
10
E
sch
eric
hia
Ph
age
vB
_E
coP
_S
U10
77327
42.1
127
P
odovi
ridae
N/A
-
Kura
viru
s*
KM
044272.1
Eco
S_95
Esc
her
ichia
phag
e vB
_E
coS
-95
50910
44.8
87
Sip
hovi
ridae
Uncl
assi
fied
A
2
Uncl
assi
fied
M
F564201.1
Jahat
_M
G145
Esc
her
ichia
phag
e Ja
hat
_M
G145
50984
45.7
87
Sip
hovi
ridae
Tunavi
rinae
A3
Uncl
assi
fied
M
K552105.1
Lam
bd
a E
nte
rob
acte
ria
ph
age
lam
bd
a
48502
49.9
71
Sip
hovi
ridae
Tu
navi
rinae
- L
am
bdavi
rus
J02459.1
T1
E
nte
rob
acte
ria
ph
age
T1
48836
45.6
76
Sip
hovi
ridae
Tu
navi
rinae
B
Tunavi
rus
AY
216660.1
T4
E
nte
rob
acte
ria
ph
age
T4
168903
35.3
266
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
AF
158101.6
T5
B
acte
riophag
e T
5
121750
39.3
172
Sip
hovi
ridae
N/A
D
T
equin
tavi
rus*
A
Y543070.1
T7
E
nte
robac
teri
a phag
e T
7
39937
48.4
52
Podovi
ridae
Auto
gra
phiv
irin
ae
- T
esep
tim
avi
rus
NC
_001604.1
P1
E
sch
eric
hia
vir
us
P1
66750
47.6
92
M
yovi
ridae
N/A
-
Punavi
rus*
M
H445381.1
P2
E
nte
rob
acte
ria
ph
age
P2
31200
52.6
49
M
yovi
ridae
Ped
uovi
rinae
- P
eduovi
rus
NC
_041848.1
P22
E
nte
robac
teri
a phag
e P
22
41724
47.1
68
Podovi
ridae
N/A
-
Led
erber
gvi
rus*
N
C_002371.2
Mu
E
nte
robac
teri
a phag
e M
u
36717
52.1
54
Myo
viri
dae
N/A
-
Muvi
rus*
N
C_000929.1
Hen
u8**
E
scher
ichia
phag
e H
enu8
49890
44.2
85
Sip
hovi
ridae
Tunavi
rinae
A1
Hanri
verv
irus
MN
055691.1
G2
9_
2*
*
Esc
her
ich
ia p
hag
e v
B_
Eco
S_G
29-2
51739
44.0
89
Sip
hovi
ridae
Tu
navi
rinae
A1
Hanri
verv
irus
MK
373798.1
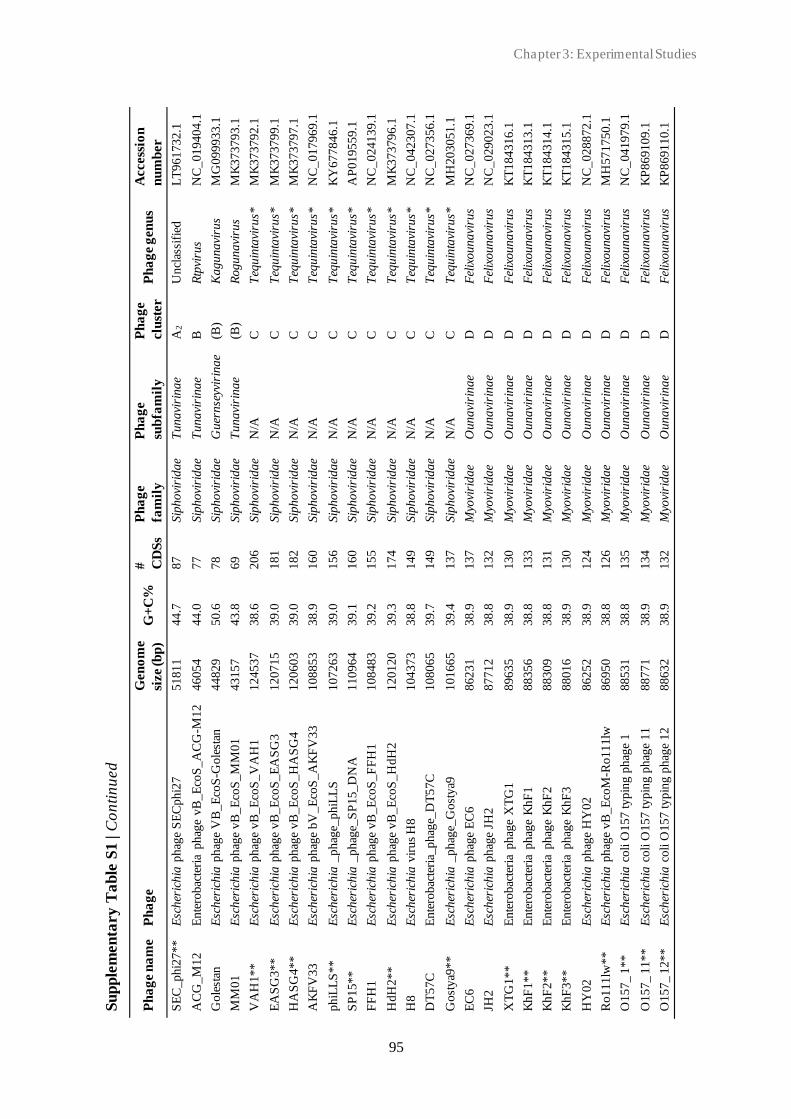
Chapter 3: Experimental Studies
95
S
up
ple
men
tary
Ta
ble
S1
| C
on
tin
ued
Ph
ag
e n
am
e P
ha
ge
Gen
om
e
size (b
p)
G+
C%
#
CD
Ss
Ph
ag
e
fam
ily
Ph
ag
e
sub
fam
ily
Ph
ag
e
clu
ster
P
ha
ge g
en
us
Access
ion
nu
mb
er
SE
C_phi2
7**
E
scher
ichia
phag
e S
EC
ph
i27
51811
44.7
87
Sip
hovi
ridae
Tuna
viri
nae
A2
Uncl
assi
fied
L
T961732.1
AC
G_M
12
Ente
robac
teri
a phag
e vB
_E
coS
_A
CG
-M12
46054
44.0
77
Sip
hovi
ridae
Tuna
viri
nae
B
Rtp
viru
s N
C_019404.1
Go
lest
an
Esc
her
ich
ia p
hag
e V
B_
Eco
S-G
ole
stan
44829
50.6
78
Sip
hovi
ridae
Guer
nse
yvir
ina
e (B
) K
agunavi
rus
MG
099933.1
MM
01
Esc
her
ich
ia p
hag
e v
B_
Eco
S_
MM
01
43157
43.8
69
Sip
hovi
ridae
Tun
avi
rina
e (B
) R
ogunavi
rus
MK
373793.1
VA
H1**
Esc
her
ichia
phag
e v
B_E
coS
_V
AH
1
124537
38.6
206
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
M
K373792.1
EA
SG
3**
Esc
her
ichia
phag
e v
B_E
coS
_E
AS
G3
120715
39.0
181
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
M
K373799.1
HA
SG
4*
*
Esc
her
ich
ia p
hag
e v
B_
Eco
S_
HA
SG
4
120603
39.0
182
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
M
K373797.1
AK
FV
33
E
sch
eric
hia
ph
age
bV
_E
coS
_A
KF
V33
108853
38.9
160
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
N
C_017969.1
ph
iLL
S*
*
Esc
her
ich
ia _
ph
age_
ph
iLL
S
107263
39.0
156
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
K
Y677846.1
SP
15**
E
scher
ichia
_ph
age_
SP
15_
DN
A
110964
39.1
160
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
A
P019559.1
FF
H1
E
scher
ichia
phag
e v
B_E
coS
_F
FH
1
108483
39.2
155
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
N
C_024139.1
Hd
H2
**
E
sch
eric
hia
ph
age
vB
_E
coS
_H
dH
2
120120
39.3
174
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
M
K373796.1
H8
Esc
her
ich
ia v
iru
s H
8
104373
38.8
149
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
N
C_042307.1
DT
57C
E
nte
robac
teri
a_phag
e_D
T57C
108065
39.7
149
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
N
C_027356.1
Gost
ya9
**
E
scher
ichia
_ph
age_
Gost
ya9
101665
39.4
137
Sip
hovi
ridae
N/A
C
T
equin
tavi
rus*
M
H203051.1
EC
6
Esc
her
ichia
phag
e E
C6
86231
38.9
137
Myo
viri
dae
Ouna
viri
nae
D
Fel
ixounavi
rus
NC
_027369.1
JH2
E
sch
eric
hia
ph
age
JH2
87712
38.8
132
M
yovi
ridae
Oun
avi
rina
e D
F
elix
ounavi
rus
NC
_029023.1
XT
G1
**
E
nte
rob
acte
ria
ph
age
XT
G1
89635
38.9
130
M
yovi
ridae
Oun
avi
rina
e D
F
elix
ounavi
rus
KT
184316.1
KhF
1**
Ente
robac
teri
a phag
e K
hF
1
88356
38.8
133
Myo
viri
dae
Ouna
viri
nae
D
Fel
ixounavi
rus
KT
184313.1
KhF
2**
Ente
robac
teri
a phag
e K
hF
2
88309
38.8
131
Myo
viri
dae
Ouna
viri
nae
D
Fel
ixounavi
rus
KT
184314.1
Kh
F3
**
E
nte
rob
acte
ria
ph
age
Kh
F3
88016
38.9
130
M
yovi
ridae
Oun
avi
rina
e D
F
elix
ounavi
rus
KT
184315.1
HY
02
Esc
her
ich
ia p
hag
e H
Y0
2
86252
38.9
124
M
yovi
ridae
Oun
avi
rina
e D
F
elix
ounavi
rus
NC
_028872.1
Ro111lw
**
E
scher
ichia
phag
e v
B_E
coM
-Ro111lw
86950
38.8
126
Myo
viri
dae
Ouna
viri
nae
D
Fel
ixounavi
rus
MH
571750.1
O157_ 1
**
Esc
her
ichia
coli
O157
typin
g p
hag
e 1
88531
38.8
135
Myo
viri
dae
Ouna
viri
nae
D
Fel
ixounavi
rus
NC
_041979.1
O157_ 1
1**
E
scher
ichia
coli
O157
typin
g p
hag
e 1
1 88771
38.9
134
Myo
viri
dae
Ouna
viri
nae
D
Fel
ixounavi
rus
KP
869109.1
O1
57
_ 1
2*
*
Esc
her
ich
ia c
oli
O1
57
ty
pin
g p
hag
e 1
2
88632
38.9
132
M
yovi
ridae
Oun
avi
rina
e D
F
elix
ounavi
rus
KP
869110.1

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
96
Su
pp
lem
en
tary
Ta
ble
S1
| C
on
tin
ued
Ph
ag
e n
am
e
Ph
ag
e
Gen
om
e
size (b
p)
G+
C%
#
CD
Ss
Ph
ag
e
fam
ily
Ph
ag
e
sub
fam
ily
Ph
ag
e
clu
ster
P
ha
ge g
en
us
Access
ion
nu
mb
er
WV
8
Ente
robac
teri
a phag
e W
V8
8
84
87
38.9
134
Myo
viri
dae
Ounavi
rinae
D
Fel
ixounavi
rus
NC
_012749.1
HX
01
Ente
robac
teri
a phag
e H
X01
1
69
15
8
37.6
268
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
NC
_018855.1
KA
W3E
185**
Esc
her
ich
ia p
hag
e
vB
_E
coM
_K
AW
3E
185
1
70
18
7
37.6
270
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
MK
373782.1
WF
bE
185**
E
scher
ichia
phag
e vB
_E
coM
_W
FbE
185
1
70
42
9
37.6
272
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
MK
373778.1
G5
3*
*
Esc
her
ich
ia p
hag
e v
B_
Eco
M_G
53
1
67
83
4
37.8
268
M
yovi
ridae
Tev
envi
rinae
E
Mosi
gvi
rus
MK
327943.1
AP
EC
c01
E
sch
eric
hia
ph
age
AP
EC
c01
1
68
77
1
37.7
267
M
yovi
ridae
Tev
envi
rinae
E
Mosi
gvi
rus
NC
_029091.1
MM
02
**
E
sch
eric
hia
ph
age
vB
_E
coM
_M
M02
1
69
20
1
37.6
269
M
yovi
ridae
Tev
envi
rinae
E
Mosi
gvi
rus
MK
373784.1
HP
3
Esc
her
ichia
phag
e H
P3
1
70
25
4
37.6
268
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
NC
_041920.1
AT
K47**
E
nte
robac
teri
a phag
e A
TK
47
1
70
02
0
37.6
270
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
KT
184309.1
AT
K4
8*
*
En
tero
bac
teri
a p
hag
e A
TK
48
1
69
72
9
37.6
270
M
yovi
ridae
Tev
envi
rinae
E
Mosi
gvi
rus
KT
184310.1
O1
57
_ 6
E
sch
eric
hia
co
li O
15
7 ty
pin
g p
hag
e 6
1
60
57
0
37.6
249
M
yovi
ridae
Tev
envi
rinae
E
Mosi
gvi
rus
NC
_041864.1
JS09
E
scher
ichia
phag
e vB
_E
coM
_JS
09
1
69
14
8
37.6
271
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
NC
_024124.2
G2285**
Esc
her
ichia
phag
e vB
_E
coM
_G
2285
1
66
67
5
37.5
262
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
MK
327933.1
G2469**
Esc
her
ichia
phag
e vB
_E
coM
_G
2469
1
70
45
2
37.6
271
Myo
viri
dae
Tev
envi
rinae
E
Mosi
gvi
rus
MK
327934.1
G2
54
0_
3*
*
Esc
her
ich
ia p
hag
e v
B_
Eco
M_G
2540
-3
16
86
54
35.3
271
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MK
327944.1
G2
9*
*
Esc
her
ich
ia p
hag
e v
B_
Eco
M_G
29
1
68
24
1
35.3
269
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MK
327940.1
G4500**
Esc
her
ichia
phag
e vB
_E
coM
_G
4500
1
68
36
3
35.3
268
Myo
viri
dae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MK
327945.1
D5505**
Esc
her
ichia
phag
e D
5505
1
68
04
9
35.4
269
Myo
viri
dae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MK
327929.1
G9
06
2*
*
Esc
her
ich
ia p
hag
e v
B_
Eco
M_G
9062
1
68
67
0
35.3
268
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MK
373779.1
CF
2
Esc
her
ich
ia p
hag
e C
F2
1
68
18
8
35.4
264
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
NC
_041919.1
AC
G_
C4
0*
*
Ente
robac
teri
a phag
e vB
_E
coM
_A
CG
-
C40
16
73
96
35.2
273
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
NC
_019399.1
OE
55
05
**
E
sch
eric
hia
ph
age
vB
_E
coM
_O
E5505
1
68
75
6
35.2
273
M
yovi
ridae
Tev
envi
rinae
F
Teq
uatr
ovi
rus
MK
373785.1
*N
o s
ubfa
mily i
s def
ined
acc
ord
ing t
o the
Inte
rnat
ional
Com
mitte
e on T
axonom
y V
iruse
s (I
CT
V).
Phag
e gen
us
is u
sed inst
ead. N
/A =
non
-applica
ble
. -
= s
ingle
ton,
no c
lust
er.
**N
ot fo
und in
IC
TV
dat
abas
e. C
lass
ific
atio
n a
ccord
ing t
o N
atio
nal
Cen
ter
for
Bio
tech
nolo
gy info
rmat
ion (
NC
BI)
.
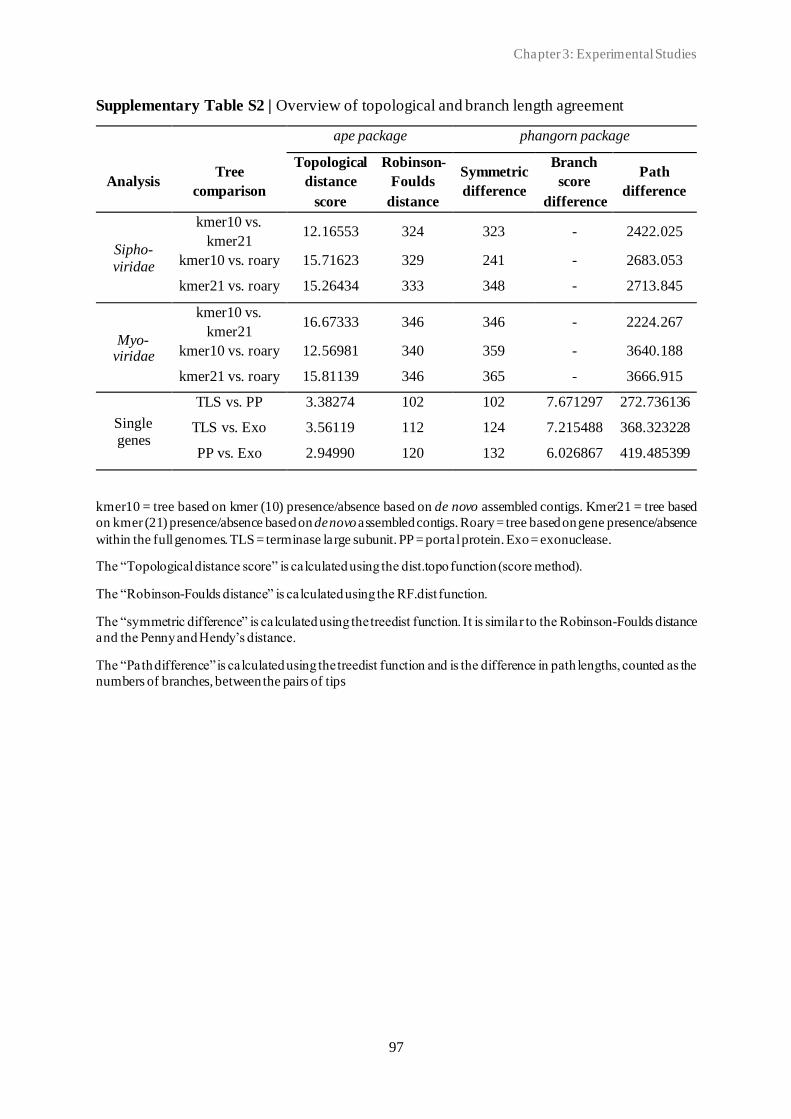
Chapter 3: Experimental Studies
97
Supplementary Table S2 | Overview of topological and branch length agreement
ape package phangorn package
Analysis Tree
comparison
Topological
distance
score
Robinson-
Foulds
distance
Symmetric
difference
Branch
score
difference
Path
difference
Sipho-
viridae
kmer10 vs.
kmer21 12.16553 324 323 - 2422.025
kmer10 vs. roary 15.71623 329 241 - 2683.053
kmer21 vs. roary 15.26434 333 348 - 2713.845
Myo-viridae
kmer10 vs.
kmer21 16.67333 346 346 - 2224.267
kmer10 vs. roary 12.56981 340 359 - 3640.188
kmer21 vs. roary 15.81139 346 365 - 3666.915
Single
genes
TLS vs. PP 3.38274 102 102 7.671297 272.736136
TLS vs. Exo 3.56119 112 124 7.215488 368.323228
PP vs. Exo 2.94990 120 132 6.026867 419.485399
kmer10 = tree based on kmer (10) presence/absence based on de novo assembled contigs. Kmer21 = tree based
on kmer (21) presence/absence based on de novo assembled contigs. Roary = tree based on gene presence/absence
within the full genomes. TLS = terminase large subunit. PP = portal protein. Exo = exonuclease.
The “Topological distance score” is calculated using the dist.topo function (score method).
The “Robinson-Foulds distance” is calculated using the RF.dist function.
The “symmetric difference” is calculated using the treedist function. It is similar to the Robinson-Foulds distance
and the Penny and Hendy’s distance.
The “Path difference” is calculated using the treedist function and is the difference in path lengths, counted as the
numbers of branches, between the pairs of tips

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
98
Su
pp
lem
en
tary
Ta
ble
S3
| O
verv
iew
of
ass
em
bly
sett
ings
an
d s
tati
stic
s
R
aw
da
ta
T
rim
min
g
Ass
em
bly
Ph
age
# o
f
rea
ds
Av
g.
len
gth
# o
f re
ad
s
aft
er t
rim
Av
g. le
ng
th
aft
er t
rim
# o
f m
atc
hed
red
s
Min
. co
nti
g
len
gth
Max. co
nti
g
len
gth
# o
f
con
tigs
N50
Phag
e 8
20854
212.9
20792
199.1
20792
51031
51031
1
51031
Phag
e 10
175980
212.0
175598
212.2
174572
380
167166
4
167166
Phag
e 1
1
23
71
28
1
97
.3
23
64
93
1
97
.6
23
58
00
5
03
169478
3
169478
Phag
e 1
5
78
81
6
18
4.6
7
83
77
1
85
.2
77
99
2
16
9392
169392
1
169392
Phag
e 17
28544
204.8
28.4
73
204.9
28300
250
44592
5
44592
Phag
e 18
122820
179.6
122040
180.3
121527
477
169391
2
169391
Phag
e 28
21654
230.5
21628
230.4
21376
254
52716
2
52716
Phag
e 3
0
16
20
94
1
87
.9
16
14
82
1
88
.3
16
06
23
2
53
169109
10
169109
Phag
e 4
7
32
58
2
19
8.2
3
24
62
1
98
.4
32
30
5
95
8
50105
2
50105
Phag
e 48
18862
211.8
18844
210.8
18665
51031
51031
1
51031
Phag
e 52
44154
208.7
44024
208.7
43542
373
50784
5
50784
Phag
e 5
3
37
15
4
21
0.5
3
70
72
2
10
.6
36
88
3
25
3
50582
2
50582
Phag
e 5
4
62
70
2
21
0.2
6
25
41
2
10
.3
62
21
3
25
1
51031
5
51031
Phag
e 55
233558
198.8
232979
199.0
231603
254
167131
8
167131
Phag
e 56
55786
209.9
55672
209.9
55010
50829
52716
2
52716
Phag
e 58
51386
209.7
51321
209.6
51069
256
44592
3
44592
Phag
e 5
9
16
27
0
23
5.4
1
62
58
2
35
.3
16
05
3
67
1
51031
2
51031
Phag
e 6
0
34
73
0
20
2.1
3
46
24
2
02
.5
34
43
5
86
237
86237
1
86237
Phag
e 61
219040
202.5
218539
202.5
217486
254
108138
6
108138
Phag
e 62
27242
201.9
27136
202.4
26952
87871
87871
1
87871
Phag
e 6
3
50
71
8
20
6.5
5
06
16
2
06
.5
50
41
6
49
132
49132
1
49132
Phag
e 6
4
44
87
2
20
9.0
4
47
58
2
09
.1
44
52
7
32
1
51031
2
51031
Phag
e 65
41890
210.1
41804
210.2
41612
51031
51031
1
51031
Phag
e 66
46548
208.5
46400
209.0
46135
257
89604
3
89604
Phag
e 68
65582
211.2
65499
211.0
65188
262
51029
2
51029

Chapter 3: Experimental Studies
99
Su
pp
lem
en
tary
Ta
ble
S3
| C
on
tin
ued
R
aw
da
ta
Tri
mm
ing
A
ssem
bly
Ph
age
# o
f
read
s
Avg.
len
gth
# o
f re
ad
s
aft
er t
rim
Avg. le
ngth
aft
er t
rim
# o
f m
atc
hed
red
s
Min
. co
nti
g
len
gth
Max. co
nti
g
len
gth
# o
f
con
tigs
N50
Phag
e 6
9
48
63
2
21
4.9
4
85
32
21
4.8
4
79
24
2
78
50777
22
50777
Phag
e 7
0
45
03
2
21
2.0
4
49
34
21
2.1
4
47
11
2
50
44290
2
44290
Phag
e 71
24156
230.6
2
41
28
230.7
23787
415
51031
2
51031
Phag
e 72
32508
222.4
3
24
42
222.5
32100
253
51031
2
51031
Phag
e 7
3
42
83
2
22
6.7
4
27
52
22
6.8
4
24
07
4
6738
46738
1
46738
Phag
e 7
4
61
36
2
21
1.3
6
12
08
21
1.5
6
07
37
2
58
45171
4
45171
Phag
e 75
44444
208.3
4
43
54
208.3
44086
50445
50445
1
50445
Phag
e 76
43978
219.5
4
39
26
219.3
43522
504
50843
3
50843
Phag
e 77
59306
215.2
5
91
83
215.3
58920
51073
51073
1
51073
Phag
e 7
8
49
21
8
20
6.4
4
90
98
20
6.6
4
86
77
2
56
89644
2
89644
Phag
e 7
9
50
82
2
24
6.4
5
08
16
24
6.4
5
03
60
2
53
87100
7
87100
Phag
e 80
46730
207.1
4
66
08
207.2
46307
52703
52703
1
52703
D
efa
ult s
ett
ings
wa
s u
sed f
or C
LC
Genom
ics
Wo
rkbench d
e n
ov
o a
ssem
bly
, ex
cept f
or m
inim
um
co
ntig le
ngth
th
at w
as
chan
ged
to
250 b
p.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
100
Su
pp
lem
en
tary F
igu
re S
1 |
Ro
ary
matr
ix b
ase
d o
n g
en
e li
st o
f S
iph
ovi
rida
e colip
hages.
Ph
ages
iso
late
d in
th
is s
tud
y a
re h
igh
ligh
ted. E
ach
co
lour re
pre
sents
a c
lust
er:
Clu
ster A
(b
lue),
clu
ster B
(gre
en
), a
nd
clu
ster
C (re
d).
Clu
ster A
su
bclu
ster
s in
clu
de
A1
(ligh
t b
lue),
A2
(b
lue),
an
d A
3 (d
ark
blu
e).

Chapter 3: Experimental Studies
101
Su
pp
lem
en
tary F
igu
re S
2 |
UP
GM
A tre
e b
ase
d o
n r
oary
matr
ix o
f S
iphovir
idae c
olip
hages.
Ph
ages
iso
late
d in
th
is s
tud
y a
re h
igh
ligh
ted. E
ach c
olo
ur re
pre
sents
a
clu
ster:
Clu
ster A
(b
lue),
clu
ster B
(gre
en
), a
nd
clu
ster C
(re
d).
Clu
ster A
su
bclu
ster
s in
clu
de
A1
(ligh
t b
lue),
A2
(b
lue),
an
d A
3 (d
ark
blu
e).

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
102
Su
pp
lem
en
tary F
igu
re
S2
| R
oary
matr
ix b
ase
d o
n g
en
e list
of
Myovir
idae c
olip
hages.
Ph
ages
iso
late
d in
th
is s
tudy a
re h
igh
ligh
ted. E
ach
colo
ur
rep
rese
nts
a c
lust
er:
Clu
ster D
(o
ran
ge),
clu
ster E
(p
urp
le),
and
clu
ster F
(bro
wn
).
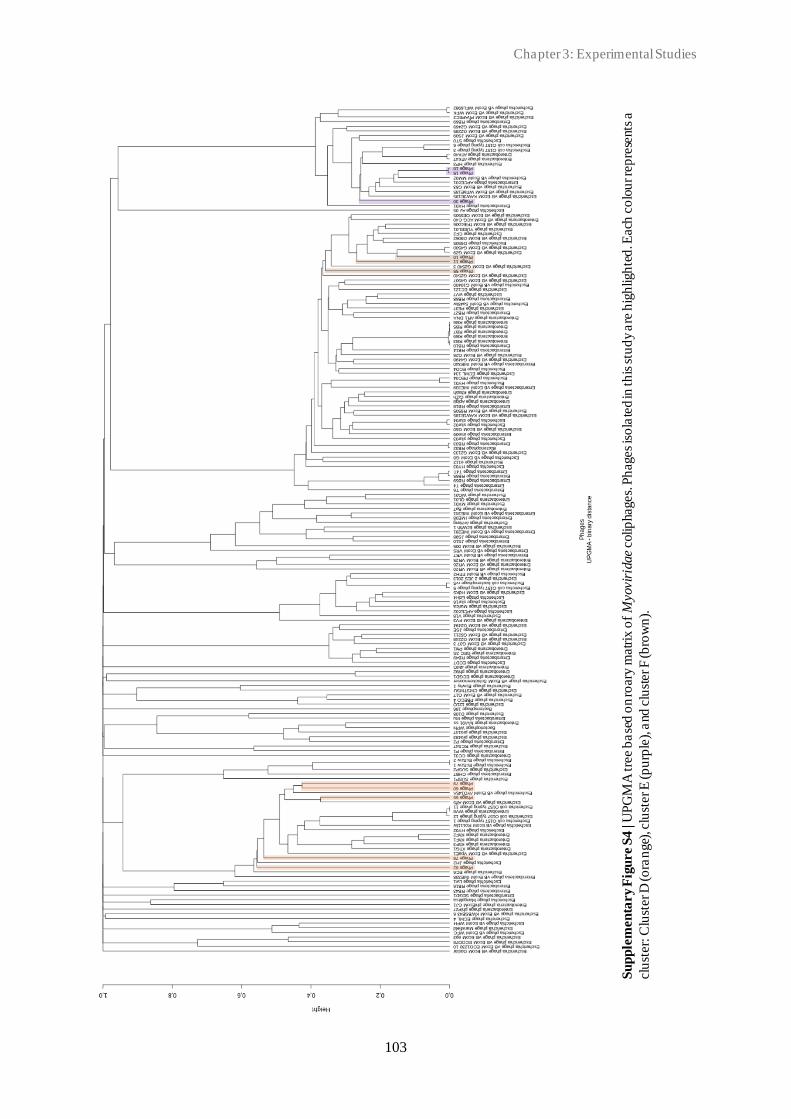
Chapter 3: Experimental Studies
103
Su
pp
lem
en
tary F
igu
re
S4
| U
PG
MA
tre
e b
ase
d o
n ro
ary
matr
ix o
f M
yovir
idae c
olip
hages.
Ph
ages
iso
late
d in
th
is s
tud
y a
re h
igh
ligh
ted
. Each
co
lou
r re
pre
sents
a
clu
ster:
Clu
ster D
(o
ran
ge)
, clu
ster E
(p
urp
le),
and
clu
ster F
(b
row
n).

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
104
Supplementary Figure S5 | Maximum likelihood tree based on the nucleotide sequence of the phage portal
protein. Phages were grouped together into six clusters: A-F, according to phage family and subfamily. Cluster A
and B: Siphoviridae, Tunavirinae, cluster C: Siphoviridae and Tequintavirus genus, cluster D: Myoviridae,
Ounavirinae, and cluster E and F: Myoviridae, Tevenvirinae. Cluster A was divided into three subclusters: A1,
A2 and A3. Subcluster A2 was divided in two. The tree was constructed using the MEGA X software. The
percentage of threes in which the associated taxa clustered together is shown next to the branches. The tree is
drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 62
nucleotide sequences.
A1
A2
A2
A3
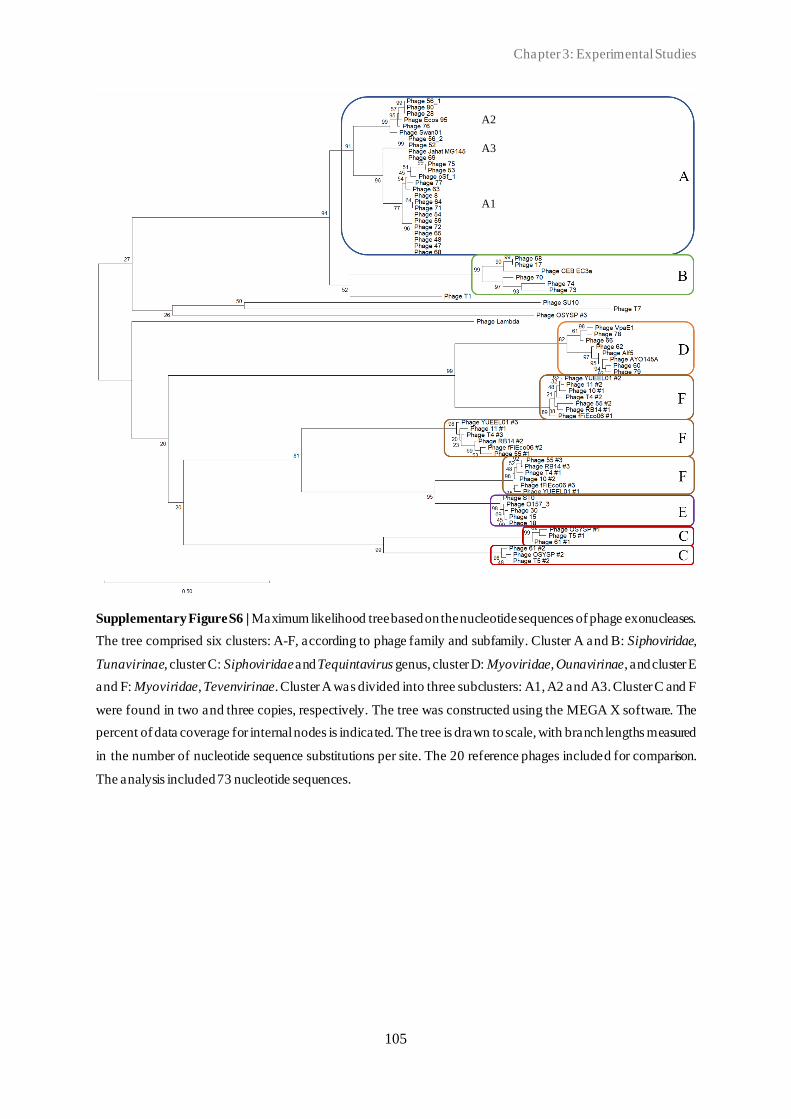
Chapter 3: Experimental Studies
105
Supplementary Figure S6 | Maximum likelihood tree based on the nucleotide sequences of phage exonucleases.
The tree comprised six clusters: A-F, according to phage family and subfamily. Cluster A and B: Siphoviridae,
Tunavirinae, cluster C: Siphoviridae and Tequintavirus genus, cluster D: Myoviridae, Ounavirinae, and cluster E
and F: Myoviridae, Tevenvirinae. Cluster A was divided into three subclusters: A1, A2 and A3. Cluster C and F
were found in two and three copies, respectively. The tree was constructed using the MEGA X software. The
percent of data coverage for internal nodes is indicated. The tree is drawn to scale, with branch lengths measured
in the number of nucleotide sequence substitutions per site. The 20 reference phages included for comparison.
The analysis included 73 nucleotide sequences.
A1
A2
A3

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
106
Comparative genomics (Supplementary Figure S7-14)
General description
Phage genome sequences were compared for each cluster (cluster A-F) using the
progressiveMauve software. Boxes with same colours represent LCBs, indicating homologous
DNA regions shared by two or more genomes without sequence rearrangements. LCBs
indicated below the horizontal black line represent reverse compliments of the reference LCB
(reference genome is marked with a blue square). The height of the similarity profile within
the LCBs corresponds to the average level of conservation in that region of the genome
sequence. White boxes below the horizontal black line represents annotated genes in the
reference sequences included. The terminase large subunit encoding genes is indicated with a
black square in each genome sequence.

Chapter 3: Experimental Studies
107
Supplementary Figure S7 | Comparative genomics of subcluster A1 phages. Genome sequences of 17
Siphoviridae subcluster A1 phages were compared. 16 LCBs were identified.
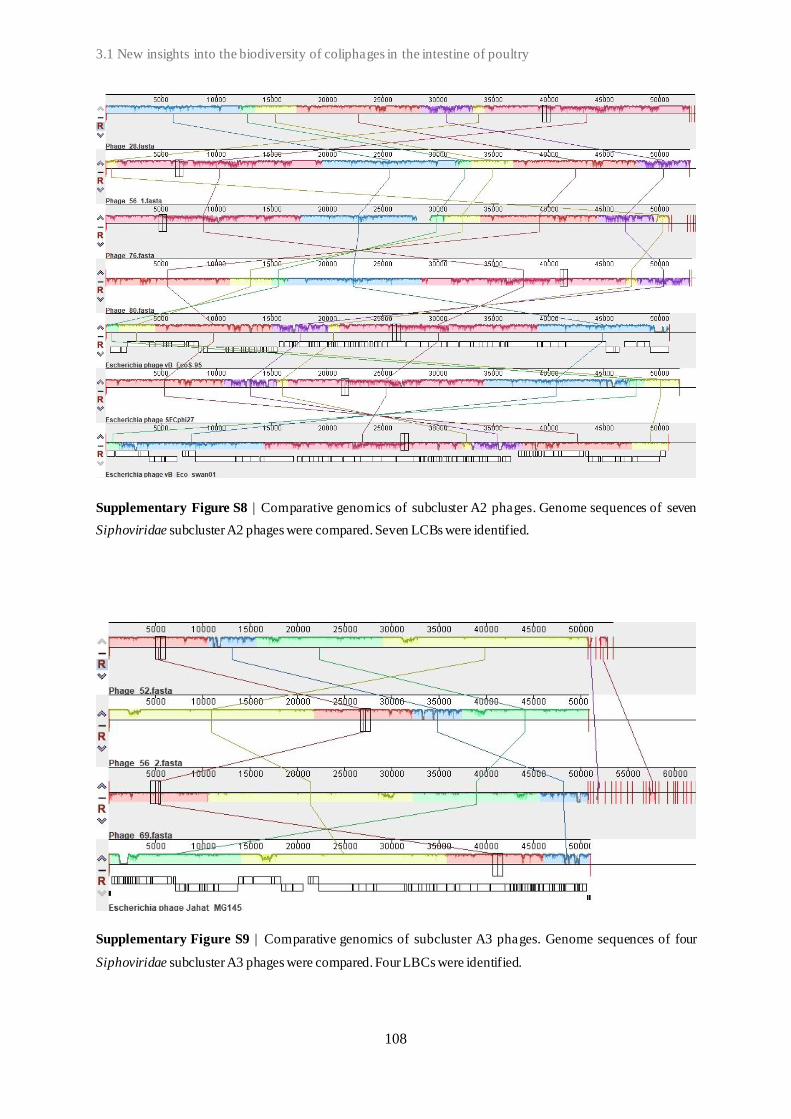
3.1 New insights into the biodiversity of coliphages in the intestine of poultry
108
Supplementary Figure S8 | Comparative genomics of subcluster A2 phages. Genome sequences of seven
Siphoviridae subcluster A2 phages were compared. Seven LCBs were identified.
Supplementary Figure S9 | Comparative genomics of subcluster A3 phages. Genome sequences of four
Siphoviridae subcluster A3 phages were compared. Four LBCs were identified.

Chapter 3: Experimental Studies
109
Supplementary Figure S10 | Comparative genomics of cluster B phages. Genome sequences of six Siphoviridae
cluster B phages were compared. Six LCBs were identified.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
110
Supplementary Figure S11 | Comparative genomics of cluster C phages. 10 Genome sequences of 10
Siphoviridae phages were compared. 6-10 LCBs were identified in each genome. Repeat-rich regions are indicated
with salmon-coloured bars next to the white annotation boxes.

Chapter 3: Experimental Studies
111
Supplementary Figure S12 | Comparative genomics of cluster D phages. Genome sequences of 19 Myoviridae
cluster D phages were compared. 14-17 LCBs were identified for each phage genome.

3.1 New insights into the biodiversity of coliphages in the intestine of poultry
112
Supplementary Figure S13 | Comparative genomics of cluster E phages. Genome sequences of 18 Myoviridae
cluster E phages were compared. 16-17 LCBs were identified for each phage genome.

Chapter 3: Experimental Studies
113
Supplementary Figure S14 | Comparative genomics of F subcluster phages. Genome sequences of 13 Myoviridae
cluster F phages were compared. Five LCBs were identified for each phage genome.

114

Chapter 3: Experimental Studies
115
3.2
Classification of in vitro phage-host population growth dynamics
Patricia E. Sørensen1,2, Duncan Y. K. Ng3, Luc Duchateau4 , Hanne Ingmer5, An
Garmyn1, and Patrick Butaye1,2
1 Department of Pathobiology, Pharmacology and Zoological Medicine, Ghent University, Merelbeke,
Belgium
2 Department of Biomedical Sciences, Ross University School of Veterinary Medicine, St. Kitts, West
Indies
3 Department of Bacteria, Parasites and Fungi, Statens Serum Institut, Denmark
4 Biometrics Research Center, Ghent University, Belgium
5 Department of Veterinary and Animal Sciences, University of Copenhagen, Denmark
Published in Microorganisms 2021, 9 (2470)
3.2 Classification of in vitro phage-host population growth dynamics

3.2 Classification of in vitro phage-host population growth dynamics
116
Abstract
The therapeutic use of bacteriophages (phage therapy) represents a promising alternative to
antibiotics to control bacterial pathogens. However, the understanding of the phage-bacterium
interactions and population dynamics seems essential for successful phage therapy
implementation. Here, we investigated the effect of three factors, phage species (18 lytic E.
coli-infecting phages), bacterial strain (10 APEC strains), and multiplicity of infection (MOI)
(MOI 10, 1, and 0.1) on the bacterial growth dynamics. All factors had a significant effect, but
the phage appeared to be the most important. The results showed seven distinct growth patterns.
The first pattern corresponded to the normal bacterial growth pattern in the absence of a phage.
The second pattern was complete bacterial killing. The remaining patterns were in -between,
characterised by delayed growth and/or variable killing of the bacterial cells. In conclusion,
this study demonstrates that the phage-host dynamics is an important factor in the capacity of
a phage to eliminate bacteria. The classified patterns show that this is an essential factor to
consider when developing a phage therapy. This methodology can be used to rapidly screen
for novel phage candidates for phage therapy. Accordingly, the most promising candidates
were phages found in Group 2, characterised by growth dynamics with high bacterial killing.

Chapter 3: Experimental Studies
117
Introduction
Bacteriophages (phages) are viruses that specifically infect bacteria. They are estimated to be
the most abundant organisms on Earth (~1031 entities) and play a major role in shaping the
microbial communities [1, 2]. Phages are unable to replicate independently of a susceptible
bacterial host, and their host range is determined by a combination of various factors, including
host-binding protein specificity and bacterial phage-resistance mechanisms [3, 4]. Bacteria can
readily evolve resistance to phage infections though different mechanisms, which can result in
distinct resistance phenotypes [5]. These can differ in whether the resistance is partial or
complete, in the level of fitness cost associated with resistance, and in whether the mutation
can be countered by the infecting phage. Consequently, these differences determine the effect
of the phage infection on the bacterial population dynamics and the resulting community
structure [6, 7].
The therapeutic use of phages (phage therapy) represents an urgently needed alternative or
supportive antibacterial agent to antibiotics to control bacterial pathogens [8–11]. However,
the outcome of phage therapy is still difficult to predict [12]. Several factors can be involved,
both animal host and bacterial host related as well as phage related. Whether phages are able
to infect and kill a susceptible target bacterial population at a specific site depends on the
change in phage densities in different tissues of the host (pharmacokinetics (PK)) and the
population dynamics of the phage-bacterial interaction (pharmacodynamics (PD)) [7].
Understanding the phage-bacterium interactions and population dynamics has been shown to
be essential for more reliable in vitro and/or in vivo outcomes, and thereby, essential for
successful phage therapy development and application [7, 13, 14]. Several models have been
developed to predict the behaviour and dynamics of phage-bacteria populations [15–18]. These
models include parameters such as bacterial growth and mutation rate, phage adsorption rate,
burst size, latent period, and virulence, as well as multiplicity of infection (MOI) [7, 18]. While
no single model to date has been able to capture all aspects of the complex phage -host
interactions, together, suitable models can be selected to predict and explain basic behaviours
of the population dynamics, as well as identify the dominant factors that contribute the
dynamics [7, 15, 17, 18]. The dynamics of in vitro phage-host interactions may differ from
those observed in vivo. This is similar to what is seen also with antibiotics [19, 20]. Still, in
vitro observations constitute a necessary initial step in understanding and predicting phage -
host population dynamics in in vivo settings [7]. Several models focus on dynamics of only an
individual phage and single host [17, 18], and can fail to report what defines the dynamic(s).

3.2 Classification of in vitro phage-host population growth dynamics
118
This study investigates the in vitro growth dynamics of a group of Escherichia coli (E. coli)-
infecting phages (coliphages) and multi-drug resistant avian pathogenic E. coli (APEC). We
define the specific patterns observed using a multiple parameter-based approach and estimate
the parameter(s) (phage type, APEC strains, and MOI) that are key to each pattern. APEC (with
O-serogroups O1, O2, or O78) was chosen as the bacterial model due to the significant problem
it represents to poultry worldwide [21, 22]. This pathogen causes a large range of extraintestinal
infections, collectively referred to as colibacillosis, which are becoming harder to treat with the
increasing resistance of APEC to different classes of antibiotics [23, 24].
Methods
Bacterial strains and growth conditions
The 10 APEC strains are part of an in-house collection that were isolated from clinical poultry
faeces samples suspected of APEC infection. Samples were collected in Belgium during 2013-
2014 by the Animal Health Care Flanders (Torhout, Belgium). Strains were grown in LB broth
(Miller) (Sigma-Aldrich, Saint Louis, MO, USA) or on LB agar supplemented with 1.5%
bacteriological agar no. 1 (w/v) (Oxoid, Basingstoke, UK) overnight (16-18 h) at 37 °C unless
stated otherwise. Broth cultures were incubated with shaking (120 rpm). Strains were stored at
-80 °C in LB broth supplemented with 15% glycerol (Sigma-Aldrich, Saint Louis, MO, USA).
Genomic DNA extraction and sequencing
Bacterial genomic DNA was extracted using Qiagen’s DNeasy Blood and Tissue Kit (Qiagen,
Hilden, Germany), with the subsequent library construction using the Nextera XT Kit
(Illumina, Little Chesterford, UK), and sequenced using a 300-cycle kit on the Illumina
NextSeq platform according to the manufacturer’s instructions.
Bacterial genome analysis
The bifrost platform (https://github.com/ssi-dk/bifrost) (accessed on 15 January 2021), v1.1.0,
was used for quality control validation of the raw reads data. The raw reads were de novo
assembled using SPAdes v3.12.1 [25] and MLST typed with the MLST command-line tool
(https://github.com/tseemann/mlst) (accessed on 15 January 2021), v2.19. The serotypes were
predicted using SerotypeFinder, v2.0 [26]. ABRicate v1.0.1

Chapter 3: Experimental Studies
119
(https://github.com/tseemann/abricate) (accessed on 18 June 2021) with default options was
used to screen the assembled contigs for antimicrobial resistance genes with ResFinder [27]
and the Comprehensive Antibiotic Resistance Database (CARD) [28]. Virulence genes were
identified using ABRicate with Ecoli_VF database data. Prophage regions were identified
using the PHAge Search Tool Enhanced Release (PHASTER) tool [29]. The Rapid Annotation
using Subsystem Technology (RAST) server and the SEED viewer, v2.0 [30], were used to
identify coding sequences (CDSs) and for initial annotation of the APEC draft genomes.
Bacteriophage isolation, purification, and enumeration
A total of 18 lytic coliphages were used in this study. These were selected from the collection
based on their genomic diversity. Phages were isolated from poultry faecal material using E.
coli laboratory strain K514, sequenced and identified as described before [31]. Phage stocks
were stored at titers of 1.2x107 to 4.5x1010 plaque forming units (PFU)/ml at 4 °C. Working
stocks used for phage infectivity and phage-host growth experiments were kept at titers of
~1010 PFU/ml.
Phage infectivity and phage-host growth dynamics
Bacterial overnight cultures were used, and the cell concentration was adjusted to ~108 CFU/ml
for each experiment. Bacterial solutions were inoculated with phage, yielding initial MOIs of
10, 1, or 0.1. All bacterial reduction curves were generated using 96-well plates with working
volumes of 200 µl. Experiments were performed in triplicate. For the experiments of the
susceptible combinations, the experiment was performed on a duplicate plate at another time.
A well of phage-free bacterial culture and a well of bacteria-free phage culture were included
on every plate as control experiments in addition to one media blank for reference. The optical
density (OD) was measured for a wavelength of 600 nm (OD600) with the Thermo Fisher
Scientific Multiskan GO Microplate Spectrophotometer, v1.01.12, and the data were recorded
using SkanIt software, v6.0.2.3. The OD600 was measured with fast measurement mode and
no pathlength correction or use of transmittance, and the measurements were taken
immediately after inoculation and then at regular intervals of 30 minutes afterward for 22 hours.
The incubation temperature was 37 °C and shaking was continuous at a medium speed.
Growth curves were obtained by plotting OD600 values after baseline adjustment against time.
Phage infectivity was defined based on endpoint measurements. Successful phage infection

3.2 Classification of in vitro phage-host population growth dynamics
120
was defined as OD600: <0.2, somewhat successful infection was defined as OD600: 0.2 -0.5,
and failed phage infection as OD600: >0.5. The phage-host growth dynamics were assessed
based on measurements throughout the experiment.
Assessing the effect of phage species, APEC strain and MOI
The two different response variables, PhageScore [32] and local virulence score [16] were
derived to assess the effect of phage species, APEC strain, and MOI on the growth dynamics.
For the PhageScore method, the area under the growth curve (AUC) was determined for the
complete study period for each treatment combination and for the corresponding control, i.e.,
without phage, and the ratio of the difference between the control and the treatment over the
control (multiplied with 100) was calculated. For the local virulence score, the same was done,
but only measurements until the stationary phase in the control group were taken into
consideration, i.e., until the timepoint before the maximum OD value.
A fixed effects model with a normally distributed error term was used, and phage, bacterium,
and MOI, as well as all two-way interactions, were included in the model. F-tests were
performed at the 5% significance level to assess the effects of the different factors. Finally, the
Pearson correlation coefficient between the PhageScore and the virulence score was calculated.
Classification of phage-bacterium growth dynamics
To classify the growth dynamics of phage-host interactions based on the OD measurements,
we applied two statistical data mining techniques: non-metric multidimensional scaling
(NMDS) ordination and principal component analysis (PCA), with hierarchical agglomerative
clustering. A NMDS ordination plot (Euclidean distance) using the vegan package in R
(http://www.R-project.org/) (accessed on 18 April 2021) [33] was applied to quantify and
visualise the pairwise dissimilarity between samples of each timepoint. The stress value was
determined to access how well the data were transformed. A stress value between 0.02 and
0.01 was considered an acceptable fit, and <0.01 was considered a good fit. Metadata was
included using the envfit function to determine the effect of each factor (phage species,
bacterial strain, and MOI). Using the factoextra package the right number of groups (of the
growth dynamics pattern) was determined using bootstrap values = 100 and hierarchical
agglomerative clustering (“ward.D” method). The results were visualised using the ggplot2
package. A PCA was performed as verification (sanity check) of how much of the variability
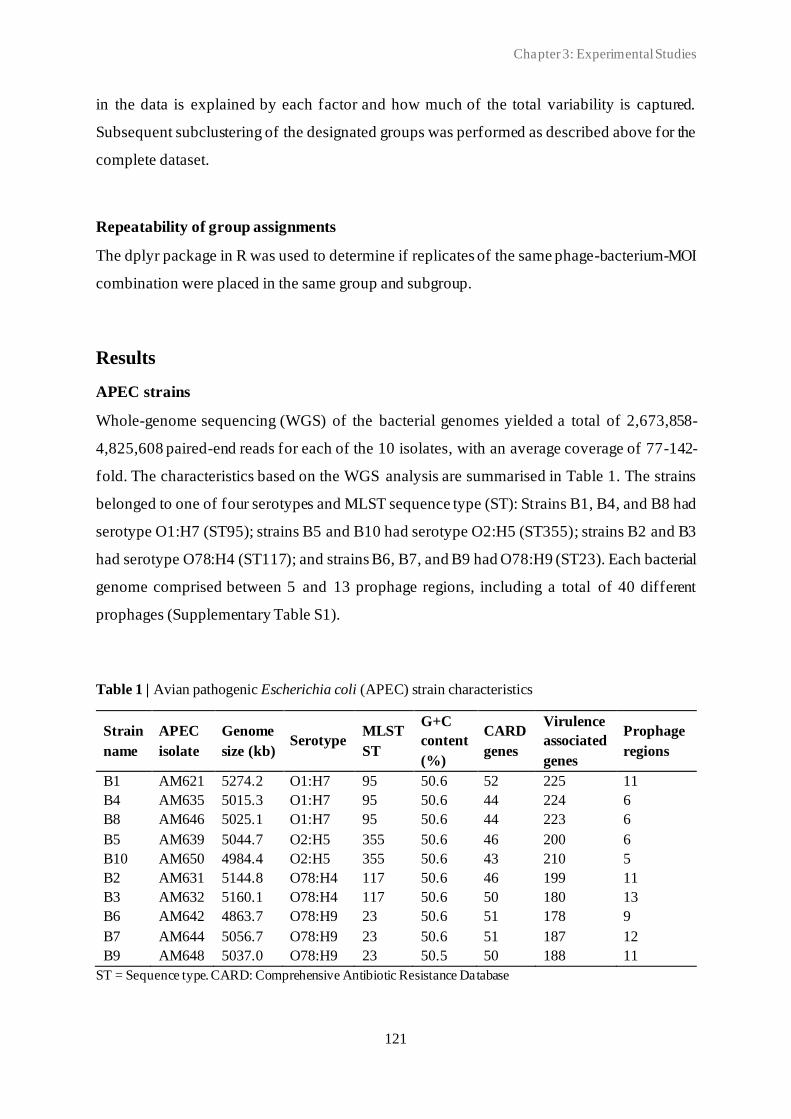
Chapter 3: Experimental Studies
121
in the data is explained by each factor and how much of the total variability is captured.
Subsequent subclustering of the designated groups was performed as described above for the
complete dataset.
Repeatability of group assignments
The dplyr package in R was used to determine if replicates of the same phage-bacterium-MOI
combination were placed in the same group and subgroup.
Results
APEC strains
Whole-genome sequencing (WGS) of the bacterial genomes yielded a total of 2,673,858-
4,825,608 paired-end reads for each of the 10 isolates, with an average coverage of 77-142-
fold. The characteristics based on the WGS analysis are summarised in Table 1. The strains
belonged to one of four serotypes and MLST sequence type (ST): Strains B1, B4, and B8 had
serotype O1:H7 (ST95); strains B5 and B10 had serotype O2:H5 (ST355); strains B2 and B3
had serotype O78:H4 (ST117); and strains B6, B7, and B9 had O78:H9 (ST23). Each bacterial
genome comprised between 5 and 13 prophage regions, including a total of 40 different
prophages (Supplementary Table S1).
Table 1 | Avian pathogenic Escherichia coli (APEC) strain characteristics
Strain
name
APEC
isolate
Genome
size (kb) Serotype
MLST
ST
G+C
content
(%)
CARD
genes
Virulence
associated
genes
Prophage
regions
B1 AM621 5274.2 O1:H7 95 50.6 52 225 11
B4 AM635 5015.3 O1:H7 95 50.6 44 224 6
B8 AM646 5025.1 O1:H7 95 50.6 44 223 6
B5 AM639 5044.7 O2:H5 355 50.6 46 200 6
B10 AM650 4984.4 O2:H5 355 50.6 43 210 5
B2 AM631 5144.8 O78:H4 117 50.6 46 199 11
B3 AM632 5160.1 O78:H4 117 50.6 50 180 13
B6 AM642 4863.7 O78:H9 23 50.6 51 178 9
B7 AM644 5056.7 O78:H9 23 50.6 51 187 12
B9 AM648 5037.0 O78:H9 23 50.5 50 188 11
ST = Sequence type. CARD: Comprehensive Antibiotic Resistance Database

3.2 Classification of in vitro phage-host population growth dynamics
122
Coliphages
The coliphages used in this study were previously characterised [30]. The characteristics are
summarised in Table 2. The phages belonged to one of seven different genera. Tequatrovirus
phages included P1, P2, and P9. Mosigvirus phages included P3, P5, and P6. Guelphvirus
phages included P4, P13, and P14. Hanrivervirus phages included P7, P8, and P16.
Felixounavirus phages included P10, P12, P17, and P18. Tequintavirus phages included P11.
Warwickvirus phages included P15.
Table 2 | Coliphage characteristics
Na
me
Phage
name
Genome
size (kb) Phage family
Phage
subfamily Phage genus Accession no.
P10 Phage 60 86.2 Myoviridae Ounavirinae Felixounavirus SRX8360069
P12 Phage 62 87.9 Myoviridae Ounavirinae Felixounavirus SRX8360072
P17 Phage 78 89.9 Myoviridae Ounavirinae Felixounavirus SRX8360088
P18 Phage 79 89.7 Myoviridae Ounavirinae Felixounavirus SRX8360089
P4 Phage 17 45.9 Drexlerviridae Braunvirinae Guelphvirus SRX8360091
P13 Phage 70 44.5 Drexlerviridae Braunvirinae Guelphvirus SRX8360079
P14 Phage 74 46.7 Drexlerviridae Braunvirinae Guelphvirus SRX8360084
P7 Phage 53 50.8 Drexlerviridae Tempevirinae Hanrivervirus SRX8360063
P8 Phage 54 52.6 Drexlerviridae Tempevirinae Hanrivervirus SRX8360064
P16 Phage 77 51.1 Drexlerviridae Tempevirinae Hanrivervirus SRX8360087
P3 Phage 15 169.6 Myoviridae Tevenvirinae Mosigvirus SRX8360082
P5 Phage 18 169.9 Myoviridae Tevenvirinae Mosigvirus SRX8360092
P6 Phage 30 173.4 Myoviridae Tevenvirinae Mosigvirus SRX8360094
P1 Phage 10 169.0 Myoviridae Tevenvirinae Tequatrovirus SRX8360061
P2 Phage 11 171.4 Myoviridae Tevenvirinae Tequatrovirus SRX8360071
P9 Phage 55 170.0 Myoviridae Tevenvirinae Tequatrovirus SRX8360065
P11 Phage 61
109.9 Demerecvirida
e
Markadamsvir
inae Tequintavirus SRX8360070
P15 Phage 76 51.9 Drexlerviridae Tempevirinae Warwickvirus SRX8360086
Phage classification according to current (16 September 2021) International Committee on Taxonomy of
Viruses (ICTV) taxonomy.
Phage infectivity
The infectivity of the 18 coliphages against each of the 10 APEC strains at MOI 10, 1 , and 0.1
is shown in Figure 1. The levels of infectivity of the tested coliphages were highly variable,
varying between 0% and 100% of the tested APEC strains. Variations in the degree of infection
(successful, somewhat successful, or failed) were detected, with the most successful infections
at MOI 10 and least at MOI 0.1. Phage P1 was shown to be the most infective , as it was the

Chapter 3: Experimental Studies
123
only one able to infect all APEC strains at all MOI tested, except for B7-MOI 0.1. Phages P2,
P3, and P9 were able to infect nine of the APEC strains, excluding B4, B4, and B1, respectively.
Phage P5 was able to infect eight of the APEC strains, excluding B1 and B4. Phage P8 was
able to infect six of the APEC strains, not including B1, B4, B6, and B7. Phages P4, P6, and
P7 were able to infect five of the APEC strains, excluding B1, B4, B6, B7, and B8. The nine
phages P10-18 were not able to infect any of the 10 APEC strains.
Figure 1 | Infectivity of the tailed coliphages based on the endpoint OD. P = phage. B = bacterium. MOI =
multiplicity of infection. Ratio = phage:APEC. OD was measured at 600 nm. (OD600). Dark blue = final OD600:
<0.2, light blue = final OD600: 0.2-0.5, and red = final OD600: >0.5. The final OD was determined based on the
average of three-nine replicates.
Assessing the effect of phage species, APEC strain, and MOI
A total of 2869 phage-host combination experiments were performed. The average local
virulence score and its standard error for each phage-bacterium combination are shown in
Figure 2. All factors were significant, i.e., MOI (F2,2495 = 938.8, p < .0001), Phage
(F17,2495=1095.8, p < .0001) and Bacterium (F9,2495=459.5, p < .0001), and the interactions too
but to a lesser extent, i.e., Phage-Bacterium (F153,2495=42.8, p < .0001), Phage-MOI
(F34,2495=10.7, p < .0001) and Bacterium-MOI (F18,2495=9.7, p < .0001). The phage effect was
thus more pronounced than the bacterium effect. Based on the virulence scores, the greatest

3.2 Classification of in vitro phage-host population growth dynamics
124
reduction in bacterial growth of the six strains B1, B2, B4, B6, B7, and B8 was observed with
phage P1 and with phage P5 for the four strains B3, B5, B9, and B10.
Figure 2 | Virulence score average by bacteria and phage. A higher virulence score correlates with a higher
virulence of the phage and higher bacterial growth reductions. The standard error is shown. A total of 2869 phage-
host combination experiments were included.
The average PhageScore and its standard error for each phage-bacterium combination are
shown in Figure 3. All factors were significant, i.e., MOI (F2,2495 = 447.9, p < .0001), Phage
(F17,2495 = 693.6, p < .0001) and Bacteria (F9,2495 = 364.6, p < .0001), and the interactions too
but to a lesser extent, i.e., Phage-Bacteria (F153,2495 = 38.1, p < .0001), Phage-MOI (F34,2495 =
13.0, p < .0001) and Bacteria-MOI (F18,2495 = 12.5, p < .0001). The phage effect was therefore
also more pronounced than the bacterium effect when looking at the PhageScore. Based on the
PhageScore, the greatest reduction in bacterial growth of the four strains B1, B4, B6, and B8
was observed with phage P1, and with phage P5 for the four strains B5, B7, B9, and B10. The
greatest reduction of B2 and B3 growth was observed for phages P9 and P6, respectively.

Chapter 3: Experimental Studies
125
Figure 3 | PhageScore average by bacteria and phage. A higher PhageScore correlates with a higher efficiency of
the phage and higher bacterial growth reduction. A total of 2869 phage-host combination experiments were
included. The standard error is shown.
The Pearson’s correlation coefficient between the local virulence score and the PhageScore
was equal to 0.94.
Classification of phage-bacterium growth dynamics
Phage-host growth dynamics were classified based on the OD measurements from a total of
2,729 phage-host combination experiments. The NMDS analysis grouped the data into three
different groups (Figure 4). Group 1 comprised resistant phage-host combinations
characterised by bacterial growth, Group 2 comprised fully susceptible combinations
characterised by bacterial killing, and Group 3 comprised in-between combinations. A two-
dimensional plot was considered appropriate, as the generated stress value was below 0.01
(0.007). Growth dynamics curves associated with each combination experiment and phage-free
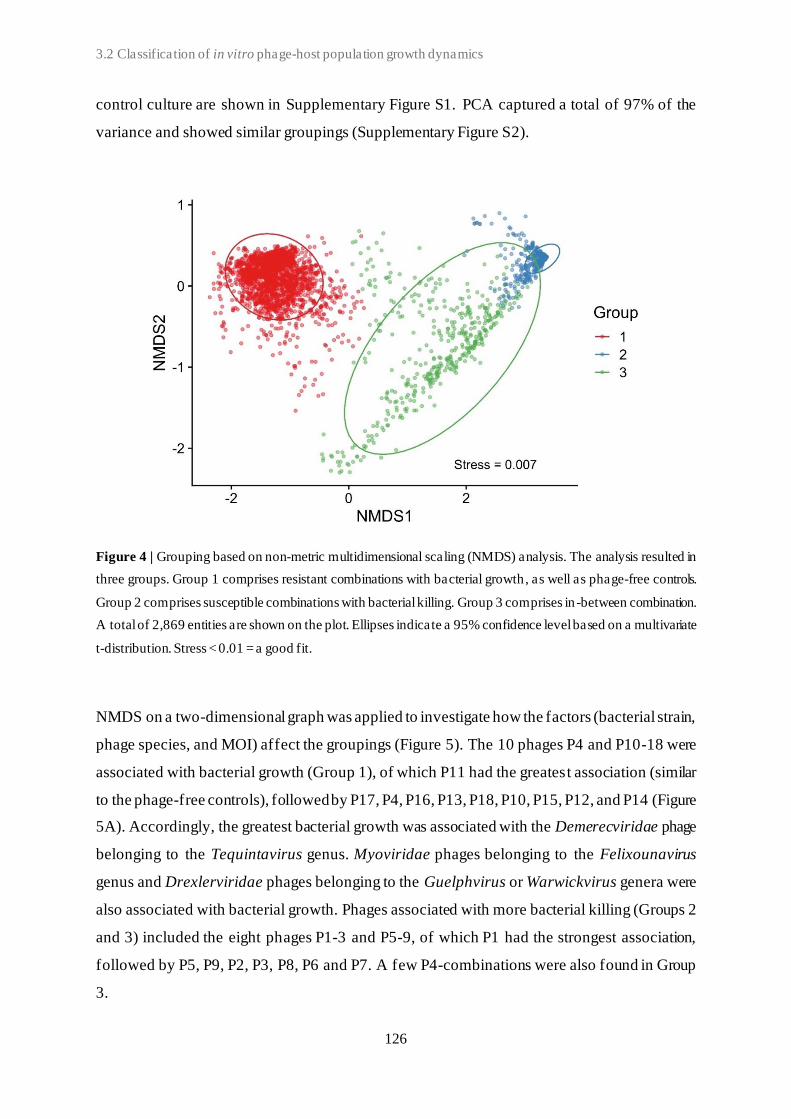
3.2 Classification of in vitro phage-host population growth dynamics
126
control culture are shown in Supplementary Figure S1. PCA captured a total of 97% of the
variance and showed similar groupings (Supplementary Figure S2).
Figure 4 | Grouping based on non-metric multidimensional scaling (NMDS) analysis. The analysis resulted in
three groups. Group 1 comprises resistant combinations with bacterial growth, as well as phage-free controls.
Group 2 comprises susceptible combinations with bacterial killing. Group 3 comprises in -between combination.
A total of 2,869 entities are shown on the plot. Ellipses indicate a 95% confidence level based on a multivariate
t-distribution. Stress < 0.01 = a good fit.
NMDS on a two-dimensional graph was applied to investigate how the factors (bacterial strain,
phage species, and MOI) affect the groupings (Figure 5). The 10 phages P4 and P10-18 were
associated with bacterial growth (Group 1), of which P11 had the greatest association (similar
to the phage-free controls), followed by P17, P4, P16, P13, P18, P10, P15, P12, and P14 (Figure
5A). Accordingly, the greatest bacterial growth was associated with the Demerecviridae phage
belonging to the Tequintavirus genus. Myoviridae phages belonging to the Felixounavirus
genus and Drexlerviridae phages belonging to the Guelphvirus or Warwickvirus genera were
also associated with bacterial growth. Phages associated with more bacterial killing (Groups 2
and 3) included the eight phages P1-3 and P5-9, of which P1 had the strongest association,
followed by P5, P9, P2, P3, P8, P6 and P7. A few P4-combinations were also found in Group
3.
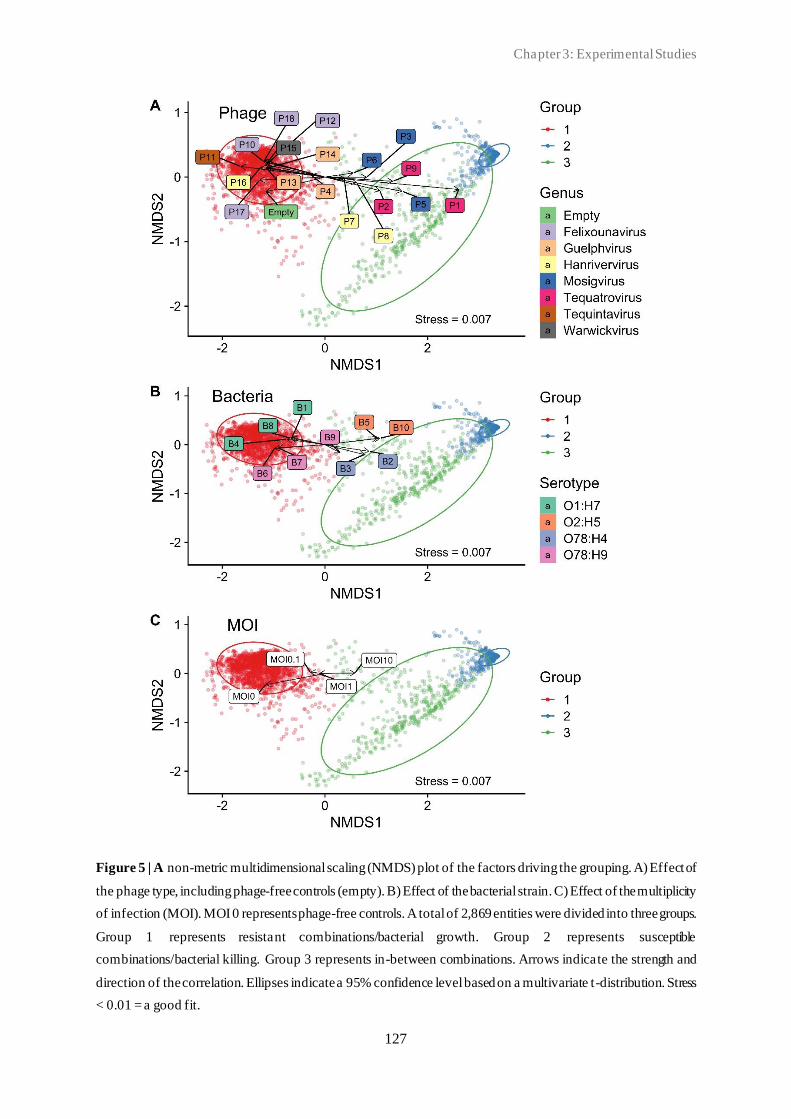
Chapter 3: Experimental Studies
127
Figure 5 | A non-metric multidimensional scaling (NMDS) plot of the factors driving the grouping. A) Effect of
the phage type, including phage-free controls (empty). B) Effect of the bacterial strain. C) Effect of the multiplicity
of infection (MOI). MOI 0 represents phage-free controls. A total of 2,869 entities were divided into three groups.
Group 1 represents resistant combinations/bacterial growth. Group 2 represents susceptible
combinations/bacterial killing. Group 3 represents in-between combinations. Arrows indicate the strength and
direction of the correlation. Ellipses indicate a 95% confidence level based on a multivariate t-distribution. Stress
< 0.01 = a good fit.

3.2 Classification of in vitro phage-host population growth dynamics
128
Accordingly, the greatest bacterial killing was associated with Myoviridae phages belonging
to the Tequatrovirus genus. Myoviridae phages belonging to the Mosigvirus genus were also
associated with bacterial killing, as well as two out of three Hanrivervirus phages
(Drexlerviridae family). The five bacterial strains B1, B4, B6, B7, and B8 were associated with
bacterial growth (Group 1). This included all three O1-serotype strains (B1, B4 and B8), as
well as two O78-serotype strains (Figure 5B). The other five strains: B2, B3, B5, B9, and B10,
were associated with more bacterial killing and included two O2-serotype strains (B5 and B10)
and three O78-serotype strains. The overall effect of the bacterial factor on the plot was less
compared to the effect of the phage factor. A high MOI (MOI 10) was associated with more
bacterial killing, and a low MOI (MOI 0.1) was associated with less killing (Figure 5C).
Based on the NMDS analysis, Group 3, including 419 entries, was divided into five subgroups
(Figure 6). Only combinations with phages P1-P9 were found. The growth curves associated
with each subgroup are shown in Supplementary Figure S3.
Figure 6 | Subgrouping of Group 3 based on non-metric multidimensional scaling (NMDS). Group 3 (n = 419)
was divided into five subgroups. Subgroups 3.1 (n = 167), 3.2 (n = 95), and 3.3 (n = 43) represent combinations
with initial bacterial killing followed by exponential bacterial growth and stationary phase. Subgroups 3.4 (n =
66) and 3.5 (n = 48) represent combinations with initial bacterial growth followed by stationary phase. Ellipses
indicate a 95% confidence level based on a multivariate t-distribution. Stress < 0.02 = an acceptable fit.
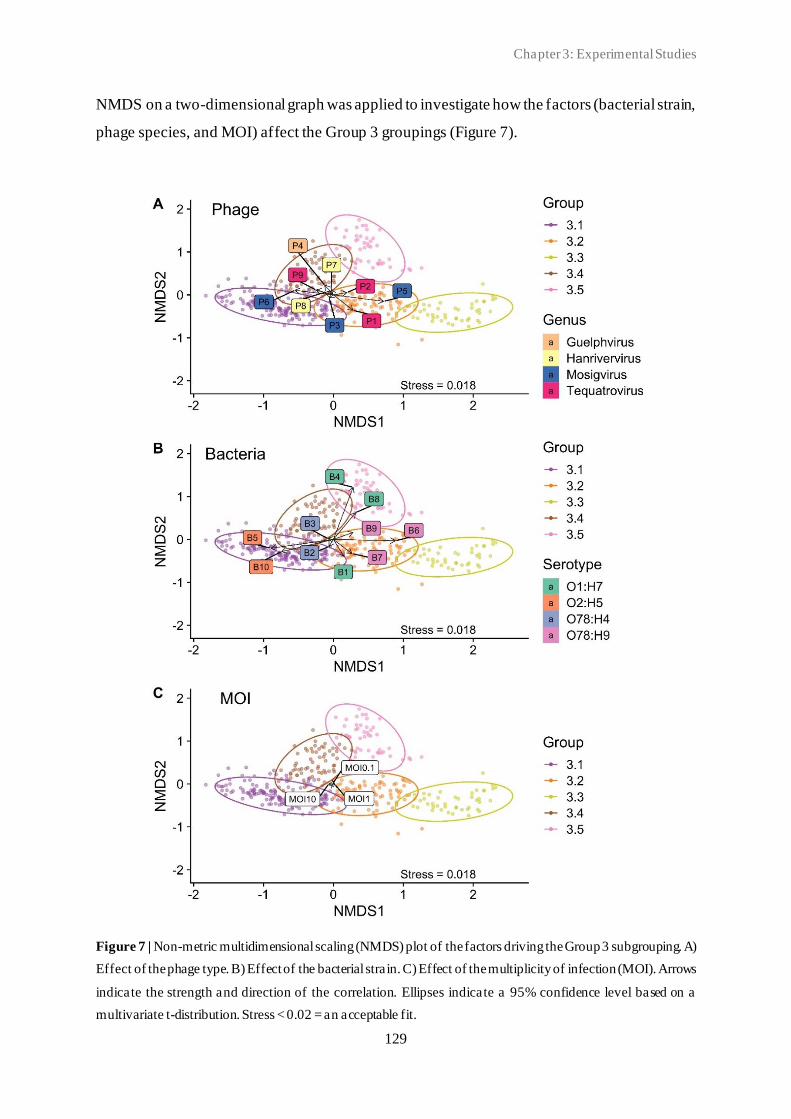
Chapter 3: Experimental Studies
129
NMDS on a two-dimensional graph was applied to investigate how the factors (bacterial strain,
phage species, and MOI) affect the Group 3 groupings (Figure 7).
Figure 7 | Non-metric multidimensional scaling (NMDS) plot of the factors driving the Group 3 subgrouping. A)
Effect of the phage type. B) Effect of the bacterial strain. C) Effect of the multiplicity of infection (MOI). Arrows
indicate the strength and direction of the correlation. Ellipses indicate a 95% confidence level based on a
multivariate t-distribution. Stress < 0.02 = an acceptable fit.

3.2 Classification of in vitro phage-host population growth dynamics
130
The association of phage P3 was in the direction of subgroup 3.1 ; however, the association was
weak. Phages P1 and P5 had a strong association with subgroup 3.2, while P2 and P3 had a
weak association. P5 association was in the direction towards subgroup 3.3. The remaining five
phages, including P4, P6, P7, P8, and P8, were associated with subgroup 3.4. P6 was the only
phage with a strong association (Figure 7A). Bacterial strains B5 and B10 (O2 serotype) had a
strong association with subgroup 3.1, and B2 (O78) had a weak association. B1 (O1), as well
as O78 strains B6, B7, and B9 had an association with subgroup 3.2. B6 was the only strain
with a strong association, as well as the only strain driving the plot towards subgroup 3.3. No
strains were driving the plot towards subgroup 3.4. B3 was found in this subgroup but showed
a weak/no association. O1 strains B4 and B8 had a strong association with subgroup 3.5 (Figure
7B). The MOIs did not have a strong association with any of the subgroups (Figure 7C).
Description of defined growth dynamics patterns
Based on the bacterial growth (OD) detected in the 2,869 phage-host combination experiments,
three different growth dynamics patterns groups, including five subgroups, were defined
(Figure 8). The groups included: 1) Combinations with a fully resistant bacterial growth
pattern; 2) Phage-host combinations with a fully susceptible pattern, showing minimal or no
bacterial growth; and 3) Combinations with one of five in-between patterns characterised by
delayed growth, lower killing, or variable killing of the bacterial cells. For all dynamics patterns
except Group 1, the phage effect on the bacterial growth kinetics was observed within only a
few hours of incubation. When the stationary phase was reached, the bacterial density remained
stable throughout all the co-culturing experiments.
Group 1 (bacterial growth): The bacterial growth continued to increase during the first 7 h of
incubation until the cultures reached the stationary phase. The final OD600 was ~0.7. The
pattern observed showed logistic growth as under standard conditions without phage present
and could be explained by the presence of naturally phage-resistant strains, where the phage is
unable to infect and has no effect on the growth. Group 2 (high level of bacterial killing): The
bacteria were lysed and never recovered. A single small peak of bacterial growth followed by
bacterial killing was observed in some cases but was not reflected in the average OD growth
curve. Group 3.1 (initial bacterial killing followed by bacterial growth): Prolongation of the
lag phase with no or low bacterial growth for ~9 hours was observed , followed by a slow
increase in bacterial growth until the stationary phase was reached. The final OD600 was ~0.45.

Chapter 3: Experimental Studies
131
The greatest variations were seen in this subgroup (Supplementary Figure S3). Group 3.2
(initial bacterial killing followed by bacterial growth): Prolongation of the lag phase with no
or low bacterial growth was observed, followed by a sharp increase in the bacterial density
after ~7 hours of incubation before reaching the stationary phase. The final OD600 was ~0.5.
Figure 8 | Growth dynamic patterns of the coliphage-APEC co-culture combinations. Patterns are defined based
on the average OD value for each timepoint for each group.
Group 3.3 was characterised in a similar way as Group 3.2, except for a higher final OD600
of ~7.5, the highest observed for the seven different patterns. Group 3.4 (impaired bacterial
growth): Increase of the bacterial growth was observed during the first ~9.5 hours of incubation
until the cultures reached the stationary phase. Impaired growth was observed compared to the
Group 1 and Group 3.5 patterns with a final OD600 of ~0.26. Group 3.5 was similarly
characterised by impaired bacterial growth. Increase of the bacterial growth was observed
during the first ~12 hours of incubation until the cultures reached the stationary phase. Impaired
growth was observed compared to the Group 1 patterns, with a final OD600 of ~0.45 (similarly
to Group 3.1).
Repeatability of group assignments
All replicates, including the phage-bacteria-MOI combinations with phage P10-P18, were
found in same NMDS group (Group 1). Some discrepancies in the group assignments were
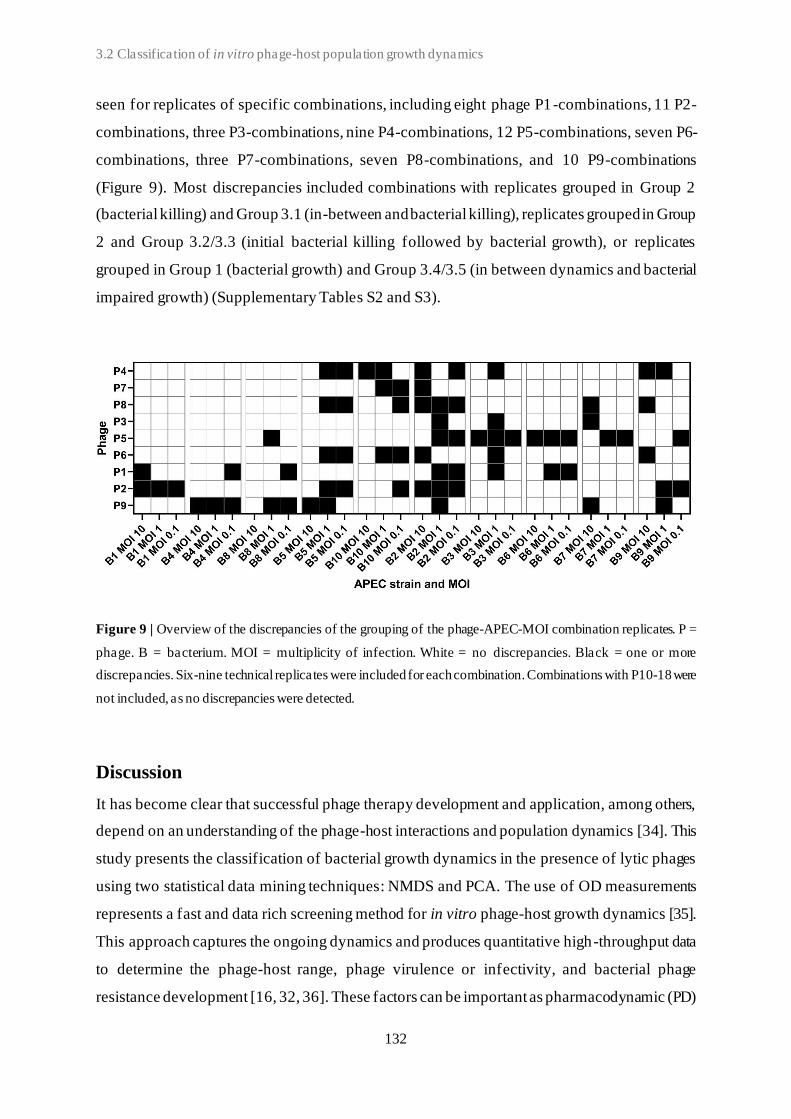
3.2 Classification of in vitro phage-host population growth dynamics
132
seen for replicates of specific combinations, including eight phage P1-combinations, 11 P2-
combinations, three P3-combinations, nine P4-combinations, 12 P5-combinations, seven P6-
combinations, three P7-combinations, seven P8-combinations, and 10 P9-combinations
(Figure 9). Most discrepancies included combinations with replicates grouped in Group 2
(bacterial killing) and Group 3.1 (in-between and bacterial killing), replicates grouped in Group
2 and Group 3.2/3.3 (initial bacterial killing followed by bacterial growth), or replicates
grouped in Group 1 (bacterial growth) and Group 3.4/3.5 (in between dynamics and bacterial
impaired growth) (Supplementary Tables S2 and S3).
Figure 9 | Overview of the discrepancies of the grouping of the phage-APEC-MOI combination replicates. P =
phage. B = bacterium. MOI = multiplicity of infection. White = no discrepancies. Black = one or more
discrepancies. Six-nine technical replicates were included for each combination. Combinations with P10-18 were
not included, as no discrepancies were detected.
Discussion
It has become clear that successful phage therapy development and application, among others,
depend on an understanding of the phage-host interactions and population dynamics [34]. This
study presents the classification of bacterial growth dynamics in the presence of lytic phages
using two statistical data mining techniques: NMDS and PCA. The use of OD measurements
represents a fast and data rich screening method for in vitro phage-host growth dynamics [35].
This approach captures the ongoing dynamics and produces quantitative high-throughput data
to determine the phage-host range, phage virulence or infectivity, and bacterial phage
resistance development [16, 32, 36]. These factors can be important as pharmacodynamic (PD)

Chapter 3: Experimental Studies
133
parameters and include also a part of the pharmacokinetics (PK) as it assesses the potential
increase of the treatment dose. This would not be the case when relying on a single endpoint
measurement. However, it is understood that ODs do not differentiate between viable and dead
cells, and as such, there was no exact link between the OD values and bacteria viability.
Nevertheless, it is a good proxy for estimating bacterial numbers. The repeatability of the
NMDS grouping was shown to be acceptable. Fully natural phage-resistant combinations
(Group 1) were clearly identified. Most discrepancies observed between groupings of the
replicates from the same phage-APEC-MOI combinations can be explained by grouping cut-
off values. Replicates grouped in Group 1 and Group 3.4/3.5 included dynamics characterised
by higher or lower bacterial growths. Replicates grouped in Group 2 and Group 3.1, included
dynamics characterised by the initial bacterial killing, with no or low subsequent growth.
The optimal number of clusters/groups varied depending on the method used. In this study, we
chose five clusters. However, subgroups 3.2 and 3.3 and subgroups 3.4 and 3.5 were relatively
similar and could potentially be combined, resulting in only three Group 3 subclusters, and this
would also create fewer discrepancies in the group allocations. The discrepancies due to
biological variations can be expected due to the spontaneous emergence of phage-resistant
variants after varying incubation time (Group 2 vs. Groups 3.1, 3.2, and 3.3). It is also possible
for a very small (partially) resistant sub-population of bacteria to be naturally present in the
culture at the start of the experiment [17].
Various factors affecting the phage PK/PD have been described using mathematical and
experimental models [15, 17, 18, 37, 38]. In this study, the influence of the factors (phage type,
bacterial strain, and MOI) on the observed growth patterns was determined. Previous studies
have highlighted the MOI influence on phage therapy, and recently, a fast microtiter plate assay
for determination of the optimum MOI for a coliphage was further described [39]. However,
in this study, we found the MOI to have a less significant effect on the phage -host growth
dynamics outcome compared to the phage species. Furthermore, a recent study suggested that
the description of MOI alone is not sufficient, as the concentration, particular to the bacteria,
can significantly affect the results [40]. Therefore, in this study, the MOI at all the tested values
was based on a fixed bacterial concentration.
A quantitative assessment of the phage lytic activity using the virulence score and PhageScore
across a large dataset allowed direct comparisons of individual phages. In contrast to a single
OD endpoint measurement and the well-established plaque assay, these methods (virulence

3.2 Classification of in vitro phage-host population growth dynamics
134
score and PhageScore) captured the dynamics of phage infection, including bacterial
(re)growth after prolonged growth inhibition or lysis. Additionally, compared to the overlay-
based efficiency of plating assays and direct spot testing, these methods represent an accurate
and less cumbersome and time-consuming and do not depend on the subjectivity and/or
experience of the observer [41]. However, upscaling of this approach depends on the
availability of a high-throughput plate reader. In accordance with previous findings, we found
the two methods highly comparable, showing similar properties/values for the studied phages
[32, 42]. In this study, whenever the bacteria were able to grow and reach the stationary phase,
the growth remained stable throughout all the experiments. If a second peak would have
appeared or the growth would have started to decrease (after reaching the stationary phase), the
comparability of the local virulence score and the PhageScore would be reduced. In this study,
we only analysed the growth dynamics of co-cultures of a single phage type and bacterial strain.
However, both the virulence score and PhageScore have previously been used to compare
phage combinations for use in phage therapy cocktails [32, 41]. In future studies, the inclusion
of mixed phage cultures (phage cocktails), preferably targeting different host receptors, may
provide further indications of their potential as therapeutics against pathogenic target bacteria
[17]. Accordingly, for future applications, the inclusion of phage, as well as bacterial traits,
may be required for classification.
Given their great abundance and diversity, multiple candidate phages might be available to
infect a target host; yet, we still lack a better understanding of which phage would perform best
[43]. One approach to identify the cause(s) of treatment success is to compare the
characteristics of phages with high success rates with those of phages with low success rates.
The characteristics differing between these two groups of phages become candidates for
causation. In this study, Tequintavirus phages (associated with bacterial growth) and
Tequatrovirus phages (associated with bacterial killing) would be great candidates for
comparisons. Accordingly, the inclusion of phage characteristics, such as the phage receptor,
adsorption rate, latency period, burst size, and virion size, may provide further explanation of
the phage-host dynamics and may help predict the phage therapy efficacy [44–46].
Phage therapy is, by its nature, a strongly selective treatment [17]. Accordingly, when selecting
phages for therapeutical application, the emergence of phage-resistant bacteria should be taken
into consideration [7]. Bacteria can develop resistance against phages through various
mechanisms, including the modification of phage receptor-encoding genes, innate immune
systems (such as CRISPR-Cas), and the presence of prophages in the bacterial genome [47,

Chapter 3: Experimental Studies
135
48]. Here, O1 serotype strains B1, B4, and B10 were associated with natural phage
resistance/high levels of bacterial growth, whereas the strains with serotype O2 and serogroup
O78:H4 were associated with phage susceptibility/bacterial killing. Moreover, a ll phage-host
combinations including P10-18 were found in Group 1 (fully resistant combinations). These
phages would be excluded as candidates for phage therapy targeting the selected APEC strains.
P1 was the only phage not included in any Group 1 combinations and showed the greatest
bacterial killing potential. Accordingly, phages only included in Group 2, characterised by high
bacterial killing, are considered the most promising candidates for phage therapy. Multiple
phage-host-MOI combinations were characterised by initial bacterial killing followed by
bacterial growth (subgroups 3.1, 3.2, and 3.3), suggesting the emergence of phage-resistant
bacterial variants. Whether phages found in these subgroups should be considered suitable for
phage therapy depends on their specific applications and further studies are needed to
determine if the initial inhibition of bacterial growth for ~7 hours is sufficient to clear out the
infection.
Although in vitro experiments do not capture many in vivo realities, such experiments can give
significant insights into the phage-host dynamics and lead to interesting predictions, which
could be useful in phage therapy and exploited in appropriately designed in vivo models [13].
Recently, a framework (Clinical Phage Microbiology) with recommendations for in vitro
identification and the evaluation of phages intended for treatment was published [42]. One step
of the framework pipeline includes determination of the growth kinetics of liquid cultures and
highlights the need for a standardised quantitative assessment with reproducible scoring . The
methodology applied here constitutes such an assessment and may help to improve the
standardisation of the quantitative evaluation of phage candidates.
In conclusion, our methodology assessing the host-phage interaction in vitro provides a high-
throughput method for classifying bacterial growth dynamics in the presence of virulent
coliphages using measurements of bacterial growth by OD as inputs. The established in vitro
model was not only used to gain a better understanding of the phage PK/PD but can also be
applied as a screening method for selecting new suitable phage candidates for therapeutic
applications against pathogenic target bacteria. However, to fully understand the complexity
of these phage-host dynamics, the underlying mechanisms behind these different interactions
need to be deciphered.

3.2 Classification of in vitro phage-host population growth dynamics
136
References
[1] Güemes, A. G. C.; Merry Youle; Cantú, V. A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners
in the Game of Life. Annu. Rev. Virol., 2016, 3 (1), 197–214. https://doi.org/10.1146/annurev-
virology-100114-054952.
[2] Hatfull, G. F. Dark Matter of the Biosphere: The Amazing World of Bacteriophage Diversity. J.
Virol., 2015, 89 (16), 8107–8110. https://doi.org/10.1128/jvi.01340-15.
[3] Clokie, M. R. J.; Millard, A. D.; Letarov, A. V; Heaphy, S. Phages in Nature. Bacteriophage, 2011,
1 (1), 31–45. https://doi.org/10.4161/bact.1.1.14942.
[4] Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages.
Front. Microbiol., 2016, 7, 1352. https://doi.org/10.3389/fmicb.2016.01352.
[5] Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy.
Viruses, 2018, 10 (7). https://doi.org/10.3390/v10070351.
[6] Bohannan, B. J. M.; Lenski, R. D. Linking Genetic Change to Community Evolution: Insights from
Studies of Bacteria and Bacteriophage. Ecol. Lett., 2000, 3 (4), 362–377.
https://doi.org/https://doi.org/10.1046/j.1461-0248.2000.00161.x.
[7] Levin, B. R.; Bull, J. J. Population and Evolutionary Dynamics of Phage Therapy. Nat. Rev.
Microbiol., 2004, 2 (2), 166–173. https://doi.org/10.1038/nrmicro822.
[8] Pirnay, J.; Verbeken, G.; Rose, T.; Jennes, S.; Zizi, M.; Huys, I.; Lavigne, R.; Merabishvili, M.;
Vaneechoutte, M.; Buckling, A.; et al. Introducing Yesterday’s Phage Therapy in Today’s Medicine.
Future Virol., 2012, 7 (4), 379–390. https://doi.org/10.2217/fvl.12.24.
[9] Gordillo Altamirano, F. L.; Barr, J. J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev.,
2019, 32 (2), 1–25. https://doi.org/10.1128/CMR.00066-18.
[10] Lu, T. K.; Koeris, M. S. The next Generation of Bacteriophage Therapy. Curr. Opin. Microbiol.,
2011, 14 (5), 524–531. https://doi.org/10.1016/j.mib.2011.07.028.
[11] Lin, D. M.; Koskella, B.; Lin, H. C. Phage Therapy: An Alternative to Antibiotics in the Age of
Multi-Drug Resistance. World J. Gastrointest. Pharmacol. Ther., 2017, 8 (3), 162–173.
https://doi.org/10.4292/wjgpt.v8.i3.162.
[12] Tsonos, J.; Oosterik, L. H.; Tuntufye, H. N.; Klumpp, J.; Butaye, P.; De Greve, H.; Hernalsteens, J.
P.; Lavigne, R.; Goddeeris, B. M. A Cocktail of in Vitro Efficient Phages Is Not a Guarantee for in
Vivo Therapeutic Results against Avian Colibacillosis. Vet. Microbiol., 2014.
https://doi.org/10.1016/j.vetmic.2013.10.021.
[13] Casey, E.; van Sinderen, D.; Mahony, J. In Vitro Characteristics of Phages to Guide ‘Real Life’
Phage Therapy Suitability. Viruses, 2018, 10 (4), 163. https://doi.org/10.3390/v10040163.
[14] Gill, J.; Hyman, P. Phage Choice, Isolation, and Preparation for Phage Therapy. Curr. Pharm.
Biotechnol., 2010, 11 (1), 2–14. https://doi.org/10.2174/138920110790725311.
[15] Storms, Z. J.; Sauvageau, D. Modeling Tailed Bacteriophage Adsorption: Insight into Mechanisms.
Virology, 2015, 485, 355–362. https://doi.org/10.1016/j.virol.2015.08.007.
[16] Storms, Z. J.; Teel, M. R.; Mercurio, K.; Sauvageau, D. The Virulence Index: A Metric for
Quantitative Analysis of Phage Virulence. Phage, 2020, 1 (1), 27–36.
https://doi.org/10.1089/phage.2019.0001.
[17] Cairns, B. J.; Timms, A. R.; Jansen, V. A. A.; Connerton, I. F.; Payne, R. J. H. Quantitative Models
of in Vitro Bacteriophage-Host Dynamics and Their Application to Phage Therapy. PLoS Pathog.,
2009, 5 (1), e1000253. https://doi.org/10.1371/journal.ppat.1000253.
[18] Santos, S. B.; Carvalho, C.; Azeredo, J.; Ferreira, E. C. Population Dynamics of a Salmonella Lytic
Phage and Its Host: Implications of the Host Bacterial Growth Rate in Modelling. PLoS One, 2014,

Chapter 3: Experimental Studies
137
9 (7), e102507. https://doi.org/10.1371/journal.pone.0102507.
[19] Broussou, D. C.; Toutain, P. L.; Woehrlé, F.; Garch, F. El; Bousquet -Melou, A.; Ferran, A. A.
Comparison of in Vitro Static and Dynamic Assays to Evaluate the Efficacy of an Antimicrobial
Drug Combination against Staphylococcus Aureus. PLoS One, 2019, 14 (1), e0211214.
https://doi.org/10.1371/journal.pone.0211214.
[20] He, S.; He, H.; Chen, Y.; Chen, Y.; Wang, W.; Yu, D. In Vitro and in Vivo Analysis of Antimicrobial
Agents Alone and in Combination against Multi-Drug Resistant Acinetobacter Baumannii. Front.
Microbiol., 2015, 6, 507. https://doi.org/10.3389/fmicb.2015.00507.
[21] Lutful Kabir, S. M. Avian Colibacillosis and Salmonellosis: A Closer Look at Epidemiology,
Pathogenesis, Diagnosis, Control and Public Health Concerns. Int. J. Environ. Res. Public Health,
2010, 7 (1), 89–114. https://doi.org/10.3390/ijerph7010089.
[22] Kathayat, D.; Lokesh, D.; Ranjit, S.; Rajashekara, G. Avian Pathogenic Escherichia Coli (APEC):
An Overview of Virulence and Pathogenesis Factors, Zoonotic Potential, and Control Strategies.
Pathogens, 2021, 10 (4), 467. https://doi.org/10.3390/pathogens10040467.
[23] Nhung, N. T.; Chansiripornchai, N.; Carrique-Mas, J. J. Antimicrobial Resistance in Bacterial
Poultry Pathogens: A Review. Front. Vet. Sci., 2017, 4, 126.
https://doi.org/10.3389/fvets.2017.00126.
[24] Dziva, F.; Stevens, M. P. Colibacillosis in Poultry: Unravelling the Molecular Basis of Virulence of
Avian Pathogenic Escherichia Coli in Their Natural Hosts. Avian Pathol., 2008, 37 (4), 355–366.
https://doi.org/10.1080/03079450802216652.
[25] Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A. A.; Dvorkin, M.; Kulikov, A. S.; Lesin, V. M.;
Nikolenko, S. I.; Pham, S.; Prjibelski, A. D.; et al. SPAdes: A New Genome Assembly Algorithm
and Its Applications to Single-Cell Sequencing. J. Comput. Biol., 2012, 19 (5), 455–477.
https://doi.org/10.1089/cmb.2012.0021.
[26] Joensen, K. G.; Tetzschner, A. M. M.; Iguchi, A.; Aarestrup, F. M.; Scheutz, F. Rapid and Easy in
Silico Serotyping of Escherichia Coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin.
Microbiol., 2015, 53 (8), 2410–2426. https://doi.org/10.1128/JCM.00008-15.
[27] Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.
M.; Larsen, M. V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob.
Chemother., 2012, 67 (11), 2640–2644. https://doi.org/10.1093/jac/dks261.
[28] Alcock, B. P.; Raphenya, A. R.; Lau, T. T. Y.; Tsang, K. K.; Bouchard, M.; Edalatmand, A.; Huynh,
W.; Nguyen, A.-L. V; Cheng, A. A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance
with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res., 2019, 48 (D1), D517–
D525. https://doi.org/10.1093/nar/gkz935.
[29] Arndt, D.; Grant, J. R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D. S. PHASTER: A Better,
Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res., 2016, 44, W16–W21.
https://doi.org/10.1093/nar/gkw387.
[30] Overbeek, R.; Olson, R.; Pusch, G. D.; Olsen, G. J.; Davis, J. J.; Disz, T.; Edwards, R. A.; Gerdes,
S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using
Subsystems Technology (RAST). Nucleic Acids Res., 2014, 42 (D1), 206–214.
https://doi.org/10.1093/nar/gkt1226.
[31] Sørensen, P. E.; Van Den Broeck, W.; Kiil, K.; Jasinskyte, D.; Moodley, A.; Garmyn, A.; Ingmer,
H.; Butaye, P. New Insights into the Biodiversity of Coliphages in the Intestine of Poultry. Sci. Rep.,
2020, 10, 15220. https://doi.org/10.1038/s41598-020-72177-2.
[32] Konopacki, M.; Grygorcewicz, B.; Dołęgowska, B.; Kordas, M.; Rakoczy, R. PhageScore: A Simple
Method for Comparative Evaluation of Bacteriophages Lytic Activity. Biochem. Eng. J., 2020, 161,
107652. https://doi.org/10.1016/j.bej.2020.107652.

3.2 Classification of in vitro phage-host population growth dynamics
138
[33] R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for
Statistical Computing: Vienna, Austria 2020.
[34] Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage–Host
Interactions. Viruses, 2019, 11 (6), 567. https://doi.org/10.3390/v11060567.
[35] Gayder, S.; Parcey, M.; Nesbitt, D.; Castle, A. J.; Svircev, A. M. Population Dynamics between
Erwinia Amylovora, Pantoea Agglomerans and Bacteriophages: Exploiting Synergy and
Competition to Improve Phage Cockta il Efficacy. Microorganisms, 2020, 8 (9), 1449.
https://doi.org/10.3390/microorganisms8091449.
[36] Xie, Y.; Wahab, L.; Gill, J. J. Development and Validation of a Microtiter Plate-Based Assay for
Determination of Bacteriophage Host Range and Virulence. Viruses, 2018, 10 (4).
https://doi.org/10.3390/v10040189.
[37] Qadir, M. I.; Mobeen, T.; Masood, A. Phage Therapy: Progress in Pharmacokinetics. Brazilian J.
Pharm. Sci., 2018, 54 (1), e17093. https://doi.org/10.1590/s2175-97902018000117093.
[38] Dąbrowska, K. Phage Therapy: What Factors Shape Phage Pharmacokinetics and Bioavailability?
Systematic and Critical Review. Med. Res. Rev., 2019, 39 (5), 2000–2025.
https://doi.org/10.1002/med.21572.
[39] Benala, M.; Vaiyapuri, M.; Visnuvinayagam, S.; George, J. C.; Raveendran, K.; George, I.;
Mothadaka, M. P.; Badireddy, M. R. A Revisited Two-Step Microtiter Plate Assay: Optimization of
in Vitro Multiplicity of Infection (MOI) for Coliphage and Vibriophage. J. Virol. Methods, 2021,
294, 114177. https://doi.org/10.1016/j.jviromet.2021.114177.
[40] Rajnovic, D.; Muñoz-Berbel, X.; Mas, J. Fast Phage Detection and Quantification: An Optical
Density-Based Approach. PLoS One, 2019, 14 (5), e0216292.
https://doi.org/10.1371/journal.pone.0216292.
[41] Haines, M. E. K.; Hodges, F. E.; Nale, J. Y.; Mahony, J.; van Sinderen, D.; Kaczorowska, J.;
Alrashid, B.; Akter, M.; Brown, N.; Sauvageau, D.; et al. Analysis of Selection Methods to Develop
Novel Phage Therapy Cocktails Against Antimicrobial Resistant Clinical Isolates of Bacteria. Front.
Microbiol., 2021, 12, 613529. https://doi.org/10.3389/fmicb.2021.613529.
[42] Gelman, D.; Yerushalmy, O.; Alkalay-Oren, S.; Rakov, C.; Ben-Porat, S.; Khalifa, L.; Adler, K.;
Abdalrhman, M.; Coppenhagen-Glazer, S.; Aslam, S.; et al. Clinical Phage Microbiology: A
Suggested Framework and Recommendations for the in-Vitro Matching Steps of Phage Therapy.
The Lancet Microbe, 2021, 2 (10), e555–e563. https://doi.org/10.1016/S2666-5247(21)00127-0.
[43] Monferrer, E.; Domingo-Calap, P. Virus-Host Coevolution as a Tool for Controlling Bacterial
Resistance to Phage Therapy. J. Biotechnol. Biomed., 2019, 2 (3), 96–104.
https://doi.org/10.26502/jbb.2642-91280013.
[44] Lindberg, H. M.; McKean, K. A.; Wang, I.-N. Phage Fitness May Help Predict Phage Therapy
Efficacy. Bacteriophage, 2014, 4 (4), e964081. https://doi.org/10.4161/21597073.2014.964081.
[45] Bull, J. J.; Gill, J. J. The Habits of Highly Effective Phages: Population Dynamics as a Framework
for Identifying Therapeutic Phages. Front. Microbiol., 2014, 5 (618).
https://doi.org/10.3389/fmicb.2014.00618.
[46] Koonjan, S.; Seijsing, F.; Cooper, C. J.; Nilsson, A. S. Infection Kinetics and Phylogenetic Analysis
of VB_EcoD_SU57, a Virulent T1-Like Drexlerviridae Coliphage. Front. Microbiol., 2020, 11,
565556. https://doi.org/10.3389/fmicb.2020.565556.
[47] Labrie, S. J.; Samson, J. E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev.
Microbiol., 2010, 8 (5), 317–327. https://doi.org/10.1038/nrmicro2315.
[48] Bondy-Denomy, J.; Qian, J.; Westra, E. R.; Buckling, A.; Guttman, D. S.; Davidson, A. R.; Maxwell,
K. L. Prophages Mediate Defense a gainst Phage Infection through Diverse Mechanisms. ISME J.,
2016, 10 (12), 2854–2866. https://doi.org/10.1038/ismej.2016.79.Güemes, A. G. C.; Merry Youle;

Chapter 3: Experimental Studies
139
Cantú, V. A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners in the Game of Life. Annu. Rev.
Virol., 2016, 3 (1), 197–214. https://doi.org/10.1146/annurev-virology-100114-054952.

3.2 Classification of in vitro phage-host population growth dynamics
140
Su
pp
lem
en
tary F
igu
re
S1
| G
row
th d
ynam
ics
cu
rves
for
phage-b
acte
rium
co
mbin
ati
ons
at
all M
OIs
. P
hage-f
ree c
ontr
ol cu
lture
s fo
r ea
ch A
PE
C s
train
are
inclu
ded
(E
mpty
). B
acte
rial g
row
th (O
D6
00) w
as
mea
sure
d f
or 2
2 h
.

Chapter 3: Experimental Studies
141
Supplementary Figure S2 | Principal component analysis (PCA) of the differences in OD trajectories/values of
the phage-host combinations. The analysis captures 97% of the variance and grouped the total of 2869 entities in
three groups. Group 1 comprises resistant combinations with bacterial growth. Group 2 comprises susceptible
combinations with bacterial killing. Group 3 comprises in-between combination.
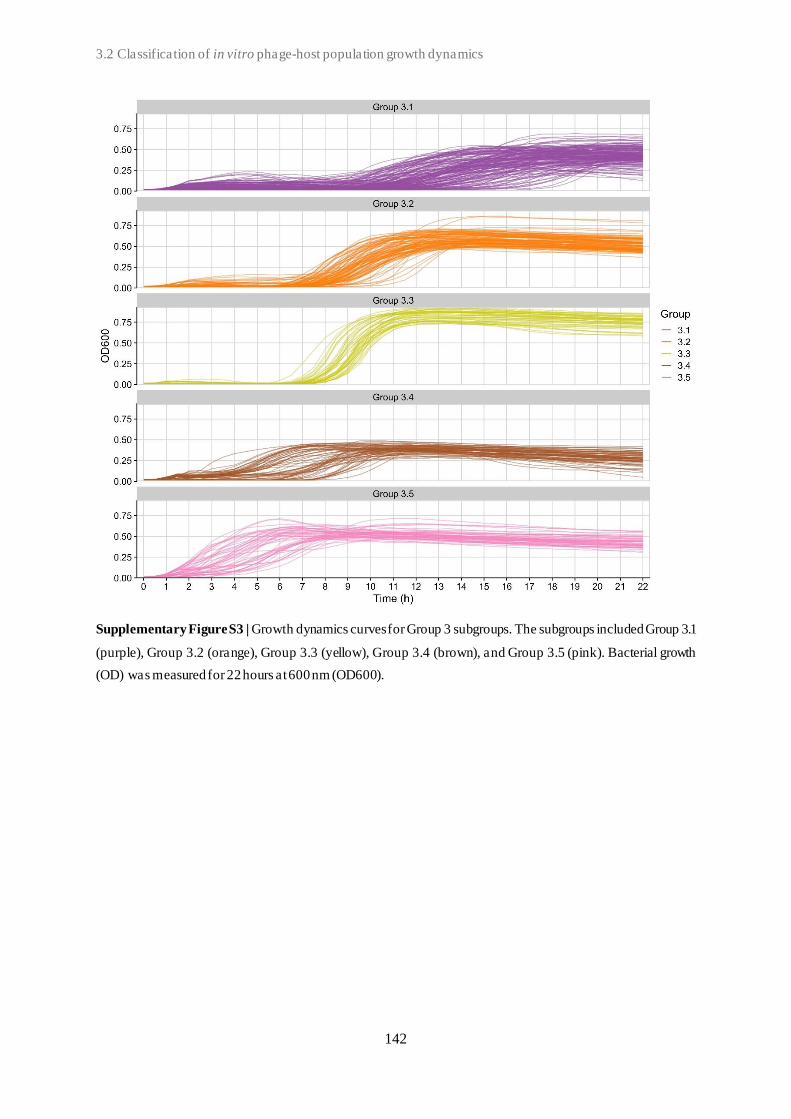
3.2 Classification of in vitro phage-host population growth dynamics
142
Supplementary Figure S3 | Growth dynamics curves for Group 3 subgroups. The subgroups included Group 3.1
(purple), Group 3.2 (orange), Group 3.3 (yellow), Group 3.4 (brown), and Group 3.5 (pink). Bacterial growth
(OD) was measured for 22 hours at 600 nm (OD600).
Chapter 3: Experimental Studies
143
Supplementary Table S1 | Prophages identified in the APEC genomes
Prophage hit GenBank Length (Kb)
Completeness Score # Total Proteins
APEC Strain
Sero-group
Entero_BP_4795 NC_004813 6 incomplete 40 7 B2 O78:H4
Entero_BP_4795 NC_004813 8.2 incomplete 20 16 B8 O1:H7
Entero_cdtI NC_009514 31.1 incomplete 60 28 B3 O78:H4
Entero_cdtI NC_009514 6.7 incomplete 30 7 B9 O78:H9
Entero_DE3 NC_042057 37 incomplete 60 46 B10 O2:H5
Entero_DE3 NC_042057 12.1 incomplete 60 15 B10 O2:H5
Entero_DE3 NC_042057 9.2 incomplete 20 11 B3 O78:H4
Entero_DE3 NC_042057 10.5 incomplete 50 20 B6 O78:H9
Entero_DE3 NC_042057 10.5 incomplete 50 20 B9 O78:H9
Entero_fiAA91_ss NC_022750 25.9 intact 150 32 B2 O78:H4
Entero_HK629 NC_019711 17.1 questionable 70 27 B5 O2:H5
Entero_HK629 NC_019711 8.5 incomplete 20 11 B2 O78:H4
Entero_HK630 NC_019723 15.3 incomplete 20 18 B3 O78:H4
Entero_HK630 NC_019723 13 incomplete 60 16 B2 O78:H4
Entero_JenP1 NC_029028 5.3 incomplete 30 6 B1 O1:H7
Entero_lambda NC_001416 28.2 incomplete 10 26 B2 O78:H4
Entero_lambda NC_001416 26.3 incomplete 10 26 B3 O78:H4
Entero_lambda NC_001416 20.7 incomplete 30 27 B5 O2:H5
Entero_lambda NC_001416 22.9 incomplete 60 21 B7 O78:H9
Entero_lambda NC_001416 10.8 questionable 70 15 B1 O1:H7
Entero_mEp460 NC_019716 14 incomplete 40 19 B6 O78:H9
Entero_mEp460 NC_019716 14 incomplete 40 19 B7 O78:H9
Entero_mEp460 NC_019716 28.1 intact 150 41 B3 O78:H4
Entero_mEp460 NC_019716 23 incomplete 40 39 B2 O78:H4
Entero_mEp460 NC_019716 21 intact 150 29 B1 O1:H7
Entero_mEp460 NC_019716 27.7 intact 140 38 B5 O2:H5
Entero_mEp460 NC_019716 33.8 intact 150 51 B10 O2:H5
Entero_mEp460 NC_019716 6.8 incomplete 50 17 B4 O1:H7
Entero_mEp460 NC_019716 23.2 incomplete 50 10 B5 O2:H5
Entero_mEp460 NC_019716 10.2 incomplete 60 20 B6 O78:H9
Entero_mEp460 NC_019716 9 incomplete 50 17 B7 O78:H9
Entero_mEp460 NC_019716 8.4 incomplete 40 21 B9 O78:H9
Entero_mEp460 NC_019716 23.2 incomplete 50 8 B10 O2:H5
Entero_mEp460 NC_019716 8.7 incomplete 50 15 B6 O78:H9
Entero_mEp460 NC_019716 7.8 incomplete 60 20 B8 O1:H7
Entero_mEp460 NC_019716 8.9 incomplete 30 16 B9 O78:H9
Entero_N15 NC_001901 20.2 incomplete 20 31 B7 O78:H9
Entero_Sf101 NC_027398 38.7 intact 120 61 B1 O1:H7
Entero_SfI NC_027339 9.8 incomplete 30 18 B4 O1:H7
Entero_SfI NC_027339 9.4 incomplete 20 17 B8 O1:H7
Entero_SfI NC_027339 32 incomplete 30 31 B1 O1:H7
Entero_SfV NC_003444 14.1 incomplete 40 9 B9 O78:H9
Entero_SfV NC_003444 10.5 incomplete 60 20 B7 O78:H9
Entero_Wphi NC_005056 23.3 intact 110 28 B7 O78:H9
Entero_YYZ_2008 NC_011356 10.4 incomplete 30 12 B2 O78:H4
Erwini_PEp14 NC_016767 9.1 incomplete 40 14 B9 O78:H9
Escher_500465_1 NC_049342 18.5 incomplete 60 12 B6 O78:H9
Escher_500465_1 NC_049342 13.6 incomplete 60 14 B7 O78:H9
Escher_500465_1 NC_049342 19.1 incomplete 50 22 B9 O78:H9
Escher_500465_1 NC_049342 23.8 questionable 86 29 B1 O1:H7
Escher_500465_1 NC_049342 8.1 incomplete 60 11 B5 O2:H5
3.2 Classification of in vitro phage-host population growth dynamics
144
Supplementary Table S1 | Prophages identified in the APEC genomes
Prophage hit GenBank Length
(Kb) Completeness
Scor
e
# Total
Proteins
APEC
Strain
Sero-
group
Escher_Stx1 NC_004913 13 incomplete 60 14 B3 O78:H4
Escher_phiV10 NC_007804 45.5 intact 98 43 B2 O78:H4
Escher_phiV10 NC_007804 48.5 intact 100 45 B3 O78:H4
Escher_pro483 NC_028943 31.2 intact 150 32 B1 O1:H7
Escher_RCS47 NC_042128 54 incomplete 50 52 B9 O78:H9
Escher_SH2026Stx1 NC_049919 11.1 questionable 70 18 B2 O78:H4
Escher_SH2026Stx1 NC_049919 7 incomplete 40 11 B3 O78:H4
Escher_SH2026Stx1 NC_049919 5 questionable 70 10 B4 O1:H7
Escher_SH2026Stx1 NC_049919 5 incomplete 60 9 B8 O1:H7
Escher_SH2026Stx1 NC_049919 14.4 incomplete 10 18 B5 O2:H5
Flavob_FCL_2 NC_027125 7 incomplete 50 10 B1 O1:H7
Klebsi_4LV2017 NC_047818 15.4 incomplete 10 24 B9 O78:H9
Klebsi_ST437_
OXA245phi4.2 NC_049449 22.2 incomplete 10 13 B6 O78:H9
Klebsi_ST437_
OXA245phi4.2 NC_049449 22.2 incomplete 10 13 B7 O78:H9
Klebsi_ST437_
OXA245phi4.2 NC_049449 13.7 incomplete 10 10 B9 O78:H9
Marino_P12026 NC_018269 9.1 incomplete 40 14 B1 O1:H7
Mycoba_Gaia NC_026590 7 incomplete 50 11 B4 O1:H7
Pectob_ZF40 NC_019522 29.8 incomplete 40 25 B1 O1:H7
Pectob_ZF40 NC_019522 31.7 incomplete 60 30 B4 O1:H7
Pectob_ZF40 NC_019522 30.5 incomplete 40 23 B8 O1:H7
Pseudo_phiPSA1 NC_024365 20 intact 100 25 B3 O78:H4
Rhodoc_RGL3 NC_016650 9.1 incomplete 40 15 B7 O78:H9
Salmon_118970_sal
3 NC_031940 24.5 intact 150 36 B3 O78:H4
Salmon_118970_sal
3 NC_031940 14.9 incomplete 30 28 B7 O78:H9
Salmon_118970_sal
3 NC_031940 8.9 incomplete 60 14 B7 O78:H9
Salmon_SJ46 NC_031129 45 questionable 70 68 B9 O78:H9
Salmon_SJ46 NC_031129 7.6 incomplete 10 6 B6 O78:H9
Salmon_SSU5 NC_018843 9.1 incomplete 40 13 B2 O78:H4
Shigel_POCJ13 NC_025434 24.4 incomplete 20 21 B3 O78:H4
Shigel_SfII NC_021857 26.2 intact 150 37 B1 O1:H7
Shigel_SfII NC_021857 10.2 incomplete 60 13 B6 O78:H9
Staphy_SPbeta_like NC_029119 17.5 incomplete 40 8 B3 O78:H4
Stx2_c_1717 NC_011357 3.4 incomplete 60 7 B10 O2:H5
Stx2_c_Stx2a_F451 NC_049924 6.7 incomplete 60 10 B3 O78:H4
Vibrio_X29 NC_024369 26.7 intact 150 32 B2 O78:H4
Vibrio_X29 NC_024369 30 intact 150 34 B7 O78:H9
Yersin_L_413C NC_004745 23.3 intact 110 28 B6 O78:H9
Yersin_L_413C NC_004745 27.9 intact 120 29 B4 O1:H7
Yersin_L_413C NC_004745 7.3 incomplete 30 7 B8 O1:H7
Supplementary Table S2: Non-metric multidimensional scaling (NMDS) grouping summary and
Supplementary Table S3: Non-metric multidimensional scaling (NMDS) Group 3 subgrouping
summary are not included in this thesis due to their large size. Both tables are available upon request.

Chapter 3: Experimental Studies
145
3.3
Spontaneous phage resistance in avian pathogenic Escherichia
coli
Patricia E. Sørensen1,2, Sharmin Baig3, Marc Stegger3, Hanne Ingmer5, An Garmyn1,
and Patrick Butaye1,2
1 Department of Pathology, Bacteriology and Poultry diseases, Ghent University, Belgium
2 Department of Biomedical Sciences, Ross University School of Veterinary Medicine, St. Kitts, West
Indies
3 Department of Bacteria, Parasites and Fungi, Statens Serum Institut, Copenhagen, Denmark
4 Department of Veterinary and Animal Sciences, University of Copenhagen, Denmark
Published in Frontiers in Microbiology 2021, 12 (782757)
3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
146
Abstract
Avian pathogenic Escherichia coli (APEC) is one of the most important bacterial pathogens
affecting poultry worldwide. The emergence of multidrug-resistant pathogens has renewed the
interest in the therapeutic use of bacteriophages (phages). However, a major concern for the
successful implementation of phage therapy is the emergence of phage-resistant strains. The
understanding of the phage-host interactions, as well as underlying mechanisms of resistance,
have shown to be essential for the development of a successful phage therapy. Here, we
demonstrate that the strictly lytic Escherichia phage vB_EcoM-P10 rapidly selected for
resistance in the APEC ST95 O1 strain AM621. Whole-genome sequence analysis of 109
spontaneous phage-resistant mutant strains revealed 41 mutants with single-nucleotide
polymorphisms (SNPs) in their core genome. In 32 of these, a single SNP was detected while
two SNPs were identified in a total of nine strains. In total, 34 unique SNPs were detected. In
42 strains, including 18 strains with SNP(s), gene losses spanning 17 different genes were
detected. Affected by genetic changes were genes known to be involved in phage resistance
(outer membrane protein A, lipopolysaccharide-, O-antigen-, or cell wall-related genes) as well
as genes not previously linked to phage resistance, including two hypothetical genes. In several
strains, we did not detect any genetic changes. Infecting phages were not able to overcome the
phage resistance in host strains. However, interestingly the initial infection was shown to have
a great fitness cost for several mutant strains, with up to ~65% decrease in overall growth. In
conclusion, this study provides valuable insights into the phage-host interaction and phage
resistance in APEC. Although acquired resistance to phages is frequently observed in
pathogenic E. coli, it may be associated with loss of fitness, which could be exploited in phage
therapy.

Chapter 3: Experimental Studies
147
Introduction
Bacteriophages (phages) are viruses that specifically infect bacteria, and are estimated to be
the most abundant organisms on Earth with more than 1031 entities [1]. Phages are unable to
replicate independently of a susceptible bacterial host, and their host range is determined by a
combination of various factors, including specificity of host-binding phage proteins and
bacterial phage-resistance mechanisms [2, 3]. Virulent phages are strict parasites of their host
and confer a selective pressure on their host population through host cell lysis [4]. In response,
bacteria can evolve resistance to phage infection through various mechanisms, such as
spontaneous mutations, acquisition of restriction-modification (R-M) systems, and adaptive
immunity via Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Cas
system(s). These mechanisms can be used to target dif ferent steps of the phage life cycle,
including phage attachment, adsorption, replication, and host cell lysis [5, 6]. The different
resistance mechanisms result in distinct resistance phenotypes. These can differ in whether the
resistance is partial or complete, in the fitness cost associated with resistance, and in whether
the mutation can be countered by a mutation in the infecting phage [7, 8]. Although various
antiviral defense systems are found in bacteria, the emergence of phage resistance as well as
phage-bacterium co-evolution are often driven by spontaneous mutations [6, 9], which may
confer phage resistance by modifying phage-associated receptors on the bacterial surface.
However, such changes have also been associated with reduced fitness relative to non-resistant
strains [10]. Phage-resistant bacteria may become less virulent as in case when mutations occur
in their lipopolysaccharides (LPS), or may experience impaired growth in case of mutations in
genes involved in essential cell functions [11]. Additionally, maintenance of defense systems
such as R-M enzymes and CRISPR-Cas, also has its own costs associated with enzyme
production and expression [12–14].
Avian pathogenic Escherichia coli (APEC) is one of the most important bacterial pathogens
affecting poultry. These pathogens cause a large range of extra-intestinal infections, which
collectively are referred to as colibacillosis. These infections can result in high morbidity and
mortality, and hereby, significant economic loses to the poultry industry worldwide [15–18].
Here, the APEC with O-serogroups O1, O2 and O78 constitute more than 80% of the infection
cases [19]. As current antimicrobials become increasingly inadequate to treat bacterial
infections and a global focus to reduce conventional antimicrobial usage in general, alternative
treatment strategies, such as the therapeutic use of phages (phage therapy), are urgently needed
[20–22]. However, being able to understand phage-host interactions as well as the underlying

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
148
mechanisms of resistance is essential for successful phage therapy application [23]. Here, we
investigate the phage-host interactions and resistance through isolation and characterisation of
spontaneous phage-resistant mutants of APEC.
Materials and methods
Bacterial strains and growth conditions
The APEC ST95 O1:H7 strain, AM621, is part of the in-house collection that was isolated from
clinical material suspected of APEC infection from Belgium collected during 2013-2014 by
Animal Health Care Flanders (Torhout, Belgium). The E. coli K-12 derived laboratory strain
K514 [24] was included as a phage-susceptible control and host strain. Bacterial strains were
grown in LB broth (Miller) or on LB agar supplemented with 1.5% bacteriological agar no. 1
(w/v) (Oxoid, Thermo Fisher Scientific, USA) overnight (16-18 h) at 37°C unless stated
otherwise. Broth cultures were incubated with shaking (120 rpm). Strains were stored at -80°C
in LB broth supplemented with 15% glycerol.
Bacteriophage isolation and propagation
The strictly virulent Escherichia phage vB_EcoM-P10 (SRA accession no. SRX8360061) used
in this study is a part of the in-house phage collection (Phage P1 from Chapter 3.2). The phage
was isolated from poultry feces and processed as previously described [25]. Phage lysates were
stored at 4°C, at titers ranging from ~1.2 x 108 to 1.4 x 109 PFU/ml. Escherichia phage
vB_EcoM-P10 was classified (according to the International Committee on Taxonomy of
Viruses (ICTV) taxonomy) as a tailed Myoviridae phage belonging to the Tevenvirinae
subfamily and Tequatrovirus genus.
Isolation of phage-resistant mutant strains
Phage-resistant APEC strains were obtained using the agar plate (AP) [26] and the secondary
culture (SC) technique [27] with minor modifications (Supplementary Figure S1). Briefly,
overnight culture of WT strain AM621 was inoculated in LB broth supplemented with CaCl2
(final concentration of 10 mM) and then infected with suspension of virulent phage vB_EcoM-
P10, at a multiplicity of infection (MOI) of 0.1, 1, 10, and 100. For the AP technique,
suspensions were streaked directly onto LB agar plates supplemented with CaCl2 (final

Chapter 3: Experimental Studies
149
concentration of 10 mM) and incubated for 48 h at 37°C. After incubation of 24 h and 48 h,
individual colonies were selected from each MOI suspension and cultured in LB broth. Isolates
were purified by three consecutive streakings on LB agar (in the absence of phage) and
recovered as presumptive phage-resistant mutants. Remaining MOI cultures that were not
streaked on agar plates were subjected to the SC technique. Cultures were incubated at 37°C
with shaking (120 rpm) for ~5 hours. Cultures exhibiting complete or partial lysis and
subsequent (secondary) growth after an additional incubation of 24 h were selected and
streaked on LB agar plates. Remaining “SC-T24” solutions were stored at 4°C until required.
Presumptive phage-resistant mutants were recovered as described for the AP technique and
stored at 4°C until required. An experiment with phage-susceptible E. coli laboratory strain
K514 was performed in parallel as control. The AP/SC experiments were repeated six times.
Presumptive phage-resistant mutants were infected with phage vB_EcoM-P10 using the fitness
test experimental set-up (described below). Mutants that displayed normal bacterial growth or
increased growth compared to the phage-sensitive AM621 WT strain were defined as true
phage-resistant mutants and stored at -80°C in LB broth supplemented with 15% glycerol (v/v).
Efficiency of the phage-resistant mutant recovery was calculated according to the formula
presented by Capra et al. (2011): (number of true phage-resistant mutants ⁄ number of
presumptive phage-resistant mutants) * 100.
Isolation and enumeration of potential phage mutants
To isolate potential phage mutants, the SC-T24 solutions were centrifuged and filtered using a
0.2 µm filter (Whatman, GE Healthcare, Germany). The filtrated SC-T24-phage suspensions
were enumerated and tested for lytic activity on the host bacteria, E. coli K-12 derived
laboratory strain K514, using the double-layer agar (DLA) technique [29]. Briefly, phage
suspensions were serial diluted and spotted on an overlay of the host bacteria on LB agar
supplemented with 0.7% agar and 0.5 mM CaCl2. A clear zone in the plate, a plaque, resulting
from the lysis of host bacterial cells, indicated the presence of virulent phage. Phage lysates
were stored at 4°C until required.
Bacterial fitness
Bacterial reduction experiments were performed as described previously [30, 31], with minor
modifications. Bacterial overnight cultures were used, and the cell concentration was adjusted

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
150
to ~108 CFU/ml for every experiment. Bacterial suspensions were inoculated with phage,
yielding MOIs of 0.1, 1, 10, and 100. All bacterial reduction curves were generated using 96-
well plates with working volumes of 200 µl. The experiment was carried out in duplicates and
repeated three times. Two wells of phage-free bacterial cultures and two wells of bacteria-free
phage culture were included on every plate as control experiments in addition to one media
blank for reference. Optical density (OD) for the wavelength of 600 nm was measured with the
Thermo Fisher Scientific Multiskan GO Microplate Spectrophotometer and the data were
recorded using the SkanIt Software, v6.0.2.3. OD600 measurements were taken immediately
after inoculation and then at 30 minutes intervals afterwards for 22 h. The protocol parameters
included incubation temperature of 37°C and continuous shaking with medium speed.
Reduction curves were obtained by plotting OD600 values after baseline adjustments against
time. For each reduction curve, area under the curve (AUC) was calculated using GraphPad
Prism v9.1.0.221 with default settings. AUC was calculated as average of four replicates.
Strains were defined as truly resistant when % of decrease in AUC in the presence of phage
was minimum 20% less relative to the WT strain. Fitness cost associated with acquired
mutations in true resistant strains was defined as decrease in AUC compared to WT strain in
the absence of phage.
Genomic DNA extraction and sequencing
Genomic DNA was extracted from true phage-resistant bacterial strains using Qiagen’s
DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), with subsequent library
construction using the Nextera XT Kit (Illumina, Little Chesterford, UK) using a 300-cycle kit
on the Illumina NextSeq 550 platform according to the manufacturer’s instructions.
Phage DNA was extracted and purified using Phage DNA Isolation Kit (Norgen Biotek Corp.,
Canada), as indicated by the instructions provided by the manufacturer. The DNA yield was
quantified using the QuantiFluor dsDNA System (Promega) and Quantus Fluorometer. The
DNA purity (OD 260/280 ratio of ~1.7-1.8) was measured using NanoDrop (Isogen Life
Science). Libraries were constructed using the Nextera XT Kit (Illumina, Little Chesterford,
UK) using a 300-cycle kit on the Illumina NextSeq platform according to the manufacturer’s
instructions.

Chapter 3: Experimental Studies
151
Bacterial genome analysis
The open-source bifrost software (https://github.com/ssi-dk/bifrost), v1.1.0, was used for
quality control of the WGS data. The raw reads were de novo assembled using SPAdes v3.11.1
[32], and contigs with less than 200 bp were excluded. APEC serotype was predicted for each
of the strains using SerotypeFinder, v2.0 [33]. Genomes were annotated using Prokka, v1.12
[34], and pan genome analysis was carried out with Roary, v.3.12.0 [35], with minimum 90%
similarity on protein level. Gene presence was subsequent confirmed using Mykrobe predictor,
v0.5.6 [36]. Genes classified as present were further filtered for coverage (c>70) and depth
(d>3). When inconsistencies were observed, manual BLASTn searches were performed. Cases
where a gene was detected in a mutant strain but not in the WT strain were excluded from
further analysis, as this was assumed to be sequencing error or contamination (a false-positive).
PlasmidFinder 2.1 with default settings was used to screen assembled genomes for plasmids in
the Enterobacteriaceae database. Plasmid replicons with less than 90% identity and 60%
coverage were excluded. ABRicate v1.0.1 (https://github.com/tseemann/abricate) with default
options was used to screen assembled genomes for antimicrobial resistance genes with
ResFinder database [37], NCBI Bacterial Antimicrobial Resistance Reference Gene Database
[38], and the Comprehensive Antibiotic Resistance Database (CARD) [39]. Virulence genes
were identified using ABRicate with sequences from the Ecoli_VF database. CRISPR systems
were identified using the Geneious Prime v2020.1.1 Crispr Recognition Tool Wrapper v1.1.
and CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index) [40] with
default settings. A quality score was automatically given to CRISPR arrays consisting of
repeats and spacer sequences in the form of “evidence level”, rated 1-4, where 1 includes small
CRISPRs (with three or less spaces) and 2-4 are classified based on repeat and spacer similarity.
BLAST analysis was performed to determine if identified CRISPR spacer sequences matched
the invading Escherichia phage vB_EcoM-P10 genome.
Bacterial core genome SNP analysis
To assess the relationship between strains, a single nucleotide polymorphism (SNP)-based
phylogeny was obtained using SNPs identified by the Northern Arizona SNP Pipeline (NASP),
v1.2.0 [41], with the Burrows-Wheeler Aligner (BWA) algorithm, v0.7.17-r1188 [42].
Illumina reads from all individual strains were aligned against the AM621 WT scaffold genome
obtained as described with a cut-off of all contigs <500 bp above after removal of duplicated
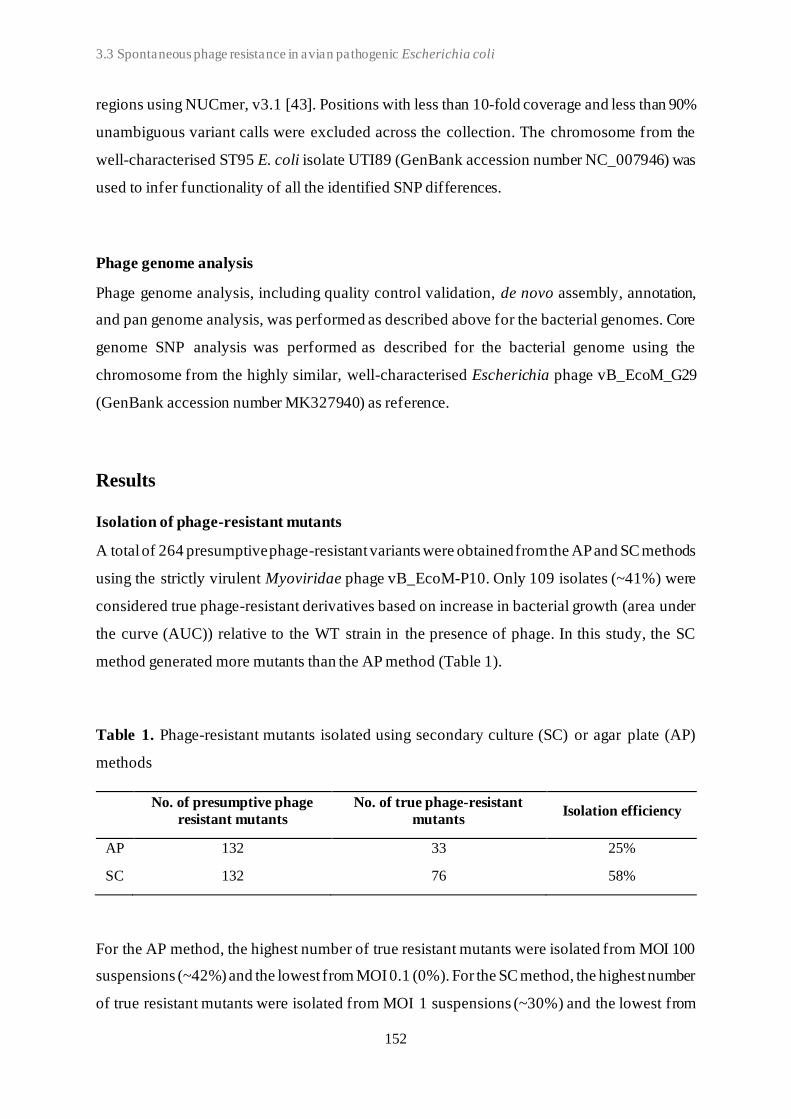
3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
152
regions using NUCmer, v3.1 [43]. Positions with less than 10-fold coverage and less than 90%
unambiguous variant calls were excluded across the collection. The chromosome from the
well-characterised ST95 E. coli isolate UTI89 (GenBank accession number NC_007946) was
used to infer functionality of all the identified SNP differences.
Phage genome analysis
Phage genome analysis, including quality control validation, de novo assembly, annotation,
and pan genome analysis, was performed as described above for the bacterial genomes. Core
genome SNP analysis was performed as described for the bacterial genome using the
chromosome from the highly similar, well-characterised Escherichia phage vB_EcoM_G29
(GenBank accession number MK327940) as reference.
Results
Isolation of phage-resistant mutants
A total of 264 presumptive phage-resistant variants were obtained from the AP and SC methods
using the strictly virulent Myoviridae phage vB_EcoM-P10. Only 109 isolates (~41%) were
considered true phage-resistant derivatives based on increase in bacterial growth (area under
the curve (AUC)) relative to the WT strain in the presence of phage. In this study, the SC
method generated more mutants than the AP method (Table 1).
Table 1. Phage-resistant mutants isolated using secondary culture (SC) or agar plate (AP)
methods
No. of presumptive phage
resistant mutants
No. of true phage-resistant
mutants Isolation efficiency
AP 132 33 25%
SC 132 76 58%
For the AP method, the highest number of true resistant mutants were isolated from MOI 100
suspensions (~42%) and the lowest from MOI 0.1 (0%). For the SC method, the highest number
of true resistant mutants were isolated from MOI 1 suspensions (~30%) and the lowest from
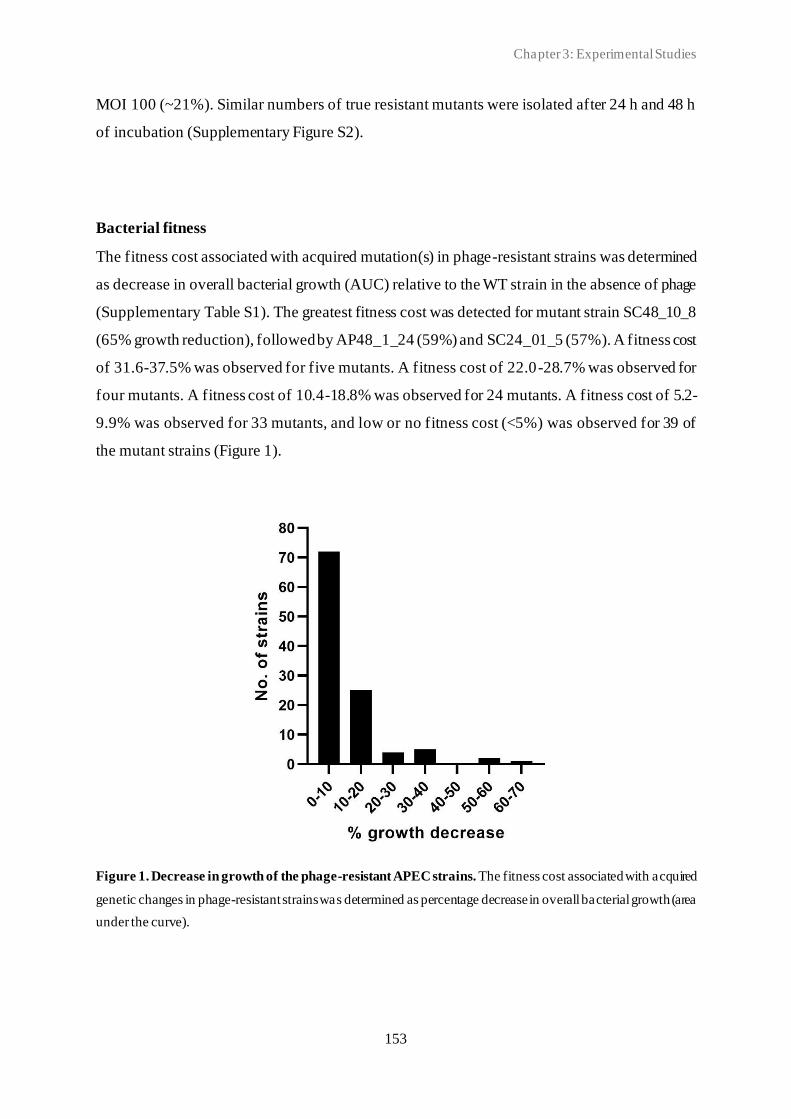
Chapter 3: Experimental Studies
153
MOI 100 (~21%). Similar numbers of true resistant mutants were isolated after 24 h and 48 h
of incubation (Supplementary Figure S2).
Bacterial fitness
The fitness cost associated with acquired mutation(s) in phage-resistant strains was determined
as decrease in overall bacterial growth (AUC) relative to the WT strain in the absence of phage
(Supplementary Table S1). The greatest fitness cost was detected for mutant strain SC48_10_8
(65% growth reduction), followed by AP48_1_24 (59%) and SC24_01_5 (57%). A fitness cost
of 31.6-37.5% was observed for five mutants. A fitness cost of 22.0-28.7% was observed for
four mutants. A fitness cost of 10.4-18.8% was observed for 24 mutants. A fitness cost of 5.2-
9.9% was observed for 33 mutants, and low or no fitness cost (<5%) was observed for 39 of
the mutant strains (Figure 1).
Figure 1. Decrease in growth of the phage-resistant APEC strains. The fitness cost associated with acquired
genetic changes in phage-resistant strains was determined as percentage decrease in overall bacterial growth (area
under the curve).

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
154
Bacterial genome analysis
WGS of the bacterial genomes yielded a total of 1,934,298- 6,753,240 paired-end reads for
each isolate with an average coverage of 51-177-fold. De novo assembly resulted in 192-353
contigs and an N50 value from between 51,335- 189,445 bp.
The bacterial strains were subjected to WGS analysis. All 109 resistant strains showed similar
genetic characteristics as the AM621 WT, including a genome size between ~5.27 and ~5.40
Mbp and G+C content between 50.2 and 50.6%. Gene absence/presence analysis identified a
total of 17 different accessory genes (after exclusion of false positives), that were lost (partial
or complete) in one or more of mutant strains (Figure 2 and Table 2). A full overview of the
genes lost in phage-resistant mutants is shown in Supplementary Table S2. None of the mutant
strains lost any plasmid replicons compared to the WT. The six plasmid replicons detected
included Col(MG828), IncFIA, IncFIB(AP001918), IncFIC(FII), IncI1-I(Alpha), and IncX1.
Figure 2. Genes affected by genetic changes in phage-resistant APEC strains. In total, 44 different genes were
affected by genetic change(s). Genetic changes included complete gene loss, partial gene loss, or point mutations
(nonsense, missense, or synonymous). Full circle = complete gene loss in few mutants or nonsense mutation in
one mutant. Striped circle = missense mutation in one mutant. * = protein name is shown as gene name is
unknown.

Chapter 3: Experimental Studies
155
Ta
ble
2 | S
um
mary
of
gen
eti
c c
hanges
an
d a
ffecte
d g
en
es
in p
hage
-res
ista
nt E
. co
li s
train
s
Ch
an
ge
Aff
ecte
d
gen
e
An
no
tati
on
F
un
cti
on
N
o. o
f
stra
ins
Ref(
s).
Gen
e lo
ss
neu
E
Poly
sial
ic a
cid b
iosy
nth
esis
pro
tein
, N
euE
E
. co
li K
1 s
iali
c ac
id c
apsu
le s
ynth
esis
24
[4
4]
Gen
e lo
ss
gro
up_67
H
ypo
thet
ical
pro
tein
U
nknow
n
24
-
Gen
e lo
ss
gro
up
_2
37
H
yp
oth
etic
al p
rote
in
Un
kn
ow
n
10
-
Gen
e lo
ss
gro
up
_3
10
T
etra
tric
op
epti
de
rep
eat (T
PR
) p
rote
in
Med
iati
on
of
pro
tein
-pro
tein
inte
ract
ions
8
[45
]
SN
P
ack
A
Ace
tate
kin
ase
Phosp
hory
lati
on o
f ac
etat
e to
ace
tyl phosp
hat
e 7
[46
]
SN
P
SN
P1
*
Hy
po
thet
ical
pro
tein
U
nk
no
wn
6
-
Gen
e lo
ss
mp
rA_
1
Mar
R f
amil
y t
ran
scri
pti
on
al r
egu
lato
r R
egu
lati
on
of
num
erous
cell
ula
r pro
cess
es
5
[47
]
Gen
e lo
ss
rfbX
O
-anti
gen
tra
nsp
ort
er
Tra
nsp
ort
of
O-p
oly
sacc
har
ide
mole
cule
s 4
[48
]
SN
P
gly
S
Gly
cyl-
tRN
A s
yn
thet
ase
bet
a ch
ain
tR
NA
rec
og
nit
ion
4
[49
]
Gen
e lo
ss
fad
E
Acy
l-C
oA
deh
yd
rog
enas
e D
ehy
dro
gen
atio
n o
f ac
yl-
coen
zym
es A
3
[50
]
Gen
e lo
ss
yafV
Y
afV
(2
-oxoglu
tara
mat
e am
idas
e)
Met
aboli
te r
epai
r en
zym
e 3
[51
]
Gen
e lo
ss
ivy
Ver
tebra
te lyso
som
e in
hib
itor
Pro
tect
ion
ag
ainst
lyso
zym
e-m
edia
ted c
ell w
all
hy
dro
lysi
s 3
[52
]
SN
P
gln
D
Pro
tein
-PII
uri
dyly
ltra
nsf
eras
e N
itro
gen
reg
ula
tion
3
[53
]
SN
P
pta
P
hosp
hat
e ac
etylt
ransf
eras
e A
ceta
te m
etab
oli
sm
3
[46
]
Gen
e lo
ss
epsM
_1
A
cety
ltra
nsf
eras
e / N
euD
pro
tein
E
. co
li K
1 s
iali
c ac
id c
apsu
le s
yn
thes
is
2
[54
]
Gen
e lo
ss
wzy
O
1 f
amil
y O
-anti
gen
poly
mer
ase
Synth
esis
of
the
LP
S B
-ban
d O
anti
gen
2
[55
]
SN
P
om
pA
O
ute
r m
embra
ne
pro
tein
A (
Om
pA
)
Key
E. co
li v
irule
nce
fac
tor
2
[56
, 5
7]
SN
P
relA
G
TP
py
rop
ho
sph
ok
inas
e S
yn
thes
is o
f p
pG
pp f
rom
GT
P
2
[58
]
SN
P
pyk
F
Pyru
vat
e kin
ase
Reg
ula
tion o
f th
e gly
coly
tic
pat
hw
ay
2
[59
]
Gen
e lo
ss
wek
M
Gly
cosy
ltra
nsf
eras
e fa
mil
y 4
P
epti
dogly
can b
iosy
nth
esis
1
[48
, 6
0]
Gen
e lo
ss
gro
up
_2
71
H
yp
oth
etic
al p
rote
in
Un
kn
ow
n
1
-
Gen
e lo
ss
hokA
H
okA
T
oxin
of
a ty
pe
I to
xin
-anti
toxin
(T
A)
syst
em
1
[61
]
Gen
e lo
ss
ydfO
D
UF
139
8 f
amil
y p
rote
in, Y
dfO
U
nknow
n
1
[62
]
Gen
e lo
ss
neu
A
Acy
lneu
ram
inat
e cy
tid
yly
ltra
nsf
eras
e E
. co
li K
1 s
iali
c ac
id c
apsu
le s
yn
thes
is
1
[54
]
Gen
e lo
ss
splE
S
erin
e pro
teas
e S
plE
In
volv
ed i
n v
ario
us
bio
logic
al p
roce
sses
1
[63
]
SN
P
dsb
B
Per
ipla
smic
thio
l:dis
ulf
ide
oxid
ore
duct
ase
Dsb
B
Ele
ctro
n tra
nsf
er c
atal
yst
1
[64
]
SN
P
wzz
B
O-a
nti
gen
ch
ain
len
gth
det
erm
inan
t p
rote
in W
zzB
L
ipo
po
lysa
cchar
ide
(LP
S)
bio
synth
esis
1
[65
]
SN
P
met
G
Met
hio
ny
l-tR
NA
synth
etas
e P
rote
in b
iosy
nth
esis
1
[66
]
SN
P
SN
P11*
tR
NA
-Val
-GA
C
Tra
nsf
er o
f am
ino a
cids
to t
he
riboso
me
1
[67
]
SN
P
na
rP
Nit
rate
/nit
rite
res
po
nse
reg
ula
tor
pro
tein
Nar
P
Gen
e ex
pre
ssio
n r
egula
tion
1
[68
]

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
156
Ta
ble
2 | C
on
tin
ued
Ch
an
ge
Aff
ecte
d
gen
e
An
no
tati
on
F
un
cti
on
N
o. o
f
stra
ins
Ref(
s).
SN
P
glt
X
Glu
tam
yl-
tRN
A s
ynth
etas
e P
rote
in b
iosy
nth
esis
1
[69
]
SN
P
hcp
T
6S
S c
om
pon
ent H
cp
Bac
teri
al inte
ract
ion w
ith h
ost
cel
ls
1
[70
]
SN
P
rap
Z
RN
ase
adap
ter
pro
tein
Rap
Z
Cel
l en
vel
op
e pre
curs
or
sensi
ng a
nd s
ignal
ing
1
[71
–7
3]
SN
P
rbsA
M
on
osa
cch
arid
e-tr
ansp
ort
ing
AT
Pas
e T
ran
sfer
of
solu
tes
acro
ss m
embra
nes
1
[74
]
SN
P
xylB
X
ylu
lose
kin
ase
Phosp
hory
lati
on o
f D
-xylu
lose
to D
-xylu
lose
5-
ph
osp
hat
e 1
[75
]
SN
P
wa
aC
L
ipo
po
lysa
cch
arid
e co
re h
epto
sylt
ran
sfer
ase
I L
ipo
po
lysa
cchar
ide
(LP
S)
bio
synth
esis
1
[60
]
SN
P
waaY
L
ipop
oly
sacc
har
ide
core
hep
tose
(II
) kin
ase
Rfa
Y
Lip
opoly
sacc
har
ide
(LP
S)
bio
synth
esis
1
[60
]
SN
P
fur
Fer
ric
up
tak
e re
gu
lati
on
pro
tein
FU
R
Tra
nsc
rip
tio
nal
reg
ula
tion o
f ir
on m
etab
oli
sm
1
[76
]
SN
P
SN
P3
1*
5
'-n
ucl
eoti
das
e H
yd
roly
sis
of
the
phosp
hat
e gro
up o
f 5′-
nucl
eoti
des
1
[77
]
SN
P
phnD
P
ho
sph
on
ate
AB
C tra
nsp
ort
er s
ub
stra
te-b
ind
ing
pro
tein
Phn
D
Phosp
honat
e upta
ke
and u
tili
sati
on p
athw
ay
1
[78
]
SN
P
ybjT
U
nch
arac
teri
sed
pro
tein
Yb
jT
E. co
li L
PS
bio
synth
esis
and o
ther
core
cel
l en
vel
op
e co
mp
onen
ts
1
[79
]
SN
P
rne
Rib
on
ucl
ease
E
RN
A p
roce
ssin
g a
nd m
RN
A d
egra
dat
ion
1
[80
]
SN
P
sgcR
sg
c re
gio
n tra
nsc
ripti
onal
reg
ula
tor
Tra
nsc
ripti
onal
reg
ula
tion
1
[81
]
SN
P
SN
P37*
H
ypo
thet
ical
pro
tein
U
nknow
n
1
-

Chapter 3: Experimental Studies
157
All but one mutant strain encoded the same resistance genes as the WT strain. Only one mutant
strain, AP24_100_8, had lost qnrS1, a quinolone resistance gene. A total of 226 different
virulence genes were all identified in both the WT strain and all the mutants. Two different
type I-F CRISPR systems (evidence level 4) were detected in the AM621 WT strain. The first
system comprised seven repeat units of 20 bp and six CRISPR spacers, including five spacers
of 40 bp and one spacer of 41 bp. The second system comprised six repeat units of 28 bp and
five spacers of 32 bp. Moreover, two additional small CRISPR-like structures (evidence level
1); one with only two CRISPR repeats (44 bp) and one spacer (52 bp) and another with only
two repeats (36 bp) and one spacer (59 bp) were separately identified in the genome. The same
two CRISPR systems and two small CRISPR-like elements were found in all 109 mutant
strains. Additionally, between one and eight evidence level one CRISPR-like structures, which
were not in the WT strain, were detected in 102 of the mutants (Supplementary Table S3). Only
three mutant strains, AP24_10_14, AP48_1_24, and SC24_01_5, had acquired a CRISPR-like
element spacer of 53 bp that matched the invading phage genome.
Bacterial core genome SNP analysis
SNP analysis identified between 0-2 SNP difference(s) in the core genome between AM621
and the mutants. Of the 109 mutants, 66 showed no SNP differences, 33 mutants showed one
SNP difference and 10 mutants showed two SNP differences (Figure 3). A summary of SNPs
identified in the mutants is shown in Table 2 and Figure 2. A total of 37 unique SNPs were
identified, five of which resulted in a nonsense mutation, 21 in a missense mutation, six in a
synonymous mutation, and five of which were found in non-coding regions when analysed
against the annotation of the UTI89 genome. Nonsense mutations were found in five different
genes, including acetate kinase (ackA), outer membrane protein A (ompA), phosphate
acetyltransferase (pta), LPS core heptosyltransferase I (waaC), and LPS core heptose (II)
kinase (waaY) (Figure 2). Missense mutations were found in 19 different genes (Supplementary
Table S2).
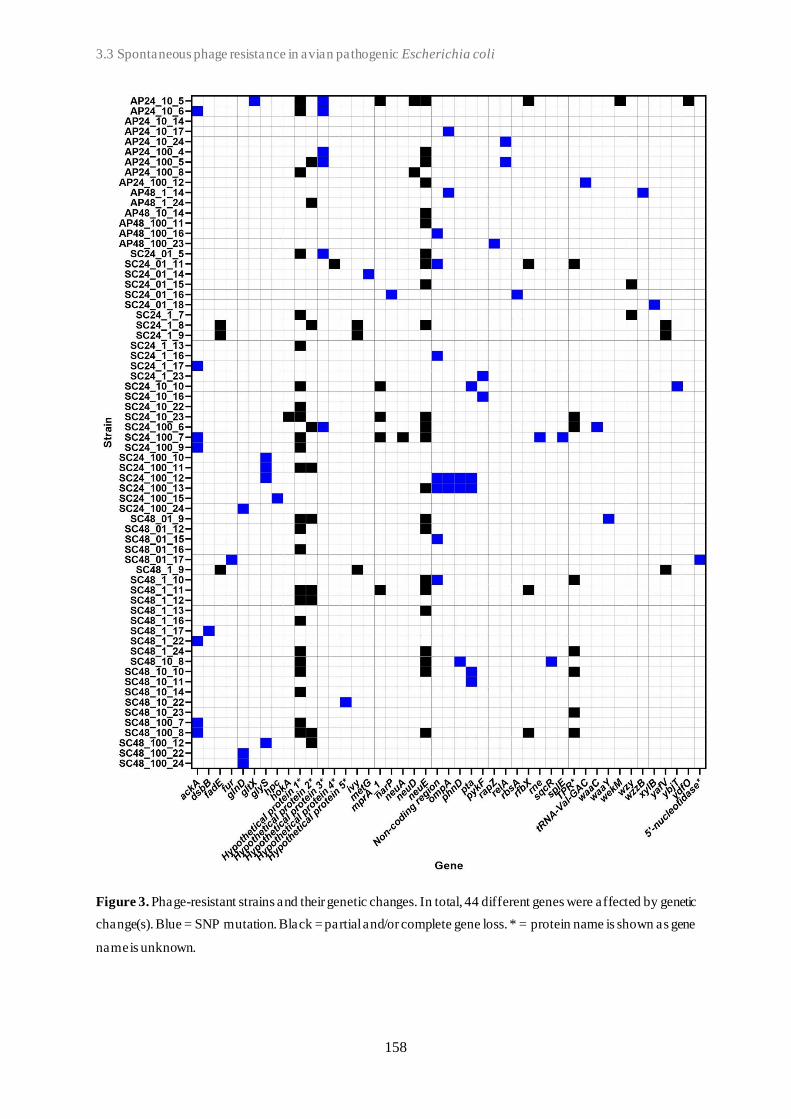
3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
158
Figure 3. Phage-resistant strains and their genetic changes. In total, 44 different genes were affected by genetic
change(s). Blue = SNP mutation. Black = partial and/or complete gene loss. * = protein name is shown as gene
name is unknown.
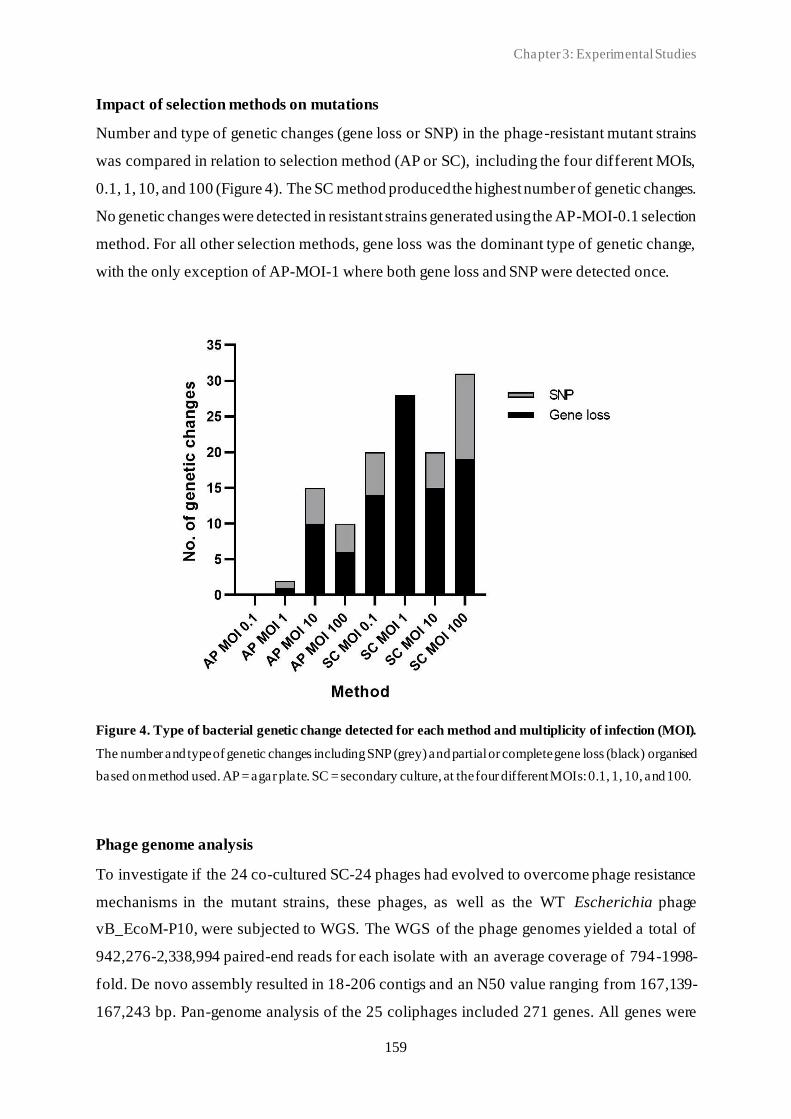
Chapter 3: Experimental Studies
159
Impact of selection methods on mutations
Number and type of genetic changes (gene loss or SNP) in the phage-resistant mutant strains
was compared in relation to selection method (AP or SC), including the four different MOIs,
0.1, 1, 10, and 100 (Figure 4). The SC method produced the highest number of genetic changes.
No genetic changes were detected in resistant strains generated using the AP-MOI-0.1 selection
method. For all other selection methods, gene loss was the dominant type of genetic change,
with the only exception of AP-MOI-1 where both gene loss and SNP were detected once.
Figure 4. Type of bacterial genetic change detected for each method and multiplicity of infection (MOI).
The number and type of genetic changes including SNP (grey) and partial or complete gene loss (black) organised
based on method used. AP = agar plate. SC = secondary culture, at the four different MOIs: 0.1, 1, 10, and 100.
Phage genome analysis
To investigate if the 24 co-cultured SC-24 phages had evolved to overcome phage resistance
mechanisms in the mutant strains, these phages, as well as the WT Escherichia phage
vB_EcoM-P10, were subjected to WGS. The WGS of the phage genomes yielded a total of
942,276-2,338,994 paired-end reads for each isolate with an average coverage of 794-1998-
fold. De novo assembly resulted in 18-206 contigs and an N50 value ranging from 167,139-
167,243 bp. Pan-genome analysis of the 25 coliphages included 271 genes. All genes were

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
160
detected in all potential mutant phages using BLAST. SNP analysis identified no SNP
differences in the core genome between WT Escherichia phage vB_EcoM-P10 and the
potential phage mutants.
Discussion
In this study, we selected and characterised phage-resistant mutant strains of O1 APEC strain
AM621. Using a combined approach of the SC and AP method resulted in an overall mutant
isolation efficiency of ~41%. Previous studies using this approach found an isolation efficiency
of true resistant Lactobacillus paracasei isolates of 56% [82] and an average isolation
efficiency of 36.5% (ranging between 29.5-50%) of true resistant Lactobacillus delbrueckii
isolates [83]. We found an SC method isolation efficiency of 57.6%, while the AP method
efficiency was much lower (25.0%). The higher efficiency of the SC method has been reported
before though with similar, smaller or larger differences [82–84]. The lower AP efficiency
(especially at low MOIs) could be explained by a low selection pressure for phage resistance.
When comparing the specific isolation percentages, one must take into consideration the
differences in how “true resistance” was defined as well as the differences of the bacterial WT
strains used. In our study, the resistant mutants were quantitatively defined (increased AUC
relative to the WT strain in the presence of phage) whereas previous studies used a qualitative
approach (visual comparison of turbidity between phage-host co-cultures and control culture)
to define true resistance. As opposed to the qualitative approach, defining true resistant mutants
based on AUC provide high-throughput assessment based on fixed cut-off values, which can
easily be compared, and do not depend on experience and/or subjectivity of the observer.
However, one must be aware of the potential pitf alls related to the AUC as selection criterion.
If the AUC increase percentage cut-off value is too high, true resistant mutants may wrongfully
be excluded. If the cut-off value is too low, this approach could select both resistant mutants
and non-resistant strains.
Bacteria have been shown to evolve resistance to phage infection through mechanisms of
adsorption inhibition, including loss or modification of phage receptors [6, 7, 85]. There is a
great diversity reported in coliphage receptors, which include bacterial oute r membrane
proteins (OMPs), porins, capsule and LPS [56, 72, 86]. OMPs participate in outer membrane
functionality, including diffusion and transport mechanisms, cell shape as well as virulence
[87]. Also, the OmpA protein is a key virulence factor of pathogenic E. coli playing a role in

Chapter 3: Experimental Studies
161
conjugation, adhesion, immune system evasion, resistance to environmental stress [57].
Therefore, mutation in such gene, while conferring resistance, may decrease bacterial adhesion
and immune system evasion, and hereby, the overall strain virulence in vivo/in situ. In addition,
phage resistance may also have a fitness cost [11]. In this study, we observed up to 65%
decrease in in vitro fitness (bacterial growth) in mutant strains that had acquired resistance
through genetic mutations and/or gene loss. However, such fitness cost may vary in in vivo/in
situ environments, as the magnitude has been shown to depend on the genetic basis of the
resistance as well as on the environmental context [88].
Recently, Maffei et al. (2021) investigated the coliphage-host interaction and identified phage
receptors. In accordance with previous findings, Myoviridae coliphages belonging to the
Tequatrovirus genus were found to use the OMP, Tsx (T6-like phages), FadL (T2-like phages),
OmpA, OmpC (T4-like phages), or OmpF as primary receptor. A recent study similarly
identified the OmpA protein as a Myoviridae coliphage receptor and reported that all phage-
resistant strains had acquired mutations in just two pathways, the LPS biosynthesis and the
OmpA expression [90]. LPS are known to play an essential role in the OMP folding and
placement in the cell wall [91]. Accordingly, loss or changes in the structure of LPS could
prevent OmpA from being properly positioned in the outer membrane, and thereby, making
the phage receptor unavailable. In our study, we detected SNPs in the ompA gene, encoding
the OmpA protein, suggesting this could act as receptor for phage vB_EcoM-P10. However,
further studies are needed to confirm if OmpA is the primary receptor as well as determine the
indirect effects on infection due to LPS changes.
While for some phages the absence of the primary receptor results in complete absence of
infection, other phages, including those utilizing several receptors, are still able to infect [89,
92, 93]. The specificity for the second receptor depends on the short tail fibers of which two
variants have been described to date [89]. The first variant (encoded by phages such as T2, T4,
and T6) targets the lipid A Kdo region deep in the LPS core, and a second variant targets the
upper part(s) of the LPS core, which requires an intact inner LPS core for infectivity. The
Myoviridae phage used in this study clusters with the latter group [25]. We found genetic
changes in the gene encoding glycosyltransferase required for the assembly of the LPS as well
as in the genes encoding LPS inner core heptose (II) kinase (waaY) and heptosyltransferase I
(waaC). Accordingly, as both waaY and waaC are essential for the LPS inner core, the
nonsense mutations detected in these genes will most likely have an effect on the infectivity of
an infecting phage. Either a direct effect as shown for phages utilising the LPS as a receptor

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
162
[94] or an indirect effect where waa mutation(s) interfere with the recognition of outer
membrane protein phage receptors [95]. At the same time, mutants with truncated LPS at the
inner core have been shown to have attenuated in vivo virulence and to be more sensitive to
antimicrobials [86, 90].
The O-antigen biosynthesis operon has been shown to play a major role in E. coli phage
resistance against Myoviridae phage T4 [48] and Demerecviridae (previous Siphoviridae)
phage T5 [56]. In accordance with these previous observations, we found genetic changes
(missense mutation, partial or complete gene loss) in four O-antigen operon genes encoding a
glycosyltransferase, the O-antigen polymerase (wzy), a chain length determinant protein (wzzB
gene) [65], and the O antigen flippase (wzx gene / rbfX gene), all of which could potentially
confer phage resistance. These findings could support the LPS as a potential binding site for
our phage.
In both Gram-negative and Gram-positive bacteria, the RNase adaptor protein RapZ plays a
central role in regulatory pathway of glucosamine-6-phosphate (GlcN6P), an early and
essential precursor in the synthesis of the bacterial cell envelope components, including
peptidoglycan, LPS and colanic acid [96]. Recent studies have demonstrated that phage
resistance in E. coli and Staphylococcus aureus can be acquired through mutation(s) in the
rapZ gene, encoding RapZ [71, 72, 97]. Zhou et al. (2021) reported that mutation in the rapZ
gene conferred E. coli phage resistance by inhibiting 93.5% phage adsorption. In this study, we
similarly detected a missense mutation in the rapZ gene supporting its involvement in phage
resistance against lytic Myoviridae coliphages. Moreover, in according with finding of Zhou et
al. (2021), no in vitro fitness cost (measured by bacterial growth) was associated with the
acquired resistance.
The polysaccharide capsule of pathogen E. coli K1 is an essential virulence factor and consist
of polymers of sialic acid (NeuNAc). The kps gene cluster encodes six proteins, NeuDBACES,
required for synthesis, activation, and polymerisation of NeuNAc [54, 98, 99]. In this study,
we detected partial gene loss of neuD (involved in the synthesis of sialic acid) [54], neuA
(synthase involved in activation the sugar prior to polymerisation) [54], and neuE (involved in
synthesis and export of NeuAc) [44]. The capsule is recognised as a receptor by some phages,
such as K-specific coliphages and the Myoviridae coliphage phi92, which have virion-
associated polysaccharide-degrading enzymes [100, 101]. Contrary, Scholl and colleagues
(2005) showed that the expression of the E. coli K1 capsule physically blocks infection by

Chapter 3: Experimental Studies
163
phage T7, a phage that recognise LPS core as the primary receptor. Whether or not our
Myoviridae phage can utilise the capsule as receptor needs to be investigated further.
Nevertheless, as polysaccharide capsule is a key virulence factor, the interesting finding that
~23% of the phage resistant isolates have lost part of one of the neu genes could add to the
phage therapy potential of the infecting phage. Being as the infection could result in reduced
virulence as well as reduced competitiveness. Accordingly, (partial) loss of neuE may be
associated with great fitness cost as up to ~65% growth decrease was observed for the phage-
resistant mutant strains. However, in all affected strains two or more other genetic changes
were detected, strongly implying that further studies are needed to determine the exact effect
of neuE loss alone and in combination with the other affected genes.
Even though we were able to connect some of the genetic changes in the mutant strains to
known phage resistance mechanisms, most SNPs (n = 23) and gene losses (partial or complete)
(n = 11) were found in a gene not previously linked to phage resistance. Among others, these
genes encode acetate kinase (essential for bacterial growth) [46], Acyl-CoA dehydrogenase
(involved in the beta-oxidation cycle of fatty acid degradation) [50], the MarR family
transcriptional regulator (involved in numerous cellular processes, including stress responses,
virulence, and efflux of harmful chemicals and antimicrobials) [47], pyruvate kinases (essential
for the regulation of the glycolytic pathway) [59], a tetratricopeptide repeat (TPR) protein
(involved in various biological processes and mediates protein-protein interactions) [45],
uridylyltransferase (involved in nitrogen regulation) [53] as well as several hypothetical
proteins. We found loss of the gene or mutation in an acetate kinase, pyruvate kinase, TPR
protein and uridylyltransferase as the sole genetic change indicating that the phage -host
interaction might be more complex that previous thought. Interestingly, partial or complete loss
of one of two genes (group_67 and group_237) encoding hypothetical proteins was detected
in a great number of phage-resistant mutant strains, and as sole resistance mechanisms in some.
Loss of group_67 gene as sole resistance mechanism resulted in an average fitness cost (growth
reduction) of only 6.3%. Similarly, loss of the neuE gene as sole resistance mechanism resulted
in an average fitness cost of only ~3.9%. However, the greatest fitness cost was observed for
the mutant strain that had lost both the group_67 and neuE (65.2%) or both genes in
combination with a point mutation in the phnD gene (57.0%), indicating that a combination
loss of group_67 and neuE might have an additive effect on the fitness cost. The point mutation
in phnD was only observed in one mutant and only in combination with group_67 and neuE
gene loss. Only one mutant had lost the group_237 gene as sole resistance mechanisms and

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
164
suffered a great fitness cost of 59.1%. Moreover, an average fitness cost of 23.2% was observed
for the 10 mutant strains with group_237 gene loss, suggesting that while mutation in this gene
might confer phage resistance, the resistance comes with a cost for the host bacterium.
Furthermore, one mutant had lost genes encoding both group_237 and group_67 and suffered
a fitness cost (34.6%), supporting the essential role of group_237 and the potential additive
effect of group_67 gene loss. However, as additional genetic changes (potentially related to
phage resistance) were detected in most of both the group_67 and group_237 mutant strains.
We tried to decipher the potential function of the hypothetical proteins, using PANDA [103]
and LocTree3 [104], however we could not find any motifs that could give an indication (data
not shown). Also, the role of these proteins in E. coli phage resistance needs to be further
investigated.
A nonsense mutation was detected in the gene encoding the YbjT protein. This protein has
been shown to be physically tethered to the inner membrane of E. coli and part of the metabolic
pathway involved in the biogenesis of the bacterial cell envelope [105]. However, as this
genetic change was not the only one detected in the affected strain, its potential involvement
in phage resistance remains to be investigated. Finally, six different synonymous SNPs were
identified in this study. Although unlikely, these mutations may still play a role in phage
resistance as synonymous mutations can affect cellular processes such as translation efficiency
or mRNA structures, depending on the gene affected [106].
CRISPR-Cas systems are found among ~36% of bacteria and confer a sequence specific
adaptive immunity against invading foreign DNA, including phages [107]. Previous studies
have reported varying findings when it comes to phage resistance conferred by acquired
CRISPR spacer(s). As opposed to findings of Denes et al. (2015) where no CRISPR immunity
was observed in any of the spontaneous phage-resistant Listeria mutant strains, in most of the
phage-resistant Streptococcus mutant strains one or two CRISPR spacer(s) were acquired
[109]. In this study, we found three phage-resistant strains with a newly acquired CRISPR
spacer sequence that matched the invading phage genome. This spacer was found in a short
CRISPR array, only consisting of this one spacer (evidence level 1), which makes it difficult
to determine if this array is a false CRISPR-like element or a true CRISPR. However, the lack
of similar repeats in larger CRISPR arrays, associated cas genes and leader sequence upstream
of the CRISPR array, are indications that the detected CRISPR spacer in the three phage-
resistant mutant strains most likely is a false positive [40]. Moreover, two out of the three
mutant strains had acquired one or three genetic changes in addition to the CRISPR-like spacer

Chapter 3: Experimental Studies
165
acquisition, including partial loss of the group_237 gene or partial loss of neuE, complete loss
of the group_67 gene and a silent point mutation in a hypothetical protein. As discussed earlier,
the partial and/or complete gene loss(es) are more likely to explain the resistance observed.
Phages can evolve to counteract bacterial antiviral mechanisms, such as inhibition of phage
adsorption, R-M systems, CRISPR-Cas systems and phage escape strategies [9, 110]. Such
adaptation can be conferred by point mutations in specific genes, such as receptor binding
proteins (RBPs) and/or tail fibers, genome rearrangement, and genetic exchange with other
viral or bacterial genomes to new traits [110]. Phage genes involved in host recognition are
among the fastest evolving phage genes due to the selection pressures conferred by the phage-
bacterium co-evolution [95, 110]. Meyer et al. (2012) showed that a lytic coliphage was able
to evolve as such that it could use an alternative receptor after eight days of co-culture with a
resistant bacterial host. Similarly, Wandro et al. (2019) showed that after eight days of co-
culture the lytic Enterococcus Phage EfV12-phi1was able to combat phage-resistance through
adaptation of the tail fiber. Hall et al. (2011) were able to detect adaptation in Pseudomonas
phage SBW25Φ2 tail fiber protein and structural protein after only two and four days of co -
culture, respectively. As opposed to these findings, in this study we did not detect any genetic
changes in phages co-cultured with phage-resistant strains. However, this is most likely a
reflection of a too short co-culture incubation period (<24 hours) rather than the ability of the
phage to co-evolve to bypass the phage resistance.
Understanding the phage-host interactions provides insight into the phage-host interaction and
dynamics and may lead to new strategies for the development and application of successful
phage therapy [114, 115]. Furthermore, the understanding of the interactions makes it possible
adapt to phage selection towards the desired outcome [116]. This includes selecting optimal
phage(s) that can overcome host phage-resistance mechanisms, select for attenuated virulence,
for impaired fitness/growth, and/or select for increased susceptibility to antimicrobials. Further
studies comparing how different phages select for resistant bacteria may also lead to better
understanding on how bacteria react on phage infection. Although the full complexity of the
interactions cannot be captured, in vitro experiments can still provide essential information
needed for further application in a therapeutic setting (in vivo/in situ) [117].
For 44 phage-resistant strains no detected genomic changes differentiated them from the WT
strain. This could be caused by both laboratory issues, such as non-resistant strains were
erroneously defined as true resistant mutants based on AUC values, or actual variations that

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
166
were missed due to genetic variation within discarded repetitive regions identified by NUCmer
or partly loss off genes of which the consequence on the overall gene function were not
investigated. Our experiments were conducted in vitro and thus caution should be used when
interpreting our findings for in vivo applications. The co-evolutionary interactions, including
phage resistance, observed in laboratory experiments can differ from the highly complex
interactions found in natural environments, which may influence the ecology and evolution of
both phages and their hosts [118].
In conclusion, under selective pressure of virulent phages, bacterial strains of E. coli can
acquire one or more spontaneous mutations or gene losses that confer phage resistance in vitro.
The majority of detected phage-resistant mutant strains from this study were shown to resist
phage infection through mechanisms related to phage adsorption inhibition. Interestingly, we
also found several new genes, including two encoding hypothetical proteins, that could
potentially play a role in E. coli phage resistance. There were no indications that the infecting
phages were able to overcome the phage resistance. Nevertheless, as the initial infection
targeted known E. coli virulence factors, such as OMPs and the LPS, and thus, potentially
decreased the APEC virulence, the infecting phage still possessed desirable traits for phage
therapy application. Furthermore, in many cases phage resistance was associated with fitness
cost for the affected mutant strain resulting in up to ~65% decrease in growth. Thus, this study
provides valuable information about the interactions between virulent coliphages and their
host, which may aid prediction of the phage-host interaction outcome and future development
of a successful phage therapy.

Chapter 3: Experimental Studies
167
References
[1] Güemes, A. G. C.; Merry Youle; Cantú, V. A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners
in the Game of Life. Annu. Rev. Virol., 2016, 3 (1), 197–214. https://doi.org/10.1146/annurev-
virology-100114-054952.
[2] Clokie, M. R. J.; Millard, A. D.; Letarov, A. V; Heaphy, S. Phages in Nature. Bacteriophage, 2011,
1 (1), 31–45. https://doi.org/10.4161/bact.1.1.14942.
[3] Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages.
Front. Microbiol., 2016, 7, 1352. https://doi.org/10.3389/fmicb.2016.01352.
[4] Buckling, A.; Rainey, P. B. Antagonistic Coevolution between a Bacterium and a Bact eriophage.
Proc. R. Soc. B Biol. Sci., 2002, 269 (1494), 931–936. https://doi.org/10.1098/rspb.2001.1945.
[5] Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D. a;
Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science (80-. ).,
2007, 315 (5819), 1709–1712. https://doi.org/10.1126/science.1138140.
[6] Labrie, S. J.; Samson, J. E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev.
Microbiol., 2010, 8 (5), 317–327. https://doi.org/10.1038/nrmicro2315.
[7] Bohannan, B. J. M.; Lenski, R. D. Linking Genetic Change to Community Evolution: Insights from
Studies of Bacteria and Bacteriophage. Ecol. Lett., 2000, 3 (4), 362–377.
https://doi.org/https://doi.org/10.1046/j.1461-0248.2000.00161.x.
[8] Weissman, J. L.; Holmes, R.; Barrangou, R.; Moineau, S.; Fagan, W. F.; Levin, B.; Johnson, P. L.
F. Immune Loss as a Driver of Coexistence during Host-Phage Coevolution. ISME J., 2018, 12 (2),
585–597. https://doi.org/10.1038/ismej.2017.194.
[9] Koskella, B.; Brockhurst, M. A. Bacteria –Phage Coevolution as a Driver of Ecological and
Evolutionary Processes in Microbial Communities. FEMS Microbiol. Rev., 2014, 38 (5), 916–931.
https://doi.org/10.1111/1574-6976.12072.
[10] Azam, A. H.; Tanji, Y. Bacteriophage-Host Arm Race: An Update on the Mechanism of Phage
Resistance in Bacteria and Revenge of the Phage with the Perspective for Phage Therapy. Appl.
Microbiol. Biotechnol., 2019, 103, 2121–2131. https://doi.org/10.1007/s00253-019-09629-x.
[11] Burmeister, A. R.; Turner, P. E. Trading-off and Trading-up in the World of Bacteria –Phage
Evolution. Curr. Biol., 2020, 30 (19), R1120–R1124. https://doi.org/10.1016/j.cub.2020.07.036.
[12] Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to
Cellular Defense. Microbiol. Mol. Biol. Rev., 2013, 77 (1), 53–72.
https://doi.org/10.1128/mmbr.00044-12.
[13] Vale, P. F.; Lafforgue, G.; Gatchitch, F.; Gardan, R.; Moineau, S.; Gandon, S. Costs of CRISPR-
Cas-Mediated Resistance in Streptococcus Thermophilus. Proc. R. Soc. B Biol. Sci., 2015, 282
(1812), 20151270. https://doi.org/10.1098/rspb.2015.1270.
[14] Bradde, S.; Mora, T.; Walczak, A. M. Cost and Benefits of Clustered Regularly Interspaced Short
Palindromic Repeats Spacer Acquisition. Philos. Trans. R. Soc. B Biol. Sci., 2019, 374 (1772).
https://doi.org/10.1098/rstb.2018.0095.
[15] Zhao, S.; Maurer, J. J.; Hubert, S.; De Villena, J. F.; McDermott, P. F.; Meng, J.; Ayers, S.; English,
L.; White, D. G. Antimicrobial Susceptibility and Molecular Characterization of Avian Pathogenic
Escherichia Coli Isolates. Vet. Microbiol., 2005, 107 (3–4), 215–224.
https://doi.org/10.1016/j.vetmic.2005.01.021.
[16] Dho-Moulin, M.; Fairbrother, J. M. Avian Pathogenic Escherichia Coli (APEC). Vet. Res., 1999, 30
(2–3), 299–316.
[17] Lutful Kabir, S. M. Avian Colibacillosis and Salmonellosis: A Closer Look at Epidemiology,

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
168
Pathogenesis, Diagnosis, Control and Public Health Concerns. Int. J. Environ. Res. Public Health,
2010, 7 (1), 89–114. https://doi.org/10.3390/ijerph7010089.
[18] Nolan, L. K.; Barnes, H. J.; Vaillancourt, J.; Abdul-aziz, T.; Logue, C. M. Colibacillosis. In Diseases
of Poultry; Swayne, D. E., Ed.; John Wiley & Sons, Inc., 2013; pp 751–805.
https://doi.org/10.1002/9781119421481.
[19] Kathayat, D.; Helmy, Y. A.; Debla is, L.; Rajashekara, G. Novel Small Molecules Affecting Cell
Membrane as Potential Therapeutics for Avian Pathogenic Escherichia Coli. Sci. Rep., 2018, 8 (1),
1–16. https://doi.org/10.1038/s41598-018-33587-5.
[20] Centers for Disease Control and Prevention (CDC). Antibiotic Resistance Threats in the United
States; 2013.
[21] World Health Organization (WHO). Antimicrobial Resistance: Global Report on Surveillance;
Geneva, Switzerland, 2014. https://doi.org/10.1007/s13312-014-0374-3.
[22] Lin, D. M.; Koskella , B.; Lin, H. C. Phage Therapy: An Alternative to Antibiotics in the Age of
Multi-Drug Resistance. World J. Gastrointest. Pharmacol. Ther., 2017, 8 (3), 162–173.
https://doi.org/10.4292/wjgpt.v8.i3.162.
[23] Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy.
Viruses, 2018, 10 (7). https://doi.org/10.3390/v10070351.
[24] Colson, C.; Glover, S. W.; Symonds, N.; Stacey, K. A. The Location of the Genes for Host -
Controlled Modification and Restriction in Escherichia Coli K-12. Genetics, 1965, 52 (5), 1043–
1050.
[25] Sørensen, P. E.; Van Den Broeck, W.; Kiil, K.; Jasinskyte, D.; Moodley, A.; Garmyn, A.; Ingmer,
H.; Butaye, P. New Insights into the Biodiversity of Coliphages in the Intestine of Poultry. Sci. Rep.,
2020, 10, 15220. https://doi.org/10.1038/s41598-020-72177-2.
[26] Reinheimer, J. A.; Morelli, L.; Bottazzi, V.; Suárez, V. Phenotypic Variability among Cells of
Lactobacillus Helveticus ATCC 15807. Int. Dairy J., 1995, 5, 97–103.
[27] Carminati, D.; Zennaro, R.; Neviani, E.; Giraffa, G. Selezione e Caratteristiche Di Mutanti Fago-
Resistenti Di Lactobacillus Helveticus. Sci. e Tec. Latt., 1993, 44 (1), 33–48.
[28] Capra, M. L.; Mercanti, D. J.; Rossetti, L. C.; Reinheimer, J. A.; Quiberoni, A. Isolation and
Phenotypic Characterization of Lactobacillus Casei and Lactobacillus Paracasei Bacteriophage-
Resistant Mutants. J. Appl. Microbiol., 2011, 111 (2), 371–381. https://doi.org/10.1111/j.1365-
2672.2011.05056.x.
[29] Kropinski, A. M.; Mazzocco, A.; Waddell, T. E.; Lingohr, E.; Johnson, R. P. Enumeration of
Bacteriophages by Double Agar Overlay Plaque Assay. In Bacteriophages. Methods in Molecular
Biology; Clokie, M. R., Kropinski, A. M., Eds.; Humana Press, 2009; Vol. 501, pp 69 –76.
https://doi.org/10.1007/978-1-60327-164-6_7.
[30] Xie, Y.; Wahab, L.; Gill, J. J. Development and Validation of a Microtiter Plate-Based Assay for
Determination of Bacteriophage Host Range and Virulence. Viruses, 2018, 10 (4).
https://doi.org/10.3390/v10040189.
[31] Storms, Z. J.; Teel, M. R.; Mercurio, K.; Sauvageau, D. The Virulence Index: A Metric for
Quantitative Analysis of Phage Virulence. Phage, 2020, 1 (1), 27–36.
https://doi.org/10.1089/phage.2019.0001.
[32] Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A. A.; Dvorkin, M.; Kulikov, A. S.; Lesin, V. M.;
Nikolenko, S. I.; Pham, S.; Prjibelski, A. D.; et al. SPAdes: A New Genome Assembly Algorithm
and Its Applications to Single-Cell Sequencing. J. Comput. Biol., 2012, 19 (5), 455–477.
https://doi.org/10.1089/cmb.2012.0021.
[33] Joensen, K. G.; Tetzschner, A. M. M.; Iguchi, A.; Aarestrup, F. M.; Scheutz, F. Rapid and Easy in
Silico Serotyping of Escherichia Coli Isolates by Use of Whole-Genome Sequencing Data. J. Clin.

Chapter 3: Experimental Studies
169
Microbiol., 2015, 53 (8), 2410–2426. https://doi.org/10.1128/JCM.00008-15.
[34] Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics, 2014, 30 (14), 2068–
2069. https://doi.org/10.1093/bioinformatics/btu153.
[35] Page, A. J.; Cummins, C. A.; Hunt, M.; Wong, V. K.; Reuter, S.; Holden, M. T. G.; Fookes, M.;
Falush, D.; Keane, J. A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis.
Bioinformatics, 2015, 31 (22), 3691–3693. https://doi.org/10.1093/bioinformatics/btv421.
[36] Bradley, P.; Gordon, N. C.; Walker, T. M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.
J.; Anson, L.; De Cesare, M.; et al. Rapid Antibiotic-Resistance Predictions from Genome Sequence
Data for Staphylococcus Aureus and Mycobacterium Tuberculosis. Nat. Commun., 2015, 6, 1–14.
https://doi.org/10.1038/ncomms10063.
[37] Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.
M.; Larsen, M. V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob.
Chemother., 2012, 67 (11), 2640–2644. https://doi.org/10.1093/jac/dks261.
[38] Feldgarden, M.; Brover, V.; Haft, D. H.; Prasad, A. B.; Slotta, D. J.; Tolstoy, I.; Tyson, G. H.; Zhao,
S.; Hsu, C. H.; McDermott, P. F.; et al. Validating the AMRFINder Tool and Resistan ce Gene
Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of
Isolates. Antimicrob. Agents Chemother., 2019, 63 (11), 1–19. https://doi.org/10.1128/AAC.00483-
19.
[39] Alcock, B. P.; Raphenya, A. R.; Lau, T. T. Y.; Tsang, K. K.; Bouchard, M.; Edalatmand, A.; Huynh,
W.; Nguyen, A.-L. V; Cheng, A. A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance
with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res., 2019, 48 (D1), D517–
D525. https://doi.org/10.1093/nar/gkz935.
[40] Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E. P.
C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder,
Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic
Acids Res., 2018, 46 (W1), W246–W251. https://doi.org/10.1093/nar/gky425.
[41] Sahl, J. W.; Lemmer, D.; Travis, J.; Schupp, J. M.; Gillece, J. D.; Aziz, M.; Driebe, E. M.; Drees, K.
P.; Hicks, N. D.; Williamson, C. H. D.; et al. NASP: An Accurate, Rapid Method for the
Identification of SNPs in WGS Datasets That Supports Flexible Input and Output Formats. Microb.
genomics, 2016, 2 (8), e000074. https://doi.org/10.1099/mgen.0.000074.
[42] Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform.
Bioinformatics, 2009, 25 (14), 1754–1760. https://doi.org/10.1093/bioinformatics/btp324.
[43] Marçais, G.; Delcher, A. L.; Phillippy, A. M.; Coston, R.; Salzberg, S. L.; Zimin, A. MUMmer4: A
Fast and Versatile Genome Alignment System. PLoS Comput. Biol., 2018, 14 (1), e1005944.
https://doi.org/10.1371/journal.pcbi.1005944.
[44] Steenbergen, S. M.; Vimr, E. R. Biosynthesis of the Escherichia Coli K1 Group 2 Polysialic Acid
Capsule Occurs within a Protected Cytoplasmic Compartment. Mol. Microbiol., 2008, 68 (5), 1252–
1267. https://doi.org/10.1111/j.1365-2958.2008.06231.x.
[45] Cerveny, L.; Straskova, A.; Dankova, V.; Hartlova, A.; Ceckova, M.; Staud, F.; Stulik, J.
Tetratricopeptide Repeat Motifs in the World of Bacterial Pathogens: Role in Virulence Mechanisms.
Infect. Immun., 2013, 81 (3), 629–635. https://doi.org/10.1128/IAI.01035-12.
[46] Schütze, A.; Benndorf, D.; Püttker, S.; Kohrs, F.; Bettenbrock, K. The Impact of AckA, Pta, and
AckA-Pta Mutations on Growth, Gene Expression and Protein Acetylation in Escherichia Coli K-
12. Front. Microbiol., 2020, 11 (February), 1–13. https://doi.org/10.3389/fmicb.2020.00233.
[47] Deochand, D. K.; Grove, A. MarR Family Transcription Factors: Dynamic Variations on a Common
Scaffold. Crit. Rev. Biochem. Mol. Biol., 2017, 52 (6), 595–613.
https://doi.org/10.1080/10409238.2017.1344612.

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
170
[48] Cowley, L. A.; Low, A. S.; Pickard, D.; Boinett, C. J.; Dallman, T. J.; Day, M.; Perry, N.; Gally, D.
L.; Parkhill, J.; Jenkins, C.; et al. Transposon Insertion Sequencing Elucidates Novel Gene
Involvement in Susceptibility and Resistance to Phages T4 and T7 in Escherichia Coli O157. MBio,
2018, 9 (4), e00705-18. https://doi.org/10.1128/mBio.00705-18.
[49] Nagel, G. M.; Cumberledge, S.; Johnson, M. S.; Petrella, E.; Weber, B. H. The β Subunit of E. Coil
Glycyl-TRNA Synthetase Plays a Major Role in TRNA Recognition. Nucleic Acids Res., 1984, 12
(10), 4377–4384. https://doi.org/10.1093/nar/12.10.4377.
[50] Campbell, J. W.; Cronan, J. E. The Enigmatic Escherichia Coli FadE Gene Is YafH. J. Bacteriol.,
2002, 184 (13), 3759–3764. https://doi.org/10.1128/JB.184.13.3759-3764.2002.
[51] Peracchi, A.; Veiga-Da-Cunha, M.; Kuhara, T.; Ellens, K. W.; Paczia, N.; Stroobant, V.; Seliga, A.
K.; Marlaire, S.; Jaisson, S.; Bommer, G. T.; et al. Nit1 Is a Metabolite Repair Enzyme That
Hydrolyzes Deaminated Glutathione. Proc. Natl. Acad. Sci. U. S. A., 2017, 114 (16), E3233–E3242.
https://doi.org/10.1073/pnas.1613736114.
[52] Deckers, D.; Vanlint, D.; Callewaert, L.; Aertsen, A.; Michiels, C. W. Role of the Lysozyme Inhibitor
Ivy in Growth or Survival of Escherichia Coli and Pseudomonas Aeruginosa Ba cteria in Hen Egg
White and in Human Saliva and Breast Milk. Appl. Environ. Microbiol., 2008, 74 (14), 4434–4439.
https://doi.org/10.1128/AEM.00589-08.
[53] Zhang, Y.; Pohlmann, E. L.; Serate, J.; Conrad, M. C.; Roberts, G. P. Mutagenesis and Functional
Characterization of the Four Domains of GlnD, a Bifunctional Nitrogen Sensor Protein. J. Bacteriol.,
2010, 192 (11), 2711–2721. https://doi.org/10.1128/JB.01674-09.
[54] Daines, D. A.; Wright, L. F.; Chaffin, D. O.; Rubens, C. E.; Silver, R. P. NeuD Plays a Role in the
Synthesis of Sialic Acid in Escherichia Coli K1. FEMS Microbiol. Lett., 2000, 189 (2), 281–284.
https://doi.org/10.1111/j.1574-6968.2000.tb09244.x.
[55] Wright, R. C. T.; Friman, V. P.; Smith, M. C. M.; Brockhurst, M. A. Resistance Evolution against
Phage Combinations Depends on the Timing and Order of Exposure. MBio, 2019, 10 (5), e01652-
19. https://doi.org/10.1128/mBio.01652-19.
[56] Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host Receptors for Bacteriophage Adsorption. FEMS
Microbiol. Lett., 2016, 363 (4), 1–11. https://doi.org/10.1093/femsle/fnw002.
[57] Smith, S. G. J.; Mahon, V.; Lambert, M. A.; Fagan, R. P. A Molecular Swiss Army Knife: OmpA
Structure, Function and Expression. FEMS Microbiol. Lett., 2007, 273 (1), 1–11.
https://doi.org/10.1111/j.1574-6968.2007.00778.x.
[58] Korch, S. B.; Henderson, T. A.; Hill, T. M. Characterization of the HipA7 Allele of Escherichia Coli
and Evidence That High Persistence Is Governed by (p)PpGpp Synthesis. Mol. Microbiol., 2003, 50
(4), 1199–1213. https://doi.org/10.1046/j.1365-2958.2003.03779.x.
[59] Valentini, G.; Chiarelli, L.; Fortin, R.; Speranza, M. L.; Galizzi, A.; Mattevi, A. The Allosteric
Regulation of Pyruvate Kinase. J. Biol. Chem., 2000, 275 (24), 18145–18152.
https://doi.org/10.1074/jbc.M001870200.
[60] Wang, Z.; Wang, J.; Ren, G.; Li, Y.; Wang, X. Influence of Core Oligosaccharide of
Lipopolysaccharide to Outer Membrane Behavior of Escherichia Coli. Mar. Drugs, 2015, 13 (6),
3325–3339. https://doi.org/10.3390/md13063325.
[61] Pedersen, K.; Gerdes, K. Multiple Hok Genes on the Chromosome of Escherichia Coli. Mol.
Microbiol., 1999, 32 (5), 1090–1102. https://doi.org/10.1046/j.1365-2958.1999.01431.x.
[62] Frederix, M.; Hütter, K.; Leu, J.; Batth, T. S.; Turner, W. J.; Rüegg, T. L.; Blanch, H. W.; Simmons,
B. A.; Adams, P. D.; Keasling, J. D.; et al. Development of a Native Escherichia Coli Induction
Dystem for Ionic Liquid Tolerance. PLoS One, 2014, 9 (7), e101115.
https://doi.org/10.1371/journal.pone.0101115.
[63] Stach, N.; Kalinska, M.; Zdzalik, M.; Kitel, R.; Karim, A.; Serwin, K.; Rut, W.; Larsen, K.; Jabaiah,

Chapter 3: Experimental Studies
171
A.; Firlej, M.; et al. Unique Substrate Specificity of SplE Serine Protease from Staphylococcus
Aureus. Structure, 2018, 26 (4), 572-579.e4. https://doi.org/10.1016/j.str.2018.02.008.
[64] Grauschopf, U.; Fritz, A.; Glockshuber, R. Mechanism of the Electron Transfer Catalyst DsbB from
Escherichia Coli. EMBO J., 2003, 22 (14), 3503–3513. https://doi.org/10.1093/emboj/cdg356.
[65] Stenberg, F.; Chovanec, P.; Maslen, S. L.; Robinson, C. V.; Ilag, L. L.; von Heijne, G.; Daley, D. O.
Protein Complexes of the Escherichia Coli Cell Envelope. J. Biol. Chem., 2005, 280 (41), 34409–
34419. https://doi.org/10.1074/jbc.M506479200.
[66] Deniziak, M. A.; Barciszewski, J. Methionyl-TRNA Synthetase. Acta Biochim. Pol., 2001, 48 (2),
337–350. https://doi.org/10.18388/abp.2001_3919.
[67] Shepherd, J.; Ibba, M. Bacterial Transfer RNAs. FEMS Microbiol. Rev., 2015, 39 (3), 280–300.
https://doi.org/10.1093/femsre/fuv004.
[68] Noriega, C. E.; Lin, H.-Y.; Chen, L.-L.; Williams, S. B.; Stewart, V. Asymmetric Cross-Regulation
between the Nitrate-Responsive NarX-NarL and NarQ-NarP Two-Component Regulatory Systems
from Escherichia Coli K-12. Mol. Microbiol., 2010, 75 (2), 394–412. https://doi.org/10.1111/j.1365-
2958.2009.06987.x.
[69] Breton, R.; Sanfacon, H.; Papayannopoulos, I.; Biemann, K.; Lapointe, J. Glutamyl-TRNA
Synthetase of Escherichia Coli. Isolation and Primary Structure of the GltX Gene and Homology
with Other Aminoacyl-TRNA Synthetases. J. Biol. Chem., 1986, 261 (23), 10610–10617.
https://doi.org/10.1016/s0021-9258(18)67429-0.
[70] Navarro-Garcia, F.; Ruiz-Perez, F.; Cataldi, Á.; Larzábal, M. Type VI Secretion System in
Pathogenic Escherichia Coli: Structure, Role in Virulence, and Acquisition. Front. Microbiol., 2019,
10, 1965. https://doi.org/10.3389/fmicb.2019.01965.
[71] Zhou, Y.; Bao, H.; Zhang, H.; Pang, M.; Zhu, S.; Wang, R. A Spontaneous RapZ Mutant Impairs
Infectivity of Lytic Bacteriophage VB_EcoM_JS09 against En terotoxigenic Escherichia Coli.
mSphere, 2021, 6, e01286-20. https://doi.org/10.1128/mSphere.01286-20.
[72] Mutalik, V. K.; Adler, B. A.; Rishi, H. S.; Piya, D.; Zhong, C.; Koskella, B.; Kutter, E. M.; Calendar,
R.; Novichkov, P. S.; Price, M. N.; et al. High-Throughput Mapping of the Phage Resistance
Landscape in E. Coli. PLOS Biol., 2020, 18 (10), e3000877.
https://doi.org/10.1371/journal.pbio.3000877.
[73] Khan, M. A.; Durica‐Mitic, S.; Göpel, Y.; Heermann, R.; Görke, B. Small RNA‐binding Protein
RapZ Mediates Cell Envelope Precursor Sensing and Signaling in Escherichia Coli. EMBO J., 2020,
39, e103848. https://doi.org/10.15252/embj.2019103848.
[74] Keseler, I. M.; Mackie, A.; Santos-Zavaleta, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.;
Fulcher, C.; Gama-Castro, S.; Kothari, A.; Krummenacker, M.; et al. The EcoCyc Database:
Reflecting New Knowledge about Escherichia Coli K-12. Nucleic Acids Res., 2017, 45 (D1), D543–
D550. https://doi.org/10.1093/nar/gkw1003.
[75] Di Luccio, E.; Petschacher, B.; Voegtli, J.; Chou, H.; Stahlberg, H.; Nidetzky, B.; Wilson, D. K.
Structural and Kinetic Studies of Induced Fit in Xylulose Kinase from Escherichia Coli. J. Mol. Biol.,
2007, 365 (3), 783–798. https://doi.org/10.1016/j.jmb.2006.10.068.
[76] Seo, S. W.; Kim, D.; Latif, H.; O’Brien, E. J.; Szubin, R.; Palsson, B. O. Deciphering Fur
Transcriptional Regulatory Network Highlights Its Complex Role beyond Iron Metabolism in
Escherichia Coli. Nat. Commun., 2014, 5, 4910. https://doi.org/10.1038/ncomms5910.
[77] Terakawa, A.; Natsume, A.; Okada, A.; Nishihata, S.; Kuse, J.; Tanaka, K.; Takenaka, S.; Ishikawa,
S.; Yoshida, K. Bacillus Subtilis 5′-Nucleotidases with Various Functions and Substrate
Specificities. BMC Microbiol., 2016, 16, 249. https://doi.org/10.1186/s12866-016-0866-5.
[78] Alicea, I.; Marvin, J. S.; Miklos, A. E.; Ellington, A. D.; Looger, L. L.; Schreiter, E. R. Structure of
the Escherichia Coli Phosphonate Binding Protein PhnD and Rationally Optimized Phosphonate

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
172
Biosensors. J. Mol. Biol., 2011, 414 (3), 356–369. https://doi.org/10.1016/j.jmb.2011.09.047.
[79] Al Mamun, A. A. M.; Lombardo, M.-J.; Shee, C.; Lisewski, A. M.; Gonzalez, C.; Lin, D.; Nehring,
R. B.; Saint-Ruf, C.; Gibson, J. L.; Frisch, R. L.; et al. Identity and Function of a Large Gene Network
Underlying Mutagenic Repair of DNA Breaks. Science (80-. )., 2012, 338 (6112), 1344–1348.
https://doi.org/10.1126/science.1226683.
[80] Khemici, V.; Poljak, L.; Luisi, B. F.; Carpousis, A. J. The RNase E of Escherichia Coli Is a
Membrane-Binding Protein. Mol. Microbiol., 2008, 70 (4), 799–813. https://doi.org/10.1111/j.1365-
2958.2008.06454.x.
[81] Magnet, S.; Courvalin, P.; Lambert, T. Activation of the Cryptic Aac(6’)-Iy Aminoglycoside
Resistance Gene of Salmonella by a Chromosomal Deletion Generating a Transcriptional Fusion. J.
Bacteriol., 1999, 181 (21), 6650–6655. https://doi.org/10.1128/JB.181.21.6650-6655.1999.
[82] Sunthornthummas, S.; Doi, K.; Rangsiruji, A.; Krajangsung, S.; Sarawaneeyaruk, S.; Pringsulaka,
O. Isolation and Cha racterization of Spontaneous Phage-Resistant Mutants of Lactobacillus
Paracasei. Food Control, 2019, 99 (September 2018), 114–123.
https://doi.org/10.1016/j.foodcont.2018.12.037.
[83] Guglielmotti, D. M.; Reinheimer, J. A.; Binetti, A. G.; Giraffa, G.; Carminati, D.; Quiberoni, A.
Characterization of Spontaneous Phage-Resistant Derivatives of Lactobacillus Delbrueckii
Commercial Strains. Int. J. Food Microbiol., 2006, 111 (2), 126–133.
https://doi.org/10.1016/j.ijfoodmicro.2006.04.035.
[84] Binetti, A. G.; Bailo, N. B.; Reinheimer, J. A. Spontaneous Phage-Resistant Mutants of
Streptococcus Thermophilus: Isolation and Technological Characteristics. Int. Dairy J., 2007, 17 (4),
343–349. https://doi.org/10.1016/j.idairyj.2006.05.002.
[85] Rostøl, J. T.; Marraffini, L. (Ph)Ighting Phages: How Bacteria Resist Their Parasites. Cell Host
Microbe, 2019, 25 (2), 184–194. https://doi.org/10.1016/j.chom.2019.01.009.
[86] Hantke, K. Compilation of Escherichia Coli K-12 Outer Membrane Phage Receptors – Their
Function and Some Historical Remarks. FEMS Microbiol. Lett., 2020, 367 (2), fnaa013.
https://doi.org/10.1093/femsle/fnaa013.
[87] Wang, Y. The Function of OmpA in Escherichia Coli. Biochem. Biophys. Res. Commun., 2002, 292
(2), 396–401. https://doi.org/10.1006/bbrc.2002.6657.
[88] Mangalea, M. R.; Duerkop, B. A. Fitness Trade-Offs Resulting from Bacteriophage Resistance
Potentiate Synergistic Antibacterial Strategies. Infect. Immun., 2020, 88 (7), e00926-19.
https://doi.org/10.1128/IAI.00926-19.
[89] Maffei, E.; Shaidullina, A.; Burkolter, M.; Heyer, Y.; Estermann, F.; Druelle, V.; Sauer, P.; Willi,
L.; Michaelis, S.; Hilbi, H.; et al. Systematic Exploration of Escherichia Coli Phage–Host
Interactions with the BASEL Phage Collection. PLOS Biol., 2021, 19 (11), e3001424.
https://doi.org/10.1371/journal.pbio.3001424.
[90] Salazar, K. C.; Ma, L.; Green, S. I.; Zulk, J. J.; Trautner, B. W.; Ramig, R. F.; Clark, J. R.;
Terwilliger, A. L.; Maresso, A. W. Antiviral Resistance and Phage Counter Adaptation to Antib iotic-
Resistant Extraintestinal Pathogenic Escherichia Coli. MBio, 2021, 12 (2), e00211-21.
https://doi.org/10.1128/mBio.00211-21.
[91] Bulieris, P. V.; Behrens, S.; Holst, O.; Kleinschmidt, J. H. Folding and Insertion of the Outer
Membrane Protein OmpA Is Assisted by the Chaperone Skp and by Lipopolysaccharide. J. Biol.
Chem., 2003, 278 (11), 9092–9099. https://doi.org/10.1074/jbc.M211177200.
[92] Islam, M. Z.; Fokine, A.; Mahalingam, M.; Zhang, Z.; Garcia -Doval, C.; Van Raaij, M. J.; Rossmann,
M. G.; Rao, V. B. Molecular Anatomy of the Receptor Binding Module of a Bacteriophage Long
Tail Fiber. PLoS Pathog., 2019, 15 (12), e1008193. https://doi.org/10.1371/journal.ppat.1008193.
[93] Chen, P.; Sun, H.; Ren, H.; Liu, W.; Li, G.; Zhang, C. LamB, OmpC, and the Core

Chapter 3: Experimental Studies
173
Lipopolysaccharide of Escherichia Coli K-12 Function as Receptors of Bacteriophage Bp7. J. Virol.,
2020, 94 (12), e00325-20. https://doi.org/10.1128/JVI.00325-20.
[94] Pagnout, C.; Sohm, B.; Razafitianamaharavo, A.; Caillet, C.; Offroy, M.; Leduc, M.; Gendre, H.;
Jomini, S.; Beaussart, A.; Bauda, P.; et al. Pleiotropic Effects of Rfa -Gene Mutations on Escherichia
Coli Envelope Properties. Sci. Rep., 2019, 9 (1), 9696. https://doi.org/10.1038/s41598-019-46100-3.
[95] Borin, J. M.; Avrani, S.; Barrick, J. E.; Petrie, K. L.; Meyer, J. R. Coevolutionary Phage Training
Leads to Greater Bacterial Suppression and Delays the Evolution of Phage Resistance. Proc. Natl.
Acad. Sci. U. S. A., 2021, 118 (23), e2104592118. https://doi.org/10.1073/pnas.2104592118.
[96] Gonzalez, G. M.; Durica -Mitic, S.; Hardwick, S. W.; Moncrieffe, M. C.; Resch, M.; Neumann, P.;
Ficner, R.; Görke, B.; Luisi, B. F. Structural Insights into RapZ-Mediated Regulation of Bacterial
Amino-Sugar Metabolism. Nucleic Acids Res., 2017, 45 (18), 10845–10860.
https://doi.org/10.1093/nar/gkx732.
[97] Azam, A. H.; Hoshiga, F.; Takeuchi, I.; Miyanaga, K.; Tanji, Y. Analysis of Phage Resistance in
Staphylococcus Aureus SA003 Revea ls Different Binding Mechanisms for the Closely Related
Twort-like Phages ɸSA012 and ɸSA039. Appl. Microbiol. Biotechnol., 2018, 102 (20), 8963–8977.
https://doi.org/10.1007/s00253-018-9269-x.
[98] Vimr, E. R.; Kalivoda, K. A.; Deszo, E. L.; Steenbergen, S. M. Diversity of Microbial Sialic Acid
Metabolism. Microbiol. Mol. Biol. Rev., 2004, 68 (1), 132–153.
https://doi.org/10.1128/MMBR.68.1.132-153.2004.
[99] Silver, R. P.; Prior, K.; Nsahlai, C.; Wright, L. F. ABC Transporters and the Export of Capsular
Polysaccharides from Gram-Negative Bacteria. Res. Microbiol., 2001, 152 (3–4), 357–364.
https://doi.org/10.1016/S0923-2508(01)01207-4.
[100] Schwarzer, D.; Buettner, F. F. R.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.;
Bowman, V. D.; Stummeyer, K.; Muhlenhoff, M.; et al. A Multivalent Adsorption Apparatus
Explains the Broad Host Range of Phage Phi92: A Comprehensive Genomic and Structural Analysis.
J. Virol., 2012, 86 (19), 10384–10398. https://doi.org/10.1128/jvi.00801-12.
[101] Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-
Encoded Virion-Associated Enzymes to Overcome the Carbohydrate Barriers during the Infection
Process. Appl. Microbiol. Biotechnol., 2017, 101 (8), 3103–3119. https://doi.org/10.1007/s00253-
017-8224-6.
[102] Scholl, D.; Adhya, S.; Merril, C. Escherichia Coli K1’s Capsule Is a Barrier to Bacteriophage T7.
Appl. Environ. Microbiol., 2005, 71 (8), 4872–4874. https://doi.org/10.1128/AEM.71.8.4872-
4874.2005.
[103] Wang, Z.; Zhao, C.; Wang, Y.; Sun, Z.; Wang, N. PANDA: Protein Function Prediction Using
Domain Architecture and Affinity Propagation. Sci. Rep., 2018, 8 (1), 3484.
https://doi.org/10.1038/s41598-018-21849-1.
[104] Goldberg, T.; Hecht, M.; Hamp, T.; Karl, T.; Yachdav, G.; Ahmed, N.; Altermann, U.; Angerer, P.;
Ansorge, S.; Balasz, K.; et al. LocTree3 Prediction of Localization. Nucleic Acids Res., 2014, 42
(W1), W350–W355. https://doi.org/10.1093/nar/gku396.
[105] Hu, P.; Janga, S. C.; Babu, M.; Díaz-Mejía, J. J.; Butland, G.; Yang, W.; Pogoutse, O.; Guo, X.;
Phanse, S.; Wong, P.; et al. Global Functional Atlas of Escherichia Coli Encompassing Previously
Uncharacterized Proteins. PLoS Biol., 2009, 7 (4), e1000096.
https://doi.org/10.1371/journal.pbio.1000096.
[106] Plotkin, J. B.; Kudla, G. Synonymous but Not the Same: The Causes and Consequences of Codon
Bias. Nat. Rev. Genet., 2011, 12 (1), 32–42. https://doi.org/10.1038/nrg2899.
[107] Pourcel, C.; Touchon, M.; Villeriot, N.; Vernadet, J.-P.; Couvin, D.; Toffano-Nioche, C.; Vergnaud,
G. CRISPRCasdb a Successor of CRISPRdb Containing CRISPR Arrays and Cas Genes from
Complete Genome Sequences, and Tools to Download and Query Lists of Repeats and Spacers.

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
174
Nucleic Acids Res., 2020, 48 (D1), D535–D544. https://doi.org/10.1093/nar/gkz915.
[108] Denes, T.; Den Bakker, H. C.; Tokman, J. I.; Guldimann, C.; Wiedmann, M. Selection and
Characterization of Phage-Resistant Mutant Strains of Listeria Monocytogenes Reveal Host Genes
Linked to Phage Adsorption. Appl. Environ. Microbiol., 2015, 81 (13), 4295–4305.
https://doi.org/10.1128/AEM.00087-15.
[109] Levin, B. R.; Moineau, S.; Bushman, M.; Barrangou, R. The Population and Evolutionary Dynamics
of Phage and Bacteria with CRISPR–Mediated Immunity. PLoS Genet., 2013, 9 (3), e1003312.
https://doi.org/10.1371/journal.pgen.1003312.
[110] Samson, J. E.; Magadán, A. H.; Sabri, M.; Moineau, S. Revenge of the Phages: Defeating Bacterial
Defences. Nat. Rev. Microbiol., 2013, 11 (10), 675–687. https://doi.org/10.1038/nrmicro3096.
[111] Meyer, J. R.; Dobias, D. T.; Weitz, J. S.; Barrick, J. E.; Quick, R. T.; Lenski, R. E. Repeatability and
Contingency in the Evolution of a Key Innovation in Phage Lambda. Science (80-. )., 2012, 335
(6067), 428–432. https://doi.org/10.1126/science.1214449.
[112] Wandro, S.; Oliver, A.; Gallagher, T.; Weihe, C.; England, W.; Martiny, J. B. H.; Whiteson, K.
Predictable Molecular Adaptation of Coevolving Enterococcus Faecium and Lytic Phage EfV12 -
Phi1. Front. Microbiol., 2019, 9, 3192. https://doi.org/10.3389/fmicb.2018.03192.
[113] Hall, A. R.; Scanlan, P. D.; Buckling, A. Bacteria‐phage Coevolution and the Emergence of
Generalist Pathogens. Am. Nat., 2011, 177 (1), 44–53. https://doi.org/10.1086/657441.
[114] Chaturongakul, S.; Ounjai, P. Phage-Host Interplay: Examples from Tailed Phages and Gram -
Negative Bacterial Pa thogens. Front. Microbiol., 2014, 5, 442.
https://doi.org/10.3389/fmicb.2014.00442.
[115] Federici, S.; Nobs, S. P.; Elinav, E. Phages and Their Potential to Modulate the Microbiome and
Immunity. Cell. Mol. Immunol., 2021, 18 (4), 889–904. https://doi.org/10.1038/s41423-020-00532-
4.
[116] Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage–Host
Interactions. Viruses, 2019, 11 (6), 567. https://doi.org/10.3390/v11060567.
[117] Casey, E.; van Sinderen, D.; Mahony, J. In Vitro Characteristics of Phages to Guide ‘Real Life’
Phage Therapy Suitability. Viruses, 2018, 10 (4), 163. https://doi.org/10.3390/v10040163.
[118] Laanto, E.; Hoikkala, V.; Ravantti, J.; Sundberg, L.-R. Long-Term Genomic Coevolution of Host-
Parasite Interaction in the Natural Environment. Nat. Commun., 2017, 8, 111.
https://doi.org/10.1038/s41467-017-00158-7.

Chapter 3: Experimental Studies
175
Su
pp
lem
en
tary F
igu
re
S1
. E
xperi
menta
l se
tup o
f th
e is
ola
tio
n o
f p
hag
e r
esi
stant
AP
EC
str
ain
s b
y s
eco
ndary
cu
lture
(S
C)
or
agar
pla
te (
AP
) m
eth
ods.
At
tim
e 0
h,
susc
ep
tib
le A
M621
WT
cu
lture
s w
ere
infe
cte
d w
ith
phage a
t fo
ur d
iffe
rent m
ult
iplicit
ies
of
infe
ctio
n (M
OIs
): M
OI
0.1
, MO
I 1
, M
OI
10
, and M
OI
100
. An
un
-infe
cted
co
ntr
ol (A
M6
21)
and n
ega
tiv
e co
ntr
ols
(P
hage a
nd L
B)
were
in
clu
ded. A
P a
pp
roach
: su
spen
sion
s w
ere
str
eaked d
irect
ly o
nto
LB
aga
r p
late
s a
nd in
cubate
d f
or
48 h
.
Resi
sta
nt
co
lon
ies
were
pic
ked
aft
er 2
4 h
and
48 h
of
incu
batio
n, gro
wn
in
LB
bro
th a
nd p
uri
fied b
y t
hre
e c
onse
cutiv
e s
trea
kin
gs
on
LB
agar
pla
tes.
SC
appro
ach
:
Rem
ain
ing “
0h” s
usp
ensi
ons
were
incu
bate
d f
or 5
h. S
usp
en
sion
s ex
hib
itin
g ly
sis
were
in
cu
bate
d f
or a
ddit
ional 2
4 h
. If
sub
seq
uent (s
econdary
) gro
wth
wa
s o
bse
rved
,
susp
en
sio
ns
were
str
eak
ed o
nto
pla
tes
an
d p
roces
sed
as
desc
rib
ed f
or th
e A
P a
pp
roa
ch. P
hage resi
stant st
rain
s w
ere
nam
ed a
cco
rdin
g to
ex
peri
menta
l setu
p (
AP
or SC
an
d M
OI.
Ex
per
imen
tal s
teps
follo
win
g th
e is
ola
tion a
re o
utlin
ed in
gre
en
.

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
176
Supplementary Figure S2. Phage-resistant strains isolated from pathogenic E. coli by secondary culture (SC) or
agar plate (AP) methods. (A) Number of resistant strains for each method and multiplicity of infection (MOI)
using the AP or SC method, including MOI 0.1 (black), MOI 1 (dark grey), MOI 10 (light grey), and MOI 100
(white). (B) Number of resistant strains isolated for each timepoint (24 hours or 48 hours of incubation) for the
AP method (black) and SC method (grey). The number of resistant strains is indicated for each combination. The
numbers were obtained based on six repeated isolation experiments.
(A)
(B)

Chapter 3: Experimental Studies
177
Su
pp
lem
en
tary
Ta
ble
S1
| A
rea u
nd
er
the c
urv
e (
AU
C)
an
d A
UC
decre
ase f
or
ph
age
-res
ista
nt st
rain
s re
lati
ve
to th
e W
T s
train
Mu
tan
t A
UC
D
ecrease
D
ecrea
se %
M
uta
nt
AU
C
Decrease
D
ecrea
se %
M
uta
nt
AU
C
Decrease
D
ecrea
se %
SC
48
_10
_8
5,7
1
0,6
6
5,2
S
C4
8_
01
_9
14
,7
1,6
9
,9
SC
24
_01
_11
15
,2
0,7
4
,4
AP
48
_1
_24
6
,0
8,7
5
9,1
A
P4
8_
10_2
4
13
,2
1,4
9
,8
SC
24
_1_
7
15
,6
0,7
4
,4
SC
24
_01
_5
6,8
9
,0
57
,0
SC
48
_1_
22
13
,2
1,4
9
,8
AP
48
_1
0_2
2
14
,1
0,5
3
,7
SC
48
_1_
9
10
,1
6,1
3
7,6
S
C2
4_
10
0_9
14
,8
1,5
9
,3
SC
24
_10
0_15
1
3,2
0
,5
3,7
S
C4
8_
1_
12
10
,4
5,5
3
4,6
A
P2
4_
100_
4
14
,4
1,4
8
,7
SC
24
_10
_14
14
,4
0,5
3
,6
AP
24
_1
0_1
4
9,2
4
,5
33
,0
SC
48
_10
0_7
14
,9
1,4
8
,5
SC
24
_10
_13
14
,4
0,5
3
,5
SC
24
_1_
9
11
,1
5,2
3
1,8
S
C2
4_
01
_15
13
,7
1,3
8
,4
AP
24
_1
0_2
4
14
,1
0,5
3
,5
SC
24
_1_
8
11
,1
5,1
3
1,6
S
C2
4_
1_
24
13
,5
1,2
8
,2
AP
48
_1
0_1
4
14
,5
0,5
3
,4
SC
48
_10
_23
10
,4
4,2
2
8,7
S
C2
4_
01
_13
13
,8
1,2
8
,1
AP
24
_1
00_
16
14
,5
0,5
3
,3
SC
24
_10
0_6
11
,5
4,3
2
7,1
S
C2
4_
10
_15
13
,8
1,2
7
,9
SC
48
_1_
10
15
,4
0,5
3
,2
SC
48
_10
_22
11
,1
3,5
2
4,0
S
C4
8_
10
_17
13
,8
1,2
7
,8
SC
24
_1_
16
14
,5
0,5
3
,2
AP
24
_1
00_
5
12
,3
3,5
2
2,0
S
C2
4_
01
_18
13
,8
1,2
7
,7
SC
24
_10
_16
14
,5
0,5
3
,0
SC
48
_10
_11
12
,9
3,0
1
8,8
S
C2
4_
1_
23
13
,6
1,1
7
,4
AP
48
_1
00_
11
15
,4
0,5
3
,0
SC
48
_10
_10
13
,0
2,9
1
8,1
S
C4
8_
1_
15
13
,9
1,1
7
,4
SC
24
_10
0_13
1
4,5
0
,4
2,9
AP
24
_1
0_5
1
3,1
2
,7
17
,1
SC
48
_10
_15
13
,9
1,1
7
,4
SC
48
_1_
16
14
,6
0,4
2
,3
SC
48
_1_
11
13
,2
2,6
1
6,7
S
C4
8_
10
_13
13
,9
1,1
7
,3
SC
48
_10
0_16
1
4,7
0
,3
1,9
SC
24
_10
_24
12
,2
2,4
1
6,6
S
C4
8_
01
_16
13
,9
1,1
7
,3
SC
24
_10
_17
14
,7
0,3
1
,9
SC
24
_01
_16
12
,5
2,4
1
6,2
S
C2
4_
01
_17
13
,9
1,1
7
,2
AP
24
_1
0_2
2
14
,4
0,3
1
,8
SC
24
_10
_10
13
,3
2,5
1
5,9
S
C4
8_
10
0_8
15
,1
1,2
7
,1
AP
24
_1
00_
23
14
,4
0,2
1
,6
SC
48
_01
_12
13
,3
2,5
1
5,9
A
P2
4_
10_2
3
13
,6
1,0
7
,0
SC
48
_10
_14
13
,5
0,1
1
,1
SC
24
_10
_23
12
,4
2,2
1
5,1
A
P2
4_
10_6
1
4,7
1
,1
6,8
A
P4
8_
100_
24
14
,5
0,1
1
,0
SC
48
_10
_24
12
,5
2,2
1
4,9
S
C2
4_
1_
13
14
,0
1,0
6
,8
AP
24
_1
_14
1
4,8
0
,2
1,0
AP
24
_100_18
12
,8
2,2
1
4,7
A
P2
4_
100_
8
15
,2
1,1
6
,5
SC
48
_10
0_22
1
4,5
0
,1
0,8
S
C2
4_
01
_14
11
,8
1,9
1
4,0
S
C4
8_
1_
13
14
,0
0,9
6
,2
AP
24
_1
00_
15
13
,6
0,1
0
,8
SC
24
_10
0_7
14
,0
2,2
1
3,8
A
P4
8_100_16
14
,0
0,9
6
,2
AP
24
_1
0_1
7
14
,9
0,1
0
,7
SC
24
_10
_22
12
,6
2,0
1
3,8
S
C2
4_100_18
14
,0
0,9
6
,1
AP
24
_1
0_1
3
13
,6
0,1
0
,6
SC
48
_01
_13
11
,8
1,8
1
3,4
S
C2
4_100_24
13
,8
0,9
5
,9
AP
48
_1
00_
14
13
,6
0,1
0
,6
AP
24
_100_12
13
,8
2,1
1
3,3
S
C2
4_
1_
14
14
,1
0,9
5
,7
SC
48
_01
_15
13
,6
0,1
0
,4
SC
48
_100_12
13
,9
2,0
1
2,4
A
P4
8_100_22
13
,8
0,8
5
,7
AP
48
_1
_13
1
4,9
0
,1
0,4
S
C2
4_100_12
13
,9
2,0
1
2,3
S
C4
8_
01
_14
14
,1
0,8
5
,6
SC
48
_10
0_24
1
4,6
0
,0
0,1
SC
24
_100_10
13
,9
1,9
1
2,3
S
C4
8_
01
_17
14
,1
0,8
5
,5
SC
48
_1_
14
13
,7
0,0
0
,1
SC
24
_1_
17
13
,2
1,8
1
1,9
S
C2
4_
1_
15
14
,1
0,8
5
,5
SC
48
_01
_23
14
,7
0,0
-0
,1
SC
24
_100_11
14
,0
1,8
1
1,6
S
C4
8_
1_
17
14
,2
0,8
5
,2
AP
48
_1
_22
1
4,7
-0
,1
-0,4
S
C2
4_
1_
22
13
,0
1,7
1
1,4
A
P4
8_
1_14
1
3,0
0
,7
4,9
S
C4
8_
01
_22
14
,8
-0,2
-1
,2
SC
48
_1_
24
13
,1
1,5
1
0,6
S
C4
8_
01
_18
14
,2
0,7
4
,8
AP
24
_1
_23
1
5,0
-0
,4
-2,4
SC
48
_1_
23
13
,1
1,5
1
0,5
A
P4
8_100_23
14
,0
0,7
4
,7
AP
24
_1
0_1
1
14
,2
1,6
1
0,4
A
P4
8_
1_15
1
4,3
0
,7
4,5

3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
178
Supplementary Table S2 | Overview of affected genes in APEC phage resistant strains
Change
Change name
Affected gene
Annotation Function n Strain(s) Ref
Gene loss (P)
Group_25
neuE
NeuE (polysialic
acid biosynthesis protein)
E. coli K1 sialic
acid capsule synthesis
24
AP24_100_12,AP48_100_11, AP48_10_14, AP24_100_4, AP24_100_5, AP24_10_5,
SC24_100_7, SC24_100_12, SC24_100_13, SC24_10_23
SC24_1_8, SC24_01_11 SC24_01_15, SC48_100_8 SC48_10_8, SC48_10_10
SC48_1_10, SC48_1_11 SC48_1_13, SC48_1_24 SC48_01_9, SC48_01_12
SC24_01_5, SC24_100_6
[44,
54, 98, 99]
Gene
loss (P/C)
Group_
67
group_
67
Hypothetical
protein (1) Unknown
2
4
AP24_100_8, AP24_10_5 AP24_10_6, SC24_100_7 SC24_100_9, SC24_100_11
SC24_10_10, SC24_10_22 SC24_10_23, SC24_1_7 SC24_1_13, SC48_100_7
SC48_100_8, SC48_10_8 SC48_10_10, SC48_10_14
SC48_1_11, SC48_1_12 SC48_1_16, SC48_1_24 SC48_01_9, SC48_01_12
SC48_01_16, SC24_01_5
-
Gene loss (P/C)
Group_237
group_237
Hypothetical protein (2)
Unknown 10
AP24_100_5, AP48_1_24
SC24_100_6, SC24_1_8 SC24_100_11, SC48_01_9 SC48_1_11, SC48_1_12
SC48_100_12, SC48_100_8
-
Gene loss (P)
Group_310
group_310
Tetratricopeptide repeat (TPR) protein
Mediation of protein-protein interactions
8
SC24_10_23, SC24_01_11 SC48_100_8, SC48_10_10 SC48_10_23, SC48_1_10
SC48_1_24, SC24_100_6
[45]
SNP (AA/ST)
SNP 17/18/19
ackA Acetate kinase Phosphorylation of acetate to acetyl phosphate
7
AP24_10_6, SC24_100_7
SC24_100_9, SC24_1_17 SC48_100_7, SC48_100_8 SC48_1_22
[46]
SNP (s) SNP 1 SNP1* Hypothetical
protein (3) Unknown 6
AP24_100_4, AP24_100_5
AP24_10_5, AP24_10_6 SC24_100_6, SC24_01_5
-
Gene loss (P)
mprA_1
mprA_1
MarR family transcriptional regulator
Regulation of numerous cellular processes
5 AP24_10_5, SC24_100_7 SC24_10_10, SC24_10_23 SC48_1_11
[47]
Gene
loss (P/C)
rfbX wzx /
rfbX
Oligosaccharide flippase protein /
O-antigen transporter
Transport of O-
polysaccharides molecules
4 AP24_10_5, SC24_01_11
SC48_100_8, SC48_1_11 [48]
SNP (AA)
SNP 25 glyS Glycyl-tRNA synthetase beta
chain
tRNA recognition 4 SC24_100_10, SC24_100_11 SC24_100_12, SC48_100_12
[49]
Gene
loss (C) fadE
fadE /
yafH
Acyl-CoA
dehydrogenase
Dehydrogenation
of acyl-coenzymes A
3 SC24_1_8, SC24_1_9
SC48_1_9 [50]
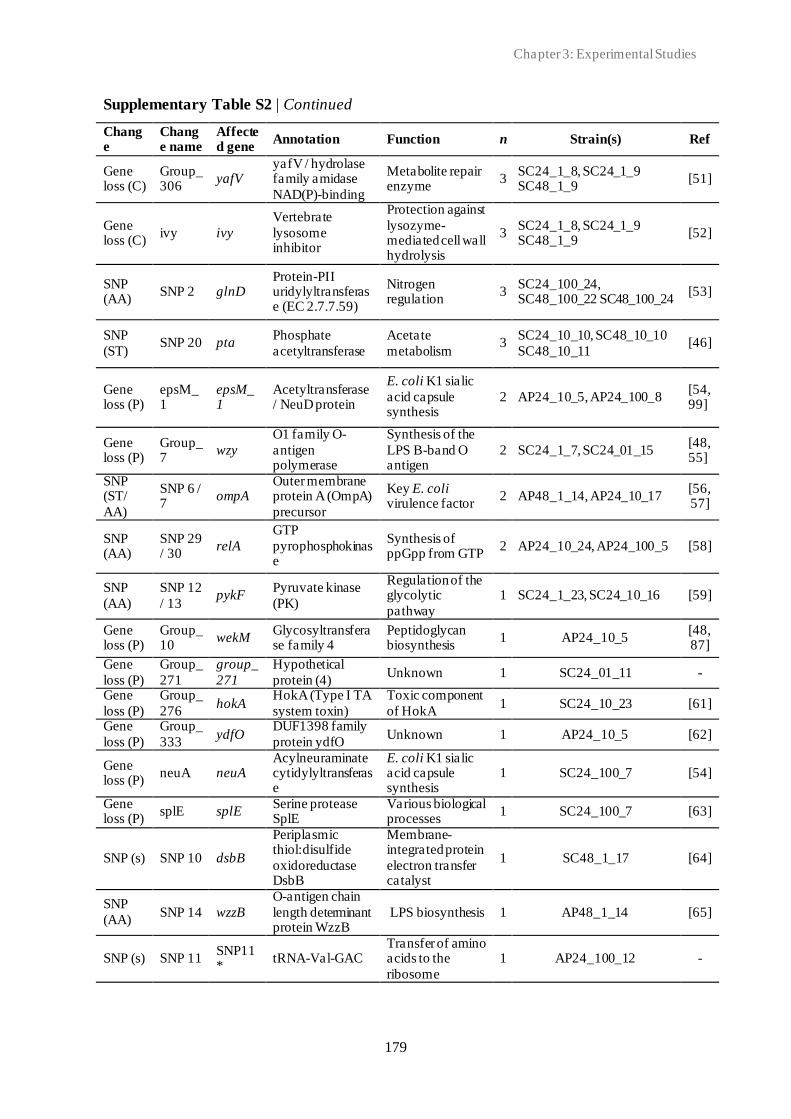
Chapter 3: Experimental Studies
179
Supplementary Table S2 | Continued
Change
Change name
Affected gene
Annotation Function n Strain(s) Ref
Gene loss (C)
Group_306
yafV yafV / hydrolase family amidase
NAD(P)-binding
Metabolite repair enzyme
3 SC24_1_8, SC24_1_9 SC48_1_9
[51]
Gene loss (C)
ivy ivy
Vertebrate
lysosome inhibitor
Protection against
lysozyme-mediated cell wall hydrolysis
3 SC24_1_8, SC24_1_9 SC48_1_9
[52]
SNP (AA)
SNP 2 glnD Protein-PII uridylyltransferase (EC 2.7.7.59)
Nitrogen regulation
3 SC24_100_24, SC48_100_22 SC48_100_24
[53]
SNP
(ST) SNP 20 pta
Phosphate
acetyltransferase
Acetate
metabolism 3
SC24_10_10, SC48_10_10
SC48_10_11 [46]
Gene loss (P)
epsM_1
epsM_1
Acetyltransferase / NeuD protein
E. coli K1 sialic
acid capsule synthesis
2 AP24_10_5, AP24_100_8 [54, 99]
Gene loss (P)
Group_7
wzy
O1 family O-
antigen polymerase
Synthesis of the
LPS B-band O antigen
2 SC24_1_7, SC24_01_15 [48, 55]
SNP (ST/
AA)
SNP 6 / 7
ompA Outer membrane protein A (OmpA)
precursor
Key E. coli virulence factor
2 AP48_1_14, AP24_10_17 [56, 57]
SNP (AA)
SNP 29 / 30
relA
GTP
pyrophosphokinase
Synthesis of ppGpp from GTP
2 AP24_10_24, AP24_100_5 [58]
SNP
(AA)
SNP 12
/ 13 pykF
Pyruvate kinase
(PK)
Regulation of the glycolytic
pathway
1 SC24_1_23, SC24_10_16 [59]
Gene loss (P)
Group_10
wekM Glycosyltransferase family 4
Peptidoglycan biosynthesis
1 AP24_10_5 [48,87]
Gene
loss (P)
Group_
271
group_
271
Hypothetical
protein (4) Unknown 1 SC24_01_11 -
Gene
loss (P)
Group_
276 hokA
HokA (Type I TA
system toxin)
Toxic component
of HokA 1 SC24_10_23 [61]
Gene
loss (P)
Group_
333 ydfO
DUF1398 family
protein ydfO Unknown 1 AP24_10_5 [62]
Gene loss (P)
neuA neuA Acylneuraminate cytidylyltransferase
E. coli K1 sialic acid capsule synthesis
1 SC24_100_7 [54]
Gene loss (P)
splE splE Serine protease SplE
Various biological processes
1 SC24_100_7 [63]
SNP (s) SNP 10 dsbB
Periplasmic thiol:disulfide
oxidoreductase DsbB
Membrane-integrated protein
electron transfer catalyst
1 SC48_1_17 [64]
SNP
(AA) SNP 14 wzzB
O-antigen chain
length determinant protein WzzB
LPS biosynthesis 1 AP48_1_14 [65]
SNP (s) SNP 11 SNP11*
tRNA-Val-GAC Transfer of amino acids to the
ribosome
1 AP24_100_12 -
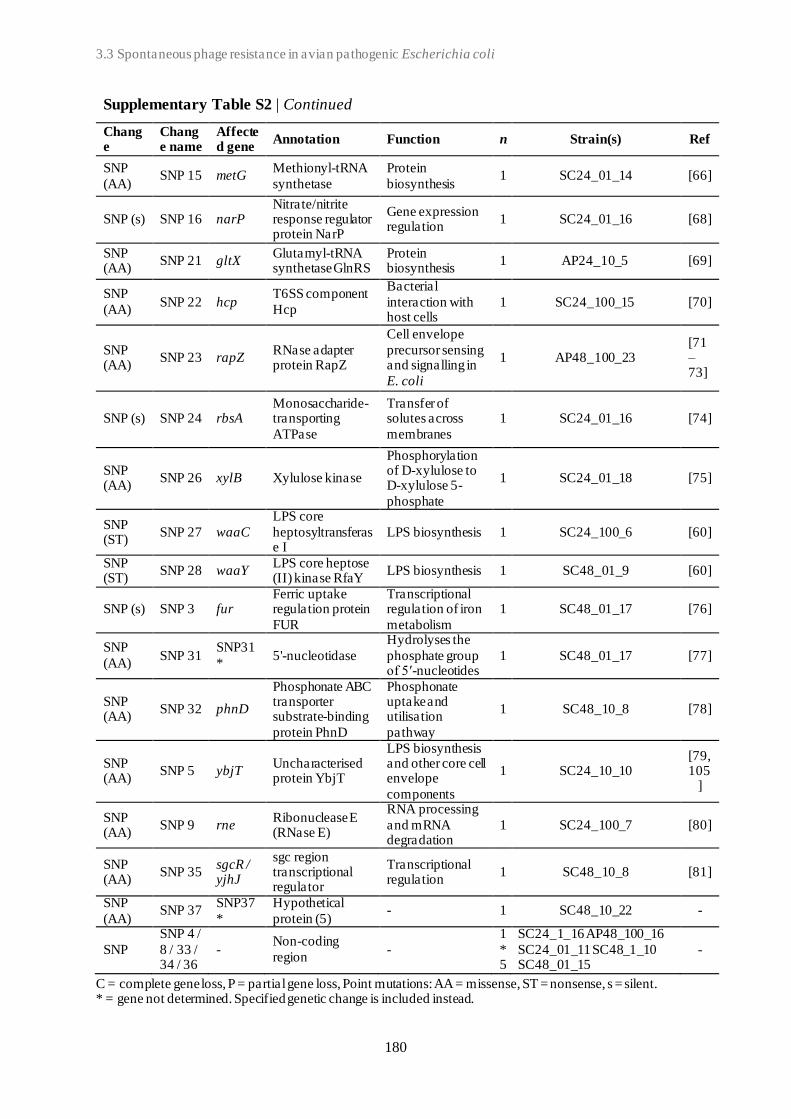
3.3 Spontaneous phage resistance in avian pathogenic Escherichia coli
180
Supplementary Table S2 | Continued
Change
Change name
Affected gene
Annotation Function n Strain(s) Ref
SNP
(AA) SNP 15 metG
Methionyl-tRNA
synthetase
Protein
biosynthesis 1 SC24_01_14 [66]
SNP (s) SNP 16 narP Nitrate/nitrite response regulator protein NarP
Gene expression regulation
1 SC24_01_16 [68]
SNP (AA)
SNP 21 gltX Glutamyl-tRNA synthetase GlnRS
Protein biosynthesis
1 AP24_10_5 [69]
SNP
(AA) SNP 22 hcp
T6SS component
Hcp
Bacterial
interaction with host cells
1 SC24_100_15 [70]
SNP (AA)
SNP 23 rapZ RNase adapter protein RapZ
Cell envelope
precursor sensing and signalling in
E. coli
1 AP48_100_23 [71–73]
SNP (s) SNP 24 rbsA Monosaccharide-transporting
ATPase
Transfer of solutes across
membranes
1 SC24_01_16 [74]
SNP (AA)
SNP 26 xylB Xylulose kinase
Phosphorylation of D-xylulose to D-xylulose 5-
phosphate
1 SC24_01_18 [75]
SNP (ST)
SNP 27 waaC
LPS core
heptosyltransferase I
LPS biosynthesis 1 SC24_100_6 [60]
SNP (ST)
SNP 28 waaY LPS core heptose (II) kinase RfaY
LPS biosynthesis 1 SC48_01_9 [60]
SNP (s) SNP 3 fur Ferric uptake regulation protein
FUR
Transcriptional regulation of iron
metabolism
1 SC48_01_17 [76]
SNP
(AA) SNP 31
SNP31
* 5'-nucleotidase
Hydrolyses the
phosphate group of 5′-nucleotides
1 SC48_01_17 [77]
SNP (AA)
SNP 32 phnD
Phosphonate ABC transporter substrate-binding
protein PhnD
Phosphonate uptake and utilisation
pathway
1 SC48_10_8 [78]
SNP (AA)
SNP 5 ybjT Uncharacterised protein YbjT
LPS biosynthesis and other core cell envelope
components
1 SC24_10_10 [79, 105
]
SNP (AA)
SNP 9 rne Ribonuclease E (RNase E)
RNA processing
and mRNA degradation
1 SC24_100_7 [80]
SNP (AA)
SNP 35 sgcR / yjhJ
sgc region transcriptional regulator
Transcriptional regulation
1 SC48_10_8 [81]
SNP
(AA) SNP 37
SNP37
*
Hypothetical
protein (5) - 1 SC48_10_22 -
SNP
SNP 4 /
8 / 33 / 34 / 36
- Non-coding
region -
1
*5
SC24_1_16 AP48_100_16
SC24_01_11 SC48_1_10 SC48_01_15
-
C = complete gene loss, P = partial gene loss, Point mutations: AA = missense, ST = nonsense, s = silent. * = gene not determined. Specified genetic change is included instead.

Chapter 3: Experimental Studies
181
Su
pp
lem
en
tary
Ta
ble
S3
| O
verv
iew
of
ev
idence lev
el 1
-CR
ISP
R s
pacers
dete
cte
d in
ph
age-r
esi
stan
t st
rain
s
Sp
ace
r
#
Len
gth
(bp
) S
pace
r se
qu
ence
B
LA
ST
hit
G
enB
an
k
An
nota
tion
1
40
G
CG
CT
GC
GG
GT
CA
TT
TT
TG
AA
AT
TA
CC
CC
C
GC
TG
TG
CT
GT
E
sch
eri
ch
ia c
oli
str
ain
SB
02
58
h1
C
P0
71
95
4.1
G
en
era
l st
ress
pro
tein
2
54
G
CC
GT
TG
CC
GA
AT
GT
AG
GC
CG
GA
TA
AG
GC
GT
TC
AC
GC
CG
CA
TC
CG
GC
AA
CC
AG
C
Esc
heri
ch
ia c
oli
str
ain
EcP
F5
C
P0
54
23
6.1
-
3
34
C
TG
TA
AT
TT
TC
AT
GA
AA
GG
TG
GA
TG
GC
TG
C
GC
AC
Esc
heri
ch
ia c
oli
str
ain
CP
8-
3_
Sic
hu
an
pla
smid
pC
P8
-3-I
ncX
1
CP
05
37
40
.1
Pla
smid
(p
CP
8-3
-
IncX
1)
4
38
C
GG
AC
GC
AG
GA
TG
GT
GC
GT
TC
AA
TT
GG
AC
T
CG
AA
CC
AA
E
sch
eri
ch
ia c
oli
str
ain
LW
Y6
C
P0
72
20
4.1
tR
NA
-Va
l
5
58
G
GA
GC
CA
GA
AG
AA
CA
GA
TT
GA
TC
CG
CG
CA
AA
GC
CG
CC
GT
CG
AA
GC
TG
CT
AT
TG
CC
CG
T
Esc
heri
ch
ia c
oli
str
ain
SC
U-4
87
C
P0
54
45
4.1
Ele
ctr
on
tra
nsp
ort
co
mp
lex
su
bu
nit
Rsx
C
6
53
T
TT
CA
AG
TA
TT
GT
AA
AA
CA
TT
TG
AT
GC
AA
T
CG
CT
TA
TA
TT
GC
CG
AA
TC
TT
TT
G
Esc
heri
ch
ia p
ha
ge v
B_
Eco
M_G
29
M
K3
27
94
0.1
P
inA
pep
tid
ase
inh
ibit
or
7
24
G
GG
GG
GG
GG
GG
GG
GG
GG
GG
GT
TT
G
- -
-
8
27
C
CC
CC
CC
CC
CC
CC
CC
CC
CC
CC
CC
CC
CC
-
- -

3.4 Schematic overview of the experimental studies and main findings
182
3.4
Schematic overview of the experimental studies and main findings
3.4 Schematic overview of the experimental studies and main findings

Chapter 3: Experimental Studies
183
The main findings of the experimental studies (Chapter 3.1, 3.2, and 3.3) are summarised in
Figure 1 and are discussed in the following pages (Chapter 4).
Figure 1 | Overview of the experimental studies (scientific chapters) and the main findings.

184

Chapter 4: General Discussion
185
Chapter 4: General Discussion
Chapter 4
General Discussion
4.1 Avian pathogenic E. coli (APEC) - The need for alternative treatment options
APEC is one of the most important pathogens affecting poultry worldwide, with infections,
collectively referred to as colibacillosis, resulting in increased morbidity and mortality [1].
Various APEC serotypes have been associated with cases of colibacillosis, though, the three
serotypes O1, O2 and O78 account for most of the cases [1–3]. Antibiotics are commonly used
to control APEC infection in poultry, however the emergence of antimicrobial resistant
bacterial strains and antibiotic failures and restrictions have led to a growing interest in much-
needed complement treatment strategies, such as the prophylactic and therapeutic application
of phages (phage therapy) [1, 4–7]. Multiple studies evaluating the preventative and therapeutic
efficacy of phages against APEC in poultry have been conducted [reviewed by 1, 8]. These
studies suggest that phage application can be a valuable approach to prevent and control APEC
infections, and hereby, highly beneficial to both animals and humans in the framework of
animal welfare, farmers income, higher productivity, food security, and the environment as
well as reduced antimicrobial resistance. However, no phage treatment has yet advanced into

Chapter 4: General Discussion
186
field application, possibly due to challenges in large-scale production and application as well
as regulatory hurdles [1, 8]. Considering the specific poultry sector economics, with large
numbers of animals with each a low intrinsic value and relatively short lifespan, phage
application (cost, route, etc.), like application of antibiotics, should be economically viable.
Due to the significant problem it represents to poultry welfare and industry, multi-drug resistant
APEC was chosen as the bacterial model organism in this work.
4.2 The remarkable diversity of E. coli-infecting phages
Phages are viruses that specifically infect bacteria. An opening statement such as “Phages are
the most abundant and diverse organisms on Earth” has frequently been used, when describing
phages [9–11]. In recent years, the understating of phage diversity has expanded enormously
through the increased availability of phage sequences [9, 12, 13]. Moreover, high-throughput
screening methods enable fast isolation and identification of hundreds of diverse phages [14].
However, the advancements have also revealed that we have only scratched the surface in the
discovery of novel viruses. The recent progress within the understanding of phage diversity is
also reflected in the extensive update of phage taxonomy in 2020 carried out by the ICTV [15].
The majority of phages isolated to date belong to the Caudovirales order of tailed double-
stranded DNA phages [16], possibly due to isolation biases and dedicated work of phage-
isolation programmes, such as the Science Education Alliance-Phage Hunters Advancing
Genomics and Evolutionary Science (SEA-PHAGES) [17–19]. In chapter 3.1, we investigated
the diversity of coliphages in the intestines of poultry as well as the overall coliphage genomic
diversity. Using the enrichment culture approach combined with WGS, we were able to isolate
and characterise Caudovirales coliphages belonging to the Myoviridae, Demerecviridae
(previously Siphoviridae), or the Drexlerviridae (previously Siphoviridae) families. The
classification was done by combining, morphological and WGS analysis (including whole
genome and phage marker gene analysis). We were able to isolate new phage species, some of
which belong to a newly created phage genus (Warwickvirus). Unravelling the enormous
diversity of phages and their genomes might be of great benefit. Exploring their diverse mode
of host interaction as well as structure-function correlation will contribute to research going on
to find new ways to efficiently exploit phages for application in various scientific and
therapeutic fields [16, 20]. Comparative genomics of phages allows for better understanding of
phage adhesion factors and the affinity between a bacterial receptor and a phage RBPs. Such

Chapter 4: General Discussion
187
knowledge may be used to develop new adhesins, new ways to target bacterial pathogens, or
new ways to modulate the microbiome (such as probiotics) [21].
In this work we used E. coli K-12 derived laboratory strains (C600 and K514), as preliminary
studies (unpublished data) showed higher susceptibility compared to APEC strains for the
isolation of phages. Both laboratory strains were modified in their restriction, which most likely
promotes phage infection, and hereby, allows for isolation of more diverse phages [22]. Still,
one should be aware of the biases that the use of any strain creates and limits the possibility to
have a full picture of the coliphage diversity. Novel synthetic biology strategies may allow for
the creation of bacterial strains encoding pluripotent receptors but no phage resistance
mechanisms (R-M systems, Abi systems, CRISPR-Cas etc.), enabling phage isolation with
reduced bias. A metagenomic isolation bias-free approach is not suitable as individual phages
were needed for in vitro characterisation. However, for future work it would be interesting to
compare the findings of this current work with metagenomic analysis of the poultry phage
microbiome to determine the degree of diversity and evolution of coliphages that is reflected
in our collection.
4.2.1 Phage host spectrum
Phages differ in their host-specificity and show very diverse lytic behavior ranging from very
specifically lysing only one strain to infecting numerous strains [23]. In chapter 3.2, variation
in infectivity spectrum of tested coliphages was similarly observed with phages able to infect
all APEC strains while others not able to infect any of the tested APEC strains. The infectivity
of phages depends on their ability to successfully bind to the phage receptor of the host cells
and to overcome or evade strain-specific bacterial immunity [18, 24]. Caudovirales phages
bind to host surface receptors using phage RBPs, such as tail fibers, tail spikes or central tail
tips [22, 25]. Recently, Maffei et al. 2021 highlighted the patterns of phage receptor specificity
and showed that Siphoviridae and Myoviridae phages only target a very limited number of all
the E. coli OMPs. However, for most studied phages, including the ones used in this work, the
host receptor is unknown. RBPs are thought to be the primary determinant of phage host range,
and phages are believed to move between related hosts at least in part through genetic exchange
of or modifications in RBP-encoding genes [25–27]. For expanding the host spectrum, the
RBPs can also be engineered, based on extended sequence variation analysis and detailed
interaction studies, allowing to determine the importance of each of the RBPs in the adhesion

Chapter 4: General Discussion
188
or go through a host co-evolutionary process [27–29]. Phage host range has been highlighted
as a crucial element for phage therapy application as broader/adapted infectivity, natural,
trained or engineered enable phages to be used against different target strains (thus different
phage receptor molecules) expanding the phage application spectrum [22, 27]. Also, the
different aspects of phage (extended) host spectrum (ability to infect vs. ability to replicate)
should be considered when choosing treatment strategy: “passive” treatment with initial
sufficient phage titer to reduce bacterial numbers (depends on phage bactericidal activity) or
“active” treatment requiring on-going in situ phage replication to reach/maintain numbers
sufficient to control the target bacteria [30, 31].
4.2.2 Hypothetical proteins of unknown function
Sequenced phage genomes are often annotated for gene identification and assignment of
putative gene function [32, 33]. As no single programme consistently outperforms the others,
most often multiple programmes are combined with manual interpretation of the finding to
achieve the highest accuracy [32]. In chapter 3.1, a similar approach combing automatic and
manual annotation was used. Still, in accordance with findings from various other studies, the
function of the majority of phage-encoded gene products are still not known and are referred
to as hypothetical proteins of unknown function [34–36]. Many phage genome annotations
include false positives (e.g., when every open reading frame longer than specific length is
annotated as a protein-coding gene) and/or false negatives (e.g., when genes shorter than 100
bp is falsely excluded as this is a commonly used cut-off length) [33]. Several approaches have
been put forth to overcome the major challenge of assigning function to hypothetical proteins,
including in silico structure/function prediction bioinformatic tools and comparative genomic
combined with functional genetic studies [35, 37, 38]. In this study, gene function of unknown
genes encoding hypothetical proteins was predicted using a comparative genomics approach,
comparing identified genes to homologue genes with defined functions in other related phage
genomes. Continuous advances in genome sequencing technologies, bioinformatics, and
proteomics will play a major role in unravelling the functions of hypothetical phage proteins
and provide a promising approach to identify more and novel targets of pathogenic bacteria
and to exploit phages therapeutic potential as well as their application in other scientific fields
[32, 34, 36].

Chapter 4: General Discussion
189
4.3 Phage-host population growth dynamics
In the presence of sensitive bacteria, phage PK (phage movement and persistence in animal
body) and PD (phage host range and bacterial resistance) are fundamentally different from
those of antibiotics/chemical drugs as phages have the ability to self -replicate, co-evolve, and
elicit immune responses [31, 39–43]. Successful treatment is dependent on the suitable phages
reaching the targeted bacteria in sufficient numbers [31]. Accordingly, PK/PD are essential
parameters for better understanding the success of phage therapy [39, 44], and the sparse
knowledge is limiting its clinical applications [31, 41, 42].
Studies of in vitro phage-host interactions in liquid culture are commonly used to assess
bacterial growth dynamics in response to different phage exposure, using OD as input [45–49].
This method is fast and can be used for high-throughput comparison using 96-well plate
incubating spectrophotometers [50], similar to the experimental setup described in chapters
3.2 and 3.3. In our study, the growth dynamics of well-characterised virulent coliphages and
multidrug-resistant APEC strains at different MOIs were assessed. Using OD as input we
classified dynamic patters and determined the effect of phage species, APEC strain and MOI
on the observed dynamics. Out of the three variables the phage species had the most significant
effect of the dynamics outcome in the established phage-host model. Myoviridae phages
belonging to the Tevenvirinae subfamily (Tequatrovirus genus) exhibited the broadest host
range infecting both O1, O2, and O78 APEC strains. Similar findings have recently been
reported highlighting the great efficiency against target strains [22, 23, 51]. The specific
evolution of the phage-host population growth dynamics, including bacterial reduction and the
emergence of a phage-resistant bacterial population, depends on the various interactions of the
specific phage-host combination during the course of infection [52]. The binding affinity of the
phage to the bacterial host has been identified as one of the key parameters for the reduction of
the bacterial population size. Future studies looking at different phage and bacteria l gene
expression patterns might be able to identify additional crucial parameters related to the
different dynamics.
The co-evolution between phage and host bacterium has been described as antagonistic in
which a continuous arms race takes place [53–55]. However, similar to findings of Holguín et
al. (2019), no evidence of antagonistic co-evolution between phage and host strain was found
in the model used in chapter 3.2 and 3.3. Instead, an asymmetric dynamic in favour of the
bacteria was observed. Phages were not able to overcome existing resistance mechanisms nor
counteract phage resistance development. However, the (experimental) time factor could be of

Chapter 4: General Discussion
190
great importance. Given enough time, co-cultures phages have the ability to evolve to
overcome the host phage resistance [57–59]. Phages applied therapeutically most likely do not
have a long time to adapt to overcome phage resistance mechanisms present in target bacteria.
Thus, potential adaptation of the phage(s) should happen prior to the application (using
engineering, training or other approaches described earlier). Some phages might be able to
overcome the resistance fast enough for direct application. Through studies with different
phage-host combinations, these fast-evolving phages could be identified along with the
underlying mechanisms conferring this ability.
In this work, the model employed to understand the coliphage-host population dynamics
involved in vitro co-cultured phage-host combination comprising a single phage (monophage)
and target strain. However, in practice the use of multiple phages (phage cocktail) is usually
favoured [60]. Nevertheless, the individual patterns may guide the composition of the cocktail
though the individual profiles are also important for the understanding of the dynamics. We
can potentially increase the host range, a more efficient suppression of bacterial growth can be
achieved, and phage resistance may be hampered [50, 51, 60, 61]. The inclusion of phage
cocktails as well as mixed cultures of target bacteria could help capture more complex
dynamics [62]. The model established in chapter 3.2 can be applied as an in vitro screening
approach for phage candidates for phage therapy, and may aid a more standardised and
quantitative evaluation [49]. The approach captures the ongoing phage-host population
dynamics and produces quantitative high-throughput data to determine phage host range, phage
virulence/infectivity, and emergence of bacterial phage-resistance, which facilitates a rational
design of phage therapies [50, 52]. However, in order to quantify and individually tract multiple
phages and bacteria in mixed cultures, alternative approaches such as real-time quantitative
PCR (RT-qPCR) might be needed [50, 63]. The effect of each defined dynamics pattern on the
course of infections needs to be determined in vivo.
4.4 Phage resistance in APEC
The ubiquity of phage predation has driven the evolution of various bacterial immune systems
targeting any step of the phage infection process [39, 64–66]. These mechanism include (but
are not limited to) R-M systems that target specific sequences on the invading phage genome,
Abi systems that lead to cell death or metabolic arrest of the bacterial cell upon phage infection,
and CRISPR-Cas, which provides acquired adaptive immunity through memorisation of past

Chapter 4: General Discussion
191
phage attach [24, 67–69]. Moreover, new anti-phage strategies are still being discovered and
continues to expand the known arsenal of bacterial defence systems [70, 71].
One of the major hurdles to a successful phage therapy implementation is the presence and/or
emergence of phage-resistant bacteria [72, 73]. Whereas the first limits the applicability of
phage candidates for presumptive use and makes it harder to keep standardised phage cocktail
up to date, the latter may impede favourable treatment outcomes. Several in vitro studies have
reported the emergence of phage-resistant variants within a short period of time after phage
exposure [51, 73–75]. In our study, we detected bacterial growth after as early as ~7 hours with
exposure to a phage in chapter 3.2 and within 48 hours in the phage-host model of chapter
3.3. Contrary to more complex natural environments where mechanisms such as CRISPR-Cas
evolve rapidly [76, 77], in laboratory conditions, bacterial phage resistance is typically
conferred by mutations or loss of phage receptor-encoding genes, blocking phage adsorption
[76, 78]. Previously, E. coli knockout libraries, such as the Keio collection [79] (single-gene
knockout mutants of all non-essential genes of E. coli K-12) have been used to identify specific
genes involved in coliphage infection [22, 80]. However, this method can be laborious,
involving the screening of individual phage samples on very large numbers of mutant bacterial
strains if genes for phage receptors are unknown. Transposon mutant libraries have been used
to identify bacterial resistant mutants [81, 82]. While this approach is less laborious, it is limited
by the number of transposon mutants chosen for follow-up study, making it potentially
challenging to identify the receptors(s) used by a single phage [82]. Transposon mutagenesis
produces randomised gene mutations/insertions in the bacterial genome, not reflecting the
selective pressure conferred by the presence of phage. As we wanted to mimic natural
selections/patterns of targeted genes involved in phage resistance, natural mutation
development was used. This approach, however, necessitates comparing WGS of phage -
resistant mutants with a well-annotated reference genome to identify mutations. Spontaneous
mutation(s) can, but are not guaranteed to, arise in cell surface structures that are required for
initial phage adsorption to bacteria [78]. These mutations include both one or more single point
mutation(s) as well as partial and/or complete gene loss. In this study, gene loss and/or
mutations in genes encoding the OmpA and LPS were detected in the phage-resistant variants
and that alone was sufficient for bacterial resistance. However, in accordance with previous
studies we also found that multiple linked mutations may be necessary for full phage resistance
[78, 82, 83]. In some of our phage-resistant mutant strains from chapter 3.3 we detected point
mutations and/or gene loss in two or three genes. Affected genes included genes known to be

Chapter 4: General Discussion
192
involved in phage resistance as well as genes not previously linked to resistance, indicating
that the phage-host interaction might be more complex than previously thought. The latter
group included, among others, genes encoding hypothetical proteins with unknow function.
However, the potential involvement in phage resistance remains to be confirmed. Further
characterisation of the hypothetical proteins, including gene function assignment, will also be
necessary to fully understand the involvement of this genetic changes in phage resistance.
Some phages have been shown to be more able to circumvent the bacterial resistance, and the
mechanisms they use for this may help overcome these resistance mechanisms [58, 84]. It has
been shown that (Myoviridae) Tevenvirinae phages exhibited broad resistance to R-M systems
due to cytosines modification in their genomes [22, 85]. Maffei et al. (2021) found
(Myoviridae) Vequintavirinae phages to have an exceptional host range, possibly due to their
ability to bypass the O-antigen barrier. However, they also reported a high sensitivity to various
R-M systems. Moreover, (Myoviridae) Ounavirinae phages belonging to the Felixounavirus
genus showed remarkable sensitivity to several tested R-M systems. Engineering these R-M-
sensitive phages by inserting the cytosine modification mechanisms may prevent this
resistance, however engineering of phages is still problematic [86], but technologies are
evolving and may become available in the future. This would lead to engineered phages with
a wanted host spectrum and less sensitivity towards bacterial phage resistance mechanisms. In
our study, we found the Tevenvirinae phages able to circumvent these natural immune systems
and infect most of the O1, O2, and O78 APEC strains. Very recent, Maffei et al. (2021) showed
a correlation between Drexlerviridae (previously Siphoviridae) phylogeny and
sensitivity/resistance to bacterial immunity, though the mechanisms behind still need to be
unravelled. However, while it might be an interesting target for phage engineering, high
resistance to bacterial immunity is not a guarantee for successful treatment outcome (chapter
3.2). Another hurdle to be considered is the fact that these engineered phages fall under the
regulations of genetically modified organisms (GMO). Such phages can be subjected to
additional regulations compared to natural phages and this may cause problems for in filed
acceptance of the therapies [87]. The safety and consequences of such genetically engineered
phages should be well determined [73].
Understanding the structural and molecular mechanisms of phage-host interaction is thus
crucial for the application of phages [25]. The use of phage cocktails, composed of phages
using different host receptors, has proved to be advantageous against several bacterial
pathogens and less likely to select for phage-resistant strains [51, 88]. Though it remains no

Chapter 4: General Discussion
193
guarantee for successful application [51, 89–91]. In this work only single phage single host-
combinations were used when studying growth dynamics (chapter 3.2) and phage resistance
development (chapter 3.3). In future experiments, carefully selected phages (see below), based
on the single reactions, can be combined in a cocktail, and exposed to target APEC strains.
However, more work is necessary to define what properties would be necessary in a good
cocktail. Our work contributed to the variables of interest in composing such cocktail.
4.4.1 The cost of phage resistance
Emergence of phage-resistant bacteria is, with our current knowledge and capacities to
engineer phages, unavoidable, and resistance confers a clear selective advantage of the
bacterium in the presence of phage. However, the resistance often comes with a cost for the
bacterium as demonstrated in our results, and the magnitude of the trade-off depends on the
genetic basis of the resistance as well as the environmental context [31, 39, 66, 72, 78, 92].
Immune systems such as R-M systems and CRISPR-Cas is associated with fitness cost as these
can be extremely energy consuming and may use resources that would otherwise be invested
in cell growth [66, 93, 94]. Resistance conferred by modifications to surface LPS, membrane
porins, siderophores, efflux pumps, pili, and flagella may come with fitness cost as well,
including reduced virulence, decreased resistance to environmental pressures, colonisation
defects, reduced growth rate, reduced motility, and re-sensitisation to antibiotics or the host
immune system [51, 66, 78, 95–98]. The cost of losing surface receptors, is likely to be lower
in laboratory settings, where nutrients are readily available and competition is less, than in
natural environments [66, 78, 99]. Depending on the risk/frequency of phage infection, either
CRISPR systems (low risk) or surface modification (high risk) is favoured as resistance
mechanisms by the bacterium [66, 100]. Inducible resistance mechanisms (triggered upon
phage infection) such as CRISPR is associated with an induced cost of resistance, whereas
constitutive mechanisms (always active), such as loss of cell surface receptors, are associated
with a fixed cost. The overall cost of the inducible resistance will depend on the infection
frequency and will determine the most favourable defence strategy. The phage-resistant
variants obtained in chapter 3.3 did not show changes their CRISPR loci, however, several
genes associated with phage adoption were affected by point mutation or gene loss.
Interestingly, the greatest fitness cost, measured by decrease in growth relative to the
susceptible WT strain, was conferred by mutation in genes with unknown function. These
genes can serve as potential targets for new therapeutic applications. By inhibiting the function

Chapter 4: General Discussion
194
of these genes using for example chemicals, the growth of these bacteria can be inhibited, and
the infection can be overcome by the susceptible host. Deletions or mutations in these genes
can also be used in vaccine developments if shown that these reduce the virulence to an
acceptable level.
While phages, antibiotics, or animal host immunity may not be sufficient for clearing bacterial
infections singlehandedly, combination therapies with phage(s) and antibiotics are clinically
promising as they are showing synergistic effects [88, 101–105]. Phages can utilise critical
bacterial surface molecules that provide either defence from antibiotics via efflux, uptake of
nutrients in resource-limited host environments, or general cell wall maintenance [78].
Accordingly, by understanding and exploiting phage resistance, phage therapy can “steer”
pathogenic bacteria toward deleterious surface mutations that allow for more favourable
treatment outcomes and extend the useful lifespan of antibiotics that otherwise would have
been discarded [95, 104]. Since not all mutations in surface factors will lead to desirable
treatment outcome, and given the complexity of phage–bacteria interactions, special
considerations to context-dependent (in vitro vs. in vivo) fitness cost are required when
developing effective phage steering therapies for clinical use [39, 78, 95].
4.5 Acquisition and selection of suitable phages for phage therapy
Phages can readily be isolated from various environments, and a much larger set of phages is
often available than can be used in any application [10, 18, 106]. When excess phages are
available, how to select the best phage(s)? Before administration, phage therapy candidate
phages undergo careful examination of their safety and interaction with target bacteria [18],
currently best done by WGS and determination of the host range and dynamics. Recently, a
framework for a standardised in vitro evaluation of phage candidates for phage therapy was
suggested [49]. A key first step is to develop a large collection of well-characterised candidate
phages followed by matching of phage(s) to target bacteria, including an investigation of the
phage-host growth kinetics in liquid cultures. Subsequently, the interactions between target
bacteria and phage(s) should be examined as well as the effect of the host immune system on
the phage(s) to obtain optimal synergistic results [20]. Seen the current difficulties in
engineering phages, such phages are not yet available, but may represent another solution to
the therapeutic use of phages as discussed above.

Chapter 4: General Discussion
195
In chapter 3.1, a well-characterised collection of virulent coliphages was established. All
phage comprised genetic properties desirable for phage therapy, including no genes encoding
any known toxins, virulence factors, or resistance to antibiotics [18, 107]. However, when in
vitro phage-host growth kinetics were determined (chapter 3.2), half of the phages were not
able to infect any of the target bacteria (O1, O2, and O78 APEC strains). One possible
explanation for lack of infectivity might be found in the choice of bacterial strain(s) used to
isolate the phages. In this work, phages were initially isolated on non-pathogenic E. coli
laboratory strains. We hypothesised that these would provide a broad diversity of phages as
opposed to WT AEPC strains, which most likely are better equipped to resist a wide array of
phage infections. For phage therapy application, the phage lytic potential on relevant
pathogenic bacterial strains is needed to determine their clinical coverage and importance for
inclusion in phage cocktails [108]. Using multiple host strains during isolation can more
reliably select for broader host range phages (able to inf ect multiple bacterial species),
compared to phages isolated using a single bacterial host strain [109]. In accordance with
previous findings [51, 56, 106, 110], we found the phage species, the APEC strain, and the
MOI all significantly influenced the observed dynamics, of which the phage species factor had
the most significant influence. Bull and Gill (2014) recommended a comparative approach to
predict efficacy. By comparing phages with differences in their dynamic properties and
treatment efficacy, properties associated with success can be identified. When combining
multiple phages in a well composed cocktail, it is possible to obtain a synergistic effect against
target bacteria, including faster killing and/or delay/hindrance of resistance development [51,
56, 61, 101, 111]. However, Holguín et al. (2019) and Pinto et al. (2021) showed that the
combination of any two phages may not have the desired effect. Selecting suitable phage(s) for
therapeutic application require sufficient understanding of phage-host interaction and can be
the difference between application success or failure. Still, a high in vitro efficiency is not
necessarily able to simulate the complex in vivo interplay of phage, bacteria and animal host,
and therefore, not a guarantee for successful therapy application [51, 89–91].
The use of standard growth conditions (LB medium, 37 °C, regular aeration), like for most
phage work of the last decades, was applied in this work to generate systematic, reproducible
data. However, such conditions mimic only a small part (if any) of the diverse environments
and physiological conditions that add up to the complex habitats of E. coli and its phages.
Safety and efficacy data obtained from preliminary in vitro testing of host range, efficiency of
plating, killing curves, phage kinetics, phage receptor(s) determination, cocktail optimisation

Chapter 4: General Discussion
196
and phage/antibiotic interactions can be combined with data obtained from complex ex vivo
infection models mimicking the microenvironment [108, 112]. Also, stability data of the phage
(cocktail) at various temperatures, pH ranges, and oxygen and nutrient levels, mimicking both
the environment of the animal host and the application route (feed, water, injection, spray), is
essential to determine the potential for therapeutic use as well as shelf -life expectancy [113].
These steps will provide robust preclinical data to support the translation of in vitro data to in
vivo application. Before in vivo application, candidate phage (cocktail) with most desirable in
vitro properties, should be produced at high titre and meet regulatory criteria, purified form
toxins and produced under GMP conditions [108]. Companies may however be reluctant to
carry out large-scale production, until overall cost, including infrastructure and equipment
costs, have decreased enough to make it profitable [114]. Several complex external factors may
influence the application success in vivo, such as rapid clearance of the phages by the animal
host immune system and interactions of phages with other microorganisms [108, 113]. Future
evaluation of phages in vivo will include steps to determine phage safety and [108, 115]. Safety
data should provide information on the impact on other components of the poultry microbiome
and the eventual immunopathological responses. Efficacy data should provide information on
the phages impact on the colonisation of the target bacteria and on the curing effect.
The increase of sequence data of phages, and the improvement of sequence analysis tools as
well as the increasing knowledge on phage properties and phage-host interactions will aid a
more refined translation of in vitro data to in vivo outcomes in future phage therapy
applications.
4.6 Conclusions and future perspectives
This thesis contributes to a better understanding of the coliphage diversity as well as the phage -
host interaction and population dynamics. First, we provided new insight into the diversity of
coliphages in the intestines of poultry, where they live together with the target APEC, and have
established a well-characterised collection of lytic coliphages, some of which could be
candidates for therapeutic application against APEC infections. The coliphage collection can
advantageously be extended, including phages from currently unrepresented genera, such as
Vequintavirinae. Secondly, a growth dynamics in vitro model was combined with two data
mining techniques and quantitative scoring algorithms. This methodology can be used to
rapidly screen for novel phage candidates as part of the initial steps of phage therapy

Chapter 4: General Discussion
197
development. Moreover, future research using this methodology could benefit from linking
specific phage and/or bacterial properties to specific growth dynamics and therapeutic
outcomes. Finally, we identified underlying mechanisms of in vitro phage resistance in APEC
by identifying genes affected by genetic change(s). Among others, these genes included two
genes encoding hypothetical proteins with unknown function, which have never been described
as involved in phage resistance. Considering the prevalence of genetic change(s), the great
associated fitness cost as well as possible involvement in phage-resistance, these genes may be
promising targets for future investigations and targets for therapies against APEC. Finally, the
therapeutic potential of multiple phages (phage cocktail) should be determined and potential
synergistic effects with antibiotics should be explored in vitro as well as in vivo.
The process form phage isolation to successful therapeutic application comprises many steps.
It is clear that currently, we are not able to translate well the in silico to the in vitro data, though
progress is made in the essential first steps in this thesis, namely characterising candidate
phages and their interactions with target pathogenic bacteria. There is still a large need to
extend this work. The phage adhesins need to be better characterised as well as the relevance
of the mutations found in target APEC strains. However, if enough phage receptors are
identified for sequenced phages, bioinformatic approaches might be extended to allow for high-
throughput identification of the phage receptor(s) and bacterial host(s) based on the genomes
of the phage alone. Similarly, essential knowledge on the phage-host interactions may be
obtained using machine learning models and be applied to predict PK/PD in suitable in vivo
models. Continuous model refinement can be applied if the experimental data do not fit the
predicted ones. With increasing well-characterised phage collections housed in reference phage
banks that can rapidly be matched with target bacterium, knowledge gaps in phage research are
being filled and continuous advanced in the field of phage research facilitate future effective
translation into promising therapeutic application.

Chapter 4: General Discussion
198
References
[1] Kathayat, D.; Lokesh, D.; Ranjit, S.; Rajashekara, G. Avian Pathogenic Escherichia Coli (APEC):
An Overview of Virulence and Pathogenesis Factors, Zoonotic Potential, and Control Strategies.
Pathogens, 2021, 10 (4), 467. https://doi.org/10.3390/pathogens10040467.
[2] Huja, S.; Oren, Y.; Trost, E.; Brzuszkiewicz, E.; Biran, D.; Blom, J.; Goesmann, A.; Gottschalk, G.;
Hacker, J.; Ron, E. Z.; et al. Genomic Avenue to Avian Colisepticemia. MBio, 2015, 6 (1), 1–13.
https://doi.org/10.1128/mBio.01681-14.
[3] Zhao, S.; Maurer, J. J.; Hubert, S.; De Villena, J. F.; McDermott, P. F.; Meng, J.; Ayers, S.; English,
L.; White, D. G. Antimicrobial Susceptibility and Molecular Characterization of Avian Pathogenic
Escherichia Coli Isolates. Vet. Microbiol., 2005, 107 (3–4), 215–224.
https://doi.org/10.1016/j.vetmic.2005.01.021.
[4] Wernicki, A.; Nowaczek, A.; Urban-Chmiel, R. Bacteriophage Therapy to Combat Bacterial
Infections in Poultry. Virol. J., 2017, 14 (1), 179. https://doi.org/10.1186/s12985-017-0849-7.
[5] Tang, K. L.; Caffrey, N. P.; Nóbrega, D. B.; Cork, S. C.; Ronksley, P. E.; Barkema, H. W.; Polachek,
A. J.; Ganshorn, H.; Sharma, N.; Kellner, J. D.; et al. Restricting the Use of Antibiotics in Food -
Producing Animals and Its Associations with Antibiotic Resistance in Food-Producing Animals and
Human Beings: A Systematic Review and Meta -Analysis. Lancet Planet. Heal., 2017, 1 (8), e316–
e327. https://doi.org/10.1016/S2542-5196(17)30141-9.
[6] Lin, D. M.; Koskella, B.; Lin, H. C. Phage Therapy: An Alternative to Antibiotics in the Age of
Multi-Drug Resistance. World J. Gastrointest. Pharmacol. Ther., 2017, 8 (3), 162–173.
https://doi.org/10.4292/wjgpt.v8.i3.162.
[7] Brives, C.; Pourraz, J. Phage Therapy as a Potential Solution in the Fight against AMR: Obstacles
and Possible Futures. Palgrave Commun., 2020, 6, 100.
[8] Żbikowska, K.; Michalczuk, M.; Dolka, B. The Use of Bacteriophages in the Poultry Industry.
Animals, 2020, 10 (5), 872. https://doi.org/10.3390/ani10050872.
[9] Dion, M. B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev.
Microbiol., 2020, 12. https://doi.org/10.1038/s41579-019-0311-5.
[10] Clokie, M. R. J.; Millard, A. D.; Letarov, A. V; Heaphy, S. Phages in Nature. Bacteriophage, 2011,
1 (1), 31–45. https://doi.org/10.4161/bact.1.1.14942.
[11] Barylski, J.; Enault, F.; Dutilh, B. E.; Schuller, M. B.; Edwards, R. A.; Gillis, A.; Klumpp, J.;
Knezevic, P.; Krupovic, M.; Kuhn, J. H.; et al. Analysis of Spounaviruses as a Case Study for the
Overdue Reclassification of Tailed Phages. Syst. Biol., 2019, 69 (1), 110–123.
https://doi.org/10.1093/sysbio/syz036.
[12] Hayes, S.; Mahony, J.; Nauta, A.; Van Sinderen, D. Metagenomic Approaches to Assess
Bacteriophages in Various Environmental Niches. Viruses, 2017, 9 (6), 127.
https://doi.org/10.3390/v9060127.
[13] Olsen, N. S.; Kot, W.; Junco, L. M. F.; Hansen, L. H. Exploring the Remarkable Diversity of
Escherichia Coli Phages in the Danish Wastewater Environment, Including 91 Novel Phage Species.
bioRxiv, 2020.
[14] Olsen, N. S.; Hendriksen, N. B.; Hansen, L. H.; Kot, W. A New High-Throughput Screening Method
for Phages: Enabling Crude Isolation and Fast Identification of Diverse Phages with Therapeutic
Potential. Phage, 2020, 1 (3), 137–148. https://doi.org/10.1089/phage.2020.0016.
[15] Krupovic, M.; Turner, D.; Morozova, V.; Dyall, M.; Hanna, S.; Edwards, R.; Dutilh, B. E.; Lehman,
S. M.; Reyes, A.; Baquero, D. P. Bacterial Viruses Subcommittee and Archaeal Viruses
Subcommittee of the ICTV : Update of Taxonomy Changes in 2021. Arch. Virol., 2021, 1–6.
https://doi.org/10.1007/s00705-021-05205-9.

Chapter 4: General Discussion
199
[16] Zrelovs, N.; Dislers, A.; Kazaks, A. Motley Crew: Overview of the Currently Available Phage
Diversity. Front. Microbiol., 2020, 11. https://doi.org/10.3389/fmicb.2020.579452.
[17] Pope, W. H.; Bowman, C. A.; Russell, D. A.; Jacobs-Sera, D.; Asai, D. J.; Cresawn, S. G.; Jacobs,
W. R.; Hendrix, R. W.; Lawrence, J. G.; Hatfull, G. F. Whole Genome Comparison of a Large
Collection of Mycobacteriophages Reveals a Continuum of Phage Genetic Diversity. Elife, 2015, 4,
e06416. https://doi.org/10.7554/elife.06416.
[18] Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth.
Pharmaceuticals, 2019, 12 (1), 35. https://doi.org/10.3390/ph12010035.
[19] Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D. J.; Hobman, J.;
Jones, M. A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-
Scale Biases in the Current Collection of Cultured Phage Genomes. Phage, 2021, 1–10.
https://doi.org/10.1089/phage.2021.0007.
[20] Rohde, C.; Wittmann, J. Phage Diversity for Research and Application. Antibiotics, 2020, 9 (11),
734. https://doi.org/10.3390/antibiotics9110734.
[21] Szymczak, P.; Rau, M. H.; Monteiro, J. M.; Pinho, M. G.; Filipe, S. R.; Vogensen, F. K.; Zeidan, A.
A.; Janzen, T. A Comparative Genomics Approach for Identifying Host -Range Determinants in
Streptococcus Thermophilus Bacteriophages. Sci. Rep., 2019, 9, 7991.
https://doi.org/10.1038/s41598-019-44481-z.
[22] Maffei, E.; Shaidullina, A.; Burkolter, M.; Heyer, Y.; Estermann, F.; Druelle, V.; Sauer, P.; Willi,
L.; Michaelis, S.; Hilbi, H.; et al. Systematic Exploration of Escherichia Coli Phage–Host
Interactions with the BASEL Phage Collection. PLOS Biol., 2021, 19 (11), e3001424.
https://doi.org/10.1371/journal.pbio.3001424.
[23] Korf, I. H. E.; Meier-Kolthoff, J. P.; Adriaenssens, E. M.; Kropinski, A. M.; Nimtz, M.; Rohde, M.;
van Raaij, M. J.; Wittmann, J. Still Something to Discover: Novel Insights into Escherichia Coli
Phage Diversity and Taxonomy. Viruses, 2019, 11 (5), 1–29. https://doi.org/10.3390/v11050454.
[24] Hyman, P.; Abedon, S. T. Bacteriophage Host Range and Bacterial Resistance. In Advances in
Applied Microbiolog; Elsevier Inc., 2010; Vol. 70, pp 217–248. https://doi.org/10.1016/S0065-
2164(10)70007-1.
[25] Nobrega, F. L.; Vlot, M.; de Jonge, P. A.; Dreesens, L. L.; Beaumont, H. J. E.; Lavigne, R.; Dutilh,
B. E.; Brouns, S. J. J. Targeting Mechanisms of Tailed Bacteriophages. Nat. Rev. Microbiol., 2018,
16 (12), 760–773. https://doi.org/10.1038/s41579-018-0070-8.
[26] Grose, J. H.; Casjens, S. R. Understanding the Enormous Diversity of Bacteriophages: The Tailed
Phages That Infect the Bacterial Family Enterobacteriaceae. Virology, 2014, 468, 421–443.
https://doi.org/10.1016/j.virol.2014.08.024.
[27] Abdelsattar, A.; Dawooud, A.; Rezk, N.; Makky, S.; Safwat, A.; Richards, P.; El-Shibiny, A. How
to Train Your Phage: The Recent Efforts in Phage Training. Biologics, 2021, 1 (2), 70–88.
https://doi.org/10.3390/biologics1020005.
[28] Laanto, E.; Hoikkala, V.; Ravantti, J.; Sundberg, L.-R. Long-Term Genomic Coevolution of Host-
Parasite Interaction in the Natural Environment. Nat. Commun., 2017, 8, 111.
https://doi.org/10.1038/s41467-017-00158-7.
[29] Yehl, K.; Lemire, S.; Yang, A. C.; Ando, H.; Mimee, M.; Torres, M. D. T.; de la Fuente-Nunez, C.;
Lu, T. K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail
Fiber Mutagenesis. Cell, 2019, 179 (2), 459-469.e9. https://doi.org/10.1016/j.cell.2019.09.015.
[30] Payne, R. J. H.; Jansen, V. A. A. Pharmacokinetic Principles of Bacteriophage Therapy. Clin.
Pharmacokinet., 2003, 42 (4), 315–325. https://doi.org/10.2165/00003088-200342040-00002.
[31] Dąbrowska, K.; Abedon, S. T. Pharmacologically Aware Phage Therapy: Pharmacodynamic and
Pharmacokinetic Obstacles to Phage Antibacterial Action in Animal and Human Bodies. Microbiol.

Chapter 4: General Discussion
200
Mol. Biol. Rev., 2019, 83 (4), e00012-19. https://doi.org/10.1128/mmbr.00012-19.
[32] Lazeroff, M.; Ryder, G.; Harris, S. L.; Tsourkas, P. K. Phage Commander, an Application for Rapid
Gene Identification in Bacteriophage Genomes Using Multiple Programs. PHAGE, 2021, 00 (00),
1–10. https://doi.org/10.1089/phage.2020.0044.
[33] Mcnair, K.; Zhou, C.; Dinsdale, E. A.; Souza, B.; Edwards, R. A. PHANOTATE: A Novel Approach
to Gene Identification in Phage Genomes. Bioinformatics, 2019, 35 (22), 4537–4542.
https://doi.org/10.1093/bioinformatics/btz265.
[34] Mohanraj, U.; Wan, X.; Spruit, C. M.; Skurnik, M.; Pajunen, M. I. A Toxicity Screening Approach
to Identify Bacteriophage-Encoded Anti-Microbial Proteins. Viruses, 2019, 11 (11), 1057.
https://doi.org/10.3390/v11111057.
[35] Klumpp, J.; Fouts, D. E.; Sozhamannan, S. Bacteriophage Functional Genomics and Its Role in
Bacterial Pathogen Detection. Brief. Funct. Genomics, 2013, 12 (4), 354–365.
https://doi.org/10.1093/bfgp/elt009.
[36] Kasurinen, J.; Spruit, C. M.; Wicklund, A.; Pajunen, M. I.; Skurnik, M. Screening of Bacteriophage
Encoded Toxic Proteins with a Next Generation Sequencing-Based Assay. Viruses, 2021, 13 (5),
750. https://doi.org/10.3390/v13050750.
[37] Ijaq, J.; Malik, G.; Kumar, A.; Das, P. S.; Meena, N.; Bethi, N.; Sundararajan, V. S.; Suravajhala, P.
A Model to Predict the Function of Hypothetical Proteins through a Nine-Point Classification
Scoring Schema. BMC Bioinformatics, 2019, 20 (1), 14. https://doi.org/10.1186/s12859-018-2554-
y.
[38] Zhou, C. L. E.; Kimbrel, J.; Edwards, R.; McNair, K.; Souza, B. A.; Malfatti, S. MultiPhATE2: Code
for Functional Annotation and Comparison of Phage Genomes. G3 Genes, Genomes, Genet., 2021,
11 (5), jkab074. https://doi.org/10.1093/g3journal/jkab074.
[39] Dąbrowska, K. Phage Therapy: What Factors Shape Phage Pharmacokinetics and Bioavailability?
Systematic and Critical Review. Med. Res. Rev., 2019, 39 (5), 2000–2025.
https://doi.org/10.1002/med.21572.
[40] Qadir, M. I.; Mobeen, T.; Masood, A. Phage Therapy: Progress in Pharmacokinetics. Brazilian J.
Pharm. Sci., 2018, 54 (1), e17093. https://doi.org/10.1590/s2175-97902018000117093.
[41] Dhungana, G.; Nepal, R.; Regmi, M.; Malla, R. Pharmacokinetics and Pharmacodynamics of a Novel
Virulent Klebsiella Phage Kp_Pokalde_002 in a Mouse Model. Front. Cell. Infect. Microbiol., 2021,
11, 684704. https://doi.org/10.3389/fcimb.2021.684704.
[42] Lin, Y. W.; Chang, R. Y.; Rao, G. G.; Jermain, B.; Han, M. L.; Zhao, J. X.; Chen, K.; Wang, J. P.;
Barr, J. J.; Schooley, R. T.; et al. Pharmacokinetics/Pharmacodynamics of Antipseudomonal
Bacteriophage Therapy in Rats: A Proof-of-Concept Study. Clin. Microbiol. Infect., 2020, 26 (9),
1229–1235. https://doi.org/10.1016/j.cmi.2020.04.039.
[43] Levin, B. R.; Bull, J. J. Population and Evolutionary Dynamics of Phage Therapy. Nat. Rev.
Microbiol., 2004, 2 (2), 166–173. https://doi.org/10.1038/nrmicro822.
[44] Stone, E.; Campbell, K.; Grant, I.; McAuliffe, O. Understanding and Exploiting Phage–Host
Interactions. Viruses, 2019, 11 (6), 567. https://doi.org/10.3390/v11060567.
[45] Konopacki, M.; Grygorcewicz, B.; Dołęgowska, B.; Kordas, M.; Rakoczy, R. PhageScore: A Simple
Method for Comparative Evaluation of Bacteriophages Lytic Activity. Biochem. Eng. J., 2020, 161,
107652. https://doi.org/10.1016/j.bej.2020.107652.
[46] Storms, Z. J.; Teel, M. R.; Mercurio, K.; Sauvageau, D. The Virulence Index: A Metric for
Quantitative Analysis of Phage Virulence. Phage, 2020, 1 (1), 27–36.
https://doi.org/10.1089/phage.2019.0001.
[47] Xie, Y.; Wahab, L.; Gill, J. J. Development and Validation of a Microtiter Plate-Based Assay for
Determination of Bacteriophage Host Range and Virulence. Viruses, 2018, 10 (4).

Chapter 4: General Discussion
201
https://doi.org/10.3390/v10040189.
[48] Santos, S. B.; Carvalho, C.; Azeredo, J.; Ferreira, E. C. Population Dynamics of a Salmonella Lytic
Phage and Its Host: Implications of the Host Bacterial Growth Rate in Modelling. PLoS One, 2014,
9 (7), e102507. https://doi.org/10.1371/journal.pone.0102507.
[49] Gelman, D.; Yerushalmy, O.; Alkalay-Oren, S.; Rakov, C.; Ben-Porat, S.; Khalifa, L.; Adler, K.;
Abdalrhman, M.; Coppenhagen-Glazer, S.; Aslam, S.; et al. Clinical Phage Microbiology: A
Suggested Framework and Recommendations for the in-Vitro Matching Steps of Phage Therapy.
The Lancet Microbe, 2021, 2 (10), e555–e563. https://doi.org/10.1016/S2666-5247(21)00127-0.
[50] Gayder, S.; Parcey, M.; Nesbitt, D.; Castle, A. J.; Svircev, A. M. Population Dynamics between
Erwinia Amylovora, Pantoea Agglomerans and Bacteriophages: Exploiting Synergy and
Competition to Improve Phage Cocktail Efficacy. Microorganisms, 2020, 8 (9), 1449.
https://doi.org/10.3390/microorganisms8091449.
[51] Korf, I. H. E.; Kittler, S.; Bierbrodt, A.; Mengden, R.; Rohde, C.; Rohde, M.; Kroj, A.; Lehnherr, T.;
Fruth, A.; Flieger, A.; et al. In Vitro Evaluation of a Phage Cocktail Controlling Infect ions with
Escherichia Coli. Viruses, 2020, 12 (12), 1470. https://doi.org/10.3390/v12121470.
[52] Loessner, H.; Schlattmeier, I.; Anders-Maurer, M.; Bekeredjian-Ding, I.; Rohde, C.; Wittmann, J.;
Pokalyuk, C.; Krut, O.; Kamp, C. Kinetic Fingerprinting Links Bacteria-Phage Interactions with
Emergent Dynamics: Rapid Depletion of Klebsiella Pneumoniae Indicates Phage Synergy.
Antibiotics, 2020, 9 (7), 408. https://doi.org/10.3390/antibiotics9070408.
[53] Buckling, A.; Rainey, P. B. Antagonistic Coevolution between a Bacterium and a Bacteriophage.
Proc. R. Soc. B Biol. Sci., 2002, 269 (1494), 931–936. https://doi.org/10.1098/rspb.2001.1945.
[54] Poullain, V.; Gandon, S.; Brockhurst, M. A.; Buckling, A.; Hochberg, M. E. The Evolution of
Specificity in Evolving and Coevolving Antagonistic Interactions between a Bacteria and Its Phage.
Evolution (N. Y)., 2008, 62 (1), 1–11. https://doi.org/10.1111/j.1558-5646.2007.00260.x.
[55] Perry, E. B.; Barrick, J. E.; Bohannan, B. J. M. The Molecular and Genetic Basis of Repeatable
Coevolution between Escherichia Coli and Bacteriophage T3 in a Laboratory Microcosm. PLoS One,
2015, 10 (6), e0130639. https://doi.org/10.1371/journal.pone.0130639.
[56] Holguín, A.; Cárdenas, P.; Prada-Peñaranda, C.; Rabelo Leite, L.; Buitrago, C.; Clavijo, V.; Oliveira,
G.; Leekitcharoenphon, P.; Møller Aarestrup, F.; Vives, M. Host Resistance, Genomics and
Population Dynamics in a Salmonella Enteritidis and Phage System. Viruses, 2019, 11 (2), 188.
https://doi.org/10.3390/v11020188.
[57] Meyer, J. R.; Dobias, D. T.; Weitz, J. S.; Barrick, J. E.; Quick, R. T.; Lenski, R. E. Repeatability and
Contingency in the Evolution of a Key Innovation in Phage Lambda. Science (80-. )., 2012, 335
(6067), 428–432. https://doi.org/10.1126/science.1214449.
[58] Wandro, S.; Oliver, A.; Gallagher, T.; Weihe, C.; England, W.; Martiny, J. B. H.; Whiteson, K.
Predictable Molecular Adaptation of Coevolving Enterococcus Faecium and Lytic Phage EfV12 -
Phi1. Front. Microbiol., 2019, 9, 3192. https://doi.org/10.3389/fmicb.2018.03192.
[59] Hall, A. R.; Scanlan, P. D.; Buckling, A. Bacteria‐phage Coevolution and the Emergence of
Generalist Pathogens. Am. Nat., 2011, 177 (1), 44–53. https://doi.org/10.1086/657441.
[60] Chan, B. K.; Abedon, S. T.; Loc-Carrillo, C. Phage Cocktails and the Future of Phage Therapy.
Future Microbiol., 2013, 8 (6), 769–783. https://doi.org/10.2217/fmb.13.47.
[61] Naghizadeh, M.; Torshizi, M. A. K.; Rahimi, S.; Dalgaard, T. S. Synergistic Effect of Phage Therapy
Using a Cocktail Rather than a Single Phage in the Control of Severe Colibacillosis in Quails. Poult.
Sci., 2019, 98 (2), 653–663. https://doi.org/10.3382/ps/pey414.
[62] Betts, A.; Gifford, D. R.; MacLean, R. C.; King, K. C. Parasite Diversity Drives Rapid Host
Dynamics and Evolution of Resistance in a Bacteria-Phage System. Evolution (N. Y)., 2016, 70 (5),
969–978. https://doi.org/10.1111/evo.12909.

Chapter 4: General Discussion
202
[63] Duyvejonck, H.; Merabishvili, M.; Pirnay, J.; Vos, D. De; Simaey, L. Van; Vermeulen, S.; Mechelen,
E. Van; Vaneechoutte, M. Development of a QPCR Platform for Quantification of the Five
Bacteriophages Within. Sci. Rep., 2019, 9, 13893. https://doi.org/10.1038/s41598-019-50461-0.
[64] Hampton, H. G.; Watson, B. N. J.; Fineran, P. C. The Arms Race between Bacteria and Their Phage
Foes. Nature, 2020, 577 (7790), 327–336. https://doi.org/10.1038/s41586-019-1894-8.
[65] Dy, R. L.; Richter, C.; Salmond, G. P. C.; Fineran, P. C. Remarkable Mechanisms in Microbes to
Resist Phage Infections. Annu. Rev. Virol., 2014, 1 (1), 307–331. https://doi.org/10.1146/annurev-
virology-031413-085500.
[66] van Houte, S.; Buckling, A.; Westra, E. R. Evolutionary Ecology of Prokaryotic Immune
Mechanisms. Microbiol. Mol. Biol. Rev., 2016, 80 (3), 745–763.
https://doi.org/10.1128/mmbr.00011-16.
[67] Lopatina, A.; Tal, N.; Sorek, R. Abortive Infection: Bacterial Suicide as an Antiviral Immune
Strategy. Annu. Rev. Virol., 2020, 7 (1), 371–384. https://doi.org/10.1146/annurev-virology-011620-
040628.
[68] Azam, A. H.; Tanji, Y. Bacteriophage-Host Arm Race: An Update on the Mechanism of Phage
Resistance in Bacteria and Revenge of the Phage with the Perspective for Phage Therapy. Appl.
Microbiol. Biotechnol., 2019, 103, 2121–2131. https://doi.org/10.1007/s00253-019-09629-x.
[69] Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D. a;
Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science (80-. ).,
2007, 315 (5819), 1709–1712. https://doi.org/10.1126/science.1138140.
[70] Picton, D. M.; Luyten, Y. A.; Morgan, R. D.; Nelson, A.; Smith, D. L.; Dryden, D. T. F.; Hinton, J.
C. D.; Blower, T. R. The Phage Defence Island of a Multidrug Resistant Plasmid Uses Both BREX
and Type IV Restriction for Complementary Protection from Viruses. Nucleic Acids Res., 2021,
gkab906. https://doi.org/10.1093/nar/gkab906.
[71] Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R.
Systematic Discovery of Antiphage Defense Systems in the Microbial Pangenome. Science (80-. ).,
2018, 359 (6379), eaar4120. https://doi.org/10.1126/science.aar4120.
[72] Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy.
Viruses, 2018, 10 (7). https://doi.org/10.3390/v10070351.
[73] Pires, D. P.; Costa, A. R.; Pinto, G.; Meneses, L.; Azeredo, J. Current Challenges and Future
Opportunities of Phage Therapy. FEMS Microbiol. Rev., 2020, fuaa017, 1–17.
https://doi.org/10.1093/femsre/fuaa017.
[74] Oechslin, F.; Piccardi, P.; Mancini, S.; Gabard, J.; Moreillon, P.; Entenza, J. M.; Resch, G.; Que, Y.
A. Synergistic Interaction between Phage Therapy and Antibiotics Clears Pseudomonas Aeruginosa
Infection in Endocarditis and Reduces Virulence. J. Infect. Dis., 2017, 215 (5), 703–712.
https://doi.org/10.1093/infdis/jiw632.
[75] Labrie, S. J.; Samson, J. E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev.
Microbiol., 2010, 8 (5), 317–327. https://doi.org/10.1038/nrmicro2315.
[76] Alseth, E. O.; Pursey, E.; Luján, A. M.; McLeod, I.; Rollie, C.; Westra, E. R. Bacterial Biodiversity
Drives the Evolution of CRISPR-Based Phage Resistance. Nature, 2019, 574 (7779), 549–552.
https://doi.org/10.1038/s41586-019-1662-9.
[77] Andersson, A. F.; Banfield, J. F. Virus Population Dynamics and Acquired Virus Resistance in
Natural Microbial Communities. Science (80-. )., 2008, 320 (5879), 1047–1050.
https://doi.org/10.1126/science.1157358.
[78] Mangalea, M. R.; Duerkop, B. A. Fitness Trade-Offs Resulting from Bacteriophage Resistance
Potentiate Synergistic Antibacterial Strategies. Infect. Immun., 2020, 88 (7), e00926-19.
https://doi.org/10.1128/IAI.00926-19.

Chapter 4: General Discussion
203
[79] Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K. A.; Tomita, M.;
Wanner, B. L.; Mori, H. Construction of Escherichia Coli K-12 in-Frame, Single-Gene Knockout
Mutants: The Keio Collection. Mol. Syst. Biol., 2006, 2, 2006.0008.
https://doi.org/10.1038/msb4100050.
[80] Qimron, U.; Marintcheva, B.; Tabor, S.; Richardson, C. C. Genomewide Screens for Escherichia
Coli Genes Affecting Growth of T7 Bacteriophage. Proc. Natl. Acad. Sci. U. S. A., 2006, 103 (50),
19039–19044. https://doi.org/10.1073/pnas.0609428103.
[81] Maynard, N. D.; Birch, E. W.; Sanghvi, J. C.; Chen, L.; Gutschow, M. V.; Covert, M. W. A Forward-
Genetic Screen and Dynamic Analysis of Lambda Phage Host-Dependencies Reveals an Extensive
Interaction Network and a New Anti-Viral Strategy. PLoS Genet., 2010, 6 (7), e1001017.
https://doi.org/10.1371/journal.pgen.1001017.
[82] Kortright, K. E.; Chan, B. K.; Turner, P. E. High-Throughput Discovery of Phage Receptors Using
Transposon Insertion Sequencing of Bacteria. Proc. Natl. Acad. Sci. U. S. A., 2020, 117 (31), 18670–
18679. https://doi.org/10.1073/pnas.2001888117.
[83] Borin, J. M.; Avrani, S.; Barrick, J. E.; Petrie, K. L.; Meyer, J. R. Coevolutionary Phage Training
Leads to Greater Bacterial Suppression and Delays the Evolution of Phage Resistance. Proc. Natl.
Acad. Sci. U. S. A., 2021, 118 (23), e2104592118. https://doi.org/10.1073/pnas.2104592118.
[84] Samson, J. E.; Magadán, A. H.; Sabri, M.; Moineau, S. Revenge of the Phages: Defeating Bacterial
Defences. Nat. Rev. Microbiol., 2013, 11 (10), 675–687. https://doi.org/10.1038/nrmicro3096.
[85] Weigele, P.; Raleigh, E. A. Biosynthesis and Function of Modified Bases in Bacteria and Their
Viruses. Chem. Rev., 2016, 116 (20), 12655–12687. https://doi.org/10.1021/acs.chemrev.6b00114.
[86] Łobocka, M.; Dąbrowska, K.; Górski, A. Engineered Bacteriophage Therapeutics: Rationale,
Challenges and Future. BioDrugs, 2021, 35 (3), 255–280. https://doi.org/10.1007/s40259-021-
00480-z.
[87] Bretaudeau, L.; Tremblais, K.; Aubrit, F.; Meichenin, M.; Arnaud, I. Good Manufacturing Practice
(GMP) Compliance for Phage Therapy Medicinal Products. Front. Microbiol., 2020, 11, 1161.
https://doi.org/10.3389/fmicb.2020.01161.
[88] Loponte, R.; Pagnini, U.; Iovane, G.; Pisanelli, G. Phage Therapy in Veterinary Medicine.
Antibiotics, 2021, 10 (4), 421. https://doi.org/10.3390/antibiotics10040421.
[89] Tsonos, J.; Oosterik, L. H.; Tuntufye, H. N.; Klumpp, J.; Butaye, P.; De Greve, H.; Hernalsteens, J.
P.; Lavigne, R.; Goddeeris, B. M. A Cocktail of in Vitro Efficient Phages Is Not a Guarantee for in
Vivo Therapeutic Results against Avian Colibacillosis. Vet. Microbiol., 2014.
https://doi.org/10.1016/j.vetmic.2013.10.021.
[90] Kittler, S.; Mengden, R.; Korf, I. H. E.; Bierbrodt, A.; Wittmann, J.; Plötz, M.; Jung, A.; Lehnherr,
T.; Rohde, C.; Lehnherr, H.; et al. Impact of Bacteriophage-Supplemented Drinking Water on the E.
Coli Population in the Chicken Gut. Pathogens, 2020, 9 (4), 293.
https://doi.org/10.3390/pathogens9040293.
[91] Chibani-Chennoufi, S.; Sidoti, J.; Bruttin, A.; Kutter, E.; Sarker, S.; Brüssow, H. In Vitro and in
Vivo Bacteriolytic Activities of Escherichia Coli Phages: Implications for Phage Therapy.
Antimicrob. Agents Chemother., 2004, 48 (7), 2558–2569. https://doi.org/10.1128/AAC.48.7.2558-
2569.2004.
[92] Jessup, C. M.; Bohannan, B. J. M. The Shape of an Ecological Trade-off Varies with Environment.
Ecol. Lett., 2008, 11 (9), 947–959. https://doi.org/10.1111/j.1461-0248.2008.01205.x.
[93] Vale, P. F.; Lafforgue, G.; Gatchitch, F.; Gardan, R.; Moineau, S.; Gandon, S. Costs of CRISPR -
Cas-Mediated Resistance in Streptococcus Thermophilus. Proc. R. Soc. B Biol. Sci., 2015, 282
(1812), 20151270. https://doi.org/10.1098/rspb.2015.1270.
[94] Bradde, S.; Mora, T.; Walczak, A. M. Cost and Benefits of Clustered Regularly Interspaced Short

Chapter 4: General Discussion
204
Palindromic Repeats Spacer Acquisition. Philos. Trans. R. Soc. B Biol. Sci., 2019, 374 (1772).
https://doi.org/10.1098/rstb.2018.0095.
[95] Gurney, J.; Brown, S. P.; Kaltz, O.; Hochberg, M. E. Steering Phages to Combat Bacterial Pathogens.
Trends Microbiol., 2020, 28 (2), 85–94. https://doi.org/10.1016/j.tim.2019.10.007.
[96] Ricci, V.; Piddock, L. J. V. Exploiting the Role of TolC in Pathogenicity: Identificatio n of a
Bacteriophage for Eradication of Salmonella Serovars from Poultry. Appl. Environ. Microbiol.,
2010, 76 (5), 1704–1706. https://doi.org/10.1128/AEM.02681-09.
[97] Chan, B. K.; Sistrom, M.; Wertz, J. E.; Kortright, K. E.; Narayan, D.; Turner, P. E. Phage Selection
Restores Antibiotic Sensitivity in MDR Pseudomonas Aeruginosa. Sci. Rep., 2016, 6, 26717.
https://doi.org/10.1038/srep26717.
[98] León, M.; Bastías, R. Virulence Reduction in Bacteriophage Resistant Bacteria. Front. Microbiol.,
2015, 6 (343), 1–7. https://doi.org/10.3389/fmicb.2015.00343.
[99] Meaden, S.; Paszkiewicz, K.; Koskella, B. The Cost of Phage Resistance in a Plant Pathogenic
Bacterium Is Context-Dependent. Evolution (N. Y)., 2015, 69 (5), 1321–1328.
https://doi.org/10.1111/evo.12652.
[100] Westra, E. R.; Van houte, S.; Oyesiku-Blakemore, S.; Makin, B.; Broniewski, J. M.; Best, A.; Bondy-
Denomy, J.; Davidson, A.; Boots, M.; Buckling, A. Parasite Exposure Drives Selective Evolution of
Constitutive versus Inducible Defense. Curr. Biol., 2015, 25 (8), 1043–1049.
https://doi.org/10.1016/j.cub.2015.01.065.
[101] Tagliaferri, T. L.; Jansen, M.; Horz, H. P. Fighting Pathogenic Bacteria on Two Fronts: Phages and
Antibiotics as Combined Strategy. Front. Cell. Infect. Microbiol., 2019, 9, 22.
https://doi.org/10.3389/fcimb.2019.00022.
[102] He, S.; He, H.; Chen, Y.; Chen, Y.; Wang, W.; Yu, D. In Vitro and in Vivo Analysis of Antimicrobial
Agents Alone and in Combination against Multi-Drug Resistant Acinetobacter Baumannii. Front.
Microbiol., 2015, 6, 507. https://doi.org/10.3389/fmicb.2015.00507.
[103] Liu, C. G.; Green, S. I.; Min, L.; Clark, J. R.; Salazar, K. C.; Terwilliger, A. L.; Kaplan, H. B.;
Trautner, B. W.; Ramig, R. F.; Maresso, A. W. Phage-Antibiotic Synergy Is Driven by a Unique
Combination of Antibacterial Mechanism of Action and Stoichiometry. MBio, 2020, 11 (4), e01462-
20. https://doi.org/10.1128/mBio.01462-20.
[104] Torres-Barceló, C.; Hochberg, M. E. Evolutionary Rationale for Phages as Complements of
Antibiotics. Trends Microbiol., 2016, 24 (4), 249–256. https://doi.org/10.1016/j.tim.2015.12.011.
[105] Segall, A. M.; Roach, D. R.; Strathdee, S. A. Stronger Together? Perspectives on Phage-Antibiotic
Synergy in Clinical Applications of Phage Therapy. Curr. Opin. Microbiol., 2019, 51, 46–50.
https://doi.org/10.1016/j.mib.2019.03.005.
[106] Bull, J. J.; Gill, J. J. The Habits of Highly Effective Phages: Population Dynamics as a Framework
for Identifying Therapeutic Phages. Front. Microbiol., 2014, 5 (618).
https://doi.org/10.3389/fmicb.2014.00618.
[107] Fernández, L.; Gutiérrez, D.; García, P.; Rodríguez, A. The Perfect Bacteriophage for Therapeutic
Applications—A Quick Guide. Antibiotics, 2019, 8 (3), 126.
https://doi.org/10.3390/antibiotics8030126.
[108] Nale, J. Y.; Clokie, M. R. Preclinical Data and Safety Assessment of Phage Therapy in Humans.
Curr. Opin. Biotechnol., 2021, 68, 310–317. https://doi.org/10.1016/j.copbio.2021.03.002.
[109] Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages.
Front. Microbiol., 2016, 7, 1352. https://doi.org/10.3389/fmicb.2016.01352.
[110] Benala, M.; Vaiyapuri, M.; Visnuvinayagam, S.; George, J. C.; Raveendran, K.; George, I.;
Mothadaka, M. P.; Badireddy, M. R. A Revisited Two-Step Microtiter Plate Assay: Optimization of
in Vitro Multiplicity of Infection (MOI) for Coliphage and Vibriophage. J. Virol. Methods, 2021,

Chapter 4: General Discussion
205
294, 114177. https://doi.org/10.1016/j.jviromet.2021.114177.
[111] Pinto, A. M.; Faustino, A.; Pastrana, L. M.; Bañobre-López, M.; Sillankorva, S. Pseudomonas
Aeruginosa PAO 1 In Vitro Time–Kill Kinetics Using Single Phages and Phage Formulations—
Modulating Death, Adaptation, and Resistance. Antibiotics, 2021, 10 (7), 877.
https://doi.org/10.3390/antibiotics10070877.
[112] Bryson, K. J.; Garrido, D.; Esposito, M.; McLachlan, G.; Digard, P.; Schouler, C.; Guabiraba, R.;
Trapp, S.; Vervelde, L. Precision Cut Lung Slices: A Novel Versatile Tool to Examine Host -
Pathogen Interaction in the Chicken Lung. Vet. Res., 2020, 51, 2. https://doi.org/10.1186/s13567-
019-0733-0.
[113] Jończyk-Matysiak, E.; Łodej, N.; Kula, D.; Owczarek, B.; Orwat, F.; Międzybrodzki, R.; Neuberg,
J.; Bagińska, N.; Weber-Dąbrowska, B.; Górski, A. Factors Determining Phage Stability/Activity:
Challenges in Practical Phage Application. Expert Rev. Anti. Infect. Ther., 2019, 17 (8), 583–606.
https://doi.org/10.1080/14787210.2019.1646126.
[114] García, R.; Latz, S.; Romero, J.; Higuera, G.; García, K.; Bastías, R. Bacteriophage Production
Models: An Overview. Front. Microbiol., 2019, 10, 1187.
https://doi.org/10.3389/fmicb.2019.01187.
[115] Mosimann, S.; Desiree, K.; Ebner, P. Efficacy of Phage Therapy in Poultry: A Systematic Review
and Meta-Analysis. Poult. Sci., 2021, 100 (12), 101472. https://doi.org/10.1016/j.psj.2021.101472.

206

Summary
207
Summary
Excess use of antimicrobials and release into the environment for over half a century have
generated a constant selective pressure for resistant bacterial strains. Consequently, we are
facing a worldwide antibiotic resistance challenge with increasing numbers of bacterial
infection becoming difficult to treat once again. Avian pathogenic Escherichia coli (APEC) is
one of the leading pathogens affecting poultry worldwide, and various multi-drug resistant
strains have been isolated. APEC strains with O-serogroup O1, O2, or O78 have been shown
to cause the majority of infections. The advances in the therapeutic use of the bacterial viruses,
bacteriophages (phages), have highlighted their potential use as an alternative or supplement
treatment against bacterial pathogens. However, we are only just beginning to understand the
diversity of phages, and the use of phages in therapy requires a detailed characterisation of the
candidate phages prior to their application to ensure that they have the expected potential to
kill pathogenic bacteria and have therapeutic effects, while minimising negative environmental
modifications. The understanding of the phage-host interactions has shown to be essential for
the development and application of a successful phage therapy. Th is PhD dissertation
contributes to our understanding of E. coli-infecting phage (coliphage)-host interaction for
phage therapy against APEC by determine in vitro growth dynamics as well as underlying
mechanism of phage resistance.

Summary
208
Phages are the most abundant organisms on Earth and can be found in every explored
ecosystem. In recent years, advances in sequencing and bioinformatics have broadened our
understanding of phage diversity, taxonomy, host-specificity, population structure and
genomic evolution. In chapter 3.1, we characterised coliphages isolated from poultry faeces.
The characterisation included phenotypic characterisation of the phage morphology and
genetic analysis of the phage genome. All isolated coliphages belonged to the Caudovirales
order (tailed phages), which comprise ~96% of the phages isolated to date. Phylogenetic
analysis based on conserved “marker” genes as well as full draft genomes was performed and
grouped phages according to genome size, G+C content and, phage subfamily. While the
phages were isolated from an otherwise similar ecosystem, a great diversity was observed
amongst them highlighting the mosaic nature of the phage genomes as well as the continuous
evolution. In accordance with previous studies, most of the phage genes products were
annotated as hypothetical proteins of unknown function. However, for all phages no genes
encoding known virulence- or antibiotic resistance- associated genes as well as other undesired
characteristics (for phage therapy) were detected.
Phage therapy represents a supportive to antibiotics to control bacterial pathogens. However,
current research indicates that there are still shortcomings in our understanding of phage
therapy as the in vitro results do not always correlate with the in vivo results. It has become
clear that knowledge on the phage-host interaction is essential to select and/or construct phages
with the desired host spectrum and activity, and hereby a more reliable in vitro and in vivo
outcome. Therefore, in chapter 3.2, we established an in vitro model to determine the
coliphage-host interaction and population growth dynamics. The coliphages (characterised in
chapter 3.1) were co-cultured with each of 10 APEC strains with O-serogroup O1, O2, or O78.
Growth dynamics were classified based on optical density (OD) of bacterial growth using a
combined exploratory and statistical approach. Growth dynamic patterns were defined as
resistant, susceptible, or in-between. Various factors affecting the phage pharmacokinetics
(PK) and pharmacodynamics (PD) have been described using mathematical and experimental
models. In this study, the influence of the three parameters: phage type, APEC strain, and
multiplicity of infection (MOI) were determined, identifying the MOI as the factor with the
least significant effect. In accordance with previous finding, different phage types showed
different host range, Tevenvirinae exhibiting the broadest and Ounavirinae not able to infect
any of the APEC strains. The established in vitro model was not only used to gain a better

Summary
209
understanding of the phage PK/PD but also provided a fast quantitative screening method for
candidate phages against a target bacterial pathogen.
One of the major concerns of phage therapy is the emergence of phage-resistant bacterial
mutants. Bacteria can develop resistance against phage through various mechanisms, including
modification of phage receptor-encoding genes and innate immune systems (such as CRISPR-
Cas), each associated with a different level of fitness cost for the bacterial strain. In chapter
3.3 we determined the factors involved in APEC in vitro phage resistance. Spontaneous phage
resistant strains were obtained from liquid co-cultures of the susceptible combination of
virulent Tevenvirinae coliphages and O1 APEC strain (determined in chapter 3.2). Whole-
genome sequence (WGS) analysis revealed that one or more single nucleotide polymorphisms
(SNPs) were detected in the bacterial core genome and/or that one or more genes were lost
(partial or complete). Genes affected by these genetic changes included genes known to be
involved in phage resistance through adsorption inhibition, including outer membrane protein
A (OmpA), lipopolysaccharide (LPS)-, O-antigen-, or cell wall-related genes as well as genes
not previously linked to phage resistance, including several hypothetical genes. Using bacterial
growth as an indicator, we determined the fitness cost associated with the genetic change(s)
detected. For several phage resistant mutants decrease (up to 65%) in overall growth was
detected. However, the magnitude of such fitness cost may vary in more complex in vivo
environment. Interestingly, genetic changes in genes encoding hypothetical proteins with
unknown function were one of the most prevalent and associated with a great fitness cost. As
such, these genes could serve as targets in future studies and potentially be exploited in phage
therapy.
In conclusion, this thesis provided novel insights into the coliphage diversity in the intestine
of poultry as well as the overall coliphage diversity. Moreover, a well-characterised collection
of coliphages was established, some of which with desired properties for phage therapy. By
studying the phage-host (APEC) interaction, much needed knowledge essential for a better
understanding of growth dynamics and the underlying mechanisms of phage resistance was
obtained. Although the full complexity of the interactions cannot be captured in vitro, this
knowledge is essential for the development of a more reliable, and hereby, a future successful
phage therapy against APEC.

210

Samenvatting
211
Samenvatting
Overmatig gebruik van antimicrobiële middelen en het vrijkomen van deze producten in de
omgeving gedurende meer dan een halve eeuw, hebben geleid tot een constante selectiedruk
voor resistente bacteriën. Bijgevolg worden we wereldwijd steeds vaker geconfronteerd met
de uitdagingen die met antibioticaresistentie gepaard gaan en waarbij een toenemend aantal
bacteriële infecties moeilijk te behandelen wordt. Aviaire pathogene Escherichia coli (APEC)
is wereldwijd een van de belangrijkste bacteriële pathogenen in pluimvee en er zijn reeds
verschillende multiresistente stammen geïsoleerd. De meerderheid van deze infecties zou
veroorzaakt worden door APEC-stammen met O-serogroep O1, O2 of O78. De vooruitgang in
het therapeutisch gebruik van bacteriële virussen of bacteriofagen (fagen), hebben duidelijk
potentieel als alternatieve of aanvullende behandeling tegen bacteriële pathogenen. We
beginnen de diversiteit van deze fagen echter nog maar net ten volle te begrijpen. Voorafgaand
aan hun toepassing, vereist het gebruik van fagen in therapie een gedetailleerde karakterisering
van de kandidaat-fagen. Dit is nodig om ervoor te zorgen dat ze hun potentieel om pathogene
bacteriën te doden en therapeutisch gunstige effecten te veroorzaken, ten volle kunnen benutten
en negatieve effecten op het milieu worden beperkt. Het verstaan van de faag-gastheer
interacties is essentieel gebleken voor de ontwikkeling en toepassing van een succesvolle

Samenvatting
212
faagtherapie. Deze doctoraatsstudie vergroot ons inzicht in de interactie tussen de E. coli-
infecterende faag (colifaag) en de gastheer voor faagtherapie tegen APEC, met behulp van in
vitro groeidynamieken, alsook door het onderliggende mechanisme van faagresistentie te
bepalen.
Fagen zijn de meest voorkomende organismen op Aarde en zijn te vinden in elk ecosysteem.
In de afgelopen jaren hebben vooruitgangen in sequenering en bio-informatica ervoor gezorgd
dat we faagdiversiteit, taxonomie, gastheerspecificiteit, populatiestructuur en genomische
evoluties nog beter verstaan. In hoofdstuk 3.1 werden colifagen gekarakteriseerd die werden
geïsoleerd uit uitwerpselen van pluimvee. De karakterisering omvatte fenotypische
karakterisering van de faagmorfologie en genetische analyse van het faaggenoom. Alle
geïsoleerde colifagen behoorden tot de orde van de Caudovirales (staartfagen), die ~96% van
de tot nu toe geïsoleerde fagen omvat. Met behulp van fylogenetische analyse op basis van
geconserveerde merker-genen en volledige conceptgenomen werden fagen gegroepeerd
volgens genoomgrootte, G+C-gehalte en faagsubfamilie. Hoewel de fagen werden geïsoleerd
uit een gelijkaardig ecosysteem, werd er een grote diversiteit onder hen waargenomen, wat
wijst op de mozaïek-structuur van de faaggenomen en een continue evolutie. In
overeenstemming met eerdere studies werden de meeste faag genenproducten geannoteerd als
hypothetische eiwitten met onbekende functie. Bij geen enkele van de bestudeerde fagen
werden echter genen gedetecteerd die coderen voor bekende virulentie- of
antibioticaresistentie-geassocieerde genen, evenals andere ongewenste kenmerken (voor
faagtherapie).
Faagtherapie ondersteunt antibiotica om bacteriële pathogenen onder controle te houden.
Huidig onderzoek geeft echter aan dat we faagtherapie nog altijd niet ten volle begrijpen,
aangezien de in vitro resultaten niet altijd correleren met de in vivo resultaten. Kennis over de
faag-gastheer interactie is essentieel om fagen met het gewenste gastheerspectrum en -activiteit
te selecteren en/of te construeren, en hiermee een betrouwbaarder in vitro en in vivo resultaat
te genereren. Daarom werd er in hoofdstuk 3.2 een in vitro model opgesteld om de interactie
tussen colifaag en gastheer en de populatiegroei dynamiek te bepalen. De colifagen (besproken
in hoofdstuk 3.1) werden samen opgekweekt met elk van de 10 APEC-stammen met O-
serogroep O1, O2 of O78. De groeidynamiek werd geclassificeerd op basis van optische
dichtheid (OD) van bacteriegroei met behulp van een gecombineerde verkennende en
statistische benadering. Dynamische groeipatronen werden gedefinieerd als resistent, vatbaar
of daartussenin. Verschillende factoren die de farmacokinetiek van faag (PK) en

Samenvatting
213
farmacodynamiek (PD) beïnvloeden, werden beschreven met behulp van wiskundige en
experimentele modellen. In deze studie werd de invloed van de drie parameters: faagtype,
APEC-stam en multipliciteit van infectie (MOI) bepaald, waarbij de MOI werd geïdentificeerd
als de factor met slechts weinig of geen effect. In overeenstemming met eerdere bevindingen
vertoonden verschillende faagtypen verschillende mogelijke gastheren. Tevenvirinae
vertoonde het grootste gastheerbereik en Ounavirinae waren niet in staat om één van de APEC-
stammen te infecteren. Het gebuikte in vitro model werd niet alleen gebruikt om een beter
begrip te verkrijgen van de faag PK/PD, maar bood ook een snelle kwantitatieve
screeningsmethode voor kandidaat-fagen tegen een bacteriële doelwitpathogeen.
Opkomst van faagresistente bacteriële mutanten is momenteel één van de grootste zorgen.
Bacteriën kunnen resistentie tegen fagen ontwikkelen via verschillende mechanismen,
waaronder modificatie van genen die faagreceptoren coderen en via hun aangeboren
immuunsysteem (zoals CRISPR-Cas), elke aanpassing resulteert in een eigen fitnesskost voor
de bacteriestam. In hoofdstuk 3.3 werden de factoren bepaald die betrokken zijn bij de in vitro
faagresistentie van APEC. Spontane faagresistente stammen werden verkregen uit vloeibare
co-culturen van de gevoelige combinatie van virulente Tevenvirinae-colifagen en O1 APEC-
stam (bepaald in hoofdstuk 3.2). Via whole-genome sequence (WGS)-analyse werden een of
meer single nucleotide polymorphisms (SNP's) gedetecteerd in het bacteriële kerngenoom
en/of werd ontdekt dat een of meerdere genen verloren gingen (gedeeltelijk of volledig). Genen
die bij deze veranderingen betrokken zijn, omvatten genen waarvan bekend is dat ze betrokken
zijn bij faagresistentie door middel van adsorptieremming, waaronder buitenmembraaneiwit A
(OmpA), lipopolysaccharide (LPS)-, O-antigeen- of celwandgerelateerde genen, evenals genen
die voorheen niet gelinkt werden aan faagresistentie, waaronder verschillende hypothetische
genen. Met behulp van bacteriële groei als indicator werd de “fitnesscost” bepaald die gelinkt
is aan deze gedetecteerde genetische verandering(en). Voor verschillende faagresistente
mutanten werd een verminderde (tot 65%) totale groei gedetecteerd. De omvang van dergelijke
“fitnesscost” kan echter verschillen in een complexere in vivo-omgeving. Interessant is dat
veranderingen in genen die coderen voor hypothetische eiwitten met onbekende functie, een
van de meest voorkomende waren en dat deze ook geassocieerd werden met een hoge
“fitnesscost”. Deze genen kunnen daarom verder onderzocht worden in toekomstige studies en
mogelijks ook benut worden bij faagtherapie.
Ter conclusie kan er gesteld worden dat er nieuwe inzichten zijn verschaft in de
colifagendiversiteit in de darmen van pluimvee en de algehele colifagendiversiteit. Bovendien

Samenvatting
214
werd een goed gekarakteriseerde collectie colifagen bekomen, waarvan sommige met gewenste
eigenschappen voor faagtherapie. Door de faag-gastheer (APEC) interactie te bestuderen, werd
de nodige kennis verkregen die essentieel is voor een beter begrip van de groeidynamiek en de
onderliggende mechanismen van faagresistentie. Hoewel de volledige complexiteit van de
interacties niet in vitro kan worden vastgelegd, is deze kennis essentieel voor de ontwikkeling
van een meer betrouwbare en daarmee een toekomstige succesvolle faagtherapie tegen APEC.

Curriculum Vitae
215
Curriculum Vitae
Patricia Espenhain Sørensen was born on July 4, 1992 in Gentofte, Denmark. In 2018, she
obtained a Master of Science degree in Biology-Biotechnology from University of
Copenhagen, Denmark. From June 2018, she joined the Department of Pathology,
Bacteriology and Poultry Diseases at the Faculty of Veterinary Medicine as a doctoral student,
where she spent three years researching the interactions of bacteriophages and avian pathogenic
Escherichia coli (APEC). She is the first author of three publications in international journals
and has presented her research results at international conferences.
The research project is a part of the EU’s ambitious Horizon 2020 Marie Skłodowska -Curie
Initial Training Network: Combatting Antimicrobial Resistance Training Network
(CARTNET), and is conducted in a collaboration between Ghent University, University of
Copenhagen, Denmark, Ross University School of Veterinary Medicine (RUSVM), St. Kitts,
and Statens Serum Institut, Denmark.

216

Bibliography
217
Bibliography
Sørensen, P.E., Van Den Broeck, W., Kiil, K., Jasinskyte, J., Moodley, A., Garmyn, A.,
Ingmer, H., and P. Butaye. 2020. New insights into the biodiversity of coliphages in the
intestine of poultry. Sci Rep 10, 15220. DOI: 10.1038/s41598-020-72177-2.
Sørensen, P. E., Ng, D., Duchateau, L., Ingmer, H., Garmyn, A., and P. Butaye. 2021.
Classification of in vitro phage–host population growth dynamics. Microorganisms 9, 2470.
DOI: 10.3390/microorganisms9122470
Sørensen, P. E., Baig, S., Stegger, M., Ingmer, H., Garmyn, A., and P. Butaye. 2021.
Spontaneous phage resistance in avian pathogenic Escherichia coli. Front. Microbiol. 12,
782757. DOI: 10.3389/fmicb.2021.782757

218

Conference contributions
219
Conference contributions
ASM Microbe 2020 Online
Sørensen, P.E., Garmyn, A., Ingmer, H., and P. Butaye. Dynamics of Bacteriophage-Host
Interactions. ePoster presentation. July 20, 2020.
Sørensen, P.E., Garmyn, A., Kiil, K., Jasinskyte, D., Moodley, A., Ingmer, H., and P. Butaye.
Novel Insights into the Biodiversity of Coliphages in the Intestine of Poultry. ePoster
presentation. July 20, 2020.
World Microbe Forum 2021 (online)
Sørensen, P.E., Baig, S., Stegger, M., Garmyn, A., Ingmer, H., and P. Butaye. Phage-Host
Interactions: In Vitro Generated E. coli Phage Resistance . iPoster presentation. June 20, 2021.

220

Acknowledgements
221
Acknowledgements
The past three and a half years have been quite a journey, which ultimately resulted in this
dissertation. All of this would not have been possible without the help and support of many
different people. I would like to thank everyone that contributed, directly or indirectly, to me
achieving my doctoral degree.
First and foremost, I would like to thank my three supervisors. Prof. dr. Patrick Butaye, thank
you for giving me the opportunity to do this PhD, and for all your guidance, patience, and
encouragement. Prof. dr. An Garmyn. Thank you for always making time for my questions and
for reading my reports and manuscripts quickly and thoroughly and for the valuable feedback.
Prof. dr. Hanne Ingmer, despite the physical distance during my PhD, you have always showed
interest in my work. Thank you for this, and for giving me support when I needed it.
I would also like to thank the members of my examination committee, chair Prof. dr. Niek
Sanders, Prof. dr. Gunther Antonissen, Dr. Ilias Chantziaras, dr. Steven Van Borm, Prof. dr.
Rob Lavigne, and Prof. dr. Felix Toka. Thank you for taking your time to read this thesis, for

Acknowledgements
222
your genuine interest, constructive questions, and valuable suggestions to improve this
dissertation.
A special thanks to the co-authors of my publications, Dziuginta, Arshnee, Luc, Marc, Sharmin,
Duncan, and Kristoffer. Thank you for all your assistance and good collaborations.
I want to thank the CARTNET consortium for the great training and network meetings and for
staying connected even when a worldwide pandemic made it challenging. Especially Duncan.
Thanks for all the crash courses in bioinformatics and for helping me even as you yourself were
busy and out of time in your own PhD. To my WP3 fellows, Frida, Helena, Anaëlle, and Emilia.
Our professional collaboration might have been limited, but I am truly grateful for all the nice
moments and conversations we have shared.
My gratitude also goes to current and past members from the Department of Pathobiology,
Pharmacology and Zoological Medicine for providing such a pleasant environment in and
outside of the lab. I am grateful to Serge for his cooperation during the COVID-19 lock-down
and for assisting my lab work when restrictions kept me from going. To Marleen, thank you
for always being so accommodating and helpful whenever needed. I acknowledge Liesbeth for
performing the electron microscopy (TEM) reported in chapter 3.1, and Kristof, for assisting
with a translation when my Dutch skills were insufficient. To my office members, Jasmien,
Evelien, Yani, and Katrien, thank you for the nice chats in-between lab experiments. Evy, thank
you for always making time, even though your schedule is always full. To Jill, Tessa, Ilse,
Martina, Alessandra, Lore, Silvio, and Svieta, you have all been a special part of my journey
in Belgium and I am forever grateful for all the great moments we have shared.
Thank you to the members of the RUSVM phage group. Especially Andreas for his guidance
and in the beginning of the PhD and Jake for assisting with the phage work on St. Kitts.
Furthermore, I would like to thank my St. Kitts Island family for making my stay in St. Kitts
truly special. Thank you for the many hours of Monday beach volley, snorkel tours, bonfires
and the great conversations and moments we spent together.
I want to thank my family and my closest friends for their unconditional love and support in
whatever I do. Thank you for being there when needed and for taking your time to come visit.
Finally, I would like to thank my partner Cecilie. Thank you for your love, patience, and
support during this rollercoaster ride and for always believing in me. Thank you for following
me across the world (twice) and being my person through it all.