hdac1 and hdac2 collectively regulate intestinal stem cell homeostasis
TRANSCRIPT

The FASEB Journal • Research Communication
HDAC1 and HDAC2 collectively regulate intestinal stemcell homeostasis
Cheryl D. Zimberlin,* Cesare Lancini,† Rachel Sno,† Sanne L. Rosekrans,‡
Chelsea M. McLean,† Hanneke Vlaming,† Gijs R. van den Brink,‡ Michael Bots,*Jan Paul Medema,*,§,1 and Jan-Hermen Dannenberg†,1,2,3
*Laboratory for Experimental Oncology and Radiobiology, Center for Experimental MolecularMedicine, Academic Medical Center, Amsterdam, The Netherlands; †Division of Gene Regulation,Netherlands Cancer Institute, Amsterdam, The Netherlands; ‡Tygat Institute for Liver and IntestinalResearch and Department of Gastroenterology and Hepatology, Academic Medical Center, Amsterdam,The Netherlands; and §Cancer Genomics Center, Utrecht, The Netherlands
ABSTRACT Histone deacetylases (HDACs) are post-translational modifiers that deacetylate proteins. Despitetheir crucial role in numerous biological processes, the useof broad-range HDAC inhibitors (HDACi), has shown clin-ical efficacy. However, undesired side effects highlight thenecessity to better understand the biology of differentHDACs and target the relevant HDACs. Using a novelmouse model, in which HDAC1 and HDAC2 can be simul-taneouslydeleted in the intestineof adultmice,we show thatthe simultaneous deletion of HDAC1 and HDAC2 leads toa rapid loss of intestinal homeostasis. Importantly, this de-letion cannot be sustained, and 8 days after initial ablation,stem cells that have escaped HDAC1 or HDAC2 deletionswiftly repopulate the intestinal lining. In vitro ablation ofHDAC1 and HDAC2 using intestinal organoid culturesresulted in a down-regulation ofmultiple intestinal stem cellmarkers and functional loss of clonogenic capacity. Impor-tantly, treatment of wild-type organoids with class I–specificHDACi MS-275 also induced a similar loss of stemness,providing a possible rationale for the gastrointestinal sideeffects often observed in HDACi-treated patients. In con-clusion, these data show that HDAC1 and HDAC2 havea redundant function and are essential tomaintain intestinalhomeostasis.—Zimberlin, C. D., Lancini, C., Sno, R.,Rosekrans, S. L., McLean, C. M., Vlaming, H., van denBrink, G. R., Bots, M., Medema, J. P., Dannenberg, J.-H.HDAC1andHDAC2collectively regulate intestinal stemcellhomeostasis. FASEB J. 29, 000–000 (2015). www.fasebj.org
Key Words: organoids • histone deacetylase inhibitor • histonedeacetylase • MS-275
POSTTRANSLATIONAL MODIFICATIONS (PTMs) are importantprotein modifications that contribute to a dynamic stateof the cell. PTMs such as phosphorylation, methylation,ubiquitination, and acetylation are involved in proteinfunction, structure, and turnover. Protein acetylation and
deacetylation of lysine residues is controlled by the oppo-site functions of histone acetyltransferases (HATs) andhistone deacetylases (HDACs), respectively (1, 2). Thereare 18 known HDAC family members, grouped in 4 mainclasses: classes I (HDAC1, -2, -3, and -8), II (HDAC4, -5, -6,-7, -9, and -10), III (sirtuins), and IV (HDAC11) (3).HDACs govern the interplay between acetylation anddeacetylation of histones and nonhistone proteins such astranscription factors (4). Herewith, HDACs regulate tran-scription through modulation of chromatin structure andby direct regulation of transcription factor activity.
Despite their critical role in cellular proliferation andmaintenance, the inhibitionofHDACsusingsmallmolecules(HDACi) has proven successful in the treatment of sub-cutaneous and peripheral T-cell lymphoma, with 2 HDACi,suberanilohydroxamic acid (SAHA) and FK228, being ap-proved by the U.S. Food and Drug Administration (5, 6).Currently$12 differentHDACi are used in ongoing clinicaltrials for both single and combination therapies in solid andhematologic malignancies (6). As HDACs are involved innumerous biologic processes, current HDACi treatmentcauses many undesired side effects, including; bonemarrowdepression, diarrhea, weight loss, taste disturbances, electro-lyte changes, disordered clotting, fatigue, and cardiacarrhythmias (7). A widely accepted rational for the sideeffects caused by HDACi is that the currently availableinhibitors targetmultipleHDACs.Alternatively, broad-actingHDACimay cause side effects by interferingwithnon-HDACproteins. To improve therapeutic targeting, it is thereforeessential to understand the specific function of the targetedHDACs.HDACclass I (HDAC1, -2, -3, and -8)members havebeen of particular interest for therapeutic targeting becausethese have often been linked to cell proliferation and celldeath (5). In addition, HDACs 1–3 have been shown to be
Abbreviations: ALDH, aldehyde dehydrogenase; BrdU,bromodeoxyuridine; EdU, 5-ethynyl-2´-deoxyuridine; HAT,histone acetylase; HDAC, histone deacetylase; HDACi, histonedeacetylase inhibitor; IP, intraperitoneal; ISC, intestinal stemcell; b-NF, b-naphthoflavone; PMT, posttranslational modifi-cation; WT, wild-type
1 These authors share senior authorship.2 Current affiliation: Genmab B.V., Antibody Sciences,
Utrecht, The Netherlands.3 Correspondence: Division of Gene Regulation, Netherlands
Cancer Institute, Plesmanlaan 121, 1066 CX, Amsterdam, TheNetherlands. E-mail: [email protected]: 10.1096/fj.14-257931This article includes supplemental data. Please visit http://
www.fasebj.org to obtain this information.
0892-6638/15/0029-0001 © FASEB 1
The FASEB Journal article fj.14-257931. Published online February 3, 2015.

recruited by oncogenic fusion proteins and are up-regulatedin a variety of tumor types, including colon cancer (7–9).
Despite this connection to colon cancer, the funda-mental role of HDACs in normal intestinal homeostasisremains poorly defined. The intestinal lining itself isa tightly controlled fast cycling system, where almost allepithelial cells are replaced on aweekly basis (10). This fastcycling tissue is highly organized, and the different in-testinal cell types are well defined (11). Intestinal homeo-stasis ismaintainedat thebase of the crypts by the intestinalstem cells (ISCs), which can be identified by severalmarkers, including Ascl2, Lgr5, Bmi1, Tnfrsf19, and Olfm4(12). ISCs are thought to maintain intestinal homeostasisregulated by a plethora of essential morphogenetic signalsincluding Wnt, Notch, bone morphogenetic proteins, andHedgehog (12, 13). Although all these pathways are criticalin maintaining the crypt villi organization, a lot of attentionhas gone to theWntpathway,whichhasbeen shown toact asa functionalmarker that identifies ISCs inhealthy, adenoma,and carcinoma tissue (13, 14). Furthermore, overexpressionof the Wnt pathway activator R-spondin leads to increasedproliferation in vivo (15) and prevents Wnt signaling,resulting in lossof crypts inadultmice(16–18). Interestingly,HDAC2 has been linked to theWnt signaling cascade as it isthought to promote b-catenin–T-cell transcription factor(TCF)–c-MYC (MYC) signaling, thereby preventing apo-ptosis (9). However, the precise effect of HDAC deletion inISC homeostasis still remains to be established.
Deletion of HDAC1 has previously been shown to resultin embryonic lethality before E10.5 (19). Hdac2-null miceare smaller than wild-type (WT) littermates and displayshortened crypts and villi in their intestine. When Hdac2-nullmice are crossedwithApcminmice, fewer adenomas areformed, pointing to an important role of this HDAC inintestinal homeostasis and pathology (20–22). Althoughthese results suggest specific functions for HDAC1 andHDAC2,many tissues and cell types are not affected by lossofHDAC1orHDAC2. In agreement,HDAC1andHDAC2share 85% amino acid homology and appear to have re-dundant functions, with loss of HDAC1 resulting in in-creased HDAC2 protein expression, and vice versa, loss ofHDAC2 in resulting in increased HDAC1 protein ex-pression (23–25). Indeed, previously we showed thatsimultaneous deletion of HDAC1 and HDAC2 in the he-matopoietic system, in contrast to single deficiencies,resulted in anemia and thrombocytopenia. HDAC1 andHDAC2deficiency in the liverdidnot result in a detectablephenotype, indicating that HDAC1 and HDAC2 havetissue-specific functions, which might be linked to theproliferative state of a tissue (25). With respect to the in-testinal lining in mice, a recent study by Turgeon et al.showed the phenotypic implications of constitutive de-letion ofHDAC1and “patchy”deletionofHDAC2at E15.5ofembryonicdevelopment(26,27). In this study,HDAC1/2deletion in the intestine, early during development,resulted in loss of differentiation into different intestinalcell lineages, increased proliferation, and altered barrierfunctioning (26). However, the reported patchy HDAC2deletion leaves room for interpretation of these data,especially as recent studies have indicated that HDAC1/2may have dose-dependent effects. These results implicatethat complete deletion of HDAC1/2may cause a differentphenotype compared with partially deleted HDAC1/2
(28–30). In addition, acute loss of HDAC1 and/orHDAC2 in the intestinal lining of adult mice in contrastto deletion in the embryonic developing intestinemimicsmore closely HDACi treatment in patients and may thusyield clinically relevant data. To study the effects ofcomplete and partial deletion of HDAC1 and HDAC2in the intestine of adult mice, we combined the b-naphthoflavone (b-NF) inducible Ah-cre transgene (31,32) with our Hdac1 and Hdac2 conditional knockoutalleles (22, 25). Remarkably, using this in vivomodel, weshow that simultaneous deletion of HDAC1 and HDAC2is not sustainable within the intestinal lining, in contrastto deletion of only HDAC1 or HDAC2. Moreover, loss ofHDAC1 and HDAC2 in organoid cultures leads to a di-rect loss of stemness and clonogenic potential, indicatingthat thepresenceofHDAC1andHDAC2 is a prerequisitefor normal intestinal stem cell function.
MATERIALS AND METHODS
Mice
The Hdac1 and Hdac2 conditional knockout mice have beenpreviously described (22, 25). TheHdac2-null mice (HDAC2KO)used were previously described (22). Intestinal specific deletionof Hdac1 and Hdac2 was obtained using Ah-cre transgenic mice(31, 32) in combination with Hdac1 and/or Hdac2 conditionalknockout alleles. To induce Cre expression via the Ah promoter,the Ah-cre–containing mice each received 3 intraperitoneal (IP)injections of 80.0 mg/kg b-NF (Sigma-Aldrich, Zwijndrecht, TheNetherlands) dissolved in corn oil at intervals of 3 h, as describedpreviously (31). All cohorts were in a mixed FVB/n, C57BL/6,and 129/Sv background. All mice used for analysis were 8 weeksof age. Mice used to analyze in vivo bromodeoxyuridine (BrdU)incorporation were killed 24 hours after receiving 1 mg BrdU(Sigma-Aldrich) by intraperitoneal injection. All experimentswere approved by a local ethical committee and performedaccording to national guidelines.
Organoid culturing
Organoid cultures were generated from the first 5 cm of theduodenum.The intestinewasfirstflushedwith coldPBSand thencut longitudinally with the villi facing upward. After careful re-moval of villi using a glass coverslip, the intestine was sliced intosmall pieces, washed 20 times with cold PBS and dissociated with2 mMEDTA at 4°C for 30minutes. For plating,;100–200 cryptswere resuspended in 25 ml matrigel (BD Bioscience, Breda, TheNetherlands) per well in a 48-well plate. Crypts were maintainedin Advanced DMEM/F12 medium (Life Technologies, Bleiswijk,TheNetherlands) supplementedwith 100ng/mlmurine-Noggin(Prospec, Rehovot, Israel), 50 ng/ml murine endothelial growthfactor (Peprotech, London, United Kingdom), and 500 ng/mlR-Spondin (R&D Systems, Abingdon, United Kingdom). Treat-ments of organoid cultures were done 3 days after plating. Tenmicrograms per milliliter b-NF in vitro stimulations were done inthe medium for 72 hours before analysis. Treatments with 5 mMMS-275 (Sigma-Aldrich) was done in medium for 24 (assessingproliferation and stemness) and 72 hours (assessing clonogeniccapacity). Thymidine (Sigma-Aldrich) treatments were done for24 hours using 2.5 mM thymidine to block proliferation.
Organoid RNA isolation and quantitative RT-PCR
According to manufacturer protocols, organoids were first re-leased frommatrigel (BD Biosciences, San Jose, CA, USA) using
2 Vol. 29 May 2015 ZIMBERLIN ET AL.The FASEB Journal x www.fasebj.org

cell recovery solution (BD Biosciences), and total RNA wasextracted using the NucleoSpin RNA kit (Macherey-Nagel,Duren, Germany). RNA quantity and quality were assessed us-ing Thermo-Scientific nanodrop 1000 Spectrophotometer(Thermo-Fischer Scientific, Landsmeer, The Netherlands). Fol-lowing DNAse treatment using DNAseI Amplification Grade(Life Technologies, Carlsbad, CA, USA), cDNA was synthesizedusing SuperScript III reverse transcriptase (Life Technologies)with random primers (Life Technologies). Quantitative RT-PCRwas performed using 23 SYBR Green Master Mix (Roche,Almere, TheNetherlands), and transcript levels were normalizedto that of b-2-microglobulin (B2M). Primers used in this studywere as follows—B2M: forward, 59-CTTCAGTCGTCAGCATGG-39; reverse, 59-GTTCTTCAGCATTTGGATTTC-39; Olfm4: for-ward, 59-GCCACTTTCCAATTTCAC-39; reverse, 59-GAGCCTCT-TCTCATACAC-39; Bmi1: forward, 59-TATAACTGATGATGAGA-TAATAAGC-39; reverse, 59-CTGGAAAGTATTGGGTATGTC-39;Lgr5: forward, 59-TTCGTAGGCAACCCTTCTCT-39; reverse, 59-TCCTGTCAAGTGAGGAAATTCA-39; Ascl2: forward, 59-GGAAG-CACACCTTGACTGGT-39; reverse, 59- GAAGTGGACGTTTG-CACCTT-39;Tnfrsf19: forward, 59-ATTCTCTTCCTACTCCACCTG-39; reverse, 59-CATAGCCGAAGCCACATTC-39.
Flow cytometry analysis
Before analysis, organoids were released from matrigel (BDBiosciences) using cell recovery solution (BD Biosciences)and treated with TrypleExpress (Life Technologies) for3 minutes at 37°C to obtain single cells. To measure aldehydedehydrogenase (ALDH) activity, the ALDEFLUOR fluores-cent reagent system (STEMCELL Technologies, Grenoble,France) was used according to the manufacturer’s protocol.Organoid proliferation by 5-ethynyl-29-deoxyuridine (EdU)incorporation was measured using the standard protocol forthe Click-IT EdU Alexa Fluor 488 Flow Cytometry Assay Kit(Life Technologies). For propidium iodide analysis, cells werefixed with 4% paraformaldehyde, washed with PBS, and trea-ted with propidium iodide (Sigma-Aldrich) according to thestandard protocol. All staining’s were analyzed on the FACS-Canto (BD Biosciences).
Immunohistochemical, Immunofluorescent, and in situhybridization stainings
For immunohistochemical analysis, the first 3 cm of the smallintestinewasfixed in ethanol-acetic acid-formol saline fixative for24 hours and subsequently embedded in paraffin. Periodicacid–Schiff and hematoxylin and eosin stainings were conductedusing standardized protocols. For immunohistochemistry, sec-tions were preincubated with goat serum (Sanquin, Amsterdam,The Netherlands) for 30 minutes and subsequently incubatedovernight with the following antibodies: HDAC1 (IMG-337;Imgenex, San Diego, CA, USA), HDAC2 (SC-7899; Santa CruzBiotechnology, Santa Cruz, CA, USA), lysozyme (EC 3.2.1.17;DAKO, Heverlee, Belgium), and cleaved Caspase3 (9661L; CellSignaling Technology, BIOKE, Leiden, The Netherlands). Sec-ondary antibody poly-horseradish peroxidase-anti-rabbit IgG(Immunologic, Duiven, The Netherlands) was then applied for30 minutes. The slides were washed with PBS, incubated withdiaminobenzidine substrate chromogen system (DAKO), andcounterstained with hematoxylin (Merck Millipore, Amsterdam,The Netherlands). For HDAC2/BrdU double immunofluores-cenceof the small intestinesections, thesampleswerepreparedasdescribed before. For antigen unmasking, samples were micro-waved and then pretreated for BrdU immunodetection, afterwhich they were incubated with a mixture of rabbit anti-HDAC2
(Santa Cruz Biotechnology) andmonoclonal mouse anti-BrdU(DAKO)primary antibodies overnight at 4°C. This was followedby addition of secondary antibodies Alexa Fluor 488 goat anti-rabbit IgG (H+L) (A11013; Molecular Probes, Life Technolo-gies), Alexa Fluor 568 goat anti-mouse IgG (H+L) (A11004;Molecular Probes, Life Technologies), and DAPI (Sigma-Aldrich). Organoids were fixed overnight at 4°C in 4% para-formaldehyde (157-8; Electron Microscopy Sciences, Hatfield,PA, USA) and passed through a series of ethanol steps beforeparaffin embedding. Four micrometer sections were collectedand analyzed by immunohistochemistry using standard tech-niques. Primary antibodies used were HDAC1 antibody (IMG-337; Imgenex/Novus Biologicals, Littleton, CO,USA), HDAC2(SC-7899; Santa Cruz Biotechnology), and Ki67 (Abcam,Cambridge, United Kingdom). For in situ hybridizations, 10 mmethanol-acetic acid-formol saline-fixed paraffin sections were cutand processed as previously described (33).
Western blot analysis
Total protein lysate from small intestine tissue was prepared asdescribed previously (28). To collect lysates from organoids, theorganoids were released from matrigel using cell recovery solu-tion (BD Biosciences) and lysed in Cell Lysis buffer (Cell Signal-ing Technology, Danvers, MA, USA) containing protease/phosphatase inhibitor cocktail (Cell Signaling Technology).Protein quantification was done using the Pierce BCA ProteinAssayKit, and5mgwas loadedperwell of 4–15%precast gels (Bio-Rad, Veenendaal, The Netherlands), transferred to a Hybond-Pmembrane (GE Healthcare Life Sciences, formally AmershamBiosciences, Diegem, Belgium), and blocked for 1 hour in 5%bovine serum albumin in Tris-buffered saline and Tween 20(0.1%). Primary antibodies for overnight 4°C incubation were1:1000 HDAC1 (PA1-860; Thermo Fisher Scientific, Waltham,MA, USA) and 1:1000 HDAC2 (ab19862; Cell Signaling Tech-nology). After three 10 minute washes with TBS-T (0.1%), thesecondary antibody (1:2000, anti-rabbit-horseradish peroxidase,#7074;Cell SignalingTechnology)wasapplied for 1hourat roomtemperature. After washing, the membrane was developed usingthe ImageQuanti LAS4000 (GE Healthcare Life Sciences). Pro-teinwasnormalized toextracellular signal-regulatedkinase1/2asa control (#9107; Cell Signaling Technology).
RESULTS
Acute deletion of HDAC1 and HDAC2 in adultintestinal epithelium
To identify the role of HDAC1 and HDAC2 in intestinalhomeostasis, an in vivo mouse model was generated thatallows acute loss of HDAC1 and HDAC2 in the adult in-testinal epitheliumby combining theb-NF inducibleAh-cretransgene (31, 32) with Hdac1 or/and Hdac2 condi-tional knockout alleles (22, 25). First, Ah-cre;Hdac1L/L
(HDAC1KO) mice were generated to study the effect ofHDAC1 loss in the intestine, whereas loss ofHDAC2wasstudied in the viable, constitutive germ-line HDAC2knockout (HDAC2KO) mice (22). Considering thepossibility that HDAC1 and HDAC2 have redundantfunctions in the intestine, double knockout Ah-cre;Hdac1L/L;Hdac2L/L (DKO) mice were generated to studycombined loss of HDAC1 and HDAC2. Furthermore, aswe previously observed a dose-dependent function forHDAC1 and HDAC2 in thymocytes (28), the Ah-cre;
INTESTINAL STEM CELLS REQUIRE HDAC1 AND HDAC2 3

Hdac1L/L;Hdac2L/+ mice (HOM;HET) mice were gen-erated to test whether HDAC1 and HDAC2 dosage isalso critical for intestinal development.
HDAC1 and HDAC2 have previously been shown to befunctionally redundant in a variety of tissues (24). Inagreement, deletion of either HDAC1 or HDAC2 aloneshowed no distinct intestinal phenotype, despite effectivedeletion of the genes as evidenced by loss of protein ex-pression (Supplemental Fig. S1A). Similarly, 4 days afterthe last b-NF injection, a significant loss of HDAC1 andHDAC2 protein expression was observed in DKO andHOM;HET mice by Western blot (Fig. 1A) and immuno-histochemical staining (Fig. 1B). However, in the cryptbase, rare cells maintaining HDAC1 or HDAC2 could beobserved (black arrows; Fig. 1B). From these observations,we can conclude that this novelAh-cre;Hdac1;Hdac2mousemodel allows for acute and efficient deletion of HDAC1and/or HDAC2 in the intestine of adult mice.
Simultaneous loss of HDAC1 and HDAC2 cannot besustained in vivo
Recently, Turgeon et al. (26) described a constitutive de-letion ofHDAC1 andHDAC2, using Villin-cre-recombinasetransgenic mice. In this genetic model, HDAC1 andHDAC2 are initially deleted in the intestine during em-bryonic development. However, in contrast to the DKOmice generated using the Ah-cre background, the HDAC1and HDAC2 double knockout mice from the Villin-cre-recombinase background appear to retain patchy HDAC2expression (26). Nevertheless, similar phenotypes wereobserved in both mouse models. As in the Villin-cre back-ground, the intestinal epithelium of HOM;HET and DKOmice displayed a bulgy appearance of the crypt pockets,suggesting a loss in polarization of epithelial cells (Fig. 1B).In addition, loss of differentiated cell linages in both DKOandHOM;HET intestines was observed as evidencedby lossof both Paneth and Goblet cell lineages (SupplementalFig. S1B, C). Increased cell death was also detected usingimmunohistochemical staining for activated Caspase-3 inDKOmice (Supplemental Fig. S1D)
Although these observations suggest an overlap in phe-notypes conferred by inactivation of HDAC1 and HDAC2during embryonic development or in the adult stage ofintestinal development, acute deletion of HDAC1 andHDAC2 in adult mice appeared to have a more extremephenotype, whereas the HOM;HET mice, which retainsome HDAC2 expression, closely resembled the HDAC1and HDAC2 deletion mice from Turgeon et al. (26). Thiswas most evident when proliferation was studied. TheHOM;HET mice showed a significant increase in in vivolabeling of BrdU 4 days after the last b-NF injection, in-dicative of increased proliferative capacity in the cryptcompartment compared withWT mice (Fig. 1C). In starkcontrast, the capacity to proliferate in the crypt compart-ment in DKO mice was almost completely lost upon si-multaneous HDAC1 and HDAC2 deletion in adult mice,demonstrated by a significant decrease in in vivo BrdUlabeling (Fig. 1C). This indicates that the complete de-letion of HDAC1 and HDAC2 is incompatible with pro-liferation in the crypt, whereas the presence of 1 Hdac2allele, in the absence of HDAC1, enhanced proliferation.
In agreement, immunofluorescence for HDAC2 ex-pression and BrdU labeling revealed that all cells in thecrypt base of DKO mice that maintained BrdU labelingalso maintained HDAC2 expression (Fig. 1D). In con-clusion, whereas complete loss of HDAC1 and HDAC2resulted in cessation of proliferation in the intestinalcrypt, low levels of HDAC2 in the absence of HDAC1enhanced proliferation, suggesting a dosage effect asreported before (28).
To test whether loss of proliferation, associated withcomplete loss of HDAC1 and HDAC2 in the intestine ofDKOmice, also affected intestinal function at the organismlevel, the body weight of DKO and control mice was mon-itored. Body weight measurements revealed a drastic re-duction in weight of DKOmice during the first 6 days afterb-NF stimulation, after which these mice regain bodyweight (Fig. 1E). This recovery coincided with the re-population of nonrecombined intestinal epithelial cells,forming a mosaic pattern of both HDAC1 and HDAC2expression along the intestinal tract (Fig. 1F). The pres-ence of HDAC1/HDAC2-expressing cells suggestsa highly selective advantage of nonrecombined cellsthat maintain HDAC1 and/or HDAC2 expression. Thisselective advantage is likely responsible for the rescue ofintestinal function and regained body weight. In con-clusion, these results indicate that HDAC1 and/orHDAC2 collectively play an essential role for intestinalhomeostasis and that full deletion is not sustainable andtherefore rapidly competed out.
Deletion of HDAC1 and HDAC2 leads to loss ofstemness in vivo
As cells maintaining HDAC1 and/or HDAC2 quicklyrepopulate the entire intestinal lining, HDAC1 andHDAC2 are likely to play critical roles in the maintenanceof intestinal homeostasis. LGR5+ stem cells, situated at thebase of the intestinal crypts, are at the apex of the intestinalhierarchy, and the observed phenotype could be consis-tent with a role for HDAC1 and HDAC2 in stem cell ho-meostasis. To directly test this hypothesis, we analyzed theexpression of well-defined intestinal stem markers Olfm4and Lgr5, using in situ hybridizations, 4 days after b-NF–induced deletion of HDAC1/2. Indeed, this analysisrevealed a significant decrease in stem cell marker ex-pression inDKO intestines, suggesting lossof stemness (Fig.2A,B).Todirectly assess the impactofHDAC1andHDAC2loss on ISC function, the clonogenic capacity of ex vivointestinal organoid cultures from WT, HOM;HET, andDKO mice was determined. The same number of cryptswere isolated from the small intestine of the WT, HOM;HET, and DKOmice, and the outgrowth of organoids wascounted. Quantification revealed no significant differ-ence in outgrowth betweenWT and HOM;HET organoids.However, a significant reduction in DKO organoidnumbers was observed compared with WT organoids(Fig. 2C). Further immunohistochemical analysis of thefew organoids that managed to grow from the DKOintestines showed that these organoids maintainedHDAC1 and/or HDAC2 expression, indicating thatthey originated from nonrecombined cells that hadescaped HDAC1/2 deletion (Supplemental Fig. S2).
4 Vol. 29 May 2015 ZIMBERLIN ET AL.The FASEB Journal x www.fasebj.org

Figure 1. Simultaneous loss of HDAC1 and HDAC2 cannot be sustained. Protein levels of HDAC1 and HDAC2 in the smallintestine of WT (n $ 3), HOM;HET (n $ 2), and DKO (n $ 3) mice, 4 days after stimulation with b-NF, can be observed by (A)Western blot and (B) immunohistochemistry. Black arrows indicate residual HDAC1 and HDAC2 expression in DKO mice, andinsets show close-up of the crypt base. C) Sections of the small intestine, from WT (n = 6), HOM;HET (n = 6), and DKO (n = 6)
(continued on next page)
INTESTINAL STEM CELLS REQUIRE HDAC1 AND HDAC2 5

These data confirm the hypothesis that HDAC1 and/orHDAC2 are essential to maintain ISCs and consequentlyintestinal homeostasis.
HDAC1 and HDAC2 regulate intestinal stemness ina cell autonomous fashion
To examine whether HDAC1 and HDAC2 have a cell au-tonomous function in the intestinal epithelium, organoidswere derived from noninducedWT and DKO mice. West-ern blot analysis showed that both HDAC1 and HDAC2could be successfully deleted in vitro in DKO organoidcultures after 72 hours of b-NF treatment (Fig. 3A). Theloss of HDAC1 and HDAC2 in organoids was further veri-fied by immunohistochemistry, which revealed loss ofnuclear HDAC1 andHDAC2 expression (Fig. 3B). Similarto the in vivo results, simultaneous deletion of HDAC1and HDAC2 resulted in a dramatic reduction in pro-liferative capacity as indicated by decreased expression of
proliferation marker Ki67 (Fig. 3B). Moreover, quantita-tive RT-PCR 72 hours after b-NF treatment revealed a sig-nificant reduction in both proliferative and quiescentstem cell-associated markers, Olfm4, Lgr5, Bmi1, Ascl2, andTnfrsf19 (Fig. 3C), pointing to a loss of stemness. Inagreement, combined HDAC1 and HDAC2 deletion inorganoids resulted in a significant inhibition of functionalstem cell-associated ALDH1 activity, indicating that notonly mRNA expression levels are altered, but also, proteinlevels associated with stemness are not maintained (Fig.3D). In contrast, this loss of ALDH1 activity was not ob-served in WT and HOM;HET crypts treated with b-NF(Supplemental Fig. S3A).To testwhether lossofHDAC1/2also resulted in a functional stemness defect, weperformedclonogenic assays bypassagingWT andDKOorganoids thatwere treatedwithb-NF for 72hours before being passaged.As it was previously shown that stem cells have the ability torecapitulate an entire organoid from a single cell, onewould expect no organoid growth in case of a stem celldefect. Consistent with our data showing loss of stem cell
mice were subjected to immunohistochemical staining for BrdU. Insets show close-up of the crypt base. Positive cells in the cryptwere quantified and are depicted to the right. D) Immunofluorescent costainings of the intestine of WT (n = 3) and DKO (n = 3)mice were stained for BrdU (red), HDAC2 (green), and DAPI (blue); white arrows in the DKO section show BrdU/HDAC2double positive cells. E) Impact of HDAC1 and HDAC2 loss on body weight is shown in WT (n = 3) and DKO (n = 3) mice aretreated with b-NF at day 0 and monitored for 8 days. F) Immunohistochemical stainings for WT (n = 3) and DKO (n = 3) mice atday 8 shows a selection for HDAC1- and HDAC2-positive cells. Original magnification:320 panel (B, C, F). *P# 0.05; **P# 0.01;***P # 0.005 (t test).
Figure 2. Deletion of HDAC1 and HDAC2 leads to loss of stem cell markers in vivo and loss of clonogenic capacity ex vivo.Representative in situ hybridizations on small intestine sections on WT (n = 3) and DKO (n = 3) mice using antisense probes forstem cell markers Lgr5 (A) and Olfm4 (B) reveal characteristic signals at the crypt base in WT mice and reduced signal for DKOmice. Original magnification is 320, and magnified regions are 340. C) Four days after b-NF stimulation, crypts were isolatedfrom the small intestine of WT (n = 3), HOM;HET (n = 3), and DKO (n = 3) mice; 100 crypts were plated per well, and outgrowthwas quantified after 7 days. ***P # 0.005 (t test).
6 Vol. 29 May 2015 ZIMBERLIN ET AL.The FASEB Journal x www.fasebj.org

markers in DKO organoids, we observed no growth uponpassaging DKO organoids, indicating a complete loss infunctional stem cell activity (Fig. 3E). In conclusion, theseobservations indicate that HDAC1 and HDAC2 are in-trinsically required to sustain ISC capacity and therebyserve a crucial function in maintaining the intestinalepithelium.
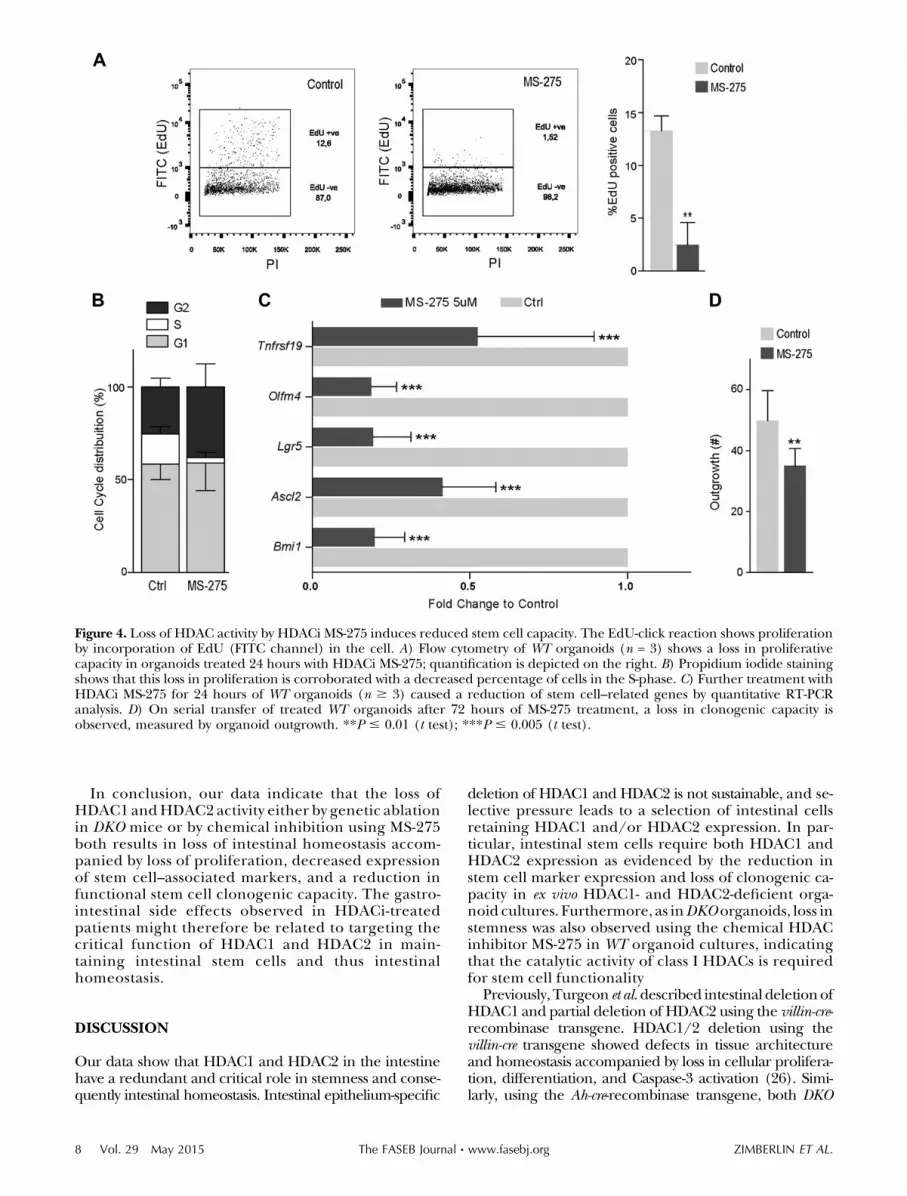
Chemical HDAC inhibitor MS-275 mimics loss ofstemness by genetic ablation of HDAC1/2
Our data, obtained from in vivo and in vitro experiments inDKO mice, reveal a critical function for HDAC1 andHDAC2 in maintaining intestinal homeostasis by regulat-ing stemness. These results could have significant impli-cations for the toxicity of HDACi in clinical settings, as allapproved HDACi or those currently tested in clinical trialsare relatively nonspecific, targeting multiple HDACs ofsingle or distinct HDAC classes. To test whether chemicalinhibition of HDAC1/2 using an HDACi is comparableto genetic inactivation of HDAC1 and HDAC2, WTorganoids were treated with the class I–specific HDACi
MS-275 and assayed for stemness. Similar to genetic de-letion of HDAC1/2 in DKO mice and organoids, short-term MS-275 treatment of WT organoids for 24 hoursresulted in a significant loss in proliferation capacity asindicated by the reduction in DNA synthesis, measuredby loss of EdU incorporation (Fig. 4A) and the numberof cells in S-phase of the cell cycle (Fig. 4B). mRNAanalysis by quantitative RT-PCR revealed a significantreduction in all proliferative and quiescent stem cell-associated markers tested, including Olfm4, Lgr5, Bmi1,Ascl2, and Tnfrsf19 (Fig. 4C), suggesting a loss of stemness.Indeed, similar to our observations in DKO mice andorganoids, inhibition of HDAC activity in WT organoidsusing MS-275 for 72 hours resulted in a significant loss inorganoid outgrowth compared with control-treatedorganoids (Fig. 4D). Interestingly, inhibition of pro-liferation using thymidine treatment did not result inloss of stem cell-associated markers (Supplemental Fig.S3B, C) suggesting that proliferation alone is not suffi-cient to induce a loss in stemness. Combined, these datasuggest that the loss of clonogenic capacity observed ondeletion of HDAC1 and HDAC2 primarily effects stem-ness of the ICS and not merely proliferation.
Figure 3. Inherent loss of stemness observed on HDAC1 and HDAC2 deletion in vitro. Intestinal crypts were isolated fromWT (n = 3)and noninduced DKO (n = 3) mice and grown in vitro. A) Protein expression of HDAC1 and HDAC2, after 72 hours in vitrob-NF stimulation, was determined by immunoblot. B) Representative immunohistochemical stainings of control vs. b-NF–treatedDKO mouse organoids showed decreased proliferative capacity (Ki67), as well as decreased HDAC1 and HDAC2 levels. Scale baris 100 mM. C) Quantitative RT-PCR of intestinal stem cell–related genes in control vs. b-NF–treated organoids (n = 3). D) Asmeasured by the ALDEFLUOR assay, a functional loss of stem cell activity, mean fluorescent intensity, is observed in DKOorganoids on b-NF treatment (n $ 3). E) Representative picture of WT and DKO organoids following serial passage after beingtreated with b-NF for 72 hours. In contrast toWT organoids, no outgrowth was ever observed in b-NF–treated DKO organoids (n$ 3).Original magnification: 203 (B, E). *P # 0.05, **P # 0.01, ***P # 0.005 (t test).
INTESTINAL STEM CELLS REQUIRE HDAC1 AND HDAC2 7
In conclusion, our data indicate that the loss ofHDAC1 andHDAC2 activity either by genetic ablationin DKO mice or by chemical inhibition using MS-275both results in loss of intestinal homeostasis accom-panied by loss of proliferation, decreased expressionof stem cell–associated markers, and a reduction infunctional stem cell clonogenic capacity. The gastro-intestinal side effects observed in HDACi-treatedpatients might therefore be related to targeting thecritical function of HDAC1 and HDAC2 in main-taining intestinal stem cells and thus intestinalhomeostasis.
DISCUSSION
Our data show that HDAC1 and HDAC2 in the intestinehave a redundant and critical role in stemness and conse-quently intestinal homeostasis. Intestinal epithelium-specific
deletion of HDAC1 and HDAC2 is not sustainable, and se-lective pressure leads to a selection of intestinal cellsretaining HDAC1 and/or HDAC2 expression. In par-ticular, intestinal stem cells require both HDAC1 andHDAC2 expression as evidenced by the reduction instem cell marker expression and loss of clonogenic ca-pacity in ex vivo HDAC1- and HDAC2-deficient orga-noid cultures. Furthermore, as inDKO organoids, loss instemness was also observed using the chemical HDACinhibitor MS-275 in WT organoid cultures, indicatingthat the catalytic activity of class I HDACs is requiredfor stem cell functionality
Previously, Turgeon et al.described intestinal deletionofHDAC1 and partial deletion of HDAC2 using the villin-cre-recombinase transgene. HDAC1/2 deletion using thevillin-cre transgene showed defects in tissue architectureand homeostasis accompanied by loss in cellular prolifera-tion, differentiation, and Caspase-3 activation (26). Simi-larly, using the Ah-cre-recombinase transgene, both DKO
Figure 4. Loss of HDAC activity by HDACi MS-275 induces reduced stem cell capacity. The EdU-click reaction shows proliferationby incorporation of EdU (FITC channel) in the cell. A) Flow cytometry of WT organoids (n = 3) shows a loss in proliferativecapacity in organoids treated 24 hours with HDACi MS-275; quantification is depicted on the right. B) Propidium iodide stainingshows that this loss in proliferation is corroborated with a decreased percentage of cells in the S-phase. C) Further treatment withHDACi MS-275 for 24 hours of WT organoids (n $ 3) caused a reduction of stem cell–related genes by quantitative RT-PCRanalysis. D) On serial transfer of treated WT organoids after 72 hours of MS-275 treatment, a loss in clonogenic capacity isobserved, measured by organoid outgrowth. **P # 0.01 (t test); ***P # 0.005 (t test).
8 Vol. 29 May 2015 ZIMBERLIN ET AL.The FASEB Journal x www.fasebj.org

and HOM;HET mice also demonstrated a loss of cellulardifferentiation into Paneth and Goblet cell lineages, whichwas not detected in single HDAC1 or HDAC2 knockoutmice.Therefore, we conclude that a critical level ofHDAC1or HDAC2 is required to maintain ISCs and to generatedifferentiated intestinal cells. In addition, HDAC1 andHDAC2 levels appear to be critical for proliferative capacityof the intestinal lining.Accordingly, intestinal cellsdeficientfor HDAC1 and HDAC2 in DKO mice were unable toproliferate as evidenced by a lack of BrdU incorporation.Moreover this phenotype correlated with a severe re-duction in body weight of these mice. Surprisingly, HOM;HET intestinal cells (similar to the villin-cre HDAC1/2deleted intestinal cells) showed increased BrdU in-corporation, suggesting increased proliferation in theintestinal crypts. Interestingly, similar observations weremade in immature CD8+ thymocytes only expressing1HDAC2 allele (Lck-cre;Hdac12/2;Hdac2+/2 thymocytes),which showed a dramatic increase in proliferation andultimately transformed into lymphoma (28). Increasedexpression of oncogenes (such as c-Myc and Jdp2) wasattributed to the increased proliferation and trans-formation. Indeed, increased expression of the c-Mycproto-oncogene was also observed in premalignantHdac1-deficient skin (30). Whether increased expres-sion of genes involved in proliferation is also observed inHOM;HET intestinal cells remains to be investigated. Theincreased proliferation in Villin-cre DKOmice as observedby Turgeon et al.might be caused by the mosaic/patchyHDAC2 expression, suggesting a selection against com-plete loss of this HDAC1/2 activity (26).
Stem cells residing at the base of the crypt are at theapex of renewal of the intestinal lining, maintainingcell renewal and differentiation. After stem cell di-vision, the daughter cells (transient amplifying cells)are highly proliferative, differentiating after 2 daysbefore exiting the crypt (34). Stem cells thus preservethe crypt-villus axis. DKO mice showed both pro-liferative and cellular differentiation capacity to begrossly diminished by simultaneous loss of HDAC1and HDAC2. Furthermore, the deletion of HDAC1and HDAC2 in DKOmice was highly disadvantageous,suggesting that not only the transient amplifying zonebut also the source, stem cells, were affected. Indeed,on loss of expression of HDAC1 and HDAC2 in in vitroorganoid cultures, a significant loss in stem cell–related markers Lgr5, Bmi1, Tnfrsf19, Olfm4, andAscl2 was observed. More importantly, a complete lossof clonogenic capacity was seen, where no singleorganoid survived in the absence of HDAC1 andHDAC2. We therefore conclude that HDAC1 andHDAC2 are required to maintain stem cell function-ality. The mechanism by which HDAC1 and HDAC2could regulate intestinal stemness has not beenaddressed in the current study. However, the Wntpathway, an important morphologic pathway crucialto stemness maintenance (12, 13), is a promisingcandidate. The Wnt pathway has been shown to beenhanced in both healthy and cancer stem cells,where high Wnt activity can be used to define stemcells (12, 13). Interestingly, Wnt activity has also beenpreviously linked to HDAC2 functioning, whereHDAC2 was induced on Wnt pathway activation (9),
suggesting that stem cells may have a higher HDACpresence and thus be more sensitive to genetic abla-tion of HDAC1 and HDAC2.
HDACi have entered the clinic to treat cutaneous andperipheral T-cell lymphoma.However, currently availableHDACi are broad-spectrum inhibitors, promiscuouslytargeting multiple HDACs, thereby causing undesirableside effects. Therefore, it is important to uncoverwhether these side effects are caused by off-target(targeting non-HDAC proteins) or on-target (targetingHDACs) HDACi activity. Our results demonstrate thatablation of HDAC1 and HDAC2 results in serious in-testinal malfunction. As this phenotype is mimickedby chemical inhibition of HDACs using the HDACiMS-275, it is very likely that thegastrointestinal sideeffects,often observed in patients, are caused by inhibition ofHDAC1 and HDAC2, which results in a (temporal)blockade in intestinal homeostasis, caused by inhibit-ing proliferation, differentiation, and stem cell func-tioning in the intestinal lining.
Because inhibition ofHDAC1 andHDAC2 seem to becritical for the antitumor activity of HDACi, it will bedifficult if not impossible to prevent on-target sideeffects. Therefore, the efficacy of HDACi will de-pendent on the therapeutic window of these anticancerdrugs. The increased dependency of tumors cells onHDACs may determine the width of this therapeuticwindow. In colorectal lesions, a therapeutic windowmaybe present, as HDAC1 and HDAC2 have been found tobe up-regulated during transformation (7–9). More-over, HDAC2 has been linked to the Wnt pathway,a pathway whose alteration is considered the key initia-tor in colorectal lesion (9). More importantly, HDAC2seems to be crucial as the deletion of HDAC2 can curbadenoma formation in APCmin mice (21). Herewith,colorectal lesions may be more dependent on HDACactivity and potentially more susceptible to HDAC1 andHDAC2 inhibition. It is therefore vital to assess whethertumor tissues have a different sensitivity compared withtheir healthy counterparts. It is noteworthy that in anunbiased drug screen, it was found that leukemias car-rying mutations in FBXW7 showed increased sensitivitytoward MS-275, suggesting that defined genetic alter-ations may increase the therapeutic window of HDACi(35). In addition, to increase the therapeutic windowand possibly decrease the amount of HDACi needed,combining HDACi with a standard chemotherapeuticcould be an option.
Partial inhibition of HDAC1 and HDAC2, like in ourHOM;HET mice, may still cause antitumor activity,providing a possibility to reduce side effects by fine-tuning HDACi doses and dosing schemes. However,HDAC1/2 dose-dependent effects have previously beenshown to induce lymphomas in which the tumor in-cidence and latency inversely correlated with theremaining HDAC activity (28). It is also interesting tonote that the function of HDACs may vary at differentstages of carcinogenesis, as HDAC1 was recently shownto act as a tumor suppressor during tumorgenesis, butto act as an oncogene in tumor maintenance (29).Therefore, to establish the crucial therapeutic windowto target tumor cells and not the healthy intestinalcompartment, HDAC1/2 inhibition should be carefully
INTESTINAL STEM CELLS REQUIRE HDAC1 AND HDAC2 9

assessed to establish the critical dosage, timing, andcombination required.
CONCLUSIONS
In this study, we show for the first time that the simulta-neous deletion of HDAC1 and HDAC2 in the murine in-testine leads to a loss of intestinal integrity, which is notsustainable on a long-term basis. Furthermore, we showthat HDAC1 and HDAC2 have a critical and cell autono-mous function in maintaining intestinal stem cells. Nota-bly, loss in stem cell functionality is also observed upontreatment of WT organoid cultures with the HDAC in-hibitorMS-275, indicating that chemical inhibitionof classI HDACs also negatively affects stem cell functionality,providing a rationale for the intestinal side effects oftenobserved in patients treated with HDAC inhibitors.
The authors thank Martijn Koppens for providing the invitro b-NF protocol. The authors thank Ton Schrauwers,Corine van Langen, Auke Zwerver, Cor Spaan, and DienkeJonkers for excellent animal care. This work was supported bygrants from the Dutch Cancer Society (KWF-2007-3978) andNetherlands Organization for Scientific Research (NWO-VIDI864.07.008) (to J.H.D.). H.V. and C.M.M. are supported bya grant from the Dutch Cancer Society (KWF NKI 2009-4511).J.P.M. is supported by the Dutch Cancer Society (UvA2009-4416 and UvA2012-5735), the Netherlands Organization forScientific Research (Gravitation; Zwaartekracht), Maag, Leveren Darm Stichting (MLDS) (FP13-07), and Alpe d’Huzes/Dutch Cancer Society (CONNECTION).
REFERENCES
1. Berger, S. L. (2007) The complex language of chromatinregulation during transcription. Nature 447, 407–412
2. Kouzarides, T. (2007) Chromatin modifications and theirfunction. Cell 128, 693–705
3. Gregoretti, I. V., Lee, Y. M., and Goodson, H. V. (2004)Molecular evolution of the histone deacetylase family:functional implications of phylogenetic analysis. J. Mol. Biol.338, 17–31
4. Glozak, M. A., Sengupta, N., Zhang, X., and Seto, E. (2005)Acetylation and deacetylation of non-histone proteins. Gene 363,15–23
5. Khan, O., and La Thangue, N. B. (2012) HDAC inhibitors incancer biology: emerging mechanisms and clinical applications.Immunol. Cell Biol. 90, 85–94
6. Wagner, J. M., Hackanson, B., Lubbert, M., and Jung, M. (2010)Histone deacetylase (HDAC) inhibitors in recent clinical trialsfor cancer therapy. Clin Epigenetics 1, 117–136
7. Witt, O., Deubzer, H. E., Milde, T., and Oehme, I. (2009) HDACfamily: What are the cancer relevant targets? Cancer Lett. 277,8–21
8. Weichert, W., Roske, A., Niesporek, S., Noske, A., Buckendahl,A. C., Dietel, M., Gekeler, V., Boehm, M., Beckers, T., andDenkert, C. (2008) Class I histone deacetylase expression hasindependent prognostic impact in human colorectal cancer:specific role of class I histone deacetylases in vitro and in vivo.Clin. Cancer Res. 14, 1669–1677
9. Zhu, P., Martin, E., Mengwasser, J., Schlag, P., Janssen, K. P.,and Gottlicher, M. (2004) Induction of HDAC2 expressionupon loss of APC in colorectal tumorigenesis. Cancer Cell 5,455–463
10. Heath, J. P. (1996) Epithelial cell migration in the intestine. CellBiol. Int. 20, 139–146
11. Radtke, F., and Clevers, H. (2005) Self-renewal and cancer of thegut: two sides of a coin. Science 307, 1904–1909
12. Medema, J. P., and Vermeulen, L. (2011) Microenvironmentalregulation of stem cells in intestinal homeostasis and cancer.Nature 474, 318–326
13. Prasetyanti, P. R., Zimberlin, C. D., Bots, M., Vermeulen, L.,Melo, Fde. S., and Medema, J. P. (2013) Regulation of stem cellself-renewal and differentiation by Wnt and Notch are conservedthroughout the adenoma-carcinoma sequence in the colon. Mol.Cancer 12, 126
14. Vermeulen, L., De Sousa E Melo, F., van der Heijden, M.,Cameron, K., de Jong, J. H., Borovski, T., Tuynman, J. B.,Todaro, M., Merz, C., Rodermond, H., Sprick, M. R.,Kemper, K., Richel, D. J., Stassi, G., and Medema, J. P.(2010) Wnt activity defines colon cancer stem cells and isregulated by the microenvironment. Nat. Cell Biol. 12,468–476
15. Kim, K. A., Kakitani, M., Zhao, J., Oshima, T., Tang, T., Binnerts,M., Liu, Y., Boyle, B., Park, E., Emtage, P., Funk, W. D., andTomizuka, K. (2005) Mitogenic influence of human R-spondin1on the intestinal epithelium. Science 309, 1256–1259
16. Korinek, V., Barker, N., Moerer, P., van Donselaar, E., Huls, G.,Peters, P. J., and Clevers, H. (1998) Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4.Nat. Genet. 19, 379–383
17. Pinto, D., Gregorieff, A., Begthel, H., and Clevers, H. (2003)Canonical Wnt signals are essential for homeostasis of theintestinal epithelium. Genes Dev. 17, 1709–1713
18. Kuhnert, F., Davis, C. R., Wang, H. T., Chu, P., Lee, M., Yuan, J.,Nusse, R., and Kuo, C. J. (2004) Essential requirement for Wntsignaling in proliferation of adult small intestine and colonrevealed by adenoviral expression of Dickkopf-1. Proc. Natl. Acad.Sci. USA 101, 266–271
19. Lagger, G., O’Carroll, D., Rembold, M., Khier, H., Tischler, J.,Weitzer, G., Schuettengruber, B., Hauser, C., Brunmeir, R.,Jenuwein, T., and Seiser, C. (2002) Essential function of histonedeacetylase 1 in proliferation control and CDK inhibitorrepression. EMBO J. 21, 2672–2681
20. Trivedi, C. M., Luo, Y., Yin, Z., Zhang, M., Zhu, W., Wang, T.,Floss, T., Goettlicher, M., Noppinger, P. R., Wurst, W., Ferrari,V. A., Abrams, C. S., Gruber, P. J., and Epstein, J. A. (2007)Hdac2 regulates the cardiac hypertrophic response by modulatingGsk3 beta activity. Nat. Med. 13, 324–331
21. Zimmermann, S., Kiefer, F., Prudenziati, M., Spiller, C., Hansen,J., Floss, T., Wurst, W., Minucci, S., and Gottlicher, M. (2007)Reduced body size and decreased intestinal tumor rates inHDAC2-mutant mice. Cancer Res. 67, 9047–9054
22. Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H.,Joseph, N., Gao, J., Nieland, T. J., Zhou, Y., Wang, X., Mazitschek,R., Bradner, J. E., DePinho, R. A., Jaenisch, R., and Tsai, L. H.(2009) HDAC2 negatively regulates memory formation andsynaptic plasticity. Nature 459, 55–60
23. Haberland, M., Montgomery, R. L., and Olson, E. N. (2009) Themany roles of histone deacetylases in development andphysiology: implications for disease and therapy. Nat. Rev.Genet. 10, 32–42
24. Montgomery, R. L., Davis, C. A., Potthoff, M. J., Haberland, M.,Fielitz, J., Qi, X., Hill, J. A., Richardson, J. A., and Olson, E. N.(2007) Histone deacetylases 1 and 2 redundantly regulatecardiac morphogenesis, growth, and contractility. Genes Dev. 21,1790–1802
25. Wilting, R. H., Yanover, E., Heideman, M. R., Jacobs, H.,Horner, J., van der Torre, J., DePinho, R. A., and Dannenberg,J. H. (2010) Overlapping functions of Hdac1 and Hdac2 incell cycle regulation and haematopoiesis. EMBO J. 29,2586–2597
26. Turgeon, N., Blais, M., Gagne, J. M., Tardif, V., Boudreau, F.,Perreault, N., and Asselin, C. (2013) HDAC1 and HDAC2restrain the intestinal inflammatory response by regulatingintestinal epithelial cell differentiation. PLoS ONE 8, e73785
27. Turgeon, N., Gagne, J. M., Blais, M., Gendron, F. P., Boudreau,F., and Asselin, C. (2014) The acetylome regulators Hdac1 andHdac2 differently modulate intestinal epithelial cell dependenthomeostatic responses in experimental colitis. Am. J. Physiol.Gastrointest. Liver Physiol. 306, G594–G605
28. Heideman, M. R., Wilting, R. H., Yanover, E., Velds, A., de Jong, J.,Kerkhoven, R. M., Jacobs, H., Wessels, L. F., and Dannenberg, J. H.
10 Vol. 29 May 2015 ZIMBERLIN ET AL.The FASEB Journal x www.fasebj.org

(2013) Dosage-dependent tumor suppression by histone deacety-lases 1 and 2 through regulation of c-Myc collaborating genes andp53 function. Blood 121, 2038–2050
29. Santoro, F., Botrugno, O. A., Dal Zuffo, R., Pallavicini, I.,Matthews, G. M., Cluse, L., Barozzi, I., Senese, S., Fornasari, L.,Moretti, S., Altucci, L., Pelicci, P. G., Chiocca, S., Johnstone,R. W., andMinucci, S. (2013) A dual role for Hdac1: oncosuppressorin tumorigenesis, oncogene in tumor maintenance. Blood 121,3459–3468
30. Winter, M., Moser, M. A., Meunier, D., Fischer, C., Machat,G., Mattes, K., Lichtenberger, B. M., Brunmeir, R.,Weissmann, S., Murko, C., Humer, C., Meischel, T., Brosch,G., Matthias, P., Sibilia, M., and Seiser, C. (2013)Divergent roles of HDAC1 and HDAC2 in the regulation ofepidermal development and tumorigenesis. EMBO J. 32,3176–3191
31. Ireland, H., Kemp, R., Houghton, C., Howard, L., Clarke,A. R., Sansom, O. J., and Winton, D. J. (2004) InducibleCre-mediated control of gene expression in the murine gastro-intestinal tract: effect of loss of beta-catenin. Gastroenterology 126,1236–1246
32. Kemp, R., Ireland, H., Clayton, E., Houghton, C., Howard, L.,and Winton, D. J. (2004) Elimination of backgroundrecombination: somatic induction of Cre by combinedtranscriptional regulation and hormone binding affinity. NucleicAcids Res. 32, e92
33. Heijmans, J., van Lidth de Jeude, J. F., Koo, B. K., Rosekrans,S. L., Wielenga, M. C., van de Wetering, M., Ferrante, M., Lee,A. S., Onderwater, J. J., Paton, J. C., Paton, A. W., Mommaas,A. M., Kodach, L. L., Hardwick, J. C., Hommes, D. W., Clevers,H., Muncan, V., and van den Brink, G. R. (2013) ER stress causesrapid loss of intestinal epithelial stemness through activation ofthe unfolded protein response. Cell Reports 3, 1128–1139
34. Clevers, H. (2013) Stem Cells: A unifying theory for the crypt.Nature 495, 53–54
35. Garnett, M. J., Edelman, E. J., Heidorn, S. J., Greenman, C. D.,Dastur, A., Lau, K. W., Greninger, P., Thompson, I. R., Luo, X.,Soares, J., Liu, Q., Iorio, F., Surdez, D., Chen, L., Milano, R. J.,Bignell, G. R., Tam, A. T., Davies, H., Stevenson, J. A.,Barthorpe, S., Lutz, S. R., Kogera, F., Lawrence, K.,McLaren-Douglas, A., Mitropoulos, X., Mironenko, T., Thi, H.,Richardson, L., Zhou, W., Jewitt, F., Zhang, T., O’Brien, P.,Boisvert, J. L., Price, S., Hur, W., Yang, W., Deng, X., Butler, A.,Choi, H. G., Chang, J. W., Baselga, J., Stamenkovic, I.,Engelman, J. A., Sharma, S. V., Delattre, O., Saez-Rodriguez,J., Gray, N. S., Settleman, J., Futreal, P. A., Haber, D. A.,Stratton, M. R., Ramaswamy, S., McDermott, U., and Benes,C. H. (2012) Systematic identification of genomic markers ofdrug sensitivity in cancer cells. Nature 483, 570–575
Received for publication August 13, 2014.Accepted for publication January 9, 2015.
INTESTINAL STEM CELLS REQUIRE HDAC1 AND HDAC2 11