enzyme structure, function, and evolution in flavonoid
TRANSCRIPT

Enzyme Structure, Function, and Evolution in Flavonoid Biosynthesis
by
Geoffrey Liou
B.A. Molecular and Cell Biology University of California, Berkeley, 2013
Submitted to the Department of Biology
in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2019
© 2019 Massachusetts Institute of Technology. All rights reserved. Signature of Author _____________________________________________________________
Geoffrey Liou Department of Biology
May 24, 2019 Certified by ___________________________________________________________________
Jing-Ke Weng Assistant Professor of Biology
Thesis Supervisor Accepted by __________________________________________________________________
Amy E. Keating Professor of Biology
Co-Director, Biology Graduate Committee
1

2

Enzyme Structure, Function, and Evolution in Flavonoid Biosynthesis
by
Geoffrey Liou
Submitted to the Department of Biology on May 24, 2019 in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Biology Abstract
Plant specialized metabolism is a key evolutionary adaptation that has enabled plants to migrate from water onto land and subsequently spread throughout terrestrial environments. Flavonoids are one particularly important class of plant specialized metabolites, playing a wide variety of roles in plant physiology including UV protection, pigmentation, and defense against herbivores and pathogens. Flavonoid diversity has increased in conjunction with land plant evolution over the past 470 million years. This dissertation examines the structure, function, and evolution of enzymes in the flavonoid biosynthetic pathway. First, we structurally and biochemically characterized orthologs of chalcone synthase (CHS), the enzyme that catalyzes the first step of flavonoid biosynthesis, from diverse plant lineages. By doing so, we gained insight into the sequence changes that gave rise to increased reactivity of the catalytic cysteine residue in CHS orthologs in euphyllophytes compared to basal land plants. We then developed methods and transgenic plant lines to study the in vivo function of these CHS orthologs, as well as whether their functional differences play a role in redox-based regulation of flavonoid biosynthesis. Finally, we examined enzymes involved in the biosynthesis of galloylated catechins, a highly enriched class of flavonoids in tea that are thought to have health benefits in humans. These findings contribute to an understanding of the evolution of enzyme structure and function in flavonoid biosynthesis, and how it has facilitated the adaptation of plants to a wide variety of terrestrial habitats. Thesis Supervisor: Jing-Ke Weng Title: Assistant Professor of Biology
3

4

Acknowledgements First and foremost, I would like to thank my thesis advisor, Jing-Ke Weng. I feel very fortunate to have joined the lab as the first group of students and to have watched it grow over the years. I remember being instantly captivated by the research when I saw your presentation to the first-year graduate students. It was a perfect alignment of my interests at the time, structural biology and plant biology, but my experience has turned out to be more than I could ever have imagined. Your enthusiasm for and breadth of knowledge in all aspects of biology has been inspiring and has taught me to keep learning and discover my passions. Your insight and support has been invaluable in helping me think more broadly and work past the difficult parts of my research. To the members of the Weng Lab, I can’t imagine a better group of people to work with. Tim Fallon, you have been a great classmate, seat neighbor, labmate, roommate, and most of all friend throughout the years. I’ll never forget the first time I saw fireflies when we went to New Jersey to collect Photinus pyralis. Olesya Levsh, I’m so glad to have joined the lab together with you. Your kindness set the tone of the lab from the beginning and has left a mark in the form of many lab traditions. Joe Jacobowitz, thanks for all the fun game nights and putting up with our teasing. Sophia Xu, it has been great sharing Asian snacks and bonding as baymates despite our differences (Go Bears!). Bena Chan, thank you for warmly welcoming me to the lab during my rotation, and for staying in touch and all your help in your current position in the Metabolomics Core. Valentina Carballo, thank you for everything you do to keep the lab running and making everyone feel like part of a family. Fu-Shuang Li, it has been inspiring to see your dedication to your family and your work. Mike Spence, thanks for imparting your wisdom in biochemistry and life experience over the years. Bastien Christ, thank you for sharing your knowledge of plants, fondue, and sense of adventure. Tomáš Pluskal, I have learned so much about metabolomics, mind-bending films, and life from you, and I aspire to be an international man of mystery like you. Roland Kersten, thanks for your help over the years and bringing some California spirit to the lab. Chengchao Xu, Andrew Mitchell, Yasmin Chau, Matthew Hill, Chris Glinkerman, Menglong Xu, Anastassia Bobokalonova, Amy Zhang, Sheena Vazquez, Brian Levine, Jack Liu, Michael Gutierrez, Naoki Wada, Colin Kim, and others: thank you for making this lab a fun place to work. It has been a pleasure and privilege to get to know such an interesting, diverse group of people. Thanks to Biograd 2013 for all the memories. In particular, Chetan, Aneesha, Rachit, Emir, Nicole, and Amelie, it’s been great sharing our journey through grad school and getting together to relax and unwind. Thank you to my friends at MIT Japanese Lunch Table, in the Cal Alumni Club of New England, in the Boston area, in New York, back home in California, and in Japan, and also my extended family in Taiwan, for all the fun times and keeping me connected to the things I love outside of school. Special thanks to MIT Japan Program Director Chris Pilcavage, Miyuki-san, Masako-san, the Baber family, Joey, Kristine, Matthew, Mark, Diana, Sherman, Roger, Yuzo, Heechan, Willie, Sam, David, Alex, Daniel, Tomoya, Moka, Chihhi, Yuihan, and Dahyun, among many others.
5

Finally, I want to thank my family. Kerry and Zachary, it’s been great to watch you follow your own paths as we become independent adults. To my mother and father, thank you for everything you have done for us, first and foremost valuing our education, which has brought us to where we are today. We are very fortunate to have so much opportunity here in the United States, and I am grateful for all your sacrifice and hard work that has brought us here.
6

Table of Contents
Abstract 3 Acknowledgements 5 Table of Contents 7 Chapter 1. Introduction 9 Overview of land plant evolution 9 Plant specialized metabolism 11 Flavonoid biosynthesis and diversity 13 Type III polyketide synthases 18 Applications of plant metabolic and enzyme engineering 21 Concluding remarks 24 References 26 Chapter 2. Mechanistic basis for the evolution of chalcone synthase catalytic cysteine
reactivity in land plants 31 Abstract 32 Introduction 33 Results 37
Basal-plant CHSs contain reduced catalytic cysteine in their crystal structures 37 Basal-plant CHSs only partially complement the Arabidopsis CHS-null mutant 41 The pKa of the catalytic cysteine is higher in basal-plant CHSs than in euphyllophyte
CHSs 42 Residues near the active-site cavity affect the pKa and reactivity of the catalytic
cysteine 43 Molecular dynamics simulations reveal differences in active-site interactions between
basal-plant and euphyllophyte CHSs 49 Discussion 54 Materials and Methods 58 References 65 Supporting Information 68 Chapter 3. Regulation of chalcone synthase activity in vivo by oxidation of the catalytic
cysteine 81 Abstract 82 Introduction 83 Results 87
tt5 mutant Arabidopsis thaliana accumulates naringenin and can be used for metabolic tracing to measure CHS activity in vivo 87
tt5 mutant Arabidopsis thaliana accumulates both enantiomers of naringenin 89 Generation of and metabolic tracing with tt5 and mbs1-1 mutant Arabidopsis crosses 89
7

The catalytic cysteine in AtCHS is more sensitive to in vitro oxidation than in SmCHS 91 FLAG-tag purification and western blotting of CHS 95 Generation and characterization of transgenic Arabidopsis thaliana lines expressing
FLAG-tagged CHS orthologs 98 Discussion and Future Directions 101 Materials and Methods 103 References 111 Appendix. Investigation of galloylated catechin biosynthetic enzymes in tea 113 Abstract 114 Introduction 115 Results 119
CsUGGT expression in Nicotiana benthamiana produces β-glucogallin 119 Identification of ECGT candidate genes 119 Nicotiana benthamiana leaf protein extraction fails to show ECGT activity 121
Discussion and Future Directions 124 Materials and Methods 126 References 131
8

Chapter 1 Introduction Overview of land plant evolution
Terrestrial life as it exists today was seeded and shaped by the transition of plants from
water to land. Embryophytes, or land plants, evolved from the charophycean green algae
approximately 470 million years ago (Kenrick & Crane, 1997). Life on land brought numerous
challenges to plants previously ameliorated by an aquatic environment: ultraviolet radiation,
desiccation, lack of structural support, and gas exchange. As other clades of life also adapted to
land, plants also needed to fend off pathogens and herbivores. To adapt to these previously
unencountered stresses, plants have evolved an extraordinary array of physiological innovations.
Departure from aquatic life required plants to develop a way to prevent desiccation on
land. The evolution of the cuticle, a thin layer of lipids and waxes on the epidermal cells of
plants, allowed for the control of water loss from transpiration and gas exchange (Riederer &
Muller, 2008). This innovation also allowed plants to concentrate the resources necessary for
photosynthesis, enabling more efficient carbon fixation and energy production. Sporopollenin, an
extremely chemically inert polymer, was developed to protect spores and pollen, allowing plants
to reproduce without dependence on water and to disseminate their progeny long distances,
further facilitating their spread across land (Li, Phyo, Jacobowitz, Hong, & Weng, 2019).
Without the buoyant forces provided by water, plants needed to develop new systems of
structural support. Lignin, a hydrophobic polymer, provided this rigidity and also enabled the
development of vascular tissues to transport water long distances (Weng & Chapple, 2010).
9

These physical features combined to allow plants to grow much larger in size than previously
possible, furthering their dominance over the terrestrial landscape. Lignin is now the second
most abundant organic polymer, surpassed only by cellulose, illustrating the outsized importance
of this metabolic innovation in shaping life on Earth (Boerjan, Ralph, & Baucher, 2003). The rise
of vascular plants further shaped the biosphere: lignin provided a stable sink for carbon, and the
development of roots led to increased weathering of Ca-Mg rocks (Berner, 1993). These
processes both contributed to an 8 to 20-fold decrease in atmospheric CO2 (Harrison & Morris,
2018). The subsequent increase in atmospheric O2 enabled the diversification of many clades of
animals, as well as the evolution of physiological features such as flight in insects and megaphyll
leaves in plants (Beerling, Osborne, & Chaloner, 2001; Graham, Aguilar, Dudley, & Gans,
1995).
The evolution of seeds, which occurred roughly 320 million years ago, granted a variety
of advantages that allowed embryophytes to reproduce successfully even in difficult
environments: the seed coat protects the embryo from physical damage and herbivores, the
endosperm provides a source of nutrients, and dormancy allows germination to be delayed until
environmental conditions are favorable (Linkies, Graeber, Knight, & Leubner-Metzger, 2010).
The emergence of flowers approximately 130 million years ago provided a more efficient
method of pollination. During the Cretaceous, angiosperms subsequently underwent
extraordinary diversification and became the dominant flora on Earth, outnumbering all other
land plants in species abundance. The majority of angiosperms today are pollinated by insects,
serving as a prominent example of symbiosis and its far-reaching impact on evolution (Friis,
Crane, & Pedersen, 2011).
10

Plant specialized metabolism
Evolutionary adaptation to new ecological niches was facilitated by not only anatomical
adaptations but also metabolic innovations. As sessile organisms, plants cannot move away from
stressors and instead have developed a vast chemical arsenal to adapt to their ecological niches.
In contrast to primary metabolism conserved across all kingdoms of life, these biosynthetic
pathways are often restricted to particular lineages of plants. These metabolites are also not
strictly essential for survival, although they can greatly enhance the fitness of organisms in a
particular niche. This limited distribution and essentiality led to these pathways initially being
dubbed secondary metabolism, but as scientists came to better understand the roles of these
diverse chemical compounds, they were renamed specialized metabolism (Moghe & Last, 2015).
Specialized metabolites act as pigments, flavors, scents, defense compounds, and so on to
mitigate biotic or abiotic stresses, attract pollinators or seed dispersers, deter herbivores, and
defend against pathogens, among many other functions. Many of these compounds also show
evidence of therapeutic effects in humans, giving rise to various traditions of herbal medicine
around the world. A prominent example is artemisinin from sweet wormwood, or Artemisia
annua; Tu Youyou was awarded the 2015 Nobel Prize in Physiology or Medicine for her work in
identifying the active compound of this traditional Chinese medicine. Even modern medicine
relies heavily on plant natural products: at least 25% of drugs used today are derived from plant
specialized metabolites (Schmidt, Ribnicky, Lipsky, & Raskin, 2007).
Terpenoids, encompassing over 36,000 different compounds, are the largest class of plant
specialized metabolites (Roberts, 2018). They exhibit enormous structural diversity in both the
basic carbon skeleton—from relatively simple branched alkyl chains to polycyclic, bridged
11

compounds—and subsequent decorations such as hydroxylation and oxidation. Due to their
hydrophobic and volatile nature, terpenoids often function in signaling, which encompasses
defense against herbivores, attraction of pollinators, growth inhibition of nearby plants, and more
(Roberts, 2018).
Terpenoids are terpenes modified with additional functional groups. Common among all
these varied structures is the basic building block of C5 isoprene units, which are synthesized by
the mevalonic acid pathway or the methylerythritol phosphate pathway. These isoprene units are
joined together by various prenyltransferases to form a linear intermediate, which is then
cyclized by terpene cyclases that fold the compound into a conformation to facilitate cyclization.
The resulting products are classified by their size: monoterpene (C10), sesquiterpene (C15), or
diterpene (C20). Longer products such as triterpenes (C30) are synthesized from the linear
intermediate squalene and cyclized by oxidosqualene cyclase (Thimmappa, Geisler, Louveau,
O’Maille, & Osbourn, 2014). Subsequent modifications of terpenoids are performed by many
classes of enzymes, such as cytochrome P450 enzymes and 2-oxoglutarate-dependent
dioxygenases (Roberts, 2018).
Alkaloids are another large class of plant specialized metabolites, totaling about 12,000
compounds (Ziegler & Facchini, 2008). They are highly diverse in structure, with a heterocyclic
nitrogen being the only common functional group. Accordingly, the various classes of alkaloids
have diverse biosynthetic origins, such as the benzylisoquinolines derived from tyrosine, or
purine alkaloids derived from purine nucleotides. Many alkaloids show pharmacological effects
in humans, from anticancer compounds like vinblastine to stimulants like caffeine and nicotine.
12

Phenylpropanoids are another major class of plant specialized metabolites. They are
found in all land plants and function in mediating many biotic and abiotic stresses and
interactions (Vogt, 2010). Phenylpropanoid biosynthesis derives from the shikimate pathway,
which produces the aromatic amino acid phenylalanine (Chapter 2, Figure 1). Phenylalanine is
then deaminated by phenylalanine ammonia lyase (PAL) to form cinnamic acid. Cinnamic acid
4-hydroxylase (C4H) then produces 4-coumarate (or p-coumarate), which is conjugated to
coenzyme A by 4-coumarate:CoA ligase (4CL) to produce 4-coumaroyl-CoA (or
p-coumaroyl-CoA).
A key branching point occurs at this step, where p-coumaroyl-CoA can be used by
chalcone synthase to feed into flavonoid biosynthesis. The other branch is catalyzed by
hydroxycinnamoyl-CoA:shikimate hydroxycinnamoyl transferase (HCT), which reacts
p-coumaroyl-CoA with shikimic acid to form p-coumaroyl shikimate (Levsh et al., 2016). This
compound serves as the precursor for many important phenylpropanoid metabolites, including
monolignols, the monomeric units of lignin. Other phenylpropanoids function as pigments,
flavor compounds, phytoalexins, and so on (Vogt, 2010).
Flavonoid biosynthesis and diversity
Flavonoids are a diverse class of plant specialized metabolites found in all extant land
plants. They serve many of aforementioned roles important for plants’ survival on land: UV
protection, pigmentation, defense, and communication with symbiotic microbes (Winkel-Shirley,
2001). Flavonoids have also garnered considerable interest for numerous potential health benefits
in humans, including antioxidant, anticancer, cardioprotective, and anti-aging effects, which
13

have been observed in a wide variety of studies ranging from cell culture experiments to mouse
models to epidemiological studies. (Yao et al., 2004).
Flavonoids consist of a C6-C3-C6 core structure, formally called a phenylbenzopyran
moiety, with the three rings named A, B, and C and carbon atoms numbered as shown in Figure
1. Flavonoid biosynthesis derives from phenylpropanoid biosynthesis. Chalcone synthase (CHS)
catalyzes the first step by reacting p-coumaroyl-CoA with three molecules of malonyl-CoA to
form naringenin chalcone (Heller & Hahlbrock, 1980). Chalcone isomerase (CHI) then rapidly
and stereospecifically performs a ring closure to form (2S)-naringenin, the precursor to
downstream flavonoid biosynthesis.
Over 6000 flavonoids have been identified in plants; this enormous diversity is achieved
through various enzymatic modifications including hydroxylation, O-methylation, prenylation,
glycosylation, oxidation, and reduction (Austin & Noel, 2003). In particular, different classes of
flavonoids are named based on the degree of oxidation and saturation at the C-3, C-4, and C-5
positions of the C ring (Figure 1). These various tailoring enzymes can be expressed either
constitutively or in response to developmental or environmental cues, and many are restricted to
certain plant lineages that have evolved a class of flavonoids as a specific adaptation (R. A.
Dixon & Paiva, 1995).
Naringenin, a flavanone, can be hydroxylated by flavanone 3-hydroxylase (F3H) to
produce dihydrokaempferol (DHK), a dihydroflavonol. These first three enzymatic steps of CHS,
CHI, and F3H likely evolved early in land plants, namely in bryophytes, liverworts, and
hornworts (Markham, 1988). These early flavonoids likely acted as sunscreens against UV
radiation damage, because they can absorb UV wavelengths, and because they are found in the
14
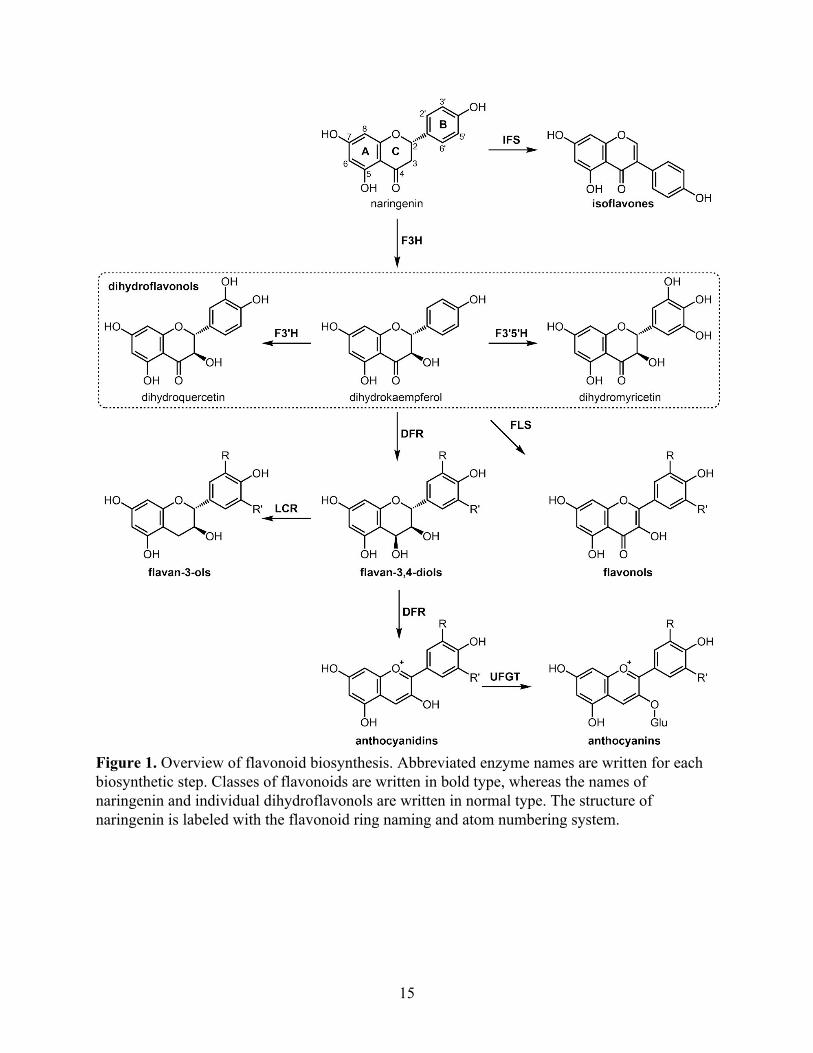
Figure 1. Overview of flavonoid biosynthesis. Abbreviated enzyme names are written for each biosynthetic step. Classes of flavonoids are written in bold type, whereas the names of naringenin and individual dihydroflavonols are written in normal type. The structure of naringenin is labeled with the flavonoid ring naming and atom numbering system.
15

upper leaf surface in the epicuticular wax due to their lipophilicity (Harborne & Williams, 2000).
Another possible function, evolved even before UV protection, is that of regulating or
chaperoning plant hormones, because the low amounts of flavonoids produced by early enzymes
could have been sufficient for this role (Stafford, 1991).
Another important step in flavonoid diversification likely evolved in these early land
plants as well: the three major branches of flavonoid modification by differential B-ring
hydroxylation (Rausher, 2006). The B-ring of DHK can be hydroxylated at the 3′ and/or 5′
position by flavonoid 3′-hydroxylase (F3′H) or flavonoid 3′,5′-hydroxylase (F3′5′H) to form
dihydroquercetin (DHQ) or dihydromyricetin (DHM). These dihydroflavonols can then be
converted by flavonol synthase (FLS) into flavonols, which are also found in all land plants
(Rausher, 2006).
Vascular plants, beginning with pteridophytes, produce flavan-3,4-diols from
dihydroflavonols using dihydroflavonol-4-reductase (DFR). Flavan-3,4-diols can polymerize to
form condensed tannins (also known as proanthocyanidins, because their depolymerization
produces anthocyanidins). Tannins function primarily as defense against bacterial and fungal
pathogens, and their astringency also deters herbivores (Feeny, 1970).
In seed plants, flavan-3,4-diols (also known as leucoanthocyanidins) can also be oxidized
by anthocyanidin synthase (ANS) to form anthocyanidins. Glycosylation of these aglycones,
usually at the 3-O position by anthocyanin 3-O-glucosyltransferase (UFGT) using UDP-glucose,
results in anthocyanins, the major red-blue pigments found usually in fruits and flowers but also
in other parts of the plant. Different combinations of hydroxylation and methylation of the 3′ and
5′ positions, as well as the identity of the glycosyl group(s), determines the color of the final
16

anthocyanin. Anthocyanins are stored in vacuoles, where the pH and complex formation with
metals, malonic acid, or flavones may also modify the color (Austin & Noel, 2003). Flower color
is critical in mediating interactions with pollinators, and evolutionary changes in color, underlain
by evolution of flavonoid biosynthetic enzymes, often coincide with changes in flower
morphology (Rausher, 2006).
Isoflavonoids are a class of flavonoids restricted mostly to legumes, except for a few
gymnosperm lineages and one moss species (Dewick, 1988). Their structure differs from other
flavonoids in that the B ring is shifted from C-2 to C-3 on the lactone ring, the result of an
oxidative rearrangement catalyzed by isoflavone synthase (IFS) (Austin & Noel, 2003).
Isoflavonoids function in communication with symbiotic rhizobia, a specialized feature of
legumes.
As is the case with many specialized metabolic pathways, the enzymes catalyzing these
various biosynthetic steps evolved from progenitor enzymes in primary metabolism (Weng,
Philippe, & Noel, 2012). Most of the tailoring enzymes in flavonoid biosynthesis are members of
one of three enzyme families: 2-oxoglutarate-dependent dioxygenases (2OGD, such as F3H and
ANS), cytochrome P450 reductases (P450, such as F3′H and F3′5′H), or NADPH-dependent
reductases (such as DFR and LCR) (Richard A. Dixon & Steele, 1999; Winkel-Shirley, 2001).
Catalytic promiscuity is also a common feature of specialized metabolic enzymes, and this
feature is also critical for the diversity of flavonoids (Weng et al., 2012). For example, DHK,
DHQ, and DHM can all be reduced by DFR or converted into flavonols by FLS, forming a
metabolic grid. This extensive flavonoid diversity, produced by the myriad combinations of
17

modifications by specialized metabolic enzymes, has enabled plants to adapt to a wide range of
ecological niches.
Type III polyketide synthases
CHS is no different from the aforementioned specialized metabolic enzymes in
possessing evolutionary origins in primary metabolic enzymes. CHS is a member of the type III
polyketide synthase (PKS) superfamily, which evolved from β-ketoacyl-acyl carrier protein
synthase III (KAS III), a type of fatty acid synthase in bacterial fatty acid biosynthesis (Austin &
Noel, 2003). Type III PKS enzymes and KAS III share a conserved structural fold and an
analogous catalytic mechanism. Both are homodimers, in which each monomer consists of an
αβαβα thiolase fold domain and a bottom domain that together form the active-site cavity, and
both contain the same catalytic triad residues (Ferrer, Jez, Bowman, Dixon, & Noel, 1999). Both
enzymes perform the same basic catalysis of adding a two-carbon acetate unit to an acyl thioester
starter molecule: KAS III uses malonyl-acyl carrier protein as the donor to lengthen a 2-carbon
fatty acid thioester (e.g. acetyl-CoA) to a 4-carbon fatty acid in the first step of fatty acid
biosynthesis in bacteria, whereas CHS uses three molecules of malonyl-CoA to iteratively extend
p-coumaroyl-CoA by a total of six carbons to form a polyketide.
The CHS catalytic triad consists of C164, H303, and N336 (Chapter 2, Figure 1), as
numbered in Medicago sativa CHS (MsCHS), the first CHS ortholog to be structurally
characterized by X-ray crystallography (Ferrer et al., 1999). The catalytic cysteine is conserved
in all thiolase-fold enzymes and is located at the N-terminus of an α-helix. This cysteine
performs the first step of the CHS catalytic mechanism, a nucleophilic attack on the
18

p-coumaroyl-CoA substrate to generate an acyl-enzyme intermediate. To perform this reaction,
the cysteine must be present in the deprotonated thiolate state, suggesting that the active-site
environment has evolved to lower the pKa of cysteine from 8.8 of free cysteine in solution to a
value below physiological pH. The helix dipole moment lowers the pKa of cysteine from 8.8 to
7.2 in model peptides, suggesting a mechanistic role for this conserved structural feature
(Kortemme & Creighton, 1995). The Nε of H303 of the catalytic triad forms a stable
imidazolium-thiolate ion pair with C164, and mutations of H303 to glutamate and alanine shift
the cysteine pKa from 5.5 to 6.6 and 7.6, respectively (Jez & Noel, 2000; Suh, Kagami, Fukuma,
& Sankawa, 2000).
The protonated nitrogens of H303 and N336 form an oxyanion hole that stabilizes
multiple steps of the CHS catalytic mechanism (Austin & Noel, 2003). First, the tetrahedral
transition state formed after Cys nucleophilic attack is stabilized. Second, the enol tautomer of
malonyl-CoA is stabilized, promoting its decarboxylation and subsequent condensation with the
p-coumaroyl moiety. A conserved phenylalanine residue (position 215 in MsCHS) also promotes
the formation of a neutral CO2. These loading, decarboxylation, and condensation steps are
performed three times until a linear tetraketide intermediate is formed. An intramolecular Claisen
condensation then occurs between C-1 and C-6, followed by aromatization to form naringenin
chalcone.
Many details of the CHS catalytic mechanism have been elucidated by comparing CHS
to other members of the type III PKS superfamily that utilize different acyl donors and acceptors
to produce a wide variety of polyketides. The number of malonyl-CoA units incorporated, which
determines the length of the linear polyketide intermediate and thus the size of the final product,
19

depends upon the volume of the active-site cavity. The enzyme 2-pyrone synthase (2-PS) from
Gerbera hybrida, for example, uses a smaller acetyl-CoA starter molecule and performs only
two malonyl-CoA additions to produce triacetic acid lactone. The overall structures of CHS and
2-PS are highly similar, except for a two-thirds reduction in active-site volume in 2-PS resulting
from three key active-site residue substitutions compared to CHS (Jez et al., 2000). In a striking
example, octaketide synthase (OKS) from Aloe arborescens, which uses eight molecules of
malonyl-CoA to produce SEK4 and SEK4b, was subjected to site-directed mutagenesis to
thoroughly investigate the effect of active-site volume on product profile. A series of
substitutions of a key glycine residue, ranging from a small alanine to a large tryptophan,
generated correspondingly smaller products ranging from heptaketides to tetraketides (Abe,
Oguro, Utsumi, Sano, & Noguchi, 2005). Together, these examples illustrate how the steric bulk
of residues lining the active site are responsible for limiting the iterative extension steps and
directing the subsequent cyclization step of the type III PKS mechanism.
The cyclization step is another avenue for diversification in type III PKS enzymes.
Stilbene synthase (STS) catalyzes the formation of resveratrol, a compound that has garnered
significant attention for its potential contribution to the health benefits of red wine consumption
(Frémont, 2000). STS uses the same substrates as CHS but differs in its cyclization mechanism,
which involves a C-2 to C-7 intramolecular aldol condensation. STS and CHS are 60-90%
identical in amino acid sequence, and the difference in function is due to a few substitutions near
a buried loop, which causes a subtle change in the hydrogen-bonding network of an active-site
threonine. This small difference in the electronic environment of the active site is enough to
favor one cyclization mechanism over another (Austin, Bowman, Ferrer, Schröder, & Noel,
20

2004). In summary, subtle changes in side chain positioning, even if caused by a mutation distant
from the residue involved in the catalytic mechanism, can lead to large differences in type III
PKS function.
Applications of plant metabolic and enzyme engineering
Metabolic engineering is the modification of existing biosynthetic pathways to change
the amounts of metabolites produced in a particular organism, or to create new pathways and
metabolites altogether. Metabolic flux of existing chemistries can be altered by deleting or
overexpressing biosynthetic genes in an existing pathway, or by expressing biosynthetic genes in
a heterologous host. Novel chemical reactions, however, require engineering of individual
enzymes to perform new catalysis (Erb, Jones, & Bar-Even, 2017). To accomplish this,
understanding the structure-function relationships of enzymes is critical.
Plant metabolic engineering has many potential applications in energy, pharmaceuticals,
food, and agriculture. Biofuels have emerged as a fossil fuel alternative for transportation and
other energy needs, but the use of food crops such as corn and sugarcane can conflict with the
rising demand for food, especially in developing countries (Tyner, 2012). Second-generation
biofuels, which use non-food crop plants (e.g. switchgrass or miscanthus) or residual biomass
from food crops, may be an alternative source (Evans, Ramage, DiRocco, & Potts, 2015). The
presence of lignin in plant cell walls, however, inhibits access to polysaccharides by enzymes
used to produce fermentable sugars.
Given the important role of lignin in structural support and water transport, there have
been extensive efforts to reduce or alter lignin content without causing growth defects. One
21

recent effort in Arabidopsis thaliana involved engineering lignin biosynthesis to occur only in
vessels, while increasing the thickness of cell walls in secondary cells to provide structural
support (Yang et al., 2013). Lignin consisting of syringyl units (S lignin) is less condensed than
lignin containing p-hydroxyphenyl or guaiacyl units (H or G lignin), resulting in enhanced
chemical and enzymatic digestibility of S-rich lignocellulosic biomass (Renault,
Werck-Reichhart, & Weng, 2019). There have been engineering efforts to increase S lignin
content in several angiosperm species by overexpressing enzymes in the biosynthetic pathway
(Franke et al., 2000; Meyer, Shirley, Cusumano, Bell-Lelong, & Chapple, 1998; Stewart,
Akiyama, Chapple, Ralph, & Mansfield, 2009).
As the world population continues to grow, combined with the effects of climate change,
food security will become a greater challenge. Even in a low global warming scenario of 1.5 °C,
crop yields and nutritional composition are predicted to change, and engineered crops with
elevated stress tolerance or nutrient levels could be an important climate change mitigation
strategy (Intergovernmental Panel on Climate Change, 2018). Golden Rice is a well known
example of a crop engineered to address nutritional deficiencies; three genes were heterologously
expressed in rice endosperm to increase the content of beta-carotene, a precursor of vitamin A
(Ye et al., 2000). Flavonoids, given their numerous potential health benefits, could be another
target for metabolic engineering in food crops. Recently, fruit-specific expression of the A.
thaliana transcription factor MYB12 was shown to increase phenylpropanoid content, including
flavonols, in tomatoes (Y. Zhang et al., 2015).
Many small molecule plant hormones control abiotic stress responses, such as auxin,
brassinosteroids, salicylic acid, and jasmonates. Abscisic acid (ABA), a terpenoid, is a
22

particularly important secondary messenger in stress responses like reducing transpiration during
drought stress and enhancing root growth under nitrogen deficiency (Wani, Kumar, Shriram, &
Sah, 2016). As such, many attempts at engineering ABA-mediated stress tolerance have been
made. Overexpression of zeaxanthin epoxidase, an enzyme in ABA biosynthesis, in A. thaliana
conferred elevated tolerance to drought and salt stress (Park et al., 2008). Transgenic
overexpression of A. thaliana LOS5, which activates a key cofactor in the last step of ABA
biosynthesis, in maize led to greater biomass accumulation under salt stress (J. Zhang et al.,
2016).
Enzymes can also be expressed, purified, and used in large-scale industrial reactions as
biocatalysts. This process, also known as chemoenzymatic synthesis, has the advantages of
chemo-, regio-, stereo-, and enantiospecificity over traditional chemical synthesis. Enzyme
engineering has also improved biocatalysis by expanding the substrate range, catalytic rate, and
stability of enzymes (Bornscheuer et al., 2012). Initially, knowledge of an enzyme’s structure
and/or catalytic mechanism allowed for rational design of mutations that could accommodate a
new substrate, for example. With the advent of new biotechnological tools to allow for rapid
DNA synthesis and high-throughput screening of enzyme activity, directed evolution enabled the
identification of beneficial mutations in enzymes whose structures or mechanisms are unknown,
or simple amino acid substitutions that are difficult to rationalize due to epistasis (Tracewell &
Arnold, 2009). Rational design and directed evolution can also be combined to perform
smaller-scale saturating mutagenesis screening of amino acid positions thought to be important
for the desired function (Strohmeier, Pichler, May, & Gruber-Khadjawi, 2011).
23

The majority of biocatalysts are bacterial or fungal enzymes, but one class of plant
enzymes that has been particularly useful in biocatalysis is hydroxynitrile lyases (HNLs). HNLs
from Manihot esculenta (cassava) and Prunus amygdalus (almond) have been subjected to
rational design and saturating mutagenesis to generate enzymes with improved specificity in
producing the correct enantiomer of intermediates in the syntheses of the antiplatelet drug
Clopidogrel, vitamin B5, and other compounds (Strohmeier et al., 2011). Rational mutations have
also been engineered to improve expression of plant HNLs in microbial hosts.
In addition to novel chemistry, biocatalysis also enables more efficient and
environmentally friendly industrial chemical manufacturing. Enzymes are made from renewable
sources and are biodegradable; their higher product purities lead to less waste production; and
enzymatic reactions usually operate at ambient temperature, pressure, and pH, requiring less
energy use (Bornscheuer et al., 2012). Recently, a novel CO2-fixation pathway was designed in
vitro using engineered enzymes from all three kingdoms of life, surpassing the efficiency of the
Calvin cycle used by plants (Schwander, Schada von Borzyskowski, Burgener, Cortina, & Erb,
2016). This metabolic pathway could be engineered into an organism to produce valuable
downstream chemicals using CO2 as the carbon feedstock. Plant metabolism, which has helped
shape Earth’s biosphere, climate, and rich species diversity, could play a key role in creating a
sustainable future for the planet.
Concluding remarks
Plants have evolved diverse specialized metabolism, facilitating their successful
colonization of all but the most extreme corners of land on Earth. Flavonoids play particularly
24

important roles in plant physiological adaptation, and derived plant clades grew their flavonoid
arsenals as they diversified into increasingly challenging niches. To accommodate this increased
demand for flavonoid production, the key enzyme chalcone synthase has also evolved. Chapter 2
investigates the structural features that enabled increased reactivity of the catalytic cysteine
residue by comparison of CHS orthologs from five diverse plant lineages. Chapter 3 establishes
methods for investigating the in vivo consequences of this differential cysteine reactivity toward
oxidation and its possible role in a redox regulation system to control flavonoid biosynthesis. In
the appendix, I explore the function of enzymes in biosynthesis galloylated catechins, major
flavonoids found in tea. Altogether, this thesis investigates the relationships among structure,
function, and evolution of enzymes in flavonoid biosynthesis.
25

References
Abe, I., Oguro, S., Utsumi, Y., Sano, Y., & Noguchi, H. (2005). Engineered biosynthesis of plant polyketides: chain length control in an octaketide-producing plant type III polyketide synthase. Journal of the American Chemical Society, 127(36), 12709–12716.
Austin, M. B., Bowman, M. E., Ferrer, J.-L., Schröder, J., & Noel, J. P. (2004). An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chemistry & Biology, 11(9), 1179–1194.
Austin, M. B., & Noel, J. P. (2003). The chalcone synthase superfamily of type III polyketide synthases. Natural Product Reports, 20(1), 79–110.
Beerling, D. J., Osborne, C. P., & Chaloner, W. G. (2001). Evolution of leaf-form in land plants linked to atmospheric CO2 decline in the Late Palaeozoic era. Nature, 410(6826), 352–354.
Berner, R. A. (1993). Paleozoic Atmospheric CO2: Importance of Solar Radiation and Plant Evolution. Science, 261(5117), 68–70.
Boerjan, W., Ralph, J., & Baucher, M. (2003). Lignin biosynthesis. Annual Review of Plant Biology, 54, 519–546.
Bornscheuer, U. T., Huisman, G. W., Kazlauskas, R. J., Lutz, S., Moore, J. C., & Robins, K. (2012). Engineering the third wave of biocatalysis. Nature, 485(7397), 185–194.
Dewick, P. M. (1988). Isoflavonoids. The Flavonoids. https://doi.org/10.1007/978-1-4899-2913-6_5
Dixon, R. A., & Paiva, N. L. (1995). Stress-Induced Phenylpropanoid Metabolism. The Plant Cell, 7(7), 1085–1097.
Dixon, R. A., & Steele, C. L. (1999). Flavonoids and isoflavonoids – a gold mine for metabolic engineering. Trends in Plant Science. https://doi.org/10.1016/s1360-1385(99)01471-5
Erb, T. J., Jones, P. R., & Bar-Even, A. (2017). Synthetic metabolism: metabolic engineering meets enzyme design. Current Opinion in Chemical Biology, 37, 56–62.
Evans, S. G., Ramage, B. S., DiRocco, T. L., & Potts, M. D. (2015). Greenhouse gas mitigation on marginal land: a quantitative review of the relative benefits of forest recovery versus biofuel production. Environmental Science & Technology, 49(4), 2503–2511.
Feeny, P. (1970). Seasonal Changes in Oak Leaf Tannins and Nutrients as a Cause of Spring Feeding by Winter Moth Caterpillars. Ecology, 51(4), 565–581.
Ferrer, J. L., Jez, J. M., Bowman, M. E., Dixon, R. A., & Noel, J. P. (1999). Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nature Structural Biology, 6(8), 775–784.
Franke, R., McMichael, C. M., Meyer, K., Shirley, A. M., Cusumano, J. C., & Chapple, C. (2000). Modified lignin in tobacco and poplar plants over-expressing the Arabidopsis gene encoding ferulate 5-hydroxylase. The Plant Journal: For Cell and Molecular Biology, 22(3), 223–234.
Frémont, L. (2000). Biological effects of resveratrol. Life Sciences, 66(8), 663–673. Friis, E. M., Crane, P. R., & Pedersen, K. R. (2011). Early Flowers and Angiosperm Evolution.
Cambridge University Press. Graham, J. B., Aguilar, N. M., Dudley, R., & Gans, C. (1995). Implications of the late
Palaeozoic oxygen pulse for physiology and evolution. Nature, 375(6527), 117–120. Harborne, J. B., & Williams, C. A. (2000). Advances in flavonoid research since 1992.
26

Phytochemistry, 55(6), 481–504. Harrison, C. J., & Morris, J. L. (2018). The origin and early evolution of vascular plant shoots
and leaves. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 373(1739). https://doi.org/10.1098/rstb.2016.0496
Heller, W., & Hahlbrock, K. (1980). Highly purified “flavanone synthase” from parsley catalyzes the formation of naringenin chalcone. Archives of Biochemistry and Biophysics. https://doi.org/10.1016/0003-9861(80)90395-1
Intergovernmental Panel on Climate Change. (2018). Global Warming of 1.5°C: An IPCC Special Report on the Impacts of Global Warming of 1.5°C Above Pre-industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty.
Jez, J. M., Austin, M. B., Ferrer, J., Bowman, M. E., Schröder, J., & Noel, J. P. (2000). Structural control of polyketide formation in plant-specific polyketide synthases. Chemistry & Biology, 7(12), 919–930.
Jez, J. M., & Noel, J. P. (2000). Mechanism of Chalcone Synthase: pKa of the Catalytic Cysteine and the Role of the Conserved Histidine in a Plant Polyketide Synthase. The Journal of Biological Chemistry, 275(50), 39640–39646.
Kenrick, P., & Crane, P. R. (1997). The origin and early evolution of plants on land. Nature, 389(6646), 33–39.
Kortemme, T., & Creighton, T. E. (1995). Ionisation of Cysteine Residues at the Termini of Model α-Helical Peptides. Relevance to Unusual Thiol pKaValues in Proteins of the Thioredoxin Family. Journal of Molecular Biology, 253(5), 799–812.
Levsh, O., Chiang, Y.-C., Tung, C. F., Noel, J. P., Wang, Y., & Weng, J.-K. (2016). Dynamic Conformational States Dictate Selectivity toward the Native Substrate in a Substrate-Permissive Acyltransferase. Biochemistry, 55(45), 6314–6326.
Li, F.-S., Phyo, P., Jacobowitz, J., Hong, M., & Weng, J.-K. (2019). The molecular structure of plant sporopollenin. Nature Plants, 5(1), 41–46.
Linkies, A., Graeber, K., Knight, C., & Leubner-Metzger, G. (2010). The evolution of seeds. The New Phytologist, 186(4), 817–831.
Markham, K. R. (1988). Distribution of flavonoids in the lower plants and its evolutionary significance. In J. B. Harborne (Ed.), The Flavonoids: Advances in Research since 1980 (pp. 427–468). Boston, MA: Springer US.
Meyer, K., Shirley, A. M., Cusumano, J. C., Bell-Lelong, D. A., & Chapple, C. (1998). Lignin monomer composition is determined by the expression of a cytochrome P450-dependent monooxygenase in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 95(12), 6619–6623.
Moghe, G. D., & Last, R. L. (2015). Something Old, Something New: Conserved Enzymes and the Evolution of Novelty in Plant Specialized Metabolism. Plant Physiology, 169(3), 1512–1523.
Park, H.-Y., Seok, H.-Y., Park, B.-K., Kim, S.-H., Goh, C.-H., Lee, B.-H., … Moon, Y.-H. (2008). Overexpression of Arabidopsis ZEP enhances tolerance to osmotic stress. Biochemical and Biophysical Research Communications, 375(1), 80–85.
Rausher, M. D. (2006). The Evolution of Flavonoids and Their Genes. In E. Grotewold (Ed.), The Science of Flavonoids (pp. 175–211). New York, NY: Springer New York.
27

Renault, H., Werck-Reichhart, D., & Weng, J.-K. (2019). Harnessing lignin evolution for biotechnological applications. Current Opinion in Biotechnology, 56, 105–111.
Riederer, M., & Muller, C. (2008). Annual Plant Reviews, Biology of the Plant Cuticle. John Wiley & Sons.
Roberts, J. A. (Ed.). (2018). Biochemistry of Terpenoids: Monoterpenes, Sesquiterpenes and Diterpenes. In Annual Plant Reviews online (Vol. 202, pp. 258–303). Chichester, UK: John Wiley & Sons, Ltd.
Schmidt, B. M., Ribnicky, D. M., Lipsky, P. E., & Raskin, I. (2007). Revisiting the ancient concept of botanical therapeutics. Nature Chemical Biology, 3(7), 360–366.
Schwander, T., Schada von Borzyskowski, L., Burgener, S., Cortina, N. S., & Erb, T. J. (2016). A synthetic pathway for the fixation of carbon dioxide in vitro. Science, 354(6314), 900–904.
Stafford, H. A. (1991). Flavonoid evolution: an enzymic approach. Plant Physiology, 96(3), 680–685.
Stewart, J. J., Akiyama, T., Chapple, C., Ralph, J., & Mansfield, S. D. (2009). The Effects on Lignin Structure of Overexpression of Ferulate 5-Hydroxylase in Hybrid Poplar1. PLANT PHYSIOLOGY. https://doi.org/10.1104/pp.109.137059
Strohmeier, G. A., Pichler, H., May, O., & Gruber-Khadjawi, M. (2011). Application of designed enzymes in organic synthesis. Chemical Reviews, 111(7), 4141–4164.
Suh, D.-Y., Kagami, J., Fukuma, K., & Sankawa, U. (2000). Evidence for Catalytic Cysteine–Histidine Dyad in Chalcone Synthase. Biochemical and Biophysical Research Communications, 275(3), 725–730.
Thimmappa, R., Geisler, K., Louveau, T., O’Maille, P., & Osbourn, A. (2014). Triterpene biosynthesis in plants. Annual Review of Plant Biology, 65, 225–257.
Tracewell, C. A., & Arnold, F. H. (2009). Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Current Opinion in Chemical Biology, 13(1), 3–9.
Tyner, W. E. (2012). Biofuels and agriculture: a past perspective and uncertain future. International Journal of Sustainable Development and World Ecology, 19(5), 389–394.
Vogt, T. (2010). Phenylpropanoid biosynthesis. Molecular Plant, 3(1), 2–20. Wani, S. H., Kumar, V., Shriram, V., & Sah, S. K. (2016). Phytohormones and their metabolic
engineering for abiotic stress tolerance in crop plants. The Crop Journal, 4(3), 162–176. Weng, J.-K., & Chapple, C. (2010). The origin and evolution of lignin biosynthesis. The New
Phytologist, 187(2), 273–285. Weng, J.-K., Philippe, R. N., & Noel, J. P. (2012). The rise of chemodiversity in plants. Science,
336(6089), 1667–1670. Winkel-Shirley, B. (2001). Flavonoid biosynthesis. A colorful model for genetics, biochemistry,
cell biology, and biotechnology. Plant Physiology, 126(2), 485–493. Yang, F., Mitra, P., Zhang, L., Prak, L., Verhertbruggen, Y., Kim, J.-S., … Loqué, D. (2013).
Engineering secondary cell wall deposition in plants. Plant Biotechnology Journal, 11(3), 325–335.
Yao, L. H., Jiang, Y. M., Shi, J., Tomás-Barberán, F. A., Datta, N., Singanusong, R., & Chen, S. S. (2004). Flavonoids in food and their health benefits. Plant Foods for Human Nutrition , 59(3), 113–122.
Ye, X., Al-Babili, S., Klöti, A., Zhang, J., Lucca, P., Beyer, P., & Potrykus, I. (2000). Engineering the Provitamin A (β-Carotene) Biosynthetic Pathway into (Carotenoid-Free)
28

Rice Endosperm. Science, 287(5451), 303–305. Zhang, J., Yu, H., Zhang, Y., Wang, Y., Li, M., Zhang, J., … Li, Z. (2016). Increased abscisic
acid levels in transgenic maize overexpressing AtLOS5 mediated root ion fluxes and leaf water status under salt stress. Journal of Experimental Botany, 67(5), 1339–1355.
Zhang, Y., Butelli, E., Alseekh, S., Tohge, T., Rallapalli, G., Luo, J., … Martin, C. (2015). Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nature Communications, 6, 8635.
Ziegler, J., & Facchini, P. J. (2008). Alkaloid biosynthesis: metabolism and trafficking. Annual Review of Plant Biology, 59, 735–769.
29

30

Chapter 2 Mechanistic basis for the evolution of chalcone synthase catalytic cysteine reactivity in land plants Authors Geoffrey Liou1,2, Ying-Chih Chiang3, Yi Wang3, and Jing-Ke Weng1,2
Author Affiliations 1. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 2. Whitehead Institute for Biomedical Research, Cambridge, MA 02142, USA 3. Department of Physics, The Chinese University of Hong Kong, Shatin, NT, Hong Kong Published As Liou, G., Chiang, Y.-C., Wang, Y., & Weng, J.-K. (2018). Mechanistic basis for the evolution of chalcone synthase catalytic cysteine reactivity in land plants. Journal of Biological Chemistry 293: 18601-18612. Author Contributions G.L. and J.-K.W. performed crystallography, seed complementation, and phylogenetic analysis. G.L. performed enzyme assays. Y.-C.C. and Y.W. performed molecular dynamics simulations and wrote the relevant results and discussion. G.L. wrote the remaining sections together with supervision from J.-K.W.
31

Abstract
Flavonoids are important polyphenolic natural products, ubiquitous in land plants, that
play diverse functions in plants’ survival in their ecological niches, including UV protection,
pigmentation for attracting pollinators, symbiotic nitrogen fixation, and defense against
herbivores. Chalcone synthase (CHS) catalyzes the first committed step in plant flavonoid
biosynthesis and is highly conserved in all land plants. In several previously reported crystal
structures of CHSs from flowering plants, the catalytic cysteine is oxidized to sulfinic acid,
indicating enhanced nucleophilicity in this residue associated with its increased susceptibility to
oxidation. In this study, we report a set of new crystal structures of CHSs representing all five
major lineages of land plants (bryophytes, lycophytes, monilophytes, gymnosperms, and
angiosperms), spanning 500 million years of evolution. We reveal that the structures of CHS
from a lycophyte and a moss species preserve the catalytic cysteine in a reduced state, in contrast
to the cysteine sulfinic acid seen in all euphyllophyte CHS structures. In vivo complementation,
in vitro biochemical and mutagenesis analyses, and molecular dynamics simulations identified a
set of residues that differ between basal-plant and euphyllophyte CHSs and modulate catalytic
cysteine reactivity. We propose that the CHS active-site environment has evolved in
euphyllophytes to further enhance the nucleophilicity of the catalytic cysteine since the
divergence of euphyllophytes from other vascular plant lineages 400 million years ago. These
changes in CHS could have contributed to the diversification of flavonoid biosynthesis in
euphyllophytes, which in turn contributed to their dominance in terrestrial ecosystems.
32

Introduction
In their transition from aquatic domains to terrestrial environments, early land plants
faced several major challenges, including exposure to damaging UV-B radiation once screened
by aquatic environments, lack of structural support once provided by buoyancy in water,
drought, and novel pathogens and herbivores. To cope with many of these stresses, land plants
have evolved a series of specialized metabolic pathways, among which phenylpropanoid
metabolism was probably one of the most critical soon after the transition from water to land
(Weng & Chapple, 2010).
Flavonoids are a diverse class of plant phenolic compounds found in all extant land
plants, with important roles in many aspects of plant life, including UV protection, pigmentation
for attracting pollinators and seed dispersers, defense, and signaling between plants and microbes
(Winkel-Shirley, 2001). Some flavonoids are also of great interest for their anti-cancer and
antioxidant activities as well as other potential health benefits to humans (Yao et al., 2004). After
the core flavonoid biosynthetic pathway was established in early land plants, new branches of the
pathway continued to evolve over the history of plant evolution, producing structurally and
functionally diverse flavonoids to cope with changing habitats, co-evolving pathogens and
herbivores, and other aspects of plants’ ecological niches. Basal bryophytes biosynthesize the
three main classes of flavonoids, namely flavanones, flavones, and flavonols, which likely
emerged as UV sunscreens (Rausher, 2006). The lycophyte Selaginella biosynthesizes a rich
diversity of biflavonoids, many of which were shown to be cytotoxic and may function as
phytoalexins (Weng & Noel, 2013). The ability to synthesize the astringent, polyphenolic
tannins, which defend against bacterial and fungal pathogens, seems to have evolved in
33

euphyllophytes (Rausher, 2006). Finally, seed plants, including gymnosperms and angiosperms,
developed elaborate anthocyanin biosynthetic pathways to produce the vivid colors used to
attract pollinators or ward off herbivores.
Chalcone synthase (CHS), a highly conserved plant type III polyketide synthase (PKS), is
the first committed enzyme in the plant flavonoid biosynthetic pathway. CHS synthesizes
naringenin chalcone from a molecule of p-coumaroyl-CoA and three molecules of malonyl-CoA
(Weng & Noel, 2012) (Figure 1A). The proposed catalytic mechanism of CHS involves loading
of the starter molecule p-coumaroyl CoA onto the catalytic cysteine, which also serves as the
attachment site of the growing polyketide chain during the iterative elongation steps (Austin &
Noel, 2003). This initial reaction step requires the cysteine to be present as a thiolate anion
before loading of the starter molecule (Figure 1B). Using thiol-specific inactivation and the pH
dependence of the malonyl-CoA decarboxylation reaction, the pKa of the catalytic cysteine (Cys
164) of Medicago sativa CHS (MsCHS) was measured to be 5.5, a value significantly lower than
8.7 for free cysteine (Jez & Noel, 2000).
Interestingly, we observed that the catalytic cysteine residues in the previously reported
MsCHS crystal structures appear to be oxidized to sulfinic acid (PDB ID 1BI5 and 1BQ6)
(Ferrer et al. 1999). Furthermore, the same phenomenon was observed in the crystal structures
for several other plant type III PKSs evolutionarily derived from CHS, including Gerbera
hybrida 2-pyrone synthase (PDB ID 1QLV) (Jez et al., 2000) (Figure S1). The other
non-catalytic cysteines in these proteins do not appear to be oxidized. These findings suggest that
the oxidation of the catalytic cysteine observed in several type III PKS crystal structures may not
simply be an artifact of X-ray crystallography, but rather reflects the intrinsic redox potential and
34

reactivity of the catalytic cysteine evolved in this family of enzymes. Indeed, the propensity for a
particular cysteine residue to undergo oxidation has been previously indicated to correlate with
low pKa (Reddie & Carroll, 2008).
Here, we present a set of new crystal structures of orthologous CHSs representing five
major lineages of land plants, namely bryophytes, lycophytes, monilophytes, gymnosperms, and
angiosperms, spanning 500 million years of land plant evolution. Through comparative structural
analysis, in vivo complementation, in vitro biochemistry, mutagenesis studies, and molecular
dynamics simulations, we reveal that CHSs of basal land plants, i.e. bryophytes and lycophytes,
contain a catalytic cysteine less reactive than that of the CHSs from higher plants, i.e.
euphyllophytes. We probe into the structure-function relationship of a set of residues that
modulate the reactivity of the catalytic cysteine, which leads us to propose that euphyllophytes
may have evolved a more catalytically efficient CHS to enhance flavonoid biosynthesis relative
to their basal plant relatives.
35
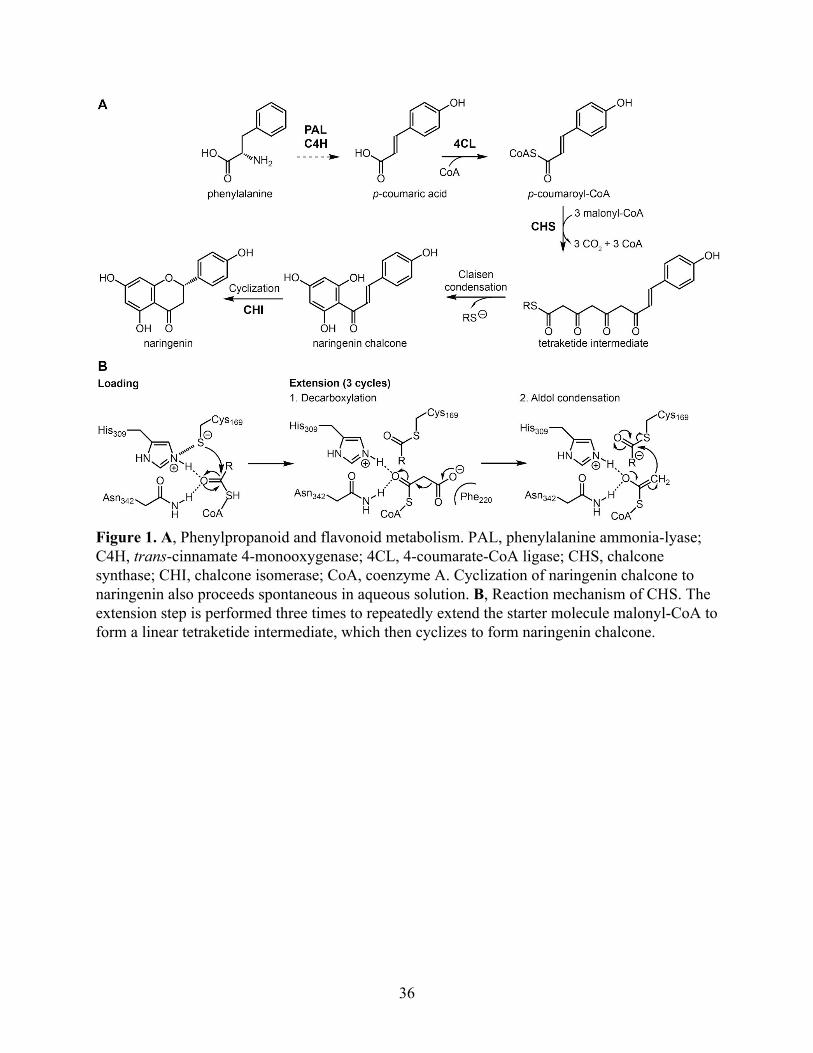
Figure 1. A, Phenylpropanoid and flavonoid metabolism. PAL, phenylalanine ammonia-lyase; C4H, trans-cinnamate 4-monooxygenase; 4CL, 4-coumarate-CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; CoA, coenzyme A. Cyclization of naringenin chalcone to naringenin also proceeds spontaneous in aqueous solution. B, Reaction mechanism of CHS. The extension step is performed three times to repeatedly extend the starter molecule malonyl-CoA to form a linear tetraketide intermediate, which then cyclizes to form naringenin chalcone.
36

Results
Basal-plant CHSs contain reduced catalytic cysteine in their crystal structures
To examine the structural basis for the evolution of CHS across major land plant
lineages, we cloned, expressed, and solved the crystal structures of the five CHS orthologs from
the bryophyte Physcomitrella patens (PpCHS), the lycophyte Selaginella moellendorffii
(SmCHS), the monilophyte Equisetum arvense (EaCHS), the gymnosperm Pinus sylvestris
(PsCHS), and the angiosperm Arabidopsis thaliana (AtCHS) (Figure 2, Table 1). Like
previously reported crystal structures of type III polyketide synthases, all five CHS orthologs
form symmetric homodimers and share the same αβαβα thiolase fold, suggesting a common
evolutionary origin (Ferrer, Jez, Bowman, Dixon, & Noel, 1999). The catalytic triad of cysteine,
histidine, and asparagine is found in a highly similar conformation to other PKS and related fatty
acid biosynthetic β-ketoacyl-(acyl-carrier-protein) synthase III (KAS III) enzymes, suggesting
that they share a similar general catalytic mechanism (Figure 2B).
Based on the previously proposed reaction mechanism for MsCHS, the catalytic cysteine
is C169 in AtCHS and C159 in SmCHS. This residue initiates the reaction mechanism by
performing nucleophilic attack on p-coumaroyl-CoA (Figure 1B). The other two members of the
catalytic triad consist of H309 and N342 in AtCHS, and H302 and N335 in SmCHS. The
catalytic histidine contributes to the lowered pKa of the catalytic cysteine by forming a stable
imidazolium-thiolate ion pair (Jez & Noel, 2000). The histidine and asparagine also form the
oxyanion hole that stabilizes the tetrahedral transition states formed during the initial
nucleophilic attack by cysteine on p-coumaroyl-CoA and after malonyl-CoA decarboxylation
(Figure 1B).
37
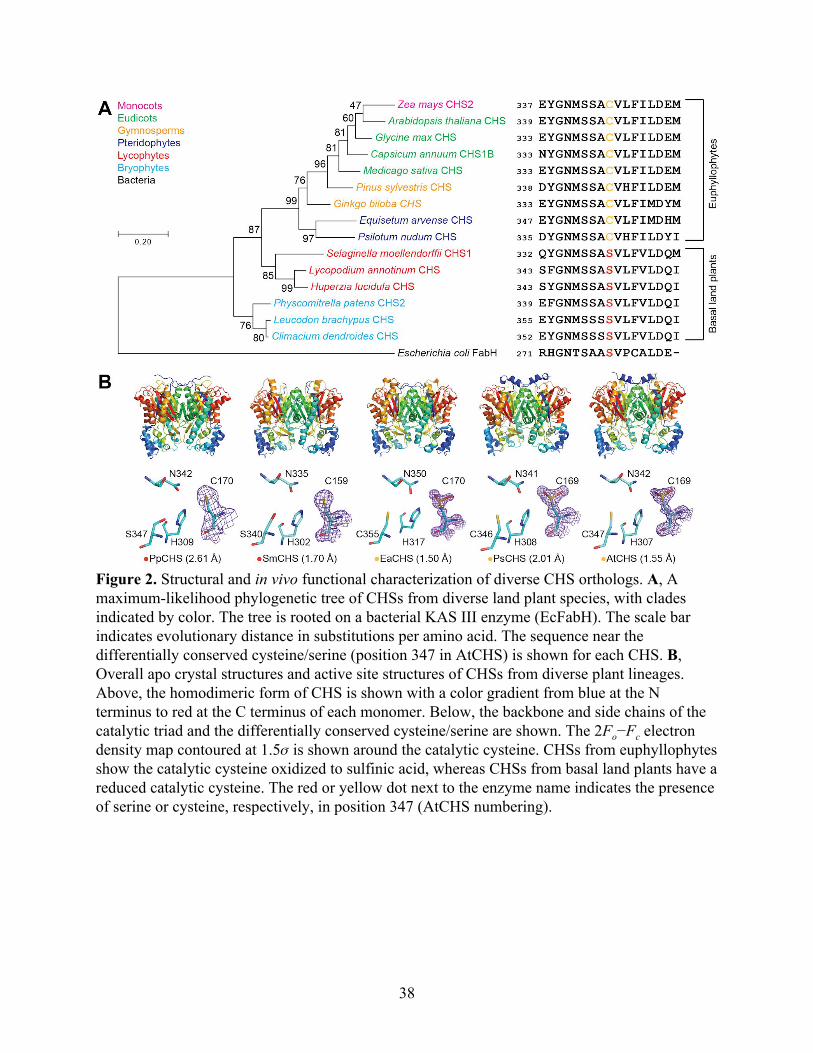
Figure 2. Structural and in vivo functional characterization of diverse CHS orthologs. A, A maximum-likelihood phylogenetic tree of CHSs from diverse land plant species, with clades indicated by color. The tree is rooted on a bacterial KAS III enzyme (EcFabH). The scale bar indicates evolutionary distance in substitutions per amino acid. The sequence near the differentially conserved cysteine/serine (position 347 in AtCHS) is shown for each CHS. B, Overall apo crystal structures and active site structures of CHSs from diverse plant lineages. Above, the homodimeric form of CHS is shown with a color gradient from blue at the N terminus to red at the C terminus of each monomer. Below, the backbone and side chains of the catalytic triad and the differentially conserved cysteine/serine are shown. The 2Fo−Fc electron density map contoured at 1.5𝜎 is shown around the catalytic cysteine. CHSs from euphyllophytes show the catalytic cysteine oxidized to sulfinic acid, whereas CHSs from basal land plants have a reduced catalytic cysteine. The red or yellow dot next to the enzyme name indicates the presence of serine or cysteine, respectively, in position 347 (AtCHS numbering).
38
PpCHS SmCHS
PDB ID 6DX7 6DX8
Data collection
Total reflections 161385 (13620) 493910 (50217)
Unique reflections 82406 (7763) 81737 (6434)
Multiplicity 2.0 (1.8) 6.0 (6.2)
Completeness (%) 98.95 (94.60) 90.60 (79.18)
Mean I/sigma(I) 14.31 (1.57) 12.32 (1.62)
R-merge 0.03961 (0.4442) 0.1427 (1.194)
CC1/2 0.998 (0.747) 0.995 (0.51)
Refinement
Resolution range (Å) 36.24 - 2.61
(2.703 - 2.61) 39.01 - 1.7
(1.761 - 1.7)
Space group P 2 21 21 P 1 21 1
Unit cell (Å) 71.6 192.83 195.51 55.1702 66.6703 102.55
Unit cell (°) 90 90 90 90 91.35 90
R-work 0.1810 (0.3187) 0.1927 (0.3111)
R-free 0.2627 (0.3951) 0.2362 (0.3567)
Non-hydrogen protein atoms 17592 5776
Water molecules 59 549
RMSD bonds (Å) 0.014 0.012
RMSD angles (°) 1.5 1.32
Ramachandran favored (%) 94.07 97.74
Ramachandran allowed (%) 5.58 1.99
Ramachandran outliers (%) 0.35 0.27
Average B-factor 63.61 22.24
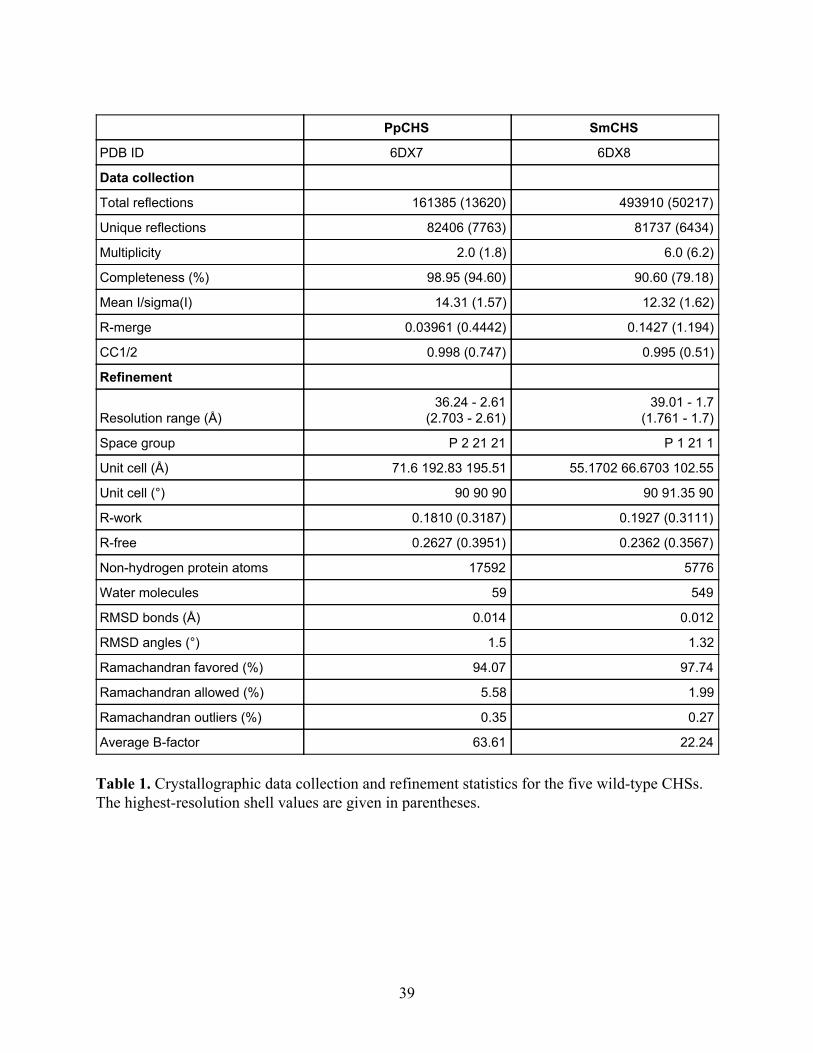
Table 1. Crystallographic data collection and refinement statistics for the five wild-type CHSs. The highest-resolution shell values are given in parentheses.
39
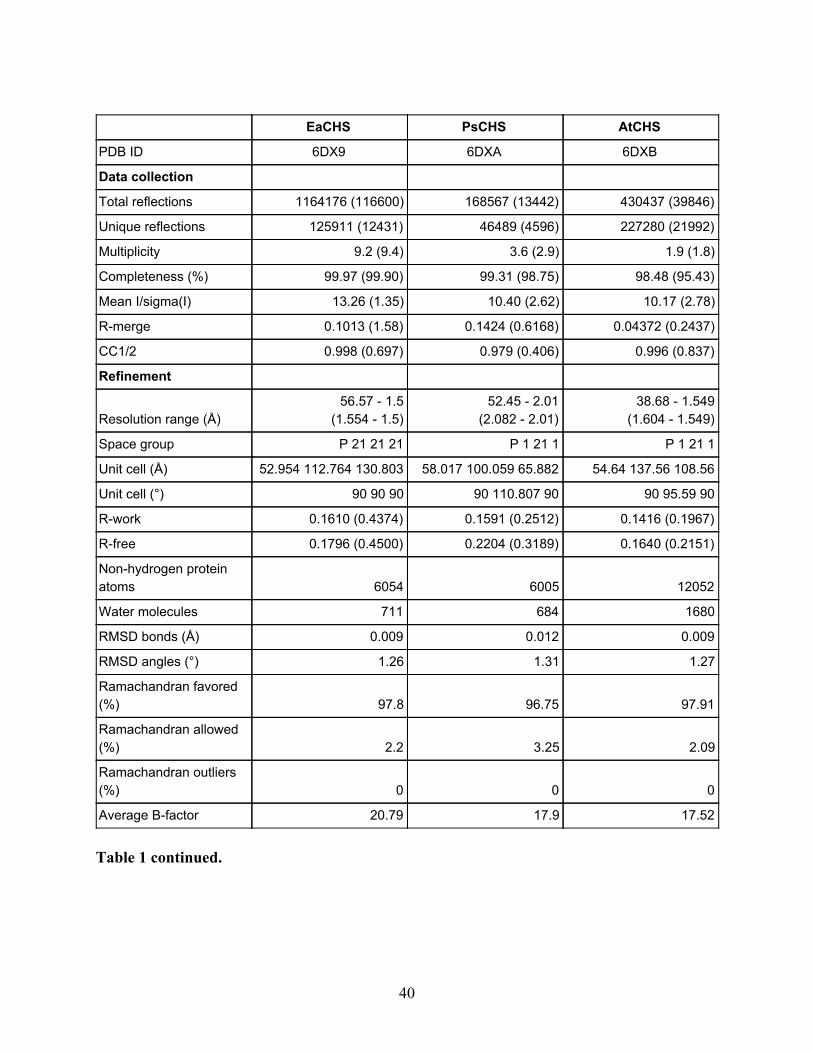
EaCHS PsCHS AtCHS
PDB ID 6DX9 6DXA 6DXB
Data collection
Total reflections 1164176 (116600) 168567 (13442) 430437 (39846)
Unique reflections 125911 (12431) 46489 (4596) 227280 (21992)
Multiplicity 9.2 (9.4) 3.6 (2.9) 1.9 (1.8)
Completeness (%) 99.97 (99.90) 99.31 (98.75) 98.48 (95.43)
Mean I/sigma(I) 13.26 (1.35) 10.40 (2.62) 10.17 (2.78)
R-merge 0.1013 (1.58) 0.1424 (0.6168) 0.04372 (0.2437)
CC1/2 0.998 (0.697) 0.979 (0.406) 0.996 (0.837)
Refinement
Resolution range (Å) 56.57 - 1.5
(1.554 - 1.5) 52.45 - 2.01
(2.082 - 2.01) 38.68 - 1.549
(1.604 - 1.549)
Space group P 21 21 21 P 1 21 1 P 1 21 1
Unit cell (Å) 52.954 112.764 130.803 58.017 100.059 65.882 54.64 137.56 108.56
Unit cell (°) 90 90 90 90 110.807 90 90 95.59 90
R-work 0.1610 (0.4374) 0.1591 (0.2512) 0.1416 (0.1967)
R-free 0.1796 (0.4500) 0.2204 (0.3189) 0.1640 (0.2151)
Non-hydrogen protein atoms 6054 6005 12052
Water molecules 711 684 1680
RMSD bonds (Å) 0.009 0.012 0.009
RMSD angles (°) 1.26 1.31 1.27
Ramachandran favored (%) 97.8 96.75 97.91
Ramachandran allowed (%) 2.2 3.25 2.09
Ramachandran outliers (%) 0 0 0
Average B-factor 20.79 17.9 17.52
Table 1 continued.
40

Notably, SmCHS and PpCHS are the first CHSs for which a reduced catalytic cysteine
has been observed in the crystal structure (Figure 2B). The catalytic cysteine in SmCHS can still
become oxidized to sulfenic acid when the crystal is soaked in hydrogen peroxide, indicating that
it is still susceptible to oxidation at a lower rate (Figure S2). Like most other euphyllophyte type
III PKS crystal structures solved to date, AtCHS, PsCHS, and EaCHS contain doubly oxidized
catalytic cysteine sulfinic acid (Figure 2B). This interesting observation suggests a functional
divide between basal-plant and euphyllophyte CHSs. Despite shared orthology, the redox
potential of the catalytic cysteine in PpCHS and SmCHS may differ from that of the
euphyllophyte CHSs, resulting in different levels of sensitivity to oxidation under similar
crystallization conditions. This could be due to the evolution of some novel molecular features in
euphyllophyte CHSs not present in the lower-plant CHSs.
Basal-plant CHSs only partially complement the Arabidopsis CHS-null mutant
CHS orthologs have been identified in all land plant species sequenced to date,
suggesting a highly conserved biochemical function. To test whether the five CHSs from the five
major plant lineages are functionally equivalent, we generated transgenic Arabidopsis thaliana
lines expressing each of the five different CHSs driven by the Arabidopsis CHS promoter in the
CHS-null mutant transparent testa 4-2 (tt4-2) background (Shirley et al., 1995) (Figure S3).
Twenty independent T1 plants were selected for each construct. The phenotypes of the
transgenic plants described below were represented by the majority of independent transgenic
events for each unique construct. As the name indicates, the tt4-2 mutant is devoid of flavonoid
biosynthesis and therefore lacks the accumulation of the brown condensed tannin pigments in
41

seed coats, revealing the pale yellow color of the underlying cotyledons (Shirley et al., 1995).
Whereas AtCHS, PsCHS and EaCHS fully complement the tt phenotype of tt4-2, PpCHS and
SmCHS only partially rescue the seed tt phenotype of tt4-2 (Figure S3), suggesting that PpCHS
and SmCHS are likely less active than their higher-plant counterparts in vivo. This result also
correlates with the crystallographic observation where the catalytic cysteine of basal plant and
euphyllophyte CHSs exhibit differential susceptibility to oxidation.
The pKa of the catalytic cysteine is higher in basal-plant CHSs than in euphyllophyte CHSs
To perform nucleophilic attack on the p-coumaroyl-CoA substrate, the catalytic cysteine
must be present in the thiolate anion form. As shown previously in MsCHS, the pKa of the
catalytic cysteine is lowered to 5.5, well below physiological pH, in order to stabilize this
deprotonated state (Jez & Noel, 2000). Two factors could contribute to the depressed pKa of
C164. First, H303, one of the catalytic triad of CHS in vicinity of C164, provides an ionic
interaction with C164 that can further stabilize the cysteine thiolate anion. Second, C164 is
positioned at the N-terminus of the MsCHS α-9 helix (Ferrer et al., 1999), which provides a
stabilizing effect on the cysteine thiolate anion through the partial positive charge of the helix
dipole (Kortemme & Creighton, 1995). The acidic pKa of the catalytic cysteine in CHS ensures
the presence of a cysteine thiolate anion in the enzyme active site at physiological pH to serve as
the nucleophile for starter molecule loading.
To measure the pKa of the catalytic cysteine in the five land plant CHS orthologs, we
performed pH-dependent inactivation of CHS using iodoacetamide, a thiol-specific compound
that reacts with sulfhydryl groups that are sufficiently nucleophilic, followed by a CHS activity
42

assay at the usual reaction pH. At pH values above the pKa, the catalytic cysteine is deprotonated
and able to react with iodoacetamide, thus inactivating CHS. At pH values below the pKa, the
catalytic cysteine is protonated and protected from iodoacetamide modification, thus retaining
CHS activity in the subsequent enzyme assay. The amount of CHS activity remaining after
iodoacetamide treatment was expressed as a ratio compared to the CHS activity of a control
treatment at the same pH but without iodoacetamide. The pKa was calculated using nonlinear
regression to fit a log(inhibitor) vs. response equation, which gave the pH at which 50% of
maximal inhibition was obtained.
The pKa for AtCHS was measured to be 5.428, which is close to the 5.5 measured for
MsCHS (Figure 3A). The pKa for SmCHS was measured to be 6.468, approximately 1 pH unit
higher than that of the two angiosperm CHS orthologs. This elevated pKa measured for SmCHS
is consistent with the observation of a catalytic cysteine that is less reactive and less prone to
oxidation. Also consistent with the crystallographic and plant complementation results, pKa
values around 5.5 were measured for euphyllophyte orthologs PsCHS and EaCHS, and around
6.5 for the basal-plant orthologs PpCHS (Figure S4).
Residues near the active-site cavity affect the pKa and reactivity of the catalytic cysteine
We next examined the sequence and structural differences between basal-plant and
euphyllophyte CHSs that could play a role in modulating catalytic cysteine reactivity. This led us
to first identifying a residue near the active site that is conserved as C347 (AtCHS numbering) in
AtCHS and other euphyllophyte sequences, and as S340 (SmCHS numbering) in SmCHS and
other lycophyte and bryophyte sequences (Figure 2A).
43
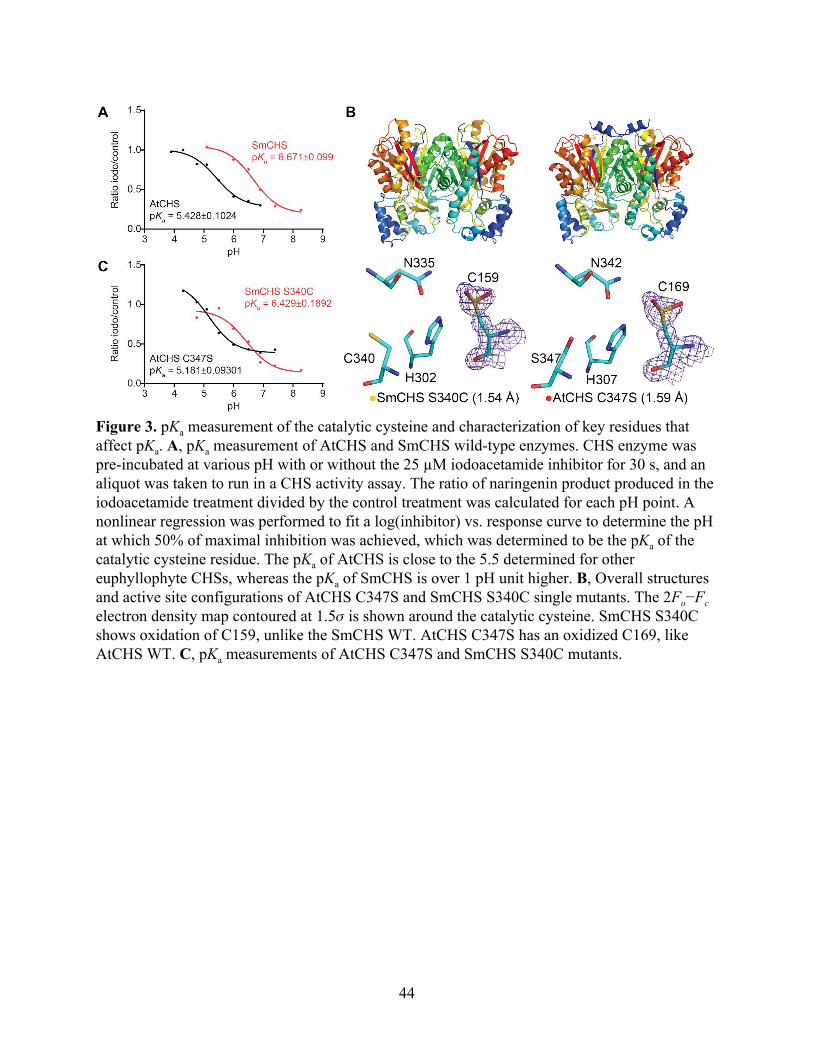
Figure 3. pKa measurement of the catalytic cysteine and characterization of key residues that affect pKa. A, pKa measurement of AtCHS and SmCHS wild-type enzymes. CHS enzyme was pre-incubated at various pH with or without the 25 µM iodoacetamide inhibitor for 30 s, and an aliquot was taken to run in a CHS activity assay. The ratio of naringenin product produced in the iodoacetamide treatment divided by the control treatment was calculated for each pH point. A nonlinear regression was performed to fit a log(inhibitor) vs. response curve to determine the pH at which 50% of maximal inhibition was achieved, which was determined to be the pKa of the catalytic cysteine residue. The pKa of AtCHS is close to the 5.5 determined for other euphyllophyte CHSs, whereas the pKa of SmCHS is over 1 pH unit higher. B, Overall structures and active site configurations of AtCHS C347S and SmCHS S340C single mutants. The 2Fo−Fc electron density map contoured at 1.5𝜎 is shown around the catalytic cysteine. SmCHS S340C shows oxidation of C159, unlike the SmCHS WT. AtCHS C347S has an oxidized C169, like AtCHS WT. C, pKa measurements of AtCHS C347S and SmCHS S340C mutants.
44
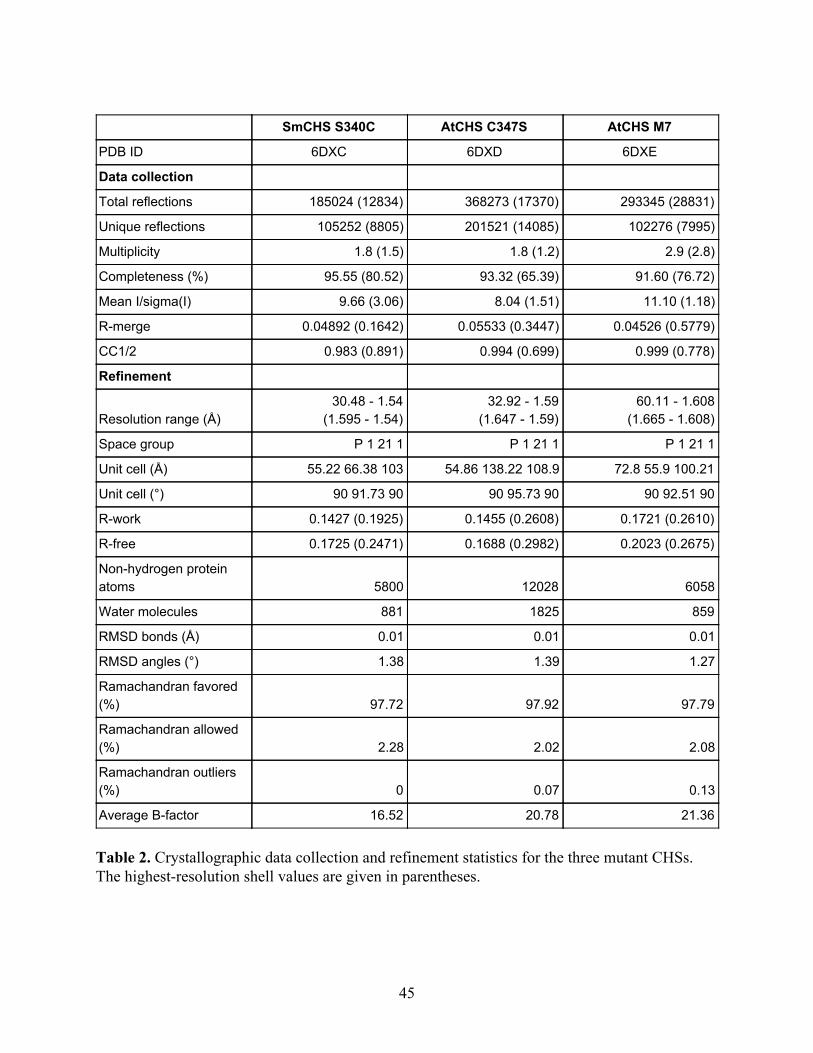
SmCHS S340C AtCHS C347S AtCHS M7
PDB ID 6DXC 6DXD 6DXE
Data collection
Total reflections 185024 (12834) 368273 (17370) 293345 (28831)
Unique reflections 105252 (8805) 201521 (14085) 102276 (7995)
Multiplicity 1.8 (1.5) 1.8 (1.2) 2.9 (2.8)
Completeness (%) 95.55 (80.52) 93.32 (65.39) 91.60 (76.72)
Mean I/sigma(I) 9.66 (3.06) 8.04 (1.51) 11.10 (1.18)
R-merge 0.04892 (0.1642) 0.05533 (0.3447) 0.04526 (0.5779)
CC1/2 0.983 (0.891) 0.994 (0.699) 0.999 (0.778)
Refinement
Resolution range (Å) 30.48 - 1.54
(1.595 - 1.54) 32.92 - 1.59
(1.647 - 1.59) 60.11 - 1.608
(1.665 - 1.608)
Space group P 1 21 1 P 1 21 1 P 1 21 1
Unit cell (Å) 55.22 66.38 103 54.86 138.22 108.9 72.8 55.9 100.21
Unit cell (°) 90 91.73 90 90 95.73 90 90 92.51 90
R-work 0.1427 (0.1925) 0.1455 (0.2608) 0.1721 (0.2610)
R-free 0.1725 (0.2471) 0.1688 (0.2982) 0.2023 (0.2675)
Non-hydrogen protein atoms 5800 12028 6058
Water molecules 881 1825 859
RMSD bonds (Å) 0.01 0.01 0.01
RMSD angles (°) 1.38 1.39 1.27
Ramachandran favored (%) 97.72 97.92 97.79
Ramachandran allowed (%) 2.28 2.02 2.08
Ramachandran outliers (%) 0 0.07 0.13
Average B-factor 16.52 20.78 21.36
Table 2. Crystallographic data collection and refinement statistics for the three mutant CHSs. The highest-resolution shell values are given in parentheses.
45

To investigate the role of this residue in modulating catalytic cysteine reactivity, we
generated the reciprocal mutations in SmCHS and AtCHS respectively and first characterized
these mutant proteins using X-ray crystallography (Figure 3B, Table 2). Under identical
crystallization conditions as wild-type SmCHS, the SmCHS S340C mutant exhibits a partially
oxidized catalytic cysteine in its crystal structure, suggesting that the residue does play some role
in determining cysteine reactivity. The AtCHS C347S mutant, however, still retains an oxidized
catalytic cysteine in its crystal structure.
We then measured the pKa of the catalytic cysteine in both SmCHS S340C and AtCHS
C347S mutants (Figure 3C). The pKa for SmCHS S340C decreases by about 0.25 pH units
compared to wild-type SmCHS, consistent with the observation that the SmCHS S340C crystal
structure contained a partially oxidized catalytic cysteine. The pKa for AtCHS C347S decreases
by about 0.25 pH units compared to wild-type AtCHS, also consistent with the observation that
the AtCHS C347S crystal structure retained an oxidized catalytic cysteine. Taken together, the
crystallographic and pKa measurement results suggest that the reciprocal mutation at this position
is not sufficient to act as a simple switch between the active-site environments of euphyllophyte
and basal-plant CHSs to modulate catalytic cysteine reactivity. Additional sequence and
structural features likely contribute to an active-site environment that lowers the pKa of the
catalytic cysteine in AtCHS.
To identify these features, we examined a multiple sequence alignment of CHS orthologs
from diverse plant species and identified residues that show conserved variations between
euphyllophytes and basal-plant lineages (Figure 4A and Figure S5). Two residues, F170 and
G173 in euphyllophyte CHSs, were found to be substituted as serine and alanine, respectively, in
46
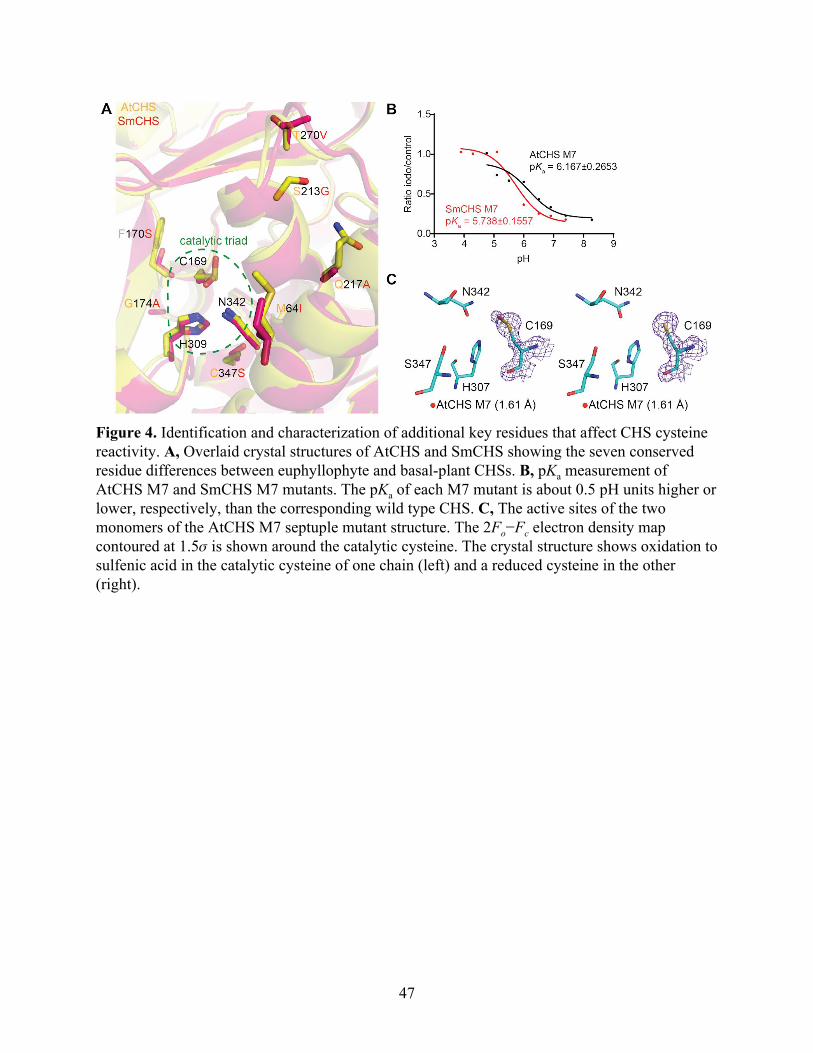
Figure 4. Identification and characterization of additional key residues that affect CHS cysteine reactivity. A, Overlaid crystal structures of AtCHS and SmCHS showing the seven conserved residue differences between euphyllophyte and basal-plant CHSs. B, pKa measurement of AtCHS M7 and SmCHS M7 mutants. The pKa of each M7 mutant is about 0.5 pH units higher or lower, respectively, than the corresponding wild type CHS. C, The active sites of the two monomers of the AtCHS M7 septuple mutant structure. The 2Fo−Fc electron density map contoured at 1.5𝜎 is shown around the catalytic cysteine. The crystal structure shows oxidation to sulfenic acid in the catalytic cysteine of one chain (left) and a reduced cysteine in the other (right).
47

basal-plant lineages. Because of their positions in the alpha helix immediately C-terminal to the
catalytic C169, we postulated that these two residues could play a role in determining the
structure of the helix, which would have an effect on the electronic environment of the active
site, due to the helix dipole’s contribution to lowering the catalytic cysteine pKa (Ferrer et al.,
1999). Four additional residues near the active-site opening of CHS were also identified as
differentially conserved between euphyllophytes and basal plants. We postulated that these
positions might affect the dynamics of the active-site tunnel and solvent access to the active site.
The six aforementioned residues were mutated in the SmCHS S340C background to their
corresponding residues in AtCHS to generate the SmCHS I54M S160F A163G G203S A207Q
V258T S340C septuple mutant, termed SmCHS M7. Likewise, the reciprocal mutations were
also made in the AtCHS C347S background to generate AtCHS M7.
Compared to SmCHS S340C, the six additional mutations in SmCHS M7 lower the pKa
by nearly 0.7 pH units from 6.429 to 5.738 (Figure 4B). Similarly, the six mutations of AtCHS
M7 raise the pKa by almost 1 pH unit from 5.181 to 6.167 compared to AtCHS C347S.
Consistent with the pKa observation, the dimeric crystal structure of AtCHS M7 has one
monomer with a catalytic cysteine singly oxidized to sulfenic acid and one monomer with a
reduced cysteine (Figure 4C). Sulfenic acid is more reduced than the doubly oxidized sulfinic
acid seen in other euphyllophyte crystal structures, indicating that these six mutations decreased
the reactivity of the catalytic cysteine. These mutations represent a part of a possible
evolutionary path from ancestral basal-plant CHSs toward the stronger pKa-lowering properties
of euphyllophyte CHSs. Any further attempts at engineering CHS to fully swap the pKa-lowering
properties between AtCHS and SmCHS would likely require different methods of searching for
48

conserved sequence differences, beyond visual observation of structural differences. An analysis
of the CHS multiple sequence alignment using ancestral sequence reconstruction with FastML
(Ashkenazy et al., 2012) identified eight additional positions that are differently conserved
between euphyllophytes and basal plants and could affect CHS function based on their position
in the CHS crystal structure (Figure S6).
Molecular dynamics simulations reveal differences in active-site interactions between
basal-plant and euphyllophyte CHSs
Our crystal structures revealed a correlation between the pKa of the catalytic cysteine and
a set of residues near the active site. To further investigate the mechanisms underlying these
conserved differences between euphyllophyte and basal-plant CHSs, we employed molecular
dynamics (MD) simulations to examine the interactions between these residues. We first
surveyed the potential role of the C347S substitution (AtCHS numbering) in affecting the active
site environment in wild-type AtCHS and SmCHS (Figure 5A). In wild-type AtCHS, where the
largest cluster represents 70.3% of all structures sampled in this simulation, the thiol group of
C347 points away from the active site and cannot form any stable interaction with the catalytic
H309 (distance 6.5 Å). In contrast, the corresponding S340 in SmCHS is 2.8 Å away from the
histidine in the largest cluster, representing 98.7% of all structures sampled in the SmCHS
simulation.
Next, we determined the inter-residue distances between the ionic pair C169-H309 as in
AtCHS or C159-H302 as in SmCHS and between residue C347 (AtCHS)/S340 (SmCHS) and
the catalytic histidine (Figure 5B). For wild-type SmCHS simulation, we observe a sharp peak at
49
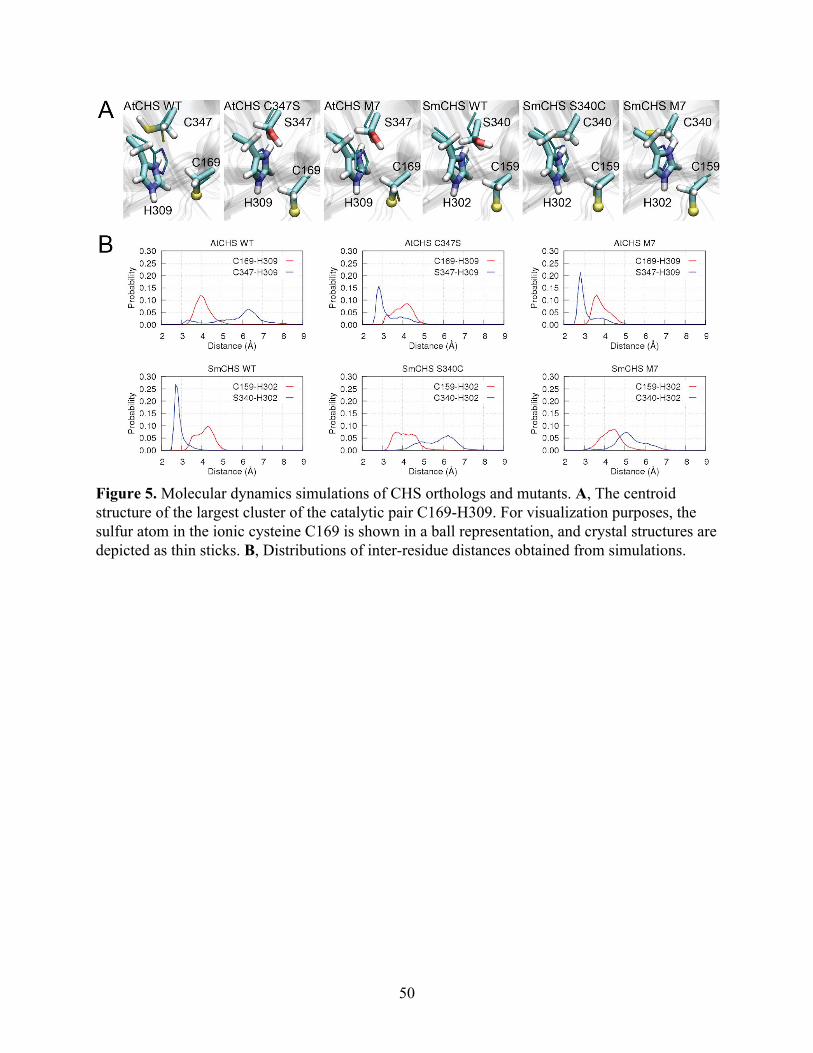
Figure 5. Molecular dynamics simulations of CHS orthologs and mutants. A, The centroid structure of the largest cluster of the catalytic pair C169-H309. For visualization purposes, the sulfur atom in the ionic cysteine C169 is shown in a ball representation, and crystal structures are depicted as thin sticks. B, Distributions of inter-residue distances obtained from simulations.
50

around 2.8 Å between S340 and H302, reflecting a stable hydrogen bond between the two
residues. On the contrary, no such short-distance peak is observed for wild- type AtCHS. These
results suggest that the catalytic histidine is stabilized upon forming a hydrogen bond with S340
in SmCHS, but such an interaction is relatively loose in AtCHS. Similar differences between the
other euphyllophyte and basal-plant CHSs are also seen for PsCHS, EaCHS, and PpCHS (Figure
S7).
To further investigate the motion of the catalytic histidine in various mutant enzyme
active-site environments, we also performed MD simulations of AtCHS C347S, SmCHS S340C,
AtCHS M7, and SmCHS M7 (Figure 5A). The largest cluster sizes were 86.0%, 66.6%, and
96.7%, and 71.0%, respectively. S347 and H309 in AtCHS mutants adopt similar conformation
to the corresponding residues in wild-type SmCHS. In contrast, no stable hydrogen bond
between C340 and H302 is formed in the largest cluster of the SmCHS mutant simulations.
Introducing point mutations dramatically changes the distributions of those key inter-residue
distances. In the AtCHS C347S mutant, the S347-H309 distance dramatically shortens to a peak
around 2.8 Å, and introducing the six additional mutations in AtCHS M7 further increases the
height of the peak. This suggests that mutating these seven positions in AtCHS to the
corresponding residues in SmCHS can allow the active-site residues to approximate the
interactions of wild-type SmCHS. The opposite effect is seen in SmCHS S340C and SmCHS M7
mutants, which recapitulate the weak interaction between C347 and H309 seen in wild-type
AtCHS.
Based on these results, we hypothesize that the strong S340-H302 interaction facilitated
by the SmCHS active site environment may weaken the stabilizing effect of H302 on the
51

catalytic cysteine thiolate compared to that in AtCHS, thus contributing to the higher pKa.
Meanwhile, the inter-residue distance of the catalytic cysteine-histidine ionic pair is rather stable
in all CHS simulations, ranging from 3 to 5 Å and centered at around 4.1 Å. This suggests that
the C347S substitution (AtCHS numbering) does not directly break this ionic interaction but may
subtly influence the charge distribution on the histidine imidazole ring to perturb the catalytic
cysteine pKa (Figure 6). In addition, the presence of a cysteine appears to decrease solvent
content in the active site compared to serine, which would increase the pKa-lowering effect of the
ionic interaction between histidine and the catalytic cysteine (Figure S8 and Supporting Note).
Taken together, our results suggest that euphyllophyte CHSs have evolved to enhance the
reactivity of the catalytic cysteine through the modification of specific interactions between
active-site residues to allow for stronger stabilization of the thiolate.
52
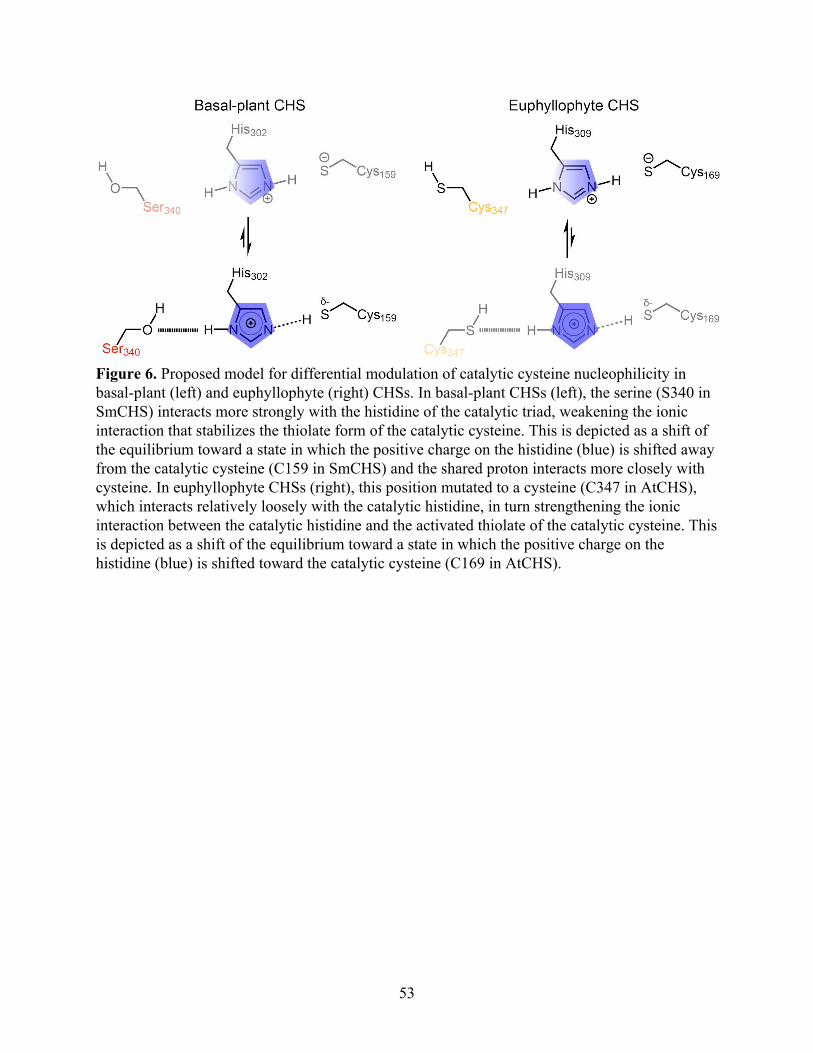
Figure 6. Proposed model for differential modulation of catalytic cysteine nucleophilicity in basal-plant (left) and euphyllophyte (right) CHSs. In basal-plant CHSs (left), the serine (S340 in SmCHS) interacts more strongly with the histidine of the catalytic triad, weakening the ionic interaction that stabilizes the thiolate form of the catalytic cysteine. This is depicted as a shift of the equilibrium toward a state in which the positive charge on the histidine (blue) is shifted away from the catalytic cysteine (C159 in SmCHS) and the shared proton interacts more closely with cysteine. In euphyllophyte CHSs (right), this position mutated to a cysteine (C347 in AtCHS), which interacts relatively loosely with the catalytic histidine, in turn strengthening the ionic interaction between the catalytic histidine and the activated thiolate of the catalytic cysteine. This is depicted as a shift of the equilibrium toward a state in which the positive charge on the histidine (blue) is shifted toward the catalytic cysteine (C169 in AtCHS).
53

Discussion
As early plants initially migrated from water to land and further radiated to occupy
diverse terrestrial environmental niches, they continuously encountered new challenges from
biotic and abiotic stresses. The greatly expanded diversity and increased abundance of flavonoids
in certain plant lineages could have increased the demand for metabolic flux into flavonoid
biosynthesis. One adaptive strategy to meet this demand, among many others, is to increase the
enzymatic efficiency of chalcone synthase, the first committed enzyme of flavonoid biosynthesis
that gates flux from general phenylpropanoid metabolism. One property of CHS that affects its
enzymatic efficiency is the reactivity of the first step of nucleophilic attack on p-coumaroyl-
CoA. To investigate this, we performed structural, biochemical, mutagenesis, and molecular
dynamics experiments on CHS orthologs from five major plant lineages. Our results suggest that
euphyllophyte CHSs have indeed evolved new structural features to increase the reactivity of
their catalytic cysteine compared to basal-plant CHSs.
To identify sequence and structural features between euphyllophyte and basal-plant CHSs
that lead to this difference in enzymatic properties, we generated mutants in the background of
AtCHS and SmCHS at various positions with conserved sequence differences segregating
euphyllophyte and basal-plant CHSs. AtCHS M7 and SmCHS M7 had pKa values raised by
about 0.7 pH units and lowered by about 1 pH unit from the wild-type enzymes, respectively.
Furthermore, AtCHS M7 also exhibits a less oxidized catalytic cysteine in its crystal structure
than in wild-type AtCHS. These results indicate that we were able to identify residue changes
that partially traced the evolutionary path from SmCHS to AtCHS that increased the reactivity of
the catalytic cysteine. In the type III PKS family, the introduction of a large number of mutations
54

to yield subtle changes in enzyme activity is not unprecedented. Stilbene synthase (STS)
produces resveratrol, a tetraketide product whose biosynthetic mechanism differs from that of
naringenin chalcone in only the final cyclization step. In a previous study, a total of 18 point
mutations were required to convert CHS activity to STS activity, through small changes in the
hydrogen-bonding network in the active site (Austin, Bowman, Ferrer, Schröder, & Noel, 2004).
To examine in detail the intramolecular interactions that lead to enhanced cysteine
reactivity, we performed molecular dynamics simulations on CHS. In comparing different CHS
orthologs and point mutants, we observed that the presence of a cysteine in position 347 (AtCHS
numbering) leads to a weak interaction between that cysteine and histidine, as indicated by the
broad distribution of inter-residue distances centered at a distance greater than 5 Å, too long for a
stable hydrogen bond. In contrast, when a serine is present, the sharp peak of serine-histidine
inter-residue distance around 2.75 Å suggests the presence of a strong hydrogen bond. This
hydrogen bonding likely shifts the electron density of the histidine away from the catalytic
cysteine, weakening the imidazoline-thiolate ion pair. This weakened ionic interaction would
lead to less pKa depression compared to CHS orthologs and mutants containing a cysteine in the
nearby position, where the histidine is able to maintain a stronger ion pair with the catalytic
cysteine and lower the pKa to a greater degree. This is reminiscent of the role of aspartate 158 in
papain, a cysteine protease that also uses a cysteine-histidine-asparagine catalytic triad for
nucleophilic attack on its substrates (Storer & Ménard, 1994). Although D158 is not essential for
papain activity, its side chain affects the pH-activity profile by forming a hydrogen bond with the
backbone amide of the catalytic histidine. This interaction stabilizes the catalytic ionic pair and
maintains an optimal orientation of active-site residues. A D158E mutant papain had a
55

pH-activity profile shifted by 0.3 pH units, about the same magnitude of the effect we observed
on pKa for CHS cysteine/serine mutants.
We propose a model of the role of position 347 in enhancing CHS reactivity (Figure 6).
In the basal example of SmCHS, the serine interacts more strongly with the histidine of the
catalytic triad, weakening the ionic interaction that stabilizes the thiolate form of the catalytic
cysteine. In euphyllophyte CHSs, this position mutated to a cysteine, which interacts more
poorly with the histidine, strengthening the ionic interaction and stabilizing the activated thiolate
of the catalytic cysteine.
While the mechanism of how the other six mutations in the M7 mutants affect the
catalytic cysteine is not entirely clear, we noticed that, possibly due to the smaller side chains of
the S213G and Q217A mutations, AtCHS M7 has a surface helix in a slightly different
conformation than wild-type AtCHS, leading to a slightly wider active-site opening. There is
also a newly solvent-accessible cavity as determined by a computational cavity-finding software
(Figure S9). These structural differences could lead to subtle changes in the amino acid backbone
dynamics near the active site and thus alter the active-site volume or electronic environment,
which could alter the pKa of the catalytic cysteine (Jez & Noel, 2000).
Although cysteine sulfenic and sulfinic acid have been thought of as crystallographic
artifacts, an increasing number of studies have shown that this type of cysteine oxidation can
play an important functional role. In particular, cysteine sulfinic acid has been shown to play a
regulatory role in reversible inhibition of the activity of enzymes such as protein tyrosine
phosphatase 1B and glyceraldehyde-3-phosphate dehydrogenase, suggesting that cysteine redox
56

potential can be an evolved trait (Peralta et al., 2015; van Montfort, Congreve, Tisi, Carr, &
Jhoti, 2003).
Our results demonstrate that euphyllophytes could have evolved a CHS enzyme that is
intrinsically more active, with increased cysteine reactivity as a component, as one adaptation to
produce the larger suite of flavonoids needed to counter the various environmental stresses they
face. Although it may seem counterintuitive for euphyllophytes, which encounter more oxidative
environments than do basal plants, to rely on a CHS enzyme that is more susceptible to
oxidation, this susceptibility may be an unavoidable trade-off resulting from the chemical nature
of a more nucleophilic cysteine: a catalytic cysteine more reactive toward substrate is also more
reactive toward oxidants like hydrogen peroxide. To compensate for this increased susceptibility
to oxidation, euphyllophytes may have evolved other systems to better maintain the redox
environment inside the cell, one of those systems being the antioxidant flavonoids themselves.
57

Materials and Methods
Cloning and site-directed mutagenesis of CHSs
Total RNA was obtained from Arabidopsis thaliana, Pinus sylvestris, Equisetum arvense,
Selaginella moellendorffii, and Physcomitrella patens. Reverse transcription was performed to
obtain cDNA. The open reading frames (ORFs) of five CHS orthologs were amplified via PCR
from cDNA, digested with NcoI and XhoI, and ligated into NcoI- and XhoI-digested pHis8-3 or
pHis8-4B Escherichia coli expression vectors. Site-directed mutagenesis was performed
according to the QuikChange II Site-Directed Mutagenesis protocol (Agilent Technologies).
Transgenic Arabidopsis
The AtCHS promoter (defined as 1328 bp of sequence upstream of the CHS transcription
start site) was amplified via PCR from Arabidopsis genomic DNA, digested with HindIII and
XhoI, and ligated into HindIII- and XhoI-digested pCC 1136, a promoterless Gateway cloning
binary vector containing a BAR resistance gene marker, to generate pJKW 0152. The five CHS
ORFs described above were then PCR amplified from cDNA and cloned into pCC 1155, an
ampicillin-resistant version of the pDONR221 Gateway cloning vector, with BP clonase in the
Gateway cloning method (Thermo-Fisher). The resulting vectors were recombined with pJKW
0152 using LR clonase in the Gateway cloning method to generate the final binary constructs.
Agrobacterium tumefaciens-mediated transformation of Arabidopsis was performed using the
floral dipping method (Weigel & Glazebrook, 2002).
58

Recombinant protein expression and purification
CHS genes were cloned into pHis8-3 or pHis8-4B, bacterial expression vectors
containing an N-terminal 8×His tag followed by a thrombin or tobacco etch virus (TEV)
cleavage site, respectively, for recombinant protein production in E. coli. Proteins were
expressed in the BL21(DE3) E. coli strain cultivated in terrific broth (TB) and induced with 0.1
mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 18 °C. E. coli cells were
harvested by centrifugation, resuspended in 150 mL lysis buffer (50 mM Tris pH 8.0, 500 mM
NaCl, 30 mM imidazole, 5 mM DTT), and lysed with five passes through an M-110L
microfluidizer (Microfluidics). The resulting crude protein lysate was clarified by centrifugation
(19,000 g, 1 h) prior to QIAGEN nickel–nitrilotriacetic acid (Ni–NTA) gravity flow
chromatographic purification. After loading the clarified lysate, the Ni–NTA resin was washed
with 20 column volumes of lysis buffer and eluted with 1 column volume of elution buffer (50
mM Tris pH 8.0, 500 mM NaCl, 300 mM imidazole, 5 mM DTT). 1 mg of His-tagged thrombin
or TEV protease was added to the eluted protein, followed by dialysis at 4 °C for 16 h in dialysis
buffer (50 mM Tris pH 8.0, 500 mM NaCl, 5 mM DTT). After dialysis, the protein solution was
passed through Ni–NTA resin to remove uncleaved protein and His-tagged TEV. The
recombinant proteins were further purified by gel filtration on an ÄKTA Pure fast protein liquid
chromatography (FPLC) system (GE Healthcare Life Sciences). The principal peaks were
collected, verified by SDS–PAGE, and dialyzed into a storage buffer (12.5 mM Tris pH 8.0, 50
mM NaCl, 5 mM DTT). Finally, proteins were concentrated to >10 mg/mL using Amicon
Ultra-15 Centrifugal Filters (Millipore).
59

Protein crystallization
All protein crystals were grown by hanging drop vapor diffusion at 4 °C, except for
EaCHS at 20 °C. For AtCHS wild-type and C347S crystals, 1 µL of 10 mg/mL protein was
mixed with 1 µL of reservoir solution containing 0.1 M HEPES (pH 7.5), 0.3 M ammonium
acetate, 14% (v/v) PEG 8000, and 5 mM DTT. For AtCHS M7 crystals, 1 µL of 16.33 mg/mL
protein was mixed with 1 µL of reservoir solution containing 0.125 NaSCN, 20% (v/v) PEG
3350, and 5 mM DTT; 0.2 µL of a crystal seed stock from previous rounds of crystal
optimization was also added. For SmCHS wild-type and S340C crystals, 1 µL of 10 mg/mL
protein was mixed with 1 µL of reservoir solution containing 0.1 M MOPSO (pH 6.6), 0.3 M
Mg(NO3)2, 19% (v/v) PEG 4000, and 5 mM DTT. For EaCHS, 1 µL of 16.92 mg/mL protein
was mixed with 1 µL of reservoir solution containing 0.15 M LiCl, 8% PEG 6000, and 5 mM
DTT. For PsCHS, 1.66 µL of 14.65 mg/mL protein was mixed with 0.67 µL of reservoir solution
containing 0.14 M NH4Cl, 20% (v/v) PEG 3350, and 5 mM DTT; 0.2 µL of a crystal seed stock
from previous rounds of crystal optimization was also added. For PpCHS, 1 µL of 10 mg/mL
protein was mixed with 1 µL of reservoir solution containing 0.1 M MES (pH 6.9), 18% (v/v)
PEG 20000, and 5 mM DTT. Crystals were harvested within 1 week and transferred to a
cryoprotection solution of 17% glycerol and 83% reservoir solution. H2O2 soaking of SmCHS
crystals was performed by adding H2O2 to 1 mM to the cryoprotection solution and incubating at
4 °C for 75 min. Single crystals were mounted in a cryoloop and flash-frozen in liquid nitrogen.
60

X-ray diffraction and structure determination
X-ray diffraction data were collected at beamlines 8.2.1 and 8.2.2 of the Advanced Light
Source at Lawrence Berkeley National Laboratory on ADSC Quantum 315 CCD detectors for
AtCHS wild-type, AtCHS C347S, and SmCHS S340C crystals. X-ray diffraction data were
collected at beamlines 24-ID-C and 24-ID-E of the Advanced Photon Source at Argonne
National Laboratory on an ADSC Quantum 315 CCD detector, Eiger 16M detector, or Pilatus
6M detector for SmCHS wild-type, EaCHS, PsCHS, and AtCHS M7 crystals. Diffraction
intensities were indexed and integrated with iMosflm (Battye, Kontogiannis, Johnson, Powell, &
Leslie, 2011) and scaled with Scala under CCP4 (Evans, 2006; Winn et al., 2011). The phases
were determined with molecular replacement using Phaser under Phenix (Adams et al., 2010).
Further structural refinement utilized Phenix programs. Coot was used for manual map
inspection and model rebuilding (Emsley & Cowtan, 2004). Crystallographic calculations were
performed using Phenix.
Comparative sequence and structure analyses
CHS protein sequences were derived from NCBI and the 1000 Plants (1KP) Project
(Matasci et al., 2014; NCBI Resource Coordinators, 2016). In all cases, AtCHS was used as the
search query. Amino acid alignment of CHS orthologs was created using MUSCLE with default
settings (Edgar, 2004). UCSF Chimera and ESPript were used to display the multiple-sequence
alignments shown in Figure 2, Figure S5, and Figure S6 (Pettersen et al., 2004; Robert & Gouet,
2014). Phylogenetic analysis was performed using MEGA7 (Kumar, Stecher, & Tamura, 2016).
All structural figures were created with the PyMOL Molecular Graphics System, version 1.3
61

(Schrödinger, LLC) (DeLano, 2016). Active site cavity measurements for the AtCHS and AtCHS
M7 structures were determined using KVFinder (Oliveira et al., 2014).
Enzyme assays and pKa measurement
A 4CL-CHS coupled assay was used for kinetic analysis. A 4CL reaction master mix was
made by incubating 917 nM Arabidopsis thaliana 4CL1 (NCBI accession number NP_175579.1)
in 100 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 5 mM ATP, 100 µM p-coumaric acid, 100 µM
coenzyme A, and 10 or 50 µM malonyl-CoA for 30 min at room temperature to generate
p-coumaroyl-CoA at a final concentration of 70 µM. This 4CL was divided into individual
aliquots of 196 µL in Eppendorf tubes. CHS enzyme was incubated for 30 or 60 s in 16 µL
volumes using a triple buffer system (50 mM AMPSO, 50 mM sodium phosphate, 50 mM
sodium pyrophosphate, various pH) (Ellis & Morrison, 1982) (Schlegel, Jez, & Penning, 1998) at
room temperature in the presence of 25 µM iodoacetamide for the inactivation sample or water
for the control sample. Aliquots (4 µL) were withdrawn from the incubation mixture and added
to the standard coupled CHS assay system. The CHS reaction was run for 10 min at room
temperature and stopped by addition of 200 µL methanol.
The assay samples were centrifuged and analyzed directly by liquid
chromatography−mass spectrometry (LC−MS). LC was conducted on a Dionex UltiMate 3000
UHPLC system (Thermo Fisher Scientific), using water with 0.1% formic acid as solvent A and
acetonitrile with 0.1% formic acid as solvent B. Reverse phase separation of analytes was
performed on a Kinetex C18 column, 150 × 3 mm, 2.6 μm particle size (Phenomenex). The
column oven was held at 30 °C. Samples were eluted with a gradient of 5–60% B for 9 min, 95%
62

B for 3 min, and 5% B for 3 min, with a flow rate of 0.7 mL/min. MS analysis was performed on
a TSQ Quantum Access Max mass spectrometer (Thermo Fisher Scientific) operated in negative
ionization mode with a SIM scan centered at 271.78 m/z to detect naringenin chalcone.
The pH profiles (pH on the X-axis, ratio of naringenin chalcone produced with
iodoacetamide-treatment to control on the Y-axis) were determined by fitting raw data to the
log(inhibitor) vs. response equation using nonlinear regression in Prism, version 6.0f (GraphPad
Software).
Molecular dynamics
All MD simulations were performed using the GROMACS 5.1.4 package (Abraham et
al., 2015) and CHARMM force field (Best et al., 2012). The catalytic residues were modeled as
protonated histidine (H309 in AtCHS number) and deprotonated cysteine (C169 in AtCHS
numbering). All CHSs were constructed as dimers and were pre-aligned to the wild-type AtCHS
crystal structure using the Multiseq plugin of VMD (Roberts, Eargle, Wright, &
Luthey-Schulten, 2006). All CHS dimers were solvated with 0.1 M NaCl in a dodecahedron box.
Before the production runs, all systems were submitted to a minimization, followed by a 500-ps
NVT and a 500-ps NPT run with heavy atoms constrained. This was followed by another 5-ns
NPT simulation with protein backbone constrained. In all simulations, an integration time step of
2 fs was used, with bonds involving hydrogens constrained using LINCS (Hess, 2008; Hess,
Bekker, Berendsen, Fraaije, & Others, 1997). The van der Waals interaction was smoothly
switched off starting from 10 Å, with a cut-off distance of 12 Å. The neighboring list was
updated every 10 steps with Verlet cutoff-scheme. The electrostatic interaction was evaluated
63

using Particle-Mesh-Ewald (PME) summation (Darden, York, & Pedersen, 1993) with a grid
spacing of 1.5 Å to account for the long-range interaction, while its short-range interaction in
real space had a cut-off distance of 12 Å. The velocity-rescaling thermostat (Bussi, Donadio, &
Parrinello, 2007) and Parrinello-Rahman barostat (Nosé & Klein, 1983; Parrinello & Rahman,
1981) were employed to maintain the temperature at 300 K and the pressure at 1 bar.
For each CHS, three copies of 200-ns production runs were performed. The aggregated
simulation time of all CHS wildtype and mutant systems is 5.4 μs. The two monomers of a given
CHS were treated equivalently in the analysis; i.e., the three copies of trajectories of each
monomer were combined after they were aligned to chain A of the associated crystal structure,
resulting in a total of 1.2-μs trajectory for analysis of a given CHS system. Clustering analysis
was carried out with GROMACS gmx cluster with a RMSD cutoff of 0.1 nm. The inter-residue
distance was measured using the tcl scripting abilities provided by VMD (Humphrey, Dalke, &
Schulten, 1996). The minimum distance between the two nitrogen atoms of the catalytic histidine
and the associated hydroxyl, thiol, or thiolate group of its serine or cysteine partener was taken as
the inter-residue distance. Water occupancy calculation was performed using the volmap plugin
of VMD (Humphrey et al., 1996).
64

References
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19–25.
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., … Zwart, P. H. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica. Section D, Biological Crystallography, 66(Pt 2), 213–221.
Ashkenazy, H., Penn, O., Doron-Faigenboim, A., Cohen, O., Cannarozzi, G., Zomer, O., & Pupko, T. (2012). FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Research, 40(Web Server issue), W580–W584.
Austin, M. B., Bowman, M. E., Ferrer, J.-L., Schröder, J., & Noel, J. P. (2004). An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chemistry & Biology, 11(9), 1179–1194.
Austin, M. B., & Noel, J. P. (2003). The chalcone synthase superfamily of type III polyketide synthases. Natural Product Reports, 20(1), 79–110.
Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R., & Leslie, A. G. W. (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallographica. Section D, Biological Crystallography, 67(Pt 4), 271–281.
Best, R. B., Zhu, X., Shim, J., Lopes, P. E. M., Mittal, J., Feig, M., & Mackerell, A. D., Jr. (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. Journal of Chemical Theory and Computation, 8(9), 3257–3273.
Bussi, G., Donadio, D., & Parrinello, M. (2007). Canonical sampling through velocity rescaling. The Journal of Chemical Physics, 126(1), 014101.
Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98(12), 10089–10092.
DeLano, W. L. (2016). The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA: 2002. There Is No Corresponding Record for This Reference.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797.
Ellis, K. J., & Morrison, J. F. (1982). Buffers of constant ionic strength for studying pH-dependent processes. Methods in Enzymology, 87, 405–426.
Emsley, P., & Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallographica. Section D, Biological Crystallography, 60(Pt 12 Pt 1), 2126–2132.
Evans, P. (2006). Scaling and assessment of data quality. Acta Crystallographica. Section D, Biological Crystallography, 62(Pt 1), 72–82.
Ferrer, J. L., Jez, J. M., Bowman, M. E., Dixon, R. A., & Noel, J. P. (1999). Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nature Structural Biology, 6(8), 775–784.
Harris, T. K., & Turner, G. J. (2002). Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life, 53(2), 85–98.
Hess, B. (2008). P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation.
65

Journal of Chemical Theory and Computation, 4(1), 116–122. Hess, B., Bekker, H., Berendsen, H. J. C., Fraaije, J. G., & Others. (1997). LINCS: a linear
constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463–1472.
Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: visual molecular dynamics. Journal of Molecular Graphics, 14(1), 33–38, 27–28.
Jez, J. M., Austin, M. B., Ferrer, J., Bowman, M. E., Schröder, J., & Noel, J. P. (2000). Structural control of polyketide formation in plant-specific polyketide synthases. Chemistry & Biology, 7(12), 919–930.
Jez, J. M., & Noel, J. P. (2000). Mechanism of Chalcone Synthase: pKa of the Catalytic Cysteine and the Role of the Conserved Histidine in a Plant Polyketide Synthase. The Journal of Biological Chemistry, 275(50), 39640–39646.
Kortemme, T., & Creighton, T. E. (1995). Ionisation of Cysteine Residues at the Termini of Model α-Helical Peptides. Relevance to Unusual Thiol pKa Values in Proteins of the Thioredoxin Family. Journal of Molecular Biology, 253(5), 799–812.
Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution, 33(7), 1870–1874.
Matasci, N., Hung, L.-H., Yan, Z., Carpenter, E. J., Wickett, N. J., Mirarab, S., … Wong, G. K.-S. (2014). Data access for the 1,000 Plants (1KP) project. GigaScience, 3, 17.
NCBI Resource Coordinators. (2016). Database resources of the National Center for Biotechnology Information. Nucleic Acids Research, 44(D1), D7–D19.
Nosé, S., & Klein, M. L. (1983). Constant pressure molecular dynamics for molecular systems. Molecular Physics, 50(5), 1055–1076.
Oliveira, S. H. P., Ferraz, F. A. N., Honorato, R. V., Xavier-Neto, J., Sobreira, T. J. P., & de Oliveira, P. S. L. (2014). KVFinder: steered identification of protein cavities as a PyMOL plugin. BMC Bioinformatics, 15, 197.
Parrinello, M., & Rahman, A. (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52(12), 7182–7190.
Peralta, D., Bronowska, A. K., Morgan, B., Dóka, É., Van Laer, K., Nagy, P., … Dick, T. P. (2015). A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nature Chemical Biology, 11(2), 156–163.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612.
Rausher, M. D. (2006). The Evolution of Flavonoids and Their Genes. In E. Grotewold (Ed.), The Science of Flavonoids (pp. 175–211). New York, NY: Springer New York.
Reddie, K. G., & Carroll, K. S. (2008). Expanding the functional diversity of proteins through cysteine oxidation. Current Opinion in Chemical Biology, 12(6), 746–754.
Roberts, E., Eargle, J., Wright, D., & Luthey-Schulten, Z. (2006). MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics, 7, 382.
Robert, X., & Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Research, 42(Web Server issue), W320–W324.
Schlegel, B. P., Jez, J. M., & Penning, T. M. (1998). Mutagenesis of 3α-Hydroxysteroid Dehydrogenase Reveals a “Push- Pull” Mechanism for Proton Transfer in Aldo- Keto
66

Reductases. Biochemistry, 37(10), 3538–3548. Shirley, B. W., Kubasek, W. L., Storz, G., Bruggemann, E., Koornneef, M., Ausubel, F. M., &
Goodman, H. M. (1995). Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. The Plant Journal: For Cell and Molecular Biology, 8(5), 659–671.
Storer, A. C., & Ménard, R. (1994). Catalytic mechanism in papain family of cysteine peptidases. In Methods in Enzymology (Vol. 244, pp. 486–500). Academic Press.
van Montfort, R. L. M., Congreve, M., Tisi, D., Carr, R., & Jhoti, H. (2003). Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature, 423(6941), 773–777.
Weigel, D., & Glazebrook, J. (2002). Arabidopsis: a laboratory manual. CSHL Press. Weng, J.-K., & Chapple, C. (2010). The origin and evolution of lignin biosynthesis. The New
Phytologist, 187(2), 273–285. Weng, J.-K., & Noel, J. P. (2012). Structure-function analyses of plant type III polyketide
synthases. Methods in Enzymology, 515, 317–335. Weng, J.-K., & Noel, J. P. (2013). Chemodiversity in Selaginella: a reference system for parallel
and convergent metabolic evolution in terrestrial plants. Frontiers in Plant Science, 4, 119. Winkel-Shirley, B. (2001). Flavonoid biosynthesis. A colorful model for genetics, biochemistry,
cell biology, and biotechnology. Plant Physiology, 126(2), 485–493. Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., … Wilson,
K. S. (2011). Overview of the CCP4 suite and current developments. Acta Crystallographica. Section D, Biological Crystallography, 67(Pt 4), 235–242.
Yao, L. H., Jiang, Y. M., Shi, J., Tomás-Barberán, F. A., Datta, N., Singanusong, R., & Chen, S. S. (2004). Flavonoids in food and their health benefits. Plant Foods for Human Nutrition , 59(3), 113–122.
67
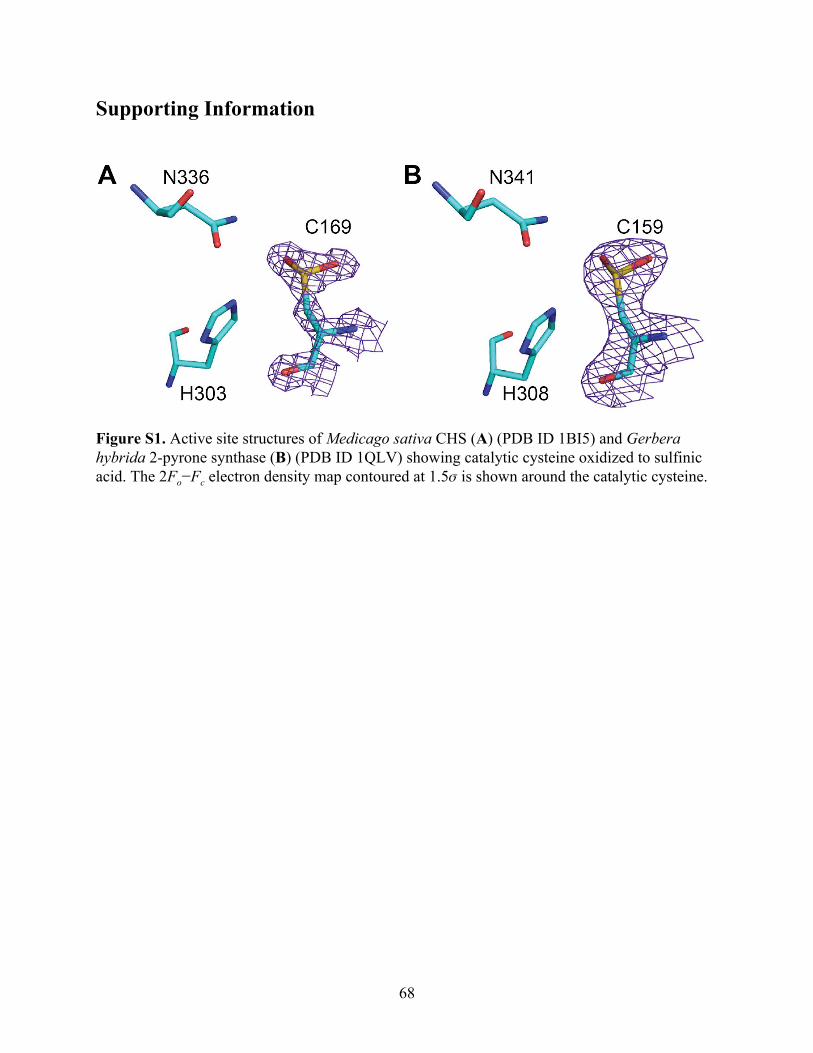
Supporting Information
Figure S1. Active site structures of Medicago sativa CHS (A) (PDB ID 1BI5) and Gerbera hybrida 2-pyrone synthase (B) (PDB ID 1QLV) showing catalytic cysteine oxidized to sulfinic acid. The 2Fo−Fc electron density map contoured at 1.5𝜎 is shown around the catalytic cysteine.
68
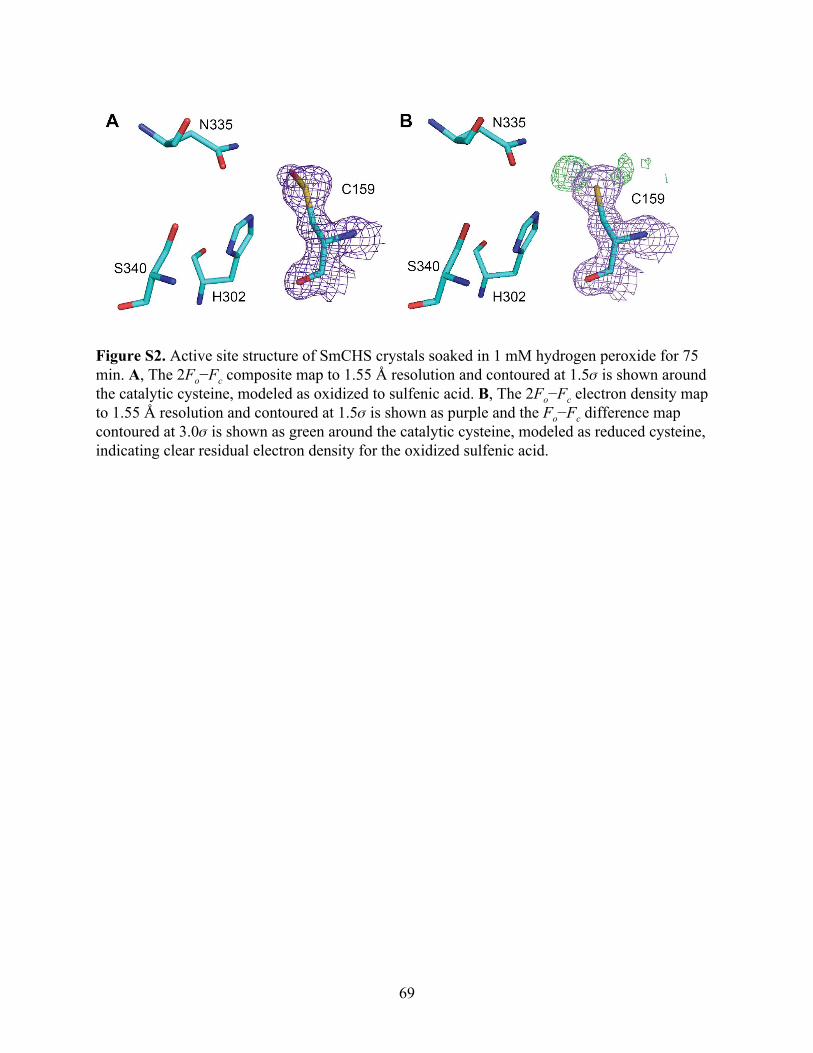
Figure S2. Active site structure of SmCHS crystals soaked in 1 mM hydrogen peroxide for 75 min. A, The 2Fo−Fc composite map to 1.55 Å resolution and contoured at 1.5𝜎 is shown around the catalytic cysteine, modeled as oxidized to sulfenic acid. B, The 2Fo−Fc electron density map to 1.55 Å resolution and contoured at 1.5𝜎 is shown as purple and the F o−F c difference map contoured at 3.0𝜎 is shown as green around the catalytic cysteine, modeled as reduced cysteine, indicating clear residual electron density for the oxidized sulfenic acid.
69
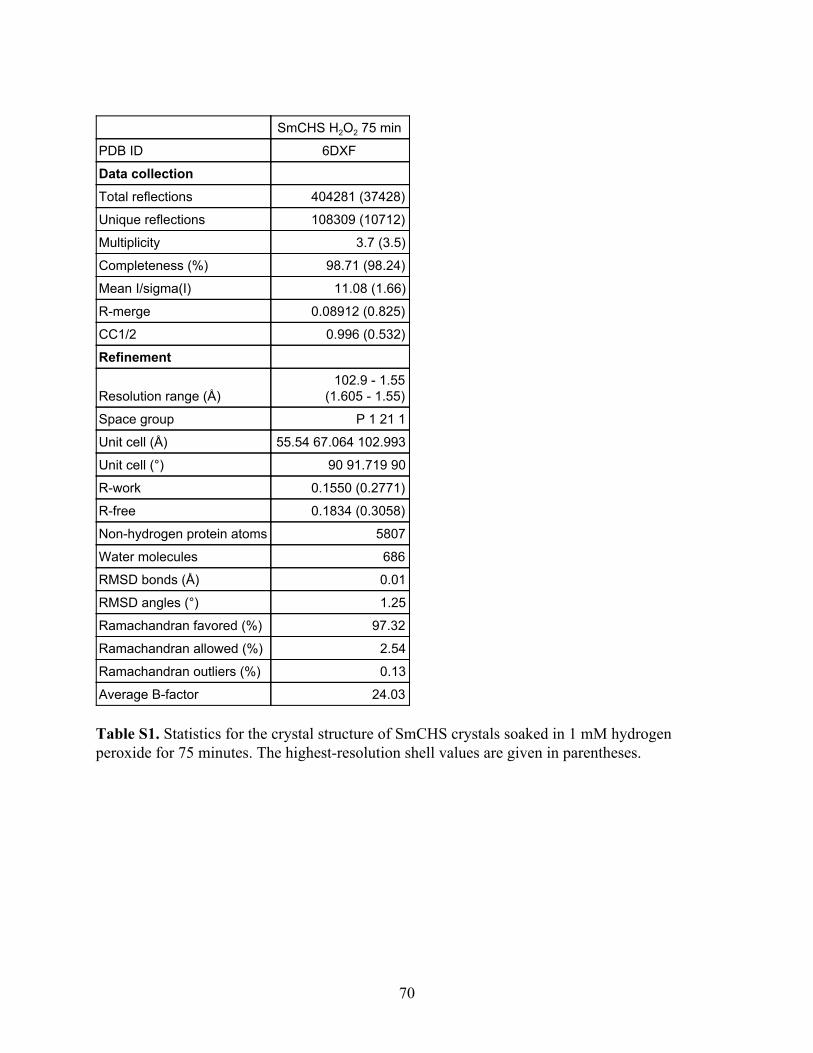
SmCHS H2O2 75 min
PDB ID 6DXF
Data collection
Total reflections 404281 (37428)
Unique reflections 108309 (10712)
Multiplicity 3.7 (3.5)
Completeness (%) 98.71 (98.24)
Mean I/sigma(I) 11.08 (1.66)
R-merge 0.08912 (0.825)
CC1/2 0.996 (0.532)
Refinement
Resolution range (Å) 102.9 - 1.55
(1.605 - 1.55)
Space group P 1 21 1
Unit cell (Å) 55.54 67.064 102.993
Unit cell (°) 90 91.719 90
R-work 0.1550 (0.2771)
R-free 0.1834 (0.3058)
Non-hydrogen protein atoms 5807
Water molecules 686
RMSD bonds (Å) 0.01
RMSD angles (°) 1.25
Ramachandran favored (%) 97.32
Ramachandran allowed (%) 2.54
Ramachandran outliers (%) 0.13
Average B-factor 24.03 Table S1. Statistics for the crystal structure of SmCHS crystals soaked in 1 mM hydrogen peroxide for 75 minutes. The highest-resolution shell values are given in parentheses.
70

Figure S3. Complementation of the transparent testa seed phenotype of tt4-2 mutant Arabidopsis thaliana. CHS orthologs were expressed under the AtCHS promoter. CHS from euphyllophytes (AtCHS, PsCHS, EaCHS) fully complement the mutant phenotype, whereas CHS from basal land plants (SmCHS, PpCHS) only partially complement.
71
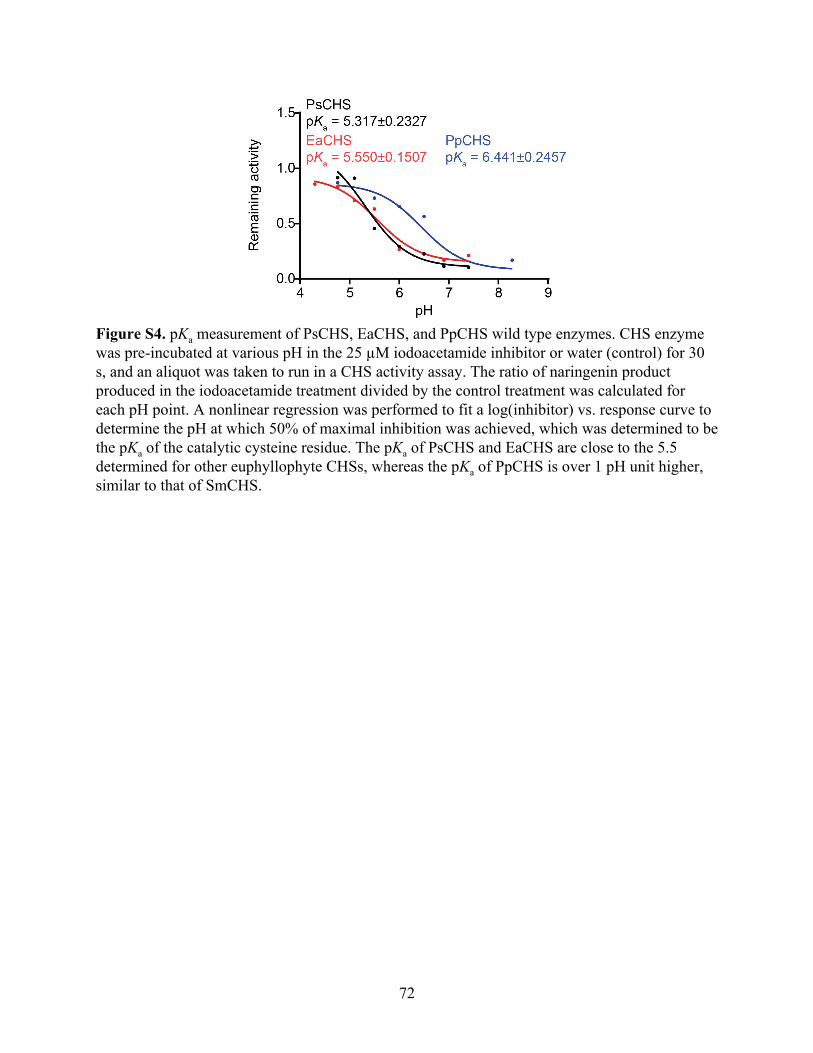
Figure S4. pKa measurement of PsCHS, EaCHS, and PpCHS wild type enzymes. CHS enzyme was pre-incubated at various pH in the 25 µM iodoacetamide inhibitor or water (control) for 30 s, and an aliquot was taken to run in a CHS activity assay. The ratio of naringenin product produced in the iodoacetamide treatment divided by the control treatment was calculated for each pH point. A nonlinear regression was performed to fit a log(inhibitor) vs. response curve to determine the pH at which 50% of maximal inhibition was achieved, which was determined to be the pKa of the catalytic cysteine residue. The pKa of PsCHS and EaCHS are close to the 5.5 determined for other euphyllophyte CHSs, whereas the pKa of PpCHS is over 1 pH unit higher, similar to that of SmCHS.
72

Figure S5. Multiple sequence alignment of CHSs. Sequence numbers of the beginning of each block for each CHS sequence are indicated. Residues outlined in thin black boxes are conserved with > 70% similarity across all sequences. Residues with 100% conservation are in white text with a black background. Red boxes indicate the seven positions mutated in the AtCHS M7 and SmCHS M7 constructs; these positions are differentially conserved between euphyllophyte and basal-plant CHSs, which are divided by the horizontal red line.
73

Figure S5 continued.
74
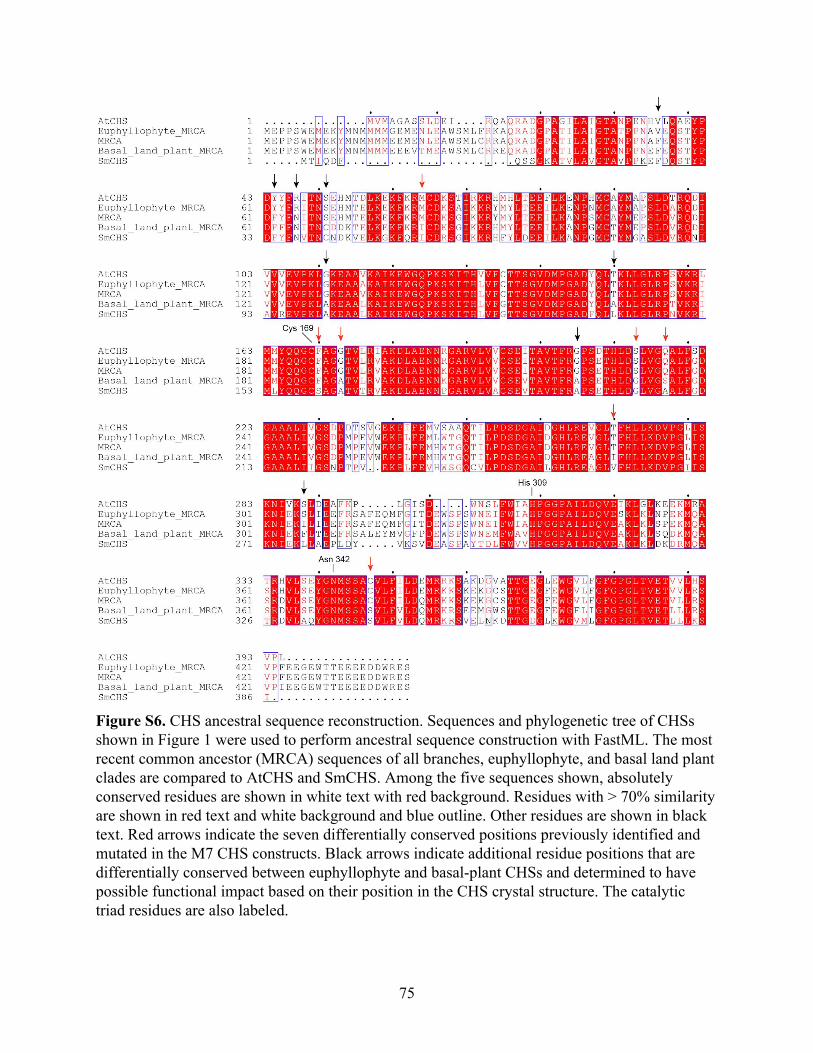
Figure S6. CHS ancestral sequence reconstruction. Sequences and phylogenetic tree of CHSs shown in Figure 1 were used to perform ancestral sequence construction with FastML. The most recent common ancestor (MRCA) sequences of all branches, euphyllophyte, and basal land plant clades are compared to AtCHS and SmCHS. Among the five sequences shown, absolutely conserved residues are shown in white text with red background. Residues with > 70% similarity are shown in red text and white background and blue outline. Other residues are shown in black text. Red arrows indicate the seven differentially conserved positions previously identified and mutated in the M7 CHS constructs. Black arrows indicate additional residue positions that are differentially conserved between euphyllophyte and basal-plant CHSs and determined to have possible functional impact based on their position in the CHS crystal structure. The catalytic triad residues are also labeled.
75
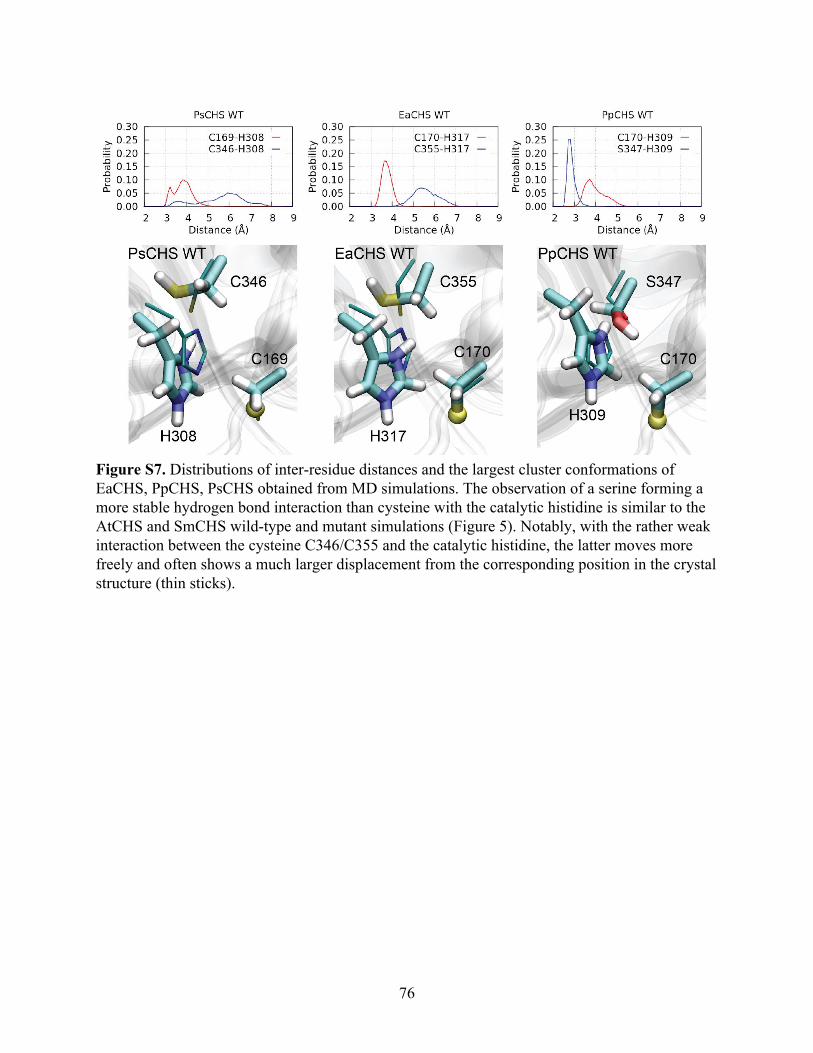
Figure S7. Distributions of inter-residue distances and the largest cluster conformations of EaCHS, PpCHS, PsCHS obtained from MD simulations. The observation of a serine forming a more stable hydrogen bond interaction than cysteine with the catalytic histidine is similar to the AtCHS and SmCHS wild-type and mutant simulations (Figure 5). Notably, with the rather weak interaction between the cysteine C346/C355 and the catalytic histidine, the latter moves more freely and often shows a much larger displacement from the corresponding position in the crystal structure (thin sticks).
76
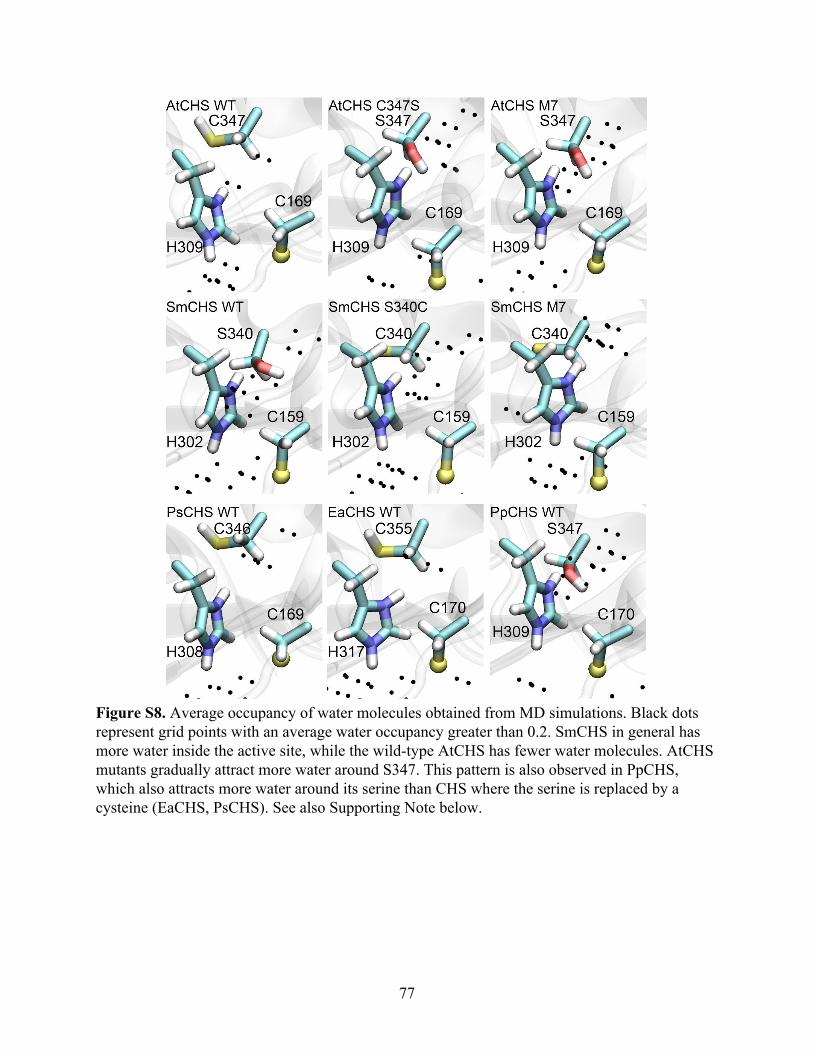
Figure S8. Average occupancy of water molecules obtained from MD simulations. Black dots represent grid points with an average water occupancy greater than 0.2. SmCHS in general has more water inside the active site, while the wild-type AtCHS has fewer water molecules. AtCHS mutants gradually attract more water around S347. This pattern is also observed in PpCHS, which also attracts more water around its serine than CHS where the serine is replaced by a cysteine (EaCHS, PsCHS). See also Supporting Note below.
77
Figure S9. Comparison of wild-type AtCHS (yellow) and AtCHS M7 (yellow) crystal structures. The catalytic triad residues and two of the seven mutations from wild-type to M7 are modelled as sticks and labeled. The yellow and magenta surfaces represent the solvent-accessible cavities measured using the cavity-finding program KVFinder. The helix containing the two marked mutations is shifted in AtCHS M7 compared to wild type, leading to a larger active-site cavity.
78

Supporting Note
Our MD calculations show that the C347S substitution (AtCHS numbering) can
significantly affect active-site solvation. The occupancy of water molecules within the active site
was measured with a resolution of 1 Å3 (Figure S8). Interestingly, S347 in AtCHS C347S and
M7 mutants attracts more water toward itself and H309. Similarly, the wild-type SmCHS is also
considerably wetter than the wild-type AtCHS: employing a cylinder with a radius of 9 Å and a
height of 13 Å to enclose the catalytic residues, we found that the average number of water
molecules enclosed was 40.0 for SmCHS and 31.4 for AtCHS. The ability of a serine to attract
more water is also observed in simulations of EaCHS, PpCHS, and PsCHS, although in SmCHS
mutants the active site remains rather wet despite the mutation of serine to cysteine (Figure S8).
AtCHS M7 also showed a wider active-site opening than wild-type AtCHS, which may
also affect solvent access to the active site, as shown by the large cavity found in cavity analysis.
In addition to changing the hydrogen bonding network, the decreased solvation in euphyllophyte
CHSs would enhance the pKa-lowering effect of the histidine, because ionic effects are enhanced
as the dielectric constant decreases along with solvent polarity (Harris & Turner, 2002).
79

80

Chapter 3 Regulation of chalcone synthase activity in vivo by oxidation of the catalytic cysteine Authors Geoffrey Liou1,2 and Jing-Ke Weng1,2
Author Affiliations 1. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 2. Whitehead Institute for Biomedical Research, Cambridge, MA 02142, USA
81

Abstract
Oxidation of the cysteinyl side chain in proteins occurs widely in all living organisms,
often as a result of increased levels of reactive oxygen species (ROS). However, the molecular
mechanisms and biochemical consequences of cysteine oxidation are not well understood. We
have previously observed that chalcone synthase (CHS), the first committed enzyme in plant
flavonoid biosynthesis, has evolved a catalytic cysteine that is both more reactive and sensitive
to oxidation in euphyllophytes than in basal land plants. We aim to gain a better understanding of
how CHS activity and flavonoid metabolism is controlled by oxidative inactivation at the
systems level. Furthermore, we seek to understand how such an intricate molecular mechanism
could have arisen evolutionarily, and what functional significance this trait might have
contributed to the adaptation of land plants to the challenging terrestrial environments in the past
450 million years. We have begun to develop several experimental methods that will allow us to
address these questions in vivo.
82

Introduction
Post-translational modification is a process that changes the properties of proteins and
extends their functions beyond those defined simply by their amino acid sequences. Compared to
other post-translational modifications, oxidative thiol modifications of cysteine residues have
received little attention and have often been disregarded as in vitro artifacts (Brandes, Schmitt, &
Jakob, 2009). More recently, however, the role of cysteine oxidation as a dynamic post-
translational modification to regulate protein function in signal transduction and other important
physiological processes has become apparent (Kettenhofen & Wood, 2010). These advances in
understanding have been facilitated by the advent of chemical probes and omics tools for
trapping and detecting specific oxidized forms of cysteine (Seo & Carroll, 2011).
Reactive oxygen species (ROS), including hydrogen peroxide (H2O2), superoxide (O2• −),
and the hydroxyl radical (•OH), are generally considered toxic byproducts of aerobic metabolism
that must be eradicated to maintain cellular homeostasis (Paulsen & Carroll, 2010). Recent
findings, however, suggest that ROS can act as secondary messengers to mediate cellular
signaling and metabolism (D’Autréaux & Toledano, 2007). The cysteinyl side chain in proteins
can undergo a series of oxidative transformations upon reaction with ROS: the cysteine thiol
(Cys-SH) is reversibly oxidized to sulfenic acid (Cys-SOH), which can then be successively
oxidized irreversibly to sulfinic (Cys-SO2H) and sulfonic acid (Cys-SO3H) (Kettenhofen &
Wood, 2010).
These oxidative modifications of cysteine can alter the properties of proteins and regulate
their activity in various ways, as has been studied in a few systems. For example, protein
tyrosine phosphatases (PTPs) were among the first family of proteins found to be regulated by
83

intracellular redox state through cysteine oxidation (Denu & Tanner, 1998). The catalytic
mechanism of PTPs involves nucleophilic attack by a conserved catalytic cysteine on a
phosphotyrosine substrate to remove the phosphate group, and PTP activity can be reversibly
inhibited by oxidation of the catalytic cysteine to sulfenic acid in vivo (van Montfort, Congreve,
Tisi, Carr, & Jhoti, 2003). PTP inactivation by oxidation has also been shown to occur in plants,
which correlates with the previous observation of the activation by H2O2 of the downstream
mitogen-activated protein (MAP) kinase (Gupta & Luan, 2003). Similar to PTPs,
glyceraldehyde-3-phosphate dehydrogenase (GAPDH), an enzyme in glycolysis, contains a
catalytic cysteine that is prone to oxidation both in vitro and in vivo (Butterfield, Allan
Butterfield, Hardas, & Bader Lange, 2010; Ishii et al., 1999). Interestingly, oxidative
modification of this catalytic cysteine to sulfenic acid, but not other forms, grants a novel acyl
phosphatase activity to GAPDH while inhibiting its dehydrogenase activity (Schmalhausen,
Nagradova, Boschi-Muller, Branlant, & Muronetz, 1999).
Although cysteine sulfinic and sulfonic acid are generally considered irreversible
oxidative modifications, recent studies have discovered a family of eukaryotic enzymes, known
as sulforedoxins (Srx), that can specifically reduce the sulfinic acid form of the catalytic cysteine
in the 2-Cys peroxiredoxins (Prx) back to the thiol form in an ATP-dependent manner (Basu &
Koonin, 2005). Srx binds to Prx proteins by specific interactions with several critical
surface-exposed residues of the Prx proteins and transfers the γ-phosphate of ATP to sulfinic
acid, using its conserved cysteine as the phosphate carrier. The resulting sulfinic phosphoryl
ester is then reduced to cysteine thiol after oxidation of four thiol equivalents (Rhee, Jeong,
Chang, & Woo, 2007). Although it is largely unknown whether similar enzyme-mediated
84

reduction of cysteine sulfinic and sulfonic acid in proteins occurs more widely using protein
substrates other than Prxs or involves alternative classes of reductases other than Srx,
reactivation of enzymes inactivated by cysteine oxidation could be a critical component of a
system to regulate their activity.
A recent study on singlet oxygen responses in Chlamydomonas reinhardtii and
Arabidopsis thaliana identified a conserved small zinc finger protein, MBS1, that is required for
proper ROS signaling in response to oxidative stress caused by singlet oxygen (Shao, Duan, &
Bock, 2013). Intriguingly, under high-light stress, which elicits singlet oxygen overproduction by
photosystem II, the MBS1-overexpression lines showed a much stronger accumulation of
anthocyanins than the wild type, while the mbs1-1 mutant lacked visible accumulation of
anthocyanins (Shao et al., 2013). The CHS transcript and protein levels were unaltered in both
the mbs1-1 mutant and the 35S:MBS1 lines compared to wild-type (Ning Shao, personal
communication). We therefore postulate that the decreased anthocyanin accumulation in the
mbs1-1 mutant could be due to CHS inhibition through catalytic cysteine oxidation, as the
mbs1-1 mutant may fail to maintain proper redox homeostasis under high-light stress. On the
other hand, the enhanced ROS signaling in the MBS1 overexpression lines may prevent CHS
from being oxidized, resulting in enhanced flux into flavonoid biosynthesis, and consequently,
the hyperaccumulation of anthocyanins. MBS1 may be part of a redox regulatory network that
has evolved to modulate flavonoid biosynthesis. We have obtained these mbs1-1 mutant and
MBS1 overexpression lines, and they can be crossed with our CHS transgenic lines to examine
the differential effect of altered redox signaling on various CHS orthologs, and with tt5 mutant
lines to measure flavonoid biosynthetic flux under different redox stress and light conditions.
85

In this chapter, we aimed to develop transgenic Arabidopsis thaliana lines and various
methods to measure CHS activity and CHS redox state in vivo. To measure CHS activity and
flavonoid production, we developed a metabolic tracing method using Arabidopsis seedlings
grown in d6-p-coumaric acid and measured the production of labeled naringenin using a mutant
that accumulates naringenin. We also crossed this mutant with the mbs1-1 mutant and performed
metabolic tracing as a preliminary test of whether altered redox regulation affected flavonoid
metabolic flux. To measure CHS redox state, we developed a proteomic mass spectrometric
method to detect peptides containing different oxidized forms of cysteine, and this method was
first tested with heterologously expressed CHS. To develop a purification workflow for isolating
CHS from plant tissues, we generated transgenic Arabidopsis lines expressing 3×FLAG-tagged
CHS and characterized their CHS gene expression by qRT-PCR. We also piloted a FLAG-based
purification and detection method with heterologously expressed 3×FLAG-CHS.
86

Results
tt5 mutant Arabidopsis thaliana accumulates naringenin and can be used for metabolic
tracing to measure CHS activity in vivo
Chalcone isomerase (CHI) is the enzyme immediately downstream of CHS in the
flavonoid biosynthetic pathway. CHI catalyzes the intramolecular cyclization of naringenin
chalcone to form (2S)-naringenin, which serves as the precursor to all downstream flavonoids
(Jez, Bowman, Dixon, & Noel, 2000). In the absence of CHI, naringenin chalcone can
spontaneously cyclize in aqueous solution to form a mixture of both enantiomers, (2S)- and
(2R)-naringenin.
The tt5 mutant of Arabidopsis thaliana contains a knockout of the CHI gene and
consequently lacks flavonoids in all tissues, resulting in the namesake transparent testa
phenotype of pale yellow seeds lacking brown colored flavonoids in the seed coat, as well as the
lack of hypocotyl flavonoid accumulation (Shirley et al., 1995). tt5 has been reported to
accumulate naringenin chalcone (Peer, 2001). We observed an accumulation of naringenin,
possibly due to spontaneous cyclization during metabolite extraction and LC/MS analysis. This
accumulation serves as a useful readout for CHS activity in vivo.
tt5 Arabidopsis thaliana seedlings were incubated in Murashige-Skoog media containing
100 µM d6-p-coumaric acid for 2 to 24 hours, and the abundances of unlabeled and
deuterium-labeled downstream phenylpropanoids and flavonoids were measured. Naringenin
showed an increase in label incorporation that plateaued at 8 hours, whereas the endpoint
phenylpropanoid sinapoyl malate and the endpoint flavonoid kaempferol 3-O-glucoside-
87
Figure 1. Incorporation of deuterium labeling into flavonoid (A, B) and phenylpropanoid (C) metabolites over time during Arabidopsis thaliana seedling incubation in d6-p-coumaric acid. The peak areas of the unlabeled (M) and d6- or d4-labeled compounds (M+6 or M+4, respectively) are plotted in blue and red, respectively. The percentage of labeled compound peak area as a percentage of the sum of the labeled compound peak area and unlabeled compound peak area is plotted in green.
88

7-O-rhamnoside showed a linear incorporation rate (Figure 1). This suggests that d6-p-coumaric
acid tracing could be used as a measurement of flavonoid metabolic flux through CHS.
tt5 mutant Arabidopsis thaliana accumulates both enantiomers of naringenin
In addition to the pale seed color common to all transparent testa mutants, tt5 exhibits
slightly stunted growth; male sterility; and shortened, curled siliques. This is somewhat
surprising because one would expect that naringenin chalcone produced by CHS would
spontaneously cyclize to form (2S)-naringenin, which could serve as the precursor to
downstream flavonoid biosynthesis. We hypothesized that the accumulation of the incorrect R
enantiomer of naringenin may somehow interfere with downstream flavonoid biosynthesis,
possibly through competitive inhibition of the biosynthetic enzymes. To examine the metabolic
changes that may lead to this phenotype, we prepared metabolic extracts from tt5 leaves and
siliques. Chiral chromatography revealed that these tissues indeed accumulate both the R and S
enantiomers of naringenin in approximately equal abundance (Figure 2).
Generation of and metabolic tracing with tt5 and mbs1-1 mutant Arabidopsis crosses
After observing that metabolic tracing with tt5 could be used to measure flavonoid
metabolic flux in vivo, we wanted to examine whether mutants in MBS1-mediated redox
homeostasis would affect flavonoid metabolism in a way that could be measured by tracing. An
mbs1-1 individual was used as the female and a tt5 individual was used as the male in a genetic
cross (mbs1-1 × tt5). The F1 seeds from this cross were collected and allowed to germinate. The
F2 seeds were collected and allowed to germinate; notably, only 64 F2 seeds germinated out of
89
Figure 2. Chiral chromatography shows that tt5 Arabidopsis accumulates both enantiomers of naringenin. Metabolic extracts from tt5 Arabidopsis silique and leaf were compared to a racemic naringenin standard. The total ion current (TIC) of the mass window to detect the [M−H]− ion of naringenin is shown.
90

270 total planted. The F2 individuals that exhibited lack of hypocotyl flavonoids and/or yellow
seeds were assumed to be tt5/tt5 homozygous, and leaf tissue samples were collected for
genotyping the MBS1 locus. Of these, approximately 75% were confirmed to have at least one
wild-type copy of MBS1, indicating that they were not homozygous for mbs1-1/mbs1-1. Because
the genotyping reaction for the T-DNA insertion in mbs1-1 failed to produce a PCR product
despite extensive efforts to optimize the reaction, we assumed that the lack of a PCR product in
the MBS1 wild-type reaction indicated mbs1-1/mbs1-1 homozygosity. Double homozygous
tt5/mbs1-1 lines were carried forward to the F3 generation.
Metabolic tracing with d6-p-coumaric acid was performed with F3 seedlings. Two of the
lines, 51 and 54, turned out not to be tt5 homozygous based on their accumulation of hypocotyl
and cotyledon flavonoids (Figure 3A and 3B). Only one line, 14, showed the same naringenin
accumulation as the tt5 control, whereas the remaining mbs1-1 × tt5 lines failed to accumulate
naringenin despite lacking hypocotyl flavonoids (Figure 3C). We also did not observe any
difference in labeled or unlabeled naringenin abundance between tt5 and mbs1-1 × tt5 line 14,
since the seedlings were grown under normal light conditions and not high-light stress.
The catalytic cysteine in AtCHS is more sensitive to in vitro oxidation than in SmCHS
To develop a method of detecting oxidized cysteine species in CHS isolated from plant
tissue samples, we first performed a pilot in vitro oxidation assay with CHS followed by
proteomic mass spectrometry. Recombinantly expressed and purified AtCHS and SmCHS were
incubated in aqueous buffer with one of the following oxidizing and reducing agents added: 5
mM or 1 mM hydrogen peroxide, 5 mM oxidized glutathione, water (no additional redox agent),
91
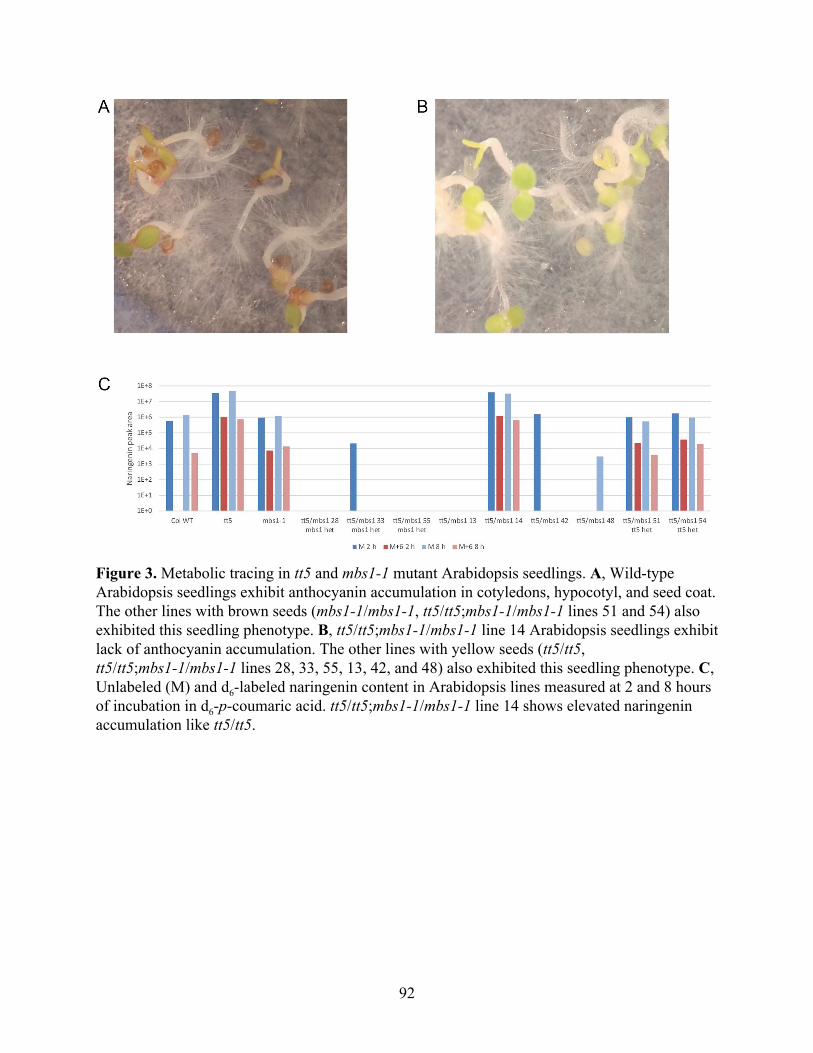
Figure 3. Metabolic tracing in tt5 and mbs1-1 mutant Arabidopsis seedlings. A, Wild-type Arabidopsis seedlings exhibit anthocyanin accumulation in cotyledons, hypocotyl, and seed coat. The other lines with brown seeds (mbs1-1/mbs1-1, tt5/tt5;mbs1-1/mbs1-1 lines 51 and 54) also exhibited this seedling phenotype. B, tt5/tt5;mbs1-1/mbs1-1 line 14 Arabidopsis seedlings exhibit lack of anthocyanin accumulation. The other lines with yellow seeds (tt5/tt5, tt5/tt5;mbs1-1/mbs1-1 lines 28, 33, 55, 13, 42, and 48) also exhibited this seedling phenotype. C, Unlabeled (M) and d6-labeled naringenin content in Arabidopsis lines measured at 2 and 8 hours of incubation in d6-p-coumaric acid. tt5/tt5;mbs1-1/mbs1-1 line 14 shows elevated naringenin accumulation like tt5/tt5.
92

5 mM reduced glutathione, or 5 mM dithiothreitol (DTT). The protein samples were then run on
a non-reducing SDS-PAGE to isolate the bands corresponding to CHS, which were then
submitted for proteomic mass spectrometric analysis. We searched for the peptides containing
the catalytic cysteine and determined the redox modification by the difference in mass from the
peptide containing reduced cysteine (Figure 4A and 4B). Peptide abundance was determined
using spectral counting.
Both AtCHS and SmCHS contain five non-catalytic cysteine residues located at various
positions across the protein. In these ten non-catalytic cysteines, the cysteinyl side chains are all
buried and therefore inaccessible to the solvent. Consistent with this notion, these noncatalytic
cysteines in both AtCHS and SmCHS were detected predominantly as carbamidomethylated at
the cysteine residues (>99.9%), indicating that these non-catalytic cysteines existed as reduced
thiols prior to iodoacetamide treatment. This ratio is invariable across samples incubated in the
six different redox conditions.
In contrast, the peptide containing the catalytic cysteine shows large variations in the
abundances of cysteine oxidized states across samples incubated in different redox conditions
(Figure 4C). Under reducing conditions, the catalytic cysteine exists primarily as a reduced thiol.
Under increasingly oxidizing conditions, however, the oxidative states of cysteine sulfenic,
sulfinic, and sulfonic acid were all detected in increasing ratios. Interestingly, this pilot
proteomic experiment comparing recombinant AtCHS and SmCHS indicates that the catalytic
cysteine in AtCHS is more sensitive to oxidation than that of SmCHS (Figure 4C). In the most
oxidizing condition (5 mM H2O2, condition A in Figure 4C), less than 10% of the catalytic
cysteine in AtCHS remained reduced and over 85% was oxidized to sulfonic acid. In contrast,
93
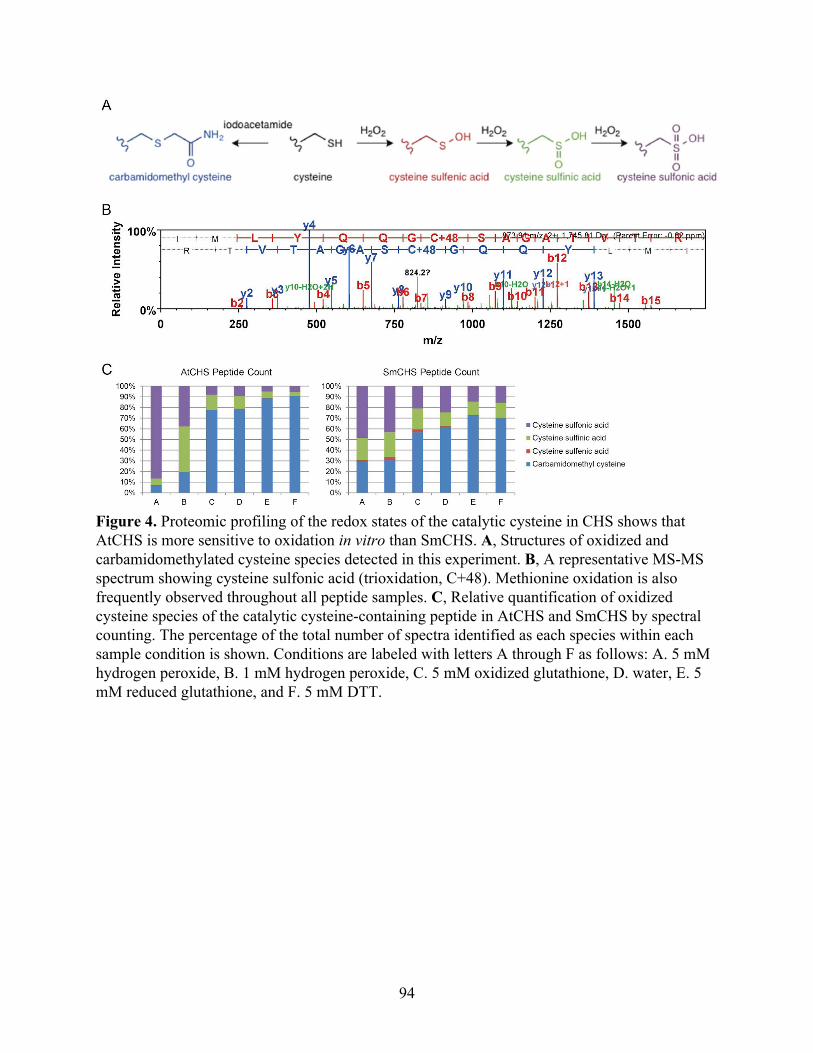
Figure 4. Proteomic profiling of the redox states of the catalytic cysteine in CHS shows that AtCHS is more sensitive to oxidation in vitro than SmCHS. A, Structures of oxidized and carbamidomethylated cysteine species detected in this experiment. B, A representative MS-MS spectrum showing cysteine sulfonic acid (trioxidation, C+48). Methionine oxidation is also frequently observed throughout all peptide samples. C, Relative quantification of oxidized cysteine species of the catalytic cysteine-containing peptide in AtCHS and SmCHS by spectral counting. The percentage of the total number of spectra identified as each species within each sample condition is shown. Conditions are labeled with letters A through F as follows: A. 5 mM hydrogen peroxide, B. 1 mM hydrogen peroxide, C. 5 mM oxidized glutathione, D. water, E. 5 mM reduced glutathione, and F. 5 mM DTT.
94

about 30% of the catalytic cysteine in SmCHS remained reduced together with a significant
portion of sulfinic acid not fully oxidized to sulfonic acid. This observation is consistent with the
our previous observations, made by X-ray crystallography and enzyme assays, that the catalytic
cysteine is more reactive and nucleophilic in euphyllophyte CHSs than basal land plant CHSs
(Liou, Chiang, Wang, & Weng, 2018).
Although the catalytic cysteine in CHS could theoretically be oxidized through forming
mixed disulfides with glutathione or DTT in some of the aforementioned incubation conditions,
none of these species were detected in our analysis. This suggests that the CHS active site
environment may preclude the entry of DTT or glutathione in effective orientations, and/or
kinetically favors other oxidative products over mixed disulfide formation.
FLAG-tag purification and western blotting of CHS
To develop a method for affinity purification of StrepII-tagged CHS from Arabidopsis
tissue for proteomic profiling of the catalytic cysteine redox state in vivo, we first attempted to
develop a protocol for affinity purification and western blotting of StrepII-tagged CHS. We used
both Arabidopsis protein extracts and CHS recombinantly expressed in E. coli. We attempted to
use both a StrepTactin-HRP conjugate (StrepTactin is a modified streptavidin protein that binds
specifically to the StrepII tag) and an anti-StrepII-tag mouse primary antibody followed by
detection with an anti-mouse IgG goat secondary antibody-HRP conjugate, but neither method
specifically detected the StrepII-tagged CHS from either Arabidopsis or E. coli sources. We thus
decided to try a different protein tag on CHS, an N-terminal 3×FLAG tag.
95

A 3×FLAG tag sequence was introduced onto the 5′ end of the AtCHS and SmCHS
sequences, and the combined 3×FLAG-CHS insert was cloned into an E. coli expression vector
to also introduce an N-terminal 8×His tag at the N-terminus of the 3×FLAG sequence, separated
by a tobacco etch virus (TEV) protease cleavage site. This His tag allows for an independent
affinity purification method to first purify 3×FLAG-CHS for pilot testing of FLAG affinity
purification and anti-FLAG western blotting. The presence of both His and FLAG tags could
also be useful for tandem affinity purification from plant tissue.
3×FLAG-tagged AtCHS and SmCHS were expressed in BL21 E. coli. Cell lysis was
performed as in our standard protein purification protocol (Liou et al., 2018). TEV protease
digestion to remove the His tag was performed on half of the cleared protein lysate in order to
assess whether the His tag would interfere with FLAG purification or western blotting. FLAG
purification was then performed using ANTI-FLAG M2 magnetic beads following the
manufacturer’s instructions (Sigma-Aldrich). SDS-PAGE of the bead-purified CHS samples
showed approximately equal enrichment of the TEV treated and untreated CHS, indicating that
the His tag does not interfere with the FLAG tag for purification of CHS (Figure 5A). Proteomic
mass spectrometric analysis of the excised gel pieces also confirmed the presence of AtCHS and
SmCHS in their respective protein bands (data not shown).
Anti-FLAG western blotting was then performed on the purified 3×FLAG-CHS samples,
as well as samples collected from various steps in the purification process. A monoclonal
ANTI-FLAG M2 mouse antibody (Sigma-Aldrich) was used as the primary antibody, and an
anti-mouse IgG goat antibody-HRP conjugate was used as the secondary antibody.
Chemiluminescence detection was performed using the Pierce ECL substrate (Thermo Fisher).
96
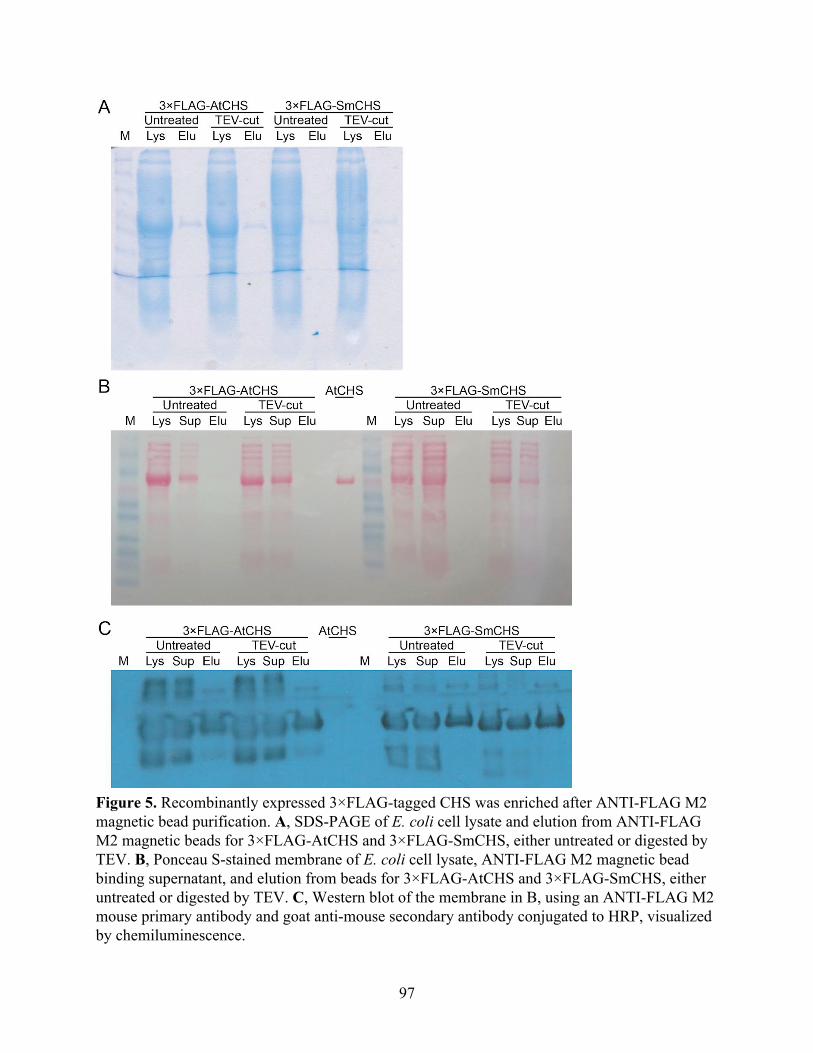
Figure 5. Recombinantly expressed 3×FLAG-tagged CHS was enriched after ANTI-FLAG M2 magnetic bead purification. A, SDS-PAGE of E. coli cell lysate and elution from ANTI-FLAG M2 magnetic beads for 3×FLAG-AtCHS and 3×FLAG-SmCHS, either untreated or digested by TEV. B, Ponceau S-stained membrane of E. coli cell lysate, ANTI-FLAG M2 magnetic bead binding supernatant, and elution from beads for 3×FLAG-AtCHS and 3×FLAG-SmCHS, either untreated or digested by TEV. C, Western blot of the membrane in B, using an ANTI-FLAG M2 mouse primary antibody and goat anti-mouse secondary antibody conjugated to HRP, visualized by chemiluminescence.
97

Although some nonspecific blotting was observed in the E. coli lysate and ANTI-FLAG bead
supernatant samples, the bead elution samples show mostly specific detection of 3×FLAG-CHS,
indicating that ANTI-FLAG purification did indeed enrich for 3×FLAG-CHS, and that the
ANTI-FLAG M2 antibody is suitable for western blot detection (Figure 5B and 4C).
Generation and characterization of transgenic Arabidopsis thaliana lines expressing
FLAG-tagged CHS orthologs
We have previously generated transgenic Arabidopsis thaliana lines containing
C-terminal StrepII-tagged CHS orthologs from different species, under the control of the
Arabidopsis CHS promoter in the CHS-null mutant tt4-2 background. These lines were used to
observe the capability of different CHS orthologs to complement in vivo the flavonoid-deficient
phenotypes of tt4-2 (Chapter 2, Figure S3).
Because the FLAG tag-based purification and western blotting worked better than the
StrepII-tag based system, we generated new transgenic Arabidopsis lines containing N-terminal
3×FLAG-AtCHS or SmCHS under the control of the Arabidopsis CHS promoter in the tt4-2
background using Agrobacterium tumefaciens floral dipping. For each construct, 16 to 20 T1
individuals were selected based on hypocotyl purple coloration, indicating flavonoid
accumulation and complementation of the CHS-null phenotype. Seeds and leaf tissue were
collected from these individuals for propagation and gene expression analysis, respectively.
CHS gene expression analysis was performed by quantitative RT-PCR (qRT-PCR), with
At1g13320 as a reference gene control. New gene-specific primer pairs were designed for both
AtCHS and SmCHS, with efficiencies of 50.52% and 45.87%, respectively (Figure 6A).
98

Although their amplification efficiencies are quite low, the primers can still be used to measure
the relative expression levels of the different CHS independent lines. The T1 individuals for both
constructs exhibited a wide range of CHS expression levels (Figure 6B).
The T2 seeds from the T1 lines were planted and allowed to germinate, then subject to
Basta selection. The percentage of T2 seedlings surviving Basta selection was counted for each
T1 line, and those with close to 75% survival were taken forward as single-insertion lines. T3
seeds were planted and subject to Basta selection, and those that had 100% seedling survival
were confirmed as homozygous in the T2 generation. These were further carried forward into the
T4 generation, but some lines were discarded after discovering that a narrow, curled leaf
phenotype had emerged. The remaining T4 individuals had their CHS expression characterized
by qRT-PCR. Two lines each for AtCHS and SmCHS were carried forward based on the
consistency of CHS expression between T4 individuals and their overall health and morphology
(Figure 6C).
99
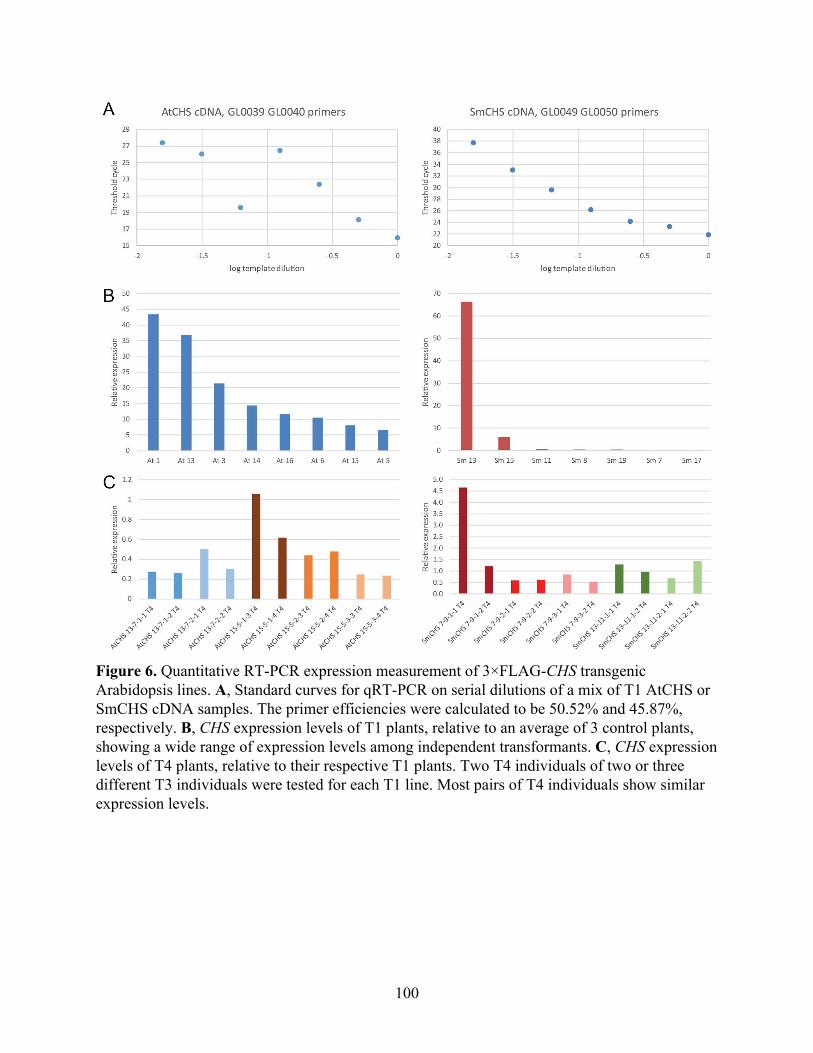
Figure 6. Quantitative RT-PCR expression measurement of 3×FLAG-CHS transgenic Arabidopsis lines. A, Standard curves for qRT-PCR on serial dilutions of a mix of T1 AtCHS or SmCHS cDNA samples. The primer efficiencies were calculated to be 50.52% and 45.87%, respectively. B, CHS expression levels of T1 plants, relative to an average of 3 control plants, showing a wide range of expression levels among independent transformants. C, CHS expression levels of T4 plants, relative to their respective T1 plants. Two T4 individuals of two or three different T3 individuals were tested for each T1 line. Most pairs of T4 individuals show similar expression levels.
100

Discussion and Future Directions
We have developed several transgenic lines and experimental methods to examine the in
vivo activity and oxidation of CHS. Metabolic tracing with d6-p-coumaric acid in tt5 Arabidopsis
showed linear accumulation of deuterium-labeled naringenin. This system could be used to
compare Arabidopsis tt4 mutants complemented with CHS orthologs from different plant
lineages to confirm whether the in vitro biochemical differences we observed previously also
affect their in vivo activities. The mbs1-1 × tt5 line can be used to examine the effect of redox
homeostasis on CHS activity and flavonoid biosynthesis. The various CHS transgenic lines could
be crossed with tt5, and the plants could be grown under different light cycle or oxidative stress
conditions to see whether their flavonoid biosynthetic flux is affected.
Like all biological processes, flavonoid metabolism is subject to regulation at many
different levels. To observe changes in transcriptional regulation, we developed qRT-PCR
primers to measure CHS expression, although their efficiencies could be improved by using a
different method to design them, such as QuantPrime. We also generated multiple independent
lines for each 3×FLAG-CHS ortholog to examine the effect of expression level on flavonoid
biosynthesis, since each line has the transgene inserted randomly so that transcription may be
affected by the surrounding genomic sequence.
To observe the effect of post-translational oxidative modifications on cysteine, we aimed
to develop a method of isolating CHS and measuring the different cysteine oxidized states. We
were able to detect peptides containing these cysteine residues that were oxidized in an in vitro
redox treatment. We have also preliminarily showed that the 3×FLAG-tagged CHS can be
101

isolated and detected specifically from biological samples using protein heterologously
expressed in E. coli, although this protocol has yet to be tested from plant tissue.
Our proteomic method can be further optimized to facilitate quantitative profiling of
cysteine oxidation in CHSs in vivo. For example, dimedone (5,5-dimethyl-1,3-cyclohexane-
dione) has been used to derivatize cysteine sulfenic acid, which is the least stable cysteine
hyperoxidative product (Nelson et al., 2010). Our pilot experiment did not involve derivatization
of any of the oxidized cysteine states, so an unstable state like cysteine sulfenic acid may have
been underrepresented in our proteomic quantitation data. Addition of dimedone as soon as
possible during the process of protein isolation from plant tissue could help us better quantify the
redox state of CHS in vivo.
In plant cells, ROS are produced in the photosynthetic reaction centers of the chloroplast
in excess light conditions. The chloroplast contains redox-active enzymes and small molecules to
quench ROS, but some species like H2O2 can diffuse long distances out of the chloroplast
(Asada, 2006). ROS are also produced as primary signaling molecules in response to pathogens
and other biotic and abiotic stresses (Møller, Jensen, & Hansson, 2007). Flavonoids likely serve
as a second line of ROS scavengers in plants, and flavonoid biosynthesis increases in oxidative
stress conditions (Fini, Brunetti, Di Ferdinando, Ferrini, & Tattini, 2011). Although it may seem
counterintuitive that euphyllophyte CHSs have evolved to become more sensitive to oxidation,
this may be an unavoidable consequence of selection for higher enzyme activity by increase
reactivity of the catalytic cysteine. These plants may have instead evolved other regulatory
systems to subsequently increase the expression of CHS and other flavonoid biosynthetic genes
to compensate for an initial inactivation by ROS.
102

103

Materials and Methods
Arabidopsis thaliana metabolic tracing
1 mg of tt5 Arabidopsis thaliana seeds were plated on top of a round piece of filter paper
on a Murashige-Skoog (Caisson Labs) 1% agarose plate (6 cm diameter). Seeds were vernalized
at 4 °C for 96 hours, then transferred to a plant growth chamber set to 25 °C and 16 hour light
and 8 hour dark cycle. After 5 days, the filter paper with germinated seedlings was removed and
placed into a new petri dish (6 cm diameter) with 1 mL of liquid Murashige-Skoog media with
100 µM d6-p-coumaric acid and incubated in the growth chamber at 25 °C. Samples were taken
at 2, 4, 8, and 24 hours of incubation by scraping the seedlings off the filter paper and patting
them dry with a Kimwipe. Seedlings were then placed in a microcentrifuge tube, weighed, flash
frozen in liquid nitrogen, and stored at -80 °C. Each seedling sample had a fresh weight of about
30 mg on average.
Metabolite extraction was performed by adding 5 µL of 50% methanol per mg of
seedling tissue, then incubating for 2 hours at 50 °C. Samples were centrifuged at 21,000 g for
15 min, and the supernatant was taken for analysis by liquid chromatography−high resolution
mass spectrometry (LC−HRMS). LC was performed on a Dionex UltiMate 3000 UHPLC system
(Thermo Fisher Scientific) using water with 0.1% formic acid (Solvent A) and acetonitrile with
0.1% formic acid (Solvent B) and a Kinetex 2.6 μm C18 100 Å column (Phenomenex) at 30 °C.
The elution gradient gradient was 5% B for 2 min, 5-95% B for 23 min, 95% B for 3 min, 95-5%
B for 0.1 min, 5% B for 2.9 min, at a flow rate of 0.8 mL/min. Compounds were detected on a
high-resolution Q-Exactive benchtop Orbitrap mass spectrometer (Thermo Fisher Scientific)
using a full scan range of 100−1250 m/z in negative ionization mode. The amount of label
104

incorporation for each metabolite was calculated as the peak area of the deuterium-labeled
metabolite (M+6 for naringenin; M+4 for sinapoyl malate and kaempferol 3-O-glucoside-7-O-
rhamnoside) divided by the sum of the peak areas of the deuterium-labeled and unlabeled
compounds.
Chiral chromatographic analysis of tt5 metabolic extracts
Approximately 10 mg each of tt5 leaf and silique tissue were incubated in 1 mL 50%
methanol at 50 °C for 2 hours. The supernatant was taken for chiral LC−MS analysis. LC was
performed on a Dionex UltiMate 3000 UHPLC system (Thermo Fisher Scientific) using water
with 0.1% formic acid (Solvent A) and acetonitrile with 0.1% formic acid (Solvent B) and a Lux
3 µm Cellulose-4 column (Phenomenex) at 30 °C. The elution gradient gradient was 5% B for 2
min, 5−95% B for 23 min, 95% B for 3 min, 95−5% B for 0.1 min, 5% B for 2.9 min, at a flow
rate of 0.8 mL/min. MS was performed on a TSQ Quantum Access MAX mass spectrometer
(Thermo Fisher Scientific) using a full scan range of 100−800 m/z in negative ionization mode.
Genotyping
2 to 3 cauline leaves from each plant were placed in a microcentrifuge tube and ground
with a pestle. 500 µL of extraction buffer (200 mM Tris-HCl pH 8, 250 mM NaCl, 25 mM
EDTA, 0.5% SDS) was added, and the tube was vortexed. The sample was incubated at 50 °C
for 10 min, vortexed, and centrifuged at max speed for 10 min in a tabletop centrifuge at room
temperature. 300 µL of the supernatant was transferred to a new tube containing 300 µL
isopropanol, and the tube was vortexed at low speed, then placed at -20 °C for 15 to 30 min. The
105

samples were centrifuged at max speed for 10 min in a tabletop centrifuge at room temperature,
and the supernatant was removed using a vacuum line. 500 µL of ice-cold 70% ethanol was
added, and the sample was centrifuged at max speed for 10 min at 4 °C. The supernatant was
removed using a vacuum line, and the pellet was dried on the benchtop or in a speed vac. The
pellet was resuspended in 100 µL of TE Buffer (10 mM Tris pH 7, 1 mM EDTA) by pipetting
and centrifuged at max speed for 1 min at 4 °C. The supernatant was transferred to a new tube,
and 1 µL was used as a template for genotyping PCR. Genotyping primers were designed using
the SIGnAL Salk T-DNA primer design tool (http://signal.salk.edu/tdnaprimers.2.html) (Table
1).
CHS in vitro redox treatment and proteomic mass spectrometry
About 10 µg of purified recombinant AtCHS and SmCHS protein was incubated in an
assay buffer [6.25 mM Tris pH 8, 25 mM NaCl, and 0.2 mM dithiothreitol (DTT)],
supplemented with one of the six following redox conditions: A. 5 mM hydrogen peroxide, B. 1
mM hydrogen peroxide, C. 5 mM oxidized glutathione, D. water (no additional redox agent), E.
5 mM reduced glutathione, and F. 5 mM DTT. After incubation for 15 min at room temperature,
10 µL of 2× non-reducing SDS sample buffer (250 mM Tris-HCl pH 6.8, 8% SDS, 40%
glycerol, 0.02% bromophenol blue) was added to 10 µL of the protein samples. Samples were
run on 12% SDS-PAGE. The gel was fixed for 15 min in 40% methanol/10% acetic acid, stained
with BioSafe Coomassie (Bio-Rad), and destained with distilled water overnight. Gel bands were
cut out with a razor blade and submerged in 100 µL 50% methanol. After dehydration, the
samples were incubated in 100 mM iodoacetamide for 30 min at room temperature to label
106
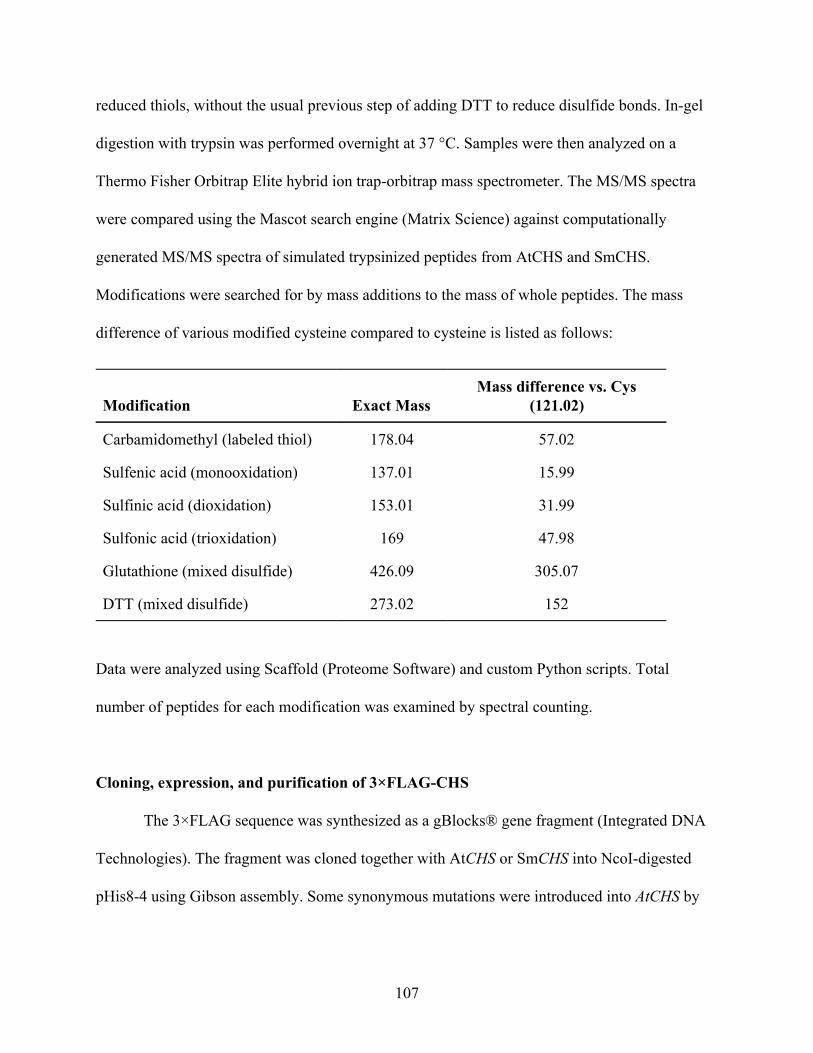
reduced thiols, without the usual previous step of adding DTT to reduce disulfide bonds. In-gel
digestion with trypsin was performed overnight at 37 °C. Samples were then analyzed on a
Thermo Fisher Orbitrap Elite hybrid ion trap-orbitrap mass spectrometer. The MS/MS spectra
were compared using the Mascot search engine (Matrix Science) against computationally
generated MS/MS spectra of simulated trypsinized peptides from AtCHS and SmCHS.
Modifications were searched for by mass additions to the mass of whole peptides. The mass
difference of various modified cysteine compared to cysteine is listed as follows:
Modification Exact Mass Mass difference vs. Cys
(121.02)
Carbamidomethyl (labeled thiol) 178.04 57.02
Sulfenic acid (monooxidation) 137.01 15.99
Sulfinic acid (dioxidation) 153.01 31.99
Sulfonic acid (trioxidation) 169 47.98
Glutathione (mixed disulfide) 426.09 305.07
DTT (mixed disulfide) 273.02 152
Data were analyzed using Scaffold (Proteome Software) and custom Python scripts. Total
number of peptides for each modification was examined by spectral counting.
Cloning, expression, and purification of 3×FLAG-CHS
The 3×FLAG sequence was synthesized as a gBlocks® gene fragment (Integrated DNA
Technologies). The fragment was cloned together with AtCHS or SmCHS into NcoI-digested
pHis8-4 using Gibson assembly. Some synonymous mutations were introduced into AtCHS by
107

PCR to fix a mis-annealing error encountered in Gibson assembly: in the AtCHS coding
sequence, they are T15A, T18A, T19A and C20G. Proteins were expressed in the BL21(DE3) E.
coli strain cultivated in terrific broth (TB) and induced with 0.1 mM isopropyl
β-D-1-thiogalactopyranoside (IPTG) overnight at 18 °C. E. coli cells were harvested by
centrifugation, resuspended in 150 mL lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 30 mM
imidazole, 5 mM DTT), and lysed with five passes through an M-110L microfluidizer
(Microfluidics). The resulting crude protein lysate was clarified by centrifugation (19,000 g, 1 h).
TEV digestion was performed on half of the cleared lysate overnight at 4 °C.
ANTI-FLAG M2 magnetic bead purification was performed according to the manufacturer’s
instructions for the batch format and elution with FLAG peptide (Sigma-Aldrich). Proteomic
mass spectrometry to identify AtCHS and SmCHS from SDS-PAGE bands was performed as
described above.
Western blotting of 3×FLAG-CHS
Tris-glycine SDS-PAGE was performed with the cleared E. coli lysate (1 µL),
supernatant after ANTI-FLAG M2 bead binding (1 µL), and the bead elution (7.5 µL) for
AtCHS and SmCHS, untreated or TEV cleaved. Electrophoretic transfer was performed using
the Bio-Rad Criterion Blotter system using a transfer buffer consisting of 25 mM Tris, 192 mM
glycine, and 20% analytical grade methanol adjusted to pH 8.3, at a constant current of 300 mA
for 3 hours. Ponceau S staining was performed on the membrane for 5 min, followed by
destaining with distilled water. Blocking was performed with 3% nonfat dry milk in TBS.
Primary blotting was performed with the ANTI-FLAG M2 antibody (Sigma-Aldrich, Lot number
108

SLBJ7864V) at a concentration of 1 µg/mL at room temperature for 30 min. The membrane was
washed with TBS, and then secondary blotting was performed with a goat anti-mouse IgG
antibody-HRP conjugate (Sigma Aldrich) at 1:10,000 dilution with 3% nonfat dry milk.
Chemiluminescence detection was performed with the Pierce ECL substrate (Thermo Fisher).
Generation of transgenic Arabidopsis lines
The AtCHS promoter (defined as 1328 bp of sequence upstream of the CHS transcription
start site) was amplified via PCR from Arabidopsis genomic DNA, digested with HindIII and
XhoI, and ligated into HindIII- and XhoI-digested pCC 1136, a promoterless Gateway cloning
binary vector containing a BAR resistance gene marker, to generate pJKW 0152. The
3×FLAG-AtCHS and SmCHS ORFs were then PCR amplified from previously generated
plasmid constructs and cloned into pCC 1155, an ampicillin-resistant version of the pDONR221
Gateway cloning vector, with BP clonase in the Gateway cloning method (Thermo-Fisher). The
resulting vectors were recombined with pJKW 0152 using LR clonase in the Gateway cloning
method to generate the final binary constructs. Agrobacterium tumefaciens-mediated
transformation of Arabidopsis was performed using the floral dipping method (19).
Basta selection was performed on T1 seedlings. T1 individuals exhibiting purple
hypocotyl coloration were chosen to carry forward for gene expression analysis and seed
collection: 16 for AtCHS, 20 for SmCHS. Among each set of transformants, 2 or 3 individuals
that did not exhibit purple hypocotyl coloration were chosen as control plants for gene
expression analysis.
109

Seed planting and Basta selection was performed for subsequent generations. The
percentage of resistant T2 seedlings was counted for each T1 line, and those with close to 75%
survival were taken forward as single-insertion lines. T3 seeds that had 100% seedling survival
were confirmed as homozygous in the T2 generation.
Quantitative RT-PCR
Total RNA was extracted using a Qiagen RNeasy Plant Mini Kit according to the
manufacturer’s instructions, with on-column DNase treatment. The concentration and purity of
RNA were determined by absorbance at 260/280 nm. First-strand cDNA was synthesized from
1 µg of RNA using SuperScript III Reverse Transcriptase with Oligo dT primers (Thermo
Fisher). Reactions were run on a QuantStudio 6 system (Thermo Fisher) using SYBR Green
Master Mix (Thermo Fisher) and primers listed in Table 1. Gene expression values were
calculated using CT values and normalized using the reference gene At1g13320.
110
Table 1. Primers used in this study.
Name Sequence (5′ to 3′) Purpose
GL0039 TGACTGGAACTCCCTCTTCT AtCHS qRT-PCR forward
GL0049 GCCCTCATCTTCTCTTCCTTTAG AtCHS qRT-PCR reverse
GL0049 CTCTCATCATCGGCTCCAATC SmCHS qRT-PCR forward
GL0050 TCCCAGAATTGCTCCATCAC SmCHS qRT-PCR reverse
GL0053 TTCCATTTTCTCACCGACCAA MBS1 genotyping LP
GL0054 TTCTTCAAGCTTCCCCTGAT MBS1 genotyping RP
JKW0054 GCCTTTTCAGAAATGGATAAATAGCCTTGCTTCC MBS1 genotyping BP (SAIL LB1)
JKW0444 TAACGTGGCCAAAATGATGC At1g13320 genotyping LP and qRT-PCR
JKW0445 GTTCTCCACAACCGCTTGGT At1g13320 genotyping RP and qRT-PCR
111

References
Asada, K. (2006). Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiology, 141(2), 391–396.
Basu, M. K., & Koonin, E. V. (2005). Evolution of Eukaryotic Cysteine Sulfinic Acid Reductase, Sulfiredoxin (Srx), from Bacterial Chromosome Partitioning Protein ParB. Cell Cycle. https://doi.org/10.4161/cc.4.7.1786
Brandes, N., Schmitt, S., & Jakob, U. (2009). Thiol-based redox switches in eukaryotic proteins. Antioxidants & Redox Signaling, 11(5), 997–1014.
Butterfield, D. A., Allan Butterfield, D., Hardas, S. S., & Bader Lange, M. L. (2010). Oxidatively Modified Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) and Alzheimer’s Disease: Many Pathways to Neurodegeneration. Journal of Alzheimer’s Disease. https://doi.org/10.3233/jad-2010-1375
D’Autréaux, B., & Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews. Molecular Cell Biology, 8(10), 813–824.
Denu, J. M., & Tanner, K. G. (1998). Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry, 37(16), 5633–5642.
Fini, A., Brunetti, C., Di Ferdinando, M., Ferrini, F., & Tattini, M. (2011). Stress-induced flavonoid biosynthesis and the antioxidant machinery of plants. Plant Signaling & Behavior. https://doi.org/10.4161/psb.6.5.15069
Gupta, R., & Luan, S. (2003). Redox control of protein tyrosine phosphatases and mitogen-activated protein kinases in plants. Plant Physiology, 132(3), 1149–1152.
Ishii, T., Sunami, O., Nakajima, H., Nishio, H., Takeuchi, T., & Hata, F. (1999). Critical role of sulfenic acid formation of thiols in the inactivation of glyceraldehyde-3-phosphate dehydrogenase by nitric oxide. Biochemical Pharmacology, 58(1), 133–143.
Jez, J. M., Bowman, M. E., Dixon, R. A., & Noel, J. P. (2000). Structure and mechanism of the evolutionarily unique plant enzyme chalcone isomerase. Nature Structural Biology, 7(9), 786–791.
Kettenhofen, N. J., & Wood, M. J. (2010). Formation, reactivity, and detection of protein sulfenic acids. Chemical Research in Toxicology, 23(11), 1633–1646.
Liou, G., Chiang, Y.-C., Wang, Y., & Weng, J.-K. (2018). Mechanistic basis for the evolution of chalcone synthase catalytic cysteine reactivity in land plants. Journal of Biological Chemistry, 293, 18601–18612.
Møller, I. M., Jensen, P. E., & Hansson, A. (2007). Oxidative modifications to cellular components in plants. Annual Review of Plant Biology, 58, 459–481.
Nelson, K. J., Klomsiri, C., Codreanu, S. G., Soito, L., Liebler, D. C., Rogers, L. C., … Poole, L. B. (2010). Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods in Enzymology, 473, 95–115.
Paulsen, C. E., & Carroll, K. S. (2010). Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chemical Biology, 5(1), 47–62.
Peer, W. A. (2001). Flavonoid Accumulation Patterns of Transparent Testa Mutants of Arabidopsis. PLANT PHYSIOLOGY. https://doi.org/10.1104/pp.126.2.536
112

Rhee, S. G., Jeong, W., Chang, T.-S., & Woo, H. A. (2007). Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney International. Supplement, (106), S3–S8.
Schmalhausen, E. V., Nagradova, N. K., Boschi-Muller, S., Branlant, G., & Muronetz, V. I. (1999). Mildly oxidized GAPDH: the coupling of the dehydrogenase and acyl phosphatase activities. FEBS Letters, 452(3), 219–222.
Seo, Y. H., & Carroll, K. S. (2011). Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angewandte Chemie, International Edition, 50(6), 1342–1345.
Shao, N., Duan, G. Y., & Bock, R. (2013). A mediator of singlet oxygen responses in Chlamydomonas reinhardtii and Arabidopsis identified by a luciferase-based genetic screen in algal cells. The Plant Cell, 25(10), 4209–4226.
Shirley, B. W., Kubasek, W. L., Storz, G., Bruggemann, E., Koornneef, M., Ausubel, F. M., & Goodman, H. M. (1995). Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. The Plant Journal: For Cell and Molecular Biology, 8(5), 659–671.
van Montfort, R. L. M., Congreve, M., Tisi, D., Carr, R., & Jhoti, H. (2003). Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature, 423(6941), 773–777.
113

Appendix Investigation of galloylated catechin biosynthetic enzymes in tea Authors Geoffrey Liou1,2 and Jing-Ke Weng1,2
Author Affiliations 1. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 2. Whitehead Institute for Biomedical Research, Cambridge, MA 02142, USA
114

Abstract
Galloylated catechins, such as epigallocatechin gallate (EGCG), are highly abundant
flavan-3-ols found in tea, Camellia sinensis. They function in pathogen defense in the tea plant,
and the health benefits of tea consumption, such as antioxidative and anti-inflammatory effects,
are attributed to EGCG and related flavonoid compounds. The final step of galloylated catechin
biosynthesis, the addition of the galloyl moiety onto unmodified flavan-3-ols, was recently
shown to involve two enzymatic steps: UDP-glucosyl:galloyl-1-O-β-D-glucosyltransferase
(UGGT) and epicatechin:galloyl-1-O-β-D-glucose O-galloyltransferase (ECGT). The gene
encoding the enzyme performing the UGGT step has been identified and biochemically
characterized, but not yet for the ECGT step. In this chapter, we investigated candidate genes
from C. sinensis and developed a heterologous expression system in Nicotiana benthamiana to
test for ECGT activity both in vivo and in vitro.
115

Introduction
Tea is the second most widely consumed beverage worldwide, surpassed only by water
(Brody, 2019). It is prepared from the leaves of two varieties of Camellia sinensis, var. sinensis
and var. assamica. Tea has been consumed for around 4000 years, originating in China, where it
has been valued for its stimulative properties and other health benefits for millennia. Modern
research has begun to shed light on the metabolites in tea that may contribute to these
traditionally attested health effects, such as stress relief and improved memory.
The stimulative effect of tea is well known, and so is the mechanism of caffeine, the
compound in tea that provides this effect. Caffeine, which functions as a toxic deterrent to
insects and herbivores in plants, inhibits the adenosine A2A receptor in humans (Huang et al.,
2005). Adenosine promotes sleep in the sleep-wake cycle, so caffeine promotes wakefulness by
acting as an antagonist. Consumption of caffeinated beverages has also been associated with
reduced risk of Parkinson’s disease (Pluskal & Weng, 2018). Some of the beneficial effects of
tea may result from the complex mixture of natural products found in the plant and the prepared
beverage. Theanine, an amino acid, is highly abundant in tea and contributes to its umami flavor.
It has been shown to improve memory and reaction time in combination with caffeine, more so
than either theanine or caffeine alone (Haskell, Kennedy, Milne, Wesnes, & Scholey, 2008).
Flavanols are an extremely abundant class of polyphenols in tea, making up 25% of the
dry weight of tea leaves (Balentine, Wiseman, & Bouwens, 1997). The majority of these
flavanols are catechins, a subclass of flavan-3-ols that have di- or trihydroxy substitution on the
B ring and dihydroxy substitution on the A ring. Tea contains mostly (−)-epicatechin (EC),
(−)-epicatechin gallate (ECG), (−)-epigallocatechin (EGC), and (−)-epigallocatechin gallate
116

(EGCG). Catechins, particularly EGCG, have been of interest for many potential health benefits
in humans, such as antioxidant, anticancer, and anti-inflammatory properties (Cabrera, Artacho,
& Giménez, 2006).
The biosynthetic pathways of the non-galloylated catechins epicatechin and
epigallocatechin are well understood. The flavan-3,4-diols leucocyanidin and leucodelphinidin
are catalyzed by anthocyanidin synthase (ANS) to form cyanidin and delphinidin, respectively.
These are subsequently reduced by anthocyanidin reductase to form EC and EGC, respectively
(Punyasiri et al., 2004). The enzymatic activities involved in galloylation of EC and EGC
remained unsolved for a long time, until they were recently elucidated by activity-guided
fractionation of C. sinensis crude protein extract (Liu et al., 2012). First, a UDP-glucosyl:galloyl-
1-O-β-D-glucosyltransferase (UGGT) step uses gallic acid and UDP-glucose to form
β-glucogallin. This compound serves as an activated intermediate for transfer of the galloylation
of a variety of phenolic compounds, such as gallotannins in Quercus robur (pedunculate oak)
(Mittasch, Böttcher, Frolova, Bönn, & Milkowski, 2014). The second enzymatic step,
epicatechin:galloyl-1-O-β-D-glucose O-galloyltransferase (ECGT), uses β-glucogallin and EC or
EGC to form their respective galloylated versions, ECG and EGCG.
The UGGT gene was later identified as CsUGT84A22 (Cui et al., 2016), but the ECGT
gene has yet to be completely characterized. The original study identifying the two enzymatic
activities from tea protein extract identified several properties of the ECGT enzyme that
suggested that it belongs to the serine carboxypeptidase-like (SCPL) family (Liu et al., 2012).
SDS-PAGE analysis of ECGT activity-containing fractions indicated that the basic ECGT is a 58
or 60 kDa heterodimeric protein consisting of a 34 or 36 kDa and a 28 kDa subunit. These
117

subunits, or larger assemblies of the 58 or kDa units, may be held together by disulfide bonds,
because β-mercaptoethanol inhibited ECGT activity. PMSF, an inhibitor of serine proteases, also
inhibited ECGT activity, suggesting that it shares a similar reaction mechanism. The uncertain
mass of the protein came from possible post-translational modifications; SCPL enzymes are
often glycosylated. Proteomic mass spectrometry analysis also identified a peptide sequence
from the ECGT sample that matched to a protein that the researchers called SCPL1199,
translated from the C. sinensis genome. A different group later cloned a gene they called
CsSCPL and showed that it is highly expressed in leaf buds and young leaves; its expression
increases in response to heat stress and decreases in response to cold, salt, or drought stress; and
galloylated catechin content correlated with CsSCPL expression (Chiu, Chen, Tzen, & Yang,
2016). This gene had the same sequence as a candidate gene that we had identified from a C.
sinensis EST database.
SCPL enzymes are a relatively recently identified class of acyltransferases. As their name
implies, they share sequence homology to serine carboxypeptidases, and they share the same
catalytic triad of serine, histidine, and aspartic acid residues (Milkowski & Strack, 2004). The
first SCPL to be cloned and characterized was sinapoylglucose:malate sinapoyltransferase from
Arabidopsis thaliana (AtSMT) (Lehfeldt et al., 2000). AtSMT is the gene mutated in Arabidopsis
sng1 (sinapoylglucose accumulator 1) mutants, which cannot synthesize sinapoylmalate, a major
phenylpropanoid. Other SCPL enzymes have since been identified in Arabidopsis and other plant
species, such as sinapoylglucose:choline sinapoyltransferase (SCT) in A. thaliana, Brassica
napus, and Avena strigosa (lopsided oat) (Milkowski, Baumert, Schmidt, Nehlin, & Strack,
2004; Mugford et al., 2009; Shirley, McMichael, & Chapple, 2001);
118

sinapoylglucose:anthocyanin acyltransferase in A. thaliana (Fraser et al., 2007); and enzymes
that form acyl sugars in Lycopersicon pennellii (Li & Steffens, 2000).
SCPL enzymes often undergo various post-translational modifications. There are
N-glycosylation sites found in the amino acid sequence of AtSMT that may explain the
differences between the observed mass in several studies and the predicted mass from the
sequence alone (Ciarkowska, Ostrowski, Starzyńska, & Jakubowska, 2019). SCPL enzymes also
possess an N-terminal signal sequence for translocation to the endoplasmic reticulum during
translation, eventually directing them to the vacuole. AtSCT was also observed as a heterodimer
formed by proteolytic cleavage of an internal loop; many SCPL sequences have this loop that
must be removed for the enzyme to exhibit proper activity (Ciarkowska et al., 2019). Homology
modeling also suggested that AtSMT, BnSCT, and AtSCT require disulfide bonds to hold the
subunits of the heterodimer together (Stehle, Brandt, Milkowski, & Strack, 2006).
The genomes and transcriptomes of C. sinensis var. assamica and var. sinensis were
sequenced recently (Wei et al., 2018; Xia et al., 2017). The C. sinensis var. sinensis genome
paper also investigated galloylated catechin biosynthesis, showing that tea has 22 SCPL genes
and comparative expression analysis of these genes across various tissues. To identify and
characterize the SCPL from C. sinensis responsible for galloylation of EC and EGC, we took a
candidate gene approach using these new genomic resources. We searched the C. sinensis var.
sinensis young leaf transcriptome using the peptide sequence provided by Liu et al. 2012. We
then attempted to express these candidate genes in the heterologous hosts E. coli, Saccharomyces
cerevisiae, and Nicotiana benthamiana.
119

Results
CsUGGT expression in Nicotiana benthamiana produces β-glucogallin
CsUGGT (CsUGT84A22) was cloned into the pEAQ-HT vector, and the resulting
plasmid was used to transform Agrobacterium tumefaciens strain LBA4404. Transient protein
expression was performed in Nicotiana benthamiana leaves by Agrobacterium-mediated
transformation. Gallic acid and/or β-glucogallin substrate was also co-infiltrated with
Agrobacterium. Protein (mGFP) and no-substrate controls were also performed. Expression of
CsUGGT with gallic acid infiltration, either with or without UDP-glucose infiltration, led to high
amounts of β-glucogallin production (Figure 1). A small amount of β-glucogallin (not visible at
the scale of Figure 1) was also detected when CsUGGT was expressed and no substrate was
co-infiltrated, suggesting that there may be a small amount of gallic acid present natively in
Nicotiana benthamiana.
Other peaks were also detected in the same mass window as β-glucogallin but at different
retention times when mGFP was expressed and gallic acid was infiltrated, either with or without
UDP-glucose infiltration (Figure 1). This suggests that N. benthamiana possesses a UGT enzyme
that can use gallic acid and UDP-glucose to produce an isomer of β-glucogallin.
Identification of ECGT candidate genes
The peptide sequence published in Liu et al. 2012 was used as the query in a tblastn
search of a Camellia sinensis EST database. Blastp searches of the Arabidopsis thaliana and
Vitis vinifera proteomes were also performed. A neighbor-joining tree was generated for the
combined
120
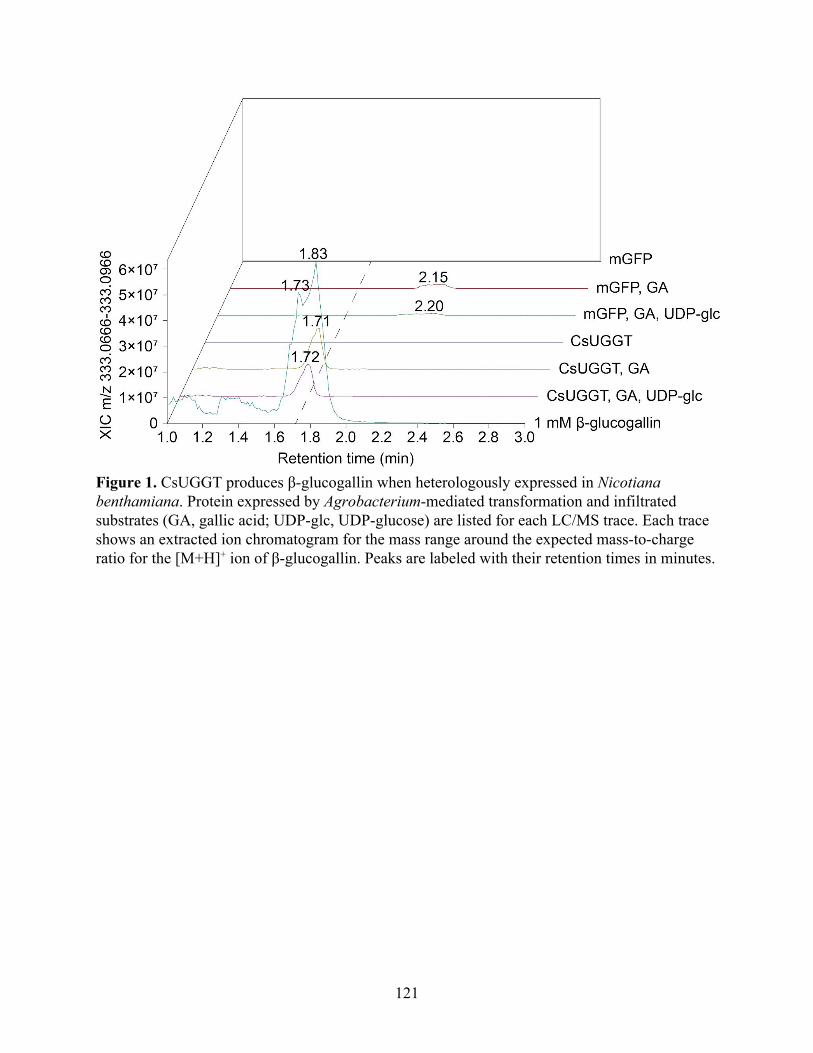
Figure 1. CsUGGT produces β-glucogallin when heterologously expressed in Nicotiana benthamiana. Protein expressed by Agrobacterium-mediated transformation and infiltrated substrates (GA, gallic acid; UDP-glc, UDP-glucose) are listed for each LC/MS trace. Each trace shows an extracted ion chromatogram for the mass range around the expected mass-to-charge ratio for the [M+H]+ ion of β-glucogallin. Peaks are labeled with their retention times in minutes.
121

list of top hits from each search. Three closely clustered C. sinensis sequences were designated
CsECGT1, CsECGT2, and CsECGT3. Primers were designed to amplify these genes from C.
sinensis cDNA. These were the first candidate ECGT genes investigated.
Additional candidate genes were identified later after the publication of the Camellia
sinensis var. sinensis genome and transcriptome. This transcriptome was queried with the same
peptide sequence. Out of the resulting hits, 10 of the most highly and differentially expressed
genes, as listed in the supplemental data of Wei et al. 2018, were selected for further analysis.
Sequence alignment of these 10 sequences showed that they clustered into 7 groups with
identical sequences at the extreme 5′ and 3′ ends (the first and last 15 nucleotides), so 7 pairs of
primers were designed to amplify these genes from C. sinensis leaf cDNA. Only two of the most
highly expressed genes were successfully cloned; they were named CsSCPL1 and CsSCPL3.
The RNA and cDNA samples were prepared from relatively old leaf tissue samples, so the
expression of these SCPL candidate genes was likely lower than they would have been in young
leaves, making it difficult to amplify the more lowly expressed genes for cloning.
Nicotiana benthamiana leaf protein extraction fails to show ECGT activity
Transient overexpression of CsECGT1 was attempted in N. benthamiana leaves along
with co-infiltration of β-glucogallin and EGC substrates, but EGCG was not detected in the leaf
metabolite extracts. We suspected that even if EGCG product formation had occurred, enzymes
in N. benthamiana may be consuming the product and rendering it undetectable. To test this
hypothesis and to search for any potential downstream metabolites by untargeted metabolomics,
we infiltrated ECG and EGCG into N. benthamiana leaves with or without co-infiltration of
122

mGFP or CsUGGT enzymes. Leaf samples were collected for metabolite extraction 1 day post
infiltration. ECG and EGCG were not detected, and there were no obvious flavonoid-derived
compounds enriched in the samples. These results suggested that metabolic extraction of leaves
overexpressing CsECGT would not be the best way to detect EGCG formation.
We then tried crude protein extraction from leaves overexpressing CsECGT, and then
using the protein extract for in vitro enzyme assays to measure ECG or EGCG formation from
β-glucogallin and EC or EGC. CsSCPL1 and CsSCPL3 were also tested, and mGFP and
CsUGGT were used as negative controls. Leaf tissue was frozen and ground in a mortar and
pestle, then extracted with a pH 6 sodium citrate buffer, without protease inhibitors for these
initial experiments. A boiled protein control was also performed. Two distinct peaks on the
EGCG SRM trace, with different retention times from the EGCG standard, were detected for
boiled, but not unboiled, N. benthamiana leaf protein extract samples, regardless of which
enzyme was expressed (Figure 2). This was a very unexpected result; it suggests both that there
is enzymatic activity in N. benthamiana that can use β-glucogallin and EGC to make a
compound with the same mass and similar fragmentation pattern as EGCG, and that the activity
requires activation by boiling of the protein extract. No activity was detected in any samples
when EC was used as the substrate.
123
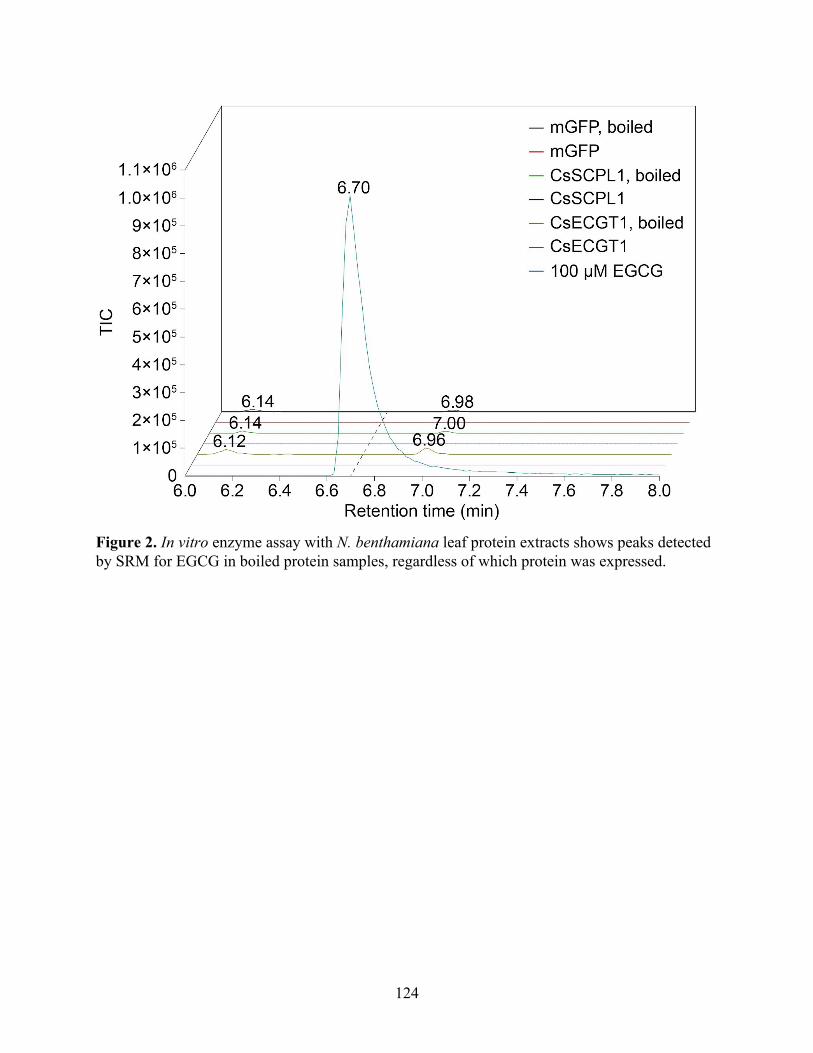
Figure 2. In vitro enzyme assay with N. benthamiana leaf protein extracts shows peaks detected by SRM for EGCG in boiled protein samples, regardless of which protein was expressed.
124

Discussion and Future Directions
To identify the gene responsible for catechin galloylation in tea, we found candidates for
both the UGGT and ECGT enzymatic steps and attempted to heterologously express them. We
were able to reconstitute UGGT activity in Nicotiana benthamiana. We thus far have failed to
observe ECGT activity when any of the candidate genes were expressed in N. benthamiana or S.
cerevisiae (data not shown), and attempts at CsECGT1 expression in E. coli also failed to
produce detectable amounts of protein. The various post-translational modifications required for
SCPL enzymes is the likely cause of these failures.
Our protein extraction methods from N. benthamiana and S. cerevisiae may also have
been insufficient to detect activity even if the enzyme was being expressed and modified
properly. We used mostly simple whole-cell lysis techniques and use the protein lysate with
minimal purification steps. It may be helpful to use a previously studied SCPL enzyme, such as
AtSMT or AtSCT, as a positive control and follow the published expression and purification
methods. Some groups have optimized expression in S. cerevisiae by changing the N-terminal
signal sequence to that of a yeast protein, for example (Stehle, Stubbs, Strack, & Milkowski,
2008). All published heterologous expression methods also seem to rely on activity-guided
fractionation of the protein lysate (Mugford & Milkowski, 2012).
Because of the high sequence similarity between the ECGT and SCPL candidates we
have investigated, it is possible that they have overlapping function in planta. Thus, to show both
necessity and sufficiency of these genes in catechin galloylation, a combinatorial approach to
heterologous expression may be optimal. Others in our group have attempted this strategy, which
involves expressing all candidate genes in N. benthamiana by infiltrating a mixture of
125

Agrobacterium strains that each harbor one gene. Formation of the target compound is detected
in N. benthamiana plants infiltrated with the mixture, the experiment can be repeated with one
strain can be dropped out at a time to identify whether any single strain is necessary for activity.
To clone additional SCPL candidates that have not yet been successfully cloned, new cDNA can
be prepared from young leaf samples of C. sinensis.
126

Materials and Methods
RNA extraction and cDNA template preparation
Total RNA was extracted from C. sinensis var. sinensis adult leaf tissue using the RNeasy
Plant Mini Kit (QIAGEN). First-strand cDNAs were synthesized by RT-PCR from the total
RNA samples as templates using the SuperScript III First-Strand Synthesis System with the
oligo(dT)20 primer (Thermo Fisher Scientific).
Cloning of candidate genes from cDNA
Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific) was used for PCR
amplifications from C. sinensis var. sinensis cDNA. Gibson assembly was used to clone the
amplified genes into target vectors. Restriction enzymes and Gibson assembly reagents were
purchased from New England Biolabs. Oligonucleotide primers were purchased from Integrated
DNA Technologies. All primers used for cloning are listed in Table 1.
Transcriptome assembly
RNA-seq datasets for Camellia sinensis var. assamica stem and young leaf (NCBI SRA
accessions SRR5421032 and SRR5421035) were de novo assembled into a transcriptome using
Trinity (Grabherr et al., 2011). Camellia sinensis var. assamica and var. sinensis genomes were
also downloaded from their respective genome project websites (Wei et al., 2018; Xia et al.,
2017). Transcriptome and genome mining were performed on a local BLAST server (Priyam et
al., 2015).
127

Sequence alignment and phylogenetic analysis
Sequence alignment and phylogenetic analyses. Sequence alignments were performed
using the MUSCLE algorithm (Edgar, 2004) in MEGA7 (Kumar, Stecher, & Tamura, 2016).
Evolutionary histories were inferred by using the Maximum Likelihood method based on the
JTT matrix-based model (Jones, Taylor, & Thornton, 1992). Bootstrap values were calculated
using 1,000 replicates. All phylogenetic analyses were conducted in MEGA7 (Kumar et al.,
2016).
Transient expression in Nicotiana benthamiana
Candidate genes were cloned into the pEAQ-HT vector (Peyret & Lomonossoff, 2013;
Sainsbury, Thuenemann, & Lomonossoff, 2009) and transformed into the ElectroMAX
Agrobacterium tumefaciens strain LBA4404 (Invitrogen). Bacteria were cultivated at 30 °C to
OD600 of 1.5 in 50 mL of YM medium (0.4 g/L yeast extract, 10 g/L mannitol, 0.1 g/L NaCl, 0.2
g/L MgSO4·7H2O, 0.5 g/L K2HPO4·3H2O), washed with 0.5× PBS (68 mM NaCl, 1.4 mM KCl,
5 mM Na2HPO4, 0.9 mM KH2PO4), and resuspended in 0.5× PBS to OD600 of 0.8.
Approximately 1 mL of the final culture was used to infiltrate the underside of 5-6 week old N.
benthamiana leaves. Leaves were harvested 3 days post infiltration for protein extraction, or 5
days post infiltration for metabolite extraction.
Plant metabolite extraction
Approximately 100 mg of plant leaf tissue was dissected, transferred into grinding tubes
containing approximately 15 zirconia/silica disruption beads (2 mm diameter; Research Products
128

International), and snap-frozen in liquid nitrogen. The frozen samples were homogenized twice
on a TissueLyser II (QIAGEN). Metabolites were extracted using 5 to 10 volumes (w/v) of 50%
methanol at 50 °C for 1 hour. Extracts were centrifuged twice (13,000 g, 20 min) and
supernatants were collected for LC−MS analysis.
Plant protein extraction
Whole leaves (550 to 750 mg wet weight) were collected from N. benthamiana and
frozen in liquid nitrogen. Leaves were pulverized to a fine powder with a mortar and pestle, and
10× w/v 100 mM sodium citrate pH 6 buffer (40.9 mM sodium citrate dihydrate, 59.03 mM
citric acid, 0.1% Tween 20) was added and allowed to incubate at room temperature for 10 to 20
min. Samples were centrifuged in a JA-20 rotor at 19,000 rpm for 40 min at 4 °C. The
supernatant was transferred to a 30,000 Da MWCO concentrator tube and concentrated to about
50% of the starting volume. Finally, the concentrated sample was centrifuged at 13,000 g for 10
min, and the supernatant was stored at 4 °C for later use in enzyme assays.
In vitro ECGT enzyme activity assay
40 µL of N. benthamiana protein extract was added to a 210 µL reaction mix consisting
of 50 mM sodium phosphate buffer pH 6, 1 mM β-glucogallin, and 0.4 mM EC or EGC. As a
negative control, the protein extract was heated at 95 °C for 15 min and then centrifuged at
13,000 g for 3 min before being added to the reaction. The reaction was stopped after
approximately 18 hours by addition of 250 µL methanol, and the reaction mix was centrifuged at
129

13,000 g for 20 min. The supernatant was transferred to new tubes and taken for LC−MS
analysis.
LC−MS analysis
LC was conducted on a Dionex UltiMate 3000 UHPLC system (Thermo Fisher
Scientific), using water with 0.1% formic acid as solvent A and acetonitrile with 0.1%formic
acid as solvent B. Reverse phase separation of analytes was performed on a Kinetex C18
column, 150 × 3 mm, 2.6 μm particle size (Phenomenex). The column oven was held at 30 °C.
Injections were eluted with 5% B for 2 min, a gradient of 5–36.3% B for 8 min, 95% B for 3
min, and 5% B for 2 min, with a flow rate of 0.7 mL/min. MS analyses for the in vitro enzyme
assays were performed on a TSQ Quantum Access Max mass spectrometer (Thermo Fisher
Scientific) operated in positive ionization mode with single reaction monitoring for
β-glucogallin, EGC, and EGCG. MS analyses for plant metabolic extracts were performed on a
high-resolution Q-Exactive benchtop Orbitrap mass spectrometer (Thermo Fisher Scientific)
operated in positive ionization mode with full scan range of 100−1250 m/z and top 5
data-dependent MS/MS scans. Raw LC−MS data were analyzed using XCalibur (Thermo Fisher
Scientific).
130
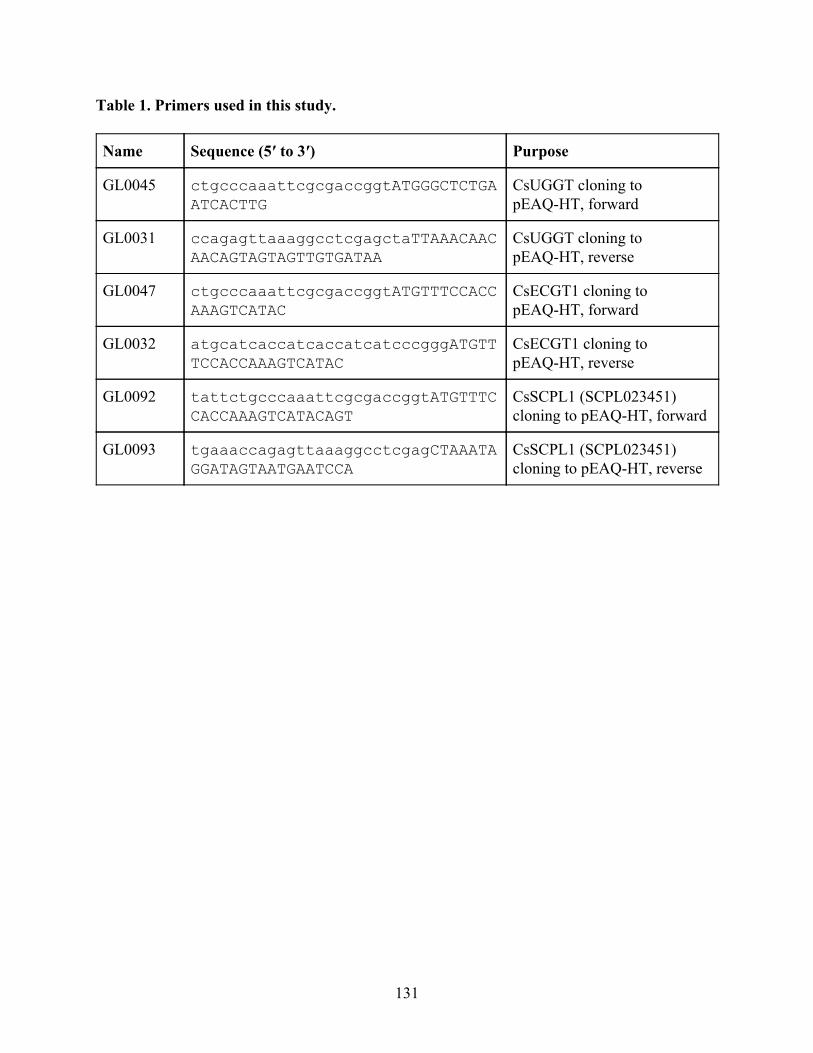
Table 1. Primers used in this study.
Name Sequence (5′ to 3′) Purpose
GL0045 ctgcccaaattcgcgaccggtATGGGCTCTGA
ATCACTTG
CsUGGT cloning to pEAQ-HT, forward
GL0031 ccagagttaaaggcctcgagctaTTAAACAAC
AACAGTAGTAGTTGTGATAA
CsUGGT cloning to pEAQ-HT, reverse
GL0047 ctgcccaaattcgcgaccggtATGTTTCCACC
AAAGTCATAC
CsECGT1 cloning to pEAQ-HT, forward
GL0032 atgcatcaccatcaccatcatcccgggATGTT
TCCACCAAAGTCATAC
CsECGT1 cloning to pEAQ-HT, reverse
GL0092 tattctgcccaaattcgcgaccggtATGTTTC
CACCAAAGTCATACAGT
CsSCPL1 (SCPL023451) cloning to pEAQ-HT, forward
GL0093 tgaaaccagagttaaaggcctcgagCTAAATA
GGATAGTAATGAATCCA
CsSCPL1 (SCPL023451) cloning to pEAQ-HT, reverse
131

References
Balentine, D. A., Wiseman, S. A., & Bouwens, L. C. (1997). The chemistry of tea flavonoids. Critical Reviews in Food Science and Nutrition, 37(8), 693–704.
Brody, H. (2019). Tea. Nature, 566(7742), S1. Cabrera, C., Artacho, R., & Giménez, R. (2006). Beneficial Effects of Green Tea—A Review.
Journal of the American College of Nutrition, 25(2), 79–99. Chiu, C.-H., Chen, G.-H., Tzen, J. T. C., & Yang, C.-Y. (2016). Molecular identification and
characterization of a serine carboxypeptidase-like gene associated with abiotic stress in tea plant, Camellia sinensis (L.). Plant Growth Regulation. https://doi.org/10.1007/s10725-015-0138-7
Ciarkowska, A., Ostrowski, M., Starzyńska, E., & Jakubowska, A. (2019). Plant SCPL acyltransferases: multiplicity of enzymes with various functions in secondary metabolism. Phytochemistry Reviews, 18(1), 303–316.
Cui, L., Yao, S., Dai, X., Yin, Q., Liu, Y., Jiang, X., … Xia, T. (2016). Identification of UDP-glycosyltransferases involved in the biosynthesis of astringent taste compounds in tea (Camellia sinensis). Journal of Experimental Botany, 67(8), 2285–2297.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797.
Fraser, C. M., Thompson, M. G., Shirley, A. M., Ralph, J., Schoenherr, J. A., Sinlapadech, T., … Chapple, C. (2007). Related Arabidopsis serine carboxypeptidase-like sinapoylglucose acyltransferases display distinct but overlapping substrate specificities. Plant Physiology, 144(4), 1986–1999.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., … Regev, A. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652.
Haskell, C. F., Kennedy, D. O., Milne, A. L., Wesnes, K. A., & Scholey, A. B. (2008). The effects of L-theanine, caffeine and their combination on cognition and mood. Biological Psychology, 77(2), 113–122.
Huang, Z.-L., Qu, W.-M., Eguchi, N., Chen, J.-F., Schwarzschild, M. A., Fredholm, B. B., … Hayaishi, O. (2005). Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nature Neuroscience, 8(7), 858–859.
Jones, D. T., Taylor, W. R., & Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences: CABIOS, 8(3), 275–282.
Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular Biology and Evolution, 33(7), 1870–1874.
Lehfeldt, C., Shirley, A. M., Meyer, K., Ruegger, M. O., Cusumano, J. C., Viitanen, P. V., … Chapple, C. (2000). Cloning of the SNG1 gene of Arabidopsis reveals a role for a serine carboxypeptidase-like protein as an acyltransferase in secondary metabolism. The Plant Cell, 12(8), 1295–1306.
Li, A. X., & Steffens, J. C. (2000). An acyltransferase catalyzing the formation of diacylglucose
132

is a serine carboxypeptidase-like protein. Proceedings of the National Academy of Sciences of the United States of America, 97(12), 6902–6907.
Liu, Y., Gao, L., Liu, L., Yang, Q., Lu, Z., Nie, Z., … Xia, T. (2012). Purification and characterization of a novel galloyltransferase involved in catechin galloylation in the tea plant (Camellia sinensis). The Journal of Biological Chemistry, 287(53), 44406–44417.
Milkowski, C., Baumert, A., Schmidt, D., Nehlin, L., & Strack, D. (2004). Molecular regulation of sinapate ester metabolism inBrassica napus: expression of genes, properties of the encoded proteins and correlation of enzyme activities with metabolite accumulation. The Plant Journal. https://doi.org/10.1111/j.1365-313x.2004.02036.x
Milkowski, C., & Strack, D. (2004). Serine carboxypeptidase-like acyltransferases. Phytochemistry, 65(5), 517–524.
Mittasch, J., Böttcher, C., Frolova, N., Bönn, M., & Milkowski, C. (2014). Identification of UGT84A13 as a candidate enzyme for the first committed step of gallotannin biosynthesis in pedunculate oak (Quercus robur). Phytochemistry, 99, 44–51.
Mugford, S. T., & Milkowski, C. (2012). Serine carboxypeptidase-like acyltransferases from plants. Methods in Enzymology, 516, 279–297.
Mugford, S. T., Qi, X., Bakht, S., Hill, L., Wegel, E., Hughes, R. K., … Osbourn, A. (2009). A serine carboxypeptidase-like acyltransferase is required for synthesis of antimicrobial compounds and disease resistance in oats. The Plant Cell, 21(8), 2473–2484.
Peyret, H., & Lomonossoff, G. P. (2013). The pEAQ vector series: the easy and quick way to produce recombinant proteins in plants. Plant Molecular Biology, 83(1-2), 51–58.
Pluskal, T., & Weng, J.-K. (2018). Natural product modulators of human sensations and mood: molecular mechanisms and therapeutic potential. Chemical Society Reviews, 47(5), 1592–1637.
Priyam, A., Woodcroft, B. J., Rai, V., Munagala, A., Moghul, I., Ter, F., … Wurm, Y. (2015). Sequenceserver: a modern graphical user interface for custom BLAST databases. bioRxiv. https://doi.org/10.1101/033142
Punyasiri, P. A. N., Abeysinghe, I. S. B., Kumar, V., Treutter, D., Duy, D., Gosch, C., … Fischer, T. C. (2004). Flavonoid biosynthesis in the tea plant Camellia sinensis: properties of enzymes of the prominent epicatechin and catechin pathways. Archives of Biochemistry and Biophysics, 431(1), 22–30.
Sainsbury, F., Thuenemann, E. C., & Lomonossoff, G. P. (2009). pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnology Journal, 7(7), 682–693.
Shirley, A. M., McMichael, C. M., & Chapple, C. (2001). The sng2 mutant of Arabidopsis is defective in the gene encoding the serine carboxypeptidase-like protein sinapoylglucose:choline sinapoyltransferase. The Plant Journal. https://doi.org/10.1046/j.1365-313x.2001.01123.x
Stehle, F., Brandt, W., Milkowski, C., & Strack, D. (2006). Structure determinants and substrate recognition of serine carboxypeptidase-like acyltransferases from plant secondary metabolism. FEBS Letters. https://doi.org/10.1016/j.febslet.2006.10.046
Stehle, F., Stubbs, M. T., Strack, D., & Milkowski, C. (2008). Heterologous expression of a serine carboxypeptidase-like acyltransferase and characterization of the kinetic mechanism. FEBS Journal. https://doi.org/10.1111/j.1742-4658.2007.06244.x
Wei, C., Yang, H., Wang, S., Zhao, J., Liu, C., Gao, L., … Wan, X. (2018). Draft genome
133

sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proceedings of the National Academy of Sciences of the United States of America, 115(18), E4151–E4158.
Xia, E.-H., Zhang, H.-B., Sheng, J., Li, K., Zhang, Q.-J., Kim, C., … Gao, L.-Z. (2017). The Tea Tree Genome Provides Insights into Tea Flavor and Independent Evolution of Caffeine Biosynthesis. Molecular Plant, 10(6), 866–877.
134