enhanced electrokinetic treatment of marine sediments contaminated by heavy metals and pahs
TRANSCRIPT

Chemosphere 81 (2010) 46–56
Contents lists available at ScienceDirect
Chemosphere
journal homepage: www.elsevier .com/locate /chemosphere
Enhanced electrokinetic treatment of marine sediments contaminatedby heavy metals and PAHs
Antonio Colacicco a, Giorgia De Gioannis b, Aldo Muntoni b, Emmanuela Pettinao b, Alessandra Polettini a,*,Raffaella Pomi a
a University of Rome ‘‘La Sapienza”, Department of Hydraulics, Transportation and Roads, Via Eudossiana 18, 00184 Rome, Italyb University of Cagliari, Department of Geoengineering and Environmental Technologies, Piazza d’Armi, 09123 Cagliari, Italy
a r t i c l e i n f o
Article history:Received 8 April 2010Received in revised form 7 July 2010Accepted 8 July 2010Available online 5 August 2010
Keywords:Chemically aided electrokineticsDredged sedimentsHeavy metalsPAHs
0045-6535/$ - see front matter � 2010 Elsevier Ltd. Adoi:10.1016/j.chemosphere.2010.07.004
* Corresponding author. Tel./fax: +39 06 44 585 03E-mail address: [email protected] (
a b s t r a c t
Dredged sediments contaminated by heavy metals and PAHs were subjected to both unenhanced andenhanced electrokinetic remediation under different operating conditions, obtained by varying theapplied voltage and the type of conditioning agent used at the electrode compartments in individualexperiments. While metals were not appreciably mobilized as a result of the unenhanced process, metalremoval was found to be significantly improved when both the anodic and cathodic reservoirs were con-ditioned with the chelating agent EDTA, with removal yields ranging from 28% to 84% depending on thecontaminant concerned. As for the effect on organic contaminants, under the conditions tested the elec-trokinetic treatment displayed a poor removal capacity towards PAHs, even when a surfactant (Tween80) was used to promote contaminant mobilization, indicating the need for further investigation on thisissue. Further research on organics removal from this type of materials through electrokinetic remedia-tion is thus required. Furthermore, a number of technical and environmental issues will also require acareful evaluation with a view to full-scale implementation of electrokinetic sediment remediation. Theseinclude controlling side effects during the treatment (such as anodic precipitation, oxidation of the con-ditioning agent, and evolution of toxic gases), as well as evaluating the potential ecotoxicological effectsof the chemical agents used.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Since surface waters receive discharges of various liquid and so-lid wastes often containing hazardous compounds, these may sorbonto sediment particles, which thus become a reservoir of contam-inants having the potential of re-dissolving or migrating into thewater column. The treatment processes for contaminated sedi-ments have mainly been developed from soil remediation tech-niques, however in most cases their efficacy can be impaired byspecific sediment properties such as the high water, salt and organ-ic contents as well as the presence of significant amounts of fineparticles. In addition, the need for combined application of differ-ent treatment processes to deal with different contaminant types,and the huge volumes of materials with relatively low pollutantconcentrations, imply higher treatment costs compared to thoseexperienced for soils. Up to now, the above mentioned reasonshave prevented sediment treatment from being extensively prac-ticed, therefore very few proved sediment cleanup cases and de-fined performance standards are currently available.
ll rights reserved.
7.A. Polettini).
Among the available treatment processes, electrokinetic remedi-ation deserves particular attention due to its peculiar advantages,including the capability of treating fine and low-permeability mate-rials, and achieving consolidation, dewatering and removal of saltsand inorganic contaminants in a single stage. Furthermore, the pro-cess can be applied in situ (and may thus be adopted where decon-tamination is required but dredging is not).
Several studies have investigated the electrokinetic removal ofmetals from soils (mostly spiked model soils). Reddy andChinthamreddy (2004) performed unenhanced electrokineticremediation tests on high acid buffering spiked glacial till soils. Anegligible removal of cationic contaminants due to precipitationphenomena under the high pH conditions was observed; similarresults were also obtained by De Gioannis et al. (2008) in unen-hanced tests on marine sediments. Grundl and Reese (1997) ob-served that the presence of calcite buffers the system preventingpH from shifting to the acidic range, which would be more favour-able to metal detachment from the solid matrix. As observed byseveral authors (see Ottosen et al., 2009 and references therein),to improve metal removal and reduce the remediation time,enhancement methods are recommended, which are aimed atproperly controlling pH by promoting the movement of the acidic

A. Colacicco et al. / Chemosphere 81 (2010) 46–56 47
front, detaching metals from solid particles and favouring theirmigration within the system. Removal efficiencies of up to 80%for the above mentioned glacial till soils spiked with Cr(VI), Niand Cd were achieved by Reddy and Chinthamreddy (2004) whenpurging solutions were adopted; the highest removal of Ni and Cdwas obtained using 1.0 M acetic acid at a 1.0 V cm�1 voltage gradi-ent; using 0.1 M NaCl as the anolyte and 0.1 M EDTA as the catho-lyte resulted in significant contaminant migration towards the soilregions adjacent to the electrodes, although the net removal waspoor. In other experiments on removal of the same target metalsfrom spiked kaolin, Reddy et al. (2004) indicated the lower capabil-ity of metal removal through EDTA-enhanced electrokinetics(where the catholyte was conditioned with EDTA and the anolytewith NaOH) if compared to batch washing tests; this was relatedto the poor electromigration of EDTA towards the anode causedby its relatively large size and low mobility as well as the opposingeffect of the electroosmotic flow and the pH-dependant speciationof EDTA complexes. To this regard, Yeung and Hsu (2005) provedthat, when the electroosmotic flow direction was reversed uponthe addition of a high pH buffer solution, the removal of Cd fromspiked kaolinite through EDTA-assisted electrokinetics was signif-icantly improved. Chung and Kang (1999) applied an electrokinetictreatment to a marine clay spiked with Pb using nitric acid, EDTAor acetic acid to enhance the process yield; the removal yieldwas higher than 88% (up to 94% using acetic acid).
Electrokinetics has also been applied to remove organic contam-inants (including PAHs, chlorinated organic compounds, pesticides,herbicides) from soils. With specific reference to PAHs, a number ofhybrid technologies which involve coupling electrokinetics withultrasonic and Fenton processes have been investigated. Ultra-sound-coupled electrokinetics has been found to increase the elec-troosmotic flow, the current intensity and the PAH removal rate;Pazos et al. (2010) found that this was increased by 2–9% uponultrasonic enhancement. Electro-Fenton processes are capable ofenhancing oxidant delivery and activation of oxidizing radicalsand can induce oxidative/reductive reactions (Pazos et al., 2010).Alcántara et al. (2008) found 99% PAH destruction in 14 d. Enhanc-ing agents including surfactants, cosolvents, cyclodextrins and bio-surfactants have also been tested for electrokinetic PAH removal,the use of which is widely acknowledged as mandatory to achieveadequate remediation yields (Badr et al., 2004) due to the low aque-ous solubility of non-polar PAHs and their strong adsorption ontosoil and sediment particles. Surfactants are capable of loweringthe interfacial tension and to collect the PAHs by forming micelles;anionic surfactants are generally preferred over the cationic onessince these can sorb onto negatively charged particles; however,anionic surfactants are typically less soluble and have higher criti-cal micelles concentrations (CMC), thus requiring higher additions.Park et al. (2007) reported that, when added to the anodic chamber,sodium dodecyl sulphate (SDS) was dragged by the electroosmoticflow towards the cathode and effectively removed phenanthrene.However, other investigators (Yang et al., 2005) reported a lowPAH removal caused by the surfactant being present mainly inthe anodic region. When SDS was added to the cathodic compart-ment, it was found to electromigrate towards the anode and toproduce high PAH removal yields (Yang and Lee, 2009). The use ofnon-ionic surfactants (mainly Tween 80, Brij 30, APG, IgepalCA-720 and Tergitol (Yang and Lee, 2009)), which have low CMCsand toxicities as well as relatively high solubilities, is consideredto be promising and cost-effective. Among several non-ionic andanionic surfactants, Park et al. (2007) found APG to attain the bestremoval of phenanthrene from kaolinite. Reddy and Ala (2006) usedTween 80 for PAH removal from dredged sediments and showedthat it may not be effective for high organic matter contents, requir-ing either increased surfactant concentrations or pH control to en-sure adequate remediation. Similar results were obtained by
Saichek (2002) using Igepal CA-720; 90% PAH removal efficiencieswere attained with this agent using high voltage gradients (Reddyand Saichek, 2004). The application of non-ionic and readily biode-gradable Tergitol series was shown to yield >95% phenanthreneremoval at high dosages, which decreased to about 25% at lowersurfactant concentrations (Yang and Lee, 2009). Co-solvents suchas lower alcohols, ketones and alkyl amines have also been testedsince they are capable of reducing the net polarity of the solutionas well as enhancing the solubility and decreasing the adsorptionof non-ionic organic molecules. Maturi and Reddy (2008) showedappreciable phenanthrene removal from soil using n-butylamine.Ethanol was reported to be less effective in phenanthrene removalthan Tween 80 (Saichek and Reddy, 2003). The use of cyclodextrinshas also been recently proposed due to their biodegradability, ab-sence of toxicity and poor sorption onto soil particles over a widerange of pH values; however, they are less effective than traditionaladditives, and some are also expensive (Saichek and Reddy, 2005).Hydropropyl b-cyclodextrin (HPCD) at high dosages was shown toyield a 75% removal of phenanthrene from clay (Yang and Lee,2009). Reddy and Ala (2006) compared the removal of PAHs fromlake sediments by 5% Igepal CA-720 surfactant, 20% n-butylamineco-solvent and 10% HPCD, showing that while HPCD was almostineffective, the other two agents were capable of yielding a partialPAH mobilization. Biosurfactants (including such species as fattyacids, phospho- and glyco-lipids, lipopeptides produced by bacteriaor fungi) have also raised some interest for use in electrokineticprocesses, however very few studies have been performed so farand research is still necessary for process optimization (Yang andLee, 2009).
Notwithstanding the studies available on remediation of low-permeability porous media contaminated by PAHs, enhanced elec-trokinetics is considered under-researched and less understood incomparison with metal removal (Yang and Lee, 2009): adequatefield studies are missing and further research is still necessary inorder to improve the understanding of many key factors, includingthe assessment of the influence of enhancing agents on pH, viscos-ity, dielectric constant of the system (Yang and Lee, 2009), and, inturn, on the electroosmotic flow.
It should also be mentioned that most of the studies of electro-kinetic remediation are related to applications to (often spiked)soils, whilst relatively few deal with contaminated field sediments(Doering et al., 2002; Chung and Kamon, 2005; De Gioannis et al.,2009; Li et al., 2009, 2010; Andreottola et al., 2010).
Within this framework, the main objective of the present studyis to assess the effectiveness of enhanced electrokinetics for reme-diation of real dredged sediments contaminated by heavy metalsand PAHs.
2. Materials and methods
2.1. Analytical methods
The sediments were dredged from the Northern canal of PortoMarghera (Venice, Italy). After dredging, the material was homog-enized and stored under controlled conditions. Homogenization in-volved settling and separation of water, air-drying for 72 h andsieving to remove the 4-mm oversize fraction (mainly consistingof wood and shell particles).
The particle size distribution and moisture content were mea-sured according to the ISO 3310-1:2000 (wet sieving) and ASTMD2216-05 methods, respectively. The ISO 10390:2005 procedurewas applied for pH measurement, while a total organic carbon(TOC) analyzer (TOC-VCHS and SSM-5000 module, Shimadzu,Japan) was used to measure the TOC content.
48 A. Colacicco et al. / Chemosphere 81 (2010) 46–56
The elemental composition was determined by energy disper-sive X-ray fluorescence (Spectro X-LAB 2000), while the heavy me-tal content was measured using ICP-OES after acid digestion inaccordance with the US EPA 3050b method.
The PAH content of sediment was determined using the US EPASW 3541, SW 3620 and SW 8270B methods for extraction, purifi-cation and measurement, respectively. PAHs were extracted fromthe aqueous solutions following the US EPA SW 3510 standard,while purification and quantification were accomplished usingthe previously reported US EPA methods.
All the analytical procedures were applied to four replicatesamples.
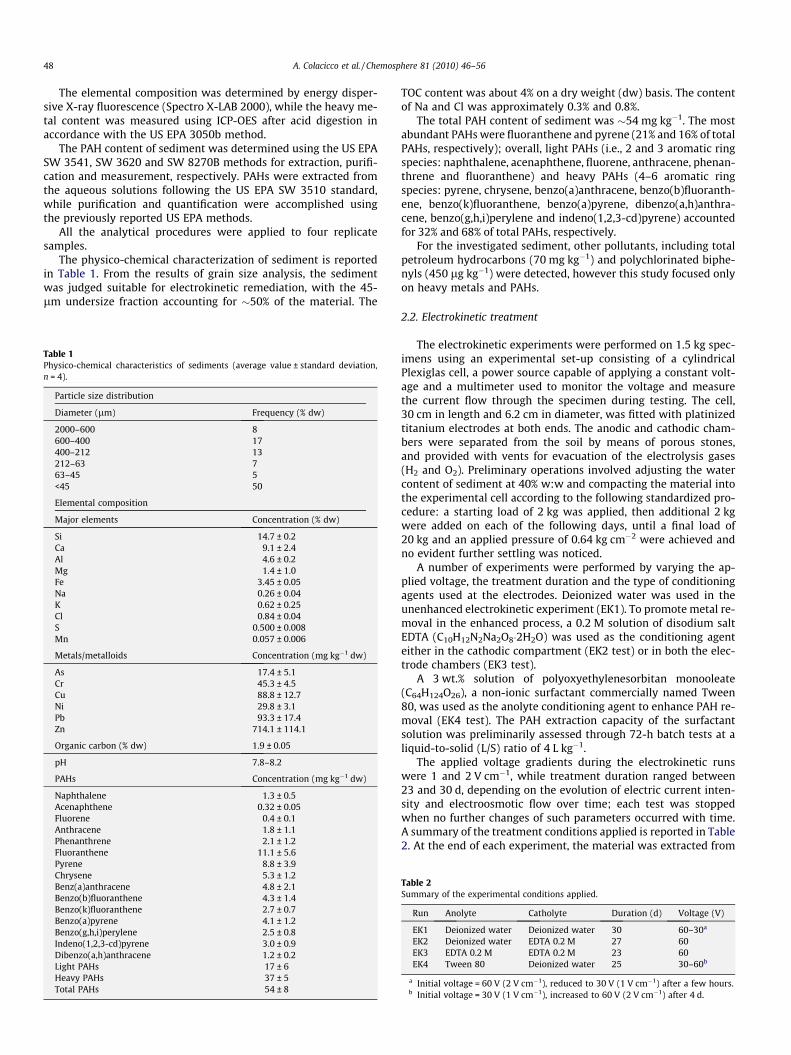
The physico-chemical characterization of sediment is reportedin Table 1. From the results of grain size analysis, the sedimentwas judged suitable for electrokinetic remediation, with the 45-lm undersize fraction accounting for �50% of the material. The
Table 1Physico-chemical characteristics of sediments (average value ± standard deviation,n = 4).
Particle size distribution
Diameter (lm) Frequency (% dw)
2000–600 8600–400 17400–212 13212–63 763–45 5<45 50
Elemental composition
Major elements Concentration (% dw)
Si 14.7 ± 0.2Ca 9.1 ± 2.4Al 4.6 ± 0.2Mg 1.4 ± 1.0Fe 3.45 ± 0.05Na 0.26 ± 0.04K 0.62 ± 0.25Cl 0.84 ± 0.04S 0.500 ± 0.008Mn 0.057 ± 0.006
Metals/metalloids Concentration (mg kg�1 dw)
As 17.4 ± 5.1Cr 45.3 ± 4.5Cu 88.8 ± 12.7Ni 29.8 ± 3.1Pb 93.3 ± 17.4Zn 714.1 ± 114.1
Organic carbon (% dw) 1.9 ± 0.05
pH 7.8–8.2
PAHs Concentration (mg kg�1 dw)
Naphthalene 1.3 ± 0.5Acenaphthene 0.32 ± 0.05Fluorene 0.4 ± 0.1Anthracene 1.8 ± 1.1Phenanthrene 2.1 ± 1.2Fluoranthene 11.1 ± 5.6Pyrene 8.8 ± 3.9Chrysene 5.3 ± 1.2Benz(a)anthracene 4.8 ± 2.1Benzo(b)fluoranthene 4.3 ± 1.4Benzo(k)fluoranthene 2.7 ± 0.7Benzo(a)pyrene 4.1 ± 1.2Benzo(g,h,i)perylene 2.5 ± 0.8Indeno(1,2,3-cd)pyrene 3.0 ± 0.9Dibenzo(a,h)anthracene 1.2 ± 0.2Light PAHs 17 ± 6Heavy PAHs 37 ± 5Total PAHs 54 ± 8
TOC content was about 4% on a dry weight (dw) basis. The contentof Na and Cl was approximately 0.3% and 0.8%.
The total PAH content of sediment was �54 mg kg�1. The mostabundant PAHs were fluoranthene and pyrene (21% and 16% of totalPAHs, respectively); overall, light PAHs (i.e., 2 and 3 aromatic ringspecies: naphthalene, acenaphthene, fluorene, anthracene, phenan-threne and fluoranthene) and heavy PAHs (4–6 aromatic ringspecies: pyrene, chrysene, benzo(a)anthracene, benzo(b)fluoranth-ene, benzo(k)fluoranthene, benzo(a)pyrene, dibenzo(a,h)anthra-cene, benzo(g,h,i)perylene and indeno(1,2,3-cd)pyrene) accountedfor 32% and 68% of total PAHs, respectively.
For the investigated sediment, other pollutants, including totalpetroleum hydrocarbons (70 mg kg�1) and polychlorinated biphe-nyls (450 lg kg�1) were detected, however this study focused onlyon heavy metals and PAHs.
2.2. Electrokinetic treatment
The electrokinetic experiments were performed on 1.5 kg spec-imens using an experimental set-up consisting of a cylindricalPlexiglas cell, a power source capable of applying a constant volt-age and a multimeter used to monitor the voltage and measurethe current flow through the specimen during testing. The cell,30 cm in length and 6.2 cm in diameter, was fitted with platinizedtitanium electrodes at both ends. The anodic and cathodic cham-bers were separated from the soil by means of porous stones,and provided with vents for evacuation of the electrolysis gases(H2 and O2). Preliminary operations involved adjusting the watercontent of sediment at 40% w:w and compacting the material intothe experimental cell according to the following standardized pro-cedure: a starting load of 2 kg was applied, then additional 2 kgwere added on each of the following days, until a final load of20 kg and an applied pressure of 0.64 kg cm�2 were achieved andno evident further settling was noticed.
A number of experiments were performed by varying the ap-plied voltage, the treatment duration and the type of conditioningagents used at the electrodes. Deionized water was used in theunenhanced electrokinetic experiment (EK1). To promote metal re-moval in the enhanced process, a 0.2 M solution of disodium saltEDTA (C10H12N2Na2O8�2H2O) was used as the conditioning agenteither in the cathodic compartment (EK2 test) or in both the elec-trode chambers (EK3 test).
A 3 wt.% solution of polyoxyethylenesorbitan monooleate(C64H124O26), a non-ionic surfactant commercially named Tween80, was used as the anolyte conditioning agent to enhance PAH re-moval (EK4 test). The PAH extraction capacity of the surfactantsolution was preliminarily assessed through 72-h batch tests at aliquid-to-solid (L/S) ratio of 4 L kg�1.
The applied voltage gradients during the electrokinetic runswere 1 and 2 V cm�1, while treatment duration ranged between23 and 30 d, depending on the evolution of electric current inten-sity and electroosmotic flow over time; each test was stoppedwhen no further changes of such parameters occurred with time.A summary of the treatment conditions applied is reported in Table2. At the end of each experiment, the material was extracted from
Table 2Summary of the experimental conditions applied.
Run Anolyte Catholyte Duration (d) Voltage (V)
EK1 Deionized water Deionized water 30 60–30a
EK2 Deionized water EDTA 0.2 M 27 60EK3 EDTA 0.2 M EDTA 0.2 M 23 60EK4 Tween 80 Deionized water 25 30–60b
a Initial voltage = 60 V (2 V cm�1), reduced to 30 V (1 V cm�1) after a few hours.b Initial voltage = 30 V (1 V cm�1), increased to 60 V (2 V cm�1) after 4 d.
1000
1500
2000
2500
rent
inte
nsity
(mA)
EK1EK2EK3EK4
(a)
A. Colacicco et al. / Chemosphere 81 (2010) 46–56 49
the apparatus, cut into five slices (S1–S5, from anode to cathode)and characterized for pH, TOC and concentration of residual con-taminants. The cathodic and anodic solutions were also analyzedfor the same parameters.
The energy consumption (Eu) per unit volume of sediment (vs)was calculated on the basis of the measures of electric currentintensity as a function of time (I(t)) and the applied voltage (V)according to the following equation:
EuðtÞ ¼1v s
Z t
0VIðtÞdt
0
200
400
600
800
1000
1200
0 5 10 15 20 25 30
Elec
trosm
otic
flow
(mL)
EK1EK2EK3EK4
0
500
0 2 4 6 8 10
Cur
(b)
(c)
0
5
10
15
20
25
0 5 10 15 20 25 30Time (days)
TOC
(g L
-1)
EK1EK2EK3EK4
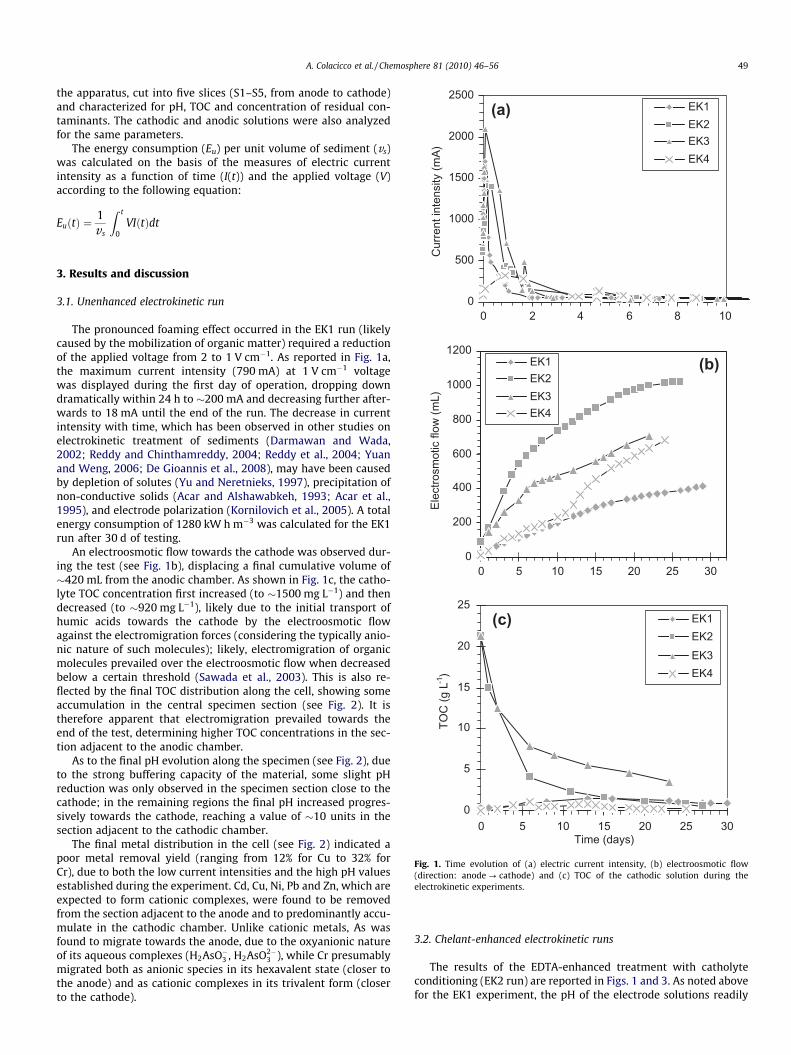
Fig. 1. Time evolution of (a) electric current intensity, (b) electroosmotic flow(direction: anode ? cathode) and (c) TOC of the cathodic solution during theelectrokinetic experiments.
3. Results and discussion
3.1. Unenhanced electrokinetic run
The pronounced foaming effect occurred in the EK1 run (likelycaused by the mobilization of organic matter) required a reductionof the applied voltage from 2 to 1 V cm�1. As reported in Fig. 1a,the maximum current intensity (790 mA) at 1 V cm�1 voltagewas displayed during the first day of operation, dropping downdramatically within 24 h to �200 mA and decreasing further after-wards to 18 mA until the end of the run. The decrease in currentintensity with time, which has been observed in other studies onelectrokinetic treatment of sediments (Darmawan and Wada,2002; Reddy and Chinthamreddy, 2004; Reddy et al., 2004; Yuanand Weng, 2006; De Gioannis et al., 2008), may have been causedby depletion of solutes (Yu and Neretnieks, 1997), precipitation ofnon-conductive solids (Acar and Alshawabkeh, 1993; Acar et al.,1995), and electrode polarization (Kornilovich et al., 2005). A totalenergy consumption of 1280 kW h m�3 was calculated for the EK1run after 30 d of testing.
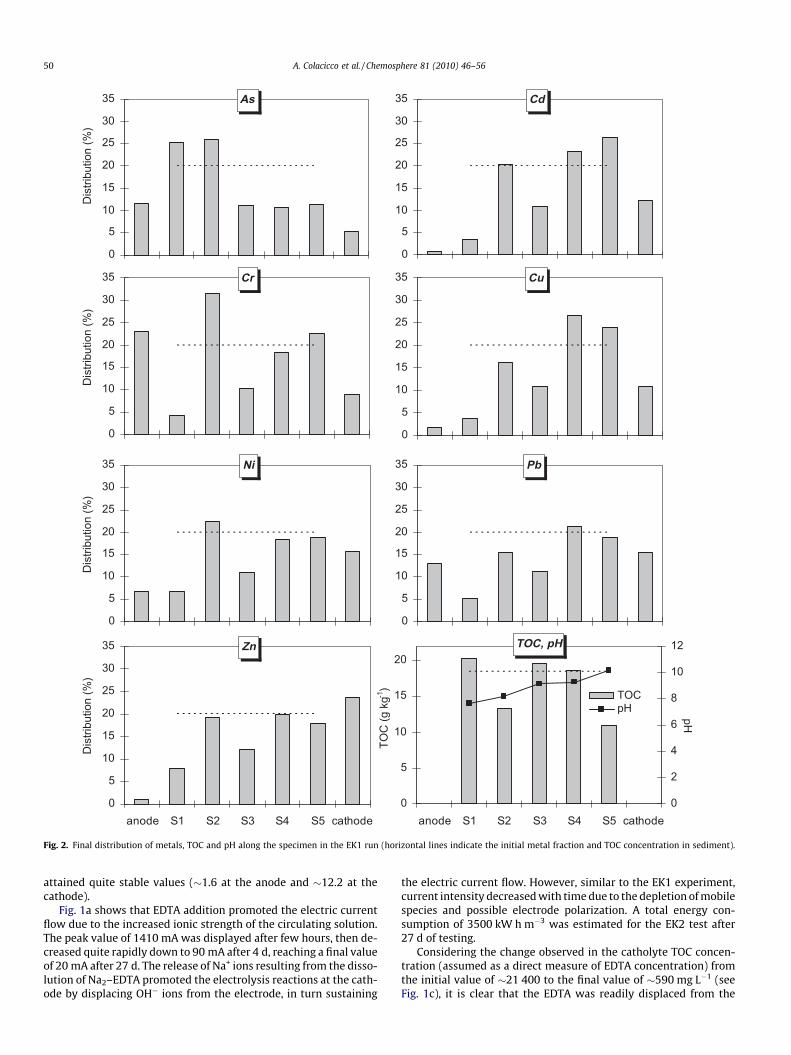
An electroosmotic flow towards the cathode was observed dur-ing the test (see Fig. 1b), displacing a final cumulative volume of�420 mL from the anodic chamber. As shown in Fig. 1c, the catho-lyte TOC concentration first increased (to �1500 mg L�1) and thendecreased (to �920 mg L�1), likely due to the initial transport ofhumic acids towards the cathode by the electroosmotic flowagainst the electromigration forces (considering the typically anio-nic nature of such molecules); likely, electromigration of organicmolecules prevailed over the electroosmotic flow when decreasedbelow a certain threshold (Sawada et al., 2003). This is also re-flected by the final TOC distribution along the cell, showing someaccumulation in the central specimen section (see Fig. 2). It istherefore apparent that electromigration prevailed towards theend of the test, determining higher TOC concentrations in the sec-tion adjacent to the anodic chamber.
As to the final pH evolution along the specimen (see Fig. 2), dueto the strong buffering capacity of the material, some slight pHreduction was only observed in the specimen section close to thecathode; in the remaining regions the final pH increased progres-sively towards the cathode, reaching a value of �10 units in thesection adjacent to the cathodic chamber.
The final metal distribution in the cell (see Fig. 2) indicated apoor metal removal yield (ranging from 12% for Cu to 32% forCr), due to both the low current intensities and the high pH valuesestablished during the experiment. Cd, Cu, Ni, Pb and Zn, which areexpected to form cationic complexes, were found to be removedfrom the section adjacent to the anode and to predominantly accu-mulate in the cathodic chamber. Unlike cationic metals, As wasfound to migrate towards the anode, due to the oxyanionic natureof its aqueous complexes (H2AsO�3 , H2AsO2�
3 ), while Cr presumablymigrated both as anionic species in its hexavalent state (closer tothe anode) and as cationic complexes in its trivalent form (closerto the cathode).
3.2. Chelant-enhanced electrokinetic runs
The results of the EDTA-enhanced treatment with catholyteconditioning (EK2 run) are reported in Figs. 1 and 3. As noted abovefor the EK1 experiment, the pH of the electrode solutions readily
Cd
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathodeCu
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathodePb
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathode
As
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Cr
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Ni
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Zn
0
5
10
15
20
25
30
35
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
TOC, pH
0
5
10
15
20
anode S1 S2 S3 S4 S5 cathode
TOC
(g k
g-1)
0
2
4
6
8
10
12
pH
TOCpH
Fig. 2. Final distribution of metals, TOC and pH along the specimen in the EK1 run (horizontal lines indicate the initial metal fraction and TOC concentration in sediment).
50 A. Colacicco et al. / Chemosphere 81 (2010) 46–56
attained quite stable values (�1.6 at the anode and �12.2 at thecathode).
Fig. 1a shows that EDTA addition promoted the electric currentflow due to the increased ionic strength of the circulating solution.The peak value of 1410 mA was displayed after few hours, then de-creased quite rapidly down to 90 mA after 4 d, reaching a final valueof 20 mA after 27 d. The release of Na+ ions resulting from the disso-lution of Na2–EDTA promoted the electrolysis reactions at the cath-ode by displacing OH� ions from the electrode, in turn sustaining
the electric current flow. However, similar to the EK1 experiment,current intensity decreased with time due to the depletion of mobilespecies and possible electrode polarization. A total energy con-sumption of 3500 kW h m�3 was estimated for the EK2 test after27 d of testing.
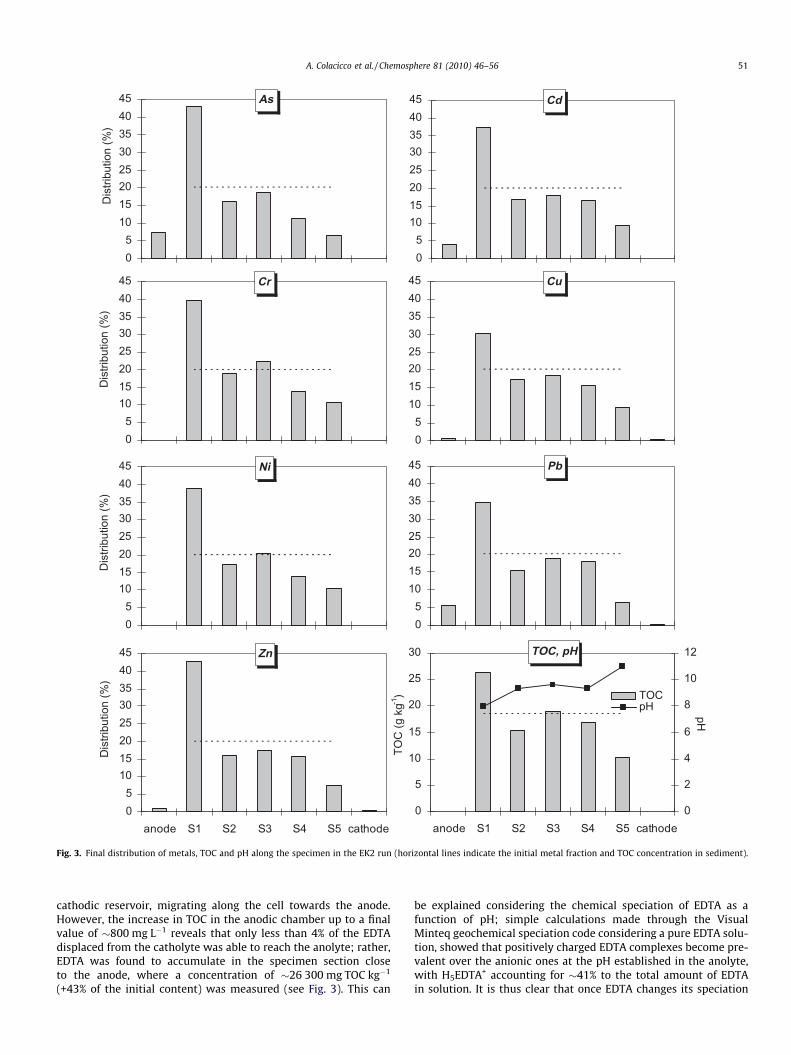
Considering the change observed in the catholyte TOC concen-tration (assumed as a direct measure of EDTA concentration) fromthe initial value of �21 400 to the final value of �590 mg L�1 (seeFig. 1c), it is clear that the EDTA was readily displaced from the
Cd
05
1015202530354045
anode S1 S2 S3 S4 S5 cathodeCu
05
1015202530354045
anode S1 S2 S3 S4 S5 cathodePb
05
1015202530354045
anode S1 S2 S3 S4 S5 cathode
As
05
1015202530354045
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Cr
05
1015202530354045
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Ni
05
1015202530354045
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Zn
05
1015202530354045
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
TOC, pH
0
5
10
15
20
25
30
anode S1 S2 S3 S4 S5 cathode
TOC
(g k
g-1)
0
2
4
6
8
10
12
pH
TOCpH
Fig. 3. Final distribution of metals, TOC and pH along the specimen in the EK2 run (horizontal lines indicate the initial metal fraction and TOC concentration in sediment).
A. Colacicco et al. / Chemosphere 81 (2010) 46–56 51
cathodic reservoir, migrating along the cell towards the anode.However, the increase in TOC in the anodic chamber up to a finalvalue of �800 mg L�1 reveals that only less than 4% of the EDTAdisplaced from the catholyte was able to reach the anolyte; rather,EDTA was found to accumulate in the specimen section closeto the anode, where a concentration of �26 300 mg TOC kg�1
(+43% of the initial content) was measured (see Fig. 3). This can
be explained considering the chemical speciation of EDTA as afunction of pH; simple calculations made through the VisualMinteq geochemical speciation code considering a pure EDTA solu-tion, showed that positively charged EDTA complexes become pre-valent over the anionic ones at the pH established in the anolyte,with H5EDTA+ accounting for �41% to the total amount of EDTAin solution. It is thus clear that once EDTA changes its speciation
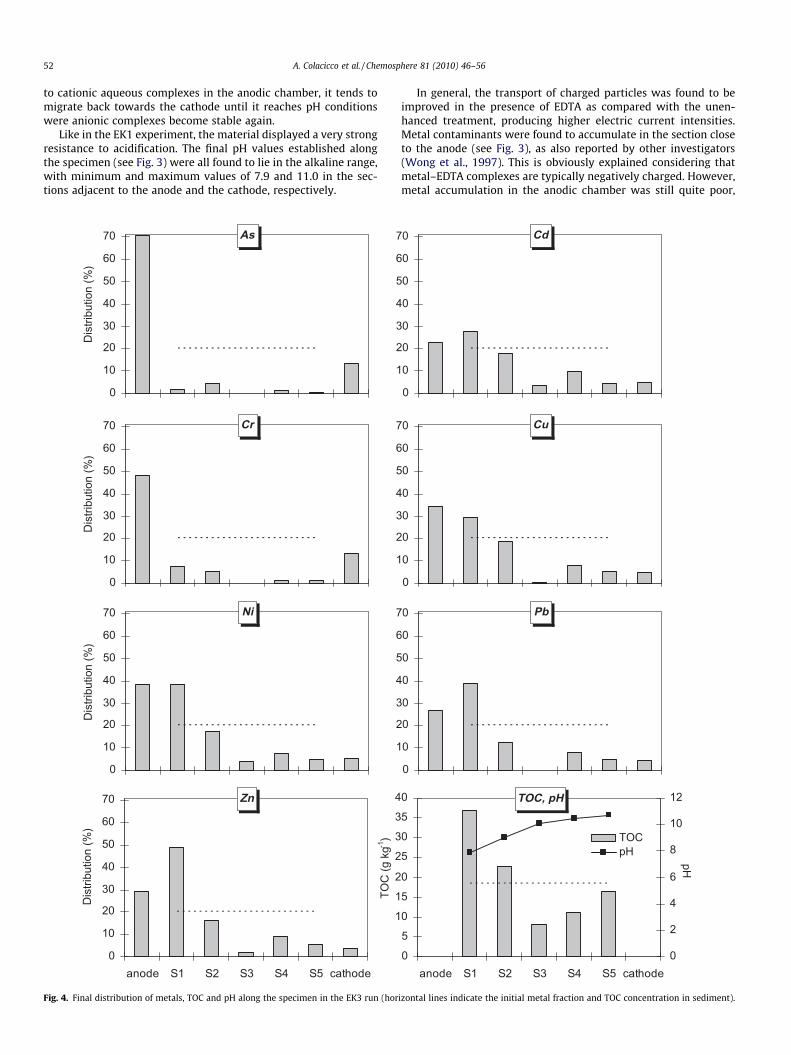
52 A. Colacicco et al. / Chemosphere 81 (2010) 46–56
to cationic aqueous complexes in the anodic chamber, it tends tomigrate back towards the cathode until it reaches pH conditionswere anionic complexes become stable again.
Like in the EK1 experiment, the material displayed a very strongresistance to acidification. The final pH values established alongthe specimen (see Fig. 3) were all found to lie in the alkaline range,with minimum and maximum values of 7.9 and 11.0 in the sec-tions adjacent to the anode and the cathode, respectively.
1
2
3
4
5
6
7
1
2
3
4
5
6
7
1
2
3
4
5
6
7
As
0
10
20
30
40
50
60
70
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Cr
0
10
20
30
40
50
60
70
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Ni
0
10
20
30
40
50
60
70
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
Zn
0
10
20
30
40
50
60
70
anode S1 S2 S3 S4 S5 cathode
Dis
tribu
tion
(%)
1
1
2
2
3
3
4
TOC
(g k
g-1)
Fig. 4. Final distribution of metals, TOC and pH along the specimen in the EK3 run (hor
In general, the transport of charged particles was found to beimproved in the presence of EDTA as compared with the unen-hanced treatment, producing higher electric current intensities.Metal contaminants were found to accumulate in the section closeto the anode (see Fig. 3), as also reported by other investigators(Wong et al., 1997). This is obviously explained considering thatmetal–EDTA complexes are typically negatively charged. However,metal accumulation in the anodic chamber was still quite poor,
Cd
0
0
0
0
0
0
0
0
anode S1 S2 S3 S4 S5 cathodeCu
0
0
0
0
0
0
0
0
anode S1 S2 S3 S4 S5 cathodePb
0
0
0
0
0
0
0
0
anode S1 S2 S3 S4 S5 cathodeTOC, pH
0
5
0
5
0
5
0
5
0
anode S1 S2 S3 S4 S5 cathode0
2
4
6
8
10
12
pH
TOCpH
izontal lines indicate the initial metal fraction and TOC concentration in sediment).
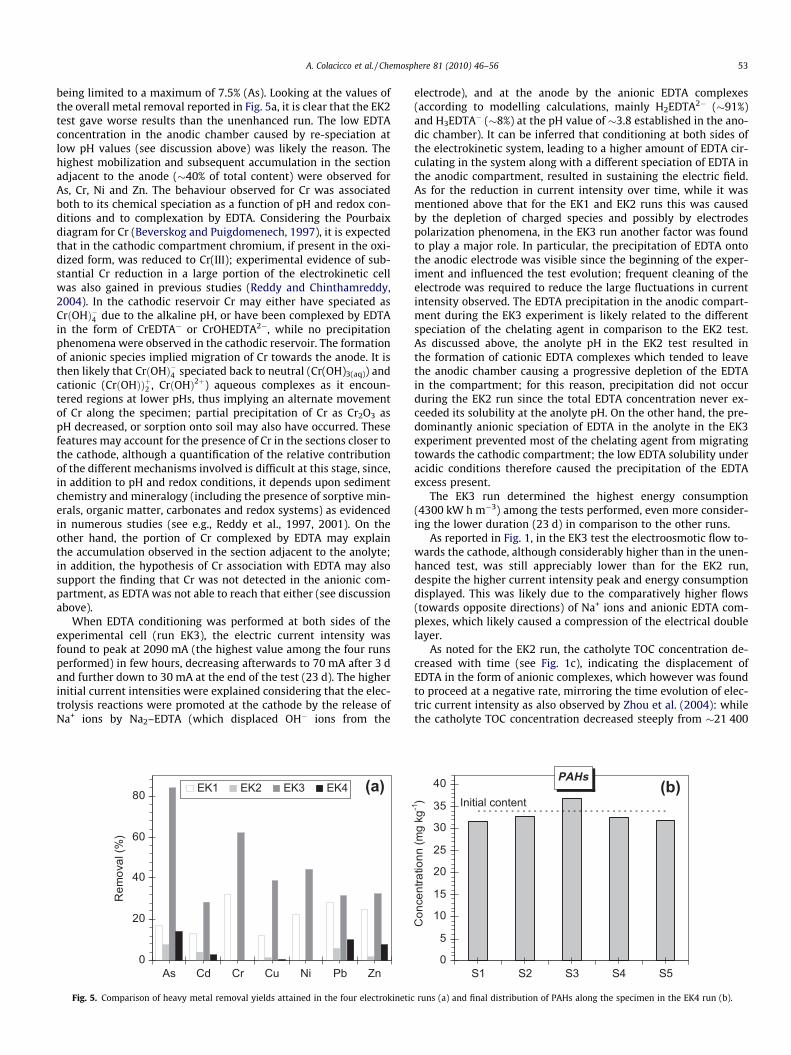
A. Colacicco et al. / Chemosphere 81 (2010) 46–56 53
being limited to a maximum of 7.5% (As). Looking at the values ofthe overall metal removal reported in Fig. 5a, it is clear that the EK2test gave worse results than the unenhanced run. The low EDTAconcentration in the anodic chamber caused by re-speciation atlow pH values (see discussion above) was likely the reason. Thehighest mobilization and subsequent accumulation in the sectionadjacent to the anode (�40% of total content) were observed forAs, Cr, Ni and Zn. The behaviour observed for Cr was associatedboth to its chemical speciation as a function of pH and redox con-ditions and to complexation by EDTA. Considering the Pourbaixdiagram for Cr (Beverskog and Puigdomenech, 1997), it is expectedthat in the cathodic compartment chromium, if present in the oxi-dized form, was reduced to Cr(III); experimental evidence of sub-stantial Cr reduction in a large portion of the electrokinetic cellwas also gained in previous studies (Reddy and Chinthamreddy,2004). In the cathodic reservoir Cr may either have speciated asCrðOHÞ�4 due to the alkaline pH, or have been complexed by EDTAin the form of CrEDTA� or CrOHEDTA2�, while no precipitationphenomena were observed in the cathodic reservoir. The formationof anionic species implied migration of Cr towards the anode. It isthen likely that CrðOHÞ�4 speciated back to neutral (Cr(OH)3(aq)) andcationic (CrðOHÞÞþ2 , CrðOHÞ2þ) aqueous complexes as it encoun-tered regions at lower pHs, thus implying an alternate movementof Cr along the specimen; partial precipitation of Cr as Cr2O3 aspH decreased, or sorption onto soil may also have occurred. Thesefeatures may account for the presence of Cr in the sections closer tothe cathode, although a quantification of the relative contributionof the different mechanisms involved is difficult at this stage, since,in addition to pH and redox conditions, it depends upon sedimentchemistry and mineralogy (including the presence of sorptive min-erals, organic matter, carbonates and redox systems) as evidencedin numerous studies (see e.g., Reddy et al., 1997, 2001). On theother hand, the portion of Cr complexed by EDTA may explainthe accumulation observed in the section adjacent to the anolyte;in addition, the hypothesis of Cr association with EDTA may alsosupport the finding that Cr was not detected in the anionic com-partment, as EDTA was not able to reach that either (see discussionabove).
When EDTA conditioning was performed at both sides of theexperimental cell (run EK3), the electric current intensity wasfound to peak at 2090 mA (the highest value among the four runsperformed) in few hours, decreasing afterwards to 70 mA after 3 dand further down to 30 mA at the end of the test (23 d). The higherinitial current intensities were explained considering that the elec-trolysis reactions were promoted at the cathode by the release ofNa+ ions by Na2–EDTA (which displaced OH� ions from the
0
20
40
60
80
As Cd Cr Cu Ni Pb Zn
Rem
oval
(%)
EK1 EK2 EK3 EK4 (a)
Fig. 5. Comparison of heavy metal removal yields attained in the four electrokinetic
electrode), and at the anode by the anionic EDTA complexes(according to modelling calculations, mainly H2EDTA2� (�91%)and H3EDTA� (�8%) at the pH value of �3.8 established in the ano-dic chamber). It can be inferred that conditioning at both sides ofthe electrokinetic system, leading to a higher amount of EDTA cir-culating in the system along with a different speciation of EDTA inthe anodic compartment, resulted in sustaining the electric field.As for the reduction in current intensity over time, while it wasmentioned above that for the EK1 and EK2 runs this was causedby the depletion of charged species and possibly by electrodespolarization phenomena, in the EK3 run another factor was foundto play a major role. In particular, the precipitation of EDTA ontothe anodic electrode was visible since the beginning of the exper-iment and influenced the test evolution; frequent cleaning of theelectrode was required to reduce the large fluctuations in currentintensity observed. The EDTA precipitation in the anodic compart-ment during the EK3 experiment is likely related to the differentspeciation of the chelating agent in comparison to the EK2 test.As discussed above, the anolyte pH in the EK2 test resulted inthe formation of cationic EDTA complexes which tended to leavethe anodic chamber causing a progressive depletion of the EDTAin the compartment; for this reason, precipitation did not occurduring the EK2 run since the total EDTA concentration never ex-ceeded its solubility at the anolyte pH. On the other hand, the pre-dominantly anionic speciation of EDTA in the anolyte in the EK3experiment prevented most of the chelating agent from migratingtowards the cathodic compartment; the low EDTA solubility underacidic conditions therefore caused the precipitation of the EDTAexcess present.
The EK3 run determined the highest energy consumption(4300 kW h m�3) among the tests performed, even more consider-ing the lower duration (23 d) in comparison to the other runs.
As reported in Fig. 1, in the EK3 test the electroosmotic flow to-wards the cathode, although considerably higher than in the unen-hanced test, was still appreciably lower than for the EK2 run,despite the higher current intensity peak and energy consumptiondisplayed. This was likely due to the comparatively higher flows(towards opposite directions) of Na+ ions and anionic EDTA com-plexes, which likely caused a compression of the electrical doublelayer.
As noted for the EK2 run, the catholyte TOC concentration de-creased with time (see Fig. 1c), indicating the displacement ofEDTA in the form of anionic complexes, which however was foundto proceed at a negative rate, mirroring the time evolution of elec-tric current intensity as also observed by Zhou et al. (2004): whilethe catholyte TOC concentration decreased steeply from �21 400
(b) Initial content
PAHs
0
5
10
15
20
25
30
35
40
S1 S2 S3 S4 S5
Con
cent
ratio
nn (m
g kg
-1)
runs (a) and final distribution of PAHs along the specimen in the EK4 run (b).

54 A. Colacicco et al. / Chemosphere 81 (2010) 46–56
to �7900 mg L�1 within the first 6 d of the test, the residual decayto the final value of �3600 mg L�1 after 23 d was considerablysmoother. The TOC profile in the anodic chamber was forced to re-main at approximately the same level during the experiment, dueto the periodical EDTA additions required to replace the volume ofsolution displaced by the electroosmotic flow towards the cathode.Nevertheless, electromigration towards the anode prevailed overelectroosmotic dragging, as indicated by the TOC profile alongthe specimen (see Fig. 4) and also observed by Acar et al. (1995),with EDTA accumulation in the two specimen sections closer tothe anode (S1 and S2) and a general TOC depletion in the remain-ing portions of the experimental cell.
As already noted for the previous experiments, the final pHalong the specimen (see Fig. 4) was still in the alkaline range, vary-ing from 7.9 to 10.7 from the anode to the cathode, very similarlyto the pattern observed in the EK2 run.
As for metal removal, a significant improvement compared tothe EK1 and EK2 runs was observed (see Fig. 5a). In particular,the best removal yield (84%) was observed for As, followed by Cr(62%). When looking at the final distribution along the specimen(see Fig. 4), it is clear that a considerable portion of As and Crwas found to migrate towards the anode, accumulating in the ano-dic chamber (As: �71%; Cr: �48%). The discussion presented aboveupon the behaviour of Cr in the EK2 run is thought to hold for theEK3 test as well. The higher accumulation of Cr in the anodic com-partment observed in this case can be explained by the differentanolyte pH established, which allowed EDTA complexes to enterthe anodic compartment. Since, according to the Pourbaix diagram,Cr oxidization is expected to occur in the anodic reservoir, it can beassumed that the Cr(III) portion reaching the anolyte with EDTAwas converted here to Cr(VI), which then remained in the anodiccompartment due to its typically anionic speciation (mainlyHCrO�4 in the conditions existing in the anolyte).
In the case of As, the mobilization effect observed in the pres-ence of EDTA, rather than directly resulting from chelation mech-anisms, is believed to have been mostly caused by an indirecteffect of the chelating agent upon sustaining the electric field inthe system, as mirrored by the higher electric current intensitiesobserved in this experiment.
For heavy metal cations the amount accumulated in the anodicchamber was appreciably higher than for runs EK1 and EK2, withan associated improved mobilization from all the specimen sec-tions. It is assumed that the anionic EDTA complexes migratingfrom the cathode were capable of detaching heavy metals from soilcausing the formation of anionic metal–EDTA complexes whichwere then attracted by the anode. The removal yield was in therange 28–44% for Cd, Cu, Ni, Pb and Zn. For all the investigated hea-vy metals the amount accumulated in the specimen section adja-cent to the anode was also significant, varying from 28% (Cd) to49% (Zn). This is also a positive feature of the treatment, sincethe residual contamination in sediment was confined in a reducedportion of the initial volume. The observed accumulation of metalsin the specimen section adjacent to the anodic compartment how-ever suggests that upon prolonging the treatment duration, the re-moval yield may be further improved and the build-up of metals inpart of the treated volume avoided.
When interpreting the positive results obtained in the EK3 test,it may be argued that the higher anolyte pH compared to the EK1and EK2 runs improved the overall energetic performance, sinceless energy was devoted to the transport of the acidic front.
3.3. Surfactant-enhanced electrokinetic run
The PAH removal attained in the unenhanced electrokinetictreatment was, as expected, quite poor, although a certain mobili-zation effect leading to a slight accumulation of PAHs near the
anode (58 vs. the initial concentration of 54 mg kg�1) was ob-served. Since PAHs are non-polar species, they are not subject toelectromigration, being transported by electroosmotic draggingonly (Saichek and Reddy, 2003). The potential removal is thereforeproportional to the number of pore volumes displaced by the elec-troosmotic flow. However, considering that PAHs are hydrophobicand their solubility decreases with molecular weight, the prevail-ing presence of heavy PAHs in the sediment under study was likelythe cause of the low removal attained under unenhanced condi-tions. In addition, the contrasting effect of electroosmotic transportand electromigration on the movement of PAHs displaced fromsediment by humic substances (which have been shown to playa role in the mobilization of organic contaminants in electrokineticprocesses; William and Amy, 1995) may explain the slight PAHaccumulation observed near the anode in the EK1 experiment.
As expected, enhancing the electrokinetic process by EDTAaddition did not cause any appreciable improvement in PAH re-moval, since the chelation process does not involve non-polarmolecules.
The need was thus recognized for dedicated electrokineticexperiments aimed at PAH removal (EK4 test). In the preliminarybatch washing tests a 3% Tween 80 solution yielded a PAH removalof �18%. Higher surfactant concentrations were not considereddue to the increasing viscosity of the resulting solution whichwould have impaired the electrokinetic process.
The applied voltage gradient during the EK4 test was initiallykept at 1 V cm�1 to prevent possible foam formation caused bythe presence of the surfactant, and increased to 2 V cm�1 after4 d. The electric current evolution differed from the previous tests,with significantly lower intensities as well as less rapid changesover time. A current peak of 318 mA was observed after 18 h fromthe onset of the experiment, decreasing afterwards down to 66 mAon the fourth d; when the voltage was increased to 2 V cm�1, thecurrent intensity increased again up to 130 mA, then progressivelydecreased down to the final value of 20 mA after 25 d. An energyconsumption of 1884 kW h m�3 over 25 d. was estimated. The evo-lution of electric current was likely affected by the presence ofTween 80, considering possible mechanisms including surfacecharging of the initially neutral surfactant molecule and deproto-nation mechanisms (Ko et al., 1998; Saichek and Reddy, 2003), aswell as adsorption onto the electrodes and formation of micellaraggregates above the CMC (0.012 mM (Holmberg et al., 2002) asopposed to the initial concentration in the anolyte of �23 mM),which can hinder the transport of charges throughout the system(Szymula and Narkiewicz-Michalek, 2006).
Assuming that Tween 80 gave the major contribution to the organ-ic matter in the system, the final TOC distribution along the cell and itsconcentration in the cathodic chamber (see Fig. 1c) reveal the absenceof any significant transport of the surfactant towards the cathode, anda prevalent accumulation in the section close to the anode. The negli-gible transport may have been caused by adsorption onto sedimentparticles (as also reported by Reddy and Ala (2006)), by the high vis-cosity of the Tween 80 solution and also by possible deprotonation,resulting in electromigration towards the anode as opposed to elec-troosmotic dragging.
As for PAHs, although the surfactant concentration in the initialsolution was much higher than its CMC, a very poor mobilizationfrom sediment was observed, with an increase in PAH concentra-tion in the middle section of the specimen by 8% only after 25 dof treatment (Fig. 5b), and a mobilization of �4–7% from the othersections. The treatment efficacy towards PAHs was therefore fullyinadequate, despite the positive results of the batch tests, a re-duced performance also observed, among the others, by Yangand Lee (2009). The poor transport of Tween 80 and the low cumu-lative L/S ratio attained (1.7 L kg�1, calculated from the measuredcumulative electroosmotic flow of �1.2 pore volumes) were

A. Colacicco et al. / Chemosphere 81 (2010) 46–56 55
certainly the main causes of the low PAH removal achieved. Theachieved L/S ratio was an obvious consequence of the electroos-motic flow, which in turn is affected by the pH and is directly pro-portional to the dielectric constant of the circulating solution, thezeta potential and the voltage gradient, and inversely proportionalto the solution viscosity. Although the strong acid buffering capac-ity of sediment should have sustained the electroosmotic flow, thesurfactant solution had a lower dielectric constant and a higherviscosity than water. Sorption of the surfactant onto the solid ma-trix may have even caused PAHs to get more stably adsorbed to thesolid phase; this may be supported by the findings of some authors(Yang and Lee, 2009), which indicated that adsorbed surfactantscan act as PAH sorbents in a much more effective way than thesoil/sediment organic matter itself can do. However, other mecha-nisms including changes in pH, ion concentration, electric gradient,pore pressure and particle surface charge have also been advocatedas possible causes of low removal yields of organic pollutants bysurfactants during electroremediation (Saichek and Reddy, 2003),and may have played a role during the present experiment.
4. Conclusions
The experimental study revealed that the electrokinetic remedi-ation treatment was affected by both the nature of contaminants/sediment interactions and the buffering capacity of the material.Without any appropriate conditioning agent, the process was notcapable of producing appreciable mobilization of either organicor inorganic contaminants. The use of EDTA significantly improvedheavy metal removal only when added at both sides of the electro-kinetic cell, which increased the chelant availability to the wholespecimen, at the same time improving the electrochemical charac-teristics of the system and the energy efficiency. It should howeverbe mentioned that the positive results obtained in terms of metalremoval from sediment may be somehow outbalanced by the in-crease in ecotoxicity of the treated material caused by the use ofEDTA; to this regard, the use of non-toxic and biodegradableextracting agents in electrokinetic remediation should deserve ma-jor attention in view of environmental sustainability of the process.
The effect of the electrokinetic process on PAH removal was notsatisfactory, even when the surfactant Tween 80 was applied; thelow L/S ratio achieved, the high viscosity of Tween 80 and the pos-sible surfactant deprotonation were likely the reasons. Differenttreatment conditions such as enhancement of the electroosmoticflow and improvement in the electrochemical properties of thesystem (e.g., using saline solutions), or addition of other surfac-tants may be investigated to promote PAH removal.
With a view to full-scale implementation of enhanced electroki-netics the practical implications of this work in terms of both thetechnical and the environmental issues involved should also beevaluated. In this respect, proper control of side effects includinganodic precipitation, oxidation of the conditioning agent and evo-lution of toxic gases (e.g., Cl2) is mandatory to ensure adequateremediation efficiencies. Furthermore, the ecotoxicological effectsresulting from large additions of chemicals should be thoroughlyconsidered, and specific methods aimed at removing (and recover-ing) the reagent excess after treatment should be studied anddeveloped. In this context, the use of alternative inorganic condi-tioning agents capable of ensuring adequate electrochemical char-acteristics of the system is worth to be investigated (Wong et al.,1997; Reddy and Chinthamreddy, 2004).
Acknowledgements
This work was financially supported by the Italian Ministry ofEducation, University and Research. The Italian Institute for the
Scientific and Technological Research Applied to the Sea (ISPRA,formerly ICRAM) is gratefully acknowledged for the assistance inproviding the sediment samples.
The authors wish to thank Orietta Masala and the IGAG – CNR(Environmental Geology and Geoengineering Institute of the Na-tional Research Council, Italy) of Cagliari for the precious coopera-tion in the chemical–physical analyses.
References
Acar, Y.B., Alshawabkeh, A.N., 1993. Principles of electrokinetic remediation.Environ. Sci. Technol. 27, 2638–2647.
Acar, Y.B., Gale, R.J., Alshawabkeh, A.N., Marks, R.E., Puppala, S., Bricka, M., Parker, R.,1995. Electrokinetic remediation: basics and technology status. J. Hazard.Mater. 40, 117–137.
Alcántara, T., Pazos, M., Gouveia, S., Cameselle, C., Sanromán, M.A., 2008. Remediationof phenanthrene from contaminated kaolinite by electroremediation – fentontechnology. J. Environ. Sci. Health A 43, 901–906.
Andreottola, G., Bonomo, L., De Gioannis, G., Ferrarese, E., Muntoni, A., Polettini, A.,Pomi, R., Saponaro, S., 2010. Lab-scale feasibility tests for sediment treatmentusing different physico-chemical techniques. J. Soils Sediments 10, 142–150.
Badr, T., Hanna, K., de Brauer, C., 2004. Enhanced solubilisation and removal ofnaphthalene and phenanthrene by cyclodextrins from two contaminated soils.J. Hazard. Mater. 112, 215–223.
Beverskog, B., Puigdomenech, I., 1997. Revised Pourbaix diagrams from chromiumat 25–300 �C. Corros. Sci. 39, 43–57.
Chung, H.I., Kamon, M., 2005. The coupled effect of electrokinetic and ultrasonicremediation for the treatment of contaminated sediments. In: Chun, J.S., Hong,S.W., Koo, J., Komai, T., Koterayama, W. (Eds.), Proceedings InternationalOffshore and Polar Engineering Conference. Seoul, Korea, pp. 652–657.
Chung, H.I., Kang, B.H., 1999. Lead removal from contaminated marine clay byelectrokinetic soil decontamination. Eng. Geol. 53, 139–150.
Darmawan, S., Wada, I., 2002. Effect of clay mineralogy on the feasibility ofelectrokinetic soil decontamination technology. Appl. Clay Sci. 20, 283–293.
De Gioannis, G., Muntoni, A., Polettini, A., Pomi, R., 2008. Enhanced electrokinetictreatment of different marine sediments contaminated by heavy metals. J.Environ. Sci. Health A 43, 852–865.
De Gioannis, G., Muntoni, A., Polettini, A., Pomi, R., 2009. Electrokinetic treatment ofcontaminated marine sediments. In: Cameselle, C., Reddy, K.R. (Eds.),Electrochemical Remediation Technologies for Polluted Soils, Sediments andGroundwater. John Wiley, New York, pp. 149–177.
Doering, F., Doering, N., Hill, D.G., McIlvride, W.A., Iovenitti, J.L., Wittle, J.K., 2002.Electrochemical remediation technologies for metals remediation in soil,sediment, and ground water. In: Proceedings 3rd International Conference onRemediation of Chlorinated and Recalcitrant Compounds. Battelle Press, pp.2095–2102.
Grundl, T., Reese, C., 1997. Laboratory study of electrokinetic effects in complexnatural sediments. J. Hazard. Mater. 55, 187–201.
Holmberg, K., Jönsson, B., Kronberg, B., Lindman, B., 2002. Surfactants and Polymersin Aqueous Solution. John Wiley, Chichester, UK.
Ko, S., Schlautman, M.A., Carraway, E.R., 1998. Effects of solution chemistry on thepartitioning of phenanthrene to sorbed surfactants. Environ. Sci. Technol. 32,3542–3548.
Kornilovich, B., Mishchuk, N., Abbruzzese, K., Pshinko, G., Klishchenko, R., 2005.Enhanced electrokinetic remediation of metals-contaminated clay. ColloidsSurf. A 265, 114–123.
Li, T., Yuan, S., Wan, J., Lin, L., Long, H., Wu, X., Lu, X., 2009. Pilot-scale electrokineticmovement of HCB and Zn in real contaminated sediments enhanced withhydroxypropyl-b-cyclodextrin. Chemosphere 76, 1226–1232.
Li, T., Yuan, S., Wan, J., Lu, X., 2010. Hydroxypropyl-b-cyclodextrin enhancedelectrokinetic remediation of sediment contaminated with HCB and heavymetals. J. Hazard. Mater. 176, 306–312.
Maturi, K., Reddy, K.R., 2008. Cosolvent-enhanced desorption and transport ofheavy metals and organic contaminants in soils during electrokineticremediation. Water Air Soil Pollut. 189, 199–211.
Ottosen, L.M., Hansen, H.K., Jensen, P.E., 2009. Electrokinetic removal of heavymetals. In: Cameselle, C., Reddy, K.R. (Eds.), Electrochemical RemediationTechnologies for Polluted Soils, Sediments and Groundwater. John Wiley, NewYork, pp. 97–126.
Park, J.Y., Lee, H.H., Kim, S.J., Yang, J.W., 2007. Surfactant-enhanced electrokineticremoval of phenanthrene from kaolinite. J. Hazard. Mater. 140, 230–236.
Pazos, M., Rosales, E., Alcántara, T., Gómez, J., Sanromán, M.A., 2010.Decontamination of soils containing PAHs by electroremediation: a review. J.Hazard. Mater. 177, 1–11.
Reddy, K.R., Ala, P.R., 2006. Electrokinetic remediation of contaminated dredgedsediment. J. ASTM Int. 3, 254–267.
Reddy, K.R., Chinthamreddy, S., 2004. Enhanced electrokinetic remediation of heavymetals in glacial till soils using different electrolyte solutions. J. Environ. Eng.130, 442–455.
Reddy, K.R., Saichek, R.E., 2004. Enhanced electrokinetic removal of phenanthrenefrom clay soil by periodic electric potential application. J. Environ. Sci. Health A39, 1189–1212.

56 A. Colacicco et al. / Chemosphere 81 (2010) 46–56
Reddy, K.R., Parupudi, U.S., Devulapalli, S.N., Xu, C.Y., 1997. Effects of soilcomposition on the removal of chromium by electrokinetics. J. Hazard. Mater.55, 135–158.
Reddy, K.R., Xu, C.Y., Chinthamreddy, S., 2001. Assessment of electrokinetic removalof heavy metals from soils by sequential extraction analysis. J. Hazard. Mater.84, 279–296.
Reddy, K.R., Danda, S., Saichek, R.E., 2004. Complicating factors of usingethylenediamine tetraacetic acid to enhance electrokinetic remediation ofmultiple heavy metals in clayey soils. J. Environ. Eng. 130, 1357–1366.
Saichek, R.E., 2002. Electrokinetically Enhanced In-situ Flushing for HOC-contaminated Soils. Ph.D. Thesis, University of Illinois, Chicago, USA.
Saichek, R.E., Reddy, K.R., 2003. Effect of control at the anode for the electrokineticremoval of phenanthrene from kaolin soil. Chemosphere 51, 273–287.
Saichek, R.E., Reddy, K.R., 2005. Electrokinetically enhanced remediation ofhydrophobic organic compounds in soils: a review. Crit. Rev. Environ. Sci.Technol. 35, 115–192.
Sawada, A., Tanaka, S., Fukushima, M., Tatsumi, K., 2003. Electrokinetic remediationof clayey soils containing copper(II)-oxinate using humic acid as a surfactant. J.Hazard. Mater. 96, 145–154.
Szymula, M., Narkiewicz-Michalek, J., 2006. The effect of surfactant adsorption at aglassy carbon electrode on electrochemical oxidation of propyl gallate. J. Appl.Electrochem. 36, 455–462.
William, P.J., Amy, G.L., 1995. Facilitated transport and enhanced desorption ofpolycyclic aromatic hydrocarbons by natural organic matter in aquifersediments. Environ. Sci. Technol. 29, 807–817.
Wong, J.S.H., Hicks, R.E., Probstein, R.F., 1997. EDTA-enhanced electroremediation ofmetal-contaminated soils. J. Hazard. Mater. 55, 61–79.
Yang, J.W., Lee, Y.J., 2009. Electrokinetic removal of PAHs. In: Cameselle, C., Reddy,K.R. (Eds.), Electrochemical Remediation Technologies for Polluted Soils,Sediments and Groundwater. John Wiley, New York, pp. 197–217.
Yang, J.W., Lee, Y.J., Park, J.Y., Kim, S.J., Lee, J.Y., 2005. Application of APG and Calfax16L-35 on surfactant-enhanced electrokinetic removal of phenanthrene fromkaolinite. J. Eng. Geol. 77, 243–251.
Yeung, A.T., Hsu, C.N., 2005. Electrokinetic remediation of cadmium-contaminatedclay. J. Environ. Eng. 131, 298–304.
Yu, J.W., Neretnieks, I., 1997. Theoretical evaluation of a technique for electrokineticdecontamination of soils. J. Contam. Hydrol. 26, 291–299.
Yuan, C., Weng, C.-H., 2006. Electrokinetic enhancement removal of heavy metalsfrom industrial wastewater sludge. Chemosphere 65, 88–96.
Zhou, D.M., Deng, C.F., Cang, L., 2004. Electrokinetic remediation of a Cucontaminated red soil by conditioning catholyte pH with different enhancingchemical reagents. Chemosphere 56, 265–273.