enantiomeric separations by means of nano-lc
TRANSCRIPT

J. Sep. Sci. 2013, 00, 1–24 1
Anna Rocco1,2
Audrius Maruska1∗Salvatore Fanali2
1Department of Biochemistry andBiotechnologies, VytautasMagnus University, Kaunas,Lithuania
2Institute of ChemicalMethodologies, ConsiglioNazionale delle Ricerche, Rome,Italy
Received September 19, 2012Revised October 24, 2012Accepted October 26, 2012
Review Article
Enantiomeric separations by meansof nano-LC
Enantiomers represent a class of compounds extensively investigated since they can showtotally different behaviors when they interact with a chiral environment. Because of theiridentical chemical structure (they differ only in the spatial arrangement of the atoms in themolecule), the separation of optical isomers is a challenging task of analytical chemistry. Sofar employed methods for the separation of enantiomers are mainly based on chromatog-raphy. CE as well was considered as an analytical technique suitable for chiral separations,characterized by high efficiency and low consumption of reagent. Recently, miniaturizationwas introduced in LC to answer the needs to perform analyses in the minimum time, touse the smallest amount of samples and to reduce environmental pollution. Nano-LC repre-sents nowadays a valid alternative to the abovementioned conventional analytical techniques,and can be advantageously exploited for enantiomeric separation especially because it needsminute amounts of the chiral material necessary to carry out enantiomeric separations. Thisreview describes the development and applications of nano-LC in the field of chiral sepa-rations. The data reported in literature show its relevance for the study enantiomers-chiralselectors interaction, as well as for application in pharmaceutical and clinical research.
Keywords: Capillary column / Chiral separation / Enantiomer / Nano-LCDOI 10.1002/jssc.201200886
1 Introduction
Enantiomer separations represent an important topic of re-search in analytical chemistry, since the bioactivity of manymolecules (drugs, pesticides, proteins, etc.) is related to theirchirality.
In the pharmaceutical field, i.e. where a wide number ofdrugs has one or more than one asymmetric centre, only oneisomer can be responsible for the desired activity, while theother may exhibit no therapeutic effects and may be the onlyaccountable for adverse effects, as a consequence of differentstereoselective interactions with biological matrices, such asreceptors. Metabolism of two enantiomers of a certain drugmay be different as well and thus analytical methods are
Correspondence: Dr. Salvatore Fanali, Institute of ChemicalMethodologies, Consiglio Nazionale delle Ricerche, Via SalariaKm 29.300, 00015 Monterotondo, Rome, ItalyE-mail: [email protected]: +390690672269
Abbreviations: CD, cyclodextrin; CDCPC, cellulose tris(3,5-dichlorophenylcarbamate); CDMPC, cellulose tris(3,5-dimethylphenylcarbamate); CLC, capillary LC; CMPA, chiralmobile phase additive; CS, chiral selector; CSP, chiral sta-tionary phase; FITC, fluorescein isothiocyanate; HP-�-CD,hydroxypropyl-�-cyclodextrin; MA, macrocyclic antibiotic;MIP, molecular imprinted polymer; OT, open tubular;Ph-�-CD, perphenylcarbamoylated �-CD; PPAR�, perox-isome proliferator-activated receptor; SFC, supercriticalfluid chromatography; THP, tetrahydropalmatine; TM-�-CD,heptakis(2,3,6-tri-O-methyl)-�-cyclodextrin
required for chiral purity control of pharmaceuticals, phar-macokinetic, and metabolism studies, etc. [1–3].
Analytical separation techniques so far employed to dis-criminate chiral compounds are GC, LC, supercritical fluidchromatography, and electrokinetic techniques ones (mostlyCE) [4–7]. In order to discriminate chiral compounds twostrategies can be used: so called direct and indirect methods.In the second approach, analytes are derivatized with an enan-tiomerically pure reagent resulting in the formation of a pairof diastereomers, which can be afterwards separated in anachiral environment. This approach is less common becauseit requires additional reactions and derivatization can affectthe results of quantitative analysis. To the best of our knowl-edge, no works are reported in literature, concerning the sep-aration in nano-LC of enantiomers by the indirect method.
The direct approach is based on interactions with a chiralselector and results in the formation of transient, noncova-lent diastereoisomer complexes. The chiral selector can beeither bound (by a covalent bond, absorption, or entrapped)to the stationary phase contained in the column, directlybound to the wall of the column, or dissolved in the mobilephase/background electrolyte as chiral additive. The enan-tiomer resolution process is based on different interactions,between analyte enantiomers and the chiral selector (CS),which depend on chiral selector functional groups, asym-metric centers, and in many cases, on steric repulsion. Theinteraction between the analyte and the CS should be fast
∗Additional Correspondence: Professor Audrius Maruska,E-mail: [email protected]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

2 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
and the equilibrium constant for each enantiomer should bediverse to result in different migration time [2, 6, 8].
Recently, miniaturization, a trend of modern analyticalchemistry, was successfully introduced in conventional LC,resulting in the development of micro-LC and later on ofcapillary- and nano-LC (CLC and nano-LC, respectively). Ini-tially, there was no a clear distinction among these minia-turized chromatographic systems. Currently micro-LC, CLC,and nano-LC refer to chromatographic separations performedin capillary columns with internal diameter (id) of 0.5–1 mm,100–500 �m, and 10–100 �m, respectively. Nano-LC offersseveral advantages over conventional LC, e.g. use of smallamount of reagent (i.e. mobile and stationary phases) with aconsequent low environmental pollution, low sample require-ment, good efficiency, short analysis time, and easy couplingwith MS [9–12]. Furthermore, compared with other minia-turized techniques, such as CE and CEC, nano-LC is char-acterized by an easier reliability, reproducibility, universalitythat make it more adequate for industries ruled by regulatoryissues [13].
In case of chiral separations, where expensive chiralstationary phases (CSPs) or chiral mobile phase additives(CMPA) have to be employed, nano-LC results very usefulsince it allows to perform analysis with a very small amountof this costly material.
The aim of this review was to reveal general features ofnano-LC, outline the main aspects of chiral separation, andreport potential and applications of nano-LC for enantiosep-arations.
2 Nano-LC
Nowadays, nano-LC has established itself as a complemen-tary and/or competitive separation technique to conventionalLC, providing a wide number of important applications par-ticularly in pharmaceutical field and “omics” branches of bi-ology [14, 15].
Even if currently different apparatus for nano-LC are com-mercially available, in the past great efforts have been madeto solve technical problems such as nano-flow rate control,gradient performance, required injection volume, and detec-tion [11, 16, 17].
As nano-LC is performed in capillary columns with an idof 10–100 �m, the working flow rate usually is in the orderof a few hundred nanoliters-per-minute. The low flow ratemakes nano-LC suitable for the coupling with MS, and thisaspect markedly contributed to its success in the analyticalfield. Fused silica capillaries are mostly used for fabricationof nano-LC columns.
Capillary columns used in nano-LC can be distinguishedinto three different types: packed, open tubular (OT), andmonolithic columns [17–19].
Packed capillary columns are made with the particulatestationary phases widely used in HPLC. Usually 3–5 �m par-ticles are employed, however recently, particles of smaller size(sub-2 �m), developed to obtain higher efficiency and selec-
tivity, have been also successfully tested in nano-LC. Severalpacked capillary columns are commercially available for bothchiral and achiral separations. They can be prepared in lab-oratory as well, however, the packing procedure requires aconsiderable experience. The entrapment of the stationaryphases in the capillary column is also a challenging task andis obtained in the most of cases with retaining frits at the ex-tremity of the packed section of the column. Inlet and outletfrits are usually prepared in situ by sintering the packed ma-terial with a heated wire. This procedure causes the removalof polyimide coating at the frit zone and makes the columnfragile. Furthermore, a bad sintering can compromise theflow through the column and change the properties of thepacking material where the heating has been applied [20,21].
In OT-columns, the stationary phase is directly boundor adsorbed to the inner wall of the capillary, and for thisreason their id is lower, usually between 10 and 60 �m. Theadsorption of the stationary phase, or modifier, can occur dy-namically (weak interactions, the inner modifier is added tothe mobile phase) or physically (modifier strongly adsorbed).The layer of stationary phase can be also covalently attachedand/or cross-linked. OT-columns are characterized by a lowconvective dispersion, good efficiency, and high permeabil-ity, however, they suffer of poor selectivity and low sampleloading capacity, both due to the very low phase ratio [22,23].
The introduction of non-particulate monolithic stationaryphases in LC and then in miniaturized systems, resulted innew advantages for these techniques. In the capillary format,monolithic column can be easily prepared by in situ polymer-ization and it does not require frits, thus avoiding problemsrelated to the stationary phase packing procedures, frits per-meability, fragility, and manufacture. This kind of columns ischaracterized by high permeability and high efficiency. Thewide availability of monomers permits to obtain columnswith specific chromatographic properties [24–26].
Monolithic columns can be obtained by polymerizationof organic monomers or by sol-gel process in case of silica-based monoliths. Water-soluble comonomers in the presenceof salt as phase separation catalyzing agent, or organic solventsoluble comonomers in the presence of porogen cosolvent,can be used for the polymerization process.
Polymeric mixtures are useful for the preparation ofmolecularly imprinted monoliths, which have been used forseveral applications in the analytical field, including chiralseparations. Molecular imprinted stationary phases are car-ried out by the addition of a template (imprint) moleculein the polymeric mixture. Before starting polymerization,functional groups on the template interact with those onthe monomer(s), and when the reaction begins monomersresult arranged around the template. During the polymer-ization the template, depending on its structure, can react,forming covalent bonds or give rise to secondary interac-tions with other monomers. If the imprinting is success-ful, after the removal of the imprint molecule, the poly-mer network will posses cavities having shape, size, andfunctional groups complementary to those of the template[27, 28].
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 3
2.1 Microdevices
Currently, one of the main requirements in the analyticalfield is the possibility to analyze as many as possible samples,in the minimum quantity and in the shortest time. Further,a great effort is being performed in order to achieve samplepretreatment and analytical separation with the same instru-ments (so-called �-total analysis system). This has broughtto the advent of microdevices, consisting of, e.g. plastic chipthat showed high potential in the analytical field, especially for“omics” sciences [29]. They offer good peak capacity and sen-sitivity, low sample handling, minute sample requirementsand are portable, giving the opportunity to perform field anal-ysis. Mostly, microdevices are used for electrophoretic anal-ysis, since the conditions to realize this kind of separations(liquid media, application of a voltage) are easy to realizefor such small devices [30, 31]. However, the introduction ofmonolithic materials has given a new impetus to the prepa-ration of microdevices for chromatographic purposes. In situpreparation instead of packing procedure and high perme-ability of the media can be considered as the main advantagesthat should make easier to realize chromatographic systemin chip dimensions. At the moment, promising results wereachieved with microchip CEC [32–35]. As an example, Zeng etal. obtained the enantioresolution of FITC-labeled dansyl-d,l-threonine in about 100 s, employing a microdevice based ona monolithic polydimethylsiloxane (PDMS) column deriva-tized with allyl-�-CD [36]. In a more recent work [37], thesame group developed a chip-based enantioselective open-tubular CEC employing BSA-gold nanoparticle conjugates asa CSP. The simultaneous enantioresolution of norephedrineand ephedrine was achieved in less than 300 s.
3 Chiral separations
Chiral compounds are classically defined as not superposableon their mirror image. Chirality does not affect chemico-physical properties of these molecules, with exception ofpolarized light rotation in opposite direction, however, itcan strongly influence activities of enantiomers in a chiralenvironment. For this reason, the stereochemistry of com-pounds is an important tool with regard to interaction be-tween stereoisomers and biological targets, such as receptorsand enzymes, which are at molecular level homochiral [38].
This has resulted in the requirements for chiralcompounds-based products and over the last decades, therehas been a growing interest in the separation of chiral drugs.
The determination of the stereochemical composition orstereochemical purity control are required in pharmaceuti-cal industry by regulatory authorities, such as US Food andDrug Administartion and the European Agency for the Eval-uation of Medicinal Products, which promote those drugsdeveloped as single enantiomer. This kind of inspection iscrucial not only during chiral drug synthesis, but also duringthe determination and monitoring of the drug metabolites fortherapy optimization or toxicological studies. Further, stereo-
chemical studies involve chemical and cosmetic industry, aswell as biological, agrochemical, and environmental analyses[39, 40].
In all these fields, analytical separative methods arerequired, e.g. for the quantitative assessment of the exactcomposition of a racemate or determining the enantiomericexcess (ee) of an enantiomeric pure substance. The latter typeof verification gains in importance since, in 2006, The Inter-national Conference on Harmonisation of Technical Require-ments for Registration of Pharmaceuticals for Human Use(ICH) guidelines included the noneffective enantiomer (dis-tomer) as impurity in case of single enantiomer drug (www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002823.pdf), [41].
Considering that already at a content of 0.05% the enan-tiomeric impurity should be reported, and identified at 0.1%,it results evident the difficulty to unequivocally evaluate theee, when the content of minor enantiomer has to be evaluatedin the presence of the major enantiomer.
4 Chiral selectors
Enantioseparations in nano-LC are mostly performed em-ploying the same CS already known from HPLC and CE,which have been thoroughly reviewed previously [4,42]. Con-sequently, in this paragraph a short overview of the mainfeatures of the principal classes of CSs used in nano-LC isgiven. It should be again emphasized, instead, that nano-LCallows to perform chiral separations either with a CSP oradding the CS to the mobile phase. The last mode is consid-ered prohibitive for HPLC, where huge volumes of solventare consumed, even if some applications are reported in lit-erature, while is largely exploited in CE. In nano-LC, bothapproaches result advantageous since nano-LC need smallamounts of CSP and offers low mobile phase and selectorconsumption.
Considering this aspect, cyclodextrins (CDs) and CDderivatives represent the ideal chiral selector that can beused as CSP or in the free form as CMPA. CDs form tran-sient diastereomeric inclusion complexes with enantiomersby means of the cavity, while the hydroxyl groups of glu-copyranose units favor the enantiodiscrimination process byhydrogen-bonding interactions. CDs offer high efficiency andselectivity toward a large numbers of molecules. The deriva-tization of the hydroxyl groups on the external rims leads toCD derivatives with higher solubility, different depth of cav-ity, different interaction sites, etc. The use of CDs as CMPAis also justified by the fact that they are no UV-absorbingand they have good solubility in aqueous/polar organic mo-bile phase [43–49]. Another class of CSs, related to CD andconsidered as the most powerful for chiral separations, isrepresented by polysaccharide derivatives (e.g. cellulose oramylose based). Their enantiorecognition capability is deter-mined by the higher order structure of the polymer as well asby functional groups naturally or synthetically occurring onit. Among all polysaccharides derivatives, phenylcarbamate
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
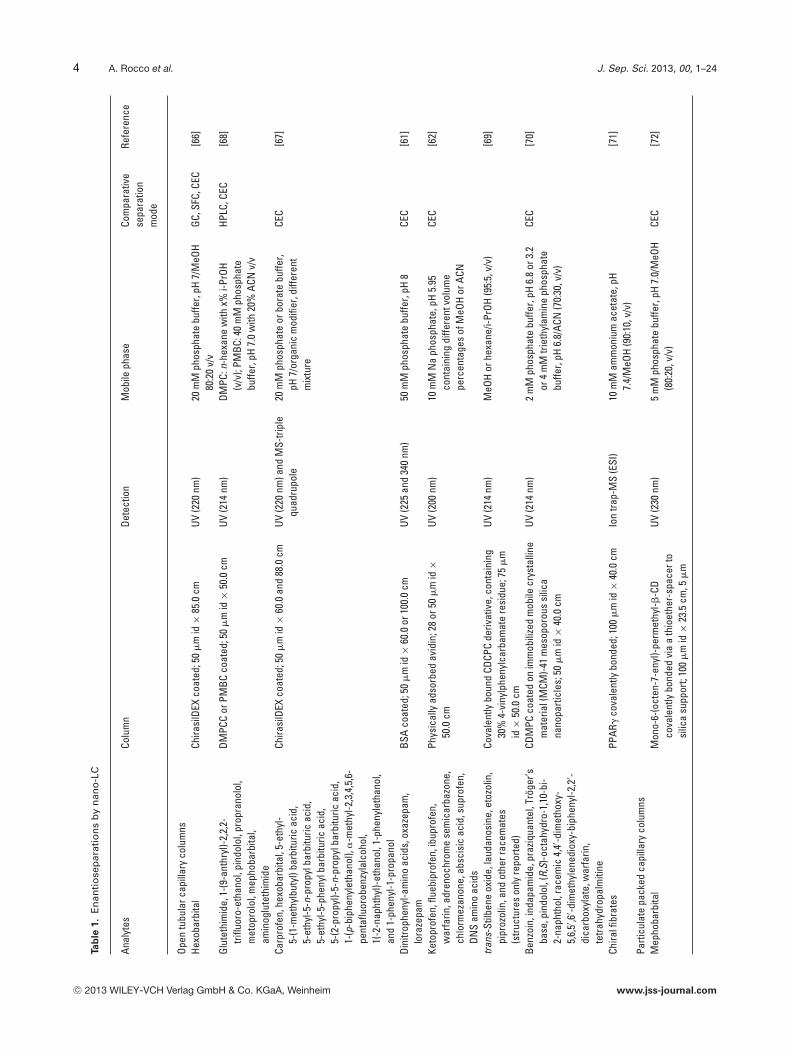
4 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Ta
ble
1.
En
anti
ose
par
atio
ns
by
nan
o-L
C
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
Open
tubu
larc
apill
ary
colu
mns
Hexo
barb
ital
Chira
silD
EXco
ated
;50
�m
id×
85.0
cmUV
(220
nm)
20m
Mph
osph
ate
buffe
r,pH
7/M
eOH
80:2
0v/
vGC
,SFC
,CEC
[66]
Glut
ethi
mid
e,1-
(9-a
nthr
yl)-2
,2,2
-tri
fluor
o-et
hano
l,pi
ndol
ol,p
ropr
anol
ol,
met
opro
lol,
mep
hoba
rbita
l,am
inog
lute
thim
ide
DMPC
Cor
PMBC
coat
ed;5
0�
mid
×50
.0cm
UV(2
14nm
)DM
PC:n
-hex
ane
with
x%i-P
rOH
(v/v
);PM
BC:4
0m
Mph
osph
ate
buffe
r,pH
7.0
with
20%
ACN
v/v
HPLC
,CEC
[68]
Carp
rofe
n,he
xoba
rbita
l,5-
ethy
l-5-
(1-m
ethy
lbut
yl)b
arbi
turic
acid
,5-
ethy
l-5-n
-pro
pylb
arbi
turic
acid
,5-
ethy
l-5-p
heny
lbar
bitu
ricac
id,
5-(2
-pro
pyl)-
5-n-
prop
ylba
rbitu
ricac
id,
1-(p
-bip
heny
leth
anol
),�
-met
hyl-2
,3,4
,5,6
-pe
ntafl
uoro
benz
ylal
coho
l,1(
-2-n
apht
hyl)-
etha
nol,
1-ph
enyl
etha
nol,
and
1-ph
enyl
-1-p
ropa
nol
Chira
silD
EXco
ated
;50
�m
id×
60.0
and
88.0
cmUV
(220
nm)a
ndM
S-tri
ple
quad
rupo
le20
mM
phos
phat
eor
bora
tebu
ffer,
pH7/
orga
nic
mod
ifier
,diff
eren
tm
ixtu
re
CEC
[67]
Dini
troph
enyl
-am
ino
acid
s,ox
azep
am,
lora
zepa
mBS
Aco
ated
;50
�m
id×
60.0
or10
0.0
cmUV
(225
and
340
nm)
50m
Mph
osph
ate
buffe
r,pH
8CE
C[6
1]
Keto
prof
en,fl
uebi
prof
en,i
bupr
ofen
,w
arfa
rin,a
dren
ochr
ome
sem
icar
bazo
ne,
chlo
rmez
anon
e,ab
scis
icac
id,s
upro
fen,
DNS
amin
oac
ids
Phys
ical
lyad
sorb
edav
idin
;28
or50
�m
id×
50.0
cmUV
(200
nm)
10m
MN
aph
osph
ate,
pH5.
95co
ntai
ning
diffe
rent
volu
me
perc
enta
ges
ofM
eOH
orAC
N
CEC
[62]
trans
-Stil
bene
oxid
e,la
udan
osin
e,et
ozol
in,
pipr
ozol
in,a
ndot
herr
acem
ates
(stru
ctur
eson
lyre
porte
d)
Cova
lent
lybo
und
CDCP
Cde
rivat
ive,
cont
aini
ng30
%4-
viny
lphe
nylc
arba
mat
ere
sidu
e;75
�m
id×
50.0
cm
UV(2
14nm
)M
eOH
orhe
xane
/i-Pr
OH(9
5:5,
v/v)
[69]
Benz
oin,
inda
pam
ide,
praz
iqua
ntel
,Tro
ger’s
base
,pin
dolo
l,(R
,S)-o
ctah
ydro
-1,1
0-bi
-2-
naph
thol
,rac
emic
4,4′
-dim
etho
xy-
5,6,
5′,6
′ -dim
ethy
lene
diox
y-bi
phen
yl-2
,2′ -
dica
rbox
ylat
e,w
arfa
rin,
tetra
hydr
opal
miti
ne
CDM
PCco
ated
onim
mob
ilize
dm
obile
crys
talli
nem
ater
ial(
MCM
)-41
mes
opor
ous
silic
ana
nopa
rticl
es;5
0�
mid
×40
.0cm
UV(2
14nm
)2
mM
phos
phat
ebu
ffer,
pH6.
8or
3.2
or4
mM
triet
hyla
min
eph
osph
ate
buffe
r,pH
6.8/
ACN
(70:
30,v
/v)
CEC
[70]
Chira
lfibr
ates
PPAR
�co
vale
ntly
bond
ed;1
00�
mid
×40
.0cm
Ion
trap-
MS
(ESI
)10
mM
amm
oniu
mac
etat
e,pH
7.4/
MeO
H(9
0:10
,v/v
)[7
1]
Parti
cula
tepa
cked
capi
llary
colu
mns
Mep
hoba
rbita
lM
ono-
6-(o
cten
-7-e
nyl)-
perm
ethy
l-�-C
Dco
vale
ntly
bond
edvi
aa
thio
ethe
r-sp
acer
tosi
lica
supp
ort;
100
�m
id×
23.5
cm,5
�m
UV(2
30nm
)5
mM
phos
phat
ebu
ffer,
pH7.
0/M
eOH
(80:
20,v
/v)
CEC
[72]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
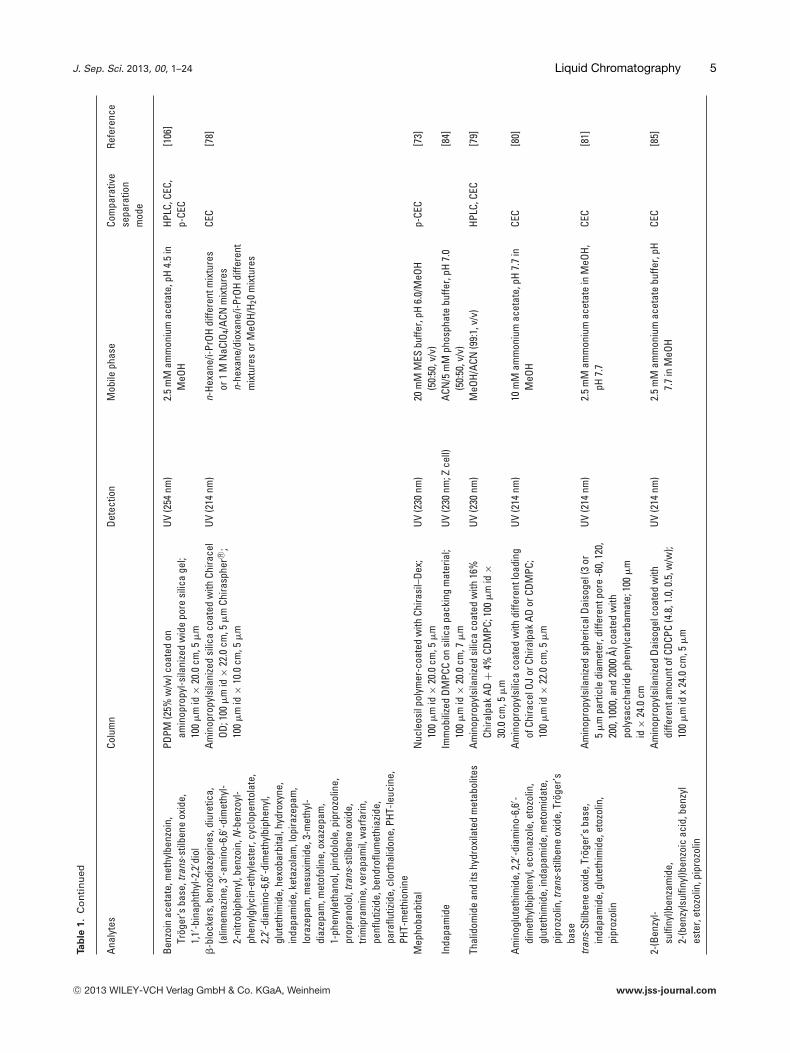
J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 5Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
Benz
oin
acet
ate,
met
hylb
enzo
in,
Trog
er’s
base
,tra
ns-s
tilbe
neox
ide,
1,1′
-bin
apht
hyl-2
,2′ di
ol
PDPM
(25%
w/w
)coa
ted
onam
inop
ropy
l-sila
nize
dw
ide
pore
silic
age
l;10
0�
mid
×20
.0cm
,5�
m
UV(2
54nm
)2.
5m
Mam
mon
ium
acet
ate,
pH4.
5in
MeO
HHP
LC,C
EC,
p-CE
C[1
06]
�-b
lock
ers,
benz
odia
zepi
nes,
diur
etic
a,(a
limem
azin
e,3′
-am
ino-
6,6′
-dim
ethy
l-2-
nitro
biph
enyl
,ben
zoin
,N-b
enzo
yl-
phen
ylgl
ycin
-eth
yles
ter,
cycl
open
tola
te,
2,2′
-dia
min
o-6,
6′-d
imet
hylb
iphe
nyl,
glut
ethi
mid
e,he
xoba
rbita
l,hy
drox
yne,
inda
pam
ide,
keta
zola
m,l
opira
zepa
m,
lora
zepa
m,m
esux
imid
e,3-
met
hyl-
diaz
epam
,met
ofol
ine,
oxaz
epam
,1-
phen
ylet
hano
l,pi
ndol
ole,
pipr
ozol
ine,
prop
rano
lol,
trans
-stil
bene
oxid
e,tri
mip
ram
ine,
vera
pam
il,w
arfa
rin,
penfl
utizi
de,b
endr
oflum
ethi
azid
e,pa
raflu
tizid
e,cl
orth
alid
one,
PHT-
leuc
ine,
PHT-
met
hion
ine
Amin
opro
pyls
ilani
zed
silic
aco
ated
with
Chira
cel
OD;1
00�
mid
×22
.0cm
,5�
mCh
irasp
herR©
;10
0�
mid
×10
.0cm
,5�
m
UV(2
14nm
)n-
Hexa
ne/i-
PrOH
diffe
rent
mix
ture
sor
1M
NaC
lO4/
ACN
mix
ture
sn-
hexa
ne/d
ioxa
ne/i-
PrOH
diffe
rent
mix
ture
sor
MeO
H/H 2
0m
ixtu
res
CEC
[78]
Mep
hoba
rbita
lN
ucle
osil
poly
mer
-coa
ted
with
Chira
sil–
Dex;
100
�m
id×
20.0
cm,5
�m
UV(2
30nm
)20
mM
MES
buffe
r,pH
6.0/
MeO
H(5
0:50
,v/v
)p-
CEC
[73]
Inda
pam
ide
Imm
obili
zed
DMPC
Con
silic
apa
ckin
gm
ater
ial;
100
�m
id×
20.0
cm,7
�m
UV(2
30nm
;Zce
ll)AC
N/5
mM
phos
phat
ebu
ffer,
pH7.
0(5
0:50
,v/v
)[8
4]
Thal
idom
ide
and
itshy
drox
ilate
dm
etab
olite
sAm
inop
ropy
lsila
nize
dsi
lica
coat
edw
ith16
%Ch
iralp
akAD
+4%
CDM
PC;1
00�
mid
×30
.0cm
,5�
m
UV(2
30nm
)M
eOH/
ACN
(99:
1,v/
v)HP
LC,C
EC[7
9]
Amin
oglu
teth
imid
e,2,
2′-d
iam
ino-
6,6′
-di
met
hylb
iphe
nyl,
econ
azol
e,et
ozol
in,
glut
ethi
mid
e,in
dapa
mid
e,m
etom
idat
e,pi
proz
olin
,tra
ns-s
tilbe
neox
ide,
Trog
er’s
base
Amin
opro
pyls
ilica
coat
edw
ithdi
ffere
ntlo
adin
gof
Chira
celO
Jor
Chira
lpak
ADor
CDM
PC;
100
�m
id×
22.0
cm,5
�m
UV(2
14nm
)10
mM
amm
oniu
mac
etat
e,pH
7.7
inM
eOH
CEC
[80]
trans
-Stil
bene
oxid
e,Tr
oger
’sba
se,
inda
pam
ide,
glut
ethi
mid
e,et
ozol
in,
pipr
ozol
in
Amin
opro
pyls
ilani
zed
sphe
rical
Dais
ogel
(3or
5�
mpa
rticl
edi
amet
er,d
iffer
entp
ore
-60,
120,
200,
1000
,and
2000
A)co
ated
with
poly
sacc
harid
eph
enyl
carb
amat
e;10
0�
mid
×24
.0cm
UV(2
14nm
)2.
5m
Mam
mon
ium
acet
ate
inM
eOH,
pH7.
7CE
C[8
1]
2-(B
enzy
l-su
lfiny
l)ben
zam
ide,
2-(b
enzy
lsul
finyl
)ben
zoic
acid
,ben
zyl
este
r,et
ozol
in,p
ipro
zolin
Amin
opro
pyls
ilani
zed
Dais
ogel
coat
edw
ithdi
ffere
ntam
ount
ofCD
CPC
(4.8
,1.0
,0.5
,w/w
);10
0�
mid
x24
.0cm
,5�
m
UV(2
14nm
)2.
5m
Mam
mon
ium
acet
ate
buffe
r,pH
7.7
inM
eOH
CEC
[85]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
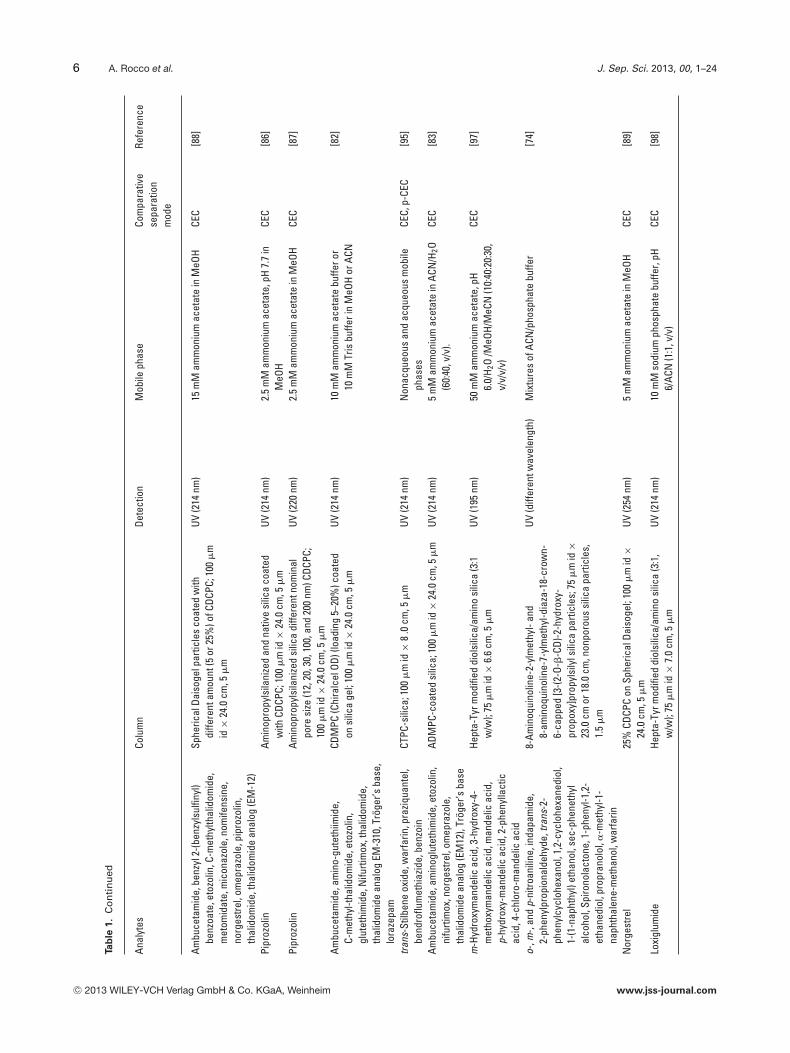
6 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
Ambu
ceta
mid
e,be
nzyl
2-(b
enzy
lsul
finyl
)be
nzoa
te,e
tozo
lin,C
-met
hylth
alid
omid
e,m
etom
idat
e,m
icon
azol
e,no
mife
nsin
e,no
rges
trel,
omep
razo
le,p
ipro
zolin
,th
alid
omid
e,th
alid
omid
ean
alog
(EM
-12)
Sphe
rical
Dais
ogel
parti
cles
coat
edw
ithdi
ffere
ntam
ount
(5or
25%
)ofC
DCPC
;100
�m
id×
24.0
cm,5
�m
UV(2
14nm
)15
mM
amm
oniu
mac
etat
ein
MeO
HCE
C[8
8]
Pipr
ozol
inAm
inop
ropy
lsila
nize
dan
dna
tive
silic
aco
ated
with
CDCP
C;10
0�
mid
×24
.0cm
,5�
mUV
(214
nm)
2.5
mM
amm
oniu
mac
etat
e,pH
7.7
inM
eOH
CEC
[86]
Pipr
ozol
inAm
inop
ropy
lsila
nize
dsi
lica
diffe
rent
nom
inal
pore
size
(12,
20,3
0,10
0,an
d20
0nm
)CDC
PC;
100
�m
id×
24.0
cm,5
�m
UV(2
20nm
)2.
5m
Mam
mon
ium
acet
ate
inM
eOH
CEC
[87]
Ambu
ceta
mid
e,am
ino-
gute
thiim
ide,
C-m
ethy
l-tha
lidom
ide,
etoz
olin
,gl
utet
him
ide,
Nifu
rtim
ox,t
halid
omid
e,th
alid
omid
ean
alog
EM-3
10,T
roge
r’sba
se,
lora
zepa
m
CDM
PC(C
hira
lcel
OD)(
load
ing
5–20
%)c
oate
don
silic
age
l;10
0�
mid
×24
.0cm
,5�
mUV
(214
nm)
10m
Mam
mon
ium
acet
ate
buffe
ror
10m
MTr
isbu
fferi
nM
eOH
orAC
N[8
2]
trans
-Stil
bene
oxid
e,w
arfa
rin,p
raziq
uant
el,
bend
roflu
met
hiaz
ide,
benz
oin
CTPC
-sili
ca;1
00�
mid
×8
.0cm
,5�
mUV
(214
nm)
Non
acqu
eous
and
acqu
eous
mob
ileph
ases
CEC,
p-CE
C[9
5]
Ambu
ceta
mid
e,am
inog
lute
thim
ide,
etoz
olin
,ni
furti
mox
,nor
gest
rel,
omep
razo
le,
thal
idom
ide
anal
og(E
M12
),Tr
oger
’sba
se
ADM
PC-c
oate
dsi
lica;
100
�m
id×
24.0
cm,5
�m
UV(2
14nm
)5
mM
amm
oniu
mac
etat
ein
ACN
/H2O
(60:
40,v
/v).
CEC
[83]
m-H
ydro
xym
ande
licac
id,3
-hyd
roxy
-4-
met
hoxy
man
delic
acid
,man
delic
acid
,p-
hydr
oxy-
man
delic
acid
,2-p
heny
llact
icac
id,4
-chl
oro-
man
delic
acid
Hept
a-Ty
rmod
ified
diol
silic
a/am
ino
silic
a(3
:1w
/w);
75�
mid
×6.
6cm
,5�
mUV
(195
nm)
50m
Mam
mon
ium
acet
ate,
pH6.
0/H 2
O/M
eOH/
MeC
N(1
0:40
:20:
30,
v/v/
v/v)
CEC
[97]
o-,m
-,an
dp-
nitro
anili
ne,i
ndap
amid
e,2-
phen
ylpr
opio
nald
ehyd
e,tra
ns-2
-ph
enyl
cycl
ohex
anol
,1,2
-cyc
lohe
xane
diol
,1-
(1-n
apht
hyl)
etha
nol,
sec-
phen
ethy
lal
coho
l,Sp
irono
lact
one,
1-ph
enyl
-1,2
-et
hane
diol
,pro
pran
olol
,�-m
ethy
l-1-
naph
thal
ene-
met
hano
l,w
arfa
rin
8-Am
inoq
uino
line-
2-yl
met
hyl-
and
8-am
inoq
uino
line-
7-yl
met
hyl-d
iaza
-18-
crow
n-6-
capp
ed[3
-(2-O
-�-C
D)-2
-hyd
roxy
-pr
opox
y]pr
opyl
sily
lsili
capa
rticl
es;7
5�
mid
×23
.0cm
or18
.0cm
,non
poro
ussi
lica
parti
cles
,1.
5�
m
UV(d
iffer
entw
avel
engt
h)M
ixtu
res
ofAC
N/p
hosp
hate
buffe
r[7
4]
Nor
gest
rel
25%
CDCP
Con
Sphe
rical
Dais
ogel
;100
�m
id×
24.0
cm,5
�m
UV(2
54nm
)5
mM
amm
oniu
mac
etat
ein
MeO
HCE
C[8
9]
Loxi
glum
ide
Hept
a-Ty
rmod
ified
diol
silic
a/am
ino
silic
a(3
:1,
w/w
);75
�m
id×
7.0
cm,5
�m
UV(2
14nm
)10
mM
sodi
umph
osph
ate
buffe
r,pH
6/AC
N(1
:1,v
/v)
CEC
[98]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
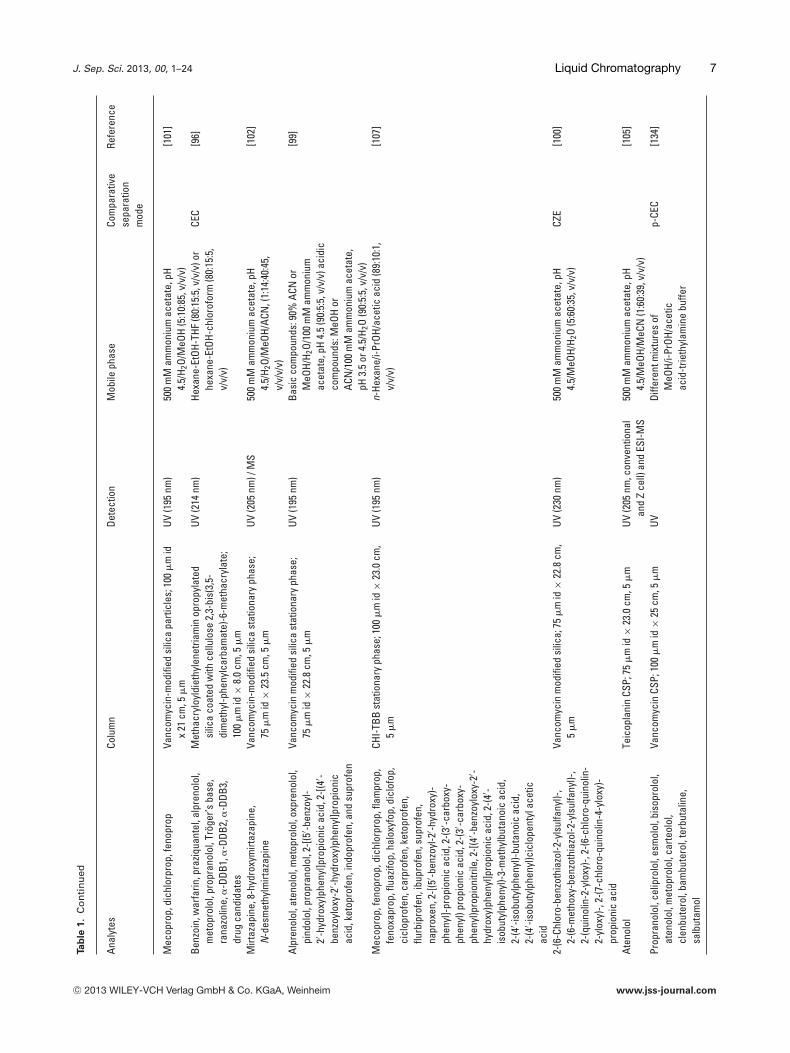
J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 7Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
Mec
opro
p,di
chlo
rpro
p,fe
nopr
opVa
ncom
ycin
-mod
ified
silic
apa
rticl
es;1
00�
mid
x21
cm,5
�m
UV(1
95nm
)50
0m
Mam
mon
ium
acet
ate,
pH4.
5/H 2
O/M
eOH
(5:1
0:85
,v/v
/v)
[101
]
Benz
oin,
war
farin
,pra
ziqua
ntel
,alp
reno
lol,
met
opro
lol,
prop
rano
lol,
Trog
er’s
base
,ra
nazo
line,
�-D
DB1,
�-D
DB2,
�-D
DB3,
drug
cand
idat
es
Met
hacr
yloy
ldie
thyl
enet
riam
inop
ropy
late
dsi
lica
coat
edw
ithce
llulo
se2,
3-bi
s(3,
5-di
met
hyl-p
heny
lcar
bam
ate)
-6-m
etha
cryl
ate;
100
�m
id×
8.0
cm,5
�m
UV(2
14nm
)He
xane
-EtO
H-TH
F(8
0:15
:5,v
/v/v
)or
hexa
ne-E
tOH-
chlo
rofo
rm(8
0:15
:5,
v/v/
v)
CEC
[96]
Mirt
azap
ine,
8-hy
drox
ymirt
azap
ine,
N-d
esm
ethy
lmirt
azap
ine
Vanc
omyc
in-m
odifi
edsi
lica
stat
iona
ryph
ase;
75�
mid
×23
.5cm
,5�
mUV
(205
nm)/
MS
500
mM
amm
oniu
mac
etat
e,pH
4.5/
H 2O/
MeO
H/AC
N,(
1:14
:40:
45,
v/v/
v/v)
[102
]
Alpr
enol
ol,a
teno
lol,
met
opro
lol,
oxpr
enol
ol,
pind
olol
,pro
pran
olol
,2-[(
5′-b
enzo
yl-
2′-h
ydro
xy)p
heny
l]pro
pion
icac
id,2
-[(4′
-be
nzoy
loxy
-2′ -h
ydro
xy)p
heny
l]pro
pion
icac
id,k
etop
rofe
n,in
dopr
ofen
,and
supr
ofen
Vanc
omyc
inm
odifi
edsi
lica
stat
iona
ryph
ase;
75�
mid
×22
.8cm
,5�
mUV
(195
nm)
Basi
cco
mpo
unds
:90%
ACN
orM
eOH/
H 2O/
100
mM
amm
oniu
mac
etat
e,pH
4.5
(90:
5:5,
v/v/
v)ac
idic
com
poun
ds:M
eOH
orAC
N/1
00m
Mam
mon
ium
acet
ate,
pH3.
5or
4.5/
H 2O
(90:
5:5,
v/v/
v)
[99]
Mec
opro
p,fe
nopr
op,d
ichl
orpr
op,fl
ampr
op,
feno
xapr
op,fl
uazif
op,h
alox
yfop
,dic
lofo
p,ci
clop
rofe
n,ca
rpro
fen,
keto
prof
en,
flurb
ipro
fen,
ibup
rofe
n,su
prof
en,
napr
oxen
,2-[(
5′-b
enzo
yl-2
′ -hyd
roxy
)-ph
enyl
]-pro
pion
icac
id,2
-(3′ -c
arbo
xy-
phen
yl)p
ropi
onic
acid
,2-(3
′ -car
boxy
-ph
enyl
)pro
pion
itrile
,2-[(
4′-b
enzo
ylox
y-2′
-hy
drox
y)ph
enyl
]pro
pion
icac
id,2
-(4′ -
isob
utyl
phen
yl)-3
-met
hylb
utan
oic
acid
,2-
(4′ -is
obut
ylph
enyl
)-but
anoi
cac
id,
2-(4
′ -isob
utyl
phen
yl)c
iclo
pent
ylac
etic
acid
CHI-T
BBst
atio
nary
phas
e;10
0�
mid
×23
.0cm
,5
�m
UV(1
95nm
)n-
Hexa
ne/i-
PrOH
/ace
ticac
id(8
9:10
:1,
v/v/
v)[1
07]
2-(6
-Chl
oro-
benz
othi
azol
-2-y
lsul
fany
l)-,
2-(6
-met
hoxy
-ben
zoth
iazo
l-2-y
lsul
fany
l)-,
2-(q
uino
lin-2
-ylo
xy)-,
2-(6
-chl
oro-
quin
olin
-2-
ylox
y)-,
2-(7
-chl
oro-
quin
olin
-4-y
loxy
)-pr
opio
nic
acid
Vanc
omyc
inm
odifi
edsi
lica;
75�
mid
×22
.8cm
,5
�m
UV(2
30nm
)50
0m
Mam
mon
ium
acet
ate,
pH4.
5/M
eOH/
H 2O
(5:6
0:35
,v/v
/v)
CZE
[100
]
Aten
olol
Teic
opla
nin
CSP;
75�
mid
×23
.0cm
,5�
mUV
(205
nm,c
onve
ntio
nal
and
Zce
ll)an
dES
I-MS
500
mM
amm
oniu
mac
etat
e,pH
4.5/
MeO
H/M
eCN
(1:6
0:39
,v/v
/v)
[105
]
Prop
rano
lol,
celip
rolo
l,es
mol
ol,b
isop
rolo
l,at
enol
ol,m
etop
rolo
l,ca
rteol
ol,
clen
bute
rol,
bam
bute
rol,
terb
utal
ine,
salb
utam
ol
Vanc
omyc
inCS
P;10
0�
mid
×25
cm,5
�m
UVDi
ffere
ntm
ixtu
res
ofM
eOH/
i-PrO
H/ac
etic
acid
-trie
thyl
amin
ebu
ffer
p-CE
C[1
34]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
8 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
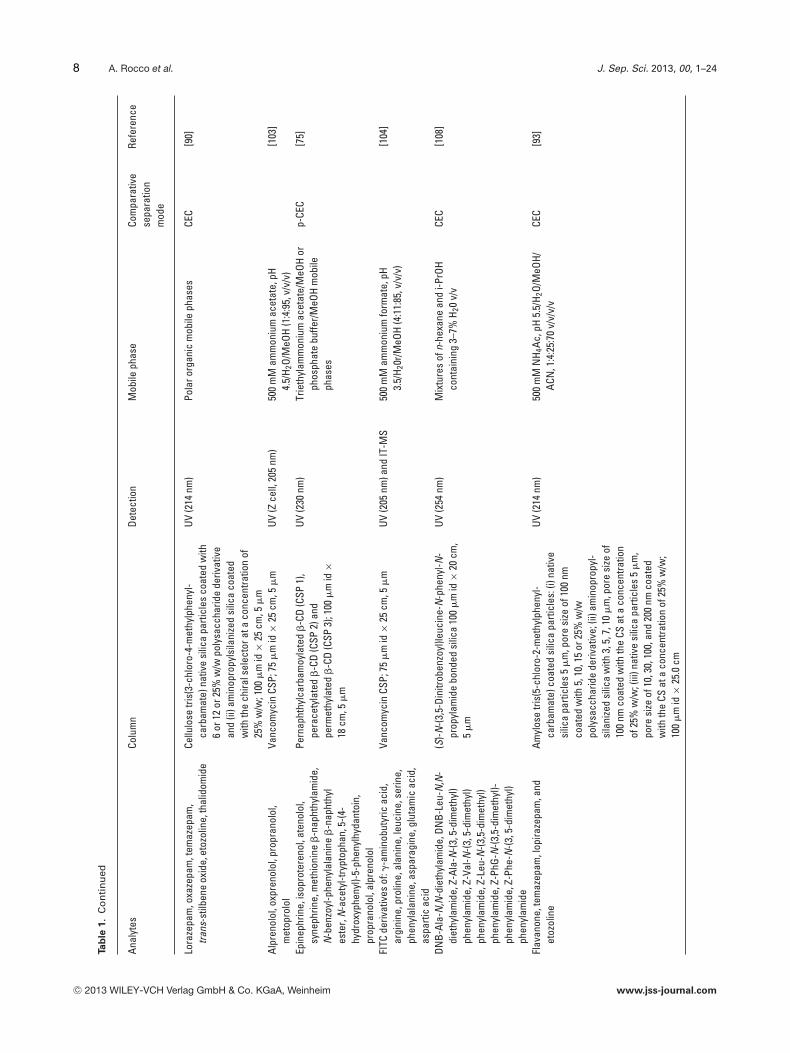
Lora
zepa
m,o
xaze
pam
,tem
azep
am,
trans
-stil
bene
oxid
e,et
ozol
ine,
thal
idom
ide
Cellu
lose
tris(
3-ch
loro
-4-m
ethy
lphe
nyl-
carb
amat
e)na
tive
silic
apa
rticl
esco
ated
with
6or
12or
25%
w/w
poly
sacc
harid
ede
rivat
ive
and
(ii)a
min
opro
pyls
ilani
zed
silic
aco
ated
with
the
chira
lsel
ecto
rata
conc
entra
tion
of25
%w
/w;1
00�
mid
×25
cm,5
�m
UV(2
14nm
)Po
laro
rgan
icm
obile
phas
esCE
C[9
0]
Alpr
enol
ol,o
xpre
nolo
l,pr
opra
nolo
l,m
etop
rolo
lVa
ncom
ycin
CSP;
75�
mid
×25
cm,5
�m
UV(Z
cell,
205
nm)
500
mM
amm
oniu
mac
etat
e,pH
4.5/
H 2O/
MeO
H(1
:4:9
5,v/
v/v)
[103
]
Epin
ephr
ine,
isop
rote
reno
l,at
enol
ol,
syne
phrin
e,m
ethi
onin
e�
-nap
hthy
lam
ide,
N-b
enzo
yl-p
heny
lala
nine
�-n
apht
hyl
este
r,N
-ace
tyl-t
rypt
opha
n,5-
(4-
hydr
oxyp
heny
l)-5-
phen
ylhy
dant
oin,
prop
rano
lol,
alpr
enol
ol
Pern
apht
hylc
arba
moy
late
d�
-CD
(CSP
1),
pera
cety
late
d�
-CD
(CSP
2)an
dpe
rmet
hyla
ted
�-C
D(C
SP3)
;100
�m
id×
18cm
,5�
m
UV(2
30nm
)Tr
ieth
ylam
mon
ium
acet
ate/
MeO
Hor
phos
phat
ebu
ffer/M
eOH
mob
ileph
ases
p-CE
C[7
5]
FITC
deriv
ativ
esof
:�-a
min
obut
yric
acid
,ar
gini
ne,p
rolin
e,al
anin
e,le
ucin
e,se
rine,
phen
ylal
anin
e,as
para
gine
,glu
tam
icac
id,
aspa
rtic
acid
Vanc
omyc
inCS
P;75
�m
id×
25cm
,5�
mUV
(205
nm)a
ndIT
-MS
500
mM
amm
oniu
mfo
rmat
e,pH
3.5/
H 20r
/MeO
H(4
:11:
85,v
/v/v
)[1
04]
DNB-
Ala-
N,N
-die
thyl
amid
e,DN
B-Le
u-N
,N-
diet
hyla
mid
e,Z-
Ala-
N-(3
,5-d
imet
hyl)
phen
ylam
ide,
Z-Va
l-N-(3
,5-d
imet
hyl)
phen
ylam
ide,
Z-Le
u-N
-(3,5
-dim
ethy
l)ph
enyl
amid
e,Z-
PhG-
N-(3
,5-d
imet
hyl)-
phen
ylam
ide,
Z-Ph
e-N
-(3,5
-dim
ethy
l)ph
enyl
amid
e
(S)-N
-(3,5
-Din
itrob
enzo
yl)le
ucin
e-N
-phe
nyl-N
-pr
opyl
amid
ebo
nded
silic
a10
0�
mid
×20
cm,
5�
m
UV(2
54nm
)M
ixtu
res
ofn-
hexa
nean
di-P
rOH
cont
aini
ng3–
7%H 2
0v/
vCE
C[1
08]
Flav
anon
e,te
maz
epam
,lop
iraze
pam
,and
etoz
olin
eAm
ylos
etri
s(5-
chlo
ro-2
-met
hylp
heny
l-ca
rbam
ate)
coat
edsi
lica
parti
cles
:(i)
nativ
esi
lica
parti
cles
5�
m,p
ore
size
of10
0nm
coat
edw
ith5,
10,1
5or
25%
w/w
poly
sacc
harid
ede
rivat
ive;
(ii)a
min
opro
pyl-
sila
nize
dsi
lica
with
3,5,
7,10
�m
,por
esi
zeof
100
nmco
ated
with
the
CSat
aco
ncen
tratio
nof
25%
w/w
;(iii
)nat
ive
silic
apa
rticl
es5
�m
,po
resi
zeof
10,3
0,10
0,an
d20
0nm
coat
edw
ithth
eCS
ata
conc
entra
tion
of25
%w
/w;
100
�m
id×
25.0
cm
UV(2
14nm
)50
0m
MN
H 4Ac
,pH
5.5/
H 2O/
MeO
H/AC
N,1
:4:2
5:70
v/v/
v/v
CEC
[93]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 9
Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
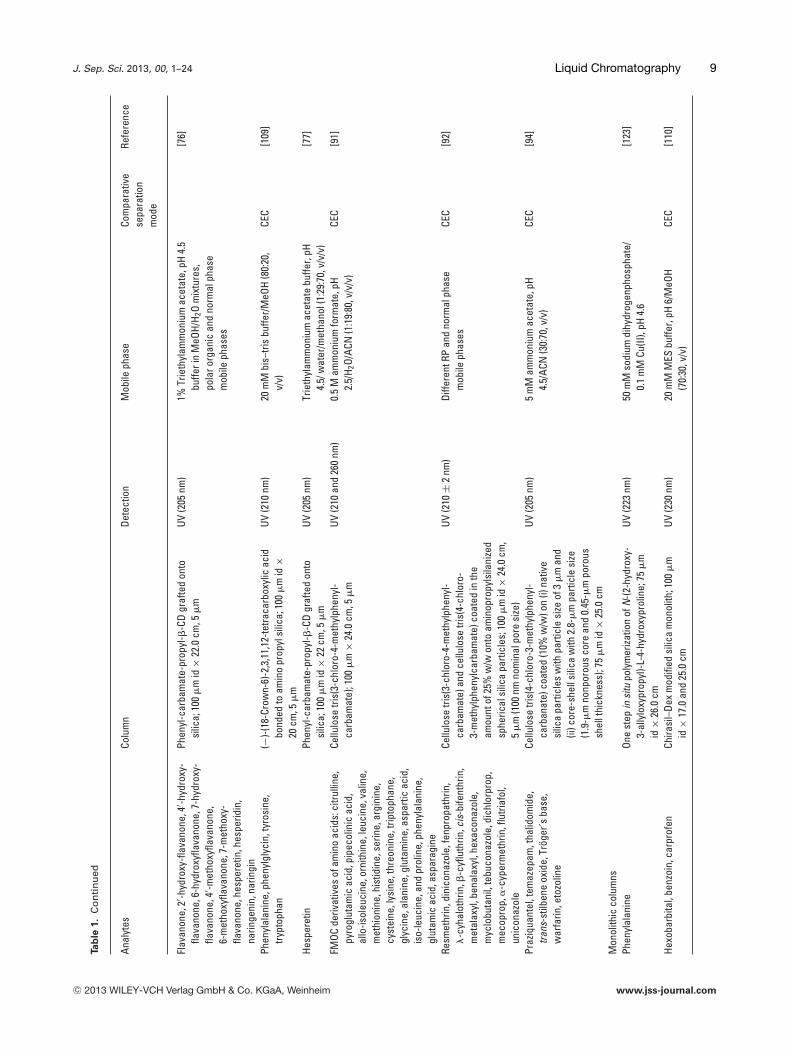
Flav
anon
e,2′
-hyd
roxy
-flav
anon
e,4′
-hyd
roxy
-fla
vano
ne,6
-hyd
roxy
flava
none
,7-h
ydro
xy-
flava
none
,4′ -m
etho
xyfla
vano
ne,
6-m
etho
xyfla
vano
ne,7
-met
hoxy
-fla
vano
ne,h
espe
retin
,hes
perid
in,
narin
geni
n,na
ringi
n
Phen
yl-c
arba
mat
e-pr
opyl
-�-C
Dgr
afte
don
tosi
lica;
100
�m
id×
22.0
cm,5
�m
UV(2
05nm
)1%
Trie
thyl
amm
oniu
mac
etat
e,pH
4.5
buffe
rin
MeO
H/H 2
Om
ixtu
res,
pola
rorg
anic
and
norm
alph
ase
mob
ileph
ases
[76]
Phen
ylal
anin
e,ph
enyl
glyc
in,t
yros
ine,
trypt
opha
n(−
)-(18
-Cro
wn-
6)-2
,3,1
1,12
-tetra
carb
oxyl
icac
idbo
nded
toam
ino
prop
ylsi
lica;
100
�m
id×
20cm
,5�
m
UV(2
10nm
)20
mM
bis–
tris
buffe
r/MeO
H(8
0:20
,v/
v)CE
C[1
09]
Hesp
eret
inPh
enyl
-car
bam
ate-
prop
yl-�
-CD
graf
ted
onto
silic
a;10
0�
mid
×22
cm,5
�m
UV(2
05nm
)Tr
ieth
ylam
mon
ium
acet
ate
buffe
r,pH
4.5/
wat
er/m
etha
nol(
1:29
:70,
v/v/
v)[7
7]
FMOC
deriv
ativ
esof
amin
oac
ids:
citru
lline
,py
rogl
utam
icac
id,p
ipec
olin
icac
id,
allo
-isol
euci
ne,o
rnith
ine,
leuc
ine,
valin
e,m
ethi
onin
e,hi
stid
ine,
serin
e,ar
gini
ne,
cyst
eine
,lys
ine,
thre
onin
e,tri
ptop
hane
,gl
ycin
e,al
anin
e,gl
utam
ine,
aspa
rtic
acid
,is
o-le
ucin
e,an
dpr
olin
e,ph
enyl
alan
ine,
glut
amic
acid
,asp
arag
ine
Cellu
lose
tris(
3-ch
loro
-4-m
ethy
lphe
nyl-
carb
amat
e);1
00�
m×
24.0
cm,5
�m
UV(2
10an
d26
0nm
)0.
5M
amm
oniu
mfo
rmat
e,pH
2.5/
H 2O/
ACN
(1:1
9:80
,v/v
/v)
CEC
[91]
Resm
ethr
in,d
inic
onaz
ole,
fenp
ropa
thrin
,�
-cyh
alot
hrin
,�-c
yflut
hrin
,cis
-bife
nthr
in,
met
alax
yl,b
enal
axyl
,hex
acon
azol
e,m
yclo
buta
nil,
tebu
cona
zole
,dic
hlor
prop
,m
ecop
rop,
�-c
yper
met
hrin
,flut
riafo
l,un
icon
azol
e
Cellu
lose
tris(
3-ch
loro
-4-m
ethy
lphe
nyl-
carb
amat
e)an
dce
llulo
setri
s(4-
chlo
ro-
3-m
ethy
lphe
nylc
arba
mat
e)co
ated
inth
eam
ount
of25
%w
/won
toam
inop
ropy
lsila
nize
dsp
heric
alsi
lica
parti
cles
;100
�m
id×
24.0
cm,
5�
m(1
00nm
nom
inal
pore
size
)
UV(2
10±
2nm
)Di
ffere
ntRP
and
norm
alph
ase
mob
ileph
ases
CEC
[92]
Praz
iqua
ntel
,tem
azep
am,t
halid
omid
e,tra
ns-s
tilbe
neox
ide,
Trog
er´s
base
,w
arfa
rin,e
tozo
line
Cellu
lose
tris(
4-ch
loro
-3-m
ethy
lphe
nyl-
carb
anat
e)co
ated
(10%
w/w
)on
(i)na
tive
silic
apa
rticl
esw
ithpa
rticl
esi
zeof
3�
man
d(ii
)cor
e-sh
ells
ilica
with
2.8-
�m
parti
cle
size
(1.9
-�m
nonp
orou
sco
rean
d0.
45-�
mpo
rous
shel
lthi
ckne
ss);
75�
mid
×25
.0cm
UV(2
05nm
)5
mM
amm
oniu
mac
etat
e,pH
4.5/
ACN
(30:
70,v
/v)
CEC
[94]
Mon
olith
icco
lum
nsPh
enyl
alan
ine
One
step
insi
tupo
lym
eriza
tion
ofN
-(2-h
ydro
xy-
3-al
lylo
xypr
opyl
)-L-4
-hyd
roxy
prol
ine;
75�
mid
×26
.0cm
UV(2
23nm
)50
mM
sodi
umdi
hydr
ogen
phos
phat
e/0.
1m
MCu
(II),
pH4.
6[1
23]
Hexo
barb
ital,
benz
oin,
carp
rofe
nCh
irasi
l–De
xm
odifi
edsi
lica
mon
olith
;100
�m
id×
17.0
and
25.0
cmUV
(230
nm)
20m
MM
ESbu
ffer,
pH6/
MeO
H(7
0:30
,v/v
)CE
C[1
10]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
10 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
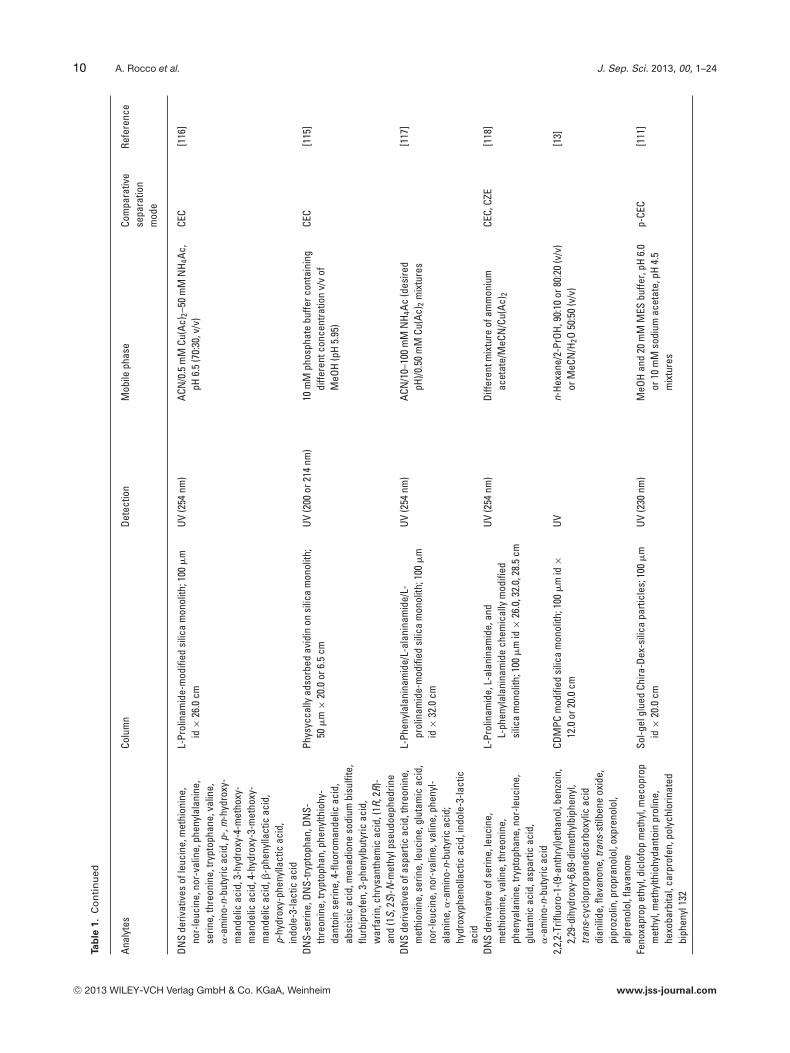
DNS
deriv
ativ
esof
leuc
ine,
met
hion
ine,
nor-
leuc
ine,
nor-
valin
e,ph
enyl
alan
ine,
serin
e,th
reon
ine,
trypt
opha
ne,v
alin
e,�
-am
ino-
n-bu
tyric
acid
,p-,
m-h
ydro
xy-
man
delic
acid
,3-h
ydro
xy-4
-met
hoxy
-m
ande
licac
id,4
-hyd
roxy
-3-m
etho
xy-
man
delic
acid
,�-p
heny
llact
icac
id,
p-hy
drox
y-ph
enyl
lact
icac
id,
indo
le-3
-lact
icac
id
L-Pr
olin
amid
e-m
odifi
edsi
lica
mon
olith
;100
�m
id×
26.0
cmUV
(254
nm)
ACN
/0.5
mM
Cu(A
c)2–
50m
MN
H 4Ac
,pH
6.5
(70:
30,v
/v)
CEC
[116
]
DNS-
serin
e,DN
S-try
ptop
han,
DNS-
thre
onin
e,try
ptop
han,
phen
ylth
iohy
-da
ntoi
nse
rine,
4-flu
orom
ande
licac
id,
absc
isic
acid
,men
adio
neso
dium
bisu
lfite
,flu
rbip
rofe
n,3-
phen
ylbu
tyric
acid
,w
arfa
rin,c
hrys
anth
emic
acid
,(1R
,2R)
-an
d(1
S,2S
)-N-m
ethy
lpse
udoe
phed
rine
Phys
ycca
llyad
sorb
edav
idin
onsi
lica
mon
olith
;50
�m
×20
.0or
6.5
cmUV
(200
or21
4nm
)10
mM
phos
phat
ebu
fferc
onta
inin
gdi
ffere
ntco
ncen
tratio
nv/
vof
MeO
H(p
H5.
95)
CEC
[115
]
DNS
deriv
ativ
esof
aspa
rtic
acid
,thr
eoni
ne,
met
hion
ine,
serin
e,le
ucin
e,gl
utam
icac
id,
nor-
leuc
ine,
nor-
valin
e,va
line,
phen
yl-
alan
ine,
�-a
min
o-n-
buty
ricac
id;
hydr
oxyp
heno
llact
icac
id,i
ndol
e-3-
lact
icac
id
L-Ph
enyl
alan
inam
ide/
L-al
anin
amid
e/L-
prol
inam
ide-
mod
ified
silic
am
onol
ith;1
00�
mid
×32
.0cm
UV(2
54nm
)AC
N/1
0–10
0m
MN
H 4Ac
(des
ired
pH)/0
.50
mM
Cu(A
c)2
mix
ture
s[1
17]
DNS
deriv
ativ
eof
serin
e,le
ucin
e,m
ethi
onin
e,va
line,
thre
onin
e,ph
enya
lani
ne,t
rypt
opha
ne,n
or-le
ucin
e,gl
utam
icac
id,a
spar
ticac
id,
�-a
min
o-n-
buty
ricac
id
L-Pr
olin
amid
e,L-
alan
inam
ide,
and
L-ph
enyl
alan
inam
ide
chem
ical
lym
odifi
edsi
lica
mon
olith
;100
�m
id×
26.0
,32.
0,28
.5cm
UV(2
54nm
)Di
ffere
ntm
ixtu
reof
amm
oniu
mac
etat
e/M
eCN
/Cu(
Ac) 2
CEC,
CZE
[118
]
2,2,
2-Tr
ifluo
ro-1
-(9-a
nthr
yl)e
than
ol,b
enzo
in,
2,29
-dih
ydro
xy-6
,69-
dim
ethy
lbip
heny
l,tra
ns-c
yclo
prop
aned
icar
boxy
licac
iddi
anili
de,fl
avan
one,
trans
-stil
bene
oxid
e,pi
proz
olin
,pro
pran
olol
,oxp
reno
lol,
alpr
enol
ol,fl
avan
one
CDM
PCm
odifi
edsi
lica
mon
olith
;100
�m
id×
12.0
or20
.0cm
UVn-
Hexa
ne/2
-PrO
H,90
:10
or80
:20
(v/v
)or
MeC
N/H
2O50
:50
(v/v
)[1
3]
Feno
xapr
opet
hyl,
dicl
ofop
met
hyl,
mec
opro
pm
ethy
l,m
ethy
lthio
hyda
ntoi
npr
olin
e,he
xoba
rbita
l,ca
rpro
fen,
poly
chlo
rinat
edbi
phen
yl13
2
Sol-g
elgl
ued
Chira
-Dex
-sili
capa
rticl
es;1
00�
mid
×20
.0cm
UV(2
30nm
)M
eOH
and
20m
MM
ESbu
ffer,
pH6.
0or
10m
Mso
dium
acet
ate,
pH4.
5m
ixtu
res
p-CE
C[1
11]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 11
Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
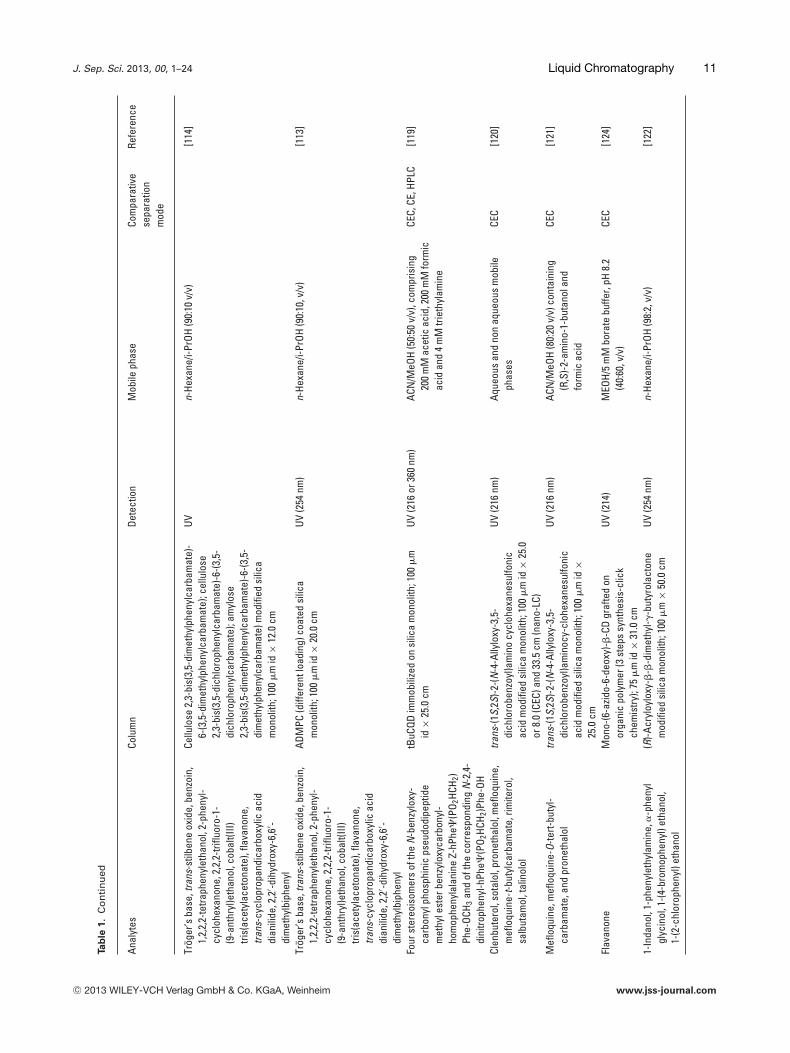
Trog
er’s
base
,tra
ns-s
tilbe
neox
ide,
benz
oin,
1,2,
2,2-
tetra
phen
ylet
hano
l,2-
phen
yl-
cycl
ohex
anon
e,2,
2,2-
triflu
oro-
1-(9
-ant
hryl
)eth
anol
,cob
alt(I
II)tri
s(ac
etyl
acet
onat
e),fl
avan
one,
trans
-cyc
lopr
opan
dica
rbox
ylic
acid
dian
ilide
,2,2
′ -dih
ydro
xy-6
,6′ -
dim
ethy
lbip
heny
l
Cellu
lose
2,3-
bis(
3,5-
dim
ethy
lphe
nylc
arba
mat
e)-
6-(3
,5-d
imet
hylp
heny
lcar
bam
ate)
;cel
lulo
se2,
3-bi
s(3,
5-di
chlo
roph
enyl
carb
amat
e)-6
-(3,5
-di
chlo
roph
enyl
carb
amat
e);a
myl
ose
2,3-
bis(
3,5-
dim
ethy
lphe
nylc
arba
mat
e)-6
-(3,5
-di
met
hylp
heny
lcar
bam
ate)
mod
ified
silic
am
onol
ith;1
00�
mid
×12
.0cm
UVn-
Hexa
ne/i-
PrOH
(90:
10v/
v)[1
14]
Trog
er’s
base
,tra
ns-s
tilbe
neox
ide,
benz
oin,
1,2,
2,2-
tetra
phen
ylet
hano
l,2-
phen
yl-
cycl
ohex
anon
e,2,
2,2-
triflu
oro-
1-(9
-ant
hryl
)eth
anol
,cob
alt(I
II)tri
s(ac
etyl
acet
onat
e),fl
avan
one,
trans
-cyc
lopr
opan
dica
rbox
ylic
acid
dian
ilide
,2,2
′ -dih
ydro
xy-6
,6′ -
dim
ethy
lbip
heny
l
ADM
PC(d
iffer
entl
oadi
ng)c
oate
dsi
lica
mon
olith
;100
�m
id×
20.0
cmUV
(254
nm)
n-He
xane
/i-Pr
OH(9
0:10
,v/v
)[1
13]
Four
ster
eois
omer
sof
the
N-b
enzy
loxy
-ca
rbon
ylph
osph
inic
pseu
dodi
pept
ide
met
hyle
ster
benz
ylox
ycar
bony
l-ho
mop
heny
lala
nine
Z-hP
he�
(PO 2
HCH 2
)Ph
e-OC
H 3an
dof
the
corr
espo
ndin
gN
-2,4
-di
nitro
phen
yl-h
Phe�
(PO 2
HCH 2
)Phe
-OH
tBuC
QDim
mob
ilize
don
silic
am
onol
ith;1
00�
mid
×25
.0cm
UV(2
16or
360
nm)
ACN
/MeO
H(5
0:50
v/v)
,com
pris
ing
200
mM
acet
icac
id,2
00m
Mfo
rmic
acid
and
4m
Mtri
ethy
lam
ine
CEC,
CE,H
PLC
[119
]
Clen
bute
rol,
sota
lol,
pron
etha
lol,
mefl
oqui
ne,
mefl
oqui
ne-t-
buty
lcar
bam
ate,
rimite
rol,
salb
utam
ol,t
alin
olol
trans
-(1S,
2S)-2
-(N-4
-Ally
loxy
-3,5
-di
chlo
robe
nzoy
l)am
ino
cycl
ohex
anes
ulfo
nic
acid
mod
ified
silic
am
onol
ith;1
00�
mid
×25
.0or
8.0
(CEC
)and
33.5
cm(n
ano-
LC)
UV(2
16nm
)Aq
ueou
san
dno
naq
ueou
sm
obile
phas
esCE
C[1
20]
Mefl
oqui
ne,m
efloq
uine
-O-te
rt-bu
tyl-
carb
amat
e,an
dpr
onet
halo
ltra
ns-(1
S,2S
)-2-(N
-4-A
llylo
xy-3
,5-
dich
loro
benz
oyl)a
min
ocy-
cloh
exan
esul
foni
cac
idm
odifi
edsi
lica
mon
olith
;100
�m
id×
25.0
cm
UV(2
16nm
)AC
N/M
eOH
(80:
20v/
v)co
ntai
ning
(R,S
)-2-a
min
o-1-
buta
nola
ndfo
rmic
acid
CEC
[121
]
Flav
anon
eM
ono-
(6-a
zido-
6-de
oxy)
-�-C
Dgr
afte
don
orga
nic
poly
mer
(3st
eps
synt
hesi
s-cl
ick
chem
istry
);75
�m
id×
31.0
cm
UV(2
14)
MEO
H/5
mM
bora
tebu
ffer,
pH8.
2(4
0:60
,v/v
)CE
C[1
24]
1-In
dano
l,1-
phen
ylet
hyla
min
e,�
-phe
nyl
glyc
inol
,1-(4
-bro
mop
heny
l)et
hano
l,1-
(2-c
hlor
ophe
nyl)
etha
nol
(R)-A
cryl
oylo
xy-�
-�-d
imet
hyl-�
-but
yrol
acto
nem
odifi
edsi
lica
mon
olith
;100
�m
×50
.0cm
UV(2
54nm
)n-
Hexa
ne/i-
PrOH
(98:
2,v/
v)[1
22]
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
12 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Ta
ble
1.
Co
nti
nu
ed
Anal
ytes
Colu
mn
Dete
ctio
nM
obile
phas
eCo
mpa
rativ
eRe
fere
nce
sepa
ratio
nm
ode
13ra
cem
icco
mpo
unds
(stru
ctur
eon
lyre
porte
d)Ph
-�-C
D-si
lica
hybr
idm
onol
ith;7
5�
mid
×30
.0cm
UV(2
14nm
)He
xane
/i-Pr
OH(9
0:10
v/v)
orM
eOH/
triet
hyla
mm
oniu
mac
etat
e,pH
4.2
(60:
40v/
v)
[112
]
Keto
prof
en,f
enop
rofe
n,flu
rbip
rofe
n,su
prof
enPG
Aim
mob
ilize
don
achi
rale
poxy
-sili
cam
onol
ith;1
00�
mid
×7.
0or
14.0
cmUV
(200
nm)
50m
Mph
osph
ate
buffe
r,pH
7.0
CEC
[65]
Mol
ecul
arim
prin
ted
poly
mer
icco
lum
nsDN
S-ph
enyl
alan
ine
DNS-
L-ph
enyl
alan
ine
mol
ecul
arim
prin
tpol
ymer
;25
�m
id×
85.0
cm,o
pen
tubu
lar
UV(2
80nm
)M
eCN
/ace
ticac
id(9
9.5:
0.5,
v/v)
CEC
[125
]
Bupi
vaca
ine,
mep
ivac
aine
,and
ropi
vaca
ine
Bupi
vaca
ine,
mep
ivac
aine
,orS
-rop
ivac
aine
tem
plat
efo
rgra
fting
poly
mer
izatio
non
orga
nic
poly
mer
icm
onol
ith;1
00�
mid
×5.
0cm
UV(2
15nm
)AC
N[1
28]
THP,
Trog
er’s
base
L-TH
Pan
d(5
S,11
S)-(-
)-Tro
ger’s
base
impr
inte
dsi
lica
base
dm
onol
ith;7
5�
mid
×8.
5,20
.0,a
nd24
.5cm
UV(2
14nm
)AC
N/5
mM
acet
ate
buffe
r,pH
6.0
(80:
20,v
/v)
CEC
[129
]
Keto
prof
en,n
apro
xen,
ibup
rofe
n,fe
nopr
ofen
S-Ke
topr
ofen
mol
ecul
arim
prin
tpol
ymer
;100
�m
id×
30.0
cm,o
pen
tubu
lar
UV(2
14nm
)AC
N/a
ceta
tebu
ffer,
pH4.
5(3
0:70
,v/v
)CE
C[1
30]
Chira
lmob
ileph
ase
addi
tive
Nap
roxe
nOD
S(C
18,H
yper
sil)
pack
ed;7
5�
mid
×22
.0cm
UV(2
32nm
)20
mM
met
hyl-�
-CD
in50
mM
sodi
umac
etat
e,pH
3.0/
ACN
(80:
20,v
/v)
HPLC
[131
]
Nap
roxe
n,in
dopr
ofen
,ket
opro
fen,
flurb
ipro
fen,
carp
rofe
n,ci
clop
rofe
n,flu
noxa
prof
en,s
upro
fen,
2-[(5
′ -ben
zoyl
-2′ -
hydr
oxy)
-phe
nyl]-
prop
ioni
cac
id,
2-[(4
′ -ben
zoyl
oxy-
2′-h
ydro
xy)
phen
yl]p
ropi
onic
acid
LiCh
rosp
her1
00RP
185
�m
parti
cles
,Cog
entR©
Bide
ntat
eC1
8ty
peC
4.2
�m
silic
aph
ase,
Chro
mSp
herC
183
�m
,Nuc
leos
ilR©
C85
�m
,Pi
nnac
leII
Cyan
o3
�m
;100
�m
id×
25.0
,10
.0cm
UV(2
05nm
)30
mM
TM-�
-CD
in50
mM
sodi
umac
etat
e,pH
3/AC
N(7
0:30
,v/v
)[1
32]
Oflox
acin
Pinn
acle
IIPh
enyl
;100
�m
id×
15.0
cm,3
�m
UV(2
90nm
)5
mM
hept
akis
-(2,3
-dia
cety
l-6-s
ulfo
)-�
-CD
in50
mM
sodi
umac
etat
e,pH
3.0/
ACN
(70:
30v/
v)
[133
]
Ibup
rofe
n,ke
topr
ofen
,flur
bi-p
rofe
n,su
prof
en,i
ndop
rofe
n,ci
clop
rofe
n,ca
rpro
fen,
napr
oxen
Chro
mSp
herC
183
�m
pack
edan
dCa
pRod
RP-1
8em
onol
ith;1
00�
mid
×10
.0cm
UV(2
00nm
)30
mM
TM-�
-CD
in50
mM
sodi
umac
etat
e,pH
3/AC
N(7
0:30
v/v)
,15
mM
HP-�
-CD
in25
mM
sodi
umac
etat
e,pH
3/M
eOH
(90:
10v/
v)
[49]
AC
N,
acet
on
itri
le;
AD
MP
C,
amyl
ose
tris
(3,5
-dim
ethy
lph
enyl
carb
amat
e);
CH
I-T
BB
,te
rt-b
uty
lben
zoyl
ated
tart
ard
iam
ide;
CT
PC
,ce
llulo
setr
isp
hen
ylca
rbam
ate;
DM
PC
C,
3,5-
dim
ethy
lph
enyl
carb
amo
ylce
llulo
se;
�-D
DB
,�
-dim
ethy
ld
icar
box
ylb
iph
enyl
der
ivat
ives
;D
NB
,3,
5-d
init
rob
enzo
yl;
DN
S,
dan
syl;
EtO
H,
eth
ano
l;FM
OC
,fl
uo
reny
lmet
hylo
xyca
rbo
nyl;
i-Pr
OH
,iso
pro
pan
ol;
MeO
H,m
eth
ano
l;M
ES
,2-(
N-m
orp
ho
lino
)eth
anes
ulf
on
icac
id;P
MB
C,p
-met
hylb
enzo
ylce
llulo
se;P
HT,
ph
thal
yl;P
DP
M,p
oly
(dip
hen
yl-2
-pyr
idyl
met
hylm
eth
acry
late
);P
GA
,pen
icill
inG
acyl
ase;
TH
F,te
trah
ydro
fura
n;t
Bu
CQ
D,O
-9-(
tert
-bu
tylc
arb
amo
yl)q
uin
idin
e;Z
,ben
zylo
xyca
rbo
nyl.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 13
derivative has been demonstrated as highly effective for enan-tiomer separations. In addition, the selectivity of this class ofCS can be further modulated by the introduction in the phenylring of electron-donating and electron-withdrawing groups,suggesting that the most important adsorbing sites for chi-ral discrimination on phenylcarbamate derivatives may bethe polar carbamate groups. Initially, their performance wasoptimized in normal phase conditions, even due to the sol-ubility of polysaccharides derivatives in some solvent. How-ever, later a relatively high enantiorecognition capability wasshown in aqueous organic mobile phases and recently, theiremployment with polar organic mobile phases was demon-strated to be successful [50–53]. Polysaccharides-based CSswere deeply investigated in miniaturized techniques as well,namely nano-LC and CEC, as demonstrated by the large num-ber of works reported in Table 1, and very interesting resultswere achieved (see below).
Even if present in a lower number of research papers,macrocyclic antibiotics (MAs) assume important implica-tions for nano-LC enantioseparations, too. MAs possess manystereogenic centres and functional groups, which are respon-sible of multiple interactions, i.e. hydrogen bonds, ionic-,�–� interactions. Furthermore, glycopeptides antibiotics,such as vancomycin, teicoplanin, and ristocetin A, are char-acterized by an aglycon portion with basket shape made byfused macrocyclic rings that allows the formation of inclu-sion complexes [54–56]. All these features are responsible oftheir high enantioselectivity toward several chiral compoundsand it is noteworthy mentioning, among all the applicationsof enantioseparative nano-LC to real samples, the most arerelated with the use of MAs (see Section 5.2).
Proteins and glycoproteins result instead less employedin nano-LC, with only five works present in literature (see Ta-ble 1). They are well known to bind stereoselectively drugs andcan be considered as polymers composed of several chiral sub-units (amino acids), characterized by a 3D structure, wherebyhydrophobic, electrostatic interactions, and hydrogen bondsguarantee chiral recognition. The high specificity of some pro-tein reduces their spectrum of selectivity and, further, whena protein has to be incorporated into a matrix to create a CSP,special attention has to be paid during immobilization in or-der to maintain natural conformation of the protein, essentialfor chiral discrimination. [57–60]. These drawbacks can be re-sponsible of their limited use in nano-LC. When they wereused in OT-LC, low retention factors, efficiency and resolutionwere observed [61,62]. However, proteins immobilization onsilica monoliths seems to be promising since higher coverageof proteins, and consequently higher resolution factors, thanthose obtained with silica particles or the organic polymer-based monolith were achieved [63, 64]. The work proposedby Gotti et al. [65], and discussed later on, go as well in thisdirection, showing high stability of the protein-based chiralselector and improved enantioselectivity and efficiency, dueto the covalent immobilization on monolithic silica capillarycolumn.
Finally, a quite large number of works is related withthe use of low molecular mass selectors (mainly with ligand
exchange properties), especially in combination with mono-lithic matrix, as described in the following sections.
5 Enantioseparations by means
of nano-LC
Considering the aforementioned advantages of nano-LC, theemploy of this miniaturized technique for enantiomeric as-sessment is furthermore gainful, since a minute amount ofexpensive CSP or CMPA is needed. Most of applications ofchiral nano-LC relate with the pharmaceutical field. In thisreview, works will be classified considering the type of chiralcolumn used (OT, monolithic, particulate packed or molecu-lar imprinted polymer, MIP), the employ of a chiral selectoras mobile phase additive and eventually the use of indirectmethod for enantioseparation. In Table 1 all the researchpapers concerning chiral separation by means of nano-LCare reported. Since many works used the same column (i.e.CEC), the same CSP (i.e. HPLC), or the same CS (i.e. CZE)for comparative studies, the table column entitled “Compar-ative separation mode” takes into account this evaluation.Furthermore, when reviewed research papers gave detailedinformation concerning, e.g. particles and pore size, amountof coating, etc., we reported those data in the Table 1, sincewe consider them useful for a critical estimation of the works.
5.1 OT chiral separation
The first chiral separation in nano-LC was obtained bySchurig et al. [66] employing an OT column (50 �m id) coatedwith ChirasilDEX, a permethylated-�-CD chemically linked todimethylpolysiloxane. Considering hexobarbital as test com-pounds, it was demonstrated the versatility of the same col-umn toward different techniques, as GC, supercritical fluidchromatography, and CEC (see Fig. 1). The same type of col-umn was evaluated in OT-LC and OT-CEC, coupled with MS,for a series of chiral drugs and alcohols [67]. In this regards,it should be underlined that several works are based on com-parative studies between nano-LC and CEC, where the samecolumn and, quite often, the same instrumentation was usedfor both techniques.
Francotte et al. separated both acidic and basiccompounds of pharmaceutical interest employing cap-illaries coated with cellulose derivative, 3,5-dimethyl-phenylcarbamoyl cellulose or p-methylbenzoyl cellulose [68].Even if both CSPs showed high enantioselectivity, they werecharacterized by short lifetime. To increase the stability of thecoating of the column, Wakita et al. [69] proposed a copoly-merization reaction of another cellulose derivative, cellulosetris(3,5-dichlorophenylcarbamate) (CDCPC) and carried outthe separation of several racemates.
Recently, Zou et al. proposed the use of nanoparti-cles to improve phase ratio and consequently chiral reso-lution in OT-LC [70]. Mobile crystalline material (MCM-41)type silica nano-particles were immobilized on a bare fused
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

14 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Figure 1. Enantiomer separation of hexo-barbital on a 50-�m id × 1 m fused sil-ica column coated with Chirasil-DEX byGC, SFC, nano-LC, and CEC. Effective col-umn length in nano-LC and CEC, 85.0 cm.Buffer, borate-phosphate, pH 7 (the arrowindicates the dead volume). From [66] withpermission.
silica capillary and successively coated with cellulose tris(3,5-dimethylphenyl carbamate) (CDMPC) at different concen-trations. The capillary column obtained with this procedureshowed higher enantioselectivity than a bare-fused silica cap-illary coated with the same amount of CS (0.3% w/v CDMPC).
In addition to polysaccharides-based CSP, proteins aswell were used as CS in OT-LC. BSA, chemically boundto the capillary wall, was used for the enantioseparation ofdinitrophenyl-amino acids, oxazepam, and lorazepam [61],while with avidin (a glycoprotein), physically adsorbed on thecapillary wall, the enantioseparation of dansyl-amino acidsand some drugs was achieved [62].
In an interesting work, OT-LC was also proposed to per-form enantioselective studies by frontal affinity chromatog-raphy. The gamma isoform ligand-binding domain of perox-isome proliferator-activated receptor (PPAR�), immobilizedinto a capillary column, was used to characterize, in combi-nation with MS, the enantioselective interaction of some an-tagonist for this biological target. Different binding affinitiesof enantiomers to PPAR� resulted in a characteristic biphasicfrontal profile as the result of the two saturation events [71].Despite of the promising result showed, the number of re-search papers related to OT-LC chiral separations is quitemodest mainly due to their poor selectivity arising from thelow phase ratio [23].
5.2 Particulate packed chiral capillary columns
A large number of works is present in literature whereenantioseparations were carried out with packed columns.Permethylated-�-CD-modified silica was used to performenantiomeric separation and to compare the perfor-mance of CEC and nano-LC [72, 73]. As expected, effi-ciency in CEC was higher than nano-LC. Gong et al.tested two bonded CSPs, 8-aminoquinoline-2-ylmethyl- and8-aminoquinoline-7-ylmethyl-diaza-18-crown-6-capped [3-(2-O-�-CD)-2-hydroxypropoxy] propylsilyl silica particles (non-porous, 1.5 �m), where the combination of crown ether with�-CD was exploited to obtain better selectivity toward po-sitional isomers and chiral compounds [74]. Analyses were
performed in a capillary column (75 �m id), working at highpressure (>8000 p.s.i.) and high column efficiencies wereachieved. Three other different �-CD-based CSP, containingpernaphthylcarbamoylated �-CD, peracetylated �-CD, andpermethylated �-CD, respectively, already used in HPLC,were also studied in nano-LC and CEC. It was noted thatthe presence of carbonyl groups as well as aromatic ringincreased the enantioseparation capability. Concerning thefirst examined CSP, it showed better enantioseparation of �-blockers than HPLC, and this phenomenon was attributed toimprovement of separation efficiency due to different columnfabrications [75].
A CSP based on �-CD was also employed by Si-Ahmedet al. in nano-LC to perform the enantioseparation of severalflavanoids and diastereoisomeric separation of two flavanoneglycosides [76]. Good eanantioresolutions were achieved, inshorter analysis time compared to the results reported in lit-erature, for the same compounds, with conventional HPLC.The same CSP, phenyl-carbamate-propyl-�-CD stationaryphase, was used to perform analysis of hesperetin, a chiral fla-vanone abundant in citrus fruits with antioxidant, anticancer,anti-inflammatory, and analgesic activities, in human urineafter ingestion of blood orange juice [77]. Precisely, after thevalidation of the methods, in terms of sensitivity (LOD andLOQ, values 0.1 and 0.5 �g/mL, respectively), linearity, pre-cision, accuracy, and recovery of the extraction procedure,the excretion of R- and S-hesperitin in urine was monitoredwithin 24 h and it was observed that S-hesperetin was ex-creted at higher concentrations than R-hesperetin, while theflavanoid was almost completely eliminated in about 12 h.
CSPs widely employed are based on polysaccharidederivatives. In particular, those derived from cellulose andamylose, were extensively investigated and compared innano-LC as well as in CEC by Chankvetadze’s group. Ina preliminary study, polyacrylamide, and polysaccharidederivatives, namely Chiraspher R© (prepared by radical copoly-merisation of N-acryloyl-L-phenyl-alanineethylester with sil-ica gel) and CDMPC, were tested as CSP for the separa-tion of several racemic drugs, working in both normal andreversed phase conditions [78]. While the polyacrilamide-based SP was covalently bonded to silica particles, cellulose
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 15
derivative was coated. The feasibility of miniaturizationfrom HPLC to nano-LC and CEC for chiral separation wasdemonstrated, underlining the importance to avoid columnoverloading in downscaled separation in order to not fade theperformance.
In a following study, CDMPC (also called ChiracelOD) enantio-resolving capability was compared withthose ones of Chiralpak AD, consisting of amylose-tris(3,5-dimethylphenylcarbamate) and cellulose-tris(4-methylbenzoate) (Chiralcel OJ) for the separation ofthalidomide and its hydroxilated metabolites in convectionalHPLC column, nano-LC, and CEC [79]. In nano-LC, evenif Chiralpak AD showed higher enantioselectivity, thebaseline resolution of all racemic compounds in one run wasobtained only with a CSP composed by a combination of twopolysaccharide derivatives (16% AD plus 14% OD) coatedonto aminopropylsilica.
Later on, the same CPSs were evaluated considering theloading of the chiral selector on aminopropyl derivatized silicagel, the pore, and particle size of silica gels, temperature and,obviously, the mobile phase composition [80–84].
A CSP based on CDCPC was also studied in terms of load-ing of the chiral selector, particle, pore size, and type of the sil-ica support, and optimization of separative medium [85–88].Employing capillary columns packed with CDCPC, nano-LC,and CEC techniques were compared for the determination ofenantiomeric purity of the contraceptive drug levonorgestrelin a commercial sample [89]. Despite of higher efficiencyoffered by the electrochromatographic method, the minorenantiomeric impurity ((+)-norgestrel) of a drug formulationwas better detected with nano-LC, due to its minor baselinenoise. Even if the method did not allow to clearly detect anyenantiomeric impurity of (+)-norgestrel in the samples of lev-onorgestrel used in the study, the standard addition methodclearly indicated that the impurity on the level below 0.1%w/w might be present. Consequently, 0.1% w/w was consid-ered as the apparent LOD of detection by the authors, beingthe levonorgestrel sample at the concentration of 5 mg/mL.
The effect of the presence of different electron-donatingand electron-withdrawing groups on polysaccharide deriva-tives was investigated introducing cellulose tris(3-chloro-4-methylphenylcarbamate) (Sepapak-2) [90], which was laterproposed for the enantiomeric separation of fluorenylmethy-loxycarbonyl derivatives of amino acids in nano-LC andCEC, and the determination of the content and the enan-tiomeric purity of the non-protein amino acid citrullinein food supplements by CEC [91]. Sepapak-2 has been re-cently compared with Sepapak-4 ((cellulose tris(4-chloro-3-methylphenylcarbamate)) for the enantioseparation of 16 pes-ticides [92]. Sepapak-4 provided better resolution toward highnumber of compounds and was selected for further studiesin CEC.
Amylose tris(5-chloro-2-methylphenylcarbamate)-basedCSP was also subject of an accurate testing in both nano-LCand CEC, and showed good enantio-resolving capability for aseries of racemic drugs [93]. Recently, cellulose tris(4-chloro-3-methylphenylcarbamate) was also coated on core-shell silica
particles and slightly better results, in terms of chiral selec-tivity and resolution, were obtained compared with totallyporous material [94].
A bonded type of cellulose derivative CSP, cellulosetrisphenylcarbamate, was also proposed by Chen et al. [95] forenantioseparations in nano-LC and CEC and good stability ofthe column was demonstrated. The same group proposedafter a positively charged cellulose derivative to be used inCEC and nano-LC [96]. Working in normal phase conditions,the enantiosepareation of some drug candidates was achievedwith a capillary column packed for 8 cm only.
Another class of chiral selectors widely employed in nano-LC are MAs. Fanali et al. obtained the enantioseparation ofseveral hydroxy acids employing a capillary column packedfor only 7 cm with silica stationary phase modified with MDL63 246 (Hepta-Tyr), a glycopeptide antibiotic from the te-icoplanin family [97]. The same type of column was selectedfor a study of enantiopurity of a pharmaceutical formula-tion of a new chiral drug (loxiglumide) under investigationfor the treatment of gastrointestinal diseases [98]. The assayof the pharmaceutical sample (ampoule) declared to containonly D-loxiglumide (0.5%, w/v) gave a recovery of the drugin agreement with the labeled content (recovery = 99.88 ±2.16%), while L-loxiglumide was not detected because absentor at concentration lower than the LOD value (0.5 �g/mL forL-loxiglumide).
Several works are related with the use of a stationaryphase derivatized with vancomycin. Enantioseparation ofacid and basic drugs, such as no steroidal anti-inflammatorydrugs, �-blockers, and clofibric acid [99, 100], chlorophenoxyacid herbicides [101] were obtained in nano-LC. Validatedmethods were applied for the determination of metopro-lol in pharmaceutical formulation [99] and to detect simul-taneously mirtazapine, a second-generation antidepressantdrug, and its metabolites in serum samples as shown inFig. 2 [102]. In this work, coupling the nano-LC system withan ESI-MS interface, it was possible to obtain about oneorder lower LOD and LOQ than those obtained with UV.LOD and LOQ values for mirtazapine, 8-hydroxymirtazapine
Figure 2. Separation of mirtazapine, 8-hydroxymirtazapine, andN-desmethylmirtazapine with (R)-1-(2-naphthyl)ethylamine as in-ternal standard. Chromatographic conditions: capillary column,75 �m id × 23.5 cm packed length; mobile phase, ammoniumacetate 500 mM pH 4.5/H2O/MeOH/ACN (1:14:40:45, v/v/v/v), flowrate: 200 nL/min; injected volume, about 60 nL. From [101] withpermission.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

16 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
and N-desmethylmirtazapine, with UV detection were in therange 5–15 and 10–40 �g/mL, respectively, while with MSdetection were 0.3–5 and 0.6–35 �g/mL, in that order. Forsome �-blockers, a study concerning on-column focusingwas carried out in order to improve the sensitivity and thedeveloped method was applied to determination of alprenololin human plasma [103]. Dissolving the analyte in methanol,which demonstrated a lower eluting capability than that of themobile phase used in the experiments, it was possible to in-ject a sample volume such high as 1500 nL, resulting in LODand LOQ values for both alprenolol enantiomers of 9.0 and15.6 ng/mL, respectively.
A capillary column packed with vancomycin-modified sil-ica particles was selected to carry out enantiomeric separationof several D- and L-amino acids derivatized with FITC, bymeans of nano-LC coupled with MS. The method was ap-plied to verify the quality of orange juice samples, since thepresence of D-amino acids can be attributed to, e.g. micro-biological contamination and adulteration [104]. In order toachieve detection limits suitable for this kind of analysis, an-alytes were focused on a C18 cartridge, employing a column-switching system. This apparatus allowed the injection of5-�L sample volume. Further, the chromatographic separa-tion system was coupled with an ion-trap mass spectrometerthrough a nano spray interface and finally it provided LODvalues as low as 8 ng/mL. Figure 3 shows nano-LC–MS chro-matograms of the analysis of a fresh (a) and a commercial (b)orange juice, respectively.
A CSP based on teicoplanin, another glicopeptidic antibi-otic, was also used for the determination of racemic atenololin human urine by nano-LC hyphenated with MS [105]. Aftercomparing the sensitivity of the nano-LC method obtainedby using a conventional on-column UV detector for CE, or azeta cell (3 nL volume), the ion-trap MS detection was chosen
since offered the highest sensitivity (LOD, 50 ng/mL, andLOQ, 400 ng/mL for each atenolol enantiomer).
Less common CSPs to carry out enantioseparations bymeans of nano-LC are also reported in literature. Widepore aminopropyl silica gel coated with helically chiralpoly(diphenyl-2-pyridylmethyl methacrylate) was used as CSPfor a comparative study among conventional HPLC, nano-LC, pressure-assisted CEC, and CEC. Enantioseparations ob-tained in nano-LC showed higher peak efficiency, and con-sequently higher resolutions, than those achieved with acommon-size HPLC column. Comparing with CEC, in nano-LC similar efficiencies were unexpectedly observed [106].Fanali et al. investigated the use of tert-butylbenzoylatedtartardiamide chiral stationary phase for the separation ofracemic acidic compounds of pharmaceutical and environ-mental interest, working in normal phase mode [107]. Tert-butylbenzoylated tartardiamide chiral stationary phase be-longs to a series of CSP based on optically active �-amino acidsor tartaric acid diamides able to form weak diasteromericcomplexes with analytes based on hydrogen bonding. A studyconcerning the influence of capillary temperature on enan-tioresolution was also carried out demonstrating that reten-tion of analytes was an exothermic process. It should be un-derlined that this kind of study can be easily performed withcapillary columns, since they are rapidly conditioned at thedesired temperature.
In normal phase, chiral separations of amino acid amideswere also obtained using (S)-N-(3,5-dinitrobenzoyl)leucine-N-phenyl-N-propylamide-bonded silica as CSP. The chiralrecognition mechanism of this CSP utilizes the �−� donor–acceptor and hydrogen-bonding interactions. The data ob-tained in nano-LC were compared with those from CEC andit was concluded that enantioselectivities were similar, butresolution was better in CEC. However, the authors stated
Figure 3. Comparison of two differ-ent nano-LC-MS profiles from orangejuice: fresh squeezed and commercialjuice. Capillary chiral column, 75 �mid × 25.0 cm packed length; mobilephase, 500 mM ammonium formatepH 3.5/water/MeOH, 4:11:95 v/v/v; MSconditions: positive ion mode; capil-lary voltage: 30 V; ion-spray voltage:2.0 kV; capillary temperature: 170C.From [104] with permission.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 17
that nano-LC separation was not optimized for injection vol-ume and extra-column band broadening contributions [108].Another comparison between nano-LC and CEC, was re-cently reported by Lee et al. [109]. (−)-(18-Crown-6)-2,3,11,12-tetracarboxylic acid-bonded silica as CSP was employed forenantioseparation of some �-amino acids. Chiral recognitionmechanism involves the tripodal complexation of the proto-nated primary amino group (R-NH3
+) inside the cavity ofthe 18-crown-ether ring via three +N–H···O hydrogen bondsand additional roles are played by the two free carboxylic acidgroups acting as chiral barriers, enantioselective hydrogen-bonding sites, or ionic interaction sites. Generally, CEC gavebetter enantioselectivity and resolutions than nano-LC, as aconsequence of higher efficiency due to the flat EOF profilepresent CEC.
5.3 Chiral monolithic columns
Monolithic media are widely used for capillary columns sincethey can be easily prepared in situ and do not require frits toretain the stationary phase. The latter aspect is particularly ad-vantageous when CSPs have to be employed, because whensilica particles have been derivatized with a CS, quite oftenthey are no more suitable for frit preparation or new appro-priate conditions of sintering have to be found. Two maincategories of polymeric material can be distinguished: thoseobtained by polymerization of organic monomers and thoseconsisting of an inorganic silica network. The polymeriza-tion of silica monoliths can be performed by sol-gel processor by fusion of porous silica packing material in a capillaryvia sintering, entrapping in a silica network or by sol-gel pro-cess. In the last case, a sort of hybrid between monolithicand packed bed is obtained. Wistuba et al. prepared a chi-ral silica monolith, by sintering a bare silica bed followed byon-column modification with permethylated �-CD (Chirasil-Dex) [110]. The column was successfully tested in nano-LC,pressure assisted–CEC (p-CEC) and CEC for the enantiomerseparation of several chiral compounds. Later, the same groupentrapped silica particles into a silica matrix by sol-gel pro-cess, creating a so-called particle-glued monolithic columnby means of immobilization of Chira-Dex-silica (permethyl-�-CD-silica) [111]. Also in this work, the same column wasutilized for chiral separation in nano-LC, p-CEC, and CEC aswell. Considering hexobarbital as test compound and apply-ing a pressure of only 10 bar in nano-LC and 10 bar plus 20 kVin p-CEC, retention time in nano-LC was four time longer,which demonstrated that in p-CEC the EOF is the main driv-ing force. When the applied pressure was 150 bar for bothtechniques (carprofen was used as test compound), reten-tion time was slightly higher in CEC, since electrophoreticmobility of carprofen (negatively charged) was in the oppo-site direction of EOF. A small improvement of enantiores-olution and efficiency was also observed. When the polarityof the applied voltage was inverted, a reduction of analysistime was observed, but resolution decreased as well. Em-ploying another CD derivative, perphenylcarbamoylated �-
CD (Ph-�-CD), as chiral selector, a silica hybrid monolith wasproposed by Zou et al. [112]. The capillary column was pre-pared by a “one-pot” approach. First, mono (6A-N-allylamino-6A-deoxy)-Ph-�-CD was synthesized starting from mono-6A-deoxy-6A-(p-tolylsulfonyl) �-CD. After, the polycondensationof alkoxysilanes and in situ copolymerization of mono (6A-N-allylamino-6A-deoxy)-Ph-�-CD and vinyl group on the pre-condensed siloxanes took place in a pretreated fused silicacapillary. The influence of several parameters on the synthe-sis of the chiral monolith were evaluated, such as polyconden-sation temperature, amount of chiral monomer, the ratio ofN,N-dimethylformamide/methanol, and the content of waterin the polymeric mixture. Finally, the capability of the col-umn for enantioseparation was demonstrated resolving 13racemates in normal- and reversed phase modes.
Monolithic silica columns were derivatized with polysac-charide derivatives, exploiting the high permeability and ef-ficiency offered by monolithic silica materials as well asavoiding frits fabrication. In a preliminary study, nativesilica-based monolith was modified in situ with CDMPC[13]. Working with mobile phases composed by n-hexane/2-propanol mixtures, enantioseparation of analytes as 2,2,2-trifluoro-1-(9-anthryl)ethanol, benzoin, 2,2’-dihydroxy-6,6’-dimethylbiphenyl, flavanone, and trans-stilbene oxide, wereobtained in less than 4 min, while for several �-blockers enan-tioresolution was achieved at most within 12 min (see Fig. 4).The same column showed enantiorecognition capability alsooperating with reversed phase conditions. Moreover, a studywas carried out in order to further decrease analysis time and,as an example, it was reported the chiral separation of 2,2,2-trifluoro-1-(9-anthryl)-ethanol obtained in less than 30 s, witha column of 12 cm packed length. However, for this columnthe coating procedure with the cellulose derivative was re-peated three times. The obtained positive results promptedthe authors to modify silica monolith with amylose tris(3,5-dimethylphenylcarbamate) [113]. The effect of the amountof amylose tris(3,5-dimethylphenylcarbamate) coated ontomonolithic silica on separation characteristics was also stud-ied and it was noted that, even if increasing the concentrationof the CS during the coating procedure brought to higherresolutions, it negatively affected efficiency. In situ modifica-tion of monolithic-fused silica capillary columns was realizedcovalently bonding 3,5-disubstituted phenylcarbamate deriva-tives of cellulose and amylase as well, to increase the stabilityof these CSP toward different solvents [114]. Although, as ex-pected, polysaccharide derivatives coated onto silica supportshowed fairly higher enantiorecognition compared to the ma-terials covalently attached onto the surface, covalent immobi-lization of polysaccharide derivatives allowed multiple coat-ing and immobilization steps with a consequent increasedretention and separation factors, with few exceptions.
A monolithic silica capillary column was used also for thephysical adsorption of avidin and tested for nano-LC and CECchiral separation [115]. The effect of type and concentrationof activating capillary reagent (sodium hydroxide solutionsor ammonia solutions) was investigated and optimized in or-der to increase the adsorbed amount of avidin. Reducing the
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
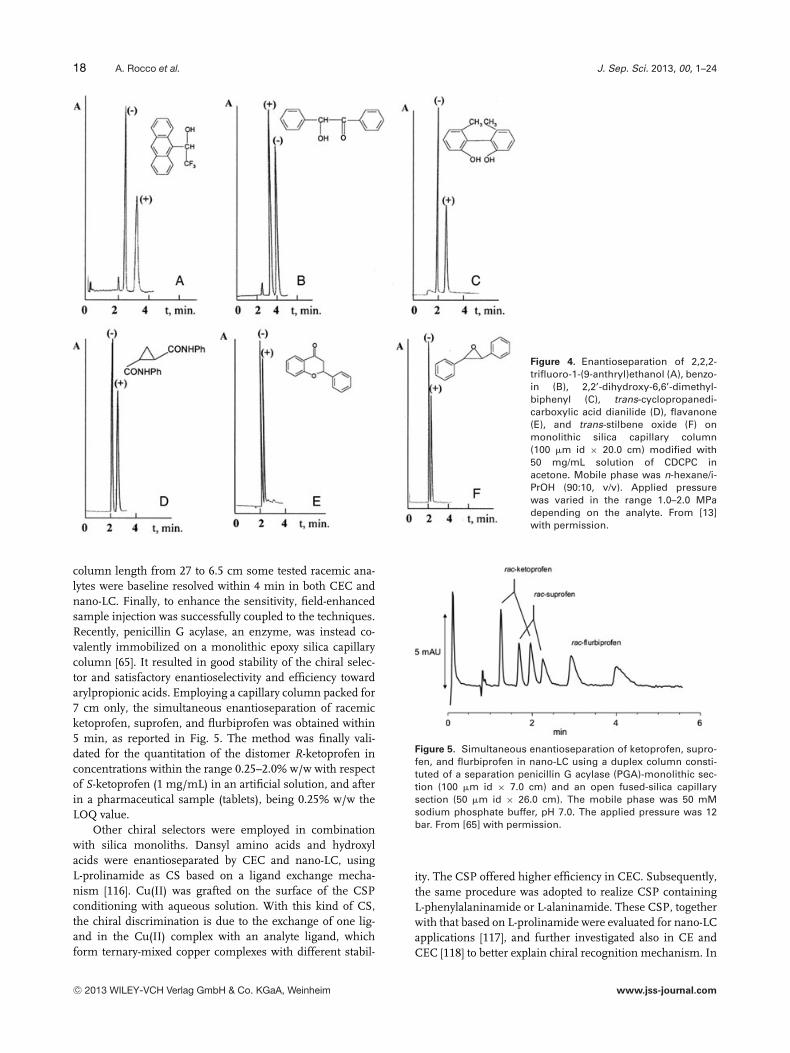
18 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Figure 4. Enantioseparation of 2,2,2-trifluoro-1-(9-anthryl)ethanol (A), benzo-in (B), 2,2’-dihydroxy-6,6’-dimethyl-biphenyl (C), trans-cyclopropanedi-carboxylic acid dianilide (D), flavanone(E), and trans-stilbene oxide (F) onmonolithic silica capillary column(100 �m id × 20.0 cm) modified with50 mg/mL solution of CDCPC inacetone. Mobile phase was n-hexane/i-PrOH (90:10, v/v). Applied pressurewas varied in the range 1.0–2.0 MPadepending on the analyte. From [13]with permission.
column length from 27 to 6.5 cm some tested racemic ana-lytes were baseline resolved within 4 min in both CEC andnano-LC. Finally, to enhance the sensitivity, field-enhancedsample injection was successfully coupled to the techniques.Recently, penicillin G acylase, an enzyme, was instead co-valently immobilized on a monolithic epoxy silica capillarycolumn [65]. It resulted in good stability of the chiral selec-tor and satisfactory enantioselectivity and efficiency towardarylpropionic acids. Employing a capillary column packed for7 cm only, the simultaneous enantioseparation of racemicketoprofen, suprofen, and flurbiprofen was obtained within5 min, as reported in Fig. 5. The method was finally vali-dated for the quantitation of the distomer R-ketoprofen inconcentrations within the range 0.25–2.0% w/w with respectof S-ketoprofen (1 mg/mL) in an artificial solution, and afterin a pharmaceutical sample (tablets), being 0.25% w/w theLOQ value.
Other chiral selectors were employed in combinationwith silica monoliths. Dansyl amino acids and hydroxylacids were enantioseparated by CEC and nano-LC, usingL-prolinamide as CS based on a ligand exchange mecha-nism [116]. Cu(II) was grafted on the surface of the CSPconditioning with aqueous solution. With this kind of CS,the chiral discrimination is due to the exchange of one lig-and in the Cu(II) complex with an analyte ligand, whichform ternary-mixed copper complexes with different stabil-
Figure 5. Simultaneous enantioseparation of ketoprofen, supro-fen, and flurbiprofen in nano-LC using a duplex column consti-tuted of a separation penicillin G acylase (PGA)-monolithic sec-tion (100 �m id × 7.0 cm) and an open fused-silica capillarysection (50 �m id × 26.0 cm). The mobile phase was 50 mMsodium phosphate buffer, pH 7.0. The applied pressure was 12bar. From [65] with permission.
ity. The CSP offered higher efficiency in CEC. Subsequently,the same procedure was adopted to realize CSP containingL-phenylalaninamide or L-alaninamide. These CSP, togetherwith that based on L-prolinamide were evaluated for nano-LCapplications [117], and further investigated also in CE andCEC [118] to better explain chiral recognition mechanism. In
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 19
nano-LC, among all the studied CSP, L-phenylalaninamide-modified monolithic column showed higher selectivity thanalaninamide-modified one, while L-prolinamide- and L-phenylalaninamide-modified CSPs showed a different enan-tioselectivity. Preinerstorfer et al. modified a monolithicsilica column with O-9-(tert-butylcarbamoyl)quinidine, cin-chona alkaloid-derived anion-exchange-type chiral selector,for the stereoselective analysis of some phosphinic acid pseu-dodipeptides in CEC. The same column was tested in nano-LC as well, but the lower efficiency gave a worse separation.However, the same negative outcome was observed in con-ventional HPLC, too [119]. A silica monolith functionalizedby a cation exchange type chiral selector, trans-(1S,2S)-2-(N-4-allyloxy-3,5-dichlorobenzoyl)aminocyclohexanesulfonic acid,was studied by the same team for the enantioseparation ofvarious chiral bases in nano-LC and CEC. The influence ofCS coverage was considered and it was concluded that nei-ther the second immobilization cycle (29 nmol/cm of CSinstead of 22 nmol/cm with first immolization) nor the dy-namic flow-through immobilization improved enantiomer-resolving ability [120]. In a following work, considering meflo-quine, mefloquine-O-tert-butylcarbamate, and pronethalol astest solutes, individual migration contributions in CEC (elec-trokinetic and chromatographic) were better investigated toexplain the behavior of the silica-based monolithic capil-lary column under CEC and nano-LC conditions and theseparation mechanism of charged solutes in stereoselec-tive ion-exchange CEC. Comparing the performance of bothtechniques in terms of kinetic plots, it was noted that theefficiency gain in CEC was mainly due to a greatly reducedA-term contribution to peak dispersion, while apparently thecolumn gave comparable efficient mass-transfer character-istics, with a C-term slightly smaller in CEC [121]. Recently,(R)-acryloyloxy-�-�-dimethyl-�-butyrolacton, a low-molecularmass CS was employed to create a brush type CSP. Afterthe derivatization of native-free OH silica monolith with 3-methacrylamidopropyl-triethoxysilane, the resulting hybridmonolith was in situ copolymerized with the CS. A set ofracemic secondary alcohols and a chiral amine were sepa-rated and the main mechanism of enantiorecognition wasascribed to the H-bond acceptor and the hydrophobic interac-tion with the gem dimethyl group of the butyrolacton [122].
Organic polymer monolith materials were also used toprepare chiral media. Generally, they offer easier and fasterprocedures of preparation than silica-based monolith, butlower efficiency and reproducibility. Schmid et al. prepared achiral ligand exchange stationary phase by a one-step in situpolymerization of acrylamide based continuous bed, utiliz-ing N-(2-hydroxy-3-allyloxypropyl)-L-4-hydroxyproline as CS[123]. The column was projected to be used in CEC, however,for D, L-phenylalanine enantioresolution was demonstratedin nano-LC, too.
Guerrouache et al. synthesized an organic polymer basedon N-acryloxysuccinimide and ethylene dimethacrylate, byUV-initiated free radical polymerization [124]. In a secondstep, succinimide groups were derivatized with propargy-lamine to insert alkine moieties, and finally an azido deriva-
tive of �-CD (mono-(6-azido-6-deoxy)-�-CD) was grafted onthe monolithic bed, exploiting a click reaction. As prelimi-nary result, the enantioseparation of flavanone was achievedin both CEC and nano-LC.
5.4 MIP stationary phases
Monolithic matrices give the possibility to prepare MIP, bythe addition of a template (imprint) molecule in the poly-meric mixture. Monomers arrange around the template, be-cause of secondary interactions (e.g. hydrogen bonds) or theformation of covalent bonds between functional groups ofthe template and those of the monomers. Once polymeriza-tion is completed, the imprint molecule is extracted and thepolymer network will posses cavities having shape, size, andfunctional groups complementary to those of the template.As a consequence it shows a remarkable affinity toward thetemplate. For this characteristic, MIP has been widely stud-ied for synthetic chemistry, catalysis, solid-phase extractions,sensor design, and as CSP for chromatographic separations.For conventional LC, MIPs are synthesized in bulk and af-ter ground, sieved and packed into columns. However, thisprocedure presents some drawbacks, such as wide particlesize distribution and irregular particles shape, which nega-tively affect efficiency. The introduction of capillary columnsallowed the synthesis in situ of MIPs as well [125–127].
Dansyl-L-phenylalanine was used as template by Tan andRemcho, to prepare 25-�m id capillary columns for chiral OT-LC and OT-CEC [125]. The chiral separation of a mixture of D-and L-dansyl phenylalanine was obtained applying a pressurein the range of 100–500 mbar. At 350 mbar enantiomers wereresolved in less than 8 min, with a column with an effectivelength of 85 cm.
A limitation when using one-step polymerizations to pre-pare MIPs is the need to optimize the conditions to obtainthe desired porosity for each used polymeric mixture and tem-plate. A way to manage surface chemistry without stronglychange the permeability of a given material, consist of graft-ing procedure. If a porogenic agent is added to the graftingmixture, it is possible to graft a mesoporous layer with aconsequent increase of the surface area and the number offunctional groups. Courtois et al. proposed this expedientto prepare capillary columns for low abundance sample en-richment [128]. Mepivacaine, bupivacaine, and enantiopureS-ropivacaine were alternatively used as imprint molecules.During their evaluation, authors observed that the columnprepared with S-ropivacaine showed high selectivity for thesame enantiomer, and a split peak for mepivacaine, charac-teristic of racemic compounds. This phenomenon could befurther investigated to prepare analytical column by meansof the same procedure. A similar approach was developed byOu et al. to anchor MIP onto the surface of a monolithic sil-ica capillary column [129]. L-Tetrahydropalmatine (THP) and(5S,11S)-(–)Troger’s base were used as template, respectively.L-THP was also used as imprint molecule to prepare an or-ganic monolithic capillary column in a one-step procedure.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

20 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Both L-THP-based columns, composite monolith and organicmonolith, were tested in nano-LC and it was noted that theretention of THP enantiomers was stronger on the organiccolumn, as a consequence of large number of recognitionsites on its surface, but concerning column efficiency andseparation time the composite monolith gave better perfor-mance.
Ziadi et al. reported in situ preparation of an OT MIPmonolithic column where S-ketoprofen was used as tem-plate [130]. To secure formation of the MIP layer into a silicacapillary of 100-�m id, the reaction mixture was rinsed intothe capillary and let to react in a water bath at 50C for 3 h.Then the capillary was flushed with a 0.5 MPa nitrogen flowfor 5 min, and was again placed in the water bath for 2 h tocomplete MIP formation. The column showed high enantios-electivity not only toward ketoprofen, but for ibuprofen andnaproxen as well, demonstrating high cross-selectivity.
5.5 CMPA
As already reported in this manuscript, miniaturized tech-niques offer the advantages to use the chiral selector dissolvedin the mobile phase in its free form, since very few amountof it is needed to perform analysis. The use of a CMPA can beconsidered convenient compared to a stationary phase con-taining it, when CMPA results less expensive, the synthesisof the CSP is difficult, accessibility of the CS present in freesolution for interaction with analytes is easier. Furthermore,the combination of achiral stationary phases with CMPAs orof different chiral additives can provide higher flexibility forthe selection of experimental conditions or improve chiralrecognition.
Healy et al. employed methyl-�-CD as CMPA in combina-tion with a C18-RP capillary column for the enantioseparationof the anti-inflammatory drug naproxen. They demonstratedthat, working with the same experimental conditions used inconventional HPLC, in nano-LC the consumption of mobilephase was reduced more than 1000 folds [131].
Later on, Rocco and Fanali carried out a study onthe enantioseparation of several racemic nonsteroidal anti-inflammatory drugs employing different achiral stationaryphases in combination with heptakis (2,3,6-tri-O-methyl)-�-CD (TM-�-CD) [132]. Among all investigated stationaryphases, i.e. cyano, C8 and different types of C18 (5- and3-�m particle size, bidentate 4.2-�m particle size), C18, 3 �m,reversed phase gave best results in terms of achieved enan-tioresolution. The chromatographic method was also appliedfor the determination of the association constant betweenracemic analyte and CD. An analogous work about the com-bination of CD with an appropriate stationary phase, regardedthe development and validation of an analytical methodfor the determination of a chiral fluoroquinolone antibiotic,ofloxacin. In this case, enantioresolution was achieved em-ploying heptakis-(2,3-diacetyl-6-sulfo)-�-CD as CMPA and anachiral reversed phase capillary column packed with Pinna-cle II Phenyl 3 �m [133]. The method performance was eval-
uated thorough the following analytical parameters: LOD,LOQ, linear range, accuracy, and precision. Satisfactory datawere achieved such as LOD values of 1.94 and 3.75 �g/mLfor the R-(+) and S-(–) enantiomer, respectively, and intradayand interday precision of peak areas, expressed as RSD, aslow as 4.74%.
The same group accomplished a research about the com-parison of the performance of a RP-C18 monolithic columnand a RP-C18-packed column of the same dimensions, usedin combination with TM-�-CD or hydroxypropyl-�-CD (HP-�-CD) [49]. As expected, the monolithic column showed highpermeability but lower selectivity compared to the packedone. For this reason, in case of TM-�-CD, only when enan-tioresolutions were high enough with the packed column,compounds could be still baseline resolved with the mono-lithic column and in reduced analysis time. The monolithiccolumn resulted particularly advantageous when HP-�-CDwas selected as CMPA. In fact with this CS, enantiodiscrimi-nation occurs with very low percentage of organic modifier oreven in a completely aqueous mobile phase. With the packedcolumn in those conditions (0–10% organic modifier), mostof studied compounds not eluted in reasonable time, whilewith the monolithc column they were resolved within 30 minat most.
6 Conclusions
The downscaling of chromatographic technique, up tonanoscale level, aroused lively interest among scientists in-volved in separation science, since this miniaturization in-troduced advantages as low consumption of reagent, smallamount of sample needed, reduced analysis time, good effi-ciency, and easy coupling with MS. For these reasons, nowa-days, nano-LC represents a valid alternative to classical chro-matographic techniques in many analytical fields, includingchiral analysis.
The main characteristic of nano-LC that determined itssuccess in the field of enantioseparation is related to the lowrequirement of CSP or CMPA, since this material can be veryexpensive. Nano-LC, e.g. is suitable for the study of new CSP,especially when small amount is available.
Several chiral separations by means of nano-LC are re-ported in literature. A large number of works is related tothe study of selectand/selector interactions, however, applica-tions in the pharmaceutical, clinical, environmental, and foodareas have been carried out as well. Among chiral selectors,polysaccharides, CDs, and MAs were widely employed. Thereduced flow rate and the consequent limited consumptionof mobile phase, allows the use of chiral additives. Most ofresearch papers involve the use of particulate-packed capillarycolumns. However, as a common trend in other applicationfields, the synthesis of chiral monolithic columns is continu-ously investigated. In fact, due to the capillary format of thecolumns employed in nano-LC, they can be easily preparedin situ, offer high permeability and do not need frits to re-tain the stationary phase. Furthermore, they allow to realize
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 21
highly selective material as MIP and seem to represent a goodopportunity to realize nano-LC in microdevices.
The authors have declared no conflict of interest.
7 References
[1] Waldeck, B., Chirality 1993, 5, 350–355.
[2] Szymura-Oleksiak, J., Bojarski, J., Aboul-Enein, H. Y.,Chirality 2002, 14, 417–435.
[3] Van Eeckhaut, A., Michotte, Y., Chiral Separations byCapillary Electrophoresis, Taylor and Francis Group,Boca Raton 2010.
[4] Gubitz, G., Schmid, M. G., Biopharm. Drug Dispos.2001, 22, 291–336.
[5] Fanali, S., Catarcini, P., Blaschke, G., Chankvetadze, B.,Electrophoresis 2001, 22, 3131–3151.
[6] Scriba, G. K. E., Electrophoresis 2003, 24, 2409–2421.
[7] Chankvetadze, B., Capillary Electrophoresis in ChiralAnalysis, John Wiley and Sons, New York 1997.
[8] Lammerhofer, M., Mayer, N. M., Lindner, W., in: Sny-der, L. R., Kirkland, J. J., Dolan, J. W. (Eds.), Introduc-tion to Modern Liquid Chromatography, John Wileys& Sons, Hoboken 2010, pp. 665–724.
[9] Vissers, J. P. C., Claessens, H. A., Cramers, C. A., J.Chromatogr. A 1997, 779, 1–28.
[10] Ishii, D., Takeuchi, T., TrAC, Trends Anal. Chem. 1990,9, 152–157.
[11] Hernandez-Borges, J., Aturki, Z., Rocco, A., Fanali, S.,J. Sep. Sci. 2007, 30, 1589–1610.
[12] Abian, J., Oosterkamp, A. J., Gelpı, E., J. Mass Spec-trom. 1999, 34, 244–254.
[13] Chankvetadze, B., Yamamoto, C., Tanaka, N., Nakan-ishi, K., Okamoto, Y., J. Sep. Sci. 2004, 27, 905–911.
[14] Ishihama, Y., J. Chromatogr. A 2005, 1067, 73–83.
[15] Mechref, Y., Novotny, M. V., J. Chromatogr. B 2006, 841,65–78.
[16] Chervet, J. P., Ursem, M., Salzmann, J. P., Anal. Chem.1996, 68, 1507–1512.
[17] Unger, K. K., Skudas, R., Schulte, M. M., J. Chromatogr.A 2008, 1184, 393–415.
[18] Saito, Y., Jinno, K., Greibrokk, T., J. Sep. Sci. 2004, 27,1379–1390.
[19] Zotou, A., Cent. Eur. J. Chem. 2012, 10, 554–569.
[20] Chen, J. R., Zare, R. N., Peters, E. C., Svec, F., Frechet,J. J., Anal. Chem. 2001, 73, 1987–1992.
[21] Rocco, A., Fanali, S., J. Chromatogr. A 2008, 1191,263–267.
[22] Dong, X., Wu, R., Dong, J., Wu, M., Zhu, Y., Zou, H.,Electrophoresis 2009, 30, 141–154.
[23] Lammerhofer, M., Svec, F., Frechet, J. M. J., Lindner,W., Trends Anal. Chem. 2000, 19, 676–698.
[24] Maruska, A., Kornysova, O., J. Biochem. Biophys.Methods 2004, 59, 1–48.
[25] Svec, F., J. Sep. Sci. 2004, 27, 1419–1430.
[26] Guillarme, D., Ruta, J., Rudaz, S., Veuthey, J.-L., Anal.Bioanal. Chem. 2010, 397, 1069–1082.
[27] Maier, N.M., Lindner, W., Anal. Bioanal. Chem. 2007,389, 377–397.
[28] Lammerhofer, M., Gargano, A., J. Pharm. Biomed.Anal. 2010, 53, 1091–1123.
[29] Gauthier, G. L., Grimm, R., Drug Discov. Today 2006, 3,59–66.
[30] Nagl, S., Schulze, P., Ludwig, M., Belder, D., Elec-trophoresis 2009, 30, 2765–2772.
[31] Nagl, S., Schulze, P., Ohla, S., Beyreiss, R., Gitlin, L.,Belder, D., Anal. Chem. 2011, 83, 3232–3238.
[32] Zeng, H.-L., Li, H.-F., Lin, J.-M., Anal. Chim. Acta 2005,551, 1–8.
[33] Bi, H., Weng, X., Qu, H., Kong, J., Yang, P., Liu, B., J.Proteome Res. 2005, 4, 2154–2160.
[34] Weng, X., Bi, H., Liu, B., Kong, J., Electrophoresis 2006,27, 3129–3135.
[35] Qu, P., Lei, J., Sheng, J., Zhang, L., Ju, H., Analyst 2011,136, 920–926.
[36] Zeng, H.-L., Li, H.-F., Wang, X., Lin, J.-M., Talanta 2006,69, 226–231.
[37] Li, H.-F., Zeng, H., Chen, Z., Lin, J.-M., Electrophoresis2009, 30, 1022–1029.
[38] Van Eeckhaut, A., Michotte, Y., in Van Eeckhaut, A., Mi-chotte, Y. (Eds.), Chiral Separations by Capillary Elec-trophoresis, Taylor & Francis Group, Boca Raton 2010,pp. 1–22.
[39] Pevarello, P., Future Med. Chem. 2009, 1, 35–48.
[40] Ahuja, S., Chiral Separations: Applications and Tech-nology, American Chemical Society, Washington1997.
[41] European Medicines Agency, CPMP/ICH/367/96, EMA,London 2000.
[42] Wistuba, D., Schurig, V., J. Chromatogr. A 2000, 875,255–276.
[43] Han, S. M., Biomed. Chromatogr. 1997, 11, 259–271.
[44] Fanali, S., J. Chromatogr. A 2000, 875, 89–122.
[45] Schneiderman, E., Stalcup, A. M., J. Chromatogr. B2000, 745, 83–102.
[46] Guan, J., Yang, J-, Bi, Y., Shi, S., Yan, F., Li, F., J. Sep.Sci. 2008, 31, 288–293.
[47] Berta, R., Szakacs, Z., Babjak, M., Gazdag, M., Chro-matographia 2010, 71, S35–S42.
[48] Ye, J., Yu, W., Chen, G., Shen, Z., Zeng, S., Biomed.Chromatogr. 2010, 24, 799–807.
[49] Rocco, A., Maruska, A., Fanali, S., Anal. Bioanal. Chem.2012, 402, 2935–2943.
[50] Okamoto, Y., Kaida, Y., J. Chromatogr. A 1994, 666,403–419.
[51] Okamoto, Y., Yashima, E., Angew. Chem. Int. Ed. 1998,37, 1020–1043.
[52] Yashima, E., J. Chromatogr. A 2001, 906, 105–125.
[53] Peng, L., Jayapalan, S., Chankvetadze, B.,Farkas, T., J. Chromatogr. A 2010, 1217,6942–6955.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

22 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
[54] Gasper, M. P., Berthod, A., Nair, U. B., Armstrong, D.W., Anal. Chem. 1996, 68, 2501–2514.
[55] Ward, T. J., Farris III, A. B., J. Chromatogr. A 2001, 906,73–89.
[56] Fanali, S., Aturki, Z., D’Orazio, G., Rocco, A., in: VanEeckhaut, A., Michotte, Y. (Eds.), Chiral Separations byCapillary Electrophoresis, Taylor and Francis Group,Boca Raton 2010, pp. 109–137.
[57] Loun, B., Hage, D. S., Anal. Chem. 1994, 66, 3814–3822.
[58] Haginaka, J., J. Chromatogr. A 2001, 906, 253–273.
[59] Millot, M. C., J. Chromatogr. B 2003, 797, 131–159.
[60] Machtejevas, E., Maruska, A., J. Sep. Sci. 2002, 25,1303–1309.
[61] Hofstetter, H., Hofstetter, O., Schurig, V., J. Microcol-umn Sep. 1998, 10, 287–291.
[62] Liu, Z., Otsuka, K., Terabe, S., J. Sep. Sci. 2001, 24,17–26.
[63] Wistuba, D., J. Chromatogr. A 2010, 1217, 941–952.
[64] Calleri, E., Massolini, G., Lubda, D., Temporini, C.,Loiodice, F., Caccialanza, C., J. Chromatogr. A 2004,1031, 93–100.
[65] Gotti, R., Fiori, J., Calleri, E, Temporini, C., Lubda, D.,Massolini, G., J. Chromatogr. A 2012, 1234, 45–49.
[66] Schurig, V., Jung, M., Mayer, S., Fluck, M., Ne-gura, S., Jakubetz H., J. Chromatogr. A 1995, 694,119–128.
[67] Jakubetz, H., Czesla, H., Schurig, V., J. MicrocolumnSep. 1997, 9, 421–431.
[68] Francotte, E., Jung, M., Chromatographia 1996, 42,521–527.
[69] Wakita, T., Chankvetadze, B., Yamamoto, C., Okamoto,Y., J. Sep. Sci. 2002, 25, 167–169.
[70] Dong, X., Wu, R., Dong, J., Wu, M., Zhu, Y., Zou, H.,Electrophoresis 2008, 29, 3933–3940.
[71] Calleri, E., Fracchiolla, G., Montanari, R., Pochetti, G.,Lavecchia, A., Loiodice, F., Laghezza, A., Piemontese,L., Massolini, G., Temporini, C., J. Chromatogr. A 2012,1232, 84–92.
[72] Wistuba, D., Czesla, H., Roeder, M., Schurig, V., J. Chro-matogr. A 1998, 815, 183–188.
[73] Wistuba, D., Schurig, V., Electrophoresis 1999, 20,2779–2785.
[74] Gong, Y., Xiang, Y., Yue, B., Xue, G., Bradshaw, J. S.,Lee, H. K., Lee, M. L., J. Chromatogr. A 2003, 1002,63–70.
[75] Lin, B., Ng, S.-C., Feng, Y.-Q., Electrophoresis 2008, 29,4045–4054.
[76] Si-Ahmed, K., Tazerouti, F., Badjah-Hadj-Ahmed, A. Y.,Aturki, Z., D’Orazio, G., Rocco, A., Fanali, S., J. Chro-matogr. A 2010, 1217, 1175–1182.
[77] Si-Ahmed, K., Tazerouti, F., Badjah-Hadj-Ahmed, A. Y.,Aturki, Z., D’Orazio, G., Rocco, A., Fanali, S., J. Pharm.Biomed. Anal. 2010, 51, 225–229.
[78] Krause, K., Girod, M., Chankvetadze, B., Blaschke, G.,J. Chromatogr. A 1999, 837, 51–63.
[79] Meyring, M., Chankvetadze, B., Blaschke, G., J. Chro-matogr. A 2000, 876, 157–167.
[80] Girod, M., Chankvetadze, B., Blaschke, G., J. Chro-matogr. A 2000, 887, 439–455.
[81] Girod, M., Chankvetadze, B., Blaschke, G., Elec-trophoresis 2001, 22, 1282–1291.
[82] Chankvetadze, L., Kartozia, I., Yamamoto, C., Chankve-tadze, B., Blaschke, G., Okamoto, Y., Electrophoresis2002, 23, 486–493.
[83] Chankvetadze, L., Kartozia, I., Yamamoto, C., Chankve-tadze, B., Blaschke, G., Okamoto, Y., J. Sep. Sci. 2002,25, 653–660.
[84] Mayer, S., Briand, X., Francotte, E., J. Chromatogr. A2000, 875, 331–339.
[85] Girod, M., Chankvetadze, B., Okamoto, Y., Blaschke, G.,J. Sep. Sci. 2001, 24, 27–34.
[86] Chankvetadze, B., Kartozia, I., Breitkreutz, J., Girod, M.,Knobloch, M., Okamoto, Y., Blaschke, G., J. Sep. Sci.2001, 24, 251–257.
[87] Chankvetadze, B., Kartozia, I., Okamoto, Y., Blaschke,G., J. Sep. Sci. 2001, 24, 635–642.
[88] Chankvetadze, B., Kartozia, I., Breitkreutz, J,Okamoto, Y., Blaschke, G., Electrophoresis 2001, 22,3327–3334.
[89] Chankvetadze, B., Kartozia, I., Yamamoto, C., Okamoto,Y., Blaschke, G., J. Pharm. Biomed. Anal. 2003, 30,1897–1906.
[90] Fanali, S., D’Orazio, G., Lomsadze, K., Chankvetadze,B., J. Chromatogr. B 2008, 875, 296–303.
[91] Domınguez-Vega, E., Crego, A. L., Lomsadze, K.,Chankvetadze B., Marina, M. L., Electrophoresis 2011,32, 2700–2707.
[92] Perez-Fernandez, V., Dominguez-Vega, E., Chankve-tadze, B., Crego, A. L., Garcıa, M. A., Marina, M. L.,J. Chromatogr. A 2012, 1234, 22–31.
[93] Fanali, S., D’Orazio, G., Lomsadze, K., Samakashvili,S., Chankvetadze, B., J. Chromatogr. A 2010, 1217,1166–1174.
[94] Fanali, S., D’Orazio, G., Farkas, T., Chankvetadze, B., J.Chromatogr. A 2012, 1269, 136–142.
[95] Chen, X., Zou, H., Ye, M., Zhang, Z., Electrophoresis2002, 23, 1246–1254.
[96] Chen, X., Qin, F., Liu, Y., Kong, L., Zou, H., Electrophore-sis 2004, 25, 2817–2824.
[97] Fanali, S., Catarcini, P., Presutti, C., Stancanelli, R.,Quaglia, M. G., J. Chromatogr. A 2003, 990, 143–151.
[98] Fanali, S., D’Orazio, G., Quaglia, M. G., Rocco, A., J.Chromatogr. A 2004, 1051, 247–252.
[99] D’Orazio, G., Aturki, Z., Cristalli, M., Quaglia, M. G.,Fanali, S., J. Chromatogr. A 2005, 1081, 105–113.
[100] Fantacuzzi, M., Bettoni, G., D’Orazio, G., Fanali, S., Elec-trophoresis 2006, 27,1227–1236.
[101] Rosales-Conrado, N., Leon-Gonzalez, M. E., D’Orazio,G., Fanali, S., J. Sep. Sci. 2004, 27, 1303–1308.
[102] Fanali, S., Aturki, Z., Kasicka, V., Raggi, M. A., D’Orazio,G., J. Sep. Sci. 2005, 28, 1719–1728.
[103] D’Orazio, G., Fanali, S., J. Sep. Sci. 2008, 31, 2567–2571.
[104] D’Orazio, G., Cifuentes, A., Fanali, S., Food Chem. 2008,108, 1114–1121.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 00, 1–24 Liquid Chromatography 23
[105] D’Orazio, G., Fanali, S., J. Pharm. Biomed. Anal. 2006,40, 539–544.
[106] Krause, K., Chankvetadze, B., Okamoto, Y., Blaschke,G., Electrophoresis 1999, 20, 2772–2778.
[107] Fanali, S., D’Orazio, G., Rocco, A., J. Sep. Sci. 2006, 29,1423–1431.
[108] Kim, I. W., Hyun, M. H., Gwon, J., Jin, J., Park, J. H.,Electrophoresis 2009, 30, 1015–1021.
[109] Lee, T., Lee, W., Hyun, M. H., Park, J. H., J. Chromatogr.A 2010, 1217, 1425–1428.
[110] Wistuba, D., Schurig, V., Electrophoresis 2000, 21,3152–3159.
[111] Wistuba, D., Banspach, L., Schurig, V., Electrophoresis2005, 26, 2019–2026.
[112] Zhang, Z., Wu, M., Wu, R., Dong, J., Ou, J., Zou, H.,Anal. Chem. 2011, 83, 3616–3622.
[113] Chankvetadze, B., Yamamoto, C., Kamigaito, M.,Tanaka, N., Nakanishi, K., Okamoto, Y., J. Chromatogr.A 2006, 1110, 46–52.
[114] Chankvetadze, B., Kubota, T., Ikai, T., Yamamoto, C.,Kamigaito, M., Tanaka, N., Nakanishi, K., Okamoto, Y.,J. Sep. Sci. 2006, 29, 1988–1995.
[115] Liu, Z., Otsuka, K., Terabe, S., Motokawa, M., Tanaka,N., Electrophoresis 2002, 23, 2973–2981.
[116] Chen, Z., Hobo, T., Electrophoresis 2001, 22, 3339–3346.
[117] Chen, Z., Uchiyama, K., Hobo, T., J. Chromatogr. A2002, 942, 83–91.
[118] Chen, Z., Niitsuma, M., Uchiyama, K., Hobo, T., J. Chro-matogr. A 2003, 990, 75–82.
[119] Preinerstorfer, B., Lubda, D., Mucha, A., Kafarski, P.,Lindner, W., Lammerhofer, M., Electrophoresis 2006,27, 4312–4320.
[120] Preinerstorfer, B., Hoffmann, C., Lubda, D.,Lammerhofer, M., Lindner, W., Electrophoresis2008, 29, 1626–1637.
[121] Preinerstorfer, B., Lammerhofer, M., Hoffmann, C. V.,Lubda, D., Lindner, W., J. Sep. Sci. 2008, 31, 3065–3078.
[122] Ghanem, A., Ikegami, T., Tanaka, N., Chirality 2011, 23,887–890.
[123] Schmid, M.G., Grobuschek, N., Tuscher, C., Gubitz, G.,Vegvari, A., Machtejevas, E., Maruska, A., Hjerten, S.,Electrophoresis 2000, 21, 3141–3144.
[124] Guerrouache, M., Millot, M.-C., Carbonnier, B., Macro-mol. Rapid Commun. 2009, 30, 109–113.
[125] Tan, Z. J., Remcho, V. T., Electrophoresis 1998, 19,2055–2060.
[126] Liu, H., Row, K. H., Yang, G., Chromatographia 2005,61, 429–432.
[127] Haupt, K., Mosbach, K., Chem. Rev. 2000, 100, 2495–2504.
[128] Courtois, J., Fischer, G., Sellergren, B., Irgum, K., J.Chromatogr. A 2006, 1109, 92–99.
[129] Ou, J., Li, X., Feng, S., Dong, J., Dong, X., Kong, L., Ye,M., Zou, H., Anal. Chem. 2007, 79, 639–646.
[130] Zaidi, S. A., Cheong, W.J., J. Sep. Sci. 2008, 31, 2962–2970.
[131] Healy, L. O., Murrihy, J. P., Tan, A., Cocker, D., McEnery,M., Glennon, J. D., J. Chromatogr. A 2001, 924, 459–464.
[132] Rocco, A., Fanali, S., J. Sep. Sci. 2009, 32, 1696–1703.
[133] Rosales-Conrado, N., Leon-Gonzalez, M. E., Rocco, A.,Fanali, S., Curr. Anal. Chem. 2010, 6, 209–216.
[134] Chen, Z., Zeng, S., Yao, T., Pharmazie 2007, 62, 585–592.
Anna Rocco graduated in Pharmaceutical Chemistry and Technologies from the Univer-sity “La Sapienza”, Rome, in 2003. Since 2004 she is working at the Institute of ChemicalMethodologies CNR, in Rome. In 2008 she started her Ph.D. studies at the Faculty ofNatural Science, Vytautas Magnus University, Kaunas. She has experience in analysis ofsamples of interest in pharmaceutical, environmental and food fields by CE, CEC andn-LC. She has also practice in preparing packed and monolithic capillary columns.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

24 A. Rocco et al. J. Sep. Sci. 2013, 00, 1–24
Audrius Maruska graduated from Kaunas University of Technology (1985), obtainedthere PhD degree in synthesis and evaluation of cellulose adsorbents (1990) and Habili-tated Dr. degree at Vilnius University in the field of stationary phases and techniques formicroseparations (2002), spent long-term scientific stays at Mainz, Marburg and UppsalaUniversities. Currently he is a head of research group at Vytautas Magnus Universityactive in the field of development of microseparations, synthesis of stationary phases,coupling of methods and phytochemical analysis.
Salvatore Fanali achieved the title of Doctor in Chemistry at the University of Rome “LaSapienza”. His research include either chromatography and electromigration techniquesin conventional and miniaturized modes. Special attention was paid in coupling nano-LC, CE and CEC with mass spectrometry. He is head of a research group at Instituteof Chemical Methodologies active in the development of miniaturized instrumentationand applications in the field of food analysis, enantiomers separation, new columntechnology, environmental analysis etc.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com