domain swapping in g-protein coupled receptor dimers
TRANSCRIPT

Protein Engineering vol.11 no.12 pp.1181–1193, 1998
Domain swapping in G-protein coupled receptor dimers
Paul R.Gouldson, Christopher R.Snell1,Robert P.Bywater2, Christopher Higgs andChristopher A.Reynolds3
Department of Biological Sciences, University of Essex, Wivenhoe Park,Colchester, Essex CO4 3SQ,1Novartis Institute for Medical Sciences,5 Gower Place, London WC1E 6BN, UK and2Biostructure Department,Novo Nordisk A/S, Novo Nordisk Park DK-2760 Måløv, Denmark
3To whom correspondence should be addressed
Computer simulations were performed on models of theβ2-adrenergic receptor dimer, including 5,6-domainswapped dimers which have been proposed as the active,high affinity form (here the dimer interface lies betweenhelices 5 and 6). The calculations suggest that the domainswapped dimer is a high energy structure in both the apodimer and in the presence of propranolol. In the presenceof agonist the energy of the domain swapped dimer issignificantly lowered. Analysis of the dimer structure sug-gests that the agonist-induced conformational changeoptimizes the helix–helix interactions at the 5-6 interface.An antagonist on the other hand has little effect on theseinteractions. These observations are consistent with thehypothesis that the agonist functions by shifting the equilib-rium in favour of the domain swapped dimer. Indirectsupport for the domain swapping hypothesis was obtainedfrom the correlated mutations amongst the external res-idues of the known β2-adrenergic receptors. These occurmainly at the 5-6 interface at precisely the locationspredicted by the simulations; site-directed mutagenesisdata in support of a functional role for these lipid-facingcorrelated residues is presented. The article includes areview of the experimental evidence for G-protein coupledreceptor dimerization. Many other aspects of G-proteincoupled receptor activation are discussed in terms of thisdomain swapping hypothesisKeywords: adrenergic receptor agonist/correlated mutations/domain swapping/G-protein coupled receptors/dimers
Introduction
Domain swapping is an important mechanism which may beused for extending the functional repertoire of dimeric proteins(Bennett et al., 1994, 1995; Tegoniet al., 1996; Cameronet al., 1997; Gouldsonet al., 1997a,b,c,d; Schluneggeret al.,1997; Larsenet al., 1998). Although domain swapping hasnot to our knowledge been explicitly mentioned by others inthe context of G-protein coupled receptors (Baldwin, 1994;Strader, 1994; Watkinson, 1994; Guddermannet al., 1997),recent experiments (Maggioet al., 1993a, 1996; Ciruelaet al.,1995; Hebertet al., 1996; Romanoet al., 1996; Cvejic andDevi, 1997) have suggested that dimerization is important.Some of the more compelling evidence for dimerization isrelated to the role of dimers in functional rescue (Maggioet al., 1993a, 1996; Monnotet al., 1996), but as yet there is
© Oxford University Press 1181
no definitive evidence on the role of dimers in activation.Since there is currently much debate on the role of dimers inG-protein coupled receptors, we present here a preliminarycorrelated mutation analysis (Go¨bel et al., 1994; Singeret al.,1995; Pazoset al., 1997) and molecular dynamics (Karplus andPetsko, 1990) investigation of various possibleβ2-adrenergicreceptor (Main, 1990; Timmermanset al., 1990) dimers todetermine whether such models are consistent with currentexperimental understanding of the activation process.
There have been many illuminating studies of the conforma-tional changes involved in activation of the G-protein coupledreceptors. These have included computational simulations(Zhang et al., 1993; Luo et al., 1994; Sheeret al., 1996;Gouldsonet al., 1997a), site-directed spin labelling studies(Farabakhshet al., 1995; Altenbachet al., 1996; Farrenset al., 1996), substituted cysteine accessibility studies onconstitutionally active receptors (Javitchet al., 1997) andfluorescence studies on constitutive and agonist induced activa-tion (Getheret al., 1995, 1997a,b). Many of these studieshighlight changes in helices six, but also changes in helicesthree and five. Kobilkaet al. (1988) and Maggioet al. (1993b)observed reassociation and subsequent activity when receptorswere split between helices five and six and then coexpressed.These functional rescue studies involved the receptors domainsin much larger relative movements. Similar results confirmingthe dynamic nature of the receptor domains were obtained bySchoneberget al. (1995) and Monnotet al. (1996).
All this work must be seen against a background wherethere is no widely accepted understanding of the activationprocess at the molecular level. Consequently, many phenomenaassociated with G-protein coupled receptors, such as partialagonism, inverse agonism, high and low affinity states of thereceptor, autoactivation and constitutive activation have noclear explanation.
The dimerization theory may shed light on many of theseprocesses. For example, our knowledge of high and low affinityforms is indicative of a conformational change in the receptor,but when Potteret al. (1991) observed that only 50% ofdopamineD2 receptors were in their high affinity forms at anyone time, this pointed towards dimerization. Indeed, for awhile there has been much circumstantial evidence for dimeriz-ation. Here we will simply list some of the evidence but adiscussion of how this may relate to dimerization will bedeferred until the discussion. The evidence includes the auto-activation of G-protein coupled receptors when overexpressedin transgenic mice (Bondet al., 1995), the activation ofreceptors by both bivalent antibodies (Leiberet al., 1984) andby antagonists coupled back to back (Hazum and Keinan,1981), the particularly strong activation of G-proteins obtainedby coupling together two copies of peptides from intracellularloop three (Wadeet al., 1994), the observation of dimers inthe two-dimensional projections maps of rhodopsin (Corlesset al., 1982; Schertleret al., 1993; Unger and Schertler,1995), bell-shaped dose response curves (Ja¨rv, 1995), the

P.R.Gouldsonet al.
Fig. 1. A schematic diagram of the proposed domain swappingrearrangement of G-protein coupled receptors. The figure shows Maggioet al.’s (1993a) inactive chimeric receptors forming a 1,7-dimer. Also shownare two intermediates on the domain swapping pathway and the active 5,6-domain swapped dimer.
immunological studies of Ciruelaet al. (1995), the identifica-tion of SDS-resistant dimers by Hebertet al. (1996) and theobservation that peptides derived from transmembrane helixsix (Hebert et al., 1996) can inhibit G-protein activation;peptides derived from helix seven have been shown to inhibitdimerization (Nget al., 1996). The most illuminating evidencehowever comes from the observations of Maggioet al. (1993a,1996) who prepared chimeric receptors where the N-terminusand helix one through to helix five was taken from themuscarinicM3 receptor while helices six and seven throughto the C-terminus were taken from theα2-adrenergic receptor.The receptor was found to be inactive, as was the alternativeadrenergic–muscarinic chimera (in both chimeras, intracellularloop three was taken from the muscarinic receptor). However,when the two chimeras were coexpressed, they were able toactivate G-proteins but no precise molecular explanation wasgiven. Elsewhere (Gouldson and Reynolds, 1997; Gouldsonet al., 1997a,c,d) we have proposed that this may be explainedby the domain swapping mechanism shown in Figure 1(discussed below) and here we analyse this mechanism toidentify areas where it may be compatible with our understand-ing of G-protein coupled receptor activation derived fromexperiment.
Correlated mutation analysis (Go¨bel et al., 1994; Singeret al., 1995; Pazoset al., 1997) is a method of probing forstructurally significant residues in proteins with a similar foldand function. It involves systematically searching for highlycorrelated patterns of change through a set of aligned sequences.It therefore highlights those positions in the sequence concernedwith the finer details of intermolecular interactions such asthose involved in specificity. In particular, it indicates wherea mutation in one area may be compensated by a mutation inanother, as shown for example in the ligand binding studieson serotonin (Kuiperset al., 1994), dopamine (Bywateret al.,
1182
1995) glucagon-like receptors (Hornet al., 1998) and in ananalysis of subtype specificity in the adrenergic, muscarinicand dopamine receptors (Gouldsonet al., 1997e). In theadrenergic receptors, if the receptor only made non-specificinteractions with the lipid we would expect no externalcorrelated mutations. However, one of the exciting observationsto emerge is that a large number of external correlatedmutations were observed (Gouldsonet al., 1997d). Here, it isimportant to note that gene sequences are experimental dataand so these data are complementary to that from the proteinmodelling.
Methods
Theβ2-adrenergic receptor model
In order to carry out the computer simulations and to interpretthe correlated mutation analysis, we have constructed a modelof theβ2-adrenergic receptor. The model is based on a multiplesequence alignment involving rhodopsin in theβ1-, β2- andβ3-adrenergic receptors and a rhodopsin template based on theelectron cryomicroscopy projection structure of rhodopsin(Unger and Schertler, 1995). The receptor model containingpropranolol is in agreement with the recent rhodopsin electroncryomicroscopy projection structure (Ungeret al., 1997)—theroot mean square deviation from the corresponding transmem-brane alpha carbon coordinates (Baldwinet al., 1997) is 2.5 Åin the central regions, increasing to about 3.8 Å when thehelix ends are included. This agreement is within the resolutionof the experimental structure, particularly bearing in mind thatthe two receptors are different. The rotational orientation ofeach helix in our model is in agreement with Baldwin’s towithin about 10°. The vertical displacement of the alphacarbons of the conserved residues is in agreement to withinapproximately 2Å. The agreement with the agonist-boundmodel is, as expected, not as good. The r.m.s. deviation ofalpha carbons is 5.6Å and there are probably two additionalreasons for this. Firstly, the experimental rhodopsin structureis an inactive form and secondly large helical movementsof up to 8Å have been observed on activation (Gouldsonet al., 1997).
In constructing this model, we followed the lead of Herzykand Hubbard (1995, 1996) by incorporating a range of experi-mental and other data including information from the helixprediction algorithm of Rost (1996) and from site-directedmutagenesis data on both rhodopsin and theβ2-adrenergicreceptor. The latter was largely used to determine the inwardfacing residues, but clearly this approach has been followedcautiously as one of our main messages in this article is thatexternal transmembrane residues may have a genuine function.The model was refined by minimization and moleculardynamics using the AMBER software (Singhet al., 1988).The antagonist (propranolol) and the agonist (norepinephrine)were docked into the receptor using our novel domain-baseddocking strategy (Gouldsonet al., 1997a) and the results werefound to conform with the site-directed mutagenesis results.The results of Suryanarayanaet al. (1991, 1993) in particularsuggest that the antagonist binds between the conservedaspartate on helix three, the hydrophobic pocket betweenhelices one, two (and three) and a key residue on helix seven,namely Asn312. This site is referred to as the antagonist bindingsite. The agonist binding site on the other hand lies betweenthe conserved Asp113 on helix three and conserved serines onhelix five (Ser204 and Ser207) while making contact with helix

Domain swapping in G-protein coupled receptor dimers
Fig. 2. (a) The 1,2-contact dimer, (b) the 1,7-contact dimer and (c) the 5,6-domain swapped dimer.
seven (Leu311) and an alternative hydrophobic pocket formedby residues on helix six (Trp286and Phe290) and three (Gouldsonet al., 1997a). The observation that mutation of residueshomologous to Asn312 in the antagonist binding site of relatedreceptors only affects some antagonist binding (Wesset al.,1991) may be an indication that not all antagonists bind to the‘antagonist’ binding site. Moreover, mutation of residues inthe agonist binding site may affect antagonist binding (Wesset al., 1993; Blumlet al., 1994). Thus, it is possible that someantagonists bind in the agonist binding site but this is probablynot the case here.
The receptor model and docking methodology are fullydescribed elsewhere and the coordinates have been depositedas supporting information (Gouldsonet al., 1997a—this articlecontains 115 references).
Molecular dynamics simulations on the dimersThree dimers were selected for further study: (i) the 1,2-dimer,because this appears to be the structure observed in the two-dimensional electron diffraction maps of rhodopsin (Corlesset al., 1982; Schertleret al., 1993); (ii) the 5,6-domain swappeddimer, which we have proposed to explain the results ofMaggio et al. (1993a); and (iii) the 1,7-dimer, which we haveproposed as a possible intermediate on the domain swappingpathway which could be formed by a slight rearrangement ofthe 1,2-dimer. The 1,2-dimer is best referred to as a contactdimer since the contact point between the two receptor mon-omers involves helices one and two of the first receptorcontacting helices two and one, respectively, of the othermonomers (Figure 2a). The 1,7-dimer is similar in that helicesone and seven contact helices seven and one respectively ofthe other monomer, as shown in Figure 1 (top left) andFigure 2b. In both of these contact dimers there is no covalentlink across the dimer interface. The 5,6-domain swapped dimershown in Figure 1 (bottom right) and Figure 2c is more
1183
complicated. A superficial glance at only the transmembranehelices would suggest that the dimer interface is betweenhelices five and six. However, since the B domain (comprisedof helices six and seven and the associated loops) of eachmonomer is swapped out and replaced by the B domain of theother monomer, the dimer interface is actually much larger, asshown in Figure 2c and the bottom right of Figure 1. To allintents and purposes, the transmembrane bundle of the domainswapped dimer has the same interactions as the monomer—but the loops have new interactions and these will be differentin the contact dimer and the domain swapped dimer. (Here wenote that our simulations only consider the interactions betweenthe transmembrane helices so the 5,6-domain swapped dimerreferred to here is essentially the same as the 5,6-contact dimer.)
The simulations on the dimers (or dimer plus one ligandmolecule) were performed using the all-atom AMBER forcefield (Weineret al., 1984, 1986) within the AMBER 4.1 suiteof programs (Singhet al., 1988). The simulations were run at298 K using a non-bonded cut-off of 10 Å and a time step of0.0005 ps. A distance-dependent dielectric constant was usedsince the environment within the receptor is intermediatebetween bulk water and the interior of a protein (Gouldsonet al., 1995). The dimer structures were generated by releasingthe system in the appropriate orientation with the monomersinitially separated by 9 Å. A similar strategy was used fordocking the ligands into the GPCR. This involved docking theligands onto the A domain and simulating the reassociation ofthe B domain from a distance of about 6 Å during the courseof a molecular dynamics simulation; this strategy yieldedcomplexes which are very well supported by experiment(Gouldsonet al., 1997a).
The systems were gently heated and equilibrated for 27 psaccording to the protocol described by Gouldsonet al. (1997a)and then equilibrated for a further 40 ps at 298 K. (The finalstructure was essentially formed during the first 20 ps but theenergies required at least 50 ps to settle down.) The simulationswere then run for at least 400 ps.
Free energy simulationsTo assess whether our dimer models are consistent with theknown experimental site-directed mutagenesis information onresidues believed to lie at the dimer interface (Huanget al.,1994; Hebertet al., 1996), we carried out the correspondingmutations during the course of a free energy differencesimulation (Beveridge and DiCapua, 1989; Reynoldset al.,1992) on a 5,6-6,5 four helix bundle taken from the 5,6-dimerinterface. The helix bundle was equilibrated for 20 ps at 10 K,20 ps at 100 K and 100 ps at 298 K using SHAKE (vanGunsteren and Berendsen, 1977) and a time step of 0.001 ps.The G276A, G280A and L284A mutations (Hebertet al.,1996) were carried out concurrently and the free energy changewas computed using the windowing approach which is basedon the exponential formula (Zwanzig, 1954):
∆GAB 5 –RT ln , exp(–∆HAB/RT).A (1)
Here,∆GAB is the free energy difference between states Aand B,∆HAB is the corresponding molecular mechanics energydifference, and,....A means that the average of exp(–∆HAB/RT) is evaluated over the configuration of state A. In practice,∆HAB must be small to ensure convergence and so the overallmutation was split into 21 ‘windows’ with each subsequentwindow corresponding to an extra 5% of the mutation. Eachwindow consisted of 10 000 steps of equilibration followed

P.R.Gouldsonet al.
by 10 000 steps of data collection; the mutation thereforeoccurred over a period of 420 ps which is comparable to thelength of the simulations described above. This simulationwas also run on the fully equilibrated 14 helix 5,6-dimer butonly 500 steps of equilibration and 500 steps data collectionwere carried out for each window. The Y209A mutation(Huanget al., 1994) was much more difficult as it representsa significant reduction in size and severe difficulties wereencountered in evaluating the derivatives for the dummy atomsused to represent the disappearing atoms (Reynoldset al.,1992). Such mutations are usually carried out using MonteCarlo (MC) methods as MC does not require derivatives.However, MC methods are not so useful in protein simulations.Consequently, the mutation was carried out in three stages:(i) Tyr → Phe, (ii) Phe→ Ala* and (iii) Ala* → Ala whereAla* represents an alanine with an unusually large Cβ suchthat the Phe→ Ala* mutation results in essentially no volumechange; dummy atoms not required in the next stage wereeliminated to minimize the number of dummy atom derivativesrequired [thus the dummy atoms formed from the Tyr OHgroup during the Tyr→ Phe mutation were not required forstage (ii)]. Each mutation was carried out in 21 windows, asdescribed above, and so the total duration of the mutation was1.25 ns. Free energy is a state function and so the total freeenergy change is still meaningful even though the mutationwas carried out via the hypothetical amino acid Ala*. Theadditional force field parameters used in the free energysimulations are available from the authors by request.
Correlated mutation analysis
The adrenergic receptor sequences used were obtained fromthe GPCRDB database (Vriend); this is a G-protein coupledreceptor database website. The alignments were obtained usingthe profile alignment method in the protein modelling programWHATIF (Vriend, 1990) which is similar to that of Sanderand Schneider (1991). The GPCRDB mnemonics for thesequences are given in Table I. Pazoset al. (1997) warn thatat least 15 sequences with reasonable diversity are usuallyrequired for a reliable CMA and here we have used 52sequences spanning all the adrenergic receptor subtypes. CMAmay be more appropriate for GPCRs than for many otherproteins because (i) so many sequences are available; (ii) theyhave a similar fold, particularly in the transmembrane regionwhich is the focus of this study; and (iii) the ligand bindingsite has the characteristics of a domain boundary becauseGPCRs consist of two domains, as shown in Figures 1 and 2(see Oliveiraet al., 1993; Singeret al., 1995; Hornet al.,1998 for interesting applications of CMA to GPCRs). Thesignificance level for reporting the correlated mutations corre-sponded to a correlation coefficient of 0.975, though somecorrelations occurring below this level are discussed. (Thismeans that a small percentage of the reported residues maybe statistical anomalies rather than residues that have genuinelymutated in a correlated manner. For this reason, it is beneficialto couple a correlated mutation analysis with other techniquessuch as molecular modelling or site-directed mutagenesis.)The correlation coefficients,rij, for the pairwise comparisonof sequence positionsi andj is defined fully elsewhere (Singeret al., 1995). This equation is based on sequence alone and isvery similar to other implementations of correlated mutationanalysis which have yielded positive results on problemsrelated to protein folding (Go¨bel et al., 1994; Brenner 1995;Pazoset al., 1997; Ortizet al., 1998); an alternative approach,
1184
Table I. Mnemonics of the adrenergic sequences used in the correlatedmutation analysis
Sequence Mnemonic
1 a1aa-human2 a1aa-bovin3 a1aa-oryla4 a1aa-rabbit5 a1aa-rat6 a1ab-human7 a1ab-mesau8 a1ab-mouse9 a1ab-rat
10 a1ad-human11 a1ad-mouse12 a1ad-rabbit13 a1ad-rat14 a2aa-cavpo15 a2aa-human16 a2aa-mouse17 a2aa-pig18 a2aa-rat19 a2ab-cavpo20 a2ab-human21 a2ab-mouse22 a2ab-rat23 a2ac-cavpo24 a2ac-didma25 a2ac-human26 a2ac-mouse27 a2ac-rat28 a2ad-human29 a2ar-carau30 a2ar-labos31 b1ar-canfa32 b1ar-human33 b1ar-macmu34 b1ar-melga35 b1ar-mouse36 b1ar-rat37 b1ar-xenla38 b2ar-bovin39 b2ar-canfa40 b2ar-human41 b2ar-macmu42 b2ar-mesau43 b2ar-mouse44 b2ar-pig45 b2ar-rat46 b3ar-bovin47 b3ar-canfa48 b3ar-human49 b3ar-macmu50 b3ar-mouse51 b3ar-rat52 b4ar-melga
as followed by Tayloret al. (1994, 1997), is to build in aminoacid properties such as volume or hydrophobicity which maybe related to the underlying physical properties in correlatedmutations which also have a compensatory nature. However,this alternative approach has not been as successful and thismay be because compensatory mutations do not necessarilyoccur in the protein core as alternative mechanisms foralleviating strain are possible—see for example Lesk andChothia (1980), van Gunsteren and Mark (1992), Hubbardet al. (1994), Hornet al. (1998). Correlated mutations becameparticularly useful when it was realized by Oliveiraet al.(1997) and Hornet al. (1998) that above all it is the functionthat must be considered, and that to achieve this mutations atdomain boundaries (where binding and catalytic sites typically

Domain swapping in G-protein coupled receptor dimers
reside), rather than in protein cores, must be compensated for.Interestingly, mutations related to function have also beendetected at protein–protein interfaces which are similar todomain interfaces (Hornet al., 1998).
Elsewhere, Pazoset al. (1997) has shown that correlationmutation analysis can be used to single out the right inter-domain docking solution amongst many alternatives for twodomain proteins. Consequently, we have implemented theirharmonic average (Xd) according to their formula
i5nPic – PiaXd 5 Σ (2)di 3 ni51
wherePic is the percentage ofinter-monomercorrelated pairswith distance betweendi anddi–1 and Pia is the same percentagefor all pairs of positions. Heredi represents the upper limit ofone of n distance bins and each bin has a span of 4 Å. Thedifference between the two percentages is weighted by a factorinversely proportional to the distance of the corresponding binto increase the weight of closer distances. The position of eachamino acid is determined from the coordinates of the Cβ exceptin the case of glycine where the Cα coordinates were used.
The correlated mutation analysis, the determination ofchanges in surface accessible area and much of the interactivemodelling were performed using the WHATIF protein model-ling software (Vriend, 1990). The residue numbers reportedare for the humanβ2-adrenergic receptor.
ResultsThe energies obtained from the molecular dynamics simulationsare shown in Figure 3. Figure 3a shows the potential energyfor the three dimer structures in the absence of ligand. Figure 3bshows the potential energy for the alternative dimer structurescontaining one mole of antagonist (propranolol) and Figure 3cshows the corresponding energies in the presence of one moleof agonist (norepinephrine). For clarity, the energies weredumped every 0.5 ps and displayed as the average over theprevious 10 ps.
Figure 4 shows the correlated residues displayed on a snakediagram. It also shows the residue–residue interactions at the5-6 interface, identified either from their interaction energy orusing the WHATIF molecular graphics software. The helix–helix packing angles at the interface are generally around theoptimum value of123° proposed by Chothiaet al. (1981)and indeed the interactions at the 5-6 interface generally belongto the class 3-4 set of interactions where sidechains separatedby three residues form a series of ridges which lie in thegrooves formed between ridges separated by four residues;this pattern is most readily observed by viewing only the Cβatoms of the interfacial sidechains. The same general patternis observed in all three 5-6 domain swapped dimers but ineach case the packing is asymmetric. Moreover, the agonistinduces rotation and a displacement perpendicular to themembrane plane in both helices 5 and 6 and this is observedin both the monomer (Gouldsonet al., 1997a) and the dimer.Consequently, the precise identity of the residue comprisingthe ridges and grooves at the interface is different in all three5,6-domain swapped dimers, as recorded in Table II. Thestructures are however related by translation along the grooves.
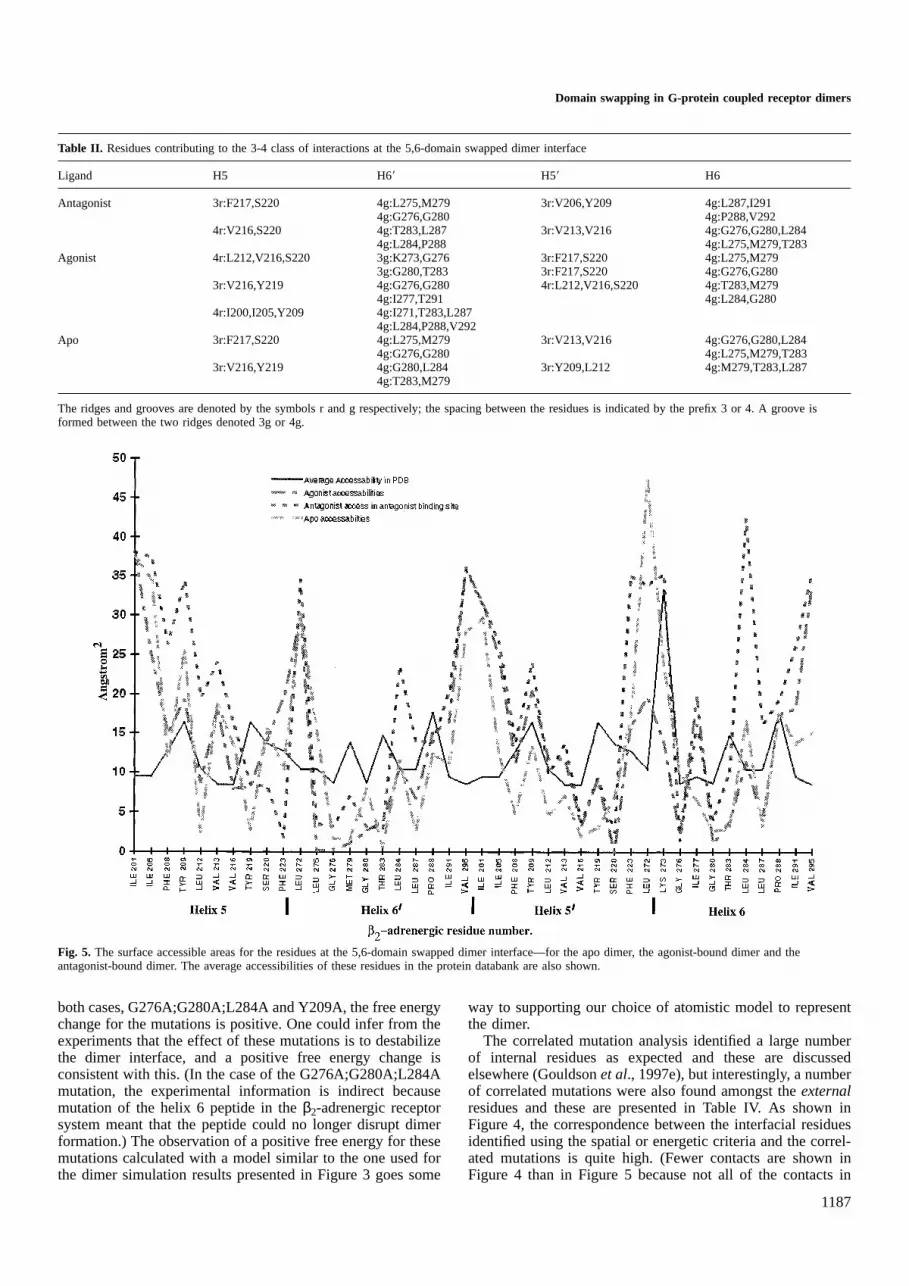
The accessible surface area of each interfacial residue inthe various 5,6-domain swapped dimer structures is shown inFigure 5 and so this figure may be used to identify the residuesmaking contact at the interface. Here it is apparent that the
1185
Fig. 3. The variation of potential energy with time during the moleculardynamics simulations for (a) the apo-receptor dimers, (b) the dimer–antagonist complexes and (c) the dimer–agonist complexes. (The dimerstructures are shown in Figure 2.)

P.R.Gouldsonet al.
Fig. 4. (a) A snake diagram of theβ2-adrenergic receptor displaying the correlated residues which occur at a correlation cut-off value of 0.975 or higher (see alsoTable IV). The diagram also shows the key interactions at the 5-6 interface of the domain swapped dimer, as identified using spatial and energetic criteria. Theconserved residues are denoted by black circles, with an uppercase letter to denote residues identified as external; the correlated residues are denoted by grey circleswith a white letter to denote residues identified as external. (b) The corresponding results for a correlation cut-off value of 0.95 or higher.
residues are generally less accessible at the intracellular endof helices 5 and 6 and indeed the closer contacts in this regionare apparent from a molecular graphics analysis. The interfacialresidues in the antagonist-bound dimer generally have the
1186
highest accessibilities. The results in Figures 4 and 5 weredetermined by averaging the structures generated at 50 psintervals.
The free energy differences are reported in Table III. In
Domain swapping in G-protein coupled receptor dimers
Table II. Residues contributing to the 3-4 class of interactions at the 5,6-domain swapped dimer interface
Ligand H5 H69 H59 H6
Antagonist 3r:F217,S220 4g:L275,M279 3r:V206,Y209 4g:L287,I2914g:G276,G280 4g:P288,V292
4r:V216,S220 4g:T283,L287 3r:V213,V216 4g:G276,G280,L2844g:L284,P288 4g:L275,M279,T283
Agonist 4r:L212,V216,S220 3g:K273,G276 3r:F217,S220 4g:L275,M2793g:G280,T283 3r:F217,S220 4g:G276,G280
3r:V216,Y219 4g:G276,G280 4r:L212,V216,S220 4g:T283,M2794g:I277,T291 4g:L284,G280
4r:I200,I205,Y209 4g:I271,T283,L2874g:L284,P288,V292
Apo 3r:F217,S220 4g:L275,M279 3r:V213,V216 4g:G276,G280,L2844g:G276,G280 4g:L275,M279,T283
3r:V216,Y219 4g:G280,L284 3r:Y209,L212 4g:M279,T283,L2874g:T283,M279
The ridges and grooves are denoted by the symbols r and g respectively; the spacing between the residues is indicated by the prefix 3 or 4. A groove isformed between the two ridges denoted 3g or 4g.
Fig. 5. The surface accessible areas for the residues at the 5,6-domain swapped dimer interface—for the apo dimer, the agonist-bound dimer and theantagonist-bound dimer. The average accessibilities of these residues in the protein databank are also shown.
both cases, G276A;G280A;L284A and Y209A, the free energychange for the mutations is positive. One could infer from theexperiments that the effect of these mutations is to destabilizethe dimer interface, and a positive free energy change isconsistent with this. (In the case of the G276A;G280A;L284Amutation, the experimental information is indirect becausemutation of the helix 6 peptide in theβ2-adrenergic receptorsystem meant that the peptide could no longer disrupt dimerformation.) The observation of a positive free energy for thesemutations calculated with a model similar to the one used forthe dimer simulation results presented in Figure 3 goes some
1187
way to supporting our choice of atomistic model to representthe dimer.
The correlated mutation analysis identified a large numberof internal residues as expected and these are discussedelsewhere (Gouldsonet al., 1997e), but interestingly, a numberof correlated mutations were also found amongst theexternalresidues and these are presented in Table IV. As shown inFigure 4, the correspondence between the interfacial residuesidentified using the spatial or energetic criteria and the correl-ated mutations is quite high. (Fewer contacts are shown inFigure 4 than in Figure 5 because not all of the contacts in

P.R.Gouldsonet al.
Table III. Calculated free energy differences/kcal mol–1
Mutation Total simulation time/ps ∆G
G276A;G280A;L294A 420 1.16 0.2A276G;A280G;A284L 420 –2.506 0.01G276A;G280A;L284A 11a 2.2 6 0.8Y209F 420 –0.06456 0.0005F209A* 420 5.76 0.4A*209A 420 –1.36 0.3b
The simulations were performed on the four helix bundle representing the5,6-dimer interface unless indicated otherwise. The error bars givenrepresent the difference between forward and reverse sampling (Reynoldset al., 1992).aSimulation performed on full 5,6-dimer.bThe total free energy change for the Y209A mutation is 3.760.5 kcal mol–1.
Table IV. The full network of correlated mutations, identified using acorrelation level of 0.975 or higher
Both theβ2 and the universal numbering scheme of Oliveiraet al. (1993)are used.
the apo or antagonist-bound dimers are preserved in theagonist-bound dimer. Likewise, many of the residues whichcontribute to the interfacial ridges or grooves do not makeserious contacts in the dimer because the side chain pointsaway and these do not appear in Figure 4.)
The Xd values (Pazoet al., 1997) are –0.23 and –0.26 forthe 1,2- and 1,7-dimer in the presence of antagonist—anantagonist has essentially no effect on the structure (Gouldsonet al., 1997) and10.13 for the 5,6-dimer in the absence ofligand; this goes up to10.38 in the presence of agonist. The
1188
value of Xd is relatively insensitive to the correlation cut-offvalue chosen; moreover the same qualitative picture is retainedif the conserved residues are included in theXd analysis.
Discussion
Energetics, agonism and domain swapping
The main evidence for domain swapping comes from thechimeric studies of Maggio (1993a) and the double mutantstudies of Monnot (1996). If we assume that the 5,6-domainswapped dimer is the high affinity active form, the results inFigures 1 and 3 give us a new framework for discussingreceptor activation. In Figures 3a and b the mean energy ofthe 5,6-domain swapped dimer from 50 to 100 ps onwards ishigher than the other two systems by almost 300 kJ mol–1,and this is consistent with the general requirement that anagonist is required for activation since the large energydifference would ensure that only a small proportion of thereceptors were in the active form. Figure 3c shows that theenergy of the 5,6-domain swapped dimer is significantlylowered (relative to the other structures) in the presence ofagonist. This is consistent with the idea that the agonistfunctions by shifting the equilibrium in favour of the activeform, here proposed to be the 5,6-domain swapped dimer. Itis dangerous to assume that these energies have any quantitativesignificance because molecular mechanics energies are relativerather than absolute and because the treatment of solvation isonly partial. Because the energies are relative, it is possible tocompare energies within a figure (e.g. within Figure 3a) but itis not possible to compare energies between figures—thus itis not valid to compare energies of the antagonist and agonist-bound dimers in Figures 3b and c. However, a number ofgeneral conclusions may be made and here we will discusshow these relate to our understanding of G-protein coupledreceptor activation.
For example, the results are not inconsistent with the ideathat the low level of intrinsic activity in the absence of ligandcould arise from the small equilibrium concentration of the5,6-domain swapped dimer. Indeed, the autoactivation ofG-protein coupled receptors when overexpressed in transgenicmice (Bondet al., 1995) can be interpreted by assuming thatthe basal activity of G-protein coupled receptors is related tothe rate of dimer formation and that dimer formation is greatlyassisted by agonist binding or a higher concentration ofreceptors. While there is as yet no definitive proof that dimerformation is essential for activation, our results and hypothesisare certainly consistent with Bond’s results.
Given the view that agonism follows dimer formation, it isrelatively straightforward to explain the activation of receptorsby both bivalent antibodies (Leiberet al., 1984) and byantagonistscoupled back to back (Hazum and Keinan, 1985)since they will assist in bringing two monomers together.Again, the activation of G-proteins obtained by couplingtogether two copies of peptides from intracellular loop three(Wade et al., 1994) is consistent with this view of receptoractivation as it suggests that the G-protein requires two copiesof this loop. [Intracellular loop three connects helices five andsix and is essential for activity (Straderet al., 1994).] Moreover,this latter observation supports the idea that the active dimeris the 5,6-domain swapped dimer rather than say the 1,2-dimeras observed in the rhodopsin two-dimensional projection maps.The two copies of intracellular loop three are about 70 Å apartin the projection maps while the linker is merely a disulphide

Domain swapping in G-protein coupled receptor dimers
bridge. However, the two copies of intracellular loop threemove much closer together in the 5,6-domain swapped dimer(by about 40 Å; Gouldson and Reynolds, 1997b). Ciruelaraised antibodies against the second extracellular loop and thethird intracellular loop of the A1-adenosine receptor (Ciruelaet al., 1995), and so unwittingly provided further evidence forthe 5,6-domain swapped dimer. He described identification ofboth monomers and dimers and their interconversion by agonistor antagonist binding. However, the antibody against the thirdintracellular loop did not bind to the dimer. If the physiologicaldimer is indeed a 5,6-domain swapped dimer, then the reasonsfor this are apparent in the scheme shown in Figure 1, sincethe two adjacent copies of intracellular loop three wouldsterically hinder each other and prevent access by the antibody.The hinge loop between the two domains, i.e. intracellularloop three, is frequently the largest intracellular loop and thiswill facilitate domain swapping. Maggioet al. (1996) hasshown that shortening this loop results in a loss of activityand such behaviour is characteristic of domain swapping.
Structural changes
It is well known throughout computational chemistry thatenergy changes are difficult to predict to high accuracy butthat structural changes can be predicted with more certainty(Williams, 1993). Indeed, in our monomer–agonist simulations(Gouldsonet al., 1997a) we observe very similar structuralchanges to those of Zhang and Weinstein (1993) and Louet al.(1994), namely a twist and vertical displacement of helicesfive and six resulting in a modest change in their tilt angle.These computational results are consistent with the experiment-ally derived information on the nature of the agonist-inducedconformational change (see for example Getheret al., 1997a,1995; Javitchet al., 1997). The ligand induced conformationalchanges observed here in the dimer are essentially thoseobserved in the monomer (Gouldsonet al., 1997a). Con-sequently, analysis of the structural changes during the simula-tions shows that the agonist induced conformational changeoptimizes the helix–helix packing at the 5,6-dimer interface.Thus, in the 5,6-domain swapped dimer–agonist complex theinterfacial helices most readily adopt the optimal packing angleof about 23° (Cohenet al., 1979; Chothiaet al., 1981;Sternberget al., 1982), though all three dimer structuresinteract via the required 3-4 interactions. The binding of asecond agonist molecule would also affect the tilt angles ofhelices five and six in the second receptor. However, thiswould be less favourable as the helix–helix packing at the5-6 interface would no longer be optimal. This second site inthe second receptor could therefore correspond to the lowaffinity form. These structural changes may also explainthe origin of bell-shaped dose response curves which areoccasionally observed in G-protein coupled receptors (Ja¨rv,1995) and which are characteristic of dimerization in othersystems (De Meytset al., 1995) since they explain why bindingof additional agonist may destabilize the active receptor andresult in lower activity. (Bell-shaped dose response curves mayalso arise for other reasons.)
The simulations show that the antagonist essentially doesnot affect the helix–helix packing since the antagonist causesonly minor conformational changes in both the monomer andthe dimer. Consequently, our simulations are consistent withthe observation that there is no distinction between high andlow affinity forms for antagonist binding. In the monomersimulations (Gouldsonet al., 1997a), the partial agonist pindo-
1189
lol causes a change in helix five but not helix six. Consequently,the corresponding 5,6-domain swapped dimer would be stabil-ized by only one out of a possible two optimal helix packinginteractions. Such partially optimized interactions may underliepartial agonism. The high energy of the 1,7-dimer–agonistcomplex (Figure 3c) suggests that this species is unlikely tobe formed. Consequently, to form the 5,6-domain swappeddimer, the agonist may bind directly to the open form shownin Figure 1 and indeed since the extracellular loops form atight canopy over the receptor this may be the kineticallypreferred route. In addition, the high energy of the 1,7-dimer–agonist complex may help to increase the concentration of theactive 5,6-domain swapped dimer by preventing the 1,2-dimer(or monomer) from re-forming.
Inspection of Figure 1 and the various dimer structures alsoallows us to propose a distinction between inverse agonistsand antagonists. An inverse agonist would shift the equilibriumaway from the 5,6-domain swapped dimer. As such it is likelyto favour the formation of monomers, here most closelyrepresented by the 1,2-dimer. Support for this idea comes fromthe experimental observations of Hebertet al. (1996) for theβ2-adrenergic receptor which have shown inverse agonists tofavour reversal of dimer formation. There is little evidencefrom Figure 3 that propranolol does this. However, Figure 5does show that the propranolol-bound dimer residues generallyhave the highest accessibility.
Buried surface area is often taken as a measure of thestrength of a protein–protein interfacial interaction (Stites,1997). Figure 5 indicates the residues with the greatest changein buried surface area: residues whose accessibility is less thanthe average in the 5,6-domain swapped dimer interface includeLeu212, Val216, Ser220, Leu275, Gly276, Met279, Gly280, Thr283
and Leu287. It is interesting that Leu212, Val216, Gly276, Met279,Thr283 and Leu287 were all identified as external correlatedresidues at a level of 0.8 or higher and that Leu275, Gly280 andGly284 are conserved or essentially conserved in the adrenergicreceptors. Tyr209 is not in this list, but here the interaction islargely electrostatic rather than due to dispersion/hydrophobicinteractions, as discussed below.
For many years, scientists have been puzzled as to whysmall structural changes to a ligand can cause a switch inbehaviour from that of an agonist to that of an antagonist (orvice versa). However, Murrayet al. (1995) have shownthat the energy difference between monomers and a domainswapped dimer maybe small and that single mutations can havea noticeable effect on this equilibrium. This latter observationprobably underlies constitutive activation which can arise inmutated G-protein coupled receptor—see for example thework by Paschkeet al. (1994), Spaldinget al. (1997) andLaue (1995).
Correlated mutation analysis
It may be significant that many of the external correlatedmutations (Table IV) lie at the 5-6 interface and that there isa high correspondence with the external residues identifiedusing the spatial, energetic or surface area criteria, as shownin Figures 4 and 5. The work of Pazoset al. (1997) on over20 proteins has certainly shown that correlated residues tendto accumulate at inter-domain and inter-protein interfaces.According to Pazos, theXd descriptor can be used to singleout the right inter-domain docking solution from many wrongalternatives. Pazoset al. found (for nine proteins) that a higherXd was associated with a good docked structure while a more

P.R.Gouldsonet al.
negativeXd was likely to indicate an incorrect structure. Herewe have only considered two ‘wrong’ alternatives (the 1,2-and 1,7-dimers) to the 5,6-domain swapped dimer but sincethese both have negativeXd values while the 5,6-dimer has apositive Xd value, it appears that a preference for the 5,6-domain dimer is consistent with this analysis. (This analysisis unable to distinguish between a 5,6-contact dimer and a5,6-domain swapped dimer.) We have also carried out a CMAon the major histocompatibility complex (MHC) class IIreceptors of the human leucocyte antigen (HLA) and againhave observed an accumulation of correlated residues at theprotein interface—65% of all correlated residues in HAL DR(23 sequences for theβ chains), HLA DP (23 sequences) andHLA DQ (24 sequences) occur at the domain and dimerinterfaces (Nilsson,A,. Ferna´ndez,N. and Reynolds,C.A., per-sonal communication). For the human MHC class II receptorthere is a crystal structure of the dimer of heterodimers(Jardetzky et al., 1996) but more recently Cherryet al.(1998) have used single particle imaging on living cells andimmunochemical techniques on detergent-lysates to followreceptor dimerization on the cell surface and have also shownthat the MHC class II receptors form temperature dependentdimers. The evidence is therefore growing that correlatedmutations are found at protein interfaces, as suggested byOliveira et al. (1993) and Hornet al. (1998).
Some of the adrenergic correlated mutations can be explainedby considering the packing effects of the 5-6 interface. Themost interesting correlated mutation involves Tyr209 which hasbeen shown by the calculations to make a strong interactionwith helix six through a hydrogen bond to the backbonecarbonyl group of Leu284. It appears that where Tyr209 isreplaced by Phe, aromatic stacking from the extra Phe atposition 287 (which seems to require a smaller valine atposition 284) compensates for the loss of hydrogen bonding.
Some researchers may question whether hydrophobic res-idues alone are able to provide a highly specific dimer interface.Certainly Larsenet al. (1998) in a study of 136 homodimericproteins found that most protein interfaces were not entirelyhydrophobic—unless the dimer was domain swapped (10cases). In a study of helix–helix interactions in globularproteins, Chothiaet al. (1981) observed that half of theseinteractions were mediated by hydrophobic interactions alone.More significantly, in the glycophorin A dimer, which involvestwo transmembrane helices, theL75IXXGVXXGVXXT87 motifis responsible for dimerization. This motif is very sensitiveto minor changes, such as mutation of Val84 to Leu or Ile76 toAla (Lemmonet al., 1992, 1994; MacKenzieet al., 1997) butthe authors were still able to generate a good model usingmolecular dynamics. Moreover, these are precisely the kindof changes observed in the external correlated mutations.
The evolutionary trace (ET) method (Lichtage, 1996a,b,1997) has many similarities to correlated mutation analysisand may be used to identify active sites or functional interfaces,as has been shown for G-proteins, SH2 and SH3 domainsand for the zinc binding domains of intracellular receptors.Inspection of the sequence alignment and the phylogenetictree for the adrenergic receptors suggests that a number ofconserved and class specific positions lie at the 5–6 interface.For example Phe208, Leu275 and Gly280 are conserved whilePhe223 and His269 are essentially class-specific positions at aposition identity cut-off (PIC) of 30% while Tyr209 is a class-specific position at a PIC of 40%. Many of the correlatedresidues at this interface become class-specific positions at
1190
PICs of about 50–60%. The concurrence between these twomethods, CMA and ET, provides further evidence that residuesat the 5–6 interface have an important function.
The significance of some of the external conserved andcorrelated residues has been probed by site-directed mutagen-esis: mutation of Tyr205 in the neurokinin NK-1 receptor (Tyr209
in β2-AR) receptor, results in loss of activity (Huanget al.,1994) (this is thought to be an external residue though theauthors do not identify it as such). Similar results were obtainedfor the corresponding Tyr206 in the neurokinin NK-2 receptor(Huang et al., 1995). Neither of these mutations affectedantagonist binding, and this is consistent with, but not proofof, the idea that the mutation affects helix–helix packing atthe dimer interface. Similarly, mutation of Gly276, Gly280 andLeu284 to alanine in Hebertet al.’s helix six peptide (1996)significantly reduced its ability to inhibit dimerization; again,these correspond to external correlated residues. Because therotational orientation of each helix in our model is in agreementwith much experimental data, including Baldwin’s coordinates(Baldwin et al., 1997) which are based on Unger’s recentexperimental projection map (Ungeret al., 1997), it is likelythat the assignment of these external residues is correct. Thepositive free energy differences recorded in Table III lendsupport to our receptor model in which these residues lie atthe dimer interface.
The correlated mutations on helices one, two and sevenmay be involved in assisting in the formation of the 1,7-intermediate. There may be data to suggest that helix seven isimportant in dimerization since mutation of the conservedTrp313 on the external face of helix seven does result in asmall loss of activity (Wesset al., 1993)—though Trp313 isintermediate between internal and external (Gouldsonet al.,1997a) and its precise location is currently unclear. Nget al.(1996) have shown that peptides derived from transmembranehelix seven can inhibit dimerization. We propose that the mostlikely explanation of Ng’s observations is that helix sevenblocks the activation process by preventing formation of the1,7-intermediate rather than by binding to the dimer interface.In this context it must be remembered that the helix one–helixseven interaction is probably the weakest as it involves parallelhelix dipoles; this is probably an important factor in initiatingthe domain swapping process.
The correlated mutations were derived independently fromthe structural model and so the agreement between the twoapproaches—correlated mutation analysis and homology mod-elling with simulation—is encouraging, particularly as the roleof key residues is supported by site-directed mutagenesis(Huanget al., 1994, 1995; Hebertet al., 1996), free energysimulations and the evolutionary trace method.
Elsewhere we have investigated correlated mutations in anumber of other systems, including the dopaminergic andmuscarinic receptors and we have again found correlatedmutations amongst the external residues of each helix(Gouldsonet al., 1997d). Singeret al. (1995) also found acorrelated mutation amongst the external residues of theolfactory receptors (but the significance was not discussed).
Many of the internal correlated residues in the adrenergic,dopaminergic and muscarinic receptors correlate with subtypespecificity and indeed mutation of these residues has beenshown to switch the ligand binding properties to that of thealternative subtype (Ferenczyet al., 1997; Gouldsonet al.,1997e). Such observations show that correlated mutationanalysis does indeed give results which are closely related to

Domain swapping in G-protein coupled receptor dimers
Fig. 6. Formation of a 4,5-domain swapped dimer. This mechanism mayexplain the recovery of binding because domain swapping regenerates one‘intact’ receptor from two defective receptors. (The helices with fatalmutations are shown in black.) The failure to recover activity may arisebecause the two copies of intracellular loops three are not in the correctrelative orientation—see Figure 1.
experiment, at least in this family of receptors, and givesconfidence that the external residues predicted by this techniquemay indeed have a functional role.
Possible advantages of domain swappingGiven the evidence in favour of the 5,6-domain swappeddimer, it is probably important to ask what are the possibleadvantages of forming a 5,6-domain swapped dimer as opposedto say a 5,6-contact dimer? Firstly, domain swapping is a veryefficient method of forming oligomers since the interactionswithin the monomer are reused in the oligomer. This principlehas been elegantly elucidated by Bennettet al. (1994, 1995).Moreover, Tegoniet al. (1996) has shown how domainswapping can be part of a normal activation mechanism.Secondly, the results of Monnotet al. (1996) suggest thatdomain swapping may also result in the formation of a 4,5-domain swapped dimer. The mechanism in Figure 6 showshow coexpression of an angiotensin II receptor with a fatalmutation on helix three with one containing a fatal mutationon helix five can result in formation of a dimer which regainsits ability to bind ligands because the two fatal mutations canbe swapped out to generate a seven helix bundle which isfully functional in terms of binding. (The additional energyfrom the ligand binding process may be the reason why thisunusual domain swapped dimer is formed.) However, thisreceptor is not able to activate G-proteins and a possibleexplanation for this is that the two copies of the intercellularloops are in the wrong relative orientation (compare Figures1 and 6). Thus, a 5,6-domain swapped dimer may be requiredto present the two copies of intracellular loop three in theorientation required for G-protein activation. (Thus, we wouldsuggest that coexpression of receptors with fatal mutations onsay helix three and helix six would result in receptors able tobind ligands and activate G-proteins.) Finally, domain swappingmay be a mechanism for minimizing the effects of loss offunction mutations in important receptors provided that twocopies of the gene are expressed and that each copy is mutatedin a different domain. The phenomena of regaining activityby mixing defective multimeric proteins has been known fora long time (Fincham and Pateman, 1957; Fincham, 1962;Garen and Garen, 1963; Schlesinger and Levinthal, 1963) and
1191
so it is quite plausible that similar mechanisms may occur fordomain swapped multidomain proteins.
ConclusionsIn summary, a fusion of theoretical and experimental data hasbeen used to investigate the hypothesis that G-protein coupledreceptor activation involves receptor dimers. In particular, wehave investigated the possibility that the activation processmay involve domain swapped dimers. This domain swappingmodel has provided a useful framework for discussing themolecular events associated with receptor activation. In particu-lar, the results are consistent with the idea that the active, highaffinity form of the receptor is a 5,6-domain swapped dimerand that the agonist functions by favouring dimer formation.Our simulations suggest that this is achieved by an agonist-induced conformational change which optimizes the helix–helix interactions at the 5-6 interface. Within this model anantagonist would have no effect on the helix–helix interactionsat the dimer interface. An inverse agonist would generatehelix–helix interactions which are even less favoured than inthe apo-dimer, and would thus shift the equilibrium towardsthe monomers. Indeed the results for propranolol are notinconsistent with this idea.
The molecular nature of the 5,6-domain swapped dimer hasbeen supported by a correlated mutation analysis through theidentification of statistically significant and highly correlatedpatterns of change amongst the external residues at the 5-6dimer interface at precisely the locations predicted by thesimulations. Site-directed mutagenesis experiments and freeenergy difference simulations on these residues provides furtherevidence for the mechanism discussed here, as does theevolutionary trace method. At present the importance of domainswapping in the wider context is not yet known. For example,it is not yet clear whether dimerization is important for allclasses of G-protein coupled receptors. For example, the resultsof Cvejic and Devi (1997) on the opioid receptor suggest thatagonist decreases the concentration of dimers. This is incontrast to the work of Hebertet al. (1996) on theβ2-adrenergicreceptor which showed that agonist increased the dimer concen-tration. There are a number of possible explanations for thesediffering conclusions. For example, the opioid receptor mostlikely couples to Gi/Go whereas theβ2-adrenergic receptorcouples to Gs. Alternatively, since Cvejic and Devi did notcarry out any dose response experiments, we cannot rule outthe possibility that they are observing the tail end of a bell-shaped dose response curve (see Figure 2 of their article)Either way, both groups observe that dimerization is important.It is possible that domain swapped dimers only becomeimportant when functional rescue is required and some receptordimers could perhaps be the substrates for auxiliary enzymessuch as kinases (Palczewski and Benovic, 1991). However,taken together with the other evidence for receptor dimerizationpresented here, this study offers evidence that dimerizationand domain swapping are important events in G-proteincoupled receptor activation which certainly merit furtherexperimental and theoretical investigation.
AcknowledgementsThis work was supported by Novo Nordisk, the BBSRC (B/06081) and theEPSRC (94309861). We wish to thank Novo Nordisk for supporting inpresenting aspects of this work at the GPCR meeting in Uppsala, 1996. Wealso wish to thank Jon Essex for helpful discussions relating to free energysimulations.

P.R.Gouldsonet al.
ReferencesAltenbach,C., Yang,K., Farrens,D.L., Farahbakhsh,Z.T., Khorana,G. and
Hubbell,W.L. (1996)Biochemistry, 35, 12470–12478.Baldwin,J.M. (1994)Curr. Opin. Struct. Biol., 6, 180–190.Baldwin,J.M., Schertler,G.F.X. and Unger,V.M. (1997)J. Mol. Biol., 272,
144–164.Beveridge,D.L. and DiCapua,F.M. (1989)Annu. Rev. Biophys. Biophys. Chem.,
18, 431–492.Bennett,M.J., Choe,S. and Eisenberg,D. (1994)Proc. Natl Acad. Sci. USA,
91, 3127–3311.Bennett,M.J., Schlunegger,M.P. and Eisenberg,D. (1995)Protein Sci., 4,
2455–2468.Bluml,K., Mutschler,E. and Wess,J. (1994)J. Biol. Chem., 269, 18870–18876.Bond,R.A., Leff,P., Johnson,T.D., Milano,C.A., Rockman,H.A., McMinn,T.R.,
Apparsundaram,S., Hyek,M.F., Kenakin,T.P., Allen,L.F. and Lefkowitz,R.J.(1995)Nature, 374, 272–276.
Bywater,R.P., Vriend,G., Oliveira,L. and van Alten,D. (1995) In Hohr,H. andBrunak,S. (eds)Protein Folds, A Distance Based Approach. CRC Press,Boca Raton, pp. 174–185.
Cameron,A.D., Olin,B., Ridderstro¨m,M., Mannervik,B. and Jones,T.A. (1997)EMBO J., 16, 3386–3395.
Cherry,R.J., Wilson,K.M., Triantafilou,K., O’Toole,P., Morrison,I.G.,Smith,P.R. and Ferna´ndez,N. (1998)J. Cell Biol., 140, 71–79.
Chothia,C., Levitt,M. and Richardson,D. (1981)J. Mol. Biol., 145, 215–250.Ciruela,F., Casado,V., Mallol,J., Canela,E.I., Lluis,C. and Franco,R. (1995)
J. Neurosci. Res., 2, 818–828.Cohen,F.E., Richmond,T.J. and Richards,F.M. (1979)J. Mol. Biol., 132,
275–288.Corless,J.M., McCaslin,D.R. and Scott,B.L. (1982)Proc. Natl Acad. Sci. USA,
79, 1116–1120.Cvejic,S. and Devi,L.A. (1997)J. Biol. Chem., 272, 26959–26964.De Meyts,P., Ursø,B., Christoffersen,C.T. and Shymko,R.M. (1995)Ann. NY
Acad. Sci., 766, 388–401.Farahbakhsh,Z.T., Ridge,K.D., Khorana,H.G. and Hubbell,W.L. (1995)
Biochemistry, 34, 8812–8819.Farrens,D.L., Altenbach,C., Yang,K., Hubbell,W.L. and Khorana,H.G. (1996)
Science, 274, 768–770.Ferenczy,G.G., Winn,P.J. and Reynolds,C.A. (1997)J. Phys. Chem., 101,
5446–5455.Fincham,J.R.S. (1962)J. Mol. Biol., 4, 257–274.Fincham,J.R.S. and Pateman,J.A. (1957)Nature, 179, 741–742.Garen,A. and Garen,S. (1963)J. Mol. Biol., 7, 13–22.Gether,U., Lin,S. and Kobilka,B.K. (1995)J. Biol. Chem., 270, 28268–28275.Gether,U., Lin,S., Ghanouni,P., Ballesteros,J.A., Weinstein,H. and
Kobilka,B.K. (1997a)EMBO J., 16, 6737–6747.Gether,U., Ballesters,J.A., Seifert,R., Sanders-Bush,E., Weinstein,H. and
Kobilka,B.K. (1997b)J. Biol. Chem., 272, 2587–2590.Gobel,U., Sander,C., Schneider,R. and Valencias,A. (1994)Proteins, 18,
309–317.Gouldson,P.R., Winn,P.J. and Reynolds,C.A. (1995)J. Med. Chem., 38,
4080–4086.Gouldson,P.R. and Reynolds,C.A. (1997b)Biochem. Soc. Trans., 25,
1066–1071.Gouldson,P.R., Snell,C.R. and Reynolds,C.A. (1997a)J. Med. Chem., 40,
4871–3886.Gouldson,P.R., Snell,C.R., Bywater,R.P. and Reynolds,C.A. (1997c)Biochem.
Soc. Trans., 25, 429S.Gouldson,P.R., Bywater,R.P. and Reynolds,C.A. (1997d)Biochem. Soc. Trans.,
25, 529S.Gouldson,P.R., Bywater,R.P., Snell,C.R. and Reynolds,C.A. manuscript in
preparation.Gouldson,P.R., Bywater,R.P., and Reynolds,C.A. (1997e)Biochem. Soc. Trans.,
25, 434S.Gouldson,P.R., Bywater,R.P., and Reynolds,C.A. manuscript in preparation.Guddermann,T., Scho¨neberg,T. and Schultz,G. (1997)Annu. Rev. Neurosci.,
20, 399–427.Hazum,E. and Keinan,D. (1985)Biochem. Biophys. Res. Commun., 133,
449–456.Hebert,T.E., Moffett,S., Morello,J.-P., Loisel,T.P., Bichet,D.G. and Bouvier,M.
(1996)J. Biol. Chem., 271, 16384–16392.Herzyk,P. and Hubbard,R.E. (1995)Biophys. J., 69, 2419–2442.Herzyk,P. and Hubbard,R.E. (1996)Biophys. J., 70, 169.Horn,F., Bywater,R.P., Krause,G., Kuipers,W., Oliveira,L., Paiva,A.C.M.,
Sander,C. and Vriend,G. (1998)Receptors Channels, 5, 305–314.Huang,R.-R.C., Yu,H., Strader,C.D. and Fong,T.M. (1994)Biochemistry, 33,
3007–3013.
1192
Huang,R.R.C., Vicario,P.P., Strader,C.D. and Fong,T.M. (1995)Biochemistry,34, 10048–10055.
Hubbard,S.J., Gross,K.-H. and Argos,P. (1994)Protein Engng,, 7, 613–626.Jarv,J. (1995)J. Theor. Biol., 175, 577–582.Javitch,J.A., Fu,D., Liapakis,G. and Chen,J. (1997)J. Biol. Chem., 272,
18546–18549.Karplus,M. and Petsko,G.A. (1990)Nature, 347, 631–639.Kobilka,B.K., Kobilka,T.S., Daniel,K., Regan,J.W., Caron,M.G. and
Lefkowtiz,R.J. (1988)Science, 240, 1310–1316.Kristiansen,K., Dahl,S.G. and Edvardsen,X. (1996)Proteins Struct. Funct.
Genet., 26, 81–94.Kuipers,W., Oliveira,L., Paiva,A.C.M., Rippmann,F., Sander,C., Vriend,G.,
Krause,C.G., van Wijngaarden,I. and Ijzerman,A.P. (1994)Proceedings ofthe Molecular Graphics Society, Leeds.
Larsen,T.A., Olson,A.J. and Goodsell,D.S. (1998)Structure, 6, 421–427.Laue,L., Chan,W.Y., Hsueh,A.J.W., Kudo,M., Hsu,S.Y., Wu,S.M.,
Blomberg,L.A. and Cutler,G.B. (1995)Proc. Natl Acad. Sci. USA, 92,1906–1910.
Leiber,D., Harbon,S., Guillet,J.G., Andre,C. and Strosberg,A.D. (1984)Proc.Natl Acad. Sci. USA, 81, 4331–4334.
Lemmon,M.A., Flanagan,J.M., Treutlein,H.R., Zhang,J. and Engelmann,D.M.(1992)Biochemistry, 31, 12719–12725.
Lemmon,M.A., Treutlein,H.R., Adams,P.D., Brunger,A.T. andEngelmann,D.M. (1994)Nature Struct Biol., 1, 157–163.
Lesk,A.M. and Chothia,C. (1980)J. Mol. Biol., 136, 225–270.Lichtarge,O., Bourne,H.R. and Cohen,F.E. (1996a)J. Mol. Biol., 257, 342–358.Lichtarge,O., Bourne,H.R. and Cohen,F.E. (1996b)Proc. Natl Acad. Sci. USA,
93, 7507–7511.Lichtarge,O., Yamamoto,K.R. and Cohen,F.E. (1997)J. Mol. Biol., 274,
325–337.Luo,X.C., Zhang,D.Q. and Weinstein,H. (1994)Protein Engng, 7, 1441–1448.MacKenzie,K.R., Prestegard,J.H. and Engelmann,D.M. (1997)Science, 276,
131–133.Maggio,R., Vogel,Z. and Wess,J. (1993a)Proc. Natl Acad. Sci. USA, 90,
3103–3107.Maggio,R., Vogel,Z. and Wess,J. (1993b)FEBS Lett., 319, 195–200.Maggio,R., Barbier,P., Cornai,F. and Corsini,G.U. (1996)J. Biol. Chem., 271,
31055–31060.Main,B.G. (1990) β2-Adrenergic Receptors. In Emmett,J.C. (ed.),
Comprehensive Medicinal Chemistry, Vol. 3. Pergamon, Oxford, pp. 188–228.Monnot,C., Bihoreau,C., Conchon,S., Curnow,K.M., Corvol,P. and Clauser,E.
(1996)J. Biol. Chem., 271, 1507–1513.Murray,A.J., Lewis,S.J., Barclay,A.N. and Brady,R.L. (1995)Proc. Natl Acad.
Sci. USA, 92, 7337–7341.Ng,G.Y.K., O’Dowd,B.F., Lee,S.P., Chung,H.T., Brann,M.R., Seeman,P. and
George,S.R. (1996)Biochem. Biophys. Res. Commun., 227, 200–204.Oliveira,L., Paiva,A. and Vriend,G. (1993)J. Comp. Aided Mol. Des., 7,
649–658.Palczewski,K. and Benovic,K. (1991)TIPS, 16, 387–391.Paschke,R., Tonacchera,M., Vansande,J., Parma,J. and Vassart,G. (1994)
J. Clin. Endocrinol. Metab., 79, 1785–1789.Pazos,F., Helmer-Citterich,M., Ausiello,G. and Valencia,A. (1997)J. Mol.
Biol., 271, 511–523.Potter,L.T., Ballesteros,L.A., Bichajian,L.H., Ferrendelli,C.A., Fisher,A.,
Hanchett,H.E. and Zhang,R. (1991)Mol. Pharmacol., 39, 211–221.Reynolds,C.A., King,P.M. and Richards,W.G. (1992)Mol. Phys., 76, 251–276.Romano,C., Yang,W.-L. and O’Malley,K.L. (1996)J. Biol. Chem., 271,
28612–28616.Rost,B. (1996)Methods Enzymol., 266, 525–539.Sander,C. and Schneider,R. (1991)Proteins Struct. Funct. Genet., 9, 56–68.Scheer,A., Fanelli,F., Costa,T., De Benedetti,P.G. and Cotecchia,S. (1996)
EMBO J., 15, 3566–3578.Schertler,G.F.X., Villa,C. and Henderson,R. (1993)Nature, 362, 770–772.Schlesinger,M.J. and Levinthal,C. (1963)J. Mol. Biol., 7, 1–12.Schlunegger,M.P., Bennett,M.J. and Eisenberg,D. (1997)Adv. Protein Chem.,
50, 61–122.Schoneberg,T., Liu,J. and Wess,J. (1995)J. Biol. Chem., 270, 18000–18006.Singh,U.C., Weiner,P.K., Caldwell,J.W. and Kollman,P.A. (1988) AMBER
(Version 4.1), Department of Pharmaceutical Chemistry, University ofCalifornia, San Francisco.
Spalding,T.A., Burstein,E.S., Wells,J.W. and Brann,M.R. (1997)Biochemistry,36, 10109–10116.
Sternberg,M.J.E., Cohen,F.E. and Taylor,W.R. (1982)Biochem. Soc. Trans.,10, 299–301.
Stites,W.E. (1997)Chem. Rev., 97, 1233–1250.Strader,C.D., Fong,T.M., Tota,M.R. and Underwood,D. (1994)Annu. Rev.
Biochem., 63, 101–132.

Domain swapping in G-protein coupled receptor dimers
Suryanarayana,S. and Kobilka,B.K. (1993)Mol. Pharmacol., 44, 11–114.Suryanarayana,S., Daunt,D.A., von Zastrow,M. and Kobilka,B.K. (1991)
J. Biol. Chem., 266, 15488–15492.Tegoni,M., Ramoni,R., Bignetti,E., Spinelli,S. and Cambillau,C. (1996)Nature
Struct. Biol., 3, 863–867.Timmermans,P.B.M.W.N., Chui,A., Hoolen,A. and Main,B.G. (1990)
β2-Adrenergic receptors. In Emmett,J.C. (ed.),Comprehensive MedicinalChemistry, Vol. 3. Pergamon, Oxford, pp. 133–187.
Unger,V.M. and Schertler,G.F.X. (1995)Biophys. J., 68, 1776–1786.Unger,V.M., Hargrave,P.A., Baldwin,J.M. and Schertler,G.F.X. (1997)Nature,
389, 203–206.van Gunsteren,W.F. and Berendsen,H.J.C. (1977)Mol. Phys., 34, 1311–1327.van Gunsteren,W.F. and Mark,A.E. (1992)J. Mol. Biol., 227, 389–395.Vriend,G. (1990)J. Mol. Graph., 8, 52–56.Vriend,G. (1990) The G-Protein Coupled Receptor Database, EMBL,
Heidelberg, Germany (http://ww.sander.embl-heidelberg.de/7tm).Wade,S.M., Dalman,H.M., Yang,S.Z. and Neubig,R.R. (1994)Mol.
Pharmacol., 45, 1191–1197.Watkinson,S. and Arkinstall,S. (1994)The G-protein Linked Receptor Facts
Book. Academic Press, London.Weiner,S.J., Kollman,P.A., Case,D.A., Singh,U.C., Ghio,C., Alagona,G.,
Profeta,S., Jr and Weiner,P. (1984)J. Am. Chem. Soc., 106, 765–784.Weiner,S.J., Kollman,P.A., Nguyen,D.T. and Case,D.A. 91986)J. Comput.
Chem., 7, 230–252.Wess,J., Gdula,D. and Brann,M.R. (1991)EMBO J., 10, 3729–3734.Wess,J., Nanavati,S., Vogel,Z. and Maggio,R. (1993)EMBO J., 12, 331–338.Williams,I.H. (1993)Chem. Soc. Rev., 22, 277–283.Zhang,D.Q. and Weinstein,H. (1993)J. Med. Chem., 36, 934–938.Zwanzig,R.W. (1954)J. Chem. Phys., 22, 1420–1426.
Received November 24, 1997; revised July 27, 1998; accepted August 5, 1998
1193