differential effect of ccl4 on renal function in cirrhotic and non-cirrhotic rats
TRANSCRIPT

Exp Toxic Pathol 1999; 51: 199-205 URBAN & FISCHER http://www.urbanfischer.de/joumals/exptoxpath
llnstitute of Molecular Biology in Medicine and Hospital Civil de Belen, CUCS. University of Guadalajara, Jalisco, Mexico, 2Department of Biology, Facultad de Qufmica UN AM, 3Department of Gastroenterology, Instituto Nacional de la Nutrici6n S.Z. Mexico, D.F.
Differential effect of CCl4 on renal function in cirrhotic and non-cirrhotic rats
ANA ROSA RINc6Ni, AMADOR COVARRUBIAS', JOSE PEDRAZA-CHAVERRf2, JORGE LUIS P003, JUAN ARMENDARIZBORUNDAl, and ARTURO PANDURO'
With 2 figures and 4 tables
Received: February 4, 1998; Accepted: February 24, 1998
Address for correspondence: Dr. ANA ROSA RINC6N-SANCHEZ. Institute of Molecular Biology in Medicine, University of Guadalajara, P.O. Box: 2-500, Guadalajara, Jalisco Mexico, c.P. 44281 ; Fax: 52 (3) 617- 41- 59, 614-77-43, e-mail: [email protected]
Key words: Liver regeneration; Renal function; Cirrhosis, liver; CCI4 ; Nephrotoxicity.
Abbreviations: AST: aspartate aminotransferase. ALT: alanine aminotransferase. BUN: blood urea nitrogen. GFR: glomerular filtration rate. HRS: hepatorenal syndrome. MAP: mean arterial pressure. PP: portal pressure. PRA: plasma renin activity. PRe: plasma renin concentration.
Summary
The pathogenesis of renal function alteration associated with liver disease remains to be elucidated. Although different experimental animal models have been utilized in order to explain such pathophysiological state, none of them have completely explained the mechanisms involved. In this study we performed differential hemodynamic, hepatic and renal function alteration studies after induction of acute liver damage via intragastric administration of a single dose of CCl4 to cirrhotic and non-cirrhotic rats . Cirrhotic rats with acute liver damage exhibited a significant decrease in mean arterial pressure followed by a decreased glomerular filtration rate, urinary sodium concentration and an induction of plasma renin concentration and activity. At the same time, a significant association between oliguria and mortality was observed. The renal histopathological studies revealed glomeruli with mesangial hypercellularity and thickening of capillary wall, but not tubular epithelial injury. All these alterations were not detected in the control group, i.e. by non-cirrhotic rats with acute liver damage. This study suggests that the effect of CCl4 on kidney structure and function depends on the functional state of the liver. Since this experimental model of acute liver damage in cirrhotic rats presents hemodynamics and renal function alterations similar to those observed in the hepatorenal syndrome in man, it could be utilized to study the pathogenesis of renal function alterations associated with liver damage.
Introduction
CC14 induced cirrhosis in rats represents an adequate experimental model of cirrhosis in man (PEREZ-T AMA YO 1983; WENSING et al. 1990a; L6PEZ-NovOA et al. 1977). In 1936, CAMERON and KARUNARATNE (1936) described the morphological changes in liver after repeated administration ofCC14 to rats. Thereafter, many techniques to administer CCl4 in animal species have been used (PROCTOR and CHATAMRA 1982; PEREZ-TAMAYO 1983). However, for induction of experimental cirrhosis, it is necessary to give repeated doses of CC14 regardless of the animal species and route of administration (CAMERON and KARUNARATNE 1936; PROCTOR and CHATAMRA 1982; PEREZTAMAYO 1983; WENSING et al. 1990a; L6PEZ-NovOA et al. 1977). Intraperitoneal administration of CCl4 to rats for 8 weeks, results in cirrhosis (EHRINPREIS et al. 1980; PEREZTAMAYO 1983; PANDURO et al. 1988) and functional alterations similar to "compensated cirrhosis" in man (EHRINPREIS et al. 1980; PEREZ-TAMA YO 1983; ELIAS et al. 1990).
Changes in renal function are associated with hepatic function alterations (WENSING et al. 1990a, b). For example, the peripheral arterial vasodilatation hypothesis emphasizes a functional, rather than mechanical role of the liver in salt retention and ascites (SCHRIER et aI. 1988), and suggests that decreased hepatic function is associated
0940-2993/99/51/03-199 $ 12.00/0 199

with increased vascular capacitance leading to peripheral arterial vasodilatation. Subsequently, hormonal and neural changes initiate renal sodium retention followed by ascites when compensatory mechanisms become insufficient (SCHRIER 1983). WENSING et al. (1990a), have shown that once liver function falls below a critical threshold, sodium retention occurs as an acute phenomenon followed by ascites. This effect was not observed in all CC14 treated rats because of interanimal variability in development of decompensated cirrhosis (WENSING et al. 1990a, b) and regeneration.
Liver regeneration has been widely studied after intragastric administration of a single dose of CC14 to non-cirrhotic rats (PEREZ-TAMAYO 1983; FAUSTO 1984; PANDURO et al.1986). After CC14 administration, rats manifest acute liver damage characterized by steatosis and cell necrosis followed by regeneration (PEREZ-TAMAYO 1983; PANDURO et al.1986). Therefore, induction of acute liver damage by intragastric administration of a single dose of CCl4 to cirrhotic rats may provide a better definition of the association between liver and renal function alterations.
No nephrotoxic effects in rats made cirrhotic after 12 weeks of treatment with repeated small doses of CCl4,
were detected (WENSING et al. 1990a, b). However, it is not known if a single intragastric administration of CCl4
to cirrhotic rats induces tubular necrosis, which is the typical nephrotoxic effect of CCl4 in non-cirrhotic rats (STRIKER et al. 1968), and associates to renal function alterations.
In a previous study, liver cirrhotic regeneration after induction of acute damage was observed in cirrhotic rats treated with a single intragastric dose of CCl4 (PANDURO et al. 1988). During and after the regenerative process, the rats had ascites and oliguria for a short period of time but the renal function was not analyzed. In this study, we investigated whether cirrhotic rats with acute liver damage induced by a single intragastric administration of CCl4
manifest necrosis and hemodynamic and renal function alterations.
Material and methods
Induction ofliver cirrhosis: Liver cirrhosis was produced in 100 male Wistar (CUCS farm-U. de G.) rats (40-60 g initial weight) by intraperitoneal injection of 0.20 ml of a mixture of CCl4 (Merck Company, Darmstadt, FRG) with mineral oil (Sigma Chemical Company, St. Louis MO, USA) 3 times per week during 8 weeks, progressively increasing the concentration of CCl4 until the fourth week of treatment (EHRINPREIS et al. 1980). The percentages of CCl4 in mineral oil (v/v) were as follows: week 1, 13 %;week 2, 16 %; week 3, 20 %; and weeks 4 to 8, 25 %. Age and sex matched control animals were included. The rats were maintained under day-night cycles of 12 h, fed ad libitum with rodent lab diet (PMI-500l).
Induction of acute liver damage in cirrhotic and age matched control rats: At the end of the CCl4 treatment, cirrhotic and age matched control rats were kept in individual metabolic cages. To avoid cumulative chronic effect of
200 Exp Toxic Pathol51 (1999) 3
CC14 in cirrhotic rats, the induction of acute liver damage was carried out 6 days later. Cirrhotic and age-matched control rats received a single intragastric dose of a 1: 1 (v/v) mixture of CClicorn oil (0.5 ml per 100 g body weight) (PANDURO et a1.1986). Twenty four hour urine-samples at indicated times were collected from the different experimental groups of rats and the total urinary volume of each rat was measured and tested for electrolytes and creatinine.
Hemodynamic studies: Rats were deprived of food but not water overnight and were anesthetized with ether (BAER et at. 1993) during hemodynamic evaluation. The portal vein was exposed by median laparotomy and a polyethylene catheter was placed alongside the femoral artery for arterial pressure and heart rate measurements. Another catheter was inserted in an ileocolic vein for portal pressure measurement. Heart rate, arterial pressure, and portal pressure (PP) were recorded on an square wave electromagnetic manometer (PPG Medical Electronics, RM-300, The Netherlands). Pressure recording was performed every 5 minutes until 3 continuous stable measurements were obtained. Liver and kidney were quickly removed and a small fragment of each tissue was fixed in formalin for further histopathological analysis, as described below.
Renal and liver function tests: To perform renal and liver function tests, including renin activity, we used 35 rats that were decapitated. Serum and urine sodium were determined by flame photometry (Flame photometer model 403. Instrumentation Laboratory, Lexington, MA.). Blood urea nitrogen (BUN) and creatinine were measured using an autoanalyzer (BUN analyzer 2 and creatinine analyzer 2, Beckman Instruments Inc., Fullerton CA, USA) (PEDRAZACHEV ARRI et al. 1993). Creatinine clearance was used to estimate glomerular filtration rate (GFR). Plasma renin activity (PRA) was measured by radioimmunoassay (RIA) of angiotensin I (AI) (Du Pont New England Nuclear, Boston MA, USA) produced for endogenous renin substrate after incubation of plasma at pH 6.0 for 1 h at 37°C, as described elsewhere (IBARRA-RUBIO et al. 1990). Plasma renin concentration (PRC) was measured by RIA of Al after incubation of plasma (1 h at 37°C) with an excess of renin substrate (Plasma from 24 h nephrectomized rats) (IBARRA-RUBIO et al. 1990).
Liver damage was histologically and biochemically assessed by determining serum albumin, total proteins, bilirubin, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities.
Histological samples examination: Slices of liver and kidney were used for histological examination. Tissue blocks for light microscopy were fixed in 10 % formaldehyde in 0.1 M phosphate (pH 7.2). All samples were embedded in paraffin, cut in sections of 4 !lm thickness, and stained with hematoxylin and eosin. Optical microscopy evaluation was performed by the same researcher who was blinded to the experimental tissue to be studied. At least three animals were used at each experimental time point and fifteen glomerulus were analyzed (40 X) in each kidney for the counting of mesangial cells.
Plasma Volume: Plasma volume was measured by the Evan's Blue dilution method (WANG 1959). A catheter was placed into the femoral artery of anesthetized rats. 0.2 ml of Evan's blue dye solution (5 % of Evan's blue dye WN in sterile isotonic saline) were injected and a blood sample was taken 15 minutes later. Plasma dye concentration was read at 610 nm on a Spectrophotometer (Beckman 650) and plasma volume determined as reported by KAUFMAN et al. (1981).
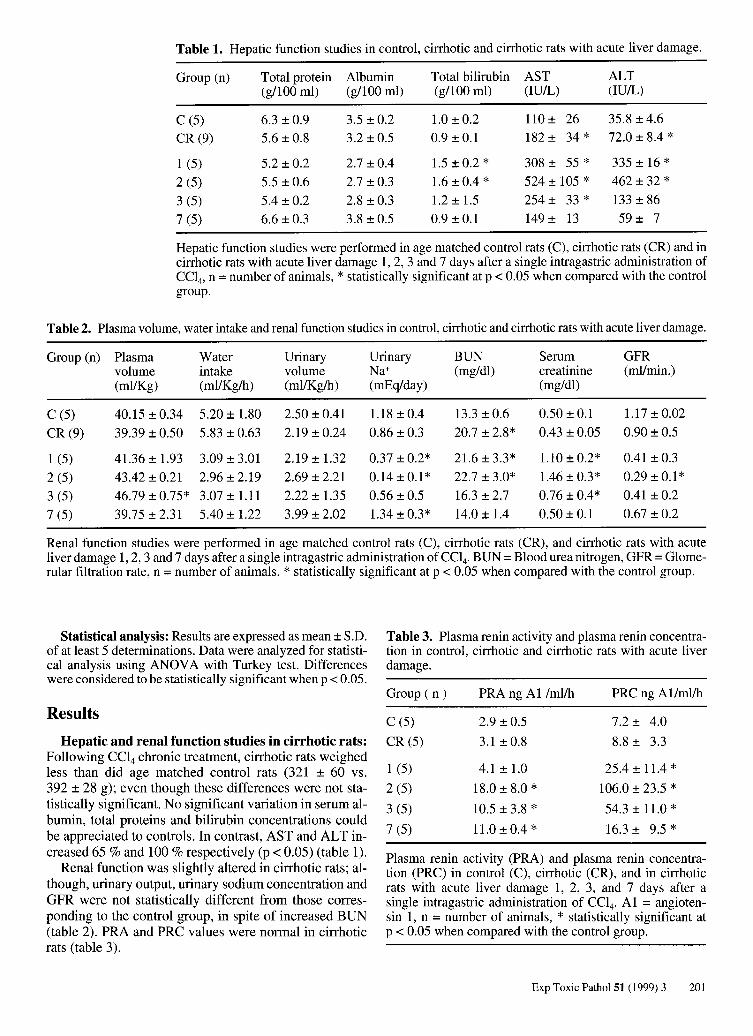
Table 1. Hepatic function studies in control, cirrhotic and cirrhotic rats with acute liver damage.
Group (n) Total protein Albumin Total bilirubin AST ALT (g/100 ml) (gllOO m!) (g/100 ml) (lUlL) (lUlL)
C (5) 6.3 ±0.9 3.5 ±0.2 1.0 ± 0.2 110± 26 35.8 ± 4.6 CR(9) 5.6 ±0.8 3.2 ±0.5 0.9 ± 0.1 182 ± 34 * 72.0 ± 8.4 * 1 (5) 5.2 ±0.2 2.7 ± 0.4 1.5 ± 0.2 * 308 ± 55 * 335 ± 16 * 2 (5) 5.5 ±0.6 2.7 ± 0.3 1.6 ± 0.4 * 524± 105 * 462 ± 32 * 3 (5) 5.4 ± 0.2 2.8 ±0.3 1.2 ± 1.5 254± 33 * 133 ± 86 7 (5) 6.6 ± 0.3 3.8 ±0.5 0.9 ± 0.1 149 ± 13 59 ± 7
Hepatic function studies were performed in age matched control rats (C), cirrhotic rats (CR) and in cirrhotic rats with acute liver damage 1,2,3 and 7 days after a single intragastric administration of CCI4 , n = number of animals, * statistically significant at p < 0.05 when compared with the control group.
Table 2. Plasma volume, water intake and renal function studies in control, cirrhotic and cirrhotic rats with acute liver damage.
Group (n) Plasma Water Urinary Urinary BUN Serum GFR volume intake volume Na+ (mg/dl) creatinine (mllmin.) (mllKg) (mllKg/h) (mllKg/h) (mEq/day) (mg/dl)
C (5) 40.15 ± 0.34 5.20 ± 1.80 2.50 ± 0.41 1.18 ± 0.4 13.3 ± 0.6 0.50 ± 0.1 1.17 ± 0.02
CR (9) 39.39 ± 0.50 5.83 ± 0.63 2.19 ± 0.24 0.86 ± 0.3 20.7 ± 2.8* 0.43 ± 0.05 0.90 ± 0.5
1 (5) 41.36 ± 1.93 3.09 ± 3.01 2.19 ± 1.32 0.37 ± 0.2* 21.6 ± 3.3* 1.10 ± 0.2* 0.41 ± 0.3 2 (5) 43.42 ± 0.21 2.96 ± 2.19 2.69 ± 2.21 0.14 ± 0.1 * 22.7 ± 3.0* 1.46 ± 0.3* 0.29 ± 0.1 * 3 (5) 46.79 ± 0.75* 3.07 ± 1.11 2.22 ± 1.35 0.56 ± 0.5 16.3 ± 2.7 0.76 ± 0.4* 0.41 ± 0.2 7 (5) 39.75 ± 2.31 5.40 ± 1.22 3.99 ± 2.02 1.34 ± 0.3* 14.0± 1.4 0.50 ± 0.1 0.67 ± 0.2
Renal function studies were performed in age matched control rats (C), cirrhotic rats (CR), and cirrhotic rats with acute liver damage 1, 2, 3 and 7 days after a single intragastric administration ofCC14 • BUN = Blood urea nitrogen, GFR = Glome-rular filtration rate, n = number of animals, * statistically significant at p < 0.05 when compared with the control group.
Statistical analysis: Results are expressed as mean ± S.D. of at least 5 determinations. Data were analyzed for statistical analysis using ANOV A with Turkey test. Differences were considered to be statistically significant when p < 0.05.
Results
Hepatic and renal function studies in cirrhotic rats: Following CCl4 chronic treatment, cirrhotic rats weighed less than did age matched control rats (321 ± 60 vs. 392 ± 28 g); even though these differences were not statistically significant. No significant variation in serum albumin, total proteins and bilirubin concentrations could be appreciated to controls. In contrast, AST and ALT increased 65 % and 100 % respectively (p < 0.05) (table 1).
Renal function was slightly altered in cirrhotic rats; although, urinary output, urinary sodium concentration and GFR were not statistically different from those corresponding to the control group, in spite of increased BUN (table 2). PRA and PRC values were normal in cirrhotic rats (table 3).
Table 3. Plasma renin activity and plasma renin concentration in control, cirrhotic and cirrhotic rats with acute liver damage.
Group (n) PRA ng Al /ml/h PRC ng Allmllh
C (5) 2.9 ±0.5 7.2± 4.0
CR (5) 3.1 ± 0.8 8.8 ± 3.3
1 (5) 4.1 ± 1.0 25.4 ± 11.4 *
2 (5) 18.0 ± 8.0 * 106.0 ± 23.5 *
3 (5) 10.5 ± 3.8 * 54.3 ± 11.0 *
7 (5) 11.0 ± 0.4 * 16.3 ± 9.5 *
Plasma renin activity (PRA) and plasma renin concentration (PRC) in control (C), cirrhotic (CR), and in cirrhotic rats with acute liver damage 1, 2, 3, and 7 days after a single intragastric administration of CCI4. Al = angiotensin 1, n = number of animals, * statistically significant at p < 0.05 when compared with the control group.
Exp Toxic Pathol51 (1999) 3 201
Table 4. Mesangial cells in kidneys of control, cirrhotic, and cirrhotic rats with acute liver damage.
Group (n)
Control (3)
Cirrhotic (0 d) (3)
1 d (7)
2 d (3)
3 d (6)
5 d (5)
7 d (3)
Number of mesangial cells 1
50 ± 3 66±4
81 ± 9 * 90 ±4 *
104±6*
78 ±7 * 85 ± 3 *
Values are means ± S.D; 1,2,3,5 and 7 represents the days after acute liver damage. n = number of animals studied; I number of mensangial cells per 15 glomerulus; * statistically significant at p<0.05 when compared with the control group.
202 Exp Toxic Pathol 51 (1999) 3
Fig. 1. Histology of kidney of cirrhotic rats without (A) and with (B) acute liver damage 48 hours after CC14 administration. As shown in B, morphological changes were evident in the glomerulus displaying hypercellularity of messangial cells with respect to A. Hematoxylin and eosin 900 X. C shows a higher magnification (1300 X) of B in order to clearly identify the mesangiocapillar hypercellularity and mild thickening of the capillary loops (arrows).
Hemodynamic studies in cirrhotic rats: Portal hypertension in cirrhotic rats was observed, 19.0 ± 2.1 nun Hg compared to 7.7 ± 0.5 mm Hg in the age matched control group (p < 0.05), whereas MAP was lower than in controls (92.0 ± 11 mm Hg vs. 98.0 ± 2 mm Hg) (p = NS).
Histopathologic studies of cirrhotic rats with acute liver damage: Extensive liver fibrosis following chronic CC14 treatment was manifested (P ANDURO et al. 1988). At the same experimental time point, the kidneys of cirrhotic rats were histologically normal (fig. lA). On the other hand, 48 hours after cirrhotic rats received acute CC14
treatment, when liver regeneration was supposed to be completed (PANDURO et al. 1986), the kidneys of rats presenting acute liver damage showed glomerular mesangial hypercellularity (fig. 1B and table 4), and mild thickening of the capillary wall (fig. 1 C). Tubules appeared normal.
These histological changes were reversed 7 days after acute liver damage.
Hemodynamic studies in cirrhotic rats with acute liver damage: Portal hypertension persisted in cirrhotic rats with acute liver damage. Portal pressure in this group of animals was 13.0 ± 3 mm Hg 24 hours after CCl4 treatment, (control group 7.7 ± 0.5 mm Hg), showing a significant decrease at this experimental time point when compared to cirrhotic rats (19.0 ± 2.1 mm Hg). However, after 48 hours, there was a further increase to 16.0 ± 2 mm Hg (p < 0.05). MAP decreased significantly from 92.0 ± 11 to 69.0 ± 2 mm Hg (p < 0.05) 24 hours after administration of CCl4 reaching almost normal levels after 48 hours (82 ± 10 mm Hg).
Hepatic and renal function studies in cirrhotic rats with acute liver damage: Maximum liver damage of cirrhotic rats occurred 24 and 48 hours after acute administration of CCl4 followed by replication of parenchymal cells (PANDURO et al. 1988). In this study, liver function tests revealed no significant reduction in serum albumin concentration (table 1). Neither significant changes in total serum protein were detected post CCl4 treatment. However, a statistically significant increase in total bilirubin concentration and aminotransferases (AST and ALT) activity were detected during acute liver damage (table 1). Total bilirubin returned to control values 7 days post-CCl4 treatment, whereas AST and ALT remained elevated (table O.
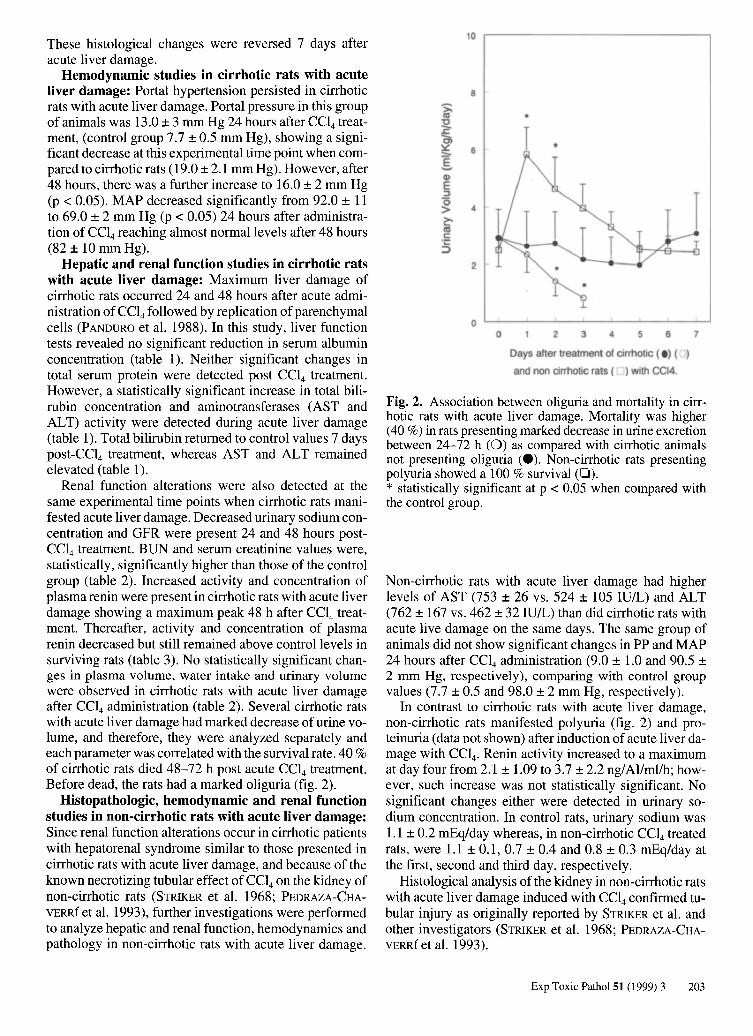
Renal function alterations were also detected at the same experimental time points when cirrhotic rats manifested acute liver damage. Decreased urinary sodium concentration and GFR were present 24 and 48 hours postCCl4 treatment. BUN and serum creatinine values were, statistically, significantly higher than those of the control group (table 2). Increased activity and concentration of plasma renin were present in cirrhotic rats with acute liver damage showing a maximum peak 48 h after CCl4 treatment. Thereafter, activity and concentration of plasma renin decreased but still remained above control levels in surviving rats (table 3). No statistically significant changes in plasma volume, water intake and urinary volume were observed in cirrhotic rats with acute liver damage after CCl4 administration (table 2). Several cirrhotic rats with acute liver damage had marked decrease of urine volume, and therefore, they were analyzed separately and each parameter was correlated with the survival rate. 40 % of cirrhotic rats died 48-72 h post acute CCl4 treatment. Before dead, the rats had a marked oliguria (fig. 2).
Histopathologic, hemodynamic and renal function studies in non-cirrhotic rats with acute liver damage: Since renal function alterations occur in cirrhotic patients with hepatorenal syndrome similar to those presented in cirrhotic rats with acute liver damage, and because of the known necrotizing tubular effect of CC14 on the kidney of non-cirrhotic rats (STRIKER et al. 1968; PEDRAZA-CHAVERRf et al. 1993), further investigations were performed to analyze hepatic and renal function , hemodynamics and pathology in non-cirrhotic rats with acute liver damage.
~ tG
~ ~ :::0-
S CD E ::J 0 > ?: CIS c:: 0t: :>
10 ,----------------,
8
•
• 8
"
2
o ~-~---~-------~ o 2 3 " 5
Days alter treatment 01 CIrrhotIC ( e) (
and non arrho\Jc ralS ( ) With CCI4
7
Fig. 2. Association between oliguria and mortality in cirrhotic rats with acute liver damage. Mortality was higher (40 %) in rats presenting marked decrease in urine excretion between 24-72 h (0) as compared with cirrhotic animals not presenting oliguria (e). Non-cirrhotic rats presenting polyuria showed a 100 % survival (0). * statistically significant at p < 0.05 when compared with the control group.
Non-cirrhotic rats with acute liver damage had higher levels of AST (753 ± 26 vs. 524 ± 105 lUlL) and ALT (762 ± 167 vs. 462 ± 32 lUlL) than did cirrhotic rats with acute live damage on the same days. The same group of animals did not show significant changes in PP and MAP 24 hours after CCl4 administration (9.0 ± 1.0 and 90.5 ± 2 mm Hg, respectively), comparing with control group values (7.7 ± 0.5 and 98.0 ± 2 mm Hg, respectively).
In contrast to cirrhotic rats with acute liver damage, non-cirrhotic rats manifested polyuria (fig. 2) and proteinuria (data not shown) after induction of acute liver damage with CCI4• Renin activity increased to a maximum at day fourfrom 2.1 ± 1.09 to 3.7 ± 2.2 ng/Allml/h; however, such increase was not statistically significant. No significant changes either were detected in urinary sodium concentration. In control rats, urinary sodium was 1.1 ± 0.2 mEq/day whereas, in non-cirrhotic CCl4 treated rats, were 1.1 ± 0.1,0.7 ± 0.4 and 0.8 ± 0.3 mEq/day at the first, second and third day, respectively.
Histological analysis of the kidney in non-cirrhotic rats with acute liver damage induced with CCl4 confirmed tubular injury as originally reported by STRIKER et al. and other investigators (STRIKER et al. 1968; PEDRAZA-CHAVERRf et al. 1993).
Exp Toxic Patho151 (1999) 3 203

Discussion
In this study we observed that hemodynamic, renal and pathological alterations exhibited by cirrhotic rats with an additional CC14 -induced acute liver damage, were different from those of non-cirrhotic rats similarly treated.
Maximal liver damage occurred 24 and 48 hours after CC14 administration to cirrhotic rats followed by replication of parenchymal cells, as evidenced by the increased incorporation of 3H-thymidine around 72 hours after intoxication (PANDURO et a1.l988). The increase of serum total bilirubin and AST and ALT activity at 48 hours, reflected acute liver damage in CC14-treated cirrhotic rats. In agreement with previous observations no changes in total serum proteins were detected following the CC14
administration (PANDURO et al. 1986, 1988). Association between liver and renal function altera
tions was detected in all CC14 treated cirrhotic rats. When acute liver damage occurred, hemodynamic and renal function alterations were present. Twenty-four hours post CC14-treatment, a significant decrease in MAP was registered followed by a decreased GFR, and urinary sodium concentration and induction of plasma renin concentration and activity. This order of events is in agreement with other reports showing that decrease in MAP precedes onset of sodium retention (WENSING et al. 1990a). Decreased MAP in cirrhotic dogs and rats is attributed to decreased peripheral vascular resistance in the presence of normal or increased cardiac output (FERNANDEZMUNOz et al. 1985; BOSCH et at. 1993). Induction of plasma renin in decompensated cirrhosis is probably due to equilibrating mechanisms to avoid a fall in MAP (SCHRIER et al. 1988).
Cirrhotic rats presented portal hypertension which persisted after administration of CC14• However, a small decrease in portal hypertension observed 24 h after acute intoxication may be explained by blood sequestration in liver and spleen, since an increase in wet weight of both organs was detected (data not shown). To our knowledge, no such alteration has been reported in human or animal models before, most probably because such measurements are not routinely performed in patients with decompensated cirrhosis.
Repeated small doses of CC14 do not produce nephrotoxicity (L6PEZ-NovOA et al. 1980; PROCTOR and CHATAMRA 1982; JIMENEZ et a1.l985; WENSING et al. 1990a), as it was observed in the kidney of cirrhotic rats; however, we notice that the acute doses of CC14 used to produce tubular necrosis in non-cirrhotic rats did not produce tubular morphological changes in cirrhotic rats. Intragastric administration of a single dose of CC14 to cirrhotic rats did not cause polyuria, proteinuria or tubular necrosis. In contrast, glomeruli with mesangial hypercellularity and mild thickening of the capillary wall were observed with normal tubuli.
CC14 is mediated through cytochrome P450-dependent metabolism (WOLF et al. 1980). The initial metabolite is the trichloromethyl free radical, which is believed to in-
204 Exp Toxic Pathol 51 (1999) 3
itiate biochemical events ultimately translated as liver cell necrosis (WOLF et al. 1980). A decrease of cytochrome p450 gene expression occurs during liver regeneration in CC14 treated rats (PANDURO et al. 1986); therefore, decreased activity or concentration of cytochrome p450 in cirrhotic rats could lead to a reduced nephrotoxicity and explain, at least partially, why tubular morphological changes are not present in cirrhotic rats. Besides, renal excretion capacity drops after hepatic failure if a toxic substance is biotransformed in the liver before its renal excretion can take place (BARTH et al. 1996; WESTPHAL and BROGARD 1997).
No significant changes in urine volume were detected in cirrhotic rats with acute liver damage because of a large standard variation. However, when oliguric rats were analyzed separately, a close correlation was observed between oliguria and mortality. 40 % of the animals died during the first 72 hours after CC14 administration and, before dead, manifested statistically significant decrease in urine volume (p < 0.05).
Since the maximum peak of thymidine incorporation in CC14 treated cirrhotic rats occurs at 72 hours (P ANDURO et al. 1986), we infer that mortality is related to failure of the cirrhotic liver to regenerate in the presence of decreased liver function (WENSING et al. 1990a).
Such findings could also explain why decompensated cirrhosis occurs only when repeated small doses of CC14
were administered for prolonged periods of time or in the presence of cytochrome p450 system inducers (RECKNAGEL and GLENDE 1973; WOLF et al. 1980; BRATTIN et at. 1995). But, when acute liver damage is produced in cirrhotic rats with a single dose of CC14, decompensated cirrhosis is observed with hemodynamic and renal function alterations similar to those observed in the hepatorenal syndrome in man (GINES et al. 1993; ARROYO et al. 1996; BATALLER et al. 1997).
The hemodynamic and renal function alterations detected in CC14-treated cirrhotic rats such as decrease in MAP preceded by sodium retention and induction of plasma renin activity at a time when liver function suddenly deteriorates, may explain the rapid formation of ascites in all experimental animals and are consistent with the peripheral arterial vasodilatation hypothesis (SCHRIER et al. 1988).
Acknowledgments: We are grateful to Prof. IRWIN M. ARIAS, Massachusetts and M.D., Ph.D. MARCOS ROJKIND, NY for kindly revising the manuscript.
This work was supported by grants from CONACYT No. l726-PN, 05l5PM and Secretarfa de Salud (Molecular Biology in Medicine Program) to A.P.
References
ARROYO V, GINES P, GERBES AL, et al.: Definition and diagnostic criteria of refractary ascites and hepatorenal syndrome in cirrhosis. Hepatology 1996; 23:164-176.

BAER HU, GUASTELLA T, WHEATLEY AM, et al.: Acute effects of partial hepatectomy on liver blood flow in the jaundice rats. J Hepatol1993; 19: 377-382.
BARTH A, FLECK C, KLINGER W: Development organic anion transport in the liver. Exp Toxic Pathol 1996; 48: 421-432.
BATALLER R, GINES P, GUEVARA M, ARROYO V. Hepatorenal syndrome. Sem Liver Dis 1997; 17: 233-247.
BOSCH J, ENRIQUEZ R, GROSZMANN RJ, STORER EH: Chronic bile duct ligation in the dog: hemodynamic characterization of a portal hypertensive model. Hepatology 1983;3: 1002-1007.
BRATTIN WJ, GLENDE EA JR., RAGHOW R, KANG AH: Pathological mechanisms in carbon tetrachloride hepatotoxicity. J Free Rad BioI Med 1985; 1: 27-38.
CAMERON GR, KARUNARATNE W AE: Carbon tetrachloride cirrhosis in relation to liver regeneration. J Pathol Bacteriol1936; 42: 1-2l.
EHRINPREIS MN, GIAMBRONE MA, ROJKIND M: Liver proline oxidase activity and collagen synthesis in rats with cirrhosis induced by carbon tetrachloride. Biochim Biophys Acta 1980; 629: 184-193.
ELIAS AN, V AZIRI ND, DOMURAT ES, et al.: Atrial natriuretic peptide, arginine vasopressin, aldosterone and plasma renin activity in carbon tetrachloride-induced cirrhosis in rats. J Pharm Exp Ther 1990; 252: 438-441.
FAUSTO N: Messenger RNA in regenerating liver: implications for the understanding of regulated growth. Mol Cell Biochem 1984; 59: 131-147.
FERNANDEZ-MuNOZ D, CARAMELO C, SANTOS JC, et al.: Systemic and splanchnic hemodynamic disturbances in conscious rats with experimental liver cirrhosis without ascites. Am J Physiol1985; 249: G316-G320.
GINES A, ESCORSELL A, GINES P, et al.: Incidence, predictive factors and prognosis of hepatorenal syndrome in cirrhosis with ascites. Gastroenterology 1993; 105: 229-236.
IBARRA-RuBIO ME, CRUZ C, TAPIA E, et al.: Serum angiotensin converting activity and plasma renin activity in experimental models of rats. Clin Exp Pharmacol Physiol. 1990; 17: 391-399.
JIMENEZ W, MARTINEZ-PARDO A, ARROYO V et al.: Temporal relationship between hyperaldosteronism, sodium retention and ascites formation in rats with experimental cirrhosis. Hepatology 1985; 5: 245-250.
KAUFMAN S, MACKAy BJ, SCOTT JZ: Daily water and electrolyte balance in chronically hyperprolactinaemic rats. J Physiol (London) 1981; 321: 11-19.
L6PEZ C, JIMENEZ W, ARROYO V, et al.: Temporal relationship between the decrease in arterial pressure and sodium retention in conscious spontaneously hypertensive rats with carbon tetrachloride-induced cirrhosis. Hepatology 1991; 13: 585-589.
L6PEZ-NovOA JM, RENGEL MA, RODICIO JL, HERNANDO L: A micropuncture study of salt and water retention in chronic experimental cirrhosis. Am J Physiol1977; 232: F315-F318.
L6PEZ-NovOA JM, RENGEL MA, HERNANDO L: Dynamics of ascites formation in rats with experimental cirrhosis. Am J Physiol1980; 7: F353-F357.
MICHAEL UF, BARENBERG RL, LEVI D, et al.: Abnormal sodium regulation in rats following chronic intermittent exposure to carbon tetrachloride PSEBM 1971; 138: 796-799.
PANDURO A, SHALABY F, WEINER FR, et al.: Transcriptional switch from albumin to (X-fetoprotein and changes in transcription of other genes during carbon tetrachloride induced liver regeneration. Biochemistry 1986; 25: 1414-1420.
PANDURO A, SHALABY F, BIEMPICA L, SHAFRITZ DA: Changes in Albumin, alfa -fetoprotein and collagen gene transcription in CC14- induced hepatic fibrosis. Hepatology 1988; 8: 259-266.
PEDRAZA-CHAVERRf J, CRUZ C, HERNANDEZ-PANDO R, et al.: Angiotensin I-converting enzyme activity in rats with carbon tetrachloride-induced acute renal failure. Renal Failure 1993; 15: 19-26.
PEREZ-TAMAYO R: Is cirrhosis of the liver experimentally produced by CC14 an adequate model of human cirrhosis? Hepatology 1983; 3: 112-120.
PROCTOR E, CHATAMRA K: High yield micronodular cirrhosis in the rat. Gastroenterology 1982; 83: 1183-1190.
RECKNAGEL RO, GLENDE E JR.: Carbon tetrachloride hepatotoxicity: an example of lethal cleavage. CRC Critical Rev Toxicol1973; 2: 263-297.
SCHRIER RW: Pathogenesis of sodium retention in highoutput and low-output cardiac failure, nephrotic syndrome, cirrhosis and pregnancy. N Eng J Med 1983: 319: 1127-1134.
SCHRIER RW, ARROYO V, BERNARDI M, et al.: Peripheral arterial vasodilatation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology 1988; 8: 1151-1157.
STRIKER GE, SMUCKLER EA, KOHNEN PW, NAGLE RB: Structural and functional changes in rat kidney during CC14 intoxication. Am J Pathol1968; 53: 769-789.
VOROBIOFF J, BREDFELDT JE, GROSZMANN RJ: Increased blood flow through the portal system in cirrhotic rats. Gastroenterology 1984; 87: 1120-1126.
WANG L: Plasma volume, cell volume, total blood volume and F cells factor in the normal and splenectomized Sherman rat. Am J Physiol1959; 196: 188-192.
WENSING G, SABRA R, BRANCH RA: The onset of sodium retention in experimental cirrhosis in rats is related to a critical threshold of liver function. Hepatology 1990; 11: 779-786.
WENSING G, SABRA R, BRANCH RA: Renal and systemic hemodynamics in experimental cirrhosis in rats: relation to hepatic function. Hepatology 1990; 12: 13-19.
WESTPHAL JF and BROGARD JM. Drug administration in chronic liver disease. Drug Saf 1997; 17: 47-73.
WOLF CR, HARRELSON WG, NASTAINCZYK WM, et al.: Metabolism of carbon tetrachloride in hepatic microsomes and reconstituted monooxigenase systems and its relationship to lipid peroxidation. Mol Pharmaco1.l980; 18: 553-558.
Exp Toxic Pathol 51 (1999) 3 205