dicationic states of benzene dimer: benzene dimer cation and benzene dication parenthood patterns
TRANSCRIPT

Dicationic States of Benzene Dimer:Benzene Dimer Cation and BenzeneDication Parenthood Patterns
EUGENE S. KRYACHKODepartment of Chemistry, Bat. B6c, University of Liege, Sart-Tilman, B-4000 Liege 1, Belgium andBogoliubov Institute for Theoretical Physics, Kiev-143, 03680 Ukraine
Received 25 January 2007; accepted 24 April 2007Published online 1 June 2007 in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/qua.21432
ABSTRACT: High density functional computational levels, B3LYP/cc-pVDZ andB3LYP/cc-pVTZ, are invoked to study the structural patterns exhibiting between a varietyof the lower-energy conformers of the benzene dimer cation and benzene dication, on onehand, and the obtuse and acute ground-state conformers of the benzene cation radical, onthe other. In particular, these “obtuse versus acute” patterns for the lowest-energy stablesandwich structures of the benzene dimer cation, formed due to a strong charge-transferresonance interaction between the benzene cation radical as a hole donor and theground-state benzene molecule as a hole acceptor benzene dimer cation, are examinedusing the molecular orbital analysis and a two-state model. It is shown the existence ofhomodimers of the benzene dimer cation composed of equivalent benzene rings,mimicking either the obtuse or acute conformers of the benzene radical cation, andrepresenting saddle points on the potential energy surface. The computed potential energysurface of ionization of the benzene radical cation demonstrates a slight preference of thetriplet state of the benzene dication originated from the obtuse benzene cation over thesinglet which, in contrast, arises from the acute one. The parenthood features of thebenzene dimer cation and benzene dication that appear in the dicationic states of benzenedimer are discussed. © 2007 Wiley Periodicals, Inc. Int J Quantum Chem 107: 2741–2755, 2007
Key words: dicationic state; benzene dimer; benzene dimer cation; benzene dication;two-state model
Correspondence to: E. S. Kryachko; e-mail: [email protected]
The Proceedings of the XI-th Conference “Quantum Systemsin Chemistry and Physics”
Contract grant sponsor: Limburgs Universitair Centrum(Bijzonder Onderzoeksfonds).
Contract grant sponsor: Region Wallonne.Contract grant number: RW.115012.Contract grant sponsor: EC FET-OPEN STREP Project MOL-
DYNLOGIC.
International Journal of Quantum Chemistry, Vol 107, 2741–2755 (2007)© 2007 Wiley Periodicals, Inc.

KRYACHKO
1. Introduction
B enzene is the most fundamental aromatic mol-ecule. Its clusters are the simplest prototype
systems with π/π interactions that are important inmany areas of chemistry and biology [1]. The neutralbenzene clusters and the benzene dimer in particularwere the focus of numerous experimental and the-oretical studies [2] (see also note [3]1). The benzenedimer admits the two most stable and nearly isoen-ergetic slipped-parallel and T-shape π -structures. Incontrast, much less studies have been conducted oncharged clusters of benzene (C6H6)
Z+n (n ≥ 1) [4].2
The simplest systems in this series are the benzenecation radical (n = 1, Z = 1, n + Z = 2) recentlydrawn a particular attention as a typical Jahn-Tellersystem [5], benzene dimer cation (n = 2, Z = 1), andbenzene dication (n = 1, Z = 2).
The benzene dimer cation and benzene dica-tion are the basic elements in the studies ofdoubly-charged benzene clusters which are origi-nated from multiple ionization processes and oftenundergo the energetic dissociation that is well-known as Coulomb explosion [6]. It has recentlybeen demonstrated that in contrast to monomers,doubly-charged clusters behave fundamentally dif-ferent under inner-valence ionization. This behavior,referred to as the intermolecular Coulomb decay[6], is simply explained by that the double ion-ization threshold in clusters is lowered relative tothe isolated monomer since two holes in the for-mer can be spatially separated via localizing ondifferent monomers, thus reducing their hole–holeCoulomb repulsion and decreasing the double ion-ization threshold. The hole–hole Coulomb repulsionis usually rather strong and therefore, the dicationic
1The neutral benzene dimer is predominantly a van der Waalscomplex. As though, the B3LYP/cc-pVDZ and B3LYP/cc-pVTZcomputational levels used in the present work also predict twostable and nearly isoenergetic parallel displaced and T-shapedstructures of the neutral benzene dimer (precisely, the former has atiny imaginary frequency of 3 ı cm−1 which is within the computa-tional margin), we cannot be confident with their estimates of thecorresponding interaction energies. This is due to the well-knowndeficiency of many density functionals to significantly under-estimate the dispersion interaction, even to predict it as beingrepulsive. However, for the charged dimers, electrostatic andcovalent interactions are the dominant contributions to the inter-action energy, and therefore, the aforementioned computationallevels are rather accurate.
2There is no evidence of an existence of negatively chargedbenzene clusters in the gas phase. Notice that in the gas-phaseelectron affinity of the ground-state benzene molecule is negative,EAa(C6H6) = −1.12 eV.
state of a given small cluster lies higher the corre-sponding monocation species. That is why a doubly-charged cluster is “Coulombically” exploded.
It is intuitively clear that there might exist somecritical size nc(Z+) of a multiply charged cluster ofcharge Z+, composed of n molecules, such that ifn(Z+) < nc(Z+), the fission into two charged frag-ments is more favorable and, on the contrary, ifn(Z+) > nc(Z+), a rather high Coulomb repulsionbarrier renders a given cluster rather substantial life-time [7].Asimple estimation of the critical size nc(Z+)
of a given cluster is provided by the liquid dropmodel (LDM) which resembles the model of theCoulomb instability of liquid droplets proposed in1882 by Lord Rayleigh [8] and developed by Meitnerand Frisch [9], and Bohr and Wheeler [10].Accordingto the LDM [11–13], nLDM
c (Z+) is obtained by equatingthe electrostatic energy Z2/R of a charged sphere ofradius R with Z positive charges to its surface energyγ R2 where γ is the surface tension. This yields thecritical radius RLDM
c (Z+) equal to (Z2/γ )1/3 and henceimplies that nLDM
c (Z+), as being proportional to thevolume of the sphere, scales as Z2.
Despite the crudeness of the LDM, its estimatenLDM
c (Z+)provides a quite reasonable lower bound tothe experimental value nexpt
c (Z+) available for manymultiply charged van der Waals (vdW) clusters: agiven vdW Z+-charged cluster of size n that obeysthe inequality n � nc is intrinsically unstable and itsfragmentation is exothermic [11–13].
For benzene, the LDM predicts nLDMc (C6H6; 2+) =
18 (see Table II of Ref. 12a) which is comparable withnexpt
c (C6H6; 2+) = 23 [14] (see particularly Fig. 2of Refs. 13 and 15). nexpt
c (C6H6; 2+) suggests thatthe benzene dimer dication may exist in the gasphase as the vdW cluster of 21 benzene moleculesmediating two terminal benzene cation radicals.These 21 benzene molecules “soften” the mutualCoulomb repelling of two positively charged ben-zene cations and prevent the entire cluster to beCoulomb exploded.
The validity of the LDM, which only accounts forelectrostatic effects to explain the relative stability ofmultiply charged atomic or molecular clusters andrather crudely assumes that, first, the excess charge(hole) spreads over the entire cluster surface and sec-ond, that the constituting molecules are arranged ina manner to minimize the total surface energy, hasbeen debated for a long time. The recent work [16]has provided a strong computational evidence forthe existence of a novel class of the kinetically sta-ble, long-lived dicationic states of benzene clusters(C6H6)
+2n (n ≥ 2; see Fig. 1 for n = 2) and small dica-
2742 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14

DICATIONIC STATES OF BENZENE DIMER
FIGURE 1. The B3LYP/T lower-energy portion of the potential energy surface (PES) of (C6H6)2+2 (right) and the
structure (left) of the long-lived dicationic state of the benzene dimer with a fused interring C C bond. The low-energyasymptote is 2 C6H+
6 (2B2g ) and the upper one is C6H6 + C6H2+6 (3B1g ).
tionic assemblies of benzene, hexafluorobenzene,and naphthalene. Their existence that lies beyondthe LDM’s grasp unequivocally demonstrates thata newly formed C(sp3) C(sp3) interring bond at theexpense of cyclic π -conjugation surpasses the strongCoulomb repulsion, normally prevailing betweencharged monomers, and produces a rather highCoulomb barrier (Fig. 1) to confer a substan-tial kinetic stability to covalently fused dicationicclusters.
The parent species of the dicationic states of ben-zene dimer, the benzene dimer cation (C6H6)
+2 , and
benzene dication C6H2+6 are the main theme of the
present study. It is demonstrated that their struc-tural patterns originate from the obtuse and acuteground-state conformers of the benzene cation rad-ical. The computational methodology invoked forthis purpose is outlined in the next section.
2. Computational Framework
All computations reported in the present workwere performed with the hybrid density functional
B3LYP potential using the GAUSSIAN 98 pack-age of programs [17]. The Dunning’s correlationconsistent polarized valence bases of double- andtriple-zeta quality cc-pVDZ (≡ D) and cc-pVTZ (≡ T)[18] were employed to perform all geometry opti-mizations with the default integration grid. Thesymmetry constraints were not imposed. The com-putational levels B3LYP/cc-pVDZ (B3LYP/D) andB3LYP/cc-pVTZ (B3LYP/T) are known to be ade-quate for the geometry optimization and vibrationalfrequencies of such species as benzene cation andbenzene dimer cation (vide Refs. 3, 5e and refer-ences therein). The analytical harmonic vibrationalfrequencies and corresponding zero-point vibra-tional energies (ZPVE) were calculated at the bothcomputational levels in order to topologically dis-tinguish the stationary structures. Enthalpies andentropies were also estimated from the partitionfunctions calculated at room temperature under apressure of 1 atm, using Boltzmann thermostatisticsand the rigid-rotor-harmonic-oscillator approxima-tion. The expectation value of the S2 operator forradicals was obtained equal to 0.750. The naturalbond orbital (NBO) analysis was performed for some
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2743

KRYACHKO
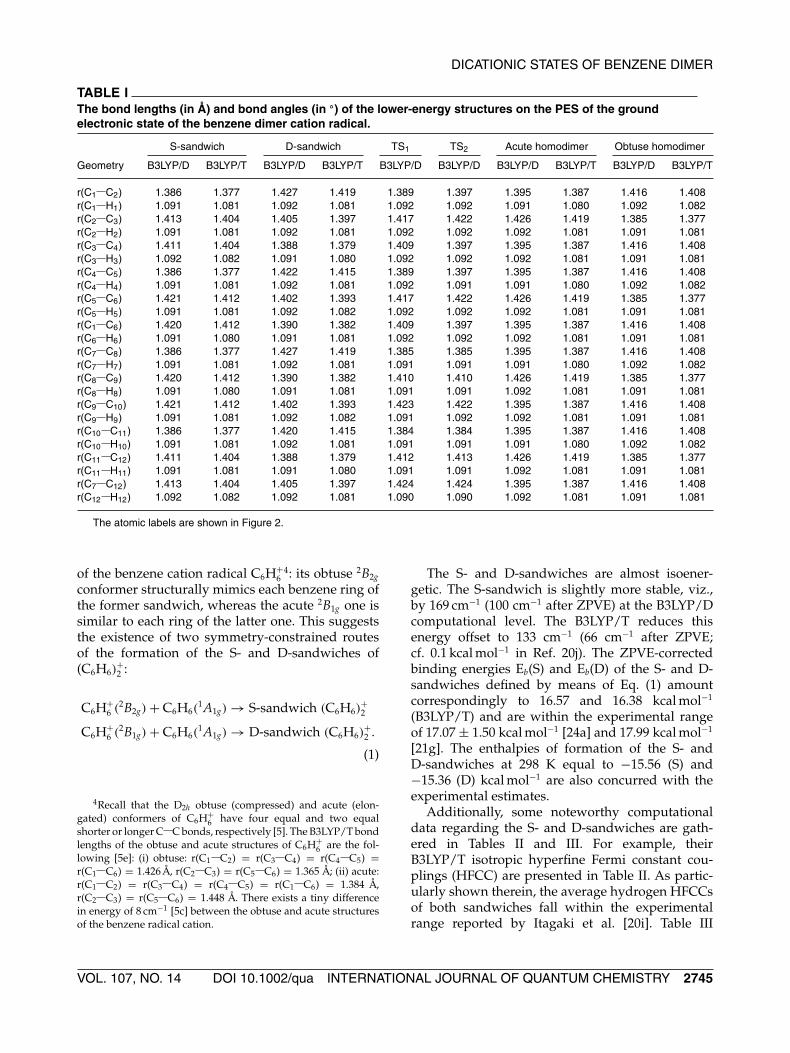
FIGURE 2. The S- and D-sandwiches of (C6H6)+2 in its ground electronic state. Their selected B3LYP/T distances are
given in Å. Their optimized geometrical parameters are collected in Table I. Notice that the benzene rings of the S- andD-sandwiches are skewed relative to each other, viz., in the S-sandwich by ≈8◦ (∠C3C6C9C12 = 172.2◦ and in the D-oneby ≈11◦ (∠C5C2C8C11 = 11.2◦).
selected lower-energy conformers of the benzenedimer cation.
3. Benzene Dimer Cation - Ionizationof Benzene Dimer
3.1. GROUND-STATE SANDWICH STRUCTURES
The benzene dimer cation (C6H6)+2 has been exten-
sively studied during the last four decades, mainlyon the experimental side [19–24]. The enthalpy �Hf
of its formation from the ground-state benzene mol-ecule and benzene cation radical estimated by meansof several experimental techniques ranges from −8to −21 kcal mol−1 [19a–e].
The structure of (C6H6)+2 has been for a long time a
rather controversial issue [19–21]. Initially, i has beensuggested that its ground state represents a symmet-ric sandwich of a D6h symmetry with two paralleland perfectly superimposed benzene ring [11a, 12c].This structure has been later ruled out as the globalenergy minimum in favor of the displaced sand-wich [21d] and T-shape [19f] ones. The displaced-sandwich model of (C6H6)
+2 relies on the existence
of at least two sandwich-type structures [21c] max-imizing the charge-transfer resonance interactionC6H+
6 · C6H6 ⇔ C6H6 · C6H+6 [19(a, d), 20(c, e)]. It has
also been invoked to interpret its photodissociation
spectra revealing four peaks at 2.82, 2.14, 1.35, and1.07 eV [21]. On the other hand, pump-probe hole-burning experiments have demonstrated that if theT-shape structure of (C6H6)
+2 would exist, its rela-
tive population is smaller compared to that of thesandwich [21f]. The full geometry optimization ofrelaxed benzene rings, recently carried out by meansof the CASSCF, MRSDCI, and MRCPAmethods [20j],reveals the existence of two different sandwichlikestructures, one corresponds to the slipped (S-) C2h-sandwich and the other to the distorted (D-) C2v one(Fig. 2).
The S- and D-sandwiches are characterized byanticlockwise ordering of the C C bond lengthswithin their two rings (vide Table I and Fig. 2). Struc-turally, each ring of the S-sandwich has four longer(≥1.40 Å) and two shorter (1.377 Å) C C bonds com-pared to the neutral benzene molecule3. In contrast,each ring of the D-sandwich has four C C bondswhose lengths are smaller than 1.40 Å and the twolonger ones (≥1.41 Å). These structural patterns ofthe S- and D-sandwiches resemble those pertainingto the two nearly energetically equivalent isomers
3The ground-state neutral benzene molecule is characterizedby the following B3LYP/T geometry [5e]: r(C C) = 1.391 Å,r(C H) = 1.082 Å, and ∠C6C1C2 = ∠C1C2H2 = 120◦.
2744 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14
DICATIONIC STATES OF BENZENE DIMER
TABLE IThe bond lengths (in Å) and bond angles (in ◦) of the lower-energy structures on the PES of the groundelectronic state of the benzene dimer cation radical.
S-sandwich D-sandwich TS1 TS2 Acute homodimer Obtuse homodimer
Geometry B3LYP/D B3LYP/T B3LYP/D B3LYP/T B3LYP/D B3LYP/D B3LYP/D B3LYP/T B3LYP/D B3LYP/T
r(C1 C2) 1.386 1.377 1.427 1.419 1.389 1.397 1.395 1.387 1.416 1.408r(C1 H1) 1.091 1.081 1.092 1.081 1.092 1.092 1.091 1.080 1.092 1.082r(C2 C3) 1.413 1.404 1.405 1.397 1.417 1.422 1.426 1.419 1.385 1.377r(C2 H2) 1.091 1.081 1.092 1.081 1.092 1.092 1.092 1.081 1.091 1.081r(C3 C4) 1.411 1.404 1.388 1.379 1.409 1.397 1.395 1.387 1.416 1.408r(C3 H3) 1.092 1.082 1.091 1.080 1.092 1.092 1.092 1.081 1.091 1.081r(C4 C5) 1.386 1.377 1.422 1.415 1.389 1.397 1.395 1.387 1.416 1.408r(C4 H4) 1.091 1.081 1.092 1.081 1.092 1.091 1.091 1.080 1.092 1.082r(C5 C6) 1.421 1.412 1.402 1.393 1.417 1.422 1.426 1.419 1.385 1.377r(C5 H5) 1.091 1.081 1.092 1.082 1.092 1.092 1.092 1.081 1.091 1.081r(C1 C6) 1.420 1.412 1.390 1.382 1.409 1.397 1.395 1.387 1.416 1.408r(C6 H6) 1.091 1.080 1.091 1.081 1.092 1.092 1.092 1.081 1.091 1.081r(C7 C8) 1.386 1.377 1.427 1.419 1.385 1.385 1.395 1.387 1.416 1.408r(C7 H7) 1.091 1.081 1.092 1.081 1.091 1.091 1.091 1.080 1.092 1.082r(C8 C9) 1.420 1.412 1.390 1.382 1.410 1.410 1.426 1.419 1.385 1.377r(C8 H8) 1.091 1.080 1.091 1.081 1.091 1.091 1.092 1.081 1.091 1.081r(C9 C10) 1.421 1.412 1.402 1.393 1.423 1.422 1.395 1.387 1.416 1.408r(C9 H9) 1.091 1.081 1.092 1.082 1.091 1.092 1.092 1.081 1.091 1.081r(C10 C11) 1.386 1.377 1.420 1.415 1.384 1.384 1.395 1.387 1.416 1.408r(C10 H10) 1.091 1.081 1.092 1.081 1.091 1.091 1.091 1.080 1.092 1.082r(C11 C12) 1.411 1.404 1.388 1.379 1.412 1.413 1.426 1.419 1.385 1.377r(C11 H11) 1.091 1.081 1.091 1.080 1.091 1.091 1.092 1.081 1.091 1.081r(C7 C12) 1.413 1.404 1.405 1.397 1.424 1.424 1.395 1.387 1.416 1.408r(C12 H12) 1.092 1.082 1.092 1.081 1.090 1.090 1.092 1.081 1.091 1.081
The atomic labels are shown in Figure 2.
of the benzene cation radical C6H+6
4: its obtuse 2B2g
conformer structurally mimics each benzene ring ofthe former sandwich, whereas the acute 2B1g one issimilar to each ring of the latter one. This suggeststhe existence of two symmetry-constrained routesof the formation of the S- and D-sandwiches of(C6H6)
+2 :
C6H+6 (2B2g) + C6H6(
1A1g) → S-sandwich (C6H6)+2
C6H+6 (2B1g) + C6H6(
1A1g) → D-sandwich (C6H6)+2 .
(1)
4Recall that the D2h obtuse (compressed) and acute (elon-gated) conformers of C6H+
6 have four equal and two equalshorter or longer C C bonds, respectively [5]. The B3LYP/T bondlengths of the obtuse and acute structures of C6H+
6 are the fol-lowing [5e]: (i) obtuse: r(C1 C2) = r(C3 C4) = r(C4 C5) =r(C1 C6) = 1.426 Å, r(C2 C3) = r(C5 C6) = 1.365 Å; (ii) acute:r(C1 C2) = r(C3 C4) = r(C4 C5) = r(C1 C6) = 1.384 Å,r(C2 C3) = r(C5 C6) = 1.448 Å. There exists a tiny differencein energy of 8 cm−1 [5c] between the obtuse and acute structuresof the benzene radical cation.
The S- and D-sandwiches are almost isoener-getic. The S-sandwich is slightly more stable, viz.,by 169 cm−1 (100 cm−1 after ZPVE) at the B3LYP/Dcomputational level. The B3LYP/T reduces thisenergy offset to 133 cm−1 (66 cm−1 after ZPVE;cf. 0.1 kcal mol−1 in Ref. 20j). The ZPVE-correctedbinding energies Eb(S) and Eb(D) of the S- and D-sandwiches defined by means of Eq. (1) amountcorrespondingly to 16.57 and 16.38 kcal mol−1
(B3LYP/T) and are within the experimental rangeof 17.07 ± 1.50 kcal mol−1 [24a] and 17.99 kcal mol−1
[21g]. The enthalpies of formation of the S- andD-sandwiches at 298 K equal to −15.56 (S) and−15.36 (D) kcal mol−1 are also concurred with theexperimental estimates.
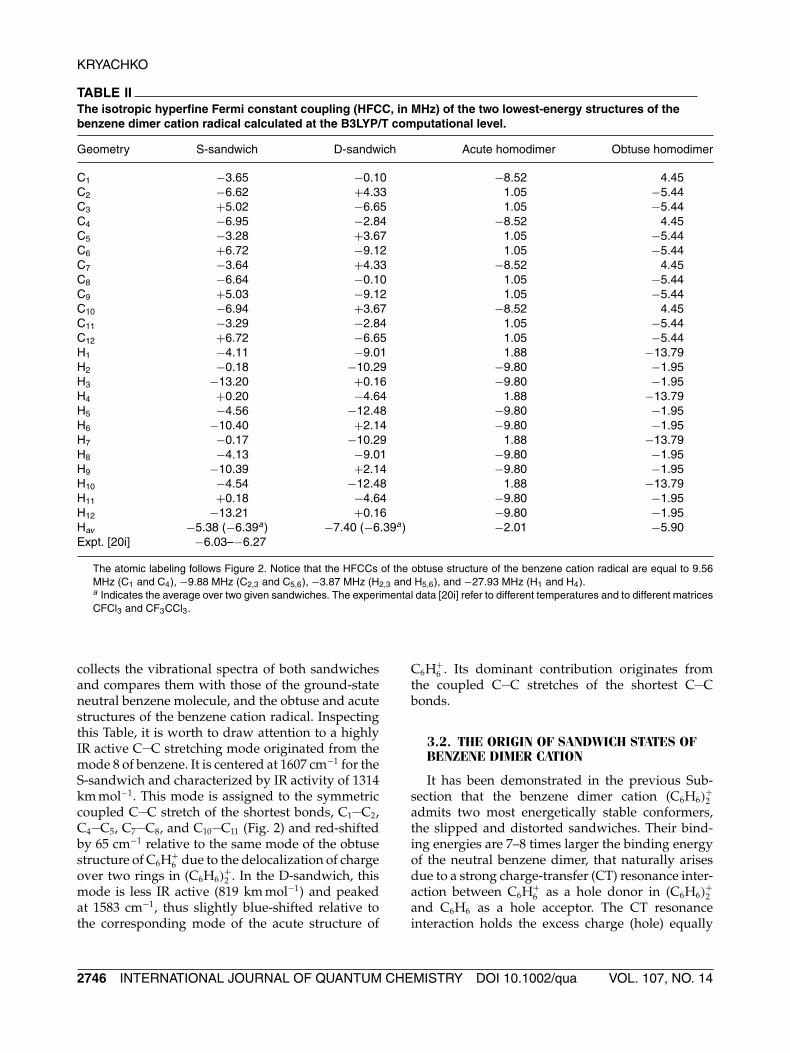
Additionally, some noteworthy computationaldata regarding the S- and D-sandwiches are gath-ered in Tables II and III. For example, theirB3LYP/T isotropic hyperfine Fermi constant cou-plings (HFCC) are presented in Table II. As partic-ularly shown therein, the average hydrogen HFCCsof both sandwiches fall within the experimentalrange reported by Itagaki et al. [20i]. Table III
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2745
KRYACHKO
TABLE IIThe isotropic hyperfine Fermi constant coupling (HFCC, in MHz) of the two lowest-energy structures of thebenzene dimer cation radical calculated at the B3LYP/T computational level.
Geometry S-sandwich D-sandwich Acute homodimer Obtuse homodimer
C1 −3.65 −0.10 −8.52 4.45C2 −6.62 +4.33 1.05 −5.44C3 +5.02 −6.65 1.05 −5.44C4 −6.95 −2.84 −8.52 4.45C5 −3.28 +3.67 1.05 −5.44C6 +6.72 −9.12 1.05 −5.44C7 −3.64 +4.33 −8.52 4.45C8 −6.64 −0.10 1.05 −5.44C9 +5.03 −9.12 1.05 −5.44C10 −6.94 +3.67 −8.52 4.45C11 −3.29 −2.84 1.05 −5.44C12 +6.72 −6.65 1.05 −5.44H1 −4.11 −9.01 1.88 −13.79H2 −0.18 −10.29 −9.80 −1.95H3 −13.20 +0.16 −9.80 −1.95H4 +0.20 −4.64 1.88 −13.79H5 −4.56 −12.48 −9.80 −1.95H6 −10.40 +2.14 −9.80 −1.95H7 −0.17 −10.29 1.88 −13.79H8 −4.13 −9.01 −9.80 −1.95H9 −10.39 +2.14 −9.80 −1.95H10 −4.54 −12.48 1.88 −13.79H11 +0.18 −4.64 −9.80 −1.95H12 −13.21 +0.16 −9.80 −1.95Hav −5.38 (−6.39a) −7.40 (−6.39a) −2.01 −5.90Expt. [20i] −6.03–−6.27
The atomic labeling follows Figure 2. Notice that the HFCCs of the obtuse structure of the benzene cation radical are equal to 9.56MHz (C1 and C4), −9.88 MHz (C2,3 and C5,6), −3.87 MHz (H2,3 and H5,6), and −27.93 MHz (H1 and H4).a Indicates the average over two given sandwiches. The experimental data [20i] refer to different temperatures and to different matricesCFCl3 and CF3CCl3.
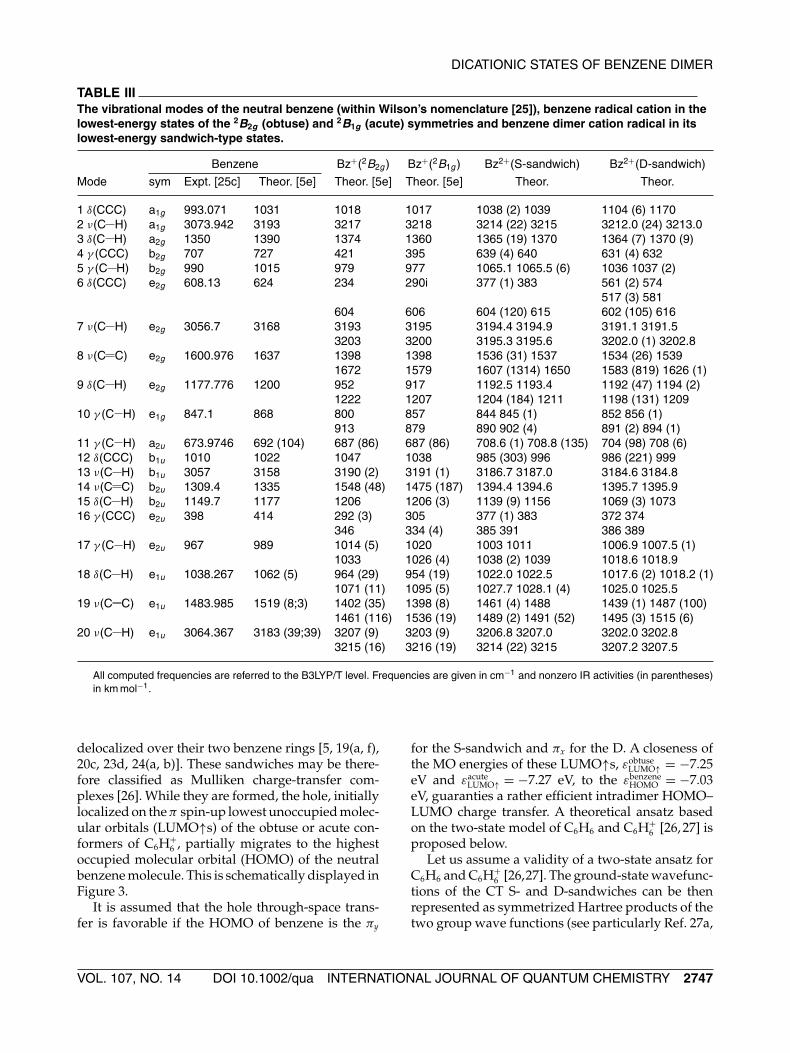
collects the vibrational spectra of both sandwichesand compares them with those of the ground-stateneutral benzene molecule, and the obtuse and acutestructures of the benzene cation radical. Inspectingthis Table, it is worth to draw attention to a highlyIR active C C stretching mode originated from themode 8 of benzene. It is centered at 1607 cm−1 for theS-sandwich and characterized by IR activity of 1314km mol−1. This mode is assigned to the symmetriccoupled C C stretch of the shortest bonds, C1 C2,C4 C5, C7 C8, and C10 C11 (Fig. 2) and red-shiftedby 65 cm−1 relative to the same mode of the obtusestructure of C6H+
6 due to the delocalization of chargeover two rings in (C6H6)
+2 . In the D-sandwich, this
mode is less IR active (819 km mol−1) and peakedat 1583 cm−1, thus slightly blue-shifted relative tothe corresponding mode of the acute structure of
C6H+6 . Its dominant contribution originates from
the coupled C C stretches of the shortest C Cbonds.
3.2. THE ORIGIN OF SANDWICH STATES OFBENZENE DIMER CATION
It has been demonstrated in the previous Sub-section that the benzene dimer cation (C6H6)
+2
admits two most energetically stable conformers,the slipped and distorted sandwiches. Their bind-ing energies are 7–8 times larger the binding energyof the neutral benzene dimer, that naturally arisesdue to a strong charge-transfer (CT) resonance inter-action between C6H+
6 as a hole donor in (C6H6)+2
and C6H6 as a hole acceptor. The CT resonanceinteraction holds the excess charge (hole) equally
2746 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14
DICATIONIC STATES OF BENZENE DIMER
TABLE IIIThe vibrational modes of the neutral benzene (within Wilson’s nomenclature [25]), benzene radical cation in thelowest-energy states of the 2B2g (obtuse) and 2B1g (acute) symmetries and benzene dimer cation radical in itslowest-energy sandwich-type states.
Benzene Bz+(2B2g ) Bz+(2B1g ) Bz2+(S-sandwich) Bz2+(D-sandwich)
Mode sym Expt. [25c] Theor. [5e] Theor. [5e] Theor. [5e] Theor. Theor.
1 δ(CCC) a1g 993.071 1031 1018 1017 1038 (2) 1039 1104 (6) 11702 ν(C H) a1g 3073.942 3193 3217 3218 3214 (22) 3215 3212.0 (24) 3213.03 δ(C H) a2g 1350 1390 1374 1360 1365 (19) 1370 1364 (7) 1370 (9)4 γ (CCC) b2g 707 727 421 395 639 (4) 640 631 (4) 6325 γ (C H) b2g 990 1015 979 977 1065.1 1065.5 (6) 1036 1037 (2)6 δ(CCC) e2g 608.13 624 234 290i 377 (1) 383 561 (2) 574
517 (3) 581604 606 604 (120) 615 602 (105) 616
7 ν(C H) e2g 3056.7 3168 3193 3195 3194.4 3194.9 3191.1 3191.53203 3200 3195.3 3195.6 3202.0 (1) 3202.8
8 ν(C C) e2g 1600.976 1637 1398 1398 1536 (31) 1537 1534 (26) 15391672 1579 1607 (1314) 1650 1583 (819) 1626 (1)
9 δ(C H) e2g 1177.776 1200 952 917 1192.5 1193.4 1192 (47) 1194 (2)1222 1207 1204 (184) 1211 1198 (131) 1209
10 γ (C H) e1g 847.1 868 800 857 844 845 (1) 852 856 (1)913 879 890 902 (4) 891 (2) 894 (1)
11 γ (C H) a2u 673.9746 692 (104) 687 (86) 687 (86) 708.6 (1) 708.8 (135) 704 (98) 708 (6)12 δ(CCC) b1u 1010 1022 1047 1038 985 (303) 996 986 (221) 99913 ν(C H) b1u 3057 3158 3190 (2) 3191 (1) 3186.7 3187.0 3184.6 3184.814 ν(C C) b2u 1309.4 1335 1548 (48) 1475 (187) 1394.4 1394.6 1395.7 1395.915 δ(C H) b2u 1149.7 1177 1206 1206 (3) 1139 (9) 1156 1069 (3) 107316 γ (CCC) e2u 398 414 292 (3) 305 377 (1) 383 372 374
346 334 (4) 385 391 386 38917 γ (C H) e2u 967 989 1014 (5) 1020 1003 1011 1006.9 1007.5 (1)
1033 1026 (4) 1038 (2) 1039 1018.6 1018.918 δ(C H) e1u 1038.267 1062 (5) 964 (29) 954 (19) 1022.0 1022.5 1017.6 (2) 1018.2 (1)
1071 (11) 1095 (5) 1027.7 1028.1 (4) 1025.0 1025.519 ν(C C) e1u 1483.985 1519 (8;3) 1402 (35) 1398 (8) 1461 (4) 1488 1439 (1) 1487 (100)
1461 (116) 1536 (19) 1489 (2) 1491 (52) 1495 (3) 1515 (6)20 ν(C H) e1u 3064.367 3183 (39;39) 3207 (9) 3203 (9) 3206.8 3207.0 3202.0 3202.8
3215 (16) 3216 (19) 3214 (22) 3215 3207.2 3207.5
All computed frequencies are referred to the B3LYP/T level. Frequencies are given in cm−1 and nonzero IR activities (in parentheses)in km mol−1.
delocalized over their two benzene rings [5, 19(a, f),20c, 23d, 24(a, b)]. These sandwiches may be there-fore classified as Mulliken charge-transfer com-plexes [26]. While they are formed, the hole, initiallylocalized on the π spin-up lowest unoccupied molec-ular orbitals (LUMO↑s) of the obtuse or acute con-formers of C6H+
6 , partially migrates to the highestoccupied molecular orbital (HOMO) of the neutralbenzene molecule. This is schematically displayed inFigure 3.
It is assumed that the hole through-space trans-fer is favorable if the HOMO of benzene is the πy
for the S-sandwich and πx for the D. A closeness ofthe MO energies of these LUMO↑s, εobtuse
LUMO↑ = −7.25eV and εacute
LUMO↑ = −7.27 eV, to the εbenzeneHOMO = −7.03
eV, guaranties a rather efficient intradimer HOMO–LUMO charge transfer. A theoretical ansatz basedon the two-state model of C6H6 and C6H+
6 [26, 27] isproposed below.
Let us assume a validity of a two-state ansatz forC6H6 and C6H+
6 [26,27]. The ground-state wavefunc-tions of the CT S- and D-sandwiches can be thenrepresented as symmetrized Hartree products of thetwo group wave functions (see particularly Ref. 27a,
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2747

KRYACHKO
FIGURE 3. The MO picture of the formation of the S-and D-sandwiches of (C6H6)
+2 . The upper MO is the
HOMO of C6H6, whereas the lower is the LUMO↑ of theobtuse and acute structures of C6H+
6 , correspondingly,for the S- and D-sandwiches.
Section 2–16 and also Refs. 21c, 21h for a singlesandwich),
�S+({ri}2N−1
i=1
) = 1√2(1 + SS)
× [φ+obtuse
1
({ri}N−1i=1
)φo
2
({ri}2N−1i=N
)
+ φo1
({ri}Ni=1
)φ+obtuse
2
({ri}2N−1i=N+1
)]
�D+({ri}2N−1
i=1
) = 1√2(1 + SD)
× [φ+acute
1
({ri}N−1i=1
)φo
2
({ri}2N−1i=N
)
+ φo1
({ri}Ni=1
)φ+acute
2
({ri}2N−1i=N+1
)]. (2)
In Eq. (2), φo1({ri}N
i=1) and φo2({ri}N
i=1) are the ground-and first-excited state normalized wave functionsof C6H6 with the corresponding eigenenergies εo
1and εo
2. φ+x1 ({ri}N−1
i=1 ) and φ+x2 ({ri}N−1
i=1 ) are those of thex-structure of C6H+
6 (x = obtuse, acute) character-ized by the eigenenergies ε+x
1 and ε+x2 . SS and SD
are the overlap integrals defined correspondinglyas 〈φ+obtuse
1 φo2 | φo
1φ+obtuse2 〉 and 〈φ+acute
1 φo2 | φo
1φ+acute2 〉;
N = 42, and the spins are henceforth omitted. Thecontributions of the wave functions of the obtuseconformer of C6H+
6 to �S+ and that of the acute one
to �D+ are worth noticed.
The total electronic Hamiltonian H of (C6H6)+2
casts as
H = H+1 (1, . . . , N − 1) + Ho
2(N, . . . , 2N) + V
= Ho1(1, . . . , N) + H+
2 (N + 1, . . . , 2N − 1) + V ′ (3)
depending on a partition of the studied (2N − 1)-electron dimer into the cationic and neutral partsgoverned by the sub-Hamiltonians H+
1 (H+2 ) and Ho
2(Ho
1), respectively. V and V ′ are the total intradimerCoulomb interaction operators whose difference (inatomic units), �V ≡ V−V ′ = [N−1
i=1 −2N−1i=N+1]1/riN −
[Mα=1 − 2M
α=M+1]Zα/rNα , describes the electron-holeand hole-nuclear Coulomb interactions (the sub-script α = 1, . . . , M = 12 refers to the α-th nucleuscharacterized by the radius vector Rα and the nuclearcharge Zα ; rij = | ri − rj |, and riα = |ri − Rα|).
Within the ansatz (2), the corresponding bindingenergies of the S- and D-sandwiches are explicitlygiven as
−Eb(S) ≡ ⟨�S
+|H|�S+⟩−[⟨⟨
φ+obtuse1
∣∣H+1
∣∣φ+obtuse1
⟩⟩1,2,...,N−1
+ ⟨⟨φo
1
∣∣Ho2
∣∣φo1
⟩⟩N,N+1,...,2N−1
]
= 12(1 + SS)
[Iobtuse + IIobtuse + IIIobtuse
+ IVobtuse] + 12�εobtuse (4)
and
− Eb(D) ≡ ⟨�D
+ |H|�D+⟩ − [⟨⟨
φ+acute1
∣∣H+1
∣∣φ+acute1
⟩⟩1,2,...,N−1
+ ⟨⟨φo
1
∣∣Ho2
∣∣φo1
⟩⟩N,N+1,...,2N−1
]
= 12(1 + SD)
[Iacute + IIacute + IIIacute
+ IVacute] + 12�εacute (5)
where 〈〈. . .〉〉L,L+1,...,L+K indicates the integration overthe L, L+1, . . . , L+K electrons and �εx ≡ (εo
2 − εo1)−
(ε+x2 − ε+x
1 ). In Eqs. (4) and (5), the matrix elementsof the Coulomb interaction operators V and V ′ aredesignated as:
Ix ≡ ⟨φ+x
1 φo2 | V | φ+x
1 φo2
⟩
IIx ≡ ⟨φo
1φ+x2 | V ′ | φo
1φ+x2
⟩
IIIx ≡ ⟨φ+x
1 φo2 | V | φo
1φ+x2
⟩
IVx ≡ ⟨φ+x
1 φo2 | V ′ | φo
1φ+x2
⟩ = IIIx + �εx. (6)
The charge-resonance energy of the X-sandwich(X = S or D) is defined as the energy difference,�ECT
X , between the first excited and ground statesof the X-sandwich. Within the ansatz (2), the first-excited state wave function is written as (see in
2748 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14

DICATIONIC STATES OF BENZENE DIMER
particular Ref. 27a, Section 2–16 and also Refs. 20c,20h for a single sandwich)
�X−({ri}2N−1
i=1
) = 1√2(1 − SX)
[φ+x
1
({ri}N−1i=1
)φo
2
({ri}2N−1i=N
)
− φo1
({ri}Ni=1
)φ+x
2
({ri}2N−1i=N+1
)]
(x = obtuse (X = S), acute (X = D)). (7)
Obviously, �X− is orthogonal to �X
+. One then explic-itly obtains
�ECTX = 1
1 − S2X
[(Ix + IIx)SX − (IIIx + IVx)]. (8)
�ECTX is the energy separation between the peaks
of the �X+ → �X
− CT absorption transition [26, 27].Two CT resonance bands at 920 nm (1.35 eV) andat 1,160 nm (1.07 eV) were experimentally observedfor the benzene dimer cation [20(b–d), 22(a)] (cf.Eq. (5) in Ref. 20c). Interestingly, if the overlap inte-grals SX � 1, �ECT
X is strongly determined by the CTintegrals,
�ECTX = −(IIIx + IVx) = −(2IIIx + �εx). (9)
Nevertheless, the above assumption of smallness ofthe overlap integrals SS and SD is not fully validin the present case of the S- and D-sandwiches asrather strong CT dimers whose interring distancesare shorter (especially in the contact regions) thanthe sum of van der Waals radii for C6H6 and C6H+
6 .Finally, neglecting �εx, one obtains from Eq. (9) that
�ECTX −2IIIx. (10)
Within the assumption SX � 1, one readily derivesfrom Eqs. (4) and (5) that
Eb(X) = −IIIx − �εx − 12(Ix + IIx)
= 12
(�ECT
X − �εx) − 1
2(Ix + IIx). (11)
Comparing �ECT,exptS = 1.35 eV and �ECT,expt
D = 1.07eV with Eb(S) = 0.72 eV and Eb(D) = 0.71 eV,one concludes that the first term, �ECT
X /2 on therhs of the last equation in (11) provides the domi-nant contribution to the binding energies of the S-and D-sandwiches. Therefore, within the ansatz (2),the difference between their CT resonance bands,estimated experimentally as being equal to 0.28 eV,
arises due to the difference of the last three termsfor the S- and D-sandwiches, particularly, to that ofthe Coulomb integrals Ix and IIx given by Eq. (6).Concluding, this ansatz provides the explicit for-mulas for Eb(S) and Eb(D), and also some estimatesof the energy differences, �ECT
X , between the firstexcited and ground states of each sandwich, thusdetermining their charge-resonance energies.
3.3. LOW-ENERGY PORTION OF PESOF BENZENE DIMER CATION
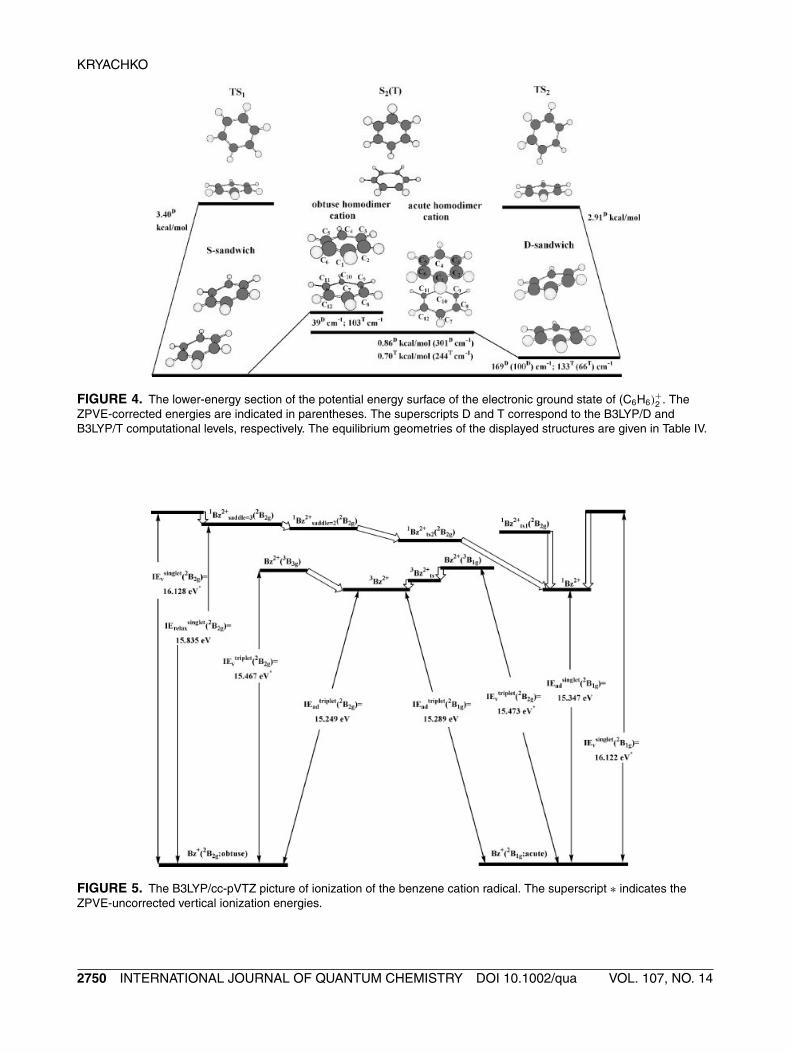
In Figure 4 we picture the lower-energy portionof the PES of (C6H6)
+2 which include the obtuse and
acute cationic homodimers having an approximateD2h symmetry. By a direct analogy with the corre-sponding parent structures of the benzene cationradical [5], the former is a first-order saddle struc-ture with the imaginary transition frequency of 29 ı
cm−1, whereas the latter is a second-order one withtwo imaginary transition frequencies of 186 ı and26 ı cm−1. Only 244 cm−1 (B3LYP/T; 301 cm−1 atB3LYP/D) separate the acute homodimer from theS-sandwich. The obtuse homodimer lies only 3 cm−1
(4 cm−1; precisely speaking these energy differ-ences are within the error margins of the employedcomputational methods) below the acute one at theB3LYP/D (B3LYP/T) level, although the inclusion ofZPVE correction reverses this order and places theacute one below by 39 cm−1 (103 cm−1).
Structurally, the four shorter C C bonds of eachbenzene ring in the acute homodimer are slightlyelongated (by 0.003 Å), whereas the two longer onesare contracted quite substantially (by 0.029 Å), ascompared to the acute C6H+
6 benzene cation radical(see footnote 4) (vide Table 1). A similar geometri-cal pattern holds for the obtuse homodimer, viz., itsfour longer C C bonds are contracted by 0.010 Åwhereas the two shorter are elongated by 0.010 Å.Both benzene rings in the obtuse and acute cationichomodimers are nearly parallel to each other. In theacute one, for example, the B3LYP/cc-pVTZ inter-ring distances between the carbon atoms bonded viatwo short C C bonds are slightly larger (3.572 Å)compared to that between the carbon atoms sharingthe short and long C C bonds (3.546 Å). Moreover,the hydrogen atoms in this homodimer are slightlypulled out (by 0.022–0.023 Å) of the benzene interiorregion. It is worth mentioning that the NBO chargesof both homodimers indicate that the hole is equallydelocalized over two rings. Their transition modesdistort such interring disposition.
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2749
KRYACHKO
FIGURE 4. The lower-energy section of the potential energy surface of the electronic ground state of (C6H6)+2 . The
ZPVE-corrected energies are indicated in parentheses. The superscripts D and T correspond to the B3LYP/D andB3LYP/T computational levels, respectively. The equilibrium geometries of the displayed structures are given in Table IV.
FIGURE 5. The B3LYP/cc-pVTZ picture of ionization of the benzene cation radical. The superscript ∗ indicates theZPVE-uncorrected vertical ionization energies.
2750 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14

DICATIONIC STATES OF BENZENE DIMER
TABLE IVThe B3LYP/T electronic energy E (in hartree), ZPVE (in kcal mol−1), enthalpy H (in hartree), and entropy S(in kcal mol−1 K−1) of the benzene cation and dicationic structures.
Bz+(2B1g ) 1Bz2+ 3Bz2+ 3Bz2+ts Bz+(2B2g ) 1Bz2+
saddle=31Bz2+
saddle=21Bz2+
ts11Bz2+
ts2
E + 231 −0.998846 −0.434007 −0.437225 −0.436961 −0.998995 −0.413117 −0.413311 −0.418875 −0.424546ZPVE 60.72 60.20 60.43 60.28 61.30 58.83 58.86 60.23 59.15H + 231 −0.897373 −0.333041 −0.334661 −0.336022 −0.896119 −0.315163 −0.314607 −0.317812 −0.325438S 68.91 71.41 73.57 70.76 70.68 66.09 70.60 71.19 69.59
It is worth finally mentioning the existence of twoT-skewed first-order saddle structures, TS1 and TS2,and the perfect T-shape second-order saddle struc-ture S2(T) of C2 symmetry on the PES of (C6H6)
+2 .
The transition-state linker TS1 to the S-sandwich isplaced 3.40 kcal mol−1 higher (B3LYP/D). The othersaddle structure TS2 related to the D-one lies 2.91kcal mol−1 above it. The B3LYP/D optimized geome-tries of these saddle structures are collected in Table I.Notice that by analogy with the S-sandwich, bothrings of TS1 have two shorter and four longer C Cbonds compared to those of benzene. While a similarC C bond pattern holds for the upper ring of TS2,its lower one resembles the D-sandwich.
4. Benzene Dication - DoubleIonization of Benzene Molecule
The benzene dication C6H2+6 is the simplest dica-
tion in the series of doubly charged benzene clusters.True, it has the important and longstanding practicalvalue as itself (see, e.g., Refs. 28–31 and referencestherein). Experimentally, C6H2+
6 has been detectedunder ionization of benzene in intense laser fields[32–34]. In the charge-separation processes [29], thebenzene dication fragments into the cations C5H+
3and CH+
3 [35] (see also Refs. 36, 37). The correspond-ing kinetic energy release amounts to 2.6–2.8 eV[35] implying that the intercharge distance reaches≈5.1–5.5 Å [38], that is almost twice as large as theC C diameter (≈2.9 Å) of the ground-state neutralbenzene molecule.
Almost four decades ago [38], the benzene dica-tion was treated as an open structure. Regrettably, theexperiments on charge stripping and charge strip-ping/charge exchange were unable to provide anystructural information on this molecular species [37](see also Ref. 39)—this was actually the reason whyit has been for a long time considered as a “diffi-cult system” whose structure and properties haveremained “considerably less known” [37]. Later, in
the eighties [31,37,39], contrary to the early view, thebenzene dication was regarded as being structurally“the same as neutral benzene” [37]. This is one sideof this story.
The other one is that the benzene molecule is char-acterized by at least 226 doubly ionized states whichspan within an electron binding energy range of 23–40 eV [41]. In contrast to its first ionization that yieldsthe benzene cation radical C6H+
6 which exists eitheras the 2B2g (obtuse) state or the 2B1g (acute) one (see,e.g., Ref. 5 and references therein), its double oneexhibits a rather complex pattern first reported inthe present work and displayed in Figure 5 (see alsoTable IV). As shown in particular in Figure 5, thevertical ionization of each ground-state conformerof C6H+
6 leads to two structures, singlet or triplet(B3LYP/T):
IEsingletv (Bz+ : 2B2g(obtuse) ⇒ 1B2g)
= 16.13 eV, IEtripletv (Bz+ : 2B2g(obtuse) ⇒ 3B2g)
= 15.47 eV;
IEsingletv (Bz+ : 2B1g(acute) ⇒ 1B1g)
= 16.12 eV, IEtripletv (Bz+ : 2B1g(acute) ⇒ 3B1g)
= 15.47 eV.
The above values of the vertical ionization ener-gies fairly agree with the experimental IEexpt
v (Bz+) =16.2 eV [42], that was determined from the chargestripping data, and the early computational ones,IEtheor
v (Bz+) = 16.10 (singlet) and 15.47 (triplet) eV5
(B3LYP/6-311G(d,p) calculations [28]; cf. with theupper boundary of 17.16 eV [43]).
A simple comparison of the reported vertical ion-ization energies of C6H+
6 indicates that the tripletstates of C6H2+
6 are energetically favorable by about
5Similarly to Ref. [30], it is worthwhile mentioning that a smalldiscrepancy between the experimental and theoretical ionizationenergies might arise due to Franck-Condon effects in verticalionization.
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2751

KRYACHKO
TABLE VThe B3LYP/T structures of the benzene dication.
Bz+(2B1g ) 1Bz2+ 3Bz2+ 3Bz2+ts Bz+(2B2g ) 1Bz2+
saddle=31Bz2+
saddle=21Bz2+
ts11Bz2+
ts2
r(C1 C2) 1.384 1.388 1.424 1.426 1.426 1.470 1.468 1.381 1.432r(C2 C3) 1.448 1.457 1.424 1.426 1.365 1.351 1.351 1.535 1.373r(C1 H1) 1.080 1.084 1.087 1.087 1.083 1.091 1.092 1.084 1.099r(C2 H2) 1.082 1.097 1.087 1.087 1.081 1.085 1.085 1.088 1.089∠C6C1C2 118.4 107.2 119.7 120.0 121.6 123.3 123.1 117.0 125.2∠C1C2H2 120.6 119.1 120.1 120.0 119.4 118.8 119.1 121.6 122.3
The atomic labels are indicated in Figure 6. Bond lengths are given in Å and bond angles in ◦ (Ref. 23 for the geometry of the groundelectronic state of the neutral benzene molecule).
0.5 eV over the singlets (see also Refs. 28, 44). Tak-ing into account the first vertical ionization energyof benzene of 9.24 eV [5] evaluated at the B3LYP/Tlevel, the double vertical ionization energy thusamounts to 24.72 eV (triplet) and 25.27–25.36 eV(singlet). Note that the B3LYP/6-311G(d,p) yieldscorrespondingly 25.55 and 24.50 eV [28] and thephotoionizationation threshold value is 25.3 eV [44]and 26.0 eV [30] (the B3LYP/6-31G(d,p) estimate is25.4 eV [45]).
However, as we observe in Figure 5, four verti-cally ionized states of C6H2+
6 are relaxed in a dras-tically different way. While the singlet state, thatis originated from C6H+
6 (2B2g; obtuse), transformsadiabatically into the planar third-order saddle
structure 1Bz2+saddle =3(2B2g), characterized by the tran-
sition frequencies of 1159 ı, 509 ı, and 79 ı cm−1
(see Table V), the other, arised from the acutebenzene cation radical, directly relaxes to the sin-glet energy minimum structure 1Bz2+ characterizedby the ZPVE-corrected adiabatic ionization energyIEsinglet
a (Bz+ : 2B1g(acute) ⇒ 1B1g) = 15.35 eV (theB3LYP/6-311G(d,p) method yields 15.36 eV [30]).1Bz2+ has a planar carbon ring with four short C Cbonds (1.388 Å) and the two longer ones (1.457Å; vide Figure 6 and Table V) implying that thissinglet state of C6H2+
6 is structurally close to theacute benzene cation radical rather than to the neu-tral benzene. The longer C C bonds have longerC H bonds (1.097 Å) lying out of the ring plane
FIGURE 6. The B3LYP/cc-pVTZ singlet- and triplet-state structures, 1Bz2+ and 3Bz2+ of the benzene dication C6H2+6 .
Their bond lengths are given in Å and bond angles in ◦ (some geometrical parameters of the parent obtuse C6H+6 (2B2g )
and acute C6H+6 (2B1g ) structures of the benzene cation radical, calculated at the same computational level [5e], are
presented in Table V).
2752 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14

DICATIONIC STATES OF BENZENE DIMER
(the dihedral angle ∠H2C2C3H3 = 48.0o) in orderto reduce the mutual Coulomb repulsions. Interest-ingly, the obtuse-type branch of the relaxation routealso ends at the acute-like structure 1Bz2+ of thesinglet-state benzene dication (vide Fig. 6).
The triplet structure 3Bz2+ resulted from ioniza-tion of Bz+(2B2g; obtuse) is the energy minimumof C6H2+
6 . The relevant ZPVE-corrected adiabaticionization energy IEtriplet
a (Bz+ : 2B2g(obtuse) ⇒3B2g) amounts to 15.25 eV (cf. with the B3LYP/6-311G(d,p) value of 15.28 eV [28]). Figure 6 andTable IV demonstrate that 3Bz2+ is characterizedby almost equal C C bonds (1.424 Å). Its nonpla-narity arises due to a small out-of-ring dispositionof the four hydrogen atoms bonded to C2, C3, C5,and C6 (the dihedral angle ∠H2C2C3H3 = 9.8◦).The triplet ionization route started from the acutebenzene cation radical involves the transition-statelinker 3Bz2+
ts (2B1g) with the transition frequency of1941ı cm−1 and ends at the same triplet state 3Bz2+
(IEtripleta (Bz+ : 2B1g(acute) ⇒ 3B2g) = 15.29 eV) as the
ionization route originated from the obtuse struc-ture of C6H+
6 . 0.06 eV determines a differencebetween the adiabatic ionization energies corre-sponding to the singlet and triplet ionization routesof the benzene cation radical and thus indicates aslight preference of the triplet-state benzene dicationover its singlet state. However, this small differencefalls within the range of chemical computationalaccuracy [46].
5. Summary and Conclusions
In the present work, invoking the B3LYP/D andB3LYP/T computational methods, we have demon-strated that the benzene dimer cation only admitstwo lowest-energy sandwiches of the S- and D-types.Their origins have been explained in terms of the par-ent obtuse and acute structures of the benzene cationradical. Two novel acute and obtuse homodimersof (C6H6)
+2 have been found in a close proximity
of the S- and D-sandwiches. It will be shown else-where that they determine the barrier shown inFigure 1 that confers a substantial kinetic stability tocovalently fused dicationic cluster of benzene dimer.All studied T-shaped-like structures of the benzenedimer cation, including the perfect T-shaped one,lie on a rather flat portion of the PES that preventsthe precise identification of their saddle structurefeatures.
We have thoroughly investigated the computedPES of ionization of the benzene cation radical in
all its structural motifs. It has been demonstratedthat the ionization route of the obtuse benzene cationradical directly yields the lowest triplet state of thebenzene dication, whereas the singlet state originatesfrom that of the acute conformer. A small preferenceof 0.06 eV (see also Ref. 31d, p. 1050 and Ref. 31g, p.1194) predicted at the B3LYP/T computational levelfor the benzene dication to finally occupy the moststable triplet state rather than the singlet one hasbeen reported. In contrast to the longstanding belief,neither the singlet state nor the triplet one of thebenzene dication structurally resemble the neutralground-state benzene molecule.
In conclusion, we have unveiled the importanceof the “obtuse versus acute” structural motif of thebenzene cation radical underlying the structural pat-terns that are exhibited on the low-energy portionsof the PESs of the benzene dimer cation and benzenedication.
ACKNOWLEDGMENTS
The author gratefully thanks Lenz Cederbaum forlively and fruitful discussions, Francoise Remacle,Jean-Pierre Francois, and Michael Deleuze for warmhospitality.
References
1. (a) Burley, S. K.; Petsko, G. A. Science 1985, 229, 23; (b) Rebek,J., Jr. Chem Soc Rev 1996, 25, 255; (c) Fyfe, M. C. T.; Stoddart,J. F. Acc Chem Res 1997, 10, 393; (d) McGaughey, G. B.; Gagné,M.; Rappe, A. K. J Biol Chem 1998, 273, 15458; (e) Tsuzuki, S.;Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. J Am ChemSoc 2002, 124, 104; (f) Sumathy, R.; Kryachko, E. S. J Phys ChemA 2002, 106, 510; (g) Kryachko, E. S.; Nakatsuji, H. J. Ibid A2002, 106, 731; (h) Nguyen, M. T.; Kryachko, E. S.; Vanquick-enborne, L. in The Chemistry of Phenols; Rappoport, Z., Ed.;Wiley: New York, 2003; Chapter 1, pp 1–198; and referencestherein.
2. (a) Hobza, P.; Selzle, H. L.; Schlag, E. W. Chem Rev 1994, 94,1767; (b) Steed, J. M.; Dixon, T. A.; Klemperer, W. J ChemPhys 1979, 70, 4940; (c) Karlstrom, G.; Linse, P.; Wallqvist, A.;Jonsson, B. J Am Chem Soc 1983, 105, 3777; (d) Krause, H.;Ernstberger, B.; Neusser, H. J. Chem Phys Lett 1991, 184, 411;(e) Jaffe, R. L.; Smith, G. D. J Chem Phys 1996, 105, 2780; (f)Hobza, P.; Selzle, H. L.; Schlag, E. W. J Phys Chem 1996, 100,18790; (g) Hobza, P.; Špirko, V.; Selzle, H. L.; Schlag, E. W.J Phys Chem A 1998, 102, 2501; (h) Špirko, V.; Engkvist, O.;Soldán, P.; Selzle, H. L.; Schlag, E. W.; Hobza, P. J Chem Phys1999, 111, 572; (i) Kim, K. S.; Tarakeshwar, P.; Lee, J. Y. ChemRev 2000, 100, 4145; (j) Tsuzuki, S.; Uchimaru, T.; Matsumura,K.; Mikami, T.; Tanabe, K. Chem Phys Lett 2000, 319, 547; (k)Tsuzuki, S.; Uchimaru, T.; Sugawara, K.-i.; Mikami, M. J ChemPhys 2002, 117, 11216; (l) Gonzalez, C.; Lim, E. C. J Phys ChemA 2001, 105, 1904; (m) Iimori, T.; Aoki, Y.; Ohshima, Y. J Chem
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2753

KRYACHKO
Phys 2002, 117, 3675; (n) Iimori, T.; Ohshima, Y. Ibid 2002,117, 3656; (o) DiStasio, R. A., Jr.; Helden, G. v.; Steele, R. P.;Head-Gordon, M. Chem Phys Lett 2007, 437, 277.
3. (a) Kohn, W.; Meir, Y.; Makarov, D. E. Phys Rev Lett 1998, 80,4153; (b) Tuma, C.; Boese, A. D.; Handy, N. C. Phys ChemChem Phys 1999, 1, 3939, and references therein.
4. (a) Frasinsky, L. J.; Codling, K.; Hatherly, P.; Barr, J.; Ross, I. N.;Toner, W. T. Phys Rev Lett 1987, 58, 2424.
5. (a) Huang, M.; Lunell, S. J Chem Phys 1990, 92, 6081; (b)Müller-Dethlefs, K. Ibid 1999, 111, 10550; (c) Lindner, R.;Müller-Dethlefs, K.; Wedum, E.; Haber, K.; Grant, E. R. Science1996, 271, 1698; (d) Applegate, B. E.; Miller, T. A. J Chem Phys2002, 117, 10654; (e) Deleuze, M. S.; Claes, L.; Kryachko, E. S.;Francois, J.-P. Ibid 2003, 119, 3106 and references therein.
6. (a) Cederbaum, L. S.; Zobeley, J.; Tarantelli, F. Phys Rev Lett1997, 79, 4778; (b) Zobeley, J.; Cederbaum, L. S.; Tarantelli,F. J Chem Phys 1998, 108, 9737; (c) Rühl, E.; Schmale, C.;Schmelz, H. C.; Baumgärtel, H. Chem Phys Lett 1992, 191,430; (d) Rühl, E. Int J Mass Spectrom 2003, 229, 117; (e) Santra,R.; Cederbaum, L. S. Phys Rep 2002, 368, 1.
7. (a) Sattler, K.; Mühlbach, J.; Echt, O.; Pfau, P.; Recknagel, E.Phys Rev Lett 1981, 47, 160; (b) Bréchignac, C.; Cahuzac, Ph.;Carliez, F.; de Frutos, M. Ibid 1990, 64, 2893; (c) Echt, O.; Märk,T. D. In Clusters of Atoms and Molecules; Haberland, H., Ed.;Springer: Berlin, 1994; p 183; (d) Mühlbach, J.; Sattler, K.; Pfau,P.; Recknagel, E. Phys Lett A 1982, 87, 415; (e) Pfau, P.; Sattler,K.; Pfaum, P.; Recknagel, E. Ibid 1984, 104A, 262.
8. Lord Rayleigh, Phil Mag 1882, 14, 184.9. Meitner, L.; Frisch, O. R. Nature 1939, 143, 239.
10. Bohr, N.; Wheeler, J. A. Phys Rev 1939, 56, 426.11. (a) Casero, R.; Sáenz, J. J.; Soler, J. M. Phys Rev A1988, 37, 1401;
(b) Kreisle, D.; Echt, O.; Knapp, M.; Recknagel, E.; Leiter, K.;Märk, T. D.; Sáenz, J. J.; Soler, J. M. Phys Rev Lett 1986, 56,1551.
12. (a) Echt, O.; Kreisle, D.; Recknagel, E.; Sáenz, J. J.; Casero,R.; Soler, J. M. Phys Rev A 1988, 38, 3236; (b) Rauth, T.;Grill, V.; Märk, T. D. Int J Mass Spectrom Ion Proc 1994, 139,147.
13. Märk, T. D.; Dcheier, P. Nucl Instrum Methods Phys Res B1995, 98, 469.
14. (a) Schriver, K. E.; Hahn, M. Y.; Whetten, R. L. Phys Rev Lett1987, 59, 1906; (b) Hahn, M. Y.; Schriver, K. E.; Whetten, R. L.J Chem Phys 1988, 88, 4242; (c) Stace, A. J.; Bernard, D. M.;Crooks, J. J.; Reid, K. L. Mol Phys 1987, 60, 671.
15. (a) Geiger, J.; Rühl E. Int J Mass Spectrom 2002, 220, 99; (b)Inokuchi, Y.; Naitoh, Y.; Ohashi, K.; Saitow, K.-I.; Yoshihara,K.; Nishi, N. Chem Phys Lett 1997, 269, 298.
16. Deleuze, M. S.; Francois, J.-P.; Kryachko, E. S. J Am Chem Soc2005, 127, 16824.
17. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Mont-gomery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.;Millan, J. M.; Daniels,A. D.; Kudin, K. N.; Strain, M. C.; Farkas,O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci,B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Peter-son, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.;Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski,J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz,P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith,T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez,
C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.;Wong, M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.;Pople, J. A. GAUSSIAN 98; Gaussian, Inc.: Pittsburgh, PA,1998.
18. (a) Dunning, T. H., Jr. J Chem Phys 1989, 90, 1007; (b) Kendall,R. A.; Dunning, T. H., Jr.; Harrison, R. J. Ibid 1992, 96, 6796.
19. (a) Field, F. H.; Hamlet, P.; Libby, W. F. J Am Chem Soc 1969,91, 2839; (b) Wexter, S.; Polo, L. G. J Phys Chem 1970, 74, 257;(c) Jones, E. G.; Bhattacharya, A. K.; Tiernan, T. O. Int J MassSpectrom Ion Phys 1975, 17, 147; (d) Meot-Ner (Mautner), M.;Hamlet, P.; Hunter, E. P.; Field, F. H. J Am Chem Soc 1978, 100,5466; (e) Rusyniak, M.; Ibrahim, Y.; Alsharaeh, E.; Meot-Ner(Mautner), M.; El-Shall, M. S. J Phys Chem A 2003, 107, 7656;(f) Hiraoka, K.; Fujimaki, S.; Aruga, K.; Yamabe, S. J ChemPhys 1991, 95, 8413; (g) Grover, J. P.; Walters, E. A.; Hui, E. T.J Phys Chem 1987, 91, 3233.
20. (a) Edlund, O.; Kinell, P. O.; Lund, A. J Chem Phys 1967, 96,3679; (b) Edlund, O.; Kinell, P. O.; Lund, A.; Shimizu, AdvChem Ser 1968, 82, 311; (c) Badger, B.; Brocklehurst, B. Nature1968, 219, 263; (d) Milosevich, S. A.; Saichek, K.; Hinchey, L.;England, W. B.; Kovacic, P. J Am Chem Soc 1983, 105, 1088; (e)Chandra, A. K.; Bhanuprakash, K.; Jyoti Bhasu, V. C.; Srikan-than, D. Mol Phys 1984, 52, 733; (f) Eberson, L.; Hartshorn,M. P.; Persson, O. J Chem Soc, Perkin Trans 2 1995, 409;(g) Miyoshi, E.; Ichikawa, T.; Sumi, T.; Sakai, Y.; Shida, N.Chem Phys Lett 1997, 275, 404; (h) Kadam, R. M.; Erickson,R.; Komaguchi, K.; Shiotani, M.; Lund, A. Chem Phys Lett1998, 290, 371; (i) Itagaki, Y.; Benetis, N. P.; Kadam, R. M.;Lund, A. Phys Chem Chem Phys 2000, 2, 2683; (j) Miyoshi,E.; Yamamoto, N.; Sekiya, M.; Tanaka, K. Mol Phys 2003, 101,227.
21. (a) Ohashi, K.; Nishi, N. J Chem Phys 1991, 95, 4002;(b) Ohashi, K.; Nishi, N. J Phys Chem 1992, 96, 2931; (c) Ohashi,K.; Nakai, Y.; Shibata, T.; Nishi, N. Laser Chem 1994, 14, 3;(d) Shibata, T.; Ohashi, K.; Nakai, Y.; Nishi, N. Chem PhysLett 1994, 229, 604; (e) Nakai, Y.; Ohashi, K.; Nishi, N. Ibid1995, 233, 36; (f) Ohasi, K.; Inokuchi, Y.; Nishi, N. Ibid 1996,263, 167; (g) Shinohara, H.; Nishi, N. J Chem Phys 1989, 91,6743; (h) Inokuchi, Y.; Nishi, N. Ibid 2001, 114, 7059.
22. (a) Shida, T. Electronic Absorption Spectra of Radical Ions;Elsevier: Amsterdam, 1988; p 12; (b) Bühler, R. E.; Funk, W.J Phys Chem 1975, 79, 2098; (c) Badger, B.; Brocklehurst, B.Trans Faraday Soc 1969, 65, 2582; (d) Shida, T.; Hamill, W. H.J Chem Phys 1966, 44, 4372; (e) Miller, J. H.; Andrews, L.;Lund, P. A.; Schatz, P. N. Ibid 1980, 73, 4932; (f) Badger, B.;Brocklehurst, B. Trans Faraday Soc 1969, 65, 2588; (g) Kira, A.;Arai, S.; Imamura, M. J Phys Chem 1972, 76, 1119; (h) Badger,B.; Brocklehurst, B.; Russell, R. D. Chem Phys Lett 1976, 1, 122;(i) Snodgrass, J. T.; Dunbar, R. C.; Bowers, M. T. J Phys Chem1990, 94, 3648.
23. (a) Inokuchi, Y.; Ohashi, K.; Nishi, N. Chem Phys Lett 1997,279, 73; (b) Inokuchi, Y.; Naitoh, Y.; Ohashi, K.; Saitow, K.-i.;Yoshihara, K.; Nishi, N. Ibid 1997, 269, 298. (c) Misaizu, F.;Shinohara, H.; Nishi, N.; Kondow, T.; Kinoshita, M. Int J MassSpectrom Ion Proc 1990, 102, 99; (d) Börnsen, K. O.; Selzle,H. L.; Schlag, E. W. Chem Phys Lett 1992, 190, 497; (e) Ohashi,K.; Nakai, Y.; Shibata, T.; Nishi, N. Surf Rev Lett 1996, 3, 601;(f) Tagawa, S.; Beck, G.; Schnabel, W. Z Naturforsch 1982, 37a,982.
24. (a) Krause, H.; Ernstberger, B.; Neusser, H. J. Chem Phys Lett1991, 184, 411; (b) Jarzeba, W.; Schlief, R. E.; Barabara, P. E.J Phys Chem 1994, 98, 9102; (c) Gould, I. R.; Farid, S. J Am
2754 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 107, NO. 14

DICATIONIC STATES OF BENZENE DIMER
Chem Soc 1993, 115, 4814; (d) Ojima, S.; Miyasaka, H.; Mataga,N. J Phys Chem 1990, 94, 5834; (e) Ojima, S.; Miyasaka, H.;Mataga, N. Ibid 1990, 94, 4147; (f) Bockman, T. M.; Karpinski,Z. J.; Sankararaman, S.; Kochi, J. K. J Am Chem Soc 1986, 114,1970; (g) Rusyniak, M.; Ibrahim, Y.; Wright, D. L.; Khanna,S. N.; El-Shall, M. S. Ibid 2003, 125, 12001.
25. (a) Wilson, E. B. Phys Rev 1934, 45, 706; (b) Varsanyi, G.Assignment for Vibrational Spectra of Seven Hundred Ben-zene Derivatives; Adam Hilger: London, 1974; (c) Goodman,L.; Ozkabak, A. G.; Thakur, S. N. J Phys Chem 1991, 95, 9044;(d) Palafox, M. A. Int J Quantum Chem 2000, 77, 61; (e) Forcurrent work see, e. g., Ref. [1f] and references therein.
26. (a) Mulliken, R. S. J Am Chem Soc 1952, 74, 811; (b) Mulliken,R. S. J Phys Chem 1952, 56, 801; (c) Mulliken, R. S.; Person,W. B. Molecular Complexes; Wiley: New York, 1969.
27. (a) Mataga, N.; Kubota, T. Molecular Interactions and Elec-tronic Spectra; Marcel Dekker: New York, 1970; (b) Birks,J. B. Photophysics of Aromatic Molecules; Wiley-Interscience:London, 1970 and references therein.
28. Schröder, D.; Loos, J.; Schwarz, H.; Thissen, R.; Preda, D. V.;Scott, L. T.; Caraiman, D.; Frach, M. V.; Böhme, D. K. HelvChim Acta 2001, 84, 1625.
29. Prakash, G. K. S. Pure Appl Chem 1998, 70, 2001.30. Olah, G. A.; Shamma, T.; Burrichter, A.; Rasul, G.; Prakash,
G. K. S. J Am Chem Soc 1997, 119, 3407.31. (a) Hall, R. I.; Avaldi, L.; Dawber, G.; McConkey, A. G.;
MacDonald, M. A.; King, G. C. Chem Phys 1994, 187,125; (b) Griffiths, W. J.; Harris, F. M. Ibid 1991, 157, 299;(c) Krogh-Jespersen, K. J Am Chem Soc 1991, 113, 417 andreferences therein. (d) Lammertsma, K.; Schleyer, P. v. R. J AmChem Soc 1983, 105, 1049; (e) Dewar, M. J. S.; Holloway,M. K. Ibid 1984, 106, 6619; (f) Jonkman, H. T.; Nieuwpoort,W. C. Tetrahedron Lett 1973, 1671; (g) See also Rosi, M.;Bauschlicher, Jr., C. W.; Bakes, E. L. O. Astrophys J 2004, 609,1192.
32. Ledingham, K. W. D.; Smith, D. J.; Singhal, R. P.; McCanny, T.;Graham, P.; Kilic, H. S.; Peng, W. X.; Langley, A. J.; Taday, P. F.;Kosmidis, C. J Phys Chem A 1999, 103, 2952.
33. Smith, D. J.; Ledingham, K. W. D.; Singhal, R. P.; Kilic, H. S.;McCanny, T.; Langley, A. J.; Taday, P. F.; Kosmidis, C. RapidCommun Mass Spectrom 1998, 12, 813.
34. Itakura, R.; Watanabe, J.; Hishikawa, A.; Yamanouchi, K.J Chem Phys 2001, 114, 5598.
35. (a) Higgins, W.; Jennings, K. R. Chem Commun 1965, 99; (b)Higgins, W.; Jennings, K. R. Trans Faraday Soc 1966, 62, 97;(c) Engel, R.; Halpern, D.; Funk, B.-A. Org Mass Spectrom1973, 7, 177; (d) Ast, T. Adv Mass Spectrom 1980, 8A, 555.
36. Field, F. H.; Franklin, J. L. Electron Impact Phenomena;Academic: New York, 1957.
37. (a) Appling, J. R.; Musier, K. M.; Moran, T. F. Org Mass Spec-trom 1984, 19, 412; (b) Vékey, K.; Brenton, A. G.; Beynon,J. H. J Phys Chem 1986, 90, 3569; (c) Hayward, M. J.; Mabud,M. A.; Cooks, R. G. J Am Chem Soc 1988, 110, 1343; (d) Vékey,K; Brenton, A. G.; Beynon, J. H. Org Mass Spectrom 1989,24, 31.
38. (a) Dorman, F. H.; Morrison, J. D. J Chem Phys 1961, 35, 575;(b) Beynon, J. H.; Fontaine, A. E. Chem Commun 1966, 717;(c) Spohr, R.; Bergmark, T.; Magnusson, N.; Werme, L. O.;Nordling, C.; Siegbahn, K. Phys Scr 1970, 2, 31; (d) Holmes,J. L.; Osborne, A. D.; Weese, G. M. Org Mass Spectrom 1975,10, 867; (e) Derrick, P. J. Ibid 1975, 10, 1171; (f) Tsai, B. P.; Eland,J. H. D. Int J Mass Spectrom Ion Phys 1980, 36, 143; (g) Grif-fiths, I. W.; Mukhtar, E. S.; Harris, F. M.; Beynon, J. H. Ibid1981, 39, 257 and references therein.
39. (a) Richardson, P. J.; Eland, J. H. D.; Lablanquie, P. Org MassSpectrom 1986, 21, 289; (b) Guilhaus, M.; Kingston, R. G.;Brenton, A. G.; Beynon, J. H. Ibid 1985, 20, 565.
40. (a) Rosenstock, H. M.; Dannacher, J.; Liebman, J. F. Radiat PhysChem 1982, 20, 7; (b) Kingston, R. G.; Guilhaus, M.; Brenton,A. G.; Beynon, J. H. Org Mass Spectrom 1985, 20, 406.
41. Tarantelli, F.; Sgamellotti, A.; Cederbaum, L. S.; Schirmer, J.J Chem Phys 1987, 86, 2201.
42. Tobita, S.; Leach, S.; Jochims, H. J.; Rühl, E.; Illenberger, E.;Baumgärtel, H. Can J Phys 1994, 72, 1060.
43. Leach, S. J. Electron Spectrosc Rel Phenomena 1986, 41,427.
44. Ghergel, L.; Brand, J. D.; Baumgarten, M.; Müllen, K. J AmChem Soc 1999, 121, 8104.
45. Zyubina, T. S.; Kim, G.-S.; Lin, S. H.; Mebel, A. M.; Bandrauk,A. D. Chem Phys Lett 2002, 359, 253.
46. Morokuma, K. Phil Trans Proc Roy Soc London A 2002, 360,1149.
VOL. 107, NO. 14 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 2755