combination of donor characters in a donor–acceptor–donor (dad) type polymer containing...
TRANSCRIPT

Organic Electronics 11 (2010) 1877–1885
Contents lists available at ScienceDirect
Organic Electronics
journal homepage: www.elsevier .com/locate /orgel
Combination of donor characters in a donor–acceptor–donor (DAD)type polymer containing benzothiadiazole as the acceptor unit
Merve Sendur a, Abidin Balan a, Derya Baran a, Baris Karabay a, Levent Toppare a,b,c,⇑a Middle East Technical University, Department of Chemistry, 06531 Ankara, Turkeyb Department of Biotechnology, Middle East Technical University, 06531 Ankara, Turkeyc Department of Polymer Science and Technology, Middle East Technical University, 06531 Ankara, Turkey
a r t i c l e i n f o
Article history:Received 3 August 2010Received in revised form 2 September 2010Accepted 2 September 2010Available online 16 September 2010
Keywords:Conjugated polymersElectrochromismDonor–acceptor theoryCopolymersp- and n-doping
1566-1199/$ - see front matter � 2010 Elsevier B.Vdoi:10.1016/j.orgel.2010.09.001
⇑ Corresponding author at: Middle East Technicament of Chemistry, 06531 Ankara, Turkey. Tel.: +90 33122103200.
E-mail address: [email protected] (L. Toppare
a b s t r a c t
A benzothiadiazole bearing donor–acceptor–donor (D–A–D) type monomer (M3) was syn-thesized using the combination of 3, 4-ethylenedioxythiophene (EDOT) and thiophenedonor units to understand the effect of donor strength on the optoelectronic and electro-chemical properties. The resulting monomer was polymerized electrochemically (P3) andcompared with its symmetrical thiophene (P1) and EDOT (P2) bearing homologues whetherthere exists a combination of the electrochemical and optical characteristics. Also, copoly-mer studies were performed with symmetrical thiophene (M1) and EDOT (M2) containingmonomers in order to compare the results with P3. Cyclic voltammetry (CV) and spectro-electrochemistry results revealed that P3 is a low band gap polymer (1.18 eV) having bothp-and n-type property which is superior to the copolymers synthesized using M1 and M2.
� 2010 Elsevier B.V. All rights reserved.
1. Introduction
Due to their favorable electronic, optical and mechanicalproperties conductive polymers (CPs) are used in a widerange of applications such as photovoltaics [1], light-emit-ting diodes [2], field-effect transistors [3], sensors [4], andelectrochromic devices [5]. CPs as a class of electrochromicmaterials have attracted great interest due to several supe-rior properties over their inorganic counterparts such ascoloration efficiency [6], fast switching ability [7], multiplecolors with the same material [8], and fine-tuning of theband gap with small structural modifications [5b,6–9].Due to the fact that electrochromic and non emissiveapplications of CPs provide stimuli effect (viewing angle,light intensity or sunlight exposure) free, light-weight andflexible displays, their development is considered to beimportant for future display technologies [10].
. All rights reserved.
l University, Depart-122103251; fax: +90
).
In order to use CPs in electrochromic devices, there aretwo issues to be resolved; to have a low band gap to max-imize the conductivity of the polymers and satisfy threeadditive colors in the neutral state to obtain full color space[11]. Approaches to achieve RGB (red, green, blue) colorswith CPs have become the subject of many studies; how-ever, only handful of them revealed true red [12], blue[13] or green [14] neutral state polymers. Up to date, do-nor–acceptor–donor (DAD) route has been considered asthe most effective way to obtain low band gap polymerswith desired neutral state colors. DA structured monomersare usually obtained with electropolymerizable heterocy-cles as the end group in order to achieve the necessarypolymers electrochemically [15].
Electrochemical copolymerization is also a facile meth-od to obtain desired neutral state colors for these types ofmaterials. In copolymerization, electroactive monomersare polymerized from a bulk containing the co-monomers.Resulting polymer generally shows unique optical andelectronic properties with enhanced mechanical and ki-netic properties compared to those of the homo-polymers[16]. Fine tuning in the band gap and neutral state color

1878 M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885
can be achieved by tailoring the co-monomer feed ratio aswell as the working potential of copolymerization [17].However, exact structures of electrochemical copolymersare hard to determine due to the insolubility of the depos-ited films which hinders the solution characterizations.
Recently, poly(4,7-di(2,3-dihydro-thieno[3,4-b][1,4]di-oxin-5-yl)benzo[1,2,5] thiadiazole) [14d] (P2) was charac-terized in terms of its electrochromic properties in ourgroup. It was shown to be the first green to transmissiveelectrochromic material with two simultaneous maximumabsorptions placed at around 400 and 750 nm. Its homo-logue with lower electron density, poly(4,7-dithien-2-yl-2,1,3-benzothiadiazole) [18] (P1) has an absorptionmaximum at around 560 nm where its second transitionis in UV region. At the first glance, it seems that a copolymerobtained from these two monomers should have an absorp-tion covering the entire visible region, but the copolymersreported in this manuscript revealed neutral state colorsin between the two, since absorption maximum of thecopolymer usually lies between the two homo-polymers.
Herein, we report our approach to explain the effect ofdifferent donor groups on the electronic and optical prop-erties of DAD type polymers. An asymmetric monomerhaving both 3,4-ethylenedioxytiophene and thiophene asthe donor groups was synthesized and electrochemicallypolymerized. Since the monomer contains two differentdonor units, we anticipated that its polymer may behaveas a copolymer of the symmetric monomer having thesame donor groups. Hence, the properties of copolymersachieved with the co-monomers having either thiopheneor 3,4-ethylenedioxythiophene as the donor group werealso reported.
2. Results and discussion
2.1. Synthesis
Donor–acceptor–donor type materials 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (M1) [18,19], 4,7-di(2,3-dihydro-thieno[3,4-b][1,4]dioxin-5-yl)benzo[1,2,5]thia-
Scheme 1. Synthesis scheme of the
diazole (M2) [14d,20] and 4-(2,3-di hydrothieno[3,4-b][1,4]dioxin-5-yl)-7-(thiophen-2-yl)benzo[c][1,2,5]thia-diazole (M3) were synthesized as shown in Scheme 1 accord-ing to previously reported methods. In order to synthesize theasymmetric monomer (M3) 4,7 dibromobenzothiadiazolewas first coupled with tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)stannane and then the isolatedcompound was coupled with tributyl(thiophen-2-yl)stannane.
2.2. Cyclic voltammetry
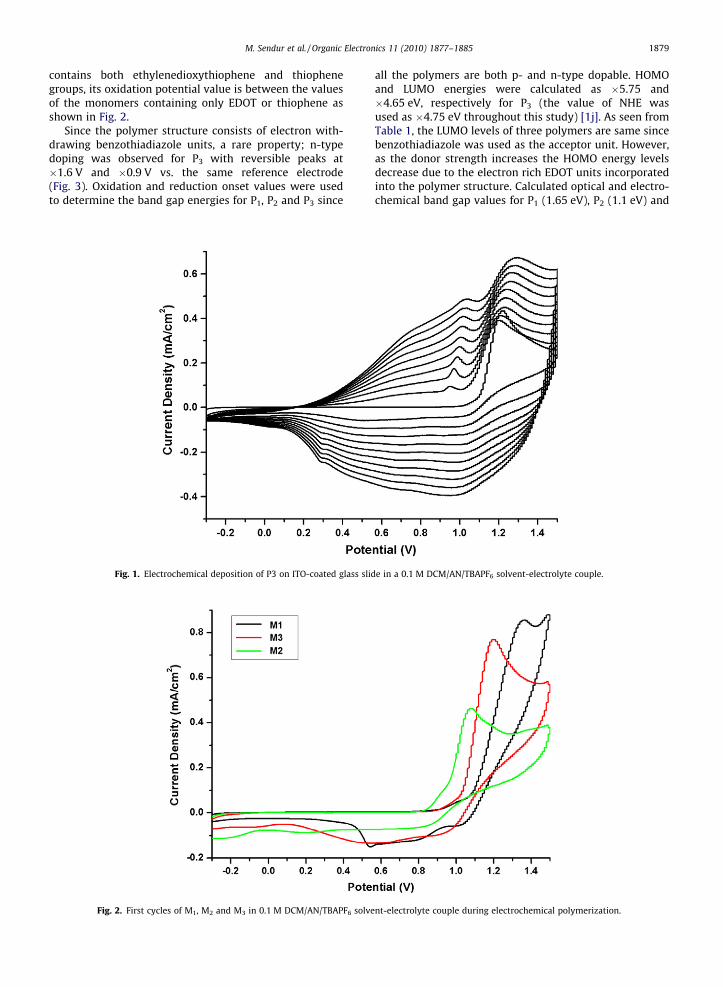
M3 was polymerized potentiodynamically between�0.3 V and 1.5 V vs. Ag wire pseudo reference electrode(0.3 V vs. Fc/Fc+) in a 0.1 M dichloromethane (DCM)/aceto-nitrile (AN) (5:95, v:v) / tetrabutylammonium hexafluoro-phosphate (TBAPF6) solution onto ITO-coated glass slidesin order to observe the polymer characteristics. Duringpolymerization, a characteristic oxidation peak of mono-mer was observed at 1.2 V accompanied by a reversible re-dox couple of the polymer. In Fig. 1, the increase in currentdensity with the number of scans clearly shows the depo-sition of an electroactive film of P3 on ITO. The resultantpolymer revealed reversible p-type doping propertieswhere the p doping/de-doping was indicated by the peaksat 1.06 V and 0.85 V, respectively. The P3 film is blue in itsneutral state and reveals light blue color when oxidized.
Generally, EDOT bearing DAD type molecules have low-er monomer oxidation potentials than thiophene bearingones under identical experimental conditions owing tothe electron rich ethylenedioxythiophene group. As re-ported M1, revealed a monomer oxidation at 0.85 V in0.1 M DCM/AN (5:95, v:v)/TBAClO4 vs. the Ag/Ag+ refer-ence electrode and its EDOT bearing homologue M2 has amonomer oxidation at 0.95 V in DCM/TBAPF6. In order tohave a reliable comparison with M3, we performed allexperiments in DCM/AN/TBAPF6 system vs. Ag wire pseudoreference electrode. As a result, M3 revealed a monomeroxidation at 1.2 V vs. Ag wire pseudo reference electrodewhich was lying between the two others. Since M3
monomers; M1, M2 and M3.
M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885 1879
contains both ethylenedioxythiophene and thiophenegroups, its oxidation potential value is between the valuesof the monomers containing only EDOT or thiophene asshown in Fig. 2.
Since the polymer structure consists of electron with-drawing benzothiadiazole units, a rare property; n-typedoping was observed for P3 with reversible peaks at�1.6 V and �0.9 V vs. the same reference electrode(Fig. 3). Oxidation and reduction onset values were usedto determine the band gap energies for P1, P2 and P3 since
Fig. 1. Electrochemical deposition of P3 on ITO-coated glass slid
Fig. 2. First cycles of M1, M2 and M3 in 0.1 M DCM/AN/TBAPF6 solve
all the polymers are both p- and n-type dopable. HOMOand LUMO energies were calculated as �5.75 and�4.65 eV, respectively for P3 (the value of NHE wasused as �4.75 eV throughout this study) [1j]. As seen fromTable 1, the LUMO levels of three polymers are same sincebenzothiadiazole was used as the acceptor unit. However,as the donor strength increases the HOMO energy levelsdecrease due to the electron rich EDOT units incorporatedinto the polymer structure. Calculated optical and electro-chemical band gap values for P1 (1.65 eV), P2 (1.1 eV) and
e in a 0.1 M DCM/AN/TBAPF6 solvent-electrolyte couple.
nt-electrolyte couple during electrochemical polymerization.
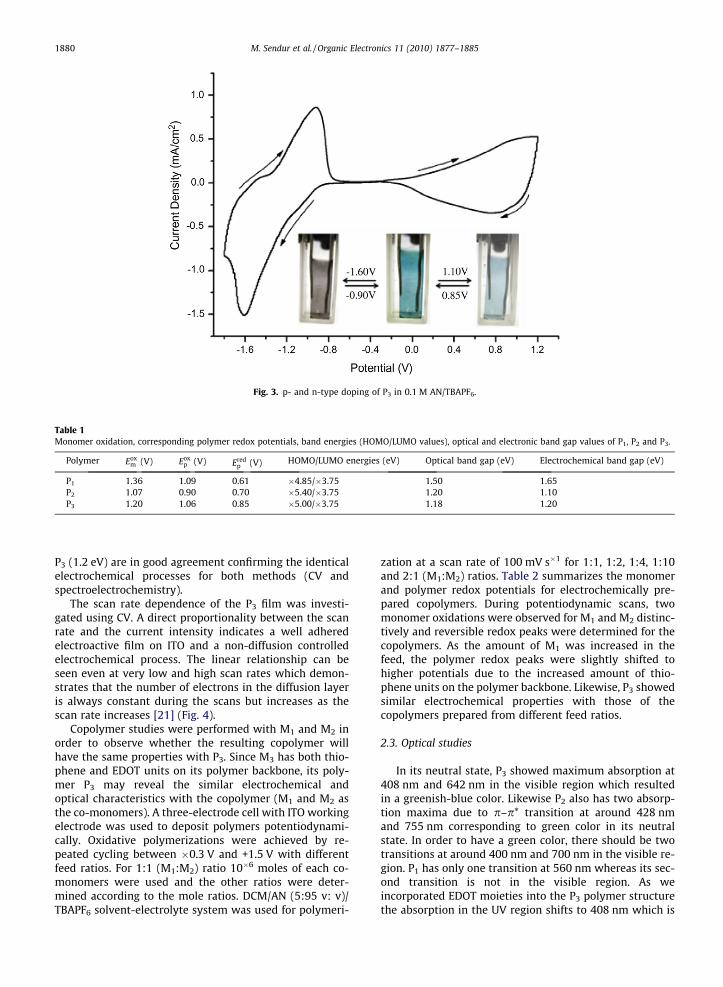
Fig. 3. p- and n-type doping of P3 in 0.1 M AN/TBAPF6.
Table 1Monomer oxidation, corresponding polymer redox potentials, band energies (HOMO/LUMO values), optical and electronic band gap values of P1, P2 and P3.
Polymer Eoxm (V) Eox
p (V) Eredp (V) HOMO/LUMO energies (eV) Optical band gap (eV) Electrochemical band gap (eV)
P1 1.36 1.09 0.61 �4.85/�3.75 1.50 1.65P2 1.07 0.90 0.70 �5.40/�3.75 1.20 1.10P3 1.20 1.06 0.85 �5.00/�3.75 1.18 1.20
1880 M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885
P3 (1.2 eV) are in good agreement confirming the identicalelectrochemical processes for both methods (CV andspectroelectrochemistry).
The scan rate dependence of the P3 film was investi-gated using CV. A direct proportionality between the scanrate and the current intensity indicates a well adheredelectroactive film on ITO and a non-diffusion controlledelectrochemical process. The linear relationship can beseen even at very low and high scan rates which demon-strates that the number of electrons in the diffusion layeris always constant during the scans but increases as thescan rate increases [21] (Fig. 4).
Copolymer studies were performed with M1 and M2 inorder to observe whether the resulting copolymer willhave the same properties with P3. Since M3 has both thio-phene and EDOT units on its polymer backbone, its poly-mer P3 may reveal the similar electrochemical andoptical characteristics with the copolymer (M1 and M2 asthe co-monomers). A three-electrode cell with ITO workingelectrode was used to deposit polymers potentiodynami-cally. Oxidative polymerizations were achieved by re-peated cycling between �0.3 V and +1.5 V with differentfeed ratios. For 1:1 (M1:M2) ratio 10�6 moles of each co-monomers were used and the other ratios were deter-mined according to the mole ratios. DCM/AN (5:95 v: v)/TBAPF6 solvent-electrolyte system was used for polymeri-
zation at a scan rate of 100 mV s�1 for 1:1, 1:2, 1:4, 1:10and 2:1 (M1:M2) ratios. Table 2 summarizes the monomerand polymer redox potentials for electrochemically pre-pared copolymers. During potentiodynamic scans, twomonomer oxidations were observed for M1 and M2 distinc-tively and reversible redox peaks were determined for thecopolymers. As the amount of M1 was increased in thefeed, the polymer redox peaks were slightly shifted tohigher potentials due to the increased amount of thio-phene units on the polymer backbone. Likewise, P3 showedsimilar electrochemical properties with those of thecopolymers prepared from different feed ratios.
2.3. Optical studies
In its neutral state, P3 showed maximum absorption at408 nm and 642 nm in the visible region which resultedin a greenish-blue color. Likewise P2 also has two absorp-tion maxima due to p–p* transition at around 428 nmand 755 nm corresponding to green color in its neutralstate. In order to have a green color, there should be twotransitions at around 400 nm and 700 nm in the visible re-gion. P1 has only one transition at 560 nm whereas its sec-ond transition is not in the visible region. As weincorporated EDOT moieties into the P3 polymer structurethe absorption in the UV region shifts to 408 nm which is
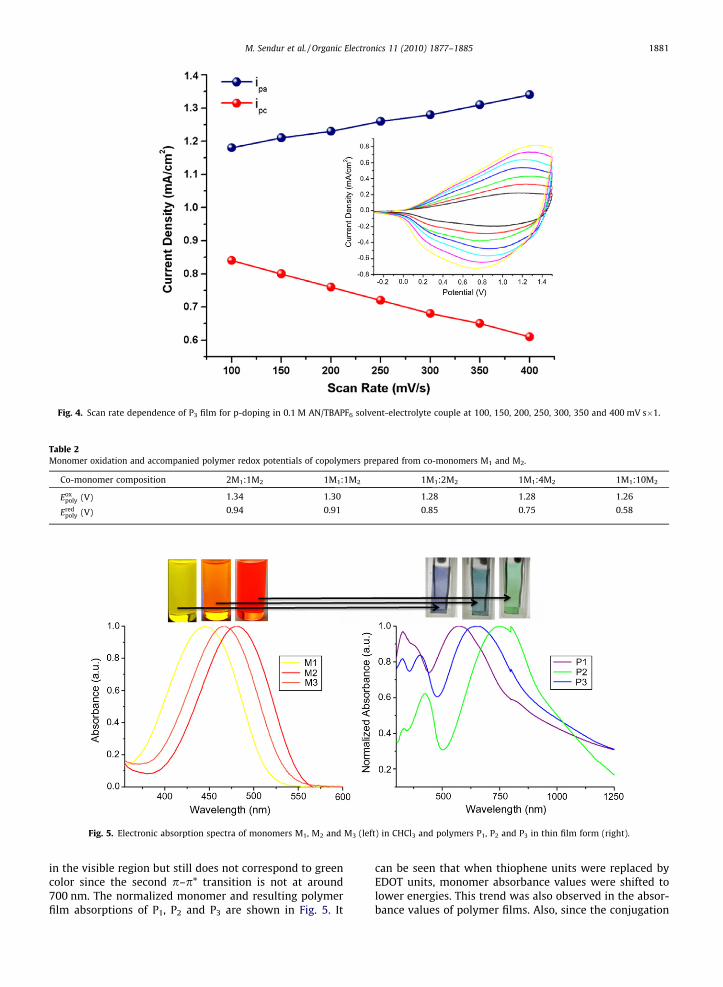
Fig. 4. Scan rate dependence of P3 film for p-doping in 0.1 M AN/TBAPF6 solvent-electrolyte couple at 100, 150, 200, 250, 300, 350 and 400 mV s�1.
Table 2Monomer oxidation and accompanied polymer redox potentials of copolymers prepared from co-monomers M1 and M2.
Co-monomer composition 2M1:1M2 1M1:1M2 1M1:2M2 1M1:4M2 1M1:10M2
Eoxpoly (V) 1.34 1.30 1.28 1.28 1.26
Eredpoly (V) 0.94 0.91 0.85 0.75 0.58
Fig. 5. Electronic absorption spectra of monomers M1, M2 and M3 (left) in CHCl3 and polymers P1, P2 and P3 in thin film form (right).
M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885 1881
in the visible region but still does not correspond to greencolor since the second p–p* transition is not at around700 nm. The normalized monomer and resulting polymerfilm absorptions of P1, P2 and P3 are shown in Fig. 5. It
can be seen that when thiophene units were replaced byEDOT units, monomer absorbance values were shifted tolower energies. This trend was also observed in the absor-bance values of polymer films. Also, since the conjugation
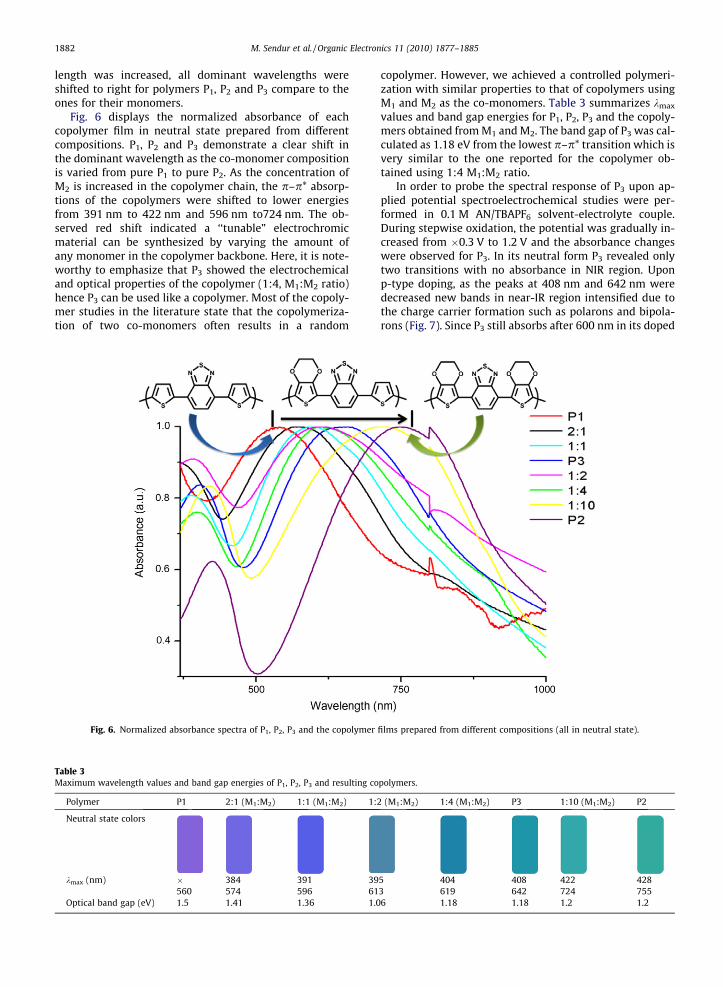
1882 M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885
length was increased, all dominant wavelengths wereshifted to right for polymers P1, P2 and P3 compare to theones for their monomers.
Fig. 6 displays the normalized absorbance of eachcopolymer film in neutral state prepared from differentcompositions. P1, P2 and P3 demonstrate a clear shift inthe dominant wavelength as the co-monomer compositionis varied from pure P1 to pure P2. As the concentration ofM2 is increased in the copolymer chain, the p–p* absorp-tions of the copolymers were shifted to lower energiesfrom 391 nm to 422 nm and 596 nm to724 nm. The ob-served red shift indicated a ‘‘tunable” electrochromicmaterial can be synthesized by varying the amount ofany monomer in the copolymer backbone. Here, it is note-worthy to emphasize that P3 showed the electrochemicaland optical properties of the copolymer (1:4, M1:M2 ratio)hence P3 can be used like a copolymer. Most of the copoly-mer studies in the literature state that the copolymeriza-tion of two co-monomers often results in a random
Fig. 6. Normalized absorbance spectra of P1, P2, P3 and the copolymer
Table 3Maximum wavelength values and band gap energies of P1, P2, P3 and resulting co
Polymer P1 2:1 (M1:M2) 1:1 (M1:M2) 1:
Neutral state colors
kmax (nm) � 384 391 39560 574 596 61
Optical band gap (eV) 1.5 1.41 1.36 1.0
copolymer. However, we achieved a controlled polymeri-zation with similar properties to that of copolymers usingM1 and M2 as the co-monomers. Table 3 summarizes kmax
values and band gap energies for P1, P2, P3 and the copoly-mers obtained from M1 and M2. The band gap of P3 was cal-culated as 1.18 eV from the lowest p–p* transition which isvery similar to the one reported for the copolymer ob-tained using 1:4 M1:M2 ratio.
In order to probe the spectral response of P3 upon ap-plied potential spectroelectrochemical studies were per-formed in 0.1 M AN/TBAPF6 solvent-electrolyte couple.During stepwise oxidation, the potential was gradually in-creased from �0.3 V to 1.2 V and the absorbance changeswere observed for P3. In its neutral form P3 revealed onlytwo transitions with no absorbance in NIR region. Uponp-type doping, as the peaks at 408 nm and 642 nm weredecreased new bands in near-IR region intensified due tothe charge carrier formation such as polarons and bipola-rons (Fig. 7). Since P3 still absorbs after 600 nm in its doped
films prepared from different compositions (all in neutral state).
polymers.
2 (M1:M2) 1:4 (M1:M2) P3 1:10 (M1:M2) P2
5 404 408 422 4283 619 642 724 7556 1.18 1.18 1.2 1.2
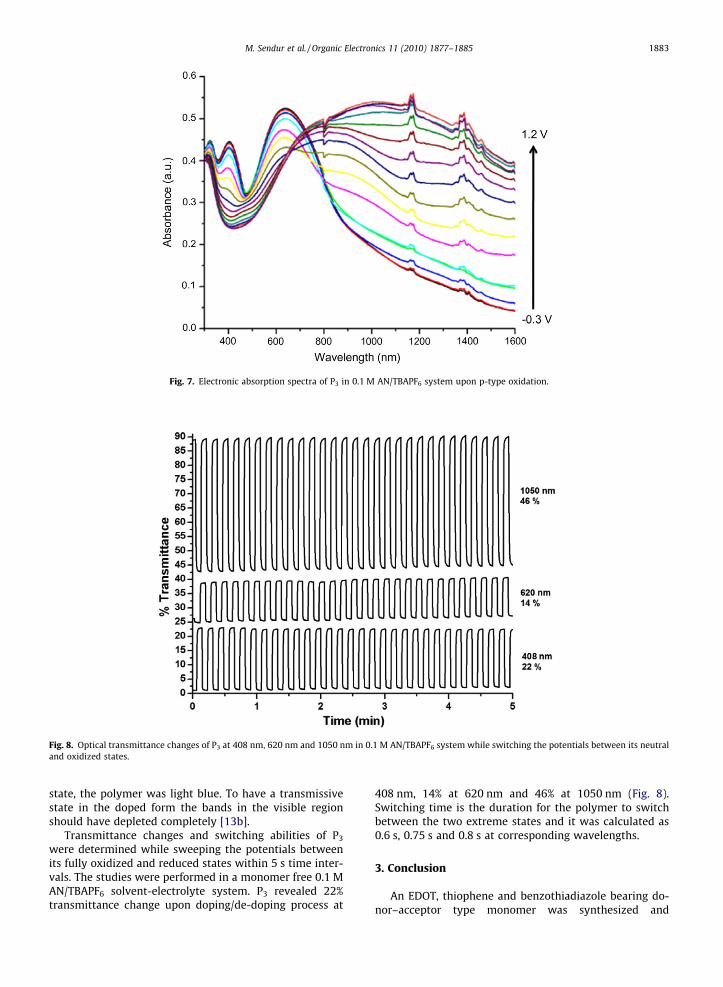
Fig. 7. Electronic absorption spectra of P3 in 0.1 M AN/TBAPF6 system upon p-type oxidation.
Fig. 8. Optical transmittance changes of P3 at 408 nm, 620 nm and 1050 nm in 0.1 M AN/TBAPF6 system while switching the potentials between its neutraland oxidized states.
M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885 1883
state, the polymer was light blue. To have a transmissivestate in the doped form the bands in the visible regionshould have depleted completely [13b].
Transmittance changes and switching abilities of P3
were determined while sweeping the potentials betweenits fully oxidized and reduced states within 5 s time inter-vals. The studies were performed in a monomer free 0.1 MAN/TBAPF6 solvent-electrolyte system. P3 revealed 22%transmittance change upon doping/de-doping process at
408 nm, 14% at 620 nm and 46% at 1050 nm (Fig. 8).Switching time is the duration for the polymer to switchbetween the two extreme states and it was calculated as0.6 s, 0.75 s and 0.8 s at corresponding wavelengths.
3. Conclusion
An EDOT, thiophene and benzothiadiazole bearing do-nor–acceptor type monomer was synthesized and

1884 M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885
polymerized electrochemically as an alternative to copoly-mer studies. The polymer was characterized by electro-chemically and optically. The resulting polymer is both p-and n-type dopable and showed the characteristics of acopolymer where two monomers are required. The resultsrevealed that P3 can be synthesized and its polymerizationcan be controlled to avoid the formation of a randomcopolymer.
4. Experimental
4.1. General
All chemicals were purchased from Aldrich except THF(tetrahydrofuran) which was purchased from Acros. Tribu-tyl(thiophene-2-yl)stannane [22] and tributyl(2,3-dihy-drothieno[3,4-b][1,4]dioxin-5-yl)stannane [23] weresynthesized according to a previously described method.All reactions were carried out under argon atmosphere un-less otherwise mentioned. All electrochemical studies wereperformed under ambient conditions using a Voltalab 50potentiostat. Electropolymerizations were performed in athree-electrode cell consisting of an indium tin oxide dopedglass slide (ITO) as the working electrode, platinum wire asthe counter electrode, and a silver wire pseudo referenceelectrode. After each measurement the reference electrodewas calibrated with ferrocene. The potential of quasi refer-ence electrode was determined as 50 mV vs. the normalhydrogen electrode (NHE).
1H and 13C NMR spectra were recorded in CDCl3 on aBruker Spectrospin Avance DPX-400 Spectrometer. Chemi-cal shifts are given in ppm downfield from tetramethylsil-ane. A Varian Cary 5000 UV–Vis spectrophotometer wasused to perform the spectroelectrochemical studies of thepolymer at a scan rate of 2000 nm/min. Column chroma-tography of all products was performed using Merck SilicaGel 60 (particle size: 0.040–0.063 mm, 230–400 meshASTM). Reactions were monitored by thin layer chroma-tography using silica gel coated aluminum sheets. Solventsused for spectroscopy experiments were spectrophotomet-ric grade.
4.2. Synthesis of 4,7-dibromobenzo[c] [1,2,5] thiadiazole
A mixture of hydrobromic acid (16 ml) and bromine(2 ml) was added slowly to a mixture containing hydrobro-mic acid (36 ml) and benzothiadiazole (2.0 g, 0.015 mol)via continuous stirring at 150 �C. The reaction was left forovernight reflux. Then, the mixture was kept in ice bath;orange crystals were produced in 3–4 h. The crystals werefiltered, and dissolved in dichloromethane. The organicphase was extracted by two portions of sodium bisulfidesolution, and then with saturated sodium chloride solu-tion. After the washing process, mixture was dried overMgSO4 and the solvent DCM was evaporated by rotaryevaporator. The yellowish solid was collected after dryingin oven for overnight. 1H NMR (400 MHz, CDCl3): d(ppm)7.65 (s, 2H). 13C NMR (100 MHz, CDCl3): d(ppm) 152.75,132.13, 113.70.
4.3. Synthesis of 4-bromo-7-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)benzo[c][1,2,5]thiadiazole
4,7-Dibromobenzo[c] [1,2,5] thiadiazole (1.5 g, 5.1 mmol)and tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)stann-ane (3.3 g, 7.7 mmol) were dissolved in dry tetrahydrofuran(THF) (60 ml) and set for refluxing under argon atmosphere.The catalyst, dichlorobis(triphenylphosphine)-palladium(II)(60 mg, 0.085� 10�3 mmol) was added and the mixturewas stirred at 100 �C under argon atmosphere for 16 h whilemonitoring the reaction with TLC. At the end of 16 h, the mix-ture was cooled and THF was removed from the mixture byrotary evaporator. Then the residue was subjected to columnchromatography (Chloroform: Hexane, 3:1) to afford a purplesolid, 4-bromo-7-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)benzo[c][1,2,5]thiadiazole. 1H NMR (400 MHz, CDCl3):d(ppm) 7.77 (d, 1H), 8.16 (d, 1H), 6.54 (s, 1H), 4.24 (m, 2H),4.34 (m, 2H). 13C NMR (100 MHz, CDCl3): d(ppm) 61.95,62.73, 100.63, 105.36, 115.65, 123.47, 124.02, 127.67,127.84, 130.21, 139.34, 151.15.
4.4. Synthesis of 4-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-7-(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (M3)
4-Bromo-7-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)benzo[c][1,2,5]thiadiazole (1 g, 2.8 mmol), and tribu-tyl(thiophen-2-yl)stannane (2.1 g, 5.6 mmol) weredissolved in THF (60 ml) and dichlorobis(triphenyl-phosphine)-palladium(II) (50 mg, 0.071 � 10�3 mmol) wasadded at room temperature. The mixture was refluxed for12 h under argon atmosphere. Solvent was evaporated un-der vacuum and the crude product was purified by columnchromatography (Cholorform: Hexane, 3:1) to obtain a darkred solid, 4-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-7-(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (M3) . 1H NMR(400 MHz, CDCl3): d(ppm) 6.5 (s, 1H), 7.8 (d, 1H), 6.9 (s,2H), 8.3 (d, 1H), 8.0 (m, 1H), 7.1 (m, 1H), 7.4 (m, 1H), 4.3(m, 2H), 4.2 (m, 2H); 13C NMR (100 MHz, CDCl3): d(ppm)151.56, 151.38, 140.70, 140.66, 140.37, 139.21, 138.32,126.64, 125.90, 125.11, 124.92, 124.85, 123.51, 101.13,63.75, 63.04.
4.5. Electrochemical polymerization of M3
For electrochemical polymerization, anodic electrode-position of the polymer was performed from a dichloro-methane (DCM) and acetonitrile (AN) mixture (5/95, v/v)with 0.1 M TBAPF6 (tetrabutylammonium hexafluorophos-phate) solvent-electrolyte couple at a scan rate of 100 mV s�1.ITO-coated glass slides, Pt wire and Ag wire were used asworking, counter and pseudo reference electrodes, respec-tively. The potentiodynamic scans were carried out between�0.3 and 1.5 V. After polymerization, the film was washedwith ACN to remove the unreacted monomers and the sup-porting electrolyte.
4.6. Synthesis of copolymers
A three-electrode cell containing ITO-coated glass slideas the working electrode was used to deposite copolymerfilms potentiodynamically. 4,7-Di(thiophen-2-yl)benzo[c]-

M. Sendur et al. / Organic Electronics 11 (2010) 1877–1885 1885
[1,2,5]thiadiazole (M1) and 4,7-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)benzo[c][1,2,5]thiadiazole (M2) weresynthesized according to the previously reported method[14d,18,20b]. The oxidative electrochemical copolymeriza-tions were achieved by repeated potential cycling indifferent solutions. M1 and M2 were used as the co-mono-mers in 0.1 M tetrabutylammonium hexafluorophosphate(TBAPF6)/acetonitrile (AN)/dichloromethane (DCM) (95:5,v: v) solvent-electrolyte couple at a scan rate of100 mV s�1. All polymerizations were done under the sameconditions for comparison purposes. For 1:1 (M1:M2) ratio10�6 moles of each co-monomer were used and the otherratios; 1:2, 2:1, 1:4 and 1:10 (M1:M2) were determinedaccordingly to mimic M3. Potentiodynamic runs were be-tween �0.3 and 1.5 V for all copolymer depositions witha scan rate of 100 mV s�1. After electrolysis, the films werewashed with ACN to remove the supporting electrolyteand the unreacted monomers.
Acknowledgement
The authors thank TUBA for financial support.
References
[1] (a) F. Huang, K.S. Chen, H.L. Yip, S.K. Hau, O. Acton, Y. Zhang, J. Luo,A.K.Y. Jen, J. Am. Chem. Soc. 131 (2009) 13886;(b) J.C. Bijleveld, A.P. Zoombelt, S.G.J. Mathijssen, M.M. Wienk, M.Turbiez, D.M. de Leeuw, R.A.J. Janssen, J. Am. Chem. Soc. 131 (2009)16616;(c) K.M. Coakley, M.D. McGehee, Chem. Mater. 16 (2004) 4533;(d) H. Hoppe, N.S. Sariciftci, J. Mater. Res. 19 (2004) 1924;(e) C.J. Brabec, N.S. Sariciftci, J.C. Hummelen, Adv. Funct. Mater. 11(2001) 15;(f) S. Gunes, D. Baran, G. Gunbas, F. Ozyurt, A. Fuchsbauer, N.S.Sariciftci, L. Toppare, Sol. Energy Mater. Sol. Cells. 92 (2008) 1162;(g) A.P. Zoombelt, M. Fonrodona, M.M. Wienk, A.B. Sieval, J.C.Hummelen, R.A.J. Janssen, Org. Lett. 11 (2009) 903;(h) K. Colladet, S. Fourier, T.J. Cleij, L. Lutsen, J. Gelan, D.Vanderzande, L.H. Nguyen, H. Neugebauer, S. Sariciftci, A. Aguirre,G. Janssen, E. Goovaerts, Macromology 40 (2007) 65;(i) G. Dennler, N.S. Sariciftci, Proc. IEEE 93 (2005) 1429;(j) D. Baran, A. Balan, S. Celebi, B.M. Esteban, H. Neugebauer, N.S.Sariciftci, L. Toppare, Chem. Mater. 22 (2010) 2978.
[2] (a) A. Kraft, A.C. Grimsdale, A.B. Holmes, Angew. Chem. Intl. Ed. 37(1998) 402;(b) N.R. Evans, L.S. Devi, C.S.K. Mak, S.E. Watkins, S.I. Pascu, A.Kohler, R.H. Friend, C.K. Williams, A.B. Holmes, J. Am. Chem Soc. 128(2006) 6647;(c) Q. Huang, G.A. Evmenenko, P. Dutta, P. Lee, N.R. Armstrong, T.J.Marks, J. Am. Chem. Soc. 127 (2005) 10227.
[3] (a) H.H. Fong, V.A. Pozdin, A. Amassian, G.G. Malliaras, D.M.Smilgies, M. He, S. Gasper, F. Zhang, M. Sorensen, J. Am. Chem. Soc.130 (2008) 13202;(b) M.H. Hoang, M.J. Cho, D.C. Kim, K.H. Kim, J.W. Shin, M.Y. Cho, J.Joo, D.H. Choi, Org. Elec. 10 (2009) 607;(c) P.M. Beaujuge, W. Pisula, H.N. Tsao, S. Ellinger, K. Mullen, J.R.Reynolds, J. Am. Chem. Soc. 131 (2009) 7514;(d) S. Pang, H.N. Tsao, X. Feng, K. Mullen, Adv. Mater. 21 (2009)3488;(e) S. Cho, K. Lee, A.J. Heeger, Adv. Mater. 21 (2009) 1941;
(f) N. Marjanovic, T.B. Singh, G. Dennler, S. Günes, H. Neugebauer,N.S. Sariciftci, R. Schwödiauer, S. Bauer, Org. Elec. 7 (2006) 188;(g) I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. MacDonald,M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M.L.Chabinyc, R.J. Kline, M.D. McGehee, M.F. Toney, Nat. Mater. 5 (2006)328.
[4] D.T. McQuade, A.E. Pullen, T.M. Swager, Chem. Rev. 100 (2000) 2537.[5] (a) A. Argun, A. Cirpan, J.R. Reynolds, Adv. Mater. 15 (2003) 1338;
(b) P.M. Beaujuge, J.R. Reynolds, Chem. Rev. 110 (2010) 268.[6] G. Sonmez, H. Meng, F. Wudl, Chem. Mater. 16 (2004) 574.[7] (a) S.A. Sapp, G.A. Sotzing, J.R. Reynolds, Chem. Mater. 10 (1998)
2101;(b) A. Kumar, D.M. Welsh, M.C. Morvant, F. Piroux, K.A. Abboud, J.R.Reynolds, Chem. Mater. 10 (1998) 896;(c) Y. Coskun, A. Cirpan, L. Toppare, J. Mater. Sci. 42 (2007) 368;(d) L. Groenendaal, G. Zotti, P.H. Aubert, S.M. Waybright, J.R.Reynolds, Adv. Mater. 15 (2003) 855;(e) J.M. Nadeau, T.M. Swager, Tetrahedron 60 (2004) 7141;(f) P. Schottland, K. Zong, C.L. Gaupp, B.C. Thompson, C.A. Thomas, I.Giurgiu, R. Hickman, K.A. Abboud, J.R. Reynolds, Macromology 33(2000) 7051;(g) C.L. Gaupp, K. Zong, P. Schottland, B.C. Thompson, C.A. Thomas,J.R. Reynolds, Macromology 33 (2000) 1132.
[8] (a) A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt, L. Toppare,Chem. Commun. 44 (2009) 6768;(b) F. Ozyurt, E.G. Gunbas, A. Durmus, L. Toppare, Org. Elec. 9 (2008)296.
[9] E. Kose Unver, S. Tarkuc, Y. Arslan Udum, C. Tanyeli, L. Toppare, J.Polym. Sci. Polym. Chem. 48 (2010) 1714.
[10] F.C. Krebs, Nature Mater. 7 (2008) 766.[11] G. Sonmez, H.B. Sonmez, C.K.F. Shen, F. Wudl, Adv. Mater. 16 (2004)
1905.[12] R.M. Walczak, J.R. Reynolds, Adv. Mater. 18 (2006) 1121.[13] (a) M. Amb, P.M. Beaujuge, J.R. Reynolds, Adv. Mater. 22 (2010) 724;
(b) A. Balan, G. Gunbas, A. Durmus, L. Toppare, Chem. Mater. 20(2008) 7510.
[14] (a) A. Durmus, G.E. Gunbas, P. Camurlu, L. Toppare, Chem. Commun.31 (2007) 3246;(b) A. Durmus, G.E. Gunbas, L. Toppare, Chem. Mater. 19 (2007)6247;(c) G.E. Gunbas, A. Durmus, L. Toppare, Adv. Mater. 20 (2008) 691;(d) G.E. Gunbas, A. Durmus, L. Toppare, Adv. Funct. Mater. 18 (2008)2026;(e) P.M. Beaujuge, S. Ellinger, J.R. Reynolds, Adv. Mater. 20 (2008)2772.
[15] C.A. Thomas, K. Zong, K.A. Abboud, P.J. Steel, J.R. Reynolds, J. Am.Chem. Soc. 126 (2004) 16440.
[16] (a) S. Celebi, D. Baran, A. Balan, L. Toppare, Electrochimi. Acta 55(2010) 2373;A.T. Taskin, A. Balan, Y. Arslan Udum, L. Toppare, Smart Mater.Struct. 19 (2010) 065005;(c) G.A. Cetin, A. Balan, A. Durmus, G. Gunbas, L. Toppare, Org. Elec.10 (2009) 34.
[17] (a) C.L. Gaupp, J.R. Reynolds, Macromology 36 (2003) 6305;(b) E. Yildiz, P. Camurlu, C. Tanyeli, I. Akhmedov, L. Toppare, J.Electroanal. Chem. 612 (2008) 247.
[18] O. Atwani, C. Baristiran, A. Erden, G. Sonmez, Synth. Met. 158 (2008)83.
[19] C. Kitamura, S. Tanaka, Y. Yamashita, Chem. Mater. 8 (1996) 570.[20] (a) D. Aldakov, M.A. Palacios, P.Jr. Anzenbacher, Chem. Mater. 17
(2005) 5238;(b) P. Blanchard, J.M. Raimundo, J. Roncali, Synth. Met. 119 (2001)527.
[21] G. Sonmez, I. Schwendeman, P. Schottland, K. Zong, J.R. Reynolds,Macromology 36 (2003) 639.
[22] S.S. Zhu, T.M. Swager, J. Am. Chem. Soc. 119 (1997) 12568.[23] J.T. Pinhey, E.G. Roche, J. Chem. Soc. Perkin Trans. 1 (1988) 2415.