catalytic mechanism of dna-(cytosine-c5)-methyltransferases revisited: covalent intermediate...
TRANSCRIPT

Catalytic Mechanism of DNA-(cytosine-C5)-methyltransferases Revisited: Covalent IntermediateFormation is not Essential for Methyl Group Transferby the Murine Dnmt3a Enzyme
Sabine Reither, Fuyang Li, Humaira Gowher and Albert Jeltsch*
Institut fur Biochemie, FB 8Justus-Liebig UniversitatHeinrich-Buff-Ring 58, 35392Giessen, Germany
Co-transfections of reporter plasmids and plasmids encoding the catalyticdomain of the murine Dnmt3a DNA methyltransferase lead to inhibitionof reporter gene expression. As Dnmt3a mutants with C ! A and E ! Aexchanges in the conserved PCQ and ENV motifs in the catalytic centerof the enzyme also cause repression, we checked for their catalytic activityin vitro. Surprisingly, the activity of the cysteine variant and of the corre-sponding full-length Dnmt3a variant is only two to sixfold reduced withrespect to wild-type Dnmt3a. In contrast, enzyme variants carryingE ! A, E ! D or E ! Q exchanges of the ENV glutamate are catalyticallyalmost inactive, demonstrating that this residue has a central function incatalysis. Since the glutamic acid residue contacts the flipped base, itsmain function could be to hold the target base at a position that supportsmethyl group transfer. Whereas wild-type Dnmt3a and the ENV variantsform covalent complexes with 5-fluorocytidine modified DNA, the PCNvariant does not. Therefore, covalent complex formation is not essentialin the reaction mechanism of Dnmt3a. We propose that correct positioningof the flipped base and the cofactor and binding to the transition state ofmethyl group transfer are the most important roles of the Dnmt3a enzymein the catalytic cycle of methyl group transfer.
q 2003 Elsevier Science Ltd. All rights reserved
Keywords: DNA methylation; 5-methylcytosine; enzyme mechanism; site-directed mutagenesis; covalent complex*Corresponding author
Introduction
In mammals, methylation of DNA at the C5-pos-ition of cytosine residues is the only knowncovalent modification of DNA. As an importantepigenetic mark on the DNA, DNA methylationleads to the repression of gene expression. It isinvolved in regulation of gene expression, develop-ment, X chromosome inactivation, parentalimprinting and protection of the genome againstparasitic DNA elements like transposons (for gen-eral reviews on DNA methyltransferases andDNA methylation see Refs 1–8). DNA methyl-ation is functionally connected to different kindsof histone modification (reviews9,10). AberrantDNA methylation is involved in cancer pro-gression and aging (reviews11 – 14). There are three
known active DNA methyltransferases (MTases)in mammals, Dnmt1, Dnmt3a and Dnmt3b, whichall are essential for proper development.15,16
Dnmt3a is a de novo DNA MTase in the sensethat it does not prefer hemimethylated DNAsubstrates17,18 as opposed to Dnmt1, the classicalmaintenance MTase.19 The enzyme comprises 908amino acid residues, whilst a second, shorter iso-form has been detected recently.20 Dnmt3a is activein vivo21 – 23 and in vitro17,18,24 – 26 and methylatesDNA at CG and non-CG sites18,21,24 with a prefer-ence for CG sites in a YNCGY context.26 All aminoacid motifs characteristic for cytosine-C5 MTasesare located in the C-terminal part of Dnmt3a(Figure 1 and Supplementary Material Figure S1),which shows high amino acid sequence similarityto prokaryotic DNA MTases and is an active DNAMTase without the N-terminal part.27 The N-term-inal part of Dnmt3a contains a PWWP domain,which forms an independent DNA binding site,28
and a Cys-rich domain. It is involved in
0022-2836/03/$ - see front matter q 2003 Elsevier Science Ltd. All rights reserved
E-mail address of the corresponding author:[email protected]
Abbreviations used: MTase, DNA methyltransferase.
doi:10.1016/S0022-2836(03)00509-6 J. Mol. Biol. (2003) 329, 675–684

interactions to other proteins and subcellular tar-geting of the enzyme.20,29 –31
The commonly accepted mechanism of cytosine-C5 MTases is shown in Figure 2 (see for exampleRefs 1,4,6,7,8). The catalytic center of theseenzymes comprises two highly conserved aminoacid motifs (reviews7,32), one PCQ motif (motif IV)and one ENV motif (motif VI) (SupplementaryMaterial Figure S1B and C). The reaction mechan-ism resembles a Michael-addition.33 – 35 Initiallythe cysteine–SH group of the PCQ motif performsa nucleophilic attack at position 6 of the cytosinering. The enzyme assists the nucleolytic attack onC6 by a transient protonation of the cytosine ringat the endocyclic nitrogen atom N3 that ismediated by the glutamic acid residue within theENV motif with the help of an arginine located inmotif VIII of most MTases.36 Upon deprotonationof N3, position 5 of the cytosine ring is stronglyactivated and performs a nucleophilic attack onthe methyl group. The covalent complex betweenenzyme and methylated DNA is a stable inter-mediate in the methylation reaction. It is resolvedby deprotonation at position 5, which leads to theelimination of the enzyme–SH and re-establishesaromaticity. To follow this mechanism, the enzymerequires access to both sides of the aromatic ringsystem of the cytosine, because the initial attack ofthe sulfhydryl group on the cytosine and thenucleophilic attack of the cytosine ring system onthe S-adenosyl methionine (AdoMet) methylgroup must occur at different sides of the ring sys-
tem. Therefore, the catalytic mechanism ofC-MTases involves flipping the target base com-pletely out of the DNA helix.36,37
The catalytic mechanism described above is sup-ported by overwhelming experimental evidence.The existence of a covalent reaction intermediatebetween methylated DNA and the active sitecysteine has been demonstrated for many DNAMTases (see for example Refs 33,38–42) and it hasbeen seen in all structures of DNA-(cytosine-C5)-MTase in complex with DNA known so far.32,43
Covalent complex formation is dependent on thecysteine residue in the PCQ motif.40,41 In addition,the importance of the cysteine residue for catalysishas been shown by site-directed mutagenesis insome cases.40,41,44 – 46 However, site-directed muta-genesis has not been employed for functionalstudies of eukaryotic DNA methyltransferases sofar.
During the course of our functional studies withDnmt3a, we prepared different enzyme variantscarrying amino acid exchanges in the active site ofthe enzyme and analyzed their respective catalyticproperties. Surprisingly, we found that the activesite cysteine is not essential for catalysis by thisenzyme.
Results
It has been known for decades that methylationin the promotor regions of genes leads to transcrip-tional silencing (see Ref. 47 and references therein,see for some recent examples Refs 48–55). In anattempt to follow these experimental approaches,we investigated whether co-transfection of a reportergene plasmid together with plasmids encodingactive DNA methyltransferases leads to genesilencing in vivo. We used the catalytic domain ofthe Dnmt3a DNA methyltransferase as model sys-tem (CD-Dnmt3a), because it has similar activityto wild-type Dnmt3a and it can be easilyexpressed.27 The reporter gene was luciferaseunder the control of TK promotor. As shown inTable 1, approximately fourfold repression ofreporter gene expression was observed in HEK293cells as compared with vector controls. As anadditional control, we used CD-Dnmt3a variants,which carry a Cys ! Ala exchange in the PCNmotif, corresponding to the PCQ motif in the
Figure 1. Primary structure of the Dnmt3a DNAMTase. The enzyme comprises an N-terminal part con-taining a PWWP and a Cys-rich domain and a C-terminalpart containing all motifs characteristic for DNA-(cyto-sine-C5)-MTases. The protein fragment used in thiswork comprises the complete C-terminal part and anadditional His6-tag located at the N-terminal end of thefragment.
Figure 2. Conventional catalytic mechanism of DNA-(cytosine-C5)-MTases. For details consult the text. The Glu inthe Figure refers to the ENV-glutamate, the Cys to the PCN-cysteine residue.
676 Catalytic Mechanism of the Dnmt3a DNA MTase

catalytic center of most cytosine-C5 MTases (seeSupplementary Material Figure S1B) (PCN-A vari-ant) or a glutamic acid ! alanine exchange in theconserved ENV motif (see Supplementary MaterialFigure S1C). The first exchange removes thecysteine residue that is a central element in thecommonly accepted catalytic mechanism of cyto-sine-C5 MTases; the second one removes the gluta-mic acid residue that is highly conserved amongcytosine-C5 MTases and contacts the flipped base.Surprisingly, we repeatedly found, that co-transfec-tion of the reporter plasmid with these variantsalso result in approximately twofold inhibition ofthe luciferase expression (Table 1). One explanationfor this result could be that contrary to what weexpected the PCN-A and ENV-A variants ofDnmt3a could have catalytic activity. To investigatethis possibility, we started an in vitro study toexamine the catalytic properties of the PCN-A andENV-A variants.
In vitro catalytic activity of CD-Dnmt3a variants
The CD-Dnmt3a, PCN-A and ENV-A variantswere cloned into the pET28 Escherichia coliexpression system for in vitro studies of the methyl-ation activity. In addition, we prepared two vari-ants of CD-Dnmt3a, which carry Glu ! Gln andGlu ! Asp exchanges in the ENV motif (ENV-Q,ENV-D)†. Restriction enzyme cleavage and DNAsequencing confirmed cloning and successfulmutagenesis as well as the absence of additionalmutations. The proteins were overexpressed inRosetta E. coli cells and purified to .95% purity asdetermined from Coomassie-stained SDS-gels(Supplementary Material Figure S2). All proteinscould be produced at concentrations in the highmM range, indicating that they are properly folded.Interestingly, the total yields were about three to
fourfold higher with the ENV variants, whichmight be correlated with the finding that thesevariants are catalytically almost inactive (seebelow) and expression of active cytosine-C5MTases is toxic in the Rosetta host cells, becausethis cell line is mcrBC positive. This conclusion issupported by the observation that the expressionstrains are genetically unstable and the cells diesome hours after induction of expression of anactive Dnmt3a variant.
The catalytic activities of the variants were deter-mined using a biotinylated 558 bp PCR fragmentamplified from pAT153 and radio-labeledS-[methyl-3H]AdoMet. Methylation kinetics werecarried out under single turnover conditions,because multiple turnover reactions of enzymesinteracting with DNA often are limited by productrelease, which would obscure changes in the rateof the chemical step. All kinetics were carried outat least in triplicate with different protein prep-arations; the results are summarized in Table 2,representative time-courses are shown in Figure 3.In each individual experiment, we observed thatthe PCN-A variant shows catalytic activity that isreduced only five to sixfold when compared withwild-type CD-Dnmt3a. In contrast, all variants car-rying an exchange in the ENV motif were inactivein the tritium transfer assay, even at higher enzymeconcentrations of up to 10 mM. Since 5% residualactivity is easily detectable in the methylationassay, and the ENV-variants were used in a tenfoldhigher concentration than wild-type, their catalyticactivities are reduced 200-fold at least. Since thefinding that the PCN-A variant shows a catalyticactivity was unpredicted, we repeated DNAsequencing of the PCN-A variant using the sameDNA preparation that was used for proteinexpression to confirm the presence of the mutation(Supplementary Material Figure S3).
The methylation activities of the enzyme vari-ants were also investigated by the restriction pro-tection assay. To this end, the PCR fragment wasradioactively labeled using [a-32P]dATP in thePCR reaction. The fragment contains a single Cla Irestriction site (ATCGAT) almost in a central posi-tion. Methylation of the CG sequence within this
Table 1. Inhibition of luciferase expression by co-trans-fection of the luciferase reporter plasmid withpcDNA3.1 plasmids encoding CD-Dnmt3a and CD-Dnmt3a variants
Inhibition of gene expression (fold)
CD-Dnmt3a 3.9PCN-A 1.6ENV-A 2.0
Inhibition is given in fold in comparison to experimentswhere identical amounts of pcDNA3.1 vector were co-trans-fected with the reporter plasmid. All values given are averagesof .4 independent experiments, data were reproducible within^25%.
Figure 3. Catalytic activities of wild-type CD-Dnmt3aand the PCN-A and ENV-A variants as determined bythe tritium transfer methylation assay. Experimentswere carried out as described in Experimental Pro-cedures using 800 nM enzyme and 10 nM DNA. Theresults of four to five independent experiments witheach enzyme are summarized in Table 1.
† The active site cysteine residue corresponds to aminoacid residue 706 in SPDB entry o88508, the glutamic acidresidue in the ENV motif corresponds to residue number752. Since the mutants were investigated in the context ofthe catalytic domain of DNmt3a and in the context of thefull-length enzyme, we use the abbreviations PCN-A andENV-A, D or Q throughout this work.
Catalytic Mechanism of the Dnmt3a DNA MTase 677

site inhibits Cla I cleavage. As shown in Figure 4,incubation of the PCR fragment with wild-typeCD-Dnmt3a or PCN-A variants leads to an almostcomplete methylation of the Cla I site. This resultconfirms our conclusion that the PCN-A variantdisplays catalytic activity. In contrast, the ENVvariants were almost inactive (Figure 4). It shouldbe noticed that the restriction protection assay ismuch more sensitive than the tritium transferassay, because a higher enzyme/substrate ratio isused and the AdoMet concentration in the assay ishigher. Therefore, the small residual activity of theENV variants in the restriction protection assay isnot in disagreement with the absence of detectableactivity observed with the tritium transfer assay.
Several lines of evidence confirmed that theactivity observed with the PCN-A variants is notdue to a contamination. (i) Activity was reproduci-bly obtained in several independent enzyme prep-arations. (ii) Rosetta E. coli cells only contain thedam MTase, which is a DNA-(adenine-N6)-MTasespecific for GATC sites. However, the substrateused in our studies does not contain GATC sitessuch that a contamination with dam MTase cannotbe the reason for the catalytic activity detectedwith the PCN-A preparations†. (iii) We observedno significant methylation activity with controlpurifications prepared from Rosetta andRosetta(pET28aþ ) cells either in the tritium trans-fer or in the restriction protection assay (data notshow). (iv) The ENV variants prepared followingthe same expression and purification protocol asthe PCN-A variant from the same E. coli strainshow a much lower level of catalytic activity thanthe PCN-A variant.
These data confirm that the inhibition of geneexpression observed with wild-type CD-Dnmt3a isdue to the DNA methylation activity of theenzyme. However, the residual repression activityobserved with the PCN-A and ENV-A variantsmost likely is not due to DNA methylation, sincethe ENV-A variant, which is almost inactive,caused a slightly higher level of repression thanthe PCN-A variant. The unexpected finding that aDnmt3a variant in which the active site cysteineresidue is removed still shows catalytic activityprompted us for a more detailed characterizationof the biochemical properties of these variants.
Preparation of a DCys CD-Dnmt3a variant
RNA-(cytosine-C5)-MTases contain a conservedPCQ motif that strongly resembles that of DNAMTases.56 In spite of this, these enzymes do notuse the PCQ cysteine residue for covalent complexformation, but a second cysteine located close tothe ENV motif, which is conserved only amongRNA MTases.57 We, therefore, investigated ifanother cysteine residue could act as catalyticnucleophile in Dnmt3a. The protein contains fouradditional cysteine residues. According to struc-tural model building based on the M.Hha Istructure in complex with DNA (PDB code:3MHT),36 none of them approaches the flippedtarget base closer than by 10 A (data not shown).Nevertheless, by site-directed mutagenesis we pre-pared a CD-Dnmt3a variant in which all these fouradditional cysteine residues are exchanged byserine (DCys), purified the protein and determinedits catalytic activity in the tritium transfer assay.The DCys variant showed 13(^3)% activity withrespect to wild-type CD-Dnmt3a, demonstratingthat none of the other cysteine residues has anessential function in catalysis.
DNA binding and covalent complex formation
DNA binding of all variants was investigated bythe nitrocellulose filter-binding assay (Figure 5).The DNA binding constants of all enzymes are
Table 2. Compilation of the catalytic activities and DNA binding properties of CD-Dnmt3a wild-type and variants
CD-Dnmt3a PCN-A ENV-A ENV-D ENV-Q
Catalytic activity (tritium transfer) (molCH3 h21 molenzyme21 ) 0.507 (1.0) 0.093 (0.18) 0 (,0.005) 0 (,0.005) 0 (,0.005)
Relative catalytic activity (restriction protection) þþþ þþ (þ ) (þ ) (þ)DNA binding ðKassÞ (M21) 1.7 £ 107 7.6 £ 106 2.2 £ 107 2.5 £ 107 1.7 £ 107
Covalent complex formation þ 2 þ n.a. n.a.
All experiments were repeated at least in triplicate. Rate constants are valid by ^15%, DNA binding constants by ^30%. n.a., notanalyzed.
Figure 4. Catalytic activities of wild-type CD-Dnmt3aand the variants as determined by the restriction protec-tion assay. The lane labelled with n shows the 558mernot cleaved by Cla I. The lanes labelled with c showunmethylated DNA cleaved by Cla I. The lanes labelledwith CD-Dnmt3a, PCN-A, ENV-A, ENV-Q and ENV-Dshow DNA incubated with the corresponding Dnmt3avariant and subsequently cleaved with Cla I. Protectionof Cla I cleavage indicates methylation of the Cla I siteby the MTase variant.
† We observed similar relative catalytic activities of thePCN-A mutants also in initial experiments with asubstrate containing GATC sites (data not shown).
678 Catalytic Mechanism of the Dnmt3a DNA MTase
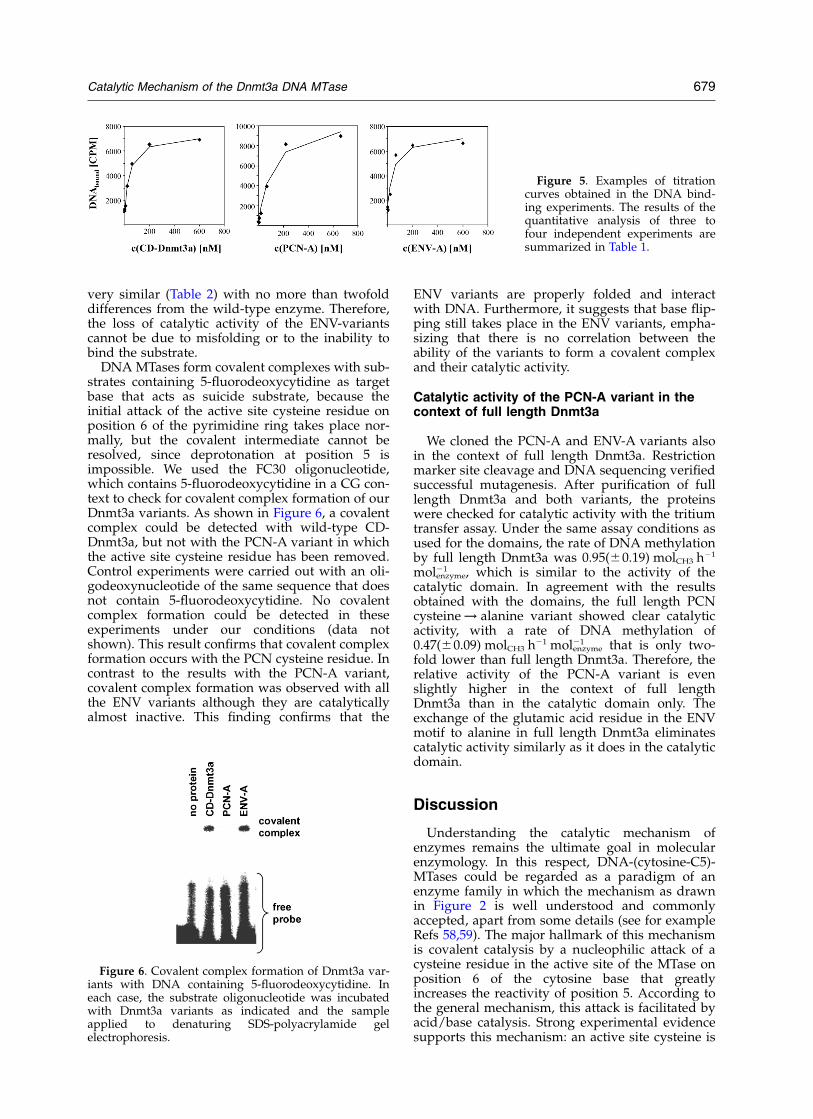
very similar (Table 2) with no more than twofolddifferences from the wild-type enzyme. Therefore,the loss of catalytic activity of the ENV-variantscannot be due to misfolding or to the inability tobind the substrate.
DNA MTases form covalent complexes with sub-strates containing 5-fluorodeoxycytidine as targetbase that acts as suicide substrate, because theinitial attack of the active site cysteine residue onposition 6 of the pyrimidine ring takes place nor-mally, but the covalent intermediate cannot beresolved, since deprotonation at position 5 isimpossible. We used the FC30 oligonucleotide,which contains 5-fluorodeoxycytidine in a CG con-text to check for covalent complex formation of ourDnmt3a variants. As shown in Figure 6, a covalentcomplex could be detected with wild-type CD-Dnmt3a, but not with the PCN-A variant in whichthe active site cysteine residue has been removed.Control experiments were carried out with an oli-godeoxynucleotide of the same sequence that doesnot contain 5-fluorodeoxycytidine. No covalentcomplex formation could be detected in theseexperiments under our conditions (data notshown). This result confirms that covalent complexformation occurs with the PCN cysteine residue. Incontrast to the results with the PCN-A variant,covalent complex formation was observed with allthe ENV variants although they are catalyticallyalmost inactive. This finding confirms that the
ENV variants are properly folded and interactwith DNA. Furthermore, it suggests that base flip-ping still takes place in the ENV variants, empha-sizing that there is no correlation between theability of the variants to form a covalent complexand their catalytic activity.
Catalytic activity of the PCN-A variant in thecontext of full length Dnmt3a
We cloned the PCN-A and ENV-A variants alsoin the context of full length Dnmt3a. Restrictionmarker site cleavage and DNA sequencing verifiedsuccessful mutagenesis. After purification of fulllength Dnmt3a and both variants, the proteinswere checked for catalytic activity with the tritiumtransfer assay. Under the same assay conditions asused for the domains, the rate of DNA methylationby full length Dnmt3a was 0.95(^0.19) molCH3 h21
molenzyme21 , which is similar to the activity of the
catalytic domain. In agreement with the resultsobtained with the domains, the full length PCNcysteine ! alanine variant showed clear catalyticactivity, with a rate of DNA methylation of0.47(^0.09) molCH3 h21 molenzyme
21 that is only two-fold lower than full length Dnmt3a. Therefore, therelative activity of the PCN-A variant is evenslightly higher in the context of full lengthDnmt3a than in the catalytic domain only. Theexchange of the glutamic acid residue in the ENVmotif to alanine in full length Dnmt3a eliminatescatalytic activity similarly as it does in the catalyticdomain.
Discussion
Understanding the catalytic mechanism ofenzymes remains the ultimate goal in molecularenzymology. In this respect, DNA-(cytosine-C5)-MTases could be regarded as a paradigm of anenzyme family in which the mechanism as drawnin Figure 2 is well understood and commonlyaccepted, apart from some details (see for exampleRefs 58,59). The major hallmark of this mechanismis covalent catalysis by a nucleophilic attack of acysteine residue in the active site of the MTase onposition 6 of the cytosine base that greatlyincreases the reactivity of position 5. According tothe general mechanism, this attack is facilitated byacid/base catalysis. Strong experimental evidencesupports this mechanism: an active site cysteine is
Figure 5. Examples of titrationcurves obtained in the DNA bind-ing experiments. The results of thequantitative analysis of three tofour independent experiments aresummarized in Table 1.
Figure 6. Covalent complex formation of Dnmt3a var-iants with DNA containing 5-fluorodeoxycytidine. Ineach case, the substrate oligonucleotide was incubatedwith Dnmt3a variants as indicated and the sampleapplied to denaturing SDS-polyacrylamide gelelectrophoresis.
Catalytic Mechanism of the Dnmt3a DNA MTase 679

conserved among all cytosine-C5 MTases(reviews7,32) and a covalent complex betweenMTases and DNA has been detected biochemicallyand by structural analysis (see for example Refs32,33,38–43). It has been shown biochemically andby structural analysis that the cross-link betweenthe enzyme and the DNA occurs at the active sitecysteine.32,40,41,43 Finally, MTase variants in whichthe active site cysteine is removed show a stronglyreduced catalytic activity.40,41,44 – 46 Similar resultshave been obtained for bacteriophage T4 thymi-dylate synthase60 and T4 deoxycytidylatehydroxymethylase,61 which both also follow acovalent catalysis mechanism.
We show here that the catalytic activities ofDnmt3a variants that do not have an active sitecysteine residue are reduced only by a factor of 2–6with respect to the wild-type enzyme, a verysmall reduction in activity given that the residuepresumed to have the most important catalyticfunction has been exchanged. This is in apparentdisagreement with results obtained with prokaryo-tic MTases and also with Dnmt3a by anothergroup.22,26 Hsieh22 could not detect inhibition ofgene expression with a PCN-A variant of Dnmt3a.However, the total inhibition observed by Hsieh etal. was weak and a different target sequence wasused than in our study such that these findingsmay be consistent with each other. The lack ofactivity of the PCN-A variant in vitro reported byLin26 is in clear disagreement with our findings. Itmay be due to the usage of a GST-tag in the Lin26
paper or problems with the preparation of thevariant in that work which may have led toinactivation of the enzyme. With respect to theprokaryotic enzymes, one should note that thecatalytic activity of variants in which the activesite cysteine is altered has been analyzed for onlya few prokaryotic MTases in quantitative termsand it is possible that after studying moreexamples more enzymes of the Dnmt3a type willbe described. Moreover, it is possible that MTaseswith a PCN motif in the active site, like Dnmt3a,which is very rare among cytosine-C5 MTases thatmostly have PCQ (see Supplementary MaterialFigure S1B), behave differently from those havinga PCQ active site. So far, no other PCN C-MTasehas been investigated by site-directed mutagenesis.We infer from our data that covalent complex for-mation between Dnmt3a and the DNA is notessential for methylation of cytosine residues atposition 5. This conclusion does not contradict thegeneral observation that a covalent complex isformed between the enzyme and the DNA (evenDnmt3a shows a covalent complex). However, theformation of a covalent complex is not the onlypathway for catalysis and the covalent complex isnot an obligate intermediate of methyl group trans-fer. Therefore, the general catalytic mechanism ofDNA-(cytosine-C5)-MTases has to be extended.
In addition, we have shown that three variantsin which the glutamic acid residue in the ENVmotif is altered have almost completely lost cata-
lytic activity, demonstrating that this residue iscritically important for catalysis. The finding thatthe ENV variants form a covalent complex withDNA containing 5-fluorodeoxycytidine indicatesthat base flipping still takes place. It also showsthat acid/base catalysis by the glutamic acid resi-due as drawn in Figure 2 is not required forcovalent complex formation. However, we cannotrule out that the transient protonation of the cyto-sine base at position N3 might facilitate this step,because we did not investigate the time-course ofcovalent complex formation. Since in the crystalstructures of cytosine-C5 MTases in complex withDNA,32,43 the glutamic acid residue forms a hydro-gen bond to the flipped base, it most likely isresponsible for correct positioning of the cytosinewith respect to the AdoMet into a conformationthat supports methyl group transfer. Our findingthat even variants carrying very conservativeexchanges at this position (glutamic acid !aspartic acid, glutamic acid ! glutamine) do notshow catalytic activity underscores the delicatenature of this function. Taken together, our resultssuggest that arranging the flipped base and theAdoMet in a conformation that allows methylgroup transfer is the most important role of theenzyme.
Our data show that both central elements in thecommonly accepted mechanism of DNA-(cyto-sine-C5)-MTases as shown in Figure 2 (acid basecatalysis by the glutamic acid in the ENV motifand covalent complex formation by the cysteineresidue in the PCN/PCQ motif) are not essentialfor Dnmt3a. Therefore, the catalytic mechanism ofcytosine-C5 MTases must be reconsidered andextended. In general, enzyme catalysis is based ontwo universal principles, which are likely to beoperative also in the case of DNA MTases: sub-strate and cofactor binding and positioning, whichreduces the entropic costs of transition state for-mation, and selective binding of the transitionstate, which reduces the enthalpy of transitionstate formation.62,63 The efficiency of positioningand transition state binding in catalysis is illus-trated by the 105-fold rate enhancement observedwith catalytic antibodies.64 In addition, covalentcomplex formation may occur and speed up cata-lysis to a different degree in different MTases.However, our data show that methyl group trans-fer can also take place without formation of acovalent complex. There are two possible path-ways for cytosine-C5 methylation in the absenceof covalent catalysis: either the cytosine ringdirectly attacks the AdoMet, which would generatea positive charge delocalized at C6 and N1. Thecationic transition state could be stabilized by thedeprotonated Glu from the ENV motif. Alterna-tively, an OH2 from water might replace thecysteine and serve as a nucleophile. Interestingly,correct positioning of the flipped base and theAdoMet and preferential binding of the cationictransition state of methyl group transfer alsoare the most important factors for catalysis by
680 Catalytic Mechanism of the Dnmt3a DNA MTase

DNA-(adenine-N6)-MTases.65,66 Thus, there aremechanistic parallels between these two classes ofenzymes in addition to the known structuralsimilarities.4,7
Our results resemble classical results from muta-genesis experiments with proteases, which perhapsare the best-characterized family of enzymes thatmake use of covalent catalysis. For example, a vari-ant of chymotrypsin in which the aspartic acid resi-due of the catalytic triad is changed to asparagineshowed clear residual activity67 and even afterremoval of all three amino acid residues from thecatalytic triad of subtilisin the enzyme still showedresidual catalytic activity.68 Similar results havebeen obtained with many more enzymes formdifferent families (review69), demonstrating thatenzymes generally use more than one way toenhance the rate of chemical reactions. However,there is one clear difference between our resultsand the examples cited above: whereas in the for-mer cases catalytic activity always is severelyreduced by the amino acid exchange in the activesite, this is not the case with Dnmt3a, demonstrat-ing that covalent catalysis plays a minor role inthe catalytic mechanism of wild-type Dnmt3a. Inthis context, it is interesting to note, that Dnmt3apurified from E. coli but also from insect cellsshows only poor catalytic activity.17,18,24 This indi-cates that the active site of Dnmt3a is not in anideal conformation and this could be the reasonwhy the cysteine residue is not positioned to per-form the nucleophilic attack on the C6 position.This would explain why the covalent catalysis bythe active site cysteine is not efficient and the cata-lytic mechanism changes to a much slower, alterna-tive pathway that only depends on the orientationof substrate and cofactor and transition state bind-ing. Removal of the active site cysteine could notimpede catalysis by this “backup” mechanism,because the cysteine residue does not have animportant role in it. However, it might be possiblethat a covalent modification of the enzyme or aninteraction with another protein could induce aconformational change of the catalytic site that acti-vates the enzyme and switches the catalytic mech-anism to the classical scheme. This model couldexplain why the catalytic cysteine residue is con-served even in the Dnmt3 family, which would bedifficult to understand if it had no important func-tion in catalysis. The mammalian Dnmt1 enzymemight be a precedent for this kind of activation,because although the full length enzyme is highlyactive, its catalytic domain is not active in an iso-lated form, although it is properly folded, whichimplies that an interaction of the catalytic domainwith the rest of the protein is essential for the cata-lytic domain to adopt a catalytically competentconformation (19 and references therein).
Conclusions
We have shown that a Dnmt3a variant in which
the active site cysteine residue is removed still hascatalytic activity. Therefore, the classical covalentcatalysis mechanism of cytosine-C5 methylationneeds to be extended, because methyl group trans-fer is possible without covalent catalysis. Othercatalytic mechanisms must support methyl grouptransfer by Dnmt3a, and most likely also by otherMTases, which could be preferential binding ofenzyme to the transition state of the reaction andpositioning of the substrate and cofactor. Boththese mechanisms are well established in enzymol-ogy and are central in the catalytic mechanism ofadenine-N6 MTases. The relative contributions ofcovalent catalysis and orientation/transition statebinding may be different for different cytosine-C5MTases, in the case of Dnmt3a the contribution ofcovalent catalysis is obviously low. This could bedue to an imperfect conformation of the active siteof the enzyme.
Experimental Procedures
Mutagenesis, protein expression andprotein purification
The catalytic domain of Dnmt3a (SwissProt data baseentry: o88508) used in this work comprises the 300C-terminal amino acid residues of the enzyme (aminoacid residues 608–908 in o88508) and carries an N-term-inal His6-tag.27 PCR-mutagenesis was carried out asdescribed.70,71 Briefly, the mutation, together with arestriction enzyme marker site used for screening, wasintroduced with a PCR primer in a first PCR reaction.The first PCR product was used to amplify the wholeplasmid in a second PCR. Mutagenesis was confirmedby restriction marker site analysis as well as sequencingof both strands of the DNA. Protein expression was car-ried out in Rosetta(DE3)pLysS [F2 ompT hsdSB (rB
2 mB2)
gal dcm lacY1(DE3) pLysSRARE (CmR)] (Novagene) andpurification were performed as described.18,27 For the co-transfection assay, the catalytic domain of Dnmt3a wascloned into pcDNA3.1-His (Invitrogen) at the Eco RI andNot I sites.
Reporter gene assays
HEK293 cells were obtained from the Institute of Gen-etics (Justus-Liebig Universitat, Giessen). Cells were cul-tured in DMEM (Invitrogen) supplemented with 10%(v/v) fetal bovine serum (FBS, Invitrogen). The cultureplate was coated with polylysine by incubating with thepolylysine solution (10 mg/ml, Sigma) for ten minutesand rinsed with pure water. At 24 hours before transfec-tion, cells were seeded to 48-well plate at a density of4 £ 104 cells/well. The luciferase reporter plasmid encod-ing the luciferase gene under the control of TK promoterwas obtained from N. Landsberger (Universita degliStudi dell’Insubria, Busto Arsizio, Italy).55 Plasmids forco-transfection were diluted with 12.5 ml serum-freeDMEM culture medium per well, mixed with 2 ml Plusreagent (Invitrogen) and incubated at room temperaturefor 15 minutes. Lipofectamine (0.5 ml/well, Invitrogen)was diluted in 12.5 ml of serum-free medium, mixedwith the plasmids by vortexing, and put at room tem-perature for 15 minutes. A volume of 100 ml of full
Catalytic Mechanism of the Dnmt3a DNA MTase 681

culture medium (DMEM supplemented with 10% FBS)was added. The old culture medium of the cells wasdiscarded and the fresh culture medium containingplasmid–lipofectamine complex added to the wells. Thecells were incubated at 37 8C at 5% CO2 for three hours,then 200 ml/well full culture medium was added andthe cells cultured for 36 hours. For the detection of theluciferase activity, 200 ml culture medium supernatantwere removed and discarded. Then 100 ml luciferase sub-strate solution (LucLite plus kit, Packkard) were addedto the cells, the mixture transferred to white polystyrenemicroplate (Packard) and luciferase activity was deter-mined by a TopCount NXT Mircoplate Scintillation andLuminescence Counter (Packard). Transfection efficiencywas monitored by green fluorescent protein (GFP) signalcount under fluorescence microscope after co-transfec-tion with a GFP reporter plasmid.
Tritium transfer methylation assay
For the in vitro methylation experiments, a biotinyl-ated 558mer PCR product derived from pAT153 wasused. The sequence was selected not to contain anyGATC site. It contains 34 CG sites and 188 additionalcytosine residues in a non-CG context. The PCR productwas purified using Concert Rapid PCR Purification kit(Gibco). The concentration of the purified DNA wasdetermined from the A260 nm using an e260 nm of7.36 £ 106 M21 cm21. DNA (10 nM) and enzyme (0.5–1 mM) were incubated in methylation buffer (20 mMHepes (pH 7.5), 1 mM EDTA, 25 mg/ml BSA) containing3.7 mM S-[methyl-3H]AdoMet, specific activity0.555 £ 1015 Bq/mol (Movarek Biochemicals). At definedtimes, aliquots of 2 ml were removed and processed asdescribed.72 Methylation reactions were started byaddition of the enzyme. The yield of the recovery ofDNA was determined using identical amounts of 32P-labelled PCR product, which was prepared by PCRusing [a-32P]dATP (Hartmann Analytic) and purifiedusing Concert Rapid PCR Purification kit (Gibco). Allturnover numbers were corrected for incomplete recov-ery of the DNA.
Restriction protection methylation assay
The 558mer PCR fragment contains one Cla I site(ATCGAT) at position 281. Methylation of the centralcytosine of this site prevents cleavage by Cla I. For themethylation reaction, 1 nM radioactively labelled558mer was incubated with 0.8 mM of the differentenzyme variant for one hour at 37 8C in methylation buf-fer supplemented with 1 mM AdoMet (Sigma). Then, theDNA was precipitated with ethanol, re-dissolved in 1 £Cla I buffer (50 mM potassium acetate, 20 mM Tris–acet-ate (pH 7.9), 10 mM magnesium acetate, 1 mM DTT,100 mg/ml BSA), incubated with five units Cla I (NEB)for two hours at 37 8C and applied to a 6% (w/v) poly-acrylamide gel run in TPE (80 mM Tris–phosphate,20 mM EDTA, pH 8.0). Radioactivity was analyzedusing an Instant Imager (Amersham Bioscience).
DNA binding analysis
DNA binding was analyzed using the Nitrocellulosefilter binding method as described.18,73,74 For the experi-ments, 1 nM 32P-labelled 558mer was used. Protein con-centrations were varied between 0 nM and 700 nM. Thebinding reactions were carried out in methylation buffer
at ambient temperature. Data analysis was performed asdescribed.74
Covalent complex formation
DNA containing 5-fluorodeoxycytidine instead ofcytosine as target base forms stable covalent complexeswith DNA MTases. We used an oligonucleotide (FU30:GAA GCT GGG ACT TC5FC GGG AGG AGA GTG CAA=TTG CAC TCT CCT CCC GGA AGT CCC AGC TTC; 5FC:5-fluorodeoxycytidine) purchased from Thermo Hybaid(Ulm, Germany). 5 nM DNA radioactively labelledusing T4-PNK (NEB) and [g-32P]dATP (Hartmann Ana-lytic) were incubated with 50 nM enzyme in methylationbuffer containing 100 mM AdoMet (Sigma) for one hourat 37 8C in a total volume of 20 ml. Then, 5 ml LAP(160 mM Tris–HCl (pH 6.8), 2% (w/v) SDS, 5% (v/v)b-mercaptoethanol, 40% (v/v) glycerol, 0.1% (w/v) bro-mophenol blue) were added, the samples heated to92 8C for ten minutes and applied to denaturing proteingel (stacking gel: 5% polyacrylamide, main gel 15% poly-acrylamide) containing 10% SDS. After the gel run, theradioactivity was analyzed using an Instant Imager. Con-trol experiments were carried out with an oligodeoxyn-cucleotide of the same sequence that does not contain5FC.
Acknowledgements
The gift of a luciferase reporter plasmid by DrNicolleta Landsberger (Universita degli Studidell’Insubria, Busto Arsizio, Italy) is gratefullyacknowledged. Thanks are due to A. Hermann,M. Roth, V. Pingoud and A. Pingoud for discus-sions. This work was supported by the DFG (JE252/1-2, 1-3) and the BMBF BioFuture program.
References
1. Jeltsch, A. (2002). Beyond Watson and Crick: DNAmethylation and molecular enzymology of DNAmethyltransferases. ChemBioChem, 3, 274–293.
2. Bird, A. (2002). DNA methylation patterns and epi-genetic memory. Genes Dev. 16, 6–21.
3. Jones, P. A. & Takai, D. (2001). The role of DNAmethylation in mammalian epigenetics. Science, 293,1068–1070.
4. Cheng, X. & Roberts, R. J. (2001). AdoMet-dependentmethylation, DNA methyltransferases and base flip-ping. Nucl. Acids Res. 29, 3784–3795.
5. Robertson, K. D. & Wolffe, A. P. (2000). DNA methyl-ation in health and disease. Nature Rev. Genet. 1,11–19.
6. Cheng, X. & Blumenthal, R. M. (1999). S-adenosyl-methionine-dependent Methyltransferases: Structures andFunctions, World Scientific Publishing, Singapore.
7. Cheng, X. (1995). Structure and function of DNAmethyltransferases. Annu. Rev. Biophys. Biomol.Struct. 24, 293–318.
8. Bestor, T. H. & Verdine, G. L. (1994). DNA methyl-transferases. Curr. Opin. Cell Biol. 6, 380–389.
9. Ng, H. H. & Bird, A. (1999). DNA methylation andchromatin modification. Curr. Opin. Genet. Dev. 9,158–163.
682 Catalytic Mechanism of the Dnmt3a DNA MTase

10. Bird, A. (2002). Methylation talk between histonesand DNA. Science, 294, 2113–2115.
11. Warnecke, P. M. & Bestor, T. H. (2000). Cytosinemethylation and human cancer. Curr. Opin. Oncol.12, 68–73.
12. Baylin, S. B. & Herman, J. G. (2000). DNA hyper-methylation in tumorigenesis: epigenetics joins gen-etics. Trends Genet. 16, 168–174.
13. Jones, P. A. & Vogt, P. K. (2000). DNA methylationand cancer. In Current Topics in Microbiology andImmunology (Compans, R. W., Cooper, M., Ito, Y.,Koprowski, H., Melchers, F., Oldstone, M., et al.),Springer Verlag, Heidelberg.
14. Jones, P. A. & Baylin, S. B. (2002). The fundamentalrole of epigenetic events in cancer. Nature Rev. Genet.3, 415–428.
15. Li, E., Bestor, T. H. & Jaenisch, R. (1992). Targetedmutation of the DNA methyltransferase gene resultsin embryonic lethality. Cell, 69, 915–926.
16. Okano, M., Bell, D. W., Haber, D. A. & Li, E. (1999).DNA methyltransferases Dnmt3a and Dnmt3b areessential for de novo methylation and mammaliandevelopment. Cell, 99, 247–257.
17. Okano, M., Xie, S. & Li, E. (1998). Cloning andcharacterization of a family of novel mammalianDNA (cytosine-5) methyltransferases. Nature Genet.19, 219–220.
18. Gowher, H. & Jeltsch, A. (2001). Enzymatic proper-ties of recombinant Dnmt3a DNA methyltransferasefrom mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpGsites. J. Mol. Biol. 309, 1201–1208.
19. Fatemi, M., Hermann, A., Pradhan, S. & Jeltsch, A.(2001). The activity of the murine DNA methyltrans-ferase Dnmt1 is controlled by interaction of the cata-lytic domain with the N-terminal part of theenzyme leading to an allosteric activation of theenzyme after binding to methylated DNA. J. Mol.Biol. 309, 1189–1199.
20. Chen, T., Ueda, Y., Xie, S. & Li, E. (2002). A novelDnmt3a isoform produced from an alternative pro-moter localizes to euchromatin and its expressioncorrelates with active de novo methylation. J. Biol.Chem. 277, 38746–38754.
21. Ramsahoye, B. H., Biniszkiewicz, D., Lyko, F., Clark,V., Bird, A. P. & Jaenisch, R. (2000). Non-CpG methyl-ation is prevalent in embryonic stem cells and maybe mediated by DNA methyltransferase 3a. Proc.Natl Acad. Sci. USA, 97, 5237–5242.
22. Hsieh, C.-L. (1999). In vivo activity of murine de novomethyltransferases Dnmt3a and Dnmt3b. Mol. Cell.Biol. 19, 8211–8218.
23. Lyko, F., Ramsahoye, B. H., Kashevsky, H., Tudor, M.,Mastrangelo, M. A., Orr-Weaver, T. L. & Jaenisch, R.(1999). Mammalian (cytosine-5) methyltransferasescause genomic DNA methylation and lethality inDrosophila. Nature Genet. 23, 363–366.
24. Aoki, A., Suetake, I., Miyagawa, J., Fujio, T., Chijiwa,T., Sasaki, H. & Tajima, S. (2001). Enzymatic proper-ties of de novo-type mouse DNA (cytosine-5) methyl-transferases. Nucl. Acids Res. 29, 3506–3512.
25. Yokochi, T. & Robertson, K. D. (2002). Preferentialmethylation of unmethylated DNA by Mammaliande novo DNA methyltransferase Dnmt3a. J. Biol.Chem. 277, 11735–11745.
26. Lin, I. G., Han, L., Taghva, A., O’Brien, L. E. & Hsieh,C. L. (2002). Murine de novo methyltransferaseDnmt3a demonstrates strand asymmetry and site
preference in the methylation of DNA in vitro. Mol.Cell. Biol. 22, 704–723.
27. Gowher, H. & Jeltsch, A. (2002). Molecular enzymol-ogy of the catalytic domains of the Dnmt3a andDnmt3b DNA methyltransferases. J. Biol. Chem. 277,20409–20414.
28. Qiu, C., Sawada, K., Zhang, X. & Cheng, X. (2002).The PWWP domain of mammalian DNA methyl-transferase Dnmt3b defines a new family of DNA-binding fold. Nature Struct. Biol. 9, 217–224.
29. Fuks, F., Burgers, W. A., Godin, N., Kasai, M. &Kouzarides, T. (2001). Dnmt3a binds deacetylasesand is recruited by a sequence-specific repressor tosilence transcription. EMBO J. 20, 2536–2544.
30. Bachman, K. E., Rountree, M. R. & Baylin, S. B.(2001). Dnmt3a and Dnmt3b are transcriptionalrepressors that exhibit unique localization propertiesto heterochromatin. J. Biol. Chem. 276, 32282–32287.
31. Fuks, F., Hurd, P. J., Wolf, D., Nan, X., Bird, A. P. &Kouzarides, T. (2003). The methyl-CpG-binding pro-tein MeCP2 links DNA methylation to histone meth-ylation. J. Biol. Chem. 278, 4035–4040.
32. Klimasauskas, S., Kumar, S., Roberts, R. J. & Cheng,X. (1994). Hhal methyltransferase flips its targetbase out of the DNA helix. Cell, 76, 357–369.
33. Santi, D. V., Norment, A. & Garrett, C. E. (1984).Covalent bond formation between a DNA-cytosinemethyltransferase and DNA containing 5-azacyto-sine. Proc. Natl Acad. Sci. USA, 81, 6993–6997.
34. Wu, J. C. & Santi, D. V. (1987). Kinetic and catalyticmechanism of HhaI methyltransferase. J. Biol. Chem.262, 4778–4786.
35. Carreras, C. W. & Santi, D. V. (1995). The catalyticmechanism and structure of thymidylate synthase.Annu. Rev. Biochem. 64, 721–762.
36. O’Gara, M., Klimasauskas, S., Roberts, R. J. & Cheng,X. (1996). Enzymatic C5-cytosine methylation ofDNA: mechanistic implications of new crystal struc-tures for HhaI methyltransferase-DNA-AdoHcycomplexes. J. Mol. Biol. 261, 634–645.
37. Roberts, R. J. & Cheng, X. (1998). Base flipping. Annu.Rev. Biochem. 67, 181–198.
38. Osterman, D. G., DePillis, G. D., Wu, J. C., Matsuda,A. & Santi, D. V. (1988). 5-Fluorocytosine in DNA isa mechanism-based inhibitor of HhaI methylase. Bio-chemistry, 27, 5204–5210.
39. Chen, L., MacMillan, A. M., Chang, W., Ezaz-Nikpay,K., Lane, W. S. & Verdine, G. L. (1991). Direct identi-fication of the active-site nucleophile in a DNA(Cytosine-5)-methyltransferase. Biochemistry, 30,11018–11025.
40. Wyszynski, M. W., Gabbara, S., Kubareva, E. A.,Romanova, E. A., Oretskaya, T. S., Gromova, E. S.et al. (1993). The cysteine conserved among DNAcytosine methylases is required for methyl transfer,but not for specific DNA binding. Nucl. Acids Res.21, 295–301.
41. Hanck, T., Schmidt, S. & Fritz, H. J. (1993). Sequence-specific and mechanism-based crosslinking of DcmDNA cytosine-C5 methyltransferase of E. coli K-12to synthetic oligonucleotides containing 5-fluoro-20-deoxycytidine. Nucl. Acids Res. 21, 303–309.
42. Yoder, J. A., Soman, N. S., Verdine, G. L. & Bestor,T. H. (1997). DNA (cytosine-5)-methyltransferases inmouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol. 270, 385–395.
43. Reinisch, K. M., Chen, L., Verdine, G. L. & Lipscomb,W. N. (1995). The crystal structure of Hae III methyl-transferase covalently complexed to DNA: an
Catalytic Mechanism of the Dnmt3a DNA MTase 683

extrahelical cytosine and rearranged base pairing.Cell, 82, 143–153.
44. Gabbara, S. & Bhagwat, A. S. (1995). The mechanismof inhibition of DNA (cytosine-5-)-methyltransfer-ases by 5-azacytosine is likely to involve methyltransfer to the inhibitor. Biochem. J. 307, 87–92.
45. Wilke, K., Rauhut, E., Noyer-Weidner, M., Lauster,R., Pawlek, B., Behrens, B. & Trautner, T. A. (1988).Sequential order of target-recognizing domains inmultispecific DNA-methyltransferases. EMBO J. 7,2601–2609.
46. Mi, S. & Roberts, R. J. (1993). The DNA binding affi-nity of HhaI methylase is increased by a singleamino acid substitution in the catalytic center. Nucl.Acids Res. 21, 2459–2464.
47. Doerfler, W. (1983). DNA methylation and geneactivity. Annu. Rev. Biochem. 52, 93–124.
48. Siegfried, Z., Eden, S., Mendelsohn, M., Feng, X.,Tsuberi, B. Z. & Cedar, H. (1999). DNA methylationrepresses transcription in vivo. Nature Genet. 22,203–206.
49. Campanero, M. R., Armstrong, M. I. & Flemington,E. K. (2000). CpG methylation as a mechanism forthe regulation of E2F activity. Proc. Natl Acad. Sci.USA, 97, 6481–6486.
50. Chen, C., Yang, M. C. & Yang, T. P. (2001). Evidencethat silencing of the HPRT promoter by DNAmethylation is mediated by critical CpG sites. J. Biol.Chem. 276, 320–328.
51. Jackson-Grusby, L., Beard, C., Possemato, R., Tudor,M., Fambrough, D., Csankovszki, G. et al. (2001).Loss of genomic methylation causes p53-dependentapoptosis and epigenetic deregulation. Nature Genet.27, 31–39.
52. Suzuki, H., Gabrielson, E., Chen, W., Anbazhagan,R., van Engeland, M., Weijenberg, M. P. et al. (2002).A genomic screen for genes upregulated bydemethylation and histone deacetylase inhibition inhuman colorectal cancer. Nature Genet. 31, 141–149.
53. Uhl, J., Klan, N., Rose, M., Entian, K. D., Werz, O. &Steinhilber, D. (2002). The 5-lipoxygenase promoteris regulated by DNA methylation. J. Biol. Chem. 277,4374–4379.
54. Yoon, B. J., Herman, H., Sikora, A., Smith, L. T., Plass,C. & Soloway, P. D. (2002). Regulation of DNAmethylation of Rasgrf1. Nature Genet. 30, 92–96.
55. Curradi, M., Izzo, A., Badaracco, G. & Landsberger,N. (2002). Molecular mechanisms of gene silencingmediated by DNA methylation. Mol. Cell. Biol. 22,3157–3173.
56. Reid, R., Greene, P. J. & Santi, D. V. (1999). Expositionof a family of RNA m(5)C methyltransferases fromsearching genomic and proteomic sequences. Nucl.Acids Res. 27, 3138–3145.
57. Liu, Y. & Santi, D. V. (2000). m5C RNA m5C DNAmethyl transferases use different cysteine residuesas catalysts. Proc. Natl Acad. Sci. USA, 97, 8263–8265.
58. Voet, D. & Voet, J. G. (1995). Biochemistry, 2nd edit.,Wiley, New York.
59. Mathews, C. K. & van Holde, K. E. (1996). Biochemis-try, The Benjamin/Cummings Publishing Company,Inc, Menlo Park, CA.
60. LaPat-Polasko, L., Maley, G. F. & Maley, F. (1990).Properties of bacteriophage T4 thymidylate synthasefollowing mutagenic changes in the active site andfolate binding region. Biochemistry, 29, 9561–9572.
61. Graves, K. L., Butler, M. M. & Hardy, L. W. (1992).Roles of Cys148 and Asp179 in catalysis by deoxy-
cytidylate hydroxymethylase from bacteriophage T4examined by site-directed mutagenesis. Biochemistry,31, 10315–10321.
62. Page, M. I. & Jencks, W. P. (1971). Entropic contri-butions to rate accelerations in enzymic and intra-molecular reactions and the chelate effect. Proc. NatlAcad. Sci. USA, 68, 1678–1683.
63. Kraut, J. (1988). How do enzymes work? Science, 242,533–540.
64. Yu, J., Choi, S. Y., Moon, K. D., Chung, H. H., Youn,H. J., Jeong, S. et al. (1998). A glycosidase antibodyelicited against a chair-like transition state analog byin vitro immunization. Proc. Natl Acad. Sci. USA, 95,2880–2884.
65. Schluckebier, G., Labahn, J., Granzin, J. & Saenger, W.(1998). M.TaqI: possible catalysis via cation–pi inter-actions in N-specific DNA methyltransferases. Biol.Chem. 379, 389–400.
66. Newby, Z. E., Lau, E. Y. & Bruice, T. C. (2002). Atheoretical examination of the factors controlling thecatalytic efficiency of the DNA-(adenine-N6)-methyl-transferase from Thermus aquaticus. Proc. Natl Acad.Sci. USA, 99, 7922–7927.
67. Craik, C. S., Roczniak, S., Largman, C. & Rutter, W. J.(1987). The catalytic role of the active site asparticacid in serine proteases. Science, 237, 909–913.
68. Carter, P. & Wells, J. A. (1988). Dissecting the catalytictriad of a serine protease. Nature, 332, 564–568.
69. Peracchi, A. (2001). Enzyme catalysis: removingchemically ‘essential’ residues by site-directed muta-genesis. Trends Biochem. Sci. 26, 497–503.
70. Jeltsch, A. & Lanio, T. (2002). Site-directed muta-genesis by polymerase chain reaction. Methods Mol.Biol. 182, 85–94.
71. Roth, M., Helm-Kruse, S., Friedrich, T. & Jeltsch, A.(1998). Functional roles of conserved amino acid resi-dues in DNA methyltransferases investigated bysite-directed mutagenesis of the Eco RV adenine-N6-methyltransferase. J. Biol. Chem. 273, 17333–17342.
72. Roth, M. & Jeltsch, A. (2000). Biotin-Avidin micro-plate assay for the quantitative analysis of enzymaticmethylation of DNA by DNA methyltransferases.Biol. Chem. 381, 269–272.
73. Jeltsch, A., Friedrich, T. & Roth, M. (1998). Kinetics ofmethylation and binding of DNA by the Eco RV ade-nine-N6 methyltransferase. J. Mol. Biol. 275, 747–758.
74. Pingoud, A., Urbanke, C., Hogget, J. & Jeltsch, A.(2002). Quantitative analysis of biochemical data. Bio-chemical Methods, Wiley-VCH, Weinheim.
Edited by J. Karn
(Received 18 February 2003; received in revised form1 April 2003; accepted 3 April 2003)
Supplementary Material comprising threeFigures is available on Science Direct
684 Catalytic Mechanism of the Dnmt3a DNA MTase