an exploration of silsesquioxanes and zeolites using high-speed experimentation
TRANSCRIPT
Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/27351603
AnExplorationofSilsesquioxanesandZeolitesusingHigh-SpeedExperimentation
ARTICLE
Source:OAI
CITATIONS
2
READS
30
1AUTHOR:
PaoloPescarmona
UniversityofGroningen
72PUBLICATIONS1,362CITATIONS
SEEPROFILE
Availablefrom:PaoloPescarmona
Retrievedon:04February2016

An Exploration of Silsesquioxanes and Zeolites using High-Speed Experimentation
Paolo Prospero Pescarmona

Cover: drawing by Gianpiero Pescarmona.

An Exploration of Silsesquioxanes and Zeolites using High-Speed Experimentation
Proefschrift
ter verkrijging van de graad van doctor
aan de Technische Universiteit Delft,
op gezag van de Rector Magnificus prof. dr. ir. J.T. Fokkema,
voorzitter van het College voor Promoties,
in het openbaar te verdedigen
op vrijdag 19 september 2003 om 10.30 uur
door
Paolo Prospero PESCARMONA
Dottore in Chimica, Università di Torino (Italië)
geboren te Torino (Italië)

Dit proefschrift is goedgekeurd door de promotor:
Prof. dr. Th. Maschmeyer
Adviseur: Dr. ir. J.C. van der Waal
Samenstelling promotiecomissie:
Rector Magnificus voorzitter
Prof. dr. Th. Maschmeyer Technische Universiteit Delft, promotor
Prof. dr. ir. H. van Bekkum Technische Universiteit Delft
Prof. dr. ir. D.E. De Vos Katholieke Universiteit Leuven, België
Prof. L. Marchese Università del Piemonte Orientale, Italië
Prof. A.F. Masters University of Sydney, Australië
Prof. Sir J.M. Thomas University of Cambridge, Verenigd Koninkrijk
Dr. ir. J.C. van der Waal Technische Universiteit Delft - Avantium
Technologies Amsterdam, adviseur
ISBN 90-9017266-1

Better being strange than stranger.
to all the persons in whose eyes I met a smile


Preface __________________________________________________________________________________________________________
I
Preface and thesis outline
This thesis describes the study of silsesquioxanes and zeolites by means of
Combinatorial Chemistry and High-Speed Experimentation. As it often happens with
specialised work, this dissertation includes topics that probably are not familiar to most
of the readers. In order to overcome this hurdle, introductory chapters providing general
information over the subjects of this thesis have been included. The information
supplied in these chapters will help readers in possession of chemical knowledge to
understand the content of the rest of the thesis.
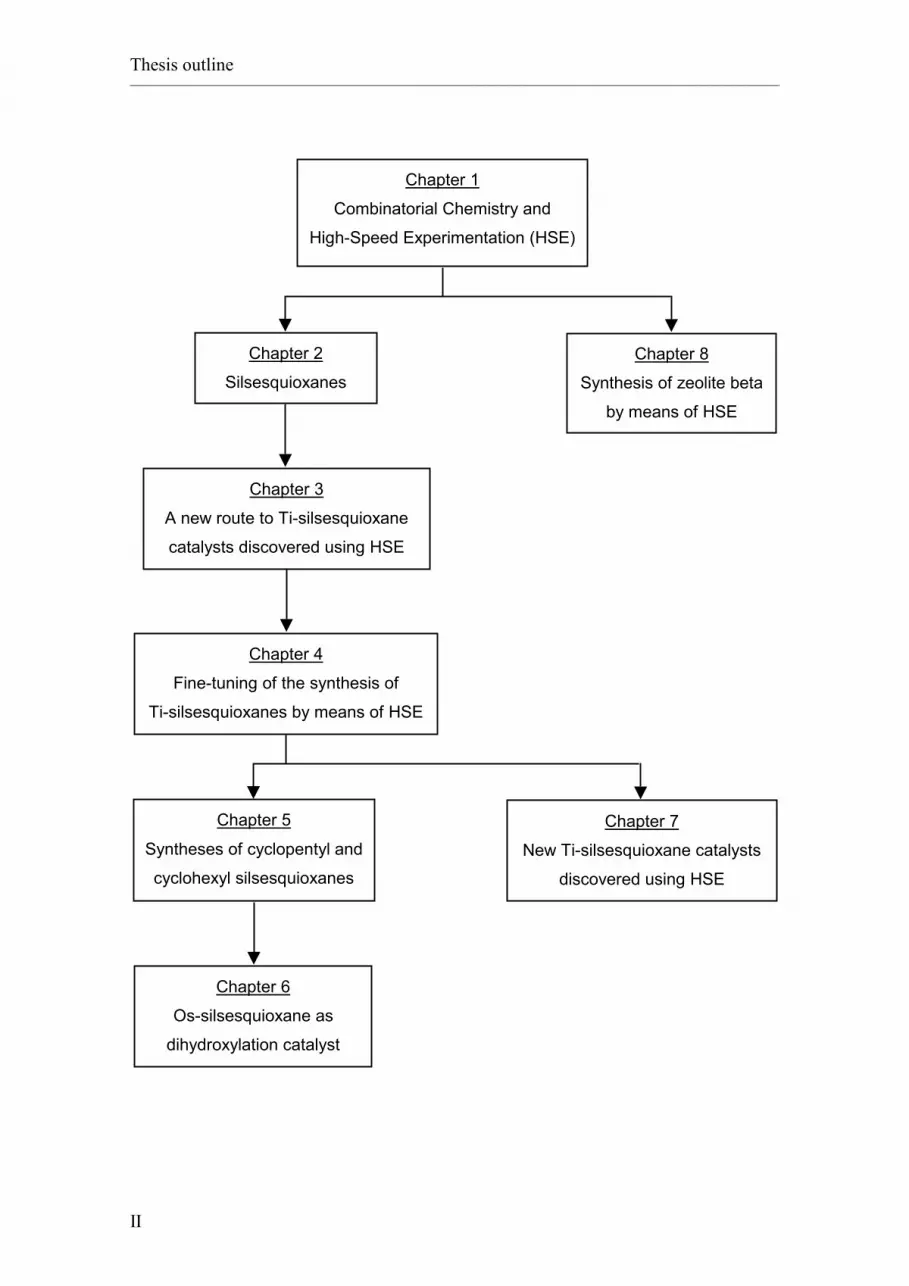
Thesis outline:
• Chapter 1 gives an introduction to Combinatorial Chemistry and High-Speed
Experimentation. These recently developed methods for chemical research have been
widely applied in this thesis.
• Chapter 2 presents an overview about silsesquioxanes. The investigation of this
family of compounds constitutes the major topic of this thesis.
• Chapter 3 describes a first optimisation of the synthesis of titanium silsesquioxanes as
epoxidation catalysts by means of High-Speed Experimentation techniques.
• Chapter 4 is a continuation of Chapter 3 and describes a fine-tuning of the synthesis
of titanium silsesquioxanes using High-Speed Experimentation techniques.
• Chapter 5 reports new synthetic methods for cyclopentyl and cyclohexyl
silsesquioxanes based on the High-Speed Experimentation work described in
Chapters 3 and 4.
• Chapter 6 presents an application of the cyclopentyl silsesquioxane discussed in
Chapter 5 as ligand for an osmium catalyst for the dihydroxylation of alkenes.
• Chapter 7 describes new tert-butyl and phenyl silsesquioxane precursors for
titanium-based epoxidation catalysts identified by exploring the synthesis of silsesquioxanes
in highly polar solvents by means of High-Speed Experimentation techniques.
• Chapter 8, after a concise introduction to zeolites, reports the application of
High-Speed Experimentation techniques to the study of the synthesis of zeolite beta.
• Appendix A describes the High-Speed Experimentation equipment.
• Appendix B consists of a glossary of concepts and abbreviations used in this thesis.
Thesis outline __________________________________________________________________________________________________________
II
Chapter 2
Silsesquioxanes Chapter 8
Synthesis of zeolite beta
by means of HSE
Chapter 1
Combinatorial Chemistry and
High-Speed Experimentation (HSE)
Chapter 3
A new route to Ti-silsesquioxane
catalysts discovered using HSE
Chapter 4
Fine-tuning of the synthesis of
Ti-silsesquioxanes by means of HSE
Chapter 5
Syntheses of cyclopentyl and
cyclohexyl silsesquioxanes
Chapter 7
New Ti-silsesquioxane catalysts
discovered using HSE
Chapter 6
Os-silsesquioxane as
dihydroxylation catalyst

Contents __________________________________________________________________________________________________________
III
Contents
Preface and thesis outline
Contents
1. Combinatorial Chemistry and High-Speed Experimentation techniques applied to
catalysis.
1.1. Introduction.
1.2. Methods.
1.1.1. Mix&split synthesis
1.1.2. Masking strategies
1.1.3. Parallel synthesis and High-Speed Screening (HSE techniques).
1.3. Applications.
1.4. High-Speed Experimentation techniques applied to catalysis.
2. Silsesquioxanes.
2.1. Introduction and nomenclature.
2.2. Synthesis of oligomeric silsesquioxanes.
2.2.1. Hydrolytic condensation of RSiX3.
2.2.2. Cleavage of Si-O-Si bonds.
2.2.3. Reaction of the R-group.
2.2.4. Corner-capping reactions.
2.3.Characterisation.
2.3.1. NMR spectroscopy.
2.3.2. Mass spectrometry.
2.3.3. IR spectroscopy.
2.3.4. X-ray diffraction.
2.4. Completely condensed silsesquioxanes (RSiO1.5)a.
2.4.1. R4Si4O6 (a4b0).
2.4.2. R6Si6O9 (a6b0).
2.4.3. R8Si8O12 (a8b0).
I
III
1
2
3
4
6
7
8
10
13
14
17
17
20
20
21
22
22
23
24
24
24
25
25
26

Contents __________________________________________________________________________________________________________
IV
2.4.4. R10Si10O15 (a10b0).
2.4.5. R12Si12O18 (a12b0)
2.4.6. Higher completely condensed silsesquioxanes (a > 12).
2.5. Incompletely condensed silsesquioxanes
2.5.1. RSi(OH)3 (a1b3).
2.5.2. R2Si2O(OH)4 (a2b4).
2.5.3. R4Si4O4(OH)4 (a4b4).
2.5.4. R6Si6O7(OH)4 (a6b4).
2.5.5. R7Si7O9(OH)3 (a7b3).
2.5.5.1 Metallasilsesquioxane catalysts for the epoxidation of
alkenes.
2.5.5.2 Metallasilsesquioxane catalysts for the polymerisation of
alkenes.
2.5.5.3 Other metallasilsesquioxanes.
2.5.6. R8Si8O11(OH)2 (a8b2).
2.5.7. Other incompletely condensed structures (a6b2, a7b1, a8b4).
2.6. Conclusions.
3. A new, efficient route to titanium-silsesquioxane epoxidation catalysts
developed by using High-Speed Experimentation.
3.1. Introduction.
3.2. The High-Speed Experimentation approach.
3.3. Results and discussion.
3.3.1. Characterisation of the HSE lead.
3.4. Conclusions.
3.5. Experimental.
4. Fine-tuning of the synthesis of titanium-silsesquioxane epoxidation catalysts by
means of High-Speed Experimentation.
4.1. Introduction.
4.2. The High-Speed Experimentation approach.
4.3. Results and discussion.
4.3.1. Synergetic effect of mixtures of silanes.
28
29
29
29
31
31
31
32
33
34
36
37
38
39
39
45
46
48
49
52
56
57
61
62
62
63
63

Contents __________________________________________________________________________________________________________
V
4.3.2. Effect of the reaction conditions.
4.3.3. Effect of the nature of the titanium centre.
4.4. Conclusions.
4.5. Experimental.
5. Fast and high-yield syntheses of cyclopentyl and cyclohexyl
silsesquioxanes using acetonitrile as reactive solvent.
5.1. Introduction.
5.2. Results and discussion.
5.2.1. Synthesis of cyclopentyl silsesquioxane a7b3.
2.5.5.4 Monitoring by means of mass spectrometry.
2.5.5.5 Monitoring by means of infrared spectroscopy.
5.2.2. Synthesis of cyclohexyl silsesquioxanes.
5.3. Conclusions.
5.4. Experimental.
5.4.1. Synthesis of cyclopentyl silsesquioxane a7b3.
5.4.2. Synthesis of cyclohexyl silsesquioxanes.
6. Osmium silsesquioxane as model compound and homogeneous catalyst for
the dihydroxylation of alkenes.
6.1. Introduction.
6.2. Results and discussion.
6.3. Conclusions.
6.4. Experimental.
7. New tert-butyl and phenyl silsesquioxane precursors for epoxidation
titanium catalysts discovered by means of High-Speed Experimentation.
7.1. Introduction.
7.2. Results and discussion.
7.2.1. The High-Speed Experimentation screening.
7.2.2. Phenyl silsesquioxanes synthesised in H2O.
7.2.3. tert-butyl silsesquioxanes synthesised in H2O.
7.2.4. tert-butyl silsesquioxanes synthesised in DMSO.
64
69
73
73
77
78
79
79
82
88
93
98
99
100
101
105
106
107
112
112
117
118
118
118
120
122
132

Contents __________________________________________________________________________________________________________
VI
7.3. Conclusions.
7.4. Experimental.
7.4.1. The High-Speed Experimentation screening.
7.4.2. Phenyl silsesquioxanes synthesised in H2O.
7.4.3. tert-butyl silsesquioxanes synthesised in H2O.
7.4.4. tert-butyl silsesquioxanes synthesised in DMSO.
8. Study of the synthesis of zeolite beta using High-Speed Experimentation.
8.1. Introduction.
8.1.1. Zeolite beta.
8.2. The High-Speed Experimentation approach.
8.3. Results and discussion.
8.3.1. Up-scaling of the HSE lead.
8.4. Conclusions.
8.5. Experimental.
Appendix A. High-Speed Experimentation equipment.
A.1. The HSE automated workstation.
A.2. The heating blocks.
A.3. The vacuum centrifuge.
Appendix B. Glossary.
B.1. Concepts.
B.2. Abbreviations.
Summary.
Samenvatting.
Acknowledgements.
Publications and oral presentations.
Curriculum vitae.
135
136
136
136
137
139
143
144
145
146
147
150
151
152
155
155
157
158
159
159
160
161
165
169
171
174



1
1 Combinatorial Chemistry and High-Speed Experimentation techniques applied to catalysis
Abstract
Combinatorial Chemistry and High-Speed Experimentation techniques are methods that
have been developed recently and that allow the fast preparation and analysis of large
numbers of samples. These techniques, initially developed for the discovery of
pharmaceuticals, are being increasingly applied to various fields of chemical research,
particularly to catalysis and materials science.
____________________ The contents of this chapter have been published in:
P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Catal. Lett., 1999, 63, 1.
P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, Catal. Today, 2003, 81, 347.

Chapter 1 __________________________________________________________________________________________________________
2
1.1 Introduction
Combinatorial Chemistry (CC) and High-Speed Experimentation (HSE)
techniques are research methods that allow the preparation and the testing of large
numbers of samples in a short time.1 The fascinating idea behind the first applications of
Combinatorial Chemistry methods was to mimic nature’s evolutionary approach in the
search of pharmaceutical active compounds. The concept was to combine many
synthetic parameters to produce a vast number of different compounds and to test them
for pharmaceutical activity.2 Just the active species would ‘survive’ this screening and
would be further studied as candidates for active drugs. Starting at the end of the
nineteen-eighties, the field of Combinatorial Chemistry rapidly developed both from the
methodological and technical point of view, in such a way that these methods are
becoming a standard tool for the discovery of novel drugs. The success in the
pharmaceutical area has stimulated application of these techniques to other fields of
chemical research and particularly to catalysis1,3-8 (both homogeneous9-11 and
heterogeneous12-14) and to materials science (e.g. superconducting, optical, magnetic,
dielectric, ferroelectric or polymeric materials).4,15-17 Combinatorial methods were
developed and modified in different ways to adapt them to each field of application and
started to be often associated with High-Speed Experimentation techniques. The term
combinatorial indicates the methodology of combining different experimental
parameters and to test the so-obtained combinations for selected properties, while the
term High-Speed Experimentation, alternatively known as High-Throughput
Experimentation (HTE), refers to the automated equipment that, allowing fast
preparation and screening of samples, is often necessary to perform a combinatorial
approach. HSE automated workstations can perform operations rapidly (and for 24
hours a day) and can also cope with very small amounts of reactants with high
precision. Besides being in many cases faster and cheaper per experiment,
Combinatorial Chemistry and High-Speed Experimentation methods are also safer and
have a lower environmental impact, since they use only small quantities of reactants. A
further advantage of the experiments carried out with these techniques is the high
reproducibility: since the operations are largely performed by robotic equipment, the
experimental errors due to different preparation conditions or different operators are
reduced to systematic ones and the reproducibility of the experiments is notably increased.

Combinatorial Chemistry and High-Speed Experimentation __________________________________________________________________________________________________________
3
The incentive to adopt Combinatorial Chemistry and High-Speed
Experimentation is provided by the fact that by these techniques it is possible to
synthesise and screen tens or hundreds of samples in a much shorter time compared to
traditional methods. Clearly, this approach has the potential for substantial time-savings
in research and development, allowing faster testing of hypotheses as well as reducing
considerably the time-to-market of new commercially relevant developments. Industrial
interest in CC and HSE technology is demonstrated by the increasing number of small,
dedicated new companies and joint ventures in this field. Besides their use in applied
research for the identification of new leads, CC and HSE techniques can result in a
powerful tool for academic scientific research. A correct application of these techniques
can provide a new approach for fundamental research: by means of a rational and
scientific design of the experiments these techniques can lead to the identification of
trends that give a much better first understanding/appreciation of the system under
study. Moreover, by performing a large number of experiments in a short time, more
opportunities may be offered to serendipity.
1.2 Methods
In order better to understand what Combinatorial Chemistry is, it is instructive to
consider, as an example, the reaction of a compound of class A (e.g. a Lewis acid) with
a compound of class B (e.g. a Lewis base) in a solvent S to give a product P. Combining
n different compounds of class A with m different compounds of class B and l different
solvents S, a parameter space constituted of n×m×l different combinations would be
defined, where each combination could lead to a different product or to a different yield
for a particular product. Such an approach can be useful if one were trying, e.g., to
obtain product P with the highest yield. Once the collection of products (usually
referred to as a library18) based on the selected parameter space has been synthesised, it
is necessary to subject it to a screening test regarding the performance or physical
properties of the products. The mode of screening depends on the type of product and
on its properties (e.g. catalyst selectivity or product yield).
When performing a synthetic Combinatorial Chemistry experiment, a number of
basically different strategies may be followed to create a library of compounds. The
Chapter 1 __________________________________________________________________________________________________________
4
most commonly used are: mix&split (or split and pool) synthesis, masking strategies
and parallel synthesis.
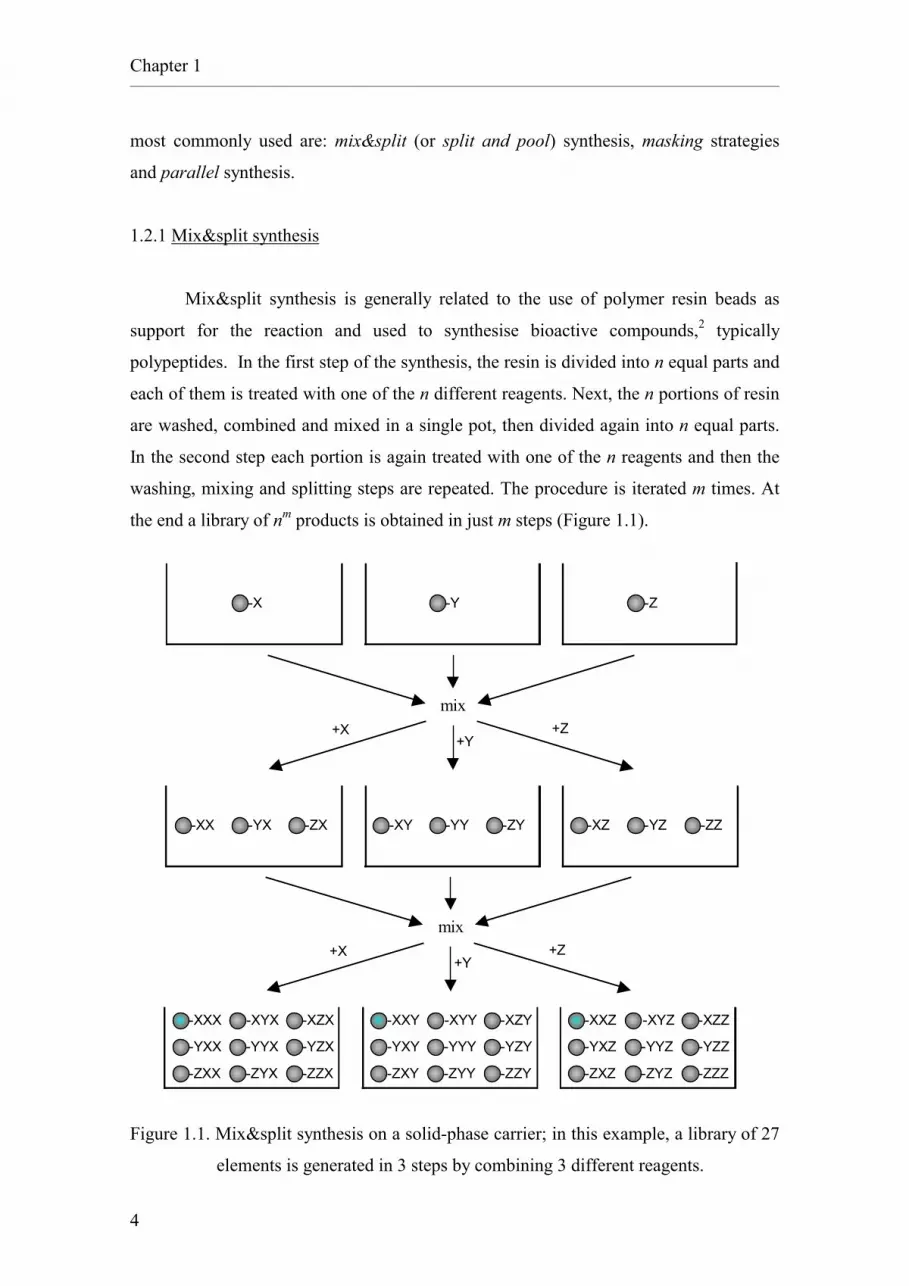
1.2.1 Mix&split synthesis
Mix&split synthesis is generally related to the use of polymer resin beads as
support for the reaction and used to synthesise bioactive compounds,2 typically
polypeptides. In the first step of the synthesis, the resin is divided into n equal parts and
each of them is treated with one of the n different reagents. Next, the n portions of resin
are washed, combined and mixed in a single pot, then divided again into n equal parts.
In the second step each portion is again treated with one of the n reagents and then the
washing, mixing and splitting steps are repeated. The procedure is iterated m times. At
the end a library of nm products is obtained in just m steps (Figure 1.1).
Figure 1.1. Mix&split synthesis on a solid-phase carrier; in this example, a library of 27
elements is generated in 3 steps by combining 3 different reagents.
-X -Y -Z
-XX -YX -ZX -XY -YY -ZY -XZ -YZ -ZZ
mix +X +Z
+Y
-YXX -YYX -YZX
-XXX -XYX -XZX
-ZXX -ZYX -ZZX
-YXY -YYY -YZY
-XXY -XYY -XZY
-ZXY -ZYY -ZZY
-YXZ -YYZ -YZZ
-XXZ -XYZ -XZZ
-ZXZ -ZYZ -ZZZ
mix +X +Z
+Y

Combinatorial Chemistry and High-Speed Experimentation __________________________________________________________________________________________________________
5
Once such a library has been created, it has to be tested to determine, e.g., the bioactive
compound(s). A problem that arises when the mix&split synthesis method is used, is
how to identify the compound(s) giving positive results during the screening among the
set of the components of the library.19 One way to overcome this problem is by a
process known as deconvolution.2 In the first step, the elements of the library are
divided into the n vessels and screened for activity: the active vessel is identified and
the others are eliminated. The compounds of this vessel are resynthesised in smaller
libraries and screened again for activity. The process of elimination goes on until the
active compound is established (Figure 1.2).
Figure 1.2. Example of deconvolution process for a library of 27 elements.
- YXX - YYX - YZX - XXX - XYX - XZX
- ZXX - ZYX - ZZX -YXY -YYY - YZY -XXY -XYY - XZY
-ZXY -ZYY - ZZY -YXZ - YYZ - YZZ -XXZ - XYZ - XZZ
-ZXZ - ZYZ - ZZZ
- XX - YX - ZX -XY -YY - ZY - XZ - YZ -ZZ
- XXY - YXY - ZXY -XYY -YYY - ZYY - XZY -YZY - ZZY
Active vessel
Step 1
Step 2
- XZ -YZ - ZZ
- XZY -YZY -ZZY
Step 3
Active compound
+Y +Y +Y
+Y +Y +Y
Active vessel

Chapter 1 __________________________________________________________________________________________________________
6
Although deconvolution has proved to be useful, it presents some drawbacks: besides
being a quite time-consuming process, when the active vessel is identified, there is no
certainty that the activity is due to the presence of a single active product rather than
resulting from the presence of many weakly active compounds.
Another technique used to identify the compounds that were active during screening is
that of encoding. The method consists in tagging each compound of the library during
the synthesis with a different tag; suitable tags show particular chemical or physical
properties that can be easily detected.20,21 Common chemical tagging methods make use
of tags that can be cleaved to liberate compounds (e.g. amines), which can then be
detected by standard analytical techniques (e.g. HPLC, MS).19
1.2.2 Masking strategies
Masking strategies are used for the combinatorial synthesis of thin films of solid
materials.15,16 These strategies are based on the successive deposition of thin films of solids
over a support. By means of a set of masks, the deposition of each solid precursor is limited
to a specific region of the support, creating a library of solids with different compositions.
Figure 1.3. Examples of binary and quaternary masks.
quaternary masking
binary masking

Combinatorial Chemistry and High-Speed Experimentation __________________________________________________________________________________________________________
7
Mainly two types of masking are reported in the literature: binary and quaternary
(Figure 1.3). When using binary masking, 2n compositions can be obtained with n
masks in n steps; with quaternary masking, 4n compositions are produced using n masks
in 4n steps (Figure 1.4). Binary masking allows the synthesis of the highest number of
different solids with a given number of deposition steps; quaternary masking allows
greater flexibility in the design of the library, since elements added in the four steps
used for each mask will not overlap.
The analytical technique employed for the screening of the library depends on the
desired property of the materials (e.g. resistance measurements for superconductors,
photoluminescence measurements for phosphors).4
Figure 1.4. Binary and quaternary masking strategies for the synthesis of materials libraries.
1.2.3 Parallel synthesis and High-Speed Screening (HSE techniques)
In the parallel synthesis approach the various reactions take place in separate
vessels; typically, robotic equipment is used to pick and mix the reactants in different
miniature vessels or wells, so that an array of distinct products is obtained. The library
of products has to be screened to determine the active compound(s). In contrast to the
mix&split approach, the identification of the active species is not a problem, since
A A B
D C
AE BE
CE DE
90° rotation + B90° rotation + C90° rotation + Dmask 1 + A mask 2 + E
AB A
B
A ABC AC AB A
C BCB
+A +B +C
binary masking
quaternary masking

Chapter 1 __________________________________________________________________________________________________________
8
separate wells are used. However, finding a suitable way to test all the isolated products
at high-throughput speeds presents a principal challenge. If the screening process of the
library is performed one well at a time, it can be very expensive and time-consuming
and would become a bottleneck nullifying some of the speed advantages of
combinatorial synthesis. To avoid this, fast and affordable analytical methods, usually
referred to as High-Speed Screening (HSS) or High-Throughput Screening (HTS)
techniques,22 are being developed by many groups. Current HSS technology is based on
the use of miniaturised, automated and parallel versions of tools conventionally
employed to screen the compounds under study, such as chromatographic,
spectroscopic or thermographic techniques, and in some cases of new specifically
developed methods.1,4,11,13,23 (e.g. a mass spectrometer equipped with an autosampler
has been used to assay the enantioselectivity of catalytic reactions of isotopically
labelled substrates, with a throughput of about 1000 samples per day).24 The choice of
the type of HSS is related to the particular feature that has to be detected and, therefore,
has to be tailored for any system under investigation.
The use of parallel synthesis associated to High-Speed Screening is presently referred to
as High-Speed Experimentation (or High-Throughput Experimentation).
1.3 Applications
To illustrate how and in which cases Combinatorial Chemistry and High-Speed
Experimentation techniques can be applied advantageously, a general case of the
discovery/optimisation of a catalytic system will be considered. Given a chemical reaction
for which a catalyst is wanted, the first step in primary screening is conventional, i.e.
consists of gathering all the knowledge already available in the literature about the
subject. In many cases this search provides basic information about the characteristics of
the most suitable catalytic system for the chosen chemical reaction (e.g. an acid rather
than a basic catalyst, a homogeneous rather than a heterogeneous catalyst). If this
information is scarce or absent, the issue becomes how to find a way to get basic knowledge
of the system under investigation. This could be achieved with an experimental approach
and/or by means of computational modelling. If an experimental approach is chosen, this
second step in primary screening should cover a broad range of parameters in order to gain

Combinatorial Chemistry and High-Speed Experimentation __________________________________________________________________________________________________________
9
very general information about the system. This usually requires an extremely large
number of experiments to be performed, for which purpose Combinatorial Chemistry and
High-Speed Experimentation techniques are particularly helpful.
Comparing the three synthetic strategies described in Paragraph 1.2, it is clear
that the mix&split and the masking approaches might be advantageous for primary
screening since they allow the fast synthesis and screening of very large libraries of
compounds (thousands per week). High-Speed Experimentation parallel methods have a
lower throughput (hundreds of samples per week) than mix&split and masking
methods, but one that is still very high when compared with traditional, serial
techniques. The lower throughput of HSE parallel techniques might be a drawback for a
primary screening, which requires gathering a broad set of information in a short time.
This problem can be overcome by using Design of Experiments (DOE) methods and
search algorithms, that allow decreasing the number of experiments to be performed by
means of statistical data-handling.12,13,17,25 Moreover, parallel methods are more
versatile than mix&split and masking methods: with the latter, only the composition of
a target compound can be optimised, while parallel methods also allow the variation of
other synthetic parameters (solvent, pH, concentrations, temperature etc.). Besides, HSE
parallel equipment can be designed to handle solid, liquid and gas phases, while
mix&split and masking strategies are limited to liquid and solid phases, respectively.
Finally, parallel and masking approaches allow for the correlation of each sample with
its properties (i.e. its activity as a catalyst), while this is not possible, prior to
deconvolution, using mix&split methods where the samples are screened as a whole. In
this sense, parallel and masking techniques are more suitable when the goal of
identifying new leads is coupled with that of a fundamental understanding of the system
under study. Given all these considerations it is clear why HSE parallel techniques are
becoming the most common approach for primary screening.
Typically, the primary screening provides a set of lead compounds and some
basic correlation between the properties of the leads and their structural and synthetic
characteristics. In the secondary screening one or more of these leads are further
optimised: the information obtained from the primary screening is the basis to focus
further research on a smaller experimental parameter space. Dealing with a more
focussed parameter space means that more detailed and specific information can be
obtained in this screening phase. The fact that the parameter space screened is smaller in

Chapter 1 __________________________________________________________________________________________________________
10
the secondary than in the primary screening implies that the number of experiments
performed in the secondary screening is lower while the information gathered from each
experiment should be more detailed. Therefore, HSE parallel techniques are particularly
useful to speed up the secondary screening. The applicability of HSE techniques to
secondary screening is determined by the technical limitations of the automated
workstations employed. These limitations may range from the preparation of the
samples (e.g. inefficient mixing of reaction mixtures in the small HSE vessels) to their
analysis (e.g. equipment for high-throughput screening of the samples is not yet
available for every spectroscopic technique). The quality of the HSE equipment is
continuously improving, nevertheless at this stage not every chemical synthesis can be
studied by means of HSE techniques.
At the end of the secondary screening a restricted number of leads will be
identified and the correlation between their properties and their structural and synthetic
characteristics will be much more defined than at the end of the primary screening. It is
at this point that the lead(s) can be studied in detail at a conventional laboratory scale:
this typically includes the characterisation of the lead(s) with different spectroscopic
and/or diffractive techniques. If Combinatorial Chemistry and High-Speed
Experimentation techniques have been used during the earlier screening stages,
repeating the experiment in a conventional manner is an important check of the
reproducibility of the results on a larger-volume scale. Results should never be
published without such a check, since the ability to independently validate a particular
result should not be dependent on the HSE-equipment used.26
1.4 High-Speed Experimentation techniques applied to catalysis
The development of new catalysts is a challenging task. In many cases the
correlation between their features (structural, electronic) and their performance
(activity, selectivity, lifetime) is not easily established, especially for supported
heterogeneous catalysts. Therefore, an iterative process of ‘design’, synthesis and
testing is usually followed to improve catalyst performance. HSE techniques can
accelerate this process considerably, allowing for the simultaneous evaluation of a large
number of candidates (Figure 1.5). All operations involved in the development of a

Combinatorial Chemistry and High-Speed Experimentation __________________________________________________________________________________________________________
11
catalyst lend themselves to the application of HSE techniques. The synthesis of both
homogeneous and heterogeneous catalysts, their screening for activity and selectivity in
test reactions, and also the determination of the optimal process parameters for a
specific reaction, can all be conducted with much greater speed and efficiency
employing the miniaturised, automated combinatorial/HSE procedures than by the use
of conventional methods.
It is important to realise that HSE techniques have to be considered as a powerful tool to
increase the number of samples to be studied, rather than as an alternative to rational
methods for the development of catalysts: a multi-dimensional and scientific approach –
based, e.g., on literature data, computational modelling, personal chemical knowledge
and intuition - is essential to determine which parameter space has to be investigated to
gain the desired information from the experiment.
Figure 1.5. Comparison between classic and High-Speed Experimentation approach.
In this sense, HSE techniques have to be seen as a new and useful tool for chemical
research in the same way as, say, spectroscopic techniques or quantomechanical
calculations. Therefore, they can be very suitable to study some systems and inefficient
for others. The range of chemical systems that can be studied using HSE techniques will
probably get broader with the technical improvement of the automated equipment.
Testing
Design
Synthesis Testing
Design
Synthesis
Classic HSE

Chapter 1 __________________________________________________________________________________________________________
12
References
1 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Catal. Lett., 1999, 63, 1. 2 N.K. Terret, M. Gardner, D.W. Gordon, R.J. Kobylecki, J. Steele Tetrahedron, 1995, 30, 8135. 3 J.M. Newsam, F. Schüth, Biotechnol. Bioeng. (Comb. Chem.), 1998/1999, 61, 203. 4 B. Jandeleit, D.J. Schaefer, T.S. Powers, H.W. Turner, W.H. Weinberg, Angew. Chem. Int. Ed., 1999,
38, 2494. 5 R. Schlögl, Angew,. Chem. Int. Ed., 1998, 37, 2333. 6 T. Bein, Angew. Chem. Int. Ed., 1999, 38, 323. 7 W.F. Maier, Angew. Chem. Int. Ed., 1999, 38, 1216. 8 J.M. Thomas, Angew. Chem. Int. Ed., 1999, 38, 3589. 9 Shimizu, K.D., Snapper, M.L., Hoveyda, A.H., Chem. Eur. J., 1998, 4, 1885. 10 R.H. Crabtree, Chem. Commun., 1999, 1611. 11 M.T. Reetz, Angew. Chem. Int. Ed., 2001, 40, 284. 12 A. Holzwarth, P. Denton, H. Zanthoff, C. Mirodatos, Catal. Today, 2001, 67, 309. 13 S. Senkan, Angew. Chem. Int. Ed., 2001, 40, 312. 14 J.M. Newsam, T. Bein, J. Klein, W.F. Maier, W. Stichert, Micropor. Mesopor. Mater., 2001, 48, 355. 15 T.X. Sun, Biotechnol. Bioeng. (Comb. Chem.), 1998/1999, 61, 193. 16 X.-D. Xiang, Biotechnol. Bioeng. (Comb. Chem.), 1998/1999, 61, 227. 17 J.M Cawse, Acc. Chem. Res., 2001, 34, 213. 18 See Appendix B for a general definition of library in CC and HSE. 19 C. Barnes, S. Balasubramanian, Curr. Opin. Chem. Biol., 2000, 4, 346. 20 J.J. Baldwin, J.J. Burbaum, I. Henderson, M.H.J. Ohlmeyer, J. Am. Chem. Soc., 1995, 117, 5588. 21 E.J. Moran, S. Sarshar, J.F. Cargill, M.M. Shahbaz, A. Lio, A.M.M. Mjalli, W.W. Armstrong, J. Am.
Chem. Soc., 1995, 117, 10787. 22 M.F. Asaro, R.B. Wilson, Chemistry & Industry, 1998, 19, 777. 23 M.T. Reetz, Angew. Chem. Int. Ed., 2002, 41, 1335. 24 M.T. Reetz, M.H. Becker, H.-W. Klein, D. Stöckigt, Angew. Chem. Int. Ed., 1999, 38, 1758. 25 D.C. Montgomery, Design and Analysis of Experiments, J. Wiley & Sons, 1996, 4th edition. 26 M. Baerns, C. Mirodatos, NATO Science Series, 2002, Ser. II Vol. 69, 469.

13
2
Silsesquioxanes
Abstract
Silsesquioxanes are a family of inorganic-organic hybrid compounds with applications
in the fields of catalysis, materials science and coordination chemistry. This Chapter
presents a review of their synthesis and characterisation as well as selected applications. ____________________
The contents of this chapter have been published in:
P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583.

Chapter 2 __________________________________________________________________________________________________________
14
2.1 Introduction and nomenclature
The importance of silsesquioxanes as chemical structures of interest to a large
variety of chemists has grown steadily since their discovery as a ‘curious white
precipitate’ during silane polymerisations by Sprung and Guenther in 1955. Their
structures are manifold and usually crystallographically characterised. Their synthetic
accessibility, once, a major drawback due to synthesis times of months, has much
improved and many types of silsesquioxanes can now be synthesised in a matter of
hours or days. Applications of silsesquioxanes can be found in the areas of catalysis,
materials science (polymers, photo-resists) and coordination chemistry.
The etymology of the term silsesquioxane indicates a family of compounds
characterised by a ratio of 1.5 (sesqui, from the Latin semisque, ‘and a half’) between
silicon and oxygen atoms. Silsesquioxanes are compounds of the general formula
(RSiO1.5)a(H2O)0.5b, or, rearranging it, RaSiaO(1.5a-0.5b)(OH)b, where R is an hydrogen
atom or an organic group and a and b are integer numbers (a = 1,2,3…; b = 0,1,2,3,…)
related according to:
a + b = 2n, where n is an integer (n = 1,2,3,…)
b ≤ a + 2
Given this formula, a silsesquioxane with a particular degree of condensation can be
described by the values of a and b, e.g. a7b3 refers to a silsesquioxane containing seven
Si atoms and three -OH groups (Figure 2.2, compound 13). However, it is important to
notice that, for any set of a and b values, more than one structure might be drawn: for
instance, two a8b2 silsesquioxane structures are reported in the literature (Figure 2.2,
compounds 14 and 15).1,2
Structurally, silsesquioxane frameworks consist of tetrahedral units in which a silicon is
bound to three oxygens and one R-group. The oxygen atoms can act as bridges between
two silicon atoms, belonging to different tetrahedral units, or between a silicon and a
hydrogen atom. A unit where a silicon is connected to three oxygens is commonly
referred to as T unit (an M unit describes silicon bound to one oxygen, D when bound to
two oxygens and Q when bound to four oxygens, as is the case in a silicate).3 An

Silsesquioxanes __________________________________________________________________________________________________________
15
alternative nomenclature for silsesquioxanes may be based on the number of T units
they contain (Ta).
Figure 2.1. Completely condensed silsesquioxanes (these structures are simplified
representations as to, e.g., SiOSi angles).
Silsesquioxanes can be synthesised either as oligomeric species, i.e. discrete structures
constituted of a limited number of tetrahedral units, or as polymeric species. The
properties and applications of these two types of silsesquioxanes are rather different and
their fields of research separated. Here, the attention will be focussed on oligomeric
silsesquioxanes (oligosilsesquioxanes).
On the basis of the general formula reported above, silsesquioxanes can be
subdivided in two main groups: completely condensed and incompletely condensed
silsesquioxanes, according to b = 0 or b ≠ 0, respectively. In the case of completely
condensed silsesquioxanes, the general formula can be reduced to (RSiO1.5)a, where a is
now an even number greater than 2. In these compounds, all oxygen atoms act as
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
R
Si
Si
Si
Si
Si
OO
O
OO
O
R
R
R
R R
O
O
R
O
Si
SiSiOO
O O
OO
Si
Si
RR
R
R
Si SiO
O
R
RO
R
O
O
R R
O
O
O
R
Si
Si
O
R
R
Si
Si
O
R
SiSi
Si Si
SiSi
RR R
O O
OO
O
O O
OO
O
SiOOO
Si
R
O
O
R R
O
OR
O
R
SiSi
Si Si
Si
R
O
SiO
O
O
Si
O
SiO
R R
R
R
ROSi
R
RO
R
OR R
O
O
O
R
Si
Si
O
R
RSi
SiSi
SiSi
SiSi
R
R
OO
O
O
OO
Si
O
O
R
Si
O
O
O
21 3
54 6
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
RSiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
R
Si
Si
Si
Si
Si
OO
O
OO
O
R
R
R
R R
O
O
R
O
Si
Si
Si
Si
Si
Si
OO
O
OO
O
R
R
R
R R
O
O
R
O
Si
SiSiOO
O O
OO
Si
Si
RR
R
R
SiSiOO
O O
OO
Si
Si
RR
R
R
Si SiO
O
R
RO
R
O
O
R R
O
O
O
R
Si
Si
O
R
R
Si
Si
O
R
SiSi
Si Si
SiSi
RR R
O O
OO
O
O O
O
Si SiO
O
R
RO
R
O
O
R R
O
O
O
R
Si
Si
O
R
R
Si
Si
O
R
SiSi
Si Si
SiSi
RR R
O O
OO
O
O O
OO
O
SiOOO
Si
R
O
O
R R
O
OR
O
R
SiSi
Si Si
Si
R
O
SiO
O
O
Si
O
SiO
R R
R
R
O
O
SiOOO
Si
R
O
O
R R
O
OR
O
R
SiSi
Si Si
Si
R
O
SiO
O
O
Si
O
SiO
R R
R
R
ROSi
R
RO
R
OR R
O
O
O
R
Si
Si
O
R
RSi
SiSi
SiSi
SiSi
R
R
OO
O
O
OO
Si
O
O
R
Si
O
O
ORO
Si
R
RO
R
OR R
O
O
O
R
Si
Si
O
R
RSi
SiSi
SiSi
SiSi
R
R
OO
O
O
OO
Si
O
O
R
Si
O
O
O
21 3
54 6
Chapter 2 __________________________________________________________________________________________________________
16
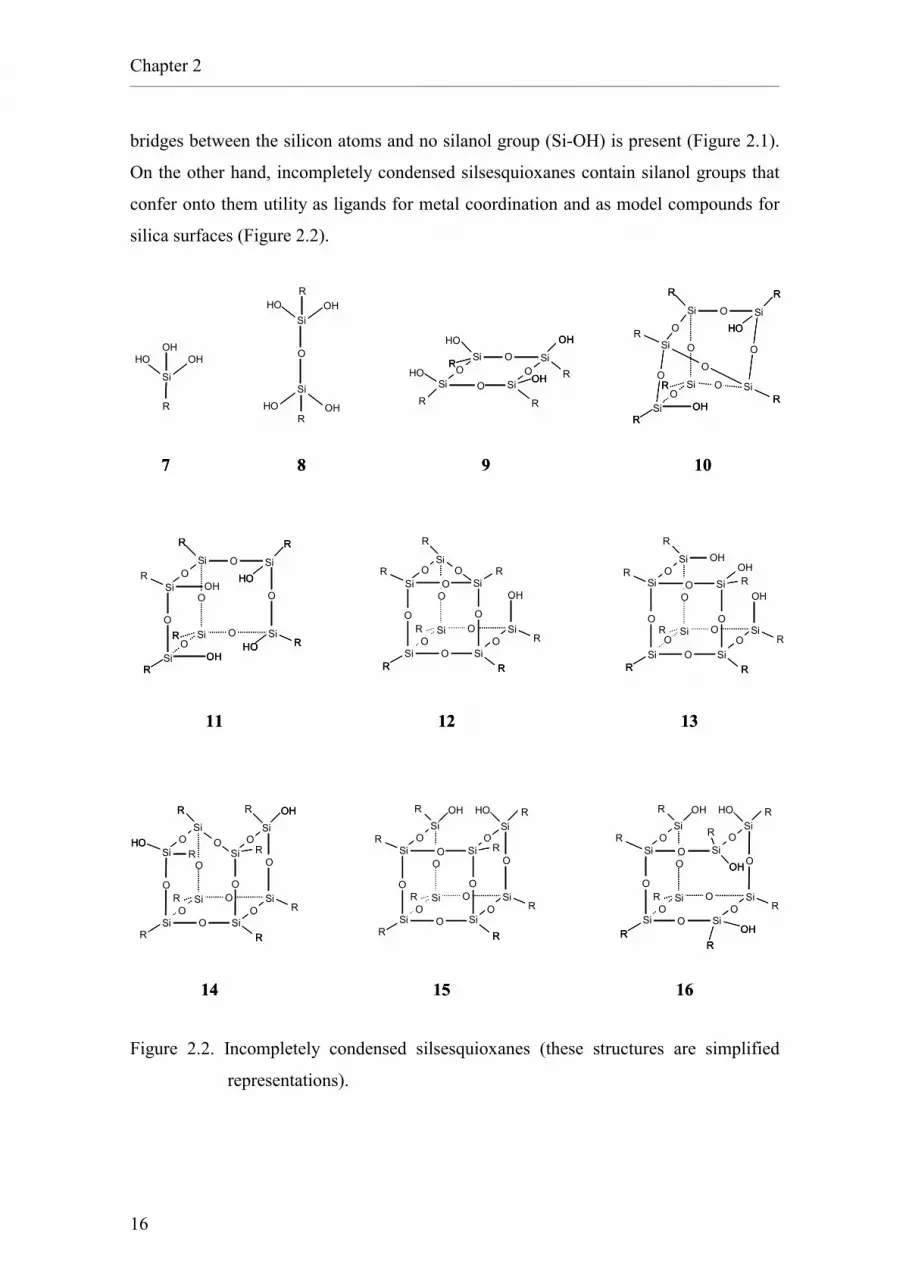
bridges between the silicon atoms and no silanol group (Si-OH) is present (Figure 2.1).
On the other hand, incompletely condensed silsesquioxanes contain silanol groups that
confer onto them utility as ligands for metal coordination and as model compounds for
silica surfaces (Figure 2.2).
Figure 2.2. Incompletely condensed silsesquioxanes (these structures are simplified
representations).
Si Si
SiSi
Si
Si
Si
Si
OH
OO
O
O
OO
O
OO
O
R
R
R
R
R
R R
RO
HOSi Si
Si
OH
O O
O
O
R
R
R
R
RR
Si O
O
RO
HO
Si Si
SiSi
O
O
OOH
OH
R
R
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
OH
R
R
R
HO
R
RO
R
R
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OH
OO
O
R
R
R
R
R R
R
O
O
Si Si
Si
Si Si
OO
OHHO
OH
O
O
OHO
O
R
R
R
R
R
RO
Si
O
Si Si
Si
Si Si
OO HO
OH
O
O
O
R
R
R
R
R
RO
Si
OSi
OHHO
R
OH
Si
OHHOR
O
SiOHHO
R
SiSiO
Si SiO
R
OH
OO
OHHO
R
HO
RR
87 10
1211 13
9
1514 16
Si Si
SiSi
Si
Si
Si
Si
OH
OO
O
O
OO
O
OO
O
R
R
R
R
R
R R
RO
HOSi Si
SiSi
Si
Si
Si
Si
OH
OO
O
O
OO
O
OO
O
R
R
R
R
R
R R
RO
HOSi Si
Si
OH
O O
O
O
R
R
R
R
RR
Si O
O
RO
HO
Si Si
SiSi
O
O
OOH
OH
RSi Si
Si
OH
O O
O
O
R
R
R
R
RR
Si O
O
RO
HO
Si Si
SiSi
O
O
OOH
OH
R
R
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
OH
R
R
R
HO
R
RO
R
R
R
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
OH
R
R
R
HO
R
RO
R
R
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OH
OO
O
R
R
R
R
R R
R
O
O
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OH
OO
O
R
R
R
R
R R
R
O
O
Si Si
Si
Si Si
OO
OHHO
OH
O
O
OHO
O
R
R
R
R
R
RO
Si
Si Si
Si
Si Si
OO
OHHO
OH
O
O
OHO
O
R
R
R
R
R
RO
Si
O
Si Si
Si
Si Si
OO HO
OH
O
O
O
R
R
R
R
R
RO
Si
O
O
Si Si
Si
Si Si
OO HO
OH
O
O
O
R
R
R
R
R
RO
Si
OSi
OHHO
R
OH
Si
OHHO
R
OH
Si
OHHOR
O
SiOHHO
R
Si
OHHOR
O
SiOHHO
R
SiSiO
Si SiO
R
OH
OO
OHHO
R
HO
RR
SiSiO
Si SiO
R
OH
OO
OHHO
R
HO
RR
87 10
1211 13
9
1514 16

Silsesquioxanes __________________________________________________________________________________________________________
17
2.2 Synthesis of oligomeric silsesquioxanes
A number of different ways are currently used to synthesise oligomeric silsesquioxanes.
2.2.1 Hydrolytic condensation of RSiX3
The most common synthetic route to obtain oligomeric silsesquioxanes is the
hydrolytic condensation of monosilanes RSiX3 (X = Cl, OMe, OEt,…):4,5
RSiX3 + 3 H2O RSi(OH)3 + 3 HX [1]
a RSi(OH)3 (RSiO1.5)a(H2O)0.5b + (1.5a – 0.5b) H2O [2]
The first step is the hydrolysis of the monosilane to give the corresponding trisilanol.
This reaction is generally very fast.6 The second step is the condensation of the trisilanol
yielding different silsesquioxane species (see Figures 2.1 and 2.2 for some structures
reported in literature). The trisilanol itself is very reactive and in most of the cases
cannot be isolated; the only reported exceptions are in the case of rather large organic
groups.7-10 The condensation reaction is a multistep process involving the formation of
many different intermediate structures. Sprung and Guenther11,12 and later Brown and
Vogt6,13 studied the hydrolytic condensation of different organomonosilanes RSiX3
(R = methyl, ethyl, phenyl, cyclohexyl). They hypothesised various mechanisms of
formation, involving the consecutive condensation of the monomeric trisilanol to give
linear, cyclic and, finally, polycyclic and polyhedral silsesquioxanes. For instance, in
the case of the synthesis of completely condensed phenyl silsesquioxanes 3 (Ph8Si8O12,
a8b0 or T8), they proposed the reaction to proceed by the consecutive formation of the
dimer, of the cyclic tetramer and, finally, of the cubic silsesquioxane (Figure 2.3).13
Lavrent’yev et al. followed the hydrolytic condensation of ethyltrichlorosilane in
aqueous butanol by GC-MS: the identification of intermediates and products allowed
them to outline the complex mechanism of formation.4 Recently, Kudo and Gordon
performed a theoretical study of the mechanism for the synthesis of
hydrosilsesquioxanes.14,15 Using ab initio quantum mechanical methods, they calculated
the energy barriers for the initial hydrolysis of trichlorosilane HSiCl3 to HSi(OH)3 and

Chapter 2 __________________________________________________________________________________________________________
18
the successive formation of the dimer H2Si2O(OH)4 [a2b4], cyclic trimer H3Si3O3(OH)3
[a3b3] and cyclic tetramer H4Si4O4(OH)4 [a4b4]. They found that the presence of a
water molecule strongly reduced the energy barriers that have to be overcome to form
the selected species, suggesting that the solvent may play a relevant role in influencing
the synthesis.
Figure 2.3. Proposed mechanism of formation for the completely condensed
silsesquioxane 3.
In fact, many factors are known to influence the hydrolytic condensation, determining
which silsesquioxanes structures are formed and in which ratios,4,5 they are namely:
- the nature of the R-group
- the nature of the X-group
- the solvent
- the concentration of the monosilane RSiX3
- the rate of addition and quantity of H2O
- the temperature
- the pH
- the reaction time
All these factors influence the hydrolytic condensation mutually and, therefore, they
cannot be studied independently. Nevertheless, some general conclusions about the
effect of any of them can be drawn.
The nature of the R-group might influence the thermodynamics and kinetics of
formation of silsesquioxanes via both steric and electronic effects. For example, for
bulky R-groups like cyclohexyl1 or cyclopentyl,16 the formation of incompletely
condensed silsesquioxanes is known to be favoured, while the hydrolytic condensation
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
R
SiOHHO
R
OH
Si
OHHOR
O
SiOHHO
R
Si
SiO R
R
Si
Si
O
R
O
O
RHO
HO
HO
HO
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
R
SiOHHO
R
OH
SiOHHO
R
OH
Si
OHHOR
O
SiOHHO
R
Si
OHHOR
O
SiOHHO
R
Si
SiO R
R
Si
Si
O
R
O
O
RHO
HO
HO
HO
Si
SiO R
R
Si
Si
O
R
O
O
RHO
HO
HO
HO

Silsesquioxanes __________________________________________________________________________________________________________
19
of monosilanes with smaller groups like methyl11,17 or hydrogen18 yields more
frequently completely condensed structures. This trend indicates that a steric effect of
the R-group determines to a significant extent the level of condensation of the products.
Moreover, the nature of the R-group determines, together with the nature of the solvent,
the solubility of the silsesquioxanes species, thereby influencing the equilibria and the
rate of the condensation reactions. In this context, it is interesting to note that the
solubility of the incompletely condensed silsesquioxane a7b3 in many organic solvents
is substantially lower when R is cyclopentyl or cycloheptyl than when it is cyclohexyl.16
This difference could account for the higher selectivity as well as for the higher yields
reported for cyclopentyl and cycloheptyl silsesquioxane a7b3.19
The nature of the X-group does not have a major influence on the synthesis of
silsesquioxanes, since the group reacts during the first, usually fast, step of the process.
When X is a halide, the hydrolysis is faster than when X is an alkoxy group. Moreover,
the hydrolysis of trihalidesilanes produces acids (HX) that may catalyse successive
condensation reactions. Trichlorosilanes are usually chosen as starting material for the
synthesis of silsesquioxanes.
The solvent plays a strong role in influencing the synthesis of silsesquioxanes, since
solvent molecules interact with the silsesquioxanes species present in the reaction
solution. Polar molecules form hydrogen bonds with the silanol groups (Si-OH) and,
therefore, stabilise incompletely condensed species. Recently, the favourable effect of
an high polarity solvent on the synthesis of incompletely condensed silsesquioxanes was
reported.20 As mentioned above, the solvent also determines the solubility of the
silsesquioxane species, influencing the rate and the products of the reaction. Finally, the
solvent can affect the kinetics of the synthesis by interacting with the transition-state
intermediates of the condensation reactions: theoretical calculations14,15 showed that the
interaction of a transition-state intermediate with a polar solvent molecule can stabilise
the system and, therefore, reduce the activation barrier for the formation of higher
silsesquioxane structures.
The starting concentration of the monosilane RSiX3 influences the reaction kinetics.
Since it has been impossible, so far, to obtain a satisfactory kinetic equation for the
synthesis of silsesquioxanes, it is not possible to preview which can be the effect of the
starting concentration on the reaction. It has been suggested that a high concentration
facilitates the formation of polymeric silsesquioxanes.4

Chapter 2 __________________________________________________________________________________________________________
20
The quantity and the rate of addition of H2O influence the kinetics of the hydrolytic
condensation (water acts both as a reactive compound and as a solvent). However, for the
reasons mentioned previously, the precise effect of this parameter has not yet been elucidated.
The temperature of the reaction influences the reaction kinetics and the solubility of the
silsesquioxanes species present in the reaction solution. It has been reported that a high
reaction temperature favours the formation of highly condensed polymeric species.5
The hydrolysis and condensation reactions of silsesquioxanes are catalysed by both acid
and basic medium. Low pH values have been reported to favour the formation of
oligosilsesquioxanes, while at high pH values polymeric species are favoured.12
The synthesis of silsesquioxanes species can be very slow and periods of several
months, even years, can be required before the reaction goes to completion.1 If the
reaction is stopped after a set reaction time and the silsesquioxane species forced to
condense by drying the reaction solution, the silsesquioxane structures that are formed
are different from those that would have been formed by letting the synthesis proceed.
Usually, the silsesquioxanes so obtained are of lower molecular weight, since they are
generated by the rapid condensation of intermediate species.
2.2.2 Cleavage of Si-O-Si bonds
Recently, Feher and coworkers developed new methods for the synthesis of
incompletely condensed silsesquioxanes by the cleavage of Si-O-Si bonds of
completely condensed silsesquioxanes.21 The reaction of the readily available R6Si6O9
and R8Si8O12 (Figure 2.1, compounds 2 and 3) with a strong acid (HBF4/BF3, CF3SO3H
or CH3SO3H)2,22,23 or with a base (NEt4OH)24,25 produces various incompletely
condensed structures (e.g. compounds 10, 11, 13, 14, 15, 16) in good yields and often
with good selectivity. These methods offer a fast, though somewhat complex,
alternative to the hydrolytic condensation of organomonosilanes to obtain various
incompletely condensed silsesquioxanes in synthetically useful amounts.
2.2.3 Reaction of the R-group
Different types of reaction involving the conversion of the R-group of
silsesquioxanes to another group are known. These reactions can be useful if the desired

Silsesquioxanes __________________________________________________________________________________________________________
21
silsesquioxane were difficult to obtain via the hydrolytic condensation. These types of
synthesis methods have been applied to completely condensed silsesquioxanes and
particularly to the cubic structure R8Si8O12 (Figure 2.1, compound 3). The
octahydrosilsesquioxane H8Si8O12 has been reacted with a number of 1,2-unsaturated
hydrocarbons in the presence of H2PtCl6 as the catalyst to get the addition of the organic
group on the Si atom (Figure 2.4).26 This hydrosilylation reaction is rather general and
can be used to produce a variety of octasilsesquioxanes a8b0, from mono- to completely
substituted.27 The cyclohexyl silsesquioxane (c-C6H11)8Si8O12 can be synthesised via the
catalytic hydrogenation of the corresponding phenyl silsesquioxane (C6H5)8Si8O12.28
(2-C4H3S)8Si8O12 can be brominated with Br2 in the presence of HBr to give
(2-C4Br3S)8Si8O12.29 Modification of the R-group with various functionalities has been
reported fo R = C6H530 and p-ClCH2C6H4.31
Figure 2.4. Hydrosilylation reaction of 1,2-unsaturated hydrocarbons with H8Si8O12.
2.2.4 Corner-capping reactions
This method has been used by Feher and coworkers to obtain the cubic
silsesquioxane a8b0 from the incompletely condensed structure a7b3 (Figure 2.5).1 The
reaction of the cyclohexyl silsesquioxane (c-C6H11)7Si7O9(OH)3 with an
organotrichlorosilane RSiCl3 in the presence of an amine offers a straightforward route
to various monosubstituted octasilsesquioxanes.
Figure 2.5. Corner-capping reaction (Chex = c-C6H11).
Si H + H2C CH Si CH2CH2
H2PtCl6Si H + H2C CH Si CH2CH2
H2PtCl6
SIOR
OSi
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
ORSiCl3
Si
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
OHOH
OH
SIOR
OSi
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
O
SIOR
OSi
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
ORSiCl3
Si
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
OHOH
OH
Si
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
OHOH
OH

Chapter 2 __________________________________________________________________________________________________________
22
2.3 Characterisation
2.3.1 NMR spectroscopy
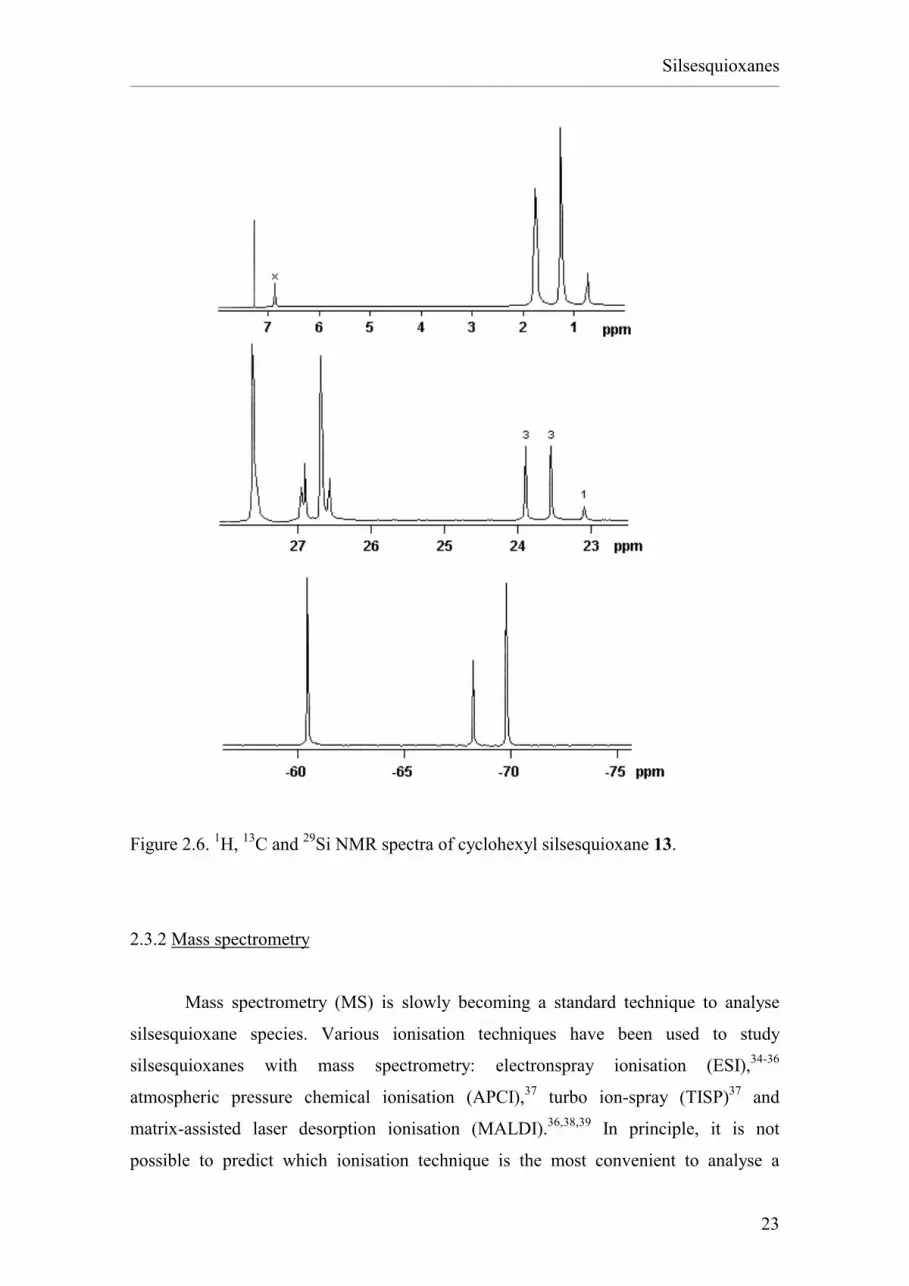
Silsesquioxane structures have at least three groups of nuclei that can be
analysed by NMR techniques: 1H, 13C and 29Si. The NMR spectrum of each of these
three provides useful information for the characterisation of silsesquioxane species. 1H NMR spectroscopy is used to determine the number of silanol groups present in
incompletely condensed silsesquioxanes and to establish if they are hydrogen bonded to
other silanol groups (δ 5 / 7.5 ppm, e.g. the peak marked with × in Figure 2.6) or if they
are isolated (δ 2 / 4 ppm). 13C NMR spectroscopy often allows to distinguish the ipso carbon atoms, i.e. those
bound to a silicon atom, from the other carbons of the R-group. For instance, in the
a7b3 silsesquioxane (c-C6H11)7Si7O9(OH)3 the ipso carbons are in a different region
(δ 22 / 25 ppm) from the remaining CH2 groups (δ 26.5 / 30 ppm) (Figure 2.6).32 29Si NMR spectroscopy is the most useful, though also the most time-consuming, NMR
technique to characterise silsesquioxanes. The 29Si NMR spectrum of a silsesquioxane
provides information about the symmetry of the structure and about the coordination
sphere of the Si atoms. For example, the 29Si NMR spectrum of the a7b3 silsesquioxane
(c-C6H11)7Si7O9(OH)3 displays three peaks with integral ratios 3:1:3, in agreement with
the C3v symmetry of the molecule (Figure 2.6).32 The position of the peaks accounts for
the presence of silanol groups: peaks at lower shifts are due to Si exclusively connected
to other Si atoms through oxygen bridges (δ -68 / -70 ppm), while peaks of Si atoms
connected to an -OH group are at higher shifts (around -60 ppm). More generally, the
position of the peaks also depends on the nature of the R-group connected to the silicon,
while the shift between peaks from silicons with a different number of -OH groups is
approximately constant (~10 ppm per -OH group).33 The presence of geometrically
strained structures also causes the Si peak position to be moved towards higher shifts:
for example, the completely condensed silsesquioxane (c-C6H11)6Si6O9 gives a single
peak at ~-56 ppm, while the less strained (c-C6H11)8Si8O12 gives a single peak at
~-69 ppm.1
Silsesquioxanes __________________________________________________________________________________________________________
23
Figure 2.6. 1H, 13C and 29Si NMR spectra of cyclohexyl silsesquioxane 13.
2.3.2 Mass spectrometry
Mass spectrometry (MS) is slowly becoming a standard technique to analyse
silsesquioxane species. Various ionisation techniques have been used to study
silsesquioxanes with mass spectrometry: electronspray ionisation (ESI),34-36
atmospheric pressure chemical ionisation (APCI),37 turbo ion-spray (TISP)37 and
matrix-assisted laser desorption ionisation (MALDI).36,38,39 In principle, it is not
possible to predict which ionisation technique is the most convenient to analyse a

Chapter 2 __________________________________________________________________________________________________________
24
certain silsesquioxane sample. Advantages of mass spectrometry are its rapidity and the
low amount of sample needed for the analysis (10-50 µg/ml). A drawback is the
impossibility to have an absolute intensity scale, limiting the results to a qualitative
level. For this reason MS is often coupled to other analytic techniques, amongst which
is usually NMR. The applications of MS range from the assignment of structures to
unknown silsesquioxane species1,37 to the study of the species formed during the
synthesis of silsesquioxanes.34,35
2.3.3 IR spectroscopy
Infrared spectroscopy has been applied to study many silsesquioxane structures.
Characteristic vibrations present in the IR spectra of silsesquioxanes are:4 the Si-O-Si
asymmetric stretching, at 1100-1140 cm-1 or at 1057-1085 cm-1 in case of strained
geometry; deformational vibrations of the silicon-oxygen framework, in the region
between 360 and 600 cm-1; the Si-R vibrations, which are strongly influenced by the
nature of the R-group. 6,17,18,40 Recently, the information provided by IR spectroscopy
was used as the basis of a theoretical study of the structure of an organometallic
silsesquioxane complex.41
2.3.4 X-ray diffraction
X-ray single-crystal diffraction is used to obtain a structural characterisation of
silsesquioxane species that can be synthesised in crystalline form, providing information
about bond distances and angles on the basis of which plots of the silsesquioxane
structures can be drawn. Crystallisation of silsesquioxanes is usually difficult and even
apparently suitable crystals often show pronounced disorder.
2.4 Completely condensed silsesquioxanes (RSiO1.5)a
These small, discrete cage compounds are usually referred to as polyhedral
oligomeric silsesquioxanes (POSS). No structure is known with a = 2, probably because
the compound would be geometrically too strained. For a ≥ 4, completely condensed

Silsesquioxanes __________________________________________________________________________________________________________
25
silsesquioxanes with various R-groups have been synthesised and characterised
(Figure 2.1).5
2.4.1 R4Si4O6 (a4b0)
The structure of this silsesquioxane can be represented as a distorted triangular
pyramid (compound 1 in Figure 2.1); the Si-O framework has a Td symmetry. This
structure is very geometrically strained and this makes the silsesquioxane very unlikely
to be formed. In fact, R4Si4O6 silsesquioxane has only been claimed in a very early
publication42 in the case of R = tert-butyl.
2.4.2 R6Si6O9 (a6b0)
The completely condensed silsesquioxanes R6Si6O9 have the shape of a prism,
with two distorted-triangular faces and three distorted-square faces (structure 2, Figure
2.1). The Si-O framework presents a D3h symmetry. The distorted triangular faces
contain three Si atoms, each bridged by three O atoms: this arrangement is somewhat
strained and this accounts for the fact that this silsesquioxane structure is usually found
just as a by-product. This structure has been reported for R = methyl,11,17 ethyl,12 heptyl,
octyl, isononyl,43 phenyl44 and cyclohexyl.6,1.
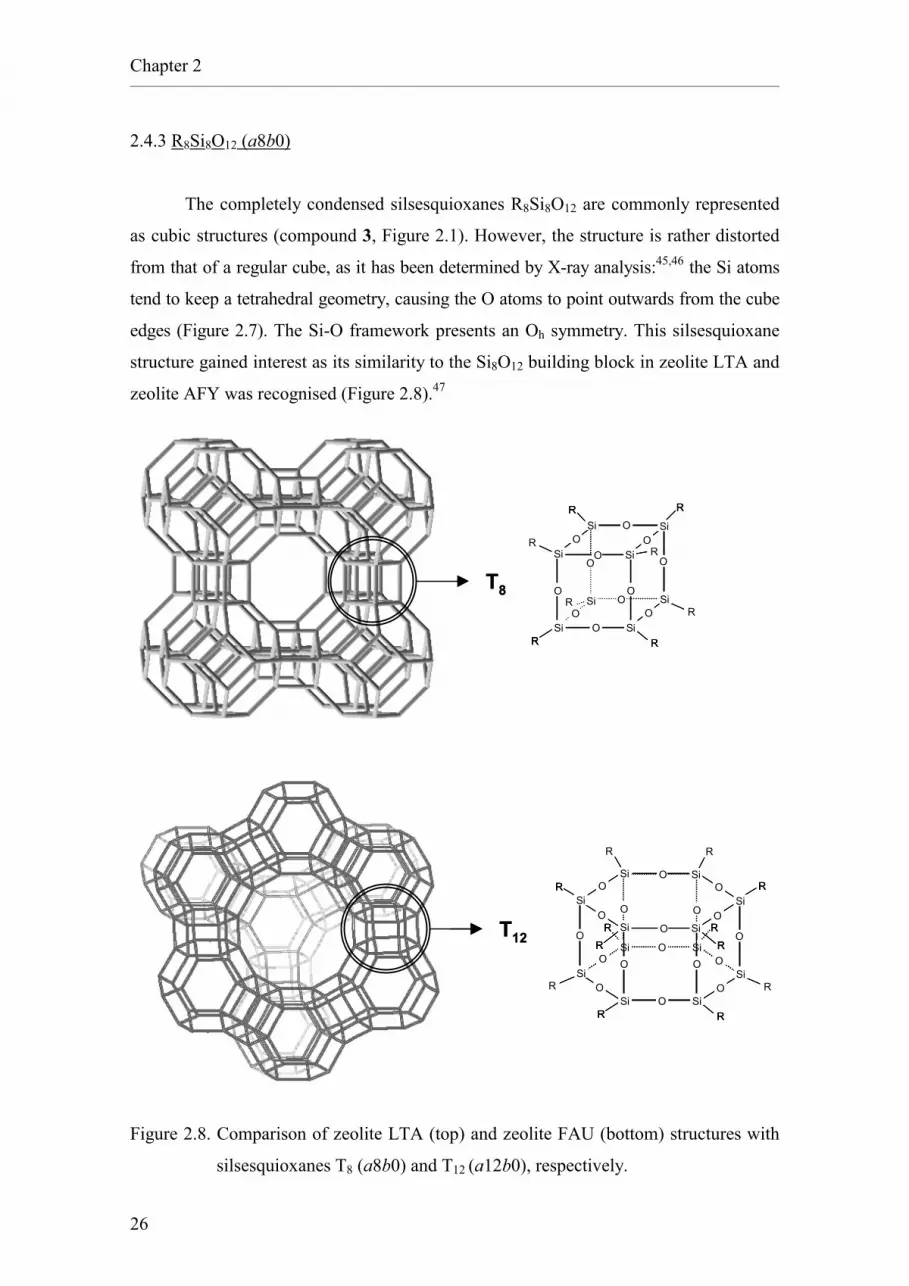
Figure 2.7. X-ray structure of methyl silsesquioxane a8b0.
Chapter 2 __________________________________________________________________________________________________________
26
2.4.3 R8Si8O12 (a8b0)
The completely condensed silsesquioxanes R8Si8O12 are commonly represented
as cubic structures (compound 3, Figure 2.1). However, the structure is rather distorted
from that of a regular cube, as it has been determined by X-ray analysis:45,46 the Si atoms
tend to keep a tetrahedral geometry, causing the O atoms to point outwards from the cube
edges (Figure 2.7). The Si-O framework presents an Oh symmetry. This silsesquioxane
structure gained interest as its similarity to the Si8O12 building block in zeolite LTA and
zeolite AFY was recognised (Figure 2.8).47
Figure 2.8. Comparison of zeolite LTA (top) and zeolite FAU (bottom) structures with
silsesquioxanes T8 (a8b0) and T12 (a12b0), respectively.
T8
T12Si SiO
O
R
RO
R
O
O
R R
O
O
O
R
Si
Si
O
R
R
Si
Si
O
R
SiSi
Si Si
SiSi
RR R
O O
OO
O
O O
O
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
R
T8
T12Si SiO
O
R
RO
R
O
O
R R
O
O
O
R
Si
Si
O
R
R
Si
Si
O
R
SiSi
Si Si
SiSi
RR R
O O
OO
O
O O
O
Si SiO
O
R
RO
R
O
O
R R
O
O
O
R
Si
Si
O
R
R
Si
Si
O
R
SiSi
Si Si
SiSi
RR R
O O
OO
O
O O
O
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
RSiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Si
Si Si
O
O
OO
R
OO
O
R

Silsesquioxanes __________________________________________________________________________________________________________
27
The fact that silsesquioxanes R8Si8O12 could be seen as model compounds for zeolites
encouraged many theoretical calculations of the energies and structural features of these
silsesquioxanes and particularly of H8Si8O12.47-50 Due to the increase in computational
power, the most recent calculations could be performed on an ab initio quantum
mechanical level and were, therefore, more rigorous than the previous ones.49,50 The
total energy of H8Si8O12 was compared to that of other completely condensed
hydrosilsesquioxanes (HSiO1.5)a, showing that hydrosilsesquioxanes with lower values
of a are less stable than H8Si8O12, while those with higher a values have a similar
energy. This same trend was found using different calculation methods (HF49 and
DFT50).
The completely condensed silsesquioxane R8Si8O12 has been obtained either as a main
or as a by-product from the hydrolytic condensation of various monosilanes:
R = hydrogen,18,51 linear alkyl groups (methyl,11,52,53 ethyl,12,52 n-propyl,52 n-butyl,52
n-pentyl,54 n-hexyl43), vinyl,55 cyclohexyl,52 phenyl,13,40,54 benzyl,28 m-tolyl,28
3,5-dimethylphenyl28 and para-substituted phenyl.31 Alternative synthetic methods are
reported in Paragraphs 2.2.3 and 2.2.4. Monosubstituted octasilsesquioxanes RIR7Si8O12
with different R- and RI-groups have also been synthesised.27 Octasilsesquioxanes with
R being a saturated alkyl group are rather stable. More reactive is the Si-H group in the
octahydrosilsesquioxane H8Si8O12, even if still not so reactive when compared to Si-H
groups in non-cyclic silanes.5 H8Si8O12 can be obtained in relatively good yields
(17.5%) by the hydrolytic condensation of HSiCl3 in a biphasic aqueous HCl/hexane
system, using partially hydrated FeCl3 as water source.51 H8Si8O12 can react with
different substrates to give a complete substitution of the H atoms with other groups, i.e.
Cl,56 OCH3,56 OSi(CH3)3,57 OSn(CH3)3,58 OSb(CH3)458 and a number of organic groups
by hydrosilylation reactions (see Paragraph 2.2.3). Another reactive octasilsesquioxane
is (CH2=CH)8Si8O12, where the reactive moiety is the double bond of the vinyl groups.
H8Si8O12 and (CH2=CH)8Si8O12 have been used as precursors for hybrid
organic-inorganic polymers: a detailed description of these polymeric species is outside
the purposes of this dissertation and can be found elsewhere.5,59 H8Si8O12,60-62
(CH2=CH)8Si8O1263-65 and (p-ICH2C6H4)8Si8O12
66 have been used as precursors for
dendrimers with silsesquioxane cores (Figure 2.9). Among these highly-branched
molecules, particularly interesting for their application to catalysis are those containing
phosphine groups that can act as ligands for metal centres.64-68 The dendrimer 17 with
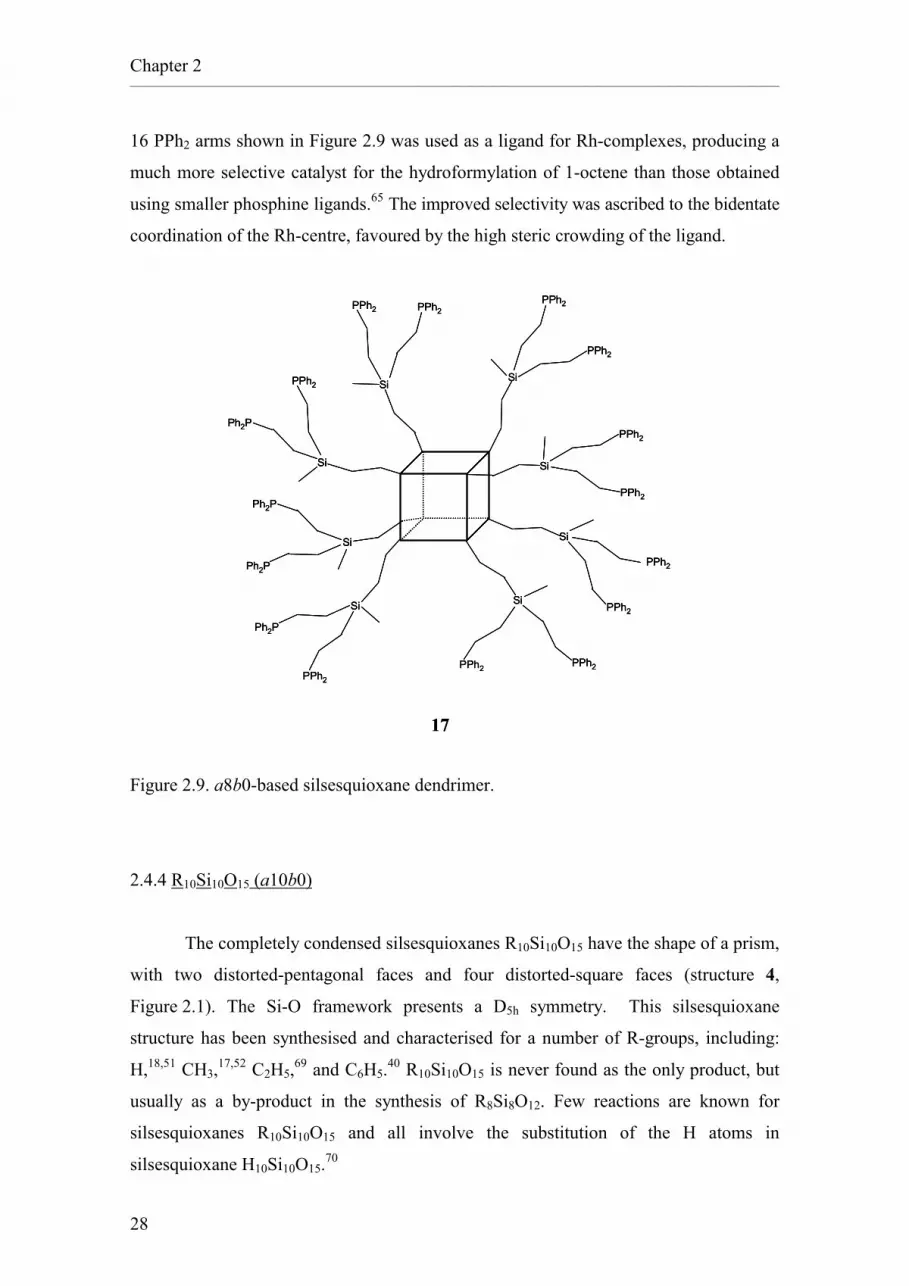
Chapter 2 __________________________________________________________________________________________________________
28
16 PPh2 arms shown in Figure 2.9 was used as a ligand for Rh-complexes, producing a
much more selective catalyst for the hydroformylation of 1-octene than those obtained
using smaller phosphine ligands.65 The improved selectivity was ascribed to the bidentate
coordination of the Rh-centre, favoured by the high steric crowding of the ligand.
Figure 2.9. a8b0-based silsesquioxane dendrimer.
2.4.4 R10Si10O15 (a10b0)
The completely condensed silsesquioxanes R10Si10O15 have the shape of a prism,
with two distorted-pentagonal faces and four distorted-square faces (structure 4,
Figure 2.1). The Si-O framework presents a D5h symmetry. This silsesquioxane
structure has been synthesised and characterised for a number of R-groups, including:
H,18,51 CH3,17,52 C2H5,69 and C6H5.40 R10Si10O15 is never found as the only product, but
usually as a by-product in the synthesis of R8Si8O12. Few reactions are known for
silsesquioxanes R10Si10O15 and all involve the substitution of the H atoms in
silsesquioxane H10Si10O15.70
Si
PPh2PPh2
Si
PPh2
Ph2P
Si
Ph2P
Ph2P
SiSi
Si
Si
Si
PPh2
Ph2P
PPh2 PPh2PPh2
PPh2
PPh2
PPh2
PPh2
PPh2
17
Si
PPh2PPh2
Si
PPh2
Ph2P
Si
Ph2P
Ph2P
SiSi
Si
Si
Si
PPh2
Ph2P
PPh2 PPh2PPh2
PPh2
PPh2
PPh2
PPh2
PPh2Si
PPh2PPh2
Si
PPh2
Ph2P
Si
Ph2P
Ph2P
SiSi
Si
Si
Si
PPh2
Ph2P
PPh2 PPh2PPh2
PPh2
PPh2
PPh2
PPh2
PPh2
17

Silsesquioxanes __________________________________________________________________________________________________________
29
2.4.5 R12Si12O18 (a12b0)
For the completely condensed silsesquioxanes R12Si12O18, two unstrained
structures can be drawn: one has the shape of a prism, with two distorted-hexagonal
faces and six distorted-square faces (structure 5, Figure 2.1), the other consists of four
distorted-pentagonal faces and four distorted-square faces (structure 6, Figure 2.1). The
Si-O framework of 5 has a D6h symmetry, while that of 6 has a D2d symmetry.
Theoretical calculations showed that the two isomeric structures 5 and 6 have very
similar energies.49,50 R12Si12O18 silsesquioxanes have been obtained in small amounts
from the hydrolytic condensation of monosilanes, for R = H18,71, CH317 and C6H5.40
R12Si12O18 structure 5 is interesting since it can be seen as a model compound for a
building block present in zeolite FAU (Figure 2.8).
2.4.6 Higher completely condensed silsesquioxanes (a > 12)
Completely condensed structures with a > 12 are uncommon, though, some
examples have been reported.4,71
2.5 Incompletely condensed silsesquioxanes
Incompletely condensed silsesquioxanes (RSiO1.5)a(H2O)0.5b contain Si-OH
groups (the number of silanol groups is given by the value of b). The presence of these
silanol groups makes these silsesquioxanes suitable as soluble model compounds for
surfaces of silica and silicate-based materials.72,73 Particularly, incompletely condensed
silsesquioxanes with an extensive silicon-oxygen framework (Figure 2.2, structures with
a ≥ 6) should have electronic properties similar enough to those of silica.32 As their
homologues on silica surfaces, the silanol groups on incompletely condensed
silsesquioxanes such as a7b3 can bind metal centres (Figure 2.10). The so-obtained
complexes can be used as model compounds to study active metal sites at the surface of
heterogeneous catalysts at a molecular level.74 Additionally, they may themselves result
in homogeneous catalysts with comparable or even higher activity than their
heterogeneous counterparts.75,76 Analogously to what happens on silica surfaces, silanol

Chapter 2 __________________________________________________________________________________________________________
30
groups on incompletely condensed silsesquioxanes with a rigid framework will retain
their geometry after the insertion of a metal centre, thereby determining the
coordination geometry around the metal atom.32 It can be concluded that incompletely
condensed silsesquioxanes as ligands for metal centres can be excellent models for
silica-supported systems, given their chemical, electronic and geometric similarity.
These silsesquioxanes are usually obtained by the hydrolytic condensation of
monosilanes: the factors that favour the formation of incompletely rather than
completely condensed species are manifold, as discussed in Paragraph 2.2.1. An
alternative synthetic path is the selective cleavage of Si-O-Si bonds described in
Paragraph 2.2.2.
Figure 2.10. Reaction of silsesquioxane 13 and its trimethylsilylated derivatives with
metal centres.
MOL
OL
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si M
Si Si
O
O
OO
L
OO
O
RML4
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OSiMe3
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
Me3SiCl
Me3SiCl
5% Et3N
5% Et3N
ML4
ML4
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OO
O
OSiMe3
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
ML3
MOL
OLMO
L
OL
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OH
OO
O
R
R
R
R
R R
RO
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si M
Si Si
O
O
OO
L
OO
O
RML4
Si
SiSi
Si
Si
Si
Si
OHO
O
OH
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OHO
O
OSiMe3
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
Me3SiCl
Me3SiCl
5% Et3N
5% Et3N
ML4
ML4
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
Si
SiSi
Si
Si
Si
Si
OO
O
OSiMe3
O
OO
OSiMe3
OO
O
R
R
R
R
R R
RO
ML3

Silsesquioxanes __________________________________________________________________________________________________________
31
2.5.1 RSi(OH)3 (a1b3)
This incompletely condensed silsesquioxane (Figure 2.2, compound 7) is
extremely reactive and tends to condense to give higher silsesquioxane species.
Nevertheless, in the case of some rather bulky R-groups, the isolation and
characterisation of the trisilanol monomer has been reported (R = tert-butyl,7 phenyl,8
cyclohexyl,9,77 tris(trimethylsilyl)silyl and tris(trimethylsilyl)methyl10). Typically, the
synthesis of RSi(OH)3 is performed by careful hydrolysis of the corresponding
trichlorosilane RSiCl3 in the presence of aniline as hydrochloric acid acceptor.8
2.5.2 R2Si2O(OH)4 (a2b4)
When silsesquioxanes are synthesised via the hydrolytic condensation of
monosilanes, the first step after the formation of the trisilanol monomer RSi(OH)3 is
supposed to be its condensation to give the dimer R2Si2O(OH)4 (Figure 2.2, compound 8).
As the monomer, in general, this compound is not very stable and tends to condense
further to give higher silsesquioxanes. Just two reports of R2Si2O(OH)4 are known, for
R = tert-butyl78 and cyclohexyl,6 two bulky substituents. In the case of R = tert-butyl, just
two other silsesquioxanes have been reported: the monomer ButSi(OH)37 and the completely
condensed But4Si4O6.42 In the case of R = cyclohexyl, many incompletely and completely
condensed silsesquioxanes have been reported, 1,6,9,52 ranging from the monomer
(c-C6H11)Si(OH)3 to the cubic (c-C6H11)8Si8O12: the preferential formation of any of them
is strongly dependent on the reaction conditions, as described in Paragraph 2.2.1.
2.5.3 R4Si4O4(OH)4 (a4b4)
This incompletely condensed silsesquioxane has been synthesised and isolated
for R = phenyl,13,79 cyclopentyl80 and isopropyl81 as a square planar structure
(Figure 2.2, compound 9). (C6H5)4Si4O4(OH)4 and (c-C5H9)4Si4O4(OH)4 are obtained
via the hydrolytic condensation of the respective trichlorosilanes, while the method for
the synthesis of (i-C3H7)4Si4O4(OH)4 is somewhat more complex:
(isopropyl)diphenylchlorosilane first undergoes a hydrolytic condensation, followed by
the substitution of the phenyl groups with chlorine atoms and, finally, by a second

Chapter 2 __________________________________________________________________________________________________________
32
hydrolytic condensation. In principle, different isomers of the R4Si4O4(OH)4 structure
are possible, depending on the relative position of the four OH groups. A detailed
characterisation by NMR and X-ray allowed to determine that the actual structure is the
cis-cis-cis one, with all the OH groups on the same side of the plane of the
molecule.79,81 This result is supported by quantum mechanical calculations which
suggest that such structure is stabilised by the hydrogen bonding among the four OH
groups.15 Both (C6H5)4Si4O4(OH)4 and (i-C3H7)4Si4O4(OH)4 have been used as
precursors to synthesise other silsesquioxanes, including completely condensed and
polymeric species.79,81
2.5.4 R6Si6O7(OH)4 (a6b4)
Three incompletely condensed silsesquioxanes corresponding to the formula
R6Si6O7(OH)4 have been isolated and characterised, with R = cyclohexyl,24
cycloheptyl16 and norbornyl.82 Like for all the other mentioned incompletely condensed
silsesquioxanes, the organic groups are rather bulky. The framework of these
silsesquioxane structures presents a C2v symmetry and can be described as two Si4O4
planes sharing an edge or as an R6Si6O9 structure with two hydrolysed Si-O-Si bonds
(Figure 2.2, compound 11). (c-C6H11)6Si6O7(OH)4 is obtained by the base-catalysed
cleavage of silsesquioxane (c-C6H11)Si6O9.24 Both the cycloheptyl and the norbornyl
structures are obtained via the hydrolytic condensation of the corresponding
trichlorosilanes. R6Si6O7(OH)4 is not the only silsesquioxane product: in the case of
cycloheptyl, (c-C7H13)6Si6O7(OH)4 [~40%] is obtained together with
(c-C7H13)7Si7O9(OH)3 [~60%]; in the case of norbornyl, many other silsesquioxanes
besides (C7H11)6Si6O7(OH)4 are synthesised. (c-C7H13)6Si6O7(OH)4 has recently found
application as ligand for a water-stable titanium-catalyst that is claimed to be active in
the epoxidation of alkenes with both TBHP and H2O2.72 The catalyst is obtained by
reacting TiCl4 with (c-C7H13)6Si6O7(OH)4; the resulting cluster contains four titanium
atoms and three silsesquioxane units, as determined by X-ray analysis.

Silsesquioxanes __________________________________________________________________________________________________________
33
2.5.5 R7Si7O9(OH)3 (a7b3)
This incompletely condensed silsesquioxane has been synthesised in significant
quantities, isolated and characterised for three bulky, cyclic R-groups: cyclopentyl,16
cyclohexyl1,6 and cycloheptyl.16 The framework of this silsesquioxane is characterised
by a C3v symmetry and can be described as a cubic R8Si8O12 structure with a missing
corner (Figure 2.2, compound 13).
R7Si7O9(OH)3 is the silsesquioxane structure with the largest number of applications as
a ligand for metal centres and as a model compound for silica and silicate-based
materials surfaces. The compound with R = cyclohexyl, (c-C6H11)7Si7O9(OH)3, was first
synthesised and studied by Brown and Vogt.6 It was prepared by the hydrolytic
condensation of the trichlorosilane (c-C6H11)SiCl3 in aqueous acetone. The reaction was
extremely slow and proceeded for approximately 3 years to give a final yield in
(c-C6H11)7Si7O9(OH)3 of 60-70%. Given the interest of this silsesquioxane structure,
Feher et al. studied in detail the same reaction for cyclohexyl as well as for cyclopentyl
and cycloheptyl as R-groups,1,16 and were able to isolate and identify some other
silsesquioxane structures formed in smaller amounts together with the main product
R7Si7O9(OH)3. In the case of R = cyclopentyl, the a7b3 silsesquioxane was almost the
only product and the synthesis was optimised by refluxing the reaction mixture to give a
yield of 29% in pure (c-C5H9)7Si7O9(OH)3 after 3 days.16 A further optimisation of the
synthesis of (c-C5H9)7Si7O9(OH)3 has been realised on the basis of High-Speed
Experimentation results20 by using acetonitrile instead of acetone as a solvent, thus
getting to a yield of 64% in 18 hours.19
An alternative way to synthesise R7Si7O9(OH)3 is by the base-mediated cleavage of
R8Si8O12.25
Silsesquioxane R7Si7O9(OH)3 contains three adjacent silanol groups that can react as a
tridentate ligand with a metal centre to give a complex (Figure 2.10). In some cases, it is
preferred to have the silsesquioxane doubly or even just singly coordinated to the metal
centre. For this purpose, it can be useful to let one or two of the silanol groups react
beforehand with trimethylchlorosilane [TMSCl, (CH3)3SiCl] (Figure 2.10). The
silylation of R7Si7O9(OH)3 was studied by Feher and Newman, providing also a
mechanistic study for the silylation of silica surfaces.83 It was found that silylation is
favoured for mutually hydrogen-bonded silanol groups with respect to isolated, not

Chapter 2 __________________________________________________________________________________________________________
34
hydrogen-bonded silanols. Moreover, it was possible to selectively silylate just one of
the three silanols of R7Si7O9(OH)3, particularly if the reaction was carried out in the
presence of an amine base like Et3N. Successive silylations were not as selective and
afforded a mixture of the di- and trisilylated silsesquioxanes.
An increasing number of reactions involving the reaction of R7Si7O9(OH)3
silsesquioxane and its silylated derivatives with metal centres have been reported in the
last years. Several of these metallasilsesquioxanes find applications as model
compounds for metal centres grafted on the surface of silica and silicate-based
heterogeneous catalysts and as homogeneous catalysts themselves.
2.5.5.1 Metallasilsesquioxane catalysts for the epoxidation of alkenes
In 1994 Maschmeyer et al.84 reported the synthesis of (c-C6H11)7Si7O12Ti(η5-C5Ph5)
(Figure 2.11, 18a) from (η5-C5Ph5)2TiCl2 and the trisilanol (c-C6H11)7Si7O9(OH)3 which
led in 1995 to the highly successful grafting procedure of Ti(η5-C5H5)2Cl2 onto silica
MCM-41, yielding the most active (per mole of titanium) heterogeneous epoxidation
catalyst of alkenes with organic hydroperoxides to date.85
Similarly, when Ti(η5-C5H5)Cl3 was reacted with the trisilanol (c-C6H11)7Si7O9(OH)3 the
titanium silsesquioxane (c-C6H11)7Si7O12Ti(η5-C5H5) was produced (Figure 2.11, 18b).86,87
In 1997 Abbenhuis et al.88 tested this complex as catalyst for the epoxidation of alkenes
with tert-butyl hydroperoxide (TBHP) as the oxidant, obtaining good conversions and
selectivities for a number of substrates. The catalyst was reported not to leach the titanium
centre nor the cyclopentadienyl ligand during the epoxidation reaction and has a remarkable
stability towards hydrolysis even in 1M HCl solution. In order to heterogenise this active
homogeneous catalyst, (c-C6H11)7Si7O12Ti(η5-C5H5) was immobilised on the channels of
MCM-41 via a straightforward adsorption.89 The heterogeneous catalyst was tested for
epoxidation showing a good activity, even if lower than that of the homogeneous
(c-C6H11)7Si7O12Ti(η5-C5H5). Leaching of the Ti-silsesquioxane occurred just for Al-containing
MCM-41 samples. The adsorption of (c-C6H11)7Si7O12Ti(η5-C5H5) on all-silica MCM-41
was investigated by spectroscopic techniques, demonstrating that the Ti-silsesquioxane is
unaltered as adsorbed species and suggesting that the catalytically-active Ti-centre points
towards the centre of the MCM-41 pore.90 The interaction between the silsesquioxane
complex and the MCM-41 surface takes place through the cyclohexyl groups.
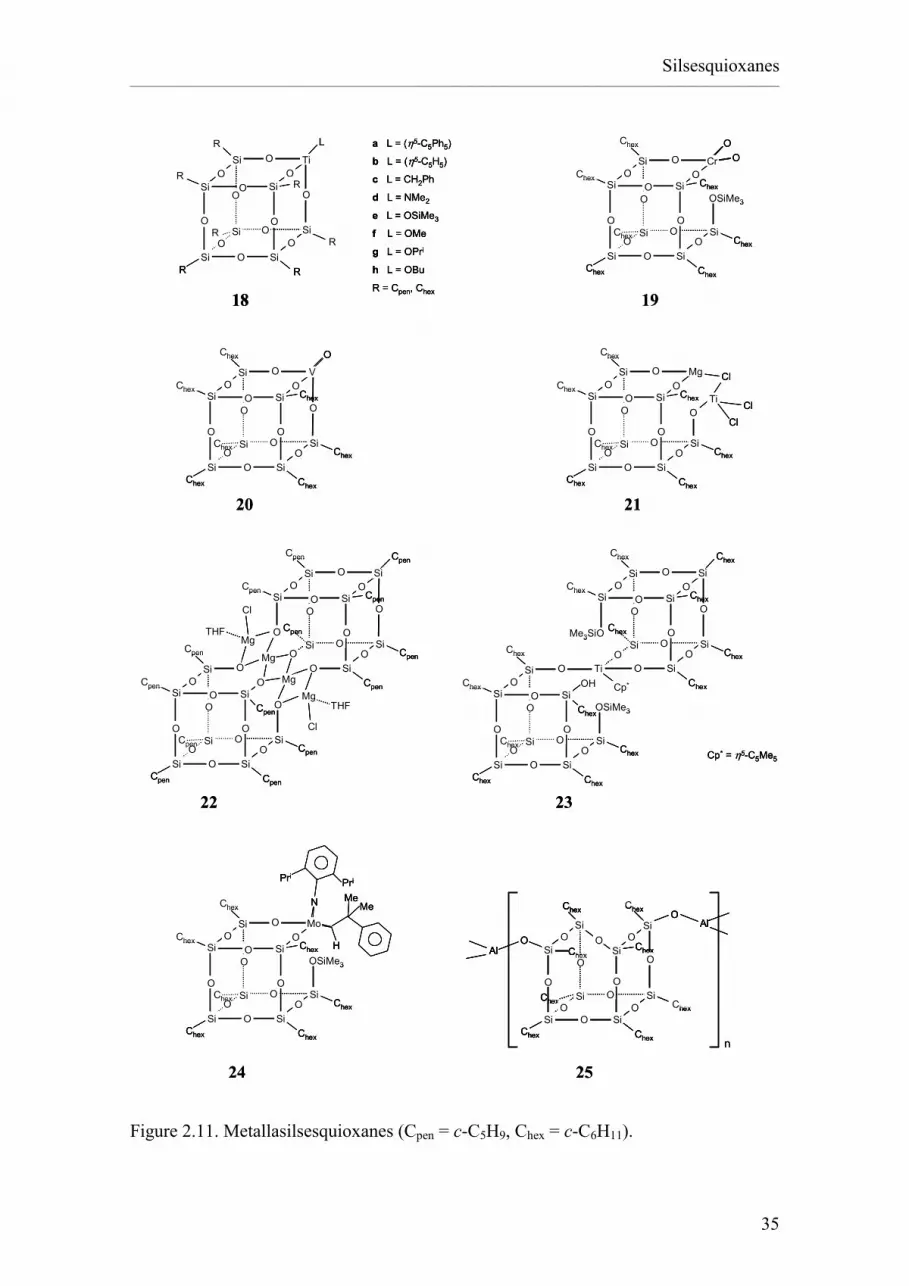
Silsesquioxanes __________________________________________________________________________________________________________
35
Figure 2.11. Metallasilsesquioxanes (Cpen = c-C5H9, Chex = c-C6H11).
CrOO
OOSi
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
VOO
OSi
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
O
MgOO
Si
SiSi
Si
Si
Si
Si
OO
O
OO
O
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
Ti
Cl
ClCl
MoOO
N
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
H
MeMe
PriPri
Si
Si
O
OO
OSi
SiO
OO
Cpen
Cpen
Cpen
OCpen
OSi
Si
SiO
O Cpen
Cpen
Cpen
O
SiOO
Si
SiSi
Si
Si
Si
OO
O
OOO
O
O
Cpen
Cpen
Cpen
Cpen
Cpen
Cpen
O
Cpen
O
Mg
Mg
Mg
Mg
Cl
THF
THF
Cl
OOH
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
SiOO
Si
SiSi
Si
Si
Si
OO
O
OO
O
O
Chex
Chex
Chex
Chex
Chex
Chex
O
Chex
O
Ti
Me3SiO
Cp*
Cp* = η5-C5Me5
Chex
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
O
Chex
Chex
Chex
O
Chex
ChexO
Chex
Chex
Al
Al
n
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Ti
Si Si
O
O
OO
L
OO
O
R
a L = (η5-C5Ph5)
b L = (η5-C5H5)
c L = CH2Ph
d L = NMe2
e L = OSiMe3
f L = OMe
g L = OPri
h L = OBu
R = Cpen, Chex18 19
20 21
22 23
24 25
CrOO
OOSi
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
CrOO
OOSi
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
VOO
OSi
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
O
VOO
OSi
SiSi
Si
Si
Si
Si
OO
O
OOO
O
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
O
MgOO
Si
SiSi
Si
Si
Si
Si
OO
O
OO
O
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
Ti
Cl
ClCl
MgOO
Si
SiSi
Si
Si
Si
Si
OO
O
OO
O
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
Ti
Cl
ClCl
MoOO
N
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
H
MeMe
PriPri
MoOO
N
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
H
MeMe
PriPri
Si
Si
O
OO
OSi
SiO
OO
Cpen
Cpen
Cpen
OCpen
OSi
Si
SiO
O Cpen
Cpen
Cpen
O
SiOO
Si
SiSi
Si
Si
Si
OO
O
OOO
O
O
Cpen
Cpen
Cpen
Cpen
Cpen
Cpen
O
Cpen
O
Mg
Mg
Mg
Mg
Cl
THF
THF
Cl
Si
Si
O
OO
OSi
SiO
OO
Cpen
Cpen
Cpen
OCpen
OSi
Si
SiO
O Cpen
Cpen
Cpen
O
SiOO
Si
SiSi
Si
Si
Si
OO
O
OOO
O
O
Cpen
Cpen
Cpen
Cpen
Cpen
Cpen
O
Cpen
O
Mg
Mg
Mg
Mg
Cl
THF
THF
Cl
OOH
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
SiOO
Si
SiSi
Si
Si
Si
OO
O
OO
O
O
Chex
Chex
Chex
Chex
Chex
Chex
O
Chex
O
Ti
Me3SiO
Cp*
Cp* = η5-C5Me5
OOH
Si
SiSi
Si
Si
Si
Si
OO
O
OO
OSiMe3
OO
O
Chex
Chex
Chex
Chex
Chex Chex
OChex
SiOO
Si
SiSi
Si
Si
Si
OO
O
OO
O
O
Chex
Chex
Chex
Chex
Chex
Chex
O
Chex
O
Ti
Me3SiO
Cp*
Cp* = η5-C5Me5
Chex
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
O
Chex
Chex
Chex
O
Chex
ChexO
Chex
Chex
Al
Al
nChex
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
O
Chex
Chex
Chex
O
Chex
ChexO
Chex
Chex
Al
Al
Chex
O
Si Si
SiSi
Si
Si
Si
Si
O O O
O
OO
O
O
O
O
Chex
Chex
Chex
O
Chex
ChexO
Chex
Chex
Al
Al
n
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Ti
Si Si
O
O
OO
L
OO
O
R
a L = (η5-C5Ph5)
b L = (η5-C5H5)
c L = CH2Ph
d L = NMe2
e L = OSiMe3
f L = OMe
g L = OPri
h L = OBu
R = Cpen, Chex18
SiSi
Si Si
O
O
O
O R
R
R
R R
RO
Si Ti
Si Si
O
O
OO
L
OO
O
R
a L = (η5-C5Ph5)
b L = (η5-C5H5)
c L = CH2Ph
d L = NMe2
e L = OSiMe3
f L = OMe
g L = OPri
h L = OBu
R = Cpen, Chex18 19
20 21
22 23
24 25

Chapter 2 __________________________________________________________________________________________________________
36
Silsesquioxanes (c-C6H11)7Si7O9(OH)3 and (c-C5H9)7Si7O9(OH)3 were also reacted with
a variety of TiL4 centres (L = CH2Ph, NMe2, OSiMe3, OMe, OPri, OBu) to give
R7Si7O12TiL complexes that were studied as homogeneous epoxidation catalysts and as
model compounds for isolated titanium centres on silicate materials like TS-1 and
MCM-41 (Figure 2.11, 18c-h).74,76,91 All the catalysts were tested for the epoxidation of
1-octene with TBHP: the highest activities were found for L = alkoxy group. The
activities of these catalysts are reported to be comparable to or higher than those of
Ti-MCM-41 (on the basis of turnover frequencies). (c-C5H9)7Si7O12TiOPri is found to
exist as an equilibrium between two forms in solution: a monomer with
tetra-coordinated titanium and a dimer with penta-coordinated titanium.91 In order to
investigate also monodentate and bidentate Ti-silsesquioxanes as models for analogous
species on silicate surfaces, the mono- and disilylated silsesquioxanes
R7Si7O9(OSiMe3)(OH)2 and R7Si7O9(OSiMe3)2OH (R = cyclohexyl, cyclopentyl) were
reacted with Ti-centres.76,92 Studies of the activity of these complexes allowed to
determine that tridentate Ti-silsesquioxanes are more active catalysts than complexes
with lower coordination number of siloxy groups. Moreover, it was shown that
OSi(CH3)3 can weakly coordinate to the Ti-centre, diminishing the access to the active
site and consequently lowering the activity.92 A heterogeneous derivative of a
Ti-complex of (c-C6H11)7Si7O9(OH)3 was prepared by modifying MCM-41 with
(3-glycidyloxypropyl)trimethoxysilane followed by reaction of the oxirane ring with
deprotonated (c-C6H11)7Si7O9(OH)3 and finally by reaction with Ti(OBu)4.93 The
heterogeneous material so-obtained was characterised and tested as catalyst for the
epoxidation of cyclooctene, showing promising results when using H2O2 as oxidant.
2.5.5.2 Metallasilsesquioxane catalysts for the polymerisation of alkenes
The reaction of the disilanol (c-C6H11)7Si7O9(OSiMe3)(OH)2 with CrO3 in the presence
of MgSO4 as dehydrating agent yields the chromium silsesquioxane
(c-C6H11)7Si7O9(OSiMe3)(O2CrO2) (Figure 2.11, 19).94 This complex was found to
catalyse the polymerisation of ethene when activated with Al(CH3)3.
(c-C6H11)7Si7O9(OSiCH3)(O2CrO2) was also proposed as a model compound for the
Phillips catalyst (CrO3 supported on silica or alumina), which is industrially used for the
polymerisation of ethene.

Silsesquioxanes __________________________________________________________________________________________________________
37
Vanadium silsesquioxane (c-C6H11)7Si7O12VO (Figure 2.11, 20) is obtained by the
reaction of (c-C6H11)7Si7O9(OH)3 with (C3H7O)3VO.95 In the presence of Al(CH3)3 or
Al(CH2SiMe3)3 as an activating agent, (c-C6H11)7Si7O12VO catalyses the polymerisation
of ethene and, to a lesser extent, of other alkenes.95,96 A study of the catalytically active
site involving the coordination of the alkylaluminium to the vanadium silsesquioxane
was performed by means of 1H, 13C, 17O, 29Si and 51V NMR spectroscopy.97
The reaction of (c-C6H11)7Si7O9(OH)3 with BuMgEt produces
[(c-C6H11)7Si7O9(OH)O2Mg]n (n = 1,2), which further reacts with TiCl4 to give
[(c-C6H11)7Si7O12MgTiCl3]n (n = 1,2) as a mixture of monomer and dimer (Figure 2.11,
21).75 [(c-C6H11)7Si7O12MgTiCl3]n was studied as a model compound for a
silica-supported catalyst having the same magnesium and titanium content. Remarkably,
the silsesquioxane-based homogeneous catalyst showed a higher activity (per gram of
titanium) than its Ti/Mg/SiO2 heterogeneous counterpart for the polymerisation of
ethene in the presence of Al(CH3)3.
Reaction of (c-C5H9)7Si7O9(OH)3 and its silylated derivatives with Group 4 (Ti, Zr, Hf)
metal centres produced a series of silsesquioxane-based complexes with different
coordination number between the metal and the silsesquioxane structure(s).98-100 These
compounds were used as models for silica-grafted homogeneous alkene-polymerisation
catalysts. Nearly all the prepared silsesquioxane catalysts were active in the
polymerisation of ethene when using methylalumoxane as co-catalyst. Nevertheless, the
metal centre on the silsesquioxane is easily substituted by methylalumoxane and the
so-obtained complex results in the active catalyst.100 When using B(C6F5)3 as milder
cocatalyst, no leaching of the metal centre was registered; still, some of the
metallasilsesquioxanes showed good polymerisation activities.100,101
2.5.5.3 Other metallasilsesquioxanes
The reaction of (c-C5H9)7Si7O9(OH)3 with CH3MgCl in THF gives a complex
containing four Mg atoms and two silsesquioxane ligands (Figure 2.11, 22).102 From
X-ray analysis it was found that the magnesium-chlorine bonds are unusually short,
indicating highly electron deficient magnesium atoms. The complex has been subjected
to transmetalation reactions, showing a low activity.
Aluminosilsesquioxanes as model compounds for acidic sites in zeolites or on silica
surfaces have been obtained by reacting AlMe3 or AlEt3 with (c-C5H9)7Si7O9(OH)3 and

Chapter 2 __________________________________________________________________________________________________________
38
(c-C5H9)7Si7O9(OSiR3)(OH)2, where SiR3 = SiMe3 or SiMe2Ph.103-105 Products have
been characterised by spectroscopic and X-ray techniques.
A series of titanium silsesquioxanes were obtained by the reaction of
(c-C6H11)7Si7O9(OH)3 and its mono- and disilylated derivatives with a number of Ti(III)
and Ti(IV)-centres.106-108 Particularly interesting seems to be the complex obtained by
reacting (c-C6H11)7Si7O9(OSiMe3)(OH)2 with (η5-C5Me5)Ti(η5,η1-C5Me4CH2).107 This
structure contains a Ti-centre coordinated to two silsesquioxane ligands and to a vicinal
Si-OH group (Figure 2.11, 23): this arrangement makes the complex an interesting
model compound for a silica surface Ti-site. However, no catalytic test has been yet
reported for this system.
A number of iron silsesquioxanes has been synthesised by reacting
(c-C5H9)7Si7O9(OH)3 and (c-C5H9)7Si7O9(OSiMe3)(OH)2 with FeCl3P(c-C6H11)3 and
FeCl2(dcpe), where dcpe = bis(dicyclohexylphosphino)ethane.109 Phosphine ligands are
used to prevent oligomerisation of the Fe-centres. These iron silsesquioxanes could be
seen as models for iron zeolites catalysts. Nevertheless, the first attempts to check the
activity of these complexes in the oxidation of benzene were not successful.
The reaction of (c-C6H11)7Si7O9(OSiMe3)(OH)2 with two equivalents of TlOEt affords
the complex (c-C6H11)7Si7O9(OSiMe3)(OTl)2. This compound was then used as
transmetalating agent to obtain (c-C6H11)7Si7O9(OSiMe3)(O2Mo)(CHCMe2Ph)(NAr),
where Ar = 2,6-di-isopropylphenyl) (Figure 2.11, 24).110 The molybdenum
silsesquioxane complex turned out to be a very efficient catalyst for the metathesis of
various alkenes.
Finally, (c-C6H11)7Si7O9(OH)3, (c-C5H9)7Si7O9(OH)3 and related structures have been
reacted with a number of other metal centres,32,86 including Cu,111 Rh,112 W,113 Os,114
Pt,115 Nd and Y116. None of these complexes has yet been tested for catalytic
performance.
2.5.6 R8Si8O11(OH)2 (a8b2)
Two different incompletely condensed silsesquioxane structures correspond to
the general formula R8Si8O11(OH)2 (Figure 2.2, compounds 14 and 15).
Silsesquioxane 14 has been obtained with low yields for R = cyclohexyl as a by-product
of the synthesis of (c-C6H11)7Si7O9(OH)3.1 Since the two silanol groups of this

Silsesquioxanes __________________________________________________________________________________________________________
39
(c-C6H11)8Si8O11(OH)2 silsesquioxane point in opposite directions, the compound can
be used as a bifunctional monodentate ligand for metal species to obtain polymeric
complexes with bridging metal units (Figure 2.11, 25). Reaction of compound 14 with
Al(CH3)3 produced a gel that results in an active heterogeneous catalyst for Diels-Alder
reactions of enones.117 A similar polymeric species is obtained by reacting
compound 14 with TiCl4 or Ti(CH2Ph)4; when tested as catalyst for the epoxidation of
alkenes, the titanium-containing gel undergoes hydrolysis resulting in the reformation of
silsesquioxane 14 and catalytically active Ti-species.88
Silsesquioxane 15 can be obtained by the selective cleavage of a Si-O-Si bond of
R8Si8O12 structures, as described in Paragraph 2.2.2.2,22,25 R8Si8O11(OH)2
silsesquioxane 15 with R = cyclopentyl, cyclohexyl has been used as a ligand for
Al-complexes104 and Ce-complexes.118
2.5.7 Other incompletely condensed structures (a6b2, a7b1, a8b4)
Incompletely condensed silsesquioxanes R6Si6O8(OH)2 and R8Si8O10(OH)4
(Figure 2.2, 10 and 16) have been synthesised by the cleavage of Si-O bonds of,
respectively, R6Si6O923 and R8Si8O12.25 No application of these two structures have been
reported so far. R7Si7O10(OH) silsesquioxanes (Figure 2.2, 12) can be obtained by
dehydration of R7Si7O9(OH)3.1 Few applications of this silsesquioxane, with
R = cyclopentyl or cyclohexyl, have been reported.21,86,92
2.6 Conclusions
The field of oligosilsesquioxane chemistry has expanded significantly over the
last 10 years yielding exciting developments in their synthesis as well as in their
application. It can be expected that these compounds, having left their ‘niche’ existence
(as demonstrated above), will make an increasingly large contribution to new
developments in the fundamental understanding of chemical phenomena (when used as
models for, e.g., zeolites or silica surface chemistry) and to novel applications which are
based on them, either as catalysts or composite material building blocks operating at the
mesostructure level.

Chapter 2 __________________________________________________________________________________________________________
40
References
1 F.J. Feher, D.A. Newman, J.F. Walzer, J. Am. Chem. Soc., 1989, 111, 1741. 2 F.J. Feher, D. Soulivong, A.G. Eklund, Chem. Commun., 1998, 399. 3 R.H. Baney, M. Itoh, A. Sakakibara, T. Suzuki, Chem. Rev., 1995, 95, 1409. 4 M.G. Voronkov, V.I. Lavrent’yev, Top. Curr. Chem., 1982, 102, 199. 5 P.G. Harrison, J. Organomet. Chem., 1997, 542, 141. 6 J.F. Brown, L.H. Vogt, J. Am. Chem. Soc., 1965, 87, 4313. 7 N. Winkhofer, H.W. Roesky, M. Noltemeyer, W.T. Robinson, Angew. Chem. Int. Ed., 1992, 31, 599. 8 T. Takiguchi, J. Am. Chem. Soc., 1959, 81, 2359. 9 H. Ishida, J.L. Koenig, J. Polym. Sci. Polym. Phys. Ed., 1979, 17, 1807. 10 S.S. Al-Juaid, N.H. Buttrus, R.I. Damja, Y. Derouiche, C. Eaborn, P.B. Hitchcock, P.D. Lickiss, J.
Organomet. Chem., 1989, 371, 287. 11 M.M. Sprung, F.O. Guenther, J. Am. Chem. Soc., 1955, 77, 3990. 12 M.M. Sprung, F.O. Guenther, J. Am. Chem. Soc., 1955, 77, 3996. 13 J.F. Brown, J. Am. Chem. Soc., 1965, 87, 4317. 14 T. Kudo, M.S. Gordon, J. Am. Chem. Soc., 1998, 120, 11432. 15 T. Kudo, M.S. Gordon, J. Phys. Chem. A, 2000, 104, 4058. 16 F.J. Feher, T.A. Budzichowski, R.L. Blanski, K.J. Weller, J.W. Ziller, Organometallics, 1991, 10,
2526. 17 L.H. Vogt, J.F. Brown, Inorg. Chem., 1963, 2, 189. 18 C.L. Frye, W.T. Collins, J. Am. Chem. Soc., 1970, 92, 5586. 19 See Chapter 5. 20 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Angew. Chem. Int. Ed., 2001, 40,
740. 21 F.J. Feher, D. Soulivong, G.T. Lewis, J. Am. Chem. Soc., 1997, 119, 11323. 22 F.J. Feher, D. Soulivong, F. Nguyen, Chem. Commun., 1998, 1279. 23 F.J. Feher, F. Nguyen, D. Soulivong, J.W. Ziller, Chem. Commun., 1999, 1705. 24 F.J. Feher, R. Terroba, J.W. Ziller, Chem. Commun., 1999, 2153. 25 F.J. Feher, R. Terroba, J.W. Ziller, Chem. Commun., 1999, 2309. 26 D. Herren, H. Bürgy, G. Calzaferri, Helv. Chim. Acta, 1991, 74, 24. 27 C. Marcolli, G. Calzaferri, Appl. Organometal., 1999, 13, 213. 28 F.J. Feher, T.A. Budzichowski, J. Organomet. Chem., 1989, 373, 153. 29 K. Olsson, C. Axen, Arkiv. Kemi., 1964, 22, 237. 30 K. Olsson, C. Gronwall, Arkiv. Kemi., 1961, 17, 529. 31 F.J. Feher, T.A. Budzichowski, J. Organomet. Chem., 1989, 379, 33. 32 F.J. Feher, T.A. Budzichowski, Polyhedron, 1995, 14, 3239. 33 M.P. Besland, C. Guizard, N. Hovnanian, A. Larbot, L. Cot, J. Sanz, I. Sobrados, M. Gregorkiewitz, J.
Am. Chem. Soc., 1991, 113, 1982.

Silsesquioxanes __________________________________________________________________________________________________________
41
34 P. Bussian, F. Sobott, B. Brutschy, W. Scrader, F. Schüth, Angew. Chem. Int. Ed., 2000, 39, 3901. 35 L. Matejka, O. Dukh, J. Brus, W.J. Simonsick jr., B. Meissner, J. Non-Cryst. Solids, 2000, 270, 34. 36 D.P. Fasce, R.J.J. Williams, R. Erra-Balsells, Y. Ishikawa, H. Nonami, Macromolecules, 2001, 34,
3534. 37 R. Bakhtiar, F.J. Feher, Rapid Commun. Mass Spectrom., 1999, 13, 687. 38 W.E. Wallace, C.M. Guttman, J.M. Antonucci, J. Am. Soc. Mass Spectrom., 1999, 10, 224. 39 P. Eisenberg, R. Erra-Balsells, Y. Ishikawa, J.C. Lucas, A.N. Mauri, H. Nonami, C.C. Riccardi, R.J.J.
Williams, Macromolecules, 2000, 33, 1940. 40 J.F. Brown, L.H. Vogt, P.I. Prescott, J. Am. Chem. Soc., 1964, 86, 1120. 41 I.E. Davidova, L.A. Gribov, I.V. Maslov, V. Dufaud, G.P. Niccolai, F. Bayard, J.M. Basset, J. Mol.
Struct., 1998, 443, 67. 42 E. Wiberg, W. Simmler, Z. Anorg. Allg. Chem., 1955, 282, 330. 43 K.A. Adrianov, B.A. Izmaylov, J. Organomet. Chem., 1967, 8, 435. 44 K.A. Adrianov, V.A. Odinets, Izv. Akad. Nauk SSSR Otd. Khim. Nauk, 1959, 460. 45 M.A. Hossain, M.B. Hursthouse, K.M.A. Malik, Acta Cryst., 1979, B35, 2258. 46 G. Koellner, U. Müller, Acta Cryst., 1989, C45, 1106. 47 G. Calzaferri, R. Hoffmann, J. Chem. Soc., Dalton Trans., 1991, 917. 48 C.W. Earley, Inorg. Chem., 1992, 31, 1250. 49 C.W. Earley, J. Phys. Chem., 1994, 98, 8693. 50 K.-H. Xiang, R. Pandey, U.C. Pernisz, C. Freeman, J. Phys. Chem. B, 1998, 102, 8704. 51 P.A. Agaskar, Inorg. Chem., 1991, 30, 2707. 52 A.J. Barry, W.H. Daudt, J.J. Domicone, J.W. Gilkey, J. Am. Chem. Soc., 1955, 77, 4248. 53 M.M. Sprung, F.O. Guenther, J. Am. Chem. Soc., 1955, 77, 6045. 54 M.M. Sprung, F.O. Guenther, J. Polym. Sci., 1958, 28, 17. 55 T.N. Martynova, V.P. Korchkov, Zh. Obshch. Khim., 1982, 52, 1585. 56 V.W. Day, W.G. Klemperer, V.V. Mainz, D.M. Millar, J. Am. Chem. Soc., 1985, 107, 8262. 57 F.J. Feher, K.J. Weller, Organometallics, 1990, 9, 2638. 58 F.J. Feher, K.J. Weller, Inorg. Chem., 1991, 30, 880. 59 J.D. Lichtenhan, Comments Inorg. Chem., 1995, 17, 115. 60 A.R. Bassindale, T.E. Gentle, J. Mater. Chem., 1993, 3, 1319. 61 A.R. Bassindale, T.E. Gentle, J. Organomet. Chem., 1996, 521, 391. 62 F.J. Feher, K.D. Wyndham, Chem. Commun., 1998, 323. 63 P.-A. Jaffrès, R.E. Morris, J. Chem. Soc., Dalton Trans., 1998, 2767. 64 L. Ropartz, R.E. Morris, G.P. Schwarz, D.F. Foster, D.J. Cole-Hamilton, Inorg. Chem. Commun., 2000,
3, 714. 65 L. Ropartz, R.E. Morris, , D.F. Foster, D.J. Cole-Hamilton, Chem. Commun., 2001, 361. 66 F.J. Feher, J.J. Schwab, S.H. Phillips, A. Eklund, E. Martinez, Organometallics, 1995, 14, 4452. 67 B. Hong, T.P.S. Thomas, H.J. Murfee, M.J. Lebrun, Inorg. Chem., 1997, 36, 6146. 68 H.J. Murfee, T.P.S. Thomas, B. Hong, J. Greaves, Inorg. Chem., 2000, 39, 5209.

Chapter 2 __________________________________________________________________________________________________________
42
69 M.G. Voronkov, V.I. Lavrent’yev, V.M. Kovrigin, J. Organomet. Chem., 1981, 220, 285. 70 P.A.. Agaskar, Synth. React. Inorg. Met. Org. Chem., 1990, 20, 483. 71 P.A.. Agaskar, V.W. Day, W.G. Klemperer, J. Am. Chem. Soc., 1987, 109, 5554. 72 H.C.L. Abbenhuis, Chem. Eur. J., 2000, 6, 25. 73 S. Krijnen, R.W.J.M. Hanssen, H.C.L. Abbenhuis, J.H.C. van Hooff, R.A. van Santen, Chem.
Commun., 1999, 501. 74 J.M. Thomas, G. Sankar, M.C. Klunduk, M.P. Attfield, T. Maschmeyer, B.F.G. Johnson, R.B. Bell, J.
Phys. Chem. B, 1999, 103, 8809. 75 J.-C. Liu, Chem. Commun., 1996, 1109. 76 M. Crocker, R.H.M. Herold, A.G. Orpen, Chem. Commun., 1997, 241. 77 H. Ishida, J.L. Koenig, K.C. Gardner, J. Chem. Phys., 1982, 77, 5748. 78 P.D. Lickiss, S.A. Litster, A.D. Redhouse, C.J. Wisener, Chem. Commun., 1991, 173. 79 F.J. Feher, J.J. Schwab, D. Soulivong, J.W. Ziller, Main Group Chem., 1997, 2, 123. 80 T.S. Haddad, B.M. Moore, S.H. Phillips, Polymer Preprints, 2001, 42, 196. 81 M.Unno, K. Takada, H. Matsumoto, Chem. Lett., 1998, 489. 82 T.W. Hambley, T. Maschmeyer, A.F. Masters, Appl. Organomet. Chem., 1992, 6, 253. 83 F.J. Feher, D.A. Newman, J. Am. Chem. Soc., 1990, 112, 1931. 84 L.D. Field, C.M. Lindall, T. Maschmeyer, A.F. Masters, Aust. J. Chem., 1994, 47, 1127. 85 T. Maschmeyer, F. Rey, G. Sankar and J.M. Thomas, Nature, 1995, 378, 159. 86 F.J. Feher, T.A. Budzichowski, K. Rahimian, J.W. Ziller, J. Am. Chem. Soc., 1992, 114, 3859. 87 I.E. Buys, T.W. Hambley, D.J. Houlton, T. Maschmeyer, A.F. Masters, A.K. Smith, J. Mol. Catal.,
1994, 86, 309. 88 H.C.L. Abbenhuis, S. Krijnen, R.A. van Santen, Chem. Commun., 1997, 311. 89 S. Krijnen, H.C.L. Abbenhuis, R.W.J.M. Hanssen, J.H.C. van Hooff, R.A. van Santen, Angew. Chem.
Int. Ed., 1998, 37, 356. 90 S. Krijnen, B.L. Mojet, H.C.L. Abbenhuis, J.H.C. van Hooff, R.A. van Santen, Phys. Chem. Chem.
Phys., 1999, 1, 361. 91 T. Maschmeyer, M.C. Klunduk, C.M. Martin, D.S. Shephard, J.M. Thomas, B.F.G. Johnson, Chem.
Commun., 1997, 1847. 92 M.C. Klunduk, T. Maschmeyer, J.M. Thomas, B.F.G. Johnson, Chem. Eur. J., 1999, 5, 1481. 93 P. Smet, J. Riondato, T. Pauwels, L. Moens, L. Verdonck, Inorg. Chem. Commun., 2000, 3, 557. 94 F.J. Feher, R.L. Blanski, Chem. Commun., 1990, 1614. 95 F.J. Feher, J.F. Walzer, Inorg. Chem., 1991, 30, 1689. 96 F.J. Feher, J.F. Walzer, R.L. Blanski, J. Am. Chem. Soc., 1991, 113, 3618. 97 F.J. Feher, R.L. Blanski, J. Am. Chem. Soc., 1992, 114, 5886. 98 R. Duchateau, H.C.L. Abbenhuis, R.A. van Santen, S.K.-H. Thiele, M.F.H. van Tol, Organometallics,
1998, 17, 5222. 99 R. Duchateau, H.C.L. Abbenhuis, R.A. van Santen, A. Meetsma, S.K.-H. Thiele, M.F.H. van Tol,
Organometallics, 1998, 17, 5663.

Silsesquioxanes __________________________________________________________________________________________________________
43
100 R. Duchateau, U. Cremer, R.J. Harmsen, S.I. Mohamud, H.C.L. Abbenhuis, R.A. van Santen, A.
Meetsma, S.K.-H. Thiele, M.F.H. van Tol, M. Kranenburg, Organometallics, 1999, 18, 5447. 101 R. Duchateau, R.A. van Santen, G.P.A. Yap, Organometallics, 2000, 19, 809. 102 R.W.J.M. Hanssen, A. Meetsma, R.A. van Santen, H.C.L. Abbenhuis, Inorg. Chem., 2001, 40, 4049. 103 R. Duchateau, R.J. Harmsen, H.C.L. Abbenhuis, R.A. van Santen, A. Meetsma, S.K.-H. Thiele, M.
Kranenburg, Chem. Eur. J.,1999, 5, 3130. 104 M.D. Skowronska-Ptasinska, R. Duchateau, R.A. van Santen, G.P.A. Yap, Eur. J. Inorg. Chem., 2001,
133. 105 M.D. Skowronska-Ptasinska, R. Duchateau, R.A. van Santen, G.P.A. Yap, Organometallics, 2001, 20,
3519. 106 F.J. Feher, S.L. Gonzales, J.W. Ziller, Inorg. Chem., 1988, 27, 3440. 107 F.T. Edelmann, S. Giessmann, A. Fischer, Chem. Commun., 2000, 2153. 108 F.T. Edelmann, S. Giessmann, A. Fischer, J. Organomet. Chem., 2001, 620, 80. 109 F. Liu, K.D. John, B.L. Scott, R.T. Baker, K.C. Ott, W. Tumas, Angew. Chem. Int. Ed., 2000, 39,
3127. 110 F.J. Feher, T.L. Tajima, J. Am. Chem. Soc., 1994, 116, 2145. 111 F.T. Edelmann, S. Giessmann, A. Fischer, Inorg. Chem. Commun., 2000, 3, 658. 112 M. Nowotny, T. Maschmeyer, B.F.G. Johnson, P. Lahuerta, J.M. Thomas, J.E. Davies, Angew. Chem.
Int. Ed., 2001, 40, 955. 113 P. Smet, B. Devreese, F. Verpoort, T. Pauwels, I. Svoboda, S. Foro, J. van Beeumen, L. Verdonck,
Inorg. Chem., 1998, 37, 6583. 114 J.-C. Liu, S.R. Wilson, J.R. Shapley, F.J. Feher, Inorg. Chem., 1990, 29, 5138. 115 H.C.L. Abbenhuis, A.D. Burrows, H. Kooijman, M. Lutz, M.T. Palmer, R.A. van Santen, A.L. Spek,
Chem. Commun., 1998, 2627. 116 W.A. Herrmann, R. Anwander, V. Dufaud, W. Scherer, Angew. Chem., 1994, 106, 1338. 117 H.C.L. Abbenhuis, H.W.G. van Herwijnen, R.A. van Santen, Chem. Commun., 1996, 1941. 118 Y.K. Gun’ko, R. Reilly, F.T. Edelmann, H.-G. Schmidt, Angew. Chem. Int. Ed., 2001, 40, 1279.

44

45
3
A new, efficient route to titanium-silsesquioxane epoxidation catalysts developed by using High-Speed Experimentation
Abstract
High-Speed Experimentation techniques have been applied to the synthesis and testing
of silsesquioxane-based titanium catalysts for the epoxidation of alkenes. In the
optimisation of the hydrolytic condensation of silanes to incompletely condensed
silsesquioxane structures, different solvents and organotrichlorosilanes were employed.
The HSE approach allowed the identification of a new, fast and straightforward method
to synthesise a silsesquioxane precursor for a Ti-catalyst the performance of which is
comparable to the best silsesquioxane-based titanium catalysts reported in literature. At
the same time, some knowledge was gained about the effects of the solvent and of the
organic group R on the silanes on the synthesis of silsesquioxanes.
____________________ The contents of this chapter have been published in:
P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Angew. Chem. Int. Ed., 2001, 40, 740.
P.P. Pescarmona, J.J.T. Rops, J.C. van der Waal, J.C. Jansen, T. Maschmeyer, J. Mol. Catal. A, 2002, 182-183, 319.
P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, Catal. Today, 2003, 81, 347.

Chapter 3 __________________________________________________________________________________________________________
46
3.1 Introduction
Titanium centres dispersed on, and supported by, various forms of silica are
catalysts with remarkable properties for partial oxidations.1 The character of the active
site varies with the silica support used. Although it has been established that in the best -
on the basis of activity per gram of titanium - heterogeneous catalysts the Ti-centres are
all tetra-coordinated,2-4 it is still unclear whether, for optimum performance, the active
catalytic species requires the specific configuration of four siloxy groups, or if being
partially hydrolysed to, say, one hydroxy and three siloxy ligands is equally good.
Chemical modelling studies by various groups5-8 have shown that Ti-catalysts with
fewer than four siloxy groups should also be active as catalysts. These considerations
indicate that there is still some residual uncertainty regarding the active site, despite the
many experimental and computational studies carried out to resolve it.9-13 Indeed,
several mechanisms, or variations of one underlying mechanism, might operate
depending on the precise Ti-site configuration. Hence, different types of Ti-centres
might exhibit the desired characteristics.
For these heterogeneous catalysts, which are difficult to characterise, hybrid
inorganic-organic compounds have proved to be useful models. Especially
silsesquioxanes14 (RSiO1.5)a(H2O)0.5b have been a focus of attention as model
compounds for silica - as described in Chapter 2. Particularly interesting for
applications in catalysis are the incompletely condensed silsesquioxanes (b ≠ 0), which
contain Si-OH groups and therefore are able to complex catalytically active centres.14-16
The incompletely condensed silsesquioxane a7b3 has found applications as ligand for a
number of metal centres. Particularly, titanium complexes of silsesquioxane a7b3
resulted in very active homogeneous catalysts for the epoxidation of alkenes
(Figure 3.1).5-8 There is considerable interest in these catalysts, but commercial
introduction is severely hindered by the long and expensive preparation method of the
silsesquioxane precursor a7b3.17,18 Therefore, the identification of a new, faster and
efficient way to synthesise silsesquioxane precursors for titanium catalysts active in the
epoxidation of alkenes was set as the first goal of this research.
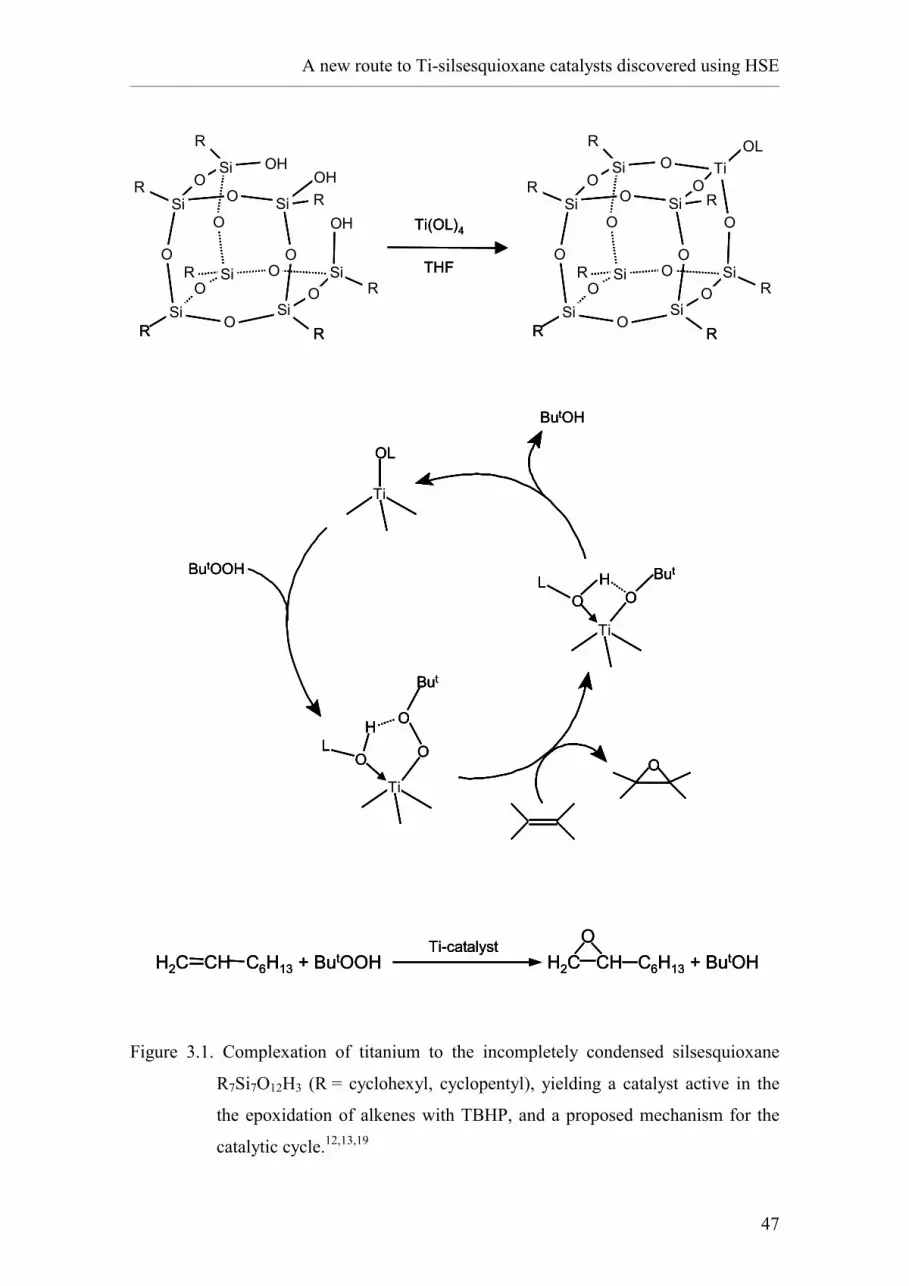
A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
47
Figure 3.1. Complexation of titanium to the incompletely condensed silsesquioxane
R7Si7O12H3 (R = cyclohexyl, cyclopentyl), yielding a catalyst active in the
the epoxidation of alkenes with TBHP, and a proposed mechanism for the
catalytic cycle.12,13,19
OHOH
OH
R
R
R
R
R R
ROO
SiO
Si
O
O
OSiSi
OSiOSi
O
SiTi(OL)4
THF
OL
R
R
R
R
R R
ROO
SiO
Si
O
O
OSiSi
OSiOSi
O
SiO
OO Ti
ButOOH
OL
Ti
But
LO
Ti
O
OH
O
ButL
O
Ti
OH
ButOH
H2C CH C6H13 + ButOOH H2C CH C6H13 + ButOHTi-catalyst O
OHOH
OH
R
R
R
R
R R
ROO
SiO
Si
O
O
OSiSi
OSiOSi
O
SiTi(OL)4
THF
OL
R
R
R
R
R R
ROO
SiO
Si
O
O
OSiSi
OSiOSi
O
SiO
OO TiOH
OH
OH
R
R
R
R
R R
ROO
SiO
Si
O
O
OSiSi
OSiOSi
O
SiTi(OL)4
THF
OL
R
R
R
R
R R
ROO
SiO
Si
O
O
OSiSi
OSiOSi
O
SiO
OO Ti
ButOOH
OL
Ti
But
LO
Ti
O
OH
O
ButL
O
Ti
OH
ButOH
ButOOH
OL
Ti
OL
Ti
But
LO
Ti
O
OH
But
LO
Ti
O
OH
OO
ButL
O
Ti
OH But
LO
Ti
OH
ButOH
H2C CH C6H13 + ButOOH H2C CH C6H13 + ButOHTi-catalyst O
H2C CH C6H13 + ButOOH H2C CH C6H13 + ButOHTi-catalyst O

Chapter 3 __________________________________________________________________________________________________________
48
3.2 The High-Speed Experimentation approach
On the basis of a primary screening consisting of a survey of the literature about
silsesquioxanes and of chemical knowledge of the subject, some general conclusions
can be drawn:
• Silsesquioxanes are usually synthesised by the hydrolytic condensation of
organosilanes (RSiX3): this is a multiple-step reaction for which an overall
mechanism is not available.
• Many parameters influence the hydrolytic condensation and determine which
silsesquioxane species are formed and in which amounts.
• The hydrolytic condensation of organosilanes usually produces a mixture of
completely and incompletely condensed silsesquioxanes.
• Incompletely condensed silsesquioxanes different from a7b3 may also act as
precursors for titanium complexes catalytically active in the epoxidation of alkenes.
Given all this information, High-Speed Experimentation techniques20 were chosen as
the most suitable method to investigate the system. Since a good knowledge about
silsesquioxanes was already available, no experimental primary screening was
necessary. In the secondary screening, the synthesis of silsesquioxanes was optimised as
a function of the activity of the catalysts obtained after titanium coordination to the
silsesquioxane structures. Therefore, this approach aimed at producing any incompletely
condensed silsesquioxane that would result in active catalysts after titanium
coordination rather than a specific structure (e.g. silsesquioxane a7b3). The epoxidation
of 1-octene with tert-butyl hydroperoxide (TBHP) as the oxidant was chosen as test
reaction for the activity of the catalysts.6
The first step of this secondary screening was to decide which parameter space had to
been screened. It was hypothesised that the solvent and the R-group have the most
relevant role among the parameters influencing the hydrolytic condensation of
organosilanes (see Paragraph 2.2.1). Therefore, the approach was to study a parameter
space defined by the combination of these two parameters. The choice of extensively
studying a small set of parameters rather than investigating a more varied parameter
space also gives the possibility of more clearly identifying trends concerning the effects
of these parameters on the reaction. It is important that each of the parameters is
sufficiently varied within the parameter space in order to minimise the risk of

A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
49
identifying a local maximum or of missing local trends.
3.3 Results and discussion
The parameter space was defined by the combination of 4 solvents and 10
R-groups (or mixtures of them). The set of R-groups consisted of 8 trichlorosilanes
RSiCl3 (R = cyclohexyl, cyclopentyl, phenyl, methyl, ethyl, tert-butyl, n-octyl and allyl)
and 2 equimolar mixtures of n-octyl- and methyltrichlorosilane and of cyclohexyl- and
phenyltrichlorosilane. Since the reaction includes water among the reagents, only
water-miscible solvents were selected: acetone, acetonitrile, methanol and
tetrahydrofuran (THF). The solvents were chosen on a varying scale of polarity to check
the influence of this property on the synthesis of silsesquioxanes. The parameter space
was screened for the activity of the catalysts obtained by coordination of titanium
isopropoxide, Ti(OPri)4, to the silsesquioxane precursors. Since the goal was to identify
a more efficient way to synthesise silsesquioxane structures, the reaction time for the
hydrolytic condensation was set at 18 hours, a much shorter time than that commonly
required for the synthesis of silsesquioxane a7b3.17,18 Concerning the other parameters,
they were chosen as follows: reaction temperature = 50°C, X = Cl, solvent : water = 4 : 1,
trichlorosilane concentration = 0.136M.
The epoxidation activity of the titanium catalysts as a function of the different
solvents and R-groups varied in the synthesis of the silsesquioxanes precursors is
reported in Figure 3.2. The values are normalised to the activity of the complex
obtained by reacting Ti(OPri)4 with the pure cyclopentyl silsequioxane a7b3 in THF.
The results show some general trends:
• Catalysts derived from silsesquioxane structures which were synthesised in
acetonitrile as solvent show the highest catalytic activity, followed, in decreasing
order of activity, by those from acetone, methanol and from tetrahydrofuran
(Figure 3.2). This trend applies for all the R-groups except ethyl and tert-butyl for
which silsesquioxanes synthesised in THF generate more active catalysts than those
synthesised in acetone and methanol. Interestingly, acetonitrile is more effective
than acetone, the solvent commonly reported for the synthesis of incompletely
condensed silsesquioxane a7b3.17,18 The fact that acetonitrile gives the best results
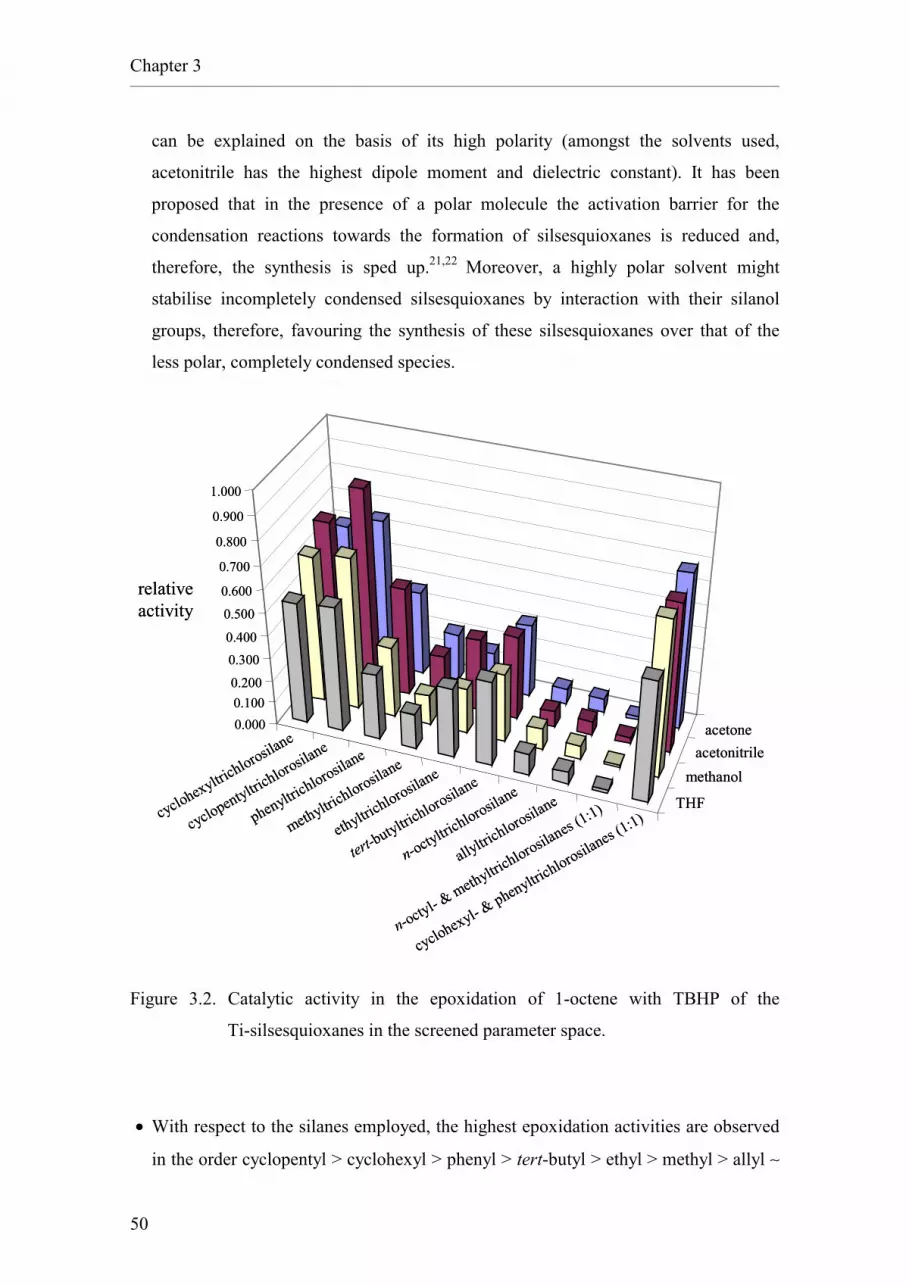
Chapter 3 __________________________________________________________________________________________________________
50
can be explained on the basis of its high polarity (amongst the solvents used,
acetonitrile has the highest dipole moment and dielectric constant). It has been
proposed that in the presence of a polar molecule the activation barrier for the
condensation reactions towards the formation of silsesquioxanes is reduced and,
therefore, the synthesis is sped up.21,22 Moreover, a highly polar solvent might
stabilise incompletely condensed silsesquioxanes by interaction with their silanol
groups, therefore, favouring the synthesis of these silsesquioxanes over that of the
less polar, completely condensed species.
Figure 3.2. Catalytic activity in the epoxidation of 1-octene with TBHP of the
Ti-silsesquioxanes in the screened parameter space.
• With respect to the silanes employed, the highest epoxidation activities are observed
in the order cyclopentyl > cyclohexyl > phenyl > tert-butyl > ethyl > methyl > allyl ∼
THF
methanolacetonitrile
acetone0.0000.1000.200
0.3000.400
0.500
0.600
0.700
0.800
0.900
1.000
relativeactivity
cyclohexyltrichlorosila
ne
cyclopentyltrichlorosila
ne
phenyltrichlorosila
ne
methyltrichlorosila
ne
ethyltrichlorosila
ne
tert-butyltric
hlorosilane
n-octyltrichlorosila
ne
allyltrichlorosila
ne
n-octyl- & methyltrichlorosila
nes (1:1)
cyclohexyl- & phenyltrichlorosila
nes (1:1)THF
methanolacetonitrile
acetone0.0000.1000.200
0.3000.400
0.500
0.600
0.700
0.800
0.900
1.000
relativeactivity
cyclohexyltrichlorosila
ne
cyclopentyltrichlorosila
ne
phenyltrichlorosila
ne
methyltrichlorosila
ne
ethyltrichlorosila
ne
tert-butyltric
hlorosilane
n-octyltrichlorosila
ne
allyltrichlorosila
ne
n-octyl- & methyltrichlorosila
nes (1:1)
cyclohexyl- & phenyltrichlorosila
nes (1:1)

A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
51
n-octyl ~ 0, for all solvents apart from THF for which tert-butyl- and
ethyltrichlorosilanes yield more active catalysts than phenyltrichlorosilanes.
(Figure 3.2). This trend is in good agreement with the literature, where cyclopentyl-
and cyclohexyltrichlorosilanes are reported to form incompletely condensed
silsesquioxanes in high yields.17,18 In principle, the nature of the organic group might
influence which silsesquioxane structures are produced and in which ratios,14 as well
as the electronic and spatial properties of the actual catalyst. Considering that the
organic groups are linked to the titanium centre through a Si-O unit, electronic
effects should have a negligible influence on the catalytic properties and, since the
organic groups point away from the titanium centre, steric effects are probably also
not relevant. The principal effect of the organic group is, therefore, considered to be
its role in determining which silsesquioxane structures are formed during the
hydrolytic condensation and in which amounts. The trend identified seems to be
related to the size of the organic substituent: bulky groups probably hinder the
formation of completely condensed silsesquioxanes, which are not able to bind to
titanium centres and therefore to generate active catalysts.
From a methodological point of view, the fact that different points in the parameter
space screened show very different activities confirms the hypothesis that the chosen
parameters have a relevant influence on the hydrolytic condensation: this means that a
representative parameter space was studied. An important question that arose from this
experiment was whether the parameter space was narrowed too much using literature
and experience as the starting point. In this respect, the coincidence of finding only
cyclopentyl and cyclohexyl as good candidates while they are also the only ones
reported in the literature for the synthesis of silsesquioxane a7b3,17,18 might be due to
bias for the synthesis condition particularly suited for these silanes. This remark
together with the trend indicating a beneficial effect of polarity suggest that it could be
interesting in further research to broaden the screened parameter space to other polar
solvents (see Chapter 7).
The two 1:1 molar mixtures, of n-octyl- and methyltrichlorosilane and of
cyclohexyl- and phenyltrichlorosilane, were used to investigate possible synergetic
effects between two different organotrichlorosilanes in the formation of incompletely
condensed structures (Figure 3.2). The n-octyl/methyl mixture gave a very low

Chapter 3 __________________________________________________________________________________________________________
52
epoxidation activity. As in the case of the pure n-octyl compound, the hydrolytic
condensation produced a gel, generated by micelle formation (probably due to the
slower hydrolysis rate of long-chain alkylsilanes).23 Water retained in this gel would be
inimical to the Ti-complexation. The cyclohexyl/phenyl mixture showed considerable
activity, suggesting the possibility of synergy between trichlorosilanes with different
organic R-groups (see Chapter 4).
The activities of the catalysts synthesised using High-Speed Experimentation
techniques were compared with those of Ti(OPri)4 and of the
Ti-(cyclopentyl silsesquioxane) catalyst produced by the reaction of pure cyclopentyl
silsesquioxane a7b3 with Ti(OPri)4. The High-Speed Experimentation catalysts with
R = cyclohexyl, cyclopentyl, phenyl, tert-butyl, ethyl, methyl and the 1:1 mixture of
cyclohexyl and phenyl, show an activity higher than Ti(OPri)4, confirming the formation
of Ti-silsesquioxanes as the active species. The Ti-catalyst derived from the
silsesquioxanes synthesised by the hydrolytic condensation of
cyclopentyltrichlorosilane in acetonitrile presents the highest catalytic activity
(Figure 3.2). This activity is 87% of that of the Ti-catalyst obtained using pure
silsesquioxane a7b3 as precursor. The relevance of this result lies in the fact that the
synthesis of these silsesquioxane precursors does not require any purification process
and is much less time-consuming than the synthesis of silsesquioxane a7b3.
3.3.1 Characterisation of the HSE lead
It is important to notice that with this HSE approach it was possible to identify a
lead but that no information was obtained regarding the actual nature of the
silsesquioxane precursor produced by the hydrolytic condensation of
cyclopentyltrichlorosilane in acetonitrile. It was thus interesting to characterise this lead
on a conventional laboratory scale. First, the synthesis and testing of the
Ti-silsesquioxane was repeated on a 125-ml scale (50 times up-scaling) yielding a
catalyst with an equal activity and, therefore, confirming the applicability of HSE
techniques to the synthesis of silsesquioxanes. Then, the silsesquioxane precursor was
characterised prior to reaction with the titanium centre. The reaction of cyclopentyl
trichlorosilane with water in acetonitrile, for 18 hours at 50°C, yields two
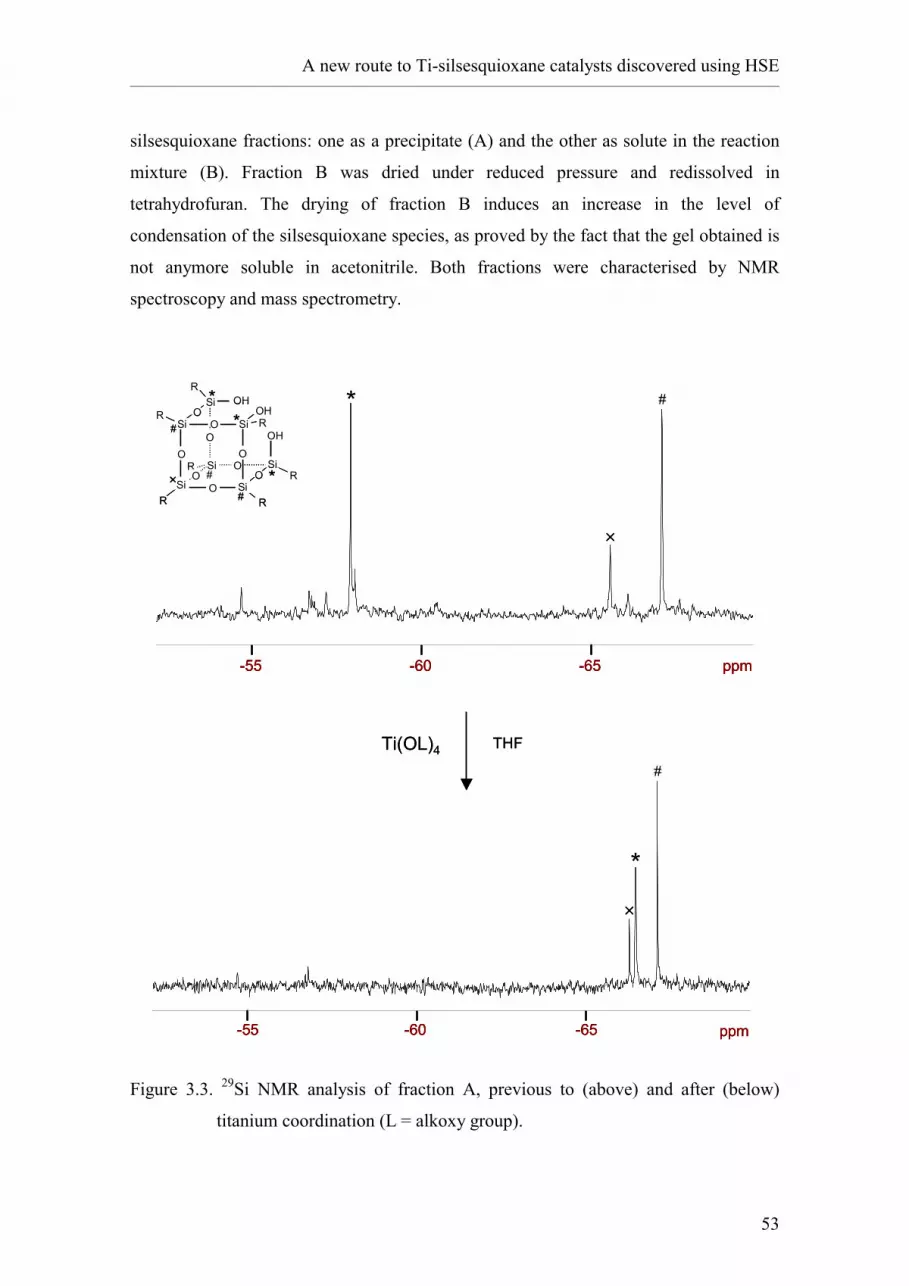
A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
53
silsesquioxane fractions: one as a precipitate (A) and the other as solute in the reaction
mixture (B). Fraction B was dried under reduced pressure and redissolved in
tetrahydrofuran. The drying of fraction B induces an increase in the level of
condensation of the silsesquioxane species, as proved by the fact that the gel obtained is
not anymore soluble in acetonitrile. Both fractions were characterised by NMR
spectroscopy and mass spectrometry.
Figure 3.3. 29Si NMR analysis of fraction A, previous to (above) and after (below)
titanium coordination (L = alkoxy group).
-55 -60 -65 ppm
-55 -60 -65 ppm
Ti(OL)4 THF
* #
××××
Si
SiSi
Si
Si
Si
Si
OHO
OOH
O
OO
OH
OO
O
R
R
R
R
R R
RO
#
#
#
*
*
*
××××
*
#
××××
-55 -60 -65 ppm
-55 -60 -65 ppm
Ti(OL)4 THF
-55 -60 -65 ppm
-55 -60 -65 ppm
Ti(OL)4 THFTi(OL)4 THF
* #
××××
Si
SiSi
Si
Si
Si
Si
OHO
OOH
O
OO
OH
OO
O
R
R
R
R
R R
RO
#
#
#
*
*
*
××××
Si
SiSi
Si
Si
Si
Si
OHO
OOH
O
OO
OH
OO
O
R
R
R
R
R R
RO
#
#
#
*
*
*
××××
*
#
××××
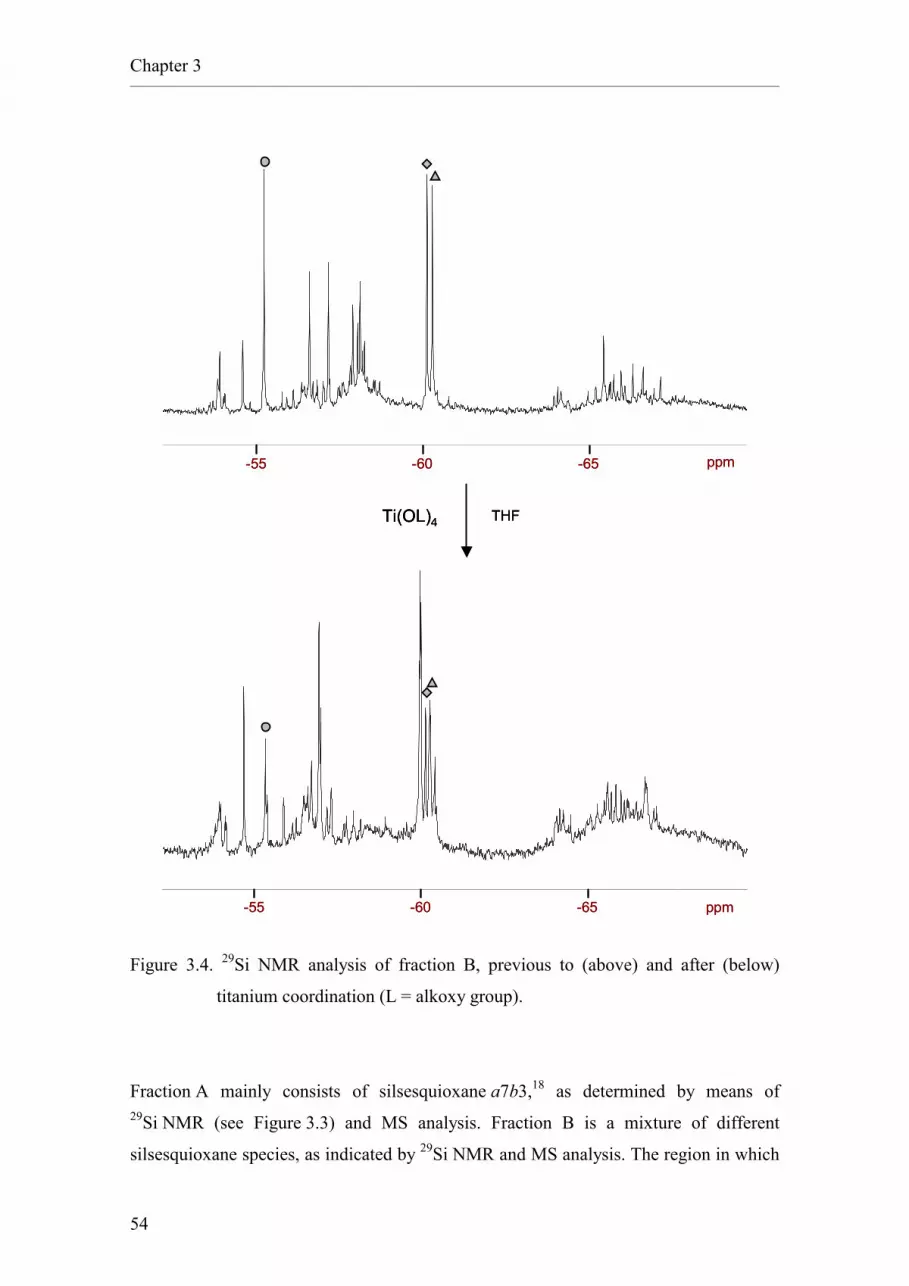
Chapter 3 __________________________________________________________________________________________________________
54
Figure 3.4. 29Si NMR analysis of fraction B, previous to (above) and after (below)
titanium coordination (L = alkoxy group).
Fraction A mainly consists of silsesquioxane a7b3,18 as determined by means of 29Si NMR (see Figure 3.3) and MS analysis. Fraction B is a mixture of different
silsesquioxane species, as indicated by 29Si NMR and MS analysis. The region in which
-55 -60 -65 ppm
-55 -60 -65 ppm
Ti(OL)4 THF
-55 -60 -65 ppm
-55 -60 -65 ppm
Ti(OL)4 THFTi(OL)4 THF

A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
55
most of the peaks in the 29Si NMR spectrum of fraction B are found is that of silanol
groups belonging to incompletely condensed silsesquioxanes (see Figure 3.4).24 It is
equally difficult to assign all the species present in the mixture or to separate them.
However, both NMR and MS data suggest that the main species is the silsesquioxane
structure a6b2 shown in Figure 3.5; this structure is in agreement with the three 29Si NMR peaks marked in Figure 3.4 and with the main set of MS peaks,
corresponding to silsesquioxanes with 6 Si atoms. This assignment is supported by the
similarity of the 29Si NMR spectrum with that of cyclohexyl silsesquioxane a6b2, which
can be obtained by cleavage of the completely condensed a6b0 structure.25
Figure 3.5. Schematic representation of cyclopentyl silsesquioxane a6b2
[R = cyclopentyl].
Finally, both fractions were reacted with a titanium alkoxide. The coordination of the
titanium centre to the silsesquioxane structures was confirmed by 29Si NMR
(Figures 3.3 and 3.4). The catalytic activity in the epoxidation of 1-octene of the HSE
lead and of the two fractions in which the latter can be divided (all the three catalysts
are homogeneous) were studied as a function of the reaction time (Figure 3.6). The
activity of fraction A (TOF = 0.97 molepo·molTi-1·min-1) is comparable to that of the
Ti-catalyst obtained with pure silsesquioxane a7b3, which after 4 hours of reaction
gives complete conversion of TBHP towards the epoxide. Fraction B shows a much
lower, but still significant, activity (TOF = 0.16 molepo·molTi-1·min-1): this result confirms
Si
Si
Si
O
O
O
O
R
Si
O
O
Si
Si OH
HO
O
O
R R
R R
R
Si
Si
Si
O
O
O
O
R
Si
O
O
Si
Si OH
HO
O
O
R R
R R
R
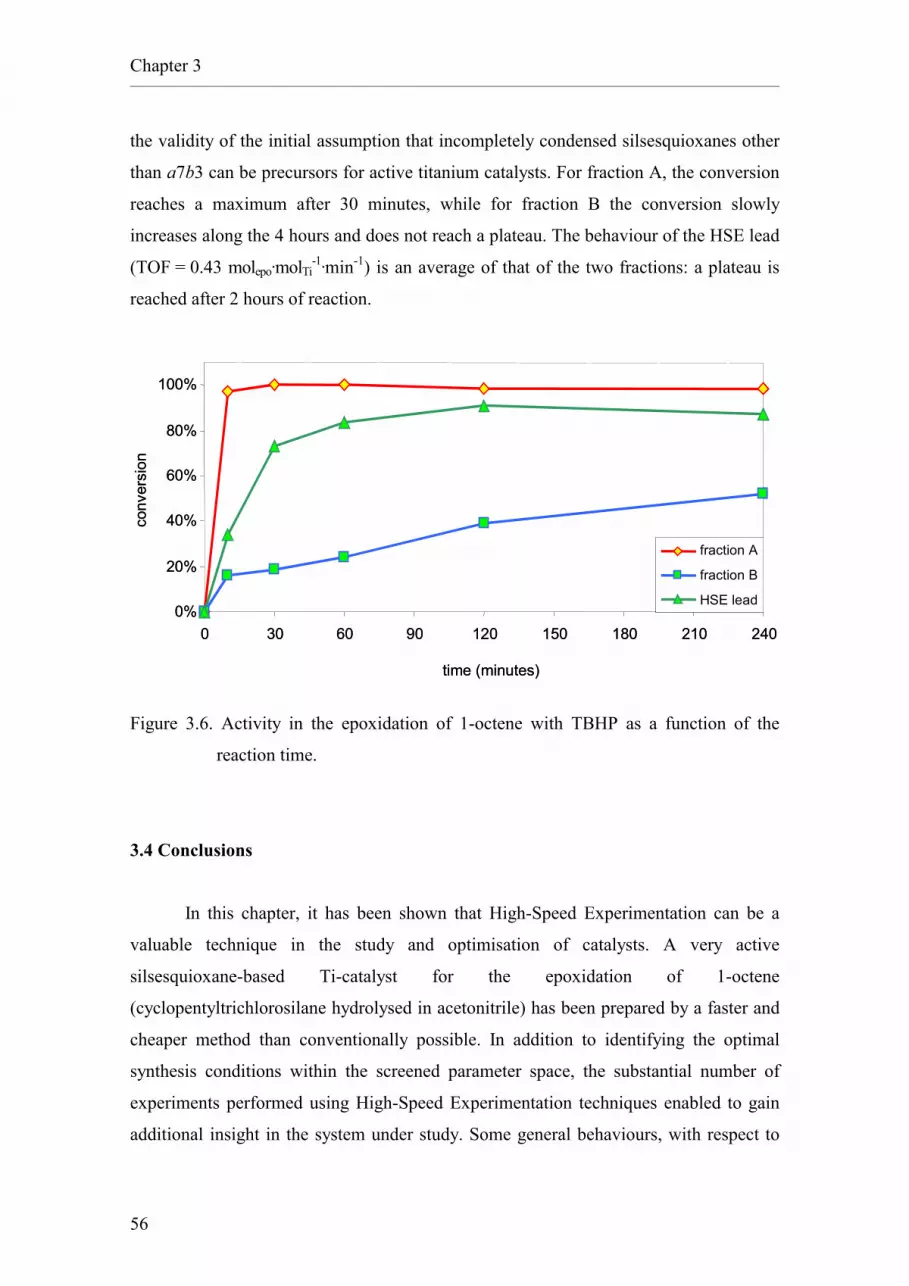
Chapter 3 __________________________________________________________________________________________________________
56
the validity of the initial assumption that incompletely condensed silsesquioxanes other
than a7b3 can be precursors for active titanium catalysts. For fraction A, the conversion
reaches a maximum after 30 minutes, while for fraction B the conversion slowly
increases along the 4 hours and does not reach a plateau. The behaviour of the HSE lead
(TOF = 0.43 molepo·molTi-1·min-1) is an average of that of the two fractions: a plateau is
reached after 2 hours of reaction.
Figure 3.6. Activity in the epoxidation of 1-octene with TBHP as a function of the
reaction time.
3.4 Conclusions
In this chapter, it has been shown that High-Speed Experimentation can be a
valuable technique in the study and optimisation of catalysts. A very active
silsesquioxane-based Ti-catalyst for the epoxidation of 1-octene
(cyclopentyltrichlorosilane hydrolysed in acetonitrile) has been prepared by a faster and
cheaper method than conventionally possible. In addition to identifying the optimal
synthesis conditions within the screened parameter space, the substantial number of
experiments performed using High-Speed Experimentation techniques enabled to gain
additional insight in the system under study. Some general behaviours, with respect to
0%
20%
40%
60%
80%
100%
0 30 60 90 120 150 180 210 240
time (minutes)
conv
ersi
on
fraction A
fraction B
HSE lead0%
20%
40%
60%
80%
100%
0 30 60 90 120 150 180 210 240
time (minutes)
conv
ersi
on
fraction A
fraction B
HSE lead
fraction A
fraction B
HSE lead

A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
57
the roles in the synthesis of silsesquioxanes of both the solvent and the R-group of the
organotrichlorosilane have been identified.
The synthesis of the HSE lead was repeated on a conventional lab-scale and the
silsesquioxane precursor was characterised by means of MS and NMR spectroscopy.
3.5 Experimental
Experiments were performed on an automated parallel synthesis workstation26
coupled with a personal computer supplied with software enabling to program the
workstation. Reagents: cyclohexyl-, cyclopentyl-, phenyl-, methyl-, ethyl-, tert-butyl-,
n-octyl-, and allyltrichlorosilanes (RSiCl3); acetone, acetonitrile, methanol and
tetrahydrofuran (THF) as solvents; deionised water. In a typical experiment, 6 times
2-ml aliquots of each of the 4 solvents were dispensed in a rack containing an 6×4 array
of glass tubes, followed by the addition of 340 µmol of each of 6 silanes to the
solvent-containing reaction vessels in such a way that 24 individually different silane
solutions were prepared. Hydrolysis of the silane was started by the addition of 0.5 ml
of water to the reaction vessel and placing the vessel array on an orbital shaker26 at
50°C for 18 hours. After removal of the solvent, and of the excess water and
hydrochloric acid, in a vacuum centrifuge,26 the samples were stored under argon.
Titanium complexation was performed by dissolving the crude silsesquioxane mixture
in 2 ml of THF under argon, followed by the addition of 54 µmol of titanium
isopropoxide (as a solution in isopropanol) to each sample. After 5 hours at 60°C, the
tetrahydrofuran was removed by means of the vacuum centrifuge and the samples were
stored under argon.
The catalytic activity of the materials obtained was determined by the
epoxidation of 1-octene with tert-butyl hydroperoxide (TBHP). The reaction was
performed by adding to each dried sample ~0.5 mmol of TBHP as a ~40%wt solution in
cyclohexane (TBHP : catalyst ratio ≈ 10) and 0.0144 mol of 1-octene, that therefore
acted both as reactant and solvent (1-octene : catalyst ratio = 300). The employed
1-octene contained 2%vol of decane as an internal standard for the following GC
analysis. Samples were taken after reacting for 4 hours at 80°C and analysed on a
UNICAM Pro GC using a CP-Sil-5B column. The epoxidation reaction did not proceed

Chapter 3 __________________________________________________________________________________________________________
58
further at room temperature. The reported activities were obtained by normalising the
1,2-epoxyoctane GC peak area by means of the internal standard. The values for the
activities are the averages of the results from different experiments. The experimental
average error for these values is under 3%, as determined by calibration and averaging
of the results of three identical experimental runs. Since 1-octene was used both as
reactant and solvent, it has not been possible to realise a mass balance. The reaction was
completely selective towards 1,2-epoxyoctane: no other products were detected. The
conversion selectivity of TBHP towards 1,2-epoxyoctane lies between 80 and 94%.
The other catalysts (Ti(OPri)4 and Ti-(cyclopentyl silsesquioxane a7b3) were
tested using the same amount of titanium and the same experimental conditions as
described above.
The 125-ml scale synthesis of the HSE lead was performed by carefully adding
25 ml of deionised water into a round-bottom flask containing a solution of 2.8 ml of
cyclopentyltrichlorosilane (c-C5H9)SiCl3 (97% purity) in 100 ml of acetonitrile
(CH3CN:H2O = 4:1 in volume). The solution was then heated to 50°C while stirring for
18 hours: a white precipitate was formed (fraction A: 1.025 g, 50% yield). The
remaining solution (fraction B) was dried under reduced pressure to afford a dense gel,
which was then dissolved in 10 ml of tetrahydrofuran: an insoluble NH4Cl residue
(produced by the hydrolysis of CH3CN) was removed. Consecutive drying of the
solution afforded a silsesquioxane gel that was characterised by NMR and MS.
Titanium complexation was performed by dissolving each fraction in 50 ml of
tetrahydrofuran, followed by the addition of 0.4 ml of titanium butoxide, Ti(OBu)4, to
each solution. Both samples were then heated to 60°C for 5 hours and finally dried
under reduced pressure. To test the catalytic activity in epoxidation reactions, the two
solid samples were dissolved each in 56.5 ml of 1-octene (with 2%vol of decane as
internal standard for GC analysis), followed by addition of 3 ml of TBHP solution
(~40%wt solution in cyclohexane), stirred and heated to 80°C. Samples were taken at
different times (see Figure 3.6) and analysed with a UNICAM Pro GC using a
CP-Sil-5B column. The activity data were obtained by normalising the 1,2-epoxyoctane
GC peak area by means of the internal standard. For all the three catalysts the reaction
was completely selective towards 1,2-epoxyoctane: no other products were found.

A new route to Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
59
29Si NMR spectra were measured on a Varian VXR-400S (79.5 MHz, 1H decoupled,
deuterated THF as solvent, 25°C). Main peaks for fraction A, δ: -57.91, -65.70, -67.29
(3:1:3); fraction B, δ: -55.13, -60.16, -60.33 (1:1:1).
Mass spectrometry analysis was performed using a Micromass Quattro LC-MS with
ESI+ as ionisation technique. The samples were prepared by dissolving ~0.05 g of crude
product in 4 ml of tetrahydrofuran, 1 ml of acetonitrile and 0.1 ml of formic acid 0.1M.
From the MS data it is possible to determine the number of Si atoms (the value of a in
(RSiO1.5)a(H2O)0.5b) of the detected species but not the level of condensation (the value
of b). The reason for this is that in the ionisation process silsesquioxanes can lose water
molecules, as confirmed by the fact that the ratio among the peaks for a given value of a
changes as a function of the cone voltage applied in the MS analysis.
Selected MS data:
Fraction A (cone voltage = 65V). For a = 7, m/z: 875.04 (H+, 100%), 839.00 (H+, 41%),
897.08 (Na+, 16%), 911.09 (H+, 16%), 857.03 (H+, 11%). For a = 8, m/z: 969.03 (H+,
4%), 987.05 (H+, 2%).
Fraction B (cone voltage = 30V). For a = 4, m/z: 543.60 (Na+, 14%), 557.56 (H+,
+ H2O, 13%). For a = 5, m/z: 655.53 (Na+, 30%), 633.61 (H+, 28%). For a = 6, m/z:
763.41 (H+, 100%), 785.39 (Na+, 55%), 745.41 (H+, 38%). For a = 9, m/z: 1157.23
(Na+, 22%), 1175.24 (Na+, 17%), 1135.12 (H+, 18%). For a = 10, m/z: 1287.03 (Na+,
29%), 1305.09 (Na+, 27%), 1265.11 (H+, 25%). For a = 11, m/z: 1417.97 (Na+, 36%),
1396.11 (H+, 26%). For a = 12, m/z: 1547.83 (Na+, 39%), 1529.95 (Na+, 26%).

Chapter 3 __________________________________________________________________________________________________________
60
References
1 B. Notari, Ad. Catal., 1996, 41, 253. 2 G. Bellussi, M.S. Rigutto, Stud. Surf. Sci. Catal., 1994, 85, 177. 3 T. Maschmeyer, F. Rey, G. Sankar, J.M. Thomas, Nature, 1995, 378, 159. 4 M.C. Klunduk, T. Maschmeyer, J.M. Thomas, B.F.G. Johnson, Chem. Eur. J., 1999, 5 (5), 1481. 5 T. Maschmeyer, M.C. Klunduk, C.M. Martin, D.S. Shephard, J.M. Thomas, B.F.G. Johnson, Chem.
Commun., 1997, 1847. 6 M. Crocker, R.H.M. Herold, A.G. Orpen, Chem. Commun., 1997, 2411. 7 H.C.L. Abbenhuis, S. Krijnen, R.A. van Santen, Chem. Commun., 1997, 331. 8 S. Krijnen, H.C.L. Abbenhuis, R.W.J.M. Hanssen, J.H.C. van Hooff, R.A. van Santen, Angew. Chem.
Int. Ed., 1998, 37, 356. 9 L. Marchese, T. Maschmeyer, E. Gianotti, S. Coluccia, J.M. Thomas, J. Phys. Chem. B, 1997, 101,
8836. 10 P.E. Sinclair, G. Sankar, C.R.A. Catlow, J.M. Thomas, T. Maschmeyer, J. Phys. Chem. B, 1997, 101,
4232. 11 D. Tantanak, M.A. Vincent, I.H. Hiller, Chem. Commun., 1998, 1031. 12 P.E. Sinclair, C.R.A. Catlow, J. Phys. Chem. B, 1999, 103, 1084. 13 C.M. Barker, D. Gleeson, N. Kaltsoyannis, C.R.A. Catlow, G. Sankar, J.M. Thomas, Phys. Chem.
Chem. Phys., 2002, 4, 1228. 14 P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583. 15 T. Maschmeyer, J.M. Thomas, A.F. Masters, NATO ASI Ser., New Trends in Materials Chemistry,
1997, 498, 461. 16 H.C.L. Abbenhuis, Chem. Eur. J., 2000, 6, 25. 17 F.J. Feher, D.A. Newman, J.F. Walzer, J. Am. Chem. Soc., 1989, 111, 1741. 18 F.J. Feher, T.A. Budzichowski, R.L. Blanski, K.J. Weller, J.W. Ziller, Organometallics, 1991, 10,
2526. 19 K.A. Jørgensen, Chem. Rev., 1989, 89, 431. 20 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Catal. Lett., 1999, 63, 1, see also
Chapter 1. 21 T. Kudo, M.S. Gordon, J. Am. Chem. Soc., 1998, 120, 11432. 22 T. Kudo, M.S. Gordon, J. Phys. Chem. A, 2000, 104, 4058. 23 P.G. Harrison, J. Organomet. Chem., 1997, 542, 141. 24 See Paragraph 2.3.1. 25 F.J. Feher, F. Nguyen, D. Soulivong, J.W. Ziller, Chem. Commun., 1999, 1705. 26 See Appendix A.

61
4
Fine-tuning of the synthesis of titanium-silsesquioxane epoxidation catalysts by means of High-Speed Experimentation
Abstract
High-Speed Experimentation techniques have been applied to a fine-tuning of the
preparation of titanium silsesquioxane catalysts for the epoxidation of alkenes. The first
part of this optimisation led to an adjustment of the reaction conditions for the synthesis
of the silsesquioxane precursors. Next, the effect of the nature of the titanium centres
bound to the silsesquioxane structures on the catalytic activity was explored: the
coordination of diols to the titanium centres resulted in an improvement of the activity
when using H2O2 as the oxidant.
____________________ The contents of this chapter have been published in:
P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Angew. Chem. Int. Ed., 2001, 40, 740.
P.P. Pescarmona, T. Maschmeyer, NATO Science Series, 2002, Ser. II Vol. 69, 173.
P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, Catal. Today, 2003, 81, 347.
P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Chem. Eng. Commun., accepted.

Chapter 4 __________________________________________________________________________________________________________
62
4.1 Introduction
Besides leading to the identification of a new route to synthesise a promising
Ti-catalyst and to an additional knowledge about the synthesis of silsesquioxane
structures, the HSE exploration described in Chapter 3 provided a starting point for
further investigation. One of the directions that can be taken is that of studying other
parameters influencing the catalytic activity of titanium silsesquioxanes. The initial
choice of investigating the effect of the nature of the solvent and of the R-group on the
synthesis of silsesquioxane precursors for Ti-catalysts was based on the assumption that
these two parameters were the most relevant in determining which silsesquioxane
structures were formed and in which amounts. The validity of this hypothesis can be
verified (or falsified)1,2 by studying the effect of the other parameters affecting the
synthesis of silsesquioxanes.3 This study would also provide an adjustment of the
method to prepare silsesquioxane precursors for epoxidation catalysts. After having
completed the optimisation of the synthesis of the silsesquioxane precursors, the next
step of the preparation of the Ti-catalysts can be investigated by varying the nature of
the titanium centre bound to the silsesquioxane structures.
4.2 The High-Speed Experimentation approach
Different HSE approaches can be used to investigate the system. In the case of
the effect of pH variation, the procedure was similar to that reported in Chapter 3. For
all other parameters a different approach was used. The parameter set was reduced from
12 (8 R-groups + 4 solvents) to 4 (3 R-groups + 1 solvent) elements: cyclopentyl-,
cyclohexyl- and phenyltrichlorosilane with acetonitrile as solvent, i.e. the combinations
that led to the highest catalytic activities (see Paragraph 3.3). This reduced parameter
space was then used as a basis to create larger parameter spaces by varying the other
parameters influencing the synthesis. Finally, for the exploration of the nature of the
titanium centres, two silsesquioxane precursors were used to bind to a set of different
titanium compounds. These Ti-silsesquioxanes were then tested for the epoxidation of
1-octene with both TBHP and H2O2.
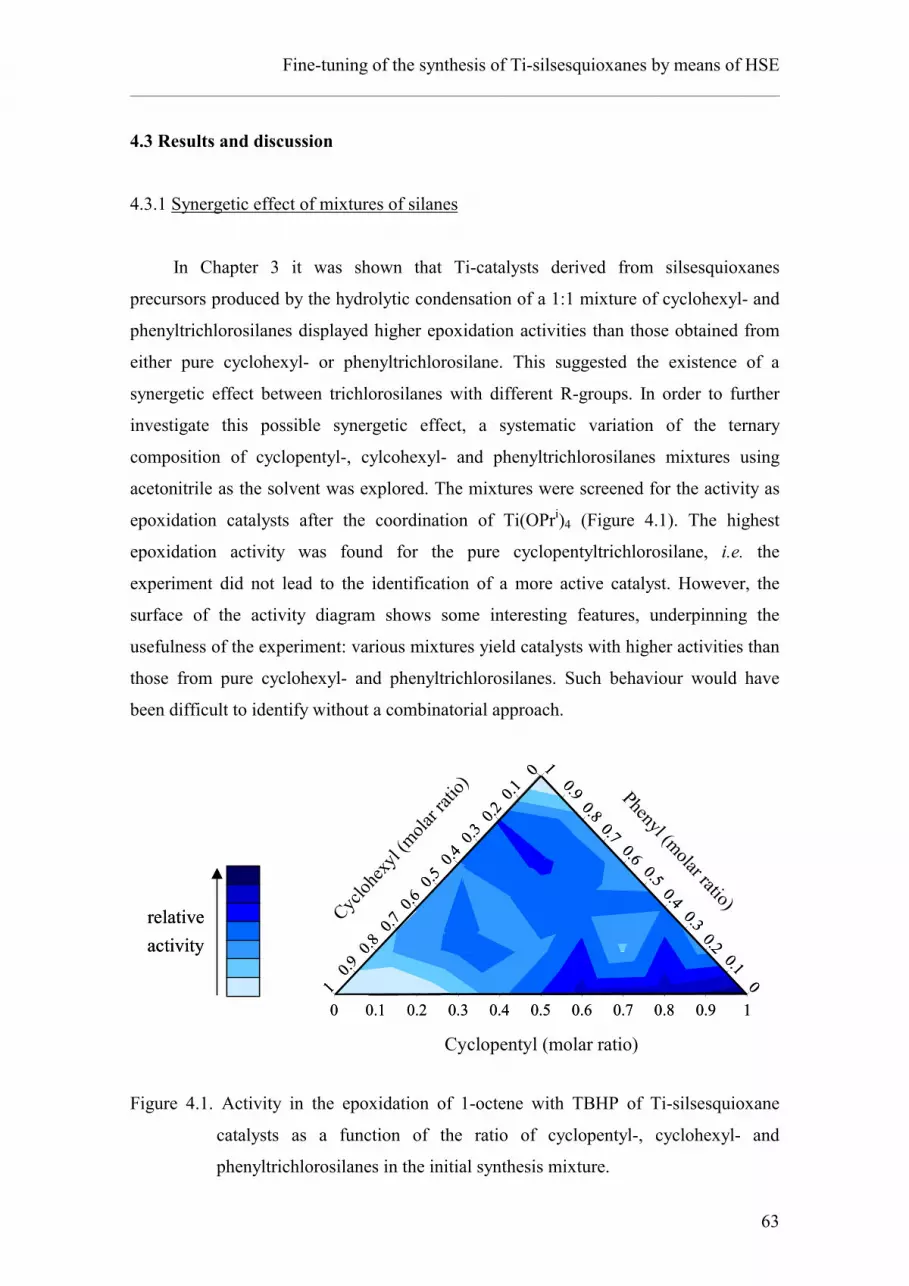
Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
63
4.3 Results and discussion
4.3.1 Synergetic effect of mixtures of silanes
In Chapter 3 it was shown that Ti-catalysts derived from silsesquioxanes
precursors produced by the hydrolytic condensation of a 1:1 mixture of cyclohexyl- and
phenyltrichlorosilanes displayed higher epoxidation activities than those obtained from
either pure cyclohexyl- or phenyltrichlorosilane. This suggested the existence of a
synergetic effect between trichlorosilanes with different R-groups. In order to further
investigate this possible synergetic effect, a systematic variation of the ternary
composition of cyclopentyl-, cylcohexyl- and phenyltrichlorosilanes mixtures using
acetonitrile as the solvent was explored. The mixtures were screened for the activity as
epoxidation catalysts after the coordination of Ti(OPri)4 (Figure 4.1). The highest
epoxidation activity was found for the pure cyclopentyltrichlorosilane, i.e. the
experiment did not lead to the identification of a more active catalyst. However, the
surface of the activity diagram shows some interesting features, underpinning the
usefulness of the experiment: various mixtures yield catalysts with higher activities than
those from pure cyclohexyl- and phenyltrichlorosilanes. Such behaviour would have
been difficult to identify without a combinatorial approach.
Figure 4.1. Activity in the epoxidation of 1-octene with TBHP of Ti-silsesquioxane
catalysts as a function of the ratio of cyclopentyl-, cyclohexyl- and
phenyltrichlorosilanes in the initial synthesis mixture.
relativeactivity
Phenyl (molar ratio)
Cycloh
exyl
(mola
r rati
o)
Cyclopentyl (molar ratio)
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
00.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.91
0.8
0.9
1
0.5
0.6
0.7
0.2
0.3
0.4
00.1
relativeactivity
Phenyl (molar ratio)
Cycloh
exyl
(mola
r rati
o)
Cyclopentyl (molar ratio)
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
00.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.91
0.8
0.9
1
0.5
0.6
0.7
0.2
0.3
0.4
00.1
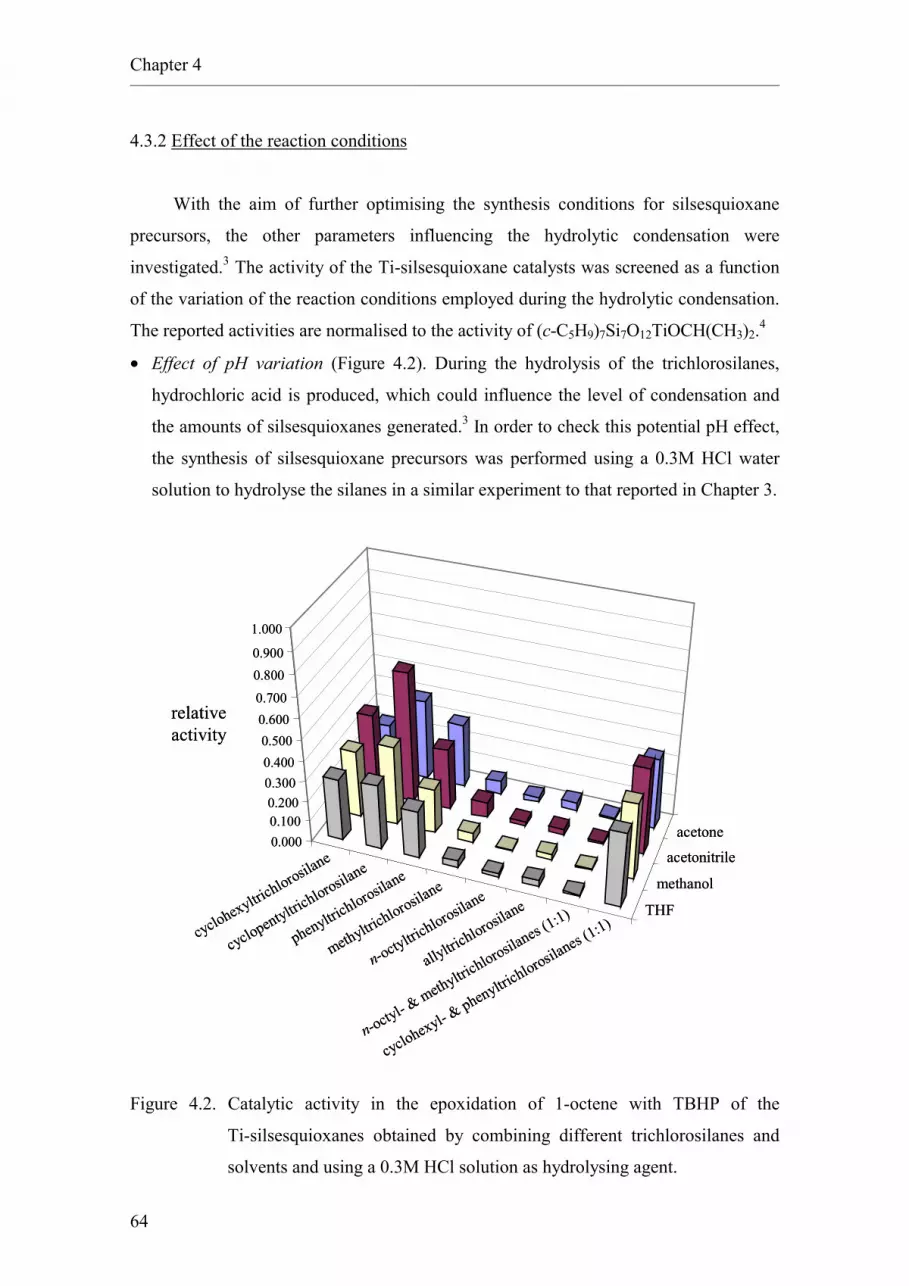
Chapter 4 __________________________________________________________________________________________________________
64
4.3.2 Effect of the reaction conditions
With the aim of further optimising the synthesis conditions for silsesquioxane
precursors, the other parameters influencing the hydrolytic condensation were
investigated.3 The activity of the Ti-silsesquioxane catalysts was screened as a function
of the variation of the reaction conditions employed during the hydrolytic condensation.
The reported activities are normalised to the activity of (c-C5H9)7Si7O12TiOCH(CH3)2.4
• Effect of pH variation (Figure 4.2). During the hydrolysis of the trichlorosilanes,
hydrochloric acid is produced, which could influence the level of condensation and
the amounts of silsesquioxanes generated.3 In order to check this potential pH effect,
the synthesis of silsesquioxane precursors was performed using a 0.3M HCl water
solution to hydrolyse the silanes in a similar experiment to that reported in Chapter 3.
Figure 4.2. Catalytic activity in the epoxidation of 1-octene with TBHP of the
Ti-silsesquioxanes obtained by combining different trichlorosilanes and
solvents and using a 0.3M HCl solution as hydrolysing agent.
THF
methanol
acetonitrile
acetone0.0000.1000.2000.3000.4000.5000.6000.700
0.8000.900
1.000
cyclohexyltrichlorosila
ne
cyclopentyltrichlorosila
ne
phenyltrichlorosila
ne
methyltrichlorosila
ne
n-octyltrichlorosila
ne
allyltrichlorosila
ne
n-octyl- & methyltrichlorosila
nes (1:1)
cyclohexyl- & phenyltrichlorosila
nes (1:1)
relativeactivity
THF
methanol
acetonitrile
acetone0.0000.1000.2000.3000.4000.5000.6000.700
0.8000.900
1.000
cyclohexyltrichlorosila
ne
cyclopentyltrichlorosila
ne
phenyltrichlorosila
ne
methyltrichlorosila
ne
n-octyltrichlorosila
ne
allyltrichlorosila
ne
n-octyl- & methyltrichlorosila
nes (1:1)
cyclohexyl- & phenyltrichlorosila
nes (1:1)
relativeactivity
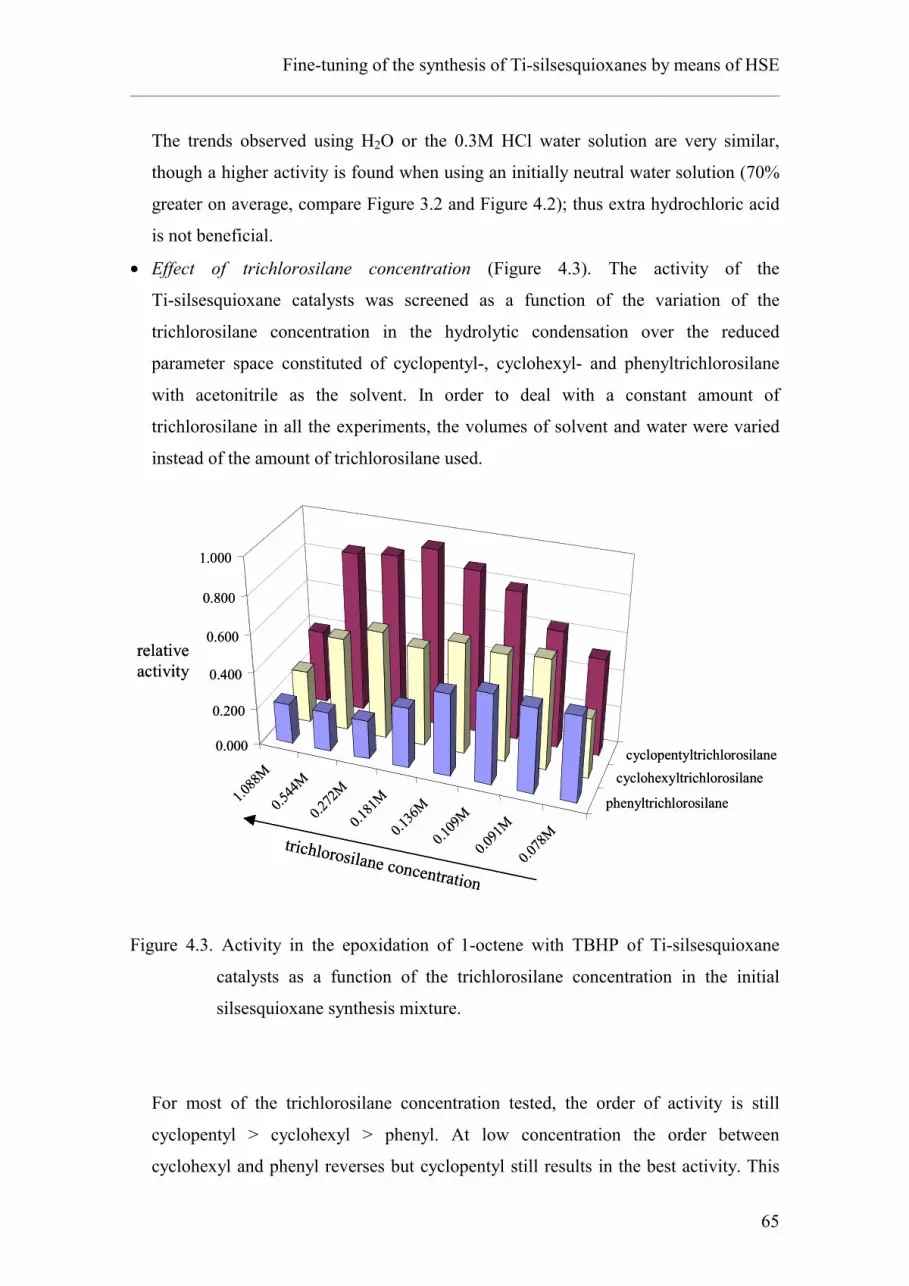
Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
65
The trends observed using H2O or the 0.3M HCl water solution are very similar,
though a higher activity is found when using an initially neutral water solution (70%
greater on average, compare Figure 3.2 and Figure 4.2); thus extra hydrochloric acid
is not beneficial.
• Effect of trichlorosilane concentration (Figure 4.3). The activity of the
Ti-silsesquioxane catalysts was screened as a function of the variation of the
trichlorosilane concentration in the hydrolytic condensation over the reduced
parameter space constituted of cyclopentyl-, cyclohexyl- and phenyltrichlorosilane
with acetonitrile as the solvent. In order to deal with a constant amount of
trichlorosilane in all the experiments, the volumes of solvent and water were varied
instead of the amount of trichlorosilane used.
Figure 4.3. Activity in the epoxidation of 1-octene with TBHP of Ti-silsesquioxane
catalysts as a function of the trichlorosilane concentration in the initial
silsesquioxane synthesis mixture.
For most of the trichlorosilane concentration tested, the order of activity is still
cyclopentyl > cyclohexyl > phenyl. At low concentration the order between
cyclohexyl and phenyl reverses but cyclopentyl still results in the best activity. This
1.088
M
0.544
M
0.272
M
0.181
M
0.136
M
0.109
M
0.091
M
0.078
M
phenyltrichlorosilane
cyclohexyltrichlorosilanecyclopentyltrichlorosilane0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
trichlorosilane concentration
1.088
M
0.544
M
0.272
M
0.181
M
0.136
M
0.109
M
0.091
M
0.078
M
phenyltrichlorosilane
cyclohexyltrichlorosilanecyclopentyltrichlorosilane0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
trichlorosilane concentration

Chapter 4 __________________________________________________________________________________________________________
66
result confirms the starting hypothesis that the R-group is a more relevant factor than
the concentration of the silane in influencing the hydrolytic condensation. The
maximum of activity corresponds to a different silane concentration for each of the
three R-groups. The maximum for cyclopentyl as R-group - corresponding to a
concentration of silane of 0.181M - displays an activity that is 95% of that of pure
Ti-(cyclopentyl silsesquioxane a7b3): this is a relevant improvement compared to the
optimum obtained previously (87%).5 These results show that the experiment
worked as a fine-tuning of the synthesis of silsesquioxane precursors described in
Chapter 3. From a methodological point of view it is important to notice that in the
wide concentration range applied, no activity low enough to reject any of the three
R-groups - if such trichlorosilane concentration would have been chosen in the first
experiment - was obtained. This significantly strengthens the assumption that the
conditions of the first experiment were sufficiently discriminative towards the
R-groups screened.
• Effect of the H2O concentration (Figure 4.4). The activity of the Ti-silsesquioxane
catalysts was screened as a function of the variation of the amount of H2O used in the
hydrolytic condensation over the reduced parameter space constituted of
cyclopentyl-, cyclohexyl- and phenyltrichlorosilane with acetonitrile as solvent. To
separate the effect of the water concentration from the effect of the trichlorosilane
concentration, the total amount of liquid (solvent + water) in which the synthesis was
carried out was kept constant (2.5 ml). The acetonitrile/water ratio was varied from
32 to 0.25. For any acetonitrile/water ratio, the order of activity is still
cyclopentyl > cyclohexyl > phenyl, apart from an inversion between cyclohexyl and
phenyl at a ratio ≤ 0.5. As for the trichlorosilane concentration experiment, this result
confirms the hypothesis that the nature of the R-group has a directing role in the
formation of silsesquioxanes. Similarly, the position of the maximum of the activity
is different for each R-group: the maximum activity for phenyl is at high water
content while for cyclopentyl and cyclohexyl it is at lower water content. The highest
activity is found for cyclopentyl at an acetonitrile/water ratio of 2: this activity
corresponds to 95% of that of Ti-(cyclopentyl silsesquioxane a7b3), so a relevant
improvement of activity is obtained by tuning this parameter.
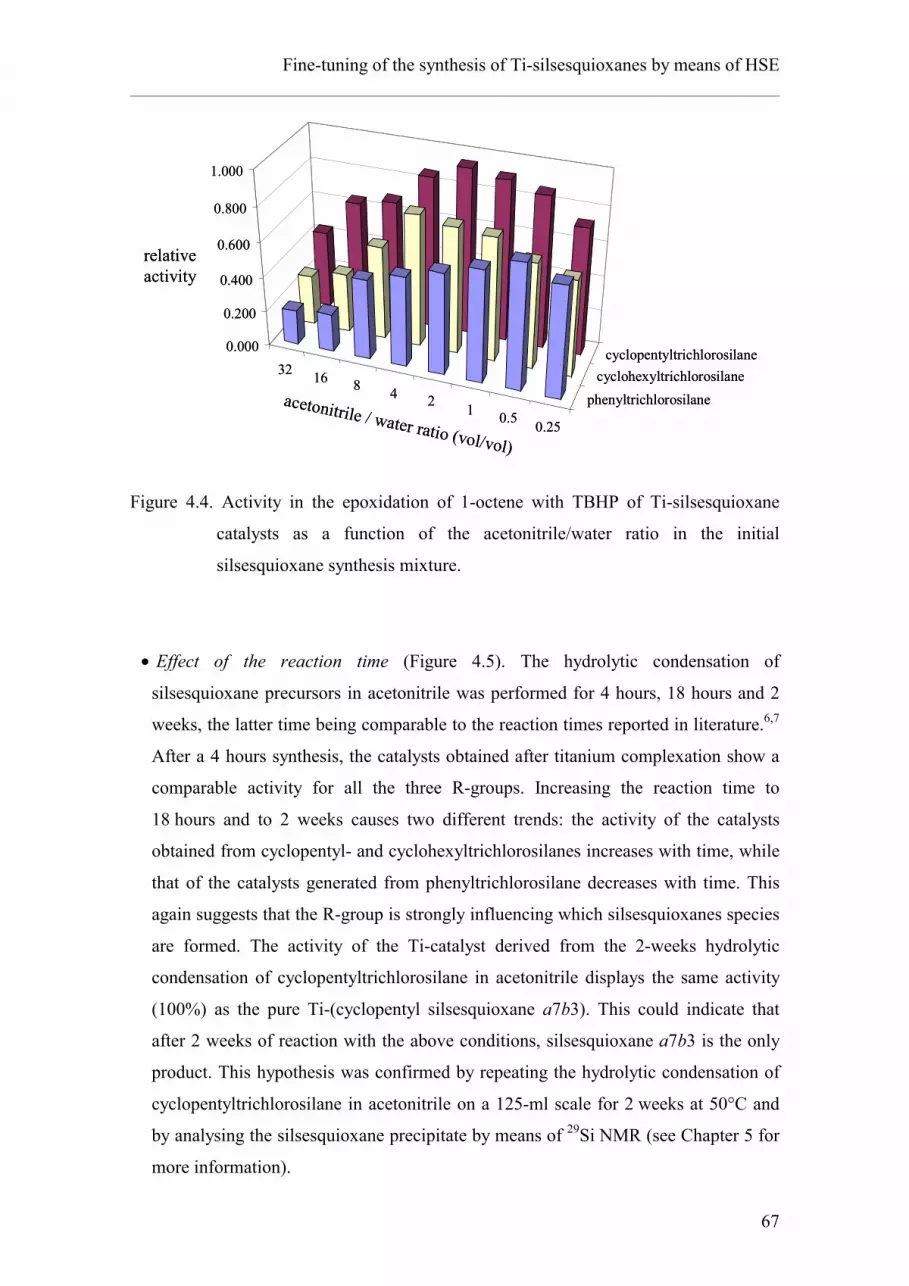
Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
67
Figure 4.4. Activity in the epoxidation of 1-octene with TBHP of Ti-silsesquioxane
catalysts as a function of the acetonitrile/water ratio in the initial
silsesquioxane synthesis mixture.
• Effect of the reaction time (Figure 4.5). The hydrolytic condensation of
silsesquioxane precursors in acetonitrile was performed for 4 hours, 18 hours and 2
weeks, the latter time being comparable to the reaction times reported in literature.6,7
After a 4 hours synthesis, the catalysts obtained after titanium complexation show a
comparable activity for all the three R-groups. Increasing the reaction time to
18 hours and to 2 weeks causes two different trends: the activity of the catalysts
obtained from cyclopentyl- and cyclohexyltrichlorosilanes increases with time, while
that of the catalysts generated from phenyltrichlorosilane decreases with time. This
again suggests that the R-group is strongly influencing which silsesquioxanes species
are formed. The activity of the Ti-catalyst derived from the 2-weeks hydrolytic
condensation of cyclopentyltrichlorosilane in acetonitrile displays the same activity
(100%) as the pure Ti-(cyclopentyl silsesquioxane a7b3). This could indicate that
after 2 weeks of reaction with the above conditions, silsesquioxane a7b3 is the only
product. This hypothesis was confirmed by repeating the hydrolytic condensation of
cyclopentyltrichlorosilane in acetonitrile on a 125-ml scale for 2 weeks at 50°C and
by analysing the silsesquioxane precipitate by means of 29Si NMR (see Chapter 5 for
more information).
32 16 8 4 2 1 0.5 0.25
phenyltrichlorosilanecyclohexyltrichlorosilane
cyclopentyltrichlorosilane0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
acetonitrile / water ratio (vol/vol)
32 16 8 4 2 1 0.5 0.25
phenyltrichlorosilanecyclohexyltrichlorosilane
cyclopentyltrichlorosilane0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
acetonitrile / water ratio (vol/vol)
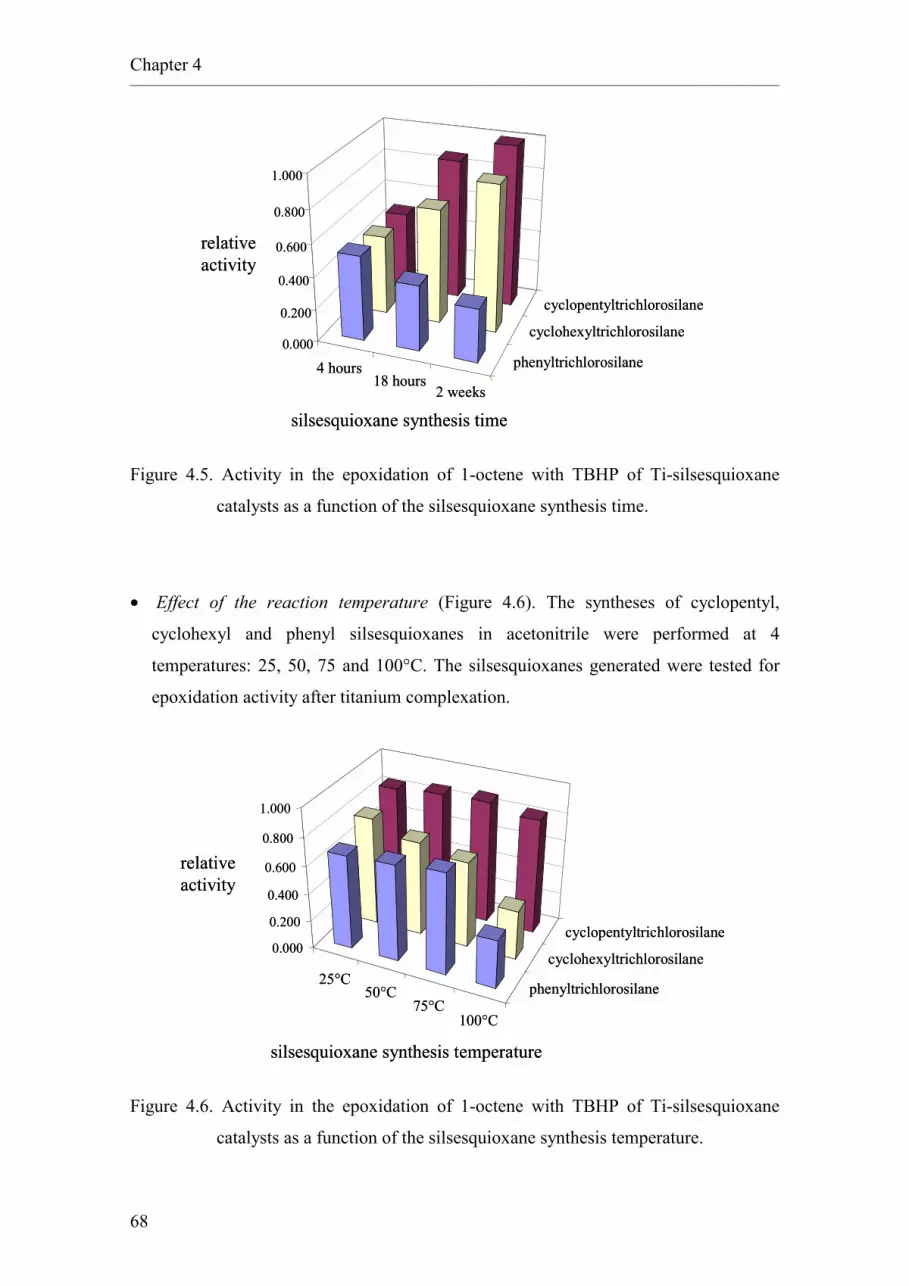
Chapter 4 __________________________________________________________________________________________________________
68
Figure 4.5. Activity in the epoxidation of 1-octene with TBHP of Ti-silsesquioxane
catalysts as a function of the silsesquioxane synthesis time.
• Effect of the reaction temperature (Figure 4.6). The syntheses of cyclopentyl,
cyclohexyl and phenyl silsesquioxanes in acetonitrile were performed at 4
temperatures: 25, 50, 75 and 100°C. The silsesquioxanes generated were tested for
epoxidation activity after titanium complexation.
Figure 4.6. Activity in the epoxidation of 1-octene with TBHP of Ti-silsesquioxane
catalysts as a function of the silsesquioxane synthesis temperature.
4 hours18 hours
2 weeks
phenyltrichlorosilane
cyclohexyltrichlorosilane
cyclopentyltrichlorosilane
0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
silsesquioxane synthesis time
4 hours18 hours
2 weeks
phenyltrichlorosilane
cyclohexyltrichlorosilane
cyclopentyltrichlorosilane
0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
silsesquioxane synthesis time
25°C50°C
75°C100°C
phenyltrichlorosilane
cyclohexyltrichlorosilane
cyclopentyltrichlorosilane0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
silsesquioxane synthesis temperature
25°C50°C
75°C100°C
phenyltrichlorosilane
cyclohexyltrichlorosilane
cyclopentyltrichlorosilane0.000
0.200
0.400
0.600
0.800
1.000
relativeactivity
silsesquioxane synthesis temperature

Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
69
Some technical problems were discovered in retrospect, i.e. the leaking of the
reaction vessels at high temperatures. This slightly affected the reliability of the
results for T ≥ 75°C and the data have to be interpreted with this in mind. However,
there seems to be a trend showing that temperature does not have a major effect on
the performance of the silsesquioxanes as precursors for titanium catalysts.
Some general conclusions can be drawn from these experiments. The activity of
the Ti-catalysts based on phenyl silsesquioxanes decreases when increasing the
concentration of the trichlorosilane and with longer reaction times for the hydrolytic
condensation. For cyclohexyl and cyclopentyl the trend concerning the reaction time
seems to be opposite. Further, the dependence on the trichlorosilane and water
concentrations and on the reaction time suggests that the formation of silsesquioxanes is
kinetically controlled. It can be hypothesised that the nature of the R-group influences
the energy of intermediates in the synthesis of silsesquioxanes, determining which
silsesquioxane structure is favoured. If the hypothesis is correct, this means that the
favoured phenyl silsesquioxanes are completely condensed and/or consist of other
structures that are not suitable for complexating titanium. On the other hand, the favoured
cyclohexyl and cyclopentyl silsesquioxane would be silsesquioxane a7b3 and/or another
incompletely condensed structure (see Chapter 5) suitable for complexating titanium in
a way that leads to a very active catalyst. The hypothesis is in agreement with the
characterisation of the HSE lead described in Paragraph 3.3.1 and with the literature,
where the formation of completely condensed phenyl silsesquioxanes8 and of
incompletely condensed cyclohexyl and cyclopentyl silsesquioxane a7b36,7,9 are reported.
4.3.3 Effect of the nature of the titanium centre
After the optimisation of the synthesis of the silsesquioxane precursors, it is
interesting to further study the titanium-silsesquioxane epoxidation catalysts by varying
the nature of the active titanium centre. Different ligands on the titanium could
influence the activity of the catalyst both through steric and electronic effects.10,11 It
would be particularly useful to identify a titanium silsesquioxane that could be used
with aqueous hydrogen peroxide (H2O2) as the oxidant. The advantage of using
hydrogen peroxide rather than TBHP or other organic hydroperoxides, is that hydrogen

Chapter 4 __________________________________________________________________________________________________________
70
peroxide would yield H2O as the by-product with evident environmental and
economical benefits for the process. So far, the use of H2O2 as the oxidant with
homogeneous titanium catalysts has been hindered either by the strong adsorption of
H2O on the titanium centre or by the rapid deactivation of the catalyst due to the
leaching/hydrolysis of the titanium centre and subsequent formation of inactive
oligomeric TiOx species. Few examples of epoxidation of alkenes with H2O2 using
heterogeneous titanium catalysts with a hydrophobic environment have been
reported.12,13
Here, HSE techniques were used for a further secondary screening of the titanium
silsesquioxanes. As silsesquioxane precursors, both the pure cyclopentyl
silsesquioxane a7b3 and the lead mixture obtained from the previous HSE
screenings5,14,15 (cyclopentyltrichlorosilane hydrolysed for 18 hours in acetonitrile) were
used. As oxidants, TBHP in cyclohexane and H2O2 (aq) were used. As titanium
precursors, 5 different titanium alkoxides Ti(OL)4 (L = Me, Et, Pr, Pri, Bu) and
cyclopentadienyltitanium trichloride were chosen. Besides, a series of diols (ethylene
glycol, 2,3-butanediol, 1,2-cyclohexanediol (mixture of cis and trans), pinacol and
benzopinacol) were used as ligands for the Ti-centre, with Ti(OBu)4 as titanium
precursor. The purpose of having a diol coordinating to the Ti-centre (Figure 4.7) is to
reduce its accessibility for polar molecules by increasing the hydrophobicity around the
catalytic centre:13 this may prevent the reaction with water and consequent catalyst
deactivation when using H2O2 as oxidant.
Figure 4.7. Possible interactions between a diol and the Ti-centre on a silsesquioxane.
The resulting 44-element parameter space was screened for activity in the epoxidation
of 1-octene (Figure 4.8). The values are normalised to the activity of the complex
OL
Ti
O
Ti
LO
Ti
OHHO
RIV
RIIIRII
RI
OH OH
HO
RIV
RIII
RIIRI
RIV
RIII
RIIRI
or/and
OL
Ti
O
Ti
LO
Ti
OHHO
RIV
RIIIRII
RI
OH OH
HO
RIV
RIII
RIIRI
RIV
RIII
RIIRI
or/and
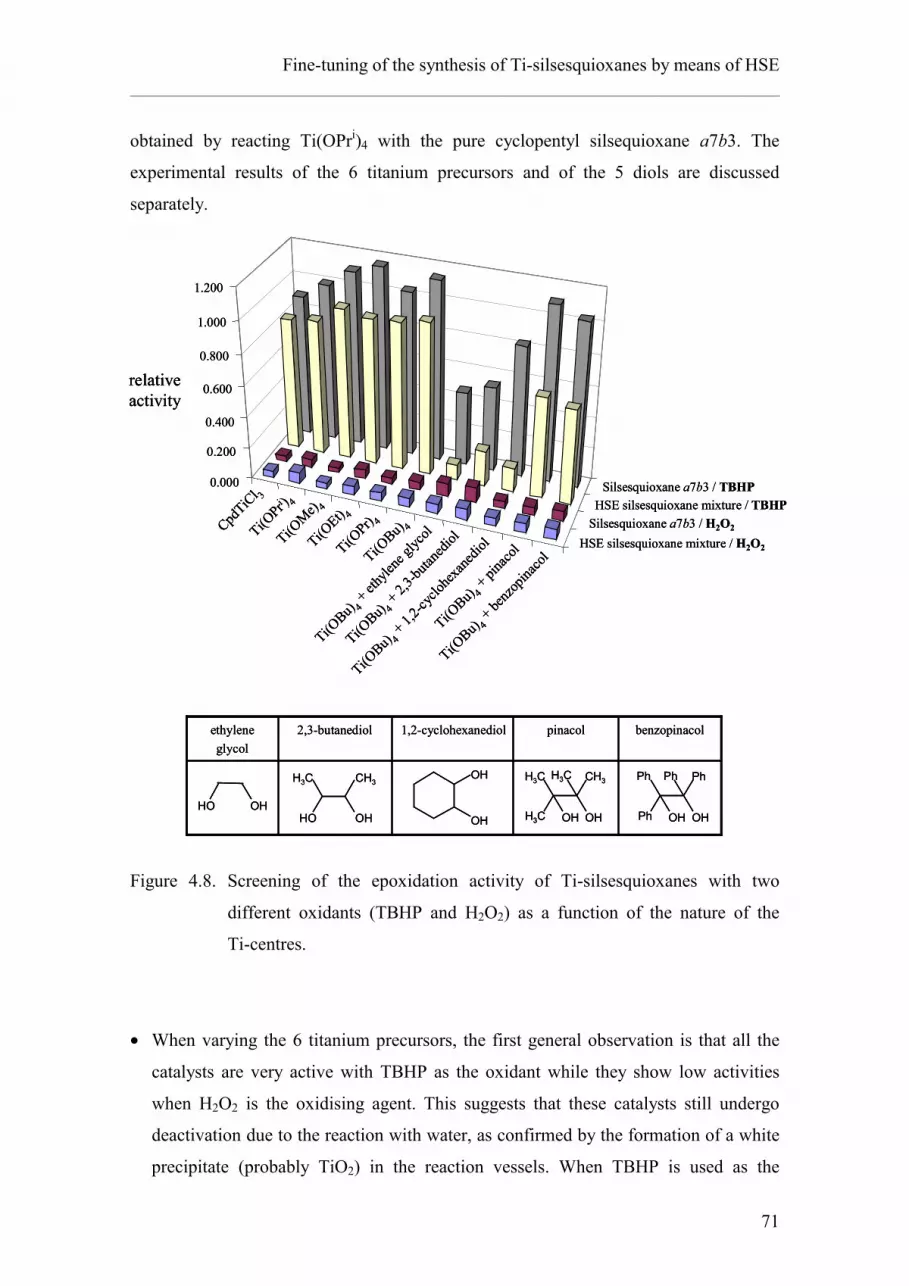
Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
71
obtained by reacting Ti(OPri)4 with the pure cyclopentyl silsequioxane a7b3. The
experimental results of the 6 titanium precursors and of the 5 diols are discussed
separately.
Figure 4.8. Screening of the epoxidation activity of Ti-silsesquioxanes with two
different oxidants (TBHP and H2O2) as a function of the nature of the
Ti-centres.
• When varying the 6 titanium precursors, the first general observation is that all the
catalysts are very active with TBHP as the oxidant while they show low activities
when H2O2 is the oxidising agent. This suggests that these catalysts still undergo
deactivation due to the reaction with water, as confirmed by the formation of a white
precipitate (probably TiO2) in the reaction vessels. When TBHP is used as the
benzopinacolpinacol1,2-cyclohexanediol2,3-butanediolethyleneglycol
OHHOOHHO
H3C CH3OH
OH OH
CH3
OH
H3CH3C
H3C OH
Ph
OH
PhPh
Ph
CpdTiC
l 3
Ti(OPri ) 4
Ti(OMe) 4
Ti(OEt) 4
Ti(OPr) 4
Ti(OBu) 4
Ti(OBu) 4
+ ethy
lene g
lycol
Ti(OBu) 4
+ 2,3-b
utane
diol
Ti(OBu) 4
+ 1,2-c
ycloh
exan
ediol
Ti(OBu) 4
+ pinaco
l
Ti(OBu) 4
+ benz
opina
col
HSE silsesquioxane mixture / H2O2
Silsesquioxane a7b3 / H2O2
HSE silsesquioxane mixture / TBHPSilsesquioxane a7b3 / TBHP0.000
0.200
0.400
0.600
0.800
1.000
1.200
relativeactivity
benzopinacolpinacol1,2-cyclohexanediol2,3-butanediolethyleneglycol
OHHOOHHO
H3C CH3OH
OH OH
CH3
OH
H3CH3C
H3C OH
Ph
OH
PhPh
Ph
benzopinacolpinacol1,2-cyclohexanediol2,3-butanediolethyleneglycol
OHHOOHHO
H3C CH3OH
OH OH
CH3
OH
H3CH3C
H3C OH
Ph
OH
PhPh
Ph
CpdTiC
l 3
Ti(OPri ) 4
Ti(OMe) 4
Ti(OEt) 4
Ti(OPr) 4
Ti(OBu) 4
Ti(OBu) 4
+ ethy
lene g
lycol
Ti(OBu) 4
+ 2,3-b
utane
diol
Ti(OBu) 4
+ 1,2-c
ycloh
exan
ediol
Ti(OBu) 4
+ pinaco
l
Ti(OBu) 4
+ benz
opina
col
HSE silsesquioxane mixture / H2O2
Silsesquioxane a7b3 / H2O2
HSE silsesquioxane mixture / TBHPSilsesquioxane a7b3 / TBHP0.000
0.200
0.400
0.600
0.800
1.000
1.200
relativeactivity
CpdTiC
l 3
Ti(OPri ) 4
Ti(OMe) 4
Ti(OEt) 4
Ti(OPr) 4
Ti(OBu) 4
Ti(OBu) 4
+ ethy
lene g
lycol
Ti(OBu) 4
+ 2,3-b
utane
diol
Ti(OBu) 4
+ 1,2-c
ycloh
exan
ediol
Ti(OBu) 4
+ pinaco
l
Ti(OBu) 4
+ benz
opina
col
HSE silsesquioxane mixture / H2O2
Silsesquioxane a7b3 / H2O2
HSE silsesquioxane mixture / TBHPSilsesquioxane a7b3 / TBHP0.000
0.200
0.400
0.600
0.800
1.000
1.200
relativeactivity

Chapter 4 __________________________________________________________________________________________________________
72
oxidant, the pure silsesquioxane a7b3 turns out being a slightly better precursor than
the HSE mixture for any Ti-centre. On average, the ratio between the activities of the
HSE mixture and the pure a7b3 catalysts is 0.87. Considering the various Ti-centres,
hardly any variation in activity was observed among the titanium alkoxides with
L = Me, Et, Pr, Bu: most of the differences fall within the same range, given that the
experimental error of these HSE data is ± 3%. The activity with Ti(OPri)4 and
cyclopentadienyltitanium trichloride are slightly, but noticeably, lower. This trend
can be explained by considering that the isopropoxy and cyclopentadienyl groups are
bulkier than the linear alkoxides. Therefore, the accessibility to the titanium site is
reduced, causing a decrease in catalytic activity.16 On the basis of literature data,16 a
trend in activity among the linear alkoxides was expected (the shorter the alkyl chain,
the higher the activity). The experimental data reported here were not conclusive in
confirming this trend, due to the level of inaccuracy of the HSE techniques. From a
methodological point of view this indicates a limit of HSE techniques, which may
not be able to discern between activities within a very narrow range of values.
• For what concerns the addition of a diol to the Ti-centre, the expected effect should
be a lower catalytic activity when using TBHP as oxidant due to the lower
accessibility of the site (similarly to what is described above for isopropoxy and
cyclopentadienyl groups). On the other hand, a higher activity with H2O2 may occur
if the diol ligand were protecting the titanium site from reaction with water. The data
reported in Figure 4.8 show the expected decrease in activity with TBHP when
ethylene glycol, 2,3-butanediol and 1,2-cyclohexanediol are used, while the effect is
less relevant with the two bulkiest diols, pinacol and benzopinacol. This indicates
that the coordination of pinacol and benzopinacol to the titanium centres on the
silsesquioxanes is probably sterically hindered, particularly for the Ti-centre on the
pure silsesquioxane a7b3. The same effect to a minor extent can explain the
intermediate activity obtained with 1,2-cyclohexanediol. The fact that the presence of
a diol causes a much higher decrease in activity with the HSE mixture of
silsesquioxanes than with pure silsesquioxane a7b3 points to the different nature of
the Ti-complexes present in the HSE mixture, some of which probably become
completely inaccessible for catalysis in the presence of one or more diol ligands.
When H2O2 is used as oxidant, a positive effect of the diols can be seen mainly with
ethylene glycol and 2,3-butanediol, in agreement with the result for TBHP, where for

Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
73
these two diols the biggest decrease in activity is observed. Unfortunately, the
activity is still rather low, with the best result being 8% of conversion of H2O2 into
1,2-epoxyoctane in the case of 2,3-butanediol with pure silsesquioxane a7b3. This
indicates that the effect of the diols, although detectable – thus, the concept works, is
not sufficient to produce a good and robust catalyst for the epoxidation of 1-octene
with hydrogen peroxide.
4.4 Conclusions
The effect of different variables on the preparation of Ti-silsesquioxane catalysts
was explored by means of HSE techniques. Fine-tuning the parameters influencing the
synthesis of the silsesquioxane precursors led to slight improvements in the activity of
the Ti-catalysts and to some knowledge in regards to how these parameters affect the
hydrolytic condensation of trichlorosilanes. The initial hypothesis that, among these
parameters, the nature of the solvent and of the R-group were dominant in influencing
the formation of silsesquioxanes was confirmed. The optimisation of the features of the
catalytic titanium centre anchored to the silsesquioxane structures evidenced the
importance of the accessibility of the titanium site in determining the activity of the
catalyst.
4.5 Experimental
All the experiments described in this chapter were performed by means of the
HSE automated parallel synthesis workstation described in Appendix A. The
experimental procedure followed for the fine-tuning of the parameters influencing the
synthesis of silsesquioxane precursors is analogous to that of the first HSE study
described in Chapter 3. For the HSE study of the Ti-centres, titanium complexation was
performed by dissolving either 4.7·10-5 mol of the pure cyclopentyl silsesquioxane a7b3
or a corresponding amount of the HSE-optimised cyclopentyl silsesquioxane mixture in
2 ml of THF, followed by the addition of 54 µmol of one of the titanium precursors as a
solution in an organic solvent (the corresponding alcohol for the titanium alkoxides,

Chapter 4 __________________________________________________________________________________________________________
74
dichloromethane for cyclopentadienyltitanium trichloride). Samples were stirred in an
orbital shaker for 5 hours at 60°C. In the case of the experiment with diols, THF
solutions, each containing 0.1 mmol of a diol, were added to the appropriate vials.
Samples were then stirred in the orbital shaker for 10 hours at 50°C. Finally, THF was
removed by means of a vacuum centrifuge and the samples were stored under argon.
The activity of the Ti-silsesquioxanes in the epoxidation of 1-octene was determined by
adding to each dried sample 2.26 ml of 1-octene (with 2%vol of decane as internal
standard), that acted both as reactant and as solvent, and 121 µl of TBHP solution
(~45%wt solution in cyclohexane) or 70 µl of H2O2 (35%wt solution in H2O). H2O2 and
1-octene are not miscible: to obtain a monophasic system, the reactions were
alternatively performed with 1-propanol as solvent (1.13 ml of 1-propanol, 1.13 ml of
1-octene and 70 µl of aqueous H2O2). However, the activities obtained in these
conditions were lower than with the biphasic system, probably because of faster catalyst
deactivation caused by reaction with water. Samples were taken after having been
stirred in the orbital shaker for 4 hours at 80°C (TBHP) or for 6 hours at 60°C (H2O2)
and analysed on the UNICAM Pro GC. The reported activities were obtained by
normalising the 1,2-epoxyoctane GC peak area by means of the internal standard. As a
reference, a blank reaction with 1-octene and H2O2 and without catalyst was performed,
giving no epoxidation activity.

Fine-tuning of the synthesis of Ti-silsesquioxanes by means of HSE __________________________________________________________________________________________________________
75
References
1 K.R. Popper, The Logic of Scientific Discovery, Hutchinson of London, 1959. 2 K.R. Popper, Conjectures and Refutations. The Growth of Scientific Knowledge, Routledge and Kegan
Paul, 1969, 3rd edition. 3 P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583, see also Paragraph 2.2.1. 4 See Paragraph 3.3. 5 P. P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Angew. Chem. Int. Ed., 2001, 40,
740, see also Chapter 3. 6 F. J. Feher, D. A. Newman, J. F. Walzer, J. Am. Chem. Soc., 1989, 111, 1741. 7 F. J. Feher, T. A. Budzichowski, R. L. Blanski, K. J. Weller, J. W. Ziller, Organometallics, 1991, 10,
2526. 8 J.F. Brown, L.H. Vogt, P.I. Prescott J. Am. Chem. Soc., 1964, 86, 1120. 9 J.F. Brown, L.H. Vogt, J. Am. Chem. Soc., 1965, 87, 4313. 10 M.C. Klunduk, T. Maschmeyer, J.M. Thomas, B.F.G. Johnson, Chem. Eur. J., 1999, 5, 1481. 11 J.M. Thomas, G. Sankar, M.C. Klunduk, M.P. Attfield, T. Maschmeyer, B.F.G. Johnson, R.G. Bell, J.
Phys. Chem. B, 1999, 103, 8809. 12 F. Figueras, H. Kochkar, S. Caldarelli, Micropor. Mesopor. Mat., 2000, 39, 249. 13 J.M. Fraile, J.I. García, J.A. Mayoral, E. Vispe, J. Catal., 2000, 189, 40. 14 P.P. Pescarmona, T. Maschmeyer, NATO Science Series, 2002, Ser. II Vol. 69, 173. 15 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Chem. Eng. Commun., accepted
for publication. 16 T. Maschmeyer, M.C. Klunduk, C.M. Martin, D.S. Shephard, J.M. Thomas, B.F.G. Johnson, Chem.
Commun., 1997, 1847.

76

77
5
Fast and high-yield syntheses of cyclopentyl and cyclohexyl silsesquioxanes using acetonitrile as reactive solvent
Abstract
New and efficient methods for the synthesis of cyclopentyl and cyclohexyl
silsesquioxanes are described. In the case of cyclopentyl silsesquioxanes, structure a7b3
was obtained selectively with an isolated yield of 64% within 18 hours. The synthesis
was monitored by means of mass spectrometry and in-situ infrared spectroscopy in
combination with chemometric analysis methods. This study allowed to identify the
silsesquioxane species present in the reaction mixture, to explain the high selectivity of
the reaction towards silsesquioxane a7b3 and to propose a mechanism for its formation.
In the case of cyclohexyl silsesquioxanes, the synthesis gave high yields but was not
selective towards a particular structure type. The main products were: silsesquioxanes
a6b0, a6b2, a7b1 and a7b3. The crucial role of acetonitrile as a reactive solvent in the
syntheses is discussed.
____________________ The contents of this chapter have been submitted to:
P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, Eur. J. Inorg. Chem.
P.P. Pescarmona, M.E. Raimondi, J. Tetteh, B. McKay, T. Maschmeyer, J. Phys. Chem. B, accepted.

Chapter 5 __________________________________________________________________________________________________________
78
5.1 Introduction
Chapter 3 and 4 described a study of the synthesis of silsesquioxanes1 as
precursors for Ti-catalysts active in the epoxidation of alkenes, realised by means of
High-Speed Experimentation (HSE) techniques.2,3 The best catalysts were obtained with
cyclopentyl and cyclohexyl silsesquioxanes synthesised by the hydrolytic condensation
of the corresponding trichlorosilanes with acetonitrile as solvent. Here, a follow-up
study of these two HSE leads by means of other laboratory techniques is reported, with
the aim to fully characterise the silsesquioxane intermediates and products and to
understand the role of the solvent.
The hydrolytic condensation of trichlorosilanes in acetone to produce incompletely
condensed silsesquioxanes was first reported by Brown and Vogt4 (for R = cyclohexyl)
and more recently it was optimised and studied in detail by Feher et al. (for
R = cyclopentyl and cyclohexyl).5,6 In the case of R = cyclopentyl, silsesquioxane a7b3
(Figure 5.1) was the only product and was obtained with a yield of 29% after 3 days of
reaction.6 When R = cyclohexyl, the hydrolytic condensation proceeded more slowly
and produced a mixture of three silsesquioxanes: a6b0, a7b3 and a8b2.5 Silsesquioxane
a7b3 was the main product, but with a low yield (8% after 12 weeks of reaction).
Figure 5.1. Silsesquioxanes a7b3, R7Si7O9(OH)3 [R = cyclopentyl, cyclohexyl].
OHOH
OH
R
R
OO
SiO
Si
O
O
OSiSi
OSiOSi
O
Si
a7b3
RR
R
R
R
OHOH
OH
R
R
OO
SiO
Si
O
O
OSiSi
OSiOSi
O
Si
a7b3
RR
R
R
R

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
79
More information about the hydrolytic condensation of organosilanes can be found in
Paragraph 2.2.1. The reaction scheme is reproduced below since it will be useful to refer
to it throughout this chapter:
RSiCl3 + 3 H2O RSi(OH)3 + 3 HCl [1]
a RSi(OH)3 (RSiO1.5)a(H2O)0.5b + (1.5a – 0.5b) H2O [2]
5.2 Results and discussion
5.2.1 Synthesis of cyclopentyl silsesquioxane a7b3
The hydrolytic condensation of cyclopentyltrichlorosilane in acetonitrile in the
presence of an excess of water led to the formation of a white precipitate. After 18 hours
of reaction at 50°C, a crude precipitate was obtained in ~50 % yield.I It mainly
contained silsesquioxane a7b3 [(c-C5H9)7Si7O9(OH)3], as determined on the basis of 29Si NMR and MS analyses.7 Small amounts of impurities, predominantly the cubic
completely condensed silsesquioxane a8b0, (c-C5H9)8Si8O12, were present in the
precipitate. If the reaction time was extended from 18 hours to 2 weeks,3 the yield of
precipitated silsesquioxanes was nearly quantitative with the main fraction still being
silsesquioxane a7b3. The synthesis was further optimised according to the fine-tuning of
the reaction conditions described in Paragraph 4.3.2 and on the basis of the observation of
Feher et al., that the yield of the synthesis of cyclopentyl silsesquioxane a7b3 in acetone
can be significantly improved by refluxing the reaction mixture.5 Therefore, the synthesis
method was modified by increasing the reaction temperature from 50°C to reflux
conditions. This led to an increase of the yield to 81% after 18 hours of reaction, with the
high selectivity towards silsesquioxane a7b3 being preserved. Silsesquioxane a7b3 was
then purified by pyridine extraction5,6 and the pure compound (as determined by
I Relative to silsesquioxane a7b3 (the yield is calculated by dividing the grams of product by the grams of
silsesquioxane a7b3 that would be obtained if the reaction quantitatively gave silsesquioxane a7b3).
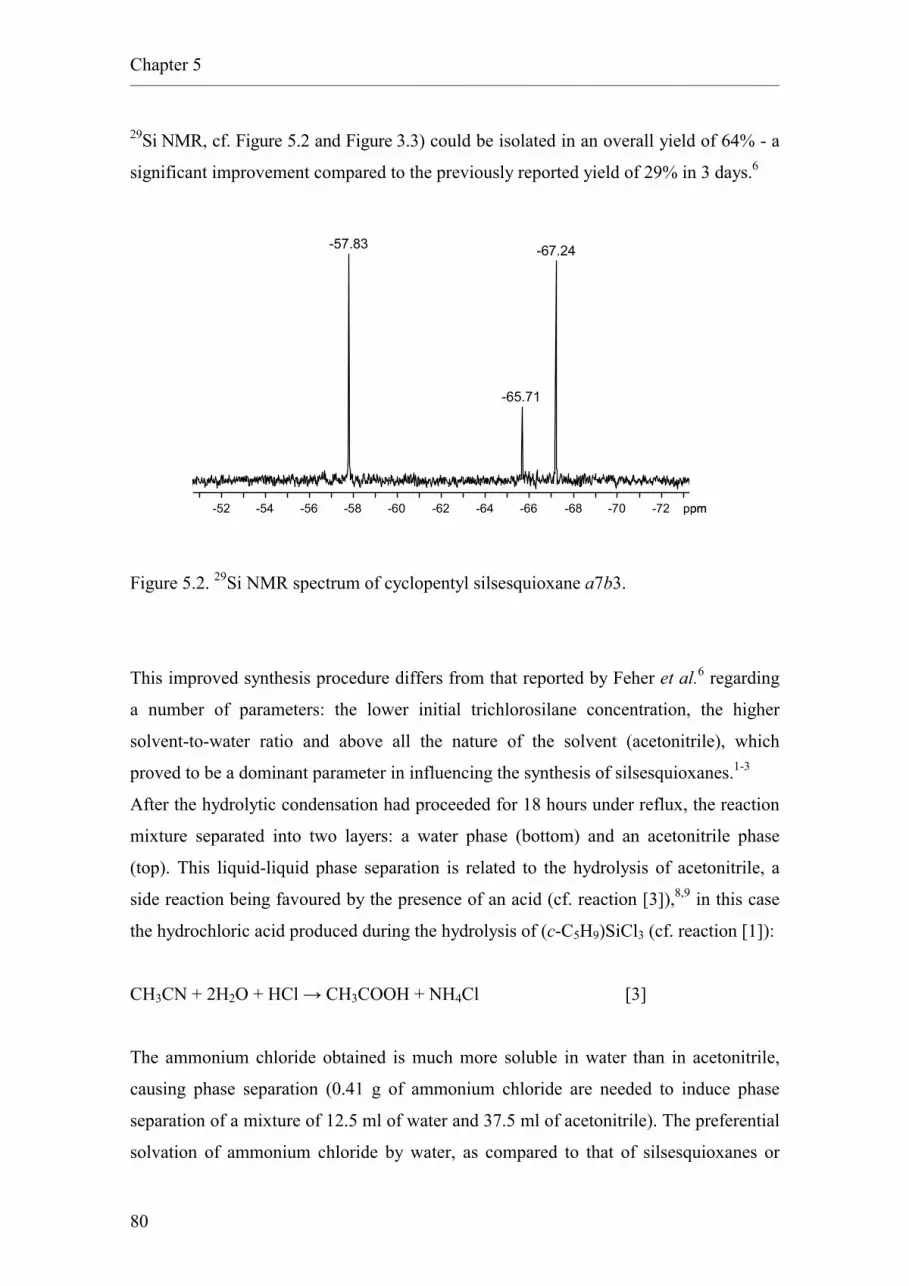
Chapter 5 __________________________________________________________________________________________________________
80
29Si NMR, cf. Figure 5.2 and Figure 3.3) could be isolated in an overall yield of 64% - a
significant improvement compared to the previously reported yield of 29% in 3 days.6
Figure 5.2. 29Si NMR spectrum of cyclopentyl silsesquioxane a7b3.
This improved synthesis procedure differs from that reported by Feher et al.6 regarding
a number of parameters: the lower initial trichlorosilane concentration, the higher
solvent-to-water ratio and above all the nature of the solvent (acetonitrile), which
proved to be a dominant parameter in influencing the synthesis of silsesquioxanes.1-3
After the hydrolytic condensation had proceeded for 18 hours under reflux, the reaction
mixture separated into two layers: a water phase (bottom) and an acetonitrile phase
(top). This liquid-liquid phase separation is related to the hydrolysis of acetonitrile, a
side reaction being favoured by the presence of an acid (cf. reaction [3]),8,9 in this case
the hydrochloric acid produced during the hydrolysis of (c-C5H9)SiCl3 (cf. reaction [1]):
CH3CN + 2H2O + HCl → CH3COOH + NH4Cl [3]
The ammonium chloride obtained is much more soluble in water than in acetonitrile,
causing phase separation (0.41 g of ammonium chloride are needed to induce phase
separation of a mixture of 12.5 ml of water and 37.5 ml of acetonitrile). The preferential
solvation of ammonium chloride by water, as compared to that of silsesquioxanes or
-57.83
-65.71
-67.24
ppm-68 -70-66 -72-64-62-60-54 -58-52 -56
-57.83
-65.71
-67.24
ppm-68 -70-66 -72-64-62-60-54 -58-52 -56

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
81
acetonitrile, drives reaction [2] towards the formation of more condensed species. This
effect is very pronounced towards the end of the experiment, when actual phase
separation is observed. At the same time, the hydrolysis of acetonitrile in presence of an
acid plays an important role due to the fact that the formation of ammonium chloride
(partially) neutralises the reaction mixture. A detrimental effect of the acidity of the
solution was already observed in the synthesis of silsesquioxane as Ti-catalyst
precursors.2 Since under acidic conditions silsesquioxanes undergo fast
hydrolysis/condensation reactions,1 the species formed might be partially decomposed
or further condensed before they would start to precipitate from the solution. The partial
neutralisation of the reaction mixture could slow down the hydrolysis/condensation
reactions, favouring the precipitation of the species that are only poorly soluble in this
medium, like the incompletely condensed silsesquioxane a7b3.II
A further contributing factor regarding the improved yield when using acetonitrile as the
solvent is its higher polarity compared to acetone, the solvent used in literature for the
synthesis of silsesquioxane a7b3.6 A highly polar solvent can better stabilise
incompletely condensed species by interaction with their silanol groups, therefore,
favouring the synthesis of these silsesquioxanes over completely condensed structures.
Moreover, it has been proposed that in the presence of a polar molecule the activation
barrier for the condensation reactions towards the formation of silsesquioxanes is
reduced and, therefore, the synthesis is sped up.10,11
In this context, it is also relevant to note that acetonitrile has a higher boiling point than
acetone (82°C against 56°C). Since the reaction is performed under reflux, the synthesis
is accelerated.
High-Speed Experimentation was an efficient means to determine the optimum
conditions for the high selectivity synthesis of silsesquioxane a7b3,2 but could not
directly be used to elucidate the mechanism of formation. In order to shed light on this
mechanism, to identify the species formed during the process and to try to explain the II While the partial neutralisation of the HCl formed during the hydrolysis of trichlorosilanes might have a
role in accelerating the formation of silsesquioxane precipitates, the complete neutralisation is
detrimental. A decrease of the yield (from 79% to 44%, as crude precipitate) was observed by performing
the synthesis of cyclohexyl silsesquioxanes in the presence of triethylamine (C2H5)3N. This indicates that
without HCl the hydrolysis/condensation reactions are significantly slowed down from the beginning of
the synthesis.
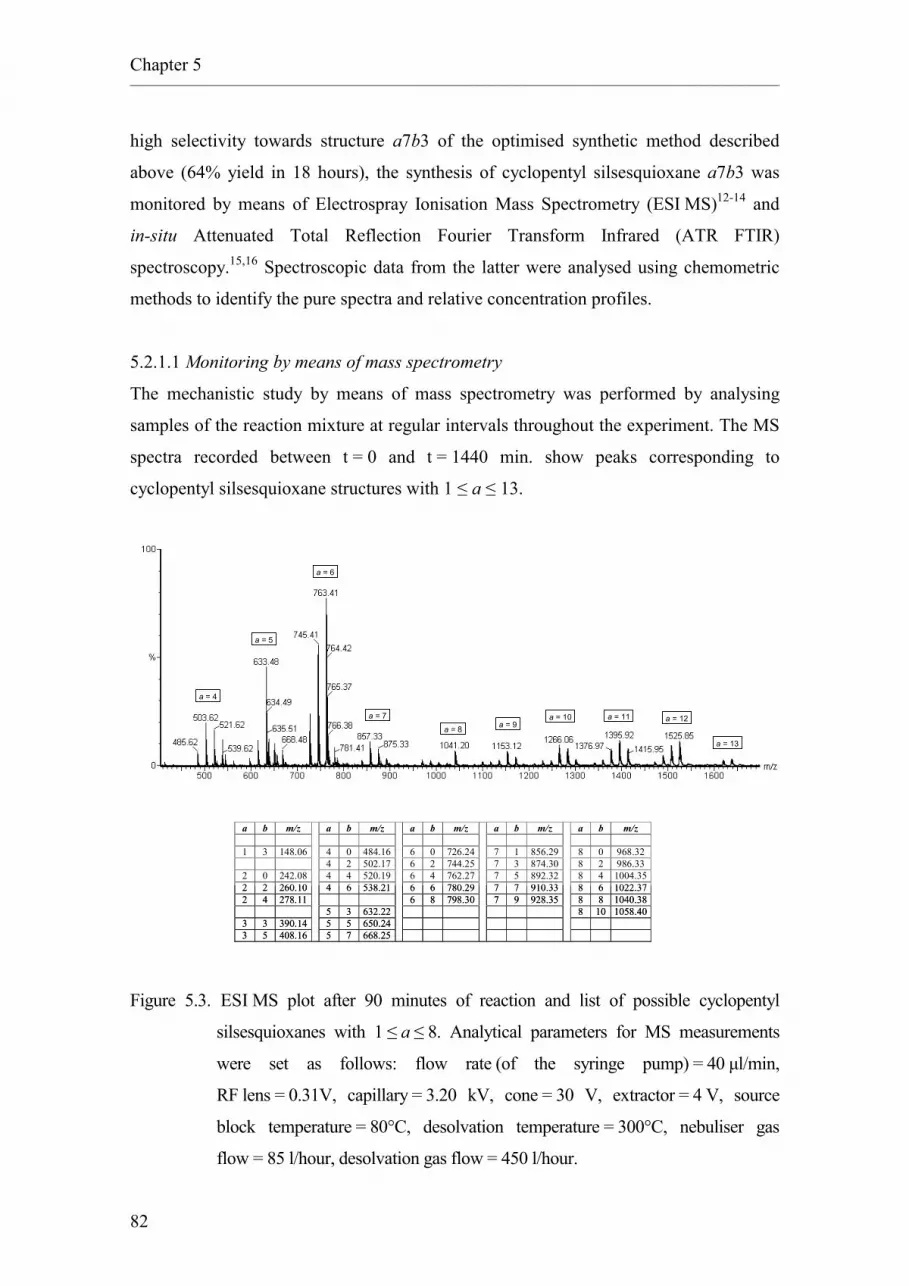
Chapter 5 __________________________________________________________________________________________________________
82
high selectivity towards structure a7b3 of the optimised synthetic method described
above (64% yield in 18 hours), the synthesis of cyclopentyl silsesquioxane a7b3 was
monitored by means of Electrospray Ionisation Mass Spectrometry (ESI MS)12-14 and
in-situ Attenuated Total Reflection Fourier Transform Infrared (ATR FTIR)
spectroscopy.15,16 Spectroscopic data from the latter were analysed using chemometric
methods to identify the pure spectra and relative concentration profiles.
5.2.1.1 Monitoring by means of mass spectrometry
The mechanistic study by means of mass spectrometry was performed by analysing
samples of the reaction mixture at regular intervals throughout the experiment. The MS
spectra recorded between t = 0 and t = 1440 min. show peaks corresponding to
cyclopentyl silsesquioxane structures with 1 ≤ a ≤ 13.
Figure 5.3. ESI MS plot after 90 minutes of reaction and list of possible cyclopentyl
silsesquioxanes with 1 ≤ a ≤ 8. Analytical parameters for MS measurements
were set as follows: flow rate (of the syringe pump) = 40 µl/min,
RF lens = 0.31V, capillary = 3.20 kV, cone = 30 V, extractor = 4 V, source
block temperature = 80°C, desolvation temperature = 300°C, nebuliser gas
flow = 85 l/hour, desolvation gas flow = 450 l/hour.
a = 4
a = 5
a = 6
a = 8a = 7
a = 9a = 10 a = 11 a = 12
a = 13
a b m/z a b m/z a b m/z a b m/z a b m/z
1 3 148.06 4 0 484.16 6 0 726.24 7 1 856.29 8 0 968.32 4 2 502.17 6 2 744.25 7 3 874.30 8 2 986.33
2 0 242.08 4 4 520.19 6 4 762.27 7 5 892.32 8 4 1004.352 2 260.10 4 6 538.21 6 6 780.29 7 7 910.33 8 6 1022.372 4 278.11 6 8 798.30 7 9 928.35 8 8 1040.38 5 3 632.22 8 10 1058.40
3 3 390.14 5 5 650.24 3 5 408.16
5 7 668.25
a = 4
a = 5
a = 6
a = 8a = 7
a = 9a = 10 a = 11 a = 12
a = 13
a = 4
a = 5
a = 6
a = 8a = 7
a = 9a = 10 a = 11 a = 12
a = 13
a b m/z a b m/z a b m/z a b m/z a b m/z
1 3 148.06 4 0 484.16 6 0 726.24 7 1 856.29 8 0 968.32 4 2 502.17 6 2 744.25 7 3 874.30 8 2 986.33
2 0 242.08 4 4 520.19 6 4 762.27 7 5 892.32 8 4 1004.352 2 260.10 4 6 538.21 6 6 780.29 7 7 910.33 8 6 1022.372 4 278.11 6 8 798.30 7 9 928.35 8 8 1040.38 5 3 632.22 8 10 1058.40
3 3 390.14 5 5 650.24 3 5 408.16
5 7 668.25

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
83
Peaks due to a = 7 species (m/z = 839.3, 857.3, 875.3, 893.3) are present in all the
recorded spectra (see Figure 5.3 for an example). The relative abundance of these peaks
compared to that of the peaks of other silsesquioxane species is rather low at any
reaction time, indicating a low concentration in solution. Knowing that the precipitate
produced by the synthesis is mainly silsesquioxane a7b3, it can be inferred that this
compound has a low solubility in the reaction mixture and tends to precipitate as it gets
formed. Its lower solubility with respect to other cyclopentyl silsesquioxanes is
probably due to the tendency of silsesquioxane a7b3 to form dimers,5,17 which are
insoluble in polar solvents such as acetonitrile. This is in agreement with the observed
formation of a white precipitate after 1 hour of reaction.
The concentration of the a = 7 species in solution can, therefore, be considered
approximately constant during the course of the reaction. This assumption allowed to
normalise the intensity of the peaks of the other silsesquioxanes relative to the a = 7
peaks, making it possible to compare the spectra measured at the various reaction
times.III The relative concentrations of the principal silsesquioxane species with
increasing a values are plotted against reaction time in Figure 5.4.
This plot shows how various silsesquioxane species formed and disappeared over time.
At t = 0, silsesquioxane species with 1 ≤ a ≤ 6 were present in considerable amounts.
No cyclopentyltrichlorosilane was detected, indicating that the hydrolysis step [1] was
complete before the first sample was injected in the mass spectrometer.17 The presence
of various species at t = 0 shows that the process of condensation of silsesquioxane
a1b3, (c-C5H9)Si(OH)3, (step [2]) leading to the formation of larger silsesquioxane
structures is also very fast. At t = 0, species with a = 4 were the most abundant.
Silsesquioxanes with 1 ≤ a ≤ 4 can be seen as the building blocks for larger
silsesquioxane structures (see Figure 5.5 for a proposed synthesis scheme). They are
likely to be reactive species that tend to condense further, as confirmed by the fact that
their concentrations diminished quickly as the reaction proceeded. The smaller the
structures, the faster they disappeared to form larger ones. At t = 30 minutes,
silsesquioxane a1b3 had already disappeared from the reaction mixture. Silsesquioxanes
III A normalisation of the MS data is necessary, because the mass spectrometer produces plots in which
the intensities of the peaks are not absolute but normalised to the most intense peak, the value of which is
set at 100%.
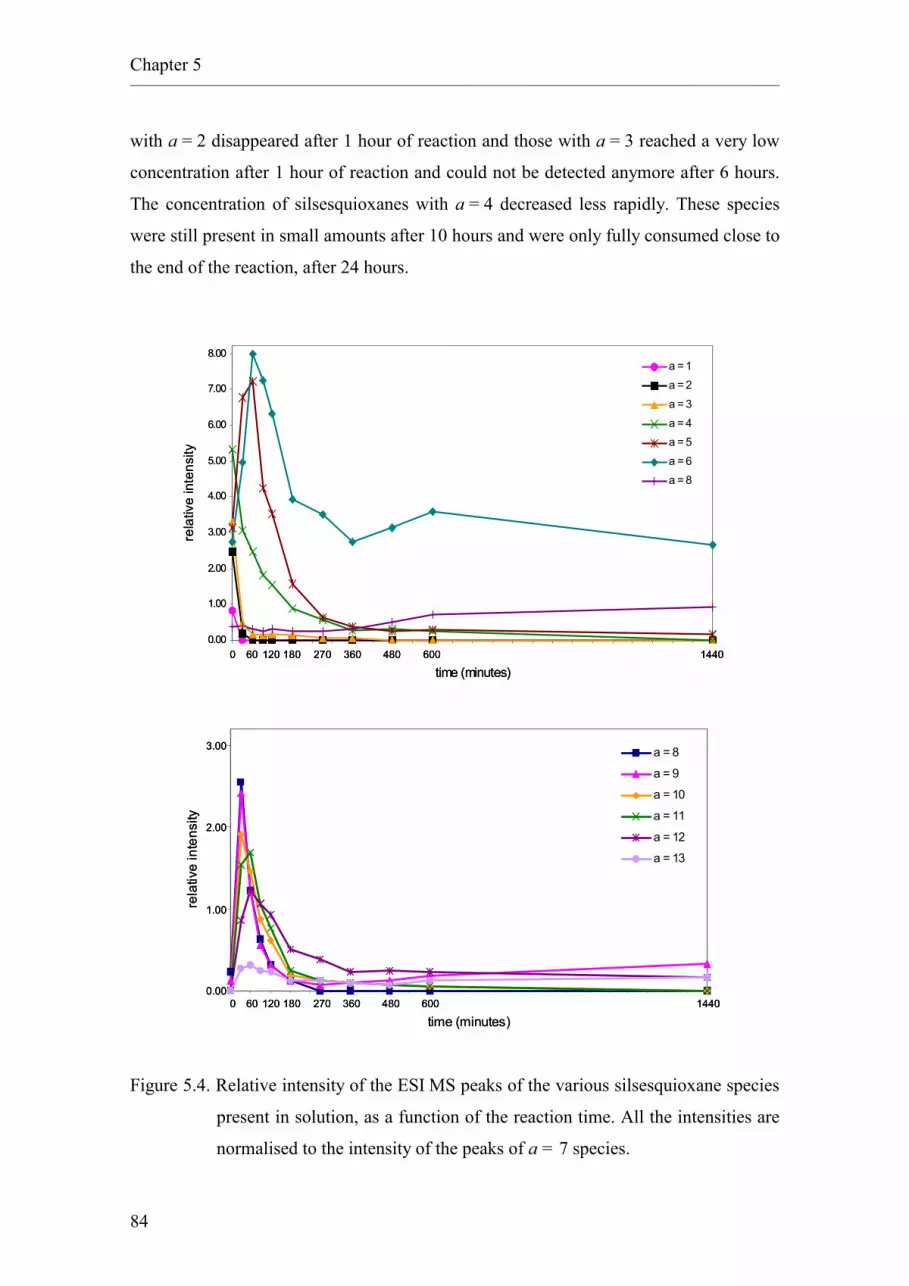
Chapter 5 __________________________________________________________________________________________________________
84
with a = 2 disappeared after 1 hour of reaction and those with a = 3 reached a very low
concentration after 1 hour of reaction and could not be detected anymore after 6 hours.
The concentration of silsesquioxanes with a = 4 decreased less rapidly. These species
were still present in small amounts after 10 hours and were only fully consumed close to
the end of the reaction, after 24 hours.
Figure 5.4. Relative intensity of the ESI MS peaks of the various silsesquioxane species
present in solution, as a function of the reaction time. All the intensities are
normalised to the intensity of the peaks of a = 7 species.
0.00
1.00
2.00
3.00
0 60 120 180 240 300 360 420 480 540 600 660 720 780 840 900 960 1020 1080 1140 1200 1260 1320 1380 1440
time (minutes)
rela
tive
inte
nsity
a = 8
a = 9
a = 10
a = 11
a = 12
a = 13
0.00
1.00
2.00
3.00
4.00
5.00
6.00
7.00
8.00
0 60 120 180 240 300 360 420 480 540 600 660 720 780 840 900 960 1020 1080 1140 1200 1260 1320 1380 1440
time (minutes)
rela
tive
inte
nsity
a = 1a = 2a = 3a = 4a = 5a = 6a = 8
0 60 120 180 270 360 480 600 1440
0 60 120 180 270 360 480 600 1440
3.00
2.00
1.00
0.000.00
1.00
2.00
3.00
0 60 120 180 240 300 360 420 480 540 600 660 720 780 840 900 960 1020 1080 1140 1200 1260 1320 1380 1440
time (minutes)
rela
tive
inte
nsity
a = 8
a = 9
a = 10
a = 11
a = 12
a = 13
0.00
1.00
2.00
3.00
4.00
5.00
6.00
7.00
8.00
0 60 120 180 240 300 360 420 480 540 600 660 720 780 840 900 960 1020 1080 1140 1200 1260 1320 1380 1440
time (minutes)
rela
tive
inte
nsity
a = 1a = 2a = 3a = 4a = 5a = 6a = 8
0 60 120 180 270 360 480 600 14400 60 120 180 270 360 480 600 1440
0 60 120 180 270 360 480 600 14400 60 120 180 270 360 480 600 1440
3.00
2.00
1.00
0.00
3.00
2.00
1.00
0.00

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
85
For a = 1, the only possible structure is the trisilanol a1b3.1 For a = 2 and a = 3,
more structures are possible but a2b4 and a3b5 are the only two that do not present a large
geometrical strain. For a = 4, two structures are the most likely: a4b4 and a4b6, the first
being a ring and the second a linear structure. The ring structure is part of the structure of
the final product a7b3, and so it can be assumed that a4b418 is the precursor species. These
structures are schematised in Figure 5.5, together with their most likely paths of reaction.
For most of the reaction, silsesquioxanes with a = 5 and a = 6 were the two
major species in solution. Their concentrations increased in the beginning of the
reaction to reach a maximum after 1 hour and then gradually decreased. This happened
more rapidly for species with a = 5 that were only present in very small amounts at the
end of the reaction, than for silsesquioxanes with a = 6, which were still present in
relevant amounts after 24 hours of reaction. The two most likely structures with a = 5
and a = 6 are silsesquioxanes a5b5 and a6b4, which are represented in Figure 5.5,
together with their possible pathways of formation.
For silsesquioxanes with a = 8, two groups of peaks with different relative
concentration profiles exist. The different behaviour as a function of the reaction time
suggests that the two groups of peaks belong to two different species with a = 8. The
first group presented a main peak at m/z = 987.15, which corresponds to an a8b2
silsesquioxane. This species has a low concentration during the entire reaction, with
only a slightly positive slope towards the end (see Figure 5.4, top plot). The second
group of peaks presented a main peak at m/z = 1041.20, its intensity reached a
maximum at t = 30 minutes and then decreased rapidly to disappear after 3 hours of
reaction (see Figure 5.4, bottom plot). The intensity profile for these species is very
similar to those of species with 9 ≤ a ≤ 13, indicating a maximum species concentration
at t = 30 minutes for a = 9, 10 and at t = 60 minutes for 11 ≤ a ≤ 13. Given the short
lifetime of the species with 8 ≤ a ≤ 13 and the fact that they were present in the early
stages of the reaction, it can be suggested that these species are instable aggregates of
two smaller silsesquioxanes.IV For example, species with a = 8 are probably dimers of
a = 4 species, those with a = 9 are constituted by a = 4 and a = 5 species, and so on.
IV A similar effect can be seen measuring a mass spectrum of pure cyclopentyl silsesquioxane a7b3 with
similar experimental conditions but at two different cone voltages: with cone = 30V two main peaks are
present, at m/z = 875.33 and at m/z = 1749.89; with cone = 65V, the peak at m/z = 1749.89, due to the
a7b3 dimer, disappears.
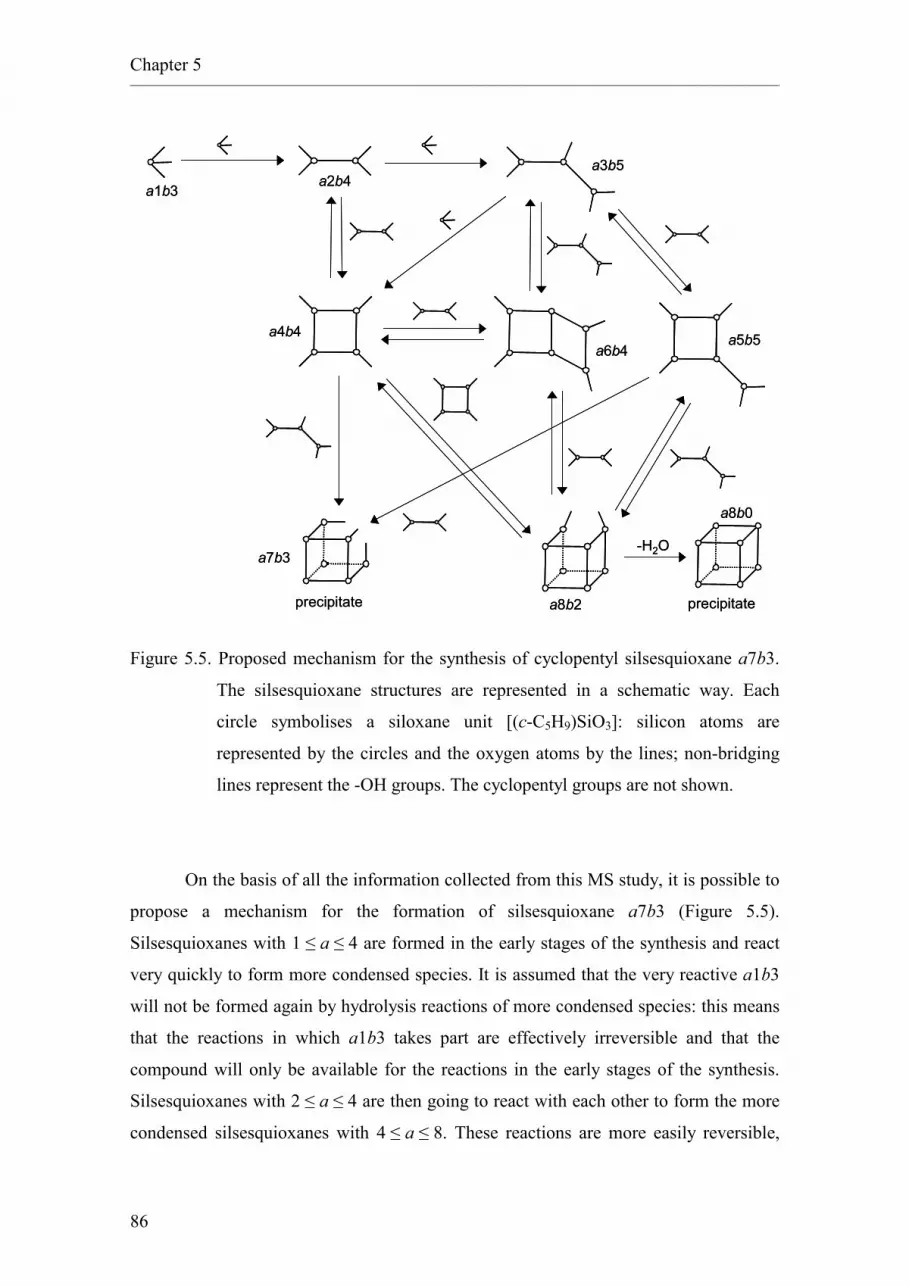
Chapter 5 __________________________________________________________________________________________________________
86
Figure 5.5. Proposed mechanism for the synthesis of cyclopentyl silsesquioxane a7b3.
The silsesquioxane structures are represented in a schematic way. Each
circle symbolises a siloxane unit [(c-C5H9)SiO3]: silicon atoms are
represented by the circles and the oxygen atoms by the lines; non-bridging
lines represent the -OH groups. The cyclopentyl groups are not shown.
On the basis of all the information collected from this MS study, it is possible to
propose a mechanism for the formation of silsesquioxane a7b3 (Figure 5.5).
Silsesquioxanes with 1 ≤ a ≤ 4 are formed in the early stages of the synthesis and react
very quickly to form more condensed species. It is assumed that the very reactive a1b3
will not be formed again by hydrolysis reactions of more condensed species: this means
that the reactions in which a1b3 takes part are effectively irreversible and that the
compound will only be available for the reactions in the early stages of the synthesis.
Silsesquioxanes with 2 ≤ a ≤ 4 are then going to react with each other to form the more
condensed silsesquioxanes with 4 ≤ a ≤ 8. These reactions are more easily reversible,
-H2O
precipitate precipitate
a1b3 a2b4a3b5
a4b4a6b4
a5b5
a7b3
a8b2
a8b0
-H2O
precipitate precipitate
a1b3 a2b4a3b5
a4b4a6b4
a5b5
a7b3
a8b2
a8b0

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
87
meaning that the structures that are formed by condensation can be hydrolysed back to
the original compounds or to other less condensed species.
As mentioned above, silsesquioxane a7b3 is less soluble than other
silsesquioxanes present in the reaction mixture and precipitates as a white solid. This
will influence the equilibrium composition and consequently drive the reactions that
involve its formation towards the product; therefore, these reactions are considered
irreversible. Silsesquioxanes with a = 6 exhibit a maximum of concentration in solution
after 1 hour of reaction; they are then rapidly consumed up until 3 hours of reaction,
after which the reaction proceeds more slowly. Since no a = 6 silsesquioxane is present
in the precipitate, it is inferred that the compound gets slowly hydrolysed to smaller
species which then recombine to give the product a7b3.6,19 The absence of an a = 6
silsesquioxane among the products is, therefore, not due to the instability of the
compound but rather to its higher solubility in the reaction mixture. The completely
condensed silsesquioxane a8b0, obtained by condensation of a8b2, is only present in
traces in the precipitate. This means that, although silsesquioxane a8b0 might be
expected to be the most thermodynamically favoured structure, its formation is
unfavourable compared to that of silsesquioxane a7b3, suggesting that the reaction is
kinetically controlled.
An alternative way to represent the results of the MS analysis is by plotting the
fraction of the total silsesquioxane structures for each value of a (with 1 ≤ a ≤ 8), as a
function of the reaction time (Figure 5.6). This graph gives a measure of the distribution
of the various species in solution at various stages of the reaction. This plot does not,
however, provide any information about the total concentration of silsesquioxane
species in solution (the value of which is always set at 100%).III(page 83) This graph nicely
shows how the smaller silsesquioxane species constitute the main fraction in the early
stages of the synthesis and then tend to gradually be consumed in favour of bigger
structures as the reaction proceeds. Towards the end of the synthesis, silsesquioxanes
with a = 6 are the major fraction in solution, though the relative fractions of species
with a = 7 and a = 8 are increasing. This is in agreement with the hypothesis of the
concentrations of silsesquioxanes a = 7 and a = 8 being constant in solution and that of
silsesquioxanes a = 6 slowly decreasing upon hydrolysis and consecutive condensation
towards silsesquioxane a7b3.
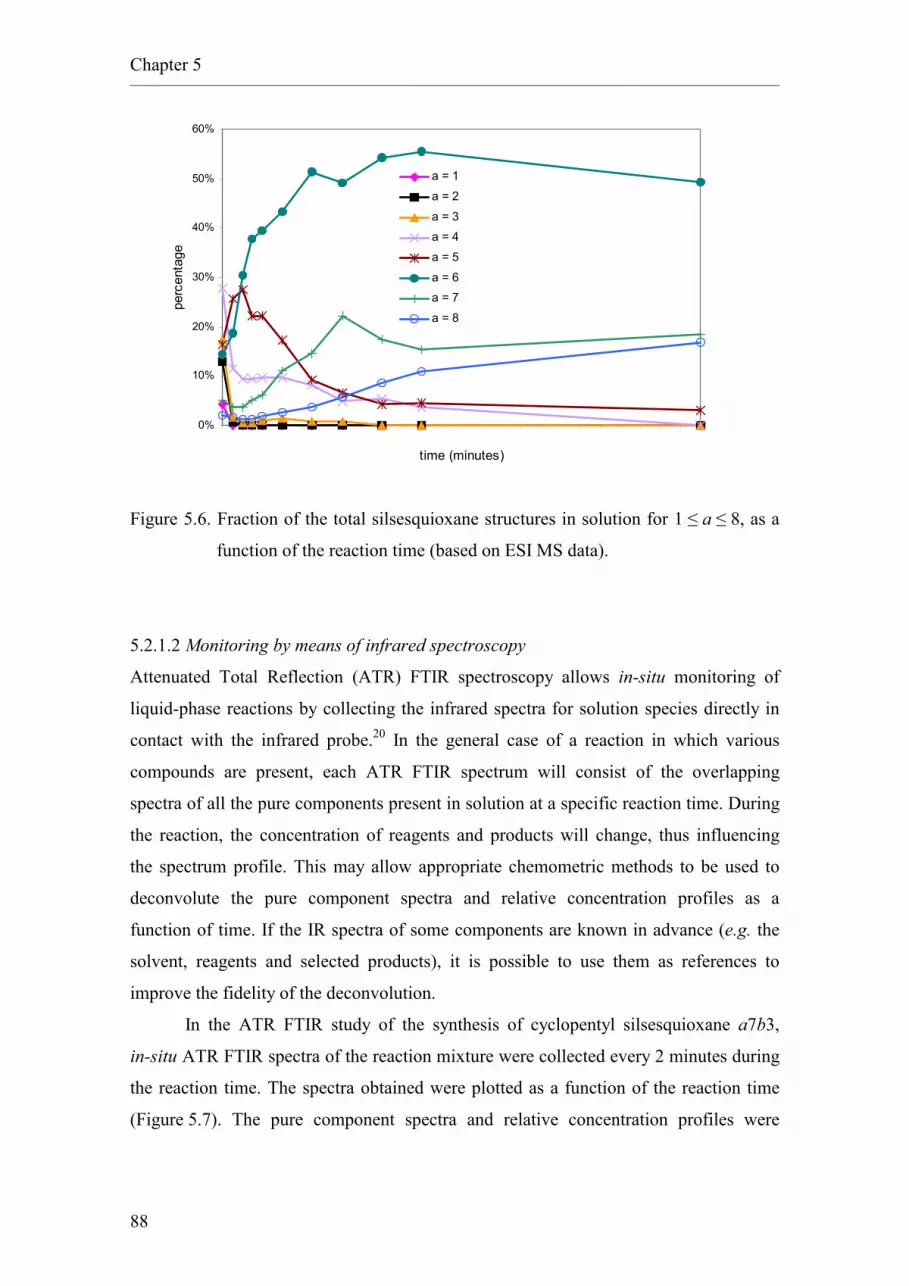
Chapter 5 __________________________________________________________________________________________________________
88
Figure 5.6. Fraction of the total silsesquioxane structures in solution for 1 ≤ a ≤ 8, as a
function of the reaction time (based on ESI MS data).
5.2.1.2 Monitoring by means of infrared spectroscopy
Attenuated Total Reflection (ATR) FTIR spectroscopy allows in-situ monitoring of
liquid-phase reactions by collecting the infrared spectra for solution species directly in
contact with the infrared probe.20 In the general case of a reaction in which various
compounds are present, each ATR FTIR spectrum will consist of the overlapping
spectra of all the pure components present in solution at a specific reaction time. During
the reaction, the concentration of reagents and products will change, thus influencing
the spectrum profile. This may allow appropriate chemometric methods to be used to
deconvolute the pure component spectra and relative concentration profiles as a
function of time. If the IR spectra of some components are known in advance (e.g. the
solvent, reagents and selected products), it is possible to use them as references to
improve the fidelity of the deconvolution.
In the ATR FTIR study of the synthesis of cyclopentyl silsesquioxane a7b3,
in-situ ATR FTIR spectra of the reaction mixture were collected every 2 minutes during
the reaction time. The spectra obtained were plotted as a function of the reaction time
(Figure 5.7). The pure component spectra and relative concentration profiles were
0%
10%
20%
30%
40%
50%
60%
0 60 120 180 240 300 360 420 480 540 600 660 720 780 840 900 960 1020 1080 1140 1200 1260 1320 1380 1440
time (minutes)
perc
enta
gea = 1a = 2a = 3a = 4a = 5a = 6a = 7a = 8
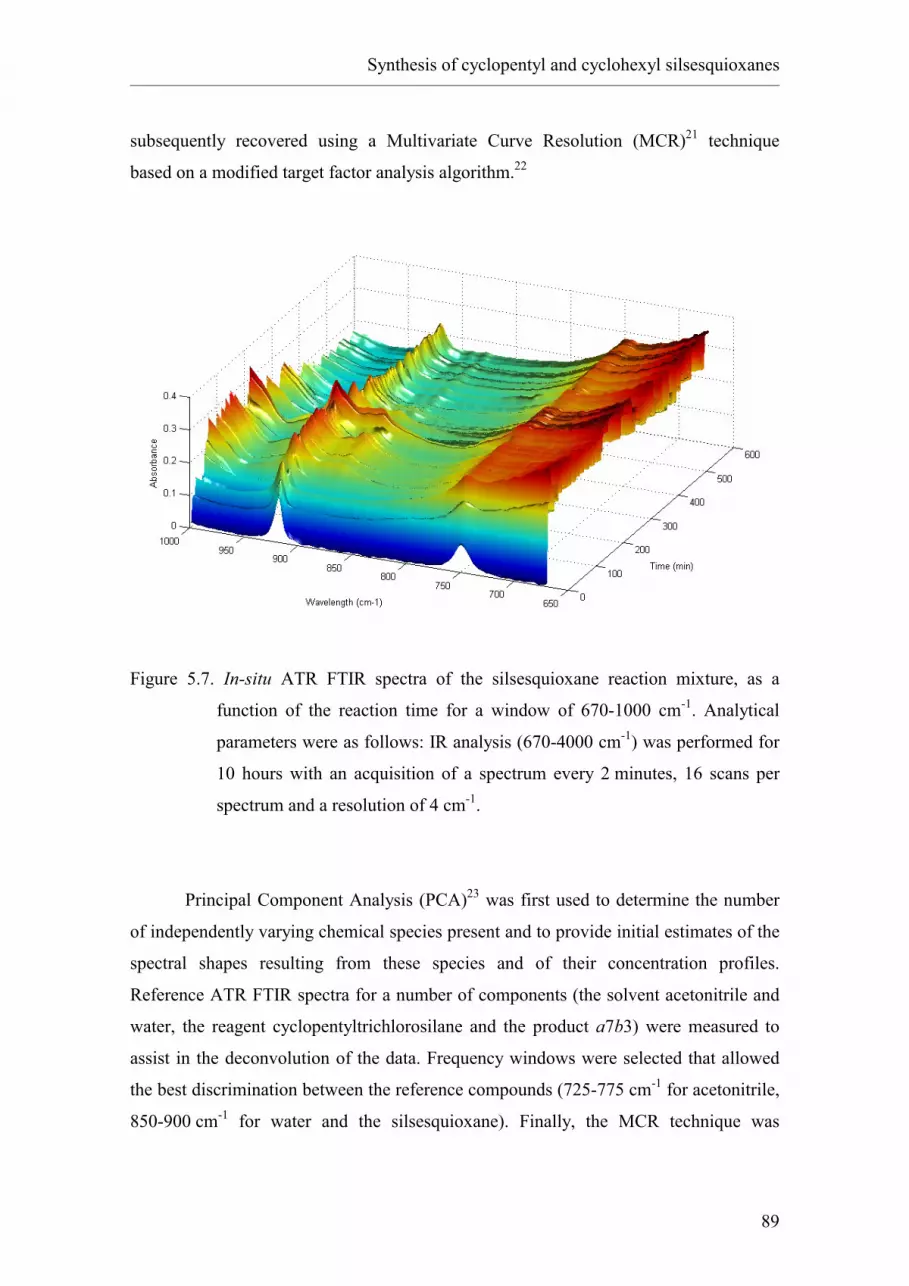
Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
89
subsequently recovered using a Multivariate Curve Resolution (MCR)21 technique
based on a modified target factor analysis algorithm.22
Figure 5.7. In-situ ATR FTIR spectra of the silsesquioxane reaction mixture, as a
function of the reaction time for a window of 670-1000 cm-1. Analytical
parameters were as follows: IR analysis (670-4000 cm-1) was performed for
10 hours with an acquisition of a spectrum every 2 minutes, 16 scans per
spectrum and a resolution of 4 cm-1.
Principal Component Analysis (PCA)23 was first used to determine the number
of independently varying chemical species present and to provide initial estimates of the
spectral shapes resulting from these species and of their concentration profiles.
Reference ATR FTIR spectra for a number of components (the solvent acetonitrile and
water, the reagent cyclopentyltrichlorosilane and the product a7b3) were measured to
assist in the deconvolution of the data. Frequency windows were selected that allowed
the best discrimination between the reference compounds (725-775 cm-1 for acetonitrile,
850-900 cm-1 for water and the silsesquioxane). Finally, the MCR technique was
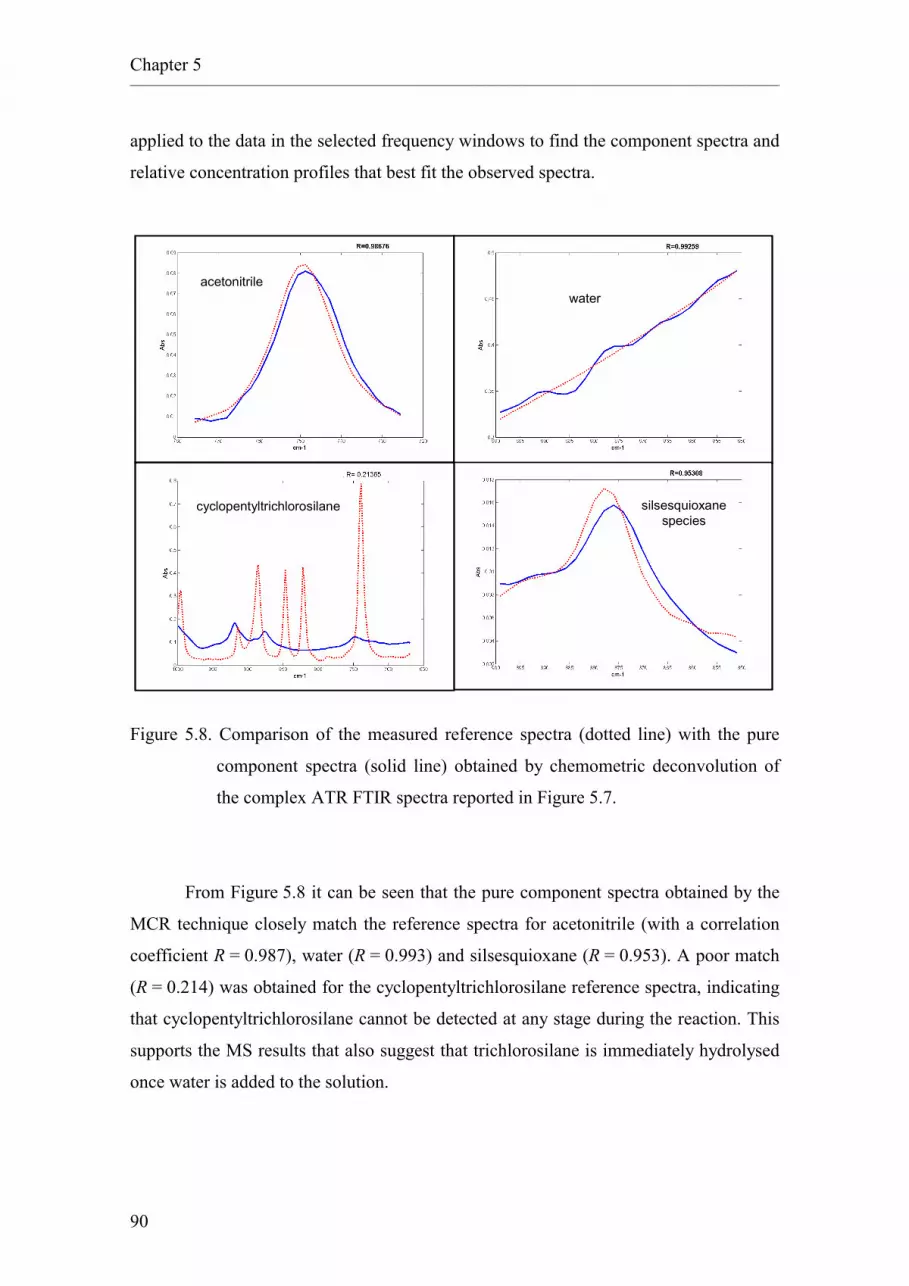
Chapter 5 __________________________________________________________________________________________________________
90
applied to the data in the selected frequency windows to find the component spectra and
relative concentration profiles that best fit the observed spectra.
Figure 5.8. Comparison of the measured reference spectra (dotted line) with the pure
component spectra (solid line) obtained by chemometric deconvolution of
the complex ATR FTIR spectra reported in Figure 5.7.
From Figure 5.8 it can be seen that the pure component spectra obtained by the
MCR technique closely match the reference spectra for acetonitrile (with a correlation
coefficient R = 0.987), water (R = 0.993) and silsesquioxane (R = 0.953). A poor match
(R = 0.214) was obtained for the cyclopentyltrichlorosilane reference spectra, indicating
that cyclopentyltrichlorosilane cannot be detected at any stage during the reaction. This
supports the MS results that also suggest that trichlorosilane is immediately hydrolysed
once water is added to the solution.
acetonitrile
silsesquioxanespecies
water
cyclopentyltrichlorosilane
acetonitrile
silsesquioxanespecies
water
cyclopentyltrichlorosilane
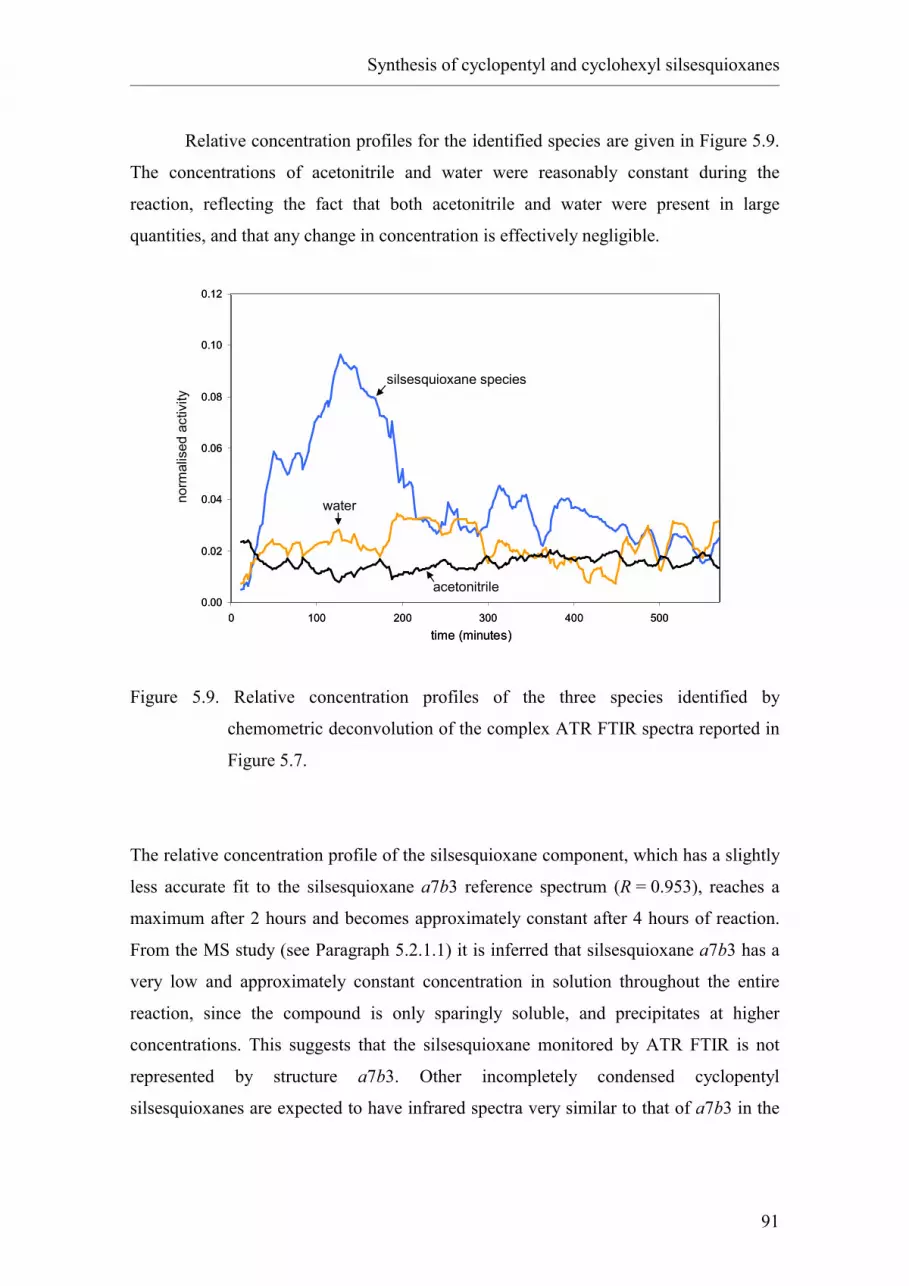
Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
91
Relative concentration profiles for the identified species are given in Figure 5.9.
The concentrations of acetonitrile and water were reasonably constant during the
reaction, reflecting the fact that both acetonitrile and water were present in large
quantities, and that any change in concentration is effectively negligible.
Figure 5.9. Relative concentration profiles of the three species identified by
chemometric deconvolution of the complex ATR FTIR spectra reported in
Figure 5.7.
The relative concentration profile of the silsesquioxane component, which has a slightly
less accurate fit to the silsesquioxane a7b3 reference spectrum (R = 0.953), reaches a
maximum after 2 hours and becomes approximately constant after 4 hours of reaction.
From the MS study (see Paragraph 5.2.1.1) it is inferred that silsesquioxane a7b3 has a
very low and approximately constant concentration in solution throughout the entire
reaction, since the compound is only sparingly soluble, and precipitates at higher
concentrations. This suggests that the silsesquioxane monitored by ATR FTIR is not
represented by structure a7b3. Other incompletely condensed cyclopentyl
silsesquioxanes are expected to have infrared spectra very similar to that of a7b3 in the
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0 100 200 300 400 500time (minutes)
norm
aliz
ed in
tens
ity
silsesquioxane species
acetonitrile
waternorm
alis
ed a
ctiv
ity
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0 100 200 300 400 500time (minutes)
norm
aliz
ed in
tens
ity
silsesquioxane species
acetonitrile
waternorm
alis
ed a
ctiv
ity

Chapter 5 __________________________________________________________________________________________________________
92
studied spectral regions.12,17,V Therefore, it is proposed that the species identified by
MCR is an a = 5 or an a = 6 structure, which presents an MS concentration profile
similar to that obtained by ATR FTIR, or a mixture of more silsesquioxane structures.
The hypothesis of an a = 6 structure is supported by the fact that the ATR FTIR
concentration profile for the silsesquioxane species as a function of time is rather
similar to the MS concentration profile obtained for the a = 6 silsesquioxane (compare
Figures 5.4 and 5.9). The slightly different position of the maximum in the two plots is
considered to be due to the longer time required in the ATR FTIR experiment to reach
the reflux temperature.VI
The combined MS and ATR FTIR results point to the fact that the concentration
of the silsesquioxane species in solution becomes almost constant after 4 hours of
reaction. This might imply that a reaction time shorter than 18 hours could be sufficient
to obtain silsesquioxane a7b3 in high yield. Therefore, the synthesis was repeated with
the same conditions but with a reaction time of 6 hours: silsesquioxane a7b3 was
isolated with a yield of 54%. After collection of the precipitate, the reaction mixture was
allowed to react for 12 more hours yielding more precipitate and the total yield of 64%
was achieved. Although the yield after 6 hours is lower than that obtained after 18 hours
of reaction, the experiment confirmed that most of the yield of the desired a7b3
structure is generated within the first hours of reaction. This finding would certainly be
of practical importance for a potential up-scaling of silsesquioxane a7b3 production.
V This similarity was confirmed by measuring and comparing the transmission IR spectra (KBr pellet) of
cyclopentyl silsesquioxane a7b3 with that of the mixture of silsesquioxanes (mainly containing a = 5 and
a = 6 species) obtained by removing the solvent from the reaction mixture after 18 hours of reaction at
50°C (instead of reflux conditions). The two spectra present corresponding sets of peaks (within 6 cm-1)
in the region 670-1000 cm-1.
VI This explanation was confirmed by repeating part of the MS study of the synthesis of
silsesquioxane a7b3 at 50°C (instead of reflux temperature). In these conditions the hydrolytic
condensation is slower and, though the same silsesquioxane species were present in solution, species with
1 ≤ a ≤ 4 disappeared more slowly and those with a = 5 and a = 6 reached the maximum of the
concentration profile after a longer reaction time.
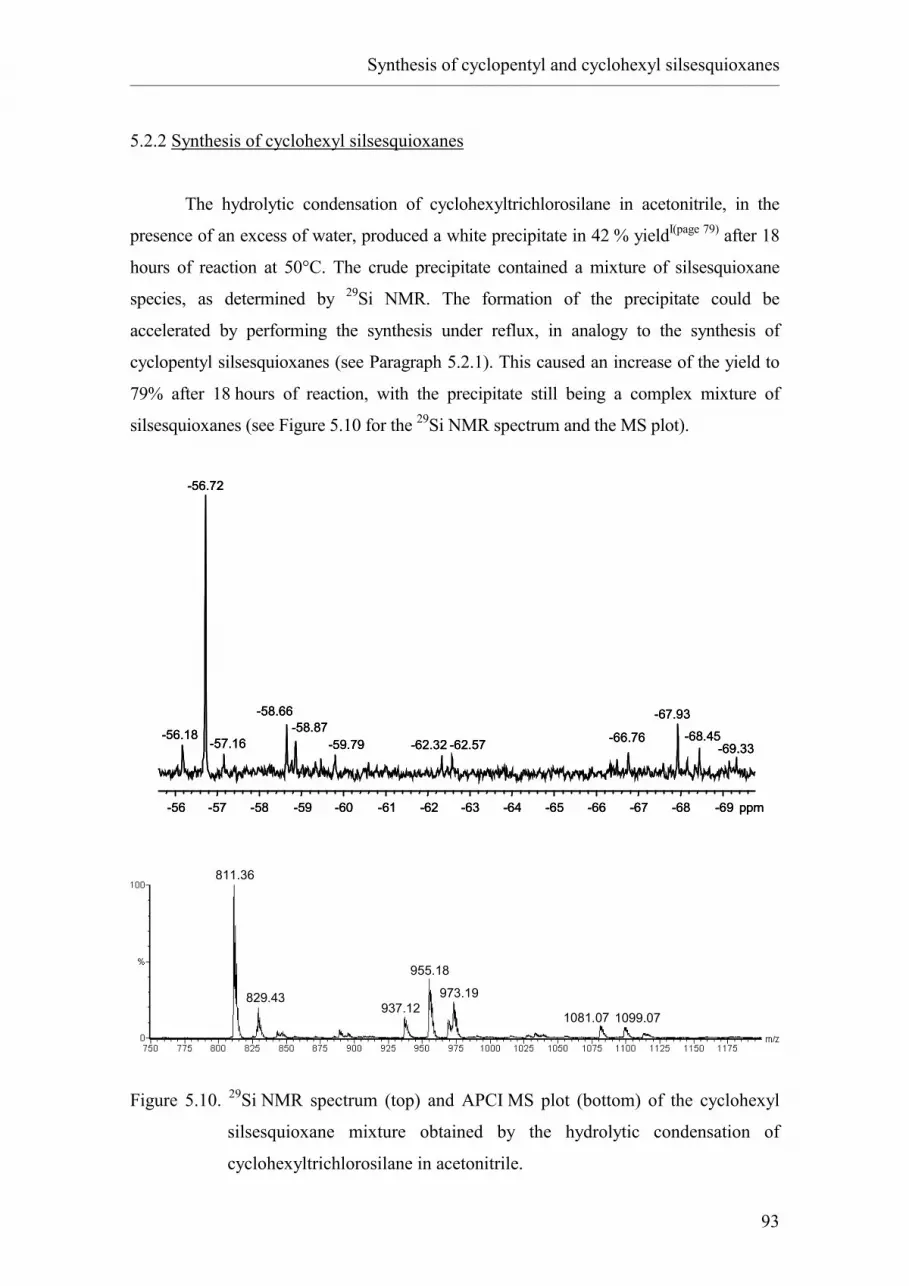
Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
93
5.2.2 Synthesis of cyclohexyl silsesquioxanes
The hydrolytic condensation of cyclohexyltrichlorosilane in acetonitrile, in the
presence of an excess of water, produced a white precipitate in 42 % yieldI(page 79) after 18
hours of reaction at 50°C. The crude precipitate contained a mixture of silsesquioxane
species, as determined by 29Si NMR. The formation of the precipitate could be
accelerated by performing the synthesis under reflux, in analogy to the synthesis of
cyclopentyl silsesquioxanes (see Paragraph 5.2.1). This caused an increase of the yield to
79% after 18 hours of reaction, with the precipitate still being a complex mixture of
silsesquioxanes (see Figure 5.10 for the 29Si NMR spectrum and the MS plot).
Figure 5.10. 29Si NMR spectrum (top) and APCI MS plot (bottom) of the cyclohexyl
silsesquioxane mixture obtained by the hydrolytic condensation of
cyclohexyltrichlorosilane in acetonitrile.
ppm-69-68-67-66-65-64-63-62-61-60-59-58-57-56
-56.18
-56.72
-57.16
-58.66-58.87
-59.79 -62.32 -62.57 -66.76
-67.93
-68.45-69.33
811.36
829.43937.12
955.18
973.19
1081.07 1099.07
ppm-69-68-67-66-65-64-63-62-61-60-59-58-57-56
-56.18
-56.72
-57.16
-58.66-58.87
-59.79 -62.32 -62.57 -66.76
-67.93
-68.45-69.33
ppm-69-68-67-66-65-64-63-62-61-60-59-58-57-56
-56.18
-56.72
-57.16
-58.66-58.87
-59.79 -62.32 -62.57 -66.76
-67.93
-68.45-69.33
811.36
829.43937.12
955.18
973.19
1081.07 1099.07
811.36
829.43937.12
955.18
973.19
1081.07 1099.07
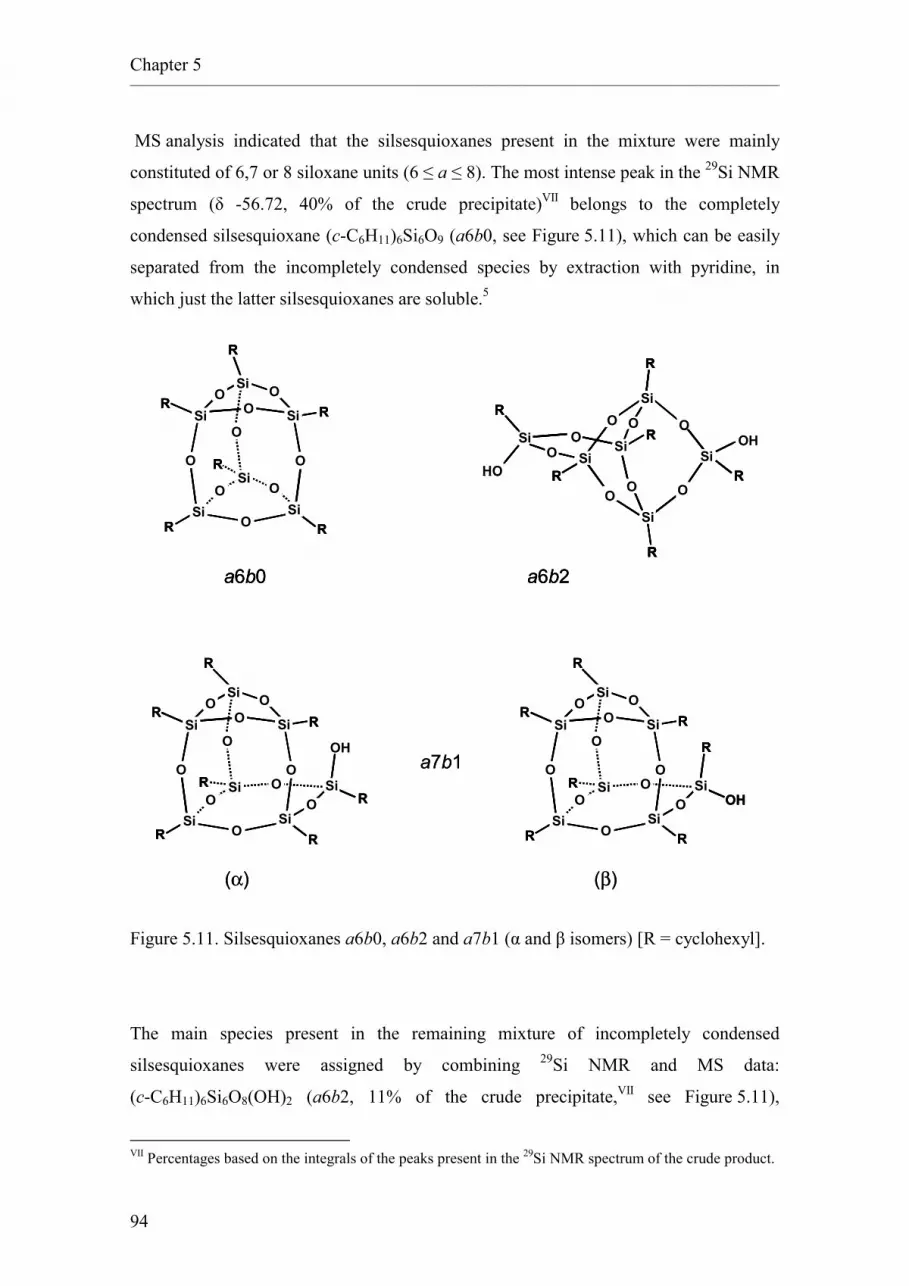
Chapter 5 __________________________________________________________________________________________________________
94
MS analysis indicated that the silsesquioxanes present in the mixture were mainly
constituted of 6,7 or 8 siloxane units (6 ≤ a ≤ 8). The most intense peak in the 29Si NMR
spectrum (δ -56.72, 40% of the crude precipitate)VII belongs to the completely
condensed silsesquioxane (c-C6H11)6Si6O9 (a6b0, see Figure 5.11), which can be easily
separated from the incompletely condensed species by extraction with pyridine, in
which just the latter silsesquioxanes are soluble.5
Figure 5.11. Silsesquioxanes a6b0, a6b2 and a7b1 (α and β isomers) [R = cyclohexyl].
The main species present in the remaining mixture of incompletely condensed
silsesquioxanes were assigned by combining 29Si NMR and MS data:
(c-C6H11)6Si6O8(OH)2 (a6b2, 11% of the crude precipitate,VII see Figure 5.11),
VII Percentages based on the integrals of the peaks present in the 29Si NMR spectrum of the crude product.
O
OHOO
SiO
Si
OO
OSiSi
OSiOSi
O
Si
R
RR
R R
RR
O
OH
OO
SiO
Si
OO
OSiSi
OSiOSi
O
Si
R
RR
R R
R
Ra7b1
O
OO
SiO
Si
OO
SiSiO O
Si
O
Si
R
R
RR
R
R
a6b0
OH
RO
OSi
O
Si
Si
O
SiSi
O
O
Si OO
HO
R
R
R
R
R
a6b2
(α) (β)
O
OHOO
SiO
Si
OO
OSiSi
OSiOSi
O
Si
R
RR
R R
RR
O
OHOO
SiO
Si
OO
OSiSi
OSiOSi
O
Si
R
RR
R R
RR
O
OH
OO
SiO
Si
OO
OSiSi
OSiOSi
O
Si
R
RR
R R
R
R
O
OH
OO
SiO
Si
OO
OSiSi
OSiOSi
O
Si
R
RR
R R
R
Ra7b1
O
OO
SiO
Si
OO
SiSiO O
Si
O
Si
R
R
RR
R
R
a6b0
O
OO
SiO
Si
OO
SiSiO O
Si
O
Si
R
R
RR
R
R
a6b0
OH
RO
OSi
O
Si
Si
O
SiSi
O
O
Si OO
HO
R
R
R
R
R
a6b2
OH
RO
OSi
O
Si
Si
O
SiSi
O
O
Si OO
HO
R
R
R
R
R
OH
RO
OSi
O
Si
Si
O
SiSi
O
O
Si OO
HO
R
R
R
R
R
a6b2
(α) (β)
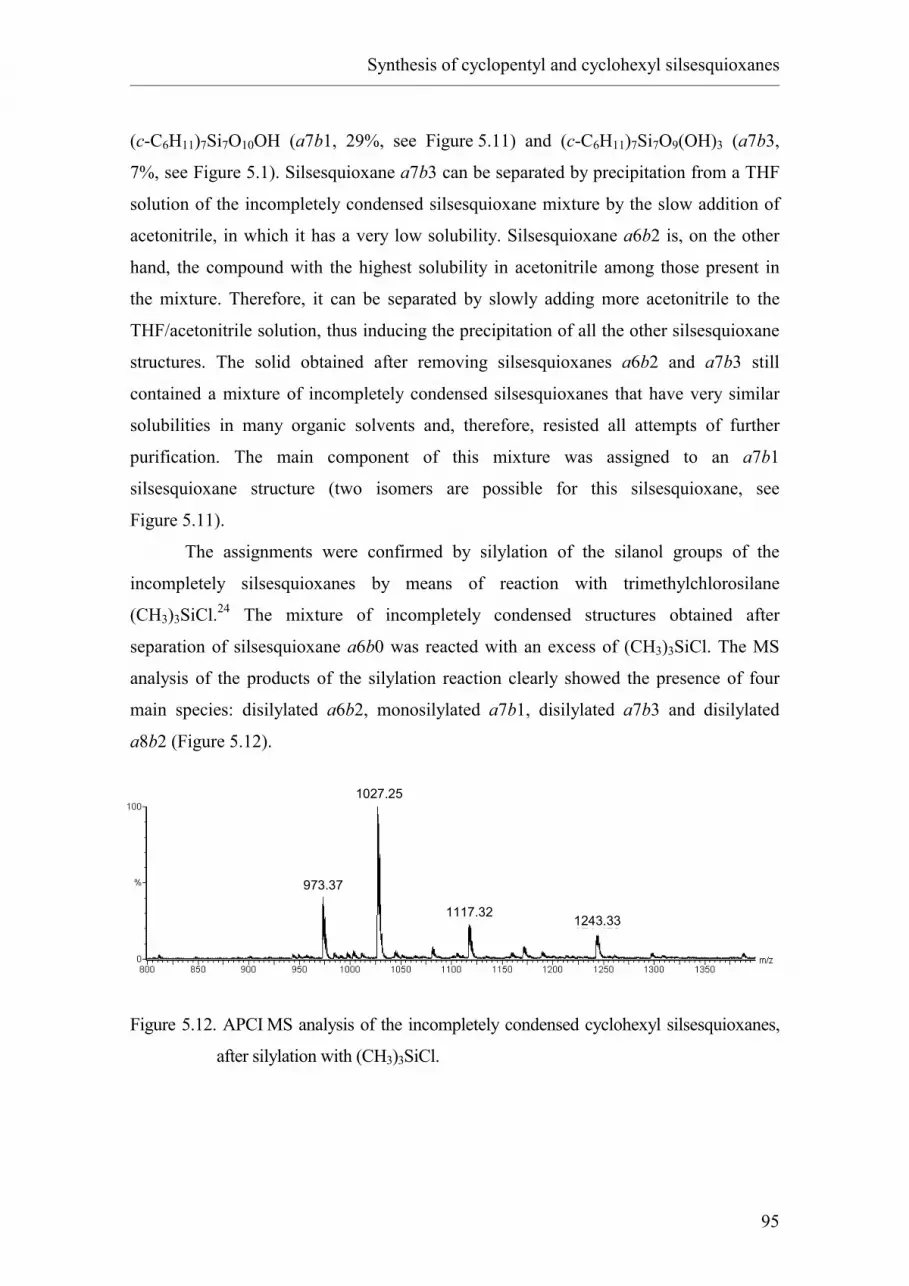
Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
95
(c-C6H11)7Si7O10OH (a7b1, 29%, see Figure 5.11) and (c-C6H11)7Si7O9(OH)3 (a7b3,
7%, see Figure 5.1). Silsesquioxane a7b3 can be separated by precipitation from a THF
solution of the incompletely condensed silsesquioxane mixture by the slow addition of
acetonitrile, in which it has a very low solubility. Silsesquioxane a6b2 is, on the other
hand, the compound with the highest solubility in acetonitrile among those present in
the mixture. Therefore, it can be separated by slowly adding more acetonitrile to the
THF/acetonitrile solution, thus inducing the precipitation of all the other silsesquioxane
structures. The solid obtained after removing silsesquioxanes a6b2 and a7b3 still
contained a mixture of incompletely condensed silsesquioxanes that have very similar
solubilities in many organic solvents and, therefore, resisted all attempts of further
purification. The main component of this mixture was assigned to an a7b1
silsesquioxane structure (two isomers are possible for this silsesquioxane, see
Figure 5.11).
The assignments were confirmed by silylation of the silanol groups of the
incompletely silsesquioxanes by means of reaction with trimethylchlorosilane
(CH3)3SiCl.24 The mixture of incompletely condensed structures obtained after
separation of silsesquioxane a6b0 was reacted with an excess of (CH3)3SiCl. The MS
analysis of the products of the silylation reaction clearly showed the presence of four
main species: disilylated a6b2, monosilylated a7b1, disilylated a7b3 and disilylated
a8b2 (Figure 5.12).
Figure 5.12. APCI MS analysis of the incompletely condensed cyclohexyl silsesquioxanes,
after silylation with (CH3)3SiCl.
1027.25
1117.32 1243.33
973.37
1027.25
1117.32 1243.33
973.37

Chapter 5 __________________________________________________________________________________________________________
96
The monosilylated a7b1 was the species present in the highest concentration. On the
basis of these experiments, the assignment of a7b1 as the main incompletely condensed
species appears unequivocal. However, the 29Si NMR peaks belonging to this
silsesquioxane a7b1, both silylated or not, did not correspond to those of the analogous
silsesquioxane reported in the literature.5,VIII The peaks corresponding to the silylated
a7b1 reported by Feher were present in the 29Si NMR spectrum just as the second main
species. This suggests that the two isomers of silsesquioxane a7b1 (Figure 5.11) were
formed with the synthetic method reported here, with the one known from the literature
being the less abundant (α:β ≈ 1:2). The two isomers differ only in the spatial
orientation of the silanol group. They are both possible products of the hydrolytic
condensation of cyclohexyltrichlorosilane, while only the α-isomer of silsesquioxane
a7b1 is obtained by the dehydration of silsesquioxane a7b3 reported by Feher.5
After 18 hours of hydrolytic condensation under reflux and removal of the
precipitate, the reaction was left to continue at room temperature for 20 weeks: more
white precipitate was formed (additional 10% yield).I(page 79) The crude product was a
mixture of three silsesquioxane species, with some impurities, as determined by 29Si NMR: a6b0 (21%),VII(page 94) a6b2 (27%) and a7b3 (51%). A similar increase of the
ratio of silsesquioxane a7b3 among the products as the reaction time becomes longer
had already been reported by Feher et al..5
The synthesis procedure described here is the first reported method to obtain
silsesquioxanes a6b2 and a7b1 (both isomers) via the hydrolytic condensation of
cyclohexyltrichlorosilane. Cyclohexyl silsesquioxane a6b2 had previously been VIII Peaks corresponding exactly to the 29Si{1H} NMR spectrum of the α-isomer of a7b1 reported by
Feher5 (assumed to have been measured in C6D6 although reported to be in CDCl3) were found as the
second main species in the 29Si{1H} NMR spectrum of the mixture of incompletely condensed
silsesquioxanes measured in C6D6 [δ -55.12, -57.05, -57.15, -66.06, -66.40 (1:1:2:1:2)]. In the 29Si{1H} NMR spectra measured in THF or CDCl3, only the two most intense peaks due to the α-isomer
of a7b1 could be assigned [in THF, δ -59.47, -68.45; in CDCl3, δ -57.76, -67.12], whilst the three less
intense peaks could not be unambiguously assigned among the remaining peaks of the silsesquioxane
mixture.

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
97
synthesised via the acid-mediated cleavage and rearrangement of silsesquioxane a6b0.25
The α-isomer of cyclohexyl silsesquioxane a7b1 was formerly obtained by dehydration
of silsesquioxane a7b3.5 The synthesis also afforded cyclohexyl silsesquioxane a7b3 in
significant yields, although its separation from the rest of the mixture is not
straightforward. This new method presents a number of differences from that reported
by Feher et al. for the hydrolytic condensation of cyclohexyltrichlorosilane,5 namely:
the solvent acetonitrile, the lower initial trichlorosilane concentration and the higher
solvent-to-water ratio. Additionally, performing the reaction under reflux caused a
noticeable increase in precipitate yield in this synthesis, while the rate of silsesquioxane
formation could not be improved for the reaction reported in literature. Comparing the
two synthetic methods, it is evident that the formation of silsesquioxanes is much faster
with the method reported here, albeit less selective. Such an increase of the rate of
formation of silsesquioxanes suggests that the conditions for the precipitation of
silsesquioxanes are met more readily and for a larger number of different structures.
This fact points to the relevance of using acetonitrile as a solvent, similarly to what is
observed for the synthesis of cyclopentyl silsesquioxanes (see Paragraph 5.2.1).
Analogously, after 18 hours of reaction under reflux, the hydrolysis of acetonitrile and
the consequent formation of NH4Cl caused a liquid-liquid phase separation of the
reaction mixture. In order to further study the effect of the presence of NH4Cl, two
additional experiments were performed:
1) Some NH4Cl (0.073M) was added to the starting synthesis mixture, leading, after
18 hours of reaction under reflux, to a silsesquioxane precipitate with a yield of 82%.I(page 79) 29Si NMR analysis showed that the ratio of a6b0 increased compared to the other
(incompletely condensed) silsesquioxanes. Thus, the addition of NH4Cl increased the
yield but also drove the reaction towards completely condensed species, confirming the
influence of the preferential solvation of NH4Cl by water on the silsesquioxane
synthesis.
2) The hydrolytic condensation of cyclohexyltrichlorosilane was performed in acetone
(the solvent used by Brown4 and Feher5) in presence of NH4Cl (0.15M) as additive:
after 18 hours of reaction under reflux no precipitate was formed (as in the case with no
NH4Cl). This indicates that the preferential solvation of NH4Cl by water alone is not
sufficient to accelerate the formation of silsesquioxanes.

Chapter 5 __________________________________________________________________________________________________________
98
The lower selectivity of the synthesis of cyclohexyl silsesquioxanes compared to that of
cyclopentyl silsesquioxanes seems to be connected to the nature of the organic group.1
The larger steric hindrance of cyclohexyl might account for the formation of the
completely condensed silsesquioxane a6b0. The Si-O framework of this molecule is
more strained than in the case of silsesquioxane a7b3, therefore its formation might be
expected to be unfavourable. On the other hand, in this structure the organic groups on
the silicon atoms point away from each other: this may favour the formation of this
molecule for bulkier organic groups like cyclohexyl. Another effect of the organic
group is to influence the solubility of the silsesquioxanes.1 Cyclohexyl
silsesquioxane a7b3 has been reported to be more soluble in organic solvents than
cyclopentyl silsesquioxane a7b3.5,6 The low solubility of the cyclopentyl structure can
account for its selective formation as precipitate, in agreement with what discussed in
Paragraph 5.2.1.1.
5.3 Conclusions
New methods for the synthesis of cyclopentyl and cyclohexyl silsesquioxanes
have been developed. In both cases, acetonitrile plays a strong role in accelerating the
formation of a silsesquioxane precipitate. The influence of the hydrolysis of acetonitrile
on the synthesis has been ascertained.
In the case of cyclopentyl silsesquioxanes, the synthesis is very selective towards the
formation of structure a7b3, which can be obtained in 64% yield after 18 hours of
reaction. This method provides a fast and high-yield way to prepare silsesquioxane
a7b3, therefore making this compound more readily available for its many applications.
By coupling MS and ATR FTIR analytical techniques, it was possible to obtain detailed
information on the time-resolved mechanism of silsesquioxane formation, with the ATR
FTIR results being consistent with the mechanism based on the interpretation of the MS
data. The study has shown that the formation of the desired silsesquioxane a7b3 follows
a complex pathway, via initial formation of short-lived smaller oligomers. The final,
high selectivity for the a7b3 species is ascribed to its lower solubility, which impedes
re-dissolution and equilibrium reactions yielding other silsesquioxanes.

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
99
In the case of the synthesis of cyclohexyl silsesquioxanes, four silsesquioxane structures
are produced in significant yields: a6b0, a6b2, a7b1 (both isomers) and a7b3. This
method offers a facile way to synthesise the non-easily accessible silsesquioxane
structures a6b2 and a7b1, though their purification proved to be difficult. The lower
selectivity of this reaction seems to be connected to the higher solubility of cyclohexyl
silsesquioxanes as compared to that of cyclopentyl silsesquioxanes.
5.4 Experimental
29Si NMR characterisation of the silsesquioxanes was performed on a Varian
VXR-400S (79.5 MHz, 1H decoupled, 25°C) and on a Varian Inova-300 (59.6 MHz, 1H
decoupled, 25°C). The position of the silsesquioxanes peaks in the 29Si NMR spectra is
influenced by the solvent in which the silsesquioxanes are dissolved. Spectra were
recorded in THF and CDCl3, with TMS as a reference.
Mass spectrometry characterisation of the silsesquioxanes was performed on a
Micromass Quattro LC-MS with ESI+ (electrospray ionisation) and APCI+
(atmospheric pressure chemical ionisation) as ionisation techniques. Mass spectrometry
analysis allows identifying the number of siloxane units constituting the silsesquioxane
structures (i.e. the value of a) but not their level of condensation (i.e. the value of b).
The reason for this is that during the ionisation process silsesquioxanes can lose water
molecules, as confirmed by the fact that the ratio of peak intensities for a given value of
a is influenced by the cone voltage applied during the MS analysis.
In-situ infrared analysis was performed by means of an ASI ReactIR™1000 Reaction
Analysis System, based on Attenuated Total Reflection (ATR) Fourier Transform
Infrared (FTIR) spectroscopy. The spectrometer was equipped with a zinc selenide
(ZnSe) crystal cell, a diamond probe and a mercury cadmium telluride (MCT) detector.
During the analysis, the IR diamond probe had to be positioned just below the surface
of the reaction mixture in order to avoid coating of the probe by the silsesquioxane
precipitate formed during the synthesis. The reaction was carried out under reflux
conditions, which led to bubble formation at the surface of the solution. This affected

Chapter 5 __________________________________________________________________________________________________________
100
the contact between the probe surface and the reaction mixture, causing some noise in
the recorded spectra. Noisy or spiky data may also be caused by impact on the ATR
probe of the precipitate formed during the reaction.
5.4.1 Synthesis of cyclopentyl silsesquioxane a7b3
Typically, 37.5 ml of deionised water were carefully added into a 500-ml
round-bottom flask containing a solution of 5.6 ml of cyclopentyltrichlorosilane
((c-C5H9)SiCl3, 97% purity, 3.3·10-2 moles) in 150 ml of acetonitrile
(CH3CN:H2O = 4:1 in volume). This homogeneous solution was then heated to 50°C or
under reflux while vigorously stirring for 18 hours: a white precipitate was formed. The
crude precipitate was dissolved in 20 times its weight of pyridine and stirred overnight.
Next, the solution was filtered to remove the insoluble completely condensed species
(traces) and poured into an equal volume of ice-cold aqueous HCl (37%): a white
precipitate formed. The solid was filtered, carefully washed with water and dried
overnight at 45°C: NMR-pure cyclopentyl silsesquioxane a7b3 was obtained.
Selected spectroscopic data. 29Si{1H} NMR (THF as solvent), δ -57.83, -65.71, -67.24 (3:1:3).
MS (ESI+, cone voltage = 60 V, THF 78%, CH3CN 20%, HCO2H 0.1M 2%), m/z:
875.04 ([a7b3 + H]+, 100%), 839.00 ([a7b3 - 2H2O + H]+, 41%), 897.08 ([a7b3 + Na]+,
16%), 911.09 ([a7b3 + 2H2O + H]+, 16%), 857.03 ([a7b3 - H2O + H]+, 11%).
For the mechanistic study by mass spectrometry and in-situ ATR FT-IR spectroscopy,
the synthesis of cyclopentyl silsesquioxane a7b3 was performed on a smaller-volume
scale: 9.4 ml of deionised water were carefully added to a 100-ml round-bottomed flask
containing a solution of 1.4 ml of cyclopentyltrichlorosilane ((c-C5H9)SiCl3, 97%
purity) in 37.5 ml of acetonitrile. The homogeneous solution was vigorously stirred and
heated under reflux conditions in an oil bath set at 90°C. A white precipitate was
already visible after 1 hour. At the end of the reaction, the white precipitate (mainly
composed of silsesquioxane a7b3) was present on the walls of the flask and a
liquid-liquid phase separation of the reaction solution was observed.
For the mechanistic study by means mass spectrometry, ESI+ was used as ionisation
technique. At regular intervals throughout the experiment 200-µl samples were taken

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
101
from the reaction supernatant liquid, ensuring no aspiration of solids into the syringe,
and directly injected into the mass spectrometer by means of a syringe pump. The first
sample (t = 0) was taken straight after adding water to the acetonitrile solution of
cyclopentyltrichlorosilane, and subsequent samples were taken after 30, 60, 90, 120,
180, 270, 360, 480, 600, 1440 minutes. The presence of water and acids (hydrochloric
and acetic) in the reaction mixture yielded favourable ionisation conditions for MS.
A list of possible cyclopentyl silsesquioxane structures and their masses, based on the
equations that define the a and b values in the formula (RSiO1.5)a(H2O)0.5b (see
Paragraph 2.1), is shown in Figure 5.3, together with a MS plot of the silsesquioxane
synthesis mixture.
Main MS peaks for the different values of a: For a = 1, m/z: 257.55 (H+, a1b3 + 6H2O),
275.61 (H+, a1b3 + 7H2O), 293.62 (H+, a1b3 + 8H2O), 316.67 (H+, 275.61 + CH3CN).
For a = 2, m/z: 261.59 (H+), 279.59 (H+), 302.65 (H+, 261.59 + CH3CN), 320.65 (H+,
279.59 + CH3CN). For a = 3, m/z: 373.64 (H+, 391.64 - H2O), 391.64 (H+), 414.64 (H+,
373.64 + CH3CN), 432.64 (H+, 391.64 + CH3CN). For a = 4, m/z: 485.57 (H+), 503.57
(H+), 521.57 (H+), 544.56 (H+, 503.57 + CH3CN). For a = 5, m/z:. For a = 5, m/z:
597.49 (H+, 633.49 – 2H2O), 615.49 (H+, 633.49 – H2O), 633.49 (H+), 638.48 (H+,
597.49 + CH3CN), 651.43 (H+). For a = 6, m/z: 727.42 (H+), 745.36 (H+), 763.36 (H+),
781.36 (H+). For a = 7, m/z: 839.28 (H+, 857.28 – H2O), 857.28 (H+), 875.28 (H+),
893.28 (H+), 911.22 (H+). For a = 8, m/z: 969.20 (H+), 987.20 (H+), 1005.19 (H+),
1023.20 (H+), 1041.20 (H+). For a = 9, m/z: 1099.06 (H+), 1117.12 (H+), 1135.19 (H+),
1153.12 (H+), 1171.13 (H+). For a = 10, m/z: 1211.05 (H+), 1229.00 (H+), 1247.05 (H+),
1265.05 (H+), 1282.99 (H+), 1301.00 (H+). For a = 11, m/z: 1340.91 (H+), 1358.91 (H+),
1376.97 (H+), 1394.91 (H+), 1412.91 (H+). For a = 12, m/z: 1470.90 (H+), 1488.90 (H+),
1506.84 (H+), 1524.84 (H+), 1542.84 (H+). For a = 13, m/z:1600.82 (H+), 1618.76 (H+),
1636.76 (H+), 1654.76 (H+).
5.4.2 Synthesis of cyclohexyl silsesquioxanes
Typically, 125 ml of deionised water were carefully added into a 1-l
round-bottom flask containing a solution of 15 ml of cyclohexyltrichlorosilane
((c-C6H11)SiCl3, 97% purity, 8.2·10-2 moles) in 500 ml of acetonitrile (CH3CN:H2O = 4:1
in volume). This homogeneous solution was then heated to 50°C or under reflux while

Chapter 5 __________________________________________________________________________________________________________
102
vigorously stirring for 18 hours.IX The crude precipitate formed was dissolved in 20
times its weight of pyridine and stirred overnight. The solution was then filtered to
remove the insoluble completely condensed species (a6b0) and poured into an equal
volume of ice-cold aqueous HCl (37%) to precipitate the incompletely condensed
silsesquioxanes. The solid mixture of silsesquioxanes was filtered, carefully washed
with water and dried overnight at 45°C. Silsesquioxane a7b3 was separated by
dissolving the mixture of incompletely condensed species in 5 times its weight of
tetrahydrofuran (THF) and then by slowly layering an equal weight of acetonitrile over
the THF solution. Upon these conditions, a white precipitate mainly constituted of
silsesquioxane a7b3 was obtained. The remaining solution was partially dried under
reduced pressure and more acetonitrile was slowly added until a precipitate deposited.
The solid was separated by filtration and the solution dried under reduced pressure to
afford silsesquioxane a6b2.
The silylation of the mixture of incompletely condensed silsesquioxanes was performed
by adding 200 µl of trimethylchlorosilane (CH3)3SiCl to a solution of 0.3 g of the
silsesquioxanes in 30 ml of toluene and 1.5 ml of triethylamine (C2H5)3N. The turbid
solution was stirred overnight at room temperature. Next, the solvent and the
triethylamine were removed under reduced pressure to afford a white solid. The solid
was extracted with pentane, the solution was filtered to remove the insoluble
(C2H5)3N·HCl and dried under reduced pressure to give a solid containing the silylated
silsesquioxanes.
Selected spectroscopic data. 29Si{1H} NMR: a6b0 (in THF), δ -56.72; a6b2 (in THF) δ -57.16, -62.32, -62.57 (2:2:2),
(in CDCl3) δ -55.20, -61.72, -61.92 (2:2:2); a7b1 (in THF) δ -56.18, -58.66, -58.87,
-66.76, -67.93 (1:2:1:1:2), (in CDCl3) δ -56.23, -57.95, -58.58, -66.82, -67.66 (1:1:2:1:2);
silylated a7b1 isomers (in CDCl3): (α) δ 9.06 ((CH3)3Si), -56.32, -59.02, -66.67, -68.74,
-68.83 (1:2:1:1:2, (c-C6H11)Si) and (β) δ 8.20 ((CH3)3Si), -56.28, -58.70, -66.89, -67.35,
-68.14 (1:2:1:1:2, (c-C6H11)Si); a7b3 (in THF) δ -59.79, -67.86, -69.33 (3:1:3).
IX The synthesis under reflux was performed both on a 100-ml scale and on a 1000-ml scale, yielding to
the formation of the same silsesquioxane mixture.

Synthesis of cyclopentyl and cyclohexyl silsesquioxanes __________________________________________________________________________________________________________
103
MS: crude precipitate (APCI+, cone voltage = 15 V, CH2Cl2, see Figure 5.10), for a = 6,
m/z: 811.36 ([a6b0 + H]+, 100%), 829.43 ([a6b2 + H]+, 20%); for a = 7, m/z: 937.12
([a7b1 – H2O + H]+, 14%), 955.18 ([a7b1 + H]+, 38%), 973.19 ([a7b3 + H]+, 23%); for
a = 8, m/z: 1081.07 ([a8b0 + H]+, 8%), 1099.07 ([a8b2 + H]+, 7%); silylated
incompletely condensed silsesquioxanes (APCI+, cone voltage = 15 V,
toluene/acetonitrile (2:1), few drops of CH3CO2H; see Figure 5.12), m/z: 973.37
([disilylated a6b2 + H]+, 40%), 1027.25 ([monosilylated a7b1 + H]+, 100%), 1117.32
([disilylated a7b3 + H]+, 22%), 1243.33 ([disilylated a8b2 + H]+, 15%).

Chapter 5 __________________________________________________________________________________________________________
104
References 1 P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583, see also Chapter 2. 2 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Angew. Chem. Int. Ed., 2001, 40,
740, see also Chapter 3. 3 P.P. Pescarmona, T. Maschmeyer, NATO Science Series, 2002, Ser. II Vol. 69, 173, see also Chapter 4. 4 J.F. Brown, L.H. Vogt, J. Am. Chem. Soc., 1965, 87, 4313. 5 F. J. Feher, D. A. Newman, J. F. Walzer, J. Am. Chem. Soc., 1989, 111, 1741. 6 F. J. Feher, T. A. Budzichowski, R. L. Blanski, K. J. Weller, J. W. Ziller, Organometallics, 1991, 10,
2526. 7 See Paragraph 3.3.1. 8 V.K. Krieble, C.I. Noll, J. Am. Chem. Soc., 1939, 61, 560. 9 A.J. Belsky, T.B. Brill, J. Phys. Chem. A, 1999, 103, 3006. 10 T. Kudo, M.S. Gordon, J. Am. Chem. Soc., 1998, 120, 11432. 11 T. Kudo, M.S. Gordon, J. Phys. Chem. A, 2000, 104, 4058. 12 M.G. Voronkov, V.I. Lavrent’yev, Top. Curr. Chem., 1982, 102, 199. 13 P. Bussian, F. Sobott, B. Brutschy, W. Schrader, F. Schüth, Angew. Chem. Int. Ed., 2000, 39, 3901. 14 R. Bakhtiar, F.J. Feher, Rapid Commun. Mass Spectrom., 1999, 13, 687. 15 N.J. Harrick, J. Phys. Chem., 1960, 64, 1110. 16 J. Fahrenfort, Spectrochim. Acta, 1961, 17, 698. 17 J.F. Brown, L.H. Vogt, J. Am. Chem. Soc., 1965, 87, 4313. 18 T.S. Haddad, B.M. Moore, S.H. Phillips, Polymer Preprints, 2001, 42, 196. 19 F.J. Feher, R. Terroba, J.W. Ziller, Chem. Commun., 1999, 2153. 20 N.J. Harrick, Internal Reflection Spectroscopy, John Wiley & Sons, New York, 1967. 21 R. Tauler , B. Kowalski, S. Fleming, Anal. Chem., 1993, 65, 2040. 22 E. Metcalfe, J. Tetteh, Polym. Mater. Sci. Eng., 2000, 83, 88. 23 J.E. Jackson, A users guide to principal components, John Wiley & Sons, New York, 1991. 24 F.J. Feher, D.A. Newman, J. Am. Chem. Soc., 1990, 112, 1931. 25 F. J. Feher, F. Nguyen, D. Soulivong, J. W. Ziller, Chem. Commun., 1999, 1705.

105
6
Osmium silsesquioxane as model compound and homogeneous catalyst for the dihydroxylation of alkenes
Abstract
A homogeneous dihydroxylation catalyst was synthesised by complexation of OsO4
with a silsesquioxane ligand containing a tetrasubstituted olefin moiety. The advantage
of this procedure consists in avoiding the presence of the highly toxic and volatile OsO4
in solution during the dihydroxylation reaction. The Os-silsesquioxane complex was
used as a homogeneous catalyst and as a model of the catalytic site of the heterogeneous
analogue. The homogeneous catalyst gave 84% conversion with 99% selectivity for the
dihydroxylation of cyclopentene and 99% conversion with 99% selectivity for the
dihydroxylation of cyclohexene.
____________________ The contents of this chapter have been submitted to:
P.P. Pescarmona, A. F. Masters, J.C. van der Waal, T. Maschmeyer, Angew. Chem. Int. Ed.
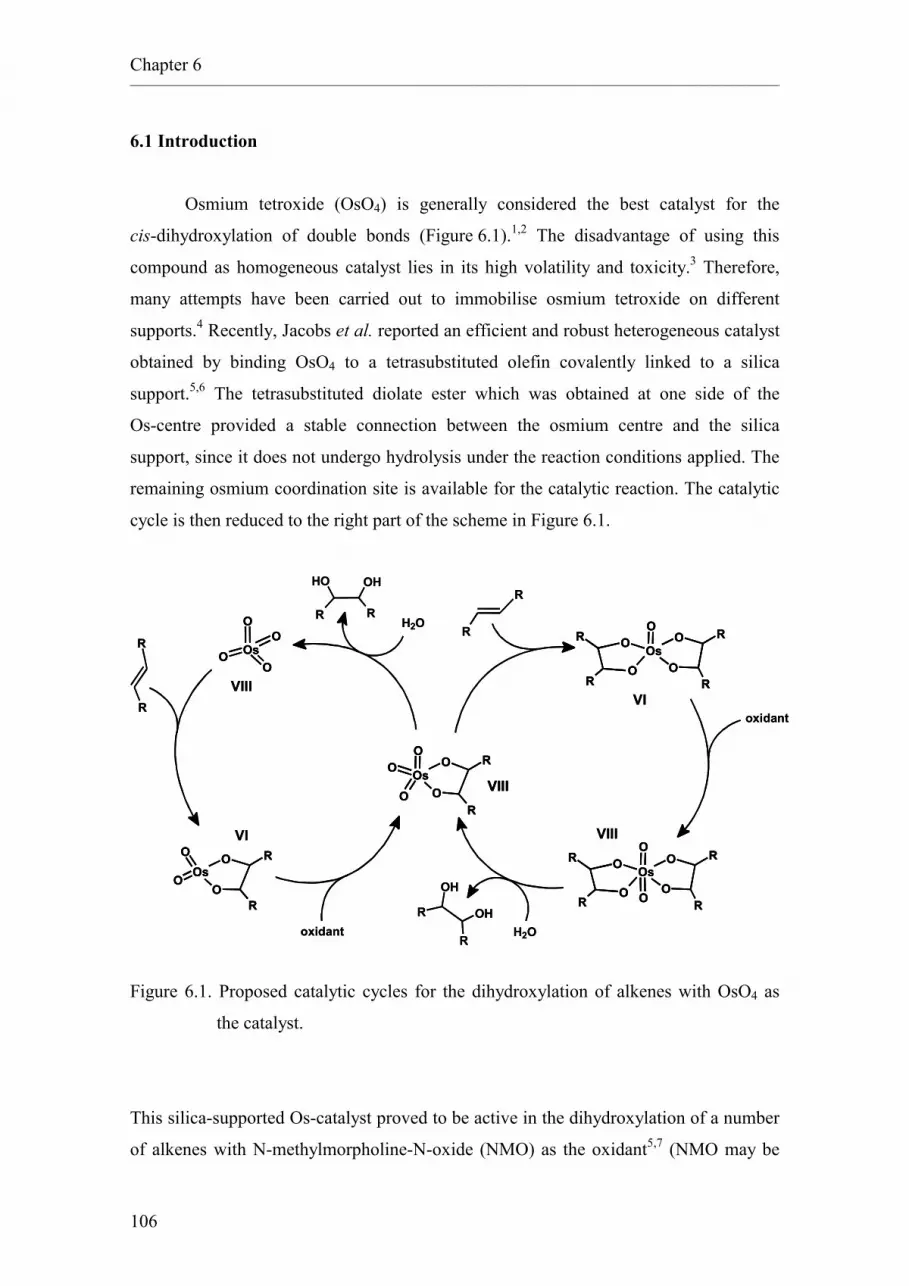
Chapter 6 __________________________________________________________________________________________________________
106
6.1 Introduction
Osmium tetroxide (OsO4) is generally considered the best catalyst for the
cis-dihydroxylation of double bonds (Figure 6.1).1,2 The disadvantage of using this
compound as homogeneous catalyst lies in its high volatility and toxicity.3 Therefore,
many attempts have been carried out to immobilise osmium tetroxide on different
supports.4 Recently, Jacobs et al. reported an efficient and robust heterogeneous catalyst
obtained by binding OsO4 to a tetrasubstituted olefin covalently linked to a silica
support.5,6 The tetrasubstituted diolate ester which was obtained at one side of the
Os-centre provided a stable connection between the osmium centre and the silica
support, since it does not undergo hydrolysis under the reaction conditions applied. The
remaining osmium coordination site is available for the catalytic reaction. The catalytic
cycle is then reduced to the right part of the scheme in Figure 6.1.
Figure 6.1. Proposed catalytic cycles for the dihydroxylation of alkenes with OsO4 as
the catalyst.
This silica-supported Os-catalyst proved to be active in the dihydroxylation of a number
of alkenes with N-methylmorpholine-N-oxide (NMO) as the oxidant5,7 (NMO may be
R
R
R
R
OHOH
R ROH2
oxidant
oxidant OH2
OH
OH
R
R
OsO
OO
O
R
R
VI
OsO
OO
O
R
R
O
R
R
VI
OsO
OO
O
R
R
OR
R O
VIIIR
OsO
OO
O
RO
VIII
OsO
OO
O
VIII
R
R
R
R
OHOH
R ROH2
oxidant
oxidant OH2
OH
OH
R
R
OsO
OO
O
R
R
OsO
OO
O
R
R
VI
OsO
OO
O
R
R
O
R
ROs
O
OO
O
R
R
O
R
R
VI
OsO
OO
O
R
R
OR
R O
OsO
OO
O
R
R
OR
R O
VIIIR
OsO
OO
O
RO
VIIIR
OsO
OO
O
RO
VIII
OsO
OO
O OsO
OO
O
VIII

Os-silsesquioxane as dihydroxylation catalyst __________________________________________________________________________________________________________
107
regenerated by oxidation with H2O2, see Figure 6.2).6 Careful heterogeneity tests
confirmed the absence of OsO4 leaching. Given the relevance of these results, the
preparation, characterisation and catalytic test of a silsesquioxane8-based homogeneous
analogue of this heterogeneous silica-supported catalyst becomes interesting in order to
establish the surface chemistry unequivocally and to broaden the scope for
immobilisation (e.g. silsesquioxane complexes of titanium have been successfully
immobilised on MCM-41, yielding highly active, non-leaching heterogeneous
catalysts).9-11
Figure 6.2. NMO regeneration by oxidation of N-methylmorpholine (NMM) with H2O2,
using vanadyl acetylacetonate as peroxide activator.
6.2 Results and discussion
The synthesis of the silsesquioxane ligand for the Os-catalyst was performed
analogously to that of the functionalised silica support described by Severeyns et al..5
Cyclopentyl silsesquioxane a7b3 (1)12 was used as the precursor for the synthesis of
silsesquioxane 3, which contains a tetrasubstituted olefin moiety to which OsO4 can be
anchored (Figure 6.3). In the first step, silsesquioxane 1 was reacted with
3-aminopropyltrimethoxysilane to obtain the amino-functionalised silsesquioxane 2 as
the only product (as assessed by means of ESI MS). In the second step, the amino group
of silsesquioxane 2 was reacted with 3,4-dimethylcyclohex-3-enylcarbonyl chloride, C,
to produce silsesquioxane 3. The synthesis was performed under a N2 flow and in the
presence of a stoichiometric amount of triethylamine to remove HCl formed during the
reaction. The formation of silsesquioxane 3 was confirmed by means of ESI MS and 13C NMR analysis.
The acid chloride, C, was prepared by means of a three-steps synthesis (Figure 6.4):
1) Diels-Alder reaction of 2,3-dimethyl-1,3-butadiene with ethyl acrylate to give ethyl
NMO
NMM
Voxidised
Vreduced
H2O2
H2O
NMO
NMM
Voxidised
Vreduced
H2O2
H2O
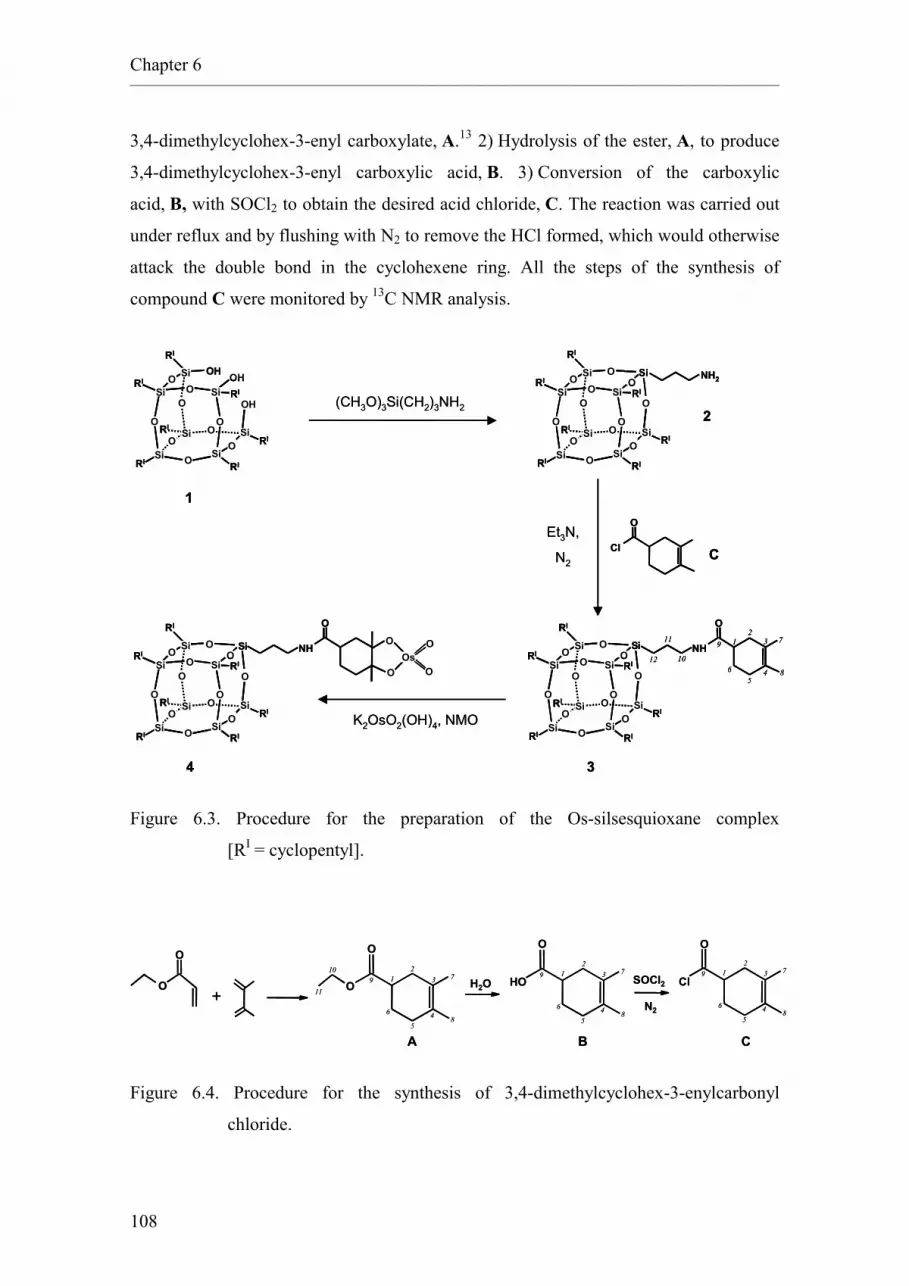
Chapter 6 __________________________________________________________________________________________________________
108
3,4-dimethylcyclohex-3-enyl carboxylate, A.13 2) Hydrolysis of the ester, A, to produce
3,4-dimethylcyclohex-3-enyl carboxylic acid, B. 3) Conversion of the carboxylic
acid, B, with SOCl2 to obtain the desired acid chloride, C. The reaction was carried out
under reflux and by flushing with N2 to remove the HCl formed, which would otherwise
attack the double bond in the cyclohexene ring. All the steps of the synthesis of
compound C were monitored by 13C NMR analysis.
Figure 6.3. Procedure for the preparation of the Os-silsesquioxane complex
[RI = cyclopentyl].
Figure 6.4. Procedure for the synthesis of 3,4-dimethylcyclohex-3-enylcarbonyl
chloride.
+
O
O
O
O
O
HO
O
ClSOCl2
N2
H2O
A B C
12
3
45
6
7
8
910
11
1 3
45
6
7
8
9 1 3
45
6
7
8
92 2
+
O
O
O
O
O
HO
O
ClSOCl2
N2
H2O
A B C
12
3
45
6
7
8
910
11
1 3
45
6
7
8
9 1 3
45
6
7
8
92 2
(CH3O)3Si(CH2)3NH2
K2OsO2(OH)4, NMO
Et3N,
N2
O
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
SiNHSi
RI
RI RI
RIRI
RI
O
OOs
O
O
4
OH OH
OH
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
Si
RI
RI RI
RIRI
RI
1
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
Si
Si
RI
RI RI
RIRI
RI
NH2
2
O
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
SiNHSi
RI
RI RI
RIRI
RI
3
12
3
45
6
7
8
9
10
11
12
O
Cl C
(CH3O)3Si(CH2)3NH2
K2OsO2(OH)4, NMO
Et3N,
N2
O
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
SiNHSi
RI
RI RI
RIRI
RI
O
OOs
O
O
4
O
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
SiNHSi
RI
RI RI
RIRI
RI
O
OOs
O
O
4
OH OH
OH
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
Si
RI
RI RI
RIRI
RI
1
OH OH
OH
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
Si
RI
RI RI
RIRI
RI
1
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
Si
Si
RI
RI RI
RIRI
RI
NH2
2
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
Si
Si
RI
RI RI
RIRI
RI
NH2
2
O
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
SiNHSi
RI
RI RI
RIRI
RI
3
12
3
45
6
7
8
9
10
11
12
O
OO
O
RI
OO
SiO Si
OO
OSiSi
OSiOSi
O
SiNHSi
RI
RI RI
RIRI
RI
3
12
3
45
6
7
8
9
10
11
12
O
Cl C
O
Cl C

Os-silsesquioxane as dihydroxylation catalyst __________________________________________________________________________________________________________
109
The next step of the synthesis of the catalytic complex is the addition of OsO4 to the
double bond of the functionalised silsesquioxane 3 (in 1:1 molar ratio). To avoid the
risks connected with the handling of OsO4, potassium osmate K2OsO2(OH)4 was used
as osmium source.14 A solution of potassium osmate was oxidised to OsO4 by means of
N-methylmorpholine-N-oxide (NMO). In the case of the synthesis of the Os-silsesquioxane
catalyst, care must be taken to avoid the reaction of the Os-centre with two silsesquioxane
ligands 3, since this would lead to the formation of a stable, inactive bis-chelate complex.
The reaction of compound 3 with OsO4 prepared by oxidation of potassium osmate with
NMO using stoichiometric amounts of the reagents, did not lead to the desired
Os-silsesquioxane 4. It was found that the oxidation of potassium osmate with NMO did
not proceed if a stoichiometric amount of the oxidant was used. It was then inferred that
an excess of NMO should be used to oxidise the potassium osmate (20:1).
In order to prevent the formation of the bis-chelate while using an excess of NMO,
compound 3 was added to the reaction mixture together with an excess of cyclohexene.
The double bond of the cyclohexene would then compete with that of compound 3 for
binding to the osmium centre. Being less sterically hindered and being in large excess
(19:1) compared to compound 3, cyclohexene will be favoured in reacting with the
osmium centre and will tend to occupy both catalytic sites. Cyclohexene is not a
tetrasubstituted alkene, therefore its bond with the osmium centre is labile and in the
presence of H2O and of NMO it undergoes oxidative hydrolysis, yielding
1,2-cyclohexanediol and restoring the catalytic site on the osmium. This process proceeds
according to the catalytic cycle for the dihydroxylation of alkenes (Figure 6.1). As the
concentration of cyclohexene in solution decreases, it becomes more likely that
compound 3 binds to the osmium centre to produce complex 4 and the
silesquioxane-Os-cyclohexane bisdiolate complex 5 (Figure 6.5). This latter complex
presents an osmium centre bound to one side to the tetrasubstituted olefin of compound 3
and on the other side to cyclohexene. The cyclohexane diolate can be hydrolysed
providing the catalytic site for dihydroxylation reactions, while the tetrasubstituted diolate
is stable under the employed reaction conditions and, therefore, prevents the formation of
free OsO4 during the catalytic process. The synthesis was monitored by means of ESI
MS: after 3 days of reaction, compound 5 is the main product, with small amounts of
compound 4 and of unreacted compound 3 (Figure 6.6). A mixture of H2O, ButOH and
CH2Cl2 was used as solvent during the synthesis: H2O was needed to dissolve the
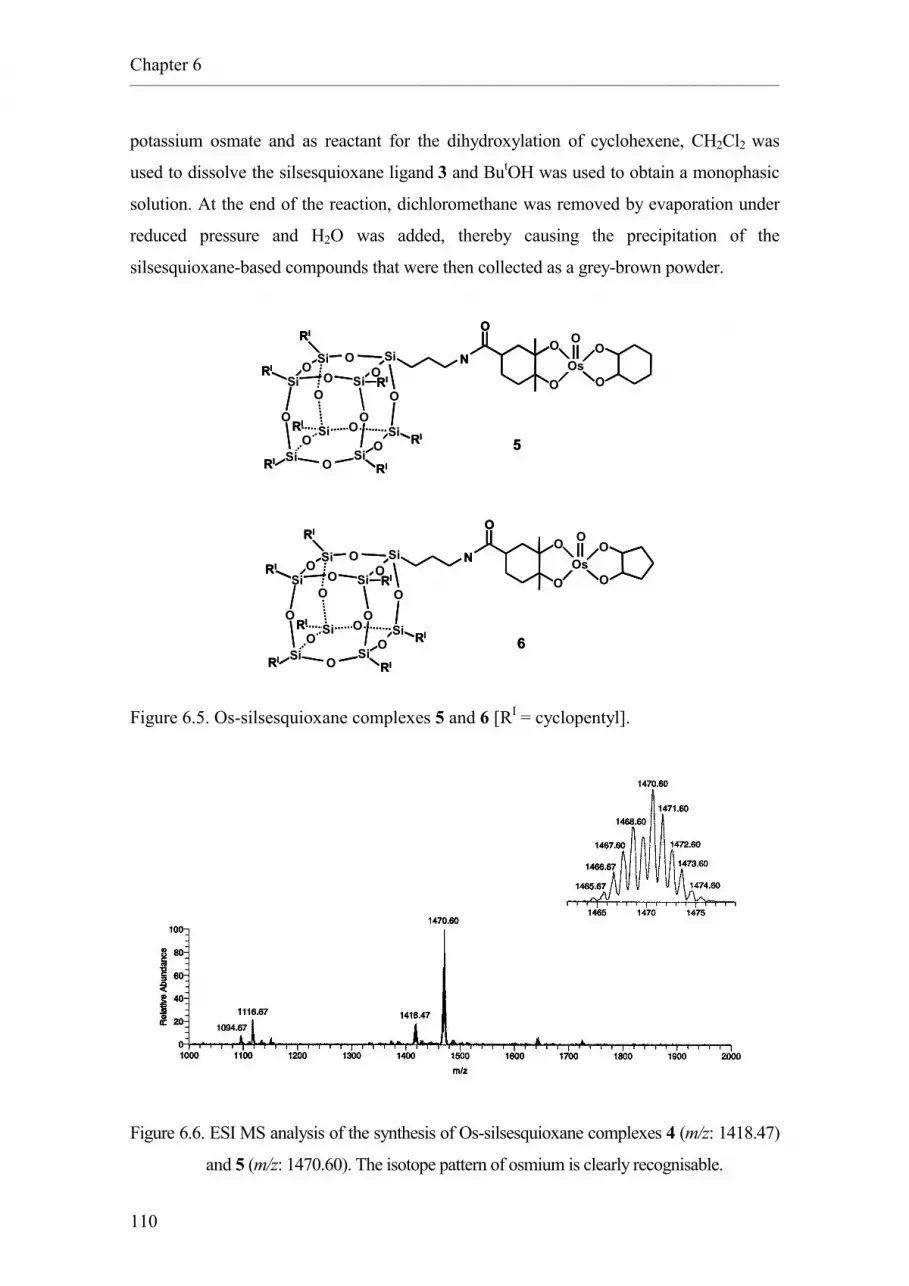
Chapter 6 __________________________________________________________________________________________________________
110
potassium osmate and as reactant for the dihydroxylation of cyclohexene, CH2Cl2 was
used to dissolve the silsesquioxane ligand 3 and ButOH was used to obtain a monophasic
solution. At the end of the reaction, dichloromethane was removed by evaporation under
reduced pressure and H2O was added, thereby causing the precipitation of the
silsesquioxane-based compounds that were then collected as a grey-brown powder.
Figure 6.5. Os-silsesquioxane complexes 5 and 6 [RI = cyclopentyl].
Figure 6.6. ESI MS analysis of the synthesis of Os-silsesquioxane complexes 4 (m/z: 1418.47)
and 5 (m/z: 1470.60). The isotope pattern of osmium is clearly recognisable.
OO
OOO
SiO Si
OO
OSiSi
O SiOSi
O
Si
O
NO
OOs
O
O
OSi
5RI
RI
RI
RI
RI
RI
RI
OO
OOO
SiO Si
OO
OSiSi
O SiOSi
O
Si
O
NO
OOs
O
O
OSi
6RI
RI
RI
RI
RI
RI
RI
OO
OOO
SiO Si
OO
OSiSi
O SiOSi
O
Si
O
NO
OOs
O
O
OSi
5RI
RI
RI
RI
RI
RI
RIO
O
OOO
SiO Si
OO
OSiSi
O SiOSi
O
Si
O
NO
OOs
O
O
OSi
5RI
RI
RI
RI
RI
RI
RI
OO
OOO
SiO Si
OO
OSiSi
O SiOSi
O
Si
O
NO
OOs
O
O
OSi
6RI
RI
RI
RI
RI
RI
RIO
O
OOO
SiO Si
OO
OSiSi
O SiOSi
O
Si
O
NO
OOs
O
O
OSi
6RI
RI
RI
RI
RI
RI
RI

Os-silsesquioxane as dihydroxylation catalyst __________________________________________________________________________________________________________
111
A catalytic test of complex 5 was performed under the same reaction conditions used for
testing the heterogeneous silica-supported analogue of the Os-silsesquioxane complex.5
Two different substrates were used: cyclopentene and cyclohexene. The results of the
catalytic test for the dihydroxylation of these two substrates are reported in Table 6.1.
Conversions and selectivities were determined by GC and GC-MS analysis. The
catalyst was active and very selective in the dihydroxylation of both substrates: after 3
hours reaction a TONcyclopentene of 130 and a TONcyclohexene of 414 were observed. The
conversion was higher for cyclohexene than for cyclopentene, probably because the
complex between the osmium centre and cyclopentene ring is geometrically strained
and its formation is, therefore, less favourable than for the complex with cyclohexene.
The homogeneous Os-silsesquioxane showed similar trends but higher activity than its
silica-supported analogue.5 For the dihydroxylation of cyclopentene, a conversion of
84% was achieved after 24 hours of reaction, while with the silica-supported catalyst a
conversion of 83% was obtained after 48 hours. The difference in performance is even
more evident for cyclohexene, for which total conversion is achieved after 48 hours
with the heterogeneous catalyst and after just 3 hours with the Os-silsesquioxane
catalyst.
Table 6.1. Catalytic test of the Os-silsesquioxane in the dihydroxylation of cyclopentene
and cyclohexene.
After the dihydroxylation of cyclopentene proceeded for 24 hours, the catalyst was
analysed by means of ESI MS: complex 6 (Figure 6.5) was the only Os-complex present
substrate time conversion selectivitycyclopentene 3 h 36% 99%
6 h 50% 99%24 h 84% 99%
cyclohexene 3 h 99% 99%
OH
OH
OH
OH
NMO, H2O
Os-silsesquioxaneOs-silsesquioxane
NMO, H2O
substrate time conversion selectivitycyclopentene 3 h 36% 99%
6 h 50% 99%24 h 84% 99%
cyclohexene 3 h 99% 99%
OH
OH
OH
OH
NMO, H2O
Os-silsesquioxaneOs-silsesquioxane
NMO, H2O OH
OH
OH
OH
NMO, H2O
Os-silsesquioxaneOs-silsesquioxane
NMO, H2O

Chapter 6 __________________________________________________________________________________________________________
112
in solution. This finding is in agreement with the proposed mechanism for the
dihydroxylation reaction: the cyclohexane diolate of complex 5 was hydrolysed
restoring the catalytic site, which was then active in the dihydroxylation of
cyclopentene. The silsesquioxane ligand, bound to the osmium centre via a
tetrasubstituted diolate, did not undergo hydrolysis and, therefore, prevented formation
of the undesired OsO4 in solution.
6.3 Conclusions
An Os-silsesquioxane complex was synthesised, characterised and tested for
activity in dihydroxylation reactions. The complex provided a model for a
silica-supported Os-catalyst. Characterisation of the homogeneous Os-silsesquioxane
confirmed the proposed nature of the heterogeneous catalytic site, where the osmium
centre is on one face bound in a stable fashion to the silsesquioxane ligand and on the
other face is available for catalysing the dihydroxylation of olefins. The
dihydroxylations of cyclopentene and cyclohexene were used as test reactions. In both
cases, the homogeneous catalyst displayed higher turnover frequencies than the
heterogeneous one, while keeping the same selectivity (99%).
6.4 Experimental
13C NMR spectra were measured at 25°C in CDCl3 as solvent on a Varian
Inova-300 (75.5 MHz, 1H decoupled) and on a Bruker DPX-300 (75.5 MHz, 1H
decoupled).
Mass spectrometry analysis was performed on a Micromass Quattro LC-MS with ESI as
ionisation technique and on a ThermoQuest Finnigan LCQ Deca equipped with ESI
source and controlled by Xcalibur software. The desolvation temperature was set to
300°C, the cone voltage to 20 V. The samples had a concentration of ~0.01 mg of
silsesquioxane-based compound diluted in 1 ml of methanol (or of a
methanol/dichloromethane 1:1 mixture) with addition of a drop of acetic acid if
necessary.

Os-silsesquioxane as dihydroxylation catalyst __________________________________________________________________________________________________________
113
GC analysis was performed on a Hewlett-Packard 5890 equipped with a split/splitless
capillary injector, FID detector and a Phenomenex Zebron ZB-5 column (30 m). In the
temperature program, the temperature was set at 30°C for 1 minute, and then it was
raised to 250°C with a rate of 10°C/min and hold at that temperature for 5 minutes.
GC-MS analysis was performed on a Finnigan Polaris Q ion trap mass spectrometer
with a Trace GC. The GC was equipped with a HP1-MS column (25 m). The GC
method started with a temperature of 40°C for 2 minutes, then the temperature was
raised to 200°C at a rate of 10°C/min and hold for 1 minute. The ionisation technique
for MS analysis was EI (70 eV).
Synthesis of compound 2. 2.0·10-3 moles of cyclopentyl silsesquioxane a7b3 1 were
dissolved in 70 ml of tetrahydrofuran (THF) and 2.2·10-3 moles of
3-aminopropyltrimethoxysilane, (CH3O)3Si(CH2)3NH2, were dissolved in 50 ml of
THF. The two solutions were mixed and allowed to react for two days at room
temperature while stirring. The solvent was then removed under reduced pressure and
the residue was washed with acetonitrile to remove any 3-aminopropyltrimethoxysilane
that might still be present. The residue, in the form of a white gel, was redissolved in
dichloromethane and dried again under reduced pressure yielding compound 2 as a
white solid (1.484g, corresponding to a yield of 78%). MS data, m/z: 958.22
(compound 2 + H+, 100%), other peaks belonging to impurities with intensities < 3%.
Synthesis of compound B. 0.15 moles of 3,4-dimethylcyclohex-3-enyl ethyl
carboxylate A were added to 250 ml of 10%wt NaOH solution in H2O. ~0.30g of
tetrabutylammonium bromide were added to the biphasic system to favour mixing
between the ester A and water. The reaction mixture was stirred overnight under reflux
to give a monophasic solution. The ethanol formed in the hydrolysis of compound A
was removed from the solution under reduced pressure. H2SO4 1N was added to the
solution until an acidic pH was reached: the carboxylic acid B precipitated as a white
solid that was then dried overnight under reduced pressure. Total conversion of
compound A to compound B was achieved. 13C{1H} NMR data: for compound A,
δ 14.32 (11), 18.87 (7), 19.01 (8), 25.96 (6), 31.12 (5), 33.84 (2), 40.33 (1), 60.20 (10),
124.02 (4), 125.29 (3), 176.00 (9); for compound B, δ 19.05 (7), 19.17 (8), 25.80 (6),
31.05 (5), 33.64 (2), 40.14 (1), 123.94 (4), 125.59 (3), 182.04 (9).

Chapter 6 __________________________________________________________________________________________________________
114
Synthesis of compound C. 4.0·10-2 moles of thionyl chloride (SOCl2) were added to
1.6·10-2 moles of the carboxylic acid B and the solution obtained was stirred for 3 hours
and 30 minutes under reflux, while the surface of the reaction mixture was flushed with
N2. Gaseous HCl leaving the reaction mixture through the reflux cooler was redirected
into an aqueous solution of NaOH. The reaction mixture was dried under reduced
pressure to remove the thionyl chloride still present. The remaining liquid contained the
acid chloride C (~70%) together with some carboxylic acid B (~30%), as determined on
the basis 13C NMR analysis. The sample was stored under N2. 13C{1H} NMR data for
compound C, δ 19.04 (7), 19.33 (8), 25.38 (6), 30.74 (5), 33.15 (2), 41.31 (1), 123.51
(4), 125.58 (3), 171.81 (9).
Synthesis of compound 3. 1.6·10-3 moles of compound 2 were dissolved in a solution of
1.6·10-3 moles of triethylamine in 30 ml of dichloromethane. Subsequently, the solution
was added to the mixture of acid chloride C and carboxylic acid B and stirred overnight
at room temperature under N2 flow. The solution was then concentrated to a volume of
5 ml under reduced pressure and 60 ml of acetonitrile were added: a white solid
precipitated. The solid was washed with 0.8%wt NaOH(aq) to remove B and C that might
still be present. The yield in compound 3 was ~68%. MS data, m/z: 1094.15
(compound 3 + H+, 13%), 1116.11 (compound 3 + Na+, 100%), 1132.00 (compound 3 +
K+, 8%). 13C{1H} NMR data for compound 3, δ 9.36 (12), 18.84 (7), 18.98 (8), 23.26
(11) 26.56 (6), 31.14 (5), 34.54 (2), 41.47 (1), 42.33 (10), 124.09 (4), 125.48 (3), 175.76
(9); δ 22.23 (ipso carbons of the cyclopentyl groups on the silsesquioxane), 27.03, 27.30
(multiplets; remaining carbons of the cyclopentyl groups on the silsesquioxane).
Synthesis of complexes 4 and 5. 4.0·10-5 moles of potassium osmate, K2OsO2(OH)4,
were dissolved in 2 ml of H2O to give a dark-red solution. 8.0·10-4 moles of
N-methylmorpholine-N-oxide (NMO) were dissolved in 0.5 ml of H2O. The two
aqueous solutions were mixed together with 4 ml of tert-butanol and the obtained pink
solution was stirred overnight at room temperature. Upon addition of 0.1 ml of a 0.42M
aqueous solution of acetic acid the solution turned colourless. Next, 0.044 g (~4·10-5
moles) of compound 3 and 7.6·10-4 moles of cyclohexene, previously dissolved in 5 ml
of tert-butanol and 3 ml of dichloromethane, were added to the Os-containing solution:
the solution immediately turned dark brown, indicating the formation of the Os-diolate.
After stirring the reaction mixture for 3 days at room temperature, a sample was taken
and analysed by MS. Then, dichloromethane was removed under reduced pressure from

Os-silsesquioxane as dihydroxylation catalyst __________________________________________________________________________________________________________
115
the homogeneous dark brown solution. Next, 15 ml of H2O were added to the slightly
turbid solution and a precipitate was formed. The solution was filtered on a folded paper
filter and the grey-brown residue was collected (0.028 g, corresponding to ~1.7·10-5
moles and, therefore, to a yield of ~42% in Os-silsesquioxane complexes). MS data,
m/z: 1470.60 (complex 5 + Na+, 100%), 1418.47 (complex 4 + 3Na+, 18%), 1116.67
(compound 3 + Na+, 21%), 1094.67 (compound 3 + H+, 8%).
Catalytic test. 0.014 g of the grey-brown solid (~8.4·10-6 moles of Os-silsesquioxane
complexes) were dissolved in 2.5 ml of dichloromethane and 5 ml of tert-butanol. To
this solution, 4.0·10-3 moles of alkene (cyclopentene or cyclohexene), 2.0·10-3 moles of
n-nonane (internal standard for GC analysis), 4.0·10-3 moles of NMO and 500 µl of H2O
were added. The yellow-brown solution was stirred at room temperature for 24 hours.
Samples for GC and GC-MS analysis were taken after 3, 6 and 24 hours of reaction.
MS analysis of the catalyst after the dihydroxylation of cyclopentene, m/z: 1456.53
(complex 6 + Na+).
Safety precautions: all handling of osmium compounds was performed in a fume hood,
while wearing a class P2 (particulate) respirator. The reactions in which OsO4 was
formed were carried out in sealed flasks or, if any gas was leaving the synthesis flask, it
was led through a bottle containing oil, rich in unsaturated fatty acids, in order to trap
any OsO4 that might have been developed from the reaction mixture.

Chapter 6 __________________________________________________________________________________________________________
116
References
1 M. Schroeder, Chem. Rev., 1980, 80, 187. 2 H.C. Kolb, M.S. VanNieuwenhze, K.B. Sharpless, Chem. Rev., 1994, 94, 2483. 3 R.J. Lewis, Sax’s Dangerous Properties of Industrial Materials, Wiley-Interscience, 10th edition, 2000. 4 A. Severeyns, D.E. De Vos, P.A. Jacobs, Top. Catal., 2002, 19, 125. 5 A. Severeyns, D.E. De Vos, L. Fiermans, F. Verpoort, P.J. Grobet, P.A. Jacobs, Angew. Chem. Int. Ed.,
2001, 40, 586. 6 A. Severeyns, D.E. De Vos, P.A. Jacobs, Green Chem., 2002, 4, 380. 7 V. VanRheenen, R.C. Kelly, D.Y. Cha, Tetrahedron Lett., 1976, 23, 1973. 8 P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583, see also Chapter 2. 9 S. Krijnen, H.C.L. Abbenhuis, R.W.J.M. Hanssen, J.H.C. van Hooff, R.A. van Santen, Angew. Chem.
Int. Ed., 1998, 37, 356. 10 S. Krijnen, B.L. Mojet, H.C.L. Abbenhuis, J.H.C. van Hooff, R.A. van Santen, Phys. Chem. Chem.
Phys., 1999, 1, 361. 11 P. Smet, J. Riondato, T. Pauwels, L. Moens, L. Verdonck, Inorg. Chem. Commun., 2000, 3, 557. 12 See Chapter 5. 13 J. Monnin, Helv. Chim. Acta, 1958, 225, 2112. 14 W.D. Lloyd, B.J. Navarette, M.F. Shaw, Synthesis, 1972, 610.

117
7
New tert-butyl and phenyl silsesquioxane precursors for epoxidation titanium catalysts discovered by means of High-Speed Experimentation
Abstract
A set of new titanium-silsesquioxane epoxidation catalysts was discovered by exploring
the hydrolytic condensation of a series of trichlorosilanes in highly-polar solvents by
means of High-Speed Experimentation techniques. The three most promising
silsesquioxane leads were resynthesised in a larger-volume scale and characterised. The
lead obtained from the hydrolytic condensation of ButSiCl3 in water consisted of a
single silsesquioxane structure: But2Si2O(OH)4. This is the first reported example of the
use of this silsesquioxane as precursor for active titanium catalysts. The Ti-complexes
prepared with But2Si2O(OH)4 were supported on silica to produce an active
heterogeneous epoxidation catalyst. The lead generated by the hydrolytic condensation
of ButSiCl3 in DMSO consisted of a set of incompletely condensed silsesquioxane
structures. The lead obtained by the hydrolytic condensation of PhSiCl3 in water was
constituted of polymeric silsesquioxanes.
____________________ The contents of this chapter have been submitted to:
P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, Chem. Eur. J.

Chapter 7 __________________________________________________________________________________________________________
118
7.1 Introduction
Chapter 3 and 4 reported the successful application of High-Speed
Experimentation (HSE) techniques to the optimisation of silsesquioxane precursors for
titanium catalysts active in the epoxidation of alkenes.1,2 This work resulted in a new
and efficient way to synthesise silsesquioxane precursors and generated knowledge
about the effect of the various parameters involved in the synthesis. Particularly, one of
the observed trends indicated a favourable effect induced by high-polarity solvents. This
result stimulated further investigation: here, the application of High-Speed
Experimentation techniques to the optimisation of the synthesis of silsesquioxanes in
very polar solvents is presented. The experimental approach is similar to that used in
previous studies1,2 and it is thoroughly described in Paragraph 3.2.
7.2 Results and discussion
7.2.1 The High-Speed Experimentation screening
The synthesis of silsesquioxane precursors for titanium catalysts via the
hydrolytic condensation of 6 different trichlorosilanes (R = cyclohexyl, cyclopentyl,
phenyl, methyl, ethyl and tert-butyl) in 3 highly-polar solvents (dimethyl sulfoxide
(DMSO), water and formamide) was performed by means of High-Speed
Experimentation techniques. Previous work showed that the R-group on the
organosilane and the solvent in which the hydrolytic condensation is carried out are the
most relevant parameters influencing the synthesis of silsesquioxanes.3 The parameter
space studied was defined by the full-factorial combination of the 6 trichlorosilanes and
the 3 solvents. This parameter space was screened as a function of the activity in the
epoxidation of 1-octene with tert-butyl hydroperoxide (TBHP)4 displayed by the
catalysts which were obtained after coordination of Ti(OBu)4 to the silsesquioxane
structures. In Figure 7.1, the relative activities of the titanium silsesquioxanes are shown
together with those of the titanium silsesquioxanes obtained from silsesquioxanes
synthesised in acetonitrile, which gave the best results among the solvents tested in
previous work.1,2 The values are normalised to the activity of the most active titanium
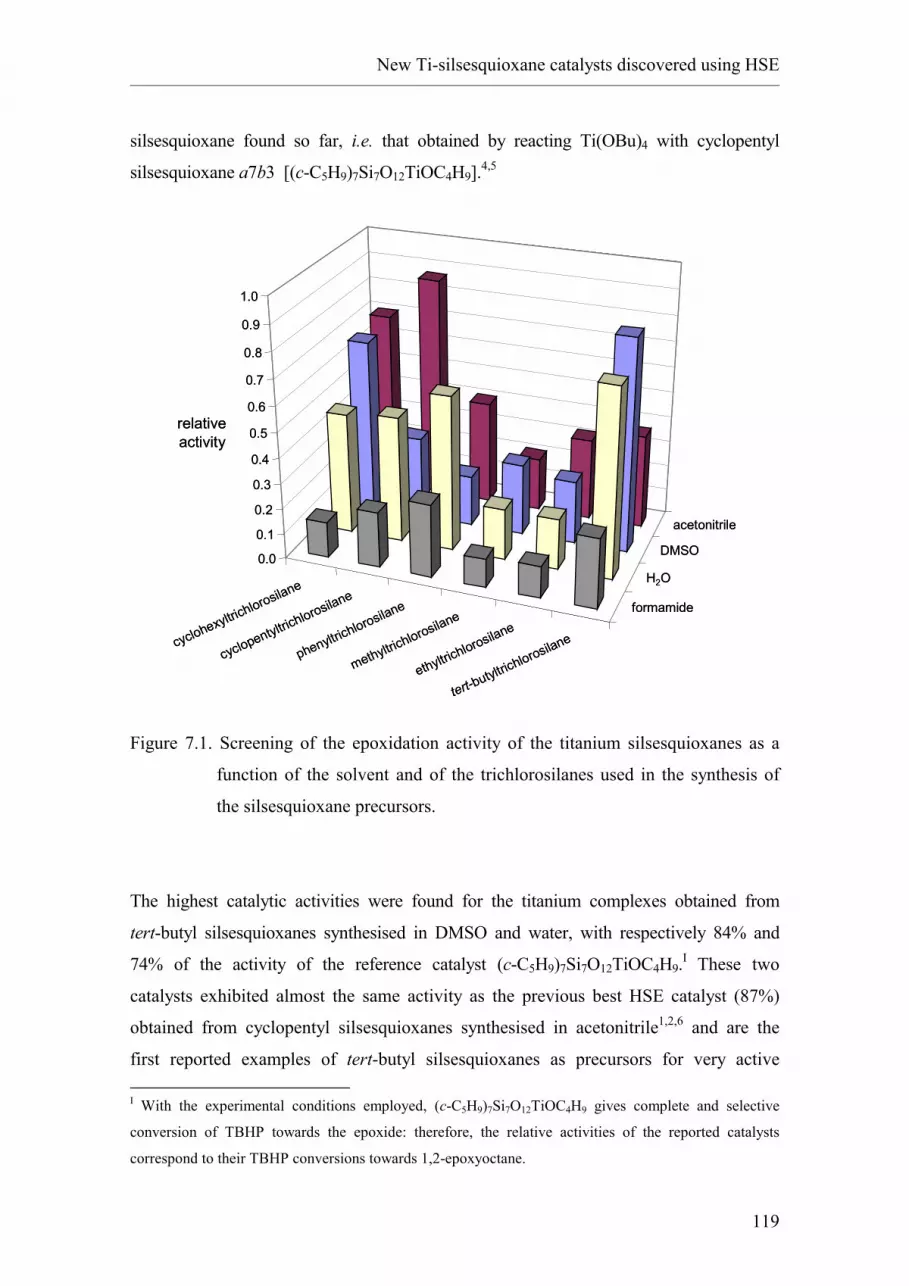
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
119
silsesquioxane found so far, i.e. that obtained by reacting Ti(OBu)4 with cyclopentyl
silsesquioxane a7b3 [(c-C5H9)7Si7O12TiOC4H9].4,5
Figure 7.1. Screening of the epoxidation activity of the titanium silsesquioxanes as a
function of the solvent and of the trichlorosilanes used in the synthesis of
the silsesquioxane precursors.
The highest catalytic activities were found for the titanium complexes obtained from
tert-butyl silsesquioxanes synthesised in DMSO and water, with respectively 84% and
74% of the activity of the reference catalyst (c-C5H9)7Si7O12TiOC4H9.I These two
catalysts exhibited almost the same activity as the previous best HSE catalyst (87%)
obtained from cyclopentyl silsesquioxanes synthesised in acetonitrile1,2,6 and are the
first reported examples of tert-butyl silsesquioxanes as precursors for very active I With the experimental conditions employed, (c-C5H9)7Si7O12TiOC4H9 gives complete and selective
conversion of TBHP towards the epoxide: therefore, the relative activities of the reported catalysts
correspond to their TBHP conversions towards 1,2-epoxyoctane.
cyclohexyltrichlorosilane
cyclopentyltrichlorosilane
phenyltrichlorosilane
methyltrichlorosilane
ethyltrichlorosilane
tert-butyltrichlorosilane
formamide
H2O
DMSO
acetonitrile
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
relativeactivity
cyclohexyltrichlorosilane
cyclopentyltrichlorosilane
phenyltrichlorosilane
methyltrichlorosilane
ethyltrichlorosilane
tert-butyltrichlorosilane
formamide
H2O
DMSO
acetonitrile
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
relativeactivity

Chapter 7 __________________________________________________________________________________________________________
120
titanium catalysts. Relevant catalytic activities were also obtained with cyclohexyl
silsesquioxanes synthesised in DMSO (67%) and with phenyl silsesquioxanes
synthesised in H2O (61%).
Besides the identification of these leads, the High-Speed Experimentation screening
provided some more information about the system under study. No regular trend can be
identified, i.e. for each solvent the order of activity as a function of the R-groups is
different. This can be explained on the basis of the different nature of the solvents
employed. Water acts both as solvent and reagent, and the fact that silsesquioxanes with
only a very low level of condensation are soluble in water7 causes the fast precipitation
of condensed structures from solution. Formamide has a high boiling point (220°C),
requiring high temperatures to remove the solvent from the silsesquioxane products. At
these high temperatures, the silsesquioxanes tend to condense further to produce
completely condensed structures, which are not suitable for binding metal centres.8 This
can explain the weak activity observed for the catalysts obtained from silsesquioxanes
synthesised in formamide.
Finally, all the methyl and ethyl silsesquioxanes are poor precursor for titanium-based
epoxidation catalysts, in agreement with what found in previous work.1,2
Although High-Speed Experimentation techniques proved to be a powerful tool
to identify leads and gain knowledge about the system under study, they did not provide
any information about the actual silsesquioxane structures that were formed.6 To gain
such information, selected HSE leads can be synthesised on a conventional laboratory
scale and characterised by appropriate analytical techniques (e.g. spectroscopy and
chromatography). Here, the phenyl silsesquioxanes synthesised in water and the
tert-butyl silsesquioxanes synthesised in DMSO and water (i.e. the silsesquioxane
precursors that gave more active catalysts than those synthesised in acetonitrile,1,2 see
Figure 7.1) were selected for further investigation.
7.2.2 Phenyl silsesquioxanes synthesised in H2O
The hydrolytic condensation of phenyltrichlorosilane in H2O was performed in a
25-fold up-scaling of the HSE synthesis. The white precipitate obtained from the reaction
was characterised by NMR Spectroscopy. The liquid-phase 13C NMR spectrum presents
two sets of partially overlapping peaks due to the phenyl rings, indicating that two
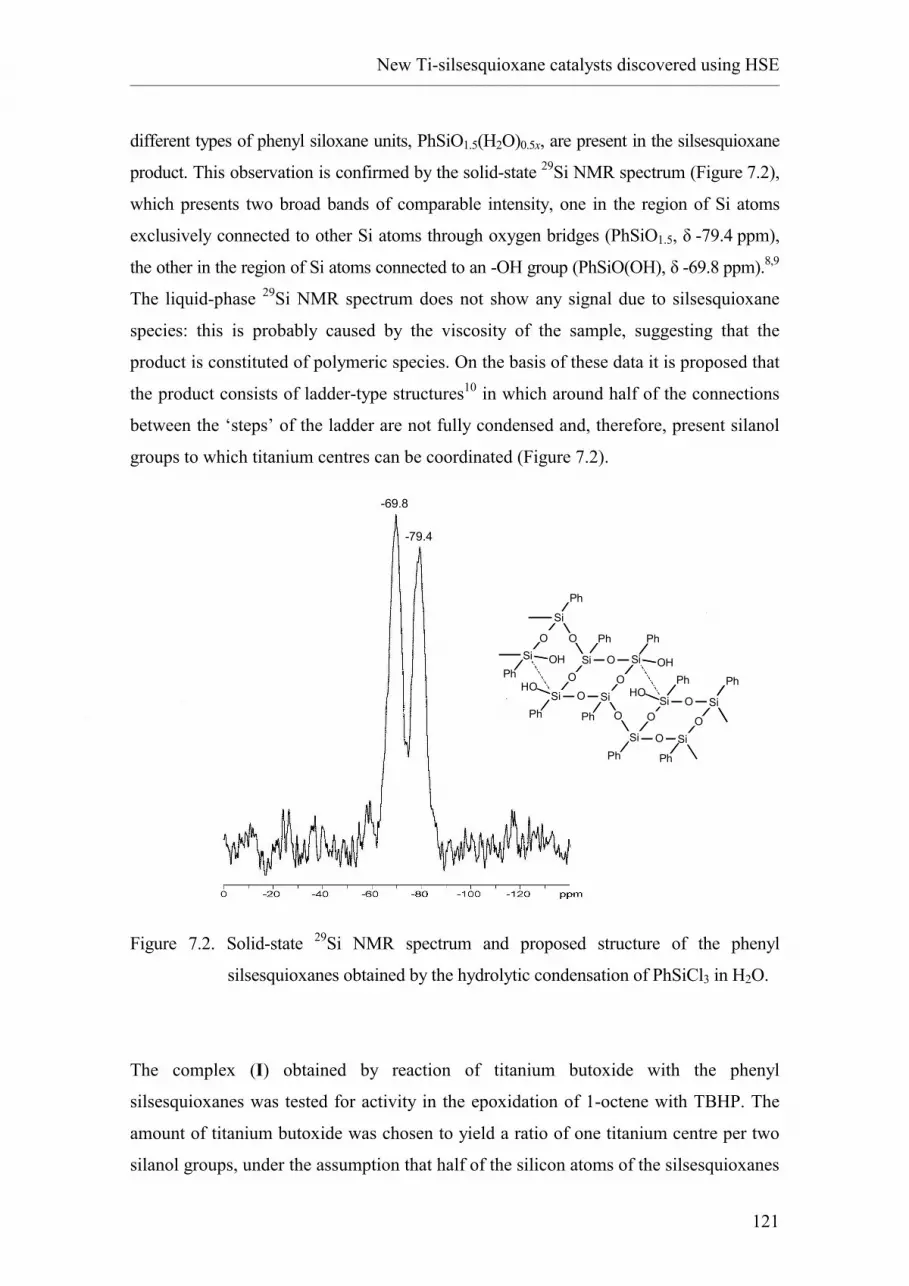
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
121
different types of phenyl siloxane units, PhSiO1.5(H2O)0.5x, are present in the silsesquioxane
product. This observation is confirmed by the solid-state 29Si NMR spectrum (Figure 7.2),
which presents two broad bands of comparable intensity, one in the region of Si atoms
exclusively connected to other Si atoms through oxygen bridges (PhSiO1.5, δ -79.4 ppm),
the other in the region of Si atoms connected to an -OH group (PhSiO(OH), δ -69.8 ppm).8,9
The liquid-phase 29Si NMR spectrum does not show any signal due to silsesquioxane
species: this is probably caused by the viscosity of the sample, suggesting that the
product is constituted of polymeric species. On the basis of these data it is proposed that
the product consists of ladder-type structures10 in which around half of the connections
between the ‘steps’ of the ladder are not fully condensed and, therefore, present silanol
groups to which titanium centres can be coordinated (Figure 7.2).
Figure 7.2. Solid-state 29Si NMR spectrum and proposed structure of the phenyl
silsesquioxanes obtained by the hydrolytic condensation of PhSiCl3 in H2O.
The complex (I) obtained by reaction of titanium butoxide with the phenyl
silsesquioxanes was tested for activity in the epoxidation of 1-octene with TBHP. The
amount of titanium butoxide was chosen to yield a ratio of one titanium centre per two
silanol groups, under the assumption that half of the silicon atoms of the silsesquioxanes
-69.8
-79.4
PhPh
Ph
Ph
Ph
Si
SiO
OH
Si Si Si
Si
Si
Si
Si Si
OO
O
O O
O
O
O
OOHO
Ph
PhPh
Ph
Ph
OH
HO
-69.8
-79.4
-69.8
-79.4
PhPh
Ph
Ph
Ph
Si
SiO
OH
Si Si Si
Si
Si
Si
Si Si
OO
O
O O
O
O
O
OOHO
Ph
PhPh
Ph
Ph
OH
HO
PhPh
Ph
Ph
Ph
Si
SiO
OH
Si Si Si
Si
Si
Si
Si Si
OO
O
O O
O
O
O
OOHO
Ph
PhPh
Ph
Ph
OH
HO

Chapter 7 __________________________________________________________________________________________________________
122
contain -OH groups. This titanium to silsesquioxane ratio is almost double than the one
employed in the HSE experiment. The catalyst (I) showed a similar activity (per mole
of titanium) to the corresponding HSE-catalyst (61% TBHP conversion and >99%
selectivity towards 1,2-epoxyoctane after 4 hours of reaction, see Table 7.1 on
page 130). Differently from other reported titanium catalysts containing phenyl
silsesquioxanes,11 these titanium silsesquioxanes are not soluble in the epoxidation
reaction mixture and remain a distinct powder. Finally, the catalyst was tested for the
epoxidation of 1-octene with aqueous H2O2 as the oxidant, resulting in negligible
activity accompanied by catalyst deactivation.
7.2.3 tert-butyl silsesquioxanes synthesised in H2O
The hydrolytic condensation of tert-butyltrichlorosilane ButSiCl3 in water was
performed in a 25-fold up-scaling of the HSE synthesis. ButSiCl3 is a solid at room
temperature: since the HSE workstation employed can only handle liquids, the
trichlorosilane was dissolved in a minimum of acetonitrile. The same procedure was
used for the up-scaled synthesis. After 18 hours of reaction at 50°C, the reaction
mixture contained a white precipitate, which was isolated by filtration (fraction A).
Upon drying of the filtrate, a white solid was obtained (fraction B). The two fractions
were exsiccated in an oven at 100°C. Liquid-phase NMR characterisation showed that both
fractions contained the same single silsesquioxane structure constituted of equivalent
ButSiO1.5(H2O)0.5x units (Figure 7.3). The position of the 29Si NMR peak (δ -49.55 ppm)
indicates that this silsesquioxane contains one or more silanol groups (x ≥ 1).8 The
structure was assigned to silsesquioxane But2Si2O(OH)4 (a2b4, Figure 7.4)8 on the basis
of these NMR data and of single-crystal X-ray diffraction analysis, which provided the
same cell parameters of the tert-butyl silsesquioxane a2b4 reported in literature.12 The
synthetic procedure reported here allows complete and selective conversion of ButSiCl3
into silsesquioxane But2Si2O(OH)4. This method is more straightforward and results in a
higher isolated yield (90%) than the literature method (65%) in which the compound is
synthesised by addition of ButSiCl3 in diethyl ether to a mixture of KOH, water and
methanol.12 The selectivity of the synthesis is ascribed to the bulkiness of the tert-butyl
group, which can hinder further reaction of But2Si2O(OH)4, and to the use of water as
the solvent, which disfavours condensation.
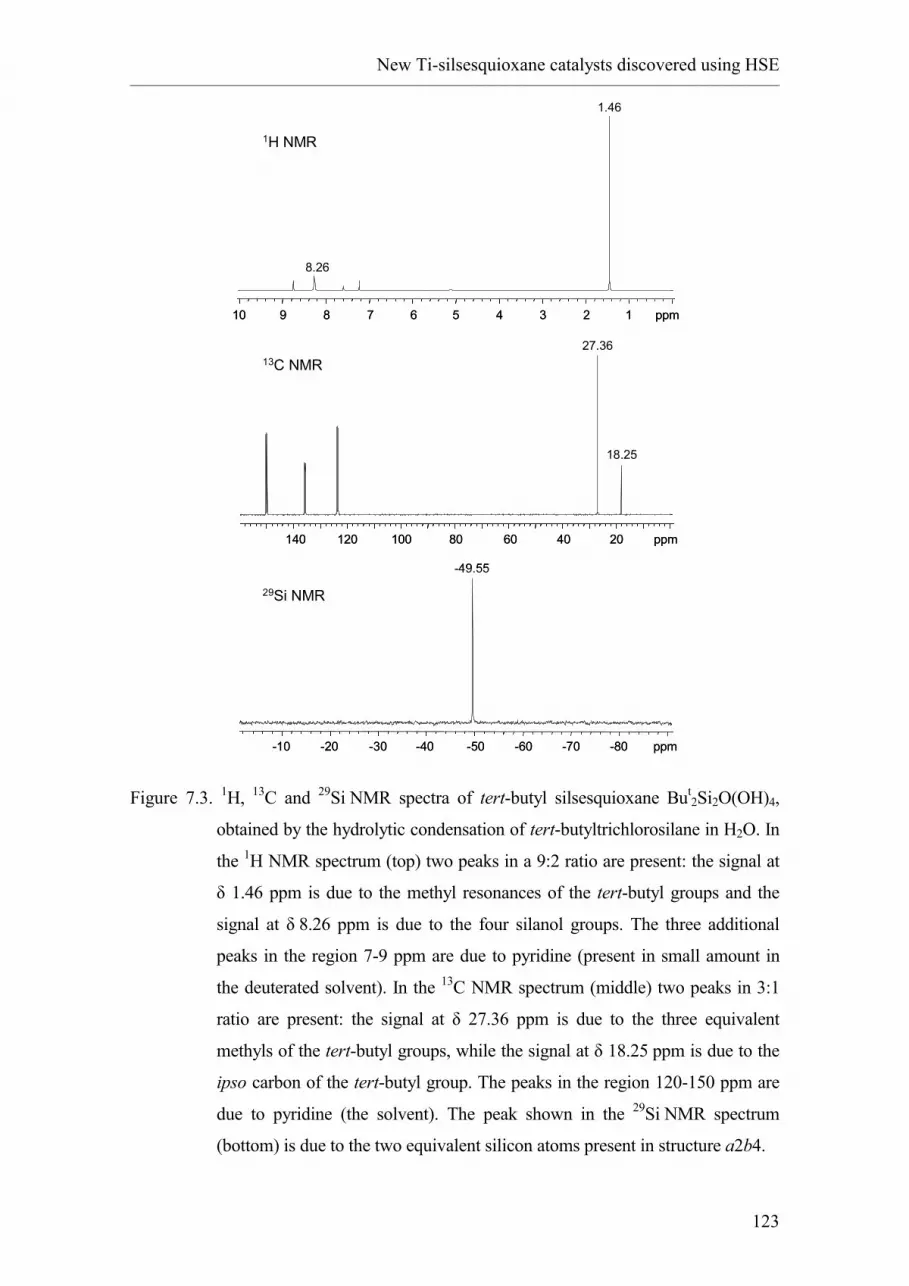
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
123
Figure 7.3. 1H, 13C and 29Si NMR spectra of tert-butyl silsesquioxane But2Si2O(OH)4,
obtained by the hydrolytic condensation of tert-butyltrichlorosilane in H2O. In
the 1H NMR spectrum (top) two peaks in a 9:2 ratio are present: the signal at
δ 1.46 ppm is due to the methyl resonances of the tert-butyl groups and the
signal at δ 8.26 ppm is due to the four silanol groups. The three additional
peaks in the region 7-9 ppm are due to pyridine (present in small amount in
the deuterated solvent). In the 13C NMR spectrum (middle) two peaks in 3:1
ratio are present: the signal at δ 27.36 ppm is due to the three equivalent
methyls of the tert-butyl groups, while the signal at δ 18.25 ppm is due to the
ipso carbon of the tert-butyl group. The peaks in the region 120-150 ppm are
due to pyridine (the solvent). The peak shown in the 29Si NMR spectrum
(bottom) is due to the two equivalent silicon atoms present in structure a2b4.
0
1 0 0 0
2 0 0 0
3 0 0 0
4 0 0 0
5 0 0 0
6 0 0 0
29Si NMR
13C NMR
1H NMR
ppm20406080100120140
ppm-70-50 -80-60-40-20-10 -30
ppm2468 79 510 3 1
8.26
1.46
27.36
18.25
-49.55
0
1 0 0 0
2 0 0 0
3 0 0 0
4 0 0 0
5 0 0 0
6 0 0 0
29Si NMR
13C NMR
1H NMR
ppm20406080100120140
ppm-70-50 -80-60-40-20-10 -30
ppm2468 79 510 3 1
8.26
1.46
27.36
18.25
-49.55

Chapter 7 __________________________________________________________________________________________________________
124
Figure 7.4. tert-butyl silsesquioxane a2b4, But2Si2O(OH)4.
The fact that silsesquioxane a2b4 was obtained as the only product both as a precipitate
and by drying the remaining reaction mixture prompted an investigation of whether a
shorter reaction time would lead to the same product. Therefore, the hydrolytic
condensation was performed for 20 minutes at room temperature. Next, the solvent was
removed by filtration to afford a white solid that was then exsiccated in an oven at
100°C. 13C and 29Si NMR analysis showed that, besides silsesquioxane a2b4, the
product contained a second species assigned to silsesquioxane ButSi(OH)3 (a1b3)13 on
the basis of the position of the 29Si NMR peak (δ -40.06 ppm).8 The molecular ratio
between the a1b3 and a2b4 structures was ~2:1. This experiment indicates that the
formation of tert-butyl silsesquioxane a2b4 takes place by the slow condensation of
tert-butyl silsesquioxane a1b3. The silsesquioxane a2b4 does not react further and it
starts to precipitate as its concentration in solution increases. Further investigation
showed that 7 hours of hydrolytic condensation at 50°C are necessary to obtain
silsesquioxane a2b4 as the only product.
The nature of the solvent in which the ButSiCl3 is dissolved before being added
to the water does not seem to influence the reaction: the same product was obtained if
acetone was used instead of acetonitrile. On the other hand, the amount of this
co-solvent has a relevant role: when ButSiCl3 was dissolved in a 5-fold smaller volume
of acetone, after 18 hours at 50°C the hydrolytic condensation produced large amounts
of precipitate containing viscous polymeric silsesquioxanes together with structure a2b4
(as determined by NMR analysis). After removing the precipitate by filtration, the
filtrate afforded silsesquioxane a2b4 upon drying. The lower selectivity and the
a2b4
OH
But
OHSiSi O
HO
HO
But
a2b4
OH
But
OHSiSi O
HO
HO
ButOH
But
OHSiSi O
HO
HO
But

New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
125
increased amount of precipitate indicate that when less co-solvent is present in solution,
the formation and the precipitation of silsesquioxanes takes place more rapidly,
probably as a consequence of the reduced solubility of the silsesquioxanes in the
reaction mixture. This behaviour is similar to that reported above for the hydrolytic
condensation of phenyltrichlorosilane in H2O, where no co-solvent was used.
These observations confirm the complexity of the mechanism of formation of
silsesquioxanes: small changes in the reaction conditions result in significantly different
amounts and types of structures.
Besides the intrinsic value of the identification of a new, selective and high yield
method to synthesise silsesquioxane But2Si2O(OH)4, this experiment proved that
tert-butyl silsesquioxane a2b4 is a suitable precursor for titanium catalysts. Thus,
silsesquioxane structures different from the known precursor silsesquioxane a7b3
[R7Si7O9(OH)3]4,5,14 can effectively coordinate titanium centres to yield almost equally
active epoxidation catalysts. In order to investigate how the titanium centre coordinates
to tert-butyl silsesquioxane a2b4 and which is the optimum number of titanium centres
that the structure can accommodate, catalysts with different titanium to silsesquioxane
ratios were prepared, characterised and tested. The HSE lead had a titanium to
silsesquioxane ratio of ~ 1:3, the ratio having been tuned for more condensed
silsesquioxanes (e.g. structure a7b3) which present a low average number of -OH
groups per silicon atom. Silsesquioxane a2b4 has four -OH groups that may react with
titanium alkoxide complexes. Coordination of a titanium centre to two -OH groups on
the same silicon would produce a geometrically strained complex and is, therefore,
unlikely. In principle, each titanium could coordinate to two -OH groups on two
different silicons to form a polymeric chain with alternate titanium centres and
silsesquioxanes (Figure 7.5). This chain presents a 1:1 titanium to silsesquioxane molar
ratio. Less ordered structures with a titanium to silsesquioxane molar ratio > 1 are also
possible. Therefore, complexes were prepared by reacting Ti(OBu)4 with tert-butyl
silsesquioxane a2b4 in 1:1 (catalyst II) and 2:1 (catalyst III) molar ratios. The two
catalysts were characterised and tested for epoxidation activity.

Chapter 7 __________________________________________________________________________________________________________
126
Figure 7.5. Ideal structure of a polymeric chain obtained from the reaction of Ti(OBu)4
with But2Si2O(OH)4 in a 1:1 molar ratio.
The liquid-phase 29Si NMR spectrum of catalyst II shows a single peak at -49.55 ppm
due to the unreacted tert-butyl silsesquioxane a2b4, while the spectrum of catalyst III
presents a very weak and broad signal around -56 ppm. These data indicate that a
titanium to silsesquioxane ratio > 1 is needed to react all the silsesquioxane a2b4 and
suggest that both catalysts consist of polymeric titanium complexes (which are difficult
to detect in liquid-phase 29Si NMR). The solid-state 29Si NMR spectrum of catalyst II
(Figure 7.6, top) shows the peak due to tert-butyl silsesquioxane a2b4 (already visible
in the liquid-phase spectrum) and two broad signals centred at -56 ppm and -66 ppm
(integral ratio ~ 1:2:1). The complexation of titanium causes the silicon peak position
to move towards higher shifts,6,15 suggesting that the signals around -56 ppm originate
from siloxy groups to which one titanium centre is coordinated, while the signals at
-66 ppm derive from siloxy groups to which two titanium centres are coordinated. The
solid-state 29Si NMR spectrum of catalyst III (Figure 7.6, bottom) presents two broad
and partially overlapping signals. These two peaks are in the same position as those
found for catalyst II, but exhibit a different ratio (4:1 as compared to 2:1), consistent
with catalyst III containing proportionally more siloxy groups to which only one
titanium centre is coordinated.
But
Si
Si
But
O Ti
O
O
O
O
m
But
Si
Si
But
O Ti
O
O
O
O
m
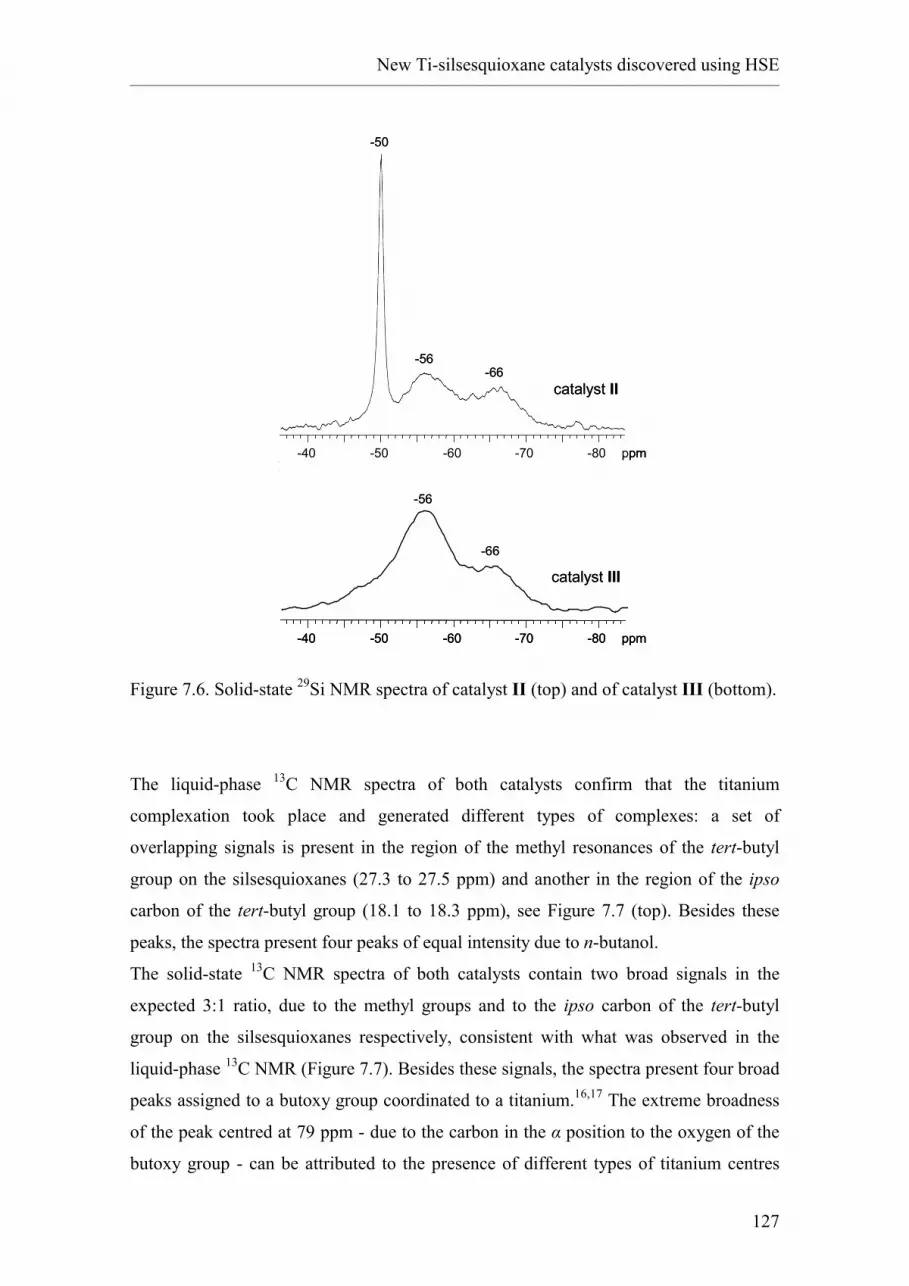
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
127
Figure 7.6. Solid-state 29Si NMR spectra of catalyst II (top) and of catalyst III (bottom).
The liquid-phase 13C NMR spectra of both catalysts confirm that the titanium
complexation took place and generated different types of complexes: a set of
overlapping signals is present in the region of the methyl resonances of the tert-butyl
group on the silsesquioxanes (27.3 to 27.5 ppm) and another in the region of the ipso
carbon of the tert-butyl group (18.1 to 18.3 ppm), see Figure 7.7 (top). Besides these
peaks, the spectra present four peaks of equal intensity due to n-butanol.
The solid-state 13C NMR spectra of both catalysts contain two broad signals in the
expected 3:1 ratio, due to the methyl groups and to the ipso carbon of the tert-butyl
group on the silsesquioxanes respectively, consistent with what was observed in the
liquid-phase 13C NMR (Figure 7.7). Besides these signals, the spectra present four broad
peaks assigned to a butoxy group coordinated to a titanium.16,17 The extreme broadness
of the peak centred at 79 ppm - due to the carbon in the α position to the oxygen of the
butoxy group - can be attributed to the presence of different types of titanium centres
-40 -50 -60 -70 -80 ppm
-40 -50 -60 -70 -80 ppm
-56
-56
-66
-66
-50
catalyst II
catalyst III
-40 -50 -60 -70 -80 ppm
-40 -50 -60 -70 -80 ppm
-56
-56
-66
-66
-50
catalyst II
catalyst III
-40 -50 -60 -70 -80 ppm
-40 -50 -60 -70 -80 ppm
-56
-56
-66
-66
-50
catalyst II
catalyst III
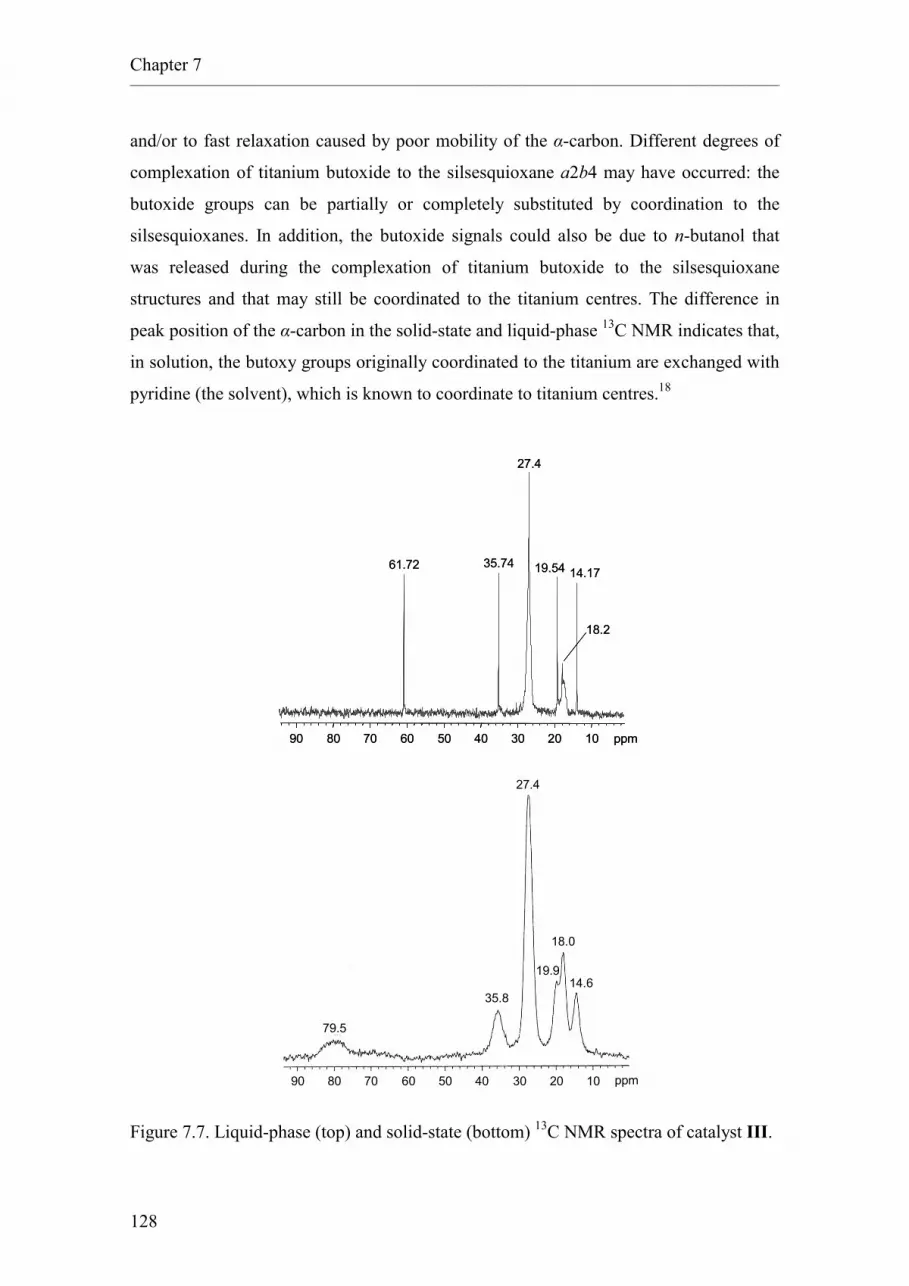
Chapter 7 __________________________________________________________________________________________________________
128
and/or to fast relaxation caused by poor mobility of the α-carbon. Different degrees of
complexation of titanium butoxide to the silsesquioxane a2b4 may have occurred: the
butoxide groups can be partially or completely substituted by coordination to the
silsesquioxanes. In addition, the butoxide signals could also be due to n-butanol that
was released during the complexation of titanium butoxide to the silsesquioxane
structures and that may still be coordinated to the titanium centres. The difference in
peak position of the α-carbon in the solid-state and liquid-phase 13C NMR indicates that,
in solution, the butoxy groups originally coordinated to the titanium are exchanged with
pyridine (the solvent), which is known to coordinate to titanium centres.18
Figure 7.7. Liquid-phase (top) and solid-state (bottom) 13C NMR spectra of catalyst III.
ppm102030405060708090
61.72 35.74
27.4
19.54
18.2
14.17
ppm102030405060708090
79.5
35.8
27.4
19.9
18.0
14.6
ppm102030405060708090
61.72 35.74
27.4
19.54
18.2
14.17
ppm102030405060708090
79.5
35.8
27.4
19.9
18.0
14.6
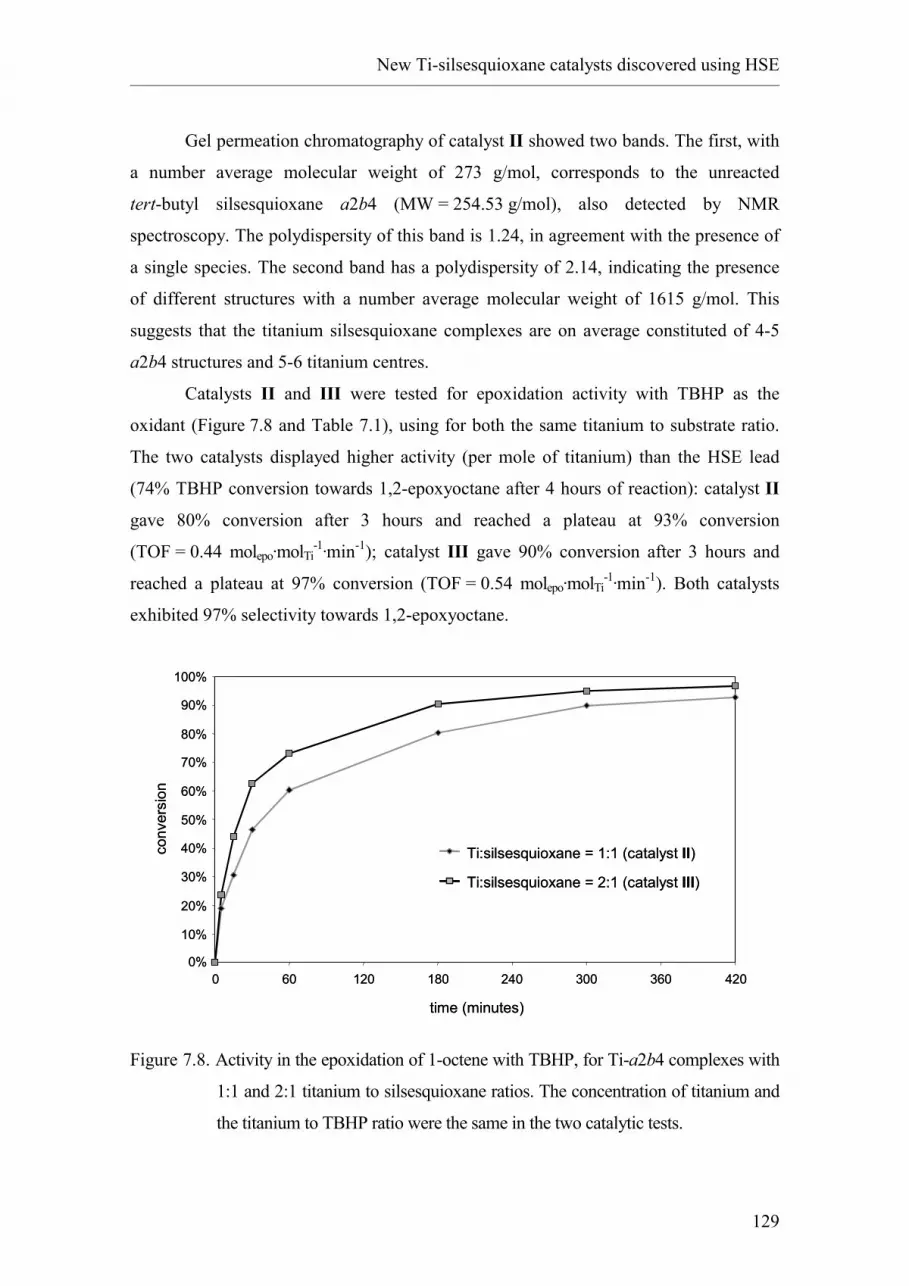
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
129
Gel permeation chromatography of catalyst II showed two bands. The first, with
a number average molecular weight of 273 g/mol, corresponds to the unreacted
tert-butyl silsesquioxane a2b4 (MW = 254.53 g/mol), also detected by NMR
spectroscopy. The polydispersity of this band is 1.24, in agreement with the presence of
a single species. The second band has a polydispersity of 2.14, indicating the presence
of different structures with a number average molecular weight of 1615 g/mol. This
suggests that the titanium silsesquioxane complexes are on average constituted of 4-5
a2b4 structures and 5-6 titanium centres.
Catalysts II and III were tested for epoxidation activity with TBHP as the
oxidant (Figure 7.8 and Table 7.1), using for both the same titanium to substrate ratio.
The two catalysts displayed higher activity (per mole of titanium) than the HSE lead
(74% TBHP conversion towards 1,2-epoxyoctane after 4 hours of reaction): catalyst II
gave 80% conversion after 3 hours and reached a plateau at 93% conversion
(TOF = 0.44 molepo·molTi-1·min-1); catalyst III gave 90% conversion after 3 hours and
reached a plateau at 97% conversion (TOF = 0.54 molepo·molTi-1·min-1). Both catalysts
exhibited 97% selectivity towards 1,2-epoxyoctane.
Figure 7.8. Activity in the epoxidation of 1-octene with TBHP, for Ti-a2b4 complexes with
1:1 and 2:1 titanium to silsesquioxane ratios. The concentration of titanium and
the titanium to TBHP ratio were the same in the two catalytic tests.
60 120 180 240 300 360 420
time (minutes)
conv
ersi
on
Ti:silsesquioxane = 1:1 (catalyst II)
Ti:silsesquioxane = 2:1 (catalyst III)
00%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
60 120 180 240 300 360 420
time (minutes)
conv
ersi
on
Ti:silsesquioxane = 1:1 (catalyst II)
Ti:silsesquioxane = 2:1 (catalyst III)
Ti:silsesquioxane = 1:1 (catalyst II)
Ti:silsesquioxane = 2:1 (catalyst III)
00%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
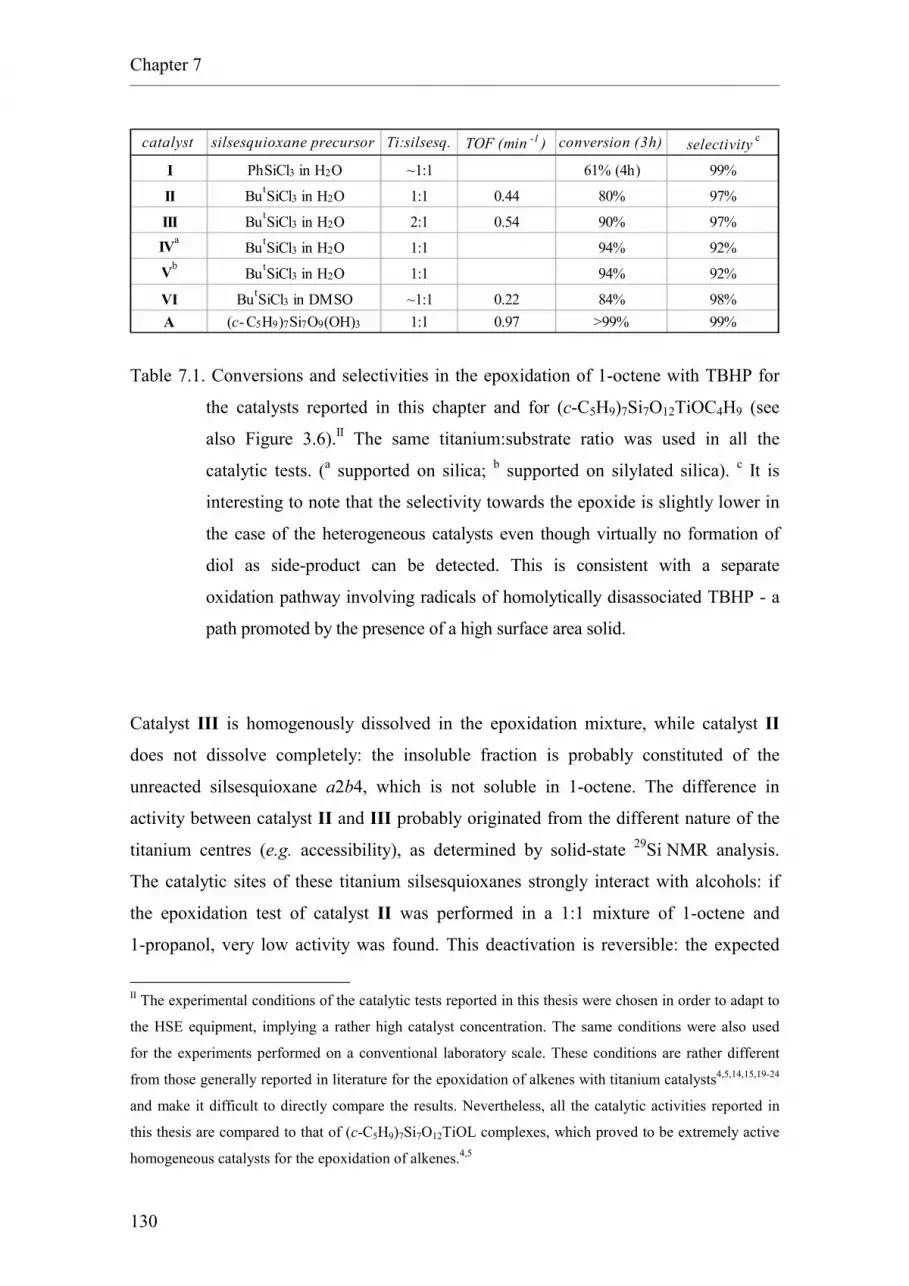
Chapter 7 __________________________________________________________________________________________________________
130
Table 7.1. Conversions and selectivities in the epoxidation of 1-octene with TBHP for
the catalysts reported in this chapter and for (c-C5H9)7Si7O12TiOC4H9 (see
also Figure 3.6).II The same titanium:substrate ratio was used in all the
catalytic tests. (a supported on silica; b supported on silylated silica). c It is
interesting to note that the selectivity towards the epoxide is slightly lower in
the case of the heterogeneous catalysts even though virtually no formation of
diol as side-product can be detected. This is consistent with a separate
oxidation pathway involving radicals of homolytically disassociated TBHP - a
path promoted by the presence of a high surface area solid.
Catalyst III is homogenously dissolved in the epoxidation mixture, while catalyst II
does not dissolve completely: the insoluble fraction is probably constituted of the
unreacted silsesquioxane a2b4, which is not soluble in 1-octene. The difference in
activity between catalyst II and III probably originated from the different nature of the
titanium centres (e.g. accessibility), as determined by solid-state 29Si NMR analysis.
The catalytic sites of these titanium silsesquioxanes strongly interact with alcohols: if
the epoxidation test of catalyst II was performed in a 1:1 mixture of 1-octene and
1-propanol, very low activity was found. This deactivation is reversible: the expected
II The experimental conditions of the catalytic tests reported in this thesis were chosen in order to adapt to
the HSE equipment, implying a rather high catalyst concentration. The same conditions were also used
for the experiments performed on a conventional laboratory scale. These conditions are rather different
from those generally reported in literature for the epoxidation of alkenes with titanium catalysts4,5,14,15,19-24
and make it difficult to directly compare the results. Nevertheless, all the catalytic activities reported in
this thesis are compared to that of (c-C5H9)7Si7O12TiOL complexes, which proved to be extremely active
homogeneous catalysts for the epoxidation of alkenes.4,5
catalyst silsesquioxane precursor Ti:silsesq. TOF (min -1 ) conversion (3h) selectivity c
I PhSiCl3 in H2O ~1:1 61% (4h) 99%
II ButSiCl3 in H2O 1:1 0.44 80% 97%
III ButSiCl3 in H2O 2:1 0.54 90% 97%IVa ButSiCl3 in H2O 1:1 94% 92%Vb ButSiCl3 in H2O 1:1 94% 92%
VI ButSiCl3 in DMSO ~1:1 0.22 84% 98%A (c- C5H9)7Si7O9(OH)3 1:1 0.97 >99% 99%

New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
131
epoxidation activity was recovered when reusing the catalyst without 1-propanol as
co-solvent. The same activity loss was observed when methanol was used as co-solvent
instead of 1-propanol. This reversible coordination of alcohols to the titanium is in
agreement with the results of the 13C NMR analysis of the titanium-silsesquioxane a2b4
complexes. The catalyst deactivation is ascribed to the interaction between the hydroxyl
group and the titanium centre,5 which would reduce the accessibility to the catalytic site.
The deactivation caused by coordination of alcohols to the titanium centre is not
encountered when using (c-C5H9)7Si7O12TiOC4H9 as catalyst, hence pointing to a
different nature of the catalytic centre.
The main drawback of homogeneous catalysts lies in the difficulty to recover
them from the reaction mixture. In order to overcome this downside, the
titanium-silsesquioxane a2b4 complex was supported on high surface area silica and the
materials obtained were tested as heterogeneous catalysts for the epoxidation of
1-octene.14,19-24 Titanium silsesquioxane a2b4 was supported by adsorption on two types
of silica: one untreated and the other dehydroxylated by derivatisation with (CH3)2SiCl2
(catalysts IV and V, respectively). The larger size of the titanium-silsesquioxane
complexes compared to that of the other molecules present in the epoxidation mixture
should favour the adsorption of the complexes as well as prevent their leaching from the
silica supports (entropic driving force). After adsorption of the complexes on the silica
supports, possible leaching species were removed by prolonged Soxhlet extraction in
tetrahydrofuran. The epoxidation test gave a 94% TBHP conversion and 92% selectivity
towards 1,2-epoxyoctane after 3 hours of reaction for both catalysts. Leaching of the
titanium silsesquioxanes from the silica support was checked by filtering the solid from
the epoxidation mixture and by measuring the catalytic activity of the possible soluble
titanium silsesquioxanes: the soluble fractions presented negligible epoxidation activity
(equal to that of a blank sample), hence proving that no leaching of active species
occurred. The heterogeneous catalyst supported on untreated silica (IV) was reused for
3 cycles and yielded comparable epoxidation activity (± 4% of the activity obtained in
the first catalytic test).
The titanium-silsesquioxane a2b4 complexes were supported on both untreated and
silylated silica in order to investigate the nature of the adsorption. The similar activity
and selectivity obtained with the two supports suggests that the adsorption of the

Chapter 7 __________________________________________________________________________________________________________
132
complexes takes place by physical interaction with the silica surface rather than through
chemical anchoring on the -OH groups (totally or almost absent on the silica derivatised
with (CH3)2SiCl2).
Remarkably, the heterogeneous catalysts displayed an epoxidation activity (per mole
of titanium) similar to that of the homogeneous titanium-silsesquioxane a2b4
complexes, although with a lower selectivity towards 1,2-epoxyoctane (see Table 7.1
on page 130).
Neither the homogeneous titanium-silsesquioxane a2b4 complexes nor the
silica-supported heterogeneous catalysts showed good activity in the epoxidation of
1-octene with aqueous H2O2 as the oxidant.
7.2.4 tert-butyl silsesquioxanes synthesised in DMSO
The hydrolytic condensation of tert-butyltrichlorosilane in DMSO was
performed in a 25-fold up-scaling of the HSE synthesis. Similarly to what has been
described for the synthesis of tert-butyl silsesquioxane a2b4, the
tert-butyltrichlorosilane was first dissolved in acetonitrile and then added to the DMSO.
After 18 hours of reaction at 50°C, the clear solution obtained was distilled under
reduced pressure to give a yellow gel that still contained DMSO.III Adding water to the
gel caused the precipitation of the silsesquioxane product that could then be separated
from the DMSO/H2O solution. 13C and 29Si NMR analysis showed that the sample
consisted of a number of oligomeric silsesquioxane species. The 29Si NMR spectrum
(Figure 7.9) presents two sets of peaks, one in the region of silicons connected to three
other silicons via oxygen bridges (-55 ppm < δ < -61 ppm), and the other of silicons
connected to one or more -OH groups (-45 ppm < δ < -51 ppm).8 The latter set of peaks
has a much higher intensity than the former, indicating that most of the silsesquioxanes
are incompletely condensed structures with a low level of condensation.
III Removing DMSO from the silsesquioxanes is necessary to prevent competition between the oxidation
of DMSO to dimethyl sulfone and the epoxidation of 1-octene to 1,2-epoxyoctane. Complete removal of
DMSO was attained in the HSE experiment by drying the samples in a vacuum centrifuge that, therefore,
proved to be more effective than a distillation under vacuum.
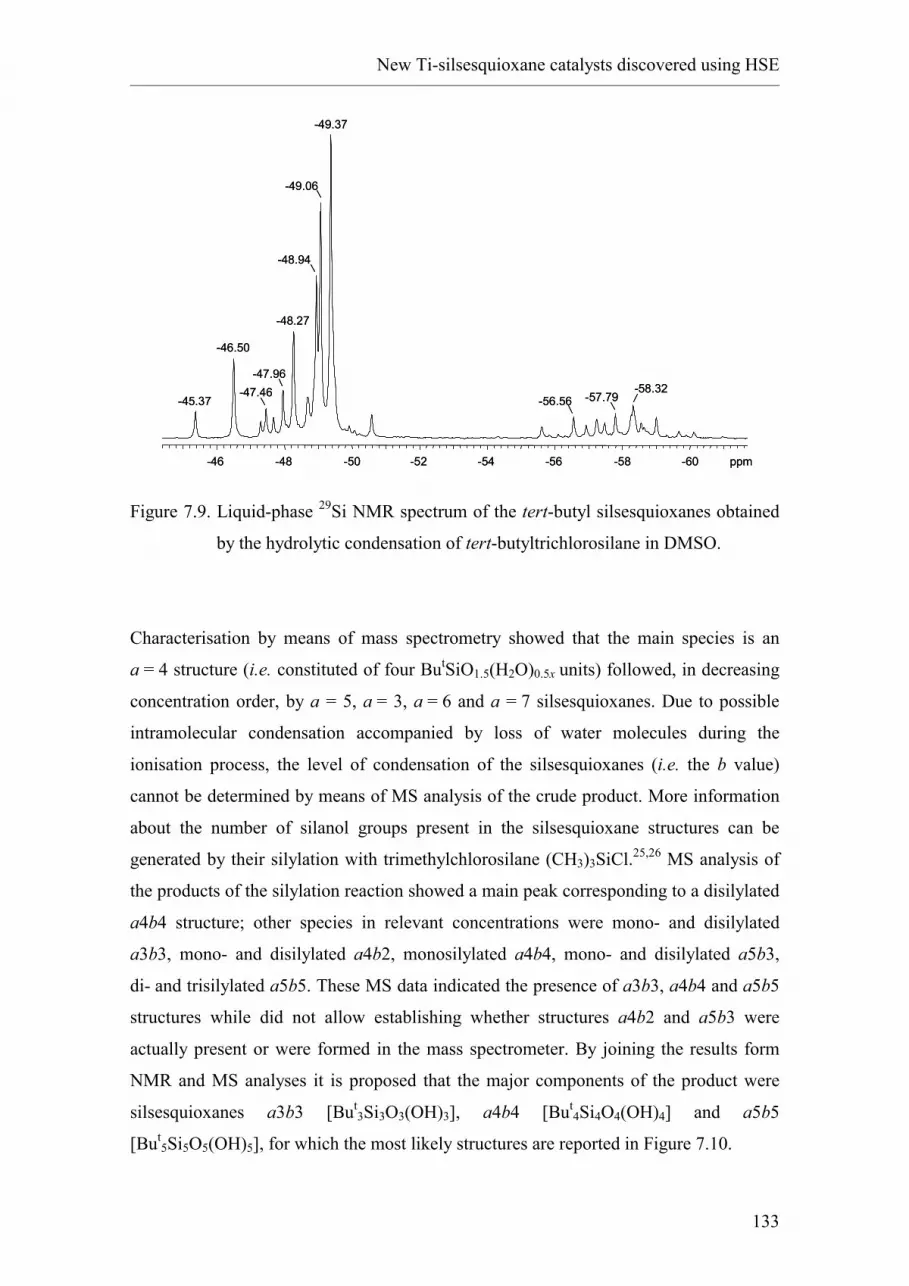
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
133
Figure 7.9. Liquid-phase 29Si NMR spectrum of the tert-butyl silsesquioxanes obtained
by the hydrolytic condensation of tert-butyltrichlorosilane in DMSO.
Characterisation by means of mass spectrometry showed that the main species is an
a = 4 structure (i.e. constituted of four ButSiO1.5(H2O)0.5x units) followed, in decreasing
concentration order, by a = 5, a = 3, a = 6 and a = 7 silsesquioxanes. Due to possible
intramolecular condensation accompanied by loss of water molecules during the
ionisation process, the level of condensation of the silsesquioxanes (i.e. the b value)
cannot be determined by means of MS analysis of the crude product. More information
about the number of silanol groups present in the silsesquioxane structures can be
generated by their silylation with trimethylchlorosilane (CH3)3SiCl.25,26 MS analysis of
the products of the silylation reaction showed a main peak corresponding to a disilylated
a4b4 structure; other species in relevant concentrations were mono- and disilylated
a3b3, mono- and disilylated a4b2, monosilylated a4b4, mono- and disilylated a5b3,
di- and trisilylated a5b5. These MS data indicated the presence of a3b3, a4b4 and a5b5
structures while did not allow establishing whether structures a4b2 and a5b3 were
actually present or were formed in the mass spectrometer. By joining the results form
NMR and MS analyses it is proposed that the major components of the product were
silsesquioxanes a3b3 [But3Si3O3(OH)3], a4b4 [But
4Si4O4(OH)4] and a5b5
[But5Si5O5(OH)5], for which the most likely structures are reported in Figure 7.10.
-45.37
-46.50
-47.96
-48.27
-49.06
-48.94
-49.37
-56.56 -57.79-58.32
ppm-60-58-56-54-52-50-48-46
-47.46-45.37
-46.50
-47.96
-48.27
-49.06
-48.94
-49.37
-56.56 -57.79-58.32
ppm-60-58-56-54-52-50-48-46
-47.46
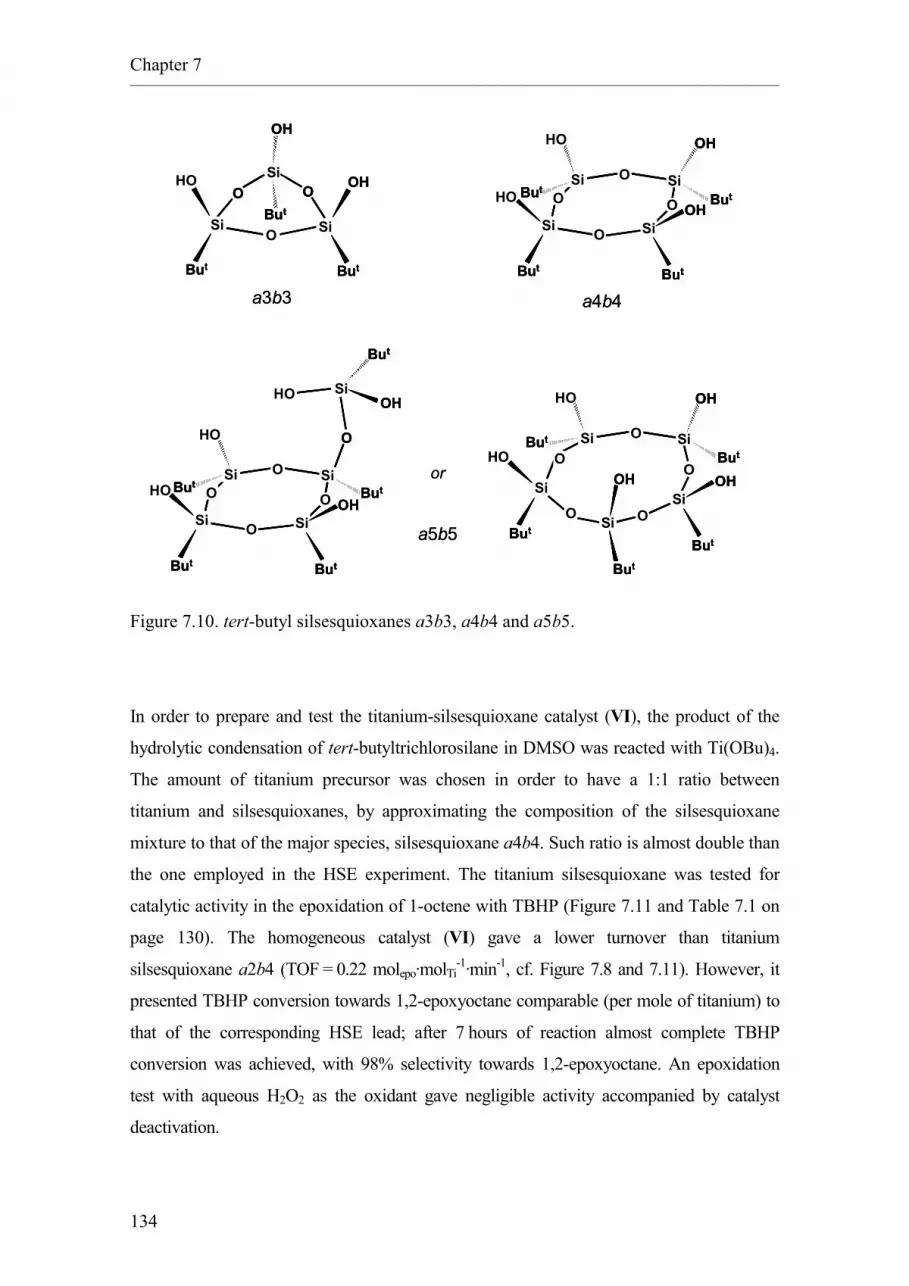
Chapter 7 __________________________________________________________________________________________________________
134
Figure 7.10. tert-butyl silsesquioxanes a3b3, a4b4 and a5b5.
In order to prepare and test the titanium-silsesquioxane catalyst (VI), the product of the
hydrolytic condensation of tert-butyltrichlorosilane in DMSO was reacted with Ti(OBu)4.
The amount of titanium precursor was chosen in order to have a 1:1 ratio between
titanium and silsesquioxanes, by approximating the composition of the silsesquioxane
mixture to that of the major species, silsesquioxane a4b4. Such ratio is almost double than
the one employed in the HSE experiment. The titanium silsesquioxane was tested for
catalytic activity in the epoxidation of 1-octene with TBHP (Figure 7.11 and Table 7.1 on
page 130). The homogeneous catalyst (VI) gave a lower turnover than titanium
silsesquioxane a2b4 (TOF = 0.22 molepo·molTi-1·min-1, cf. Figure 7.8 and 7.11). However, it
presented TBHP conversion towards 1,2-epoxyoctane comparable (per mole of titanium) to
that of the corresponding HSE lead; after 7 hours of reaction almost complete TBHP
conversion was achieved, with 98% selectivity towards 1,2-epoxyoctane. An epoxidation
test with aqueous H2O2 as the oxidant gave negligible activity accompanied by catalyst
deactivation.
a3b3 a4b4
a5b5
or
SiO
HO
SiSiO
O
But But
OH
OH
But
SiO
O
OSiSi O
Si
But
OHHO
But
But
OHHO
But
SiO
O
O
SiSi
Si
Si
But
OHHO
But
But
OHHO
But
OO
But
OHSiO
O
OSiSi O
Si
But
OHHO
But
But
OHO
But
Si
But
OHHO
a3b3 a4b4
a5b5
or
SiO
HO
SiSiO
O
But But
OH
OH
But
SiO
HO
SiSiO
O
But But
OH
OH
But
SiO
O
OSiSi O
Si
But
OHHO
But
But
OHHO
ButSi
OO
OSiSi O
Si
But
OHHO
But
But
OHHO
But
SiO
O
O
SiSi
Si
Si
But
OHHO
But
But
OHHO
But
OO
But
OH
SiO
O
O
SiSi
Si
Si
But
OHHO
But
But
OHHO
But
OO
But
OHSiO
O
OSiSi O
Si
But
OHHO
But
But
OHO
But
Si
But
OHHO
SiO
O
OSiSi O
Si
But
OHHO
But
But
OHO
But
Si
But
OHHO
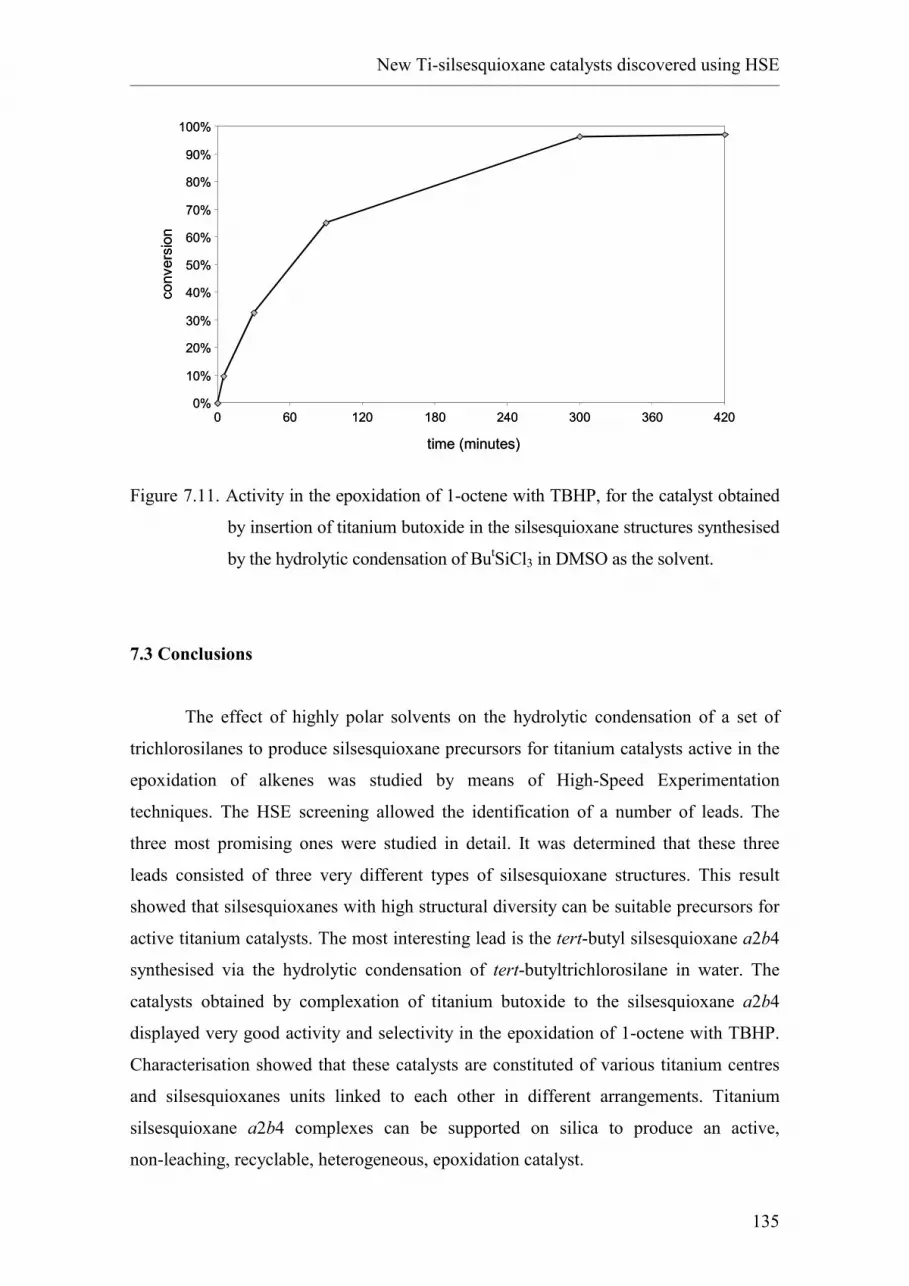
New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
135
Figure 7.11. Activity in the epoxidation of 1-octene with TBHP, for the catalyst obtained
by insertion of titanium butoxide in the silsesquioxane structures synthesised
by the hydrolytic condensation of ButSiCl3 in DMSO as the solvent.
7.3 Conclusions
The effect of highly polar solvents on the hydrolytic condensation of a set of
trichlorosilanes to produce silsesquioxane precursors for titanium catalysts active in the
epoxidation of alkenes was studied by means of High-Speed Experimentation
techniques. The HSE screening allowed the identification of a number of leads. The
three most promising ones were studied in detail. It was determined that these three
leads consisted of three very different types of silsesquioxane structures. This result
showed that silsesquioxanes with high structural diversity can be suitable precursors for
active titanium catalysts. The most interesting lead is the tert-butyl silsesquioxane a2b4
synthesised via the hydrolytic condensation of tert-butyltrichlorosilane in water. The
catalysts obtained by complexation of titanium butoxide to the silsesquioxane a2b4
displayed very good activity and selectivity in the epoxidation of 1-octene with TBHP.
Characterisation showed that these catalysts are constituted of various titanium centres
and silsesquioxanes units linked to each other in different arrangements. Titanium
silsesquioxane a2b4 complexes can be supported on silica to produce an active,
non-leaching, recyclable, heterogeneous, epoxidation catalyst.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 60 120 180 240 300 360 420
time (minutes)
conv
ersi
on
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 60 120 180 240 300 360 420
time (minutes)
conv
ersi
on

Chapter 7 __________________________________________________________________________________________________________
136
7.4 Experimental
7.4.1 The High-Speed Experimentation screening
The preparation of the HSE samples was performed by means of a Hamilton
Dual-Arm liquid-handling robotic workstation27 coupled with a personal computer
supplied with software enabling to program the workstation. Reagents for the hydrolytic
condensation: cyclohexyl-, cyclopentyl-, phenyl-, methyl-, ethyl- and
tert-butyltrichlorosilanes; the latter was dissolved beforehand in acetonitrile (340 µmol
in 0.5 ml of CH3CN); acetonitrile, dimethyl sulfoxide (DMSO), deionised water and
formamide as solvents; deionised water. The silsesquioxane precursors were prepared
by dispensing 2-ml aliquots of each of the solvents in racks containing 6x4 arrays of
glass tube reactors (3 ml working volume), followed by the addition of 340 µmol of
each of the trichlorosilanes and by the addition of 0.5 ml of deionised water to each
reaction tube. Subsequently, the racks were placed in a steel heater block mounted on an
orbital shaker27 and heated to 50°C for 18 hours. After evaporation of the liquids in a
vacuum centrifuge27 overnight, the silsesquioxane samples were dissolved in
tetrahydrofuran (THF), 54 µmol of titanium butoxide, [Ti(OBu)4, 98% purity] were
added to each sample and the rack was stirred for 6 hours at 60°C. After titanium
complexation, THF was removed by vacuum centrifugation. Each sample was then
tested for epoxidation activity by adding 2.26 ml of 1-octene (with 2%vol of decane as
internal standard) and 121 µl of TBHP (~45%wt solution in cyclohexane). After having
been stirred in the orbital shaker for 4 hours at 80°C, samples were analysed on an
automated Unicam Pro GC27 under isothermal conditions. The activities were obtained
by normalising the 1,2-epoxyoctane GC peak area by means of the internal standard.
The reported results are the average of multiple runs.
7.4.2 Phenyl silsesquioxanes synthesised in H2O
1.35 ml of phenyltrichlorosilane [PhSiCl3, 97% purity] were carefully added to
62.5 ml of deionised water. As soon as the trichlorosilane was added, a white solid
started to precipitate. After having been stirred for 18 hours at 50°C, the solution was
filtered. The filtrate was evaporated under reduced pressure to afford negligible amounts

New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
137
of solid. The precipitate was dried overnight in an oven at 100°C to afford 1.005g of
white solid (92% yield on the hypothesis of a silsesquioxane structure containing 50%
of PhSiO1.5 units and 50% of PhSiO2H units). The solid is soluble in acetone, THF and
pyridine; dissolution is slow.
The titanium complexation and the catalytic test were performed analogously to what is
described for the HSE study. The molar ratio between titanium and TBHP and the
concentration of TBHP in the epoxidation mixture were the same in all the catalytic
tests reported in this chapter.
NMR characterisation of the silsesquioxane product was performed on a Varian
VXR-400S (1H decoupled, 25°C). Selected data: 13C{1H} NMR (liquid phase, deuterated
acetone as the solvent), two sets of partially overlapping peaks due to the phenyl groups:
δ 128.77 ppm, 128.85 ppm (carbons meta to the ipso carbons), multiplet around
δ 131.53 ppm (ipso carbons and carbons para to it), δ 134.93 ppm, 135.07 ppm (carbons
ortho to the ipso carbons); 29Si{1H} NMR (solid state), δ -69.8 ppm, -79.4 ppm.
7.4.3 tert-butyl silsesquioxanes synthesised in H2O
1.63 g of tert-butyltrichlorosilane [ButSiCl3, >95% purity] were dissolved in
12.5 ml of acetonitrile. This solution was added to 50 ml of deionised water. After 18
hours of reaction at 50°C upon stirring, a white, fine precipitate was formed. The
precipitate was collected by filtration (fraction A) and the filtrate solution was dried
under reduced pressure to yield a white, fine powder (fraction B). To remove water that
might still be present, the two fractions were dried for 24 hours in an oven at 100°C:
after drying, 0.335 g of fraction A and 1.502 g of fraction B were present. Both
fractions were constituted of tert-butyl silsesquioxane a2b4, (t-C4H9)2Si2O(OH)4
[MW = 254.43 g/mol]. The yield in the isolated silsesquioxane a2b4 is 90%, with >99%
selectivity. The compound melts between 192 and 204°C and is soluble in pyridine,
DMSO and, to a lesser extent, in THF. In order to obtain suitable crystals for
single-crystal X-ray diffraction analysis, 0.1 g of the compound were dissolved in 10 ml
of THF at 50°C and recrystallised by carefully adding 1-ml aliquots of acetonitrile (5 ml
in total) to the THF solution.

Chapter 7 __________________________________________________________________________________________________________
138
The synthesis of catalyst II and III was performed by titanium complexation to the
silsesquioxane structure with similar experimental conditions to those described for the
HSE study.
CAB-O-SIL fumed silica EH-5 [surface area: 380 m2/g, ~4 hydroxyl groups/nm2] was
used as support for the heterogenisation of the titanium-silsesquioxane a2b4 complexes.
Dehydroxylation of the silica was performed by adding 1 ml of (CH3)2SiCl2 to a
suspension of 1 g of silica in 30 ml of diethyl ether and by stirring the suspension for
1 hour at room temperature. The volatiles were removed under reduced pressure. The
titanium-silsesquioxane a2b4 complexes (catalyst II) were supported on the silica (both
untreated and dehydroxylated) by adding a 150-ml THF solution of the complexes
(containing 5·10-4 mol of titanium) to a suspension of 1 g of silica in 100 ml of THF and
by stirring the suspension for 3 hours at 60°C. After isolation of the solid by filtration,
non-adsorbed species were removed by Soxhlet extraction with THF for 7 hours.
Finally, the silica-supported catalysts (IV and V) were dried overnight at 120°C under
reduced pressure.
The catalytic tests were performed analogously to what is described for the HSE study.
The same ratio between titanium and TBHP and the same concentration of TBHP in the
reaction mixture were employed in all the catalytic tests reported in this chapter.
For single-crystal X-ray diffraction of But2Si2O(OH)4, the crystal was mounted along an
arbitrary axis on an Enraf-Nonius CAD-4 diffactometer. Lattice constants were determined
from 25 reflections with 10° < θ < 17º (standard deviations are within parentheses):
a = 6.226(6), b = 6.223(6), c = 9.956(4); α = 85.06(5), β = 80.72(5), γ = 70.51(9).
NMR spectra were measured on a Varian VXR-400S (1H decoupled, 25°C) and on a
Varian Inova-300 (1H decoupled, 25°C).
Selected data for silsesquioxane a2b4, liquid-phase NMR (deuterated pyridine as the
solvent): 1H NMR, δ 1.46 ppm, (But; relative integral = 9), 8.26 ppm (Si-OH; relative
integral = 2); 13C{1H} NMR, δ 18.25 ppm (ipso carbon of But; relative integral = 1)
δ 27.36 ppm (CH3 groups of But; relative integral = 3); 29Si{1H} NMR, δ -49.55 ppm
(ButSi(OH)2O0.5).
Selected data for the silsesquioxane attributed to the a1b3 structure, liquid-phase NMR
(deuterated pyridine as the solvent): 13C{1H} NMR, δ 18.32 ppm (ipso carbon of But;

New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
139
relative integral = 1) δ 27.51 ppm (CH3 groups of But; relative integral = 3); 29Si{1H} NMR, δ -40.06 ppm (ButSi(OH)3).
Selected data for catalyst II: solid-state 29Si{1H} NMR, δ -50 ppm (relative integral
~ 1), δ -56 ppm, (broad signal; relative integral ~ 2), δ -66 ppm (broad signal; relative
integral ~ 1).
Selected data for catalyst III: liquid-phase 13C{1H} NMR (deuterated pyridine as the
solvent), δ 14.17 ppm (δ), 19.54 ppm (γ), 35.74 ppm (β), 61.72 ppm (α) (1:1:1:1; δCH3
γCH2βCH2
αCH2OH), δ 18.2 ppm, 27.4 ppm (~1:3; multiplets; respectively: ipso
carbon and CH3 groups of the But-group on the silsesquioxane species); solid-state 13C{1H} NMR, δ 14.6 ppm (δ), 19.9 ppm (γ), 35.8 ppm (β), 79.5 ppm (α) (~1:1:1:1;
broad peaks, α very broad; δCH3γCH2
βCH2αCH2OTi), δ 18.0 ppm, 27.4 ppm (~1:3;
broad peaks; respectively: ipso carbon and CH3 groups of the But-group on the
silsesquioxane species); solid-state 29Si{1H} NMR, δ -56 ppm, (broad band; relative
integral ~ 4), δ -66 ppm (broad signal; relative integral ~ 1).
Gel permeation chromatography of catalyst II was performed on a Waters 410
differential refractometer gel permeation chromatographer equipped with a Waters
Styragel HT 6E column calibrated on polystyrene standards. THF was used as the
eluent.
7.4.5 tert-butyl silsesquioxanes synthesised in DMSO
1.63 g of tert-butyltrichlorosilane [ButSiCl3, >95% purity] were dissolved in
12.5 ml of acetonitrile. The solution was added to 37.5 ml of DMSO, followed by
addition of 12.5 ml of deionised water. The transparent, colourless solution was stirred
for 18 hours at 50°C and then distilled under reduced pressure to remove volatile
species and to isolate the silsesquioxanes. The yellow gel obtained still contained
DMSO, which was removed by adding 50 ml of H2O: the silsesquioxanes precipitated
as a sticky, soft, solid that glued to the glass walls of the reaction flask, from which the
water/DMSO solution was easily removed. The silsesquioxane mixture is soluble in
THF and pyridine.
The silylation of the silsesquioxane mixture was performed by adding 172 µl of
trimethylchlorosilane (CH3)3SiCl to a solution containing 0.04 g of silsesquioxanes in

Chapter 7 __________________________________________________________________________________________________________
140
30 ml of toluene and 1 ml of triethylamine (C2H5)3N. The turbid solution was stirred
overnight at room temperature. Next, the toluene and the triethylamine were removed
under reduced pressure and a white solid was obtained. The solid was extracted with
pentane; the solution was filtered to remove the insoluble (C2H5)3N·HCl and dried under
reduced pressure to yield a solid containing the silylated silsesquioxanes.
The titanium complexation and the catalytic test were performed similarly to what is
described for the HSE study. The molar ratio between titanium and TBHP and the
concentration of TBHP in the reaction mixture were the same in all the catalytic tests
reported in this chapter. The NMR spectrum of the silsesquioxane mixture was collected on a Varian Inova-300
(1H decoupled, 25°C). Selected data: 29Si{1H} NMR (liquid phase, deuterated pyridine
as the solvent), δ -45.37 ppm (relative integral ~ 1), -46.50 ppm (~3), -47.46 ppm (~1),
-47.96 ppm (~2), -48.27 ppm (~4.5), -48.94 ppm (~6.5), -49.06 ppm (~8.5), -49.37 ppm
(~11), -56.56 ppm (~1), -57.79 ppm (~1), -58.32 ppm (~1).
Mass spectrometry analysis was performed on a Micromass Quattro LC-MS with
APCI+ as ionisation technique. For the analysis, ~0.03 g of the sample were dissolved
in 5 ml of CH2Cl2/THF (4:1) and few drops of CH3CO2H were added.
Silsesquioxane product (cone voltage = 45 V): for a = 3, m/z: 337.50 ([a3b1 + H]+,
20%), 355.54 ([a3b3 + H]+, 17%), 409.50 ([a3b5 + 2H2O + H]+, 22%); for a = 4, m/z:
437.47 ([a4b0 + H]+, 100%), 455.52 ([a4b2 + H]+, 99%), 509.54 ([a4b6 + H2O + H]+,
32%), 527.39 ([a4b6 + 2H2O + H]+, 28%); for a = 5, m/z: 537.45 ([a5b1 – H2O + H]+,
40%), 555.56 ([a5b1 + H]+, 25%), 573.48 ([a5b3 + H]+, 52%), 627.43 ([a5b7 + H2O +
H]+, 14%); for a = 6, m/z: 655.35 ([a6b0 + H]+, 15%), 673.39 ([a6b2 + H]+, 33%),
691.25 ([a6b4 + H]+, 11%); for a = 7, m/z: 773.37 ([a7b1 + H]+, 14%), 791.17 ([a7b3 +
H]+, 7%), 809.34 ([a7b5 + H]+, 9%); for a = 8, m/z: 873.34 ([a8b0 + H]+, 4%), 891.20
([a8b2 + H]+, 6%), 909.06 ([a8b4 + H]+, 6%), 927.17 ([a8b6 + H]+, 5%); for a = 9, m/z:
991.30 ([a9b1 + H]+, 7%), 1027.15 ([a9b5 + H]+, 9%), 1045.32 ([a9b7 + H]+, 6%); for
a = 10, m/z: 1145.23 ([a10b6 + H]+, 11%).
Silylated silsesquioxanes (cone voltage = 15 V): for a = 3, m/z: 427.66 ([silylated a3b3
+ H]+, 37%), 499.61 ([disilylated a3b3 + H]+, 22%); for a = 4, m/z: 455.64 ([a4b2 +
H]+, 16%), 527.53 ([silylated a4b2 + H]+, 51%), 545.53 ([silylated a4b4 + H]+, 38%),

New Ti-silsesquioxane catalysts discovered using HSE __________________________________________________________________________________________________________
141
599.53 ([disilylated a4b2 + H]+, 22%), 617.60 ([disilylated a4b4 + H]+, 100%), for
a = 5, m/z: 645.52 ([silylated a5b3 + H]+, 37%), 717.46 ([disilylated a5b3 + H]+, 17%),
735.40 ([disilylated a5b5 + H]+, 19%), 807.41 ([trisilylated a5b5 + H]+, 11%); for a = 6,
m/z: 763.45 ([silylated a6b4 + H]+, 7%), 835.33 ([disilylated a6b4 + H]+, 7%); for a = 7,
m/z: 953.32 ([disilylated a7b5 + H]+, 7%).

Chapter 7 __________________________________________________________________________________________________________
142
References
1 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Angew. Chem. Int. Ed., 2001, 40,
740, see also Chapter 3. 2 P.P. Pescarmona, J.J.T. Rops, J.C. van der Waal, J.C. Jansen, T. Maschmeyer, J. Mol. Cat. A, 2002,
182-183, 319, see also Chapter 3. 3 P.P. Pescarmona, T. Maschmeyer, NATO Science Series, 2002, Ser. II Vol. 69, 173, see also Chapter 4. 4 M. Crocker, R.H.M. Herold, A.G. Orpen, Chem. Commun., 1997, 2411. 5 T. Maschmeyer, M.C. Klunduk, C.M. Martin, D.S. Shephard, J.M. Thomas, B.F.G. Johnson, Chem.
Commun.,1997, 1847. 6 P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, Catal. Today, 2003, 81, 347, see also Chapter 3. 7 J.F. Brown, L.H. Vogt, J. Am. Chem. Soc., 1965, 87, 4313. 8 P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583, see also Chapter 2. 9 F.J. Feher, J.J. Schwab, D. Soulivong, J.W. Ziller, Main Group Chem., 1997, 2, 123. 10 J.F. Brown, L.H. Vogt, P.I. Prescott, J. Am. Chem. Soc., 1964, 86, 1120. 11 G. Blanco-Brieva, J.M. Campos-Martín, M.P. de Frutos, J.L.G. Fierro, Chem. Commun., 2001, 2228. 12 P.D. Lickiss, S.A. Litster, A.D. Redhouse, C.J. Wisener, Chem. Commun., 1991, 173. 13 N. Winkhofer, H.W. Roesky, M. Noltemeyer, W.T. Robinson, Angew. Chem. Int. Ed., 1992, 31, 599. 14 S. Krijnen, H.C.L. Abbenhuis, R.W.J.M. Hanssen, J.H.C. van Hooff, R.A. van Santen, Angew. Chem.
Int. Ed., 1998, 37, 356. 15 H.C.L. Abbenhuis, S. Krijnen, R.A. van Santen, Chem. Commun., 1997, 331. 16 E. Albizzati, L. Abis, E. Pettenati, E. Giannetti, Inorg. Chim. Acta, 1986, 120, 197. 17 S.I. Chervina, E.G. Maksimenko, R.S. Barshtein, N.V. Shabanova, A.K. Bulai, Y.I. Kotov, I.Y. Slonim,
Kinet. Catal., 1987, 28, 947. 18 M. Altaf Hossain, M.B. Hursthouse, M.A. Mazid, A.C. Sullivan, Chem. Commun., 1988, 1305. 19 T. Maschmeyer, F. Rey, G. Sankar, J.M. Thomas, Nature, 1995, 378, 159. 20 S. Thorimbert, S. Klein, W.F. Maier, Tetrahedron, 1995, 51, 3787. 21 R. Hutter, T. Mallat, A. Baiker, J. Catal., 1995, 153, 177. 22 A. Corma, U. Díaz, V. Fornés, J.L. Jordá, M. Domine, F. Rey, Chem. Commun., 1999, 779. 23 Z. Shan, E. Gianotti, J.C. Jansen, J.A. Peters, L. Marchese, T. Maschmeyer, Chem. Eur. J., 2001, 7,
1437. 24 J. Jarupatrakorn, T.D. Tilley, J. Am. Chem. Soc., 2002, 124, 8380. 25 F.J. Feher, D.A. Newman, J. Am. Chem. Soc., 1990, 112, 1931. 26 P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, submitted to Eur. J. Inorg. Chem., see also
Chapter 5. 27 See Appendix A.

143
8
Study of the synthesis of zeolite beta using High-Speed Experimentation
Abstract
Zeolites are a well-known family of crystalline microporous materials with broad
applications as heterogeneous catalysts. The application of HSE techniques to the study
of the synthesis of aluminium-rich zeolite beta (Si/Al ratio from 2.5 to 5) allowed the
investigation of the effect of the Si/Al and TEA+/Al ratios on the formation of such
zeolite. A Si/Al = 5 was found as the lowest ratio to produce pure zeolite beta. Lower
Si/Al ratios led to mixtures of zeolites (mainly beta and NaP1).
____________________
The contents of this chapter have been published in:
P.P. Pescarmona, J.J.T. Rops, J.C. van der Waal, J.C. Jansen, T. Maschmeyer, J. Mol. Catal. A, 2002, 182-183, 319.
P.P. Pescarmona, T. Maschmeyer, NATO Science Series, 2002, Ser. II Vol. 69, 173.

Chapter 8 __________________________________________________________________________________________________________
144
8.1 Introduction
In 1756, Crønstedt reported the discovery of the first natural zeolite.1,2 Upon
heating, the zeolite released occluded water: this property gave the material its general
name, zeolite, after the Greek zeo (to boil) and lithos (stone). Since then, many zeolite
structures - both natural and synthetic - have been discovered and studied.
Zeolites3 are crystalline aluminosilicates characterised by a framework consisting of Si
and Al atoms connected by oxygen bridges. Each Si4+ and Al3+ atom is tetrahedrally
surrounded by four O2- atoms (Figure 8.1). The presence of Al3+ results in a negative
charge into the framework, which is compensated by the presence of cations (e.g. H+,
Na+, Ca2+). In the case these counterions are protons, the zeolite exhibits Brønsted
acidity. Since Al-O-Al bonds are not encountered in zeolites (Löwenstein rule), the
Si/Al molar ratio is ≥ 1. By varying the Si/Al ratio of a zeolite, the amount and strength
of acid sites can be changed: the number of acid sites increases with increasing amount
of Al atoms while their strength decreases. This decrease in acidity is caused by the
lower electronegativity of AlO4-tetrahedral units compared to SiO4-tetrahedral units.
Figure 8.1. Schematic representation of a zeolite in the H-form.
The SiO4- and AlO4-tetrahedral units can be connected in many different ways, thus
generating a variety of possible zeolite structures. The arrangement of the tetrahedral
units generates frameworks characterised by three-dimensional networks of cages and
channels. The pore openings are in the 0.3-1.0 nm range: a comparable size to that of
small molecules. For these characteristics, zeolites are referred to as crystalline
microporous materials with uniform pore dimensions.
The properties described above account for the many applications of zeolites, and
OSi
OO
OAl
O O
OSi
OSi
O OO O
OAl
OSi
O
O OO O
H H
OSi
OO
OAl
O O
OSi
OSi
O OO O
OAl
OSi
O
O OO O
H H

Synthesis of zeolite beta by means of HSE __________________________________________________________________________________________________________
145
particularly: size and shape selective heterogeneous catalysts, ion-exchangers and
molecular sieves.3
8.1.1 Zeolite beta
Zeolite beta (BEA) is a so-called large pore zeolite characterised by three
families of mutually perpendicular channels with 12-membered ring apertures
(Figure 8.2),4,5 which make it suitable as heterogeneous catalyst for numerous organic
reactions6 (e.g. incorporation of titanium into zeolite beta leads to a versatile oxidation
and Lewis acid catalyst).7,8 Synthetic zeolite beta can be prepared with a wide range of
Si/Al ratios (from 5 to ∞).9,10 The control of the Si/Al ratio is important since it allows
tuning the number and strength of acid sites as well as the hydrophobicity of the zeolite.
Here, the optimisation of the synthesis of aluminium-rich zeolite beta in order to reach
the lowest possible Si/Al ratio is described. The synthesis of an aluminium-rich zeolite
beta is particularly interesting also because Tschernichite, the natural form of beta, has
Si/Al = 3.11
Figure 8.2. Three-dimensional structure of zeolite beta viewed along [100].
0.76 nm0.76 nm

Chapter 8 __________________________________________________________________________________________________________
146
The preparation of zeolites involves three main constituents: the source of the
framework elements (Si and Al), the mobiliser or mineraliser (OH- or F-), which
provides the appropriate pH for the reaction mixture, and the template (typically an
organic or inorganic cation), which directs the synthesis towards the formation of a
specific zeolite structure. Zeolite beta is commonly synthesised by hydrothermal
crystallisation from alkaline media, using tetraethylammonium (C2H5)4N+ (TEA+) as the
template.12,13
8.2 The High-Speed Experimentation approach
Similarly to what has been described for the formation of silsesquioxanes,14 the
synthesis of zeolites consists of a multiple-step reaction for which an overall mechanism
is not available. Various parameters determine which structures are formed and in
which amounts:12,15-17
- the nature of the silica and alumina sources and the ratio between the two
- the nature and the concentration of the template
- the presence, type and concentration of alkali cations
- the water content
- the pH
- the temperature
- the reaction time.
Given the complexity of the system, a High-Speed Experimentation18 approach
allowing the fast screening of broad parameter spaces is particularly suitable to study
and optimise the synthesis zeolite beta. Taking into account the good level of
knowledge available about the synthesis of zeolite beta, it was decided that no primary
screening was necessary. The HSE workstation19 employed in the project can only
handle low-viscosity liquids: this influenced the choice of the silica and alumina sources
used in the experiments. As silica and alumina sources, the cheap and easy to handle
Ludox-HS 40 colloidal silica and an aqueous solution of sodium aluminate were chosen.
Hydrothermal synthesis using tetraethylammonium ions (TEA+) as templates in an
alkaline environment was chosen among the methods commonly used for the
preparation of aluminium-rich zeolite beta for its adaptability to the HSE workstation.
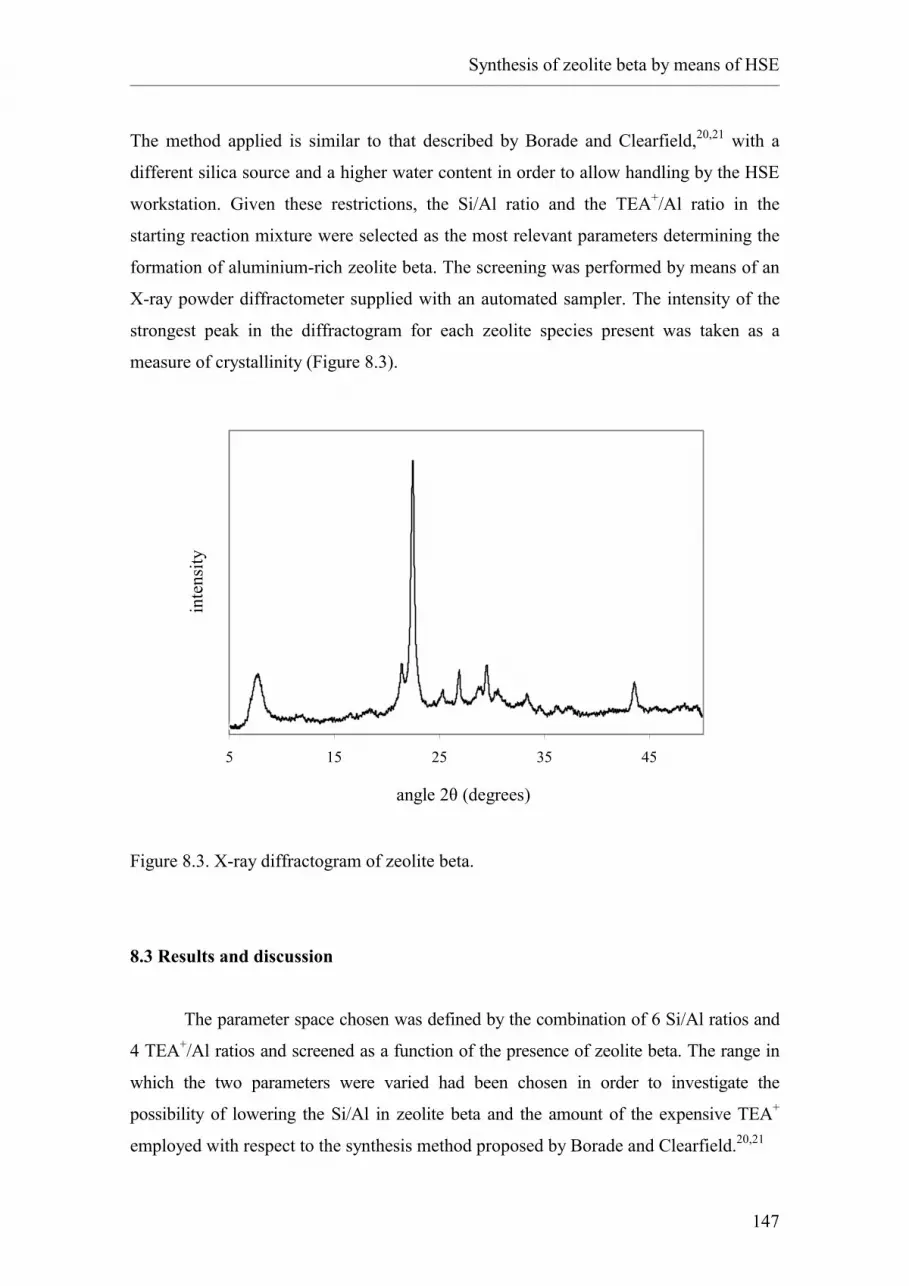
Synthesis of zeolite beta by means of HSE __________________________________________________________________________________________________________
147
The method applied is similar to that described by Borade and Clearfield,20,21 with a
different silica source and a higher water content in order to allow handling by the HSE
workstation. Given these restrictions, the Si/Al ratio and the TEA+/Al ratio in the
starting reaction mixture were selected as the most relevant parameters determining the
formation of aluminium-rich zeolite beta. The screening was performed by means of an
X-ray powder diffractometer supplied with an automated sampler. The intensity of the
strongest peak in the diffractogram for each zeolite species present was taken as a
measure of crystallinity (Figure 8.3).
Figure 8.3. X-ray diffractogram of zeolite beta.
8.3 Results and discussion
The parameter space chosen was defined by the combination of 6 Si/Al ratios and
4 TEA+/Al ratios and screened as a function of the presence of zeolite beta. The range in
which the two parameters were varied had been chosen in order to investigate the
possibility of lowering the Si/Al in zeolite beta and the amount of the expensive TEA+
employed with respect to the synthesis method proposed by Borade and Clearfield.20,21
5 15 25 35 45
angle 2θ (degrees)
inte
nsity
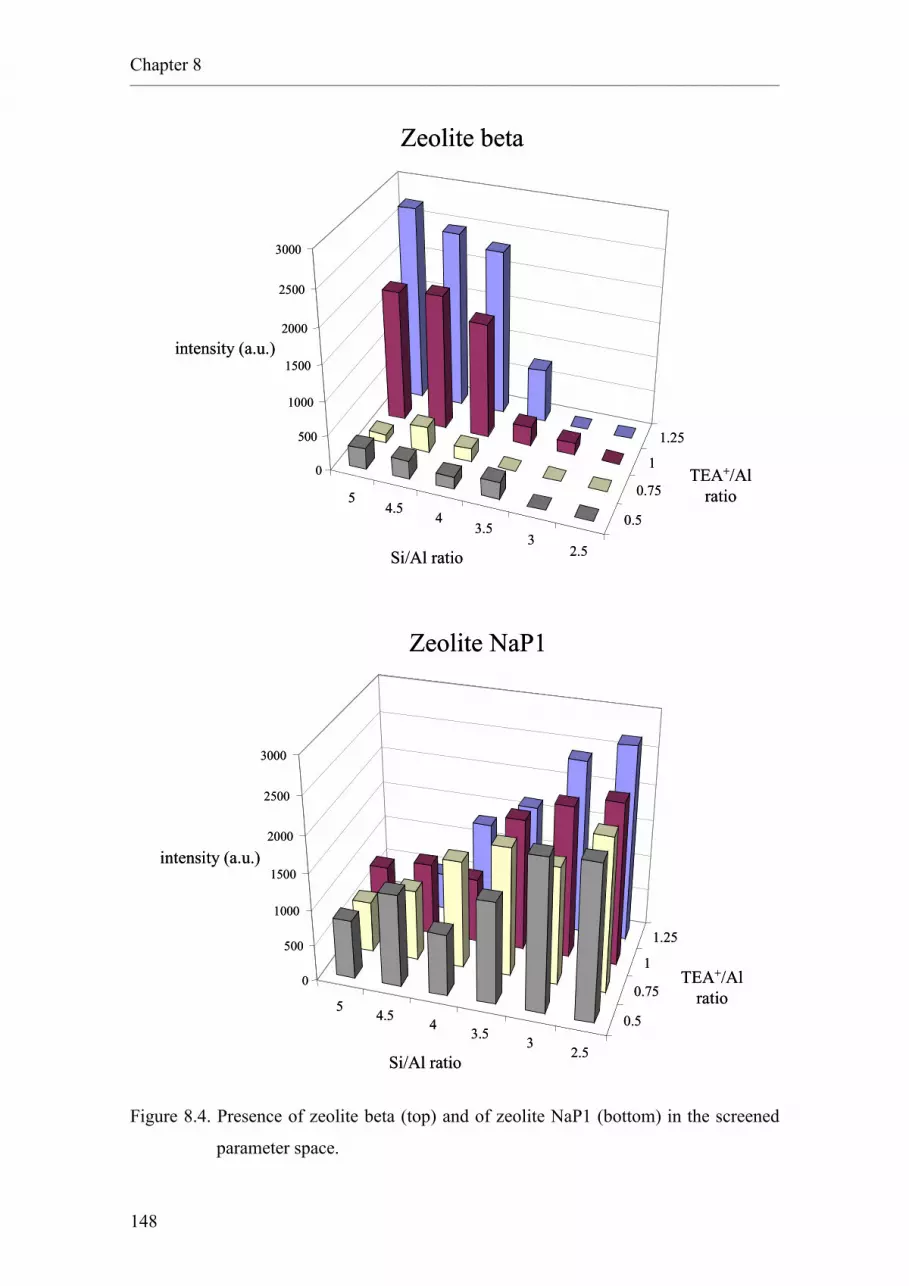
Chapter 8 __________________________________________________________________________________________________________
148
Figure 8.4. Presence of zeolite beta (top) and of zeolite NaP1 (bottom) in the screened
parameter space.
54.5
43.5
32.5
1.25
1
0.75
0.5
0
500
1000
1500
2000
2500
3000
Si/Al ratio
TEA+/Al ratio
intensity (a.u.)
Zeolite beta
5 4.5 4 3.5 32.5
1.25
1
0.75
0.5
0
500
1000
1500
2000
2500
3000
Si/Al ratio
TEA+/Al ratio
Zeolite NaP1
intensity (a.u.)
54.5
43.5
32.5
1.25
1
0.75
0.5
0
500
1000
1500
2000
2500
3000
Si/Al ratio
TEA+/Al ratio
intensity (a.u.)
Zeolite beta
5 4.5 4 3.5 32.5
1.25
1
0.75
0.5
0
500
1000
1500
2000
2500
3000
Si/Al ratio
TEA+/Al ratio
Zeolite NaP1
intensity (a.u.)

Synthesis of zeolite beta by means of HSE __________________________________________________________________________________________________________
149
The products of the 24 syntheses mainly consist of a mixture of two zeolites: zeolite
beta and zeolite NaP1 (Figure 8.4). NaP1 is a gismondine-type of zeolite (GIS), with a
Si/Al ratio of 1.5 and Na+ ions as template ions. Hence, NaP1 is found as a product at
low values of Si/Al ratio in the starting reaction mixture, while beta is formed at higher
values of Si/Al ratio. Pure zeolite beta was obtained at a Si/Al ratio of 5 and
TEA+/Al ratio of 1.25 in the starting reaction mixture, while pure NaP1 was obtained at
a Si/Al ratio of 2.5 for the entire screened TEA+/Al ratio. In between, mixtures of both
zeolites were observed. Some samples also contained amorphous silica and small
amounts of crystalline impurities (consistent with analcime (ANA)). A higher value of
TEA+/Al ratio causes a positive trend towards the formation of zeolite beta, as
competition takes place between the two templates (Na+ for NaP1 and TEA+ for beta).
This experiment suggests that a Si/Al ratio of 5 and a TEA+/Al ratio of 1.25 in the
original reaction mixture are the limiting values to obtain pure zeolite beta under
hydrothermal synthesis conditions and using TEA+ as the template. The corresponding
sample was further characterised to determine the actual Si/Al in the zeolite framework.
From ICP OES elemental analysis, a Si/Al ratio of 4.7 and an Al/Na ratio of 0.8 were
determined. 27Al NMR was used to check if all the aluminium species in the sample
were part of the zeolite framework. The 27Al NMR spectrum shows a high-intensity
peak at 59 ppm corresponding to tetrahedral aluminium species and a low-intensity
peak at 4 ppm corresponding to octahedral aluminium species (Figure 8.5). Tetrahedral
aluminium species are assigned to aluminium present in the zeolite structure, while
octahedral species are likely to be due to non-zeolitic aluminium or extra-framework
species. The ratios between the integrals of the two peaks is 92/8. This implies that a
maximum of 8% of the total aluminium is not part of the zeolite framework.
Considering just the tetrahedral aluminium as part of the zeolite framework, the
Si/Al ratio has then to be corrected to a value of 5.1. This value is slightly higher than
the Si/Al ratio of 4.5 reported by Borade and Clearfield20,21 and is obtained using a
higher TEA+/Al ratio (1.25 against 0.8). The highest TEA+/Al ratio needed can be a
consequence of the higher water content used in this method. A relevant advantage of
the synthesis method described here is the use of Ludox HS-40 as silica source instead
of fumed silica, since the former is cheaper, easier and safer to handle and forms a less
dense synthesis gel.

Chapter 8 __________________________________________________________________________________________________________
150
Figure 8.5. 27Al NMR of the pure zeolite beta obtained with Si/Al = 5 and
TEA+/Al = 1.25 in the starting reaction mixture.
8.3.1 Up-scaling of the HSE lead
Up-scaling of the synthesis of zeolite beta with a Si/Al ratio of 5 and a TEA+/Al
ratio of 1.25 in the original reaction mixture to a 50-ml autoclave led to a similar result
though small amounts of NaP1 were present as by-products.
To further study the effects of differences in scale on the crystallisation of zeolites, a
synthesis gel for zeolite beta was crystallised in a 50-ml autoclave and in 3-ml
autoclaves in a HSE 24-array. Zeolite beta with a Si/Al ratio ~ 11 was synthesised using
the method proposed by Pérez-Pariente et al.,12 which has been verified by the
International Zeolite Association (IZA). Analysing the synthesis gel after only 20 hours
of crystallisation results in amorphous material for the 50-ml autoclaves, while in the
3-ml autoclaves the formation of zeolite beta already started, as can be seen in
Figure 8.6. After 40 hours of crystallisation in the 50-ml autoclaves, pure crystalline
zeolite beta is obtained using this method.
Differences in results using HSE 3-ml autoclaves and conventional larger-volume
autoclaves are probably due to the higher heating rate of the small autoclaves, which is
mainly caused by their lower thermal conductance (due to the thinner walls of the
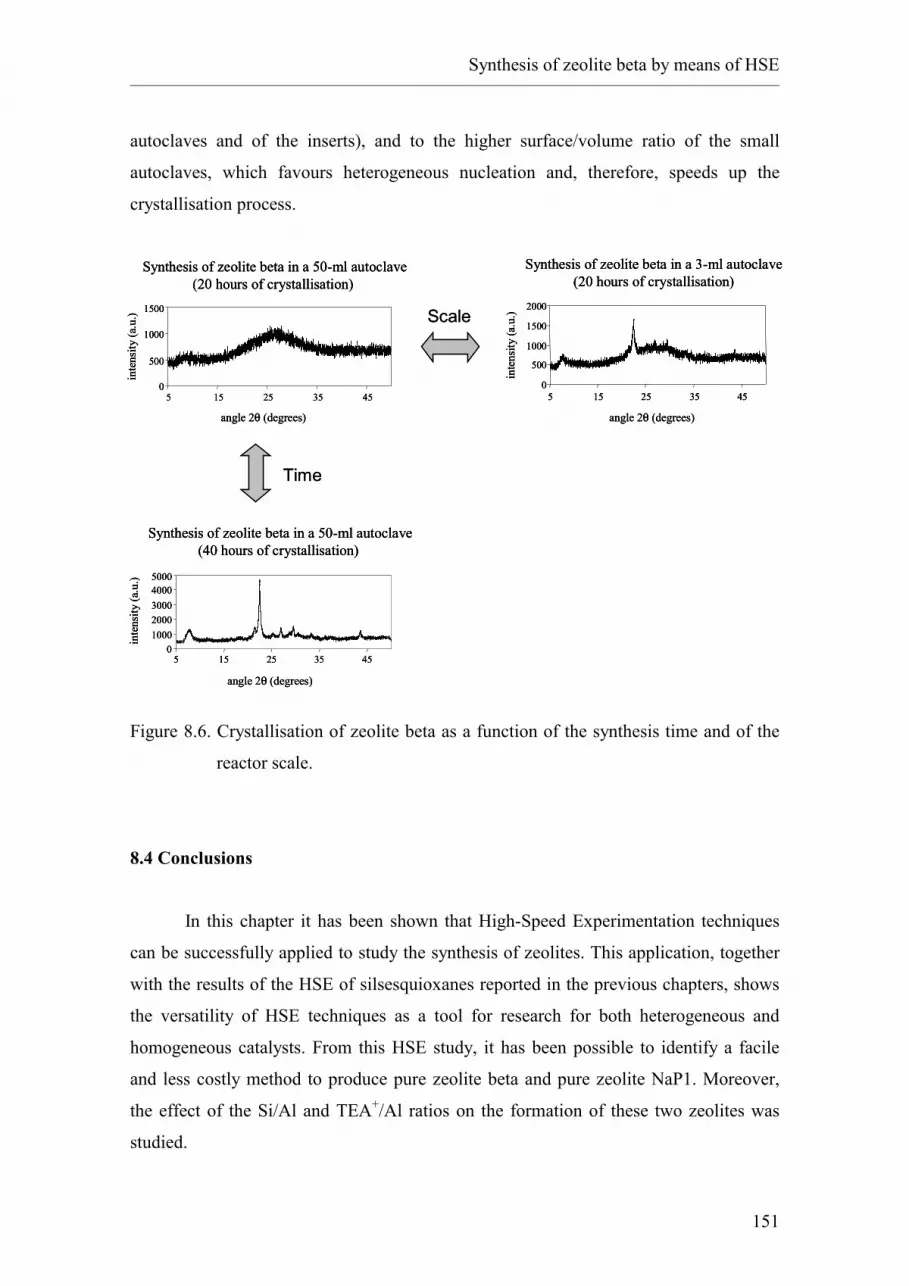
Synthesis of zeolite beta by means of HSE __________________________________________________________________________________________________________
151
autoclaves and of the inserts), and to the higher surface/volume ratio of the small
autoclaves, which favours heterogeneous nucleation and, therefore, speeds up the
crystallisation process.
Figure 8.6. Crystallisation of zeolite beta as a function of the synthesis time and of the
reactor scale.
8.4 Conclusions
In this chapter it has been shown that High-Speed Experimentation techniques
can be successfully applied to study the synthesis of zeolites. This application, together
with the results of the HSE of silsesquioxanes reported in the previous chapters, shows
the versatility of HSE techniques as a tool for research for both heterogeneous and
homogeneous catalysts. From this HSE study, it has been possible to identify a facile
and less costly method to produce pure zeolite beta and pure zeolite NaP1. Moreover,
the effect of the Si/Al and TEA+/Al ratios on the formation of these two zeolites was
studied.
Time
Scale
Synthesis of zeolite beta in a 50-ml autoclave(20 hours of crystallisation)
0
500
1000
1500
5 15 25 35 45
angle 2θ (degrees)
inte
nsity
(a.u
.)
0
500
1000
1500
2000
5 15 25 35 45
inte
nsity
(a.u
.)
Synthesis of zeolite beta in a 3-ml autoclave(20 hours of crystallisation)
angle 2θ (degrees)
010002000300040005000
5 15 25 35 45
Synthesis of zeolite beta in a 50-ml autoclave(40 hours of crystallisation)
angle 2θ (degrees)
inte
nsity
(a.u
.)
Time
Scale
Synthesis of zeolite beta in a 50-ml autoclave(20 hours of crystallisation)
0
500
1000
1500
5 15 25 35 45
angle 2θ (degrees)
inte
nsity
(a.u
.)
Synthesis of zeolite beta in a 50-ml autoclave(20 hours of crystallisation)
0
500
1000
1500
5 15 25 35 45
angle 2θ (degrees)
inte
nsity
(a.u
.)
0
500
1000
1500
2000
5 15 25 35 45
inte
nsity
(a.u
.)
Synthesis of zeolite beta in a 3-ml autoclave(20 hours of crystallisation)
angle 2θ (degrees)
0
500
1000
1500
2000
5 15 25 35 45
inte
nsity
(a.u
.)
Synthesis of zeolite beta in a 3-ml autoclave(20 hours of crystallisation)
angle 2θ (degrees)
010002000300040005000
5 15 25 35 45
Synthesis of zeolite beta in a 50-ml autoclave(40 hours of crystallisation)
angle 2θ (degrees)
inte
nsity
(a.u
.)
010002000300040005000
5 15 25 35 45
Synthesis of zeolite beta in a 50-ml autoclave(40 hours of crystallisation)
angle 2θ (degrees)
inte
nsity
(a.u
.)

Chapter 8 __________________________________________________________________________________________________________
152
8.5 Experimental
Experiments were performed on an automated parallel synthesis workstation19
coupled with a personal computer supplied with software enabling to program the
workstation. Reactants: Ludox-HS 40 colloidal silica (40%wt suspension of SiO2 in
water) as silica source; sodium aluminate (40-45%wt Na2O, 50-56%wt Al2O3) as alumina
source; tetraethylammonium hydroxide (TEAOH) (35% solution in water) as template
source; deionised water. Appropriate amounts of reactants for the zeolite synthesis were
dispensed by the D-arm of the workstation in a 6×4 matrix of small 3-ml teflon-lined
stainless steel autoclaves. An equal amount of alumina source was dispensed in each
autoclave in order to set the pH of all the samples at ~ 14. In a typical experiment, after
dispensing the alumina, the template and the silica sources, the autoclaves were filled up
to 75% of their volume with water. Next, the obtained aluminosilicate gels were stirred
in an ultrasonic bath (T ≈ 60°C) for 1 hour. The autoclaves were then closed and heated
statically at 170 °C. After 48 hours, heating was stopped and the autoclaves were cooled
down to room temperature. Finally, the autoclaves with the formed solids were washed
twice with deionised water, centrifuged at 1500 r.p.m. for 1 hour and dried in a vacuum
centrifuge.19 After the experiment, the teflon inserts were cleaned overnight with NaOH
(4M) at reaction temperature or with HF (10%) at room temperature.
Powder diffraction data were collected on a Philips PW1840 diffractometer generating
Cu Kα radiation. Data were obtained from 5° to 50° 2θ with a step of 0.01° and a
counting time of 0.5 s/step. The receiving slit had a width of 0.3 mm. The tube voltage
was 40 kV and the tube current was 50 mA. In order to reduce the analysis time, an
automatic sampler was installed, thus allowing X-ray analysis of 24 samples in
15 hours. Elemental analyses were performed on a Perkin-Elmer 3000 DV ICP OES.
After dissolution of the zeolites in a 1% HF (40%)/1.3% H2SO4 solution, the
concentrations of silicon, aluminium and sodium were measured with induced coupled
plasma optical emission spectroscopy (ICP OES). The samples were analysed twice as
independent duplicates to get an indication of the precision of the analysis. The
accuracy in the analysis of Si and Al is estimated at about ± 5%, for Na at ±10%.
High-resolution solid-state 27Al MAS NMR spectra were recorded on a Varian
VXR-400S spectrometer.

Synthesis of zeolite beta by means of HSE __________________________________________________________________________________________________________
153
All the HSE results reported in this chapter are averages of the values obtained in
different sets of experiments. The deviation of the values of each set of experiments
from the average is larger than what was found for the HSE study of the synthesis of
silsesquioxanes.22 This means that while the identified trends are reliable, the absolute
values suffer of a level of uncertainity. For example, considering the results reported in
Figure 8.4, for Si/Al ratio = 2.5, the yield in NaP1 is not constant for all the 4 TEA+/Al
ratios: this is more likely to be due to small experimental fluctuations rather than being
related to the different TEA+/Al ratio. The experimental error of these HSE results is
bound to be connected to technical problems that occurred during the synthesis of
zeolites, particularly those caused by the leaking of the autoclaves.

Chapter 8 __________________________________________________________________________________________________________
154
References
1 A.F. Crønstedt, Akad. Handl. Stockholm, 1756, 18, 120. 2 R.W. Tschernich, Zeolites of the world, Geoscience, 1992. 3 H. van Bekkum, E.M. Flanigen, P.A. Jacobs, J.C. Jansen (editors), Introduction to Zeolite Science and
Practice, Elsevier, 2001, 2nd edition. 4 M.M.J. Treacy, J. M. Newsam, Nature, 1988, 332, 249. 5 J.M. Newsam, M.M.J. Treacy, W. T. Koetsier, C. B. De Gruyter, Proc. R. Soc. Lond. A, 1988, 420, 375. 6 P.B. Venuto, Microporous Mater., 1994, 2, 297. 7 J.C van der Waal, M.S. Rigutto, H. van Bekkum, Appl. Catal. A-Gen., 1998, 167, 331. 8 J.C van der Waal, P.J. Kunkeler, K. Tan, H. van Bekkum, J. Catal., 1998, 173, 74. 9 R.L. Wadlinger, G.T. Kerr, E.J Rosinski, US Pat. Appl. 3.308.069, 1967. 10 J.C van der Waal, M.S. Rigutto, H. van Bekkum, J. Chem. Soc., Chem. Commun., 1994, 1241. 11 J.V. Smith, J.J. Pluth, R.C. Boggs, D.G. Howard, J. Chem. Soc., Chem. Commun., 1991, 363. 12 J. Pérez-Pariente, J.A. Martens, P.A. Jacobs, Appl. Catal., 1987, 31, 35. 13 F. Vaudry, F. Di Renzo, P. Espiau, F. Fajula, P. Schulz, Zeolites, 1997, 19, 253. 14 P.P. Pescarmona, T. Maschmeyer, Aust. J. Chem., 2001, 54, 583, see also Chapter 2. 15 M.A. Camblor, J. Pérez-Pariente, Zeolites, 1991, 11, 202. 16 M.A. Camblor, A. Misfud, J. Pérez-Pariente, Zeolites, 1991, 11, 792. 17 M.J. Eapen, K.S.N. Reddy, V.P. Shiralkar, Zeolites, 1994, 14, 294. 18 P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, Catal. Lett., 1999, 63, 1, see also
Chapter 1. 19 See Appendix A. 20 R.B. Borade, A. Clearfield, Microporous Mater., 1996, 5, 289. 21 R.B. Borade, A. Clearfield, Chem. Commun., 1996, 625. 22 See Paragraph 3.5.

The HSE equipment __________________________________________________________________________________________________________
155
Appendix A: High-Speed Experimentation equipment
A.1 The HSE automated workstation
A Hamilton Dual Arm liquid-handling robotic workstation was used for the HSE
synthesis of samples described and discussed in this thesis (Figure A.1). The robot is
suited for dispensing and transferring chemicals in the liquid phase by means of two
types of devices: the single needle arm and the D-arm.
Figure A.1. The Hamilton robot.
1) Two kind of operations can be performed using the single stainless steel needle arm:
A. Transfer set amounts of liquids from a rack of vials to another.
B. Dispense desired amounts of liquids to a rack of vials from an array of 24 bottles
containing different liquids, connected to the needle through 3 teflon valves each
with 8 exits (Figure A.2).
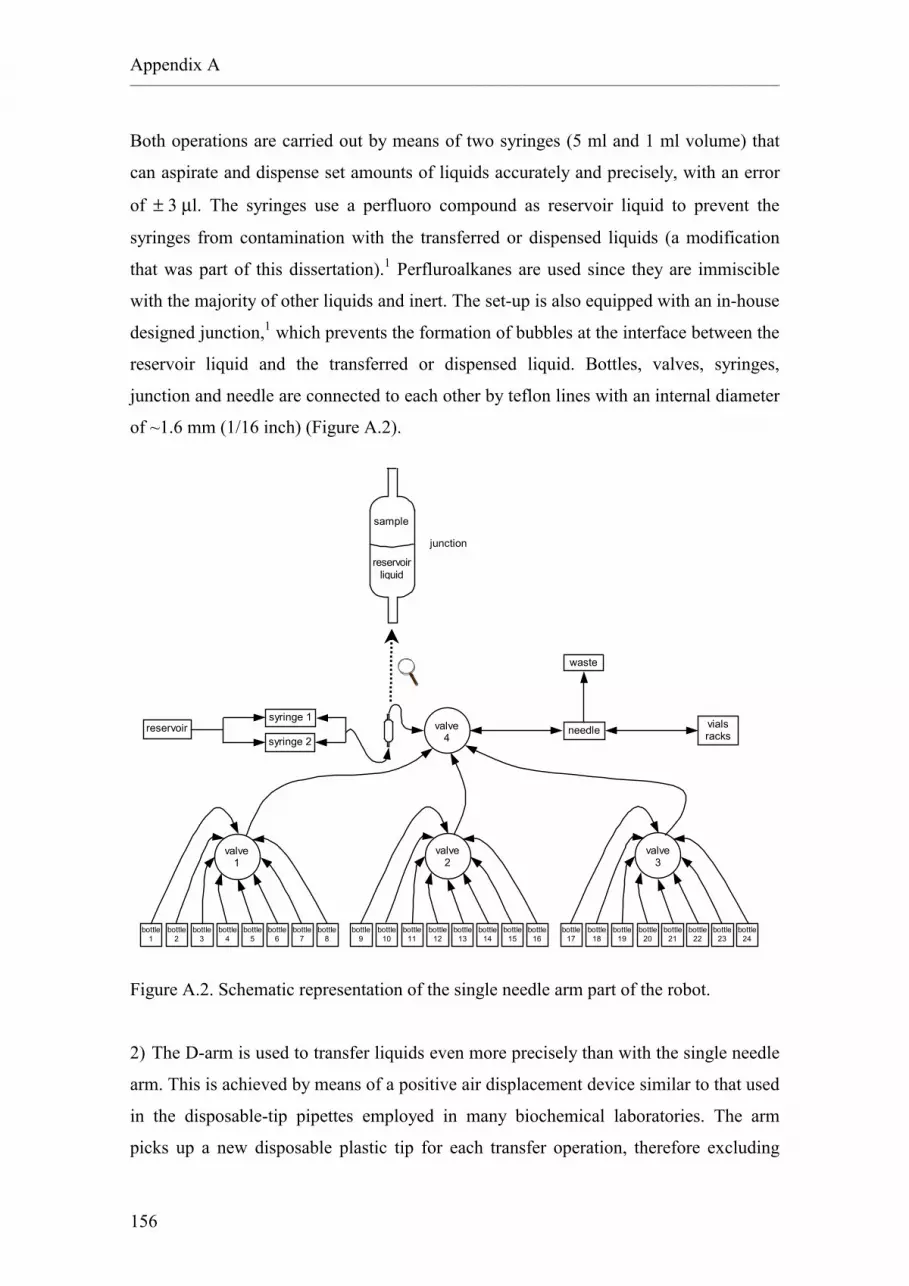
Appendix A __________________________________________________________________________________________________________
156
Both operations are carried out by means of two syringes (5 ml and 1 ml volume) that
can aspirate and dispense set amounts of liquids accurately and precisely, with an error
of ± 3 µl. The syringes use a perfluoro compound as reservoir liquid to prevent the
syringes from contamination with the transferred or dispensed liquids (a modification
that was part of this dissertation).1 Perfluroalkanes are used since they are immiscible
with the majority of other liquids and inert. The set-up is also equipped with an in-house
designed junction,1 which prevents the formation of bubbles at the interface between the
reservoir liquid and the transferred or dispensed liquid. Bottles, valves, syringes,
junction and needle are connected to each other by teflon lines with an internal diameter
of ~1.6 mm (1/16 inch) (Figure A.2).
Figure A.2. Schematic representation of the single needle arm part of the robot.
2) The D-arm is used to transfer liquids even more precisely than with the single needle
arm. This is achieved by means of a positive air displacement device similar to that used
in the disposable-tip pipettes employed in many biochemical laboratories. The arm
picks up a new disposable plastic tip for each transfer operation, therefore excluding
reservoirsyringe 1
bottle13
bottle 14
bottle 12
bottle 9
bottle 10
bottle 11
bottle 16
bottle15
valve 4
needle
bottle 21
bottle22
bottle20
bottle 17
bottle18
bottle 19
bottle 24
bottle 23
bottle 5
bottle 6
bottle 4
bottle 1
bottle 2
bottle 3
bottle 8
bottle 7
vials racks
waste
reservoirliquid
sample
valve2
valve 3
valve 1
syringe 2
junction

The HSE equipment __________________________________________________________________________________________________________
157
contamination risks. Another advantage is that the D-arm can dispense more viscous
liquids than the single needle arm. On the other hand, differently from the single needle
arm, it cannot dispense liquids through septa and is therefore not adequate for
experiments that require a closed environment (e.g. air-sensitive compounds that require
an inert atmosphere).
Various kinds of racks have been used in this thesis, with variable number of vessels.
For silsesquioxane experiments, the most common reaction racks consisted of 6×4
arrays of 24 glass tubes, each with a working volume of 3.5 ml. The tubes can be
closed with a set of teflon septa, therefore allowing reactions in a closed environment
(Figure A.3). For the synthesis of zeolites, the racks consisted of a 6×4 matrix of 24
small 3-ml teflon-lined stainless steel autoclaves, each supplied with a safety valve to
avoid pressure to exceed the value of 30 bar (Figure A.3).
All the operations performed by the robot are controlled by a computer supplied with a
Hamilton software. An in-house written module for this software allows programming
of the robot by writing simple files in a Microsoft Excel format.
Figure A.3. 6×4 glass-tube rack (left) and 6×4 autoclave rack (right).
A.2 The heating blocks
Steel heating blocks able to arrange the 6×4 type of racks were used to heat the
samples for a selected time to a desired temperature, with a maximum allowed value of

Appendix A __________________________________________________________________________________________________________
158
500°C. The blocks are placed on an orbital shaker that can provide a gentle stirring of
the liquid samples contained in the racks, with a maximum allowed rotation speed of
525 rpm.
A.3 The vacuum centrifuge
A centrifuge connected to a vacuum pump was used to remove solvents from
samples. This vacuum centrifuge can arrange up to 4 6×4 racks. The chamber of the
centrifuge is also connected to a gas-dispensing line, allowing setting the samples under
argon (inert) atmosphere.
Note: all the equipment described in this appendix was kindly provided by Avantium
Technologies, Amsterdam-Delft.
References
1 P. J. van den Brink, P.P. Pescarmona, J.C. van der Waal, Patent EPA99310598, WO0148443,
AU2389701, 1999.

Glossary __________________________________________________________________________________________________________
159
Appendix B: Glossary
B.1 Concepts
Parameter space: the space defined by the combination of each element of a chosen set
of experimental parameters with each element of each of the other chosen sets of
experimental parameters (see Chapter 1). The dimension of the parameter space is
given by the number of sets of experimental parameters; the number of elements
contained in a parameter space is given by the product of the number of elements
presents in each of the sets of experimental parameters. As an example, in the
optimisation of the synthesis of the silsesquioxane precursors reported in
Chapter 3, two parameters were investigated: the solvent and the R-group. The
set of solvents consisted of 4 elements, while the set of R-groups consisted of 10
elements. Therefore, the parameter space had dimension 2 and was constituted of
40 elements.
Library: a collection of compounds related to each other by a common feature (e.g. a
library of catalysts, a library of products). Combinatorial Chemistry and High-Speed
Experimentation techniques allow fast preparation and screening of libraries of products
(see Chapter 1).
Primary screening: a first, general investigation of the system under study (see
Paragraph 1.3).
Secondary screening: a successive, more focussed investigation of the system under
study (see Paragraph 1.3).

Appendix B __________________________________________________________________________________________________________
160
B.2 Abbreviations
APCI: Atmospheric Pressure Chemical Ionisation. ATR FTIR: Attenuated Total Reflection Fourier Transform Infrared. Bu: butyl. But: tert-butyl. CC: Combinatorial Chemistry. DMSO: dimethyl sulfoxide. ESI: Electrospray Ionisation. Et: ethyl. GC: Gas Chromatography. HSE: High-Speed Experimentation. IR: Infrared. MCR: Multivariate Curve Resolution. Me: methyl. MS: Mass Spectrometry. NMO: N-methylmorpholine-N-oxide. NMR: Nuclear Magnetic Resonance. PCA: Principal Component Analysis. Ph: phenyl. Pr: propyl. Pri: isopropyl. TBHP: tert-butyl hydroperoxide. TEA+/TEAOH: tetraethylammonium (ion)/tetraethylammonium hydroxide. THF: tetrahydrofuran.

Summary __________________________________________________________________________________________________________
Paolo P. Pescarmona: “An Exploration of Silsesquioxanes and Zeolites using
High-Speed Experimentation”
Summary
This thesis describes the most interesting results of the Ph.D. research of the
author. As stated in the title, the subject of this research is the exploration of
silsesquioxanes (synthesis and catalysis) and zeolites (synthesis) using High-Speed
Experimentation (HSE).
Combinatorial Chemistry and High-Speed Experimentation techniques are recently
developed methods with an increasing number of applications to many fields of
research: originally introduced in the search of pharmaceutical active compounds, these
techniques have now found useful applications in other fields such as catalysis and
materials science. One of the basic ideas behind Combinatorial Chemistry and
High-Speed Experimentation is to enable an evolutionary process in order to identify
suitable compounds or conditions for a chosen purpose. This means that with these
techniques many parameters are combined to produce large numbers of samples that are
then tested for the desired property. Automated workstations are normally employed to
prepare and screen these samples. In this dissertation, the attention is focussed on the
application of High-Speed Experimentation techniques to catalysis. (Chapter 1).
The study and optimisation of the synthesis of silsesquioxanes is one of the main
subjects of this thesis. Silsesquioxanes are a family of compounds of the general
formula (RSiO1.5)a(H2O)0.5b. They are constituted of tetrahedral units in which a silicon
is connected to three oxygens and to an organic group R (or to a hydrogen). Therefore,
silsesquioxanes are characterised by a Si-O framework similar to that of silica or
zeolites. The presence of the organic groups makes these compounds soluble in a
number of organic solvents. Here, particular attention is given to incompletely
condensed silsesquioxanes, which contain free silanol groups on which metal centres
can be anchored. Incompletely condensed silsesquioxanes have found applications as
model compounds for silica surfaces and as ligands for homogeneous catalysts.
(Chapter 2).
161

Summary __________________________________________________________________________________________________________
Complexes of titanium with silsesquioxane a7b3 have been used as models for
heterogeneous silica-based catalysts and as homogenous catalysts for the epoxidation of
alkenes. Here, the synthesis of silsesquioxane precursors for Ti-catalysts was studied by
means of High-Speed Experimentation with the aim to either improve the otherwise
long and expensive preparation method of silsesquioxane a7b3 or to generate other
silsesquioxane structures suitable for forming active epoxidation Ti-catalysts. For this
purpose, the hydrolytic condensation of trichlorosilanes to produce silsesquioxanes was
optimised as a function of the epoxidation activity of the catalysts obtained after
reaction of the silsesquioxane precursors with a titanium centre. In a first stage, the two
parameters that were assumed to have the most relevant influence on determining the
selectivity and the yield of the synthesis of silsesquioxanes were studied: the R-group of
the organotrichlorosilanes RSiCl3 and the solvent in which the hydrolytic condensation
occurs. The High-Speed Experimentation approach allowed the rapid screening of a
large number of trichlorosilanes and solvents and, consequently, the identification of
some trends concerning these two experimental parameters. The best silsesquioxane
precursor for an epoxidation catalyst was obtained by the hydrolytic condensation of
cyclopentyltrichlorosilane in acetonitrile. Characterisation by means of NMR and MS
showed that the lead contained a mixture of silsesquioxanes, with silsesquioxane a7b3
as the main component. (Chapter 3).
In a second stage, the best synthesis methods identified were further optimised by
varying the other parameters influencing the hydrolytic condensation. This fine-tuning,
also performed by means of High-Speed Experimentation, allowed adjusting the
conditions for the synthesis of the silsesquioxane precursors and to gain knowledge
about the effect of the various parameters. Next, the effect of the nature of the titanium
centre bound to the silsesquioxane was investigated using High-Speed Experimentation.
(Chapter 4).
The two best Ti-catalysts identified by means of HSE were obtained with cyclopentyl
and cyclohexyl silsesquioxanes synthesised in acetonitrile. The synthesis of the two
precursors was further improved and the silsesquioxane products were fully
characterised. In the case of cyclopentyl silsesquioxanes, structure a7b3 was selectively
obtained in 64% yield after 18 hours of reaction – a significant improvement compared
to the formerly known methods. The synthesis was monitored by MS and in-situ IR in
order to identify the silsesquioxanes present in the reaction mixture, to explain the high
162

Summary __________________________________________________________________________________________________________
selectivity of the reaction towards structure a7b3 and to propose a mechanism for its
formation. In the case of cyclohexyl silsesquioxanes, the synthesis had a high yield but
was less selective than in the case of cyclopentyl. The main products were:
silsesquioxanes a6b0, a6b2, a7b1 and a7b3. The role of acetonitrile as a reactive
solvent in the syntheses was ascertained. (Chapter 5).
Cyclopentyl silsesquioxane a7b3, synthesised with the new, improved method, was
used to prepare an Os-complex in which the poisonous OsO4 is rendered stable. This
Os-silsesquioxane complex was used both as model compound for a silica-supported,
heterogeneous Os-catalyst and as homogeneous catalyst for the dihydroxylation of
alkenes. The Os-silsesquioxane catalyst displayed higher turnover frequencies than the
heterogeneous one, while keeping the same selectivity (99%). (Chapter 6).
The first screening of the synthesis of silsesquioxane precursors for epoxidation
Ti-catalysts using High-Speed Experimentation showed a positive effect of the polarity
of the solvent (Chapter 3). This trend encouraged the investigation of the effect of other
highly polar solvents on the synthesis of the silsesquioxane precursors. The results of
this HSE study did not show clear trends but allowed the identification of a number of
very active epoxidation Ti-catalysts, the most promising of which were fully
characterised: each lead consisted of a different type of silsesquioxane structures. The
lead synthesised by the hydrolytic condensation of ButSiCl3 in water was exclusively
constituted of silsesquioxane a2b4. This was the first reported example of the use of this
silsesquioxane structure to prepare active Ti-catalysts. These complexes were supported
on silica to produce an active, non-leaching, heterogeneous, epoxidation catalyst. The
lead obtained by the hydrolytic condensation of ButSiCl3 in DMSO consisted of a
number of incompletely condensed structures (mainly with 3 < a < 5). The lead
produced by the hydrolytic condensation of PhSiCl3 in water consisted of polymeric
silsesquioxanes. (Chapter 7).
After the successful application of High-Speed Experimentation to the synthesis of
silsesquioxanes, these techniques were used to study the synthesis of zeolite beta.
Zeolites are well-known microporous crystalline materials with many applications as
heterogeneous catalysts, ion-exchangers and molecular sieves. Here, the synthesis of
aluminium-rich zeolite beta was investigated by performing the synthesis with different
Si/Al and TEA+/Al ratios (where TEA+ is the template). It was found that Si/Al = 5 and
TEA+/Al = 1.25 in the starting reaction mixture are the lowest ratios to obtain pure
163

Summary __________________________________________________________________________________________________________
zeolite beta with the employed hydrothermal method. Mixtures of zeolites (mainly beta
and NaP1) were observed at lower Si/Al and TEA+/Al ratios. (Chapter 8).
Altogether, it was shown that High-Speed Experimentation techniques, by coupling
discovery and understanding, are a powerful research tool for applied and fundamental
research in the field of catalysis.
164

Samenvatting __________________________________________________________________________________________________________
Paolo P. Pescarmona: “Een Exploratie van Silsesquioxanen en Zeolieten door middel
van High-Speed Experimentation”.
Samenvatting
In dit proefschrift worden de meest interessante resultaten van het
promotieonderzoek van de auteur weergegeven. Zoals al blijkt uit de titel, is het
onderwerp van het promotieonderzoek de exploratie van silsesquioxaanchemie en
zeolietsynthese met behulp van High-Speed Experimentation (HSE) technieken.
Combinatorial Chemistry en High-Speed Experimentation technieken zijn recent
ontwikkelde methoden met toenemend aantal toepassingen binnen verschillende
weteschappenlijke disciplines. Oorspronklijk zijn ze ontwikkeld om farmaceutisch
actieve verbindingen te ontdekken, maar ze worden tegenwoordig ook toegepast in
andere vakgebieden, met name in de katalyse en de materiaalkunde. Het idee achter
Combinatorial Chemistry en High-Speed Experimentation is het vinden van de voor een
bepaald doel optimale verbinding of condities, door middel van een evolutie proces.
Met deze technieken, worden door systematische combinatie van veel parameters grote
aantallen monsters geproduceerd, die dan voor de gewenste eigenschap getest kunnen
worden. Geautomatiseerde werkstations worden gewoonlijk gebruikt om deze grote
aantallen monsters te bereiden en testen. Binnen dit promotie onderzoek is de aandacht
gericht op de toepassing van High-Speed Experimentation in de katalyse. (Hoofdstuk 1).
De bestudering en optimalisatie van de synthese van silsesquioxanen is een van de
belangrijkste onderwerpen van dit proefschrift. Silsesquioxanen zijn een familie van
organosilicium verbindingen met de algemene structuurformule (RSiO1.5)a(H2O)0.5b. Ze
bestaan uit tetraëders waarin een siliciumatoom gebonden is aan drie zuurstofatomen en
een organische groep (of een waterstofatoom). Ze bezitten daardoor een met silica en
zeolieten vergelijkbare Si-O-structuur. Dankzij de organische groepen zijn
silsesquioxanen in een aantal organische oplosmiddelen oplosbaar. In het
promotieonderzoek is de aandacht gericht op incompleet gecondenseerde
silsesquioxanen: structuren die nog silanol groepen bevatten, waardoor ze aan
metaalcentra verankerd kunnen worden. Dankzij deze eigenschappen zijn incompleet
gecondenseerde silsesquioxanen uitermate geschikt als modelverbindingen voor
silica-oppervlakken en als liganden voor homogene katalysatoren. (Hoofdstuk 2).
165

Samenvatting __________________________________________________________________________________________________________
Complexen van titanium met silsesquioxaan a7b3 zijn als model voor heterogene
silica-gedragen titaniumkatalysatoren en homogene titaniumkatalysatoren voor de
epoxidatie van alkenen bekend. In dit promotieonderzoek is vooral de synthese van
silsesquioxaan precursors voor titaniumkatalysatoren door middel van High-Speed
Experimentation bestudeerd. Het doel was de lange en dure synthesemethode voor
silsesquioxaan a7b3 te verbeteren of andere geschikte structuren te vinden om actieve
epoxidatie Ti-katalysatoren te genereren. Hiertoe werd de hydrolytische condensatie van
trichloorsilanen die silsesquioxanen produceert als functie van de epoxidatie activiteit
van de katalysatoren, verkregen door de reactie van het ruwe silsesquioxaanmengsel
met een titaniumcentrum, geoptimaliseerd. In de eerste fase zijn de twee parameters met
naar verwachting de grootste invloed op de selectiviteit en opbrengst van de
silsesquioxaansynthese bestudeerd: de organische R-groep van het organotrichloorsilaan
RSiCl3 en het oplosmiddel waarin de hydrolytische condensatie plaatsvindt. De
High-Speed Experimentation aanpak maakte een snelle test mogelijk van een groot
aantal trichloorsilanen en oplosmiddelen en, derhalve, de identificatie van belangrijke
trends over de twee bestudeerd experimenteel parameters. De beste
silsesquioxaanprecursor voor een epoxidatiekatalysator is door de hydrolytische
condensatie van cyclopentyltrichloorsilaan in acetonitril verkregen. Karakterisering
door NMR en MS toonde aan dat de beste lead uit een mengsel van silsesquioxanen
bestaat, waarvan structuur a7b3 de belangrijkste component is. (Hoofdstuk 3).
In een volgende fase werden de beste synthesemethoden verder geoptimaliseerd door
het variëren van de overige parameters die een invloed op de hydrolytische condensatie
hebben. High-Speed Experimentation leidde tot een verfijning van de synthesecondities
en tot nieuwe kennis over de effecten van de verschillende parameters. Tevens is het
effect van het aan het silsesquioxaan gebonden titaniumcentrum onderzocht.
(Hoofdstuk 4).
De twee beste katalysatoren, welke door middel van HSE geïdentificeerd waren, waren
de in acetonitril gesynthetiseerde cyclopentyl- en cyclohexylsilsesquioxanen. De
synthese van de twee silsesquioxanen werd verder verbeterd en de silsesquioxaan
producten werden volledig gekarakteriseerd. Voor cyclopentylsilsesquioxanen werd
structuur a7b3 selectief verkregen in 64% opbrengst na 18 uur reactie; een relevante
verbetering ten opzichte van de vroeger bekende methoden. Om de hoge selectiviteit
naar structuur a7b3 te verklaren en een synthetisch mechanisme voor te kunnen stellen,
166

Samenvatting __________________________________________________________________________________________________________
werd de synthese met MS en in-situ IR gevolgd om de verschillende silsesquioxaan
species in de reactiemengsel te identificeren. Ook voor cyclohexyltrichloorsilaan heeft
de synthese een hoge opbrengst maar de selectiviteit is lager dan die voor
cyclopentyltrichloorsilaan. De belangrijkste producten zijn silsesquioxanen a6b0, a6b2,
a7b1 en a7b3. Daarnaast is de rol van acetonitril als reagerend oplosmiddel
opgehelderd. (Hoofdstuk 5).
De cyclopentylsilsesquioxaan a7b3 werd daarna gebruikt om een Os-complex te
bereiden waarin OsO4 is geimmobiliseerd. Het Os-silsesquioxaan-complex is als model
voor een silica-gedragen heterogene katalysator en als homogene katalysator voor de
dihydroxylatie van alkenen gebruikt. De Os-silsesquioxaan katalysator toont hogere
turnover frequenties dan de heterogenene katalysator bij gelijke, hoge selectiviteit
(99%). (Hoofdstuk 6).
Een positief effect van de polariteit van het oplosmiddel was een belangrijke conclusie
uit eerder onderzoek van de synthese van silsesquioxaan precursors voor epoxidatie
Ti-katalysatoren door middel van High-Speed Experimentation (Hoofdstuk 3).
Aangemoedigd door de gevonden trend, richtte het onderzoek zich op het effect van
andere zeer polaire oplosmiddelen op de synthese van silsesquioxanen. De resultaten
van deze HSE-bestudering toonden geen duidelijke trends maar leverden wel een aantal
zeer actieve epoxidatie-katalysatoren op. De meest interessante katalysatoren zijn
gekarakteriseerd: elke lead bestaat uit verschillende soorten silsesquioxaanstructuren.
De lead verkregen uit de hydrolytische condensatie van ButSiCl3 in water, bestaat
uitsluitend uit silsesquioxaan a2b4. Dit is het eerste bekende voorbeeld van het gebruik
van deze silsesquioxaanstructuur om een actieve Ti-katalysator te bereiden. Het
complex werd op silica gezet om zo een actieve, niet-uitlogende heterogene katalysator
voor epoxidatie van alkenen te krijgen. De lead verkregen uit de hydrolytische
condensatie van ButSiCl3 in DMSO bestaat uit een aantal incompleet gecondenseerde
structuren (vooral met 3 < a < 5). De lead verkregen uit de hydrolytische condensatie
van PhSiCl3 in water als oplosmiddel bestaat uit een polymeer silsesquioxaan.
(Hoofdstuk 7).
Na de positieve resultaten van de toepassing van High-Speed Experimentation in de
synthese van silsesquioxanen werden de technieken gebruikt om de synthese van
zeolieten te bestuderen. Zeolieten zijn microporeuze materialen met toepassingen als
heterogene katalysatoren, als ionen-wisselaars en als moleculaire zeven. In dit
167

Samenvatting __________________________________________________________________________________________________________
onderzoek werd de synthese van aluminium-rijk zeoliet beta met verschillende Si/Al en
TEA+/Al verhoudingen bestudeerd met het TEA+ kation als het templaat. De laagste
gevonden verhoudingen in het reactiemengsel die met de gebruikte hydrothermale
methode nog tot zuiver puur zeoliet beta leidden waren Si/Al = 5 en TEA+/Al = 1.25.
Bij lagere Si/Al en TEA+/Al verhoudingen werden mengsels van zeolieten (vooral beta
en NaP1) gevonden. (Hoofdstuk 8).
Een belangrijke conclusie van het promotieonderzoek is dat High-Speed
Experimentation een krachtig hulpmiddel blijkt te zijn voor toegepast en fundamenteel
katalyse-onderzoek.
168

Acknowledgements __________________________________________________________________________________________________________
Acknowledgements
Here is the end of this thesis, and with it comes the time to take a look back at
the four and a half nice years of my Ph.D.. Through this experience I met a lot of
persons who helped me with my work and, more important, who created a lively,
multicultural environment that made the days spent together fine and pleasant. I want to
express my gratitude to this people.
First of all I would like to thank my two supervisors. Thomas, for having given
me the opportunity to come to Delft for my Ph.D., for your advices, for your friendly
attitude, for always having a good word to encourage and motivate me and for the
excellent balance between independence and guidance that you gave me in my research
during these years. Jan Kees, for your advices, for the nice and useful scientific
discussions (‘is it only one peak?’), for sharing with me your knowledge, for your
positive mood and overall for the good time we had together in the lab. Thomas and
Jan Kees, it was a great pleasure to work with the two of you and I hope that we will
have the chance to collaborate again in the future.
Next, I am grateful to all the staff of TOCK/BOC and more in general of
DelftChemTech (Bibliotheek, Technische dienst, Facilitaire dienst, IT-groep) for the
kind help provided. Adrie Knol, Anton van Estrik, Ben Norder, Ernst Wurtz, Lars
Könemann, Mieke Jacobs, Mieke van der Kooij, Niek van de Pers: hartelijk dank! A
special acknowledgement goes to Anton Sinnema and Kristina Djanashvili for the
measurement and help in the interpretation of numerous NMR spectra and for the nice
talking in between. Moreover, I would like to thank Greet de Loos, Henk van
Koningsveld, Herman van Bekkum, Joop Peters, Koos Jansen, Leen Maat and Ulf
Hanefeld for having always been available for fruitful discussions.
In these years spent in the lab I met many Ph.D.’s, post-docs and students: with
some of them I shared many good moments, with others the contact was short but
nevertheless pleasant. I know it is not going to be easy to keep in contact with all of
you; anyhow, I will be happy whenever I meet you again. Thanks to all of you: Frank,
mijn Nederlandse spreektaal leraar. Inma, por tu cariño. Lars, for your sincerity.
Luca G., perché nonostante ci sia di mezzo il Po, si è buoni compagni. Martijn, for
seldom agreeing but still liking to discuss. Paulo, por tu amistad. Rute, for all the strong
feelings and the many good and few bad times we shared. Silvia P., for your sensitivity.
169

Acknowledgements __________________________________________________________________________________________________________
Hans, it was a very positive experience to have you as my student and to know you as
the kind person you are. The ‘Combi’ group: Leon, Silvia G., SP and JK; the good days
spent together at the congress in Ischia (and surroundings) will always be a special
memory. Abdel, Allard, Andrea, Angel, Aniel, Anne B., Anne P., Annemieke, Arné,
Bruno, Carla, Carlos G., Carlos P., César, Chrétien, Cindy, Dean, Delia, Elena, Eva,
Ewald, Fatma, Filip, Gema, Göran, Guillaume, Heidi, Hilda, Jan, José Miguel, Leszek,
Lingqiu, Luca F., Luka, Luuk, Maikel, Manuela, Margreth, Marina, Mathias, Maxim,
Menno, Mette, Michel P., Michel V., Michiel H., Michiel v.V., Mike, Mireia, Moira,
Naseem, Nazely, Nina, Paloma, Pedro, Petra, Raffaella, Sameh, Sandrine, Scoob,
Stefan, Stephan, Tanya, Teresa, Ton, Weiwen Ma, Xavier, Yuxin Li, Zbigniew,
Zhiping: it was nice to meet all of you!
I would like to thank all the people from Avantium with whom I have been in
touch and particularly: Maria, for our rare but nice meetings, Arie, Bas, Ben, Chris,
François, John, Lisa, René, Roger.
During my short and intense stay at the University of Sydney I had the pleasure
to meet and work with a number of persons: Tony, thanks for the good period I had
there and for coming all the way from Down Under for my Ph.D. defence, Jim Beattie,
Keith Fisher, Kelvin Picker, Ian Luck, Dave, John and Ricky.
Finally, I am grateful to all the members of the Examination Committee for
accepting to take part to my Ph.D. defence.
There are many other persons who do not have any direct relationship with my
Ph.D. but to whom I am happy to be close (although often not geographically): my
parents and family, friends from Italy, friends met in the Netherlands and many others I
met around the world.
170

Publications and oral presentations __________________________________________________________________________________________________________
Publications and oral presentations
Publications
1. G. Pacchioni, P. Pescarmona, “Structure and stability of oxygen vacancies on
sub-surface, terraces, and low-coordinated surface sites of MgO: an ab initio study”,
Surf. Sci., 1998, 412/413, 657.
2. P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, “Combinatorial
Chemistry, High-Speed Screening and catalysis”, Catal. Lett., 1999, 63, 1.
3. P. Pescarmona, M. Chiesa, M.C. Paganini, E. Giamello, “OH-D2 (OD-H2) isotopic
exchange at the surface of CaO monitored by electron paramagnetic resonance”,
Magn. Reson. Chem., 2000, 38, 833.
4. P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, “A new,
efficient route to titanium-silsesquioxane epoxidation catalysts developed by using
High-Speed Experimentation techniques”, Angew. Chem. Int. Ed., 2001, 40, 740.
5. P.P. Pescarmona, T. Maschmeyer, “Oligomeric silsesquioxanes: synthesis,
characterization and selected applications”, Aust. J. Chem., 2001, 54, 583.
6. P.P. Pescarmona, J.T.T. Rops, J.C. van der Waal, J.C. Jansen, T. Maschmeyer,
“High-Speed Experimentation techniques applied to the study of zeolites and
silsesquioxanes”, J. Mol. Cat. A, 2002, 182-183, 319.
7. P.P. Pescarmona, T. Maschmeyer, “Serial and parallel ways to enhance and
accelerate catalyst testing”, NATO Science Series, 2002, Ser. II Vol. 69, 173.
8. P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, “Application of Combinatorial
Chemistry and High-Speed Experimentation concepts to the study of
titanium-silsesquioxane catalysts”, Catal. Today, 2003, 81, 347.
9. P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, “High-Speed
Experimentation techniques applied to the synthesis of titanium-silsesquioxane
epoxidation catalysts”, Chem. Eng. Comm., accepted.
10. P.P. Pescarmona, M.E. Raimondi, J. Tetteh, B. McKay, T. Maschmeyer, “A
mechanistic study of silsesquioxane synthesis by mass spectrometry and in-situ
ATR FT-IR spectroscopy”, J. Phys. Chem. B, accepted.
171

Publications and oral presentations __________________________________________________________________________________________________________
11. P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, “Fast and high-yield
silsesquioxane syntheses using acetonitrile as reactive solvent”, Eur. J. Inorg.
Chem., submitted.
12. P.P. Pescarmona, A. F. Masters, J.C. van der Waal, T. Maschmeyer, “Osmium
silsesquioxane as model compound and homogeneous catalyst for the
dihydroxylation of alkenes”, Angew. Chem. Int. Ed., submitted.
13. P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, “New silsesquioxane-based
homogeneous and heterogeneous epoxidation catalysts developed using
High-Speed Experimentation”, Chem. Eur. J., submitted.
Patents
1. P. J. van den Brink, P.P. Pescarmona, J.C. van der Waal, “Liquid dispensing device”,
Patent EPA99310598, WO0148443, AU2389701, 1999.
Oral presentations and posters
1. P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, “Development
of a new, efficient route to titanium-silsesquioxane epoxidation catalysts using
High-Speed Experimentation techniques”, Frontiers in Homogeneous Catalysis,
Joint meeting of the NIOK and the Katalyseverbund NRW, Eindhoven University
of Technology, Netherlands, October 9, 2000: poster.
2. P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, “Development
of titanium-silsesquioxane epoxidation catalysts using High-Speed Experimentation
techniques”, 2000 Annual Meeting of the American Institute of Chemical
Engineers (AIChE), Los Angeles, United States of America, November 12-17,
2000: oral presentation.
3. P.P. Pescarmona, J.C. van der Waal, I.E. Maxwell, T. Maschmeyer, “Development
of new titanium-silsesquioxane epoxidation catalysts using High-Speed
Experimentation techniques”, NCCC2, Noordwijkerhout, Netherlands, March 5-7,
2001: oral presentation.
172

Publications and oral presentations __________________________________________________________________________________________________________
4. P.P. Pescarmona, J.T.T. Rops, J.C. van der Waal, J.C. Jansen, T. Maschmeyer,
“Synthesis of zeolites and silsesquioxanes using High-Speed Experimentation
techniques”, 10th International Symposium on Relations between Homogeneous and
Heterogeneous Catalysis, Lyon, France, July 2-6, 2001: oral presentation.
5. P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, “Study of the synthesis of
incompletely condensed silsesquioxanes by means of High-Speed Experimentation
techniques”, NCCC3, Noordwijkerhout, Netherlands, March 4-6, 2002: poster.
6. P.P. Pescarmona, “Study of the synthesis of silsesquioxanes by means of
High-Speed Experimentation techniques”, University of Torino, Italy, May 29, 2002:
oral presentation.
7. P.P. Pescarmona, J.C. van der Waal, T. Maschmeyer, “Study of the synthesis of
incompletely condensed silsesquioxanes by means of High-Speed Experimentation
techniques”, EuroCombiCat, European Workshop on Combinatorial Catalysis, Ischia,
Italy, June 2-5, 2002: poster.
8. P.P. Pescarmona, “High-Speed Experimentation techniques applied to the synthesis
of Ti-silsesquioxanes as epoxidation catalysts”, University of Sydney, Australia,
October 25, 2002: oral presentation.
9. P.P. Pescarmona, J.K. Beattie, A. F. Masters, J.C. van der Waal, T. Maschmeyer,
“An osmium silsesquioxane as a catalyst for the dihydroxylation of alkenes”,
Conference of the Inorganic Chemistry Division of the Royal Australian Chemical
Institute, Melbourne, Australia, February 2-6, 2003: poster.
10. P.P. Pescarmona, J.C. van der Waal, M.E. Raimondi, J. Tetteh, T. Maschmeyer,
“Improved synthesis of silsesquioxane a7b3 [(c-C5H9)7Si7O9(OH)3]”, NCC4
(Netherlands Catalysis & Chemistry Conference) Noordwijkerhout, Netherlands,
March 10-12, 2003: oral presentation and poster.
11. P.P. Pescarmona, J.K. Beattie, A. F. Masters, J.C. van der Waal, T. Maschmeyer,
“An osmium silsesquioxane as a catalyst for the dihydroxylation of alkenes”, 6th
International Symposium on Catalysis Applied to Fine Chemicals (CAFC 6), Delft,
Netherlands, April 6-10, 2003: poster.
173

Curriculum vitae __________________________________________________________________________________________________________
Curriculum vitae Paolo Pescarmona was born on the 2nd of November 1973 in Torino, Italy. He
obtained his secondary education diploma in 1992 at the Liceo Scientifico Statale
Galileo Ferraris, Torino, Italy, with a grade of 60/60. In October 1992 he started his
university studies in Chemistry at the Università di Torino, Italy. He received his Laurea
in Chimica (Master of Science in Chemistry, 5 years) on the 4th of December 1997 with
a grade of 110/110 cum laude et mentione. His final project, “Defective centres at the
surface of alkaline-earth oxides: spectroscopic studies (EPR) and quantum mechanical
calculations”, was carried out under the supervision of Prof. Elio Giamello (Università
di Torino) and of Prof. Gianfranco Pacchioni (Università di Milano). From December
1997 till October 1998 he fulfilled his Civil Service at the Foreigners Office of the City
of Torino, Italy. In February 1999 he started his Ph.D. research at the Laboratory of
Applied Organic Chemistry and Catalysis of the Technische Universiteit Delft,
Netherlands, under the supervision of Prof. dr. Thomas Maschmeyer and Dr. ir. Jan
Kees van der Waal on the “Exploration of silsesquioxanes and zeolites using
High-Speed Experimentation”. The results of this research are described in this thesis.
174

Propositions
belonging to the thesis:
“An Exploration of Silsesquioxanes and Zeolites using High-Speed Experimentation”
by Paolo P. Pescarmona
1. The term ‘polarity’, although being one of the most commonly used in chemistry, is still rather undefined.
2. The capability of pronouncing the word ‘silsesquioxanes’ is an effective criterion
to recognise people that worked with these compounds. 3. Although colour blindness may generate some difficulties for the experimental
chemist, it can favour a critical attitude towards observation. 4. A multidisciplinary approach to scientific research can be compared to a
multiethnic society: once a common language is found and the reciprocal differences are understood and accepted, the blend will produce great benefits.
5. (Scientific and technological) progress should be driven by its social value rather
than by its economic potential. 6. One of the risks of economic endeavours is to confuse the well-being of companies
with that of people. 7. Nice would be a world where all the persons are equal, but not uniform; alas,
uniformity is growing whilst equality is decreasing. 8. Ties, depilation and hijab (headscarf) have a common feature: making use of them
should be a free individual choice and not an imposition. 9. One of the most reliable methods to recognise whether somebody is Dutch, is to
ask her/him to write an ‘8’.
10. A rather underestimated skill often acquired in University and Ph.D. studies is the interpretation of the handwriting of professors.

Stellingen
behorende bij het proefschrift:
“An Exploration of Silsesquioxanes and Zeolites using High-Speed Experimentation”
van Paolo P. Pescarmona
1. De term ‘polariteit’, hoewel een van de meest gebruikte in de scheikunde, is nog niet goed gedefinieerd.
2. De manier waarop het woord ‘silsesquioxanen’ wordt uitgesproken, is een goed
criterium om mensen die met deze verbindingen werken te herkennen. 3. Hoewel kleurenblindheid bepaalde problemen voor de experimentele chemicus
genereert, kan het een kritische houding voor het waarnemen begunstigen. 4. Een multi-disciplinaire aanpak voor wetenschappelijk onderzoek is met een
multi-etnische maatschappij vergelijkbaar: als een gemeenschappelijke taal gevonden is en wederzijdse verschillen begrepen en geaccepteerd zijn, zal het mengsel grote voordelen opleveren.
5. (Wetenschappelijke en technologische) vooruitgang behoort geleid te worden door
zijn sociale waarde eerder dan door het economische potentieel. 6. Een van de risico’s van de moderne economie is het welzijn van bedrijven met die
van mensen te verwarren. 7. Mooi zou een wereld zijn waarin alle mensen gelijk waren maar niet uniform;
helaas groeit tegenwoordig de uniformiteit terwijl de gelijkheid vermindert. 8. Dassen, ontharing en hijab (hoofddoek) hebben een gemeenschappelijke
eigenschap: het gebruik ervan behoort een vrije keuze te zijn en niet een verplichting.
9. Een van de meest betrouwbare manieren om een Nederlander te herkennen is
haar/hem te vragen een ‘8’ te schrijven.
10. Een doorgaans onderschatte kennis die vaak gedurende een universitaire of Ph.D.-studie wordt opgedaan is de interpretatie van het handschrift van professoren.