acetylcholinesterase is required for neuronal and muscular development in the zebrafish embryo
TRANSCRIPT

ACh is the major neurotransmitter of the cholinergic system.ACh is secreted from the presynaptic nerve terminal and bindsto AChRs, which are clustered in the postsynaptic membrane.After release, ACh is rapidly removed from the synaptic cleft byAChE, which belongs to the family of type B carboxylesterasesand which cleaves acetylcholine into choline and acetate (forreview, see refs 1, 2). The importance of AChEs in body homeo-stasis is underscored by the fact that they are targets of some ofthe most potent toxins including insecticides, snake venom andchemical weapons3. However, inhibitors of AChE activity are alsoused as treatment of Alzheimer’s disease and the autoimmunedisorder myasthenia gravis4,5.
AChE is expressed in a number of tissues, including the cen-tral nervous system and muscle. In zebrafish embryos, expres-sion of ache commences in an anterior-to-posterior wave inthe presomitic mesoderm at the six-somite stage, significantlyearlier than the onset of body movement6. Expression of AChEcan also be detected in many primary neurons of the zebrafishembryo, including both motor neurons and sensory neurons,which differentiate in the nascent central nervous system dur-ing early somitogenesis stages6–8. Drawing on the timing andpattern of expression in the vertebrate embryo, so-called non-classical functions of AChE have been postulated that may notentail hydrolysis of ACh (for review, see refs. 1, 9). AChE sharesstructural similarity with cell adhesion proteins (for review,see ref. 1). A number of experiments suggest that AChE canpromote neurite outgrowth, an effect that does not necessari-ly require catalytic activity1,10.
Mice carrying targeted knockouts of ache show a mild phe-notype. Homozygous mutant mice are slightly smaller and show
Acetylcholinesterase is required forneuronal and muscular developmentin the zebrafish embryo
Martine Behra1, Xavier Cousin2, Christelle Bertrand2, Jean-Luc Vonesch1, DominiqueBiellmann1, Arnaud Chatonnet2 and Uwe Strähle1
1 Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS/INSERM/ULP, BP 163, 67404 Illkirch Cedex, C.U. de Strasbourg, France2 Unite Différenciation Cellulaire et Croissance—INRA, 2 place Viala, 34060 Montpellier, France
Correspondence should be addressed to U.S. ([email protected])
Published online: 2 January 2002, DOI: 10.1038/nn788
The neurotransmitter acetylcholine (ACh) has a crucial role in central and neuromuscular synapsesof the cholinergic system. After release into the synaptic cleft, ACh is rapidly degraded byacetylcholinesterase (AChE). We have identified a mutation in the ache gene of the zebrafish, whichabolishes ACh hydrolysis in homozygous animals completely. Embryos are initially motile but subse-quently develop paralysis. Mutant embryos show defects in muscle fiber formation and innervation,and primary sensory neurons die prematurely. The neuromuscular phenotype in ache mutants is sup-pressed by a homozygous loss-of-function allele of the α-subunit of the nicotinic acetylcholine recep-tor (nAChR), indicating that the impairment of neuromuscular development is mediated byactivation of nAChR in the mutant. Here we provide genetic evidence for non-classical functions ofAChE in vertebrate development.
increased sensitivity to organophosphate inhibitors of AChEbut are viable for months (ref. 11 and A.C., unpublished data).The related esterase butyrylcholinesterase (BuChE) also hydrol-yses acetylcholine, albeit at slower rate12. This and otheresterase-like activities may compensate for the lack of AChEactivity in the knockout mice11.
We report here the identification and phenotypic characteri-zation of a mutation in zebrafish ache. The mutation abolishedAChE enzymatic activity in the embryo completely, caused severeimpairment of motility and was lethal at early larval stages. Aszebrafish do not contain a functional BuChE6,8, this mutant pro-vided a unique opportunity to investigate AChE function dur-ing vertebrate embryogenesis. We showed that AChE activity wasrequired for development and maintenance of the axial muscleapparatus and for survival of primary sensory neurons.
RESULTSA mutation that abolishes AChE activityWe identified chemically induced, recessive mutations in thezebrafish that cause motility defects in embryos and young lar-vae (data not shown). To identify mutations affecting AChE activ-ity, mutant lines were re-screened histochemically for alteredAChE activity13. One mutation, which impaired motility in one-quarter of embryos (26.6%, n = 856), abolished AChE activityboth in primary neurons of the central nervous system (Fig. 1aand b) and in somites (Fig. 1c and d). To confirm this seeminglack of AChE activity in quantitative terms, extracts were pre-pared from mutant and wild-type embryos, and AChE activitywas determined spectrophotometrically. In contrast to proteinextracts from wild-type embryos, extracts from mutant embryos
articles
nature neuroscience • volume 5 no 2 • february 2002 111
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om
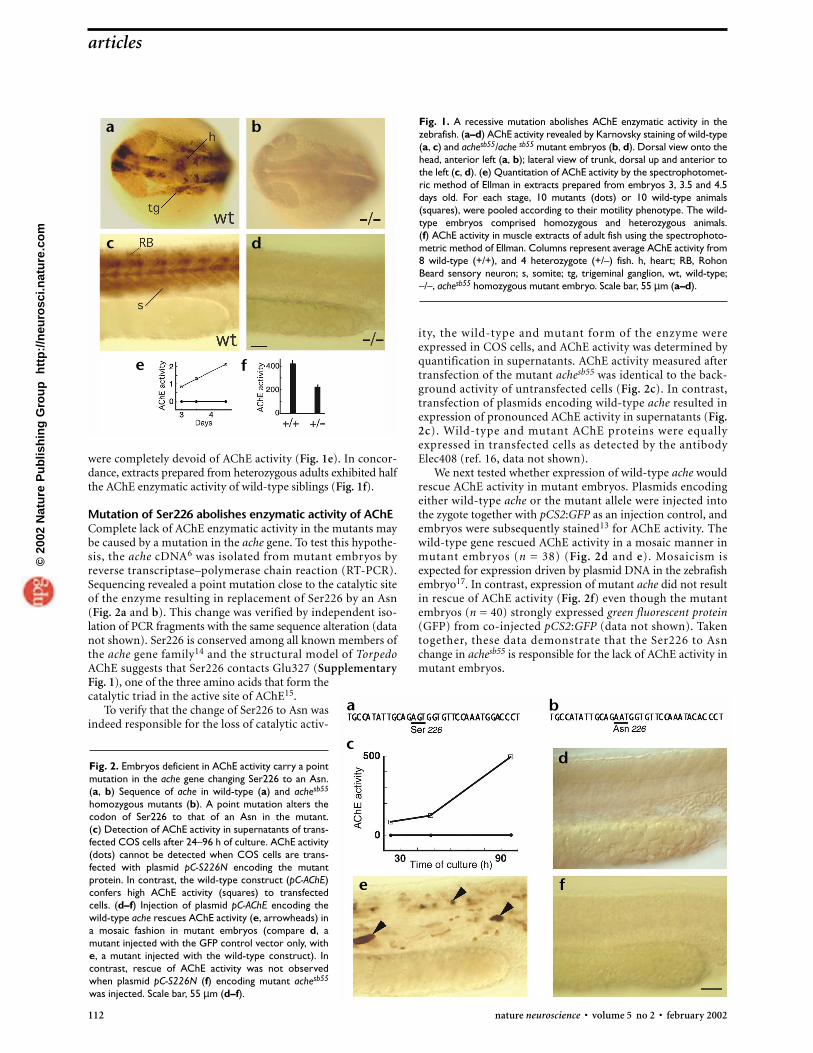
were completely devoid of AChE activity (Fig. 1e). In concor-dance, extracts prepared from heterozygous adults exhibited halfthe AChE enzymatic activity of wild-type siblings (Fig. 1f).
Mutation of Ser226 abolishes enzymatic activity of AChEComplete lack of AChE enzymatic activity in the mutants maybe caused by a mutation in the ache gene. To test this hypothe-sis, the ache cDNA6 was isolated from mutant embryos byreverse transcriptase–polymerase chain reaction (RT-PCR).Sequencing revealed a point mutation close to the catalytic siteof the enzyme resulting in replacement of Ser226 by an Asn(Fig. 2a and b). This change was verified by independent iso-lation of PCR fragments with the same sequence alteration (datanot shown). Ser226 is conserved among all known members ofthe ache gene family14 and the structural model of TorpedoAChE suggests that Ser226 contacts Glu327 (SupplementaryFig. 1), one of the three amino acids that form thecatalytic triad in the active site of AChE15.
To verify that the change of Ser226 to Asn wasindeed responsible for the loss of catalytic activ-
Fig. 2. Embryos deficient in AChE activity carry a pointmutation in the ache gene changing Ser226 to an Asn.(a, b) Sequence of ache in wild-type (a) and achesb55
homozygous mutants (b). A point mutation alters thecodon of Ser226 to that of an Asn in the mutant. (c) Detection of AChE activity in supernatants of trans-fected COS cells after 24–96 h of culture. AChE activity(dots) cannot be detected when COS cells are trans-fected with plasmid pC-S226N encoding the mutantprotein. In contrast, the wild-type construct (pC-AChE)confers high AChE activity (squares) to transfectedcells. (d–f) Injection of plasmid pC-AChE encoding thewild-type ache rescues AChE activity (e, arrowheads) ina mosaic fashion in mutant embryos (compare d, amutant injected with the GFP control vector only, withe, a mutant injected with the wild-type construct). Incontrast, rescue of AChE activity was not observedwhen plasmid pC-S226N (f) encoding mutant achesb55
was injected. Scale bar, 55 µm (d–f).
112 nature neuroscience • volume 5 no 2 • february 2002
ity, the wild-type and mutant form of the enzyme wereexpressed in COS cells, and AChE activity was determined byquantification in supernatants. AChE activity measured aftertransfection of the mutant achesb55 was identical to the back-ground activity of untransfected cells (Fig. 2c). In contrast,transfection of plasmids encoding wild-type ache resulted inexpression of pronounced AChE activity in supernatants (Fig.2c). Wild-type and mutant AChE proteins were equallyexpressed in transfected cells as detected by the antibodyElec408 (ref. 16, data not shown).
We next tested whether expression of wild-type ache wouldrescue AChE activity in mutant embryos. Plasmids encodingeither wild-type ache or the mutant allele were injected intothe zygote together with pCS2:GFP as an injection control, andembryos were subsequently stained13 for AChE activity. Thewild-type gene rescued AChE activity in a mosaic manner inmutant embryos (n = 38) (Fig. 2d and e). Mosaicism is expected for expression driven by plasmid DNA in the zebrafishembryo17. In contrast, expression of mutant ache did not resultin rescue of AChE activity (Fig. 2f) even though the mutantembryos (n = 40) strongly expressed green fluorescent protein(GFP) from co-injected pCS2:GFP (data not shown). Takentogether, these data demonstrate that the Ser226 to Asn change in achesb55 is responsible for the lack of AChE activity inmutant embryos.
articles
Fig. 1. A recessive mutation abolishes AChE enzymatic activity in thezebrafish. (a–d) AChE activity revealed by Karnovsky staining of wild-type(a, c) and achesb55/ache sb55 mutant embryos (b, d). Dorsal view onto thehead, anterior left (a, b); lateral view of trunk, dorsal up and anterior tothe left (c, d). (e) Quantitation of AChE activity by the spectrophotomet-ric method of Ellman in extracts prepared from embryos 3, 3.5 and 4.5days old. For each stage, 10 mutants (dots) or 10 wild-type animals(squares), were pooled according to their motility phenotype. The wild-type embryos comprised homozygous and heterozygous animals. (f) AChE activity in muscle extracts of adult fish using the spectrophoto-metric method of Ellman. Columns represent average AChE activity from8 wild-type (+/+), and 4 heterozygote (+/–) fish. h, heart; RB, RohonBeard sensory neuron; s, somite; tg, trigeminal ganglion, wt, wild-type;–/–, achesb55 homozygous mutant embryo. Scale bar, 55 µm (a–d).
a b
c d
e
a b
cd
fe
f
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om

ache mutants lose motilityHomozygous ache mutant embryos are motile at 24 hours post-fertilization (hpf). The first movements of wild-type zebrafishembryos, which begins around 18 hpf, is characterized by spon-taneous twitching of the body axis. First signs of impaired motil-ity became apparent at 27 hpf; mutants started to exhibitspasmodic movements instead of the spontaneous smooth bend-ing, seen in wild-type embryos. At 48 hpf, spontaneous andtouch-provoked motility were severely impaired in the mutants asshown by slow-motion videomicroscopy (Fig. 3). Wild-typeembryos exposed to a tactile stimulus at the side of the trunkresponded with a strong bend of the body axis away from thesource of stimulation, followed by an extended episode of swim-ming (Fig. 3, left column). In contrast, ache mutant embryosreacted only weakly with a short episode of alternating left-rightbends of the posterior tip of the tail (Fig. 3, right column). More-over, mutants returned rapidly to a motionless state. By threedays of development, mutant embryos were completely para-lyzed. These results demonstrate that AChE activity is requiredfor maintenance of activity of the axial musculature.
AChE activity is required for development of axial muscleIn view of the strong impairment of motility in the 48 hour–oldmutant, differentiation of the axial musculature was investigated.We examined embryos under polarized light to compare the bire-fringence of the axial muscles. In contrast to that of wild-typeembryos (n = 20) at 48 hpf, the axial musculature of ache mutants(n = 20) displayed a reduced birefringence (Fig. 4a and b), sug-gesting that lack of AChE activity affects either the arrangementor the integrity of myofibers.
We thus investigated the organization of myofibers by elec-tron microscopy. In 48 hpf wild-type embryos, long myofibrilswere arranged in parallel and occupied most of the intracellularspace of the muscle cells (Fig. 4c). In contrast, in mutant embryos,patches of short myofibrils were interrupted by extensive regionsof vacuolated sarcoplasm (Fig. 4d). Moreover, the nuclear
envelopes had irregular shapes and the nuclear membranes wereseparated, forming vacuole-like structures (Fig. 4c and d). Thus,lack of AChE activity profoundly disrupted the subcellular orga-nization of muscle fibers in 48 hpf embryos. In contrast, the ultra-tructure of 27-hour myoblasts was indistinguishable from thatof wild-type embryos (Supplementary Fig. 2).
On morphological inspection, somite structure appeared nor-mal in 27 hpf mutant embryos. In situ hybridization with probesdirected against the mRNAs of the muscle-specific mef2A, mef2C,mef2D18 and myoD19 mRNAs did not reveal obvious differencesbetween wild-type and mutant somitic tissue (data not shown).Immunohistochemical staining with the A4.1025 antibody, whichbinds myosin of fast and slow muscle fibers in zebrafishembryos20, was also similar in mutant (n = 49) and wild-type
articles
nature neuroscience • volume 5 no 2 • february 2002 113
Fig. 3. ache mutants have severely impaired motility at 48 hpf. Slowmotion video-microscopy of wild-type sibling (left column) and achesb55/ache sb55 homozygous mutant embryos (ache, right column).Embryos were touched on the side of the trunk. Frames were takenunder stroboscope illumination at the times (t, ms) indicated to theright. wt, wild-type; ache, achesb55 homozygous mutant embryo. Scalebar, 0.3 mm.
a b
c d
e f
g h
Fig. 4. Lack of AChE activity disrupts organization of myofibers. (a, b)Birefringence of wild-type (a) and achesb55 mutant embryos (b) at 48hpf. Embryos are in lateral view at the level of the hindgut extension.Anterior left, dorsal up. (c, d) Electron micrographs of longitudinal sec-tions through axial muscles of the trunk of wild-type (c) and achesb55
mutant (d) at 48 hpf. Myofibers (arrowhead with asterisk) are disruptedin the mutant. The nuclear envelop forms large vacuoles in the mutant(arrowhead). (e, f) Immunofluorescence staining with antibody A4.1025reveals slow and fast fiber myosin in somitic muscles of wild-type (e)and achesb55 mutant (f) embryos at 27 hpf. (g, h) Immunofluorescencestaining with antibody F59 reveals slow fiber myosin in somitic musclesof wild-type (g) and mutant (h) at 27 hpf. Expression of slow musclemyosin is strongly reduced in the mutant. (e–h) Lateral views (dorsalup, anterior left), generated from optical sections taken with a confocalmicroscope through the entire width of the embryo. n, nucleus, wt,wild-type; –/– achesb55 homozygous mutant embryo. Scale bars, 40 µm(a, b), 0.2 µm (c, d), 30 µm (e–h).
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om

vertical lines, Fig. 5), the axons cover anarrower area of the somite in themutant (28.6 ± 4.1 µm, n = 30 somitesfrom 10 ache mutants). This area isapproximately twice larger in wild-typeembryos (65.6 ± 11.9 µm, n = 30somites from 10 wild-type embryos,Supplementary Fig.5, Animations 1 and 2). Moreover, the extending axonsappeared frequently thinner in themutant (Fig. 5a and b).
To assess whether lack of AChE activ-ity affects development of the neuro-muscular junctions, embryos wereincubated with fluorescent α-bungaro-toxin, which specifically binds to nAChR.In 27 hpf embryos, α -bungarotoxinstaining allows visualization of motorendplates28, which are arranged in a cres-cent-shaped band of lines in the centerof each somite (Fig. 5c–f). nAChRs arealso enriched in the vertical myosepta,separating adjacent somites (Fig. 5c–f).α-bungarotoxin staining at the somiteboundary was barely detectable in themutant (Fig. 5d). Moreover, the cluster-ing of nAChRs in the center of the somite(ep) is reduced in ache mutants (n = 36).Instead, diffuse staining throughout thesomite was observed, suggesting thatmutant embryos fail to cluster nAChRsas efficiently as wild-type embryos (Fig. 5c and d, Supplementary Fig. 4,Animations 1 and 2). Motor axons andthe reduced clusters of nAChRs colocal-ized similarly in the mutant as in thewild-type embryo (Fig. 5e and f).
Primary sensory neurons die in ache mutantsRohon Beard (RB) sensory neurons are located at the dorsal aspectof the spinal cord and, like primary motor neurons, express highlevels of AChE6,8. A marked reduction of dendritic extensions ofRB sensory neurons was noticed when mutant embryos 27 hoursold were stained with an anti-acetylated tubulin antibody (Fig.6a and b, arrowheads). In the posterior trunk over the hindgutextension and tail, wild-type embryos (n = 4) had, on average,22.5 ± 4.3 dendrites, which were longer than 120 µm. In contrast,only 8 ± 1.42 dendrites were longer than 120 µm in the same areaof ache mutants (n = 4). Instead, a large number of RB dendrites(on average, 14 ± 4.69 dendrites) were shorter than 60 µm in theposterior trunk and tail of mutants. Wild-type siblings (n = 4)did not show these short dendrites (Supplementary Fig. 6). Thissuggests that neurite outgrowth of RB cells requires ache.
We assayed apoptosis in 27 hpf embryos29. ache mutants (7/7embryos) showed a high number of large TUNEL-positive cells inthe dorsal neural tube (Fig. 6c and d) indicating that RB neuronsdie in the mutant. RB identity of TUNEL-positive cells was con-firmed by re-staining with an RB-specific antibody (Supple-mentary Fig. 8). A fraction of RB neurons of the 27 hpf embryosare destined to death also in wild-type embryos30. At 20 hpf, wild-type and ache mutant embryos showed the same frequency ofdying RB cells (Fig. 6e). However at 24 hpf, 27 hpf and 48 hpf,the number of apoptotic RB cells was consistently higher in ache
Fig. 5. Development of neuromuscular junctions is impaired in achesb55 mutant embryos. (a, b)Primary motor axons of wild-type (a) and achesb55 mutant (b) embryos stained immunofluores-cently with antibody znp1. (c, d) Alexa green–α-bungarotoxin staining of AChRs in wild-type (c)and mutant (d) embryos shown in (a) and (b), respectively. (e, f) Overlay of the pictures (a, c) and(b, d) show the neuromuscular junctions (yellow) of the wild type (e) and the mutant (f), respec-tively. Axons of the 3 primary motor neurons RoP, MiP and CaP appear thinner in the mutant (com-pare a and e with b and f). Clustering of nAChRs at the somite boundaries (sb) and in the motorendplates (ep) is reduced in the mutant (c, d). Also in comparison to wild-type embryos, higher dif-fuse α-bungarotoxin staining was noted throughout the somites in mutant embryos, suggesting amore ubiquitous distribution of the nAChRs (compare c and d). In wild-type embryos, the CaP andMiP axons form a crescent following roughly the center of the somite. As outlined by thin verticallines (compare a and e with b and f), CaP and MiP axons form a shallower arc in the mutant. Largepanels in (a) to (f) are lateral views (anterior left and dorsal up), made of projections of all opticalsections taken with a confocal microscope through the half width of the embryos . The inserts aretransverse views (median left, dorsal up), of the same sections, restricted to the regions spanned bythe vertical lines, after a –90° rotation along the body axis, showing the growth of motor axonsalong the median surface of the somites. (g) The primary motor axon defect of wild-type (red) andache mutant (blue) embryos. Embryos are 27 h old and are shown at the level of the hindgut exten-sion. ep, motor endplates; nmj, neuromuscular junction; N, notochord; NT, neural tube; sb, somiteboundary, wt, wild-type; –/–, achesb55 homozygous mutant embryo. Scale bar, 30 µm.
114 nature neuroscience • volume 5 no 2 • february 2002
embryos (n = 50, Fig. 4e and f). Strikingly, however, immunore-activity with antibody F59, which predominantly recognizes slowmuscle myosin and only weakly the myosin of fast fibers20,21 wasstrongly reduced in the mutants (n = 40, Fig. 4g and h). Thus,slow muscle myosin seems to be selectively impaired in 27 hpfache mutants (Supplementary Fig. 3), whereas fast muscle myosinseems to be expressed normally.
Neuromuscular junction is impaired in ache mutantsAChE has been implicated in the regulation of neurite out-growth22–24. We examined motor neuron projections with theantibody znp1 (ref. 25), which labels the extending axons ofall primary motor neurons. The somitic muscles are initiallyinnervated by three primary cholinergic motor neurons perhemisegment in a stereotypic fashion26. Concomitantly withthe onset of spontaneous locomotion, the ventral projections ofthe primary neuron CaP can be detected in the anterior, most-mature somites, followed slightly later by the axons of RoP andMiP, the two other primary neurons that innervate the middleand dorsal part of each somite, respectively26,27. Axon exten-sion of primary motor neurons was not significantly affectedby the ache mutation (Fig. 5a and b). However, the mutant CaPand MiP axons followed a more straight ventral and dorsal tra-jectory, respectively, when compared to the deep arc of thewild-type axons (Fig. 5a, b and e–g). As a consequence (thin
articles
a b
c d
e f
g
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om
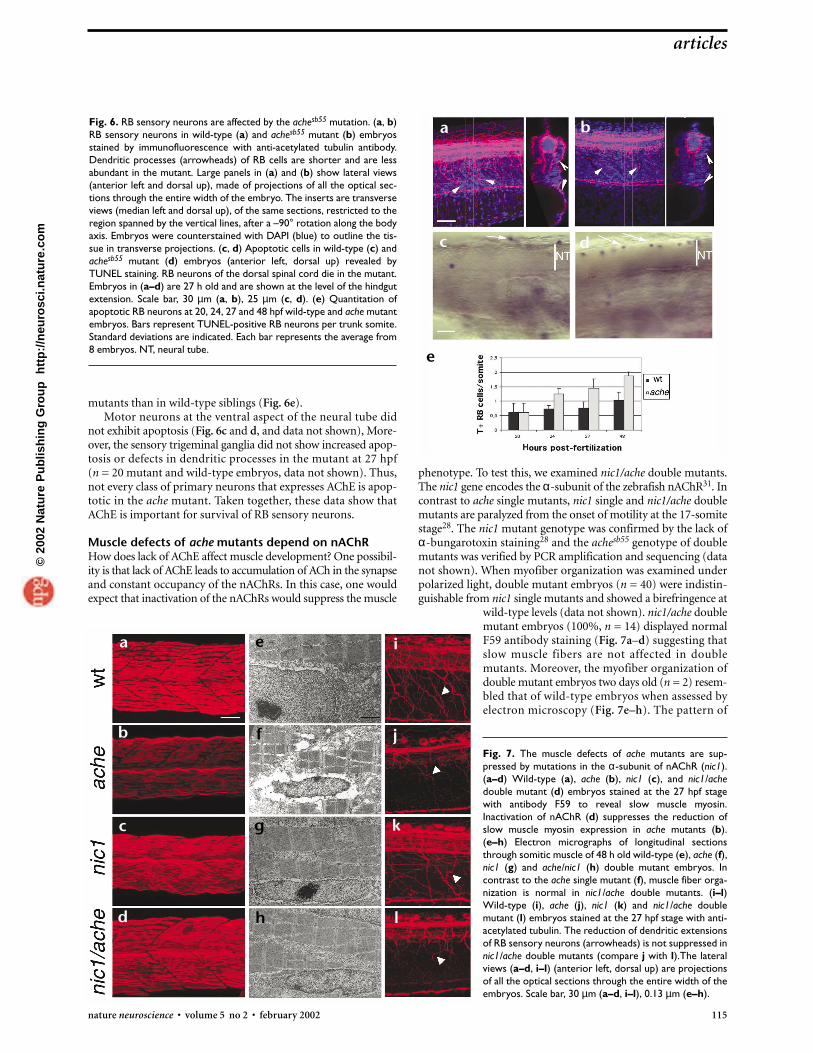
phenotype. To test this, we examined nic1/ache double mutants.The nic1 gene encodes the α-subunit of the zebrafish nAChR31. Incontrast to ache single mutants, nic1 single and nic1/ache doublemutants are paralyzed from the onset of motility at the 17-somitestage28. The nic1 mutant genotype was confirmed by the lack ofα-bungarotoxin staining28 and the achesb55 genotype of doublemutants was verified by PCR amplification and sequencing (datanot shown). When myofiber organization was examined underpolarized light, double mutant embryos (n = 40) were indistin-guishable from nic1 single mutants and showed a birefringence at
wild-type levels (data not shown). nic1/ache doublemutant embryos (100%, n = 14) displayed normalF59 antibody staining (Fig. 7a–d) suggesting thatslow muscle fibers are not affected in doublemutants. Moreover, the myofiber organization ofdouble mutant embryos two days old (n = 2) resem-bled that of wild-type embryos when assessed byelectron microscopy (Fig. 7e–h). The pattern of
Fig. 7. The muscle defects of ache mutants are sup-pressed by mutations in the α-subunit of nAChR (nic1).(a–d) Wild-type (a), ache (b), nic1 (c), and nic1/achedouble mutant (d) embryos stained at the 27 hpf stagewith antibody F59 to reveal slow muscle myosin.Inactivation of nAChR (d) suppresses the reduction ofslow muscle myosin expression in ache mutants (b).(e–h) Electron micrographs of longitudinal sectionsthrough somitic muscle of 48 h old wild-type (e), ache (f),nic1 (g) and ache/nic1 (h) double mutant embryos. Incontrast to the ache single mutant (f), muscle fiber orga-nization is normal in nic1/ache double mutants. (i–l)Wild-type (i), ache (j), nic1 (k) and nic1/ache doublemutant (l) embryos stained at the 27 hpf stage with anti-acetylated tubulin. The reduction of dendritic extensionsof RB sensory neurons (arrowheads) is not suppressed innic1/ache double mutants (compare j with l).The lateralviews (a–d, i–l) (anterior left, dorsal up) are projectionsof all the optical sections through the entire width of theembryos. Scale bar, 30 µm (a–d, i–l), 0.13 µm (e–h).
mutants than in wild-type siblings (Fig. 6e).Motor neurons at the ventral aspect of the neural tube did
not exhibit apoptosis (Fig. 6c and d, and data not shown), More-over, the sensory trigeminal ganglia did not show increased apop-tosis or defects in dendritic processes in the mutant at 27 hpf (n = 20 mutant and wild-type embryos, data not shown). Thus,not every class of primary neurons that expresses AChE is apop-totic in the ache mutant. Taken together, these data show thatAChE is important for survival of RB sensory neurons.
Muscle defects of ache mutants depend on nAChRHow does lack of AChE affect muscle development? One possibil-ity is that lack of AChE leads to accumulation of ACh in the synapseand constant occupancy of the nAChRs. In this case, one wouldexpect that inactivation of the nAChRs would suppress the muscle
Fig. 6. RB sensory neurons are affected by the achesb55 mutation. (a, b)RB sensory neurons in wild-type (a) and achesb55 mutant (b) embryosstained by immunofluorescence with anti-acetylated tubulin antibody.Dendritic processes (arrowheads) of RB cells are shorter and are lessabundant in the mutant. Large panels in (a) and (b) show lateral views(anterior left and dorsal up), made of projections of all the optical sec-tions through the entire width of the embryo. The inserts are transverseviews (median left and dorsal up), of the same sections, restricted to theregion spanned by the vertical lines, after a –90° rotation along the bodyaxis. Embryos were counterstained with DAPI (blue) to outline the tis-sue in transverse projections. (c, d) Apoptotic cells in wild-type (c) andachesb55 mutant (d) embryos (anterior left, dorsal up) revealed byTUNEL staining. RB neurons of the dorsal spinal cord die in the mutant.Embryos in (a–d) are 27 h old and are shown at the level of the hindgutextension. Scale bar, 30 µm (a, b), 25 µm (c, d). (e) Quantitation ofapoptotic RB neurons at 20, 24, 27 and 48 hpf wild-type and ache mutantembryos. Bars represent TUNEL-positive RB neurons per trunk somite.Standard deviations are indicated. Each bar represents the average from8 embryos. NT, neural tube.
articles
nature neuroscience • volume 5 no 2 • february 2002 115
a
b
c
d
e
f
g
h
i
j
k
l
a b
c d
e
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om
innervation by primary motor neurons also appeared normal inthe double mutant (data not shown). These data demonstrate thatlack of nAChRs suppresses the neuromuscular phenotype of achemutant embryos and underscore a downstream involvement ofnAChRs in the development of the muscle phenotype in achemutant.
However, the dendritic phenotype of RB neurons was not sup-pressed in nic1/ache double mutants (n = 7; Fig. 7i–l). This is like-ly a reflection of the fact that nAChR is not expressed in RB cells(data not shown). To test whether the RB dendritic phenotypedepends on electrical activity, we bathed wild-type and mutantembryos in 10 µM tetrodotoxin (TTX). Motility of embryos wascompletely abolished by this concentration of TTX. Wild-typeembryos (n = 47) treated between 16 and 48 hpf recovered motil-ity, indicating that 10 µM TTX did not have a general toxic effecton the embryos. Mutant embryos (100%, n = 5) bathed in TTXfrom 16 hpf onward showed in all cases the dendritic defects ofuntreated, 27 hour–old ache mutants: TTX-treated ache mutantshad a large number of short dendrites (21.2 ± 6.6 dendrites below60 µm), and the number of long dendrites (6 ± 1.7 dendrites above120 µm) was reduced as in ache mutants. Dendritic extensions ofwild-type siblings (100%, n = 15) treated with TTX developed asin untreated wild-type embryos (Supplementary Fig. 6). Takentogether, TTX did not suppress this aspect of the ache mutant phe-notype, suggesting that the defects in dendritic extensions is notthe result of perturbance of electrical activity in the mutant.
achesb55 causes a null phenotypeFunctions of ache in neurite extension and cell adhesion have beenproposed that do not seem to require catalytic activity1. Even thoughthe mutation in achesb55 causes loss of catalytic activity, the mutantprotein bound the conformation-sensitive antibody Elec408 (datanot shown) suggesting that the overall conformation of the mutantprotein is similar to that of the wild-type protein. The mutant pro-tein may have thus retained non-catalytic activities. To assess thenull-mutant phenotype, we used a knock-down approach with
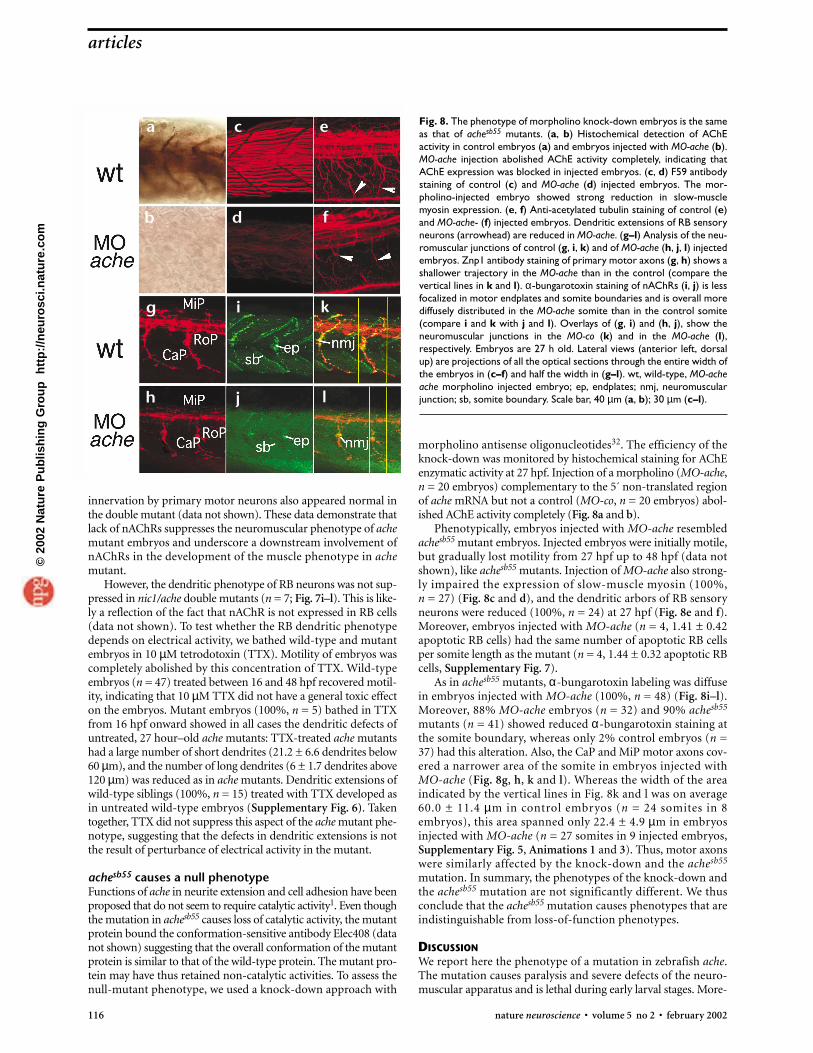
Fig. 8. The phenotype of morpholino knock-down embryos is the sameas that of achesb55 mutants. (a, b) Histochemical detection of AChEactivity in control embryos (a) and embryos injected with MO-ache (b).MO-ache injection abolished AChE activity completely, indicating thatAChE expression was blocked in injected embryos. (c, d) F59 antibodystaining of control (c) and MO-ache (d) injected embryos. The mor-pholino-injected embryo showed strong reduction in slow-musclemyosin expression. (e, f) Anti-acetylated tubulin staining of control (e)and MO-ache- (f) injected embryos. Dendritic extensions of RB sensoryneurons (arrowhead) are reduced in MO-ache. (g–l) Analysis of the neu-romuscular junctions of control (g, i, k) and of MO-ache (h, j, l) injectedembryos. Znp1 antibody staining of primary motor axons (g, h) shows ashallower trajectory in the MO-ache than in the control (compare thevertical lines in k and l). α-bungarotoxin staining of nAChRs (i, j) is lessfocalized in motor endplates and somite boundaries and is overall morediffusely distributed in the MO-ache somite than in the control somite(compare i and k with j and l). Overlays of (g, i) and (h, j), show theneuromuscular junctions in the MO-co (k) and in the MO-ache (l),respectively. Embryos are 27 h old. Lateral views (anterior left, dorsalup) are projections of all the optical sections through the entire width ofthe embryos in (c–f) and half the width in (g–l). wt, wild-type, MO-acheache morpholino injected embryo; ep, endplates; nmj, neuromuscularjunction; sb, somite boundary. Scale bar, 40 µm (a, b); 30 µm (c–l).
morpholino antisense oligonucleotides32. The efficiency of theknock-down was monitored by histochemical staining for AChEenzymatic activity at 27 hpf. Injection of a morpholino (MO-ache,n = 20 embryos) complementary to the 5´ non-translated regionof ache mRNA but not a control (MO-co, n = 20 embryos) abol-ished AChE activity completely (Fig. 8a and b).
Phenotypically, embryos injected with MO-ache resembledachesb55 mutant embryos. Injected embryos were initially motile,but gradually lost motility from 27 hpf up to 48 hpf (data notshown), like achesb55 mutants. Injection of MO-ache also strong-ly impaired the expression of slow-muscle myosin (100%, n = 27) (Fig. 8c and d), and the dendritic arbors of RB sensoryneurons were reduced (100%, n = 24) at 27 hpf (Fig. 8e and f).Moreover, embryos injected with MO-ache (n = 4, 1.41 ± 0.42apoptotic RB cells) had the same number of apoptotic RB cellsper somite length as the mutant (n = 4, 1.44 ± 0.32 apoptotic RBcells, Supplementary Fig. 7).
As in achesb55 mutants, α-bungarotoxin labeling was diffusein embryos injected with MO-ache (100%, n = 48) (Fig. 8i–l).Moreover, 88% MO-ache embryos (n = 32) and 90% achesb55
mutants (n = 41) showed reduced α-bungarotoxin staining atthe somite boundary, whereas only 2% control embryos (n =37) had this alteration. Also, the CaP and MiP motor axons cov-ered a narrower area of the somite in embryos injected withMO-ache (Fig. 8g, h, k and l). Whereas the width of the areaindicated by the vertical lines in Fig. 8k and l was on average60.0 ± 11.4 µm in control embryos (n = 24 somites in 8embryos), this area spanned only 22.4 ± 4.9 µm in embryosinjected with MO-ache (n = 27 somites in 9 injected embryos,Supplementary Fig. 5, Animations 1 and 3). Thus, motor axonswere similarly affected by the knock-down and the achesb55
mutation. In summary, the phenotypes of the knock-down andthe achesb55 mutation are not significantly different. We thusconclude that the achesb55 mutation causes phenotypes that areindistinguishable from loss-of-function phenotypes.
DISCUSSIONWe report here the phenotype of a mutation in zebrafish ache.The mutation causes paralysis and severe defects of the neuro-muscular apparatus and is lethal during early larval stages. More-
116 nature neuroscience • volume 5 no 2 • february 2002
articles
a
b
c
d
e
f
g
h
i
j
k
l
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om

over, mutants show reduction in dendritic extensions and sur-vival of primary sensory neurons. Our data demonstrate crucialfunctions of AChE activity during early vertebrate development.
The identified point mutation in ache causes a replacement ofSer226 by an Asn in the mutant protein. This replacement abol-ishes enzymatic activity of AChE. The model of AChE structureand catalytic mechanism does not predict a direct participationof Ser226 in the catalytic triad of the active site of the enzyme15.Ser226 is, however, conserved among all AChE proteins identi-fied to date14 and the structural model suggests that Ser226 canestablish a hydrogen bond with Glu327, one of the three aminoacids that form the catalytic triad (Supplementary Fig. 1).Replacement of Ser226 by a bulky Asn is likely to disrupt thisinteraction and may disturb the conformation of the active site.
Targeted mutation of ache in mouse embryos resulted in a verymild phenotype. Homozygous mutant mice were motile and sur-vived for months after birth11 (A.C., unpublished data). Howev-er, the animals developed spasms with increasing age, were smallerthan their littermates and showed hypersensitivity to organophos-phate poisoning. This phenotype is in striking contrast to thedefects observed in the zebrafish ache mutant reported here. Itwas proposed that other esterases such as BuChE compensate forthe loss of AChE activity11. Zebrafish were shown to lack a buchegene6. The complete absence of ACh hydrolyzing activity inhomozygous ache mutant embryos demonstrates that AChE isthe only ACh-cleaving esterase in the zebrafish embryo. The moresevere mutant phenotype in the zebrafish can thus be explainedby the total lack of ACh hydrolyzing activity. The zebrafish achesb55
mutant represents a unique vertebrate model, with which to studythe function of AChE during development. This property mayalso be useful to study off-target effects of AChE inhibitors.
Presence of AChE in non-cholinergic neurons is well docu-mented. This is notably the case of sensory neurons of the dorsalroot ganglia in mammals and birds and was the primary obser-vation leading to the suggestion of a non-cholinergic role ofAChE33. Previously, experiments with active site inhibitors and/ormutant AChE variants, which carry point mutations in the activesite, pointed to functions of AChE other than hydrolysis of AChE.Stimulation of neurite outgrowth by AChE has repeatedly beenreported in studies using different in vitro systems1,10. We didnot detect a significant effect on the rate of primary motor axonextension in achesb55 mutant embryos. Primary motor axonsshowed, however, abnormal navigation. In zebrafish, AChE isdetected in RB sensory neurons6. In contrast to motor axons,dendrites of RB neurons are significantly shorter in the mutant.Moreover, RB cells die at an higher rate in the mutant than inwild-type embryos. There were no phenotypic differencesbetween the achesb55 mutants and MO-injected embryos, whichare impaired in AChE translation, suggesting that the defectsobserved represent the null phenotype. In contrast to the neu-romuscular phenotype, the sensory defects are not suppressedby the nic1 mutation, but nAChR is not expressed in RB sensoryneurons (M.B., unpublished data). The inability of the Na+ chan-nel inhibitor TTX to suppress the RB phenotype of achesb55
mutants suggests, however, that the defects are not caused byelectrical overstimulation. Dependence of neuronal survival onache is highly selective as other neurons in the CNS that alsoexpress ache at very high levels6 do not die in the mutant.
The routing of primary motor neurons is altered in the achesb55
mutant. However, axon growth and spinal cord exit occur correct-ly. It seems that defects of axon trajectories concern late phases oftargeting. Among numerous factors, neurotransmitter concentra-tion influences these latter phases (for review, see ref. 34). Mis-
routing of primary neurons may thus be due to lack of ACh hydrol-ysis in the mutant. Accumulation of ACh in the mutant may alsoexplain why AChRs are much less focalized in the achesb55 mutantmuscles. In zebrafish embryos, clustering of nAChR is tightly cor-related with the emerging motor axons and does not occur in theabsence of innervation35. Current knowledge of formation of theneuromuscular junctions in mice indicates that nAChRs are clus-tered in a broad band in the center of muscle fibers before the nerveencounters the muscles36. After the nerve has contacted the mus-cle, clusters of nAChRs enlarge at synapses and extrasynapticnAChRs are eliminated. The mechanism of elimination of extrasy-naptic AChRs is unclear and could include nerve-evoked electricalactivity and short-range inhibitory signals. The more diffuse dis-tribution of AChRs in the achesb55 mutant depends on a function-al nAChR and may be due to persistent activation of theextrasynaptic nAChRs by accumulated ACh.
Homozygous mutant embryos are initially motile. Lack ofAChE is likely to lead to a progressive accumulation of ACh in theneuromuscular junction, which will, with increasing and focalizedmuscle stimulation during further development, cause the spasmsin embryos older than 27 hpf and the myopathy observed at laterstages. Necrosis was also noted in mammals after acute exposure toAChE inhibitors37–39. Double inhibitor experiments suggested thatincreased calcium influx and activation of AChR caused the necrot-ic symptoms38. Moreover, mice with targeted mutations of collagen Q (ColQ), which is required to assemble and cluster theasymmetric forms of AChE and BuChE in the neuromuscularjunction, had severely reduced ACh cleaving activity at the neuro-muscular junction and showed symptoms of necrosis in approxi-mately two-thirds of subsynaptic sites at postnatal day 20 (ref. 40).
Whereas maintenance of muscle cell integrity clearly dependson AChE activity in older embryos and larvae, muscle fiber orga-nization at the ultrastructural level seems normal in 27 hpfembryos. There are, however, muscle defects already in theseyoung embryos, such as impaired expression of slow musclemyosin. Electrical activity has well-established morphogeneticfunctions in both neurons and muscle, where it refines andmaintains patterns of innervation, thus assuring survival of neu-rons and expression of muscle fiber-type in adult muscle (forreview, see ref. 41). The selective impairment of slow musclemyosin expression is intriguing, as the frequency of motor nerveactivity modulates expression of fast and slow muscle specifictraits in the adult41. In the achesb55 mutant, impaired slow mus-cle myosin expression depends on nAChR, in line with thenotion that stimulation of the muscle by ACh is involved indevelopment of the phenotype. In summary, AChE activity isrequired for the correct development of the muscle apparatusand for protection against damage in subsequent stages.
METHODSFish stocks. Fish were bred and raised as previously described42. Chem-ical mutagenesis with ethyl-nitroso-urea has been previouslydescribed43,44. Animal care and experimentation were done in compli-ance with French and European law (Authorization B67-218-5 from23.02.1999).
Cloning of achesb55 and genotyping. RT-PCR and all other cloning wasdone following standard procedures45. For genotyping the achesb55 mutantembryos, genomic DNA was prepared followed by PCR amplification ofa 300 bp fragment, surrounding codon Ser226 and sequencing.
Microinjection. Injections were performed as described46. Morpholinos(Genetools LLC, Philomath, Oregon), were injected at 1 mM32. Thesequence of MO-ache is CTGAGGTCTTCATGGCTTCTTTTCA and ofMO-co is CCATATCGGAGTATACGATCTCCAT.
articles
nature neuroscience • volume 5 no 2 • february 2002 117
©20
02 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://n
euro
sci.n
atu
re.c
om

118 nature neuroscience • volume 5 no 2 • february 2002
In situ analysis and microscopy. In situ hybridization18,19,47, TUNEL (TdT-mediated dUTP nick-end labeling)29 and immunohistochemistry25,48,49
was described. nAChR was stained with Alexa green–conjugated α-bun-garotoxin42. AChE enzymatic activity was revealed as described 13,50.
Optical sections were taken with a Leica TCS 4D confocal microscope.Stacks of images were reconstructed in three dimensions to generate lat-eral and transverse projections with programs tcstk and timt (unpub-lished data).
Note: Further details on the experimental design and supplementary data are
available on the Nature Neuroscience web site (http://neurosci.nature.com/web_
specials).
AcknowledgementsWe thank V. Korzh, H.G. Frohnhöfer, B. Ticho, J. Grassi, E. Krejci, H. Blau,
B. Trevarrow, M. Westerfield and the Developmental Studies Hybridoma Bank
(University of Iowa) for mutants and materials. We thank A. Gansmüller and
M. Digelmann for help with electron microscopy, and P. Blader, F. Müller and
M. König for critically reading the manuscript. We also thank A. Karmin and
O. Nkundwa, for care of the fish and to D. Hentsch, M. Bogelin for help with the
confocal microscope. We thank N. Fischer for technical assistance and INRA,
INSERM, CNRS, HUS, AFM, ARC and ACI for support.
Competing interests statement. The authors declare that they have no competing financial interests.
RECEIVED 17 OCTOBER; ACCEPTED 29 NOVEMBER 2001
1. Soreq, H. & Seidman, S. Acetylcholinesterase—new roles for an old actor. Nat.Rev. Neurosci. 2, 294–302 (2001).
2. Massoulie, J., Pezzementi, L., Bon, S., Krejci, E. & Vallette, F. M. Molecular andcellular biology of cholinesterases. Prog. Neurobiol. 41, 31–91 (1993).
3. Millard, C. B. & Broomfield, C. A. Anticholinesterases: medical applications ofneurochemical principles. J. Neurochem. 64, 1909–1918 (1995).
4. Parnetti, L. Clinical pharmacokinetics of drugs for Alzheimer’s disease. Clin.Pharmacokinet. 29, 110–129 (1995).
5. Linton, D. M. & Philcox, D. Myasthenia gravis. Dis. Mon. 36, 593–637 (1990).6. Bertrand, C. et al. Zebrafish acetylcholinesterase is encoded by a single gene
localized on linkage group 7. Gene structure and polymorphism; molecularforms and expression pattern during development. J. Biol. Chem. 276,464–474 (2001).
7. Hanneman, E., Trevarrow, B., Metcalfe, W. K., Kimmel, C. B. & Westerfield,M. Segmental pattern of development of the hindbrain and spinal cord of thezebrafish embryo. Development 103, 49–58 (1988).
8. Hanneman, E. & Westerfield, M. Early expression of acetylcholinesteraseactivity in functionally distinct neurons of the zebrafish. J. Comp. Neurol. 284,350–361 (1989).
9. Layer, P. G. Cholinesterases preceding major tracts in vertebrate neurogenesis.Bioessays 12, 415–420 (1990).
10. Layer, P. G. & Willbold, E. Novel functions of cholinesterases in development,physiology and disease. Prog. Histochem. Cytochem. 29, 1–94 (1995).
11. Xie, W. et al. Postnatal developmental delay and supersensitivity toorganophosphate in gene-targeted mice lacking acetylcholinesterase. J.Pharmacol. Exp. Ther. 293, 896–902 (2000).
12. Radic, Z., Pickering, N. A., Vellom, D. C., Camp, S. & Taylor, P. Three distinctdomains in the cholinesterase molecule confer selectivity for acetyl- andbutyrylcholinesterase inhibitors. Biochemistry 32, 12074–12084 (1993).
13. Karnovsky, M. J. & Roots, L. A direct colouring thiocholine method forcholinesterases. J. Histochem. Cytochem. 12, 219–221 (1964).
14. Cousin, X., Hotelier, T., Giles, K., Toutant, J. P. & Chatonnet, A. aCHEdb: thedatabase system for ESTHER, the α/β fold family of proteins and theCholinesterase gene server. Nucleic Acids Res. 26, 226–228 (1998).
15. Sussman, J. L. et al. Atomic structure of acetylcholinesterase from Torpedocalifornica: a prototypic acetylcholine-binding protein. Science 253, 872–879(1991).
16. Simon, S., Le Goff, A., Frobert, Y., Grassi, J. & Massoulie, J. The binding sites ofinhibitory monoclonal antibodies on acetylcholinesterase. Identification of anovel regulatory site at the putative “back door.” J. Biol. Chem. 274,27740–27746 (1999).
17. Westerfield, M., Wegener, J., Jegalian, B. G., DeRobertis, E. M. & Püschel, A.W. Specific activation of mammalian Hox promoters in mosaic transgeniczebrafish. Genes Dev. 6, 591–598 (1992).
18. Ticho, B. S., Stainier, D. Y., Fishman, M. C. & Breitbart, R. E. Three zebrafishMEF2 genes delineate somitic and cardiac muscle development in wild-typeand mutant embryos. Mech. Dev. 59, 205–218 (1996).
19. Weinberg, E. S. et al. Developmental regulation of zebrafish MyoD in wild-type, no tail and spadetail embryos. Development 122, 271–280 (1996).
20. Devoto, S. H., Melancon, E., Eisen, J. S. & Westerfield, M. Identification ofseparate slow and fast muscle precursor cells in vivo, prior to somiteformation. Development 122, 3371–3380 (1996).
21. Miller, J. B., Teal, S. B. & Stockdale, F. E. Evolutionarily conserved sequences ofstriated muscle myosin heavy chain isoforms. Epitope mapping by cDNAexpression. J. Biol. Chem. 264, 13122–13130 (1989).
22. Sternfeld, M. et al. Acetylcholinesterase enhances neurite growth and synapsedevelopment through alternative contributions of its hydrolytic capacity, coreprotein, and variable C termini. J. Neurosci. 18, 1240–1249 (1998).
23. Koenigsberger, C., Chiappa, S. & Brimijoin, S. Neurite differentiation ismodulated in neuroblastoma cells engineered for altered acetylcholinesteraseexpression. J. Neurochem. 69, 1389–1397 (1997).
24. Coronas, V., Durand, M., Chabot, J. G., Jourdan, F. & Quirion, R.Acetylcholine induces neuritic outgrowth in rat primary olfactory bulbcultures. Neuroscience 98, 213–219 (2000).
25. Trevarrow, B., Marks, D. L. & Kimmel, C. B. Organization of hindbrainsegments in the zebrafish embryo. Neuron 4, 669–679 (1990).
26. Myers, P. Z., Eisen, J. S. & Westerfield, M. Development and axonal outgrowthof identified motoneurons in the zebrafish. J. Neurosci. 6, 2278–2289 (1986).
27. Eisen, J. S., Myers, P. Z. & Westerfield, M. Pathway selection by growth cones ofidentified motoneurones in live zebra fish embryos. Nature 320, 269–271(1986).
28. Westerfield, M., Liu, D. W., Kimmel, C. B. & Walker, C. Pathfinding andsynapse formation in a zebrafish mutant lacking functional acetylcholinereceptor. Neuron 4, 867–874 (1990).
29. Hensey, C. & Gautier, J. A developmental timer that regulates apoptosis at theonset of gastrulation. Mech. Dev. 69, 183–195 (1997).
30. Williams, J. A. et al. Programmed cell death in zebrafish rohon beard neuronsis influenced by TrkC1/NT-3 signaling. Dev. Biol. 226, 220–230 (2000).
31. Sepich, D. S., Wegner, J., O’Shea, S. & Westerfield, M. An altered introninhibits synthesis of the acetylcholine receptor α-subunit in the paralyzedzebrafish mutant nic1. Genetics 148, 361–372 (1998).
32. Nasevicius, A. & Ekker, S. C. Effective targeted gene ‘knockdown’ in zebrafish.Nat. Genet. 26, 216–220 (2000).
33. Silver, A. The Biology of Cholinesterases (North Holland, Amsterdam, 1974).34. Tessier-Lavigne, M. Axon guidance by diffusible repellants and attractants.
Curr. Opin. Genet. Dev. 4, 596–601 (1994).35. Liu, D. W. & Westerfield, M. Clustering of muscle acetylcholine receptors
requires motoneurons in live embryos, but not in cell culture. J. Neurosci. 12,1859–1866 (1992).
36. Lin, W. et al. Distinct roles of nerve and muscle in postsynaptic differentiationof the neuromuscular synapse. Nature 410, 1057–1064 (2001).
37. Laskowski, M. B., Olson, W. H. & Dettbarn, W. D. Initial ultrastructuralabnormalities at the motor end plate produced by a cholinesterase inhibitor.Exp. Neurol. 57, 13–33 (1977).
38. Leonard, J. P. & Salpeter, M. M. Agonist-induced myopathy at theneuromuscular junction is mediated by calcium. J. Cell Biol. 82, 811–819(1979).
39. Wecker, L., Mrak, R. E. & Dettbarn, W. D. Evidence of necrosis in humanintercostal muscle following inhalation of an organophosphate insecticide.Fundam. Appl. Toxicol. 6, 172–174 (1986).
40. Feng, G. et al. Genetic analysis of collagen Q: roles in acetylcholinesterase andbutyrylcholinesterase assembly and in synaptic structure and function. J. CellBiol. 144, 1349–1360 (1999).
41. Buonanno, A. & Fields, R. D. Gene regulation by patterned electrical activityduring neural and skeletal muscle development. Curr. Opin. Neurobiol. 9,110–120 (1999).
42. Westerfield, M. The Zebra Fish Book (University of Oregon Press, Eugene,1993).
43. Solnica-Krezel, L., Schier, A. & Driever, W. Efficient recovery of ENU-inducedmutations from the zebrafish germline. Genetics 136, 1401–1420 (1994).
44. Mullins, M. C., Hammerschmidt, M., Haffter, P. & Nüsslein-Volhard, C.Large-scale mutagenesis in the zebrafish: in search of genes controllingdevelopment in a vertebrate. Curr. Biol. 4, 189–202 (1994).
45. Sambrock, J. & Russel, D. W. Molecular Cloning: A Laboratory Manual (ColdSpring Harbor Press, New York, 2001).
46. Müller, F. et al. Intronic enhancers control expression of zebrafish sonichedgehog in floor plate and notochord. Development 126, 2103–2116 (1999).
47. Oxtoby, E. & Jowett, T. Cloning of the zebrafish krox-20 gene (krx-20) and itsexpression during hindbrain development. Nucl. Acids Res. 21, 1087–1095(1993).
48. Crow, M. T. & Stockdale, F. E. Myosin expression and specialization among theearliest muscle fibers of the developing avian limb. Dev. Biol. 113, 238–254(1986).
49. Piperno, G. & Fuller, M. T. Monoclonal antibodies specific for an acetylatedform of α-tubulin recognize the antigen in cilia and flagella from a variety oforganisms. J. Cell Biol. 101, 2085–2094 (1985).
50. Ellman, G. L., Courtney, K. D., Andres, V. & Featherstone, R. M. A new andrapid colorimetric dtermination of acetylcholinesterase activity. Biochem.Pharmacol. 7, 88–95 (1961).
articles©
2002
Nat
ure
Pu
blis
hin
g G
rou
p
htt
p:/
/neu
rosc
i.nat
ure
.co
m