a small-molecule inhibitor of type iii secretion inhibits different stages of the infectious cycle...
TRANSCRIPT

A small-molecule inhibitor of type III secretioninhibits different stages of the infectious cycleof Chlamydia trachomatisSandra Muschiol*†, Leslie Bailey‡, Åsa Gylfe‡, Charlotta Sundin§, Kjell Hultenby¶, Sven Bergstrom‡, Mikael Elofsson�,Hans Wolf-Watz‡, Staffan Normark*†, and Birgitta Henriques-Normark*†**
*Swedish Institute for Infectious Disease Control, SE-171 82 Solna, Sweden; †Department of Microbiology, Tumor Biology, and Cell Biology, KarolinskaInstitutet, SE-171 77 Stockholm, Sweden; ‡Department of Molecular Biology, Umeå University, SE-901 87 Umeå, Sweden; §Innate Pharmaceuticals, UmestanForetagspark, SE-903 47 Umeå, Sweden; ¶Department of Laboratory Medicine, Karolinska Institutet, SE-141 86 Huddinge, Sweden; and �Organic Chemistry,Department of Chemistry, Umeå University, SE-901 87 Umeå, Sweden
Communicated by Rino Rappuoli, Chiron Corporation, Siena, Italy, July 27, 2006 (received for review July 5, 2006)
The intracellular pathogen Chlamydia trachomatis possesses a typeIII secretion (TTS) system believed to deliver a series of effectorproteins into the inclusion membrane (Inc-proteins) as well as intothe host cytosol with perceived consequences for the pathogenic-ity of this common venereal pathogen. Recently, small moleculeswere shown to block the TTS system of Yersinia pseudotubercu-losis. Here, we show that one of these compounds, INP0400,inhibits intracellular replication and infectivity of C. trachomatis atmicromolar concentrations resulting in small inclusion bodies fre-quently containing only one or a few reticulate bodies (RBs).INP0400, at high concentration, given at the time of infection,partially blocked entry of elementary bodies into host cells. Earlytreatment inhibited the localization of the mammalian protein14-3-3� to the inclusions, indicative of absence of the early inducedTTS effector IncG from the inclusion membrane. Treatment withINP0400 during chlamydial mid-cycle prevented secretion of theTTS effector IncA and homotypic vesicular fusions mediated by thisprotein. INP0400 given during the late phase resulted in thedetachment of RBs from the inclusion membrane concomitant withan inhibition of RB to elementary body conversion causing amarked decrease in infectivity.
type III secretion system
Screening for novel antimicrobials has traditionally been done byscoring for growth inhibition in vitro on artificial media. This
approach has over the years led to a limited number of antimicrobialclasses. To combat increasing antibacterial resistance, developmenthas focused on modifying compounds within the existing classesrather than identifying small molecules with a completely novelmode of action. Targeting growth and virulence under in vivo-likeconditions will likely identify completely new sets of molecules, asrecently shown for Vibrio cholerae where a small-molecule inhibitorof the transcriptional activator ToxT, virstatin, prevented both toxinand pili expression, protecting infant mice from colonization (1).Small molecules belonging to a class of acylated hydrazones ofsalicyl aldehydes were recently identified (2, 3) that inhibited typeIII secretion (TTS)-dependent delivery of Yersinia pseudotubercu-losis Yop effectors into target cells without inducing a measurabletoxicity on the host cells. Neither virstatin nor the Yersinia TTSinhibitors affected bacterial growth in vitro. It is likely that also invivo, the bacterial multiplication rates are not affected by thesenovel classes of antivirulence drugs but only their ability to causepathophysiology and�or to evade the host innate immune response.
Chlamydia trachomatis is the most common sexually transmittedbacterial disease and the leading cause of preventable blindnessworldwide (4). Chlamydia are Gram-negative, obligate, intracellu-lar bacteria that share a unique biphasic developmental cycle (5).Infection is initiated by attachment of elementary bodies (EBs) toeukaryotic host cells. A few hours after internalization, infectiousbut metabolically inactive EBs differentiate into reticulate bodies
(RBs), the metabolically active form of Chlamydia. RBs will thenreplicate within a cytoplasmic vacuole termed inclusion before theyredifferentiate to EBs. Upon EB release from the infected host cell,a new round of infection can begin. Throughout their entire time inthe host cell, Chlamydia remain within the confinements of theparasitophorous vacuole, which very early during infection exits theendocytic pathway and becomes instead fusiogenic with a subset ofexocytic vesicles originating from the ER�Golgi network and lateendosomes (6, 7).
Like many other Gram-negative pathogenic bacteria, Chlamydiapossess a TTS system that enables them to deliver effector proteinsinto the host cell (8, 9). C. trachomatis EBs rapidly induces its ownentry into host cells, through an internalization process believed tobe promoted by the TTS effector protein TARP (10). During theearly phase of infection, C. trachomatis induces and secretes a set ofputative type III effectors (Inc-proteins) (11), of which IncG hasbeen shown to specifically interact with the mammalian signaltransducer protein 14-3-3� at the inclusion membrane (12). Littleis known about the function of these early-phase proteins that aredisplayed at the interface of intravacuolar Chlamydia and host cell.IncA, a protein induced and secreted during chlamydial mid-cycle,has been demonstrated to be involved in homotypic fusion betweenChlamydia-containing vesicles at high multiplicities of infection(13). In addition to the family of Inc-proteins, other chlamydialproteins secreted into the inclusion membrane have been identified(14, 15). Moreover, proteins targeted into the host-cell cytosol havebeen described (16–18).
In the absence of a genetic system to modify Chlamydia, thefunction of most of the Inc proteins still remains unknown, and ithas not been possible to elucidate whether Inc-proteins or othertype III effector proteins secreted into the inclusion membrane orthe host cytosol are required for Chlamydia to undergo a normalinfectious cycle.
In this article, we demonstrate that INP0400, a small moleculeidentified in a TTS inhibitor screen of Y. pseudotuberculosis, withoutaffecting in vitro multiplication, causes a dose- and growth phase-dependant inhibition of C. trachomatis RB multiplication. Drugtreatment at different stages in the chlamydial developmental cycle
Author contributions: S.M., S.N., and B.H.-N. designed research; S.M., L.B., Å.G., and K.H.performed research; C.S. and M.E. contributed new reagents�analytic tools; S.M., S.B.,H.W.-W., S.N., and B.H.-N. analyzed data; and S.M., S.N., and B.H.-N. wrote the paper.
Conflict of interest statement: C.S. is employed by and M.E., H.W.-W., and S.N. areassociated with Innate Pharmaceuticals.
Freely available online through the PNAS open access option.
Abbreviations: EB, elementary body; MOI, multiplicity of infection; p.i., postinfection; RB,reticulate body; SIB, small inclusion bodies; TTS, type III secretion.
**To whom correspondence should be addressed at: Department of Bacteriology, SwedishInstitute for Infectious Disease Control, Nobels vag 18, SE-171 82 Solna, Sweden. E-mail:[email protected].
© 2006 by The National Academy of Sciences of the USA
14566–14571 � PNAS � September 26, 2006 � vol. 103 � no. 39 www.pnas.org�cgi�doi�10.1073�pnas.0606412103

reveals a partial block of entry, an inhibition of the translocation ofthe TTS effectors IncG and IncA during the early and middlephase, respectively, and a bacterial detachment from the inclusionmembrane during the late stage concomitant with an inhibition ofterminal differentiation from RBs to infectious EBs.
ResultsThe Small-Molecule TTS Inhibitor INP0400 Given at the Time ofInfection Inhibits RB Multiplication Resulting in Small Inclusion Bodies(SIB). C. trachomatis serovar L2 was used to infect McCoy cells at alow multiplicity of infection (MOI 0.5–1.0). At the time of infection,cells were treated with INP0400, a compound isolated to specificallyblock TTS of Y. pseudotuberculosis. INP0400 (Fig. 1B) was found toinhibit bacterial replication as shown by immunofluorescence mi-croscopy (Fig. 1A). Inclusion size was affected at concentrations aslow as 10 �M, without a detectable effect on the number of infectedcells. At a concentration of 20 �M, inclusions were barely visible(Fig. 1A). However, the number of infected cells was only 5–10%lower as compared with untreated control (data not shown).Electron microscopy, after 30 h of infection in the presence ofINP0400, showed a dose-dependant reduction in the size of theinclusion bodies and a concomitant reduction in the number ofintracellular bacteria (Fig. 1C). At 20 �M or higher of INP0400, theSIB contained only one or few RBs (Fig. 1C).
Reinfection in the absence of drug after 48 h treatment withINP0400 revealed a dose-dependant reduction in the number ofinclusions (Fig. 2). A 20-fold reduction was seen with 20 �M andan impressive 3-log reduction was observed when using 25 �M (Fig.2). Cytotoxicity assays performed on the McCoy cells were negativefor INP0400 at concentrations �70 �M (data not shown).
Partial Inhibition of EB Entry. C. trachomatis EBs have been shownto be equipped with a TTS apparatus able to secrete one protein,TARP, thought to interact with the actin cytoskeleton, therebyactivating the host cell to internalize the organism (10). To seewhether INP0400 had an effect on uptake, McCoy cells wereinfected by Chlamydia at a MOI of 0.5 by centrifugation of bacteriaonto the cells or by a temperature shift. INP0400 at differentconcentrations was given at the time of infection and removed after3 h by washing (Fig. 3). Both methods of infection resulted in adose-dependant decrease in the number of cells containing detect-able inclusion bodies after 40 h growth in the absence of the drug.
At the highest concentration used (40 �M), inhibition of entry was�40%.
Early Drug Treatment Prevents the IncG-Interacting Host Protein14-3-3� from Decorating Chlamydia-Containing Inclusion Bodies. TheTTS effector protein IncG is synthesized already 2 h postinfection(p.i.) (19). It localizes to the inclusion membrane and there interactswith the mammalian protein 14-3-3� (12). To see whether INP0400inhibits IncG translocation, we performed immunofluorescencewith antibodies to 14-3-3� 24 h after infection with Chlamydia andtreatment with INP0400 at 50 �M (Fig. 4). Whereas untreatedinfected cells accumulated most of their cellular content of 14-3-3�to the inclusion membrane, there was no detectable decoration ofSIB, leaving 14-3-3� evenly distributed in the cytosol as in nonin-fected cells (Fig. 4).
INP0400 Treatment During the Chlamydial Mid-Cycle Blocks IncATranslocation and Homotypic Fusion Without Affecting Inclusion Mem-brane Targeting of the Early-Phase Effector Protein IncG. A numberof putative chlamydial TTS effector proteins have been identifiedby screenings in heterologous secretion systems like that of Shigellaflexneri or Salmonella typhimurium (16, 20). However, the functionof most of these effector proteins has still to be assigned. ClinicalC. trachomatis isolates, mutated in the gene encoding the putativeTTS effector protein IncA, have been shown to give rise to multipleinclusions after infection with a high MOI in contrast to the singleinclusions typically seen in IncA-producing wild-type isolates (21,22). However, intracellular replication of C. trachomatis was notinhibited in the absence of homotypic fusions. IncA transcript canbe detected 10–12 h p.i. and continue to be produced during the midand late phase of the infectious cycle (13). We therefore infectedMcCoy cells with C. trachomatis at a MOI of 5 and treated the cellswith INP0400 at 50 �M after 8 h of infection. As seen in Fig. 5, thisconcentration of drug inhibited IncA-mediated homotypic fusion,resulting in a number of distinctly separated SIB, each decorated by14-3-3� as evidence for a successful translocation of IncG, anearly-phase TTS effector. In accordance with this finding, we wereunable to detect IncA in the inclusion membrane of SIB whenfollowing the same treatment protocol (Fig. 6). Intermediateconcentrations of INP0400 were also tested, showing that concen-trations around 50 �M INP0400 are required to observe thesephenotypes (data not shown).
Fig. 1. The small molecule INP0400 inhibits RB multi-plication. (A) Dose-dependent growth inhibition of C.trachomatis serovar L2 by INP0400. McCoy cells wereinfected with C. trachomatis serovar L2 (MOI 0.5–1) andcultured in the presence of INP0400 at the concentra-tions indicated. At 24 h p.i., Chlamydia were labeledwith anti-Chlamydia-LPS antibody (green) and ana-lyzed by immunofluorescence microscopy. (Scale bar,25 �m.) Cells were counterstained with Evan’s blue(red). (B) Chemical structure of INP0400. (C) Electronmicrographs of chlamydial inclusions treated withINP0400. McCoy cells were infected with C. trachomatisserovar L2 (MOI 0.5) and cultured in the presence ofINP0400 at the concentrations indicated. At 30 h p.i.,infected cells were analyzed by electron microscopy.Electron micrographs reveal a dose-dependent reduc-tion of intracellular bacteria, resulting in inclusion bod-ies with a gradual decrease in size. Chlamydial inclu-sions in INP0400-treated cultures contain primarily RBs,whereas in untreated controls, RBs have already startedto redifferentiate to infectious EBs (arrows indicate RBsand EBs of untreated cells).
Muschiol et al. PNAS � September 26, 2006 � vol. 103 � no. 39 � 14567
MIC
ROBI
OLO
GY
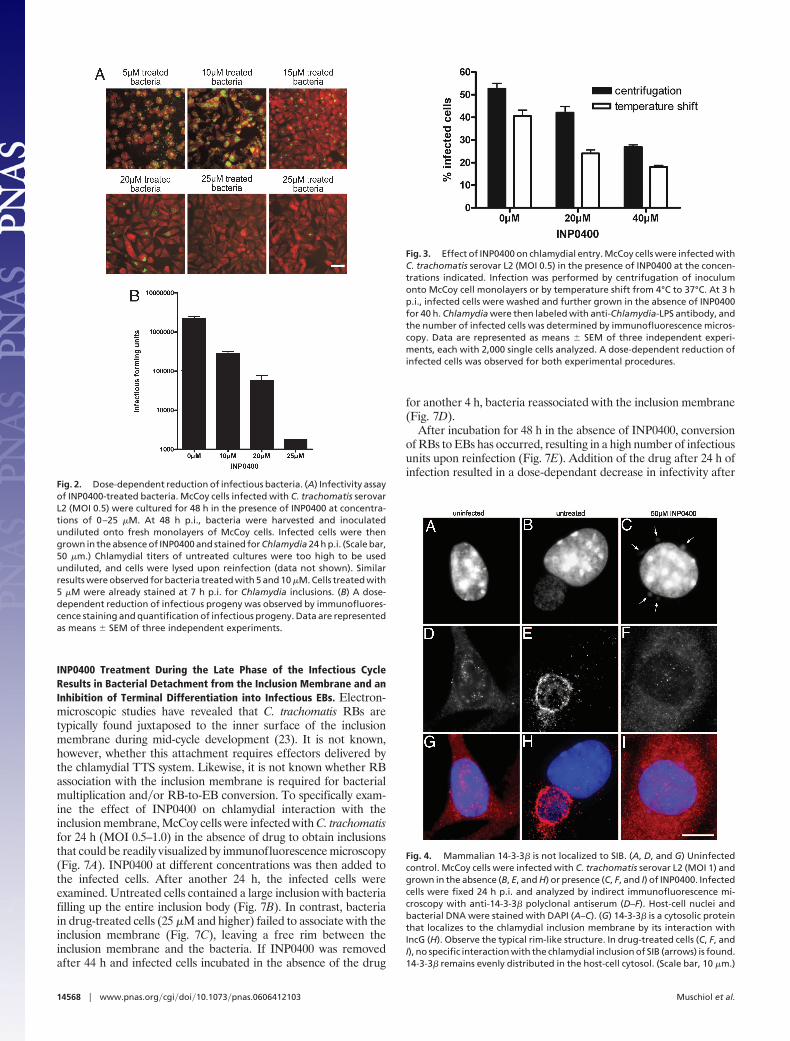
INP0400 Treatment During the Late Phase of the Infectious CycleResults in Bacterial Detachment from the Inclusion Membrane and anInhibition of Terminal Differentiation into Infectious EBs. Electron-microscopic studies have revealed that C. trachomatis RBs aretypically found juxtaposed to the inner surface of the inclusionmembrane during mid-cycle development (23). It is not known,however, whether this attachment requires effectors delivered bythe chlamydial TTS system. Likewise, it is not known whether RBassociation with the inclusion membrane is required for bacterialmultiplication and�or RB-to-EB conversion. To specifically exam-ine the effect of INP0400 on chlamydial interaction with theinclusion membrane, McCoy cells were infected with C. trachomatisfor 24 h (MOI 0.5–1.0) in the absence of drug to obtain inclusionsthat could be readily visualized by immunofluorescence microscopy(Fig. 7A). INP0400 at different concentrations was then added tothe infected cells. After another 24 h, the infected cells wereexamined. Untreated cells contained a large inclusion with bacteriafilling up the entire inclusion body (Fig. 7B). In contrast, bacteriain drug-treated cells (25 �M and higher) failed to associate with theinclusion membrane (Fig. 7C), leaving a free rim between theinclusion membrane and the bacteria. If INP0400 was removedafter 44 h and infected cells incubated in the absence of the drug
for another 4 h, bacteria reassociated with the inclusion membrane(Fig. 7D).
After incubation for 48 h in the absence of INP0400, conversionof RBs to EBs has occurred, resulting in a high number of infectiousunits upon reinfection (Fig. 7E). Addition of the drug after 24 h ofinfection resulted in a dose-dependant decrease in infectivity after
Fig. 2. Dose-dependent reduction of infectious bacteria. (A) Infectivity assayof INP0400-treated bacteria. McCoy cells infected with C. trachomatis serovarL2 (MOI 0.5) were cultured for 48 h in the presence of INP0400 at concentra-tions of 0–25 �M. At 48 h p.i., bacteria were harvested and inoculatedundiluted onto fresh monolayers of McCoy cells. Infected cells were thengrown in the absence of INP0400 and stained for Chlamydia 24 h p.i. (Scale bar,50 �m.) Chlamydial titers of untreated cultures were too high to be usedundiluted, and cells were lysed upon reinfection (data not shown). Similarresults were observed for bacteria treated with 5 and 10 �M. Cells treated with5 �M were already stained at 7 h p.i. for Chlamydia inclusions. (B) A dose-dependent reduction of infectious progeny was observed by immunofluores-cence staining and quantification of infectious progeny. Data are representedas means � SEM of three independent experiments.
Fig. 3. Effect of INP0400 on chlamydial entry. McCoy cells were infected withC. trachomatis serovar L2 (MOI 0.5) in the presence of INP0400 at the concen-trations indicated. Infection was performed by centrifugation of inoculumonto McCoy cell monolayers or by temperature shift from 4°C to 37°C. At 3 hp.i., infected cells were washed and further grown in the absence of INP0400for 40 h. Chlamydia were then labeled with anti-Chlamydia-LPS antibody, andthe number of infected cells was determined by immunofluorescence micros-copy. Data are represented as means � SEM of three independent experi-ments, each with 2,000 single cells analyzed. A dose-dependent reduction ofinfected cells was observed for both experimental procedures.
Fig. 4. Mammalian 14-3-3� is not localized to SIB. (A, D, and G) Uninfectedcontrol. McCoy cells were infected with C. trachomatis serovar L2 (MOI 1) andgrown in the absence (B, E, and H) or presence (C, F, and I) of INP0400. Infectedcells were fixed 24 h p.i. and analyzed by indirect immunofluorescence mi-croscopy with anti-14-3-3� polyclonal antiserum (D–F). Host-cell nuclei andbacterial DNA were stained with DAPI (A–C). (G) 14-3-3� is a cytosolic proteinthat localizes to the chlamydial inclusion membrane by its interaction withIncG (H). Observe the typical rim-like structure. In drug-treated cells (C, F, andI), no specific interaction with the chlamydial inclusion of SIB (arrows) is found.14-3-3� remains evenly distributed in the host-cell cytosol. (Scale bar, 10 �m.)
14568 � www.pnas.org�cgi�doi�10.1073�pnas.0606412103 Muschiol et al.

48 h despite the fact that drug treatment between 24 and 48 h in theinfectious cycle had no apparent effect on bacterial growth (Fig.7C). Hence, INP0400 inhibits terminal RB-to-EB differentiationduring the late cycle. This effect of the drug was reversible (Fig. 7F).Thus, compared with cells treated from 24 to 48 h in the infectiouscycle, infectivity increased by 1-log when INP0400 added at 24 h wasremoved after 12 h and infected cells were allowed to incubate foranother 12 h in the absence of drug (Fig. 7F).
DiscussionA class of acylated hydrazones was recently identified that inhibitedTTS of Y. pseudotuberculosis without affecting the in vitro growthrate (2, 3). The same class of compounds has also been shown toinhibit TTS of Pseudomonas aeruginosa as well as S. typhimurium(unpublished data), suggesting that these compounds target thesecretory apparatus itself rather than individual effector molecules.The obligate intracellular pathogen C. trachomatis also possesses aTTS system believed to deliver a large number of chlamydialproteins into either the inclusion membrane or the host cytosol. Sofar, only one protein, Tarp, has been shown to be delivered fromextracellular EBs upon contact with target cells, whereas the otherputative TTS effectors are produced at specific stages during thechlamydial infectious cycle. The differential expression pattern ofTTS effectors suggests that they play an instrumental role atdifferent stages in the chlamydial developmental cycle. However,lack of genetic tools has made it difficult to determine whether TTSeffectors are needed for Chlamydia to undergo its different devel-opmental phases. There are, however, evidences that TTS effectorspromote entry (TARP) and modulate properties of the inclusion
membrane to escape from the endosomal pathways, and to promoteheterotypic as well as homotypic interactions.
Here, we initiate a chemical genetic approach to dissect a role forChlamydia TTS during different stages in the infectious cycle ofChlamydia. TTS inhibitors have been screened such that they do notsignificantly affect the in vitro growth rate of Yersinia. In contrast,in the obligate intracellular pathogens C. trachomatis and C. pneu-moniae (L.B., Å.G., C.S., S.M., M.E., P. Nordstrom, B.H.-N., A.Waldenstrom, H.W.-W., and S.B., unpublished data), those TTSinhibitors tested exhibited a dose-dependant antibacterial activity,suggesting that the TTS system of Chlamydia is required for theorganism to undergo a productive infectious cycle.
The small molecule INP0400 partially inhibited uptake of EBs,suggesting an inhibitory effect on the internalization process. Thisinhibition, however, was not complete, meaning either that thecompound did not completely inhibit translocation of Tarp fromEBs or that some internalization may proceed also in the absenceof this effector protein. Internalized EBs in the presence of drugconverted to RBs, but RB multiplication was inhibited in a dose-dependant fashion, resulting in ultimately smaller inclusion bodies(SIB) containing just one or a few RBs.
We demonstrate that early drug treatment prevented the hostprotein 14-3-3� from localizing to the inclusion membrane of SIB,a process known to depend on translocation of the TTS effectorIncG into the inclusion membrane. IncG is expressed already 2 hafter infection (19). This correlates well with our finding thatinfected cells treated 8 h in the infectious cycle result in SIB able torecruit 14-3-3� to the membrane, indicative of a normal IncGtranslocation. Whether early treatment with INP0400 affects trans-location of all early TTS effectors is not known. The antibacterialactivity of INP0400, however, can not be dependent on an inhibitionof TTS translocation in the early phase, because inhibition of RB
Fig. 5. INP0400 inhibits homotypic vesicle fusion of SIB. Infected McCoy cells(MOI 5) were grown in the absence of drug for 8 h (A, C, and E). At 8 h p.i., 50�M INP0400 was added, and cells were allowed to grow for an additional 22h (B, D, and F). Homotypic vesicle fusion was then analyzed by indirectimmunofluorescence staining with anti-14-3-3� polyclonal antibodies (C andD). Host-cell nuclei and bacterial DNA were stained with DAPI (A and B). Inuntreated cultures, 14-3-3� localizes to the inclusion membrane (E). In drug-treated cultures, single SIB were identified, indicative for impaired vesiclefusion due to the absence of IncA (F). (Scale bar, 10 �m.) 14-3-3� decorates theindividual SIB, suggesting that IncG is localized to the inclusion membranes ofSIB when INP0400 is added at 8 h p.i.
Fig. 6. IncA is absent on SIB. McCoy cells were infected with C. trachomatisserovar L2 (MOI 5) and grown in the absence of INP0400 for 8 h (A, C, and E).At 8 h p.i., 50 �M INP0400 was added and cells were allowed to grow for anadditional 22 h before analysis by indirect immunofluorescence microscopy (B,D, and F) using IncA polyclonal antibodies (C and D). Host-cell nuclei andbacterial DNA were stained with DAPI (A and B). Although IncA is localized tothe chlamydial inclusion membrane of untreated cells (E), no specific IncAstaining pattern was observed for SIB in cultures treated with 50 �M INP0400(F). (Scale bar, 10 �m.)
Muschiol et al. PNAS � September 26, 2006 � vol. 103 � no. 39 � 14569
MIC
ROBI
OLO
GY
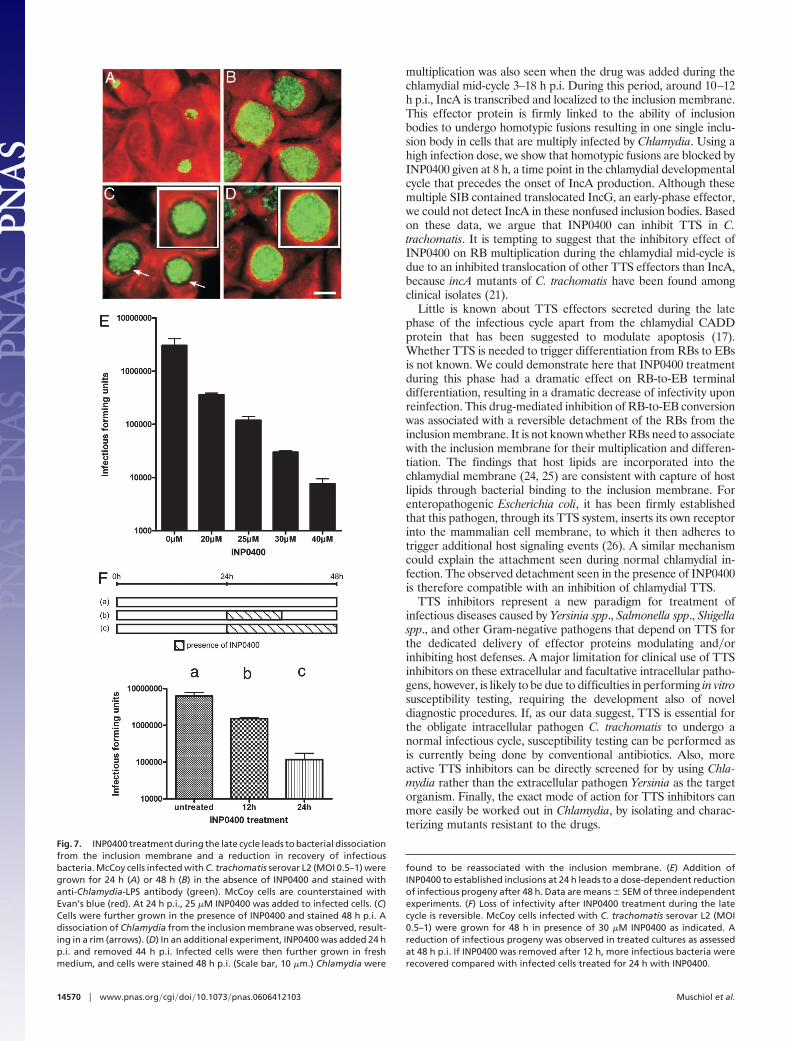
multiplication was also seen when the drug was added during thechlamydial mid-cycle 3–18 h p.i. During this period, around 10–12h p.i., IncA is transcribed and localized to the inclusion membrane.This effector protein is firmly linked to the ability of inclusionbodies to undergo homotypic fusions resulting in one single inclu-sion body in cells that are multiply infected by Chlamydia. Using ahigh infection dose, we show that homotypic fusions are blocked byINP0400 given at 8 h, a time point in the chlamydial developmentalcycle that precedes the onset of IncA production. Although thesemultiple SIB contained translocated IncG, an early-phase effector,we could not detect IncA in these nonfused inclusion bodies. Basedon these data, we argue that INP0400 can inhibit TTS in C.trachomatis. It is tempting to suggest that the inhibitory effect ofINP0400 on RB multiplication during the chlamydial mid-cycle isdue to an inhibited translocation of other TTS effectors than IncA,because incA mutants of C. trachomatis have been found amongclinical isolates (21).
Little is known about TTS effectors secreted during the latephase of the infectious cycle apart from the chlamydial CADDprotein that has been suggested to modulate apoptosis (17).Whether TTS is needed to trigger differentiation from RBs to EBsis not known. We could demonstrate here that INP0400 treatmentduring this phase had a dramatic effect on RB-to-EB terminaldifferentiation, resulting in a dramatic decrease of infectivity uponreinfection. This drug-mediated inhibition of RB-to-EB conversionwas associated with a reversible detachment of the RBs from theinclusion membrane. It is not known whether RBs need to associatewith the inclusion membrane for their multiplication and differen-tiation. The findings that host lipids are incorporated into thechlamydial membrane (24, 25) are consistent with capture of hostlipids through bacterial binding to the inclusion membrane. Forenteropathogenic Escherichia coli, it has been firmly establishedthat this pathogen, through its TTS system, inserts its own receptorinto the mammalian cell membrane, to which it then adheres totrigger additional host signaling events (26). A similar mechanismcould explain the attachment seen during normal chlamydial in-fection. The observed detachment seen in the presence of INP0400is therefore compatible with an inhibition of chlamydial TTS.
TTS inhibitors represent a new paradigm for treatment ofinfectious diseases caused by Yersinia spp., Salmonella spp., Shigellaspp., and other Gram-negative pathogens that depend on TTS forthe dedicated delivery of effector proteins modulating and�orinhibiting host defenses. A major limitation for clinical use of TTSinhibitors on these extracellular and facultative intracellular patho-gens, however, is likely to be due to difficulties in performing in vitrosusceptibility testing, requiring the development also of noveldiagnostic procedures. If, as our data suggest, TTS is essential forthe obligate intracellular pathogen C. trachomatis to undergo anormal infectious cycle, susceptibility testing can be performed asis currently being done by conventional antibiotics. Also, moreactive TTS inhibitors can be directly screened for by using Chla-mydia rather than the extracellular pathogen Yersinia as the targetorganism. Finally, the exact mode of action for TTS inhibitors canmore easily be worked out in Chlamydia, by isolating and charac-terizing mutants resistant to the drugs.
Fig. 7. INP0400 treatment during the late cycle leads to bacterial dissociationfrom the inclusion membrane and a reduction in recovery of infectiousbacteria. McCoy cells infected with C. trachomatis serovar L2 (MOI 0.5–1) weregrown for 24 h (A) or 48 h (B) in the absence of INP0400 and stained withanti-Chlamydia-LPS antibody (green). McCoy cells are counterstained withEvan’s blue (red). At 24 h p.i., 25 �M INP0400 was added to infected cells. (C)Cells were further grown in the presence of INP0400 and stained 48 h p.i. Adissociation of Chlamydia from the inclusion membrane was observed, result-ing in a rim (arrows). (D) In an additional experiment, INP0400 was added 24 hp.i. and removed 44 h p.i. Infected cells were then further grown in freshmedium, and cells were stained 48 h p.i. (Scale bar, 10 �m.) Chlamydia were
found to be reassociated with the inclusion membrane. (E) Addition ofINP0400 to established inclusions at 24 h leads to a dose-dependent reductionof infectious progeny after 48 h. Data are means � SEM of three independentexperiments. (F) Loss of infectivity after INP0400 treatment during the latecycle is reversible. McCoy cells infected with C. trachomatis serovar L2 (MOI0.5–1) were grown for 48 h in presence of 30 �M INP0400 as indicated. Areduction of infectious progeny was observed in treated cultures as assessedat 48 h p.i. If INP0400 was removed after 12 h, more infectious bacteria wererecovered compared with infected cells treated for 24 h with INP0400.
14570 � www.pnas.org�cgi�doi�10.1073�pnas.0606412103 Muschiol et al.

Materials and MethodsAntibodies and Reagents. INP0400, a small-molecule inhibitor iso-lated to block TTS of Y. pseudotuberculosis, was provided by InnatePharmaceuticals. Rabbit polyclonal antibodies against mammalian14-3-3� were obtained from Santa Cruz Biotechnology (SantaCruz, CA). Rabbit polyclonal antiserum specific to IncA wasgenerated by Innovagen (Lund, Sweden), using a peptide contain-ing the following amino acids: CSQIRETLSSPRKSA (correspond-ing to amino acids 252–266). Cy3-conjugated goat anti-rabbitsecondary antibodies were purchased from Jackson ImmunoRe-search Laboratories (West Grove, PA). The Pathfinder Chlamydiaculture confirmation kit, containing genus-specific fluorescein-conjugated murine monoclonal antibodies to Chlamydia-LPS, wasobtained from Bio-Rad (Hercules, CA). The nucleic acid stainDAPI (4�,6�-diamino-2-phenylindole) was purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture and Propagation of Chlamydia. C. trachomatis serovar L2(ATCC VR-902B) was propagated in McCoy cells (ATCC CRL-1696). Cells were grown at 37°C with 5.0% CO2 in RPMI medium1640 supplemented with 10% (vol�vol) FBS, 25 mM Hepes, 2 mML-glutamine and 10 �g�ml gentamicin. For propagation, McCoycells were infected with serovar L2 and grown for 48 h in mediumadditionally containing 2 �g�ml cycloheximide. At 48 h p.i., cellswere scraped off into medium and rigorously vortexed with glassbeads to release bacteria from the cells. Whole lysates werecentrifuged at 500 � g to pellet cellular debris. Supernatantcontaining Chlamydia was stored in aliquots at �70°C for laterinfection. If not otherwise stated, McCoy cell monolayers wereinfected with L2 at a MOI of 0.5–1 and centrifuged at 1,000 � g for45 min at 35°C. Cells were subsequently washed twice with Hank’sbalanced salt solution (HBSS) and incubated in medium containingcycloheximide (see above) for the times indicated.
Cytotoxicity Assay. Cytotoxicity of INP0400 was assessed by using acalceinAM assay (L.B., Å.G., C.S., S.M., M.E., P. Nordstrom,B.H.-N., A. Waldenstrom, H.W.-W., and S.B., unpublished data).Briefly, McCoy cells were grown in the presence of INP0400 atdifferent concentrations and times. Cytotoxicity was then analyzedby the ability of living cells to convert nonfluorescent calceinAM togreen fluorescent calcein. Fluorescence intensities were measuredin a microplate reader, and the relative amount of living cellscompared with dead cells was calculated.
Inhibitors and Chlamydia. McCoy cells were infected as describedabove and grown in the presence of TTS inhibitor INP0400 for theconcentrations and times indicated. Effects of inhibitors on chla-mydial growth were determined by immunofluorescence staining,infectivity assays, and electron microscopy.
Infectivity Assay. Infected McCoy cells were cultured in the pres-ence of INP0400 dissolved in medium at different concentrations
(0–50 �M). At 48 h p.i., cells were washed twice with HBSS andharvested as described above. Bacterial suspensions obtained wereused to reinfect fresh monolayers of McCoy cells. Infected cellswere grown for 24 h in the absence of INP0400 and immunolabeledwith anti-Chlamydia-LPS antibody. Immunofluorescence micros-copy was performed to determine the number of infection-formingunits. Data are represented as means � SEM of three independentexperiments, each with 2,000 single cells analyzed.
Immunofluorescence Microscopy. McCoy cells were grown on sterileglass coverslips and infected as described above. For indirectimmunofluorescence staining, coverslips were fixed in ice-coldmethanol for 10 min at the times indicated. Coverslips wereair-dried and washed three times with PBS. Permeabilization wasperformed with PBS�0.5% Triton X-100 for 5 min. Cells werewashed three times with PBS and blocked overnight in 10%goat-serum�PBS. Incubation with first antibody (1:100–1:300) wasperformed for 45 min at 37°C in the dark. Coverslips were washedthree times with PBS, and cells were subsequently incubated withsecondary antibody (1:300) for 45 min at 37°C. Host-cell nuclei andintracellular bacteria were stained with the DNA-specific fluoro-chrome DAPI (1 �g�ml) for 2 min. Coverslips were mounted onMowiol containing 10% wt�vol 1,4-diazabicyclo[2.2.2]octane(DABCO). When using the Pathfinder Chlamydia culture confir-mation kit, coverslips were fixed in methanol as described aboveand directly stained for Chlamydia. Images were acquired by usinga Leica (Vienna, Austria) DMRE fluorescence microscope. Ran-dom areas were selected for analysis. Pictures were processed byusing Photoshop 7 software (Adobe Systems, San Jose, CA).
Transmission Electron Microscopy. McCoy cells infected with C.trachomatis L2 (MOI 0.5–1) were fixed at the indicated time pointsin 2% glutaraldehyde and 0.5% paraformaldehyde in 0.1 M sodiumcacodylate buffer containing 0.1 M sucrose and 3 mM CaCl2 (pH7.4) at room temperature for 30 min and stored at 4°C untilembedding. After fixation, cells were rinsed in 0.15 M sodiumcacodylate buffer containing 3 mM CaCl2 (pH 7.4) and centrifuged.The pellets were resuspended and postfixed in 2% osmium tetrox-ide in 0.07 M sodium cacodylate buffer containing 1.5 mM CaCl2(pH 7.4) at 4°C for 2 h, dehydrated in ethanol followed by acetone,and embedded in LX-112 (Ladd, Burlington, VT). Sections werecontrasted with uranyl acetate followed by lead citrate and exam-ined in a Tecnai 10 transmission electron microscope (FEI, Hills-boro, OR) at 80 kV. Digital images were captured by using aMegaView III digital camera (Soft Imaging System, Munster,Germany).
This work was partly funded by VINNOVA, the Swedish Foundation forStrategic Research, the Swedish Research Council and Torsten andRagnar Soderbergs Foundation, and the European Marie Curie programEuropean Initiative for basic research in Microbiology and InfectiousDiseases.
1. Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ (2005) Science 310:670–674.2. Kauppi AM, Nordfelth R, Uvell H, Wolf-Watz H, Elofsson M (2003) Chem Biol
10:241–249.3. Nordfelth R, Kauppi AM, Norberg HA, Wolf-Watz H, Elofsson M (2005) Infect Immun
73:3104–3114.4. Schachter J (1999) in Chlamydia: Intracellular Biology, Pathogenesis, and Immunity, ed
Stephens RS (Am Soc Microbiol, Washington, DC), pp 139–169.5. AbdelRahman YM, Belland RJ (2005) FEMS Microbiol Rev 29:949–959.6. Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA (1996) EMBO J 15:964–977.7. Beatty WL (2006) J Cell Sci 119:350–359.8. Hsia RC, Pannekoek Y, Ingerowski E, Bavoil PM (1997) Mol Microbiol 25:351–359.9. Hueck CJ (1998) Microbiol Mol Biol Rev 62:379–433.
10. Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA,Hackstadt T (2004) Proc Natl Acad Sci USA 101:10166–10171.
11. Rockey DD, Scidmore MA, Bannantine JP, Brown WJ (2002) Microbes Infect 4:333–340.12. Scidmore MA, Hackstadt T (2001) Mol Microbiol 39:1638–1650.13. Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER (1999) Cell Microbiol
1:119–130.14. Fling SP, Sutherland RA, Steele LN, Hess B, D’Orazio SE, Maisonneuve J, Lampe MF,
Probst P, Starnbach MN (2001) Proc Natl Acad Sci USA 30:1160–1165.
15. Fields KA, Hackstadt T (2000) Mol Microbiol 38:1048–1060.16. Subtil A, Delevoye C, Balana ME, Tastevin L, Perrinet S, Dautry-Varsat A (2005) Mol
Microbiol 56:1636–1647.17. Stenner-Liewen F, Liewen H, Zapata JM, Pawlowski K, Godzik A, Reed JC (2002) J Biol
Chem 277:9633–9636.18. Zhong G, Fan P, Ji H, Dong F, Huang Y (2001) J Exp Med 193:935–942.19. Scidmore-Carlson MA, Shaw EI, Dooley CA, Fischer ER, Hackstadt T (1999) Mol
Microbiol 33:753–765.20. Ho TD, Starnbach MN (2005) Infect Immun 73:905–911.21. Suchland RJ, Rockey DD, Bannantine JP, Stamm WE (2000) Infect Immun 68:360–361.22. Rockey DD, Viratyosin W, Bannantine JP, Suchland RJ, Stamm WE (2002) Microbiology
148:2497–2505.23. Hackstadt T, Fischer ER, Scidmore MA, Rockey DD, Heinzen RA (1997) Trends
Microbiol 5:305–306.24. Wylie JL, Hatch GM, McClarty G (1997) J Bacteriol 179:7233–7242.25. Newhall WJ (1988) in Microbiology of Chlamydia, ed Barron AL (CRC, Boca Raton, FL),
pp 47–70.26. Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB (1997) Cell
91:511–520.
Muschiol et al. PNAS � September 26, 2006 � vol. 103 � no. 39 � 14571
MIC
ROBI
OLO
GY