a muc1/il-18 dna vaccine induces anti-tumor immunity and increased survival in muc1 transgenic mice
TRANSCRIPT

Vaccine 24 (2006) 3340–3352
A MUC1/IL-18 DNA vaccine induces anti-tumor immunity andincreased survival in MUC1 transgenic mice
Linda A. Snyder ∗, Theresa J. Goletz, George R. Gunn, Frank F. Shi, Michael C. Harris,Karyn Cochlin, Christine McCauley, Stephen G. McCarthy,
Patrick J. Branigan, David M. KnightCentocor, Inc., 145 King of Prussia Road, Radnor, PA 19087, United States
Received 7 July 2005; received in revised form 4 December 2005; accepted 9 January 2006Available online 19 January 2006
Abstract
MUC1 (mucin 1) is a tumor-associated antigen that is overexpressed in many adenocarcinomas. Active immunotherapy targeting tumorsexpressing MUC1 could have great treatment value. MUC1 DNA vaccines were evaluated in MUC1 transgenic (MUC1.Tg) mice challengedwcamMAtf©
K
1
eebaspapi
0d
ith MC38/MUC1+ tumor cells. Vaccination with MUC1 plasmid DNA (pMUC1) alone was insufficient to induce tumor protection. However,o-administration of pMUC1 with a plasmid encoding murine interleukin-18 (pmuIL-18) resulted in significant tumor protection and survivalfter tumor challenge. Protection was durable in the absence of additional vaccination, as demonstrated by continued protection of vaccinatedice following tumor rechallenge. Mice surviving challenges with MC38/MUC1+ cells showed significant protection after challenge withUC1− MC38 tumor cells, suggesting that these mice had developed immune responses to epitopes shared between the tumor cell lines.ntibody-mediated depletion of lymphocyte subsets demonstrated that protection was due largely to CD4+ T cells. This work demonstrates
hat a naked DNA vaccine can break tolerance to MUC1 and induce an immune response capable of mediating both significant protectionrom tumor challenge and increased survival.
2006 Elsevier Ltd. All rights reserved.
eywords: MUC1; IL-18; DNA vaccine; Immunotherapy; Cancer; Tolerance
. Introduction
MUC1 is a transmembrane glycoprotein normallyxpressed on the apical surface of ductal epithelia [1]. How-ver, epithelial adenocarcinomas of several tissues, includingreast, prostate, lung, colon, ovary and pancreas, express anberrant form of MUC1 [2,3]. On tumors, polarized expres-ion of MUC1 is lost and the normally heavily glycosylatedrotein is over-expressed in hypoglycosylated forms. Suchlterations are recognized by the immune system of canceratients with MUC1+ tumors. Low titer antibodies, primar-ly IgM, and low frequency cellular responses, that are often
∗ Corresponding author. Tel.: +1 610 651 6231; fax: +1 610 651 6915.E-mail address: [email protected] (L.A. Snyder).
major histocompatilibity complex (MHC)-unrestricted, havebeen detected [4–10]. These studies indicated that immunerecognition of MUC1 could be directed to epitopes in thetandem repeat domain (TRD). The TRD is comprised of avariable number (20–100) of repeats of a 20 amino acid (aa)sequence and makes up the majority of the extracellular por-tion of the protein. In tumor cells expressing MUC1, abnor-mal glycosylation exposes novel epitopes within the TRD,making this an immunodominant region. However, in someexperimental systems, epitopes outside the TRD have beenidentified [11–13]. Despite evidence of immune recognition,responses in humans are weak and inadequate in mediat-ing clinical effects. Thus, immunotherapies that qualitativelyand quantitatively enhance immune recognition of tumorcells expressing aberrant forms of MUC1 would be clinicallyuseful.
264-410X/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.vaccine.2006.01.014

L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352 3341
Many approaches have been studied for the inductionof MUC1-specific immune responses, both in mice and inhumans. These include vaccination with peptides [14–17],proteins/fusion proteins [18], MUC1 recombinant viruses[18–20], dendritic cells (DC) and DC/tumor cell fusions[21–25], and DNA vaccines [26–28]. In clinical studies,MUC1-specific antibodies and/or cellular responses canoften be detected after using these strategies, but the inductionof clinically effective anti-tumor immune responses remainsto be achieved.
This study reports on the design and testing of a MUC1/IL-18 DNA vaccine. DNA vaccines have been extensivelystudied for many antigens, in both preclinical and clini-cal settings, and they offer certain advantages over othervaccines and immunotherapies [29]. First, DNA vaccinesinduce MHC-restricted and human leukocyte antigen (HLA)-restricted CD4+ and CD8+ T cells, as well as antibodyresponses. This is due to cellular uptake of DNA vac-cines, which results in antigen expression, and is fol-lowed by processing and presentation in the context ofMHC Class I and/or Class II molecules. Second, DNAvaccines have a flexible coding capacity, which enablesa single plasmid to encode and express multiple genes.These genes could include antigen(s) as well as cytokineadjuvant(s) that can optimize the vaccine and enhance theimmune response. Adjuvants are often required to aug-mvagC1csiih
a[tfteatTevgioctip
2. Materials and methods
2.1. Plasmid construction and manufacturing
The pMUC1 plasmid encodes a human MUC1 cDNA(kindly provided by Donald Kufe) cloned in the Vical 1055plasmid backbone at the SalI and BamHI sites [36] (Fig. 1A).The MUC1 cDNA encodes 508 amino acids, and it con-tains a truncated TRD comprised of 6.5 repeats. An opti-mized Kozak sequence [37] was engineered into the startcodon region. The MUC1 cDNA is identical otherwise to thelonger MUC1 splice variant reported elsewhere [38]. Tran-sient transfection and flow cytometry experiments demon-strated that pMUC1 induced expression of human MUC1 inmultiple human and non-human tissue culture cells (data notshown).
The pmuIL-18 plasmid contains the mature mouse IL-18cDNA sequence linked to a mouse immunoglobulin heavychain (HC) signal sequence (Fig. 1B and C). A genomicfragment encoding a mouse immunoglobulin HC signalsequence was cloned into pUC19 at the XbaI site. Thegenomic fragment was modified at its BstZ17 I site locatedat the 3′ end of the signal sequence, by ligation to a linkerencoding the first three residues of mature mouse IL-18(NGR; Fig. 1C) and a SalI site. Mouse IL-18 was cloned bypolymerase chain reaction (PCR) from mouse kidney polyA+
R(TGteAm(LC5(sHc(
psfsmt
rPtd7
ent immune responses to vaccines, particularly when theaccine is targeting weak antigens or self-antigens, suchs MUC1. Cytokines such as IL-2, IL-12, IL-18, andranulocyte-macrophage colony-stimulating factor (GM-SF) are particularly strong choices to enhance T helper(Th1) cellular immune responses induced by DNA vac-
ines expressing a variety of antigens. Finally, results ofome clinical trials have demonstrated that DNA vaccinesnduce antigen-specific immune responses [30,31], confirm-ng that this mode of vaccination merits further study inumans.
Several groups have investigated MUC1 DNA vaccinesnd reported induction of anti-tumor protection in mice26–28,32–34]. While the vaccines were effective, they wereested in model systems in which human MUC1 was aoreign antigen and, as such, should readily elicit a pro-ective immune response. In the studies reported here, thefficacy of a MUC1/IL-18 DNA vaccine was evaluated in
more physiologic and clinically relevant model, usingransgenic mice expressing human MUC1 (MUC1.Tg) [35].hese mice are tolerant to MUC1 protein and exhibit anxpression profile and tissue distribution of human MUC1ery similar to that observed in humans. In addition, syn-eneic tumors expressing human MUC1 can readily grown the MUC1.Tg mice. Thus, this model enables rigor-us evaluation of whether a MUC1/IL-18 DNA vaccinean break tolerance to MUC1 and induce efficacious anti-umor immune responses to MUC1+ tumors, as well asdentification of immune mechanisms responsible for tumorrotection.
NA (Clontech, Palo Alto, CA) by reverse transcriptaseRT)-PCR, using the forward and reverse primers 5′-TTGTCGACTTCACTGTACAACCGCA-3′ and 5′-TTTTCGACACTTTGATGTAAGTTAGTGAGAG-3′, respec-
ively. The primers incorporated SalI sites (bold) thatnabled in-frame cloning with the HC signal sequence.
stop codon was incorporated after codon 157 in theature mouse IL-18 sequence by site-directed mutagenesis
Quikchange® Site-Directed Mutagenesis kit, Stratagene,a Jolla, CA) using complementary oligonucleotides 5′-TAACTTACATCAAAGTTAGGACCCTGGTACC-3′ and′ -GGTACCAGGGTCCTAACTTTGATGTAAGTTAG-3′stop codon is underlined on sense oligonucleotide; KpnIite shown in bold on both oligonucleotides). The correctedC-IL-18 fragment was excised with XbaI/KpnI for sub-
loning into the final plasmid expression vector pmuIL-18Fig. 1B and C).
Transient transfection and ELISA assay verified thatmuIL-18 could induce secretion of mouse IL-18 (data nothown). Mouse IL-18 was shown to be functional by inter-eron gamma (IFN�) induction experiments with mouseplenocytes (data not shown). The control empty vector plas-id is similar to the plasmid backbone for pmuIL-18, except
hat it does not encode any eukaryotic genes (not shown).All plasmids were grown in E. coli DH10BTM strain (Invit-
ogen), and were manufactured by Puresyn, Inc. (Malvern,A) to a specification of >90% supercoiled DNA and lesshan 1 EU/mg of endotoxin. Plasmids were diafiltered to theesired concentration in 0.15 M sodium phosphate buffer, pH.0.

3342 L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352
Fig. 1. Schematics of pMUC1 and pmuIL-18 (A and B), and sequence of mouse IL-18 gene insert (C). Each plasmid contains the human cytomegalovirusimmediate early (HCMV) promoter to drive expression of either human MUC1 or mouse IL-18, with a polyadenylation signal (polyA), and contains a bacterialorigin of replication (ori) and kanamycin resistance gene (kanr) to enable plasmid propagation in bacteria. (A) pMUC1; (B) pmuIL-18; (C) nucleotide sequenceof HC genomic fragment linked to mature mouse IL-18. Nucleotides in upper case represent coding or untranslated sequence; nucleotides in lower case representthe HC intron. Amino acid residues are shown above the DNA sequence, and numbering of the polypeptide begins with the N (in bold) at the amino terminusof mature mouse IL-18. Locations of relevant restriction sites used for cloning are indicated.

L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352 3343
2.2. Mice and tumor cells
The human MUC1 transgenic (MUC1.Tg) mice on aC57Bl/6 background have been described previously [35]and were bred at Taconic Farms (Germantown, NY). FemaleMUC1.Tg mice (8–14 weeks of age) were used in all experi-ments. The MC38 (C57Bl/6-derived adenocarcinoma) [39]and MC38/MUC1 (MC38 cells transfected with humanMUC1) [40] tumor cells were kindly provided by Don-ald Kufe. Cells were maintained in Dulbecco’s ModifiedEagle Medium (DMEM) containing GlutaMAXTM, 110 mg/lsodium pyruvate, 10% heat-inactivated fetal bovine serum(FBS), and 0.1 mM nonessential amino acids (Invitrogen).The MC38/MUC1 tumor cells highly overexpress MUC1as measured by flow cytometry (data not shown). MUC1expressed on the MC38/MUC1 tumor cells can bind to anti-bodies that can detect normal or tumor-specific MUC1 epi-topes (data not shown).
2.3. Vaccination protocol and tumor challenge studies
Mice received three biweekly immunizations (days 1, 15,and 29). Plasmids were prepared in 0.15 M sodium phosphatebuffer for intradermal (i.d.) administration in a total volumeof 100 �l. Mice were anesthetized with ketamine/xylazineadministered intraperitoneally (i.p.) prior to each immuniza-tbactdml(tiwwdw
Mflwtcat
aer
ra
Care and Use Committee, and were conducted in accordancewith “The Guide for the Care and Use of Laboratory Ani-mals”.
2.4. Antibody-mediated depletion studies
Mice were immunized i.d. with pMUC1/pmuIL18(30 �g/plasmid) or empty vector (60 �g) on days 1, 15 and29. Immunized mice were randomly assigned to seven treat-ment groups (n = 14–15/group) and depleting antibodies orPBS controls were administered by i.p. injection. One dayprior to tumor challenge (day 35), mice received anti-CD4[41] (GK1.5, Harlan Bioproducts, Indianapolis, IN), anti-CD8 [42] (53-6.72, Harlan Bioproducts) or anti-natural killer(NK) [43,44] (anti-asialo GM1 (Rabbit), Wako Chemicals,Inc., Richmond, VA) depleting antibodies alone, or in combi-nations. GK1.5 and 53-6.72 were administered (1 mg/mouseper injection) weekly for 4 weeks. Anti-asialo GM1 wasadministered twice weekly for 4 weeks to maintain NKdepletion (20 �l/mouse). Mice not receiving anti-asialo GM1were treated with PBS concurrent with the second weeklyadministration of anti-asialo GM1. This depletion regimenmaintained animals in a depleted state as determined by flowcytometry (>99% depletion for CD4+ and CD8+ T cells,>94% depletion of NK cells; data not shown). One weekafter the last immunization (day 36), mice were challengedsdms
2
bmMCtpatw(oD4et
2
pad
ion. Mice received 50 �l injections of DNA or phosphateuffer vehicle in each ear, using a 28 gauge needle. One weekfter the final immunization (day 36 or week 5), mice werehallenged subcutaneously (s.c.) with 2 × 105 MC38/MUC1umor cells on the right flank, and monitored daily for tumorevelopment. Once palpable tumors were detected, measure-ents were made twice weekly with calipers for width and
ength. Mean tumor diameter was calculated by the formulaW + L)/2. In some experiments, mice were challenged s.c. onhe flank with 3 × 105 MC38 tumor cells. Mice were mon-tored for approximately 4 weeks after tumor challenge, athich time mice with tumors were sacrificed and the tumorseighed. In survival studies, animals were monitored for 91ays after tumor challenge. Mice were sacrificed when theidth dimension of the tumor reached 15 mm.In rechallenge studies, mice protected from the first
C38/MUC1 challenge were rechallenged on the oppositeank at week 13 (week 8 post-first challenge). These miceere monitored for 8 more weeks. Those mice protected from
he first and second MC38/MUC1 tumor challenges werehallenged on the original flank a third time with MC38 cellst week 21 (week 16 post-first challenge). These mice werehen monitored to week 25.
Mice were monitored for signs of ill health (possibly due toutoimmune effects of the vaccine) during all studies. How-ver, all remained healthy during the survival study and theechallenge experiments.
All animal studies were conducted at least twice, withepresentative results shown in the figures. All protocols fornimal experiments were approved by the Centocor Animal
.c. with 2 × 105 MC38/MUC1 cells. Mice were monitoredaily until tumors became palpable. Tumor diameters wereeasured twice weekly for the duration of the study. These
tudies were conducted twice with similar results obtained.
.5. Anti-MUC1 antibody detection
Serum samples were obtained from mice by retro-orbitalleeding, prior to study start, on day 35, and at study ter-ination. Samples were analyzed for the presence of anti-UC1 antibodies by ELISA. Microtiter plates (Costar, Inc.,orning, NY) were coated with 50 ng/well MUC1 pro-
ein (DF3 antigen; Centocor, Inc.) at 4 ◦C overnight. Thelates were blocked (0.5% porcine gelatin, 4% BSA in PBS)nd serum samples were assayed at four 1:10 serial dilu-ions (1:10–1:10,000). Anti-MUC1 antibodies were detectedith horseradish peroxidase (HRP)-labeled anti-mouse IgG
H + L) (Pierce Biotechnology, Inc., Rockford, IL) and devel-ped with tetramethylbenzidine-stable (TMB-S; Researchiagnostics, Inc., Flanders, NJ). Absorbance was read at50 nm. 1/titer values were scored as positive for the pres-nce of MUC1 antibodies if the OD readings were at leasthree-fold over PBS control wells.
.6. Proliferation assays
Mice were vaccinated with pMUC1/pmuIL-18 or�gal/pmuIL-18 plasmids (100 �g each plasmid) three timest biweekly intervals. Splenocytes were harvested 7 or 14ays after the third immunization. Splenocytes were isolated
3344 L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352
and samples were pooled and resuspended in RPMI completemedia (RPMI 1640, 100 U/ml penicillin, 100 �g/ml strepto-mycin, 10% FBS and 50 �M 2-mercaptoethanol). 2 × 105
cells from each group were set up in triplicate in 200 �l ofmedia alone, 10 or 1 �g/ml �gal protein (Sigma, St. Louis,MO), 25 or 3 U/ml MUC1 protein (Cortex Biochem, SanLeandro, CA) or 5 �g/ml concanavalin A (Sigma). Cellswere incubated in a humidified chamber at 37 ◦C at 5%CO2 for 4 days. 1 �Ci/well 3H-thymidine (Amersham Phar-macia Biotech, Piscataway, NJ) was added for an addi-tional 18 h. Cultures were harvested onto Unifilter plates(Packard Instrument, Meriden, CT) and counted on the Top-Count Scintillation Counter (Packard Instrument). Stimula-tion index was determined from the formula: experimentalcount/spontaneous count.
2.7. Statistical analysis
To determine if differences in tumor incidence betweentreatment groups were significant, a two-sided Fisher’s exacttest was used. For each study, the tumor diameter data wereanalyzed separately for each day. To assess differences inmean tumor diameter between groups on a given day, anoverall F-test was used to assess any differences between alltreatment groups. If the F-test was significant, then compar-isons were made between the treatment groups and the controlgvtsso
3
3M
iMfIpchsib
3s
a
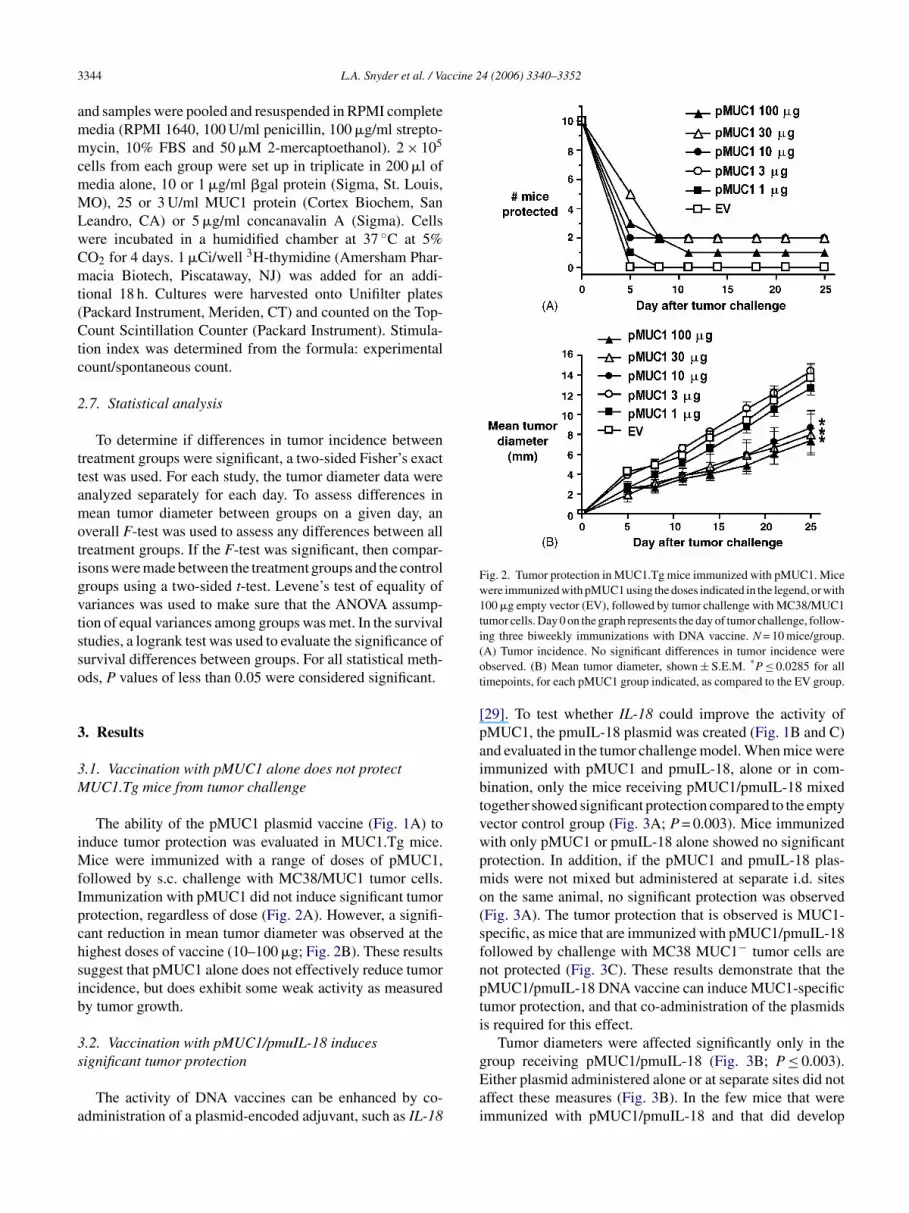
Fig. 2. Tumor protection in MUC1.Tg mice immunized with pMUC1. Micewere immunized with pMUC1 using the doses indicated in the legend, or with100 �g empty vector (EV), followed by tumor challenge with MC38/MUC1tumor cells. Day 0 on the graph represents the day of tumor challenge, follow-ing three biweekly immunizations with DNA vaccine. N = 10 mice/group.(A) Tumor incidence. No significant differences in tumor incidence wereobserved. (B) Mean tumor diameter, shown ± S.E.M. *P ≤ 0.0285 for alltimepoints, for each pMUC1 group indicated, as compared to the EV group.
[29]. To test whether IL-18 could improve the activity ofpMUC1, the pmuIL-18 plasmid was created (Fig. 1B and C)and evaluated in the tumor challenge model. When mice wereimmunized with pMUC1 and pmuIL-18, alone or in com-bination, only the mice receiving pMUC1/pmuIL-18 mixedtogether showed significant protection compared to the emptyvector control group (Fig. 3A; P = 0.003). Mice immunizedwith only pMUC1 or pmuIL-18 alone showed no significantprotection. In addition, if the pMUC1 and pmuIL-18 plas-mids were not mixed but administered at separate i.d. siteson the same animal, no significant protection was observed(Fig. 3A). The tumor protection that is observed is MUC1-specific, as mice that are immunized with pMUC1/pmuIL-18followed by challenge with MC38 MUC1− tumor cells arenot protected (Fig. 3C). These results demonstrate that thepMUC1/pmuIL-18 DNA vaccine can induce MUC1-specifictumor protection, and that co-administration of the plasmidsis required for this effect.
Tumor diameters were affected significantly only in thegroup receiving pMUC1/pmuIL-18 (Fig. 3B; P ≤ 0.003).Either plasmid administered alone or at separate sites did notaffect these measures (Fig. 3B). In the few mice that wereimmunized with pMUC1/pmuIL-18 and that did develop
roups using a two-sided t-test. Levene’s test of equality ofariances was used to make sure that the ANOVA assump-ion of equal variances among groups was met. In the survivaltudies, a logrank test was used to evaluate the significance ofurvival differences between groups. For all statistical meth-ds, P values of less than 0.05 were considered significant.
. Results
.1. Vaccination with pMUC1 alone does not protectUC1.Tg mice from tumor challenge
The ability of the pMUC1 plasmid vaccine (Fig. 1A) tonduce tumor protection was evaluated in MUC1.Tg mice.
ice were immunized with a range of doses of pMUC1,ollowed by s.c. challenge with MC38/MUC1 tumor cells.mmunization with pMUC1 did not induce significant tumorrotection, regardless of dose (Fig. 2A). However, a signifi-ant reduction in mean tumor diameter was observed at theighest doses of vaccine (10–100 �g; Fig. 2B). These resultsuggest that pMUC1 alone does not effectively reduce tumorncidence, but does exhibit some weak activity as measuredy tumor growth.
.2. Vaccination with pMUC1/pmuIL-18 inducesignificant tumor protection
The activity of DNA vaccines can be enhanced by co-dministration of a plasmid-encoded adjuvant, such as IL-18

L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352 3345
Fig. 3. Tumor protection in MUC1.Tg mice immunized with pMUC1−/+pmuIL-18. Mice were immunized with either vehicle, or the plasmids asindicated in the legends, followed by challenge with MC38/MUC1 tumorcells (A and B) or MC38 tumor cells (C). In the pMUC1/pmuIL-18 group,the plasmids were mixed first, and then injected i.d. at two sites. In thepMUC1 + pmuIL-18 group, each plasmid was kept separate and injectedat separate intradermal sites. Each plasmid was administered at 30 �gdoses. N = 10 mice/group. (A) Tumor incidence in mice challenged withMC38/MUC1 tumor cells. *P = 0.003. (B) Mean tumor diameter in micechallenged with MC38/MUC1 tumor cells, shown ± S.E.M. *P ≤ 0.0003when compared to either the EV or vehicle control groups. (C) Tumor inci-dence in mice challenged with MC38 tumor cells.
tumors, the tumors were significantly smaller than in theother groups (data not shown). These results demonstrate thatpMUC1/pmuIL-18 vaccination breaks tolerance to MUC1 inMUC1.Tg mice, and it induces significant protection fromtumor development. In addition, these results suggest thatIL-18 can act as an effective adjuvant for MUC1, but onlythrough co-administration with the antigen-expressing plas-mid.
3.3. Efficacy of pMUC1/pmuIL-18 vaccine is dependenton vaccine dose
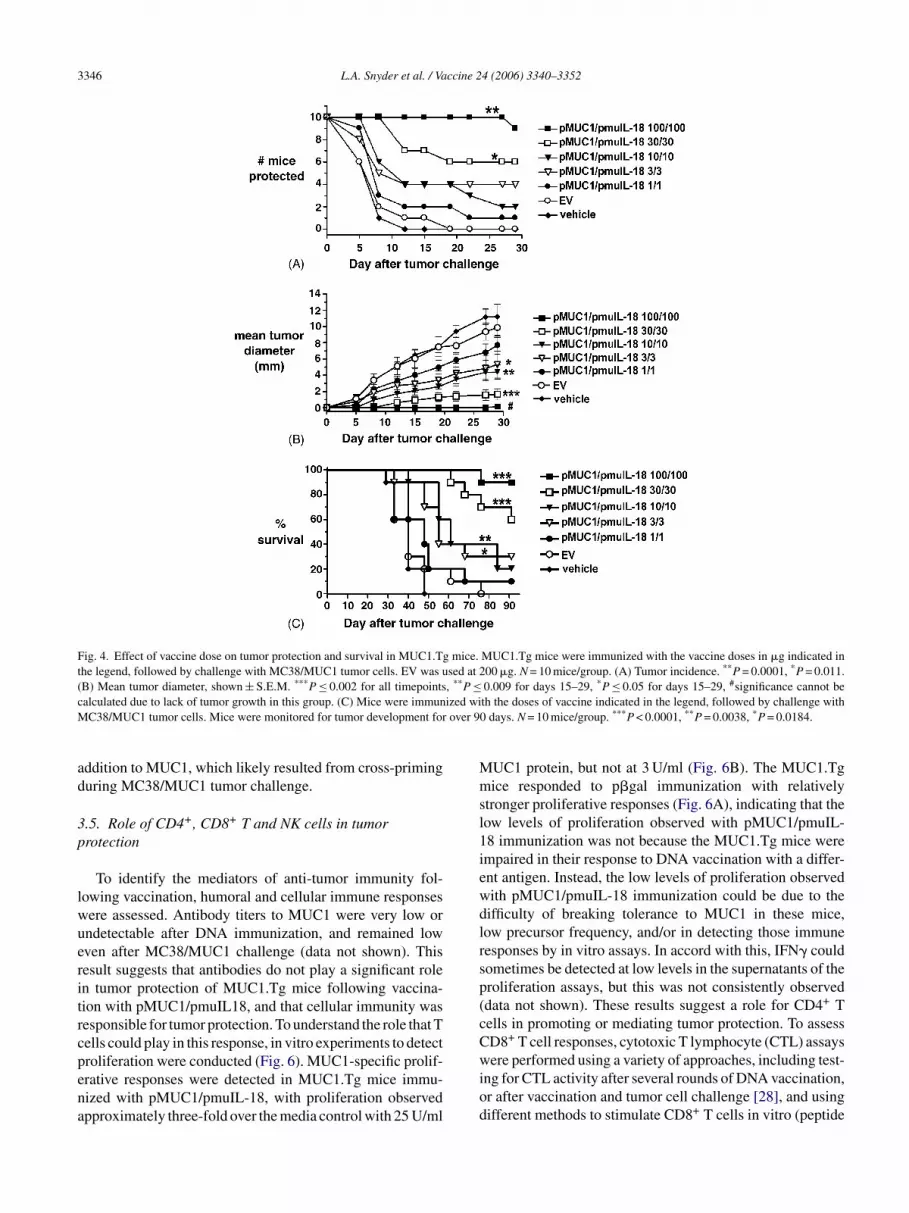
To determine the impact of dose of pMUC1/pmuIL-18on protection and tumor growth, mice were immunized witha range of doses of the vaccine (1–100 �g; Fig. 4). Theseresults show that higher doses (30–100 �g of each plas-mid) of the vaccine induce significant protection, while lower
doses (1–10 �g) were not as effective in inducing protection(Fig. 4A). Higher doses of the vaccine also control tumorgrowth significantly better than do lower doses (Fig. 4B).
Long-term survival of pMUC1/pmuIL-18-vaccinatedmice was assessed. Immunized mice were tumor-challengedand monitored for tumor growth for over 90 days (Fig. 4C).Higher doses (100 or 30 �g) of pMUC1/pmuIL-18 inducedsignificantly greater survival (P < 0.0001) than the con-trol empty vector group. Interestingly, significant survivalwas still observed with lower doses of the vaccine (10 �g:P < 0.0038; or 3 �g: P < 0.0184), though the effect was notas dramatic as with the higher doses. Thus, these resultsshow that pMUC1/pmuIL-18 DNA vaccine induces signifi-cant dose-dependent tumor protection and survival benefit.
3.4. pMUC1/pmuIL-18 induces a memory response andepitope spreading
A strong vaccine can induce antigen-specific memoryimmune responses that can protect animals from a tumorrechallenge. To assess whether the pMUC1/pmuIL-18 DNAvaccine could initiate a memory immune response, the exper-imental scheme shown in Fig. 5A was used. MUC1.Tg micewere immunized with 30 �g doses of pMUC1/pmuIL-18, fol-lowed by challenge with MC38/MUC1 tumor cells. Micethat rejected the first tumor challenge were rechallengedwttmtlMc
Dtbctsiirvltramtmc(rm
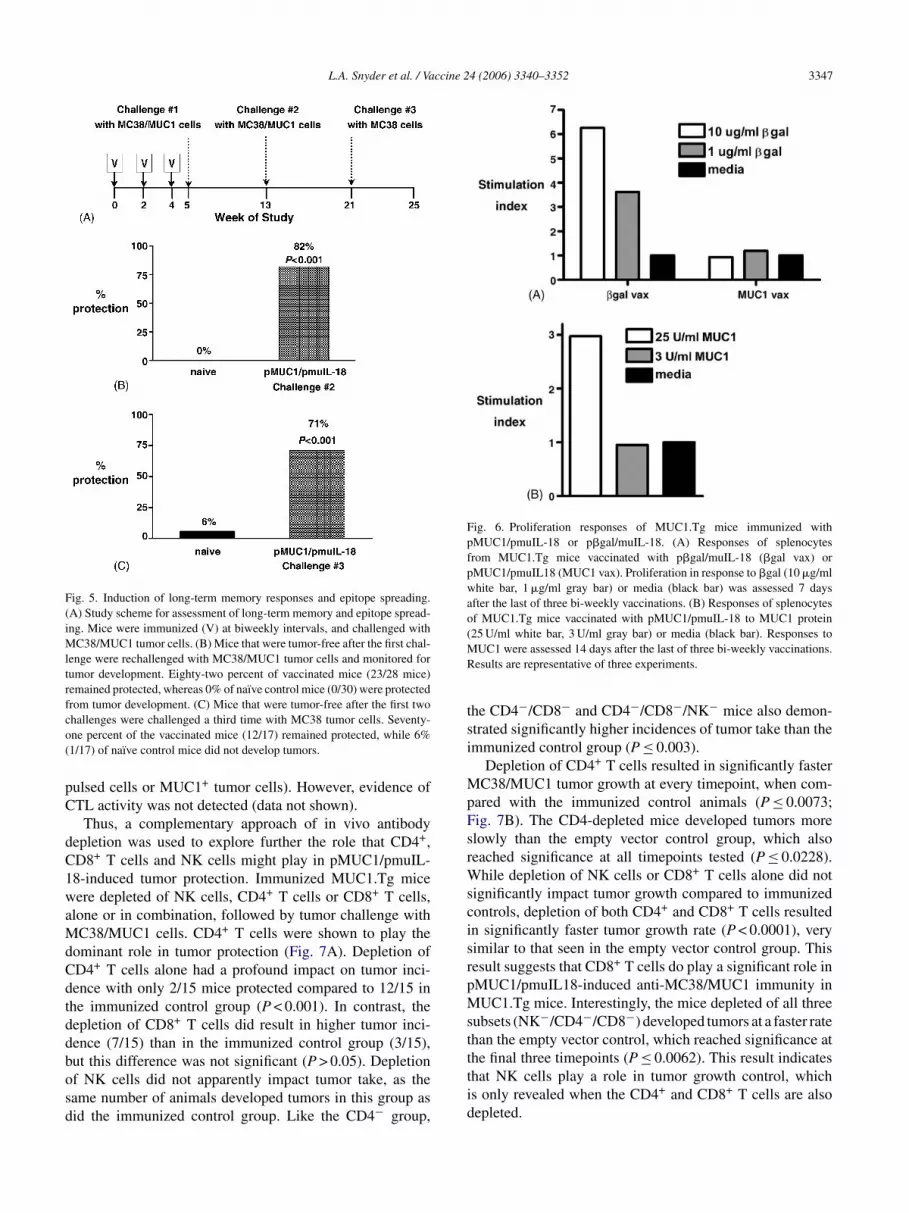
ith MC38/MUC1 tumor cells, approximately 8 weeks afterhe first challenge. Of the rechallenged mice, 82% rejectedhe second challenge (P < 0.001), though none of the naı̈ve
ice remained tumor-free (Fig. 5B). This result indicateshat the vaccinated mice, in conjunction with tumor chal-enge, developed a durable, long-term memory response to
UC1 that could be recalled upon later challenge with tumorells.
The above results indicate that the pMUC1/pmuIL-18NA vaccine induces an antigen-specific immune response
hat results in the elimination of the first tumor challengey MC38/MUC1 tumor cells. During the process of tumorell destruction, cross-priming may occur, in which otherumor-associated antigens could be presented to the immuneystem. This could result in subsequent induction of anmmune response to these epitopes, or determinant spread-ng [45]. To determine if the mice developed immuneesponses to non-MUC1 epitopes, MUC1.Tg mice that sur-ived two MC38/MUC1 tumor cell challenges were chal-enged a third time with MC38 tumor cells (Fig. 5A). Rejec-ion of MC38 by the mice would indicate that an immuneesponse had developed to epitopes shared between MC38nd MC38/MUC1 tumor cells. As shown in Fig. 5C, 71% ofice remained protected after the third challenge (P < 0.001),
hough nearly all naı̈ve mice developed tumors. In contrast,ice that are immunized with the vaccine and immediately
hallenged with MC38 tumor cells rapidly develop tumorsFig. 3C), demonstrating that the initial protective immuneesponse is MUC1-specific. These results show that over timeice developed protective immune responses to epitopes in
3346 L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352
Fig. 4. Effect of vaccine dose on tumor protection and survival in MUC1.Tg mice. MUC1.Tg mice were immunized with the vaccine doses in �g indicated inthe legend, followed by challenge with MC38/MUC1 tumor cells. EV was used at 200 �g. N = 10 mice/group. (A) Tumor incidence. **P = 0.0001, *P = 0.011.(B) Mean tumor diameter, shown ± S.E.M. ***P ≤ 0.002 for all timepoints, **P ≤ 0.009 for days 15–29, *P ≤ 0.05 for days 15–29, #significance cannot becalculated due to lack of tumor growth in this group. (C) Mice were immunized with the doses of vaccine indicated in the legend, followed by challenge withMC38/MUC1 tumor cells. Mice were monitored for tumor development for over 90 days. N = 10 mice/group. ***P < 0.0001, **P = 0.0038, *P = 0.0184.
addition to MUC1, which likely resulted from cross-primingduring MC38/MUC1 tumor challenge.
3.5. Role of CD4+, CD8+ T and NK cells in tumorprotection
To identify the mediators of anti-tumor immunity fol-lowing vaccination, humoral and cellular immune responseswere assessed. Antibody titers to MUC1 were very low orundetectable after DNA immunization, and remained loweven after MC38/MUC1 challenge (data not shown). Thisresult suggests that antibodies do not play a significant rolein tumor protection of MUC1.Tg mice following vaccina-tion with pMUC1/pmuIL18, and that cellular immunity wasresponsible for tumor protection. To understand the role that Tcells could play in this response, in vitro experiments to detectproliferation were conducted (Fig. 6). MUC1-specific prolif-erative responses were detected in MUC1.Tg mice immu-nized with pMUC1/pmuIL-18, with proliferation observedapproximately three-fold over the media control with 25 U/ml
MUC1 protein, but not at 3 U/ml (Fig. 6B). The MUC1.Tgmice responded to p�gal immunization with relativelystronger proliferative responses (Fig. 6A), indicating that thelow levels of proliferation observed with pMUC1/pmuIL-18 immunization was not because the MUC1.Tg mice wereimpaired in their response to DNA vaccination with a differ-ent antigen. Instead, the low levels of proliferation observedwith pMUC1/pmuIL-18 immunization could be due to thedifficulty of breaking tolerance to MUC1 in these mice,low precursor frequency, and/or in detecting those immuneresponses by in vitro assays. In accord with this, IFN� couldsometimes be detected at low levels in the supernatants of theproliferation assays, but this was not consistently observed(data not shown). These results suggest a role for CD4+ Tcells in promoting or mediating tumor protection. To assessCD8+ T cell responses, cytotoxic T lymphocyte (CTL) assayswere performed using a variety of approaches, including test-ing for CTL activity after several rounds of DNA vaccination,or after vaccination and tumor cell challenge [28], and usingdifferent methods to stimulate CD8+ T cells in vitro (peptide
L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352 3347
Fig. 5. Induction of long-term memory responses and epitope spreading.(A) Study scheme for assessment of long-term memory and epitope spread-ing. Mice were immunized (V) at biweekly intervals, and challenged withMC38/MUC1 tumor cells. (B) Mice that were tumor-free after the first chal-lenge were rechallenged with MC38/MUC1 tumor cells and monitored fortumor development. Eighty-two percent of vaccinated mice (23/28 mice)remained protected, whereas 0% of naı̈ve control mice (0/30) were protectedfrom tumor development. (C) Mice that were tumor-free after the first twochallenges were challenged a third time with MC38 tumor cells. Seventy-one percent of the vaccinated mice (12/17) remained protected, while 6%(1/17) of naı̈ve control mice did not develop tumors.
pulsed cells or MUC1+ tumor cells). However, evidence ofCTL activity was not detected (data not shown).
Thus, a complementary approach of in vivo antibodydepletion was used to explore further the role that CD4+,CD8+ T cells and NK cells might play in pMUC1/pmuIL-18-induced tumor protection. Immunized MUC1.Tg micewere depleted of NK cells, CD4+ T cells or CD8+ T cells,alone or in combination, followed by tumor challenge withMC38/MUC1 cells. CD4+ T cells were shown to play thedominant role in tumor protection (Fig. 7A). Depletion ofCD4+ T cells alone had a profound impact on tumor inci-dence with only 2/15 mice protected compared to 12/15 inthe immunized control group (P < 0.001). In contrast, thedepletion of CD8+ T cells did result in higher tumor inci-dence (7/15) than in the immunized control group (3/15),but this difference was not significant (P > 0.05). Depletionof NK cells did not apparently impact tumor take, as thesame number of animals developed tumors in this group asdid the immunized control group. Like the CD4− group,
Fig. 6. Proliferation responses of MUC1.Tg mice immunized withpMUC1/pmuIL-18 or p�gal/muIL-18. (A) Responses of splenocytesfrom MUC1.Tg mice vaccinated with p�gal/muIL-18 (�gal vax) orpMUC1/pmuIL18 (MUC1 vax). Proliferation in response to �gal (10 �g/mlwhite bar, 1 �g/ml gray bar) or media (black bar) was assessed 7 daysafter the last of three bi-weekly vaccinations. (B) Responses of splenocytesof MUC1.Tg mice vaccinated with pMUC1/pmuIL-18 to MUC1 protein(25 U/ml white bar, 3 U/ml gray bar) or media (black bar). Responses toMUC1 were assessed 14 days after the last of three bi-weekly vaccinations.Results are representative of three experiments.
the CD4−/CD8− and CD4−/CD8−/NK− mice also demon-strated significantly higher incidences of tumor take than theimmunized control group (P ≤ 0.003).
Depletion of CD4+ T cells resulted in significantly fasterMC38/MUC1 tumor growth at every timepoint, when com-pared with the immunized control animals (P ≤ 0.0073;Fig. 7B). The CD4-depleted mice developed tumors moreslowly than the empty vector control group, which alsoreached significance at all timepoints tested (P ≤ 0.0228).While depletion of NK cells or CD8+ T cells alone did notsignificantly impact tumor growth compared to immunizedcontrols, depletion of both CD4+ and CD8+ T cells resultedin significantly faster tumor growth rate (P < 0.0001), verysimilar to that seen in the empty vector control group. Thisresult suggests that CD8+ T cells do play a significant role inpMUC1/pmuIL18-induced anti-MC38/MUC1 immunity inMUC1.Tg mice. Interestingly, the mice depleted of all threesubsets (NK−/CD4−/CD8−) developed tumors at a faster ratethan the empty vector control, which reached significance atthe final three timepoints (P ≤ 0.0062). This result indicatesthat NK cells play a role in tumor growth control, whichis only revealed when the CD4+ and CD8+ T cells are alsodepleted.

3348 L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352
Fig. 7. Mechanism of tumor protection. Mice were immunized either withempty vector (60 �g) or with pMUC1/pmuIL-18 (30 �g doses of eachplasmid), at weeks 0, 2, and 4. At week 5, all mice were challengedwith MC38/MUC1 tumor cells. The groups designated “empty vector” and“immunized” received PBS injections instead of depleting antibodies, andrepresent negative and positive controls for tumor take, respectively. Theremaining groups were immunized with pMUC1/pmuIL-18, but were thendepleted of one or more lymphocyte subsets as indicated in the legend.N = 14–15 mice/group. (A) Tumor incidence. (B) Mean tumor diameter,shown ± S.E.M. Differences in mean tumor diameter were calculated fromdata collected on day 12 and thereafter.
4. Discussion
These studies demonstrate that a MUC1/IL-18 DNAvaccine breaks tolerance to the MUC1 self-antigen andinduces significant tumor protection in MUC1.Tg mice. Dif-ferent types of MUC1 vaccination strategies have succeededin eliciting significant MUC1-specific tumor protection inhuMUC1 mice, including viral vectors encoding MUC1/IL-2 and dendritic cell immunotherapies [18,21–23]. However,this is the first example of a MUC1 naked DNA vaccinethat has afforded significant tumor protection in a huMUC1-tolerant setting. There are reports of MUC1 DNA vaccinesthat induce tumor protection, using a variety of tumor modelsystems [26–28,32–34]. However, these studies were con-ducted in nontransgenic mouse strains in which humanMUC1 would be a foreign antigen. Thus, these MUC1 DNAvaccines would be expected to induce a robust protectiveimmune response, without the requirement for breaking tol-erance. Similar to the cited studies, pMUC1 alone protectedC57Bl/6 mice that were challenged with MC38/MUC1 tumorcells (data not shown). However, pMUC1 alone was insuf-ficient to induce significant tumor protection in MUC1.Tgmice (Fig. 2). Only when pMUC1 was co-administered withpmuIL-18 could the vaccine break tolerance in MUC1.Tgmice and induce significant tumor protection. This outcomedemonstrates the importance of an adjuvant molecule in cre-
ating a vaccine capable of breaking tolerance, as would berequired of the vaccine in a clinical setting.
To improve the efficacy of pMUC1, adjuvants that wouldenhance the induction of strong cellular immune responseswere evaluated. DNA vaccines themselves tend to skewtoward a Th1 response, at least in part because of the pres-ence of natural CpG motifs in the plasmid backbone [29].These motifs bind to toll-like receptor 9 (TLR-9) with sub-sequent induction of IFN�, which in turn upregulates MHCclass I and class II expression on antigen-presenting cellsand induces their maturation. However, the presence of nat-ural CpG motifs in the current constructs was insufficient toenhance the activity of pMUC1, as shown by the inability ofpMUC1 alone to protect mice from tumor challenge (Fig. 2).Thus, many adjuvant molecules were evaluated in combi-nation with pMUC1, among which were plasmids encodingGM-CSF, IL-12 (data not shown) or IL-18. These cytokineswere chosen because each can enhance induction of strongTh1 cellular immune responses from DNA vaccines directedtoward infectious disease and tumor targets [29]. However,only pmuIL-18 synergized with pMUC1 to induce significanttumor protection in this system.
It is interesting to speculate why IL-18 is an effective adju-vant in this setting. IL-18 was identified based on its abilityto synergize with IL-12 to increase induction of IFN� [46].In the presence of IL-12, IL-18 stimulates the differentia-tiasTrttcmeicidcocIwaaooui
afv
ion of naı̈ve T cells to Th1 cells, which may be criticallymportant for an adjuvant molecule. IL-18 induces NK cellctivation, which can lead to destruction of tumor cells andubsequent induction of a tumor-specific immune response.hese IL-18 activities bridge the innate and adaptive immune
esponses, and are the basis for the use of IL-18 protein inreatment of cancer in preclinical models [47,48], as well as inhe clinic. These properties make IL-18 attractive for use in aancer DNA vaccine. IL-18 has other distinct properties thatay make it a strong adjuvant molecule. For example, IL-18
xpression from an injected IL-18 plasmid has been shown tonduce a localized inflammatory infiltrate, which was asso-iated with subsequent development of an antigen-specificmmune response [49]. IL-18 is a chemoattractant for den-ritic cells [50,51] and T cells [52], and induces Langerhansell (LC) migration to draining lymph nodes [53]. If thesebservations can be applied to the pMUC1/pmuIL-18 vac-ine, injection of pmuIL-18 would result in locally producedL-18 that could recruit DC and enhance their migration. Thisould indirectly enhance pMUC1 plasmid/antigen uptake
nd presentation. In conjunction with increased NK activitynd T cell activation, this may lead to more effective inductionf a protective immune response in a tolerant setting. Basedn our results, it is possible that IL-18 is uniquely suited forse as an adjuvant molecule in oncology vaccines, as well asn infectious disease vaccines [29,54].
In the context of a DNA vaccine, activities of IL-18re likely to be exerted in a localized rather than systemicashion. Relatively small amounts of the pMUC1/pmuIL-18accine are used for immunization, and regional uptake and

L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352 3349
expression of MUC1 antigen and IL-18 would be expected atthe injection site. Consistent with this, systemic increases inneither IL-18 protein nor IFN� were observed after pmuIL-18 administration (data not shown). Although other studieshave shown that IL-18 protein in large amounts can inducetumor protection [47,48], it is unlikely to be responsible forprotection in this model, as mice immunized with pmuIL-18 alone show no protection (Fig. 3). In addition, the factthat tumor protection requires that pMUC1 and pmuIL-18 beadministered in a mixture, rather than at separate sites (Fig. 3),supports the idea that localized co-expression of antigen andadjuvant is required for the generation of a systemic and pro-tective anti-MUC1 immune response.
The MUC1 immunotherapy literature indicates that cel-lular immune responses are required for tumor protection[55–57], while antibody responses to MUC1 play littlerole, if any [58]. Consistent with this, anti-MUC1 antibodyresponses are low or undetectable in pMUC1/pmuIL-18 vac-cinated mice, and therefore appear unimportant in mediatingtumor protection induced by the vaccine. In vitro assays indi-cated that the vaccine induced MUC1-specific proliferativeresponses, most likely by CD4+ T cells, but CTL activitycould not be detected. This latter result could be ascribed tolow precursor frequency in a tolerant setting, as well as thatCTL assays are conducted under conditions that are nonphys-iologic and thus may not mimic the microenvironment at thetdctcicTooII(ebpsGbl
tpiuo[dit
tumor challenge, without any vaccination, only CD4+ T cellsmediated tumor growth control, with no apparent role forCD8+ T cells [56,59]. Further, the soluble factors that medi-ated tumor growth control included FasL, consistent witha role for lytic CD4+ T cells, and probably IFN�, whichcould be secreted by CD4+ T cells to stimulate the NK cellinvolvement in tumor growth control [59]. The cellular medi-ators in that model differ from those defined here, in thatthe pMUC1/pmuIL-18 vaccine induces an immune responsedominated by CD4+ T cells, but with a significant role forCD8+ T cells and a small but detectable role for NK cells.The differences in the effectors seen in these two modelscould arise because of differences in how antigen is processedand presented after vaccination versus after tumor challengealone. Our results do indicate that in order to be able to controlthe growth of MC38/MUC1 tumors, the activity of multiplecell types is required, whereas the activation of a single celltype might be inadequate. Future experiments to assess therelevant in vivo mediators of tumor growth control used by thecellular effectors generated by the vaccine, such as Fas/FasL,perforin, IL-12, etc., could include the generation of the rele-vant knockout mice on the MUC1.Tg background, followedby vaccination and tumor challenge.
As a soluble factor, IFN� may be of particular interest, asit is known to enhance immune and nonimmune mechanismswhich can aid tumor regression, including direct cytotoxic-iiabiitwdcattpsaItco
ttM(ttatg
umor site. However, the results of in vivo depletion studiesemonstrate clearly that CD4+ T cells play a dominant role inontrolling tumor initiation, while tumor progression is con-rolled by CD4+ and to a lesser degree by CD8+ T cells. NKells appear to play a minor role in tumor growth control thats only revealed when T cells are co-depleted. The CD4+ Tell function could be exerted indirectly, through providing
cell help to CD8+ T cells, and/or activation of NK cellsr other effectors, as well as directly, by CD4-mediated lysisf tumor cells. The MC38/MUC1 tumor cells express Class, which is upregulated by incubation with IFN�, but ClassI expression cannot be detected even with IFN� treatmentdata not shown). The fact that Class II molecules are notxpressed by the tumor cells would argue against the possi-ility that CD4+ T cells are lytic in this model. However, it isossible that the in vivo expression of Class II at the tumorite differs from what can be recapitulated in tissue culture.iven the expression of Class I molecules by the tumor cells,efore and after IFN� treatment, and the depletion data, aytic role for CD8+ T cells is likely.
The MUC1 immunotherapy literature varies with respecto the roles that CD4+ and CD8+ T cells play in tumorrotection in MUC1.Tg mice. This variability may be duen part to different tumor model systems, as some studiesse subcutaneous or lung metastasis tumor challenge, whilethers measure immune responses to autochthonous tumors55–57]. It has been observed that CD4+ T cells can play aominant role in mediating protection from MUC1+ tumorsn experimental systems. For example, in a B16.MUC1 modelhat assessed anti-MUC1 immune responses generated after
ty in combination with tumor necrosis factor alpha (TNF�),nduction of nitric oxide production, slowed cellular prolifer-tion and inhibition of angiogenesis [60–62]. IFN� is secretedy antigen-specific T cells, induced through the synergisticnteractions between IL-12 and IL-18, or secreted by NK cellsn direct response to activation by IL-18. The fact that deple-ion of NK cells had a small impact on tumor growth thatas only detected when CD4+ and CD8+ T cells were alsoepleted suggests that NK cells or NK-derived IFN� directlyontributed little to anti-tumor responses. This may not besurprising result, because vaccination with pmuIL-18 is
emporally separate from tumor cell challenge, and becausehere is no detectable systemic increase in IL-18 or IFN� aftermuIL-18 vaccination (data not shown). There may not beufficient systemic IL-18 present at the right time to stimulaten NK response that plays a major role in tumor protection.f IFN� plays a significant role in this model, it is more likelyo be derived from antigen-specific T cells. The role of IFN�ould be evaluated through antibody depletion experiments,r through generation of IFN� KO/MUC1.Tg mice.
The induction of a memory response is a desired fea-ure of a cancer vaccine. These results demonstrate clearlyhat the pMUC1/pmuIL-18 vaccine, in conjunction with
C38/MUC1 tumor challenge, induces a memory responseof at least 8 weeks) to MUC1 that afforded significant pro-ection from secondary tumor challenge. To our knowledge,his study is the first to report long-term protection in thebsence of additional vaccination in a huMUC1 tolerant set-ing. As it is important to be able to control the immediaterowth of MUC1+ tumors by the vaccine treatment, as well

3350 L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352
as for the life of the patient if tumors recur, it is encourag-ing to see that the pMUC1/pmuIL-18 vaccine can induce amemory response that continues to protect the animal duringsubsequent tumor challenge.
The rechallenge results demonstrate that the mice devel-oped immunity to MC38/MUC1− cells after immunizationand challenge with MC38/MUC1+ tumor cells. This find-ing suggests that the mice were able to develop immuneresponses to other epitopes expressed in common on thetumor cell lines, which is known as determinant spreading[45]. Determinant spreading is most likely achieved throughantigen cross-priming during lysis and clearance of tumorcells. This is an important observation for the utility of thevaccine in the clinic, as it is known that human tumors canbe heterogeneous with respect to MUC1 antigen expression[3], and that escape mechanisms for tumors include selec-tive downregulation/expression of surface antigens [63]. Avaccine that induces immune responses limited to MUC1epitopes could offer disease control for a period of time, butonly until escape variants lacking MUC1 expression arise.For more sustained control of tumor growth in patients,the ideal vaccine will either include multiple antigens, orinduce immune responses or determinant spreading to multi-ple tumor epitopes. It is encouraging therefore that the micevaccinated with pMUC1/pmuIL-18 develop such a multi-epitope response.
vtahdvsspTdthrep
1ttfvtovsaDM
Acknowledgements
We thank Dr. Donald Kufe for providing reagents andhelpful advice, Drs. Mani Lakshminarayanan and LilianeKim for performing the statistical analyses, and Dr. BernieScallon for advice on pmuIL-18 construction. We thank Dr.Deborah Marshall for assistance with flow cytometry experi-ments, and Eva Emmell and George Treacy for their supportwith animal studies. We thank Patti Sassoli for assistancewith manuscript preparation.
References
[1] Gendler SJ, Spicer AP. Epithelial mucin genes. Annu Rev Physiol1995;57:607–34.
[2] Girling A, Bartkova J, Burchell J, Gendler S, Gillett C, Taylor-Papadimitriou J. A core protein epitope of the polymorphic epithe-lial mucin detected by the monoclonal antibody SM-3 is selec-tively exposed in a range of primary carcinomas. Int J Cancer1989;43(6):1072–6.
[3] Ho SB, Niehans GA, Lyftogt C, et al. Heterogeneity of mucingene expression in normal and neoplastic tissues. Cancer Res1993;53(3):641–51.
[4] Kotera Y, Fontenot JD, Pecher G, Metzgar RS, Finn OJ. Humoralimmunity against a tandem repeat epitope of human mucin MUC-1in sera from breast, pancreatic, and colon cancer patients. CancerRes 1994;54(11):2856–60.
[
[
[
[
The results obtained with the pMUC1/pmuIL-18 DNAaccine demonstrate that a DNA vaccine can break toleranceo the MUC1 self-antigen and induce tumor protection insubcutaneous tumor challenge model. The next importanturdle is to evaluate whether the vaccine can treat pre-existingisease, which is how it would be used in patients. Otheraccination strategies, such as DC/tumor cell fusions, havehown the ability to prevent tumors in tolerant settings, and inome instances, to treat pre-existing tumor burden in similarreclinical models to the one used in this report [21,25,55].o address whether pMUC1/pmuIL-18 can treat pre-existingisease, the vaccine has been tested in a lung metastasisumor treatment model with MC38/MUC1 tumor cells, whichas yielded positive results (manuscript in preparation). Theesults indicate that the pMUC1/pmuIL-18 DNA vaccine isfficacious in multiple tumor settings, which also lends sup-ort to the idea that it could be efficacious clinically.
In summary, these results show that the pMUC1/pmuIL-8 DNA vaccine is a unique immunotherapy that breaksolerance to the MUC1 self-antigen and induces significantumor protection. The IL-18 adjuvant plasmid was requiredor induction of tumor protection, whereas many other adju-ants failed, suggesting that properties of IL-18 may makehis cytokine especially well suited as an adjuvant for this andther vaccines. Though high doses of the pMUC1/pmuIL-18accine induced the most profound tumor protection, evenmall doses induced significant survival benefit in vaccinatednimals. The results suggest strongly that a MUC1/IL-18NA vaccine may offer clinical benefit to patients withUC1+ tumors.
[5] von Mensdorff-Pouilly S, Petrakou E, Kenemans P, et al. Reactiv-ity of natural and induced human antibodies to MUC1 mucin withMUC1 peptides and n-acetylgalactosamine (GalNAc) peptides. Int JCancer 2000;86(5):702–12.
[6] Barnd DL, Lan MS, Metzgar RS, Finn OJ. Specific, major histo-compatibility complex-unrestricted recognition of tumor-associatedmucins by human cytotoxic T cells. Proc Natl Acad Sci USA1989;86(18):7159–63.
[7] Ioannides CG, Fisk B, Fan D, Biddison WE, Wharton JT, O’BrianCA. Cytotoxic T cells isolated from ovarian malignant ascites rec-ognize a peptide derived from the HER-2/neu proto-oncogene. CellImmunol 1993;151(1):225–34.
[8] Jerome KR, Barnd DL, Bendt KM, et al. Cytotoxic T-lymphocytesderived from patients with breast adenocarcinoma recognize anepitope present on the protein core of a mucin molecule pref-erentially expressed by malignant cells. Cancer Res 1991;51(11):2908–16.
[9] Domenech N, Henderson RA, Finn OJ. Identification of an HLA-A11-restricted epitope from the tandem repeat domain of the epithe-lial tumor antigen mucin. J Immunol 1995;155(10):4766–74.
10] Hiltbold EM, Ciborowski P, Finn OJ. Naturally processed class IIepitope from the tumor antigen MUC1 primes human CD4+ T cells.Cancer Res 1998;58(22):5066–70.
11] Brossart P, Heinrich KS, Stuhler G, et al. Identification of HLA-A2-restricted T-cell epitopes derived from the MUC1 tumor antigen forbroadly applicable vaccine therapies. Blood 1999;93(12):4309–17.
12] Carmon L, El-Shami KM, Paz A, et al. Novel breast-tumor-associated MUC1-derived peptides: characterization inDb−/− × beta2 microglobulin (beta2m) null mice transgenic for achimeric HLA-A2.1/Db-beta2 microglobulin single chain. Int J Can-cer 2000;85(3):391–7.
13] Heukamp LC, van der Burg SH, Drijfhout JW, Melief CJ, Taylor-Papadimitriou J, Offringa R. Identification of three non-VNTRMUC1-derived HLA-A*0201-restricted T-cell epitopes that induceprotective anti-tumor immunity in HLA-A2/K(b)-transgenic mice.Int J Cancer 2001;91(3):385–92.

L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352 3351
[14] Ding L, Lalani EN, Reddish M, et al. Immunogenicity of syntheticpeptides related to the core peptide sequence encoded by the humanMUC1 mucin gene: effect of immunization on the growth of murinemammary adenocarcinoma cells transfected with the human MUC1gene. Cancer Immunol Immunother 1993;36(1):9–17.
[15] Zhang S, Graeber LA, Helling F, et al. Augmenting the immuno-genicity of synthetic MUC1 peptide vaccines in mice. Cancer Res1996;56(14):3315–9.
[16] Gilewski T, Adluri S, Ragupathi G, et al. Vaccination of high-risk breast cancer patients with mucin-1 (MUC1) keyhole limpethemocyanin conjugate plus QS-21. Clin Cancer Res 2000;6(5):1693–701.
[17] Soares MM, Mehta V, Finn OJ. Three different vaccines based onthe 140-amino acid MUC1 peptide with seven tandemly repeatedtumor-specific epitopes elicit distinct immune effector mechanismsin wild-type versus MUC1-transgenic mice with different potentialfor tumor rejection. J Immunol 2001;166(11):6555–63.
[18] Acres B, Apostolopoulos V, Balloul JM, et al. MUC1-specificimmune responses in human MUC1 transgenic mice immunizedwith various human MUC1 vaccines. Cancer Immunol Immunother2000;48(10):588–94.
[19] Scholl SM, Balloul JM, Le Goc G, et al. Recombinant vaccinia virusencoding human MUC1 and IL2 as immunotherapy in patients withbreast cancer. J Immunother 2000;23(5):570–80.
[20] Rochlitz C, Figlin R, Squiban P, et al. Phase I immunotherapy with amodified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advancedcancer. J Gene Med 2003;5(8):690–9.
[21] Gong J, Chen D, Kashiwaba M, et al. Reversal of tolerance tohuman MUC1 antigen in MUC1 transgenic mice immunized withfusions of dendritic and carcinoma cells. Proc Natl Acad Sci USA
[
[
[
[
[
[
[
[
[
[
[
the same region greatly enhanced MUC1-specific antitumor immu-nity in a murine model. Cancer Gene Ther 2002;9(4):330–7.
[33] Kamata M, Denda-Nagai K, Kubota N, Aida S, Takeda K, Irimura T.Vaccination of mice with MUC1 cDNA suppresses the developmentof lung metastases. Clin Exp Metastasis 2002;19(8):689–96.
[34] Vasilevko V, Ghochikyan A, Sadzikava N, et al. Immunization witha vaccine that combines the expression of MUC1 and B7 co-stimulatory molecules prolongs the survival of mice and delaysthe appearance of mouse mammary tumors. Clin Exp Metastasis2003;20(6):489–98.
[35] Rowse GJ, Ritland SR, Gendler SJ. Genetic modulation ofneu proto-oncogene-induced mammary tumorigenesis. Cancer Res1998;58(12):2675–9.
[36] Hartikka J, Sawdey M, Cornefert-Jensen F, et al. An improved plas-mid DNA expression vector for direct injection into skeletal muscle.Hum Gene Ther 1996;7(10):1205–17.
[37] Kozak M. The scanning model for translation: an update. J Cell Biol1989;108(2):229–41.
[38] Ligtenberg MJ, Kruijshaar L, Buijs F, van Meijer M, LitvinovSV, Hilkens J. Cell-associated episialin is a complex containingtwo proteins derived from a common precursor. J Biol Chem1992;267(9):6171–7.
[39] Fox BA, Spiess PJ, Kasid A, et al. In vitro and in vivo antitumorproperties of a T-cell clone generated from murine tumor-infiltratinglymphocytes. J Biol Response Mod 1990;9(5):499–511.
[40] Akagi J, Hodge JW, McLaughlin JP, et al. Therapeutic antitu-mor response after immunization with an admixture of recombi-nant vaccinia viruses expressing a modified MUC1 gene and themurine T-cell costimulatory molecule B7. J Immunother 1997;20(1):38–47.
[41] Dialynas DP, Wilde DB, Marrack P, et al. Characterization of
[
[
[
[
[
[
[
[
[
[
1998;95(11):6279–83.22] Koido S, Kashiwaba M, Chen D, Gendler S, Kufe D, Gong J.
Induction of antitumor immunity by vaccination of dendritic cellstransfected with MUC1 RNA. J Immunol 2000;165(10):5713–9.
23] Tanaka Y, Koido S, Chen D, Gendler SJ, Kufe D, Gong J. Vaccina-tion with allogeneic dendritic cells fused to carcinoma cells inducesantitumor immunity in MUC1 transgenic mice. Clin Immunol2001;101(2):192–200.
24] Pecher G, Haring A, Kaiser L, Thiel E. Mucin gene (MUC1) trans-fected dendritic cells as vaccine: results of a phase I/II clinical trial.Cancer Immunol Immunother 2002;51(11/12):669–73.
25] Chen D, Xia J, Tanaka Y, et al. Immunotherapy of spontaneousmammary carcinoma with fusions of dendritic cells and mucin 1-positive carcinoma cells. Immunology 2003;109(2):300–7.
26] Graham RA, Burchell JM, Beverley P, Taylor-Papadimitriou J. Intra-muscular immunisation with MUC1 cDNA can protect C57 micechallenged with MUC1-expressing syngeneic mouse tumour cells.Int J Cancer 1996;65(5):664–70.
27] Johnen H, Kulbe H, Pecher G. Long-term tumor growth suppressionin mice immunized with naked DNA of the human tumor antigenmucin (MUC1). Cancer Immunol Immunother 2001;50(7):356–60.
28] Plunkett T, Graham R, Correa I, et al. Protection against MUC1expressing mouse tumours by intra-muscular injection of MUC1cDNA requires functional CD8+ and CD4+ T cells but doesnot require the MUC1 tandem repeat domain. Int J Cancer2004;109(5):691–7.
29] Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology,application, and optimization. Annu Rev Immunol 2000;18:927–74.
30] Wang R, Doolan DL, Le TP, et al. Induction of antigen-specificcytotoxic T lymphocytes in humans by a malaria DNA vaccine.Science 1998;282(5388):476–80.
31] Calarota S, Bratt G, Nordlund S, et al. Cellular cytotoxic responseinduced by DNA vaccination in HIV-1-infected patients. Lancet1998;351(9112):1320–5.
32] Kontani K, Taguchi O, Ozaki Y, et al. Novel vaccination protocolconsisting of injecting MUC1 DNA and nonprimed dendritic cells at
the murine antigenic determinant, designated L3T4a, recognized bymonoclonal antibody GK1.5: expression of L3T4a by functional Tcell clones appears to correlate primarily with class II MHC antigen-reactivity. Immunol Rev 1983;74:29–56.
42] Ledbetter JA, Herzenberg LA. Xenogeneic monoclonal antibod-ies to mouse lymphoid differentiation antigens. Immunol Rev1979;47:63–90.
43] Kasai M, Iwamori M, Nagai Y, Okumura K, Tada T. A glycol-ipid on the surface of mouse natural killer cells. Eur J Immunol1980;10(3):175–80.
44] Habu S, Fukui H, Shimamura K, et al. In vivo effects of anti-asialoGM1. I. Reduction of NK activity and enhancement of transplantedtumor growth in nude mice. J Immunol 1981;127(1):34–8.
45] Ribas A, Timmerman JM, Butterfield LH, Economou JS. Deter-minant spreading and tumor responses after peptide-based cancerimmunotherapy. Trends Immunol 2003;24(2):58–61.
46] Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol2001;19:423–74.
47] Micallef MJ, Yoshida K, Kawai S, et al. In vivo antitumor effectsof murine interferon-gamma-inducing factor/interleukin-18 in micebearing syngeneic Meth A sarcoma malignant ascites. CancerImmunol Immunother 1997;43(6):361–7.
48] Osaki T, Peron JM, Cai Q, et al. IFN-gamma-inducing factor/IL-18administration mediates IFN-gamma- and IL-12-independent antitu-mor effects. J Immunol 1998;160(4):1742–9.
49] Kremer L, Dupre L, Wolowczuk I, Locht C. In vivo immunomodu-lation following intradermal injection with DNA encoding IL-18. JImmunol 1999;163(6):3226–31.
50] Kaser A, Kaser S, Kaneider NC, Enrich B, Wiedermann CJ, TilgH. Interleukin-18 attracts plasmacytoid dendritic cells (DC2s) andpromotes Th1 induction by DC2s through IL-18 receptor expression.Blood 2004;103(2):648–55.
51] Gutzmer R, Langer K, Mommert S, Wittmann M, Kapp A, WerfelT. Human dendritic cells express the IL-18R and are chemoattractedto IL-18. J Immunol 2003;171(12):6363–71.

3352 L.A. Snyder et al. / Vaccine 24 (2006) 3340–3352
[52] Komai-Koma M, Gracie JA, Wei XQ, et al. Chemoattraction ofhuman T cells by IL-18. J Immunol 2003;170(2):1084–90.
[53] Cumberbatch M, Dearman RJ, Antonopoulos C, Groves RW, Kim-ber I. Interleukin (IL)-18 induces Langerhans cell migration by atumour necrosis factor-alpha- and IL-1beta-dependent mechanism.Immunology 2001;102(3):323–30.
[54] Zhu M, Xu X, Liu H, et al. Enhancement of DNA vaccine potencyagainst herpes simplex virus 1 by co-administration of an interleukin-18 expression plasmid as a genetic adjuvant. J Med Microbiol2003;52:223–8.
[55] Gong J, Chen D, Kashiwaba M, Kufe D. Induction of antitumoractivity by immunization with fusions of dendritic and carcinomacells. Nat Med 1997;3(5):558–61.
[56] Tempero RM, VanLith ML, Morikane K, Rowse GJ, GendlerSJ, Hollingsworth MA. CD4+ lymphocytes provide MUC1-specifictumor immunity in vivo that is undetectable in vitro and is absentin MUC1 transgenic mice. J Immunol 1998;161(10):5500–6.
[57] Mukherjee P, Ginardi AR, Madsen CS, et al. Mice with sponta-neous pancreatic cancer naturally develop MUC-1-specific CTLsthat eradicate tumors when adoptively transferred. J Immunol2000;165(6):3451–60.
[58] Tempero RM, Rowse GJ, Gendler SJ, Hollingsworth MA. Passivelytransferred anti-MUC1 antibodies cause neither autoimmune disor-ders nor immunity against transplanted tumors in MUC1 transgenicmice. Int J Cancer 1999;80(4):595–9.
[59] VanLith ML, Kohlgraf KG, Sivinski CL, Tempero RM,Hollingsworth MA. MUC1-specific anti-tumor responses: molec-ular requirements for CD4-mediated responses. Int Immunol2002;14(8):873–82.
[60] Revel M, Chebath J. Interferon-activated genes. Trends Biochem Sci1986;11(4):166–70.
[61] Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encod-ing calcium-independent nitric oxide synthase confers inducibilityby interferon gamma and bacterial lipopolysaccharide. J Exp Med1993;177(6):1779–84.
[62] Coughlin CM, Salhany KE, Wysocka M, et al. Interleukin-12and interleukin-18 synergistically induce murine tumor regressionwhich involves inhibition of angiogenesis. J Clin Invest 1998;101(6):1441–52.
[63] Gilboa E. The promise of cancer vaccines. Nat Rev Cancer 2004;4(5):401–11.