6 pathogenesis of copd - de gruyter
TRANSCRIPT

Michael T. Borchers, PhD, Gregory Motz, PhD6 Pathogenesis of COPDKey Points1. The pathogenesis of COPD is a complex, multifactorial process that includes
genetics, proteolytic imbalance, oxidative stress, inflammation, occupational and environmental exposures, and innate and adaptive immune function.
2. Excessive secretion of proteases and inhibition of antiproteases contribute to the degradation of lung extracellular matrix.
3. Oxidative stress can directly injure alveolar epithelial cells and induce mucus secretion of airway epithelial cells.
4. The chronic inflammation that defines COPD is a strong correlate of disease severity in patients and is critically involved in disease development experimentally.
5. The pathogenesis of COPD includes extrapulmonary manifestations including systemic inflammation, cardiovascular disease, nutritional abnormalities and skeletal muscle dysfunction.
6.1 Introduction
Pathologically, COPD is defined as a combination of emphysema, small airway disease (fibrosis, scarring, increases in smooth muscle surrounding the airways), and chronic bronchitis (inflammation and mucus hypersecretion). The extent of each condition varies within individual patients (Barnes, 2003; Chung, 2008; Cosio, 2009). These pathological conditions affect both the large and small airways, as well as the lung parenchyma. The reduction in lung elasticity caused by the destruction of alveoli (emphysema) produces a loss of support and closure of the small airways during expiration (Barnes, 2004). Additionally, the small airways are physically narrowed by fibrotic scarring and an increase in the surrounding smooth muscle mass. This obstruction to airflow diminishes expiratory alveolar emptying and produces air trapping and hyperinflation (Barnes, 2004). Finally, a mucus‐rich inflammatory exudate clogs the airways augmenting airflow resistance (Barnes, 2004). Together, these changes contribute to airflow obstruction that causes a reduction in normal lung function, measured as FEV1 (forced expiratory volume in 1 second). In addition, COPD is associated with multiple other pulmonary diseases such as fibrosis, asthma, and lung cancer.

77 Pathogenesis of COPD
6.2 Chronic Bronchitis
Chronic bronchitis is defined clinically as a productive cough of greater than 3 months
duration for more than two successive years, and is characterized by inflammation and excessive mucus production. The role of airway inflammation in COPD forms the basis of our current understanding of the pathogenesis of COPD and is elaborated upon in detail below. In COPD, mucus overproduction can physically plug the airways, and mucus hypersecretion is associated with a decline in FEV1 (Prescott, 1995; Vestbo, 1996). Further, a role for mucus production in COPD is supported by the specific observation that accumulation of inflammatory exudates and mucus in the small airways increases with disease severity (Hogg, 2004). Mechanistically, it is believed that mucus/goblet cell hyperplasia and cellular phenotype alterations caused by cigarette smoking are the major causes of mucus hypersecretion in COPD (Innes, 2006).
6.3 Emphysema
Emphysema is characterized by a loss of lung elasticity (increased pulmonary compliance) caused by parenchymal lung destruction. Smoking causes both panacinar (destruction of parenchymal alveoli) and centriacinar (destruction around respiratory bronchioles) emphysema (Kim, 1991). Emphysema is a significant contributor to airflow obstruction in COPD and several studies have demonstrated correlations between the severity of macroscopic emphysema and measurable lung function decline (Hogg, 1994; Nakano, 2000). The causes of emphysema are multifactorial, but the best studied hypothesis for the development of emphysema is a protease:antiprotease imbalance leading to the destruction of lung tissue (Chung, 2008). Another major mechanism causing emphysema is lung epithelial and endothelial cell apoptosis. Apoptosis may by caused by oxidative stress or the cytotoxic functions of activated lymphocytes (e.g. CD8+ T cells, NK cells) that release perforin and granzymes directly killing lung epithelial cells. Finally, increased cellular senescence and a failure of lung maintenance and repair after chronic cigarette smoke exposure may also contribute to the development of emphysema (Chung, 2008).
6.4 Smooth Muscle
The amount of smooth muscle surrounding the airways, defined by total area, is increased in COPD patients and the amount of smooth muscle inversely correlates with lung function (FEV1) (Bosken, 1990; Cosio, 1980; Saetta, 1998). In patients with severe COPD (GOLD stages 3 and 4), the amount of smooth muscle may be increased as much as 50% (Hogg, 2004). It is not entirely known how, or whether, increases in

Pathogenesis of COPD 78
smooth muscle contribute to the development of COPD. However, it has been speculated that the release of smooth musclederived inflammatory mediators in response to cigarette smoke or increased contractility may be involved (Chung, 2008).
6.5 Fibrosis
In addition to increases in periairway smooth muscle mass in COPD patients, there is also a significant increase in collagen deposition and fibrotic scarring around the small airways (Chung, 2008; Hogg, 2004). The fibrotic process is believed to further restrict proper contractility of the airways during breathing, thus contributing to airflow obstruction (Hogg, 2004). It is not entirely known how the development of fibrosis proceeds, both transforming growth factor β (TGFβ) and connective tissue growth factor (CTGF) levels are elevated in the airways of COPD patients (de Boer, 1998; Takizawa, 2001). TGFβ may cause fibrosis by inducing the expression of CTGF, which in turn drives the production and deposition of collagen (Ihn, 2002).
6.6 Pathogenesis of COPD
6.6.1 Genetics: Gene-association Studies
Early studies estimated that only 10 to 15% of smokers develop COPD (Rennard, 2006). However, as early COPD is often asymptomatic, it is believed that COPD is significantly underdiagnosed (Coultas, 2001). More recent studies estimate the prevalence of COPD is as high as 20% and that up to 50% of smokers may have spirometric evidence of airflow obstruction (Buist, 2008; Halbert, 2006; Stang, 2000). Most lifelong smokers will develop COPD eventually, provided they do not die from nonpulmonary smokingrelated diseases first (e.g. cardiovascular disease, metabolic disease, cancer) (Rennard, 2006). However, a small proportion of individuals will develop very severe COPD even after only smoking for a relatively short period of time (or as lifelong never smokers). The appreciation that the development of COPD in individuals with similar smoking histories is heterogenious has led to fervent investigation into genetic risk factors that contribute to disease development. The first gene associated with the development of COPD was the α1antitrypsin gene. Individuals who smoke and are homozygous for mutations of this gene develop severe COPD at a very early age. However, only about 1–2% of all COPD patients have this particular deficiency (Wan & Silverman, 2009). Studies involved in the identification of additional genes that confer susceptibility to COPD have been difficult due to the extreme phenotypic heterogeneity among COPD patients (Wan, 2009). Therefore, nonreplication of genetic association studies is extremely commonplace in COPD studies (Wan, 2009). However, there are a number of genetic associations that have been sufficiently rep

79 Pathogenesis of COPD
licated across several labs. Replicated geneassociation studies have identified proteases (MMP9, MMP12), detoxification/oxidative stress genes (EPHX1, HMOX1, GSTP1), genes involved in the immune response (TNFα, GC), and genes regulating apoptosis (TGFβ) as genes associated with development of COPD (Wan, 2009).
6.6.2 Epigenetics
In addition to classical geneassociation studies, epigenetic control of gene expression is thought to be involved in COPD. However, as the field itself is relatively new, there are few studies that address this issue in COPD. Histone acetylation typically enhances gene transcriptional activity, and acetylation status is controlled by an enzyme that removes acetyl groups called histone deacetylase. With increasing disease severity, there is a corresponding decrease in overall histone deacetylase activity (increased histone acetylation) in the lungs of COPD patients (Ito, 2005). Therefore, this increased acetylation is believed to increase the transcriptional activity of proinflammatory genes in COPD patients. Supporting this hypothesis, COPD patients exhibit increased levels of histone acetylation and IL‐8 expression (Ito, 2005). These observations are supported by animal studies in smoke exposed rats that demonstrate decreased histone deacetylase activity, increased histone acetylation, and enhanced NF‐κB binding (Marwick, 2004). It is believed that histone deacetylase activity is reduced due to ubiquitination and subsequent degradation after cigarette smoke exposure (Adenuga, 2009).
6.6.3 Protease: Antiprotease Imbalance
The most well‐studied mechanism of COPD pathogenesis is an imbalance in pulmonary protease and antiprotease levels. This hypothesis developed from the observation that some smokers with reduced levels of α1‐antitrypsin develop emphysema at a very early age. However, these individuals only represent a very small minority of COPD patients (1–2%). Many COPD patients develop a protease:antiprotease imbalance through the overproduction of proteases by activated neutrophils and macrophages (Barnes, 2003). Proteases are important for the development of emphysema because these enzymes, particularly elastin, physically digest the extracellular matrix. A role for proteases in COPD development is supported by the demonstration that the instillation of a number of different proteases into the lungs of animals produces significant emphysema (Barnes, 2003). Elastase derived from neutrophils has received significant attention because it is the molecular target of α1‐antitrypsin. Levels of neutrophil elastase are increased in COPD patients and instillation of neutrophil elastase into rodent lungs induces emphysema (Barnes, 2003). In addition to physical destruction of the airways, neutrophil elastase is also believed to be involved in inflammatory

Pathogenesis of COPD 80
cell recruitment and mucus production (Barnes, 2003). Further, chemical inhibition of neutrophil elastase prevents inflammation and emphysema in guinea pigs exposed to cigarette smoke (Table 6.1) (Wright, 2002). Although neutrophil elastase has received a significant amount of attention, a broad range of proteases are activated in COPD. Increased concentrations of cysteine proteases (cathepsins) have been observed in COPD patients, and chemical inhibition in rodent models prevents the development of a number of COPD phenotypes (Table 6.1) (Barnes, 2003). Further, matrix metalloproteinases (MMPs) may also be involved in disease development as a number of MMPs are upregulated in COPD patients and MMP knockout mice exposed to cigarette smoke are protected from the development of COPD phenotypes (Table 6.1) (Barnes, 2003).
6.6.4 Oxidative Stress
Cigarette smoke contains 1017 oxidant molecules per puff and is a significant source of reactive oxygen and reactive nitrogen species (ROS and RNS) (MacNee, 2000). There is considerable evidence of elevated oxidative stress in the lungs of cigarette smokers and there is an even greater increase in individuals with COPD (MacNee, 2005). The evidence in COPD patients includes increased levels of exhaled H2O2, 8isopostane, and NO, all indirect measures of oxidative stress (MacNee, 2005). Additionally, COPD patients exhibit a decrease in plasma antioxidants, and an increase of oxidized lipids and proteins in plasma and tissues (MacNee, 2005). Based upon this evidence, it has been proposed that oxidative stress may be critical to the development and progression of COPD. Mechanistically, oxidative stress is believed to contribute to a number of important COPD phenotypes. Inactivation of α1‐antitrypsin occurs by oxidation and a decrease in antiprotease activity is observed in bronchoalveolar lavage (BAL) fluid obtained after smoke exposure. Therefore, it is believed that physical inactivation of antiproteases by oxidative stress may significantly contribute to the protease:antiprotease imbalance in COPD patients (MacNee, 2005). Oxidative metabolites can induce the secretion of mucus from airway epithelial cells, and therefore, may be involved in mucus hypersecretion in COPD (Adler, 1990). Also, direct cell death of alveolar epithelial cells induced by oxidative stress has been proposed as a significant mechanism in the development of emphysema (MacNee, 2005; Petrache, 2005). Because oxidative stress can induce the expression of proinflammatory cytokines as well as chemotactic proteins, oxidative stress caused by cigarette smoke exposure may also be involved in the inflammatory response in COPD patients (MacNee, 2005).
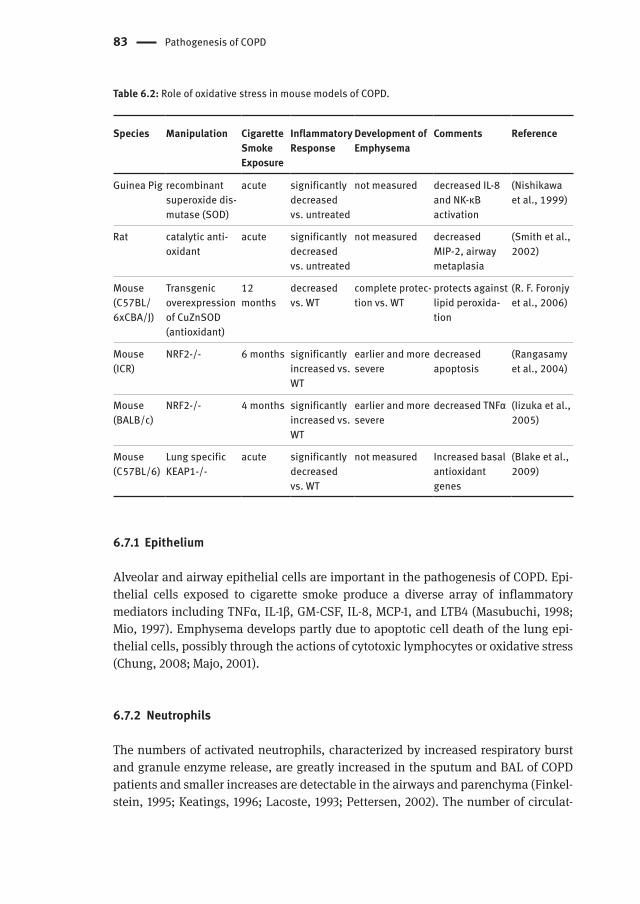
A role for oxidative stress in COPD is strongly supported by animal models of COPD (Table 6.2). Therapeutic administration of antioxidants or transgenic overexpression of antioxidant genes decreases both the inflammatory response as well as the development of emphysema following cigarette smoke exposure (Foronjy, 2006;
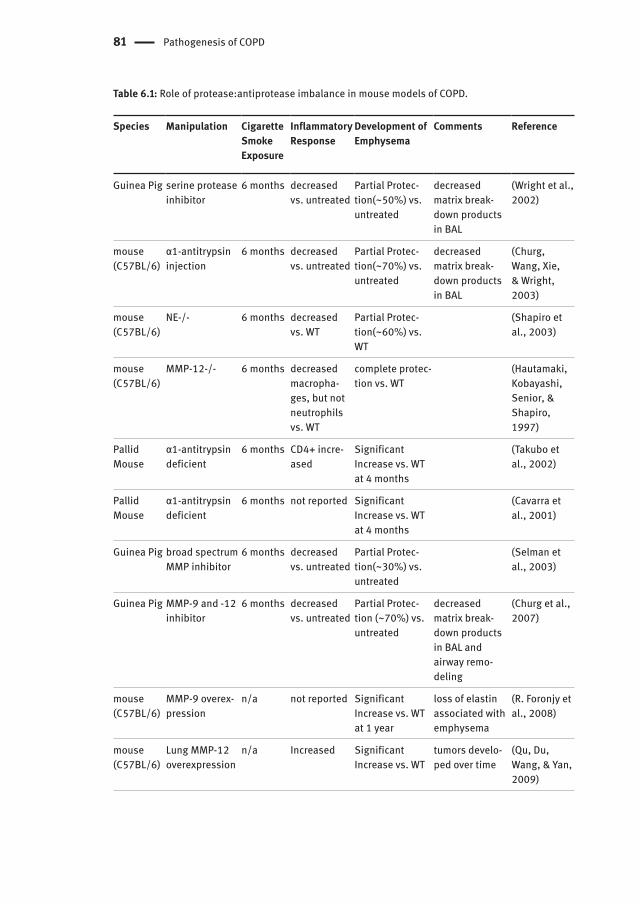
81 Pathogenesis of COPD
Table 6.1: Role of protease:antiprotease imbalance in mouse models of COPD.
Species Manipulation Cigarette Smoke Exposure
Inflammatory Response
Development of Emphysema
Comments Reference
Guinea Pig serine protease inhibitor
6 months decreased vs. untreated
Partial Protec-tion(~50%) vs. untreated
decreased matrix break-down products in BAL
(Wright et al., 2002)
mouse (C57BL/6)
α1-antitrypsin injection
6 months decreased vs. untreated
Partial Protec-tion(~70%) vs. untreated
decreased matrix break-down products in BAL
(Churg, Wang, Xie, & Wright, 2003)
mouse (C57BL/6)
NE-/- 6 months decreased vs. WT
Partial Protec-tion(~60%) vs. WT
(Shapiro et al., 2003)
mouse (C57BL/6)
MMP-12-/- 6 months decreased macropha-ges, but not neutrophils vs. WT
complete protec-tion vs. WT
(Hautamaki, Kobayashi, Senior, & Shapiro, 1997)
Pallid Mouse
α1-antitrypsin deficient
6 months CD4+ incre-ased
Significant Increase vs. WT at 4 months
(Takubo et al., 2002)
Pallid Mouse
α1-antitrypsin deficient
6 months not reported Significant Increase vs. WT at 4 months
(Cavarra et al., 2001)
Guinea Pig broad spectrum MMP inhibitor
6 months decreased vs. untreated
Partial Protec-tion(~30%) vs. untreated
(Selman et al., 2003)
Guinea Pig MMP-9 and -12 inhibitor
6 months decreased vs. untreated
Partial Protec-tion (~70%) vs. untreated
decreased matrix break-down products in BAL and airway remo-deling
(Churg et al., 2007)
mouse (C57BL/6)
MMP-9 overex-pression
n/a not reported Significant Increase vs. WT at 1 year
loss of elastin associated with emphysema
(R. Foronjy et al., 2008)
mouse (C57BL/6)
Lung MMP-12 overexpression
n/a Increased Significant Increase vs. WT
tumors develo-ped over time
(Qu, Du, Wang, & Yan, 2009)

Airway Inflammation in COPD 82
Nishikawa, 1999; Smith, 2002). In mice, cigarette smoke exposure activates the NRF2 protein, a redox sensitive protein critical to the expression of detoxification and antioxidant genes (Rangasamy, 2004). In addition, a broad panoply of antioxidant genes is activated in a NRF2 dependent manner after cigarette smoke exposure (Rangasamy, 2004). Important to the pathogenesis of COPD, NRF2 deficient mice exposed to cigarette smoke have a significantly enhanced inflammatory response, develop more severe emphysema and increased lung apoptosis, and have significantly enhanced levels of oxidative stress relative to wildtype mice (Iizuka, 2005; Rangasamy, 2004). In agreement with this observation, lung specific deletion of KEAP1, an inhibitor of NRF2 activation, leads to an increased basal antioxidant status and decreased pulmonary inflammation after cigarette smoke exposure (Blake, 2009). In conclusion, oxidative stress likely contributes to the development of COPD through multiple mechanisms.
6.7 Airway Inflammation in COPD
The significance of chronic inflammation observed in COPD has been demonstrated experimentally (Table 6.3) (Cosio, 2009; Hogg, 2004; Rennard, 2006). Increased inflammation occurs throughout the lungs in the large and small airways, as well as in the parenchyma (Hogg, 2004). It is believed that the inflammation surrounding the small airways is the most significant contributor to airflow limitation (Hogg, 2004). Additionally, patients with severe disease who quit smoking exhibit sustained inflammation and lung function decline, further implicating inflammation as a critical process in COPD development and progression (Gamble, 2007; Turato, 1995). Detailed below are the known functions of specific inflammatory cells in the development of COPD. Although individual cell types contribute to disease development in a number of ways, numerous types of immune cells likely act in a collective, reciprocal manner to cause disease. In addition, identification of the specific functions and roles of each cell type in the pathogenesis of COPD will provide new therapeutic targets.
Species Manipulation Cigarette Smoke Exposure
Inflammatory Response
Development of Emphysema
Comments Reference
mouse (C57BL/6)
cathepsin S inhibitor used on lung IFNγ overexpressing mouse
n/a not reported decreased vs. untreated
reduced apop-tosis
(Zheng et al., 2005)
continued Table 6.1: Role of protease:antiprotease imbalance in mouse models of COPD.
83 Pathogenesis of COPD
6.7.1 Epithelium
Alveolar and airway epithelial cells are important in the pathogenesis of COPD. Epithelial cells exposed to cigarette smoke produce a diverse array of inflammatory mediators including TNFα, IL1β, GMCSF, IL8, MCP1, and LTB4 (Masubuchi, 1998; Mio, 1997). Emphysema develops partly due to apoptotic cell death of the lung epithelial cells, possibly through the actions of cytotoxic lymphocytes or oxidative stress (Chung, 2008; Majo, 2001).
6.7.2 Neutrophils
The numbers of activated neutrophils, characterized by increased respiratory burst and granule enzyme release, are greatly increased in the sputum and BAL of COPD patients and smaller increases are detectable in the airways and parenchyma (Finkelstein, 1995; Keatings, 1996; Lacoste, 1993; Pettersen, 2002). The number of circulat
Table 6.2: Role of oxidative stress in mouse models of COPD.
Species Manipulation Cigarette Smoke Exposure
Inflammatory Response
Development of Emphysema
Comments Reference
Guinea Pig recombinant superoxide dis-mutase (SOD)
acute significantly decreased vs. untreated
not measured decreased IL-8 and NK-κB activation
(Nishikawa et al., 1999)
Rat catalytic anti-oxidant
acute significantly decreased vs. untreated
not measured decreased MIP-2, airway metaplasia
(Smith et al., 2002)
Mouse (C57BL/ 6xCBA/J)
Transgenic overexpression of CuZnSOD (antioxidant)
12 months
decreased vs. WT
complete protec-tion vs. WT
protects against lipid peroxida-tion
(R. F. Foronjy et al., 2006)
Mouse (ICR)
NRF2-/- 6 months significantly increased vs. WT
earlier and more severe
decreased apoptosis
(Rangasamy et al., 2004)
Mouse (BALB/c)
NRF2-/- 4 months significantly increased vs. WT
earlier and more severe
decreased TNFα (Iizuka et al., 2005)
Mouse (C57BL/6)
Lung specific KEAP1-/-
acute significantly decreased vs. WT
not measured Increased basal antioxidant genes
(Blake et al., 2009)

Airway Inflammation in COPD 84
ing and lung neutrophils correlates with the decline in FEV1 and neutrophil numbers in induced sputum correlate with COPD severity, thus implicating them in disease progression (Baraldo, 2004; Keatings, 1996; Sparrow, 1984). It is likely these changes are induced by IL8. IL8 is chemotactic for neutrophils, activates neutrophils, and expression is increased in COPD patients (Barnes, 2003; Pettersen, 2002). It is unknown how neutrophils specifically contribute to disease progression, but neutrophils secrete a number of proteases important in COPD pathogenesis such as neutrophil elastase, cathepsin G, proteinase3, as well as matrix metalloproteinase MMP8 and MMP9 (Barnes, 2003). Thus, neutrophils are believed to be crucially involved in tipping the protease:antiprotease balance to a more proteolytic environment within the lungs of individuals with COPD. This hypothesis is further supported by the observation that neutrophil elastase knockout mice are partially protected from disease development after cigarette smoke exposure (Table 6.1) (Shapiro, 2003). Interestingly, although neutrophils can cause an increase in proteases that can cause experimental emphysema, similar emphysematous destruction is not observed in other chronic pulmonary diseases dominated by neutrophils like cystic fibrosis and bronchiectasis (Barnes, 2003).
6.7.3 Macrophages
Macrophages are significant contributors to the pathogenesis of COPD. There is an overwhelming increase in the number of macrophages in the BAL and sputum, airways, and parenchyma in COPD patients (Barnes, 2003). Further, macrophages are localized to sites of tissue destruction in COPD (Finkelstein, 1995; Meshi, 2002) and there is a correlation of macrophages with the severity of disease (Di Stefano, 1998). Macrophages secrete a number of inflammatory mediators after exposure to cigarette smoke that may activate and recruit additional cell types (Barnes, 2003). Additionally, alveolar macrophages taken from COPD patients are intrinsically altered, and secrete more inflammatory mediators and have more proteolytic activity at baseline (Barnes, 2003). Macrophages are also significant sources of destructive proteases MMP2,9, and 12, and cathepsin K, L, and S, that have key functions in the development of COPD in various mouse models (Table 6.1).
6.7.4 Eosinophils
In COPD patients, the number of eosinophils is increased in the sputum, BAL, and airway walls and there are elevated levels of eosinophil cationic protein in the BAL and sputum (Fujimoto, 1999; Lacoste, 1993; Lams, 1998; Linden, 1993; Panzner, 2003). Cytokines that are associated with eosinophilia in asthma, IL‐4 and IL‐5, are also upregulated in COPD (Zhu, 2007). During COPD exacerbations, the number of eosin

85 Pathogenesis of COPD
ophils is increased and IL5 production is enhanced (Saetta, 1996; Zhu, 2001). It is unclear whether these increases in eosinophil numbers in COPD represents a distinct subgroup of patients with concomitant COPD and asthma, and it is equally unclear whether eosinophilia in COPD correlates to severity of disease. However, patients with enhanced numbers of eosinophils typically respond well to inhaled corticosteroids (Chanez, 1997; Pizzichini, 1998).
6.7.5 Mast Cells
Mast cells are present in the airways and alveoli of healthy individuals (Andersson, 2009b; Carroll, 2002) and have critical roles in innate immunity, T cell regulation, and antigen presentation. Despite the fact that these essential functions suggest that mast cells might have a pivotal role in COPD pathogenesis (Dawicki, 2007; Jawdat, 2006; Metz, 2008), very few studies have examined mast cell function in the context of COPD. Although increases in mast cells have been reported in the airways of COPD patients (Grashoff, 1997; Pesci, 1994), a more recent study demonstrated a reduction in mast cells in COPD patients, and that fewer numbers of mast cells correlated with worse lung function suggesting a protective, regulatory role for mast cells in COPD (Gosman, 2008). An additional study corroborates this finding and demonstrated that mast cell numbers decreased with increasing disease severity (Andersson, 2009a). In addition, these authors found that as COPD worsened, the mast cell populations in the lung underwent changes in density, distribution, and molecular phenotype implicating them in COPD pathogenesis (Andersson, 2009a).
6.7.6 γδ T Cells
Very little is known about the function of γδ T cells in COPD. γδ T cells are increased in the airways, bronchoalveolar lavage, and peripheral blood of smokers, regardless of whether they have emphysema (EkbergJansson, 2000; Majo, 2001; Pons, 2005; Richmond, 1993). In some cases, the number of γδ T cells is reduced in patients with COPD compared with their levels in smokers (Pons, 2005). γδ T cells are important regulators of tissue repair and mucosal homeostasis (Jameson, 2003). Consistent with this role, the absence of γδ T cells caused enhanced caspasedependent epithelial cell death in a mouse model of COPD, although inflammation was not affected (Borchers , 2008). Based upon these observations, not only are γδ T cell numbers reduced in COPD but their function may also be impaired.

Airway Inflammation in COPD 86
6.7.7 Natural Killer (NK) Cells
There are few studies examining NK cells in COPD, and therefore the role of NK cells in COPD is relatively unknown. One study has shown no increases in NK cells in the lung parenchyma in COPD patients relative to controls, but this study was limited by small sample sizes (Majo, 2001). In contrast, two additional studies using a greater number of samples demonstrated an increase of NK cells in the lungs of COPD patients (Di Stefano, 1998; Elliott, 2009). Importantly, NK cell numbers correlated with disease severity (Di Stefano, 1998; Elliott, 2009). A major function of NK cells is their capacity for cell specific, directed cytotoxicity mediated through the release of perforin and granzymes that induce target apoptosis. Localized inflammatory sites in the lungs of COPD patients are rich with granzyme A+ and granzyme B+ NK cells (Vernooy, 2007). Additionally, the expression of NK cell perforin and granzyme B, as well as functional NK cell cytotoxic effector function, was shown to be increased in the sputum of COPD patients (Urbanowicz, 2009). In a corollary study, perforin and granzyme B expression was increased in peripheral blood NK cells of COPD patients and smokers (n=60) (Hodge, 2006), but this observation was not reproduced by two separate groups using a small number of patients (n=10) (Morissette, 2007). Therefore, it is likely that NK cells contribute to apoptotic cell death and the development of emphysema in COPD. Recent data demonstrate an additional role for NK cells in COPD. In COPD patients, there is increased expression of MICA on the lung epithelium, a stressinducible ligand for the NK cell activating receptor NKG2D (Borchers, 2009). Importantly, expression of MICA on the lung epithelium correlates with disease severity. Experimentally, chronic cigarette smoke exposure also induced expression of NKG2D ligands on the mouse airway and alveolar epithelium. These observations provide strong support for a role of NKG2D activation and NK cells, in particular, in the development of COPD. In contrast, there is some data suggesting that NK cells may be functionally suppressed in COPD.
6.7.8 Natural Killer T (NKT) Cells
A role for NKT cells in COPD is illdefined and is limited by the overall paucity of studies. CD56+CD3+ NKTlike cell number and function is reduced in the peripheral blood of COPD patients (Urbanowicz, 2009). However, this report is severely limited by the use of very few patients (n=10), the majority of whom were using inhaled corticosteroids, a variable known to interfere with functional assays. In one study, no differences in the numbers of Type1, invariant NKT (iNKT) cells were detected in the sputum of COPD patients with stable disease or during COPD exacerbations (Vijayanand, 2007). However, in a more recent study using immunohistochemistry, increased numbers of iNKT cells were found in the lungs of COPD patients. iNKT cells stimulate the production of IL‐13 which drives alternative activation of macrophages

87 Pathogenesis of COPD
and leads to macrophage generated IL‐13 in a feed‐forward loop. Persistent IL‐13 production is believed to contribute to protease activation, mucus hypersecretion, and mucus cell hyperplasia, all important phenotypic changes in COPD.
6.7.9 T Cells
In COPD patients, there are increased numbers of T cells in the lung parenchyma and peripheral and central airways, and, in severe COPD, T cells are located within lymphoid follicles adjacent to airways (Cosio, 2002; Finkelstein, 1995; Hogg, 2004; Majo, 2001; O’Shaughnessy, 1997; Retamales, 2001; Saetta, 1999; Saetta, 1998). Similar to COPD patients, CD3+ T cells are organized in lymphoid follicles in mice chronically exposed to cigarette smoke, suggesting a common mechanism (Bracke, 2006). Although both CD4+ and CD8+ T cell numbers increase, the greatest elevation is in the CD8+ T cell population (Hogg, 2004; Majo, 2001; Saetta, 1999; Saetta, 1998). Significantly, the increase in CD8+ T cells directly correlates with lung function decline (Saetta, 1999; Saetta, 1998). There is also an increase in the number of CD8+ T cells in the peripheral blood of COPD patients who don’t smoke (de Jong, 1997; Kim, 2002). Further, peripheral T cells are more activated in COPD patients, and activation correlates with the loss of lung function (Zhu, 2009). Mouse models exhibit the hallmark features of COPD including mucus cell metaplasia, airspace enlargement and peribronchial/perivascular infiltration of neutrophils, macrophages, and T cells. In these models, CD8deficient mice (but not CD4deficient mice) fail to develop these key features of COPD, including macrophage accumulation and airspace enlargement (Borchers, 2007; Maeno, 2007). While evidence mounts supporting a causal role for T cells in COPD, the initial stimulus remains to be identified. The most likely mechanism driving T cell‐mediated pathology is antigen‐specific clonal expansion. In support of this theory, oligoclonal expansions of CD4+ and CD8+ T cells are detected in the lungs of COPD patients (Korn, 2005; Sullivan, 2005). Although the antigen specificity or function of T cells in COPD remains wholly uninvestigated, it is probable that oligoclonal expansions reflect recognition of self antigens. In support of this idea, T cells reactive with elastin have recently been demonstrated in COPD patients. It is unclear what role autoreactive T cells may play, but T cell recognition of antigens present on the lung epithelium may cause robust inflammation and the production of cytokines and chemokines in COPD (Enelow, 1998; Zhao, 2000). Presently, mechanistic data supporting a causal role for T cells in the development or progression of COPD are limited. Derangement of the alveolar architecture may be caused by multiple processes including matrix breakdown as well as epithelial and endothelial cell destruction. The prevailing theory is that T cells are aberrantly activated in COPD patients and contribute to tissue destruction through both direct and indirect effector mechanisms. The direct mechanisms likely involve CD8+ T cell‐mediated apoptosis of alveolar cells via the elaboration of cytotoxic granules (perforin‐

Airway Inflammation in COPD 88
granzyme) or Fas molecules (Henkart, 1994). Along these lines, alveolar epithelial cell apoptosis correlates with CD8+ T cell numbers in emphysema (Majo, 2001) and this theory is supported by evidence that CD8+ T cells in patients with COPD express high levels of perforin with a concomitant enhanced lytic capacity (Chrysofakis, 2004). Indirect mechanisms whereby lymphocyte activation contributes to COPD are more complex and difficult to assess. However, a role for the broad inflammatory cell recruitment and activation in COPD patients and experimental models seems likely. For example, many studies indicate that the production of Th1 cytokines such as IFNγ (Di Stefano, 2004; Grumelli, 2004; Hodge, 2007) and TNFα (Aaron, 2001; Keatings, 1996) are increased in T cells of COPD patients. Di Stefano et al. reported that the number of STAT4positive cells (a transcription factor critical fohr the development of Th1 lineage cytotoxic T cells and IFNγ production) are increased compared to healthy smokers and nonsmokers (Di Stefano, 2004). Furthermore, the number of STAT4+ lymphocytes correlated with increased airflow obstruction and IFNγ+ lymphocytes. In a more detailed study, Grumelli et al. demonstrated that lung function decline in patients with COPD correlated with Th1 cell markers on both CD4+ and CD8+ T cells, and these cells secreted increased amounts of IFNγ, but not IL4, relative to control patients (Grumelli, 2004). IFNγ induction may amplify inflammation and increase macrophage metalloproteinase secretion which can lead to matrix degradation and alveolar destruction (Grumelli, 2004; Wang, 2000). Grumelli et al. (Grumelli, 2004) also provided a mechanistic link between increased T cell activation and emphysema by demonstrating enhanced production of CXCR3 ligands (i.e., IP10) which leads to increased MMP12 activation (elastindegrading protease linked to emphysema) by alveolar macrophages. In addition to their proinflammatory effector function, subpopulations of CD4+ T cells (i.e., regulatory T cells) may also serve to limit the adaptive immune responses. In the context of COPD, a breakdown in the function of these cells may lead to increased expansion of pathogenic T cells.
6.7.10 B Cells
CD20+ B cells are increased in the lungs of COPD patients (Gosman, 2006; Hogg, 2004). Importantly, the number or presence of CD20+ B cells in airways and periphery correlates with disease severity (Gosman, 2006; Hogg, 2004). Most often, B cells are organized in lymphoid follicles in COPD patients that are interspersed with CD4+ and CD8+ T cells (Hogg, 2004; Kelsen, 2009; van der Strate, 2006). Interestingly, lymphoid follicles containing high numbers of B cells also develop in mice chronically exposed to cigarette smoke (Bracke, 2006; D’Hulst, 2005; van der Strate, 2006). Currently, it is unknown whether B cells are specifically required for the development of COPD. A primary function of B cells is the generation of antigenspecific antibodies. Differentiated antibody producing B cells, known as plasma B cells, also are increased in the lungs of COPD patients (J. Zhu, 2007), and B cells organized in lymphoid follicles were

89 Pathogenesis of COPD
identified as being oligoclonal, suggesting specific, antigendirected antibody production (van der Strate, 2006). B cells are likely responding to selfantigens because, in addition to elastinreactive T cells, elastinreactive antibodies are present in COPD patients (Lee, 2007). Further evidence for a selfantigen comes from the observation
that COPD patients have pulmonary epithelial cellreactive antibodies (FeghaliBostwick, 2008).
6.7.11 Dendritic Cells
Dendritic cells (DCs) are key cells in antigen presentation and bridge the gap between the innate and adaptive immune system. Increases in DCs occur in patients and mouse models of COPD (D’Hulst, 2005; Demedts, 2007; Freeman, 2009) and the DCs frequently have an activated phenotype (D’Hulst, 2005; Freeman, 2009). Maturation of DCs correlates with disease severity, strongly suggesting a mechanistic role for DCs in COPD (Freeman, 2009). Further, the expression of maturation markers correlates with the activation of CD4+ T cells in COPD patients (Freeman, 2009). However, evidence has accumulated for roles of DCs in COPD independent of their antigen presentation capabilities. Myeloid dendritic cells isolated from the lungs of COPD patients were able to induce Th1 and Th17 responses in the absence of antigen presentation (Shan, 2009). Additionally, in vitro exposure of dendritic cells to cigarette smoke extract induced the expression of inflammatory mediators leading to the activation and proliferation of cocultured CD8+ T cells, providing a potential link between DCs and CD8+ T cell accumulation in COPD (Mortaz, 2009).
6.8 Cytokines and Chemokines in COPD
Cytokines and chemokines are involved in a diverse array of COPD phenotypes including the recruitment and activation of macrophages, neutrophils, T cells, B cells, and other immune cells. Cytokines and chemokines are also believed to be involved in the processes of airway remodeling, mucus cell hyperplasia, and emphysema. Therefore, the roles of cytokines and chemokines in COPD are multitudinous. CCL1/I‐309, CCL2/MCP‐1, CCL3/MIP‐1α, CCL4/MIP‐1β, CCL11/eotaxin, CXCL1/GROα, CXCL5/ENA‐78, CXCL8/IL‐8, IL‐1β, IL‐6, IL‐18, IFNγ, IP‐10, GM‐CSF, and TNFα are among the many cytokines and chemokines that are increased in individuals with COPD (Chung, 2008). These increases are detected in the lung tissue, sputum, or the BAL fluid, and the levels of cytokines and chemokines frequently correlate with disease severity (Chung, 2008). These cytokines and chemokines are frequently derived from a number of resident inflammatory cells as well as the airway epithelium (Chung, 2008). In addition to the cytokines mentioned here, a number of additional cytokines and chemokines are undoubtedly increased in COPD but have yet to be studied. The study of cytokine and

COPD as an Autoimmune Disease 90
chemokines in the development of COPD phenotypes has been aided using mouse models of COPD. Studies utilizing mice that have had either cytokines/chemokines or their receptors knocked out are almost always protected from the development of emphysema, and have reduced inflammation after smoke exposure. Conversely, a number of studies have utilized mice that overexpress proinflammatory cytokines/chemokines explicitly in the lung, and these mice develop COPD‐like disease (emphysema and inflammation).
6.9 COPD as an Autoimmune Disease
Autoimmune diseases are chronic inflammatory conditions caused by failure of self‐tolerance mechanisms. Although it is not completely understood how this breakdown occurs, it likely results from a complex interplay of genetics, failure of natural tolerance, and environmental triggers. The defining criteria for autoimmune diseases are classically defined by Witebsky’s postulates (Rose, 1993) a) circumstantial evidence consisting of lymphocytic infiltration of the target organ or association of the disease with a particular MHC haplotype, b) indirect evidence consisting of the isolation of autoantibodies or self‐reactive T cells from the organ or identification of human antigen and using immunization processes to reproduce the disease in animals, and finally, c) direct proof consisting of reproducing the disease in a normal recipient by the direct transfer of autoantibodies or T cells. Based on the associations of T cell numbers with declining lung function in COPD patients and evidence that inflammation persists in COPD patients upon smoking cessation (Gamble, 2007; Turato, 1995), Cosio and colleagues proposed the existence of an autoimmune component in COPD (Cosio, 2002; Cosio, 2009). Since then, multiple lines of evidence have accumulated supporting this hypothesis. As mentioned above, oligoclonal T cells and B cells that can react with selfantigens have been identified. Recent evidence that maturation of dendritic cells correlates with T cell activation COPD further supports this hypothesis (Freeman, 2009). In addition, single nucleotide polymorphisms in multiple HLA loci associate with the severity of COPD (DeMeo, 2008). Therefore, since the early hypothesis, a significant body of evidence has accumulated suggesting that COPD has an autoimmune component. Moreover, the possibility exists that the cigarette smokeinduced autoreactivity may have broader implications for systemic manifestations of COPD as cigarette smoking has been identified as a risk factor for established autoimmune diseases such as rheumatoid arthritis (Papadopoulos, 2005), Graves disease (Prummel, 1993), and biliary cirrhosis (Gershwin, 2005).

91 Pathogenesis of COPD
6.10 Systemic Inflammation
The presence of lowgrade systemic inflammation is a wellknown characteristic of COPD (Agusti, 2003; Gan, 2004). Patients with stable COPD have an increased numbers of circulating leukocytes occasionally present with an activated phenotype (Noguera, 2001; Noguera, 1998). Further, activation of circulating lymphocytes correlates with lung function (Zhu, 2009). Further, COPD patients have increased circulating levels of inflammatory mediators such as Creactive protein, fibrinogen, IL6, and TNFa (Agusti, 2003). Additionally, the severity of this inflammation is increased during exacerbations of COPD (Hurst, 2006). Because COPD is overwhelmingly associated with an abnormal inflammatory response in the lungs, it has been proposed that the systemic inflammatory response is simply the result of a spilling over effect from the lungs (Agusti, 2007). However, several investigators were unable to find relationships between inflammation using specific measured variables in the lungs and blood of individual patients (Hurst, 2005; Vernooy, 2002). Therefore, it’s really unknown whether a spillover is really a sufficient explanation. Interestingly, several extrapulmonary diseases frequently associated with COPD are also associated with a systemic inflammatory response (Agusti, 2007).
6.10.1 Cardiovascular Disease
COPD and cardiovascular disease (CVD) share the common risk factor of cigarette smoking. Additionally, COPD and CVD patients share commonalities such as advanced age and decrease in physical activity. However, several lines of evidence have established that, independent of known risk factors including smoking, COPD correlates as an independent risk factor for CVD (RodriguezRoisin, 2008; Sin, 2006). COPD is a significant risk factor for a broad spectrum of CVD manifestations such as atherosclerosis, ischemic heart disease, stroke and sudden cardiac death (Sin, 2006). In patients with mild COPD, complications from CVD are overwhelming the cause (42%) for first time hospitalization (RodriguezRoisin, 2008; Sin, 2006). In contrast, respiratory complications are the cause for only 14% of hospitilzations. Most important, CVD is extremely important in COPD as between 25–50% (depending on the study) of all COPD deaths are due to complications of cardiovascular disease (RodriguezRoisin, 2008; Sin, 2006). The exact mechanisms involved in cardiac disease in COPD patients are unknown. As described above, patients with COPD have significant levels of systemic inflammation. Systemic inflammation is a key risk factor for the development of CVD (Buffon, 2002). Because systemic inflammation is a common feature in patients with COPD, even in mild to moderate stages, it has been suggested by multiple investigators as a key component of CVD risk. Particularly, increased levels of Creactive protein is significant risk factor for combined COPD and CVD (Sin, 2003). More work is needed in this area.

Systemic Inflammation 92
6.10.2 Nutritional Abnormalities
Divergent Weight Loss and Obesity in COPD. Numerous nutritional abnormalities have been detected in patients with COPD (Nici, 2006; Schols, 2000). Changes in the basal metabolic rate, caloric intake, and body composition are common in patients with COPD (Nici, 2006; Schols, 2000). Weight loss with no apparent explanation occurs in about half of patients with severe COPD, but is also detected in about 15% of COPD patients with mild to modeate disease (Creutzberg, 1998; Nici, 2006). It is believed that weight loss is not due to loss of fat, rather the majority of loss is in skeletal muslcle mass (Schols, 2000). Further, recent weight loss is a significant predictor of future mortality and morbidity (Nici, 2006). Although these changes may be responsible for changes in caloric intake, increases in basal metabolic rate from medications, systemic inflammation, and/or tissue hypoxia have been proposed (Agusti, 2005).
Briefly, metabolic syndrome is characterized by a multitude of factors including abdominal obesity, atherogenic dyslipidemia, hypertension, and insulin resistance (Watz, 2009). In contrast to the description of weight loss above, a signicant number of COPD patients have coexisting metabolic syndrome or obesity (Watz, 2009; Franssen, 2008). Further, obesity is more prevalent in patients with COPD than in the general population (Franssen, 2008). Interestingly, the metabolic syndrome and obesity in COPD patients was associated with increases in markers of systemic inflammation CRP and IL6 (Watz, 2009). Further, in this study there was a correlation between an increase in severity of COPD and a lower BMI (Watz, 2009). Studies have demonstrated that patients with a predominance of chronic bronchitis are more likely to have a higher BMI, whereas patients with predominantly emphysema will have a lower BMI (Ogawa, 2009). Whatever the mechanism behind the obsesityCOPD relationship, obesity nonetheless is a major problem for COPD patients as obese patients are frequently exercise intolerant, a major problem for exercisebased pulmonary rehabilitation therapies (Franssen, 2008).
6.10.3 Skeletal Muscle Dysfunction
Skeletal muscle dysfunction is extremely common in COPD patients (Nici, 2006). There are specific anatomic changes as well as functional changes (Nici, 2006). The precise mechanism underlying muscle wasting is unknown, but could be due to inactivityinduced deconditioning, systemic inflammation, use of medications, oxidative stress or reductions in muscle mass (Nici, 2006). Although little is known in on this topic, these changes significantly contribute to loss of exercise capacity critical for pulmonary rehabilitation and a reduction in the quality of life (Nici, 2006).

93 Pathogenesis of COPD
6.10.4 Osteoporosis
The incidence of osteoporosis is also increased in patients with COPD. The specific causes are unknown, but have been speculated to caused by malnutrition, smoking, steroid use, lack of exercise, and systemic inflammation (Agusti, 2003). However, little is known about osteoporosis in COPD beyond that.
6.10.5 Neurological Disorders
There are significant alterations in the nervous system in patients with COPD. The actual bioenergeentic metabolism of the brain is altered in patients with COPD (Mathur, 1999). Importantly, COPD patients frequently suffer depression (Light, 1985; Xu, 2008; Maurer, 2008). The depression and anxiety in COPD have been found to be important and serious risk factors for disease progression (Xu, 2008). It’s likely that depression develops in response to having a chronic disease. However, it’s been proposed that systemic inflammation may also be involved (Agusti, 2003).
6.11 COPD Exacerbations
The underlying COPD pathologies are punctuated by exacerbations of symptoms. Standard criteria for COPD exacerbations do not exist, but they are generally characterized by increased dyspnea, enhanced sputum, enhanced inflammation, and decline in lung function (Bhowmik, 2000; Sapey, 2006), (see Chapters 16, COPD Exacerbations Outpatient Management, and 17, COPD Exacerbations Inpatient Management). Consequently, exacerbations accelerate disease progression and are an integral part of disease pathogenesis. The toll of COPD exacerbations is also psychologically destructive; patients with frequent exacerbations have a lower quality of life and decreased mobility, leading to increased depression (Quint, 2008). Most importantly, exacerbations that require hospitalization result in death 8–11% of the time, and the remaining patients have a mortality rate of 23–43% one year following admission (Connors, 1996; Groenewegen, 2003). Between 40 and 60% of exacerbations are believed to be caused by viral infections (Celli, 2007; Sapey, 2006). The remaining exacerbations are likely the result of nonviral infections and pollution, although roughly 20% have an unknown etiology (Celli, 2007). Exacerbations associated with viral infections are severe; viral exacerbations lead to hospitalization more often than other causes, and have a longer recovery time (Wedzicha, 2004). The connection between viruses and COPD exacerbations remained mostly associative until, in a recent landmark study, Kang et al. experimentally demonstrated that exacerbations of symptoms following cigarette smoke (CS) exposure can be caused by viral infection (Kang, 2008). Key to the progression of COPD, viral infection following CS

References 94
exposure led to enhanced pulmonary inflammation, increased alveolar apoptosis, accelerated emphysema, and airway fibrosis (Kang, 2008). However, the underlying cellular mechanisms responsible for these effects remain undiscovered.
6.12 Summary Points
1. The pathophysiology of COPD is incompletely understood, but is thought to involve chronic inflammation, oxidative stress, and increased elastolytic potential in the lung
2. Increased numbers of macrophages, neutrophils, lymphocytes and eosinophils in and around the airways, parenchyma, and vasculature characterize the inflammation. Pathogenesis of COPD is examined mechanistically. Current and future research into the pathways that lead to COPD will provide insight into much needed therapeutic targets.
3. COPD also manifests systemically including systemic inflammation, skeletal muscle dysfunction, metabolic syndrome, cardiovascular disease, osteoporosis, and neurological defects
4. Enhanced understanding of the role of the immune system in acute exacerbations in COPD is critical in the future identification of susceptible patients and the development of new therapeutic strategies.
ReferencesAaron, S.D., Angel, J.B., Lunau, M., Wright, K., Fex, C., Le Saux, N., & Dales, R.E. (2001). Granulocyte
inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 163(2), 349–355.
Adenuga, D., Yao, H., March, T.H., Seagrave, J., & Rahman, I. (2009). Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol, 40(4), 464–473.
Adler, K.B., Holden-Stauffer, W.J., & Repine, J.E. (1990). Oxygen metabolites stimulate release of high-molecular-weight glycoconjugates by cell and organ cultures of rodent respiratory epithelium via an arachidonic acid-dependent mechanism. J Clin Invest, 85(1), 75–85.
Agusti, A. (2007). Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should). Proc Am Thorac Soc, 4(7), 522–525.
Agusti, A.G. (2005). Systemic effects of chronic obstructive pulmonary disease. Proc Am Thorac Soc, 2(4), 367–370; discussion 371–362.
Agusti, A.G., Noguera, A., Sauleda, J., Sala, E., Pons, J., & Busquets, X. (2003). Systemic effects of chronic obstructive pulmonary disease. Eur Respir J, 21(2), 347–360.
Andersson, C.K., Mori, M., Bjermer, L., Lofdahl, C.G., & Erjefalt, J.S. (2009a). Alterations in Lung Mast Cell Populations in Patients with COPD. Am J Respir Crit Care Med.
Andersson, C.K., Mori, M., Bjermer, L., Lofdahl, C.G., & Erjefalt, J.S. (2009b). Novel site-specific mast cell subpopulations in the human lung. Thorax, 64(4), 297–305.

95 Pathogenesis of COPD
Baraldo, S., Turato, G., Badin, C., Bazzan, E., Beghe, B., Zuin, R., Saetta, M. (2004). Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax, 59(4), 308–312.
Barnes, P.J. (2004). Small airways in COPD. N Engl J Med, 350(26), 2635–2637. Barnes, P.J., Shapiro, S.D., & Pauwels, R.A. (2003). Chronic obstructive pulmonary disease:
molecular and cellular mechanisms. Eur Respir J, 22(4), 672–688. Bhowmik, A., Seemungal, T.A. R., Sapsford, R.J., & Wedzicha, J.A. (2000). Relation of sputum
inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax, 55(2), 114–120.
Blake, D.J., Singh, A., Kombairaju, P., Malhotra, D., Mariani, T.J., Tuder, R.M., Biswal, S. (2009). Deletion of Keap1 in the Lung Attenuates Acute Cigarette Smoke-induced Oxidative Stress and Inflammation. Am J Respir Cell Mol Biol.
Borchers, M.T., Wesselkamper, S.C., Curull, V., Ramirez-Sarmiento, A., Sánchez-Font, A., Garcia-Aymerich, J., Orozco-Levi, M. (2009). Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J Clin Invest, 119(3), 636–649.
Borchers, M.T., Wesselkamper, S.C., Eppert, B.L., Motz, G.T., Sartor, M.A., Tomlinson, C.R., Tichelaar, J.W. (2008). Nonredundant Functions of {alpha}{beta} and {gamma}{delta} T Cells in Acrolein-Induced Pulmonary Pathology. Toxicol Sci, 105(1), 188–199.
Borchers, M.T., Wesselkamper, S.C., Harris, N.L., Deshmukh, H., Beckman, E., Vitucci, M., Leikauf, G.D. (2007). CD8+ T cells contribute to macrophage accumulation and airspace enlargement following repeated irritant exposure. Exp Mol Pathol, 83(3), 301–310.
Bosken, C.H., Wiggs, B.R., Pare, P.D., & Hogg, J.C. (1990). Small airway dimensions in smokers with obstruction to airflow. Am Rev Respir Dis, 142(3), 563–570.
Bracke, K.R., D’Hulst, A.I., Maes, T., Moerloose, K.B., Demedts, I.K., Lebecque, S., Brusselle, G.G. (2006). Cigarette Smoke-Induced Pulmonary Inflammation and Emphysema Are Attenuated in CCR6-Deficient Mice. J Immunol, 177(7), 4350–4359.
Buffon, A., Biasucci, L.M., Liuzzo, G., D’Onofrio, G., Crea, F., & Maseri, A. (2002). Widespread coronary inflammation in unstable angina. N Engl J Med, 347(1), 5–12.
Buist, A.S., Vollmer, W.M., & McBurnie, M.A. (2008). Worldwide burden of COPD in high- and low-income countries. Part I. The burden of obstructive lung disease (BOLD) initiative. Int J Tuberc Lung Dis, 12(7), 703–708.
Carroll, N.G., Mutavdzic, S., & James, A.L. (2002). Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur Respir J, 19(5), 879–885.
Cavarra, E., Bartalesi, B., Lucattelli, M., Fineschi, S., Lunghi, B., Gambelli, F., Lungarella, G. (2001). Effects of cigarette smoke in mice with different levels of alpha(1)-proteinase inhibitor and sensitivity to oxidants. Am J Respir Crit Care Med, 164(5), 886–890.
Celli, B.R., & Barnes, P.J. (2007). Exacerbations of chronic obstructive pulmonary disease. Eur Respir J, 29(6), 1224–1238.
Chanez, P., Vignola, A.M., O’Shaugnessy, T., Enander, I., Li, D., Jeffery, P.K., & Bousquet, J. (1997). Corticosteroid reversibility in COPD is related to features of asthma. Am J Respir Crit Care Med, 155(5), 1529–1534.
Chrysofakis, G., Tzanakis, N., Kyriakoy, D., Tsoumakidou, M., Tsiligianni, I., Klimathianaki, M., & Siafakas, N.M. (2004). Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest, 125(1), 71–76.
Chung, K.F., & Adcock, I.M. (2008). Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J, 31(6), 1334–1356.
Churg, A., Wang, R., Wang, X., Onnervik, P.O., Thim, K., & Wright, J.L. (2007). Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax, 62(8), 706–713.
Churg, A., Wang, R.D., Xie, C., & Wright, J.L. (2003). alpha-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med, 168(2), 199–207.

References 96
Connors, A.F., Jr., Dawson, N.V., Thomas, C., Harrell, F.E., Jr., Desbiens, N., Fulkerson, W.J., Knaus, W.A. (1996). Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am J Respir Crit Care Med, 154(4 Pt 1), 959–967.
Cosio, M.G., Hale, K.A., & Niewoehner, D.E. (1980). Morphologic and morphometric effects of prolonged cigarette smoking on the small airways. Am Rev Respir Dis, 122(2), 265–221.
Cosio, M.G., Majo, J., & Cosio, M.G. (2002). Inflammation of the Airways and Lung Parenchyma in COPD. Chest, 121(5 suppl), 160S-165S.
Cosio, M.G., Saetta, M., & Agusti, A. (2009). Immunologic Aspects of Chronic Obstructive Pulmonary Disease. N Engl J Med, 360(23), 2445–2454.
Coultas, D.B., Mapel, D., Gagnon, R., & Lydick, E. (2001). The health impact of undiagnosed airflow obstruction in a national sample of United States adults. Am J Respir Crit Care Med, 164(3), 372–377.
Creutzberg, E.C., Schols, A.M., Bothmer-Quaedvlieg, F.C., & Wouters, E.F. (1998). Prevalence of an elevated resting energy expenditure in patients with chronic obstructive pulmonary disease in relation to body composition and lung function. Eur J Clin Nutr, 52(6), 396–401.
D’Hulst A, I., Maes, T., Bracke, K.R., Demedts, I.K., Tournoy, K.G., Joos, G.F., & Brusselle, G.G. (2005). Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respir Res, 6, 147.
D’Hulst, A.I., Vermaelen, K.Y., Brusselle, G.G., Joos, G.F., & Pauwels, R.A. (2005). Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J, 26(2), 204–213.
Dawicki, W., & Marshall, J.S. (2007). New and emerging roles for mast cells in host defence. Curr Opin Immunol, 19(1), 31–38.
de Boer, W.I., van Schadewijk, A., Sont, J.K., Sharma, H.S., Stolk, J., Hiemstra, P.S., & van Krieken, J.H. (1998). Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 158(6), 1951–1957.
de Jong, J.W., van der Belt-Gritter, B., Koeter, G.H., & Postma, D.S. (1997). Peripheral blood lymphocyte cell subsets in subjects with chronic obstructive pulmonary disease: association with smoking, IgE and lung function. Respir Med, 91(2), 67–76.
Demedts, I.K., Bracke, K.R., Van Pottelberge, G., Testelmans, D., Verleden, G.M., Vermassen, F.E., Brusselle, G.G. (2007). Accumulation of Dendritic Cells and Increased CCL20 Levels in the Airways of Patients with Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med, 175(10), 998–1005.
DeMeo, D., Campbell, E.J., Rennard, S.I., Delorey, T.M., Barker, A.F., Brantly, M.L., Silverman, E.K. (2008). Association of MHC region SNPs with airflow obstruction in severe Alpha 1-Antitrypsin Deficiency, Philadelphia, PA.
Di Stefano, A., Capelli, A., Lusuardi, M., Balbo, P., Vecchio, C., Maestrelli, P., Saetta, M. (1998). Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med, 158(4), 1277–1285.
Di Stefano, A., Caramori, G., Capelli, A., Gnemmi, I., Ricciardolo, F.L., Oates, T., Adcock, I.M. (2004). STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J, 24(1), 78–85.
Ekberg-Jansson, A., Andersson, B., Avra, E., Nilsson, O., & Lofdahl, C.G. (2000). The expression of lymphocyte surface antigens in bronchial biopsies, bronchoalveolar lavage cells and blood cells in healthy smoking and never-smoking men, 60 years old. Respir Med, 94(3), 264–272.
Elliott, W., McDonough, J., Wladichuck, A., Buzatu, L., Utokaparch, S., Javadifard, A., Hogg, J. (2009). The Nature of the Mucosal Immune Response in COPD. Am. J. Respir. Crit. Care Med., 179(1_MeetingAbstracts), A2837-.

97 Pathogenesis of COPD
Enelow, R.I., Mohammed, A.Z., Stoler, M.H., Liu, A.N., Young, J.S., Lou, Y.H., & Braciale, T.J. (1998). Structural and functional consequences of alveolar cell recognition by CD8(+) T lymphocytes in experimental lung disease. J Clin Invest, 102(9), 1653–1661.
Feghali-Bostwick, C.A., Gadgil, A.S., Otterbein, L.E., Pilewski, J.M., Stoner, M.W., Csizmadia, E., Duncan, S.R. (2008). Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 177(2), 156–163.
Finkelstein, R., Fraser, R.S., Ghezzo, H., & Cosio, M.G. (1995). Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med, 152(5 Pt 1), 1666–1672.
Foronjy, R., Nkyimbeng, T., Wallace, A., Thankachen, J., Okada, Y., Lemaitre, V., & D’Armiento, J. (2008). Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol, 294(6), L1149–1157.
Foronjy, R.F., Mirochnitchenko, O., Propokenko, O., Lemaitre, V., Jia, Y., Inouye, M., D’Armiento, J.M. (2006). Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am J Respir Crit Care Med, 173(6), 623–631.
Franssen, F.M., O’Donnell, D.E., Goossens, G.H., Blaak, E.E., & Schols, A.M. (2008). Obesity and the lung: 5. Obesity and COPD. Thorax, 63(12), 1110–1117.
Freeman, C.M., Martinez, F.J., Han, M.K., Ames, T.M., Chensue, S.W., Todt, J.C., Curtis, J.L. (2009). Lung Dendritic Cell Expression of Maturation Molecules Increases with Worsening COPD. Am J Respir Crit Care Med, 180, 1179–1188.
Fujimoto, K., Kubo, K., Yamamoto, H., Yamaguchi, S., & Matsuzawa, Y. (1999). Eosinophilic inflammation in the airway is related to glucocorticoid reversibility in patients with pulmonary emphysema. Chest, 115(3), 697–702.
Gamble, E., Grootendorst, D.C., Hattotuwa, K., O’Shaughnessy, T., Ram, F.S., Qiu, Y., Barnes, N.C. (2007). Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J.
Gan, W.Q., Man, S.F., Senthilselvan, A., & Sin, D.D. (2004). Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax, 59(7), 574–580.
Gershwin, M.E., Selmi, C., Worman, H.J., Gold, E.B., Watnik, M., Utts, J., Vierling, J.M. (2005). Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology, 42(5), 1194–1202.
Gosman, M.M., Postma, D.S., Vonk, J.M., Rutgers, B., Lodewijk, M., Smith, M., Timens, W. (2008). Association of mast cells with lung function in chronic obstructive pulmonary disease. Respir Res, 9, 64.
Gosman, M.M., Willemse, B.W., Jansen, D.F., Lapperre, T.S., van Schadewijk, A., Hiemstra, P.S., Kerstjens, H.A. (2006). Increased number of B-cells in bronchial biopsies in COPD. Eur Respir J, 27(1), 60–64.
Grashoff, W.F., Sont, J.K., Sterk, P.J., Hiemstra, P.S., de Boer, W.I., Stolk, J., van Krieken, J.M. (1997). Chronic obstructive pulmonary disease: role of bronchiolar mast cells and macrophages. Am J Pathol, 151(6), 1785–1790.
Groenewegen, K.H., Schols, A.M. W.J., & Wouters, E.F. M. (2003). Mortality and Mortality-Related Factors After Hospitalization for Acute Exacerbation of COPD. Chest, 124(2), 459–467.
Grumelli, S., Corry, D.B., Song, L.Z., Song, L., Green, L., Huh, J., Kheradmand, F. (2004). An Immune Basis for Lung Parenchymal Destruction in Chronic Obstructive Pulmonary Disease and Emphysema. Plos Med, 1(1), e8.
Halbert, R.J., Natoli, J.L., Gano, A., Badamgarav, E., Buist, A.S., & Mannino, D.M. (2006). Global burden of COPD: systematic review and meta-analysis. Eur Respir J, 28(3), 523–532.
Hautamaki, R.D., Kobayashi, D.K., Senior, R.M., & Shapiro, S.D. (1997). Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science, 277(5334), 2002–2004.

References 98
Henkart, P.A. (1994). Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity, 1(5), 343–346.
Hodge, G., Nairn, J., Holmes, M., Reynolds, P.N., & Hodge, S. (2007). Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin Exp Immunol, 150(1), 22–29.
Hogg, J.C., Chu, F., Utokaparch, S., Woods, R., Elliott, W.M., Buzatu, L., Pare, P.D. (2004). The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N Engl J Med, 350(26), 2645–2653.
Hogg, J.C., Wright, J.L., Wiggs, B.R., Coxson, H.O., Opazo Saez, A., & Pare, P.D. (1994). Lung structure and function in cigarette smokers. Thorax, 49(5), 473–478.
Hurst, J.R., Perera, W.R., Wilkinson, T.M., Donaldson, G.C., & Wedzicha, J.A. (2006). Systemic and upper and lower airway inflammation at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 173(1), 71–78.
Hurst, J.R., Wilkinson, T.M., Perera, W.R., Donaldson, G.C., & Wedzicha, J.A. (2005). Relationships among bacteria, upper airway, lower airway, and systemic inflammation in COPD. Chest, 127(4), 1219–1226.
Ihn, H. (2002). Pathogenesis of fibrosis: role of TGF-beta and CTGF. Curr Opin Rheumatol, 14(6), 681–685.
Iizuka, T., Ishii, Y., Itoh, K., Kiwamoto, T., Kimura, T., Matsuno, Y., Sekizawa, K. (2005). Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells, 10(12), 1113–1125.
Innes, A.L., Woodruff, P.G., Ferrando, R.E., Donnelly, S., Dolganov, G.M., Lazarus, S.C., & Fahy, J.V. (2006). Epithelial mucin stores are increased in the large airways of smokers with airflow obstruction. Chest, 130(4), 1102–1108.
Ito, K., Ito, M., Elliott, W.M., Cosio, B., Caramori, G., Kon, O.M., Barnes, P.J. (2005). Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med, 352(19), 1967–1976.
Jameson, J., Witherden, D., & Havran, W.L. (2003). T-cell effector mechanisms: gammadelta and CD1d-restricted subsets. Curr Opin Immunol, 15(3), 349–353.
Jawdat, D.M., Rowden, G., & Marshall, J.S. (2006). Mast cells have a pivotal role in TNF-independent lymph node hypertrophy and the mobilization of Langerhans cells in response to bacterial peptidoglycan. J Immunol, 177(3), 1755–1762.
Kang, M.J., Lee, C.G., Lee, J.Y., Dela Cruz, C.S., Chen, Z.J., Enelow, R., & Elias, J.A. (2008). Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest, 118(8), 2771–2784.
Keatings, V.M., Collins, P.D., Scott, D.M., & Barnes, P.J. (1996). Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med, 153(2), 530–534.
Kelsen, S.G., Aksoy, M.O., Georgy, M., Hershman, R., Ji, R., Li, X., Kim, V. (2009). Lymphoid follicle cells in chronic obstructive pulmonary disease overexpress the chemokine receptor CXCR3. Am J Respir Crit Care Med, 179(9), 799–805.
Kim, W.D., Eidelman, D.H., Izquierdo, J.L., Ghezzo, H., Saetta, M.P., & Cosio, M.G. (1991). Centri-lobular and panlobular emphysema in smokers. Two distinct morphologic and functional entities. Am Rev Respir Dis, 144(6), 1385–1390.
Kim, W.D., Kim, W.S., Koh, Y., Lee, S.D., Lim, C.M., Kim, D.S., & Cho, Y.J. (2002). Abnormal peripheral blood T-lymphocyte subsets in a subgroup of patients with COPD. Chest, 122(2), 437–444.
Korn, S., Wiewrodt, R., Walz, Y.C., Becker, K., Mayer, E., Krummenauer, F., & Buhl, R. (2005). Charac-terization of the interstitial lung and peripheral blood T cell receptor repertoire in cigarette smokers. Am J Respir Cell Mol Biol, 32(2), 142–148.

99 Pathogenesis of COPD
Lacoste, J.Y., Bousquet, J., Chanez, P., Van Vyve, T., Simony-Lafontaine, J., Lequeu, N., Michel, F.B. (1993). Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary disease. J Allergy Clin Immunol, 92(4), 537–548.
Lams, B.E., Sousa, A.R., Rees, P.J., & Lee, T.H. (1998). Immunopathology of the small-airway submucosa in smokers with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 158(5 Pt 1), 1518–1523.
Lee, S.H., Goswami, S., Grudo, A., Song, L.Z., Bandi, V., Goodnight-White, S., Kheradmand, F. (2007). Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med, 13(5), 567–569.
Linden, M., Rasmussen, J.B., Piitulainen, E., Tunek, A., Larson, M., Tegner, H., Brattsand, R. (1993). Airway inflammation in smokers with nonobstructive and obstructive chronic bronchitis. Am Rev Respir Dis, 148(5), 1226–1232.
MacNee, W. (2000). Oxidants/antioxidants and COPD. Chest, 117(5 Suppl 1), 303S-317S. MacNee, W. (2005). Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive
pulmonary disease. Proc Am Thorac Soc, 2(1), 50–60. Maeno, T., Houghton, A.M., Quintero, P.A., Grumelli, S., Owen, C.A., & Shapiro, S.D. (2007). CD8+ T
Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol, 178(12), 8090–8096.
Majo, J., Ghezzo, H., & Cosio, M.G. (2001). Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J, 17(5), 946–953.
Marwick, J.A., Kirkham, P.A., Stevenson, C.S., Danahay, H., Giddings, J., Butler, K., Rahman, I. (2004). Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol, 31(6), 633–642.
Masubuchi, T., Koyama, S., Sato, E., Takamizawa, A., Kubo, K., Sekiguchi, M., Izumi, T. (1998). Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol, 153(6), 1903–1912.
Mathur, R., Cox, I.J., Oatridge, A., Shephard, D.T., Shaw, R.J., & Taylor-Robinson, S.D. (1999). Cerebral bioenergetics in stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 160(6), 1994–1999.
Maurer, J., Rebbapragada, V., Borson, S., Goldstein, R., Kunik, M.E., Yohannes, A.M., & Hanania, N.A. (2008). Anxiety and depression in COPD: current understanding, unanswered questions, and research needs. Chest, 134(4 Suppl), 43S-56S.
Meshi, B., Vitalis, T.Z., Ionescu, D., Elliott, W.M., Liu, C., Wang, X.D., Hogg, J.C. (2002). Emphysematous lung destruction by cigarette smoke. The effects of latent adenoviral infection on the lung inflammatory response. Am J Respir Cell Mol Biol, 26(1), 52–57.
Metz, M., Siebenhaar, F., & Maurer, M. (2008). Mast cell functions in the innate skin immune system. Immunobiology, 213(3–4), 251–260.
Mio, T., Romberger, D.J., Thompson, A.B., Robbins, R.A., Heires, A., & Rennard, S.I. (1997). Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med, 155(5), 1770–1776.
Morissette, M.C., Parent, J., & Milot, J. (2007). Perforin, granzyme B, and FasL expression by peripheral blood T lymphocytes in emphysema. Respir Res, 8, 62.
Mortaz, E., Kraneveld, A.D., Smit, J.J., Kool, M., Lambrecht, B.N., Kunkel, S.L., Folkerts, G. (2009). Effect of cigarette smoke extract on dendritic cells and their impact on T-cell proliferation. PLoS ONE, 4(3), e4946.
Nakano, Y., Muro, S., Sakai, H., Hirai, T., Chin, K., Tsukino, M., Mishima, M. (2000). Computed tomographic measurements of airway dimensions and emphysema in smokers. Correlation with lung function. Am J Respir Crit Care Med, 162(3 Pt 1), 1102–1108.
Nici, L., Donner, C., Wouters, E., Zuwallack, R., Ambrosino, N., Bourbeau, J., Troosters, T. (2006). American Thoracic Society/European Respiratory Society statement on pulmonary rehabi-litation. Am J Respir Crit Care Med, 173(12), 1390–1413.

References 100
Nishikawa, M., Kakemizu, N., Ito, T., Kudo, M., Kaneko, T., Suzuki, M., Okubo, T. (1999). Superoxide mediates cigarette smoke-induced infiltration of neutrophils into the airways through nuclear factor-kappaB activation and IL-8 mRNA expression in guinea pigs in vivo. Am J Respir Cell Mol Biol, 20(2), 189–198.
Noguera, A., Batle, S., Miralles, C., Iglesias, J., Busquets, X., MacNee, W., & Agusti, A.G. (2001). Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax, 56(6), 432–437.
Noguera, A., Busquets, X., Sauleda, J., Villaverde, J.M., MacNee, W., & Agusti, A.G. (1998). Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 158(5 Pt 1), 1664–1668.
O’Shaughnessy, T.C., Ansari, T.W., Barnes, N.C., & Jeffery, P.K. (1997). Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med, 155(3), 852–857.
Ogawa, E., Nakano, Y., Ohara, T., Muro, S., Hirai, T., Sato, S., Mishima, M. (2009). Body mass index in male patients with COPD: correlation with low attenuation areas on CT. Thorax, 64(1), 20–25.
Panzner, P., Lafitte, J.J., Tsicopoulos, A., Hamid, Q., & Tulic, M.K. (2003). Marked up-regulation of T lymphocytes and expression of interleukin-9 in bronchial biopsies from patients With chronic bronchitis with obstruction. Chest, 124(5), 1909–1915.
Papadopoulos, N.G., Alamanos, Y., Voulgari, P.V., Epagelis, E.K., Tsifetaki, N., & Drosos, A.A. (2005). Does cigarette smoking influence disease expression, activity and severity in early rheumatoid arthritis patients? Clin Exp Rheumatol, 23(6), 861–866.
Pesci, A., Rossi, G.A., Bertorelli, G., Aufiero, A., Zanon, P., & Olivieri, D. (1994). Mast cells in the airway lumen and bronchial mucosa of patients with chronic bronchitis. Am J Respir Crit Care Med, 149(5), 1311–1316.
Petrache, I., Natarajan, V., Zhen, L., Medler, T.R., Richter, A.T., Cho, C., Tuder, R.M. (2005). Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med, 11(5), 491–498.
Pettersen, C.A., & Adler, K.B. (2002). Airways inflammation and COPD: epithelial-neutrophil interactions. Chest, 121(5 Suppl), 142S-150S.
Pizzichini, E., Pizzichini, M.M., Gibson, P., Parameswaran, K., Gleich, G.J., Berman, L., Hargreave, F.E. (1998). Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitis. Am J Respir Crit Care Med, 158(5 Pt 1), 1511–1517.
Pons, J., Sauleda, J., Ferrer, J.M., Barcelo, B., Fuster, A., Regueiro, V., Agusti, A.G. N. (2005). Blunted {gamma}{delta} T-lymphocyte response in chronic obstructive pulmonary disease. Eur Respir J, 25(3), 441–446.
Prescott, E., Lange, P., & Vestbo, J. (1995). Chronic mucus hypersecretion in COPD and death from pulmonary infection. Eur Respir J, 8(8), 1333–1338.
Prummel, M.F., & Wiersinga, W.M. (1993). Smoking and risk of Graves’ disease. Jama, 269(4), 479–482.
Qu, P., Du, H., Wang, X., & Yan, C. (2009). Matrix metalloproteinase 12 overexpression in lung epithelial cells plays a key role in emphysema to lung bronchioalveolar adenocarcinoma transition. Cancer Res, 69(18), 7252–7261.
Quint, J.K., Baghai-Ravary, R., Donaldson, G.C., & Wedzicha, J.A. (2008). Relationship between depression and exacerbations in COPD. Eur Respir J, 32(1), 53–60. doi: 10.1183/09031936.00120107
Rangasamy, T., Cho, C.Y., Thimmulappa, R.K., Zhen, L., Srisuma, S.S., Kensler, T.W., Biswal, S. (2004). Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest, 114(9), 1248–1259.
Rennard, S.I., & Vestbo, J. (2006). COPD: the dangerous underestimate of 15%. Lancet, 367(9518), 1216–1219.

101 Pathogenesis of COPD
Retamales, I., Elliott, W.M., Meshi, B., Coxson, H.O., Pare, P.D., Sciurba, F.C., Hogg, J.C. (2001). Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med, 164(3), 469–473.
Richmond, I., Pritchard, G.E., Ashcroft, T., Corris, P.A., & Walters, E.H. (1993). Distribution of gamma delta T-cells in the bronchial tree of smokers and non-smokers. J Clin Pathol, 46(10), 926–930.
Rodriguez-Roisin, R., & Soriano, J.B. (2008). Chronic obstructive pulmonary disease with lung cancer and/or cardiovascular disease. Proc Am Thorac Soc, 5(8), 842–847.
Rose, N.R., & Bona, C. (1993). Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today, 14(9), 426–430.
Saetta, M., Baraldo, S., Corbino, L., Turato, G., Braccioni, F., Rea, F., Fabbri, L.M. (1999). CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 160(2), 711–717.
Saetta, M., Di Stefano, A., Maestrelli, P., Turato, G., Mapp, C.E., Pieno, M., Fabbri, L.M. (1996). Airway eosinophilia and expression of interleukin-5 protein in asthma and in exacerbations of chronic bronchitis. Clin Exp Allergy, 26(7), 766–774.
Saetta, M., Di Stefano, A., Turato, G., Facchini, F.M., Corbino, L., Mapp, C.E., Fabbri, L.M. (1998). CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 157(3 Pt 1), 822–826.
Sapey, E., & Stockley, R.A. (2006). COPD exacerbations . 2: aetiology. Thorax, 61(3), 250–258. Schols, A.M. (2000). Nutrition in chronic obstructive pulmonary disease. Curr Opin Pulm Med, 6(2),
110–115. Selman, M., Cisneros-Lira, J., Gaxiola, M., Ramirez, R., Kudlacz, E.M., Mitchell, P.G., & Pardo, A.
(2003). Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in Guinea pigs. Chest, 123(5), 1633–1641.
Shan, M., Cheng, H.-F., Song, L.-z., Roberts, L., Green, L., Hacken-Bitar, J., Kheradmand, F. (2009). Lung Myeloid Dendritic Cells Coordinately Induce TH1 and TH17 Responses in Human Emphysema. Science Translational Medicine, 1(4), 4ra10–14ra10.
Shapiro, S.D., Goldstein, N.M., Houghton, A.M., Kobayashi, D.K., Kelley, D., & Belaaouaj, A. (2003). Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol, 163(6), 2329–2335.
Sin, D.D., Anthonisen, N.R., Soriano, J.B., & Agusti, A.G. (2006). Mortality in COPD: Role of comorbidities. Eur Respir J, 28(6), 1245–1257.
Sin, D.D., & Man, S.F. (2003). Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation, 107(11), 1514–1519.
Smith, K.R., Uyeminami, D.L., Kodavanti, U.P., Crapo, J.D., Chang, L.Y., & Pinkerton, K.E. (2002). Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med, 33(8), 1106–1114.
Sparrow, D., Glynn, R.J., Cohen, M., & Weiss, S.T. (1984). The relationship of the peripheral leukocyte count and cigarette smoking to pulmonary function among adult men. Chest, 86(3), 383–386.
Stang, P., Lydick, E., Silberman, C., Kempel, A., & Keating, E.T. (2000). The prevalence of COPD: using smoking rates to estimate disease frequency in the general population. Chest, 117(5 Suppl 2), 354S-359S.
Sullivan, A.K., Simonian, P.L., Falta, M.T., Mitchell, J.D., Cosgrove, G.P., Brown, K.K., Fontenot, A.P. (2005). Oligoclonal CD4+ T Cells in the Lungs of Patients with Severe Emphysema. Am J Respir Crit Care Med, 172(5), 590–596.
Takizawa, H., Tanaka, M., Takami, K., Ohtoshi, T., Ito, K., Satoh, M., Umeda, A. (2001). Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med, 163(6), 1476–1483.

References 102
Takubo, Y., Guerassimov, A., Ghezzo, H., Triantafillopoulos, A., Bates, J.H., Hoidal, J.R., & Cosio, M.G. (2002). Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med, 166(12 Pt 1), 1596–1603.
Turato, G., Di Stefano, A., Maestrelli, P., Mapp, C.E., Ruggieri, M.P., Roggeri, A., Saetta, M. (1995). Effect of smoking cessation on airway inflammation in chronic bronchitis. Am J Respir Crit Care Med, 152(4 Pt 1), 1262–1267.
Urbanowicz, R., Lamb, J., Corne, J., & Fairclough, L. (2009). Cytotoxic Cells in the Induced Sputum of Smokers with COPD. Am. J. Respir. Crit. Care Med., 179(1_MeetingAbstracts), A4297-.
Urbanowicz, R., Lamb, J., Todd, I., Corne, J., & Fairclough, L. (2009). Altered effector function of peripheral cytotoxic cells in COPD. Respiratory Research, 10(1), 53.
van der Strate, B.W., Postma, D.S., Brandsma, C.A., Melgert, B.N., Luinge, M.A., Geerlings, M., Kerstjens, H.A. (2006). Cigarette smoke-induced emphysema: A role for the B cell? Am J Respir Crit Care Med, 173(7), 751–758.
Vernooy, J.H., Kucukaycan, M., Jacobs, J.A., Chavannes, N.H., Buurman, W.A., Dentener, M.A., & Wouters, E.F. (2002). Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med, 166(9), 1218–1224.
Vernooy, J.H., Moller, G.M., van Suylen, R.J., van Spijk, M.P., Cloots, R.H., Hoet, P.H., Wouters, E.F. (2007). Increased granzyme A expression in type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 175(5), 464–472.
Vestbo, J., Prescott, E., & Lange, P. (1996). Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am J Respir Crit Care Med, 153(5), 1530–1535.
Vijayanand, P., Seumois, G., Pickard, C., Powell, R.M., Angco, G., Sammut, D., Djukanovic, R. (2007). Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med, 356(14), 1410–1422.
Wan, E.S., & Silverman, E.K. (2009). Genetics of COPD and emphysema. Chest, 136(3), 859–866. Wang, Z., Zheng, T., Zhu, Z., Homer, R.J., Riese, R.J., Chapman, H.A., Jr., Elias, J.A. (2000). Interferon
gamma induction of pulmonary emphysema in the adult murine lung. J Exp Med, 192(11), 1587–1600.
Watz, H., Waschki, B., Kirsten, A., Muller, K.C., Kretschmar, G., Meyer, T., Magnussen, H. (2009). The metabolic syndrome in patients with chronic bronchitis and COPD: frequency and associated consequences for systemic inflammation and physical inactivity. Chest, 136(4), 1039–1046.
Wedzicha, J.A. (2004). Role of Viruses in Exacerbations of Chronic Obstructive Pulmonary Disease. Proc Am Thorac Soc, 1(2), 115–120.
Wright, J.L., Farmer, S.G., & Churg, A. (2002). Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigs. Am J Respir Crit Care Med, 166(7), 954–960.
Xu, W., Collet, J.P., Shapiro, S., Lin, Y., Yang, T., Platt, R.W., Bourbeau, J. (2008). Independent effect of depression and anxiety on chronic obstructive pulmonary disease exacerbations and hospitali-zations. Am J Respir Crit Care Med, 178(9), 913–920.
Zhao, M.Q., Stoler, M.H., Liu, A.N., Wei, B., Soguero, C., Hahn, Y.S., & Enelow, R.I. (2000). Alveolar epithelial cell chemokine expression triggered by antigen-specific cytolytic CD8(+) T cell recognition. J Clin Invest, 106(6), R49–58.
Zheng, T., Kang, M.J., Crothers, K., Zhu, Z., Liu, W., Lee, C.G., Elias, J.A. (2005). Role of Cathepsin S-Dependent Epithelial Cell Apoptosis in IFN-{gamma}-Induced Alveolar Remodeling and Pulmonary Emphysema. J Immunol, 174(12), 8106–8115.
Zhu, J., Qiu, Y., Valobra, M., Qiu, S., Majumdar, S., Matin, D., Jeffery, P.K. (2007). Plasma cells and IL-4 in chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 175(11), 1125–1133.

103 Pathogenesis of COPD
Zhu, X., Gadgil, A.S., Givelber, R., George, M.P., Stoner, M.W., Sciurba, F.C., & Duncan, S.R. (2009). Peripheral T Cell Functions Correlate with the Severity of Chronic Obstructive Pulmonary Disease. J Immunol, 182(5), 3270–3277.
Zhu, Z., Lee, C.G., Zheng, T., Chupp, G., Wang, J., Homer, R.J., Elias, J.A. (2001). Airway inflammation and remodeling in asthma. Lessons from interleukin 11 and interleukin 13 transgenic mice. Am J Respir Crit Care Med, 164(10 Pt 2), S67–70.