2005 anal chim acta glycerol
TRANSCRIPT

Analytica Chimica Acta 549 (2005) 140–150
Properties and analytical application of PQQ-dependent glyceroldehydrogenase fromGluconobactersp. 33
I. Lapenaitea,c,∗, B. Kurtinaitieneb, J. Razumieneb, V. Laurinaviciusb,L. Marcinkevicieneb, I. Bachmatovab, R. Meskysb, A. Ramanaviciusa,c,∗∗
a Department of Analytical and Environmental Chemistry, Faculty of Chemistry, Vilnius University,Naugarduko 24, 03225 Vilnius, Lithuania
b Institute of Biochemistry, Mokslininku 12, 08662 Vilnius, Lithuaniac Sector of Immunoanalysis and Informatics, Institute of Immunology of Vilnius University, Moletu pl. 29, 08409 Vilnius, Lithuania
Received 3 January 2005; received in revised form 27 May 2005; accepted 8 June 2005Available online 14 July 2005
Abstract
++ ++
i 9.5. It wasd turesh erald selectivityo branes ofG ethanola ne( nd it waso enase( erola©
K
1
cofoP
(
ent
rate,zymei-
, glu-
, alco-,l
lentlyw in
0d
PQQ-dependent glycerol dehydrogenase (GlyDH) was purified fromGluconobactersp. 33. Activation of this enzyme by Caand Mgons and PQQ was analysed. The purified GlyDH displayed an optimum activity at pH 7.0–7.5 and was the most stable at pH 8.5–etermined that the rate of glycerol oxidation had a maximum at 33–37◦C. However, a rapid enzyme inactivation began at the temperaigher than 35◦C. The calculatedKM values for glycerol,d-sorbitol, andd-mannitol were 0.83, 1.0, and 2.4 mM, respectively. Sevifferent designs of enzymatic electrodes were constructed and involved into glycerol biosensor design. Investigation of substratef immobilised (cross-linked by glutaraldehyde) GlyDH shows, that immobilisation procedure decreases the activity of the memluconobactersp. 33 cells (mGlyDH) for glucose, sorbitol, mannitol, and ethanol, but increases the activity of mGlyDH for mnd dulcitol. Soluble phenazine methosulphate (PMS) and insoluble 4-ferrocenylphenol (FP),N-(4-hydroxybenzylidene)-4-ferrocenylaniliHBFA) were applied for construction of GlyDH-based biosensors. Inactivation of GlyDH by heavy metal ions was investigated abserved that biosensors based on membranes ofGluconobactersp. 33 cells (mGlyDH) and purified PQQ-dependent glycerol dehydrogpGlyDH) were sensitive to Pb++, Cd++, Co++, Cu++, Ni++, and Hg++. Possibility to apply GlyDH-based biosensors for detection of glycnd triglycerides in real wine samples was shown.2005 Elsevier B.V. All rights reserved.
eywords:PQQ; Quinoproteins; Biosensor; Glycerol dehydrogenase; Bioelectrochemistry
. Introduction
Quinoproteins are attractive components for the appli-ation to biosensor design due to their insensibility toxygen. Besides, some of them exhibit direct electron trans-
er between the active site and gold[1], carbon [2,3]r polypyrrole-modified[4] electrode. However, wiring ofQQ-dependent enzymes by red–ox mediators makes them
∗ Corresponding author. Tel.: +370 5 2469242, fax: +370 5 2469210.∗∗ Corresponding author. Tel.: +370 5 2336310; fax: +370 5 2330987.
E-mail addresses:[email protected] (I. Lapenaite), [email protected]. Ramanavicius).
more effective[5,6], especially in the cases if PQQ-dependenzyme has no hemes incorporated into its structure[7]. PQQis directly responsible for a redox conversion of the substand thus it has been evaluated as the next major coenfollowing pyridine nucleotide and flavin in biological oxdoreduction[8].
Several quinoproteins are soluble, such as sorbosecose, methanol, alcohol (type II) dehydrogenases[8] whileothers are membrane-bound, such as glucose, glycerolhol (type III) [8], fructose [9], aldehyde[10], quinated-sorbitol, d-arabitol, meso-erythritol, and cyclic alcoho[11,12] dehydrogenases bearing a PQQ as a non-covabound prosthetic group and have been found until no
003-2670/$ – see front matter © 2005 Elsevier B.V. All rights reserved.oi:10.1016/j.aca.2005.06.025

I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150 141
various acetic acid bacteria ofGluconobacterstrains. Nat-ural/physiological electron acceptors of PQQ enzymes arecytochromec, cytochromeb or ubiquinone. Various syn-thetic electron acceptors can be used to oxidize the reducedenzymes[7].
Due to low stability of the purified glycerol dehydroge-nase only a few analytical systems using this enzyme havebeen reported yet[13]. A few reports dealing with biocon-version of glycerol to dihydroxyacetone by the immobilisedGluconobacter oxydanscells containing membrane-boundglycerol dehydrogenase or describing the properties of PQQ-dependent glycerol dehydrogenases have been published[14–17]. Glycerol dehydrogenase was defined as an enzymethat shows broad substrate specificity toward many kindsof polyhydroxyl alcohols including glycerol,d-mannitol,d-sorbitol, d-arabitol, adonitol, propylene glycol, andmeso-erythritol, but not ethanol, aliphatic aldehydes,d-glucose,d-fructose,d-gluconate or 2-keto-d-gluconate[16]. Recentstudies of two different membrane-bound quinoproteinsd-arabitol (ArDH) andd-sorbitol (SlDH) dehydrogenases puri-fied from Gluconobacter suboxydansIFO 3257 and IFO3255, respectively, showed that these quinoproteins are iden-tical [18] and responsible for almost all sugar alcohol oxida-tion inGluconobacterspecies.
Here, we describe an application of the PQQ-dependentglycerol dehydrogenase purified from the mutant strain ofG
2
2
n-d ri-fi1 lu-c2 A).
anal( A),a ulci-t ereoC MS),2m vedfb ),N A)we /dl( uis,U iono mR
2.2. Purification of glycerol dehydrogenase fromGluconobacter sp. 33
PQQ-dependent glycerol dehydrogenase (pGlyDH) waspurified by a modified scheme from biomass ofGluconobac-ter sp. 33[22]. The collected cells were suspended in 50 mMTris–maleate buffer, pH 9.0, and disrupted by sonication. Celldebris was removed by centrifugation (7500×g, 10 min).Membranes were collected by centrifugation (125,000×g,3 h) suspended in 5 mM Tris–maleate buffer, pH 9.0, con-taining 0.2% MEGA-10 and incubated at 4◦C for 1 h. Aninsoluble material was removed by centrifugation (7500×g,1 h) and supernatant was applied on a DEAE-Toyopearl col-umn (2.5 cm× 6 cm), equilibrated with 5 mM Tris–maleatebuffer, pH 9.0. The enzyme was eluted by a linear gradi-ent of 50–200 mM of NaCl in 75 mM Tris–maleate buffer,pH 9.0, containing 0.2% of MEGA-10. Active fractionswere collected, concentrated by ultrafiltration and dial-ysed against 5 mM Tris–maleate buffer, pH 9.5, containing1 mM CaCl2 and 1 mM MgCl2. The sample was applied ona DEAE–toyopearl column (1.5 cm× 4.5 cm), equilibratedwith the same buffer. The enzyme was simultaneously elutedby the linear gradient of 50–200 mM of NaCl in 75 mMTris–maleate buffer, pH 9.5, containing 0.2% of MEGA-10,1 mM CaCl2 and 1 mM MgCl2. Active fractions were col-lected, concentrated by ultrafiltration and dialysed against5 ardsa -d otas-sM
)p erec s-p
me-t uri,UM ingS e ona ianC e ofB
2
reda pol-i llar,S ste,f tera phitee
d-i d ares
luconobactersp. 33.
. Materials and methods
.1. Chemicals and enzymes
Microbial lipase (EC 3.1.1.3) fromPseudomonas meocina, 3121 with activity 1000 U/ml was isolated and pued using previously published method[19]. Catalase (EC.11.1.6) from bovine liver (2000 U/mg of protein) and gose oxidase fromAspergillus nigerof specific activity45.9 U/g of solid was received from Sigma (St. Louis, US
Glutaraldehyde (GA, 25%) was purchased from ReBudapest, Hungary). Bovine serum albumin (BSluminum paste, acetone, methanol, ethanol, d
ol, d-sorbitol, d-mannitol, and heavy metal salts wbtained from Reachim (Kiev, Ukraine),d-glucose fromeretor (Germany). Phenazine methosulphate (P,6-dichlorophenolindophenol (DCIP), N-decanoyl-N-ethylglucamine (MEGA-10), and glycerol were recei
rom Sigma (St. Louis, USA). Osmium-bis-N,N′-(2,2′-ipiridyl)-bichloride (Os-BP), 4-ferrocenylphenol (FP-(4-hydroxybenzylidene)-4-ferrocenylaniline (HBFere obtained as previously described[6,20,21]. Triglyc-ride solution in 0.1 M KCl containing surfactant, 300 mg3.6 mM) was purchased from Sigma Diagnostic (St. LoSA). Commercial test kit TG GPO-PAP for determinatf glycerol with NAD-dependent GlyDH was obtained frooche, France.
mM potassium phosphate buffer, pH 9.3 and afterwpplied on HAP column (1.5 cm× 4 cm). The glycerol dehyrogenase was eluted by a linear gradient of 5–100 mM pium phosphate buffer, containing 5 mM Mg++ and 0.2%EGA-10.The membranes ofGluconobactersp. 33 cells (mGlyDH
ossessing glycerol dehydrogenase activity (25 U/ml) wollected as previously described[23]. Membranes were suended in 10 mM Tris–HCl buffer, pH 8.0.
Graphite rod electrodes (99.999% purity) 3 mm in diaer were obtained from Sigma–Aldrich (St. Louis, MissoSA). MINICO M 7000 G isolating and silver MINICO
4200 pastes were obtained from Emerson & Cumpecialty Polymers (Westerlo, Belgium). Carbon pastbase of carbon black RAVEN-M obtained from Columbhemicals (Atlanta, US) was developed at the Institutiochemistry (Vilnius, Lithuania)[24].
.3. Preparation of modified enzyme electrodes
Electrode preparation. Graphite electrodes were prepas follows: rods of spectroscopic graphite were cut and
shed on fine emery paper (Tufback, Durite P1200, Aterling Heights, MI, USA) and then with aluminum pa
ollowed by rinsing the electrode surface with distilled wand drying at room temperature. The working area of gralectrodes was of 0.071 cm2.Construction of the sensors. Several methods for mo
fication of prepared carbon electrodes were used anummarised inTable 1.

142 I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150
Table 1Construction of glycerol biosensors
Biosensor abbreviation Enzyme preparate Mediator Immobilisation method Support
pGlyDH/C 3�l pGlyDH – Adsorbed GraphitemGlyDH/C 3�l mGlyDH – Adsorbed GraphitepGlyDH/Cp/C 3�l pGlyDH – Adsorbed Graphite coated
1–1.5 mm of carbon pastemGlyDH/Cp/C 3�l mGlyDH – Adsorbed Graphite coated
1–1.5 mm of carbon pastepGlyDH/GA/C 3�l pGlyDH – Immobilised over 25%
glutaraldehyde (GA) solution(10 min)
Graphite
mGlyDH/GA/C 3�l mGlyDH – Immobilised over 25% GA(10 min)
Graphite
mGlyDH/BSA/GA/C 3�l of mixture (1:1) ofthe mGlyDH and 10%BSA
– Immobilised over 3% GA (24 h) Graphite
Os-BP/mGlyDH/C 2�l mGlyDH 2�l (5 mg/ml) Os-BP in acetone Adsorbed GraphiteOs-BP/mGlyDH/GA/C 2�l mGlyDH 2�l (5 mg/ml) Os-BP in acetone Immobilised over 3% GA (24 h) GraphiteFP/mGlyDH/C 2�l mGlyDH 2�l (2 mg/ml) FP in acetone Adsorbed GraphiteFP/mGlyDH/GA/C 2�l mGlyDH 2�l (2 mg/ml) FP in acetone Immobilised over 3% GA (24 h) GraphiteHBFA/pGlyDH/GA/C 3�l pGlyDH 2�l (1 mg/ml) HBFA in acetone Immobilised over 25% GA
(5 min)Graphite
HBFA/pGlyDH/GA/CE 3�l pGlyDH 2�l (1 mg/ml) HBFA in acetone Immobilised over 25% GA(30 min)
Screen-printed electrode
The screen-printed electrode (CE) was made according tothe method described previously[24]. The electrode workingarea of 0.04 cm2 was coated with appropriate mediator solu-tion and dried. Then enzyme was deposited on the electrodeand the electrode held over glutaraldehyde water solution.Finally the electrode was covered with semi-permeable tery-lene film (10�m thickness, pore diameter 0.38�m, poredensity 106/cm2) [25].
All the electrodes were dried, washed with distilled waterand kept in the refrigerator at +4◦C after the modification.
2.4. Instrumentation
Construction of the sensors. Electrochemical measure-ments were performed on polarographic analyzer PA-2 Lab-oratory Pristroje (Czech Republic) with XY recorder Endim622.01 (USA) in three electrode mode, consisting of a work-ing carbon electrode, a saturated Ag/AgCl reference electrodeand a Pt wire (0.5 mm diameter and 2.5 cm length) as an aux-iliary electrode.
Electrochemical measurements with screen-printedHBFA modified electrodes were performed using theflow-through three-electrode amperometric cell[24], whereworking electrode forms the bottom of the flow cell. As areference electrode we used a silver wire (1 mm diameter and2 cm length) in saturated KCl. The titanium tube (diameter2 rode.T 15,Z adys lyzerO
-m or
and standard glycerol trioleate solution was used. Preliminaryhydrolysis of triglycerides was achieved after 1 h of incuba-tion at 25◦C with addition of 100 U of lipase. The biosensorresponses to gradual glycerol addition were measured. Thetrioleate concentration was calculated using calibration graphobtained by gradual addition of glycerol into hydrolysed tri-oleate solution. Measurements were performed in 0.05 Macetate buffer, pH 6.5, with 1 mM PMS, at 0.3 V electrodepotential versus Ag/AgCl reference electrode.
Electrochemical measurements of heavy metal ionswereperformed with pGlyDH/GA/C and mGlyDH/GA/C modi-fied electrodes in 0.05 M acetate buffer, pH 6.0, with 1 mMPMS and 10 mM glycerol at 0.25 V. Initially the steady statecurrent response of the biosensor to 10 mM of glycerol wasmeasured. Then, the decrease of steady state current followedby gradual heavy metal addition is recorded.
The determination of the glycerol in wines. Biosen-sor HBFA/pGlyDH/CE was installed into the flow-throughthree-electrode amperometric cell[24]. The biosensor wasdesigned with a purified PQQ-dependent glycerol dehydro-genase, which possessed glucose dehydrogenase activity.The glucose was eliminated from wine samples by treat-ment with glucose oxidase and catalase mixture. The sampleswere assayed in the flow-through biosensor system. Standard1 mM glycerol solution after appropriate dilution (up to 100times) with 0.05 M acetate buffer, pH 6.0, was supplied to thec
ys nm.T ye-l te( P)a s
mm and 2 cm length) was used as an auxiliary electhe flow cell was supplied with a peristaltic pump (type 3ALIMP, Warsaw, Poland). Flow rate was 5 ml/min. Stetate currents were recorded on the polarographic anaH-105 (Radelkis, Hungary).Electrochemical triglyceride determination. For the deter
ination of triglycerides, mGlyDH/BSA/GA/C biosens
ell during the measurements.Spectrophotometrical measurementswere carried out b
pectrophotometer Specord UV VIS (Germany) at 600he pGlyDH and mGlyDH activity was assayed in a d
inked system containing 50�M phenazine methosulphaPMS) and 100�M 2,6-dichlorophenol indophenol (DCIs described previously[22]. One unit of enzyme activity wa

I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150 143
defined as the amount of enzyme that catalyses the reductionof 1�mol of DCIP per minute.
Spectrophotometrical wine measurementswere per-formed using commercial test-kit TG GPO-PAP (Roche,France) atλ = 340 nm.
3. Results and discussion
3.1. Purification of glycerol dehydrogenase fromGluconobacter sp. 33
pGlyDH from Gluconobactersp. 33 was purified asdescribed in Section2. The purification procedure is sum-marised inTable 2. The usual yield of complete purificationis 14% of initial enzyme activity. The modified protocol forGlyDH purification fromGluconobactersp. 33 allow achiev-ing of specific activity of 20.45 U/mg of protein which is fivetimes higher comparing with previously reported[22]. More-over, the application of hydroxyappatite-based chromatogra-phy allowed separation of GlyDH and glucose dehydrogenaseand preparation of a homogenous enzyme according to theSDS-PAGE electrophoresis (Fig. 1). A significant stabiliza-tion of GlyDH was achieved by using more alkaline buffersduring the purification. According to the SDS-PAGE analy-sis, the molecular mass of GlyDH is about 72 kDa (Fig. 1). Iti asep( i-l rolda dentG
3
ret mMT Qc ymei glu-c movedbt ation1 ep , them d the
Fig. 1. SDS–PAGE (12% gel) analysis of purified glycerol dehydrogenasefrom Gluconobactersp. 33. The gel was stained with Coomassie brilliantblue R-250. Lane 1, prestained molecular mass marker, lane 2, the purifiedglycerol dehydrogenase.
activity of GlyDH was analysed (Table 3). The results pre-sented demonstrate that GlyDH fromGluconobactersp. 33is a PQQ-dependent enzyme and Ca++ ions are necessary forthe binding of the PQQ cofactor to the GlyDH apo-enzymesince the addition of just PQQ did not restore the apo-enzymeto holo-enzyme after 4 h incubation. The optimal concentra-tions of Ca++ and PQQ were approximately 8–10 mM and
TS fuconobactersp. 33 culture
S l protein (mg) Specific activity (U/mg of protein) Yield (%)
M 1.47 100E 2.25 81D 8.2 66D 16.8 52H 20.45 14
s similar to the molecular mass of glycerol dehydrogenurified fromGluconobactersp. (80 kDa)[18]. TheKM value0.83 mM) calculated for the purified GlyDH was very simar to theKM value (0.8 mM) of the NAD-dependent glyceehydrogenase purified fromArthrobacter sp. MS-7 [26]nd almost 40 times lower if compared with PQQ-depenlyDH fromGluconobacter industrius[17].
.2. Properties of GlyDH
Cofactor requirement. The purified enzyme lost mohan 70% of its activity after 3 days dialysis against 50ris–HCl buffer, pH 8.0, containing 5 mM EDTA. The PQofactor is not usually covalently bound to an apo-enzn majority of the PQQ-dependent enzymes (glycerol,ose, alcohol and other dehydrogenases) and can be rey dialysis in the presence of chelators[27,28]. To prove
hose presumptions, an excess of PQQ (final concentr�M) with 8 mM Ca++ and/or 8 mM Mg++ was added to throbes of the dialysed enzyme in various combinationsixtures were incubated at room temperature for 4 h an
able 2ummary of a typical purification of glycerol dehydrogenase from 1 l oGl
tep Total activity (U) Tota
embrane fraction 312 212xtraction with MEGA-10 252 112EAE chromatography I 208 25EAE chromatography II 163 9.7ydroxyapatite 45 2.2
144 I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150
Table 3Reconstitution of the GlyDH holoenzyme in presence of divalent cations
Component Immediatelyafter mixing
1 h 2 h 4 h
None 100 104 95 908 mM Ca++ 106 106 92 908 mM Mg++ 108 124 124 1248 mM Ca++, 8 mM Mg++ 114 114 114 1141�M PQQ 119 119 114 1141�M PQQ, 8 mM Ca++ 119 188 545 5571�M PQQ, 8 mM Mg++ 120 118 102 1201�M PQQ, 8 mM Ca++,
8 mM Mg++154 335 800 789
A relative activity (%) of enzyme is shown. One hundred percent correspondsto remaining activity of GlyDH after dialysis against EDTA.
0.05�M, respectively. The 20-fold excess of PQQ was usedfor the most rapid and complete reactivation of apo-enzyme toholo-enzyme. Notwithstanding that Mg++ ions alone showeda little effect the most effective and rapid reactivation wasachieved when Ca++, Mg++ ions, and PQQ were includedinto the mixture simultaneously. It was concluded that theMg++ ions played the supporting role in binding of PQQ andshowed some effect on stabilization of the enzyme (data notshown), too. In respect of the PQQ cofactor binding to apo-enzyme the described GlyDH isolated fromGluconobactersp. 33 differs from an analogous enzyme fromG. industrius,which needs only Mg++ for the binding of PQQ to the apo-enzyme[17].
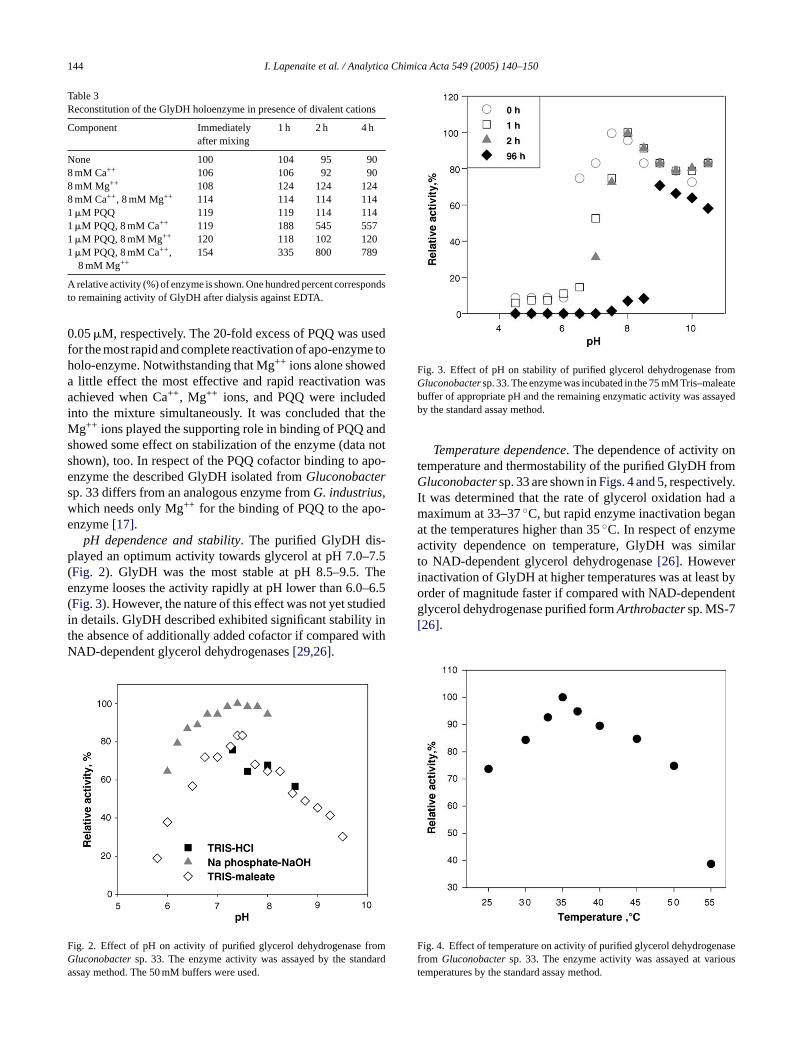
pH dependence and stability. The purified GlyDH dis-played an optimum activity towards glycerol at pH 7.0–7.5(Fig. 2). GlyDH was the most stable at pH 8.5–9.5. Theenzyme looses the activity rapidly at pH lower than 6.0–6.5(Fig. 3). However, the nature of this effect was not yet studiedin details. GlyDH described exhibited significant stability inthe absence of additionally added cofactor if compared withNAD-dependent glycerol dehydrogenases[29,26].
F romG darda
Fig. 3. Effect of pH on stability of purified glycerol dehydrogenase fromGluconobactersp. 33. The enzyme was incubated in the 75 mM Tris–maleatebuffer of appropriate pH and the remaining enzymatic activity was assayedby the standard assay method.
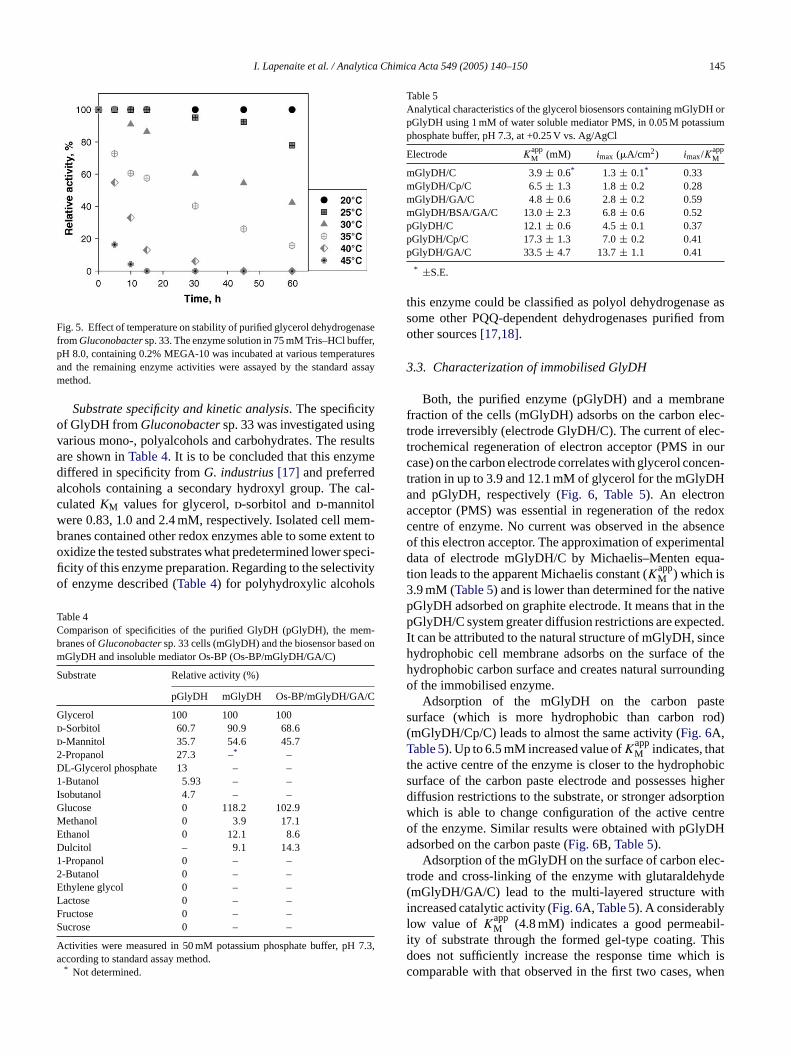
Temperature dependence. The dependence of activity ontemperature and thermostability of the purified GlyDH fromGluconobactersp. 33 are shown inFigs. 4 and 5, respectively.It was determined that the rate of glycerol oxidation had amaximum at 33–37◦C, but rapid enzyme inactivation beganat the temperatures higher than 35◦C. In respect of enzymeactivity dependence on temperature, GlyDH was similarto NAD-dependent glycerol dehydrogenase[26]. Howeverinactivation of GlyDH at higher temperatures was at least byorder of magnitude faster if compared with NAD-dependentglycerol dehydrogenase purified formArthrobactersp. MS-7[26].
F nasef ioust
ig. 2. Effect of pH on activity of purified glycerol dehydrogenase fluconobactersp. 33. The enzyme activity was assayed by the stanssay method. The 50 mM buffers were used.
ig. 4. Effect of temperature on activity of purified glycerol dehydrogerom Gluconobactersp. 33. The enzyme activity was assayed at varemperatures by the standard assay method.
I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150 145
Fig. 5. Effect of temperature on stability of purified glycerol dehydrogenasefromGluconobactersp. 33. The enzyme solution in 75 mM Tris–HCl buffer,pH 8.0, containing 0.2% MEGA-10 was incubated at various temperaturesand the remaining enzyme activities were assayed by the standard assaymethod.
Substrate specificity and kinetic analysis. The specificityof GlyDH fromGluconobactersp. 33 was investigated usingvarious mono-, polyalcohols and carbohydrates. The resultsare shown inTable 4. It is to be concluded that this enzymediffered in specificity fromG. industrius[17] and preferredalcohols containing a secondary hydroxyl group. The cal-culatedKM values for glycerol,d-sorbitol andd-mannitolwere 0.83, 1.0 and 2.4 mM, respectively. Isolated cell mem-branes contained other redox enzymes able to some extent tooxidize the tested substrates what predetermined lower speci-ficity of this enzyme preparation. Regarding to the selectivityof enzyme described (Table 4) for polyhydroxylic alcohols
Table 4Comparison of specificities of the purified GlyDH (pGlyDH), the mem-branes ofGluconobactersp. 33 cells (mGlyDH) and the biosensor based onmGlyDH and insoluble mediator Os-BP (Os-BP/mGlyDH/GA/C)
Substrate Relative activity (%)
pGlyDH mGlyDH Os-BP/mGlyDH/GA/C
Glycerol 100 100 100d-Sorbitol 60.7 90.9 68.6d-Mannitol 35.7 54.6 45.72-Propanol 27.3 –* –DL-Glycerol phosphate 13 – –1-Butanol 5.93 – –Isobutanol 4.7 – –Glucose 0 118.2 102.9MED12ELFS
A 7.3,a
Table 5Analytical characteristics of the glycerol biosensors containing mGlyDH orpGlyDH using 1 mM of water soluble mediator PMS, in 0.05 M potassiumphosphate buffer, pH 7.3, at +0.25 V vs. Ag/AgCl
Electrode KappM (mM) imax (�A/cm2) imax/K
appM
mGlyDH/C 3.9± 0.6* 1.3 ± 0.1* 0.33mGlyDH/Cp/C 6.5± 1.3 1.8± 0.2 0.28mGlyDH/GA/C 4.8± 0.6 2.8± 0.2 0.59mGlyDH/BSA/GA/C 13.0± 2.3 6.8± 0.6 0.52pGlyDH/C 12.1± 0.6 4.5± 0.1 0.37pGlyDH/Cp/C 17.3± 1.3 7.0± 0.2 0.41pGlyDH/GA/C 33.5± 4.7 13.7± 1.1 0.41
* ±S.E.
this enzyme could be classified as polyol dehydrogenase assome other PQQ-dependent dehydrogenases purified fromother sources[17,18].
3.3. Characterization of immobilised GlyDH
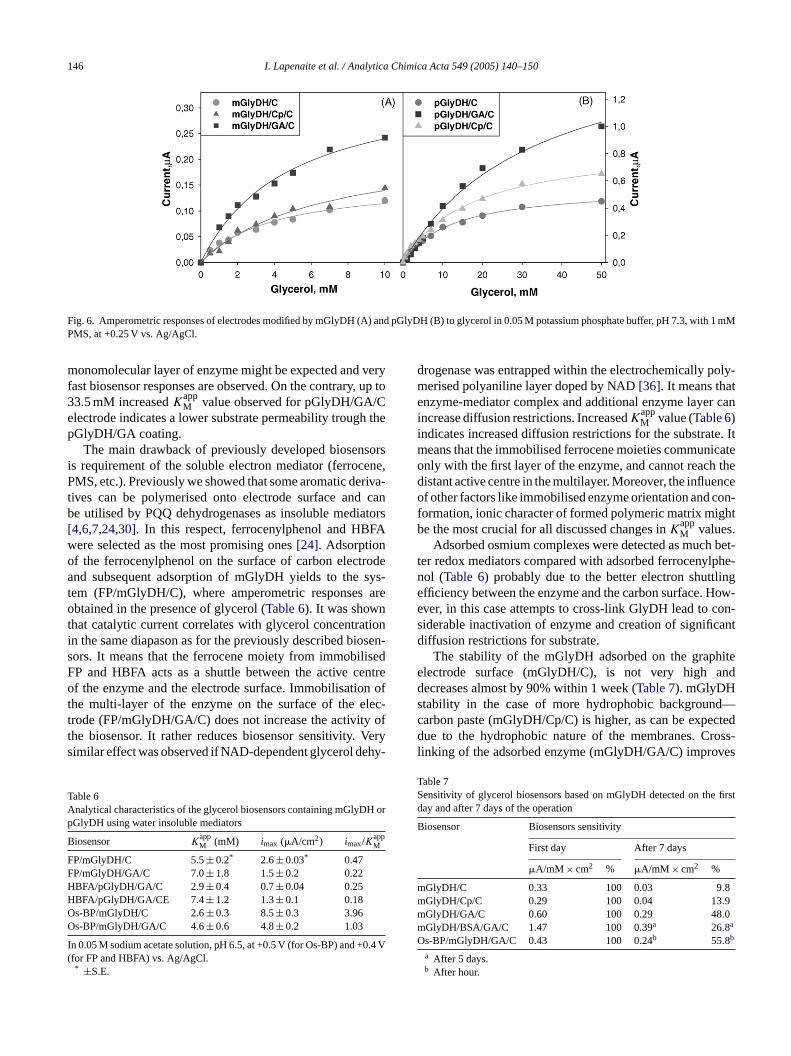
Both, the purified enzyme (pGlyDH) and a membranefraction of the cells (mGlyDH) adsorbs on the carbon elec-trode irreversibly (electrode GlyDH/C). The current of elec-trochemical regeneration of electron acceptor (PMS in ourcase) on the carbon electrode correlates with glycerol concen-tration in up to 3.9 and 12.1 mM of glycerol for the mGlyDHand pGlyDH, respectively (Fig. 6, Table 5). An electronacceptor (PMS) was essential in regeneration of the redoxcentre of enzyme. No current was observed in the absenceof this electron acceptor. The approximation of experimentaldata of electrode mGlyDH/C by Michaelis–Menten equa-tion leads to the apparent Michaelis constant (K
appM ) which is
3.9 mM (Table 5) and is lower than determined for the nativepGlyDH adsorbed on graphite electrode. It means that in thepGlyDH/C system greater diffusion restrictions are expected.It can be attributed to the natural structure of mGlyDH, sincehydrophobic cell membrane adsorbs on the surface of thehydrophobic carbon surface and creates natural surroundingof the immobilised enzyme.
stes rod)(T tt obics higherd ptionw ntreo DHa
lec-t yde( ithil il-i hisd h isc hen
ethanol 0 3.9 17.1thanol 0 12.1 8.6ulcitol – 9.1 14.3-Propanol 0 – –-Butanol 0 – –thylene glycol 0 – –actose 0 – –ructose 0 – –ucrose 0 – –
ctivities were measured in 50 mM potassium phosphate buffer, pHccording to standard assay method.* Not determined.
Adsorption of the mGlyDH on the carbon paurface (which is more hydrophobic than carbonmGlyDH/Cp/C) leads to almost the same activity (Fig. 6A,able 5). Up to 6.5 mM increased value ofK
appM indicates, tha
he active centre of the enzyme is closer to the hydrophurface of the carbon paste electrode and possessesiffusion restrictions to the substrate, or stronger adsorhich is able to change configuration of the active cef the enzyme. Similar results were obtained with pGlydsorbed on the carbon paste (Fig. 6B, Table 5).
Adsorption of the mGlyDH on the surface of carbon erode and cross-linking of the enzyme with glutaraldehmGlyDH/GA/C) lead to the multi-layered structure wncreased catalytic activity (Fig. 6A, Table 5). A considerablyow value of K
appM (4.8 mM) indicates a good permeab
ty of substrate through the formed gel-type coating. Toes not sufficiently increase the response time whicomparable with that observed in the first two cases, w
146 I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150
Fig. 6. Amperometric responses of electrodes modified by mGlyDH (A) and pGlyDH (B) to glycerol in 0.05 M potassium phosphate buffer, pH 7.3, with 1 mMPMS, at +0.25 V vs. Ag/AgCl.
monomolecular layer of enzyme might be expected and veryfast biosensor responses are observed. On the contrary, up to33.5 mM increasedKapp
M value observed for pGlyDH/GA/Celectrode indicates a lower substrate permeability trough thepGlyDH/GA coating.
The main drawback of previously developed biosensorsis requirement of the soluble electron mediator (ferrocene,PMS, etc.). Previously we showed that some aromatic deriva-tives can be polymerised onto electrode surface and canbe utilised by PQQ dehydrogenases as insoluble mediators[4,6,7,24,30]. In this respect, ferrocenylphenol and HBFAwere selected as the most promising ones[24]. Adsorptionof the ferrocenylphenol on the surface of carbon electrodeand subsequent adsorption of mGlyDH yields to the sys-tem (FP/mGlyDH/C), where amperometric responses areobtained in the presence of glycerol (Table 6). It was shownthat catalytic current correlates with glycerol concentrationin the same diapason as for the previously described biosen-sors. It means that the ferrocene moiety from immobilisedFP and HBFA acts as a shuttle between the active centreof the enzyme and the electrode surface. Immobilisation ofthe multi-layer of the enzyme on the surface of the elec-trode (FP/mGlyDH/GA/C) does not increase the activity ofthe biosensor. It rather reduces biosensor sensitivity. Verysimilar effect was observed if NAD-dependent glycerol dehy-
TA H orp
B
FFHHOO
I .4 V(
drogenase was entrapped within the electrochemically poly-merised polyaniline layer doped by NAD[36]. It means thatenzyme-mediator complex and additional enzyme layer canincrease diffusion restrictions. IncreasedK
appM value (Table 6)
indicates increased diffusion restrictions for the substrate. Itmeans that the immobilised ferrocene moieties communicateonly with the first layer of the enzyme, and cannot reach thedistant active centre in the multilayer. Moreover, the influenceof other factors like immobilised enzyme orientation and con-formation, ionic character of formed polymeric matrix mightbe the most crucial for all discussed changes inK
appM values.
Adsorbed osmium complexes were detected as much bet-ter redox mediators compared with adsorbed ferrocenylphe-nol (Table 6) probably due to the better electron shuttlingefficiency between the enzyme and the carbon surface. How-ever, in this case attempts to cross-link GlyDH lead to con-siderable inactivation of enzyme and creation of significantdiffusion restrictions for substrate.
The stability of the mGlyDH adsorbed on the graphiteelectrode surface (mGlyDH/C), is not very high anddecreases almost by 90% within 1 week (Table 7). mGlyDHstability in the case of more hydrophobic background—carbon paste (mGlyDH/Cp/C) is higher, as can be expecteddue to the hydrophobic nature of the membranes. Cross-linking of the adsorbed enzyme (mGlyDH/GA/C) improves
TS firstd
B
mmmmO
able 6nalytical characteristics of the glycerol biosensors containing mGlyDGlyDH using water insoluble mediators
iosensor KappM (mM) imax (�A/cm2) imax/K
appM
P/mGlyDH/C 5.5± 0.2* 2.6± 0.03* 0.47P/mGlyDH/GA/C 7.0± 1.8 1.5± 0.2 0.22BFA/pGlyDH/GA/C 2.9± 0.4 0.7± 0.04 0.25BFA/pGlyDH/GA/CE 7.4± 1.2 1.3± 0.1 0.18s-BP/mGlyDH/C 2.6± 0.3 8.5± 0.3 3.96s-BP/mGlyDH/GA/C 4.6± 0.6 4.8± 0.2 1.03
n 0.05 M sodium acetate solution, pH 6.5, at +0.5 V (for Os-BP) and +0for FP and HBFA) vs. Ag/AgCl.* ±S.E.
able 7ensitivity of glycerol biosensors based on mGlyDH detected on theay and after 7 days of the operation
iosensor Biosensors sensitivity
First day After 7 days
�A/mM × cm2 % �A/mM × cm2 %
GlyDH/C 0.33 100 0.03 9.8GlyDH/Cp/C 0.29 100 0.04 13.9GlyDH/GA/C 0.60 100 0.29 48.0GlyDH/BSA/GA/C 1.47 100 0.39a 26.8a
s-BP/mGlyDH/GA/C 0.43 100 0.24b 55.8b
a After 5 days.b After hour.
I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150 147
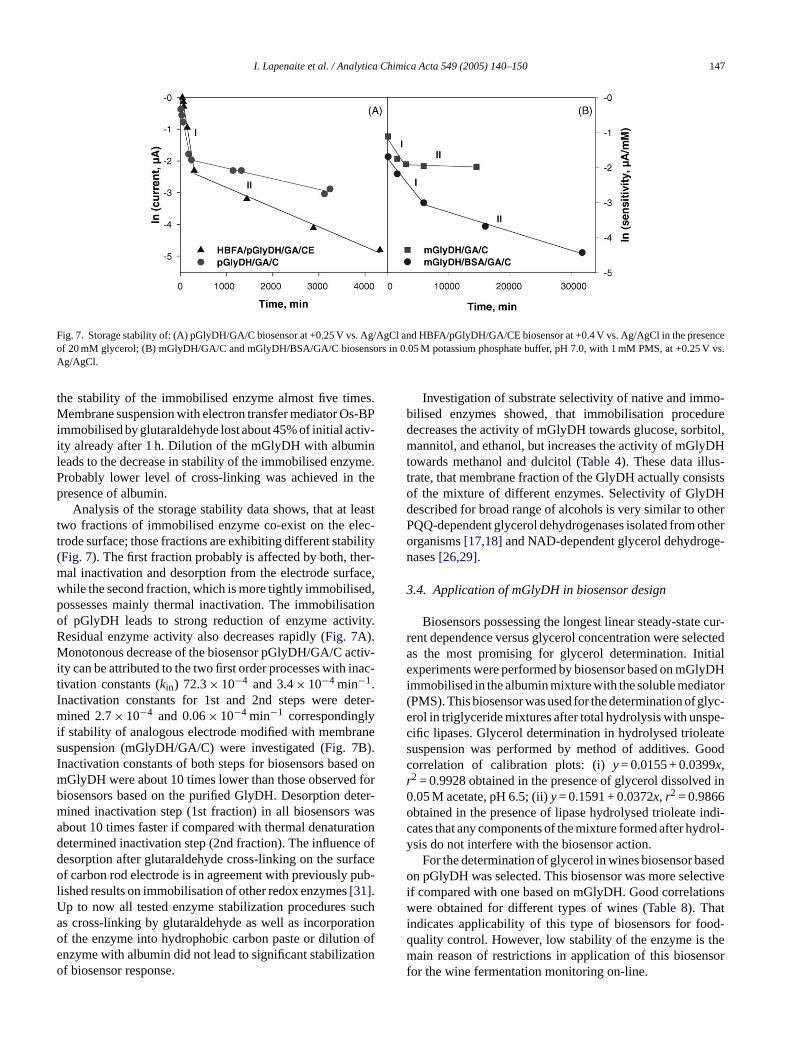
Fig. 7. Storage stability of: (A) pGlyDH/GA/C biosensor at +0.25 V vs. Ag/AgCl and HBFA/pGlyDH/GA/CE biosensor at +0.4 V vs. Ag/AgCl in the presenceof 20 mM glycerol; (B) mGlyDH/GA/C and mGlyDH/BSA/GA/C biosensors in 0.05 M potassium phosphate buffer, pH 7.0, with 1 mM PMS, at +0.25 V vs.Ag/AgCl.
the stability of the immobilised enzyme almost five times.Membrane suspension with electron transfer mediator Os-BPimmobilised by glutaraldehyde lost about 45% of initial activ-ity already after 1 h. Dilution of the mGlyDH with albuminleads to the decrease in stability of the immobilised enzyme.Probably lower level of cross-linking was achieved in thepresence of albumin.
Analysis of the storage stability data shows, that at leasttwo fractions of immobilised enzyme co-exist on the elec-trode surface; those fractions are exhibiting different stability(Fig. 7). The first fraction probably is affected by both, ther-mal inactivation and desorption from the electrode surface,while the second fraction, which is more tightly immobilised,possesses mainly thermal inactivation. The immobilisationof pGlyDH leads to strong reduction of enzyme activity.Residual enzyme activity also decreases rapidly (Fig. 7A).Monotonous decrease of the biosensor pGlyDH/GA/C activ-ity can be attributed to the two first order processes with inac-tivation constants (kin) 72.3× 10−4 and 3.4× 10−4 min−1.Inactivation constants for 1st and 2nd steps were deter-mined 2.7× 10−4 and 0.06× 10−4 min−1 correspondinglyif stability of analogous electrode modified with membranesuspension (mGlyDH/GA/C) were investigated (Fig. 7B).Inactivation constants of both steps for biosensors based onmGlyDH were about 10 times lower than those observed forbiosensors based on the purified GlyDH. Desorption deter-m wasa tiond e ofd rfaceo ub-lU sucha tiono n ofe tiono
Investigation of substrate selectivity of native and immo-bilised enzymes showed, that immobilisation proceduredecreases the activity of mGlyDH towards glucose, sorbitol,mannitol, and ethanol, but increases the activity of mGlyDHtowards methanol and dulcitol (Table 4). These data illus-trate, that membrane fraction of the GlyDH actually consistsof the mixture of different enzymes. Selectivity of GlyDHdescribed for broad range of alcohols is very similar to otherPQQ-dependent glycerol dehydrogenases isolated from otherorganisms[17,18]and NAD-dependent glycerol dehydroge-nases[26,29].
3.4. Application of mGlyDH in biosensor design
Biosensors possessing the longest linear steady-state cur-rent dependence versus glycerol concentration were selectedas the most promising for glycerol determination. Initialexperiments were performed by biosensor based on mGlyDHimmobilised in the albumin mixture with the soluble mediator(PMS). This biosensor was used for the determination of glyc-erol in triglyceride mixtures after total hydrolysis with unspe-cific lipases. Glycerol determination in hydrolysed trioleatesuspension was performed by method of additives. Goodcorrelation of calibration plots: (i)y= 0.0155 + 0.0399x,r2 = 0.9928 obtained in the presence of glycerol dissolved in0 2
o indi-c drol-y
sedo ctivei ionswi od-q them sorf
ined inactivation step (1st fraction) in all biosensorsbout 10 times faster if compared with thermal denaturaetermined inactivation step (2nd fraction). The influencesorption after glutaraldehyde cross-linking on the suf carbon rod electrode is in agreement with previously p
ished results on immobilisation of other redox enzymes[31].p to now all tested enzyme stabilization proceduress cross-linking by glutaraldehyde as well as incorporaf the enzyme into hydrophobic carbon paste or dilutionzyme with albumin did not lead to significant stabilizaf biosensor response.
.05 M acetate, pH 6.5; (ii)y= 0.1591 + 0.0372x, r = 0.9866btained in the presence of lipase hydrolysed trioleateates that any components of the mixture formed after hysis do not interfere with the biosensor action.
For the determination of glycerol in wines biosensor ban pGlyDH was selected. This biosensor was more sele
f compared with one based on mGlyDH. Good correlatere obtained for different types of wines (Table 8). That
ndicates applicability of this type of biosensors for fouality control. However, low stability of the enzyme isain reason of restrictions in application of this biosen
or the wine fermentation monitoring on-line.
148 I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150
Table 8Application of HBFA/pGlyDH/GA/CE glycerol biosensor for detection ofglycerol in wine samples
Wines Glycerol (mM)
Biosensor data(HBFA/pGlyDH/GA/CE)
Spectrophotometrictest kit data
Tokaji aszu (Hungary) 292± 12 240± 13Gluwein (Germany) 177± 8 180± 6Kadarka (Germany) 423± 16 405± 20Apple wine (Lithuania) 62± 3 50 ± 2
The presented concentration values are the means of three measurements.*±S.E.
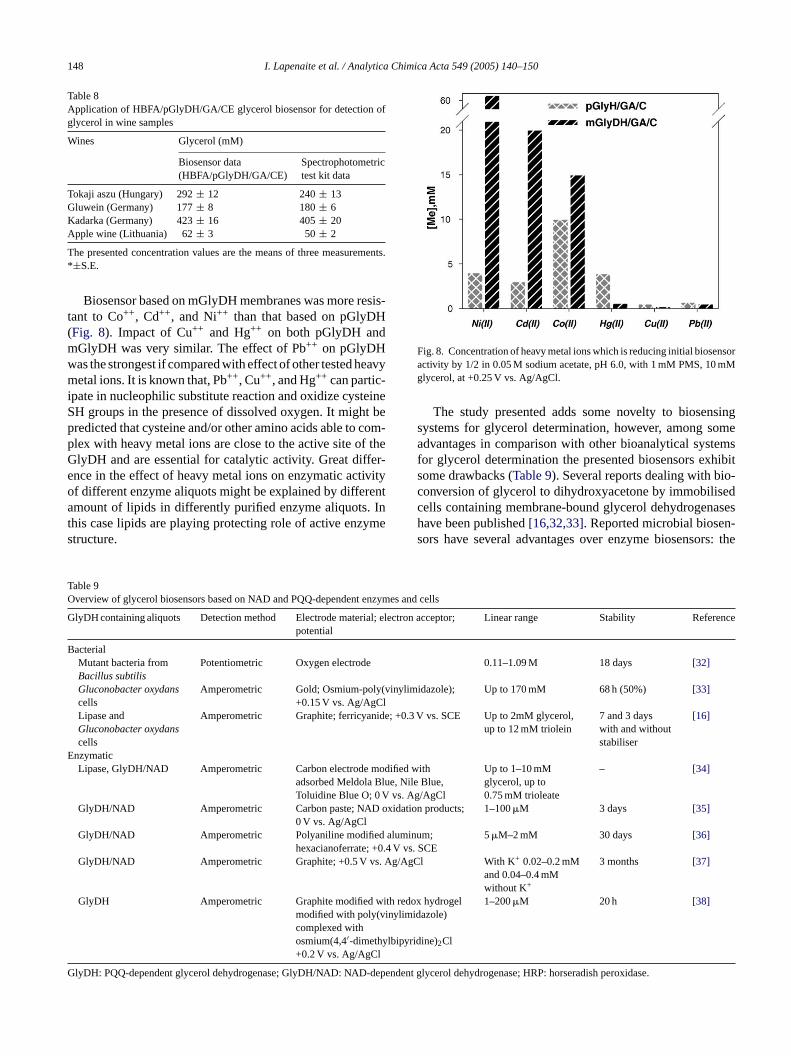
Biosensor based on mGlyDH membranes was more resis-tant to Co++, Cd++, and Ni++ than that based on pGlyDH(Fig. 8). Impact of Cu++ and Hg++ on both pGlyDH andmGlyDH was very similar. The effect of Pb++ on pGlyDHwas the strongest if compared with effect of other tested heavymetal ions. It is known that, Pb++, Cu++, and Hg++ can partic-ipate in nucleophilic substitute reaction and oxidize cysteineSH groups in the presence of dissolved oxygen. It might bepredicted that cysteine and/or other amino acids able to com-plex with heavy metal ions are close to the active site of theGlyDH and are essential for catalytic activity. Great differ-ence in the effect of heavy metal ions on enzymatic activityof different enzyme aliquots might be explained by differentamount of lipids in differently purified enzyme aliquots. Inthis case lipids are playing protecting role of active enzymestructure.
Fig. 8. Concentration of heavy metal ions which is reducing initial biosensoractivity by 1/2 in 0.05 M sodium acetate, pH 6.0, with 1 mM PMS, 10 mMglycerol, at +0.25 V vs. Ag/AgCl.
The study presented adds some novelty to biosensingsystems for glycerol determination, however, among someadvantages in comparison with other bioanalytical systemsfor glycerol determination the presented biosensors exhibitsome drawbacks (Table 9). Several reports dealing with bio-conversion of glycerol to dihydroxyacetone by immobilisedcells containing membrane-bound glycerol dehydrogenaseshave been published[16,32,33]. Reported microbial biosen-sors have several advantages over enzyme biosensors: the
Table 9Overview of glycerol biosensors based on NAD and PQQ-dependent enzymes and cells
GlyDH containing aliquots Detection method Electrode material; electron acceptor;potential
Linear range Stability Reference
BacterialMutant bacteria fromBacillus subtilis
Potentiometric Oxygen electrode 0.11–1.09 M 18 days [32]
Gluconobacter oxydans Amperometric Gold; Osmium-poly(vinylimidazole);l
Up to 170 mM 68 h (50%) [33]
ide; +0.3 V vs. SCE Up to 2mM glycerol,up to 12 mM triolein
7 and 3 dayswith and withoutstabiliser
[16]
Emodified withlue, Nile Blue,
V vs. Ag/AgCl
Up to 1–10 mMglycerol, up to0.75 mM trioleate
– [34]
oxidation products; 1–100�M 3 days [35]
d alum0.4 V v. Ag/Ag
with redinylimid
lbipyri
G
cells +0.15 V vs. Ag/AgCLipase andGluconobacter oxydanscells
Amperometric Graphite; ferricyan
nzymaticLipase, GlyDH/NAD Amperometric Carbon electrode
adsorbed Meldola BToluidine Blue O; 0
GlyDH/NAD Amperometric Carbon paste; NAD0 V vs. Ag/AgCl
GlyDH/NAD Amperometric Polyaniline modifiehexacianoferrate; +
GlyDH/NAD Amperometric Graphite; +0.5 V vs
GlyDH Amperometric Graphite modifiedmodified with poly(vcomplexed withosmium(4,4′-dimethy+0.2 V vs. Ag/AgCl
lyDH: PQQ-dependent glycerol dehydrogenase; GlyDH/NAD: NAD-depen
inum;s. SCE
5�M–2 mM 30 days [36]
Cl With K+ 0.02–0.2 mMand 0.04–0.4 mMwithout K+
3 months [37]
ox hydrogelazole)
dine)2Cl
1–200�M 20 h [38]
dent glycerol dehydrogenase; HRP: horseradish peroxidase.

I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150 149
enzyme does not need to be isolated, enzymes are usuallymore stable in their natural environment in the cell, andessential coenzymes and activators are already present inthe system. However, such biosensors are extremely unselec-tive because of the presence of a number of other enzymesand biologically active materials. It is the main reason whybiosensors based on purified enzymes are under higher inter-est. Among glycerol biosensors reported by other authorsmainly NAD-dependent glycerol dehydrogenases[34–37]or NAD-dependent glycerol dehydrogenases combined withlipases[34] were applied in combination with amperomet-ric detection (Table 9). However, NAD-dependent enzyme-based sensors lack of stability if cofactor NAD is not addedbefore each measurement. This drawback might be avoidedby application of PQQ-dependent enzymes[38]. In this studywe have used more homogeneous highly purified enzyme andmore simple biosensor design in comparison with that usedin glycerol biosensor reported previously[38]. The stabilityof the presented sensor was similar to the stability of pre-viously reported PQQ-dependent glycerol dehydrogenase-based biosensors but lower if compared with NAD-dependentdehydrogenases-based biosensors[35–37]. However, dur-ing operation of some NAD-dependent dehydrogenase-basedbiosensors[35,37], cofactor NAD should be added beforeeach measurement, during present study addition of cofac-tor (PQQ) before/during the measurements were avoided.H butm rox-y ali-b erolb glyc-e dwd nd/orc yme-b andm oseb utivem
3
oundP t somp en-s ifc ses ngleu ppli-c y ofG raw-b , lows zy-m htb icalf rol
dehydrogenase was achieved by incorporation of this enzymewithin conducting polymer polyaniline[36], moreover, ourpreliminary study showed that encapsulation within polypyr-role might be even more promising for application of someredox enzymes in design of amperometric biosensors[44].
Acknowledgements
This work was partially supported by the Lithuanian StateScience and Studies Foundation (programme C-02/2003BIOHEMAS, project T-71/04 and COST action P12).
References
[1] T. Ikeda, Direct redox communication between enzymes and elec-trodes, in: Frontiers in Biosensors I: Fundamental Aspects, Basel,1997, p. 243.
[2] J. Razumiene, M. Niculesku, A. Ramanavicius, V. Laurinavicius, E.Csoregi, Electroanalysis 14 (2002) 43.
[3] A. Ramanavicius, A. Kausaite, A. Ramanaviciene, Sens. ActuatorsB, in press.
[4] A. Ramanavicius, K. Habermuller, E. Csoregi, V. Laurinavicius, W.Schuhmann, Anal. Chem. 71 (1999) 3581.
[5] V. Laurinavicius, J. Razumiene, A. Ramanavicius, A.D. Ryabov,Biosens. Bioelectron. 20 (2004) 1217.
[6] K. Habermuller, A. Ramanavicius, V. Laurinavicius, W. Schuhmann,
s.
iol.
[ of
[ ina-3.
[ M.0.
[ .-
[ ch-
[ .
[ 1.[ iol.
[ N.ppl.
[ 2
[ zu-.
[bov,
[ .
[ ,5.
owever, the reported biosensor still lacks of stabilityight be applied in single use biosensors for polyhyd
lic alcohol determination or in biosensors with precration step. If compared with spectrophotometric glyciosensors that were based mainly on NAD-dependentrol dehydrogenases[39–41]and glycerol kinase combineith protein kinase, lactate dehydrogenase[42] in this studyescribed sensors might be applied in non-transparent aoloured samples (e.g. wines, juices, etc.). Single enzased biosensors are more simple during preparationore predictable during application if compared with thased on application of several enzymes acting in consecode.
.5. Conclusions and future developments
The results presented illustrate that membrane bQQ-dependent glycerol dehydrogenases have at leasotential for application in glycerol and triglyceride biosors. However, highly purified GlyDH has low stabilityompared with a partially purified GlyDH. But in this cauch GlyDH modified electrodes might be applied as sise test strips for amperometric biosensors. However, aation of a partially purified enzyme reduces selectivitlyDH-based biosensors what of course is a significant dack of such bioanalytical systems. On the other handelectivity of GlyDH might be exploited as advantage in enatic biofuel cells[43] where maximal current yield mige achieved by conversion of maximal number of biolog
uels. Significant stabilization of NAD-dependent glyce
e
Electroanalysis 12 (2000) 1383.[7] A. Malinauskas, J. Kuzmarskyte, R. Meskys, A. Ramanavicius, Sen
Actuators B 100 (2004) 387.[8] K. Matsushita, H. Toyama, M. Yamada, O. Adachi, App. Microb
Biotechnol. 58 (2002) 13.[9] J.A. Duine, J. Biosci. Bioeng. 88 (1999) 231.10] V.L. Davidson, in: V. Davidson (Ed.), Principles and Applications
Quinoproteins, Marcel Dekker, NY, 1993, p. 65.11] O. Adachi, D. Moonmangmee, H. Toyama, M. Yamada, E. Sh
gawa, K. Matsushita, Appl. Microbiol. Biotechnol. 60 (2003) 6412] O. Adachi, D. Moonmangmee, E. Shinagawa, H. Toyama,
Yamada, K. Matsushita, Biochim. Biophys. Acta 1647 (2003) 113] V. Laurinavicius, J. Razumiene, B. Kurtinaitiene, I. Lapenaite, I
Bachmatova, L. Marcinkeviciene, R. Meskys, A. Ramanavicius, Bioelectrochemistry 55 (2002) 29.
14] J. Tkac, M. Navratil, E. Sturdik, P. Gemeiner, Enzyme Microb. Tenol. 28 (2001) 383.
15] M. Navratil, J. Tkac, J. Svitel, B. Danielsson, E.Sturdik, ProcessBiochem. 36 (2001) 1045.
16] J. Tkac, J. Svitel, R. Novak, E. Sturdik, Anal. Lett. 33 (2000) 24417] M. Ameyama, E. Shinagawa, K. Matsushita, O. Adachi, Agric. B
Chem. 49 (1985) 1001.18] K. Matsushita, Y. Fujii, Y. Ano, H. Toyama, M. Shinjoh,
Tomiyama, T. Miyazaki, T. Sugisawa, T. Hoshino, O. Adachi, AEnviron. Microbiol. 69 (2003) 1959.
19] I. Bachmatova, L. Marcinkeviciene, G. Brazenas, Biologija 1–(1995) 57.
20] A.D. Ryabov, V.S. Kurova, V.N. Goral, M.D. Reshetova, J. Ramiene, R. Simkus, V. Laurinavicius, Chem. Mater. 11 (1999) 600
21] J. Razumiene, A. Vilkanauskyte, V. Gureviciene, V. Laurinavicius,N.V. Roznyatovskaya, Y.V. Ageeva, M.D. Reshetova, A.D. RyaJ. Organomet. Chem. 668 (2003) 83.
22] L. Marcinkeviciene, I. Bachmatova, R. Semenaite, I. Lapenaite, BKurtinaitiene, V. Laurinavicius, R. Meskys, Biologija 2 (2000) 39.
23] I. Lapenaite, B. Kurtinaitiene, L. Marcinkeviciene, I. BachmatovaV. Laurinavicius, A. Ramanavicius, Chem. Papers 55 (2001) 34

150 I. Lapenaite et al. / Analytica Chimica Acta 549 (2005) 140–150
[24] J. Razumiene, V. Gureviciene, V. Laurinavicius, J.V. Grazulevicius,Sens. Actuators B 78 (2001) 243.
[25] B.V. Mchedlishvily, G.N. Flerov, J. Soviet Chem. Union Mendeleev32 (1987) 641 (in Russian).
[26] M. Stibor, M. Potocky, A. Pickova, P. Karasova, N.J. Russell, B.Kralova, Enzyme Microb. Technol. 32 (2002) 532.
[27] A. Ramanavicius, R. Meskys, V. Laurinavicius, Biologija 1–2 (1995)53.
[28] A. Sato, K. Takagi, K. Kano, N. Kato, J.A. Duine, T. Ikeda, Biochem.J. 357 (2001) 893.
[29] S.N. Ruzheinikov, J. Burke, S. Sedelnikova, P.J. Baker, R. Taylor,P.A. Bullough, N.M. Muir, M.G. Gore, D.W. Rice, Structure 9 (2001)789.
[30] A. Ramanavicius, B. Kurtinaitiene, J. Razumiene, V. Laurinavicius,R. Meskys, R. Rudomanskis, I. Bachmatova, L. Marcinkeviciene,Biologija (Suppl. 1) (1998) 15.
[31] A. Ramanavicius, A. Bimbiris, V. Laurinavicius, R. Meskys, R.Rudomanskis, Biologija 1 (1997) 77.
[32] Z.H. Liu, M.L. Wen, Y. Yao, N.H. Shi, S.Q. Liu, M. Collect. Czech.Chem. Commun. 64 (1999) 1412.
[33] I. Vostiar, E.E. Ferapontova, L. Gorton, Electrochem. Commun. 6(2004) 621.
[34] V. Laurinavicius, B. Kurtinaitiene, V. Gureviciene, L. Boguslavsky,L. Geng, T. Skotheim, Anal. Chim. Acta 330 (1996)159.
[35] M.I. Alvarez-Gonzalez, S.B. Saidman, M.J. Lobo-Castanon, A.J.Miranda-Ordieres, P. Tunon-Blanco, Anal. Chem. 72 (2000)520.
[36] A. Eftekhari, Sens. Actuators B 80 (2001) 283.[37] M.I. Prodromidis, C.D. Stalikas, S.M. Tzouwara-Karayanni, M.I.
Karayannis, Talanta 43 (1996) 27.[38] M. Niculescu, R. Mieliauskiene, V. Laurinavicius, E. Csoregi, Food
Chem. 82 (2003) 481.[39] M. Masoom, P.J. Worsfold, Anal. Chim. Acta 188 (1986) 281.[40] A.O.S.S. Rangel, I.V. Toth, Anal. Chim. Acta 416 (2000) 205.[41] E.N. Fernandes, M.N. Campos Moura, J.L.F. Costa Lima, B.F. Reis,
Microchem. J. 77 (2004) 107.[42] R. Li, B. Keymeulen, E. Gerlo, Clin. Chem. Lab. Med. 39 (2001)
20.[43] A. Ramanavicius, A. Kausaite, A. Ramanaviciene, Biosens. Bioelec-
tron. 20 (2005) 1962.[44] A. Ramanavicius, A. Kausaite, A. Ramanaviciene, Polypyrrole-
coated glucose oxidase nanoparticles for biosensor design, Sens.Actuators B, in press.