2-4 ich overview
TRANSCRIPT

Satish Mallya January 20-22, 2010
1 |
2-4 ICH Quality Guidances:an overview
PQP Assessment Training
January 18-21, 2012
Satish Mallya
January 18-21, 2012

Satish Mallya January 20-22, 2010
2 |
ICH Topics Stability - Q1A – Q1F Analytical Validation – Q2 Impurities – Q3A - Q3C (Q3D – concept paper) Pharmacopoeias – Q4A - Q4B (and annexes) Quality of Biotechnological Products – Q5A – Q5E Specifications – Q6A – Q6B Good Manufacturing Practice – Q7 Pharmaceutical Development – Q8 Quality Risk Management - Q9 Pharmaceutical Quality System – Q10 Development and Manufacturing of Drug Substances – Q11
January 18-21, 2012

Satish Mallya January 20-22, 2010
3 |
Focus
Stability - Q1A, B, C, D, E & F
Validation of Analytical Methods – Q2(R1)
Impurities – Q3A, B & C
Specifications – Q6A (Chemical Substances) & Q6B (Biotechnology/Biological Products) & Q6B (Biotechnology/Biological Products)
January 18-21, 2012

Satish Mallya January 20-22, 2010
4 |
Stability Q1A(R2) Stability Testing of New Drug Substances
and Products Q1B Photostability Testing of New Drug Substances
and Products Q1C Stability Testing for New Dosage Forms Q1D Bracketing and Matrixing Designs for Stability
Testing of New Drug Substances and Products Q1E Evaluation of Stability Data Q1F Stability Data Package for Registration
Applications in Climatic Zones III & IV (withdrawn – June 2006)
January 18-21, 2012
Satish Mallya January 20-22, 2010
5 |
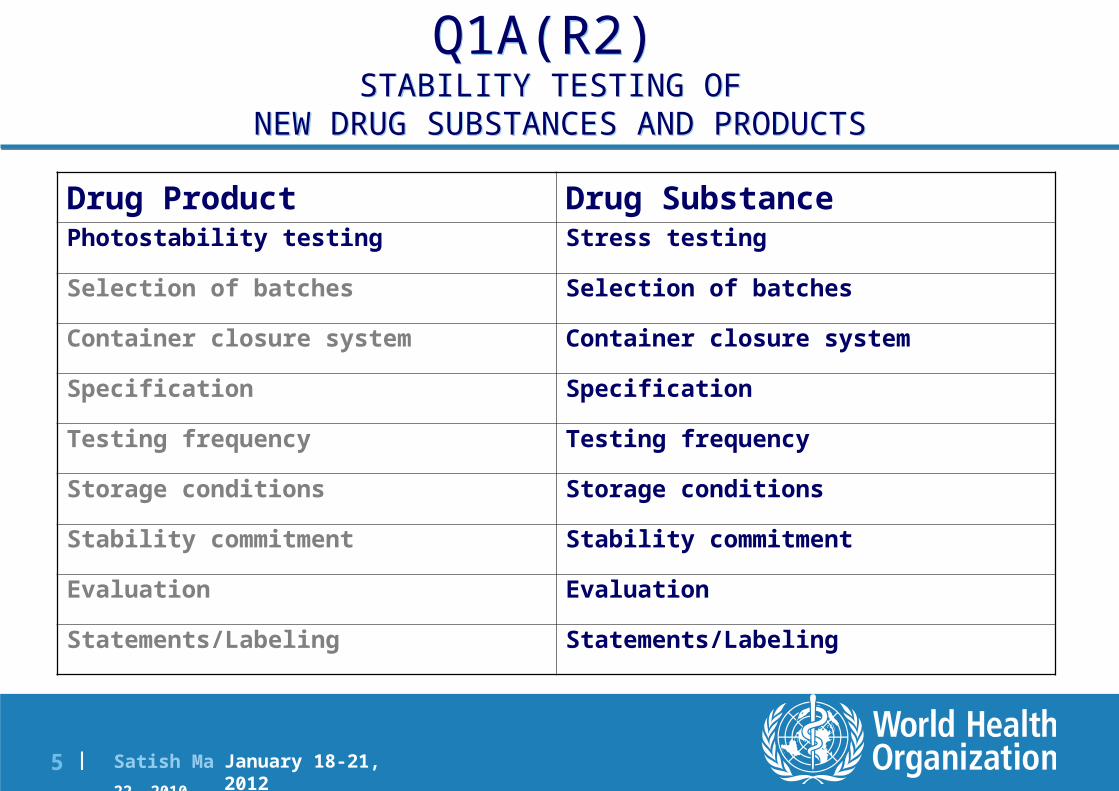
Q1A(R2) STABILITY TESTING OF
NEW DRUG SUBSTANCES AND PRODUCTS
Drug SubstanceDrug ProductStress testingPhotostability testing
Selection of batchesSelection of batches
Container closure systemContainer closure system
SpecificationSpecification
Testing frequencyTesting frequency
Storage conditionsStorage conditions
Stability commitmentStability commitment
EvaluationEvaluation
Statements/LabelingStatements/Labeling
January 18-21, 2012

Satish Mallya January 20-22, 2010
6 |
Stress Testing/Photostability
Drug Substance Drug Product
One primary batch
Effect of temperatures (in 10°C increments (e.g., 50°C, 60°C, etc.) above that for accelerated testing)
Effect of humidity (e.g., > 75%RH)
One primary batch
As in ICH Q1B:
January 18-21, 2012

Satish Mallya January 20-22, 2010
7 |
Selection of Batches
Drug Substance Drug Product
Data on at least three primary batches of minimum pilot scale manufactured by the same synthetic route as used for production batches.
Data on at least three primary batches of the drug product – two pilot and third one can be smaller - same formulation and packaged in the same container closure system as proposed for marketing.
The manufacturing process used for primary batches should simulate that to be applied to production batches
Where possible, use different batches of the drug substance.
Should be performed on each individual strength and container size of the drug product unless bracketing or matrixing is applied.
January 18-21, 2012

Satish Mallya January 20-22, 2010
8 |
Container Closure System
Drug SubstanceDrug Product
Studies to be conducted on the API packaged in a container closure system that is identical to or simulates the proposed commercial packaging
Studies to be carried out in container closure system identical to commercial packaging; studies carried out in other packaging materials can be used as supporting information
January 18-21, 2012

Satish Mallya January 20-22, 2010
9 |
Specification
Drug Substance
Drug Product
Studies to include attributes susceptible to change during storage and which can influence quality, safety and efficacy: - Physical- Chemical- Microbiological
Studies to include attributes susceptible to change during storage and which can influence quality, safety and efficacy:- Physical- chemical,- microbiological,- preservative content- functionality tests (e.g.
with delivery systems)
Validated analytical methods to be employed
Validated analytical methods to be employed
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
10
|
Testing Frequency Drug Substance
Drug Product
For API with proposed re-test period/shelf-life of at least 12 months: Every 3 months over first year, every 6 months over next 12 months and annually thereafter.
For FPP with proposed re-test period/shelf-life of at least 12 months: Every 3 months over first year, every 6 months over next 12 months and annually thereafter.
Accelerated condition: Minimum of 3 time points, including initial and final time points (e.g. 0, 3 & 6 months)
Accelerated condition: Minimum of 3 time points, including initial and final time points (e.g. 0, 3 & 6 months)
Intermediate condition (due to significant change under accelerated condition): study design should include 4 time points (e.g. 0, 6, 9 and 12 months
Intermediate condition (due to significant change under accelerated condition): study design should include 4 time points (e.g. 0, 6, 9 and 12 months)
Matrixing or Bracketing may be applied
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
11
|
Storage Conditions
General Case Drug Substance
Drug Product
StudyStorage Conditions
Minimum period covered by data at submission
StudyStorage Conditions
Minimum period covered by data at submission
Long term25⁰C+2⁰C/ 60%+5%RH or30⁰C+2⁰C /65% +5%RH
12 monthsLong term25⁰C+2⁰C /60%+5%RH or30⁰C+2⁰C /65% +5%RH
12 months
Intermediate
30⁰C+2⁰C /65% +5%RH
6 monthsIntermediate
30⁰C+2⁰C /65% +5%RH
6 months
Accelerated
40⁰C+2⁰C /75% +5%RH
6 monthsAccelerated
40⁰C+2⁰C /75% +5%RH
6 months
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
12
|
Storage Conditions
Storage in refrigerator Drug Substance
Drug Product
StudyStorage Conditions
Minimum period covered by data at submission
StudyStorage Conditions
Minimum period covered by data at submission
Long term5⁰C + 3⁰C12monthsLong term5⁰C + 3⁰C12months
Accelerated
25⁰C + 2⁰C / 60% + 5% RH
6 months Accelerated
25⁰C + 2⁰C / 60% + 5% RH
6 months
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
13
|
Storage Conditions
Storage in freezer
Storage below - 20⁰C : Case by case basis
Drug Substance
Drug Product
StudyStorage Conditions
Minimum period covered by data at submission
StudyStorage Conditions
Minimum period covered by data at submission
Long term
- 20⁰C + 5⁰C
12monthsLong term
- 20⁰C + 5⁰C
12months
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
14
|
Storage Conditions – Drug Product
Semi-permeable containers :StudyStorage
ConditionsMinimum period covered by data at submission
Long term25⁰C+2⁰C/40%+5% RH or30⁰C+2⁰C/35%+5% RH
12 months
Intermediate30⁰C+2⁰C/65%+5% RH6 months
Accelerated40⁰C+2⁰C/NMT 25% RH6 months
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
15
|
Significant ChangeDrug SubstanceDrug Product Defined as failure to meet specifications
->5% change in assay from the initial results
-Any degradation product exceeding its acceptance criterion
-Failure to meet acceptance criteria for appearance, physical attributes and functionality tests
-Failure to meet acceptance criteria for pH
-Failure to meet acceptance criteria for dissolution of 12 dosage unitsJanuary 18-21,
2012

Satish Mallya January 20-22, 2010
16
|
Evaluation
Drug Substance
Drug Product
Statistical analysis not necessary if data exhibits little or no degradation and variability
Statistical analysis not necessary if data exhibits little or no degradation and variability
Limited extrapolation of real time data permitted with justification
Limited extrapolation of real time data permitted with justification
June 2010January 18-21, 2012

Satish Mallya January 20-22, 2010
17
|
Q1B PHOTOSTABILITY TESTING OF
NEW DRUG SUBSTANCES AND PRODUCTS Provides 2 options for sources of light:
– artificial daylight fluorescent lamp combining visible and ultraviolet (UV) outputs, xenon, or metal halide lamp
– sample should be exposed to both the cool white fluorescent and near ultraviolet lamp
Test on API first – if not photosensitive then no further testing is required
If API is photosensitive then testing to be continued on (as appropriate): – Tests on the exposed drug product outside of the immediate
pack – Tests on the drug product in the immediate pack – Tests on the drug product in the marketing pack
Where appropriate, impact of light during manufacturing
January 18-21, 2012

Satish Mallya January 20-22, 2010
18
|
Q1C Annex to Q1A (R2)
Additional guidance on line extensions
Reduced requirements at time of filing: 6 months accelerated and 6 months long term
January 18-21, 2012
Satish Mallya January 20-22, 2010
19
|
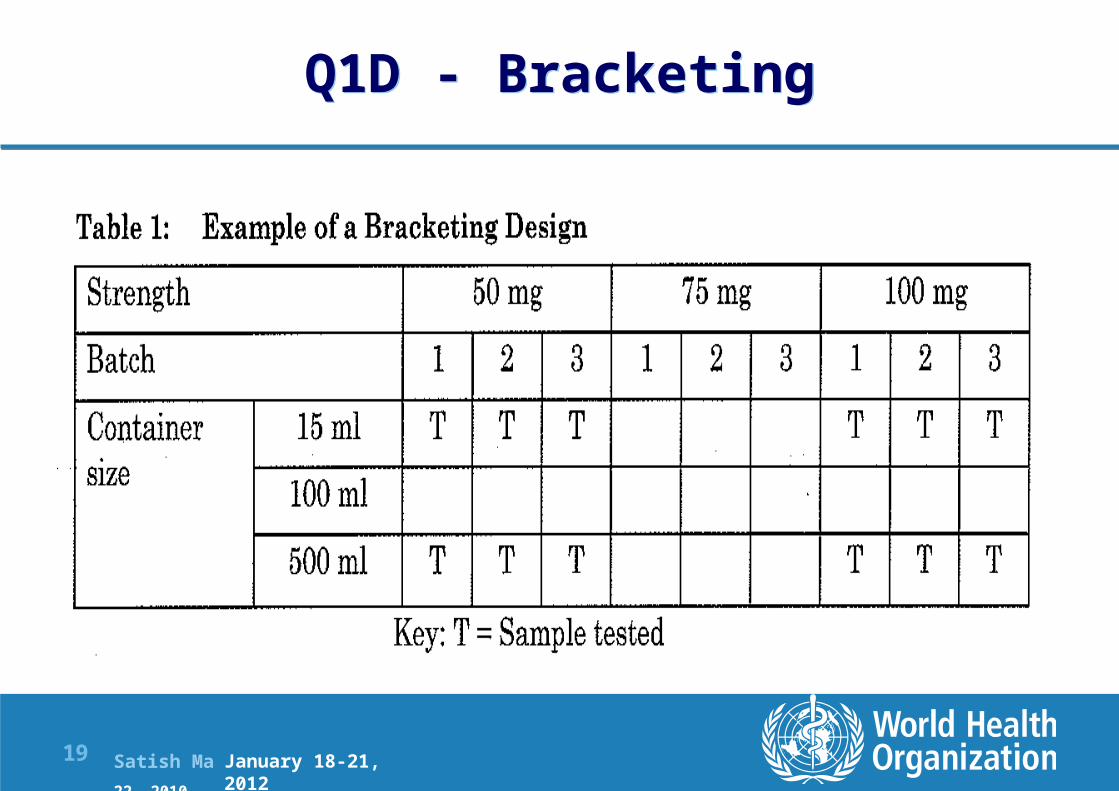
Q1D - Bracketing
January 18-21, 2012

Satish Mallya January 20-22, 2010
20
|
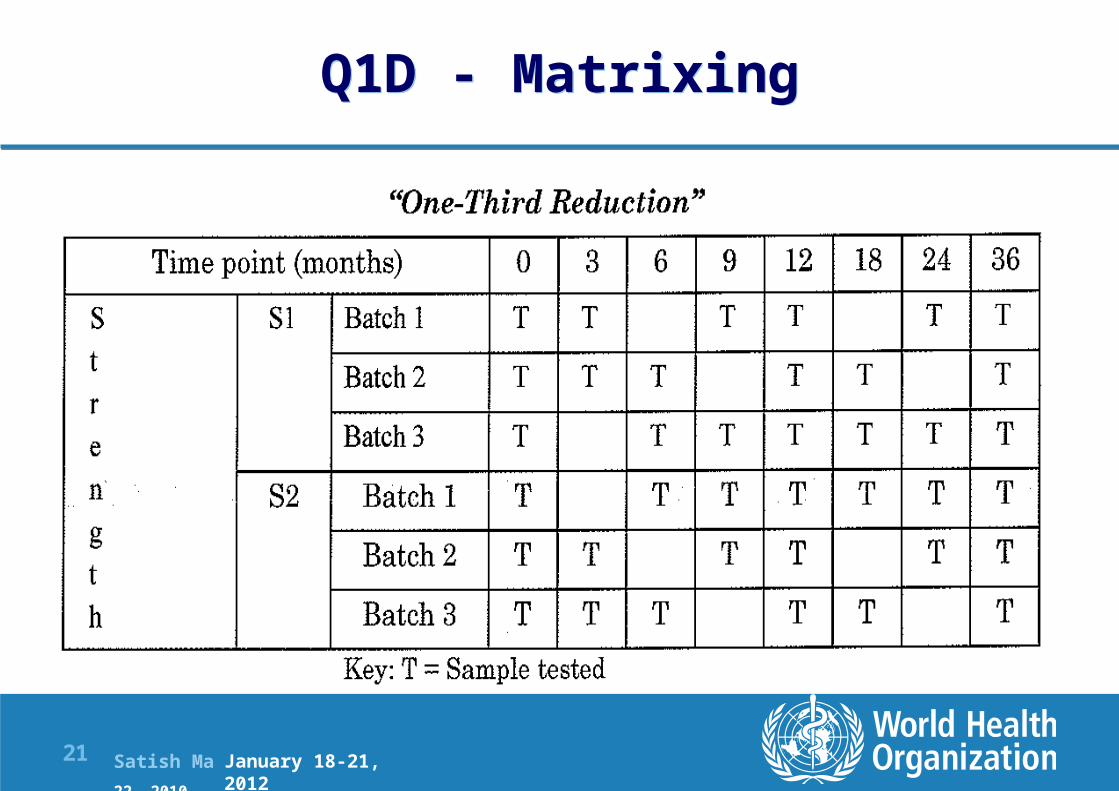
Q1D - Matrixing
January 18-21, 2012
Satish Mallya January 20-22, 2010
21
|
Q1D - Matrixing
January 18-21, 2012

Satish Mallya January 20-22, 2010
22
|
Q1E
EVALUATION OF STABILITY DATA
Provides recommendations for: (at RT, Refrigerated and Freezer storages)– treating stability data – Extending re-test period or shelf-life beyond period covered by long-term data
– Statistical approaches to analysis of stability data
Progression: – Start with data under accelerated condition – Then assess data under intermediate condition, if appropriate
– Finally evaluate trends and variability of the long-term data
January 18-21, 2012

Satish Mallya January 20-22, 2010
23
|
Outcomes
When there is no significant change under accelerated conditions (RT)Long-term and accelerated data showing little or no change over time and little or no variability
Retest period or shelf life can be up to twice, but NMT 12 months beyond the period covered by long-term data
Long-term or accelerated data showing change over time and/or variability
Data not amenable to statistical analysis, but relevant supporting data provided: Retest period or shelf life can be up to 1.5 times, but NMT 6 months beyond the period covered by long-term data
If a statistical analysis is performed: Retest period or shelf life of up to twice, but not more than 12 months beyond the period covered by long-term data
January 18-21, 2012

Satish Mallya January 20-22, 2010
24
|
Outcomes
When there is significant change under accelerated conditions (RT) but no significant change at intermediate condition:
Data not amenable to statistical analysis: Retest period or shelf life can be up to 3 months beyond the period covered by long-term data if backed by relevant documentation
If statistical analysis is performed: Retest period or shelf life can be up to 1.5 times, but NMT 6 months beyond the period covered by long-term data when backed by statistical analysis and relevant supporting data
January 18-21, 2012

Satish Mallya January 20-22, 2010
25
|
Q2(R1) VALIDATION OF ANALYTICAL PROCEDURES
Defines validation characteristics:– Accuracy – Precision
• Repeatability • Intermediate Precision
– Specificity – Detection Limit – Quantitation Limit – Linearity – Range
Robustness to be considered at appropriate stage of development of the analytical method
System suitability test parameters to be established for a particular procedure depending on the type of procedure being validated - Pharmacopoeias to be consulted for additional information
January 18-21, 2012

Satish Mallya January 20-22, 2010
26
|
VALIDATION CHARACTERISTICS
Validation characteristics
ID Impurities
Quant. limit
Assay
Accuracy
Precision
Repeatibility
Int.Precision
Specificity
LOD
LOQ
Linearity
Range
-
-
-
+
-
-
-
-
+ -
+ -
+ -
+ +
- +
+ -
+ -
+ -
+
+
+
+
-
-
+
+
January 18-21, 2012

Satish Mallya January 20-22, 2010
27
|
Q3 Impurities
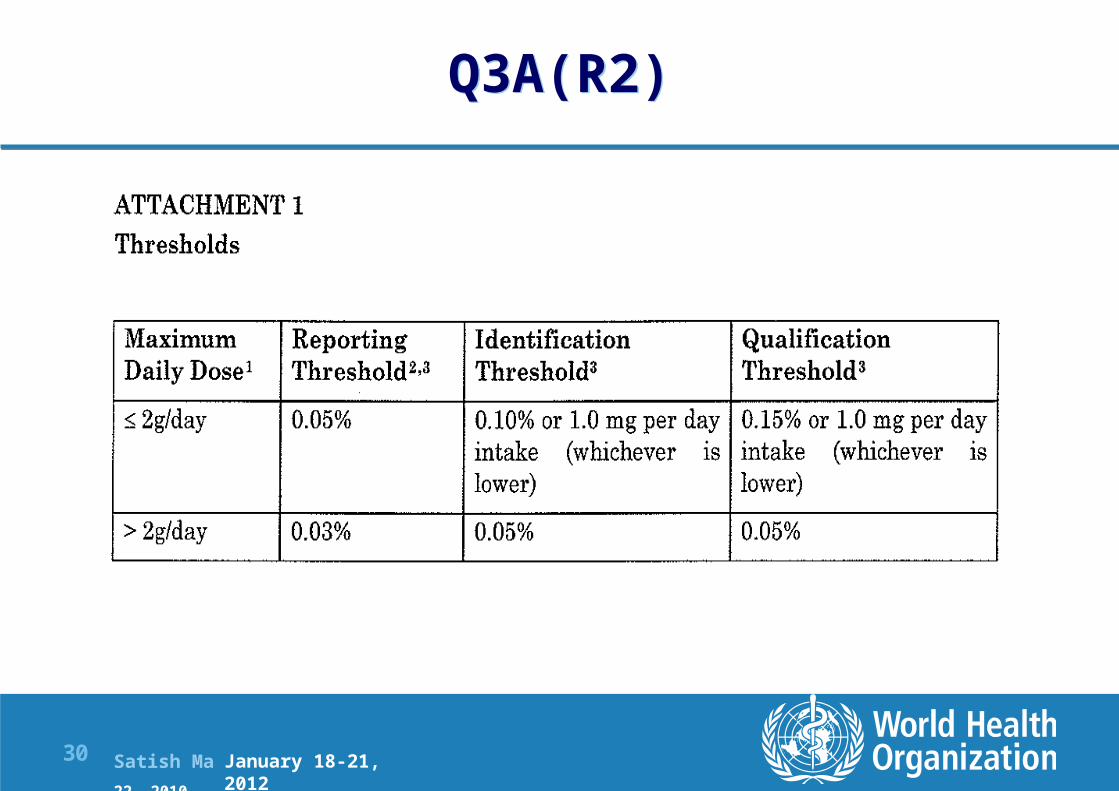
Impurities in New Drug Substances Q3A(R2): Defines thresholds for reporting, identification and qualification of impurities in DS
Impurities in New Drug Products Q3B(R2): Defines thresholds for reporting, identification and qualification of impurities in DP
Guideline for Residual Solvents Q3C (R5): Classifies residual solvents by risk assessment:– Class 1 solvents: solvents to be avoided– Class 2 solvents: solvents to be limited– Class 3 solvents: solvents with low toxic potential
Guideline for Metal Impurities Q3D (Concept paper – July 2009)
January 18-21, 2012

Satish Mallya January 20-22, 2010
28
|
Q3A(R2) CLASSIFICATION OF IMPURITIES
Organic Impurities– Starting materials– By-products– Intermediates– Degradation products– Reagents, ligands, catalysts
Inorganic Impurities– Reagents, ligands, catalysts– Heavy metals or other residual metals– Inorganic salts– Other materials (e.g., filter aids, charcoal)
Residual Solvents
January 18-21, 2012

Satish Mallya January 20-22, 2010
29
|
Q3A(R2) Definitions
Qualification: The process of acquiring and evaluating data that establishes the biological safety of an individual impurity or a given impurity profile at the level(s) specified.
Reporting Threshold: A limit above (>) which an impurity should be reported.
Specified Impurity: An impurity that is individually listed and limited with a specific acceptance criterion in the new drug substance specification. A specified impurity can be either identified or unidentified.
Unidentified Impurity: An impurity for which a structural characterisation has not been achieved and that is defined solely by qualitative analytical properties (e.g., chromatographic retention time)
Unspecified impurity: An impurity that is limited by a general acceptance criterion, but not individually listed with its own specific acceptance criterion, in the new drug substance specification
January 18-21, 2012
Satish Mallya January 20-22, 2010
30
|
Q3A(R2)
January 18-21, 2012
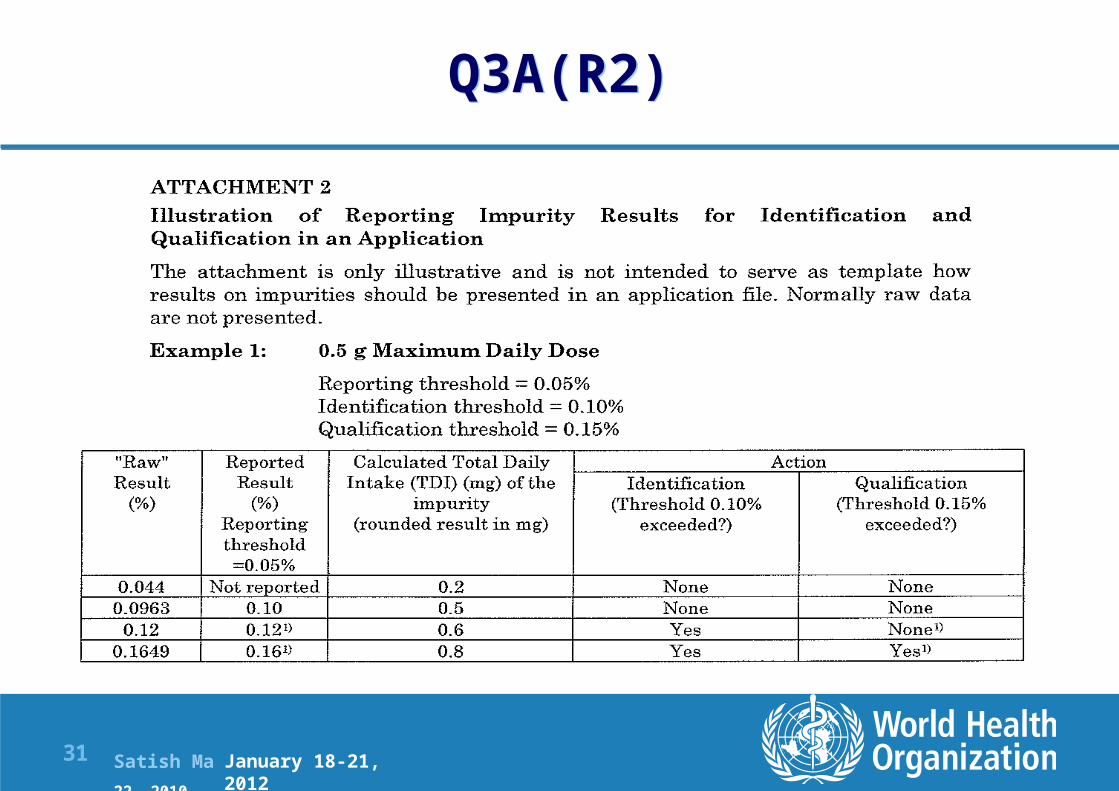
Satish Mallya January 20-22, 2010
31
|
Q3A(R2)
January 18-21, 2012

Satish Mallya January 20-22, 2010
32
|
Q3B(R2)
January 18-21, 2012
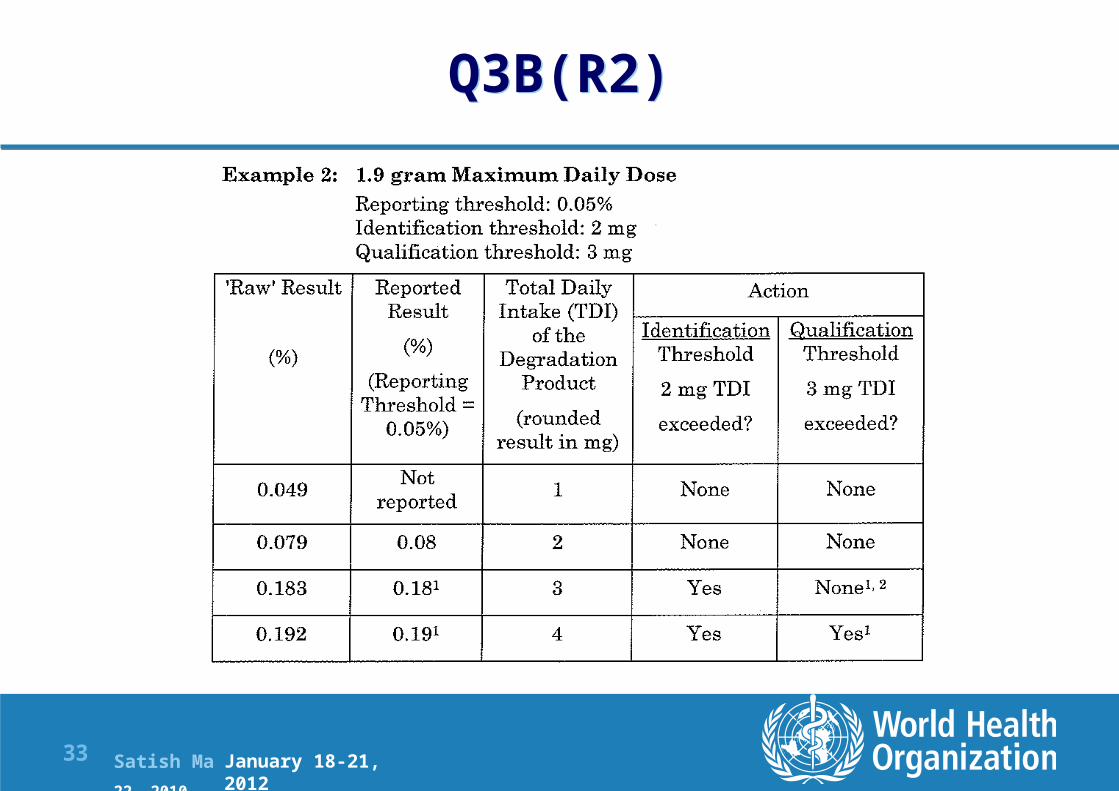
Satish Mallya January 20-22, 2010
33
|
Q3B(R2)
January 18-21, 2012
Satish Mallya January 20-22, 2010
34
|
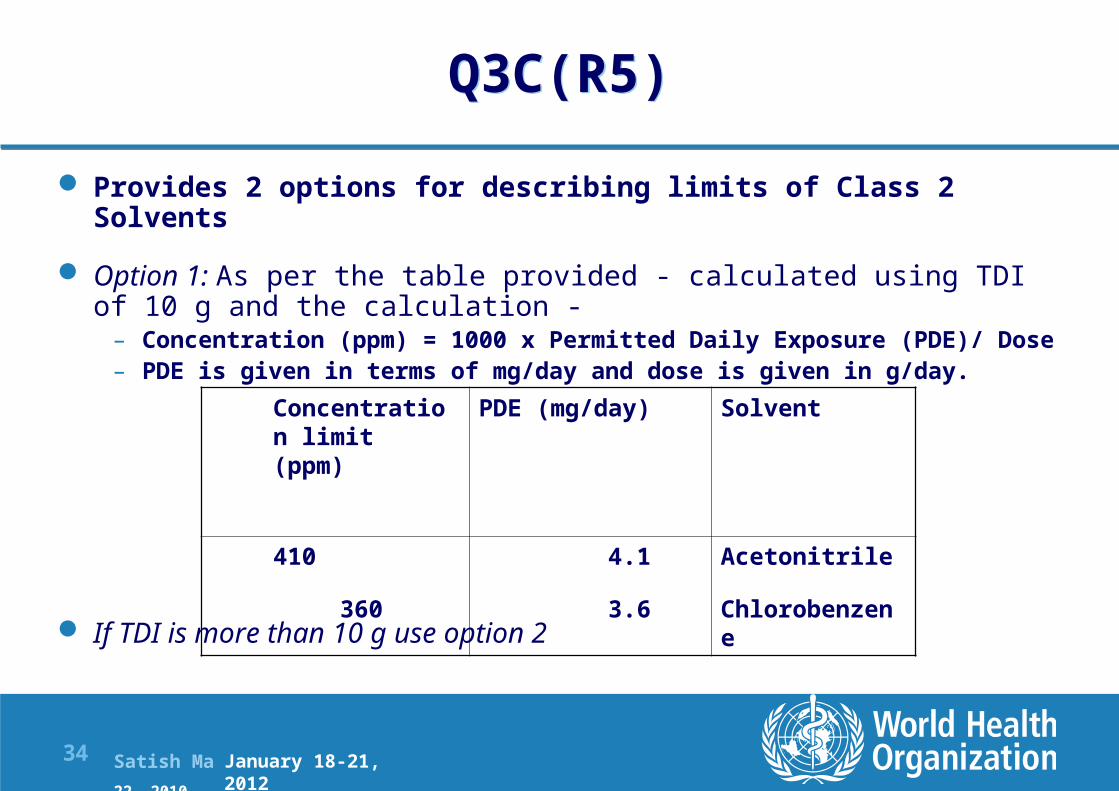
Q3C(R5) Provides 2 options for describing limits of Class 2
Solvents Option 1: As per the table provided - calculated using TDI
of 10 g and the calculation - – Concentration (ppm) = 1000 x Permitted Daily Exposure (PDE)/ Dose– PDE is given in terms of mg/day and dose is given in g/day.
If TDI is more than 10 g use option 2
SolventPDE (mg/day)Concentration limit (ppm)
Acetonitrile
Chlorobenzene
4.1
3.6
410
360
January 18-21, 2012

Satish Mallya January 20-22, 2010
35
|
Example for Option 2 Option 2: It is not considered necessary for each component of the drug
product to comply with the limits given in Option 1. The PDE in terms of mg/day can be used with the known maximum daily dose and equation (Concentration (ppm) = 1000 x PDE/ Dose) to determine the concentration of residual solvent allowed in drug productExample: PDE of acetonitrile is 4.1mg/dayComponent Amount in formulation Acetonitrile content Daily exposure
Drug substance 0.3 g 800 ppm 0.24 mg Excipient 1 0.9 g 400 ppm 0.36 mg Excipient 2 3.8 g 800 ppm 3.04 mg Drug Product 5.0 g 728 ppm 3.64 mg
The sum of the amounts of solvent per day should be less than that given by the PDE.
January 18-21, 2012

Satish Mallya January 20-22, 2010
36
|
Q6A
Addresses aspects such as:– Periodic or skip testing– Release vs shelf-life criteria– In-process tests– Design and development considerations– Limited data available at filing– Parametric release– Alternative procedures– Pharmacopoeial tests and acceptance criteria– Evolving technologies– Impact of drug substance on drug product specifications– Reference standard
January 18-21, 2012

Satish Mallya January 20-22, 2010
37
|
Q6A Decision Trees
#1 – Establishing acceptance criteria for specified impurity In DS
#2 – Establishing acceptance criteria for degradation product in DP
#3 – Establishing acceptance criteria for PSD in DS #4 – Investigating need to set acceptance criteria for
polymorphism in DS and DP #5 – Establishing ID, Assay and enantiomeric impurity
procedures for chiral DS and chiral DS in DP #6 – Microbiological Quality Attributes of DS and Excipients #7 – Setting acceptance criteria for DP dissolution #8 – Microbiological Quality Attributes of non sterile DP
January 18-21, 2012

Satish Mallya January 20-22, 2010
38
|
Periodic or Skip Testing
Should be justified.
May be applied to certain tests only (e.g. residual solvents and microbiological test for solid oral products)
Recommend that it should be applied post approval
Batch to batch retesting to be restored in the event of failure
January 18-21, 2012

Satish Mallya January 20-22, 2010
39
|
Design and Development Considerations
It may be possible to propose excluding or replacing certain tests based on experience and data accumulated: – microbiological testing for drug substances and solid dosage forms which have been shown during development not to support microbial viability or growth (Decision Trees #6 and #8)
– extractables from product containers where it has been reproducibly shown that either no extractables are found in the drug product or the levels meet accepted standards for safety
January 18-21 2012January 18-21, 2012

Satish Mallya January 20-22, 2010
40
|
Design and Development Considerations
– particle size testing may be performed as an in-process test, or may be performed as a release test, depending on its relevance to product performance
– dissolution testing for immediate release solid oral drug products made from highly water soluble drug substances may be replaced by disintegration testing, if these products have been demonstrated during development to have consistently rapid drug release characteristics (Decision Tree #7) (only accepted in exceptional circumstances and all conditions must be met including substantial development data)
January 18-21, 2012

Satish Mallya January 20-22, 2010
41
|
January 18-21, 2012