α-arylation of lithiated allylic and vinylic ureas
TRANSCRIPT

-Arylation of Lithiated Allylic and Vinylic Ureas
A thesis submitted to the University of Manchester for the degree of
Doctor of Philosophy
In the faculty of Engineering and Physical Science
2013
Michael Buchanan Tait
School of Chemistry

2

3
Contents
Abstract 7
Declaration and Copyright Statement 8
Acknowledgements 9
Abbreviations 11
Preface 14
Chapter 1 Introduction 15
1.1 -Tertiary amines 15
1.2 Synthesis of -tertiary amines 16
1.2.1 Additions to ketimines 16
1.2.2 Imine activation 17
1.2.3 Auxiliary controlled addition to ketimines 17
1.2.3.1 N-Terminus control 18
1.2.3.2 C-Terminus control 25
1.2.4 Mannich Reaction 26
1.2.5 Overman (Aza-Claisen) rearrangement 28
1.2.6 Lithiation of pyrrolidine and piperidine analogues 33
1.3 Carbolithiation 38
1.3.1 Stereoselective Carbolithiation 41
1.3.2 Enantioselective Carbolithiation 44
1.3.3 Sparteine analogues 49
1.4 Organolithium Rearrangements 53
1.4.1 Brook Rearrangement 53
1.4.2 Wittig Rearrangement 54
1.4.3 N to C Acyl migration 58

4
Chapter 2 Past work and aims of the project 60
2.1 Previous Work 60
2.1.1 Rearrangement of benzylic ureas 60
2.1.2 Rearrangement of N-pyridyl ureas 275 65
2.1.3 Rearrangement of cyclic benzylic ureas 66
2.1.4 Double -arylation of N-allyl ureas 289 68
2.1.5 Reversal of migration in N-diarylallyl ureas 69
2.2 N to C aryl migration in urea analogues 69
2.2.1 -Arylation of benzylic carbamates 70
2.2.2 -Arylation of benzylic thiocarbamates 71
2.3 Vinyl migration 71
2.4 Carbolithiation-Rearrangement 73
2.4.1 Of ureas 73
2.4.2 Of carbamates 75
2.4.3 Of thiocarbamates 76
2.5 Aims of the Project 76
Chapter 3 Enantioselective Carbolithiation-Rearrangement 78
3.1 Previous Work 78
3.1.1 Reaction optimisation 78
3.1.2 Substrate scope 83
3.2 Extension of Methodology 85
3.2.1 Extending Migrating Aryl Ring Scope 85
3.2.2 Non-commercial Organolithiums 86
3.3 Absolute Configuration 91
3.3.1 Solvolysis of ureas 91
3.3.2 Preparation of an authentic sample 93

5
Chapter 4 Synthesis of substituted piperidines 97
4.1 Part 1: 6-Membered cyclic vinylic ureas via a cyclic imine 97
4.1.1 Synthesis of 6-membered cyclic vinylic ureas 97
4.1.2 Lithiation of 6-membered cyclic vinyl ureas 338 100
4.1.2.1 Carbolithiation-Rearrangement of diphenyl urea 338a 101
4.1.2.2 Rearrangement of functionalised aryl rings 108
4.1.2.3 Stereospecificity of carbolithiation 109
4.1.2.4 LDA mediated rearrangement 114
4.1.2.5 Enantioselective Carbolithiation-Rearrangement 116
4.1.2.6 Urea solvolysis 118
4.2 Part 2: 6-Membered cyclic vinylic ureas via a ring closing metathesis approach 120
4.2.1 Ring closing metathesis 120
4.2.2 6-Membered cyclic vinylic ureas by RCM. 123
4.2.2.1 Heteroaryl systems 128
4.2.3 Carbolithiation-rearrangement of 6-membered cyclic vinylic ureas 129
4.3 Part 3: Enantioenriched 6-membered cyclic vinylic and allylic ureas 131
4.3.1 Synthesis of enantioenriched cyclic allylic and vinylic ureas 131
4.3.2 Lithiation of enantioenriched cyclic vinylic ureas 470 137
4.3.3 Lithiation of enantioenriched cyclic allylic ureas 468 140
4.3.3.1 Determination of relative configuration of rearranged cyclic allylic ureas 142
4.3.4 Urea Solvolysis 145
Chapter 5 Synthesis of substituted pyrrolidines and pyrrolines 146
5.1 Synthesis of 5-membered cyclic ureas 146
5.1.1 By the reaction of cyclic imines and aryl isocyanates 146
5.1.2 Use of RCM in synthesis of 5-membered cyclic ureas 147
5.2 Lithiation of 5-membered cyclic ureas 152
5.2.1 Lithiation of 501 152
5.2.2 Carbolithiation-rearrangement of 5-membered cyclic vinylic ureas 153
5.2.3 Lithiation of 5-membered-cyclic allylic ureas 154
5.2.4 Variable Temperature 1H NMR Spectroscopy 156

6
Chapter 6 Synthesis of ketamine analogues 159
6.1 Rearrangement of cyclohexenyl ureas 159
6.1.1 Previous Work 159
6.1.2 Synthesis of cyclohexenyl ureas 161
6.1.3 Lithiation of cyclohexenyl ureas 162
6.1.4 Solvolysis of cyclohexenyl ureas 164
6.1.5 Regioselective Oxidation 165
6.1.5.1 Epoxide formation 165
6.1.5.2 Alternative oxidation methods 166
6.2 Rearrangement of -ketoureas 169
6.2.1 Previous work 169
6.2.2 Synthesis of -ketoureas 170
6.2.3 Lithiation of -ketoureas 173
Chapter 7 Conclusions and Future work 176
7.1 Conclusions 176
7.2 Future Work 179
7.2.1 Cyclohexenyl ureas 179
7.2.2 -Ketoureas 179
Chapter 8 Experimental 181
8.1 General Information 181
8.2 General Procedures 183
8.3 Experimental Data 191
Chapter 9 References 298
Appendix 1: X-Ray crystal structure data 306
Word Count: 72536

7
-Arylation of Lithiated Allylic and Vinylic Ureas
A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy In the faculty of Engineering and Physical Science at The University of Manchester
Michael Buchanan Tait, 2013 Chiral amines are widespread in naturally occurring and synthetic bioactive molecules. However, it remains challenging to synthesise these molecules in an enantiopure form. Investigations within our group have shown that treatment of vinylic ureas with organolithium reagents results in an umpolung addition of the organolithium, followed by N to C aryl migration of the lithiated intermediate. This thesis details investigations into developing an enantioselective version of this reaction through the use of the chiral ligands (−)-sparteine and the (+)-sparteine surrogate. The
enantioenriched ureas are easily transformed into the corresponding -tertiary amines. Use of the different chiral ligands allows access to either enantiomer of the rearranged urea.
Further work has centred on the application of this methodology towards cyclic substrates; with
a view to synthesising -tertiary cyclic diaryl amines. Studies have shown that treatment of vinylic ureas bearing cyclic substituents with a number of different organolithium reagents successfully produce the desired rearranged ureas, with complete diastereoselectivity.
Subsequent solvolysis of these ureas yields hindered cyclic -tertiary amines in good yields.
During these investigations it has been shown that allylic ureas undergo a rearrangement to the least sterically hindered side of the cyclic urea. This allows for fine tuning of the migration reaction depending on where the double bond is positioned in the molecule.
A range of vinylic and allylic ureas, including substrates with interesting pharmaceutical properties, have been investigated. Their synthesis and reactivities upon lithiation are discussed within this thesis.

8
Declaration
No portion of the work referred to in the dissertation has been submitted in support of an
application for another degree or qualification of this or any other university or other institute
of learning.
Copyright Statement
i. The author of this thesis (including any appendices and/or schedules to this thesis)
owns any copyright in it (the “Copyright”) and he has given The University of
Manchester the right to use such Copyright for any administrative, promotional,
educational and/or teaching purposes.
ii. Copies of this thesis, either in full or in extracts, may be made only in accordance with
the regulations of the John Rylands University Library of Manchester. Details of these
regulations may be obtained from the Librarian. This page must form part of such
copies made.
iii. The ownership of any patents, designs, trade marks and any and all other intellectual
property rights except for the Copyright (the “Intellectual Property Rights”) and any
reproductions of copyright works, for example graphs and tables (“Reproductions”),
which may be described in this thesis, may not be owned by the author and may be
owned by third parties. Such Intellectual Property Rights and Reproductions cannot and
must not be made available for use without prior written permission of the owner(s) of
the relevant Intellectual Property Rights and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and
commercialisation of this thesis, the Copyright and any Intellectual Property Rights
and/or Reproductions described in it may take place is available in the University IP
Policy (see
http://www.campus.manchester.ac.uk/medialibrary/policies/intellectualproperty.pdf),
in any relevant Thesis restriction declarations deposited in the University Library, The
University Libraryʼs regulations (see
http://www.manchester.ac.uk/library/aboutus/regulations) and in The Universityʼs
policy on presentation of Theses.

9
Acknowledgements
This thesis is dedicated to Steve Watson and Molly Tait.
Firstly, massive thanks to Jonathan for offering me the opportunity for work within his research
group and the countless hours of help and support over the last four years, and for his
unlimited patience in answering all of my stupid questions and listening to my terrible jokes.
I’d like to also thank everyone who I have worked with during my time in Manchester: Rob,
Nadia, Dan Tetlow, Paul, James, Jemma, Julien, Anne, Edmund, Gaëlle, Simon, Rachel, Sam,
Francis, Bryden, Dan Leonard, Romina, Tony, Beckii, Abby, Simon, Matteo, Liam, Vincent, Sarah,
Thomas, Jordi, Gilles, Morgan, Bea, Nicole, Daniele, Marta, Hatice, Renzo, Phil, Richard, Julien II,
Nelson, Ole, Jon, Ugo, Josep, Erik, Marju, Stefan, Ophelie, Alistair, Dan Foley, Dean, Shamima,
Scott, Wojciech, Ross, Lucy and Jen. I hope that’s everyone. Unfortunately for you lot I do
consider you all friends and will keep in touch. Good luck trying to come up with more dingbats
for the next Christmas quiz; I was really scraping the bottom of the barrel last year (as you very
well know).
I could probably write another thesis on the laughs and stories we have shared so special
mention to Thomas’ stiff neck, Rachel and her bottle of N,N-DMF, the time Nadia ran away
giggling from Edmund and I, Rob’s pyrene artistry, the doll from Sitges and Dan’s tremendous
Kung-Fu skills. The less said about the music tastes of some of you though, the better. Many
thanks go to Dan, Liam and Paul for proof reading this thesis.
I would also like to thank the technical services at the university; to Ian and Roger for their NMR
help, Jim for X-Ray crystallography and Gareth, Val and (the sadly missed) Rehana for their help
with mass spectrometry and HPLC breakdowns.
Also thanks to everyone at AZ who made my 3 months there such an enjoyable experience:
Tom, Gary, Ryan, Radleigh, Rachel, Chris, Sam and Laura for their thought provoking lunch time
conversation topics. To Lyman, for looking after me in the lab and somehow managing to set up
a THF still in industry and to my industrial supervisor Sam for his help and guidance both at
Alderley Park and during various meet ups over the last 2 and a bit years.

10
Finally, I would not be where I am today if it was not for the continuous love and support of my
family and friends who have helped me through both the good times, and the bad. This thesis is
more for them than it is for me. Mum, thanks for finally learning it’s organic chemistry that I’ve
studied for the last four years. Dad, I hope I’ve not turned out too average for your liking, ya big
Jambo ye. To Susan and Scott, the force is strong between you two, enjoy it. And to Freda, who
saw me at my lowest and thought there’s someone I want to get to know and love, and who
has made the last year and a half the best of my life, when I thought it would be anything but. I
can’t wait to experience the adventures we will have together in Chapel and beyond, you never
know it might involve a cow or two along the way too. Cool.
All the best,
Mike

11
Abbreviations
°C Degrees Celsius
Å Angstrom
Ac Acetate
Aq Aqueous
Ar Aryl
Bn Benzyl
Boc tert-Butoxycarbonyl
Bu Butyl
n-Bu Butyl
n-BuLi n-Butyllithium
s-Bu sec-Butyl
s-BuLi sec-Butyllithium
t-Bu tert-Butyl
t-BuLi tert-Butyllithium
t-BuONO tert-Butyl Nitrite
Bz Benzoyl
c. Concentration
CAN Cerium(IV) Ammonium Nitrate
Calcd. Calculated
Cat. Catalytic
Cb Carbamate
Cbz Carboxybenzyl
CDI Carbomyldiimidazole
cm Centimetre
COP Colbalt Oxazoline Palladacycle
COSY Correlation Spectroscopy
Cy Cyclohexyl
d Doublet
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DCE 1,2-Dichloroethane
DCM Dichloromethane
DIBAL Diisobutylaluminium Hydride
DMAP 4-Dimethylaminopyridine
DME 1,2-Dimethoxyethane
DMEDA N,N′-Dimethylethylenediamine
DMF N,N-Dimethylformamide
DMPU 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone
DMS Dimethyl Sulfide
DMDO Dimethyldioxirane
DMSO Dimethyl Sulfoxide
dr Diastereomeric Ratio
E Electrophile

12
E Entgegen
ee Enantiomeric Excess
er Enantiomer Ratio
ES Electrospray
Et Ethyl
Et2O Diethyl Ether
EtOAc Ethyl acetate
EWG Electron Withdrawing Group
FIP Ferrocenyl Imidazoline Palladacycle
Fmoc Fluorenylmethyloxycarbonyl
FOP Ferrocenyl Oxazoline Palladacycle
g Grams
h Hour
HCl Hydrochloric Acid
n-Hex n-Hexyl
n-HexLi n-Hexyllithium
HMPA Hexamethylphosphoramide
HPLC High-Performance Liquid Chromatography
HRMS High Resolution Mass Spectrometry
Hz Hertz
IPA Isopropylalcohol
IR Infrared
KHMDS Potassium Bis(trimethylsilyl)amide
L Ligand
LCMS Liquid Chromatography Mass Spectrometry
LDA Lithium Diisopropylamide
LiHMDS Lithium Bis(trimethylsilyl)amide
Lit Literature
LiTMP Lithium Tetramethylpiperidide
m Multiplet
M Molar
Me Methyl
MEM β-Methoxyethoxymethyl Ether
MHz Megahertz
w Microwave
min Minute
mL Millilitres
mmol Millimole
mol Mole
MP Melting Point
MS Molecular Sieves
NaHMDS Sodium Bis(trimethylsilyl)amide
NMR Nuclear Magnetic Resonance
NOE Nuclear Overhauser Effect

13
Ns Nosyl
Nu Nucleophile
PG Protecting Group
Ph Phenyl
PMB para-Methoxybenzyl
PMP para-Methoxyphenyl
PPTS Pyridinium para-Toluenesulfonate
PTSA para-Toluenesulfonic Acid
n-Pr n-Propyl
i-Pr Isopropyl
i-PrLi Isopropyllithium
py Pyridine
q Quartet
Quant. Quantitative
Rf Retention Factor
Recryst Recrystallisation
rt Room Temperature
Sat Saturated
SM Starting Material
soln. Solution
t Triplet
T Temperature
TBAF Tetra-n-Butylammonium Fluoride
TBDMS tert-Butyldimethylsilyl
TFA Trifluoroacetic Acid
THF Tetrahydrofuran
TIPS Triisopropylsilyl
TLC Thin Layer Chromatography
TMSCN Trimethylsilyl Cyanide
Tol Toluene
Ts Tosyl
UV Ultraviolet
VT Variable Temperature
Xantphos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene
Z Zusammen

14
Preface
The author graduated from the University of Edinburgh in 2009 with a Master of Chemistry
(MChem) in Biological and Medicinal Chemistry with Industrial experience with first class
honours. This incorporated a 13 month industrial placement at Piramal Healthcare Ltd (formerly
NPIL Pharmaceuticals), Morpeth, Northumberland. His final year project was undertaken in the
laboratories of Professor Stephen Chapman. The project investigated the isolation of the
dodecaheme cytochrome GSU 1996 from Geobacter sulfurreducens and its interactions with
both single and double stranded DNA.
From 2009 the author joined the group of Professor Jonathan Clayden at the University of
Manchester working on the lithiation of N-vinyl and N-allyl ureas. This research is embodied in
this thesis. In 2012, the author completed a placement in the Oncology department at
AstraZeneca, Alderley Park under the supervision of Dr Sam Butterworth.
In 2013 the author will take up a position with Peakdale Molecular Ltd.

15
Chapter 1 Introduction
1.2 -Tertiary amines
Chiral -tertiary amines are widespread in naturally occurring and synthetic bioactive molecules
(Figure 1).1 However, even now it remains challenging to synthesise these molecules in an
enantiopure form.
Figure 1:-Tertiary amines
Compounds of general structure 1 are a very important class of compounds as they are
commonly found in a variety of natural and unnatural biologically active compounds, and it is
known that the nitrogen atom plays a key role in the biological effect of these molecules.2,3 As a
result, -tertiary amines are present in the vast majority of pharmacological drug targets such
as the analgesic and anaesthetic ketamine 2,4 ras farnesyl protein transferase (FTPase) inhibitor
3,5 poly(ADP-ribose) polymerase inhibitor veliparib (ABT-888) 46 and NK1 receptor antagonist 5.7
-Tertiary amines can also be valuable intermediates towards the synthesis of natural and
unnatural amino acid derivatives 68 (Figure 2). It is therefore surprising that despite their
prevalence in important chemical targets, there are relatively few methods available for the
direct synthesis of enantiomerically pure chiral -tertary amines.
Figure 2

16
1.3 Synthesis of -tertiary amines
Chiral -tertiary amines can be synthesised by a variety of methods, the most practical of which
will be outlined in this introduction. One such approach is nucleophilic addition to imine
derivatives, whilst molecular rearrangements offer a useful alternative. For a more exhaustive
list of routes towards -tertiary amines see a recent review on the subject, and the references
within.9
1.3.1 Additions to ketimines
The most commonly used method for the synthesis of chiral -tertiary amines 8 is the
enantioselective 1,2 addition of nucleophiles to ketimines 7 (Scheme 1).10
Scheme 1: Nucleophilic addition to ketimine
However, this addition can be problematic due to the poor electrophilicity of the ketimine
carbon in comparison to ketones. This often requires the use of reactive nucleophiles such as
organolithiums and Grignard reagents to facilitate reaction. It is therefore essential to take into
consideration the steric and electronic properties of the N-substituent to avoid undesired side
reactions such as -deprotonation and reduction (Scheme 2).11 Furthermore, N-substitution is
essential to prevent oligomerization.
Scheme 2: Side reactions of nucleophilic addition

17
1.3.2 Imine activation
The simplest method of avoiding -deprotonation is to activate the imine towards nucleophilic
attack by attaching electron withdrawing substituents to the nitrogen or through quaternization
of the nitrogen to generate an iminium ion. One of the first examples of this methodology used
trimethylsilyl triflate to generate iminium ion 12 to improve the addition of organometallic
reagents to a range of imines 11 (Scheme 3).12
Scheme 3: Imine activation and nucleophilic addition
Imines can also be activated by coordination of the imine nitrogen to a range of organometallic
reagents. For example -tertiary amine 16 is obtained by the addition of an organocerium
reagent to N-unsubstituted ketimine 14 via the N-metalloimine 15 (Scheme 4).13
Scheme 4: Organocerium addition to ketimines
1.3.3 Auxiliary controlled addition to ketimines
The use of chiral auxiliaries to promote enantioselective addition to ketimines is a powerful tool
for the synthesis of -tertiary amines. The auxiliaries can either be attached on the N- or C-
terminus of the C-N double bond (Figure 3).14
Figure 3: Auxiliary substituted ketimines

18
1.3.3.1 N-Terminus control
The stereochemistry of the amine product can be controlled through the diastereoselective
addition of a nucleophile to an imine which bears a chiral substituent on nitrogen. Subsequent
removal of the chiral auxiliary allows the isolation of the corresponding enantioenriched -
tertiary amine 19 (Scheme 5). However, this approach can suffer from poor atom economy if
the auxiliary itself cannot be recycled.
Scheme 5: N-terminus auxiliary controlled nucleophilic addition to ketimines
One such example of N-terminus control is the N-p-toluenesulfinyl group 20, which produces
imines that are both stable and activated towards nucleophilic attack (Figure 4).15 Furthermore,
due to its large steric bulk, 20 provides high diastereofacial selectivity during the nucleophilic
addition, which prevents attack on one side of the imine. An important advantage for the use of
this group is that it is also readily cleaved under mild acidic conditions.
Figure 4
Moreau and co-workers utilised the N-p-toluenesulfinyl aldimines 21 in the synthesis of -
branched amines (Scheme 6).16 The reaction involved the addition of benzyl Grignard to the
sulfinyl aldimine 21. Removal of the activating group under treatment with trifluoroacetic acid,
gave the free amines 22 in moderate to good yields and enantioselectivities, whilst also
regenerating the chiral auxiliary.

19
Scheme 6: p-Toluenesulfinyl chiral auxiliary
However, addition to sulfinyl aldimines 21 is only successful with allyl and benzyl Grignard
reagents. Treatment with more nucleophilic Grignard reagents, such as methyl Grignard, causes
attack at sulfur rather than the carbon of the aldimine to give a mixture of 23 and the aromatic
aldehyde 24 (Scheme 7).
Scheme 7: Addition to sulfur
To improve on this, a more chemoselective N-tert-butylsulfinamide auxiliary 25 has been
developed by Ellman et al. (Figure 5).17 The amine is now more nucleophilic and therefore forms
imines more readily than its tolyl analogue. The tert-butyl group is also more sterically hindered
and as a consequence nucleophilic attack at sulfur is reduced, resulting in an increased scope in
the nucleophiles used during the addition. Furthermore, 25 maintains sufficient steric bulk to
retain the diastereofacial selectivity of the addition.
Figure 5

20
The chiral auxiliary 25 is easily synthesised in one step through a vanadium-catalysed
asymmetric oxidation of the inexpensive di-tert-butyl disulfide 26, followed by treatment with
lithium amide (Scheme 8).18 The sulfinamide 25 is crystalline, making it easy to prepare in an
enantiopure form on a large scale.10 N-substituted ketimines 27 are then easily generated by
condensation with a ketone in the presence of titanium ethoxide, which is used as both a Lewis
acid and as a water scavenger.17
Scheme 8: Chiral sulfinamide synthesis
Treatment of the tert-butyl sulfinyl aldimines 29 with a range of Grignard reagents provide the
resulting -branched amines 31 in good to excellent yields and diastereoselectivities (Scheme
9).19 Aliphatic and aromatic aldimines and alkyl, aryl and vinyl Grignard reagents all react to give
the hydrochloride salts of the desired -branched amines after removal of the auxiliary.
Scheme 9: Synthesis of -branched amines
The reaction proceeds through cyclic six-membered transition state 30 with magnesium
coordinated to the oxygen of the sulfinyl group. The highest selectivities are found when the
reaction is carried out in non-coordinating solvents such as dichloromethane, an observation
that further supports the proposed chelated metal transition state.
21
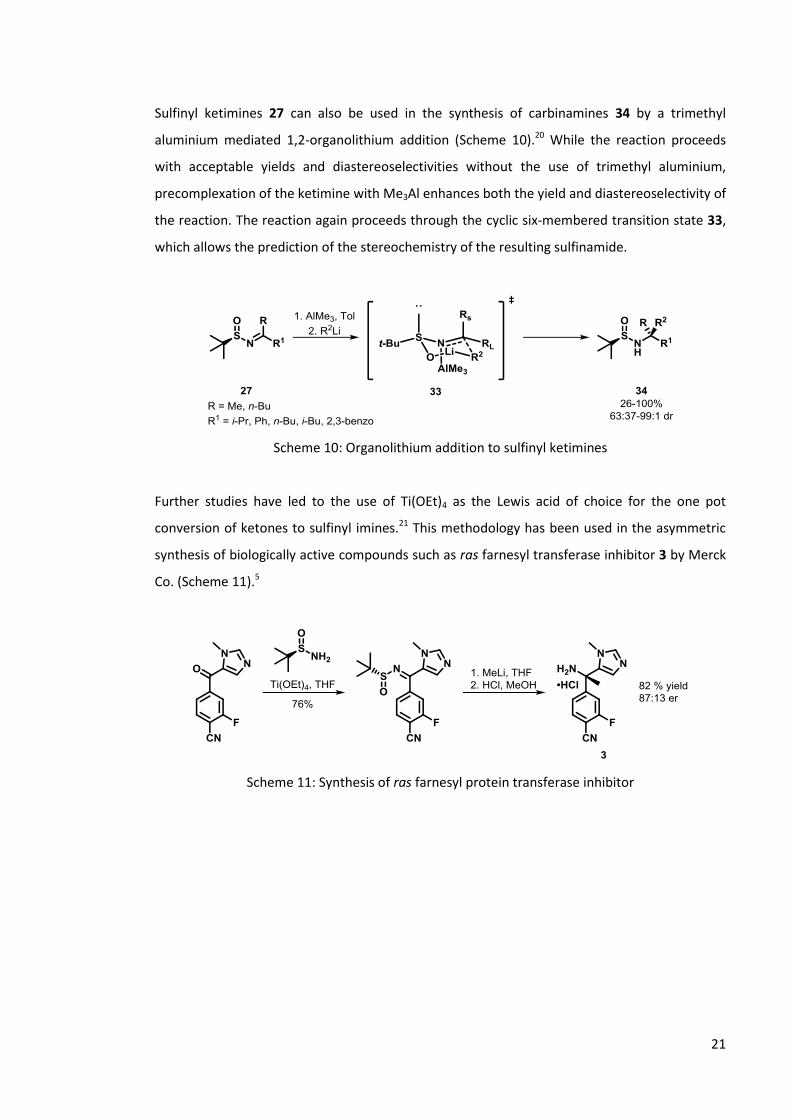
Sulfinyl ketimines 27 can also be used in the synthesis of carbinamines 34 by a trimethyl
aluminium mediated 1,2-organolithium addition (Scheme 10).20 While the reaction proceeds
with acceptable yields and diastereoselectivities without the use of trimethyl aluminium,
precomplexation of the ketimine with Me3Al enhances both the yield and diastereoselectivity of
the reaction. The reaction again proceeds through the cyclic six-membered transition state 33,
which allows the prediction of the stereochemistry of the resulting sulfinamide.
Scheme 10: Organolithium addition to sulfinyl ketimines
Further studies have led to the use of Ti(OEt)4 as the Lewis acid of choice for the one pot
conversion of ketones to sulfinyl imines.21 This methodology has been used in the asymmetric
synthesis of biologically active compounds such as ras farnesyl transferase inhibitor 3 by Merck
Co. (Scheme 11).5
Scheme 11: Synthesis of ras farnesyl protein transferase inhibitor

22
Furthermore, N-tert-butylsulfinyl ketimines 27 are used in the synthesis of highly substituted -
amino acid derivatives 36, by a Mannich-type addition of titanium enolates (Scheme 12).22 Once
more, yields and diastereoselectivities of the addition are good to excellent, even when ,-
disubstituted enolates are used to provide ,,,-tetrasubstituted -amino acid derivatives (R,
R1, R2, R3 ≠ H). The high diastereoselectivity of the reaction can be rationalised using a six-
membered Zimmerman-Traxler-type transition state 35, where both the sulfinyl nitrogen and
oxygen interact with the coordinatively unsaturated titanium (IV). This creates a high level of
preorganisation which causes attack opposite to the sterically bulky tert-butyl group, resulting
in high selectivities.
Scheme 12: Reaction of sulfinimines with substituted titanium enolates
The tert-butyl sulfinamide directing group has also been used in the diastereoselective allylation
of ketimines (Scheme 13). A number of benzylic ketimine derivatives 37 have been transformed
into their corresponding sulfinyl amines 38 in good yield and excellent diastereoselectivities.23
Scheme 13: Allylation of tert-butyl sulfinimines

23
Use of In(OTf)3 with the corresponding aldimine derivatives 39 results in a chelation controlled
transition state 42, where the Lewis acid is able to coordinate to the oxygen of 40, to yield
sulfinamide 41 (Figure 6). In contrast, the use of HMPA as an additive reverses the
stereochemical outcome, and allows for the preparation of the opposite diastereomer 40
(Scheme 14). HMPA coordinates to the allyl metal, which prevents coordination of the allyl
metal to the sulfinamide (Figure 6). This switch in stereochemistry is only applicable to
aldimines; the use of HMPA during addition to ketimines retards the reaction.
Scheme 14: Additive controlled addition to tert-butyl sulfinyl aldimines
Figure 6: Proposed transition states
N-tert-Butylsulfinyl ketimines 27 are also used in the synthesis of ,-dibranched propargyl
sulfinamides 44, through a trimethylaluminium-mediated addition of lithium acetylides
(Scheme 15).24 A range of propargyl sulfinamides have been made in good yields and with
excellent diastereoselectivity. Acidic cleavage of the tert-butylsulfinyl group yields the free ,-
dibranched propargyl amines, isolated as their hydrochloride salts 45.
Scheme 15: Lithium acetylide addition to sulfinyl ketimines

24
The tert-butylsulfinyl group cleaved under the acidic deprotection conditions can be recycled by
treating the N-tert-butylsulfinyl amines 46 with hydrochloric acid to recover the amine
hydrochloride salt 47 and tert-butylsulfinyl chloride 48 in quantitative yields.25 48 is then
treated with ethanol and catalytic quinidine as a sulfinyl transfer catalyst, to produce the
corresponding ethyl tert-butanesulfinate 49 in 88% ee. Addition of sodium amide in ammonia
yields tert-butylsulfinyl amides 25 in 99% ee and 67% yield from the starting N-tert-butylsulfinyl
amine 46 (Scheme 16).
Scheme 16: Recycling of tert-butylsulfinyl group
The use of N-terminus chiral auxiliaries to control nucleophilic additions to imines is not
exclusive to the tert-butyl sulfinamide group. An organoindium auxiliary, prepared from
stoichiometric indium, has been used in the highly diastereoselective allylation of ketimines
50.26 It is postulated that this occurs through bicyclic transition state 51 (Scheme 17).
Coordination of the allyl metal to both the chiral auxiliary and the ester group forces the ester
group into a pseudo-axial position. The auxiliary can then be removed to yield the amino acid
derivatives 54 (Scheme 18).
Scheme 17: Allylation of ketimine 50 by organoindium addition

25
Scheme 18: Amino acid derivative 54 TMSE: Trimethylsilyl ethyl
1.3.3.2 C-Terminus control
Although less common than N-terminus control, it is also possible to control the addition of
nucleophiles to imines through the C-terminus of the -system. This is best illustrated by the
diastereoselective sequential addition of nucleophiles to nitrile 55 (Scheme 19).27
Scheme 19: C-terminus controlled addition to ketimines
Once more, chelation is used to control the geometry of 55. The chiral group to the resultant
imine is then used to control the facial selectivity of the second addition (Scheme 20).27
Scheme 20: Chelation controlled addition
These amines are then easily converted to their corresponding amino acids 60 (Scheme 21). Cbz
protection of the amine and subsequent acetal removal yields diol 59. 59 then undergoes
oxidative cleavage to its corresponding aldehyde, which is then oxidised to amino acid 60.

26
Scheme 21: Amino acid synthesis
1.3.4 Mannich Reaction
Mannich type reactions are used to convert ketimines to their corresponding -amino acid
derivatives (Scheme 22). Unfortunately, the addition of enolates to ketimines is much more
difficult, and as a result less widely reported, than for the corresponding aldimines.28
Scheme 22: Mannich type reaction
The Lewis acidic chiral silane 63 is used in the synthesis of -amino acid derivatives 64.29 63 is
easily prepared from pseudoephedrine.30,31 The hydrazone derivative 61, which is used as a
ketamine analogue, is essential in order for the reaction to take place, as coordination of the
Lewis acid to the hydrazide moiety determines the stereochemical outcome of the reaction
(Scheme 24). A range of -amino acid derivatives 64 have thus been synthesised in good yields
and excellent enantioselectivities (Scheme 23). The hydrazine group can be easily removed by
treatment with samarium iodide.32
Scheme 23: Chiral silane Lewis acid mediated addition

27
Scheme 24: Chiral silane Lewis acid mediated addition
The high enantioselectivity found in this reaction is all the more remarkable considering the
relatively low stereochemical purity in the hydrazone derivative 61 and silane 63. This implies
that all three components of the reaction are reacting through one single common transition
state complex, presumably 65, with the stereochemistry of the product controlled by the
pseudoephedrine group.29
Shibasaki et al. have demonstrated that it is possible to carry out the Mannich reaction
catalytically.33 A copper catalyst is used along with large diphosphinyl ligands, such as 69 or 70,
to provide the chiral environment for the reaction. This allows the silyl ketene acetal 67 to
attack with a high degree of stereocontrol in excellent yields (Scheme 25). The ligand used
through the course of the reaction is dependent on whether an aryl or alkyl ketimine is used.
The phosphinoyl group can be easily removed under acidic conditions (Scheme 26) to yield the
corresponding -amino acid derivatives 71.
Scheme 25: Copper catalysed asymmetric Mannich reaction. Xy = 3,5-Xylyl
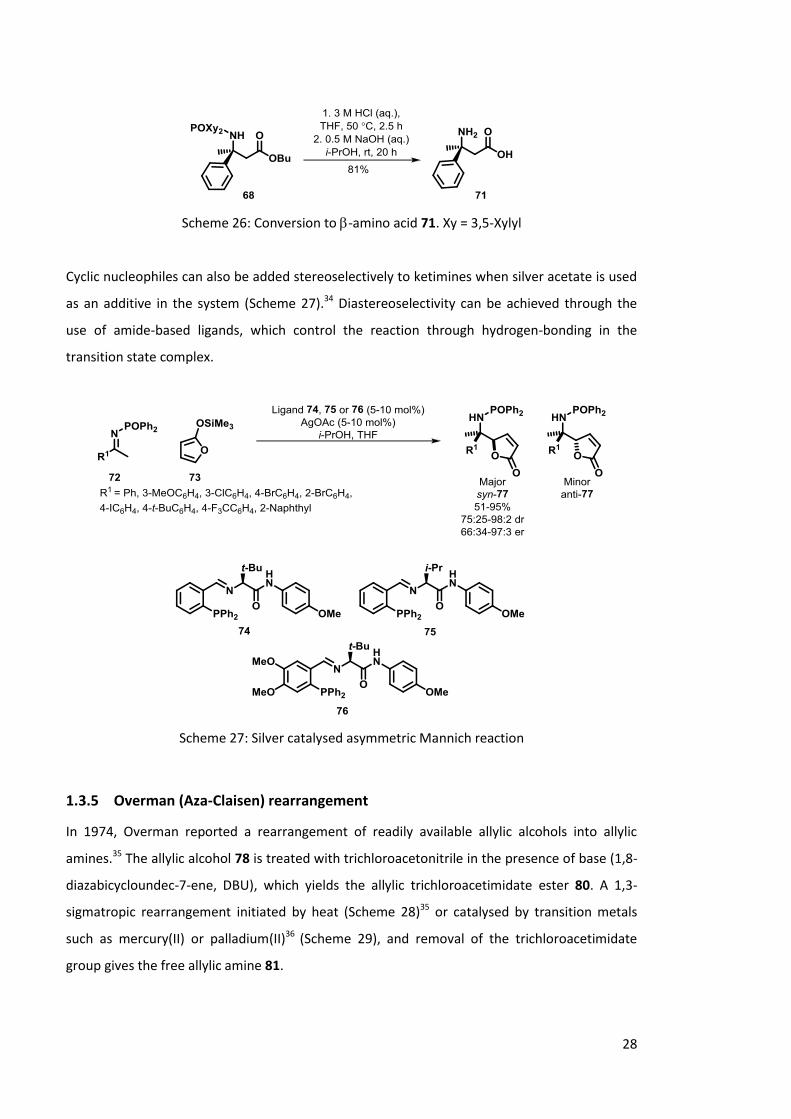
28
Scheme 26: Conversion to -amino acid 71. Xy = 3,5-Xylyl
Cyclic nucleophiles can also be added stereoselectively to ketimines when silver acetate is used
as an additive in the system (Scheme 27).34 Diastereoselectivity can be achieved through the
use of amide-based ligands, which control the reaction through hydrogen-bonding in the
transition state complex.
Scheme 27: Silver catalysed asymmetric Mannich reaction
1.3.5 Overman (Aza-Claisen) rearrangement
In 1974, Overman reported a rearrangement of readily available allylic alcohols into allylic
amines.35 The allylic alcohol 78 is treated with trichloroacetonitrile in the presence of base (1,8-
diazabicycloundec-7-ene, DBU), which yields the allylic trichloroacetimidate ester 80. A 1,3-
sigmatropic rearrangement initiated by heat (Scheme 28)35 or catalysed by transition metals
such as mercury(II) or palladium(II)36 (Scheme 29), and removal of the trichloroacetimidate
group gives the free allylic amine 81.

29
Scheme 28: The Overman rearrangement
Scheme 29: Metal catalysed Overman rearrangement
The stereochemistry of the allylic trichloroacetimidate esters 82 is retained through the
sigmatropic rearrangement into the allylic amide 84. The rearrangement proceeds in a
suprafacial manner through the chair-like transition state 83 (Scheme 30).37
Scheme 30: Overman rearrangement
The Overman rearrangement has proved invaluable in a variety of total synthesis of natural
products and pharmaceutical drug targets. For example, an Overman rearrangement was
utilised in the synthesis of the natural product tetrodotoxin 87, a potent toxin found in puffer
fish.38,39 Reaction of the allylic alcohol with trichloroacetonitrile gives the acetimidate 85, which
undergoes rearrangement to allyic amine 86. Due to the steric constraints of the ring system,
the favoured conformer 88 dictates the stereochemistry of the rearrangement. Further
chemical transformations yield the natural product tetrodotoxin 57 (Scheme 31).
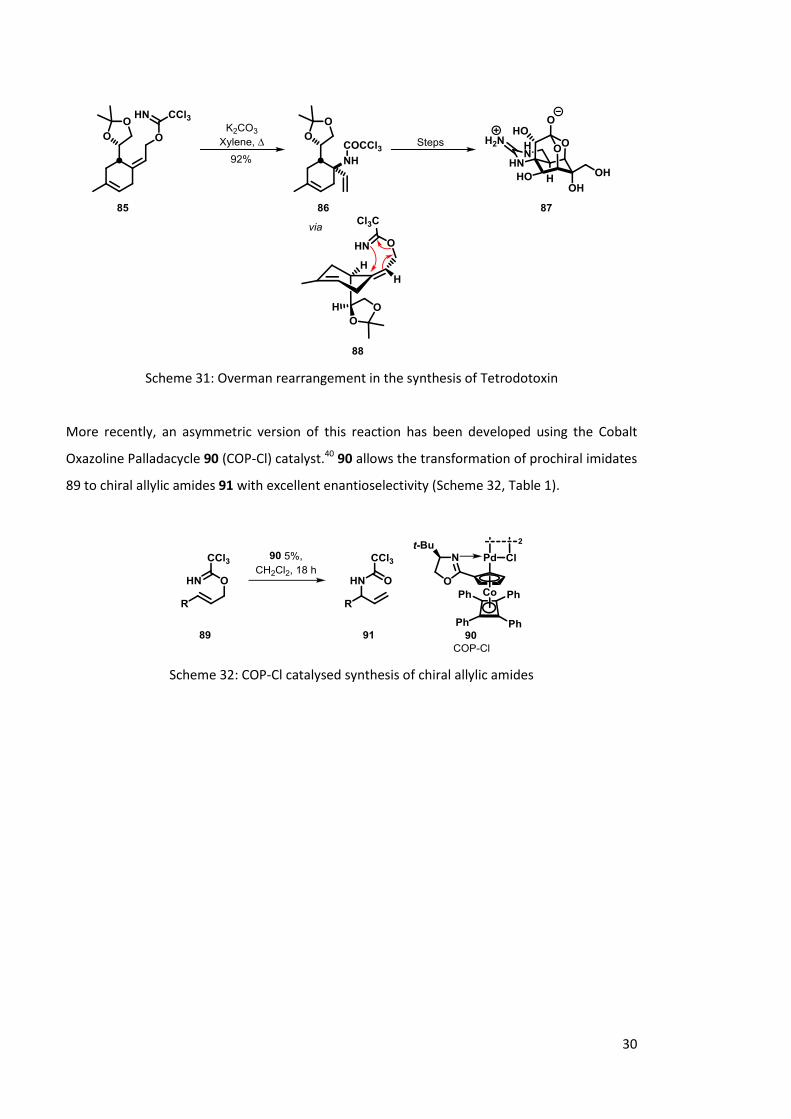
30
Scheme 31: Overman rearrangement in the synthesis of Tetrodotoxin
More recently, an asymmetric version of this reaction has been developed using the Cobalt
Oxazoline Palladacycle 90 (COP-Cl) catalyst.40 90 allows the transformation of prochiral imidates
89 to chiral allylic amides 91 with excellent enantioselectivity (Scheme 32, Table 1).
Scheme 32: COP-Cl catalysed synthesis of chiral allylic amides

31
Entry R E/Z Temp (°C) Yield 91 (%) ee 91 (%)
1 n-Pr E rt 80 94 (S)
2 n-Pr E 38 99 95 (S)
3 n-Pr Z 38 17 71 (R)
4 i-Bu E 38 95 96 (S)
5 i-Bu E 38 92 98 (S)
6 i-Bu Z 38 8 73 (R)
7 Me E rt 85 92 (S)
8 Cy E 38 82 96 (S)
9 CH2CH2Ph E rt 83 96 (S)
10 CH2CH2Ph E 38 93 93 (S)
11 Ph E rt 13 nd
12 t-Bu E 38 7 nd
Table 1: COP-Cl catalysed synthesis of chiral allylic amides
However, use of 90 in the synthesis of amines bearing quaternary stereocenters has proved
problematic due to steric clash between the disubstituted allylic alcohols and the catalyst. To
overcome this problem, Peters and co-workers have developed a similar ferrocenyl imidazoline
palladacycle 94 (FIP-Cl) catalyst to efficiently rearrange a variety of p-methoxyphenyl
trifluoroacetamides 95 (Scheme 33, Table 2).41
Scheme 33: FIP-Cl catalysed synthesis of chiral trifluoroacetamides
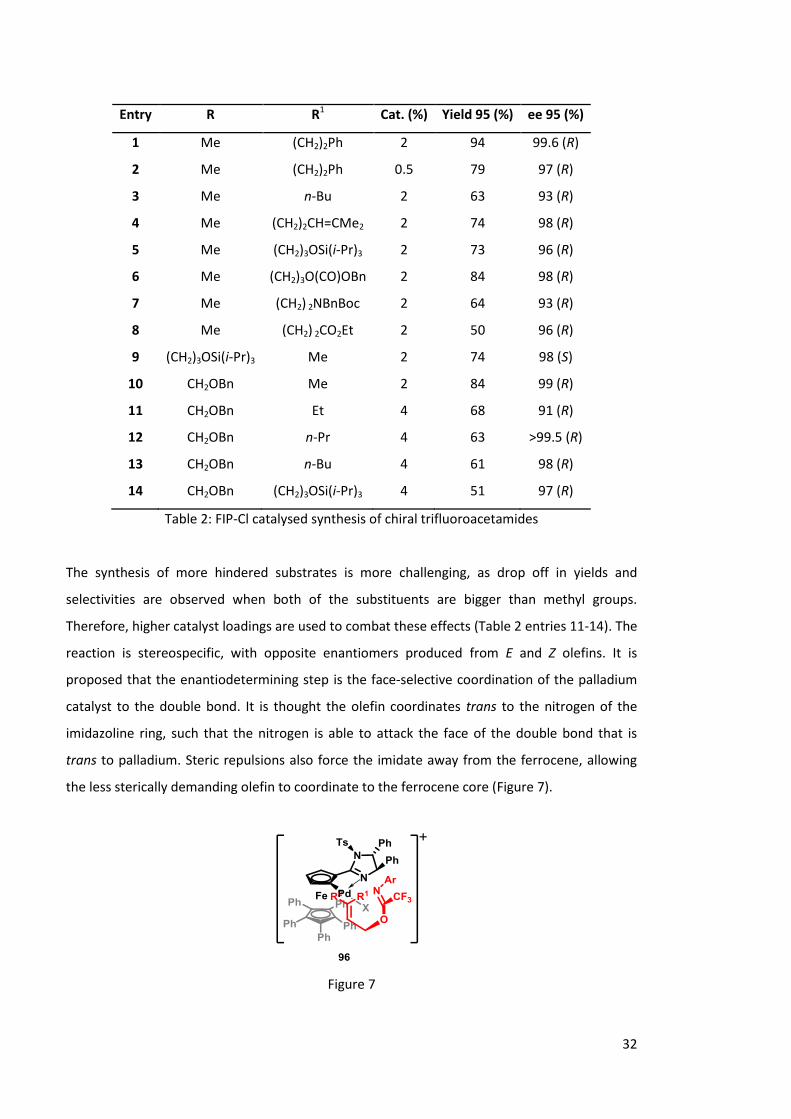
32
Entry R R1 Cat. (%) Yield 95 (%) ee 95 (%)
1 Me (CH2)2Ph 2 94 99.6 (R)
2 Me (CH2)2Ph 0.5 79 97 (R)
3 Me n-Bu 2 63 93 (R)
4 Me (CH2)2CH=CMe2 2 74 98 (R)
5 Me (CH2)3OSi(i-Pr)3 2 73 96 (R)
6 Me (CH2)3O(CO)OBn 2 84 98 (R)
7 Me (CH2) 2NBnBoc 2 64 93 (R)
8 Me (CH2) 2CO2Et 2 50 96 (R)
9 (CH2)3OSi(i-Pr)3 Me 2 74 98 (S)
10 CH2OBn Me 2 84 99 (R)
11 CH2OBn Et 4 68 91 (R)
12 CH2OBn n-Pr 4 63 >99.5 (R)
13 CH2OBn n-Bu 4 61 98 (R)
14 CH2OBn (CH2)3OSi(i-Pr)3 4 51 97 (R)
Table 2: FIP-Cl catalysed synthesis of chiral trifluoroacetamides
The synthesis of more hindered substrates is more challenging, as drop off in yields and
selectivities are observed when both of the substituents are bigger than methyl groups.
Therefore, higher catalyst loadings are used to combat these effects (Table 2 entries 11-14). The
reaction is stereospecific, with opposite enantiomers produced from E and Z olefins. It is
proposed that the enantiodetermining step is the face-selective coordination of the palladium
catalyst to the double bond. It is thought the olefin coordinates trans to the nitrogen of the
imidazoline ring, such that the nitrogen is able to attack the face of the double bond that is
trans to palladium. Steric repulsions also force the imidate away from the ferrocene, allowing
the less sterically demanding olefin to coordinate to the ferrocene core (Figure 7).
Figure 7

33
1.3.6 Lithiation of pyrrolidine and piperidine analogues
The formation of quaternary stereocenters by the lithiation of cyclic amine precursors has been
well studied.42,43 O’Brien and Coldham et al. have recently used in situ IR spectroscopy to
monitor the lithiation of N-Boc-2-phenylpyrrolidine 97 and –piperidine 100 for the development
of optimum conditions for the stereospecific lithiation and electrophilic quenching of these
substrates (Scheme 34).42
Scheme 34: Lithiation-substitution of N-Boc-2-phenylpyrrolidines and -piperidines
Two factors had to be taken into consideration in order for this to be a synthetically useful
transformation. First, as the lithiation of 97 and 100 is known to be directed by the carbonyl of
the Boc group,44,45 slow rotation of this group would adversely affect the yield of the reaction.
Second, the organolithium intermediates 98 and 101 have to be configurationally stable on the
timescale of the reaction.46
Use of in situ IR spectroscopy showed that the rotation of the Boc group was found to be
significantly slower in lithiated 97 when compared to 100, as treatment of 97 at −78 °C led to
approximately only 40% lithiation, as determined by in situ IR and Variable Temperature 1H
NMR spectroscopy. However, complete lithiation of 97 was observed at 0 °C. This is in contrast
to the lithiation of 100, which is complete within two minutes, even at −78 °C.
The configurational stability of 97 was determined through a series of experiments at various
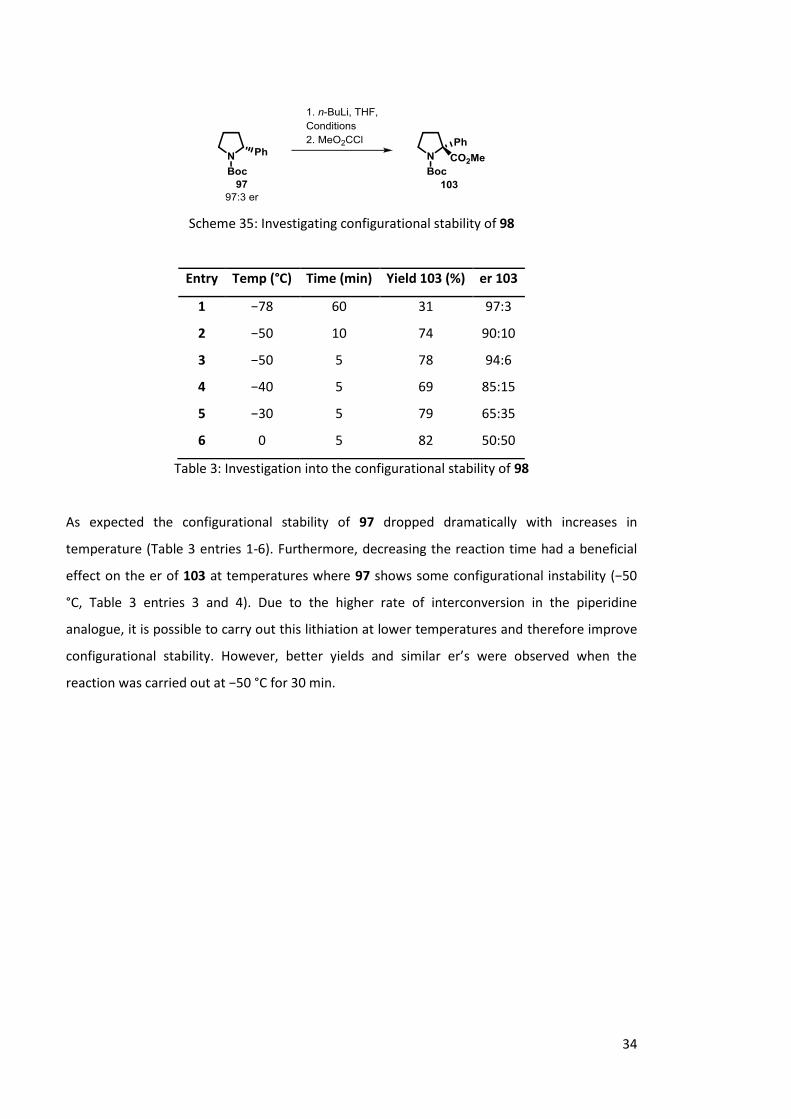
temperatures and reaction times (Scheme 35, Table 3).
34
Scheme 35: Investigating configurational stability of 98
Entry Temp (°C) Time (min) Yield 103 (%) er 103
1 −78 60 31 97:3
2 −50 10 74 90:10
3 −50 5 78 94:6
4 −40 5 69 85:15
5 −30 5 79 65:35
6 0 5 82 50:50
Table 3: Investigation into the configurational stability of 98
As expected the configurational stability of 97 dropped dramatically with increases in
temperature (Table 3 entries 1-6). Furthermore, decreasing the reaction time had a beneficial
effect on the er of 103 at temperatures where 97 shows some configurational instability (−50
°C, Table 3 entries 3 and 4). Due to the higher rate of interconversion in the piperidine
analogue, it is possible to carry out this lithiation at lower temperatures and therefore improve
configurational stability. However, better yields and similar er’s were observed when the
reaction was carried out at −50 °C for 30 min.

35
With optimised conditions in hand, a range of electrophiles were investigated (Scheme 36). In
each case the 2,2-disubstituted heterocycles were obtained in good yield and excellent ers.
Scheme 36: Lithiation-substitution of N-Boc-2-phenylpyrrolidines and -piperidines
Tertiary benzylic organolithiums often suffer from poor configurational stability, with the free
energy barrier to inversion of configuration generally found to be in the range of 9-14 kcal/mol
(t½ ≤ 10 min) at temperatures between −80 and +25 °C.47 With such low barriers, the use of
tertiary benzylic organolithiums in the synthesis of quaternary stereocenters is extremely
challenging. However, this problem can be overcome if the electrophilic trap is intramolecular
(See Section 2.1 and 3.1). To compensate for these high levels of configurational instability,
chiral auxiliaries48 or ligands49-51 are often used during lithiation to give a greater degree of
stability in the ground state or generate a diastereomeric bias in the transition state
respectively. Building on this, Gawley et al. have investigated the configurational stability of
lithiated 2-arylpyrrolidines and –piperidines.43
N-Boc-2-phenylpiperidine 100 was treated with 1 equivalent of s-BuLi in Et2O or THF at −80 °C in
the presence or absence of TMEDA to generate 101, which was immediately trapped with
MeOD (Scheme 37, Table 4).

36
Scheme 37: Investigating the configurational stability of 101
Entry Solvent Ligand (equiv.) er 106 (R:S)
1 Et2O TMEDA (1) 96:4
2 Et2O TMEDA (4) 96:4
3a Et2O TMEDA (4) 85:15
4 Et2O Noneb 91:9
5 THF TMEDA (1) 94:6
6 THF TMEDA (4) 95:5
7 THF Noneb 92:8
Table 4: Investigating the configurational stability of 101. a Transferred to a bath at −55 °C and
stirred for 1 h. b Lithiated for 1 h.
The organolithium intermediate 101 was found to racemise slowly in both solvents at −80 °C in
the absence of TMEDA, with TMEDA slightly improving its configurational stability. A slight loss
of enantiopurity was found when using THF when compared to Et2O. Furthermore, 101 was
found to be less stable with an increase in temperature of the reaction. A range of other 2-
arylpiperidines and electrophiles have since been studied, and shown to be configurationally
stable, with the corresponding products formed in good yield and with excellent retention of
configuration (Scheme 38, Table 5).
Scheme 38: Lithiation/substitution of enantioenriched 107
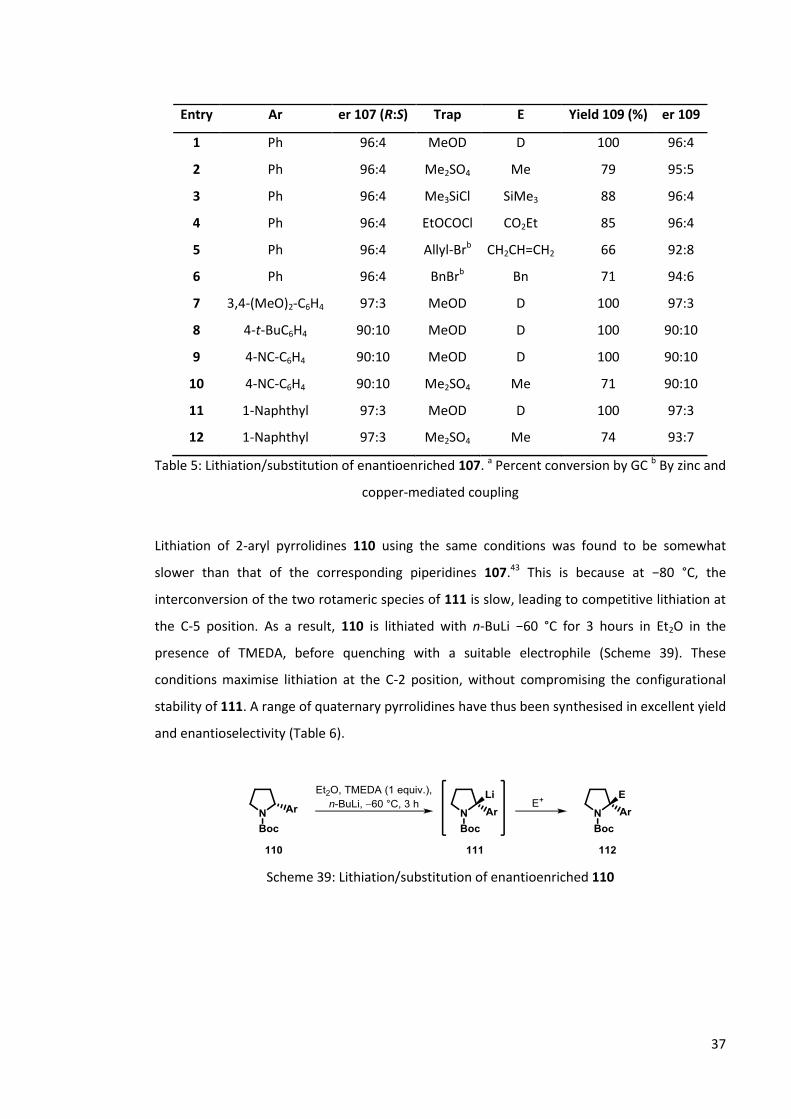
37
Entry Ar er 107 (R:S) Trap E Yield 109 (%) er 109
1 Ph 96:4 MeOD D 100 96:4
2 Ph 96:4 Me2SO4 Me 79 95:5
3 Ph 96:4 Me3SiCl SiMe3 88 96:4
4 Ph 96:4 EtOCOCl CO2Et 85 96:4
5 Ph 96:4 Allyl-Brb CH2CH=CH2 66 92:8
6 Ph 96:4 BnBrb Bn 71 94:6
7 3,4-(MeO)2-C6H4 97:3 MeOD D 100 97:3
8 4-t-BuC6H4 90:10 MeOD D 100 90:10
9 4-NC-C6H4 90:10 MeOD D 100 90:10
10 4-NC-C6H4 90:10 Me2SO4 Me 71 90:10
11 1-Naphthyl 97:3 MeOD D 100 97:3
12 1-Naphthyl 97:3 Me2SO4 Me 74 93:7
Table 5: Lithiation/substitution of enantioenriched 107. a Percent conversion by GC b By zinc and
copper-mediated coupling
Lithiation of 2-aryl pyrrolidines 110 using the same conditions was found to be somewhat
slower than that of the corresponding piperidines 107.43 This is because at −80 °C, the
interconversion of the two rotameric species of 111 is slow, leading to competitive lithiation at
the C-5 position. As a result, 110 is lithiated with n-BuLi −60 °C for 3 hours in Et2O in the
presence of TMEDA, before quenching with a suitable electrophile (Scheme 39). These
conditions maximise lithiation at the C-2 position, without compromising the configurational
stability of 111. A range of quaternary pyrrolidines have thus been synthesised in excellent yield
and enantioselectivity (Table 6).
Scheme 39: Lithiation/substitution of enantioenriched 110

38
Entry Ar er 110 (R:S) Trap E Yield 112 (%) er 112
1 Ph 96:4 MeOD D 100a,b 96:4
2 Ph 96:4 MeOD D 100a 96:4
3 Ph 96:4 Me2SO4 Me 86c 94:6
4 Ph 96:4 DMF CHO 83d (88a) >99:1f
5 Ph 96:4 EtOCOCl CO2Et 70d (79a) 94:6
6 Ph 96:4 2-Bromotoluene 2-MeC6H4 8d,e 92:8
7 Ph 50:50 2-Bromotoluene 2-MeC6H4 12b,c,e 50:50
8 2-MeC6H4 90:10 MeOD D 100a 90:10
9 2-Pyridyl 90:10 MeOD D 100a 90:10
10 1-Naphthyl 95:5 MeOD D 100a 95:5
11 1-Naphthyl 95:5 Me2SO4 Me 91c 95:5
12 1-Naphthyl 95:5 PhBr Ph <5a,b,e 95:5
13 4-NC-C6H4 7:93 MeOD D 100a 93:7
Table 6: Lithiation/substitution of enantioenriched 110. a Percent conversion by GC b Lithiated in
the absence of TMEDA c Isolated yield d Isolated yield of C-2 and C-5 substitution products after
deprotonation by s-BuLi/TMEDA e Via Pd-catalysed coupling f Increase in er possibly due to self-
disproportionation of enantiomers on the achiral stationary phase52
1.4 Carbolithiation
Carbolithiation is the formation of a C-C bond from the reaction of a C=C double bond with an
organolithium reagent.53 Organolithiums are one of the most versatile reagents in chemistry,
due to the strong polarisation of the carbon-lithium bond. This leads to their use as strong
bases as well as highly reactive nucleophiles.54 It is possible to carry out carbolithiations both
inter- and intramolecularly, however only intermolecular reactions will be discussed in this
thesis. For more information on intramolecular carbolithiation see a recent review on the
subject.55

39
Cason and Brooks published one of the first examples of carbolithiation in 1952.56 They treated
triphenylvinylsilane 113 with phenyl lithium to produce phenethyltriphenylsilane 115 (R = Ph,
Scheme 40). In order to confirm their hypothesis, different organolithium reagents were tested
under the same reaction conditions, with addition of the organolithium to the double bond
found in each case. The reaction has since been well studied and can be carried out with many
different organolithiums on a variety of alkenes.57-60
Scheme 40: Carbolithiation of triphenylvinylsilane
The intermediate organolithium species produced as a result of carbolithiation reactions needs
to be stabilised either by conjugation or through coordination to a heteroatom in order to
prevent polymerisation (Scheme 41).61-65
Scheme 41: Stabilised organolithiums
For example, the organolithium species 123 formed as a result of carbolithiation of 122 is
stabilised by the formation of a 5-membered chelate to the oxygen atom present in 122
(Scheme 42).66
Scheme 42

40
The use of additives such as TMEDA is another common strategy to stabilise and increase the
reactivity of organolithium intermediates.67,68 Here, the ligand is able to chelate to the lithium
through the two nitrogens present within it (Scheme 43).
Scheme 43: Stabilisation of organolithiums through the use of TMEDA
Carbolithiation of trisubstituted double bonds is much more challenging and only a few
examples have been published.69 Benzopyran derivative 127 was treated with methyl lithium to
give the corresponding alkylated product 128 in 32% yield (Scheme 44). It was postulated that
the double bond in 127 was activated by the cyano-substituted aromatic ring, and that the
carbonyl group was key to assisting in the addition by coordination to the lithium atom.
Scheme 44: Carbolithiation of a tri-substituted double bond
The umpolung carbolithiation of 2,3-disubstituted ene-carbamates 129 has also been used as a
tool in the synthesis of N-heterocycles of various sizes.70 The cyclic ene-carbamates 129 were
easily accessed through a palladium catalysed Suzuki coupling.71 129 was then submitted to a
small range of organolithium reagents to yield the alkylated products 131 in good to excellent
yields (Scheme 45). The carbolithiation was found to be regioselective to the C-3 position of
129, whilst the allylic protons were not affected by the addition. However, the reaction showed
no diastereoselectivity.

41
Scheme 45: Carbolithiation of ene-carbamates
The anionic intermediate species 130 can be quenched with a variety of electrophiles in order
to obtain differently substituted azepino-derivatives. 2,3-Disubstituted ene-carbamates 133
were therefore synthesised by bromination of 130, before dehydrobromination in a one-pot
sequence (Scheme 46). The carbolithiation was carried out with an excess of the organolithium
reagent to act as a base in order to facilitate the elimination of hydrogen bromide; thereby
giving the desired 2,3-disubstituted ene-carbamates 133 in moderate to good yields.
Scheme 46: Synthesis of 2,3-disubstituted ene-carbamates
1.4.1 Stereoselective Carbolithiation
It is often found that when the organolithium species produced after carbolithiation is stabilised
by coordination to a heteroatom, that the addition is stereoselective if this heteroatom is
adjacent to an existing stereocenter. For example, the stereochemistry of the product obtained
from carbolithiation of 134 can be predicted by the transition state 135 (Scheme 47). The syn
product 137 is obtained as a single diastereomer, in excellent yield.72
Scheme 47: Diastereoselective carbolithiation of vinyl alcohol

42
A similar result is found in the carbolithiation of cinnamyl alcohol 138, which preferentially
forms the syn diastereomer 140 or 141 after quenching with different electrophiles (Scheme
48).73,74 Furthermore, both the E- and Z-isomers of cinnamyl alcohol form the syn diastereomer,
with excellent diastereoselectivity (de = 96%). In this case it appears that the syn intermediate
139 is thermodynamically more stable than the corresponding anti intermediate (Scheme 49).
Scheme 48: Diastereoselective carbolithiation of cinnamyl alcohol
Scheme 49: Proposed transition state for syn-carbolithiation
Interestingly, when the reaction was carried out using the phenylthio analogue 143 of cinnamyl
alcohol, the opposite diastereoselectivity was observed and the anti product 145 was obtained
(Scheme 50).74 It is proposed that this reactivity is due to 144 taking on a pyramidal structure
146, with both lithium ions coordinated on the same face of the molecule. This is followed by a
retentive quench, as one would expect for an organolithium at a sp3 carbon, to give the anti-
product 147 (Figure 8).74,75
Scheme 50: Diastereoselective carbolithiation of phenylthio-cinnamyl alcohol

43
Figure 8: Transition state of the carbolithiation of cinnamyl and phenylthio-cinnamyl alcohol
This anti diastereoselectivity was also observed with other derivatives of cinnamyl alcohol such
as cinnamyl ether 148 and amine 150 (Scheme 51).76,77
Scheme 51
Normant also compared the effects of altering the geometry of the double bond of amine 150.
Switching the geometry of the olefin from E to Z had no effect on the stereochemistry of the
product, with anti-amine 151 isolated on both occasions. This is due to both starting materials
sharing the common intermediate, 152, which is stabilised by coordination of the lithium to
nitrogen forming a 5-membered ring (Scheme 52).76
Scheme 52: Carbolithiation of E and Z cinnamyl amine 150

44
1.4.2 Enantioselective Carbolithiation
Carbolithiations can be made enantioselective through the use of chiral ligands in order to
discriminate between the two faces of the olefin, the most common of which is the diamine (−)-
sparteine 153 (Figure 9). However, this enantiofacial discrimination is particularly difficult for
non-activated alkenes, as a result, only a few examples have been reported.78
Figure 9: (−)-Sparteine 153
(−)-Sparteine 153 is a strong activator of organolithiums and can be used to promote
carbolithiation of unfunctionalised styrenic double bonds, such as that found in 154 in good
yield and enantioselectivity (Scheme 53 Table 7).79 It was also possible to use (−)-sparteine 153
catalytically in these systems (Table 7 entry 4), but lower ee’s were obtained.
Scheme 53: Carbolithiation of 154
Entry RLi Yield 156 (%) ee 156 (%)
1 n-Bu 83 85
2 n-Pr 92 76
3 n-Hex 86 84
4 n-Bu 83 70a
Table 7: Carbolithiation of 154 a using 10 mol% of (−)-sparteine used
Normant et al. have also demonstrated the use of asymmetric carbolithiation in the kinetic
resolution of -ethylstyrene 157.79 Treatment of both isomers of 157 (9:1 E:Z) with n-BuLi in the
presence of (−)-sparteine 153 results in the formation of carbolithiated product 158 in excellent
yield, whilst Z-157 is recovered in 7% yield. Both isomers of 157 are able to undergo
carbolithiation; however Z-157 reacts much more slowly and with a lower ee (Scheme 54).
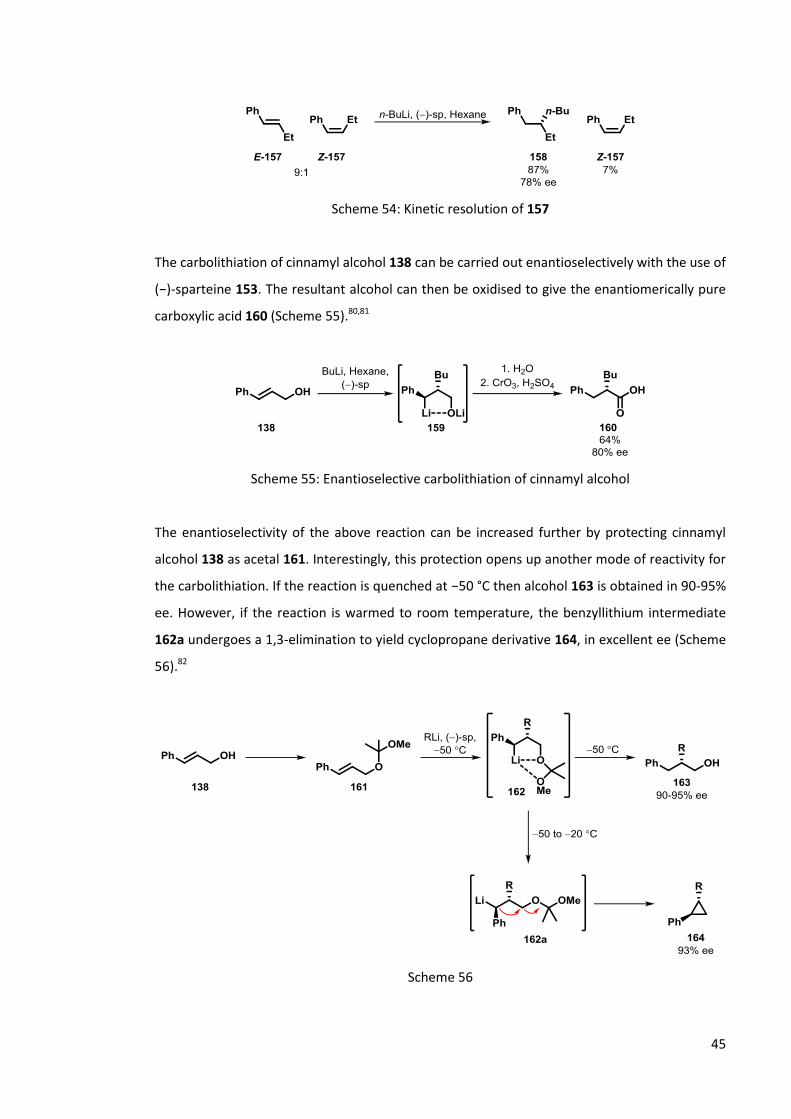
45
Scheme 54: Kinetic resolution of 157
The carbolithiation of cinnamyl alcohol 138 can be carried out enantioselectively with the use of
(−)-sparteine 153. The resultant alcohol can then be oxidised to give the enantiomerically pure
carboxylic acid 160 (Scheme 55).80,81
Scheme 55: Enantioselective carbolithiation of cinnamyl alcohol
The enantioselectivity of the above reaction can be increased further by protecting cinnamyl
alcohol 138 as acetal 161. Interestingly, this protection opens up another mode of reactivity for
the carbolithiation. If the reaction is quenched at −50 °C then alcohol 163 is obtained in 90-95%
ee. However, if the reaction is warmed to room temperature, the benzyllithium intermediate
162a undergoes a 1,3-elimination to yield cyclopropane derivative 164, in excellent ee (Scheme
56).82
Scheme 56

46
Subsequent studies have focused on the carbolithiation of unactivated conjugated alcohols,
which lack the extra stabilisation of the aromatic group, albeit with limited success (Scheme
57).83 Reacting alcohol 165 with n-BuLi and (−)-sparteine 153 only led to a regioisomeric mixture
of Z-167 and E-167. 167 is formed by the addition of n-BuLi to the terminal olefin followed by
subsequent elimination of Li2O to form diene 166. 166 can then undergo a second
carbolithiation with n-BuLi to give 167 after protonation.
Scheme 57: Carbolithiation of unactivated diene 165
Protection of the alcohol as acetal 168 one more opens up a potential 1,3-elimination of the
benzyllithium intermediate to yield vinylcyclopropanes 170.84 It is also possible to carry out this
transformation with sub-stoichiometric amounts of (−)-sparteine 153 to give 170 in moderate
to good ee’s (Scheme 58).
Scheme 58: Asymmetric carbolithiation of unactivated diene 168
By turning the terminal olefin in 165 into a trisubstituted one, its reactivity can be attenuated
and only carbolithiation at the C-2 position is found (Scheme 59).83 However, protonation of
this substrate is not regioselective. Subsequent hydrogenation of the remaining olefin after
carbolithiation results in the formation of 173 in good yield and moderate ee.

47
Scheme 59: Carbolithiation of 171
Substituted styrene derivatives also undergo efficient enantioselective carbolithiation in the
presence of (−)-sparteine 153. However in order for good ee’s to be obtained, there must be
electron donating substituents in the ortho- or para-position of the aromatic ring. This
deactivates the double bond towards organolithium addition and prevents polymerisation from
occurring. For example, styrene derivative 174 undergoes effective enantioselective
carbolithiation with trapping of the organolithium intermediate with a range of electrophiles.
This is followed by in situ ring closure and dehydration to yield indole 176 in excellent ee’s
(Scheme 60). Varying the electrophile used allows for a range of different functional groups to
be introduced at the C-2 position of 176.85,86
Scheme 60: Asymmetric carbolithiation of styrene derivatives

48
-Carbamoyloxy-substituted styrenes 177 also undergo asymmetric carbolithiation, but with
limited enantiofacial discrimination when compared to 174. A range of different diamine
ligands have been trialled to improve the enantiofacial discrimination, with the best results
found with (−)--isosparteine 181 (Scheme 61).67 The enantiofacial discrimination can be
explained by the coordination of the organolithium to the diamine to generate two conformers
of the benzyllithium intermediate 179 upon carbolithiation. 179 is configurationally stable on
the timescale of the reaction, and can be trapped. The low ee’s obtained from the reaction are
believed to be a result of the interconversion of the two conformers 178a and 178b being too
slow and that the energetic barrier of this interconversion is of a similar size to the activation
energy of the competing diastereomeric carbolithiation.
Scheme 61: Asymmetric carbolithiation of vinyl carbamates 177
Ferrocene derivatives 188 can also be synthesised through the asymmetric carbolithiation of 6-
(N,N-dimethylamino)fulvene 186 with aryllithiums in the presence of (−)-sparteine 153 (Scheme
62).87 Reaction of 186 with an aryllithium, generated from lithium and the corresponding aryl
bromide, produces chiral cyclopentadienyllithium 187, which is easily transformed to 188
through treatment with either FeCl2 or Fe(acac)2. Enantioselectivities for the reaction are
excellent, with greater than 99% ee found when two chiral side chains are introduced, one on
each cyclopentadienyl ring. 188 are themselves valuable synthetic intermediates and are often
used in diastereoselective ortho-lithiation reactions.88

49
Scheme 62: Asymmetric carbolithiation in the synthesis of ferrocene derivatives 186
1.4.3 Sparteine analogues
Sparteine is only readily available as one isomer. As a result, many different groups have tried
to synthesise (+)-sparteine, with the first asymmetric synthesis published by Aubé et al. in 2002
(Scheme 63).89 The synthesis was completed in 15 steps starting from 2,5-norbornadione, with
an overall yield of 16%. The two key steps of the synthesis were two nitrogen ring expansions,
an intramolecular Schmidt reaction and a variant of the photo-Beckmann rearrangement.
Scheme 63: Asymmetric synthesis of (+)-sparteine

50
Many (+)-sparteine analogues have also been synthesised, with the (+)-sparteine surrogate 195
developed by the O’Brien group now regarded as the best alternative to (+)-sparteine 192. 195
is easily synthesised in four steps from Laburnum anagyroides cytisus seeds (Scheme 64).90,91
Extraction of the Laburnum anagyroides cytisus seeds yields (−)-cytisine 193 which is
subsequently N-protected as its methyl carbamate derivative 194. Diastereoselective
hydrogenation of the pyridone moiety followed by reduction of the carbamate generates the
(+)-sparteine surrogate 195.
Scheme 64: Synthesis of (+)-sparteine surrogate 195
Many promising results have been obtained using (+)-sparteine surrogate 195 in
enantioselective carbolithiations and deprotonations instead of (−)-sparteine 153.92 Treatment
of N-Boc-pyrrolidine 196 with s-BuLi in the presence of (−)-sparteine 153, followed by reaction
with chlorotrimethylsilane, yields (S)-197 in excellent enantioselectivity. Carrying out the
reaction with (+)-sparteine surrogate 195 instead of (−)-sparteine 153 generates the opposite
enantiomer of the product, (R)-197, with no loss in enantioselectivity (Scheme 65).90
Scheme 65: Comparison of (−)-sparteine 153 and (+)-sparteine surrogate 195
The relative rates of lithiation of 196 with both (−)-sparteine 153 and (+)-sparteine surrogate
195 have been studied through a competition experiment (Scheme 66).93 Treatment of 196
with 2.6 equivalents of s-BuLi and 1.3 equivalents of each of (−)-sparteine 153 and (+)-sparteine
surrogate 195 followed by reaction with chlorotrimethylsilane generates (R)-197 with the same
sense of induction as that shown by the surrogate. This indicates that the s-BuLi-195 complex
lithiates 196 faster than the s-BuLi-153 complex.

51
Scheme 66: Competition experiment
(+)-Sparteine surrogate 195 has also been utilised within the total synthesis of (−)-kainic acid
(Scheme 67).94 Lithiation-carboxylation of N-Boc pyrrolidine 198 was originally trialled using s-
BuLi in THF at −78 °C, however this led to poor regio- and stereocontrol after electrophilic
trapping with carbon dioxide. Addition of (+)-sparteine surrogate 195 to the reaction did
however yield a 4:1 regioisomeric mixture in favour of the desired product. Both 199A and
199B were obtained as single diastereomers. Although this mixture was inseparable, the methyl
ester derivative 200 could be isolated as a single stereo- and regioisomer which was then used
to complete the synthesis.
Scheme 67: (+)-Sparteine surrogate 195 in the total synthesis of (−)-kainic acid

52
The use of (+)-sparteine surrogate 195 is not limited to asymmetric deprotonation reactions.
The benzylic lithiation and trapping of N-pivaloyl-ortho-anilide 202 is known to proceed by
dynamic thermodynamic resolution.95 As a result the dianion generated by treatment of 202
with excess s-BuLi is equilibrated in the presence of (−)-sparteine 153 at −25 °C. The mixture is
then rapidly cooled to −78 °C and trapped with chlorotrimethylsilane to yield (R)-203 in
excellent enantioselectivity. Use of (+)-sparteine surrogate 195 instead of (−)-sparteine 153
generates (S)-203 in similar er (Scheme 68).96
Scheme 68: Dynamic thermodynamic resolution of N-pivaloyl-ortho-anilide 202
(+)-Sparteine surrogate 195 has also been used in the asymmetric carbolithiation of cinnamyl
alcohols 138 with similar results to (−)-sparteine 153 (Scheme 69).97 Treatment of 138 with a n-
BuLi-195 complex in cumene at 0 °C generates (R)-205 in good yield and in 71% ee (c.f. 83% ee
is found when using (−)-sparteine 153, See Scheme 55). Use of (+)-sparteine surrogate 195
generates 205 with the opposite enantioselectivity when compared to (−)-sparteine 153.
Scheme 69: Asymmetric carbolithiation of cinnamyl alcohol using (+)-sparteine surrogate 195
(+)-Sparteine surrogate 195 is also utilised in a variety of other reactions that fall outwith the
scope of this thesis. For a more comprehensive analysis of the different reactions (+)-sparteine
surrogate 195 is used in, including asymmetric alkylations, dynamic resolution of lithiated
boranes, Grignard reactions, and the Pd(II)-mediated oxidative kinetic resolution of benzylic
alcohols, see a recent review on its use in synthesis.92

53
1.5 Organolithium Rearrangements
1.5.1 Brook Rearrangement
The 1,2-Brook rearrangement is an intramolecular [1,2]-anionic migration of a silyl group from
carbon to oxygen in the presence of a catalytic amount of base (Scheme 70).98 The Brook
rearrangement has since been applied to a range of silyl carbinol analogues such that a [1,n]-
migration is now known. The rearrangement is reversible, and this reverse process is termed
the retro-Brook rearrangement.
Scheme 70: [1,2]- and [1,n]-Brook and Retro-Brook rearrangements
The mechanism of the Brook rearrangement involves the formation of a cyclic pentavalent
silicon species 212 as a result of deprotonation of 210. Ring opening and protonation of the
carbanion either by the starting alcohol or its conjugate base produces the silyl enol ether 214
(Scheme 71).
Scheme 71: Mechanism of the Brook rearrangement
The driving force of the reaction is the generation of a stronger oxygen-silicon bond, in
comparison to a carbon-silicon bond. Furthermore, if R1 is an electron withdrawing substituent,
the carbanion is stabilised facilitating the kinetics of the reaction. The equilibrium can be
further shifted towards silyl ether formation by the use of polar solvents, such as THF, which
destabilises alkoxide 211. The retro-Brook process can be favoured through the use of a
stoichiometric amount of base and counterions that form strong ion pairs with oxygen (for
example lithium).

54
1.5.2 Wittig Rearrangement
Wittig rearrangements are often used in the synthesis of -tertiary alcohols,99 and entails the
[1,2]-migration of an alkyl group from oxygen to carbon on treatment with base (Scheme 72).
Original mechanistic studies indicated that reaction proceeded with formation of a radical
pair.100,101
Scheme 72: [1,2]-Wittig rearrangement
More, recently a [2,3]-analogue of this rearrangement has been discovered, and has been
shown to be extremely useful for the synthesis of homoallyl alcohols (Scheme 73).102
Scheme 73: [2,3]-Wittig rearrangement
The nitrogen analogues of these rearrangements are also known, and are termed the [1,2]- and
[2,3]-aza-Wittig rearrangements respectively (Scheme 74).99 Eisch and Kovacs were the first to
study these rearrangements extensively and showed that these migrations are generally less
selective and much slower than their oxygen-based counterparts.103
Scheme 74: [2,3]-Aza-Wittig rearrangement
Much of the early work in this field utilised the relief of ring strain in cyclic systems to promote
the [2,3]-aza-Wittig rearrangement. For example, azetidinone 225 is opened to azepinone 226,
whist vinyl aziridines 227 are transformed to tetrahydropyridines 228 in excellent yield and
stereoselectivity (Scheme 75).104-110

55
Scheme 75
However, application of this methodology to acyclic systems remains challenging, limiting its
use for the synthesis of -tertiary amines. The Anderson group were the first to attempt such
transformations in 1995, utilising crotyl amine derivatives 229 (Scheme 76).109 Here, the
electron withdrawing Boc group is specifically included to promote the rearrangement through
stabilisation of the N-centred anion formed during the reaction. Rearranged amine 230 is
isolated in good yield but with minimal diastereoselectivity.
Scheme 76: [2,3]-Aza-Wittig rearrangement of 229
Good diastereoselectivities can be achieved however with the use of silylated olefins 231,
allowing the synthesis of -tertiary amines 232 (Scheme 77). The high diastereoselectivity is
thought to be governed by transition state 234, where the steric clash between the bulky silyl
group and the R groups favour the formation of 234A over 234B (Scheme 78). This forces the
largest of the R groups to have an anti relationship with the methyl group of the cis double
bond.111

56
Scheme 77: Synthesis of -tertiary amine 232 through [2,3]-aza-Wittig rearrangement
Scheme 78: Proposed transition states
As a result, the methyl esters 236 were also investigated and showed good diastereoselectivity.
A similar resulted was obtained for the isopropyl substituted esters, whilst the benzyl
substituted esters showed no stereoselectivity whatsoever (Scheme 79).111 Here the alkyl group
is considered the larger group based on A-value analysis.112
Scheme 79

57
More recent work has centred on an enantioselective variation of this reaction using
enantioenriched allyl amines, however with limited success.110 This is also the case for studies
using alkyllithium-sparteine complexes. This is thought to be due to the rate of epimerisation of
the organolithium intermediate being faster than that of the [2,3]-aza-Wittig rearrangement. As
a result more focus is being placed on the use of chiral auxiliaries and Lewis acids in order to
promote a sense of chiral induction.113,114 However, this has not yet been applied to the
synthesis of -tertiary amines.
This methodology has also since been applied to a [2,3]-aza-Wittig rearrangement that is
subsequently followed by an unusual 5-endo-trig cyclisation (Scheme 80).115 The substituted
proline derivatives 238 synthesised are isolated in good yields and as single diastereomers,
indicating that the minor diastereomer is unable to undergo the cyclisation step. No cyclisation
to the proline derivative was found if the -position was only mono-substituted (R1 = H), with
only the product from [2,3]-aza-Wittig rearrangement found. The geminal substitution pattern
is therefore key to promoting cyclisation, probably due to the Thorpe-Ingold effect.116
Scheme 80: Diastereoselective synthesis of substituted prolines

58
1.5.3 N to C Acyl migration
Rouden et al. published an unexpected N to C acyl migration in 2002.117 During investigations
into the use of (−)-cystisine derivative 239 as a chiral inductor in enantioselective alkylations,
the rearranged product 241 was obtained exclusively as opposed to the expected product 240
(Scheme 81).
Scheme 81: N to C acyl migration
The proposed mechanism involves deprotonation to nitrogen followed by intramolecular
cyclisation with the carbonyl of the other amide. Subsequent rearrangement generates the
anion 244 which can react with benzyl bromide to form the rearranged product 241 (Scheme
82).
Scheme 82: Proposed mechanism for N to C acyl migration

59
A similar rearrangement has been published by Coudert et al.,118 where a N to C migration of a
Boc group was found during investigations into the carbolithiation of acyclic ene-carbamates
255 (Scheme 83). The reaction proceeds in moderate to good yields, with a variety of
commercial organolithiums and with substrates bearing different aromatic substituents.
Scheme 83: N to C migration of the Boc group
The reaction proceeds by umpolung carbolithiation of the ene-carbamate 255, followed by
rearrangement upon warming of the reaction mixture from −78 to 0 °C. Quenching of the
mixture generates the -tertiary -amino esters 256 (Scheme 84).
Scheme 84: Mechanism of the Boc migration

60
Chapter 2 Past work and aims of the project
2.1 Previous Work
2.1.1 Rearrangement of benzylic ureas
During investigations within the Clayden group in probing the site of lithiation of benzylic ureas
257 (Figure 10), a unique aryl migration reaction was discovered.119 257 underwent benzylic
lithiation, which was followed by N to C transfer of the distal aryl group. Subsequent quenching
with iodomethane yielded alkylated urea 258 (Scheme 85). However, this product was unstable
and isolated in poor yield. Replacement of the iodomethane quench with aqueous ammonium
chloride resulted in urea 259 being isolated in excellent yield (Scheme 86).
Figure 10: Possible sites of lithiation of benzylic ureas
Scheme 85: Rearrangement of 257
Scheme 86: Rearrangement of 257
61
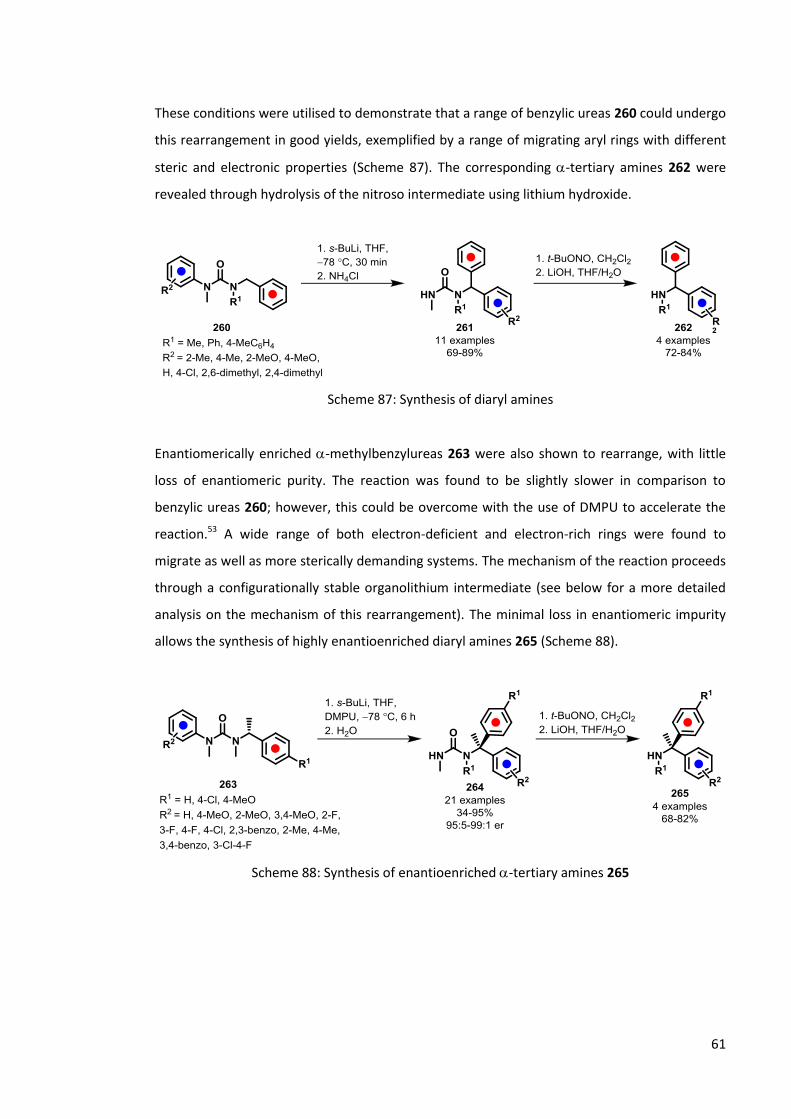
These conditions were utilised to demonstrate that a range of benzylic ureas 260 could undergo
this rearrangement in good yields, exemplified by a range of migrating aryl rings with different
steric and electronic properties (Scheme 87). The corresponding -tertiary amines 262 were
revealed through hydrolysis of the nitroso intermediate using lithium hydroxide.
Scheme 87: Synthesis of diaryl amines
Enantiomerically enriched -methylbenzylureas 263 were also shown to rearrange, with little
loss of enantiomeric purity. The reaction was found to be slightly slower in comparison to
benzylic ureas 260; however, this could be overcome with the use of DMPU to accelerate the
reaction.53 A wide range of both electron-deficient and electron-rich rings were found to
migrate as well as more sterically demanding systems. The mechanism of the reaction proceeds
through a configurationally stable organolithium intermediate (see below for a more detailed
analysis on the mechanism of this rearrangement). The minimal loss in enantiomeric impurity
allows the synthesis of highly enantioenriched diaryl amines 265 (Scheme 88).
Scheme 88: Synthesis of enantioenriched -tertiary amines 265

62
It was originally postulated that the rearrangement proceeded through dearomatised
intermediate 267, which ring opened upon quenching (Scheme 89). 267 has been trapped out
under oxidative conditions to the spirocyclic enone 268 when the migrating group was a 1-
naphthyl ring. Crystallisation of 268 has proved that the aryl migration proceeds with retention
of configuration. However, it should be noted that this is the only case where a derivative of
267 is observed.
Scheme 89: Proposed reaction mechanism

63
The mechanism of this rearrangement has since been more extensively studied by in situ IR and
NMR spectroscopy.120 Benzylic urea 269 was dissolved in THF in an NMR tube and cooled to −78
°C before addition of s-BuLi and the solution immediately transferred to an NMR probe at −40
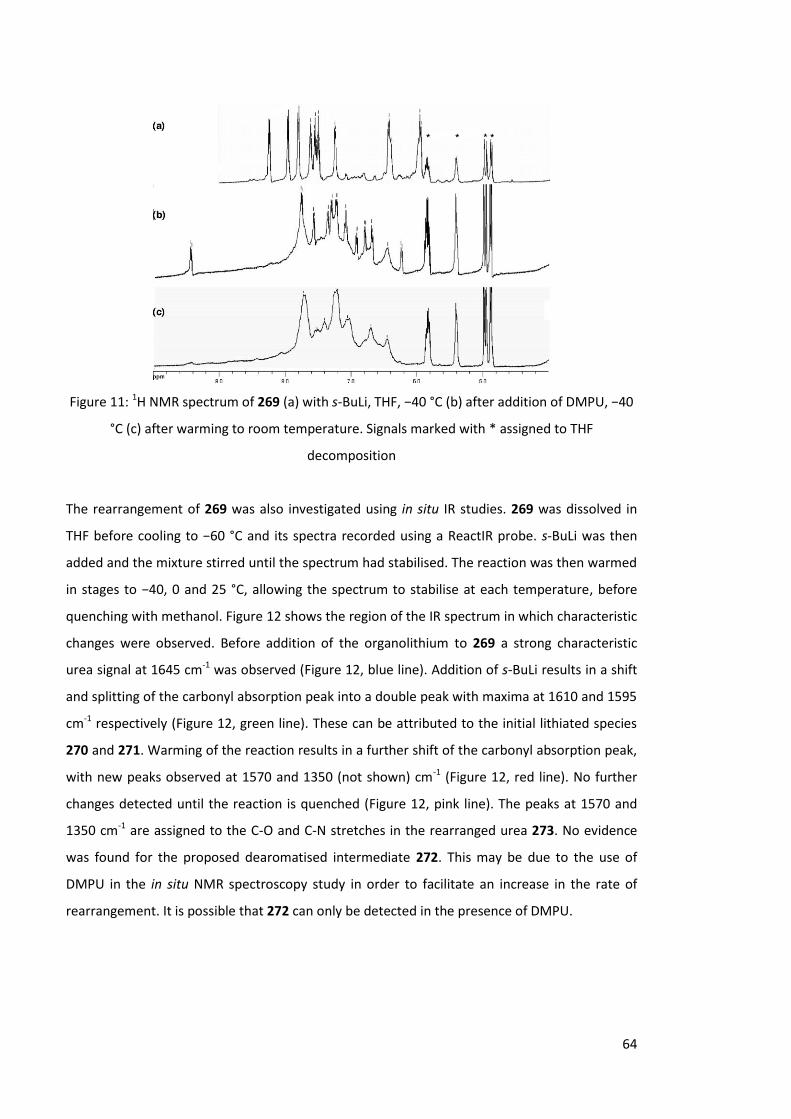
°C (Scheme 90). Figure 11a shows a characteristic shift upfield for the protons in the aromatic
region, indicating complete benzylic lithiation had taken place. Upon addition of DMPU to the
reaction mixture, eleven distinct sharp signals were observed between 6.2 and 9.5 ppm (Figure
11b). These signals are overlaid upon a much broader set of signals between 6.5 and 8.0 ppm.
Warming of the reaction mixture to room temperature resulted in the disappearance of the
sharp signals, with only the broad signals detected (Figure 11c). It is assumed that the broad
signals are due to lithiated product 273, with the sharp signals attributed to an intermediate en
route to 273 (presumably 272).
Scheme 90: Rearrangement of 269
64
Figure 11: 1H NMR spectrum of 269 (a) with s-BuLi, THF, −40 °C (b) after addition of DMPU, −40
°C (c) after warming to room temperature. Signals marked with * assigned to THF
decomposition
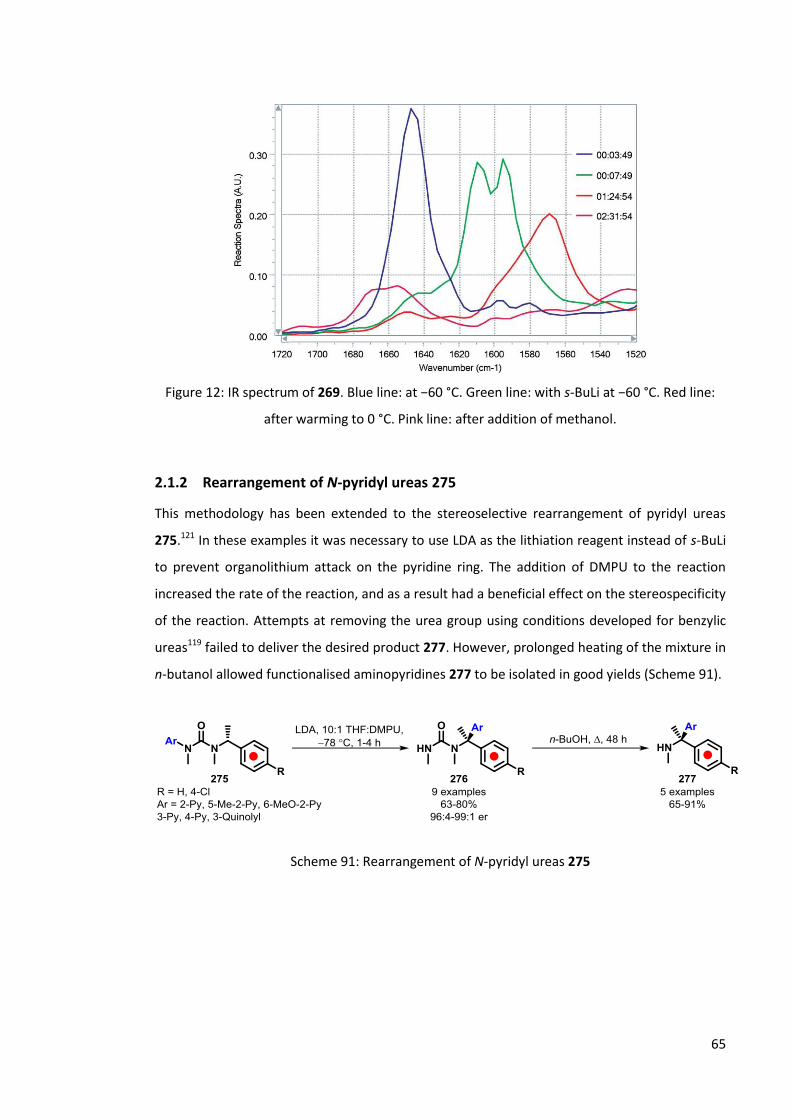
The rearrangement of 269 was also investigated using in situ IR studies. 269 was dissolved in
THF before cooling to −60 °C and its spectra recorded using a ReactIR probe. s-BuLi was then
added and the mixture stirred until the spectrum had stabilised. The reaction was then warmed
in stages to −40, 0 and 25 °C, allowing the spectrum to stabilise at each temperature, before
quenching with methanol. Figure 12 shows the region of the IR spectrum in which characteristic
changes were observed. Before addition of the organolithium to 269 a strong characteristic
urea signal at 1645 cm-1 was observed (Figure 12, blue line). Addition of s-BuLi results in a shift
and splitting of the carbonyl absorption peak into a double peak with maxima at 1610 and 1595
cm-1 respectively (Figure 12, green line). These can be attributed to the initial lithiated species
270 and 271. Warming of the reaction results in a further shift of the carbonyl absorption peak,
with new peaks observed at 1570 and 1350 (not shown) cm-1 (Figure 12, red line). No further
changes detected until the reaction is quenched (Figure 12, pink line). The peaks at 1570 and
1350 cm-1 are assigned to the C-O and C-N stretches in the rearranged urea 273. No evidence
was found for the proposed dearomatised intermediate 272. This may be due to the use of
DMPU in the in situ NMR spectroscopy study in order to facilitate an increase in the rate of
rearrangement. It is possible that 272 can only be detected in the presence of DMPU.
65
Figure 12: IR spectrum of 269. Blue line: at −60 °C. Green line: with s-BuLi at −60 °C. Red line:
after warming to 0 °C. Pink line: after addition of methanol.
2.1.2 Rearrangement of N-pyridyl ureas 275
This methodology has been extended to the stereoselective rearrangement of pyridyl ureas
275.121 In these examples it was necessary to use LDA as the lithiation reagent instead of s-BuLi
to prevent organolithium attack on the pyridine ring. The addition of DMPU to the reaction
increased the rate of the reaction, and as a result had a beneficial effect on the stereospecificity
of the reaction. Attempts at removing the urea group using conditions developed for benzylic
ureas119 failed to deliver the desired product 277. However, prolonged heating of the mixture in
n-butanol allowed functionalised aminopyridines 277 to be isolated in good yields (Scheme 91).
Scheme 91: Rearrangement of N-pyridyl ureas 275

66
2.1.3 Rearrangement of cyclic benzylic ureas
The scope of the rearrangement has also been broadened to include cyclic starting materials.122
Aryl migration was observed in -aryl pyrrolidines 278 with the quaternary arylated pyrrolidines
279 obtained in excellent yield (Scheme 92). This methodology was used in the synthesis of
nicotine regioisomer 280.
Scheme 92: Rearrangement of pyrrolidine derivatives
Substrates derived from tetrahydroisoquinoline 281 were also found to rearrange successfully.
Rearrangement of 282 under the standard lithiation conditions was found to proceed in only
37% yield. The yield was dramatically increased to 78% if the lithiation was carried out in the
absence of DMPU, with the migration promoted by an increase in the temperature of the
reaction. The methylated derivative 284 nonetheless underwent clean rearrangement with
DMPU as a co-solvent (Scheme 93).
Scheme 93: Rearrangement of tetrahydroisoquinoline derivatives

67
Carbocyclic substrates 286 with the nitrogen in an exocyclic benzylic position were also found
to successfully rearrange in good to excellent yields. The corresponding amines 288 were
obtained by refluxing the rearranged urea 287 in n-butanol for 48 hours (Scheme 94, Table 8).
Scheme 94: Rearrangement of carbocyclic substrates
Entry n R Lithiation reagent Yield 287 (%) Yield 288 (%)
1 2 H LDA 81 71
2 1 H s-BuLi 66 -
3 1 2,3-Benzo s-BuLi 70 -
4 1 2,6-dimethyl s-BuLi 78 -
5 1 4-Cl LDA 55 61
Table 8: Rearrangement of carbocyclic substrates
Rearrangement of the related acyclic ureas 263 was shown to be stereospecific, resulting in
enantiomerically enriched products. Unfortunately, no stereospecificity was found in the
rearrangement of cyclic starting materials 286. All reactions carried out with enantiomerically
enriched ureas resulted in racemic products. This can be explained by the rearrangement
competing poorly against racemisation of the organolithium intermediate, possibly a direct
result of the extended reaction times needed to ensure acceptable yields of the rearranged
ureas 287.

68
2.1.4 Double -arylation of N-allyl ureas 289
N-Allyl ureas 289 are also suitable substrates for the rearrangement, where lithiation with LDA
generates allyl lithium intermediate 290.123 Quenching of the reaction yields rearranged vinyl
urea 291, which is presumably generated by -protonation of the allylic anion. Although
theoretically possible, no migration of the aryl ring to the -position is observed. It is possible to
carry out a second -arylation in these systems once the N-aryl urea is reformed using a
Buchwald-Hartwig amination (Scheme 95). Solvolysis of the urea group in these systems by
standard methods119 leads to the isolation of diarylallyl ureas 294.
Scheme 95: Double -arylation of N-vinyl ureas
The second migration in this series could be made enantioselective through the use of chiral
lithium amides such as 295. This allows the construction of highly enantioenriched diarylallyl
ureas 296 (Scheme 96). The stereochemistry of these products is determined by the
stereoselective formation of a configurationally stable organolithium intermediate and not a
stereoselective reaction of an unstable intermediate.

69
Scheme 96: Enantioselective -arylation
2.1.5 Reversal of migration in N-diarylallyl ureas
Attempts at removing the urea group in rearranged ureas 293 by standard solvolysis
methods,119 failed to give the corresponding allylic amine 294. Instead, C to N migration was
found in quantitative yield (Scheme 97).124 This C to N migration is general for a range of
electron deficient aromatic rings, although it is possible to rearrange a phenyl ring if the other
ring is electron-rich. Almost complete conversion is found after a few minutes when sodium
hydride is used as the base. Electron-rich rings do not undergo C to N aryl migration.
Scheme 97: C to N aryl migration
2.2 N to C aryl migration in urea analogues
The N to C rearrangement is not limited to ureas; recent work within the group has shown that
this methodology can be applied to carbamates and thiocarbamates. This allows the synthesis
of -tertiary alcohols 297 and thiols 298 respectively (Figure 13).125,126
Figure 13:-Tertiary alcohols and thiols

70
2.2.1 -Arylation of benzylic carbamates
The lithiation of benzylic carbamates 299 has been investigated.125 As with their urea
counterparts 263, a wide range of aryl rings can be migrated, regardless of their electronic or
steric nature (Scheme 98).
Scheme 98: Aryl migration with benzylic carbamates
However, the benzyllithium intermediate formed during the course of the reaction is less
configurationally stable, when compared to the urea analogues 263. After lithiation of
enantioenriched -methyl benzyl carbamates such as 301, the er of the isolated products 302
were generally much lower than that of the corresponding urea analogues 263. This indicates
that the benzyllithium intermediate is racemising over the course of the reaction. The best
results were found when less co-ordinating solvents, such as Et2O, were used. Use of THF or
DMPU during the reaction led to complete racemisation of 301. In contrast to ureas,
carbamates undergo an N to C aryl migration with overall inversion of stereochemistry. This was
assigned based on comparison of optical rotation data with an authentic sample of 302.127
Further investigations are being carried out into why carbamates proceed with inversion of
configuration. This methodology has since been utilised in the first enantioselective synthesis of
the antihistamine agent clemastine, as its (S,S)-isomer (Scheme 99).128
Scheme 99: Synthesis of (S,S)-clemastine

71
2.2.2 -Arylation of benzylic thiocarbamates
Upon lithiation benzylic thiocarbamates 304 undergo the rearrangement reaction.126 Once
more; the benzyl lithium intermediate is less configurationally stable than in the urea analogues
263 (cf. carbamates). As a result lithiation with s-BuLi or LDA resulted in poor ers when
enantioenriched thiocarbamates were lithiated. However, replacing the lithiation agent with a
more bulky base such as LiTMP resulted in minimal loss of the enantiomeric purity of the
starting material. Unlike the carbamates however, thiocarbamates rearrange with retention of
configuration. The rearranged thiocarbamates 305 can be easily converted to the-tertiary
thiols 306 by cleavage with NaOEt in ethanol. It is also possible to carry out this solvolysis in situ
by quenching the rearrangement with methanol instead of propionic acid (Scheme 100).
Scheme 100: Synthesis of -tertiary thiols
2.3 Vinyl migration
More recent work within the Clayden group has shown that it is possible to migrate N-alkenyl
substituents as well as aromatic rings (Scheme 101).129 This method was found to be applicable
to ureas, carbamates and thiocarbamates.
Scheme 101: N-alkenyl migration. Conditions: For X = NMe, s-BuLi, THF:DMPU 10:1, −78 °C, 1 h.
For X = S, LDA, THF, −60 °C, 2 min. For X = O, LDA, THF:DMPU 4:1, −45 °C, 1-4 h.
72
The mechanism of this transformation has been studied more closely using in situ IR
experiments (Scheme 102). Upon dissolution of urea 309 in THF, the IR spectrum shows two
absorptions at 1660 cm-1 and 1620 cm-1, which rapidly disappear upon organolithium addition.
This results in a single peak at 1575 cm-1, which is characteristic of rearranged urea 313, the
same spectrum is recorded when product 314 is treated with s-BuLi). The migration was found
to be much slower in Et2O, allowing a more comprehensive analysis of the reaction pathway.
Addition of s-BuLi to 309 resulted in a short-lived intermediate 310 with an absorption peak at
1646 cm-1 which gave way to a second intermediate 311 with absorption peaks at 1608 cm-1 and
1593 cm-1. Slow warming of the reaction to room temperature resulted in the gradual
disappearance of the two peaks at 1608 cm-1 and 1593 cm-1 and the appearance of a peak at
1575 cm-1, already assigned to rearranged urea 313. No evidence was found for the formation
of a cyclic intermediate such as 312.
Scheme 102: In situ IR monitoring of vinyl migration
73
2.4 Carbolithiation-Rearrangement
2.4.1 Of ureas
During investigations into the rearrangement of N-vinyl urea 315, an unusual reactivity pattern
was discovered (Scheme 103).130 Instead of the expected allylic rearrangement to form 316,
carbolithiated product 317 was isolated in moderate yield.
Scheme 103: Carbolithiation of 315
It was subsequently found that prolonged reaction times or the addition of DMPU could be
used to initiate rearrangement in these systems. Initial studies focused on the tandem
carbolithiation-rearrangement of terminal N-vinyl ureas 318 (Scheme 104).131 The reaction was
found to be generally applicable, with a range of aryl rings and organolithiums partaking in the
reaction in good to excellent yields.
Scheme 104: Tandem carbolithiation-rearrangement of N-vinyl ureas 318
The scope of the reaction was broadened further to include ureas with trisubstituted double
bonds 320.131 The rearrangement was applicable to a wide range of substrates in good to
excellent yields with complete diastereospecificity (Scheme 105). As expected, inverting the
configuration of the double bond in the starting material or exchanging the two aryl rings
inverted the configuration of the product. Therefore, both the addition and rearrangement
steps are stereospecific. If the reaction is carried out in toluene in the absence of DMPU, the
carbolithiated product can be isolated, with no aryl migration observed.
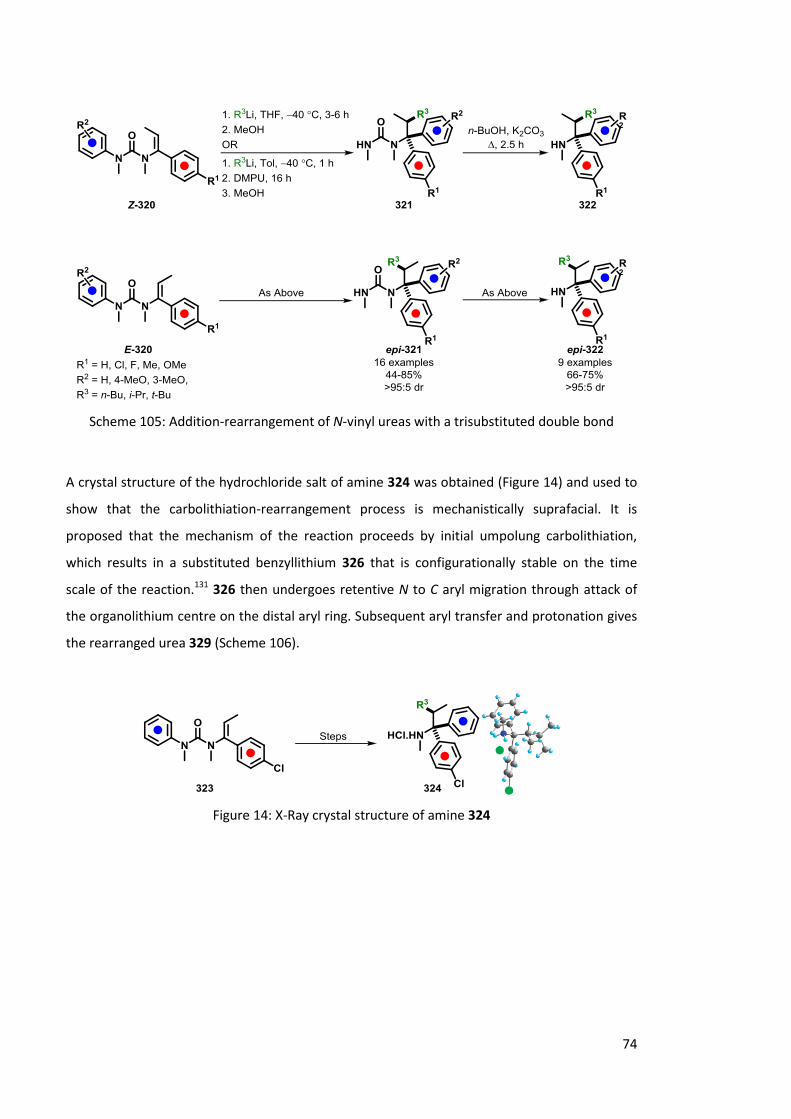
74
Scheme 105: Addition-rearrangement of N-vinyl ureas with a trisubstituted double bond
A crystal structure of the hydrochloride salt of amine 324 was obtained (Figure 14) and used to
show that the carbolithiation-rearrangement process is mechanistically suprafacial. It is
proposed that the mechanism of the reaction proceeds by initial umpolung carbolithiation,
which results in a substituted benzyllithium 326 that is configurationally stable on the time
scale of the reaction.131 326 then undergoes retentive N to C aryl migration through attack of
the organolithium centre on the distal aryl ring. Subsequent aryl transfer and protonation gives
the rearranged urea 329 (Scheme 106).
Figure 14: X-Ray crystal structure of amine 324
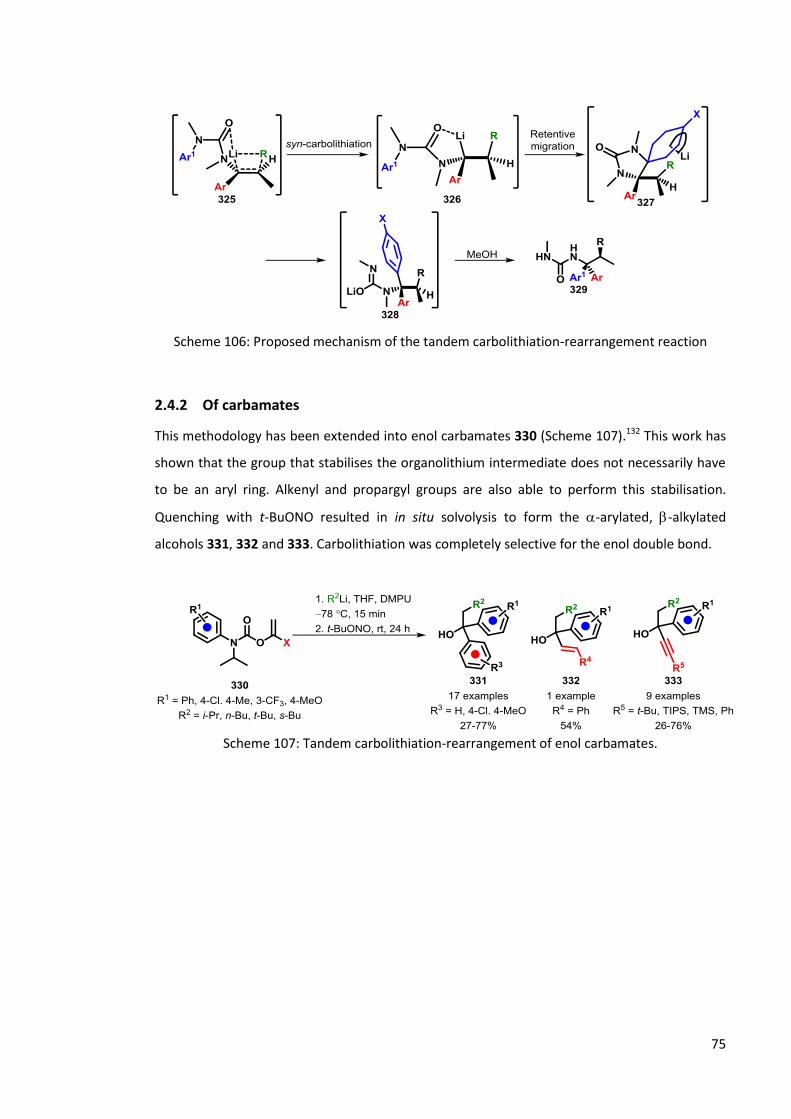
75
Scheme 106: Proposed mechanism of the tandem carbolithiation-rearrangement reaction
2.4.2 Of carbamates
This methodology has been extended into enol carbamates 330 (Scheme 107).132 This work has
shown that the group that stabilises the organolithium intermediate does not necessarily have
to be an aryl ring. Alkenyl and propargyl groups are also able to perform this stabilisation.
Quenching with t-BuONO resulted in in situ solvolysis to form the -arylated, -alkylated
alcohols 331, 332 and 333. Carbolithiation was completely selective for the enol double bond.
Scheme 107: Tandem carbolithiation-rearrangement of enol carbamates.

76
2.4.3 Of thiocarbamates
More recently, the tandem carbolithiation-rearrangement of S-alkenyl thiocarbamates has been
investigated (Scheme 108).133 Treatment of a range of thiocarbamates 334 with a variety of
organolithium reagents afforded the corresponding -arylated, -alkylated thiocarbamates
335, when the reaction was quenched with acid at low temperatures. These can then be easily
transformed into their corresponding thiol derivatives 336 in excellent yield.
Scheme 108: Tandem carbolithiation-rearrangement in S-alkenyl thiocarbamates.
2.5 Aims of the Project
The aim of the project is to develop an enantioselective version of the tandem carbolithiation-
rearrangement reaction of N-vinyl ureas 318 pioneered by the Clayden group. The effect of a
variety of chiral ligands on the reaction is to be investigated with a view to synthesising
enantiomerically pure -tertiary amines 337 (Scheme 109). The use of non-commercial
organolithium reagents is also to be investigated.
Scheme 109: Synthesis of enantiomerically pure -tertiary amines
Furthermore, it is hoped that the scope of the addition-migration can be expanded to include
cyclic substrates such as 338 and 341. This could potentially allow access towards synthetically
challenging cyclic -tertiary amines 340 and 343, a class of compounds which show interesting
pharmaceutical properties (Scheme 110).42 The diastereoselectivity of the reaction will also
need to be confirmed, such that the reaction still proceeds with syn-carbolithiation followed by
retentive aryl migration as found in the acyclic analogues 320.
77
Scheme 110: Tandem carbolithiation-rearrangement of cyclic substrates
In order to obtain the desired starting materials for investigations into the tandem
carbolithiation-rearrangement reaction, a simple and efficient synthesis has to be developed
that allows for the preparation of a wide range of substituted ureas.
Investigations will also centre on applying the N to C aryl transfer reaction to synthesise
pharmaceutically interesting targets.

78
Chapter 3 Enantioselective Carbolithiation-Rearrangement
As part of the tandem addition-migration methodology reported within the Clayden group, an
enantioselective variation has been investigated through the use of chiral ligands. If successful,
this would give access to enantiomerically enriched, hindered -tertiary amines 337 that would
be difficult to synthesise by any other method (Scheme 111).
Scheme 111: Synthesis of enantiomerically enriched -tertiary amines
3.1 Previous Work
3.1.1 Reaction optimisation
All work on the optimisation of reaction conditions using (−)-sparteine 153 was carried out by
Dr Alberto Minassi and Dr Morgan Donnard. All examples of applying this methodology using
(−)-sparteine 153 was also carried out by Dr Alberto Minassi and Dr Morgan Donnard, except
where noted.
Vinylic urea 344 was chosen for a series of trial reactions to determine if this enantioselective
reaction could take place (Scheme 112, Table 9). Rapid racemic carbolithiation was found when
344 was treated with n-BuLi in ethereal solvents at −78 °C. In Et2O only the carbolithiated
product 346 was found after one hour (Table 9 entry 1). However, in THF the reaction yielded a
mixture of 346 and the carbolithiated-rearranged product 348 (Table 9 entry 2). Only 35% of
346 was isolated when the reaction was carried out in toluene, indicating a much slower
reaction. Addition of the chiral ligand (−)-sparteine 153 in Et2O (Table 9 entry 4) and toluene
promoted the rearrangement, resulting in a mixture of 346 and 348. DMPU was added to
further promote the rearrangement.119,121-123,131 By adding DMPU after one hour of asymmetric
carbolithiation in the presence of (−)-sparteine 153 at −78 °C in toluene, 348 was isolated in
moderate yield but in low er. By delaying this DMPU addition to 6 hours the er could be
significantly improved (Table 9 entry 6), implying that the carbolithiation was incomplete at 1
hour, with DMPU promoting racemic carbolithiation of any unreacted starting material.

79
Scheme 112: Optimisation of solvent and conditions *
Conditions were required that would allow faster asymmetric carbolithiation to ensure the
reaction was complete before addition of DMPU, such that the possibility of any racemisation
or epimerisation of the organolithium intermediate could be avoided. A large excess of
(−)-sparteine 153 allowed a rapid asymmetric reaction to take place in toluene with an
encouraging er (Table 9 entry 7). Use of cumene or t-BuOMe allowed 348 to be obtained with
similar er, but without the need to use a large excess of chiral ligand, with cumene also giving a
better yield of 348. These solvents also allowed the reaction to take place at elevated
temperatures without any significant erosion of er, but with an increase in yield (Table 9 entries
9 and 10). This can be explained by the possible increased rate of racemisation being balanced
by an increase in the rate of asymmetric carbolithiation. These optimised conditions allowed
carbolithiation-rearrangement of 344 with i-PrLi in cumene in the presence of (−)-sparteine 153
to take place with excellent yield and er (Table 9 entry 12). The beneficial effects of cumene are
attributed to its resistance to deprotonation by the organolithium at temperatures sufficiently
high that still allow complete carbolithiation of the urea before DMPU addition. Furthermore,
as cumene is a non-coordinating solvent, the organolithium intermediate 345 has high
configurational stability. Unfortunately, using (−)-sparteine 153 in sub-stoichiometric amounts
was not possible without a statistical erosion to the er of the reaction (Table 9 entry 13).
* All carried out by Dr Alberto Minassi and Dr Morgan Donnard
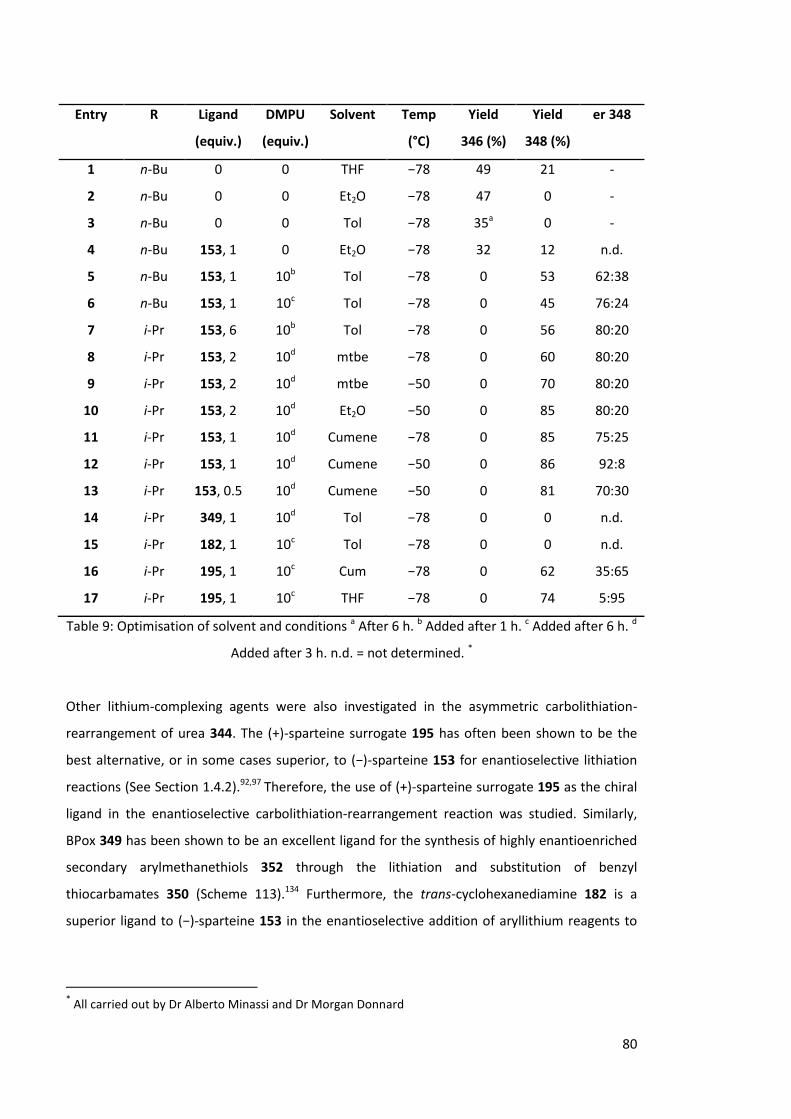
80
Entry R Ligand
(equiv.)
DMPU
(equiv.)
Solvent Temp
(°C)
Yield
346 (%)
Yield
348 (%)
er 348
1 n-Bu 0 0 THF −78 49 21 -
2 n-Bu 0 0 Et2O −78 47 0 -
3 n-Bu 0 0 Tol −78 35a 0 -
4 n-Bu 153, 1 0 Et2O −78 32 12 n.d.
5 n-Bu 153, 1 10b Tol −78 0 53 62:38
6 n-Bu 153, 1 10c Tol −78 0 45 76:24
7 i-Pr 153, 6 10b Tol −78 0 56 80:20
8 i-Pr 153, 2 10d mtbe −78 0 60 80:20
9 i-Pr 153, 2 10d mtbe −50 0 70 80:20
10 i-Pr 153, 2 10d Et2O −50 0 85 80:20
11 i-Pr 153, 1 10d Cumene −78 0 85 75:25
12 i-Pr 153, 1 10d Cumene −50 0 86 92:8
13 i-Pr 153, 0.5 10d Cumene −50 0 81 70:30
14 i-Pr 349, 1 10d Tol −78 0 0 n.d.
15 i-Pr 182, 1 10c Tol −78 0 0 n.d.
16 i-Pr 195, 1 10c Cum −78 0 62 35:65
17 i-Pr 195, 1 10c THF −78 0 74 5:95
Table 9: Optimisation of solvent and conditions a After 6 h. b Added after 1 h. c Added after 6 h. d
Added after 3 h. n.d. = not determined. *
Other lithium-complexing agents were also investigated in the asymmetric carbolithiation-
rearrangement of urea 344. The (+)-sparteine surrogate 195 has often been shown to be the
best alternative, or in some cases superior, to (−)-sparteine 153 for enantioselective lithiation
reactions (See Section 1.4.2).92,97 Therefore, the use of (+)-sparteine surrogate 195 as the chiral
ligand in the enantioselective carbolithiation-rearrangement reaction was studied. Similarly,
BPox 349 has been shown to be an excellent ligand for the synthesis of highly enantioenriched
secondary arylmethanethiols 352 through the lithiation and substitution of benzyl
thiocarbamates 350 (Scheme 113).134 Furthermore, the trans-cyclohexanediamine 182 is a
superior ligand to (−)-sparteine 153 in the enantioselective addition of aryllithium reagents to
* All carried out by Dr Alberto Minassi and Dr Morgan Donnard

81
aromatic imines 353 (Scheme 114)135 and its effects on the carbolithiation-rearrangement
reaction were also examined.
Scheme 113: Use of 349 as a chiral ligand in the lithiation of benzyl thiocarbamates134
Scheme 114: 182 as a chiral ligand in the enantioselective addition of aryllithium reagents to
aromatic imines135
No carbolithiation was found to take place using 349 or 182 in toluene (Table 9 entries 14 and
15); however, excellent results were obtained with (+)-sparteine surrogate 195 in THF (Table 9
entries 16 and 17). This is in concordance with previous results.136 The solution structures of (+)-
sparteine surrogate 195 and (−)-sparteine 153 complexed to i-PrLi in d10-Et2O and d8-THF have
previously been deduced through detailed 6Li and 13C NMR analysis (Figure 15). In Et2O the (−)-
sparteine 153 i-PrLi mixture exists as a solvent-complexed heterodimer 355; in contrast the
complex with 195 is a head-to-tail homodimer 356. However, in THF no complexation between
(−)-sparteine 153 and the organolithium is observed until more than 3 equivalents of 153 is
added, whilst monomer 357 was characterised when 6 equivalents were used, whereas
complete formation of monomer 358 was observed using only 1 equivalent of (+)-sparteine
surrogate 195. This explains the stark contrast in enantioselectivities found in the asymmetric
deprotonation of N-Boc pyrrolidine 196 with (−)-sparteine 153 and (+)-sparteine surrogate 195
in THF (Scheme 115, Table 10),136 and the excellent result found for the carbolithiation-
rearrangement reaction (Table 9 entry 17). The use of (+)-sparteine surrogate 195 in the
asymmetric carbolithiation-rearrangement of vinylic ureas will be discussed in more detail in
Section 3.2.1.

82
Figure 15: Diamine-organolithium complexes in Et2O and THF136
Scheme 115: Comparison of (−)-sparteine 153 and (+)-sparteine surrogate 195 in Et2O and THF
for the asymmetric deprotonation of N-Boc pyrrolidine 196136
Entry Ligand Solvent Yield syn-359 (%) er syn-359 Yield anti-359 (%) er anti-359
1 153 Et2O 64 97:3 22 95:5
2 195 Et2O 68 98:2 23 95:5
3 153 THF 65 63:37 22 60:40
4 195 THF 66 97:3 21 97:3
Table 10: Comparison of (−)-sparteine 153 and (+)-sparteine surrogate 195 in Et2O and THF for
the asymmetric deprotonation of N-Boc pyrrolidine 196136

83
3.1.2 Substrate scope
With optimised conditions in hand, a range of vinylic ureas 318 were subjected to the reaction
conditions using a range of commercial organolithium reagents and (−)-sparteine 153 as the
chiral ligand (Scheme 116). Both electron-poor and electron-rich aromatic rings underwent
carbolithiation-rearrangement in good yields and good to excellent enantioselectivities. Only
the reactions using phenyllithium and methyllithium resulted in racemic products being
isolated. This is despite several examples in the literature of both methyl-87 and phenyllithium137
being used in enantioselective syntheses. This could be due to a problem of (−)-sparteine 153
complexation. However, the solid state structure of methyllithium-(−)-sparteine 153 complex
has been shown to have a dimeric structure exactly like the corresponding i-PrLi-(−)-sparteine
153 complex 355.138 The crystal structure of the phenyllithium-(−)-sparteine 153 complex 361
has also been determined, and exits as an unusual 4:2 ladder structure where each lithium is
capped by two (−)-sparteine 153 molecules (Figure 16).139 Nonetheless, there are also other
examples where the use of methyl- and phenyllithium have had a detrimental effect on the
enantioselectivity of a reaction.81 The use of t-BuLi resulted in only the carbolithiated product
360 being observed, with no rearrangement found, probably due to the highly hindered nature
of the organolithium intermediate. This is unsurprising as previous attempts to carbolithiate
and rearrange using t-BuLi in similar systems also resulted in only the corresponding
carbolithiated product being isolated.131
Figure 16: Phenyllithium-(−)-sparteine 153 complex

84
Scheme 116: Asymmetric carbolithiation-rearrangement using (−)-sparteine 153. For the
determination of absolute stereochemistry see Section 3.3 *‡
* Carried out by Dr Alberto Minassi and Dr Morgan Donnard
‡ This example only (319h) carried out by Michael Tait

85
3.2 Extension of Methodology
The next stage of the work explored in the current study increasing the scope of the reaction
through modification of the migrating aryl ring to include ortho- and meta-substituted rings and
use of non-commercial organolithiums. The excellent earlier result (Table 9 entry 17) found
when (+)-sparteine surrogate 195 was used in the asymmetric carbolithiation-rearrangement of
344 will also be built upon and further investigated.
3.2.1 Extending Migrating Aryl Ring Scope
Two ureas 362a and 362b were prepared (by Dr Morgan Donnard) that contained ortho- and
meta-substituents so that their effects on the tandem enantioselective carbolithiation-
rearrangement reaction could be studied (Scheme 117).
Scheme 117: Asymmetric carbolithiation-rearrangement using (+)-sparteine surrogate 195 a
Approximately 90:10, unable to fully separate peaks b At 0.1 M. c At 0.3 M.

86
The desired products were obtained in good to excellent yields but with varying degrees of
enantioenrichment. In general good ers were obtained when i-PrLi was used. However, with n-
BuLi the products 363b and 363d were close to or fully racemic. However, a slight increase in er
of 363d was found when the concentration of the reaction was increased from 0.1 M to 0.3 M.
This suggests that the complexation of (+)-sparteine surrogate 195 and n-BuLi in THF may be
concentration-dependent. Nonetheless, 6Li and 13C NMR studies have shown that complete
coordination between (+)-sparteine surrogate 195 and n-BuLi is observed at 0.07 M.140
Attempts to solve this problem through changing the solvent to cumene or toluene failed to
yield the intended urea, with only starting material isolated. Better results were obtained using
(+)-sparteine surrogate 195 that was synthesised in house (using the procedure described by
O’Brien et al.91 Scheme 64), rather than commercially available material.
3.2.2 Non-commercial Organolithiums
A further extension of this methodology was attempted through the use of non-commercial
organolithiums (Scheme 118, Table 11). Non-commercial organolithiums were made by either:
lithium-halogen exchange in THF using 2.2 equivalents of t-BuLi (Table 11 entries 1, 3 and 6)141,
directed metallation (Table 11 entries 4 and 5)142,143 or through the use of cyclopentyl chloride
and lithium metal (Table 11 entry 2).144
Organolithium species synthesised by lithium-halogen exchange were formed at −78 °C before
warming to room temperature. The organolithium was then added to a cooled (−78 °C) solution
of (+)-sparteine surrogate 195 in THF, before addition of a solution of urea 362a in THF. More
than two equivalents of t-BuLi are used as the first equivalent is used for the exchange whilst
the second equivalent reacts with the t-butyl iodide produced to generate lithium iodide,
isobutene and isobutane. This process was chosen as t-BuLi is known to undergo rapid
decomposition in THF at room temperature (a half-life of 40 minutes at −20 °C).145 Any excess t-
BuLi present in the reaction mixture is subsequently destroyed and can no longer take part in a
competing carbolithiation reaction. The organolithium formed in entry 6 was not warmed to
room temperature due to the possibility of a Brook-rearrangement occurring.98,146

87
Scheme 118: Asymmetric carbolithiation-rearrangement using non-commercial organolithiums
Entry RLi Yield 364 (%) e.r. 364
1
78 50:50
2
88 47:53
3
- -
4
- -
5
- -
6 - -
Table 11: Asymmetric carbolithiation-rearrangement using non-commercial organolithiums
Unfortunately, only the reaction of the substrates with the organolithiums derived from entries
1 and 2 produced any carbolithiated-rearranged product. Furthermore entries 1 and 2 failed to
produce significantly enantioenriched products with racemic product found from the addition
of 4-methoxyphenyllithium (as was found when phenyllithium was used in the presence of (−)-
sparteine 153) and only a slight increase in er was found for cyclopentyllithium. However, these
products were formed in excellent yield.
An unexpected reaction occurred during attempts to apply this methodology to iodide 365
(Scheme 119). It was expected that 365 would undergo rapid lithium-halogen exchange to
generate the corresponding organolithium 368147 which could then undergo the tandem
carbolithiation-rearrangement process to yield 367 (Scheme 120). However, urea 366 was
instead isolated.
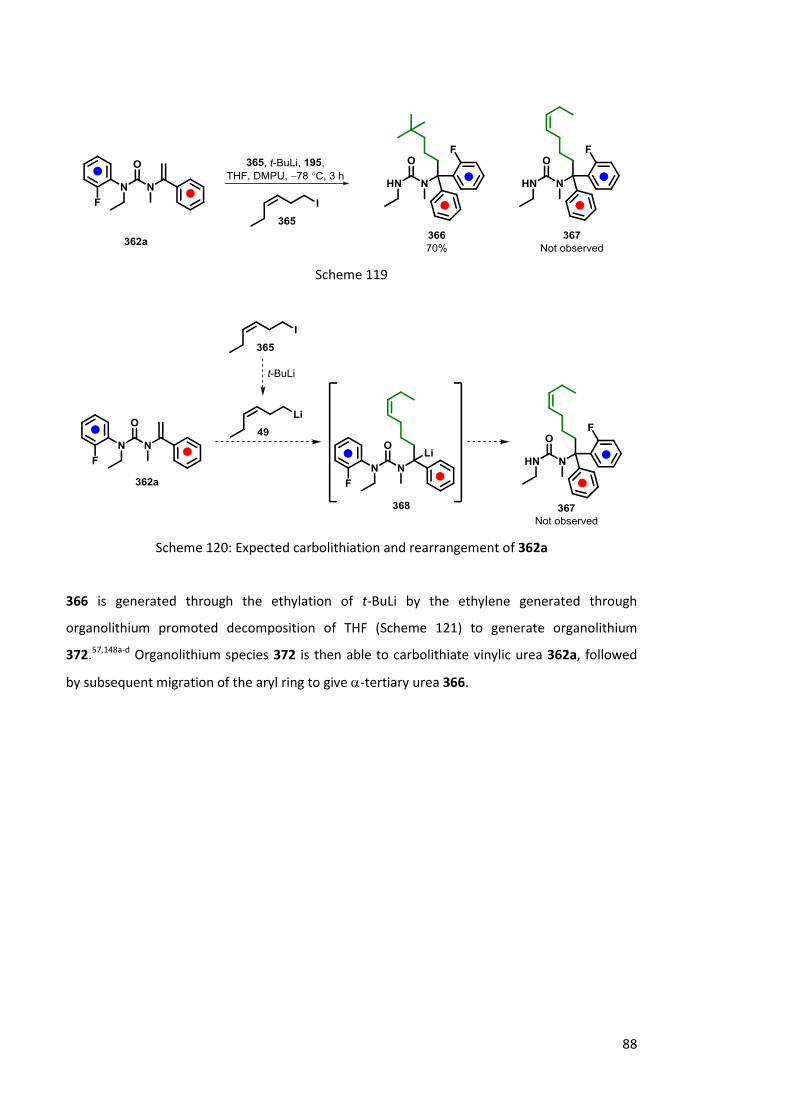
88
Scheme 119
Scheme 120: Expected carbolithiation and rearrangement of 362a
366 is generated through the ethylation of t-BuLi by the ethylene generated through
organolithium promoted decomposition of THF (Scheme 121) to generate organolithium
372.57,148a-d Organolithium species 372 is then able to carbolithiate vinylic urea 362a, followed
by subsequent migration of the aryl ring to give -tertiary urea 366.

89
Scheme 121: Decomposition and ethylation of THF
It should be noted that the presence of lithium complexing additives, such as HMPA, can alter
the pathway of THF decomposition by organolithiums.148a Addition of HMPA to the reaction
mixture can result in the isolation of ester 378. 378 can only be generated by acylation of
organolithium 376, which in turn can only be generated by one of two methods. Either by -
elimination of 370 or by a reverse 5-endo-trig (Baldwin disfavoured)149 decomposition of THF
(Scheme 122). It is not entirely clear why HMPA promotes this alternative decomposition
pathway. Perhaps HMPA’s ability as a strong deaggregating agent promotes the -elimination
pathway, or possibly helps direct deprotonation to the C-3 position of THF. It is possible that
other lithium complexing agents such as DMPU could have similar effects on the decomposition
of THF.

90
Scheme 122: Alternative THF decomposition pathway
Attempts were made to try to repeat this reaction deliberately by carrying out the reaction in
the absence of (+)-sparteine surrogate 195 in cumene with 2 equivalents of THF; but no
reaction was observed (Scheme 123).
Scheme 123

91
3.3 Absolute Configuration
3.3.1 Solvolysis of ureas
As (−)-sparteine 153 and (+)-sparteine surrogate 195 give opposite enantiomers, a method was
sought to determine the absolute stereochemistry of the products of the enantioselective
carbolithiation-rearrangement reaction. A crystalline derivative which contained a heavy atom
would provide the required information through analysis of its absolute structure parameter. As
none of the products of the enantioselective carbolithiation-rearrangement reaction were
crystalline, derivatives were synthesised. Work previously within the group has shown that the
corresponding -tertiary amines (or their hydrochloride salts) are mostly crystalline.131 A small
range of -tertiary amines 337 were thus synthesised by solvolysis of the urea group. Attempts
to remove the urea moiety from 363a under standard solvolysis conditions131 failed to give the
intended product 337a. Instead only the product 379 arising from elimination of the urea group
was isolated (Scheme 124).
Scheme 124: Deprotection of ureas under solvolysis conditions a As a 1:1 mix of E and Z isomers
New conditions were therefore developed where the rearranged ureas 363 were treated with 2
M NaOH solution in EtOH under microwave irradiation.130 The corresponding -tertiary amines
were isolated after work up of the reaction in near quantitative yields (Scheme 125, Table 12).
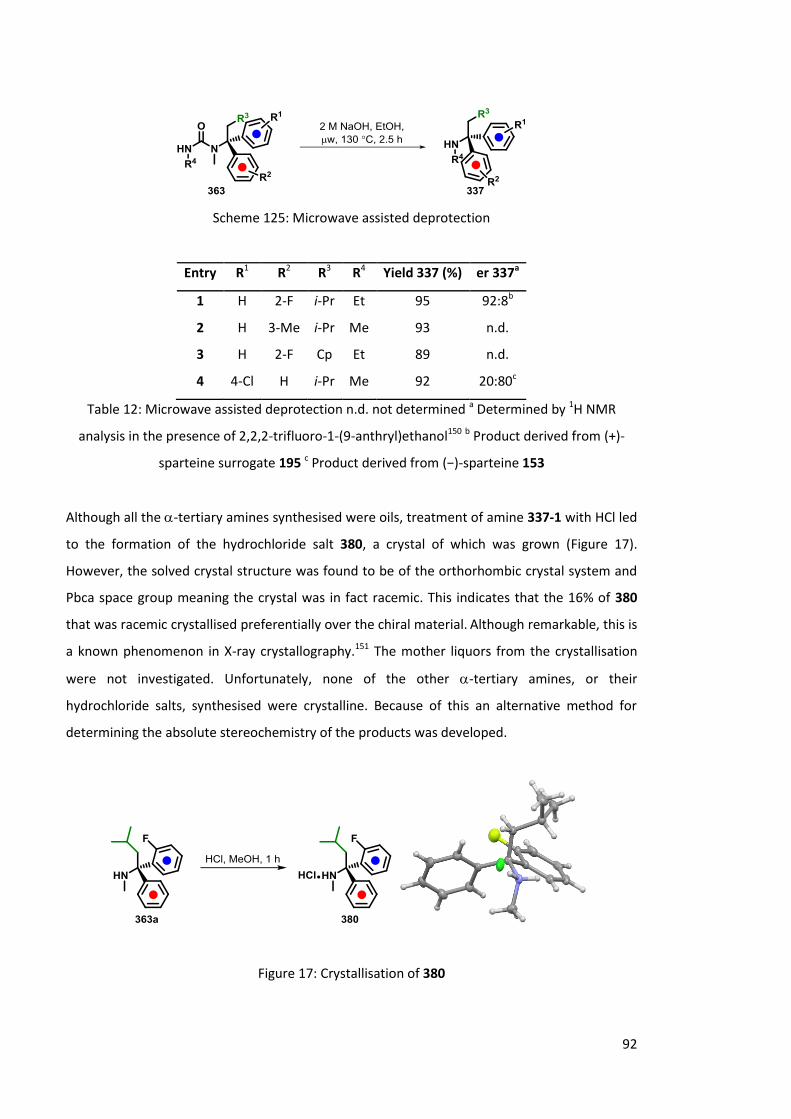
92
Scheme 125: Microwave assisted deprotection
Entry R1 R2 R3 R4 Yield 337 (%) er 337a
1 H 2-F i-Pr Et 95 92:8b
2 H 3-Me i-Pr Me 93 n.d.
3 H 2-F Cp Et 89 n.d.
4 4-Cl H i-Pr Me 92 20:80c
Table 12: Microwave assisted deprotection n.d. not determined a Determined by 1H NMR
analysis in the presence of 2,2,2-trifluoro-1-(9-anthryl)ethanol150 b Product derived from (+)-
sparteine surrogate 195 c Product derived from (−)-sparteine 153
Although all the -tertiary amines synthesised were oils, treatment of amine 337-1 with HCl led
to the formation of the hydrochloride salt 380, a crystal of which was grown (Figure 17).
However, the solved crystal structure was found to be of the orthorhombic crystal system and
Pbca space group meaning the crystal was in fact racemic. This indicates that the 16% of 380
that was racemic crystallised preferentially over the chiral material. Although remarkable, this is
a known phenomenon in X-ray crystallography.151 The mother liquors from the crystallisation
were not investigated. Unfortunately, none of the other -tertiary amines, or their
hydrochloride salts, synthesised were crystalline. Because of this an alternative method for
determining the absolute stereochemistry of the products was developed.
Figure 17: Crystallisation of 380

93
3.3.2 Preparation of an authentic sample
The product 381 resulting from asymmetric carbolithiation of 344 could be isolated in good
yield when no DMPU was added to the reaction (Scheme 126). 381 was formed in 65:35 er
under these conditions and found to give an []D25 of +12.8. It was postulated therefore, that if
an authentic sample of one enantiomer of 381 could be synthesised, comparison of the optical
rotation of the two products would allow the determination of the absolute stereochemistry.
Scheme 126: Asymmetric carbolithiation of 344
As discussed earlier (See Section 1.3.3.1) the tert-butylsulfinamide group 25 has been shown to
give excellent diastereocontrol during the Grignard addition to the corresponding tert-
butylsulfinimine.152 Careful selection of conditions and additives allows the diastereoselectivity
of the reaction to be chosen. It was therefore envisaged that this group could be used to
provide an authentic sample of 381.
Condensation of benzaldehyde with tert-butylsulfinamide 25 under standard conditions gave
the sulfinimine 382 in excellent yield. Attack on 382 by pentylmagnesium bromide yielded the
desired product 384 in good yield and as a single diastereomer by 1H NMR spectroscopy. As
shown previously (See Section 1.3.3.1), these Grignard additions are known to proceed through
the 6-membered transition state 383, allowing the diastereomeric configuration of 384 to be
determined.152 Acidic hydrolysis of the tert-butylsulfinyl group yielded the amine hydrochloride
salt 385, which was acylated with 4-chlorophenylisocyanate. Methylation produced urea 381
with (S)- absolute configuration, which had an []D25 of −59.7 (Scheme 127).

94
Scheme 127: Preparation of an authentic sample of 381
As the two samples give opposite signs in the optical rotation we can deduce that they are of
opposite absolute configuration. Therefore, (−)-sparteine 153 induces the formation of 348 with
(R)-absolute configuration. As (−)-sparteine 153 and (+)-sparteine surrogate 195 give opposite
enantiomers we can therefore infer that the (+)-sparteine surrogate 195 would give the (S)-
enantiomer (Scheme 128).
Scheme 128: Asymmetric carbolithiation-rearrangement

95
The first step of the reaction is presumably an asymmetric carbolithiation, where the ligand-
complexed organolithium attacks one enantiotopic face of the alkene (Scheme 129). This results
in the production of a stereodefined organolithium 386, formed under kinetic control, which is
configurationally stable on the time scale of the reaction.53,119,121-123 It is therefore improbable
that any enantioenrichment is a result of any equilibration of the organolithium
intermediate.46,49d,153 Retentive migration of the aryl ring yields the carbolithiated-rearranged
product 348. This migration takes place through an intramolecular nucleophilic aromatic
substitution mechanism as discussed previously (See Section 2.1.1). Since both protonation and
aryl migration are stereochemically retentive in ureas,119,154 and carbolithiation is known to be a
syn-selective reaction,131 we rationalise that (−)-sparteine 153 leads to aryl migration to the Si
face as drawn of alkene 344. The selectivity observed in the asymmetric carbolithiation-
rearrangement reaction is consistent with other asymmetric carbolithiations found in the
literature.80,85,86,97,155
Scheme 129: Mechanism of enantioselective carbolithiation-rearrangement
In the case of the 1-naphthyl migration, a dearomatised product can be trapped by bubbling dry
air through the reaction (Scheme 130).*156 The mechanism of the reaction for the migration of a
1-naphthyl ring proceeds through intermediate allyllithium 390, allowing the oxidation to take
place. This is not possible in the migration of other aryl rings.
* Performed by Dr Julien Lefranc
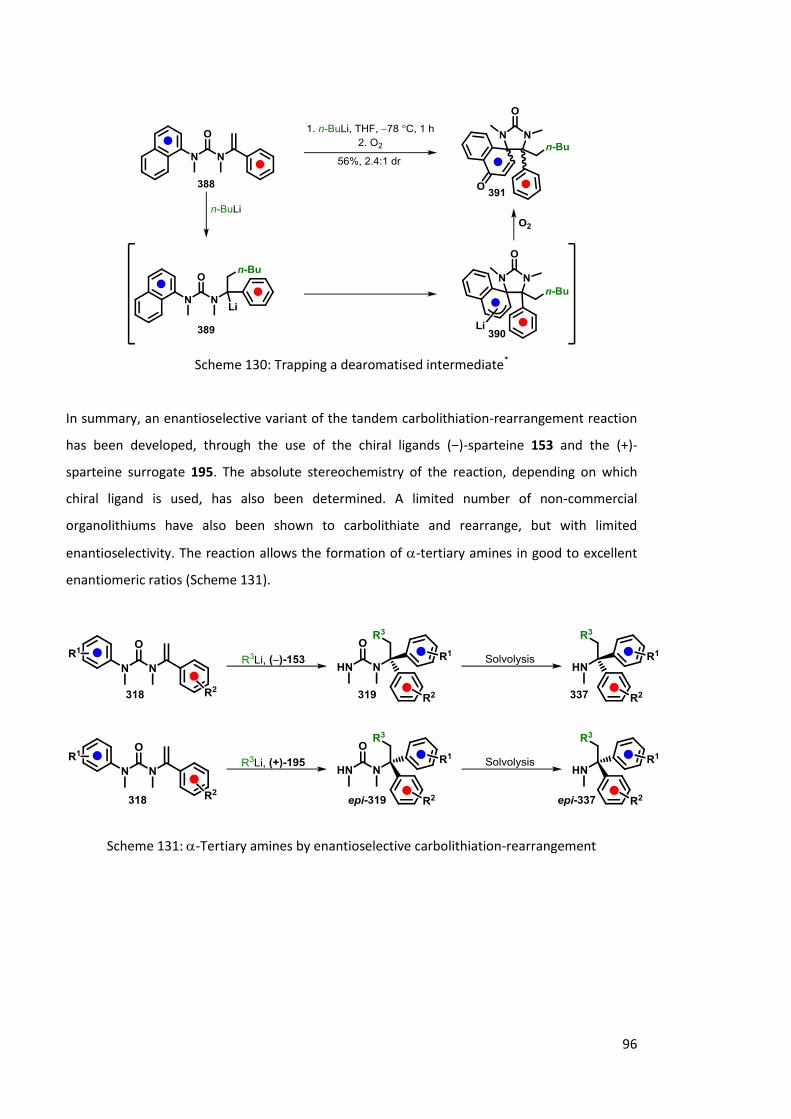
96
Scheme 130: Trapping a dearomatised intermediate*
In summary, an enantioselective variant of the tandem carbolithiation-rearrangement reaction
has been developed, through the use of the chiral ligands (−)-sparteine 153 and the (+)-
sparteine surrogate 195. The absolute stereochemistry of the reaction, depending on which
chiral ligand is used, has also been determined. A limited number of non-commercial
organolithiums have also been shown to carbolithiate and rearrange, but with limited
enantioselectivity. The reaction allows the formation of -tertiary amines in good to excellent
enantiomeric ratios (Scheme 131).
Scheme 131: -Tertiary amines by enantioselective carbolithiation-rearrangement

97
Chapter 4 Synthesis of substituted piperidines
4.1 Part 1: 6-Membered cyclic vinylic ureas via a cyclic imine
The excellent results found for the tandem carbolithiation-rearrangement of acyclic vinylic
ureas 318 (See Section 2.4.1) led us to investigate the application of this methodology to cyclic
systems (Scheme 132). If successful, this would allow the synthesis of substituted piperidine
(Chapter 4) or pyrrolidine (Chapter 5) analogues. The diastereoselectivity of the reaction has
also been studied.
Scheme 132: 6-Membered cyclic vinylic ureas
4.1.1 Synthesis of 6-membered cyclic vinylic ureas
A route was therefore desired for the synthesis of cyclic vinylic ureas 338. The use of cyclic
imine 395 was a logical starting point for the synthesis as it could potentially be transformed
into a wide range of cyclic vinylic ureas 338 using methodology already established within the
group.157 Cyclic imine 395 was synthesised through a procedure developed by Buchwald et al.
by reacting 5-bromovaleronitrile 394 with phenyllithium.158 This is a fairly remarkable reaction
as it appears that the phenyllithium is carbolithiating the CN triple bond quicker than it
exchanges with bromine. However, as the lithium-halogen exchange process is reversible,
formation of 397 may be favoured (Scheme 134). 395 was then acylated using a range of aryl
isocyanates, and methylated to yield the 6-membered cyclic vinylic ureas 338. A slight change in
reaction conditions were required from that used in the reaction of an isocyanate with an
acyclic imine.157 The initial addition failed when THF was used as the solvent; instead it was
necessary to change the solvent to CH2Cl2 and for DMAP to be added to the reaction.
Methylation of the ureas also required a change in solvent from THF to DMF. This is due to DMF
being a polar aprotic solvent and as a result is poor at solvating the intermediate anion of the
reaction. A range of ureas 338 were synthesised by this method in moderate yields (Scheme
133), consistent with those found in the acyclic counterparts 320.175 No starting material or any
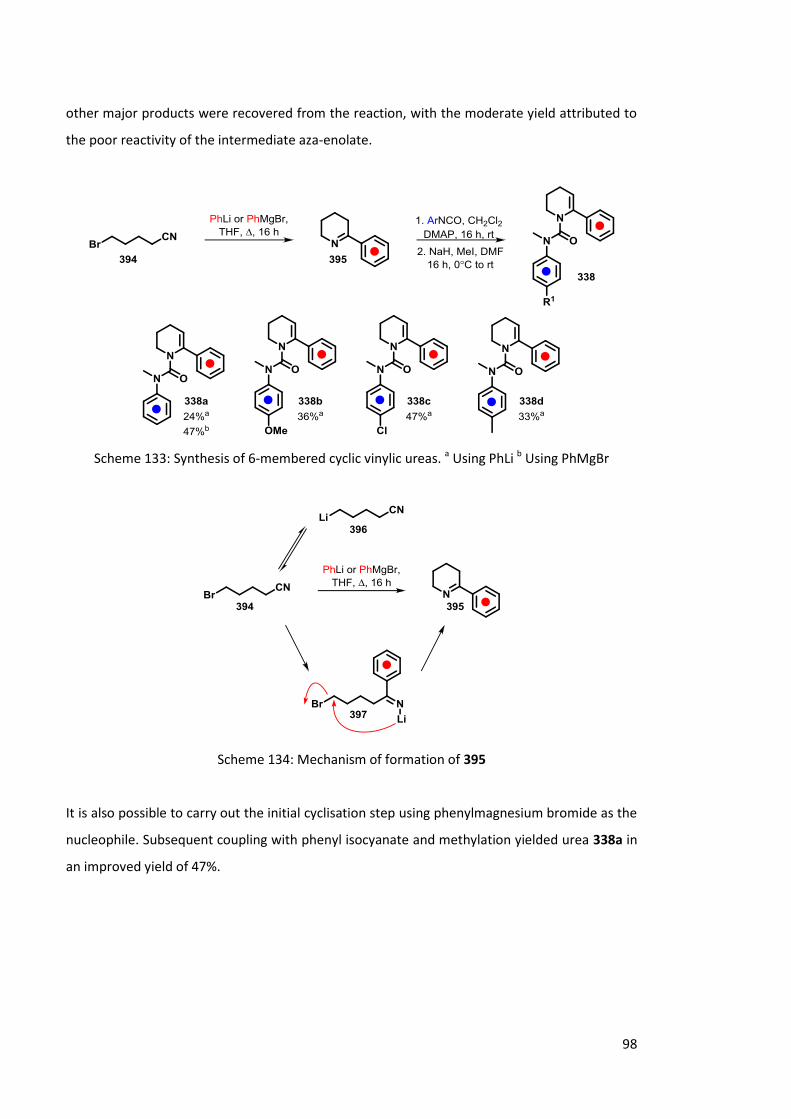
98
other major products were recovered from the reaction, with the moderate yield attributed to
the poor reactivity of the intermediate aza-enolate.
Scheme 133: Synthesis of 6-membered cyclic vinylic ureas. a Using PhLi b Using PhMgBr
Scheme 134: Mechanism of formation of 395
It is also possible to carry out the initial cyclisation step using phenylmagnesium bromide as the
nucleophile. Subsequent coupling with phenyl isocyanate and methylation yielded urea 338a in
an improved yield of 47%.

99
Synthesis of 6-membered cyclic vinylic ureas with a functionalised aryl ring to nitrogen was
also attempted. The 6-membered cyclic imine was synthesised by initial lithium-halogen
exchange using t-butyllithium to give the corresponding aryllithium. This could then be used in
the attack and cyclisation of 394 to yield cyclic imines with functionalised aryl rings to
nitrogen 399 in moderate to good yields (Scheme 135).
Scheme 135: Synthesis of aryl cyclic imines 399
Unfortunately, attempts to acylate 399 with phenyl isocyanate failed to produce the
corresponding urea 400. Use of a strong base such as NaHMDS or LDA to deprotonate to
nitrogen and generate an azaenolate also failed to yield the desired urea 400 (Scheme 136). It is
not known why this reaction fails as the imine nitrogen does not add to phenylisocyanate in the
presence of either electron withdrawing or donating aryl rings. It is possible that there may be
some salts present in the commercial organolithium and Grignard reagents that are aiding the
reactivity. These impurities may not be present in the organolithiums made by lithium-halogen
exchange. A similar problem was encountered when attempting to synthesise 5-membered
cyclic vinylic ureas (See Section 5.1.1 for further details).

100
Scheme 136: Attempts to form urea 400
4.1.2 Lithiation of 6-membered cyclic vinyl ureas 338
Previous work within the group has shown that vinylic ureas can undergo two different types of
lithiation, depending on the type of organolithium used (Scheme 95 and Scheme 104, Section
2.1). If an alkyl- or aryllithium is used, the organolithium is able to act as a nucleophile and the
tandem carbolithiation-rearrangement process can occur.131 However, as LDA is less
nucleophilic due to the steric hindrance of its two isopropyl substituents,53 the product formed
is a result of deprotonation and rearrangement.123 Use of either method would allow
alternative connective routes to the target substituted piperidines with either an olefin or
substituent at the 3-position of the ring. The potential diastereoselectivity of the carbolithiation
reaction has been investigated (Scheme 137).
Scheme 137: Possible products arising from the lithiation of 338

101
4.1.2.1 Carbolithiation-Rearrangement of diphenyl urea 338a
Initial investigations employed conditions developed within the group131 that were found to be
optimal for acyclic vinylic ureas 320 in an attempt to carbolithiate and rearrange the diphenyl
urea 338a using i-PrLi (Scheme 138). A solution of the urea in THF was cooled to −78 °C before
dropwise addition of 2.5 equivalents of the organolithium. These conditions gave only an
isolated yield of 12% of carbolithiated-rearranged urea 405. The majority of the material
obtained was unreacted starting material, whilst a moderate amount of the product 403 (35%)
resulting from carbolithiation and no rearrangement was also found. 403 was isolated as a
single diastereomer (as determined by 1H NMR spectroscopy). This initial result indicates that
carbolithiation of 338a to 402 is a slow process, whilst the rearrangement to form 404 is even
slower. The relative stereochemistry was determined through nOe studies (See Section 4.1.2.3),
which has allowed us to prove that the carbolithiation of 338a is stereospecific and is a syn-
process. Subsequent studies have also proved that the migration of the distal aryl ring in these
systems is also stereospecific (See Section 4.1.2.3). These results are consistent with those
found for the acyclic vinylic ureas 320.131
Scheme 138: Lithiation of 338a. Relative stereochemistry determined by nOe studies (See
Section 4.1.2.3)

102
In order to probe the reaction further an experiment was carried out where the reaction was
carried out at a higher temperature and quenched after 30 minutes (Scheme 139). These
conditions have been used within the group to promote the carbolithiation of acyclic vinylic
ureas 320.156 Indeed, TLC analysis showed that the majority of 338a had been consumed, and
the product of carbolithiation but no rearrangement 403 was isolated in good yield. This result
highlights that the rearrangement of the intermediate organolithium 402, not the initial
carbolithiation of 338a, is the problematic step in the tandem carbolithiation-rearrangement
reaction.
Scheme 139: Carbolithiation of 338a. Relative stereochemistry determined by nOe studies
(See Section 4.1.2.3)
In order to try and promote the rearrangement, DMPU (added as a co-solvent relative to the
amount of THF used in the reaction; 4:1 THF:DMPU) was added to the reaction mixture after
the addition of the organolithium. However, this had no significant effect on the yield of the
reaction (Table 13 entry 2). Therefore a range of conditions were screened in a bid to optimise
the carbolithiation-rearrangement reaction (Scheme 140, Table 13).
Scheme 140: Lithiation of 338a. Relative stereochemistry determined by nOe studies (See
Section 4.1.2.3)

103
Entry Temp (°C) Time (h) Solvent Yield 405 (%) Yield 403 (%)
1 −78 3 THF 12 35
2 −78 1 + 2 THF + DMPUa,b 12 24
3 −78 16 THF 15 22
4 −78 6 + 10 THF + DMPUa,c 14 23
5 −40 3 THF 40 20
6 −40 3 THF + DMPUa,d 70 -
7 0 3 THF + DMPUa,d 64 -
8 −40 3 DMPU 56 -
Table 13: Rearrangement conditions for 338a a 4:1 THF:DMPU b DMPU added 1 hour after
addition of i-PrLi c DMPU added 1 hour after addition of i-PrLi d DMPU added straight after
addition of i-PrLi
Raising the temperature of the reaction from −78 °C to −40 °C was essential in order to give
good yields of the rearranged urea 405 (Table 13 entries 1 and 5). It was hoped that the
addition of DMPU to the reaction after an initial period of carbolithiation could improve these
yields. Unfortunately, addition of DMPU 1 to 6 hours after organolithium addition had taken
place had little effect on the yield of 405 (Table 13 entries 2 and 4). However, yields could be
markedly improved by adding DMPU at the start of the reaction immediately following
organolithium addition (Table 13 entries 6 and 7). Using DMPU as the sole solvent however,
resulted in diminished yields (Table 13 entry 8).
The optimised conditions (Table 13 entry 6) were then applied to the carbolithiation-
rearrangement of 338a with n-, s- and t-butyllithium (Scheme 141, Table 14).
Scheme 141: Lithiation of 338a

104
Entry R Temperature (°C) Yield 406 (%) Yield 407 (%)
1 n-Bu −40 16 32
2 s-Bu −40 42a -
3 n-Bu 0 23 46
4 s-Bu 0 58a -
5 t-Bu 0 21 42
Table 14: Rearrangement conditions for 338a a 2:1:0:0 dr
Unfortunately, use of primary and tertiary organolithiums resulted in an inseparable mixture of
carbolithiated-rearranged urea 406 and diaryl allylic urea 407 being isolated in a 2:1 ratio in
favour of 407. 407 arises from an allyllithium intermediate formed by deprotonation to
nitrogen, followed by migration of the aromatic ring (Scheme 142). This result highlights the
importance in the balance between the nucleophilicity and basicity of the organolithium; with
both the more basic t-BuLi and the more nucleophilic n-BuLi resulting in a mixture of products
being isolated (Table 14 entries 1, 3 and 5). However, secondary organolithiums such as i-PrLi
(Table 13 entry 6) and s-BuLi (Table 14 entries 2 and 4) gave only the carbolithiated-rearranged
products 405 and 406 respectively.
The yields of the reaction were found to be slightly lower when n- or s-BuLi were used at −40 °C
when compared to the use of i-PrLi (Table 14 entries 1 and 2). Use of s-BuLi resulted in the
isolation of 406 as a 2:1:0:0 diastereomeric mixture at the exocyclic centre. In order to improve
the yield of the carbolithiated-rearranged products 406, the temperature of the reaction was
raised to 0 °C (Table 14 entries 3 and 4). This led to a slight improvement in the yield of 406;
however allylic urea 407 was also isolated when n-BuLi was used, again in a 2:1 ratio in favour
of 407. These conditions were then applied to the reaction with t-BuLi (Table 14 entry 5). It was
not anticipated that the intermediate organolithium 402 resulting from carbolithiation of t-
butyllithium would rearrange as only carbolithiation and no rearrangement was found in the
use of t-BuLi in acyclic vinylic ureas 318 (See Section 2.4.1).131 However, carbolithiated-
rearranged urea 406 was isolated, along with allylic urea 407.

105
Scheme 142: Mechanism of formation of 407
Several avenues were explored in an attempt to prevent the deprotonation reaction from
occurring. It was initially thought the outcome of the reaction could be dependent on solvent
concentration. A series of reactions were therefore carried out to test this hypothesis (Table
15).
Entry RLi Conc (M) Yield 406 (%) Yield 407 (%)
1 n-Bu 2.0 23 46
2 n-Bu 1.0 19 38
3 n-Bu 0.05 22 44
4 t-Bu 0.3 21 42
Table 15: Solvent concentration screen
It can be seen that solvent concentration had no effect on the relative composition of the two
products obtained. Therefore, the effect of different lithium salts that may be present in the
commercial solution of organolithium reagents was also investigated. Reactions were carried
out in the presence of 2.5 equivalents (as 2.5 equivalents of organolithium are used in the
reaction) of different lithium salts with freshly opened bottles of organolithium solution
(Scheme 143, Table 16).

106
Scheme 143: Effect of lithium salts on the carbolithiation-rearrangement reaction
Entry Xa Yield 410 (%) Yield 407 (%)
1 Cl 24 48
2 Oi-Pr 19 38
3 OH 18b 36
Table 16: Effect of lithium salts on the carbolithiation-rearrangement reaction a 2.5 equivalents
of LiX used b 1:1 dr
Once more, the reaction resulted in a mixture of products being isolated. Furthermore, the use
of LiOH as an additive resulted in complete destruction of the diastereospecificity of the
reaction with both diastereomers of the addition formed in a 1:1 ratio (Table 16 entry 3).
Therefore it is presumed that LiOH is epimerising the benzyl lithium intermediate 402. It is
somewhat surprising that LiOH epimerises 402 whilst LiOi-Pr does not. Previous studies have
shown that the lithium halide and lithium alkoxide salts can build up during the course of many
electrophilic substitution reactions, and as a result facilitate the inversion of some
organolithium compounds.159,160 It is postulated that this is a result of the formation of mixed
aggregates during the formation of the reactions’ transition state.161
This problem could be overcome for secondary and tertiary organolithium additions by carrying
out the initial carbolithiation in toluene, followed by addition of DMPU (4:1 toluene:DMPU) to
promote the rearrangement.119,121-123 As toluene is a non-coordinating solvent deprotonation is
minimised,123 ensuring only carbolithiation occurs whilst simultaneously preventing any
rearrangement.131,156 DMPU was then added to promote the rearrangement once complete
carbolithiation was observed by TLC (Scheme 144). Use of toluene as the solvent did however
require longer reaction times in order for acceptable yields to be obtained, with the reaction
left to warm to room temperature overnight; once more highlighting the rearrangement step
was the slower of the two steps.

107
It was not possible to carry out this reaction using primary organolithiums as these
preferentially deprotonate toluene over vinylic ureas.156 However, use of cumene as the solvent
allows for primary organolithiums to be used in the tandem carbolithiation-rearrangement
reaction. This is because cumene is not deprotonated by primary organolithiums and therefore
has a longer half-life under the conditions of the reaction.53
Scheme 144: Carbolithiation-rearrangement of 338a. DMPU added as a cosolvent once
complete carbolithiation was observed (4:1 solvent:DMPU) a Cumene was used as solvent
instead of toluene

108
4.1.2.2 Rearrangement of functionalised aryl rings
These newly optimised conditions (toluene/cumene at −40 °C, DMPU, stirring overnight with
warming to room temperature) were then applied to the carbolithiation-rearrangement of 6-
membered cyclic vinylic ureas bearing functionalised aryl rings (Scheme 145).
Scheme 145: Carbolithiation-Rearrangement of functionalised aryl rings. Relative
stereochemistry determined by nOe studies (See Section 4.1.2.3)
The rearrangement was found to be general with moderate to good yields observed for the
migration of three different functionalised aryl rings and with four organolithium reagents.
Yields were found to be comparable with that observed for the phenyl migration. In all cases
only one diastereomer was observed in the 1H NMR spectrum of the crude reaction mixture,
except for when s-BuLi was used, where the products were isolated in a 2:1:0:0 diastereomeric
ratio at the exocyclic centre.
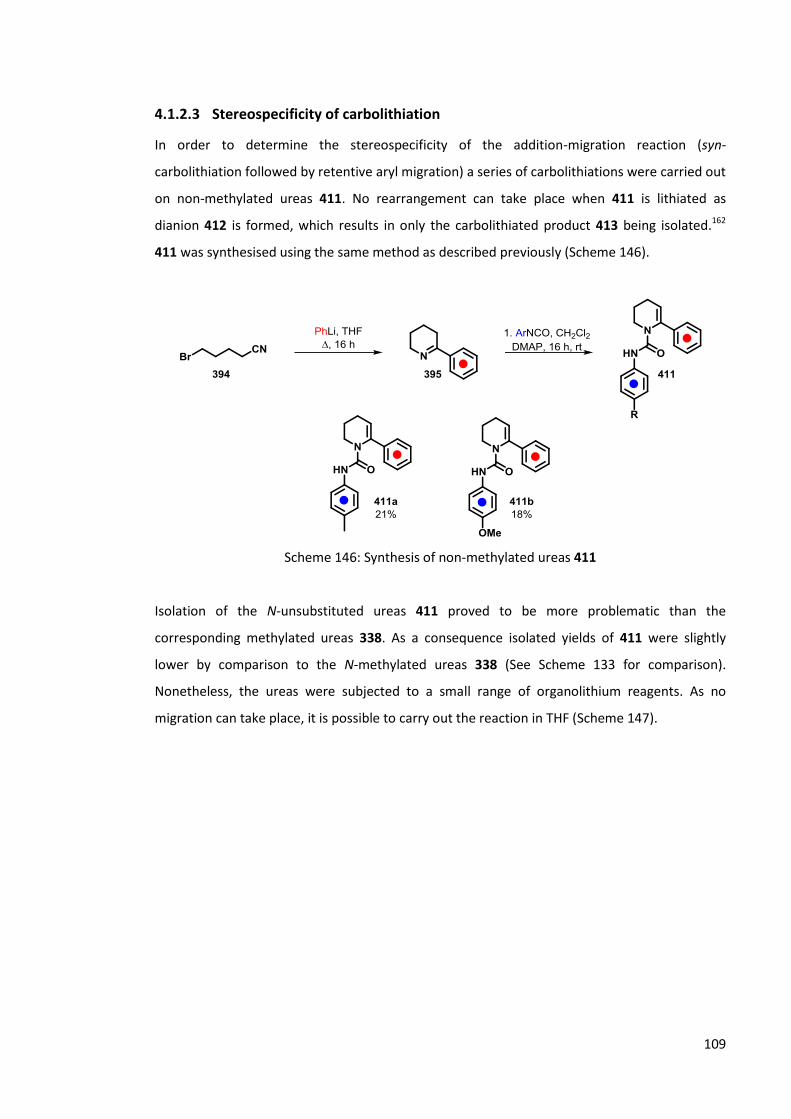
109
4.1.2.3 Stereospecificity of carbolithiation
In order to determine the stereospecificity of the addition-migration reaction (syn-
carbolithiation followed by retentive aryl migration) a series of carbolithiations were carried out
on non-methylated ureas 411. No rearrangement can take place when 411 is lithiated as
dianion 412 is formed, which results in only the carbolithiated product 413 being isolated.162
411 was synthesised using the same method as described previously (Scheme 146).
Scheme 146: Synthesis of non-methylated ureas 411
Isolation of the N-unsubstituted ureas 411 proved to be more problematic than the
corresponding methylated ureas 338. As a consequence isolated yields of 411 were slightly
lower by comparison to the N-methylated ureas 338 (See Scheme 133 for comparison).
Nonetheless, the ureas were subjected to a small range of organolithium reagents. As no
migration can take place, it is possible to carry out the reaction in THF (Scheme 147).
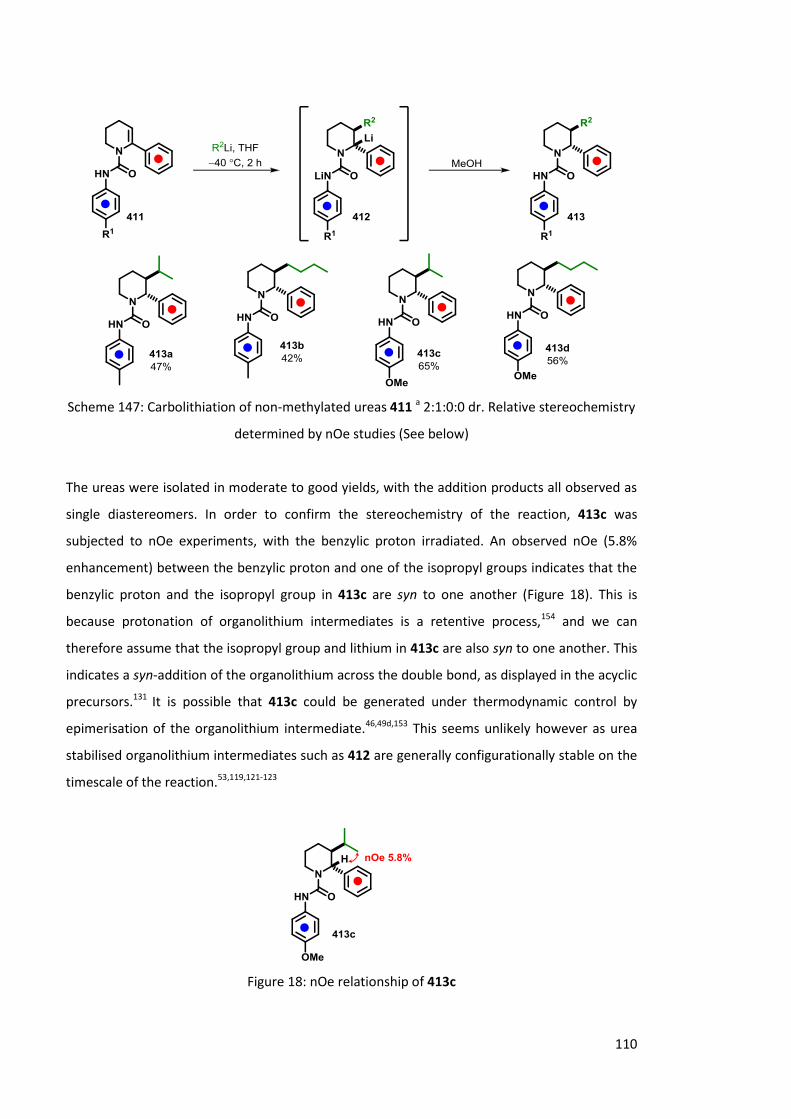
110
Scheme 147: Carbolithiation of non-methylated ureas 411 a 2:1:0:0 dr. Relative stereochemistry
determined by nOe studies (See below)
The ureas were isolated in moderate to good yields, with the addition products all observed as
single diastereomers. In order to confirm the stereochemistry of the reaction, 413c was
subjected to nOe experiments, with the benzylic proton irradiated. An observed nOe (5.8%
enhancement) between the benzylic proton and one of the isopropyl groups indicates that the
benzylic proton and the isopropyl group in 413c are syn to one another (Figure 18). This is
because protonation of organolithium intermediates is a retentive process,154 and we can
therefore assume that the isopropyl group and lithium in 413c are also syn to one another. This
indicates a syn-addition of the organolithium across the double bond, as displayed in the acyclic
precursors.131 It is possible that 413c could be generated under thermodynamic control by
epimerisation of the organolithium intermediate.46,49d,153 This seems unlikely however as urea
stabilised organolithium intermediates such as 412 are generally configurationally stable on the
timescale of the reaction.53,119,121-123
Figure 18: nOe relationship of 413c

111
In order for the benzylic proton and isopropyl group in 413c to be syn to one another, then we
would expect the benzylic proton to be in an axial position if 413c is adopting chair
conformation 414 (Figure 19). However, the benzylic proton has a coupling constant of only 3.8
Hz indicating that the proton may be equatorial. As the isopropyl substituent would not be
expected to be in an axial position, 413c may therefore be adopting boat conformation 415. It is
known that 1,3-allylic strain in N-acylpiperidines can force large substituents in the 2-position to
adopt an axial position over an equatorial one.163 As a result it is highly likely that 413c is
adopting boat conformation 415 with the phenyl group equatorial and the benzylic proton syn
to the axial isopropyl group.
Figure 19: Possible conformations of 413c
The stereospecificity of the rearrangement was analysed through the methylation and
rearrangement of the carbolithiated urea 413a. This produced the same diastereomer of urea
339e as that from the tandem carbolithiation-rearrangement reaction on methylated urea 338d
(Scheme 148). As the same diastereomer was obtained from both reactions we can deduce that
the rearrangement is stereospecific.
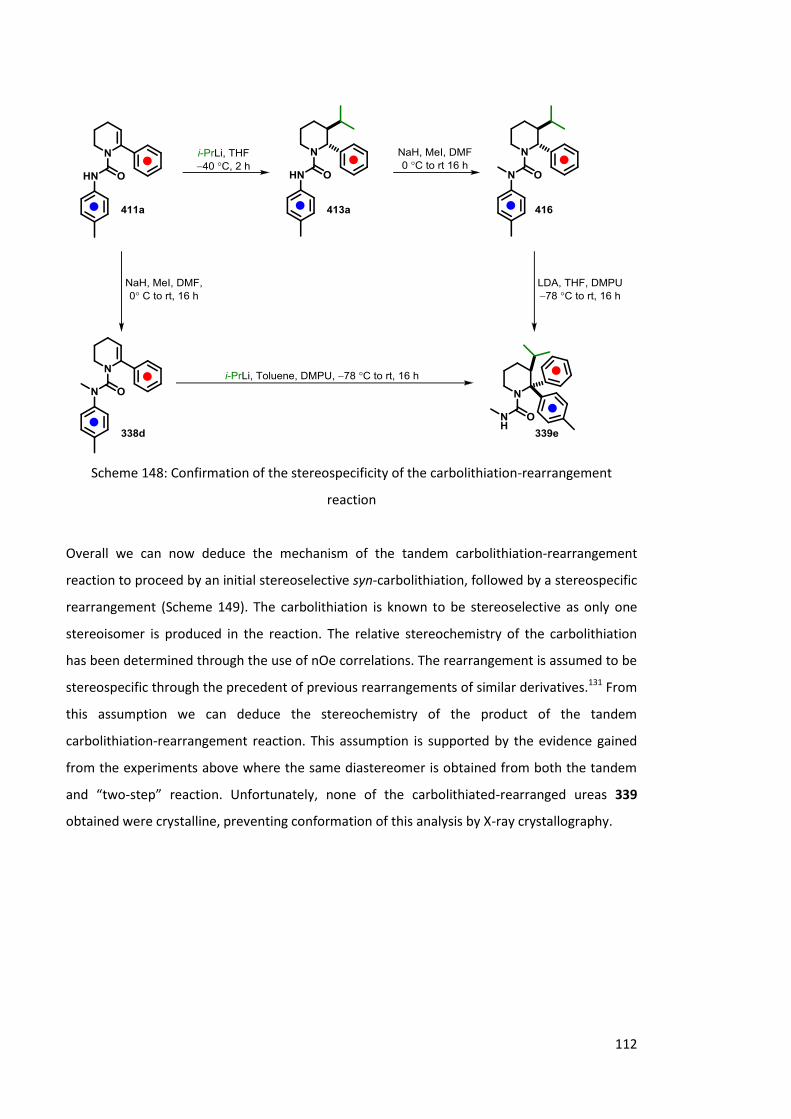
112
Scheme 148: Confirmation of the stereospecificity of the carbolithiation-rearrangement
reaction
Overall we can now deduce the mechanism of the tandem carbolithiation-rearrangement
reaction to proceed by an initial stereoselective syn-carbolithiation, followed by a stereospecific
rearrangement (Scheme 149). The carbolithiation is known to be stereoselective as only one
stereoisomer is produced in the reaction. The relative stereochemistry of the carbolithiation
has been determined through the use of nOe correlations. The rearrangement is assumed to be
stereospecific through the precedent of previous rearrangements of similar derivatives.131 From
this assumption we can deduce the stereochemistry of the product of the tandem
carbolithiation-rearrangement reaction. This assumption is supported by the evidence gained
from the experiments above where the same diastereomer is obtained from both the tandem
and “two-step” reaction. Unfortunately, none of the carbolithiated-rearranged ureas 339
obtained were crystalline, preventing conformation of this analysis by X-ray crystallography.
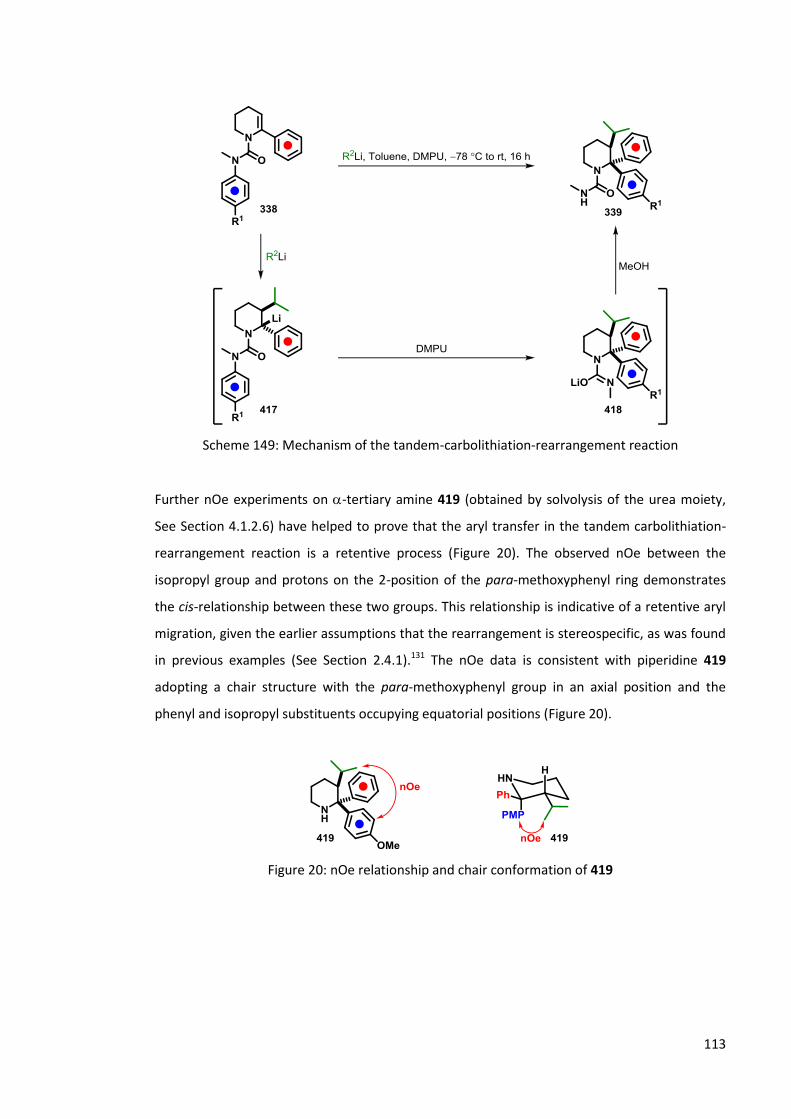
113
Scheme 149: Mechanism of the tandem-carbolithiation-rearrangement reaction
Further nOe experiments on -tertiary amine 419 (obtained by solvolysis of the urea moiety,
See Section 4.1.2.6) have helped to prove that the aryl transfer in the tandem carbolithiation-
rearrangement reaction is a retentive process (Figure 20). The observed nOe between the
isopropyl group and protons on the 2-position of the para-methoxyphenyl ring demonstrates
the cis-relationship between these two groups. This relationship is indicative of a retentive aryl
migration, given the earlier assumptions that the rearrangement is stereospecific, as was found
in previous examples (See Section 2.4.1).131 The nOe data is consistent with piperidine 419
adopting a chair structure with the para-methoxyphenyl group in an axial position and the
phenyl and isopropyl substituents occupying equatorial positions (Figure 20).
Figure 20: nOe relationship and chair conformation of 419

114
4.1.2.4 LDA mediated rearrangement
As part of our research into developing new routes to piperidine derivatives, we have also
investigated the use of LDA as the base. Previous work in this area has shown that LDA initiates
a deprotonation in vinylic ureas 292 to form an allyllithium intermediate which undergoes
rearrangement to give diaryl allyl ureas 293 (See Section 2.1.4).123 This is made possible as LDA
is less nucleophilic than other organolithiums as the lithium is bound to nitrogen rather than
carbon. Furthermore, due to the steric hindrance of its two isopropyl substituents, LDA is less
nucleophilic when compared to other organolithium reagents where the lithium is bound to
nitrogen.53 Application of this methodology to 6-membered cyclic vinylic ureas 338 would result
in the synthesis of 1,2,3,6-tetrahydropyridine derivative 401 (Scheme 150).
Scheme 150: LDA mediated rearrangement
As a result, 6-membered cyclic vinylic ureas 338 were subjected to conditions where LDA was
used as the base. Based on the results found for the tandem carbolithiation-rearrangement
reaction (See Section 4.1.2.2), reactions were left to run overnight with slow warming to room
temperature, as it was anticipated that the initial deprotonation would be fast, but the
rearrangement would be slow. DMPU was also added to promote the rearrangement (Scheme
151).121-123,131

115
Scheme 151: LDA mediated migration
Yields were found to be comparable with those for the tandem carbolithiation-rearrangement
reaction, giving the diaryl cyclic allylic ureas 401 in moderate to good yields. Reactions were left
to warm to room temperature overnight to ensure good yields were obtained. This was
because of the low yields observed previously for the migration of functionalised aryl rings at
low temperatures and short reaction times. The allylic double bond contained within 401 could
serve as a potential synthetic handle for further functionalisation.
In acyclic vinylic ureas 292, chiral lithium amides such as 295 induce an asymmetric
deprotonation which is followed by a retentive migration to produce enantioenriched allylic
ureas 296 (Scheme 96, Section 2.1.4).123 We set out to use such amides in the lithiation of 6-
membered cyclic vinylic ureas in an attempt to synthesise -tertiary amines in an
enantioselective manner (Scheme 152).
Scheme 152: Possible enantioselective migration
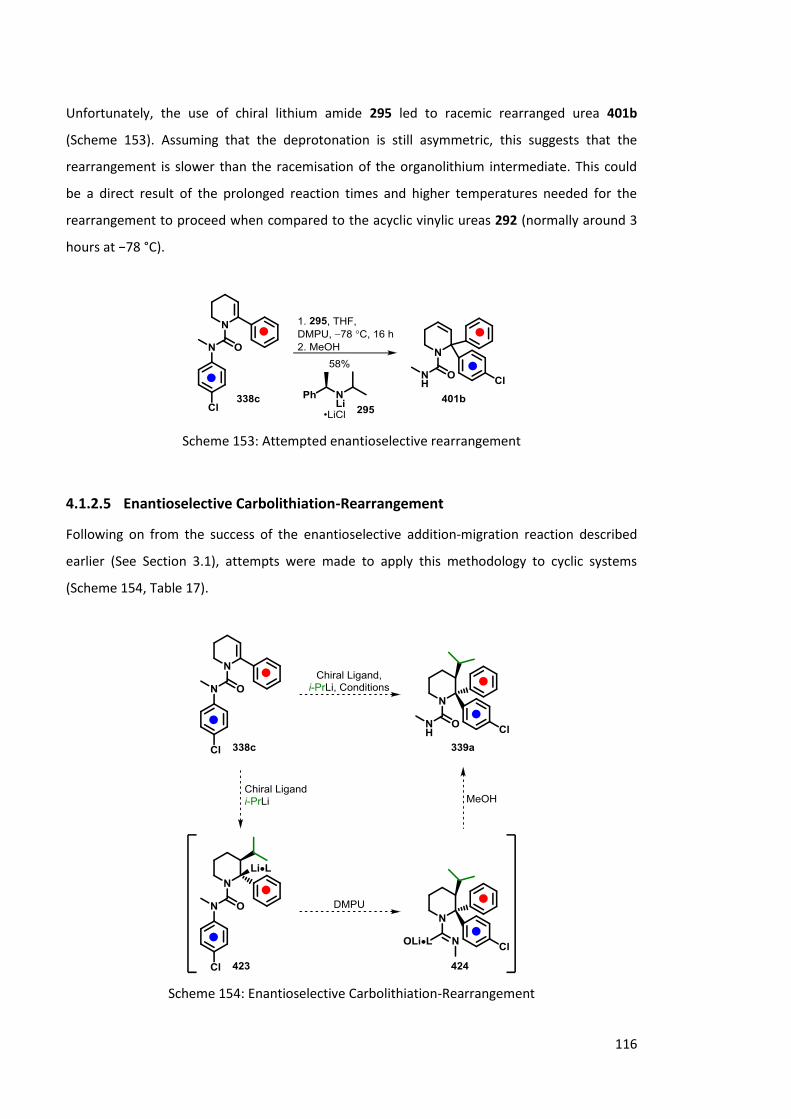
116
Unfortunately, the use of chiral lithium amide 295 led to racemic rearranged urea 401b
(Scheme 153). Assuming that the deprotonation is still asymmetric, this suggests that the
rearrangement is slower than the racemisation of the organolithium intermediate. This could
be a direct result of the prolonged reaction times and higher temperatures needed for the
rearrangement to proceed when compared to the acyclic vinylic ureas 292 (normally around 3
hours at −78 °C).
Scheme 153: Attempted enantioselective rearrangement
4.1.2.5 Enantioselective Carbolithiation-Rearrangement
Following on from the success of the enantioselective addition-migration reaction described
earlier (See Section 3.1), attempts were made to apply this methodology to cyclic systems
(Scheme 154, Table 17).
Scheme 154: Enantioselective Carbolithiation-Rearrangement
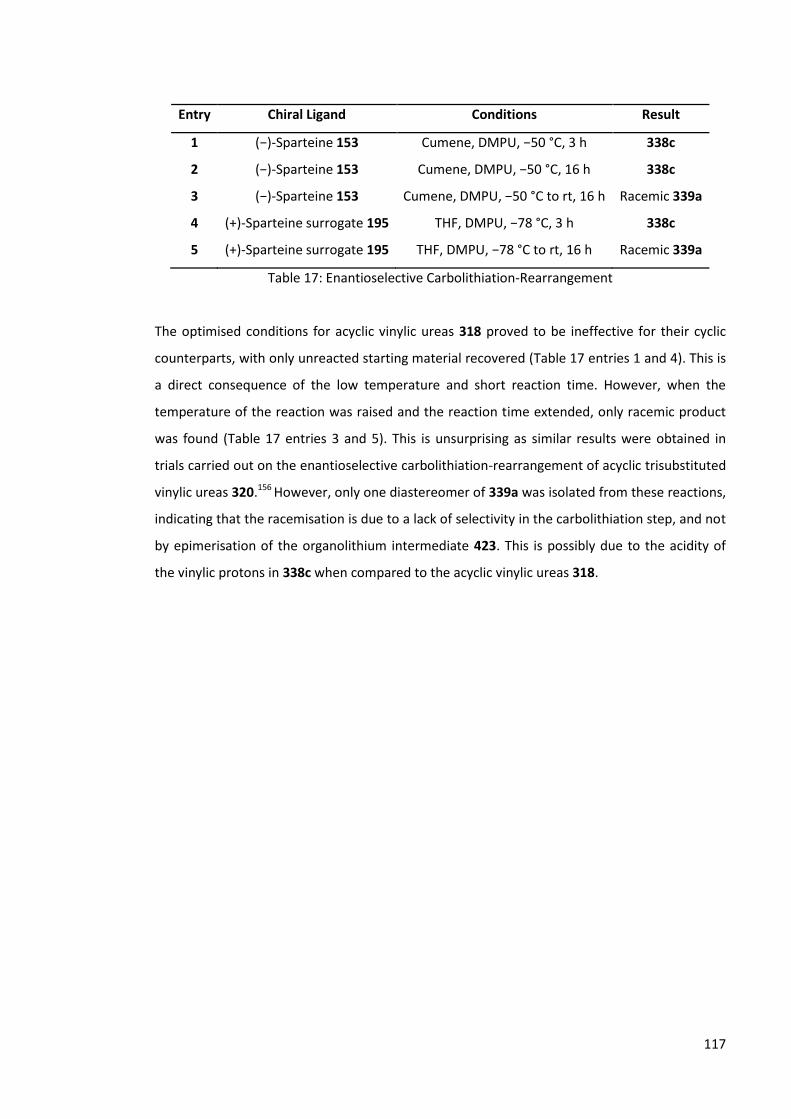
117
Entry Chiral Ligand Conditions Result
1 (−)-Sparteine 153 Cumene, DMPU, −50 °C, 3 h 338c
2 (−)-Sparteine 153 Cumene, DMPU, −50 °C, 16 h 338c
3 (−)-Sparteine 153 Cumene, DMPU, −50 °C to rt, 16 h Racemic 339a
4 (+)-Sparteine surrogate 195 THF, DMPU, −78 °C, 3 h 338c
5 (+)-Sparteine surrogate 195 THF, DMPU, −78 °C to rt, 16 h Racemic 339a
Table 17: Enantioselective Carbolithiation-Rearrangement
The optimised conditions for acyclic vinylic ureas 318 proved to be ineffective for their cyclic
counterparts, with only unreacted starting material recovered (Table 17 entries 1 and 4). This is
a direct consequence of the low temperature and short reaction time. However, when the
temperature of the reaction was raised and the reaction time extended, only racemic product
was found (Table 17 entries 3 and 5). This is unsurprising as similar results were obtained in
trials carried out on the enantioselective carbolithiation-rearrangement of acyclic trisubstituted
vinylic ureas 320.156 However, only one diastereomer of 339a was isolated from these reactions,
indicating that the racemisation is due to a lack of selectivity in the carbolithiation step, and not
by epimerisation of the organolithium intermediate 423. This is possibly due to the acidity of
the vinylic protons in 338c when compared to the acyclic vinylic ureas 318.
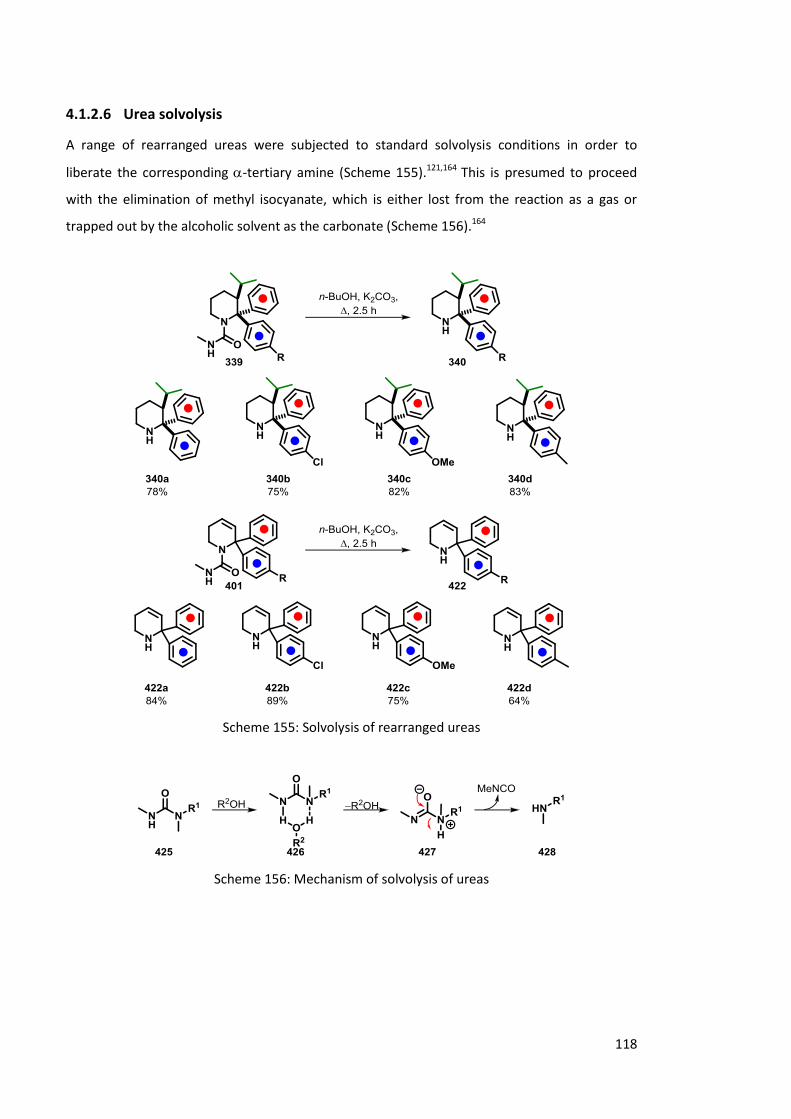
118
4.1.2.6 Urea solvolysis
A range of rearranged ureas were subjected to standard solvolysis conditions in order to
liberate the corresponding -tertiary amine (Scheme 155).121,164 This is presumed to proceed
with the elimination of methyl isocyanate, which is either lost from the reaction as a gas or
trapped out by the alcoholic solvent as the carbonate (Scheme 156).164
Scheme 155: Solvolysis of rearranged ureas
Scheme 156: Mechanism of solvolysis of ureas

119
The solvolysis conditions were found to be applicable to both ureas derived from
carbolithiation-rearrangement 339, and those derived from deprotonation-rearrangement 401.
The corresponding -tertiary amines 340 and 422 were isolated in good to excellent yields. As
noted previously, these amines would be difficult to synthesise by any other method. Indeed,
there are only 3 reported examples of 2,2-diaryl-piperidines in the literature.165-167 -Tertiary
amines are often pharmacologically active,9 with over 70% of the 200 top selling drugs
containing an amine of some description.168 The carbolithiation-rearrangement and
deprotonation-rearrangement reactions discussed within this chapter offer viable alternative
routes towards such systems.
In summary, both the tandem carbolithiation-rearrangement and aryl transfer using LDA have
proved viable routes towards the synthesis of a range of -tertiary amines (Scheme 157). The
mechanism of the tandem addition-migration reaction of 6-membered cyclic vinylic ureas has
proven to be a two-step process of syn-carbolithiation, followed by retentive rearrangement of
the aryl ring to yield the corresponding -arylated -alkylated urea.
Scheme 157: Synthesis of substituted piperidine analogues

120
Chapter 4 Synthesis of substituted piperidines
4.2 Part 2: 6-Membered cyclic vinylic ureas via a ring closing metathesis approach
As the initial method (the reaction of cyclic imines 399 with aryl isocyanates (Scheme 133,
Section 4.1.1) used to synthesise 6-membered cyclic vinylic ureas 400 failed to deliver any
functionality in the aryl ring to nitrogen, an alternative method for preparing 400 was sought.
Furthermore, problems were also encountered in using cyclic imines in the synthesis of 5-
membered cyclic vinylic ureas (See Section 5.1.1). It was envisaged that Ring Closing Metathesis
(RCM) could be used as a potential solution to these problems (Scheme 158).
Scheme 158: Metathesis as a route to cyclic vinylic ureas
4.2.1 Ring closing metathesis
Ever since the discovery of the first well defined ruthenium catalysts for olefin metathesis,169
they have played a pivotal role in the development of carbon-carbon bond forming reactions.
The original catalyst 432 is easily prepared through the reaction of 3,3-diphenylcyclopropene
431 and RuCl2(PPh3)4 430. 432 was then used in the Ring-Opening Metathesis Polymerisation
(ROMP) of norbornene 433 (Scheme 159).169 However, 432 suffered from poor functional group
tolerance and improved catalysts have been developed, although the core structure of the
catalyst (ruthenium alkylidene with 2 neutral and 2 anionic ligands) has been maintained
(Figure 21).170

121
Scheme 159: ROMP of norbornene
Figure 21 Olefin metathesis catalysts
RCM offers an attractive option for the formation of medium and large (5-8 membered) rings.
The driving force for the reaction is entropic as one molecule of substrate affords two
molecules of product, one of which is generally small and volatile, driving the reaction towards
completion.171 The first example of RCM using a ruthenium catalyst was published in 1993
(Scheme 160).172
Scheme 160: First ruthenium catalysed RCM
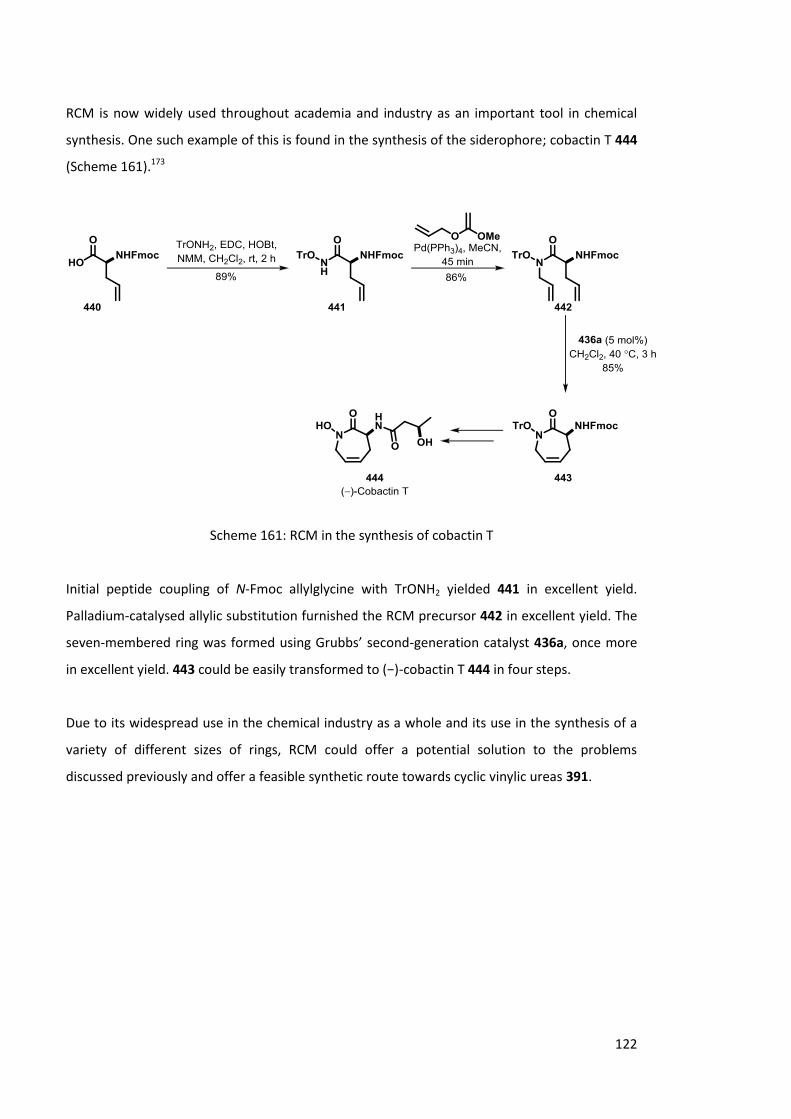
122
RCM is now widely used throughout academia and industry as an important tool in chemical
synthesis. One such example of this is found in the synthesis of the siderophore; cobactin T 444
(Scheme 161).173
Scheme 161: RCM in the synthesis of cobactin T
Initial peptide coupling of N-Fmoc allylglycine with TrONH2 yielded 441 in excellent yield.
Palladium-catalysed allylic substitution furnished the RCM precursor 442 in excellent yield. The
seven-membered ring was formed using Grubbs’ second-generation catalyst 436a, once more
in excellent yield. 443 could be easily transformed to (−)-cobactin T 444 in four steps.
Due to its widespread use in the chemical industry as a whole and its use in the synthesis of a
variety of different sizes of rings, RCM could offer a potential solution to the problems
discussed previously and offer a feasible synthetic route towards cyclic vinylic ureas 391.

123
4.2.2 6-Membered cyclic vinylic ureas by RCM.
In order to investigate whether RCM was a viable alternative for the synthesis of 6-membered
cyclic vinylic ureas 400, secondary amine 447 had to be synthesised (Scheme 162), which could
then be used to prepare metathesis precursor 449. The synthesis of the metathesis precursor
449 started with condensation of 4-chlorobenzaldehyde 445 with 3-buten-1-amine to give the
corresponding imine 446. 4-Chlorobenzaldehyde was chosen to allow the diastereospecificity of
the tandem carbolithiation-rearrangement reaction to be proven through comparison of the
results obtained with urea 338c. Exchange of the aryl rings involved in the tandem
carbolithiation-rearrangement reaction leads to the synthesis of the opposite diastereomer of
the rearranged urea (Scheme 163).131 446 was activated using diethyl zinc and subsequent
addition of vinylmagnesium bromide solution yielded the alkylated secondary amine 447 (for a
detailed analysis on the optimisation of these conditions, see Section 5.1.2).174 Diene 447 was
acylated with N-methyl-N-phenylcarbamoyl chloride 448 to give the metathesis precursor 449
in good yield over 3 steps (Scheme 162). A carbamoyl chloride was chosen to acylate the urea
instead of an isocyanate to deliver the urea in a single step. Furthermore, the urea would
require methylation if an isocyanate was used, and it was known that acyclic urea 504
underwent isomerisation of the allyl group into conjugation with the phenyl ring under basic
conditions (See Scheme 194, Section 5.1.2 for details).
Scheme 162: Synthesis of metathesis precursor 449

124
Scheme 163: Diastereospecificity of the tandem carbolithiation-rearrangement reaction
Several attempts were made to synthesise cyclic allylic urea 452 from the metathesis precursor
449 (Scheme 164, Table 18).
Scheme 164: Ring closing metathesis of 449
Entry Conditions Yield 452 (%)
1 CH2Cl2, Grubbs I (5 mol%), rt, 4 h 15
2 Toluene, Hoveyda-Grubbs II (5 mol%), , 16 h 21
3 CH2Cl2, Grubbs I (5 mol%),, 16 h 22
4 CH2Cl2, Grubbs 1 (5 mol%), Ti(i-PrO)4 (10 mol%), rt, 16 h 28
5 CH2Cl2, Grubbs 1 (5 mol%), PTSA (1 equiv.), rt, 16 h 44
Table 18: Ring closing metathesis of 449

125
Conditions that had proved effective for the synthesis of 5-membered cyclic vinylic ureas (See
Section 5.1.2) delivered the desired product in much lower yield (Table 18 entry 1), with the
majority of the remaining material found to be unreacted starting material. The more air- and
water-stable175 Hoveyda-Grubbs 2nd generation catalyst was also used, but failed to give any
marked improvement (Table 18 entry 2). Heating the reaction and prolonged reaction times
only resulted in a slight increase in yield (Table 18 entry 3). It seemed possible that this lack of
reactivity is due to coordination of the ruthenium to the urea carbonyl (Figure 22). In order to
try and overcome this, titanium isopropoxide was added, as this is known to compete with
ruthenium for coordination to basic carbonyl groups (Table 18 entry 4).176 However, this
resulted in only a small increase in the yield of 452, perhaps indicating that the reason for the
poor reactivity of 452 is more subtle. p-Toluenesulfonic acid has also been shown to increase
the reactivity of metathesis substrates by protonating basic nitrogens,177 and this did result in a
slight increase in the yield of the reaction (Table 18 entry 5).
Figure 22: Ruthenium-carbonyl coordination
With the cyclic allylic urea 452 synthesised, efforts were made to isomerise the double bond
into its vinylic counterpart 450. It was envisaged that reported ruthenium hydride transfer
catalysts could be used to do this.178-180 These catalysts are generally used to isomerise allylic
amides and amines preferentially to their enamine-(E)-isomer. This (E)-selectivity is thought to
arise due to the steric clash between the ligands on the ruthenium metal and the terminal
substituent of the double bond (shown in Scheme 165 for N-allyl and N-allyl-N-aryl amides). The
adoption of (Z)-geometry in these instances would result in steric clash between the ligands on
the ruthenium metal centre and the methyl group of the alkene.
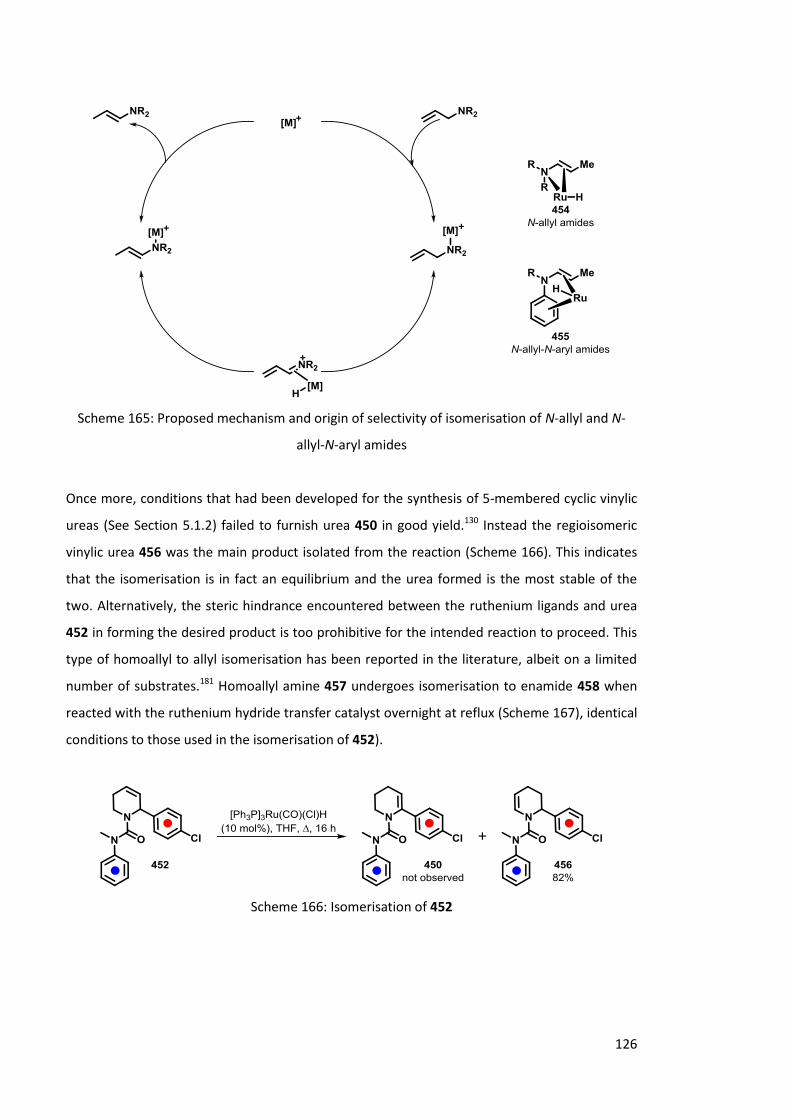
126
Scheme 165: Proposed mechanism and origin of selectivity of isomerisation of N-allyl and N-
allyl-N-aryl amides
Once more, conditions that had been developed for the synthesis of 5-membered cyclic vinylic
ureas (See Section 5.1.2) failed to furnish urea 450 in good yield.130 Instead the regioisomeric
vinylic urea 456 was the main product isolated from the reaction (Scheme 166). This indicates
that the isomerisation is in fact an equilibrium and the urea formed is the most stable of the
two. Alternatively, the steric hindrance encountered between the ruthenium ligands and urea
452 in forming the desired product is too prohibitive for the intended reaction to proceed. This
type of homoallyl to allyl isomerisation has been reported in the literature, albeit on a limited
number of substrates.181 Homoallyl amine 457 undergoes isomerisation to enamide 458 when
reacted with the ruthenium hydride transfer catalyst overnight at reflux (Scheme 167), identical
conditions to those used in the isomerisation of 452).
Scheme 166: Isomerisation of 452

127
Scheme 167: Homoallyl to allyl isomerisation
Therefore, alternative routes to urea 450 were investigated (Scheme 168). It was known that
acyclic allyl urea 504 would isomerise to vinyl urea 507 upon treatment with base during
attempts to alkylate 504 (Scheme 194, Section 5.1.2), presumably by -protonation of the allylic
anion. It was thought that this could be used to isomerise 452, providing rearrangement of the
intermediate anion could be avoided.
Scheme 168: Isomerisation of 452
Entry Conditions Result
1 NaH (10 equiv.), DMF, 16 h 452
2 LDA, THF, −78 °C, 1 h
3 LDA, Toluene, −78 °C, 1 h 452
4 LDA, Et2O, −78 °C, 1 h 452
5 KHMDS, THF, −78 °C, 1 h 450 58%
Table 19: Isomerisation of 452

128
However, treatment of 452 with an excess of sodium hydride only returned starting material
(Table 19 entry 1) presumably as it is not a strong enough base to deprotonate the benzylic
hydrogen. LDA was able to deprotonate 452; however, carrying out the reaction in THF yielded
the rearranged product 422b’ even at low temperatures and after short reaction times (Table
19 entry 2). This is unsurprising as these conditions have previously been shown to rearrange N-
allyl ureas (See Section 2.1.4).123 It was therefore thought that the use of less coordinating
solvents such as toluene or Et2O could slow down the rearrangement whilst promoting
isomerisation (Table 19 entries 3 and 4), as both solvents have previously demonstrated this
capability in the rearrangement of N-allyl ureas 289.123 Unfortunately, only decomposition of
the starting material was found and no product was isolated. KHMDS did however yield the
desired product in moderate yield (Table 19 entry 5).
4.2.2.1 Heteroaryl systems
In order to try to improve the scope of the rearrangement reaction, attempts were made to
synthesise 6-membered cyclic vinylic ureas bearing a heteroaryl substituent. Urea 462 was
made using the same method as described previously (Scheme 162), by condensation of
benzaldehyde and 3-buten-1-amine, Grignard attack and urea formation through coupling with
methyl(pyridin-2-yl)carbamic chloride 461 (Scheme 169). 461 is made in one step from the
corresponding aniline through reaction with triphosgene.182
Scheme 169: Synthesis of 462
RCM furnished the urea 463 in a much improved yield when compared to urea 452. Efforts to
isomerise the double bond once more afforded the regioisomeric vinylic urea 464 in excellent
yield, as opposed to the intended product 465 (Scheme 170).

129
Scheme 170: Synthesis of 464
4.2.3 Carbolithiation-rearrangement of 6-membered cyclic vinylic ureas
Urea 450 was subjected to standard (Scheme 145, Section 4.1.2.2) carbolithiation-
rearrangement conditions (Scheme 171).
Scheme 171: Carbolithiation-Rearrangement of 450
Urea 451 was obtained in a slightly lower yield when compared to the regioisomeric urea 339a,
48% vs. 63%. Due to the transposition of the aryl rings in the two vinylic ureas 338c and 450,
the opposite diastereomer was produced. This new route therefore allows access to either
diastereomer of the tandem addition-migration reaction by exchanging the two aryl rings.
We expected that pyridyl urea 463 would not undergo carbolithiation due to the preference of
the organolithium to attack the pyridine ring.121 Previous work within the group has shown that
it is possible to migrate a pyridine ring as long as LDA is used as the lithiation agent (Scheme 91,
Section 2.1.2).121 Both the allylic 463 and vinylic ureas 464 were therefore subjected to standard
lithiation conditions (Scheme 151, Section 4.1.2.4) in efforts to produce the corresponding
rearranged ureas 466 and 467 respectively (Scheme 172).

130
Scheme 172: Pyridyl rearrangement
Unfortunately, neither attempt at rearrangement furnished the desired urea. Lithiation of 464
only returned starting material, whilst lithiation of 463 yielded an unidentified compound which
still contained the pyridyl ring intact and three olefinic carbons.
In summary, a new synthetic route to 6-membered cyclic vinylic ureas 450 has been developed.
This new route now allows access to either diastereomer of the tandem carbolithiation-
rearrangement reaction, through exchange of the aryl rings in the starting vinylic urea (Scheme
173). However, attempts at the aryl migration of heteroaromatic rings have so far proved
unsuccessful.
Scheme 173: 6-Membered cyclic vinylic ureas by RCM and subsequent carbolithiation-
rearrangement

131
Chapter 4 Synthesis of substituted piperidines
4.3 Part 3: Enantioenriched 6-membered cyclic vinylic and allylic ureas
As part of our investigations into the synthesis of substituted piperidine analogues, a route was
desired that could be used to synthesise enantioenriched cyclic allylic 468 or vinylic ureas 470.
An aryl transfer reaction in these systems could possibly result in the production of
enantioenriched -tertiary piperidine analogues, providing the rearrangement is faster than the
epimerisation of the resultant organolithium intermediate (Scheme 174).
Scheme 174: Routes towards enantioenriched substituted piperidine analogues
4.3.1 Synthesis of enantioenriched cyclic allylic and vinylic ureas
It was postulated that the tert-butylsulfinamide chiral auxiliary 25152 could be utilised in the
synthesis of enantioenriched cyclic allylic and vinylic ureas (Scheme 175). The key step in this
synthetic plan was a diastereoselective Grignard addition to tert-butylsulfinimine 473, an area
where 25 has previously shown excellent results (See Section 1.3.3.1).19 The sulfonamide 474
could then be transformed into enantioenriched cyclic ureas using well established
methodology. Auxiliary 25 was chosen due to both enantiomers being commerically available,
meaning either enantiomer of the enantioenriched ureas could be synthesised if so desired.

132
Scheme 175: Proposed synthesis of enantioenriched cyclic allylic and vinylic ureas 470
Condensation of 25 with a range of benzaldehyde derivatives 472 using standard conditions152
developed by the Ellman group gave the corresponding tert-butylsulfinimines 473 in good to
excellent yields (Scheme 176).
Scheme 176: Synthesis of tert-butylsulfinimines 473 a Synthesised from rac-25
473 underwent diastereoselective Grignard additions to produce sulfonamides 474 in excellent
yields and diastereocontrol (Scheme 177, 474 were isolated as single diastereomers by flash
column chromatography). Again, conditions developed by the Ellman group were used.152 These
Grignard additions have been shown to proceed via the transition state 477 when these
conditions are used, allowing the relative configuration of the two chiral centres to be
determined (See Section 1.3.3.1).
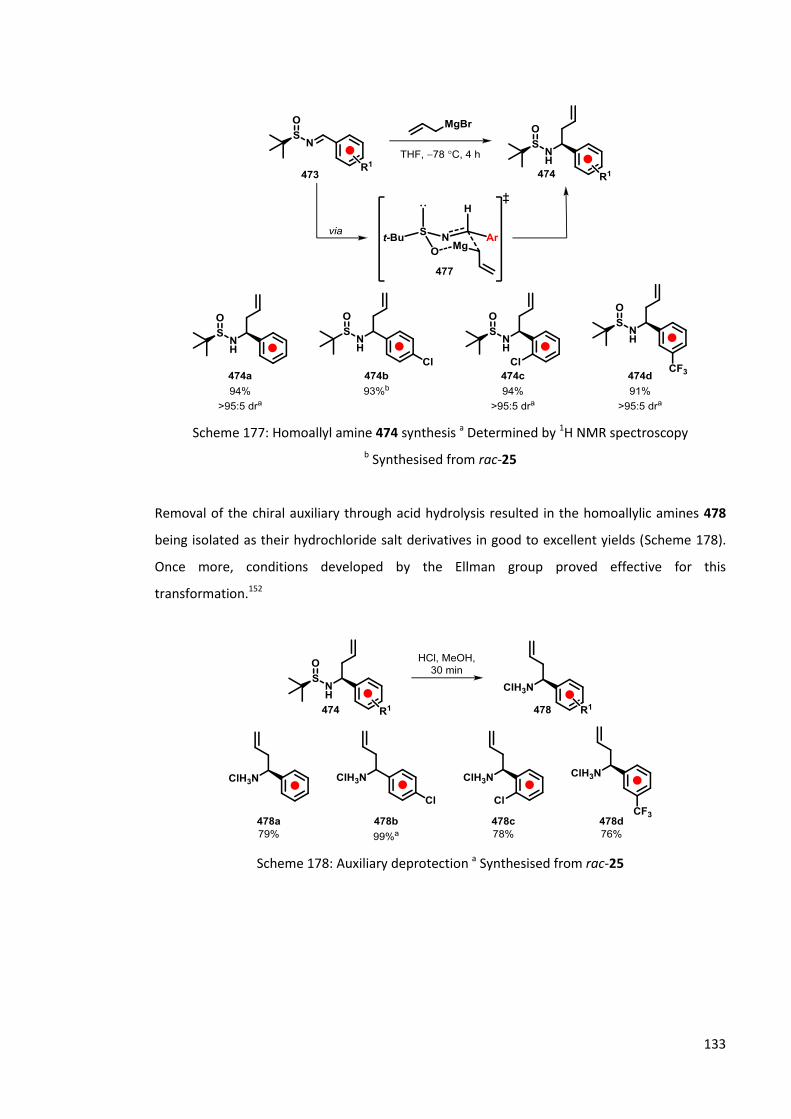
133
Scheme 177: Homoallyl amine 474 synthesis a Determined by 1H NMR spectroscopy
b Synthesised from rac-25
Removal of the chiral auxiliary through acid hydrolysis resulted in the homoallylic amines 478
being isolated as their hydrochloride salt derivatives in good to excellent yields (Scheme 178).
Once more, conditions developed by the Ellman group proved effective for this
transformation.152
Scheme 178: Auxiliary deprotection a Synthesised from rac-25

134
Initial trials into the alkylation of 478 used 1.5 equivalents of allyl bromide and an excess
amount of base (5 equivalents) at 0 °C, with warming to room temperature overnight. These
conditions were found to be highly effective and the mono-alkylated amines 475 were obtained
in good to excellent yields (Scheme 179). None of the product resulting from disubstitution was
observed in the crude NMR spectrum.
Scheme 179: Amine alkylation a Synthesised from rac-25
Amines 475 underwent efficient urea formation through acylation with a range of aryl
isocyanates and methylation (Scheme 180). It was not necessary to extend the reaction time for
the initial urea formation (the reaction of imines and aryl isocyanates was left overnight, See
Section 4.1.1) due to the increased reactivity of the amine moiety over an imine.

135
Scheme 180: Acyclic urea formation a Synthesised from rac-25
RCM of the acyclic ureas 476 afforded the equivalent enantioenriched 6-membered cyclic allylic
urea 468 in good yields (Scheme 181). Conditions that had proved successful in the synthesis of
5-membered cyclic allylic ureas 494 (See Section 5.1.2) proved to be effective for this
transformation. This is in stark contrast to the reactivity found during the synthesis of the
regioisomeric 6-membered allylic urea 452 where these conditions failed to deliver substantial
yields of urea 452 (See Section 4.2.2 for further discussion).

136
Scheme 181: RCM to furnish enantioenriched cyclic allylic ureas 468 a Synthesised from rac-25
The corresponding enantioenriched cyclic vinylic ureas 470 could be synthesised through
ruthenium-hydride mediated isomerisation of the double bond.130 A range of enantioenriched
cyclic vinylic ureas have thus been synthesised by this method in good yields and excellent
enantioenrichment (Scheme 182). None of the regioisomeric vinylic urea 338 was ever
observed in the crude NMR spectrum, possibly indicating that 470 is the more stable of the two
regioisomers.

137
Scheme 182: Enantioenriched cyclic vinylic ureas 470 a Synthesised from rac-25
4.3.2 Lithiation of enantioenriched cyclic vinylic ureas 470
A range of cyclic vinylic ureas 470 were treated under standard lithiation conditions:
deprotonation with an either LDA or an organolithium at −78 °C followed by the addition of
DMPU to promote rearrangement. 470 were treated with either LDA or an organolithium
reagent to investigate whether 470 were susceptible to deprotonation and/or carbolithiation
(Scheme 183).

138
Scheme 183: Lithiation of cyclic vinylic ureas a Using LDA b Using s-BuLi c See Scheme 184
In most cases the corresponding rearranged diaryl ureas 471 were isolated in moderate to good
yields. No issues were found in the regioselectivity of the reaction, with none of the product
resulting from allyl lithium formation observed in the crude reaction mixture. Instead, only the
urea formed by rearrangement of the benzyl lithium intermediate 480 was isolated. This is
consistent with the pKa data (benzylic pKa ~40183 vs allylic pKa ~45184) for these systems where
the more acidic benzylic proton is deprotonated preferentially over the allyl moiety.
Rearrangement in ureas where the benzylic ring bore a 2-substituent proved to be more
challenging, resulting in poor yields of the rearranged product 471. Indeed, when attempting to
migrate a 2-substituted aryl ring onto such a system no reaction took place (471e). This is
presumably due to the increase in the steric encumbrance of intermediate 480, retarding the
reaction.

139
In each case where an organolithium was used to initiate lithiation, no competing
carbolithiation of the vinylic double bond was found. It is possible to carry out carbolithiation
on the regioisomeric vinylic urea 338 (See Section 4.1.2.2), where the organolithium species
402 produced as a result of carbolithiation is stabilised through conjugation with the aromatic
ring (Scheme 145). This conjugation is no longer present and explains why no carbolithiation is
observed. Rearrangements were generally much faster than the tandem carbolithiation-
rearrangement reaction, with complete consumption of the starting material usually observed
after a few hours compared with the overnight reaction times found for carbolithiation-
rearrangement (See Section 4.1.2.2). Whilst trying to transfer a 2-fluorophenyl substituent in
urea 470b, tricycle 484 was the major product. This presumably arises from nucleophilic
aromatic substitution at the ortho-position of the aromatic ring, rather than the standard ipso-
attack (Scheme 184). Unfortunately, all products were isolated as racemic mixtures, indicating
that the rate of racemisation of the benzyllithium intermediate is faster than the rate of
rearrangement in cyclic systems.122
Scheme 184: Lithiation of 470b

140
4.3.3 Lithiation of enantioenriched cyclic allylic ureas 468
The lithiation of a range of enantioenriched cyclic allylic ureas 468 were also investigated. 468
were once more deprotonated with either LDA or an organolithium using the same conditions
as for the corresponding enantioenriched cyclic vinylic ureas 470 (Scheme 185).
Scheme 185: Lithiation of enantioenriched cyclic allylic ureas 468
None of the expected diaryl urea 469 was obtained. Instead urea 489 was isolated with
migration of the aryl ring to the less sterically hindered side of the tetrahydropyridine scaffold.
489 can be rationalised by the formation of allyl lithium intermediate 487 which is then able to
rearrange to form 2,6-disubstituted cyclic allylic urea 489 (Scheme 186). This is despite the
benzylic position being more acidic and therefore more prone to deprotonation. This
presumably means in the deprotonation of 468 kinetic acidity has a greater effect, due to a lack
of steric hindrance, than thermodynamic acidity, whilst the opposite is true for the vinylic
counterparts 470. It is unlikely that there is an equilibrium between the two possible
organolithium intermediates 485 and 487 with only 487 rearranging, as we know 485 is prone
to rearrangement when the double bond is in the vinylic position.
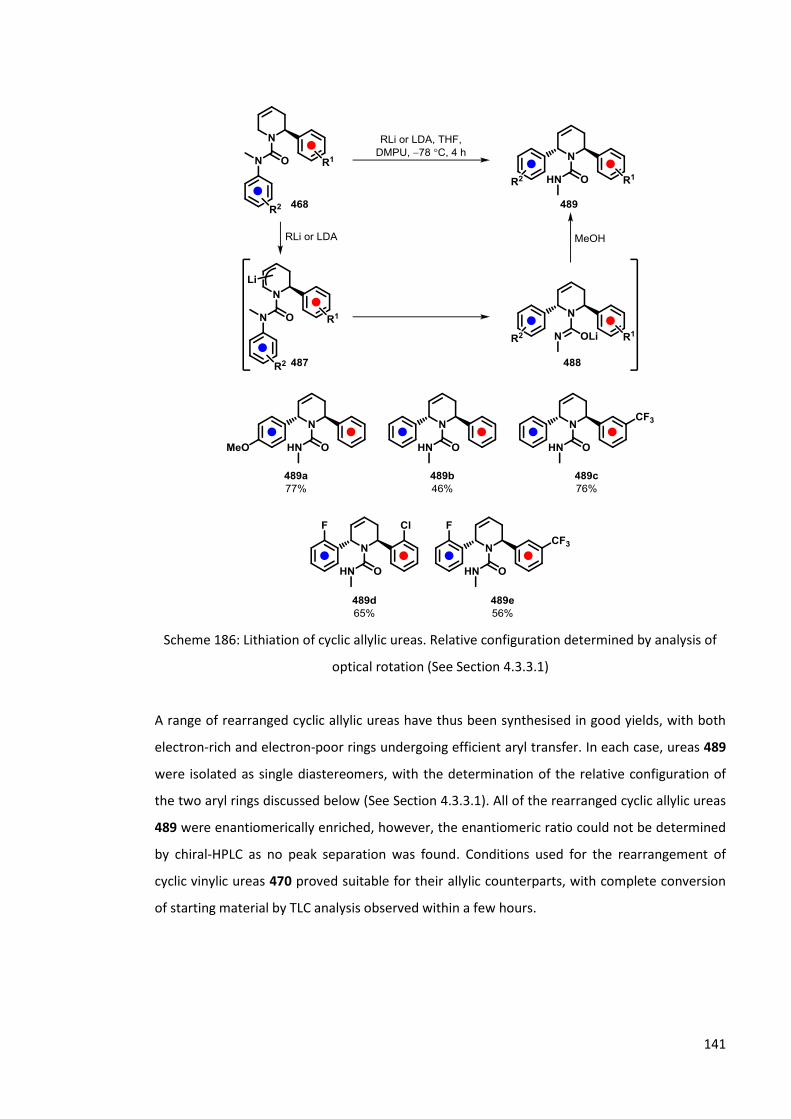
141
Scheme 186: Lithiation of cyclic allylic ureas. Relative configuration determined by analysis of
optical rotation (See Section 4.3.3.1)
A range of rearranged cyclic allylic ureas have thus been synthesised in good yields, with both
electron-rich and electron-poor rings undergoing efficient aryl transfer. In each case, ureas 489
were isolated as single diastereomers, with the determination of the relative configuration of
the two aryl rings discussed below (See Section 4.3.3.1). All of the rearranged cyclic allylic ureas
489 were enantiomerically enriched, however, the enantiomeric ratio could not be determined
by chiral-HPLC as no peak separation was found. Conditions used for the rearrangement of
cyclic vinylic ureas 470 proved suitable for their allylic counterparts, with complete conversion
of starting material by TLC analysis observed within a few hours.

142
4.3.3.1 Determination of relative configuration of rearranged cyclic allylic ureas
In order to establish the relative configuration of the two aryl rings in 489, it was envisaged that
hydrogenation of the allylic double bond in a system where the two aryl rings were identical
could yield information on the conformation of piperidine 490 (Scheme 187). NMR analysis
could then be used to determine if the aromatic substituents are occupying an axial or
equatorial position, and from this we would then be able to determine the relative
configuration of the aryl rings in piperidine 490, and by analogy, urea 489.
Scheme 187: Hydrogenation of 489b
Urea 489b was subjected to a range of hydrogenation conditions with a view to synthesising
piperidine 490 (Table 20).
Entry Conditions Result (Yield, %)
1 H2 (1 atm), Pd/C (10 mol%), MeOH, 16 h
(72%)
2 H2 (1 atm), Crabtree’s cat (10 mol%), CH2Cl2, 16 h 489b
3 H2 (1 atm), RhCl(PPh3)3 (10 mol%), IPA, 16 h 489b
4 NBSH,a MeCN, 16 h 489b
5 H2 (1 atm), Pd/C (10 mol%), IPA, 2 h 490 (66%)
Table 20: Hydrogenation of 489b a NBSH: 2-nitrobenzenesulfonylhydrazide

143
Standard hydrogenation conditions successfully reduced the allylic double bond but also
resulted in C-N hydrogenolysis (Table 20 entry 1). Crabtree’s catalyst was therefore chosen due
to its known ability to reduce olefinic double bonds in the presence of Cbz protecting groups.185
However, this only returned unreacted starting material even with prolonged reaction times
(Table 20 entry 2). Similar observations were found when the hydrogenation was attempted
using Wilkinson’s catalyst (Table 20 entry 3) or through the use of a diimide reduction (Table 20
entry 4).186 As a result a change in the reaction conditions rather than the catalyst or reduction
source was investigated. A change in solvent to IPA was studied as it is known that IPA can slow
down hydrogenation reactions and it was hoped that IPA could also slow down the
hydrogenolysis that was occurring during the reaction.187 Similarly, previous work within the
group has also shown that degassing of reaction solvent could prevent hydrogenolysis from
occurring during the hydrogenation of acyclic vinylic ureas 318.188 Pleasingly, use of these
conditions, along with careful monitoring of the reaction by TLC analysis led to the isolation of
piperidine 490 in good yield (Table 20 entry 5).
Detailed analysis of the 1H NMR spectrum has led to some interesting conclusions on the
conformation and relative configuration of 490 (Figure 23).
Figure 23: 1H NMR spectrum of 490

144
Assuming 490 is adopting a chair conformation, the broad triplet at 5.3 ppm for the two
benzylic protons would appear to indicate the molecule is adopting a configuration where the
phenyl rings are cis to one another (Figure 24A). This is because if the molecule adopted a
configuration where the aryl rings were trans to one another then one would expect this signal
to be split more into a doublet of doublets with one large axial-axial coupling and one smaller
axial-equatorial coupling (Figure 24B). Furthermore, as the triplet has a relatively small coupling
constant of 3.8 Hz, it would appear that both phenyl rings are in an axial position as neither
proton is experiencing any axial-axial coupling. This is consistent with the information gained
from carbolithiated urea 413c where 1,3-allylic strain forced the large phenyl group in the 2-
position to adopt an axial position (See Section 4.1.2.3).163 However, the symmetry of the signal
at 1.45 ppm would seem to indicate the two protons in the 4-position of the piperidine are not
diastereotopic, contradicting the information gained from the benzylic protons.
Figure 24: Possible configurations and conformations of 490
The relative configuration of the two aryl rings was confirmed to be trans by optical rotation. If
the two aryl rings in 490 were cis to one another, the compound would be meso and therefore
not give an optical rotation. The molecule does however give an []D25 with a value of −40.4.
The triplet signal for the benzylic protons in the 1H NMR spectrum is therefore rationalised by
the system rapidly ring flipping between two chair conformations, on the NMR timescale. This
averages out the two double doublets into one relatively broad triplet (Figure 25). This has
established the relative configuration of the two phenyl rings in 490, and from this we assume
the configuration of other cyclic allylic ureas 489 to be the same.
Figure 25: Configuration of 490

145
4.3.4 Urea Solvolysis
In order to prove that ureas bearing two different aryl groups also have a trans-configuration,
urea 489a was deprotected to its corresponding amine 492 using standard solvolysis conditions.
492 was then converted into its hydrochloride salt 493, and crystallisation studies are currently
on-going for definitive proof of the relative configuration of cyclic allylic ureas 489 (Scheme
188).
Scheme 188: Urea solvolysis of 489a
In summary, enantioenriched cyclic allylic 470 and vinylic 468 ureas undergo distinctly different
aryl transfer reactions. Allylic ureas 470 participate in a sterically less demanding
rearrangement through allyl lithium intermediate 487. The relative configuration of the
resultant urea 489 has been determined through its optical rotation. Vinylic ureas 468 undergo
a benzylic deprotonation and migration of the aryl ring to give diaryl urea 469. No
enantioenrichment was found in rearrangement products 469 resulting from lithiation of cyclic
vinylic ureas 468. The products 489 arising from rearrangement of cyclic allylic urea 470 are
enantiomerically enriched, however their er’s have not been determined (Scheme 189).
Scheme 189: Rearrangement of enantioenriched 6-membered cyclic ureas

146
Chapter 5 Synthesis of substituted pyrrolidines and pyrrolines
As part of our research into the synthesis of cyclic -tertiary amines, it was also decided to
investigate the lithiation of 5-membered systems. Both the carbolithiation-rearrangement and
deprotonation-rearrangement reactions will be studied (Scheme 190). The product distribution
of the deprotonation-rearrangement reaction will also be analysed to assess whether the
pyrroline analogues react in a similar fashion to 6-membered allylic and benzylic ureas 338 (to
give 495) or 6-membered allylic and non-benzylic ureas 470 (to give 496). If successful this
would allow the synthesis of substituted pyrrolidine (via carbolithiation-rearrangement) or
pyrroline (via deprotonation-rearrangement) analogues.
Scheme 190: Pyrrolines 342, 495 and 496
5.1 Synthesis of 5-membered cyclic ureas
5.1.1 By the reaction of cyclic imines and aryl isocyanates
Synthesis of 5-membered cyclic vinylic ureas 341 was originally attempted using the
methodology developed previously for 6-membered analogues (See Section 4.1.1). Initial
cyclisation to form cyclic imine 498 was successful,158 but attempts to acylate 498 with a range
of aryl isocyanates were unsuccessful (Scheme 191).
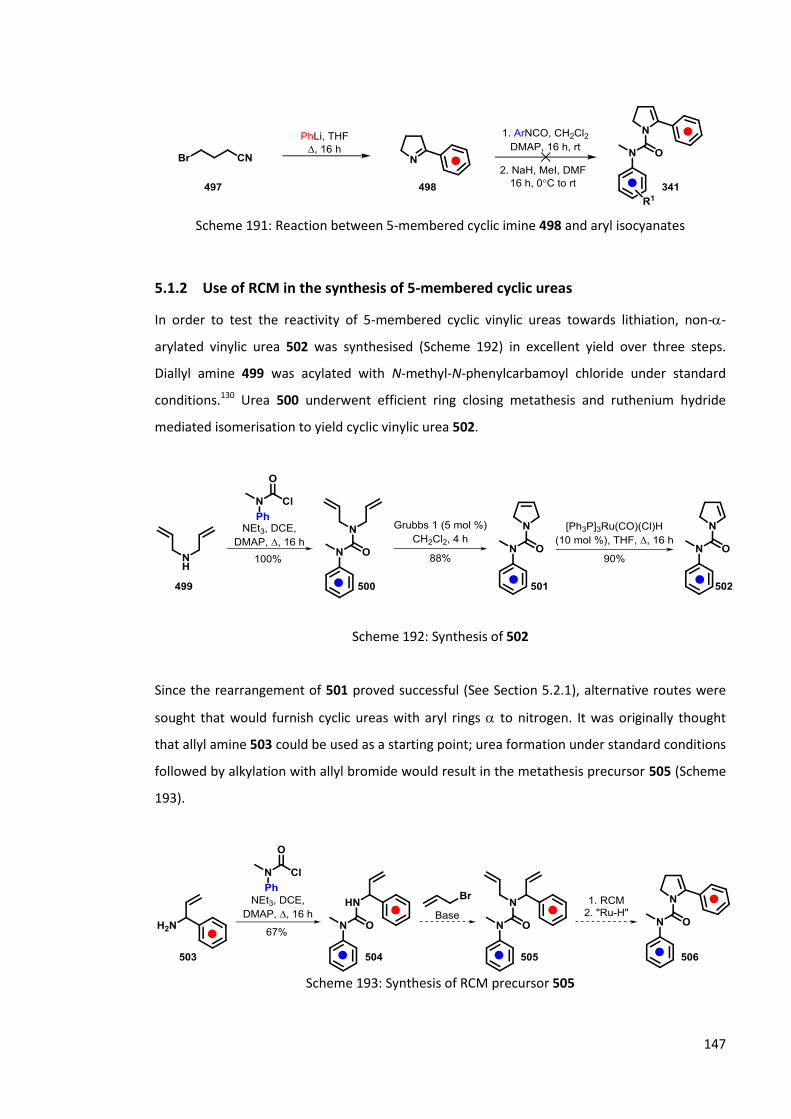
147
Scheme 191: Reaction between 5-membered cyclic imine 498 and aryl isocyanates
5.1.2 Use of RCM in the synthesis of 5-membered cyclic ureas
In order to test the reactivity of 5-membered cyclic vinylic ureas towards lithiation, non--
arylated vinylic urea 502 was synthesised (Scheme 192) in excellent yield over three steps.
Diallyl amine 499 was acylated with N-methyl-N-phenylcarbamoyl chloride under standard
conditions.130 Urea 500 underwent efficient ring closing metathesis and ruthenium hydride
mediated isomerisation to yield cyclic vinylic urea 502.
Scheme 192: Synthesis of 502
Since the rearrangement of 501 proved successful (See Section 5.2.1), alternative routes were
sought that would furnish cyclic ureas with aryl rings to nitrogen. It was originally thought
that allyl amine 503 could be used as a starting point; urea formation under standard conditions
followed by alkylation with allyl bromide would result in the metathesis precursor 505 (Scheme
193).
Scheme 193: Synthesis of RCM precursor 505

148
However, alkylation of 504 with allyl bromide failed to produce the desired allyl urea 505 under
a range of conditions (Scheme 194, Table 21). Instead the vinylic analogue 507 was obtained
exclusively as the Z-isomer, with the double bond isomerised into conjugation with the aryl ring.
A range of different bases was tested (NaH, NaHMDS, LiHMDS), but each yielded 507 in
excellent yields.
Scheme 194: Alkylation of 504
Entry Base Yield 505 (%) Yield 507 (%)
1 NaH - 93
2 NaHMDS - 85
3 LiHMDS - 88
Table 21: Alkylation of 507
As a result an alternative route was developed for the synthesis of 5-membered cyclic vinylic
341 and allylic 494 ureas. 341 and 494 were easily synthesised by a method analogous to the
metathesis route developed for 6-membered cyclic ureas (See Section 4.2.2), with allyl amine
instead of 3-buten-1-amine used in the initial imine formation step (Scheme 195).
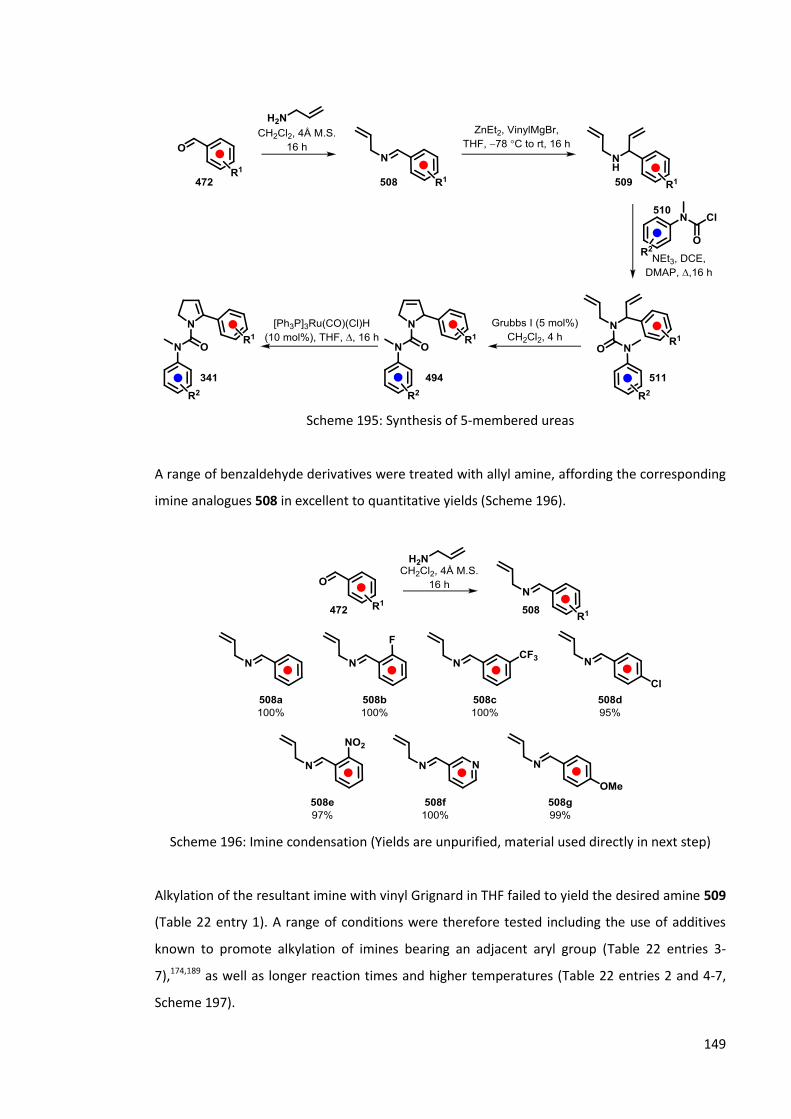
149
Scheme 195: Synthesis of 5-membered ureas
A range of benzaldehyde derivatives were treated with allyl amine, affording the corresponding
imine analogues 508 in excellent to quantitative yields (Scheme 196).
Scheme 196: Imine condensation (Yields are unpurified, material used directly in next step)
Alkylation of the resultant imine with vinyl Grignard in THF failed to yield the desired amine 509
(Table 22 entry 1). A range of conditions were therefore tested including the use of additives
known to promote alkylation of imines bearing an adjacent aryl group (Table 22 entries 3-
7),174,189 as well as longer reaction times and higher temperatures (Table 22 entries 2 and 4-7,
Scheme 197).

150
Scheme 197: Imine alkylation
Entry Conditions Yield 509 (%) Yield 511 (%, 2 steps)
1 No additive, rt, 3 h - -
2 No additive, rt, 16 h - 15
3 ZnEt2, −78 °C, 3 h - 34
4 ZnEt2, −78 °C to rt, 3 h - 43
5 ZnEt2, −78 °C to rt, 16 h - 57
6 LaCl3.2LiCl, 0 °C, 3 h - 29
7 ZnEt2, −78 °C to rt, 16 h 23 -
Table 22: Optimisation of Grignard addition
As yields for the reaction were found to be much lower if intermediate amine 509 was isolated
(Table 22 entry 7), the crude reaction mixture was taken through to an acylation with a range of
carbamoyl chlorides 510 (Scheme 198).130 Low yields of urea 511 were found when no additive
was used during the alkylation or when the reaction was carried out at low temperatures for
short reaction times (Table 22 entries 1-3). Better yields were found when diethyl zinc (Table 22
entry 4)174 was used as an additive when compared to LaCl3.2LiCl (Table 22 entry 6).189 As a
result, optimal conditions were found where the addition of the additive and Grignard were
carried out at low temperatures before allowing the mixture to warm to room temperature
overnight (Table 22 entry 5). The crude mixture could then be acylated with a carbamoyl
chloride using standard conditions.130 A small library of RCM precursors 511 were synthesised in
moderate yields over two steps using these optimised conditions (Table 22 entry 5).
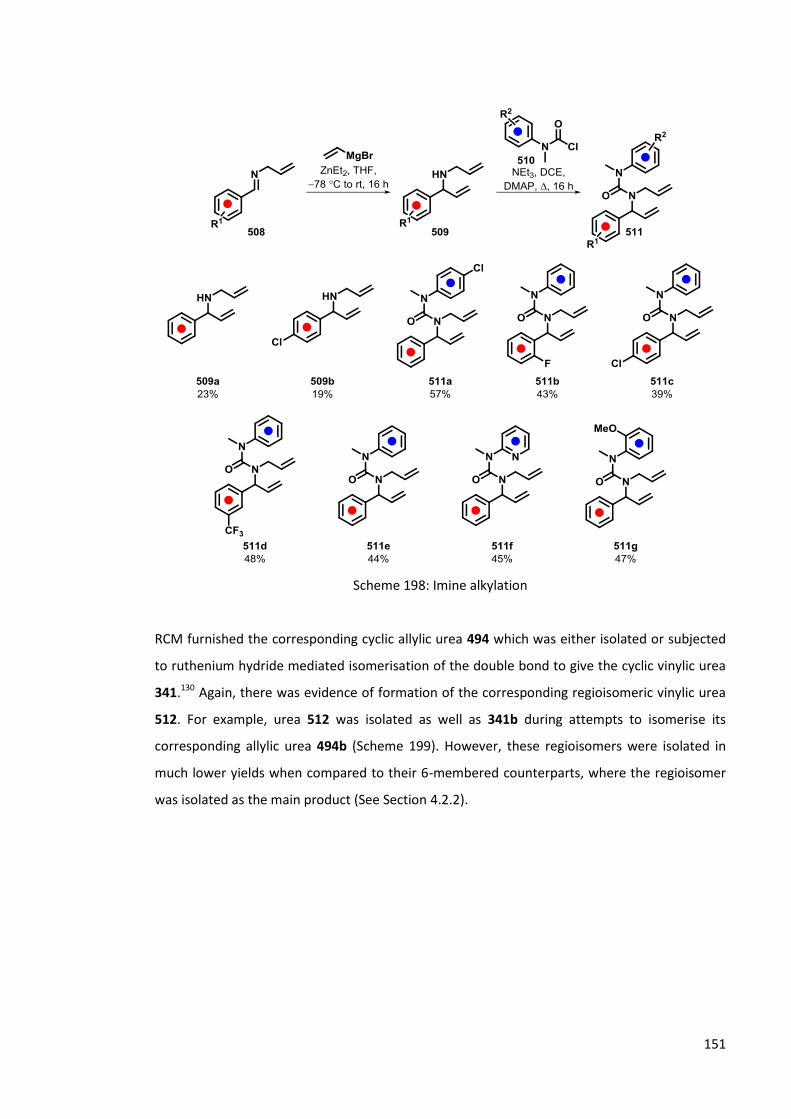
151
Scheme 198: Imine alkylation
RCM furnished the corresponding cyclic allylic urea 494 which was either isolated or subjected
to ruthenium hydride mediated isomerisation of the double bond to give the cyclic vinylic urea
341.130 Again, there was evidence of formation of the corresponding regioisomeric vinylic urea
512. For example, urea 512 was isolated as well as 341b during attempts to isomerise its
corresponding allylic urea 494b (Scheme 199). However, these regioisomers were isolated in
much lower yields when compared to their 6-membered counterparts, where the regioisomer
was isolated as the main product (See Section 4.2.2).

152
Scheme 199: Synthesis of cyclic vinylic ureas 341 a Yield over two steps
5.2 Lithiation of 5-membered cyclic ureas
5.2.1 Lithiation of cyclic allylic urea 501
Initial investigations were centred on the cyclic allylic urea 501 to ensure the rearrangement
worked on 5-membered systems. 501 was treated with LDA at −78 °C in the presence of DMPU
(Scheme 200). The rearranged urea 514 was obtained in good yield, with concomitant migration
of the double bond into conjugation with the aryl ring. A small amount of the product 515
resulting from electrocyclic opening of the ring was also found as a side product. There has only
been one previous report of a similar electrocyclic ring opening; where treatment of pyrroline
salt 516 with phenyl lithium results in the formation of amine 517 as a mixture of E and Z
isomers (Scheme 201).190
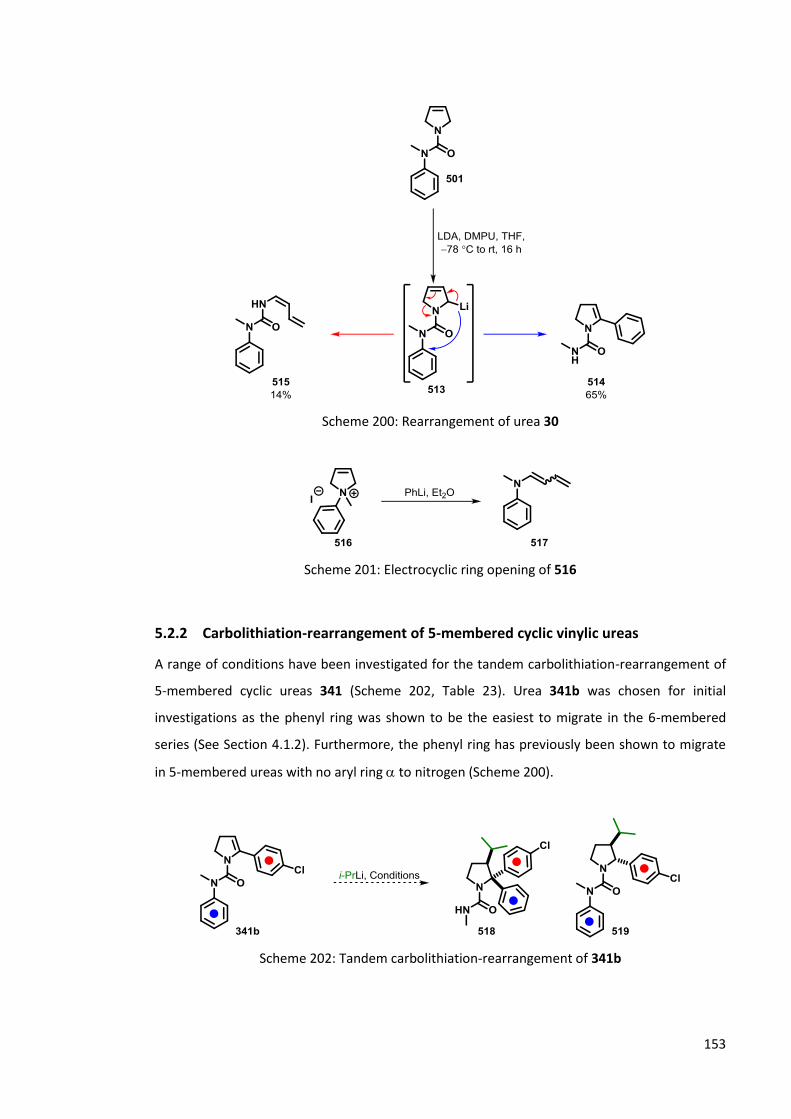
153
Scheme 200: Rearrangement of urea 30
Scheme 201: Electrocyclic ring opening of 516
5.2.2 Carbolithiation-rearrangement of 5-membered cyclic vinylic ureas
A range of conditions have been investigated for the tandem carbolithiation-rearrangement of
5-membered cyclic ureas 341 (Scheme 202, Table 23). Urea 341b was chosen for initial
investigations as the phenyl ring was shown to be the easiest to migrate in the 6-membered
series (See Section 4.1.2). Furthermore, the phenyl ring has previously been shown to migrate
in 5-membered ureas with no aryl ring to nitrogen (Scheme 200).
Scheme 202: Tandem carbolithiation-rearrangement of 341b

154
Entry Conditions Yield 518 (%) rear Yield 519 (%) carb
1 THF, −78 °C, 3 h 0 0
2 THF, DMPU, −78 °C, 3 h 0 0
3 Toluene, DMPU, −40 °C, 3 h Traces 0
4 THF, DMPU, −78 to 0 °C, 4.5 h Traces 0
5 THF, DMPU, −78 to 0 °C, 16 h Traces 0
6 THF, DMPU, −78 °C to rt, 4.5 h Decomposition Decomposition
7 Toluene, DMPU, −78 °C to rt, 16 h Decomposition Decomposition
Table 23: Tandem carbolithiation-rearrangement of 341b
Despite testing a range of conditions, including ones optimised for the tandem carbolithiation-
rearrangement of 6-membered cyclic vinylic ureas (Table 23 entry 7), no appreciable amounts
of either the carbolithiated urea 519 or the -alkylated -arylated urea 518 were isolated.
Initial tests resulted in only the return of unreacted starting material (Table 23 entries 1 and 2).
Change of the solvent to toluene or allowing the reaction to warm to 0 °C did result in small
amounts (<10 %) of a rearranged compound being observed in the crude reaction mixture
(Table 23 entries 3-5). However, warming the reaction to room temperature resulted in
complete decomposition of the reaction mixture. The fact that no carbolithiation was observed
in these reactions would seem to indicate that there is an inherent problem in the initial
organolithium addition of these systems. Indeed there are no reports of carbolithiation of 2-
pyrroline systems in the literature.
5.2.3 Lithiation of 5-membered-cyclic allylic ureas
6-Membered cyclic allylic ureas 468 undergo an aryl transfer to the least sterically encumbered
side of the cyclic urea (See Section 4.3.3). Because of this, the deprotonation of their 5-
membered counterparts was also investigated. It was thought that lithiation of 494 could
highlight some useful mechanistic insights as to whether the rearrangement prefers to proceed
through the least sterically hindered allyl lithium intermediate 520 or the benzylic and allylic
lithium intermediate 521 (Scheme 203).

155
Scheme 203: Lithiation of 5-membered cyclic allylic ureas
A range of ureas was subjected to the standard (See Section 4.3.3) lithiation conditions (Scheme
204). Urea 495 was the favoured product, with none of urea 496 ever found in the crude
reaction mixture, indicating that electronics can play a more important role than sterics in the
lithiation of cyclic systems.
Scheme 204: Lithiation of 5-membered cyclic allylic ureas
Yields varied quite substantially depending on the system being lithiated, with a phenyl ring
being migrated in each case. Unfortunately, the migration of a 2-pyridyl ring failed to give the
rearranged product 495d, with only decomposition of the starting material observed.
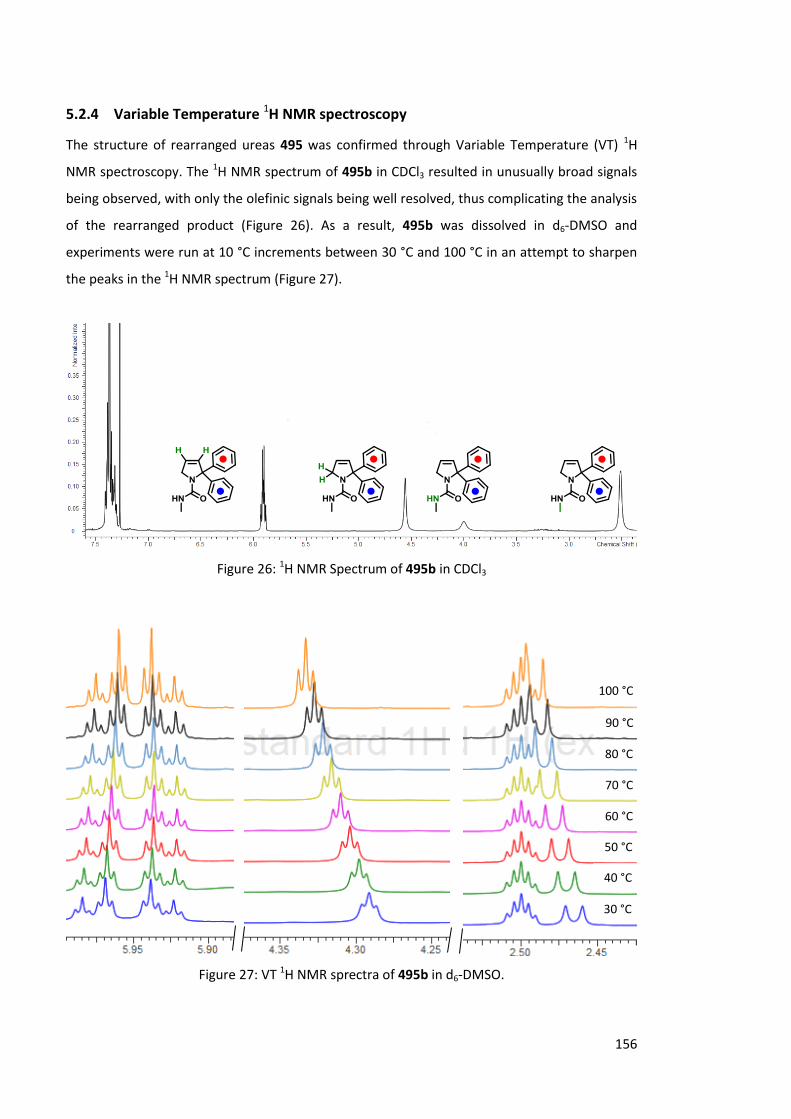
156
5.2.4 Variable Temperature 1H NMR spectroscopy
The structure of rearranged ureas 495 was confirmed through Variable Temperature (VT) 1H
NMR spectroscopy. The 1H NMR spectrum of 495b in CDCl3 resulted in unusually broad signals
being observed, with only the olefinic signals being well resolved, thus complicating the analysis
of the rearranged product (Figure 26). As a result, 495b was dissolved in d6-DMSO and
experiments were run at 10 °C increments between 30 °C and 100 °C in an attempt to sharpen
the peaks in the 1H NMR spectrum (Figure 27).
Figure 26: 1H NMR Spectrum of 495b in CDCl3
Figure 27: VT 1H NMR sprectra of 495b in d6-DMSO.
100 °C
90 °C
80 °C
70 °C
50 °C
60 °C
40 °C
30 °C
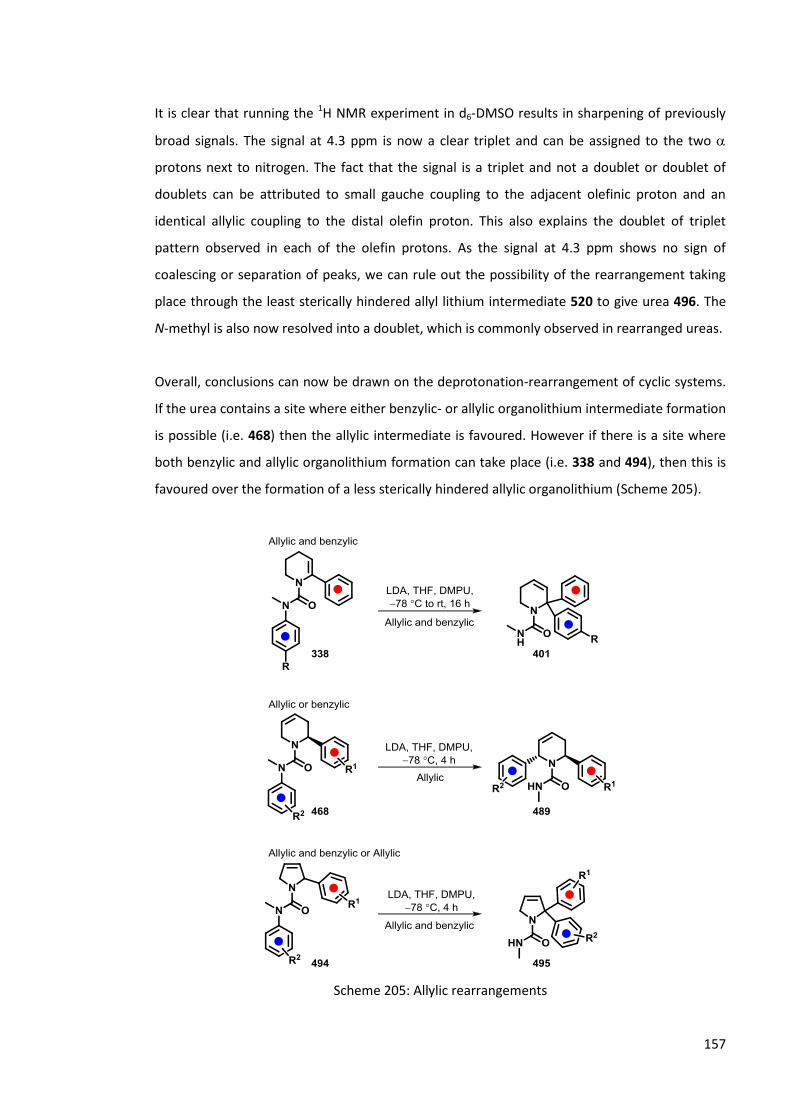
157
It is clear that running the 1H NMR experiment in d6-DMSO results in sharpening of previously
broad signals. The signal at 4.3 ppm is now a clear triplet and can be assigned to the two
protons next to nitrogen. The fact that the signal is a triplet and not a doublet or doublet of
doublets can be attributed to small gauche coupling to the adjacent olefinic proton and an
identical allylic coupling to the distal olefin proton. This also explains the doublet of triplet
pattern observed in each of the olefin protons. As the signal at 4.3 ppm shows no sign of
coalescing or separation of peaks, we can rule out the possibility of the rearrangement taking
place through the least sterically hindered allyl lithium intermediate 520 to give urea 496. The
N-methyl is also now resolved into a doublet, which is commonly observed in rearranged ureas.
Overall, conclusions can now be drawn on the deprotonation-rearrangement of cyclic systems.
If the urea contains a site where either benzylic- or allylic organolithium intermediate formation
is possible (i.e. 468) then the allylic intermediate is favoured. However if there is a site where
both benzylic and allylic organolithium formation can take place (i.e. 338 and 494), then this is
favoured over the formation of a less sterically hindered allylic organolithium (Scheme 205).
Scheme 205: Allylic rearrangements

158
In summary, a range of 5-membered cyclic allylic ureas 494 undergo an aryl transfer reaction
through a benzylic and allylic lithium intermediate 521 to form -tertiary ureas 495.
Additionally, attempts to extend the scope of the tandem carbolithiation-rearrangement
reaction to 5-membered vinylic ureas 341 to give 342 were carried out, but with no success
(Scheme 206).
Scheme 206: Rearrangement of pyrroline ureas
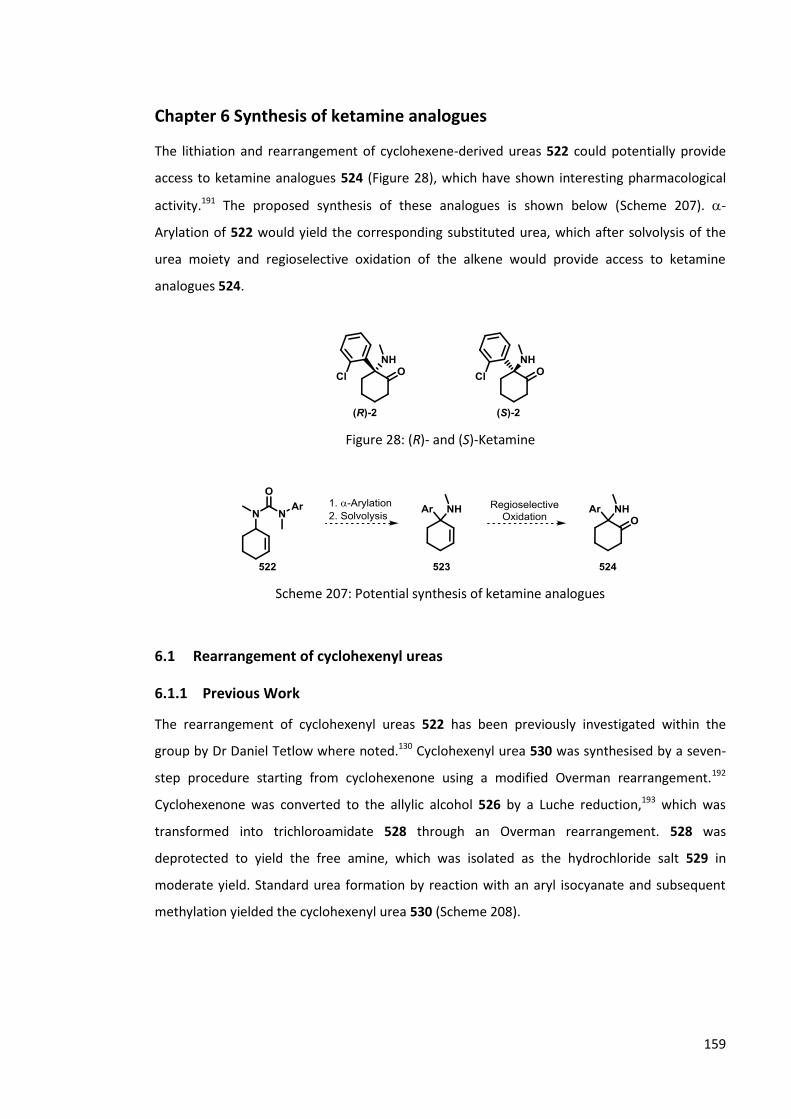
159
Chapter 6 Synthesis of ketamine analogues
The lithiation and rearrangement of cyclohexene-derived ureas 522 could potentially provide
access to ketamine analogues 524 (Figure 28), which have shown interesting pharmacological
activity.191 The proposed synthesis of these analogues is shown below (Scheme 207). -
Arylation of 522 would yield the corresponding substituted urea, which after solvolysis of the
urea moiety and regioselective oxidation of the alkene would provide access to ketamine
analogues 524.
Figure 28: (R)- and (S)-Ketamine
Scheme 207: Potential synthesis of ketamine analogues
6.1 Rearrangement of cyclohexenyl ureas
6.1.1 Previous Work
The rearrangement of cyclohexenyl ureas 522 has been previously investigated within the
group by Dr Daniel Tetlow where noted.130 Cyclohexenyl urea 530 was synthesised by a seven-
step procedure starting from cyclohexenone using a modified Overman rearrangement.192
Cyclohexenone was converted to the allylic alcohol 526 by a Luche reduction,193 which was
transformed into trichloroamidate 528 through an Overman rearrangement. 528 was
deprotected to yield the free amine, which was isolated as the hydrochloride salt 529 in
moderate yield. Standard urea formation by reaction with an aryl isocyanate and subsequent
methylation yielded the cyclohexenyl urea 530 (Scheme 208).

160
Scheme 208: Synthesis of Cyclohexenyl urea 530 *
The N-PMP analogue 534 was also synthesised, using a method described in the literature.194
Cyclohexenyl amine 533 was converted to the desired urea 534 by reaction with phenyl
isocyanate and methylation in excellent yield (Scheme 209). 534 was synthesised as the scope
of the rearrangement of acyclic allylic ureas 289 was increased when the methyl group was
replaced with a PMP group (See Section 2.1.4).
Scheme 209: Synthesis of N-PMP cyclohexenyl urea 534 *
Initial lithiation studies were carried out on both N-Me 530 and N-PMP 534 cyclohexenyl ureas.
Lithiation of 530 with LDA followed by DMPU addition resulted in the recovery of unreacted
starting material. However, lithiation with s-BuLi gave regioisomer 536, indicating
deprotonation had taken place with the allyl lithium intermediate forced to adapt a trans-
conformation due to the constraints of the cyclic system. Lithiation of 534 with lithium amide
537 gave slight conversion to the desired product 538 only after the reaction was warmed to
−50 °C (Scheme 210).
* By Dr Daniel Tetlow

161
Scheme 210: Lithiation of 530 and 534 *
6.1.2 Synthesis of cyclohexenyl ureas
We have since set about developing an improved synthesis of cyclohexenyl ureas 530.
Cyclohexenyl amine 540 was synthesised in one step from commerically available 3-
bromocyclohexene 539.195 Amine 540 was then converted to the corresponding urea 541 using
the same methodology described previously (See Section 6.1.1). A range of cyclohexenyl ureas
have thus been made in good to excellent yields over 3 steps (Scheme 211).
* By Dr Daniel Tetlow

162
Scheme 211: Synthesis of cyclohexenyl ureas 541
6.1.3 Lithiation of cyclohexenyl ureas
Due to the low yields found earlier in the rearrangement of 530 which contained a phenyl ring
(See Section 6.1.1), substrate 541d bearing a 4-chlorophenyl ring was chosen for initial
investigations. The 4-chlrophenyl ring was chosen such that a ketamine analogue could be
prepared, without synthesising actual ketamine for a number of legal and health and safety
reasons (Scheme 212).
Scheme 212: Lithiation of 541d

163
Lithiation of 541d with s-BuLi followed by DMPU addition with prolonged stirring and warming
of the reaction to room temperature yielded the desired product 544 in excellent yield.
Lithiation with LDA instead of s-BuLi also led to the formation of 544, albeit in poorer yield. It
was also found that control of the internal temperature of the reaction was critical to the
success of the reaction. The organolithium intermediate was found to be unstable at
temperatures exceeding −60 °C, with no starting urea 541d found and only decomposition
products isolated. It was necessary therefore to ensure this temperature was not exceeded
during both organolithium and DMPU addition. These optimised conditions were used to
synthesise a range of cyclohexenyl ureas 545. In most cases TLC analysis showed complete
consumption of starting material within two hours of DMPU addition, avoiding the need for
prolonged reaction times or elevated reaction temperature (Scheme 213).
Scheme 213: Synthesis of rearranged cyclohexenyl ureas 545 a 16 h b 4 h c Quenched by
bubbling air through the reaction mixture d 1 g scale reaction e 2 g scale reaction

164
Yields were found to be good to excellent for most ureas, with only poor yields found for the
migration of electron-rich and neutral aryl rings (545c and 545f). Additionally, the reaction
could be performed on a multigram scale, with minimal loss in yield. A dearomatised
intermediate of the reaction was trapped out by bubbling air through the reaction medium
when the migrating ring was 1-naphthyl.119 Spirocyclic enone 545b is formed through the
oxidation of allyl lithium intermediate 547 (Scheme 214). It should be noted that a 1-naphthyl
ring is the only migrating group that allows the isolation of 545b, the only system that
generates allyl lithium intermediate 547, which is susceptible to oxidation (See Section 2.1).
Scheme 214: Mechanism of formation of 545b
6.1.4 Solvolysis of cyclohexenyl ureas
A range of rearranged cyclohexenyl ureas 545 were then subjected to microwave assisted
deprotection conditions (Scheme 215), with the corresponding cyclohexenyl amines 548
isolated in excellent yields. This allows the synthesis of highly hindered allylic -tertiary amines
that would be difficult to synthesise by other methods.
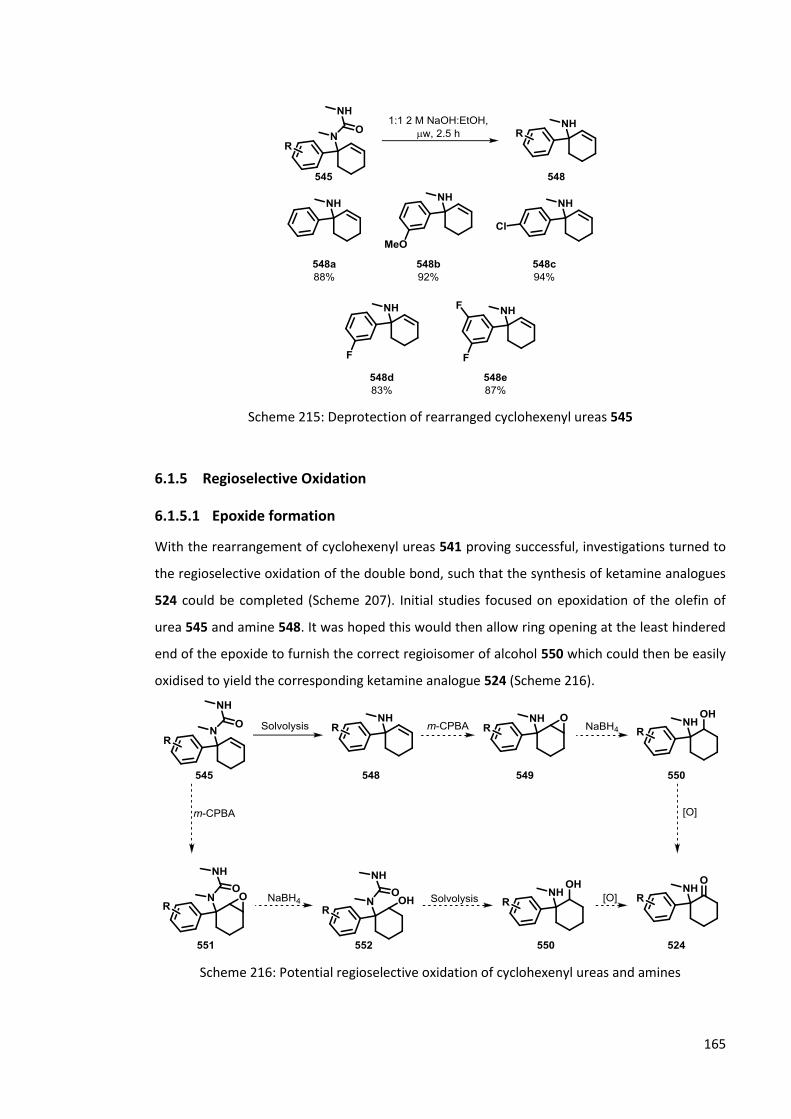
165
Scheme 215: Deprotection of rearranged cyclohexenyl ureas 545
6.1.5 Regioselective Oxidation
6.1.5.1 Epoxide formation
With the rearrangement of cyclohexenyl ureas 541 proving successful, investigations turned to
the regioselective oxidation of the double bond, such that the synthesis of ketamine analogues
524 could be completed (Scheme 207). Initial studies focused on epoxidation of the olefin of
urea 545 and amine 548. It was hoped this would then allow ring opening at the least hindered
end of the epoxide to furnish the correct regioisomer of alcohol 550 which could then be easily
oxidised to yield the corresponding ketamine analogue 524 (Scheme 216).
Scheme 216: Potential regioselective oxidation of cyclohexenyl ureas and amines
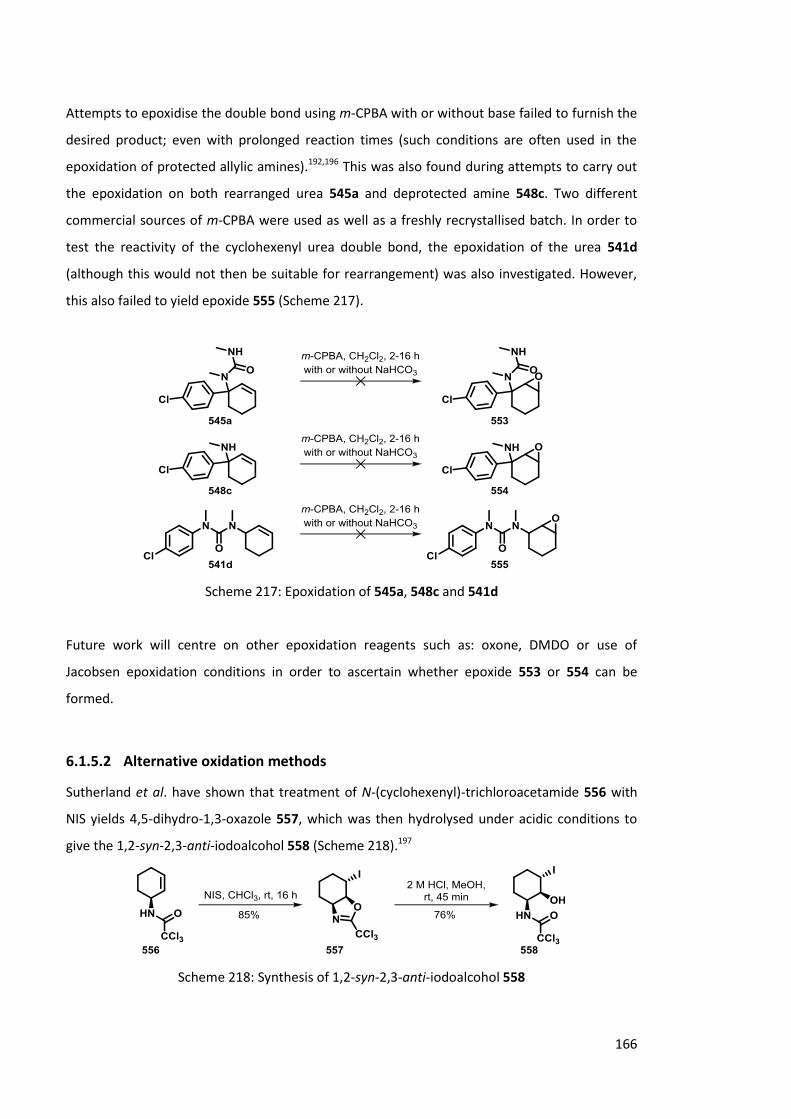
166
Attempts to epoxidise the double bond using m-CPBA with or without base failed to furnish the
desired product; even with prolonged reaction times (such conditions are often used in the
epoxidation of protected allylic amines).192,196 This was also found during attempts to carry out
the epoxidation on both rearranged urea 545a and deprotected amine 548c. Two different
commercial sources of m-CPBA were used as well as a freshly recrystallised batch. In order to
test the reactivity of the cyclohexenyl urea double bond, the epoxidation of the urea 541d
(although this would not then be suitable for rearrangement) was also investigated. However,
this also failed to yield epoxide 555 (Scheme 217).
Scheme 217: Epoxidation of 545a, 548c and 541d
Future work will centre on other epoxidation reagents such as: oxone, DMDO or use of
Jacobsen epoxidation conditions in order to ascertain whether epoxide 553 or 554 can be
formed.
6.1.5.2 Alternative oxidation methods
Sutherland et al. have shown that treatment of N-(cyclohexenyl)-trichloroacetamide 556 with
NIS yields 4,5-dihydro-1,3-oxazole 557, which was then hydrolysed under acidic conditions to
give the 1,2-syn-2,3-anti-iodoalcohol 558 (Scheme 218).197
Scheme 218: Synthesis of 1,2-syn-2,3-anti-iodoalcohol 558
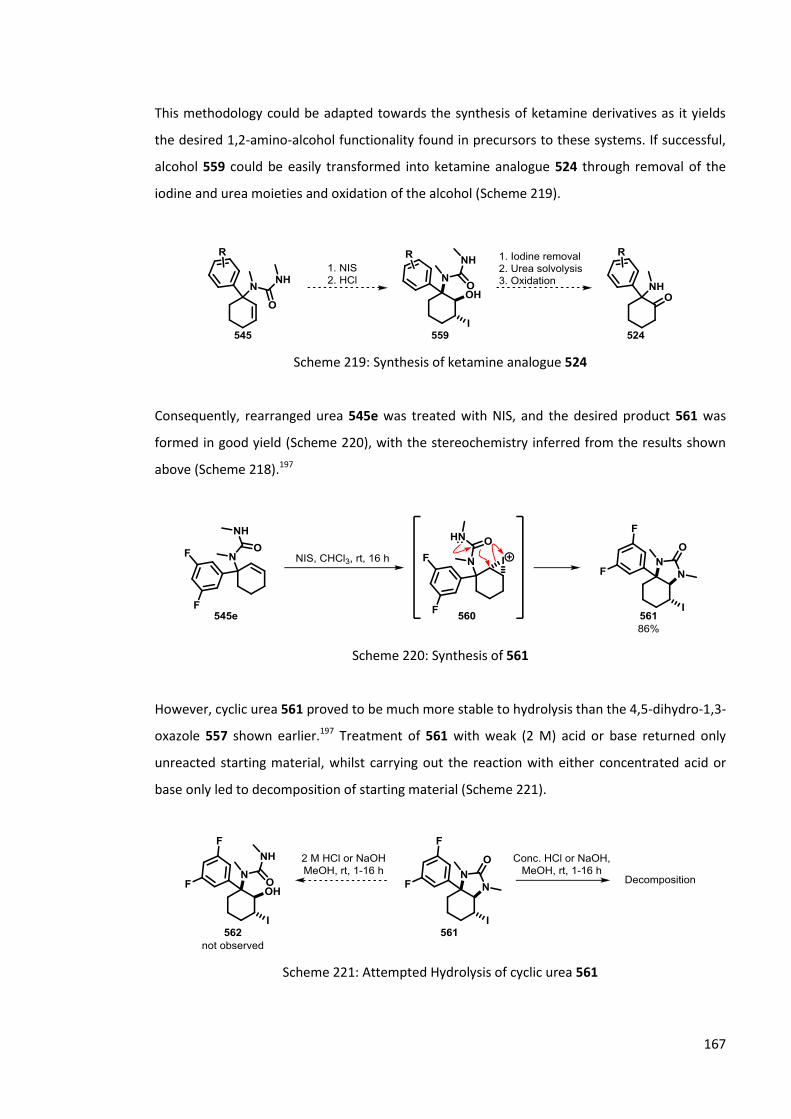
167
This methodology could be adapted towards the synthesis of ketamine derivatives as it yields
the desired 1,2-amino-alcohol functionality found in precursors to these systems. If successful,
alcohol 559 could be easily transformed into ketamine analogue 524 through removal of the
iodine and urea moieties and oxidation of the alcohol (Scheme 219).
Scheme 219: Synthesis of ketamine analogue 524
Consequently, rearranged urea 545e was treated with NIS, and the desired product 561 was
formed in good yield (Scheme 220), with the stereochemistry inferred from the results shown
above (Scheme 218).197
Scheme 220: Synthesis of 561
However, cyclic urea 561 proved to be much more stable to hydrolysis than the 4,5-dihydro-1,3-
oxazole 557 shown earlier.197 Treatment of 561 with weak (2 M) acid or base returned only
unreacted starting material, whilst carrying out the reaction with either concentrated acid or
base only led to decomposition of starting material (Scheme 221).
Scheme 221: Attempted Hydrolysis of cyclic urea 561
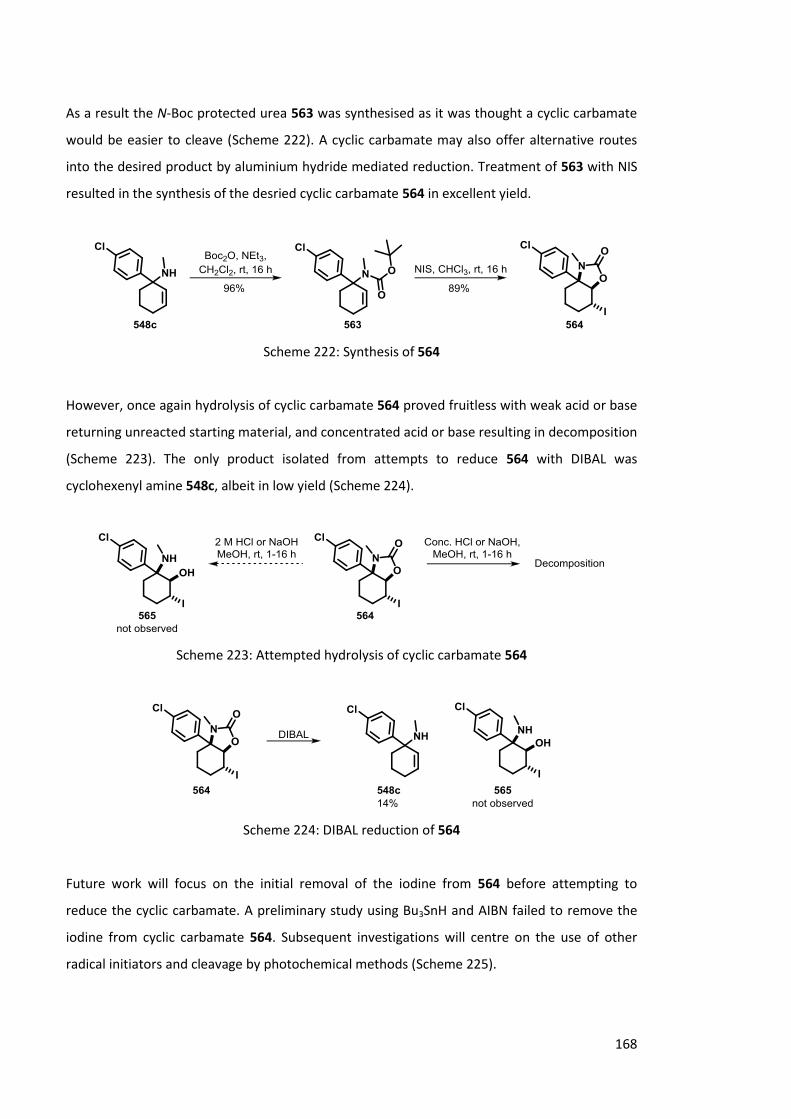
168
As a result the N-Boc protected urea 563 was synthesised as it was thought a cyclic carbamate
would be easier to cleave (Scheme 222). A cyclic carbamate may also offer alternative routes
into the desired product by aluminium hydride mediated reduction. Treatment of 563 with NIS
resulted in the synthesis of the desried cyclic carbamate 564 in excellent yield.
Scheme 222: Synthesis of 564
However, once again hydrolysis of cyclic carbamate 564 proved fruitless with weak acid or base
returning unreacted starting material, and concentrated acid or base resulting in decomposition
(Scheme 223). The only product isolated from attempts to reduce 564 with DIBAL was
cyclohexenyl amine 548c, albeit in low yield (Scheme 224).
Scheme 223: Attempted hydrolysis of cyclic carbamate 564
Scheme 224: DIBAL reduction of 564
Future work will focus on the initial removal of the iodine from 564 before attempting to
reduce the cyclic carbamate. A preliminary study using Bu3SnH and AIBN failed to remove the
iodine from cyclic carbamate 564. Subsequent investigations will centre on the use of other
radical initiators and cleavage by photochemical methods (Scheme 225).

169
Scheme 225: Alternative routes to 565
6.2 Rearrangement of -ketoureas
Alternatively, the lithiation of -ketourea 567 has also been investigated as a potential route
into ketamine analogues. This would prevent the need for any further oxidation of the urea
post-rearrangement, avoiding any possible regioselectivity problems that could be envisaged
for the oxidation of cyclohexenyl ureas 522. A simple solvolysis of the urea moiety would
directly yield ketamine analogues 524 (Scheme 226).
Scheme 226: Potential synthesis of ketamine analogues
6.2.1 Previous work
Recent work within the group has shown that is possible to carry out rearrangement reactions
on less reactive substrates such as nitriles and acids 569.198 Deprotonation of these substrates
results in the formation of enolate 570, which can then perform a new enolate-arylation
reaction. However, the anion 571 that forms as a result of the rearrangement is then able to
attack the nitrile or acid carbonyl, producing imino-hydantoin or hydantoin 572 respectively
(Scheme 227). Imino-hydantoins can be easily transformed into the corresponding hydantoin
through acid hydrolysis. For a more detailed discussion of this rearrangement, including
thorough mechanistic analysis by in situ IR spectroscopy, see a recently published article on the
subject.198
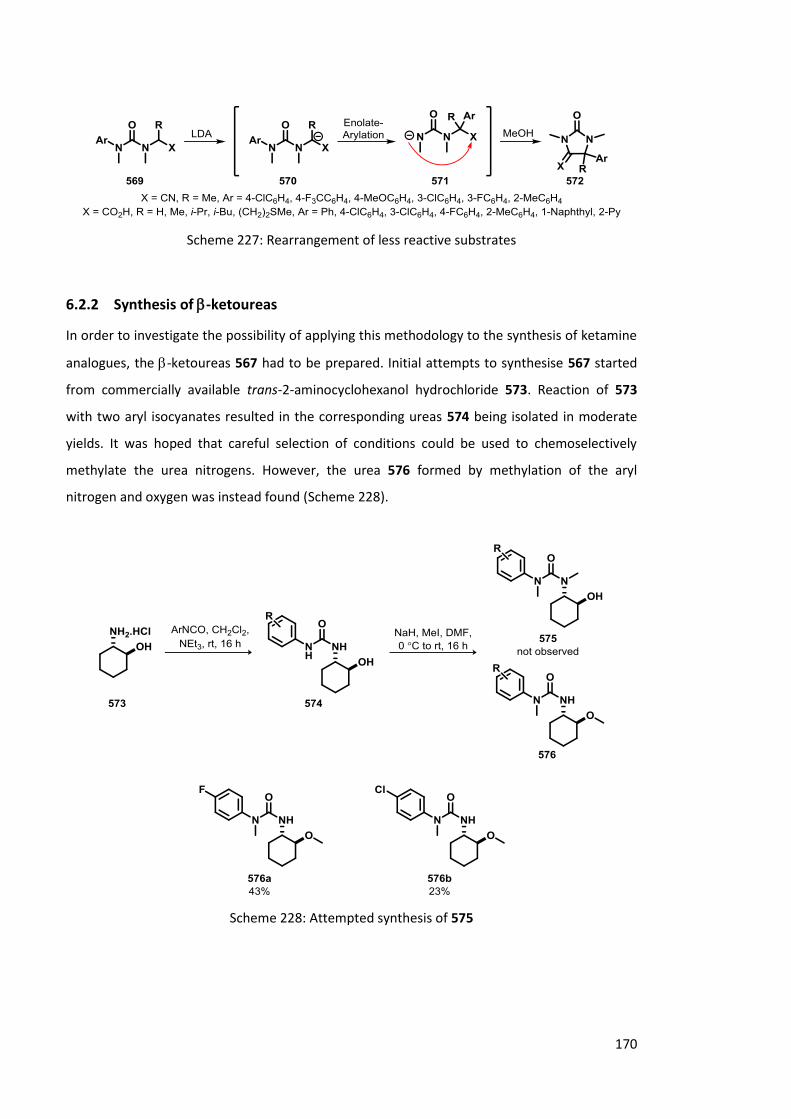
170
Scheme 227: Rearrangement of less reactive substrates
6.2.2 Synthesis of -ketoureas
In order to investigate the possibility of applying this methodology to the synthesis of ketamine
analogues, the -ketoureas 567 had to be prepared. Initial attempts to synthesise 567 started
from commercially available trans-2-aminocyclohexanol hydrochloride 573. Reaction of 573
with two aryl isocyanates resulted in the corresponding ureas 574 being isolated in moderate
yields. It was hoped that careful selection of conditions could be used to chemoselectively
methylate the urea nitrogens. However, the urea 576 formed by methylation of the aryl
nitrogen and oxygen was instead found (Scheme 228).
Scheme 228: Attempted synthesis of 575
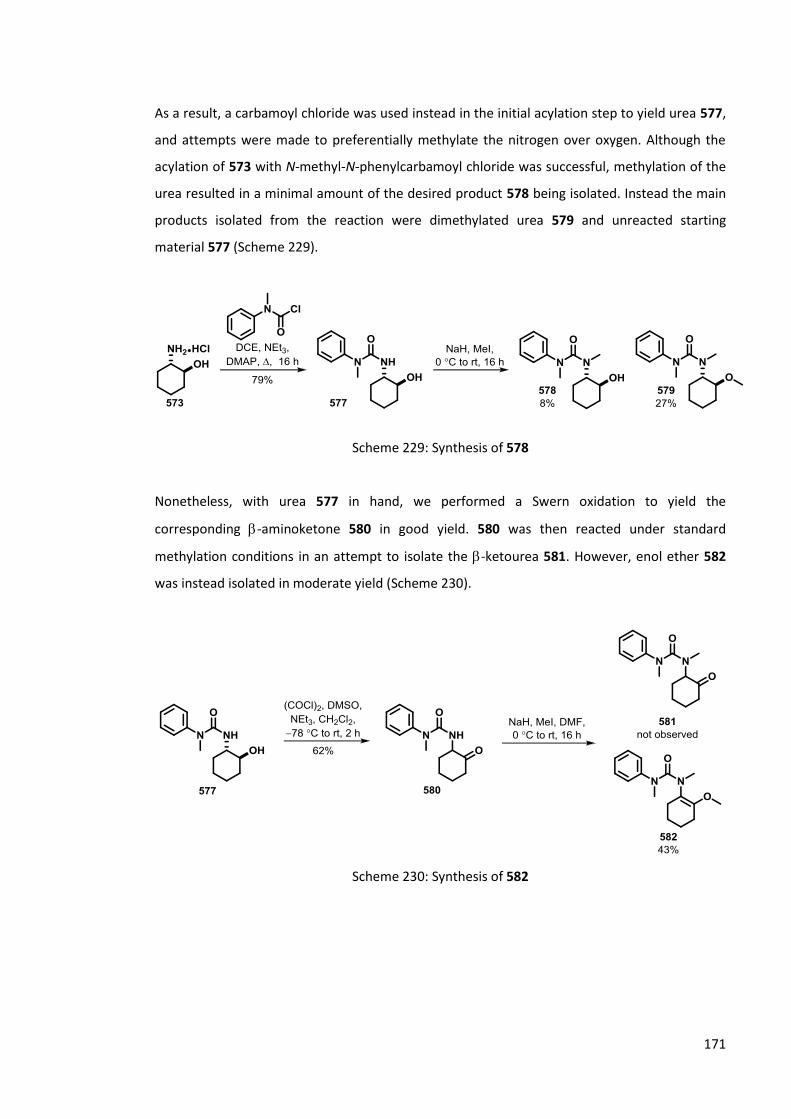
171
As a result, a carbamoyl chloride was used instead in the initial acylation step to yield urea 577,
and attempts were made to preferentially methylate the nitrogen over oxygen. Although the
acylation of 573 with N-methyl-N-phenylcarbamoyl chloride was successful, methylation of the
urea resulted in a minimal amount of the desired product 578 being isolated. Instead the main
products isolated from the reaction were dimethylated urea 579 and unreacted starting
material 577 (Scheme 229).
Scheme 229: Synthesis of 578
Nonetheless, with urea 577 in hand, we performed a Swern oxidation to yield the
corresponding -aminoketone 580 in good yield. 580 was then reacted under standard
methylation conditions in an attempt to isolate the -ketourea 581. However, enol ether 582
was instead isolated in moderate yield (Scheme 230).
Scheme 230: Synthesis of 582
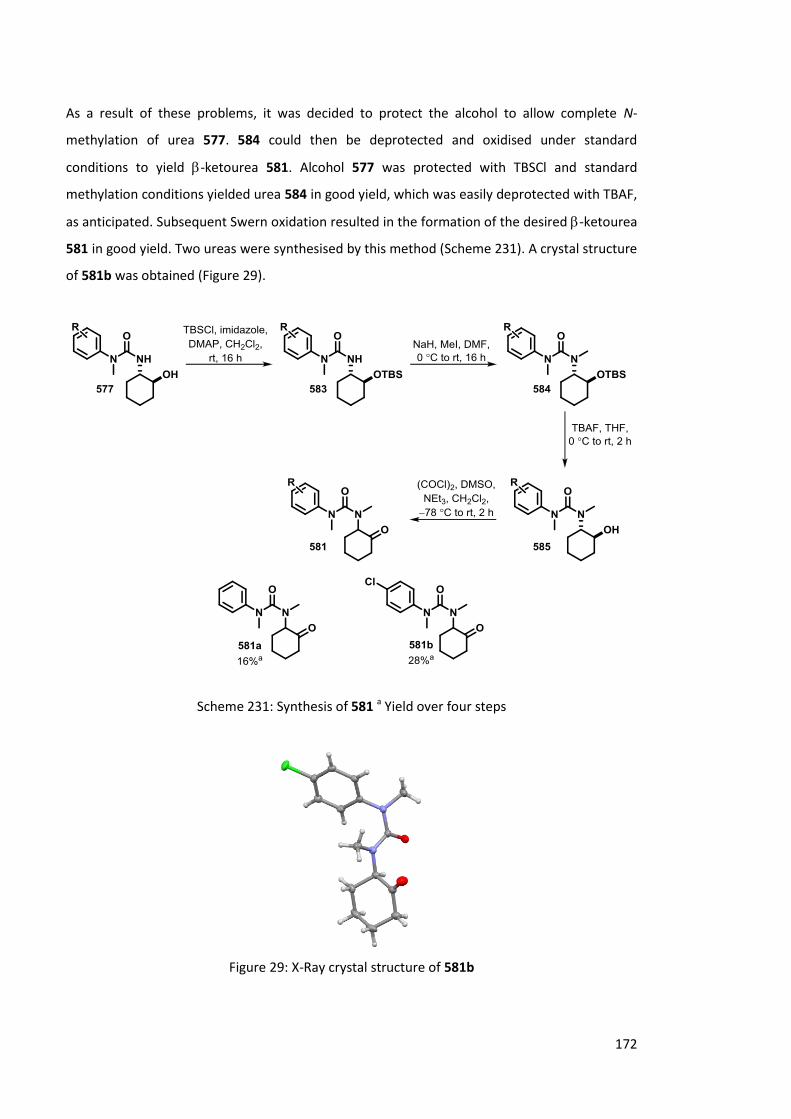
172
As a result of these problems, it was decided to protect the alcohol to allow complete N-
methylation of urea 577. 584 could then be deprotected and oxidised under standard
conditions to yield -ketourea 581. Alcohol 577 was protected with TBSCl and standard
methylation conditions yielded urea 584 in good yield, which was easily deprotected with TBAF,
as anticipated. Subsequent Swern oxidation resulted in the formation of the desired -ketourea
581 in good yield. Two ureas were synthesised by this method (Scheme 231). A crystal structure
of 581b was obtained (Figure 29).
Scheme 231: Synthesis of 581 a Yield over four steps
Figure 29: X-Ray crystal structure of 581b
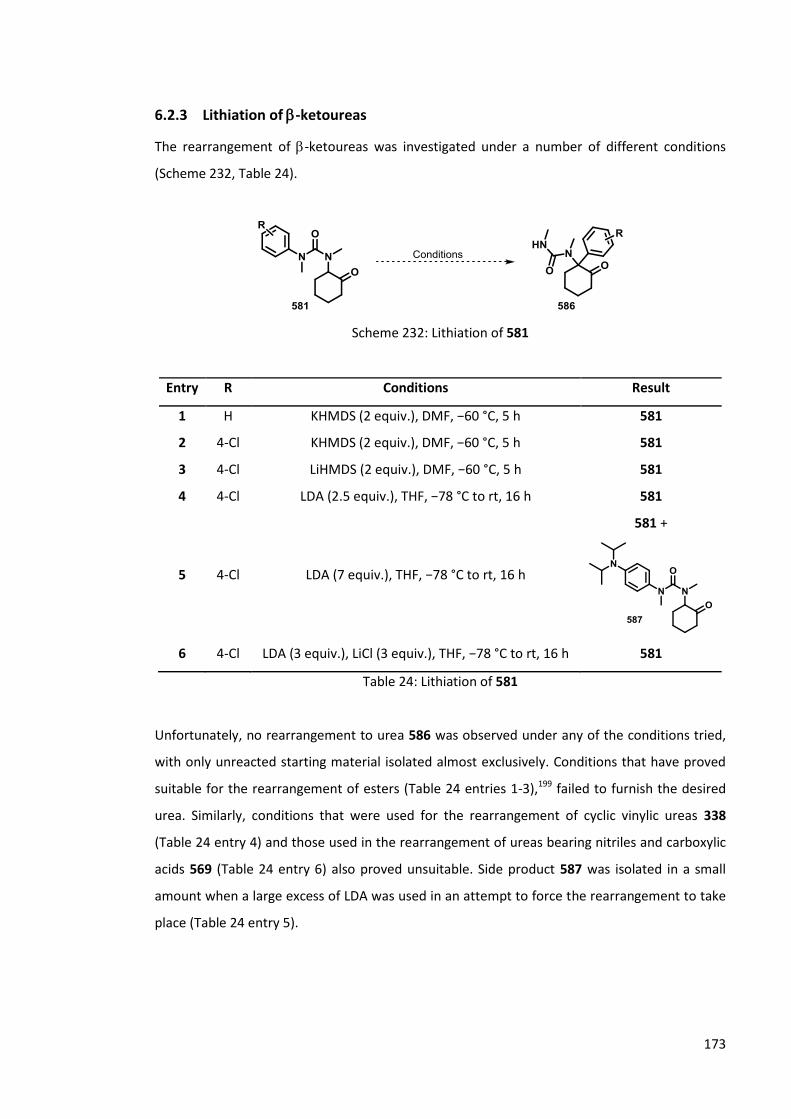
173
6.2.3 Lithiation of -ketoureas
The rearrangement of -ketoureas was investigated under a number of different conditions
(Scheme 232, Table 24).
Scheme 232: Lithiation of 581
Entry R Conditions Result
1 H KHMDS (2 equiv.), DMF, −60 °C, 5 h 581
2 4-Cl KHMDS (2 equiv.), DMF, −60 °C, 5 h 581
3 4-Cl LiHMDS (2 equiv.), DMF, −60 °C, 5 h 581
4 4-Cl LDA (2.5 equiv.), THF, −78 °C to rt, 16 h 581
5 4-Cl LDA (7 equiv.), THF, −78 °C to rt, 16 h
581 +
6 4-Cl LDA (3 equiv.), LiCl (3 equiv.), THF, −78 °C to rt, 16 h 581
Table 24: Lithiation of 581
Unfortunately, no rearrangement to urea 586 was observed under any of the conditions tried,
with only unreacted starting material isolated almost exclusively. Conditions that have proved
suitable for the rearrangement of esters (Table 24 entries 1-3),199 failed to furnish the desired
urea. Similarly, conditions that were used for the rearrangement of cyclic vinylic ureas 338
(Table 24 entry 4) and those used in the rearrangement of ureas bearing nitriles and carboxylic
acids 569 (Table 24 entry 6) also proved unsuitable. Side product 587 was isolated in a small
amount when a large excess of LDA was used in an attempt to force the rearrangement to take
place (Table 24 entry 5).

174
587 presumably arises from a formation of a benzyne intermediate 589 generated by excess
organolithium addition. The excess LDA is then able to add across the benzyne double bond,
forming 587 upon quenching with methanol (Scheme 233). The regioselectivity of the addition
of LDA across benzyne intermediate 589 is presumably governed by sterics, with the bulky
diisopropylamine group placed in the para-position.
Scheme 233: Formation of 587
There are two explanations for the lack of reactivity of these substrates. One is the decrease in
the activity of the ketone enolate in comparison to carboxylic acids or esters and nitriles. Also
the increased steric bulk of trying to carry out the reaction on a cyclic system could be having a
detrimental effect on the rearrangement. Ongoing work within the group is investigating ways
of overcoming this lack of reactivity (See Section 7.2.2).

175
In summary two different routes have been investigated with a view to synthesising ketamine
analogues. N-Cyclohexenyl ureas 522 have been shown to be susceptible to rearrangement
resulting in the synthesis of N-aryl-N-cyclohexenyl ureas 545. Attempts are currently ongoing to
regioselectively oxidise the cyclohexenyl double bond (Scheme 234). -Ketoureas 567 have also
been synthesised with a view to isolating ketamine analogues. However, they have proven
stubborn to a range of rearrangement conditions, although work is currently ongoing within the
group to surmount this problem (Scheme 235).
Scheme 234: Rearrangement of cyclohexenyl ureas 522
Scheme 235: Synthesis of -ketoureas 567

176
Chapter 7 Conclusions and Future work
7.1 Conclusions
In summary, this thesis describes the sequential transformations of a range of vinylic and allylic
ureas into their diaryl counterparts. Two routes have been developed for the production of
these ureas (Scheme 236). Vinylic ureas were synthesised by the addition of aryl isocyanates to
N-aryl substituted imines (See Chapter 3 and Chapter 4 Part 1), a method applicable to both
acyclic 318 and cyclic vinylic ureas 338. Alternatively, cyclic vinylic ureas 391 are made by a ring
closing metathesis approach (See Chapter 4 Parts 2 and 3 and Chapter 5). Cyclic allylic ureas 593
are only made through the metathesis route (See Chapter 4 Parts 2 and 3 and Chapter 5).
Scheme 236: Synthesis of vinylic and allylic ureas
Vinylic ureas 318 can be converted into highly substituted diaryl ureas 319 through a tandem
carbolithiation-rearrangement reaction. It is possible to carry out this transformation
enantioselectively with the use of chiral ligands, such as (−)-sparteine 153 or (+)-sparteine
surrogate 195, when the urea is vinylic and acyclic (Scheme 237, See Chapter 3).
Scheme 237: Enantioselective carbolithiation-rearrangement
The corresponding 6-membered cyclic vinylic ureas 338 also undergo this tandem
carbolithiation-rearrangement reaction to yield -arylated, -alkylated ureas 339. Alternatively,
the use of LDA instead of an aryl- or alkyllithium results in the isolation of ureas 401, formed by
deprotonation and rearrangement of 338 (Scheme 238, See Chapter 4 Part 1).

177
Scheme 238: Synthesis of cyclic -tertiary ureas 339 and 401
During investigations into the lithiation of 6-membered cyclic allylic 468 and vinylic 470 ureas it
was found that the position of the double bond is key to the type of rearrangement that would
take place. Vinylic ureas 470 underwent a benzylic deprotonation and rearrangement, whilst
allylic ureas 468 preferred to rearrange through a less sterically hindered allyl lithium
intermediate (Scheme 239, See Chapter 4 Part 3).
Scheme 239: Rearrangement of 468 and 470
5-Membered cyclic vinylic ureas 341 fail to undergo the tandem carbolithiation-rearrangement
reaction; however, their allylic 494 counterparts do undergo rearrangement upon lithiation
with LDA to diaryl allyl ureas 495 (Scheme 240, See Chapter 5). Analysing these allylic
rearrangements together allows us to determine that when cyclic systems are given a choice
between rearrangement of an allylic or benzylic organolithium, the allylic one is preferred.
However, if the system contains a site where both allylic and benzylic organolithium formation
can take place, then this is favoured over the formation of a less sterically hindered allylic
organolithium.
Scheme 240: Rearrangement of 494
178
Solvolysis of the diaryl ureas 594 allows the isolation of -tertiary amines bearing highly
substituted centres (Scheme 241, see Chapters 3 and 4). These amines would be difficult to
synthesise by many other methods.
Scheme 241: Deprotection of rearranged ureas 594
N-Cyclohexenyl ureas 541 are also capable of undergoing rearrangement (Scheme 242, See
Chapter 6). These have been synthesised with a view to making ketamine analogues 524.
Investigations within the group are currently concentrating on the regioselective oxidation of
the cyclohexenyl double bond. Alternative synthetic routes to 524 are also being studied (see
below).
Scheme 242: Rearrangement of N-cyclohexenyl ureas 541

179
7.2 Future Work
7.2.1 Cyclohexenyl ureas
Future work will look into the regioselective oxidation of N-aryl-N-cyclohexenyl ureas 545;
either through epoxidation of the olefin or by hydrolysis or reduction of cyclic carbamate
derivative 564 (Scheme 243). These substrates bear a resemblance to ketamine (See Chapter 6),
and as a result may have interesting biological properties.
Scheme 243: Regioselective oxidation of N-aryl-N-cyclohexenyl amines 548
7.2.2 -Ketoureas
-Ketoureas 581 have also been synthesised with a view to making ketamine analogues,
however, they have proved resistant to a range of conditions that have promoted
rearrangement in similar systems (See Chapter 6). Therefore, more reactive allyllithium
derivatives of these compounds are to be investigated for future lithiation studies. It is
envisaged that enol ether 582 or silyl enol ethers 596 or 597 could be used as substrates for aryl
migration (Scheme 244). Enol ether 582 was isolated as a side product when attempting to
methylate -aminoketone 580 and could be used as a starting point for these investigations.
Treatment of 581 with base during the synthesis of silyl enol ethers 596 or 597 could in theory
lead to the production of either regioisomer. However, both regioisomers could be susceptible
to rearrangement. If the rearrangement was to proceed, deprotection of the enol ether
protecting group should yield a rearranged -ketourea which could in turn be deprotected to
form ketamine analogues 524.
180
Scheme 244: Possible rearrangement of enol ether 582 or silyl enol ethers 596 or 597

181
Chapter 8 Experimental
8.1 General Information
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Ultrashield
300, 400 or 500 spectrometer (300, 400 or 500 MHz respectively) with residual non-deuterated
solvent as the internal standard. Carbon nuclear magnetic resonance (13C NMR) spectra were
recorded on a Varian Bruker Ultrashield 300, 400 or 500 spectrometer (75, 100 or 125 MHz
respectively). NMR data are presented as follows: chemical shift δ (in parts per million (ppm)
downfield from tetramethylsilane), integration, multiplicity, coupling constant J (in Hz) and
assignment (based on chemical shift, integration, coupling pattern and COSY, DEPT and HMQC
NMR experiments when necessary). Splitting patterns are abbreviated as follows: singlet (s),
doublet (d), triplet (t), quartet (q), multiplet (m), broad (br), or a combination of these. The
solvent used was deuterated chloroform (δ H: CDCl3 7.27 ppm; δ C: CDCl3 77.0 ppm).
Low and high resolution mass spectra were recorded by staff at the University of Manchester.
ES spectra were recorded on a Micromass Platform II; High resolution mass spectra (accurate
mass measurement) were recorded on a Thermo Finnigan MAT 95XP mass spectrometer, and
are accurate to 0.001 Da.
Infrared spectra were recorded on an ATi Perkin Elmer Spectrum RX1 FTIR spectrometer and
only absorption maxima (max) of interest are reported and quoted as wavenumbers (cm–1). All
samples were run as evaporated films.
Melting points were determined on a Kofler microscope melting point machine and are
uncorrected.
Optical rotations [α]DT were measured with a Perkin-Elmer 241 Polarimeter using a cell with a
pathlength of 0.25 dm. Concentrations (c) are given in grams per 100 mL.
Analytical TLC was carried out on pre-coated UV254 plates (Macherey-Nagel alugram. Sil G/UV254
or Machery-Nagel polygram. Alox N/UV254), with visualisation by UV light at 254 nm,
phosphomolybdic acid dip (5% in ethanol) or Seebach’s dip (2.5 g phosphomolybdic acid, 1 g
Ce(SO4)2, 6 mL conc H2SO4, 94 mL H2O).

182
Flash column chromatography was carried out using Fluorochem Davisil 40 – 63 µm 60 Å silica
or Fluka Basic 0.05-0.15 mm Aluminium Oxide, under a positive pressure by means of
compressed air, followed by removal of the solvent under reduced pressure after purification.
Reagents and solvents were purified by standard means. Tetrahydrofuran (THF) was distilled
from sodium wire and benzophenone under nitrogen. Dichloromethane (DCM) was obtained by
distillation from calcium hydride under nitrogen. Triethylamine was distilled from and stored
over potassium hydroxide. DMPU and cumene were distilled under reduced pressure from
calcium hydride and stored over molecular sieves. Toluene and diethyl ether (Et2O) were
obtained from a Solvent Purification System machine. All other chemicals were used as
received, except where otherwise noted in the experimental text. (−)-Sparteine 153 and (+)-
sparteine surrogate 195 were both distilled using a kugelrohr just before use. When stored
under argon in a −32 °C freezer, enantiomeric ratios appeared to decrease significantly after a
few days. (+)-Sparteine surrogate 195 has been synthesised following O’Brien’s procedure91
starting from Laburnum anagyroides seeds.
Enantiomeric ratios were determined by HPLC on a Hewlett-Packard system with UV detection
at 214 and 254 nm. using either a ChiralPak AD-H or Chiralcel OD-H column and hexane-IPA as
the eluent for all separations unless otherwise stated.
Petroleum ether (PE) refers to the fraction of light petroleum ether boiling between 40 C and
60 C. All other solvents and commercially obtained reagents were used as received or purified
using standard procedures. n-Butyllithium was used as a solution in hexanes (2.5 M),
isopropyllithium as a solution in pentane (0.7 M), sec-butyllithium as a solution in
cyclohexane/hexane (92/8) (1.3 M), t-butyllithium as a solution in pentane (1.7 M) and
phenyllithium as a solution in dibutylether (2.0 M). All the above organolithium solutions were
titrated prior to use against a solution of N-benzyl benzamide. Cooling baths used are
acetone/dry ice for –78 °C and acetonitrile/dry ice for –40 °C.
All experiments were performed in anhydrous conditions under an atmosphere of argon, unless
otherwise noted in the experimental text. Apparatus was flame-dried and standard techniques
were employed in handling air-sensitive materials.

183
8.2 General Procedures
General Procedure 1 – Acyclic urea formation from isocyanates
Using the method of Clayden et al.131 CH3NH2 (8 M in EtOH, 4 equiv.), the ketone (1 equiv.) and
4Å powdered molecular sieves (250 mg/mmol) were combined and heated under microwave
irradiation at 125 °C for 60 min. The mixture was filtered over diatomaceous earth and washed
with CH2Cl2 and the organic phases combined and concentrated under reduced pressure to give
the imine (quantitatively) as a pale yellow oil. To a solution of the imine (1 equiv.) in THF (0.3
M), aryl isocyanate (1 equiv.) was added dropwise. After stirring for 24 h at room temperature,
to the mixture NaH (2 equiv.) and MeI (2 equiv) were added sequentially at 0 °C. The reaction
was stirred for 24 h at room temperature, quenched with H2O and extracted twice with EtOAc.
The organic phases were combined, dried over MgSO4, concentrated under reduced pressure
and purified by flash column chromatography (PE:EtOAc 9:1 + 1% NEt3) to give the urea.
General Procedure 2 – Enantioselective Carbolithiation-Rearrangement
A – Using (−)-sparteine 153
To a cooled solution of freshly distilled (−)-sparteine 153 (1 equiv.) in cumene (1.5 mL, 0.1 M) at
−50 °C, organolithium (2 equiv.) was added resulting in a pale yellow solution. The reaction was
stirred at the same temperature for 15 min before the urea 318 (0.17 mmol, 1 equiv.) in
solution in cumene (1 mL) was slowly added dropwise resulting in a red-orange solution. The
reaction was stirred at the same temperature for 1.5 h, DMPU (10 equiv.) was added dropwise
and the mixture left to stir for an additional 1.5 h. The reaction was quenched slowly with
MeOH, warmed to room temperature and NH4Cl (sat. aq. sol.) added, and extracted twice with
EtOAc. The organic phases were combined, dried over MgSO4 and concentrated under reduced
pressure. The residue was purified by flash column chromatography (PE:EtOAc 8:2 + 1% NEt3) to
afford the rearranged product.

184
B – Using (+)-sparteine surrogate 195
To a cooled solution of freshly distilled (+)-sparteine surrogate 195 (2 equiv.) in THF (1.5 mL, 0.1
M) at −78 °C, organolithium (2 equiv.) was added resulting in a pale yellow solution. The
reaction was stirred at the same temperature for 15 min. before the urea 318 (0.17 mmol, 1
equiv.) in solution in THF (1 mL) was slowly added dropwise resulting in a red-orange solution.
The reaction was stirred at the same temperature for 1.5 h, DMPU (10 equiv.) was added
dropwise and the mixture left to stir for an additional 1.5 h. The reaction was quenched slowly
with MeOH, warmed to room temperature and NH4Cl (sat. aq. sol.), and extracted twice with
EtOAc. The organic phases were combined, dried over MgSO4 and concentrated under reduced
pressure. The residue was purified by flash column chromatography (PE:EtOAc 8:2 + 1% NEt3) to
afford the rearranged product.
General Procedure 3 – Solvolysis of ureas under reflux conditions
Urea (1 equiv.) and K2CO3 (1 equiv. w/w) were dissolved in n-BuOH (0.1 M) and heated under
reflux for 2 h. After cooling, the mixture was quenched with H2O, extracted twice with EtOAc,
washed with H2O, dried over MgSO4 and the solvent removed under reduced pressure. The
crude mixture was purified by flash column chromatography (PE/EtOAc 9:1 + 1% NEt3) to yield
the amine.
General Procedure 4 – Solvolysis of ureas under microwave conditions
To urea (1 equiv.) was added a 1:1 v/v mixture of EtOH and 2 M NaOH solution (10 equiv.). The
resultant mixture was heated to 130 °C under microwave irradiation for 2.5 h. After cooling, the
mixture was extracted twice with EtOAc, washed with brine, dried over MgSO4 and the solvent
removed under reduced pressure to yield the amine without any further purification.
General Procedure 5 – Formation of cyclic imine 395
Using the method of Buchwald et al.158 to a solution of bromo-nitrile (1 equiv.) in THF (0.4 M)
was added phenyl lithium (1.4 equiv.) dropwise. The resulting solution was heated at reflux for
16 h. The solution was cooled to room temperature and quenched with H2O. The mixture was
extracted with Et2O and washed with several portions of brine. The combined organic layers
were dried over MgSO4 and the solvent removed under reduced pressure to yield the imine as a
brown oil, which was used without further purification.

185
General Procedure 6 – Formation of cyclic imine 399
To a cooled solution of aryl bromide (1.4 equiv.) in THF (0.3 M) at −78 °C, t-BuLi (2.8 equiv.) was
added dropwise and the mixture held for 1 h. The resulting aryl lithium solution was added
dropwise to a solution of bromo-nitrile (1 equiv.) in THF (0.3 M) at −78 °C. The resulting solution
was heated to reflux and stirred for 16 h. The solution was then cooled to 0 °C and quenched
slowly with H2O. The mixture was extracted with Et2O and washed with brine. The organic layer
was dried over MgSO4 and concentrated under reduced pressure to yield the imine, which was
used without further purification.
General Procedure 7 – Cyclic urea formation from isocyanates
To a solution of cyclic imine (1 equiv.) in CH2Cl2 (0.3 M), isocyanate (1.1 equiv.) and DMAP (cat.)
were added and the solution stirred overnight at room temperature. H2O was added and the
mixture extracted with CH2Cl2 and dried over MgSO4. The solvent was evaporated under
reduced pressure and the resulting crude urea used without further purification unless
otherwise stated.
General Procedure 8 – Methylation of ureas
To a cooled solution of urea (1 equiv.) in DMF (0.2 M) at 0 °C was added NaH (2 equiv.) and the
mixture stirred for 1 h. MeI (2 equiv.) was added dropwise and the reaction mixture stirred until
1H NMR spectroscopy showed complete methylation. H2O was added carefully and the crude
mixture extracted three times with Et2O, washed thoroughly with brine, dried over MgSO4 and
the solvent removed under reduced pressure. The crude mixture was purified by flash column
chromatography (PE/EtOAc 9:1 + 1% NEt3) to yield the methylated urea.
General Procedure 9 – Carbolithiation-Rearrangement of cyclic ureas
To a cooled solution of urea (1 equiv.) in the desired solvent (0.1 M) at −78 °C organolithium
(2.5 equiv.) was added dropwise followed by DMPU (4:1 Solvent:DMPU) dropwise. The solution
was then held at the desried temperature and held for the relevant number of hours (Scheme
144 or Scheme 145). The reaction was quenched slowly with MeOH, warmed to room
temperature and NH4Cl (sat. aq. soln.) added. The mixture was extracted three times with Et2O,
washed with NH4Cl (sat. aq. soln.), dried over MgSO4 and concentrated under reduced pressure.
The crude mixture was purified by flash column chromatography (PE/EtOAc 2:1 + 1% NEt3) to
yield the rearranged urea.

186
General Procedure 10 – Preparation of LDA
To a cooled solution of diisopropylamine (1 equiv.) in THF (0.6 M) at 0 °C was added n-BuLi (1
equiv.). The reaction mixture was stirred at 0 °C for 20 min. before immediate use.
General Procedure 11 – Rearrangement of ureas using LDA
To a cooled solution of urea (1 equiv.) in THF (0.1 M), at −78 °C was added LDA (2 equiv.,
prepared by general procedure 10) dropwise. The mixture was stirred for 2 h before dropwise
addition of DMPU (4:1 THF:DMPU). The reaction was stirred for a further 2 h or until complete
consumption of starting material by TLC analysis. MeOH was added slowly and the mixture
allowed to warm to room temperature. NH4Cl (sat. aq. soln.) was added and the mixture
extracted twice with Et2O. The organic phases were combined and washed with brine and dried
over MgSO4 and concentrated under reduced pressure. The crude product was purified by flash
column chromatography to give the -arylated urea.
General Procedure 12 – Carbolithiation of NH-ureas
To a cooled solution of urea (1 equiv.) in THF (0.1 M) at −40 °C, organolithium (3 equiv.) was
added dropwise and the mixture held for 2 h. The reaction was slowly quenched with MeOH
and NH4Cl (sat. soln.), extracted twice with Et2O, washed twice with NH4Cl (sat. soln.), dried
over MgSO4 and the solvent removed under reduced pressure. The crude mixture was purified
by flash column chromatography (PE/EtOAc 9:1 + 1% NEt3) to yield the carbolithiated urea.
General Procedure 13 – Imine formation
To aldehyde (1 equiv.) and 4Å molecular sieves (250 mg/mmol) in CH2Cl2 (0.2-0.4 M) was added
amine (2 equiv.) and the reaction stirred overnight at room temperature. The reaction mixture
was filtered through diatomaceous earth, and the solvent removed under reduced pressure.
The resulting crude imine was used without any further purification unless otherwise stated.

187
General Procedure 14 – Grignard alkylation of imines
Using the method of Ghelfi et al.174 to a cooled solution of imine (1 equiv.) in THF (0.1 M) at −78
°C, ZnEt2 solution (2 equiv., 1 M in hexanes) was added dropwise. The mixture was stirred at
−78 °C for 2 h before dropwise addition of vinyl magnesium bromide solution (2 equiv., 1M in
THF). The reaction was warmed to room temperature overnight before and quenched with H2O.
The mixture was extracted four times with Et2O, washed twice with brine, dried over MgSO4
and the solvent removed under reduced pressure to yield the crude amine, which was used
without further purification.
General Procedure 15 – Urea formation from carbamoyl chlorides
To a solution of amine (1 equiv.) and NEt3 (1.6 equiv.) in DCE (0.3 M), carbamoyl chloride (1.3
equiv.) and DMAP (cat) were added. The mixture was heated at reflux overnight. After cooling,
the mixture was quenched with NaHCO3 (sat. aq. soln.), extracted twice with CH2Cl2, washed
with brine, dried over MgSO4 and the solvent removed under reduced pressure. The crude
mixture was purified by flash column chromatography (PE/EtOAc 9:1 + 1% NEt3) to yield the
urea.
General Procedure 16 – Ring closing metathesis of ureas
To a solution of urea (1 equiv) in CH2Cl2 (0.05 M), Grubbs 1st generation catalyst (0.05 equiv.)
was added. The mixture was stirred for 4 h at room temperature or until complete consumption
of starting material was observed by TLC. The solvent was removed under reduced pressure
and the crude mixture purified by flash column chromatography (PE/EtOAc 19:1 + 1% NEt3) to
yield the cyclic urea.
General Procedure 17 – Isomerisation of allyl ureas
To a solution of cyclic allyl urea (1 equiv.) in THF (0.1 M),
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.1 equiv) was added. The mixture
was heated under reflux overnight. The solvent was removed under reduced pressure. The
crude mixture was purified by flash column chromatography (PE/EtOAc 9:1 + 1% NEt3) to yield
the cyclic vinyl urea.

188
General Procedure 18 – Formation of tert-butyl sulfinimines
Using the method of Ellman et al.152 to a solution of aldehyde (3 equiv.) in CH2Cl2 (0.5 M) was
added MgSO4 (10 equiv.), PPTS (0.05 equiv.) and sulfinamide 25 (1 equiv.) and the reaction
stirred overnight at room temperature. The reaction mixture was filtered through
diatomaceous earth, and the solvent removed under reduced pressure. The crude mixture was
purified by flash column chromatography (CH2Cl2:MeOH 95:5) to yield the sulfinimine
General Procedure 19 – Grignard addition to tert-butyl sulfinimines
Using the method of Ellman et al.152 to a cooled solution of sulfinimine (1 equiv.) in CH2Cl2 (0.2
M) at −78 °C was added allyl magnesium bromide solution (2 equiv., 1 M in Et2O) dropwise. The
reaction was held at −78 °C until complete consumption of starting material was observed by
TLC analysis. The reaction was quenched slowly with NH4Cl (sat. soln.), extracted twice with
CH2Cl2, washed with brine, dried over MgSO4 and the solvent removed under reduced pressure.
The crude mixture was purified by flash column chromatography (PE:EtOAC 4:1 + 1% NEt3) to
yield the sulfinamide.
General Procedure 20 – Deprotection of sulfinimines
Using the method of Ellman et al.152 to a solution of sulfinamide (1 equiv.) in MeOH (0.2 M) was
added HCl solution (10 equiv., 2 M in Et2O) dropwise and the mixture stirred for 1 h at room
temperature. The solvent was removed under reduced pressure and the resultant precipitate
filtered and washed with Et2O to give the amine hydrochloride salt which was used without
further purification unless otherwise stated.
General Procedure 21 – Alkylation of amine hydrochloride salts
To a cooled solution of amine hydrochloride salt (1 equiv.) in DMF (0.2 M) at 0 °C was added
NaH (5 equiv.) and the mixture stirred for 30 min. Allyl bromide (1 equiv.) was added dropwise
and the reaction slowly warmed to room temperature overnight. The reaction was quenched
slowly with H2O, extracted three times with Et2O, washed three times with brine, dried over
MgSO4 and the solvent removed under reduced pressure. The crude mixture was purified by
flash column chromatography (PE:EtOAc 4:1 + 1% NEt3) to yield the alkylated amine.

189
General Procedure 22 – Urea formation from amine and isocyanates
To a solution of amine (1 equiv.) in CH2Cl2 (0.2 M) was added NEt3 (1.5 equiv.) and aryl
isocyanate (1 equiv.). The reaction was stirred overnight at room temperature. The solvent was
removed under reduced pressure and the residue dissolved in DMF (0.2 M) and cooled to 0 °C.
NaH (2 equiv.) was added and the mixture stirred for 30 min. MeI (2 equiv.) was added
dropwise and the reaction mixture stirred until 1H NMR analysis showed complete methylation.
H2O was added carefully and the crude mixture extracted three times with Et2O, washed
thoroughly with brine, dried over MgSO4 and the solvent removed under reduced pressure. The
crude mixture was purified by flash column chromatography (PE/EtOAc 9:1 + 1% NEt3) to yield
the urea.
General Procedure 23 – Rearrangement of ureas using alkyl lithiums
To a cooled solution of urea (1 equiv.) in THF (0.1 M), at −78 °C was added organolithium (2.5
equiv.) dropwise. The mixture was stirred for 2 h before the dropwise addition of DMPU (4:1
THF:DMPU). The reaction was stirred for a further 2 h or until complete consumption of starting
material by TLC analysis. MeOH was added slowly and the mixture allowed to warm to room
temperature. NH4Cl (sat. soln.) was added and the mixture extracted twice with Et2O. The
organic phases were combined and washed with brine and dried over MgSO4 and concentrated
under reduced pressure. The crude product was purified by flash column chromatography
(PE/EtOAc 4:1 + 1% NEt3) to give the -arylated urea.
General Procedure 24 – Boc protection of amines
To a solution of amine (1 equiv.) in CH2Cl2 (0.3 M) was added NEt3 (2 equiv.) and Boc2O (1.1
equiv.). The mixture was stirred overnight at room temperature. The reaction was quenched
with brine, extracted with CH2Cl2, dried over MgSO4 and the solvent removed under reduced
pressure. The crude mixture was purified by flash column chromatography (PE/EtOAc 4:1 + 1%
NEt3) to yield the carbamate.
General Procedure 25 – NIS promoted cyclisation of carbamates
To a solution of carbamate (1 equiv.) in CHCl3 (0.1 M) was added NIS (1.5 equiv.). The mixture
was stirred overnight at room temperature. The solvent was removed under reduced pressure
and the residue dissolved in EtOAc and washed four times with H2O. After drying over MgSO4
the solvent was removed under reduced pressure and the crude cyclic carbamate used without
any further purification unless otherwise stated.

190
General Procedure 26 – TBS protection of alcohols
To a solution of alcohol (1 equiv.) in CH2Cl2 (0.1 M) was added imidazole (1.1 equiv.), TBSCl (1.1
equiv.) and DMAP (0.1 equiv.). The mixture was stirred overnight at room temperature. The
reaction was quenched with H2O, extracted twice with CH2Cl2, washed with brine, dried over
MgSO4 and the solvent removed under reduced pressure. The crude mixture was purified by
flash column chromatography (PE/EtOAc 2:1 + 1% NEt3) to yield the silyl protected alcohol.
General Procedure 27 – TBS deprotection
To a cooled solution of silyl protected alcohol (1 equiv.) in THF (0.1 M) at 0 °C was added TBAF
(1.5 equiv., 1 M in THF). The mixture was stirred for 1 h at 0 °C before warming to room
temperature. Once TLC analysis showed complete consumption of starting material, the
reaction was filtered through a short silica plug and washed with Et2O. The resultant crude
alcohol was used without any further purification unless otherwise stated.
General Procedure 28 – Swern oxidation of alcohols
To a cooled solution of oxalyl chloride (2.2 equiv.) in CH2Cl2 (0.1 M) at −78 °C was added a
solution of DMSO (2.8 equiv.) in CH2Cl2 (1 M) dropwise. After five min. a solution of alcohol (1
equiv.) in CH2Cl2 (1 M) was added dropwise. After further stirring for 1.5 h, NEt3 (5 equiv.) was
added dropwise and the mixture allowed to slowly warm to room temperature. The reaction
was held at room temperature for 30 min. before quenching with NaHCO3 (sat. soln.), extracted
three times with CH2Cl2, washed three times with brine, dried over MgSO4 and the solvent
removed under reduced pressure. The crude mixture was purified by flash column
chromatography (PE/EtOAc 1:1 + 1% NEt3) to yield the silyl protected alcohol.

191
8.3 Experimental Data
(R)-1,3-Dimethyl-1-(3-methyl-1-(naphthalen-1-yl)-1-phenylbutyl)urea 319h
Following general procedure 2A. To a solution of freshly distilled (−)-
sparteine 153 (0.071 g, 0.30 mmol) in cumene (1.5 mL) at −50 °C was added
i-PrLi (0.38 M, 0.80 mL, 0.30 mmol) dropwise and the mixture stirred for 15
min. To the reaction mixture was added a solution of 1,3-dimethyl-3-
(naphthalen-1-yl)-1-(1-phenylvinyl)ureaurea (0.048 g, 0.15 mmol) in cumene
(1.5 mL) dropwise and the mixture stirred for 1.5 h. DMPU (0.18 mL, 1.52 mmol) was added and
the mixture stirred for a further 1.5 h. Purification by flash column chromatography yielded the
title compound 319h as a brown oil (0.030 g, 0.08 mmol, 55%). RF 0.32 (1:1 PE:EtOAc); 1H NMR
(400 MHz, MeOD) 8.45-8.38 (1H, m, ArH), 7.84-7.79 (1H, m, ArH), 7.73 (1H, d, J = 8.3, ArH),
7.59 (2H, d, J = 8.1, ArH), 7.43-7.36 (2H, m, ArH), 7.34-7.21 (4H, m, ArH), 7.13 (1H, d, J = 6.8,
ArH), 3.15 (1H, d, J = 15.1, CCHHCH(CH3)2), 2.82 (3H, s, NMe), 2.71 (1H, dd, J = 15.1, 6.3,
CCHHCH(CH3)2), 2.51 (3H, s, NMe), 1.32-1.23 (2H, m, CH(CH3)2 + NH), 0.81 (3H, d, J = 6.8,
CH(CH3)2), 0.03 (3H, d, J = 6.6, CH(CH3)2); 13C NMR (100 MHz, MeOD) 162.1 (C=O), 146.1 (4°
ArC), 140.9 (4° ArC), 136.1 (4° ArC), 134.8 (4° ArC), 130.4 (ArC), 129.9 (ArC), 129.7 (ArC), 128.7
(ArC), 128.4 (ArC), 127.9 (ArC), 127.6 (ArC), 126.6 (ArC), 126.3 (ArC), 125.6 (ArC), 79.6 (4° C),
46.9 (CCH2CH), 35.8 (NMe), 27.7 (NMe), 27.4 (CH(CH3)2), 25.6 (CH(CH3)2), 24.2 (CH(CH3)2); IR max
(film)/cm-1 3349 (NH), 2982 (CH), 1645 (C=O); MS (ES) m/z 283 (M + Na+); HRMS m/z calcd
261.2274 for C24H29N2O (M + H+) found 261.2278; HPLC 88:12 er: ChiralPak AD-H, 90:10
Hexane:IPA 1mL/min. tr 23.8 major, 21.8 minor.

192
3-Ethyl-1-[(1S)-1-(2-fluorophenyl)-3-methyl-1-phenyl-butyl]-1-methyl-urea 363a
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.078 g, 0.40 mmol) in THF (1.5 mL) at −78 °C was
added i-PrLi (0.45 M, 0.89 mL, 0.40 mmol) dropwise and the mixture stirred
for 15 min. To the reaction mixture was added a solution of 1-(2-
fluorophenyl)-1,3-dimethyl-3-(1-phenylvinyl)urea 362a (0.060 g, 0.20 mmol) in THF (1.5 mL)
dropwise and the mixture stirred for 1.5 h. DMPU (0.24 mL, 2.00 mmol) was added and the
mixture stirred for 1.5 h. Purification by flash column chromatography yielded the title
compound 363a as a pale yellow oil (0.047 g, 0.14 mmol, 69%). RF 0.29 (2:1 PE:EtOAc); [D25
+5.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) 7.48-7.42 (2H, m, ArH), 7.34-7.28 (3H, m, ArH)
7.27-7.21 (2H, m, ArH), 7.08 (1H, ddd, J = 7.6, 7.6, 1.4, ArH), 7.00 (1H, ddd, J = 12.6, 8.1, 1.3,
ArH), 4.19 (1H, t, J = 5.3, NH), 3.11-3.02 (2H, m, NCH2CH3), 3.00 (3H, d, J = 1.0, NMe), 2.60 (1H,
d, J = 10.1, A of AB, CHABCH), 2.60 (1H, d, J = 5.4, B of AB, CHABCH), 1.65-1.53 (1H, m, CH(CH3)2),
0.89 (3H, t, J = 7.3, NCH2CH3), 0.75 (3H, d, J = 14.5, CH(CH3)2), 0.75 (3H, d, J = 6.7, CH(CH3)2); 13C
NMR (100 MHz, CDCl3) 160.6 (d, JF = 248.3, ArCF), 158.6 (C=O), 143.2 (4° ArC), 131.0 (d, JF =
10.2, 4° ArC), 129.4 (d, JF = 4.6, ArC), 128.8 (d, JF = 9.2, ArC), 127.8 (ArC), 127.8 (ArC), 126.9
(ArC), 123.5 (d, JF = 2.8, ArC), 116.5 (d, JF = 24.9, ArC), 69.7 (d, JF = 3.7, 4 °C), 46.6 (d, JF = CH2CH),
35.4 (NHCH2CH3), 34.4 (d, JF = 1.9, NMe), 25.1 (CH(CH3)2), 24.5 (CH(CH3)2), 24.4 (CH(CH3)2), 15.2
(NHCH2CH3); IR max (film)/cm-1 3423 (NH), 1659 (C=O); MS (ES) m/z 365 (M + Na+); HRMS m/z
calcd 343.2180 for C21H28N2OF (M + H+) found 343.2194; HPLC 92:8 er: Chiracel OD-H, 95:5
Hexane:IPA 1mL/min. tr 14.9 major, 10.2 minor.

193
3-Ethyl-1-[(1S)-1-(2-fluorophenyl)-1-phenyl-hexyl]-1-methyl-urea 363b
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.072 g, 0.37 mmol) in THF (1.5 mL) at −78 °C was
added n-BuLi (1.8 M, 0.20 mL, 0.37 mmol) dropwise and the mixture stirred
for 15 min. To the reaction mixture was added a solution of 1-(2-
fluorophenyl)-1,3-dimethyl-3-(1-phenylvinyl)urea 362a (0.055 g, 0.18 mmol) in THF (1.5 mL)
dropwise and the mixture stirred for 1.5 h. DMPU (0.22 mL, 1.84 mmol) was added and the
mixture stirred for 1.5 h. Purification by flash column chromatography yielded the title
compound 363b as a colourless oil (0.039 g, 0.11 mmol, 61%). RF 0.25 (2:1 PE:EtOAc); 1H NMR
(400 MHz, CDCl3) 7.39-7.19 (7H, m, ArH), 7.10 (1H, ddd, J = 8.1, 8.1, 1.3, ArH), 7.04 (1H, ddd, J
= 12.4, 8.1, 1.3, ArH), 4.11 (1H, t, J = 5.3, NH), 3.08-2.94 (5H, m, NMe + NHCH2CH3), 2.72-2.53
(2H, m, CH2(CH2)3CH3), 1.33-1.20 (4H, m, CH2CH2CH2CH2CH3), 1.18-1.08 (2H, m, (CH2)3CH2CH3),
0.83 (3H, t, J = 7.1, (CH2)4CH3), 0.81 (3H, t, J = 7.3, NHCH2CH3); 13C NMR (100 MHz, CDCl3) 160.4
(d, JF = 248.3, ArCF), 158.7 (C=O), 143.6 (4° ArC), 130.6 (d, JF = 11.1, 4° ArC), 129.3 (d, JF = 4.6,
ArC), 129.0 (d, JF = 9.2, ArC), 128.1 (ArC), 127.5 (ArC), 127.0 (ArC), 123.8 (d, JF = 2.8, ArC), 116.5
(d, JF = 24.0, ArC), 69.5 (d, JF = 3.7, 4 °C), 39.9 (d, JF = 3.7 CH2(CH2)3CH3), 35.4 (NHCH2CH3), 34.1
(d, JF = 1.9, NMe), 32.1 (CH2CH2(CH2)2CH3), 25.9 ((CH2)2CH2CH2CH3), 22.4 ((CH2)3CH2CH3), 15.0
(NHCH2CH3), 14.0 ((CH2)4CH3); IR max (film)/cm-1 1638 (C=O); MS (ES) m/z 379 (M + Na+); HRMS
m/z calcd 379.2157 for C22H29N2OFNa (M + Na+); found 379.2167; HPLC 51:49 er: Chiracel OD-H,
95:5 Hexane:IPA 1mL/min. tr 15.2 major, 8.9 minor.

194
1,3-Dimethyl-1-[(1S)-3-methyl-1-(m-tolyl)-1-phenyl-butyl]urea 363c
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.056 g, 0.29 mmol) in THF (1.5 mL) at −78 °C was
added i-PrLi (0.45 M, 0.63 mL, 0.37 mmol) dropwise and the mixture stirred
for 15 min. To the reaction mixture was added a solution of 1,3-dimethyl-3-
(1-phenylvinyl)-1-(m-tolyl)urea 362b (0.040 g, 0.14 mmol) in THF (1.5 mL) dropwise and the
mixture stirred for 1.5 h. DMPU (0.17 mL, 1.43 mmol) was added and the mixture stirred for 1.5
h. Purification by flash column chromatography yielded the title compound 363c as a pale
brown oil (0.037 g, 0.11 mmol, 82%). RF 0.29 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.47-
7.42 (2H, m, ArH), 7.34-7.28 (2H, m, ArH), 7.26-7.17 (4H, m, ArH), 7.03 (1H, d, J = 7.1, ArH), 4.14
(1H, q, J = 4.3, NH), 2.98 (3H, s, NMe), 2.55 (3H, d, J = 4.5, NHMe), 2.52 (2H, d, J = 5.6, CH2), 2.33
(3H, s, ArMe), 1.75-1.64 (1H, m, CH(CH3)2), 0.77 (3H, d, J = 6.7, CH(CH3)2), 0.77 (3H, d, J = 3.3,
CH(CH3)2); 13C NMR (100 MHz, CDCl3) 160.0 (C=O), 143.9 (4° ArC), 143.8 (4° ArC), 137.6 (4°
ArC), 128.3 (ArC), 128.0 (ArC), 127.9 (ArC), 127.7 (ArC), 127.5 (ArC), 126.7 (ArC), 124.8 (ArC),
70.4 (4 °C), 48.2 (CH2), 35.4 (NMe), 27.4 (NHMe), 24.7 (CH(CH3)2), 24.6 (CH(CH3)2), 24.6
(CH(CH3)2), 21.7 (ArMe); IR max (film)/cm-1 2984 (NH), 1663 (C=O); MS (ES) m/z 347 (M + Na+);
HRMS m/z calcd 347.2094 for C21H28N2ONa (M + Na+) found 347.2104; HPLC approx. 90:10 er:
Chiracel OD-H, 97:3 Hexane:IPA 1mL/min. tr 9.8 major, 9.5 minor.

195
1,3-Dimethyl-1-[(1S)-1-(m-tolyl)-1-phenyl-hexyl]urea 363d
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.057 g, 0.29 mmol) in THF (1.5 mL) at −78 °C was
added n-BuLi (1.8 M, 0.16 mL, 0.29 mmol) dropwise and the mixture stirred
for 15 min. To the reaction mixture was added a solution of 1,3-dimethyl-3-
(1-phenylvinyl)-1-(m-tolyl)urea 362b (0.041 g, 0.15 mmol) in THF (1.5 mL) dropwise and the
mixture stirred for 1.5 h. DMPU (0.18 mL, 1.46 mmol) was added and the mixture stirred for 1.5
h. Purification by flash column chromatography yielded the title compound 363d as a pale
yellow oil (0.042 g, 0.12 mmol, 83%). RF 0.26 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.40-
7.36 (2H, m, ArH), 7.34-7.29 (2H, m, ArH), 7.25-7.16 (4H, m, ArH), 7.06-7.02 (1H, m, ArH), 4.07
(1H, q, J = 4.5, NH), 2.96 (3H, s, NMe), 2.56-2.50 (5H, m, NHMe + CH2(CH2)3CH3), 2.33 (3H, s,
ArMe), 1.34-1.24 (6H, m, CH2(CH2)3CH3), 0.86 (3H, t, J = 6.3, (CH2)4CH3); 13C NMR (100 MHz,
CDCl3) 160.1 (C=O), 144.1 (4° ArC), 144.0 (4° ArC), 137.7 (4° ArC), 128.1 (ArC), 128.1 (ArC),
128.0 (ArC), 127.6 (ArC), 127.6 (ArC), 126.8 (ArC), 124.6 (ArC), 69.9 (4 °C), 41.1 (CH2(CH2)3CH3),
35.2 (NMe), 32.3 (CH2CH2(CH2)2CH3), 27.5 (NHMe), 25.2 ((CH2)2CH2CH2CH3), 21.8 ((CH2)3CH2CH3),
21.8 (ArMe), 14.0 ((CH2)4CH3); IR max (film)/cm-1 1654 (C=O); MS (ES) m/z 361 (M + Na+); HRMS
m/z calcd 339.2431 for C22H31N2O (M + H+) found 339.2420; HPLC 51:49 er: Chiracel AD-H, 97:3
Hexane:IPA 1mL/min. tr 9.6 major, 9.2 minor.

196
3-Ethyl-1-(1-(2-fluorophenyl)-2-(4-methoxyphenyl)-1-phenylethyl)-1-methylurea 364-1
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.070 g, 0.36 mmol) in THF (1.5 mL) at −78 °C was
added freshly prepared 4-methoxyphenyl lithium solutionA (0.2 M, 1.81 mL,
0.36 mmol) dropwise and the mixture stirred for 15 min. To the reaction
mixture was added a solution of 1-(2-fluorophenyl)-1,3-dimethyl-3-(1-
phenylvinyl)urea 362a (0.054 g, 0.18 mmol) in THF (1.5 mL) dropwise and the mixture stirred for
1.5 h. DMPU (0.22 mL, 1.81 mmol) was added and the mixture stirred for a further 1.5 h.
Purification by flash column chromatography yielded the title compound 364-1 as a brown oil
(0.057 g, 0.14 mmol, 78%). RF 0.18 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.48 (1H, td, J =
8.1, 1.8, ArH), 7.29-7.16 (6H, m, ArH), 6.99 (1H, m, ArH), 6.88-6.82 (2H, m, ArH), 6.70-6.65 (2H,
m, ArH), 4.22 (1H, t, J = 5.3, NH), 4.20 (1H, d, J = 13.9, A of AB, CHAHPMP), 4.03 (1H, d, J = 13.9,
B of AB, CHHBPMP), 3.76 (3H, s, OMe), 3.21-3.09 (2H, m, NHCH2CH3), 2.71 (3H, d, J = 1.5, NMe),
0.99 (3H, t, J = 7.1, NHCH2CH3); 13C NMR (100 MHz, CDCl3) 160.0 (d, JF = 248.3, ArCF), 159.0
(C=O), 158.1 (ArCOMe), 142.2 (4° ArC), 132.3 (d, JF = 10.2, 4° ArC), 132.1 (4° ArC), 129.5 (ArC),
129.2 (d, JF = 3.7, ArC), 128.6 (d, JF = 8.3, ArC), 128.0 (d, JF = 1.9, ArC), 127.6 (ArC), 126.8 (ArC),
123.6 (d, JF = 2.8, ArC), 116.7 (d, JF = 24.9, ArC), 113.0 (ArC), 70.8 (d, JF = 2.8, 4 °C), 55.1 (OMe),
42.0 (d, JF = 3.7, CH2PMP), 36.0 (d, JF = 3.7, NMe), 35.5 (NHCH2CH3), 15.4 (NHCH2CH3); IR max
(film)/cm-1 1653 (C=O); MS (ES) m/z 429 (M + Na+); HRMS m/z calcd 429.1949 for
C25H27N2O2FNa (M + Na+) found 429.1939; HPLC 50:50 er: Chiracel AD-H, 90:10 Hexane:IPA
1mL/min. tr 25.0 major, 30.0 minor.
A Prepared by the following method: To a cooled solution of 4-bromoanisole (0.299 g, 0.20 mL, 1.60 mmol) in THF (1.6 mL, 1 M) was added t-BuLi (1.88 mL, 3.20 mmol) dropwise and the mixture stirred for 1 h. The solution was then warmed to room temperature and used without any further purification.

197
1-[(1S)-2-Cyclopentyl-1-(2-fluorophenyl)-1-phenyl-ethyl]-3-ethyl-1-methyl-urea 364-2
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.060 g, 0.31 mmol) in THF (1.5 mL) at −78 °C was
added cyclopentyl lithium solution (0.51 M, 0.60 mL, 0.31 mmol, prepared
by the method of Rapport and Marek144) dropwise and the mixture stirred
for 15 min. To the reaction mixture was added a solution of 1-(2-fluorophenyl)-1,3-dimethyl-3-
(1-phenylvinyl)urea 362a (0.046 g, 0.15 mmol) in THF (1.5 mL) dropwise and the mixture stirred
for 1.5 h. DMPU (0.19 mL, 1.54 mmol) was added and the mixture stirred for a further 1.5 h.
Purification by flash column chromatography yielded the title compound 364-2 as a colourless
oil (0.049 g, 0.13 mmol, 88%). RF 0.09 (4:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.48-7.40 (2H,
m, ArH), 7.36-7.19 (5H, m, ArH), 7.08 (1H, ddd, J = 7.6, 7.6, 1.4, ArH), 7.01 (1H, ddd, J = 11.5, 8.1,
1.1, ArH), 4.17 (1H, t, J = 5.3, NH), 3.11-2.99 (2H, m, NCH2CH3), 3.03 (3H, s, NMe), 2.77 (2H, d, J =
4.9, CH2CH), 1.72-1.23 (7H, m, -CHCH2CH2CH2CH2-), 1.10-0.94 (2H, m, -CHCH2CH2CH2CH2-), 0.86
(3H, t, J = 7.2, NCH2CH3); 13C NMR (75 MHz, CDCl3) 160.7 (d, JF = 248.5, ArCF), 158.6 (C=O),
143.3 (4° ArC), 131.0 (d, JF = 10.9, 4° ArC), 129.6 (d, JF = 4.4, ArC), 128.9 (d, JF = 9.3, ArC), 127.9
(ArC), 127.9 (ArC), 126.9 (ArC), 123.6 (d, JF = 3.3, ArC), 116.5 (d, JF = 24.5, ArC), 69.6 (d, JF = 3.3, 4
°C), 45.0 (d, JF = 2.7, CCH2), 37.7 (CH), 35.5 (NCH2CH3), 34.3 (CH2), 34.2 (CH2), 34.1 (d, JF = 2.7,
NMe), 24.8 (CH2), 24.8 (CH2), 15.1 (NCH2CH3); IR max (film)/cm-1 3032 (NH), 1648 (C=O); MS (ES)
m/z 391 (M + Na+); HRMS m/z calcd 391.2157 for C23H29N2OFNa (M + Na+) found 391.2159;
HPLC 53:47 er: Chiracel OD-H, 95:5 Hexane:IPA 1mL/min. tr 15.6 major, 46.8 minor.
(Z)-1-Iodohex-3-ene200 365
To a solution of PPh3 (1.335 g, 5.09 mmol), I2 (1.290 g, 5.09 mmol) and imidazole
(0.347 g, 5.09 mmol) in CH2Cl2 (8 mL, 0.5 M) was added cis-3-hexen-1-ol (0.50 mL,
4.24 mmol) dropwise and the mixture stirred for 1 h. Solids were removed by filtration and the
solvent removed from the filtrate under reduced pressure. The residue was purified by flash
column chromatography (4:1 PE:EtOAc) to yield the title compound 365 as an orange oil (0.660
g, 3.14 mmol, 74%) RF 0.75 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 5.59-5.50 (1H, m,
CH=CH(CH2)2I), 5.34-5.25 (1H, m, CH=CH(CH2)2I), 3.15 (2H, t, J = 7.3, CH2I), 2.68-2.60 (2H, m,
CH2CH2I), 2.10-2.01 (2H, m, CH2CH3), 0.99 (3H, t, J = 7.6, CH3); 13C NMR (100 MHz, CDCl3) 134.3
(CH=CH(CH2)2I), 127.1 (CH=CH(CH2)2I), 31.4 (CH2CH2I), 20.7 (CH3CH2), 14.2 (CH3), 5.7 (CH2I); IR
max (film)/cm-1 2997 (CH=CH), 1645 (C=C); MS (ES) m/z 210 (M + H+); HRMS m/z calcd 210.9984
for C6H12I (M + H+) found 210.9989. Data corresponds to the reported literature.200

198
3-Ethyl-1-(1-(2-fluorophenyl)-5,5-dimethyl-1-phenylhexyl)-1-methylurea 366
Following general procedure 2B. To a solution of freshly distilled (+)-
sparteine surrogate 195 (0.087 g, 0.45 mmol) in THF (1.5 mL) at −78 °C was
added freshly prepared cis-3-hexen-1yl lithium solutionB (0.2 M, 2.25 mL,
0.45 mmol) dropwise and the mixture stirred for 15 min. To the reaction
mixture was added a solution of 1-(2-fluorophenyl)-1,3-dimethyl-3-(1-
phenylvinyl)urea 362a (0.067 g, 0.22 mmol) in THF (1.5 mL) dropwise and the mixture stirred for
1.5 h. DMPU (0.27 mL, 2.25 mmol) was added and the mixture stirred for a further 1.5 h.
Purification by flash column chromatography yielded the title compound 366 as a brown oil
(0.067 g, 0.15 mmol, 70%). RF 0.26 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.31-7.12 (7H, m,
ArH), 7.03 (1H, t, J = 7.6, ArH), 6.98 (1H, dd, J = 12.5, 8, ArH), 4.04 (1H, t, J = 5.0, NH), 3.02-2.88
(5H, m, NMe + NCH2CH3), 2.60-2.44 (2H, m, C(Ar)2CH2), 1.15-0.98 (4H, m, CH2CH2CH2C(CH3)3),
0.73 (3H, t, J = 7.3, NCH2CH3), 0.70 (9H, s, C(CH3)3); 13C NMR (125 MHz, CDCl3) 160.4 (d, JF =
248.0, ArCF), 158.7 (C=O), 143.6 (4° ArC), 130.5 (d, JF = 11.8, 4° ArC), 129.3 (d, JF = 3.6, ArC),
129.1 (d, JF = 8.2, ArC), 128.1 (ArC), 127.5 (ArC), 127.1 (ArC), 123.8 (d, JF = 2.7, ArC), 116.5 (d, JF =
24.5, ArC), 69.6, (d, JF = 3.6, 4 °C), 44.4 (CH2CH2C(CH3)3), 40.7 (d, JF = 3.6, C(Ar)2CH2), 35.4
(NCH2CH3), 34.0 (NMe), 30.2 (C(CH3)3), 29.2 (C(CH3)3), 21.3 (CH2C(CH3)3), 15.0 (NCH2CH3); IR max
(film)/cm-1 1638 (C=O); MS (ES) m/z 407 (M + Na+); HRMS m/z calcd 407.2577 for C24H33N2ONa
(M + Na+) found 407.2570.
1-Fluoro-2-[3-methyl-1-phenyl-but-1-enyl]benzene 379
Following general procedure 3. To 3-ethyl-1-[(1S)-1-(2-fluorophenyl)-3-methyl-1-
phenyl-butyl]-1-methyl-urea 363a (0.030 g, 0.09 mmol) in n-BuOH (1 mL) was
added potassium carbonate (0.030 g) and the mixture heated at reflux for 2.5 h.
Purification by flash column chromatography yielded the title compound 379 as a colourless oil
(0.17 g, 0.07 mmol, 83%) as a 1:1 mix of E:Z isomers. RF 0.56 (2:1 PE:EtOAc); 1H NMR (300 MHz,
CDCl3) 7.47-7.23 (6H, m, ArH), 7.21-7.13 (2H, m, ArH), 7.07-6.99 (1H, m, ArH), 5.91 (1H, dd, J =
10.3, 8.0 CH=CAr2), 2.50-2.34 (1H, m, CH(CH3)2), 1.03 (6H, dd, J = 6.6, 3.8, CH(CH3)2); IR max
(film)/cm-1 3020 (C=CH); MS (ES) m/z 263 (M + Na+); HRMS m/z calcd 263.1212 for C18H17FNa (M
+ Na+) found 263.1215. Decomposition in NMR tube prevented full analysis.
B Prepared by the following method: To a cooled solution of (Z)-1-Iodohex-3-ene 365 (0.220 g, 1.05 mmol) in THF (1 mL, 1 M) was added t-BuLi (1.23 mL, 2.09 mmol) dropwise and the mixture stirred for 1 h. The solution was then warmed to room temperature and used without any further purification.

199
(1S)-1-(2-Fluorophenyl)-N,3-dimethyl-1-phenyl-butan-1-amine 337-1
Following general procedure 4. To 3-ethyl-1-[(1S)-1-(2-fluorophenyl)-3-methyl-1-
phenyl-butyl]-1-methyl-urea 363a (0.030 g, 0.09 mmol) in EtOH (1 mL) was
added 2 M NaOH solution (1 mL) and the mixture heated to 130 °C for 2.5 h
under microwave irradiation. Work up yielded the title compound 337-1 as a
colourless oil (0.23 g, 0.09 mmol, 95%) which was used without further purification. RF Flame
(2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.65 (1H, td, J = 8.1, 1.7, ArH), 7.32-7.14 (7H, m,
ArH), 6.90 (1H, ddd, J = 12.4, 7.9, 1.5, ArH), 2.25 (1H, dd, J = 13.6, 5.7, A of ABX, CHAHCH(CH3)2),
2.11 (3H, s, NMe), 2.03, (1H, dd, J = 13.6, 5.1, B of ABX, CHHBCH(CH3)2), 1.68-1.54 (1H, m, X of
ABX, CHX(CH3)2), 0.75 (3H, d, J = 6.8, CH(CH3)2), 0.64 (3H, d, J = 6.6, CH(CH3)2); 13C NMR (75 MHz,
CDCl3) 160.6 (d, JF = 247.4, ArCF), 146.8 (4° ArC), 134.5 (d, JF = 10.4, 4° ArC), 128.7, (d, JF = 3.8,
ArC), 128.4 (d, JF = 8.7, ArC), 127.7 (ArC), 126.4 (d, JF = 1.6, ArC), 126.2 (ArC), 123.3 (d, JF = 3.3,
ArC), 116.2 (d, JF = 22.3, ArC), 63.9 (4 °C), 43.6 (CH2), 29.4 (NMe), 24.7 (CH(CH3)2), 24.4
(CH(CH3)2), 23.2(CH(CH3)2); IR max (film)/cm-1 3240 (NH); MS (ES) m/z 272 (M + H+); HRMS m/z
calcd 272.1810 for C18H23NF (M + H+) found 272.1805; HPLC 92:8 er: determined by 1H NMR
using the chemical shift reagent (R)-2,2,2-trifluoro-1-anthracen-9-yl-ethanol.150
(1S)-N,3-Dimethyl-1-(m-tolyl)-1-phenyl-butan-1-amine 337-2
Following general procedure 4. To 1,3-dimethyl-1-[(1S)-3-methyl-1-(m-tolyl)-1-
phenyl-butyl]urea 363c (0.030 g, 0.09 mmol) in EtOH (1 mL) was added 2 M
NaOH solution (1 mL) and the mixture heated to 130 °C for 2.5 h under
microwave irradiation. Work up yielded the title compound 337-2 as a pale
yellow oil (0.23 g, 0.09 mmol, 93%) which was used without further purification. RF Flame (2:1
PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.41-7.00 (9H, m, ArH), 2.33 (3H, s, NMe), 2.23-2.17 (2H,
m, CH2), 1.63 (1H, brs, NH), 0.92-0.82 (1H, m, CH(CH3)2), 0.69 (3H, d, J = 7.1, (CH(CH3)2), 0.69
(3H, d, J = 7.1, (CH(CH3)2); 13C NMR (75 MHz, CDCl3) 128.3 (ArC), 127.9 (ArC) , 127.7 (ArC),
124.9 (ArC), 44.8 (NCH2), 29.2 (NMe), 24.6 (CH(CH3)2), 24.6 (CH(CH3)2), 23.5 (CH(CH3)2), 21.6
(ArMe) + 3 missing 4° C; IR max (film)/cm-1 3154 (NH); MS (ES) m/z 268 (M + H+); HRMS m/z
calcd 268.2060 for C19H26N (M + H+) found 268.2058.

200
(1S)-2-Cyclopentyl-1-(2-fluorophenyl)-N-methyl-1-phenyl-ethanamine 337-3
Following general procedure 4. To 1-[(1S)-2-cyclopentyl-1-(2-fluorophenyl)-1-
phenyl-ethyl]-3-ethyl-1-methyl-urea 364-2 (0.030 g, 0.08 mmol) in EtOH (1 mL)
was added 2 M NaOH solution (1 mL) and the mixture heated to 130 °C for 2.5
h under microwave irradiation. Work up yielded the title compound 337-3 as a
pale yellow oil (0.22 g, 0.07 mmol, 89%) which was used without further purification. RF Flame
(2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.65 (1H, td, J = 7.9, 1.6, ArH), 7.32-7.15 (7H, m,
ArH), 6.90 (1H, ddd, J = 12.3, 8.2, 1.0, ArH), 2.42 (1H, dd, J = 14.0, 5.8, A of ABX, CHAHCH), 2.26
(1H, dd, J = 14.2, 5.4, B of ABX, CHHBCH), 2.12 (3H, s, NMe), 1.88 (1H, brs, NH), 1.77-1.68 (1H, m,
X of ABX CH2CHX), 1.52-1.30 (5H, m, CH-CH2CHHCH2CH2-), 0.96-0.81 (3H, m, CH-CH2CHHCH2CH2-
); IR max (film)/cm-1 3209 (NH); MS (ES) m/z 298 (M + H+); HRMS m/z calcd 298.1971 for
C20H25NF (M + H+) found 298.1977. Broad signals in 13C NMR prevented full analysis.
(1S)-1-(4-Chlorophenyl)-N,3-dimethyl-1-phenyl-butan-1-amine 337-4
Following general procedure 4. To (S)-1-(1-(4-chlorophenyl)-3-methyl-1-
phenylbutyl)-1,3-dimethylurea 319c (0.030 g, 0.09 mmol) in EtOH (1 mL) was
added 2 M NaOH solution (1 mL) and the mixture heated to 130 °C for 2.5 h
under microwave irradiation. Work up yielded the title compound 337-4 as a
pale yellow oil (0.023 g, 0.08 mmol, 92%) which was used without further purification. RF Flame
(2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.33-7.20 (9H, m, ArH), 2.15 (2H, t, J = 5.7 Hz, CH2),
2.10 (3H, s, NMe), 1.68 (1H, bs, NH), 1.56 (1H, m, CH(CH3)2), 0.73 (6H, d, J = 6.6 Hz, CH(CH3)2);
13C NMR (125 MHz, CDCl3) 147.1 (4° ArC), 146.3 (4° ArC), 131.8 (4° ArC), 129.0 (ArC), 127.8
(ArC), 127.8(ArC), 127.4(ArC), 126.2 (ArC), 65.1 (4° C), 44.5 (CH2), 29.4 (NMe), 24.7 (CH(CH3)2),
24.6 (CH(CH3)2), 23.5 (CH(CH3)2); IR max (film)/cm-1 3189 (NH); MS (ES) m/z 288 (M + H+); HRMS
m/z calcd 288.1519 for C18H23N35Cl (M + H+) found 288.1527; HPLC 92:8 er: determined by 1H
NMR using the chemical shift reagent (R)-2,2,2-trifluoro-1-anthracen-9-yl-ethanol.150

201
(R)-2-Methyl-N-((S)-1-phenylhexyl)propane-2-sulfinamide201 384
Following general procedure 19. To (R,E)-N-benzylidene-2-methylpropane-2-
sulfinamide 382 (0.686 g, 3.28 mmol) in CH2Cl2 (16 mL) at −78 °C was added
pentylmagnesium bromide solution (2 M, 3.28 mL, 6.55 mmol) dropwise.
Purification by flash column chromatography yielded the title compound 384
as a pale yellow oil (0.599 g, 0.21 mmol, 65%) as a single diastereomer. RF 0.23 (2:1 PE:EtOAc);
1H NMR (400 MHz, CDCl3) 7.33-7.22 (5H, m, ArH), 4.36-4.28 (1H, m, NHCH), 3.37 (1H, d, J = 2
Hz, NH), 1.81-1.69 (2H, m, CHCH2), 1.27-1.19 (6H, m, CHCH2CH2CH2CH2), 1.15 (9H, s, C(CH3)3),
0.84-0.78 (3H, m, CH2CH3); 13C NMR (100 MHz, CDCl3) 142.1 (4° ArC), 128.3 (ArC), 127.6 (ArC),
127.5 (ArC), 59.1 (NHCH), 55.4 (C(CH3)3), 38.8 (CHCH2), 31.5 (CHCH2CH2), 25.6 (CHCH2CH2CH2),
22.5 (C(CH3)3), 22.4 (CH2CH3), 13.9 (CH2CH3); IR max (film)/cm-1 1603 (S=O); MS (ES) m/z 304 (M
+ Na+); HRMS m/z calcd 304.1706 for C16H27NONaS (M + Na+) found 304.1693. Data corresponds
to the reported literature.201
(S)-1-Phenylhexan-1-aminium chloride 385
Following general procedure 20. To (R)-2-methyl-N-((S)-1-phenylhexyl)propane-
2-sulfinamide 364 (0.549 g, 1.95 mmol) in MeOH (10 mL) was added HCl
solution (2 M, 9.75 mL, 19.51 mmol). Purification by filtration yielded the title
compound 385 as a white salt (0.412 g, 1.93 mmol, 99%). 1H NMR (500 MHz,
CDCl3) 8.75 (2H, brs, NH2), 7.46-7.39 (2H, m, ArH), 7.37-7.32 (3H, m, ArH), 4.12-4.07 (1H, m,
NH2CH), 2.12-2.02 (1H, m, CHCHH), 2.00-1.88 (1H, m, CHCHH), 1.29-1.07 (6H, m,
CHCH2CH2CH2CH2CH3), 0.85-0.80 (3H, m, CH3); 13C NMR (125 MHz, CDCl3) 136.4 (4° ArC), 129.1
(ArC), 128.9 (ArC), 127.3 (ArC), 56.4 (NH2CH), 34.5 (CHCH2), 31.1 (CHCH2CH2), 25.3
(CHCH2CH2CH2), 22.3 (CH2CH3), 13.9 (CH3); IR max (film)/cm-1 3359 (NH); MS (ES) m/z 177 (M +
H+); HRMS m/z calcd 177.1517 for C12H20N (M + H+) found 177.1521.

202
(S)-1-(4-Chlorophenyl)-1,3-dimethyl-3-(1-phenylhexyl)urea 381
Following a modification to general procedures 22 and 8.To (S)-1-
phenylhexan-1-aminium chloride 385 (0.460 g, 2.15 mmol) in CH2Cl2
(11 mL) was added NEt3 (1.50 mL, 10.75 mmol) and 4-chlorophenyl
isocyanate (0.364 g, 2.37 mmol). After which NaH (0.172 g, 4.30
mmol) and MeI (0.27 mL, 4.30 mmol) were added in DMF (11 mL) at 0 °C. Purification by flash
column chromatography yielded the title compound 381 as a yellow oil (0.583 g, 1.62 mmol,
75%). Could alternatively be synthesised following a modification to general procedure 2A. To a
solution of freshly distilled (−)-sparteine 153 (0.039 g, 0.17 mmol) in cumene (1.5 mL) at −50 °C
was added n-BuLi (1.9 M, 0.17 mL, 0.33 mmol) dropwise and the mixture stirred for 15 min. To
the reaction mixture was added a solution of 1-(4-chlorophenyl)-1,3-dimethyl-3-(1-
phenylvinyl)urea 344 (0.050 g, 0.17 mmol) in cumene (1.5 mL) was added dropwise and the
mixture stirred for 1.5 h. Purification by flash column chromatography yielded the title
compound 381 as a yellow oil (0.032 g, 0.09 mmol, 54%). RF 0.61 (1:1 PE:EtOAc); [D25 Product
from general procedures 22 and 8 gave +12.8 (c 1.0, CHCl3), Product from general procedures
2A gave −59.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) 7.32-7.15 (7H, m, ArH), 6.95-6.89 (2H,
m, ArH), 5.35 (1H, dd, J = 8.8, 7.1 Hz, PhCH), 3.15 (3H, s, NMe), 2.26 (3H, s, NMe), 1.92-1.81 (1H,
m, CHCHH), 1.77-1.67 (1H, m, CHCHH), 1.35-1.17 (6H, m, CHCH2CH2CH2CH2CH3), 0.86 (3H, t, J =
6.9 Hz, CH2CH3); 13C NMR (100 MHz, CDCl3) 162.0 (C=O), 145.6 (4° ArC), 139.9 (4° ArC), 129.6
(4° ArC), 129.4 (ArC), 128.3 (ArC), 127.8 (ArC), 127.3 (ArC), 125.2 (ArC), 58.5 (PhCH), 40.1 (NMe),
31.7 (CHCH2CH2), 30.8 (NMe), 30.3 (CHCH2), 26.4 (CHCH2CH2CH2), 22.6 (CH2CH3), 14.0 (CH2CH3);
IR max (film)/cm-1 2963 (CH), 1641 (C=O); MS (ES) m/z 381 (M + Na+); HRMS m/z calcd 381.1704
for C21H27N2O35ClNa (M + Na+) found 381.1710; HPLC Product from general procedure 2A gave
65:35 er: ChiralPak AD-H, 95:5 Hexane:IPA 1mL/min. tr 11.4 major, 10.3 minor.

203
6-Phenyl-2,3,4,5-tetrahydropyridine158 395
Following general procedure 5. To 5-bromovaleronitrile 394 (0.67 mL, 3.09 mmol)
in THF (8 mL) was added PhLi (2.16 mL, 4.32 mmol) and the mixture heated to
reflux overnight. Work up yielded the title compound 395 as a brown oil (0.354 g,
2.22 mmol, 72%) which was used without further purification. 1H NMR (400 MHz, CDCl3) 7.76-
7.70 (2H, m, ArH), 7.36-7.31 (3H, m, ArH), 3.84-3.77 (2H, m, NCH2), 2.62-2.54 (2H, m, PhNCCH2),
1.83-1.74 (2H, m, NCH2CH2), 1.74-1.60 (2H, m, N=CCH2CH2); 13C NMR (100 MHz, CDCl3) 165.3
(C=N), 140.1 (4° ArC), 129.3 (ArC), 128.0 (ArC), 125.7 (ArC), 49.8 (NCH2), 26.8 (N=CCH2), 21.7
(NCH2CH2), 19.6 (N=CCH2CH2); IR max (film)/cm-1 1635 (C=N); MS (ES) m/z 160 (M + H+). Data
corresponds to the reported literature.158
N-Methyl-N,6-diphenyl-3,4-dihydro-2H-pyridine-1-carboxamide 338a
Following general procedures 7 and 8. To 6-phenyl-2,3,4,5-tetrahydropyridine
395 (1.200 g, 7.54 mmol) in CH2Cl2 (25 mL) was added phenyl isocyanate (0.90
mL, 8.29 mmol) and DMAP (cat.). After which NaH (0.603 g, 15.07 mmol) and
MeI (0.94 mL, 15.07 mmol) were added in DMF (38 mL). Purification by flash
column chromatography yielded the title compound 338a as a brown oil (0.529 g, 1.81 mmol,
24%). RF 0.26 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.25-7.15 (4H, m, ArH), 7.10-6.95 (4H,
m, ArH), 6.69-6.62 (2H, m, ArH), 5.01 (1H, t, J = 4.0, C=CHCH2), 3.72-3.67 (2H, m, NCH2), 3.01
(3H, s, NMe), 2.18-2.10 (2H, m, C=CHCH2), 2.00-1.90 (2H, m, NCH2CH2); 13C NMR (100 MHz,
CDCl3) 158.7 (C=O), 144.4 (NCPh), 140.8 (4° ArC), 139.2 (4° ArC), 128.3 (ArC), 127.6 (ArC),
127.3 (ArC), 125.5 (ArC), 124.3 (ArC), 124.2 (ArC), 113.9 (C=CH), 45.8 (NCH2), 38.1 (NMe), 23.2
(NCH2CH2), 23.1 (C=CHCH2); IR max (film)/cm-1 1652 (C=O), 1530 (C=C); MS (ES) m/z 315 (M +
Na+); HRMS m/z calcd 315.1468 for C19H20N2ONa (M + Na+) found 315.1465.

204
N-(4-Methoxyphenyl)-N-methyl-6-phenyl-3,4-dihydro-2H-pyridine-1-carboxamide 338b
Following general procedures 7 and 8. To 6-phenyl-2,3,4,5-tetrahydropyridine
395 (0.250 g, 1.57 mmol) in CH2Cl2 (5 mL) was added 4-methoxyphenyl
isocyanate (0.22 mL, 1.73 mmol) and DMAP (cat.). After which NaH (0.125 g,
3.14 mmol) and MeI (0.19 mL, 3.14 mmol) were added in DMF (8 mL).
Purification by flash column chromatography yielded the title compound 338b
as a yellow oil (0.182 g, 0.57 mmol, 36%). RF 0.23 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3)
7.28-7.22 (3H, m, ArH), 7.10-7.05 (2H, m, ArH), 6.76-6.71 (2H, m, ArH), 6.62-6.57 (2H, m, ArH),
5.03 (1H, t, J = 4.0, C=CHCH2), 3.80 (3H, s, OMe), 3.68-3.63 (2H, m, NCH2), 2.98 (3H, s, NMe),
2.16-2.10 (2H, m, C=CHCH2), 1.96-1.88 (2H, m, NCH2CH2); 13C NMR (100 MHz, CDCl3) 159.0
(C=O), 156.6 (ArCOMe), 140.8 (NCPh), 139.2 (4° ArC), 137.6 (4° ArC), 127.6 (ArC), 127.2 (ArC),
126.1 (ArC), 125.5 (ArC), 113.6 (ArC), 113.6 (C=CH), 55.4 (OMe), 45.9 (NCH2), 38.8 (NMe), 23.3
(NCH2CH2), 23.2 (C=CHCH2); IR max (film)/cm-1 1652 (C=O), 1510 (C=C); MS (ES) m/z 323 (M +
H+); HRMS m/z calcd 323.1754 for C20H23N2O2 (M + H+) found 323.1754.
N-(4-Chlorophenyl)-N-methyl-6-phenyl-3,4-dihydro-2H-pyridine-1-carboxamide 338c
Following general procedures 7 and 8. To 6-phenyl-2,3,4,5-tetrahydropyridine
395 (1.100 g, 6.91 mmol) in CH2Cl2 (23 mL) was added 4-chlorophenyl
isocyanate (1.167 g, 7.60 mmol) and DMAP (cat.). After which NaH (0.553 g,
13.82 mmol) and MeI (0.86 mL, 13.82 mmol) were added in DMF (35 mL).
Purification by flash column chromatography yielded the title compound 338 as
a luminous yellow oil (1.061 g, 3.25 mmol, 47%). RF 0.24 (2:1 PE:EtOAc); 1H NMR (500 MHz,
CDCl3) 7.29-7.23 (3H, m, ArH), 7.19-7.13 (2H, m, ArH), 7.05-7.00 (2H, m, ArH), 6.60-6.53 (2H,
m, ArH), 5.05 (1H, t, J = 3.9, C=CHCH2), 3.73 (2H, t, J = 5.3, NCH2), 2.96 (3H, s, NMe), 2.19-2.14
(2H, m, C=CHCH2), 1.99-1.93 (2H, m, NCH2CH2); 13C NMR (125 MHz, CDCl3) 158.3 (C=O), 142.8
(NCPh), 140.5 (4° ArC), 138.9 (4° ArC), 128.5 (ArC), 128.2 (ArC), 128.0 (ArC), 127.7 (ArC), 127.4
(ArC), 126.7 (ArC), 114.3 (C=CH), 45.7 (NCH2), 39.6 (NMe), 23.2 (NCH2CH2), 23.2 (C=CHCH2); IR
max (film)/cm-1 1653 (C=O), 1511 (C=C); MS (ES) m/z 349 (M + Na+); HRMS m/z calcd 327.1259
for C19H20N2O35Cl (M + H+) found 327.1249.

205
N-Methyl-6-phenyl-N-(p-tolyl)-3,4-dihydro-2H-pyridine-1-carboxamide 338d
Following general procedures 7 and 8. To 6-phenyl-2,3,4,5-tetrahydropyridine
395 (1.200 g, 7.54 mmol) in CH2Cl2 (25 mL) was added p-tolyl isocyanate (1.05
mL, 8.29 mmol) and DMAP (cat.). After which NaH (0.603 g, 15.07 mmol) and
MeI (0.94 mL, 15.07 mmol) were added in DMF (38 mL). Purification by flash
column chromatography yielded the title compound 338d as a brown oil (0.762
g, 2.49 mmol, 33%). RF 0.26 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.26-7.21 (3H, m, ArH),
7.08-7.04 (2H, m, ArH), 7.00 (2H, d, J = 7.9, ArH), 6.58 (2H, d, J = 7.9, ArH), 5.03 (1H, t, J = 3.9,
C=CHCH2), 3.66 (2H, t, J = 5.5, NCH2), 2.99 (3H, s, ArMe), 2.32 (3H, s, NMe), 2.14 (2H, m,
NCH2CH2), 1.93 (2H, m, C=CHCH2); 13C NMR (125 MHz, CDCl3) 158.9 (C=O), 141.9 (NCPh), 140.8
(4° ArC), 139.3 (4° ArC), 134.0 (4° ArC), 128.9 (ArC), 127.6 (ArC), 127.2 (ArC), 125.5 (ArC), 124.4
(ArC), 113.7 (C=CH), 45.9 (NCH2), 38.4 (NMe), 23.2 (NCH2CH2), 23.1(C=CHCH2), 20.8 (ArMe); IR
max (film)/cm-1 1650 (C=O), 1513 (C=C); MS (ES) m/z 307 (M + H+); HRMS m/z calcd 307.1805
for C20H23N2O (M + H+) found 307.1806.
6-(4-Fluorophenyl)-2,3,4,5-tetrahydropyridine 399a
Following general procedure 6. To 1-bromo-4-fluorobenzene (0.47 mL, 4.32
mmol) in THF (14 mL) was added t-BuLi (2.54 mL, 4.32 mmol) at −78 °C. After 1
h the solution was added dropwise to a solution of 5-bromovaleronitrile 394
(0.67 mL, 3.09 mmol) in THF (10 mL) at −78 °C, before heating to reflux overnight. Work up
yielded the title compound 399a as a yellow oil (0.307 g, 1.73 mmol, 56%) which was used
without further purification. 1H NMR (400 MHz, CDCl3) 7.70-7.63 (2H, d, J = 8.3, ArH), 7.35-
7.26 (2H, d, J = 8.3, ArH), 3.85-3.77 (2H, m, NCH2), 2.63-2.56 (2H, m, N=CArCH2), 1.85-1.79 (2H,
m, NCH2CH2), 1.74-1.61 (2H, m, N=CCH2CH2); 13C NMR (100 MHz, CDCl3) 164.3 (d, JF = 252.3,
ArCF), 164.1 (C=N), 130.5 (d, JF = 2.8, 4° ArC), 129.8 (d, JF = 8.3, ArC), 115.6 (d, JF = 23.1, ArC),
49.7 (NCH2), 26.7 (N=CCH2), 21.8 (NCH2CH2), 19.3 (N=CCH2CH2); IR max (film)/cm-1 1638 (C=N);
MS (ES) m/z 178 (M + H+).

206
6-(4-Chlorophenyl)-2,3,4,5-tetrahydropyridine203 399b
Following general procedure 6. To 1-bromo-4-chlorobenzene (0.83 g, 4.32
mmol) in THF (14 mL) was added t-BuLi (2.54 mL, 4.32 mmol) at −78 °C. After 1
h the solution was added dropwise to a solution of 5-bromovaleronitrile 394
(0.67 mL, 3.09 mmol) in THF (10 mL) at −78 °C, before heating to reflux overnight. Work up
yielded the title compound 399b as a yellow oil (0.383 g, 1.98 mmol, 64%) which was used
without further purification. 1H NMR (400 MHz, CDCl3) 7.72-7.64 (2H, d, J = 8.6, ArH), 7.35-
7.28 (2H, d, J = 8.6, ArH), 3.86-3.78 (2H, m, NCH2), 2.61-2.51 (2H, m, N=CArCH2), 1.84-1.76 (2H,
m, NCH2CH2), 1.74-1.60 (2H, m, N=CCH2CH2); 13C NMR (100 MHz, CDCl3) 164.4 (C=N), 138.6 (4°
ArC), 135.5 (4° ArC), 128.3 (ArC), 127.2 (ArC), 49.9 (NCH2), 26.8 (N=CCH2), 21.7 (NCH2CH2), 19.6
(N=CCH2CH2); IR max (film)/cm-1 1638 (C=N); MS (ES) m/z 194 (M + H+). Data corresponds to the
reported literature.203
6-(4-Methoxyphenyl)-2,3,4,5-tetrahydropyridine202 399c
Following general procedure 6. To 4-bromoanisole (0.54 mL, 4.32 mmol) in
THF (14 mL) was added t-BuLi (2.54 mL, 4.32 mmol) at −78 °C. After 1 h the
solution was added dropwise to a solution of 5-bromovaleronitrile 394 (0.67
mL, 3.09 mmol) in THF (10 mL) at −78 °C, before heating to reflux overnight. Work up yielded
the title compound 399c as a brown oil (0.375 g, 1.98 mmol, 64%) which was used without
further purification. 1H NMR (300 MHz, CDCl3) 7.69-7.60 (2H, d, J = 8.2, ArH), 7.35-7.26 (2H, d,
J = 8.2, ArH), 3.83-3.74 (2H, m, NCH2), 2.65-2.58 (2H, m, N=CArCH2), 1.85-1.76 (2H, m,
NCH2CH2), 1.72-1.60 (2H, m, N=CCH2CH2); 13C NMR (75 MHz, CDCl3) 159.8 (C=N), 159.6
(ArCOMe), 139.4 (4° ArC), 127.2 (ArC), 113.8 (ArC), 55.2 (OMe), 49.7 (NCH2), 26.6 (N=CCH2),
21.9 (NCH2CH2), 19.6 (N=CCH2CH2); IR max (film)/cm-1 1636 (C=N); MS (ES) m/z 190 (M + H+).
Data corresponds to the reported literature.202

207
(2R*,3S*)-3-Isopropyl-N-methyl-N,2-piperidine-1-carboxamide 403
To a cooled solution of N-methyl-N,6-diphenyl-3,4-dihydro-2H-pyridine-1-
carboxamide 338a (0.100 g, 0.34 mmol) in toluene (3.4 mL) at −78 °C was
added i-PrLi (0.5M, 1.71 mL, 0.86 mmol) dropwise. The solution was warmed to
−40 °C and held for 30 min at his temperature. The reaction was quenched
slowly with MeOH (1 mL) and NH4Cl (sat. aq. soln. 5 mL). The mixture was
extracted with Et2O (3 x 10 mL), washed NH4Cl (sat. aq. soln. 10 mL), dried over MgSO4 and
concentrated under reduced pressure. The crude mixture was purified by flash column
chromatography (PE:EtOAc 9:1 1% NEt3) to yield the title compound 403 as a yellow oil (0.074 g,
0.22 mmol, 64%) as a single diastereomer. RF 0.67 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3)
7.35-7.17 (6H, m, ArH), 7.12-7.04 (4H, m, ArH), 5.20 (1H, d, J = 2.5, CHPh), 3.70-3.60 (1H, m,
CHPhCH), 3.14 (3H, s, NMe), 3.00-2.86 (2H, m, NCH2CH2), 1.76-1.66 (2H, m, NCH2CH2), 1.49-1.36
(3H, m, CH2CHCH), 1.00 (3H, d, J = 6.2, CH(CH3)2), 0.87 (3H, d, J = 6.2, CH(CH3)2); 13C NMR 163.0
(C=O), 147.1 (4° ArC), 141.6 (4° ArC), 129.3 (ArC), 128.1 (ArC), 127.0 (ArC), 126.3 (ArC), 124.7
(ArC), 124.4 (ArC), 58.0 (CHPh), 44.7 (NCH2), 42.8 (NMe), 40.2 (CHPhCH), 26.9 (CH(CH3)2), 22.2
(CHPhCHCH2), 20.8 (CH(CH3)2), 20.6 (NCH2CH2), 19.2 (CH(CH3)2); IR max (film)/cm-1 1648 (C=O);
MS (ES) m/z 359 (100% M + Na+); HRMS m/z calcd 337.2274 for C22H29N2O (M + H+) found
337.2277.
3-Isopropyl-N-methyl-2,2-diphenyl-piperidine-1-carboxamide 406a
Following general procedure 9. To N-methyl-N,6-diphenyl-3,4-dihydro-2H-
pyridine-1-carboxamide 338a (0.100 g, 0.34 mmol) in THF (3.4 mL) at −78 °C
was added i-PrLi (0.5 M, 1.71 mL, 0.86 mmol) and DMPU (0.85 mL). Purification
by flash column chromatography yielded the title compound 406a as a pale
yellow solid (0.080 g, 0.24 mmol, 70%). MP 164-166 °C; RF 0.19 (1:1 PE:EtOAc); 1H NMR (400
MHz, CDCl3) 7.60 (4H, t, J = 6.6, ArH), 7.46-7.32 (6H, m, ArH), 4.29 (1H, ddd, J = 13.2, 6.0, 3.2,
NCHHCH2), 3.71 (1H, brm, NH), 3.60 (1H, ddd, J = 13.2, 12.8, 4.4, NCHHCH2), 2.70 (1H, dd, J =
13.2, 3.5, CAr2CHCH2), 2.25 (3H, d, J = 4.7, NMe), 2.00-1.89 (1H, m, NCH2CHH), 1.83-1.71 (2H, m,
NCH2CHH + CHCH(CH3)2), 1.63-1.53 (1H, m, CHCHHCH2), 1.10-0.99 (1H, m, CHCHHCH2), 0.74 (3H,
d, J = 6.9, CH(CH3)2), 0.38 (3H, d, J = 6.6, CH(CH3)2); 13C NMR (100 MHz, CDCl3): 159.2 (C=O) ,
137.5 (4° ArC), 130.2 (ArC), 128.3 (ArC), 127.7 (ArC), 71.6 (4° C), 50.4 (CAr2CHCH2), 40.0 (NCH2),
27.0 (NMe), 26.6 (CHCH(CH3)2), 25.5 (CH(CH3)2), 21.5 (NCH2CH2), 18.5 (CH(CH3)2), 17.7
(NCH2CH2CH2); IR max (film)/cm-1 1629 (C=O); MS (ES) m/z 359 (M + Na+); HRMS m/z calcd
337.2274 for C22H29N2O (M + H+) found 337.2275.

208
3-Butyl-N-methyl-2,2-diphenyl-piperidine-1-carboxamide 406b
Following general procedure 9. To N-methyl-N,6-diphenyl-3,4-dihydro-2H-
pyridine-1-carboxamide 338a (0.100 g, 0.34 mmol) in cumene (3.4 mL) at −78
°C was added n-BuLi (1.76 M, 0.49 mL, 0.86 mmol) and DMPU (0.85 mL).
Purification by flash column chromatography yielded the title compound 406b
as a colourless oil (0.070 g, 0.20 mmol, 58%). RF 0.17 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3)
7.49-7.13 (10H, m, ArH), 4.15 (1H, dt, J = 13.4, 5.0, NCHHCH2), 3.68-3.54 (2H, m, NH +
NCHHCH2), 2.52-2.42 (1H, m, CCH(CH2)), 2.15 (3H, d, J = 4.8, NMe), 1.77-1.61 (3H, m, NCH2CH2 +
NCH2CH2CHH), 1.22-0.66 (7H, m, NCH2CH2CHH + CHCH2CH2CH2CH3), 0.61 (3H, t, J = 7.1, CH2CH3);
13C NMR (100 MHz, CDCl3) 159.3 (C=O), 137.9 (4° ArC), 130.0 (ArC), 128.5 (ArC), 127.7 (ArC),
71.4 (4° C), 47.7 (CH(CH2)3), 40.7 (NCH2), 31.8 (CHCH2(CH2)2), 31.0 (CHCH2CH2CH2), 27.1 (NMe),
23.1 (CH(CH2)2CH2, 22.5 (NCH2CH2CH2), 21.3 (NCH2CH2), 13.9 ((CH2)3CH3); IR max (film)/cm-1 1642
(C=O); MS (ES) m/z 373 (M + Na+); HRMS m/z calcd 351.2431 for C23H31N2O (M + H+) found
351.2438.

209
N-Methyl-2,2-diphenyl-3-sec-butyl-piperidine-1-carboxamide 406c
Following general procedure 9. To N-methyl-N,6-diphenyl-3,4-dihydro-2H-
pyridine-1-carboxamide 338a (0.100 g, 0.34 mmol) in toluene (3.4 mL) at −78
°C was added s-BuLi (1 M, 0.86 mL, 0.86 mmol) and DMPU (0.85 mL).
Purification by flash column chromatography yielded the title compound 406c
as a pale yellow oil (0.070 g, 0.20 mmol, 58%). Isolated as an inseparable 2:1:0:0 mixture of
diastereomers. RF 0.20 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.51-7.41 (8H, m, ArH minor
and major dia.), 7.31-7.09 (12H, m, ArH minor and major dia.), 4.15 (2H, m, NCHHCH2 minor and
major dia.), 3.64-3.57 (2H, brm, NH minor and major dia.), 3.56-3.40 (2H, m, NCHHCH2 minor
and major dia.), 2.65-2.53 (1H, m, CCHCH2 minor and major dia.), 2.03 (3H, d, J = 4.5, NMe
major dia.), 2.01 (3H, d, J = 4.8, NMe minor dia.), 1.87-1.74 (2H, m, NCH2CHH minor and major
dia.), 1.71-1.61 (2H, m, NCH2CHH minor and major dia.), 1.47-1.32 (2H, m, CH2CH2CHH minor
and major dia.), 1.24-1.14 (2H, m, CH(CH3)CH2 minor and major dia.), 1.00-0.87 (2H, m,
CH2CH2CHH minor and major dia.), 0.62 (3H, d, J = 6.8, CHCH3 major dia.), 0.52-0.45 (8H, m,
CHHCH3 minor and major dia.), 0.44-0.31 (2H, m, CHHCH3 minor and major dia.), 0.21 (3H, d, J =
6.8, CHCH3 minor dia.); 13C NMR (100 MHz, CDCl3) 159.2 (C=O minor dia.), 159.2 (C=O major
dia.), 137.5 (4° ArC major dia.), 137.5 (4° minor dia.), 130.3 (ArC minor dia.), 130.0 (ArC major
dia.), 128.2 (ArC major dia.), 128.2 (ArC minor dia.), 127.5 (ArC minor dia.), 127.5 (ArC major
dia.), 71.6 (4° C minor dia.), 71.4 (4° C major dia.), 50.7 (CHCH(CH3)CH2 major dia.), 49.6
(CHCH(CH3)CH2 minor dia.), 39.9 (NCH2 minor dia.), 39.6 (NCH2 major dia.), 33.8 (CHCH(CH3)CH2
major dia.), 32.6 (CHCH(CH3)CH2 minor dia.), 32.2 (CHCH(CH3)CH2 minor dia.), 27.0 (NMe major
dia.), 27.0 (NMe minor dia.), 24,7 (CHCH(CH3)CH2 major dia.), 21.7 (NCH2CH2CH2 major dia.),
21.5 (NCH2CH2CH2 minor dia.), 21.2 (CHCH(CH3)CH2 major dia.), 18.5 (NCH2CH2 minor dia.), 17.3
(NCH2CH2 major dia.), 15.2 (CHCH(CH3)CH2 minor dia.), 12.5 (CH(CH3)CH2CH3 major dia.), 11.4
(CH(CH3)CH2CH3 minor dia.); IR max (film)/cm-1 1640 (C=O). MS (ES) m/z 373 (M + Na+); HRMS
m/z calcd 373.2250 for C23H30N2ONa (M + Na+) found 373.2256.

210
3-tert-Butyl-N-methyl-2,2-diphenyl-piperidine-1-carboxamide 406d
Following general procedure 9. To N-methyl-N,6-diphenyl-3,4-dihydro-2H-
pyridine-1-carboxamide 338a (0.100 g, 0.34 mmol) in toluene (3.4 mL) at −78
°C was added t-BuLi (1.1 M, 0.78 mL, 0.86 mmol) and DMPU (0.85 mL).
Purification by flash column chromatography yielded the title compound 406d
as a colourless oil (0.025 g, 0.07 mmol, 21%). RF 0.19 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3)
7.49-7.29 (10H, m, ArH), 4.38 (1H, ddd, J = 13.9, 7.3, 1.5, NCHHCH2), 3.80-3.73 (1H, brm, NH),
3.50 (1H, ddd, J = 13.9, 7.3, 1.5, NCHHCH2), 2.19 (3H, d, J = 4.8, NMe), 2.13-1.99 (1H, m,
NC8H2CHH), 1.90-1.78 (1H, m, NCH2CHH), 1.77-1.67 (1H, m, NCH2CH2CHH), 1.42-1.29 (1H, m,
NCH2CH2CHH), 0.60 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3) 159.0 (C=O), 136.6 (4° ArC),
127.8 (ArC), 127.5 (ArC), 127.1 (ArC), 72.2 (4° C), 51.4 (CHC(CH3)3), 38.5 (NCH2), 34.4 (C(CH3)3),
30.2 (C(CH3)3), 26.9 (NMe), 22.0 (NCH2CH2), 19.1 (NCH2CH2CH2); IR max (film)/cm-1 1634 (C=O);
MS (ES) m/z 373 (M + Na+); HRMS m/z calcd 373.2256 for C23H30N2ONa (M + Na+) found
373.2261.
(2R*,3S*)-2-(4-Chlorophenyl)-3-isopropyl-N-methyl-2-phenylpiperidine-1-carboxamide 339a
Following general procedure 9. To N-(4-chlorophenyl)-N-methyl-6-phenyl-
3,4-dihydro-2H-pyridine-1-carboxamide 338c (0.100 g, 0.31 mmol) in THF
(3.1 mL) at −78 °C was added i-PrLi (0.35 M, 2.19 mL, 0.76 mmol) and DMPU
(0.78 mL). Purification by flash column chromatography yielded the title
compound 339a as a yellow oil (0.070 g, 0.20 mmol, 63%) as a single diastereomer. RF 0.19 (1:1
PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.65-7.57 (4H, m, ArH), 7.48-7.28 (5H, m, ArH), 4.27 (1H,
ddd, J = 15.4, 6.0, 3.2, NCHHCH2), 3.79-3.72 (1H, brm, NH), 3.64-3.55 (1H, m, NCHHCH2), 2.71-
2.63 (1H, m, CHCHCH2), 2.22 (3H, d, J = 4.6, NMe), 1.97-1.86 (1H, m, NCH2CHH), 1.82-1.68 (2H,
m, NCH2CHH + (CH3)2CHCH), 1.62-1.53 (1H, m, CHCHHCH2), 1.09-0.98 (1H, m, CHCHHCH2), 0.72
(3H, d, J = 6.7, CH(CH3)2), 0.39 (3H, d, J = 6.5, CH(CH3)2); 13C NMR (100 MHz, CDCl3) 159.1
(C=O), 142.1 (4° ArC), 140.7 (4° ArC), 133.4 (4° ArC), 129.6 (ArC), 128.9 (ArC), 128.7 (ArC), 127.7
(ArC), 127.7 (ArC), 71.3 (4° C), 50.3 (CAr2CHCH2), 39.9 (NCH2), 27.0 (NMe), 26.6 (CHCH(CH3)2),
25.5 (CH(CH3)2), 21.5 (NCH2CH2), 18.4 (CH(CH3)2), 17.8 (NCH2CH2CH2); IR max (film)/cm-1 1633
(C=O); MS (ES) m/z 393 (M + Na+); HRMS m/z calcd 371.9235 for C22H28N2O35Cl (M + H+) found
371.9229.

211
(2R*,3R*)-3-Butyl-2-(4-chlorophenyl)-N-methyl-2-phenylpiperidine-1-carboxamide 339b
Following general procedure 9. To N-(4-chlorophenyl)-N-methyl-6-phenyl-
3,4-dihydro-2H-pyridine-1-carboxamide 338c (0.100 g, 0.31 mmol) in cumene
(3.1 mL) at −78 °C was added n-BuLi (2 M, 0.38 mL, 0.76 mmol) and DMPU
(0.78 mL). Purification by flash column chromatography yielded the title
compound 339b as a yellow oil (0.068 g, 0.18 mmol, 58%) as a single diastereomer. RF 0.21 (1:1
PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.52-7.18 (9H, m, ArH), 4.12 (1H, dt, J = 13.2, 5.1,
NCHHCH2), 3.68-3.55 (2H, m, NH + NCHHCH2), 2.55-2.44 (1H, m, CCH(CH2)), 2.16 (3H, d, J = 4.7,
NMe), 1.76-1.62 (3H, m, NCH2CH2 + NCH2CH2CHH), 1.24-0.66 (7H, m, NCH2CH2CHH +
CHCH2CH2CH2CH3), 0.61 (3H, t, J = 7.1, CH2CH3); 13C NMR (100 MHz, CDCl3) 159.1 (C=O), 142.1
(4° ArC), 140.7 (4° ArC), 133.4 (4° ArC), 129.6 (ArC), 128.9 (ArC), 128.7 (ArC), 127.7 (ArC), 127.7
(ArC), 71.5 (4° C), 47.8 (CH(CH2)3), 40.5 (NCH2), 31.6 (CHCH2(CH2)2), 31.0 (CHCH2CH2CH2), 27.3
(NMe), 23.2 (CH(CH2)2CH2, 22.4 (NCH2CH2CH2), 21.3 (NCH2CH2), 14.0 ((CH2)3CH3); IR max
(film)/cm-1 1650 (C=O); MS (ES) m/z 407 (M + Na+); HRMS m/z calcd 385.2042 for C23H30N2O35Cl
(M + H+) found 385.2042.
(2R*,3S*)-3-Isopropyl-2-(4-methoxyphenyl)-N-methyl-2-phenylpiperidine-1-carboxamide 339c
Following general procedure 9. To N-(4-methoxyphenyl)-N-methyl-6-
phenyl-3,4-dihydro-2H-pyridine-1-carboxamide 338b (0.100 g, 0.31 mmol)
in THF (3.1 mL) at −78 °C was added i-PrLi (0.35 M, 2.22 mL, 0.76 mmol) and
DMPU (0.78 mL) Purification by flash column chromatography yielded the
title compound 339c as a pale white solid (0.068 g, 0.19 mmol, 60%) as a single diastereomer.
MP 171-173 °C; RF 0.19 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.63-7.57 (2H, m, ArH),
7.53-7.48 (2H, m, ArH), 7.40-7.23 (4H, m, ArH), 4.28 (1H, ddd, J = 15.6, 6.0, 3.0, NCHHCH2), 3.87
(3H, s, OMe), 3.80-3.71 (1H, brm, NH), 3.63-3.54 (1H, m, NCHHCH2), 2.70-2.63 (1H, m,
CHCHCH2), 2.24 (3H, d, J = 4.5, NMe), 2.00-1.50 (4H, m, NCH2CH2 + (CH3)2CHCH + CHCHHCH2),
1.10-0.96 (1H, m, CHCHHCH2), 0.73 (3H, d, J = 7.1, CH(CH3)2), 0.40 (3H, d, J = 6.8, CH(CH3)2); 13C
NMR (100 MHz, CDCl3) 159.2 (C=O), 158.9 (ArCOMe), 144.4 (4° ArC), 131.4 (4° ArC), 129.0
(ArC), 128.2 (ArC), 127.4 (ArC), 127.2 (ArC), 112.9 (ArC), 71.1 (4° C), 55.2 (OMe), 50.2
(CHCH(CH3)2), 39.8 (NCH2), 27.0 (NMe), 26.5 (CHCH(CH3)2), 25.4 (CH(CH3)2), 21.5 (NCH2CH2), 18.5
(CH(CH3)2), 17.7 (NCH2CH2CH2); IR max (film)/cm-1 1634 (C=O); MS (ES) m/z 389 (M + Na+); HRMS
m/z calcd 367.2381 for C23H31N2O2 (M + H+) found 367.2376.

212
(2R*,3R*)-3-Butyl-2-(4-methoxyphenyl)-N-methyl-2-phenylpiperidine-1-carboxamide 339d
Following general procedure 9. To N-(4-methoxyphenyl)-N-methyl-6-phenyl-
3,4-dihydro-2H-pyridine-1-carboxamide 338c (0.100 g, 0.31 mmol) in
cumene (3.1 mL) at −78 °C was added n-BuLi (2 M, 0.39 mL, 0.76 mmol) and
DMPU (0.78 mL). Purification by flash column chromatography yielded the
title compound 339d as a colourless oil (0.061 g, 0.16 mmol, 52%) as a single diastereomer. RF
0.21 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.56-7.21 (9H, m, ArH), 4.15 (1H, dt, J = 13.5,
4.9, NCHHCH2), 3.87 (3H, s, OMe), 3.68-3.53 (2H, m, NH + NCHHCH2), 2.50-2.41 (1H, m,
CCH(CH2)), 2.16 (3H, d, J = 4.9, NMe), 1.75-1.60 (3H, m, NCH2CH2 + NCH2CH2CHH), 1.28-0.69 (7H,
m, NCH2CH2CHH + CHCH2CH2CH2CH3), 0.63 (3H, t, J = 7.1, CH2CH3); 13C NMR (100 MHz, CDCl3)
159.3 (C=O), 158.8 (ArCOMe), 144.2 (4° ArC), 131.3 (4° ArC), 129.1 (ArC), 128.2 (ArC), 127.4
(ArC), 127.3 (ArC), 113.0 (ArC), 71.4 (4° C), 55.1 (OMe), 48.0 (CH(CH2)3), 40.2 (NCH2), 31.5
(CHCH2(CH2)2), 30.9 (CHCH2CH2CH2), 27.3 (NMe), 23.2 (CH(CH2)2CH2, 22.3 (NCH2CH2CH2), 21.2
(NCH2CH2), 14.1 ((CH2)3CH3); IR max (film)/cm-1 1638 (C=O); MS (ES) m/z 403 (M + Na+); HRMS
m/z calcd 381.2542 for C24H33N2O2 (M + H+) found 381.2547.
(2R*,3S*)-3-Isopropyl-N-methyl-2-phenyl-2-(p-tolyl)piperidine-1-carboxamide 339e
Following general procedure 9. To N-methyl-6-phenyl-N-(p-tolyl)-3,4-dihydro-
2H-pyridine-1-carboxamide 338d (0.100 g, 0.33 mmol) in toluene (3.3 mL) at
−78 °C was added i-PrLi (0.35 M, 2.33 mL, 0.82 mmol) and DMPU (0.83 mL).
Purification by flash column chromatography yielded the title compound 339e
as a pale yellow solid (0.071 g, 0.20 mmol, 62%) as a single diastereomer. MP 167-169 °C; RF
0.18 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.63-7.58 (2H, m, ArH), 7.49-7.44 (2H, m, ArH),
7.37-7.31 (2H, m, ArH), 7.29-7.26 (1H, m, ArH), 7.24-7.19 (2H, m, ArH), 4.29 (1H, ddd, J = 15.6,
6.0, 3.3, NCHHCH2), 3.76-3.70 (1H, brm, NH), 3.65-3.56 (1H, m, NCHHCH2), 2.71-2.64 (1H, m,
CHCHCH2), 2.40 (3H, s, ArMe), 2.23 (3H, d, J = 4.8, NMe), 1.98-1.86 (1H, m, NCH2CHH), 1.83-1.66
(2H, m, NCH2CHH + (CH3)2CHCH), 1.60-1.51 (1H, m, CHCHHCH2), 1.08-0.96 (1H, m, CHCHHCH2),
0.73 (3H, d, J = 6.8, CH(CH3)2), 0.40 (3H,d, J = 6.6, CH(CH3)2); 13C NMR (100 MHz, CDCl3): 159.3
(C=O), 144.4 (4° ArC), 137.3 (4° ArC), 134.2 (4° ArC), 130.2 (ArC), 128.5 (ArC), 128.2 (ArC), 127.5
(ArC), 127.3 (ArC), 71.4 (4° C), 50.4 (CHCH(CH3)2), 39.9 (NCH2), 27.0 (NMe), 26.5 (CH(CH3)2), 25.5
(CH(CH3)2), 21.5 (NCH2CH2CH2), 20.9 (ArMe), 18.6 (CH(CH3)2), 17.8 (NCH2CH2); IR max (film)/cm-1
1635 (C=O); MS (ES) m/z 373 (M + Na+); HRMS m/z calcd 351.2431 for C23H31N2O (M + H+) found
351.2434.

213
(2R*,3R*)-3-Butyl-N-methyl-2-phenyl-2-(p-tolyl)piperidine-1-carboxamide 339f
Following general procedure 9. To N-methyl-6-phenyl-N-(p-tolyl)-3,4-dihydro-
2H-pyridine-1-carboxamide 338d (0.100 g, 0.41 mmol) in cumene (3.3 mL) at
−78 °C was added n-BuLi (2 M, 0.33 mL, 0.82 mmol) and DMPU (0.83 mL).
Purification by flash column chromatography yielded the title compound 339f
as a colourless oil (0.069 g, 0.19 mmol, 58%) as a single diastereomer. RF 0.17 (1:1 PE:EtOAc); 1H
NMR (400 MHz, CDCl3) 7.50-7.19 (9H, m, ArH), 4.14 (1H, dt, J = 13.4, 4.9, NCHHCH2), 3.70-3.57
(2H, m, NH + NCHHCH2), 2.53-2.44 (1H, m, CCH(CH2)), 2.41 (3H, s, ArMe), 2.13 (3H, d, J = 4.5,
NMe), 1.76-1.59 (3H, m, NCH2CH2 + NCH2CH2CHH), 1.26-0.70 (7H, m, NCH2CH2CHH +
CHCH2CH2CH2CH3), 0.60 (3H, t, J = 7.1, CH2CH3); 13C NMR (100 MHz, CDCl3) 159.2 (C=O), 144.3
(4° ArC), 137.5 (4° ArC), 134.1 (4° ArC), 130.2 (ArC), 128.6 (ArC), 128.1 (ArC), 127.4 (ArC), 127.3
(ArC), 71.2 (4° C), 47.8 (CH(CH2)3), 40.4 (NCH2), 31.9 (CHCH2(CH2)2), 31.1 (CHCH2CH2CH2), 27.0
(NMe), 23.3 (CH(CH2)2CH2, 22.7 (NCH2CH2CH2), 21.2 (NCH2CH2), 20.9 (ArMe), 13.8 ((CH2)3CH3); IR
max (film)/cm-1 1634 (C=O); MS (ES) m/z 387 (M + Na+).

214
(2R*,3S*)-3-(sec-Butyl)-N-methyl-2-phenyl-2-(p-tolyl)piperidine-1-carboxamide 339g
Following general procedure 9. To N-methyl-6-phenyl-N-(p-tolyl)-3,4-dihydro-
2H-pyridine-1-carboxamide 338d (0.100 g, 0.33 mmol) in toluene (3.3 mL) at
−78 °C was added s-BuLi (1 M, 0.82 mL, 0.82 mmol) and DMPU (0.83 mL).
Purification by flash column chromatography yielded the title compound 339g
as a colourless oil (0.061 g, 0.17 mmol, 51%) as an inseparable 2:1:0:0 mixture of diastereomer.
RF 0.19 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.49-7.19 (18H, m, ArH minor + major dia.),
4.20-4.12 (2H, ddd, J = 13.8, 6.2, 2.9, NCHH minor + major dia.), 3.65-3.58 (2H, brm, NH, minor +
major dia.), 3.56-3.41 (2H, m, NCHH minor + major dia.), 2.67-2.53 (2H, m, CCHCH2 minor +
major dia.), 2.41 (6H, s, ArMe minor + major dia.), 2.13-2.09 (6H, m, NMe minor + major dia.),
1.85-1.63 (4H, m, NCH2CH2 minor + major dia.), 1.48-1.33 (2H, m, CH2CH2CHH minor + major
dia.), 1.24-1.15 (2H, m, CH(CH3)CH2 minor + major dia.), 1.00-0.88 (2H, m, CH2CH2CHH minor +
major dia.), 0.62 (3H, d, J = 6.8, CHCH3 major dia.), 0.52-0.43 (8H, m, CHHCH3 minor + major
dia.), 0.42-0.29 (2H, m, CHHCH3 minor + major dia.), 0.21 (3H, d, J = 6.8, CHCH3 minor dia.); 13C
NMR (100 MHz, CDCl3) 159.3 (C=O minor dia.), 159.2 (C=O major dia.), 144.3 (4° ArC minor
dia.), 144.1 (4° ArC major dia.), 137.3 (4° ArC major dia.), 137.3 (4° ArC minor dia.), 134.2 (4° ArC
minor + major dia.), 130.3 (ArC minor dia.), 130.0 (ArC major dia.), 128.4 (ArC minor dia.), 128.3
(ArC major dia.), 128.2 (ArC major dia.), 128.2 (ArC minor dia.), 127.5 (ArC minor dia.), 127.4
(ArC major dia.), 127.2 (ArC major dia.), 127.2 (ArC minor dia.), 71.4 (4° C minor dia.), 71.2 (4° C
major dia.), 50.8 (CHCH(CH3)CH2 major dia.), 49.6 (CHCH(CH3)CH2 minor dia.), 39.9 (NCH2 minor
dia.), 39.6 (NCH2 major dia.), 33.7 (CHCH(CH3)CH2 major dia.), 32.5 (CHCH(CH3)CH2 minor dia.),
32.2 (CHCH(CH3)CH2 minor dia.) 27.0 (NMe major dia.), 27.0 (NMe minor dia.), 24.7
(CHCH(CH3)CH2 major dia.), 21.6 (NCH2CH2CH2 major dia.), 21.4 (NCH2CH2CH2 minor dia.), 21.3
(CHCH(CH3)CH2 major dia.), 20.9 (ArMe minor + major dia.), 18.5 (NCH2CH2 minor dia.), 17.3
(NCH2CH2 major dia.), 15.3 (CHCH(CH3)CH2 minor dia.), 12.5 (CH2CH3 major dia.), 11.4 (CH2CH3
minor dia.); IR max (film)/cm-1 1637 (C=O); MS (ES) m/z 387 (M + Na+); HRMS m/z calcd
365.2593 for C24H33N2O (M + H+) found 365.2599.

215
(2R*,3S*)-3-(tert-Butyl)-N-methyl-2-phenyl-2-(p-tolyl)piperidine-1-carboxamide 339h
Following general procedure 9. To N-methyl-6-phenyl-N-(p-tolyl)-3,4-dihydro-
2H-pyridine-1-carboxamide 338d (0.100 g, 0.33 mmol) in toluene (3.3 mL) at
−78 °C was added t-BuLi (1.1 M, 0.74 mL, 0.82 mmol) and DMPU (0.83 mL).
Purification by flash column chromatography yielded the title compound 339h
as a colourless oil (0.045 g, 0.12 mmol, 38%) as a single diastereomer. RF 0.17 (1:1 PE:EtOAc); 1H
NMR (400 MHz, CDCl3) 7.77-7.67 (4H, m, ArH), 7.36-7.18 (5H, m, ArH), 4.38 (1H, ddd, J = 13.9,
7.3, 1.5, NCHH), 3.80 (1H, q, J = 4.5, NH), 3.49 (1H, ddd, J = 14.1, 12.9, 5.3, NCHH), 2.95 (1H, dd,
J = 12.9, 2.5, CHC(CH3)3), 2.42 (3H, s, ArMe), 2.18 (3H, d, J = 4.5, NMe), 2.10-1.97 (1H, m,
NCH2CHH), 1.90-1.78 (1H, m, NCH2CHH), 1.76-1.66 (1H, m, NCH2CH2CHH), 1.42-1.29 (1H, m,
NCH2CH2CHH), 0.60 (9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3) 159.0 (C=O), 144.7 (4° ArC),
137.4 (4° ArC), 133.3 (4° ArC), 131.4 (ArC), 128.2 (ArC), 127.7 (ArC), 127.0 (ArC), 127.0 (ArC),
72.0 (4° C), 51.3 (CHC(CH3)3), 38.4 (NCH2), 34.3 (C(CH3)3), 30.2 (C(CH3)3), 26.9 (NMe), 22.0
(NCH2CH2), 20.9 (ArMe), 19.1 (NCH2CH2CH2); IR max (film)/cm-1 1641 (C=O); MS (ES) m/z 387 (M
+ Na+); HRMS m/z calcd 365.2593 for C24H33N2O (M + H+) found 365.2589.
6-Phenyl-N-(p-tolyl)-3,4-dihydropyridine-1(2H)-carboxamide 411a
Following general procedure 7. To To 6-phenyl-2,3,4,5-tetrahydropyridine 395
(1.380 g, 8.66 mmol) in CH2Cl2 (30 mL) was added p-tolyl isocyanate (1.20 mL,
9.53 mmol) and DMAP (cat.). Purification by flash column chromatography
yielded the title compound 411a as a yellow oil (0.589 g, 2.01 mmol, 23%). RF
0.45 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.54-7.31 (5H, m, ArH), 6.97-
6.91 (2H, m, ArH), 6.84-6.78 (2H, m, ArH), 6.10 (1H, brs, NH), 5.60 (1H, t, J = 4.3, CPh=CH), 3.91-
3.85 (2H, m, NCH2), 2.40-2.31 (2H, m, CHCH2), 2.22 (3H, s, ArMe), 1.99-1.88 (2H, m, NCH2CH2);
13C NMR (75 MHz, CDCl3) 155.6 (C=O), 141.5 (4° ArC), 136.9 (4° ArC), 138.1 (CPh=CH), 131.8
(4° ArC), 129.3 (ArC), 128.8 (ArC), 127.2 (ArC), 126.1 (ArC), 120.8 (ArC), 118.1 (CPh=CH), 44.3
(NCH2), 24.2 (CHCH2), 23.5 (NCH2CH2), 20.8 (ArMe); IR max (film)/cm-1 3319 (NH), 1631 (C=O);
MS (ES) m/z 315 (M + Na+); HRMS m/z calcd 315.1473 for C19H20N2ONa (M + Na+) found
315.1479.

216
N-(4-Methoxyphenyl)-6-phenyl-3,4-dihydro-2H-pyridine-1-carboxamide 411b
Following general procedure 7. To 6-phenyl-2,3,4,5-tetrahydropyridine 395
(0.500 g, 3.14 mmol) in CH2Cl2 (10 mL) was added 4-methoxyphenyl isocyanate
(0.45 mL, 3.45 mmol) and DMAP (cat.). Purification by flash column
chromatography yielded the title compound 411b as a yellow oil (0.174 g, 0.57
mmol, 18%). RF 0.33 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.53-7.48 (2H,
m, ArH), 7.43-7.32 (3H, m, ArH), 6.84-6.78 (2H, m, ArH), 6.72-6.66 (2H, m, ArH), 6.00 (1H, brs,
NH), 5.59 (1H, t, J = 3.8, CPh=CH), 3.91-3.84 (2H, m, NCH2), 3.72 (3H, s, OMe), 2.36 (2H, td, J =
6.8, 3.8, CHCH2), 1.98-1.89 (2H, m, NCH2CH2); 13C NMR (100 MHz, CDCl3) 155.7 (C=O), 154.0
(ArCOMe), 138.6 (4° ArC), 138.1 (CPh=CH), 131.5 (4° ArC), 129.2 (ArC), 128.7 (ArC), 125.8 (ArC),
121.3 (ArC), 118.1 (CPh=CH), 113.8 (ArC), 55.4 (OMe), 44.2 (NCH2), 24.1 (CHCH2), 23.5
(NCH2CH2); IR max (film)/cm-1 3322 (NH), 1629 (C=O); MS (ES) m/z 331 (M + Na+); HRMS m/z
calcd 331.1417 for C19H20N2O2Na (M + Na+) found 331.1409.
(2R*,3S*)-3-Isopropyl-2-phenyl-N-(p-tolyl)piperidine-1-carboxamide 413a
Following general procedure 12. To 6-phenyl-N-(p-tolyl)-3,4-dihydropyridine-
1(2H)-carboxamide 411a (0.165 g, 0.56 mmol) in THF (5.6 mL) at −40 °C was
added i-PrLi (0.58 M, 2.92 mL, 1.69 mmol). Purification by flash column
chromatography yielded the title compound 413a as a pale yellow oil (0.109 g,
0.32 mmol, 57%) as a single diastereomer. RF 0.38 (3:1 PE:EtOAc); 1H NMR (300
MHz, CDCl3) 7.44-7.28 (5H, m, ArH), 7.21-7.14 (2H, m, ArH), 7.10-7.03 (2H, m,
ArH), 6.29 (1H, s, NH), 5.16 (1H, d, J = 4.1, CHPh), 4.12 (1H, ddd, J = 14.5, 5.7, 2.6, NCHHCH2),
3.19 (1H, ddd, J = 14.5, 11.7, 4.5, NCHHCH2), 2.28 (3H, s, ArMe), 1.98-1.74 (3H, m,
CHPhCHCH(CH3)2 + NCH2CHHCH2), 1.67-1.46 (3H, m, NCH2CHHCH2), 1.07 (3H, d, J = 6.4,
CH(CH3)2), 0.97 (3H, d, J = 6.4, CH(CH3)2); 13C NMR (75 MHz, CDCl3) 156.1 (C=O), 141.3 (4° ArC),
136.6 (4° ArC), 132.4 (4° ArC), 129.3 (ArC), 128.9 (ArC), 127.1 (ArC), 126.5 (ArC), 120.0 (ArC),
57.4 (CHPh), 44.3 (CHPhCH), 39.9 (NCH2), 26.8 (CHPhCHCH(CH3)2), 22.2 (CH(CH3)2), 20.7 (ArMe),
20.7 (NCH2CH2CH2), 20.4 (NCH2CH2CH2), 19.1 (CH(CH3)2); IR max (film)/cm-1 2954 (CH), 1633
(C=O); MS (ES) m/z 337 (M + H+); HRMS m/z calcd 337.2280 for C22H29N2O (M + H+) found
337.2286.

217
(2R*,3R*)-3-Butyl-2-phenyl-N-(p-tolyl)piperidine-1-carboxamide 413b
Following general procedure 12. To 6-phenyl-N-(p-tolyl)-3,4-dihydropyridine-
1(2H)-carboxamide 411a (0.165 g, 0.56 mmol) in THF (5.6 mL) at −40 °C was
added n-BuLi (2.5 M, 0.68 mL, 1.69 mmol). Purification by flash column
chromatography yielded the title compound 413b as a pale yellow oil (0.124
g, 0.35 mmol, 63%) as a single diastereomer. RF 0.35 (3:1 PE:EtOAc); 1H NMR
(300 MHz, CDCl3) 7.44-7.25 (5H, m, ArH), 7.19-7.14 (2H, m, ArH), 7.10-7.03 (2H, m, ArH), 6.26
(1H, s, NH), 4.92 (1H, d, J = 3.6, CHPh), 4.16 (1H, dd, J = 13.4, 4.9, NCHHCH2), 3.10 (1H, td, J =
12.8, 3.2, NCHHCH2), 2.38-2.20 (1H, m, CHPhCH), 2.28 (3H, s, ArMe), 1.93-1.24 (10 H, m,
CHPhCHCH2CH2 + (CH2)3CH3), 0.90 (3H, t, J = 6.4, (CH2)3CH3); 13C NMR (75 MHz, CDCl3) 156.2
(C=O), 140.7 (4° ArC), 136.6 (4° ArC), 132.4 (4° ArC), 129.3 (ArC), 128.9 (ArC), 127.2 (ArC), 126.6
(ArC), 120.0 (ArC), 59.6 (CHPh), 40.0 (NCH2), 37.1 (CHPhCH), 31.7 (NCH2CH2), 29.8
(CH2CH2CH2CH3), 23.6 (NCH2CH2CH2), 22.8 (CH2CH2CH3), 20.7 (CH2CH3), 20.3 (ArMe), 14.1
((CH2)3CH3); IR max (film)/cm-1 3318 (NH), 2928 (CH), 1633 (C=O); MS (ES) m/z 351 (M + H+);
HRMS m/z calcd 351.2436 for C23H31N2O (M + H+) found 351.5440.
(2R*,3S*)-3-Isopropyl-N-(4-methoxyphenyl)-2-phenyl-piperidine-1-carboxamide 413c
Following general procedure 12. To N-(4-methoxyphenyl)-6-phenyl-3,4-
dihydro-2H-pyridine-1-carboxamide 411b (0.050 g, 0.16 mmol) in THF (1.6 mL)
at −40 °C was added i-PrLi (0.28 M, 1.74 mL, 0.49 mmol). Purification by flash
column chromatography yielded the title compound 413c as a colourless oil
(0.037 g, 0.10 mmol, 65%) as a single diastereomer. RF 0.28 (2:1 PE:EtOAc); 1H
NMR (400 MHz, CDCl3) 7.44-7.36 (2H, m, ArH), 7.34-7.28 (3H, m, ArH), 7.21-
7.16 (2H, m, ArH), 6.83-6.78 (2H, m, ArH), 6.26 (1H, brs, NH), 5.17 (1H, d, J = 3.8, NCHPh), 4.11
(1H, ddd, J = 13.1, 5.8, 2.3, NCHHCH2), 3.77 (3H, s, OMe), 3.18 (1H, m, NCHHCH2), 1.97-1.76 (3H,
m, NCH2CHH + CHCH(CH3)2), 1.64-1.57 (2H, m, CHCH2), 1.56-1.47 (1H, m, NCH2CHH), 1.08 (3H, d,
J = 6.3, CH(CH3)2), 0.97 (3H, d, J = 6.6, CH(CH3)2); 13C NMR (100 MHz, CDCl3) 156.4 (C=O), 155.6
(ArCOMe), 141.2 (4° ArC), 132.2 (4° ArC), 128.9 (ArC), 127.1 (ArC), 126.5 (ArC), 122.1 (ArC),
114.0 (ArC), 57.3 (CHPh), 55.5 (OMe), 44.3 (CHPhCH), 39.9 (NCH2), 26.8 (CHCH(CH3)2). 22.2
(CH(CH3)2), 20.6 (NCH2CH2), 20.4 (CHCH2), 19.1 (CH(CH3)2); IR max (film)/cm-1 3325 (NH), 1628
(C=O); MS (ES) m/z 353 (M + H+); HRMS m/z calcd 353.2224 for C22H29N2O2 (M + H+) found
353.2226; 1H NOESY NMR (400 MHz, CDCl3) Irradiation at 5.17 ppm results in peaks at 7.33,
6.26, 1.90, 1.60, 1.08 and 0.97 ppm.

218
(2R*,3R*)-3-Butyl-N-(4-methoxyphenyl)-2-phenyl-piperidine-1-carboxamide 413d
Following general procedure 12. To N-(4-methoxyphenyl)-6-phenyl-3,4-
dihydro-2H-pyridine-1-carboxamide 411b (0.050 g, 0.16 mmol) in THF (1.6
mL) at −40 °C was added n-BuLi (1.9 M, 0.26 mL, 0.49 mmol). Purification by
flash column chromatography yielded the title compound 413d as pale
yellow oil (0.033 g, 0.09 mmol, 56%) as a single diastereomer. RF 0.24 (2:1
PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.44-7.28 (5H, m, ArH), 7.22-7.15 (2H, m, ArH), 6.84-6.78
(2H, m, ArH), 6.24 (1H, brs, NH), 4.93 (1H, d, J = 3.5, NCHPh), 4.15 (1H, dd, J = 13.2, 4.7,
NCHHCH2), 3.77 (3H, s, OMe), 3.09 (1H, td, J = 13.4, 3.0, NCHHCH2), 2.31-2.22 (1H, m,
NCH2CHH), 1.98-1.30 (10H, m, NCH2CHHCH2CH(CH2)3CH3), 0.91 (3H, t, J = 7.1, (CH2)3CH3); 13C
NMR (125 MHz, CDCl3) 156.4 (C=O), 155.7 (ArCOMe), 140.7 (4° ArC), 132.3 (4° ArC), 128.9
(ArC), 127.1 (ArC), 126.6 (ArC), 122.1 (ArC), 114.0 (ArC), 59.5 (CHPh), 55.5 (OMe), 40.0
(CHPhCH), 37.1 (NCH2), 31.7 (CHPhCHCH2), 29.8 (CHCH2CH2), 23.6 (CH2CH3), 22.8 (NCH2CH2CH2),
20.3 (NCH2CH2), 14.1 (CH2CH3); IR max (film)/cm-1 3323 (NH), 1631 (C=O); MS (ES) m/z 367 (M +
H+); HRMS m/z calcd 367.2381 for C23H31N2O2 (M + H+) found 367.2380.
(2R*,3S*)-3-Isopropyl-N-methyl-2-phenyl-N-(p-tolyl)piperidine-1-carboxamide 416
Following general procedure 8. To (2R,3S)-3-isopropyl-2-phenyl-N-(p-
tolyl)piperidine-1-carboxamide 413a (0.060 g, 0.18 mmol) in DMF (0.9 mL) was
added NaH (0.014 g, 0.36 mmol) and MeI (0.02 mL, 0.36 mmol) at 0 °C.
Purification by flash column chromatography yielded the title compound 416 as
a yellow oil (0.058 g, 0.17 mmol, 93%) as a single diastereomer. RF 0.27 (3:1
PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.34-7.25 (2H, m, ArH), 7.24-7.17 (3H,
m, ArH), 7.10-7.06 (2H, m, ArH), 6.97-6.93 (2H, m, ArH), 5.22 (1H, d, J = 3.8, CHPh), 3.68-3.61
(1H, m, NCHHCH2), 3.12 (3H, s, NMe), 2.99-2.87 (1H, m, NCHHCH2), 2.30 (3H, s, ArMe), 1.79-
1.67 (2H, m, CHPhCHCH(CH3)2), 1.50-1.34 (3H, m, NCH2CHHCH2), 1.21-1.13 (1H, m,
NCH2CHHCH2), 1.00 (3H, d, J = 6.3, CH(CH3)2), 0.87 (3H, d, J = 6.3, CH(CH3)2); 13C NMR (125 MHz,
CDCl3) 163.2 (C=O), 144.6 (4° ArC), 141.7 (4° ArC), 134.6 (4° ArC), 129.9 (ArC), 128.1 (ArC),
127.0 (ArC), 126.2 (ArC), 124.6 (ArC), 57.9 (CHPh), 44.7 (CHPhCH), 42.9 (NCH2), 40.5 (NMe), 26.8
(CHPhCHCH(CH3)2), 22.2 (CH(CH3)2), 20.8 (ArMe), 20.8 (NCH2CH2CH2), 20.6 (NCH2CH2CH2), 19.3
(CH(CH3)2); IR max (film)/cm-1 2925 (CH), 1644 (C=O); MS (ES) m/z 351 (M + H+); HRMS m/z calcd
351.2431 for C23H31N2O (M + H+) found 351.2438.

219
N-Methyl-6,6-diphenyl-2,3-dihydropyridine-1-carboxamide 401a
Following general procedure 11. To N-methyl-N,6-diphenyl-3,4-dihydro-2H-
pyridine-1-carboxamide 338a (0.100 g, 3.4 mmol) in THF (3.4 mL) at −78 °C was
added LDA (0.86 mmol, prepared by general procedure 10) and DMPU (0.85
mL). Purification by flash column chromatography yielded the title compound
401a as a white solid (0.060 g, 0.20 mmol, 60%). MP 157-159 °C; RF 0.19 (1:1 PE:EtOAc); 1H NMR
(400 MHz, CDCl3) 7.44-7.29 (10H, m, ArH), 5.94 (1H, dt, J = 9.8, 1.5, C Ph2CH=CH), 5.89 (1H, dt,
J = 9.8, 3.5, C Ph2CH=CH), 3.99-3.92 (1H, brm, NH), 3.86 (2H, t, J = 5.6, NCH2), 2.39 (3H, d, J = 4.8,
NMe), 2.36-2.30 (2H, m, NCH2CH2); 13C NMR (100 MHz, CDCl3) 159.4 (C=O), 142.2 (4° ArC),
135.6 (CH=CHCH2), 128.7 (ArC), 128.0 (ArC), 127.7 (ArC), 122.6 (CH=CHCH2), 67.7 (4° C), 40.0
(NCH2), 27.4 (NMe), 25.4 (NCH2CH2); IR max (film)/cm-1 1631 (C=O) 1530 (C=C); MS (ES) m/z 315
(M + Na+); HRMS m/z calcd 293.1648 for C19H21N2O (M + H+) found 293.1638.
2-(4-Chlorophenyl)-N-methyl-2-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 401b
Following general procedure 11. To N-(4-chlorophenyl)-N-methyl-6-phenyl-
3,4-dihydro-2H-pyridine-1-carboxamide 338c (0.100 g, 0.31 mmol) in THF
(3.1 mL) at −78 °C was added LDA (0.76 mmol, prepared by general
procedure 10) and DMPU (0.78 mL). Purification by flash column
chromatography yielded the title compound 401b as a yellow oil (0.070 g, 0.20 mmol, 63%). RF
0.17 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.42-7.29 (9H, m, ArH), 5.94-5.83 (2H, m,
CH=CH), 4.00-3.90 (2H, m, NH + NCHH), 3.68 (1H, ddd, J = 13.1, 7.1, 4.5, NCHH), 2.42 (3H, d, J =
4.5, NMe), 2.37-2.25 (2H, m, NCH2CH2); 13C NMR (100 MHz, CDCl3) 159.1 (C=O), 142.0 (4° ArC),
140.6 (4° ArC), 135.2 (CH=CHCH2), 133.4 (4° ArC), 129.4 (ArC), 128.8 (ArC), 128.7 (ArC), 127.8
(ArC), 127.8 (ArC), 122.8 (CH=CHCH2), 67.3 (4° C), 40.0 (NCH2), 27.4 (NMe), 25.3 (NCH2CH2); IR
max (film)/cm-1 1642 (C=O) 1521 (C=C); MS (ES) m/z 349 (M + Na+); HRMS m/z calcd 327.1259
for C19H20N2O35Cl (M + H+) found 327.1252.

220
N-Methyl-6-phenyl-6-(p-tolyl)-2,3-dihydropyridine-1-carboxamide 401d
Following general procedure 11. To N-methyl-6-phenyl-N-(p-tolyl)-3,4-
dihydro-2H-pyridine-1-carboxamide 338d (0.100 g, 3.3 mmol) in THF (3.3 mL)
at −78 °C was added LDA (0.82 mmol, prepared by general procedure 10) and
DMPU (0.83 mL). Purification by flash column chromatography yielded the
title compound 401d as an off white solid (0.054 g, 0.18 mmol, 54%). MP 161-163 °C; RF 0.18
(1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.41-7.29 (6H, m, ArH), 7.26-7.23 (1H, m, ArH), 7.21-
7.15 (2H, m, ArH), 5.92 (1H, dt, J = 9.8, 1.2, CH=CHCH2), 5.90 (1H, dt, J = 9.8, 3.8, CH=CHCH2),
4.02-3.97 (1H, brm, NH), 3.86 (2H, t, J = 5.6, NCH2), 2.40 (3H, d, J = 4.5, NMe), 2.37 (3H, s,
ArMe), 2.35-2.29 (2H, m, NCH2CH2); 13C NMR (100 MHz, CDCl3) 159.5 (C=O), 142.3 (4° ArC),
139.0 (4° ArC), 137.4 (4° ArC), 135.7 (CH=CHCH2), 129.4 (ArC), 128.7 (ArC), 127.9 (ArC), 127.9
(ArC), 127.5 (ArC), 122.3 (CH=CHCH2), 67.5 (4° C), 39.9 (NCH2), 27.4 (NMe), 25.4 (NCH2CH2), 21.0
(ArMe); IR max (film)/cm-1 1639 (C=O) 1524 (C=C); MS (ES) m/z 329 (M + Na+); HRMS m/z calcd
329.1624 for C20H23N2ONa (M + Na+) found 329.1620.
3-Isopropyl-2,2-diphenyl-piperidine 340a
Following general procedure 3. To 3-isopropyl-N-methyl-2,2-diphenyl-piperidine-
1-carboxamide 406a (0.032 g, 0.10 mmol) in n-BuOH (1 mL) was added potassium
carbonate (0.032 g) and heated to reflux for 2.5 h. Purification by flash column
chromatography yielded the title compound 340a as a white solid (0.021 g, 0.07
mmol, 78%). MP 141-144 °C; RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.45-7.13
(10H, m, ArH), 2.83-2.76 (1H, m, NHCHHCH2), 2.71-2.62 (1H, m, NHCHHCH2), 2.27 (1H, q, J = 5.0,
CHCH(CH3)2), 2.05-1.95 (1H, brs, NH), 1.91-1.83 (2H, m, NHCH2CH2), 1.82-1.68 (2H, m, CH(CH3)2
+ CHCHH), 1.48-1.38 (1H, m, CHCHH), 1.00 (3H, d, J = 7.1, CH(CH3)2), 0.43 (3H, d, J = 6.8,
CH(CH3)2); 13C NMR (100 MHz, CDCl3) 147.9 (4° ArC), 127.9 (ArC), 127.6 (ArC), 125.9 (ArC), 66.5
(4° C), 45.9 (CHCH(CH3)2), 41.6 (NCH2), 27.6 (CH(CH3)2), 24.8 (CH(CH3)2), 24.2 (NCH2CH2CH2), 22.9
(NCH2CH2), 20.9 (CH(CH3)2); IR max (film)/cm-1 2924 (NH); MS (ES) m/z 280 (M + H+); HRMS m/z
calcd 280.2056 for C20H26N (M + H+) found 280.2056.

221
(2R*,3S*)-2-(4-Chlorophenyl)-3-isopropyl-2-phenyl-piperidine 340b
Following general procedure 3. To (2R,3S)-2-(4-chlorophenyl)-3-isopropyl-N-
methyl-2-phenylpiperidine-1-carboxamide 339a (0.050 g, 0.13 mmol) in n-
BuOH (1.3 mL) was added potassium carbonate (0.050 g) and the mixture
heated to reflux for 2.5 h. Purification by flash column chromatography
yielded the title compound 340b as a yellow oil (0.032 g, 0.10 mmol, 85%) as a single
diastereomer. RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.45-7.13 (9H, m, ArH),
2.83-2.76 (1H, m, NHCHHCH2), 2.71-2.62 (1H, m, NHCHHCH2), 2.27 (1H, q, J = 5.0, (CH3)2CHCH),
2.05-1.95 (1H, brs, NH), 1.91-1.83 (2H, m, NHCH2CH2), 1.82-1.68 (2H, m, (CH3)2CH + CHCHH),
1.48-1.38 (1H, m, CHCHH), 1.00 (3H, d, J = 7.1, CH(CH3)2), 0.43 (3H, d, J = 6.8, CH(CH3)2); 13C NMR
(100 MHz, CDCl3): 148.2 (4° ArC), 139.6 (4° ArC), 128.7 (ArC), 128.0 (ArC), 127.3 (ArC), 126.4
(ArC), 125.9 (ArC), 113.0 (ArC), 66.8 (4° C), 46.7 (CHCH(CH3)2), 41.5 (NCH2), 27.6(CHCH(CH3)2),
25.0 (CH(CH3)2), 24.1 (NCH2CH2CH2), 23.1 (NCH2CH2CH2), 20.8 (CH(CH3)2) + 1 missing 4° ArC; IR
max (film)/cm-1 3463 (NH); MS (ES) m/z 314 (M + H+); HRMS m/z calcd 314.1597 for C20H25N35Cl
(M + H+) found 314.1593.
(2R*,3S*)-3-Isopropyl-2-(4-methoxyphenyl)-2-phenyl-piperidine 340c
Following general procedure 3. To (2R,3S)-3-isopropyl-2-(4-methoxyphenyl)-N-
methyl-2-phenylpiperidine-1-carboxamide 339c (0.049 g, 0.13 mmol) in n-BuOH
(1.3 mL) was added potassium carbonate (0.049 g) and the mixture heated to
reflux for 2.5 h. Purification by flash column chromatography yielded the title
compound 340c as a yellow oil (0.034 g, 0.11 mmol, 82%) as a single diastereomer. RF Flame
(2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.42-7.36 (2H, m, ArH), 7.33-7.27 (2H, m, ArH), 7.22
(2H, d, J = 7.22, ArH), 7.18 (1H, t, J = 7.3, ArH), 6.79 (2H, d, J = 8.8, ArH), 3.78 (3H, s, OMe), 2.80-
2.74 (1H, m, NCHHCH2), 2.69-2.63 (1H, m, NCHHCH2), 2.20 (1H, m, NH), 1.89-1.67 (5H, m,
NCH2CHHCH2CHCH(CH3)2), 1.46-1.38 (1H, m, NCH2CHH), 0.98 (3H, d, J = 6.9, CH(CH3)2), 0.41 (3H,
d, J = 6.6, CH(CH3)2); 13C NMR (125 MHz, CDCl3) 157.5 (ArCOMe), 148.2 (4° ArC), 139.6 (4°
ArC), 128.7 (ArC), 128.0 (ArC), 127.3 (ArC), 125.9 (ArC), 113.0 (ArC), 66.2 (4ºC), 55.1 (OMe), 46.3
(CHCH(CH3)2), 41.6 (NCH2), 27.6(CHCH(CH3)2), 24.9 (CH(CH3)2), 24.3 (NCH2CH2CH2), 23.1
NCH2CH2CH2), 20.9 (CH(CH3)2); IR max (film)/cm-1 3425 (NH); MS (ES) m/z 310 (M + H+); HRMS
m/z calcd 310.2165 for C21H28NO (M + H+) found 310.2160; 1H NOESY NMR (400 MHz, CDCl3)
Irradiation at 0.98 ppm results in peaks at 7.22, 1.80 and 0.41 ppm. Irradiation at 0.41 ppm
results in peaks at 2.20, 1.80 and 0.98 ppm.

222
(2R*,3S*)-3-Isopropyl-2-phenyl-2-(p-tolyl)piperidine 340d
Following general procedure 3. To (2R,3S)-3-isopropyl-N-methyl-2-phenyl-2-
(p-tolyl)piperidine-1-carboxamide 339e (0.050 g, 0.14 mmol) in n-BuOH (1.4
mL) was added potassium carbonate (0.050 g) and the mixture heated to
reflux for 2.5 h. Purification by flash column chromatography yielded the title
compound 340d as a colourless oil (0.035 g, 0.12 mmol, 83%) as a single diastereomer. RF Flame
(2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.40 (2H, dd, J = 8.1, 1.5, ArH), 7.30 (2H, t, J = 7.3,
ArH), 7.23-7.17 (3H, m, ArH), 7.06 (2H, d, J = 7.8, ArH), 2.78 (1H, dt, J = 11.9, 4.8, NCHHCH2),
2.67 (1H, ddd, J = 11.9, 9.3, 4.0, NCHHCH2), 2.30 (3H, s, ArMe), 2.23 (1H, q, J = 5.0, CHCH(CH3)2),
1.98 (1H, brs NH), 1.88-1.67 (4H, m, NCH2CHHCH2CHCH(CH3)2), 1.47-1.38 (1H, m, NCH2CHH),
0.99 (3H, d, J = 7.1, CH(CH3)2), 0.43 (3H, d, J = 6.6, CH(CH3)2); 13C NMR (100 MHz, CDCl3) 148.1
(4° ArC), 144.5 (4° ArC), 135.2 (ArC), 128.5 (ArC), 128.0 (ArC), 127.5 (ArC), 125.9 (ArC), 125.3
(ArC), 66.4 (4 °C), 46.1 (CHCH(CH3)2), 41.6 (NCH2), 27.6 (CHCH(CH3)2), 24.9 (CH(CH3)2), 24.3
(NCH2CH2), 23.0 (NCH2CH2CH2), 21.0 (CH(CH3)2), 20.9 (ArMe); IR max (film)/cm-1 2945 (NH), 2355
(CH); MS (ES) m/z 294 (M + H+); HRMS m/z calcd 294.2217 for C21H28N (M + H+) found 294.2216.
6,6-Diphenyl-2,3-dihydro-1H-pyridine 422a
Following general procedure 3. To N-methyl-6,6-diphenyl-2,3-dihydropyridine-1-
carboxamide 401a (0.050 g, 0.17 mmol) in n-BuOH (1.7 mL) was added potassium
carbonate (0.050 g) and the mixture heated to reflux for 2.5 h. Purification by
flash column chromatography yielded the title compound 422a as a pale yellow oil (0.034 g,
0.14 mmol, 84%). RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.42-7.38 (4H, m, ArH),
7.34-7.28 (4H, m, ArH), 7.25-7.20 (2H, m, ArH), 6.19 (1H, dt, J = 10.1, 2.0, CH=CHCH2), 6.02 (1H,
dt, J = 10.1, 3.8, CH=CHCH2), 2.92 (2H, t, J = 5.8, NCH2), 2.14 (2H, tdd, J = 5.8, 3.7, 2.1, NCH2CH2),
1,72 (1H, brs, NH); 13C NMR (100 MHz, CDCl3) 147.7 (4° ArC), 133.6 (CH=CHCH2), 128.0 (ArC),
127.4 (ArC), 126.4 (ArC), 125.9 (CH=CHCH2), 62.8 (4 °C), 39.1 (NCH2), 25.5 (NCH2CH2); IR max
(film)/cm-1 3020 (NH), 1507 (C=C); MS (ES) m/z 236 (M + H+); HRMS m/z calcd 236.1434 for
C17H18N (M + H+) found 236.1437.

223
6-(4-Chlorophenyl)-6-phenyl-2,3-dihydro-1H-pyridine 422b
Following general procedure 3. To 2-(4-chlorophenyl)-N-methyl-2-phenyl-5,6-
dihydropyridine-1(2H)-carboxamide 401b (0.050 g, 0.15 mmol) in n-BuOH (1.5
mL) was added potassium carbonate (0.050 g) and the mixture heated to
reflux for 2.5 h. Purification by flash column chromatography yielded the title compound 422b
as a brown oil (0.037 g, 0.14 mmol, 89%). RF Flame (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3)
7.38-7.23 (6H, m, ArH), 7.15-7.00 (3H, m, ArH), 6.13 (1H, dt, J = 10.0, 2.1, CH=CHCH2), 5.96 (1H,
dt, J = 10.0, 3.8, CH=CHCH2), 2.85 (2H, t, J = 5.6, NCH2), 2.07 (2H, tdd, J = 5.6, 3.8, 2.1, NCH2CH2),
1.74 (1H, brs, NH); 13C NMR (75 MHz, CDCl3) 147.6 (4° ArC), 143.5 (4° ArC), 136.2 (4° ArC),
133.8 (CH=CHCH2), 128.5 (ArC), 128.1 (ArC), 127.4 (ArC), 127.3 (ArC), 126.1 (ArC), 125.7
(CH=CHCH2), 62.5 (4 °C), 39.0 (NCH2), 25.6 (NCH2CH2); IR max (film)/cm-1 3012 (NH), 1649 (C=C);
MS (ES) m/z 270 (M + H+); HRMS m/z calcd 270.0971 for C17H17N35Cl (M + H+) found 270.0965.
6-(4-Methoxyphenyl)-6-phenyl-2,3-dihydro-1H-pyridine 422c
Following general procedure 3. To 2-(4-methoxyphenyl)-N-methyl-2-phenyl-
5,6-dihydropyridine-1(2H)-carboxamide 401c (0.050 g, 0.16 mmol) in n-
BuOH (1.6 mL) was added potassium carbonate (0.050 g) and the mixture
heated to reflux for 2.5 h. Purification by flash column chromatography yielded the title
compound 422c as a pale yellow oil (0.031 g, 0.12 mmol, 75%). RF Flame (2:1 PE:EtOAc); 1H
NMR (400 MHz, CDCl3) 7.53-7.17 (9H, m, ArH), 6.14 (1H, dt, J = 10.4, 1.9, CH=CHCH2), 5.99
(1H, dt, J = 10.4, 3.5, CH=CHCH2), 3.75 (3H, s, OMe), 2.93 (2H, t, J = 5.5, NCH2), 2.11 (2H, tdd, J =
5.7, 3.7, 2.4, NCH2CH2), 1.75 (1H, brs, NH); 13C NMR (100 MHz, CDCl3) 157.3 (ArCOMe), 148.3
(4° ArC), 139.5 (4° ArC), 133.9 (CH=CHCH2), 128.6 (ArC), 127.8 (ArC), 127.2 (ArC), 125.8 (ArC),
125.7 (CH=CHCH2), 113.0 (ArC), 62.5 (4 °C), 55.1 (OMe), 39.7 (NCH2), 25.7 (NCH2CH2); IR max
(film)/cm-1 3024 (NH), 1510 (C=C); MS (ES) m/z 266 (M + H+); HRMS m/z calcd 266.1647 for
C18H20NO (M + H+) found 266.1653.

224
6-Phenyl-6-(p-tolyl)-2,3-dihydro-1H-pyridine 422d
Following general procedure 3. To N-methyl-6-phenyl-6-(p-tolyl)-2,3-
dihydropyridine-1-carboxamide 401d (0.050 g, 0.16 mmol) in n-BuOH (1.6 mL)
was added potassium carbonate (0.050 g) and the mixture heated to reflux for
2.5 h. Purification by flash column chromatography yielded the title compound 422d as a yellow
oil (0.026 g, 0.10 mmol, 64%). RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.38-7.33
(2H, m, ArH), 7.28-7.21 (4H, m, ArH), 7.20-7.14 (1H, m, ArH), 7.10-7.05 (2H, m, ArH), 6.13 (1H,
dt, J = 10.1, 2.0, CH=CHCH2), 5.96 (1H, dt, J = 10.1, 3.8, CH=CHCH2), 2.88 (2H, t, J = 5.8, NCH2),
2.29 (3H, s, ArMe), 2.09 (2H, tdd, J = 5.8, 3.8, 2.0, NCH2CH2), 1.71 (1H, brs, NH); 13C NMR (100
MHz, CDCl3) 147.9 (4° ArC), 144.8 (4° ArC), 136.0 (4° ArC), 133.8 (CH=CHCH2), 128.7 (ArC),
128.0 (ArC), 127.3 (ArC), 127.3 (ArC), 126.4 (ArC), 125.7 (CH=CHCH2), 62.6 (4 °C), 39.1 (NCH2),
25.5 (NCH2CH2), 21.0 (ArMe); IR max (film)/cm-1 3020 (NH), 1508 (C=C); MS (ES) m/z 250 (M +
H+); HRMS m/z calcd 250.1590 for C18H20N (M + H+) found 250.1597.
(E)-N-(4-Chlorobenzylidene)but-3-en-1-amine 446
Following general procedure 13. To a solution of 4-chlorobenzaldehyde
(0.500 g, 3.56 mmol) in CH2Cl2 (18 mL) was added but-3-en-1-amine (0.65
mL, 7.11 mmol). Filtration over diatomaceous earth yielded the title
compound 446 as a colourless oil (0.635 g, 3.28 mmol, 92%). 1H NMR (400 MHz, CDCl3) 8.23
(1H, s, N=CH), 7.66 (2H, brs, ArH), 7.39 (2H, brs, ArH), 5.85 (brs, 1H, CH=CH2), 5.20-4.90 (2H,
brm, CH=CH2), 3.68 (2H, brs, NCH2), 2.47 (2H, brs, NCH2CH2) 13C NMR (100 MHz, CDCl3) 159.9
(N=C), 136.4 (4° ArC), 136.1 (CH=CH2), 134.6 (4° ArC), 129.2 (ArC), 128.8 (ArC), 116.3 (CH=CH2),
61.1 (NCH2), 35.1 (NCH2CH2) IR max (film)/cm-1 2935 (C=CH2), 1638 (C=N) MS (ES) m/z 180 (M +
H+) HRMS m/z calcd 180.0575 for C10H11N35Cl (M + H+) found 180.0571.

225
1-(But-3-en-1-yl)-1-(1-(4-chlorophenyl)allyl)-3-methyl-3-phenylurea 449
Following general procedures 14 and 15. To a solution of (E)-N-(4-
chlorobenzylidene)but-3-en-1-amine 446 (0.604 g, 3.12 mmol) in THF (31
mL) at −78 °C was added diethyl zinc solution (1 M, 6.24 ml, 6.24 mmol)
dropwise. After 2 h vinyl magnesium bromide solution (1 M, 6.24 ml, 6.24
mmol) was added dropwise and the reaction left to warm to room
temperature overnight. After which N-methyl-N-phenylcarbamoyl chloride (0.688 g, 4.06
mmol), NEt3 (0.70 mL, 4.99 mmol) and DMAP (cat.) were added in DCE (10 mL) and the mixture
heated to reflux overnight. Purification by flash column chromatography yielded the title
compound 449 as a yellow oil (0.806 g, 2.27 mmol, 73%). RF 0.57 (2:1 PE:EtOAc) 1H NMR (500
MHz, CDCl3) 7.38-7.33 (2H, m, ArH), 7.28-7.25 (2H, m, ArH), 7.21-7.10 (5H, m, ArH), 5.96 (1H,
ddd, J = 17.2, 10.4, 6.8, CHArCH=CH2), 5.55 (1H, d, J = 6.6, CHAr), 5.46 (1H, ddt, J = 17.1, 10.3,
6.9, NCH2CH2CH=CH2), 5.23 (1H, dt, J = 10.4, 1.3, CHArCH=CHH), 4.94 (1H, dt, J = 17.0, 1.3,
CHArCH=CHH), 4.91-4.83 (2H, m, NCH2CH2CH=CH2), 3.20 (3H, s, NMe), 2.94 (1H, ddd, J = 14.3,
10.2, 5.4, NCHH), 2.80 (1H, ddd, J = 14.5, 10.1, 5.4, NCHH), 2.11-2.03 (1H, m, NCH2CHH), 1.90-
1.81 (1H, m, NCH2CHH) 13C NMR (125 MHz, CDCl3) 162.2 (C=O), 146.7 (4° ArC), 138.6 (4° ArC),
135.3 (NCH2CH2CH=CH2), 134.9 (CHArCH=CH2), 133.1 (4° ArC), 129.6 (ArC), 129.2 (ArC), 128.4
(ArC), 125.4 (ArC), 124.9 (ArC), 118.4 (NCH2CH2CH=CH2), 116.3 (CHArCH=CH2), 62.6 (CHAr), 44.8
(NCH2), 40.2 (NMe), 33.1 (NCH2CH2) IR max (film)/cm-1 2976 (C=CH2), 1649 (C=O), 1595 (C=C) MS
(ES) m/z 377 (M + Na+) HRMS m/z calcd 377.1391 for C21H23N2O35ClNa (M + Na+) found
377.1388.

226
2-(4-Chlorophenyl)-N-methyl-N-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 452
Following general procedure 16. To 1-(but-3-en-1-yl)-1-(1-(4-
chlorophenyl)allyl)-3-methyl-3-phenylurea 449 (0.466 g, 1.31 mmol) in
CH2Cl2 (26 mL) was added Grubbrs 1st generation catalyst (0.054 g, 0.07
mmol). Purification by flash column chromatography yielded the title
compound 452 as a yellow oil (0.189 g, 0.58 mmol, 44%). RF 0.38 (2:1
PE:EtOAc) 1H NMR (500 MHz, CDCl3) 7.37-7.28 (4H, m , ArH), 7.24-7.18 (2H, m, ArH), 7.09-7.04
(1H, m, ArH), 6.88-6.83 (2H, m, ArH), 5.94-5.87 (1H, m, CHArCH=CH), 5.73-5.63 (2H, m,
CHArCH=CH), 3.54-3.50 (1H, m, NCHH), 3.16 (3H, s, NMe), 2.73-2.62 (1H, m, NCHH), 1.95-1.84
(2H, m, NCH2CH2) 13C NMR (125 MHz, CDCl3) 161.1 (C=O), 146.9 (4° ArC), 139.1 (4° ArC), 133.3
(4° ArC), 129.6 (ArC), 129.4 (ArC), 128.4 (ArC), 126.8 (CHArCH=CH), 126.0 (CHArCH=CH), 124.6
(ArC), 123.8 (ArC), 54.8 (CHAr), 39.9 (NMe), 39.6 (NCH2), 24.5 (NCH2CH2) IR max (film)/cm-1 1660
(C=O), 1643 (C=C) MS (ES) m/z 327 (M + H+) HRMS m/z calcd 349.1078 for C19H20N2O35ClNa (M +
Na+) found 349.1082.
2-(4-Chlorophenyl)-N-methyl-N-phenyl-3,4-dihydropyridine-1(2H)-carboxamide 456
Following general procedure 17. To 6-(4-chlorophenyl)-N-methyl-N-phenyl-
5,6-dihydropyridine-1(2H)-carboxamide 456 (0.162 g, 0.50 mmol) in THF (5
mL) was added carbonylchlorohydridotris(triphenylphosphine)ruthenium(II)
(0.047 g, 0.05 mmol) and the mixture heated to reflux overnight. Purification
by flash column chromatography yielded the title compound 456 as a yellow oil (0.128 g, 0.39
mmol, 79%). RF 0.31 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.40-7.00 (9H, m, ArH), 6.59-
6.51 (1H, m, NCH=CHCH2), 5.24-5.17 (1H, m, CHAr), 4.66-4.58 (1H, m, NCH=CHCH2), 3.22 (3H, s,
NMe), 1.99-1.47 (4H, m, CHArCH2CH2); 13C NMR (75 MHz, CDCl3) 158.7 (C=O), 146.3 (4° ArC),
140.1 (4° ArC), 132.3 (4° ArC), 129.5 (ArC), 128.4 (ArC), 127.2 (ArC), 127.0 (NCH=CHCH2), 125.1
(ArC), 124.3 (ArC), 105.1 (NCH=CHCH2), 54.8 (CHAr), 40.1 (NMe), 27.2 (CHArCH2CH2),
17.6(CHArCH2CH2); IR max (film)/cm-1 2928 (C=CH), 1642 (C=O); MS (ES) m/z 349 (M + Na+);
HRMS m/z calcd 327.1259 for C19H20N2O35Cl (M + H+) found 327.1268.

227
6-(4-Chlorophenyl)-N-methyl-N-phenyl-3,4-dihydropyridine-1(2H)-carboxamide 450
To a cooled solution of 2-(4-chlorophenyl)-N-methyl-N-phenyl-5,6-
dihydropyridine-1(2H)-carboxamide 452 (0.152 g, 0.47 mmol) in THF (4.7
mL) at −78 °C was added potassium bis(trimethyl)silylamide solution (0.5 M
in toluene, 1.86 mL, 0.93 mmol) dropwise. The mixture was held at −78 °C
for 2 h before slow warming to room temperature overnight. The reaction
was quenched with MeOH (1 mL) and NH4Cl (sat. aq. soln. 5 mL), extracted with Et2O (3 x 10
mL), washed with brine (10 mL), dried over MgSO4 and the solvent removed under reduced
pressure. The crude mixture was purified by flash column chromatography (PE:EtOAc 9:1 + 1%
NEt3) to yield the title compound 450 as a yellow oil (0.113 g, 0.35 mmol, 74%) RF 0.25 (2:1
PE:EtOAc) 1H NMR (300 MHz, CDCl3) 7.25-7.16 (4H, m, ArH), 7.12-7.04 (1H, m, ArH), 6.98-6.91
(2H, m, ArH), 6.75-6.66 (2H, m, ArH), 4.99 (1H, t, J = 4.0, C=CHCH2), 3.71-3.59 (2H, m, NCH2),
3.04 (3H, s, NMe), 2.21-2.07 (2H, m, C=CHCH2), 1.99-1.89 (2H, m, NCH2CH2) 13C NMR (75 MHz,
CDCl3) 158.6 (C=O), 142.7 (NCPh), 141.7 (4° ArC), 139.2 (4° ArC), 128.5 (ArC), 128.2 (ArC),
127.8 (ArC), 126.8 (ArC), 126.2 (ArC), 124.4 (ArC) 114.1 (C=CH), 45.9 (NCH2), 39.7 (NMe), 23.1
(NCH2CH2), 23.1 (C=CHCH2) IR max (film)/cm-1 1653 (C=O) MS (ES) m/z 349 (M + H+) HRMS m/z
calcd 327.1259 for C19H20N2O35Cl (M + H+) found 327.1250.
(E)-N-Benzylidenebut-3-en-1-amine204 460
Following general procedure 13. To a solution of benzaldehyde (1.67 mL,
16.39 mmol) in CH2Cl2 (41 mL) was added but-3-en-1-amine (3.00 mL, 32.78
mmol). Filtration over diatomaceous earth yielded the title compound 460 as
a yellow oil (2.519 g, 15.82 mmol, 97%). 1H NMR (500 MHz, CDCl3) 8.29 (1H, s, N=CH), 7.77-
7.71 (2H, m, ArH), 7.44-7.40 (3H, m, ArH), 5.87 (1H, ddt, J = 17.0, 10.1, 6.9, CH=CH2), 5.15-5.03
(2H, m, CH=CH2), 3.70 (2H, td, J = 7.3, 1.6, NCH2CH2), 2.51-2.45 (2H, m, NCH2CH2) 13C NMR (125
MHz, CDCl3) 161.3 (N=CH), 136.2 (CH=CH2), 136.2 (4° ArC), 130.5 (ArC), 128.6 (ArC), 128.0
(ArC), 116.2 (CH=CH2), 61.1 (NCH2CH2), 35.2 (NCH2CH2) IR max (film)/cm-1 2939 (C=CH2), 1719
(C=C), 1682 (C=O) MS (ES) m/z 160 (M + H+) HRMS m/z calcd 160.1121 for C11H14N (M + H+)
found 160.1114. Data corresponds to the reported literature.204

228
1-(But-3-en-1-yl)-3-methyl-1-(1-phenylallyl)-3-(pyridin-2-yl)urea 462
Following general procedures 14 and 15. To a solution of (E)-N-
benzylidenebut-3-en-1-amine 460 (0.412 g, 2.59 mmol) in THF (26 mL) at
−78 °C was added diethyl zinc solution (1 M, 5.18 ml, 5.18 mmol) dropwise.
After 2 h vinyl magnesium bromide solution (1 M, 5.18 ml, 5.18 mmol) was
added dropwise and the reaction left to warm to room temperature overnight. After which
methyl(pyridin-2-yl)carbamic chloride (0.574 g, 3.37 mmol), NEt3 (0.58 mL, 4.14 mmol) and
DMAP (cat.) were added in DCE (9 mL) and the mixture heated to reflux overnight. Purification
by flash column chromatography yielded the title compound 462 as a yellow oil (0.446 g, 1.39
mmol, 54%). RF 0.47 (2:1 PE:EtOAc) 1H NMR (500 MHz, CDCl3) 8.26 (1H, d, J = 4.7 Hz, PyH),
7.47 (1H, td, J = 8.2, 0.6, PyH), 7.29-7.18 (5H, m, ArH), 6.91 (1H, d, J = 8.2 Hz, ArH), 6.83-6.79
(1H, m, ArH), 6.02 (1H, ddd, J = 17.3, 10.7, 6.9, CHPhCH=CH2), 5.66 (1H, d, J = 6.9, CHPh), 5.46
(1H, ddt, J = 17.0, 10.4, 6.9, CH2CH=CH2), 5.27 (1H, dd, J = 10.4, 1.3, CHPhCH=CHH), 5.11 (1H, dd,
J = 17.0, 0.9, CHPhCH=CHH), 4.86-4.77 (2H, m, CH2CH=CH2), 3.23 (3H, s, NMe), 3.08 (1H, ddd, J =
14.7, 9.8, 5.4, NCHHCH2), 2.94 (1H, ddd, J = 14.7, 10.7, 6.3, NCHHCH2), 2.18-2.08 (1H, m,
NCH2CHH), 1.96-1.87 (1H, m, NCH2CHH) 13C NMR (125 MHz, CDCl3) 161.5 (C=O), 157.1 (4°
PyC), 148.1 (PyC), 139.2 (4° ArC), 137.5 (PyC), 135.3 (CH2CH=CH2), 135.1 (CHPhCH=CH2), 128.4
(ArC), 127.9 (ArC), 127.6 (ArC), 118.5 (CHPhCH=CH2), 117.3 (PyC), 116.4 (CH2CH=CH2), 114.0
(PyC), 63.6 (CHPh), 45.0 (NCH2), 35.9 (NMe), 33.3 (NCH2CH2) IR max (film)/cm-1 2917 (C=CH2),
1657 (C=O), 1589 (C=C) MS (ES) m/z 344 (M + Na+) HRMS m/z calcd 344.1734 for C20H23N3ONa
(M + Na+) found 344.1725.

229
N-Methyl-2-phenyl-N-(pyridin-2-yl)-5,6-dihydropyridine-1(2H)-carboxamide 463
Following general procedure 16. To 1-(but-3-en-1-yl)-3-methyl-1-(1-
phenylallyl)-3-(pyridin-2-yl)urea 462 (0.397 g, 1.21 mmol) in CH2Cl2 (24 mL) was
added Grubbrs 1st generation catalyst (0.099 g, 0.12 mmol). Purification by
flash column chromatography yielded the title compound 463 as a brown oil
(0.294 g, 1.00 mmol, 84%). RF 0.23 (2:1 PE:EtOAc) 1H NMR (500 MHz, CDCl3) 8.28 (1H, dd, J =
4.7, 1.5, PyH), 7.46-7.41 (2H, m, ArH), 7.39-7.29 (4H, m, ArH + PyH), 6.80 (1H, dd, J = 6.9, 5.0,
PyH), 6.43 (1H, d, J = 8.2, PyH), 6.06-5.99 (1H, m, CHPhCH=CH), 5.86-5.78 (2H, m, CHPhCH=CH),
3.65 (1H, dd, J = 13.2, 6.0, NCHH), 3.29 (3H, s, NMe), 2.92 (1H, ddd, J = 15.8, 11.7, 4.1, NCHH),
2.33-2.22 (1H, m, NCH2CHH), 2.05-1.97 (1H, m, NCH2CHH) 13C NMR (125 MHz, CDCl3) 159.8
(C=O), 157.1 (4° ArC), 148.1 (PyC), 140.0 (4° ArC), 137.4 (PyC), 128.4 (ArC), 128.1 (ArC), 127.7
(ArC), 126.7 (CHPhCH=CH), 125.8 (CHPhCH=CH), 117.1 (PyC), 113.6 (PyC), 55.4 (CHPh), 39.4
(NCH2), 35.6 (NMe), 25.2 (NCH2CH2) IR max (film)/cm-1 2927 (C=CH), 1666 (C=O), 1647 (C=C) MS
(ES) m/z 316 (M + Na+) HRMS m/z calcd 316.1421 for C18H19N3ONa (M + Na+) found 316.1415.
N-Methyl-2-phenyl-N-(pyridin-2-yl)-3,4-dihydropyridine-1(2H)-carboxamide 464
Following general procedure 17. To N-methyl-2-phenyl-N-(pyridin-2-yl)-5,6-
dihydropyridine-1(2H)-carboxamide 463 (0.125 g, 0.43 mmol) in THF (9 mL) was
added carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.041 g,
0.04 mmol) was added and the mixture heated to reflux overnight. Purification
by flash column chromatography yielded the title compound 464 as a colourless oil (0.113 g,
0.39 mmol, 90%). RF 0.62 (1:1 PE:EtOAc) 1H NMR (500 MHz, CDCl3) 8.35 (1H, ddd, J = 4.7, 1.9,
0.6, PyH), 7.54 (1H, ddd, J = 9.1, 7.3, PyH), 7.33-7.28 (2H, m, ArH), 7.25-7.19 (3H, m, ArH), 6.94-
6.88 (2H, m, PyH), 6.60 (1H, d, J = 8.5, NCH=CH), 5.37 (1H, t, J = 4.1, CHPh), 4.84-4.78 (1H, m,
NCH=CH), 3.34 (3H, s, NMe), 2.14-2.02 (2H, m, CHPhCH2), 1.93-1.85 (1H, m, CHPhCH2CHH),
1.77-1.66 (1H, m, CHPhCH2CHH) 13C NMR (125 MHz, CDCl3) 158.1 (C=O), 157.0 (4° ArC), 148.2
(PyC), 140.9 (4° ArC), 137.5 (PyC), 128.3 (ArC), 126.7 (ArC), 126.6 (NCH=CH), 125.6 (ArC), 117.9
(PyC), 115.4 (PyC), 107.1 (NCH=CH), 55.3 (CHPh), 36.3 (NMe), 27.3 (CHPhCH2), 17.8
(CHPhCH2CH2) IR max (film)/cm-1 1674 (C=C), 1647 (C=O) MS (ES) m/z 316 (M + Na+) HRMS m/z
calcd 316.1421 for C18H19N3ONa (M + Na+) found 316.1416.

230
(2S*,3S*)-2-(4-Chlorophenyl)-3-isopropyl-N-methyl-2-phenylpiperidine-1-carboxamide 451
Following general procedure 9. To 6-(4-chlorophenyl)-N-methyl-N-phenyl-
3,4-dihydropyridine-1(2H)-carboxamide 450 (0.073 g, 0.22 mmol) in THF
(2.2 mL) at −78 °C was added i-PrLi (0.41 M, 1.36 mL, 0.56 mmol) and DMPU
(0.55 mL). Purification by flash column chromatography yielded the title
compound 451 as a yellow oil (0.039 g, 0.11 mmol, 48%). RF 0.19 (1:1 PE:EtOAc) 1H NMR (400
MHz, CDCl3) 7.65-7.58 (4H, m, ArH), 7.48-7.27 (5H, m, ArH), 4.19 (1H, ddd, J = 15.4, 6.0, 3.2,
NCHHCH2), 3.81-3.74 (1H, brm, NH), 3.74-3.64 (1H, m, NCHHCH2), 2.80-2.72 (1H, m, CHCHCH2),
2.24 (3H, d, J = 4.6, NMe), 2.00-1.89 (1H, m, NCH2CHH), 1.84-1.71 (2H, m, NCH2CHH +
(CH3)2CHCH), 1.62-1.51 (1H, m, CHCHHCH2), 1.12-0.99 (1H, m, CHCHHCH2), 0.75 (3H, d, J = 6.7,
CH(CH3)2), 0.40 (3H, d, J = 6.5, CH(CH3)2) 13C NMR (100 MHz, CDCl3) 159.3 (C=O), 142.3 (4°
ArC), 141.0 (4° ArC), 133.5 (4° ArC), 129.8 (ArC), 128.9 (ArC), 128.5 (ArC), 127.8 (ArC), 127.7
(ArC), 71.4 (4° C), 50.4 (CAr2CHCH2), 40.0 (NCH2), 27.3 (NMe), 26.8 (CHCH(CH3)2), 25.6
(CH(CH3)2), 21.5 (NCH2CH2), 18.6 (CH(CH3)2), 17.9 (NCH2CH2CH2) IR max (film)/cm-1 1634 (C=O)
MS (ES) m/z 393 (M + Na+) HRMS m/z calcd 371.9235 for C22H28N2O35Cl (M + H+) found
371.9240.
(R,E)-N-Benzylidene-2-methylpropane-2-sulfinamide152 473a
Following general procedure 18. To benzaldehyde (1.64 mL, 16.14 mmol) in
CH2Cl2 (11 mL) was added magnesium sulfate (6.475 g, 53.80 mmol), PPTS
(0.068 g, mmol), and (R)-(+)-2-methyl-2-propanesulfinamide 25 (0.652 g, 5.38
mmol). Purification by flash column chromatography yielded the title compound 473a as a
colourless oil (1.015 g, 4.85 mmol, 90%). RF 0.27 (CH2Cl2); [D25 −110.8 (c 2.0, CHCl3) lit. value
−122.0 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3) 8.60 (1H, s, N=CH), 7.90-7.80 (2H, m, ArH),
7.57-7.41 (3H, m, ArH), 1.27 (9H, s, C(CH3)3); 13C NMR (75 MHz, CDCl3) 162.7 (N=CH), 134.1 (4°
ArC), 132.4 (ArC), 129.4 (ArC), 128.9 (ArC), 57.7 (C(CH3)3), 22.6 (C(CH3)3); IR max (film)/cm-11643
(C=N), 1605 (S=O); MS (ES) m/z 210 (M + H+); HRMS m/z calcd 210.0953 for C11H16NOS (M + H+)
found 210.0961. Data corresponds to the reported literature.152

231
(E)-N-(4-Chlorobenzylidene)-2-methylpropane-2-sulfinamide205 473b
Following general procedure 18. To 4-chlorobenzaldehyde (2.244 g, 15.97
mmol) in CH2Cl2 (10 mL) was added magnesium sulfate (6.406 g, 53.22
mmol), PPTS (0.067 g, 0.27 mmol), and 2-methyl-2-propanesulfinamide 25
(0.645 g, 5.32 mmol). Purification by flash column chromatography yielded
the title compound 473b as a white solid (1.106 g, 4.54 mmol, 85%). MP 74-76 °C; RF 0.35
(CH2Cl2); 1H NMR (300 MHz, CDCl3) 8.56 (1H, s, N=CH), 7.80 (2H, d, J = 8.5 Hz, ArH), 7.46 (2H,
d, J = 8.5 Hz, ArH), 1.27 (9H, s, C(CH3)3); 13C NMR (75 MHz, CDCl3) 161.5 (N=CH), 138.6 (4° ArC),
132.5 (4° ArC), 130.5 (ArC), 129.3 (ArC), 57.9 (C(CH3)3), 22.6 (C(CH3)3); IR max (film)/cm-1 2961
(CH), 1608 (C=N), 1591 (S=O); MS (ES) m/z 266 (M + Na+); HRMS m/z calcd 244.0558 for
C11H15NOS35Cl (M + H+) found 244.0547. Data corresponds to the reported literature.205
(R,E)-N-(2-Chlorobenzylidene)-2-methylpropane-2-sulfinamide206 473c
Following general procedure 18. To 2-chlorobenzaldehyde (2.79 mL, 24.75
mmol) in CH2Cl2 (17 mL) was added magnesium sulfate (9.931 g, 82.51 mmol),
PPTS (0.104 g, 0.41 mmol), and (R)-(+)-2-methyl-2-propanesulfinamide 25 (1.00
g, 8.25 mmol). Purification by flash column chromatography yielded the title
compound 473c as a colourless oil (1.508 g, 6.19 mmol, 75%). RF 0.69 (CH2Cl2); [D25 −195.2 (c
2.0, CHCl3) lit. value −195.6 (c 4.3, CHCl3); 1H NMR (500 MHz, CDCl3) 9.05 (1H, s, N=CH), 8.06
(1H, dd, J = 7.9, 1.9, ArH), 7.48-7.41 (2H, m, ArH), 7.38-7.33 (1H, m, ArH), 1.28 (9H, s, C(CH3)3);
13C NMR (125 MHz, CDCl3) 159.8 (N=CH), 136.5 (4° ArC), 133.1 (ArC), 131.4 (4° ArC), 130.3
(ArC), 129.2 (ArC), 127.1 (ArC), 58.0 (C(CH3)3), 22.6 (C(CH3)3); IR max (film)/cm-1 2961 (CH), 1588
(C=N); MS (ES) m/z 266 (M + Na+); HRMS m/z calcd 266.0377 for C11H14NOS35ClNa (M + Na+)
found 266.0370. Data corresponds to the reported literature.206

232
(R,E)-2-Methyl-N-(3-(trifluoromethyl)benzylidene)propane-2-sulfinamide 473d
Following general procedure 18. To 3-(trifluoromethyl)benzaldehyde (3.31 mL,
24.75 mmol) in CH2Cl2 (17 mL) was added magnesium sulfate (9.931 g, 82.51
mmol), PPTS (0.104 g, 0.41 mmol), and (R)-(+)-2-methyl-2-propanesulfinamide
25 (1.00 g, 8.25 mmol). Purification by flash column chromatography yielded
the title compound 473d as a colourless oil (2.244 g, 8.09 mmol, 98%). RF 0.50 (CH2Cl2); [D25
−107.4 (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) 8.63 (1H, s, N=CH), 8.12 (1H, s, ArH), 8.01 (1H,
d, J = 7.8, ArH), 7.80-7.74 (1H, m, ArH), 7.62 (1H, t, J = 7.8, ArH), 1.28 (9H, s, C(CH3)3); 13C NMR
(100 MHz, CDCl3) 161.4 (C=O), 134.6 (4° ArC), 132.6 (ArC), 131.6 (q, JF = 33.2, ArCCF3), 129.6
(ArC), 128.7 (q, JF = 3.7, ArC), 125.6 (q, JF = 3.7, ArC), 123.6 (q, JF = 274.1, ArCCF3), 58.0 (C(CH3)3),
22.6 (C(CH3)3); IR max (film)/cm-1 1604 (C=N), 1587 (S=O); MS (ES) m/z 300 (M + Na+); HRMS m/z
calcd 300.0641 for C12H14NOSF3ClNa (M + Na+) found 300.0645.
(R)-2-Methyl-N-((S)-1-phenylbut-3-en-1-yl)propane-2-sulfinamide207 474a
Following general procedure 19. To (R,E)-N-benzylidene-2-methylpropane-2-
sulfinamide 473a (0.450 g, 2.15 mmol) in CH2Cl2 (11 mL) at −78 °C was added
allylmagnesium bromide solution (1 M, 4.30 mL, 4.30 mmol) dropwise.
Purification by flash column chromatography yielded the title compound 474a as a pale yellow
oil (0.510 g, 2.03 mmol, 94%). RF 0.17 (2:1 PE:EtOAc); [D25 −129.6 (c 1.0, CHCl3) lit. value
−119.8 (c 1.9, CHCl3); 1H NMR (300 MHz, CDCl3) 7.42-7.21 (5H, m, ArH), 5.83-5.63 (1H, m,
CH=CH2), 5.25-5.11 (2H, m, CH=CH2), 4.48 (1H, ddd, J = 7.9, 5.5, 2.4, CHPh), 3.67 (1H, s, NH),
2.70-2.40 (2H, m, CHCH2), 1.20 (9H, s, C(CH3)3); 13C NMR (75 MHz, CDCl3) 141.7 (4° ArC), 134.2
(CH=CH2), 128.4 (ArC), 127.6 (ArC), 127.5 (ArC), 119.3 (CH=CH2), 57.1 (CHPh), 55.6 (C(CH3)3),
43.4 (CHCH2), 22.6 (C(CH3)3); IR max (film)/cm-1 3201 (NH), 2958 (CH=CH2), 1639 (S=O); MS (ES)
m/z 274 (M + Na+); HRMS m/z calcd 252.1417 for C14H22NOS (M + H+) found 252.1413. Data
corresponds to the reported literature.207

233
N-(1-(4-Chlorophenyl)but-3-en-1-yl)-2-methylpropane-2-sulfinamide208 474b
Following general procedure 19. To (E)-N-(4-chlorobenzylidene)-2-
methylpropane-2-sulfinamide 473b (1.106 g, 4.54 mmol) in CH2Cl2 (23 mL)
at −78 °C was added allylmagnesium bromide solution (1 M, 9.07 mL, 9.07
mmol) dropwise. Purification by flash column chromatography yielded the title compound 474b
as a colourless oil (1.207 g, 4.22 mmol, 93%). RF 0.21 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3)
7.35-7.21 (4H, m, ArH), 5.79-5.62 (1H, m, CH=CH2), 5.23-5.12 (2H, m, CH=CH2), 4.45 (1H, ddd, J =
7.9, 5.5, 1.9, CHAr), 3.66 (1h, brs, NH), 2.63-2.37 (2H, m, CHArCH2), 1.19 (9H, s, C(CH3)3); 13C
NMR (75 MHz, CDCl3) 140.2 (4° ArC), 133.7 (CH=CH2)), 133.4 (4° ArC), 128.9 (ArC), 128.7 (ArC),
119.6 (CH=CH2), 56.4 (CHAr), 55.7 (C(CH3)3), 43.3 (CHArCH2), 22.5 (C(CH3)3); IR max (film)/cm-1
3199 (NH), 2957 (C=CH2), 1641 (C=O); MS (ES) m/z 308 (M + Na+); HRMS m/z calcd 286.1032 for
C14H21NOS35Cl (M + H+) found 286.1035. Data corresponds to the reported literature.208
(R)-N-((S)-1-(2-Chlorophenyl)but-3-en-1-yl)-2-methylpropane-2-sulfinamide 474c
Following general procedure 19. To (R,E)-N-(2-chlorobenzylidene)-2-
methylpropane-2-sulfinamide 473c (1.462 g, 6.00 mmol) in CH2Cl2 (30 mL) at
−78 °C was added allylmagnesium bromide solution (1 M, 12.00 mL, 12.00
mmol) dropwise. Purification by flash column chromatography yielded the title compound 474c
as a pale yellow oil (1.611 g, 5.63 mmol, 94%). RF 0.19 (2:1 PE:EtOAc); [D25 −108.4 (c 1.0,
CHCl3); 1H NMR (400 MHz, CDCl3) 7.42-7.35 (2H, m, ArH), 7.28-7.20 (2H, m, ArH), 5.78-5.72
(1H, m, CHArCH2CH=CH2), 5.21-5.17 (2H, m, CHArCH2CH=CH2), 5.01 (1H, ddd, J = 8.3, 5.2, 3.5,
CHAr), 3.72 (1H, d, J = 2.7, NH), 2.73-2.66 (1H, m, CHArCHH), 2.52-2.45 (1H, m, CHArCHH), 1.21
(9H, s, C(CH3)3); 13C NMR (100 MHz, CDCl3) 139.2 (4° ArC), 133.4 (4° ArC), 133.7
(CHArCH2CH=CH2), 133.1 (ArC), 129.7 (ArC), 128.5 (ArC), 126.7 (ArC), 119.4 (CHArCH2CH=CH2),
55.8 (CHAr), 53.7 (C(CH3)3), 42.4 (CHCH2), 22.5 (C(CH3)3); IR max (film)/cm-1 3219 (NH), 3073
(C=CH2); MS (ES) m/z 308 (M + Na+); HRMS m/z calcd 286.1032 for C14H21NSO35Cl (M + H+) found
286.1038.

234
(R)-2-Methyl-N-((S)-1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)propane-2-sulfinamide 474d
Following general procedure 19. To (R,E)-2-methyl-N-(3-
(trifluoromethyl)benzylidene)propane-2-sulfinamide 473d (2.162 g, 7.80 mmol)
in CH2Cl2 (40 mL) at −78 °C was added allylmagnesium bromide solution (1 M,
15.59 mL, 15.59 mmol) dropwise. Purification by flash column chromatography
yielded the title compound 474d as a yellow oil (2.276 g, 7.12 mmol, 91%). RF 0.15 (2:1
PE:EtOAc); [D25 −103.4 (c 2.0, CHCl3);
1H NMR (500 MHz, CDCl3) 7.60 (1H, s, ArH), 7.58-7.43
(3H, m, ArH), 5.78-5.67 (1H, m, CHArCH2CH=CH2), 5.23-5.17 (2H, m, CHArCH2CH=CH2), 4.58-4.52
(1H, m, CHAr), 3.73 (1H, brs, NH), 2.64-2.57 (1H, m, CHArCHH), 2.50-2.41 (1H, m, CHArCHH),
1.20 (9H, s, C(CH3)3); 13C NMR (125 MHz, CDCl3) 142.9 (4° ArC), 133.5 (CHArCH2CH=CH2), 131.0
(ArC), 130.8 (q, JF = 31.8, ArCCF3), 128.9 (ArC), 124.5 (q, JF = 3.6, ArC), 124.1 (q, JF = 4.5, ArC),
124.0 (q, JF = 272.5, ArCCF3), 119.9 (CHArCH2CH=CH2), 56.4 (C(CH3)3), 55.8 (CHAr), 43.3
(CHArCH2), 22.4 (C(CH3)3); IR max (film)/cm-1 2980 (C=CH2), 1326 (S=O); MS (ES) m/z 342 (M +
Na+); HRMS m/z calcd 342.1110 for C15H20NOSF3Na (M + Na+) found 342.1107.
(S)-1-Phenylbut-3-en-1-aminium chloride209 478a
Following general procedure 20. To (R)-2-methyl-N-((S)-1-phenylbut-3-en-1-
yl)propane-2-sulfinamide 474a (1.707 g, 6.79 mmol) in MeOH (34 mL) was added
HCl solution (2 M, 33.95 mL, 67.90 mmol). Purification by filtration yielded the
title compound 478a as a white salt (0.987 g, 5.37 mmol, 79%). [D25 −10.0 (c 1.0, CHCl3) lit.
value −11.4 (c 1.0, H2O); 1H NMR (400 MHz, CDCl3) 8.79 (2H, brs, NH2), 7.50-7.39 (2H, m, ArH),
7.40-7.31 (3H, m, ArH), 5.64-5.49 (1H, m, CHPhCH2CH=CH2), 5.16-5.00 (2H, m,
CHPhCH2CH=CH2), 4.21 (1H, brs, CHPh), 2.89-2.66 (2H, m, CHPhCH2); 13C NMR (100 MHz, CDCl3)
135.7 (4° ArC), 131.4 (CHPhCH2CH=CH2), 129.0 (ArC), 129.0 (ArC), 127.4 (ArC), 120.2
(CHPhCH2CH=CH2), 55.9 (CHPh), 38.9 (CHPhCH2); IR max (film)/cm-1 3036 (NH), 2891 (C=CH2),
1512 (C=C); MS (ES) m/z 295 (2M + H+); HRMS m/z calcd 148.1121 for C10H14N (M + H+) found
148.1118. Data corresponds to the reported literature.209

235
1-(4-Chlorophenyl)but-3-en-1-aminium chloride 478b
Following general procedure 20. To N-(1-(4-chlorophenyl)but-3-en-1-yl)-2-
methylpropane-2-sulfinamide 474b (1.207 g, 4.22 mmol) in MeOH (21 mL)
was added HCl solution (2 M, 21.11 mL, 42.23 mmol). Purification by filtration
yielded the title compound 478b as a white salt (0.909 g, 4.17 mmol, 99%). 1H NMR (300 MHz,
MeOD) 7.52-7.40 (4H, m, ArH), 5.69 (1H, ddt, J = 17.1, 10.2, 7.0, CH=CH2), 5.25-5.13 (2H, m,
CH=CH2), 4.37 (1H, t, J = 7.7, CHAr), 2.78-2.67 (2H, m, CHArCH2); 13C NMR (75 MHz, MeOD)
136.8 (4° ArC), 136.4 (4° ArC), 133.1 (CH=CH2), 130.5 (ArC), 130.2 (ArC), 120.8 (CH=CH2), 55.7
(CHAr), 40.0 (CHArCH2); IR max (film)/cm-1 3359 (NH), 2917 (C=CH2); MS (ES) m/z 182 (M + H+);
HRMS m/z calcd 182.0737 for C10H12N35Cl (M + H+) found 182.0741.
(S)-1-(2-Chlorophenyl)but-3-en-1-aminium chloride209 478c
Following general procedure 20. To (R)-N-((S)-1-(2-chlorophenyl)but-3-en-1-yl)-2-
methylpropane-2-sulfinamide 474c (1.564 g, 5.47 mmol) in MeOH (27 mL) was
added HCl solution (2 M, 27.35 mL, 54.72 mmol). Purification by filtration yielded
the title compound 478c as a white salt (0.993 g, 4.28 mmol, 78%). [D25 −16.5 (c 0.5, CHCl3) lit.
value −16.0 (c 1.0, H2O); 1H NMR (400 MHz, MeOD) 7.62-7.39 (4H, m, ArH), 5.73 (1H, ddt, J =
17.2, 10.1, 7.1, CHArCH2CH=CH2), 5.25-5.15 (2H, m, CHArCH2CH=CH2), 2.79-2.73 (2H, m,
CHArCH2); 13C NMR (100 MHz, MeOD) 135.6 (4° ArC), 134.8 (4° ArC), 132.7 (CHArCH2CH=CH2),
131.9 (ArC), 131.5 (ArC), 129.3 (ArC), 128.8 (ArC), 121.0 (CHArCH2CH=CH2), 52.3 (CHAr), 39.4
(CHArCH2); IR max (film)/cm-1 3354 (NH), 2913 (C=CH2); MS (ES) m/z 182 (M + H+); HRMS m/z
calcd 182.0732 for C10H13N35Cl (M + H+) found 182.0731. Data corresponds to the reported
literature.209

236
(S)-1-(3-(Trifluoromethyl)phenyl)but-3-en-1-aminium chloride 478d
Following general procedure 20. To (R)-2-methyl-N-((S)-1-(3-
(trifluoromethyl)phenyl)but-3-en-1-yl)propane-2-sulfinamide 474d (2.216 g, 6.94
mmol) in MeOH (35 mL) was added HCl solution (2 M, 34.69 mL, 69.38 mmol).
Purification by filtration yielded the title compound 478d as a white salt (1.328 g, 5.28 mmol,
76%). [D25 +49.6 (c 0.5, CHCl3);
1H NMR (500 MHz, MeOD) 7.81 (1H, s, ArH), 7.78-7.65 (3H,
m, ArH), 5.71 (ddt, J = 17.3, 10.1, 6.9, CHArCH2CH=CH2), 5.25-5.14 (2H, m, CHArCH2CH=CH2),
4.52 (t, J = 7.6, CHAr), 2.82-2.69 (2H, m, CHArCH2); 13C NMR (125 MHz, MeOD) 139.5 (4° ArC),
132.9 (CHArCH2CH=CH2), 132.4 (ArC), 132.6 (q, JF = 31.8, ArCCF3), 131.4 (ArC), 127.23 (q, JF = 4.5,
ArC), 125.4 (q, JF = 3.6, ArC), 125.5 (q, JF = 271.6, ArCCF3), 121.0 (CHArCH2CH=CH2), 55.8 (CHAr),
40.2 (CHArCH2); IR max (film)/cm-1 2963 (C=CH2), 2781 (CH), 2201 (CH); MS (ES) m/z 216 (M +
H+); HRMS m/z calcd 216.0995 for C11H13NF3 (M + H+) found 216.0992.
(S)-N-Allyl-1-phenylbut-3-en-1-amine210 475a
Following general procedure 21. To (S)-1-phenylbut-3-en-1-aminium chloride
478a (0.987 g, 5.37 mmol) in DMF (27 mL) at 0 °C was added NaH (1.075 g,
26.87 mmol) and allyl bromide (0.70 mL, 8.06 mmol). Purification by flash
column chromatography yielded the title compound 475a as a pale yellow oil (0.640 g, 3.42
mmol, 63%). RF Flame (2:1 PE:EtOAc); [D25 −43.0 (c 2.0, CHCl3) lit. value −27.0 (c 0.01, CHCl3);
1H NMR (500 MHz, CDCl3) 7.33-7.24 (4H, m, ArH), 7.23-7.17 (1H, m, ArH), 5.81 (1H, dddd, J =
17.0, 10.1, 6.6, 5.4, X of ABXYY’, NHCH2CHX=CH2), 5.67 (1H, dddd, J = 16.7, 10.1, 7.9, 6.0,
CHArCH2CH=CH2), 5.10-4.98 (4H, m, Y and Y’ of ABXYY’, NHCH2CH=CHYHY’ + CHPhCH2CH=CH2),
3.66 (1H, dd, J = 7.9, 6.0, CHPh), 3.08 (1H, ddt, J = 14.2, 5.4, 1.9, A of ABXYY’, NHCHAHCH=CH2),
2.97 (1H, ddt, J = 14.2, 6.6, 1.3, B of ABXYY’, NHCHHBCH=CH2), 2.43-2.31 (2H, m, CHArCH2), 1.55
(1H, brs, NH); 13C NMR (125 MHz, CDCl3) 143.6 (4° ArC), 136.8 (CHArCH2CH=CH2), 135.3
(NHCH2CH=CH2), 128.3 (ArC), 127.2 (ArC), 127.0 (ArC), 117.5 (CH=CH2), 115.7 (CH=CH2), 61.6
(CHAr), 49.9 (NHCH2), 42.9 (CHArCH2); IR max (film)/cm-1 3076 (NH), 2796 (CH), 1640 (C=C); MS
(ES) m/z 188 (M + H+); HRMS m/z calcd 188.1434 for C13H18N (M + H+) found 188.1437. Data
corresponds to the reported literature.210

237
(S)-N-Allyl-1-(2-chlorophenyl)but-3-en-1-amine 475c
Following general procedure 21. To (S)-1-(2-chlorophenyl)but-3-en-1-aminium
chloride 478c (0.993 g, 4.55 mmol) in DMF (23 mL) at 0 °C was added NaH
(0.911 g, 22.76 mmol) and allyl bromide (0.59 mL, 6.83 mmol). Purification by
flash column chromatography yielded the title compound 475c as a pale yellow oil (0.690 g,
3.11 mmol, 68%). RF Flame (2:1 PE:EtOAc); [D25 −33.6 (c 1.0, CHCl3);
1H NMR (500 MHz, CDCl3)
7.55 (1H, dd, J = 7.6, 1.9, ArH), 7.34 (1H, dd, J = 7.9, 1.6, ArH), 7.30-7.26 (1H, m, ArH), 7.18 (1H,
td, J = 7.6, 1.9, ArH), 5.88 (1H, dddd, J = 17.0, 10.1, 6.6, 5.4, X of ABXYY’, NHCH2CHX=CH2), 5.78
(1H, dddd, J = 16.4, 10.1, 8.2, 6.0, CHArCH2CH=CH2), 5.17-5.05 (4H, m, Y and Y’ of ABXYY’,
NHCH2CH=CHYHY’ + CHArCH2CH=CH2), 4.28 (1H, dd, J = 8.2, 5.0, CHAr), 3.12 (1H, ddt, J = 13.9,
5.4, 1.9, A of ABXYY’, NHCHAHCH=CH2), 3.04 (ddt, J = 13.9, 6.3, 1.3, B of ABXYY’,
NHCHHBCH=CH2), 2.56-2.48 (1H, m, CHArCHH), 2.36-2.27 (1H, m, CHArCHH), 1.73 (1H, brs, NH);
13C NMR (125 MHz, CDCl3) 140.6 (4° ArC), 136.5 (NHCH2CH=CH2), 135.0 (CHArCH2CH=CH2),
133.6 (4° ArC), 129.5 (ArC), 128.0 (ArC), 127.9 (ArC), 126.9 (ArC), 117.9 (CH=CH2), 116.0
(CH=CH2), 57.2 (CHAr), 50.0 (NHCH2), 41.1 (CHArCH2); IR max (film)/cm-1 3076 (NH), 1641 (C=C);
MS (ES) m/z 222 (M + H+); HRMS m/z calcd 222.1050 for C13H17N35Cl (M + H+) found 222.1058.
(S)-N-Allyl-1-(3-(trifluoromethyl)phenyl)but-3-en-1-amine 475d
Following general procedure 21. To (S)-1-(3-(trifluoromethyl)phenyl)but-3-en-1-
aminium chloride 478d (1.328 g, 5.28 mmol) in DMF (26 mL) at 0 °C was added
NaH (1.055 g, 26.38 mmol) and allyl bromide (0.68 mL, 7.91 mmol). Purification
by flash column chromatography yielded the title compound 475d as a
colourless oil (0.927 g, 3.63 mmol, 69%). RF 0.18 (2:1 PE:EtOAc); [D25 −19.6 (c 1.0, CHCl3);
1H
NMR (500 MHz, CDCl3) 7.60 (1H, s, ArH), 7.58-7.50 (2H, m, ArH), 7.49-7.43 (1H, m, ArH), 5.86
(1H, dddd, J = 17.0, 10.4, 6.6, 5.4, X of ABX, NHCH2CHX=CH2), 5.70 (1H, dddd, J = 17.0, 10.4, 7.9,
6.9, CHArCH2CH=CH2), 5.15-5.07 (4H, m, NHCH2CH=CH2 + CHArCH2CH=CH2), 3.80 (1H, t, J = 6.8,
CHAr), 3.14 (1H, dd, J = 14.0, 4.9, A of ABX, NHCHAHCH=CH2), 3.02 (1H, dd, J = 14.2, 6.9, B of
ABX, NHCHHBCH=CH2), 2.50-2.38 (2H, m, CHArCH2), 1.65 (1H, brs, NH); 13C NMR (75 MHz, CDCl3)
145.0 (4° ArC), 136.5 (NHCH2CH=CH2), 134.7 (CHArCH2CH=CH2), 130.7 (q, JF = 32.2, ArCCF3),
130.6 (ArC), 128.7 (ArC), 124.3 (q, JF = 272.0, ArCCF3), 124.1 (q, JF = 3.8, ArC), 123.9 (q, JF = 3.8,
ArC), 118.1 (CH=CH2), 115.9 (CH=CH2), 61.3 (CHAr), 50.0 (NHCH2), 42.9 (CHArCH2); IR max
(film)/cm-1 3075 (NH), 1642 (C=C). MS (ES) m/z 256 (M + H+); HRMS m/z calcd 256.1308 for
C14H17NF3 (M + H+) found 256.1306.

238
(S)-1-Allyl-3-(4-methoxyphenyl)-3-methyl-1-(1-phenylbut-3-en-1-yl)urea 476a
Following general procedures 22 and 8. To (S)-N-allyl-1-phenylbut-3-en-
1-amine 475a (0.756 g, 4.04 mmol) in CH2Cl2 (20 mL) was added NEt3
(0.84 mL, 6.06 mmol) and 4-methoxyphenyl isocyanate (0.52 mL, 4.04
mmol). After which NaH (0.323 g, 8.08 mmol) and MeI (0.50 mL, 8.08
mmol) were added in DMF (20 mL). Purification by flash column
chromatography yielded the title compound 476a as an orange oil (1.033 g, 2.95 mmol, 73%). RF
0.47 (2:1 PE:EtOAc); [D25 −89.6 (c 1.0, CHCl3);
1H NMR (300 MHz, CDCl3) 7.35-7.16 (5H, m,
ArH), 7.09-6.99 (2H, m, ArH), 6.88-6.80 (2H, m, ArH), 5.77-5.61 (1H, m, CHPhCH2CH=CH2), 5.36-
5.18 (2H, m, CHPh + X of ABXYY’, NCH2CH=CH2), 5.08-4.97 (2H, m, CHPhCH2CH=CH2), 4.91-4.75
(2H, m, Y + Y’ of ABXYY’, NCH2CH=CHYY’), 3.80 (3H, s, OMe), 3.41 (1H, ddt, J = 16.2, 5.5, 1.7, A of
ABXYY’, NCHAHCH=CH2), 3.29 (1H, ddt, J = 16.2, 5.5, 1.7, B of ABXYY’, NCHHBCH=CH2), 3.13 (3H,
s, NMe), 2.78-2.52 (2H, m, CHPhCH2CH=CH2); 13C NMR (75 MHz, CDCl3) 162.5 (C=O), 157.2 (4°
ArCOMe), 139.8 (4° ArC), 139.8 (4° ArC), 135.5 (CH=CH2), 135.4 (CH=CH2), 128.3 (ArC), 128.1
(ArC), 127.3 (ArC), 126.5 (ArC), 117.1 (CHPhCH2CH=CH2), 115.8 (NCH2CH=CH2), 114.6 (ArC), 59.7
(CHPh), 55.5 OMe), 47.2 (NCH2CH=CH2), 40.7 (NMe), 35.8 (CHPhCH2CH=CH2); IR max (film)/cm-1
3009 (C=CH2), 1651 (C=O), 1512 (C=C); MS (ES) m/z 373 (M + Na+); HRMS m/z calcd 351.2068
for C22H27N2O2 (M + H+) found 351.2065.

239
(S)-1-Allyl-3-methyl-3-phenyl-1-(1-phenylbut-3-en-1-yl)urea 476b
Following general procedures 22 and 8. To (S)-N-allyl-1-phenylbut-3-en-1-
amine 475a (0.214 g, 1.14 mmol) in CH2Cl2 (6 mL) was added NEt3 (0.24 mL,
1.71 mmol) and phenyl isocyanate (0.12 mL, 1.14 mmol). After which NaH
(0.091 g, 2.29 mmol) and MeI (0.14 mL, 2.29 mmol) were added in DMF (6
mL). Purification by flash column chromatography yielded the title compound 476b as a
colourless oil (0.320 g, 1.00 mmol, 88%). RF 0.61 (2:1 PE:EtOAc); [D25 −54.8 (c 2.0, CHCl3);
1H
NMR (500 MHz, CDCl3) 7.27-7.13 (7H, m, ArH), 7.08-7.00 (3H, m, ArH), 5.60 (1H, ddt, J = 17.0,
10.1, 6.9, CHArCH2CH=CH2), 5.22 (1H, ddt, J = 16.7, 10.7, 6.0, X of ABXYY’, NCH2CHX=CH2), 5.16
(1H, t, J = 7.9, CHPh), 5.00-4.92 (2H, m, CHArCH2CH=CH2), 4.82-4.72 (2H, m, Y and Y’ of ABXYY’,
NCH2CH=CHYHY’), 3.34 (1H, ddt, J = 16.4, 5.7, 1.6, A of ABXYY’, NCHAHCH=CH2), 3.21 (1H, ddt, J =
16.1, 6.3, 1.6, B of ABXYY’, NCHHBCH=CH2), 3.11 (3H, s, NMe), 2.69-2.61 (1H, m,
CHPhCHHCH=CH2), 2.58-2.48 (1H, m, CHPhCHHCH=CH2); 13C NMR (125 MHz, CDCl3) 162.2
(C=O), 146.6 (4° ArC), 139.6 (4° ArC), 135.3 (CH=CH2), 135.3 (CH=CH2), 129.3 (ArC), 128.3 (ArC),
128.2 (ArC), 127.4 (ArC), 124.9 (ArC), 124.7 (ArC), 117.2 (CHArCH2CH=CH2), 115.9
(NCH2CH=CH2), 59.6 (CHAr), 47.1 (NCH2), 40.0 (NMe), 35.7 CHArCH2); IR max (film)/cm-1 3063
(C=CH2), 1641 (C=O), 1595 (C=C); MS (ES) m/z 321 (M + H+); HRMS m/z calcd 343.1781 for
C21H24N2ONa (M + Na+) found 343.1784.

240
1-Allyl-1-(1-(4-chlorophenyl)but-3-en-1-yl)-3-methyl-3-phenylurea 476c
Following general procedures 21, 22 and 8. To 1-(4-chlorophenyl)but-3-en-
1-aminium chloride 478b (0.892 g, 4.11 mmol) in DMF (21 mL) at 0 °C was
added NaH (0.822 g, 20.55 mmol) and allyl bromide (0.53 mL, 6.16 mmol).
After which NEt3 (0.86 mL, 6.16 mmol) and phenyl isocyanate (0.45 mL, 4.11
mmol) were added in CH2Cl2 (21 mL). After which NaH (0.329 g, 8.22 mmol)
and MeI (0.51 mL, 8.22 mmol) were added in DMF (21 mL). Purification by flash column
chromatography yielded the title compound 476c as an off-white solid (1.065 g, 3.00 mmol,
73%). MP 62-64 °C; RF 0.60 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.40-7.07 (9H, m, ArH),
5.71 (1H, ddt, J = 17.0, 10.0, 6.8, CHArCH2CH=CH2), 5.36-5.15 (2H, m, CHArCH2CH=CH2 + X of
ABXYY’, NCH2CHX=CH2), 5.14-5.02 (2H, m, CHArCH2CH=CH2), 4.95-4.80 (2H, m, Y + Y’ o ABXYY’,
NCH2CH=CHYHY’), 3.44 (1H, ddt, J = 16.2, 5.5, 1.7, A of ABXYY’, NCHAHCH=CH2), 3.32 (1H, ddt, J =
16.4, 6.4, 1.3, B of ABXYY’, NCHHBCH=CH2), 3.21 (3H, s, NMe), 2.81-2.58 (2H, m,
CHArCH2CH=CH2); 13C NMR (75 MHz, CDCl3) 162.0 (C=O), 146.5 (4° ArC), 138.2 (4° ArC), 134.9
(CH2CH=CH2), 134.9 (CH2CH=CH2), 133.1 (4° ArC), 129.7 (ArC), 129.3 (ArC), 128.2 (ArC), 125.0
(ArC), 124.7 (ArC), 117.4 (CHArCH2CH=CH2), 116.2 (NCH2CH=CH2), 59.0 (CHAr), 47.4
NCH2CH=CH2), 40.0 (NMe), 35.6 (CHArCH2CH=CH2); IR max (film)/cm-1 3075 (C=CH2), 1641 (C=O),
1595 (C=C); MS (ES) m/z 377 (M + Na+); HRMS m/z calcd 355.1577 for C21H24N2O35Cl (M + H+)
found 355.1572.

241
(S)-1-Allyl-1-(1-(2-chlorophenyl)but-3-en-1-yl)-3-methyl-3-phenylurea 476d
Following general procedures 22 and 8. To (S)-N-allyl-1-(2-chlorophenyl)but-
3-en-1-amine 475c (0.200 g, 0.90 mmol) in CH2Cl2 (5 mL) was added NEt3
(0.19 mL, 1.35 mmol) and phenyl isocyanate (0.10 mL, 0.90 mmol). After
which NaH (0.072 g, 1.80 mmol) and MeI (0.11 mL, 1.80 mmol) were added
in DMF (5 mL). Purification by flash column chromatography yielded the title
compound 476d as a colourless oil (0.293 g, 0.83 mmol, 92%). RF 0.59 (2:1 PE:EtOAc); [D25
+86.8 (c 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) 7.48 (1H, dd, J = 7.6, 2.2, ArH), 7.38 (1H, dd, J =
7.6, 1.9, ArH), 7.30-7.17 (4H, m, ArH), 7.14-7.07 (3H, m, ArH), 5.59 (1H, ddt, J = 17.0, 10.4, 6.6,
CHArCH2CH=CH2), 5.44 (1H, dddd, J = 17.0, 10.1, 6.6, 5.4, X of ABXYY’, NCH2CHX=CH2), 5.36 (1H,
dd, J = 9.5, 6.3, CHAr), 5.04-4.86 (4H, m, Y and Y’ of ABXYY’, NCH2CH=CHYHY’), 3.47 (1H, ddt, J =
16.4, 5.4, 1.6, A of ABXYY’, NCHAHCH=CH2), 3.31 (1H, ddt, J = 16.4, 6.6, 1.3, B of ABXYY’,
NCHHBCH=CH2), 3.20 (3H, s, NMe), 2.86-2.78 (1H, m, CHArCHH), 2.65-2.57 (1H, m, CHArCHH);
13C NMR (125 MHz, CDCl3) 161.6 (C=O), 146.3 (4° ArC), 136.7 (4° ArC), 135.2 (4° ArC), 135.0
(NCH2CH=CH2), 134.8 (CHArCH2CH=CH2), 129.9 (ArC), 129.8 (ArC), 129.3 (ArC), 128.6 (ArC),
126.3 (ArC), 124.8 (ArC), 124.7 (ArC), 117.4 (CH=CH2), 116.2 (CH=CH2), 57.8 (CHAr), 48.4 (NCH2),
40.0 (NMe), 35.4 (CHArCH2); IR max (film)/cm-1 1646 (C=O), 1595 (C=C); MS (ES) m/z 355 (M +
H+); HRMS m/z calcd 377.1392 for C21H23N2O35ClNa (M + Na+) found 377.1380.

242
(S)-1-Allyl-1-(1-(2-chlorophenyl)but-3-en-1-yl)-3-(2-fluorophenyl)-3-methylurea 476e
Following general procedures 22 and 8. To (S)-N-allyl-1-(2-chlorophenyl)but-
3-en-1-amine 475c (0.217 g, 0.98 mmol) in CH2Cl2 (5 mL) was added NEt3
(0.20 mL, 1.47 mmol) and 2-fluorophenyl isocyanate (0.11 mL, 0.98 mmol).
After which NaH (0.078 g, 1.96 mmol) and MeI (0.12 mL, 1.96 mmol) were
added in DMF (5 mL). Purification by flash column chromatography yielded
the title compound 476e as a pale yellow oil (0.159 g, 0.43 mmol, 44%). RF 0.54 (2:1 PE:EtOAc);
[D25 +74.0 (c 0.2, CHCl3);
1H NMR (500 MHz, CDCl3) 7.51 (1H, dd, J = 7.6, 1.9, ArH), 7.33 (1H,
dd, J = 7.9, 1.9, ArH), 7.23-7.10 (4H, m, ArH), 7.08-7.02 (2H, m, ArH), 5.59 (1H, ddt, J = 17.0,
10.4, 6.6, CHArCH2CH=CH2), 5.31 (1H, ddt, J = 16.4, 10.7, 6.3, X of ABXYY’, NCH2CHX=CH2), 5.18
(1H, dd, J = 9.1, 6.6, CHAr), 5.03-4.87 (4H, m, Y and Y’ of ABXYY’, NCH2CH=CHYCHY’ +
CHArCH2CH=CH2), 3.57 (1H, ddt, J = 16.1, 5.7, 1.6, A of ABXYY’, NCHAHCH=CH2), 3.44 (1H, ddt, J =
16.1, 6.6, 1.6, B of ABXYY’, NCHHBCH=CH2), 3.14 (3H, s, NMe), 2.86-2.79 (1H, m,
CHArCHHCH=CH2), 2.57-2.50 (1H, m, CHArCHHCH=CH); 13C NMR (125 MHz, CDCl3) 161.8
(C=O), 157.3 (d, JF = 248.0, ArCF), 136.9 (4° ArC), 134.8 (CHArCH2CH=CH2), 134.5 (NCH2CH=CH2),
133.8 (d, JF = 11.8, 4° ArC), 129.8 (4° ArC), 129.7 (ArC), 128.6 (ArC), 128.4 (d, JF = 1.8, ArC), 127.5
(d, JF = 8.2, ArC), 126.4 (ArC), 124.8 (d, JF = 3.6, ArC), 117.3 (CH=CH2), 116.9 (ArC), 116.7 (ArC),
116.5 (CH=CH2), 58.2 (CHAr), 49.0 (NCH2), 38.9 (d, JF = 2.7, NMe), 35.4 (CHArCH2); IR max
(film)/cm-1 2924 (C=CH2), 1717 (C=C), 1654 (C=O); MS (ES) m/z 373 (M + H+); HRMS m/z calcd
373.1478 for C21H23N2OF35Cl (M + H+) found 373.1471.

243
(S)-1-Allyl-3-(4-chlorophenyl)-3-methyl-1-(1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)urea 476f
Following general procedures 22 and 8. To (S)-N-allyl-1-(3-
(trifluoromethyl)phenyl)but-3-en-1-amine 475d (0.200 g, 0.78 mmol) in
CH2Cl2 (4 mL) was added NEt3 (0.16 mL, 1.18 mmol) and 4-chlorophenyl
isocyanate (0.120 g, 0.78 mmol). After which NaH (0.063 g, 1.57 mmol)
and MeI (0.10 mL, 1.57 mmol) were added in DMF (4 mL). Purification by
flash column chromatography yielded the title compound 476f as a colourless oil (0.217 g, 0.51
mmol, 66%). RF 0.46 (2:1 PE:EtOAc); [D25 −38.0 (c 1.0, CHCl3);
1H NMR (500 MHz, CDCl3) 7.55-
7.49 (3H, m, ArH), 7.46-7.41 (1H, m, ArH), 7.29-7.25 (2H, m, ArH), 7.03-6.99 (2H, m, ArH), 5.74
(1H, ddt, J = 17.3, 10.7, 6.6, CHArCH2CH=CH2), 5.32-5.23 (2H, m, CHAr + NCH2CH=CH2), 5.15-5.08
(2H, m, CHArCH2CH=CH2), 4.94-4.80 (2H, m, NCH2CH=CH2), 3.40-3.31 (2H, m, NCH2), 3.15 (3H, s,
NMe), 2.82-2.68 (2H, m, CHArCH2); 13C NMR (125 MHz, CDCl3) 161.8 (C=O), 144.9 (4° ArC),
140.8 (4° ArC), 134.7 (CHArCH2CH=CH2), 134.5 (NCH2CH=CH2), 131.7 (4° ArC), 130.6 (q, JF = 31.8,
ArCCF3), 130.5 (ArC), 129.5 (ArC), 128.7 (ArC), 125.8 (ArC), 125.1 (q, JF = 3.6, ArC), 124.3 (q, JF =
3.6, ArC), 124.1 (q, JF = 272.5, ArCCF3), 117.9 (CHArCH2CH=CH2), 116.6 (NCH2CH=CH2), 59.3
(CHAr), 47.7 (NCH2), 40.0 (NMe), 35.7 (CHArCH2); IR max (film)/cm-1 1648 (C=O); MS (ES) m/z
423 (M + H+); HRMS m/z calcd 423.1451 for C22H23N2OF335Cl (M + H+) found 423.1458.

244
(S)-1-Allyl-3-(2-fluorophenyl)-3-methyl-1-(1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)urea
476g
Following general procedures 22 and 8. To (S)-N-allyl-1-(3-
(trifluoromethyl)phenyl)but-3-en-1-amine 475d (0.200 g, 0.78 mmol) in
CH2Cl2 (4 mL) was added NEt3 (0.16 mL, 1.18 mmol) and 2-fluorophenyl
isocyanate (0.09 mL, 0.78 mmol). After which NaH (0.063 g, 1.57 mmol)
and MeI (0.10 mL, 1.57 mmol) were added in DMF (4 mL). Purification by flash column
chromatography yielded the title compound 476g as a yellow oil (0.238 g, 0.59 mmol, 75%). RF
0.52 (2:1 PE:EtOAc); [D25 −41.6 (c 1.0, CHCl3);
1H NMR (500 MHz, CDCl3) 7.53-7.46 (2H, m,
ArH), 7.43-7.36 (2H, m, ArH), 7.28-7.06 (4H, m, ArH), 5.69 (1H, ddt, J = 17.0, 10.4, 6.6,
CHArCH2CH=CH2), 5.21 (1H, ddt, J = 17.0, 10.7, 6.0, NCH2CH=CH2), 5.12-4.99 (3H, m,
CHArCH2CH=CH2), 4.94-4.83 (2H, m, NCH2CH=CH2), 3.49-3.39 (2H, m, NCH2), 3.13 (3H, s, NMe),
2.83-2.76 (1H, m, CHArCHH), 2.72-2.64 (1H, m, CHArCHH); 13C NMR (125 MHz, CDCl3) 162.2
(C=O), 157.4 (d, JF = 249.8, ArCF), 140.7, (4° ArC), 134.7 (CHArCH2CH=CH2), 134.4 (NCH2CH=CH2),
133.9 (d, JF = 10.9, 4° ArC), 131.6 (ArC), 130.4 (q, JF = 32.7, ArCCF3), 128.6 (ArC), 128.3 (ArC),
127.9 (d, JF = 8.2, ArC), 125.2 (q, JF = 3.6, ArC), 124.9 (d, JF = 3.6, ArC), 124.1 (q, JF = 3.6, ArC),
124.1 (q, JF = 272.5, ARCCF3), 117.7 (CHArCH2CH=CH2), 117.0 (d, JF = 20.0, ArC), 116.8
(NCH2CH=CH2), 59.8 (CHAr), 48.2 (NCH2), 39.0 (d, JF = 1.8, NMe), 35.8 (CHArCH2); IR max
(film)/cm-1 2964 (C=CH2), 1651 (C=O); MS (ES) m/z 407 (M + H+); HRMS m/z calcd 429.1561 for
C22H22N2OF4Na (M + Na+) found 429.1562.

245
(S)-1-Allyl-3-(3-methoxyphenyl)-3-methyl-1-(1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)urea
476h
Following general procedures 22 and 8. To (S)-N-allyl-1-(3-
(trifluoromethyl)phenyl)but-3-en-1-amine 475d (0.200 g, 0.78 mmol) in
CH2Cl2 (4 mL) was added NEt3 (0.16 mL, 1.18 mmol) and 3-
methoxyphenyl isocyanate (0.10 mL, 0.78 mmol). After which NaH
(0.063 g, 1.57 mmol) and MeI (0.10 mL, 1.57 mmol) were added in DMF (4 mL). Purification by
flash column chromatography yielded the title compound 476h as a colourless oil (0.221 g, 0.53
mmol, 68%). RF 0.41 (2:1 PE:EtOAc); [D25 −8.0 (c 0.2, CHCl3);
1H NMR (500 MHz, CDCl3) 7.66-
7.39 (4H, m, ArH), 7.21 (1H, t, J = 8.2, ArH), 6.86-6.58 (3H, m, ArH), 5.71 (1H, ddt, J = 17.0, 10.4,
6.6, CHArCH2CH=CH2), 5.34 (1H, ddt, J = 17.3, 10.4, 6.0, NCH2CH=CH2), 5.23 (1H, t, J = 7.9, CHAr),
5.14-5.04 (2H, m, CHArCH2CH=CH2), 4.94-4.82 (2H, m, NCH2CH=CH2), 3.76 (3H, s, OMe), 3.45-
3.40 (2H, m, NCH2), 3.17 (3H, s, NMe), 2.80-2.66 (2H, m, CHArCH2); 13C NMR (125 MHz, CDCl3)
162.0 (C=O), 160.5 (4° ArCOMe), 147.7 (4° ArC), 140.8 (4° ArC), 134.8 (NCH2CH=CH2), 134.7
(CHArCH2CH=CH2), 131.7 (ArC), 130.5 (q, JF = 32.7, ArCCF3), 130.1 (ArC), 128.6 (ArC), 125.2 (q, JF
= 3.6, ArC), 124.2 (q, JF = 3.6, ArC), 124.1 (q, JF = 272.5, ArCCF3), 117.8 (CHArCH2CH=CH2), 117.1
(ArC), 116.5 (NCH2CH=CH2), 110.8 (ArC), 110.6 (ArC), 59.4 (4° C), 55.3 (OMe), 47.6 (NCH2), 40.1
(NMe), 35.8 (CHArCH2); IR max (film)/cm-1 1720 (C=C), 1649 (C=O); MS (ES) m/z 419 (M + H+);
HRMS m/z calcd 441.1761 for C23H25N2O2F3Na (M + Na+) found 441.1764.

246
(S)-1-Allyl-3-methyl-3-phenyl-1-(1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)urea 476i
Following general procedures 22 and 8. To (S)-N-allyl-1-(3-
(trifluoromethyl)phenyl)but-3-en-1-amine 475d (0.200 g, 0.78 mmol) in
CH2Cl2 (4 mL) was added NEt3 (0.16 mL, 1.18 mmol) and phenyl isocyanate
(0.09 mL, 0.78 mmol). After which NaH (0.063 g, 1.57 mmol) and MeI (0.10
mL, 1.57 mmol) were added in DMF (4 mL). Purification by flash column
chromatography yielded the title compound 476i as a colourless oil (0.301 g, 0.78 mmol, 99%).
RF 0.59 (2:1 PE:EtOAc); [D25 −56.8 (c 0.5, CHCl3);
1H NMR (500 MHz, CDCl3) 7.54-7.38 (4H, m,
ArH), 7.34-7.28 (2H, m, ArH), 7.18-7.12 (1H, m, ArH), 7.12-7.06 (2H, m, ArH), 5.72 (1H, ddt, J =
17.3, 10.4, 6.6, CHArCH2CH=CH2), 5.30-5.19 (2H, m, CHAr + NCH2CH=CH2), 5.13-5.05 (2H, m,
CHArCH2CH=CH2), 4.92-4.79 (2H, m, NCH2CH=CH2), 3.42-3.32 (2H, m, NCH2CH=CH2), 3.18 (3H, s,
NMe), 2.82-2.66 (2H, m, CHArCH2); 13C NMR (125 MHz, CDCl3) 162.1 (C=O), 146.4 (4° ArC),
140.8 (4° ArC), 134.7 (CH=CH2), 134.7 (CH=CH2), 131.6 (ArC), 130.4 (q, JF = 31.8, ArCCF3), 129.4
(ArC), 128.6 (ArC), 125.3 (ArC), 125.1 (q, JF = 3.6, ArC), 124.9 (ArC), 124.1 (q, JF = 3.6, ArC), 124.0
(q, JF = 272.5, ArCCF3), 117.7 (CHArCH2CH=CH2), 116.4 (NCH2CH=CH2), 59.2 (CHAr), 47.7 (NCH2),
40.1 (NMe), 35.6 (CHArCH2); IR max (film)/cm-1 3071 (C=CH2), 1646 (C=O), 1596 (C=C); MS (ES)
m/z 389 (M + H+); HRMS m/z calcd 389.1836 for C22H24N2OF3 (M + H+) found 389.1822.
(S)-N-(4-Methoxyphenyl)-N-methyl-6-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 468a
Following general procedure 16. To (S)-1-allyl-3-(4-methoxyphenyl)-3-methyl-1-
(1-phenylbut-3-en-1-yl)urea 476a (0.515 g, 1.47 mmol) in CH2Cl2 (15 mL) was
added Grubbs 1st generation catalyst (0.060 g, 0.07 mmol). Purification by flash
column chromatography yielded the title compound 468a as a brown oil (0.423
g, 1.31 mmol, 89%). RF 0.29 (2:1 PE:EtOAc); [D25 +70.4 (c 1.0, CHCl3);
1H NMR
(300 MHz, CDCl3) 7.32-7.18 (5H, m, ArH), 7.09-7.01 (2H, m, ArH), 6.86-6.78 (2H, m, ArH), 5.86-
5.77 (1H, m, CHPhCH2CH), 5.50-5.37 (2H, m, NCH2CH), 3.86-3.72 (1H, m, NCHH), 3.79 (3H, s,
OMe), 3.23 (3H, s, NMe), 2.97-2.83 (1H, m, NCHH), 2.49-2.35 (2H, m, CHPhCH2); 13C NMR (75
MHz, CDCl3) 161.7 (C=O), 156.8 (4° ArCOMe), 140.4 (4° ArC), 139.8 (4° ArC), 128.2 (ArC), 127.1
(ArC), 127.0 (ArC), 125.9 (ArC), 124.7 (NCH2CH), 123.6 (CHPhCH2CH), 114.6 (ArC), 55.4 (OMe),
52.3 (CHPh), 42.4 (NCH2), 40.4 (NMe), 26.6 (CHPhCH2); IR max (film)/cm-1 2835 (CH), 1639
(C=O); MS (ES) m/z 345 (M + Na+); HRMS m/z calcd 323.1755 for C20H23N2O2 (M + H+) found
323.1756.

247
(S)-N-Methyl-N,6-diphenyl-5,6-dihydropyridine-1(2H)-carboxamide 468b
Following general procedure 16. To (S)-1-allyl-3-methyl-3-phenyl-1-(1-
phenylbut-3-en-1-yl)urea 476b (0.308 g, 0.96 mmol) in CH2Cl2 (20 mL) was
added Grubbs 1st generation catalyst (0.040 g, 0.05 mmol). Purification by flash
column chromatography yielded the title compound 468b as a pale yellow oil
(0.275 g, 0.94 mmol, 98%). RF 0.50 (2:1 PE:EtOAc); [D25 +56.8 (c 1.0, CHCl3);
1H NMR (500 MHz,
CDCl3) 7.22-7.10 (7H, m, ArH), 7.04-6.96 (3H, m, ArH), 5.72 (1H, ddd, J = 10.1, 5.4, 2.5,
NCH2CH=CH), 5.40-5.30 (2H, m, CHPhCH2CH=CH), 3.73-3.65 (1H, m, NCHH), 3.18 (3H, s, NMe),
2.87-2.79 (1H, m, NCHH), 2.41-2.23 (2H, m, CHPhCH2); 13C NMR (125 MHz, CDCl3) 161.2 (C=O),
146.5 (4° ArC), 140.0 (4° ArC), 129.3 (ArC), 128.1 (ArC), 126.9 (ArC), 124.5 (NCH2CH=CH), 124.4
(ArC), 124.4 (ArC), 123.9 (ArC), 123.5 (NCH2CH=CH), 52.2 (CHPh), 42.2 (NCH2), 39.7 (NMe), 26.4
(CHPhCH2); IR max (film)/cm-1 3032 (C=CH), 1640 (C=O), 1595 (C=N); MS (ES) m/z 293 (M + H+);
HRMS m/z calcd 293.1649 for C19H21N2O (M + H+) found 293.1643.
6-(4-Chlorophenyl)-N-methyl-N-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 468c
Following general procedure 16. To 1-allyl-1-(1-(4-chlorophenyl)but-3-en-1-
yl)-3-methyl-3-phenylurea 476c (0.745 g, 2.10 mmol) in CH2Cl2 (21 mL) was
added Grubbs 1st generation catalyst (0.086 g, 0.10 mmol). Purification by
flash column chromatography yielded the title compound 468c as a brown
oil (0.441 g, 1.35 mmol, 64%). RF 0.40 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.26-6.98 (9H,
m, ArH) 5.79-5.68 (1H, m, NCH2CH=CH), 5.43-5.27 (2H, m, CHAr + NCH2CH=CH), 3.76-3.63 (1H,
m, NCHHCH=CH), 3.20 (3H, s, NMe), 2.86-2.75 (1H, m, NCHHCH=CH), 2.48-2.35 (1H, m,
CHArCHHCH), 2.31-2.18 (1H, m, CHArCHHCH); 13C NMR (75 MHz, CDCl3) 161.3 (C=O), 146.6 (4°
ArC), 138.8 (4° ArC), 132.8 (4° ArC), 129.5 (ArC), 128.6 (ArC), 128.4 (ArC), 124.8 (ArC), 124.7
(NCH2CH=CH), 124.1 (ArC), 123.4 (NCH2CH=CH), 51.7 (CHAr), 42.4 (NCH2CH=CH), 39.9 (NMe),
26.6 (CHArCH2CH); IR max (film)/cm-1 1638 (C=O), 1594 (C=C); MS (ES) m/z 349 (M + Na+); HRMS
m/z calcd 327.1259 for C19H20N2O35Cl (M + H+) found 327.1248.

248
(S)-6-(2-Chlorophenyl)-N-methyl-N-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 468d
Following general procedure 16. To (S)-1-allyl-1-(1-(2-chlorophenyl)but-3-en-1-
yl)-3-methyl-3-phenylurea 476d (0.269 g, 0.76 mmol) in CH2Cl2 (15 mL) was
added Grubbs 1st generation catalyst (0.031 g, 0.04 mmol). Purification by flash
column chromatography yielded the title compound 468d as a colourless oil
(0.237 g, 0.73 mmol, 95%). RF 0.41 (2:1 PE:EtOAc); [D25 +67.2 (c 2.0, CHCl3);
1H NMR (500 MHz,
CDCl3) 7.42-7.35 (2H, m, ArH), 7.26-7.11 (6H, m, ArH), 7.06-7.02 (1H, m, ArH), 5.92-5.86 (1H,
m, NCH2CH=CH), 5.82-5.78 (1H, m, CHAr), 5.61-5.55 (1H, m, NCH2CH=CH), 3.79-3.71 (1H, m,
NCHH), 3.28 (3H, s, NMe), 2.95-2.88 (1H, m, NCHH), 2.71-2.63 (1H, m, CHArCHH), 2.33-2.26 (1H,
m, CHArCHH); 13C NMR (125 MHz, CDCl3) 160.5 (C=O), 146.4 (4° ArC), 138.1 (4° ArC), 134.0 (4°
ArC), 129.8 (ArC), 129.3 (ArC), 128.5 (ArC), 128.2 (ArC), 126.3 (ArC), 124.9 (NCH2CH=CH), 124.3
(ArC), 124.3 (NCH2CH=CH), 124.1 (ArC), 49.5 (CHAr), 43.2 (NCH2), 39.6 (NMe), 27.4 (CHArCH2);
IR max (film)/cm-1 2897 (C=CH), 1640 (C=O), 1594 (C=C); MS (ES) m/z 327 (M + H+); HRMS m/z
calcd 327.1259 for C19H20N2O35Cl (M + H+) found 327.1268.
(S)-6-(2-Chlorophenyl)-N-(2-fluorophenyl)-N-methyl-5,6-dihydropyridine-1(2H)-carboxamide
468e
Following general procedure 16. To (S)-1-allyl-1-(1-(2-chlorophenyl)but-3-en-1-
yl)-3-(2-fluorophenyl)-3-methylurea 476e (0.159 g, 0.43 mmol) in CH2Cl2 (9 mL)
was added Grubbs 1st generation catalyst (0.018 g, 0.02 mmol). Purification by
flash column chromatography yielded the title compound 468e as a yellow oil
(0.112 g, 0.32 mmol, 76%). RF 0.44 (2:1 PE:EtOAc); [D25 +26.8 (c 1.0, CHCl3);
1H NMR (400 MHz,
CDCl3) 7.37-7.30 (2H, m, ArH), 7.23-7.01 (5H, m, ArH), 6.98-6.91 (1H, m, ArH), 5.90-5.82 (1H,
m, NCH2CH=CH), 5.71 (1H, brd, J = 6.1, CHAr), 5.63-5.55 (1H, m, NCH2CH=CH), 3.81-3.72 (1H, m,
NCHH), 3.16 (3H, s, NMe), 2.98-2.89 (1H, m, NCHH), 2.64-2.52 (1H, m, CHArCHH), 2.34-2.24 (1H,
m, CHArCHH); 13C NMR (100 MHz, CDCl3) 160.8 (C=O), 156.7 (d, JF = 247.3, ArCF), 138.1 (4°
ArC), 133.7 (4° ArC), 133.6 (d, JF = 11.1, 4° ArC), 129.7 (ArC), 128.4 (ArC), 128.1 (ArC), 127.7
(ArC), 127.1 (d, JF = 7.4, ArC), 126.2 (ArC), 124.7 (NCH2CH=CH), 124.6 (d, JF = 3.7, ArC), 124.0
(NCH2CH=CH), 116.5 (d, JF = 19.4, ArC), 49.6 (CHAr), 42.9 (NCH2), 38.5 (d, JF = 2.8, NMe), 27.4
(CHArCH2); IR max (film)/cm-1 1648 (C=O), 1607 (C=C), 1500 (C=C); MS (ES) m/z 345 (M + H+);
HRMS m/z calcd 367.0984 for C19H18N2OF35ClNa (M + Na+) found 367.0987.

249
(S)-N-(2-Fluorophenyl)-N-methyl-6-(3-(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-
carboxamide 468f
Following general procedure 16. To (S)-1-allyl-3-(2-fluorophenyl)-3-methyl-
1-(1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)urea 476g (0.238 g, 0.59
mmol) in CH2Cl2 (12 mL) was added Grubbs 1st generation catalyst (0.024 g,
0.03 mmol). Purification by flash column chromatography yielded the title
compound 468f as a pale yellow oil (0.167 g, 0.44 mmol, 75%). RF 0.45 (2:1 PE:EtOAc); [D25
+53.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) 7.51-7.34 (4H, m, ArH), 7.29-7.04 (4H, m, ArH),
5.87-5.80 (1H, m, NCH2CH=CH), 5.49 (1H, d, J = 5.8, CHAr), 5.46-5.38 (1H, m, NCH2CH=CH), 3.80-
3.69 (1H, m, NCHH), 3.19 (3H, s, NMe), 2.78-2.68 (1H, m, NCHH), 2.64-2.55 (1H, m, CHArCHH),
2.50-2.41 (1H, m, CHArCHH); 13C NMR (100 MHz, CDCl3) 161.4 (C=O), 157.0 (d, JF = 248.3,
ArCF), 141.1 (4° ArC), 133.6 (d, JF = 11.1, 4° ArC), 130.4 (q, JF = 32.5, ArCCF3), 130.3 (ArC), 128.6
(ArC), 127.7 (ArC), 127.7 (d, JF = 7.4, ArC), 124.9 (d, JF = 3.7, ArC), 124.6 (NCH2CH=CH), 124.1 (q,
JF = 3.7, ArC), 123.7 (q, JF = 3.7, ArC), 123.3 (NCH2CH=CH), 121.4 (q, JF = 276.9, ArCCF3), 116.9 (d,
JF = 19.4, ArC), 51.7 (CHAr), 42.5 (NCH2), 38.8 (d, JF = 1.8, NMe), 26.3 (CHArCH2); IR max
(film)/cm-1 1648 (C=O), 1500 (C=C); MS (ES) m/z 379 (M + Na+); HRMS m/z calcd 401.1248 for
C20H18N2OF4Na (M + Na+) found 401.1245.

250
(S)-N-(4-Chlorophenyl)-N-methyl-6-(3-(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-
carboxamide 468g
Following general procedure 16. To (S)-1-allyl-3-(4-chlorophenyl)-3-
methyl-1-(1-(3-(trifluoromethyl)phenyl)but-3-en-1-yl)urea 476f (0.217 g,
0.51 mmol) in CH2Cl2 (10 mL) was added Grubbs 1st generation catalyst
(0.021 g, 0.03 mmol). Purification by flash column chromatography yielded
the title compound 468g as a brown oil (0.188 g, 0.48 mmol, 93%). RF 0.40
(2:1 PE:EtOAc); [D25 +49.6 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) 7.53-7.37 (4H, m, ArH),
7.26-7.21 (2H, m, ArH), 7.07-7.00 (2H, m, ArH), 5.90-5.82 (1H, m, NCH2CH=CH), 5.54 (1H, d, J =
6.1, CHAr), 5.49-5.42 (1H, m, NCH2CH=CH), 3.79-3.68 (1H, m, NCHH), 3.24 (3H, s, NMe), 2.86-
2.76 (1H, m, NCHH), 2.70-2.58 (1H, m, CHArCHH), 2.51-2.40 (1H, m, CHArCHH); 13C NMR (100
MHz, CDCl3) 160.9 (C=O), 144.8 (4° ArC), 141.0 (4° ArC), 130.6 (q, JF = 33.2, ArCCF3), 130.3 (4°
ArC), 130.1 (ArC), 129.5 (ArC), 128.7 (ArC), 125.2 (ArC), 124.5 (NCH2CH=CH), 124.0 (d, JF = 3.7,
ArC), 124.0 (q, JF = 274.1, ArCCF3), 123.9 (d, JF = 3.7, ArC), 123.4 (NCH2CH=CH), 51.6 (CHAr), 42.8
(NCH2), 39.8 (NMe), 26.4 (CHArCH2); IR max (film)/cm-1 2931 (C=CH), 1644 (C=O), 1592 (C=C);
MS (ES) m/z 417 (M + Na+); HRMS m/z calcd 417.0952 for C20H18N2OF335ClNa (M + Na+) found
417.0952.
(S)-N-Methyl-N-phenyl-6-(3-(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide
468h
Following general procedure 16. To (S)-1-allyl-3-methyl-3-phenyl-1-(1-(3-
(trifluoromethyl)phenyl)but-3-en-1-yl)urea 476i (0.268 g, 0.69 mmol) in
CH2Cl2 (14 mL) was added Grubbs 1st generation catalyst (0.028 g, 0.03
mmol). Purification by flash column chromatography yielded the title
compound 468h as a colourless oil (0.189 g, 0.52 mmol, 76%). RF 0.48 (2:1 PE:EtOAc); [D25
+41.6 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) 7.52-7.36 (4H, m, ArH), 7.31-7.25 (2H, m, ArH),
7.14-7.07 (3H, m, Ar), 5.86-5.79 (1H, m, NCH2CH=CH), 5.54 (1H, dd, J = 6.6, 0.6, CHAr), 5.46-5.39
(1H, m, NCH2CH=CH), 3.82-3.73 (1H, m, NCHH), 3.27 (3H, s, NMe), 2.82-2.74 (1H, m, NCHH),
2.65-2.56 (1H, m, CHArCHH), 2.46-2.38 (1H, m, CHArCHH); 13C NMR (125 MHz, CDCl3) 161.2
(C=O), 146.4 (4° ArC), 141.3 (4° ArC), 130.5 (q, JF = 31.8, ArCCF3), 130.3 (ArC), 129.5 (ArC), 128.6
(ArC), 124.9 (ArC), 124.7 (NCH2CH=CH), 124.3 (ArC), 124.1 (q, JF = 270.7, ArCCF3), 124.0 (q, JF =
3.6, ArC), 123.8 (q, JF = 4.0, ArC), 123.2 (NCH2CH=CH), 51.6 (CHAr), 42.7 (NCH2), 40.0 (NMe),
26.4 (CHArCH2); IR max (film)/cm-1 1642 (C=O); MS (ES) m/z 361 (M + H+); HRMS m/z calcd
361.1523 for C20H20N2OF3 (M + H+) found 361.1522.

251
(S)-2-(2-Chlorophenyl)-N-methyl-N-phenyl-3,4-dihydropyridine-1(2H)-carboxamide 470a
Following general procedure 17. To (S)-6-(2-chlorophenyl)-N-methyl-N-phenyl-
5,6-dihydropyridine-1(2H)-carboxamide 468d (0.139 g, 0.43 mmol) in THF (9
mL) was added carbonylchlorohydridotris(triphenylphosphine)ruthenium(II)
(0.041 g, 0.04 mmol) and the mixture heated to reflux overnight. Purification by
flash column chromatography yielded the title compound 470a as a colourless oil (0.091 g, 0.28
mmol, 65%). RF 0.62 (2:1 PE:EtOAc); [D25 −24.0 (c 0.25, CHCl3);
1H NMR (300 MHz, CDCl3)
7.23-6.96 (9H, m, ArH), 6.67-6.61 (1H, m, NCH=CH), 5.46-5.41 (1H, m, CHAr), 4.70-4.61 (1H, m,
NCH=CH), 3.11 (3H, s, NMe), 2.04-1.92 (1H, m, CHArCHH), 1.82-1.67 (2H, m, CHArCHHCHH),
1.57-1.41 (1H, m, CHArCH2CHH); 13C NMR (75 MHz, CDCl3) 158.9 (C=O), 146.0 (4° ArC), 138.8
(4° ArC), 131.1 (4° ArC), 129.7 (ArC), 129.4 (ArC), 128.0 (ArC), 127.6 (ArC), 127.3 (NCH=CH),
126.4 (ArC), 125.2 (ArC), 124.6 (ArC), 105.0 (NCH=CH), 53.4 (CHAr), 40.2 (NMe), 24.8 (CHArCH2),
17.6 (CHArCH2CH2); IR max (film)/cm-1 2922 (C=CH), 1723 (CH), 1645 (C=O); MS (ES) m/z 327 (M
+ H+); HRMS m/z calcd 327.1259 for C19H20N2O35Cl (M + H+) found 327.1260; HPLC 91:9 er:
ChiralPak AD-H, 95:5 Hexane:IPA 1mL/min. tr 16.3 major, 11.2 minor.
(S)-2-(2-Chlorophenyl)-N-(2-fluorophenyl)-N-methyl-3,4-dihydropyridine-1(2H)-carboxamide
470b
Following general procedure 17. To (S)-6-(2-chlorophenyl)-N-(2-fluorophenyl)-
N-methyl-5,6-dihydropyridine-1(2H)-carboxamide 468e (0.060 g, 0.17 mmol) in
THF (2 mL) was added
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.017 g, 0.02
mmol) and the mixture heated to reflux overnight. Purification by flash column chromatography
yielded the title compound 470b as a pale brown oil (0.039 g, 0.11 mmol, 65%). RF 0.65 (2:1
PE:EtOAc); [D25 −40.0 (c 0.25, CHCl3);
1H NMR (400 MHz, CDCl3) 7.25-6.93 (8H, m, ArH), 6.72-
6.65 (1H, m, NCH=CH), 5.46-5.40 (1H, t, J = 4.3, CHAr), 4.82-4.74 (1H, m, NCH=CH), 3.15 (3H, s,
NMe), 2.13-2.05 (1H, m, CHArCHH), 1.89-1.76 (2H, m, CHArCHHCHH), 1.58-1.46 (1H, m,
CHArCH2CHH); 13C NMR (100 MHz, CDCl3) 158.9 (C=O), 156.9 (d, JF = 248.3, ArCF), 138.3 (4°
ArC), 133.2 (d, JF = 12.0, 4° ArC), 131.0 (4° ArC), 129.6 (ArC), 127.9 (d, JF = 0.9, ArC), 127.8 (ArC),
127.6 (ArC), 127.6 (NCH=CH), 127.1 (d, JF = 5.5, ArC), 126.2 (ArC), 124.8 (d, JF = 3.7, ArC), 116.7
(d, JF = 20.3, ArC), 105.6 (NCH=CH), 53.9 (CHAr), 39.4 (NMe), 24.9 (CHArCH2), 17.6
(CHArCH2CH2); IR max (film)/cm-1 2927 (C=CH), 1664 (C=O), 1501 (C=C); MS (ES) m/z 367 (M +
Na+); HRMS m/z calcd 345.1167 for C19H19N2OF35Cl (M + H+) found 345.1165; HPLC 90:10 er:
ChiralPak AD-H, 97:3 Hexane:IPA 1mL/min. tr 15.8 major, 10.8 minor.

252
(S)-N-Methyl-N-phenyl-2-(3-(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-carboxamide
470c
Following general procedure 17. To (S)-N-methyl-N-phenyl-6-(3-
(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide 468h
(0.099 g, 0.27 mmol) in THF (5 mL) was added
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.026 g, 0.03
mmol) and the mixture heated to reflux overnight. Purification by flash column chromatography
yielded the title compound 470c as a colourless oil (0.051 g, 0.14 mmol, 52%). RF 0.58 (2:1
PE:EtOAc); [D25 −32.0 (c 0.25, CHCl3);
1H NMR (300 MHz, CDCl3) 7.42-7.16 (6H, m, ArH), 7.07
- 6.95 (3H, m, ArH), 6.49 (1H, dt, J = 8.9, 1.1, NCH=CH), 5.26 (1H, t, J = 3.8, CHAr), 4.57 (1H, ddd,
J = 8.3, 5.5, 2.3, NCH=CH), 3.15 (3H, s, NMe), 1.93-1.84 (2H, m, CHArCH2), 1.80-1.68 (1H, m,
CHArCH2CHH), 1.55-1.41 (1H, m, CHArCH2CHH); 13C NMR (75 MHz, CDCl3) 158.6 (C=O), 146.2
(4° ArC), 142.6 (4° ArC), 130.5 (q, JF = 31.6, ArCCF3), 130.3 (ArC), 129.6 (ArC), 128.7 (ArC), 127.0
(NCH=CH), 125.3 (ArC), 124.5 (ArC), 124.2 (q, JF = 274.1, ArCCF3), 123.5 (q, JF = 3.8, ArC), 122.5
(q, JF = 3.8, ArC), 105.2 (NCH=CH), 55.0 (CHAr), 40.2 (NMe), 27.3 (CHArCH2), 17.5 (CHArCH2CH2);
IR max (film)/cm-1 2919 (C=CH), 1665 (C=O), 1644 (C=C); MS (ES) m/z 361 (M + H+); HRMS m/z
calcd 361.1520 for C20H20N2OF3 (M + H+) found 361.1523; HPLC 93:7 er: ChiralPak AD-H, 94:6
Hexane:IPA 1mL/min. tr 15.1 major, 10.4 minor.

253
(S)-N-(2-Fluorophenyl)-N-methyl-2-(3-(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-
carboxamide 470d
Following general procedure 17. To (S)-N-(2-fluorophenyl)-N-methyl-6-(3-
(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide 468f
(0.120 g, 0.32 mmol) in THF (3 mL) was added
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.030 g, 0.03
mmol) and the mixture heated to reflux overnight. Purification by flash column chromatography
yielded the title compound 470d as a pale brown oil (0.074 g, 0.20 mmol, 62%). RF 0.53 (2:1
PE:EtOAc); [D25 −60.8 (c 0.25, CHCl3);
1H NMR (400 MHz, CDCl3) 7.46-7.29 (4H, m, ArH), 7.14-
6.91 (4H, m, ArH), 6.56-6.48 (1H, m, NCH=CH), 5.29 (1H, t, J = 3.8, NCH=CH), 4.72-4.62 (1H, m,
NCH=CH), 3.19 (3H, s, NMe), 2.03-1.93 (2H, m, CHArCH2), 1.86-1.75 (1H, m, CHArCH2CHH), 1.58-
1.45 (1H, m, CHArCH2CHH); 13C NMR (100 MHz, CDCl3) 158.7 (C=O), 156.9 (d, JF = 249.2, ArCF),
142.2 (4° ArC), 133.5 (d, JF = 12.0, 4° ArC), 130.3 (q, JF = 31.4, ArCCF3), 129.1 (ArC), 128.5 (ArC),
127.8 (d, JF = 7.4, ArC), 127.7 (ArC), 126.4 (NCH=CH), 125.0 (d, JF = 3.7, ArC), 124.1 (q, JF = 271.3,
ArCCF3), 123.4 (q, JF = 3.7, ArC), 122.4 (q, JF = 3.7, ArC), 116.8 (d, JF = 19.4), 105.7 (NCH=CH), 55.3
(CHAr), 39.4 (NMe), 27.5 (CHArCH2), 17.5 (CHArCH2CH2); IR max (film)/cm-1 2932 (C=CH), 1669
(C=C), 1646 (C=O); MS (ES) m/z 379 (M + H+); HRMS m/z calcd 379.1429 for C20H19N2OF4 (M +
H+) found 379.1421; HPLC 86:14 er: ChiralPak AD-H, 97:3 Hexane:IPA 1mL/min. tr 14.8 major,
10.1 minor.

254
(S)-N-(4-Chlorophenyl)-N-methyl-2-(3-(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-
carboxamide 470e
Following general procedure 17. To (S)-N-(4-chlorophenyl)-N-methyl-6-(3-
(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide 468g
(0.140 g, 0.35 mmol) in THF (4 mL) was added
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.034 g, 0.04
mmol) and the mixture heated to reflux overnight. Purification by flash
column chromatography yielded the title compound 470e as a yellow oil (0.086 g, 0.22 mmol,
61%). RF 0.49 (2:1 PE:EtOAc); [D25 −80.0 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) 7.50-7.31
(4H, m, ArH), 7.28-7.21 (2H, m, ArH), 7.01-6.96 (2H, m, ArH), 6.56-6.52 (1H, m, NCH=CH), 5.32
(1H, t, J = 4.0, CHAr), 4.74-4.69 (1H, m, NCH=CH), 3.21 (3H, s, NMe), 2.05-1.91 (2H, m, CHArCH2),
1.91-1.80 (1H, m, CHArCH2CHH), 1.64-1.50 (1H, m, CHArCH2CHH); 13C NMR (100 MHz, CDCl3)
158.3 (C=O), 144.6 (4° ArC), 142.3 (4° ArC), 130.6 (4° ArC), 130.5 (q, JF = 33.2, ArCCF3), 129.7
(ArC), 129.2 (ArC), 128.7 (ArC), 126.7 (NCH=CH), 125.6 (ArC), 124.1 (q, JF = 271.3 ArCCF3), 123.6
(q, JF = 3.7, ArC), 122.4 (q, JF = 4.6, ArC), 105.9 (NCH=CH), 55.1 (CHAr), 40.1 (NMe), 27.3
(CHArCH2), 17.5 (CHArCH2CH2); IR max (film)/cm-1 2938 (C=CH), 1654 (C=O); MS (ES) m/z 417 (M
+ Na+); HRMS m/z calcd 395.1133 for C20H19N2OF335Cl (M + H+) found 395.1140; HPLC 82:18 er:
ChiralPak AD-H, 97:3 Hexane:IPA 1mL/min. tr 15.5 major, 12.8 minor.
2-(4-Chlorophenyl)-N-methyl-2-phenyl-3,4-dihydropyridine-1(2H)-carboxamide 471a
Following general procedure 11. To 2-(4-chlorophenyl)-N-methyl-N-phenyl-
3,4-dihydropyridine-1(2H)-carboxamide 470a (0.060 g, 0.18 mmol) in THF (1.8
mL) at −78 °C was added LDA (0.46 mmol, prepared by general procedure 10)
and DMPU (0.45 mL). Purification by flash column chromatography yielded
the title compound 471a as a yellow oil (0.042 g, 0.13 mmol, 70%). RF 0.18 (1:1 PE:EtOAc); 1H
NMR (300 MHz, CDCl3) 7.46-7.23 (10H, m, ArH + NCH=CHCH2), 4.84 (1H, dt, J = 8.5, 4.1,
NCH=CHCH2), 3.92 (1H, bq, J = 4.3, NH), 2.47-2.33 (2H, m, CAr2CH2CH2), 2.38 (3H, d, J = 4.7,
NHMe), 1.72-1.58 (2H, m, CAr2CH2CH2); 13C NMR (75 MHz, CDCl3) 156.3 (C=O), 141.9 (4° ArC),
140.2 (4° ArC), 133.5 (4° ArC), 129.1 (ArC), 128.9 (ArC), 128.8 (ArC), 127.9 (NCH=CHCH2), 127.4
(ArC), 127.3 (ArC), 103.4 (NCH=CHCH2), 66.8 (4° C), 40.5 (NMe), 27.3 (CAr2CH2CH2), 20.0
(CAr2CH2CH2); IR max (film)/cm-1 2923 (CH), 1713 (C=C), 1681 (C=O); MS (ES) m/z 349 (M + Na+);
HRMS m/z calcd 327.1259 for C19H20N2O35Cl (M + H+) found 327.1249.

255
N-Methyl-2-phenyl-2-(3-(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-carboxamide
471b
Following general procedure 11. To (S)-N-methyl-N-phenyl-6-(3-
(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide 470d
(0.051 g, 0.14 mmol) in THF (1.4 mL) at −78 °C was added LDA (0.35 mmol,
prepared by general procedure 10) and DMPU (0.35 mL). Purification by
flash column chromatography yielded the title compound 471b as a yellow oil (0.038 g, 0.11
mmol, 75%). RF 0.41 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.62-7.58 (2H, m, ArH), 7.55-
7.51 (2H, m, ArH), 7.45-7.29 (5H, m, ArH), 7.24 (1H, dt, J = 8.6, 1.8, NCH=CH), 4.85 (1H, dt, J =
8.6, 3.8, NCH=CH), 3.97 (1H, q, J = 4.5, NH), 2.49-2.42 (2H, m, CAr2CH2), 2.39 (3H, d, J = 4.5,
NMe), 1.79-1.57 (2H, m, CAr2CH2CH2); 13C NMR (100 MHz, CDCl3) 156.1 (C=O), 143.0 (4° ArC),
141.5 (4° ArC), 131.1 (ArC), 131.0 (q, JF = 32.3, ARCCF3), 129.2 (ArC), 129.0 (ArC), 128.0
(NCH=CH), 127.3 (ArC), 127.2 (ArC), 124.4 (q, JF = 3.7, ArC), 124.2 (q, JF = 3.7, ArC), 124.0 (q, JF =
272.3, ArCCF3), 103.6 (NCH=CH), 66.9 (4° C), 40.4 (CAr2CH2), 27.3 (NMe), 20.0 (CAr2CH2 CH2); IR
max (film)/cm-1 3340 (NH), 2930 (C=CH), 1716 (C=C), 1652 (C=O); MS (ES) m/z 361 (M + H+);
HRMS m/z calcd 361.1520 for C20H20N2OF3 (M + H+) found 361.1529.
2-(2-Chlorophenyl)-N-methyl-2-phenyl-3,4-dihydropyridine-1(2H)-carboxamide 471c
Following general procedure 11. To (S)-2-(2-chlorophenyl)-N-methyl-N-phenyl-
3,4-dihydropyridine-1(2H)-carboxamide 470b (0.052 g, 0.16 mmol) in THF (1.6
mL) at −78 °C was added LDA (0.40 mmol, prepared by general procedure 10)
and DMPU (0.40 mL). Purification by flash column chromatography yielded the
title compound 471c as a yellow oil (0.011 g, 0.03 mmol, 21%). RF 0.34 (2:1 PE:EtOAc); 1H NMR
(500 MHz, CDCl3) 7.56-6.99 (10H, m, ArH + NCH=CH), 4.84 (1H, dt, J = 8.6, 3.3, NCH=CH), 4.12-
4.02 (1H, m, NH), 2.43 (3H, d, J = 4.6, NMe), 2.41-2.29 (2H, m, CAr2CH2), 1.76-1.62 (2H, m,
CAr2CH2CH2); IR max (film)/cm-1 3295 (NH), 1648 (C=O); MS (ES) m/z 349 (M + Na+); HRMS m/z
calcd 327.1264 for C19H20N2O35Cl (M + H+) found 327.1269. Broad signals in 13C NMR prevented
full analysis.

256
2-(4-Chlorophenyl)-N-methyl-2-(3-(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-
carboxamide 471d
Following general procedure 11. To (S)-N-(4-chlorophenyl)-N-methyl-2-(3-
(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-carboxamide 470f
(0.086 g, 0.22 mmol) in THF (2.2 mL) at −78 °C was added LDA (0.54 mmol,
prepared by general procedure 10) and DMPU (0.55 mL). Purification by
flash column chromatography yielded the title compound 471d as a red-orange oil (0.054 g,
0.14 mmol, 63%). RF 0.52 (1:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.61-7.47 (4H, m, ArH),
7.40-7.36 (2H, m, ArH), 7.26-7.21 (2H, m, ArH), 7.08 (1H, dt, J = 8.5, 1.9, NCH=CH), 4.84 (1H, dt, J
= 8.5, 3.8, NCH=CH), 4.15-4.08 (1H, m, NH), 2.49 (3H, d, J = 4.7, NMe), 2.42 (2H, t, J = 6.0,
CAr2CH2), 1.73 - 1.68 (2H, m, CAr2CH2CH2); 13C NMR (125 MHz, CDCl3) 155.9 (C=O), 143.3 (4°
ArC), 140.0 (4° ArC), 133.6 (4° ArC), 131.0 (q, J = 30.0, ArCCF3), 130.9 (ArC), 129.2 (ArC), 128.9
(ArC), 128.8 (ArC), 127.2 (NCH=CH), 124.4 (q, J = 3.6, ArC), 124.0 (q, J = 276.1, ArCCF3), 123.9 (q,
J = 3.6, ArC), 103.9 (NCH=CH), 66.4 (4 ArC), 40.1 (CAr2CH2), 27.4 (NMe), 20.0 (CAr2CH2CH2); IR
max (film)/cm-1 3307 (NH), 2925 (C=CH), 1713 (C=C), 1651 (C=O); MS (ES) m/z 417 (M + Na+);
HRMS m/z calcd 395.1133 for C20H19N2OF335Cl (M + H+) found 395.1136.
5-Methyl-11a-(3-(trifluoromethyl)phenyl)-11,11a-dihydro-5H-pyrido[1,2-c]quinazolin-6(10H)-
one 484
Following general procedure 11. To (S)-N-(2-fluorophenyl)-N-methyl-2-(3-
(trifluoromethyl)phenyl)-3,4-dihydropyridine-1(2H)-carboxamide 470e (0.074 g,
0.20 mmol) in THF (2 mL) at −78 °C was added LDA (0.49 mmol, prepared by
general procedure 10) and DMPU (0.50 mL). Purification by flash column
chromatography yielded the title compound 484 as a yellow oil (0.029 g, 0.08
mmol, 39%). RF 0.38 (1:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.65 (1H, dd, J = 7.7, 1.7, ArH),
7.48-7.16 (7H, m, ArH + NCH=CH), 6.89 (1H, dd, J = 8.1, 1.3, ArH), 5.12-5.04 (1H, m, NCH=CH),
3.27 (3H, s, NMe), 2.86-2.76 (1H, m, NCH=CHCH2CHH), 2.34 (1H, td, J = 13.0, 4.9,
NCH=CHCH2CHH), 2.14-2.02 (1H, m, NCH=CHCHH), 1.68-1.53 (1H, m, NCH=CHCHH); 13C NMR
(75 MHz, CDCl3) 152.6 (C=O), 143.8 (4° ArC), 137.9 (4° ArC), 130.9 (q, JF = 31.1, ArCCF3), 128.9
(4° ArC), 128.8 (ArC), 128.6 (ArC), 127.9 (4° ArC), 125.2 (ArC), 124.0 (q, JF = 3.8, ArC), 123.6
(NCH=CH), 122.8 (q, JF = 265.4, ArCCF3), 122.7 (ArC), 122.0 (q, JF = 4.4, ArC), 113.5 (ArC), 105.8
(NCH=CH), 61.2 (4° C), 32.7 (NCH=CHCH2CH2), 30.7 (NMe), 17.9 (NCH=CHCH2); IR max (film)/cm-1
2926 (C=CH), 1660 (C=O), 1597 (C=C); MS (ES) m/z 359 (M + H+); HRMS m/z calcd 359.1371 for
C20H18N2OF3 (M + H+) found 359.1365.

257
(2S,6S)-2-(4-Methoxyphenyl)-N-methyl-6-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 489a
Following general procedure 11. To (S)-N-(4-methoxyphenyl)-N-
methyl-6-phenyl-5,6-dihydropyridine-1(2H)-carboxamide 468a (0.060
g, 0.19 mmol) in THF (1.9 mL) at −78 °C was added LDA (0.47 mmol,
prepared by general procedure 10) and DMPU (0.48 mL). Purification by flash column
chromatography yielded the title compound 489a as a pale yellow oil (0.046 g, 0.14 mmol,
77%). RF 0.19 (1:1 PE:EtOAc); [D25 −96.0 (c 0.05, CHCl3);
1H NMR (300 MHz, CDCl3) 7.40-7.17
(7H, m, ArH), 6.96-6.87 (2H, m, ArH), 5.97-5.88 (1H, m, CHPMPCH=CH), 5.74-5.65 (1H,
CHPMPCH=CH), 5.51 (1H, dd, J = 4.7, 3.6, CHPh), 5.45 (1H, d, J = 5.1, CHPMP), 4.26 (1H, q, J =
5.1, NH), 3.82 (3H, s, OMe), 2.94-2.81 (1H, m, CHPhCHH), 2.65-2.50 (1H, m, CHPhCHH), 2.57
(3H, d, J = 4.7, NH); 13C NMR (75 MHz, CDCl3) 159.1 (C=O), 159.0 (4° ArCOMe), 143.3 (4° ArC),
135.0 (4° ArC), 130.4 (CHPMPCH=CH), 128.4 (ArC), 127.3 (ArC), 126.9 (ArC), 126.2 (ArC), 121.8
(ArC) (CHPMPCH=CH), 114.5 (ArC), 57.1 (CHPMP), 55.3 (OMe), 53.5 (CHPh), 30.5 (NMe), 27.6
(CHPhCH2); IR max (film)/cm-1 3028 (NH), 1647 (C=O); MS (ES) m/z 345 (M + Na+); HRMS m/z
calcd 323.1755 for C20H23N2O2 (M + H+) found 323.1756.
(2S,6S)-N-Methyl-2,6-diphenyl-5,6-dihydropyridine-1(2H)-carboxamide 489b
Following general procedure 11. To (S)-N-methyl-N,6-diphenyl-5,6-
dihydropyridine-1(2H)-carboxamide 468b (0.050 g, 0.17 mmol) in THF (1.7
mL) at −78 °C was added LDA (0.43 mmol, prepared by general procedure
10) and DMPU (0.43 mL). Purification by flash column chromatography yielded the title
compound 489b as a colourless oil (0.023 g, 0.08 mmol, 46%). RF 0.16 (2:1 PE:EtOAc); [D25
−41.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) 7.45-7.21 (10H, m, ArH), 5.98 (1H, ddd, J = 9.8,
5.0, 2.8, NCHPhCH=CH), 5.75-5.68 (1H, m, NCHPhCH=CH), 5.54-5.49 (2H, m, 2 x CHPh), 4.21 (1H,
q, J = 5.0, NH), 2.93-2.86 (1H, m, CHPhCHH), 2.63-2.58 (1H, m, CHPhCHH), 2.57 (3H, d, J = 4.7,
NMe); 13C NMR (125 MHz, CDCl3) 158.9 (C=O), 143.3 (4° ArC), 143.2 (4° ArC), 130.1
(NCHPhCH=CH), 129.1 (ArC), 128.5 (ArC), 127.5 (ArC), 127.0 (ArC), 126.2 (ArC), 126.1 (ArC),
122.1 (NCHPhCH=CH), 57.7 (CHPhCH=CH), 53.6 (CHPhCH2), 30.6 (CHPhCH2), 27.6 (NMe); IR max
(film)/cm-1 3028 (NH), 1646 (C=O); MS (ES) m/z 293 (M + H+); HRMS m/z calcd 317.1624 for
C19H22N2ONa (M + H+) found 317.1616.

258
(2S,6S)-N-Methyl-2-phenyl-6-(3-(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-
carboxamide 489c
Following general procedure 11. To (S)-N-methyl-N-phenyl-6-(3-
(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide 468h
(0.050 g, 0.14 mmol) in THF (1.4 mL) at −78 °C was added LDA (0.34
mmol, prepared by general procedure 10) and DMPU (0.35 mL). Purification by flash column
chromatography yielded the title compound 489c as a pale yellow oil (0.038 g, 0.11 mmol,
76%). RF 0.42 (1:1 PE:EtOAc); [D25 −121.6 (c 0.25, CHCl3);
1H NMR (500 MHz, CDCl3) 7.52-7.48
(2H, m, ArH), 7.45-7.38 (6H, m, ArH), 7.36-7.30 (1H, m, ArH), 5.93 (1H, ddd, J = 9.8, 5.0, 3.2,
CHPhCH=CH), 5.80 (1H, dd, J = 6.3, 2.5, CHAr), 5.69-5.64 (1H, m, CHPhCH=CH), 5.32 (1H, d, J =
4.4, CHPh), 4.26 (1H, q, J = 4.4, NH), 3.01-2.94 (1H, m, CHArCHH), 2.58 (3H, d, J = 4.7, NMe),
2.57-2.49 (1H, m, CHArCHH); 13C NMR (125 MHz, CDCl3) 158.7 (C=O), 145.3 (4° ArC), 142.7 (4°
ArC), 130.7 (CHPhCH=CH), 130.0 (ArC), 129.6 (ArC), 129.5 (ArC), 128.7 (ArC), 128.0 (ArC), 125.7
(ArC), 123.6 (ArC), 121.3 (CHPhCH=CH), 57.9 (CHAr), 53.4 (CHPh), 30.9 (CHArCH2), 27.6 (NMe) +
1 missing ArC, unable to get F couplings; IR max (film)/cm-1 3013 (NH), 1649 (C=O); MS (ES) m/z
361 (M + H+); HRMS m/z calcd 361.1523 for C20H20N2OF3 (M + H+) found 361.1520.
(2S,6S)-6-(2-Chlorophenyl)-2-(2-fluorophenyl)-N-methyl-5,6-dihydropyridine-1(2H)-
carboxamide 489d
Following general procedure 11. To (S)-6-(2-chlorophenyl)-N-(2-
fluorophenyl)-N-methyl-5,6-dihydropyridine-1(2H)-carboxamide 468e
(0.060 g, 0.17 mmol) in THF (1.7 mL) at −78 °C was added LDA (0.44 mmol,
prepared by general procedure 10) and DMPU (0.43 mL). Purification by flash column
chromatography yielded the title compound 489d as a yellow-brown oil (0.039 g, 0.11 mmol,
65%). RF 0.18 (2:1 PE:EtOAc); [D25 +78.8 (c 2.0, CHCl3);
1H NMR (400 MHz, CDCl3) 7.42-7.35
(2H, m, ArH), 7.26-7.05 (6H, m, ArH), 6.28-6.21 (1H, m, CHFArCH=CH), 5.91 (1H, d, J = 5.8,
CHFAr), 5.77 (1H, d, J = 7.0, CHClAr), 5.65 (1H, ddd, J = 9.5, 6.5, 1.8, CHFArCH=CH), 4.19 (1H, q, J
= 4.8, NH), 2.91-2.82 (1H, m, CHClArCHH), 2.61 (3H, d, J = 4.5, NMe), 2.60-2.54 (1H, m,
CHClArCHH); 13C NMR (100 MHz, CDCl3) 159.7 (d, JF = 244.3, ArCF), 157.7 (C=O), 141.1 (4° ArC),
131.7 (4° ArC), 130.1 (d, JF = 13.2, 4° ArC), 129.7 (ArC), 128.7 (d, JF = 8.1, ArC), 128.4 (ArC), 128.1
(ArC), 127.9 (CHFArCH=CH), 126.8 (ArC), 126.3 (d, JF = 4.4, ArC), 124.7 (d, JF = 3.7, ArC), 122.3
(CHFArCH=CH), 115.7 (d, JF = 21.3, ArC), 52.1 (CHClAr), 51.6 (d, JF = 2.2, CHFAr), 28.7
(CHClArCH2), 27.7 (NMe); IR max (film)/cm-1 3367 (NH), 2925 (C=CH), 1647 (C=O); MS (ES) m/z
345 (M + H+); HRMS m/z calcd 345.1165 for C19H19N2OF35Cl (M + H+) found 345.1169.

259
(2S,6S)-2-(2-Fluorophenyl)-N-methyl-6-(3-(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-
carboxamide 489e
Following general procedure 11. To (S)-N-(2-fluorophenyl)-N-methyl-
6-(3-(trifluoromethyl)phenyl)-5,6-dihydropyridine-1(2H)-carboxamide
468f (0.055 g, 0.15 mmol) in THF (1.5 mL) at −78 °C was added LDA
(0.36 mmol, prepared by general procedure 10) and DMPU (0.38 mL). Purification by flash
column chromatography yielded the title compound 489e as a brown oil (0.031 g, 0.08 mmol,
56%). RF 0.15 (2:1 PE:EtOAc); [D25 −60.4 (c 1.0, CHCl3);
1H NMR (400 MHz, CDCl3) 7.52-7.25
(6H, m, ArH), 7.20-7.08 (2H, m, ArH), 6.16-6.08 (1H, m, CHFArCH=CH), 5.76-5.62 (3H, m,
CHFArCH=CHCH2CHCF3Ar), 4.26 (1H, q, J = 4.5, NH), 2.98-2.89 (1H, m, CHCF3ArCHH), 2.63 (3H, d,
J = 4.5, NMe), 2.58-2.49 (1H, m, CHCF3ArCHH); 13C NMR (100 MHz, CDCl3) 159.3 (d, JF = 242.8,
ArCF), 158.1 (C=O), 145.2 (4° ArC), 130.8 (ArC), 129.5 (ArC), 129.3 (ArC), 128.8 (CHFArCH=CH),
128.0 (ArC), 126.7 (ArC), 125.0 (ArC), 123.7 (m, ArC), 122.7 (m, ArC), 122.7 (CHFArCH=CH), 115.9
(d, JF = 22.0, ArC), 54.0 (CHCF3Ar), 51.5 (d, J = 1.5, CHFAr), 30.8 (CHCF3ArCH2), 27.7 (NMe) + 1
missing ArC; IR max (film)/cm-1 3347 (NH), 2926 (C=CH), 1637 (C=O); MS (ES) m/z 379 (M + H+);
HRMS m/z calcd 379.1429 for C20H19N2OF4 (M + H+) found 379.1424.
(2S,6S)-N-Methyl-2,6-diphenylpiperidine-1-carboxamide 490
To a solution of (2S,6S)-N-Methyl-2,6-diphenyl-5,6-dihydropyridine-1(2H)-
carboxamide 489b (0.042 g, 0.14 mmol) in degassed IPA (5 mL, 0.03 M)
was added Pd/C (10% wt, 0.042 g, 0.01 mmol) and the mixture placed
under an atmosphere of hydrogen. After 2 h the mixture was filtered over diatomaceous earth,
washed with IPA and the solvent removed under reduced pressure. The crude mixture was
purified by flash column chromatography (4:1 PE:EtOAc 1% NEt3) to yield the title compound
490 as a colourless oil (0.027 g, 0.09 mmol, 66%). RF 0.56 (1:2 PE:EtOAc); [D25 −40.4 (c 1.0,
CHCl3); 1H NMR (500 MHz, CDCl3) 7.40-7.35 (8H, m, ArH), 7.29-7.24 (2H, m, ArH), 5.32 (2H, t, J
= 3.8, CHPh + CHPh), 4.14 (1H, brs, NH), 2.66 (3H, d, J = 4.7, NMe), 2.23-2.14 (2H, m, CHPhCH2),
2.12-2.05 (2H, m, CHPhCH2), 1.47-1.40 (2H, m, CHPhCH2CH2); 13C NMR (125 MHz, CDCl3) 159.3
(C=O), 142.9 (4° ArC), 128.6 (ArC), 126.8 (ArC), 126.0 (ArC), 54.5 (CHPh), 27.6 (NMe), 27.1
(CHPhCH2), 14.2 (CHPhCH2CH2); IR max (film)/cm-1 3029 (NH), 1645 (C=O); MS (ES) m/z 295 (M +
H+); HRMS m/z calcd 295.1810 for C19H23N2O (M + H+) found 295.1816.

260
(2S,6S)-6-(4-Methoxyphenyl)-2-phenyl-1,2,3,6-tetrahydropyridine 492
Following general procedure 3. To a solution of (2S,6S)-2-(4-
methoxyphenyl)-N-methyl-6-phenyl-5,6-dihydropyridine-1(2H)-
carboxamide 489a (0.020 g, 0.062 mmol) in n-BuOH (1 mL) was added
potassium carbonate (0.020 g) and heated to reflux for 2.5 h. Purification by flash column
chromatography yielded the title compound 492 as a brown oil (0.008 g, 0.030 mmol, 49%). RF
Flame (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.30-7.13 (7H, m, ArH), 6.81 (2H, d, J = 8.8,
ArH), 6.09-5.99 (1H, m, CHArCH=CH), 5.83 (1H, ddt, J = 9.8, 3.8, 1.9, CHArCH=CH), 4.62-4.55 (1H,
m, CHAr), 3.86 (1H, t, J = 6.9, CHPh), 3.74 (3H, s, OMe), 2.29-2.24 (2H, m, CHPhCH2); 13C NMR
(125 MHz, CDCl3) 158.8 (4° ArCOMe), 135.7 (4° ArC), 132.0 (4° ArC), 129.0 (ArC), 128.4 (ArC),
127.6 (CHArCH=CH), 127.2 (ArC), 126.9 (CHArCH=CH), 126.8 (ArC), 113.7 (ArC), 56.7 (CHAr),
55.3 (OMe), 51.4 (CHPh), 33.8 (CHPhCH2); IR max (film)/cm-1 3075 (NH), 2794 (CH), 1642 (C=C);
MS (ES) m/z 266 (M + H+); HRMS m/z calcd 266.1540 for C18H20NO (M + H+) found 266.1534.
1,1-Diallyl-3-methyl-3-phenyl-urea 500
Following general procedure 15. To a solution of diallyl amine 499 (1.00 mL, 8.10
mmol) in DCE (27 mL), was added N-methyl-N-phenylcarbamoyl chloride (1.786 g,
10.53 mmol), NEt3 (1.81 mL, 12.96 mmol) and DMAP (cat.) and the mixture heated
to reflux overnight. Purification by flash column chromatography yielded the title
compound 500 as a brown oil (1.864 g, 8.10 mmol, 100%). RF 0.21 (2:1 PE:EtOAc); 1H NMR (500
MHz, CDCl3) 7.33 (2H, t, J = 7.9, ArH), 7.16-7.08 (3H, m, ArH), 5.49 (2H, ddt, J = 17.0, 10.1, 6.3,
2 x CH2CH=CH2), 5.07-4.98 (4H, m, 2 x CH2CH=CH2), 3.66 (4H, d, J = 6.3Hz, 2 x CH2CH=CH2), 3.17
(3H, s, NMe); 13C NMR (125 MHz, CDCl3) 161.9 (C=O), 147.0 (4° ArC), 133,5 (CH2CH=CH2),
129.5 (ArC), 124.9 (ArC), 124.6 (ArC), 117.5 (CH2CH=CH2), 49.8 (CH2CH=CH2), 40.1 (NMe); IR max
(film)/cm-1 3080 (C=CH2), 2971 (C=CH2), 1632 (C=O); MS (ES) m/z 253 (M + Na+); HRMS m/z
calcd 253.1312 for C14H18N2ONa (M + Na+) found 253.1307.

261
N-Methyl-N-phenyl-2,5-dihydropyrrole-1-carboxamide 501
Following general procedure 16. To a solution of 1,1-diallyl-3-methyl-3-phenyl-urea
500 (1.864 g, 8.10 mmol) in CH2Cl2 (162 mL) was added Grubbs 1st generation catalyst
(0.333 g, 0.40 mmol). Purification by flash column chromatography yielded the title
compound 501 as a black solid (1.436 g, 7.13 mmol, 88%). MP Decomposition; RF 0.24
(2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.35-7.30 (2H, m, ArH), 7.18-7.12 (3H, m, ArH), 5.61
(2H, brs, CH2CH=CHCH2), 3.87 (4H, brs, CH2CH=CHCH2), 3.24 (3H, s, NMe); 13C NMR (125 MHz,
CDCl3) 159.6 (C=O), 146.2 (4° ArC), 129.4 (ArC), 125.4 (CHCH2), 125.3 (ArC), 125.0 (ArC), 54.8
(CHCH2), 40.0 (NMe); IR max (film)/cm-1 3043 (C=C), 1679 (C=O); MS (ES) m/z 225 (M + Na+);
HRMS m/z calcd 203.1179 for C12H15N2O (M + H+) found 203.1188.
N-Methyl-N-phenyl-2,3-dihydropyrrole-1-carboxamide 502
Following general procedure 17. To a solution of N-methyl-N-phenyl-2,5-
dihydropyrrole-1-carboxamide 501 (0.200 g, 0.99 mmol) in THF (10 mL) was added
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (94 mg, 0.10 mmol) and
the mixture heated to reflux overnight. Purification by flash column chromatography
yielded the title compound 502 as a yellow gum (0.180 g, 0.89 mmol 90%). RF 0.15 (2:1
PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.37-7.28 (2H, m, ArH), 7.17-7.07 (3H, m, ArH), 6.28 (1H,
dt, J = 4.2, 2.0, NCH=CH), 4.85 (1H, dt, J = 4.3, 2.0, NCH=CH), 3.37 (2H, t, J = 9.1, NCH2CH2), 3.25
(3H, s, NMe), 2.43 (2H, tt, J = 8.7, 2.3, NCH2CH2); 13C NMR (75 MHz, CDCl3) 156.6 (C=O), 145.7
(4° ArC), 131.2 (NCH=CH), 129.4 (ArC), 125.1 (ArC), 124.9 (ArC), 107.3 (NCH=CH), 47.0
(NCH2CH2), 39.4 (NMe), 29.0 (NCH2CH2); IR max (film)/cm-1 2985 (C=C), 1619 (C=O); MS (ES) m/z
203 (M + H+); HRMS m/z calcd 203.1179 for C12H15N2O (M + H+) found 203.1171.

262
1-Methyl-1-phenyl-3-(1-phenylallyl)urea 504
Following general procedure 15. To a solution of 1-phenylprop-2-en-1-amine
503 (0.300 g, 2.25 mmol) in DCE (8 mL) was added N-methyl-N-
phenylcarbamoyl chloride (0.497 g, 2.93 mmol), NEt3 (0.50 mL, 3.61 mmol) and
DMAP (cat.) and the mixture heated to reflux overnight. Purification by flash
column chromatography yielded the title compound 504 as a pale brown oil (0.402 g, 1.51
mmol, 67%). RF 0.25 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.47-7.39 (2H, m, ArH), 7.35-
7.16 (8H, m, ArH), 5.93 (1H, ddd, J = 17.0, 10.4, 5.4, CH=CH2), 5.55 (1H, t, J = 5.8, NH), 5.18-5.01
(2H, m, CH=CH2), 4.64 (2H, d, J = 7.4, CHPh), 3.29 (3H, s, NMe); 13C NMR (75 MHz, CDCl3) 162.1
(C=O), 143.3 (4° ArC), 141.6 (4° ArC), 138.5 (CH=CH2), 130.1 (ArC), 128.6 (ArC), 127.5 (ArC),
127.4 (ArC), 127.3 (ArC), 126.9 (ArC), 115.2 (CH=CH2), 56.4 (CHPh), 37.3 (NMe); IR max
(film)/cm-1 3094 (NH), 2987 (C=CH2), 1632 (C=O); MS (ES) m/z 289 (M + Na+); HRMS m/z calcd
289.1419 for C17H18N2ONa (M + Na+) found 289.1414.
N-Allyl-1-phenyl-methanimine211 508a
Following general procedure 13. To a solution of benzaldehyde (2.50 mL, 24.59
mmol) in CH2Cl2 (60 mL) was added allyl amine (3.69 mL, 49.18 mmol). Filtration
over diatomaceous earth yielded the title compound 508a as a brown liquid
(3.625 g, 24.59 mmol, 100%). 1H NMR (400 MHz, CDCl3) 8.32 (1H, s, N=CH), 7.79-7.76 (2H, m,
ArH), 7.47-7.40 (3H, m, ArH), 6.18-6.05 (1H, m, CH=CH2), 5.26-5.18 (2H, m, CH=CH2), 4.29-4.26
(2H, m, NCH2); 13C NMR (100 MHz, CDCl3) 162.0 (N=CH), 136.1 (CH=CH2), 135.9 (4° ArC), 130.7
(ArC), 128.6 (ArC), 128.2 (ArC), 116.1 (CH=CH2), 63.6 (NCH2); IR max (film)/cm-1 3063 (C=CH2),
1650 (C=N); MS (ES) m/z 146 (M + H+); HRMS m/z calcd 145.0891 for C10H12N (M + H+) found
145.0899. Data corresponds to the reported literature.211

263
N-Allyl-1-(2-fluorophenyl)methanimine 508b
Following general procedure 13. To a solution of 2-fluorobenzaldehyde (1.05
mL, 10.00 mmol) in CH2Cl2 (25 mL) was added allyl amine (1.50 mL, 20.00
mmol). Filtration over diatomaceous earth yielded the title compound 508b as
a yellow oil (1.632 g, 10.00 mmol, 100%). 1H NMR (300 MHz, CDCl3) 8.62 (1H, s, N=CH), 8.02
(1H, td, J = 7.5, 1.9, ArH), 7.45-7.36 (1H, m, ArH), 7.19 (1H, t, J = 7.6, ArH), 7.09 (1H, ddd, J =
10.6, 8.3, 0.8, ArH), 6.08 (1H, ddt, J = 17.1, 10.3, 5.8, NCH2CH=CH2), 5.25 (1H, ddt, J = 17.3, 3.4,
1.7, NCH2CH=CHH), 5.18 (1H, ddt, J = 10.2, 3.0, 1.5, NCH2CH=CHH), 4.30 (2H, dq, J = 5.8, 1.4,
NCH2CH=CH2); 13C NMR (75 MHz, CDCl3) 160.3 (d, JF = 252.3, ArCF), 155.3 (d, JF = 4.9, N=CH),
135.7 (NCH2CH=CH2), 132.2 (d, JF = 8.7, 4° ArC), 127.7, (d, JF = 3.3, ArC), 124.3 (d, JF = 3.8, ArC),
123.8 (d, JF = 9.3, ArC), 116.3 (NCH2CH=CH2), 115.7 (d, JF = 20.7, ArC), 63.9 (NCH2CH=CH2); IR
max (film)/cm-1 3056 (C=CH2), 1630 (C=N); MS (ES) m/z 164 (M + H+); HRMS m/z calcd 164.0871
for C10H11NF (M + H+) found 164.0870.
N-Allyl-1-[3-(trifluoromethyl)phenyl]methanimine 508c
Following general procedure 13. To a solution of 3-
(trifluoromethyl)benzaldehyde (1.34 mL, 10.00 mmol) in CH2Cl2 (25 mL) was
added allyl amine (1.50 mL, 20.00 mmol). Filtration over diatomaceous earth
yielded the title compound 508c as a pale yellow oil (2.130 g, 10.00 mmol,
100%). 1H NMR (300 MHz, CDCl3) 8.35 (1H, s, N=CH), 8.05 (1H, s, ArH), 7.93 (1H, d, J = 7.7,
ArH), 7.69 (1H, d, J = 7.9, ArH), 7.55 (1H, t, J = 7.8, ArH), 6.15-5.99 (1H, m, NCH2CH=CH2), 5.30-
5.13 (2H, m, NCH2CH=CH2), 4.33-4.26 (2H, m, NCH2CH=CH2); 13C NMR (75 MHz, CDCl3) 160.3
(N=CH), 136.9 (4° ArC), 135.4 (NCH2CH=CH2), 131.3 (ArC), 131.2 (q, JF = 32.7, ArCCF3), 129.1
(ArC), 127.2 (q, JF = 3.8, ArC), 124.8 (q, JF = 3.8, ArC), 123.9 (q, JF = 272.5, ArCCF3), 116.5
(NCH2CH=CH2), 63.4 (NCH2CH=CH2); IR max (film)/cm-1 3069 (C=CH2), 1678 (C=N); MS (ES) m/z
214 (M + H+); HRMS m/z calcd 214.0839 for C11H1NF3 (M + H+) found 214.0835.

264
N-Allyl-1-(4-chlorophenyl)methanimine212 508d
Following general procedure 13. To a solution of 4-chlorobenzaldehyde
(1.406 g, 10.00 mmol) in CH2Cl2 (25 mL) was added allyl amine (1.50 mL,
20.00 mmol). Filtration over diatomaceous earth yielded the title compound
508d as a yellow oil (1.701 g, 9.50 mmol, 95%). 1H NMR (300 MHz, CDCl3) 8.27 (1H, s, N=CH),
7.74-7.66 (2H, m, ArH), 7.44-7.37 (2H, m, ArH), 6.15-5.83 (1H, m, CH=CH2), 5.30-5.13 (2H, m,
CH=CH2), 4.29-4.24 (2H, m, NCH2); 13C NMR (75 MHz, CDCl3) 160.6 (N=CH), 136.7 (4° ArC),
135.6 (CH=CH2), 134.7 (4° ArC), 129.3 (ArC), 128.9 (ArC), 116.3 (CH=CH2), 63.4 (NCH2); IR max
(film)/cm-1 3045 (C=CH2), 1632 (C=N); MS (ES) m/z 180 (M + H+); HRMS m/z calcd 180.0575 for
C10H11N35Cl (M + H+) found 180.0571. Data corresponds to the reported literature.212
N-Allyl-1-(2-nitrophenyl)methanimine 508e
Following general procedure 13. To a solution of 2-nitrobenzaldehyde (1.511 g,
10.00 mmol) in CH2Cl2 (25 mL) was added allyl amine (1.50 mL, 20.00 mmol).
Filtration over diatomaceous earth yielded the title compound 508e as a
luminous yellow liquid (1.845 g, 9.70 mmol, 97%). 1H NMR (300 MHz, CDCl3) 8.74 (1H, s,
N=CH), 8.07 (2H, dd, J = 7.7, 1.5, ArH), 7.72-7.54 (2H, m, ArH), 6.09 (1H, ddt, J = 17.2, 10.3, 5.7,
CH=CH2), 5.32-5.17 (2H, m, CH=CH2), 4.34 (2H, dq, J = 5.7, 1.5); IR max (film)/cm-1 3050 (C=CH2),
1687 (C=N), 1520 (NO2); MS (ES) m/z 191 (M + H+); HRMS m/z calcd 191.0185 for C10H11N2O2 (M
+ H+) found 191.0189. Decomposition of product in NMR tube prevented full analysis.
N-Allyl-1-(3-pyridyl)methanimine 508f
Following general procedure 13. To a solution of 3-pyridinecarboxaldehyde
(0.94 mL, 10.00 mmol) in CH2Cl2 (25 mL) was added allyl amine (1.50 mL, 20.00
mmol). Filtration over diatomaceous earth yielded the title compound 508f as a
brown liquid (1.461 g, 10.00 mmol, 100%). 1H NMR (300 MHz, CDCl3) 8.88 (1H, d, J = 1.9, ArH),
8.66 (1H, dd, J = 4.7, 1.7, ArH), 8.35 (1H, s, N=CH), 8.15 (1H, dt, J = 7.9, 1.9, ArH), 7.36 (1H, dd, J
= 7.8, 4.8, ArH), 6.08 (1H, ddt, J = 17.2, 10.3, 5.7, CH=CH2), 5.30-5.15 (2H, m, CH=CH2), 4.30 (2H,
dq, J = 5.8, 1.4, NCH2); IR max (film)/cm-1 3043 (C=CH2), 1634 (C=N); MS (ES) m/z 147 (M + H+);
HRMS m/z calcd 147.0844 for C9H11N2 (M + H+) found 147.0839. Decomposition of product in
NMR tube prevented full analysis.

265
N-Allyl-1-(4-methoxyphenyl)methanimine213 508g
Following general procedure 13. To a solution of p-anisaldehyde (1.22 mL,
10.00 mmol) in CH2Cl2 (25 mL) was added allyl amine (1.50 mL, 20.00
mmol). Filtration over diatomaceous earth yielded the title compound
508g as a yellow oil (1.733 g, 9.90 mmol, 99%). 1H NMR (300 MHz, CDCl3) 8.21 (1H, s, N=CH),
7.68 (2H, d, J = 8.9, ArH), 6.89 (2H, d, J = 8.9, ArH), 6.12-6.07 (1H, m, CH=CH2), 5.25-5.11 (2H, m,
CH=CH2), 4.22 (2H, d, J = 5.6, NCH2), 3.83 (3H, s, OMe); 13C NMR (75 MHz, CDCl3) 161.6
(ArCOMe), 161.2 (N=CH), 136.2 (CH=CH2), 129.7 (4° ArC), 129.1 (ArC), 115.8 (CH=CH2), 113.9
(ArC), 63.4 (NCH2), 55.2 (OMe); IR max (film)/cm-1 3065 (C=CH2), 1648 (C=N); MS (ES) m/z 176
(M + H+); HRMS m/z calcd 176.0997 for C11H14NO (M + H+) found 176.1001. Data corresponds to
the reported literature.213
N-Allyl-1-phenyl-prop-2-en-1-amine214 509a
Following general procedure 14. To a solution of N-allyl-1-phenyl-methanimine
508a (0.200 g, 1.38 mmol) in THF (14 mL) at −78 °C was added diethyl zinc
solution (1 M, 1 M, 2.76 ml, 2.76 mmol) dropwise. After 2 h vinyl magnesium
bromide solution (1 M, 2.76 ml, 2.76 mmol) was added dropwise and the reaction left to warm
to room temperature overnight. Purification by flash column chromatography yielded the title
compound 509a as a pale yellow oil (0.055 g, 0.32 mmol, 23%). RF Flame (2:1 PE:EtOAc); 1H
NMR (300 MHz, CDCl3) 7.52-7.17 (5H, m , ArH), 6.01-5.70 (2H, m, 2 x CH=CH2), 5.28-4.97 (4H,
m, 2 x CH=CH2), 4.22 (1H, d, J = 7.2, NCHPh), 3.25-3.10 (2H, m, NCH2); 13C NMR (75 MHz, CDCl3)
140.9 (4° ArC), 136.8 (CH=CH2), 135.8 (CH=CH2), 128.5 (ArC), 127.3 (ArC), 127.2 (ArC), 115.9
(CH=CH2), 115.1 (CH=CH2), 65.2 (NCHPh), 49.9 (NCH2); IR max (film)/cm-1 3023 (NH), 2975
(C=CH2), 2874 (C=CH2); MS (ES) m/z 174 (M + H+); HRMS m/z calcd 174.1278 for C12H16N (M +
H+) found 174.1274. Data corresponds to the reported literature.214

266
N-Allyl-1-(4-chlorophenyl)prop-2-en-1-amine214 509b
Following general procedure 14. To a solution of N-allyl-1-(4-
chlorophenyl)methanimine 508d (0.200 g, 1.11 mmol) in THF (11 mL) at −78
°C was added diethyl zinc solution (1 M, 2.22 ml, 2.22 mmol) dropwise.
After 2 h vinyl magnesium bromide solution (1 M, 2.22 ml, 2.22 mmol) was added dropwise and
the reaction left to warm to room temperature overnight. Purification by flash column
chromatography yielded the title compound 509b as a yellow oil (0.044 g, 0.21 mmol, 19%). RF
Flame (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.59-7.21 (4H, m , ArH), 6.08-5.83 (2H, m, 2 x
CH=CH2), 5.29-5.03 (4H, m, 2 x CH=CH2), 4.27 (1H, d, J = 7.2, NCHPh), 3.31-3.18 (2H, m, NCH2);
13C NMR (75 MHz, CDCl3) 140.5 (4° ArC), 136.8 (CH=CH2), 135.8 (CH=CH2), 134.9 (4° ArC),
129.5 (ArC), 128.8 (ArC), 115.9 (CH=CH2), 115.1 (CH=CH2), 65.2 (NCHAr), 49.9 (NCH2); IR max
(film)/cm-1 3102 (NH), 3040 (C=CH2), 2934 (C=CH2); MS (ES) m/z 208 (M + H+); HRMS m/z calcd
208.0893 for C12H15N35Cl (M + H+) found 208.0887. Data corresponds to the reported
literature.214

267
1-Allyl-3-(4-chlorophenyl)-3-methyl-1-(1-phenylallyl)urea 511a
Following general procedures 14 and 15. To a solution of N-allyl-1-phenyl-
methanimine 508a (0.200 g, 1.38 mmol) in THF (14 mL) at −78 °C was added
diethyl zinc solution (1 M, 2.76 ml, 2.76 mmol) dropwise. After 2 h vinyl
magnesium bromide solution (1 M, 2.76 ml, 2.76 mmol) was added dropwise
and the reaction left to warm to room temperature overnight. After which (4-
chlorophenyl)(methyl)carbamic chloride (0.366 g, 1.79 mmol), NEt3 (0.31 mL, 2.20 mmol) and
DMAP (cat.) were added in DCE (5 mL) and the mixture heated to reflux overnight. Purification
by flash column chromatography yielded the title compound 511a as a yellow oil (0.211 g, 0.62
mmol, 57%). RF 0.56 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.35-7.21 (7H, m, ArH), 7.06-
7.00 (2H, m, ArH), 6.08 (1H, ddd, J = 17.4, 10.3, 6.9, NCHPhCH=CH2), 5.61 (1H, d, J = 6.8,
NCHPhCH=CH2), 5.41 (1H, ddt, J = 16.7, 10.3, 6.1, X of ABXYY’, NCH2CHX=CH2), 5.27 (1H, dt, J =
10.3, 1.4, NCHPhCH=CHH), 5.05 (1H, dt, J = 17.2, 1.4, NCHPhCH=CHH), 4.92 (2H, dddd, J = 16.7,
10.1, 2.8, 1.4, Y and Y’ of ABXYY’, NCH2CH=CHYY’), 3.54 (1H, ddt, J = 16.1, 5.8, 1.4, A of ABXYY’,
NCHAHCH=CH2), 3.38 (1H, ddt, J = 16.1, 6.3, 1.4, B of ABXYY’, NCHHBCH=CH2), 3.16 (3H, s, NMe);
13C NMR (100 MHz, CDCl3) 161.8 (C=O), 145.0 (4° ArC), 139.6 (4° ArC), 135.2 (NCHPhCH=CH2),
134.8 (NCH2CH=CH2), 130.2 (4° ArC), 129.5 (ArC), 128.3 (ArC), 127.9 (ArC), 127.4 (ArC), 125.5
(ArC), 118.3 (NCHPhCH=CH2), 116.4 (NCH2CH=CH2), 63.1 (NCHPhCH=CH2), 48.3 (NCH2CH=CH2),
39.7 (NMe); IR max (film)/cm-1 3082 (C=CH2), 3030 (C=CH2), 1643 (C=O); MS (ES) m/z 341 (M +
H+); HRMS m/z calcd 341.1416 for C20H22N2O35Cl (M + H+) found 341.1411.

268
1-Allyl-1-[1-(2-fluorophenyl)allyl]-3-methyl-3-phenyl-urea 511b
Following general procedures 14 and 15. To a solution of N-allyl-1-(2-
fluorophenyl)methanimine 508b (0.200 g, 1.23 mmol) in THF (12 mL) at −78 °C
was added diethyl zinc solution (1 M, 2.46 ml, 2.46 mmol) dropwise. After 2
hours vinyl magnesium bromide solution (1 M, 2.46 ml, 2.46 mmol) was added
dropwise and the reaction left to warm overnight. After which N-methyl-N-
phenylcarbamoyl chloride (0.270 g, 1.59 mmol), NEt3 (0.27 mL, 1.96 mmol) and DMAP (cat.)
were added in DCE (4 mL) and the mixture heated to reflux overnight. Purification by flash
column chromatography yielded the title compound 511b as a yellow gum (0.171 g, 0.53 mmol,
43%). RF 0.54 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.39-7.21 (4H, m, ArH), 7.15-6.98 (5H,
m, ArH), 6.13 (1H, ddd, J = 16.8, 10.0, 6.2, NCHArCH=CH2), 5.68 (1H, d, J = 6.4, NCHAr), 5.43 (1H,
dddd, J = 16.8, 10.7, 5.9, 5.9, NCH2CH=CH2), 5.21 (1H, dt, J = 10.4, 1.4, NCHArCH=CHH), 5.05 (1H,
dt, J = 17.1, 1.3, NCHArCH=CHH), 4.97-4.87 (2H, m, NCH2CH=CH2), 3.51 (2H, dt, J = 6.0, 1.3,
NCH2), 3.18 (3H, s, NMe); 13C NMR (125 MHz, CDCl3) 161.8 (C=O), 160.9 (d, JF = 248.0, ArCF),
146.4, (4° ArC), 134.8 (NCHArCH=CH2), 134.8 (NCH2CH=CH2), 130.2 (d, JF = 3.8, 4° ArC), 129.3
(ArC), 129.2 (d, JF = 8.7, ArC), 126.9 (d, JF = 14.2, ArC), 125.0 (ArC), 124.7 (ArC), 123.8 (d, JF = 3.3,
ArC), 117.7 (NCHArCH=CH2), 116.4 (NCH2CH=CH2), 115.4 (d, JF = 22.3, ArC), 57.8 (d, JF = 2.2,
NCHAr), 49.2 (NCH2), 39.8 (NMe); IR max (film)/cm-1 3002 (C=CH2), 2943 (C=CH2), 1675 (C=O);
MS (ES) m/z 347 (M + Na+); HRMS m/z calcd 347.1638 for C20H21N2OFNa (M + Na+) found
347.1634.

269
1-Allyl-1-[1-(4-chlorophenyl)allyl]-3-methyl-3-phenyl-urea 511c
Following general procedures 14 and 15. To a solution of N-allyl-1-(4-
chlorophenyl)methanimine 508d (0.200 g, 1.11 mmol) in THF (11 mL) at −78
°C was added diethyl zinc solution (1 M, 2.22 ml, 2.22 mmol) dropwise.
After 2 h vinyl magnesium bromide solution (1 M, 2.22 ml, 2.22 mmol) was
added dropwise and the reaction left to warm to room temperature
overnight. After which N-methyl-N-phenylcarbamoyl chloride (0.245 g, 1.45 mmol), NEt3 (0.25
mL, 1.78 mmol) and DMAP (cat.) were added in DCE (4 mL) and the mixture heated to reflux
overnight. Purification by flash column chromatography yielded the title compound 511c as a
yellow oil (0.148 g, 0.43 mmol, 39%). RF 0.52 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.39-
7.19 (7H, m, ArH), 7.09-7.03 (2H, m, ArH), 6.08 (1H, ddd, J = 17.1, 10.4, 6.9, NCHArCH=CH2), 5.58
(1H, d, J = 6.9, NCHArCH=CH2), 5.37 (1H, ddt, J = 16.9, 10.4, 6.0, X of ABXYY’, NCH2CHX=CH2),
5.24 (1H, dt, J = 10.4, 1.1, NCHArCH=CHH), 5.01 (1H, dt, J = 17.1, 1.1, NCHArCH=CHH), 4.92 (2H,
dddd, J = 16.9, 10.3, 2.8, 1.1, Y and Y’ of ABXYY’, NCH2CH=CHYY’), 3.54 (1H, ddt, J = 16.0, 5.4, 1.1,
A of ABXYY’, NCHAHCH=CH2), 3.38 (1H, ddt, J = 16.0, 6.1, 1.2, B of ABXYY’, NCHHBCH=CH2), 3.16
(3H, s, NMe); 13C NMR (75 MHz, CDCl3) 161.7 (C=O), 144.9 (4° ArC), 139.7 (4° ArC), 135.1
(NCHArCH=CH2), 134.7 (NCH2CH=CH2), 130.2 (4° ArC), 129.5 (ArC), 128.4 (ArC), 128.0 (ArC),
127.6 (ArC), 125.4 (ArC), 118.1 (NCHArCH=CH2), 116.3 (NCH2CH=CH2), 62.8 (NCHArCH=CH2),
48.4 (NCH2), 39.6 (NMe); IR max (film)/cm-1 2976 (C=CH2), 1648 (C=O), 1596 (C=C); MS (ES) m/z
363 (M + Na+); HRMS m/z calcd 363.1342 for C20H21N2O35ClNa (M + Na+) found 363.1340.

270
1-Allyl-3-methyl-3-phenyl-1-(1-(3-(trifluoromethyl)phenyl)allyl)urea 511d
Following general procedures 14 and 15. To a solution of N-allyl-1-[3-
(trifluoromethyl)phenyl]methanimine 508c (0.441 g, 2.07 mmol) in THF (21 mL)
at −78 °C was added diethyl zinc solution (1 M, 4.14 ml, 4.14 mmol) dropwise.
After 2 h vinyl magnesium bromide solution (1 M, 4.14 ml, 4.14 mmol) was
added dropwise and the reaction left to warm to room temperature overnight.
After which N-methyl-N-phenylcarbamoyl chloride (0.457 g, 3.32 mmol), NEt3 (0.46 mL, 3.32
mmol) and DMAP (cat.) were added in DCE (7 mL) and the mixture heated to reflux overnight.
Purification by flash column chromatography yielded the title compound 511d as a colourless
oil (0.372 g, 0.99 mmol, 48%). RF 0.54 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.53-7.40 (4H,
m, ArH), 7.39-7.33 (2H, m, ArH), 7.22-7.17 (1H, m, ArH), 7.15-7.10 (2H, m, ArH), 6.15 (1H, ddd, J
= 17.3, 10.1, 7.3, CHArCH=CH2), 5.50 (1H, d, J = 7.6, CHAr), 5.34 (1H, dt, J = 10.1, 1.3,
CHArCH=CHH), 5.29 (1H, ddt, J = 17.0, 10.7, 6.0, X of ABXYY’, NCH2CHX=CH2), 5.13 (1H, dt, J =
17.0, 1.6, CHArCH=CHH), 4.97-4.85 (2H, m, Y and Y’ of ABXYY’, NCH2CH=CHYHY’), 3.52 (1H, ddt, J
= 16.1, 5.7, 1.6, A of ABXYY’, NCHAHCH=CH2), 3.42 (1H, ddt, J = 16.4, 6.6, 1.6, B of ABXYY’,
NCHHBCH=CH2), 3.15-3.20 (3H, s, NMe); 13C NMR (125 MHz, CDCl3) 162.0 (C=O), 146.3 (4°
ArC), 141.2 (4° ArC), 134.7 (CHArCH=CH2), 134.4 (NCH2CH=CH2), 131.2 (ArC), 130.5 (q, JF = 32.5,
ArCCF3), 129.6 (ArC), 128.7 (ArC), 125.6 (ArC), 125.1 (ArC), 124.5 (q, JF = 3.7, ArC), 124.1 (q, JF =
272.5, ArCCF3), 124.0 (q, JF = 3.6, ArC), 119.3 (CHArCH=CH2), 116.9 (NCH2CH=CH2), 62.8 (CHAr),
49.0 (NCH2), 40.1 (NMe); IR max (film)/cm-1 2926 (C=CH2), 1653 (C=O), 1596 (C=C); MS (ES) m/z
375 (M + H+); HRMS m/z calcd 375.1679 for C21H22N2OF3 (M + H+) found 375.1677.

271
1-Allyl-3-methyl-3-phenyl-1-(1-phenylallyl)urea 511e
Following general procedures 15. To a solution of N-allyl-1-phenyl-prop-2-en-
1-amine 509a (0.582 g, 3.36 mmol) in DCE (11 mL) was added N-methyl-N-
phenylcarbamoyl chloride (0.741 g, 4.37 mmol), NEt3 (0.75 mL, 5.38 mmol)
and DMAP (cat.) and the mixture heated to reflux overnight. Purification by
flash column chromatography yielded the title compound 511e as a yellow oil (0.453 g, 1.48
mmol, 44%). RF 0.64 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.52-6.99 (10H, m, ArH), 6.07
(1H, ddd, J = 17.1, 10.4, 6.8, CHPhCH=CH2), 5.59 (1H, d, J = 6.8, CHPh), 5.38 (1H, ddt, J = 17.3,
10.4, 6.4, X of ABXYY’, NCH2CHX=CH2), 5.27-5.21 (1H, m, NCHPhCH=CHH), 5.06-4.98 (1H, m,
NCHPhCH=CHH), 4.96-4.83 (2H, m, Y and Y’ of ABXYY’, NCH2CH=CHYHY’), 3.55 (1H, ddt, J = 16.0,
5.7, 1.7, A of ABXYY’, NCHAHCH=CH2), 3.37 (1H, ddt, J = 16.0, 6.2, 1.5, B of ABXYY’,
NCHHBCH=CH2), 3.19 (3H, s, NMe); 13C NMR (75 MHz, CDCl3) 161.5 (C=O), 140.9 (4° ArC), 139.7
(4° ArC), 134.9 (NCHArCH=CH2), 134.7 (NCH2CH=CH2), 129.7 (ArC), 129.5 (ArC), 128.4 (ArC),
128.3 (ArC), 127.6 (ArC), 126.9 (ArC), 118.0 (NCHArCH=CH2), 116.4 (NCH2CH=CH2), 62.7
(NCHArCH=CH2), 48.3 (NCH2), 39.6 (NMe); IR max (film)/cm-1 1648 (C=O); MS (ES) m/z 329 (M +
Na+); HRMS m/z calcd 307.1810 for C19H22N3O (M + H+) found 307.1804.
1-Allyl-3-methyl-1-(1-phenylallyl)-3-(pyridin-2-yl)urea 511f
Following general procedures 15. To a solution of N-allyl-1-phenyl-prop-2-en-
1-amine 509a (0.498 g, 2.88 mmol) in DCE (10 mL) was added methyl(pyridin-
2-yl)carbamic chloride (0.638 g, 3.74 mmol), NEt3 (0.64 mL, 4.60 mmol) and
DMAP (cat.) and the mixture heated to reflux overnight. Purification by flash
column chromatography yielded the title compound 511f as a yellow oil (0.400 g, 1.30 mmol,
45%). RF 0.35 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 8.33 (1H, ddd, J = 4.9, 1.9, 0.8, PyH),
7.55 (1H, ddd, J = 9.2, 7.2, 1.9, PyH), 7.37-7.25 (5H, m, ArH), 6.94 (1H, dt, J = 8.3, 0.9, PyH), 6.88
(1H, ddd, J = 7.3, 4.9, 1.1, PyH), 6.16 (1H, ddd, J = 17.1, 10.4, 6.8, CHPhCH=CH2), 5.78 (1H, d, J =
6.8 Hz, CHPh), 5.58 (1H, ddt, J = 17.1, 10.4, 5.8, X of ABXYY’, NCH2CH=CH2), 5.42-5.20 (2H, m,
CHPhCH=CH2), 5.02-4.85 (2H, m, Y and Y’ of ABXYY’, NCH2CH=CH2), 3.70 (1H, ddt, J = 16.0, 5.8,
1.7, A of ABXYY’, NCHAHCH=CH2), 3.55 (1H, ddt, J = 16.2, 6.2, 1.5, B of ABXYY’, NCHHBCH=CH2),
3.28 (3H, s, NMe); 13C NMR (75 MHz, CDCl3) 161.4 (C=O), 157.0 (4° ArC), 148.1 (4° PyC), 139.2
(4° ArC), 137.4 (PyC), 135.1 (CHPhCH=CH2), 134.5 (NCH2CH=CH2), 128.4 (ArC), 128.0 (ArC), 127.5
(ArC), 118.6 (CHPhCH=CH2), 117.2 (PyC), 116.6 (NCH2CH=CH2), 113.8 (PyC), 63.4 (CHPh), 48.3
(NCH2), 35.8 (NMe); IR max (film)/cm-1 2977 (C=CH2), 1656 (C=O), 1589 (C=C); MS (ES) m/z 330
(M + Na+); HRMS m/z calcd 308.1758 for C19H22N3O (M + H+) found 308.1767.

272
1-Allyl-3-(2-methoxyphenyl)-3-methyl-1-(1-phenylallyl)urea 511g
Following general procedures 14 and 15. To a solution of N-allyl-1-phenyl-
methanimine 508a (0.200 g, 1.38 mmol) in THF (14 mL) at −78 °C was added
diethyl zinc solution (1 M, 2.76 ml, 2.76 mmol) dropwise. After 2 h vinyl
magnesium bromide solution (1 M, 2.76 ml, 2.76 mmol) was added dropwise
and the reaction left to warm to room temperature overnight. After which (2-
methoxyphenyl)(methyl)carbamic chloride (0.357 g, 1.79 mmol), NEt3 (0.31 mL, 2.20 mmol) and
DMAP (cat.) were added in DCE (5 mL) and the mixture heated to reflux overnight. Purification
by flash column chromatography yielded the title compound 511g as a brown oil (0.218 g, 0.65
mmol, 47%). RF 0.54 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.35-7.29 (4H, m, ArH), 7.18-
6.91 (5H, m, ArH), 6.07 (1H, ddd, J = 17.4, 10.4, 6.7, NCHPhCH=CH2), 5.63 (1H, d, J = 6.9,
NCHPhCH=CH2), 5.40 (1H, ddt, J = 16.4, 10.2, 6.3, X of ABXYY’, NCH2CHX=CH2), 5.25 (1H, dt, J =
10.4, 1.5, NCHPhCH=CHH), 5.04 (1H, dt, J = 17.4, 1.5, NCHPhCH=CHH), 4.94 (2H, dddd, J = 16.4,
10.4, 2.8, 1.5, Y and Y’ of ABXYY’, NCH2CH=CHYY’), 3.83 (3H, s, OMe) 3.51 (1H, ddt, J = 16.4, 5.9,
1.5, A of ABXYY’, NCHAHCH=CH2), 3.38 (1H, ddt, J = 16.4, 6.0, 1.5, B of ABXYY’, NCHHBCH=CH2),
3.16 (3H, s, NMe); 13C NMR (75 MHz, CDCl3) 161.7 (C=O), 157.3 (ArCOMe), 148.3 (4° ArC),
139.5 (4° ArC), 135.0 (NCHPhCH=CH2), 134.6 (NCH2CH=CH2), 128.6 (ArC), 127.8 (ArC), 127.2
(ArC), 125.8 (ArC), 125.7 (ArC), 118.4 (NCHPhCH=CH2), 116.7 (NCH2CH=CH2), 62.9
(NCHPhCH=CH2), 55.2 (OMe), 48.4 (NCH2), 39.5 (NMe); IR max (film)/cm-1 3024 (C=CH2), 2897
(C=CH2), 1634 (C=O); MS (ES) m/z 359 (M + Na+); HRMS m/z calcd 359.1838 for C21H24N2O2Na
(M + Na+) found 359.1845.
N-Methyl-N,2-diphenyl-2,5-dihydro-1H-pyrrole-1-carboxamide 494a
Following general procedure 16. To 1-allyl-3-methyl-3-phenyl-1-(1-
phenylallyl)urea 511e (0.453 g, 1.48 mmol) in CH2Cl2 (14 mL) was added
Grubbs 1st generation catalyst (0.058 g, 0.07 mmol). Purification by flash
column chromatography yielded the title compound 494a as a colourless oil
(0.375 g, 1.35 mmol, 91%). RF 0.44 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.38-7.24 (5H, m,
ArH), 7.22-7.16 (3H, m, ArH), 7.04-6.98 (2H, m, ArH), 5.69-5.60 (3H, m, CHPhCH=CH), 3.98 (1H,
dd, J = 15.4, 2.1, A of AB, NCHAH), 3.61 (1H, dd, J = 15.3, 5.5, B of AB, NCHHB), 3.15 (3H, s, NMe);
13C NMR (75 MHz, CDCl3) 159.3 (C=O), 145.9 (4° ArC), 141.9 (4° ArC), 130.4 (CHPhCH=CH),
129.3 (ArC), 128.3 (ArC), 127.3 (ArC), 126.9 (ArC), 125.2 (ArC), 125.0 (ArC), 124.1 (CHPhCH=CH),
68.7 (CHPh), 55.9 (NCH2), 39.4 (NMe); IR max (film)/cm-1 1646 (C=O), 1593 (C=C); MS (ES) m/z
279 (M + Na+); HRMS m/z calcd 279.1492 for C18H19N2O (M + H+) found 279.1479.

273
2-(4-Chlorophenyl)-N-methyl-N-phenyl-2,5-dihydro-1H-pyrrole-1-carboxamide 494b
Following general procedure 16. To 1-allyl-1-[1-(4-chlorophenyl)allyl]-3-
methyl-3-phenyl-urea 511c (0.157 g, 0.46 mmol) in CH2Cl2 (9 mL) was
added Grubbs 1st generation catalyst (0.019 g, 0.02 mmol). Purification by
flash column chromatography yielded the title compound 494b as a brown
oil (0.120 g, 0.38 mmol, 83%). RF 0.31 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.36-7.28 (4H,
m, ArH), 7.20-7.11 (3H, m, ArH), 7.02-6.97 (2H, m, ArH), 5.70-5.66 (1H, m, CHArCH=CH), 5.66-
5.59 (2H, m, CHArCH=CH), 3.97-3.90 (1H, m, NCHHCH=CH), 3.56-3.48 (1H, m, NCHHCH=CH),
3.15 (3H, s, NMe); 13C NMR (125 MHz, CDCl3) 159.3 (C=O), 145.8 (4° ArC), 140.5 (4° ArC), 133.0
(4° ArC), 129.9 (CHArCH=CH), 129.4 (ArC), 128.5 (ArC), 128.4 (ArC), 125.4 (ArC), 125.3 (ArC),
124.7 (CHArCH=CH), 68.1 (CHAr), 55.9 (NCH2), 39.5 (NMe); IR max (film)/cm-1 1716 (C=C), 1642
(C=O); MS (ES) m/z 335 (M + Na+); HRMS m/z calcd 313.1103 for C18H18N2O35Cl (M + H+) found
313.1103.
N-Methyl-2-phenyl-N-(pyridin-2-yl)-2,5-dihydro-1H-pyrrole-1-carboxamide 494c
Following general procedure 16. To 1-allyl-3-methyl-1-(1-phenylallyl)-3-
(pyridin-2-yl)urea 511f (0.375 g, 1.22 mmol) in CH2Cl2 (12 mL) was added
Grubbs 1st generation catalyst (0.050 g, 0.06 mmol). Purification by flash
column chromatography yielded the title compound 494c as a pale yellow oil
(0.243 g, 0.87 mmol, 71%). RF 0.13 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 8.37 (1H, ddd, J =
5.0, 2.2, 0.6, PyH), 7.55 (1H, ddd, J = 8.2, 7.3, 1.9, PyH), 7.37-7.31 (2H, m, ArH), 7.29-7.24 (1H,
m, ArH), 7.23-7.18 (2H, m, ArH), 6.92 (1H, ddd, J = 7.3, 5.0. 0.6, PyH), 6.77 (1H, d, J = 8.2, PyH),
5.85-5.79 (1H, m, CHPhCH=CH), 5.77-5.73 (1H, m, CHPhCH=CH), 5.66 (1H, brs, CHPh), 4.17 (1H,
dd, J = 15.1, 1.9, A of AB, NCHAH), 4.02 (1H, brd, J = 14.5, B of AB, NCHHB), 3.18 (3H, s, NMe); 13C
NMR (125 MHz, CDCl3) 158.3 (C=O), 156.6 (4° ArC), 148.0 (4° PyC), 141.0 (PyC), 137.8 (PyC),
130.4 (CHPhCH=CH), 128.5 (ArC), 127.7 (ArC), 127.0 (ArC), 124.4 (CHPhCH=CH), 117.6 (PyC),
115.3 (PyC), 68.6 (CHPh), 55.3 (NCH2), 35.5 (NMe); IR max (film)/cm-1 2901 (C=CH), 1638 (C=O),
1594 (C=C); MS (ES) m/z 302 (M + Na+); HRMS m/z calcd 302.1264 for C17H17N3ONa (M + Na+)
found 302.1272.

274
N-Methyl-N-phenyl-2-(3-(trifluoromethyl)phenyl)-2,5-dihydro-1H-pyrrole-1-carboxamide 494d
Following general procedure 16. To 1-allyl-3-methyl-3-phenyl-1-(1-(3-
(trifluoromethyl)phenyl)allyl)urea 511d (0.164 g, 0.44 mmol) in CH2Cl2 (9
mL) was added Grubbs 1st generation catalyst (0.018 g, 0.02 mmol).
Purification by flash column chromatography yielded the title compound
494d as a pale yellow oil (0.121 g, 0.35 mmol, 80%). RF 0.26 (2:1 PE:EtOAc); 1H NMR (500 MHz,
CDCl3) 7.56-7.52 (1H, m, ArH), 7.50-7.44 (2H, m, ArH), 7.39-7.32 (3H, m, ArH), 7.23-7.16 (1H,
m, ArH), 7.04-7.00 (2H, m, ArH), 5.81-5.74 (1H, m, CHAr), 5.67 (2H, ddq, J = 22.7, 6.3, 2.2,
CHArCH=CH), 3.97 (1H, dq, J = 15.1, 2.2, NCHH), 3.54-3.46 (1H, m, NCHH), 3.15 (3H, s, NMe); 13C
NMR (125 MHz, CDCl3) 159.4 (C=O), 145.8 (4° ArC), 143.2 (4° ArC) 130.7 (ArC), 130.7 (q, J =
32.3, ArCCF3), 129.7 (ArC), 129.5 (CHArCH=CH), 128.9 (ArC), 125.5 (ArC), 125.5 (ArC), 125.1
(CHArCH=CH), 124.2 (q, J = 3.6, ArC), 124.0 (q, J = 271.6, ArCCF3), 123.6 (q, J = 3.6, ArC), 68.4
(CHAr), 56.0 (NCH2), 39.6 (NMe); IR max (film)/cm-1 1650 (C=O); MS (ES) m/z 347 (M + H+);
HRMS m/z calcd 347.1366 for C19H18N2OF3 (M + H+) found 347.1354.
N-(4-Chlorophenyl)-N-methyl-5-phenyl-2,3-dihydropyrrole-1-carboxamide 341a
Following general procedures 16 and 17. To 1-allyl-3-(4-chlorophenyl)-3-
methyl-1-(1-phenylallyl)urea 511c (0.150 g, 0.44 mmol) in CH2Cl2 (9 mL) was
added Grubbs 1st generation catalyst (0.018 g, 0.02 mmol). After which
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.042 g, 0.04
mmol) was added in THF (5 mL) and the mixture heated to reflux overnight.
Purification by flash column chromatography yielded the title compound 341a as a yellow oil
(0.123 g, 0.39 mmol, 89%). RF 0.22 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.37-7.25 (4H, m,
ArH), 7.19-7.06 (5H, m, ArH), 5.04 (1H, t, J = 3.8, C=CHCH2), 3.34 (2H, t, J = 8.4, NCH2CH2), 3.11
(NMe), 2.40 (2H, m, NCH2CH2); 13C NMR (75 MHz, CDCl3) 157.6 (C=O), 144.9 (4° ArC), 135.5
(CPh=CH), 134.8 (4° ArC), 129.4 (4° ArC), 128.3 (ArC), 127.9 (ArC), 127.5 (ArC), 127.4 (ArC),
126.2 (ArC), 116.1 (CPh=CHCH2), 39.8 (NMe), 39.3 (NCH2CH2), 29.8 (NCH2CH2); IR max (film)/cm-
1 3018 (C=CH), 1646 (C=O); MS (ES) m/z 335 (M + Na+); HRMS m/z calcd 335.1029 for
C18H17N2O35ClNa (M + Na+) found 335.1036.

275
5-(4-Chlorophenyl)-N-methyl-N-phenyl-2,3-dihydropyrrole-1-carboxamide 341b
Following general procedures 16 and 17. To 1-allyl-1-[1-(4-
chlorophenyl)allyl]-3-methyl-3-phenyl-urea 511e (0.150 g, 0.44 mmol) in
CH2Cl2 (9 mL) was added Grubbs 1st generation catalyst (0.018 g, 0.02
mmol). After which
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.042 g, 0.04 mmol) was added in
THF (5 mL) and the mixture heated to reflux overnight. Purification by flash column
chromatography yielded the title compound 341b as a colourless oil (0.127 g, 0.36 mmol, 82%).
RF 0.21 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.31-7.24 (4H, m, ArH), 7.16-7.06 (5H, m,
ArH), 5.07 (1H, t, J = 3.9, C=CHCH2), 3.34 (2H, t, J = 8.5, NCH2CH2), 3.12 (NMe), 2.35 (2H, m,
NCH2CH2); 13C NMR (75 MHz, CDCl3) 157.4 (C=O), 144.8 (4° ArC), 135.3 (CAr=CH), 134.4 (4°
ArC), 129.3 (4° ArC), 128.4 (ArC), 128.0 (ArC), 127.7 (ArC), 127.3 (ArC), 126.1 (ArC), 115.9
(CAr=CHCH2), 39.7 (NMe), 39.2 (NCH2CH2), 30.0 (NCH2CH2); IR max (film)/cm-1 3019 (C=CH),
1657 (C=O); MS (ES) m/z 335 (M + Na+); HRMS m/z calcd 335.1029 for C18H17N2O35ClNa (M +
Na+) found 335.1023.
N-(2-Methoxyphenyl)-N-methyl-5-phenyl-2,3-dihydropyrrole-1-carboxamide 341f
Following general procedures 16 and 17. To 1-allyl-3-(2-methoxyphenyl)-3-
methyl-1-(1-phenylallyl)urea 511i (0.150 g, 0.45 mmol) in CH2Cl2 (9 mL) was
added Grubbs 1st generation catalyst (0.018 g, 0.02 mmol). After which
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.042 g, 0.04
mmol) was added in THF (5 mL) and the mixture heated to reflux overnight. Purification by flash
column chromatography yielded the title compound 341f as a brown oil (0.109 g, 0.35 mmol,
79%). RF 0.18 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.25-7.18 (4H, m, ArH), 7.11-6.91 (5H,
m, ArH), 4.99 (1H, t, J = 4.1, C=CHCH2), 3.79 (3H, s, OMe), 3.29 (2H, t, J = 8.6, NCH2CH2), 3.08
(NMe), 2.43 (2H, m, NCH2CH2); 13C NMR (75 MHz, CDCl3) 160.1 (C=O), 156.8 (ArCOMe), 148.1
(4° ArC), 139.6 (4° ArC), 135.2 (CPh=CH), 128.6 (ArC), 127.9 (ArC), 126.8 (ArC), 125.8 (ArC), 125.6
(ArC), 116.1 (CPh=CHCH2), 55.4 (OMe), 39.5 (NMe), 39.2 (NCH2CH2), 29.7 (NCH2CH2); IR max
(film)/cm-1 3020 (C=CH), 1639 (C=O); MS (ES) m/z 331 (M + Na+); HRMS m/z calcd 331.1525 for
C19H20N2O2Na (M + Na+) found 331.1518.

276
2-(4-Chlorophenyl)-N-methyl-N-phenyl-2,3-dihydro-1H-pyrrole-1-carboxamide 512
Following general procedure 17. To 2-(4-chlorophenyl)-N-methyl-N-phenyl-
2,5-dihydro-1H-pyrrole-1-carboxamide 494b (0.310 g, 0.99 mmol) in THF
(10 mL) was added
carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (0.094 g, 0.10
mmol) and the mixture heated to reflux overnight. Purification by flash column chromatography
yielded the title compound 512 as a brown oil (0.053 g, 0.17 mmol, 17%). RF 0.21 (2:1
PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.38-7.28 (4H, m, ArH), 7.22-7.17 (1H, m, ArH), 7.15-7.10
(2H, m, ArH), 7.02-6.97 (2H, m, ArH), 6.32 (1H, dt, J = 4.4, 2.2, NCH=CH), 4.85 (1H, dd, J = 11.0,
5.4, CHAr), 4.81 (1H, dt, J = 4.4, 2.8, NCH=CH), 3.11 (3H, s, NMe), 3.05-2.97 (1H, m, CHArCHH),
2.39-2.32 (1H, m, CHArCHH); 13C NMR (125 MHz, CDCl3) 156.4 (C=O), 145.4 (4° ArC), 142.5 (4°
ArC), 132.9 (4° ArC), 131.5 (NCH=CH), 129.5 (ArC), 128.7 (ArC), 127.3 (ArC), 125.4 (ArC), 124.9
(ArC), 105.0 (NCH=CH), 61.0 (CHAr), 39.3 (CHArCH2), 39.2 (NMe); IR max (film)/cm-1 1645 (C=O),
1594 (C=C); MS (ES) m/z 313 (M + H+); HRMS m/z calcd 313.1103 for C18H18N2OCl (M + H+) found
313.1097.
N-Methyl-5-phenyl-2,3-dihydropyrrole-1-carboxamide 514
Following general procedure 11. To N-methyl-N-phenyl-2,3-dihydropyrrole-1-
carboxamide 501 (0.10 g, 0.49 mmol) in THF (5 mL) at −78 °C was added LDA
(1.24 mmol, prepared by general procedure 10) and DMPU (1.25 mL).
Purification by flash column chromatography yielded the title compound 514 as a pale yellow
oil (0.065 g, 0.32 mmol, 65%). RF 0.13 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.37-7.29 (2H,
m, ArH), 7.15-7.06 (3H, m, ArH), 5.57 (1H, t, J = 3.7, C=CHCH2), 3.30 (2H, t, J = 8.7, NCH2CH2),
2.44 (2H, m, NCH2CH2), 2.25 (3H, d, J = 4.7, NMe); 13C NMR (75 MHz, CDCl3) 157.8 (C=O), 135.6
(CPh=CH), 134.9 (4° ArC), 128.4 (ArC), 127.7 (ArC), 126.2 (ArC), 116.3 (CPh=CHCH2), 39.8
(NCH2CH2), 29.8 (NCH2CH2), 29.3 (NMe); IR max (film)/cm-1 3254 (NH), 3030 (C=CH), 1651 (C=O);
MS (ES) m/z 225 (M + Na+); HRMS m/z calcd 225.1106 for C12H14N2ONa (M + Na+) found
225.1100.

277
3-[(1E)-Buta-1,3-dienyl]-1-methyl-1-phenyl-urea 515
Following general procedure 11. To N-methyl-N-phenyl-2,3-dihydropyrrole-1-
carboxamide 501 (0.10 g, 0.49 mmol) in THF (5 mL) at −78 °C was added LDA
(1.24 mmol, prepared by general procedure 10) and DMPU (1.25 mL). Purification
by flash column chromatography yielded the title compound 515 as a colourless
oil (0.014 g, 0.07 mmol, 14%). RF 0.45 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.53-7.47 (2H,
m, ArH), 7.43-7.37 (1H, m, ArH), 7.33-7.28 (2H, m, ArH), 6.67 (1H, td, J = 10.3, 0.8, NHCH=CH),
6.44 (1H, d, J = 10.6, NH), 5.82, (1H, dtd, J = 16.7, 10.3, 1.0, CH=CH2), 5.19 (1H, ddd, J = 11.1, 9.1,
0.5, NHCH=CH), 5.05-4.99 (1H, m, CH=CHH) 4.86-4.81 (1H, m, CH=CHH), 3.333 (3H, s, NMe); 13C
NMR (100 MHz, CDCl3) 153.2 (C=O), 142.2 (4° ArC), 130.4 (ArC), 128.8 (CH=CH2), 128.1 (ArC),
127.3 (ArC), 123.8 (NHCH=CH), 114.8 (CH=CH2), 106.8 (NHCH=CH), 37.2 (NMe); IR max
(film)/cm-1 3332 (NH), 3043 (C=CH), 2914 (C=CH2), 1685 (C=O); MS (ES) m/z 225 (M + Na+);
HRMS m/z calcd 225.1106 for C12H14N2ONa (M + Na+) found 225.1099.
2-(4-Chlorophenyl)-N-methyl-2-phenyl-2,5-dihydro-1H-pyrrole-1-carboxamide 495a
Following general procedure 11. To 2-(4-chlorophenyl)-N-methyl-N-phenyl-
2,5-dihydro-1H-pyrrole-1-carboxamide 494b (0.080 g, 0.26 mmol) in THF (2.6
mL) at −78 °C was added LDA (0.64 mmol, prepared by general procedure 10)
and DMPU (0.65 mL). Purification by flash column chromatography yielded the
title compound 495a as a pale yellow oil (0.065 g, 0.21 mmol, 81%). RF 0.12 (1:1 PE:EtOAc); 1H
NMR (300 MHz, CDCl3) 7.42-7.25 (9H, m, ArH), 5.96-5.83 (2H, m, NCH2CH=CH), 4.51 (2H, brs,
NCH2), 3.99 (1H, brs, NH), 2.56 (3H, d, J = 5.1, NMe); 13C NMR (75 MHz, CDCl3) 156.4 (C=O),
141.0 (4° ArC), 139.8 (4° ArC), 136.9 (NCH2CH=CH), 133.4 (4° ArC), 129.3 (ArC), 128.6 (ArC),
128.5 (ArC), 128.4 (ArC), 127.7 (ArC), 122.7 (NCH2CH=CH), 54.5 (NCH2), 27.1 (NMe) + 1 missing
4° C; IR max (film)/cm-1 3330 (NH), 1719 (C=C), 1643 (C=O); MS (ES) m/z 335 (M + Na+); HRMS
m/z calcd 313.1103 for C18H18N2O35Cl (M + H+) found 313.1102.

278
N-Methyl-2,2-diphenyl-2,5-dihydro-1H-pyrrole-1-carboxamide 495b
Following general procedure 11. To N-methyl-N,2-diphenyl-2,5-dihydro-1H-
pyrrole-1-carboxamide 494a (0.043 g, 0.15 mmol) in THF (1.5 mL) at −78 °C was
added LDA (0.39 mmol, prepared by general procedure 10) and DMPU (0.38
mL). Purification by flash column chromatography yielded the title compound
495b as a pale yellow oil (0.015 g, 0.05 mmol, 35%). RF 0.13 (2:1 PE:EtOAc); 1H NMR (400 MHz,
DMSO) 7.31-7.26 (4H, m, ArH), 7.24-7.18 (6H, m, ArH), 6.04 (1H, brs, NH), 5.98 (1H, dt, J = 6.6,
1.5, 1H, NCH2CH=CH), 5.93 (1H, dt, J = 7.1, 1.8, NCH2CH=CH), 4.28 (2H, t, J = 2.0, NCH2), 2.46
(3H, d, J = 4.5, NMe); 13C NMR (100 MHz, DMSO) 155.4 (C=O), 142.8 (4° ArC), 137.1
(NCH2CH=CH), 128.0 (ArC), 127.4 (ArC), 126.3 (ArC), 122.2 (NCH2CH=CH), 77.6 (4° C), 53.7
(NCH2), 26.9 (NMe); IR max (film)/cm-1 3339 (NH), 1648 (C=O), 1538 (C=C); MS (ES) m/z 301 (M
+ Na+); HRMS m/z calcd 301.1317 for C18H18N2ONa (M + Na+) found 301.1324.
N-Methyl-2-phenyl-2-(3-(trifluoromethyl)phenyl)-2,5-dihydro-1H-pyrrole-1-carboxamide 495c
Following general procedure 11. To N-methyl-N-phenyl-2-(3-
(trifluoromethyl)phenyl)-2,5-dihydro-1H-pyrrole-1-carboxamide 494d
(0.049 g, 0.14 mmol) in THF (1.4 mL) at −78 °C was added LDA (0.35 mmol,
prepared by general procedure 10) and DMPU (0.35 mL). Purification by
flash column chromatography yielded the title compound 495c as a pale yellow oil (0.027 g,
0.08 mmol, 55%). RF 0.28 (1:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.62-7.55 (3H, m, ArH),
7.53-7.46 (1H, m, ArH), 7.43-7.37 (2H, m, ArH), 7.36-7.29 (3H, m, ArH), 5.97 (1H, dt, J = 6.0, 2.2,
NCH2CH=CH), 5.89 (1H, dt, J = 6.3, 2.2, NCH2CH=CH), 4.52 (2H, brs, NCH2), 4.02 (1H, brs, NH),
2.58 (3H, brs, NMe); 13C NMR (125 MHz, CDCl3) 156.3 (C=O), 142.5 (4° ArC), 140.8 (4° ArC),
136.7 (CH2CH=CH), 134.2 (d, J = 31.8, ArCCF3), 131.5 (ArC), 128.6 (ArC), 127.8 (ArC), 127.6 (ArC),
125.1 (ArC), 124.6 (m, ArC), 124.2 (m, ArC), 122.9 (CH2CH=CH), 54.5 (NCH2), 27.2 (NMe) + 1
missing ArCCF3 + 1 4° C; IR max (film)/cm-1 3340 (NH), 1644 (C=O), 1535 (C=C); MS (ES) m/z 369
(M + Na+); HRMS m/z calcd 369.1185 for C19H17N2OF3Na (M + Na+) found 369.1182.

279
N-Methylcyclohex-2-enamine195 540
Using the method of Steinhardt and Vanderwal;195 To a solution of methylamine (8M in
MeOH, 10.8 mL, 86.49 mmol) was added 3-bromo-1-cyclohexene 539 (1.00 mL, 8.65
mmol) and the mixture stirred overnight at room temperature. The reaction was
quenched with NaOH solution (0.7 M, 35 mL), extracted with CH2Cl2 (3 x 10 mL), washed with
brine (10 mL), dried over MgSO4 and the solvent removed under reduced pressure to yield the
title compound 540 which was used without any further purification as a colourless oil (0.840 g,
7.56 mmol, 87%). 1H NMR (400 MHz, CDCl3) 5.82-5.65 (2H, m, CH=CH), 3.13-3.06 (1H, m,
CHNH), 2.07-1.68 (4H, m, CHCH2CH2CH2), 1.61-1.40 (2H, m, CHCH2CH2CH2); 13C NMR (100 MHz,
CDCl3) 129.9 (CH=CHCH2), 128.6 (CH=CHCH2), 54.7 (CHNH), 33.6 (NMe), 29.2 (CHCH2), 25.5
(CH=CHCH2), 20.3 (CHCH2CH2); IR max (film)/cm-1 3300 (NH); MS (ES) m/z 134 (M + Na+). Data
corresponds to the reported literature.195
1-(Cyclohex-2-en-1-yl)-1,3-dimethyl-3-phenylurea 541a
Following general procedures 22 and 8. To N-methylcyclohex-2-enamine 540
(1.263 g, 11.36 mmol) in CH2Cl2 (57 mL) was added NEt3 (2.38 mL, 17.04 mmol)
and phenyl isocyanate (1.23 mL, 11.36 mmol) and the mixture stirred overnight
at room temperature. NaH (0.909 g, 22.72 mmol) and MeI (1.42 mL, 22.72
mmol) were added in DMF (57 mL) at 0 °C. Purification by flash column chromatography yielded
the title compound 541a as a pale yellow oil (1.912 g, 7.83 mmol, 69%). RF 0.44 (2:1 PE:EtOAc);
1H NMR (400 MHz, CDCl3) 7.34-7.27 (2H, m, ArH), 7.12-7.06 (3H, m, ArH), 5.83-5.75 (1H, m,
NCHCH=CH), 5.37-5.31 (1H, m, NCHCH=CH), 4.76-4.68 (1H, m, NCHCH=CH), 3.19 (3H, s, NMe),
2.38 (3H, s, NMe), 1.96-1.89 (2H, m, CH=CHCH2), 1.78-1.68 (2H, m, NCHCHHCHH), 1.62-1.38
(2H, m, NCHCHHCHH); 13C NMR (100 MHz, CDCl3) 162.0 (C=O), 147.1 (4° ArC), 130.9
(NCHCH=CH), 129.3 (ArC), 128.6 (NCHCH=CH), 124.2 (ArC), 123.9 (ArC), 53.5 (NCH), 39.8 (NMe),
31.1 (NMe), 26.0 NCHCH2), 24.5 (CH=CHCH2), 21.4 (NCHCH2CH2); IR max (film)/cm-1 2928
(C=CH), 1640 (C=O), 1594 (C=C); MS (ES) m/z 245 (M + H+); HRMS m/z calcd 245.1649 for
C15H21N2O (M + H+) found 245.1644.

280
1-(Cyclohex-2-en-1-yl)-1,3-dimethyl-3-(naphthalen-1-yl)urea 541b
Following general procedures 22 and 8. To N-methylcyclohex-2-enamine 540
(1.106 g, 9.95 mmol) in CH2Cl2 (50 mL) was added NEt3 (2.08 mL, 14.92
mmol) and 1-naphthyl isocyanate (1.43 mL, 9.95 mmol) and the mixture
stirred overnight at room temperature. NaH (0.796 g, 19.90 mmol) and MeI
(1.24 mL, 19.90 mmol) were added in DMF (50 mL) at 0 °C. Purification by flash column
chromatography yielded the title compound 541b as a brown oil (2.476 g, 8.41 mmol, 85%). RF
0.41 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 8.07-8.01 (1H, m, ArH), 7.90-7.85 (1H, m, ArH),
7.73 (1H, d, J = 8.3, ArH), 7.59-7.48 (2H, m, ArH), 7.41 (1H, dd, J = 8.3, 7.3, ArH), 7.23 (1H, dd, J =
7.6, 1.0, ArH), 5.75-5.67 (1H, m, NCHCH=CH), 5.27-5.20 (1H, m, NCHCH=CH), 4.76-4.69 (1H, m,
NCH), 3.22 (3H, s, NMe), 2.20 (3H, s, NMe), 1.89-1.81 (2H, m, CH=CHCH2), 1.68-1.52 (2H, m,
CHCHHCHH), 1.47-1.24 (2H, m, CHCHHCHH); 13C NMR (100 MHz, CDCl3) 163.1 (C=O), 143.5 (4°
ArC), 134.7 (4° ArC), 130.7 (NCHCH=CH), 129.5 (4° ArC), 128.7 (NCHCH=CH), 128.5 (ArC), 126.6
(ArC), 126.2 (ArC), 126.1 (ArC), 125.8 (ArC), 123.2 (ArC), 122.7 (ArC), 53.4 (NCH), 40.2 (NMe),
30.7 (NMe), 25.9 (NCHCH2), 24.4 (CH=CHCH2), 21.3 (NCHCH2CH2); IR max (film)/cm-1 2928
(C=CH), 1639 (C=O); MS (ES) m/z 295 (M + H+); HRMS m/z calcd 295.1805 for C19H23N2O (M + H+)
found 295.1802.
1-(Cyclohex-2-en-1-yl)-3-(3-methoxyphenyl)-1,3-dimethylurea 541c
Following general procedures 22 and 8. To N-methylcyclohex-2-enamine
540 (0.571 g, 5.14 mmol) in CH2Cl2 (26 mL) was added NEt3 (1.07 mL, 7.70
mmol) and 3-methoxyphenyl isocyanate (0.67 mL, 5.14 mmol) and the
mixture stirred overnight at room temperature. NaH (0.411 g, 10.27
mmol) and MeI (0.64 mL, 10.27 mmol) were added in DMF (26 mL) at 0 °C. Purification by flash
column chromatography yielded the title compound 541c as a yellow oil (0.729 g, 2.66 mmol,
52%). RF 0.32 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.22 (1H, t, J = 7.8, ArH), 6.73-6.62
(3H, m, ArH), 5.85-5.78 (1H, m, NCHCH=CH), 5.42-5.34 (1H, m, NCHCH=CH), 4.81-4.72 (1H, m,
NCHCH=CH), 3.79 (3H, s, OMe), 3.20 (3H, s, NMe), 2.42 (3H, s, NMe), 1.99-1.92 (2H, m,
CH=CHCH2), 1.82-1.72 (2H, m, NCHCHHCHH), 1.66-1.41 (2H, m, NCHCHHCHH); 13C NMR (100
MHz, CDCl3) 161.9 (C=O), 160.4 (4° ArCOMe), 148.3 (4° ArC), 131.0 (NCHCH=CH), 130.0 (ArC),
128.6 (NCHCH=CH), 116.1 (ArC), 109.6 (ArC), 109.6 (ArC), 55.3 (OMe), 53.4 (NCH), 39.7 (NMe),
31.1 (NMe), 26.0 (NCHCH2), 24.5 (CH=CHCH2), 21.5 (NCHCH2CH2); IR max (film)/cm-1 2929
(C=CH), 1641 (C=O), 1596 (C=C); MS (ES) m/z 275 (M + H+); HRMS m/z calcd 275.1755 for
C16H23N2O2 (M + H+) found 275.1749.

281
1-(4-Chlorophenyl)-3-(cyclohex-2-en-1-yl)-1,3-dimethylurea 541d
Following general procedures 22 and 8. To N-methylcyclohex-2-enamine 540
(0.840 g, 7.56mmol) in CH2Cl2 (38 mL) was added NEt3 (1.58 mL, 11.33 mmol)
and 4-chlorophenyl isocyanate (1.160 g, 7.56 mmol) and the mixture stirred
overnight at room temperature. NaH (0.604 g, 15.11 mmol) and MeI (0.94
mL, 15.11 mmol) were added in DMF (38 mL) at 0 °C. Purification by flash column
chromatography yielded the title compound 541d as a white solid (1.480 g, 5.31 mmol, 70%).
MP 77-79 °C; RF 0.44 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.23-7.19 (2H, m, ArH), 6.97-
6.93 (2H, m, ArH), 5.78-5.73 (1H, m, NCHCH=CH), 5.30-5.25 (1H, m, NCHCH=CH), 4.67-4.60 (1H,
m, NCHCH=CH), 3.10 (3H, s, NMe), 2.34 (3H, s, NMe), 1.92-1.86 (2H, m, CH=CHCH2), 1.72-1.65
(2H, m, NCHCHHCHH), 1.57-1.48 (1H, m, NCHCH2CHH), 1.44-1.35 (1H, m, NCHCHHCH2); 13C NMR
(125 MHz, CDCl3) 161.7 (C=O), 145.7 (4° ArC), 131.2 (NCHCH=CH), 129.4 (ArC), 129.3 (4° ArC),
128.3 (NCHCH=CH), 124.8 (ArC), 53.5 (NCH), 39.6 (NMe), 31.2 (NMe), 26.0 (NCHCH2), 24.5
(CH=CHCH2), 21.4 (NCHCH2CH2); IR max (film)/cm-1 2927 (C=CH), 1642 (C=O); MS (ES) m/z 301
(M + Na+); HRMS m/z calcd 279.1259 for C15H20N2O35Cl (M + H+) found 279.1255.
1-(Cyclohex-2-en-1-yl)-3-(3-fluorophenyl)-1,3-dimethylurea 541e
Following general procedures 22 and 8. To N-methylcyclohex-2-enamine 540
(0.690 g, 6.21 mmol) in CH2Cl2 (31 mL) was added NEt3 (1.30 mL, 9.31 mmol)
and 3-fluorophenyl isocyanate (0.70 mL, 6.21 mmol) and the mixture stirred
overnight at room temperature. NaH (0.496 g, 12.41 mmol) and MeI (0.77 mL,
12.41 mmol) were added in DMF (31 mL) at 0 °C. Purification by flash column chromatography
yielded the title compound 541e as a yellow oil (1.320 g, 5.03 mmol, 81%). RF 0.41 (2:1
PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.30-7.23 (1H, m, ArH), 6.89-6.75 (3H, m, ArH), 5.88-5.81
(1H, m, NCHCH=CH), 5.42-5.35 (1H, m, NCHCH=CH), 4.78-4.69 (1H, m, NCH), 3.21 (3H, s, NMe),
2.47 (3H, s, NMe), 2.01-1.92 (2H, m, CH=CHCH2), 1.84-1.72 (2H, m, NCHCHHCHH), 1.68-1.43
(2H, m, NCHCHHCHH); 13C NMR (100 MHz, CDCl3) 163.4 (d, JF = 244.7, ArCF), 161.5 (C=O),
148.7 (d, JF = 9.7, 4° ArC), 131.3 (NCHCH=CH), 130.4 (d, JF = 9.4, ArC), 128.3 (NCHCH=CH), 118.5
(d, JF = 2.9, ArC), 110.6 (d, JF = 21.1, ArC), 100.2 (d, JF = 23.3, ArC), 53.6 (NCH), 39.3 (NMe), 31.1
(NMe), 26.1 (NCHCH2), 24.6 (CH=CHCH2), 21.5 (NCHCH2CH2); IR max (film)/cm-1 1659 (C=O),
1606 (C=C), 1587 (C=C); MS (ES) m/z 263 (M + H+); HRMS m/z calcd 263.1560 for C15H20N2OF (M
+ H+) found 263.1568.

282
1-(Cyclohex-2-en-1-yl)-3-(3,5-difluorophenyl)-1,3-dimethylurea 541f
Following general procedures 22 and 8. To N-methylcyclohex-2-enamine 540
(0.640 g, 5.76 mmol) in CH2Cl2 (29 mL) was added NEt3 (1.61 mL, 11.51
mmol) and 3,5-difluorophenyl isocyanate (0.82 mL, 5.76 mmol) and the
mixture stirred overnight at room temperature. NaH (0.461 g, 11.52 mmol)
and MeI (0.72 mL, 11.52 mmol) were added in DMF (29 mL) at 0 °C. Purification by flash column
chromatography yielded the title compound 541f as a brown oil (1.252 g, 4.47 mmol, 78%). RF
0.39 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 6.62-6.43 (3H, m, ArH), 5.92-5.85 (1H, m,
NCHCH=CH), 5.45-5.39 (1H, m, NCHCH=CH), 4.79-4.69 (1H, m, NCH), 3.19 (3H, s, NMe), 2.55
(3H, s, NMe), 2.03-1.94 (2H, m, CH=CHCH2), 1.86-1.76 (2H, m, NCHCHHCHH), 1.70-1.50 (2H, m,
NCHCHHCHH); IR max (film)/cm-1 2934 (C=CH), 1614 (C=O), 1593 (C=C); MS (ES) m/z 281 (M +
H+); HRMS m/z calcd 281.1465 for C15H19N2OF2 (M + H+) found 281.1467. Broad signals in 13C
NMR prevented full analysis.
1-(4'-Chloro-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-yl)-1,3-dimethylurea 545a
Following general procedure 23. To 1-(4-chlorophenyl)-3-(cyclohex-2-en-1-yl)-
1,3-dimethylurea 541d (0.125 g, 0.45 mmol) in THF (4.5 mL) at −78 °C was
added s-BuLi (1 M, 1.12 mL, 1.12 mmol) dropwise. The mixture was stirred for
2 h and DMPU (1.1 mL) was added dropwise and the mixture stirred until TLC
analysis showed complete consumption of starting material. Purification by flash column
chromatography yielded the title compound 545a as a yellow oil (0.089 g, 0.32 mmol, 71%). RF
0.24 (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.35-7.28 (4H, m, ArH), 5.93 (1H, dt, J = 10.1,
3.5, CCH=CH), 5.88 (1H, dt, J = 10.1, 2.2, CCH=CH), 4.27 (1H, q, J = 4.1, NH), 3.03 (3H, s, NMe),
2.65 (3H, d, J = 4.7, NHMe), 2.48 (1H, ddd, J = 12.9, 9.1, 3.2, CCHHCH2), 2.20-2.02 (3H, m,
CCHHCH2CH2), 1.74-1.56 (2H, m, CCH2CH2CH2); 13C NMR (125 MHz, CDCl3) 159.0 (C=O), 145.7
(4° ArC), 132.5 (4° ArC), 131.5 (CCH=CH), 129.8 (CCH=CH), 128.7 (ArC), 127.5 (ArC), 62.2 (4° C),
33.2 (NMe), 32.9 (CCH2), 27.5 (NHMe), 24.9 (CH=CHCH2), 19.0 (CCH2CH2); IR max (film)/cm-1
3376 (NH), 2929 (C=CH), 2856 (CH), 1619 (C=O); MS (ES) m/z 301 (M + Na+); HRMS m/z calcd
279.1259 for C15H20N2O35Cl (M + H+) found 279.1260.

283
1,3-Dimethyl-3'H-spiro[cyclohex-2''-en-1''-yl]-4'H-spiro[imidazolidine-4,1'-naphthalene]-2,4'-
dione 545b
Following a modification of general procedure 23. To 1-(cyclohex-2-en-1-yl)-
1,3-dimethyl-3-(naphthalen-1-yl)urea 541b (0.113 g, 0.38 mmol) in THF (3.8
mL) at −78 °C was added s-BuLi (1 M, 0.96 mL, 0.96 mmol) dropwise and the
mixture stirred for 2 h. DMPU (0.95 mL) was added and the mixture stirred for
2 h. Compressed air was bubbled through the mixture for 30 min. Purification by flash column
chromatography yielded the title compound 545b as a brown oil (0.018 g, 0.06 mmol, 15%). RF
0.16 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 8.18 (1H, dd, J = 7.6, 1.8, ArH), 7.60-7.55 (1H,
m, ArH), 7.53-7.48 (1H, m, ArH), 7.39 (1H, dd, J = 7.8, 1.5, ArH), 7.03 (1H, d, J = 10.3,
CH=CHC=O), 6.54 (1H, d, J = 10.3, CH=CHC=O), 6.09 (1H, dt, J = 10.1, 3.8, CCH=CHCH2), 5.62 (1H,
dt, J = 10.3, 2.0, CCH=CHCH2), 2.78 (3H, s, NMe), 2.63 (3H, s, NMe), 1.98-1.76 (2H, m,
CH=CHCH2), 1.40-1.08 (4H, m, CCH2CH2); 13C NMR (100 MHz, CDCl3) 183.9 (CHC=O), 160.8
(NMeC=O), 150.7 (CCH=CHC=O), 140.0 (4° ArC), 134.2 (CCH=CH), 132.7 (4° ArC), 132.6 (ArC),
131.4 (CCH=CHC=O), 128.7 (ArC), 127.3 (ArC), 127.2 (ArC), 126.9 (CCH=CH), 66.9 (4° C), 66.1 (4°
C), 28.6 (CCH2), 27.7 (NMe), 27.0 (NMe), 24.2 (CH=CHCH2), 18.7 (CCH2CH2); IR max (film)/cm-1
1705 (C=O), 1668 (C=O); MS (ES) m/z 309 (M + H+); HRMS m/z calcd 309.1598 for C19H21N2O2 (M
+ H+) found 309.1596.

284
1-(3'-Methoxy-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-yl)-1,3-dimethylurea 545c
Following general procedure 23. To 1-(cyclohex-2-en-1-yl)-3-(3-
methoxyphenyl)-1,3-dimethylurea 541c (0.080 g, 0.29 mmol) in THF (2.9 mL) at
−78 °C was added s-BuLi (1 M, 0.73 mL, 0.73 mmol) dropwise. The mixture was
stirred for 2 h and DMPU (0.73 mL) was added dropwise and the mixture
stirred until TLC analysis showed complete consumption of starting material. Purification by
flash column chromatography yielded the title compound 545c as a colourless oil (0.027 g, 0.10
mmol, 34%). RF 0.17 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.20 (1H, t, J = 7.8, ArH), 6.95-
6.85 (2H, m, ArH), 6.75-6.69 (1H, m, ArH), 5.86-5.77 (2H, m, CCH=CHCH2), 4.22 (1H, q, J = 4.5,
NH), 3.73 (3H, s, OMe), 2.99 (3H, s, NMe), 2.52 (3H, d, J = 4.5, NHMe), 2.35 (1H, ddd, J = 12.9,
9.3, 3.3, CCHHCH2), 2.16-1.93 (3H, m, CCHHCH2CH2), 1.68-1.50 (2H, m, CCH2CH2); 13C NMR (100
MHz, CDCl3) 159.8 (C=O), 159.3 (4° ArCOMe), 148.4 (4° ArC), 131.5 (CCH=CH), 129.7 (ArC),
129.5 (CCH=CH), 118.5 (ArC), 112.5 (ArC), 111.7 (ArC), 62.3 (4° C), 55.2 (OMe), 33.1 (NMe), 32.8
(CCH2CH2), 27.5 (NHMe), 24.8 (CH=CHCH2), 19.0 (CCH2CH2); IR max (film)/cm-1 3372 (NH), 2928
(C=CH), 1623 (C=O); MS (ES) m/z 275 (M + H+); HRMS m/z calcd 275.1755 for C16H23N2O2 (M +
H+) found 275.1756.
1-(3'-Fluoro-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-yl)-1,3-dimethylurea 545d
Following general procedure 23. To 1-(3-fluorophenyl)-3-(cyclohex-2-en-1-yl)-
1,3-dimethylurea 541e (0.050 g, 0.19 mmol) in THF (1.9 mL) at −78 °C was
added s-BuLi (1.3 M, 0.37 mL, 0.48 mmol) dropwise. The mixture was stirred
for 2 h and DMPU (0.48 mL) was added dropwise and the mixture stirred until
TLC analysis showed complete consumption of starting material. Purification by flash column
chromatography yielded the title compound 545d as a pale yellow oil (0.038 g, 0.15 mmol,
76%). RF 0.18 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.30 (1H, td, J = 8.0, 6.1, ArH), 7.17
(1H, brd, J = 8.0, ArH), 7.09 (1H, dt, J = 10.6, 2.2, ArC), 6.92 (1H, td, J = 8.3, 2.4, ArC), 5.96-5.85
(2H, m, CCH=CH), 4.27 (1H, m, NH), 3.04 (3H, s, NMe), 2.64 (3H, d, J = 4.6, NHMe), 2.49 (1H,
ddd, J = 12.8, 9.0, 3.2, CCHHCH2), 2.21-2.02 (3H, m, CCHHCH2CH2), 1.76-1.57 (2H, m,
CCH2CH2CH2); 13C NMR (100 MHz, CDCl3) 163.1 (d, JF = 244.3, ArCF), 159.0 (C=O), 150.1 (d, JF =
6.2, 4° ArC), 131.5 (CCH=CH), 130.0 (d, JF = 8.2, ArC). 129.9 (CCH=CH), 121.6 (d, JF = 2.9, ArC),
113.7 (d, JF = 21.1, ArC), 113.2 (d, JF = 22.4, ArC), 62.4 (d, JF = 1.7, 4° C), 33.2 (NMe), 33.0
(CCH2CH2), 27.5 (NHMe), 24.9 (CH=CHCH2), 19.0 (CCH2CH2); IR max (film)/cm-1 3508 (NH), 2939
(C=CH), 1630 (C=O); MS (ES) m/z 263 (M + H+); HRMS m/z calcd 263.1560 for C15H20N2OF (M +
H+) found 263.1568.

285
1-(3',5'-Difluoro-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-yl)-1,3-dimethylurea 545e
Following general procedure 23. To 1-(cyclohex-2-en-1-yl)-3-(3,5-
difluorophenyl)-1,3-dimethylurea 541f (1.152 g, 4.11 mmol) in THF (41.1
mL) at −78 °C was added s-BuLi (1.3 M, 7.90 mL, 10.27 mmol) dropwise. The
mixture was stirred for 2 h and DMPU (10.28 mL) was added dropwise and
the mixture stirred until TLC analysis showed complete consumption of starting material.
Purification by flash column chromatography yielded the title compound 545e as a brown oil
(0.809 g, 2.89 mmol, 70%). RF 0.23 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 6.90-6.84 (2H, m,
ArH), 6.62 (1H, tt, J = 8.7, 2.3, ArH), 5.98-5.92 (1H, m, CCH=CH), 5.87-5.82 (1H, m, CCH=CH),
4.43-4.35 (1H, m, NH), 3.02 (3H, s, NMe), 2.67 (3H, d, J = 4.6, NHMe), 2.50 (2H, ddd, J = 12.5,
8.2, 3.6, CCH2), 2.13-2.05 (2H, m, CH=CHCH2) 1.71-1.59 (2H, m, CCH2CH2); 13C NMR (100 MHz,
CDCl3) 163.1 (d, JF = 246.1, ArCF), 163.0 (d, JF = 245.9, ArCF), 158.7 (C=O), 152.4, (t, JF = 7.8, 4°
ArC), 131.0 (CCH=CH), 130.4 (CCH=CH), 108.8, (d, JF = 25.7, ArC), 108.8 (d, JF = 11.8, ArC), 101.8
(t, JF = 25.6, ArC), 62.6 (t, JF = 2.0, 4° C), 33.1 (NMe), 33.1 (CCH2CH2), 27.4 (NHMe), 24.8
(CH=CHCH2), 18.9 (CCH2CH2); IR max (film)/cm-1 2935 (C=CH), 1642 (C=O); MS (ES) m/z 281 (M +
H+); HRMS m/z calcd 281.1465 for C15H19N2OF2 (M + H+) found 281.1470.
1,3-Dimethyl-1-(1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-yl)urea 545f
Following general procedure 23. To 1-(cyclohex-2-en-1-yl)-1,3-dimethyl-3-
phenylurea 541a (0.600 g, 2.46 mmol) in THF (25 mL) at −78 °C was added s-
BuLi (1.3 M, 4.72 mL, 6.14 mmol) dropwise. The mixture was stirred for 2 h
and DMPU (6.25 mL) was added dropwise and the mixture stirred until TLC analysis showed
complete consumption of starting material. Purification by flash column chromatography
yielded the title compound 545f as a brown oil (0.087 g, 0.36 mmol, 15%). RF 0.21 (1:1
PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.43-7.39 (2H, m, ArH), 7.38-7.32 (2H, m, ArH), 7.28-7.22
(1H, m, ArH), 5.95-5.87 (2H, m, CCH=CH), 4.28-4.19 (1H, brs, NH), 3.06 (3H, s, NMe), 2.59 (3H, d,
J = 4.6, NHMe), 2.46 (1H, ddd, J = 12.8, 9.3, 3.1, CCHH), 2.22 (1H, ddd, J = 13.0, 8.3, 3.0, CCHH),
2.15-2.01 (2H, m, CH=CHCH2), 1.78-1.58 (2H, m, CCH2CH2); 13C NMR (100 MHz, CDCl3) 159.3
(C=O), 146.7 (4° ArC), 131.9 (CCH=CH), 129.3 (CCH=CH), 128.8 (ArC), 127.0 (ArC), 126.2 (ArC),
62.4 (4° C), 33.2 (NMe), 32.9 (CCH2CH2), 27.5 (NHMe), 24.9 (CH=CHCH2), 19.1 (CCH2CH2); IR max
(film)/cm-1 2932 (C=CH), 1681 (C=O); MS (ES) m/z 281 (M + H+); HRMS m/z calcd 281.1465 for
C15H19N2OF2 (M + H+) found 281.1470.

286
N-Methyl-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-amine 548a
Following general procedure 4. To 1,3-dimethyl-1-(1,2,3,4-tetrahydro-[1,1'-
biphenyl]-1-yl)urea 545f (0.233 g, 0.95 mmol) in EtOH (2 mL) was added 2 M
NaOH solution (2 mL) and the mixture heated to 130 °C under microwave
irradiation for 2.5 h. Work up yielded the title compound 548a as a pale yellow oil (0.156 g, 0.83
mmol, 88%). RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.51-7.45 (2H, m, ArH), 7.37-
7.31 (2H, m, ArH), 7.26-7.20 (1H, m, ArH), 6.04-5.93 (2H, m, CCH=CH), 2.23 (3H, s, NMe), 2.10-
2.02 (2H, m, CH=CHCH2), 1.91-1.77 (2H, m, CCH2CH2), 1.68-1.58 (1H, m, CCH2CHH), 1.50-1.40
(1H, m, CCH2CHH); 13C NMR (100 MHz, CDCl3) 146.2 (4° ArC), 130.9 (CCH=CH), 129.2
(CCH=CH), 127.9 (ArC), 127.1 (ArC), 126.3 (ArC), 58.9 (4° C), 37.9 (CCH2CH2), 29.1 (NMe), 25.4
(CH=CHCH2), 19.1 (CCH2CH2); IR max (film)/cm-1 3335 (NH), 2927 (C=CH), 1622 (C=C); MS (ES)
m/z 188 (M + H+); HRMS m/z calcd 188.1434 for C13H18N (M + H+) found 188.1432.
3'-Methoxy-N-methyl-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-amine 548b
Following general procedure 4. To 1-(3'-methoxy-1,2,3,4-tetrahydro-[1,1'-
biphenyl]-1-yl)-1,3-dimethylurea 545c (0.050 g, 0.18 mmol) in EtOH (1 mL) was
added 2 M NaOH solution (1 mL) and the mixture heated to 130 °C under
microwave irradiation for 2.5 h. Work up yielded the title compound 548b as a
colourless oil (0.036 g, 0.17 mmol, 92%). RF Flame (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3)
7.28-7.23 (1H, m, ArH), 7.09-7.04 (2H, m, ArH), 6.80-6.77 (1H, m, ArH), 6.02 (1H, dt, J = 10.1,
3.8, CCH=CHCH2), 5.97-5.93 (1H, m, CCH=CHCH2), 3.82 (3H, s, OMe), 2.25 (3H, s, NMe), 2.11-
2.02 (2H, m, CCH=CHCH2), 1.94-1.80 (2H, m, CCH2CH2), 1.67-1.59 (1H, m, CCH2CHH), 1.49-1.39
(1H, m, CCH2CHH); 13C NMR (125 MHz, CDCl3) 159.5 (4° ArCOMe), 147.5 (4° ArC), 130.2
(CCH=CHCH2), 129.8 (CCH=CHCH2), 128.9 (ArC), 119.7 (ArC), 113.2 (ArC), 111.7 (ArC), 59.2 (4° C),
55.2 (OMe), 37.6 (CCH2CH2), 29.0 (NMe), 25.3 (CCH=CHCH2), 19.0 (CCH2CH2); IR max (film)/cm-1
2932 (C=CH), 1599 (C=C); MS (ES) m/z 218 (M + H+); HRMS m/z calcd 218.1540 for C14H20NO (M
+ H+) found 218.1546.

287
4'-Chloro-N-methyl-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-amine 548c
Following general procedure 4. To 1-(4'-chloro-1,2,3,4-tetrahydro-[1,1'-
biphenyl]-1-yl)-1,3-dimethylurea 545a (0.850 g, 3.05 mmol) in EtOH (3 mL) was
added 2 M NaOH solution (3 mL) and the mixture heated to 130 °C under
microwave irradiation for 2.5 h. Work up yielded the title compound 548c as a
yellow oil (0.635 g, 2.87 mmol, 94%). RF Flame (2:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.44-
7.39 (2H, m, ArH), 7.32-7.28 (2H, m, ArH), 6.02 (1H, dt, J = 10.4, 3.8, CCH=CH), 5.95-5.89 (1H, m,
CCH=CH), 2.22 (3H, s, NMe), 2.09-2.03 (2H, m, CCH=CHCH2), 1.85-1.79 (2H, m, CCH2), 1.66-1.58
(1H, m, CCH2CHH), 1.45-1.35 (1H, m, CCH2CHH); 13C NMR (125 MHz, CDCl3) 144.8 (4° ArC),
132.2 (4° ArC), 130.3 (CCH=CH), 129.8 (CCH=CH), 128.8 (ArC), 128.1 (ArC), 58.7 (4° C), 37.9
(CCH2), 29.0 (NMe), 25.3 (CCH2CH2CH2), 19.0 (CCH2CH2); IR max (film)/cm-1 2932 (C=CH), 1764
(C=C); MS (ES) m/z 222 (M + H+); HRMS m/z calcd 222.1045 for C13H17N35Cl (M + H+) found
222.1042.
3'-Fluoro-N-methyl-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-amine 548d
Following general procedure 4. To 1-(3'-fluoro-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-
yl)-1,3-dimethylurea 545d (0.275 g, 1.05 mmol) in EtOH (1 mL) was added 2 M
NaOH solution (1 mL) and the mixture heated to 130 °C under microwave
irradiation for 2.5 h. Work up yielded the title compound 548d as a brown oil
(0.179 g, 0.87 mmol, 83%). RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.32-7.18 (3H,
m, ArH), 6.94-6.88 (1H, m, ArH), 6.05-5.90 (2H, m, CCH=CH), 2.23 (3H, s, NMe), 2.10-2.04 (2H,
m, CCH=CHCH2), 1.87-1.77 (2H, m, CCH2), 1.68-1.58 (1H, m, CCH2CHH), 1.48-1.39 (1H, m,
CCH2CHH); 13C NMR (100 MHz, CDCl3) 162.9 (d, JF = 243.0, ArCF), 149.9 (d, JF = 6.2, 4° ArC),
130.5 (CCH=CH), 129.6 (CCH=CH), 129.2 (d, JF = 8.0, ArC), 122.8 (d, JF = 2.7, ArC), 114.3 (d, JF =
21.9, ArC), 113.1 (d, JF = 21.1, ArC), 58.8, (d, JF = 1.7, 4° C), 37.9 (CCH2CH2), 29.1 (NMe), 25.3
(CH=CHCH2), 19.0 (CCH2CH2); IR max (film)/cm-1 2937 (C=CH), 1612 (C=O), 1585 (C=C); MS (ES)
m/z 206 (M + H+); HRMS m/z calcd 206.1345 for C13H17NF (M + H+) found 206.1351.

288
3',5'-Difluoro-N-methyl-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-amine 548e
Following general procedure 4. To 1-(3',5'-difluoro-1,2,3,4-tetrahydro-[1,1'-
biphenyl]-1-yl)-1,3-dimethylurea 545e (0.200 g, 0.71 mmol) in EtOH (2 mL) was
added 2 M NaOH solution (2 mL) and the mixture heated to 130 °C under
microwave irradiation for 2.5 h. Work up yielded the title compound 548e as a
yellow oil (0.139 g, 0.62 mmol, 87%). RF Flame (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.05-
6.98 (2H, m, ArH), 6.65 (1H, tt, J = 8.7, 2.4, ArH), 6.05-5.85 (2H, m, CCH=CH), 2.21 (3H, s, NMe),
2.09-2.03 (2H, m, CCH=CHCH2), 1.80-1.75 (2H, m, CCH2), 1.66-1.56 (1H, m, CCH2CHH), 1.48-1.36
(1H, m, CCH2CHH); 13C NMR (100 MHz, CDCl3) 163.0 (d, JF = 245.3, ArCF), 162.9 (d, JF = 245.3,
ArCF), 151.4 (t, JF = 7.7, 4° ArC), 130.2 (CCH=CH), 129.9 (CCH=CH), 110.2 (d, JF = 25.2, ArC), 110.2
(d, JF = 11.9, ArC), 101.8 (t, JF = 25.6, ArC), 58.9 (4° C), 38.0 (CCH2CH2), 29.1 (NMe), 25.3
(CH=CHCH2), 19.0 (CCH2CH2); IR max (film)/cm-1 2939 (C=CH), 1621 (C=C), 1594 (C=C); MS (ES)
m/z 224 (M + H+); HRMS m/z calcd 224.1251 for C13H16NF2 (M + H+) found 224.1248.
3a-(3,5-Difluorophenyl)-7-iodo-1,3-dimethylhexahydro-1H-benzo[d]imidazol-2(3H)-one 561
Following general procedure 25. To 1-(3',5'-difluoro-1,2,3,4-tetrahydro-[1,1'-
biphenyl]-1-yl)-1,3-dimethylurea 545e (0.362 g, 1.29 mmol) in CHCl3 (13 mL)
was added NIS (0.436 g, 1.94 mmol). Work up yielded the title compound
561 as a dark brown oil (0.450 g, 1.11 mmol, 86%). 1H NMR (400 MHz, CDCl3)
6.94-6.79 (3H, m, ArC), 4.72 (1H, d, J = 5.5, CHCHI), 4.30 (1H, dt, J = 4.9, 9.7, CHI), 3.13 (3H, s,
NMe), 2.83 (3H, s, NMe), 2.41 (1H, ddd, J = 13.9, 9.1, 4.3, CHHCArN), 2.27-2.17 (1H, m, CHHCHI),
2.12-1.79 (3H, m, CHICHHCHHCHH), 1.72-1.61 (1H, m, CHICH2CHH); IR max (film)/cm-1 1705
(C=O); MS (ES) m/z 429 (M + Na+); HRMS m/z calcd 407.0432 for C15H18N2OF2I (M + H+) found
407.0432. Broad signals in 13C NMR prevented full analysis.

289
tert-Butyl (4'-chloro-1,2,3,4-tetrahydro-[1,1'-biphenyl]-1-yl)(methyl)carbamate 563
Following general procedure 24. To 4'-chloro-N-methyl-1,2,3,4-
tetrahydro-[1,1'-biphenyl]-1-amine 548c (0.255 g, 1.15 mmol) in CH2Cl2
(12 mL) was added NEt3 (0.32 mL, 2.30 mmol) and di-tert-butyl
dicarbonate (0.276 g, 1.27 mmol). Purification by flash column
chromatography yielded the title compound 563 as a pale yellow oil (0.352 g, 1.09 mmol, 96%).
RF 0.61 (2:1 PE:EtOAc); 1H NMR (300 MHz, CDCl3) 7.31-7.15 (4H, m, ArH), 5.94-5.80 (2H, m,
CCH=CH), 3.12 (3H, s, NMe), 2.41 (1H, ddd, J = 13.0, 9.2, 3.4, NCHH), 2.17-1.97 (3H, m,
CCHHCH2CH2), 1.74-1.50 (2H, m, CCH2CH2), 1.15 (9H, s, C(CH3)3); 13C NMR (125 MHz, CDCl3)
155.6 (C=O), 147.6 (4° ArC), 146.7 (4° ArC), 131.3 (CH=CH), 129.5 (CH=CH), 127.9 (ArC), 127.0
(ArC), 68.1 (4° C), 62.1 (4° C), 33.8 (CCH2), 33.1 (NMe), 28.2 (C(CH3)3), 24.9 (CCH2CH2CH2), 18.9
(CCH2CH2); IR max (film)/cm-1 2933 (C=CH), 1720 (C=O), 1690 (C=C); MS (ES) m/z 344 (M + Na+);
HRMS m/z calcd 344.1388 for C18H24NO235ClNa (M + Na+) found 344.1373.
3a-(4-Chlorophenyl)-7-iodo-3-methylhexahydrobenzo[d]oxazol-2(3H)-one 564
Following general procedure 25. To tert-butyl (4'-chloro-1,2,3,4-tetrahydro-
[1,1'-biphenyl]-1-yl)(methyl)carbamate 563 (0.212 g, 0.66 mmol) in CHCl3 (7
mL) was added NIS (0.222 g, 0.99 mmol). Work up yielded the title compound
564 as a dark brown oil (0.230 g, 0.59 mmol, 89%). 1H NMR (400 MHz, CDCl3)
7.42-7.37 (2H, m, ArH), 7.34-7.29 (2H, m, ArH), 4.71 (1H, d, J = 4.8, CHCHI), 4.36 (1H, dt, J = 7.0,
4.5, CHI), 2.66 (3H, s, NMe), 2.64-2.56 (1H, m, CHHCArN), 2.26-2.16 (1H, m, CHHCHI), 2.11-1.83
(3H, m, CHICHHCHHCHH), 1.73-1.62 (1H, m, CHICH2CHH); 13C NMR (100 MHz, CDCl3) 157.0
(C=O), 139.1 (4° ArC), 134.4 (4° ArC), 129.2 (ArC), 127.6 (ArC), 85.2 (CHCHI), 64.5 (4°C), 30.6
(CH2CHI), 28.4 (CH2CArN), 26.5 (NMe), 23.6 (CHI), 19.7 (CH2CH2CHI); IR max (film)/cm-1 1704
(C=O); MS (ES) m/z 391 (M + H+); HRMS m/z calcd 391.9914 for C45H16NO235ClI (M + H+) found
391.9921.

290
1-(4-Fluorophenyl)-3-((1S*,2S*)-2-methoxycyclohexyl)-1-methylurea 576a
Following a modification to general procedures 22 and 8. To trans-2-
aminocyclohexanol hydrochloride (1.083 g, 7.14 mmol) in CH2Cl2 (36 mL)
was added NEt3 (2.99 mL, 10.71 mmol) and 4-fluorophenyl isocyanate
(0.88 mL, 7.86 mmol) and the mixture stirred overnight at room
temperature. NaH (0.857 g, 21.43 mmol) and MeI (1.33 mL, 21.43 mmol) were added in DMF
(36 mL) at 0 °C. Purification by flash column chromatography yielded the title compound 576a
as a colourless oil (0.910 g, 3.25 mmol, 43%). RF 0.16 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3)
7.31-7.19 (2H, m, ArH), 7.16-7.04 (2H, m, ArH), 4.33 (1H, d, J = 6.3, NH), 3.62-3.49 (1H, m,
CHNH), 3.25 (3H, s, OMe), 3.22 (3H, s, NMe), 2.85 (1H, td, J = 9.3, 4.3, CHOMe), 2.19-2.09 (1H,
m, CHHCHNH), 1.97-1.88 (1H, m, CHHCHOMe), 1.74-1.62 (1H, m, CHHCH2CHNH), 1.53-1.43 (1H,
m, CHHCH2CHOMe), 1.37-1.01 (4H, m, CHNHCHHCHHCHHCHHCHOMe); 13C NMR (100 MHz,
CDCl3) 161.2 (d, JF = 247.3, ArCF), 157.2 (C=O), 139.6 (d, JF = 2.8, 4° ArC), 129.1 (d, JF = 8.3,
ArC), 116.7 (d, JF = 22.2, ArC), 81.3 (CHOMe), 55.7 (OMe), 53.6 (CHNH), 37.1 (NMe), 31.6
(CH2CHOMe), 29.0 (CH2CHNH), 23.8 (CH2CH2CHOMe), 23.5 (CH2CH2CHNH); IR max (film)/cm-1
2930 (CH), 2857 (CH), 1651 (C=O), 1507 (C=C); MS (ES) m/z 281 (M + H+); HRMS m/z calcd
281.1660 for C15H22N2O2F (M + H+) found 281.1672.

291
1-(4-Chlorophenyl)-3-((1S*,2S*)-2-methoxycyclohexyl)-1-methylurea 576b
Following a modification of general procedures 22 and 8. To trans-2-
aminocyclohexanol hydrochloride (1.000 g, 6.59 mmol) in CH2Cl2 (33
mL) was added NEt3 (2.76 mL, 19.78 mmol) and 4-chlorophenyl
isocyanate (1.114 g, 7.25 mmol) and the mixture stirred overnight at
room temperature. NaH (0.791 g, 19.78 mmol) and MeI (1.23 mL, 19.78 mmol) were added in
DMF (33 mL) at 0 °C. Purification by flash column chromatography yielded the title compound
576b as a pale yellow oil (0.449 g, 1.51 mmol, 23%). RF 0.16 (2:1 PE:EtOAc); 1H NMR (400 MHz,
CDCl3) 7.42-7.34 (2H, m, ArH), 7.25-7.18 (2H, m, ArH), 4.40 (1H, d, J = 6.6 , NH), 3.62-3.51 (1H,
m, CHNH), 3.26 (3H, s, OMe), 3.23 (3H, s, NMe), 2.87 (1H, td, J = 9.3, 3.8, CHOMe), 2.20-2.08
(1H, m, CHHCHNH), 2.01-1.90 (1H, m, CHHCHOMe), 1.74-1.64 (1H, m, CHHCH2CHNH), 1.57-1.44
(1H, m, CHHCH2CHOMe), 1.37-0.98 (4H, m, CHNHCHHCHHCHHCHHCHOMe); 13C NMR (100
MHz, CDCl3) 156.9 (C=O), 142.3 (4° ArC), 132.3 (4° ArC), 129.9 (ArC), 128.4 (ArC), 81.3
(CHOMe), 55.6 (OMe), 53.7 (CHNH), 37.0 (NMe), 31.7 (CH2CHOMe), 29.0 (CH2CHNH), 23.9
(CH2CH2CHOMe), 23.6 (CH2CH2CHNH); IR max (film)/cm-1 3436 (NH), 3334 (NH), 2858 (CH), 1660
(C=O); MS (ES) m/z 297 (M + H+); HRMS m/z calcd 297.1365 for C15H22N2O235Cl (M + H+) found
297.1360.
3-((1S*,2S*)-2-Hydroxycyclohexyl)-1-methyl-1-phenylurea 577a
Following a modification of general procedure 15. To a solution of trans-2-
aminocyclohexanol hydrochloride (0.978 g, 6.45 mmol) in DCE (32 mL) was
added N-methyl-N-phenylcarbamoyl chloride (1.422 g, 8.38 mmol), NEt3
(2.25 mL, 16.12 mmol) and DMAP (cat.) and the mixture heated to reflux
overnight. Purification by flash column chromatography yielded the title compound 577a as a
pale green oil (1.259 g, 5.07 mmol, 79%). RF 0.11 (1:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3)
7.45-7.40 (2H, m, ArH), 7.34-7.29 (1H, m, ArH), 7.28-7.25 (2H, m, ArH), 4.29 (1H, brs, NH), 3.89
(1H, brs, OH), 3.46 (1H, ddd, J = 12.3, 8.5, 3.2, CHNH), 3.27 (3H, s, NMe), 3.17 (1H, ddd, J = 10.4,
6.0, 0.7, CHOH), 2.04-1.97 (1H, m, CHHCHOH), 1.79-1.73 (1H, m, CHHCHNH), 1.70-1.58 (2H, m,
CHHCH2CHNH and CHHCH2CHOH), 1.34-1.19 (2H, m, CHHCHOH and CHHCH2CHNH) 1.12 (1H, qt,
J = 13.1, 3.5, CHHCH2CHOH), 0.97 (1h, qd, J = 12.7, 4.1, CHHCHNH); 13C NMR (125 MHz, CDCl3)
158.8 (C=O), 143.0 (4° ArC), 130.1 (ArC), 127.5 (ArC), 127.1 (ArC), 76.2 (CHOH), 56.6 (CHNH),
37.4 (NMe), 34.3 (CH2CHOH), 31.6 (CH2CHNH), 24.7 (CH2CH2CHNH), 23.9 (CH2CH2CHOH); IR max
(film)/cm-1 3353 (OH), 2930 (CH), 1639 (C=O); MS (ES) m/z 271 (M + Na+); HRMS m/z calcd
271.1417 for C14H20N2O2Na (M + Na+) found 271.1418.

292
1-(4-Chlorophenyl)-3-((1S*,2S*)-2-hydroxycyclohexyl)-1-methylurea 577b
Following a modification to general procedure 15. To a solution of
trans-2-aminocyclohexanol hydrochloride (1.765 g, 11.64 mmol) in DCE
(40 mL) was added (4-chlorophenyl)(methyl)carbamic chloride (3.088 g,
15.13 mmol), NEt3 (4.06 mL, 29.10 mmol) and DMAP (cat.) and the
mixture heated to reflux overnight. Purification by flash column chromatography yielded the
title compound 577b as a pale yellow oil (2.978 g, 10.53 mmol, 90%). RF 0.15 (1:1 PE:EtOAc); 1H
NMR (400 MHz, CDCl3) 7.38-7.33 (2H, m, ArH), 7.21-7.17 (2H, m, ArH), 4.29 (1H, d, J = 6.8,
NH), 4.23 (1H, d, J = 4.0, OH), 3.48-3.37 (1H, m, CHNH), 3.20 (3H, s, NMe), 3.18-3.09 (1H, m,
CHOH), 2.01-1.92 (1H, m, CHHCHOH), 1.82-1.74 (1H, m, CHHCHNH), 1.67-1.54 (2H, m,
CHHCH2CHOH + CHHCH2CHNH), 1.32-0.91 (4H, m CHOHCHHCHHCHHCHHCHNH); 13C NMR (100
MHz, CDCl3) 158.3 (C=O), 141.6 (4° ArC), 132.8 (4° ArC), 130.1 (ArC), 128.3 (ArC), 75.7 (CHOH),
56.5 (CHNH), 37.3 (NMe), 34.3 (CH2CHOH), 31.6 (CH2CHNH), 24.6 (CH2CH2CHNH), 23.8
(CH2CH2CHOH); IR max (film)/cm-1 3352 (OH), 2931 (CH), 1642 (C=O), 1519 (C=C); MS (ES) m/z
283 (M + H+); HRMS m/z calcd 283.1208 for C14H20N2O235Cl (M + H+) found 283.1206.
1-((1S*,2S*)-2-Hydroxycyclohexyl)-1,3-dimethyl-3-phenylurea 578
Following general procedure 8. To a solution of 3-((1S,2S)-2-
hydroxycyclohexyl)-1-methyl-1-phenylurea 577 (1.108 g, 4.46 mmol) in
DMF (22 mL) was added NaH (0.357 g, 8.92 mmol) and MeI (0.56 mL, 8.92
mmol) at 0 °C. Purification by flash column chromatography yielded the
title compound 578 as a yellow oil (0.089 g, 0.34 mmol, 8%). RF 0.15 (1:1 PE:EtOAc); 1H NMR
(300 MHz, CDCl3) 7.39-7.30 (2H, m, ArH), 7.16-7.05 (3H, m, ArH), 3.89-3.79 (1H, m, CHNMe),
3.50 (1H, td, J = 10.4, 4.7), CHOH), 3.26 (3H, s, NMe), 2.67 (1H, brs, OH), 2.38 (3H, s, NMe), 2.19-
2.08 (1H, m, CHHCHOH), 1.75-1.58 (3H, m, CHHCHHCHHCHNMe), 1.44-1.08 (4H, m,
OHCHCHHCHHCHHCHHCHNMe); 13C NMR (75 MHz, CDCl3) 164.2 (C=O), 146.3 (4° ArC), 129.5
(ArC), 124.8 (ArC), 124.3 (ArC), 70.4 (CHOH), 61.8 (CHNMe), 39.7 (NMe), 35.3 (CH2CHOH), 31.4
(NMe), 28.6 (CH2CHNMe), 25.1 (CH2CH2CHNMe), 24.2 (CH2CH2CHOH); IR max (film)/cm-1 3389
(OH), 2928 (CH), 2856 (CH), 1623 (C=O); MS (ES) m/z 547 (2M + Na+); HRMS m/z calcd 285.1574
for C15H22N2O2Na (M + Na+) found 285.1569.

293
1-((1S*,2S*)-2-Methoxycyclohexyl)-1,3-dimethyl-3-phenylurea 579
Following a modification to general procedure 8. To 3-((1S,2S)-2-
hydroxycyclohexyl)-1-methyl-1-phenylurea 577 (0.464 g, 1.87 mmol) in
DMF (10 mL) was added NaH (0.112 g, 2.80 mmol) and MeI (0.17 mL, 2.80
mmol) at 0 °C. Purification by flash column chromatography yielded the
title compound 579 as a colourless oil (0.138 g, 0.50 mmol, 27%). RF 0.29 (1:1 PE:EtOAc); 1H
NMR (300 MHz, CDCl3) 7.33=7.25 (2H, m, ArH), 7.20-7.13 (2H, m, ArH), 7.09-7.02 (1H, m, ArH),
3.82 (1H, td, J = 11.7, 4.0, CHNMe), 3.32 (3H, s, OMe), 3.20 (3H, s, NMe), 3.14-3.01 (3H, s,
CHOMe), 2.45 (3H, s, NMe), 2.23-2.14 (1H, m, CHHCHOMe), 1.76-1.54 (3H, m,
CHHCHHCHHCHNMe), 1.44-1.04 (4H, m, CHHCHHCHHCHHCHNMe); 13C NMR (75 MHz, CDCl3)
162.1 (C=O), 147.0 (4° ArC), 129.0 (ArC), 123.7 (ArC), 123.6 (ArC), 78.8 (CHOMe), 60.4 (CHNMe),
55.5 (OMe), 39.5 (NMe), 30.9 (NMe), 30.2 CH2CHOMe), 28.1 (CH2CHNMe), 24.9
(CH2CH2CHNMe), 24.2 (CH2CH2CHOMe); IR max (film)/cm-1 2930 (CH), 1640 (C=O); MS (ES) m/z
575 (2M + Na+); HRMS m/z calcd 277.1911 for C16H25N2O2 (M + H+) found 277.1911.
1-Methyl-3-(2-oxocyclohexyl)-1-phenylurea 580
Following general procedure 28. To a solution of oxalyl chloride (0.29 mL,
3.41 mmol) in CH2Cl2 (15 mL) at −78 °C was added a solution of dimethyl
sulfoxide (0.31 mL, 4.34 mmol) in CH2Cl2 (2 mL). After five minutes a solution
of 3-((1S,2S)-2-hydroxycyclohexyl)-1-methyl-1-phenylurea 577 (0.385 g, 1.55
mmol) in CH2Cl2 (2 mL) was added and the mixture stirred for 1.5 h before NEt3 (1.08 mL, 7.75
mmol) was added dropwise. Purification by flash column chromatography yielded the title
compound 580 as a brown oil (0.235 g, 0.95 mmol, 62%). RF 0.50 (EtOAc); 1H NMR (300 MHz,
CDCl3) 7.48-7.39 (2H, m, ArH), 7.34-7.26 (3H, m, ArH), 5.37 (1H, d, J = 6.4, NH), 4.41 (1H, ddd, J
= 11.8, 6.0, 5.1, CHNH), 3.27 (3H, s, NMe), 2.63-2.51 (1H, m, CHHCHNH), 2.48-2.28 (2H, m,
CH2C=O), 2.14-2.01 (1H, m, CHHCH2C=O), 1.89-1.68 (2H, m, CH2CH2CHNH), 1.63-1.43 (1H, m,
CHHCH2C=O), 1.36-1.20 (CHHCH2C=O); 13C NMR (75 MHz, CDCl3) 208.3 (C=O ketone), 156.5
(C=O urea), 143.1 (4° ArC), 129.9 (ArC), 127.2 (ArC), 127.0 (ArC), 59.4 (CHNH)), 41.0 (CH2C=O),
37.0 (NMe), 36.2 (CH2CHNH), 27.8 (CH2CH2C=O), 24.1(CH2CH2CHNH); IR max (film)/cm-1 3401
(NH), 2937 (CH), 1713 (C=O), 1655 (C=O), 1494 (C=C); MS (ES) m/z 515 (2M + Na+); HRMS m/z
calcd 269.1261 for C14H18N2O2Na (M + Na+) found 269.1260.

294
3-((1S*,2S*)-2-((tert-Butyldimethylsilyl)oxy)cyclohexyl)-1-methyl-1-phenylurea 583a
Following general procedure 26. To 3-((1S,2S)-2-hydroxycyclohexyl)-1-
methyl-1-phenylurea 577a (1.000 g, 4.03 mmol) in CH2Cl2 (40 mL) was
added imidazole (0.302 g, 4.43 mmol), tert-butyldimethylsilyl chloride
(0.668 g, 4.43 mmol) and DMAP (0.049 g, 0.40 mmol). Purification by
flash column chromatography yielded the title compound 583a as a pale yellow oil (1.431 g,
3.95 mmol, 98%). RF 0.65 (1:1 PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.49-7.43 (2H, m, ArH),
7.35-7.29 (3H, m ArH), 4.43 (1H, d, J = 6.6, NH), 3.57-3.50 (1H, m, CHNH), 3.39 (1H, td, J = 8.5,
3.8, CHOTBS), 3.31 (3H, s, NMe), 2.25-2.17 (1H, m, CHHCHNH), 1.79-1.65 (2H, m,
CHOTBSCHHCHH), 1.48-1.10 (5H, m, CHOTBSCHHCHHCCH2CHH), 0.98 (9H, s, SiC(CH3)3), 0.00
(3H, s, SiMe), -0.09 (3H, s, SiMe); 13C NMR (125 MHz, CDCl3) 156.9 (C=O), 143.6 (4° ArC), 129.9
(ArC), 127.3 (ArC), 127.0 (ArC), 72.7 (CHOTBS), 55.4 (CHNH), 37.1 (NMe), 33.7 (CHOTBSCH2),
30.6 (CHNHCH2), 25.7 (SiC(CH3)3), 23.6 (CH2CH2CHNH), 23.3 (CH2CH2CHOTBS), 17.9 (SiC(CH3)3) , -
4.3 (SiMe), -5.0 (SiMe); IR max (film)/cm-1 2923 (CH), 1668 (C=O), 1645 (C=C); MS (ES) m/z 363
(M + H+); HRMS m/z calcd 385.2282 for C20H34N2O2SiNa (M + Na+) found 385.2287.
3-((1S*,2S*)-2-((tert-Butyldimethylsilyl)oxy)cyclohexyl)-1-(4-chlorophenyl)-1-methylurea 583b
Following general procedure 26. To 1-(4-chlorophenyl)-3-((1S,2S)-2-
hydroxycyclohexyl)-1-methylurea 577b (2.832 g, 10.02 mmol) in
CH2Cl2 (100 mL) was added imidazole (0.750 g, 11.02 mmol), tert-
butyldimethylsilyl chloride (1.660 g, 11.02 mmol) and DMAP (0.122 g,
1.00 mmol). Purification by flash column chromatography yielded the title compound 583b as a
white solid (3.310 g, 8.34 mmol, 83%). MP 107-109 °C; RF 0.62 (1:1 PE:EtOAc); 1H NMR (500
MHz, CDCl3) 7.38-7.32 (2H, m, ArH), 7.22-7.15 (2H, m, ArH), 4.32 (1H, d, J = 6.3, NH), 3.49-3.40
(1H, m, CHNH), 3.37-3.29 (1H, m, CHOTBS), 3.21 (3H, s, NMe), 2.20-2.10 (1H, m, CHHCHNH),
1.75-1.60 (2H, m, CHOTBSCHHCH2CHH), 1.46-1.00 (5H, m, CHNHCHHCHHCH2CHHCHOTBS)),
0.79 (9H, s, SiC(CH3)3), -0.05 (3H, s, SiMe), -0.14 (3H, s, SiMe); 13C NMR (125 MHz, CDCl3) 156.6
(C=O), 142.2 (4° ArC), 132.4 (4° ArC), 129.9 (ArC), 128.5 (ArC), 72.9 (CHOTBS), 55.6 (CHNH), 37.0
(NMe), 33.8 (CH2CHOTBS), 30.7 (CH2CHNH), 25.7 (SiC(CH3)3), 23.7 (CH2CH2CHNH), 23.4
(CH2CH2CHOTBS), 17.8 (SiC(CH3)3), -4.3 (SiMe), -5.0 (SiMe); IR max (film)/cm-1 3350 (NH), 2928
(CH), 2855 (CH), 1648 (C=O); MS (ES) m/z 419 (M + Na+); HRMS m/z calcd 419.1893 for
C20H33N2O2Si35ClNa (M + Na+) found 419.1885.

295
1-((1S*,2S*)-2-((tert-Butyldimethylsilyl)oxy)cyclohexyl)-1,3-dimethyl-3-phenylurea 584a
Following a modification to general procedure 8. To 3-((1S,2S)-2-((tert-
butyldimethylsilyl)oxy)cyclohexyl)-1-methyl-1-phenylurea 583a (1.549 g,
4.27 mmol) in DMF (21 mL) at 0 °C was added NaH (0.256 g, 6.41 mmol)
and MeI (0.40 mL, 6.41 mmol). Purification by flash column
chromatography yielded the title compound 584a as a white solid (0.318 g, 0.84 mmol, 20%).
MP 127-129 °C; RF 0.74 (1:2 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.37-7.21 (2H, m, ArH),
7.12-7.03 (3H, m, ArH), 3.77-3.67 (1H, m, CHNMe), 3.53-3.38 (1H, m, CHOTBS), 3.16 (3H, s,
NMe), 2.59 (3H, s, NMe), 1.97-1.89 (1H, m, CHOTBSCHH), 1.64-1.07 (7H, m,
CHOTBSCHHCH2CH2CH2CHNMe), 0.89 (9H, s, SiC(CH3)3), 0.06 (3H, s, SiMe), 0.04 (3H, s, SiMe); 13C
NMR (100 MHz, CDCl3) 162.0 (C=O), 147.5 (4° ArC), 129.2 (ArC), 123.8 (ArC), 123.7 (ArC), 71.1
(CHOTBS), 52.9 (CHNMe), 39.6 (NMe), 36.0 (NMe), 28.0 (CHOTBSCH2), 25.9 (SiC(CH3)3), 25.7
(CHNMeCH2), 25.2 (CH2CH2CHNMe), 24.5 (CH2CH2CHOTBS), 17.9 (SiC(CH3)3), -3.8 (SiMe), -4.4
(SiMe); IR max (film)/cm-1 2928 (CH), 1620 (C=O); MS (ES) m/z 399 (M + Na+); HRMS m/z calcd
377.2619 for C21H37N2O2Si (M + H+) found 377.2618.
1-((1S*,2S*)-2-((tert-Butyldimethylsilyl)oxy)cyclohexyl)-3-(4-chlorophenyl)-1,3-dimethylurea
584b
Following a modification to general procedure 8. To 3-((1S,2S)-2-
((tert-butyldimethylsilyl)oxy)cyclohexyl)-1-(4-chlorophenyl)-1-
methylurea 583b (3.17 g, 7.98 mmol) in DMF (40 mL) was added NaH
(0.798 g, 19.96 mmol) and MeI (0.75 mL, 11.98 mmol) at 0 °C.
Purification by flash column chromatography yielded the title compound 584b as a white solid
(1.632 g, 3.97 mmol, 50%). MP 122-124 °C; RF 0.65 (2:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3)
7.29-7.22 (2H, m, ArH), 7.04-6.97 (2H, m, ArH), 3.72 (1H, brs, CHNMe), 3.50-3.35 (1H, m,
CHOTBS), 3.13 (3H, s, NMe), 2.60 (3H, s, NMe), 1.98-1.90 (1H, m, CHHCHOTBS), 1.66-1.52 (2H,
m, CHHCHNMe + CHHCH2CHOTBS), 1.50-1.35 (2H, m, CHNMeCH2CH2), 1.32-0.93 (3H, m,
CHNMeCHHCH2CHHCHHCHOTBS), 0.87 (9H, s, SiC(CH3)3), 0.05 (3H, s, SiMe), 0.03 (3H, s, SiMe);
13C NMR (100 MHz, CDCl3) 161.7 (C=O), 145.9 (4° ArC), 129.2 (4° ArC), 128.7 (ArC), 124.3 (ArC),
71.1 (CHOTBS), 39.4 (NMe), 35.9 (CH2CHOTBS), 28.0 (CH2CHNMe), 25.8 (SiC(CH3)3), 25.2
(CH2CH2CHNMe), 24.4 (CH2CH2CHOTBS), 17.9 (SiC(CH3)3), -3.9 (SiMe), -4.4 (SiMe) + 2 missing
Carbons (CHNCH3); IR max (film)/cm-1 2929 (CH), 2856 (CH), 1619 (C=O), 1592 (C=C), 1486 (C=C);
MS (ES) m/z 433 (M + Na+); HRMS m/z calcd 411.2229 for C21H36N2O2Si35Cl (M + H+) found
411.2233.

296
1-(4-Chlorophenyl)-3-((1S*,2S*)-2-hydroxycyclohexyl)-1,3-dimethylurea 585b
Following general procedure 27. To 1-((1S,2S)-2-((tert-
butyldimethylsilyl)oxy)cyclohexyl)-3-(4-chlorophenyl)-1,3-dimethylurea
584b (1.309 g, 3.18 mmol) in THF (32 mL) at 0 °C was added TBAF (4.78
mL, 4.78 mmol). Purification by flash column chromatography yielded
the title compound 585b as a white solid (0.846 g, 2.85 mmol, 90%). MP 147-149 °C; RF 0.17
(1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.33-7.26 (2H, m, ArH), 7.09-7.02 (2H, m, ArH), 3.88-
3.77 (1H, m, CHNMe), 3.50 (1H, td, J = 10.6, 5.0, CHOH), 3.22 (3H, s, NMe), 2.41 (3H, s, NMe),
2.18-2.09 (1H, m, CHHCHOH), 1.75-1.58 (3H, m, CHNMeCHHCHHCHH), 1.42-1.09 (4H, m,
CHOHCHHCHHCHHCHHCHNMe); 13C NMR (100 MHz, CDCl3) 163.8 (C=O), 144.8 (4° ArC), 129.9
(4° ArC), 129.5 (ArC), 125.2 (ArC), 70.4 (CHOH), 61.7 (CHNMe), 39.5 (NMe), 35.3 (CH2CHOH),
31.5 (NMe), 28.6 (CH2CHNMe), 25.0 (CH2CH2CHNMe), 24.2 (CH2CH2CHOH); IR max (film)/cm-1
3391 (OH), 2931 (CH), 1620 (C=O), 1484 (C=C); MS (ES) m/z 297 (M + H+); HRMS m/z calcd
297.1364 for C15H22N2O235Cl (M + H+) found 297.1366.
1,3-Dimethyl-1-(2-oxocyclohexyl)-3-phenylurea 581a
Following general procedure 28. To a solution of oxalyl chloride (0.06 mL,
0.70 mmol) in CH2Cl2 (4 mL) at −78 °C was added a solution of dimethyl
sulfoxide (0.06 mL, 0.90 mmol) in CH2Cl2 (0.5 mL). After five minutes a
solution of 1-((1S,2S)-2-hydroxycyclohexyl)-1,3-dimethyl-3-phenylurea 585a
(0.084 g, 0.32 mmol) in CH2Cl2 (0.5 mL) was added and the mixture stirred for 1.5 h before NEt3
(0.22 mL, 1.60 mmol) was added dropwise. Purification by flash column chromatography
yielded the title compound 581a as a colourless oil (0.071 g, 0.27 mmol, 85%). RF 0.31 (1:1
PE:EtOAc); 1H NMR (500 MHz, CDCl3) 7.35-7.29 (2H, m, ArH), 7.17-7.08 (3H, m, ArH), 4.68 (1H,
dd, J = 12.0, 5.7, CHNMe), 3.21 (3H, s, NMe), 2.43 (3H, s, NMe), 2.43-2.40 (1H, m, COCHH), 2.29
(1H, td, J = 14.1, 6.6, COCHH), 2.11-2.00 (2H, m, CHCHHCHH), 1.98-1.92 (1H, m, COCH2CHH),
1.86-1.68 (2H, m, (CHCHHCHH), 1.63-1.52 (1H, m, COCH2CHH); 13C NMR (125 MHz, CDCl3)
207.1 (C=O ketone), 161.6 (C=O urea), 146.5 (4° ArC), 129.4 (ArC), 124.5 (ArC), 124.1 (ArC), 64.7
(CHNMe), 41.4 (COCH2), 39.9 (NMe), 33.7 (NMe), 31.0 (CHCH2), 26.5 (COCH2CH2), 24.6
(CHCH2CH2); IR max (film)/cm-1 2938 (CH), 1716 (C=O), 1685 (C=O), 1642 (C=C); MS (ES) m/z 283
(M + Na+); HRMS m/z calcd 283.1417 for C15H20N2O2Na (M + Na+) found 283.1415.

297
1-(4-Chlorophenyl)-1,3-dimethyl-3-(2-oxocyclohexyl)urea 581b
Following general procedure 28. To a solution of oxalyl chloride (0.50 mL,
5.93 mmol) in CH2Cl2 (30 mL) at −78 °C was added a solution of dimethyl
sulfoxide (0.54 mL, 7.55 mmol) in CH2Cl2 (3 mL). After five minutes a
solution of 1-(4-chlorophenyl)-3-((1S,2S)-2-hydroxycyclohexyl)-1,3-
dimethylurea 58b (0.800 g, 2.70 mmol) in CH2Cl2 (3 mL) was added and the mixture stirred for
1.5 h and NEt3 (1.88 mL, 13.48 mmol) added. Purification by flash column chromatography
yielded the title compound 581b as a white solid (0.605 g, 2.05 mmol, 76%). MP 119-121 °C; RF
0.16 (1:1 PE:EtOAc); 1H NMR (400 MHz, CDCl3) 7.32-7.26 (2H, m, ArH), 7.13-7.07 (2H, m, ArH),
4.67 (1H, dd, J = 11.9, 5.5, CHNMe), 3.19 (3H, s, NMe), 2.51-2.43 (1H, m, CHHC=O), 2.48 (3H, s,
NMe), 2.39-2.26 (1H, m, CHHC=O), 2.15-2.02 (2H, m, CHNMeCHHCHH), 2.02-1.93 (1H, m,
CHHCHHC=O), 1.92-1.68 (2H, m, CHNMeCHHCHH), 1.67-1.53 (1H, m, CHHCHHC=O); 13C NMR
(100 MHz, CDCl3) 206.7 (ketone C=O), 161.4 (urea C=O), 145.1 (4° ArC), 129.6 (4° ArC), 129.5
(ArC), 125.1 (ArC), 64.7 (CHNMe), 41.4 (CH2C=O), 39.8 (NMe), 33.8 (NMe), 30.9 (CH2CHNMe),
26.4 (CH2CH2C=O), 24.6 (CH2CH2CHNMe); IR max (film)/cm-1 2939 (CH), 1719 (C=O), 1682 (C=O),
1644 (C=C); MS (ES) m/z 295 (M + H+); HRMS m/z calcd 295.1208 for C15H20N2O235Cl (M + H+)
found 295.1208.

298
Chapter 9 References
1. Roy, S.; Spino, C. Org. Lett. 2006, 8, 939.
2. Carlier, P.R.; Zhoa, H.; DeGuzman, J.; Lam, P.C-H. J. Am. Chem. Soc. 2003, 125, 11482.
3. Mashiko, T.; Hara, K.; Tanaka, D.S.; Fujiwara, Y.; Kumagai, N.; Shibasakai, M. J. Am. Chem.
Soc. 2007, 129, 11342.
4. Singh, S. Chem. Rev. 2000, 100, 925.
5. Shaw, A.W.; deSolms, S.J., Tetrahedron Lett. 2001, 42, 7173.
6. Penning, T.D.; Zhu, G.-D.; Gandhi, V.B.; Gong, J.; Liu, X.; Shi, Y.; Klinghofer, V.; Johnson,
E.F.; Donawho, C.K.; Frost, D.J.; Bontcheva-Diaz, V.; Bouska, J.J.; Osterling, D.J.; Olson,
A.M.; Marsh, K.C.; Luo, Y.; Giranda, V.L. J. Med. Chem. 2009, 52, 514.
7. Harrison, T.; Williams, B.J.; Swain, C.J. Bioorg. Med. Chem. Lett. 1994, 4, 2733.
8. Yokum, T.S.; Gauthier, T.J.; Hammer, R.P.; McLaughlin, M.L. J. Am. Chem. Soc. 1997, 119,
1167.
9. Kobayashi, S.; Mori, Y.; Fossey, J.; Salter, M. Chem. Rev. 2011, 111, 2626.
10. Cogan, D.A.; Liu, G.; Ellman, J. Tetrahedron. 1999, 55, 8883.
11. Stork, G.; Dowd, S.R. J. Am. Chem. Soc. 1963, 85, 2178.
12. Brook, M.A.; Jahangir. Synth. Commun. 1988, 18, 893.
13. Ciganek, E. J. Org. Chem. 1992, 57, 4521.
14. Nugent, T. C.; Editor Chiral Amine Synthesis: Methods, Developments and Applications;
Wiley-VCH Verlag GmbH & Co. KGaA, 2010.
15. Davis, F.A.; Reddy, R.E.; Szewozyk, J.M.; Venkat, R.G.; Portonovo, P.S., Zhang, H.; Faneli,
D.; Thimma Reddy, R.; Zhou, P.; Carroll, P.J. J. Org. Chem. 1997, 62, 2555.
16. Moreau, P.; Essiz, M.; Merour, J-Y.; Bouzard, D. Tetrahedron: Asymmetry 1997, 8, 591.
17. Ellman, J.A.; Owens, T.D.; Tang, T.P. Acc. Chem. Res. 2002, 35, 984.
18. Wakayama, M.; Ellman, J.A. J. Org. Chem. 2003, 68, 150.
19. (a) Pflum, D.A.; Krishnamurthy, D.; Han, Z.; Wald, S.A.; Senanayake, C. Tetrahedron Lett.
2002, 43, 923. (b) Plobeck, N.; Powell, D. Tetrahedron: Asymmetry 2002, 13, 303.
20. Cogan, D.A.; Ellman, J.A. J. Am. Chem. Soc. 1999, 121, 268.
21. Borg, G.; Cogan, D.A.; Ellman, J.A. Tetrahedron Lett. 1999, 40, 6709.
22. Tang, P.T.; Ellman, J.A. J. Org. Chem. 1999, 64, 12.
23. Sun, X.-W.; Xu, M.-H.; Lin, G.-Q. Org. Lett. 2006, 8, 4979.
24. Patterson, A.W.; Ellman, J.A. J. Org. Chem. 2006, 71, 7110.
25. Wakayama, M.; Ellman, J.A. J. Org. Chem. 2009, 74, 2646.

299
26. Min, Q.Q.; He, C.Y.; Zhou, H.; Zhang, X. Chem. Commun. 2010, 46, 8029.
27. Charette, A.B.; Mellon, C. Tetrahedron 1998, 54, 10525.
28. Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069.
29. Notte, G.T.; Leighton, J.L. J. Am. Chem. Soc. 2008, 130, 6676.
30. Leighton, J.L. Aldrichim. Acta 2010, 43, 3.
31. Xu, L.-W.; Li, L.; Lai, G.-Q.; Jiang, J.-X. Chem. Soc. Rev. 2011, 40, 1777.
32. Kobayashi, S.; Hirabayashi, R. J. Am. Chem. Soc. 1999, 121, 6942.
33. Shibasaki, M.; Suto, Y.; Kanai, M. J. Am. Chem. Soc. 2007, 129, 500.
34. Snapper, M.L.; Wieland, L.C.; Vieira, E.M.; Hoveyda, A.H. J. Am. Chem. Soc. 2009, 131, 570.
35. Overman, L.E. J. Am. Chem. Soc. 1974, 96, 597.
36. Anderson, C.E.; Donde, Y.; Douglas, C.J.; Overman, L.E. J. Org. Chem. 2005, 70, 648.
37. Yamamoto, Y.; Shimoda, H.; Oda, J.I.; Inouye, Y. Bull. Chem. Soc. Jpn. 1976, 49, 3247.
38. Nishikawa, T.; Asai, M.; Ohyabu, N.; Yamamoto, N.; Fukuda, Y.; Isobe, M. Tetrahedron
2001, 57, 3875.
39. Nishikawa, T.; Urabe, D.; Isobe, M. Angew. Chem. Int. Ed. 2004, 43, 4782.
40. Anderson, C.E.; Overman, L.E. J. Am. Chem. Soc. 2003, 125, 12412.
41. Fische, D.F.; Xin, Z-Q.; Peters, R. Angew. Chem. Int. Ed. 2007, 46, 7704.
42. Sheikh, N.S.; Leonori, D.; Barker, G.; Firth, J.D.; Campos, K.R.; Meijer, A.J.H.M.; O’Brien, P.;
Coldham, I. J. Am. Chem. Soc. 2012, 134, 5300.
43. Beng, T.K.; Woo, J.S.; Gawley, R.E. J. Am. Chem. Soc. 2012, 134, 14764.
44. Gallagher, D.J.; Beak, P. J. Org. Chem. 1995, 60, 7092.
45. Bertini Gross, K.M.; Beak, P. J. Am. Chem. Soc. 2001, 123, 315.
46. Basu, A.; Thayumanavan, S. Angew. Chem. Int. Ed. 2002, 41, 716.
47. Gawley, R.E. Top. Stereochem. 2010, 26, 93.
48. (a) Meyers, A.I. Tetrahedron 1992, 48, 2589. (b) Pippel, D.J.; Curtis, M.D.; Du, H.; Beak, P. J.
Org. Chem. 1998, 63, 2. (c) Bragg, R.A.; Clayden, J.; Bladon, M.; Ichihara, O. Tetrahedron
Lett. 2001, 42, 3411.
49. (a) Basu, A.; Beak, P. J. Am. Chem. Soc. 1996, 118, 1575. (b) Nakamura, S.; Nakagawa, R.;
Watanabe, Y.; Toru, T. Angew. Chem. Int. Ed. 2000, 39, 353. (c) Nakamura, S.; Nakagawa,
R.; Watanabe, Y.; Toru, T. J. Am. Chem. Soc. 2000, 122, 11340. (d) Beak, P.; Anderson, D.R.;
Curtis, M.D.; Laumer, J.M.; Pippel, D.J.; Weisenburger, G.A. Acc. Chem. Res. 2000, 33, 715.
(e) Lee, W.K.; Park, Y.S.; Beak, P. Acc. Chem. Res. 2009, 42, 224.
50. Schlosser, M.; Limat, D. J. Am. Chem. Soc. 1995, 117, 12343.

300
51. Lange, H.; Huenerbein, R.; Wibbeling, B.; Frölich, R.; Grimme, S.; Hoppe, D. Synthesis 2008,
2905.
52. (a) Soloshonok, V.A. Angew. Chem. Int. Ed. 2006, 45, 766. (b) Soloshonok, V.A.; Berbasov,
D.O. J. Fluorine Chem. 2006, 127, 597. (c) Trapp, O.; Schurig, V. Tetrahedron: Asymmetry
2010, 21, 1334.
53. Clayden, J. Organolithiums: Selectivity for Synthesis; Pergamon: Oxford, 2002.
54. Viktoria H.; Gessner, C.D.C.S. Chem. Eur. J. 2009, 15, 3320.
55. Mealy, M.J.; Bailey, W.F. J. Organomet. Chem. 2002, 646, 59.
56. Cason, L.F.; Brooks, H.G. J. Am. Chem. Soc. 1952, 74, 4582.
57. Bartlett, P.D.; Friedman, S.; Stiles, M. J. Am. Chem. Soc. 1953, 75, 1771.
58. Cason, L.F.; Brooks, H.G. J. Org. Chem. 1954, 19, 1278.
59. Peterson, D.J. J. Org. Chem. 1966, 31, 950.
60. Raucher, S.; Koolpe, G.A. J. Org. Chem. 1978, 43, 4252.
61. Crandall, J.K.; Clark, A.C. Tetrahedron Lett. 1969, 10, 325.
62. Crandall, J.K.; Clark, A.C. J. Org. Chem. 1972, 37, 4236.
63. Dimmel, D.R.; Huang, S. J. Org. Chem. 1973, 38, 2756.
64. Wei, X.; Taylor, R.J.K. Chem. Commun. 1996, 187.
65. Wei, X.; Taylor, R.J.K. Tetrahedron Lett. 1996, 37, 4209.
66. Veefkind, A.H.; Bickelhaupt, F.; Klumpp, G.W. Rec. Trav. Chim. Pays-Bas 1969, 88, 1058.
67. Peters, J.G.; Seppi, M.; Fröhlich, R.; Wibbeling, B.; Hoppe, D. Synthesis 2002, 381.
68. Wei, X.; Johnson, P.; Taylor, R.J.K. J. Chem. Soc. Perkin Trans 1 2000, 1109.
69. Gericke, R.; Harting, J.; Lues, I.; Schittenhelm, C. J. Med. Chem. 1991, 34, 3074.
70. Lepifre, F.; Cottineau, B.; Mousset, D.; Bouyssou, P.; Coudert, G. Tetrahedron Lett. 2004,
45, 483.
71. Lepifre, F.; Clavier, S.; Bouyssou, P.; Coudert, G. Tetrahedron. 2001, 57, 6969.
72. Felkin, H.; Swierczewski, G.; Tambuté, A. Tetrahedron Lett. 1969, 10, 707.
73. Kato, T.; Marumoto, S.; Sato, T.; Kuwajima, I. Synlett 1990, 671.
74. Marumoto, S.; Kuwajima, I. J. Am. Chem. Soc. 1993, 115, 9021.
75. Marumoto, S.; Kuwajima, I. Chem. Lett. 1992, 1421.
76. Klein, S.; Marek, I.; Normant, J.F. J. Org. Chem. 1994, 59, 2925.
77. Mück-Lichtenfeld, C.; Ahlbrecht, H. Tetrahedron 1999, 55, 2609.
78. Hoppe, D.; Hense, T. Angew. Chem. Int. Ed. 1997, 36, 2282.
79. Norsikian, S.; Marek, I.; Normant, J-F. Tetrahedron Lett. 1997, 38, 7523.
80. Klein, S.; Marek, I.; Poisson, J-F.; Normant, J-F. J. Am. Chem. Soc. 1995, 117, 8853.

301
81. Norsikian, S.; Marek, I.; Klein, S.; Poisson, J-F.; Normant, J-F. Chem. Eur. J. 1999, 5, 2055.
82. Norsikian, S.; Marek, I.; Poisson, J-F.; Normant, J-F. J. Org. Chem. 1997, 62, 4898.
83. Norsikian, S.; Baudry, M.; Normant, J.F. Tetrahedron Lett. 2000, 41, 6575.
84. Majumdar, S.; de Meijere, A.; Marek, I. Synlett 2002, 423.
85. Hogan, A-M.L.; O’Shea, D.F. J. Am. Chem. Soc. 2006, 128, 10360.
86. Hogan, A-M.L.; O’Shea, D.F. J. Org. Chem. 2008, 73, 2503.
87. Suzuka, T.; Ogasawara, M.; Hayashi, T. J. Org. Chem. 2002, 67, 3355.
88. Richards, C.J.; Locke, A.J. Tetrahedron: Asymmetry 1998, 9, 2377.
89. Smith, B.T.; Wendt, J.A.; Aubé, J. Org. Lett. 2002, 4, 2577.
90. Dearden, M.J.; Firkin, C.R.; Hermet, J-P.R.; O’Brien, P. J. Am. Chem. Soc. 2002, 124, 11870.
91. Dixon, A.J.; McGrath, M.J.; O’Brien, P. Org. Synth. 2006, 83, 141.
92. O’Brien, P. Chem. Commun. 2008, 655.
93. McGrath, M.J.; Bilke, J.L.; O’Brien, P. Chem. Commun. 2006, 2607.
94. Morita, Y.; Tokuyama, H.; Fukuyama, T. Org. Lett. 2005, 7, 4337.
95. Thayumanavan, S.; Basu, A; Beak, P. J. Am. Chem. Soc. 1997, 119, 8209.
96. Dearden, M.J.; O’Brien, P. Unpublished results.
97. Dearden, M.J.; McGrath, M.J.; O’Brien, P. J. Org. Chem. 2004, 69, 5789.
98. Brook, A.G. Acc. Chem. Res. 1974, 7, 77.
99. Vogel, C. Synthesis 1997, 497.
100. Wittig, G.; Löhmann, L. Liebigs Ann. Chem. 1942, 550, 260.
101. Tomooka, K.; Yamamoto, H.; Nakai, T. Liebigs Ann. 1997, 1275.
102. Nakai, T.; Mikami, K. Chem. Rev. 1986, 86, 885.
103. Eisch, J.J.; Kovacs, C.A. J. Organomet. Chem. 1971, 30, 97.
104. Ahman, J.; Somfai, P. J. Am. Chem. Soc. 1994, 116, 9781.
105. Somfai, P.; Blid, J.; Panknin, O.; Tuzina, P. J. Org. Chem. 2007, 72, 1294.
106. Somfai, P.; Panknin, O. Synlett 2007, 1190.
107. Coldham, I.; Collis, A. J.; Mould, R.J.; Rathmell, R.E. J. Chem. Soc. Perk. Trans. 1 1995,
2739.
108. Coldham, I.; Collis, A.J.; Mould, R.J.; Rathmell, R.E. Tetrahedron Lett. 1995, 36, 3557.
109. Anderson, J.C.; Siddons, D.C.; Smith, S.C.; Swarbrick, M.E. J. Chem. Soc. Chem. Commun.
1995, 1835.
110. Anderson, J.C.; O'Loughlina, J.M.A.; Tornos, J.A. Org. Biomol. Chem. 2005, 3, 2741.
111. Anderson, J.C.; Skerratt, S. J. Chem. Soc. Perkin Tans. 1 2002, 2871.
112. Booth, H.; Everett, J.R. J. Chem. Soc., Perkin Trans. 2 1980, 255.

302
113. Anderson, J.C.; Ford, J.G.; Whiting, M. Org. Biomol. Chem. 2005, 3, 3734.
114. Anderson, J.C.; Siddons, D.C.; Smith, S.C.; Swarbrick, M.E. J. Org. Chem. 1996, 61, 4820.
115. Anderson, J.C.; Davies, E.A. Tetrahedron 2010, 66, 6300.
116. Beesley, R.M.; Ingold, C.K.; Thorpe, J.F. J. Chem. Soc. 1915, 107, 1080.
117. Rouden J.; Ragot, A.; Gouault S.; Cahard D.; Plaquevent, J-C.; Lasne, M-C. Tetrahedron:
Asymmetry 2002, 13, 1299.
118. Cottineau, B.; Gillaizeau, I.; Farard, J.; Auclair, M-L., Coudert, G. Synlett 2007, 1925.
119. Clayden, J.; Dufour, J.; Grainger, D.M.; Helliwell, M. J. Am. Chem. Soc. 2007, 129, 7488.
120. Grainger, D.M.; Smith, A.C.; Vincent, M.A.; Hiller, I.H.; Wheatley, A.E.H.; Clayden, J. Eur.
J. Org. Chem. 2012, 4, 731.
121. Clayden, J.; Hennecke, U. Org. Lett. 2008, 10, 3567.
122. Bach, R.; Clayden, J.; Hennecke, U. Synlett 2009, 3, 421.
123. Tetlow, D.J.; Hennecke, U.; Raftery, J.; Waring, M.J.; Clarke, D.S.; Clayden, J. Org. Lett.
2010, 12, 5442.
124. Tetlow, D.J.; Vincent, M.A.; Hillier, I.H.; Clayden, J. Chem. Commun. 2013, 49, 1548.
125. Clayden, J.; Farnaby, W.; Grainger, D.M.; Hennecke, U.; Mancinelli, M.; Tetlow, D.J.;
Hillier, I.H.; Vincent, M.A. J. Am. Chem. Soc. 2009, 131, 3410.
126. MacLellan, P.; Clayden, J. Chem. Commun. 2011, 3395.
127. Aggarwal, V.K.; Stymiest, J.L.; Bagutski, V.; French, R.M. Nature 2008, 456, 778.
128. Fournier, A.M.; Brown, R.A.; Farnaby, W.; Miyatake-Ondozabal, H.; Clayden, J. Org. Lett.
2010, 12, 2222.
129. Lefranc, J.; Fournier, A.M.; Mignat, G.; Herbert, S.; Marcelli, T.; Clayden, J. J. Am. Chem.
Soc. 2012, 134, 7286.
130. Tetlow, D.J. Doctoral Thesis, University of Manchester, 2011.
131. Clayden, J.; Donnard, M.; Lefranc, J.; Minassi, A.; Tetlow, D.J. J. Am. Chem. Soc. 2010,
132, 6624.
132. Fournier, A.M.; Clayden, J. Org. Lett. 2012, 14, 142.
133. Castagnolo, D.; Foley, D.J.; Berber, H.; Luisi, R.; Clayden, J. Org. Lett. 2013, 15, 2116.
134. Lange, H.; Bergander, K.; Fröhlich, R.; Kehr, S.; Nakamura, S.; Shibata, N.; Toru, T.;
Hoppe, D. Chem. Asian J. 2008, 3, 88.
135. Cabello, N.; Kizirian, J-C.; Alexakis, A. Tetrahedron Lett. 2004, 45, 4639.
136. Carbone, G.; O’Brien, P.; Hilmersson, G. J. Am. Chem. Soc. 2010, 132, 15445.
137. Denmark, S.E.; Nakajima, N.; Nicaise, O.J.-C. J. Am. Chem. Soc. 1994, 116, 8797.

303
138. Strohmann, C.; Strohfeldt, K.; Schildbach, D.; McGrath, M.J.; O’Brien, P. Organometallics
2004, 23, 5389.
139. Vestergren, M.; Eriksson, J.; Hilmersson, G.; Håkansson, M.J. Organomet. Chem. 2003,
682, 172.
140. Carbone, G.; O’Brien, P. Unpublished results.
141. Negishi, E.-i.; Swanson, D.R.; Rousset, C.J. J. Org. Chem. 1990, 55, 5406.
142. Barker, G.; McGrath, J.L.; Klapars, A.; Stead, D.; Zhou, G.; Campos, K.R.; O’Brien, P. J.
Org. Chem. 2011, 76, 5936.
143. Vincent, M.A.; Smith, A.C.; Donnard, M.; Harford, P.J.; Haywood, J.; Hillier, I.H.; Clayden,
J.; Wheatley, A.E.H. Chem. Eur. J. 2012, 18, 11036.
144. Rappoprt, Z.Z.; Marek, I. The Chemistry of Organolithium Compounds Volume 1 and 2:
Wiley: Zurich, 2005.
145. Stanetty, P.; Koller, H.; Mihovilovic, M. J. Org. Chem. 1992, 57, 6833.
146. Johnson, D.S.; Singer, J.M.; White, A.D. Warner Lambert Co. WO2006/90273 A2, 2006.
147. Auvray, P.; Knochel, P.; Normant, J.F. Tetrahedron 1985, 26, 2329.
148. Clayden, J.; Yasin, S.A. New J. Chem. 2002, 26, 191. (b) Maercker, A.; Theysohn, W.
Liebigs Ann. Chem. 1971, 747, 70. (c) Spialter, L.; Harris, C.W. J. Org. Chem. 1966, 31, 4263.
(d) Krief, A.; Kenda, B.; Barbeaux, P.; Guittet, E. Tetrahedron 1994, 50, 7177.
149. Baldwin, J.E. J. Chem. Soc. Chem. Commun. 1976, 734.
150. Pirkle, W.H.; Sikkenga, D.L.; Pavlin, M.S. J. Org. Chem. 1977, 42, 384.
151. Brock, C.P.; Schweizer, W.B.; Dunitz, J.D. J. Am. Chem. Soc. 1991, 113, 9811.
152. Liu, G.; Cogan, D.A.; Ellman, J.A. J. Am. Chem. Soc. 1997, 119, 9913.
153. Beak, P.; Basu, A.; Gallagher, D.J.; Park, Y.S.; Thayumanavan, S. Acc. Chem. Res. 1996,
29, 552.
154. Clayden, J.; Helliwell, M.; Pink, J.H.; Westlund, N. J. Am. Chem. Soc. 2001, 123, 12449.
155. Gómez-SanJuan, A.; Sotomayor, N.; Lete, E. Beilstein J. Org. Chem. 2013, 9, 313.
156. Lefranc, J. Doctoral Thesis, University of Manchester, 2011.
157. Lefranc, J.; Tetlow, D.J.; Donnard, M.; Minassi, A.; Gàlvez, E; Clayden, J. Org. Lett. 2011,
13, 296.
158. Willoughby, C.A.; Buchwald, S.L. J. Am. Chem. Soc. 1994, 116, 8952.
159. Gawley, R.E. Topics in Stereochemistry, Volume 26: Stereochemical Aspects of
Organolithium Compounds; Wiley: Zurich, 2010.
160. Reich, H.J.; Medina, M.A.; Bowe, M.D. J. Am. Chem. Soc. 1992, 114, 11003.
161. Gawley, R.E.; Aubé, J. Principles of Asymmetric Synthesis; Elseveir: Oxford, 1996.

304
162. Lefranc, J.; Minassi, A.; Clayden, J. Beilstein J. Org. Chem. 2013, 6, 628.
163. Johnson, F. Chem. Rev. 1968, 68, 375.
164. Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.; Gagne, M.R.; Lloyd-Jones, G.C.;
Booker-Milburn, K.I. Angew. Chem. Int. Ed. 2009, 48, 8721.
165. Zezza, C.A.; Smith, M.B.; Ross, B.A.; Arhin, A.; Cronin, P.L.E. J. Org. Chem. 1984,
49, 4397.
166. Koller, W.; Schlack, P. Chem. Ber. 1963, 96, 93.
167. Desai, M.C.; Rosen, T.J. Pfizer Inc. US5232929 A1, 1993.
168. 2008 Top 200 Branded Drugs by Retail Dollars, Drug Topics, 2009:
http://drugtopics.modernmedicine.com/drugtopics/data/articlestandard//drugtopics/192
009/597083/article.pdf.
169. Nguyen, S.T.; Johnson, L.K.; Grubbs, R.H.; Ziller, J.W. J. Am. Chem. Soc. 1992, 114, 3974.
170. Vougioukalakis, G.C.; Grubbs, R.H. Chem. Rev. 2010, 110, 1746.
171. Grubbs, R.H.; Miller, S.J.; Fu, G.C. Acc. Chem. Res. 1995, 28, 446.
172. Fu, G.C.; Nguyen, S.T.; Grubbs, R.H. J. Am. Chem. Soc. 1993, 115, 9856.
173. Yang, S-M.; Lagu, B.; Wilson, L.J. J. Org. Chem. 2007, 72, 8123.
174. Ghelfi, F.; Parsons, A.F; Tommasini, D.; Mucci, A. Eur. J. Org. Chem. 2001, 10, 1845.
175. Hérisson, J.-L.; Chauvin, Y. Makromol. Chem. 1971, 141, 161.
176. Fürstner, A.; Thiel, O.R.; Lehmann, C.W. Organometallics 2002, 21, 331.
177. Compain, P. Adv. Synth. Catal. 2007, 349, 1829.
178. Krompiec, S.; Krompiec, M.; Penczek, R.; Ignasiak,H. Coord. Chem. Rev. 2008, 252, 1819.
179. Krompiec, S.; Kuznik, N.; Krompiec, M.; Penczek, R.; Mrzigod, J.; Torz, A. J. Mol. Catal. A:
Chem. 2006, 253, 132.
180. Krompiec, S.; Pigulla, M.; Szczepankiewicz, W.; Bieg, T.; Kuznik, N.; Leszczynska-Sejda K.;
Kubicki, M.; Borowiak, T. Tetrahedron Lett. 2001, 42, 7095.
181. Arisawa, M.; Terada, Y.; Takahashi, K.; Nakagawa, M.; Nishida, A. J. Org. Chem. 2006,
71, 4255.
182. Lemoucheux, L.; Rouden, J.; Ibazizene, M.; Sobrio, F.; Lasne, M-C. J. Org. Chem. 2003,
68, 7289.
183. Bordwell, F.G.; Algrim, D.; Vanier, N.R. J. Org. Chem. 1977, 42, 1817.
184. Bordwell, F.G. Acc. Chem. Res. 1988, 21, 456.
185. Crabtree, R. Acc. Chem. Res. 1979, 12, 331.
186. Marsh, B.J.; Carbery, D.R. J. Org. Chem. 2009, 74, 3186.
187. Gelder, E.A.; Jackson, S.D.; Lok, C.M. Catal. Lett. 2002, 84, 205.

305
188. Leonard, D.J. MChem Project Report, University of Manchester, 2011.
189. Krasovskiy, A.; Kopp, F.; Knochel, P. Angew. Chem. Int. Ed. 2006, 45, 497.
190. Wittig, G.; Sommer, H. Liebigs Ann. Chem. 1955, 594, 1.
191. Bergman, S.A. Anesth. Prog. 1999, 46, 10.
192. O'Brien, P.; Childs, A.C.; Ensor, G.J.; Hill, C.L.; Kirby, J.P.; Dearden, M.J.; Oxenford, S. J.;
Rosser, C. M. Org. Lett. 2003, 5, 4955.
193. Pandey, G.; Sekhar, B.B.V.S. Tetrahedron 1995, 51, 1483.
194. Lober, O.; Kawatsura, M.; Hartwig, J.F. J. Am. Chem. Soc. 2001, 123, 4366.
195. Steinhardt, S.E.; Vanderwal, C.D. J. Am. Chem. Soc. 2009, 131, 7546.
196. Raghavan, S.; Babu, V.S. Tetrahedron 2011, 67, 2044.
197. Ahmad, S.; Thomas, L.H.; Sutherland, A. Org. Biomol. Chem. 2011, 9, 2801.
198. Atkinson, R.C.; Leonard, D.J.; Maury, J.; Castagnolo, D.; Volz, N.; Clayden, J. Chem.
Commun. 2013, 49, 9734.
199. Tomohara, K. Doctoral Thesis, Kyoto University, 2012.
200. Detterbeck, R.; Guggisberg, A.; Hesse, M.; Popaj, K. Helv. Chim. Acta 2002, 85, 1742.
201. Reddy, L.R.; Liu, Y.; Gupta, A.P.; Villhauer, E. J. Org. Chem. 2012, 77, 2, 1095.
202. Boivin, J.; Pothier, J.; Zard, S.Z. Tetrahedron Lett. 1999, 40, 3701.
203. Abdel-Kader, M.S.; Bahler, B.D.; Malone, S.; Werkhoven, M.C.M.; van Troon, F.; Wisse,
D.J.H.; Bursuker, I.; Neddermann, K.M.; Mamber, S.W.; Kingston, D.G.I. J. Nat. Prod. 1998,
61, 1202.
204. Frank, K.E.; Aubé, J. J. Org. Chem. 2000, 65, 655.
205. Fan, R.; Pu, D.; Wen, F.; Ye, Y.; Wang, X. J. Org. Chem. 2008, 73, 3623.
206. Cheng, L.; Liu, L.; Sui, Y.; Wang, D.; Chen, Y-J. Tetrahedron: Asymmetry 2007, 18, 1833.
207. Medjahadi, M.; Gonzalez-Gomez, J.C.; Foubelo, F.; Yus, M. J. Org. Chem. 2009, 74, 7859.
208. Sun, X-W.; Liu, M.; Xu, M-H.; Lin, G-Q. Org. Lett. 2008, 10, 1259.
209. Ren, H.; Wulff, W.D. J. Am. Chem. Soc. 2011, 133, 5656.
210. Hietanen, A.; Kanerva, L.T.; Saloranta, T.; Rosenberg, S.; Latinen, E.; Leino, R. Eur. J. Org.
Chem. 2012, 10, 1945.
211. Seayad, A.M.; Ramalingam, B.; Yoshinaga, K.; Nagata, T.; Chai, C.L.L. Org. Lett. 2010, 12,
264.
212. Huang, J-M.; Zhang, J-F.; Gong, W. J. Org. Chem. 2011, 76, 3511.
213. Tehrani, K.A.; NguyenVan, T.; Karikomi, M.; Rottiers, M.; De Kimpe, N. Tetrahedron
2002, 58, 7145.
214. Bondzic, B.P.; Eilbracht, P. Org. Lett. 2008, 10, 3433.

306
Appendix 1: X-Ray crystal structure data
Crystal data and structure refinement for s3579m. 380
Identification code s3579m
Empirical formula C18 H23 Cl F N
Formula weight 307.82
Temperature 100(2) K
Wavelength 0.71073 A
Crystal system, space group Orthorhombic, Pbca
Unit cell dimensions a = 10.2494(14) A alpha = 90 deg.
b = 15.881(2) A beta = 90 deg.
c = 19.956(3) A gamma = 90 deg.
Volume 3248.3(8) A^3
Z, Calculated density 8, 1.259 Mg/m^3
Absorption coefficient 0.239 mm^-1
F(000) 1312
Crystal size 0.40 x 0.30 x 0.12 mm
Theta range for data collection 2.04 to 26.35 deg.
Limiting indices -12<=h<=10, -19<=k<=19, -20<=l<=24
Reflections collected / unique 17447 / 3309 [R(int) = 0.0838]
Completeness to theta = 26.35 99.9 %
Absorption correction None
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 3309 / 0 / 201
Goodness-of-fit on F^2 0.927
Final R indices [I>2sigma(I)] R1 = 0.0534, wR2 = 0.1245
R indices (all data) R1 = 0.0975, wR2 = 0.1370
Largest diff. peak and hole 0.753 and -0.471 e.A^-3
Cambridge Crystallographic Data Centre Number 977965

307
Crystal data and structure refinement for s3723na. 581b
Identification code s3723na
Empirical formula C15 H19 Cl N2 O2
Formula weight 294.77
Temperature 296(2) K
Wavelength 1.54178 A
Crystal system, space group Orthorhombic, Pna2(1)
Unit cell dimensions a = 15.2424(4) A alpha = 90 deg.
b = 7.0245(2) A beta = 90 deg.
c = 13.8159(4) A gamma = 90 deg.
Volume 1479.27(7) A^3
Z, Calculated density 4, 1.324 Mg/m^3
Absorption coefficient 2.313 mm^-1
F(000) 624
Crystal size 0.21 x 0.20 x 0.19 mm
Theta range for data collection 6.63 to 72.04 deg.
Limiting indices -18<=h<=18, -6<=k<=8, -17<=l<=14
Reflections collected / unique 4857 / 2289 [R(int) = 0.0252]
Completeness to theta = 66.60 98.2 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.6676 and 0.488209
Refinement method Full-matrix least-squares on F^2
Data / restraints / parameters 2289 / 1 / 183
Goodness-of-fit on F^2 1.057
Final R indices [I>2sigma(I)] R1 = 0.0278, wR2 = 0.0680
R indices (all data) R1 = 0.0284, wR2 = 0.0684
Absolute structure parameter -0.015(13)
Largest diff. peak and hole 0.194 and -0.236 e.A^-3
Cambridge Crystallographic Data Centre Number 977966