“dielectric spectroscopy - kfm.p.lodz.pl · dielectric spectroscopy • measures the...
TRANSCRIPT

“DIELECTRIC SPECTROSCOPY
&
Comparison With Other Techniques”

DIELECTRIC SPECTROSCOPY
• measures the dielectric and electric properties of a medium as a function of frequency (time)
• is based on the interaction of an external electric field with the electric dipole moment and charges of the medium

Dielectric properties
Complex dielectric permittivity relative to vacuum
ε*(ω)=ε’(ω)-iε’’(ω)
Complex conductivity
σ*(ω)=σ’(ω)+iσ’’(ω)
Complex electric modulus
Μ*(ω)=Μ’(ω)+iΜ’’(ω)
as a function of temperature

Medium in an external electric field
� � � �� � � � �
� � � � �Electric
Field
D=ε0εsE
•D=ε0E+P•P=ε0χsE•εs= χs+1

Medium in an external electric field
Relation between macroscopic and microscopic quantities
∑=i
ipV
P1
Dipole moments can have an induced or a permanent character
Permanent dipole moments, µInduced polarization, P
∞
∞∞ +=+= ∑ PV
NP
VP
i
i µµ1
µ Is determined by:•The interaction between the dipoles•The local electric field at the location of the dipole

Medium in an external electric field
EkT3
2µµ =
•No interaction between dipoles and local electric field equal to the outer applied fieldthermal energy �� interaction energy between the dipole and the electric field
V
N
kTs
2
03
1 µε
εε =− ∞
•Interactions between molecules, effect of polarization of environment
V
N
kTFgs
2
03
1 µε
εε =− ∞
( )( )∞
∞
++
=εε
εε
s
sF23
22
“Internal field” Onsager factor
ψµµ
cos12
2
int zg +== “Dipole interactions” Kirkwood-Froehlich factor
Onsager-Kirkwood-Froehlich equation
Langevin equation
Relation between macroscopic and microscopic quantities

Dielectric dispersion in time domain
Perturbation
Electric Field
E(t)
ε(t)
Response
Electric Displacement
D(t)
E0
0
E(t)
t
εεεεoo
εεεε0E0
0
D(t)
t
εεεε0εεεεsE0( ) ( ) 00 EttD εε=
( ) ( ) ( )tt s ϕεεεε ∞∞ −+=
( ) 00 =ϕ ( ) 1=∞ϕ
( ) ( )tt ϕ−=Φ 1

Dielectric dispersion in frequency domain
Perturbation
Electric Field
E(ω)
ε*(ω)
Response
Electric Displacement
D*(ω)
D(t)=D0sin(ϖϖϖϖt-δδδδ)
ϖϖϖϖt
E(t)=E0sin(ϖϖϖϖt)
δδδδ
( ) ( ) ( )ωωω DiDD ′′+′=*
( ) ( )ωδω cos0DD =′
( ) ( )ωδω sin0DD =′′
( ) ( )ωδε
ωε cos00
0
E
D=′
( ) ( )ωδε
ωε sin00
0
E
D=′′

Time-frequency domain
( ) [ ]dttidt
tdω
εεωε −−= ∫
∞
∞ exp)(0
*( )( ) [ ] ωωεωεπ
εdti
dt
tdexp
2
1)(
0
*∫∞
∞−=
( ) ( )[ ]tt s Φ−−=− ∞∞ 1)( εεεε
( ) ( ) [ ]dttidt
tds ωεεεωε −
Φ−−=− ∫
∞
∞∞ exp)(0
*
0)( =∞→Φ t
( ) ( ) ( ) ( )2
020
)(
+
=Φ
∑
∑ ∑∑<
i
i
i i ji
jiii tt
t
µ
µµµµ
1)0( =Φ
Time dependent dielectric functionε(t)
Complex dielectric functionε*(ω)
Relation between dielectric function and correlation function

Dielectric dispersion in frequency domain
Debye relaxation
−=Φ
D
tt
τexp)(
Diωτε
εωε+∆
+= ∞1
)(*
10-1
100
101
102
103
104
105
106
0
1
2
3
4
ε''
f(Hz)
ε'
−−∆=− ∞
D
tt
τεεε exp1)(

Dielectric dispersion in frequency domain
Kolhrauch-Williams-Watts KWW relaxation
−=Φ KWW
KWW
tt
β
τexp)(
( )[ ]γαωτ
εεωε
HNi+
∆+= ∞
1)(*
−−∆=− ∞
KWW
KWW
tt
β
τεεε exp1)(
10-1
100
101
102
103
104
105
106
0
1
2
3
4
ε''
f(Hz)
ε'
( )[ ]γαωτω
HN
HN
i+=Φ
1
1)(
*
αγβ ≈23.1
KWW( ) 387.01812.01 αγ −−=
Havrilliak-Negami relaxation

Dielectric dispersion in frequency domain
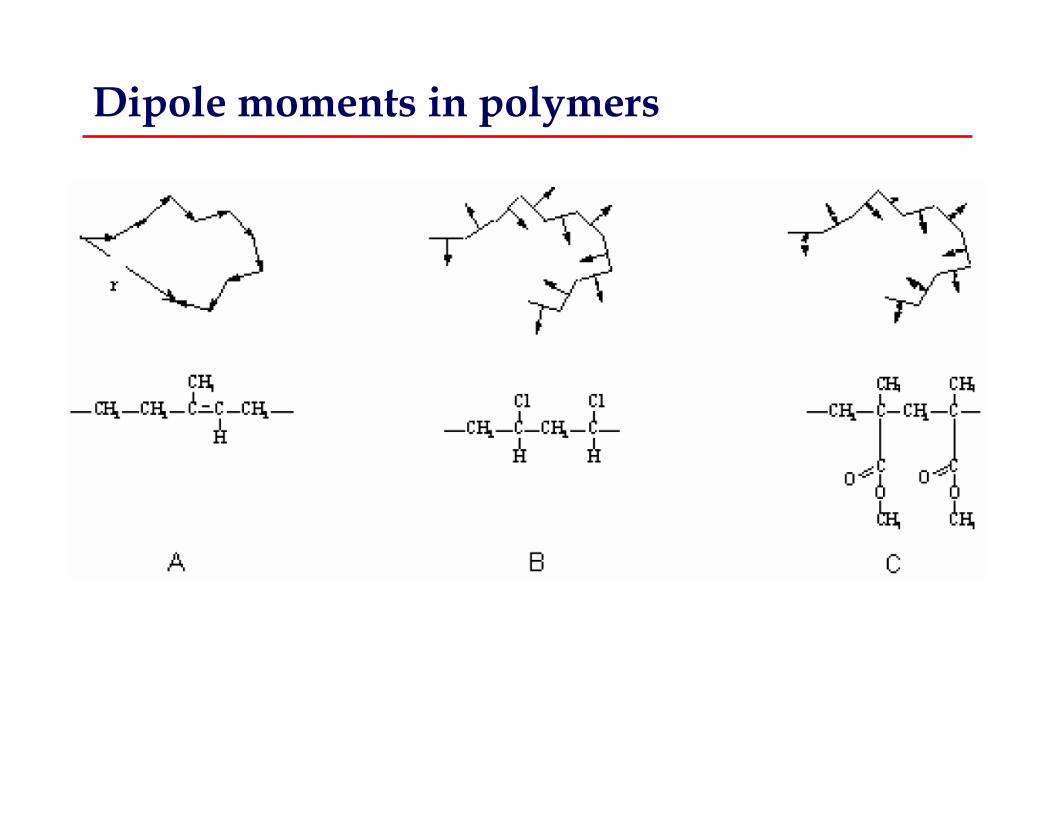
Dipole moments in polymers

Molecular motions accommodate dipole reorientation in polymers
Decreasing frequency of external field

Dielectric dispersion in frequency domain
10-1
100
101
102
103
104
105
10-1
100
d
iele
ctr
ic lo
ss
, εε εε'
'
frequency [Hz]
- 60°C - 40°C
- 120°C
- 90°C
10-1
2x10-1
3x10-1

�, � relaxations
Depends on chemical structure and local molecular environment
• Motion of groups present in side chain around
the main chain or bonds of the side chain
• Motion of groups in the main chain
• The � relaxation of Johari-Goldstein type
Glassy state (localized motions) I
Thermally activated motion
between two potential wells
separated by a potential barrier

• Temperature dependence of relaxation rate (time) is Arrhenius
• Dielectric strength, ��, increases with increasing T, more dipoles contribute
• Broad (4-6 decades) symmetric dielectric dispersion, becomes narrower with increasing T as a result of molecular environment homogenization
Glassy state (localized motions) II
Characteristics of dielectric dispersion
−= ∞kT
Eff actexpββ
V
N
kTgFOnsager
2µε ∝∆
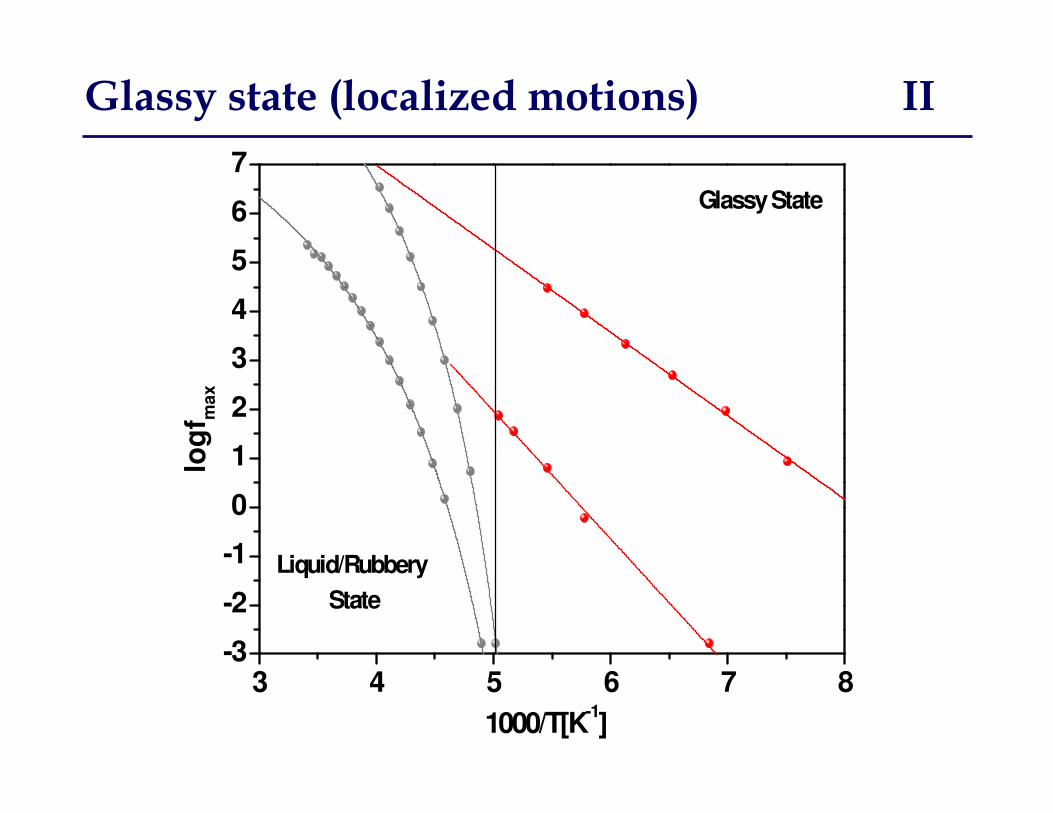
Glassy state (localized motions) II
3 4 5 6 7 8-3
-2
-1
0
1
2
3
4
5
6
7
Liquid/Rubbery
State
log
f max
1000/T[K-1]
Glassy State

� relaxation (dynamic glass transition)
• relaxation time reflects the time involved in the structural rearrangement (glass transition) of the molecules
• ττττ changes by many orders of magnitude from the liquid to the glass
• The αααα-relaxation in polymers involves cooperative micro-Brownian
segmental motions of the chains
• Segmental motions are associated with conformational transitions
taking place about the skeletal bonds
Rubbery/liquid state (segmental motions) I

Rubbery/liquid state (segmental motions) II
• Temperature dependence of relaxation rate (time) described by VTF equation
• Dielectric strength, ��, decreases with increasing T more strongly than predicted by
• Asymmetric dielectric dispersion
• High f side slope, n: local chain dynamics (n increases as T increases)
• Low f side slope, m: cooperative dynamics of environment (m increases as T increases)
Characteristics of dielectric dispersion
−−= ∞
0
expTT
Bff αα
V
N
kTgFOnsager
2µε ∝∆

3 4 5 6 7 8-3
-2
-1
0
1
2
3
4
5
6
7
Liquid/Rubbery
State
log
f max
1000/T[K-1]
Glassy State
Rubbery/liquid state (global motions) I

• Observed in polymers containing dipoles type A and AB
• Motions of the whole chain
• Observed at frequencies lower than the αααα relaxation
• The relaxation time of the process is strongly
dependent of the molecular weight
Rubbery/liquid state (global motions) I
Normal mode relaxation (nm)

• T dependence of relaxation rate (time)
same as this of � relaxation
• Dielectric strength proportional to the
mean end-to-end vector of chain
• m=1 and n=0.7 predicted by theory, in practice n considerably smaller
Rubbery/liquid state (global motions) II
Characteristics of dielectric dispersion
2
2
3
4r
kTM
FN OnsagerpA
nm
µπε =∆
3 4 5 6 7 8-3
-2
-1
0
1
2
3
4
5
6
7
Liquid/Rubbery
State
log
f max
1000/T[K-1]
Glassy State

• Motion of charge carriers
• �’ is not affected
• �’’ increases with decreasing f (slope of log�’’vs logf equal to -1)
• �’ independent of f equal to dc conductivity, �dc
• T dependence of �dcArrhenius, VTF or other gives information on the conductivity mechanism
Rubbery/liquid state (charge motions)
Conductivity
( ) ( ) ( ) ( )( )1*
0
* −=′′+′= ωεωεωσωσωσ ii
( ) ( )ωεωεωσ ′′=′0
( ) ( )( )10 −′=′′ ωεωεωσ

• Blocking of charges at interfaces inside inhomogeneous materials
• Existence of regions with different characteristics (�, �)• Relaxation rate (time) depends on:
volume fraction, shape, � and � values of regions
Separation of charges at interfaces
Maxwell-Wagner-Sillars relaxation (MWS)
Electrode Polarization
• Blocking of charges at sample/electrode interfaces
• Observed as a steep increase of �’• Magnitude and frequency position of the process depend on the conductivity of
the sample (high values of � shift polarization to high f)• Time of the process depends on the sample thickness (increase of thickness
results in an increase of time of process)

Comparison with other techniques
• Understanding the correlation between parameters of the molecular and supermolecular structures and macroscopic properties.
• Contribution to open questions regarding the � relaxation bycovering the widest possible time/frequency scale range by different experimental techniques

Material testing method & tool for studying dynamics in polymers
• Material in an external mechanical field
• insulator�elastic solid, conductor�viscous liquid
• Viscoelastic response is analogous to dielectric dispersion
• Modulus representation is used
• In order dielectric dispersion be compared to mechanical one electric modulus has to be used for data representation
Comparison with Mechanical measurements
( )( )ωε
ω*
* 1=M

Comparison with Mechanical measurements
Extension in broad f range by constructing master plots

Comparison with Mechanical measurements
Comparison of relaxation time temperature dependence

Comparison with Mechanical measurements
Selectivity of dielectric spectroscopy (block copolymers)

• Observation of the dynamics on a microscopic length and time scale
(10-7-10-11s)
• S(Q, �) contains not only temporal information but also spatial information of the particle correlation function
Comparison with Quasielastic Neutron Scattering

Comparison with Quasielastic Neutron Scattering
Different T Different T

Comparison with Quasielastic Neutron Scattering
( ) ( ) ( )QTTQ τατ =,
( )TAQ ,11−°=τ
( )Tdielτ
Comparable to
( ) ( )( )ωω
ω *Im1
, Φ−=QS

References
A. Broadband dielectric spectroscopy, F. Kremer and A. Schoenhals eds.Chapter 1Theory of Dielectric Relaxation A. Schoenhals and F. KremerChapter 7Molecular dynamics in polymer model systems, A. SchoenhalsChapter 16Dielectric and Mechanical Spectroscopy a Comparison, T. PakulaChapter 18Polymer dynamics by dielectric spectroscopy and neutron scattering-a comparison,A. Arbe, J. Colmenero and D. Richter
B. Electrical Properties of Polymers, E. Riande and R. Diaz-Calleja
C. Dielectric Spectroscopy of Polymeric materials, J. Runt and J. Fitzgerald