diagnostic flowcharts for dystonia: (1) in adults in ... · found at the networks web site . ......
TRANSCRIPT

Diagnostic flowcharts for Dystonia:
(1) In adults (2) In children & adolescents
Published by ERN-RND: 11th February 2019

This document was supported by and done in the framework of the European Reference Network for Rare Neurological Diseases (ERN-RND). ERN-RND is one of the 24 European Reference Networks (ERNs) approved by the ERN Board of Member States. The ERNs are co-funded by the European Commission (ERN-RND: 3HP 767231).
Page | 2
Introduction to the European Reference Network for Rare Neurological Diseases (ERN-RND):
ERN-RND is a European Reference Network established and approved by the European Union. ERN-RND is a healthcare infrastructure which focuses on rare neurological diseases (RND). The three main pillars of ERN-RND are (i) network of experts and expertise centres, (ii) generation, pooling and dissemination of RND knowledge, and (iii) implementation of e-health to allow the expertise to travel instead of patients and families.
ERN-RND unites 32 of Europe’s leading expert centres in 13 Member States and includes highly active patient organizations. Centres are located in Belgium, Bulgaria, Czech Republic, France, Germany, Hungary, Italy, Lithuania, Netherlands, Poland, Slovenia, Spain and the UK.
The following disease groups are covered by ERN-RND:
• Ataxias and Hereditary Spastic Paraplegias • Atypical Parkinsonism and genetic Parkinsons’ Disease • Dystonia, Paroxysmal Disorder and Neurodegeneration with Brain Ion Accumulation • Frontotemporal Dementia • Huntingtons’ Disease and other Choreas • Leukodystrophies
Specific information about the network, the expert centres and the diseases covered can be found at the networks web site www.ern-rnd.eu.
Recommendation for clinical use:
The European Reference Network for Rare Neurological Diseases developed the Diagnostic Flowcharts for Dystonia to help guide the diagnosis of Dystonia patients. The Reference Network recommends the use of these Diagnostic Flowcharts.

This document was supported by and done in the framework of the European Reference Network for Rare Neurological Diseases (ERN-RND). ERN-RND is one of the 24 European Reference Networks (ERNs) approved by the ERN Board of Member States. The ERNs are co-funded by the European Commission (ERN-RND: 3HP 767231).
Page | 3
Disclaimer:
Clinical practice guidelines, practice advisories, systematic reviews and other guidance published, endorsed or affirmed by ERN-RND are assessments of current scientific and clinical information provided as an educational service. The information (1) should not be considered inclusive of all proper treatments, methods of care, or as a statement of the standard of care; (2) is not continually updated and may not reflect the most recent evidence (new information may emerge between the time information is developed and when it is published or read); (3) addresses only the question(s) specifically identified; (4) does not mandate any particular course of medical care; and (5) is not intended to substitute for the independent professional judgement of the treating provider, as the information does account for individual variation among patients. In all cases, the selected course of action should be considered by the treating provider in the context of treating the individual patient. Use of the information is voluntary. ERN-RND provided this information on an “as is” basis, and makes no warranty, expressed or implied, regarding the information. ERN-RND specifically disclaims any warranties of merchantability or fitness for a particular use or purpose. ERN-RND assumes no responsibility for any injury or damage to persons or property arising out of or related to any use of this information or for any errors or omissions.
METHODOLOGY
The development of the Diagnostic Flowcharts for Dystonia was done by the Disease group for Dystonia, Paroxysmal Disorder and NBIA of ERN-RND.
Disease group for Dystonia, Paroxysmal Disorder and NBIA:
Disease group coordinators:
Alberto Albanese1; Thomas Klopstock2; Marie Vidailhet3
Disease group members:
Enrico Bertini4; Kailash Bhatia5; Elena Chorbadgieva6; Yaroslau Compta7; Adrian Danek2; Alejandra Darling7; Tom de Koning8; Marina de Koning-Tijssen8; Malgorazate Dec-Cwiek9; Maria Teresa Dotti10; Antonio Elia11; Antonio Federico10; Dusan Flisar12; Thomas Gasser13; Kathrin Grundmann13; Kinga Hadzsiev14; Christine Klein15; Jiri Klempir16; Maja Kojovic17; Norbert Kovacs14; Bernhard Landwehrmeier18; Ebba Lohmann13; Sebastian Löns15; Maria Jose Marti7; Maria Judit Molnar19; Alexander Münchau15; Juan Dario Ortigoza Escobar7; Damjan

This document was supported by and done in the framework of the European Reference Network for Rare Neurological Diseases (ERN-RND). ERN-RND is one of the 24 European Reference Networks (ERNs) approved by the ERN Board of Member States. The ERNs are co-funded by the European Commission (ERN-RND: 3HP 767231).
Page | 4
Osredkar12; Sebastian Paus20; Belén Pérez Dueñas21; Bart Post22; Evžen Růžička23; Sinem Tunc15; Michel Willemsen22; Giovanna Zorzi11 1 IRCCS Clinical Institute Humanitas – Rozzano, Italy; 2 Klinikum der Universität München, Germany; 3 Assistance Publique-Hôpitaux de Paris, Hôpital Pitié-Salepétrière, France: Reference Centre for Rare Diseases 'Neurogenetics'; 4 Pediatric hospital Bambino Gesù, Rome, Italy; 5 University College London Hospitals NHS Foundation Trust, United Kingdom; 6 University Neurological Hospital “St. Naum” Sofia, Bulgaria; 7 Hospital Clínic i Provincial de Barcelona y Hospital de Sant Joan de Déu, Spain; 8 University Medical Center Groningen, Netherlands; 9 University Hospital in Krakow, Poland; 10 AOU Siena, Italy; 11 Foundation IRCCS neurological institute Carlo Besta – Milan, Italy; 12 University Medical Centre Ljubljana, Slovenia; 13 Universitätsklinikum Tübingen, Germany; 14 University of Pécs, Hungary; 15 Universitätsklinikum Schleswig-Holstein, Germany; 16 General University Hospital in Prague, Czech Republic; 17 University Medical Centre Ljubljana, Slovenia; 18 Universitätsklinikum Ulm, Germany; 19 Semmelweis University, Hungary; 20 Universitätsklinikum Bonn, Germany; 21 Hospital Universitari Vall d'Hebron, Spain; 22 Stichting Katholieke Universiteit, doing business as Radboud University Medical Center Nijmegen, Netherlands; 23 Motol University Hospital, Czech Republic
Flowchart development process:
• Development of flowcharts – June – November 2017 • Discussion/Revision in ERN-RND disease group– November 2017 – June 2018 • Consent on diagnostic flowcharts during ERN-RND annual meeting 2018 –
08/06/2018 • Consent on document by whole disease group – 26/09/2018
This document was supported by and done in the framework of the European Reference Network for Rare Neurological Diseases (ERN-RND). ERN-RND is one of the 24 European Reference Networks (ERNs) approved by the ERN Board of Member States. The ERNs are co-funded by the European Commission (ERN-RND: 3HP 767231).
Page | 5
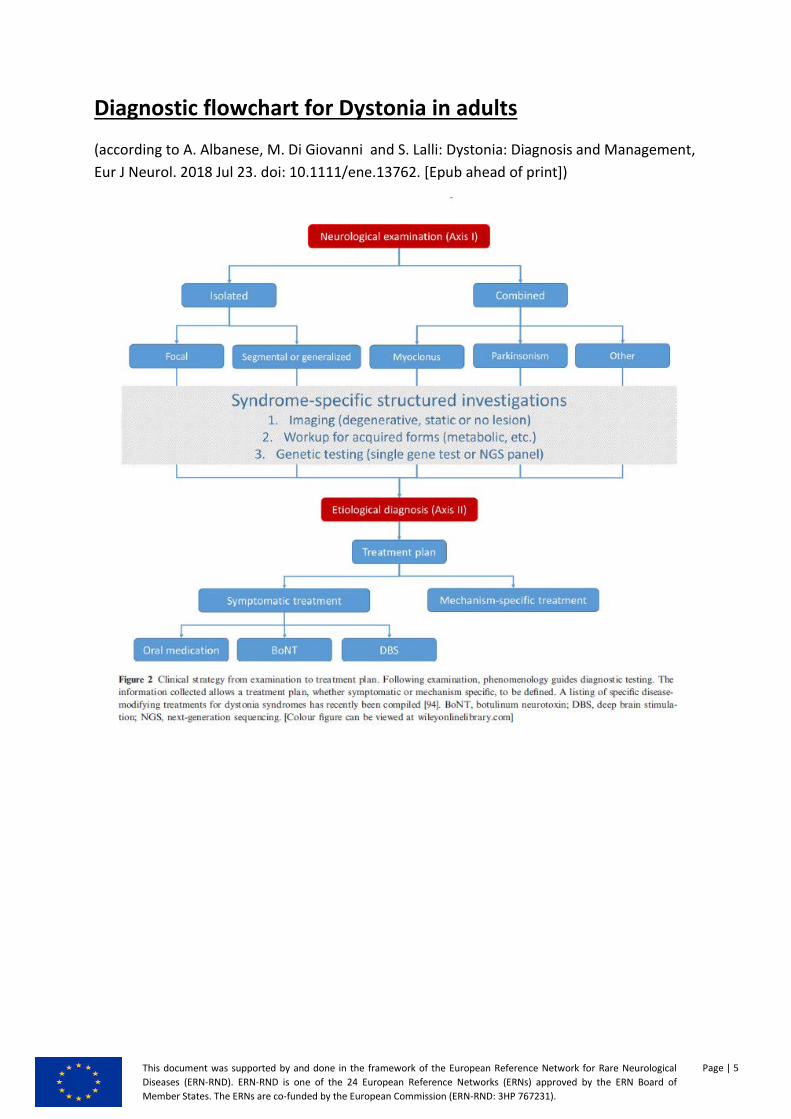
Diagnostic flowchart for Dystonia in adults
(according to A. Albanese, M. Di Giovanni and S. Lalli: Dystonia: Diagnosis and Management, Eur J Neurol. 2018 Jul 23. doi: 10.1111/ene.13762. [Epub ahead of print])

This document was supported by and done in the framework of the European Reference Network for Rare Neurological Diseases (ERN-RND). ERN-RND is one of the 24 European Reference Networks (ERNs) approved by the ERN Board of Member States. The ERNs are co-funded by the European Commission (ERN-RND: 3HP 767231).
Page | 6
Diagnostic flowchart for Dystonia in children and adolescents
(according to van Egmond ME, Kuiper A, Eggink H, et al. J Neurol Dystonia in children and adolescents: a systematic review and a new diagnostic algorithm Neurosurg Psychiatry 2015;86:774–781.)

This document was supported by and done in the framework of the European Reference Network for Rare Neurological Diseases (ERN-RND). ERN-RND is one of the 24 European Reference Networks (ERNs) approved by the ERN Board of Member States. The ERNs are co-funded by the European Commission (ERN-RND: 3HP 767231).
Page | 7
Annex - Original publications

Dystonia: diagnosis and management
A. Albanesea,b , M. Di Giovannia and S. Lallia,b
aUnit�a Operativa di Neurologia, IRCCS Istituto Clinico Humanitas, Rozzano, Milano; and bIstituto di Neurologia, Universit�a Cattolica
del Sacro Cuore, Milano, Italy
Keywords:
classification, diagnosis,
dystonia, genetics,
phenotypes
Received 28 February 2018
Accepted 20 July 2018
European Journal of
Neurology 2018, 0: 1–13
doi:10.1111/ene.13762
Clinical practice in dystonia has greatly evolved in recent years; a synthetic review
on patient management is provided here. Dystonia is a movement disorder charac-
terized by sustained or intermittent muscle contractions causing abnormal, often
repetitive, movements, postures or both. A recent classification has innovated clini-
cal practice and serves as guidance for clinical assessment: Axis I describes clinical
features, whereas Axis II indicates etiology. Dystonia presents with different syn-
dromic aggregations with varied somatic involvement and some common features.
There are five recognizable physical signs of dystonia: two main signs (dystonic
postures and movements) and three additional signs (gestes antagonistes or tricks,
mirror dystonia and overflow dystonia). There is still no validation of diagnostic
criteria for the different dystonia syndromes, and many cases with mild phe-
nomenology remain undiagnosed. Patients with dystonia also present non-motor
features that are variably combined with the movement disorder. The features of
the most common inherited and acquired dystonia syndromes are reviewed here.
There is clear evidence of genetic–environmental interaction in the determinism of
dystonia. The diagnostic process is guided by clinical examination and based on
specific laboratory examinations. Symptomatic treatments are available for dysto-
nia: botulinum neurotoxin injections are the primary choice for most focal dystonia
syndromes; deep brain stimulation is useful in some generalized and non-general-
ized syndromes. Additional treatment strategies are currently being assessed.
Introduction
More than 100 years have passed since Oppenheim first
introduced the term dystonia [1], and more than 40
since David Marsden and Stanley Fahn first attempted
to define and classify different dystonia syndromes
[2,3]. Meanwhile, the phenomenology of dystonia has
been described in great detail and several genetic forms
have been recognized. Dystonia is a movement disorder
characterized by sustained or intermittent muscle con-
tractions causing abnormal, often repetitive, move-
ments, postures or both [4]. Dystonic movements are
typically patterned, twisting and may be tremulous.
Dystonia is often initiated or worsened by voluntary
action and associated with overflow muscle activation.
The hyperkinetic disorder of dystonia is not always
easy to recognize, and it is often misdiagnosed [5].
Dystonia shares some features with parkinsonian
states: it causes bradykinesia [6], may coexist with
parkinsonism, and is observed in Parkinson’s disease
(PD) as an off-related phenomenon or a transition
dyskinesia [7,8]. The physical signs of dystonia are
easy to recognize when combined into a full-house
syndromic association, but instead more difficult when
mild or isolated.
Epidemiology
Attempts to assess the overall epidemiology of dystonia
have led to uncertain figures because of its wide heteroge-
neous expression. Overall, dystonia is not a rare disease,
but several inherited or idiopathic dystonia syndromes fall
into the current definition of rare chronic debilitating dis-
eases.1 Published epidemiological studies have probably
Correspondence: A. Albanese, Istituto Clinico Humanitas, Via A.
Manzoni, 56, 20086 Rozzano Milano, Italy (tel.: +39 02 8224-6418;
fax: +39 02 8224-2298; e-mail: [email protected]).
1See for example the definition of rare diseases provided by the
European Commission (https://ec.europa.eu/health/rare_diseases/pol
icy_en).
© 2018 EAN 1
R E V I E W A R T I C L E
EU
RO
PEA
NJO
URN
AL
OF
NEU
RO
LO
GY

underestimated the prevalence of dystonia in the general
population when accounting for 15–30 cases per 100 000
[9,10]. A classic population study in Rochester (Min-
nesota) reported a similar crude prevalence rate for all
focal dystonia syndromes [11]. In a study of a random
sample of the population over 50 years of age, the preva-
lence of isolated dystonia was estimated to be 732 per
100 000, suggesting that in the aging population dystonia
is a common neurological disorder [12]. The reported
variability amongst epidemiological studies denotes a dif-
ficulty in ascertaining the diagnosis of dystonia, given the
lack of validated criteria and the occurrence of a signifi-
cant proportion of patients with mild phenomenology
who do not request a medical consultation.
Women are affected about twice as often as men.
Of note, although the genetic causes of dystonia –especially for adult-onset focal forms – are still largely
elusive, a positive family history is reported in about
20% of dystonia sufferers [13–15]. Adult-onset focal
dystonia syndromes are by far the most frequent pre-
sentations. In two recent studies on focal syndromes,
the majority of patients had cervical dystonia (69%)
or blepharospasm (17%) [15,16], whilst other forms
were much rarer: limb dystonia (3%–7%), spasmodic
dysphonia (1%–3%), musician’s dystonia (3%) and
oromandibular dystonia (1%).
Classification
The recent classification of dystonia has greatly inno-
vated clinical practice and serves as a guide for clini-
cal assessment [4].
Axis I depicts clinical features and provides a syn-
thetic snapshot of the patient’s clinical condition at the
time of examination, whereas Axis II accommodates
etiology (Fig. 1). Five descriptors are listed under Axis
I: age at onset, body distribution, temporal pattern, co-
occurrence of other movement disorders or of other
neurological manifestations. Five age groups are distin-
guished for age at dystonia onset: infancy (birth to
2 years), childhood (3–12 years), adolescence (13–20 years), early adulthood (21–40 years) and late adult-
hood (>40 years). The body distribution can be focal,
segmental, multifocal, generalized (with or without leg
involvement) or unilateral (hemidystonia). The tempo-
ral pattern includes the disease course, which may be
Figure 1 Hierarchical organization of Axis I (clinical characteristics) and Axis II (etiology) of the dystonia classification.
© 2018 EAN
2 A. ALBANESE ET AL.

static or progressive, and the variability of symptoms,
which may be persistent, fluctuating, action specific or
paroxysmal. Associated features indicate whether dys-
tonia is combined with another movement disorder
(e.g. myoclonus dystonia) or with other neurological or
systemic manifestations.
If a patient’s condition progresses, its description
along Axis I will vary over time and the sequence of
consecutive observations will describe progression.
Instead, for the etiological classification (Axis II), the
most recent information available will be used.
Clinical features
The motor features of dystonia were originally
observed in generalized cases and later recognized to
occur also in focal syndromes with cervical or limb
involvement [17]. The features of dystonia encompass
two main physical signs that can be recognized by
expert neurological assessment (Table 1). Since the
diagnosis is based on clinical observation and there
are no supportive laboratory measures, the clinical
recognition is easier when there is a full-house phe-
nomenology, more difficult – instead – when dystonic
movements occur in isolation. Tremor may be an iso-
lated dystonic movement; therefore, isolated tremor
syndromes may be misdiagnosed as non-dystonic tre-
mor syndromes (see below). Additional clinical signs,
such as a geste, may support a diagnosis of dystonia,
but the clinical picture may still remain below a
threshold of diagnostic confidence. People with mild
focal phenomenology may have no complaint or may
not consult a doctor; hence, it is not uncommon to
recognize a focal dystonia in unaware or uncomplain-
ing subjects. The diagnosis of dystonia can be delayed
or missed more frequently than that of other hyperki-
netic movement disorders.
The physical signs listed in Table 1 are commonly
observed in patients with cervical or limb involvement.
Additional signs observed in patients with ble-
pharospasm are the presence of stereotyped, bilateral
and synchronous spasms of the orbicularis oculi mus-
cles. Spasms may be brief or sustained and may induce
narrowing or closure of the eyelids, but of course have
no torsional attitude [18]. In the case of laryngeal dysto-
nia, instrumental examination can reveal spasmodic
contractions of the vocal folds (abductor or adductor
dysphonia), also without overt torsional appearance
[19]. In muscle tension dysphonia, instead, the involun-
tary contractions affect the accessory phonation mus-
cles without necessarily involving the vocal folds.
In addition to producing the positive motor phe-
nomena listed in Table 1, dystonia may also present
failure of willed activity to occur. This has been
documented in blepharospasm (where the so-called
‘apraxia of eyelid opening’ is in fact an inability of
voluntary eyelid opening) [20], upper limb dystonia
[21] and cervical dystonia [22]. Finally, dystonia is
associated with slowing of movement (dystonic
bradykinesia) [23], to be distinguished from parkinso-
nian bradykinesia [24].
Dystonia is called ‘isolated’ when it is the sole motor
feature. The observation of tremor, representing a dys-
tonic movement, is compatible with the definition of
isolated dystonia. If additional movement disorders
occur, dystonia is called ‘combined’. Typical combined
dystonia syndromes are myoclonus dystonia or dysto-
nia parkinsonism. When dystonia is associated with
other neurological or systemic manifestations, these are
annotated as associated features (Fig. 1).
Transition from old to new terminology
The recent introduction of a new classification system
of dystonia [4] has occasionally raised uncertainties on
how to translate the old terminology. The newly intro-
duced definitions provide better clarity of language and
meaning but are not exactly synonymous with older
terms. The traditional expression ‘primary’ dystonia is
now discouraged and can be translated as ‘isolated
idiopathic’ or ‘isolated inherited’ dystonia. ‘Pure’ dys-
tonia is an obsolete denomination for isolated dysto-
nia. ‘Dystonia plus’ and ‘heredodegenerative dystonia’
can be translated, respectively, to ‘combined dystonia’
or ‘dystonia associated with neurological/systemic
manifestations’. The newer expressions are sometimes
longer; still, they hold the advantage of conveying
more detailed information than older terms.
Non-motor features of dystonia
Recent studies have revealed that, in addition to the
movement disorder, there are other, non-motor, fea-
tures in patients with isolated dystonia.
Sensory abnormalities may present months before
the movement disorder develops, as mild neck discom-
fort preceding cervical dystonia, irritation or dry eyes
before the development of blepharospasm, or throat
irritation heralding the onset of spasmodic dysphonia
[25]. Pain is reported in nearly 70% of patients with
cervical dystonia and in up to 30% of those with focal
hand dystonia or writer’s cramp [26].
Dystonia is also associated with neuropsychiatric
abnormalities, such as depressive disorders, that are
more frequent in cervical dystonia, blepharospasm,
laryngeal dystonia and focal hand dystonia compared
to healthy controls. A family history of depression,
anxiety and social anxiety is more common in
© 2018 EAN
DYSTONIA: DIAGNOSIS AND MANAGEMENT 3

dystonia than in controls [27]. Some specific inherited
dystonia syndromes have clear association with non-
motor features, as observed, for example, in aggre-
gated cohorts of patients with DYT11 dystonia [28].
In isolated dystonia syndromes, whether idiopathic
or inherited, there are usually no cognitive abnormali-
ties; by contrast, cognitive abnormalities are often
found in combined (whether inherited or idiopathic)
syndromes (Table 2).
Etiology
Classification by etiology is listed under Axis II. This is an
evolving area, particularly concerning genetic discoveries.
Evidence of degeneration, at the macroscopic,
microscopic or imaging level, provides a useful tool to
identify degenerative dystonias. Degeneration is
defined as a progressive structural abnormality, such
as neuronal loss, related to the occurrence of dystonia.
Static lesions are non-progressive neurodevelopmental
anomalies or acquired lesions. Incidental findings,
unrelated to the observed dystonia phenotype, are not
taken into account. In cases of isolated dystonia, usu-
ally there is no evidence of either degeneration or
structural lesion under Axis II.
Axis II specifies whether dystonia is acquired (due
to a known specific cause), inherited (due to a patho-
genic genetic mutation) or idiopathic (possibly related
to a yet undiscovered genetic defect).
Inherited dystonia syndromes
The number of inherited syndromes is continuously
increasing. They can present with isolated dystonia or
with a combination of dystonia plus another move-
ment disorder (Table 2). Some common syndromic
associations are highlighted hereafter.
Isolated dystonia
Dystonia is the only disease manifestation with the
possible occurrence of tremor. The best characterized
form is DYT1 dystonia, also called DYT-TOR1A
Table 1 The five physical signs of dystonia syndromes are recognized in most patients with dystonia
Physical sign Description
Main physical signs
Dystonic
postures
Muscle contractions may be continuous, forcing limbs and trunk into sustained postures (not available for blepharospasm or
laryngeal dystonia)
• A body part is flexed or twisted along its longitudinal axis
• Slowness and clumsiness for skilled movements is associated with sensation of rigidity and traction in the affected
part
Dystonic
movements
These features have to be looked for in all movement disorders, either fast or slow, also when the immediate impression is
that of a tremor, tic, chorea or myoclonus
• Tremor is a feature of dystonic movements and may appear as isolated tremor
• Movements are repetitive and patterned (i.e. consistent and predictable) or twisting
• Movements are often sustained at their peak to lessen gradually in a preferred posture (usually opposite to the
direction of movement)
Additional physical signs
Gestes
antagonistes
(‘tricks’)
Voluntary actions performed by patients that reduce or abolish the abnormal posture or the dystonic movements. They are
usually simple movements involving, or directed to, the body region affected by dystonia
• These movements are natural and graceful, not consisting of forceful opposition to the phenomenology of dystonia
• The movement does not push or pull the affected body part but simply touches it (‘sensory trick’) or accompanies it
during alleviation of dystonia
• Alleviation of dystonia occurs during the geste movement, usually soon after its start
Alleviation may last for as long as the geste or slowly reverses spontaneously before its end
To be distinguished from geste-like voluntary movements
Mirror
dystonia
It is evaluated in the upper or lower limbs. At least three different types of repetitive tasks (e.g. finger sequence, normal
writing or piano-like movements) are performed at low and fast speed in the non-affected limb
It is a unilateral posture or movement with the same or similar characteristics to the patient’s dystonia (usually postures and
some movements) that can be elicited, usually in the more severely affected side, when contralateral movements or actions
are performed
To be distinguished from non-dystonic mirror movements
Overflow
dystonia
It is observed at least once, usually ipsilaterally, in coincidence with the peak of dystonic movements
It is an unintentional muscle contraction accompanying the most prominent dystonic movement that are observed in an
anatomically distinct neighboring body region
Dystonic postures are not observed in patients with blepharospasm; mirror dystonia is only observed when the limbs are affected. Modified
from [69].
© 2018 EAN
4 A. ALBANESE ET AL.

Table 2 Common inherited dystonia syndromes, grouped according to Axis I criteria
Progressive listing (gene/protein)
Proposed
name [29] Inheritance Phenomenology
Inherited isolated dystonia syndromes
DYT1 (TOR1A/torsinA) DYT-TOR1A AD Early-onset generalized dystonia. Typically, there is limb
onset and sparing of face and neck. Alternative
phenotypes have been described
DYT4 (TUBB4/tubulin beta 4A class IVa) DYT-TUBB4A AD Rare form of dystonia presenting more commonly with
spasmodic dysphonia, with craniocervical involvement
and possible later generalization
DYT6 (THAP1/THAP domain containing
apoptosis-associated protein 1)
DYT-THAP1 AD Adolescent or young adult onset, generalized or segmental
involvement with predominance of craniocervical and
laryngeal features
DYT24 (ANO3/anoctamin 3) DYT-ANO3 AD Adult-onset tremulous craniocervical dystonia with
laryngeal involvement and upper limb tremor
DYT25 (GNAL/guanine nucleotide-binding
protein subunit alpha L)
DYT-GNAL AD Adult-onset focal craniocervical dystonia, typically
progressing to involve larynx, trunk and limbs
Inherited combined syndromes
DYT5a (GCH1/GTP cyclohydrolase 1) DYT/PARK-CGH1 AD Childhood- or young adult-onset dopa-responsive dystonia
with parkinsonism and diurnal fluctuations
DYT5b (TH/tyrosine hydroxylase) DYT/PARK-TH AR Milder form of dopa-responsive dystonia with infantile or
early childhood onset
DYT3 (TAF1/TATA box-binding
protein-associated factor 1)
DYT/PARK-TAF1 XD Segmental or generalized dystonia with marked
oromandibular involvement and parkinsonism
unresponsive to levodopa. Endemic in Panay, Philippines,
where it is known as Lubag
DYT12 (ATP1A3/ATPase Na+/K+
transporting subunit alpha 3)
DYT/PARK-
ATP1A3
AD Different phenotypes, including bridging forms, have been
described: rapid-onset dystonia parkinsonism, alternating
hemiplegia of childhood and CAPOS syndrome
PARK2 (parkin/E3 ubiquitin ligase) PARK-Parkin AR Young-onset parkinsonian syndrome with sustained
response to dopaminergic treatment and prominent leg
dystonia
DYT11 (SGCE/epsilon-sarcoglycan) DYT-SGCE AD Myoclonus dystonia with predominant neck and upper
limb involvement
N/A (NKX2.1/homeobox protein Nkx-2.1) CHOR-NKX2-1 AD Onset with chorea which can be replaced by a myoclonus
dystonia phenotype during the disease course
N/A (ADCY5/adenylate cyclase 5) CHOR-DYT-
ADCY5
AD Varied phenotype, including childhood-onset paroxysmal
or persistent chorea and dystonia
DYT10 (PRRT2/proline-rich
transmembrane protein 2)
PxMD-PRRT2 AD Paroxysmal dystonia and choreoathetosis
DYT8 (MR1/myofibrillogenesis
regulator 1)
PxMD-PNKD AD Attacks of paroxysmal non-kinesigenic dystonia, chorea,
athetosis or ballismus precipitated by specific factors such
as alcohol, caffeine, stress, hunger, fatigue or tobacco
DYT18 (SLC2A1/glucose
transporter protein type 1)
PxMD-SLC2A1 AD Paroxysmal exertion-induced chorea and dystonia in
excessively exercised body regions
Inherited combined syndromes associated
with additional neurological abnormalities
SCA3 (ATXN3/ataxin-3) SCA-ATXN3 AD Ataxic syndrome that may present with parkinsonism,
dystonia, chorea, spasticity, neuropathy or lower motor
neuron involvement
SCA17 (TBP/TATA box binding protein) SCA-TBP AD Ataxic syndrome that may present chorea and dystonia; it
may be associated with dementia and psychosis
N/A (TIMM8A/mitochondrial import inner
membrane translocase subunit Tim8 A)
DYT-TIMM8A XD Mohr–Tranebjaerg syndrome: dystonia plus additional
clinical features such as sensorineural deafness, visual or
cognitive impairment, behavioral problems, pyramidal
signs
N/A (DCAF17/nuclear
transmembrane protein)
NBIA/DYT-
DCAF17
AR Woodhouse–Sakati syndrome: dystonia and additional
clinical features such as dysarthria, deafness, seizures,
cognitive impairment, hypogonadism, alopecia, diabetes
mellitus, thyroid dysfunction
(continued)
© 2018 EAN
DYSTONIA: DIAGNOSIS AND MANAGEMENT 5

[29]. DYT1 dystonia is caused by mutations in the
TOR1A gene, encoding torsinA, a member of the
ATPases family. Almost all cases are caused by a
specific mutation, a 3-base pair deletion (delGAG) in
the coding region. This is the prototype of inherited
isolated generalized dystonia with limb onset, and it is
believed that Oppenheim’s original description
included patients with this inherited form [30]. Symp-
toms usually start in childhood with lower limb dysto-
nia and later spread to generalization. The face and
neck are typically not involved, a useful clue for clini-
cal orientation.
By contrast, there are inherited isolated syndromes
with a prominent craniocervical onset. DYT6 dystonia
(DYT-THAP1) may remain segmental or generalized,
and often has a striking laryngeal involvement.
DYT25 dystonia (DYT-GNAL) also causes adult-
onset cervical or cranial dystonia, often with promi-
nent tremor. Other generalized syndromes are still
debated and not yet confirmed, such as DYT4 (DYT-
TUBB4A), a rare generalized dystonia often with
laryngeal onset and cranial–cervical involvement, and
DYT23 dystonia (DYT-CIZ1), associated with adult-
onset autosomal dominant cervical dystonia and tre-
mor.
DYT24 (DYT-ANO3) is a cervical isolated dystonia
syndrome due to mutations of the ANO3 gene that
encodes a transmembrane protein belonging to a family
of calcium-activated chloride channels. Dystonic tre-
mor has been described as a key feature of DYT24 dys-
tonia, appearing most commonly as head or arm
tremor, and may precede the appearance of dystonic
postures. Generalization has been described in approxi-
mately 10% of cases [31].
Isolated dystonia may be the presenting feature of
combined syndromes that can remain isolated for a
significant amount of time before another movement
disorder appears. An isolated dystonia may be the
long-standing presenting feature of PARK2 parkin-
sonism (PARK-Parkin) [32], of rapid-onset dystonia
parkinsonism (DYT12, DYT-ATP1A3) [33], of Lubag
disease (DYT3, DYT/PARK-TAF1) [34] and of dopa-
responsive dystonia (DYT5a, DYT-GCH1 [35] or
DYT5b, DYT-TH [36]). In most cases, a full-house
syndrome of combined dystonia will develop in time,
whilst in some cases the phenotype will remain limited
to isolated dystonia.
Dystonia combined with myoclonus
The term ‘myoclonus dystonia’ is used to indicate the
occurrence of true myoclonus (particularly as “light-
ning jerks”) in combination with typical features of
dystonia in patients without evidence of degeneration
or structural lesion; this sets a difference with ‘my-
oclonic’ dystonia, a rather old term referring to cases
of isolated dystonia where very fast and brief dystonic
movement has a myoclonus-like appearance. Action-
induced, alcohol-responsive myoclonic jerks of subcor-
tical origin are typically combined with dystonia in
DYT11 myoclonus dystonia (DYT-SGCE). The myo-
clonic jerks typically are brief, lightning-like and often
affecting prevalently the neck, the trunk and the upper
limbs. Dystonia occurs in about two-thirds of
patients, usually in the form of mild cervical dystonia
and writer’s cramp. Some patients with myoclonus
dystonia carry no SGCE gene mutations and may
have other gene defects (e.g. ANO3, GCH1, TH,
CACNA1B, TITF1, TOR1A), with few cases remain-
ing genetically unidentified [37].
Dystonia combined with parkinsonism
In cases with childhood onset, dystonia dominates
and may be the only motor sign, whereas parkinson-
ism becomes more prominent with increasing age.
Several forms have a disorder of dopamine metabo-
lism, diurnal fluctuation of symptoms and a sustained
response to levodopa. Dopa-responsive dystonia
(DRD) syndromes include autosomal dominant
DYT5a (DYT/PARK-GCH1) and autosomal reces-
sive syndromes with more severe phenotypes, such as
DYT5b (DYT/PARK-TH) or sepiapterin reductase
deficiency (DYT/PARK-SPR).
Table 2 (Continued)
Progressive listing (gene/protein)
Proposed
name [29] Inheritance Phenomenology
NBIA1 or PKAN
(PANK2/pantothenate kinase 2)
NBIA/DYT-
PANK2
AR Dystonia with onset in childhood or adolescence,
combined dysarthria, rigidity, pyramidal signs and
cognitive impairment (previously called
Hallervorden–Spatz disease)
NBIA2, PARK14 or PLAN
(PLA2G6/A2 phospholipase)
NBIA/DYT/
PARK-PLA2G6
AR Dystonia often combined with chorea, parkinsonism,
dementia, pyramidal signs and psychiatric features
This listing is not exhaustive. AD, autosomal dominant; AR, autosomal recessive; XD, X-linked dominant; N/A, not available.
© 2018 EAN
6 A. ALBANESE ET AL.

In some patients, parkinsonism dominates; in others
dystonia prevails, particularly in the legs. The differen-
tial diagnosis is with inherited juvenile PD, particu-
larly autosomal recessive parkinsonisms, such as
PARK2 (PARK-Parkin), PARK7 (PARK-DJ1) or
PARK6 (PARK-PINK1). In the latter cases, both
dystonia and parkinsonism improve with dopaminer-
gic medication [32].
In addition, there are combined dystonia parkinson-
ism syndromes that are partially responsive to levo-
dopa, such as rapid-onset dystonia parkinsonism
(DYT12, DYT-ATP1A3), DYT3 (DYT-TAF1, Lubag
disease) and DYT16 (DYT-PRKRA) (Table 2).
Dystonia combined with ataxia
Several autosomal dominant spinocerebellar ataxias
(SCAs) can have dystonia as a part of the phenotype
and occasionally as the presenting feature. Dystonia is
most commonly observed in SCA1, SCA2, SCA3,
SCA6 and SCA17 [38,39].
Acquired dystonia syndromes
It is remarkable that dystonia syndromes indistin-
guishable from idiopathic may be caused by discrete
brain lesions [40]. Lesions causing acquired dystonia
are prevalently located in the basal ganglia, thalamus,
corticospinal tract or cerebellum and have a varied
etiology (Table 3). Frequent etiologies include vascu-
lar or traumatic lesions, perinatal brain injury and
neuroleptic usage.
Common phenotypes of acquired dystonia encom-
pass hemidystonia, which is caused by static brain
lesions in the contralateral hemisphere; Vogt’s ‘double
athetosis’ caused by cerebral palsy; and axial dystonia
caused by neuroleptics. By contrast, cervical dystonia
is most often idiopathic or inherited, although it has
been occasionally described also in acquired cases
[41]. There is no altogether reliable clinical clue to dis-
tinguish acquired from non-acquired dystonia syn-
dromes.
Genetic–environmental interplay
Dystonia syndromes are evidently influenced by both
genetic and environmental factors. This interaction is
particularly evident in isolated dystonia. An epidemio-
logical study performed in Australia showed that anx-
iety disorders, tremor, cigarette smoking and head
injuries with a loss of consciousness were associated
with increased risk of idiopathic isolated dystonia [42].
The first two factors may be either causative or sec-
ondary to dystonia, whereas the last two are probably
contributing environmental variables. Similar results
have been found in focal dystonia syndromes. Not
surprisingly, spasmodic dysphonia is associated with
several endogenous and exogenous factors [43], and
musician’s dystonia is probably caused by skilled per-
fectionist training in genetically predisposed individu-
als [13]. It has also been shown that environmental
factors may facilitate or worsen different focal dysto-
nia syndromes, such as exposure to bright light for
blepharospasm [44] or repeated skilled exercise for
task-specific dystonia [45]. Differences in prevalence,
age of onset and gender are probably correlated with
different exposures to environmental factors on a
background of genetic susceptibility.
Environmental factors play a role also in inherited
dystonia syndromes. A first evidence is provided by
the observation of incomplete penetrance: in DYT-
TOR1A penetrance is quite low, around 30%–40%[46], whereas in DYT-THAP1 it is about 60% [47].
This suggests that yet unknown environmental and
lifestyle factors can influence the expression of
pathogenic mutations even in early-onset dystonia
syndromes.
Diagnosis
The phenomenology of dystonia is a collection of
physical signs, including tremor, that require expert
neurological assessment [48,49]. The first diagnostic
criteria were proposed by Herz [50], who recognized
that dystonic movements and postures are the hall-
mark phenomenology of dystonia. They constitute the
Table 3 Main categories of acquired dystonia syndromes
Pathophysiology Etiology
Dystonic
cerebral palsy
Perinatal brain injury
Drug-induced Neuroleptics, dopamine blockers,
anticonvulsants, calcium channel blockers
Toxic Heroin inhalation, methanol, carbon monoxide,
disulfiram, cyanide, manganese, cobalt,
3-nitropropionic acid
Brain lesion Ischaemic, hemorrhagic, arteriovenous
malformation, neoplasms, radiotherapy, head
trauma, brain surgery (including ablations and
stereotactic lesions), electrical injury
Infection Viral encephalitis, subacute sclerosing
panencephalitis, human immunodeficiency virus,
encephalitis lethargica, prion disease
Immune-
mediated
Acquired disseminated encephalomyelitis
(ADEM), autoimmune or paraneoplastic
encephalitis (most frequently NMDAR
antibody-associated encephalitis)
Metabolic Hypoglycemia, hyperglycemic hyperosmolar
state, hypocalcemia, hypoparathyroidism,
hyperthyroidism, hepato-cerebral degeneration,
uremia
© 2018 EAN
DYSTONIA: DIAGNOSIS AND MANAGEMENT 7

main physical signs and are complemented by three
other physical signs: gestes antagonistes (‘tricks’), mir-
ror dystonia and overflow dystonia. Fahn first noticed
that dystonic movements are the most common type
of involuntary movements to be misdiagnosed [51],
are highly variable (either fast or slow, and irregular)
and may manifest as isolated tremor without other
clues suggesting dystonia. This may delay recognition
of dystonia in patients with an isolated tremor syn-
drome [48]. The minimal requirements for diagnosing
dystonia in specific body regions are still for the most
part undefined and, whilst the full-house phenomenol-
ogy remains unquestionable, mild or incomplete
expressions, in the past called formes frustes [52], may
remain undiagnosed.
There is no consensus on diagnostic criteria for
focal dystonia syndromes; therefore, only general
diagnostic criteria are currently available. A set of
proposed diagnostic criteria for blepharospasm [53]
still needs validation and refinement; a concerted
effort for consensus on diagnostic criteria for cervical
dystonia is currently under way.
Tremor in dystonia
Traditionally, tremor was considered a separate move-
ment disorder from dystonia, although the recent con-
sensus classification has ratified that dystonic
movements can present as isolated tremor [4]. In
patients with dystonia, tremor commonly involves the
head or the arms, where it can be postural/kinetic or
at rest [54]. Head tremor is quite specific of dystonia,
but isolated arm tremor can be mistaken for essential
or parkinsonian tremor [48].
Upper limb tremor is observed in a significant pro-
portion of patients with otherwise isolated focal dys-
tonia syndromes, such as cervical dystonia, focal
upper limb dystonia or spasmodic dysphonia [15]. It
is accepted that patients with dystonia may have a
classic essential tremor phenomenology and later may
develop dystonic postures to compose a full-house
clinical picture. It is not uncommon that patients with
isolated upper limb postural/kinetic tremor are mis-
classified as having classic essential tremor if upper
limb tremor is the presenting sign and the patients are
observed before they develop any other signs of dysto-
nia. The expression ‘isolated upper limb tremor’ is
increasingly used to describe patients who cannot be
definitely classified as having essential or instead dys-
tonic tremor.
Patients with adult-onset dystonic tremor can also
be misdiagnosed as having PD, particularly if they
have upper limb dystonic resting tremor and are not
assessed by dopaminergic imaging. Large clinical trials
on PD have shown that a percentage between 10%
and 15% of clinically diagnosed PD patients have
normal dopaminergic binding (scans without evidence
of dopaminergic deficit) [55,56]. Some of these
patients have adult-onset isolated dystonia [57] that is
particularly difficult to recognize when dystonic
bradykinesia causes reduced arm swing while walking
[58].
Investigations
The diagnosis of dystonia is primarily clinical, and
investigations are needed particularly for Axis II clas-
sification. After outlining a syndromic description
according to Axis I, syndrome-specific investigations
are performed to establish an etiological diagnosis
according to Axis II (Fig. 2). In some cases, clinical
examination may prompt specific investigations. For
example, the observation of a Kayser–Fleischer ring
may lead to a study of copper metabolism or the
observation of myoclonus dystonia may warrant
specific genetic testing.
Electromyography (EMG) mapping may provide a
complement to clinical examination, allowing some
typical neurophysiological phenomenology to be rec-
ognized. The usual EMG features observed in dysto-
nia are as follows: prolonged bursts (200–500 ms),
simultaneous contractions (cocontraction) of agonist
and antagonist muscles, involuntary activation of con-
tiguous muscles (overflow) [59].
Most patients will undergo an imaging study, usu-
ally starting with brain magnetic resonance imaging
(MRI), to identify whether there is a degenerative
condition, a static lesion or no evidence of brain
lesions. Results of imaging studies may prompt fur-
ther investigations. For example, a suspicion of neu-
rodegeneration with brain iron accumulation or a
cerebellar atrophy may lead to specific genetic testing.
In dystonia parkinsonism syndromes, by contrast,
brain imaging may be abnormal and shows accumula-
tion of metals: manganese, as in Kufor–Rakeb syn-
drome (PARK-ATP13A2) [60]; calcium, as in primary
familial brain calcifications [61]; iron, as in neurode-
generations with brain iron accumulation [62]. A brain
computed tomography scan is preferable if calcifica-
tions are suspected; otherwise MRI is the neuroimag-
ing of choice. As a rule, brain MRI should include
iron-sensitive sequences [63].
In patients where the workup has ruled out evi-
dence for an acquired or inherited dystonia syndrome,
a provisional diagnosis of idiopathic dystonia is made.
The high degree of phenotypic overlap has facilitated
the diffusion of multi-gene diagnostic dystonia panels
[64].
© 2018 EAN
8 A. ALBANESE ET AL.

Metabolic and blood testing (e.g. acanthocytes,
ceruloplasmin, serum and urinary copper, uric acid,
serum pyruvate, and lactate levels) and other comple-
mentary investigations (e.g. slit-lamp examination) are
usually performed in specific cases to reach a diagnos-
tic confirmation. DAT scan scintigraphy allows
reduced binding to dopaminergic terminals to be iden-
tified, indicating a parkinsonian syndrome.
Treatment/management
Anticholinergic medications have proved useful in
patients with generalized and focal isolated dystonia
syndromes, whereas levodopa is effective in some
patients with dopa-responsive dystonia parkinsonism.
Other agents that have been used in dystonia are
baclofen, carbamazepine and benzodiazepines, either
alone or in combination [65].
Botulinum toxin treatment
Botulinum neurotoxin (BoNT) is a potent poison pro-
duced by Clostridium botulinum that causes local mus-
cle weakness. Its efficacy is due to partial peripheral
denervation [66] with a contribution of central effects
[67].
Botulinum neurotoxin is the first-line treatment for
patients with blepharospasm and cervical dystonia
(spasmodic torticollis) [68]. It is also effective in
laryngeal and limb dystonia. Three BoNT/A sero-
types and one BoNT/B serotype are commercially
available worldwide. They have received individual
generic names and are currently considered individ-
ual products that are only partly interchangeable
[69]. In addition, other BoNT products are available
in specific countries and others are under develop-
ment [70].
Figure 2 Clinical strategy from examination to treatment plan. Following examination, phenomenology guides diagnostic testing. The
information collected allows a treatment plan, whether symptomatic or mechanism specific, to be defined. A listing of specific disease-
modifying treatments for dystonia syndromes has recently been compiled [94]. BoNT, botulinum neurotoxin; DBS, deep brain stimula-
tion; NGS, next-generation sequencing. [Colour figure can be viewed at wileyonlinelibrary.com]
© 2018 EAN
DYSTONIA: DIAGNOSIS AND MANAGEMENT 9

Cervical dystonia
There is class A evidence of efficacy for Abo BoNT/A
and Rima BoNT/B and class B evidence for Ona
BoNT/A and Inco BoNT/A in cervical dystonia [71].
BoNT/A products are commonly used as first-line
therapy for cervical dystonia.
The efficacy of BoNT products in cervical dystonia
has been confirmed by several systematic reviews.
BoNT/A is more effective than placebo [72] and anti-
cholinergic oral treatment [73]. BoNT/B has also pro-
ven efficacious in cervical dystonia [74]; a comparison
of BoNT/A and BoNT/B showed significant improve-
ment from baseline without difference between the
median duration of benefit [75].
There is no standard procedure for performing
BoNT injections in cervical dystonia: muscle selection,
BoNT dilution and dosing, and targeting techniques
(whether to use EMG or ultrasound guidance) vary sig-
nificantly amongst centers, which probably brings some
heterogeneity in outcome. Two recent consensus publi-
cations have provided a first attempt to develop prac-
tice guidelines for BoNT treatment in cervical dystonia
[76,77].
Blepharospasm
There is class B evidence on the efficacy of Ona
BoNT/A and Inco BoNT/A and class C evidence for
Abo BoNT/A in blepharospasm [71]. BoNT/A is the
first-line treatment in this condition, with an expected
success rate approaching 100% if injections are placed
in the pretarsal portion of the orbicularis oculis mus-
cle [78,79].
Other focal dystonias
Botulinum neurotoxin type A is probably effective for
the treatment of focal upper limb dystonia (including
writer’s cramp) and adductor spasmodic dysphonia
(adductor laryngeal dystonia) [80], whilst abductor
laryngeal dystonia responds less predictably [68].
Notwithstanding, BoNT/A is the treatment of choice
also for these conditions given the lack of alternative
treatments.
Most patients with dystonia have a long-term
response to BoNT after repeated treatment cycles [81].
However, in some patients the efficacy of BoNT treat-
ment may lessen to the extent that a patient may be
considered to have lost a tangible benefit. This condi-
tion, called ‘secondary non-response’, is caused almost
always by wrong muscle targeting, possibly due to
changes in the muscle activation pattern over time. The
development of neutralizing antibodies is a theoretical
possibility that is considered exceptional: after long-
term treatment with ona BoNT/A, only <2% of the
patients were positive for neutralizing antibodies [82].
Transcranial magnetic stimulation
Repetitive, low frequency transcranial magnetic stimu-
lation can enhance intracortical inhibition, a neural
process deemed to be altered in dystonia. Although
single-session studies have been reported to be ineffec-
tive, there is preliminary evidence that cumulative
effects can be obtained by repeated stimulation over
consecutive days [83].
Deep brain stimulation
Deep brain stimulation (DBS) of the internal globus
pallidus (GPi) has emerged as the surgical treatment
of choice for children and adults with disabling idio-
pathic isolated dystonia. The efficacy of DBS has been
well documented in patients with inherited generalized
dystonia, particularly DYT1 dystonia which cannot
be managed with oral medications or BoNT. This
therapeutic approach has been extended more recently
to focal dystonia syndromes, which are commonly
treated with BoNT.
Evidence of the efficacy of bilateral DBS of the
GPi on DYT1 dystonia is confirmed by several
studies [84,85]. Compared to patients with idiopathic
(not inherited) isolated dystonia, DYT1 patients had
earlier and greater improvement [86]. The efficacy of
GPi DBS on other inherited isolated dystonia syn-
dromes is less consistent than observed in DYT1.
Patients with DYT6 dystonia respond less favorably
to GPi DBS than DYT1 mutation carriers [86,87].
There is insufficient evidence of the efficacy of GPi
DBS on patients with DYT24 tremulous-prominent
cervical dystonia [31], although it is generally
believed that dystonic tremor responds to GPi DBS
implants [88].
The efficacy of GPi DBS has been consistently
reported in DYT11 (DYT-SGCE) myoclonus dystonia
and in DYT3 (DYT/PARK-TAF1 or Lubag) X-
linked dystonia parkinsonism. In DYT11, both dysto-
nia and myoclonus improve and a sustained benefit
has been reported after 10 years [89]; in Lubag,
instead, dystonia can improve more than parkinson-
ism but long-term assessments are lacking [90]. In
other inherited combined syndromes, however, there
is no clear indication of DBS efficacy. In DYT12 dys-
tonia, instead, there is evidence of inefficacy for GPi
DBS [33].
Globus pallidus internus DBS has also proved
efficacious in acquired dystonia syndromes. The
response of drug-induced, tardive, dystonia may be
very rapid and occurs within days or weeks after
bilateral GPi implants. In most cases, patients with
generalized or segmental tardive dystonia have been
© 2018 EAN
10 A. ALBANESE ET AL.

treated. The available data are mainly uncontrolled
case reports, and prospective systematic studies are
needed instead. Dystonic cerebral palsy without
prominent spasticity has also been shown to respond
to GPi DBS. Most data collected are retrospective
series or case reports that indicate a significant
potential efficacy for DBS and call attention to the
need for properly designed prospective controlled
trials [91].
The subthalamic nucleus (STN) has recently been
proposed as a potential DBS target, alternative to
GPi in patients with dystonia. A 3-year follow-up
study provided class IV evidence that STN DBS
decreases long-term severity in patients with medically
refractory isolated dystonia [92].
Overall, the available data suggest that DBS has
great potential for different dystonia syndromes,
whether idiopathic or inherited, isolated or combined.
The outcome is influenced by a number of Axis I fea-
tures, such as body distribution, age of onset, and by
the relative prevalence of dystonic movements or pos-
tures. The genetic status is also important, with two
extremes represented by DYT1 dystonia (consistently
excellent outcome) and DYT12 dystonia (consistent
lack of efficacy).
Physical treatments
A variety of physical treatments have been proposed
for dystonia, including motor learning exercises, pas-
sive or active mobilization techniques, stretching of
dystonic muscles, relaxation and electrotherapy (e.g.
EMG biofeedback or transcutaneous electrical nerve
stimulation). Different rehabilitation strategies have
been combined with BoNT to improve disability and
pain compared to BoNT treatment alone. This is still
a debated topic that has particularly regarded patients
with cervical dystonia and has not yet reached a
consensus level [93].
Disclosure of conflict of interest
The authors declare no financial or other conflict of
interests.
References
1. Oppenheim H. €Uber eine eigenartige Krampfkrankheitdes kindlichen und jugendlichen Alters (Dysbasia lordot-ica progressiva, Dystonia musculorum deformans). Neu-rologische Centralblatt 1911; 30: 1090–1107.
2. Marsden CD. Dystonia: the spectrum of the disease.Res Publ Assoc Res Nerv Ment Dis 1976; 55: 351–367.
3. Fahn S, Eldridge R. Definition of dystonia and classifi-cation of the dystonic states. Adv Neurol 1976; 14: 1–5.
4. Albanese A, Bhatia K, Bressman SB, et al. Phenomenol-ogy and classification of dystonia: a consensus update.Mov Disord 2013; 28: 863–873.
5. Lalli S, Albanese A. The diagnostic challenge of primarydystonia: evidence from misdiagnosis. Mov Disord 2010;25: 1619–1626.
6. Berardelli A, Rothwell JC, Thompson PD, Hallett M.Pathophysiology of bradykinesia in Parkinson’s disease.Brain 2001; 124: 2131–2146.
7. Del Sorbo F, Albanese A. Levodopa-induced dyskinesiasand their management. J Neurol 2008; 255(Suppl. 4):32–41.
8. Dolhun R. Dystonia and Parkinson’s disease. PractNeurol 2015; 15: 43–46.
9. Epidemiological Study of Dystonia in Europe Collabo-rative Group. A prevalence study of primary dystonia ineight European countries. J Neurol 2000; 247: 787–792.
10. Steeves TD, Day L, Dykeman J, Jette N, Pringsheim T.The prevalence of primary dystonia: a systematic reviewand meta-analysis. Mov Disord 2012; 27: 1789–1796.
11. Nutt JG, Muenter MD, Aronson A, Kurland LT, Mel-ton LJ 3rd. Epidemiology of focal and generalized dys-tonia in Rochester, Minnesota. Mov Disord 1988; 3:188–194.
12. Muller J, Kiechl S, Wenning GK, et al. The prevalenceof primary dystonia in the general community. Neurol-ogy 2002; 59: 941–943.
13. Schmidt A, Jabusch HC, Altenmuller E, et al. Etiologyof musician’s dystonia: familial or environmental? Neu-rology 2009; 72: 1248–1254.
14. Groen JL, Kallen MC, van de Warrenburg BP, et al.Phenotypes and genetic architecture of focal primarytorsion dystonia. J Neurol Neurosurg Psychiatry 2012;83: 1006–1011.
15. Williams L, McGovern E, Kimmich O, et al. Epidemio-logical, clinical and genetic aspects of adult onset iso-lated focal dystonia in Ireland. Eur J Neurol 2017; 24:73–81.
16. Wang L, Chen Y, Hu B, Hu X. Late-onset primary dys-tonia in Zhejiang province of China: a service-based epi-demiological study. Neurol Sci 2016; 37: 111–116.
17. Albanese A. How many dystonias? Clinical evidenceFront Neurol 2017; 8: 18.
18. Defazio G, Hallett M, Jinnah HA, Conte A, BerardelliA. Blepharospasm 40 years later. Mov Disord 2017; 32:498–509.
19. Hintze JM, Ludlow CL, Bansberg SF, Adler CH, LottDG. Spasmodic dysphonia: a review. Part 2: Characteri-zation of pathophysiology. Otolaryngol Head Neck Surg2017; 157: 558–564.
20. Krack P, Marion MH. ‘Apraxia of lid opening’, a focaleyelid dystonia: clinical study of 32 patients. Mov Disord1994; 9: 610–615.
21. Cohen LG, Hallett M. Hand cramps: clinical featuresand electromyographic patterns in a focal dystonia. Neu-rology 1988; 38: 1005–1012.
22. Mezaki T. Dystonia redefined as central non-paretic lossof control of muscle action: a concept including inabilityto activate muscles required for a specific movement, or‘negative dystonia’. Med Hypotheses 2007; 69: 1309–1312.
23. Berardelli A, Rothwell JC, Hallett M, Thompson PD,Manfredi M, Marsden CD. The pathophysiology of pri-mary dystonia. Brain 1998; 121(Pt 7): 1195–1212.
© 2018 EAN
DYSTONIA: DIAGNOSIS AND MANAGEMENT 11

24. Rao G, Fisch L, Srinivasan S, et al. Does this patienthave Parkinson disease? JAMA 2003; 289: 347–353.
25. Patel N, Jankovic J, Hallett M. Sensory aspects ofmovement disorders. Lancet Neurol 2014; 13: 100–112.
26. Kuyper DJ, Parra V, Aerts S, Okun MS, Kluger BM.Nonmotor manifestations of dystonia: a systematicreview. Mov Disord 2011; 26: 1206–1217.
27. Berman BD, Junker J, Shelton E, et al. Psychiatric asso-ciations of adult-onset focal dystonia phenotypes. J Neu-rol Neurosurg Psychiatry 2017; 88: 595–602.
28. Peall KJ, Dijk JM, Saunders-Pullman R, et al. Psychi-atric disorders, myoclonus dystonia and SGCE: an inter-national study. Ann Clin Transl Neurol 2016; 3: 4–11.
29. Marras C, Lang A, van de Warrenburg BP, et al.Nomenclature of genetic movement disorders: recom-mendations of the International Parkinson and Move-ment Disorder Society Task Force. Mov Disord 2016;31: 436–457.
30. Fahn S, Bressman SB, Marsden CD. Classification ofdystonia. Adv Neurol 1998; 78: 1–10.
31. Stamelou M, Charlesworth G, Cordivari C, et al. Thephenotypic spectrum of DYT24 due to ANO3 muta-tions. Mov Disord 2014; 29: 928–934.
32. Elia AE, Del Sorbo F, Romito LM, Barzaghi C, Gar-avaglia B, Albanese A. Isolated limb dystonia as pre-senting feature of Parkin disease. J Neurol NeurosurgPsychiatry 2014; 85: 827–828.
33. Albanese A, Di Giovanni M, Amami P, Lalli S. Failureof pallidal deep brain stimulation in DYT12-ATP1A3dystonia. Parkinsonism Relat Disord 2017; 45: 99–100.
34. Lee LV, Pascasio FM, Fuentes FD, Viterbo GH. Tor-sion dystonia in Panay, Philippines. Adv Neurol 1976;14: 137–151.
35. Dobricic V, Tomic A, Brankovic V, et al. GCH1 muta-tions are common in Serbian patients with dystonia-par-kinsonism: challenging previously reported prevalencerates of DOPA-responsive dystonia. Parkinsonism RelatDisord 2017; 45: 81–84.
36. Katus LE, Frucht SJ. An unusual presentation of tyrosinehydroxylase deficiency. J Clin Mov Disord 2017; 4: 18.
37. Balint B, Bhatia KP. Isolated and combined dystoniasyndromes – an update on new genes and their pheno-types. Eur J Neurol 2015; 22: 610–617.
38. Hagenah JM, Zuhlke C, Hellenbroich Y, Heide W,Klein C. Focal dystonia as a presenting sign ofspinocerebellar ataxia 17. Mov Disord 2004; 19: 217–220.
39. Manto MU. The wide spectrum of spinocerebellar atax-ias (SCAs). Cerebellum 2005; 4: 2–6.
40. Marsden CD, Obeso JA, Zarranz JJ, Lang AE. Theanatomical basis of symptomatic hemidystonia. Brain1985; 108(Pt 2): 463–483.
41. Strader S, Rodnitzky RL, Gonzalez-Alegre P. Secondarydystonia in a botulinum toxin clinic: clinical characteris-tics, neuroanatomical substrate and comparison withidiopathic dystonia. Parkinsonism Relat Disord 2011; 17:749–752.
42. Newman JR, Boyle RS, O’Sullivan JD, Silburn PA,Mellick GD. Risk factors for idiopathic dystonia inQueensland, Australia. J Clin Neurosci 2014; 21: 2145–2149.
43. Tanner K, Roy N, Merrill RM, Sauder C, Houtz DR,Smith ME. Case–control study of risk factors for spas-modic dysphonia: a comparison with other voice disor-ders. Laryngoscope 2012; 122: 1082–1092.
44. Hallett M, Evinger C, Jankovic J, Stacy M, Interna-tional Workshop. Update on blepharospasm: reportfrom the BEBRF International Workshop. Neurology2008; 71: 1275–1282.
45. Torres-Russotto D, Perlmutter JS. Task-specific dysto-nias: a review. Ann N Y Acad Sci 2008; 1142: 179–199.
46. Grundmann K, Laubis-Herrmann U, Bauer I, et al. Fre-quency and phenotypic variability of the GAG deletionof the DYT1 gene in an unselected group of patientswith dystonia. Arch Neurol 2003; 60: 1266–1270.
47. Saunders-Pullman R, Raymond D, Senthil G, et al.Narrowing the DYT6 dystonia region and evidence forlocus heterogeneity in the Amish-Mennonites. Am JMed Genet A 2007; 143A: 2098–2105.
48. Albanese A, Del Sorbo F. Dystonia and tremor: theclinical syndromes with isolated tremor. Tremor OtherHyperkinet Mov (N Y) 2016; 6: 319.
49. Albanese A. Classifying tremor: language matters. MovDisord 2018; 33: 3–4.
50. Herz E. Dystonia I. Historical review: analysis of dys-tonic symptoms and physiologic mechanisms involved.Arch Neurol Psychiatry 1944; 51: 305–318.
51. Fahn S. The varied clinical expressions of dystonia. Neu-rol Clin 1984; 2: 541–554.
52. Zeman W, Kaelbling R, Pasamanick B. Idiopathic dys-tonia musculorum deformans. II. The formes frustes.Neurology 1960; 10: 1068–1075.
53. Defazio G, Hallett M, Jinnah HA, Berardelli A. Devel-opment and validation of a clinical guideline for diag-nosing blepharospasm. Neurology 2013; 81: 236–240.
54. Gigante AF, Berardelli A, Defazio G. Rest tremor inidiopathic adult-onset dystonia. Eur J Neurol 2016; 23:935–939.
55. Whone AL, Watts RL, Stoessl AJ, et al. Slower progres-sion of Parkinson’s disease with ropinirole versus levo-dopa: the REAL-PET study. Ann Neurol 2003; 54: 93–101.
56. Marek K, Seibyl J, Eberly S, et al. Longitudinal follow-up of SWEDD subjects in the PRECEPT study. Neurol-ogy 2014; 82: 1791–1797.
57. Schneider SA, Edwards MJ, Mir P, et al. Patients withadult-onset dystonic tremor resembling parkinsonian tre-mor have scans without evidence of dopaminergic deficit(SWEDDs). Mov Disord 2007; 22: 2210–2215.
58. Albanese A, Lalli S. Distinguishing scan without evi-dence of dopaminergic depletion patients with asymmet-ric resting tremor from Parkinson’s disease: a clinicaldiagnosis of dystonia is required. Mov Disord 2010; 25:2899–2899.
59. Albanese A, Lalli S. Is this dystonia? Mov Disord 2009;24: 1725–1731.
60. Ramirez A, Heimbach A, Grundemann J, et al. Heredi-tary parkinsonism with dementia is caused by mutationsin ATP13A2, encoding a lysosomal type 5 P-typeATPase. Nat Genet 2006; 38: 1184–1191.
61. Kostic VS, Petrovic IN. Brain calcification and move-ment disorders. Curr Neurol Neurosci Rep 2017; 17: 2.
62. Amaral LL, Gaddikeri S, Chapman PR, et al. Neurode-generation with brain iron accumulation: clinicoradio-logical approach to diagnosis. J Neuroimaging 2015; 25:539–551.
63. Fung VS, Jinnah HA, Bhatia K, Vidailhet M. Assess-ment of patients with isolated or combined dystonia: anupdate on dystonia syndromes. Mov Disord 2013; 28:889–898.
© 2018 EAN
12 A. ALBANESE ET AL.

64. van Egmond ME, Lugtenberg CHA, Brouwer OF, et al.A post hoc study on gene panel analysis for the diagno-sis of dystonia. Mov Disord 2017; 32: 569–575.
65. Jankovic J. Treatment of dystonia. Lancet Neurol 2006;5: 864–872.
66. Tighe AP, Schiavo G. Botulinum neurotoxins: mecha-nism of action. Toxicon 2013; 67: 87–93.
67. Caleo M, Restani L. Direct central nervous system effectsof botulinum neurotoxin. Toxicon 2018; 147: 68–72.
68. Albanese A, Asmus F, Bhatia KP, et al. EFNS guideli-nes on diagnosis and treatment of primary dystonias.Eur J Neurol 2011; 18: 5–18.
69. Albanese A. Clinical guidelines: no more mistaken iden-tities for botulinum neurotoxins. Nat Rev Neurol 2016;12: 373–374.
70. Cocco A, Albanese A. Recent developments in clinical tri-als of botulinum neurotoxins. Toxicon 2018; 147: 77–83.
71. Simpson DM, Hallett M, Ashman EJ, et al. Practiceguideline update summary: botulinum neurotoxin forthe treatment of blepharospasm, cervical dystonia, adultspasticity, and headache: Report of the Guideline Devel-opment Subcommittee of the American Academy ofNeurology. Neurology 2016; 86: 1818–1826.
72. Costa J, Espirito-Santo C, Borges A, et al. Botulinumtoxin type A therapy for cervical dystonia. CochraneDatabase Syst Rev 2005: CD003633.
73. Costa J, Espirito-Santo C, Borges A, Ferreira JJ, CoelhoM, Sampaio C. Botulinum toxin type A versus anti-cholinergics for cervical dystonia. Cochrane DatabaseSyst Rev 2005: CD004312.
74. Costa J, Espirito-Santo C, Borges A, et al. Botulinumtoxin type B for cervical dystonia. Cochrane DatabaseSyst Rev 2005: CD004315.
75. Costa J, Borges A, Espirito-Santo C, et al. Botulinumtoxin type A versus botulinum toxin type B for cervicaldystonia. Cochrane Database Syst Rev 2005: CD004314.
76. Albanese A, Abbruzzese G, Dressler D, et al. Practicalguidance for CD management involving treatment ofbotulinum toxin: a consensus statement. J Neurol 2015;262: 2201–2213.
77. Contarino MF, Van Den Dool J, Balash Y, et al. Clini-cal practice: evidence-based recommendations for thetreatment of cervical dystonia with botulinum toxin.Front Neurol 2017; 8: 35.
78. Albanese A, Bentivoglio AR, Colosimo C, Galardi G,Maderna L, Tonali P. Pretarsal injections of botulinumtoxin improve blepharospasm in previously unresponsivepatients. J Neurol Neurosurg Psychiatry 1996; 60: 693–694.
79. Cakmur R, Ozturk V, Uzunel F, Donmez B, Idiman F.Comparison of preseptal and pretarsal injections ofbotulinum toxin in the treatment of blepharospasm andhemifacial spasm. J Neurol 2002; 249: 64–68.
80. Simpson DM, Blitzer A, Brashear A, et al. Assessment:botulinum neurotoxin for the treatment of movementdisorders (an evidence-based review): report of the Ther-apeutics and Technology Assessment Subcommittee ofthe American Academy of Neurology. Neurology 2008;70: 1699–1706.
81. Ramirez-Castaneda J, Jankovic J. Long-term efficacy,safety, and side effect profile of botulinum toxin in dys-tonia: a 20-year follow-up. Toxicon 2014; 90: 344–348.
82. Naumann M, Carruthers A, Carruthers J, et al. Meta-analysis of neutralizing antibody conversion with onabo-tulinumtoxinA (BOTOX(R)) across multiple indications.Mov Disord 2010; 25: 2211–2218.
83. Erro R, Tinazzi M, Morgante F, Bhatia KP. Non-inva-sive brain stimulation for dystonia: therapeutic implica-tions. Eur J Neurol 2017; 24: 1228–e1264.
84. Cif L, Vasques X, Gonzalez V, et al. Long-term follow-up of DYT1 dystonia patients treated by deep brainstimulation: an open-label study. Mov Disord 2010; 25:289–299.
85. Panov F, Gologorsky Y, Connors G, Tagliati M, Mira-vite J, Alterman RL. Deep brain stimulation in DYT1dystonia: a 10-year experience. Neurosurgery 2013; 73:86–93; discussion 93.
86. Vidailhet M, Jutras MF, Grabli D, Roze E. Deep brainstimulation for dystonia. J Neurol Neurosurg Psychiatry2013; 84: 1029–1042.
87. Bruggemann N, Kuhn A, Schneider SA, et al. Short-and long-term outcome of chronic pallidal neurostimula-tion in monogenic isolated dystonia. Neurology 2015; 84:895–903.
88. Fasano A, Bove F, Lang AE. The treatment of dystonictremor: a systematic review. J Neurol Neurosurg Psychia-try 2014; 85: 759–769.
89. Roze E, Vidailhet M, Hubsch C, Navarro S, Grabli D.Pallidal stimulation for myoclonus-dystonia: ten years’outcome in two patients. Mov Disord 2015; 30: 871–872.
90. Patel AJ, Sarwar AI, Jankovic J, Viswanathan A. Bilat-eral pallidal deep brain stimulation for X-linked dysto-nia-parkinsonism. World Neurosurg 2014; 82(241): e241–e244.
91. Koy A, Timmermann L. Deep brain stimulation in cere-bral palsy: challenges and opportunities. Eur J PaediatrNeurol 2017; 21: 118–121.
92. Ostrem JL, San Luciano M, Dodenhoff KA, et al.Subthalamic nucleus deep brain stimulation in isolateddystonia: a 3-year follow-up study. Neurology 2017; 88:25–35.
93. Contarino MF, Smit M, van den Dool J, Volkmann J,Tijssen MA. Unmet needs in the management of cervicaldystonia. Front Neurol 2016; 7: 165.
94. Jinnah HA, Factor SA. Diagnosis and treatment of dys-tonia. Neurol Clin 2015; 33: 77–100.
© 2018 EAN
DYSTONIA: DIAGNOSIS AND MANAGEMENT 13

REVIEW
Dystonia in children and adolescents: a systematicreview and a new diagnostic algorithmMartje E van Egmond,1 Anouk Kuiper,1 Hendriekje Eggink,1 Richard J Sinke,2
Oebele F Brouwer,1 Corien C Verschuuren-Bemelmans,2 Deborah A Sival,3
Marina A J Tijssen,1 Tom J de Koning2,3
▸ Additional material ispublished online only. To viewplease visit the journal online(http://dx.doi.org/10.1136/jnnp-2014-309106).1University of Groningen,University Medical CenterGroningen, Department ofNeurology, Groningen,The Netherlands2University of Groningen,University Medical CenterGroningen, Department ofGenetics, Groningen,The Netherlands3University of Groningen,University Medical CenterGroningen, Department ofPediatrics, Groningen,The Netherlands
Correspondence toDr Tom J de Koning,University of Groningen,University Medical CenterGroningen, Department ofGenetics, PO Box 30.001,Groningen 9700 RB,The Netherlands;[email protected]
Received 28 July 2014Revised 22 October 2014Accepted 28 October 2014Published Online First13 November 2014
To cite: van Egmond ME,Kuiper A, Eggink H, et al. JNeurol Neurosurg Psychiatry2015;86:774–781.
ABSTRACTEarly aetiological diagnosis is of paramount importancefor childhood dystonia because some of the possibleunderlying conditions are treatable. Numerous geneticand non-genetic causes have been reported, anddiagnostic workup is often challenging, time consumingand costly. Recently, a paradigm shift has occurred inmolecular genetic diagnostics, with next-generationsequencing techniques now allowing us to analysehundreds of genes simultaneously. To ensure thatpatients benefit from these new techniques, adaptationof current diagnostic strategies is needed. On the basisof a systematic literature review of dystonia with onset inchildhood or adolescence, we propose a novel diagnosticstrategy with the aim of helping clinicians determinewhich patients may benefit by applying these newgenetic techniques and which patients first require otherinvestigations. We also provide an up-to-date list ofcandidate genes for a dystonia gene panel, based on adetailed literature search up to 20 October 2014. Whilenew genetic techniques are certainly not a panacea,possible advantages of our proposed strategy includeearlier diagnosis and avoidance of unnecessaryinvestigations. It will therefore shorten the time ofuncertainty for patients and their families awaiting adefinite diagnosis.
INTRODUCTIONDystonia is a movement disorder characterised bysustained or intermittent muscle contractionscausing abnormal, often repetitive, movements,postures or both.1 For dystonia in children andadolescents, here referred to as dystonia ofchildhood (DC), the list of possible genetic andnon-genetic causes is extensive.2 3 For cliniciansencountering a young patient with dystonia, animportant practical question is how to manage thediagnostic workup, which is often challenging, timeconsuming and costly.Recently, a paradigm shift has occurred in
molecular genetic diagnostics, with next-generationsequencing (NGS) techniques now allowing us toanalyse hundreds of genes simultaneously. NGSdiagnostic strategies are particularly effective in het-erogeneous conditions, including movement disor-ders, significantly increasing the diagnostic yield atlower costs.4 5 As a significant proportion of DCcases is estimated to be genetic, a ‘genetics first’diagnostic approach for all patients with DCseems logical and appealing. However, there are
two groups of patients for whom another initialapproach should be considered. First, in childrenand adolescents who may have acquired dystonia,and second, in patients in whom the cause may bea treatable inborn error of metabolism (IEM),because for most of these IEMs biochemical investi-gations will be a faster diagnostic method thangenetic testing.We first provide a systematic literature review of
the phenomenology, classification and aetiology ofDC. We then propose a novel diagnostic strategythat will help clinicians determine which patientsmay benefit from NGS technologies and whichpatients require other initial investigations. Finally,we give an up-to-date list of dystonia gene candi-dates to enhance the development of NGS diagnos-tics for DC (see online supplement 1).
METHODSWe systematically reviewed all papers regarding DCup to 20 October 2014, both genetic and non-genetic, in three age groups (infancy, childhood andadolescence), as proposed in the latest dystoniaclassification.1 For details of our systematic search,see online supplement 2.
DYSTONIA IN CHILDREN AND ADOLESCENTS:STATE OF THE ARTPhenomenology: Is it dystonia?The first step in diagnosing DC is the identificationof a hyperkinetic movement as being ‘dystonic’.Dystonia is defined as “a movement disorder char-acterised by sustained or intermittent muscle con-tractions causing abnormal, often repetitive,movements, postures or both. Dystonic movementsare typically patterned or twisting, and may betremulous. They are often initiated or worsened byvoluntary action and associated with overflowmuscle activation”.1 This definition of dystonia isidentical for adults and children1 3 and similar tothe definition of dystonia published by theTaskforce on Childhood Movement Disorders.6 Inchildren, dystonia is more often generalised com-pared with adult-onset dystonia.Correct identification of dystonia involves both
an understanding of classification systems andvisual pattern recognition. Three important, charac-teristic, clinical features of dystonia are: (1) pat-terned, predictable contractions of the samemuscles; (2) exacerbation when performing volun-tary movements (eg, walking, running, writing) and
774 van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

(3) the so-called geste antagoniste, or sensory trick. This phe-nomenon is characterised by the relief of dystonic movementsby lightly touching the relevant or adjacent part of the body. Asensory trick is particularly frequent in cranial and cervical dys-tonia, whereas limb and trunk involvement more often predom-inate in children. Therefore, a sensory trick is not an obligatoryfeature in DC; however, when observed, it strongly favours adiagnosis of dystonia.1 6
In children, movements should be evaluated in relation totheir developmental age. For instance, a healthy toddler canhave normal overflow movements that may look like dystonia,diminishing as the child’s development progresses.3 In additionto these normal movements, abnormal movements may alsomimic dystonia (table 1). For example, children with focal,stereotyped movements of the eyelids, face or neck are morelikely to have tics than focal dystonia.7 8
Reliable diagnostic criteria for different body localisations ofdystonia are needed to help clinicians accurately differentiatedystonia from conditions mimicking dystonia. Recently, a
diagnostic guideline for diagnosing blepharospasm has beenvalidated;9 however, blepharospasm is a form of focal dystoniathat rarely occurs in childhood or adolescence. For other bodylocalisations of dystonia, specific diagnostic criteria are anunmet need.
Classification of dystoniaThe most recent general classification scheme of dystonia identi-fies two distinct axes: axis I—clinical characteristics, and axisII—aetiology.1 Axis I describes the clinical features by (1) age atonset, (2) body distribution, (3) temporal pattern, (4) coexist-ence of other movement disorders and (5) other neurological orsystemic manifestations. Axis II addresses the aetiology via twocomponents: (1) nervous system pathology and (2) whether thedystonia is inherited or acquired. Classification of aetiology intothe categories ‘inherited’ or ‘acquired’ differs from traditionalclassification schemes in which dystonia was classified intoprimary genetic dystonia or secondary dystonia.1 The reason forthis change was that primary dystonias, heredodegenerative dys-tonias and dystonia-plus syndromes are all in fact genetic disor-ders.1 These three categories are now considered together as‘inherited’. In this review, we elaborate on this recent change inaetiological classification.
Aetiology of dystoniaThere are many possible aetiologies of DC. For this review, wehighlight acquired dystonias and treatable IEMs because aninitial approach other than NGS testing needs to be consideredfor these conditions. All other genetic causes can be tested atthe same time by means of NGS diagnostics.
Acquired dystoniasWe focus on acquired forms of dystonia that are relativelycommon and/or treatable. Drugs and toxic agents that may causeDC are listed in table 2. For other causes of acquired DC, clinicalclues and recommended investigations are summarised in table 3.
Drugs and toxic agentsDC can be induced by certain drugs and toxic agents, most com-monly neuroleptics and antiemetics (table 2).7 8 Drug-induceddystonias are categorised into acute dystonic reactions andtardive (chronic use) dystonia. The latter is a well-recognised dis-order in adults, but may also occur in children.7 Acute forms ofdystonia may arise after taking a few doses or even after oneadministration or accidental ingestion.8 The dystonia usually dis-appears rapidly on withdrawing the offending drug.
Cerebral palsyDyskinetic cerebral palsy (CP) is the most common cause ofacquired DC.10 CP is a clinical diagnosis, encompassing a groupof permanent disorders that cause impairment of movement andposture, attributed to non-progressive disturbances that occurredin the developing fetal or infant brain.11 Dyskinetic CP is char-acterised by the presence of choreoathetosis and dystonia11 andpossible aetiologies are heterogeneous.8 12 It is most common inchildren, born at term, who have experienced adverse perinataleffects, since the basal ganglia are particularly vulnerable topathogenic events towards the end of gestation.12 There areguidelines to help identify whether an acute intrapartum eventwas the likely cause of any particular case of CP.13 Owing to theaggressive treatment of perinatal hyperbilirubinaemia, it is nowrare to see kernicterus as a cause of dyskinetic CP.12
In dyskinetic CP, the hyperkinetic movements are usuallybilateral and mostly begin after the first year of life, and progress
Table 1 Mimics of dystonia in children and adolescents
Type of dystonia Mimics
Mimics of facial dystonia TicsStereotypiesFunctional
Mimics of cervical dystonia(head tilt)
TicsStereotypiesTrochlear nerve palsyVestibulopathySpasmus nutansAcquired nystagmusCongenital muscular torticollisSternocleidomastoid injuriesBenign paroxysmal torticollis of infancyPosterior fossa tumoursTumours in the pineal regionChiari malformationAtlanto axial subluxation (eg, syndrome of Grisel)Cervical tumours (in cervical cord, bone or softtissue)Upper spinal cord syringomyeliaJuvenile rheumatoid arthritisSandifer syndromeKlippel-Feil syndromeFunctional
Mimics of trunk dystonia ScoliosisStiff person syndromeFunctional
Mimics of limb dystonia(posturing)
Overflow movements in toddlers (normaldevelopmental movements)StereotypiesShoulder subluxationDystonic (tonic) ticsMyotoniaNeuromyotoniaCrampSatoyoshi syndromeRigiditySpasticityFocal tonic seizuresSpasms (hypocalcaemia, hypomagnesaemia,alkalosis)Deafferentation (pseudoathetosis)Functional
Mimics of generaliseddystonia
Self-stimulationOpisthotonusStiff person syndromeFunctional
van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106 775
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

slowly for several years.7 8 In children with severe CP, dystoniamay be so profound and sustained that it manifests as hyper-tonia rather than abnormal involuntary movements.3 Brain MRI
demonstrates abnormal findings in about 80% of individualswith CP.14 Genetic analysis is recommended in those caseswhere no specific cause can be determined, as several mono-genic disorders can present with clinical features similar to CP.15
Acquired structural lesionsStructural lesions, such as stroke, neoplasms or structurallyabnormal vessels including arteriovenous malformations, mayresult in unilateral DC (focal or hemidystonia).7 8 Childhoodstroke may result in dystonia if the caudate, lenticular nucleusor thalamus are involved.7 8 In most cases, the dystonia developsmonths or even years after the incident.
Autoantibody-associated and autoimmune disordersSeveral autoantibody-associated and autoimmune disorders canlead to DC (table 3).16 We put emphasis on two autoantibody-associated disorders, as early recognition and timely therapy canimprove the outcome significantly in these conditions.16
Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis inchildren is characterised by a combination of seizures, move-ment disorders, psychiatric symptoms and encephalopathy.16
The first symptom is often non-psychiatric.17 In addition to dys-tonia, multiple movement disorders can be seen in the samepatient,16 the most characteristic being orofacial dyskinesias.17
Young children often present with temper tantrums, hyperactiv-ity or irritability, whereas in older patients anxiety, psychosisand altered personality are the main psychiatric featuresobserved.17 Recognition of the combination of symptomsshould prompt testing for anti-NMDAR antibodies, both inserum and cerebrospinal fluid (CSF).17 Brain MRI, EEG andCSF may all show non-specific abnormalities.17 18 An under-lying neoplasm is found in approximately 6% of girls youngerthan 12 years but rarely in boys, whereas the association with anovarian teratoma increases in adolescent girls.18 Treatment
Table 2 Drugs and toxic agents that may cause dystonia inchildren and adolescents
Drugs
Dopamine receptorblocking drugs
(Neuroleptics, antiemetics)
Dopamine depleting drugs (eg, Tetrabenazine)Dopamine receptorstimulants
(L-dopa, dopamine receptor agonists)
Antihistaminic drugsTricyclic antidepressantsSerotonin reuptakeinhibitorsCholinergic agonists (eg, Trihexyphenidyl)Antiepileptic drugs (Especially phenytoin and carbamazepine)Antimalarials (eg, Chloroquine, amodiaquine)Calcium channel blockersDisulfiramLithiumCocaine
Toxins Main source
Carbon monoxideCyanideManganeseMethanolOrganophosphate
Smoke inhalation, poorly functioning heatingsystems or fuel-burning devicesInhalation of smoke, ingestion of toxic householdand workplace substances or cyanogenic foodsDrinking water with a high concentration ofmanganese, long-term parenteral nutritionIngestion of certain industrial products such asantifreeze solution or cleanersExposure to or ingestion of insecticides
Table 3 Clinical clues suggesting acquired dystonia
Clinical clue Differential diagnosis Recommended initial investigations
Acute onset dystonia or rapidly progressive course Structural lesionExternal insult*Autoantibody-associated movement disorderADEMInfection
NeuroimagingNeuroimagingAutoantibodies in serum and CSFNeuroimaging, CSFNeuroimaging, serum, CSF
Unilateral dystonia† Structural lesionExternal insult*Autoantibody-associated movement disorderDemyelinating disease‡Antiphospholipid syndrome§CP
NeuroimagingNeuroimagingAutoantibodies in serum and CSFNeuroimaging, CSFSerum investigationsNeuroimaging
Psychiatric symptoms (de novo) Autoantibody-associated movement disorderInfection
Autoantibodies in serum and CSFNeuroimaging, serum, CSF
Seizures (de novo) Structural lesionAutoantibody-associated movement disorderRasmussen’s syndrome¶Infection
NeuroimagingAutoantibodies in serum and CSFNeuroimagingNeuroimaging, serum, CSF
Signs of meningo-encephalitis or encephalitis Autoantibody-associated movement disorderInfection
Autoantibodies in serum and CSFNeuroimaging, serum, CSF
Abnormal birth or perinatal history CP NeuroimagingLocal signs of autonomic disturbances and pain CRPS I Clinical diagnosis**
*External insults include head trauma and hypoxic insults caused by near-drowning, cardiac arrest or status epilepticus.†Unilateral dystonia comprises either focal or hemidystonia.‡Demyelinating diseases including ADEM, multiple sclerosis and neuromyelitis optica.§Antiphospholipid syndrome with or without associated rheumatic disease such as systemic lupus erythematosus should be considered in all children with hemidystonia of unknownorigin.¶In Rasmussen’s syndrome, dystonia can be an accompanying sign or the presenting feature.**Criteria for CRPS are described by Mersky et al, see online supplemental references (supplement 4).ADEM, acute disseminated encephalomyelitis; CP, cerebral palsy; CRPS I, complex regional pain syndrome type I.
776 van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

consists of immunotherapy and oncological treatment in thosepatients with a clinically detectable tumour.18 Outcome is goodin the majority of patients treated early enough.18
Autoimmune basal ganglia encephalitis is a syndrome charac-terised by extrapyramidal movement disorders including dys-tonia and parkinsonism, sleep disturbance, dysautonomia andpsychiatric symptoms.16 Approximately 70% of cases haveserum antidopamine-2 receptor antibodies.16 Many patientshave MRI T2 hyperintense basal ganglia abnormalities andshow signs of CSF inflammation including oligoclonal bands.16
Immune therapy is the mainstay of treatment.16 17 In the past,encephalitis with dominant involvement of the basal ganglia wasgiven a variety of names, including encephalitis lethargica and(infantile) bilateral striatal necrosis.16 These disorders and auto-immune basal ganglia encephalitis may all be part of the sameclinical entity.16
InfectionsDC caused by infection is relatively rare, but has been reportedin children with viral infections, tuberculosis, mycoplasma ortoxoplasmosis.19 Infection by flaviviruses is an important causeof DC, the most common being Japanese encephalitis.19 Otherviruses associated with DC include influenza viruses, herpesviruses (including herpes simplex and herpes zoster) andmeasles viruses, which may lead to subacute sclerosing panence-phalitis.7 8 The main bacterial infections are tuberculosis andinfection by Mycoplasma pneumoniae.8 Infection should be sus-pected in any child with dystonia and pre-existing immunodefi-ciency or signs of meningoencephalitis or encephalitis.Detecting the infectious agent may be important for the type oftherapy chosen, and therefore serum and CSF investigations areindicated in addition to neuroimaging.
Treatable IEMsIEMs are highly heterogeneous. For most clinicians who do notwork daily with IEMs, it will be virtually impossible to recog-nise all these often extremely rare conditions. Fortunately, sinceall IEMs can be detected with NGS diagnostics, early identifica-tion is only necessary for those IEMs where timely treatmentcan improve the outcome.20
In general, an important clue for an IEM is a complex clinicalpicture comprising both neurological and non-neurological fea-tures. An overview of treatable IEMs associated with DC is pro-vided in online supplement 3. We defined ‘treatable’ as theavailability of a therapy that might lead to the improvement orprevention of symptoms. We will highlight five significant sub-groups of treatable IEMs that may cause DC.
Organic aciduriasOrganic acidurias can present both acutely and intermittentlyand are associated with ‘intoxication-like’ non-specific symp-toms, such as vomiting and anorexia, progressing towardsencephalopathy. Episodes are frequently triggered by intercur-rent illness, dietary changes or prolonged fasting.21 When theunderlying enzymatic defect is severe, onset will be in thenewborn period. Milder phenotypes may present later as aslowly progressive disorder or with an intermittent course.Examples of organic acidurias associated with DC are propionicaciduria, methylmalonic aciduria, cobalamin defects and glutaricaciduria type I.22
GLUT-1 deficiencyGLUT-1 deficiency, caused by mutations in the SCL2A1 gene,can give rise to paroxysmal dystonia triggered by prolonged
exercise.23 This phenotype is also referred to as paroxysmalexertion-induced dystonia. The SCL2A1 gene encodes for theglucose transport protein 1, and mutations in this gene com-promise glucose transport to the brain. Paroxysmal dystonia canbe the sole feature, but developmental delay, spasticity, ataxiaand epilepsy can also be part of the phenotype. A ketogenic dietis the current gold standard for treatment and has proven to bebeneficial in most cases.23
Metal storageWilson’s disease (WD) and dystonia with brain manganese accu-mulation (DBMA), caused by SLC30A10 mutations, are bothmetal storage disorders in which symptoms can be fully orpartly prevented by timely treatment.24 25 In both disorders, acombination of neurological symptoms and hepatic involvementis usually present. Other manifestations are psychiatric symp-toms and a corneal Kayser-Fleischer ring in WD and parkinson-ism and polycythaemia in DBMA. Indicative biochemicalfindings include low serum copper and ceruloplasmin in WDand hypermanganesaemia in DBMA.
Lysosomal storageNiemann Pick type C is a clinically heterogeneous disorder inwhich the presenting phenotype depends on the age of onset.Infants can present with ascites and liver or pulmonary disease.The classic presentation in mid to late childhood consists ofataxia, a supranuclear vertical gaze palsy, psychiatric symptoms,dystonia and dementia, whereas the clinical picture in adults isdominated by psychiatric symptoms and cognitive decline.26
Recently, treatment with miglustat has been shown to stabilisethe progression of neurological symptoms, including in paediat-ric patients.27
Dopa-responsive dystoniasDopa-responsive dystonias (DRD) are a group of disorders witha more insidious onset, probably representing 5% of childhooddystonias.28 The autosomal dominant form, GTP-cyclohydrolasedeficiency, is most common. This form is also known asSegawa’s disease and shows an excellent and sustained responseto low doses of levodopa.29 Typically, there is a diurnal fluctu-ation of symptoms, and associated parkinsonism. Furthermore,two autosomal recessive forms of DRD have been identified:tyrosine hydroxylase deficiency and sepiapterin reductase defi-ciency, both often accompanied by intellectual disability andophthalmological problems like oculogyric crisis, upward gazeand ptosis.30
Since DRD features can be non-specific and can show consid-erable phenotypic variability, DRDs are frequently misdiagnosedas CP.30 This may result in a considerable delay in diagnosis andadequate treatment.29 30
In addition to biochemical and molecular studies, a levodopatrial can be used as a diagnostic procedure. However, it shouldbe noted that a positive response on a levodopa trial is not spe-cific for the classic DRDs, but can also be seen in other disor-ders such as ataxia telangiectasia and GLUT-1 deficiency.31 32
Classification of genetic dystoniasThe genetic forms of dystonia including IEMs may be cate-gorised into two groups. The first group consists of the mono-genetic forms of dystonia with assigned genetic loci identified asDYT1–25, formerly named ‘primary dystonias’ and ‘dystoniaplus syndromes’. These disorders are characterised by isolateddystonia, or dystonia combined with parkinsonism or myoclo-nus.1 The second group consists of genetic disorders in which
van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106 777
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

dystonia is an important feature among several other neuro-logical and systemic features. On axis I of the latest dystoniaclassification, these co-occurring neurological or systemic mani-festations are classified as ‘associated features’.1 Important asso-ciated features in children include: ataxia, epilepsy, mentalretardation, spasticity, hypotonia, abnormal eye movements,neuropathy, deafness, ophthalmological signs, hepatosplenome-galy, psychiatric and dysmorphic features. These features aredecisive for accurate phenotyping and a prerequisite for correctinterpretation of NGS results.
NGS methodologyGenetic techniques using massive parallel sequencing are calledNGS. With these new techniques, sequencing the entire genomeof a patient (whole-genome sequencing; WGS), the codingregions (exons) of every gene (whole-exome sequencing; WES)or targeting specific disease-causing genes (targeted resequen-cing; TRS) have all become a reality in DNA diagnostics.Technical details of the specific methods fall outside the scopeof this review, but are described elsewhere.33
It is important to recognise that with WGS or WESapproaches, information for all genes will become available,including those not relevant to the diagnostic question. Thesegenes need to be excluded to restrict the data analysis to a list ofknown genes that might explain the phenotype. If the pheno-type is unique and no mutation is found in the selected genes,the information about the excluded genes may be used to huntfor new disease-causing genes. The drawbacks of WGS andWES are high costs, the risk of unsolicited findings, and cover-age that is usually less than in TRS panels, compromising thediagnostic accuracy. In TRS panels, a preselected list of severalknown genes that cause dystonia are tested. By sequencing onlypreselected genes, the coverage increases significantly, contribut-ing to diagnostic accuracy, and unsolicited findings are mini-mised, at significantly lower costs.
The important benefits of NGS diagnostics compared withregular biochemical procedures are that shipping DNA to refer-ral centres is relatively cheap and straightforward, without strin-gent shipping conditions. In contrast, the costs and conditionsof shipping samples, for instance, for (CSF) biochemical testscan be a serious hurdle in the present diagnostic process.
It is to be expected that in the near future the widespread useof NGS, both in research and in clinical diagnostics, will lead tomany more reports of dystonia-associated genes, and the list ofassociated genes will grow rapidly. However, it is important thatindependent confirmation of the causal relationship betweengene variants and dystonia is performed because, in some of therecently annotated dystonia genes, variants in these genes alsooccur with high frequency in the general population.34
A NEW DIAGNOSTIC ALGORITHMOwing to the extraordinarily broad range of possible causes ofDC, several algorithms have been developed to assist cliniciansin making diagnostic decisions.2 35 36 These algorithms are notwidely applicable as they mainly focus on (rare) neurometaboliccauses and do not make use of the availability of NGS method-ologies. On the basis of our systematic literature review and ourown clinical experience, we propose a new diagnostic algorithmwith five steps (figure 1).
Step 1: Is it dystonia?The first step in the algorithm is to record a careful history andperform a physical and neurological examination to determinethat dystonia is an important feature.
Movement disorders that may be misdiagnosed as dystoniaare listed in table 1. In general, these ‘pseudodystonias’ have aknown or presumed cause that is thought to differ from thecauses of the broader dystonia group.1 Applying the algorithmand using NGS testing is not advised in these conditions.
Step 2: Could the dystonia be medication-induced or causedby toxic agents?The second step is to verify exposure to any medication or toxicagents that could be causing the dystonia (table 2). Treatmentconsists of discontinuing medication or prevention of furthertoxic exposure and, if possible, detoxification.
Step 3: Clinical clues suggesting acquired dystonia?Step 3 is to consider whether the dystonia could be acquired. Intable 3, we indicate red flags for acquired disorders with themain subgroups. These red flags are only defined to guide clini-cians to a limited number of disorders in which immediate diag-nosis and treatment is necessary to identify treatable disorders,preventing insults to the brain during the diagnostic process.
Step 4: Biochemical investigations and levodopa trialIn any child with dystonia without obvious clues for an acquiredcause, we recommend performing a laboratory workup (table 4)aimed at identifying the treatable forms, before moving on toNGS testing. Of course, this recommendation only applies forthose centres where biochemical diagnostics will provide fasterresults than NGS testing, depending on the local facilities. CSFinvestigations are only recommended in selected patients(table 4) because otherwise the diagnostic yield of CSF investi-gations is likely to be rather low.37 38
In addition to the laboratory investigations, we recommendthat all patients receive a trial of levodopa with carbidopa.30
The primary goal of the trial is diagnostic. However, an add-itional advantage is that levodopa can also give symptom reliefin non-DRD dystonia.39 The recommended starting dose oflevodopa is 1 mg/kg/day, to be gradually increased until com-plete benefit, or until dose-limiting side effects occur.7 Mostindividuals respond to 4–5 mg/kg/day in divided doses.40
Levodopa should be given for 3 months before considering thetrial a failure.39
Step 5: NGSSimultaneously with the biochemical investigations and the initi-ation of the levodopa trial, all possible genetic causes can beapproached by using NGS diagnostic technologies. To facilitatethis, we provide a list of DC-associated genes (see online supple-ment 1). For those cases that remain unsolved after NGStesting, referral to a tertiary referral centre is recommended tofurther explore the possibilities to obtain an aetiologicaldiagnosis.
DISCUSSIONWe provide a comprehensive overview of DC and propose anew diagnostic algorithm (figure 1). This five-step approach pro-vides guidance for clinicians to determine which patients maybenefit from innovative genetic tests and those for whom otherinvestigations are required first, while taking into account theimportance of early recognition of acquired and treatable causesof DC.
Our proposed flow chart (figure 1) differs from existing algo-rithms in that certain commonly used processing steps havebeen omitted, such as age at onset, temporal pattern (eg, persist-ent or paroxysmal), associated features and mode of
778 van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

inheritance.2 35 36 Indeed, ‘pattern recognition’ based on thesefeatures has been important in the delineation of dystonia disor-ders and can still be successful in identifying classical pheno-types, especially by experts in the field.1 8 However, thesefeatures were not included in our algorithm because many clini-cians will have limited experience with these rare disorders andspecific clinical patterns will easily remain unrecognised. In add-ition, recent insights from more widely applied NGS testingdemonstrate that the clinical heterogeneity of many disorders ismuch larger than expected,23 31 so clinical pattern recognitionof milder, intermediate and unusual phenotypes remainsproblematic.
Nevertheless, careful clinical phenotyping still remains indis-pensable for two reasons. First, clinicians need to define, on thebasis of these clinical parameters, the a priori risk that thepatient is indeed suffering from a genetic disorder. NGS meth-odology should not be used when the a priori risk is low,because the numerous genes being tested increase the chancethat variants will be misinterpreted as disease-causing, in genesthat are unlikely to explain the clinical phenotype. Second,closely related to the first reason, detailed phenotyping is keywhen the results of NGS diagnostic strategies are available andneed to be interpreted. As Hennekam and Biesecker41 clearly
stated, NGS and computers will not magically make patientdiagnoses for us. Instead, there will be a shift from apre-NGS-test differential diagnostic mode to a post-NGS-testdiagnostic assessment mode.41 Thus, the diagnostic skills of clin-icians will be integrated into the evaluation of NGS test resultsto make molecular diagnoses together with laboratory staff.
Notably, clinicians using NGS diagnostics should be awarethat there are some technical pitfalls in the application of NGSdiagnostics such as a limited ability to detect large structuralrearrangements. In DC, this is particularly relevant if no causa-tive mutation in a gene can be identified by NGS techniques,while at the same time the clinical picture is compatible with,for example, myoclonus dystonia or paroxysmal kinesigenic dys-kinesia, both disorders that may be caused by deletions (inSCGE and PRRT2, respectively). In these cases, additionalgenetic tests detecting deletions are still required, such as multi-plex ligation-dependent probe amplification or array-comparative genomic hybridisation (array-CGH).42
At present, we live in a period of transition between emergingNGS diagnostic tests and changing costs, budgets and availabil-ity of diagnostic procedures. In the future, NGS tools willbecome increasingly available in many areas of clinical diagnos-tics and clinical decision-making, and will be incorporated in
Figure 1 Diagnostic algorithm of dystonia in children and adolescents (IEM, inborn error of metabolism).
van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106 779
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

our daily work and change our daily routines. Although not apanacea, the advantages of this new strategy will be earlier diag-nosis, avoidance of unnecessary investigations and the possibilityof genetic counselling for family members. It will cruciallyshorten the time patients with DC and their families spend inuncertainty awaiting a definitive diagnosis.
Acknowledgements The authors thank Kate McIntyre and Jackie Senior,University Medical Center Groningen, Department of Genetics, for editing themanuscript.
Contributors MEvE was involved in the design and conceptualisation of the study,analysis and interpretation of the data, drafting and revision of the manuscript. AKwas involved in the analysis and interpretation of the data, drafting and revision of themanuscript. HE, RJS, CCV-B, DAS and OFB were involved in the revision of themanuscript. MAJT and TJdK were involved in the design and conceptualisation of thestudy, analysis and interpretation of the data, and revision of the manuscript.
Competing interests MAJT received research grants from Fonds Nuts-Ohra,Stichting wetenschapsfonds dystonie vereniging, Prinses Beatrix Foundation, STWTechnology society (NeuroSIPE). Unrestricted grants were received from IpsenPharmaceuticals, Allergan Pharmaceuticals and Medtronic for a dystonia nurse,DystonieNet and a teaching course. TJdK received a research grant from Metakidsfoundation.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES1 Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of
dystonia: a consensus update. Mov Disord 2013;28:863–73.2 Garcia-Cazorla A, Wolf NI, Serrano M, et al. Inborn errors of metabolism and motor
disturbances in children. J Inherit Metab Dis 2009;32:618–29.3 Mink JW. Special concerns in defining, studying, and treating dystonia in children.
Mov Disord 2013;28:921–5.4 de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in
persons with severe intellectual disability. N Engl J Med 2012;367:1921–9.5 Neveling K, Feenstra I, Gilissen C, et al. A post-hoc comparison of the utility of
sanger sequencing and exome sequencing for the diagnosis of heterogeneousdiseases. Hum Mutat 2013;34:1721–6.
6 Sanger TD, Chen D, Fehlings DL, et al. Definition and classification of hyperkineticmovements in childhood. Mov Disord 2010;25:1538–49.
7 Singer HS, Jankovic J, Mink JW, et al. Movement disorders in childhood.Philadelphia: Saunders Elsevier, 2010.
8 Donaldson IM, Marsden CD, Schneider SA, et al. Marsden’s book of movementdisorders. Oxford: Oxford University Press, 2012.
9 Defazio G, Hallett M, Hyder A, et al. Development and validation of a clinicalguideline for diagnosing blepharospasm. Neurology 2013;80:236–40.
10 Lin JP, Lumsden DE, Gimeno H, et al. The impact and prognosis for dystonia inchildhood including dystonic cerebral palsy: a clinical and demographic tertiarycohort study. J Neurol Neurosurg Psychiatry 2014;85:1239–44.
11 Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classificationof cerebral palsy April 2006. Dev Med Child Neurol Suppl 2007;109:8–14.
12 Himmelmann K, McManus V, Hagberg G, et al.; SCPE collaboration. Dyskineticcerebral palsy in Europe: trends in prevalence and severity. Arch Dis Child2009;94:921–6.
13 MacLennan A. A template for defining a causal relation between acute intrapartumevents and cerebral palsy: international consensus statement. BMJ 1999;319:1054–9.
14 Bax M, Tydeman C, Flodmark O. Clinical and MRI correlates of cerebral palsy: theEuropean Cerebral Palsy Study. JAMA 2006;296:1602–8.
15 Moreno-De-Luca A, Ledbetter DH, Martin CL. Genetic insights into the causes andclassification of cerebral palsies. Lancet Neurol 2012;11:283–92.
16 Dale RC, Brilot F. Autoimmune basal ganglia disorders. J Child Neurol2012;27:1470–81.
17 Dalmau J, Lancaster E, Martinez-Hernandez E, et al. Clinical experience andlaboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol2011;10:63–74.
18 Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors forlong-term outcome in patients with anti-NMDA receptor encephalitis: anobservational cohort study. Lancet Neurol 2013;12:157–65.
19 Misra UK, Kalita J. Spectrum of movement disorders in encephalitis. J Neurol2010;257:2052–8.
20 van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectualdisability: a systematic literature review. Mol Genet Metab 2012;105:368–81.
21 Saudubray JM, Sedel F, Walter JH. Clinical approach to treatable inborn metabolicdiseases: an introduction. J Inherit Metab Dis 2006;29:261–74.
22 Deodato F, Boenzi S, Santorelli FM, et al. Methylmalonic and propionic aciduria. AmJ Med Genet C Semin Med Genet 2006;142C:104–12.
23 Pearson TS, Akman C, Hinton VJ, et al. Phenotypic spectrum of glucose transportertype 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep 2013;13:342–53.
Table 4 Biochemical investigations to identify treatable inborn errors of metabolism with dystonia as an important feature
Laboratory testIn sampleof Disorder
Organic acids Urine Glutaric aciduria type I, propionic aciduria, methylmalonic aciduria, cobalamin deficienciesLactate Plasma Propionic aciduria, methylmalonic aciduria, biotin responsive basal ganglia diseasePyruvate Plasma Pyruvate dehydrogenase complex deficiencyAcylcarnitines Plasma Propionic aciduria, methylmalonic aciduria, glutaric aciduria type 1Amino acids Plasma Ornithine transcarbamylase deficiency, maple syrup urine disease, pterin defectsHomocysteine Plasma HomocystinuriaCopper, ceruloplasmin Plasma,
urineWilson’s disease
Manganese Plasma Dystonia with brain manganese accumulation
Biotinidase Plasma Biotinidase deficiencyCreatine, guanidinoaceticacid
Plasma,urine
Cerebral creatine deficiency syndrome 3 (AGAT deficiency), guanidinoacetate methyltransferase deficiency
Vitamin E (α-tocopherol) Plasma Ataxia with vitamin E deficiencyUric acid Plasma Lesch-Nyhan syndromeCholestanol Plasma Cerebrotendinous xanthomatosisGlucose CSF, plasma GLUT-1 deficiencyFolate CSF Cerebral folate deficiencyHVA, 5-HIAA CSF Tyrosine hydroxylase deficiencyPterines CSF, urine GTP-cyclohydrolase 1 deficiency, 6-pyruvoyl-tetrahydropterin synthase deficiency, aromatic l-amino acid decarboxylase
deficiencySepiapterin CSF Sepiapterin reductase deficiency
Performing this set of laboratory investigations is only recommended if obtaining the results of these tests will be faster than NGS testing.Lumbar puncture seems justified only in selected cases with a high clinical suspicion for these disorders.AADC, aromatic l-amino acid decarboxylase; AVED, ataxia with vitamin E deficiency; CSF, cerebrospinal fluid; NGS, next-generation sequencing; PDC, pyruvate dehydrogenase complex;PTPS, 6-pyruvoyl-tetrahydropterin synthase.
780 van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from

24 Taly AB, Meenakshi-Sundaram S, Sinha S, et al. Wilson disease: description of 282patients evaluated over 3 decades. Medicine (Baltimore) 2007;86:112–21.
25 Stamelou M, Tuschl K, Chong WK, et al. Dystonia with brain manganeseaccumulation resulting from SLC30A10 mutations: a new treatable disorder. MovDisord 2012;27:1317–22.
26 Patterson MC, Mengel E, Wijburg FA, et al. Disease and patient characteristics inNP-C patients: findings from an international disease registry. Orphanet J Rare Dis2013;8:12–22.
27 Lyseng-Williamson KA. Miglustat: a review of its use in Niemann-Pick disease typeC. Drugs 2014;74:61–74.
28 Jankovic J. Treatment of dystonia. Lancet Neurol 2006;5:864–72.29 Tadic V, Kasten M, Bruggemann N, et al. Dopa-responsive dystonia revisited:
diagnostic delay, residual signs, and nonmotor signs. Arch Neurol 2012;69:1558–62.30 Friedman J, Roze E, Abdenur JE, et al. Sepiapterin reductase deficiency: a treatable
mimic of cerebral palsy. Ann Neurol 2012;71:520–30.31 Saunders-Pullman R, Raymond D, Stoessl AJ, et al. Variant ataxia-telangiectasia
presenting as primary-appearing dystonia in Canadian Mennonites. Neurology2012;78:649–57.
32 Baschieri F, Batla A, Erro R, et al. Paroxysmal exercise-induced dystonia due toGLUT1 mutation can be responsive to levodopa: a case report. J Neurol2014;261:615–16.
33 Keogh MJ, Daud D, Chinnery PF. Exome sequencing: how to understand it. PractNeurol 2013;13:399–407.
34 Zech M, Gross N, Jochim A, et al. Rare sequence variants in ANO3 and GNALin a primary torsion dystonia series and controls. Mov Disord 2014;29:143–7.
35 Assmann B, Surtees R, Hoffmann GF. Approach to the diagnosis of neurotransmitterdiseases exemplified by the differential diagnosis of childhood-onset dystonia.Ann Neurol 2003;54(Suppl 6):S18–24.
36 Gouider-Khouja N, Kraoua I, Benrhouma H, et al. Movement disorders inneuro-metabolic diseases. Eur J Paediatr Neurol 2010;14:304–7.
37 Haliloglu G, Vezir E, Baydar L, et al. When do we need to perform a diagnosticlumbar puncture for neurometabolic diseases? Positive yield and retrospectiveanalysis from a tertiary center. Turk J Pediatr 2012;54:52–8.
38 Molero-Luis M, Serrano M, Ormazábal A, et al. Homovanillic acid in cerebrospinalfluid of 1388 children with neurological disorders. Dev Med Child Neurol2013;55:559–66.
39 Roubertie A, Mariani LL, Fernandez-Alvarez E, et al. Treatment for dystonia inchildhood. Eur J Neurol 2012;19:1292–99.
40 Thenganatt MA, Jankovic J. Treatment of dystonia. Neurotherapeutics2014;11:139–52.
41 Hennekam RC, Biesecker LG. Next-generation sequencing demands next-generationphenotyping. Hum Mutat 2012;33:884–6.
42 Dale RC, Grattan-Smith P, Nicholson M, et al. Microdeletions detected usingchromosome microarray in children with suspected genetic movement disorders:a single-centre study. Dev Med Child Neurol 2012;54:618–23.
van Egmond ME, et al. J Neurol Neurosurg Psychiatry 2015;86:774–781. doi:10.1136/jnnp-2014-309106 781
Movement disorderscopyright.
on 5 Septem
ber 2018 by guest. Protected by
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp-2014-309106 on 13 Novem
ber 2014. Dow
nloaded from