dft-md approach to tio /liquid interface systems for ... approach to tio 2/liquid interface systems...
TRANSCRIPT

DFT-MD approach to TiO2/liquid interface systems
for photocatalysis and dye-sensitised solar cell
Yoshitaka TATEYAMA1,2, Masato SUMITA1, and Keitaro SODEYAMA1
1Nano-System Computational Science Group,International Centre for Materials Nanoarchitectonics (WPI-MANA),
National Institute for Materials Science (NIMS),1-1 Namiki, Tsukuba, Ibaraki 305-0044
2PRESTO and CREST, Japan Science and Technology Agency (JST),4-1-8 Honcho, Kawaguchi, Saitama 333-0012
1 Introduction
Solid-liquid interface plays a crucial role inmost of energy conversion applications suchas fuel cell, Li-ion battery, solar cell and pho-tocatalysis. The efficiencies of these applica-tions have been improved for the last decadesby introducing new materials and systems inthe experimental and engineering sides, andmany commercial products have been alreadydeveloped. Nevertheless, the atomistic under-standing of phenomena at the solid-liquid in-terfaces has remained unsolved, because of thedifficulty in observing the ’hidden’ interfacialphenomena on the atomic scale. In order toachieve a breakthrough innovation over thecurrent incremental improvement, fundamen-tal elucidation is quite indispensable. In thisrespect, there is a growing expectation for com-puter simulations with high predictability.
For accurate simulations of realistic sys-tems on the atomic and electronic scales, first-principles calculations will be most appropri-ate. In particular, density functional theory(DFT) calculation is a best approach, becauseit can be applied to both condensed matterand molecular systems with reasonable accu-racy and computational cost. However, sim-ple applications of existing DFT calculationcodes to the interface systems are insufficient.Since solid-liquid interfaces are closely asso-ciated with long-established electrochemistry,one has to take the concepts in the electro-
chemistry (ex. electric double layer, flatbandpotential etc.) into account for the compati-bility.
With this point of view, we have been study-ing the solid-liquid interfaces related to energyand environmental issues, in particular semi-conductor electrode/solution interfaces. Weaddressed not only fundamental research forthe fusion of DFT and electrochemistry, butalso elucidation of microscopic mechanisms ofthe realistic applications. In fact, the semi-conductor electrode sytsems can provide vari-ous ways of energy conversion, since it may in-volves photoexcitation process as well as elec-tron transfer. Besides, the DFT approach isquite suitable since a variety of nanostructuresis to be treated on the semiconductor surfaces.
As model semiconductor electrodes, we havebeen working on DFT-based molecular dy-namics (MD) analysis of TiO2/solution in-terfaces related to photocatalysis and dye-sensitized solar cell (DSC) on the atomic andelectronic scales for the last few years. Ourfinal goal is understanding of the atomisticmechanisms of the interfacial reactions. Forthis purpose, it is inevitable to deal with elec-tron transfer (redox) processes as well as bondcleavage/formation (adsorption/desorption) atthe interfaces, taking dynamics of the solventmolecules into account. Thus, we address toestablish free energy calculation techniques forthese processes at the interfaces. Such calcula-tions require the initial, final and possible in-
1

termediate states in advance. Therefore, thefirst step should be to obtaint the equilibriumstructures and electronic states at the solid-liquid interfaces
Figure 1: Typical reaction schemes of photo-catalysis and dye-sensitised solar cell.
Our first target system was, then, theTiO2/water interface relevant to photocataly-sis. In particular, we focused on anatase poly-morph, which is believed to be more reactivethan the most popular rutile form. We ex-amined the equilibrium states of TiO2 anatase(101) and (001)/H2O interfaces. The formercrystal face is regarded as being most respon-sible for the reactivity, while the latter hasbeen recently suggested as an alternative. Sev-eral mechanisms have been already proposedfor the interfacial reactions of photocatalysis.However, most of them are based on the baresurface with submonolayer of water molecules(Fig. 1), and thus understanding of more re-alistic hydrated TiO2 surface is still lacking.Therefore, we especially examined the bulk wa-ter effects on the water adsorption on the inter-faces and the interfacial hydrogen bond (HB)network, which enables to expect how the so-lute (reactant) approaches to the interface. Wealso discussed the equilibrium electronic states
and the hydrophilicity/hydrophobicity.
The second target is a typical DSC interfaceconsisting of TiO2 anatase (101) surface andliquid acetonitrile (CH3CN(MeCN)). DSCs arebeing developed as alternatives to the con-ventional Si-based solar cells from the view-point of efficiency and cost performance. Theschematic scheme is shown in Fig. 1. Thesystems consisting of Ru(II) polypyridyl dyeswith MeCN solution usually give high effi-ciency (over 11%). This MeCN solution alsobrings about higher efficiency and durabilitywith various types of dyes. On the other hand,protic solvents like water and alcoholic solventsshow decrease of the efficiency. In general, in-troduction of the electrolyte solution is carriedout under ambient condition after the dye ad-sorption so that the involvement of ubiquitousH2O molecules in the DSC system is inevitable.Therefore, we investigated the advantage ofthe TiO2/MeCN interface and its water con-tamination effect from the viewpoint of dura-bility as well as efficiency of the DSC.
We also examined the adsorption modes ofRu(II) polypyridyl dyes. Upon the adsorptionon the TiO2 anatase (101) surface, carboxylgroups in the polypyridyl ligands act as an-chors to five-fold coordinated Ti (Ti5c) siteson the surface. For representative N3/N719dyes with two bipyridyl ligands, the adsorp-tion with two deprotonated carboxyl groupanchors was believed to be most probable.However, a three-anchors mode has been re-cently proposed. Furthermore, the protona-tion/deprotonation of the carboxyl anchors isstill controversial. Hence the number of an-chors and the proton position are still mat-ters. Since these affect the interfacial elec-tronic states and the electron injection effi-ciency, detailed investigation of the adsorptionmode is still indispensable. We then investi-gated the adsorption of Ru N749 dye, so calledblack dye, another representative dye givingthe highest efficiency.
In this article, we first discuss the charac-teristics of solid-liquid interfaces to be takeninto account in the DFT calculations in Sec. 2.Then we briefly introduce the recent threeprojects in our group: (I) TiO2 anatase(101) and (001)/ H2O interfaces for photo-
2

catalysis [1, 2], (II) TiO2 anatase (101) /MeCN interface and its water contaminationfor DSC [3], and (III) the adsorption modeof black dye on the TiO2 anatase (101) sur-face [4].
2 Calculation
Computational analysis of solid-liquid inter-face on the atomic scale is still a challengingissue. First of all, proper treatment of theliquid state is crucial. The solvent moleculesusually adsorb on the semiconductor (oxide)surface, and the solvent network near the in-terface can be different from that in the bulkliquid. Since these may affect the motion ofreactant and product species and thus the in-terfacial reactivity, the explicit solvent modelis essential. In the electron transfer and chem-ical reactions, solvent dynamics plays an im-portant role and the reaction properties canbe described as statistical ensembles. There-fore, sampling with dynamics is indispensable.Keeping realistic liquid condition is also nec-essary, because the interfacial reaction can besensitive to the liquid pressure and tempera-ture. Periodic boundary condition (PBC) ismore appropriate for this purpose.
Related to electrochemistry, description ofthe electric double layer and the space-chargelayer is crucial. Thus, the finite size effect ofsupercell with PBC should be checked. Sincethese layers are associated with the interfacialcharge and dipole, careful treatment of chargein the supercell is also necessary. The effects ofelectrolyte and counter ions are to be checked,although they are regarded not to affect thereactions in principle. When considering thebias effect, one has to take into account elec-trochemical potential of the electrode such asflatband potential and redox potential of thetarget species, with respect to the referencepotential (ex. standard hydrogen electrode).Since these potentials are originally related tothe free energy difference, not electric field, thefree energy analysis by the statistical treat-ment is essential. At the interfaces, the elec-tron transfer coupled to the adsorption shouldbe considered as well, which is closely associ-ated with the over potential.
Figure 2: (a) Atomistic model of TiO2/H2Ointerfaces used in our DFT-MD analysis. (b)Typical concepts for solid-liquid interface inelectrochemistry, and our modeling for the in-terfaces; periodic boundary condition/double-face slab model.
Toward such free energy analysis, we firstobtained the equilibrium structures and elec-tronic states of the solid-liquid interfaces.Even in the equilibrium states, proton transfer,solvation/desolvation, adsorption/desoprtioncan be involved. Therefore, first-principlestreatment of the electronic states is essen-tial, compared to the classical force field tech-niques. A promising approach is using DFTsampling with Car-Parrinello type MD. Thisuses the ’on-the fly technique’ for the dynam-ics so that the convergence problem of theelectronic states at unstable geometries can beavoided. On the other hand, keeping the calcu-lation system near the Born-Oppenheimer sur-face is necessary.
The common computational conditions inour studies are as follows. For reasonablecomputational cost and accuracy, we usedthe BLYP functional. Plane wave basisset with the cutoff energy of 70 Ry andthe norm-concerving pseudopotentials wereused. For the Ti pseudopotential, we usedthe non-linear core corrections. The valid-ity was checked with calculations of atom and
3

molecules/complexes as well as bulk solid. To-tal energies were calculated with the Γ pointsampling of the super cell approach. Forthe NVT ensemble, we used Nos-Hoover ther-mostats with the temperature of 300K. Thetime step was set to 5 a.u., and 500 a.u. wasused for the fictitious mass for electrons in theCar-Parrinello dynamics. After equilibrationwith a couple of picoseconds, we sampled theequilibrium trajectories.
In the following, we exlpain details of the in-dividual projects. There are many DFT calcu-lations for TiO2/H2O intefaces. Most of them,however, used model interfaces with some wa-ter molecules only. In contrast, we used super-cells involving TiO2 slabs and bulk H2O re-gion. TiO2/ anatase (101) and (001) slabs in-volve three or four bilayers, corresponding to(TiO2)36 thru (TiO2)48. The number of H2Omolecules are changed between 56 and 96. Inorder to achieve the ambient condition of wa-ter, we determine the number of water in thesupercell by comparing the radial distributionfunction (RDF) with the experimental results.This procedure is crucial to claim that our sim-ulations correspond to the ambient condition.To eliminate the initial structure dependence,we carried out seveal DFT-MD sampling fromdifferent structures. Check of the size effectsuggests that the interfacial water behaviouris qualitatively the same between the smallerand larger supercells, whereas the electronicstates are slightly affected within the currentsupercell sizes. Due to the computational cost,we adopted (TiO2)36 supercells (Fig. 3) for theanalysis.
For the TiO2/MeCN interfaces in theproject II, we used a monoclinic supercell in-volving (TiO2)36 slab of anatase (101) surfacewith 47 liquid MeCN molecules for the ambi-ent condition. The cell parameters are 11.2526A×10.23952 A×44.05780 A with α = 111.689degree. The number of MeCN was determinedby comparison of the RDFs between the ex-periment and the calculation. In the ’watercontamination’ system, we replaced one MeCNwith an H2O molecule. As in the project I, wecarried out seveal DFT-MD sampling from dif-ferent initial positions of the water.
In the project III for the black-dye adsorp-
tion, we calculated the relative stability amongthe structures with one and two anchors as wellas protonated and deprotonated. As the blackdye includes large terpyridyl ligand, large sur-face area is necessary. Thus we used 20.47904A× 22.7052 A× 34.0578 A monoclinic super-cell with α = 111.689 degre, correspondingto the (TiO2)96. The geometry optimisationwas done at the BLYP functional level. Asshown below, we found a structural stabilitydifferent from the conventional one. In orderto confirm this calculated stability, we carriedout the comparison of UV-Vis spectra betweenour calculation and experiment. Extractingthe adsorbed structure of black dye only fromthe supercell, we carried out TDDFT calcu-lations with the B3LYP functional. The effec-tive core potential of Stuttgart-Dresden with f-type polarized functions was used for Ru atom,while the cc-pVDZ basis sets were adopted forthe others. The solvent effects on the excita-tion spectra were included by using the CPCMtechnique.
Finally we describe the computational cost.The code used in our studies (CPMD) hasa reasonable parallel efficiency. Thus we ex-ploited 512 or 1024 MPI calculations for thelarger systems. The DFT-MD samplings ofthe supercell with moderate sizes were usuallycarried out with 128-256 MPI calculations. Onthe ISSP supercomputer, it takes several weeksto complete the sampling at each condition.This parallel efficiency is more improved by us-ing hybrid parallellisation. Therefore it couldbe possible now to carry out DFT-MD sam-pling of systems with more than 1000 atoms.
3 Results and Discussion
3.1 TiO2 anatase/liquid H2O inter-faces for photocatalysis
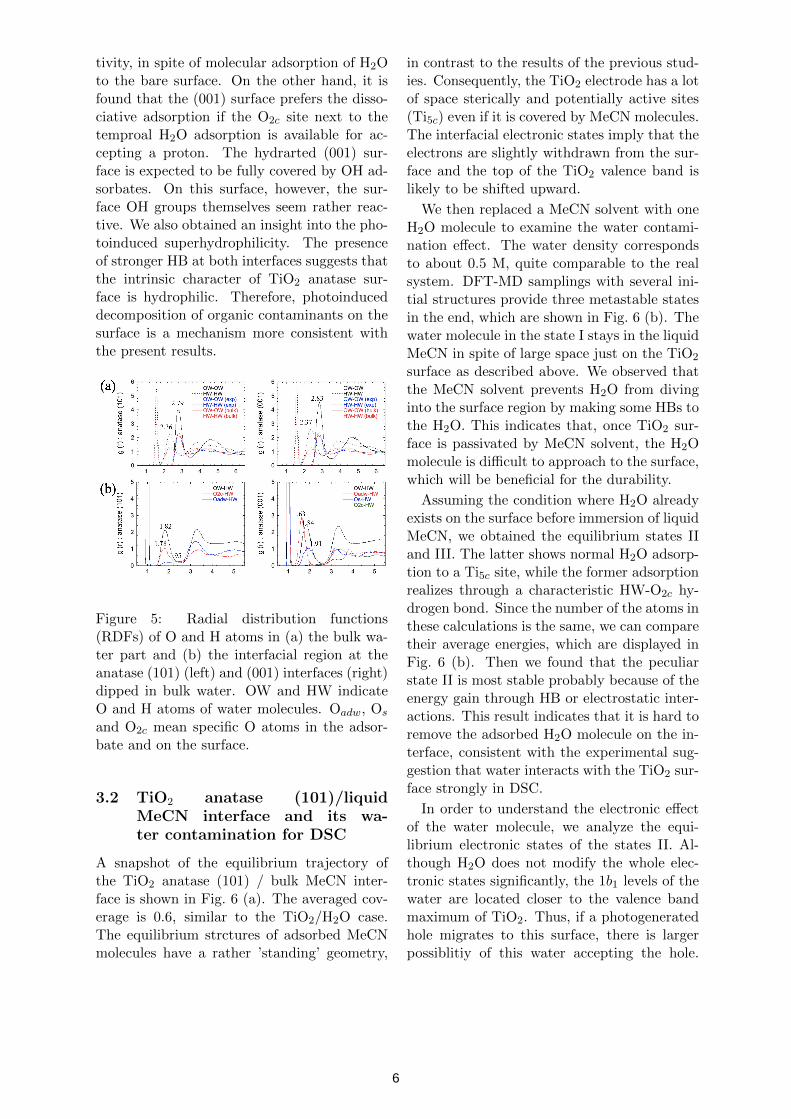
Snapshopts of equilibrium trajectories forTiO2 anatase (101) and (001) interfaces dippedin bulk water are shown in Fig. 4. Averagedstructures are analyzed with RDFs from the se-lected Ti, O and H atoms in the system. Firstof all, we examined the bulk water part in thepresent unit cells (Fig. 5 (a) ) in comparisonwith the experimental data and calculated re-
4

Figure 3: A representative supercell for theTiO2 anatase (101)/bulk H2O interface in ourstudies. Liquid water under ambient conditionis realized with the periodic boundary condi-tion.
sults of water only. These RDFs show the peakpositions consistent with those in the refer-ences, indicating that the present bulk water isunder ambient condition. We then investigatethe adsorption modes of water molecules onboth surfaces. Our calculations show that the(101) surface prefers the molecular adsorption,while the dissociative adsorption is predomi-nant on the (001) surface. These are the sameas the model proposed in the previous calcula-tions with some water adsorbates only. How-ever, the detailed structures are different. Thepresent water adsorbates on the (101) surfacehave HBs to ’the second layer water’, thoughHBs to the surface O2c sites are dominant inthe case of a few molecules. Our DFT-MDsampling with bulk water also gave informa-tion of the adsorption pathways.
More distinctive features were observed inthe interfacial HB networks. The RDFswith respect to the various types of O atoms(Fig. 5 (b)) indicate that there are stronger HBaround the interfaces than in the bulk water.They are shown in the red lines in Fig. 4. We
Figure 4: Equilibrium structures of TiO2
anatase (a) (101) and (b) (001) surfaces dippedin bulk water at room temperature. Pinkspheres represent Ti atoms. Red spheres showO atoms in TiO2 and bulk H2O. Green meanOxygen in the adsorbate, while blue and yel-low indicate Oxygen in the second layer at theinterfaces. Strong and weak hydrogen bonds(HBs) are displayed with red and green dot-ted lines. Blue dotted lines show normal HBsin the bulk water. Undercoordinated Ti (Ti5c)and O (O2c) are labeled.
also observed weaker HB on the minor (001)interfaces, which is displayed with green linesin Fig. 4. These are in good agreement withthe results of the 1H-NMR and SFG experi-ments. In addition, we found novel adsorptioncharacters as well. We then suggested a two-layer model for the interfacial water on bothsurfaces examined.
These equilibrium structures give insightsinto the reactivity of each interface as well.In contrast to naive expectation of full cover-age, the coverage of H2O adsorption is foundaround 0.6 on the anatase (101) interface. Thismeans that there are some unadsorbed Ti5csites on the surface, which can be reaction sitesof the catalysis targets. Thus the hydrated(101) interface may still have a certain reac-
5
tivity, in spite of molecular adsorption of H2Oto the bare surface. On the other hand, it isfound that the (001) surface prefers the disso-ciative adsorption if the O2c site next to thetemproal H2O adsorption is available for ac-cepting a proton. The hydrarted (001) sur-face is expected to be fully covered by OH ad-sorbates. On this surface, however, the sur-face OH groups themselves seem rather reac-tive. We also obtained an insight into the pho-toinduced superhydrophilicity. The presenceof stronger HB at both interfaces suggests thatthe intrinsic character of TiO2 anatase sur-face is hydrophilic. Therefore, photoinduceddecomposition of organic contaminants on thesurface is a mechanism more consistent withthe present results.
Figure 5: Radial distribution functions(RDFs) of O and H atoms in (a) the bulk wa-ter part and (b) the interfacial region at theanatase (101) (left) and (001) interfaces (right)dipped in bulk water. OW and HW indicateO and H atoms of water molecules. Oadw, Os
and O2c mean specific O atoms in the adsor-bate and on the surface.
3.2 TiO2 anatase (101)/liquidMeCN interface and its wa-ter contamination for DSC
A snapshot of the equilibrium trajectory ofthe TiO2 anatase (101) / bulk MeCN inter-face is shown in Fig. 6 (a). The averaged cov-erage is 0.6, similar to the TiO2/H2O case.The equilibrium strctures of adsorbed MeCNmolecules have a rather ’standing’ geometry,
in contrast to the results of the previous stud-ies. Consequently, the TiO2 electrode has a lotof space sterically and potentially active sites(Ti5c) even if it is covered by MeCN molecules.The interfacial electronic states imply that theelectrons are slightly withdrawn from the sur-face and the top of the TiO2 valence band islikely to be shifted upward.
We then replaced a MeCN solvent with oneH2O molecule to examine the water contami-nation effect. The water density correspondsto about 0.5 M, quite comparable to the realsystem. DFT-MD samplings with several ini-tial structures provide three metastable statesin the end, which are shown in Fig. 6 (b). Thewater molecule in the state I stays in the liquidMeCN in spite of large space just on the TiO2
surface as described above. We observed thatthe MeCN solvent prevents H2O from divinginto the surface region by making some HBs tothe H2O. This indicates that, once TiO2 sur-face is passivated by MeCN solvent, the H2Omolecule is difficult to approach to the surface,which will be beneficial for the durability.
Assuming the condition where H2O alreadyexists on the surface before immersion of liquidMeCN, we obtained the equilibrium states IIand III. The latter shows normal H2O adsorp-tion to a Ti5c site, while the former adsorptionrealizes through a characteristic HW-O2c hy-drogen bond. Since the number of the atoms inthese calculations is the same, we can comparetheir average energies, which are displayed inFig. 6 (b). Then we found that the peculiarstate II is most stable probably because of theenergy gain through HB or electrostatic inter-actions. This result indicates that it is hard toremove the adsorbed H2O molecule on the in-terface, consistent with the experimental sug-gestion that water interacts with the TiO2 sur-face strongly in DSC.
In order to understand the electronic effectof the water molecule, we analyze the equi-librium electronic states of the states II. Al-though H2O does not modify the whole elec-tronic states significantly, the 1b1 levels of thewater are located closer to the valence bandmaximum of TiO2. Thus, if a photogeneratedhole migrates to this surface, there is largerpossiblitiy of this water accepting the hole.
6

Figure 6: (a) Snapshot in the equilibrium tra-jectory of the TiO2 anatase (101) /bulk MeCNinterface. (b) Snapshots of metastable state I,II, and III, in the TiO2 / bulk MeCN interfacescontaminated by H2O. I: H2O molecule existsin bulk MeCN. II: the most stable structure ofH2O, which is adsorbed on the TiO2 surfacevia strong HB between HW and O2c. III: H2Omolecule is adsorbed on the TiO2 surface viathe OW-Ti5c bond. The energy diagram (inkcal mol-1) is also shown.
This means that the H2O caton radical can ap-pear on the surface more easily, which may at-tack dye molecules or other important specieson the surface. In order to make DSC robustand effective, therefore, it is better to fabricatethe DSC under a low-humidity environment, asexperimentalists have been already doing.
3.3 Adsorption mode of Ru N749black dye on the TiO2 anatase(101) surface for DSC
We also examined the adsorption of Ru N749dye, so called black dye, on the TiO2 anatase(101) surface, aiming at future investigation ofTiO2/Ru dye/MeCN interface systems. Us-ing reasonably large supercell, we examinedthe adsorption energies of several possible ad-sorption modes. Here we focus on the num-ber of carboxyl anchors (1 or 2) and their pro-
Figure 7: Protonated (left) and deprotonated(right) one-anchor structures of the black dyeadsorption to TiO2 anatase (101) surface,which are optimzed in the present DFT cal-culations. The relative energies with respectto the protonated one are also denoted.
tonation/deprotonation. As a result, the ad-sorption mode with protonated single anchoris found most stable (Fig. 7). The optimizedstructure has 1.6 A between the H atom in thecarboxyl group and the surface O2c site. Thisrather strong HB is an important source of thestability. In the two anchor structures, forma-tion of even one HB is found difficult due tothe stiffness of the terpyridine ligand. Regard-ing the proton position, the local pKa may givea reason. Typical pKa of the COOH group isaround 3-5, while the O2c site of TiO2 is re-ported to have -1. Therefore, the proton ofthe carboxyl group prefers staying in the dye.In fact, our calculations indicated significantinstability of the deprotonated anchors on thebare TiO2 anatase surface.
Since this adsorption mode is rather differ-ent from the conventional scenarios, we usedUV-vis spectra for the comparison between cal-culations and experiments. TDDFT calcula-tions at the B3LYP level were applied to theadsorption structures of N719 dye (referencedye) and the selected adsorption structures ofthe current N749 dye. Comparing the numberand relative positions of the peaks and the tail(onset) of the spectra, we demonstrated thatthe protonated single anchor, the most stablestructure in the present calculations, is mostconsistent with the experimental spectra forthe black dye. Although presence of explicitsolvent may affect the stability, our results sug-gest that the protonated anchor modes should
7

be also taken into account when consideringthe efficiency.
Figure 8: UV-visible spectra of the blackdye molecule of protonated one-anchor (red),two-anchor (orange), deprotonated one-anchor(blue), two-anchor (green), bidentate bridg-ing one-anchor (brown) structures and N719dye with the adsorbed structure, calculated byTDDFT at the B3LYP level. Experimentalspectra of black dye (black solid line) and N719dye (black dashed line) are shown on the topof the figure.
4 Summary
We have addressed DFT-MD sampling anal-ysis of solid-liquid interfaces with bulk liq-uid on the atomic and electronic scales, whichis still cutting-edge. It is demonstrated thatthe presence of bulk liquid provides a vari-ety of novel implications about phenomena at’hidden’ solid-liquid interfaces. Although thefusion of DFT and electrochemistry has notbeen achieved yet, the present findings willbe quite useful for future establishement ofelectrochemistry ’on the atomic scale’. Fromthe application point of view, we found many
atomistic features of TiO2/liquid interfaces inphotocatalysis and dye-sensitised solar cells.These studies will bring about a novel perspec-tive for the solution of the energy and environ-mental issues.
Acknowledgement
This work was partly supported by KAK-ENHI as well as the Strategic Programs forInnovative Research (SPIRE), MEXT andthe Computational Materials Science Initiative(CMSI), Japan. The calculations were carriedout on the supercomputer centers at the ISSP,The University of Tokyo and NIMS as well asT2K-Tokyo and T2K-Tsukuba.
References
[1] M. Sumita, C. Hu, and Y. Tateyama, J.Phys. Chem. C 114, 18529-18537 (2010),and references therein.
[2] Y. Tateyama, and M. Sumita, submitted.
[3] M. Sumita, K. Sodeyama, L. Han, and Y.Tateyama, J. Phys. Chem. C 115, 19849-19855 (2011), and references therein.
[4] K. Sodeyama, M. Sumita, C. O’Rourke,U. Terranova, A. Islam, L. Han, D. R.Bowler, and Y. Tateyama, J. Phys. Chem.Lett. 3, 472-477 (2012), and referencestherein.
8