development of new routes for the synthesis of
TRANSCRIPT

Illinois State University Illinois State University
ISU ReD: Research and eData ISU ReD: Research and eData
Theses and Dissertations
5-3-2017
Development Of New Routes For The Synthesis Of Development Of New Routes For The Synthesis Of
Carbaporphyrins And Carbachlorins Carbaporphyrins And Carbachlorins
Navneet Sahota Illinois State University, [email protected]
Follow this and additional works at: https://ir.library.illinoisstate.edu/etd
Part of the Chemistry Commons
Recommended Citation Recommended Citation Sahota, Navneet, "Development Of New Routes For The Synthesis Of Carbaporphyrins And Carbachlorins" (2017). Theses and Dissertations. 773. https://ir.library.illinoisstate.edu/etd/773
This Thesis is brought to you for free and open access by ISU ReD: Research and eData. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of ISU ReD: Research and eData. For more information, please contact [email protected].

DEVELOPMENT OF NEW ROUTES FOR THE SYNTHESIS OF CARBAPORPHYRINS
AND CARBACHLORINS
Navneet Sahota
250 Pages
Carbaporphyrins and carbachlorins are porphyrin analogs in which one of the nitrogens is
replaced with a carbon atom within a porphyrin-like cavity. These systems form stable
organometallic derivatives and exhibit unusual chemical reactivity. Most of the studies in this area
have been carried out with benzocarbaporphyrins, where one of the pyrrole rings is replaced by an
indene unit. Although syntheses of monocarbaporphyrins with fused benzene rings have been
widely investigated over the past 20 years, few examples of carbaporphyrins without fused rings
have been reported in the literature.
In this thesis, two projects were conducted to further explore carbaporphyrin and
carbachlorin chemistry. The first project utilized a ‘3+1’ condensation of a tripyrrane dicarboxylic
acid with dimethyl 3,5-diformylcyclopentane-1,2-dicarboxylate in the presence of trifluoroacetic
acid, followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), to yield a
novel carbaporphyrin system in 40% yield together with a 9% yield of a related carbachlorin. This
new carbaporphyrinoid system retained macrocyclic aromaticity and afforded stable silver(III) and
gold(III) complexes. In addition, alkylation with methyl iodide and potassium carbonate gave a
C-methyl derivative, and this underwent metalation with palladium(II) acetate and nickel(II)
acetate to afford related organometallic species. The second project dealt with the development
of a new route for synthesizing carbachlorins. Chlorins are a group of dihydroporphyrins and are
important biological pigments. The study of core-modified chlorins has been limited compared to

that of core modified porphyrins. Carbachlorins are chlorin analogues where carbon atom is
inserted into the chlorin core. They have interesting properties and are also promising candidates
in many practical applications. Based on a “3+1” methodology, a novel carbachlorin was
synthesized and characterized through various spectroscopic techniques, including NMR, UV-Vis
spectroscopy, and mass spectrometry. The coordination chemistry of this carbachlorin was also
investigated.
KEYWORDS: Carbaporphyrins, Carbachlorins, Porphyrins, Chlorins, Spectroscopic techniques

DEVELOPMENT OF NEW ROUTES FOR THE SYNTHESIS OF CARBAPORPHYRINS
AND CARBACHLORINS
NAVNEET SAHOTA
A Thesis Submitted in Partial
Fulfillment of the Requirements
for the Degree of
MASTER OF SCIENCE
Department of Chemistry
ILLINOIS STATE UNIVERSITY
2017

Copyright 2017 Navneet Sahota

DEVELOPMENT OF NEW ROUTES FOR THE SYNTHESIS OF CARBAPORPHYRINS
AND CARBACHLORINS
NAVNEET SAHOTA
COMMITTEE MEMBERS:
Timothy D. Lash, Chair
Shawn R. Hitchcock
Christopher C. Mulligan

i
ACKNOWLEDGMENTS
The author wishes to acknowledge all of the members of her research group for all of their
support throughout the duration of her time at Illinois State University. The author would also like
to acknowledge the faculty, staff, and fellow students in the Chemistry Department at Illinois State
University. The writer acknowledges financial support from the National Science Foundation
under grants no. CHE-1465049 and CHE-1212691 and the Petroleum Research Fund.
The author would like to thanks the members of her committee, Dr. Hitchcock and Dr.
Mulligan for all the guidance given. In addition, the author acknowledges Dr. Ferrence for
providing X-ray crystal structures for a carbaporphyrin and related metalloporphyrinoids. Great
thanks are given to her research advisor Dr. Timothy D. Lash for his encouragement, endless
support, and patience. The writer feels truly privileged and thankful to have had such an excellent
mentor for the past two years.
N.S

ii
CONTENTS
Page
ACKNOWLEDGMENTS i
CONTENTS ii
FIGURES iv
SCHEMES vii
CHAPTER
I. INTRODUCTION TO PORPHYRINS 1
Introduction 1
Structure of Porphyrins 3
Aromatic Properties 7
Biosynthesis of Heme 16
Synthesis of Porphyrins 19
Reactivity of Porphyrins 30
Macrocycles Related to Porphyrins 32
Applications 34
Carbaporphyrins 36
Carbachlorins 50
II. SYNTHESIS OF CARBAPORPHYRINS 66
Introduction 66
Results and Discussion 67
Conclusions 109
Experimental 111
III. SYNTHESIS OF A CARBACHLORIN 126
Introduction 126
Results and Discussion 129
Conclusions 141
Experimental 142
IV. SUMMARY AND CONCLUSIONS 150
REFERENCES 152
APPENDIX A: SELECTED NMR SPECTRA 156

iii
APPENDIX B: SELECTED UV-VIS SPECTRA 229

iv
FIGURES
Figure Page
1. Resonance Structures of Pyrrole 4
2. Examples of Cyclic Structures Depicting
Differences in Aromaticity 8
3. Structures of Porphyrin and [18]Annulene 9
4. Reduced Forms of Porphyrin 11
5. Induced Ring Current Effects on Porphyrin as Observed in
Nuclear Magnetic Resonance Spectroscopy 13
6. UV-Vis Spectrum of Tetraphenylporphyrin 14
7. Structures of Dipyrrolic Intermediates 23
8. Structures of Tripyrrane Intermediates Used in ‘3+1’ Condensations 29
9. Carbaporphyrins Stabilizing Metals with Rare Oxidation States 31
10. Structures of Contracted and Expanded Porphyrins 33
11. Structures of Phthalocyanine and Dicarbahemiporphyrazine 33
12. Monocarbaporphyrin Analogues 37
13. Mono-, Di-, Tri- and Tetracarbaporphyrins 38
14. Aromatic Delocalization Pathways in Porphyrin and Chlorin 51
15. Structures of Chlorin and Carbachlorin 53
16. 500 MHz 1H NMR Spectrum of Fumarate Diol in CDCl3 77
17. 500 MHz Proton NMR Spectrum of Carbaporphyrin 98 in CDCl3 80
18. 500 MHz Proton NMR Spectrum of the Carbachlorin byproduct in CDCl3 81
19. UV-Vis Spectrum of Carbaporphyrin 98 in Dichloromethane 82
20. POV-Ray generated ORTEP III Drawing of 98 83

v
21. UV-Vis Spectrum of 117 in Dichloromethane 84
22. 500 MHz Proton NMR Spectrum of Mixture of a 98H+ and 98H22+ in CDCl3 85
23. UV-Vis Spectrum of Carbaporphyrin 98H+ in CH2Cl2 86
24. 500 MHz Proton NMR Spectrum of 98H+ in CDCl3 87
25. 500 MHz 1H NMR Spectrum of 98H22+ in CDCl3 88
26. 500 MHz Proton NMR Spectra of 98 Showing Exchange of 21-H
after 10 min Exposure to d-TFA 91
27. 500 MHz proton NMR Spectra of 98 Showing Partial
Exchange of the meso-protons after 24 hr Exposure to d-TFA 91
28. 500 MHz 1H NMR Spectrum of 117H� in CDCl3 93
29. UV-Vis Spectrum of 117H+ By-product in 5% TFA-Dichloromethane. 94
30. 500 MHz 1H NMR Spectrum of 118 in CDCl3 95
31. 500 MHz 1H NMR Spectrum of 120 in CDCl3 98
32. 500 MHz 1H NMR Spectrum of 121 in CDCl3 100
33. 500 MHz 1H NMR Spectrum of 120H� in CDCl3 101
34. 500 MHz 1H NMR Spectrum of 120H22+ in CDCl3 102
35. 500 MHz 1H NMR Spectrum of 121H+ in CDCl3 103
36. 500 MHz 1H NMR Spectrum of 122 in CDCl3 105
37. UV-Vis Spectrum of Palladium(II) Carbaporphyrin in Chloroform 105
38. POV-Ray rendered ORTEP III Drawing of 122 106
39. 500 MHz 1H NMR Spectrum of 123 in CDCl3 108
40. POV-Ray rendered ORTEP III Drawing of 123 108
41. 500 MHz Proton NMR Spectrum of Carbachlorin 133 in CDCl3 133
42. UV-Vis Spectrum of Carbachlorin 133 134

vi
43. 500 MHz Proton NMR Spectrum of Carbachlorin 133H+ in CDCl3 135
44. Proton NMR Spectrum of Carbachlorin 133 in CDCl3 with Two Drops of d-TFA
after 20 min (upper spectrum) and 7 hours (lower spectrum) 138
45. 500 MHz Proton NMR Spectrum of 138 in CDCl3 140
46. UV-Vis Spectrum of Silver(III) Chlorin 138 in Dichloromethane 141

vii
SCHEMES
Scheme Page
1. Mechanism of the Knorr Pyrrole Synthesis 5
2. Mechanism for the Hantzsch Synthesis of Substituted Pyrroles 6
3. Tautomerization and Protonation of Porphyrin 7
4. Synthesis of Uroporphyrinogen III 17
5. Biosynthesis of Heme from Uroporphyrinogen III 18
6. Rothemund Synthesis of TPP 20
7. Adler-Longo synthesis of TPP 21
8. Synthesis of meso-substituted porphyrins by Lindsey’s method 22
9. Fischer Porphyrin Synthesis 23
10. MacDonald's “2+2” condensation reaction 24
11. Synthesis of Porphyrin from an Oxophlorin 25
12. Synthesis of Oxaporphyrins and Thiaporphyrins 26
13. Synthesis of the First Tripyrrane 27
14. Synthesis of Tripyrranes Reported by Sessler and Coworkers 28
15. Porphyrin Synthesis through a Tetrapyrrolic Intermediate 30
16. Bromination of Porphyrins 32
17. Synthesis of Benzocarbaporphyrin 39
18. Synthesis of Tetraaryl Benzocarbaporphyrins
from Tetraarylazuliporphyrins 40
19. Protonation of Benzocarbaporphyrin 41
20. Synthesis of Silver(III) Benzocarbaporphyrin 42
21. Synthesis of Gold(III) Benzocarbaporphyrin 42

viii
22. Formation of Ir and Rh complexes of Benzocarbaporphyrin 44
23. Alkylation of Benzocarbaporphyrin 45
24. Unexpected Alkyl Group Migration 45
25. Oxidation of Benzocarbaporphyrin with FeCl3 48
26. Synthesis of Dibenzo opp-Dicarbaporphyrin 49
27. Protonation of Dibenzo opp-Dicarbaporphyrin 50
28. Phlorin-Chlorin Conversion 52
29. Synthesis of Carbachlorins 54
30. Synthesis of Carbaporphyrins and Attempted Synthesis of a Carbachlorin 55
31. Retrosynthesis of Desired Dialdehyde 56
32. Synthesis of Carbachlorin 57
33. Protonation of Carbachlorin 58
34. Oxidation of Carbachlorin 59
35. Formation of Silver(I) Complex of Carbachlorin 61
36. Formation of N-methylated Carbachlorin 62
37. Unexpected Formation of Pd Complex 64
38. Retrosynthetic Analysis of Carbaporphyrin 98 67
39. Retrosynthetic Analysis of the Tripyrrane Intermediate 68
40. Preparation of Pyrrole Ester 101 using a Knorr-Type Synthesis 69
41. Formation of a Pyrrole Benzyl Ester 69
42. Formation of an Acetoxymethylpyrrole 70
43. Formation of Ethyl Isocyanoacetate 105 70
44. Preparation of 3,4-Diethylpyrrole via a Barton-Zard Synthesis 72

ix
45. Preparation of Tripyrrane Dicarboxylic Acid 62 73
46. Preparation of Bicyclic Adduct 111 73
47. Formation of Diol 112 74
48. Unsuccessful Attempts to Synthesize Diol 112 75
49. Formation of Bicyclic Adduct 114 76
50. Formation of Diol 115 76
51. Formation of Dialdehyde 99 77
52. Mechanism for Ring Opening 78
53. Preparation of Carbaporphyrin 98 79
54. Protonation of Carbaporphyrin 98 86
55. Protonation of Carbaporphyrin 98 88
56. Formation of Different Cationic Species in Equilibrium 90
57. Protonation of Carbachlorin byproduct 117H+ 92
58. Synthesis of Silver(III) Carbaporphyrin 118 95
59. Formation of Gold(III) Complex of Carbaporphyrin 96
60. Formation of C-methylated Carbaporphyrin 120 97
61. Formation of Dimethylated Carbaporphyrin 121 99
62. Metalation of C-methylcarbaporphyrin with Palladium(II) Acetate 104
63. Metalation of C-methylcarbaporphyrin with Nickel(II) Acetate 107
64. Reduction of Carbaporphyrin 98 110
65. New Class of Carbaporphyrin Derivatives 111
66. Retrosynthetic Analysis of Carbachlorin 133 129
67. Formation of 136 130

x
68. Formation of 135 130
69. Formation of Dialdehyde 134 131
70. Preparation of Carbachlorin 133 132
71. Protonation of 133 135
72. Proposed Equilibrium of Different Cationic Species for Carbachlorin 133 137
73. Formation of Silver(III) Chlorin 138 139

1
CHAPTER I
INTRODUCTION TO PORPHYRINS
Introduction
Porphyrins and porphyrinoids are important heterocyclic macrocycles that are widespread
in nature.1 They are usually deeply colored green, red or purple fluorescent crystalline pigments
whose origins can be either synthetic or natural.2 The parent porphyrin system is called porphin 1,
while substituted porphins are called porphyrins. The word porphyrin was derived from the Greek
word porphyra meaning purple. It was in 1873 that a German chemist named Hoppe-Seyler
isolated a purple crystalline compound from heating hemoglobin with sulfuric acid, and he
subsequently coined the name hematoporphyrin for this material from the Greek words for blood
and purple.2 Over the last 150 years, porphyrins have been widely investigated. The interesting
properties of porphyrins, which include their coordination chemistry and aromatic character, have
stimulated research into this class of organic compounds. Porphyrins and their derivatives greatly
contribute to the beauty of nature and play important roles in biological systems. For these reasons,
they have been commonly referred to as the pigments or colors of life.2

2
As mentioned above, many porphyrins and porphyrin-like compounds play significant
roles in both animals and plants. In mammals, heme (2), an iron complex of porphyrin, is a
component of hemoglobin and myoglobin. The former is involved in oxygen transportation around
the body in the blood, while the latter stores oxygen in muscle tissue. Heme is also involved in
redox reactions and electron transfer processes.2 For instance, if the external environment of the
heme is changed, it can facilitate completely different iron chemistry. Thus, in cytochrome c, it
performs the function of electron transfer in cell respiration, as the iron atom cycles through +2
and +3 oxidation states.
In vitamin B12, nature makes use of a coordinated cobalt ion within a corrin core 3. The
complex is used to transfer hydrogen atoms and to reduce organic species. Vitamin B12 is a water-
soluble vitamin and plays essential roles in the formation of red blood cells and in maintaining
healthy nerve cells. Lack of vitamin B12 leads to a disease called pernicious anemia where the
shortage of red cells and hemoglobin leads to impairment of the central nervous system. This
disease usually results from the imperfect absorption of the vitamin by the intestines.2
In plants, chlorophylls (4), magnesium complexes of porphyrin-like structures, capture
photons of light in the near-ultraviolet and red regions of the visible spectrum which therefore
make them important for photosynthesis. The magnesium ion in the reduced porphyrin macrocycle
of chlorophyll serves to modulate the energy transfer and light absorbing characteristics of
chlorophyll, while acting as a center for binding water. A number of chlorophyll molecules are
arranged into tiny molecular antennae that harvest large quantities of photons.1 The energy of the
photons is trapped as excited electrons, which are led away by a chain of electron-transporting
proteins to generate high energy molecules necessary for turning carbon dioxide into
carbohydrates during photosynthesis.2

3
Structure of Porphyrins
The macrocyclic structure of porphyrin was first proposed by Küster in 1912.1 The
macrocycle is composed of four pyrrole units connected by methine bridges at the α-carbons.1,2
The methine carbons are also referred as meso-carbons and the hydrogens attached to these carbons
are known as meso-protons. The pyrrole unit is a five-membered aromatic ring consisting of four
carbon atoms and one nitrogen atom and possesses two formal double bonds. Pyrrole is an
electron-rich aromatic system and has four dipolar resonance contributors (Figure 1) due to the
presence of an electron-donating nitrogen atom.3

4
Pyrrole is essentially non-basic since the lone pair of electrons on the nitrogen is
delocalized into the ring in order to make the structure aromatic. Due to the resonance structures
of this molecule, pyrrole is relatively electron-rich and therefore is likely to undergo electrophilic
aromatic substitution.3 The chemical and physical properties of porphyrins are very different from
the properties of the pyrrole. For instance, porphyrins are usually very deeply colored compounds
whereas pyrroles are off-white solids or colorless liquids. In addition, two of the nitrogens within
the porphyrin core are basic and the π-electrons are delocalized over the entire macrocycle.2
A large number of synthetic routes have been developed for preparing pyrroles. Among
these, the two of the most common methods used to synthesize substituted pyrroles are the Knorr
pyrrole synthesis and the Hantzsch pyrrole synthesis.2 In the Knorr pyrrole synthesis (Scheme 1),
a substituted pyrrole 5 is formed by the condensation of an α-amino ketone 6 with a β-dicarbonyl
compound 7. In order to form the reactive α-amino ketone, an oxime 8 is reduced using zinc dust
in acetic acid.2
Figure 1 Resonance Structures of Pyrrole

5
The second most common method is the Hantzsch pyrrole synthesis. In this approach, an
α-haloketone is reacted with a β-ketoester and a primary amine or ammonia.2 Nucleophilic attack
by the amine or ammonia occurs on the carbonyl carbon of a β-ketoester 9, and subsequent
elimination of water results in the formation of an enamine 10. Condensation of enamine 10 with
the α-haloketone 11, followed by ring closure, yields the substituted pyrrole 12 (Scheme 2).2
Scheme 1 Mechanism of the Knorr Pyrrole Synthesis

6
Scheme 2 Mechanism for the Hantzsch Synthesis of Substituted Pyrroles
Porphyrins can exist in several tautomeric forms, three of which are shown below
(structures 1a, 1b and 1c). Although structures 1a and 1b are equivalent and are interconverting
tautomers, tautomer 1c is less favored due to the close proximity of two adjacent internal
hydrogens that results in steric crowding effects.2 In the porphyrin core, there are two pyrrolenine
nitrogens, which can undergo protonation in the presence of acid to form mono- (1H+) or dications
(1H22+) (Scheme 3).

7
Scheme 3 Tautomerization and Protonation of Porphyrin
Aromatic Properties
Porphyrins are highly conjugated compounds, and it is the conjugated nature of these
macrocycles that is, in part, responsible for their aromatic characteristics.1,2 The concept of
aromaticity was first introduced by Kekulé in 1865.4 The term aromatic was proposed by
Hoffmann because many benzene derivatives have a sweet odor.5 As studies of aromatic
compounds progressed, it was seen that a common property for these compounds was not their
aroma but their relatively high stability. In 1931, Hückel was able to mathematically predict the
aromaticity of unsaturated cyclic systems.2 This led to the well-known Hückel’s rule for
aromaticity, according to which there needs to be [4n+2]π electrons for a molecule to be aromatic,
where n is zero or any positive integer.
Aromatic compounds are found to be more stable when compared to unsaturated
compounds that are not aromatic, and the stability is due to the delocalized electron cloud also

8
known as resonance energy. Along with having sufficient π electrons, it is also required that the
molecule is sufficiently planar, cyclic and continuously conjugated. The more planar the molecule,
the easier it is for the π electrons to interact with one another to create a delocalized system.2
Benzene is the classic system which illustrates aromaticity. It is cyclic, has 6π electrons
and is continuously conjugated, so it follows Hückel’s rule for aromaticity and is very stable. Like
benzene, porphyrin also follows Hückel’s rule for aromaticity. Although porphyrins contain 22
conjugated π electrons (26π electrons counting the lone pairs of electrons on the nitrogen atoms),
only 18 of them participate in a continuous delocalization pathway and therefore follow Hückel’s
rule where n = 4.2 Unsaturated systems can also be classified as anti-aromatic or non-aromatic
(Figure 2). Hückel described anti-aromatic species as cyclic and having a fully conjugated [4n]π
electron pathway. Anti-aromatic compounds are still planar, but are considerably less stable than
aromatic compounds (e.g. cyclobutadiene). On the other hand, non-aromatic structures are
unsaturated systems that are not fully conjugated and may be non-planar. For instance,
cyclooctatetraene is non-aromatic and adopts a tub-like shape making the structure non-planar.
Non-aromatic polyenes are quite reactive as they are relatively unstable.3
Figure 2 Examples of Cyclic Structures Depicting
Differences in Aromaticity

9
Benzene and related compounds are unusually unreactive and do not have the typical
reactivity that is associated with olefins. For example, in the case of cyclooctatetraene, which is
an unusually reactive non-aromatic species, Br2 can readily add across the double bonds giving
addition products. However, benzene is quiet stable and unreactive, and no addition reactions are
observed.3 In benzene or other aromatic compounds, due to the interaction of the π electrons to
create a delocalized π electron cloud, the bond lengths are found to be in between those of single
bonds and double bonds.
Porphyrins can be considered to be the [18]annulenes of nature.6 Annulenes are completely
conjugated hydrocarbon macrocycles with the general formula of CnHn (where n is an even
number) or CnHn+1 (where n is an odd number), n here stands for the number of carbon atoms in
the ring. Annulenes, like other cyclic systems, can be aromatic, non-aromatic or anti-aromatic.
[18]Annulene (13) is an aromatic compound with nine double bonds. The macrocycle is
reasonably planar and has 18π electrons that are fully delocalized over 18 carbon atoms. Therefore
it follows Hückel’s [4n+2]π electron rule for aromaticity, where n = 4. [18]Annulene was first
synthesized by Sondheimer et al. in 19626 (Figure 3), and its conjugation pathway closely
resembles the aromatic circuit present in porphyrins.
Figure 3 Structures of Porphyrin and [18]Annulene

10
Reduced porphyrin systems may also retain aromatic properties. Chlorins 14
bacteriochlorins 15 and isobacteriochlorins 16 all possess 18 π electron delocalization pathways
and are aromatic compounds. Porphyrins can be reduced under a variety of conditions to generate
hydroporphyrins such as chlorins, in which two or more hydrogens have been added across the β-
β double bonds of one or more pyrrole moieties to produce pyrroline rings 14 (Figure 4). Chlorins
are relatively stable compounds that can be oxidized back up to the porphyrin by high potential
quinones such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and chloranil. In chlorins,
reduction at the β-pyrrole carbons does not interrupt the continuously conjugated delocalisation
pathway. In fact, two more hydrogens may be added across another β-β double bond to give tetra-
hydroporphyrins while retaining the 18 π electron pathway. If the double bonds are reduced on
opposite pyrrole units bacterochlorins are formed, but reduction of the adjacent pyrrole units
affords isobacterochlorin.2
However, other reduced forms of porphyrin, such as phlorins 17, may not be aromatic like
chlorins. This is due to phlorin containing a reduced double bond at one of the bridges linking two
pyrrole units together.2 Without the methine bridge, continuous conjugation throughout the
macrocycle is not possible, therefore breaking one of Hückel’s rules for aromaticity.

11
Figure 4 Reduced Forms of Porphyrin
A number of factors can lead to distortions from planarity in the porphyrin macrocycle. For
instance, the presence of bulky substituents on the macrocyclic system can inhibit the planarity
due to steric interactions. In addition, distortions can arise from the binding of a metal ion within
the cavity of the macrocyclic system. When an appropriate sized metal cation binds in the cavity
of the porphyrin, it may increase the planarity of the molecule. However, if the binding metal
cation is too small or too big for the cavity, the porphyrin ring will distort itself in order to bind to
the metal and hence the planarity will decrease making the ring system potentially less aromatic.
For example, 2,3,7,8,12,13,17,18-octaethylporphyin (OEP) in its free base form is quite planar.
However, when OEP binds to nickel(II), which is a relatively small cation for the cavity of the
porphyrin, the macrocycle twists out of a planar conformation in order to coordinate with the metal.
Porphyrins can tolerate considerable distortion while retaining aromatic properties.2
The aromatic character of porphyrins can be readily assessed using proton nuclear

12
magnetic resonance (NMR) spectroscopy and UV-vis spectroscopy. In particular, the diatropic
character of these systems can easily be analyzed using proton NMR spectroscopy. When these
macrocycles are introduced into a magnetic field, their π electrons circulate to create an aromatic
ring current. The concept of aromatic ring currents was first introduced by Pauling in relation to
the anisotropic diamagnetic susceptibilities of benzenoid hydrocarbons.7 These susceptibilities
were explained in terms of an induced circulation of the delocalized π-electrons in the presence of
an applied magnetic field that produces an induced magnetic field. The strength of the induced
magnetic field generated by the ring current is dependent on the orientation of the aromatic system
with respect to the applied magnetic field. Typically, for porphyrins, the diamagnetic ring current
deshields the β-pyrrole protons and the meso-protons. Because the meso-protons are attached to
electron-deficient carbons, they are shifted even further downfield than the β-pyrrole protons and
appear near +10 ppm. However, the inner N-H protons are shifted far upfield, beyond
tetramethylsilane (TMS, the arbitrary standard for NMR measurements), showing up around -4
ppm. This is because the induced ring current, which deshields protons from the external magnetic
field outside the macrocycle, shields them if they are inside the macrocycle (Figure 5). This
shielding effect is not observed in the case of benzene as there are no internal protons. However,
in some larger aromatic molecules (e.g. [18]annulene), the internal protons may be observed
diamagnetically shielded and hence shifted upfield.

13
Figure 5 Induced Ring Current Effects on Porphyrin
as Observed in Nuclear Magnetic Spectroscopy
The diamagnetic ring currents observed in the case of porphyrins are valuable indicators
of variations in aromatic character. Upon the addition of acid, porphyrins are mono-protonated or
diprotonated, and the resulting cationic species show even stronger diamagnetic ring currents. For
example, in the proton NMR spectrum of porphyrin dications, the meso-protons are shifted slightly
further downfield, while the internal NH protons move upfield.7
Porphyrins have their own characteristic UV-visible spectra. Like benzene, and its
homologues, there are mainly two distinct regions in the porphyrin absorption spectra. However,
unlike benzene, porphyrins are deeply colored compounds due to the absorptions appearing in the
near-ultraviolet and visible regions of the electromagnetic spectrum.1 Highly conjugated systems
tend to absorb light in the near UV and visible regions of their electronic spectra because
conjugated π bonds are able to absorb light at higher wavelengths when compared to isolated π
bonds. Thus, increasing the π bond conjugation of a system increases the wavelengths for
absorption in the UV-Vis spectrum, and this leads to them being colored compounds.
Porphyrins characteristically exhibit intense peaks around 400 nm in the near ultraviolet
region.2 In 1883, Soret,8 while studying hemoglobin, noticed these intense peaks and they were

14
later named as “Soret bands” or B bands. At higher wavelengths, other less intense peaks known
as Q bands are also observed. The intensity of the Soret bands has been correlated to the
aromaticity of the system. The more aromatic the macrocycle is, the more intense the Soret band.2
In the case of tetraphenylporphyrin, an intense Soret band is observed at 417 nm and four Q bands
can be seen at 514 nm, 549 nm, 589 nm, and 646 nm (Figure 6).7
Figure 6 UV-Vis Spectrum of Tetraphenylporphyrin

15
Chlorins 14, although they have less π electron density, still exhibit a Soret band in their
UV-Vis spectra because these structures retain an 18π electron pathway. However, phlorins 17 do
not exhibit Soret bands as there is an interruption of the 18π electron conjugated system due to
reduction at one of the methine bridges. Similarly, porphyrinogens, the reduced intermediates in
the biosynthesis of porphyrins, are also not conjugated structures and in this case are colorless
compounds.2
The differences in the colors of porphyrinoid macrocycles are due to variations in their
absorptions in the visible spectrum. For example, unlike porphyrins, chlorins tend to have a
characteristic absorption near 650 nm, and this results in their characteristic green coloration. This
explains why chlorophyll, a magnesium chlorin, is excellent at absorbing red light.2
The number and intensity of the Q and B bands can give powerful clues about the
substitution pattern of porphyrins and whether they are metalated or not. Protonation of the
porphyrin also alters the UV-vis spectrum. Diprotonation of the macrocycle leads to increased
symmetry compared to the free base form, and this produces a simplification of the Q band region.
Essentially, the four Q bands collapse to two and this can lead to profound color changes.
Substitution at the β-pyrrole positions can also affect the UV-visible absorption spectra
which in turn leads to color changes. Metalloporphyrins tend to be more symmetrical macrocycles
than free-base porphyrins and for this reason they generally only have two Q bands.1 The relative
intensities of these two bands can be used as a qualitative check to see how well the metal cation
fits into the macrocyclic cavity.

16
Biosynthesis of Heme
As mentioned above, in 1912 Küster was the first to propose the structure of porphyrin.9
However, the existence of such a large macrocyclic structure was only fully accepted in 1929,
when Fischer synthesized protoheme.10 The biosynthesis of porphyrins takes place through several
enzyme-catalyzed steps, starting with 5-aminolevulinic acid (ALA). The biosynthesis of
hemoglobin, chlorophyll and vitamin B12 all begin with this precursor. The two substrates required
for the biosynthesis of ALA are succinyl coenzyme A (CoA) and the amino acid glycine. Succinyl
CoA is produced during the Krebs cycle (also known as the citric acid cycle).
In the first step of the pathway, succinyl CoA is condensed with glycine, catalyzed by ALA
synthase, to generate ALA. The condensation of two molecules of ALA, with the aid of ALA
dehydratase, forms the pyrrole intermediate porphobilinogen (PBG). Four pyrrole units then
condense together, in the third step mediated by PBG deaminase, to form the open chain
tetrapyrrolic structure hydroxymethylbilane. Then, uroporphyrinogen III cosynthetase facilitates
the cyclization of the tetrapyrrolic intermediate to form uroporphyrinogen III (Scheme 4). The
cyclization here is associated with an inversion of ring D to afford the type III arrangement of
substituents that can be observed in nearly all naturally occurring porphyrin derivatives. The
biosynthesis of vitamin B12 also branches from the uroporphyrinogen III precursor.2

17
Scheme 4 Synthesis of Uroporphyrinogen III
The enzyme uroporphyrinogen decarboxylase mediates the decarboxylation of all four
acetic acid side chains, while coproporphyrinogen oxidase subsequently oxidatively
decarboxylates only two of the propionic acid side chains. Oxidation of protoporphyrinogen IX by
the enzyme protoporphyrinogen IX oxidase then yields protoporphyrin IX. Insertion of iron(II)
into protoporphyrin IX gives the metalloporphyrin heme (Scheme 5). Heme is then inserted into
the appropriate protein to give hemoglobin or myoglobin, which are both vital moieties in many
biological processes. Protoporphyrin IX is also the biosynthetic precursor to the chlorophylls.2

18
Scheme 5 Biosynthesis of Heme from Uroporphyrinogen III

19
Synthesis of Porphyrins
Over the years, a number of routes have been developed for porphyrin synthesis. Some of
the earliest methods for porphyrin synthesis involved the degradation of naturally occurring
pigments such as heme and chlorophyll. Metalloporphyrins are abundant in nature. For example,
heme derived from blood or chlorophyll from plants, are convenient starting materials that can be
used to prepare a wide variety of porphyrins.2 Although, porphyrins can be synthesized from
different natural sources, this approach can only be used to prepare structures with similar
substitution patterns. For this reason, multistep syntheses have also been developed. The synthetic
methods for the preparation of porphyrins and analogous systems can involve the intermediacy of
monopyrrolic, dipyrrolic, tripyrrolic or tetrapyrrolic structures.2,11
Porphyrins can easily be formed by the tetramerization of monopyrroles. This technique is
mainly used for the synthesis of highly symmetrical porphyrins such as tetraphenylporphyrin or
octaethylporphyrin, and more sophisticated methods are usually required to prepare
unsymmetrical structures.11
In the 1930s, tetraphenylporphyrin was first synthesized by Rothemund, albeit in low
yields.12 He initially investigated the synthesis of porphyrins by reacting acetaldehyde with pyrrole
in methanol at different temperatures in sealed vessels. Based on the same strategy, he extended
the scope of this methodology by reacting pyrrole 18 with several other aldehydes, including
formaldehyde and propionaldehyde. When this reaction was conducted using benzaldehyde 19,
meso-tetraphenylporphyrin (TPP) 20 was isolated (Scheme 6).12 In this reaction, pyrrole undergoes
electrophilic substitution at the α positions with an aldehyde to form the macrocyclic ring.
However, this reaction has many disadvantages. For instance, the reaction time was quite long and
the setup required to do the experiment was inconvenient as it required that the reaction be carried

20
out under anaerobic conditions in a sealed tube. In addition, purification of the products was very
difficult and poor yields were commonly obtained.12
Scheme 6 Rothemund Synthesis of TPP
Improvements on this approach were not reported until the 1960s. It was in 1967, that Adler
and Longo reported an improved method for the synthesis of meso-substituted porphyrins 20.13
This was accomplished by heating benzaldehyde and pyrrole in the presence of refluxing propionic
acid for 30 minutes (Scheme 7).13 The porphyrin product precipitated from the reaction mixture in
moderate yields of approximately 20%, which was a substantial improvement over Rothemund’s
method.12 The Adler and Longo’s method also required much shorter reaction times and could be
done under aerobic conditions. However, this approach still had a few drawbacks. Firstly, many
aldehydes could not tolerate the use of acidic conditions at elevated temperatures. Secondly, in
cases where the porphyrin products did not precipitate, difficulties were encountered in removing
side products that were also formed in these reactions.2

21
Scheme 7 Adler-Longo synthesis of TPP
In 1987, Lindsey et al. introduced a new methodology that greatly improved these
syntheses.14 In Lindsey’s method, the porphyrin synthesis was done in two steps instead of one.
The first step involves reaction of equimolar quantities of pyrrole and aldehyde in the presence of
an acid catalyst such as boron trifluoride etherate or trifluoroacetic acid (TFA) in dichloromethane
at room temperature. This results in the formation of a hexahydroporphyrin or porphyrinogen 21
(Scheme 8). Subsequently, in the second step oxidation of 21 with an electron deficient quinone,
p-chloranil or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), yields meso-substituted
porphyrin 22 in approximately 40% yield (Scheme 8).14 Due to the mild conditions of this
procedure, the Lindsey method allowed for the use of aldehydes with sensitive functionality that
could otherwise not be used under the relatively harsh conditions involved in previous routes to
meso-substituted porphyrins.

22
NH
RCHO
N
NH N
HN
R
R
R
R+
NH
NH HN
HN
R
R
R
RH
O
21
22
Scheme 8 Synthesis of meso-substituted porphyrins by Lindsey’s method
The methods developed by Rothemund, Adler, Lindsey and others are only suitable for
preparing symmetrical porphyrins and fall into the category of “One Pot Syntheses”. Since 1960,
many alternative routes to asymmetrical porphyrins have been developed including the ‘2+2’
MacDonald condensation, ‘3+1’ condensations, and the cyclization of open-chain tetrapyrroles.11
The ‘2+2’ condensations rely on dipyrromethane or dipyrromethene intermediates such as
23 and 24 (Figure 7). These precursors both have two linked pyrrole units and are differentiated
by the type of carbon linkage. Dipyrromethanes 23 have a saturated bridge, while dipyrromethenes
24 have an unsaturated bridge and are usually isolated as halide salts. Fischer made extensive use
of ‘2+2’ condensations of dipyrromethenes (Scheme 9) to synthesize asymmetrical porphyrins.10

23
Figure 7 Structures of Dipyrrolic Intermediates
For example, dipyrromethenes 25 and 26 condensed together in a succinic acid melt to
form deuteroporphyrin IX 27. Deuteroporphyrin 27 was a key intermediate in the total synthesis
of heme.11
Scheme 9 Fischer Porphyrin Synthesis
The use of dipyrromethanes was initially overlooked, in part because they were considered
to be too unstable to survive condensation reactions. However, in the 1960s, MacDonald was able
to demonstrate that dipyrromethanes are reasonably stable and excellent porphyrin precursors. In

24
MacDonald's “2+2” condensation method, a 5,5’-dipyrrylmethane dialdehyde 28 was condensed
with an α-unsubstituted dipyrrylmethane dicarboxylic acid 29 in the presence of an acid catalyst
to generate a porphodimethene intermediate 30, and subsequent air oxidation afforded the final
porphyrin product 31 (Scheme 10).15 Since the original report, the MacDonald reaction has been
one of the most commonly used methods for preparing meso-unsubstituted porphyrins. In addition,
a variation on MacDonald’s condensation was a crucial step in the total synthesis of chlorophyll
a.15
Scheme 10 MacDonald's “2+2” condensation reaction
Porphyrins can also be synthesized from oxophlorins 32, which are cross-conjugated
nonaromatic compounds that are the keto tautomers of meso-hydroxyporphyrins. Oxophlorin 32

25
can be synthesized using MacDonald ‘2+2’ conditions by reacting dipyrroketone dialdehydes 33
with dipyrromethanes 34 (Scheme 11).11 Although oxophlorins favor the keto tautomer over the
aromatic hydroxy tautomer, reaction of oxophlorins with acetic anhydride and pyridine traps the
hydroxy tautomer as the corresponding acetoxyporphyrin 35. Hydrogenation of 35 over palladium
yields porphyrinogen 36, and subsequent oxidation with DDQ affords the meso-unsubstituted
porphyrin 37 in reasonable yields (Scheme 11).11
Scheme 11 Synthesis of Porphyrin from an Oxophlorin
In the synthesis of porphyrins using previously mentioned methods, the introduction and
control of symmetry remains challenging. For instance, in the ‘2+2’ approach one of the precursors
must be symmetrical or mixture of isomers will be produced. In some cases, these isomeric
mixtures can be separated by careful column chromatography but the yields of individual
porphyrins will be low. An alternative ‘3+1’ version of the MacDonald condensation has been

26
developed that enables the synthesis of structures that cannot be prepared by the “2+2” approach,
although one of the intermediates must again be symmetrical. Johnson first reported a MacDonald
type “3+1” method for porphyrin synthesis in 1971.16 In this condensation reaction (Scheme 12),
tripyrrane dicarboxylic acids were reacted with dialdehydes to afford phlorins, which on
subsequent oxidation using DDQ or chloranil generated porphyrinoid structures. Johnson initially
used this approach to prepare oxa- and thiaporphyrins, but this methodology was later extended to
the synthesis of porphyrins and porphyrin analogues. This approach makes other types of
substitution patterns accessible, thus considerably expanding the library of porphyrin structures.16
In the synthesis of heteroporphyrins 38, a furan or thiophene dialdehyde 39 was condensed with a
tripyrrane 40 under acidic conditions and following oxidation yielded monoheteroporphyrins
(Scheme 12).16
Scheme 12 Synthesis of Oxaporphyrins and Thiaporphyrins
The key precursors in the ‘3+1’ MacDonald condensation are tripyrrolic intermediates
commonly known as tripyrranes. The first example of a tripyrrane 41 was synthesized by Kenner
and coworkers using 2-acetoxymethylpyrrole 42 and dipyrromethane 43.17 (Scheme 13). Sessler
et al. later reported a more convenient method for preparing tripyrranes in which an α-unsubstituted

27
pyrrole 44 reacted with two equivalents of an acetoxymethylpyrrole 45 in the presence of an acid
catalyst to generate 46. Hydrogenation of 46 yields tripyrrane dicarboxylic acids 47 (Scheme 14).18
Scheme 13 Synthesis of the First Tripyrrane

28
Scheme 14 Synthesis of Tripyrranes Reported by Sessler and Coworkers
Tripyrranes, in general, consist of three pyrrole rings linked by two methylene units.
However, analogous structures have been prepared where the central pyrrole has been replaced
with a heterocyclic or carbocyclic ring (Figure 8). Analogs of tripyrranes with benzene19 or
azulene20 units in place of the central pyrrole ring have been prepared and have been utilized in
the synthesis of highly modified porphyrinoid systems.19,21,22 The ‘3+1’ condensation has been
used to prepare a variety of porphyrin analogs including carbaporphyrins. This method is very
powerful and many different dialdehydes can be used to introduce exotic rings into the
porphyrinoid framework.23

29
Figure 8 Structures of Tripyrrane Analogues Used in ‘3+1’ Condensations
Porphyrins can also be synthesized by making use of tetrapyrrolic systems, such as bilanes,
bilenes and biladienes. However, tetrapyrrole cyclizations are heavily conditions dependent. The
tetrapyrrolic precursors are very reactive towards acid and can easily fragment or rearrange, hence
limiting the scope of these reactions. However, when appropriate conditions are used, tetrapyrroles
can remain intact and cyclize to generate isomerically pure porphyrins. For example,
dipyrromethanes 48 and 49 condense under mild acidic conditions to generate b-bilene 50.
Following cleavage of the tert-butyl esters, the b-bilene cyclizes in the presence of a mixture of
trifluoroacetic acid and trimethyl orthoformate, and following air oxidation generates isomerically
pure porphyrin 51 in good yields (Scheme 15).11

30
Scheme 15 Porphyrin Synthesis through a Tetrapyrrolic Intermediate
Reactivity of Porphyrins
Porphyrins are very reactive towards the formation of stable metal complexes. In fact, most
of the porphyrins found in nature are present as metalated derivatives. Porphyrins have been found
to form metal complexes with almost every metal and metalloid present in the periodic table;
however, the first row transition metals, such as iron, cobalt etc. tend to form metalloporphyrins
more readily compared to others.2 Porphyrin analogs such as carbaporphyrins also coordinate with
transition metal cations and in some cases stabilize unusual oxidation states. For instance,

31
benzocarbaporphyrins were shown to react with silver(I) acetate to form stable silver(III)
complexes, and related gold(III) derivatives (Figure 9) have also been prepared from gold(III)
acetate.24 Other carbaporphyrin-like macrocycles have also been shown to give similar
organometallic derivatives, e.g. tropiporphyrin (Figure 9).37
N
N
N
MN
N
N
Me
Et
Et
EtEt
Me
M
Ar
Ar
Ar
Ar
M = Ag(III) or Au(III) M = Ag(III)
Benzocarbaporphyrin derivatives Tropiporphyrin
Figure 9 Carbaporphyrins Stabilizing Metals in Rare Oxidation States
Porphyrins and metalloporphyrins can readily undergo electrophilic substitution reactions.
Metalloporphyrins are commonly substituted on their meso-carbons. However, if the coordinating
metals used are in electrophilic oxidation states (e.g. SnIV), or the free-base porphyrin is used, the
meso-carbons are deactivated towards electrophilic attack. This leads to electrophilic substitution
being favored at the β-positions of the pyrrolic subunits. These differences are illustrated in
Scheme 16 for the bromination of porphin.2

32
Scheme 16 Bromination of Porphyrins
Macrocycles Related to Porphyrins
Through the application of previously mentioned synthetic methods, such as ‘one pot
syntheses’ and ‘3+1’ condensations, many porphyrin-type macrocycles have been synthesized.
These porphyrinoids include contracted porphyrins such as triphyrin 5225 and corrole,2 and
expanded porphyrins such as sapphyrin 5326 and texaphyrin27 (Figure 10).

33
Figure 10 Structures of Contracted and Expanded Porphyrins
Phthalocyanines 54 are also structurally related to the porphyrins. These compounds are
important synthetic pigments that are typically blue or green in color. They were first investigated
by Sir Patrick Linstead, and like many other porphyrin-type structures, were found to be aromatic.
In phthalocyanine 54, each pyrrole ring is fused to a benzene ring and the pyrrole units are no
longer connected by methine bridges but are instead linked by nitrogen atoms (Figure 11). Analogs
of phthalocyanines have also been successfully synthesized. For instance, substitution of opposite
isoindole units with benzene rings gave dicarbahemiporphyrazine 55, although this structure no
longer retains aromatic properties.28
Figure 11 Structures of Phthalocyanine and Dicarbahemiporphyrazine

34
Applications
Synthetic porphyrins have a variety of applications, including as ligands in metal catalyzed
reactions and as photosensitizers in photodynamic therapy (PDT). By changing the chromophores
in porphyrin or its analogs, the macrocycle can be tuned for use in specific applications. For
instance, in PDT (a treatment for cancer), the specific properties of the photosensitizer has great
significance. PDT is a multistage process. Photosensitizer administration to the patient is carried
out, either systemically or topically, in the absence of light. When a sufficient amount of
photosensitizer appears in the diseased tissue, it is activated by exposure to light. The light dose
supplies sufficient energy to stimulate the photosensitizer and transfer the energy to molecular
oxygen. This results in the generation of reactive oxygen species such as singlet oxygen. The
singlet oxygen thus formed is destructive, and damages the malignant tumor cells.2 Porphyrins can
be used as photosensitizers in PDT because they are good at absorbing light and transferring the
absorbed energy to oxygen. They are excellent candidates as photosensitizers for PDT, as they
tend to accumulate in malignant cells and not healthy cells.29 In PDT, porphyrins or analogues that
show strong absorptions in the 650-800 nm range are preferred as photosensitizers. N-confused
porphyrins, carbachlorins and oxidized carbaporphyrins, for example, tend to have strong
absorptions in the far red and thus can be used for this type of application.23
Metalloporphyrins have been widely used in asymmetric catalysis, including
epoxidation,30 cyclopropanation,31 and Suzuki-Miyaura cross coupling.32 Development of
metalloporphyrins as catalysts was inspired by biological studies, such as the study of the
cytochrome P450 family of monooxygenases. These enzymes contain hemes (iron porphyrins) that
act as cofactors, and have been found to exhibit a broad range of functions. One of the most
significant functions of this class of enzymes is the elimination of foreign molecules in the body

35
through biotransformation. This family of enzymes is responsible for partial metabolism of the
majority of medicines consumed by humans.33 Another example of metalloporphyrin catalysts are
manganese porphyrins absorbed on a gold surface that can react with molecular oxygen from air
to carry out epoxidation of cis-stilbene to cis-stilbenoxide.
Aside from catalyzing organic reactions or being used as medicinal agents, porphyrins in
the modern world can also be utilized as chemosensors for nuclear waste. Expanded porphyrin
analogues have been found to bind with actinides and other heavy elements better than traditional
porphyrins. These expanded macrocyclic systems can also be used for the detection of hazardous
metals such as uranium or plutonium that are often used as a source for nuclear energy.34
Phthalocyanines, which are closely related to porphyrin macrocycles, have also been
shown to have numerous applications. They are also very stable compounds. For example, one of
the first phthalocyanines characterized by Linstead was stable up to 500 °C and in the presence of
concentrated sulfuric acid. Thus, these macrocycles and their analogues can be used as dyes for
inks, paper, and paint and have applications as lubricants and semiconductors.2
Porphyrins and phthalocyanines have also been shown to exhibit phosphorescence and
fluorescence properties.28 The phosphorescence and fluorescence properties of porphyrins can be
quenched by molecular oxygen. Thus, another application of porphyrins and phthalocyanines is
monitoring of oxygen levels used in various arenas from ecological, industrial, and medicinal
standpoints. These macrocycles can therefore serve the purpose of optical sensors for molecular
oxygen.28 The concentration of molecular oxygen in the gas or liquid phase is proportional to the
decay of phosphorescence in the porphyrin and phthalocyanine.28 The most common porphyrin
optical sensors are palladium(II) and platinum(II) metalloporphyrins. In addition, ruthenium(III)
and iridium(III) metalloporphyrins have also exhibited high phosphorescence at room temperature

36
and could possibly be used as alternative oxygen sensors in the near future.2
Carbaporphyrins
In order to gain a better understanding of the reactivity and characteristics of porphyrins,
chemists began to investigate core modified porphyrins. The study of core modified porphyrins
helps to reveal the effect that modification of a conjugated system has on its properties and its
characteristics. Systems with a carbon atom inserted into the core of the porphyrin, the so-called
carbaporphyrinoid systems, have been of particular interest. These systems have been widely
investigated and have shown unique properties, such as formation of organometallic derivatives
under mild conditions.35 Related systems include N-confused porphyrin 56,36
benzocarbaporphyrins 57,35 tropiporphyrins 58,37 azuliporphyrins 5938 and benziporphyrin 6039
(Figure 12).35

37
Figure 12 Monocarbaporphyrin Analogues
These porphyrin analogues can be subdivided into “true” carbaporphyrins and “modified”
carbaporphyrins. In true carbaporphyrins, at least one of the pyrrolic units is replaced with a
cyclopentadiene or indene unit. In modified carbaporphyrins, a carbocyclic ring system other than
a cyclopentadiene unit is used to replace a pyrrole moiety.35 For instance, by replacing a pyrrolic
unit within the porphyrin framework with azulene, cycloheptatriene, or benzene, new families of
porphyrin-like macrocycles were produced. True carbaporphyrins such as benzocarbaporphyrin
57 are fully aromatic structures, while benziporphyrins 60 do not have aromatic character, and
azuliporphyrins 59 fall midway between the two extremes.35

38
In benzocarbaporphyrins 57, porphyrin-like UV−vis spectra are retained and these
compounds are highly diatropic, as judged by proton NMR spectroscopy. However,
azuliporphyrins 59 are cross-conjugated and have considerably reduced diatropic character, while
tropiporphyrins 58 have intermediary properties due to the nonplanar nature of the seven-
membered ring.35 In principle, if more than one nitrogen atom in the cavity is replaced with carbon
atoms, this results in the formation of dicarbaporphyrins, tricarbaporphyrins and
tetracarbaporphyrins (Figure 13).41
Figure 13 Mono-, Di-, Tri- and Tetracarbaporphyrins
Carbaporphyrins can easily be synthesized by the “3+1” condensation approach.16 One of
the best studied classes of carbaporphyrins are the benzocarbaporphyrins. In benzocarbaporphyrins
57, one of the pyrrole units is replaced with an indene unit. These compounds are prepared by the
acid catalyzed condensation of indene dialdehyde 61 with tripyrrane 62 (Scheme 17)40. Following

39
oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), carbaporphyrin 57 was
isolated in > 40% yield.
Scheme 17 Synthesis of Benzocarbaporphyrin
Benzocarbaporphyrins can also be prepared from azuliporphyrin 63. In this method,
azuliporphyrins 63 undergo an oxidative ring contraction when exposed to tert-butyl
hydroperoxide under basic conditions to generate the fused benzene ring. This approach provides
an indirect route to tetraaryl benzocarbaporphyrins 64 (Scheme 18).42,43
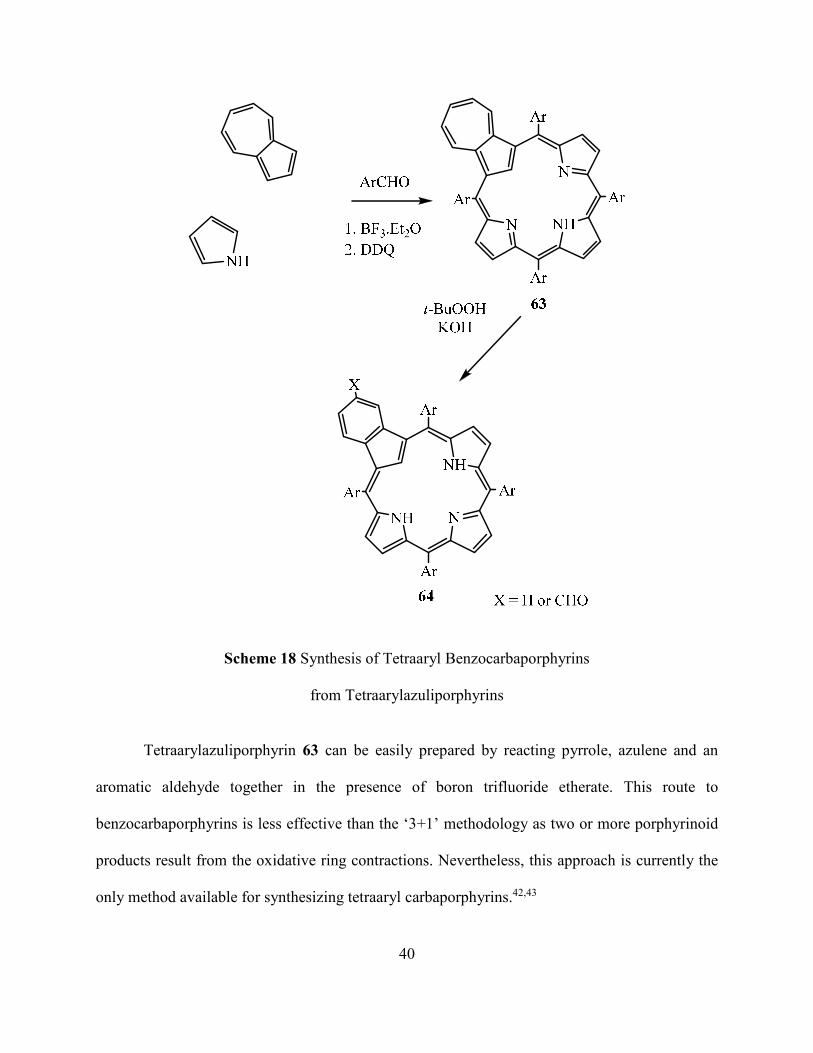
40
Scheme 18 Synthesis of Tetraaryl Benzocarbaporphyrins
from Tetraarylazuliporphyrins
Tetraarylazuliporphyrin 63 can be easily prepared by reacting pyrrole, azulene and an
aromatic aldehyde together in the presence of boron trifluoride etherate. This route to
benzocarbaporphyrins is less effective than the ‘3+1’ methodology as two or more porphyrinoid
products result from the oxidative ring contractions. Nevertheless, this approach is currently the
only method available for synthesizing tetraaryl carbaporphyrins.42,43

41
Protonation of benzocarbaporphyrin 65 can lead to the formation of both mono and
dicationic species.44,45 Upon addition of acid, protonation initially occurs on the pyrrolenine
nitrogen to give the monocation 65H+. At higher acid concentrations, further protonation takes
place on the internal carbon to give dication 65H22+ (Scheme 19).45 The second protonation goes
to completion in 50% TFA-chloroform solution, and results in the generation of a new
chromophore. Proton NMR spectroscopy in 50% TFA-CDCl3 strongly suggests that the dicationic
species retains its aromatic characteristics. In azuliporphyrins, protonation of the internal carbon
atom has not been observed.35
Scheme 19 Protonation of Benzocarbaporphyrin
Benzocarbaporphyrins 66 were found to react with silver(I) acetate to give stable silver(III)
organometallic complexes 67 (Scheme 20).46,47 The silver complex retains porphyrin-like UV-vis
spectra, showing strong a Soret band at 437 nm, along with characteristic Q bands at 482, 518, 555
and 593 nm. The meso-protons show up at 9.89 and 10.06 ppm, thereby confirming that the silver
complex has strong diatropic character. Unlike the carbaporphyrin free base, the silver complex of
benzocarbaporphyrin 67 was near planar and indene unit was only tilted by 5.08°.46

42
NH
N
HN
Me
Et
R
REt
Me
66
N
N
N
Me
Et
R
REt
Me
67
Ag
Ag(OAc)
Scheme 20 Synthesis of Silver(III) Benzocarbaporphyrin
Reaction of meso-unsubstituted benzocarbaporphyrins 66 with gold(III) acetate gave low
yields of the related gold(III) complexes 68 (Scheme 21).47 However, much better yields were
obtained in the metalation of meso-tetraaryl benzocarbaporphyrins 64.35 Gold complexes of
benzocarbaporphyrins also have strongly aromatic characteristics and gave similar UV-visible
spectra to silver complexes 67.35
Scheme 21 Synthesis of Gold(III) Benzocarbaporphyrin

43
Recently, rhodium and iridium complexes of benzocarbaporphyrin 57 have also been
obtained in good yields (Scheme 22). Reaction of 57 with 1 equiv of di-μ-
chlorotetracarbonyldirhodium(I) in refluxing dichloromethane gave the rhodium(I) dicarbonyl
complex 69 in 90% yield. The proton NMR spectrum for 69 showed the presence of strong
diamagnetic ring currents. Upon refluxing 69 in pyridine, a hexacoordinate rhodium(III) complex
70 was generated in 55% yield. In addition, when 57 was reacted with [Ir(COD)Cl]2 in refluxing
pyridine, iridium(III) derivative 71 was generated in 22% yield. The X-ray structures for these
derivatives showed that the macrocycle was near planar and the bond lengths were consistent with
the aromatic structures.48

44
Scheme 22 Formation of Ir and Rh complexes of Benzocarbaporphyrin
Benzocarbaporphyrins also readily react with methyl iodide in the presence of potassium
carbonate in refluxing acetone to give alkylated products. In this reaction, it was observed that N-
methyl carbaporphyrin 72 was the major product along with some C-alkylated by-product 73
(Scheme 23). The major product of this reaction is a chiral system, whereas the minor C-methyl
product possesses a plane of symmetry. Although the presence of internal substituents may reduce
the planarity of these structures, these derivatives retain strongly diatropic characteristics.49

45
Scheme 23 Alkylation of Benzocarbaporphyrin
Unexpectedly, reaction of N-methyl carbaporphyrin 72 with palladium(II) acetate led to
the formation of a palladium(II) complex 74 where the methyl group has migrated from the
nitrogen atom to the carbon atom (Scheme 24).49 The NMR spectrum of 74 showed that the
complex is highly aromatic and the internal methyl resonance shifted upfield to -3.21 ppm.
Scheme 24 Unexpected Alkyl Group Migration

46
Reaction of benzocarbaporphyrin 57 with ferric chloride did not generate metal complexes
but instead gave various regioselective oxidation products.50, 51 When the reaction was carried out
using 500 equivalents of ferric chloride in a refluxing alcohol solvent, ketal derivatives 75 were
generated in high yields. These ketal derivatives were isolated in the monoprotonated form,
generally as the hydrochloride salt, and exhibited strong absorptions in the far red. Further
protonation of 75 using trifluoroacetic acid generated the related dicationic species. The proton
NMR spectra for 75 showed that these ketals retain strong diatropic characteristics and have a
plane of symmetry. For this system, the meso-protons in the proton NMR spectra showed up at
9.68 and 10.93 ppm. The far-red absorptions exhibited by these derivatives indicate that
carbaporphyrin ketals could have potential applications, for instance as photosensitizers in the
photodynamic therapy (PDT). In fact, 75.HCl has been shown to be an effective agent in the
treatment of leishmaniasis.
When the oxidation reaction was attempted using an aqueous solvent system for one hour,
a chloro derivative 76 was isolated in good yields. The chloro carbaporphyrin again retains
strongly aromatic characteristics, and the proton NMR spectrum of 76 showed that the meso-
protons were shifted to below 9.5 ppm. This is interesting as the X-ray crystal structure of the
chloro derivative shows that the indene subunit is significantly tilted away from the macrocyclic
plane due to the presence of a large internal Cl atom, hence substantially disturbing the planarity
of the molecule. In addition, the UV−vis spectrum of 76 shows a split Soret band at 421 and 434
nm, and Q bands at 521, 560, 608, and 665 nm, again signifying its porphyrin-like characteristics.51
When the reaction was carried out with ferric bromide instead of ferric chloride, under the
same reaction conditions, the corresponding bromo derivative of 76 was isolated in 7% yield. Even
though the larger bromine atom will undoubtedly further distort the macrocycle, the proton NMR

47
spectrum for 76b showed that the macrocycle surprisingly retained nearly all of its diatropic
characteristics. As reactions carried out in the presence of refluxing alcohols led to the formation
of ketals, it was anticipated that reactions using aqueous ferric chloride would afford the
corresponding dihydroxy products 77 or the related ketone. When the reactions with aqueous ferric
chloride were carried out for 16 h, a polar green byproduct 77 was isolated in 22% yield (Scheme
25).51 The proton NMR spectrum for this diketone showed that this species was nonaromatic. The
two carbonyl units gave rise to resonances at 173.6 and 197.6 ppm in the carbon-13 NMR
spectrum, but the IR spectrum showed the C=O stretching peaks at unusually low wave numbers.
The low frequencies can be attributed to the vinylogous amide nature of these bonds, which greatly
reduce the bond strengths.51

48
Scheme 25 Oxidation of Benzocarbaporphyrin with FeCl3
As carbaporphyrins exhibit new and interesting chemistry, steps toward the synthesis of
doubly modified systems were also undertaken. In the first successful synthesis of this type, indane
dialdehyde 78 was reacted with one molar equivalent of diethylpyrrole 44 in the presence of a
strong acid, and subsequent oxidation with FeCl3 generated the dibenzo opp-dicarbaporphyrin 79
in moderate yields (Scheme 26).52 Dibenzo opp-dicarbaporphyrin 79 proved to be somewhat
unstable in solution. Also, the low solubility of 79 in halogenated solvents made characterization
and in-depth reactivity studies very difficult. However, spectroscopic studies did show that the

49
system has strongly diatropic character as the proton NMR spectrum showed an upfield singlet for
the internal CH proton near -6 ppm, while the NH protons appeared near -5 ppm. The meso-
protons for this compound were strongly shifted downfield to around 9.8 ppm. The
dicarbaporphyrin also gave a UV-Vis spectrum that closely resembled true porphyrins with a
strong Soret band near 450 nm and a series of weak Q bands in the visible region.52
Scheme 26 Synthesis of Dibenzo opp-Dicarbaporphyrin
When 79 was protonated using a minimal amount of acid, it was observed that C-
protonation readily occurred to give monocation 79H+ (Scheme 27),52 and this caused a significant
change in the UV-Vis spectrum of 79. The Soret band was greatly diminished. In addition, a band
developed near 500 nm, and a strong absorption emerged near 750 nm. Proton NMR spectroscopy
showed that protonation had taken place at an internal carbon position, and this addition caused
little change in the diatropic nature of the system. The internal sp3 CH2 protons of the protonated
species were observed just above -4 ppm, while the CH proton was detected above -3 ppm. This
slight reduction in the upfield shift was balanced by a slight downfield shift of the meso protons to

50
+10 ppm. Diprotonation of dibenzo opp-dicarbaporphyrin was never observed even with TFA
concentrations in excess of 50%. This may be due in part to the increased steric hindrance that
would result from placing six hydrogens within the porphyrin core.52
Scheme 27 Protonation of Dibenzo opp-Dicarbaporphyrin
Carbachlorins
Chlorophylls are the central constituents in the engine of photosynthesis by which plants
harvest their energy. The structural nucleus of the chlorophylls is a dihydroporphyrin or chlorin
14 where one of the β,β-pyrrolic double bonds has been reduced.2 Studies of chlorin chemistry are
very important due to several reasons. These include the structure elucidation and total synthesis
of naturally occurring chlorophylls. In addition, chlorins show strong absorptions in the red region
of their UV-Vis spectra and synthetic chlorins have potential applications as photosensitizers in
PDT.

51
Even though one of the double bonds found in porphyrins has been reduced in chlorins, the
system still possesses an 18 π electron delocalization pathway. For this reason, chlorins are
aromatic compounds (Figure 14).53
Figure 14 Aromatic Delocalization Pathways in Porphyrin and Chlorin
Chlorins are isomers of nonaromatic phlorins and can potentially interconvert by
prototropic tautomerization (Scheme 28). In reactions that involve the intermediacy of meso-
hydrogenated tetrapyrroles such as porphyrinogens, tautomerization followed by 4e−/4H+
oxidation leads to the formation of chlorin. This type of tautomerization may be a source of chlorin
contamination in one-flask syntheses of porphyrins.53

52
Scheme 28 Phlorin-Chlorin Conversion
Although a great deal of work has been carried out in the field of synthetic chlorin
chemistry by various groups, few studies have explored the preparation of analogous systems such
as carbachlorins that may have equally useful properties. Carbachlorins are chlorin analogues in
which one of the core nitrogen atoms are replaced with a carbon atom (Figure 15).

53
Figure 15 Structures of Chlorin and Carbachlorin
In 1998, Lash and Hayes reported the synthesis of carbachlorins 80 using a “3 + 1” variant
on the MacDonald condensation (Scheme 29).54 In this synthesis, bicyclic dialdehydes 81 were
condensed with tripyrrane 62 in the presence of TFA and subsequent oxidation with DDQ led to
the formation of propanocarbachlorin 80a and propenocarbachlorin 80b in moderate yields. It was
interesting to note that these chlorin analogues retained aromatic characteristics, and in the proton
NMR spectra the inner CH protons showed up around -7 ppm. The UV-vis spectra of these
analogues were also very porphyrin-like, showing a strong Soret band at 404 nm and a series of
small absorptions that culminated in a medium sized Q band at around 650 nm. However, although
true chlorins are easily oxidized to give porphyrins, attempts to further oxidize chlorin analogues
80a or 80b to the corresponding carbaporphyrins 82 were unsuccessful.54

54
Scheme 29 Synthesis of Carbachlorins with Fused Five-membered Rings
In order to further explore the chemistry of carbachlorins, Lash and Hayes attempted to
synthesize porphyrinoid 83 by condensing 1,3-cyclopentanedicarbaldehyde 84 with a tripyrrane
62 (Scheme 30).55 However, no chlorinoid product was formed in these experiments. It was
speculated that the presence of β-pyrrolic substituents or the additional five-membered rings in
structure 82 aids in the generation of porphyrinoid products by altering the conformation of the
intermediates and facilitating the cyclization. For instance, indene dialdehyde 85 on treatment with
tripyrrane yields benzocarbaporphyrins 86 and cyclopentadiene trialdehyde 87 similarly reacted
with tripyrrane to give carbaporphyrin aldehyde 88, albeit in low yields (Scheme 30).55

55
Scheme 30 Synthesis of Carbaporphyrins and an Attempted Synthesis of a Carbachlorin
These results indicate that conjugated dialdehydes or aromatic dialdehydes such as 85 and
87 are more effective precursors in the “3 + 1” condensation reactions than aliphatic dialdehydes

56
such as 84. Therefore, in later studies, conjugated cyclopentene dialdehydes such as 89 were
targeted as precursors for carbachlorin formation (Scheme 31). Attempts to prepare 89 from a
related acetal were unsuccessful but a similar enol ether 90 could be obtained instead (Scheme 32).
This species was very unstable and was immediately used to prepare carbachlorin 83.
Scheme 31 Retrosynthesis of Desired Dialdehyde
In the reaction, 90 was directly condensed with tripyrrane dicarboxylic acid to generate
carbachlorin 83 in 11 – 16% yield (Scheme 32).55

57
Scheme 32 Synthesis of Carbachlorin
Carbachlorin 83 was aromatic in the free base form, displaying strong diatropic character
with the internal CH proton showing up at -6.93 ppm, while the meso-protons were downfield
appearing at 9.01 and 9.68 ppm. Since there were only two signals for the four meso-protons, the
carbachlorin system must possess a plane of symmetry. When carbachlorin 83 was treated with
trifluoroacetic acid, the corresponding monocation 83H+ was generated (Scheme 33).55 The proton
NMR spectrum for 83H+ indicated that the aromatic character was increased as the internal CH
now showed up at -7.00 ppm. Protonation also split the Soret band in the UV-visible spectrum of
carbachlorin 83 to give two different peaks showing up at 409 and 426 nm. Both the proton and
carbon-13 NMR spectra of 83H+ confirmed that the cation retained a plane of symmetry.55

58
Scheme 33 Protonation of Carbachlorin
Attempts to oxidize carbachlorins 80 (Scheme 29, page 54) to the corresponding
carbaporphyrins had been unsuccessful, but this may have been due in part to the presence of a
fused five-membered ring on carbachlorin 80. This unit would not favor the presence of the sp2
hybridized carbons generated upon oxidation due to the associated angle strain.15 In contrast, when
83 was heated with DDQ in toluene, oxidation readily occurred to give the corresponding
carbaporphyrin 91 (Scheme 34).55 Carbaporphyrin 91 is also highly aromatic and the proton NMR
spectrum gave a resonance for the internal CH at -6.91 ppm, while the NHs produce a broad peak
at -3.92 ppm. The meso-protons of 91 appeared downfield as two 2H singlets at 9.77 and 9.83

59
ppm. The proton and carbon-13 NMR spectra for 91 also confirmed that the macrocycle has a
plane of symmetry, and this result is consistent with tautomer 83 (Scheme 33).55
N
N
N
Et
EtMe
Et
H
Me
Et
H
N
N
N
Et
EtMe
Et
H
Me
Et
H HN
N
N
Et
EtMe
Et
H
Me
Et
H H
DDQ
Toluene
+ H+
- H+
+ H+- H+
91
91H+91H22+
N
N
N
Et
EtMe
Et
H
Me
Et
H
Scheme 34 Oxidation of Carbachlorin
The UV-vis spectrum of carbaporphyrin 91 was less porphyrin-like, showing two broad
absorptions in the Soret region at 377 and 421 nm, and broad Q bands at higher wavelengths. This
contrasts to previously mentioned benzocarbaporphyrins and formylcarbaporphyrins, which show
strong Soret bands and better-defined Q bands in their UV-vis spectra. The corresponding
monocation 91H+, generated by the addition of trace amounts of TFA, showed a strong Soret band
at 385 nm together with a broad absorption at 409 nm and Q bands at 563, 574 and 614 nm. Further

60
addition of TFA gave rise to a third species corresponding to the C-protonated dication 91H22+
(Scheme 34).55
It is interesting to note that the formation of benzocarbaporphyrin C-protonated dications
is only complete in approximately 50% TFA solutions, whereas diprotonation of 91 was essentially
complete in 5% TFA solutions. The proton NMR spectrum of 91H22+ showed the presence of a
very strong diatropic ring current in which the internal CH2 gave a resonance at -8.27 ppm while
the external cyclopentadiene protons showed up at 11.11 ppm and the meso-protons appeared
nearby as two 2H singlets at 11.00 and 11.45 ppm. These data clearly demonstrated that the
dication has enhanced aromaticity when compared to the free base form. However, the downfield
shift of the outer cyclopentadiene protons in 91H22+ is due to the relocation of the 18π electron
delocalization pathway through the five-membered carbon ring as shown in bold (Scheme 34).55
Carbaporphyrinoids such as 56, 57 and 58 readily react with silver(I) acetate to give
silver(III) organometallic derivatives.32,33 The formation of silver complexes of carbaporphyrins
57 occurs rapidly in mixtures of dichloromethane and methanol at room temperature in the
presence of 3 equiv. of silver(I) acetate. However, in the case of carbachlorin 83 the formation of
the silver(I) complex is rather slow under these conditions. With carbachlorin 83, the silver(III)
derivative 92 was isolated in 11% yield after almost 16 hr (Scheme 35). It was interesting to
observe that when the reaction was continued for several days, under same set of conditions, the
yield was raised to 31%. However, when 7 equivalents of silver(I) acetate was used, only low
yields (6%) of impure silver(III) carbaporphyrin 93 could be isolated.55
Further attempts to react carbaporphyrin 91 with silver(I) acetate led to decomposition
without forming any metalated product. These results clearly indicate that the exposed carbocyclic
ring is prone to oxidative degradation, and the data suggest that the presence of a fused benzene

61
ring in 57 is beneficial in stabilizing these structures and hence promotes metalation. The UV-vis
spectrum for 92 was porphyrin-like, showing a strong Soret band at 411 nm and a relatively strong
Q band at 599 nm. The proton NMR spectrum for 92 indicated that the complex retained strongly
aromatic properties as the meso-protons showed up downfield at 9.23 and 9.90 ppm and the
carbachlorin CH2CH2 unit gave a 4H singlet at 5.02 ppm.55 The presence of a plane of symmetry
was evident in the proton and carbon-13 NMR spectra, and the molecular formula was confirmed
by high resolution electron impact mass spectrometry.
Scheme 35 Formation of Silver(I) Complex of Carbachlorin
Benzocarbaporphyrin 57 as such does not give any isolatable products in reactions with
palladium(II) salts. However, as mentioned previously, the N-alkyl derivatives of
benzocarbaporphyrins were found to give palladium(II) complexes when reacted with
palladium(II) acetate in refluxing acetonitrile. This chemistry is interesting because the metalation
is associated with an alkyl group migration from the nitrogen to the internal carbon atom.35 A
similar alkylation-metalation sequence was conducted on carbachlorin 83.35
Reaction of 83 with methyl iodide and potassium carbonate in refluxing acetone for 16 h
gave the N-methyl derivative 94 in 34% yield (Scheme 36). Prolonged reaction times did not

62
improve the yield as unidentified side products were formed. The site of alkylation was deduced
from the proton and carbon-13 NMR spectra as the product shows a complete loss of symmetry.
For instance, the meso-protons for 94 showed up as four 1H singlets at 8.89, 9.04, 9.37 and 9.58
ppm.35
Scheme 36 Formation of N-methylated Carbachlorin
The alkylation does introduce steric crowding within the macrocyclic cavity but the NMR
data showed that the chlorin retained most of its diatropic character. In the proton NMR spectrum
of 94, the internal methyl group gave a 3H singlet at -4.27 ppm, while the inner CH produced a
resonance at -6.31 ppm. As the methyl group is too large to pass through the cavity, carbachlorin
94 is chiral and this leads to some complexity in the proton NMR spectrum. The chirality, makes
the six methylene units present in the structure all nonequivalent and the individual protons
diastereotopic, so in principle this could lead to twelve different resonances. However, due to a
significant amount of overlap in this structure, far fewer peaks resolve and the carbachlorin
CH2CH2 unit gave rise to only three multiplets appearing at 4.40 (1H), 4.54 (1H) and 4.79 ppm
(2H).55
The UV-vis spectrum for 94 in CH2Cl2 is somewhat broadened compared to the spectrum
for 83. The alkylated carbachlorin has a Soret band at 407 nm and less well-defined Q bands

63
showing up between 500 and 664 nm. These differences in the UV-visible spectrum presumably
result from distortion of the carbachlorin chromophore due to the presence of the internal methyl
group. Addition of TFA resulted in the formation of a cation 94H+ with a split Soret band at 415
and 428 nm, and again the spectrum was somewhat broadened compared to 94.55
Methylated chlorin 94 reacted with palladium(II) acetate in refluxing acetonitrile to give a
palladium complex. Column chromatography on silica gel gave two fractions with similar
polarities corresponding to metalated products from the N-methyl carbachlorin. It was anticipated
that the product of this chemistry would be N-methyl palladium(II) complex 95. Methyl group
migration would not be expected in this case because that would interrupt the macrocyclic
conjugation pathway. Unexpectedly, the main product was identified as palladium(II)
carbaporphyrin 96 where an oxidation had occurred in addition to the methyl group migration.
Palladium complex 96 was isolated in 17% yield (Scheme 37).55

64
`
Scheme 37 Unexpected Formation of Pd Complex
The remaining fraction appeared to consist primarily of the expected palladium(II) chlorin
95, but this could not be isolated in pure form. The samples were always contaminated with 96,
and it soon became apparent that 95 was slowly converting into 96. In order to speed up the
oxidation and improve the yield of 96, the crude reaction solution was diluted with
dichloromethane and stirred vigorously with an aqueous ferric chloride solution for 24 h.
Following work up, the yield of palladium(II) complex 96 was raised to 34%. It is likely that initial
oxidation affords the N-methyl palladium(II) carbaporphyrin 97 (Scheme 37)55 and this
subsequently undergoes a rearrangement to give 96. The mechanism of the rearrangement is not

65
known, but it could be a stepwise process involving the transient formation of a Pd-methyl bond.
Palladium complex 96 has regained a plane of symmetry and retains strongly diatropic
characteristics. The proton NMR spectrum showed the internal methyl group at -4.46 ppm, while
the meso-protons appeared downfield as two 2H singlets at 10.00 and 10.42 ppm. The
cyclopentadiene protons are directly connected to the 18π electron delocalization pathway and as
a result are deshelded to give a 2H singlet at 9.60 ppm. The UV-vis spectrum is less distinctive,
giving broad bands in the Soret region at 394 and 440 nm.55 The molecular formula for 96 was
confirmed by high resolution electron impact mass spectrometry.
Although some intriguing results have been obtained for carbachlorins, these studies have
been limited due to the difficulties encountered in preparing this system. Furthermore, an effective
methodology for synthesizing carbaporphyrins without fused aromatic rings has not been reported
previously. In this thesis, a new synthetic route for preparing carbaporphyrins and carbachlorins
has been developed. The new porphyrinoids, which are stabilized by electron-withdrawing ester
functionalities, were prepared in excellent yields from cyclopentanedialdehydes. The
carbaporphyrin system proved to be an excellent organometallic ligand, forming complexes with
nickel(II), palladium(II), silver(III) and gold(III).

66
CHAPTER II
SYNTHESIS OF CARBAPORPHYRINS
Introduction to Carbaporphyrin
The synthesis of macrocyclic structures related to porphyrins has attracted considerable
attention, in part because modified structures may enhance valuable properties that are exhibited
by regular porphyrins.12 Carbaporphyrinoid systems, where one or more of the core nitrogens have
been replaced by carbon atoms, have been widely investigated over the past 20 years. Although
detailed studies on benzocarbaporphyrins have been reported, carbaporphyrins without a fused
benzene unit have only been prepared in low yields, and an improved route to carbaporphyrins of
this type is needed. Specifically, porphyrinoid 98 with two ester functionalities attached to the
carbocyclic ring is an appealing target for synthesis. This represent a new type of
carbaporphyrinoid structure. The proposed methodology for preparing this system is modeled after
the synthesis of porphyrin analogues containing bicyclooctane units previously developed in Dr.
Lash’s lab.22

67
Results and Discussion
A retrosynthetic analysis suggested that carbaporphyrin 98 and a related chlorin could be
obtained from cyclopentane dialdehyde 99 and the known tripyrrane 62. It was speculated that
acid catalyzed “3+1” condensation of dialdehyde 99 with tripyrrane 62 in presence of
trifluoroacetic acid, following oxidation with DDQ, would afford the targeted carbaporphyrin
(Scheme 38).54 In order to utilize the “3+1” methodology, it was initially necessary to synthesize
tripyrrane 62. The preparation of this tripyrrane can be carried out in 12 synthetic steps, where two
key pyrrolic intermediates 100 and 44 needed to be generated (Scheme 39). The tripyrrane was
initially formed with benzyl ester protective groups but these can be removed by hydrogenolysis
over Pd/C to afford the required dicarboxylic acid 62 (Scheme 38).
Scheme 38 Retrosynthetic Analysis of Carbaporphyrin 98

68
Scheme 39 Retrosynthetic Analysis of the Tripyrrane Intermediate
In order to synthesize pyrrole 100, it was first necessary to prepare pyrrole ester 101.
Reaction of diethyl malonate with sodium nitrite and acetic acid afforded oxime 102 and this was
reduced under hydrogen over 10% Pd/C to give diethyl aminomalonate 103. Alkylation of 2,4-
pentanedione with ethyl iodide and potassium carbonate in refluxing acetone gave the ethyl
substituted diketone 104, and this was condensed with 103 in refluxing acetic acid to produce
pyrrole ethyl ester 101 (Scheme 40).

69
Scheme 40 Preparation of Pyrrole Ester 101 using a Knorr-Type Synthesis
Pyrrole ethyl ester 101 was transesterified using benzyl alcohol in the presence of sodium
benzyloxide to give the corresponding benzyl ester 105 (Scheme 41).
Scheme 41 Formation of a Pyrrole Benzyl Ester

70
Pyrrole 105 was then selectively oxidized with lead tetraacetate in acetic acid to afford the
required acetoxymethyl pyrrole precursor 100 in good yields (Scheme 42).
Scheme 42 Formation of an Acetoxymethylpyrrole
The second pyrrolic precursor 3,4-diethylpyrrole 44, was synthesized via the Barton-Zard
methodology. Initially, glycine ethyl ester hydrochloride and triethylamine were heated to reflux
in methyl formate to afford N-formylglycinate (Scheme 43). Subsequent dehydration with
phosphorus oxychloride and triethylamine at 0 ℃ then gave ethyl isocyanoacetate 106.
Scheme 43 Formation of Ethyl Isocyanoacetate 105

71
Besides ethyl isocyanoacetate, acetoxy nitrohexane 107 was also required as a precursor to
pyrrole 44. Initially, a related nitro alcohol was prepared by reacting propionaldehyde with
1-nitropropane in the presence of potassium fluoride (Scheme 44). Subsequent treatment with
acetic anhydride under acidic conditions then afforded the required acetoxy nitro hexane 107 in
96% yield. Barton-Zard condensation of acetoxy nitrohexane 107 with ethyl isocyanoacetate 106
in the presence of two equivalents of the base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) resulted
in the formation of pyrrole ethyl ester 108. The crude pyrrole ester was then refluxed in a solution
of sodium hydroxide in ethylene glycol at 190 oC and this resulted in saponification and
decarboxylation to give diethyl pyrrole 44 in 48% yield (Scheme 44). However, the dialkyl pyrrole
was found to be moderately unstable at room temperature and was stored in the freezer for future
use.

72
Scheme 44 Preparation of 3,4-Diethylpyrrole via a Barton-Zard Synthesis
Treatment of two equivalent of 100 with 3,4-diethylpyrrole 44 and acetic acid in refluxing
ethanol gave tripyrrane dibenzyl ester in 92% yield. The benzyl ester protective groups were
cleaved by hydrogenolysis over 10% Pd/C to give the corresponding dicarboxylic acid 62 (Scheme
45).54 This product was relatively unstable and therefore had to be used within one week even
when it was stored in the freezer.

73
Scheme 45 Preparation of Tripyrrane 62
In addition to preparing the tripyrrane, it was necessary to develop a synthetic route to
cyclopentane dialdehydes such as 99. In an initial attempt, dimethyl acetylenedicarboxylate
(DMAD) 109 was reacted with cyclopentadiene 110 at room temperature to give the Diels-Alder
adduct 111 (Scheme 46).57
Scheme 46 Preparation of Bicyclic Adduct 111

74
Göksu and coworkers reported that seven bicyclo[2.2.1]heptenes, including 111, were
cleaved to cyclic dialdehydes in good yields by oxidation with KMnO4-CuSO4.5H2O.58 However,
all attempts to reproduce this procedure with norbornadiene derivative 111 failed to generate any
dialdehyde. Hence, an alternative synthetic method or a different precursor was needed. Donohoe
and coworkers reported an alternative two steps route to dialdehydes.59 In this synthetic route, a
solution of norbornadiene 111 in tert-butyl alcohol was oxidized to give 112 using KMnO4 and
NaOH in tert-butyl alcohol and water at 0 oC (Scheme 47).59 However, when we attempted to
prepare 112 using this methodology, the yields were low.
Scheme 47 Formation of Diol 112
Alternative synthetic routes for diol formation were investigated. However, all of the
conditions gave poor results. Some of the failed attempts to form diol 112 are shown in Scheme
48.58,60,61 In order to circumvent this problem, the use of an alternative norbornene intermediate
was considered.

75
Scheme 48 Unsuccessful Attempts to Synthesize Diol 112
Dimethyl fumarate (113), a readily available starting material, underwent a Diels-Alder
cycloaddition with cyclopentadiene to form the bicyclic adduct 114 (Scheme 49). Several different
procedures were attempted to carry out this transformation. Superior results were achieved using
a literature procedure by Beare and coworkers.62 In this procedure, freshly cracked
cyclopentadiene was gradually added to an aqueous suspension of dimethyl fumarate (Scheme
49).62 The mixture was stirred at room temperature for 150 minutes and then extracted with ethyl
acetate to give an excellent yield of 114. This norbornene derivative was then reacted with KMnO4
and NaOH in tert-butyl alcohol and water at 0 oC to give the related dialcohol 115 (Scheme 50).59

76
Scheme 49 Formation of Bicyclic Adduct 114
Scheme 50 Formation of Diol 115
The diol product 115 was isolated as a white crystalline solid in 73% yield. In the 1H NMR
spectrum of resulting product, two broad resonances for the OH protons could be identified (Figure
16).

77
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5 ppm
3.703.753.803.85 ppm
Figure 16 500 MHz 1H NMR Spectrum of Fumarate Diol in CDCl3
Diol 115 could be oxidatively ring opened using potassium periodate as an oxidizing agent
to give dialdehyde 99 in 55% yield (Scheme 51).54 The mechanism for ring opening of the bicyclic
adduct to give a five-membered ring dialdehyde involved the formation of a transient cyclic
periodate derivative. Elimination of an iodate ion affords the dialdehyde (Scheme 52). Dialdehyde
99 thus generated was very unstable and decomposed rapidly even at low temperatures. For this
reason, it was immediately utilized in the preparation of carbaporphyrin.
Scheme 51 Formation of Dialdehyde 99

78
Scheme 52 Mechanism for Ring Opening
Dialdehyde 99 was condensed with tripyrrane dicarboxylic acid 62 in the presence of
TFA/CH2Cl2. Initial condensation would be expected to produce a hexahydrocarbaporphyrin such
as 116. Following oxidation with DDQ in refluxing toluene, carbaporphyrin 98 was isolated as the
major product along with some carbachlorin 117 as the minor product (Scheme 53).54

79
Scheme 53 Preparation of Carbaporphyrin 98
During purification using column chromatography, three bands were collected. The first
band to elute off the column was bright red, and corresponded to a small amount of porphyrin
byproduct. The second major band corresponded to carbaporphyrin 98 and following
recrystallization using chloroform and methanol, this product was isolated as purple crystals in
40% yield. A third green band was collected that corresponded to carbachlorin 117, and following

80
recrystallization from chloroform and methanol the dihydroporphyrinoid was obtained as a green
powder in 9% yield.
As expected, carbaporphyrin 98 proved to be a fully aromatic compound. The proton NMR
spectrum for 98 (Figure 17) in CDCl3 showed the presence of a strong diamagnetic ring current.
The internal CH proton was found at -6.27 ppm, while the meso-protons were shifted downfield
to give two 2H singlets at 10.24 and 9.51 ppm. As there were only two signals for the four meso-
protons, the results indicate that the molecule is symmetrical.
-6-5-4-3-2-111 10 9 8 7 6 5 4 3 2 1 0 ppm
3.853.903.954.00 ppm
Figure 17 500 MHz proton NMR Spectrum of Carbaporphyrin 98 in CDCl3
For the carbachlorin byproduct 117, the proton NMR spectrum in CDCl3 again showed the
presence of a strong diamagnetic ring current. The internal CH proton was found further upfield
at -7.14 ppm, while the meso-protons were still deshielded to between 9.85 and 9.48 ppm (Figure

81
18). The symmetry was maintained in the carbachlorin byproduct as can be seen in the figure
below.
-7-6-5-4-3-2-111 10 9 8 7 6 5 4 3 2 1 0 ppm
-7.1 -7.2 -7.3 ppm
9.69.8 ppm
Figure 18 500 MHz proton NMR spectrum of the Carbachlorin byproduct in CDCl3
The UV-Vis spectrum for the free base carbaporphyrin had a strong absorption in the Soret
region at 436 nm, followed by a series of four weaker absorptions between 520 and 720 nm, which
is characteristic aromatic porphyrinoids (Figure 19). In addition, the structure of carbaporphyrin
98 was also confirmed using X-ray crystallography (Figure 20).

82
Figure 19 UV-Vis spectrum of Carbaporphyrin 98 in Dichloromethane
0
2
4
6
8
10
12
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

83
Figure 20 POV-Ray generated ORTEP III Drawing of 98
The UV-vis spectrum for the free base carbachlorin byproduct 117, has a strong absorption
in the Soret region at 398 nm, followed by weaker absorptions at 490 nm and 588 nm. In addition,
a strong red shifted peak was displayed at 648 nm (Figure 21), a feature that is also seen for
tetrapyrrolic chlorins.

84
Figure 21 UV-Vis spectrum of Carbachlorin 117 in Dichloromethane
In principle, the monocation of 98 can be obtained when a trace amount of TFA is added.
However, when one drop of TFA was added to an NMR tube containing a solution of 98 in CDCl3,
the resulting NMR spectrum appeared to correspond to mixture of mono 98H+ and dicationic
species 98H2+ (Figure 22).
0
5
10
15
20
25
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

85
Figure 22 500 MHz proton NMR Spectrum of a Mixture of 98H+ and 98H22+ in CDCl3
When only 4 μL of TFA was added to the solution, the proposed monocation 98H+ (Scheme
54) could be observed by proton NMR and UV-Vis spectroscopy (Figure 23). The proton NMR
spectrum of 98H+ showed that the addition of TFA to the carbaporphyrin had slightly increased
the aromatic character of carbaporphyrin, as the meso-protons were shifted slightly further
downfield to 10.40 and 9.74 ppm (Figure 24). The appearance of a broad peak at -3 ppm
corresponds to the presence of an additional NH due to protonation. In addition, there was a
significant change in the UV-Vis spectrum with the addition of the trace amounts of acid (Figure
23).
-7-6-5-4-3-2-113 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm

86
Scheme 54 Protonation of Carbaporphyrin 98
Figure 23 UV-Vis spectrum of Carbaporphyrin 98H+ in CH2Cl2
0
1
2
3
4
5
6
7
8
9
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

87
-6-5-4-3-2-112 11 10 9 8 7 6 5 4 3 2 1 0 ppm
10.0 ppm
3.23.43.63.84.04.24.4 ppm
-2.0 ppm
Figure 24 500 MHz proton NMR Spectrum of 98H+ in CDCl3
Further addition of TFA led to the formation of dication 98H22+ (Scheme 55). The proton
NMR spectrum of 98H22+ (Figure 25) showed a 2H singlet at -7.67 ppm due to the internal CH2,
and two 2H singlets at 11.80 and 11.00 ppm corresponding to the meso-protons. This not only
confirmed that diprotonation had taken place but also indicated that the aromatic character of the
conjugated system was further improved. In addition, the proton NMR spectrum for dication
98H22+ confirmed that the compound retained a plane of symmetry.

88
Scheme 55 Further Protonation of Carbaporphyrin 98H+
-7-6-5-4-3-2-113 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
-7.5 ppm
Figure 25 500 MHz 1H NMR Spectrum of 98H22+ in CDCl3.

89
In order to further understand the reactivity of carbaporphyrin 98, a few more experiments
were performed to observe how the exchange of protons takes place with the addition of acid. It
was interesting to note that addition of d-TFA to an NMR solution of 98 in CDCl3 showed rapid
exchange of the 21-H resonance and slow exchange of the meso-protons. As the solutions consisted
of a mixture of protonated species, it was necessary to extract the sample before NMR analysis
could be conducted. Two drops of d-TFA were added to the NMR tube and after 10 min the
solution was neutralized by addition of NaOD in D2O. The mixture was diluted with water,
extracted with dichloromethane and the organic solutions dried over sodium sulfate. Following
removal of the solvent, the residue was analyzed by proton NMR spectroscopy. The results showed
that the internal CH had undergone exchange but no other resonances were affected (Figure 26).
When the solution was exposed to d-TFA for 24 h, and the solution neutralized and extracted as
before, significant reduction in the intensities of the meso-proton resonances were observed. The
peak corresponding to the 5,20-protons was reduced by approximately 15%, while the 10,15-
proton resonance was diminished by >40% (Figure 27) The results indicate that meso-protonated
species such as monocations 98aH+ and 98bH+are energetically accessible and in equilibrium with
98, 98H+ and 98H22+ (Scheme 56). Similar results have previously been noted for
benzocarbaporphyrins.

90
Scheme 56 Proposed Equilibria of Different Cationic Species for Carbaporphyrin 98

91
-6-5-4-3-2-110 9 8 7 6 5 4 3 2 1 0 ppm
0.1
7
12
.29
5.6
73
.96
4.2
2
5.8
9
2.0
6
2.0
0
Figure 26 500 MHz proton NMR Spectrum of 98 Showing Exchange of the 21-H after 10 min
Exposure to d-TFA
-6-5-4-3-2-110 9 8 7 6 5 4 3 2 1 0 ppm
0.2
7
12
.84
5.8
3
4.1
2
4.2
1
6.0
0
1.1
2
1.7
0
Figure 27 500 MHz proton NMR Spectrum of 98 Showing Partial Exchange of the meso-
Protons after 24 hr Exposure to d-TFA

92
When carbachlorin 117 was exposed to a trace amount of TFA, a deep purple solution of
the corresponding monocation 117H+ (Scheme 57) could be observed by proton NMR (Figure 28)
and UV-Vis spectroscopy (Figure 29). The proton NMR spectrum for 117H+ showed that the
addition of acid to the carbachlorin system had slightly increased its aromatic nature, and although
the internal CH was still observed around -7 ppm, the meso-protons were shifted further downfield
to 10.30 and 9.84 ppm (Figure 28). The appearance of an additional broad peak at -6.0 ppm
confirmed the presence of an additional NH due to protonation. Unlike the related carbaporphyrins,
further addition of TFA to 117H+ did not give a diprotonated species as protonation at the inner
carbon would result in a disruption of macrocyclic aromatic character.
Scheme 57 Protonation of Carbachlorin byproduct 117H+

93
-7-6-5-4-3-2-112 11 10 9 8 7 6 5 4 3 2 1 0 ppm
9.810.010.2 ppm
-7.05 -7.10 ppm
-5.6 ppm
3.94.04.1 ppm
Figure 28 500 MHz 1H NMR Spectrum of 117H+ in CDCl3

94
Figure 29 UV-vis spectrum of 117H+ in 5% TFA-Dichloromethane.
Metalation of carbaporphyrin 98 was also investigated. As several carbaporphyrinoid
systems have been shown to form silver(III) derivatives, the synthesis of a silver(III) complex of
carbaporphyrin 98 was attempted. A solution of carbaporphyrin 98 in CH2Cl2 was added to a
suspension of silver(I) acetate in methanol, and the resulting mixture was stirred overnight at room
temperature. After column chromatography, a red fraction was collected and recrystallized using
chloroform and methanol to yield the organometallic complex 118 as a dark red powder in 60%
yield (Scheme 58).63 Silver complex 118 retains aromatic characteristics as indicated by proton
NMR spectroscopy (Figure 30). The meso-protons were deshielded showing up as two 2H singlets
at 9.91 and 8.79 ppm. However, due to metal complexation, there were no upfield resonances
beyond the TMS reference as the core CH and NH protons of the carbaporphyrin are no longer
present.
0
5
10
15
20
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

95
Scheme 58 Synthesis of Silver(III) Carbaporphyrin 118
Figure 30 500 MHz 1H NMR Spectrum of 118 in CDCl3.
In addition to the silver complex, the formation of a related gold(III) complex using
gold(III) acetate was attempted. Similar reaction conditions to those used for silver complex
formation were implemented. However, this reaction did not yield the gold complex. Different

96
reaction conditions were tried using the same solvent system but no metalated product could be
isolated. However, when 98 was refluxed with gold(III) acetate in pyridine, the solution rapidly
changed color from brown to dark red once it started to reflux. After 5 minutes of refluxing, it was
determined by TLC analysis that not all of the starting free base porphyrin 98 had reacted with the
gold(III) acetate. After 30 minutes of reaction time it was observed that most of the conversion has
taken place, and the reaction was stopped. Purification using column chromatography gave the
gold(III) complex 119 in 11% yield (Scheme 59). The derivative also retained aromatic
characteristics and gave a quite similar UV-vis spectrum to the silver complex.
Scheme 59 Formation of Gold(III) Complex of Carbaporphyrin
Previously, a benzocarbaporphyrin had been shown to react with methyl or ethyl iodide
and potassium carbonate in refluxing acetone to afford N- and C- alkylation products.49 Therefore,
it was of interest to see whether the new carbaporphyrin system, with two electron withdrawing
ester groups on adjacent carbons, could also be alkylated under similar conditions. It was
interesting to observe that when carbaporphyrin 98 was stirred with potassium carbonate and
methyl iodide in refluxing acetone solution for 30 minutes, an alkylation product was obtained in

97
up to 55% yield after purification (Scheme 60).64 However, unlike the alkylation of
benzocarbaporphyrin 57, only one monoalkylation product 120 was observed for this reaction.
Scheme 60 Formation of C-methylated Carbaporphyrin 120
In the proton NMR spectrum of 120 (Figure 31), the presence of singlet at -4.68 ppm (3H)
indicated that a methyl group had been introduced within the macrocyclic cavity. The symmetry
of the system was still maintained as there were only two signals for the meso-protons showing up
at 9.90 and 9.28 ppm. The spectroscopic data were consistent with a C-methyl derivative rather
than the alternative N-alkylation product.

98
-7-6-5-4-3-2-110 9 8 7 6 5 4 3 2 1 0 ppm
Figure 31 500 MHz 1H NMR Spectrum of 120 in CDCl3
When carbaporphyrin 98 was stirred with potassium carbonate using an excess of
iodomethane in acetone and the reaction was refluxed overnight, further reaction took place.
Following column chromatography, a major dialkylation product was isolated with methyl groups
on both the inner carbon and an adjacent nitrogen atom (Scheme 61).49 After recrystallization from
chloroform-methanol, the dimethylated carbaporphyrin 121 was isolated as shiny purple crystals
in 69% yield.

99
Scheme 61 Formation of Dimethylated Carbaporphyrin 121
In the proton NMR spectrum for the dimethylated carbaporphyrin, the internal methyl
groups showed up as two 3H singlets at -5.27 and -3.91 ppm. The four meso-protons appeared as
four separate singlets indicating a loss of symmetry in the structure. This loss of symmetry could
be explained if one of the methyl groups is attached to a pyrrole nitrogen that is flanking the
carbocyclic ring. In addition, as the methyl groups are too large to fit within the cavity, they could
both be placed to one side of the structure. It is also likely that the two methyl groups are placed
on opposite sides of the macrocycle. The resulting structure is chiral and would exist as a pair of
enantiomers. Although the presence of internal methyl groups may distort the structure, 121 is still
a strongly aromatic compound. As noted above, the internal methyl resonances are highly shielded,
while the meso-protons are relatively deshielded and show up at 8.90, 9.05, 9.52 and 9.54 ppm
(Figure 32).

100
Figure 32 500 MHz 1H NMR Spectrum of 121 in CDCl3
Addition of TFA to a solution of C-methylcarbaporphyrin 120 led to the formation of
monocation 120H�. The proton NMR spectrum for this species indicated a slight increase in
aromatic character is developed on protonation, as the internal methyl resonance is shifted upfield
to -5.41 ppm while the meso-protons are shifted downfield to 9.49 and 10.03 ppm (Figure 33).

101
-6-5-4-3-2-112 11 10 9 8 7 6 5 4 3 2 1 0 ppm
9.69.810.0 ppm
Figure 33 500 MHz 1H NMR Spectrum of 120H+ in CDCl3
Further addition of TFA led to the formation of a diprotonated dication 120H22+ showing
a doublet at -6.55 ppm due to protonation at the internal methyl carbon (Figure 34). The meso-
protons lie close to the TFA peak, showing up 11.08 and 11.7 ppm, indicating increased
diamagnetic character. It is worth noting that both the mono and dications maintained the plane of
symmetry.

102
Figure 34 500 MHz 1H NMR Spectrum of 120H22+ in CDCl3
Addition of TFA to a solution of dimethylcarbaporphyrin 121 led to the formation of
monocation 121H�(Figure 35). However, further addition of acid did not generate the related
dication. The proton NMR spectrum of 121H+ showed four singlets for the meso-protons at 9.92,
9.86, 9.46 and 9.45 ppm. The internal methyl resonances appeared at -3.54 and -4.91 ppm. The
results indicate that the protonated species has a slightly stronger diatropic ring current than the
free base form 121.
-6-5-4-3-2-112 11 10 9 8 7 6 5 4 3 2 1 0 ppm

103
-5-4-3-2-111 10 9 8 7 6 5 4 3 2 1 0 ppm
9.59.69.79.89.9 ppm
Figure 35 500 MHz 1H NMR Spectrum of 121H+ in CDCl3
Metalation of N-methylated benzocarbaporphyrin with palladium(II) acetate in refluxing
acetonitrile has previously been shown to give a palladium(II) organometallic derivative.64 The
metalation reaction in benzocarbaporphyrins is associated with an alkyl group migration from the
nitrogen to the inner carbon atom. However, in 120 the methyl group is already attached to the
internal carbon and no rearrangement would be necessary to form an analogous palladium
complex. In fact, reaction of 120 with palladium(II) acetate is reflexing acetonitrile gave the
organometallic derivative 122 in 63% yield (Scheme 62).49 Although, the conjugation pathway in
the metal complex has been altered, the macrocycle still remains fully aromatic.

104
Scheme 62 Metalation of C-methylcarbaporphyrin with Palladium(II) Acetate
The proton NMR spectrum of 122 (Figure 36) gave two 2H singlets at 9.85 and 10.81 ppm
in the downfield region for the four meso-protons, confirming that compound 122 has a plane of
symmetry. Moreover, the presence of a singlet at -4.12 ppm (3H) indicated that the internal methyl
group was strongly shielded. As the symmetry of the alkylation product after complexation is still
maintained, the results indicate that no rearrangement has taken place. The UV-Vis spectrum for
the Pd complex shows a strong Soret band at 306 nm and Q bands at 410, 460, 602 and 639 nm
(Figure 37) that further support its aromatic character. The structure of palladium complex 122
was also confirmed using X-ray crystallography (Figure 38).

105
-8-7-6-5-4-3-2-114 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
Figure 36 500 MHz 1H NMR Spectrum of 122 in CDCl3
Figure 37 UV-vis Spectrum of Palladium(II) Carbaporphyrin in Chloroform
0
0.5
1
1.5
2
2.5
3
3.5
4
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

106
Figure 38 POV-Ray rendered ORTEP III Drawing of 122
Nickel complexes of carbaporphyrins have not been previously reported and attempts were
made to prepare a similar complex from nickel(II) acetate. Under the same set of conditions, no
formation of a nickel complex was observed. However, when 120 was refluxed with nickel(II)
acetate in DMF, the solution rapidly turned from brown to dark green as soon as it started to reflux.
After 5 minutes of refluxing, it was determined by TLC analysis that all of the starting
carbaporphyrin 120 had reacted with the nickel(II) acetate. After column chromatography, a green
colored fraction was obtained that corresponded to nickel(II) complex 123. After recrystallization
from methanol and chloroform, the pure complex was obtained in 71% yield. (Scheme 63).

107
Scheme 63 Metalation of C-methylcarbaporphyrin with Nickel(II) Acetate
The proton NMR spectrum of 123 (Figure 39) showed two singlets at 9.59 and 10.56 ppm
in the downfield region for the four meso-protons, suggesting that compound 123 has retained the
plane of symmetry. Moreover, the presence of a singlet at -3.89 ppm (3H) indicated that an internal
methyl group was still present. Clearly, the macrocycle still retained fully aromatic characteristics.
The structure was also confirmed by X-ray crystallography (Figure 40).

108
-10-9-8-7-6-5-4-3-2-114 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
NN
N
Et
Me
MeO2C
MeO2C Me
Et
Et
Et
Ni
Me
Figure 39 500 MHz 1H NMR Spectrum of 123 in CDCl3
Figure 40 POV-Ray rendered ORTEP III Drawing of 123

109
Conclusions
A cyclopentane dialdehyde was prepared in two steps from the Diels–Alder adduct of
dimethyl fumarate and cyclopentadiene. The ‘3+1’ condensation of dialdehyde 99 and tripyrrane
dicarboxylic acid 62 led to the formation of carbaporphyrin 98 in 40% yield together with some
carbachlorin 117 byproduct. This newly synthesized carbaporphyrin system proved to be highly
aromatic. Protonation of this system can readily occur, forming both mono- and dications.
Alkylation and metalation products were also synthesized and characterized.
Carbachlorin 117 can also readily form a monocation 117H+ when TFA is added, but no
dication formation was observed. Reaction between carbaporphyrin 98 and silver(I) acetate gave
a silver(III) organometallic complex. Similarly, reaction between carbaporphyrin 98 and gold(III)
acetate afforded a gold(III) organometallic complex, albeit in low yield. Alkylation of
carbaporphyrin 98 gave a C-methylated product 120 when refluxed with methyl iodide for 1 hr,
but a dimethylated product predominated when the reaction was carried out overnight. Protonation
of the monoalkylated carbaporphyrin with TFA gave both monocationic and dicationic species,
but the dimethylated species only formed a monocation. Metalation of 120 with palladium(II)
acetate afforded palladium(II) carbaporphyrin, while reaction with nickel(II) acetate afforded a
related nickel(II) carbaporphyrin in good yields.
The straightforward synthesis of carbaporphyrin 98 will allow further studies on this
system to be conducted. It is anticipated that the ester moieties can be reduced to give
carbaporphyrin alcohols. Initial investigations with LiAlH4 led to decomposition, but reaction with
diisobutylaluminium hydride (DIBAL-H) selectively reduced one of the ester units. At -70 ℃, the
related monoaldehyde 124 was generated but at room temperature monoalcohol 125 was formed
(Scheme 64). So far attempts to reduce both ester moieties have been unsuccessful.

110
Scheme 64 Reduction of Carbaporphyrin 98
Reduction to diol 126 could enable novel cycloaddition chemistry to be investigated.
Conversion to the related dibromide 127 might be accomplished by reaction with phosphorus
tribromide. Treatment with sodium iodide may result in the formation of a transient diene 128 that
could undergo a Diels–Alder cycloaddition with suitable dienophiles such as dimethyl
acetylenedicarboxylate to generate 129 (Scheme 65). This chemistry will make new classes of
carbaporphyrin derivatives accessible.

111
Scheme 65 New Class of Carbaporphyrin Derivatives
Experimental
All chemicals were purchased from Sigma Aldrich or Acros Organic. Grade III neutral
alumina and silica gel were used to perform column chromatography. The 1H and 13C-NMR
spectra were obtained either using Bruker Avance III 500 MHz NMR spectrometer or Bukner 400
MHz NMR spectrometer at 302 K. Chemical shifts were recorded, relative to CDCl3 (residual
chloroform at δ 7.26 ppm) in proton NMR spectra and the CDCl3 triplet at δ 77.23 ppm in 13C-
NMR spectra, in parts per million (ppm). 2D experiments were performed by using standard

112
software. The UV-vis spectra for the compounds were collected by Cary 100 Bio
spectrophotometer. Melting point were gathered using a Mel-Temp apparatus. High-resolution
mass spectra (HRMS) were carried out by using a double focusing magnetic sector instrument
Mass spectrometry data was obtained from the Mass Spectral Laboratory, School of Chemical
Sciences, University of Illinois, Urbana-Champaign.
Dimethyl bicyclo[2.2.1]hept-5-ene-2-endo-3-exo-dicarboxylate (114)
Freshly distilled cyclopentadiene (6.6 ml, 78 mmol) was added to a suspension of dimethyl
fumarate (10 g, 69.3 mmol) in water (400 mL) and the suspension was stirred vigorously for 120
minutes. Ethyl acetate (150 mL) was added and the phases were separated, the organic phase was
dried over Na2SO4 and the solvent evaporated to give the title compound (10.7 g, 50.9 mmol, 73%)
as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.46 (1H, dq, J = 8.8, 1.7 Hz), 1.62 (1H, br t, J
= 8.8 Hz), 2.69 (1H, dd, J = 4.5, 1.7 Hz), 3.11-3.14 (1H, m), 3.25-3.28 (1H, m), 3.38 (1H, t, J =
4.1 Hz), 3.65 (3H, s), 3.72 (3H, s), 6.07 (1H, dd, J = 5.6, 2.8 Hz), 6.28 (1H, dd, J = 5.6, 3.1 Hz).
13C NMR (100 MHz, CDCl3): δ 45.9, 47.3, 47.5, 47.8, 48.1, 52.0, 52.3, 135.4, 137.8, 173.9, 175.1

113
Dimethyl 5,6-dihydroxybicyclo [2.2.1] heptane-2,3-dicarboxylate (115)
To a solution of norbornene 114 (10.83 g, 51.5 mmol) in tert-butyl alcohol (200 mL) and water
(50 mL) at 0 °C was added dropwise a solution of potassium permanganate (12.0 g, 76.0 mmol)
and sodium hydroxide (2.61 g, 65.3 mmol) in water (250 mL). After the addition was complete,
the reaction mixture was stirred for 20 minutes and then quenched using a saturated aqueous
solution of sodium metabisulphite (Na2S2O5) until the solution turned colorless. The mixture was
filtered and the tert-butyl alcohol was removed from the filtrate in vacuo. The solution was
extracted with ethyl acetate (4 × 100 mL) and the combined organic layers dried over MgSO4,
filtered and concentrated in vacuo to furnish the diol 115 (6.85 g, 28.0 mmol, 55%) as a white
crystalline solid, mp 84-86 oC (lit. mp59 81-84 oC). 1H NMR (400 MHz, CDCl3): δ 1.44 (1H, dp, J
= 11.0, 1.5 Hz), 1.89 (1H, dq, J = 11.0, 1.5 Hz), 2.50 (1H, br s), 2.55-2.57 (1H, m), 2.62 (1H, br),
2.73 (1H, br s overlapping with dd, J = 5.7, 1.3 Hz), 3.20 (1H, t, J = 5.1 Hz), 3.70 (3H, s), 3.71
(3H, s), 3.76-3.79 (1H, br m), 3.86-3.89 (br m). 13C NMR (100 MHz, CDCl3): δ 31.9, 45.0, 46.4,
46.7, 48.5, 52.4, 52.5, 70.6, 73.7, 173.1, 174.2. HRMS (EI): calcd for C11H16O6 + H 245.1025,
found 245.1027.

114
Dimethyl 3,5-diformylcyclopentane-1,2-dicarboxylate (99)
The foregoing diol 115 (4.15 g, 16.9 mmol) was mixed with water (5.0 ml), ether (10 ml) and
potassium periodate (4.66 g, 20.3 mmol), and the reaction was stirred at room temperature for 1
hr. The excess potassium periodate was filtered off and the two phases were separated. The water
layer was back extracted twice with ether. The ether layers were combined, dried over Na2SO4,
and the ether removed under reduced pressure to gave dialdehyde as a clear oil 99 (2.08 g, 8.59
mmol, 50%). The unstable dialdehyde was not further purified but used immediately to prepare
carbaporphyrin. 1H NMR (400 MHz, CDCl3): 2.17-2.26 (1H, m), 2.46 (1H, dt, J = 13.8, 5.9 Hz),
3.05-3.10 (1H, m), 3.26-3.32 (1H, m), 3.56-3.65 (2H, m), 3.70 (3H, s), 3.76 (3H, s), 9.65 (1H, d,
J = 0.8 Hz), 9.68 (1H, d, J = 0.9 Hz). HRMS (EI): calcd for C11H14O6 + H 243.0869, found
243.0871.

115
8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18-dimethyl-21-carbaporphyrin (98)
Tripyrrane dicarboxylic acid 62 (100 mg, 0.22 mol) was stirred with TFA (1 ml) under an
atmosphere of nitrogen for 2 minutes in a 250 ml round bottom flask. Dichloromethane (99 ml)
was added, followed immediately by dialdehyde 99 (56 mg, 0.23 mol), and the mixture was stirred
under nitrogen overnight. The mixture was neutralized by the addition of triethylamine and the
solvent removed under reduced pressure. The residue was taken up in toluene (50 ml), DDQ (150
mg, 0.66 mmol) was added and the resulting mixture was stirred under reflux for 1 hr. The solvent
was removed under reduced pressure, then the residue was dissolved in CH2Cl2 and washed with
water. The solution was concentrated under reduced pressure and the residue chromatographed on
grade III neutral alumina column eluting with CH2Cl2 and hexane in a ratio of 60:40. The product
was collected as a dark brown fraction that was the second fraction eluting from the column.
Recrystallization from chloroform and methanol gave carbaporphyrin (52 mg, 0.091 mmol, 42%)
as purple crystals, mp 234-236oC; UV-Vis (1% Et3N-CH2Cl2): λmax (log ε) 368 (4.73), 436 (5.00),
526 (4.04), 566 (4.24), 648 (3.31), 717 nm (3.43). UV-Vis (5% DBU-CH2Cl2): λmax (log ε) 361
(4.59), 450 (4.80), 530 (sh, 3.55), 572 (sh, 3.80), 625 nm (4.01). UV-Vis (100 eq TFA-CH2Cl2):
λmax (log ε) 315 (4.46), 392 (4.77), 446 (4.91), 549 nm (4.05). UV-Vis (50% TFA-CH2Cl2): λmax
(log ε) 315 (4.15), 407 (5.17), 425 (5.12), 549 (3.79), 597 (3.96), 656 nm (3.27). 1H NMR (500

116
MHz, CDCl3): δ -6.27 (1H, s), -3.12 (2H, br), 1.77-1.83 (12H, 2 overlapping triplets), 3.51 (6H,
s), 3.84 (4H, q, J = 7.6 Hz), 3.95 (4H, q, J = 7.6 Hz), 4.27 (6H, s), 9.51 (2H, s), 10.24 (2H, s). 13C
NMR (125 MHz, CDCl3): δ 11.5, 17.3, 18.4, 19.5, 19.9, 52.6, 94.6, 100.7, 107.2, 130.5, 131.6,
135.5, 136.0, 137.7, 139.4, 145.4, 156.1, 167.6. 1H NMR (500 MHz, 4µl TFA-CDCl3): δ -5.95
(1H, s), -3.14 (1H, v br), -1.97 (2H, s), 1.65 (6H, t, J = 7.6 Hz), 1.80 (6H, t, J = 7.6 Hz), 3.44 (6H,
s), 3.90-4.00 (8H, 2 overlapping quartets), 4.26 (6H, s), 9.74 (2H, s), 10.40 (2H, s). 13C NMR (125
MHz, 4µl TFA-CDCl3): δ 11.7, 16.6, 17.4, 19.78, 19.81, 53.2, 94.2, 111.8, 113.8, 132.7, 134.1,
138.5, 140.2, 140.4, 140.5, 141.5, 142.9, 167.3. 1H NMR (500 MHz, 50% TFA-CDCl3): δ -7.65
(1H, s), 1.82 (12H, t, J = 7.6 Hz), 3.79 (6H, s), 4.22-4.32 (8H, m), 4.69 (6H), 11.10 (2H, s), 11.86
(2H, s). 13C NMR (125 MHz, 50% TFA-CDCl3): δ 11.7, 16.3, 17.4, 20.6, 29.2, 55.5, 105.1, 115.0,
142.7, 144.1, 144.8, 146.1, 147.1, 148.2, 148.2, 148.3, 149.0, 149.3, 167.3. HRMS (ESI): calcd
for C35H39N3O4 + H 566.3019, found 566.3047.

117
trans–8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,8-dimethyl-2,3-dihydro-21-
carbaporphyrin (117)
A third green fraction from the previous procedure was evaporated and the residue recrystallized
from chloroform-methanol to give the carbachlorin (12 mg; 0.02 mmol, 9%) as a green powder,
mp 232-234 oC; UV-Vis (5% DBU-CH2Cl2): λmax (log ε): 398 (4.86), 437 (4.59), 450 (sh, 4.52),
464 (sh, 4.44), 569 (sh, 3.74), 587 (sh, 3.75), 619 (sh, 3.90), 645 (sh, 3.98); UV-Vis (CH2Cl2): λmax
(log ε): 398 (5.31), 490 (4.17), 588 (3.41), 645 (4.23); UV-Vis (5% TFA-CH2Cl2): λmax (log ε):
348 (4.21), 405 (5.24), 422 (5.15), 527 (3.84), 544 (sh, 3.84), 582 (3.88), 635 (3.91); 1H NMR
(500MHz, CDCl3): δ -7.14 (1H, s, 21-H), 1.82 (6H, t, J = 7.7 Hz), 1.86 (6H, t, J = 7.7 Hz), 3.53
(6H, s), 3.92 (6H, s), 3.98 (4H, q, J = 7.7 Hz), 4.06 (4H, q, J = 7.7 Hz), 6.34 (2H, s), 9.48 (2H, s),
9.85 (2H, s). 13C NMR (125 MHz CDCl3): δ 11.5, 17.6, 18.8, 19.8, 20.1, 53.0, 56.0, 96.6, 97.9,
119.7, 130.0, 133.0, 135.7, 138.0, 139.6, 143.4, 150.5, 173.7. 1H NMR (500 MHz, TFA-CDCl3):
δ -6.96 (1H, s), -6.00 (1H, br s), -5.56 (2H, br s), 1.72 (6H, t, J = 7.7 Hz), 1.82 (6H, t, J = 7.7 Hz),
3.53 (6H, s), 4.01 (6H, s), 4.12 (4H, q, J = 7.7 Hz), 4.19 (4H, q, J = 7.7 Hz), 6.34 (2H, s), 9.84
(2H, s), 10.30 (2H, s). 13C NMR (125 MHz, TFA-CDCl3): δ 11.4, 16.6, 17.5, 20.2, 20.3, 54.8, 56.9,
94.9, 103.3, 128.4, 134.8, 135.2, 136.2, 141.7, 142.3, 142.8, 143.3, 176.1. HRMS (ESI): Calcd for
C35H41N3O4 + H 568.3175, found 568.3181.

118
(8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18-dimethyl-21-carbaporphyrinato)silver(III)
(118)
A solution of carbaporphyrin 98 (10.0 mg, 0.017 mmol) in dichloromethane (10 mL) was added
to silver(I) acetate (11.0 mg, 0.066 mmol) dissolved in methanol (2.5 mL), and the mixture was
stirred at room temperature overnight. The mixture was washed with water, and the solvent was
removed under reduced pressure. The residue was chromatographed on grade III neutral alumina
eluting with dichloromethane and hexane in a ratio of 60:40. A deep red fraction was collected and
recrystallized from chloroform-methanol to give the silver(III) complex (7.2 mg, 0.011 mmol,
60%) as a red powder, mp >300oC; UV-Vis (CH2Cl2): λmax (log ε): 309 (4.06), 352 (4.29), 376
(4.29), 407 (4.44), 449 (4.64), 515 (3.63), 567 (4.13); 1H NMR (500 MHz, CDCl3): δ 1.59 (12H,
t, J = 7.7 Hz), 3.31 (6H, s), 3.59 (4H, q, J = 7.7 Hz), 3.64 ( 4H, q, J = 7.7 Hz) 4.40 (6H, s), 8.79
(2H, s), 9.91 (2H, s); 13C NMR (125 MHz, CDCl3): δ 11.5, 17.3, 18.1, 19.4, 20.0, 52.6, 96.1, 110.2,
118.3, 131.1, 134.51, 134.57, 137.2, 138.3, 139.6, 140.9, 167.5. HR MS (EI): calcd for
C35H36AgN3O4 669.1757, found 669.1752.

119
(8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18-dimethyl-21-carbaporphyrinato)gold(III)
(119)
A solution of carbaporphyrin 98 (12 mg, 0.021 mmol) in pyridine (10 mL) was added to gold(III)
acetate (12 mg, 0.032 mmol) dissolved in pyridine (2.0 mL), and the mixture was refluxed for 30
minutes. The mixture was washed with water, and the solvent removed under reduced pressure.
The residue was chromatographed on Grade III neutral alumina eluting with dichloromethane and
hexane in 60:40. A red fraction was collected and recrystallized from chloroform-methanol to give
the gold(III) complex (2.6 mg, 0.004 mmol, 16%) as a red powder, mp 260-262 oC; UV-Vis
(CH2Cl2): λmax (log ε) 348 (4.38), 373 (4.35), 403 (4.41), 441 (4.43), 474 (sh, 3.83), 513 (3.76),
557 (4.41); 1H NMR (500 MHz, CDCl3): δ 1.63-1.68 (12H, t, J = 7.7 Hz), 3.36 (6H, s), 3.69 (4H,
q, J = 7.7 Hz), 3.74 (4H, q, J = 7.7 Hz), 4.36 (6H, s), 9.02 (2H, s), 9.99 (2H, s). 13C NMR (125
MHz, CDCl3): δ 11.5, 17.3, 18.0, 19.4, 19.8, 52.5, 96.3, 111.4, 122.1, 134.07, 134.14, 135.1, 136.2,
137.3, 138.2 140.9, 167.5. HR MS (ESI): calcd for C35H36AuN3O4 760.2450, found 760.2460.

120
8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18,21-trimethyl-21-carbaporphyrin (120)
Carbaporphyrin 98 (12.0 mg, 0.021 mmol) was stirred with potassium carbonate (12 mg) and
methyl iodide (6 drops) in acetone (18 mL) under reflux for 1 h. The mixture was washed with
water and evaporated under reduced pressure. The residue was run through a grade III neutral
alumina column, eluting with dichloromethane and hexane in a ratio of 60:40, and two colored
fractions were collected. The later major red fraction was evaporated under reduced pressure and
the residue recrystallized from chloroform-methanol to yield the C-methylcarbaporphyrin (9.1 mg,
0.016 mmol, 73%) as purple powder, mp 298-300oC; UV-Vis (CH2Cl2): λmax (log ε) 312 (4.17),
428 (4.67), 537 (sh, 3.64), 580 (3.92), 647 nm (sh, 3.33). UV-Vis (1% Et3N-CH2Cl2): λmax (log ε)
408 (4.64), 431 (sh, 4.61), 468 (sh, 4.45), 576 (sh, 3.73), 649 (3.90), 701 nm (sh, 3.37). UV-Vis
(5% DBU-CH2Cl2): λmax (log ε) 405 (4.67), 432 (sh, 4.62), 467 (sh, 4.50), 564 (sh, 3.73), 648
(3.97), 700 nm (sh, 3.38). UV-Vis (5 equiv TFA-CH2Cl2): λmax (log ε) 316 (4.16), 411 (4.76), 556
(sh, 3.65), 581 (3.75), 629 nm (sh, 3.45). UV-Vis (25% TFA-CH2Cl2): λmax (log ε) 320 (3.78), 414
(5.08), 430 (sh, 4.89), 550 (3.46), 606 nm (3.79). UV-Vis (50% TFA-CH2Cl2): λmax (log ε) 316
(3.97), 413 (5.11), 432 (4.92), 529 (sh, 3.50), 549 (sh, 3.48), 598 (3.80), 620 nm (3.80). 1H NMR
(500 MHz, CDCl3): δ -4.68 (3H, s), -3.19 (2H, v br), 1.76-1.80 (12H, 2 overlapping triplets), 3.43

121
(6H, s), 3.71-3.79 (4H, m), 3.86 (4H, q, J = 7.6 Hz), 4.02 (6H, s), 9.28 (2H, s), 9.90 (22H, s). 13C
NMR (125 MHz, CDCl3): δ 10.7, 11.5, 17.2, 18.2, 19.5, 19.8, 52.3, 94.4, 101.6, 109.9, 118.3,
135.0, 135.1, 135.4, 137.8, 139.8, 145.6, 157.2, 167.0. 1H NMR (500 MHz, DBU-CDCl3): δ -4.78
(1H, s), 9.22 (2H, s), 9.83 (2H, s); 1H NMR (500 MHz, 2 equiv. TFA-CDCl3): δ -5.41 (3H, s), -
1.5 (1H, v br), -1.42 (2H, s), 1.69 (6H, t, J = 7.7 Hz), 1.80 (6H, t, J = 7.7 Hz) 3.31 (6H, s), 3.74-
3.88 (6H, m), 3.88-3.96 (2H, m), 3.98 (6H, s), 9.49 (2H, s), 10.03 (2H, s). 13C NMR (125 MHz, 2
equiv. TFA-CDCl3): δ 11.1, 11.6, 16.7, 17.3, 19.66, 19.72, 52.3, 95.0, 113.4, 115.0, 121.8, 135.4,
138.0, 138.8, 139.7, 140.3, 141.3, 142.5, 166.4. 1H NMR (500 MHz, excess TFA-CDCl3): δ -8.67
(1H, q, J = 7.6 Hz), -6.55 (3H, d, J = 7.6 Hz), 1.94 (6H, t, J = 7.6 Hz), 1.97 (6H, t, J = 7.7 Hz),
3.85 (6H, s), 4.30-4.41 (8H, m), 4.66 (6H, s), 11.16 (2H, s), 11.80 (2H, s). 13C NMR (125 MHz,
excess TFA-CDCl3): δ 11.0, 12.0, 16.6, 17.7, 20.7, 20.8, 31.9, 55.5, 105.5, 114.4, 143.3, 144.3,
144.7, 145.9, 146.3, 149.2, 149.7, 150.6, 155.3, 166.9. HRMS (ESI): calcd for C36H41N3O4 + H
580.3175, found 580.3153.

122
8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18,21,22-tetramethyl-21-carbaporphyrin (121)
Carbaporphyrin 98 (12.0 mg, 0.021 mmol) was stirred with potassium carbonate (12 mg) and
excess of methyl iodide (10 drops) in acetone (12 mL) under reflux for 16 h. The mixture was
washed with water and evaporated under reduced pressure. The residue was run through a grade
III neutral alumina column, eluting with dichloromethane and hexane in a ratio of 60:40, and a
brown colored fraction was collected. The solvent was evaporated under reduced pressure and the
residue recrystallized from chloroform-methanol to yield 121 (8.7 mg, 0.015 mmol, 69%) as purple
crystals, mp 222-224 oC; UV-Vis (CH2Cl2): λmax (log ε) 316 (4.41), 376 (4.59), 441 (5.89), 545
(3.93), 589 (4.01), 736 nm (3.30). UV-Vis (CH2Cl2): λmax (log ε) 320 (4.34), 412 (4.77), 429 (sh,
4.73), 567 (3.85), 607 nm (sh, 3.76). 1H NMR (500 MHz, CDCl3): δ -5.27 (3H, s), -3.91 (3H, s),
1.39 (3H, t, J = 7.6 Hz), 1.70 (3H, t, J = 7.7 Hz), 1.73-1.78 (6H, 2 overlapping triplets, J = 7.6 Hz),
2.95 (3H, s), 3.32 (3H, s), 3.37-3.43 (2H, m), 3.55-3.66 (3H, m), 3.68-3.76 (3H, m), 4.02 (3H, s),
4.06 (3H, s), 8.90 (1H, s), 9.05 (1H, s), 9.52 (1H, s), 9.54 (1H, s) (5,20-H). 13C NMR (125 MHz,
CDCl3): δ 9.6, 11.4, 11.7, 16.2, 17.1, 18.0, 18.1, 19.3, 19.57, 19.61, 19.8, 31.7, 52.1, 52.3, 93.7,
98.0, 108.9, 109.5, 112.5, 116.4, 123.3, 128.1, 131.2, 132.5, 134.4, 135.7, 136.1, 137.5, 141.8,
144.3, 145.8, 146.4, 150.6, 154.8, 158.2, 166.5, 167.7. 1H NMR (500 MHz, TFA-CDCl3): δ -4.91

123
(3H, s), -4.18 (1H, br s), -4.02 (1H, br s), -3.54 (3H, s), 1.46 (3H, t, J = 7.7 Hz), 1.72 (3H, t, J =
7.7 Hz), 1-80-1.85 (6H, 2 overlapping triplets, J = 7.7 Hz), 3.03 (3H, s), 3.37 (3H, s), 3.46-3.53
(2H, m), 3.82 (2H, q, J = 7.7 Hz), 3.86-3.97 (4H, m), 4.15 (3H, s), 4.19 (3H, s), 9.45 (1H, s), 9.46
(1H, s), 9.86 (1H, s), 9.92 (1H, s). 13C NMR (125 MHz, 2 µL TFA-CDCl3): δ 10.9, 11.8, 12.2,
15.8, 16.4, 17.17, 17.23, 19.6, 19.7, 19.8, 31.1, 53.0, 53.1, 92.9, 96.3, 113.3, 114.5, 117.4, 123.1,
134.3, 135.5, 136.5, 138.6, 138.9, 139.4, 141.1, 141.88, 141.94, 142.5, 144.6, 145.2, 150.6, 152.6.
HRMS (ESI): calcd for C37H43N3O4 + H 594.3332, found 594.3326.

124
(8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18,21-trimethyl-21-
carbaporphyrinato)palladium(II) (122)
Palladium(II) acetate (10 mg, 0.044 mmol) was added to a solution of C-methylcarbaporphyrin
120 (10.0 mg, 0.017 mmol) in acetonitrile (10 mL), and the solution was stirred under reflux for
30 min. The solution was cooled, diluted with chloroform, washed with water, and the organic
layer separated and then evaporated to dryness. The residue was chromatographed with grade III
neutral alumina, eluting with 30% dichloromethane-hexanes, and the product was collected as a
green band. The solvent was evaporated to dryness and the residue recrystallized from chloroform-
methanol to yield the palladium(II) complex (7.4 mg, 0.011mmol, 63%) as dark green crystals, mp
282-284 oC; UV-Vis (CHCl3): λmax (log ε) 306 (4.37), 410 (4.54), 460 (4.52), 602 (3.90), 639 nm
(sh, 3.81). 1H NMR (500 MHz, CDCl3): δ -4.12 (3H, s), 1.81 (6H, t, J = 7.7 Hz), 1.83 (6H, t, J =
7.7 Hz), 3.44 (6H, s), 3.86-3.92 (8H, 2 overlapping quartets), 4.30 (6H, s), 9.85 (2H, s), 10.81 (2H,
s). 13C NMR (125 MHz CDCl3): δ 11.9, 17.6, 18.4, 19.8, 20.1, 21.8, 36.4, 52.9, 101.7, 113.0,
131.3, 136.9, 143.4, 143.9, 145.5, 145.6, 149.2, 158.6, 167.6. HRMS (ESI): calcd for
C36H39N3O4Pd + H 683.1851, found 683.1955.

125
(8,12,13,17-Tetraethyl-2,3-di(methoxycarbonyl)-7,18,21-trimethyl-21-
carbaporphyrinato)nickel(II) (123)
Nickel(II) acetate tetrahydrate (10 mg, 0.057 mmol) was added to a solution of C-
methylcarbaporphyrin 120 (10.0 mg, 0.017 mmol) in dimethylformamide (10 mL), and the
solution was stirred under reflux for 30 min. The solution was cooled, diluted with chloroform,
washed with water, and the organic layer separated and then evaporated to dryness. The residue
was chromatographed with grade III neutral alumina, eluting with 60% dichloromethane-hexanes,
and the product was collected as a green band. The solvent was evaporated to dryness and the
residue recrystallized from chloroform-methanol to yield the nickel(II) complex (7.6 mg, 0.012
mmol, 71%) as dark green crystals, mp 267-269 oC; UV-Vis (CHCl3): λmax (log ε) 348 (4.51), 420
(4.72), 456 (sh, 4.38), 612 nm (3.96). 1H NMR (500 MHz, CDCl3): δ -3.89 (3H, s), 1.72 (6H, t, J
= 7.7 Hz), 1.77 (6H, t, J = 7.7 Hz), 3.31 (6H, s), 3.72-3.81 (8H, m), 4.26 (6H, s), 9.59 (2H, s),
10.56 (2H, s). 13C NMR (125 MHz, CDCl3): δ 11.8, 17.5, 18.2, 19.7, 19.8, 19.9, 26.7, 52.8, 99.8,
111.0, 131.3, 138.3, 144.3, 145.9, 146.1, 148.7, 150.9, 167.6. HRMS (ESI): calcd for
C36H39N3O4Ni + H 635.2294, found 635.2285.

126
CHAPTER III
SYNTHESIS OF A CARBACHLORIN
Introduction
Chlorins (2,3-dihydroporphyrins) are an important group of tetrapyrroles that provide the
parent structure for many of the chlorophylls and other biological pigments present in nature.2
Chlorins tend to have a green coloration in solution because of their strong absorptions near 650
nm in the long wavelength region of visible spectrum. It is this absorption which is in part
responsible for their potential application as photosensitizers in photodynamic therapy,29 and this
property has led to numerous investigations into their synthesis.53 However, there have been very
few studies on the preparation of analogous systems, such as carbachlorins, that may have equally
useful properties.14 It was in 1998 that the first synthesis of carbachlorins was carried out by Hayes
and Lash. For this synthesis, a “3 + 1” variant on the MacDonald condensation was used.15 Bicyclic
dialdehydes were found to condense with a tripyrrane in the presence of TFA to give, following
oxidation with DDQ, carbachlorins 130 in moderate yields. These chlorin analogues were found
to retain the aromatic properties as the proton NMR spectra showed the inner CH protons upfield
around -7 ppm.35 The UV-vis spectra were also very porphyrin-like, showing a Soret band at 404
nm and three smaller absorptions between 490 and 650 nm. The band at 650 nm was of medium
intensity and resembles the characteristic absorptions of tetrapyrrolic chlorins.15

127
In 2014, Li and Lash, in their attempts to further explore carbachlorin chemistry,
synthesized a carbachlorin 131 using 3-ethoxymethylenecyclopentene-1-carbaldehyde. This was
condensed with a tripyrrane, and following oxidation with aqueous ferric chloride solutions,
generated moderate yields of a carbachlorin 131. Also, oxidation of the carbachlorin 131 with
DDQ gave the first example of a carbaporphyrin 132 that was unsubstituted on the carbocyclic
ring.55
However, compared to studies on other core modified porphyrins, for instance
carbaporphyrins, our knowledge of carbachlorins is still very limited. Questions such as whether
these core-modified chlorins (carbachlorins) share similar properties with core-modified

128
porphyrins (carbaporphyrins) is still open. Therefore, to further investigate the chemistry of
carbachlorins, it was necessary to develop an improved synthesis of a carbachlorin.
In the previous chapter, the synthesis of a carbaporphyrin with two methyl ester groups on
adjacent carbons was performed. It was interesting to note that carbachlorin 117 was also formed
as the minor fraction in this synthesis. However, this compound was prone to oxidation and
carbaporphyrin was always the major product in these reactions. In the case of carbachlorins 130,
the presence of a fused five-membered ring appeared to inhibit further oxidation. We speculated
that carbachlorins could be generated as the major products if the site of oxidation was blocked by
geminal substitution. Carbachlorin 133 was an appealing target molecule, where the methyl esters
were placed on the same carbon rather than at vicinal positions. It was anticipated that the synthesis
and study of carbachlorin 133 would increase our understanding of the chlorin system and could
allow the formation of new porphyrinoids and their metal complexes.

129
Results and Discussion
A retrosynthetic analysis suggested that carbachlorin 133 could be obtained from
cyclopentane dialdehyde 134 and the known tripyrrane 62. A straightforward acid catalyzed ‘3+1’
condensation of dialdehyde 134 with tripyrrane 62 in the presence of trifluoroacetic acid, followed
by oxidation with DDQ, would afford the targeted carbachlorin (Scheme 66).
Scheme 66 Retrosynthetic Analysis of Carbachlorin 133
In order to accomplish this goal, a synthesis of dialdehyde 134 had to be developed. This
was accomplished using a similar strategy to the one described in Chapter 2. Initially, norbornene
diester 135 was required and this could be generated using the Diels-Alder reaction. This required
dienophile 136, which was prepared by adapting a literature procedure reported by Harada and
coworkers.63 Copper(II) acetate was added to a solution of glacial acetic acid, paraformaldehyde
and dimethyl malonate. After stirring at 100 °C for 3 h, the reaction mixture was cooled to room
temperature and the excess acetic acid was removed under reduced pressure. The residue was later
distilled under reduced pressure to afford the colorless distillate 136 (Scheme 67). This distillate
was immediately diluted with CH2Cl2 and used for the following Diels-Alder reaction.57

130
Scheme 67 Formation of 136
In order to form the bicyclic adduct 135, freshly cracked 1,3-cyclopentadiene was added
dropwise to the solution of dimethyl 2-methylenemalonate 136 dissolved in CH2Cl2 whilst stirring.
The temperature of the reaction was controlled with a water bath. The reaction mixture was stirred
at room temperature for almost 60 min and purified by column chromatography on silica gel using
hexane/EtOAc (19:1, v/v) as the eluent to give bicyclic adduct 135 as a colorless oil (Scheme 68).57
Scheme 68 Formation of 135
Once the norbornene was generated, the next step was to oxidize the compound to dialcohol
137. Several different procedures were attempted to carry out the transformation, and it was
observed that the best results were obtained using KMnO4 and NaOH for oxidation. For this
reaction, a solution of potassium permanganate and sodium hydroxide in water was added
dropwise to a solution of norbornene in tert-butyl alcohol and water while maintaining the

131
temperature at 0° C. After the addition was completed, the reaction mixture was stirred for 40
minutes and quenched with a saturated aqueous solution of sodium metabisulfite until the solution
turned colorless. The mixture was filtered and the tert-butyl alcohol was removed from the filtrate
in vacuo. The remaining solution was extracted with ethyl acetate which was filtered and
concentrated in vacuo to yield dialcohol 137 as white crystalline solid (Scheme 69).59 Compound
137 had not been prepared previously and was synthesized for the first time.
Scheme 69 Formation of Dialdehyde 134
Diol 137 could be oxidatively ring opened using potassium periodate as an oxidizing agent
to give dialdehyde 134. Dialdehyde 134 was immediately condensed with tripyrrane dicarboxylic
acid 62 in the presence of TFA/CH2Cl2 and the reaction mixture stirred overnight under the
atmosphere of nitrogen. After stirring overnight, oxidation with DDQ in refluxing toluene was
performed and carbachlorin 133 was isolated as the major product (Scheme 70).35

132
Scheme 70 Preparation of Carbachlorin 133
Carbachlorin 133, like other chlorin analogues, proved to be aromatic in the free base form.
The proton NMR spectrum for 133 (Figure 41) in CDCl3 showed the presence of a strong
diamagnetic ring current. The internal CH proton was found at -7.08 ppm, while the meso-protons
were shifted downfield to 9.86, 9.84, 9.58 and 9.25 ppm. As there were four signals for the four
meso-protons, the molecule must be asymmetrical. The UV-Vis spectrum of carbachlorin 133,
shows a strong Soret band at 398 nm along with small Q bands at 491, 588 and 646 nm. The
slightly stronger absorption at 646 nm is characteristic of chlorin-type structures (Figure 42).

133
Figure 41 500 MHz Proton NMR Spectrum of Carbachlorin 133 in CDCl3
s
-7-6-5-4-3-2-110 9 8 7 6 5 4 3 2 1 0 ppm
9.49.69.8 ppm
-7.1 -7.2 ppm
3.83.94.0 ppm

134
Figure 42 UV-Vis Spectrum of Carbachlorin 133
When carbachlorin 133 was exposed to a trace amount of TFA, a deep purple solution
corresponding to monocation 133H+ (Scheme 71) was generated. The proton NMR spectrum for
133H+ (Figure 43) showed that the addition of acid to the carbachlorin system had slightly
increased its aromatic nature, as the internal CH was shifted upfield to -7.10 ppm, while the meso-
protons were shifted further downfield to 10.23, 10.22, 9.96 and 9.71 ppm (Figure 43). The
appearance of broad singlets at -4.95 ppm (2H) and -5.96 ppm (1H) confirmed the presence of an
additional NH due to protonation. Unlike the related carbaporphyrins, further addition of TFA to
0
5
10
15
20
25
300 350 400 450 500 550 600 650 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

135
133H+ did not give a diprotonated species, as aromatic character would be disrupted if protonation
occurred at the inner carbon.
Scheme 71 Protonation of 133
-7-6-5-4-3-2-112 11 10 9 8 7 6 5 4 3 2 1 0 ppm
-5.8 -6.0 -6.2 -6.4 -6.6 -6.8 -7.0 ppm
Figure 43 500 MHz Proton NMR Spectrum of Carbachlorin 133H+ in CDCl3

136
In order to further understand the reactivity of carbachlorin 133, a few more experiments
were performed to observe how the exchange of protons takes place with addition of acid to 133.
As expected, when d-TFA was added to a solution of 133 in CDCl3 the resulting monocation did
not show any exchange of the inner CH proton, in contrast to carbaporphyrins which exhibit rapid
deuterium exchange. However, exchange of the meso-protons was evident. After 20 minutes, the
intensities for two of the meso-resonances were reduced by 15%, while the remaining two peaks
were reduced by more than 50%. After several hours, proton-deuterium exchange was virtually
100% for all four meso-positions (Figure 44). The results imply that meso-substituted monocations
such as 133a-dH+ are in equilibria with 133 and 133H+ (Scheme 72). It is also possible that related
dications could also be involved. It is noteworthy that the exchange rate for chlorin 133 is far
greater than the exchange rates noted for carbaporphyrins such as 98.

137
Scheme 72 Proposed Equilibrium of Different Cationic Species for Carbachlorin 133

138
Figure 44 Proton NMR spectra of Carbachlorin 133 in CDCl3 with Two Drops of d-TFA after
20 min (upper spectrum) and 7 hours (lower spectrum).
Metalation of carbachlorin 133 with silver acetate was also briefly investigated. A solution
of carbachlorin 133 in CH2Cl2 was added to a suspension of silver(I) acetate in methanol, and the
resulting mixture was stirred overnight at room temperature. After purification by column
chromatography, a red fraction was collected and recrystallized from chloroform and methanol to
yield organometallic complex 138 as a dark red powder in 62% yield (Scheme 73). Silver complex

139
138 retains aromatic characteristics as indicated by proton NMR spectrum (Figure 45). The UV-
Vis spectrum of 138 (Figure 46) has a strong absorptions at 386 and 410 nm in the Soret region,
along with three absorptions at 490, 522 and 546 nm. Moreover, the stronger band at 592 nm is
reminiscent of chlorin systems.
Scheme 73 Formation of Silver(III) Chlorin 138

140
-8-7-6-5-4-3-2-113 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
9.39.49.59.69.79.89.910.010.1 ppm
Figure 45 500 MHz Proton NMR spectrum of 138 in CDCl3

141
Figure 46 UV-Vis Spectrum of Silver(III) Chlorin 138 in Dichloromethane
Conclusions
A cyclopentane dialdehyde was prepared in three steps, starting with the formation of
dimethyl 2-methylenemalonate 135 from paraformaldehyde and dimethyl malonate. This
dienophile then underwent Diels-Alder chemistry with cyclopentadiene to generate the bicyclic
compound 136. Norbornene 136 was then oxidized in two steps to generate dialdehyde 134.
Although difficulties were initially encountered in preparing dialdehyde precursor 134, careful
adjustments of the reaction conditions allowed the five-memebered ring dialdehyde to be isolated
in good yields. The ‘3+1’ condensation of dialdehyde 134 and tripyrrane dicarboxylic acid 62 led
to the formation of carbachlorin 133 in 30% yield. This newly synthesized carbachlorin system
0
2
4
6
8
10
12
14
16
300 350 400 450 500 550 600 650 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

142
proved to be highly aromatic. Protonation of this system can readily occur, forming an aromatic
monocation.
Reaction between carbachlorin 133 and silver(I) acetate gave a silver(III) organometallic
complex. The straightforward synthesis of carbachlorin 133 will allow further studies on this
system to be conducted. It is anticipated that further coordination chemistry could be performed
on this system as the yields are quite good.
Experimental
All chemicals were purchased from Sigma Aldrich or Acros Organic. Grade III neutral alumina
and silica gel were used to perform column chromatography. The 1H and 13C-NMR spectra were
obtained either using Bruker Avance III 500 MHz NMR spectrometer or Bukner 400 MHz NMR
spectrometer at 302K. Chemical shifts were recorded, relative to CDCl3 (residual chloroform at δ
7.26 ppm) in proton NMR spectra and the CDCl3 triplet at δ 77.23 ppm in 13C-NMR spectra, in
parts per million (ppm). The UV-vis spectra for the compounds were collected by Cary 100 Bio
spectrophotometer. Melting point were gathered using a Mel-Temp apparatus. Mass spectrometry
data was obtained from the Mass Spectral Laboratory, School of Chemical Sciences, University
of Illinois, Urbana-Champaign.

143
Dimethyl 2-methylenemalonate (136)63
Paraformaldehyde (20 g, 133 mmol), dimethyl malonate (50 mL, 87.5 mmol), and
copper(II) acetate (3.25g, 1.64 mmol) were added to glacial acetic acid (100 ml). The reaction
mixture was stirred at 100 °C for 3 h, then cooled to room temperature, and the acetic acid removed
under reduced pressure. The residue was distilled under reduced pressure using a vacuum pump
and a colorless fraction was obtained between 90 oC to 97 oC (the external temperature was held
between 152 oC to 160 oC) corresponding to 137 (3.6 g, 0.025mmol, 28%). This product was
immediately diluted with CH2Cl2 and the solution used for the following Diels-Alder reaction. 1H
NMR (500 MHz, CDCl3): δ 6.60 (2H, s), 4.02 (6H, s).

144
5,5-Dimethoxycarbonyl-2-norbornene (135)57
A solution of dimethyl 2-methylenemalonate 137 in dichloromethane (1.37 g, 0.021mmol) was
added slowly to freshly cracked 1,3-cyclopentadiene (3.00 g, 0.021 mol) whilst stirring and
maintaining temperature at 25oC with the aid of water bath. The reaction mixture was stirred at room
temperature for 60 min and purified by chromatography on silica gel using hexane/EtOAc (19:1) to
give the Diels-Alder adduct as a colorless oil (2.97 g, 0.0141 mmol, 80%). 1H NMR (500MHz,
CDCl3): δ 1.48-1.52 (1H, m), 1.65 (1H, br d, J = 8.9 Hz), 1.99 (1H, dd, J = 12.4, 2.9 Hz), 2.10
(1H, dd, J = 12.4, 3.6 Hz), 2.90 (1H, br), 3.37-3.39 (1H, br m), 3.65 (3H, s), 3.72 (3H, s), 5.97
(1H, dd, J = 5.6, 2.9 Hz), 6.24 (1H, dd, J = 5.6, 3.1 Hz). 13C NMR (125 MHz, CDCl3): δ 36.1,
42.2, 48.9, 50.0, 52.5, 52.9, 60.4, 133.7, 140.0, 171.6, 173.2. HRMS (EI): calcd for C11H14O4
210.0892, found 210.0896.

145
Dimethyl 5,6-dihydroxybicyclo[2.2.1]heptane-2,2-dicarboxlate (137)
To a solution of norbornene 137 (10.80 g, 53.1 mmol) in tert-butyl alcohol (200 mL) and water
(50 mL) at 0 °C was added dropwise a solution of potassium permanganate (12.0 g, 76.0 mmol)
and sodium hydroxide (2.61 g, 65.3 mmol) in water (250 mL). After the addition was complete,
the reaction mixture was stirred for 20 minutes and then quenched using a saturated aqueous
solution of sodium metabisulphite (Na2S2O5) until the solution turned colorless. The mixture was
filtered and the tert-butyl alcohol was removed from the filtrate in vacuo. The solution was
extracted with ethyl acetate (4 × 100 mL) and the combined organic layers dried over MgSO4,
filtered and concentrated in vacuo to furnish the diol 138 (6.23 g, 0.025 mmol, 61%) as a white
crystalline solid, mp 86-88 oC. 1H NMR (500 MHz, CDCl3): δ 1.43 (1H, dq, J = 11.0, 1.6 Hz),
1.88 (1H, dq, J = 11.0, 1.6 Hz), 2.49-2.50 (1H, m), 2.54-2.56 (1H, m), 2.72 (1H, dd, J = 5.6, 1.5
Hz), 2.86 (1H, br), 2.96 (1H, br), 3.20 (1H, dd, J = 5.6, 4.7 Hz), 3.69 (3H, s), 3.71 (3H, s), 3.75-
3.77 (1H, m), 3.85-3.87 (1H, m). 13C NMR (125 MHz, CDCl3): δ 31.9, 45.0, 46.4, 46.7, 48.5,
52.4, 52.5, 70.5, 73.6, 173.2, 174.3. HRMS (EI): calcd for C11H16O6 + H 245.1025, found
245.1017.

146
Dimethyl 3,5-diformylcyclopentane-1,1-dicarboxylate (134)
The foregoing diol 138 (3.05 g, 0.021 mole) was mixed with water (5.0 ml), ether (10 ml) and
potassium periodate (4.16 g, 0.018 mol) and the reaction stirred at room temperature for 1 hr. The
excess potassium periodate was filtered off and the two phases were separated. The water layer
was back extracted twice with ether. The ether layers were combined, dried over sodium sulfate
and the ether removed under reduced pressure to form the dialdehyde (1.67 g, 0.021 mmol, 60%)
as a clear oil. The unstable dialdehyde was not further purified but used immediately to prepare
carbachlorin. 1H NMR (500 MHz, CDCl3): 2.17-2.25 (1H, m), 2.47 (1H, dt, J = 13.8, 5.9 Hz),
3.05-3.11 (1H, m), 3.27-3.32 (1H, m), 3.57-3.66 (2H, m), 3.70 (3H, s), 3.76 (3H, s), 9.65 (1H, d,
J = 0.8 Hz), 9.68 (1H, d, J = 0.9 Hz). HRMS (EI): calcd for C11H14O6 + H 243.0869, found
243.0870.

147
8,12,13,17-Tetraethyl-2,3-dimethoxycarbonyl-7,18-dimethyl-21-carbaporphyrin (133)
Tripyrrane dicarboxylic acid 62 (100 mg, 0.220 mmol) was stirred with TFA (1 ml) under an
atmosphere of nitrogen for 2 minutes in a 250 ml round bottom flask. Dichloromethane (99 ml)
was added, followed immediately by dialdehyde 140 (55 mg, 0.23 mmol) and the mixture was
stirred under nitrogen overnight. The mixture was neutralized by the addition of triethylamine and
the solvent removed under reduced pressure. The residue was taken up in toluene (50 ml), DDQ
(100 mg, 0.44 mmol) was added, and the resulting mixture was stirred under reflux for 1 hr. The
solvent was removed under reduced pressure, then the residue was dissolved in CH2Cl2 and washed
with water. The solution was concentrated under reduced pressure and the residue
chromatographed on grade III neutral alumina column eluting with CH2Cl2 and hexane in a ratio
of 60:40.The product was collected as a dark green fraction. Recrystallization from chloroform
and methanol gave the carbachlorin (37 mg, 0.065 mmol, 30%) as purple crystals, mp 228-230 oC.
UV-Vis (1% Et3N-CH2Cl2): λmax (log ε) 398 (5.36), 491 (4.23), 588 (3.45), 646 nm (4.31). UV-
Vis (5% TFA-CH2Cl2): λmax (log ε) 406 (5.30), 423 (5.19), 527 (3.86), 543 (3.86), 583 (3.93), 635
nm (3.99). 1H NMR (500 MHz, CDCl3): δ -7.08 (1H, s), 1.80-1.84 (6H, 2 overlapping triplets, J =
7.7 Hz), 1.85-1.88 (6H, 2 overlapping triplets, J = 7.7 Hz), 3.50 (3H, s), 3.54 (3H, s), 3.90 (6H, s),

148
3.96-4.01 (4H, 2 overlapping quartets), 4.04-4.09 (4H, 2 overlapping quartets), 5.43 (2H, s), 9.25
(1H, s), 9.58 (1H, s), 9.84 (1H, s), 9.86 (1H, s). 13C NMR (125 MHz, CDCl3): δ 11.4, 11.6, 17.6,
18.8, 19.8, 20.2, 45.3, 53.5, 96.2, 96.5, 97.7, 99.3, 120.7, 129.7, 130.2, 132.95, 133.05, 135.5,
135.8, 137.8, 137.9, 139.8, 140.6, 143.3, 143.4, 150.5, 172.2. 1H NMR (500 MHz, 5% TFA-
CDCl3): δ -7.10 (1H, s), -5.96 (1H, br s), -4.95 (2H, br s), 1.67-1.72 (6H, 2 overlapping triplets),
1.83-1.87 (2 overlapping triplets), 3.49 (3H, s), 3.51 (3H, s), 3.98 (6H, s), 4.09 (4H, q, J = 7.7 Hz),
4.16 (4H, q, J = 7.6 Hz), 9.71 (1H, s), 9.96 (1H, s), 10.226 (1H, s), 10.231 (1H, s). 13C NMR (125
MHz, 5% TFA-CDCl3): δ 11.44, 11.47, 16.6, 17.6, 20.1, 20.2, 45.7, 55.1, 68.4, 94.6, 94.7, 103.7,
103.9, 128.6, 134.5, 134.8, 134.9, 135.1, 135.6, 135.9, 141.4, 142.0, 142.1, 142.7, 142.9, 143.0,
145.0, 173.7. HRMS (ESI): calcd for C35H41N3O4 + H 568.3175, found 568.3177.

149
(8,12,13,17-Tetraethyl-2,2-dimethoxycarbonyl-7,18-dimethyl-21-carbaporphyrinato)silver(III)
(138)
A solution of carbachlorin 133 (10.0 mg, 0.017 mmol) in dichloromethane (10 mL) was added to
silver(I) acetate (12 mg, 0.019 mmol) dissolved in methanol (2.5 mL), and the mixture was stirred
at room temperature overnight. The mixture was washed with water, and the solvent was removed
under reduced pressure. The residue was chromatographed on grade III neutral alumina eluting
with dichloromethane and hexane in a ratio of 60:40. A deep red fraction was collected and
recrystallized from chloroform-methanol to give the silver(III) complex (7.3 mg, 0.011mmol,
62%) as a red powder, mp 266-268 oC. UV-Vis (CH2Cl2): λmax (log ε) 386 (sh, 4.49), 410 (5.15),
490 (3.96), 522 (3.82), 546 (3.58), 592 nm (4.43). 1H NMR (500 MHz, CDCl3): 1.81 (3H, t, J =
7.7 Hz), 1.83 (3H, t, J = 7.7 Hz), 1.88 (6H, t, J = 7.7 Hz), 3.47 (3H, s), 3.54 (3H, s), 3.93 (6H, s),
3.98-4.09 (8H, m), 9.36 (1H, s), 9.70 (1H, s), 9.93 (1H, s), 9.98 (1H, s) (10,15-H). 13C NMR (125
MHz CDCl3): δ 11.8, 12.0, 17.82, 17.83, 18.58, 18.59, 19.9, 20.48, 20.52, 45.3, 53.5, 67.5, 98.2,
98.5, 101.3, 103.0, 130.8, 133.4, 133.5, 134.2, 134.4, 136.0, 136.5, 138.7, 138.8, 140.3, 140.6,
172.4. HRMS (ESI): calcd for C35H38AgN3O4 + H 672.1992, found 672.2007.

150
CHAPTER IV
SUMMARY AND CONCLUSIONS
Carbaporphyrins and carbachlorins are interesting molecules showing strong diatropic
character and unusal coordination chemistry. In order to expand this important area, new routes to
carbaporphyrins and carbachlorins have been developed. In Chapter II, it was shown that a
carbaporphyrin could be synthesized from a cyclopentane dialdehyde in good yields and this
porphyrin analogue proved to be a versatile precursor for alkylation and coordination chemistry.
Initially, a Diels-Alder adduct was prepared from cyclopentadiene and dimethyl fumarate. This
was converted in two steps to the cyclopentane dialdehyde, and subsequent condensation with a
tripyrrane in the presence of TFA, followed by oxidation with DDQ in refluxing toluene, gave the
novel carbaporphyrin in 40% yield together with a low yield of a related carbachlorin. The
carbaporphyrin possesses macrocyclic aromaticity and afforded stable silver(III) and gold(III)
complexes. In addition, alkylation with methyl iodide and potassium carbonate gave a C-methyl
derivative and this underwent metalation with palladium(II) acetate and nickel(II) acetate to give
the related organometallic species. These reactions involved both the formation of an
organometallic species and an increase in aromaticity. On protonation, the new carbaporphyrin
also exhibited enhanced diatropic character. Mono- and diprotonated species were characterized,
and deuterium exchange studies also indicated that meso-protonated structures were present in
equilibrium. In addition to characterization by UV-Vis, proton NMR and carbon-13 NMR
spectroscopy, and mass spectrometry, the structures of the free base carbaporphyrin and two
metalated derivatives were confirmed by X-ray crystallography.
The ‘3+1’ variant of the MacDonald condensation has been widely used to prepare

151
porphyrin analogues but only one example of using aliphatic dialdehydes has previously been
reported for this synthetic strategy. Even in that case, only poor yields of porphyrinoid products
were obtained. In this work, aliphatic cyclopentane dialdehydes bearing two ester substituents
were shown to be superior precursors to carbaporphyrinoids. The efficient synthesis of a
carbaporphyrin with ester substituents lays the foundation for further investigation.
Future work entailing this carbaporphyrin could include reductions to generate dicarbinols
that could be converted into precursors for cycloaddition reactions. This strategy would provide
access to further modified structures.
In Chapter III, it was demonstrated that an equally efficient synthesis of a carbachlorin
could be facilitated using a cyclopentane dialdehyde with geminal diester substituents. The
required dialdehyde was obtained from the Diels-Alder adduct of dimethyl methylenemalonate
and cyclopentadiene. Condensation of the dialdehyde with a tripyrrane in the presence of
trifluoroacetic acid, followed by oxidation with DDQ, produced a novel carbachlorin diester in
30% yield. This macrocycle was found to react with silver(III) acetate forming a silver(III)
complex in good yields. The carbachlorin and its silver complex both exhibited strongly aromatic
characteristics. Addition of acid to the carbachlorin resulted in the formation of a monocation with
slightly enhanced diatropic character.
The methodology reported in the thesis provides the most convenient route available for
preparing carbaporphyrins and carbachlorins. The availability of the new porphyrin analogues will
allow further investigations into the reactivity of these systems. Furthermore, the presence of
absorption bands near 650 nm for carbachlorins may allow this system to be utilized as a
photosenitizers in photodynamic therapy.

152
REFERENCES
1. Porphyrins and Metalloporphyrins; Smith, K. M., ed.; Elsevier, 1975.
2. Milgrom, L. R. The Colours of Life: An Introduction to the Chemistry of Porphyrins and
Related Compounds; Oxford University Press, 1997.
3. Kekulé, A. Bull de la Societe Chimique de Paris, 1865, 3, 98–110.
4. Loudon, G. M. Organic Chemistry; Roberts and Company, 2009.
5. Hofmann, A. W. Proc. R. Soc. Lond. 1856, 8, 1–3.
6. Sondheimer, F.; Wolovsky, R.; Amiel, Y. J Am. Chem. Soc. 1962, 84, 274–284.
7. Lash, T. D. in The Porphyrin Handbook: Heteroporphyrins, Expanded Porphyrins and
Related Macrocycles; Kadish, K. M.; Smith, K. M.; Guilard, R. Academic Press, 2000;
Vol. 2, pp 125–199.
8. Soret, J. L. Compt. Rend. 1883, 97, 1267.
9. Kuster, W.; Deihle, P. Z. Physiol. Chem. 1912, 82.
10. Fischer, H.; Zeile, K. Justus Liebigs Ann. Der Chem. 1929, 468, 98–116.
11. Smith, K. M. in The Porphyrin Handbook: Synthesis and Organic Chemistry; Kadish, K.
M.; Smith, K. M.; Guilard, R Academic Press, 2000; Vol. 1, pp 2–43.
12. Rothemund, P. J. Am. Chem. Soc. 1936, 58, 625–627.
13. Adler, A. D.; Longo, F. R.; Finarelli, J. D.; Goldmacher, J.; Assour, J.; Korsakoff, L. J.
Org. Chem. 1967, 32, 476–477.
14. Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org.
Chem. 1987, 52, 827–836.
15. Arsenault, G. P.; Bullock, E.; MacDonald, S. F. J. Am. Chem. Soc. 1960, 82, 4384–4389.
16. Broadhurst, M. J.; Grigg, R.; Johnson, A. W. J. Chem. Soc. 1971, 3681–3690.
17. Cavaleiro, J. A. S.; Gonsalves, A. M. d’A R.; Kenner, G. W.; Smith, K. M. J. Chem. Soc
Perkin Trans. 1973, 1, 2471.
18. Sessler, J. L.; Johnson, M. R.; Lynch, V. J. Org. Chem. 1987, 52, 4394–4397.

153
19. Lash, T. D.; Toney, A. M.; Castans, K. M.; Ferrence, G. M. J. Org. Chem. 2013, 78,
9143–9152.
20. Graham, S. R.; Colby, D. A.; Lash, T. D. Angew. Chem. Int. Ed. 2002, 41, 1371–1374.
21. Lash, T. D.; Chaney, S. T.; Richter, D. T. J. Org. Chem. 1998, 63, 9076–9088.
22. Sessler, J. L.; Cyr, M.; Burrell, A, K. Tetrahedron, 1992, 48, 9661–9672.
23. Lash, T. D. Eur. J. Org. Chem. 2007, 33, 5461–5481.
24. Muckey, M. A.; Szczepura, L. F.; Ferrence, G. M.; Lash, T. D. Inorg. Chem. 2002, 41,
4840 – 4842.
25. Xue, Z.-L.; Shen, Z.; Mack, J.; Kuzuhara, D.; Yamada, H.; Okujima, T.; Ono, N.;
You, X.-Z.; Kobayashi, N. J. Am. Chem. Soc. 2008, 130, 16478–16479.
26. Broadhurst, M. J.; Grigg, R.; Johnson, A. W. J. Chem. Soc. Perkin Trans. 1 1972, 2111.
27. Sessler, J. L.; Murai, T.; Lynch, V.; Cyr, M. J. Am. Chem. Soc. 1988, 110, 5586–5588.
28. Amao, T.; Okura, I. J. Porphyrins Phthalocyanines 2009, 13, 1111–1122.
29. Malinski, T. in The Porphyrin Handbook: Applications Past, Present, and Future;
Kadish, K. M, Smith, K. M.; Guilard, R. Academic Press, 2000; Vol.6, pp 232–255.
30. Collman, J. P.; Brauman, J. I.; Meunier, B.; Raybuck, S. A.; Kodadek, T. Proc Natl. Acad
Sci. 1984, 81, 3245–3248.
31. Anding, B. J.; Ellern, A.; Woo, L. K. Organometallics, 2012, 31, 3628–3635.
32. Kostas, I. D.; Coutsolelos, A G; Charalambidis, G; Skondra, A. Tetrahedron Lett. 2007,
48, 6688–6691.
33. Rambo, B. M.; Sessler, J. L. Chem. –Eur. J. 2011, 17, 4946–4959.
34. Groves, J. T.; Myers, R. S. J. Am. Chem. Soc. 1983, 105, 5791–5796.
35. Lash, T. D. Chem. Rev. 2017, 117, 2313–2446.
36. Toganoh, M.; Furuta, H. Chem. Commun. 2012, 48, 937−954.
37. Bergman, K. M.; Ferrence, G. M.; Lash, T. D. J. Org. Chem. 2004, 69, 7888−7897.
38. Lash, T. D. Acc. Chem. Res. 2016, 49, 471−482.

154
39. Lash, T. D. Org. Biomol. Chem. 2015, 13, 7846−7878.
40. Lash, T. D.; Hayes, M. J. Angew. Chem., Int. Ed. Engl. 1997, 36, 840−842.
41. AbuSalim, D. I.; Lash, T. D. J. Org. Chem. 2013, 78, 11535−11548.
42. Colby, D. A.; Lash, T. D. Chem. - Eur. J. 2002, 8, 5397−5402.
43. Lash, T. D.; Colby, D. A.; Ferrence, G. M. Eur. J. Org. Chem. 2003, 2003, 4533−4548.
44. Lash, T. D.; Hayes, M. J. Angew. Chem., Int. Ed. Engl. 1997, 36, 840−842.
45. Lash, T. D.; Hayes, M. J.; Spence, J. D.; Muckey, M. A.; Ferrence, G. M.; Szczepura, L.
F. J. Org. Chem. 2002, 67, 4860−4874.
46. Muckey, M. A.; Szczepura, L. F.; Ferrence, G. M.; Lash, T. D. Inorg. Chem. 2002, 41,
4840−4842.
47. Lash, T. D.; Colby, D. A.; Szczepura, L. F. Inorg. Chem. 2004, 43, 5258−5267.
48. Adiraju, V. A. K.; Ferrence, G. M.; Lash, T. D. Dalton Trans. 2016, 45, 13691−13694.
49. Lash, T. D. Org. Lett. 2011, 13, 4632−4635.
50. Hayes, M. J.; Spence, J. D.; Lash, T. D. Chem. Commun. 1998, 34, 2409−2410.
51. Lash, T. D.; Muckey, M. A.; Hayes, M. J.; Liu, D.; Spence, J. D.; Ferrence, G. M. J. Org.
Chem. 2003, 68, 8558−8570.
52. Lash, T. D.; Romanic, J. L.; Hayes, M. J.; Spence, J. D. Chem. Commun. 1999, 35,
819−820.
53. Taniguchi, M.; Lindsey, J. S. Chem. Rev. 2017, 117, 344–535.
54. Hayes, M. J.; Lash, T. D. Chem. - Eur. J. 1998, 4, 508−511.
55. Daming, Li.; Lash, T. D. J. Org. Chem. 2014, 79, 7112–7121.
56. Barton, D. H. R.; Zard, S. Z. J. Chem. Soc. 1985, 16, 1098.

155
57. Atkins, A. J.; Fairlamb, I. J. S.; Ward, J. S.; Lynam, J. M. Organometallics 2012, 31,
5896–5902.
58. Göksu, S.; Altundas, R.; Sütbeyaz, Y. Syn. Comm. 2000, 9, 1615–1621.
59. Donohoe, T.; Jahanshahi, A.; Tucker, M. J.; Bhatti, F. L.; Roslan, I. A.; Kabeshov, M.;
Wrigley, G. Chem. Commun. 2011, 47, 5849–5851.
60. Henderson, L. C.; Li, J.; Nation, R. L.; Velkov, T.; Pfeffer, F. M.; Chem. Commun. 2010,
46, 3197–3199.
61. Hickey, S. M.; Ashton, T. D.; White, J. M.; Li, J.; Nation. R. L.; Yu, H. Y.; Elliott, A. G.;
Butler, M. S.; Huang, J. X.; Cooper, M. A.; Pfeffer, F. M. RSC Adv. 2015, 5, 28582–
28596.
62. Beare, K. D.; Yuen, A. K. L.; Masters, A. F.; Maschmeyer, T.; McErlean, C. S. P. Chem.
Commun. 2013, 49, 8347–8349.
63. Harada, M.; Asaba, K. N.; Iwai, M.; Kogure, N.; Kitajima, M.; Org. Lett., 2012, 14,
5800l.

156
APPENDIX A
SELECTED NMR SPECTRA
Figure A – 1: 400 MHz 1H NMR Spectrum of 114 in CDCl3

157
Figure A – 2: 100 MHz carbon-13 NMR spectrum of 114 in CDCl3.

158
12345678910 ppm
1.4
88
1.4
92
1.4
95
1.5
10
1.5
14
1.5
17
1.6
34
1.6
56
1.9
76
1.9
83
2.0
07
2.0
14
2.0
85
2.0
94
2.1
16
2.1
25
2.9
01
3.3
85
3.6
51
3.7
19
5.9
57
5.9
65
5.9
71
5.9
79
6.2
34
6.2
42
6.2
48
6.2
56
7.2
60
1.0
5
1.1
7
1.0
4
1.0
5
1.0
2
1.0
0
3.0
6
3.1
0
1.0
0
1.0
1
1.901.952.002.052.102.152.20 ppm
6.006.056.106.156.206.256.30 ppm
Figure A – 3: 400 MHz 1H NMR Spectrum of 135 in CDCl3

159
Figure A – 4: 100 MHz carbon-13 NMR spectrum of 135 in CDCl3.

160
Figure A – 5: 400 MHz 1H NMR Spectrum of 115 in CDCl3

161
Figure A – 6: 100 MHz carbon-13 NMR spectrum of 115 in CDCl3.

162
Figure A – 7: 400 MHz 1H NMR Spectrum of 137 in CDCl3

163
Figure A – 8: 100 MHz carbon-13 NMR spectrum of 137 in CDCl3.

164
Figure A – 9: 500 MHz 1H NMR Spectrum of Carbaporphyrin 98 in CDCl3

165
Figure A – 10: 1H-1H COSY NMR spectrum of carbaporphyrin diester 98 in CDCl3
Figure A – 11: HSQC NMR spectrum of carbaporphyrin diester 98 in CDCl3.

166
Figure A – 12: Selected nOe difference proton NMR spectra of carbaporphyrin diester 98 in
CDCl3.
2.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5 ppm

167
Figure A – 13: DEPT-135 NMR spectrum of carbaporphyrin diester 98 in CDCl3.
Figure A – 14: 125 MHz carbon-13 NMR spectrum of carbaporphyrin diester 98 in CDCl3.

168
Figure A – 15: 500 MHz 1H NMR Spectrum of C-methylated Carbaporphyrin 120 in CDCl3

169
Figure A – 16: Selected nOe difference proton NMR spectra of 120 in CDCl3
2.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.0 ppm

170
Figure A – 17: 1H-1H COSY NMR spectrum of 120 in CDCl3
Figure A – 18: HSQC NMR spectrum of 120 in CDCl3

171
Figure A – 19: DEPT-135 NMR spectrum of 120 in CDCl3.
Figure A –20: 125 MHz carbon-13 NMR spectrum of 120 in CDCl3.

172
Figure A – 21: 500 MHz 1H NMR Spectrum of 121 in CDCl3.
173
Figure A – 22: Selected nOe difference proton NMR spectra of 121 in CDCl3
2.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5 ppm

174
Figure A – 23: 1H-1H COSY NMR spectrum of 121 in CDCl3
Figure A – 24: HSQC NMR spectrum of 121 in CDCl3

175
Figure A – 25: DEPT-135 NMR spectrum of 121 in CDCl3
Figure A – 26: 125 MHz carbon-13 NMR spectrum of 121 in CDCl3

176
Figure A – 27: 500 MHz 1H NMR Spectrum of 118 in CDCl3

177
Figure A – 28: Selected nOe difference proton NMR spectra of 118 in CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

178
Figure A – 29: 1H-1H COSY NMR spectrum of 118 in CDCl3
Figure A – 30: HSQC NMR spectrum of 118 in CDCl3

179
Figure A – 31: DEPT-135 NMR spectrum of 118 in CDCl3
Figure A – 32: 125 MHz carbon-13 NMR spectrum of 118 in CDCl3
160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 ppm
1.2
3
11
.53
17
.30
18
.10
19
.39
19
.95
29
.92
31
.11
52
.61
76
.97
77
.23
77
.48
96
.13
11
0.1
6
11
8.3
2
13
1.0
9
13
4.5
2
13
4.5
5
13
4.5
8
13
7.1
6
13
8.3
1
13
9.6
4
13
9.6
6
14
0.9
0
16
7.4
5
18.018.519.019.520.0 ppm

180
Figure A – 33: 500 MHz 1H NMR Spectrum of 138 in CDCl3

181
Figure A – 34: Selected nOe difference proton NMR spectra of 138 in CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

182
Figure A – 35: 1H-1H COSY NMR spectrum of 138 in CDCl3
Figure A – 36: HSQC NMR spectrum of 138 in CDCl3

183
Figure A – 37: DEPT-135 NMR spectrum of 138 in CDCl3
Figure A – 38: 125 MHz carbon-13 NMR spectrum of 138 in CDCl3

184
-7-6-5-4-3-2-114 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
-0.0
47
0.0
27
0.0
48
0.0
65
0.0
73
0.0
80
0.0
90
0.1
08
0.1
53
0.1
89
1.2
58
1.5
39
1.6
31
1.6
47
1.6
53
1.6
62
1.6
68
1.6
84
3.3
62
3.6
82
3.6
98
3.7
31
3.7
46
4.3
66
7.2
60
9.0
16
9.9
90
12.1
3
6.2
5
4.1
9
4.0
4
6.1
2
2.0
2
2.0
0
3.33.43.53.63.73.83.94.04.14.24.34.4 ppm
NN
N
Et
Me
MeO2C
MeO2C Me
Et
Et
Et
Au
Figure A – 39: 500 MHz 1H NMR Spectrum of 119 in CDCl3

185
Figure A – 40: Selected nOe difference proton NMR spectra of 119 in CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

186
Figure A – 41: 1H-1H COSY NMR spectrum of 119 in CDCl3
Figure A – 42: HSQC NMR spectrum of 119 in CDCl3

187
Figure A – 43: DEPT-135 NMR spectrum of 119 in CDCl3
Figure A – 44: 125 MHz carbon-13 NMR spectrum of 119 in CDCl3

188
Figure A – 45: 500 MHz 1H NMR Spectrum of 133 in CDCl3

189
Figure A – 46: Selected nOe difference proton NMR spectra of 133 in CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

190
Figure A – 47: 1H-1H COSY NMR spectrum of 133 in CDCl3
Figure A – 48: HSQC NMR spectrum of 133 in CDCl3

191
Figure A – 49: DEPT-135 NMR spectrum of 133 in CDCl3
Figure A – 50: 125 MHz carbon-13 NMR spectrum of 133 in CDCl3

192
Figure A – 51: 500 MHz 1H NMR Spectrum of 117 in CDCl3

193
Figure A – 52: Selected nOe difference proton NMR spectra of 117 in CDCl3
12345678910 ppm

194
Figure A – 53: 1H-1H COSY NMR spectrum of 117 in CDCl3
Figure A – 54: HSQC NMR spectrum of 117 in CDCl3

195
Figure A – 55: DEPT-135 NMR spectrum of 117 in CDCl3
Figure A – 56: 125 MHz carbon-13 NMR spectrum of 117 in CDCl3

196
Figure A – 57: 500 MHz 1H NMR Spectrum of 122 in CDCl3

197
Figure A – 58: Selected nOe difference proton NMR spectra of 122 in CDCl3
1234567891011 ppm

198
Figure A – 59: 1H-1H COSY NMR spectrum of 122 in CDCl3
Figure A – 60: HSQC NMR spectrum of 122 in CDCl3

199
Figure A – 61: DEPT-135 NMR spectrum of 122 in CDCl3
Figure A – 62: 125 MHz carbon-13 NMR spectrum of 122 in CDCl3

200
Figure A – 63: 500 MHz 1H NMR Spectrum of 123 in CDCl3

201
Figure A – 64: Selected nOe difference proton NMR spectra of 123 in CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

202
Figure A – 65: 1H-1H COSY NMR spectrum of 123 in CDCl3
Figure A – 66: HSQC NMR spectrum of 123 in CDCl3

203
Figure A – 67: DEPT-135 NMR spectrum of 123 in CDCl3
Figure A – 68: 125 MHz carbon-13 NMR spectrum of 123 in CDCl3
204
Figure A – 69: 500 MHz 1H NMR Spectrum of 98H+ in TFA-CDCl3

205
Figure A – 70: Selected nOe difference proton NMR spectra of 98H+ in TFA-CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

206
Figure A – 71: DEPT-135 NMR spectrum of 98H+ in TFA-CDCl3
Figure A – 72: HSQC NMR spectrum of 98H+ in TFA-CDCl3

207
Figure A – 73: DEPT-135 NMR spectrum of 98H+ in TFA-CDCl3
Figure A – 74: 125 MHz carbon-13 NMR spectrum of 98H+ in TFA-CDCl3

208
Figure A – 75: 500 MHz 1H NMR Spectrum of 98H22+ in TFA-CDCl3

209
Figure A – 76: 1H-1H COSY NMR spectrum of 98H22+ in TFA-CDCl3
Figure A – 77: HSQC NMR spectrum of 98H22+ in TFA-CDCl3

210
Figure A – 78: DEPT-135 NMR spectrum of 98H22+ in TFA-CDCl3
Figure A – 79: 125 MHz carbon-13 NMR spectrum of 98H22+ in TFA-CDCl3

211
Figure A – 80: 500 MHz 1H NMR Spectrum of 120H+ in TFA-CDCl3

212
Figure A – 81: Selected nOe difference proton NMR spectra of 120H+ in TFA-CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

213
Figure A – 82: 1H-1H COSY NMR spectrum of 120H+ in TFA-CDCl3
Figure A – 83: HSQC NMR spectrum of 120H+ in TFA-CDCl3

214
Figure A – 84: 125 MHz carbon-13 NMR spectrum of 120H+in TFA-CDCl3

215
Figure A – 85: 500 MHz 1H NMR Spectrum of 120H22+ in TFA-CDCl3

216
Figure A – 86: 1H-1H COSY NMR spectrum of 120H22+ in TFA-CDCl3
Figure A – 87: HSQC NMR spectrum of 120H22+ in TFA-CDCl3

217
Figure A – 88: DEPT-135 NMR spectrum of 120H22+ in TFA-CDCl3
Figure A –89: 125 MHz carbon-13 NMR spectrum of 120H22+ in TFA-CDCl3

218
Figure A – 90: 500 MHz 1H NMR Spectrum of 133H+ in TFA-CDCl3

219
Figure A – 91: Selected nOe difference proton NMR spectra of 133H+ in TFA-CDCl3
11 10 9 8 7 6 5 4 3 2 1 ppm

220
Figure A – 92: 1H-1H COSY NMR spectrum of 133H+ in TFA-CDCl3
Figure A – 93: HSQC NMR spectrum of 133H+ in TFA-CDCl3

221
Figure A – 94: DEPT-135 NMR spectrum of 133H+ in TFA-CDCl3
Figure A – 95: 125 MHz carbon-13 NMR spectrum of 133H+ in TFA-CDCl3

222
Figure A – 96: 500 MHz 1H NMR Spectrum of 117H+ in TFA-CDCl3

223
Figure A – 97: Selected nOe difference proton NMR spectra of 117H+ in TFA-CDCl3
10 9 8 7 6 5 4 3 2 1 ppm

224
Figure A – 98: 1H-1H COSY NMR spectrum of 117H+ in TFA-CDCl3
Figure A – 99: HSQC NMR spectrum of 117H+ in TFA-CDCl3

225
Figure A – 100: DEPT-135 NMR spectrum of 117H+ in TFA-CDCl3
Figure A – 101: 125 MHz carbon-13 NMR spectrum of 117H+ in TFA-CDCl3

226
Figure A – 102: 500 MHz 1H NMR Spectrum of 121H+ in TFA-CDCl3

227
Figure A – 103: 1H-1H COSY NMR spectrum of 121H+in TFA-CDCl3
Figure A – 104: HSQC NMR spectrum of 121H+ in TFA-CDCl3

228
Figure A – 105: DEPT-135 NMR spectrum of 121H+ in TFA-CDCl3
Figure A – 106: 125 MHz carbon-13 NMR spectrum of 121H+ in TFA-CDCl3

229
APPENDIX B
SELECTED UV-VIS SPECTRA
Figure B – 1: UV-Vis Spectra of 98 in dichloromethane (red line, free base), CH2Cl2 with 100
equiv TFA (green line, monocation), 50% TFA-CH2Cl2 (blue line, dication) and 5% DBU-
CH2Cl2 (purple line, monoanion)
0
2
4
6
8
10
12
14
16
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

230
Figure B – 2: UV-vis spectra of 120 in dichloromethane (red line, free base), CH2Cl2 with 5
equiv TFA (green line, monocation), 25% TFA-CH2Cl2 (blue line, dication) and 5% DBU-
CH2Cl2 (purple line, monoanion)
0
2
4
6
8
10
12
14
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

231
Figure B – 3: UV-vis spectra of 121 in 1% Et3N-CH2Cl2 (red line, free base) and in CH2Cl2
with 50 equiv. TFA (blue line, monocation)
0
1
2
3
4
5
6
7
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

232
Figure B – 4: UV-vis spectrum of 118 in chloroform
0
1
2
3
4
5
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

233
Figure B – 5: UV-vis spectra of 123 (red line) and 122 (blue line) in chloroform
0
1
2
3
4
5
6
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

234
Figure B – 6: UV-vis spectra of 117 in dichloromethane (red line, free base), 5% TFA-
dichloromethane (green line, monocation) and 5% DBU-dichloromethane (blue line)
0
5
10
15
20
25
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

235
Figure B – 7: UV-vis spectra of carbachlorin 133 in dichloromethane (red line), carbachlorin
monocation 133H+ in 5% TFA-dichloromethane and silver(III) chlorin 138 in dichloromethane
(purple line)
0
5
10
15
20
25
300 350 400 450 500 550 600 650 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

236
Figure B – 8: UV-vis spectra of 98 in dichloromethane with 0 equiv. TFA (red line), 2 equiv.
TFA (orange line), 5 equiv. TFA (green line), 10 equiv. TFA (light blue line), 20 equiv. TFA
(dark blue line) and 50 equiv. TFA (purple line)
0
1
2
3
4
5
6
7
8
9
10
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

237
Figure B – 9: UV-vis spectrum of 98H22+ in 50% TFA-CH2Cl2
Figure B – 10: UV-vis spectrum of deprotonated carbaporphyrin 98 in 5% DBU
0
2
4
6
8
10
12
14
16
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1
0
1
2
3
4
5
6
7
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

238
Figure B – 11: UV-vis spectrum of 120 in dichloromethane
Figure B – 12: UV-vis spectrum of monoprotonated 120 in dichloromethane with 5 equiv TFA
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
300 400 500 600 700 800
Wavelength (nm)
εε εε x 1
0-4
M-1
cm
-1
0
1
2
3
4
5
6
7
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

239
Figure B – 13: UV-vis spectra of 120 in dichloromethane with 0 equiv. TFA (red line), 0.5
equiv. TFA (green line), 1 equiv. TFA (light blue line), 2 equiv. TFA (dark blue line) and 5
equiv. TFA (purple line)
0
1
2
3
4
5
6
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

240
Figure B – 14: UV-vis spectrum of diprotonated 120 in 25% TFA-CH2Cl2
Figure B – 15: UV-vis spectrum of 120 in 50% TFA-CH2Cl2
0
2
4
6
8
10
12
14
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1
0
2
4
6
8
10
12
14
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

241
Figure B – 16: UV-vis spectrum of 120 in 1% Et3N-dichloromethane showing partial
deprotonation
Figure B – 17: UV-vis spectrum of monodeprotonated 120 in 5% DBU-CH2Cl2
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
300 400 500 600 700 800
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

242
Figure B – 18: UV-vis spectrum of 121 in 1% Et3N-CH2Cl2 (free base)
Figure B – 19: UV-vis spectrum of dimethylcarbaporphyrin 121 in 5% DBU-CH2Cl2
0
1
2
3
4
5
6
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1
0
1
2
3
4
5
6
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

243
Figure B – 20: UV-vis spectrum of monoprotonated dimethylcarbaporphyrin 121 in in CH2Cl2
with 50 equiv. TFA
Figure B – 21: UV-vis spectrum of dimethylcarbaporphyrin 121 in 5% TFA-CH2Cl2
0
1
2
3
4
5
6
7
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1
0
1
2
3
4
5
6
7
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

244
Figure B – 22: UV-vis spectrum of silver(III) carbaporphyrin 118 in chloroform
0
1
2
3
4
5
300 400 500 600 700 800
Wavelength (nm)
εε εε x 1
0-4
M-1
cm
-1

245
Figure B – 23: UV-vis spectrum of gold(III) carbaporphyrin 119 in chloroform
0
0.5
1
1.5
2
2.5
3
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

246
Figure B – 24: UV-Vis spectra on silver(III) carbaporphyrin (red line) and gold(III)
carbaporphyrin (blue line) in chloroform
0
1
2
3
4
5
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

247
Figure B – 25: UV-vis spectrum of nickel(II) carbaporphyrin 123 in chloroform
Figure B – 26: UV-vis spectra of carbachlorin by-product 117 in dichloromethane with 0 equiv.
TFA (red line), 0.5 equiv. TFA (green line), 1 equiv. TFA (blue line), and 2 equiv. TFA (purple
line)
0
1
2
3
4
5
6
300 400 500 600 700 800 900
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1
0
5
10
15
20
25
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

248
Figure B – 27: UV-vis spectrum of carbachlorin by-product 133 in 5% DBU-dichloromethane
0
5
10
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

249
Figure B – 28: UV-vis spectrum of carbachlorin cation 133H+ in 1% TFA-dichloromethane
Figure B – 29: UV-vis spectrum of carbachlorin monocation 133H+ in 5% TFA-
dichloromethane
0
5
10
15
20
25
300 350 400 450 500 550 600 650 700
Wavelength (nm)
εε εε x 1
0-4
M-1
cm
-1
0
5
10
15
20
25
300 350 400 450 500 550 600 650 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1

250
Figure B – 30: UV-vis spectra of carbachlorin 133 in dichloromethane with 0 equiv. TFA (red
line), 0.5 equiv. TFA (green line), 1 equiv. TFA (blue line), and 2 equiv. TFA (purple line)
0
5
10
15
20
25
300 400 500 600 700
Wavelength (nm)
εε εεx 1
0-4
M-1
cm
-1