development and evolution of chronic lymphocytic leukemia nicholas chiorazzi karches center for cll...
TRANSCRIPT

Development and evolution
of chronic lymphocytic leukemia
Nicholas Chiorazzi
Karches Center for CLL Research
The Feinstein Institute for Medical Research
North Shore – LIJ Health System
Manhasset, NY, USA

Major messages
1. CLL develops stepwise over time, requiring accumulation of a series of genetic abnormalities. These abnormalities can complement an inherited predisposition supporting transformation. “Development”
2. Once transformation has occurred, clinical progression often results from secondary changes in protein-coding and non-protein-coding genes. “Evolution”
3. The development of primary and secondary DNA variants is facilitated by signals from the microenvironment. “Promotion”
4. Because genetic testing strategies are defining the predisposing, transforming, and progression factors, the CLL field is moving to a precise definition of patient subsets based on specific genetic changes. This will lead to unique therapies targeting defined abnormalities

Major messages – “Development”
1. CLL develops stepwise over time, requiring accumulation of a series of genetic abnormalities. These abnormalities can complement an inherited predisposition supporting transformation
A. Evidence for an inherited predisposition to CLL
B. Indications of a stepwise progression from a normal B cell to an intermediate stage to full blown CLL
C. Possible acquired genetic abnormalities involved in transformation to a CLL cell

Evidence supporting an inherited predisposition to CLL
1. Racial associations of CLL Incidence greater in white > black > yellow populations Increased incidence after irradiation exposure
• Hiroshima/Nagasaki versus Chernobyl experience
2. Familial CLL Highest familial aggregation among leukemias/lymphomas Familial and sporadic CLL are clinically and molecularly similar
3. Occurrence of other cancers in patients and family members of CLL patients Higher relative risk in family members: CLL 8.5x and NHL 1.9x Increased risk in CLL patients of solid tumors, especially epithelial (skin) cancers
• not explainable solely by prior therapy

An inherited predisposition to CLL
4. Genetic studies of CLL patients and families Inheritance of susceptibility is likely polygenic
• Genome-wide association studies (GWAS)
10 single nucleotide genetic polymorphisms (SNPs) more frequently found in CLL patients
Location Gene OR2q13 BCL2L11/Bim 1.392q37.1 SP140 1.412q37.3 FARP2 1.396q25.3 IRF4 1.548q24.21 GRAMD1B 1.2611q24.1 RFXT and NEDD4 1.4515q21.3 -- 1.3615q23 -- 1.3716q24.1 IRF8 1.2219q13.32 PRKD2 1.36
DiBernardo et al.Nat Genet 40:
1204-1210, 2008Crowther-Swanepoel et al.
Nat Genet 42:132-136, 2010

An inherited predisposition to CLL
5. Hematopoietic stem cells (HSCs) from CLL patients appear to differ functionally from most normal adults
Y. Kikushige et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 20: 246-259, 2011

An inherited predisposition to CLL
Genetic studies of CD34+CD38- CLL cells from BM
Studies carried out by injecting cells into newborn, alymphoid mice and watching their development and growth over time
CD34+ cells from CLL BM gave rise to more immature and mature B cells than CD34+ cells from BM of normal subjects
Kikushige et al. Cancer Cell 20: 246, 2011
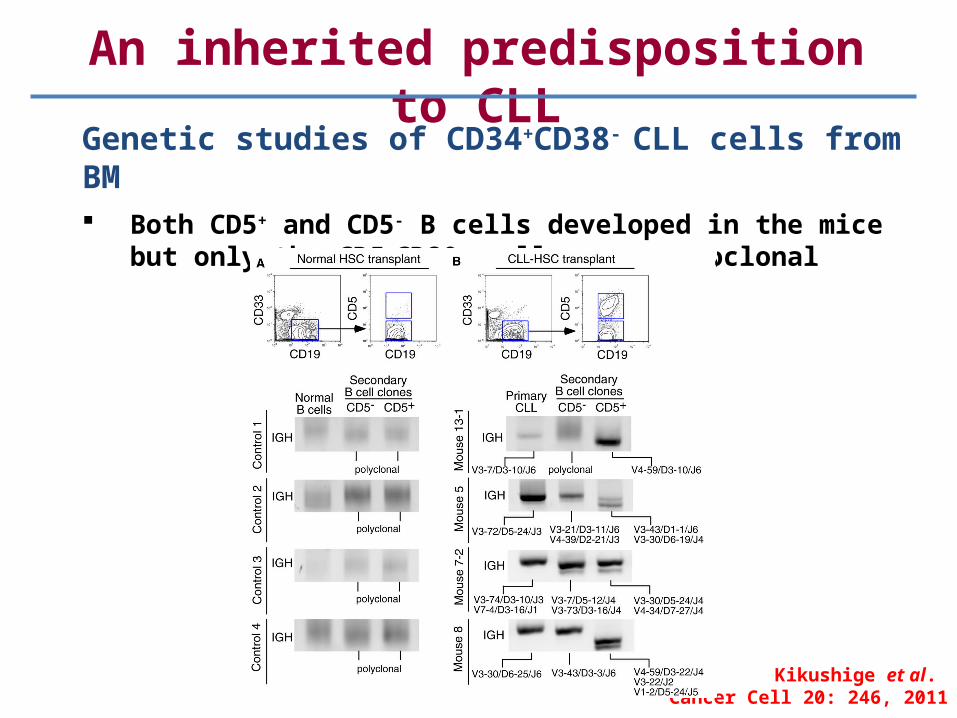
An inherited predisposition to CLL
Genetic studies of CD34+CD38- CLL cells from BM Both CD5+ and CD5- B cells developed in the mice but only
the CD5+CD20+ cells were monoclonal
Kikushige et al. Cancer Cell 20: 246, 2011

An inherited predisposition to CLL
Genetic studies of CD34+CD38- CLL cells from BM However, the CD5+ clones that developed from CLL HSCs
did not carry the IGHV-D-J rearrangement of the CLL cell donor
Kikushige et al. Cancer Cell 20: 246, 2011

An inherited predisposition to CLL
Genetic studies of CD34+CD38- CLL cells from BM Furthermore, the CD34+ cells did not exhibit the genomic
abnormality of the CLL donor, e.g., del(13q) or del(11q), etc.
Lastly, the mature CLL B-cell clones did not evolve into a “clinical CLL-like disease, although there was a suggestion that the IGHV-D-J rearrangements of the “pre-CLL” clones have the characteristics of CLL clones in general, i.e., specific gene family use.
Thus, it appears that the CD34+ cells of CLL patients are “primed” to make more B lymphocytes, often of the CD5 subtype that are more likely to be oligo/monoclonal, but presumably environmental influences (e.g., BCR selection) are required to support further transformation.
Is this monoclonal B lymphocytosis (MBL)?
Kikushige et al. Cancer Cell 20: 246, 2011

Question to consider: Are these phenomena the consequences of a somatic lesion occurring in CLL patients or representations of a genetic predisposition/polymorphism inherited in the human germline?

Major messages – “Development”
1. CLL develops stepwise over time, requiring accumulation of a series of genetic abnormalities. These abnormalities can complement an inherited predisposition supporting transformation
A. Evidence for an inherited predisposition to CLL
B. Indications of a stepwise progression from a normal B cell to an intermediate stage to full blown CLL
C. Possible acquired genetic abnormalities involved in transformation to a CLL cell

Monoclonal B lymphocytosis (MBL)
CLL-like MBL
Oligo/monoclonal expansion of circulating B cells that are L-chain restricted and express a CLL phenotype
Found in ~3-5% of normal individuals and ~20% over the ageof 65 years. ~12-15% of first-degree relatives of CLL patients
Appears to be a transition from clinical MBL to CLL
~1-2% of clinical MBL patients convert to CLL yearly• AC Rawstron et al. N Engl J Med 359: 575-583, 2008• D Rossi et al. Br J Haematol 146: 64-75, 2009• TD Shanafelt et al. J Clin Oncol 27: 3959-3963, 2009
Several immune, genetic, and prognostic parametersdiffer between clinical MBL and CLL, suggesting atransition from one to the other
Tumor burden FISH-defined genomic aberrations Lymphocyte doubling time
• Rossi et al. Br J Haematol 146: 64–75, 2009

Monoclonal B lymphocytosis (MBL)
CLL-like MBL
Transition from clinical MBL to CLL
MBL precedes CLL in the majority of instances
O Landgren et al. N Engl J Med 360: 659-667, 2009 M Frezzato et al. Am J Hematol 85: 868-871, 2010
Prospective and retrospective analyses of populations followed for another purpose indicated that almost every person who happened to develop CLL had evidence for a clonal expansion in advance

Major messages – “Development”
1. CLL develops stepwise over time, requiring accumulation of a series of genetic abnormalities. These abnormalities can complement an inherited predisposition supporting transformation
A. Evidence for an inherited predisposition to CLL
B. Indications of a stepwise progression from a normal B cell to an intermediate stage to full blown CLL
C. Acquired genetic abnormalities that may be involved in transformation to a CLL cell
Deletion of microRNAs 15a and 16-1 Overexpression of microRNAs that function as oncogenes Overexpression/activity of TCL1

Key to the 13q deletion: loss of microRNAs 15a and 16-1
Deletion at 13q
most common chromosomal abnormality in CLL
more common among IGHV mutated CLL cases
Calin, Croce, and colleagues determined that thekey elements were microRNAs 15a/16.1
15a/16.1 cluster shown to control Bcl2 levels
documentation that microRNAs can be tumor suppressors
suggestion that loss of this microRNA cluster is a key element in the development of CLL, at least for those that develop M-CLL

Klein et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40, 2010
Deleted either: only microRNAs 15a/16.1 or microRNAs 15a/16.1 and the DLeu2 gene (minimal deleted
region, MDR)
Mice can develop over time: CD5+ B-cell disorder of low penetrance: MDR faster and
more lethal clones expressing stereotyped BCRs
Targeted molecular elimination of microRNAs 15a/16.1 or these plus Leu 2
can lead to a CLL-like disease

Klein et al. Cancer Cell
17: 28–40, 2010
Mice with deletion of miR-15a/16-1 or MDRdevelop lymphoproliferations

Summary of “Development”
Development of CLL is primed by a series of incompletely defined gene variants
This genetic soil is seeded further by other somatic abnormalities acquired later in the life of the transforming B cell
The final “hit” in this cascade occurs in a B cell that has rearranged its IGHV, IGHD, and IGHJ genes to yield the signature rearrangement of the clone
These composite genetic factors (inherited and acquired) usher a normal B cell into an intermediary stage – “MBL” – and then to full-fledged CLL
Candidate acquired factors include loss (or overexpression) of microRNAs, as these can be tumor suppressors or oncogenes

Major message – “Evolution”
2. Once transformation has occurred, clinical progression often results from secondary changes in protein-coding and non-protein-coding genes - “evolution”
A. Sequential analyses in patients of: FISH abnormalities microRNA abnormalities global DNA abnormalities by comparative
genomic hybridization and SNP profiling
B. Analyses of DNA abnormalities by next generation sequencing of CLL genomes/exomes

Clonal evolution
A. Sequential analyses of FISH abnormalities, microRNA abnormalities, and global DNA abnormalities
Take home messages: ~25% of patients develop a new genetic abnormality over
time in coding or non-coding genes Occurs more frequently in:
U-CLL clones and in M-CLL clones of patients that eventually require therapy
CD38+ clones ZAP-70+ clones CD49d+ clones
Most common new lesions: del(13q) del(17p) – harbinger of accelerated disease
Greater the number of clonal aberrations the shorter the time to treatment and survival

Major messages – “Evolution”
2. Once transformation has occurred, clinical progression often results from secondary changes in protein-coding and non-protein-coding genes - “evolution”
A. Sequential analyses in patients of: FISH abnormalities microRNA abnormalities global DNA abnormalities by comparative
genomic hybridization and SNP profiling
B. Analyses of DNA abnormalities by next generation sequencing of CLL genomes/exomes

Genetic abnormalities
B. Analyses of DNA abnormalities by next generation sequencing of CLL genomes/exomes
In 2011, three exciting studies of this type:
Puente et al. Nature 475: 101-105, 2011 Fabbri et al. J Exp Med 208: 1389-1401, 2011 Wang et al. N Engl J Med 365: 2497-2506, 2011
Puente et al and Fabbri et al studied untreated patients – combined total 9 patients
Wang et al studied treated and untreated patients – total 88 patients
Placed under “evolution”, although, because some are found at diagnosis, may be initiating events.

Summary of consistent findings
B. Analyses of DNA abnormalities by next generation sequencing of CLL genomes/exomes
Genomic complexity exists in CLL but is less than that of solid tumors and DLCBL; similar to AML
Combined 18 recurrent mutations were identified. Most common abnormality was in NOTCH1
Associations of specific mutations with other known abnormalities in CLL, such as those defined by FISH, suggest links between these
Majority of mutations defined could change the structure of the encoded molecule, suggesting clonal selection and therefore functional relevance in patients

Associations between specific gene mutations and other characteristics
Wang et al.N Engl J Med 365:
2497-2506, 2011

Fabbri G. et al. J Exp Med 208: 1389–1401, 2011
NOTCH1 is mutated in a large fraction of Richter’s Syndrome cases and chemorefractory CLL

Kaplan-Meier estimates of treatment-free survival and overall survival, according to the
mutational status of NOTCH1.
Fabbri G. et al. J Exp Med 208: 1389–1401, 2011

Summary of “Evolution”
~25% of CLL clones develop new DNA aberrations over time
These are more likely to occur in patients with the more ominous prognostic markers
However, new DNA aberrations can occur without treatment, suggesting that clonal diversification is occurring continuously
The genetic changes that are recurrent among patients presumably indicate pathways that drive CLL progression, if not development
Patients that develop new abnormalities often have shorter times to treatment and survival
Therefore, halting clonal evolution by targeting specific subsets of cells within CLL clones may be a therapeutic opportunity

Major messages – “Promotion”
3. The development of primary and secondary DNA variants is facilitated by signals from the microenvironment
Novel DNA lesions occur when cells are duplicating their DNA. Therefore those signals from the microenvironment that promote cell division are most likely to promote DNA variants, e.g., those through the BCR, CD40, TLRs, etc.
Simplistically, whatever leads to DNA duplication leads to DNA mistakes.
What mechanisms within dividing cells might promote the development of new DNA?
Activation-induced deaminase (AID)? Responsible for IGHV mutations and for IG class switch recombination.

AID is expressed in tissue bound and circulating CLL cells that recently or are dividing
Patten et al. Blood 2012 in press

Patten et al. Blood 2012 in press

Summary of “Promotion”
Developing and completely transformed CLL cells owe their fate andability to evolve to receptor-ligand interactions.
Most important appear to be those delivered through the BCR, CD40, and TLRs.
Critical are the surroundings within which these signals are delivered – “the microenvironment”.
Kinase inhibitors that block stimulatory signals through the BCR and that expel CLL cells from the trophic microenvironment highlight this.
Although survival of new cellular variants is essential, the creation of DNA lesions that induce and accelerate clonal growth and progression depends on cell division.
Therefore, halting or eliminating dividing cells would block clonal evolution and possibly disease progression