d-block metal complexes: reaction...
TRANSCRIPT

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 1
Inorganic Chemistry B Inorganic Chemistry B
Chapter 26
1 Dr. Said El-Kurdi
d-Block metal complexes: reaction mechanisms
2 Dr. Said El-Kurdi
26.1 Introduction
Mechanisms of ligand substitution and electron-transfer reactions in coordination complexes.
A proposed mechanism must be consistent with all experimental facts.
A mechanism cannot be proven, since another mechanism may also be consistent with the experimental data.
26.2 Ligand substitutions: some general points
In a ligand substitution reaction: MLxX + Y MLxY + X X is the leaving group and Y is the entering group.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 2
Dr. Said El-Kurdi 3
Kinetically inert and labile complexes
Metal complexes that undergo reactions with t1/2 1min are described as being kinetically labile. If the reaction takes significantly longer than this, the complex is kinetically inert.
There is no connection between the thermodynamic stability of a complex and its lability towards substitution.
For example, values of hydGo for Cr3+ and Fe3+ are almost equal, [Cr(OH2)6]3+ (d3) undergoes substitution slowly [Fe(OH2)6]3+ (high-spin d5) undergoes substitution rapidly.
4 Dr. Said El-Kurdi
exchange of a water molecule in the first coordination sphere of [M(OH2)x]
n+ with one outside this coordination shell
average residence time (=1/k) of an H2O ligand in the first coordination sphere of a metal ion.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 3
5 Dr. Said El-Kurdi
6 Dr. Said El-Kurdi
For main group metal ions
On descending a given group, the rate of water exchange increases as:
the metal ion increases in size
the coordination number increases the surface charge density decreases
[Li(OH2)6]+ (least labile) to [Cs(OH2)8]+ (most labile).
Each group 13 M3+ forms a hexaaqua ion, and values of k range from 1 s1 for [Al(OH2)6]3+ to 107 s1 for [In(OH2)6]3+, consistent with the increase in ionic radius from 54pm (Al3+) to 80pm (In3+).

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 4
7 Dr. Said El-Kurdi
The lanthanoid M3+ complex ions are all relatively labile with k >107?????????
Stoichiometric equations say nothing about mechanism
use of H218O as solvent shows that all the oxygen in the
aqua complex is derived from carbonate
Dr. Said El-Kurdi 8

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 5
9 Dr. Said El-Kurdi
Types of substitution mechanism
In inorganic substitutions, the limiting mechanisms are dissociative (D), in which the intermediate has a lower coordination number than the starting complex and associative (A), in which the intermediate has a higher coordination number
10 Dr. Said El-Kurdi

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 6
11 Dr. Said El-Kurdi
In most metal complex substitution pathways, bond formation between the metal and entering group is thought to be concurrent with bond cleavage between the metal and leaving group. This is the interchange (I) mechanism.
In an I mechanism, there is no intermediate but various transition states are possible.
12 Dr. Said El-Kurdi
dissociative interchange (Id), in which bond breaking dominates over bond formation; the rate shows only a very small dependence on the entering group.
associative interchange (Ia), in which bond formation
dominates over bond breaking. the reaction rate shows a dependence on the entering group.
It is usually difficult to distinguish between A and Ia D and Id Ia and Id processes.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 7
13 Dr. Said El-Kurdi
Activation parameters
Dr. Said El-Kurdi 14
An Eyring plot

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 8
Dr. Said El-Kurdi 15
Values of S‡ are particularly useful in distinguishing between associative and dissociative mechanisms.
A large negative value of S‡ is indicative of an associative mechanism, i.e. there is a decrease in entropy as the entering group associates with the starting complex.
Solvent reorganization can result in negative values of S‡ even for a dissociative mechanism
The pressure dependence of rate constants leads to a measure of the volume of activation, V‡
Dr. Said El-Kurdi 16
A reaction in which the transition state has a greater volume than the initial state shows a positive V‡, whereas a negative V‡ corresponds to the transition state being compressed relative to the reactants.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 9
Dr. Said El-Kurdi 17
A large negative value of V‡ indicates an associative mechanism; a positive value suggests that the mechanism is dissociative.
Dr. Said El-Kurdi 18
26.3 Substitution in square planar complexes
Complexes with a d8 configuration often form square planar
complexes, especially when there is a large crystal field: Rh(I),
Ir(I), Pt(II), Pd(II), Au(III).
4-Coordinate complexes of Ni(II) may be tetrahedral or
square planar.
The majority of kinetic work on square planar systems has
been carried out on Pt(II) complexes because the rate of
ligand substitution is conveniently slow.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 10
Dr. Said El-Kurdi 19
The trans-influence
Consider a square planar complex which contains a trans LML’ arrangement:
Ligands L and L’ compete with each other for electron density because the formation of ML and ML’ bonds uses the same metal orbitals.
Dr. Said El-Kurdi 20
ground state trans-influence (i.e. the influence that L has on the ML’ bond)
for a series of square planar complexes trans [PtXH(PEt3)2]

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 11
Dr. Said El-Kurdi 21
for a series of square planar complexes trans [PtXH(PEt3)2]
Values of (Pt–H) show that the PtH bond is weakest for X=CN and the trans-influence of the X ligands follows the order CN > I > Br > Cl.
trans-influence may be observed wherever ligands are mutually trans, e.g. in octahedral species.
Dr. Said El-Kurdi 22
Rate equations, mechanism and the trans-effect
based on a large body of experimental work, nucleophilic substitution reactions in square planar Pt(II) complexes normally proceed by associative mechanisms (A or Ia). Negative values of S‡ and V‡ support this proposal
studied under pseudo-first order conditions, with Y (as well as the solvent, S) in vast excess.
Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 12
Dr. Said El-Kurdi 23
Dr. Said El-Kurdi 24
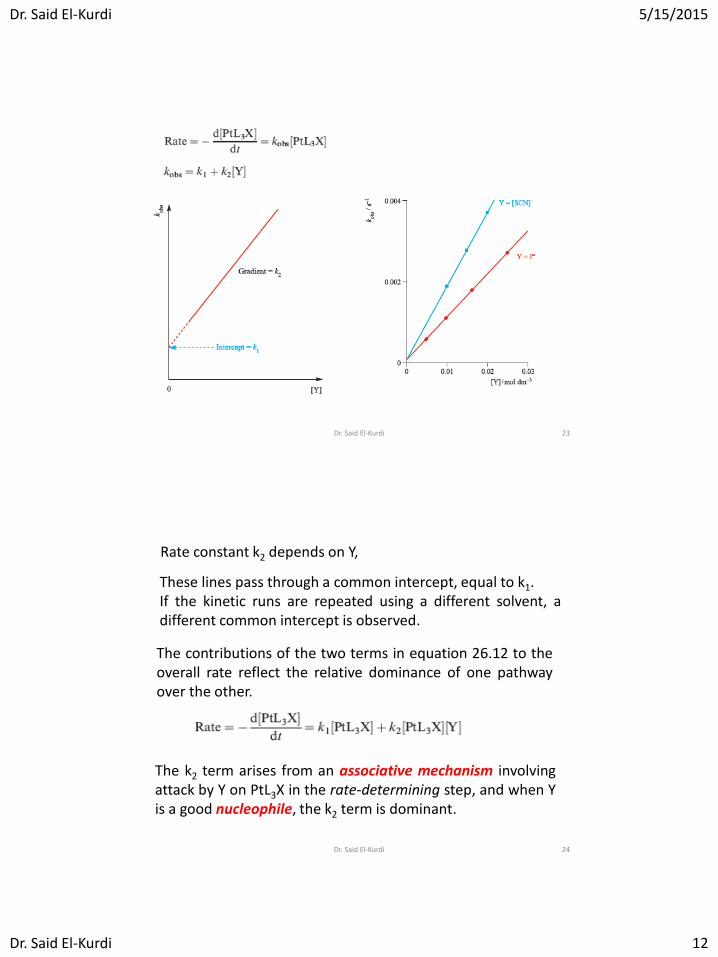
Rate constant k2 depends on Y,
These lines pass through a common intercept, equal to k1. If the kinetic runs are repeated using a different solvent, a different common intercept is observed.
The k2 term arises from an associative mechanism involving attack by Y on PtL3X in the rate-determining step, and when Y is a good nucleophile, the k2 term is dominant.
The contributions of the two terms in equation 26.12 to the overall rate reflect the relative dominance of one pathway over the other.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 13
Dr. Said El-Kurdi 25
The k1 term might appear to indicate a concurrent dissociative pathway. However, experiment shows that the k1 term becomes dominant if the reaction is carried out in polar solvents, and its contribution diminishes in apolar solvents.
When the solvent is a potential ligand (e.g. H2O), it competes with the entering group Y in the rate-determining step of the reaction, and X can be displaced by Y or S. Substitution of S by Y then occurs in a fast step, i.e. non-rate determining.
Dr. Said El-Kurdi 26
In the majority of reactions, substitution at square planar Pt(II) is stereoretentive: the entering group takes the coordination site previously occupied by the leaving group. An A or Ia mechanism involves a 5-coordinate intermediate or transition state and, since the energy difference between different 5-coordinate geometries is small, one would expect rearrangement of the 5-coordinate species
The choice of leaving group in a square planar complex is determined by the nature of the ligand trans to it;
this is the trans-effect and is kinetic in origin.
The choice of leaving group in a square planar complex is determined by the nature of the ligand trans to it;
this is the trans-effect and is kinetic in origin.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 14
Dr. Said El-Kurdi 27
Initial attack by the entering group at a square planar Pt(II) centre is from above or below the plane. Nucleophile Y then coordinates to give a trigonal bipyramidal species which loses X with retention of stereochemistry.
Dr. Said El-Kurdi 28
If L2 is a strong -acceptor (e.g. CO), it will stabilize the transition state by accepting electron density that the incoming nucleophile donates to the metal center, and will thereby facilitate substitution at the site trans to it.
The general order of the trans-effect (i.e. the ability of ligands to direct trans-substitution) spans a factor of about 106 in rates and is:

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 15
Dr. Said El-Kurdi 29
Dr. Said El-Kurdi 30

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 16
Dr. Said El-Kurdi 31
Ligand nucleophilicity
Effect of entering group
for most reactions at Pt(II), the rate constant k2 increases in the order:
This is called the nucleophilicity sequence for substitution at square planar Pt(II)
Dr. Said El-Kurdi 32
A nucleophilicity parameter, nPt, is defined by equation 26.22 where k2’ is the rate constant for reaction 26.21 with Y = MeOH (i.e. for Y = MeOH, nPt = 0).
The nucleophilicity parameter, nPt, describes the dependence of the rate of substitution in a square planar Pt(II) complex
on the nucleophilicity of the entering group.
The nucleophilicity parameter, nPt, describes the dependence of the rate of substitution in a square planar Pt(II) complex
on the nucleophilicity of the entering group.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 17
Dr. Said El-Kurdi 33
If we now consider substitution reactions of nucleophiles with other Pt(II) complexes, linear relationships are found between values of log k2 and nPt
where s is the nucleophilicity discrimination factor and k2’ is the rate constant when the nucleophile is MeOH.
Dr. Said El-Kurdi 34
For a given substrate, s can be found from the slope of a Line
Each complex has a characteristic value of s,

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 18
Dr. Said El-Kurdi 35
The relatively small value of s for [Pt(dien)(OH2)]2+ indicates that this complex does not discriminate as much between entering ligands as, for example, does trans-[PtCl2PEt3]2; i.e. [Pt(dien)(OH2)]2+ is generally more reactive towards substitution than other complexes in the table, consistent with the fact that H2O is a good leaving group.
Dr. Said El-Kurdi 36
The nucleophilicity discrimination factor, s, is a characteristic of a given square planar Pt(II) complex and describes how
sensitive the complex is to variation in the nucleophilicity of the entering ligand.
The nucleophilicity discrimination factor, s, is a characteristic of a given square planar Pt(II) complex and describes how
sensitive the complex is to variation in the nucleophilicity of the entering ligand.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 19
Dr. Said El-Kurdi 37
26.4 Substitution and racemization in octahedral complexes
The popular candidates for study have been Cr(III) (d3) and low-spin Co(III) (d6) species. These complexes are kinetically inert and their rates of reaction are relatively slow and readily followed by conventional techniques.
Water exchange
Dr. Said El-Kurdi 38
for M2+ and M3+ ions of the d-block metals, data for reaction 26.24 indicate a correlation between rate constants and electronic configuration.
The change from negative to positive values of V‡ indicates a change from associative to dissociative mechanism, and suggests that bond making becomes less (and bond breaking more) important on going from a d3 to d8 configuration

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 20
Dr. Said El-Kurdi 39
For the M3+ ions, values of V‡ suggest an associative mechanism. Where data are available, an associative process appears to operate for second and third row metal ions, consistent with the idea that larger metal centers may facilitate association with the entering ligand.
First order rate constants, k, for reaction 26.24 vary greatly among the first row d-block metals (all high-spin Mn+ in the hexaaqua ions):
Dr. Said El-Kurdi 40
The Eigen-Wilkins mechanism
Water exchange is always more rapid than substitutions with other entering ligands
for most ligand substitutions in octahedral complexes, experimental evidence supports dissociative pathways.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 21
Dr. Said El-Kurdi 41
The Eigen–Wilkins mechanism applies to ligand substitution in an octahedral complex.
An encounter complex is first formed between substrate and entering ligand in a preequilibrium step, and this is
followed by loss of the leaving ligand in the rate-determining step.
An encounter complex is first formed between substrate and entering ligand in a preequilibrium step, and this is
followed by loss of the leaving ligand in the rate-determining step.
The first step in the Eigen–Wilkins mechanism is the diffusing together of ML6 and Y to form a weakly bound encounter complex
Dr. Said El-Kurdi 42
Usually, the rate of formation of {ML6,Y} and the back reaction to ML6 and Y are much faster than the subsequent conversion of {ML6,Y} to products. Thus, the formation of {ML6,Y} is a pre-equilibrium.
The rate-determining step
estimated value
Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 22
Dr. Said El-Kurdi 43
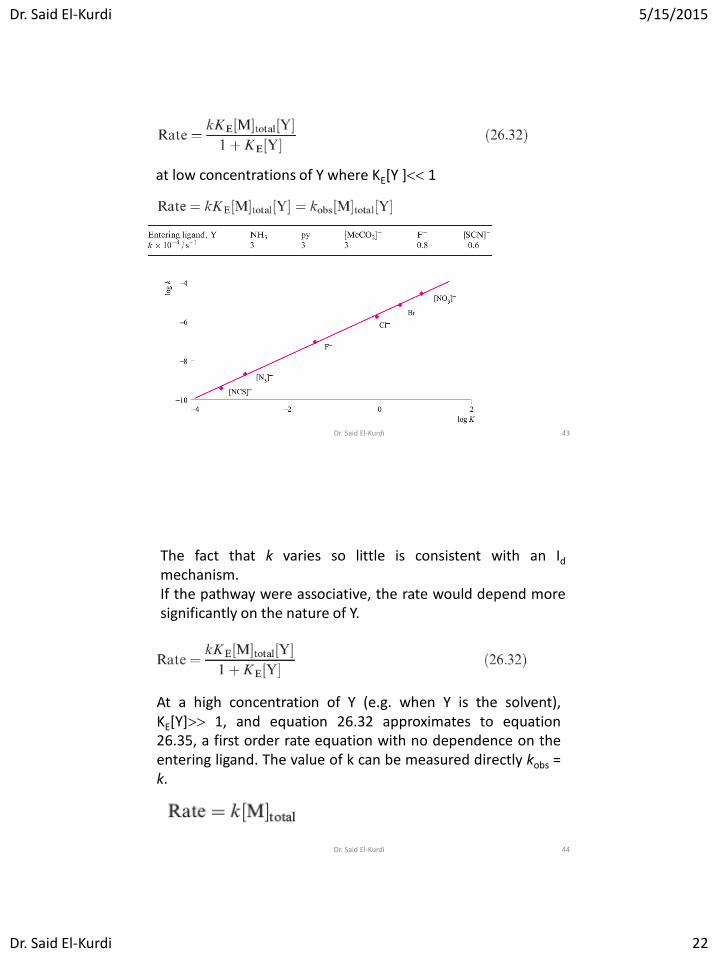
at low concentrations of Y where KE[Y ] 1
Dr. Said El-Kurdi 44
The fact that k varies so little is consistent with an Id
mechanism. If the pathway were associative, the rate would depend more significantly on the nature of Y.
At a high concentration of Y (e.g. when Y is the solvent), KE[Y] 1, and equation 26.32 approximates to equation 26.35, a first order rate equation with no dependence on the entering ligand. The value of k can be measured directly kobs = k.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 23
Dr. Said El-Kurdi 45
An Id mechanism is supported in very many instances. From experimental trends
The rate of ligand substitution usually depends on the nature of the leaving ligand.
the rate of substitution increases with X in the following order:
This trend correlates with the MX bond strength
Dr. Said El-Kurdi 46
The stronger the bond, the slower the rate, and is consistent with the rate-determining step involving bond breaking in a dissociative step

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 24
Dr. Said El-Kurdi 47
Stereochemistry of substitution
most substitutions in octahedral complexes involve D or Id pathways, consider the stereochemical implications of the D mechanism this involves a 5-coordinate species
Dr. Said El-Kurdi 48
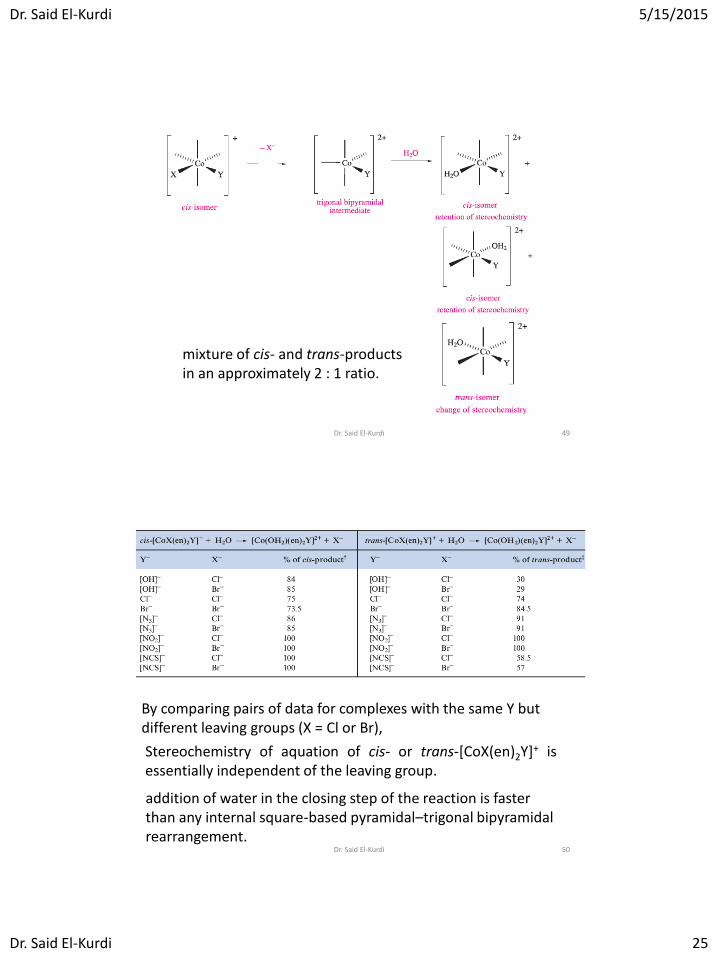
If the mechanism is limiting dissociative (D), a 5-coordinate intermediate must be involved . It follows that the stereochemistry of [Co(OH2)(en)2Y]2+ must be independent of the leaving group X, and will depend on the structure of the intermediate
Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 25
Dr. Said El-Kurdi 49
mixture of cis- and trans-products in an approximately 2 : 1 ratio.
Dr. Said El-Kurdi 50
By comparing pairs of data for complexes with the same Y but different leaving groups (X = Cl or Br),
addition of water in the closing step of the reaction is faster than any internal square-based pyramidal–trigonal bipyramidal rearrangement.
Stereochemistry of aquation of cis- or trans-[CoX(en)2Y]+ is essentially independent of the leaving group.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 26
Dr. Said El-Kurdi 51
Base-catalysed hydrolysis
because [OH] deprotonates a coordinated NH3 ligand.
pre-equilibrium is first established, followed by loss of X to give the reactive amido species
Dr. Said El-Kurdi 52
conjugate–base mechanism (Dcb or SN1cb mechanism).

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 27
Dr. Said El-Kurdi 53
Isomerization and racemization of octahedral complexes
octahedron is stereochemically rigid, loss of a ligand gives a 5-coordinate species which can undergo Berry pseudo-rotation
we discussed cases where the assumption is that such rearrangement does not occur
Dr. Said El-Kurdi 54
if the lifetime of the intermediate is long enough, it provides a mechanism for isomerization
For [Ni(bpy)3]2+ and [Ni(phen)3]2+, the rates of exchange with 14C-labelled ligands are the same as the rates of racemization.
This is consistent with a dissociative process (equation 26.47) in which the intermediate is racemic, or racemizes faster than recombination with LL.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 28
Dr. Said El-Kurdi 55
Such a dissociative mechanism is rare
Two intramolecular mechanisms are possible
Dr. Said El-Kurdi 56
(a) the Bailar twist and
(b) the Ray–Dutt twist.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 29
Dr. Said El-Kurdi 57
Cleavage and reformation of the ML bond of one end of the bidentate ligand.
Dr. Said El-Kurdi 58
26.5 Electron-transfer processes
The simplest redox reactions involve only the transfer of electrons, and can be monitored by using isotopic tracers
In an outer-sphere mechanism, electron transfer occurs without a covalent linkage being formed between the reactants. In an inner-sphere mechanism, electron transfer occurs via a covalently bound bridging ligand.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 30
Dr. Said El-Kurdi 59
Inner-sphere mechanism
All the Cr(III) produced was in the form of [Cr(OH2)5Cl]2+, and tracer experiments showed that all the chloro ligand in [Cr(OH2)5Cl]2+ originated from [Co(NH3)5Cl]2+.
Since the Co center could not have lost Cl before reduction, and Cr could not have gained Cl after oxidation, the transferred Cl must have been bonded to both metal centers during the reaction.
Dr. Said El-Kurdi 60
Cl is transferred between metal centers; such transfer is often (but not necessarily) observed.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 31
Dr. Said El-Kurdi 61
the intermediate 26.4 (which is stable enough to be precipitated as the Ba2+ salt) is slowly hydrolysed to products without transfer of the bridging ligand.
[Fe(CN)6]3 + [Co(CN)5]3 [Fe(CN)6]4 + [Co(CN)5(OH2)]2
Dr. Said El-Kurdi 62
Common bridging ligands in inner-sphere mechanisms include halides, [OH], [CN], [NCS], pyrazine and 4,4’-bipyridine.
The steps of an inner-sphere mechanism are bridge formation, electron transfer and bridge cleavage.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 32
Dr. Said El-Kurdi 63
Most inner-sphere processes exhibit second order kinetics overall.
Any one of bridge formation, electron transfer or bridge cleavage can be rate-determining.
For the following reaction with a range of ligands X,
the rate-determining step is electron transfer, and the rates of reaction depend on X
Dr. Said El-Kurdi 64
Second order rate constants for reaction 26.55 with different bridging X ligands.
The increase in k along the series F, Cl , Br , I correlates with increased ability of the halide to act as a bridge

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 33
Dr. Said El-Kurdi 65
Outer-sphere mechanism
When both reactants in a redox reaction are kinetically inert, electron transfer must take place by a tunnelling or outersphere mechanism.
In a self-exchange reaction, the left- and right-hand sides of the equation are identical; only electron transfer, and no net chemical reaction, takes place.
In a self-exchange reaction, the left- and right-hand sides of the equation are identical; only electron transfer, and no net chemical reaction, takes place.
The Franck–Condon approximation states that a molecular electronic transition is much faster than a molecular vibration.
The Franck–Condon approximation states that a molecular electronic transition is much faster than a molecular vibration.
Dr. Said El-Kurdi 66
This reductant–oxidant pair is called the encounter or precursor complex.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 34
Dr. Said El-Kurdi 67
Consider a self-exchange reaction of the type:
There is no overall reaction and therefore Go = 0, and K = 1.
Why do reactions of this type have widely differing reaction rates? Why do reactions of this type have widely differing reaction rates?
It is usually the case that the M–L bond lengths in the M(III) complex are shorter than those in the corresponding M(II) complex.
Dr. Said El-Kurdi 68
How can a reaction with Go = 0 continually lose energy as the electron is transferred between [ML6]2+ and [ML6]3+? The answer, of course, is that it cannot.
The electron transfer can only take place when the M–L bond distances in the M(II) and M(III) states are the same, i.e. the bonds in [ML6]2+ must be compressed and those in [ML6]3+ must be elongated
Franck–Condon restriction
The activation energy required to reach these vibrational excited states varies according to the system, and hence the self-exchange rate constants vary.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 35
Dr. Said El-Kurdi 69
The outer-sphere mechanism: when the reactants have differing bond lengths, vibrationally excited states with equal bond lengths must be formed in order to allow electron transfer to occur.
Dr. Said El-Kurdi 70
[Fe(bpy)3]2+ and [Fe(bpy)3]3+, low-spin Fe–N bond distances are 197 and 196 pm, respectively. Electron transfer involves only a change from t2g
5 to t2g6 (Fe2+ to
Fe3+) and vice versa. k > 106 dm3 mol 1 s1
The greater the changes in bond length required to reach the encounter complex, the slower the rate of electron transfer. The greater the changes in bond length required to reach the encounter complex, the slower the rate of electron transfer.
For example, the rate of electron transfer between [Ru(NH3)6]2+ (Ru–N= 214 pm, low-spin d6) and [Ru(NH3)6]3+ (Ru–N = 210 pm, low-spin d 5) is 104 dm3 mol 1 s1.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 36
Dr. Said El-Kurdi 71
Electron transfer between [Co(NH3)6]2+ (Co–N=211 pm) and [Co(NH3)6]3+ (Co–N=196 pm) requires not only changes in bond lengths, but also a change in spin state: [Co(NH3)6]2+ is high-spin d7 (t2g
5eg2) and
[Co(NH3)6]3+ is low-spin d6 (t2g6eg
0). Transfer of an electron between the excited states shown in Figure 26.10 leads to a configuration of t2g
5eg1 for
{[Co(NH3)6]3+}* and t2g6eg
1 for{[Co(NH3)6]2+}*. These are electronically excited states,
Dr. Said El-Kurdi 72
The activation energy for the self-exchange reaction therefore has contributions from both changes in bond lengths and changes in spin states.
self-exchange between [Co(phen)3]2+ and [Co(phen)3]3+ is much faster than between [Co(NH3)6]2+ and [Co(NH3)6]3+ or [Co(en)3]2+ and [Co(en)3]3+ (all three exchange processes are between high-spin Co(II) and low-spin Co(III)).
This is consistent with the ability of phen ligands to use their -orbitals to facilitate the intermolecular migration of an electron from one ligand to another, and phen complexes tend to exhibit fast rates of self-exchange.

Dr. Said El-Kurdi 5/15/2015
Dr. Said El-Kurdi 37
Dr. Said El-Kurdi 73
The rates of self-exchange reactions involved cationic species in aqueous solution are typically not affected by the nature and concentration of the anion present in solution.
The rate of electron transfer between anions in aqueous solution generally depends on the cation and its concentration.
Dr. Said El-Kurdi 74
For example, the self-exchange reaction between
[Fe(CN)6]3 and [Fe(CN)6]4 with K+ as the counter-ion. The
rate constant of self-exchange reaction is of the order of
104 dm3 mol1 s1
by adding the macrocyclic ligand 18-crown-6 to complex
the K+ ions, the rate constant of self-exchange is 2.4102
dm3 mol1 s1