cyp1 (hap1) regulator of oxygen-dependent gene expression in yeast
TRANSCRIPT

J. Mol. Biol. (1988) 204, 263-276
CYPl (HAPl) Regulator of Oxygen-dependent Gene Expression in Yeast
I. Overall Organization of the Protein Sequence Displays Several Novel Structural Domains
Francine Creusot, Jacqueline Verdi&e, Mauricette Gaisne and Piotr P. Slonimski
Centre de Ghne’tique Mole’culaire du C.N.R.S. Laboratoire propre associe’ ci I’Universite’ Pierre et Marie Curie
91190 Gif-sur- Yvette, France
(Received 10 February 1988, and in revised form 21 June 1988)
In the yeast Saccharomyces cerevisiae the CYPl gene that modulates the expression of isol- (CYCl) and iso2-cytochrome c (CYPS) structural genes gives rise to two classes of mutated alleles; one class, represented by CYPl-18, has opposite effects on CYCl and CYPS, it reduces the expression of CYCl while it stimulates that of CYP3. The other class, represented by cypl-23 or the related allele hapl-1, reduces the expression of both CYCl and CYP3 genes. Genetic data suggested that the CYPl product is a positive regulator of the cytochrome c genes.
The CYPl -18 allele has been cloned. We show here that the iso overproducer function of CYPl-18 is included in a 5300 base XhoI-PstI fragment. The sequence of this fragment reveals a unique, long, uninterrupted open reading frame of 4449 nucleotides able to encode a protein of 1483 amino acid residues. The predicted product of this open reading frame contains several interesting features. The N-terminal part of the protein resembles a nucleic acid-binding domain, in which two domains can be distinguished. The first is similar to a “finger” DNA binding motif, as found in TFIIIA and other regulatory proteins. The second consists of seven tandemly repeated sequences with a KCPVDH motif. Because of its structure, it is tempting to speculate that this region may act as a “redox sensor” folded around a metal atom or heme and involved in recognition of respiratory effecters. These two domains are separated by an “opa” sequence of 13 Gln residues. Implication of these domains for the function of CYPl-18 is discussed.
1. Introduction
The yeast Saccharomyces cerevisiae is a faculta- tive aerobe and the presence/absence of oxygen controls its overall cell physiology and the synthesis of many different enzymes. The apoproteins of several cytochromes and other respiratory enzymes are not synthesized under anaerobic conditions and their formation is induced by oxygen and heme (Ephrussi & Slonimski, 1950; Slonimski, 1955; Guarente & Mason, 1983).
Sherman et al., 1968; Verdi&e & Petrochilo, 1975; Downie et al., 1977; Smith et al., 1979; Montgomery et al., 1980). The expression of the two genes is differently regulated in the wild-type yeast: isol- and iso2cytochrome c account for 95 y0 and 5%, respectively, of total cellular cytochrome c and the derepression of CYCl transcription but not of CYP3 is dependent on oxygen and intracellular level of heme (Slonimski et al., 1965; Fukuhara, 1966; Guarente & Mason, 1983; Laz et al., 1984).
The regulation of cytochrome c synthesis has Catabolite derepression and oxygen/heme been used as a model system in this respect for activation of CYCl occurs through an “upstream several reasons. Two distinct molecular species of activation site” or UAS located in the 5’ non-coding this protein are found in a yeast cell; they are region of the gene. This regulatory region consists isofunctional in spite of differences in their primary of two subsites, UASl and UAS2, which are sequence and are encoded by independent genes, independent functional units and the transcription CYCl for isol-cytochrome c and CYP3 (also called dependent on UASl is derepressed by increasing CYC7) for iso2cytochrome c (Slonimski et al., 1965; the levels of intracellular heme during growth on
0022-2836/88/220263-14 $03.00/O 263
0 1988 Academic Press Limited

264 I? Creusot et al.
glucose (Guarente et al., 1984). Iso2-cytochrome c also possesses a regulatory region in the upstream 5’ non-coding sequence of the gene. This region contains both negative and positive regulatory sites and derepressed expression of CYP3 would be the result of antagonistic effects of these sites (Verdi&e & P&trochilo, 1979; Wright & Zitomer, 1984, 1985).
Genetic control of the synthesis of isocyto- chromes c and other respiratory enzymes has been discovered by the analysis of several independent genes that act in trans and modulate the expression of CYCI and CYP3 (Clavilier et al., 1969, 1976; Rothstein & Sherman, 1980). Among them, the gene CYPI is of particular interest. An intriguing class of mutations that have opposite effects on CYCI and CYP3 expression have been isolated from this locus. Such alleles, as CYPI-18, activate, at the transcriptional level, the expression of CYP3 while reducing that of CYCI. These alleles are referred to as the iso2-overproducer alleles (Clavilier et al., 1969, 1976; Montgomery et al., 1982). A second class of alleles of CYPI has been obtained that reduces both isol- and iso2-cytochrome e synthesis; these are the iso2-underproducer alleles represented by cypl-23 (Verdi&e $ Petrochilo, 1979). An indepen- dently isolated hapl-l mutation has been shown by complementation and recombination to belong to the CYPI locus. Thus CYPI and HAP1 are one and the same gene (Verdi&e et al., 1986).
Genetic data led to the proposal that the CYPl+ product would be a strong activator of CYCI and a weak activator of CYPS; CYPI-18 product would efficiently activate CYP3 and be inactive on CYCI, while the cypl-23 allele would have no effect on either CYCI or CYP3 genes (Verdi&e & Petrochilo, 1979). Consistent with that interpretation are the results of Pfeifer et al. (1987), who defined precisely the sequences bound by the CYPI’ product on the CYCI and CYP3 genes and thus uncovered the fact that the upstream target sequences present on these two cytochrome c genes show no obvious similarity. An interesting question is how one protein recognizes two different sequences in such a way that the activating function can be heme and/or oxygen-dependent for one of them (CYCI) and not for the other (CYP3).
In an approach towards understanding this regulatory system, we decided to determine the sequence of the CYPI gene. We have reported the isolation from a cosmid bank of the overproducer allele CYPI-18 (Verdi&e et al., 1985). This allele was chosen by virtue of its ability to convert an isol-cytochrome c deleted (and therefore lactate-) strain to a lactate+ phenotype. Here, we have delimited further the functional CYPI-18 gene to a shorter DNA fragment, which was subsequently sequenced. We show that CYPI-18 can encode a polypeptide chain of 164,000 Da with several interesting structural features which, we believe, are directly related to its activity as a master regulator of oxido-reduction-dependent gene expression.
In the accompanying paper (Verdikre et al.,
1988), we determine the structure of the wild-type CYPl+ gene. The comparison of the two sequences suggests a molecular model responsible for the regulatory characteristics of the CYPI protein.
2. Materials and Methods
(a) Yeast strains
All the following strains were constructed in our laboratory. VP209-7B (alpha leu2 ura3 cycl-I), FJl-32A (alpha trpl-1 ura3 cycl-1 CYPl-18), FJl-2C (alpha trpl-1 ura3 cycl-1), FJl-9B (a leu2 ura3 cycl-1 CYPl-18) and FJ21-1D (alpha trpl-1 ura3 cycl-1 cypl-m). In this last strain, the allele cypl-m cannot be stated unambiguously (see Verdi&e et aZ., 1986). This segregent is issued from a cross in which one of the parents carried the underproducer allele cypl-23 and the other the underproducer allele hapl-1 from strain BWG-7a hapl-1. As the 2 alleles have the same null phenotype, do not complement and belong to the same CYPl or HAP1 gene (Verdi&e et al., 1986), it is not possible to discriminate between the 2 underproducer alleles in the progeny.
(b) Bacterial strains
Escherichia coli HBlOl (F- Zeu- pro- Zac- gal- Hrs- recA- r- m- str-) were used for DNA cloning and JMlOl (delta lae pro) SupE thi F’ traD36 proAB lacIq ZacZdeltaM15 were used for propagating phages M13.
(c) Media
Yeast were grown on complete, minimum, glycerol or lactate media as described by Verdi&e & Petrochilo (1979). Bacteria were grown on LB or LBA or on 2 x TY medium (1 o/o (w/v) yeast extract, 1.6% (w/v) Bacto- tryptone, 0.5% (w/v) NaCI) for growth of M13. JMlOl were subcloned on minimum M9 medium (Miller, 1972).
(d) Plasmids
Plasmid clone 6 carrying the yeast ura3 marker was described by Bach et al. (1979). pG63-11 is a shuttle vector containing the yeast ura3 marker and the 2 pm origin of replication (Gerbaud et al., 1981). pMHU158 is a shuttle vector construceed by Heusterspreute et al. (1985) containing the leu2 marker and the 2 pm origin of replication. In this plasmid, the leu2 promoter is partially deleted so that the expression of the marker is reduced to 5% of the normal level. This decreases the efficiency of plasmid replication in transformed yeast. To circumvent this problem, we constructed the plasmid YEpUl by inserting the 1.2 kbt Hind111 ura3 fragment of clone 6 into the Hind111 site of pMH158. The integrative plasmid YIp2 was constructed by inserting the internal 0.9 kb NruI-SphI fragment (see Fig. 1) of CYPl-18 into the homologous sites of clone 6.
(e) DNA isolation and genetic engineering techniques
Yeast DNA and bacterial plasmid isolation, DNA fragment purification, restriction digests, ligation and bacterial transformation were carried out, &s described by Verdi&e et al. (1985).
t Abbreviations used: kb, lo3 bases or base-pairs; ORF, open reading frame.

Novel Structural Domains in the CYPl Protein 265
(f) Yeast transformation (k) Analysis of RNA
Replicative or integrative transformations were performed as described (Verdi&e et al., 1985). (Ura3+) transformants were selected on minimum medium supplemented with 60 /Ig leucine/ml or 20 pg tryptophan/ ml. The glycerol or lactate phenotypes of transformants were further tested by growth on glycerol or lactate medium.
RNA was electrophoresed through either 1 “/0 or 0.8% (w/v) agarose gels containing 2.2 M-formaldehyde and was then transferred to nitrocellulose filters as described by Maniatis et al. (1982). Filters were hybridized according to Thomas (1980).
(1) Computer unalyeis
(g) Subcloniny of CYPI-18
YEpJFM2 was digested with the appropriate enzymes (Fig, l(a)), DNA fragments were purified and inserted into the homologous sites of the vectors. The 9 kb BamHTIHindIII fragment (N.6) was subcloned in pG63-11. All the other fragments were cloned into YEpUl. For cloning the 5.3 kb PstI-XhoI fragment (N.3), YEpUl was first cut with XhoI and then partially digested with PstI in order to cut at only 1 of the 2 sites. After ligation of the fragment, recombinant plasmids were selected in E. coli HBlOl and insertion of the yeast fragment at the right location into the polylinker site of the vector was checked by restriction mapping. Subcloning of the 7 kb XbaI-XhoI fragment (N.2) was carried out in 2 steps. First, the 1 kb Xbu-XhoI fragment (N.2.1.) was cloned in the XbaI-XhoI sites of YEpUl and then the 6 kb XbaI fragment (N.2.2) was inserted into the unique XbaI site of the recombinant vector. The right orientation of the 6 kb fragment was checked by restriction analysis.
Sequence data treatments were performed using the computer facilities of CIT12 in Paris on a DPSS computer with the help of the French Minis&e de la Recherche et la Technologie (Programme mobilisateur “Essor des Biotechnologies”).
3. Results
(a) Cloning the C YPl-18 functional allele
The iso2-overproducer allele CY Pl-18 had been
(h) DNA sequencing
Shotgun cloning of the 5.3 kb Z’stI-XhoI fragment (N.3 in Fig. l(b)) was carried out into M13mp18 (Yanisch- Perron et al., 1985). Recombinant clones were selected by transformation of E. coli JMlOl. Clones were sequenced by the dideoxynucleotide chain termination method (Sanger et al., 1977) with the following modifications: DNA was primed with the 17-mer (Boehringer- Mannheim), chain termination reactions were performed with [%]dATP at 37°C for 25 min, fragments were separated on 6% (w/v) polyacrylamide gels. Clones giving band compression were resolved by using deoxyinosine instead of deoxyguanosine (Mills & Kramer, 1979). The entire sequence was determined on both strands,
(i) Preparation of RNA
Cells from strains FJl-2C (CYPl+) FJl-32A (CYPl-18) were grown on media containing 1 o/o (w/v) yeast extract, 1 y. (w/v) Bactopeptone and 2% (w/v) glucose to an A,,, of 2 (1 or 2 generations before stationary phase). Total RNA was prepared by the method of Maccechini et al. (1979), except that the LiCl precipitation step was omitted. Poly(A)+ RNA was purified on oligo(dT)- cellulose as described by Fraser (1975).
cloned by taking advantage of its ability to restore a lactate+ (let+) phenotype to an isol-cytochrome c dificient, thus (let-), yeast strain. This allele was first isolated as a rearranged DNA fragment inserted in plasmid YEpJFMl and subsequently in a non-rearranged BamHI genomic fragment of 23 kb in YEpJFM2 (Verdi&e et al., 1985). This insert was subcloned further in order to delineate the functional fragment(s). Comparison of DNA inserted in YEpJFMl and YEpJFM2 showed that the sequence located between the Hind111 and the XhoI sites at positions 14.5 and 19 kb, respectively (Fig. l(a)), displayed an identical restriction pattern. This observation suggested that the CYPl-18 function should lie in this region and prompted us to investigate more closely this part of the inserted DNA. The fragments shown in Figure l(a) were cloned in pG63-11 or in YEpUl (see Materials and Methods). Recombinant plasmids were used for transformation of a (let-) yeast strain; (ura3+) transformants were then screened for the CYPl-18 function by checking comple- mentation of the (let-) phenotype. As shown in Figure l(a), the fragments N.l (XhoI, 15 kb), N.2 (XbaI-XhoI, 7.5 kb) and N.3 (PstI-XhoI, 5.3 kb) were able to restore growth on lactate medium. None of the other fragments was able to do so; in particular, fragments N.5 and N.6 lacking about 1 kb on the right and left sides of the 5.3 kb PstI- XhoI fragment, respectively, did not have the CYPl-18 function. These results indicate that the 5.3 kb PstI-XhoI fragment is the shortest DNA sequence still carrying the complete CYPl-18 function. The recombinant plasmid carrying this insert is referred to as YEpJFM3.
( j) Preparation of radioactive probes
Double-stranded probes were nick-translated to specific activities of 5 x 10’ to 1 X 10’ cts/min per pg. Single- stranded probes were radiolabelled to approx. the same specific activity by performing DNA sequencing reactions (see above), except that no dideoxynucleotides were used and that [cr-35S]dATP was replaced by [cr-32P]dATP. The labelled DNA corresponds to the transcribed strand.
(b) Inactivation by disruption of the CYPZ gene
Different alleles of CYPl display various pheno- types with respect to cytochrome c synthesis (Clavilier et al., 1976; Verdi&e & Petrochilo, 1979). It was therefore important to check whether their inactivation leads to a common phenotype corre- sponding to the loss of all functions. Inactivation

266 F. Creusot et al.
6
5
4 ., u
2.1 ~2.r
1 :
+
s x Xb PH s Xb x
t t t ttt t t a t
I I ./ 1 I \
(b)
I ORF I
5’ 3
0.5 Kb -
Figure 1. Cloning and restriction map of the CYPl-18 region. The genomic BamHI fragment of 23 kb inserted in the plasmid YEpJFM2 (thick line) was subcloned into 7 replicative plasmids ((a), numbered 1 to 7) which were used to transform a let-) strain in order to determine the CYPl-18 function. The shortest fragment (subclone 3, PstI-XhoI fragment, 5.3 kb long), which restores growth on lactate is shown enlarged in (b). The internal SphI-NruI gene fragment cloned into the integrative plasmid YIp2, is indicated above the restriction map of the 5.3 kb fragment. The dotted lines with the numbers indicate the single-stranded DNA fragments cloned in Ml3 and used in Northern blot analysis (Fig. 5). The CYPl-18 ORF is shown under the restriction map. Abbreviations: B, BarnHI; X, XhoI; S, S&I; Bg, BgZII; Xb, XbaI; P, P&I; H, HindIII; Sp, SphI; R, EcoRI; N. NruI; Sa, SacI; and K, KpnI.
by disruption was achieved by inserting in the CYPl locus the non-replicative plasmid YIp2 by integration directed by double-strand break repair (Orr-Weaver et al., 1981) after linearization in the unique Sal1 site of the yeast insert (Fig. l(b)). CYPl+, CYPl-18 and cypl-m haploid strains (see Materials and Methods) were transformed with the linearized plasmid and (ura3+) transformants were selected. Integration into yeast DNA at the right location was checked by controlling mitotic stability of the (ura3+) phenotype and by Southern blot analysis of yeast DNA probed with 32P- labelled pBR322 (not shown). Table 1 reveals that disruption of any allele of CYPl prevents in an isol -cytochrome c-deficient (cycl-1 ) strain the growth on lactate as sole carbon source, and dramatically reduces growth on glycerol medium but allows normal growth on glucose. So, CYPl does not control any function essential for cell life. Analysis of cytochromes performed by low- temperature spectroscopy as described by Verdi&e et al. (1986) confirmed that all the disrupted
transformants behaved as an iso2- underproducer cypl-23 strain and did not synthesize any detect- able amount of cytochrome c. The fact that the null
Alleles
Table 1 Disruption of the C YYl locus
Media
GlUCO~ Glycerol Lactate
CYPl+ + + -
(‘YPI :: URA3 + +/- -
CYPI-1x f + + CYPI-18::URA3 + +/- -
cypl -In + +/- - (!YPl-m :: URA3 + +/- -
cycl-1-ura3- yeast strains carrying the CYPl+ (VP209-7B), the overproducer CYPl-18 (FJI-9B) or the underproducer cypl-m (FJZl-1D) allele were transformed with the integrative plasmid YIp2 linearized at the unique Sal1 site in the yeast insert. (ura3+) transformants were selected and tested for growth on complete, glycerol and lactate media.

Novel Structural Domains in the CYPl Protein 267
disruption or in vivo mutagenesis) have the same negative phenotype agrees well with the conclusion that both CYPl+ and CYPl-18 act as positive regulatory loci of iso-cytochrome c synthesis.
alleles of CYPI (whether obtained by gene (c) Sequence of the CYPl-18 allele
Both strands of the entire 5.3 kb P&I-XhoI fragment were sequenced by the dideoxynucleotide chain termination method (Sanger et al.. 1977). The sequence is shown in Figure 2. The sequence
1
61
121
181
241 300
7 301
27 361
47 421
67 481
87 541
107 601
127 661
147 721
167 781
187 841
207 901
227 961
247 1021
267 1081
287 1141
307 1201
327 1261
347 1321
367 1381
CTGCAGTGCTCTTTTAAGGTTTGAAGATATTGCACAAAATTGTATCTGATTTCATCTCAT
CGAACCTCATTCCATCAACTTCCTTTTATCAAAGCATCTTGTTTTGTTTTCGCAGAAAAT .
TTTTCTTCCCTTTCAATTTGGCCTTTTAGTTTTTCTCCAGTCGCAGGCAAGAAGGTAAG~
AAATAGAAGAAAAAGAAAAAAAAAAAAAGGGAACAATAGGTTAGATGTCGAATACCCCTT 1MSNTPY
ATAATTCATCTGTGCCTTCCATTGCATCCATGACCCAGTCTTCGGTCTCAAGAAGTCCTA NSSVPSIASUTQSS VSRSPN
ACATGCATACAGCAACTACGCCCGGTGCCAACACCAGCTCTAACTCTCCACCCTTGCACA N HTATTPGANTSSNSPPLHM
TGTCTTCAGATTCGTCCAAGATCAAGAGGAAGCGTAACAGAATTCCGCTCAGGTGCACCA SSDSSKI KRKRNR IPLRCTI
TTTGTCGGAAAAGGAAAGTCAAATGTGACAAACTCAGACCACACTGCCAGCAGTGCACTA CRKRKVKCDKLR P H C Q Q C T K
AAACTGGGG;AGCCCATCTCTGCCACTACATGGAACAGACCTGGGCAGA~GAGGCAGAGA T G V A Ii LCHYMEQTWAEEAEK
. AAGAATTGCTGAAGGACAACGAATTAAAGAAGCTTAGGGAGCGCGTAAAATCTTTAGAAA
ELLKDNELKKLRERV K S L E K . .
AGACTCTTTCTAAGGTGCACTCTTCTCCTTCGTCTAACTCCTTGAAAAGTTACAACACTC TLSKVHSSPSSNSLKSYNTP
CCGAGAGCAGCAACCTGTTTATGGGTAGCGATGAACACACCACCCTTGTTAATGCAAATA ESSNLFMGSDEHTTLVNANT
CAGGCTCTGCTTCCTCTGCCTCGCATATGCATCAGCAACAACAGCAACAGCAGCAACAG~ GSASSASHHHQQQQQQQQQE
AACAACAACAAGACTTTTCCAGAAGTGCGAACGCCAACGCGAATTCCTC~TCCCTTTCT~ QQQDFSRSANANANSSSLSI
. . TCTCAAATAAATATGACAACGATGAGCTGGACTTAACTAAGGACTTTGATCTTTTGCATA
S NKYDNDELDLTKDFDLLHI
TCAAAAGTAACGGAACCATCCACTTAGGTGCCACCCACTGGTTGTCTATCATGAAAGGT~ KSNGTI HLGATHWLS I Y K G D
ACCCGTACC;AAAACTTTTGTGGGGTCATATCTTCGCTATGAGGGAAAAGTTAAATGAA~ P Y L K LLWGHIFAMREKLNEW
GGTACTACCAAAAAAATTCGTACTCTAAGCTGAAGTCAAGCAAATGTCCCATCAATCAC~ Y Y Q K N S Y S K L K S S K C P I N H A
. CGCAAGCGCCGCCTTCTGCCGCTGCCGCCGCTACCAGAAAATGTCCTGTTGATCACTCC~
QAPPSAAAAATRKCPVDHSA .
CGTTTT~GTCTGG~ATGGT~G~~~~AAAGGAGGAGA~T~~T~TT~~TAGGAAATGTCCA~ FSSGMVA PKEETPL PRKCPV
TTGACCACACCATGTTCTCTTCGGGAATGATTCCTCCCAGAGAGGACACTTCGTCCCAG~ DHTMFSSGMIPPREDTSSQK
AGAGGTGTCCCGTTGACCACACCATGTATTCCGCAGGAATGATGCCGCCCAAGGACGAGA R CPVDHTM Y S A G H M P P K D E T
CACCTTCCCCATTTTCCACTAAAGCTATGATAGACCATAACAAGCATACAATGAATCCG~ P S P F S T K A M I D H N K HTMNPP
Fig. 2.
60
120
180
240
360
420
480
540
600
660
720
700
840
900
960
1020
1080
1140
1200
1260
1320
1380
1440

268 F. Creusot et al.
387 1441
407 1501
427 1561
447 1621
467 1681
487 1741
507 1801
527 1861
547 1921
567 1981
587 2041
607
2101
627 2161
647 2221
667 2281
687
2341
707 2401
727 2461
747 2521
767 2581
787 2641
807 2701
827 2761
847 2821
867 2881
CTCAGTCAAAATGTCCTGTGGACCATAGAAACTATATGAAGGATTATCCCTCTGACATGG QSKCPVDHRNYMKDYPSDHA
CAAATTCTTCTTCGAACCCGGCAAGTCGT;GCCCCATTGACCATTCAAG~ATGAAAAAT~ N S S S N P A S R c P I D H S S U K N T
CAGCGGCCT;ACCAGCTTCAACGCACAAT~CCATCCCACACCACCAACC~CAGTCCGGA~ AALPAS T Ii N T I p H H Q P Q S G S
CTCATGCTCGTTCGCATCCCGCACAAAGCAOGAAACATGATTCCTACAT~ACAGAATCT~ HARSHPAQSRK HDSYMTESE
AAGTCCTCGCAACACTTTG;GAGATGTTGCCACCAAAGCGCGTCATCGC~TTATTCATC~ V LATLCEHLPP K R V I A L F I E
AGAAATTCTTCAAACATTTATACCCTGCCATTCCAATCTTAGATGAACAGAATTTCAAAA KFFKHLYPAIPI L D E Q N F K N
ATCACGTGAATCAAATGCTTTCGTTGTCTTCGATGAATCCCACAGTTAACAACTTTGGTA H V N 0 M L S L S SHNPTVNN F G W
TGAGCATGCCATCTTCATCTACACTAGAGAACCAACCCATAACACAAATCAATCTTCCAA SMPSSSTLENQPITQIN L P K
AACTTTCCGATTCTTGTAACTTAGGTATT~TGATAATAA;CTTGAGATT~ACATGGCTA~ LSDSCNLGILIII LRLTWLS
CCATACCTTCTAATTCCTGCGAAGTCGACCTGGGAGAAGAAAGTGGCTC~TTTTTAGTG~ IPSNSCEVDLGEESGSFLVP
CCAACGAATCTAGCAATATGTCTGCATCTGCATTGACCT~GATGGCTAA~GAAGAATCA~ NESSNMSASALTSMAKEESL
. TTCTGCTAAAGCATGAGACACCGGTCGAGGCACTGGAGCTATGTCAAAA~TACTTGATT~
LLKHETPVEALELCQ K Y L I K
AATTCGATGAACTTTCTAGTATTTCCAATAACAACGTTAATTTAACCAC~GTGCAGTTT~ FDELSSI SNNNVNLTTVQFA
CCATTTTTTACAACTTCTATATGAAAAGTGCCTCTAATGATTTGACTACCTTGACAAATA I FYNFYMKSASNDLTTLTHT
. CCAACAACACTGGCATGGCCAATCCTGGTCACGATTCCGAGTCTCACCAGATCCTATTGT
N N TGHANPGHDSESHQILLS .
CCAATATTACTCAAATGGCCTTTAGTTGTGGGTTACACAGAGACCCTGATAATTTTCCTC N ITQUAFSCGLHRDPDNFPQ
. AATTAAACGCTACCATTCCAGCAACCAGCCAGGACGTGTCTAACAACGGGAGCAAAAAGG
L N A T IPATSQDVSWNGSKKA
CAAACCCTAGCACCAATCCAACTTTGAATAACAACATGTCTGCTGCCACTACCAACAGCA NPSTNPTLNNNMSAATTNSS
. GTAGCAGATCTGGCAGTGCTGATTCAAGAAGTGGTTCTAACCCTGTGAACAAGAAGGAAA
SRSGSADSRSGSNPVNKKEN . . . . . .
ATCAGGTTAGTATCGAAAGATTTAAACACACTTGGAGGAAAATTTGGTATTACATTGTTA QVSIERFKHTWRKIWYYIVS
. GCATGGATG;TAACCAATC;CTTTCCCTGGGGAGCCCTCGACTACTAAGAAATCTGAGGG
MDVNQSLSLGS PRLLRN L R D . .
ATTTCAGCGATACAAAGCTACCAAGTGCGTCAAGGATTGATTATGTTCGCGATATCAAAG FSDTKLPSASRI DYVRDIKE
. AGTTAATCATTGTGAAGAATTTTACTCTTTTTTTCCAAATTGATTTGTGTATTATTGCTG
L I I v K N F T L F F Q I DLCIIAV
TATTAAATCACATTTTGAA;GTTTCTTTAGCAAGAAGCGTGAGAAAATTTGAACTGGATT L N H ILWVSLARSVRKFELDS
CATTGATTAATTTATTGAAAAATCTGACCTATGGTACTGAGAATGTCAATGATGTAGTGA L I NLLKNLTYGTENVNDVVS
Fig. 2.
1500
1560
1620
1680
1740
1800
1860
1920
1980
2040
2100
2160
2220
2280
2340
2400
2460
2520
2580
2640
2700
2760
2820
2880
2940

Novel Structural Domains in the CYPl Protein 269
887 2941
907 3001
927 3061
947 3121
967 3181
987 3241
1007 3301
1027 3361
1047 3421
1067 3481
1087 3541
1107 3601
1127 3661
1147 3721
1167 3781
1187 3841
1207 3901
1227 3961
1247 4021
1267 4081
1287 4141
1307 4201
1327 4261
1347 4321
1367 4381
GCTCCTTGATCAACAAAGGGTTATTACCAACTTCGGAAGGTGGTTCTGTAGATTCAAATA s L I NKGLLPTSEGGSVD S N N
ATGATGAAATTTACGGTCTACCGAAACTACCCGATATTCTAAACCATGGTCAACATAACC D E I Y G L P K L P D I L N H G Q Ii N 0
. . . AAAACTTGTATGCTGATGGAAGAAATACTTCTAGTAGTGATATAGATAAGAAATTGGACC
NLYADGR NTSSSD IDKKLDL .
TTCCTCACGAATCTACAACGAGAGCTCTATTCTTTTCCAAGCATATGACAATTAGAATGT PHESTTRALF FSKHMT I R w L
. . TGCTGTACTTATTGAACTACATTTTGTTTACTCATTATGAACCAATGGGCAGTGAAGATC
LYLLNY ILFTHYEPMGSEDP . .
CTGGTACTAATATCCTAGCTAAGGAGTACGCTCAAGAGGCATTAAATTTTGCCATGGATG G T N I LAKEYAQEALNFAMDG
. . GCTACAGAAACTGCATGATTTTCTTCAACAATATCAGAAACACCAATTCACTATTCGATT
YRNCMI F F N N I R N T N S L F D Y
ACATGAATGTTATCTTGTCTTACCCTTGTTTGGACATTGGACATCGTTCTTTACAATTTA I4 N V I L S Y P C L D I GHRSLQFI
. . . . TCGTTTGTTTGATCCTGAGAGCTAAATGTGGCCCATTGACTGGTATGCGTGAATCATCGA
V c L I L R A K CGPLTGMR E S S I
TCATTACTAATGGTACATCAAGTGGATTTAATAGTTCGGTAGAAGATGAGGACGTCAAAG I TNGTSSGFNSSVEDEDVKV
. . . TTAAACAAGAATCTTCTGATGAATTGAAAAAAGACGATTTCATGAAAGATGTAAATTTGG
K Q ESSDELKKDDFMKDVNLD
ATTCAGGCGATTCATTAGCAGAGATTCTAATGTCAAGAATGCTGCTATTTCAAAAACTA~ S G D S L A E I LMSRMLLFQKLT
CAAAACAACTATCAAAGAAGTACAACTACGCTATTCGTATGAACAAATCCACTGGATTCT K Q L S K K Y N Y A I R Y NKSTGFF
TTGTCTCTTTACTAGATACACCTTCAAAGAAATCAGACTCGAAATCGGGTGGTAGTTCA~ v s LLDTPSK KSDSKSGGSSF
TCATGTTGGGTAATTGGAAACATCCAAAGGTTTCAAACATGAGCGGATTTCTTGCTGGTG N L G NWKHPKVSNMSGFLAGD
ACAAAGACCAATTACAGAAATGCCCCGTGTACCAAGATGCGCTGGGGTT~GTTAGTCCA~ K DQLQKCP v Y a DALGFVSPT
. . . CCGGTGCTAATGAAGGTTCTGCTCCGATGCAAGGCATGTCCTTACAGGGCTCTACTGCTA
GANEGSAPMQGMSLOGSTAR . . .
GGATGGGAGGGACCCAGTTGCCACCAATTAGATCATACAAACCTATCACGTACACAAGTA M G G T Q L P P I RSYKPITYTSS
. . . . GTAATCTACGTCGTATGAATGAAACGGGTGAGGCAGAAGCTAAGAGAAGAAGATTTAATG
NLRRMNETGEAEAKRRRFND . .
ATGGCTATATTGATAATAATAGTAACAACGATATACCTAGAGGAATCAGCCCAAAACCT~ GYIDNNSNNDIPRGISPKPS
CAAATGGGCTATCATCGGTGCAGCCACTA;TATCGTCAT;TTCCATGAA~CAGCTAAAC~ NGLSSVOPLLSSFSWIIQLNG
. . GGGGTACCATTCCAACGGTTCCATCGTTAACCAACATTACTTCACAAATGGGAGCTTTA~
G T I PTVPSLTN ITSQMGALP . . . .
CATCTTTAGATAGGATCACCACTAATCAAATAAATTTGCCAGACCCATCTAGAGATGAAG S L D R ITTNPINLPDPSRDEA
. . . . . CATTTGACAACTCCATCAAGCAAATGACGCCTATGACAAGTGCATTCATGAATGCTAATA
F D N S I K Q M T P N T S A F I4 N A I T . . . . .
CTACAATTCCAAGTTCAACTTTAAACGGGAATATGAACATGAATGGAGCTGGAACTGCGA T I P S S T L NGNMNMN GAGTAN
.
Fig. 2.
3000
3060
3120
3180
3240
3300
3360
3420
3480
3540
3600
3660
3720
3780
3840
3900
3960
4020
4080
4140
4200
4260
4320
4380
4440

270 F. Creusot et al.
1387 4441
1407 4501
1427 4561
1447 4621
1467 4681
4741
4801
4861
4921
4981
5041
5101
5161
5221
ATACAGATACAAGTGCCAACGGCAGTGCTTTATCGACACTGACAAGCCCACAAGGCTCAG TDTSANGSALSTLTSPOGSD
. ACTTAGCATCCAATTCTGCTACACAGTATAAACCTGACTTAGAAGACTTTTTGATGCAAA
LASNSATQYKPDLEDFLMQN
ATTCTAACTTTAATGGGCTAATGATAAATCCTTCCAGTC;GGTAGAAGT~GTTGGTGGA~ SNFNGLMI NPSSLVEVVGGY
. . ACAACGATCCTAATAACCTTGGAAGAAATGACGCGGTTGATTTTCTACCCGTTGATAATG
NDPNNLGRNDAVDFLPVDNV
TTGAAATTGATGGACTAGTAGATTTTTATAGAGCAGATTTTCCAATATGGGAGTGATGCG E I DGLVDFYRADFPIWE l 1483
GTTTGTTTATGTATAAATAGAAAGGGACAIATTTTTATTATTATTATTT~AGTTATTTA~ . . .
CATGTATGTTATTTTATCGGTGAATTGTAATGATCGAATAATAATTGGAGGTTGAAGAAG . . .
TTGCAAAAAATATTTACTTGGACGAGTCCGGAATCGAACCGGAGACCTCTCCCATGCTAA . .
GGGAGCGCGCTACCGACTACGCCACACGCCCAATTTTCTTGTTGTCTGTTGTGTTTCAGA .
ATGGGGTACGTCAGTATGATAATAATCATCCTAAACGTTCTTAAATTACATATGAAACAA .
CCTTATAGCAAAACGAACAGAATGAGCAACATGAGATGAAACTCCGCCTCCTTAACTGAA .
CTTTCCAAACGTATAAACGCCTGAAAAATTAGTTTAGATCCGAGATTCCGCGCTTCCACC .
ATCAAGTATGATCCATATTTTATATAATATATAAGATAAGTAACATTCCGTAAGCTGATA .
ATCCCTTTTGGCAACTCGTTACTTCCCAAAGACTGTTTATATTAGGATTGTCAAGGGACC . 5235
CCGGTATTGCTCGAC
4500
4560
4620
4680
4740
4800
4860
4920
4980
5040
5100
5160
5220
Figure 2. Nucleotide sequence and predicted amino acid sequence of the yeast CYPl-18 gene. The complete DNA sequence of the 5.3 kb coding strand from the 5’ P&I site to the 3’ X/WI site is presented. The supposed TATA boxes, CT block and CAAG box and termination TA(T)GT - - - - -TTT sequences are underlined. The predicted amino acid sequence, in the l-letter code, is shown below the DNA sequence. Upper and lower numbers before each line refer to the nucleotide and amino acid positions, respectively. The coding strand is the non-transcribed DNA strand identical with the mRNA. On the opposite strand we have located (Creusot et al., unpublished results) the structural gene of the alanine tRNA (positions 4819 to 4881) adjacent to a sigma element (from position 4997).
displays only one large continuous open reading frame (ORF) starting with an ATG at position 225 and ending with a TGA at position 4674. The two remaining reading frames on the same strand and the three registers on the opposite strand contain 61 to 148 stop codons and could not code for a protein longer than a few dozen amino acid residues. The fact that no TACTAAC element, indicative of a possible intron (Langford & Gallwitz, 1983; Pikielny et al., 1983), is found in the entire sequence strongly suggests that the ORF is not interrupted and can encode a large protein of 1483 amino acid residues with a predicted molecular weight of about 164,220.
(i) 5’ Upstream and 3’ downstream regions
In higher eukaryotes, TATA box sequences required for proper transcription initiation are found around 30 nucleotides upstream from the mRNA start sites (Gannon et al., 1979). In yeast, most but not all genes have TATA boxes. They usually are located further upstream and can be found as far as 150 to 200 nucleotides from the mRNA start sites (Gallwitz et aZ., 1981; Hitzeman et
al., 1981). In the CYPl-18 allele, no canonical TATA box is evident in the region upstream from
the ORF. However, a related sequence, TATCAAA located at position 87, might be an adequate TATA box. This sequence appears to be homologous to that of the MAT alpha2 promoter (Astell et al.,
1981) and is followed at position 120 to 158 by a CT block and at 168 by a CAAG box, sequences frequently found in yeast promoters (Dobson et al.,
1982). Although these sequences are generally associated with highly expressed genes, they are found in poorly expressed genes, as iso2-cyto- chrome c (Montgomery et al., 1980). A remarkable feature in the promoter region is the presence from position 169 to 213 of a long, nearly perfect homopurine stretch containing about 70% adenine. Such sequences are found in a similar location in the promoter of ADRl, a yeast regulatory protein of alcohol dehydrogenase (Hartshorne et al., 1986) and ARGRII a yeast regulatory gene of arginine metabolism (Messinguy et al., 1986). Another candidate for transcription initiation could be a TATA box present at position 240, which is located within the first seven codons of the ORF.
The sequence closest to the consensus for transcription termination (Zaret & Sherman, 1982) occurs within the first 100 nucleotides downstream from the TGA codon at 4674. It consists of three

Novel Structural Domains in the CYPl Protein 271
possible TA(T)GT sequences located at positions 4688, 4730 and 4745 and an A+T-rich region followed by TTT at position 4752. As is usual in yeast genes, there is no evident polyadenylation consensus signal AATAAA in the 562 nucleotides of the 3’ untranslated side of the gene.
detected in the structure of the CYPl-18 protein. We shall analyse them successively from the N to the C terminus.
Downstream and on the opposite strand we have located, by sequence comparisons, the structural gene of the alanine tRNA and a sigma-like element (positions 4819 and 4907, respectively; Fig. 2). This will be discussed elsewhere (Creusot et al., unpublished results).
(ii) The protein coohg region
In eukaryotes, translation is usually initiated at the most 5’ upstream ATG of the mRNA (Kozak, 1981). Even though the CYPl-I8 ORF starts with a methionine residue at position 225, a more favourable initiation codon might be the third ATG at amino acid position 27, which completely fits the consensus sequence AXXATGXXT described for yeast genes (Dobson et al., 1982). Translation of the putative protein involves all the 61 codons at least once with a bias index of 0.13. According to Bennetzen et al. (1982), codon usage in mRNA translation would be correlated to the level of expression. Although the bias index of CYPl-18 is higher than that of other little-expressed regulatory proteins, such as PPRl (Kammerer et al., 1984), PH04 (Legrain et al., 1986) or GAL4 (Laughon & Gesteland, 1984), it is similar to other regulatory proteins like ARGRTT (Messinguy et al., 1986) and ADRI (Hartshorne et al., 1986), which have an index of 0.07, and it is still indicative of a low level of expression of CYPI-18. This is again consistent with the idea of CYPl being a regulatory locus and expected not to be expressed at a high level.
The first sequence consists of a stretch of 21 amino acid residues (residues 64 to 84; Fig. 2) bordered by two C-X2-C motifs. This domain displays a high level of structural homology to the repeated units of the DNA-binding “fingers” found in the TFIIIA factor involved in the 5 S RNA gene regulation of Xenopus laevis (Miller et al., 1985) and several other regulatory proteins (see Berg, 1986; Klug & Rhodes, 1987). Figure 3 shows a com- parison of the putative DNA-binding “finger-like” region of CYPl-18 protein with six other yeast regulatory proteins; GAL4 (Laughon & Gesteland, 1984), PPRl (Kammerer et al., 1984), ARGRII (Messinguy et al., 1986), LAC9 (Breunig & Kuger, 1987), PDRl (Balzi et al., 1987) and LEU3 (Friden & Schimmel, 1987). A high level of sequence homology is evident. Besides the conserved Cys in positions corresponding to 64, 67, 81 and 84 of CYPl, three other residues of the finger are highly conserved: Arg68, Lys71 and Pro79. Homology with TFIIIA and ADRI appears to be more restricted to the C-X,-C motifs. In addition, several different proteins have been shown to contain this structural domain as, for example, the gag proteins of retroviruses, the coat protein of the double- stranded DNA cauliflower mosaic virus (Covey, 1986), the developmental genes of Drosophila (Vincent, 1986), the human glucocorticoid receptors (Weinberger et al., 1985) and, more recently, human transcription factor Spl (Kadonaga et al., 1987) and the sex-determining region of the Y chromosome (Page et al., 1987). The overall lengths of the putative finger part are similar in all these proteins, and range from 11 to 16 residues.
The CYPl-18 protein is highly hydrophilic, with The next striking feature of the N-terminal a total of 12.5% and 9.5% of basic and acidic region of the protein is the presence of an residues, respectively. Charged amino acids are not uninterrupted stretch of 12 glutamine residues and dispersed randomly throughout the sequence. The one glutamic acid residue (residues 177 to 189; N-terminal region displays two basic domains. The Fig. 2). An homologous sequence, consisting of 31 first, from residues 45 to 147, contains 29 o/o basic glutamine residues in tandem, the OPA sequence and only 12% acidic residues; the second, at was first described by Wharton et al. (1985) in the position 210 to 507, consists of 18 y. basic and 10 y. Notch locus, a gene involved in Drosophila acidic residues. The C-terminal region (from 1388 to neurogenesis and has been observed since in many 1483) is acidic, with 13% acidic and 3% basic other developmental or regulatory genes of different residues. A number of interesting features can be organisms. The function of OPA sequences is
64 67 81
Figure 3. Amino acid sequence of the N-terminal, DNA-binding finger-like domain of the CYPl-18 protein. Numbers on the left side of the sequence refer to the position of the amino acids in the corresponding protein. Homologous regions are boxed. (-), Gap alignment.

272 i? Creusot et al.
unknown. They could act as a “hinge”, separating two structural domains of the protein, a plausible interpretation, since polyglutamine stretches could easily adopt a random coil configuration, but they could be viewed as nucleic acid signals, at the DNA or RNA level, involving regular repetitions of the cytosine-purine-purine motif.
The third region of interest is found between amino acid residues 280 and 438 (Fig. 2). As shown in Figure 4, it contains seven adjacent repeat units in tandem. The most conserved amino acids in these repeats form the KCPVDH motif. A few replace- ments occurring within this motif are of con- servative nature (arginine for lysine, isoleucine for valine, asparagine for aspartic acid). This motif is repeated every 16 to 26 residues. As shown in the right panel of Figure 4, the successive KCPVDH motifs could be joined by chelating a metal, thus forming a series of finger-like domains. Although the main features of this structure are reminiscent of that of the TFIIIA finger domain, differences are observed. The order of the putative metal ligands would be Cys-His/Cys-His instead of Cys-Cys/ His-His in TFIIIA or Cys-Cys/Cys-Cys found in the &-finger motif of CYPl and other regulatory yeast proteins (see Fig. 3). The distance between metal ligands would be three amino acid residues (Pro, Val, Asp) in CYPl-18, rather than a distance of four residues in TFIIIA and two in other yeast regulatory proteins (see Fig. 3). In TFIIIA and in Drosophila proteins, the potential fingers would be connected by relatively short stretches of sequences and would protrude from a linear arrangement (Vincent, 1986), while in CYPl-18 protein the looping out regions are of comparable length on both sides of a linear arrangement (Fig. 4). These
I
II
III
Is!
YlI
Sindbir
(4
1 5 10 15 20 25
looping out regions are particularly rich in alanine, proline and serine residues (44 residues out of 102 are Ala, Pro or Ser, while only 24 are expected on a random basis, the x2 value is 10 for 1 degree of freedom). It has been noted that serine/threonine- rich segments might be sites for phosphorylation (Kadonaga et al., 1987), which could be a potential means for regulation of CYPl activity. Repeats II, III and IV are very well-conserved both in length (24 to 26 residues) and in nature of amino acids, while repeats I, V, VI and VIII have less homology. Moreover, repeat V shows a noteworthy exception, as the KCPVDH motif itself is not totally conserved: alanine and methionine residues substitute the highly conserved cysteine and proline residues, suggesting that this unit might be functionally different. It should be stressed that it is the overall structure and organization of the KCPVDH repeat but not its primary sequence that is comparable to the DNA-binding domains of other regulatory proteins (there is no sequence homology between the segments shown in Figs 3 and 4). In a computer-assisted search, we have found the KCPVDH sequence motif in the non-structural protein NS3 of Sindbis virus, a single-stranded RNA alpha virus (Strauss et al., 1984). The exact function of this protein is unknown but it is believed to be involved in the expression and/or replication of the viral genome. In this protein, the exact KCPVDH motif is present only once and the adjacent amino acid sequence displays some similarity with the repeats of the CYPl-18 protein (Fig. 4).
The C-terminal part of the CYPI-18 protein is particularly acid (the last 163 residues have 9.2% excess of acid over basic amino acids, in contrast to
P
b)
Figure 4. Amino acid sequence of the internal tandemly repeating KCPVDH motif in the CYPl-18 protein. (a) The amino acid positions within the repeats are numbered above the sequence and the repeats from I to VII. Numbers on each side of the repeats refer to the positions of the residues in the CYPl-18 sequence. Homologous regions are boxed: identical amino acids are underlined. The homologous region of the non-structural protein NS3 of the Sindbis virus
(Strauss et al., 1984) is shown below. (b) A hypothetical presentation of the successive repeats, stressing the possibility of forming alternating and looping out domains by chelating a metal. The particularly abundant residues of alanine, proline and serine are circled (44 such residues are present, while 24 are expected on a random basis).
Novel Structural Domains in the GYP1 Protein 273
the overall excess of basic over acid residues in the whole ORF). This part could be involved in the transcriptional activation following the suggestion of Hope & Struhl (1986) and Legrain et al. (1986), that properly located acidic regions may be sufficient for transcription activation.
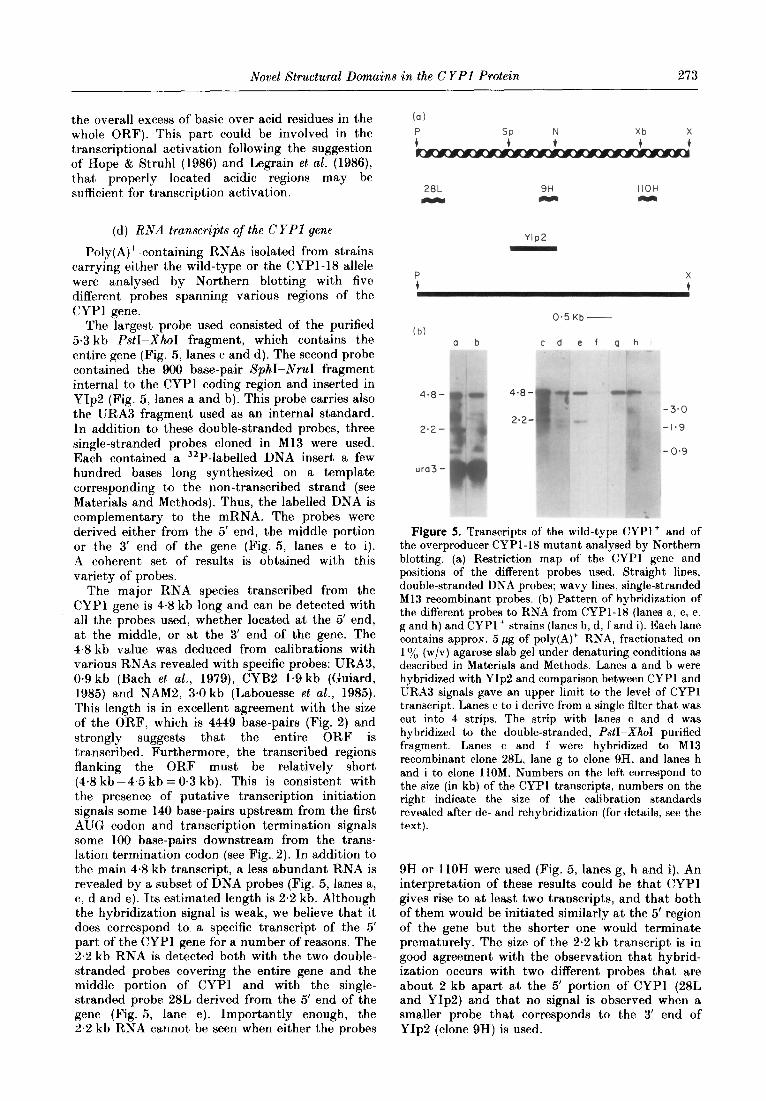
(d) RNA transcripts of the CYPl gene
Poly(A)+-containing RNAs isolated from strains carrying either the wild-type or the CYPl-18 allele were analysed by Northern blotting with five different probes spanning various regions of the CYPl gene.
The largest probe used consisted of the purified 5.3 kb PstI-XhoI fragment, which contains the entire gene (Fig. 5, lanes c and d). The second probe contained the 900 base-pair SphI-NruI fragment internal to the CYPl coding region and inserted in YIp2 (Fig. 5, lanes a and b). This probe carries also the URA3 fragment used as an internal standard. In addition to these double-stranded probes, three single-stranded probes cloned in Ml3 were used. Each contained a 32P-labelled DNA insert a few hundred bases long synthesized on a template corresponding to the non-transcribed strand (see Materials and Methods). Thus, the labelled DNA is complementary to the mRNA. The probes were derived either from the 5’ end, the middle portion or the 3’ end of the gene (Fig. 5, lanes e to i). A coherent set of results is obtained with this variety of probes.
The major RNA species transcribed from the CYPl gene is 48 kb long and can be detected with all the probes used, whether located at the 5’ end, at the middle, or at the 3’ end of the gene. The 4.8 kb value was deduced from calibrations with various RNAs revealed with specific probes: URA3, 0.9 kb (Bach et al., 1979), CYB2 1.9 kb (Guiard, 1985) and NAMZ, 3.0 kb (Labouesse et al., 1985). This length is in excellent agreement with the size of the ORF, which is 4449 base-pairs (Fig. 2) and strongly suggests that the entire ORF is transcribed. Furthermore, the transcribed regions flanking the ORF must be relatively short (4.8 kb -4.5 kb = O-3 kb). This is consistent with the presence of putative transcription initiation signals some 140 base-pairs upstream from the first AUG codon and transcription termination signals some 100 base-pairs downstream from the trans- lation termination codon (see Fig. 2). In addition to the main 4.8 kb transcript, a less abundant RNA is revealed by a subset of DNA probes (Fig. 5, lanes a, c, d and e). Its estimated length is 2.2 kb. Although the hybridization signal is weak, we believe that it does correspond to a specific transcript of the 5’ part of the CYPl gene for a number of reasons. The 2.2 kb RNA is detected both with the two double- stranded probes covering the entire gene and the middle portion of CYPl and with the single- stranded probe 28L derived from the 5’ end of the gene (Fig. 5, lane e). Importantly enough, the 2.2 kb RNA cannot be seen when either the probes
(a)
SP N Xb X
t + t t
28L 9H IIOH
WC, M Y
YIP2
(b) a b cd ef ghi
O-5 Kb -
4*8-
2*2-
ura3 -
Figure 5. Transcripts of the wild-type CYPl+ and of the overproducer CYPl-I8 mutant analysed by Northern blotting. (a) Restriction map of the CYPl gene and positions of the different probes used. Straight lines, double-stranded DNA probes; wavy lines, single-stranded Ml3 recombinant probes. (b) Pattern of hybridization of the different probes to RNA from CYPl-18 (lanes a, c, e, g and h) and CYPl+ strains (lanes b, d, f and i). Each lane contains approx. 5 pg of poly(A)+ RNA, fractionated on 1 o/0 (w/v) agarose slab gel under denaturing conditions as
described in Materials and Methods. Lanes a and b were hybridized with YIp2 and comparison between CYPl and URA3 signals gave an upper limit to the level of CYPl transcript. Lanes c to i derive from a single filter that was cut into 4 strips. The strip with lanes c and d was hybridized to the double-stranded, PatI-XhoI purified fragment. Lanes c and f were hybridized to Ml3 recombinant clone 28L, lane g to clone 9H, and lanes h and i to clone 1lOM. Numbers on the left correspond to the size (in kb) of the CYPl transcripts, numbers on the right indicate the size of the calibration standards revealed after de- and rehybridization (for details, see the text).
9H or 1lOH were used (Fig. 5, lanes g, h and i). An interpretation of these results could be that CYPl gives rise to at least two transcripts, and that both of them would be initiated similarly at the 5’ region of the gene but the shorter one would terminate prematurely. The size of the 2.2 kb transcript is in good agreement with the observation that hybrid- ization occurs with two different probes that are about 2 kb apart at the 5’ portion of CYPl (28L and YIp2) and that no signal is observed when a smaller probe that corresponds to the 3’ end of YIp2 (clone 9H) is used.

274 F. Creusot et al.
4. Discussion
The isolated 5.3 kb PstI-XhoI fragment contains the CYPl-18 function that stimulates the expression of iso2-cytochrome c structural gene CYPS. Disruption of the CYPl locus by integration of a plasmid-borne internal gene fragment, at its homologous chromosomal site, converts the CYPl’ and CYPl-18 “overproducer” alleles to a null allele that has a phenotype indistinguishable from the previously characterized “underproducer” cypl-23 allele. These results strongly support the earlier conclusion that CYPI+ and CYPI-18 are two positive regulatory alleles modulating the synthesis of iso-cytochromes c (Verdi&e & Petrochilo, 1979; Verdi&e et al., 1985).
In this paper, we present the nucleotide and predicted amino acid sequences of the entire 5.3 kb fragment and an analysis of the gene transcripts.
Northern blot analysis reveals the presence of two transcripts approximately 4.8 and 2.2 kb long. The major species is the 4.8 kb transcript. As already discussed, this size agrees well with the size of the ORF present in the CYPI-18 allele. The significance of the 2.2 kb transcript is not clear. It might reflect the existence of another gene strongly homologous to the 5’ part of CYPI or it might, result from a preferential cut in the major transcript. It might also result from premature termination due to the presence of a sequence that resembles the polyadenylation consensus signal TATG . . TTT (Zaret & Sherman, 1982) present at position 1997 and 2080 after the ATG and which would generate transcripts of about 2 kb.
The sequence displays a very large uninterrupted ORF able to encode a putative protein of 164,220 Da. Analysis of the sequence allowed us to identify several domains in the CYPl-18 sequence. An acidic domain localized in the C-terminal part of the protein is reminiscent of the short acidic sequences found in the regulatory proteins GAL4 and GCN4, which have been shown to be sufficient to confer a regulatory activity when fused to a DNA-binding region (Hope & Struhl, 1986; Ma & Ptashne, 1987). The hypothesis that the acidic C-terminal part of the CYPl-18 protein has an activating function is consistent with the observa- tion that the 6 kb XbaI fragment (fragment 5 in Fig. l), in which 145 codons at the C terminus of the ORF are missing failed to complement the lactate- phenotype of the transformed recipient cycl-I CYP3 CYPI strain. Thus, the CYPI-18 protein, which lacks only the C-terminal 10% of its total length, is unable to activate the transcription of the iso2-cytochrome c mRNA. Another argument in favour of the C-terminal region responsible for the activating function is the genetic localization (data not shown) of the null allele cypl-23 at the C-terminal part of the protein.
The most interesting features of the CYPl protein are the two conspicuous domains included in the N-terminal basic region and separated by the OPA-like sequence. The N-terminal part of the protein contains a 28-residue sequence that is
remarkably similar to the DNA binding motif that emerged from the work on TFIIIA (Miller et al., 1985). The classical structure of the repeated unit is based on the tetrahedral co-ordination of the zinc atom by either two cysteine and two histidine residues (abbreviated C-C/H-H), as in the case of TFIIIA, the yeast regulatory gene ADRl and several eukaryotic regulatory proteins (Berg, 1986) or by four cysteine residues (abbreviated C-C/C-C), as in the other known regulatory genes of yeast. Tt is assumed that this structure allows the folding of the motif into a.DNA binding finger. In yeast, the sequence of six regulatory genes PPRI, GAL4, ARGRII, PDRI, LAC9 and LEU3 has revealed the presence of one such finger (Kammerer et al., 1984; Laughon & Gesteland, 1984; Messinguy et al., 1986; Balzi et al., 1987; Breunig & Kuger, 1987; Friden & Schimmel, 1987). As shown in Figure 3, the CYPl protein conforms perfectly to a C-C/C-C consensus sequence with several additional residues (Arg, Lys, Cys, Pro) being conserved among the seven yeast regulatory proteins, and it could form an orthodox Zn finger between Cys64-Cys67 and Cys81-Cys84. However, in addition to this motif, the CYPl protein displays an additional characteristic; that is, the close presence, at positions 91 to 94, of a His-His motif. In theory, an alternative finger could be formed, either between Cys64-Cys67 and HisSl- His94, or between Cys81-Cys84 and HisSlIHis94, the two histidine residues acting as ligands for zinc instead of the four cysteine residues present in other yeast regulatory proteins. It should be emphasized that this is the first time, to our knowledge, that two pairs of cysteine and one pair of histidine residues are found adjacent in the same protein. We believe that their presence is not fortuitous but is related to the physiological role of the CYPl regulatory protein. This hypothesis is further documented and discussed in the accompanying paper (Verdiere et al., 1988).
The second domain of particular interest extends from amino acid residue 280 to 420 and consists of seven repeated units, which include the motif KCPVDH (Fig. 4). We propose two possible functions for this region. As mentioned in Results, these repeats could form a multiple finger-like structure and have potential for binding DNA and/or RNA. The motif C-H/C-H present here is different from the canonical C-C/C-C or C-C/H-H structures but has been observed in other proteins involved in nucleic acid manipulations (Berg, 1986; Herbert et aE., 1988). Another and more interesting possibility is that the KCPVDH repeated unit is not folded around a zinc atom but around a physiological “sensor”, for example heme or a metal atom, such as iron, copper, cobalt, vanadium or molybdenum, which could be either in an oxidized or in a reduced form according to the redox state of the cell. The presence in the repeated sequences of amino acid residues, particularly methionine, that could form a heme ligand reminiscent of that of some cytochrome c proteins strengthens the idea that heme could be the physiological redox sensor.

Novel Structural Domains in the C YPl Protein 275
It is thus conceivable that the CYPl protein itself would be a hemoprotein of a low redox potential.
This paper is dedicated to the memory of Marika Somlo. We thank one of the referees for underlining the presence and the interest of a conserved methionine residue in the KCVPVDH repeats. We are grateful to J. Banroques, B. Guiard, C. J. Herbert, C. Jacq, R. Labbe, M. Labouesse, E. Petrochilo for valuable suggestions; to J. Renowicki for help in the computer analysis, B. Poirer for typing the manuscript and C. Grandhamp for artwork. This work was supported by grants from the CNRS, ATP Microbiologic and Biologie Moleculaire de Gene, Ligue Nationale Francaise contre le Cancer and INSERM no. 86024.
References
Astell, C. R., Ahlstrom-Jonasson, L., Smith, M., Tatchell, K., Nasmyth, K. A. & Hall, B. D. (1981). CelE, 27, 15-24.
Bach, M. L., Lacroute, F. & Botstein, D. (1979). Proc. Nat. Acad. Sci., U.S.A. 76, 386-390.
Balzi, E., Chen, W., Vlaszewski, S., Copieaux, E. & Goffeau, A. (1987). J. Biol. Chem. 262, 16871-16879.
Bennetzen, J. L. & Hall, B. D. (1982). J. Biol. Chem. 257, 3018-3025.
Berg, ,J. M. (1986). Science, 232, 485-487.
Breunig, D. & Kuger, P. (1987). Mol. Cell. BioZ. 7, 44OC- 4406.
Clavilier, I,., P&e, G. Slonimski, P. P. (1969). MoZ. Gen.
Genet. 104, 1955218. Clavilier, L., PB&Aubert, G., Somlo, M. & Slonimski,
P. P. (1976). Biochimie, 58, 155-172. Covey, S. N. (1986). NucZ. Acids Res. 14, 623-633. Dayhoff, M. 0. (1978). Editor of Atlas of Protein Sequence
and Structure, vol. 5, suppl. 3, National Biochemical Research Foundation, Washington, DC.
Dobson, M. J., Truite, M. F., Roberts, N. A., Kingsman, A. J. & Kingsman, S. M. (1982). NucZ. Acids Res. 10, 2625-2637.
Downie, J. A., Stewart, J. W., Brockman, M., Schweingruber, A. M. & Sherman, F. (1977). J. MOE. BioZ. 133, 369-384.
Ephrussi, B. & Slonimski, P. P. (1950). Biochim. Biophys. Acta, 6, 256-267.
Fraser, R. (1975). Eur. J. Biochem. 60, 477-486.
Friden, P. & Schimmel, P. (1987). Mol. Cell. BioZ. 7, 2708-27 17.
Fukuhara, H. (1966). J. Mol. BioZ. 17, 334. Gannon, F., O’Hare, K., Perrin, F., Le Pennec, J. P.,
Benoist, C., Cachet, M., Breathnach, R., Royal, A., Garapin, A., Cami, B. & Chambon, P. (1979). Nature
(London), 278, 428-434. Gallwitz, D., Perrin, F. & Seidel, R. (1981). NucZ. Acids
Res. 9, 6339-6350. Gerbaud, C., Elmerich, C., Tandeau de Marsac, N. C.,
Chocat, P., Charpin, N., Guerineau, M. & Aubert, J. P. (1981). Curr. Genet. 3, 173-180.
Guarente, L. (1985). Current Communications in MoZecular Biology (Gluzman, Y., ed.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Guarente, L. & Mason, T. (1983). CeZZ, 32, 1279-1286. Guarente, L., Lalonde, B., Gifford, P. & Alani, E. (1984).
CeZZ, 36, 503-511. Guiard, B. (1985). EMBO J. 4, 3265-3272. Hartshorne, T. A., Blumberg, H. & Young, E. T. (1986).
,1%Lture (London), 320, 283-287.
Herbert, C. J., Labouesse, M., Dujardin, G. & Slonimski, P. P. (1988). EMBO J. 7, 473-483.
Hereford, L. & Rosbach, M. (1977). Cell, 10, 453-462. Heusterspreute, M., Oberto, J., Vinh Ha Thi & Davidson,
J. (1985). Gene, 34, 363-366. Hitzeman, R. A., Hagie, F. E., Levine, H. L., Goeddel,
D. V., Ammerer, G. & Hall, B. D. (1981). Nature (London), 293, 717-722.
Hope, I. A. & Struhl, K. (1986). Cell, 46, 885-894. Kadonaga, J. T., Carrier, K. R., Masiarz, F. R. & Tjian,
R. (1987). CeZZ, 51, 1079-1090. Kammerer, B., Guyonvarch, A. & Hubert, ,J. C. (1984).
J. Mol. BioZ. 180, 239-250. Keegan, L., Gill, G. & Ptashne, M. (1986). Science, 231,
699-704. Klug, A. $ Rhodes, D. (1987). Trends Biochem. Sci. 12,
464-469. Kozak, M. (1981). NucZ. Acids Ras. 9, 5233-5252. Labouesse, M., Dujardin, G. & Slonimski, P. P. (1985).
Cell, 41, 133-143. Langford, C. J. & Gallwitz, D. (1983). CeZZ, 33, 519-527. Laughon, A. & Gesteland, R. F. (1984). Mol. Cell. BioZ. 4,
26C-267. Laz, T. M., Pietras, D. F. & Sherman, F. (1984). Proc.
Nat. Acad. Sci., U.S.A. 81, 4475-4479. Legrain, M., De Wilde, M. & Hilger. F. (1986). NucZ.
Acids Res. 14, 3059-3073. Ma, J. & Ptashne, M. (1987). Cell, 48, 847-853. Maccechini, M. M., Rudin, Y., Blobel, G. & Shatz, G.
(1979). Proc. Nat. Acad. Sci., U.S.A. 76, 343-347. Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982).
Editors of Molecular Cloning: A Laboratory ManuaZ,
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Messinguy, F., Dubois, E. & Descamps, F. (1986). Eur. J.
Biochem. 157, 77-81. Miller, J. H. (1972). Editor of Experiments in Molecular
Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Miller, J., McLachlan, A. D. 6 Klug, A. (1985). EMBO J.
4, 1609-1614. Mills, D. R. & Kramer, F. R. (1979). Proc. Nat. Acad.
Sci., U.S.A. 76, 2232-2235. Montgomery, D. L., Leung, D. W., Smith, M., Shalit, P.,
Faye, G. & Hall, B. D. (1980). PTOC. Nat. Acad. Sci.,
U.S.A. 77, 541-545. Montgomery, D. L., Boss, J. M., McAndrew, S. J., Marr,
L., Walthall, D. A. & Zitomer, R. 8. (1982). J. BioZ.
Chem. 257, 7756-7761. Oka, M., Hayashi, T. & Nakajima, A. (1985). Z’olymer J.
17, 621-631. Orr-Weaver, T. L., Szostak, J. W. & Rothstein, R. J.
( 1981). Proc. Nat. Aead. Sci., U.S.A. 78, 6354-6358.
Page, D. C., Mosher, R., Simpson, E. R., Fisher, E. M., Mardon, G., Pollack, J., McGillivray, B., de la Chapelle, A. & Brown, L. (1987). Cell, 51, 1091-1104.
Pfeifer, K., Prezant, T. & Guarente, L. (1987). Cell, 49, 19-27.
Pikielny, C. W., Teem, ,J. L. & Rosbash, M. (1983). CeZZ, 34, 395-403.
Pinkham, L. & Guarente, L. (1985). Mol. Cell. Biol. 5, 3410-3416.
Rothstein, R. J. & Sherman, F. (1980). Genetics, 94, 87 l-889.
Sanger, F., Nicklen, S. & Coulson, A. R. (1977). Proc. Nat. Acad. Sci., U.S.A. 74, 5463-5467.
Sherman, F., Stewart, J. W., Parker, J., Inhaber, E., Shipman, M. A., Putterman, G. J., Gardisky, R. L. & Margoliash, E. (1968). J. BioZ. Chem. 243, 5446-5456.

276 F. Creusot et al.
Slonimski, P. P. (1955). In Proceeding of the Third International Congress of Biochemistry, Bruxelles (LiBbecq, C., ed.), pp. 242-252, Academic Press, New York.
Slonimski, P. P., Acher, R., PBti, G., Sels, A. & Somlo, M. (1965). In International Symposium on Mechanisms of Regulation of Cellular Activities in Microorganisms, C.N.R.S., Mareeille, pp. 435-461, Gordon and Breach, New York.
Smith, M., Leung, D. W., Gillam, S., Astell, C. R., Montgomery, D. L. & Hall, B. D. (1979). Cell, 16, 753-761.
Strauss, E. G., Rice, C. M. & Strauss, J. H. (1984). Virology, 133, 92-l 10.
Thomas, P. (1980). Proc. Nat. Acad. hi., U.S.A. 77, 5201-5205.
Verdi&e, J. & Petrochilo, E. (1975). Biochem. Biophys. Res. Commun. 67, 1451-1458.
Verdi&e, J. & Petrochilo, E. (1979). Mol. Gen. Genet. 175, 209-216.
Verdi&e, J., Creusot, F. & Guerineau, M. (1985). Mol. Gen. Genet. 199, 524-533.
Verdi&e, J., Creusot, F., Guarante, L. & Slonimski, P. P. (1986). Curr. Genet. 10, 339-342.
Verdi&e, J., Gaisne, M., Guiard, B., Deframoux, P;. & Slonimski, P. P. (1988). J. Mol. Biol. 204, 277-282.
Vincent, A. (1986). Nucl. Acids Res. 14, 4385-4391. Weinberger, C., Hollenberg, S. M., Rosenfeld, M. G. &
Evans, R. M. (1985). Nature (London), 318,670-672. Wharton, K. A., Yedvobnick, B., Finnerty, V. G. &
Artavanis-Tsakonas, S. (1985). Cell, 40, 55-62. Wright, C. F. & Zitomer, R. S. (1984). Mol. Cell. Biol. 4,
2023-2030. Wright, C. F. & Zitomer, R. S. (1985). Mol. Cell. Biol. 5,
2951-2958. Yanish-Perron, C., Vieira, J. & Messing, J. (1985). Gene,
33, 103-I 19. Zaret, K. S. & Sherman, F. (1982). CeEl, 28, 563-573. Zitomer, R. S., Sellers, J. W., McCarter, D. W., Hastings,
G. A., Wick, P. & Lowry, C. (1987). Mol. Cell. Biol. 5, 2521-2526.
Edited by A. Klug