current concepts of treatment in medical oncology: new anticancer drugs
TRANSCRIPT

J Cancer Res Clin Oncol (1996) 122:189-198 �9 Springer-Verlag 1996
Clemens Unger
Current concepts of treatment in medical oncology: new anticancer drugs
Received: 29 June 1995/Accepted: 1 October 1995
Abstract General principles for anticancer drug devel- opment include traditional drug-screening methods in biological test systems. Today, testing of a drug in a panel of selected human tumor xenografts in mice is assumed to have the best predictive value for clinical efficacy. Chemical modification of well-known anti- tumor drugs from compound groups such as purine analogs, vinca alkaloids, antifolates and platinum ana- logs are carried out to increase anticancer activity, to reduce toxic side-effects and to improve phar- macokinetic properties of the drugs. In the last decade the enormous development in molecular techniques has led to the discovery of key proteins that are inti- mately involved in the regulation of cancer growth control. Cell growth inhibitors could be developed by structure-based drug design, creating small organic molecules ("peptide mimetics") to target crucial en- zymes, oncogenes or oncogene products, tumor-sup- pressor genes and their products as well as growth factors and their corresponding receptors. Drugs rep- resenting new leading structures, like alkylphos- phocholines, topoisomerase I inhibitors, taxoids and suramin, have already entered the clinic. Novel thera- peutic approaches may provide substantial progress in cancer treatment in the very near future. Examples are the concept of high-dose chemotherapy with hema- topoietic stem cell support, the various strategies of gene therapy, the modulation of multi-drug resistance of cancer cells, and strategies to inhibit tumor an- giogenesis.
C. Unger Klinik ffir Tumorbiologie an der Albert-Ludwigs-Universitgt Freiburg, Breisacher Str. 117, D-79011 Freiburg, Germany
This review was developed on the basis of an invited lecture de- livered by the author during the Symposium "Quality Assurance and GCP in Cancer Drug Development" (see Kranich et al. 1994)
Key words Anticancer drug' Drug development" High dose chemotherapy �9 Multidrug resistance Gene therapy �9 Inhibition of angiogenesis
Patterns of cancer occurrence
Despite our best efforts, cancer mortality remains unac- ceptably high. In Germany, cancer is second only to heart disease as a cause of death and the mortality rate is currently 225000/year, whereas the rate of newly diagnosed cases of cancer is 358 000/year. Comparing the amount of newly diagnosed solid tumors and neo- plasias originating from blood, bone marrow and lymph nodes, more than 90% of these cancers are solid. Lung cancer is the commonest carcinoma in western countries, accounting for 15% of cases and 28% of deaths. Almost as many cases of colorectal cancer oc- cur as of lung cancer. The next most common are cancers of the breast and prostate. These four malig- nancies account for about 50% of the total cancer cases.
General stategies for cancer drug development
Traditional screening methods
In the past compounds were selected for clinical devel- opment on the basis of their activity in murine tumor models. The most widely used model was the "tumor panel screen" of the National Cancer Institute. This panel, however, identified only a few novel anticancer agents active in selected human tumors, mainly leukemias and lymphomas. Therefore, a few years ago the NCI tumor panel screen was revised. Since that time human tumor xenografts derived from human solid tumors have been established in nude mice and maintained by serial transfer. Human tumor xenografts

190
Table 1 General principles for anticancer drug development
1. Traditional screening methods in biological test systems to dis- cover novel compounds or new leading structures with antitumor activity
2. Chemical modification of well-known anticancer drugs to a) increase antitumor efficacy b) reduce toxic side-effects c) improve pharmacokinetic properties
3. Rational drug design: synthesis of "designer drugs" to target a) active sites of crucial enzymes b) oncogenes or oncogene products c) suppressor genes or their products d) growth factors and/or their corresponding receptors
Table 2 New drugs in medical oncology
Analogous compounds Purine analogs Vinca alkaloids Antifolates Platinum analogs Aromatase inhibitors
New leading structures Alkylphosphocholines Topoisomerase I inhibitors Taxoids Suramin
retain characteristic properties of the parental tumor, which is shown by a high correlation between tumor response to drugs in mice and man. Hence, a panel of selected human tumor xenografts seems to be a rel- evant model to predict clinical activity of a new anti- cancer drug.
Chemical modification of anticancer drugs
One promising approach to generate new anticancer drugs is chemical modification of already known com- pounds or leading structures in order to increase the antitumor activity and/or to reduce toxic side-effects of
Table 3 Clinical efficacy of pentostatin compared to interferon (INF,) in hairy-cell leukemia patients. Data from SWOG study
(January 1983 October 1991)
Response Number (%) responding
INF Pentostatin
Patients evaluable n = 129 n = 138 Complete response 24 (19%) 116 (84%) Partial response 31 (24%) 8 (6%) Total responders 55 (43%) 124 (90%)
a drug (Tables 1, 2). In this respect several new anti- metabolites have been evaluated in recent years (Fig. 1). Those with the most promising activity include the structurally related purine analogs 9-/%D-ara- binofuranosyl-2-fluoroadenine (ftudarabine), 2-chloro- 2'-deoxyadenosine (cladribine), and 2-deoxycoformycin (pentostatin). As shown for pentostatin (Table 3), these compounds have demonstrated impressive activity against a broad spectrum of indolent lympho- proliferative disorders, including hairy-cell leukemia, chronic lymphocytic leukemia, and low-grade non- Hodgkin's lymphoma (Beutler 1992; Cheson 1992). They have been generally well-tolerated in large clinical trials; however, all of them are myelosuppressive and immunosuppressive.
Vinca alkaloids remain among the most useful classes of anticancer agents used in the clinic today. Currently three vinca derivatives are in clinical trial: vinorelbine, vintripol and vinxaltine. Vinorelbine has shown consistant antitumor activity in patients with breast carcinoma and non-small-cell lung cancer with major response rates between 25% and 45% (Budman 1992). Responses have also been noted in lymphoma, esophageal carcinoma, and cervical cancer. The dose- limiting toxicity of these agents is leukopenia.
A number of promising new antifolates have been entered in clinical trials in recent years. Methotrexate-
Fig. 1A-C Chemical structures of the purine analogs pentostatin (A) cladribine (B) and fludarabine (C)
A B C
OH
""'" N H
\N)'N
H H
OH H
H2N
N CI !
HO -- CH2 O /
"7 OH
H2N
N "%
F
o
OH

191
resistant cell lines are generally sensitive to one or more of the newer antifolates, which differ from methotrexate by being either more lipophilic, more extensively poly- glutamated, or by inhibiting folate-requiring enzymes other than dihydrofolate reductase. Five of the agents are currently under clinical investigation: trimetrexate, piritrexim, edatrexate, lometrexol and D1694 (Fleming and Schilsky 1992).
Nine platinum analogs are currently in clinical devel- opment, including three that contain the diamino- cyclohexane substituent and five that contain the cyclobutane-dicarboxylato leaving group (Christian 1992). Many of them have shown activity in at least one cisplatin-resistant cell line, most commonly L1210 murine leukemia. In addition, most of them were less nephrotoxic than cisplatin in preclinical models.
Rational drug design
The traditional approach to the development of anti- cancer drugs involves mainly biological screening sys- tems. In the past, this procedure was necessary because of our limited knowledge of the molecular basis of human cancer. During the last decade, however, the enormous development of molecular genetic tech- niques has led to the discovery of key proteins that regulate normal biological functions of a cell, and, in some instances, has elucidated the pathways respon- sible for growth control in cancer cells. This informa- tion can now be utilized for the design of therapeutic drugs that specifically target these pathways. The mo- lecular targets can be categorized into three areas with regard to subcellular localization and putative func- tional properties: (a)plasma membrane receptors, (b) cytoplasmic signal-transduction enzymes and their associated binding proteins, and (c) nuclear regulators of cell cycle progression.
Three practical approaches exist for creating inhibi- tors of these signalling molecules. First, receptor-ligand binding antagonists can be designed for individual re- ceptors. Second, inhibitors of the enzymatic activity (e.g., tyrosine kinase activity), encoded by the cytoplas- mic domains of the ligand bound or activated forms of these receptors, can be developed. Third, hybrid fusion proteins, containing the specific ligand for each recep- tor joined to a mammalian cell toxin, can be created, which will selectively target and kill cells that express the particular growth factor receptors. Cytoplasmic signal-transduction proteins include a large number of oncogene products (e.g., raf, ras, src) and enzymes in- cluding protein kinases and lipid-modifying enzymes (e.g. MEK, MAP kinase, and phosphatidylinositol 3- kinase). Of the many enzymes involved in signal trans- duction, the most commonly mutated gene product associated with human tumors is the ras protein. Ras mutations impair the intrinsic GTPase activity of the
mutant ras proteins (Barbacid M 1987). Ras proteins are initially synthesized as biologically inert precursor proteins that require multiple post-translational modi- fications to achieve full biological activity, including their ability to stimulate cell growth. These modifica- tions include isoprenylation, proteolytic cleavage, carb- oxy-terminal methyl esterification, and palmitoylation reactions (Gibbs 1991). Of these reactions, the iso- prenylation of ras proteins is the most critical for at- taining biological maturity. The most common form of this reaction is catalyzed by the enzyme farnesyl pro- tein transferase (Omer 1993). Inhibitors of this enzyme have been shown to block maturation of ras proteins in cells and to inhibit the growth of ras-transformed tu- mor cells in cell culture and in animals (Gibbs et al. 1993). Other strategies for creating inhibitors of the cytoplasmic signal transduction proteins include design of specific protein (ras guanosine-triphosphatase-activ- ating protein, phosphatidylinositol 3-kinase) or lipid kinase (phospholipase j antagonists or of small-mol- ecule inhibitors of SH2 domains binding to growth factor receptors (see Fig. 2).
Intranuclear regulators of cell-cycle progression include growth-suppressor proteins such as the retino- blastoma protein (RB p105) and p53 as well as cell- growth stimulators such as f o s and E2F transcription factors. An interesting drug discovery strategy has fo- cused on restoring the biochemical defects produced by mutated or over-expressed intranuclear proteins that are only found in cancer cells (Lane 1992). This ap- proach promises exquisite tumor selectivity.
l ' { P , ~
I l r ~ ~' i T','a /" gas- "~ P-TYR [ ~ ] TYR-P <ELi
Fig. 2 Recruitment of signaling proteins to growth factor receptors. The binding of a growth factor to its receptor causes receptor dimerization and activation of the tyrosine protein kinase activity of the receptor. The autophosphorylated sites serve as binding sites for proteins with SH2 domains. Shown are the binding of p21 ras guanosine-triphosphatase-activating protein (ras-GAP), phos- pholipase 7 (PLCJ, and phosphatidylinositol 3-kinase (PI-3 kinase)

192
New leading structures
Alkylphosphocholines
Alkylphosphocholines represent a new class of mem- brane-active agents showing a relative simple chemical structure (Eibl and Unger 1990; Unger 1989). They are phosphocholine esters of aliphatic alcohols. Their physical properties are very similar to those of ly- solecithins. However, large differences in comparison to lysolecithins and generally to phospholipids exist in the metabolic fate of these molecules. For instance, substrate properties of alkylphosphocholines for me- tabolizing enzymes are markedly reduced. Thus, in contrast to lysolecithins substantial amounts of alkyl- phosphocholines accumulate in cell membranes in- fluencing cell growth and differentiation. In preclinical investigations alkylphosphocholines showed strong and selective anticancer properties. Much evidence indicates that the mode of action of alkylphosphocho- lines is related to plasma-membrane-associated phos- phoinositide metabolism. Inhibition of phospholipase C and protein kinase C are currently thought to be the main targets. Hexadecylphosphocholine (Miltefosine) was the first compound that was approved for the topical treatment of skin metastases in breast cancer patients. Although topical treatment of cutaneous breast cancer metastases has no curative intent, it can be of considerable palliative value for individual pa- tients. In various clinical trials throughout Europe, more than 200 patients were topically treated with a 6% miltefosine solution twice daily for cutaneous lesions. The overall objective response rate obtained in the different studies was about 30% (Hilgard et al. 1993). Clinical trials have been initiated with oral mil- tefosine; however, the major problem was the gastro- intestinal toxicity of the compound and, as a result, the drug could not be appropriately dosed and the re- sponse rates remained low. Therefore, new compounds with improved therapeutic indices are just entering clinical trials. One second-generation compound is an alkylphosphocholine analog without the choline moi- ety (D-20133). Another derivative under development is erucylphosphocholine, the first alkylphosphocholine that can be administered intravenously without caus- ing hemolysis.
Topoisomerase I inhibitors
induced single-strand DNA-breaks in the presence of topoisomerase I, thus identifying this enzyme as a major target of the drug. The specific effect of camptothecin and analogs is to stabilize the so-called "cleavable com- plex" between DNA and topoisomerase I. In the normal course of events, topoisomerase I senses torsional strain in the supercoiled DNA molecule and initiates a single- strand break. The topoisomerase I is covalently bound to the 3' end of the broken strand, while the intact strand is passed through the break. The enzyme then reseals the broken strand and dissociates from the DNA molecule. It has been demonstrated that qualitative or quantitative levels of topoisomerase I correlate with sensitivity or resistance to a topoisomerase I inhibitor. Furthermore, topoismerase I levels are more than tenfold higher in tissues from advanced colon carcinoma than in normal colonic mucosa (Giovanella 1989). Enzyme levels are also higher in lymphoma tissues than in normal B lymphocytes. The quantitative difference in the amount of target enzyme may lead to some selectivity for topoisomerase I inhibitors in terms of the toxic effect on tumor compared to normal tissue. Chemical modifi- cation of camptothecin has led to three compounds that are currently undergoing clinical trials. These are topotecan (Fig. 4), irinotecan (CPT-11), and 9-amino- camptothecin.
The most-studied compound, irinotecan, is a water- soluble analog of camptothecin with broad preclinical activity. In vitro activity was demonstrated in L1210 leukemia, P388 leukemia, Ehrlich ascites carcinoma, Lewis lung carcinoma, C38 colon carcinoma, and 16/C mammary adenocarcinoma among others. Clinical studies suggest activity in tumors that are poorly sensi- tive to chemotherapeutic drugs such as non-small-cell lung cancer, cervical cancer and colorectal cancer (Creemerst et al. 1994). The maximum tolerated dose seems to be 600 mg/m 2 if a 30-min i.v. infusion is used every 3 weeks. Hematological toxicity was dose-related, reversible and non-cumulative. At 350 mg/m 2, diarrhea was a significant clinical problem and required the use of high-dose loperamide, which allowed further dose escalation (Abigerges et al. 1995). Data from early clini- cal phase II trials in patients with 5-fluorouracil-refrac- tory metastatic colorectal cancer show an objective tumor response between 20% and 30%. However, fur- ther phase II trials of irinotecan are needed to confirm the remarkable antitumor activity particularly in colorectal cancer patients who become resistant to standard treatment.
In the early 1970s, camptothecin, a plant alkaloid ex- tract from the Camptotheca acuminata tree, demon- strated antineoplastic activity against gastrointestinal adenocarcinoma, melanoma, non-small-cell lung can- cer and acute myelocytic leukemia. Camptothecin is a strong inhibitor of DNA and RNA synthesis, while more recent studies revealed that camptothecin only
Taxoid compounds
As an ongoing part of the work on the isolation of novel anticancer agents from plants at the NCI, a high- ly active cytotoxic drug, taxol, was isolated from the bark of the pacific yew, Taxus brevifolia. The chemical structure of this compound is shown in Fig. 3.

Fig. 3 Chemical structure of paclitaxel (taxol)
193
0 II
CH=~C--O 0
CH, -~/ ~'(~H, o ,c., t /
HO" T H !
1 I
~ H
I
O--C ~ C H = 11 0
CH 3 I
....... N CH3 0
* HCI NCH3
Fig. 4 Chemical structure of the camtothecin water-soluble ana- logue topotecan
Taxol has been shown to possess a unique mecha- nism of action at the microtubule level, binding prefer- entially to microtubules rather than to tubulin dimers. It seems that taxol shifts the equilibrium towards microtubule assembly and stabilizes microtubules at concentrations as low as 0.05 gmol/1, which can be easily achieved in patients.
The dose-limiting toxicity of taxol is neutropenia. The major risk factor for taxol-induced neutropenia appears to be the extent of prior myelotoxic chemo- therapy or irradiation. The frequent occurrence of hypersensitivity reactions, such as hypotension, bron- chospasm, dyspnea, urticaria and pain, has been as- sumed to represent a non-immunologically mediated release of vasoactive substances induced by the drug or, more likely, by its vehicle cremophor ethanol. Premedi- cation of the patient with corticosteroids, diphenhydra- mine, and H2 antagonists decreases the incidence to less than 10%.
Clinical results show substantial activity of taxol in platin-refractory ovary carcinoma. Response rates with the standard schedule of 175 mg/m 2 were in a range between 20% and 40% (McGuire et al. 1989). This is of particular importance since patients with platin-refrac-
tory cancer of the ovary respond poorly to alternative treatment regimens. Recently the Gynecologic Oncol- ogy Group (GOG) initiated a phase III randomized trial in patients with suboptimal debulked stage III and stage IV ovarian carcinoma, comparing cisplatin and taxol (75 mg/m 2 and 135 mg/m 2) with a standard regi- men, cisplatin and cytoxan (75 mg/m 2 and 750 mg/m2). Median progression-free survival was 12.9 months for cisplatin/cytoxan and 18 months for cisplatin/taxol (P = 0.0002) and the median survival was 24.4 months for cisplatin/cytoxan and 37.5 months for cisplatin/tax- ol (P = 0.0001) (McGuire et al. 1995).
Moreover, taxol has substantial activity in meta- static breast cancer. In a multicenter randomized trial patients were allocated to two groups receiving differ- ent doses of taxol (135 or 175mg/m 2 i.v. as a 3-h infusion). All patients had previously received one or two prior regimens of chemotherapy. The overall re- sponse rate for the series was 26% with a medium duration of 8 months, whereas 43 % of the patients had stabilized disease (Nabholz et al. 1993). Taken together, even with advanced disease and heavy pretreatment in this patient population, a favorable response rate and response duration were observed.
With respect to second-generation taxoid com- pounds over 100 substances have been isolated from various taxoid plants and synthetic approaches to tax- ol-related drugs are actively being persued. Taxo- tere, NSC 628503, is a new taxoid obtained by semi- synthesis from 10-deacetylbaccatin III, a non-cytotoxic precursor extracted from the needles of the European yew Taxus baccata. Taxotere was selected for clinical development by Rhone-Poulenc Rorer and entered phase I clinical trials in 1990. Phase II studies are currently underway in Europe, the United States and Japan.
Suramin
Suramin shows diverse biological actions such as inhi- bitory effects on reverse transcriptase, adrenocortical

194
Fig. 5 Chemical structure of suramin N d O 3 - ~ HN
~ / ~ - SO3N' ~ C H 3
SO3Na
~ NH SO3Na
H3 C NaS03 NH NH S03N a
O
function, and polypeptide growth factors. The latter led to an evaluation of suramin in hormone-refractory prostate cancer. Suramin is a highly charged molecule containing six sulfate groups, which are ionized at physiological pH. The chemical structure of suramin is shown in Fig. 5. The compound inhibits the binding of, and the mitogenic action of a number of polypeptide growth factors including platelet-derived growth fac- tor, fibroblast growth factor (FGF), transforming growth factors c~ and /3 (TGFe, /3), and epidermal growth factor. During early clinical trials, some serious toxicities were identified including adrenal insuffi- ciency, coagulation abnormalities, and a peripheral neuropathy. However, with close monitoring of plasma concentrations the drug could be safely administered in further clinical trials. To date, anti tumor activity has been observed in prostate cancer, adrenocortical carci- noma, and breast and ovarian cancer (Eisenberger and Fontana 1992).
New treatment principles
New therapeutic approaches include high-dose chemo- therapy with hematopoietic stem cell support, modula- tion of multidrug resistance, gene therapy, and inhibition of tumor angiogenesis (Table 4). Some aspects of these novel approaches will be outlined in the following paragraphs.
High-dose chemotherapy
The relation between the dose of chemotherapy and treatment results exists for doses below the optimum.
Table 4 New treatment strategies in medical oncology
1. High-dose chemotherapy and hematopoietic stem cell support
2. Gene therapy 3. Modulation of multi-drug resistance 4. Inhibition of tumor angiogenesis
On the other hand high-dose chemotherapy (HDC) may be a potentially curative treatment option in some chemosensitive tumors. Dose escalation, however, is frequently limited by myelosuppression and non-hema- topoietic organ toxicity. With the combined applica- tion of hematopoietic growth factors and peripheral blood progenitor ceils it was shown that dose levels can be extended to several times the normal dose (Crown et al. 1992).
Breast cancer has been a favorite area of clinical research for this mode of treatment. Several key obser- vations were made in the first decade of clinical research in HDC of metastatic breast cancer. For instance, the median survival time of patients undergo- ing HDC is not appreciably prolonged compared to historically controlled patients treated with conven- tional dose therapy. However, a small group of patients receiving HDC achieve a disease-free survival beyond 5 years, an outcome that is extremely rare with conven- tional therapy. So far data have been derived entirely from retrospective studies and from historical controls. However, only prospective randomized trials will clar- ify the impact of HDC in stage IV breast cancer. The same holds true for patients with stage II breast cancer involving ten or more auxiliary lymph nodes. Since the outcome with conventional adjuvant treat- ment is poor and the survival rates are poor, HDC in the adjuvant setting is currently the subject of major random assignment trials in Europe and USA. Al- though HDC is a highly active and, with improvements in hematopoietic support, reasonably safe treatment, this approach is so far experimental in the clinical setting.
Gene therapy of cancer
Gene therapy can be defined as a therapeutic technique in which a functioning gene is inserted into the cell of a patient to correct an inborn genetic error or to provide a new function for the cell. A variety of tech- niques are available for introducing DNA into eu- caryotic cells. The most promising techniques are the

use of polycations or lipids complexed with DNA, encapsulation of DNA in lipid vesicles and the use of viral vectors, particularly retroviruses.
Among the different approaches for gene therapy in cancer patients immunostimulatory gene transfer is currently being investigated. Clinical work began in 1990 with Steve Rosenberg's group, who introduced cDNA for tumor necrosis factor e (TNFc~) into tumor- infiltrating lymphocytes, hoping that the transfected cells would home to the tumor site, release biologically active cytokine and thereby induce an antitumor effect. A second approach has been to take autologous tumor cells from patients and to modify them geneti- cally with cytokine genes, such as the interleukin-2 (IL-2) gene. However, some obstacles have to be over- come. For instance, tumor cells are weak antigen pres- enters and possibly some autologous tumor cells will not be immunogenic at all. What is more important, there also seems to be active induction of tolerance by tumor cells.
Many tumor cells express low levels of MHC anti- gens and/or do not provide costimulatory signals. This can result in an aborted T cell response and T cell apoptosis. Currently more than 60 gene-therapy proto- cols have been approved for cancer patients in the US. The majority of these studies employ retroviral vectors for DNA delivery. Adenoviral vectors are used in a small portion of protocols and only a few use direct in vivo injection of naked DNA into target cells. Four gene-therapy strategies have been used to date: (a) in- corporation of cytokine genes into tumor or effector cells to stimulate antitumor response, (b)antisense strategies designed to turn off oncogenes, e.g., c-myc, (c) tumor-suppressor gene modulation, i.e., introduc- tion of wild-type p53 to protect cells from converting to a tumorigenic state, and (d) drug enhancement ther- apy, i.e., the introduction of the multi-drug resistance gene (MDR) and the herpes simplex virus thymidine kinase/gancyclovir strategy.
Modulation of drug resistance
There are a number of factors that may contribute to the chemoresistance of tumor cells (Fig. 6). A major reason was found to be the expression of MDR genes. This MDR phenotype is either intrinsic to the tumor type or has been acquired after an initial response to chemotherapy following selection during treatment. The most consistent alteration in MDR cells is the increased expression of a 170-kDa membrane phos- phoglycoprotein (see Fig. 6). Transfection of P-glyco- protein cDNA sequences into drug-sensitive cells has demonstrated that increased expression of P-glycopro- tein is necessary and sufficient to generate a MDR phenotype. The MDR1 gene is part of an ATP-binding cassette superfamily of genes, which encodes different membrane transport proteins. These proteins are
195
intracellular compartition
~ ~ , transport
000
MDR / .~ efflux
repair ~ ~ reduced permeability
Fig. 6 Mechanisms of drug resistance. Most of the mechanisms of drug resistance shown in the picture have been determined in cell culture and animal models. Evidence exists that many of these mechanisms, like reduced transport or reduced cell permeability of a cytotoxic drug, operate in human cancer. The same holds true for the enhanced cellular repair capacity of DNA-strand breakage. The expression of multi-drug resistance (MDR) genes and the increased expression of a 170-kDa membrane phosphoglycoprotein in addi- tion play a major role in generation of drug resistance
capable of transporting a wide variety of substrates like peptides, inorganic ions, steroids, and cytotoxic drugs.
The P-glycoprotein is thought to function as an energy-dependent drug-efflux pump. In drug-resistant cancer cells, P-glycoprotein appears to decrease the intracellular concentration of cytotoxic drugs by bind- ing the drug and actively pumping it out of the cell before the drug reaches a critical concentration. Drugs involved in the MDR phenotype include natural prod- ucts, such as anthracyclines and the vinca alkaloids, as well as synthetic agents such as mitoxantrone and trimetrexate. Several approaches have been proposed to modulate MDR caused by the overexpression P- glycoprotein: (a) the use of cytotoxic drugs to which MDR cells exhibit collateral or increased sensitivity; (b) the use of chemosensitizing agents that are capable of inhibiting the P-glycoprotein function, such as cal- cium channel blockers and cyclosporin A; (c) the use of immunotherapeutic agents, such as monoclonal anti- bodies, to target P-glycoprotein and inhibit its func- tion; (d) the inhibition of MDR1 gene expression by use of genetic suppressor elements; and (e) the use of high-dose chemotherapy, such as that used in autologous bone marrow transplants. Several studies have used the calcium channel blocker verapamil to reverse MDR. Modest reversal of resistance has been described for patients with malignant lymphoma and myeloma. Cardiovascular side-effects associated with the high dose of verapamil were prohibitive for effective dose escalation (Miller et al. 1991). More recently, cyclosporin A has been used with some success. Again, activity was primarily reported in patients with hema- topoietic malignancies (Sonneveld et al. 1992).

196
Reduced intracellular accumulation of drugs due to reduced drug permeability of the tumor cell membrane or alterations in the transport components (see Fig. 6), namely the folate-binding protein or the reduced folate transporter, is one of the commonest mechanisms of antineoplastic drug resistance (Sirotnak et al. 1981).
The final common pathway for cytotoxic effects of many of the anticancer drugs, including the alkyl- ating agents and the topoisomerase-binding drugs, is DNA-strand breakage. In addition, other com- pounds such as the antimetabolites, produce errors in base pairing, premature chain termination, or strand breaks. The process of repair of these lesions (see Fig. 6) is poorly understood. With few excep- tions, the specific enzymes responsible for repair of various DNA lesions have not been identified. Enhanced repair capacity, which has been demon- strated for cisplatin- and alkylating-agent-resistant cells, could potentially be conferred on multiple agents (Chao et al. 1991).
Inhibition of tumor angiogenesis
The formation of new blood vessels, or angiogenesis, permits the expansion of a tumor mass in three dimen- sions. Prevascular tumors may persist as thin asympto- matic lesions, restricted by the limits of oxygen and nutrient diffusion. On the other hand, vascularized tumors expand locally and by metastases. Human tu- mors and spontaneously arising animal tumors are usually not angiogenic at the beginning of their devel- opment. In situ carcinomas may exist for months or years without neovascularization, remaining limited to a small tumor volume and growing locally. Some tu- mor cells then switch to an angiogenic phenotype and recruit new capillary blood vessels that support the growth of both angiogenic and non-angiogenic tumor cells.
The switch to the angiogenic phenotype itself de- pends on a net balance of positive and negative an- giogenic factors released by the tumor or surrounding stroma (Folkman 1995). The positive factors include FGF~, FGFfi, angiogenin, TGF~, TNF, platelet-de- rived endothelial cell growth factor, granulocyte-col- ony-stimulating factor, IL-8 and vascular endothelial growth factor (VEGF).
Although this increased production of positive an- giogenic factors is necessary, it is not sufficient for the angiogenic phenotype. Negative regulators like thrombo- spondin or angiostatin must be decreased as well. Notably, in human fibroblasts thrombospondin is nor- mally under the control of the p53 tumor-suppressor gene (Dameron et al. 1994). Fibroblasts from cancer patients with Li-Fraumeni syndrome have only one copy of p53 and, when this allele is mutated or deleted,
thrombospondin production is decreased and an- giogenic activity switched on.
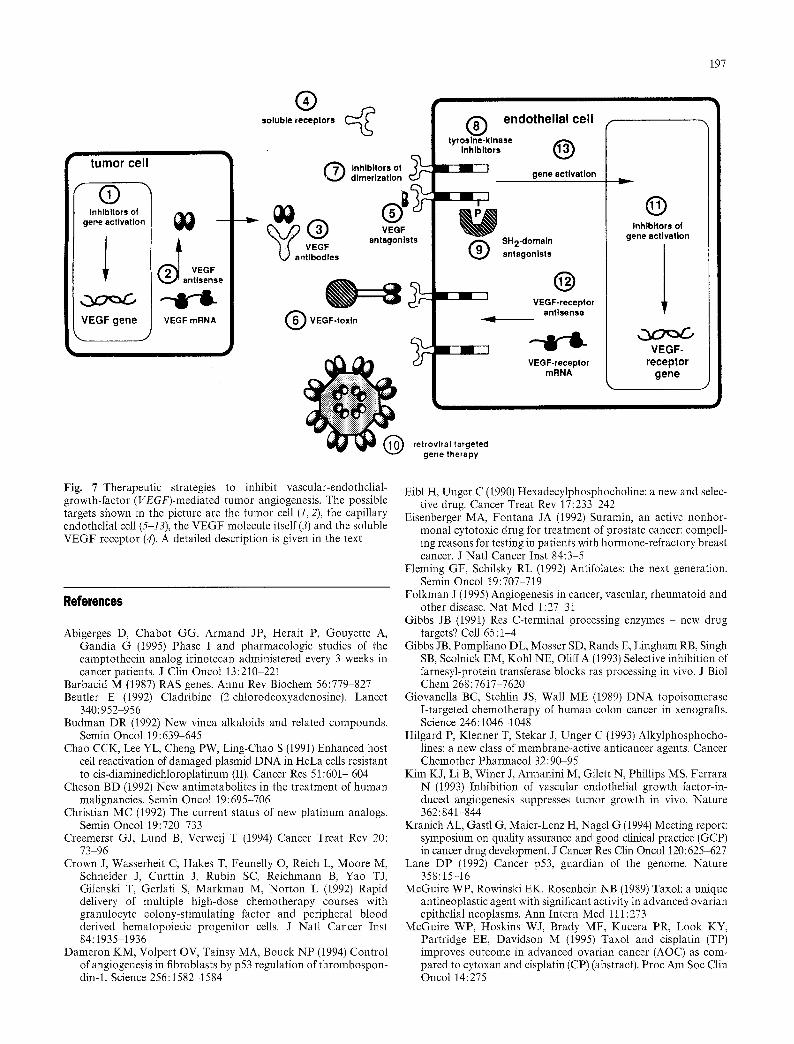
One of the most important angiogenic factors is VEGF (Kim et al. 1993). In tumor cells this angiogenic factor is expressed and secreted under the control of growth factors, oncogenes like ras and raf, protein- kinase C activators and mutated p53. Capillary endothe- lial cells possess specific receptors for VEGF. Binding of VEGF to its specific receptors causes a mitogenic signal resulting in proliferation and growth of the capil- lary endothelial cells. Current therapeutic strategies (Fig. 7, stages 1-13) are focussed on expression and secretion of VEGF in tumor cells as well as on recep- tor-mediated signal transduction of VEGF on capillary endothelial cells within tumors. The control of VEGF gene expression (1) involves growth factor receptors, GRB2, ras, raf, MAP kinase and the transcription factor AP1. The development of VEGF antisense sequences to suppress VEGF expression (2) or the application of VEGF antibodies (3) are strategies to suppress neovascularization caused by VEGF. Soluble VEGF receptors (4) may be a suit- able tool for VEGF depletion and be useful for screen- ing of VEGF antagonists. The construction of VEGF receptor antagonists (5) involves different peptides derived from epitope libraries of VEGF mutants. These peptides should compete with the binding of VEGF to KDR and/or fit-1 receptors on capillary endothelial cells.
Bifunctional VEGF toxins (6), e.g. the generation of recombinant fusion proteins between VEGF and do- mains II and II of exotoxin A of Pseudomonas aeruginosa, can promote a selective growth inhibition of the tumor by destruction of its vascular system. Inhibition of VEGF receptor dimerization (7), develop- ment of tyrosine kinase inhibitors (8) and the con- struction of SH2-domain antagonists (9) give rise to an early interference with the signal transduction of VEGF at the receptor level. A further approach aims at VEGF-receptor-targeted, retroviral gene therapy (10). This involves the generation of a recombinant retrovirus with the following characteristics: (a) the virus will only infect endothelial cells; (b) the virus carries the herpes simplex thymidine kinase gene, which allows a controllable destruction of pro- liferating cells, without affecting resting cells; (c) the virus is kept replication-defective to avoid non-desired viremia.
The search for inhibitors of VEGF receptor gene activation (11), VEGF receptor antisense sequences (12) or the search for compounds capable of interfering with the signal transduction of the KDR and/or fit-1 recep- tors at the postreceptor level (13) are further ap- proaches to inhibit the mitogenic activity of VEGF on capillary endothelial cells. Taken together, these differ- ent therapeutic strategies indicate how future clinical trials in the field of angiogenesis inhibition can be designed.
197
�9 tumor cell
Inhlbltors of gene activation
VEGF gene J
VEGF mRNA
| soluble receptors ( ~
Q Inhlbltors of dlmerlzatlon
~ o j ( ~ ) endothelial cell
rna" @ I gene activation
SH2-domaln antagonists
@ VEGF-receptor
VEGF-toxln ~ ~ antlsens~
- r ~ .
retroviral targeted gene therapy
| Inhlbltors of gene activation
VEGF- receptor
gene
Fig. 7 Therapeutic strategies to inhibit vascular-endothelial- growth-factor (VEGF)-mediated tumor angiogenesis. The possible targets shown in the picture are the tumor cell (1, 2), the capillary endothelial cell (5-13), the VEGF molecule itself (3) and the soluble VEGF receptor (4). A detailed description is given in the text
References
Abigerges D, Chabot GG, Armand JP, Herait P, Gouyette A, Gandia G (1995) Phase I and pharmacologic studies of the camptothecin analog irinotecan administered every 3 weeks in cancer patients. J Clin Oncol 13:210 221
Barbacid M (1987) RAS genes. Annu Rev Biochem 56:779-827 Beutler E (1992) Cladribine (2-chlorodeoxyadenosine). Lancet
340:952 956 Budman DR (1992) New vinca alkaloids and related compounds.
Semin Oncol 19:639-645 Chao CCK, Lee YL, Cheng PW, Ling-Chao S (1991) Enhanced host
cell reactivation of damaged plasmid DNA in HeLa cells resistant to cis-diaminedichloroplatinum (II). Cancer Res 51:601- 604
Cheson BD (1992) New antimetabolites in the treatment of human malignancies. Semin Oncol 19:695-706
Christian MC (1992) The current status of new platinum analogs. Semin Oncol 19:720-733
Creemerst GJ, Lund B, Verweij T (1994) Cancer Treat Rev 20: 73 96
Crown J, Wasserheit C, Hakes T, Feunelly O, Reich L, Moore M, Schneider J, Curttin J, Rubin SC, Reichmann B, Yao T J, Gilenski T, Gerlati S, Markman M, Norton L (1992) Rapid delivery of multiple high-dose chemotherapy courses with granulocyte colony-stimulating factor and peripheral blood derived hematopoietic progenitor cells. J Natl Cancer Inst 84:1935-1936
Dameron KM, Volpert OV, Tainsy MA, Bouck NP (1994) Control of angiogenesis in fibroblasts by p53 regulation of thrombospon- din-1. Science 256:1582-1584
Eibl H, Unger C (1990) Hexadecylphosphocholine: a new and selec- tive drug. Cancer Treat Rev 17:233 242
Eisenberger MA, Fontana JA (1992) Suramin, an active nonhor- monal cytotoxic drug for treatment of prostate cancer: compell- ing reasons for testing in patients with hormone-refractory breast cancer. J Natl Cancer Inst 84:3-5
Fleming GF, Schilsky RL (1992) Antifolates: the next generation. Semin Oncol 19:707-719
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27 31
Gibbs JB (1991) Res C-terminal processing enzymes - new drug targets? Cell 65:1-4
Gibbs JB, Pompliano DL, Mosser SD, Rands E, Lingham RB, Singh SB, Scolnick EM, Kohl NE, OtiffA (1993) Selective inhibition of farnesyl-protein transferase blocks ras processing in vivo. J Biol Chem 268:7617 7620
Giovanella BC, Stehlin JS, Wall ME (1989) DNA topoisomerase I-targeted chemotherapy of human colon cancer in xenografts. Science 246:1046-1048
Hilgard P, Klenner T, Stekar J, Unger C (1993) Alkylphosphocho- lines: a new class of membrane-active anticancer agents. Cancer Chemother Pharmacol 32:90-95
Kim KJ, Li B, Winer J, Armanini M, Gilett N, Phillips MS, Ferrara N (1993) Inhibition of vascular endothelial growth factor-in- duced angiogenesis suppresses tumor growth in vivo. Nature 362:841-844
Kranich AL, Gastl G, Maier-Lenz H, Nagel G (1994) Meeting report: symposium on quality assurance and good clinical practice (GCP) in cancer drug development. J Cancer Res Clin Oncol 120:625-627
Lane DP (1992) Cancer p53, guardian of the genome. Nature 358:15-16
McGuire WP, Rowinski EK, Rosenhein NB (1989) Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med 111:273
McGuire WP, Hoskins W J, Brady MF, Kucera PR, Look KY, Partridge EE, Davidson M (1995) Taxol and cisplatin (TP) improves outcome in advanced ovarian cancer (AOC) as com- pared to cytoxan and cisplatin (CP) (abstract). Proc Am Soc Clin Oncol 14:275

198
Miller TP, Grogan TM, Dalton E, Spier CM, Scheper R J, Salmon SE (1991) P-glycoprotein expression in malignant lymphoma and reversal of clinical drug resistance with chemotherapy plus high dose verapamil. J Clin Oncol 9:17-24
Nabholtz JM, Gelmon K, Bontenbal M, et al. (1993) Randomized trial of two doses of taxol in metastatic breast cancer: an interim analysis (abstract). Proc Am Soc Clin Oncol 12:60
Omer CA, Kral AM, Diehl RE, Prendergast GC, Powers S, Allen CM, Gibbs JB, Kohl NE (1993) Characterization of recombinant human farnesyl-protein transferase: cloning, expression, farnesyl di-phosphate binding, and functional homology with yeast prenyl-protein transferases. Biochemistry 32:516%5171
Sirotnak FM, Moccio DM, Kelleher LE, Goutsas IJ (1981) Relative frequency and kinetic properties of transport defective phenotypes among methotrexate-resistant L 1210 clonal cell lines derived in vivo. Cancer Res 41:4447-4450
Sonneveld P, Durie BGM, Lokhorst HM, Maric T, Soebu G, Sucin S, Zittonn R, Cowenberg B, Nooter K (1992) Modulation of multidrug-resistant multiple myeloma by cyclosporin. Lancet 340:255-259
Unger C (1989} Alkylphosphocholine und Analoga: Entwicklung einer neuen Substanzgruppe mit antineoplastischer Wirkung. Thieme, Stuttgart