crystal 98 1.0 february 26, 1999 v.r saunder, r. dovesi, c. roetti, m. causa, n.m. harrison, r....
Post on 20-Dec-2015
215 views
TRANSCRIPT

CRYSTAL 981.0
February 26, 1999
V.R Saunder, R. Dovesi, C. Roetti, M. Causa, N.M. Harrison, R. Orlando, C. M. Zicovish-Wilson
Oleg Sychev

Crystal 98 2
Properties of interest&Methods
Properties of interest
Equilibrium structurePhononsRelaxation around defects
Energy dispersionDensity of statesSpatial charge densityChemical bondingMagnetic interactions
Dinamical simulationsPhase boundaries
Methods
All-electronsTotal-energy methods (DFT):FLAPW, FP-LMTO, Gaussian
pseudopotencial
Methods using simplifying assumptions for the crystal potencial:
LMTO-ASA, ASW
Semiempirical methodsClassical molecular dynamics;
model Hamiltonians

Crystal 98 3
Theory Stationary Shrodinger equation:
1em,unitsatomic:followingthefor
existmustdr)r(:)r(ofopertiesPr
densityeargch),r()r(e:electronanfor
,densityprobality)r(
functionwave)r(,energypotencial)r(U),r(Um2
H
)r(E)r(H
2
2
2
22

Crystal 98 4
TheoryHartree-Fock method
LLiLi
N
1i
*i
N
1i
*i
2
)r(fa)r(
:functionsbasiseappropriatoveransionexpanbydrepresentetypicalareforsearched
orbitalsparticleonethe;yconvergencsufficienttilliterate:solutionacticalPr
\)r(rd)r(rr
)r()r(
)r(rdrr
)r()r(
)r(U2
1

Crystal 98 5
TheoryDensity functional theory
.)etcionapproximatgradient
dgeneralizeGGA,ionapproximatdensitylocalLDA(
energyncorrelatioexchangetheofFunctional:E
Edr)r(E
rdrdrr
)r()r(
2
1E
xc
xc
N
1i
xci

Crystal 98 6
Installation
Installation size is 173Mb on CD WWW Sites:
http://www.chimifm.unito.it/teorica/crystal/crystal.html
http://www.cse.clrc.ac.uk/cmg/CRYSTAL/

Crystal 98 7
Installation
CRYSTAL98 use: Unix Linux systems(all versions) Windows NT

Crystal 98 8
Introduction
The CRYSTAL package performs ab initio calculations of the ground state energy, elec-tronic wave function and properties of periodic systems. Hartree-Fock or Kohn-Sham Hamiltonians (that adopt an Exchange- Correlation potential following the postulates of Density-Functional theory) can be used. Systems periodic in 0 (molecules, 0D), 1(polymers, 1D), 2 (slabs, 2D), and 3 dimensions (crystals, 3D) are treated on an equal footing. In each case the fundamental approximation made is the expansion of the single particle wave functions ('Crystalline Orbital', CO) as a linear combination of Bloch functions (BF) defined in terms of local functions (hereafter indicated as ‘Atomic Orbitals’, AOs).

Crystal 98 9
Structure
The local functions are, in turn, linear combinations of Gaussian type functions (GTF) whose exponents and coefficients are defined by input. Functions of s, p(in the order 2z2-x2-y2; xz; yz; x2-y2; xy) symmetry can be used. Also available are sp shells (s and p shells, sharing the same set of exponents).The use of sp shells can give rise to considerable savings in CPU time.

Crystal 98 10
Structure The program can automatically handle space
symmetry: 230 space groups, 80 layer groups, 99 rod groups, 45 point groups are available (Appendix A). In the case of polymers it cannot treat helical structures (translation followed by a rotation around the periodic axis). However, when commensurate rotations are involved, a suitably large unit cell can be adopted.
Point symmetries compatible with translation symmetry are provided for molecules. Input tools allow the generation of slabs (2D system) or clusters (0D system) from a 3D crystalline structure, the elastic distortion of the lattice, the creation of a supercell with a defect and a large variety of structure editing.

Crystal 98 11
FunctionalityThe basic functionality of the code is
outlined below. The single particle potential
Restricted Hartree Fock Theory Unrestricted and Restricted Open
Shell Hartree Fock Theory Density Functional Theory for
Exchange and Correlation Effective Core Pseudopotentials

Crystal 98 12
Functionality
Algorithms Parallel processing (replicated data) Traditional SCF Direct SCF

Crystal 98 13
Functionality
Structural Editing Use of space, layer, rod and point group
symmetry Removal, insertion deletion and substitution
of atoms Displacement of atoms Rotation of groups of atoms Extraction of surface models from 3D crystal
structure Cluster generation from 3D crystals Cluster of molecules from molecular crystals

Crystal 98 14
Functionality Properties
Band structure Density of states Electronic charge density maps Electronic charge density on a 3D grid Mulliken population analysis Spherical harmonic atom and shell multipoles X-ray structure factors Electron momentum distributions Compton profiles Electrostatic potential, field and field gradients Spin polarised generalisation of properties Hyperfine electron-nuclear spin tensor A posteriori Density Functional correlation energy

Crystal 98 15
Wave function analysis and properties
Total energy Hartree-Fock wave function Hartree-Fock wave-function+DF a posteriori
correction for correlation DF SCF wave function
Band structure Density of states
Band projected DOSS AO projected DOSS
All Electron Charge Density - Spin Density Density maps Mulliken population analysis Density analytical derivatives

Crystal 98 16
Wave function analysis and properties
Atomic multipoles Electrostatic potential
Electrostatic potential maps Point charge electrostatic potential maps
Electric field Electric field gradient Structure factors Compton profiles Electron Momentum Density Fermi contact

ADEQUATE DESCRIPTION OF COPPER BAND STRUCTUREADEQUATE DESCRIPTION OF COPPER BAND STRUCTURE
Figure 3 Figure 4
M
k Z
X
K
K WL
U
Xk y
M
k X
a
our
data
b
data
from
Ref.

ADEQUATE DESCRIPTION OF MgO BAND STRUCTUREADEQUATE DESCRIPTION OF MgO BAND STRUCTURE
Figure 3 Figure 4
M
k Z
X
K
K WL
U
Xk y
M
k X
a
our
data
b
data
from
Ref.
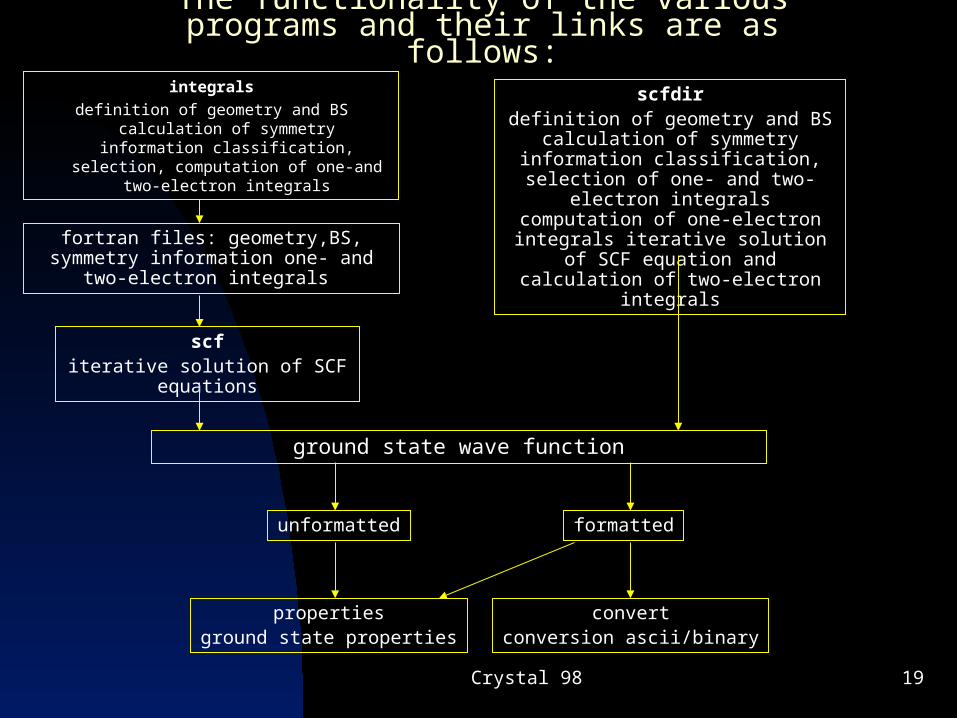
Crystal 98 19
The functionality of the various programs and their links are as follows:
integrals
definition of geometry and BS calculation of symmetry information classification,
selection, computation of one-and two-electron integrals
fortran files: geometry,BS, symmetry information one- and two-electron integrals
scfiterative solution of SCF
equations
ground state wave function
unformatted
propertiesground state properties
scfdirdefinition of geometry and BS
calculation of symmetry information classification, selection of one- and two-electron integrals computation of one-electron integrals iterative solution of SCF equation and calculation of two-
electron integrals
formatted
convertconversion ascii/binary

Crystal 98 20
Compilation
Crystal98 is written in FORTRAN 77 and is therefore easily compiled on architectures for which executibles are not provided. You may also wish to compile the code to alter the dimensions of internal arrays or to select compilation and linkage options to increase the performance of the code.

Crystal 98 21
Testing the Installation
It is very important that the installation of the code is checked by running the validation suite which is contained on the CD

Crystal 98 22
The parallel Implementation CRISTAL98 supports parallel execution on
modestly parallel hardware on computers (nodes) linked by relatively low perfomance networks (eg: Ethenet).CPU and DISK resources are shared efficiently while the memory usage is replicated on each node.
One node is chosen as the master.The master spawns the program onto other nodes (slaves) and operates dynamical load balancing of the task execution via a shared atomic counter.
During integral generation a task is defined as the calculation of a block of integrals.Thus each node computes a number of integrals which are stored to its local disk.

Crystal 98 23
Basic problems of CRYSTAL98
Optimization basis for concrete physical tasks
Value Energy Fermi is either overestimated(DFT method) or underestimated(HF-method)
Time of calculation depends from computer sizes memory (as HDD size, so Extended memory size)

Crystal 98 24
CRYSTAL 981.0
February 26, 1999
V.R Saunder, R. Dovesi, C. Roetti, M. Causa, N.M. Harrison, R. Orlando, C. M. Zicovish-Wilson
http://www.chimifm.unito.it/teorica/crystal/crystal.html
http://www.cse.clrc.ac.uk/cmg/CRYSTAL/