control of creatine metabolism by hif is an endogenous ... · and ckb, respectively) revealed...
TRANSCRIPT

Control of creatine metabolism by HIF is anendogenous mechanism of barrier regulation in colitisLouise E. Glovera,b,1, Brittelle E. Bowersa,b, Bejan Saeedia,b, Stefan F. Ehrentrauta,b, Eric L. Campbella,b,Amanda J. Baylessa,b, Evgenia Dobrinskikhb, Agnieszka A. Kendricka,b, Caleb J. Kellya,b, Adrianne Burgessa,b,Lauren Millera,b, Douglas J. Kominskya,c, Paul Jedlickad, and Sean P. Colgana,b,2
aMucosal Inflammation Program, bDepartment of Medicine, cDepartment of Anesthesiology and Perioperative Medicine, and dDepartment of Pathology,University of Colorado Anschutz Medical Campus, Aurora, CO 80045
Edited by Gregg L. Semenza, The Johns Hopkins University School of Medicine, Baltimore, MD, and approved October 18, 2013 (received for reviewFebruary 12, 2013)
Mucosal surfaces of the lower gastrointestinal tract are subject tofrequent, pronounced fluctuations in oxygen tension, particularlyduring inflammation. Adaptive responses to hypoxia are orches-trated largely by the hypoxia-inducible transcription factors (HIFs).As HIF-1α and HIF-2α are coexpressed in mucosal epithelia thatconstitute the barrier between the lumen and the underlying im-mune milieu, we sought to define the discrete contribution ofHIF-1 and HIF-2 transactivation pathways to intestinal epithelialcell homeostasis. The present study identifies creatine kinases(CKs), key metabolic enzymes for rapid ATP generation via thephosphocreatine–creatine kinase (PCr/CK) system, as a unique genefamily that is coordinately regulated by HIF. Cytosolic CKs are ex-pressed in a HIF-2–dependent manner in vitro and localize to apicalintestinal epithelial cell adherens junctions, where they are criticalfor junction assembly and epithelial integrity. Supplementationwith dietary creatine markedly ameliorated both disease severityand inflammatory responses in colitis models. Further, enzymes ofthe PCr/CK metabolic shuttle demonstrate dysregulated mucosalexpression in a subset of ulcerative colitis and Crohn disease pa-tients. These findings establish a role for HIF-regulated CK in epi-thelial homeostasis and reveal a fundamental link between cellularbioenergetics and mucosal barrier.
epithelial junctions | energy metabolism | actomyosin | IBD
Intestinal epithelia function to both facilitate nutrient transportand protect against luminal antigens. Selective permeability is
mediated by specialized anatomical features, including dynamicintercellular junctions. The apical junctional complex (AJC),comprising the tight junction (TJ) and subjacent adherens junction(AJ), is supported by a highly crosslinked cytoskeleton and is thekey determinant of paracellular permeability and barrier function.Prominent features of the perijunctional cytoskeleton include anextensive network of F-actin bundles that associate with TJs, anda dense circumferential ring of actin and myosin contiguous withAJs (1). This actomyosin ring readily copurifies with other cyto-skeletal proteins and demonstrates ATP-dependent contractilityex vivo. Such observations highlight the contractile nature ofthe apical actin network and the intimate association betweenits components (2).Based on their juxtaposition to the anoxic gut lumen, intestinal
epithelial cells (IECs) function physiologically in a low-oxygen-tension microenvironment and exhibit a uniquely adaptive oxygen-ation profile. This profile is prodigiously altered in inflammatorybowel disease (IBD) (3). Adaptive transcriptional responses tooxygen deprivation are mediated primarily through the hypoxia-inducible factor (HIF) complex, comprising a constitutive “β”subunit, and an oxygen-labile “α” component that is regulated inpart by prolyl hydroxylase (PHD) enzymes (4). Despite their con-current expression in many cell types, HIF-1α and HIF-2α playnonredundant roles (5) that appear to be highly cell specific tofacilitate both short- and long-term adaptations to hypoxia (6).Barrier dysregulation with unimpeded flux of luminal anti-
gens contributes fundamentally to the profound metabolic
shifts inherent to mucosal inflammatory lesions. Both HIF-1α andHIF-2α are expressed in inflamed mucosa from IBD patients(7) and mouse models of colitis (8). Studies of murine IBD haverevealed that loss of epithelial HIF-1α correlates with more severeclinical symptoms, whereas constitutive activation of HIF-1 andHIF-2 is protective (8). Despite this, the molecular targets of HIF-1and particularly HIF-2 in IECs are not well characterized. To de-lineate HIF-1– and HIF-2–specific target loci, we performed ChIP–chip analysis of chromatin isolated from hypoxic IECs. We iden-tified a family of genes involved in creatine (Cr) metabolism thatare coordinately regulated by HIF-2. Immunolocalization ofthe cytosolic “muscle-” and “brain-type” creatine kinases (CKMand CKB, respectively) revealed coupling to the AJC, and CKinhibition abrogated junctional assembly and barrier function.Moreover, dietary Cr supplementation ameliorated the patho-genic course of murine colitis. Collectively, these data identifya unique role for HIF in modulating the epithelial barrier throughregulation of the Cr/CK shuttle.
ResultsHIF ChIP-on-Chip Analysis Identifies Genes Involved in Cr Metabolism.ChIP was performed in human colon carcinoma Caco-2 IECsusing HIF-1α– and HIF-2α–specific polyclonal antibodies (Fig.1A). ChIP-enriched and input DNA were hybridized to a custommicroarray comprising a genome-wide set of predicted transcription
Significance
Intestinal epithelial barrier dysregulation is a hallmark of in-flammatory bowel diseases (IBDs). A central role for hypoxicsignaling has been defined in barrier modulation during in-flammation. We demonstrate that genes involved in creatinemetabolism, the creatine kinases (CKs), are coordinately regu-lated by hypoxia-inducible transcription factors (HIFs) and thatsuch regulation is critical to barrier function. Inhibition of the CKpathway abrogates apical junction assembly and barrier in-tegrity. Dietary creatine supplementation profoundly attenu-ates the pathogenic course of mucosal inflammation in mousecolitis models. Moreover, we demonstrate altered expression ofmitochondrial and cytosolic CK enzymes in IBD patient tissue.These findings highlight the fundamental contribution of crea-tine metabolism to intestinal mucosal function, homeostasis,and disease resolution.
Author contributions: L.E.G. and S.P.C. designed research; L.E.G., B.E.B., B.S., S.F.E., E.L.C.,A.J.B., A.A.K., C.J.K., A.B., L.M., and D.J.K. performed research; E.D. contributed newreagents/analytic tools; L.E.G., B.S., E.D., P.J., and S.P.C. analyzed data; and L.E.G. wrotethe paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Ex-pression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE43108).1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302840110/-/DCSupplemental.
19820–19825 | PNAS | December 3, 2013 | vol. 110 | no. 49 www.pnas.org/cgi/doi/10.1073/pnas.1302840110
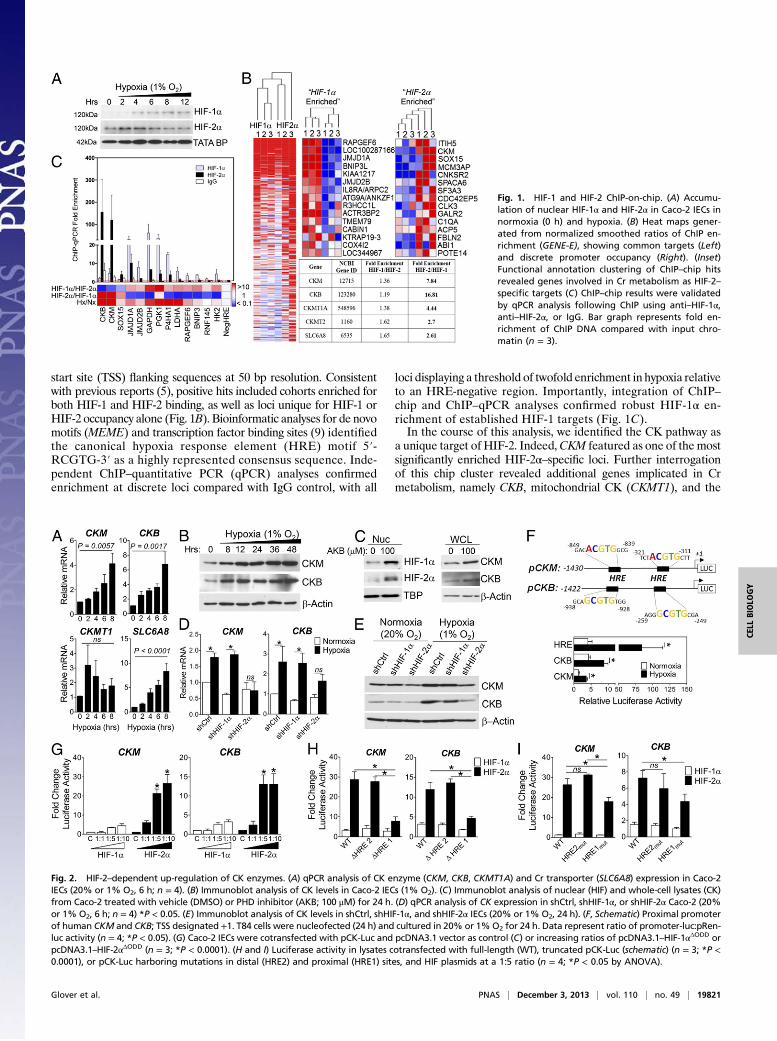
start site (TSS) flanking sequences at 50 bp resolution. Consistentwith previous reports (5), positive hits included cohorts enriched forboth HIF-1 and HIF-2 binding, as well as loci unique for HIF-1 orHIF-2 occupancy alone (Fig. 1B). Bioinformatic analyses for de novomotifs (MEME) and transcription factor binding sites (9) identifiedthe canonical hypoxia response element (HRE) motif 5′-RCGTG-3′ as a highly represented consensus sequence. Inde-pendent ChIP–quantitative PCR (qPCR) analyses confirmedenrichment at discrete loci compared with IgG control, with all
loci displaying a threshold of twofold enrichment in hypoxia relativeto an HRE-negative region. Importantly, integration of ChIP–chip and ChIP–qPCR analyses confirmed robust HIF-1α en-richment of established HIF-1 targets (Fig. 1C).In the course of this analysis, we identified the CK pathway as
a unique target of HIF-2. Indeed,CKM featured as one of the mostsignificantly enriched HIF-2α–specific loci. Further interrogationof this chip cluster revealed additional genes implicated in Crmetabolism, namely CKB, mitochondrial CK (CKMT1), and the
Fig. 1. HIF-1 and HIF-2 ChIP-on-chip. (A) Accumu-lation of nuclear HIF-1α and HIF-2α in Caco-2 IECs innormoxia (0 h) and hypoxia. (B) Heat maps gener-ated from normalized smoothed ratios of ChIP en-richment (GENE-E), showing common targets (Left)and discrete promoter occupancy (Right). (Inset)Functional annotation clustering of ChIP–chip hitsrevealed genes involved in Cr metabolism as HIF-2–specific targets (C) ChIP–chip results were validatedby qPCR analysis following ChIP using anti–HIF-1α,anti–HIF-2α, or IgG. Bar graph represents fold en-richment of ChIP DNA compared with input chro-matin (n = 3).
Fig. 2. HIF-2–dependent up-regulation of CK enzymes. (A) qPCR analysis of CK enzyme (CKM, CKB, CKMT1A) and Cr transporter (SLC6A8) expression in Caco-2IECs (20% or 1% O2, 6 h; n = 4). (B) Immunoblot analysis of CK levels in Caco-2 IECs (1% O2). (C) Immunoblot analysis of nuclear (HIF) and whole-cell lysates (CK)from Caco-2 treated with vehicle (DMSO) or PHD inhibitor (AKB; 100 μM) for 24 h. (D) qPCR analysis of CK expression in shCtrl, shHIF-1α, or shHIF-2α Caco-2 (20%or 1% O2, 6 h; n = 4) *P < 0.05. (E) Immunoblot analysis of CK levels in shCtrl, shHIF-1α, and shHIF-2α IECs (20% or 1% O2, 24 h). (F, Schematic) Proximal promoterof human CKM and CKB; TSS designated +1. T84 cells were nucleofected (24 h) and cultured in 20% or 1%O2 for 24 h. Data represent ratio of promoter-luc:pRen-luc activity (n = 4; *P < 0.05). (G) Caco-2 IECs were cotransfected with pCK-Luc and pcDNA3.1 vector as control (C) or increasing ratios of pcDNA3.1–HIF-1αΔODD orpcDNA3.1–HIF-2αΔODD (n = 3; *P < 0.0001). (H and I) Luciferase activity in lysates cotransfected with full-length (WT), truncated pCK-Luc (schematic) (n = 3; *P <0.0001), or pCK-Luc harboring mutations in distal (HRE2) and proximal (HRE1) sites, and HIF plasmids at a 1:5 ratio (n = 4; *P < 0.05 by ANOVA).
Glover et al. PNAS | December 3, 2013 | vol. 110 | no. 49 | 19821
CELL
BIOLO
GY

major Cr transporter (SLC6A8) (Fig. 1B, Table S1). The CKshuttle defines a key metabolic pathway for temporal and spatialenergy buffering. CK regulates cellular ATP through the reversibletransfer of high-energy phosphate from phospho-Cr (PCr), con-necting sites of ATP generation with compartmentalized ATPutilization (10). ChIP–qPCR quantitation of HIF binding to pro-moter regions of CKB and CKM revealed specific occupancy ofHIF-2 over HIF-1, with increased enrichment in hypoxia (Fig. 1C).To investigate correlation of HIF-2 binding to hypoxia-induced
gene expression, we evaluated expression of Cr metabolic genes byqPCR. Transcript levels of Cr transporter (SLC6A8) and cytosolic(CKM, CKB) CK isozymes were increased in a time-dependentmanner in hypoxia (Fig. 2A). CK protein levels were similarlyincreased both in hypoxic Caco-2 lysates (Fig. 2B) and in cellstreated with the PHD inhibitor AKB to stabilize HIF-1α and HIF-2α protein under normoxic conditions (Fig. 2C). These data in-dicate that CK is induced in IECs in response to both hypoxia andpharmacologic HIF stabilization, consistent with HIF-mediatedtranscription.
CK Enzymes Are Induced in Hypoxic IECs in a HIF-2–DependentManner. To determine the specificity of HIF-induced CK expres-sion, we used short hairpin RNA (shRNA) knockdown to in-dividually deplete HIF-1α and HIF-2α (Fig. S1A). qPCR analysesdemonstrated that CK mRNA levels were significantly inducedby 6 h in response to hypoxia in sh control (shCtrl) and HIF-1αknockdown cells, but not in cells with specific depletion of HIF-2α(Fig. 2D; P < 0.05). Moreover, knockdown of HIF-2α selectivelyattenuated basal levels and hypoxic induction of CK protein(Fig. 2E).Analysis of human CKM and CKB TSS-flanking gene se-
quences revealed two candidate HRE sites in each promoter(Fig. 2F). To complement our ChIP–qPCR analysis (Fig. 1C), 1.4kb fragments of CK promoter sequences were cloned upstreamof a luciferase reporter. T84 IECs were nucleofected with eitherpCK- or pHRE-Luc reporter constructs, and exposed to 20% or1% O2 for 24 h. For each construct analyzed, increased promoteractivity was observed in hypoxia (Fig. 2F). To distinguish be-tween HIF-1α– and HIF-2α–mediated influences, pCK-Luc wascotransfected into Caco-2 cells with increasing concentrationsof oxygen-stable HIF-α expression vectors. Dose-dependent in-duction of pCK-Luc was observed with HIF-2α, but not HIF-1α,consistent with ChIP–qPCR and shHIF-2α findings (Fig. 2G).Promoter activity was significantly diminished upon deletion ofthe proximal HRE (HRE1) in both constructs (Fig. 2H). Muta-tional analysis revealed selective repression of HIF-2–regulatedpromoter activity upon mutation of HRE1 in pCK-Luc (Fig. 2I).Taken together, these findings indicate that HIF-2 specificallybinds to proximal HRE sites in the promoter region ofCK genes todirectly activate their transcription in IECs.
Cytosolic CK Is Localized to Apical AJs. Differentially localized CKisozymes facilitate a high-energy PCr/CK circuit in polarizedcells, wherein mitochondria are located distantly from regions ofATP consumption (10). Previous studies defined CKB and mi-tochondrial CK as distinct terminals of this circuit in IECs (11,12). Functional coupling between CKB and myosin II (MII) atthe circumferential ring is proposed to confer a spatial energeticadvantage for myosin ATPase activity. MII isoforms localizeto cadherin junctions, where they regulate AJ development andforce generation through diverse mechanisms (13–15). In po-larized IEC, both MIIA and cytosolic CKs colocalized with actinand E-cadherin at apical junctions (Fig. S2 A and B), andcoimmunoprecipitation analyses revealed enrichment of MII infractions precipitated by an anti-CK antibody (Fig. S2C). De-tailed optical section analysis revealed that CKM and CKB arehighly enriched at AJs, with further distribution of CKM throughoutthe lateral membrane (Fig. 3A).Intercellular actomyosin forces that support mature apical
junctions are transmitted through AJs associated with actin fila-ments (16). Moreover, TJ assembly and sealing of the paracellular
space is AJ-dependent. Given that CKs accumulate at AJs, func-tionally support MII ATPase, and represent HIF-2 target genes,we speculated that apical junctions may be altered in the absenceof HIF-2 signaling. Localization of E-cadherin and ZO-1 in shCtrlmonolayers revealed linear contours of apical lateral membranes(Fig. 3B). In contrast, HIF-2α–depleted cells displayed non-uniform, undulating junctions, consistent with attenuated in-tercellular tension and junction formation (15). Transepithelialelectrical resistance (TER) measurements further indicated abro-gated barrier function (57% ± 3.5%) of HIF-2α–depleted IECscompared with control monolayers (Fig. S3B). Moreover, assess-ment of TER development following Ca2+ switch assay revealedthat barrier formation of HIF-2α knockdown IECs was significantlyattenuated compared with control cells (Fig. 3C).
CK Contributes to Junctional Assembly After Ca2+ Switch. To directlyassess the contribution of CK to AJC assembly, we monitoredthe influence of altered CK signaling on TER development fol-lowing Ca2+ switch. Cr supplementation markedly enhanced barrierrecovery (Fig. 3C), whereas inhibition of CK using dinitro-fluorobenzene (DNFB) (17) impaired recovery in a dose-dependentmanner (Fig. 3D). CK inhibition correlated with a reduction inCr and PCr metabolite levels, and increased IEC Cr/PCr ratio(Fig. S3A). To evaluate junction assembly dynamics, IECs were
Fig. 3. CK contributes to apical junction assembly. (A) Caco-2 cells werestained for CKB, CKM, and E-cadherin. (Scale bar, 20 μm.) (B) Lateral mem-brane boundaries in Caco-2 shCtrl and shHIF-2α cells stained for E-cadherinand ZO-1. (Scale bar, 10 μm.) (C) shCtrl and HIF-2α–depleted T84 cells onpermeable inserts were treated with 2 mM EDTA for 5 min and switchedto HBSS with normal Ca2+ (1.8 mM). TER was measured over time; data rep-resent percent recovery over time relative to baseline values (n = 3, P <0.0001 by ANOVA). (D) T84 cells preloaded with 10 mM Cr monohydratewere subjected to Ca2+ switch, and TER measured over time (n = 3, P <0.0001 by ANOVA). (E) T84 cells were subjected to Ca2+ switch in the pres-ence of CK inhibitor DNFB. TER was monitored over time (data relative to noDNFB; n = 3, *P < 0.05). (F) T84 IECs incubated in low Ca2+ media (16 h)before switching to normal Ca2+, with or without 10 μM DNFB, were stainedfor E-cadherin, ZO-1, CKM, and CKB. (Scale bar, 10 μm.)
19822 | www.pnas.org/cgi/doi/10.1073/pnas.1302840110 Glover et al.
cultured overnight in low-Ca2+ media to permit cellular de-polarization and protein translocation. As previously characterized(18), E-cadherin and ZO-1 localized to subapical ring-likestructures upon extended Ca2+ depletion (Fig. 3F), whereas CKlabeling was observed throughout the cytoplasm. One hour post-Ca2+ repletion, immunolabeling revealed nascent AJs and initia-tion of TJ assembly in control cells. Junctional assembly wasmarkedly retarded in cells treated with DNFB. By 24 h, controlmonolayers assumed “mature” AJC lateral membrane staining.Strikingly, CK inhibition resulted in an undulating junctionalstaining pattern reminiscent of HIF-2α–depleted cells (Fig. 3B).Previous studies have shown that a basal level of MII phosphory-lation and activity is necessary for apical junction integrity (13, 19).As such,HIF-mediated induction of CK isozymesmight temporallybuffer PCr supply and ATPase activity during periods of energeticstress. To test this hypothesis, we exposed IECs to 1% O2 in thepresence or absence of DNFB. CK inhibition markedly decreasedTERs in T84 monolayers exposed to 1% O2 (Fig. S3; P < 0.01),indicating that barrier function is compromised under hypoxicconditions upon inhibition of the CK/PCr shuttle.
Loss of HIF Signaling Results in Altered CK/PCr Circuit in IECs andPotentiates Murine Colitis. To validate the regulation of CK byHIF signaling in vivo, we evaluated expression of Ckb in IECsderived from Hif-1β IEC-specific knockout mice (Fig. S4A;Hif-1β−/−) compared with WT (Hif-1β+/+) littermates. As outlinedin Fig. S4B, Ckb expression was repressed (51.5% ± 5.6%) inHif-1β−/− IECs, whereas levels of Pgk1, a HIF-1 target gene, were
repressed by 29% ± 6.5%. Analysis of Cr and PCr metabolitelevels in epithelial isolates revealed a significant increase in theCr/PCr ratio of Hif-1β−/− IECs (Fig. S4C), consistent with CKinhibition in vitro (Fig. S3A). As an important correlative, weused the dextran sulfate sodium (DSS) and 2,4,6-trinitrobenzenesulfonic acid (TNBS) colitis models to ascertain the effect ofaltered CK metabolism on IEC permeability. Acute DSS resul-ted in significantly increased intestinal permeability in Hif-1β−/−mice compared with Hif-1β+/+ littermate controls, as determinedby translocation of FITC–dextran into serum post–oral admin-istration (Fig. S4D). Similarly, Hif-1β−/− mice demonstrated in-creased colitic disease activity in response to TNBS, resulting inattenuated survival (Fig. S4E), augmented colonic shortening(Fig. S4F), and increased serum FITC–dextran compared withHif-1β+/+ controls (Fig. S4G; 4.4- ± 1.6-fold; P = 0.0225). Fur-ther, increased inflammatory cytokine expression was deter-mined in Hif-1β+/+ mice versus controls (Fig. S4H). Collectively,these findings indicate that IEC Hif signaling mediates a pro-tective barrier effect in these IBD models (8).
Cr Supplementation Attenuates the Severity of Murine Colitis. Bar-rier maintenance requires an intricate balance between AJC andcytoskeletal rearrangements that facilitate continual IEC turn-over and transepithelial transport, both energy-dependent pro-cesses. Reduced ATP levels have been observed in inflamed IBDbiopsies (20), and noninflamed tissues from Crohn disease (CD)patients are more sensitive to uncoupling of oxidative phos-phorylation (21). Dietary Cr supplementation has been shown to
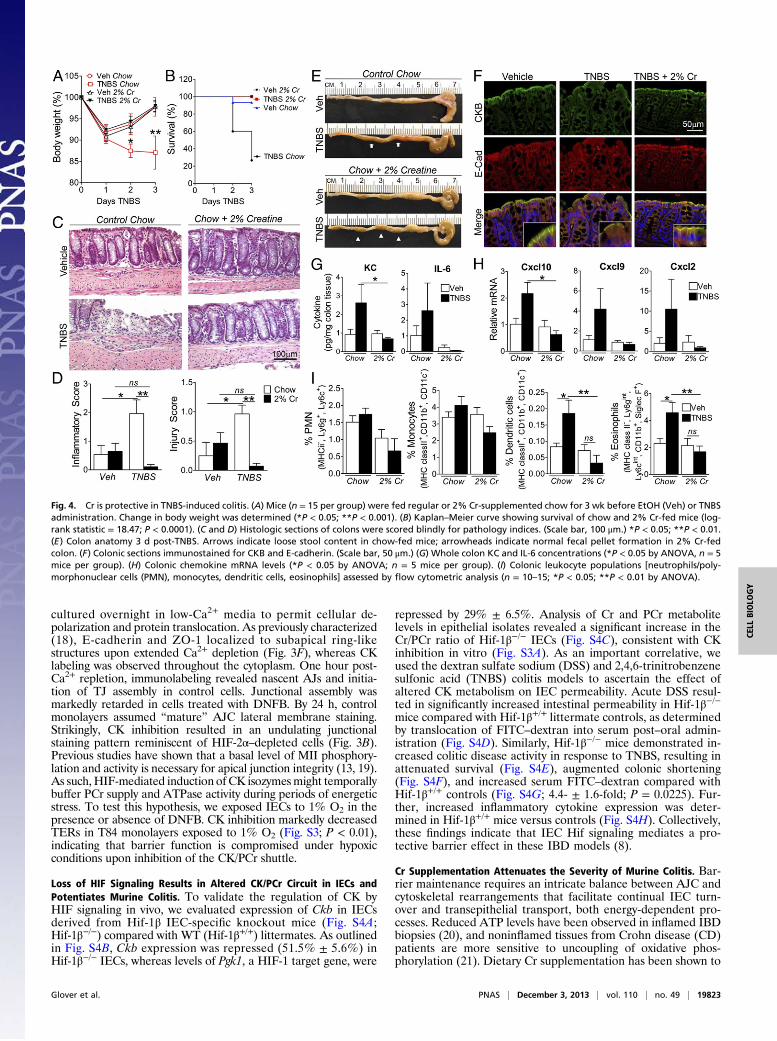
Fig. 4. Cr is protective in TNBS-induced colitis. (A) Mice (n = 15 per group) were fed regular or 2% Cr-supplemented chow for 3 wk before EtOH (Veh) or TNBSadministration. Change in body weight was determined (*P < 0.05; **P < 0.001). (B) Kaplan–Meier curve showing survival of chow and 2% Cr-fed mice (log-rank statistic = 18.47; P < 0.0001). (C and D) Histologic sections of colons were scored blindly for pathology indices. (Scale bar, 100 μm.) *P < 0.05; **P < 0.01.(E) Colon anatomy 3 d post-TNBS. Arrows indicate loose stool content in chow-fed mice; arrowheads indicate normal fecal pellet formation in 2% Cr-fedcolon. (F) Colonic sections immunostained for CKB and E-cadherin. (Scale bar, 50 μm.) (G) Whole colon KC and IL-6 concentrations (*P < 0.05 by ANOVA, n = 5mice per group). (H) Colonic chemokine mRNA levels (*P < 0.05 by ANOVA; n = 5 mice per group). (I) Colonic leukocyte populations [neutrophils/poly-morphonuclear cells (PMN), monocytes, dendritic cells, eosinophils] assessed by flow cytometric analysis (n = 10–15; *P < 0.05; **P < 0.01 by ANOVA).
Glover et al. PNAS | December 3, 2013 | vol. 110 | no. 49 | 19823
CELL
BIOLO
GY

confer protection in multiple disease models that display bio-energetic dysregulation (22, 23). We thus sought to evaluate theinfluence of Cr supplementation on mucosal inflammatory path-ogenesis in murine models of IBD.Mice were fed either normal chow or chow supplemented with
2% Cr (23), and exposed to 3% (wt/vol) DSS in drinking water.Cr-supplemented animals exhibited markedly reduced suscepti-bility to DSS-induced colitis, demonstrated by attenuated weightloss and colon shortening (Fig. S5 A and B). Histologic exami-nation of control Cr-fed mouse colons revealed no overt alterations,confirmed by blinded scoring (Fig. S5 C and D). DSS-induced co-litis in chow-fed animals resulted in extensive crypt loss, epithelialdamage (Fig. S5E) and inflammatory cell infiltrate, as well as lossof solid stool (Fig. S5E). Interestingly, Cr supplementation atten-uated histologic indices and promoted fecal pellet formation.Compared with 2% Cr-fed mice, chow-fed DSS mice displayedincreased intestinal permeability (Fig. S5F). Consistent with this,serum and colonic levels of proinflammatory mediators were re-duced in mice fed 2% Cr compared with chow alone (Fig. S6 Aand B). HPLC analysis revealed increased colonic Cr in both Cr-fed vehicle and DSS-challenged mice compared with chow-fedcohorts (Fig. S6C). Interestingly, DSS challenge itself resulted inelevated Cr levels, indicating that Cr metabolism is altered inacute mucosal inflammation.We next extended these findings to the independent TNBS
colitis model. Within 2 d of TNBS challenge, chow-fed micedisplayed progressive weight loss relative to vehicles (Fig. 4A;P < 0.05). Strikingly, Cr supplementation rendered mice re-fractory to TNBS-induced weight loss. To determine whetherattenuated wasting translates to a survival benefit in Cr-fed mice,we recorded mortality following TNBS challenge. Cr supple-mentation resulted in enhanced survival over the colitic course(Fig. 4B). Histologic analysis of colon biopsies (Fig. 4C) revealedsignificant protection conferred by Cr on both injury and in-flammatory indices (Fig. 4D), which correlated with fecal pelletformation (Fig. 4E). Vehicle IECs displayed apical enrichmentof CKB that colocalized with E-cadherin (Fig. 4F). With TNBSchallenge in chow-fed animals, patchy disruption of epithelialand crypt architecture associated with loss of E-cadherin junc-tional staining and colocalization with CKB. In contrast, bothIEC integrity and junctional staining was preserved in Cr-fed miceexposed to TNBS. To ascertain whether epithelial integrity corre-lates with abrogated inflammatory stimuli, we profiled inflammatorymediators and lamina propria leukocyte populations (Fig. S7). Crsupplementation reduced colonic expression of proinflammatorychemokines compared with chow-fed mice (Fig. 4 G and H).Moreover, we identified a significant reduction in both dendritic(P < 0.001) and eosinophil (P < 0.01) populations in Cr-treatedversus chow-fed TNBS mice (Fig. 4I). Taken together, these find-ings strongly imply that Cr supplementation exerts a beneficial ef-fect in colitis-associated mucosal inflammation.
Attenuated CK Levels Suggest Altered Cr Signaling in IntestinalBiopsies from IBD Patients. To investigate the relevance of ourfindings to human disease, we stained archived paraffin-embeddedcolon biopsy sections from IBD patients with antibodies to CKBand ZO-1. Similar to murine colonic sections, CKB exhibited dif-fuse cytoplasmic staining in non-IBD tissue, with enrichment atapical junctions colocalized with ZO-1 (Fig. 5A). Interestingly, incolon biopsies from IBD patients, CKB displayed preferentialjunctional staining relative to cytoplasmic compartments (Fig. 5A).As a corollary, we compared expression of CK enzymes in in-testinal biopsies from IBD and non-IBD subjects. A significantdecrease in transcript levels of CKM, CKB, and CKMT1 wasobserved in IBD patient tissue compared with non-IBD controls(Fig. 5B). These observations implicate marked alterations in theCK/PCr energy circuit in chronic human mucosal inflammation.
DiscussionThe cytoskeletal network that supports apical IEC junctions isamong the most highly ordered arrays of actin filaments in nature
(24). The circumferential actomyosin ring mediates selectivebarrier function in both health and disease (25), and is a primarytarget for molecular remodeling by diverse inflammatory stimuli(26). Moreover, actomyosin contraction is central to homeostaticand pathologic IEC shedding and barrier restitution (27). Epi-thelial function and barrier integrity is further regulated by lowoxygen tension, through HIF-elicited adaptive pathways (28, 29).Notable for the present work, a recent study highlighted a rolefor epithelial Hif-2α in mediating proinflammatory epithelialresponses in murine models of chemical and bacterial colitis, incontrast to the barrier protective program elicited by Hif-1α (30).As epithelial Hif1signaling has also been shown to promote in-flammation in one study using DSS colitis (31), such findingslikely reflect the context-dependent nature of these models. In-deed, the temporal kinetics of HIF-1α and HIF-2α in IBD appearto be different, with HIF-1α stabilized earlier in the diseasecourse (as early as day 1 in TNBS colitis) (8) and a more prom-inent HIF-2α signal at later time points (by day 3 in DSS colitis)(30). Nonetheless, head-to-head comparisons have yet to be done,and the relative contribution of HIF-1 versus HIF-2 to mucosalsignaling defines a key question. In the current work, we identifyCK as a unique HIF target, and define a role for the CK energyshuttle in AJC assembly and epithelial homeostasis.Initial studies of the IEC CK shuttle demonstrated functional
coupling between CKB and myosin in isolated brush borders,specifically at the circumferential actomyosin ring (11, 12). MII isthe major IEC cytoskeletal motor that converts ATP hydrolysisinto mechanical forces, mediating the static tension and contrac-tility of actin filaments. Interestingly, isoform-specific knockdownrevealed differential roles for MIIA and MIIB in coordinatingE-cadherin–based intercellular junction dynamics (14). Our obser-vations indicate that CKs localize to apical AJs, where they likelyconstitute one terminal of a functional PCr/CK energy circuit thatsupplies energy to myosin motors. In addition to PCr generationvia oxidative phosphorylation coupling, a subset of cytosolic CKhas been shown to associate with glycolytic enzymes to facilitatePCr repletion, wherein HIF-1–mediated induction of the glycolyticpathway for ATP generation represents a central adaptation forcell survival (10). Given the importance of junction integrity toepithelial homeostasis, apical ATPases associated with the AJCseem likely candidates for prioritized PCr targeting under con-ditions of energetic stress. Coupled with our findings, this posits an
Fig. 5. CK expression is reduced in chronic IBD. (A) Representative image ofcolon biopsy from a non-IBD control (Upper) and ulcerative colitis (UC) pa-tient (Lower) immunostained for CKB and apical TJ marker ZO-1. (Scale bar,20 μm.) (B) Transcript levels of CKM, CKB, and CKMT1A assessed by qPCR incDNA derived from non-IBD control (n = 5), CD (n = 21), and UC (n = 17)individuals. Data expressed as relative Ct values, calculated as 2-ΔCt (Ct target –Ct actin) and analyzed by ANOVA.
19824 | www.pnas.org/cgi/doi/10.1073/pnas.1302840110 Glover et al.

intriguing link between HIF signaling and a unique role for Cr/PCr in junctional energetics. This complex metabolic regulationunderscores the importance of balancing ATP supply andconsumption in cells with high and fluctuating energy demands.In the present studies, Cr supplementation was shown to
provide substantial benefits in murine colitis. This was mainlyattributable to enhanced cellular energetics through the PCr/CKcircuit, but may also be explained in part by Cr-mediated re-duction of oxidative stress (10). Cr has been shown to possessantioxidant properties (32), whereas mtCK activation was foundto reduce mitochondrial reactive oxygen species (ROS) (33).These properties define an intriguing overlap with HIF-2 tran-scriptional programs, as HIF-2 regulates expression of a numberof antioxidant enzymes (34). Furthermore, loss of HIF-2α resultsin increased ROS and redox imbalance in multiple cellularsettings. That HIF-mediated induction of CK may representan antioxidant pathway to buffer redox homeostasis in hypoxiccells certainly warrants further investigation.Finally, our data demonstrating attenuated expression of CK
enzymes in IBD tissue suggest that intestinal Cr metabolism andPCr/CK energetics may be compromised in at least a subset ofIBD patients. This observation is noteworthy on two levels. First,bioenergetic dysregulation resulting from impaired CK shuttlingmay contribute to the increased barrier permeability character-istic of inflamed mucosae (25). Second, chronic inflammationassociated with IBD is a major risk factor for colitis-associated
cancer. A number of studies have now identified reduced levelsof CKB in colonic tumors (35, 36), whereas studies using dom-inant negative CKB mutants have defined a correlation withmolecular markers of epithelial-to-mesenchymal transition in co-lon cancer cells (37). Taken together, these observations providea compelling argument for Cr supplementation as an adjuvanttherapy to promote epithelial restitution and ameliorate mucosalinflammation via enhanced cellular energetics.
Materials and MethodsPlease refer to SI Materials and Methods for detailed descriptions ofthe methods.
Animal Studies. Colitis models were induced as described in SI Materialsand Methods.
ChIP–Chip. Input and HIF ChIP-DNA were hybridized to a custom micro-array designed to cover a genome-wide set of human promoter regionsof ≤2 kb (Switchgear Genomics). All microarray data have been de-posited in National Center for Biotechnology Information’s Gene Ex-pression Omnibus (GSE43108).
ACKNOWLEDGMENTS. We gratefully acknowledge Frank Gonzalez (NationalCancer Institute) for his kind donation of the Hif-1β flox mice. This work wassupported by National Institutes of Health Grants DK50189, HL60569, andDK095491, and by the Crohn’s and Colitis Foundation of America.
1. Hirokawa N, Keller TC, 3rd, Chasan R, Mooseker MS (1983) Mechanism of brushborder contractility studied by the quick-freeze, deep-etch method. J Cell Biol 96(5):1325–1336.
2. Ivanov AI, Parkos CA, Nusrat A (2010) Cytoskeletal regulation of epithelial barrierfunction during inflammation. Am J Pathol 177(2):512–524.
3. Colgan SP, Taylor CT (2010) Hypoxia: An alarm signal during intestinal inflammation.Nat Rev Gastroenterol Hepatol 7(5):281–287.
4. Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol CellBiol 5(5):343–354.
5. Ratcliffe PJ (2007) HIF-1 and HIF-2: Working alone or together in hypoxia? J Clin Invest117(4):862–865.
6. Majmundar AJ, Wong WJ, Simon MC (2010) Hypoxia-inducible factors and the re-sponse to hypoxic stress. Mol Cell 40(2):294–309.
7. Giatromanolaki A, et al. (2003) Hypoxia inducible factor 1alpha and 2alpha over-expression in inflammatory bowel disease. J Clin Pathol 56(3):209–213.
8. Karhausen J, et al. (2004) Epithelial hypoxia-inducible factor-1 is protective in murineexperimental colitis. J Clin Invest 114(8):1098–1106.
9. Cartharius K, et al. (2005) MatInspector and beyond: Promoter analysis based ontranscription factor binding sites. Bioinformatics 21(13):2933–2942.
10. Wallimann T, Tokarska-Schlattner M, Schlattner U (2011) The creatine kinase systemand pleiotropic effects of creatine. Amino Acids 40(5):1271–1296.
11. Gordon PV, Keller TC, 3rd (1992) Functional coupling to brush border creatine kinaseimparts a selective energetic advantage to contractile ring myosin in intestinal epi-thelial cells. Cell Motil Cytoskeleton 21(1):38–44.
12. Keller TC, 3rd, Gordon PV (1991) Discrete subcellular localization of a cytoplasmic anda mitochondrial isozyme of creatine kinase in intestinal epithelial cells. Cell MotilCytoskeleton 19(3):169–179.
13. Ivanov AI, et al. (2007) A unique role for nonmuscle myosin heavy chain IIA in reg-ulation of epithelial apical junctions. PLoS ONE 2(7):e658.
14. Smutny M, et al. (2010) Myosin II isoforms identify distinct functional modules thatsupport integrity of the epithelial zonula adherens. Nat Cell Biol 12(7):696–702.
15. Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M (2010) alpha-Catenin asa tension transducer that induces adherens junction development. Nat Cell Biol 12(6):533–542.
16. Lecuit T, Lenne PF (2007) Cell surface mechanics and the control of cell shape, tissuepatterns and morphogenesis. Nat Rev Mol Cell Biol 8(8):633–644.
17. Linton JD, et al. (2010) Flow of energy in the outer retina in darkness and in light. ProcNatl Acad Sci USA 107(19):8599–8604.
18. Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA (2005) Differential roles for actinpolymerization and a myosin II motor in assembly of the epithelial apical junctionalcomplex. Mol Biol Cell 16(6):2636–2650.
19. Ivanov AI, McCall IC, Parkos CA, Nusrat A (2004) Role for actin filament turnover anda myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junc-tional complex. Mol Biol Cell 15(6):2639–2651.
20. Schürmann G, et al. (1999) Transepithelial transport processes at the intestinal mu-cosa in inflammatory bowel disease. Int J Colorectal Dis 14(1):41–46.
21. Söderholm JD, et al. (2002) Augmented increase in tight junction permeability byluminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 50(3):307–313.
22. Klivenyi P, et al. (1999) Neuroprotective effects of creatine in a transgenic animalmodel of amyotrophic lateral sclerosis. Nat Med 5(3):347–350.
23. Lin YS, et al. (2011) Dysregulated brain creatine kinase is associated with hearingimpairment in mouse models of Huntington disease. J Clin Invest 121(4):1519–1523.
24. Mooseker MS (1985) Organization, chemistry, and assembly of the cytoskeletal ap-paratus of the intestinal brush border. Annu Rev Cell Biol 1:209–241.
25. Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat RevImmunol 9(11):799–809.
26. Koch S, Nusrat A (2009) Dynamic regulation of epithelial cell fate and barrier functionby intercellular junctions. Ann N Y Acad Sci 1165:220–227.
27. Marchiando AM, et al. (2011) The epithelial barrier is maintained by in vivo tightjunction expansion during pathologic intestinal epithelial shedding. Gastroenterol-ogy 140(4):1208–1218.
28. Glover LE, Colgan SP (2011) Hypoxia and metabolic factors that influence in-flammatory bowel disease pathogenesis. Gastroenterology 140(6):1748–1755.
29. Mastrogiannaki M, et al. (2009) HIF-2alpha, but not HIF-1alpha, promotes iron ab-sorption in mice. J Clin Invest 119(5):1159–1166.
30. Xue X, et al. (2013) Endothelial PAS domain protein 1 activates the inflammatoryresponse in the intestinal epithelium to promote colitis in mice. Gastroenterology145(4):831–841.
31. Shah YM, et al. (2008) Hypoxia-inducible factor augments experimental colitisthrough an MIF-dependent inflammatory signaling cascade. Gastroenterology 134(7):2036–2048.
32. Lawler JM, Barnes WS, Wu G, Song W, Demaree S (2002) Direct antioxidant propertiesof creatine. Biochem Biophys Res Commun 290(1):47–52.
33. Meyer LE, et al. (2006) Mitochondrial creatine kinase activity prevents reactive oxygenspecies generation: Antioxidant role of mitochondrial kinase-dependent ADP re-cy-cling activity. J Biol Chem 281(49):37361–37371.
34. Scortegagna M, et al. (2003) Multiple organ pathology, metabolic abnormalities andimpaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet 35(4):331–340.
35. Balasubramani M, Day BW, Schoen RE, Getzenberg RH (2006) Altered expression andlocalization of creatine kinase B, heterogeneous nuclear ribonucleoprotein F, andhigh mobility group box 1 protein in the nuclear matrix associated with colon cancer.Cancer Res 66(2):763–769.
36. Friedman DB, et al. (2004) Proteome analysis of human colon cancer by two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics 4(3):793–811.
37. Mooney SM, et al. (2011) Creatine kinase brain overexpression protects colorectalcells from various metabolic and non-metabolic stresses. J Cell Biochem 112(4):1066–1075.
Glover et al. PNAS | December 3, 2013 | vol. 110 | no. 49 | 19825
CELL
BIOLO
GY