control of cardiac-specific transcription by p300 through myocyte
TRANSCRIPT

Control of Cardiac-Specific Transcription by p300
Through Myocyte Enhancer Factor-2D
Tatiana I. Slepak§, Keith A. Webster§, Jie Zang§, Howard Prentice¶, Ann O’Dowd¶, Martin N.
Hicks†, and Nanette H. Bishopric§*.
§Department of Molecular and Cellular Pharmacology, University of Miami, Miami, FL, USA;
Departments of ¶Molecular Genetics and †Medical Cardiology, Glasgow Royal Infirmary,
University of Glasgow, Glasgow, UK
Running title: p300 regulates cardiac genes through MEF-2D
Key words: CREB-binding factor, heart, differentiation, adenovirus E1A, basic helix-loop-helix
proteins, muscle-specific genes
*Address correspondence to:
Nanette H. Bishopric, M.D., F.A.C.C.University of Miami Department of Molecular and Cellular PharmacologyP.O. Box 106189 (R-189)Miami, FL 33101e-mail: [email protected] (phone)305-243-6082 (fax)
Copyright 2000 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on November 28, 2000 as Manuscript M004625200 by guest on A
pril 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
2
SUMMARY
The transcriptional integrator p300 regulates gene expression by interaction with sequence-
specific DNA binding proteins and local remodelling of chromatin. p300 is required for cardiac-
specific gene transcription, but the molecular basis of this requirement is unknown. Here we report
that the MADS-box transcription factor MEF-2D acts as the principal conduit for cardiac
transcriptional activation by p300. p300 activation of the native 2130 bp human skeletal α-actin
promoter required a single hybrid MEF-2/GATA-4 DNA motif centered at -1256bp. Maximal
expression of the promoter in cultured myocytes and in vivo correlated with binding of both MEF-2
and p300, but not GATA-4, to this AT-rich motif. p300 and MEF-2 were co-precipitated from
cardiac nuclear extracts by an oligomer containing this element. p300 was found exclusively in a
complex with MEF-2D at this and related sites in other cardiac-restricted promoters. MEF-2D, but
not other MEFs, significantly potentiated cardiac-specific transcription by p300. No physical or
functional interaction was observed between p300 and other factors implicated in skeletal actin
transcription, including GATA-4, TEF-1, or SRF. These results show that in the intact cell, p300
interactions with its protein targets are highly selective, and that MEF-2D is the preferred channel
for p300-mediated transcriptional control in the heart.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
3
INTRODUCTION
Transcriptional coactivators, or integrators, are members of a class of transcription factors
that bring about tissue- and stimulus-specific changes in gene expression by coordinating groups
of signal responsive proteins in the cell. Coactivators do not bind independently to DNA, but are
thought to stabilize the formation of specific transcription factor-DNA complexes, and to cause the
local unwinding of chromatin directly or by recruitment of histone-remodelling enymes 1;2.
Specific coactivator families have been identified for regulation of steroid hormone-responsive
genes (SRC-1, SRC-3) 3-5 and fatty acid regulatory proteins (PGC-1) 6;7. Gene targeting
experiments have confirmed that the activities of these co-activators in vivo are highly tissue-
restricted, although the molecular basis of this specificity is not well understood 4;6;8-10.
An important subgroup of coactivators is represented by the closely related transcriptional
integrators p300 and CBP (CREB-Binding Protein). These large proteins share extensive sequence
homology and many structural features, and appear to have arisen as part of an ancestral gene
duplication 11-13. The cellular levels and activities of p300 and CBP are tightly regulated through
steroid, adrenergic and growth factor signals as well as during the cell cycle 14 . Critical roles for
p300 and CBP have been identified in growth, differentiation, apoptosis and tissue-specific gene
expression, reflecting their interaction with multiple cellular regulatory proteins 13 . A particular
requirement for p300 is observed in the heart. Mice deficient in p300 have an embryonic lethal
phenotype characterized by failure of cardiac myocyte proliferation and muscle-specific gene
expression 10. In the post-natal heart, adenovirus E1A selectively inhibits cardiac muscle-specific
gene expression by binding to p300 and/or related proteins 15 16 17 . Although p300 is also required
for skeletal muscle gene transcription by the tissue-specific basic helix-loop helix protein MyoD,
the heart lacks any equivalent to this transcription factor 18 {Eckner, Yao, et al. 1996 #3207}. The
molecular partners and pathways of p300-mediated transcriptional regulation in the heart are not
known.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
4
Expression of the human skeletal α-actin gene is tightly restricted to striated muscle. In the
myocardium, skeletal actin is one of a group of "fetal" genes upregulated in response to
hypertrophic stresses such as pressure overload in vivo, and during the response to adrenergic
stimulation, growth factors and other neurohormonal effectors in cell culture models 19-22. Although
skeletal actin is the predominant sarcomeric actin isoform in the human heart, it is further
upregulated during hypertrophy 23-25, and is considered an important marker of hypertrophy in the
rat 20;26-29. Regulation of skeletal actin expression in the heart involves both extracellular signal-
responsive and cardiac-specific transcription factors; the latter have not yet been identified. The -
2130 bp human skeletal actin promoter has two major transcriptional activation domains, proximal
(-153 to -87) and distal (-2130 to -710) which are required for maximal tissue-specific expression
in both skeletal and cardiac myocytes 19 . The proximal promoter contains functional binding sites
for Sp-1, SRF, and TEF-1 30. Here, we report the sequence of the distal human skeletal actin (hSA)
promoter, and demonstrate that only one DNA motif within the entire 2130 bp transcriptional unit is
capable of transmitting the p300 activation signal. This motif, centered at -1256, binds the MADS-
box transcription factor MEF-2 as well as GATA-4, and is required for maximal expression in both
neonatal and adult rat cardiac myocytes. The motif also binds endogenous cardiac nuclear p300,
and coordinates synergistic activation of the hSA promoter by p300 and MEF-2D. Remarkably,
p300-dependent activation did not involve interaction with GATA-4, SRF, or TEF-1, indicating that
p300 selectively targets MEF-2 in the context of a native promoter. Based on these findings and
on recent data showing interactions between MEF-2 and HDAC-4 31, we propose that MEF-2
governs expression of the cardiac phenotype by acting as a primary channel for chromatin
remodelling on cardiac-specific promoters.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
5
EXPERIMENTAL PROCEDURES
Materials
Expression vectors encoding MEF-2 A-D were generously provided by Dr. Eric Olson. A
p300 expression vector (pCMVp300β) was the kind gift of Dr. R. Eckner. Polyclonal antibodies
against GATA-4, MEF-2 and p300 were obtained from Santa Cruz Biotechnology. The
monoclonal antibody against p300 (NM-11) was supplied by Pharmingen, and restriction enzymes
were from New England Biolabs. All other molecular biology reagents were purchased from
Sigma except as indicated, and were of the highest grade available.
Plasmid construction
The distal hSA promoter sequence was determined by automated DNA sequencing using an
ABI 377 DNA sequencer in the Biochemistry Core Facility, University of California at San
Francisco. The resulting information was used to construct skeletal actin promoter-luciferase
chimeras. A 2335 bp HindIII genomic fragment containing the hSA promoter sequence, comprising
2130 bp 5’ and 203 bp 3’ to the start of transcription, including all of exon 1, was cloned in sense
orientation into the HindIII site of pGL-2Basic (ProMega). Truncations and point mutations were
generated by cloning and/or PCR-mediated mutagenesis as described 32. Figure 1 shows the
structure of each construct used in this paper and the specific point mutations introduced. Primers
used for introduction of point mutations are shown in Table 1. Mutated bases are shown in bold
letters. Numbering reflects the position of hSA gene sequences relative to the transcription start site.
Our numbering reflects a systematic difference of +16 bp with the sequence previously published
by Muscat et al. as nt -1282 through -1177 33; the corresponding numbers in our sequence are -
1298 through -1193.
Internal and 5’ deletions of the distal promoter were generated by digestion of the hSA at
unique restriction sites at -1787, -1656, -1298 and –1243, and internal religation or ligation to
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
6
compatible sites in the polylinker (Figure 1B). The p1787luc 1 plasmid was created by digestion at
the hSA SpeI site and the polylinker NheI site, and re-ligating. p1656 was generated by digestion at
the hSA NdeI site and religation of the proximal end to the polylinker Bgl II site. Digestion with
NdeI and partial digestion with XbaI removed a small fragment of 358 bp resulting in the dNX
plasmid. The dNP plasmid was created by double digestion with NdeI and PstI, gel purification of
the vector fragment and religation.
Introduction of point mutations to ϕGATA and ATr sites was performed as described 34
with slight modifications. Briefly, two adjacent primers were designed on opposite DNA strands
with the mutation encoded at the 5' end of one primer. Both primers were phosphorylated before
PCR. PfuTurbo DNA polymerase (Stratagene, La Jolla, CA) was used to increase fidelity of DNA
replication and create blunt-ended PCR products. The amplified plasmid with the introduced
mutation was isolated from agarose, religated and transformed to bacteria. Mutations in the
proximal promoter were made by directional cloning of double-stranded 52 bp oligomers
containing mutations in CArG I (m8, m9), a TEF-1 site (mTEF), or sequences 5’ to the CArG box
(m6), flanked by XmaIII and XhoI sites at the 5’ and 3’ ends respectively. The parental pluc1
vector was linearized with XhoI and subjected to partial digestion with XmaIII; each oligonucleotide
was then ligated into this vector to generate m6, m8, m9 and mTEF constructs. All clones were
screened by restriction analysis and sequenced to confirm the presence of each mutation.
In vivo gene transfer to rat myocardium.
Male Sprague Dawley rats (250-400g) were premedicated with a mixture of 10-20 mg/ kg
fluanisone, 0.315-0.630 mg/kg fentanyl citrate (Hypnorm, Jansen Pharmaceuticals) and 0.5-1.0
mg/kg midazolam (Hypnovel, Roche Pharmaceuticals) given intraperitoneally. Animals were
ventilated (0.04-0.06 l/min/kg) on a small animal respirator with 0.5-1.0 cm H2O of positive end-
expiratory pressure and maintained under anaesthesia with a mixture of nitrous oxide and oxygen
in a 1:1 ratio plus 0.5-1% halothane. The chest was opened by a left thoracotomy and the
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
7
pericardium removed for DNA injection. DNA was directly injected into the apex of the left
ventricle, using 100 µl per injection in a Hamilton syringe (25 µg internal control plasmid and 50
µg hSA-luciferase plasmid suspended in phosphate buffered saline). Postoperative analgesia was
administered for at least the next 24 hours with 0.2 mg/ kg intramuscular buprenorphine
(Vetergesic, Reckitt & Coleman). Seven days after surgery animals were sacrificed with a lethal
dose of pentobarbital sodium and the heart excised for assays of reporter gene expression.
Enzyme determinations in tissue extracts were performed as described previously 35. pRSV-
luciferase and pCAT3PV (SV40 promoter linked to CAT) were used as internal controls and
were obtained from ProMega Biotech (Madison, WI). All procedures were performed under
license in accordance with NIH guidelines or in accordance with the United Kingdom Animals
(Scientific Procedures) Act, 1986.
Cell Culture and Transfection.
HeLa cells were grown in MEM with Eagle's salts, penicillin, streptomycin and 10% fetal
bovine serum. Neonatal rat myocardial cells were isolated as previously described 19 by gentle
trypsinization and mechanical dissociation over a period of 4-5 hrs, and plated at a density of 3.5-4
x 106 cells per 60-mm culture dish. Cultures were enriched for myocardial cells by preplating for
30-60 min to deplete the population of non-myocardial cells. Prior to and during transfection, cells
were maintained in MEM with Eagle's salts, penicillin, streptomycin and 5% fetal bovine serum
(MEM-FBS). Following transfection, cells were incubated in a serum-free medium consisting of
MEM supplemented with insulin, transferrin, vitamin B12, penicillin and streptomycin (MEM-
TIB). All cells were maintained in 5% CO2 atmosphere at 37˚C and transfected as described below.
Cardiac myocytes were transfected with reporter plasmids and other constructs on the day
following plating, using an adaptation of the calcium phosphate method 36. Equal numbers of
myocytes were co-transfected with 10 µg of reporter plasmid and either 5 µg of p300 expression
vector, 5 µg of MEF-2 expression plasmid or an equal amount of blank CMV expression vector.
24 hours after transfection cells were washed twice with MEM-TIB and maintained in that medium
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
8
for an additional 48 hours. Cells were then washed twice with PBS, pH 7.4, and collected in 1x
reporter lysis buffer (ProMega).
HeLa and C2C12 cells were transfected at a confluence of 50-70%, also using the calcium
phosphate method. Cells were maintained prior and during transfection in MEM (HeLa) or
DMEM (C2C12) supplemented with 10% fetal bovine serum, penicillin and streptomycin. On the
day after transfection, cells were rinsed twice with fresh media and incubated for 24-48 hr before
harvesting as described above. A commercially available kit was used to measure luciferase activity
in cell lysates (ProMega).
Gel Mobility Shift Assays
For preparation of nuclear extracts, cardiac myocytes were plated in 10 cm dishes and
cultured in MEM-FBS for 2 days, then switched to MEM-TIB for 3-5 days. Nuclear extracts were
prepared essentially as previously described 37 except that the final extract was desalted on a 1-2 ml
Sephadex G-25 chromatography mini-column instead of by dialysis. Protein concentrations were
determined using the Bio-Rad Protein assay kit (Bio-Rad Laboratories, Hercules, CA). Aliquots of
nuclear extract were frozen and stored at -80˚ C.
Double-stranded oligonucleotide gel shift probes containing the wt and mutant ϕGATA and
ATr motifs were synthesized as shown in Table 2. Gel-purified oligonucleotide pairs were
annealed and end-labeled with [32P]-ATP using T4 polynucleotide kinase (NEB) and [γ-32P]-ATP
(NEN Life Science Products). Gel mobility shift assays were performed as previously described
38(69). In brief, equal amounts of radioactive probe (1.5-2.5 x 104 cpm) were added to binding
reactions that contained 6 µg of nuclear extract protein in 20 µl of a buffer containing 4 mM Tris
(pH 7.8), 12 mM HEPES (pH 7.9), 60 mM KCl, 30 mM NaCl, 0.1 mM EDTA, 1 µg/ml of
poly(dI-dC) (Amersham Pharmacia Biotech). Reactions were incubated for 20 min at 22˚C, and
then separated on a nondenaturing 5% polyacrylamide gel at 4˚C. Where indicated, antibodies (2-4
µg/reaction) were incubated with the binding reactions for 30 min at 22˚C before addition of the
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
9
probe. For determination of sequence-specific binding, a 100-fold molar excess of unlabelled
oligonucleotides was added immediately before the probe. No DNA-antibody interaction was
observed in the absence of nuclear protein (data not shown).
Immunoprecipitation.
50 µg of nuclear extract from cardiac myocytes (prepared as described above) was mixed in
equal amounts with 2x immunoprecipitation buffer (2% Triton X-100, 300 mM NaCl, 20 mM Tris
pH 7.4, 2 mM EDTA, 2 mM EGTA pH 8.0, 0.4 mM sodium orthovanadate, 0.4 mM PMSF, 1.0%
NP-40). Subsequently, 1-5 µg of either MEF-2 or p300 antibody was added. Mixtures were
incubated for 1 hour at 4˚ C. with constant agitation. Protein A-agarose (20 µl, Santa Cruz
Biotechnology) was then added to the protein-antibody mixture and tubes were incubated for an
additional 30 min at 4˚C. At the end of the incubation, agarose beads were spun down, and the
supernatant was reserved to quantitate unbound proteins. Beads were washed three times with 1x
immunoprecipitation buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA, 1
mM EGTA pH 8.0, 0.2 mM sodium orthovanadate, 0.2 mM PMSF, 0.5% NP-40), resuspended in
30 ml of 2x electrophoresis sample buffer (250 mM Tris pH 6.8, 4% SDS, 10% glycerol, 0.006%
bromphenol blue, 2% β-mercaptoethanol) and then boiled for 5 minutes. Aliquots of the original
nuclear extract, unbound proteins and wash fractions were also mixed with 2x electrophoresis
sample buffer and boiled. All protein samples were resolved on 6% SDS-polyacrylamide gels (for
p300) or 8% SDS-polyacrylamide (for detection of MEF-2 proteins). Gels were transferred
overnight to nitrocellulose membranes using a Transblot electrophoresis transfer cell (Bio-Rad,
Hercules, Calif.). Membranes were probed with polyclonal MEF-2 antibody (SC-313, Santa Cruz
Biotechnology), polyclonal p300 (N-15) (sc-584, Santa Cruz Biotechnology) and monoclonal p300
NM11 (14991A, Pharmingen). Antigen-antibody complexes were visualized by enhanced
chemiluminescence (Pierce).
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
10
Coprecipitation of MEF-2/p300 complexes using a biotinylated probe.
A biotinylated oligonucleotide containing the antisense ATr motif (5’ biotin
TTACCAGAGCCTGCTGCAGGTTCTATTTATATCA3') was annealed to the complementary
ATr (R) sense oligonucleotide (see Figure 7A) at a final concentration of 10 µM, and linked to
Dynabeads M-280 streptavidin (Dynal Inc., Lake Success, NY) as previously described 39. For co-
precipitation of MEF-2 and p300, the beads were initially washed three times with phosphate
buffered saline (PBS, pH 7.4) containing 0.1% BSA, and twice with a buffer containing 1M NaCl,
10 mM Tris, 1 mM EDTA (TE-NaCl, pH 7.5). The beads were then mixed with 10 pmol double-
stranded biotinylated probe and incubated 20 minutes at room temperature in TE-NaCl. Sequential
washes with TE-NaCl and 1x binding buffer (10mM Tris [pH 7.5], 50 mM KCl, 1 mM MgCl2 , 1
mM EDTA, 5.5 mM DTT, 5% glycerol, 0.3% Nonidet P-40) were used to remove excess unbound
oligonucleotide and to equilibrate the beads with the binding buffer. Cardiac myocyte nuclear
extract (50 µg) was mixed with 1x binding buffer for 5’ at room temperature and incubated with the
probe-linked beads for 20’. Unbound proteins were washed from the beads three times with 1x
buffer containing 0.5 µg/ml poly(dI-dC), and aliquots of each wash were retained for analysis.
Finally, proteins specifically bound to the beads were eluted with 1x binding buffer containing 1M
NaCl.
All fractions were resolved by SDS-PAGE (6% for p300 and 12% for MEF-2) and
analyzed by Western blot using antibodies to p300 and MEF-2 as described above.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
11
RESULTS
MEF-2, SRF and TEF-1 binding sites in the hSA promoter are required for maximal
cardiac expression in vivo
p300 /CBP does not bind DNA directly, but is recruited to specific gene control regions by
transcription factors that bind to these sites. Thus, we looked for binding sites for known DNA-
binding transcription factors in the hSA promoter that could represent partners or mediators of
p300-dependent transactivation. The proximal human skeletal actin promoter contains consensus
binding sites for SRF, TEF-1, and Sp-1, similar to the chick, mouse and rat skeletal actin promoters
(41, 35, 56; see Figure 1A), and additional regulatory elements have been localized to the distal
promoter (5' to -710) 19 . To characterize these elements, we sequenced the distal human skeletal
actin promoter from -710 to -2130 (not shown; GenBank accession #AF288779). Within this
sequence are two DNA elements resembling AP-1 binding sites (centered at -1494 and –1300,
Figure 1A), as well as six variant E-boxes (CANNTG, not shown). A previously described AT-
rich motif was found centered at -1256 33 (Figures 1A and 1C). This AT-rich site differs by one
nucleotide from the consensus binding site for myocyte enhancer-binding factor-2 (MEF-2)
(CTA(A/T)4TAG, 40). We also searched for GATA binding sites by BLAST screening of the hSA
promoter with the GATA binding site from the brain natriuretic peptide promoter
(CTGATAAATCAGAGATAACC). This search revealed two potential GATA binding sites, one of
which overlapped with the MEF-2 consensus sequence. For convenience, the compound MEF-
2/GATA binding motif was called “ATr”. An additional putative GATA site was centered at -
1798 (Figures 1A, 1C), and designated ϕGATA.
To test the functional relevance of these elements, the hSA promoter was subjected to serial
5' truncations and point mutagenesis (Figures 1B and 1C). The resulting constructs were assayed
for activity in adult myocardium by direct injection as described in Methods (Figure 2). These
studies showed that the ATr site at -1256 (mATr, Figure 2), as well as the TEF-1 and SRF binding
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
12
sites in the proximal promoter (mutants mTEF, m9 respectively, Figure 2), were required for
maximal expression of the human skeletal actin promoter in the adult rat heart. Deletion of
sequences upstream of -1298 did not reduce, and in fact enhanced expression, indicating that
elements in this region (including ϕGATA) do not confer tissue-specific activation in the heart. In
contrast, point mutagenesis of ATr reduced hSA promoter activity by about 50% (Figure 2),
suggesting that this element forms the core of the previously reported distal tissue-specific element
19.
A distal AT-rich site is the major p300 responsive element
We next looked for specific DNA sequences in the hSA promoter that mediate its activation
by p300. Although the full-length hSA promoter (p2130luc1) has constitutively high basal activity
in cardiac myocytes, comparable to that of beta actin 19 , co-expression of p300 further activated it by
more than 3-fold (Figure 3). Similar to our findings in vivo, two different point mutations of the
proximal SRE (CArG I) and mutagenesis of the TEF-1 binding site each reduced basal hSA
promoter activity in cardiac myocytes by > 60% (mutants m8, m9 and mTEF, Figure 3). However,
these sites were not required for transactivation by p300 (Figure 3). Mutation of the Sp-1 site in
the proximal promoter also reduced basal activity, but did not affect p300 transactivation (data not
shown). Minimal expression of the basal promoter (truncated at -87) was increased in the presence
of p300, possibly reflecting interaction of p300 with TATAA binding factors 41 . In contrast, point
mutation of the AT-rich motif centered at -1256 (ATr) not only reduced basal hSA promoter
activity, but also abrogated transactivation by p300 (Figure 3, mutants mATr and mATrϕGA).
Although in some experiments mutation of the ϕGATA site at –1798 appeared to further reduce
p300 transactivation, this effect was not reproducible and did not achieve statistical significance (p >
0.08). Deletion or mutation of other distal sites had no significant on p300 transactivation. These
findings indicated that transcriptional activation by p300 required a single tissue-specific AT-rich
enhancer element in the hSA promoter.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
13
The ATr element is a MEF-2 binding site
The ATr site is assumed to be a MEF-2 binding site based on its homology to similar sites
in other muscle-specific promoters 42 . To identify cardiac nuclear proteins binding to the p300-
responsive ATr element, we synthesized oligonucleotides containing both the wild type and mutant
sequences used for functional assays above (Table 2). Electrophoretic mobility shift assays
(EMSAs) were performed with nuclear extracts from cardiac myocytes and from endothelial cells.
Cardiac nuclear proteins interacting with the ATr element formed at least three sequence-specific
bands (labelled 1 –3 in Figure 4A). The upper two bands required the core ATr sequence, and did
not form on a mutant ATr site (Figure 4A, lane 6), or on the GATA-like sequence at –1798,
ϕGATA (Figure 4A, lane 2). The third nucleoprotein complex formed specifically on both ATr and
mATr, suggesting that it interacts with sequences flanking the core ATATA sequence (Figure 4A,
lanes 6 and 12). All three major nucleoprotein complexes were muscle-restricted, as they were
absent in endothelial cell nuclear extracts (Figure 4A, lanes 13-14).
We next confirmed that MEF-2 proteins bind to the ATr site. The same 3 nucleoprotein
complexes identified in Figure 4A were again seen forming on the ATr site, as well as on a
synthetic MEF-2 site (Figure 4B). The synthetic MEF-2 oligonucleotide also competed effectively
for the 3 complexes binding to ATr. In both cases, bands 1 and 2 were specifically and
quantitatively supershifted by a MEF-2 antibody that recognizes MEF-2 subtypes A, C and D
(Figure 4B, lanes 5 and 10), while a control MyoD antibody did not (data not shown). Thus, MEF-
2 is present in 2 of 3 tissue-specific complexes bound to the ATr site.
The ATr site has significant homology to a GATA-4 binding site from the BNP promoter
that is required for its cardiac-specific activation 43. To determine whether GATA-4 also bound to
the ATr site, we synthesized shorter oligomers representing sequences centered at -1260 ("left"), -
1252 ("right") and -1256 ("center") (Figure 5A). All 3 previously detected complexes formed on
each of these shorter oligonucleotides (Figure 5B), but with varying efficiency. Complexes 1 and 2
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
14
preferentially interacted with the right side of ATr (Figure 5B, lane 9), while complex 3
preferentially bound to the left (Figure 5B, lane 5). For all three oligonucleotides, only complexes 1
and 2 were supershifted by the MEF-2 antibody (Figure 5B, lanes 3, 7, and 11). Thus, complex 3
binds a site adjacent to and overlapping that of complexes 1 and 2, and does not contain MEF-2. In
contrast, a polyclonal anti-GATA-4 antibody selectively depleted ATr complex 3, suggesting that it
contained GATA-4 (Figure 5C, lane 4). This depletion was accompanied by appearance or
enhancement of a band co-migrating with complex 1 (lane 4). When a synthetic GATA binding
site was used as the probe (Figure 5C lanes 5-9), we observed a single cardiac nucleoprotein
complex that co-migrated with ATr complex 3 and was effectively competed by an ATr oligomer
(Figure 5C, lane 8). Incubation with the GATA-4 antibody generated a supershifted complex that
migrated roughly in tandem with complex 1 on the ATr site (Figure 5C, compare lanes 9 and 4).
Thus, the enhanced binding in lane 4 appears to be identical with a supershifted GATA-DNA
complex, although we cannot formally exclude the possibility that this represents enhanced binding
of MEF-2. In either case, it is clear that the most rapidly migrating nucleoprotein complex on the
ATr sequence contains GATA-4.
Taken together, these data show that the distal hSA cardiac-specific element is occupied by
at least three muscle-specific protein complexes: two containing MEF-2, and one with GATA-4.
Moreover, MEF-2 and GATA-4 complexes form on distinct but overlapping sites within the ATr
motif.
p300 binds specifically to the skeletal actin MEF-2 binding site
We next looked for evidence that p300 interacts directly with the MEF-2 DNA-binding
complexes. The ATr and Atr-Left oligonucleotides shown in Figure 5A were labelled and allowed
to interact with cardiac nuclear proteins in the presence or absence of specific antibodies. As in
Figures 4B and 5B, the polyspecific MEF-2 antibody completely supershifted Bands 1 and 2
(Figure 6, lane 8). A second, MEF-2D-specific antibody reacted only with Band 1 (Figure 6, lane
7.) Band 3 on both oligonucleotides was supershifted by a GATA-4 antibody (Figure 6, lanes 3 and
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
15
10). These results show that the more rapidly migrating complex is likely to contain MEF-2A,
MEF-2C or both, while the slower-moving complex (Band 1) probably contains only the MEF-2D
isoform.
Band 1, containing MEF-2D, was also the only complex that could be shown to contain
p300. A polyclonal antibody directed against the N terminus of p300 reacted very specifically with
the MEF-2D-ATr complex (Figure 6, lane 9); bands 2 and 3 were not affected. None of the
protein-DNA complexes on the Atr-Left oligomer reacted with the p300 antibody. These data show
that p300 is specifically present in a complex with MEF-2D on the ATr, and not with GATA-4 or
other MEF-2 species. Morever, 3’ flanking sequences of the ATr are required for formation of this
complex.
A MEF-2 site in the cardiac α-myosin heavy chain (α-MHC) promoter is required for
maximal activity in cardiac myocytes 44-46. We were interested in determining whether p300 could
be identified at these sites, or at a binding site for the related MADS protein, SRF, in the proximal
hSA promoter (Figure 1A). Oligonucleotide sequences used for these studies are given in Table 2.
Figure 7 shows that the AT-rich motifs from the α-MHC and M-creatine kinase (M-CK) form
complexes similar to hSA complexes 1, 2 and 3 (Figure 7, lanes 2, 7 and 12). In each case,
complexes 1 and 2 were supershifted by a MEF-2 antibody (lanes 4, 9 and 14). Furthermore, each
complex 1 was supershifted by the p300 antibody (lanes 5, 10 and 15). No other bands were
visibly affected. Neither of two distinct sequence-specific complexes formed on the hSA SRE was
supershifted by the MEF-2 or p300 antibodies. These results suggest that p300 interacts
differentially with these two MADS proteins and is selective for MEF-2 over SRF (Figure 5, lanes
19 and 20).
Endogenous cardiac MEF-2 and p300 interact on the ATr element
We next asked whether MEF-2 and p300 bound to each other directly or via contact with
DNA. Initially we attempted to co-immunoprecipitate MEF-2 and p300 from cardiac nuclear
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
16
extracts, using antibodies against both p300 and MEF-2. The polyclonal MEF-2 antibody was
able to quantitatively immunoprecipitate at least two MEF-2 species (Figure 8A, lane 4).
However, we detected no p300 protein in the MEF-2 immunoprecipitates using either
monoclonal or polyclonal p300 antibodies (Figure 8A, lane 9). Conversely, the anti-p300
monoclonal antibody (NM-11) successfully immunoprecipitated p300 from cardiac nuclear
extracts (Figure 8A, lane 10), but did not co-precipitate detectable MEF-2 species (data not
shown). This may be attributable to inaccessibility of the interaction domains in the presence of
antibody or to the lack of high affinity interaction between the two proteins in the absence of
DNA.
To determine whether MEF-2 and p300 interacted through DNA binding, we performed
DNA “pulldown” assays using Dynal beads linked to a double-stranded oligonucleotide
containing the hSA MEF-2 site (ATr) as previously described 39. Cardiac nuclear proteins binding
specifically to this oligonucleotide were eluted from the beads and characterized by Western
analysis with MEF-2 and p300 antibodies. Two MEF-2 proteins eluted from the hSA ATr site
(Figure 8B, upper panel, lane 4). Western analysis of the same blot using a MEF-2D-specific
antibody confirmed that the upper band contains MEF-2D (data not shown). Importantly, a band
corresponding to p300 was present in the same eluate (Figure 8B, lower panel, lane 4). A second
protein of smaller size (about 140 kd) was also detected by the NM-11 antibody and may represent
a degradation product. A biotinylated mutant ATr did not bind to either MEF-2 or p300 (data not
shown). These data show that endogenous cardiac MEF-2 and p300 form a specific complex with
the ATr element, and support the results of the gel mobility retardation assays shown above.
MEF-2, but not GATA-4, is synergistic with p300
To address the functional significance of the MEF-2 - p300 interaction, we asked whether
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
17
the two proteins could activate cardiac transcription synergistically. We measured the activity of the
hSA wt promoter, and the same promoter containing a point mutation in the ATr motif (p2130luc1
and mATr, Figures 1B and C) in the presence and absence of p300 and one of the four MEF-2
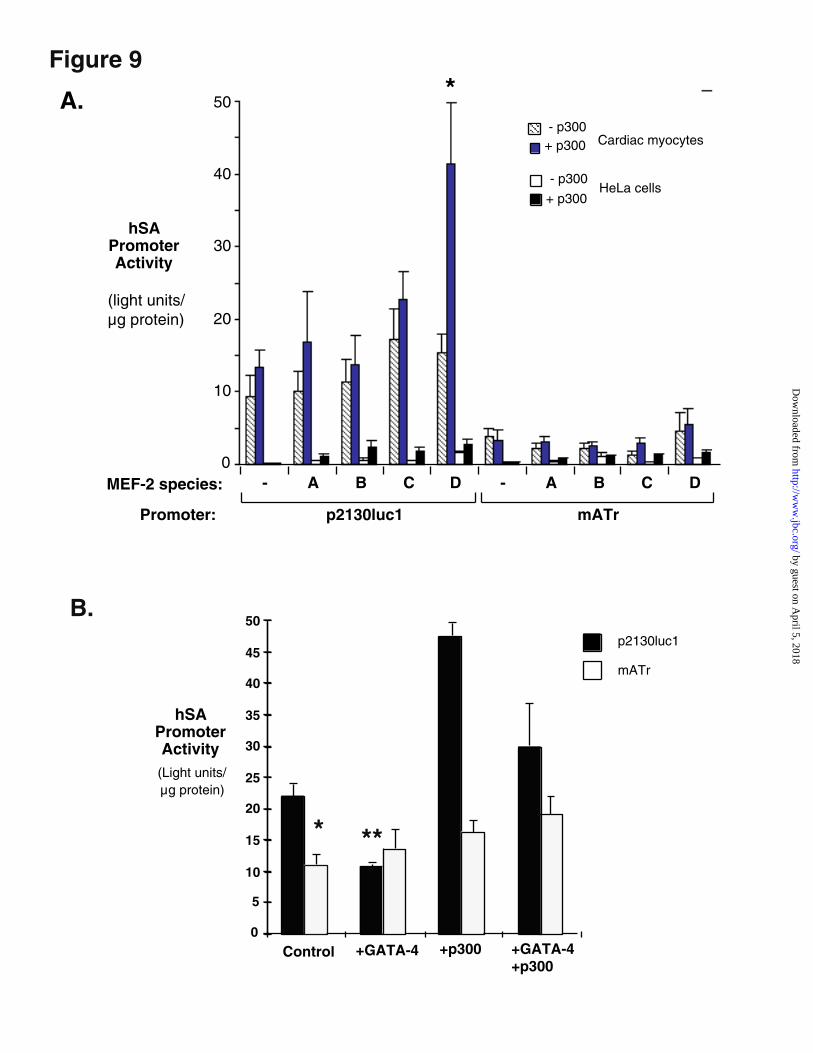
species. Parallel experiments were performed in cardiac myocytes and HeLa cells (Figure 9A).
Alone, none of the individual MEF-2 subtypes significantly activated either the wild type or mutant
hSA promoters in cardiac myocytes, although a small amount of activation was seen in HeLa cells
(Figure 9A, white bars). In cardiac myocytes, activation of the wt promoter by MEF-2A, B or C did
not significantly increase in the presence of p300 (Figure 9A, blue bars). However, the combination
of MEF-2D and p300 significantly activated hSA expression, compared with either blank vector or
p300 alone (p < 0.01, Figure 9A). Because it is not possible to establish the relative expression of
the different MEF-2 species from these vectors, it may be that the divergent behavior of MEF-2C
and MEF-2D is due to quantitative differences in MEF-2 delivery or expression. However, all
vectors contained the same CMV promoter, and were expressed at measurable levels in a HeLa cell
background, suggesting that transfer and expression were not qualitatively defective for the MEF-
2C vector (data not shown).
In HeLa cells, the combination of p300 and MEF-2D failed to activate hSA transcription
above 10% of its activity in cardiac myocytes, and this activation was largely independent of the ATr
site, suggesting that additional cell type-specific factors are required for maximal expression of this
promoter. As expected, neither p300 nor MEF-2 species activated an hSA mutant lacking the ATr
site (mATr) in cardiac myocytes. These results show that p300 and MEF-2D are synergistic for
cardiac transcription of the hSA promoter and that this synergy is exerted through the ATr site at -
1256.
The skeletal actin gene has not previously been shown to be a target for activation by
GATA-4. However, as shown above, GATA-4 binds to the hSA ATr site, flanking the MEF-2
binding sequences. Thus, GATA-4 might also transactivate the hSA promoter, or participate in
activation of hSA by p300 through the ATr site. To investigate these possibilities, a GATA-4
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
18
expression vector was co-transfected with the hSA wild type and ATr site mutants. The ATr
mutant was expressed at approximately 50% of wild type promoter levels, as reported above. Co-
expression of GATA-4 did not activate basal expression of the wild type promoter at any
concentration (Figure 9 and data not shown), and significantly reduced its activation by p300 in the
presence of an intact ATr (Figure 9, light bars). Together with the observed lack of physical
interaction in vivo, these observations suggest that GATA-4 is not directly involved in p300-
mediated activation of skeletal actin transcription.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
19
DISCUSSION
Studies described here identify the myogenic transcription factor MEF-2D as a specific
physical and functional target of p300 in the heart. Activation of the native 2180 bp human skeletal
actin promoter by p300 required a single short DNA sequence in the distal promoter, a hybrid
binding site for MEF-2 and GATA-4. Full myocardial expression of the skeletal actin promoter
required this same element, both in vitro and in vivo. p300 bound exclusively to this element, as part
of a complex with MEF-2D, and could not be identified in DNA complexes with GATA-4 or in a
structurally related DNA-SRF complex. Furthermore, p300 and MEF-2D displayed cooperative
transcriptional activation of the hSA promoter. This functional synergy was not observed in HeLa
cells, suggesting a requirement for additional cell type-specific factors. Taken together, these
findings identify MEF-2D as a dominant partner for p300 in the myocardium, and place p300 in
the regulatory hierarchy of the cardiac phenotype 15 10;16;17.
Our data show that p300 targeting of MEF-2D takes priority over its potential interactions
with several other proteins that bind to the 2130 bp hSA promoter, including GATA-4- and SRF.
This observation is remarkable, since other studies have shown that p300 can bind to both SRF and
members of the GATA family, and to activate transcription through their cognate binding sites 47 48
49 . In contrast, we did not observe physical or functional interaction between p300 and GATA-4 on
the hSA promoter, nor did we detect p300 at GATA-4 binding sites in the BNP, β-myosin heavy
chain50 or angiotensin type I receptor promoters 51 under equivalent conditions (data not shown).
Our results do not exclude the possibility that GATA-4 and p300 interact independently of DNA or
under defined conditions in vitro. Further work will be required to establish the relationship
between GATA-4- and MEF-2-dependent transcriptional activation in the heart.
The MEF-2 family of transcription factors belongs to the MADS (MCM-1, agamous,
deficiens, serum response factor) group of DNA binding proteins, characterized by their homology
within an amino terminal domain (“MADS box”). Although MEF-2 is widely expressed, its role
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
20
in cardiac-specific transcription been well documented. MEF-2C is required for normal cardiac
morphogenesis, and most if not all cardiac genes possess functionally important MEF-2 binding
sites 42 . MEF-2 species form DNA-binding homo- and heterodimers, and also bind to myogenic
helix-loop-helix proteins as co-regulators. In skeletal muscle, MEF-2 proteins collaborate with
MyoD and p300 to promote myogenesis and muscle-specific transcription, and are thought to act in
a positive autoregulatory loop that maintains myogenic differentiation 18;40;52;53. Other partners for
MEF-2 proteins in cardiac transcriptional activation remain to be identified.
Our data suggests that the MEF-2D isoform may be targeted by p300 in preference to other
MEF species. We were able to demonstrate cooperative transcriptional activation between p300 and
MEF-2D, but not with the other MEF-2 isoforms, and p300 was localized to the DNA complex
containing MEF-2D. Our data do not exclude limited functional interaction between p300 and other
MEF-2 species at this promoter, but it is also possible that the small positive interaction between the
other MEF species and p300 is mediated by heterodimerization with endogenous MEF-2D. This
finding is important because to date there are few examples of functional differences between the
four known MEF-2 species. All four mammalian MEF-2 species are expressed in the heart,
(reviewed in 42), and all subtypes recognize the same AT-rich consensus sequence
(YTA(A/T)4TAR). Differential recruitment of essential coactivator proteins may confer distinct
transcriptional activation properties on MEF-2D.
The observed lack of co-activation between MEF2C and p300 at this promoter was
unexpected. Several previous findings suggest that p300 and MEF-2C may act cooperatively in the
cardiovascular system; p300-deficient and MEF-2C deficient mice have overlapping defects in
vascularization, and both have defects in cardiac development, although these are dissimilar 54 .
MEF2C and D have closely parallel expression patterns throughout cardiac development, and
MEF2C expression actually precedes that of MEF2D in the cardiac mesoderm 42. Furthermore, co-
translated p300 and MEF2C have been shown to interact in vitro 18 . However, the physical and
functional data presented here suggests that MEF-2-p300 interactions may be subject to
modification by cell type-specific or promoter-specific factors. One possibility is that MEF2C-
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
21
p300 interactions play critical roles in mesodermal patterning and in vasculogenesis, while MEF2D-
p300 may be more important for tissue-specific gene expression in the differentiated cardiac
myocyte. Further studies of MEF2D in gene-targeted mice will help to clarify its specific roles.
The muscle-specific actions of MEF-2 cannot be explained by tissue-restricted expression,
or by differences between MEF-2 binding sites in muscle-specific and ubiquitously expressed
genes 55 . Instead, MEF-2 activity appears to be subject to significant post-transcriptional control by
phosphorylation, and by the recruitment of additional factors 40;56-59. In support of this hypothesis,
it has been suggested that MEF-2 -dependent transcription is silenced in non-muscle cell types by
recruitment of HDAC-4, a histone deacetylase 31 . We propose that the converse is also true:
activation of MEF-2 dependent genes in cardiac muscle requires recruitment of histone
acetyltransferase via p300. These findings point to a unique role for MEF-2D in channelling both
the activation and silencing signals of chromatin-remodelling enzymes to cardiac-specific
promoters.
Our studies provide the first direct evidence that MEF-2 and p300 interact to regulate
cardiac-specific transcription. Although MEF-2 proteins and p300 have been shown to interact in
vitro 52 18 , and at artificial MEF-2-dependent promoters in non-muscle cells 18 , the significance of
these findings in tissue-specific transcription has been unclear. In skeletal muscle, the MEF-2-p300
interaction may serve to stabilize critical MyoD-p300 complexes on adjacent E boxes 52 . However,
our data indicate that the MEF-2-p300 complex can activate the tissue-specific transcription of
promoters that lack essential E-boxes, and can do so in the apparent absence of bHLH proteins
analogous to MyoD or NeuroD/Beta2 60;61. It seems likely that additional, unidentified cell type-
specific factors cooperate with MEF-2 and p300 in the cardiac myocyte.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
22
REFERENCES
1. Struhl, K. (1998) Genes Dev. 12, 599-606
2. Lee, D. Y., Hayes, J. J., Pruss, D., and Wolffe, A. P. (1993) Cell 72, 73-84
3. Yao, T. P., Ku, G., Zhou, N., Scully, R., and Livingston, D. M. (1996)Proc.Natl.Acad.Sci.U.S.A. 93, 10626-10631
4. Xu, J., Liao, L., Ning, G., Yoshida-Komiya, H., Deng, C., and O'Malley, B. W. (2000) ProcNatl Acad Sci U S A :
5. Leo, C. and Chen, J. D. (2000) Gene 245. 245, 1-11
6. Lowell, B. B. and Spiegelman, B. M. (2000) Nature 404. 404, 652-60
7. Vega, R. B., Huss, J. M., and Kelly, D. P. (2000) Mol Cell Biol 20. 20, 1868-76
8. Tanaka, Y., Naruse, I., Maekawa, T., Masuya, H., Shiroishi, T., and Ishii, S. (1997)Proc.Natl.Acad.Sci.USA 94, 10215-10220
9. Kawasaki, H., Eckner, R., Yao, T. P., Taira, K., Chiu, R., Livingston, D. M., and Yokoyama, K.K. (1998) Nature 393, 284-289
10. Yao, T. P., Oh, S. P., Fuchs, M., Zhou, N. D., Ch'ng, L. E., Newsome, D., Bronson, R. T., Li,E., Livingston, D. M., and Eckner, R. (1998) Cell 93, 361-372
11. Giles, R. H., Dauwerse, H. G., van Ommen, G. J., and Breuning, M. H. (1998) Am J HumGenet 63. 63, 1240-2
12. Arany, Z., Sellers, W. R., Livingston, D. M., and Eckner, R. (1997) Cell 77, 799-800
13. Giordano, A. and Avantaggiati, M. L. (1999) J.Cell Physiol. 181, 218-230
14. Snowden, A. W. and Perkins, N. D. (1998) Biochem.Pharmacol. 55, 1947-1954
15. Bishopric, N. H., Zeng, G.-Q., Sato, B., and Webster, K. A. (1997) J.Biol.Chem. 272, 20584-20594
16. Kirshenbaum, L. A. and Schneider, M. D. (1995) J.Biol.Chem. 270, 7791-7794
17. Hasegawa, K., Meyers, M. B., and Kitsis, R. N. (1997) J.Biol Chem. 272, 20049-20054
18. Sartorelli, V., Huang, J., Hamamori, Y., and Kedes, L. (1997) Mol.Cell Biol. 17, 1010-1026
19. Bishopric, N. H. and Kedes, L. (1991) Proc.Natl.Acad.Sci.USA 88, 2132-2136
20. Bishopric, N. H., Jayasena, V., and Webster, K. A. (1992) J.Biol.Chem. 267, 25535-25540
21. Izumo, S., Nadal-Ginard, B., and Mahdavi, V. (1988) Proc.Natl.Acad.Sci.USA 85, 339-343
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
23
22. Parker, T. G., Chow, K. L., Schwartz, R. J., and Schneider, M. D. (1990)Proc.Natl.Acad.Sci.USA 87, 7066-7070
23. Boheler, K. R., Carrier, L., de la Bastie, D., Allen, P. D., Komajda, M., Mercadier, J.-J., andSchwartz, K. (1991) J.Clin.Invest. 88, 323-330
24. Adachi, S., Ito, H., Tamamori, M., Tanaka, M., Marumo, F., and Hiroe, M. (1998) Life.Sci. 63,1779-1791
25. Tanaka, M., Hiroe, M., Ito, H., Nishikawa, T., Adachi, S., Aonuma, K., and Marumo, F. (1995)J.Am.Coll.Cardiol. 26, 85-92
26. MacLellan, W. R., Lee, T. C., Schwartz, R. J., and Schneider, M. D. (1994) J.Biol.Chem. 269,16754-16760
27. Karns, L. R., Kariya, K., and Simpson, P. C. (1995) J.Biol.Chem. 270, 410-417
28. Paradis, P., MacLellan, W. R., Belaguli, N. S., Schwartz, R. J., and Schneider, M. D. (1996)J.Biol.Chem. 271, 10827-10833
29. Sugden, P. H. and Clerk, A. (1998) J.Mol.Med. 76, 725-746
30. Taylor, A., Erba, H. P., Muscat, G. E. O., and Kedes, L. (1988) Genomics 3, 323-336
31. Miska, E. A., Karlsson, C., Langley, E., Nielsen, S. J., Pines, J., and Kouzarides, T. (1999)EMBO J. 18, 5099-5107
32. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: a LaboratoryManual., 2 Ed., Cold Spring Harbor Press, New York
33. Muscat, G. E., Perry, S., Prentice, H., and Kedes, L. (1992) Gene Expr. 2, 111-126
34. Fisher, C. L. and Pei, G. K. (1997) BioTechniques 23, 570-590
35. Prentice, H., Kloner, R. A., Prigozy, T., Christensen, T., Newman, L., Li, Y., and Kedes, L.(1994) J.Mol.Cell Cardiol. 26, 1393-1401
36. Gorman, C. M., Moffat, L. F., and Howard, B. H. (1982) Mol.Cell.Biol. 2, 1044-1051
37. Dignam, J. D., Lebovitz, R. M., and Roeder, R. (1983) Nucleic Acids Res. 11, 1475-1489
38. Semenza, G. L. and Wang, G. L. (1992) Mol.Cell Biol 12, 5447-5454
39. Ebert, B. L. and Bunn, H. F. (1998) Mol.Cell Biol 18, 4089-4096
40. Molkentin, J. D., Black, B. L., Martin, J. F., and Olson, E. N. (1995) Cell 83, 1125-1136
41. Abraham, S. E., Lobo, S., Yaciuk, P., Wang, H. G., and Moran, E. (1993) Oncogene. 8, 1639-1647
42. Black, B. L. and Olson, E. N. (1998) Annu.Rev.Cell Dev.Biol 14, 167-196
43. Grepin, C., Dagnino, L., Robitaille, L., Haberstroh, L., Antakly, T., and Nemer, M. (1994)
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
24
Mol.Cell Biol. 14, 3115-3129
44. Molkentin, J. D. and Markham, B. E. (1993) J.Biol.Chem. 268, 19512-19520
45. Molkentin, J. D., Jobe, S. M., and Markham, B. E. (1996) J.Mol.Cell Cardiol. 28, 1211-1225
46. Molkentin, J. D. and Markham, B. E. (1994) Mol.Cell Biol. 14, 5056-5065
47. Ramirez, S., Ait-Si-Ali, S., Robin, P., Trouche, D., Harel-Bellan, A., and Ait Si Ali, S. A. S. A.(1997) J.Biol Chem. 272, 31016-31021
48. Blobel, G. A., Nakajima, T., Eckner, R., Montminy, M., and Orkin, S. H. (1998)Proc.Natl.Acad.Sci.U.S.A. 95, 2061-2066
49. Kakita, T., Hasegawa, K., Morimoto, T., Kaburagi, S., Wada, H., and Sasayama, S. (1999) JBiol Chem 274. 274, 34096-102
50. Hasegawa, K., Lee, S. J., Jobe, S. M., Markham, B. E., and Kitsis, R. N. (1997) Circulation.96, 3943-3953
51. Herzig, T. C., Jobe, S. M., Aoki, H., Molkentin, J. D., Cowley, A. W., Izumo, S., andMarkham, B. E. (1997) Proc.Natl.Acad.Sci.USA 94, 7543-7548
52. Eckner, R., Yao, T. P., Oldread, E., and Livingston, D. M. (1996) Genes Dev. 10, 2478-2490
53. Puri, P. L., Avantaggiati, M. L., Balsano, C., Sang, N., Graessmann, A., Giordano, A., andLevrero, M. (1997) EMBO J. 16, 369-383
54. Lin, Q., Schwarz, J., Bucana, C., and Olson, E. N. (1997) Science 276, 1404-1407
55. Naya, F. J., Wu, C., Richardson, J. A., Overbeek, P., and Olson, E. N. (1999) Development.126, 2045-2052
56. Molkentin, J. D., Kalvakolanu, D. V., and Markham, B. E. (1994) Mol.Cell Biol. 14, 4947-4957
57. Molkentin, J. D., Li, L., and Olson, E. N. (1996) J.Biol Chem. 271, 17199-17204
58. Yang, S. H., Galanis, A., and Sharrocks, A. D. (1999) Mol.Cell.Biol. 19, 4028-4038
59. Molkentin, J. D. and Olson, E. N. (1996) Proc.Natl.Acad.Sci.U.S.A. 93, 9366-9373
60. Naya, F. J., Stellrecht, C. M., and Tsai, M. J. (1995) Genes Dev 9. 9, 1009-19
61. Lee, J. E., Hollenberg, S. M., Snider, L., Turner, D. L., Lipnick, N., and Weintraub, H. (1995)Science 268, 836-844
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
25
ACKNOWLEDGEMENTS
We would like to thank Drs. R. Eckner, D. Livingston and E. Olson for the generous gift of
cDNA clones used in this paper. Dr. G.Q. Zeng assisted in construction of several of the promoter
mutants. We especially appreciate the excellent technical support of Daryl Discher and Mary
Gardiner, and the helpful comments of Dr. Gary Grotendorst. This work was supported by grants
from the National Institutes of Health HL49891 (N.H.B.) and HL44578 (K.A.W.), by an
Established Investigator Grant from the American Heart Association (N.H.B.) and by a grant from
the Miami Heart Research Institute (N.H.B.). Additional support was provided by the British Heart
Foundation (H.P.) and a Wellcome Trust Foundation Collaborative Travel Grant (H.P., K.A.W.
and N.H.B.).
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
26
FIGURE LEGENDS
Figure 1. Diagram of the human skeletal actin (hSA) promoter and mutants. A.
Schematic diagram of the hSA promoter, showing location of major restriction sites and DNA
motifs resembling binding sites for AP-1 (white boxes), GATA-4 (ϕGATA), GATA-4/MEF-2
(ATr), serum response factor (CArG), and TEF-1 as indicated. The complete sequence has been
published in GenBank (Accession # AF288779). B. Deletion and point mutants of hSA promoter
constructs. Arrow indicates transcriptional start site. Shaded box = Exon 1. Open box: start of
luciferase sequence in pGL2Basic. “X” indicates site of point mutations illustrated in C. C.
Sequence of point mutations introduced in designated hSA promoter constructs. The targeted
DNA binding motifs are boxed. Specific mutated bases are shown below the corresponding wild
type nucleotides (in bold). Details of plasmid construction are included in Methods.
Figure 2. Expression of hSA promoter in adult rat myocardium requires proximal and
distal enhancer motifs. DNA was delivered by injection into rat myocardium as described in
Methods, and luciferase activity was measured and normalized after 1 week. p2130 = p2130luc1;
p1656 = p1656luc1; p1298 = p1298luc1; p87 = p87luc1 (please refer to Figure 1B). This graph
summarizes data from a minimum of 4 different animals and at least two different plasmid
preparations per construct.
Figure 3. Identification of a single p300 responsive element in the hSA promoter. In vitro
activity of the 2130 bp hSA promoter construct and mutants in the presence and absence of co-
transfected p300 . Transfections were performed in neonatal rat cardiac myocyte cultures as
detailed in Methods, and luciferase activity was measured 40-48 hours later. Light bars: hSA-
luciferase construct alone. Dark bars: hSA luciferase construct + pCMVp300β. For all
constructs, p ≤ 0.05 for comparison between -p300 and +p300, except mATr (p = 0.3705) and
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
27
mATrϕGA (p = 1.0) (asterisks). These data summarize a minimum of 8 different transfection
experiments and 3 separate plasmid preparations per construct.
Figure 4. Muscle-specific proteins bind to the p300-responsive element. A.
Cardiomyocyte and endothelial cell nuclear protein interactions with hSA promoter.
Oligonucleotides corresponding to ϕGATA and ATr motifs (see Figure 1C and Table 1) were
synthesized and used to define nucleoprotein interactions with these sites. A mutant ATr motif
was also used as a probe (mATr). The ATr motif forms three specific protein complexes with
cardiac myocyte nuclear proteins (Cardiomyocytes; arrows labelled 1, 2 and 3). None of these
bands are seen when endothelial cell nuclear extract (ECs) is used. (-) = labelled probe only.
(NE) = probe + nuclear extract. (S) = 100x unlabelled self probe competitor. (mϕGA) =
unlabelled mutant ϕGATA oligonucleotide competitor. (ATr) = unlabelled ATr probe
competitor. (mATr) = unlabelled mutant ATr competitor. B. MEF-2 binds to the hSA p300-
responsive element. The p300 responsive element (ATr) was labelled and used as a probe in
EMSAs as in Figure 4A. For comparison, a commercially available synthetic MEF-2
oligonucleotide (MEF-2) was also used as a probe. Cardiac nuclear proteins formed three
sequence specific bands with both of these probes (labelled 1, 2 and 3 as in Figure 4A). A MEF-
2 antibody supershifted bands 1 and 2 on both oligonucleotides; three novel supershifted bands
are seen (black arrowheads). Lane designations: (-)= probe alone. (NE) = probe + nuclear
extract. (S) 100x cold self competitor. (MEF ) = unlabelled synthetic MEF-2 oligonucleotide.
(ATr) = unlabelled ATr oligonucleotide. (Ab) = antibody against MEF-2.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
28
Figure 5. Overlapping binding sites for MEF-2 and GATA-4 on the p300-responsive
element. A. Sequences of original and truncated ATr oligonucleotides. Sequences are aligned
with a GATA consensus binding site to show the relative position of the GATA (bold) and
MEF-2 consensus sequences (boxed). The position of nucleotide –1256 in the hSA promoter
sequence is shown. B. Bands 1, 2 and 3 exhibit differential affinity for truncated ATr sites. The
oligonucleotides shown in (A) were labelled and used as probes in EMSAs with cardiac myocyte
nuclear extracts as in Figure 4. A MEF-2- specific antibody was used to confirm the presence of
MEF-2 in complexes 1 and 2 (bracket), forming at least two supershifted bands (white
arrowheads). Note that complex 3 (arrow) is enhanced on the “Left” truncated oligomer and
absent or diminished from the “Right” oligomer. As in Figure 4, complex 3 is not supershifted by
the MEF-2 antibody. (-) probe alone. (NE) probe + nuclear extract. (S) 100x cold self competitor.
(ab) = probe, nuclear extract and MEF-2-specific antibody. C. GATA-4 binds to the hSA
p300-responsive element. EMSA of cardiac nuclear extracts was performed as above using the
ATr (Left) oligonucleotide and a commercially available oligonucleotide containing two GATA
binding sites (Santa Cruz Biotechnology). A band similar to complex 3 appears on the GATA
consensus probe, and is effectively competed by ATr (black arrowheads; bar labelled “3”). Both
complexes 3 are supershifted by a GATA-4 specific antibody (white arrowheads). (-) probe
alone; (NE) probe + nuclear extract; (S) 100x cold self competitor; (ATr) 100x cold ATr
oligonucleotide competitor; (ab) GATA-4 specific antibody.
Figure 6. Both MEF-2 and p300 contact the p300 -responsive element.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
29
EMSAs were performed as above, using the original ATr (ATr-Full) and ATr (Left) motifs
as probes (please refer to Figure 5A). A polyspecific antibody against MEF-2 isoforms (MEF2)
and antibodies specific for MEF-2D (MEF2D), p300 (p300) and GATA-4 (GATA) were used to
identify these proteins in DNA-protein complexes, as indicated. The three major protein-DNA
complexes are numbered (1, 2 and 3). (ATr (Full)) As in Figures 4B and 5B, the MEF-2
antibody generated three supershifted bands (black arrows) and depleted Bands 1 and 2 (lane 8).
In contrast, the MEF-2D-specific antibody supershifted only Band 1 (lane 7, black arrowhead). The
p300 antibody also selectively depleted Band 1 and generated at least 1 supershifted band (white
arrowhead). Band 3 (white arrow) is supershifted exclusively by a GATA-4 antibody. (ATr
(Left)) The Left Atr motif was examined for interaction with p300 since this oligomer maximizes
GATA-4 binding. As with the full-length ATr, Band 3 is supershifted by a GATA-4 antibody, but
not by a p300 antibody. None of the complexes formed on the ATr (Left) motif appear to interact
with p300. NE: nuclear extract.
Figure 7. p300 is present at other cardiac promoter MEF-2 sites, but not at the hSA SRF
site. Homologous AT-rich sites in the cardiac α-myosin heavy chain (α-MHC, (48)) and muscle
creatine kinase (M-CK) promoters were synthesized as shown in Table 2 and used as probes in
EMSAs. The hSA p300 responsive element (hSA ATr) and a proximal serum response element
centered at -93, CArG I (hSA-SRE,) were used for comparison. Complexes 1, 2 and 3 can be
identified on all three MEF-2 sites but not on the SRE. Black arrowheads: MEF-2 antibody-
supershifted bands. White arrowheads: p300 antibody-supershifted bands. Lanes are labelled as in
Figure 5 except (Ab) = antibody used for supershift; (M) = MEF-2 antibody; (P) = p300 antibody.
Figure 8. Cardiac MEF-2 and p300 interact in a DNA-dependent manner. A. Independent
immunoprecipitation of MEF-2 and p300. A MEF-2 antibody recognizing MEF-2A, C and D was
used to immunoprecipitate cardiac nuclear extracts. Immunoprecipitates were subjected to Western
analysis using the same MEF-2 antibody (lanes 1-4) and a polyclonal p300 antibody (lanes 5-9).
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
30
Lane 10 shows a p300 Western analysis of cardiac nuclear proteins immunoprecipitated with a
p300 antibody for comparison. MEF-2 is nearly quantitatively precipitated from extracts by this
method and appears as a doublet at around 70 kd (black arrowhead). p300 is not detectable in the
MEF-2 immunoprecipitates, but is clearly seen in the p300 immunoprecipitates (black arrow, right).
Lanes: (T) total nuclear extract. (U) unbound proteins. (W1, W2) proteins in wash fractions 1 and
2. Positions of size markers are shown on left of autoradiogram. (E) proteins specifically eluted
from beads. B. MEF-2 and p300 associate in the presence of the ATr site. A “pulldown”
assay was performed as described in Methods, using a biotinylated hSA ATr oligonucleotide linked
to streptavidin-coated beads for affinity purification of cardiac nuclear proteins. Nuclear extracts
were incubated with the DNA-linked beads for defined intervals and samples of the total input
protein (T), unbound protein (U), wash fractions (W3) and specific eluates (E) were subjected to
Western analysis with MEF-2 and p300 antibodies as in 8A. Nuclear extracts from C2C12
myoblasts were used as positive controls (T (C2)). Upper panel: MEF-2 Western. Cardiac MEF-
2 is eluted as a narrow doublet from the beads (arrows). Note the presence of additional MEF-2
species in the C2C12 extract. Lower panel: p300 Western. A band corresponding to p300 is
visible in the total cardiac nuclear extract (T) and in the specific eluate (E). Note a second band of
smaller size also recognized by the p300 antibody and specifically eluted from the affinity beads.
The identity of this protein is not known but it may represent a degradation product of p300.
Figure 9. MEF-2 but not GATA-4 is synergistic with p300. A. Functional interaction
between MEF-2D and p300. Transcriptional activities of the intact hSA promoter (p2130luc1) and
a mutant lacking the p300 responsive site (mATr) were compared in cardiac myocytes and HeLa
cells as indicated, in the presence and absence of p300, and with or without one of the four MEF-2
isoforms (A-D). 35 mm dishes of cardiac myocytes were transfected with the indicated constructs
at a ratio of reporter: p300: MEF = 2: 1: 1. Total transfected DNA was kept constant by addition of
appropriate amounts of blank CMV expression vector. This graph summarizes three independent
experiments in which substantially similar data was obtained. B. GATA-4 and p300 are not
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Slepak, T.I. et al. p300 regulates cardiac genes through MEF-2D
31
synergistic for skeletal actin promoter transcription. Cardiac myocytes were transfected with the
intact hSA promoter (p2130luc1, black bars) or the p300 responsive site mutant (mATr, light bars),
alone or in the presence of p300, GATA-4 or both. Transfections were performed as in 9A and
represent 3 independent determinations. *p < 0.05. **p < 0.01, both for comparison with control.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Table 1. Oligonucleotides used for point mutagenesis
Name Sequence
5'ϕGATA-BglII
3' ϕGATA
5'ATr-NheI
3'ATr
m6 sense
m6 antisense
m8 sense
m8 antisense
m9 sense
m9 antisense
mTEF sense
mTEF antisense
-1805 CCAGATCTACCCTATACTAGTC -1827
-1782 GAGGATAAAGTCTCCTATGGTTC -1804
-1445 GTGCTAGCAATAGAACCTGCAGC -1468
-1422 TGATATATGGGGCTATGCCTTCA -1444
-129 GGCCGAGGGAGGGGGCTCTAGTGCTTAAGACCCAAATATGGC-87
-83 TCGAGCCATATTTGGGTCTTAAGCACTAGAGCCCCCTCCCTC-125
-129 GGCCGAGGGAGGGGGCTCTAGTGCCCAACATCCGGATATGGC -87
-83 TCGAGCCATATCCGGATGTTGGGCAGTAGAGCCCCCTCCCTC -125
-129 GGCCGAGGGAGGGGGCTCTAGTGCCCAACACCCAACCATGGC -8
-83 TCGAGCCATGGTTGGGTGTTGGGCAGTAGAGCCCCCTCCCTC -125
-87 TCGAGAAGGGCAGCGAGGTACCTGCGGGGTGGCGCGGAGGGAA
TCGCCCGC -36
-38 GGGCGATTCCCTCCGCGCCACCCCGCAGGTACCTCGCTGCCCTTC
Table 2. Oligonucleotides used in EMSA assays.
Name Sequence
ϕGATA -sense
ϕGATA -antisense
ϕGATA -mutant-sense
ϕGATA -mutant-antisense
ATr-sense
ATr-antisense
ATr-mutant-sense
ATr--mutant-antisense
α-MHC-ATr sense
α-MHC-ATr antisense
MCK-ATr sense
MCK- ATr antisense
hSA-SRE sense
hSA-SRE antisense
5'ATCCTCACCGGATAGACCCTATAC3'
5'GTATAGGGTCTATCCGGTGAGGAT3'
5'ATCCTCACCAGATCTACCCTATAC3'
5'CTATAGGGTAGATCTGGTGAGGAT3'
5'CATATATCAGTGATATAAATAGAACCTGCA3'
5'TGCAGGTTCTATTTATATCACTGATATATG3'
5'CATATATCAGTGCTAGCAATAGAACCTGCA3'
5'TGCAGGTTCTATTGCTAGCACTGATATATG3'
5'GTAAGGGATATTTTTGCTTCACTTT3'
5'AAAGTGAAGCAAAAATATCCCTTAC3'
5'GCTCTAAAAATAACCCTG3'
5'CAGGGTTATTTTTAGAGC3'
5'CCAACACCCAAATATGG3'
5'GGTTGTGGGTTTATACC3'
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from


HindIII +203SstII +38
SstII -36XhoI -87
XmaIII -129SmaI -153
SalI -662XmaIII -698
PvuII -710SmaI -781PstI -1246
XbaI -1300
NdeI -1658
HindIII -2132
Ex1
GATA
SpeI-1759
ATr-2 CArG TEF-1
-2130 -1298 -628 -153 -87 +203-781-1243-1757
p2130luc1
p1757Luc1
p1656Luc1
p1298Luc1
p87Luc1
dNX
dNP
GATA
ATr
mATr GA
m6, m8, m9, mTEF
X
X
X X
X
A.
B.
Figure 1.
AP-1
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

-1810 CCTCA CCGGA TAGAC CCTAT ..A.. .CT..
GATA
-1268 TCAGT GATAT AAATA GAACC .C..G C..
ATr
m GATA
mATr
-114 CTCTA GTGCC CAACA CCCAA ATATG GCTCG T T..G. m6 T..GG m8 CC... m9
CArG I
-84 AGAAG GGCAG CGACA TTCCT GCGGG GTGGC ...GG .A...
TEF-1 Sp-1
mTEF
Figure 1C.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

0
0.1
0.2
0.3
0.4
0.5
p2130p1298
mATrp1656 m6 m8 m9
mTEF
hSA mutant
Promoter Activity
(light units/µg protein)
p87
Figure 2.
** * *
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

0 1 2 3 4 5
Fold induction
p2130luc1
p1757luc1
p1656luc1
p1298luc1
dNX
dNP
mGATA
mATr
mATrGATA
m8
m9
mTEF
p87luc1
-p300
1
1.74 ± 0.3
1.54 ± 0.25
0.6 ± 0.12
1.08 ± 0.2
0.78 ± 0.15
0.97 ± 0.15
0.71 ± 0.12
0.96 ± 0.08
0.33 ± 0.08
0.18 ± 0.06
0.15 ± 0.05
0.05 ± 0.01
+p300
2.51 ± 0.3
2.97 ± 0.88
4.01 ± 0.9
1.61 ± 0.09
2.95 ± 0.74
2.22 ± 0.45
1.67 ± 0.19
1.05 ± 0.35
0.95 ± 0.23
1.16 ± 0.26
0.92 ± 0.32
0.44 ± 0.17
0.25 ± 0.05
2.5
1.71
2.6
2.68
2.73
2.85
1.72
1.48
0.99
3.52
5.11
2.93
5
FoldConstruct
*
Figure 3.
*
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Cardiomyocytes
GATA mATr ATr
- -mGA
SNE NE NE
Probe:
S SATr mATr
ECs
NE S
ATr
12
3
Figure 4.
A.-
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ATr
1
2
3
- NE S AbMEF - NE ATr AbS
MEF-2Probe:
Figure 4.
B.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

CATATATCAGTGATATAAATAGAACCTGCAGTATATAGTCACTATATTTATCTTGGACGT
CATATATCAGTGATATAAATAGAAGTATATAGTCACTATATTTATCTT
TGATATAAATAGAACCTGCA ACTATATTTATCTTGGACGT
ATCAGTGATATAAATAGAACCT TAGTCACTATATTTATCTTGGA
CACTTGATAACAGAA GTGAACTATTGTCTT
Wild type ATr site
Left
Right
Center
Consensus GATA binding site
Figure 5.
abNE S ab- S ab- SNE NE
Center Left RightB.
A.
TruncatedATr sites
1
2
3
-1256
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

- NE ab
ATr (Left)
- NE ab
GATA consensus
S ATrS
Figure 5C.
12
3
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

1
2
3
ATr (Left) ATr (Full)
-NEGATAAntibody
+ + + + + + + +
p300- -
-
GATAp300- -
MEF2DMEF2
Figure 6.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

hSA ATr
1
2
3
- NE S M P
M-CK
- NE S M P - NE S M P
-MHC
Ab: Ab: Ab:
- NE S M P
Ab:
hSA-SRE
Figure 7.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

200 kd
120 kd
100 kd
80 kd
MEF-2 IP
MEF-2 antibody
T U W1 E T U W1 EW2
p300 antibody
p300 IP
Figure 8.
A.
T
207 kd
123 kd
86 kd
U W3 E T (C2)
p300 antibody
T U W3 E T (C2)
44 kd
123 kd86 kd
31 kdMEF-2 antibody
B.
MEF-2
p300
MEF-2
p300
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from
A.
Figure 9
hSA
ActivityPromoter
0
5
10
15
20
25
30
35
40
45
50
Control +GATA-4 +p300 +GATA-4+p300
(Light units/µg protein)
* **
hSAPromoterActivity
B.p2130luc1
mATr
(light units/µg protein)
50
40
30
20
10
0
- p300+ p300
- p300
+ p300HeLa cells
Cardiac myocytes
*
MEF-2 species: A B C D A B C D- -
p2130luc1 mATrPromoter:
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Hicks and Nanette H. BishopricTatiana I. Slepak, Keith A. Webster, Jie Zang, Howard Prentice, Ann O'Dowd, Martin N.
2-DControl of cardiac-specific transcription by p300 through myocyte enhancer factor
published online November 28, 2000J. Biol. Chem.
10.1074/jbc.M004625200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from