complex reactions/inhibition 2014 notes 6 6(inhibition)14.pdfcomplex reactions/inhibition – 2014...
TRANSCRIPT

VI–48
48
Complex Reactions/Inhibition – 2014 – Notes 6
e.g. two enzyme substrate complexes (reactant & product)
C CH
COO-
-OOC
H+ H2O C C COO-
H
OH
H
H
-OOC
fumarate (F) malate (M)
fumarase
E + F k-1
k1 E F
k2
k-2 E M
k3
k-3 E + M
Analysis gives initial rate (messy to work out, mixes rapid equilibrium & steady state, do forward reaction only, let k-3~0):
F =
][
]][[)(
3221
323121
032232
Fkkkk
kkkkkk
FEkkkkk =
]F[K
]F[V
FM
F
result appears simple, same as Michaelis Menton form:
MAX[S]/(KM+[S]) MAX = VF, but constants complex usual initial rate, experiment not separate intermediate
Aside: if do Relaxation measurement sense reverse rxn – analysis just do backwards, let "VR" be max. rev. rate
R = VR[M]/(KR
M + [M] ) from R = d[F]/dt = k-1[EF]
net rate (F-R) - did this considering Product, M
= (VFKR
M[F] – VRKR
M[M])/(KF
MKR
M + KR
M[F] + KR
M[M]) at beginning M = 0 -- gives back original form
at equilibrium: VFKR
M[F]e = VRKF
M[M]e Ke = [M]e/[F]e = VFK
RM / VRK
FM
To parse out all k1, k2 … etc. is difficult since observations combine Need to isolate/detect intermediates or vary conditions to get individual rate constants (ex. H/D exchange)

VI–49
49
Inhibition Sometimes path involves multiple intermediates
but still apparently simple (like above)
E + S k-1
k1 ES 2k ES' 3k ES'' 4k E + P
0 = kcat[E0][S]/(KM + [S])
Difference is kcat and KM combine all ki above if one step (i) is slow (rate limit) kcat = ki
turnover depends on slow step
KM always like (apparent) dissociation constant for all enzyme bound species: [E][S]/[ES] if k-1 >> ki then KM = k-1/k1 true dissociation const. Now many enzymes bind several different substrates
but only one gives product Some substrates called inhibitor, I, bind tightly
but have no reaction remove E0 - not avail. for S Competitive inhibition, S and I bind to the same site
E + S k-1
k1 ES
k2
k-2 E + P
E + I k3
k-3 EI
This agrees with books and notes, reduces amount of E0 available to create ES, depend on KI –dissoc. const. of EI
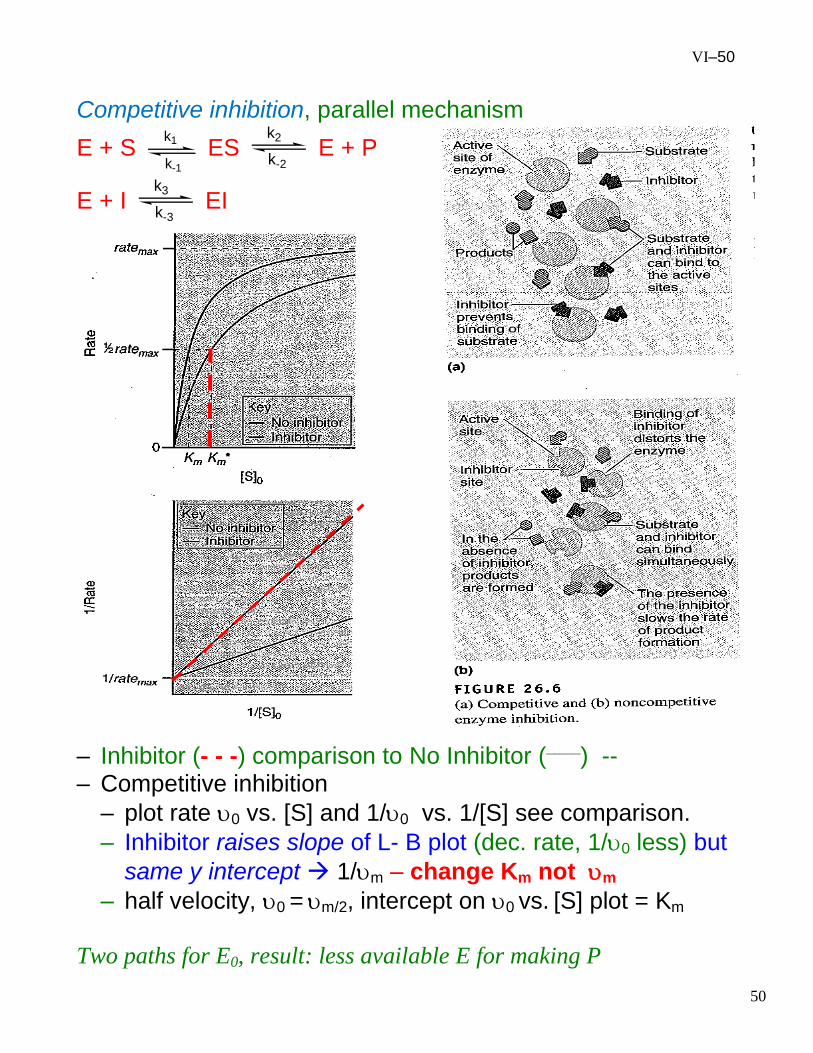
VI–50
50
Competitive inhibition, parallel mechanism
E + S k-1
k1 ES
k2
k-2 E + P
E + I k3
k-3 EI
– Inhibitor (- - -) comparison to No Inhibitor (____
) -- – Competitive inhibition
– plot rate 0 vs. [S] and 1/0 vs. 1/[S] see comparison.
– Inhibitor raises slope of L- B plot (dec. rate, 1/0 less) but
same y intercept 1/m – change Km not m
– half velocity, 0 = m/2, intercept on 0 vs. [S] plot = Km
Two paths for E0, result: less available E for making P

VI–51
51
do steady state on [ES] again -- still product only from k2 step
= k2[ES] [ES] = k1[E][S]/(k-1 + k2) = KM-1
[E][S] --as M-M but now [E0] = [E] + [ES] + [EI] reduce effect. enzyme, i.e [E] (EI ties up some of E0 - prevents formation ES, lowers E) let KI = [E][I]/[EI] KM = [E][S]/[ES] --2 dissoc const./paths [E0] = [E](1 + [I]/KI) + [ES] -- subst [EI] in [E0] eq. not substitute for ES because will solve for it
[I] reduces [E] + [ES] avail. -- consider Ek-1
k1ES equil.
Parameter for Inhibitor effect: = (1+[I]/KI) = (1+ [EI]/[E])
-- Note > 1, inc. [I] inc. ,
[E] = ([E0]–[ES])/(1+[I]/KI) = ([E0] – [ES])/ = [E]
sub [E] into [ES]: [ES] = KM-1
[E][S] = KM-1
[S]([E0]–[ES])/
rearrange - [ES](KM+[S])=[E0][S] [ES]=[E0][S]/(KM+[S])
rate like before: 0 = k2[E0][S]/(KM + [S]) = MAX/(KM/[S] + 1)
now inc. , decrease 0, but KM = K'M means
1/ vs. 1/S has diff. slope (KM/MAX) but same intercept
i.e. change Km not m - L-B plot intersect on y-axis at 1/m
- high KI, dissoc EI, = (1+[I]/KI) ~1, lots E, get same m
1/ = (K’M/ MAX)(1/[S]) + 1/MAX fit half rate K'M
As [I] increases I << KI ~ 1 ordinary M - M
I >> KI ~ [EI]/[E] >> 1, so [EI] >> [E]

VI–52
52
Three forms of inhibition are discussed in various texts: Competitive non-competitive uncompetitive E+S
k-1
k1 ES k2
k-2
E+P E+Sk-1
k1 ES k2
k-2
E+P E+Sk-1
k1 ES k2
k-2
E+P
E+I k3
k-3
EI E + I k3
k-3
EI
ES+I ESI ES+I ESI
as described above here both EI and ESI here no EI form ( = 1)
(if ’, simple case) but ESI form (' > 1) Change with inhibitor (
_____) compared to normal enzyme (
______)
The alternative to competitive are non-competitive and uncompetitive. The difference is how an inhibitor interacts with the enzyme-substrate complex, and prevents product forming Non-competitive: (derive Engel - pp 712-13)
E + S k-1
k1 ES
k2
k-2 E + P
E + I k3
k-3 EI
ES + I ESI

VI–53
53
0 = k2[E0][S]/{[S](1+[I]/KIS)+Km(1+[I]/KI)}
= MAX [S]/(’[S] + KM )
where ’ = (1+[I]/KIS), = (1+[I]/KI), KIS = [ES][I]/[EIS]
Again: [I]=0 ’=1 (MM), as [I] inc. reaction slows, less P In Lineweaver Burk, both slope and intercept change
1/ = (KM/ MAX)(1/[S]) + ’/MAX
Here - if ’>1 are independent, ≠ ’ i.e. KI≠ KIS then measured KM will be different with inhibitor
thus they do not have to intercept at x-axis at 1/ KM
However, most books show intercept on x-axis (called -1/KM )
between inhibited and no inhibitor for this, or = ’. Why? Tinoco makes comment that they describe “simplest case” – true Engel just asserts they intercept and KM is not changed
Atkins points out this is a special case, i.e. ’>1 but, = ’ Why do this? if I binds to a site away from the S binding site,
then it is likely that S does not affect the binding constant of I
if true then, KI = KIS and thus = ’ makes x-intercept be KM
Uncompetitive: drop E + I k3
k-3 EI = 1, ' > 1,
So in this case the inhibitor cannot bind enzyme alone, needs
the substrate first. Without this step KM = KM’ ( = 1)
slope not changed, but intercept change (’/MAX) Same equation, but Lineweaver-Burk plots are parallel
As Tinoco notes, this is a special case
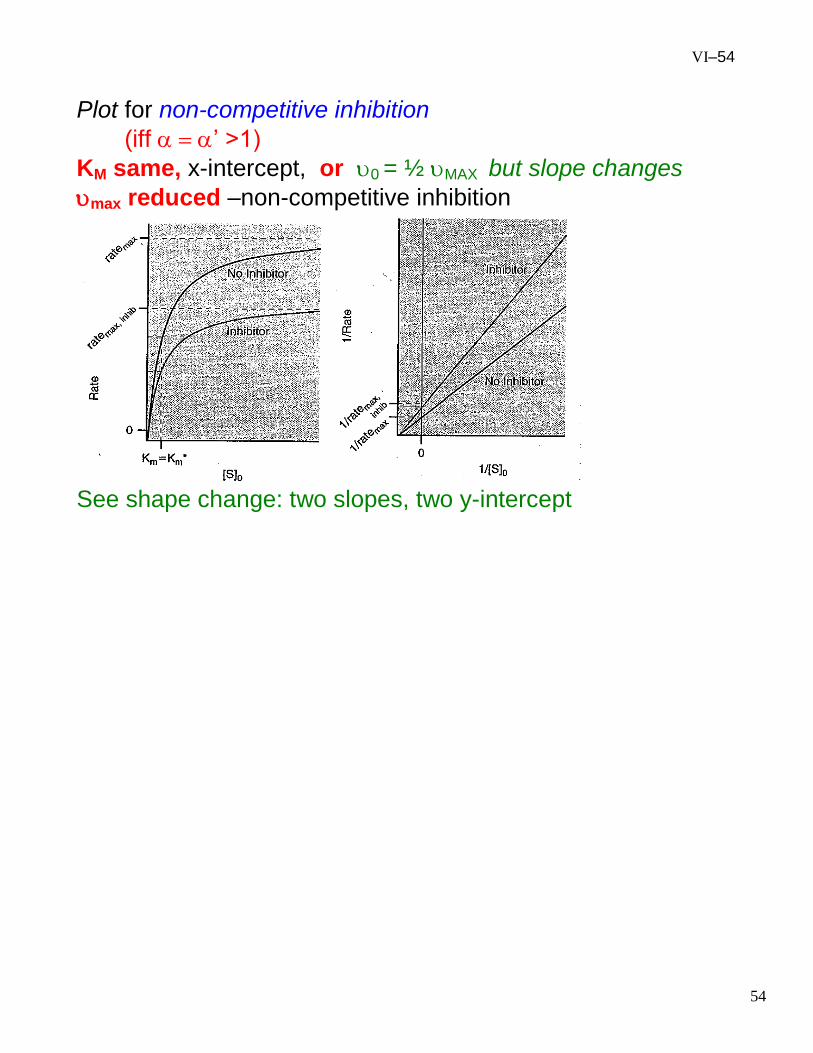
VI–54
54
Plot for non-competitive inhibition
(iff ’ >1)
KM same, x-intercept, or 0 = ½ MAX but slope changes
max reduced –non-competitive inhibition
See shape change: two slopes, two y-intercept

VI–55
55
Inhibition hugely important –
most biochemistry processes need enzymes – need specific reaction to go under control – need operate with low concentration enzyme – need fast response to S perturbation - on/off control – bio feedback/signaling– gone wrong – “sick”
Drugs/pharma intercede inhibit example dihdrofolate reductase (See Engel pp. 713-14)
DHFR reduces 7,8 dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate, a step in the biosynthesis of thymidine.
It is inhibited by methotrexate (MTX) – see structures:
Shape of MTX looks like DHF, but amine function is methylated, prevents use of N-H in reduction mechanism Used in cancer therapies, less thymidine, less cell division
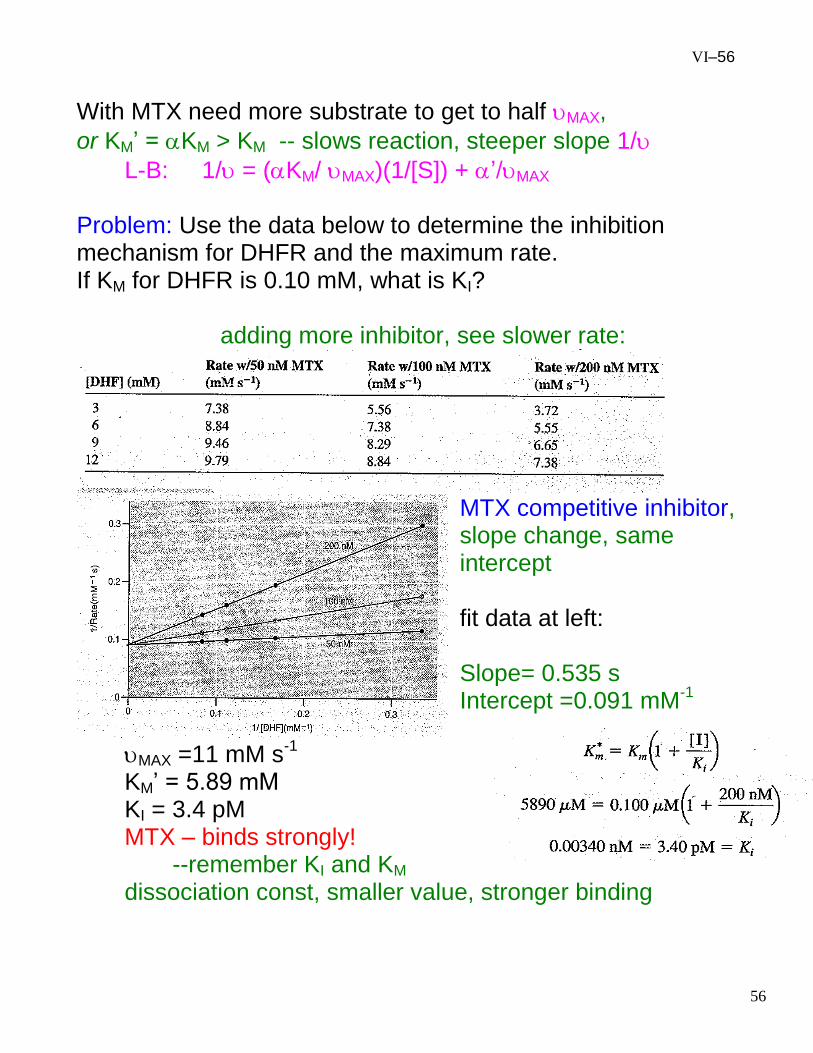
VI–56
56
With MTX need more substrate to get to half MAX,
or KM’ = KM > KM -- slows reaction, steeper slope 1/
L-B: 1/ = (KM/ MAX)(1/[S]) + ’/MAX Problem: Use the data below to determine the inhibition mechanism for DHFR and the maximum rate. If KM for DHFR is 0.10 mM, what is KI? adding more inhibitor, see slower rate:
MTX competitive inhibitor, slope change, same intercept fit data at left: Slope= 0.535 s Intercept =0.091 mM
-1
MAX =11 mM s-1
KM’ = 5.89 mM KI = 3.4 pM MTX – binds strongly!
--remember KI and KM dissociation const, smaller value, stronger binding

VI–57
57
e.g. succinic dehydrogenase Tinoco---p417-8
a) –OOC–CH2–CH2–COO
–
1HS –OOC–CH=CH–COO
–
succinate fumarate if add malonate:
–OOC–CH2–COO
–
has similar shape, 2 –COO- groups – binds active site
but cannot dehydrogenate (not form C=C) so stays on enzyme: EI forms, ties up E inhibit reaction of succinate: less ES like case (a), uses same site — Competitive inhibition b) Triosephosphate dehydrogenase - Cys in active site
if alkylate: I CH2CNH2
O
TPD S CH2C NH2
O
TPD S H +
This enzyme now lost to reaction inhibition (no reversal)
but KM same, max reduced (recall slope Km/max)
like case (c), sort of kills off the enzyme
– non-competitive ' > 1 see figure above
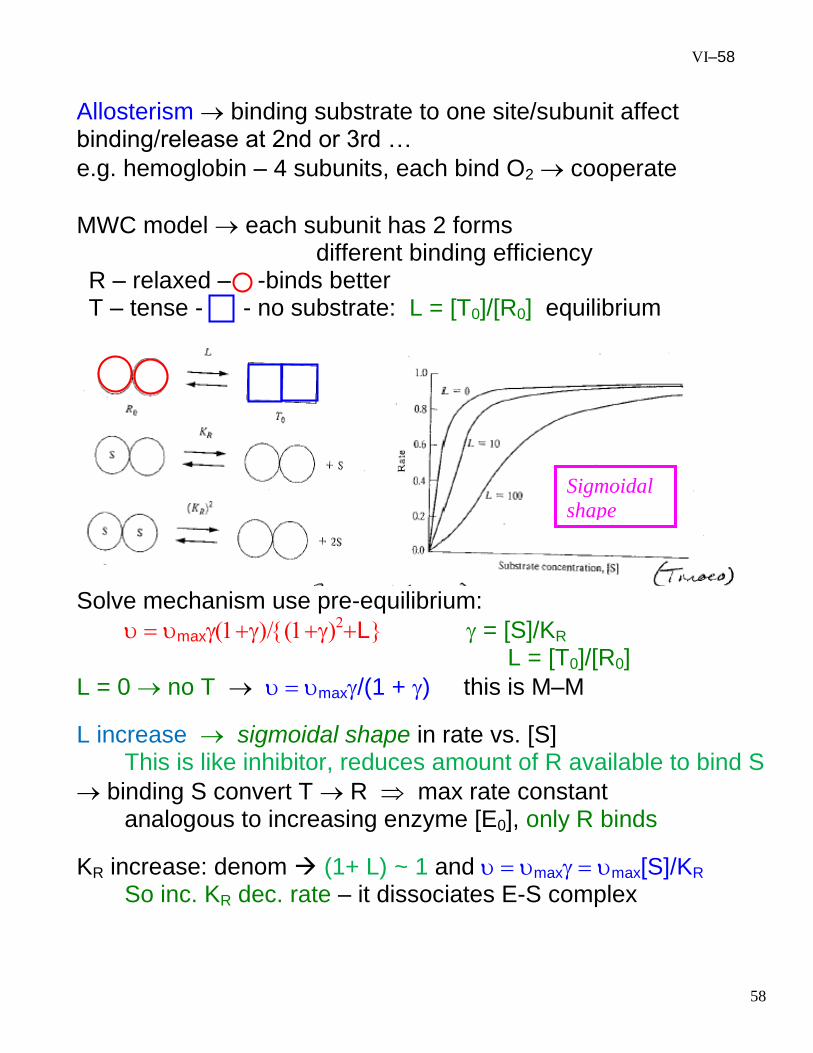
VI–58
58
Allosterism binding substrate to one site/subunit affect binding/release at 2nd or 3rd …
e.g. hemoglobin – 4 subunits, each bind O2 cooperate
MWC model each subunit has 2 forms different binding efficiency
R – relaxed – -binds better T – tense - - no substrate: L = [T0]/[R0] equilibrium
Solve mechanism use pre-equilibrium:
maxL = [S]/KR
L = [T0]/[R0]
L = 0 no T max/(1 + ) this is M–M
L increase sigmoidal shape in rate vs. [S] This is like inhibitor, reduces amount of R available to bind S
binding S convert T R max rate constant analogous to increasing enzyme [E0], only R binds
KR increase: denom (1+ L) ~ 1 and maxmax[S]/KR So inc. KR dec. rate – it dissociates E-S complex
Sigmoidal
shape

VI–59
59