combination strategy targeting vegf and hgf/c...
TRANSCRIPT

Small Molecule Therapeutics
Combination Strategy Targeting VEGF andHGF/c-met inHumanRenalCellCarcinomaModelsEric Ciamporcero1,2, Kiersten Marie Miles1, Remi Adelaiye1,3, Swathi Ramakrishnan1,3,Li Shen1, ShengYu Ku1,3, Stefania Pizzimenti2, Barbara Sennino4, Giuseppina Barrera2, andRoberto Pili1
Abstract
Alternative pathways to the VEGF, such as hepatocyte growthfactor or HGF/c-met, are emerging as key players in tumorangiogenesis and resistance to anti-VEGF therapies. The aim ofthis study was to assess the effects of a combination strategytargeting the VEGF and c-met pathways in clear cell renal cellcarcinoma (ccRCC) models. Male SCID mice (8/group) wereimplanted with 786-O tumor pieces and treated with either aselective VEGF receptor tyrosine kinase inhibitor, axitinib (36mg/kg, 2�/day); a c-met inhibitor, crizotinib (25 mg/kg, 1�/day); orcombination. We further tested this drug combination in ahuman ccRCC patient–derived xenograft, RP-R-01, in bothVEGF-targeted therapy-sensitive and -resistant models. To evalu-ate the resistant phenotype, we established an RP-R-01 sunitinib-resistant model by continuous sunitinib treatment (60 mg/kg,
1�/day) of RP-R-01–bearing mice. Treatment with single-agentcrizotinib reduced tumor vascularization but failed to inhibittumor growth in either model, despite also a significant increaseof c-met expression and phosphorylation in the sunitinib-resis-tant tumors. In contrast, axitinib treatment was effective in inhi-biting angiogenesis and tumor growth in both models, with itsantitumor effect significantly increased by the combined treat-ment with crizotinib, independently from c-met expression.Combination treatment also induced prolonged survival andsignificant tumor growth inhibition in the 786-O human RCCmodel. Overall, our results support the rationale for the clinicaltesting of combined VEGF and HGF/c-met pathway blockade inthe treatment of ccRCC, both in first- and second-line setting.MolCancer Ther; 14(1); 101–10. �2014 AACR.
IntroductionRenal cell carcinoma (RCC) strikes approximately >64,000
people and causes >13,000 deaths in a year in the United States(1). Approximately 80% of RCC cases are diagnosed as clear cellRCC (ccRCC) and the majority of them are sporadic tumors withacquired defects in both alleles of VHL (von Hippel-Landau)tumor-suppressor gene, resulting in VHL protein dysregulation(2). This defective protein is unable to bind under hypoxicconditions, and trigger proteasome-mediated degradation ofhypoxia-inducible transcription factor (HIF). The subsequenttranscriptional hyperactivation of HIF-targeted genes, such asVEGF, platelet-derived growth factor (PDGF), TGFa, hepatocyte
growth factor (HGF), and mesenchymal–epithelial transitionfactor (MET), drives tumor progression and hypervascularization(3–5).
Anti-VEGF drugs have been shown to have a great therapeuticbenefit in patients with ccRCC. The VEGF pathway does play apivotal role in tumor angiogenesis and its overactivation is oftenassociated with tumor growth and metastases (6). Among theVEGF-targeted therapies FDA approved as frontline treatment foradvanced RCC, tyrosine kinase inhibitors (TKI) represent themost common choice (7). Because of their mechanism of actionat the ATP-binding site, TKIs are selective rather than specific for asingle kinase. Sunitinib, in particular, has been shown to inhibitPDGF receptor (PDGFR), v-kit Hardy–Zuckerman 4 feline sarco-ma viral oncogene homolog (c-kit) and VEGF receptors 1 and 2(8). Albeit these multi-TKIs can be highly effective by targetingmore than one oncogenic pathway, a selective and potent VEGFRTKI may improve effectiveness and decrease the adverse eventsoften observed in patients treated with multitarget small mole-cules. Axitinib (former AG-013736) is a potent small-moleculeTKI, highly selective for VEGF receptor 1, 2, and 3 has beenapproved as a second-line treatment for RCC and is currentlybeing tested in phase II/III clinical trials for the treatment of solidtumors (9, 10). Axitinib advantages include a well-toleratedclinical safety profile and a relative short half-life (2–5 hours)that allows dose adjustment/titration (11).
VEGF-targeted therapies elicit survival benefit in RCC, butfail to produce enduring clinical responses in most patients.Indeed, inevitably, disease progresses following a transient 9- to11-month period of clinical benefit. Among the differentmechan-isms of evasive resistance to antiangiogenic therapies, the
1Genitourinary Program, Roswell Park Cancer Institute, Buffalo, NewYork. 2Department of Clinical and Biological Sciences, University ofTurin, Turin, Italy. 3Department of Cancer Pathology and Prevention,Roswell Park Cancer Institute, Buffalo, New York. 4Department ofAnatomy, Comprehensive Cancer Center, Cardiovascular ResearchInstitute, University of California-San Francisco, San Francisco,California.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
This study was previously presented at the 2013 American Association forCancer Research Annual Meeting.
Corresponding Author: Roberto Pili, Roswell Park Cancer Institute, Elm andCarlton Streets, Buffalo, NY 14263-0001. Phone: 716-845-3117; Fax: 716-845-8232; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-14-0094
�2014 American Association for Cancer Research.
MolecularCancerTherapeutics
www.aacrjournals.org 101
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

upregulation of alternative proangiogenic signals, and an increaseof the invasive and metastatic behavior of tumor cells have beenreported to play an important role (12, 13).
The HGF/MET factor (c-met) pathway has been shownto be relevant in acquired drug resistance as well as in tumorvascularization, epithelial-to-mesenchymal transition, andmetastases (14). C-met is one of the most deregulated receptortyrosine kinase (RTK) in advanced cancers and MET-activatingmutations are the genetic cause of hereditary papillary type I RCCand other cancers (15). Intriguingly, c-met is transcriptionallyactivated by hypoxia and acts as mediator of antiangiogenictherapy resistance in models of glioblastoma multiforme (16,17) and other solid tumors (18). Crizotinib (also known as PF-2341066) is an orally available, potent, and selective dual inhib-itor of anaplastic lymphoma kinase (ALK) and c-met kinase thathas been approved for the treatment of ALK-positive non–smallcell lung cancers (19, 20).
The aim of this study was to test the antitumor efficacy ofaxitinib and crizotinib combination in ccRCC models. The datasuggest that combination with crizotinib increases axitinibinduced antiangiogenic and antitumor activity in both TKIssensitive and TKIs resistant models.
Materials and MethodsCompounds
Axitinib (AG013736 or Inlyta), crizotinib (PF-02341066or Xalkori), and sunitinib (Sutent) were provided by Pfizer.For in vivo formulations, axitinib was prepared in 0.5% car-boxymethylcellulose solution and crizotinib was dissolved inwater by pH adjustment to a value between 3.5 and 4. Drugswere administered by oral gavage (per os or PO). The experi-mental groups were the following: vehicle (0.5% carboxymeth-ylcellulose, 2�/day, 5�/week, PO), axitinib (36 mg/kg, 2�/day, 5�/week, PO), crizotinib (25 mg/kg, 1�/day, 5�/week,PO), axitinib plus crizotinib combination (same schedule andconcentration as in single-agent groups). Treatments wereadministered as follows: 4 weeks (786-O 1-month endpoint),6 weeks (RP-R-01 sunitinib resistant), 10 weeks (RP-R-01sunitinib sensitive), or up to 15 weeks (786-O survival). Mousebody weight and tumor caliper measurements were takenweekly. No overt signs of toxicity were observed in any treat-ment group (i.e., significant weight loss or diarrhea).
Xenograft models and treatment protocolImmunodeficient SCID male mice purchased from Roswell
Park Cancer Institute (RPCI) were used for these studies and allprocedures were approved by the Institute Animal Care and UseCommittee.Micewere kept in a temperature controlled roomona12 of 12 hours light/dark schedule with food andwater ad libitum.Collectionof tumor sampleswasobtained via regulatory approvalat the institution.
786-O cells were purchased from the ATCC. Mice (8/group)were implanted under the right kidney capsule with approximate-ly 1-mm3 size tumor pieces derived from previously orthotopi-cally implanted, untreated 786-O tumors. Treatments beganapproximately 5 weeks later, when tumors were detectable bypalpation, and followed the schedule described above. 786-Osurvival study: 32 mice (8/group) were implanted and treated asdescribed above. Animals were monitored twice daily for healthissues, moribundmice were euthanized by CO2 asphyxiation and
deaths were recorded for eachmouse. Each animal found dead oreuthanized was necropsied. Criteria for euthanasia were based onan independent assessment by a veterinarian according to AAA-LAC guidelines and only cases in which the conditions of theanimal were considered incompatible with life were reported asdeaths. As a control of good surgical procedure, we performednecropsies onmice surviveduntil the endof treatment and tumorswere found to be present under the right kidney capsule in all ofthe cases.
RP-R-01 is a patient-derived xenograft model developedfrom a skin metastasis of a patient with sporadic ccRCCVHL�/� developed while on sunitinib treatment, as previouslydescribed (21). This model was propagated in vivo only tomaintain the heterogeneity of the primary tumor. The short-term study: mice (3/group) were implanted subcutaneously inthe flank area with approximately 4-mm2 size RP-R-01 tumorpieces. Treatment started when average tumor dimensionreached approximately 35 mm2: Mice were randomized in theabove-mentioned experimental groups and treated for either 2or 7 days. Because previous studies performed in our laboratoryshowed good antitumor efficacy of sunitinib in this model, weimplanted mice (8/group) subcutaneously with RP-R-01 tumorpieces, as described above, as models of sunitinib-sensitivehuman ccRCC. Treatment started when average tumor dimen-sion reached approximately 50 mm2. To establish a sunitinib-resistant model, we implanted 35 mice subcutaneously in theflank area with approximately 4- to 5-mm2 size RP-R-01 tumorpieces and, approximately 6 weeks later, when tumors reachedan average size of approximately 25 mm2, mice were treatedwith sunitinib (60 mg/kg, 5�/week, PO). We defined resistanttumors when they reached doubled size upon treatment (�50mm2). Thereafter, mice were divided into homogenous groups(7 mice/group) as determined by caliper measurements andrandomized to the above-mentioned experimental groups.Mice in all experiments have been sacrificed between 12 and18 hours after last treatment.
ImmunohistochemistryTissues were fixed for 24 hours in 10% neutral-buffered
formalin (c-met E-cadherin and Ki67) or zinc fixative (CD31),paraffin embedded and cut at 4 mm, placed on charged slides,and dried at 60�C for 1 hour. Slides were cooled to roomtemperature, deparaffinized in xylene, and rehydrated usinggraded alcohols. Antigen unmasking was heat mediated, incitrate buffer (pH 6.0) and followed by a 20 minutes cooldown. Endogenous peroxidases were quenched with 3% H2O2
for 10 minutes and washed with PBS-Tween20 0.1%. Slideswere then blocked for 1 hour with PBS 1% BSA and incubatedovernight in primary antibodies: Mouse CD31 (1:100, 550274;BD Pharmingen), c-met (1:300, 8198; Cell Signaling Technol-ogy), E-cadherin (1:400, 3195; Cell Signaling Technology), orKi67 (1:500; Thermo Scientific RM-9106). Sections were thenincubated in horseradish-conjugated anti-rabbit (E-cadherin,c-met, and Ki67) or anti-rat (CD31) antibody according to themanufacturer's protocol (Vector Laboratories) followed byenzymatic development in diaminobenzidine (DAB). Slideswere finally counterstained with hematoxylin, dehydrated, andmounted with cytoseal 60 (Thermo Scientific). Quantificationof the staining was performed by using ImageJ software in ablinded fashion by analyzing four randomly selected fields pertissue of six to eight samples per treatment. CD31, E-cadherin,
Ciamporcero et al.
Mol Cancer Ther; 14(1) January 2015 Molecular Cancer Therapeutics102
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

and c-met results are expressed as the average percentage ofpositive area per treatment � SE; Ki67 as the percentage ofpositive nuclei per treatment � SE calculated by Immunoratioplugin for ImageJ (22).
ImmunofluorescenceTissues were snap-frozen and stored at �80�C, 10-mm thick
sections were cut with a cryostat and placed in positively chargedslides. Sections were then fixed for 10 minutes at �20�C in PBS-4% paraformaldehyde solution and washed–permeabilized inPBS 0.3% Triton X-100. Phosphorylated c-met staining wasadapted from the protocol described by Sennino and colleagues(23): Slides were blocked for 1 hour with immunomix (PBS 0.3%Triton X-100, 5% normal horse serum, 0.2% BSA) and incubatedin primary c-met phosphorylation-specific antibody overnight atroom temperature (pYpYpY1230/1234/1235, 1:250, 44888G; Invi-trogen). Following primary incubation, sections were incubatedwith FITC-conjugated anti-rabbit antibody for 1 hour at roomtemperature in a humidified dark chamber. Cy3 goat anti-rat(Invitrogen) was used to detect the anti-CD31 antibody in thedual color fluorescence experiments. Immunocomplexes werethen briefly fixed for 5 minutes in 1% paraformaldehyde, nucleistained with DAPI, and slides mounted with vectashield mount-ing medium (Vector Laboratories). The number of phosphory-lated c-met–positive cells was counted in a blinded fashion byanalyzing at least six randomly selected 40� fields per tissue of sixsamples per treatment.
Intratumoral hypoxia detectionAt the end of treatment, mice in the RP-R-01 short-term
experimentwere injected i.p. with 60mg/kg pimonidazole hydro-chloride (Hypoxiprobe plus kit), and 1 hour later, mice wereeuthanized. To stain hypoxic areas, we followed the protocoldescribed for immunofluorescence, using 4.3.11.3 mouse-FITCMAb1 according to the manufacturer. The percentage of hypoxicarea was counted in a blinded fashion by analyzing at least sixrandomly selected 10� fields per tissue of three samples pertreatment.
Statistical analysisDifferences among experimental groups were tested by either
the Student t test or for variances by ANOVA. A P value of <0.05was considered statistically significant. The difference in tumorweight between treatment groups was statistically evaluated bynonparametric the Mann–Whitney U test.
ResultsAntitumor effect of axitinib and crizotinib in the 786-Oorthotopic model
To examine the therapeutic effect of axitinib and crizotinibin the high c-met–expressing 786-O model (SupplementaryFig. S1A), male SCID mice were orthotopically implanted underthe kidney capsule with tumor tissues. When tumors becamedetectable by palpation, mice were randomized to treatment witheither vehicle, axitinib, crizotinib, or combination. After 4 weeks,
5.0
Vehic
le
Axitini
b
Crizot
inib
Axitini
b +
crizo
tinib
Vehic
le
Axitini
b
Crizot
inib
Axitini
b +
crizo
tinib
Vehicle Axitinib
CA
D
B
Crizotinib Axitinib + crizotinib
Vehicle Axitinib
Crizotinib CombinationEO
T tu
mor
wei
ght (
g) 4.52.5
2.0
1.5
1.0
0.5
Per
cent
sur
viva
l
100
80
60
40
20
0
% C
D31
-pos
itive
are
a 8
6
4
2
0
0.0
0 30 60Days
90 120
Figure 1.Antitumor effect of axitinib andcrizotinib in the 786-O orthotopicmodel. Mice orthotopically implantedwith 786-O tumor pieces (8 mice/group) were treated for 4 weeks withvehicle, axitinib (36 mg/kg, 2�/day,5�/week), crizotinib (25 mg/kg,1�/day, 5�/week), or combination.A, endpoint tumor weight: each pointrepresents one tumor; bars, averageof each treatment group � SE;� , P < 0.05 and ��� , P < 0.001, ascompared with the combinationgroup, using two-tailed t test analysis.EOT, end of treatment. B, Kaplan–Meier survival curve of miceorthotopically implanted with 786-Otumor pieces and treated as describedabove. The vertical ticks, censoringtimes; ��, P < 0.01 calculated by thelog-rank test. C, tumors from micetreated as in A were harvested,processed, and tissue sections werestained for CD31 for visualization ofendothelial cells. D, blindedquantitative analysis of CD31,expressed as the mean percentage ofpositively stained area � SE;� , P < 0.05 and ����, P < 0.0001, ascompared with the combinationgroup, using two-tailed t test analysis.
Combination of Axitinib and Crizotinib in Human Renal Cell Carcinoma
www.aacrjournals.org Mol Cancer Ther; 14(1) January 2015 103
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

mice were sacrificed and tumors were excised and weighed.Although tumor weights in both single-agent groups were smallerthan in vehicle-treated mice (17% overall reduction in axitiniband 10% in crizotinib), no statistical differences were observed.However, the average tumorweight in the combination groupwassignificantly smaller as compared with single-agent groups (P ¼0.0292 vs. axitinib and P ¼ 0.0321 vs. crizotinib) and the vehiclegroup (P ¼ 0.0004), showing a 76% overall reduction as com-paredwith the vehicle group (Fig. 1A). Ki67 staining did not showsignificant differences in the proliferative index among groups inthis highly proliferative model (data not shown). Microvesseldensity analysis by CD31 staining revealed, as expected, a reduc-tion in tumor vascularization following treatment with eitheraxitinib (P < 0.0001) or crizotinib (Fig. 1C and D). Noteworthy,tumor vascularization was further reduced following combinedtreatment (P¼ 0.0040 vs. axitinib and P < 0.0001 vs. both vehicleand crizotinib single agent).
Then, in view of the combinatorial antitumor effect of axitiniband crizotinib, we conducted a survival experiment involving786-O tumors orthotopically implanted in mice treated asdescribed above. Kaplan–Meier survival curves show an increaseinmedian survival in both single-agent–treatedmice as comparedwith vehicle (42.5 days in vehicle, 77.5 in axitinib, 57 incrizotinib; Fig. 1B). At end of the approximately 4-month exper-iment, combination treatment resulted in a statistically significantimprovement and extension of survival (median survival, 107days; log-rank test, P ¼ 0.0045).
Axitinib and crizotinib short-term treatment in the sunitinib-sensitive RP-R-01 PDX model
To evaluate the efficacy of axitinib and crizotinib in anothersunitinib-sensitive but low c-met expression model (Supple-mentary Fig. S1A), SCID mice were implanted with the ccRCCPDX RP-R-01. Tumors were measured weekly with a caliper
Vehic
le 2d
Axitini
b 2d
Crizot
inib
2d
Axitini
b +
crizo
tinib
2d
Vehic
le 7d
Axitini
b 7d
Crizot
inib
7d
Axitini
b +
crizo
tinib
7d
Vehic
le 2d
Axitini
b 2d
Crizot
inib
2d
Axitini
b +
crizo
tinib
2d
Vehic
le 7d
Axitini
b 7d
Crizot
inib
7d
Axitini
b +
crizo
tinib
7d
C
A
D
B
Vehicle Axitinib Crizotinib Combination
Vehicle Axitinib Crizotinib Combination
% C
D31
-pos
itive
are
a
25
20
15
10
5
0
% H
ypox
ic a
rea
15
10
5
0
Figure 2.Axitinib and crizotinib short-termtreatment in the sunitinib-sensitiveRP-R-01 PDX model. SubcutaneousRP-R-01–bearing mice (3/group)were treated with vehicle, axitinib(36 mg/kg, 2�/day), crizotinib(25 mg/kg, 1�/day), or combinationfor either 2 (labeled as 2d) or 7 days(7d). Tumors were then harvested,processed, and tissue sections werestained by IHC for CD31 (A), to displayendothelial cells and pimonidazole byimmunofluorescence (green; B), toassess intratumor hypoxia (DAPIcounterstain marks nuclei in blue);scale bar, 50 mm. Blinded quantitativeanalysis of tumor vascularization (C)and intratumor hypoxia (D),expressed as the mean percentage ofpositively stained area � SE; � , P <0.05 and ��� , P < 0.001, using two-tailed t test analysis (the verticalspotted lines highlight the differencein time among groups).
Ciamporcero et al.
Mol Cancer Ther; 14(1) January 2015 Molecular Cancer Therapeutics104
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094
and, when the average tumor dimension reached approximate-ly 35 mm2, mice were randomized into eight groups andtreated with either vehicle, single agents, or combination foreither 2 or 7 days. At either time points, average tumor dimen-sion was not significantly different among groups (Supplemen-tary Fig. S1B and S1C). Despite homogenous tumor dimensionamong groups, tumor vascularization was already reduced after2 days of axitinib treatment (Fig. 2A). Crizotinib single-agenttreatment did not affect blood vessel density, but combinationtreatment displayed a stronger reduction of CD31-positive area,significantly lower than each other group after 7 days oftreatment (P ¼ 0.0159 vs. vehicle, P ¼ 0.0404 vs. axitinib, andP ¼ 0.0357 vs. crizotinib, Fig. 2C). In agreement with this
significant reduction in tumor vascularization, pimonidazolestaining (Fig. 2B) displayed a statistically significant increase inintratumor hypoxia in the combination group already at day 2(P ¼ 0.0400 vs. vehicle, Fig. 2D). Furthermore, at day 7, alsoaxitinib (P ¼ 0.0394) and crizotinib (P ¼ 0.0642) single agentsshowed an increased intratumor hypoxia compared with thevehicle group but, again, also at this time point the extent ofcombined treatment-induced hypoxia was greater (P ¼0.0009). As expected, the immunodetection of the phosphor-ylated c-met (Tyr1230/1234/1235) displayed a substantial reduc-tion in the number of positive cells in the tumors treated withcrizotinib (Supplementary Fig. S1D and S1E). CD31–pimoni-dazole dual-color immunofluorescence highlighted an obvious
Vehic
le
Axitini
b
Crizot
inib
Axitini
b +
crizo
tinib
Vehic
le
Axitini
b
Crizot
inib
Axitini
b +
crizo
tinib
Vehic
le
Axitini
b
Crizot
inib
Axitini
b +
crizo
tinib
Vehicle Axitinib
C
A
D
E
F
B
Crizotinib Axitinib + crizotinib
Vehicle Vehicle
Axitinib Axitinib
Crizotinib Crizotinib
Combination Combination
Avg
. tum
or d
imen
sion
(m
m2 ) 250
200
150
100
50
0
EO
T tu
mor
wei
ght (
g)
4
3
2
1
0
% K
i67-
posi
tive
cells 18
15
12
9
6
3
0
% C
D31
-pos
itive
are
a 12
9
6
3
0
0 7 14 21 28 35 42 49 56 63
n.s.
n.s.
Days70
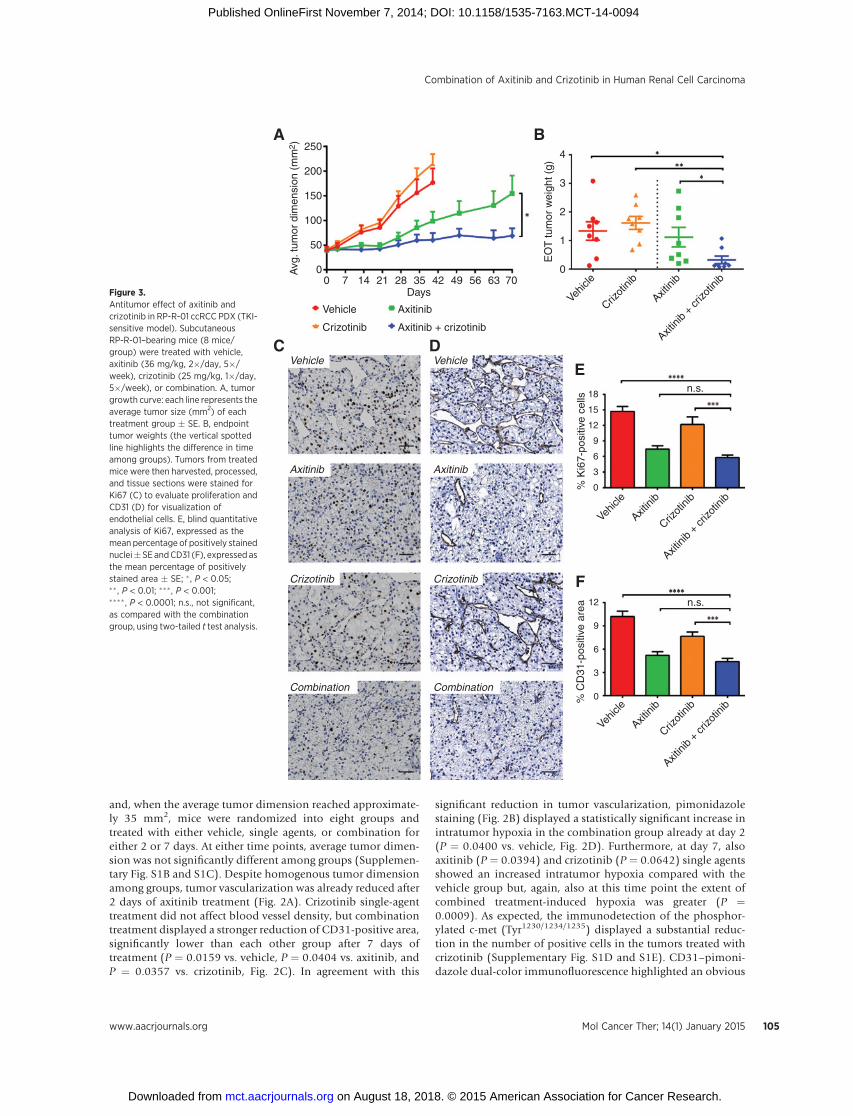
Figure 3.Antitumor effect of axitinib andcrizotinib in RP-R-01 ccRCC PDX (TKI-sensitive model). SubcutaneousRP-R-01–bearing mice (8 mice/group) were treated with vehicle,axitinib (36 mg/kg, 2�/day, 5�/week), crizotinib (25 mg/kg, 1�/day,5�/week), or combination. A, tumorgrowth curve: each line represents theaverage tumor size (mm2) of eachtreatment group � SE. B, endpointtumor weights (the vertical spottedline highlights the difference in timeamong groups). Tumors from treatedmice were then harvested, processed,and tissue sections were stained forKi67 (C) to evaluate proliferation andCD31 (D) for visualization ofendothelial cells. E, blind quantitativeanalysis of Ki67, expressed as themean percentage of positively stainednuclei�SEandCD31 (F), expressed asthe mean percentage of positivelystained area � SE; � , P < 0.05;�� , P < 0.01; ��� , P < 0.001;���� , P < 0.0001; n.s., not significant,as compared with the combinationgroup, using two-tailed t test analysis.
Combination of Axitinib and Crizotinib in Human Renal Cell Carcinoma
www.aacrjournals.org Mol Cancer Ther; 14(1) January 2015 105
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094
induction of hypoxia in all three treated groups, originatingsurprisingly from the blood vessels (with an overall morerobust effect in the combination group, SupplementaryFig. S2A). As shown in Supplementary Fig. S2B, endothelialcells of the tumor blood vessels display phosphorylated c-metstaining at least equal to RP-R-01 cancer cells, making also thema putative crizotinib target.
Antitumor effect of axitinib and crizotinib in thesunitinib-sensitive RP-R-01 PDX model
To assess the antitumor efficacy of axitinib and crizotinib inRP-R-01, we performed also a long-term treatment experiment.When the average tumor dimension reached approximately 50mm2, mice were randomized into four groups and treated witheither vehicle, single agents, or combination. Tumor growth inmice treated with either crizotinib or vehicle was similar andmice were sacrificed after 40 days of treatment (Fig. 3A).Treatment with axitinib single agent significantly decreased thegrowth of tumors, but combination with crizotinib furtherenhanced axitinib antitumor efficacy. After 70 days of treat-ment, tumors in the combination group were significantly
smaller than in the axitinib group (P ¼ 0.0474 for tumordimension and P ¼ 0.0490 for tumor weight; Fig. 3B). Ki67staining confirmed the inhibition of tumor proliferation inboth axitinib and crizotinib groups (P < 0.0001 vs.vehicle; Fig. 3C and E). Figure 3D shows representative CD31staining of tumor tissues. We observed a significant reductionin tumor vascularization in both axitinib and combinationgroups (P < 0.0001 vs. vehicle), although comparison betweenthese two groups was not significant. Blood vessel reduction incrizotinib-treated tumors as compared with the vehicle groupwas modest and not statistically significant (Fig. 3F). IHC for c-met did not show any significant difference in expressionamong the experimental groups (Supplementary Fig. S3A andS3C). Furthermore, neither c-met phosphorylation (Tyr1230/1234/1235) increase in the axitinib-treated group and decreasein the crizotinib-treated group were statistically significant(Supplementary Fig. S3B and S3D).
Establishing a sunitinib-resistant RP-R-01 ccRCC PDX modelTo mimic a TKIs resistant scenario, SCID mice were implanted
with RP-R-01 tumor tissues. Sunitinib treatment (60 mg/kg, 1�/
RP-R-01 SS RP-R-01 SR
RP-R-01 SS
RP-R-01 SS RP-R-01 SR
RP-R-01 SS RP-R-01 SR
RP-R-01 SR
1.23
0.52 1.04 1.16 1.31 0.99 2.50
0.90 1.63 1.91 2.03 2.40
c-met
Phospho c-met
β-Actin
A
C
E
F
B
D
G
18
15
12
9
6
3
0% c
-met
–pos
itive
are
a
SS SR
SS SR
SS SR
80
60
40
20
0
% P
hosp
ho c
-met
–pos
itive
cel
ls%
E-c
adhe
rin–p
ositi
ve a
rea
5
4
3
2
1
0
n.s.
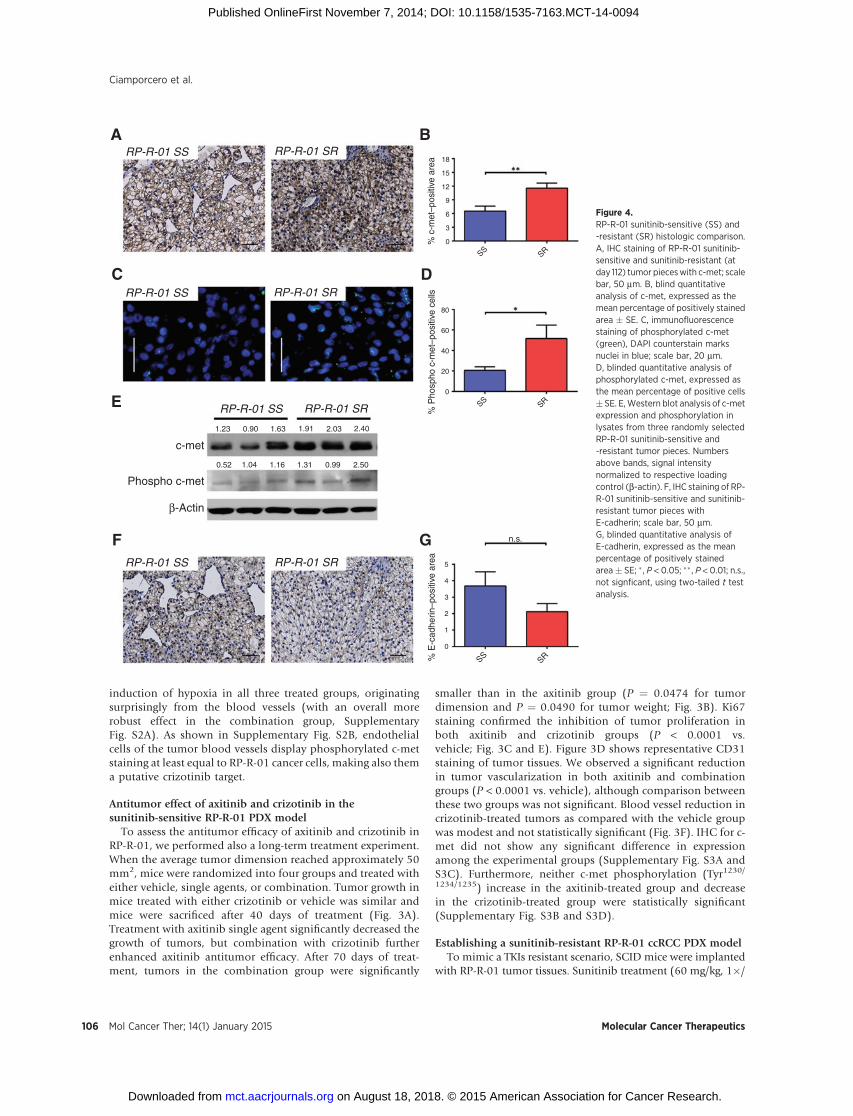
Figure 4.RP-R-01 sunitinib-sensitive (SS) and-resistant (SR) histologic comparison.A, IHC staining of RP-R-01 sunitinib-sensitive and sunitinib-resistant (atday 112) tumor pieceswith c-met; scalebar, 50 mm. B, blind quantitativeanalysis of c-met, expressed as themean percentage of positively stainedarea � SE. C, immunofluorescencestaining of phosphorylated c-met(green), DAPI counterstain marksnuclei in blue; scale bar, 20 mm.D, blinded quantitative analysis ofphosphorylated c-met, expressed asthe mean percentage of positive cells� SE. E,Western blot analysis of c-metexpression and phosphorylation inlysates from three randomly selectedRP-R-01 sunitinib-sensitive and-resistant tumor pieces. Numbersabove bands, signal intensitynormalized to respective loadingcontrol (b-actin). F, IHC staining of RP-R-01 sunitinib-sensitive and sunitinib-resistant tumor pieces withE-cadherin; scale bar, 50 mm.G, blinded quantitative analysis ofE-cadherin, expressed as the meanpercentage of positively stainedarea� SE; � , P < 0.05; �� , P < 0.01; n.s.,not signficant, using two-tailed t testanalysis.
Ciamporcero et al.
Mol Cancer Ther; 14(1) January 2015 Molecular Cancer Therapeutics106
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

day, 5�/week, PO) started when tumors were detectable bycaliper measurement (�25 mm2) and continued until theaverage tumor dimension doubled (�50 mm2), around day112. At that time, we defined tumors as sunitinib resistant andmice were then randomly distributed into five experimentalgroups: either released from treatment ("vehicle"), maintainedon sunitinib, switched to axitinib, to crizotinib or to axitinibplus crizotinib. Immunohistochemical comparison of suniti-nib-sensitive and -resistant RP-R-01 tumors (harvested at day112) indicated an increase in c-met expression (Fig. 4A and B)and a decrease in E-cadherin expression (Fig. 4F and G),suggesting a role of the HGF/c-met pathway in sunitinib-acquired resistance and a potential switch to a more mesen-chymal phenotype. Increased c-met expression was paired withan increased activity of c-met RTK in the sunitinib-resistanttumors as suggested by immunofluorescence detection of thephosphorylated Tyr1230/1234/1235 in the c-met activation loop (P¼ 0.0453, Fig. 4C and D). Western blot analysis performed onthree representative tumors per group confirmed the increase ofc-met phosphorylation in the sunitinib-resistant experimentalsetting, and suggested not only an increase in the number ofpositive cells but also an overall increase of c-met activation(Fig. 4E).
Antitumor effect of axitinib and crizotinib in thesunitinib-resistant RP-R-01 PDX model
Despite an observed increase in c-met expression and phos-phorylation, treatment with crizotinib did not result in tumorgrowth inhibition (Fig. 5A). On the other hand, mice eithertreated with axitinib or maintained on sunitinib showedsignificant inhibition of tumor growth as compared with micetaken off treatment (tumor weight at day 151, end of treat-ment: P ¼ 0.0247 vs. "vehicle" both axitinib or sunitinib,Fig. 5B). Once again, axitinib plus crizotinib combined treat-ment was more effective than continuous sunitinib treatment
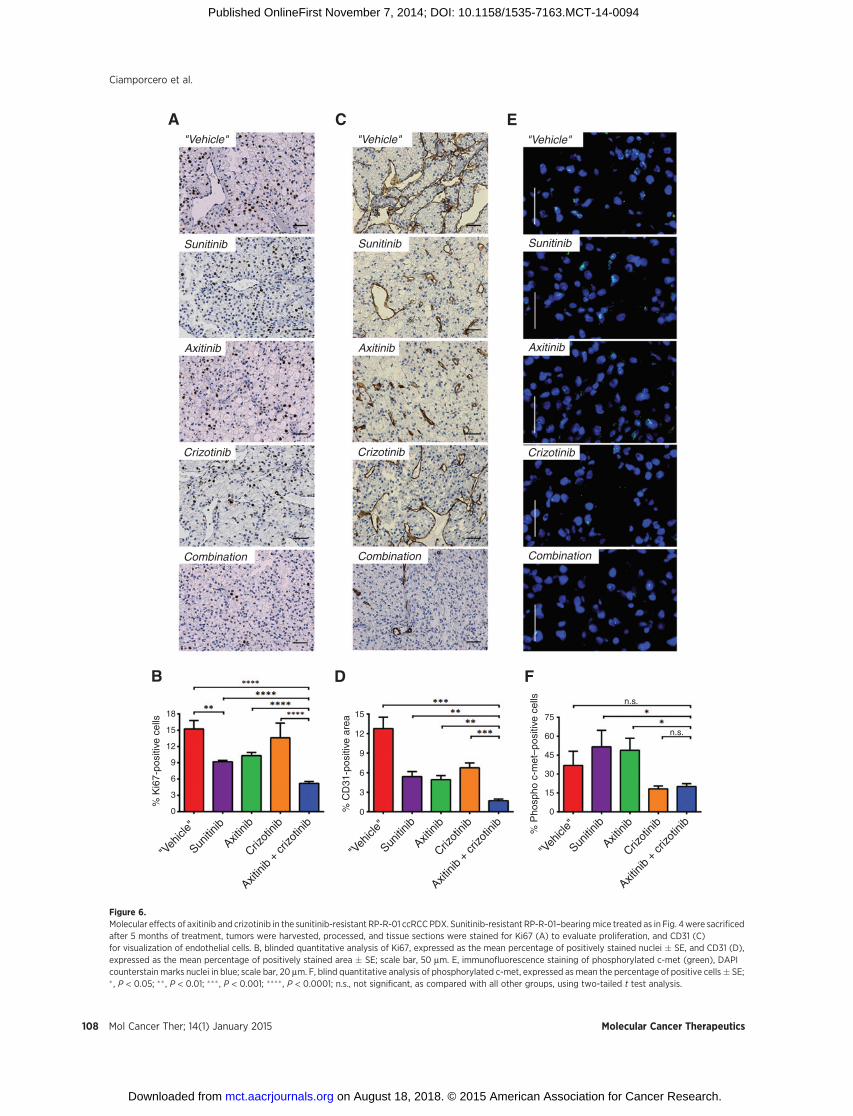
or switching to axitinib monotherapy (end of treatment tumorweight, P ¼ 0.0277 vs. sunitinib and P ¼ 0.0133 vs. axitinib).Ki67 immunostaining showed a strong increase of resistanttumor proliferation in mice taken off treatment as comparedwith tumors from mice maintained on sunitinib (P ¼0.0023, Fig. 6A and B). Despite crizotinib treatment did notshow any significant decrease in Ki67 staining, combinationwith axitinib decreased the percentage of proliferative nuclei toless than 5% (P < 0.0001). Figure 6C shows tumor blood vesselstaining with CD31. Tumors released from sunitinib treatmentwere hypervascularized (P ¼ 0.0057 "vehicle" vs. sunitinib)and this effect was reverted by both axitinib and crizotinibtreatment (P ¼ 0.0035 and P ¼ 0.0096 vs. "vehicle," respec-tively). Combination treatment reduced tumor vascularizationeven more (P ¼ 0.0013 vs. axitinib and P ¼ 0.0002 vs.crizotinib, Fig. 6D). C-met expression by IHC did not showsignificant differences among experimental groups (data notshown). Though, the strong increase in c-met phosphorylation(Tyr1230/1234/1235) in sunitinib- and axitinib-treated tumorswas dramatically reduced by concomitant crizotinib treatment(P ¼ 0.0399 vs. sunitinib and P ¼ 0.0149 vs. axitinib singleagent, Fig. 6E and F). Interestingly, single-agent crizotinibtreatment was able to significantly reduce phosphorylated c-met expression, but this biologic effect was not associated withtumor growth inhibition. In accordance with the inhibition ofthe c-met pathway activity, crizotinib also induced a decreasein E-cadherin staining. However, this reduction was not asso-ciated with significant morphologic changes (SupplementaryFig. S4).
DiscussionTreatment of patients with ccRCC with TKIs, such as suni-
tinib, has been shown to induce significant clinical benefitand represents standard of care. However, inevitably tumor
300
250
200
150
100
50
00 28 56 84 112 122 132 142 152
days
Avg
. tum
or d
imen
sion
(m
m2 )
EO
T tu
mor
wei
ght (
g)
4
3
2
1
0
"Vehicle" "Veh
icle"
Sunitinib Sunitin
ibAxit
inib
Axitini
b +
crizo
tinib
Crizot
inib
AxitinibCrizotinib Axitinib + crizotinib
A B
Figure 5.Antitumor effect of axitinib and crizotinib in the sunitinib-resistant RP-R-01 ccRCC PDX. Subcutaneous RP-R-01–bearing mice (8 mice/group) were treated withsunitinib (60 mg/Kg, 1�/day, 5�/week) until tumor dimension doubled in size. Then, mice bearing tumors defined as sunitinib resistant were randomly distributedinto five experimental groups: released from treatment ("vehicle"), maintained in sunitinib, axitinib (36 mg/kg, 2�/day, 5�/week), crizotinib (25 mg/kg, 1�/day,5�/week), or axitinib plus crizotinib. A, tumor growth curve: each line represents the average tumor size (mm2) of each treatment group � SE. B, endpointtumor weights; � , P < 0.05; ��, P < 0.01; and ��� , P < 0.001, as compared with the combination group, using two-tailed t test analysis.
Combination of Axitinib and Crizotinib in Human Renal Cell Carcinoma
www.aacrjournals.org Mol Cancer Ther; 14(1) January 2015 107
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094
"Vehicle"
"Veh
icle"
"Veh
icle"
Sunitin
ib
Sunitin
ib
Axitini
b
Axitini
b
Axitini
b +
crizo
tinib
Axitini
b +
crizo
tinib
Crizot
inib
Crizot
inib
"Veh
icle"
Sunitin
ibAxit
inib
Axitini
b +
crizo
tinib
Crizot
inib
"Vehicle" "Vehicle"
Sunitinib Sunitinib Sunitinib
Axitinib Axitinib Axitinib
Crizotinib Crizotinib Crizotinib
Combination Combination Combination
A C E
B D F
18
15
12
9
6
3
0
15
12
9
6
3
0
75
60
45
30
15
0
% K
i67-
posi
tive
cells
% C
D31
-pos
itive
are
a
% P
hosp
ho c
-met
–pos
itive
cel
ls n.s.
n.s.
Figure 6.Molecular effects of axitinib and crizotinib in the sunitinib-resistant RP-R-01 ccRCC PDX. Sunitinib-resistant RP-R-01–bearingmice treated as in Fig. 4were sacrificedafter 5 months of treatment, tumors were harvested, processed, and tissue sections were stained for Ki67 (A) to evaluate proliferation, and CD31 (C)for visualization of endothelial cells. B, blinded quantitative analysis of Ki67, expressed as the mean percentage of positively stained nuclei � SE, and CD31 (D),expressed as the mean percentage of positively stained area � SE; scale bar, 50 mm. E, immunofluorescence staining of phosphorylated c-met (green), DAPIcounterstain marks nuclei in blue; scale bar, 20 mm. F, blind quantitative analysis of phosphorylated c-met, expressed as mean the percentage of positive cells� SE;� , P < 0.05; �� , P < 0.01; ��� , P < 0.001; ���� , P < 0.0001; n.s., not significant, as compared with all other groups, using two-tailed t test analysis.
Ciamporcero et al.
Mol Cancer Ther; 14(1) January 2015 Molecular Cancer Therapeutics108
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

develops drug resistance and additional treatments are need-ed. Axitinib is a small molecule that inhibits VEGFRs activitywith high specificity. In a phase III clinical trial, axitinib hasbeen reported to significantly improve progression-free sur-vival (PFS) compared with sorafenib and has been approvedas second-line treatment in patients with advanced RCC (9,10). In our RP-R-01 sunitinib-resistant model, the comparisonbetween sunitinib and axitinib treatment did not show sig-nificant differences in terms of tumor growth and vasculari-zation. This observation suggests that, at least in this model,inhibition of additional kinases (such as PDGFR and c-kit)may not provide advantage in tumor growth inhibition ascompared with more selective VEGFR inhibition. On the otherhand, we observed early onset of tumor resistance to anti–VEGF-targeted therapy in the 786-O model as previouslyreported (24). Interestingly, axitinib treatment in the 786-Oorthotopic model inhibited tumor vascularization but nottumor growth, and did not induce significant improvementin survival compared with vehicle. However, combinationwith crizotinib not only increased axitinib inhibition oftumor microvessel density, but also tumor growth and itimproved survival. In this rapidly highly proliferative model,only when c-met inhibition and VEGFR blockade occurredconcomitantly, the treatment led to tumor growth inhibitionand improved survival.
The patient-derived xenograft developed in our laboratory,RP-R-01 (21) is a clinically relevant model of ccRCC becausethese tumors, by being passaged only in vivo, likely maintainthe original heterogeneity that is often lost in tumor cell lines,and, more importantly, retain the clear cell morphology. In theshort-term treatment experiment performed with this model,we noticed a significant reduction of blood vessels in axitinib-treated tumors in the absence of significant response in tumorgrowth. Blood vessels reduction in axitinib plus crizotinibgroup was even more significant, leading to a striking earlyinduction of hypoxia following only 2 days of treatment.Moreover, a substantial increase in intratumor hypoxia insingle-agent groups was detectable following 1-week treat-ment. HIF1a Western blot analysis also showed increasedlevels in the combination group following either short- orlong-term treatment in the sunitinib-sensitive model, butsignificant decrease in the sunitinib-resistant tumors followinglong-term treatment (Supplementary Fig. S5A, S5B, and S5C).Interestingly, pimonidazole–CD31 dual staining pointed thattreatment-induced hypoxia originates from the endothelialcells, suggesting a direct drug effect on the blood vessels.Furthermore, RP-R-01 tumors showed a strong response toVEGF-targeted therapies, and TKI resistance is a true acquiredevent that occurs after months of sunitinib treatment instead ofthe relatively short period displayed by tumor cell lines. Toestablish a TKI-resistant model, we treated RP-R-01–bearingmice with sunitinib (60 mg/kg, 1�/day, 5�/week) until, fol-lowing a period of stabilization, the average tumor size dou-bled from baseline. In these sunitinib-resistant tumors, weobserved an increase in c-met expression and activation asreported by other groups in glioblastoma, pancreatic neuro-endocrine tumors, and other solid tumors (16–18, 23). Incontrast with the sunitinib-sensitive model, in the resistant RP-R-01 tumors we noticed a significant decrease in tumor vas-cularization in the combination group as compared with theaxitinib-treated group, suggesting a role of c-met in blood
vessels homeostasis. However, regardless of c-met expressionand activity, crizotinib was not effective as single agent at thedose used, but it was able to significantly improve axitinibantitumor activity in both models. Furthermore, regardless ofthe inhibition of c-met phosphorylation following crizotinibtreatment, tumor growth rate was similar to vehicle-treatedtumors, suggesting a possible "rebound" effect when anti-VEGF therapy is halted (25), and the potential benefit ofcontinuing VEGF blockade in patients despite radiologic signsof disease progression on initial TKIs. Overall, our data suggestthat c-met inhibition is therapeutically effective in the settingof concomitant inhibition of VEGF in RCC models, withoutsignificant toxicity (Supplementary Fig. S6).
The molecular mechanisms responsible for c-met capabilityof compensating VEGF inhibition in RCC remain to be eluci-dated. One possibility is that the c-met/HGF pathway has astronger role in tumor endothelial and stromal cells, in which itacts as a potent proangiogenic trigger, supporting tumorgrowth. In fact, HGF is a well-known inducer of endothelialcell proliferation, survival and migration, and a chemoattrac-tant for proangiogenic bone marrow–derived progenitor cells(26). These changes in the tumor microenvironment may fosterangiogenesis, leading to tumor growth regardless of the statusof c-met expression in the tumor. It has been shown thattreatment with a decoy c-met not only delays the growth ofc-met–positive xenografts, but also the growth of c-met–neg-ative tumors (27). This observation could explain our findingsshowing the lack of association between the effect of crizotiniband c-met expression in cancer cells. A second hypothesis is thatanti-VEGF therapies eliminate basal c-met inhibition as dem-onstrated by Lu and colleagues (16). A model of glioblastomamultiforme showed a direct VEGFR2 physical association withc-met that led to its posttranslational inactivation. In thiscontext, VEGF blockade abrogated the suppression of c-metphosphorylation, activating the c-met/HGF pathway directly incancer cells. Finally, c-met transcriptional activation coulddirectly lead to survival benefit and prevent from apoptosis ina VEGF inhibition context, as demonstrated following EGFRinhibition by gefitinib (28).
There are emerging clinical data, suggesting that c-metrepresents a potential target for therapeutic intervention. Ina recent phase I clinical trial treatment with cabozantinib, adual c-met and VEGFR inhibitor, has been shown to induce a30% objective response rate and a PFS of 14.7 months inpatients with prior VEGF or mTOR inhibitors (29). Thesepromising results have led to the further clinical developmentof cabozantinib in patients with recurrent RCC both in first-line setting and following TKIs. Our preclinical data did notidentify an optimal setting (TKI-sensitive vs. TKI-resistantdisease) for the introduction of c-met inhibition. The challengeis represented by the fact that, at least under our experimentalconditions, tumor c-met expression does not seem to bepredictive of response to a selective inhibitor. Future preclin-ical and clinical testing of c-met inhibitors will define the roleof these agents in the armamentarium available to effectivelytreat recurrent RCC.
In conclusion, to our knowledge, this study is the first preclin-ical evidence of the key role of c-met in response to anti-VEGFtherapy in ccRCC by using different models. Overall, our resultshighlight the potential therapeutic combination of VEGF andHGF/c-met pathway inhibition in the treatment of ccRCC, both in
www.aacrjournals.org Mol Cancer Ther; 14(1) January 2015 109
Combination of Axitinib and Crizotinib in Human Renal Cell Carcinoma
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

the first- and second-line setting and independently from consti-tutively overexpressed c-met in tumor cells.
Disclosure of Potential Conflicts of InterestR. Pili reports receiving a commercial research grant and is a consultant/
advisory board member for Pfizer. No potential conflicts of interest weredisclosed by the other authors.
Authors' ContributionsConception and design: E. Ciamporcero, K.M. Miles, L. Shen, S. Pizzimenti,R. PiliDevelopment of methodology: E. Ciamporcero, K.M. Miles, S.Y. Ku, R. PiliAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): E. Ciamporcero, K.M. Miles, R. Adelaiye, R. PiliAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): E. Ciamporcero, R. Adelaiye, R. PiliWriting, review, and/or revision of the manuscript: E. Ciamporcero, L. Shen,S. Pizzimenti, B. Sennino, R. Pili
Administrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): S. Ramakrishnan, L. Shen, B. Sennino, R. PiliStudy supervision: G. Barrera, R. Pili
AcknowledgmentsThe authors thank the MTMR and Pathology Core Facilities at RPCI for
animal handling and processing the tissue samples.
Grant SupportThis research was supported in part by the National Cancer Institute,
NIH (P30CA016056; to R. Pili), and by a research grant from Pfizer (toR. Pili).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received February 5, 2014; revised October 1, 2014; accepted October 28,2014; published OnlineFirst November 7, 2014.
References1. Siegel R, NaishadhamD, Jemal A. Cancer statistics, 2013. CA Cancer J Clin
2013;63: 11–30.2. Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet 2009;373:
1119–32.3. Baldewijns MM, van Vlodrop IJH, Vermeulen PB, Soetekouw PMMB, van
Engeland M, de Bru€�ne AP. VHL and HIF signalling in renal cell carcino-genesis. J Path 2010;221: 125–38.
4. Oh RR, Park JY, Lee JH, Shin MS, KimHS, Lee SK, et al. Expression of HGF/SF and Met protein is associated with genetic alterations of VHL gene inprimary renal cell carcinomas. Acta Pathol Microbiol Immunol Scand2002;110: 229–38.
5. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Como-glio PM.Hypoxia promotes invasive growthby transcriptional activationofthe met protooncogene. Cancer Cell 2003;3: 347–61.
6. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factorpathway in tumor growth and angiogenesis. J Clin Oncol 2005;23:1011–27.
7. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumouractivity. Nature Rev Cancer 2008;8: 579–91.
8. Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinibefficacy and future clinical development. Nature Rev Drug Discov 2007;6:734–45.
9. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al.Comparative effectiveness of axitinib versus sorafenib in advanced renal cellcarcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378: 1931–9.
10. Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S,et al. Axitinib versus sorafenib as second-line treatment for advanced renalcell carcinoma: overall survival analysis and updated results from arandomised phase 3 trial. Lancet Oncol 2013;14: 552–62.
11. Hu-Lowe DD, Zou HY, Grazzini ML, HallinME,Wickman GR, AmundsonK, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib(AG-013736), an oral, potent, and selective inhibitor of vascular endo-thelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res2008;14: 7272–83.
12. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy.Nature Rev Cancer 2008;8: 592–603.
13. Sennino B, McDonald DM. Controlling escape from angiogenesis inhibi-tors. Nat Rev Cancer 2012;12: 699–709.
14. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles andfunctions in development, organ regeneration and cancer. Nat Rev MolCell Biol 2010;11: 834–48.
15. Danilkovitch-miagkova A, Zbar B. Dysregulation of Met receptor tyrosinekinase activity in invasive tumors. J Clin Invest 2002;109: 863–7.
16. Lu K V, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D,et al. VEGF inhibits tumor cell invasion and mesenchymal transitionthrough a MET/VEGFR2 complex. Cancer Cell 2012;22: 21–35.
17. Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu Y-L, Lu K, et al.Gene expression profile identifies tyrosine kinase c-Met as a targetablemediator of antiangiogenic therapy resistance. Clin Cancer Res 2013;19:1773–83.
18. Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee Pa, et al.HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistanttumors. Cancer Res 2010;70: 10090–100.
19. Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. Anorally available small-molecule inhibitor of c-Met, PF-2341066, exhibitscytoreductive antitumor efficacy through antiproliferative and antiangio-genic mechanisms. Cancer Res 2007;67: 4408–17.
20. Shaw AT, KimD-W, Nakagawa K, Seto T, Crin�o L, AhnM-J, et al. Crizotinibversus chemotherapy in advanced ALK-positive lung cancer. NEJM2013;368: 2385–94.
21. Hammers HJ, Verheul HM, Salumbides B, Sharma R, Rudek M, Jaspers J,et al. Reversible epithelial to mesenchymal transition and acquired resis-tance to sunitinib in patients with renal cell carcinoma: evidence from axenograft study. Mol Cancer Ther 2010;9: 1525–35.
22. Tuominen VJ, Ruotoistenm€aki S, Viitanen A, Jumppanen M, Isola J.ImmunoRatio: a publicly available web application for quantitative imageanalysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67.Breast Cancer Res 2010;12: R56.
23. Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW,Bhagwandin V, et al. Suppression of tumor invasion and metastasis byconcurrent inhibition of c-Met and VEGF signaling in pancreatic neuro-endocrine tumors. Cancer Discov 2012;2: 270–87.
24. Bhatt RS, Wang X, Zhang L, Collins MP, Signoretti S, Alsop DC, et al.Renal cancer resistance to antiangiogenic therapy is delayedby restoration of angiostatic signaling. Mol Cancer Ther 2010;9:2793–802.
25. MancusoMR,Davis R,Norberg SM,O'Brien S, SenninoB,Nakahara T, et al.Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J ClinInvest 2006;116: 2610–21.
26. Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L,et al. Hepatocyte growth factor is a potent angiogenic factor whichstimulates endothelial cell motility and growth. J Cell Biol 1992;119:629–41.
27. Michieli P, Mazzone M, Basilico C, Cavassa S, Sottile A, Naldini L, et al.Targeting the tumor and its microenvironment by a dual-function decoyMet receptor. Cancer Cell 2004;6: 61–73.
28. Jun HJ, Acquaviva J, Chi D, Lessard J, ZhuH,Woolfenden S, et al. AcquiredMET expression confers resistance to EGFR inhibition in amousemodel ofglioblastoma multiforme. Oncogene 2012;31: 3039–50.
29. Choueiri T, Pal SK, McDermott DF, Morrissey S, Ferguson KC, Holland J ,et al. A phase I study of cabozantinib (XL184) in patients with renal cellcancer. Ann Oncol 2014;8: 1603–8.
Mol Cancer Ther; 14(1) January 2015 Molecular Cancer Therapeutics110
Ciamporcero et al.
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094

2015;14:101-110. Published OnlineFirst November 7, 2014.Mol Cancer Ther Eric Ciamporcero, Kiersten Marie Miles, Remi Adelaiye, et al. Renal Cell Carcinoma ModelsCombination Strategy Targeting VEGF and HGF/c-met in Human
Updated version
10.1158/1535-7163.MCT-14-0094doi:
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/14/1/101.full#ref-list-1
This article cites 29 articles, 9 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/14/1/101.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/14/1/101To request permission to re-use all or part of this article, use this link
on August 18, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst November 7, 2014; DOI: 10.1158/1535-7163.MCT-14-0094