clinical cases on hemoglobin electrophoresis technologies ... · births with a pathological...
TRANSCRIPT

Clinical cases on hemoglobin Clinical cases on hemoglobin Clinical cases on hemoglobin Clinical cases on hemoglobin electrophoresis technologies electrophoresis technologies electrophoresis technologies electrophoresis technologies
ByBy YoussefYoussef MaakarounMaakaroun, MD, MDByBy YoussefYoussef MaakarounMaakaroun, MD, MDBy By YoussefYoussef MaakarounMaakaroun, MD, MDBy By YoussefYoussef MaakarounMaakaroun, MD, MD
66thth International and International and 1111thth National Congress on National Congress on Quality Improvement in Clinical LaboratoriesQuality Improvement in Clinical Laboratories66thth International and International and 1111thth National Congress on National Congress on Quality Improvement in Clinical LaboratoriesQuality Improvement in Clinical LaboratoriesQuality Improvement in Clinical LaboratoriesQuality Improvement in Clinical LaboratoriesQuality Improvement in Clinical LaboratoriesQuality Improvement in Clinical Laboratories

TopicsTopics
• Epidemiology of Hemoglobin disordersEpidemiology of Hemoglobin disorders• Available Methods used for Hb investigationsCli i l• Clinical cases
• Rules for interpretation on CE

TopicsTopics
• Epidemiology of Hemoglobin disordersEpidemiology of Hemoglobin disorders• Available Methods used for Hb investigationsCli i l• Clinical cases
• Rules for interpretation on CE
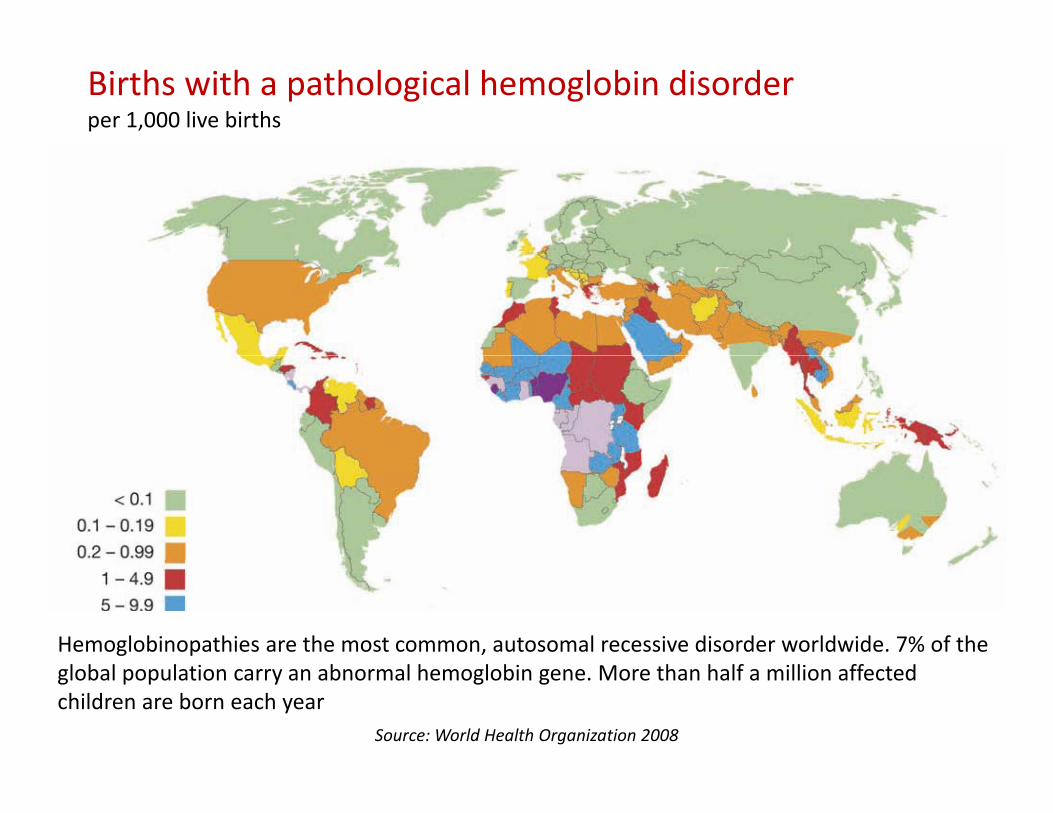
Births with a pathological hemoglobin disorderper 1,000 live births
Hemoglobinopathies are the most common, autosomal recessive disorder worldwide. 7% of the global population carry an abnormal hemoglobin gene. More than half a million affected children are born each year
Source: World Health Organization 2008

Global Epidemiology of Hemoglobin DisordersGlobal Epidemiology of Hemoglobin Disorders• Around 7% of the global population carries an
abnormal hemoglobin geneg g
• 300,000-500,000 children are born annually with clinically significant hemoglobin disorders
• About 80% of affected children are born in developing countries
Ab 30% b i h Th l i S d• About 30% are born with Thalassemia Syndromes and the rest with Sickle Cell Disease
• 50,000-100,000 children with thalassemia major die each year in low and middle income countries
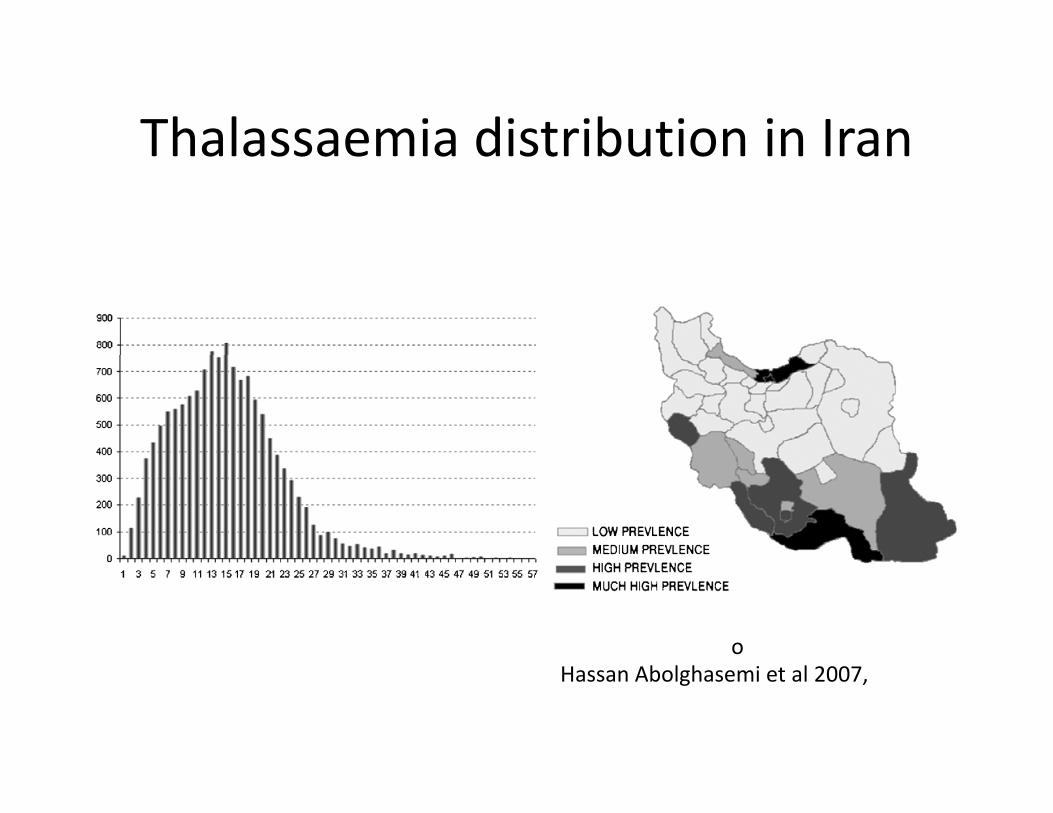
Thalassaemia distribution in IranThalassaemia distribution in Iran
oHassan Abolghasemi et al 2007,

World Health OrganizationEB118.R1 Resolution, 2006

Thalassaemia and other Haemoglobinopathies
EB118 M 2006 R l ti EB118 R1EB118, May 2006 – Resolution EB118.R1
Urges Member States: Implement and reinforce national programs on HB di d
Requests the Director-General provide technical support and advice to national programs
disordersEvaluate the impact of national programsIntensify the training of all health
expand the training and expertise of personnelsupport the further transfer of affordable technologiesy g
professionalsPromote community educationPromote international cooperation
gdrafting guidelines on prevention and managementfostering the establishment of regional groups of experts;cooperation
Develop and strengthen medical genetic servicesSupport basic and applied research
regional groups of experts;support needed research
research

TopicsTopics
• Epidemiology of Hemoglobin disordersEpidemiology of Hemoglobin disorders• Available Methods used for Hb investigationsCli i l• Clinical cases
• Rules for interpretation on CE

Separation of Hemoglobin FractionsSeparation of Hemoglobin Fractions
• Traditional methods for separation of Hemoglobin fractions include electrophoresisHemoglobin fractions include electrophoresis (alkaline and acid), isoelectric focusing (IEF) or chromatography (HPLC)chromatography (HPLC).
• Recently Capillary Electrophoresis (CE)
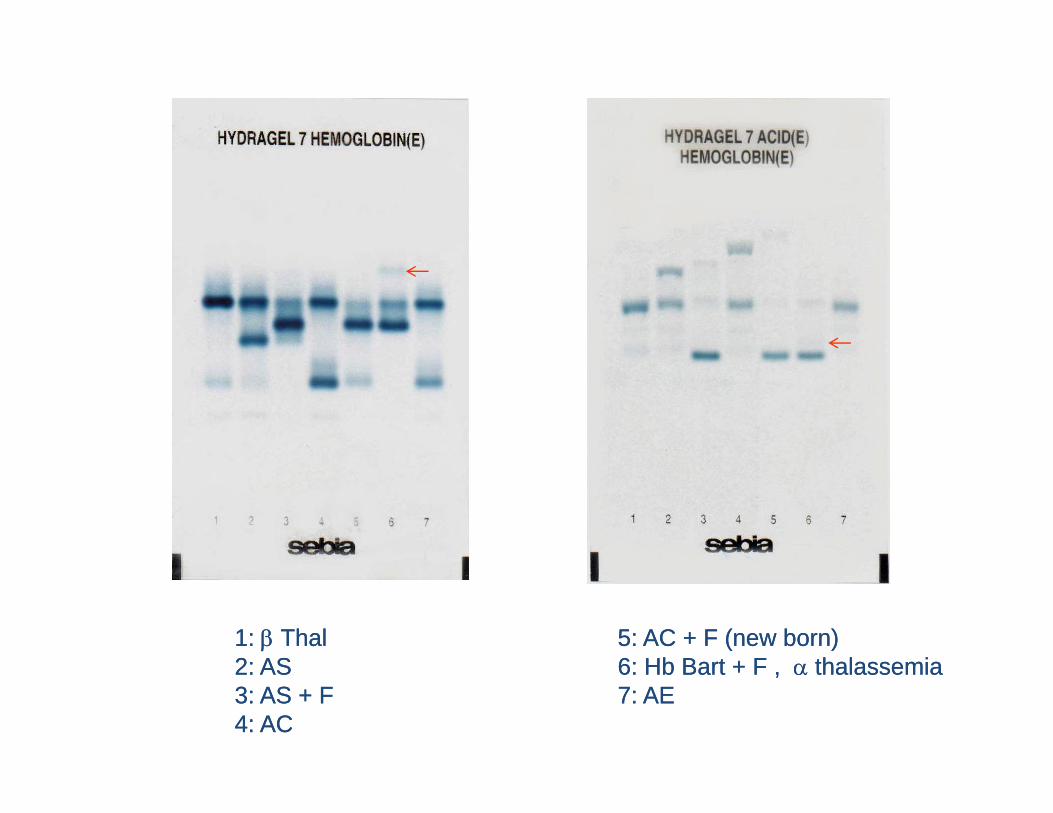
11: : ββ ThalThal 55: AC + F (new born): AC + F (new born)22: AS: AS 66: Hb Bart + F: Hb Bart + F thalassemiathalassemia22: AS: AS 66: Hb Bart + F , : Hb Bart + F , α α thalassemiathalassemia33: AS + F: AS + F 77: AE: AE44: AC : AC
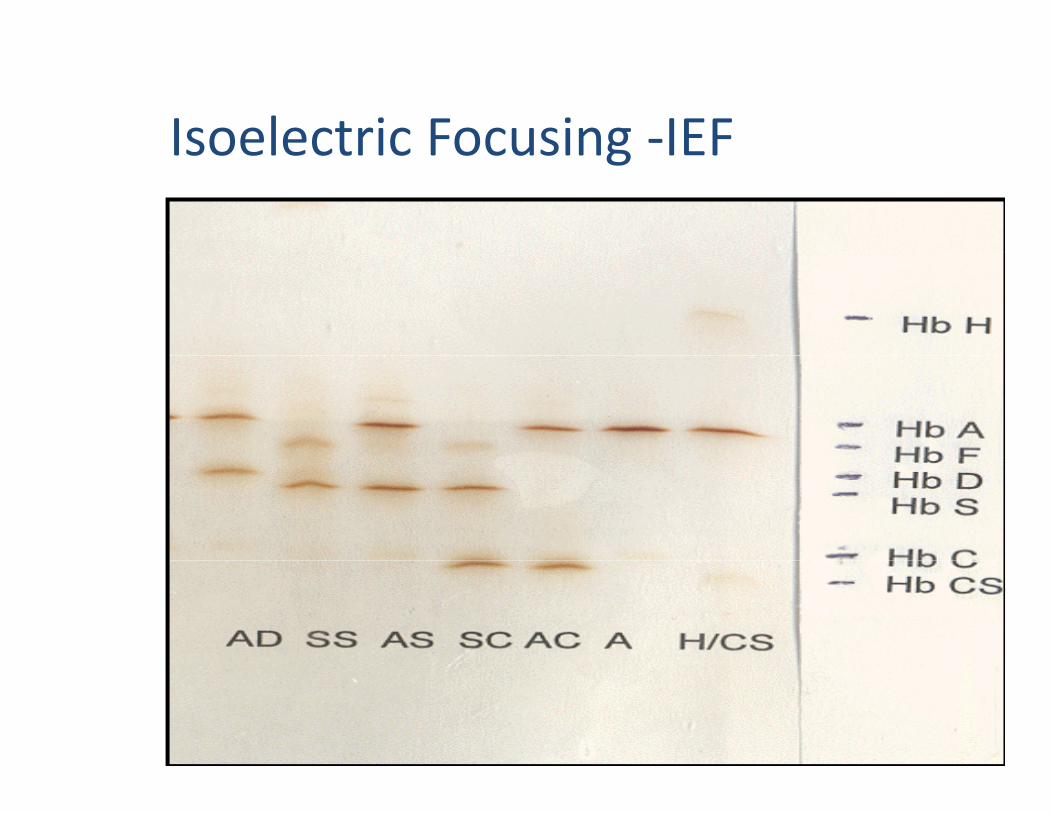
Isoelectric Focusing ‐IEFIsoelectric Focusing IEF
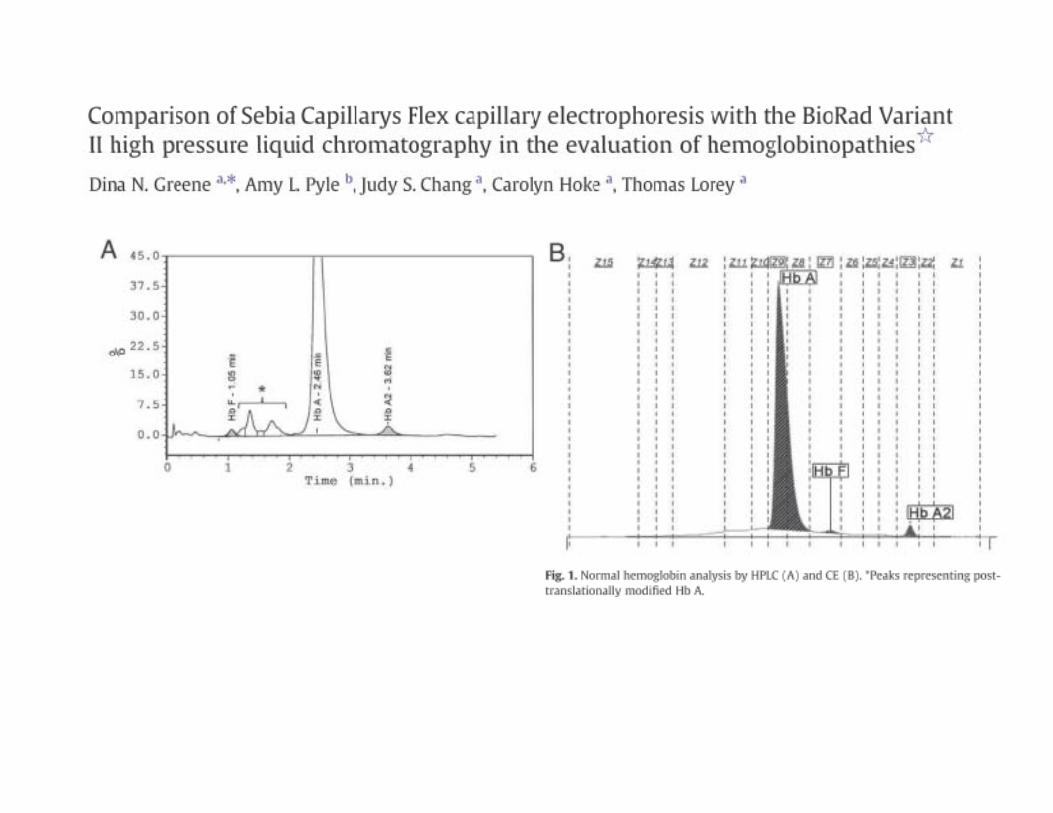
0

TopicsTopics
• Epidemiology of Hemoglobin disordersEpidemiology of Hemoglobin disorders• Available Methods used for Hb investigationsCli i l• Clinical cases
• Rules for interpretation on CE
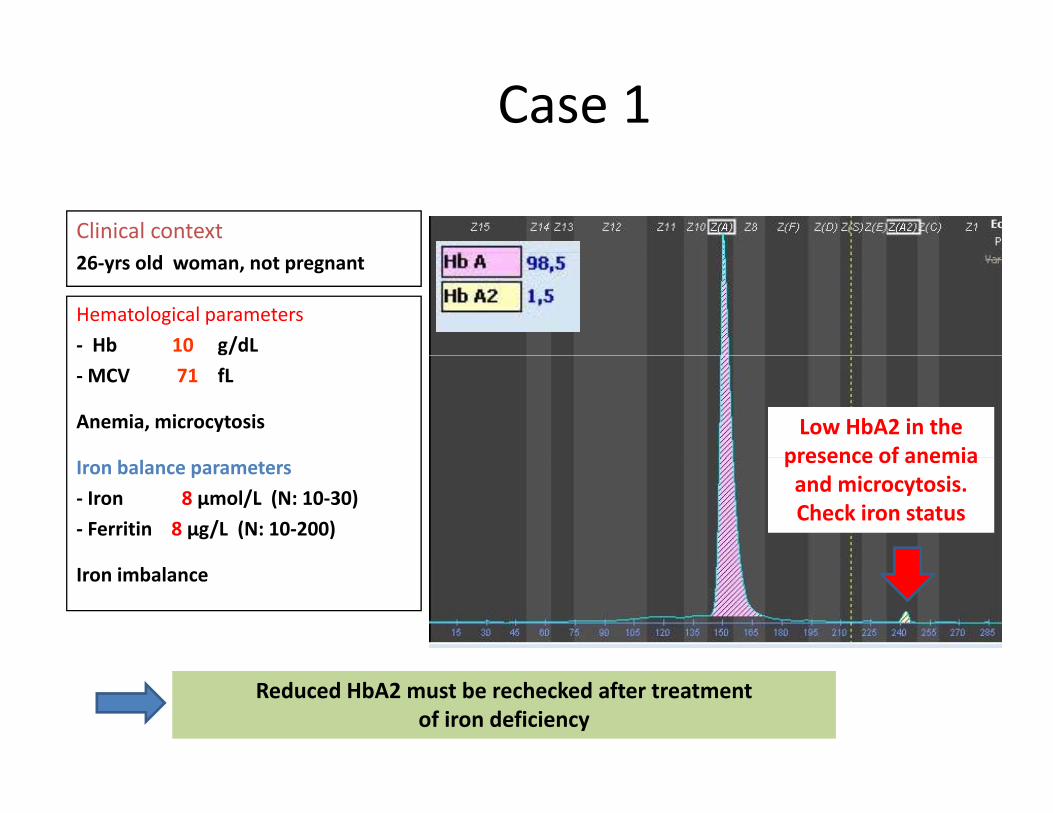
Case 1
Clinical context26‐yrs old woman, not pregnant
Hematological parameters‐ Hb 10 g/dL‐MCV 71 fL
Anemia, microcytosis Low HbA2 in the presence of anemiaIron balance parameters
‐ Iron 8 µmol/L (N: 10‐30)‐ Ferritin 8 µg/L (N: 10‐200)
presence of anemiaand microcytosis. Check iron status
Iron imbalance
Reduced HbA2 must be rechecked after treatment of iron deficiency
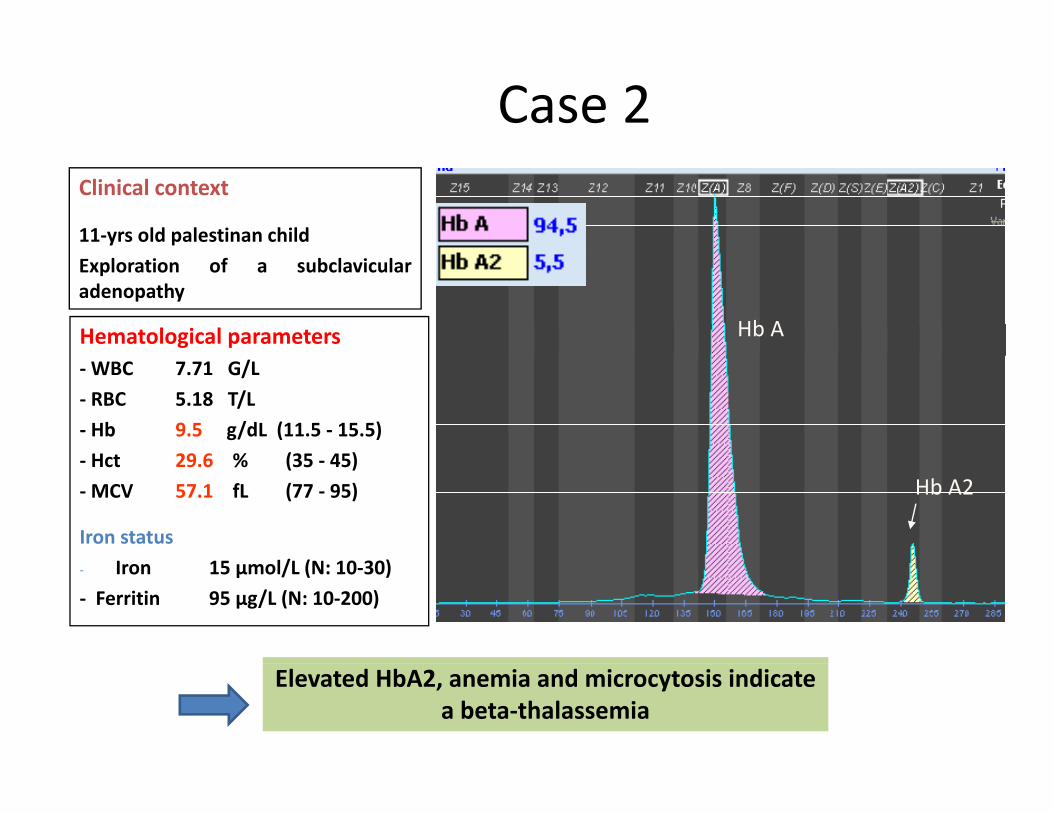
Case 2 Clinical context
11‐yrs old palestinan childExploration of a subclavicularadenopathy
Hematological parameters Hb A
‐WBC 7.71 G/L‐ RBC 5.18 T/L‐ Hb 9.5 g/dL (11.5 ‐ 15.5)Hct 29 6 % (35 45)‐ Hct 29.6 % (35 ‐ 45)
‐MCV 57.1 fL (77 ‐ 95)
Iron statusl/ ( 0 30)
Hb A2
‐ Iron 15 µmol/L (N: 10‐30) ‐ Ferritin 95 µg/L (N: 10‐200)
Elevated HbA2, anemia and microcytosis indicatea beta‐thalassemia

Case 3
Clinical context
9yrs child from Laos with9yrs child from Laos withunknown family history
Hematological parameters‐WBC 5.62 G/L Hb H 4%WBC 5.62 G/L‐ RBC 5.16 T/L‐ Hb 8 g/dL‐ Hct 27.9 %
Hb H 4%Hb A 94.7%Hb A2 1.3%
‐MCV 54.1 fL‐ CCMH 15.5 pg
Iron status
Hb A
Hb H‐ Iron 18 µmol/L (N: 10‐30) ‐ Ferritin 115 µg/L (N: 10‐200) Hb A2
Hb H(zone 15)
Presence of Hb H, reduced HbA2, anemia and microcytosis indicate an alpha‐thalassemia

Case # 4

Fessas Bodies (Hb Inclusion bodies)Fessas Bodies (Hb Inclusion bodies)
NRBC
Brilliant Cresyl Blue Stain

Case 5
Variant in the h d
β6 Glu (negative) →Val (neutral)
S zone, what to do?α αβ ββ
Hb AThe HbX value is
Hb S
Hb A
Heterozygous A/SHeterozygous A/S
The HbX value ishigher than 35%
(Hb S= ~35 to ~ 40% for heterozygote A/S)
HeterozygousA/S
βm βm
α αβm
ygyg
Hb A2
Sickle cell test or Itano test positive

Case 6
Variant in the S zone what to do?S zone, what to do?
The HbX value is α αβ ββ
below 35% (Hb S= ~35 to ~ 40% for heterozygote A/S)
b
β ββ
Hb A
Heterozygous Heterozygous A/X A/X
HeterozygousA/non S beta
i nt
Hb X non S
βm βm
α αβm
ygyg
variant
Sickle cell test or Itano test negative
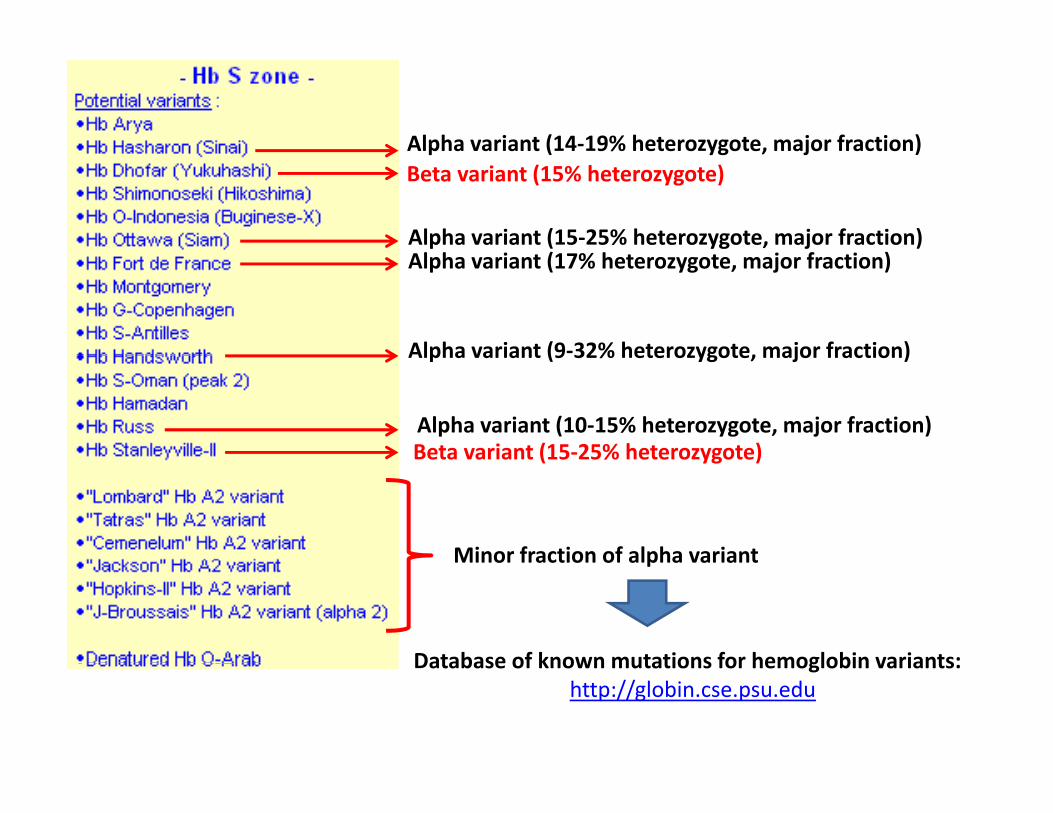
Alpha variant (14‐19% heterozygote, major fraction)Beta variant (15% heterozygote)
l h i ( % h j f i )
Alpha variant (14 19% heterozygote, major fraction)
Alpha variant (15‐25% heterozygote, major fraction)Alpha variant (17% heterozygote, major fraction)
Alpha variant (9‐32% heterozygote, major fraction)
Alpha variant (10‐15% heterozygote, major fraction)Beta variant (15‐25% heterozygote)
Alpha variant (9 32% heterozygote, major fraction)
Minor fraction of alpha variant
Beta variant (15 25% heterozygote)
Minor fraction of alpha variant
Database of known mutations for hemoglobin variants:Database of known mutations for hemoglobin variants: http://globin.cse.psu.edu

Case 7
Clinical Context
2 yrs old child with sickle cell syndromunder oracillin treatment Hb A 15.6%
Hb F 14% Hb S( S)Hematological parameters
‐WBC 10.48 G/L ‐ RBC 5.53 T/L Hb 10 6 g/dL
Hb S 64.5%Hb A2 5.9%
(zone S)
‐ Hb 10.6 g/dL‐ Hct 30.8 %‐MCV 55.7 fL Hb A Hb A2
Hb F
Biochemical parametersFerritin 177 µg/L
Itano test: Positive
Elevated HbA2, Hb S > HbA, anemia and microcytosis indicate S‐ beta‐thalassemia

Hemoglobin variant: β globin chain, heterozygoteheterozygote
α
+
β
α2β2: HbA α2β2*
+
β
bδ
α2γ2: HbF
γ α2δ2: HbA2
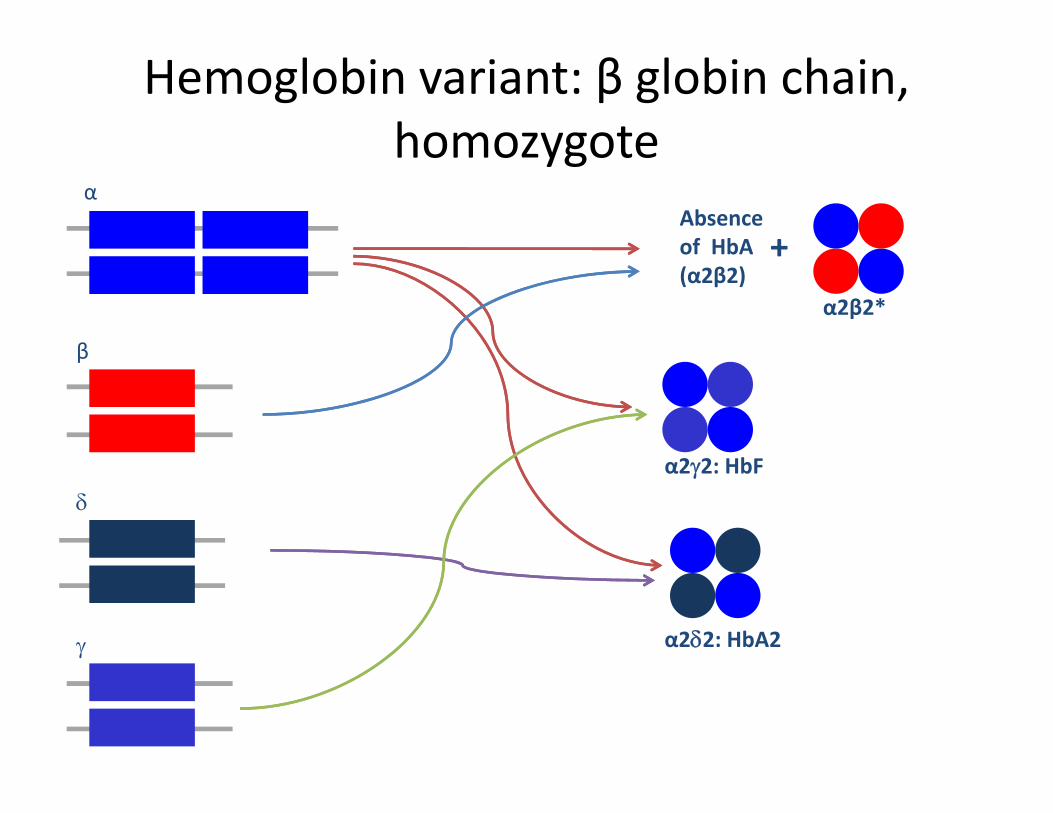
Hemoglobin variant: β globin chain, homozygotehomozygote
αAbsence of HbA +
β
of HbA(α2β2)
α2β2*
+
β
bδ
α2γ2: HbF
γ α2δ2: HbA2

Hb A + PRESENCE OF A MAJOR VARIANT (Hb S, Hb C, Hb E)
Hb A + Hb E
Hb E = ~25 ‐ 30%
Hb A + Hb C
Hb C = ~35 ‐ 40%
Hb A + Hb S
Hb S = ~35 ‐ 40%
Heterozygote A/EHeterozygote A/CHeterozygote A/S
Hb E < 25 %Hb C < 35 %Hb S < 35 %
Heterozygote A/S Heterozygote A/C Heterozygote A/EHeterozygote A/S+ α‐thalassemia orIron deficiency
Heterozygote A/C+ α‐thalassemia orIron deficiency
Heterozygote A/E+ α‐thalassemia orIron deficiency
Hb E > Hb AHb C > Hb AHb S > Hb A
DoubleHeterozygote DoubleHeterozygote DoubleHeterozygoteDoubleHeterozygoteS/β+‐thalassemia?
Vinatier I. CERBA recommandations (2010)
DoubleHeterozygoteC/β+‐thalassemia?
DoubleHeterozygoteE/β+‐thalassemia?

Case 8
Variant in the D zone, what to do?
Th HbX l iThe HbX value isbelow 35%
(Hb D= ~ 35 to ~ 40% for heterozygote A/D)
α αβ ββ
Hb A
HeterozygousA/non D beta
Hb X non DHeterozygous Heterozygous A/X A/X
A/non D beta variant
βm βm
α αβm

Frequency Frequency –– localization:localization:LL (( BB W hi )W hi ) F d i l i I liF d i l i I li f ilif iliLeporeLepore ((‐‐ Boston Boston –– Washington):Washington): Found mainly in Italian Found mainly in Italian families, families, Middle East, RomaniaMiddle East, Romania, Australia, , Australia, Mexico...Mexico...Characterization:Characterization:
β β et et δ δ chains recombination by crossing over.chains recombination by crossing over.Alkaline buffer: Decrease of the total charge→Alkaline buffer: Decrease of the total charge→ Migration slowed downMigration slowed down
Characterization:Characterization:
Alkaline buffer: Decrease of the total charge → Alkaline buffer: Decrease of the total charge → Migration slowed down Migration slowed down like like HbHb S on S on agaroseagarose gel, more anodic than S (D Zone) on gel, more anodic than S (D Zone) on CapillarysCapillarys//MinicapMinicap
Heterozygous form:Heterozygous form:Homozygous form:Homozygous form: Heterozygous form:Heterozygous form:Hb Lepore fraction: 5 – 15 %Clinical signs of minor β thalassemia
Homozygous form:Homozygous form:Hb Lepore fraction: about 30 %Clinical signs of homozygous βthalassemia
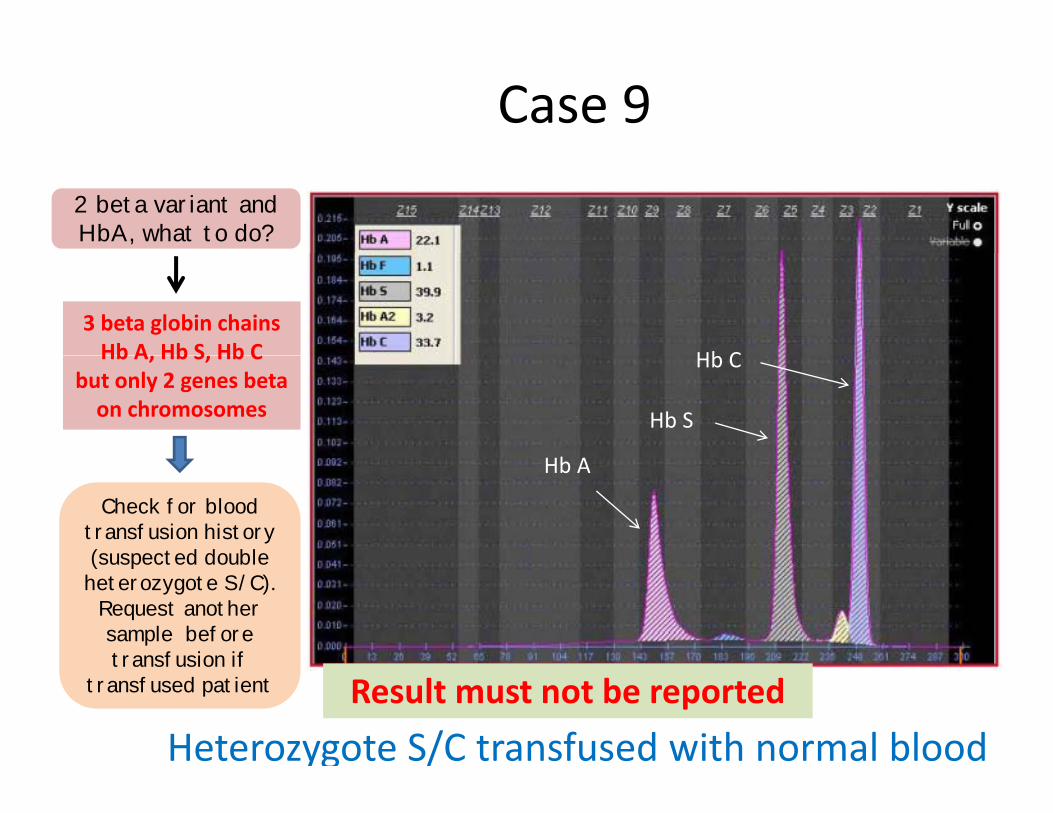
Case 9 2 beta variant and HbA, what to do?
3 beta globin chainsHb A Hb S Hb C Hb CHb A, Hb S, Hb C
but only 2 genes beta on chromosomes Hb S
Hb A
Hb C
Check for bloodtransfusion history(suspected double
Hb A
(suspected double heterozygote S/C).
Request anothersample beforetransfusion if transfusion if
transfused patient
Heterozygote S/C transfused with normal bloodResult must not be reported
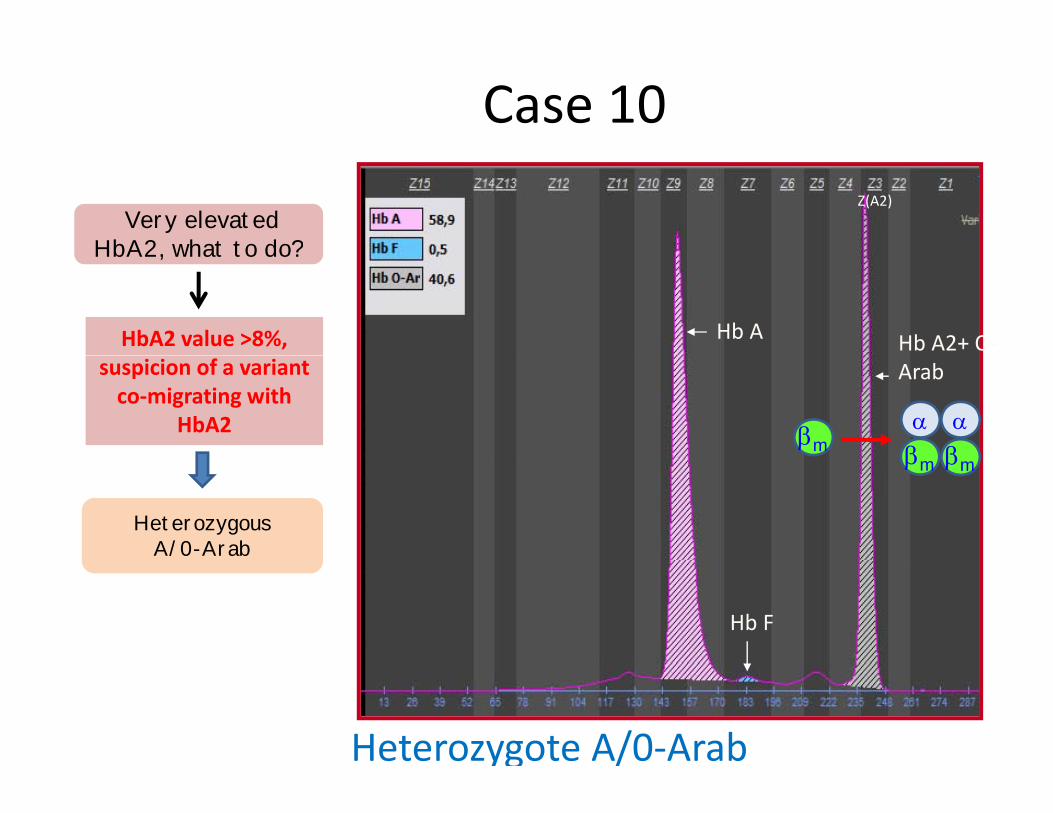
Case 10 Z(A2)
Very elevatedHbA2 what to do?
Hb A2+ O‐Hb A
HbA2, what to do?
HbA2 value >8%, Arabsuspicion of a variant
co‐migrating withHbA2
β βα αβm
HeterozygousA/0-Arab
βm βm
Hb F
Heterozygote A/0‐Arab

HbHb O O -- ArabArabββ121 121 GluGlu→→LysLys
Also Known asAlso Known as Egypt
HematologyHematology Normal in heterozygote; mild anemia in homozygotes
AgaroseAgaroseElectrophoresisElectrophoresis
Moves like Hb CMoves close to Hb S at acidic pH
Function studiesFunction studies Normal
OccurrenceOccurrence Found mainly in Gypsies and in Pomaks ( a populationOccurrenceOccurrence Found mainly in Gypsies and in Pomaks ( a population group in the Balkan countries) and also in Arabian, Egyptian and black families.Quantity in the heterozygote 30 – 40%; found in Qua t ty t e ete o ygote 30 0%; ou dcombination with Hb S, Hb C, beta-thal and alpha-thal; found in the homozygous condition in Bulgaria and Yugoslavia. Causes severe sickle cell anemia in combination with Hb S
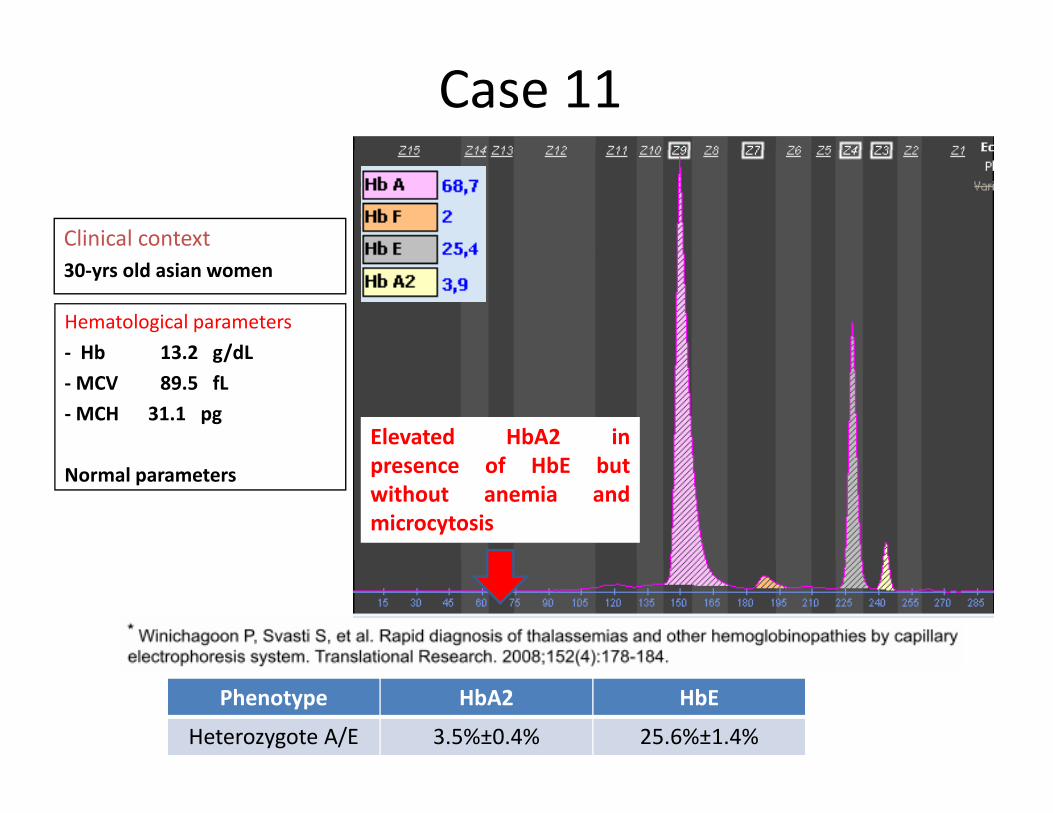
Case 11
Clinical context30‐yrs old asian women
Hematological parameters‐ Hb 13 2 g/dL
Elevated HbA2 inf HbE b
Hb 13.2 g/dL‐MCV 89.5 fL‐MCH 31.1 pg
presence of HbE butwithout anemia andmicrocytosis
Normal parameters
Phenotype HbA2 HbE
Heterozygote A/E 3.5%±0.4% 25.6%±1.4%
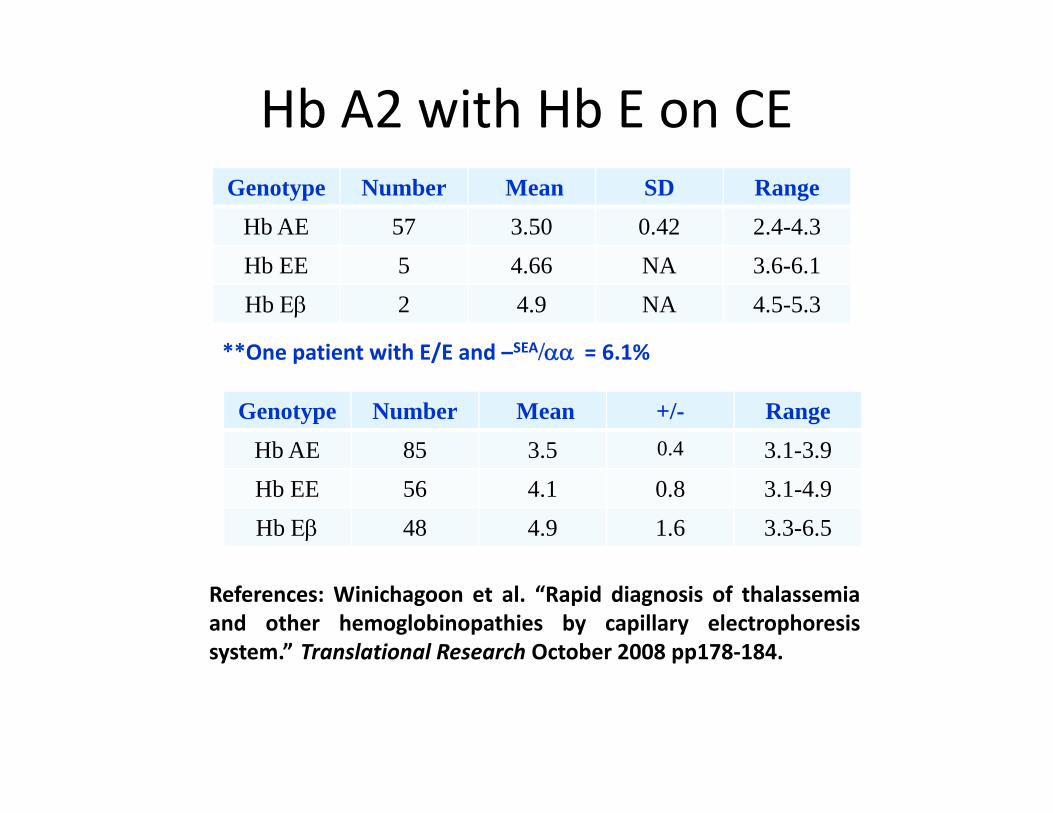
Hb A2 with Hb E on CEGenotype Number Mean SD Range
Hb AE 57 3.50 0.42 2.4-4.3Hb EE 5 4.66 NA 3.6-6.1Hb Εβ 2 4.9 NA 4.5-5.3
**One patient with E/E and –SEA/αα = 6 1%One patient with E/E and /αα = 6.1%
Genotype Number Mean +/- RangeHb AE 85 3.5 0.4 3.1-3.9b 85 3.5 3. 3.9Hb EE 56 4.1 0.8 3.1-4.9Hb Εβ 48 4.9 1.6 3.3-6.5
References: Winichagoon et al. “Rapid diagnosis of thalassemiaand other hemoglobinopathies by capillary electrophoresissystem.” Translational Research October 2008 pp178‐184.y pp
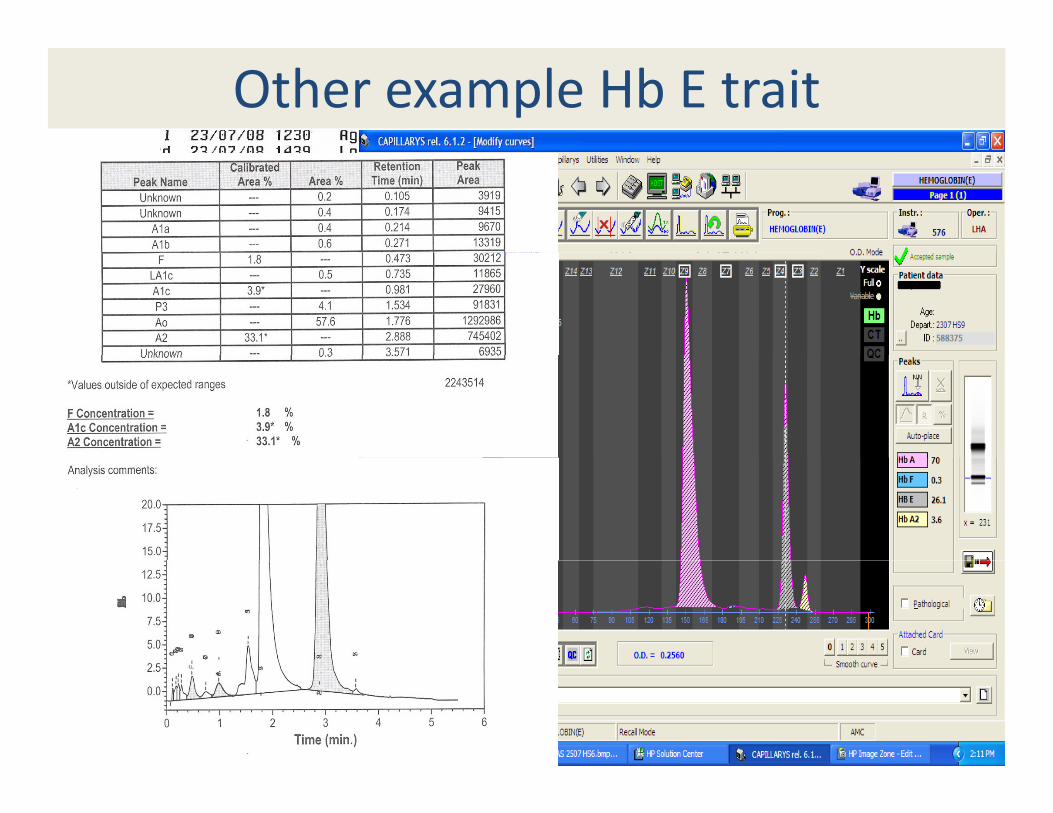
Other example Hb E trait

Heterozygote delta variantCase 12
Clinical context42 yrs old man
Heterozygote delta variant
42‐yrs old man
Hematological parameters‐ Hb 15.7 g/dL‐MCV 89 fL
Normal parameters
Iron balance parameters
Delta variantδmIron balance parameters
‐ Iron 19 µmol/L (N: 10‐30)‐ Ferritin 156 µg/L(N: 30‐300)
N l i
δm δm
α α
Hb A2Normal iron status
P f dditi l k i Z1Presence of an additional peak in zone Z1Reduced HbA2 with normal hematological and iron parameters
True HbA2 value = HbA2 value + peak in Z1
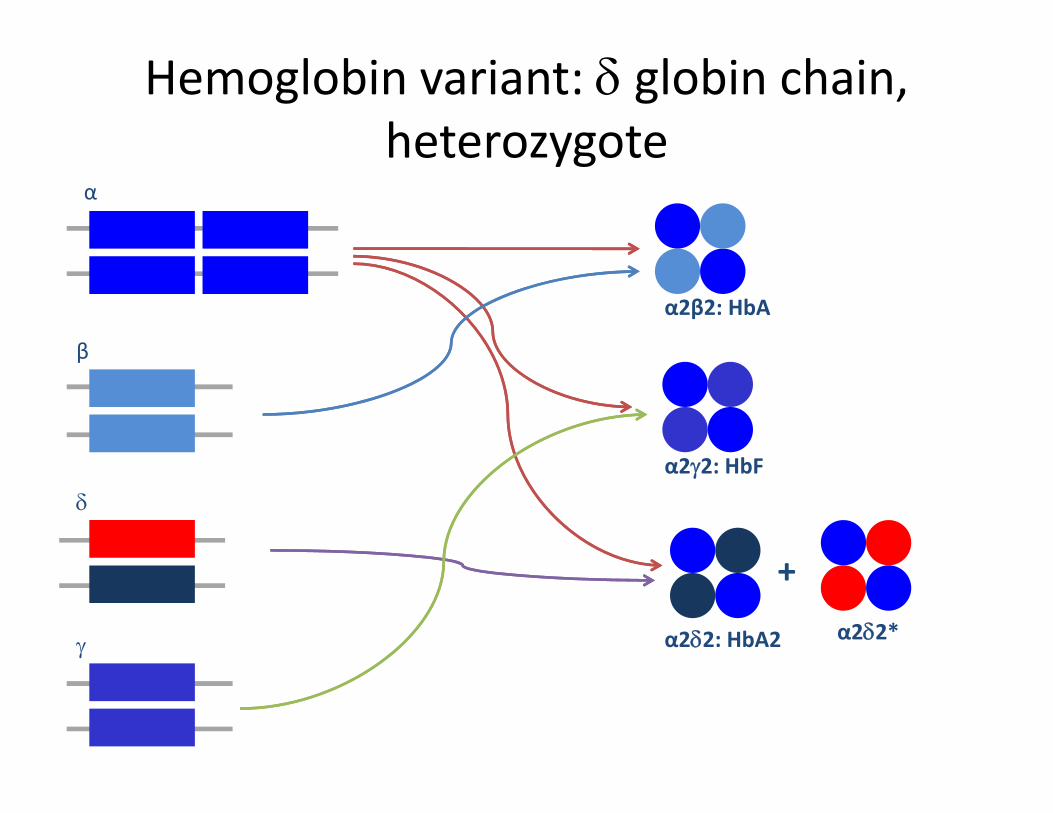
Hemoglobin variant: δ globin chain, heterozygoteheterozygote
α
β
α2β2: HbA
β
bδ
α2γ2: HbF
γ α2δ2: HbA2
+α2δ2*

Case 13
Clinical context38‐yrs old man38 yrs old man
Hematological parameters‐ Hb 15.1 g/dLMCV 90 fL
Delta variant
α α
δm
‐MCV 90 fL
Normal parameters
Iron balance parameters
δm δm
α α
DecreasedHb A2
Iron balance parameters‐ Iron 22 µmol/L (N: 10‐30)‐ Ferritin 180 µg/L (N: 30‐300)
Normal iron status
Presence of an additional peak in zone Z1 and reduced HbA2 indicated the presence of delta variant
Normal iron status
presence of delta variantHA2 value on ZA2 (Z3) < Delta variant value on Z1 with normal hematological
parameters indicated the presence of delta‐thalassemia
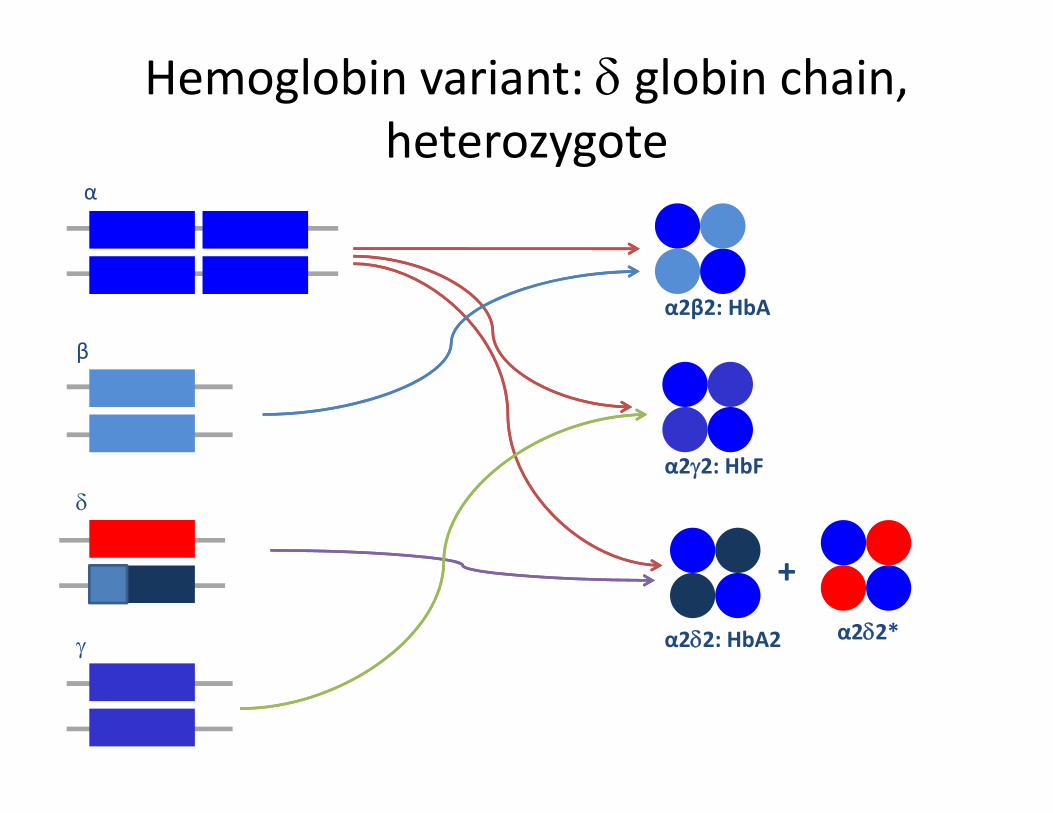
Hemoglobin variant: δ globin chain, heterozygoteheterozygote
α
β
α2β2: HbA
β
bδ
α2γ2: HbF
γ α2δ2: HbA2
+α2δ2*
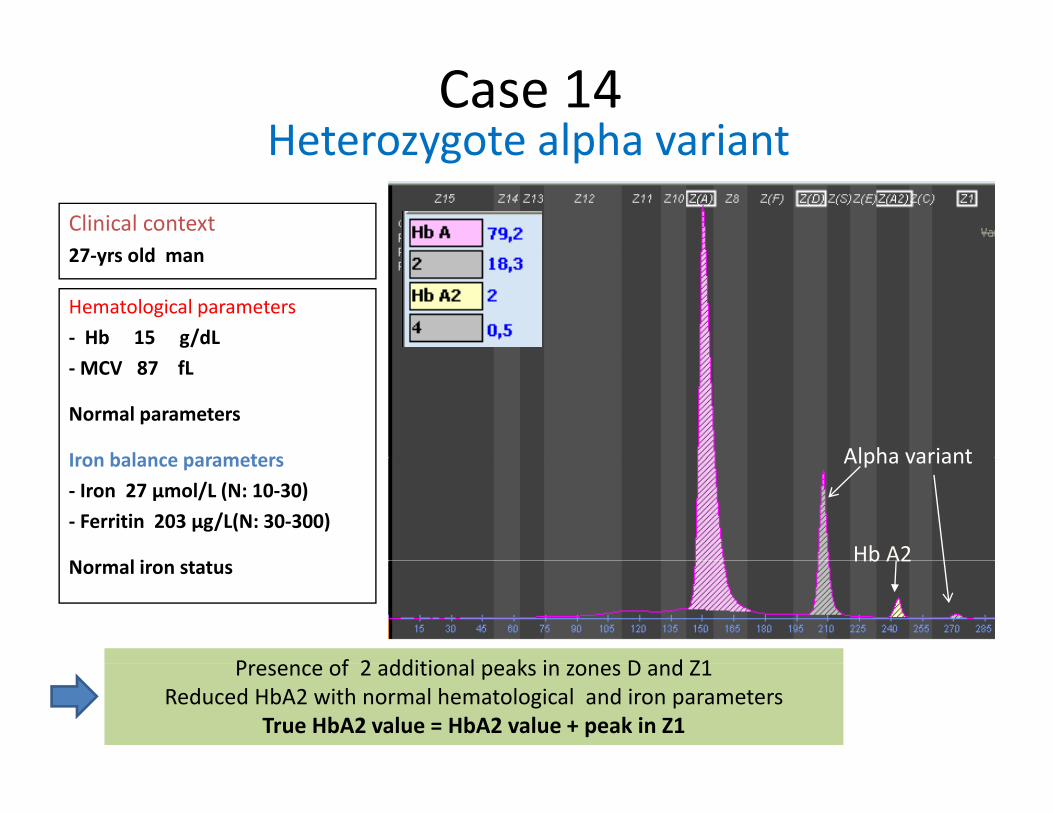
Heterozygote alpha variantCase 14
Clinical context27 yrs old man
Heterozygote alpha variant
27‐yrs old man
Hematological parameters‐ Hb 15 g/dL‐MCV 87 fL
Normal parameters
Iron balance parameters Alpha variantIron balance parameters‐ Iron 27 µmol/L (N: 10‐30)‐ Ferritin 203 µg/L(N: 30‐300)
N l i
Alpha variant
Hb A2Normal iron status
P f 2 dditi l k i D d Z1
Hb A2
Presence of 2 additional peaks in zones D and Z1Reduced HbA2 with normal hematological and iron parameters
True HbA2 value = HbA2 value + peak in Z1
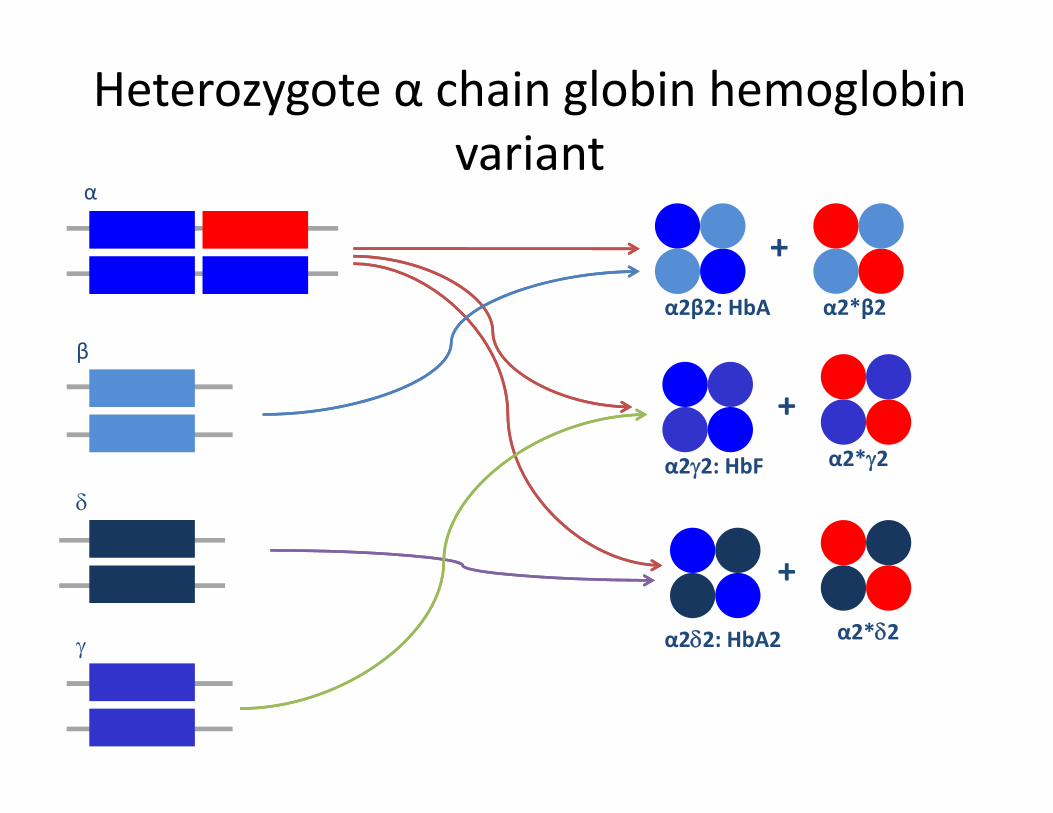
Heterozygote α chain globin hemoglobinvariantvariant
α
+
β
α2β2: HbA α2*β2
+
β
b
+α2*γ2
δα2γ2: HbF α2*γ2
γ α2δ2: HbA2
+α2*δ2

Search between zones if a similar name is found
= Major fractionof an alpha variant
= Major fractionof an alpha variant
= Minor fraction of an alpha variant
= Minor fraction of Minor fraction of an alpha variant
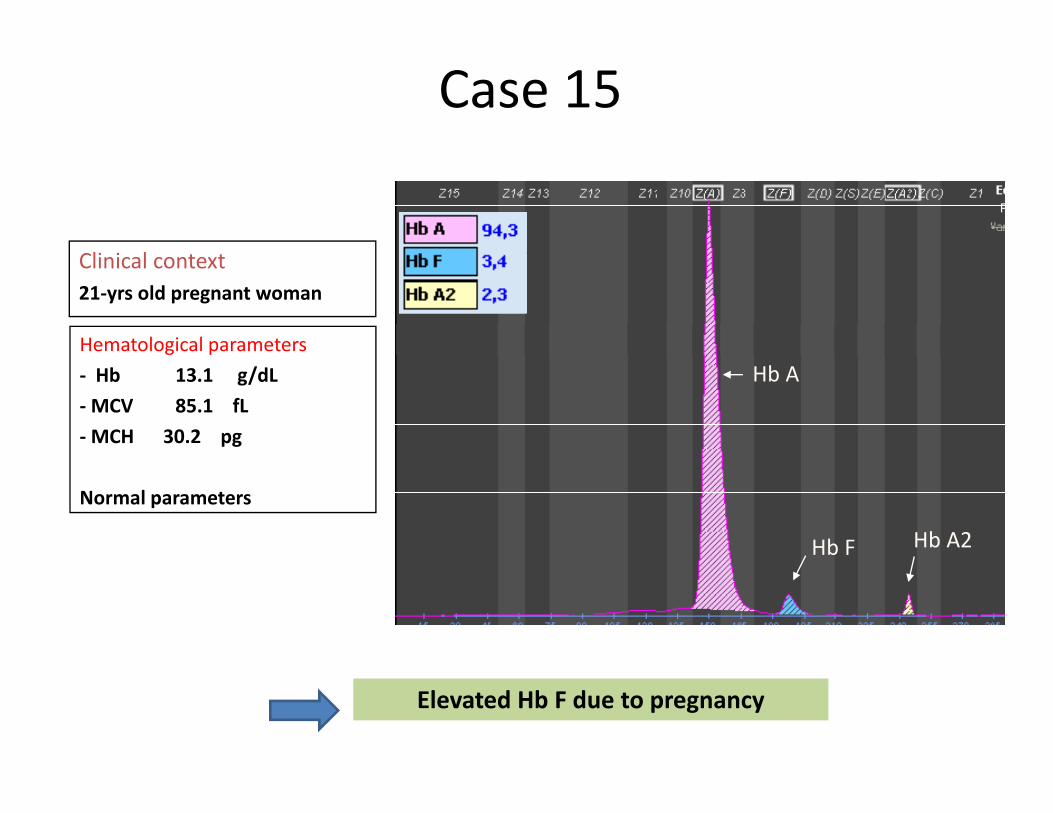
Case 15
li i lClinical context21‐yrs old pregnant woman
Hematological parametersHb A‐ Hb 13.1 g/dL
‐MCV 85.1 fL‐MCH 30.2 pg
Hb F Hb A2
Normal parameters
Elevated Hb F due to pregnancy

1<Hb F <5%Normal HbA2
5<Hb F <35%Normal or ↓ HbA2
MCH >27pg MCH >27pg
Di b tHeterozygote δβ‐thalassemiaHb F% (5‐15%), MCH<27pg
DiabetesPregnancy
Hyperthyroidism
Heterozygote hereditary persistance of Hb F (HPFH)
ChemotherapyDyserythropoiesisAnemia stress persistance of Hb F (HPFH)
Hb F% (15‐35%), normal MCHModerate hereditary persistance of Hb F
Anemia stress
persistance of Hb F
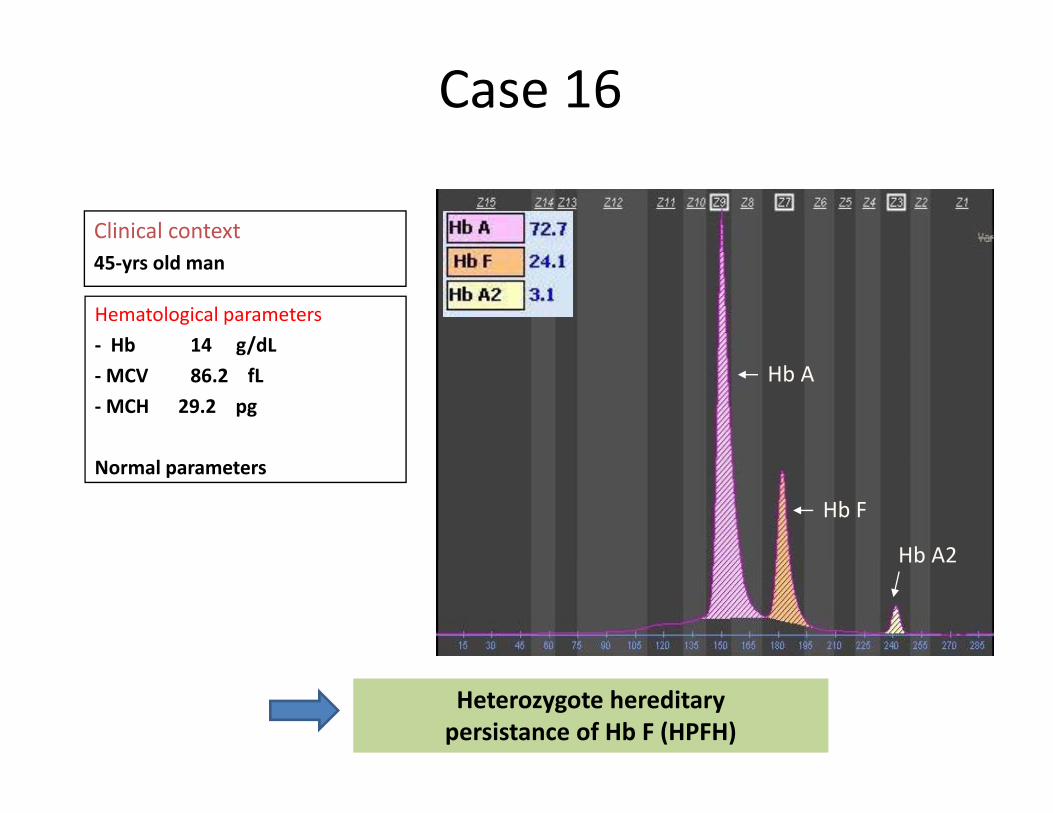
Case 16
Clinical context45‐yrs old man
Hematological parameters‐ Hb 14 g/dL‐MCV 86.2 fL‐MCH 29.2 pg
Hb A
Normal parameters
Hb F
Hb A2Hb A2
Heterozygote hereditary persistance of Hb F (HPFH)
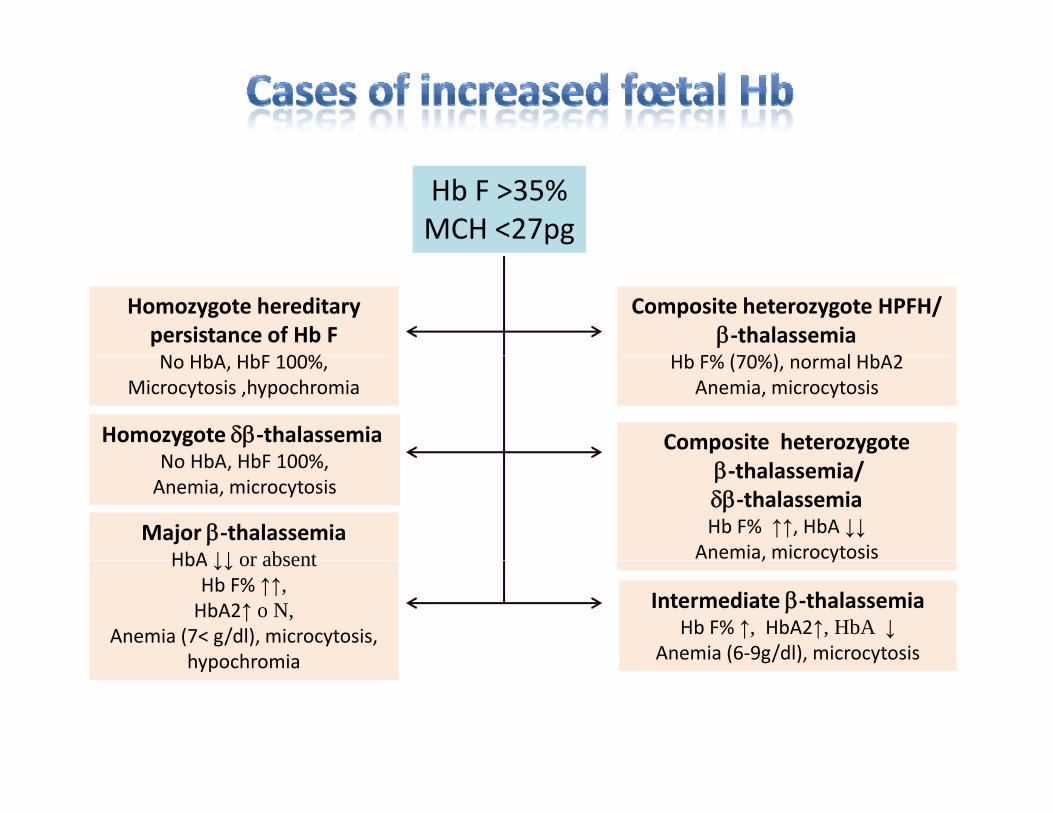
Hb F >35%MCH <27pg
Homozygote hereditary persistance of Hb FN HbA HbF 100%
Composite heterozygote HPFH/β‐thalassemia
Hb F% (70%) l HbA2No HbA, HbF 100%,Microcytosis ,hypochromia
Hb F% (70%), normal HbA2Anemia, microcytosis
Homozygote δβ‐thalassemiaNo HbA HbF 100%
Composite heterozygoteNo HbA, HbF 100%,Anemia, microcytosis
β‐thalassemia/δβ‐thalassemiaHb F% ↑↑, HbA ↓↓
Anemia, microcytosisMajor β‐thalassemia
HbA ↓↓ or absent , y
Intermediate β‐thalassemiaHb F% ↑, HbA2↑, HbA ↓
Anemia (6‐9g/dl), microcytosis
HbA ↓↓ or absentHb F% ↑↑, HbA2↑ o N,
Anemia (7< g/dl), microcytosis, hypochromia g yhypochromia
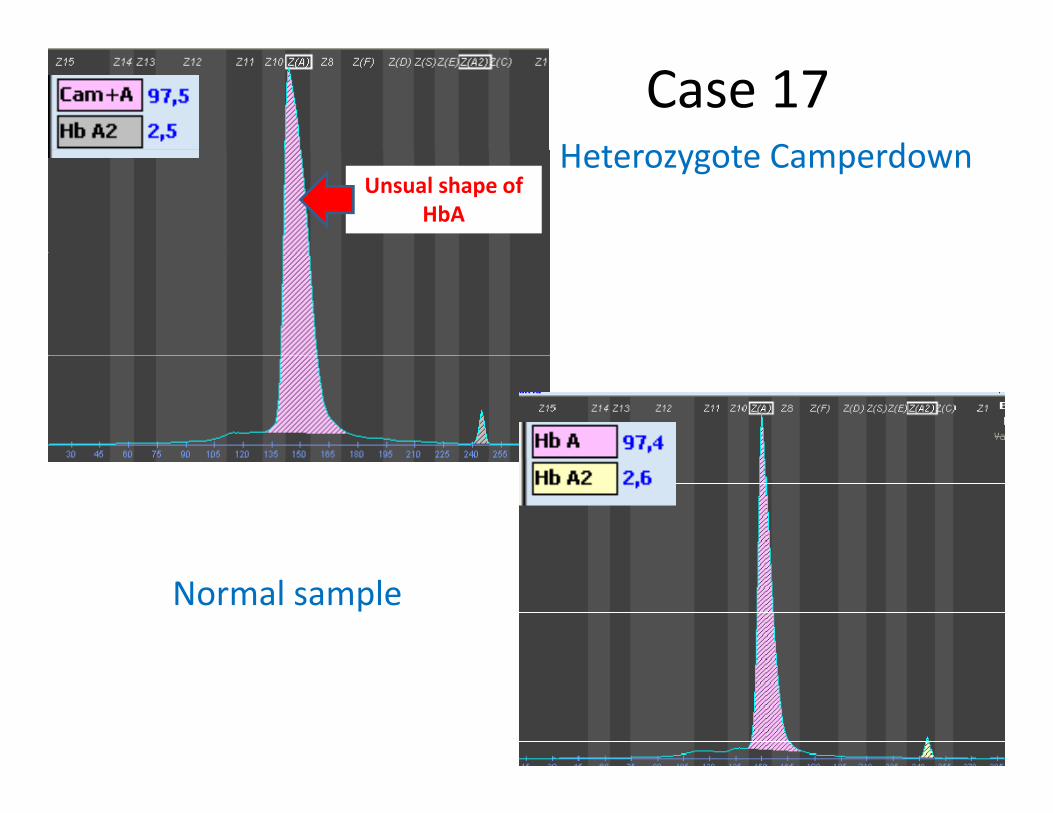
Heterozygote Camperdown
Case 17Heterozygote Camperdown
Unsual shape of HbA
Normal sample

Hb CamperdownHb Camperdown(β104 Arg→Ser)
HematologyHematology Normal in heterozygoteNormal in heterozygote
ElectrophoresisElectrophoresis Moves slightly faster than Moves slightly faster than HbHb A on A on alkalinalkalin gel.Atgel.At acidic pH, acidic pH, pp g yg y gg ppmoves between moves between HbHb A and A and HbHb F,F, close to close to HbHb F. F. Moves on Moves on HbAHbA on capillary electrophoresis on capillary electrophoresis
Function studiesFunction studies NormalNormalFunction studiesFunction studies
OccurrenceOccurrence Quantity in the heterozygote about Quantity in the heterozygote about 5050%%
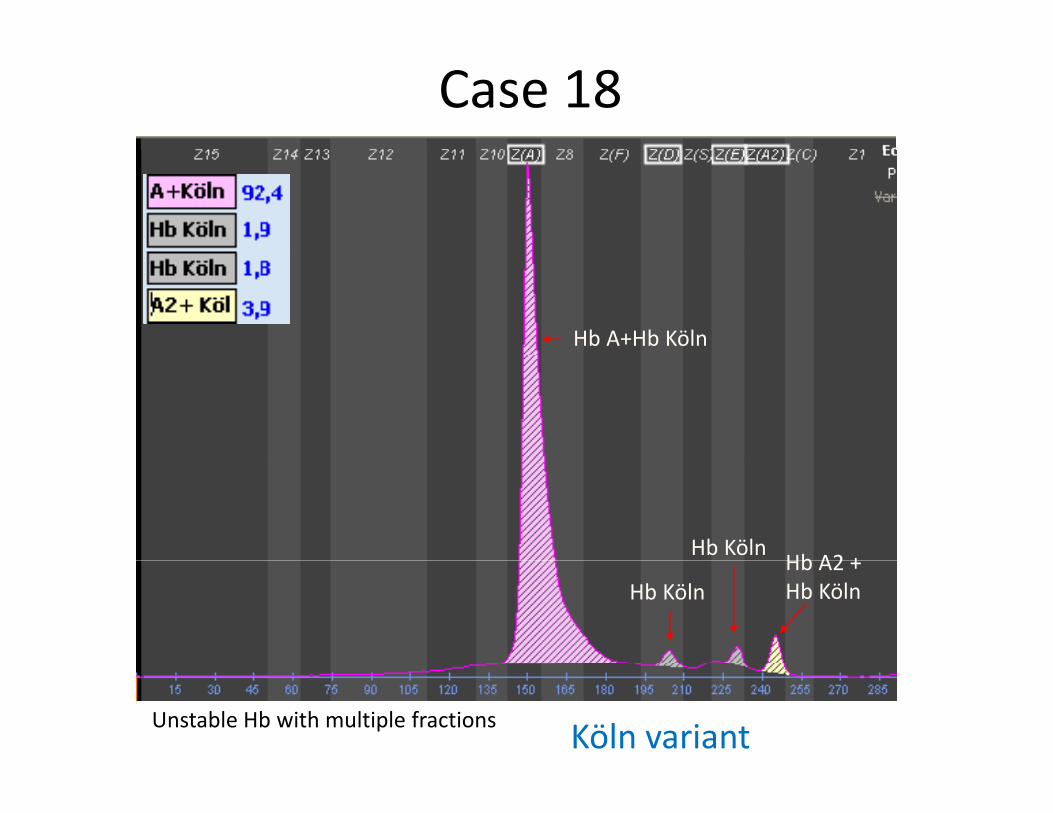
Case 18
Hb A+Hb Köln
Hb KölnHb A2 +
Hb KölnHb A2 +Hb Köln
Unstable Hb with multiple fractions Köln variant

HbHb KölnKöln ((ββ9898 ValVal→→MetMet))
ll i ( ifi ) bi ( ifi ) b
Hb Hb KölnKöln ((ββ98 98 ValVal→→MetMet))
Also Known asAlso Known as San Francisco (Pacific); UbeSan Francisco (Pacific); Ube--11
HematologyHematology Mild hemolytic anemia in the heterozygote; Mild hemolytic anemia in the heterozygote; reticulocytosis; Heinz body formationreticulocytosis; Heinz body formationreticulocytosis; Heinz body formationreticulocytosis; Heinz body formation
AgaroseAgaroseElectrophoresisElectrophoresis
Moves as a multiple Moves as a multiple HbHb component between component between HbHb A and A and HbHb AA2 2 at alkaline pHat alkaline pH
Function studiesFunction studies Increased oxygen affinityIncreased oxygen affinity
OccurrenceOccurrence Found in various racial and ethnic groups; It is the mostFound in various racial and ethnic groups; It is the mostOccurrenceOccurrence Found in various racial and ethnic groups; It is the most Found in various racial and ethnic groups; It is the most common unstable common unstable HbHb..Quantity in the heterozygote not accurately determinedQuantity in the heterozygote not accurately determined
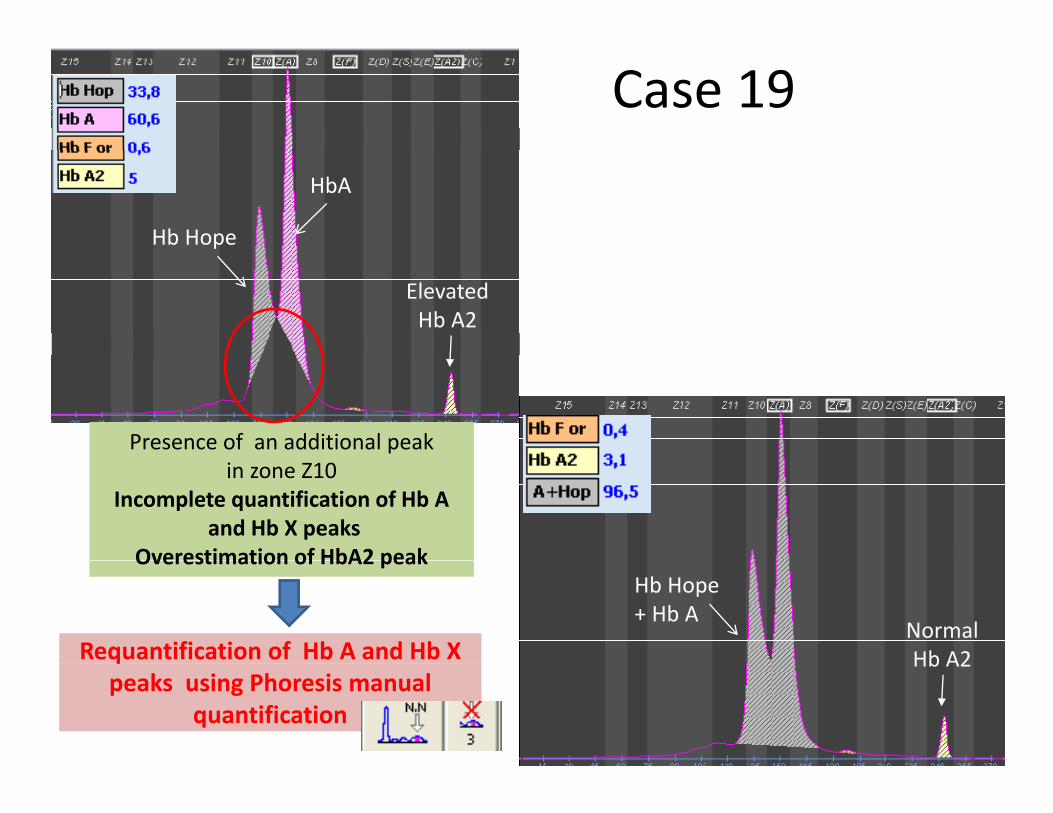
Case 19HbA
Hb Hope
ElevatedHb A2
Presence of an additional peak in zone Z10
Incomplete quantification of Hb Aand Hb X peaks
Overestimation of HbA2 peakOverestimation of HbA2 peak
Requantification of Hb A and Hb X
Hb Hope+ Hb A
NormalHb A2q
peaks using Phoresis manualquantification
Hb A2
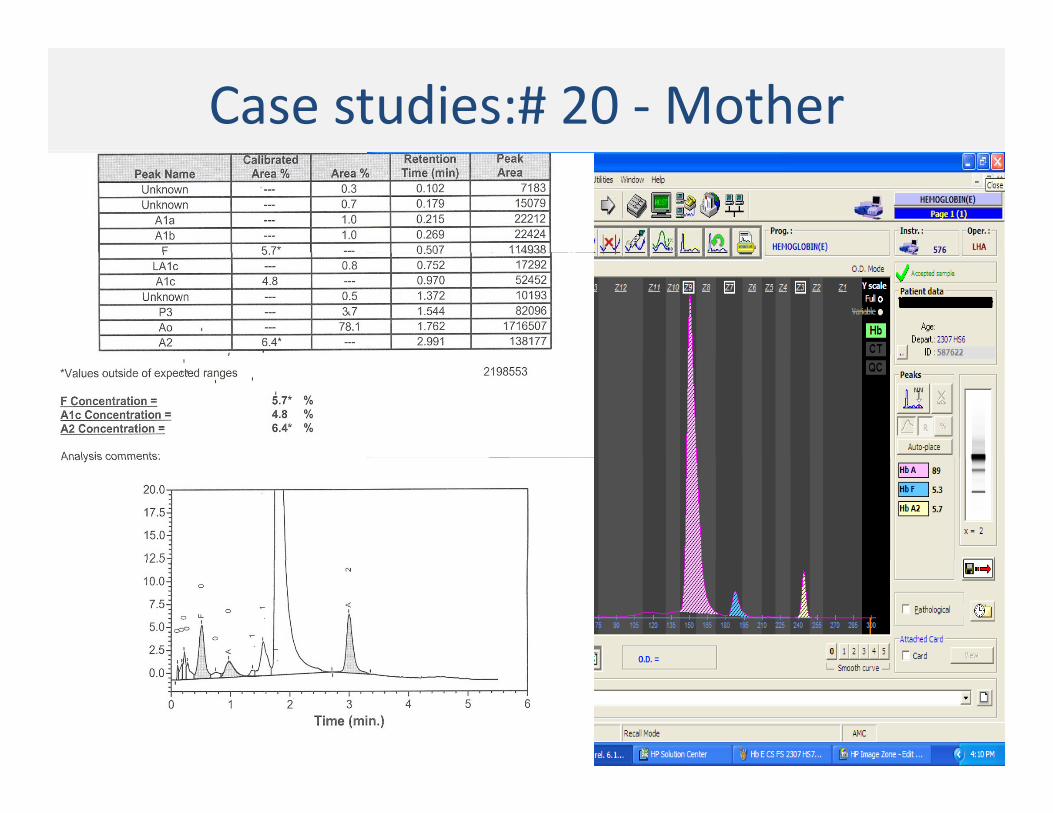
Case studies:# 20 ‐Mother
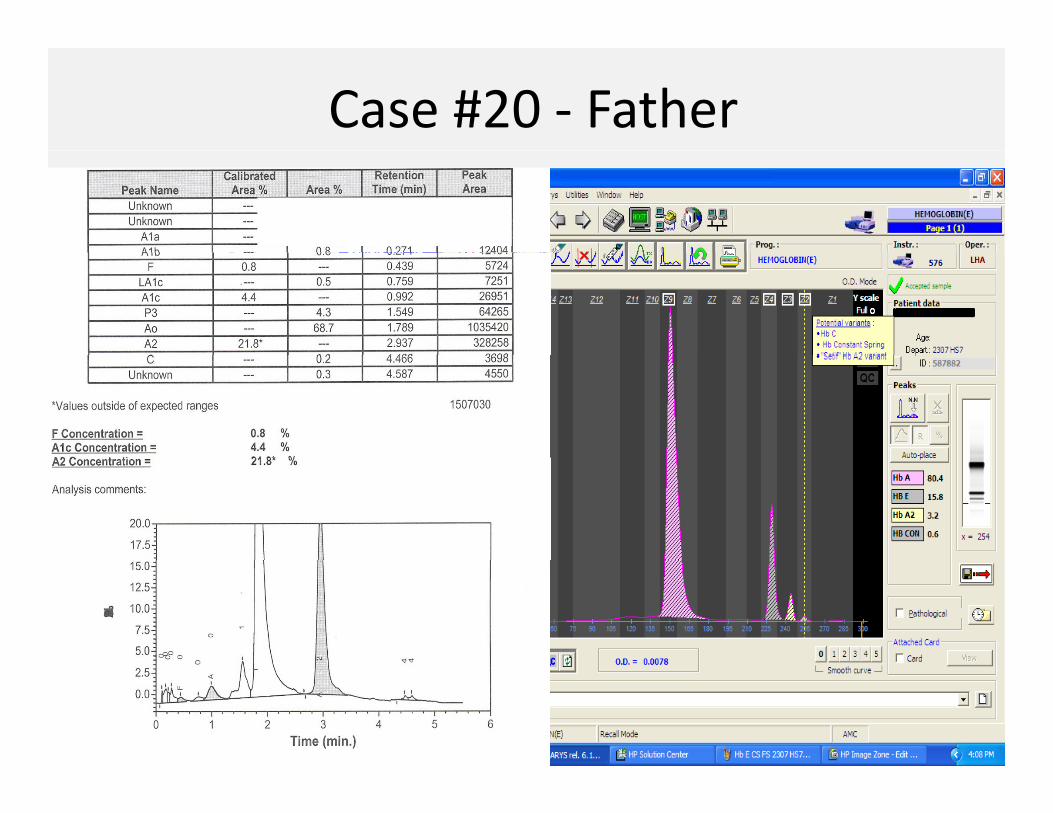
Case #20 ‐ Father
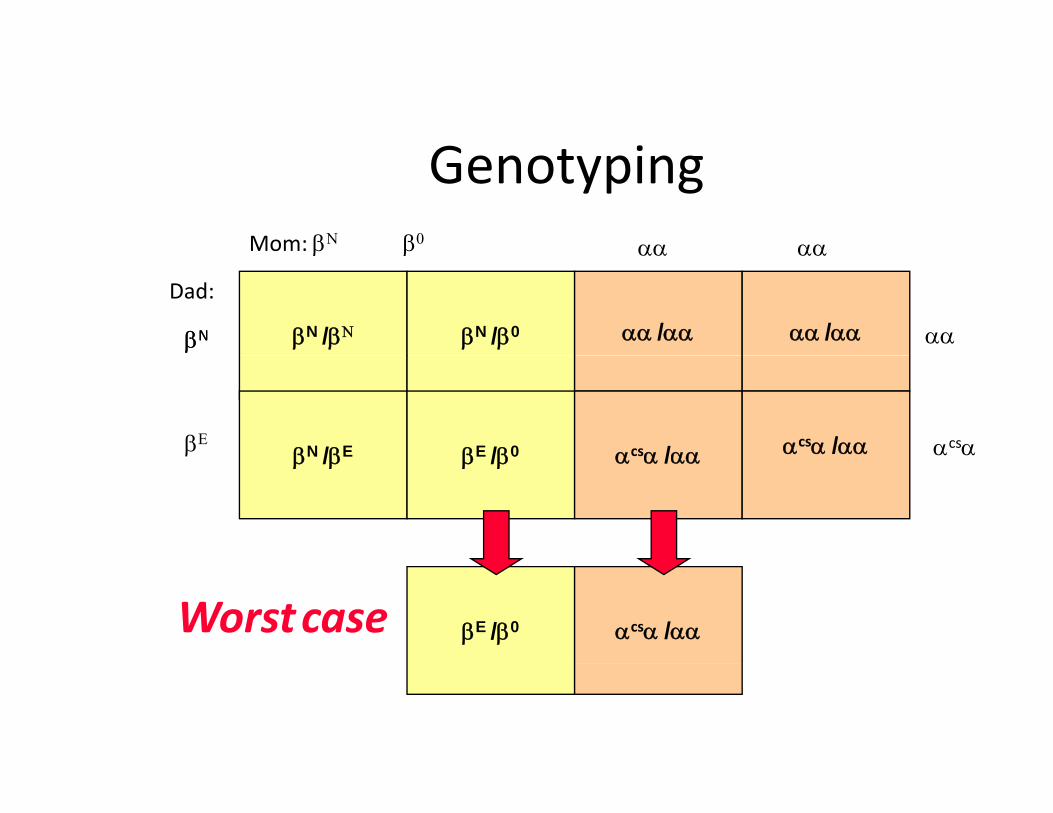
G t iGenotypingMom: βΝ β0 αα αα
βN /βΝ βN /β0 αα /αα αα /αα
β β
Dad:
βNβN
αα αα
αα
βN /βE βE /β0 αcsα /αα αcsα /ααβΕ αcsαβ /β β /β α α /αα
βE /β0 αcsα /ααWorstcase
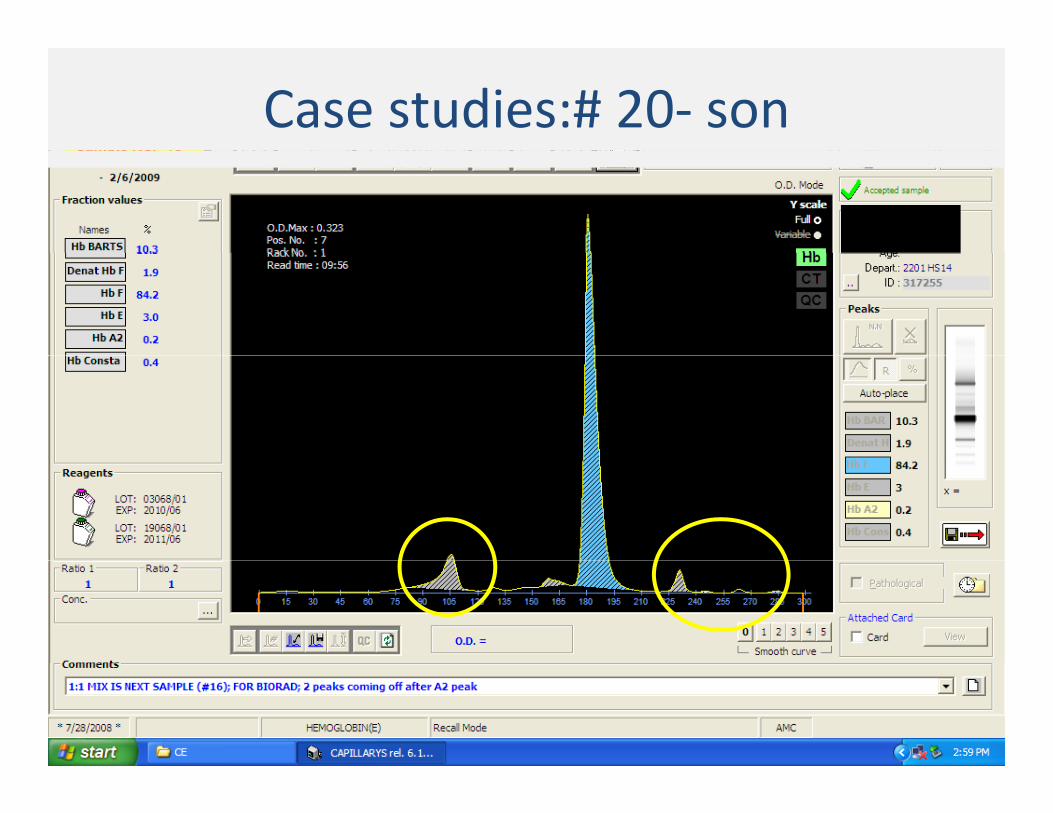
Case studies:# 20‐ son
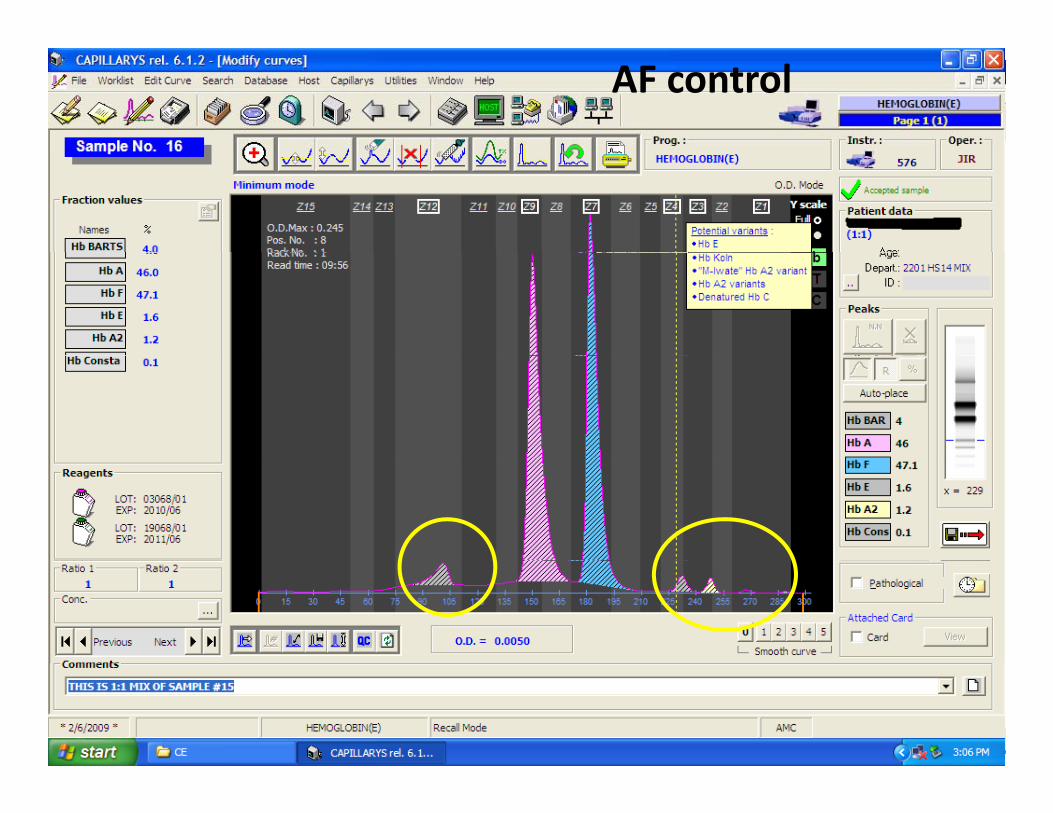
Case 4 AF control
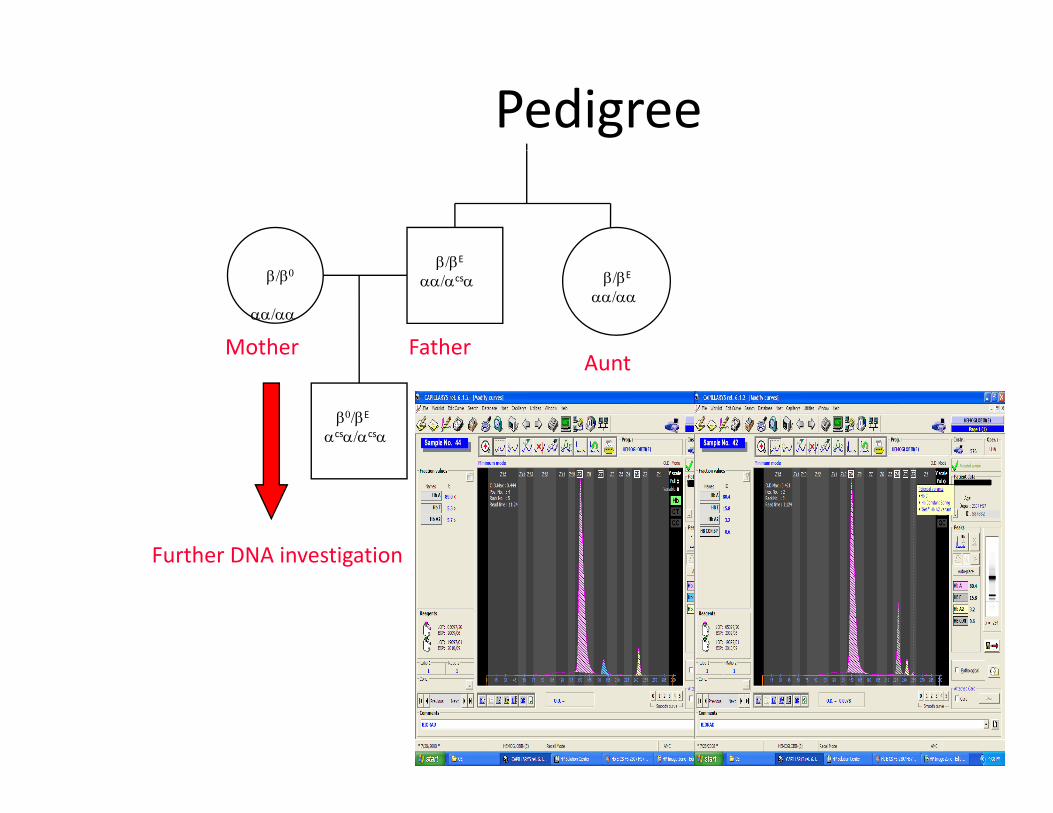
Pedigree
β/βE
αα/αα
β/βE
αα/αcsαβ/β0
αα/αα
AFatherMother
β0/βE
αcsα/αcsα
Aunt
Further DNA investigationFurther DNA investigation
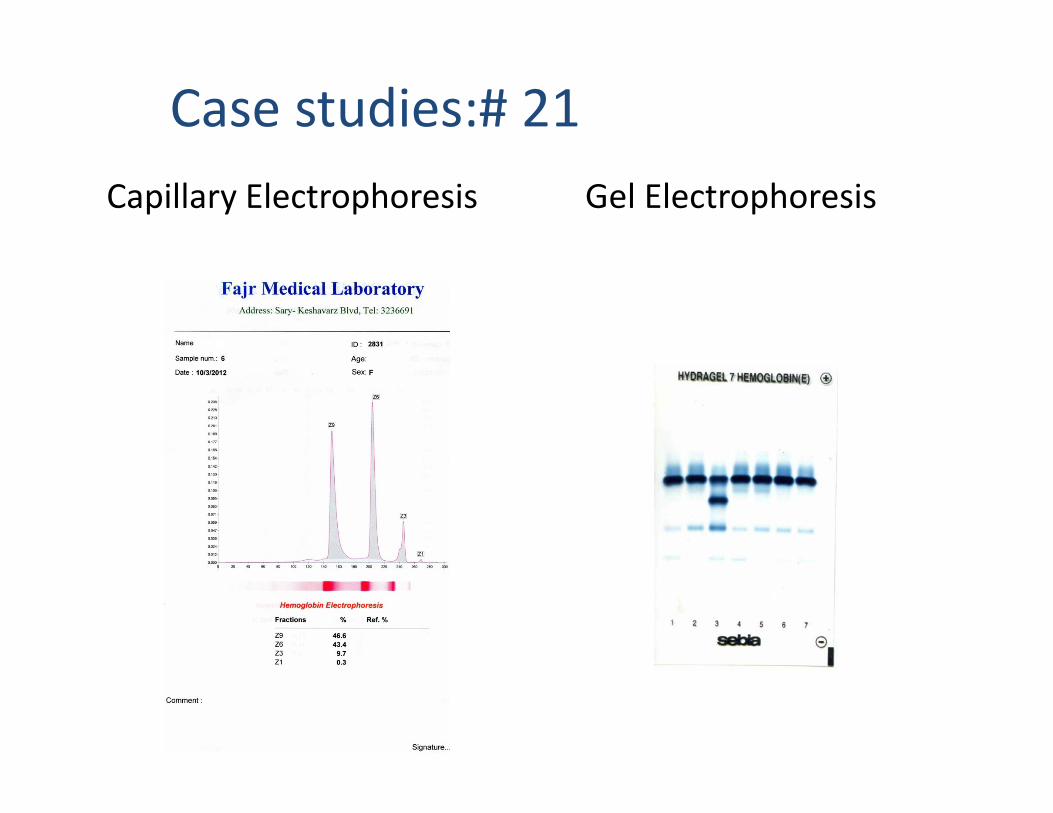
Case studies:# 21Capillary Electrophoresis Gel Electrophoresis

CBC MIXING (1 Vol. sample + 1 Vol. Hb A2 NC)

FATHER MOTHERFATHER MOTHER
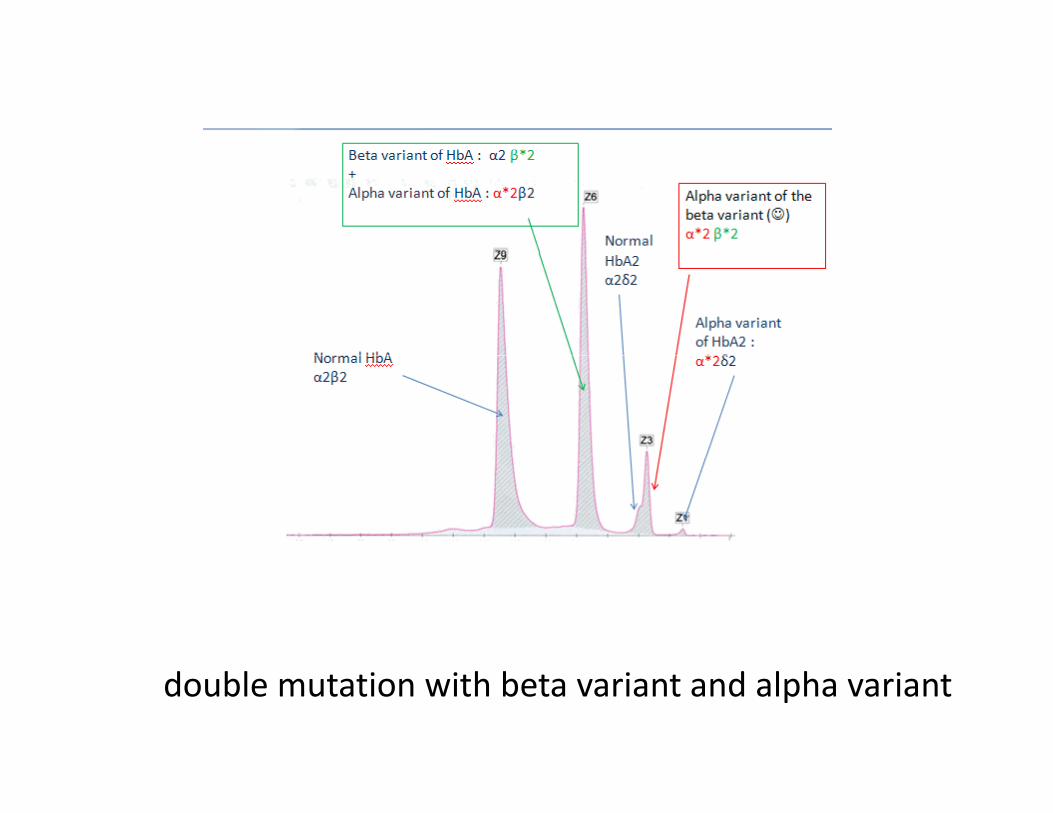
double mutation with beta variant and alpha variant
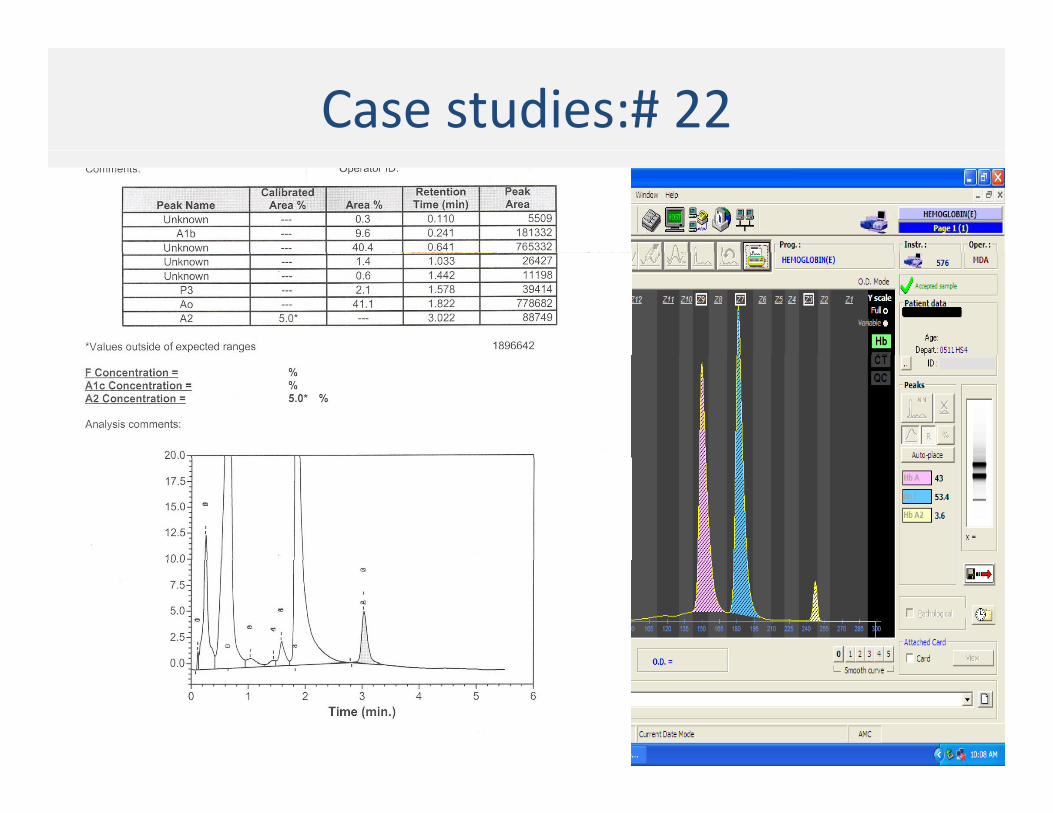
Case studies:# 22

Case # 22 : Mother and Father

Case # 22 : Sister
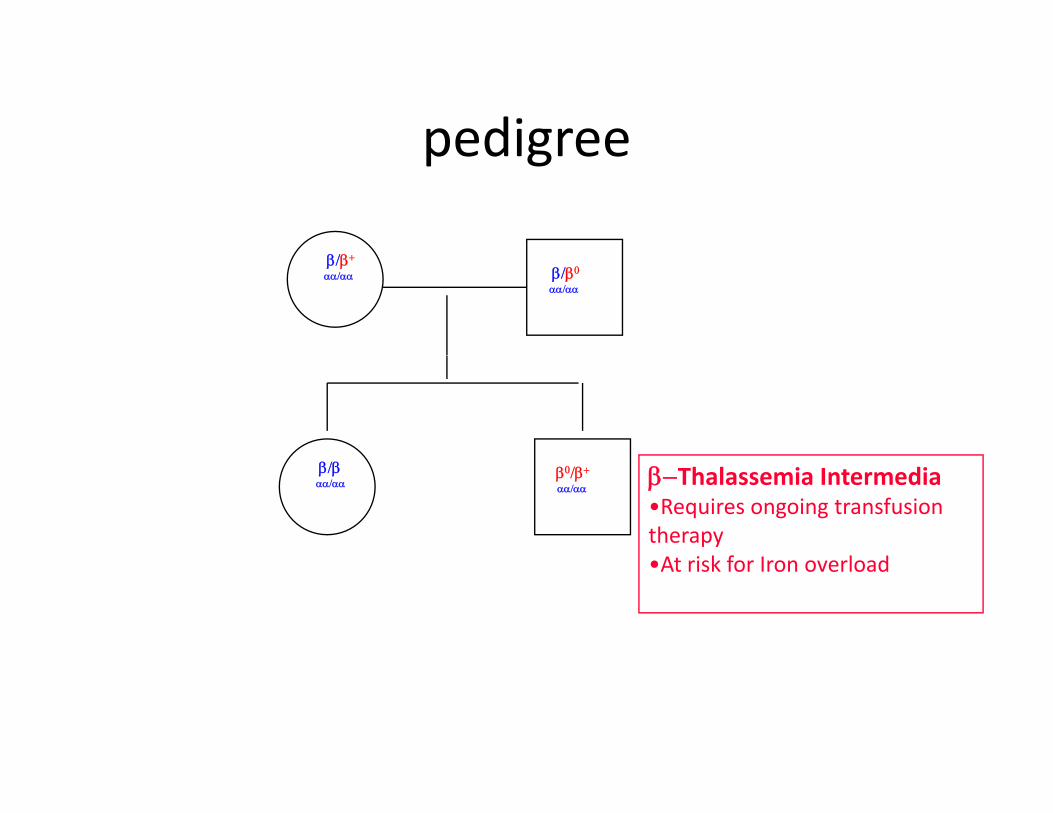
pedigreepedigree
β/β+
αα/αα β/β0
αα/αα
β/βαα/αα
β0/β+
αα/ααβ−Thalassemia Intermedia•Requires ongoing transfusion therapy•At risk for Iron overload•At risk for Iron overload
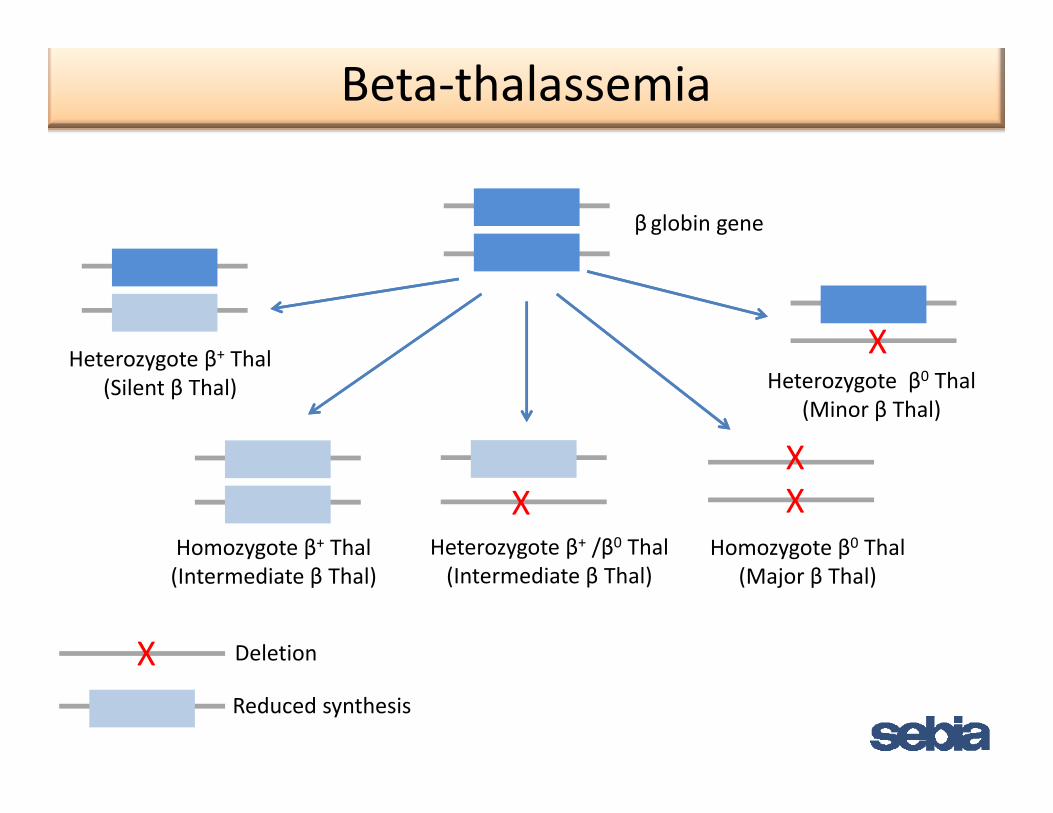
Beta‐thalassemia
β globin gene
XHeterozygote β+ Thal
X
Heterozygote β+ Thal(Silent β Thal) Heterozygote β0 Thal
(Minor β Thal)
XX
Homozygote β0 ThalHomozygote β+ Thal Heterozygote β+ /β0 Thal
X
DeletionX
(Major β Thal)(Intermediate β Thal) (Intermediate β Thal)
Reduced synthesis
X
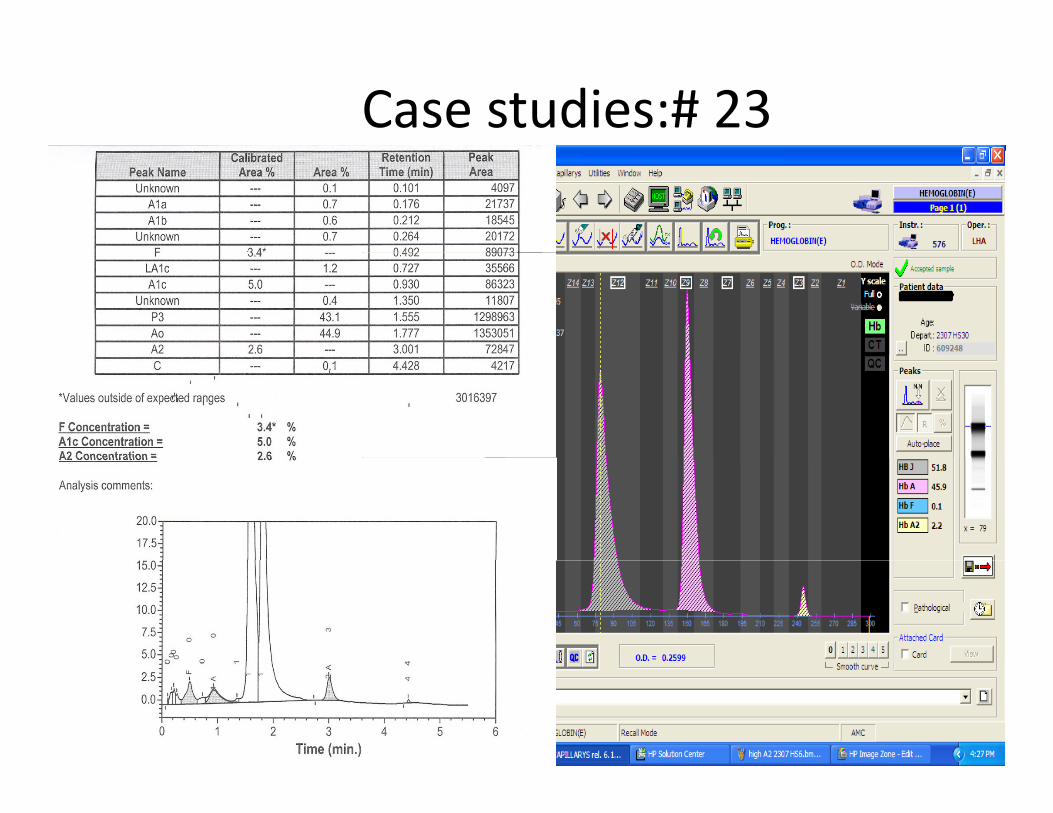
Case studies:# 23
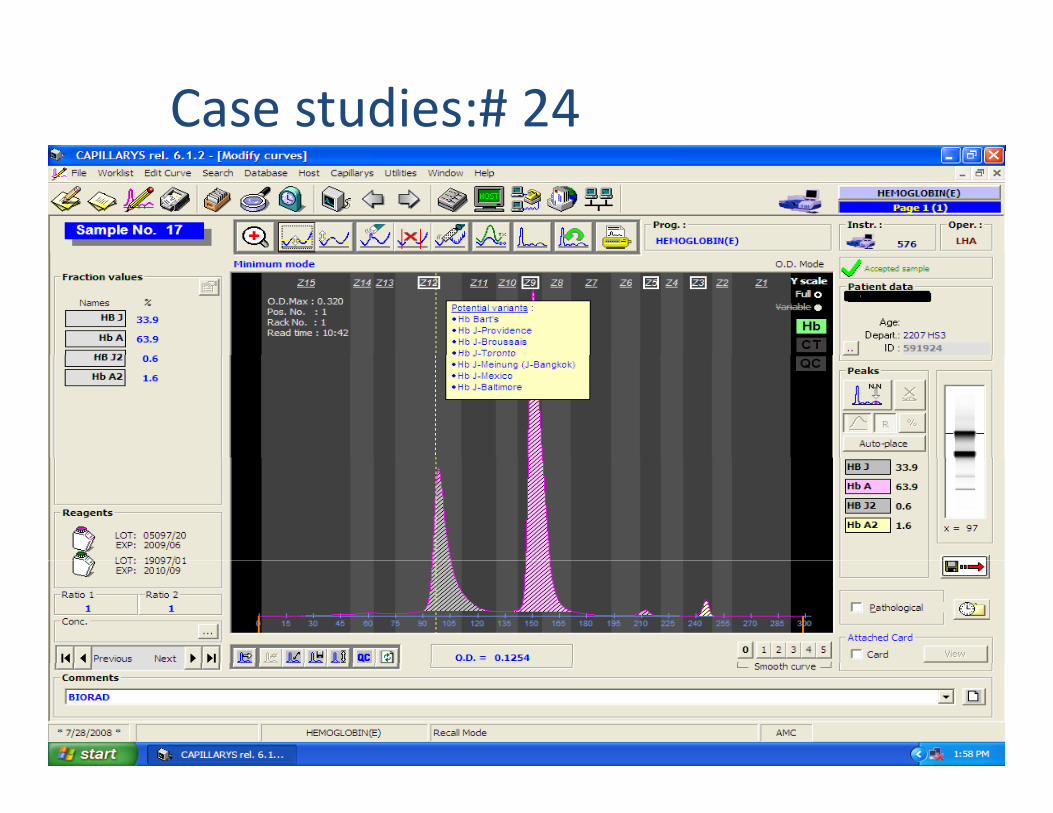
Case studies:# 24
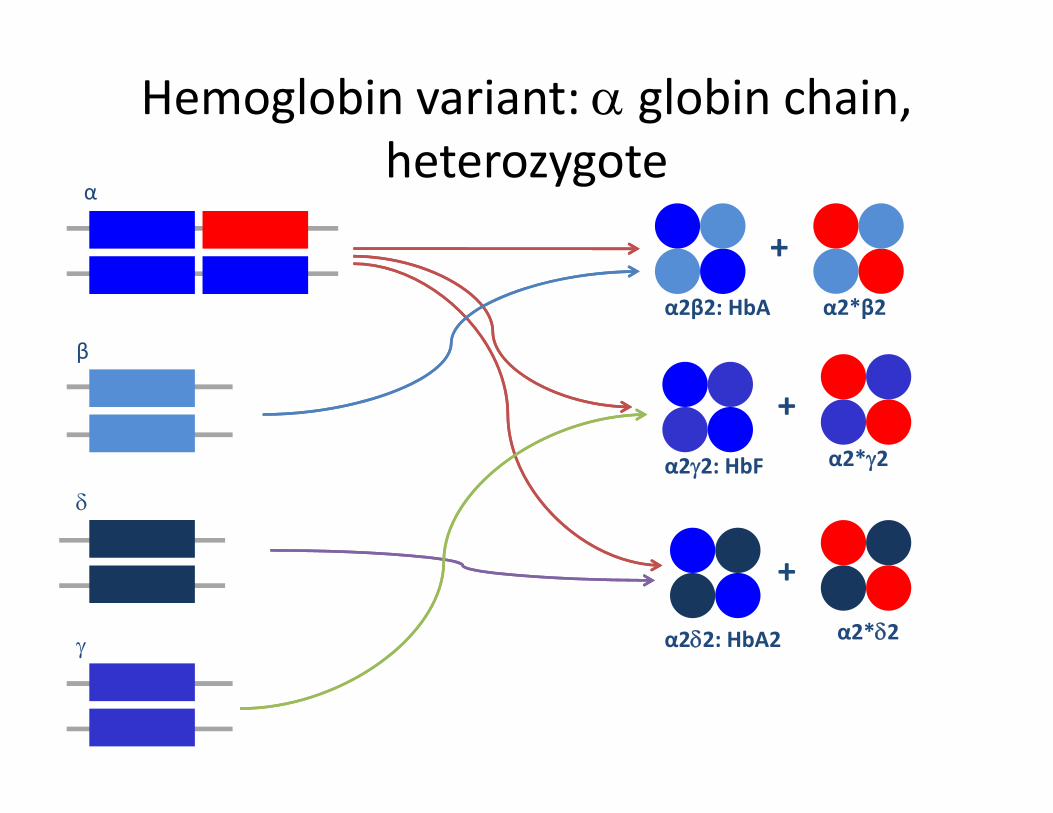
Hemoglobin variant: α globin chain, h t theterozygote
α
+
β
α2β2: HbA α2*β2
+
β
b
+α2*γ2
δα2γ2: HbF α2*γ2
γ α2δ2: HbA2
+α2*δ2
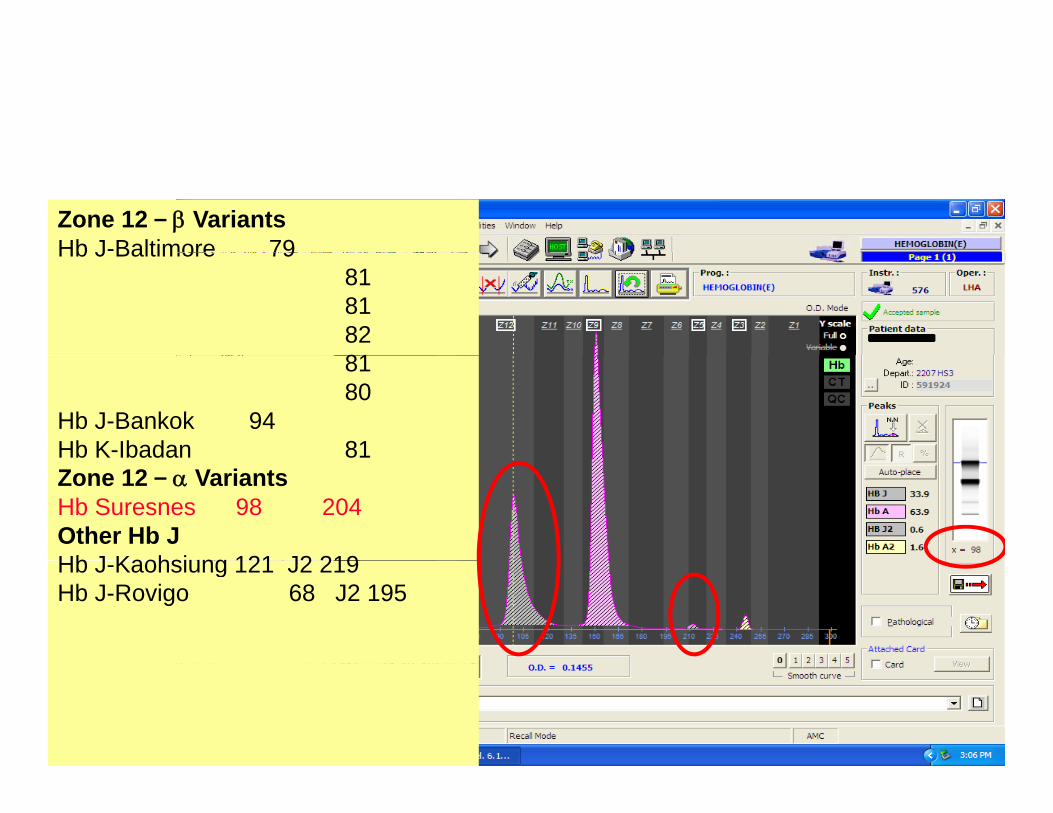
Zone 12 – β VariantsHb J-Baltimore 79Hb J Baltimore 79
818182818180
Hb J-Bankok 94Hb K-Ibadan 81Zone 12 – α VariantsHb Suresnes 98 204Other Hb JHb J Kaohsiung 121 J2 219Hb J-Kaohsiung 121 J2 219Hb J-Rovigo 68 J2 195

Case studies:#25
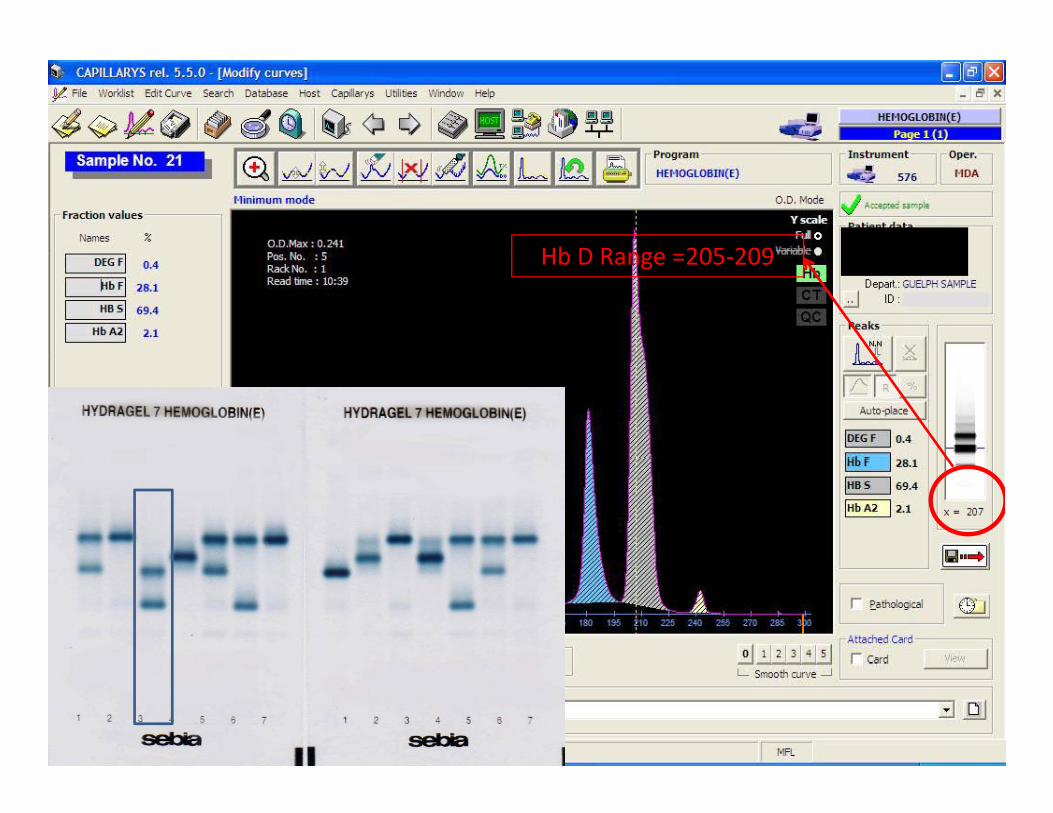
Hb D Range 205 209Hb D Range =205‐209

Case studies # 26: Baby JTy
• Baby boy born at 38 weeks gestational ageBaby boy born at 38 weeks gestational age• Parents from Vietnam, no consanguinity
h i f l h• Both parents were carriers for alpha thalassemia trait– 27 year old mother– 34 year old father

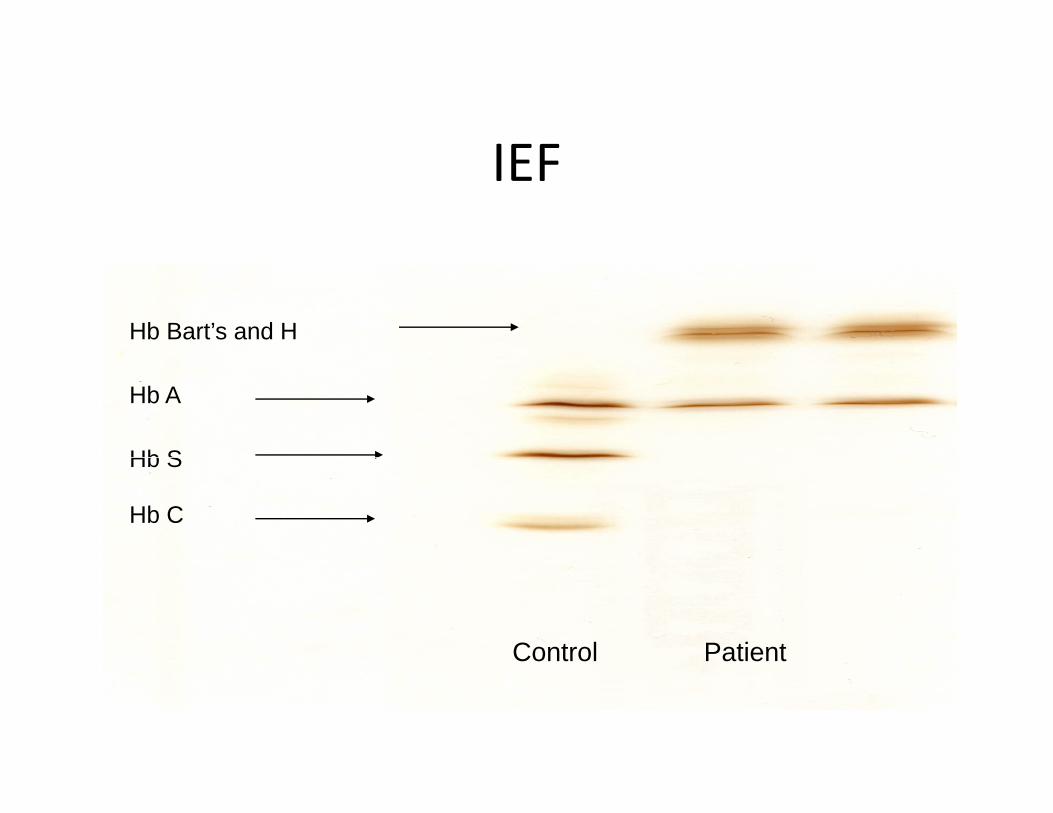
IEFIEF
Hb Bart’s and H
Hb A
Hb SHb S
Hb C
Control Patient

Single cell with H inclusion bodies: “Hemoglobin H” diseaseSingle cell with H inclusion bodies:Can be seen in any form of alpha
thalassemia
“Hemoglobin H” disease
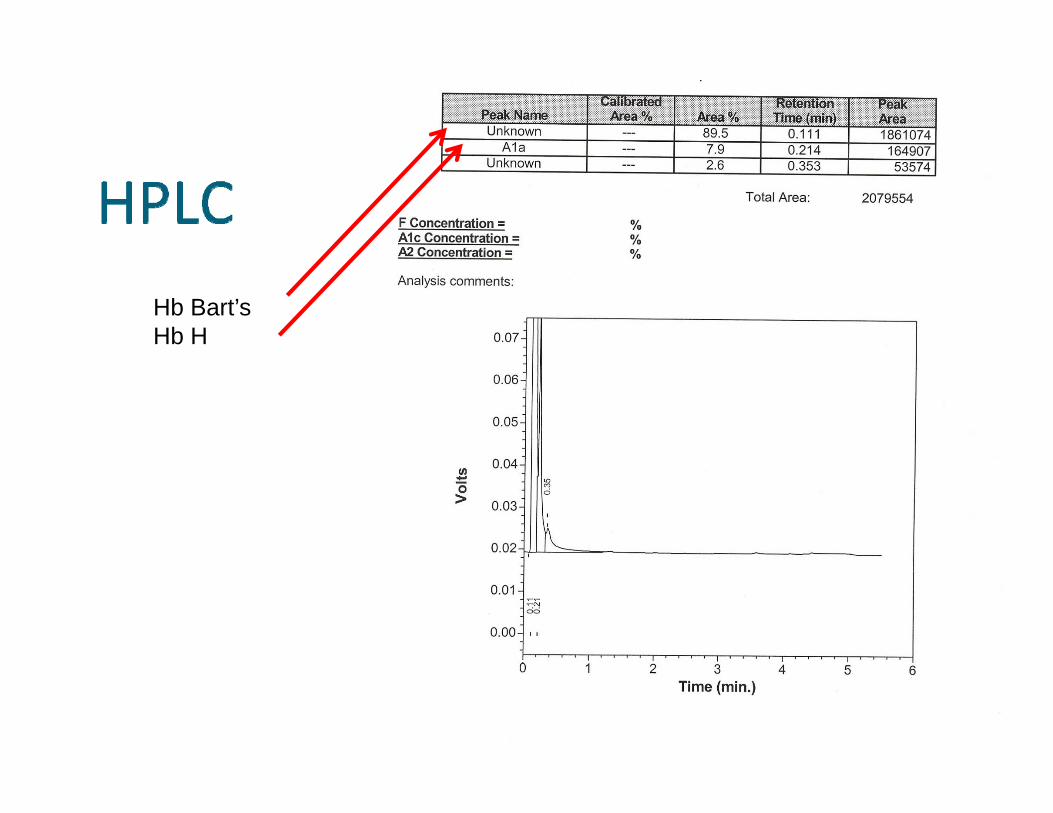
Hb Bart’s Hb H
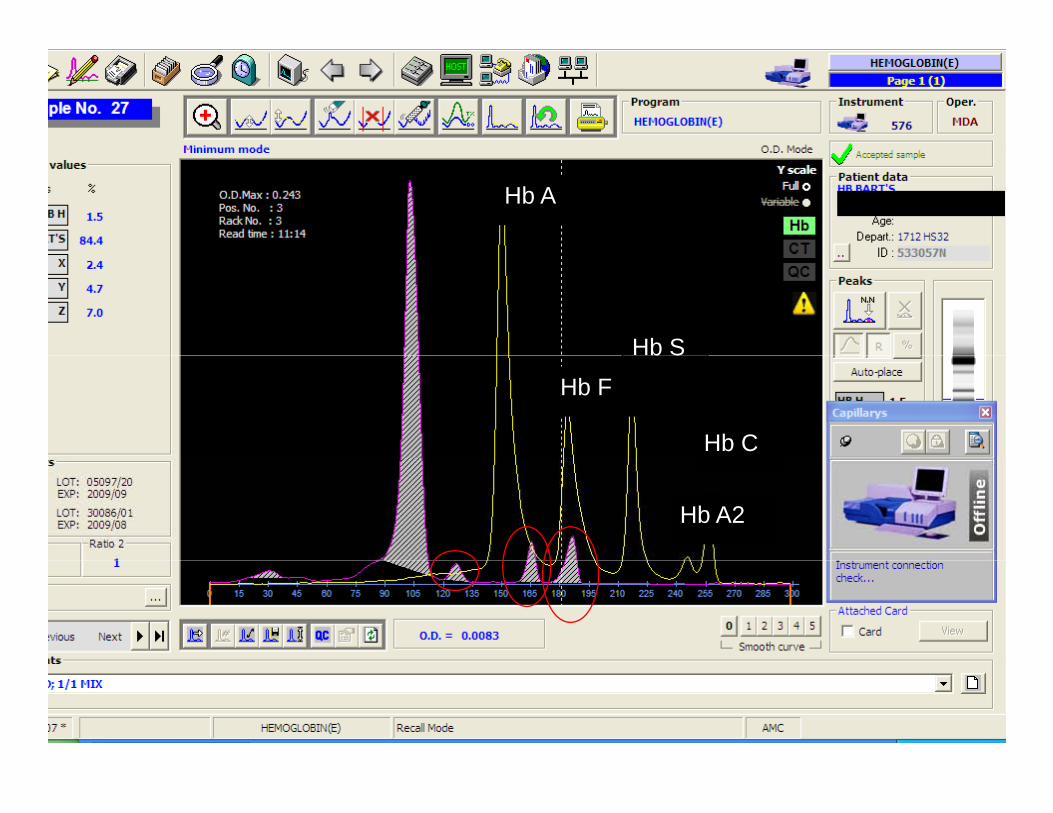
Hb A
Hb S
Hb F
b S
Hb C
Hb A2

B b JT’ Old SibliBaby JT’s Older Sibling• Ultrasound at 19 weeks GA showedUltrasound at 19 weeks GA showed unexplained fetal hydrops and limb defects
• Mother presented at 28 weeks in labour• Mother presented at 28 weeks in labour.• Baby delivered by cesarean section• Severely hydropic, died at 3 hours of age• Homozygous alpha thalassemia diagnosed on yg p gautopsy

Baby JT’s Prenatal CounsellingBaby JT s Prenatal Counselling• After first pregnancy, parents followed by…
Genetics Clinic at McMaster University Medical–Genetics Clinic at McMaster University Medical Centre referred to High Risk Obstetrics at Mount Sinai Hospital
• Both parents found to carry the Southeast Asian gene deletion (‐ ‐SEA/αα)–1 in 4 of normal baby (αα/αα)–2 in 4 chance baby carrier of deletion (‐ ‐SEA/αα)–1 in 4 chance that baby has homozygous alpha thalassemia (‐ ‐SEA/‐ ‐SEA)

B b JT’ P diBaby JT’s Pedigree
‐ ‐SEA / α α ‐ ‐SEA / α α
‐ ‐SEA / ‐ ‐SEA‐ ‐SEA / ‐ ‐SEA

Case studies # 27
HPLC seems to split Constant Spring into separate subspecies
HPLC seems to split Constant Spring into separate subspecies
Heterozygote Constant Spring
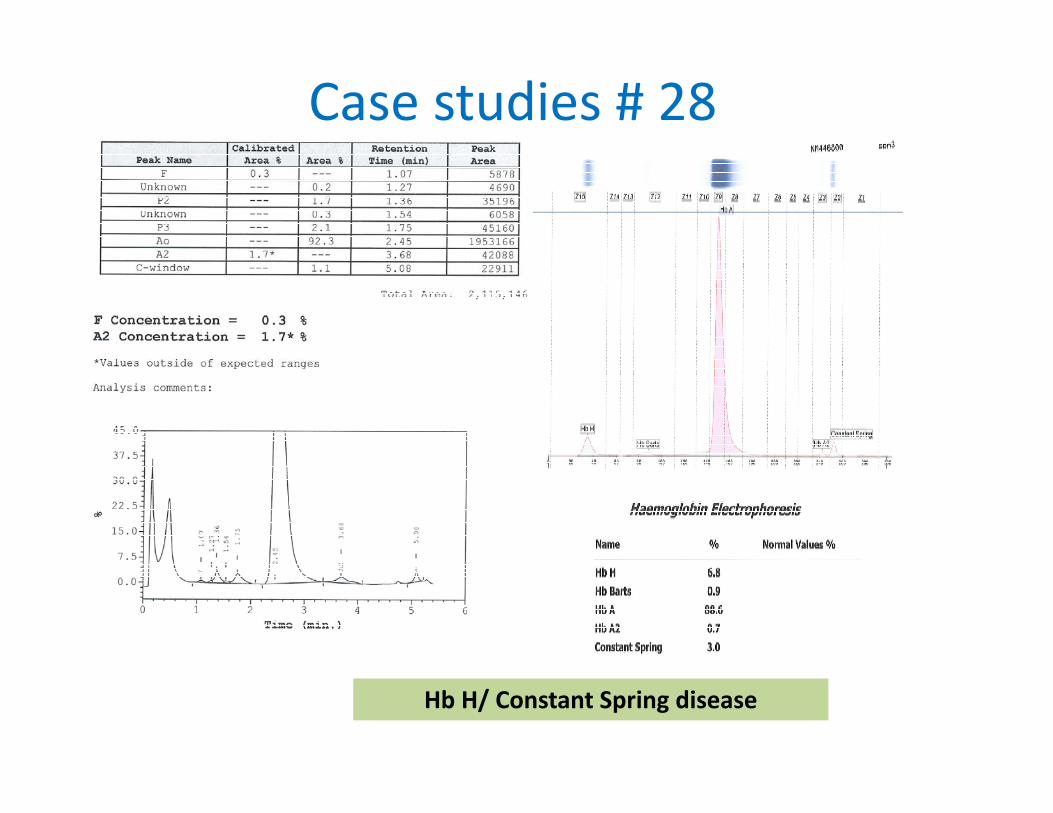
Case studies # 28
Hb H/ Constant Spring disease
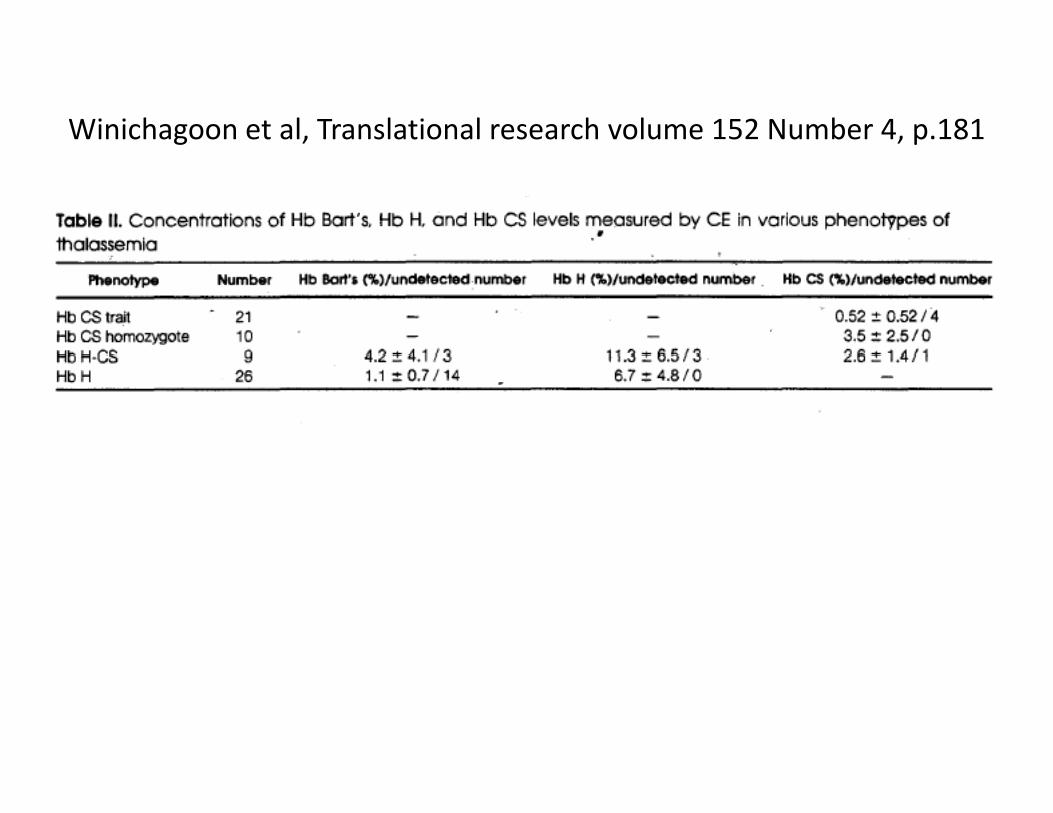
Winichagoon et al, Translational research volume 152 Number 4, p.181

Hemoglobin, 35(1):87–90, (2011). Can Liao et alG d P i S h ChiGuangdong Province‐ South China

TopicsTopics
• Epidemiology of Hemoglobin disordersEpidemiology of Hemoglobin disorders• Available Methods used for Hb investigationsCli i l• Clinical cases
• Rules for interpretation on CE
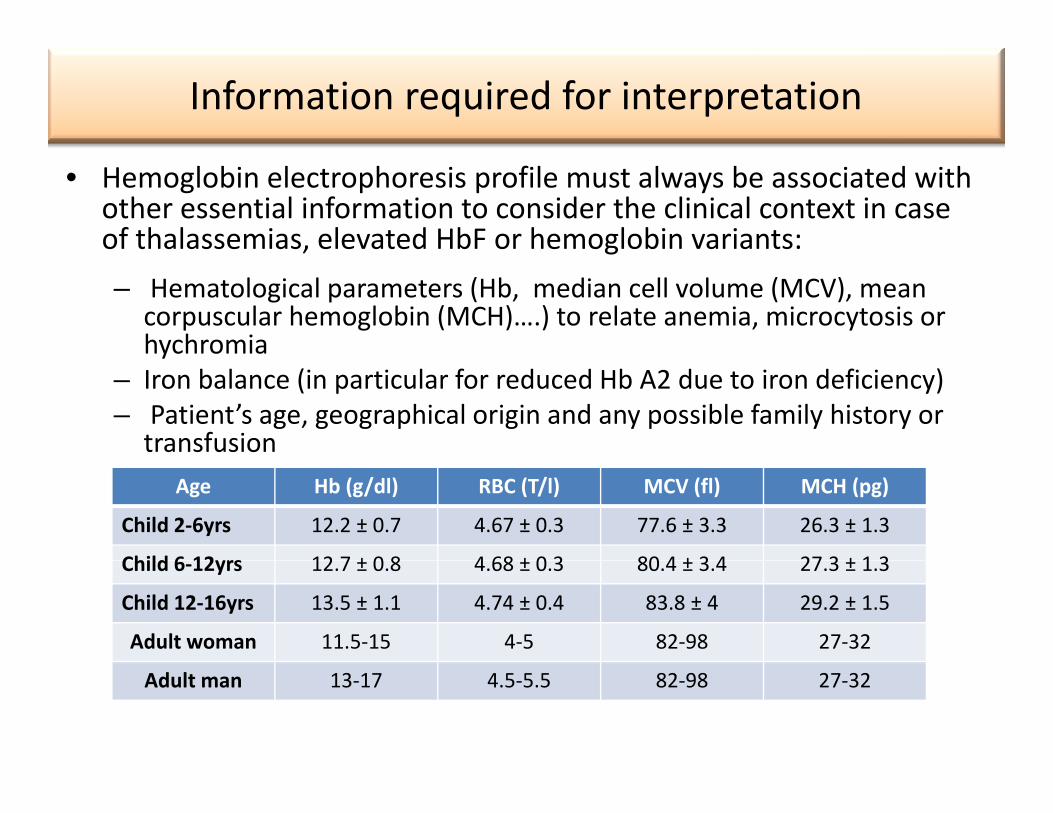
Information required for interpretation
• Hemoglobin electrophoresis profile must always be associated with other essential information to consider the clinical context in case of thalassemias, elevated HbF or hemoglobin variants:, g– Hematological parameters (Hb, median cell volume (MCV), mean
corpuscular hemoglobin (MCH)….) to relate anemia, microcytosis or hychromiay
– Iron balance (in particular for reduced Hb A2 due to iron deficiency)– Patient’s age, geographical origin and any possible family history or
transfusion Age Hb (g/dl) RBC (T/l) MCV (fl) MCH (pg)
Child 2‐6yrs 12.2 ± 0.7 4.67 ± 0.3 77.6 ± 3.3 26.3 ± 1.3
Child 6 12yrs 12 7 ± 0 8 4 68 ± 0 3 80 4 ± 3 4 27 3 ± 1 3Child 6‐12yrs 12.7 ± 0.8 4.68 ± 0.3 80.4 ± 3.4 27.3 ± 1.3
Child 12‐16yrs 13.5 ± 1.1 4.74 ± 0.4 83.8 ± 4 29.2 ± 1.5
Adult woman 11.5‐15 4‐5 82‐98 27‐32
Adult man 13‐17 4.5‐5.5 82‐98 27‐32
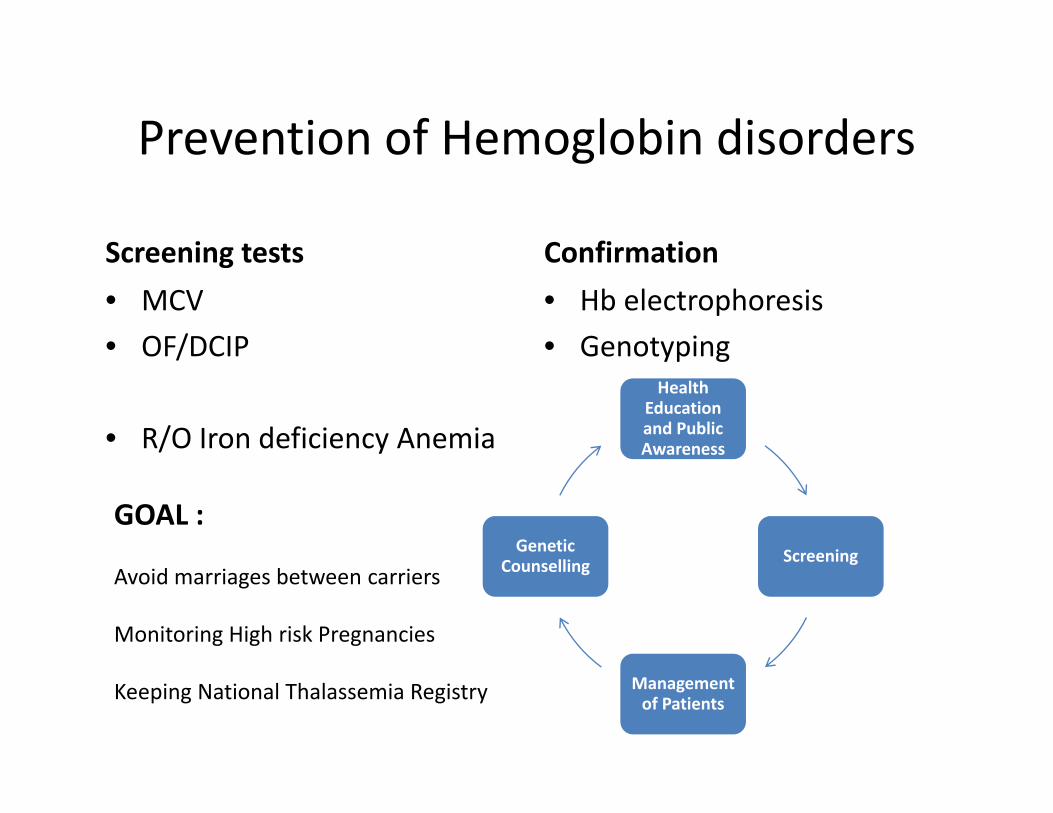
Prevention of Hemoglobin disordersPrevention of Hemoglobin disorders
Screening tests ConfirmationScreening tests• MCV• OF/DCIP
Confirmation• Hb electrophoresis• GenotypingOF/DCIP
• R/O Iron deficiency Anemia
GenotypingHealth
Education and Public Awareness
GOAL :ScreeningGenetic
C lliAvoid marriages between carriers
Monitoring High risk Pregnancies
ScreeningCounselling
Keeping National Thalassemia Registry Management of Patients
