clara camaschella anemie giovanni gromo franca pellò ... · mia emolitica il pallore è...
TRANSCRIPT

1039
88
© 2
010
ELSE
VIE
R S
.R.L
. Tu
tti
i d
irit
ti r
iser
vati
.
CAPITOLO 48
Anemie Clara Camaschella
Giovanni Gromo
Franca Pellò
Maurizio Storti
Maria D. Cappellini
Considerazioni generali* L’anemia è definita dalla diminuzione del patrimonio emoglobinico totale dell’organismo; poiché l’emoglobina (Hb) è contenuta nelle emazie, si ha anemia quando è diminuita la massa eritrocitaria. In condizioni fi siologiche si ammette che la distribuzione delle emazie nell’organismo sia omogenea, per cui la stima, nell’unità di volume di sangue periferico, della concentrazione di Hb (g/dL) e della quota del volume ematico occupato dagli eritrociti (valore ematocrito o Hct) dà valori ben correlati all’entità della massa eritrocitaria. Invece, non è signifi cativo in questo senso il numero dei globuli rossi (GR), che, in alcune for-me di anemia, come, per esempio, la talassemia minor, può essere normale o anche aumentato, con contenuto corpuscolare medio di emoglobina ridotto e quindi con diminuzione della concentrazione di emoglobina nell’unità di volume di sangue; queste emazie hanno costantemente anche volume corpuscolare medio ridotto, da cui una riduzione di Hct. Tenendo presente il concetto di massa totale eritrocitaria riuscirà più facile la comprensione di alcune situazioni di anemia o di poliglobulia solo apparenti.
Classifi cazione La massa eritrocitaria normale nell’uomo corrisponde a un volume di 30 ± 5 mL/kg; gli eritrociti hanno una vita media di circa 120 giorni, per cui si è calcolato che circa lo 0,8% della massa eritrocitaria debba essere sostituito gior-nalmente a opera del midollo osseo. La massa eritrocitaria (M) è proporzionale alla quantità di emoglobina prodotta giornalmente (H°) per la vita media dei GR in giorni (T), ossia M = H° × T. Si ha anemia quando M è inferiore alla norma. La formula permette una classifi cazione patogenetica delle anemie, per riduzione di H° o di T o di entrambi i termini; all’interno di ciascun gruppo i meccanismi responsabili possono essere diversi. • Anemie per diminuita produzione di emoglobina:
– da ridotta eritroblastogenesi per mieloftisi e per distruzione o alterazione funzionale delle cellule staminali;
– da ridotta eritrogenesi per distruzione dei precursori nel midollo con eritropoiesi ineffi cace;
– da ridotta o difettosa sintesi della globina o dell’eme. • Anemie per riduzione del tempo di sopravvivenza
dei GR:
– da perdita secondaria a emorragie acute o croniche;
– da distruzione a opera di un meccanismo emolitico.
Le due situazioni possono coesistere e la massa eritro-citaria può ridursi per il sommarsi dei due meccanismi patologici (riduzione sia di H° sia di T). In genere l’insuffi ciente produzione di GR e l’eritropoie-si ineffi cace sono caratterizzate da un basso numero di eritrociti giovani (reticolociti) nel sangue periferico; al contrario, le anemie postemorragiche e quelle emolitiche dimostrano nel sangue periferico segni di iperattività mi-dollare, con incremento del numero dei reticolociti, pre-senza di emazie con policromasia o punteggiatura basofi la o addirittura con la comparsa di qualche eritroblasto. Altre alterazioni sono pure caratteristiche delle anemie emolitiche, come l’aumento della lattato-deidrogenasi (LDH) del siero, derivato dalla lisi delle emazie, l’iperbili-rubinemia indiretta e l’urobilinuria (si veda il Capitolo 30 ), l’incremento dell’urobilinogeno fecale, la diminuzione della concentrazione di aptoglobina e l’aumento del tur-nover del ferro plasmatico. Tuttavia, l’aumento della LDH del siero è presente pure nell’eritropoiesi ineffi cace, che può anche determinare un certo aumento della bilirubinemia (iperbilirubinemia da shunt). È molto importante tenere presente questa classifi cazione patogenetica per risalire dall’anemia alla diagnosi della malattia che ne è all’origine; infatti, l’anemia è in alcuni casi un elemento di evenienza secondaria e non obbliga-toria nel corso di una forma morbosa a dignità nosografi ca ben defi nita. In altri invece costituisce il rilievo clinico più importante della malattia, che da essa prende il nome (per esempio, anemia megaloblastica, anemia aplastica ecc.). Bisogna tenere presente, però, che il primo approccio alla situazione anemica è fornito dall’esame emocro-mocitometrico, con i valori di emoglobina (11,5-16 g/dL nella donna e 12,5-17 g/dL nell’uomo), il numero di GR (4-5 milioni/mm 3 nella donna, 4,5-5,5 milioni/mm 3 nell’uomo) e l’ematocrito (36-42% nella donna e 40-45% nell’uomo) dai quali si calcolano le costanti corpuscolari, e dall’esame dello striscio del sangue periferico, che dà informazioni signifi cative sulla forma dei GR e sulla loro affi nità tintoriale. Le seguenti formule forniscono il valore dei cosiddetti parametri derivati:
MCV (80-100 m 3 ) = Hct × 10 _________________ milioni di GR per mm3 *F. Pellò
C0240.indd 1039C0240.indd 1039 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1040
MCH (27-31 pg) = Hb × 10 _________________ milioni di GR per mm3
MCHC (30-36 g/dL) = Hb × 10 _______ Hct
L’indice emocromocitometrico indiretto di anisopoichi-locitosi e anisocromia è espresso numericamente come ampiezza della distribuzione eritrocitaria (RDW, Red cell Distribution Width) . I reticolociti possono essere espressi in numero assoluto (25.000-100.000/mm 3 ) o percentuale (5-20 per mille); l’indice reticolocitario, che ha un signifi cato analogo rispetto al valore assoluto, consiste nella correzione della percentuale dei reticolo-citi per il grado di anemia:
reticolociti % × ematocrito/ematocrito normale (45).
In base ai valori di volume corpuscolare medio (MCV, Mean Corpuscular Volume) , contenuto corpuscolare medio di emoglobina (MCH, Mean Corpuscular Hemoglobin) e intensità di colorazione si costruisce la seguente classifi -cazione cosiddetta morfologica delle anemie: • anemia ipocromica microcitica (MCV < 80 fL;
MCH < 27 pg, emazie ipocolorate); • anemia normocromica normocitica (MCV: 89-94
fL; MCH 29,5 ± 2,5 pg); • anemia macrocitica (MCV > 94 fL; MCH > 32 pg).
Non vi è una correlazione precisa tra meccanismo pa-togenetico dell’anemia e tipo morfologico; tuttavia, per ciascuno di questi gruppi vi sono delle frequenze obbligate e delle frequenze preferenziali. • Anemia ipocromica microcitica :
– anemia sideropenica; – anemia sideroblastica; – talassemie, alcune emoglobinopatie; – rari casi di carenza di rame, di piridossina.
• Anemia normocitica: – anemie emolitiche acquisite; – sferocitosi ereditaria; – emoglobinuria parossistica notturna; – alcune emoglobinopatie; – anemia aplastica; – anemia da mielosostituzione; – anemia delle malattie croniche.
• Anemia macrocitica : – anemia da defi cit di vitamina B 12 o di acido folico
(anemia megaloblastica); – anemia delle epatopatie croniche; – anemia dell’ipotiroidismo; – alcuni casi di anemia aplastica; alcune fasi
in corso di anemia emolitica.
Da questa classifi cazione, in un certo senso “semeiotica”, deve partire il ragionamento diagnostico che, con l’aiuto di ulteriori esami indicati caso per caso, permett di stabilire il meccanismo patogenetico dell’anemia e di risalire alla causa determinante.
Fisiopatologia Il GR, con il suo contenuto di emoglobina, è un anello essenziale nella catena di trasporto dell’ossigeno; que-sto dipende sia dall’integrità della molecola di emoglo-bina sia dalla sua funzione, che può essere modifi cata dall’interazione con altri composti contenuti nell’ema-zia. In questa modulazione della funzione emoglobinica
è principalmente coinvolto il 2,3-difosfoglicerato (2,3-DPG) , la cui concentrazione è regolata dall’attività glico-litica dell’emazia; esso stabilizza la conformazione deossi dell’emoglobina e riduce l’affi nità per l’ossigeno; 2,3-DPG e ossigeno sono leganti competitivi per l’emoglobina. In realtà, l’effetto del 2,3-DPG è praticamente nullo alle alte pressioni parziali di ossigeno, così che non interferisce con l’assunzione di ossigeno da parte dell’emoglobina nei polmo-ni, mentre è signifi cativo alle basse pressioni parziali di ossige-no, in modo che ne venga favorita la cessione ai tessuti. Un’alterazione del trasporto di ossigeno da parte dell’ema-zia può quindi derivare da: • riduzione della massa globulare (anemia classica); • abnorme funzione dell’emoglobina per anomalie
strutturali; • concentrazione inappropriata di 2,3-DPG.
L’ipossia tissutale induce una serie di modifi cazioni com-pensatorie mediante risposte di adattamento volte ad aumentare la circolazione del sangue e l’estrazione di os-sigeno dall’Hb arteriosa, a livello soprattutto degli organi vitali (cervello, cuore, rene, fegato e ghiandole surrenali). Questo si verifi ca per: • aumento della ventilazione alveolare; • aumento della portata cardiaca; • ridistribuzione del sangue da organi non vitali (cute,
tratto gastroenterico ed estremità) a organi vitali; • diminuzione dell’affi nità dell’emazia per l’ossigeno,
mediante diminuzione del pH cellulare e aumento del 2,3-DPG;
• stimolazione dell’eritropoiesi (per quanto possibile).
La velocità con cui insorge l’anemia è molto importante ai fi ni dell’entrata in funzione di questi meccanismi di adat-tamento; infatti, un’anemia a lenta progressione permette che essi si instaurino gradualmente e raggiungano la loro massima effi cacia. È noto come l’anemia cronica consenta una vita relativamente normale, per lo meno a riposo, an-che per valori di emoglobina notevolmente ridotti. In presenza di lieve anemia entra in azione piuttosto len-tamente (circa in 10 ore) il 2,3-DPG, che riducendo, come si è visto, l’affi nità dell’emoglobina per l’ossigeno ne per-mette un’aumentata disponibilità per i tessuti; è questo un meccanismo di compenso “biochimico” che richiede un certo tempo per realizzarsi. Nell’anemia grave entra in gioco un meccanismo dinamico costituito dall’aumento della portata cardiaca; all’inizio esso è dovuto prevalentemente a un aumento della git-tata sistolica, con buon rendimento energetico. Quando l’anemia è molto grave, aumenta considerevolmente la frequenza cardiaca, con basso rapporto effi cienza/lavoro. Il livello di emoglobina in cui l’anemia diviene funzional-mente limitante e il grado di compromissione sono inver-samente proporzionali al grado di massimo adattamento e direttamente proporzionali all’intensità dell’attività fi sica, e conseguentemente, poiché il lavoro fi sico è principal-mente aerobico, alle richieste di ossigeno.
Sintomatologia Ogni tipo di anemia ha caratteristiche peculiari, trattate nei rispettivi paragrafi , mentre la sintomatologia generale, secondaria all’ipossia e all’esaltata funzione di compenso
C0240.indd 1040C0240.indd 1040 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Capitolo 48 - ANEMIE
81041
cardiorespiratorio, è comune a tutte le forme di anemia e viene qui brevemente esposta. Il sintomo più comune riferito dal paziente, che è il solo presente nella maggioranza dei casi, è l’ astenia , soprattutto sotto sforzo. In casi particolari possono sopravvenire di-sturbi da diminuita ossigenazione di vari distretti corporei (per lo più quando la loro irrorazione è già compromessa per lesioni aterosclerotiche) e si possono avere claudicatio intermittens, crampi notturni, vertigini, cefalea, episodi di sincope, dolore precordiale di tipo anginoso. Il segno più tipico dell’ipossia tissutale è il pallore cutaneo e mucoso, che va esplorato in determinate sedi quali il palmo della mano, il padiglione auricolare, la mucosa dell’interno delle labbra, la congiuntiva palpebrale; in presenza di ane-mia emolitica il pallore è accompagnato da subittero. La cute può talora essere piuttosto succulenta per la ten-denza all’edema ai malleoli e alle parti declivi, espressione di iniziale scompenso di circolo e i capelli sono opachi e fragili. Nelle anemie croniche le unghie possono avere forma “a vetrino di orologio”; si nota uno stato di scarsa ripienezza dei grossi tronchi venosi superfi ciali. L’esaltata funzione di compenso cardiorespiratorio ha come sintomi iniziali la dispnea da sforzo e le palpitazioni; l’esame obiettivo rileva polso molle e frequente, ipotensio-ne arteriosa, tachicardia, soffi funzionali a livello cardiaco e dei grossi vasi del collo, secondari all’aumentata velocità di circolo e alla ridotta viscosità del sangue. Con l’aggravarsi dell’anemia si arriva a una situazione di scompenso di circolo ad alta gittata, con ortopnea e dispnea a riposo, cardiomegalia, edemi importanti, talvolta ascite.
Le alterazioni elettrocardiografi che non sono patognomo-niche e possono riscontrarsi segni aspecifi ci di ischemia diffusa. Un altro meccanismo di compenso attivo in alcune ane-mie è costituito dall’iperplasia eritroide midollare secon-daria alla riduzione emoglobinica, da cui derivano ipossia renale e aumentata secrezione di eritropoietina (EPO). In caso di anemia di particolare gravità e di insorgenza precoce (come, per esempio, nelle anemie emolitiche congenite), l’espansione eritropoietica invade gli spazi midollari disponibili, può anche fuoriuscire dalla cavità midollare ed è causa di modifi cazioni patologiche ossee ed extraossee. Infi ne, nell’anemia si riscontra talvolta una diminuita resistenza alle infezioni, forse per un’alterata risposta immunitaria cellulo-mediata. In conclusione, va sottolineato che la probabilità di com-parsa della sintomatologia clinica dovuta ad anemia va considerata in modo dinamico, ossia come rapporto tra richieste di ossigeno e capacità di soddisfarle mediante i meccanismi di compenso. Di conseguenza, tranne che per gradi estremi di riduzione emoglobinica, si ha una grande variabilità individuale del quadro clinico, in dipendenza dalle modifi cazioni istantanee metaboliche e funzionali. In sostanza, l’attività fi sica è il fattore più importante nel determinare il fabbisogno totale di ossigeno, perché essa provoca aumenti signifi cativi nel metabolismo dei muscoli scheletrici e, in minor misura, del miocardio; pertanto, la sintomatologia può diventare manifesta solo in coincidenza di lavoro fi sico particolarmente intenso.
L’anemia aplastica è definita come una pancitopenia del sangue periferico con ipocellularità del midollo osseo, da difetto primitivo o secondario dei progenitori emato-poietici. Come si è visto dallo schema dell’emopoiesi, le cellule in grado di replicarsi sono le cellule staminali (CFU-L-M e CFU-S), quindi un loro pool adeguato e normalmente funzionante ha signifi cato critico per il mantenimento della normale crasi ematica. Queste cellule in condizioni normali si trovano per lo più in G 0 e funzionano da serba-toio di riserva per gli elementi ematici; quando si verifi ca una situazione che richiede un’esaltata emopoiesi, esse possono essere reclutate in ciclo e procedere verso l’una o l’altra delle due vie (autoreplicazione o differenziazione) insite nella loro potenzialità evolutiva. Una distruzione o un’alterazione delle cellule staminali, conseguente a danni di genesi diversa, ha come conse-guenza un progressivo impoverimento del tessuto paren-chimale midollare, perché gli elementi a valle delle CFU-S nel processo di differenziazione emopoietica, via via che si dividono e muoiono, non possono venire rimpiazzati. Si crea quindi una pancitopenia limitata alle serie di deriva-zione mieloide (eritrociti, granulociti e piastrine), perché i linfociti hanno capacità di replicarsi sino allo stadio di avanzata maturazione, quindi, il loro pool è relativamente
indipendente dal continuo rifornimento da parte delle cellule staminali. Il termine aplastic anemia (anemia aplastica) coniato dalla letteratura anglosassone ed entrato nell’uso potrebbe esse-re opportunamente sostituito da mielosi totale aplastica, secondo la scuola italiana, che meglio defi nisce l’aplasia a carico delle tre serie midollari; per mielosi parziale apla-stica si intende la citopenia a carico di una singola serie (o di due di esse). L’anemia aplastica può venire distinta in acquisita ( Tab. 48.1 ) e costituzionale .
Anemia aplastica F. Pellò
� Idiopatica � Secondaria a farmaci
– Chemioterapici – Agenti con meccanismo dose-dipendente – Agenti con meccanismo idiosincrasico
� Da agenti chimici e tossine � Da radiazioni � Da infezioni, in particolare da epatite � In rare altre circostanze (gravidanza; timoma)
Tabella 48.1 Anemia aplastica acquisita
C0240.indd 1041C0240.indd 1041 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1042
Eziopatogenesi Anemia aplastica acquisita 1. La forma idiopatica è così denominata perché
non si è trovato uno specifi co agente eziologico responsabile; sarebbe forse più esatto parlare di anemia aplastica a eziologia sconosciuta. Osservazioni cliniche e sperimentali sono a favore di un meccanismo immunologico responsabile della soppressione dell’emopoiesi. Dal punto di vista clinico conforta questa ipotesi l’osservazione che una quota notevole di trapianti di midollo tra fratelli singenici attuati per la terapia di un’anemia aplastica richiede il precondizionamento del ricevente con un’immunosoppressione per evitare il rigetto. Inoltre, in questa malattia sono stati ottenuti buoni risultati terapeutici con la globulina antitimocitica e le globuline antilinfociti del dotto toracico (che svolgono un’azione immunodepressiva). Nelle esperienze di cocoltura in cui linfociti di pazienti con anemia aplastica sono stati mescolati con cellule midollari normali istocompatibili, si osserva l’inibizione dello sviluppo di normali colonie da parte dei precursori emopoietici normali. Come in molti altri disordini autoimmuni, anche nella cosiddetta anemia aplastica idiopatica vi è, rispetto alla popolazione generale, un eccesso di un particolare antigene di istocompatibilità, l’HLA-DR 15.
2. L’ anemia aplastica da farmaci ha meccanismi e modi di insorgenza distinti. Nel caso di farmaci chemioterapici l’effetto mielosoppressivo è prevedibile ed è dose-dipendente; essi danneggiano le cellule bloccandone la replicazione e hanno azione non selettiva sulle cellule con intensa attività proliferativa, quindi anche sui progenitori e precursori ematopoietici; pertanto esercitano un’azione tossica sul midollo. Nel caso di farmaci non citostatici può esserci, in alcuni pazienti, un effetto citopenizzante dose-dipendente, che si verifi ca durante la somministrazione del farmaco, con ripristino della normale attività midollare alla sospensione. Un’azione di questo tipo si può avere, per esempio, con il CAF, la difenilidantoina, la cloropromazina, il tiouracile, la metilcillina. Pare che lo sviluppo di una soppressione eritroide reversibile sia dovuto a blocco della sintesi dell’eme; morfologicamente nell’anemia aplastica da CAF si ha spiccata vacuolizzazione del citoplasma eritroblastico. I farmaci non citostatici possono indurre anemia aplastica mediante un altro meccanismo, detto “idiosincrasico” o da reazione di ipersensibilità. L’aplasia, rara ma spesso fatale, si sviluppa settimane o mesi dopo l’inizio della terapia e per essa si sospetta una predisposizione genetica, sostenuta dal riscontro (nel caso del CAF e della difenilidantoina) di un’aumentata incidenza familiare. Lo stesso farmaco può dare aplasia sia dose-correlata sia con meccanismo idiosincrasico; le due modalità non si verifi cano nello stesso paziente.
3. Composti chimici . La maggior parte dei composti chimici in grado di provocare anemia aplastica contiene benzolo, presente in numerosi solventi, derivati dal carbone e prodotti del petrolio; anche
alcuni insetticidi sono veicolati da solventi a base di petrolio, da cui la possibilità di un duplice effetto lesivo sul midollo. Per il benzolo hanno soprattutto importanza le esposizioni lavorative negli agricoltori, nei lavoratori della pelle (calzolai), negli operai che usano collanti. L’incidenza di rischio ematologico aumenta con l’intensità e la durata dell’esposizione. Una curiosa possibilità, rara in Italia ma frequente in Gran Bretagna e negli Stati Uniti, è l’anemia aplastica dei “fi utatori di colla” (un’abitudine voluttuaria psicologicamente correlata con le tossicodipendenze). L’emopatia benzolica non si limita però all’aplasia; nella maggior parte dei casi l’azione lesiva a livello delle cellule staminali induce la formazione di precursori eritroidi difettosi, con atipie morfologiche (gigantismo cellulare, polinuclearità), incapaci di giungere a maturazione e integranti quindi un’eritropoiesi ineffi cace. L’alterazione qualitativa dei precursori predispone all’insorgenza di leucemia acuta non linfatica.
4. Radiazioni . L’esposizione a dosi massive di radiazioni, per esempio nei sopravvissuti alle esplosioni nucleari di Hiroshima e Nagasaki, ha determinato una quota rilevante di casi di anemia aplastica. Incidenti possono verifi carsi anche nei lavoratori addetti agli impianti nucleari; una ventina di anni fa alcuni fi sici jugoslavi investiti da notevole irradiazione per un errore nella lavorazione furono sottoposti al primo trapianto di midollo per anemia aplastica. Uno dei pazienti morì; negli altri, anche se il midollo non attecchì, durò un tempo suffi ciente a consentire al midollo residuo di riprendere a funzionare. I raggi Roentgen usati nella terapia antitumorale possono, per irradiazione estesa e protratta, determinare un danno alle cellule staminali e quindi anemia aplastica. Anche nei radiologi, negli anni dal 1948 al 1961, si è trovata negli Stati Uniti un’incidenza di anemia aplastica di 20 volte superiore alla popolazione controllo; attualmente, con le moderne attrezzature radiologiche, il rischio è notevolmente ridotto. Il danno indotto dalle radiazioni è a carico della replicazione del DNA, per cui le cellule staminali si dividono “a vuoto”, consumandosi; non si ha quindi un’azione lesiva diretta ma un esaurimento.
5. L’anemia aplastica è di riscontro occasionale o frequente in alcune infezioni virali (da virus epatitici, da virus di Epstein-Barr , da cytomegalovirus , da parvovirus , da virus dell’herpes zoster e varicella) , nella toxoplasmosi , nella brucellosi , raramente nella tubercolosi.
Di particolare importanza è l’anemia aplastica postepatitica , più spesso conseguente a epatite non A-non B, ma anche a epatite A o B; vi è preferenza per il sesso maschile e per l’età giovanile, sotto i 20 anni. Di solito l’anemia aplastica si riscontra entro 2 mesi dall’inizio dell’epatite, in pochi casi vi è stato un intervallo anche di mesi o anni.
6. Vengono segnalati pochi casi sicuri in cui l’anemia aplastica si è sviluppata in gravidanza ed è regredita dopo il parto; viene ipotizzato uno squilibrio tra l’azione promuovente dell’eritropoietina e l’effetto mielosoppressore degli estrogeni.
C0240.indd 1042C0240.indd 1042 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Capitolo 48 - ANEMIE
81043
Anemia aplastica costituzionale Il termine costituzio-nale indica una malattia congenita o familiare, cioè una predisposizione al defi cit midollare; lo sviluppo dell’apla-sia può essere innescato da alcuni agenti sopra ricordati (farmaci, virus, ecc.). L’anemia aplastica costituzionale comprende: • anemia di Fanconi; • anemia aplastica familiare; • discheratosi congenita; • sindrome di Shwachman-Diamond.
I pazienti con anemia di Fanconi mostrano alla nascita caratteristiche anomalie fi siche, con incidenza diversa a seconda dei casi, che includono: pollice assente o ipopla-stico, aplasia radiale, anomalie renali, ritardo mentale, microftalmia e microcefalia, strabismo, sordità, defi cit staturale. Le manifestazioni ematologiche e l’iperpigmen-tazione compaiono di solito verso i 5-10 anni e l’eredità è autosomica recessiva. Nelle famiglie di pazienti con anemia di Fanconi si possono ritrovare anomalie fi siche del tipo descritto, leucemie, tumori, diabete mellito, anche senza aplasia midollare. Il reperto di laboratorio più caratteristico è la rottura di cromosomi e il loro riarrangiamento osser-vati in colture di cellule midollari e di linfociti periferici, espressione di un defi cit nel ripristino del DNA. Le altre forme elencate sono di riscontro più raro; nella discheratosi congenita si hanno iperpigmentazione reti-colare, unghie distrofi che e leucoplachia delle mucose; può associarsi pancitopenia.
Manifestazioni cliniche I sintomi sono secondari alla pancitopenia, con anemia, piastrinopenia, granulocitopenia e diminuzione dei mo-nociti. L’entità dei sintomi dipende con buona correla-zione dal grado della citopenia; l’anemia aplastica grave è defi nita da parametri ematomidollari molto compromessi, con grave ipoplasia midollare: piastrine < 20 × 10 9 /L, gra-nulociti < 0,5 × 10 9 /L e reticolociti (con valore corretto per Hct) < 1%. Il quadro clinico varia a seconda della rapidità con la quale si manifesta la forma: forme acute, a rapida instaurazione, e forme croniche, a lenta instaurazione. Nelle forme acute prevalgono i sintomi conseguenti alla granulocitopenia (vita media dei granulociti: circa 1 giorno) e alla piastrino-penia (vita media delle piastrine: circa 9 giorni); l’anemia richiede molto più tempo per instaurarsi, data l’emivita dei GR di circa 120 giorni. La granulocitopenia molto spiccata predispone alle in-fezioni, che si sviluppano più facilmente nelle sedi ove si trovano organismi saprofi ti, come a livello dell’anello faringeo e dell’orifi zio anale; la manifestazione infettiva è frequentemente accompagnata da fenomeni ulcerativi. La piastrinopenia è causa di manifestazioni emorragiche, rappresentate da porpora, prevalente agli arti inferiori, alle mucose del cavo orale, alle congiuntive, al fondo dell’oc-chio. Si hanno inoltre ecchimosi, gengivorragie, epistassi, menorragie o metrorragie, ematuria e melena. Nelle forme croniche l’anemia ha il tempo di manifestarsi e domina quindi il quadro clinico, con i sintomi già descritti (da cui il nome di anemia aplastica, per la forma di pancitope-nia), mentre granulocitopenia e piastrinopenia di solito sono più lievi e non danno luogo a sintomi molto evidenti.
Esami di laboratorio L’esame emocromocitometrico dimostra un’anemia normo-cromica normocitica, raramente macrocitica, leucopenia con linfocitosi relativa, granulocitopenia e monocitopenia assolute, piastrinopenia. Lo striscio del sangue periferico dimostra la normocromia delle emazie con anisopoichilo-citosi nei limiti di norma. Non si osservano di regola forme immature in circolo né eritroidi né mieloidi; in sostanza lo striscio mostra l’aspetto del “sangue diluito”. Nella presunzione di anemia aplastica , l’esame del midollo va eseguito direttamente con la tecnica della biopsia con prelievo del cilindretto osteomidollare; l’esame istologico dimostra il reticolo dell’impalcatura stromale con maglie riempite principalmente da tessuto adiposo, alcuni linfociti e plasmacellule nei punti nodali ed estrema povertà della componente parenchimale. Nel caso dell’emopatia benzo-lica il quadro midollare è caratteristico, con eritroblasti di grandi dimensioni, qualche volta di tipo megaloblastico, frequentemente polinucleati; anche a carico dei precursori mieloidi si notano aumento di dimensioni, specie dei mielo-blasti e promielociti, ipergranulosità azzurrofi la e specifi ca, curva maturativa granuloblastica tendente all’inibizione. Poiché, come si è visto, l’esame del midollo è effettuato su un campione casuale di tessuto, è possibile che il prelievo cada su uno dei piccoli foci di midollo ipercellulare ancora presenti. In questo caso l’esame dà reperto di normalità e pone un quesito di diagnosi differenziale con l’anemia refrattaria; è opportuno allora eseguire, meglio se in ane-stesia generale, uno o più altri prelievi in sedi diverse, per confermare la presenza di anemia aplastica. Tra gli altri esami di laboratorio signifi cativamente alterati vanno segnalati la sideremia elevata, con saturazione quasi completa della transferrina, e le prove di emostasi ed emocoagulazione, che forniscono risultati abnormi in rapporto alla piastrinopenia.
Diagnosi La diagnosi differenziale deve tenere conto delle situazioni di pancitopenia periferica non accompagnate da ipocel-lularità midollare; questo si ha obbligatoriamente in caso di anemia mieloftisica , cioè di mielosostituzione da parte di elementi neoplastici delle serie ematologiche (leucemia acuta, mieloma multiplo) o anche, più raramente, da parte di elementi neoplastici metastatizzati nel midollo (linfo-mi, neoplasie epiteliali), in caso di anemia refrattaria con midollo iperplastico ed eccesso di mieloblasti. La pancitopenia periferica può verificarsi nell’anemia megaloblastica da carenza di vitamina B 12 o di acido foli-co, in alcuni rari casi di anemia sideropenica e di anemia sideroblastica . L’emoglobinuria parossistica notturna con midollo spes-so ipoplastico si differenzia dall’anemia aplastica per la caratteristica situazione emolitica. L’anemia aplastica va anche differenziata dalla pancito-penia da ipersplenismo , ossia da quella condizione di cospicua splenomegalia (da qualsiasi causa provocata) con sequestro degli elementi ematici circolanti; anche in questo caso l’esame del midollo è discriminante, in quanto esso si presenta polimorfo, con elementi morfo-logicamente normali e curve maturative inizialmente sti-molate, nel tentativo di sopperire alle perdite periferiche. A lungo andare, se non si interviene con la splenectomia,
C0240.indd 1043C0240.indd 1043 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1044
si determina una carenza di costituenti essenziali per l’eri-tropoiesi, in particolare folati e, infi ne, si può arrivare a una fase di esaurimento midollare, in genere transitoria, ma che può anche esitare in vera aplasia.
Decorso e prognosi Sono differenti per i diversi tipi di anemia aplastica; le forme secondarie a farmaci citostatici o agli altri farmaci ad azione dose-dipendente regrediscono di solito alla so-spensione della sostanza responsabile. In caso di citopenia da meccanismo idiosincrasico la regressione non è obbliga-toria e può anche instaurarsi un’aplasia defi nitiva. L’aplasia postepatitica è molto grave, la remissione spontanea è assai rara e la mortalità a 1 anno è intorno al 90%. La forma acquisita idiopatica ha decorso diverso in funzio-ne di alcuni fattori prognostici; sono da considerare sfavore-voli il sesso maschile, la rapidità di comparsa dei sintomi, le manifestazioni emorragiche precoci, il quadro periferico di “anemia aplastica grave”. In alcuni pazienti l’anemia apla-stica sembra essere associata allo sviluppo di cloni abnormi; l’anomalia più comune, probabilmente di natura clonale, è lo sviluppo di una popolazione di cellule che presenta le caratteristiche dell’emoglobinuria parossistica notturna. La trasformazione in leucemia acuta, osservata nel 5-10% dei casi di anemia aplastica a oltre 2 anni dalla diagnosi, sia per la forma acquisita sia per quella congenita, è probabilmente espressione di una seconda malattia clonale.
Terapia Nell’anemia aplastica secondaria a farmaci, a espo-sizione a tossici o a raggi X, il primo provvedimen-to è la sospensione del farmaco o l’allontanamento dall’agente responsabile. La terapia dell’aplasia stabilizzata, sia congenita sia acquisita, primitiva o secondaria, prende in conside-razione tre tipi di intervento: • stimolazione con androgeni; • terapia immunosoppressiva con globulina anti-
linfociti T, con ciclosporina, con farmaci immu-nosoppressori;
• trapianto di midollo.
La terapia con androgeni è basata sul loro effetto sti-molante la produzione di eritropoietina e l’induzione in ciclo delle cellule staminali, con sintesi di DNA; in questo modo si ha un maggior numero di cellule EPO-sensibili sottoposte a una più intensa attività eritropoietinica. È ovvio che questo tipo di effetto richiede una residua presenza di progenitori dispo-nibili, e quindi gli androgeni trovano indicazione nell’anemia aplastica non di estrema gravità. La terapia con globulina antilinfocitaria, associata o meno a farmaci immunosoppressori come la ciclofo-sfamide, è utilizzata nell’anemia aplastica acquisita idiopatica in cui è ipotizzato un danno alle cellule sta-minali mediato dai linfociti T; nell’aplasia eritroblasti-ca pura associata a timoma va eseguita l’asportazione del timoma e, successivamente, può essere impiegata la ciclofosfamide. Nei casi non associati a timoma, a
sospetta patogenesi autoimmunitaria, trova indicazio-ne teorica l’uso della terapia immunosoppressiva. Il trapianto di midollo, da fratello HLA identico, è la terapia di elezione dell’anemia aplastica grave, in età giovanile, in particolare dell’aplasia postepatitica.
Aplasia pura della serie eritroide (o eritroblastopenia pura)
Nel midollo può verificarsi una situazione di aplasia elettiva del compartimento eritroide, ossia una mielosi parziale aplastica, acquisita o congenita, che ha come conseguenza anemia, non accompagnata da granuloci-topenia e piastrinopenia. L’aplasia eritroide pura acquisita ha andamento cronico, si verifi ca negli adulti, molto spesso associata a timoma; si ipotizza una patogenesi autoimmune con meccanismo operante la distruzione intramidollare dei precursori eri-tropoietici e/o l’inibizione della normale differenziazione eritropoietica. Nel siero dei pazienti si sono dimostrati: • un anticorpo citotossico complemento-dipendente
che lisa elettivamente gli eritroblasti midollari; • un anticorpo o immunoglobuline (Ig) G anti-EPO; • un anticorpo che interferisce con il legame
dell’eritropoietina con i precursori eritroidi e quindi inibisce la differenziazione delle BFU-E.
L’aplasia eritroide pura acquisita , di solito transitoria, può verifi carsi in corso di anemia emolitica cronica , di lupus eritematoso sistemico , durante la terapia con alcuni far-maci (per esempio, aminopirina, CAF, clorpropamide, fenilbutazone, isoniazide ecc.), con meccanismo per lo più dose-dipendente. Una forma particolare è stata osservata in pazienti con anemia da insuffi cienza renale cronica che assumono eritropoietina a scopo terapeutico. In alcuni di essi si è osservato un aggravamento dell’anemia dovuto ad aplasia pura della serie rossa, a causa dello sviluppo di anticorpi neutralizzanti ad alta affi nità diretti contro l’EPO. Questa sostanza, impiegata a scopi terapeutici, è di natura ricombinante e differisce da quella endogena per la sua componente carboidrata. Tuttavia, gli anticorpi che si possono produrre in questo caso non sono diretti contro carboidrati che entrano nella molecola dell’EPO, ma verosimilmente contro un epitopo conformazionale della proteina. Le conseguenze di questa reazione immu-nitaria sono molto negative, perché gli anticorpi stimolati dall’eritropoietina ricombinante reagiscono anche contro l’eritropoietina endogena, determinando un’aplasia pu-ra della serie rossa. Non sono descritti casi simili negli sportivi, soprattutto ciclisti, che assumono eritropoietina come una forma di doping, ma questo evento avverso è teoricamente possibile. L’ aplasia eritroide congenita o anemia di Diamond-Blackfan è una malattia rara; è accompagnata da lievi anomalie fi siche, del tipo di quelle descritte per l’anemia di Fanconi. Il tipo di trasmissione ereditaria è autosomico recessivo; i livelli di eritropoietina sono elevati, probabilmente vi è un defi cit a livello delle BFU-E, incapaci di rispondere al normale stimolo maturativo.
C0240.indd 1044C0240.indd 1044 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Capitolo 48 - ANEMIE
81045
Viene defi nita megaloblastica un’anemia caratterizzata da presenza di eritroblasti anomali, detti megaloblasti, nel midollo e da macrovalociti nel sangue periferico. Essa è determinata da un difetto nella sintesi di DNA conseguen-te a carenza di vitamina B 12 e/o folati. L’alterazione biochi-mica alla base (difettosa sintesi di DNA) infl uenza tutte le cellule provviste di elevata attività proliferativa, tra le qua-li i precursori emopoietici hanno un posto preminente.
Fisiologia Per spiegare la genesi della megaloblastosi, che di per sé non è necessariamente accompagnata da anemia, non si può prescindere da una breve premessa sulle modalità di sintesi del DNA e sulle principali caratteristiche della vitamina B 12 e dell’acido folico, che in essa hanno impor-tanza essenziale. Per una migliore comprensione del discorso che segue, si riporta nella tabella 48.2 la nomenclatura dei costituenti degli acidi nucleici. Com’è noto, i costituenti del DNA differiscono da quelli del RNA (acido ribonucleico) per due caratteristiche: sono deossiribosidi anziché ribosidi e comprendono la timina in luogo dell’uracile. La timina differisce dall’uracile perché ha un gruppo metili-co in più; perciò la metilazione dell’uracile, e la sua conver-sione in timina costituiscono un passo fondamentale per la sintesi del DNA, che è essenziale per la divisione cellulare. In realtà la metilazione non avviene direttamente a livello dell’uracile, ma nel corso della seguente successione:
(1) Uridina → (2) Deossi-uridina → (3) Deossi-uridilato → (4) Timidilato → DNA
La metilazione avviene nel passaggio da (3) a (4), a opera dell’enzima timidilato-sintetasi. Il deossi-uridilato si tra-sforma in timidilato ricevendo un metile da un coenzima folico, il 5-10 metilene-tetraidrofolato (5-10 metilene THF), che, come si vedrà per i coenzini folici, ha una notevole capacità di trasferire unità monocarboniose. Dopo cessione della unità monocarboniosa, il 5-10 metilene THF è trasfor-mato in diidrofolato, una forma che necessita l’acquisizione
di altri due atomi di idrogeno, a opera dell’enzima diidro-folato-reduttasi, per convertirsi in tetraidrofolato (THF, Tetrahydrofuran) ed essere così in grado di rigenerare il 5-10 metilene THF, assumendo gruppi metilenici dalla serina che si trasforma in glicina ( Fig. 48.1 ). La sintesi del DNA richiede quindi che ci sia a disposizione una quantità suffi ciente di acido THF; esso però non si trova nei depositi come tale, ma come coenzima folico N5-metil-tetraidrofolato, che per trasformarsi in acido THF cede un gruppo metilico alla vitamina B 12 (questa da cobalamina diventa metilcobalamina, funzionando da accettore di metile), la quale a sua volta lo trasferisce all’omocisteina che si trasforma in metionina (reazione ca-talizzata dall’omocisteina-metionina metil-transferasi). In caso di deficit di vitamina B 12 , non può verificarsi quest’ultima reazione di trasferimento del gruppo metilico e si ha quindi “intrappolamento del metil-tetraidrofolato”; il suo accumulo riduce il pool disponibile dei coenzimi folici, ivi inclusi l’acido THF e il 5-10 metilene-tetraidrofo-lato, necessario, come si è visto, per la sintesi del DNA.
Vitamina B 12 La molecola consiste di due parti principali, un nucleotide (5,6-dimetil-benzimidazolo) e un anello cor-rinico (gruppo planare) che assomiglia alla porfi rina. L’anel-lo corrinico contiene al centro un atomo di cobalto. Nell’uomo si trovano due coenzimi principali della vi-tamina B 12 : • la deossi-adenosil-cobalamina, presente
principalmente nel fegato, in cui il gruppo anionico legato al cobalto è il 59-deossi-adenosile; è il coenzima richiesto per la conversione del metilmalonato coenzima A in succinil-coenzima A. La sua carenza porta ad aumento dell’escrezione di metilmalonato ;
• la metil-cobalamina, presente nel plasma, in cui il gruppo anionico è il metile; è il coenzima implicato nelle reazioni con i folati per la sintesi di DNA, come si è già visto. La vitamina B 12 catalizza anche la riduzione dei ribosidi, contenuti nell’RNA a desossi-ribosidi, contenuti nel DNA.
Anemie megaloblastiche F. Pellò
Acido nucleico
Base Nucleoside Nucleotide
RNA Adenina 1 Adenosina AdenilatoGuanina 1 Guanosina GuanilatoCitosina 2 Citidina CitidilatoUracile 2 Uridina Uridilato
DNA Adenina 1 Deossi-adenosina Deossi-adenilatoGuanina 1 Deossi-guanosina Deossi-guanilatoCitosina 2 Deossi-citidina Deossi-citidilatoTimina 2 Timidina Timidilato
1 Basi puriniche. 2 Basi pirimidiniche.
Acido Base Nucleoside Nucleotide
Tabella 48.2 Nomenclatura degli acidi nucleici
DNA
Deossi-uridilato
Timidilato
TIMIDILATO-SINTETASI
Diidrofolato
5-10-metilene-THF
THF
Serina
Glicina
5-metil-THF Deossi-uridina
OmocisteinaVitamina B12
Metionina
Diidrofolato-reduttasi
Figura 48.1 Sintesi del DNA a partire dal timidilato. Ruolo dei coenzimi folici e della vitamina B 12 .
C0240.indd 1045C0240.indd 1045 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1046
Le preparazioni in commercio sono l’idrossicobalamina, in cui il gruppo anionico è l’ossidrile, e la cianocobalami-na, con il gruppo -CN legato al cobalto. La vitamina B 12 si trova negli alimenti di origine animale ed è resistente alla cottura; una dieta media quotidiana ne contiene da 5 a 30 g, di cui solo una piccola parte viene assorbita attraverso un’opportuna mediazione rappresen-tata dal fattore intrinseco. Il fattore intrinseco è una glicoproteina costituita da due catene polipeptidiche, prodotta dalle cellule parietali del corpo e del fondo gastrico. La vitamina B 12 si lega a esso molto avidamente, formando un complesso che viene assorbito a livello dell’ileo, ove la membrana dei micro-villi delle cellule della mucosa ha recettori con un’elevata affi nità per questo complesso. Dopo l’assorbimento la vitamina B 12 viene staccata dal fattore intrinseco, pare a livello dell’orletto a spazzola della mucosa, ed è veicolata nel sangue dalle transcobalamine. Le transcobalamine sono indicate con i numeri I, II e III. La I e la III sono assai simili, rappresentando frazioni diverse della cobalofi llina, una glicoproteina presente nei granu-lociti normali e di leucemia mieloide cronica, nella saliva, nel latte, nel liquido amniotico, nella bile; rappresenta in un certo senso una forma di deposito, perché lega piuttosto intensamente la vitamina B 12 . Il tempo di dimezzamento della vitamina a essa legata è di circa 9-10 giorni. Il contenuto totale di vitamina B 12 nell’organismo è di 2-5 mg, di cui circa la metà è contenuta nel fegato. Dal fegato una certa quota è eliminata attraverso la bile e recuperata completamente mediante riassorbimento con il fattore intrinseco (circolo enteroepatico). Il fabbisogno giornaliero nell’adulto è inferiore a 1 g. La transcobalamina II ha un peso molecolare di circa 30.000, non è una glicoproteina, lega la vitamina B 12 con scarsa ener-gia, il suo tempo di dimezzamento è circa di un giorno e co-stituisce il veicolo che somministra la vitamina B 12 ai tessuti. Nel siero il valore normale di vitamina B 12 è 160-900 ng/L.
Acido folico (o acido pteroilglutamico) La molecola è costituita da tre parti: pteridina, acido p-amino-benzoico e acido glutamico ( Fig. 48.2 ). Questo acido è contenuto principalmente negli alimenti vegetali, nel fegato crudo, nel lievito, sotto forma di poliglutammati, costituiti da catene di sette o più residui di acido glutamico attaccati l’uno all’altro.
Notevoli quantità di folato possono essere assorbite dai poliglutammati della dieta, principalmente a livello del digiuno prossimale, previa idrolisi a opera di un enzima de-nominato “coniugasi”, presente nella mucosa intestinale. L’acido pteroilglutamico non si trova però come tale nei tes-suti in quantità signifi cativa e non è biochimicamente atti-vo; esso è il capostipite di una serie di composti (coenzimi folici), ottenuti da reazioni di riduzione (diidro o tetraidro) per aggiunta di idrogeni nelle posizioni 5, 6, 7, 8 dell’anello pteridinico e per sostituzione di un C in posizione N5 e/o N10 con gruppi chimici contenenti un solo atomo di car-bonio (formilici, metenilici, formiminici e metilici [uni-tà monocarboniose]). La riduzione enzimatica dell’acido pteroilglutamico è catalizzata dalla diidrofolico-reduttasi. L’acido folinico è l’acido 5-formil-tetraidrofolico. Il deposito normale di folati è dell’ordine di 5-10 mg; il fegato ne contiene una gran parte sotto forma di 5-metil-tetraidrofolato, che sembra essere la forma principale di folato normalmente circolante. Il fabbisogno quotidiano di folati per un adulto è di 100-200 g, la concentrazione di acido folico nel siero è di 10 g/L, mentre la concentrazione nei GR è 30 volte tanto: 300 g/L. La vitamina B 12 è di fondamentale importanza per permettere l’ingresso dell’acido folico nelle cellule; in caso di carenza di B 12 si ha aumento del rapporto acido fo-lico siero/acido folico GR. I coenzimi folici sono implicati in tutte le reazioni in cui vi è un trasferimento di un’unità monocarboniosa; esse includono: • sintesi delle purine; • biosintesi della pirimidina (mediante metilazione
dell’acido deossi-uridilico ad acido timidilico); • tre conversioni di aminoacidi: della serina a glicina,
dell’istidina ad acido glutamico, dell’omocisteina a metionina, che richiede la vitamina B 12 .
Eziopatogenesi In linea teorica una carenza può determinarsi per: • insuffi ciente introduzione, per ridotto apporto
con la dieta; • malassorbimento; • aumentato fabbisogno, per aumento delle richieste
in situazioni fi siologiche o patologiche; • inadeguata utilizzazione.
Verranno prese in considerazione separatamente le cause di defi cit di vitamina B 12 e di acido folico, anche se molte malattie accompagnate da malassorbimento sono in grado di provocare una carenza di entrambe le vitamine.
Cause di carenza di vitamina B 12 Il defi cit di apporto nutritivo è un’evenienza che non si verifi ca in pratica, date l’esiguità del fabbisogno giornaliero (meno di 1 � g), l’entità delle scorte dell’organismo (circa 5 mg) e la ridot-ta eliminazione (circa lo 0,1% giornaliero della quantità totale presente); occorrerebbero molti anni di dieta vege-tariana per esaurire le scorte. Il malassorbimento è la causa di gran lunga prevalente di carenza di vitamina B 12 . Esso può verifi carsi in una serie di condizioni morbose e precisamente: • in caso di alterazioni quantitative o qualitative del
fattore intrinseco: anemia perniciosa, gastrectomia; • nelle malattie intestinali accompagnate
da malassorbimento (sprue tropicale, malattia celiaca
Pteridina
Acido para-aminobenzoico
Acido glutamico
NH2
NH NH10
O
C CH
COOH
COOH
CH2
CH2C1
OH
N
N
N
N
Figura 48.2 Molecola di acido
folico. Gli atomi segnati con pallini
neri legano gli atomi di idrogeno nella
conversione a THF. Le linee tratteggiate
indicano gli atomi che possono
legare le unità monocarboniose C1.
C0240.indd 1046C0240.indd 1046 6/9/10 3:08:57 PM6/9/10 3:08:57 PM

Capitolo 48 - ANEMIE
81047
dell’adulto ecc.) o in alterazioni intestinali anatomiche (ileite terminale , resezione ileale, sindrome dell’ansa cieca, fi stole e anastomosi intestinali);
• per consumo della vitamina B 12 da parte di microrganismi o parassiti intestinali (come avviene, per esempio, nelle infestazioni da botriocefalo).
Esistono condizioni di malassorbimento congenito della vitamina B 12 , come nella sindrome di Imerslund-Gräsbeck , che si accompagna a proteinuria, oppure in casi di fattore intrinseco abnorme o assente e, infi ne, in caso di altera-zioni nel trasporto e immagazzinamento della vitamina B 12 per defi cit di transcobalamina I e II. Si è già detto come il punto debole nell’assorbimento della vitamina B 12 sia la disponibilità del fattore intrinseco; se esso manca, la vitamina B 12 alle dosi fisiologiche non viene assorbita (in grandi quantità è possibile che una certa quota diffonda attraverso le pareti della mucosa); in assenza di fattore intrinseco non viene neppure assorbita la vitamina che esce dal fegato, per cui si interrompe il circolo enteroepatico. A ciò consegue un aumento di escrezione, il fabbisogno quotidiano si sposta da 1 a 3 � g e le scorte si possono esaurire in tempo minore. L’ anemia perniciosa è l’anemia megaloblastica che si accompa-gna a gastrite cronica atrofi ca , con secrezione di fattore intrin-seco ridotta o assente e malassorbimento di vitamina B 12 . Non è completamente chiarita la genesi dell’atrofi a ga-strica, anche se nei pazienti con anemia perniciosa si sono trovati anticorpi anticellule parietali , con percentuali dell’85-90% nel siero, del 75% nel succo gastrico e del 60% nelle plasmacellule della mucosa gastrica; questi reperti hanno signifi cato diagnostico, ma non sono sicuramente indicativi della patogenesi autoimmune della malattia. Anche nella gastrite cronica apparentemente idiopatica, senza anemia perniciosa, l’anticorpo anticellula parietale è presente nel 30-60% dei casi. Un secondo tipo di anticorpi diretti contro il fattore intrinseco è stato trovato in un numero minore di pazienti con ane-mia perniciosa. Gli anticorpi antifattore intrinseco sono di due tipi; il tipo I (anticorpo bloccante) blocca il sito combinatorio del fattore intrinseco per la vitamina B 12 , mentre il tipo II (anticorpo legante) si lega al complesso fattore intrinseco �vitamina B 12 . L’anticorpo di tipo I è stato dimostrato nel 75% dei sieri di anemia perniciosa, l’anticorpo di tipo II in circa il 45% dei casi e di solito sempre in presenza anche dell’anticorpo di tipo I. Anticorpi di tipo I e II sono stati dimostrati nel succo gastrico e di tipo II nelle plasmacellule della mucosa gastrica. Nel siero predominano anticorpi di tipo IgG, nel succo gastrico di tipo IgA. L’ipotesi, ritenuta assai probabile, di una patogenesi autoim-mune dell’anemia perniciosa è sostenuta anche da considera-zioni di ordine clinico; per esempio, nello stesso paziente può coesistere un’altra malattia a patogenesi sicuramente (tiroidite di Hashimoto ) o presumibilmente autoimmune (malattia di Addison , ipoparatiroidismo , vitiligine ). Anche nei parenti di pazienti con anemia perniciosa è stata rilevata un’aumentata incidenza nel siero di anticorpi anticellule parietali.
Cause di carenza di acido folico L’ insuffi ciente apporto dietetico si verifi ca, per esempio, nelle persone anziane, negli alcolisti, nelle persone che assumono una dieta quasi priva di frutta e verdure fresche.
Il malassorbimento è una causa molto frequente e si ritro-vano qui alcune delle malattie già citate per il malassorbi-mento di vitamina B 12 (sprue tropicale, malattia celiaca) e in genere le malattie appartenenti al vasto capitolo della patologia dell’intestino tenue, comprendenti defi cit di secrezioni esocrine e malattie primitive della parete inte-stinale. Alcuni farmaci anticonvulsivanti (difenilidantoi-na, primidone e barbiturati) ostacolano l’assorbimento dei poliglutammati per inibizione della coniugasi inte-stinale. La carenza di acido folico da malassorbimento è evenienza più frequente della carenza di vitamina B 12 , perché le scorte di acido folico si esauriscono in un tempo relativamente breve (circa un mese), mentre le scorte di vitamina B 12 richiedono anni per esaurirsi; nelle sindromi da malassorbimento si potrà avere inizialmente anemia megaloblastica da carenza di acido folico e solo successi-vamente, dopo 1 tempo relativamente lungo, anche da carenza di vitamina B 12 . L’ aumentato fabbisogno ha luogo in tutte le condizioni in cui nell’organismo vi è una rapida proliferazione cel-lulare; ciò può verifi carsi in condizioni fi siologiche in gravidanza, perché l’acido folico è necessario per la sintesi del DNA dell’embrione e del feto, durante l’allattamento e nei bambini prematuri. In condizioni patologiche, un aumentato fabbisogno si ha nelle neoplasie caratterizzate da rapido accrescimento cellulare; il prototipo è rappre-sentato dalle leucemie . Nelle anemie emolitiche, in cui la notevole riduzione del ciclo vitale degli eritrociti che vanno incontro a rapida distruzione determina il reclu-tamento di cellule di riserva dal midollo che aumenta di 3-4 volte la sua attività eritropoietica, si ha esaltata attività sintetica di DNA e, quindi, maggior fabbisogno di folati; se questi non vengono riforniti in quantità suffi ciente, al meccanismo emolitico si somma la carenza di folati come causa di anemia. In alcune malattie infi ammatorie (artrite reumatoide , malattia di Crohn , dermatite esfoliativa ) e metaboliche (tireotossicosi , omocistinuria ) in cui si ha probabilmente un aumentato turnover di cellule, come leucociti, cellule cutanee, può crearsi, anche se non abi-tualmente, una carenza di folati. L’ inadeguata utilizzazione è tipica nel defi cit di vitamina B 12 che, come si è visto, serve ad estrarre il folato dalla trappola del metilfolato; un elevatissimo apporto di acido folico potrebbe prescindere dalla presenza di vitamina B 12 , la quale resta però sempre il fattore essenziale in caso di apporto fi siologico di folati. Un ridotto utilizzo si ha anche in corso di terapia con farmaci antifolici, somministrati intenzionalmente come chemioterapici (methotrexate), che inibiscono la diidrofolico-reduttasi, enzima che converte il diidrofolato in tetraidrofolato. Azione analoga possiedono altri farmaci (per esempio, pirimetamina, trimetoprim) come effetto collaterale in-desiderato. L’anemia megaloblastica si verifi ca, al di fuori delle si-tuazioni di carenza di vitamina B 12 e acido folico, in altre condizioni iatrogene o spontanee che hanno come con-seguenza un’alterata sintesi di DNA. Si ricorda, in primo luogo, la somministrazione, nel corso di chemioterapia antitumorale, di farmaci classifi cati come “antimetaboliti” e precisamente di antagonisti della sintesi purinica (6-mer-captopurina, azatioprina) e pirimidinica (5-fl uorouracile, 6-azauridina) e di altri, come, per esempio, la procarbazina e l’idrossiurea, tutti inibitori della sintesi del DNA.
C0240.indd 1047C0240.indd 1047 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1048
Esistono, infi ne, alcune sindromi di carenza enzimatica, come la sindrome di Lesch-Nyhan , da defi cit dell’enzima ipoxantina-guanina transferasi, implicato nella sintesi delle purine, e l’orotico-aciduria , in cui il defi cit enzi-matico si situa sulla via sintetica dell’uridina, nucleotide pirimidinico.
Fisiopatologia Nell’anemia megaloblastica, la sintesi di RNA avviene normalmente, mentre il processo di sintesi del DNA, che precede ogni divisione mitotica, avviene con difficol-tà. L’intervallo di tempo che passa tra una replicazione cellulare e l’altra, per le cellule in rapida replicazione, è allungato; si ha quindi una produzione normale di RNA e una maggiore quantità di DNA nel nucleo. In un midollo megaloblastico numerose cellule hanno una quantità intermedia di DNA tra diploide e tetraploide. Queste (megaloblasti) appariranno più grosse, a livello sia del nucleo sia del citoplasma, perché la fase di prolungata sintesi nel ciclo cellulare permette la formazione in eccesso di alcuni composti citoplasmatici, inclusa l’emoglobina che blocca la successiva replicazione, e quindi passerà più tempo tra una divisione cellulare e l’altra. Questo vale per tutte le cellule in rapida replicazione, in particolare per i precursori eritropoietici; così, mentre negli eritroblasti normali (normoblasti) il nucleo tende a rimpicciolirsi con l’evoluzione verso la cellula matura ed è tondo e compatto, nei megaloblasti il nucleo è grosso, con reticolo cromatini-co fi ne e lasso. Queste cellule spesso non arrivano neppure a dividersi, morendo nel midollo senza riuscire a maturare e realizzando così un’eritropoiesi ineffi cace. Poiché la diffi coltà di sintesi del DNA ha luogo in tutte le cellule, pur essendo l’anemia il fenomeno principale, sono presenti granulocitopenia e piastrinopenia e si ha sofferenza anche a livello delle cellule di tessuti in rapida proliferazio-ne, non dei precursori emopoietici. Ciò vale principalmente per le cellule delle mucose delle vie digerenti, del naso, dei bronchi, delle vie urinarie e genitali. Nella carenza di vitamina B 12 esiste una manifestazione patologica ulteriore (che non si ha nel defi cit di acido folico), determinata da un’alterazione nella sintesi della mielina. Si è visto che il processo di isomerizzazione del metil-malonil-coenzima A in succinilcoenzima A sia mediato dalla deossi-adenosil-cobalamina. Inoltre, il metil-malonil-coenzima A deriva dall’acido propionico, il quale a sua volta deriva dal meta-bolismo di molti aminoacidi; quindi, questa sostanza è pre-sente nell’organismo in discreta quantità e per poter essere metabolizzata completamente necessita della conversione a succinil-coenzima A da parte della vitamina B 12 . In carenza della vitamina si determina accumulo di tutti i metaboliti a monte, con incorporazione di acidi grassi anomali nella molecola della mielina. Da ciò deriva una sintomatologia neurologica caratteristica, che interessa soprattutto i cor-doni spinali e specialmente quelli posteriori.
Manifestazioni cliniche L’ anamnesi familiare è importante, in quanto possono essere riscontrate forme di anemia non necessariamente macrocitica, ma anche aplastica sideropenica o emolitica ; endocrinopatie a patogenesi autoimmune (si sono già ricordati la tiroidite , alcune forme particolari di diabete , l’ipoparatiroidismo , la malattia di Addison ), più raramente cirrosi biliare primitiva e ipo- � -globulinemia acquisita .
L’anamnesi fi siologica prenderà in considerazione l’even-tualità di una gravidanza o di un allattamento in epoca recente, le abitudini alimentari (dieta povera di verdura e frutta fresca, abuso di alcolici), la presenza di diarrea, l’as-sunzione di farmaci (antimetaboliti, anticonvulsivanti). Nell’anamnesi patologica remota verranno indagate in modo particolare malattie di tipo immunitario o a pa-togenesi autoimmune; forme precedenti di anemia (per esempio, anemia sideropenica), interventi chirurgici (di gastroresezione, di resezione ileale), malattie intestinali accompagnate da sindrome da malassorbimento. I sintomi che portano il paziente dal medico sono di tipo aspecifi co, comuni alle altre forme di anemia e anche ad altre malattie non accompagnate da anemia; il paziente accusa astenia ingravescente, sonnolenza, aumentata sensibilità al freddo, sensazione di peso epigastrico e di ral-lentamento della digestione. Solamente in caso di defi cit di vitamina B 12 si potrà avere una sindrome neurologica, con parestesie alle mani e ai piedi, diffi coltà a mantenere la stazione eretta e alla deambulazione, più raramente diffi coltà all’uso delle mani; possono aversi anche paresi spastica e disturbi visivi da neurite retrobulbare. È op-portuno ricordare che questa sindrome neurologica può essere riscontrata anche in pazienti che non presentano anemia; si tratta di pazienti in cui l’anemia megalobla-stica da carenza di vitamina B 12 è stata trattata con dosi elevate di acido folico. Come si è visto dallo schema di interreazione metabolica tra vitamina B 12 e acido folico, questo a dosi elevate può sopperire alla mancanza relativa di B 12 e innescare ugualmente la sintesi del timidilato; tuttavia, la conversione del metil-malonil-coenzima A in succinil-coenzima A richiede necessariamente la pre-senza di vitamina B 12 ; in sua assenza si ha accumulo del metil-malonato, ritenuto responsabile delle alterazioni neurologiche.
Esame obiettivo L’esame obiettivo mette in evidenza cute pallida con sfu-matura giallastra (colore “cera vecchia”), la lingua appare arrossata e lucida (glossite di Hunter ) e ha perso il normale aspetto vellutato, perché le papille sono spianate per di-fetto di rigenerazione cellulare. Sarà inoltre possibile rilevare una modesta epatosple-nomegalia per l’aumentata attività del sistema reticolo-istiocitario, secondaria all’aumento dell’eritrocateresi.
Esami di laboratorio • Quadro ematologico : l’ esame emocromocitometrico
dimostra una diminuzione del contenuto di emoglobina di entità minore rispetto alla diminuzione dei GR; una diminuzione del valore dell’ematocrito, un aumento del volume corpuscolare medio e del contenuto corpuscolare medio di emoglobina mentre la concentrazione di emoglobina corpuscolare media è normale. Non si ha ipercromia, solamente i GR sono più grandi e quindi contengono più emoglobina, ma la sua concentrazione per ogni unità di volume di GR è normale. Si ha anemia perché è notevolmente diminuito il numero dei GR. L’ esame dello striscio del sangue periferico è abbastanza patognomonico, in quanto dimostra la presenza di macrociti, talvolta a forma ovoidale (macrovalociti), policromasia
C0240.indd 1048C0240.indd 1048 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Capitolo 48 - ANEMIE
81049
diffusa o punteggiatura basofi la, eccezionalmente rari megaloblasti policromatofi li o ortocromatici. I granulociti hanno nucleo ipersegmentato; l’esistenza di tre elementi presentanti cinque lobi o anche di un solo elemento con sei lobi permette di diagnosticare quasi sicuramente un’anemia megaloblastica. Vi sono inoltre discreta leucopenia con neutropenia e modesta piastrinopenia. Solamente con l’ esame del midollo, però, è possibile documentare con certezza l’anemia megaloblastica ; nei casi tipici e conclamati all’esame a piccolo ingrandimento, si osservano una straordinaria ricchezza di elementi cellulari e una prevalenza di voluminosi elementi a citoplasma basofi lo (cosiddetto midollo blu). L’osservazione a forte ingrandimento dimostra il reperto più caratteristico, la presenza di megaloblasti, in numero variabile ma sempre considerevole, non solo in fase basofi la, ma anche policromatofi la e perfi no ortocromatica. Si tratta di elementi cellulari di grandi dimensioni, con sviluppo asincrono del nucleo e del citoplasma, con nucleo di aspetto lasso, reticolato, a cromatina delicata, citoplasma ampio, che in fase basofi la ha una basofi lia disomogenea, con fenomeno di disbasofi lismo, consistente in zone di citoplasma a sfumatura grigio rosata commiste alle zone decisamente basofi le. Come si è già detto, l’asincronismo maturativo nucleo-citoplasmatico è anch’esso conseguenza del difetto di sintesi del DNA. Va tenuto presente che la megaloblastosi non è un fenomeno “tutto o nulla”, per cui, nel midollo, accanto ad elementi francamente megaloblastici si trovano una quota di elementi con aspetto intermedio tra megaloblasti e normoblasti e anche una discreta quota di elementi francamente normoblastici; evidentemente il difetto sintetico del DNA responsabile della morfologia megaloblastica non colpisce uniformemente tutti gli elementi. Infi ne, occorre sottolineare come una dose anche piccola di vitamina B 12 , insuffi ciente per svolgere un’azione terapeutica completa, sia in grado di provocare a livello del midollo, nel volgere di poche ore, modifi cazioni della morfologia dei megaloblasti, che tendono ad assumere l’aspetto dei normoblasti; si possono quindi incontrare diffi coltà diagnostiche anche considerevoli, per cui è molto importante che il paziente esegua gli esami prima di qualsiasi intervento terapeutico e che comunque venga fatta un’anamnesi accurata per accertare se sia stata eseguita in precedenza una terapia per l’anemia cosiddetta “ricostituente”, di solito a base di estratto epatico e vitamina B 12 . Anche a carico della serie granuloblastica si osservano modifi cazioni caratteristiche; i promielociti e specialmente i mielociti presentano dimensioni più grandi che di norma e nucleo con aspetto bizzarro (“a tronco d’albero”, “a gozzo”), per cui viene a mancare quasi completamente il normale stadio a nucleo rotondo e dal promielocito si passa quasi direttamente, attraverso queste forme bizzarre, al granulocito gigante ipersegmentato, ormai defi nitivamente maturo. L’alterazione della serie granuloblastica risente in modo minore di eventuali piccole dosi di vitamina B 12 somministrate in precedenza e
costituisce spesso un dato di utile orientamento diagnostico nei casi in cui la morfologia della serie eritroide sia meno tipica, perché parzialmente modifi cata da una terapia intempestiva e inadeguata.
• Altri esami : nel siero è presente spesso lieve iperbilirubinemia, di tipo indiretto, espressione di un’accentuata attività emolitica, secondaria all’eritropoiesi ineffi cace. Nell’anemia perniciosa l’esame del succo gastrico, in condizioni basali, dimostra achilia , che rimane invariata dopo stimolazione con istamina o pentagastrina. Nei casi in cui vi sia sindrome da malassorbimento, l’esame delle feci dà il reperto caratteristico, come è stato descritto nel Capitolo 26 relativo alle malattie dell’intestino. In presenza di un’anemia megaloblastica, è importante discriminare se vi sia all’origine una carenza di vitamina B 12 o di acido folico. Il metodo più diretto consiste nel dosare la vitamina B 12 nel siero e l’acido folico nel siero e nelle emazie. Poiché la vitamina B 12 è di fatto indispensabile per permettere l’ingresso dell’acido folico nelle cellule e quindi, tra le altre, nei GR, in carenza di essa si ha aumento del rapporto acido folico del siero/acido folico dei globuli, che, come si è visto, in condizioni normali è circa 1:30. La carenza di acido folico non ha alcuna infl uenza sulla ripartizione della vitamina B 12 , a meno che non sia di grado tale da determinare atrofi a gastrica e/o intestinale e da compromettere l’assorbimento. Altri metodi diagnostici sono basati sullo studio di processi metabolici elettivamente condizionati dalla vitamina B 12 o dall’acido folico. La carenza di vitamina B 12 è responsabile di mancata conversione del metil-malonil-coenzima A in succinilcoenzima A; si avrà quindi escrezione urinaria di acido metil-malonico, di entità maggiore se sarà stato somministrato in precedenza un carico di aminoacidi (valina o isoleucina). L’acido folico è importante nel metabolismo dell’istidina ed è necessario come coenzima per la trasformazione dell’acido formominoglutammico in acido glutamico; se si somministra un carico di istidina in carenza di acido folico, si ha eliminazione di acido formominoglutammico con le urine. Un metodo molto semplice è quello della prova terapeutica, che consiste nel trattare il paziente per una decina di giorni con il fattore che si ritiene più verosimilmente in causa come responsabile dell’anemia; la prova va fatta somministrando dosi minime (1 � g/die nel caso della B 12 e 100 � g/die nel caso dell’acido folico), perché con dosi elevate si può avere una risposta positiva anche se il fattore in causa non è quello in prova. La risposta è considerata positiva se, entro 1 settimana, si verifi ca un franco incremento numerico di reticolociti. Il procedimento seguito sin qui ha portato ad accertare la presenza di anemia megaloblastica e a determinare qual è fattore carente; il successivo passaggio diagnostico è volto a ricercare la causa della carenza in termini di apporto inadeguato, malassorbimento, aumentate richieste o alterata utilizzazione più o meno variamente combinati. La carenza di vitamina B 12 è dovuta quasi esclusivamente a un difetto di assorbimento ed
C0240.indd 1049C0240.indd 1049 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1050
esiste un test specifi co, chiamato test di Schilling , che consente di verifi care le modalità dell’assorbimento stesso. In pratica vengono somministrati per via orale 2 � g di vitamina B 12 radioattiva ( 57 CoB 12 ), seguita da un’iniezione intramuscolare di 1000 � g di vitamina B 12 non radioattiva, al fi ne di saturare i depositi. Si raccolgono le urine delle 24 ore e poi si iniettano altri 1000 � g di vitamina B 12 non radioattiva e di nuovo si raccolgono le urine per 24 ore; normalmente nelle 24 ore viene escreta con le urine una quantità superiore al 5% della dose orale somministrata. Se non si ottiene un risultato normale, si ripete il test dopo aver aggiunto alla vitamina B 12 radioattiva il fattore intrinseco; nell’anemia perniciosa, in questo secondo caso, si ha normalizzazione del test. Quando invece l’alterato assorbimento non è secondario a defi cit di fattore intrinseco, ma, per esempio, a malattia intestinale con malassorbimento, l’escrezione della vitamina radioattiva è scarsa anche dopo l’aggiunta di fattore intrinseco. È di grande importanza che il test di Schilling venga eseguito solo dopo che con l’esame del midollo è stata accertata l’anemia megaloblastica e dopo che il trial terapeutico ha permesso di discriminare il fattore responsabile; infatti, la grande quantità di vitamina B 12 iniettata per saturare i depositi è in grado di alterare entrambe le prove. In tutti i casi di anemia megaloblastica va eseguito uno studio radiologico completo del tubo digerente, data l’importanza che riveste la patologia di questo apparato per il malassorbimento di B 12 e di folati. In presenza di anemia perniciosa va eseguito anche un esame gastroscopico con prelievo bioptico, per documentare la presenza e l’entità della gastrite atrofi ca; va inoltre eseguito l’esame chimico del succo gastrico prelevato mediante sondino a intervalli di tempo successivi e dopo stimolazione, per indagare la presenza o meno di achilia.
Diagnosi La diagnosi di anemia megaloblastica è basata sul rilievo di anemia macrocitica all’esame del sangue periferico e di una megaloblastosi midollare. L’anemia macrocitica può trovarsi in alcune forme di anemia emolitica ; in questi casi si hanno consistente iperbilirubinemia ed elevata reticolocitosi; nell’anemia aplastica con macrocitosi l’aspetto dei GR non è quel-lo dei macro-ovalociti, mancano i granulociti neutrofi li ipersegmentati e coesistono marcate leucopenia e piastri-nopenia; anemia macrocitica può verifi carsi nelle leuce-mie, sia prima della terapia sia in corso di chemioterapia,
nell’anemia sideroblastica, nel mixedema, nell’alcolismo cronico. Il midollo in questi casi non è di solito di tipo megaloblastico, tranne che in casi particolari di leucemia e di anemia emolitica. Se l’anemia megaloblastica è associata ad altre situazioni patologiche responsabili di anemia ipocromica, per esempio carenza di ferro, gli indici ematologici danno valori compa-tibili con anemia normocitica o anche microcitica; ciò può verifi carsi nei gastroresecati, con malassorbimento della vitamina B 12 e del ferro, nel defi cit di folato da inadeguata nutrizione in cui coesistono carenze multiple (di folati, di ferro, di proteine) o in caso di carcinoma gastrico sanguinan-te, in cui lo stillicidio cronico causa la carenza di ferro.
Decorso e prognosi Il decorso è diverso, a seconda dell’eziologia dell’anemia megaloblastica. In ogni situazione in cui la causa della carenza viene spontaneamente a cessare o può essere com-pletamente rimossa, un’opportuna terapia atta a reintegra-re i depositi riporta la situazione in condizioni di assoluta normalità. Quando non è possibile una terapia eziologica, per esempio nell’anemia perniciosa, in caso di resezione gastrica o ileale, la terapia sostitutiva con il fattore rite-nuto carente riporta alla normalità il quadro clinico ed ematologico; è ovvio che in questi casi la terapia debba essere perseguita per tutta la vita. La prognosi è buona e l’aspettativa di vita dei pazienti è nei limiti della norma.
Terapia La terapia deve essere, se possibile, eziologica, volta cioè a correggere la situazione patologica respon-sabile dell’anemia; anche se ciò è possibile, vanno reintegrati i depositi di vitamina B 12 o di acido folico. Se la condizione morbosa di base non può essere rimossa, la terapia sostitutiva va continuata indefi-nitamente. La via di somministrazione sarà parenterale per il deficit di B 12 , generalmente secondario a malassorbi-mento, e per i casi in cui il deficit di folato riconosce questo meccanismo. Negli altri casi il folato può es-sere somministrato per via orale. Nell’anemia perniciosa, che, come si è visto, rico-nosce probabilmente una patogenesi autoimmune, sono stati fatti tentativi terapeutici con prednisone, associato o meno a vitamina B 12 ; questo indirizzo terapeutico non è però codificato e viene riservato a casi particolari.
L’anemia sideropenica si manifesta quando la disponibi-lità di ferro nell’organismo è insuffi ciente per un’adegua-ta sintesi di emoglobina. La sua caratteristica distintiva è perciò la diminuzione del contenuto corpuscolare medio di Hb, che si accompagna a una riduzione del volume medio dei GR (microcitosi).
Epidemiologia La carenza di ferro è la causa di anemia oggi conosciuta più comune in tutto il mondo (Paesi sottosviluppati e non). L’Organizzazione Mondiale della Sanità (OMS) ha defi nito il concetto di anemia come un valore di Hb inferiore ai 14 g/dL nell’uomo, ai 12 g/dL nella donna e agli 11 g/dL nella donna gravida.
Anemie sideropeniche C. Camaschella, G. Gromo, M. Storti
C0240.indd 1050C0240.indd 1050 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Capitolo 48 - ANEMIE
81051
Questo criterio non precisa però lo stato clinico di sidero-penia latente (si veda oltre, Eziopatogenesi ). Nei Paesi maggiormente sviluppati l’incidenza della si-deropenia è del 3% tra gli uomini adulti, del 20% tra le donne e del 50% tra le donne gravide. Queste percentuali sono maggiori quando si prendono in esame alcuni Paesi dell’Africa o dell’Asia in cui una dieta ridotta e un’eccessiva perdita di ferro provocata dalla presenza di parassiti intestinali portano l’anemia sidero-penica a interessare più del 50% della popolazione. Tra gli adulti è soprattutto il sesso femminile a venire colpito, in particolare durante l’età fertile. Diverso l’andamento nell’uomo, nel quale si riconoscono infatti due picchi di incidenza, durante l’adolescenza e dopo i 30 anni. L’incidenza massima comunque si verifi ca tra i 6 e i 20 mesi di vita (indifferentemente tra maschi e femmine), in particolare nei prematuri. Il defi cit di ferro, infi ne, è più frequente nei ceti meno abbienti che non nelle classi medio-alte (61% versus 39%).
Metabolismo del ferro* Il ferro è un elemento essenziale per molte funzioni bio-logiche, tra cui il trasporto di ossigeno, il metabolismo ossidativo e la proliferazione cellulare. In condizioni fi -siologiche, il ferro totale dell’organismo è di circa 3-4 g, con ampie variazioni tra i sessi (35-50 mg/kg nell’uomo; 25-35 mg/kg nella donna) ( Tab. 48.3 ). Il ferro dell’organi-smo è distribuito in tre compartimenti: funzionale, di de-posito e di trasporto. Il ferro funzionale è il più importante ed è rappresentato dall’emoglobina (il 70% pari a circa 2000 mg), da mioglobina (10%, circa 300 mg) ed enzimi (in minima quantità). Il ferro di deposito è presente come ferritina ed emosiderina per un totale di circa 1000 mg, soprattutto nel fegato e nel sistema reticolo-endoteliale (milza e midollo osseo). Il ferro di trasporto è legato alla transferrina, che ne contiene 3-4 mg (0,1%), una quota minima, ma rapidamente scambiabile con i tessuti. Il metabolismo del ferro è conservativo; il ferro recuperato dall’emocateresi viene riciclato dai macrofagi e pronta-mente ceduto alla transferrina circolante (20-25 mg/die) e solo 1-2 mg di ferro, perduti per desquamazione cellulare, vengono ricambiati giornalmente ( Fig. 48.3 ).
Il mantenimento dell’omeostasi del ferro è assicurato dalla regolazione dell’assorbimento intestinale, in quanto non esiste un meccanismo di escrezione del ferro. L’assorbimen-to, che è mediato dal peptide di produzione epatica epcidi-na (si veda oltre), è incrementato in relazione alle esigenze dell’eritropoiesi e ridotto quando i depositi di ferro sono abbondanti. Le perdite di ferro nell’uomo e nella donna dopo la menopausa ammontano a circa 1 mg/die (desqua-mazione di cellule intestinali, della cute, delle vie urinarie). Nella donna in età fertile le perdite sono incrementate in considerazione del ciclo mestruale (normalmente fi no a circa 25 mg/ciclo) e delle gravidanze, in quanto, dal con-cepimento al parto, si ha una perdita di ferro aggiuntiva di circa 700 mg; la perdita dovuta ad allattamento è di circa 1 mg/die. Il fabbisogno quotidiano di ferro varia quindi in diverse condizioni fi siologiche, anche tenendo conto delle esigenze correlate all’accrescimento corporeo ( Tab. 48.4 ). Una dieta “comune” comporta l’assunzione di 10-20 mg di ferro al giorno, ma in condizioni normali solo il 5-10% (1-2 mg circa) viene assorbito. Se il fabbisogno è aumentato, l’assorbimento può anche arrivare al 10-20%. Il processo attraverso il quale il ferro è reso disponibile per le necessità dell’organismo può essere riassunto in tre fasi principali: • tipo di alimentazione, che infl uenza il trasporto
intraluminale e l’estrazione del ferro emico e non emico;
• assorbimento mucoso; • trasporto nel circolo sanguigno.
Tipo di alimentazione Esistono due sistemi di assorbi-mento, diversi a seconda che il ferro si presenti nella strut-tura porfi rinica dell’eme o sotto forma di sali inorganici di ferro. Nel primo caso (mioglobina, emoglobina, proteine di origine animale), il ferro è assorbito come eme e viene liberato nelle cellule epiteliali intestinali. Questo tipo di assorbimento, che verosimilmente coinvolge un traspor-tatore intestinale di membrana, è scarsamente sensibile alle modifi cazioni di pH e quindi poco infl uenzato dalla dieta. Nel secondo caso, invece, il ferro deve essere ridotto da ferrico, che tenderebbe a precipitare nel lume intesti-nale, a ferroso. Ai fi ni di questo processo è importante la presenza di acido cloridrico gastrico e del normale tempo di svuotamento gastrico. Infatti, l’ambiente acido libera il ferro non emico dagli alimenti e ne facilita il legame con mucopolisaccaridi o sostanze a basso peso molecolare (zuccheri, aminoacidi, acido ascorbico ecc.), direttamente assorbiti nel duodeno o digiuno. In ambiente alcalino, invece, il ferro si lega in forma indissociabile a proteine ad alto peso molecolare, non viene assorbito e può ritrovarsi come ione ferrico nelle feci. Anche la stessa alimentazione infl uisce sull’assorbimento del ferro; ossalati, fi tati, tè, fi bre, uova, fosfati lo ritardano, perché formano con il ferro composti relativamente insolubili. Viceversa, alcol e sostanze riducenti (acido ascorbico, cisteina, fruttosio ecc.) lo favoriscono.
Assorbimento mucoso Le cellule della mucosa duode-nale e digiunale presentano sulla superfi cie luminale il trasportatore di ferro DMT1 (Divalent Metal Transporter 1), che coopera con una ferroreduttasi della membrana per recuperare dal lume il ferro sotto forma di ferro ferroso.
Ferro totale corporeo (%)
Uomini Donne Compartimento funzionale Hb Mioglobina Enzimi Transferrina
72 10 0,2 0,2 82,4
73,5 13 0,4 0,2 87,1
Immagazzinamento Ferritina (65%), emosiderina (35%)
17,6
100
12,9
100
Ferro totale corporeo (%)
Tabella 48.3 Distribuzione del ferro nei suoi compartimenti
* C. Camaschella
C0240.indd 1051C0240.indd 1051 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1052
L’espressione del trasportatore e della reduttasi è incre-mentata in carenza di ferro e soprattutto in condizioni di ipossia. Nella cellula intestinale il ferro va incontro a desti-nazioni diverse a seconda delle necessità. In condizioni di sideropenia viene esportato ed entra in circolo legandosi alla transferrina circolante. Questo processo coinvolge una proteina della membrana basolaterale dell’enterocita de-nominata ferroportina e un’ossidasi (efestina) per l’ossida-zione a ferro ferrico. In condizioni di eccesso di ferro, una quota elevata viene depositata nelle cellule della mucosa sotto forma di ferritina e, come tale, verrà perduta nel lume intestinale; una piccola quota, infi ne, viene direttamente utilizzata per le esigenze delle cellule intestinali.
Regolazione dell’assorbimento intestinale È rimasto a lungo ignoto in che modo la cellula intestinale riceva l’informazione per modulare in modo intelligente l’as-sorbimento di ferro, che è aumentato nelle situazioni di sideropenia e in caso di esaltata eritropoiesi, mentre si riduce nelle situazioni di sovraccarico marziale. Recente-mente il principale regolatore dell’assorbimento intesti-nale di ferro è stato identifi cato e denominato epcidina. Si tratta di un piccolo peptide di produzione epatica, che
ha la struttura dei peptidi antimicrobici. Il peptide attivo è formato da 25 aminoacidi a partire da un precursore di 84 aminoacidi prodotto nel fegato. L’epcidina controlla l’espressione di superfi cie dell’esportatore cellulare di ferro ferroportina, legandosi alla ferroportina e inducen-done l’internalizzazione e la degradazione lisosomiale. L’epcidina è quindi in grado di bloccare l’assorbimento in caso di eccesso di ferro. La trascrizione dell’epcidina è au-mentata nel sovraccarico di ferro e nell’infi ammazione e soppressa nella sideropenia, nell’ipossia e nell’espansione eritropoietica, condizioni in cui è necessario incrementa-re l’assorbimento di ferro per le esigenze dell’eritropoiesi. La soppressione della sintesi di epcidina lascia inalterata la ferroportina, permettendo l’assorbimento di quote ingenti di ferro ( Fig. 48.4 ). A questa scoperta si è arrivati tramite lo studio di modelli animali (il topo in cui il gene di epcidina è costitutivamente inattivato sviluppa sovraccarico di ferro, mentre il topo che iperesprime epcidina muore alla nascita per anemia sideropenica grave) e dell’emocromatosi ereditaria, che è una pato-logia tipica da sregolazione dell’assorbimento. Inoltre l’epcidina è una proteina di fase acuta, indotta dalla fl o-gosi in risposta a citochine infi ammatorie, in particolare all’interleuchina 6 (IL-6). La sua aumentata produzione nell’infi ammazione blocca l’assorbimento di ferro e causa sequestro di ferro nei macrofagi, entrambe caratteristiche dell’anemia delle malattie infi ammatorie croniche, con la fi nalità di sottrarre un fattore di crescita di eventuali microrganismi.
Trasporto del ferro nel circolo sanguigno Il ferro, entrato nel torrente circolatorio, si trova in un sistema chiuso in cui viene costantemente riciclato tra plasma e tessuti. Il midollo osseo ne impiega la gran parte per l’eritropoie-si; spetta invece al sistema reticolo-endoteliale (cellule macrofagiche del midollo, e soprattutto della milza e del fegato) fagocitare le emazie al termine del loro ciclo vitale di 120 giorni circa, recuperare il ferro e ritrasmetterlo nuo-vamente agli eritroblasti attraverso due vie, la transferrina (la quota maggiore) e il meccanismo della rofeocitosi (dal
Enterocitiduodenali
Macrofagisplenici
Fegato
Muscolo e altriorgani
Midollo osseoEritrociti
1-2 mg/die
1-2 mg/die
Perditedi sangue
Desquamazionecellulare
20-25 mg/die
Figura 48.3 Schema del
ricambio del ferro nell’organismo.
Uomo adulto normale 0,5-1,0Donna mestruata 1,2-2,0Donna gravida 2,4-4,8Donna che allatta (con amenorrea) 0,5-1,0Donna postmenopausa 0,5-1,0Adolescente 1,0-2,0Bambino 0,4-1,0Neonato 0,5-1,5
N.B. Solitamente viene assorbito il 5-10% del ferro ingerito; nel caso della donna in gravidanza non è possibile raggiungere simili dosaggi con la sola dieta, è dunque necessario aggiungere ferro con uno dei preparati a disposizione.
Uomo adulto normale 0 5-1 0
Tabella 48.4 Fabbisogno quotidiano di ferro (mg/die)
C0240.indd 1052C0240.indd 1052 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Capitolo 48 - ANEMIE
81053
greco rofeo , “aspirare”), ovvero direttamente dai macrofagi agli eritroblasti (la quota minore). Nel plasma il ferro circola veicolato dalla transferrina, una glicoproteina sintetizzata dal fegato. Essa è formata da una catena polipeptidica singola, del peso molecolare di circa 80.000, provvista di due siti specifi ci di legame per il ferro, situati agli estremi N-terminale e C-terminale della molecola. La sua concentrazione plasmatica è di 240-280 mg/dL. Può essere misurata in termini di capacità ferro-legante totale del plasma (TIBC, Total Iron-Binding Capacity), in genere 300-400 � g/dL. Normalmente la si-deremia è circa 100-130 � g/dL, per cui la transferrina è saturata per circa un terzo. La transferrina esiste in forma di apotransferrina (cioè priva di ferro), transferrina mono-ferrica (con un solo legame saturo) e transferrina diferrica (con entrambi i legami saturi). La quantità di transferri-na diferrica sarebbe il segnale captato da sensori epatici che trasmettono il segnale per la produzione di epcidina (si veda Fig. 48.4 ). Questi ultimi includono le proteine mutate nell’emocromatosi ereditaria (si veda il Capitolo 34 ). Sulla superfi cie della maggior parte delle cellule sono situati recettori per la transferrina; la captazione cellulare del ferro ha inizio con lo stabilirsi di un legame tra il com-plesso transferrina-ferro e il recettore della transferrina. La differenza nel numero di recettori adatta la captazione del ferro alle necessità del singolo tessuto; i recettori tissutali mostrano un’avidità preferenziale per la transferrina di-ferrica. Le cellule con maggior numero di recettori sono i trofoblasti placentari e i precursori eritroidi del midollo
durante la loro maturazione in relazione alle necessità di produrre emoglobina. Il meccanismo di captazione del ferro avviene attraverso un fenomeno di endocitosi in cui il recettore, la transferrina e il ferro vengono internalizzati dalla cellula in un ciclo endosomico. Il ferro viene rilascia-to al pH acido dell’endosoma e, mediante il trasportatore di membrana DMT1, viene rilasciato al citoplasma per le esigenze cellulari, mentre il recettore e l’apotransferrina vengono riciclati sulla superfi cie cellulare e iniziano un nuoco ciclo. Recentemente è stato dimostrato che anche HFE , il gene mutato nell’emocromatosi ereditaria, si asso-cia al recettore della transferrina in questo ciclo endoso-mico, verosimilmente con funzione regolatoria.
Deposito di ferro Il ferro di deposito viene immagazzi-nato sotto due forme, ferritina ed emosiderina. La ferriti-na è una molecola proteica di peso molecolare 440.000, costituita da catene leggere L e pesanti H, con funzioni diverse. La proteina forma una sorta di guscio che ricopre un nucleo di sale ferrico insolubile, comprendente sino a circa 4500 atomi per molecola. Il ferro ferritinico è ra-pidamente mobilizzabile. La ferritina è presente nel siero dove rifl ette, con buona approssimazione, i depositi di ferro nell’organismo e può essere dosata con metodo RIA (Radio Immuno Assay). Esistono differenze tra i sessi per quanto riguarda i livelli di ferritina sierici, che possono anche essere infi ciati da processi infi ammatori, anche di lieve entità. L’emosiderina, costituita da aggregati di molecole di ferritina, rappresenta una forma di deposito
Macrofago
Eritrociti
FPN
FeEpatocita
Epcidina
HJV
HAMP
HFETFR2
TFR1
TF-diferrica
Enterocita
FPN
Fe
Fe
TF
? ?
Figura 48.4 Schema della regolazione dell’assorbimento intestinale e del rilascio di ferro dai macrofagi.
Il peptide epatico epidina controlla l’espressione di membrana di ferroportina dell’enterocita e del macrofago. In presenza di epcidina meno ferro viene rilasciato alla circolazione e quindi la transferrina diferrica è poco rappresentata. Al contrario, quando l’epcidina è ridotta, più ferro è disponibile per la transferrina. La transferrina diferrica a livello dell’epatocita si lega a TFR1 e internalizza il ferro, ma funge anche da segnale per il complesso delle proteine (HFE e TFR2) dell’emocromatosi. Si sa che il segnale è poi trasmesso dall’emogiuvelina (HJV) per attivare la trascrizione del gene HAMP che codifi ca per l’epcidina, anche se i meccanismi molecolari sono in parte ignoti (?).
C0240.indd 1053C0240.indd 1053 6/9/10 3:08:58 PM6/9/10 3:08:58 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1054
più stabile da cui il ferro è scarsamente mobilizzabile; in passato veniva distinta dalla ferritina per la sua insolubili-tà e la positività alla colorazione con il blu di Prussia nelle preparazioni microscopiche. La diminuzione, o assenza, di emosiderina a livello midollare rappresenta la prima spia di un defi cit di ferro nell’organismo, che può verifi -carsi con anticipo anche di parecchi mesi sulla comparsa dell’anemia. Nelle anemie emolitiche il ferro liberato dalle emazie lisate viene captato a livello del sistema reticolo-endoteliale, in grado di mobilizzarlo prontamente, e i livelli di epcidina non sono signifi cativamente variati. Nelle situazioni di eritropoiesi ineffi cace l’assorbimento di ferro aumenta soprattutto in relazione alla riduzione della produzione di epcidina, che si verifi ca anche in presenza di livelli elevati di ferro di deposito. Questo fenomeno sottolinea come le necessità dell’eritropoiesi prevalgano nell’organismo, ma in presenza di eritropoiesi ineffi cace favoriscono lo sviluppo di sovraccarico di ferro.
Eziopatogenesi Uno stato di carenza marziale può essere determinato da: • apporto inadeguato; • aumentato fabbisogno; • assorbimento inadeguato; • perdita protratta di ferro.
Un diminuito apporto senza che vi siano problemi di assor-bimento o di fabbisogno è evento raro. Questo avviene più spesso nei vegetariani, perché anche se frutta e ver-dura contengono percentuali discrete di ferro 1 è altret-tanto vero che vi si trovano nitrati, fosfati e carboidrati che tendono a chelare il ferro e a ridurre l’assorbimento (si veda in precedenza, Metabolismo del ferro ). Durante il primo anno di vita, come si è detto, possono comparire anemie sideropeniche su base alimentare, dal momento che il latte materno e, soprattutto, quello artifi ciale sono poveri di ferro. Le condizioni nelle quali si registra un fabbisogno aumen-tato di ferro sono sostanzialmente tre: infanzia, gravidanza e allattamento. Nel primo anno di vita il bambino triplica il proprio peso corporeo e ha bisogno di 150 mg di ferro per espandere la propria massa eritrocitaria circolante e produrre proteine contenenti ferro quali la mioglobina. Il latte materno di per sé non è suffi ciente; occorrono quindi alimenti ricchi di ferro come i cereali e la carne, oppure un apporto farmacologico supplementare. Più complesso invece il bilancio del ferro in gravidanza. Ciascuna gravidanza comporta una perdita da parte della madre di circa 680 mg di ferro, equivalente a 1300 mL di sangue. Considerato che il pool totale in una donna di 50-55 kg si aggira sui 2000 mg di ferro, se ne può facilmente dedurre che la perdita totale è di un terzo, in gran parte dovuta all’espansione della massa eritrocitaria (450 mg, recuperabili) e in misura minore alle necessità del feto, alle perdite fi siologiche, alla placenta, al cordone ombelicale, alle emorragie durante il parto. Subito dopo la gravidan-za, la lattazione rappresenta un ulteriore impegno per la
madre. La perdita di ferro è stimata attorno a 0,5-1 mg/die. Il fabbisogno medio legato a questa particolare fase della vita di una donna è pari circa a quello delle mestruazioni, inibite nel post partum . Questo è il motivo per cui non si assiste a un’anemizzazione ingravescente. Il diminuito assorbimento è raramente causa di tale anemia. Tuttavia, conviene ricordare, a questo proposito, il ruolo svolto dall’acido cloridrico di derivazione gastrica (si veda in precedenza Metabolismo del ferro ). Logico quindi che tutte quelle condizioni caratterizzate da achilia presto o tardi alterino l’assorbimento del ferro; è il caso della ga-strite atrofi ca o di una gastroresezione con asportazione del piloro. È da notare come la stessa anemia sideropenica possa provocare a sua volta achilia (si veda in preceden-za Manifestazioni cliniche ) per alterazione della mucosa gastrica. Infi ne, possono determinare sideropenia tutte le malattie che sono causa di malassorbimento, quali malattia celiaca, sprue tropicale, linfomi a localizzazione addominale ecc. La causa più comune di anemia sideropenica è la perdita protratta di ferro . Nell’uomo e nella donna dopo la meno-pausa la sede più frequente di sanguinamento cronico è il tratto digestivo. Infatti , l’anemia può datare da tempo senza dare segno di sé, ovvero riconoscere sovente una causa benigna non destinata a un’evoluzione progressiva. Possono così trascorrere molti anni prima che si manifesti una situazione carenziale, fatta eccezione nel caso di una pregressa gravidanza o di mestruazioni particolarmente abbondanti che abbiano già intaccato le scorte dell’orga-nismo. Le sedi e le cause più frequenti di sanguinamento sono emorroidi , varici esofagee, ernie iatali, ulcere gastriche e duodenali, poliposi, gastriti emorragiche, cancro gastrico, diverticolosi, diverticolo di Meckel, colite ulcerosa, malattia di Crohn, cancro del colon e cause iatrogene (salicilati, corticosteroidi, farmaci antinfi ammatori non steroidei). È da notare, inoltre, che i sanguinamenti dalle porzioni distali del tubo digerente (per esempio, emorroidi) vengono considerati di solito un disturbo banale e come tale sottova-lutati anche nel caso di una perdita ematica di proporzioni cospicue. Circa il 20% dei sanguinamenti dovuti a ulcere peptiche non comporta una sintomatologia dolorosa tale da richiamare l’attenzione sulla presenza o meno di una condizione emorragica. Eppure è suffi ciente una piccola perdita anche di soli 10-15 mL/die per provocare nell’indi-viduo adulto anemia sideropenica. È necessaria quindi una ricerca di sangue occulto nelle feci, dopo dieta acarnea e per un congruo numero di giorni (almeno 3); infatti, spesso i sanguinamenti sono intermittenti. Anche l’ernia diaframmatica paraesofagea (iatale) rappre-senta una condizione relativamente frequente di anemia sideropenica nell’età avanzata. Può accompagnarsi anche sintomatologia dolorosa e dispeptica. Infi ne, sono da citare le neoplasie del tratto digerente e in particolare quelle del colon destro (tratto ascendente), perché solitamente non si accompagnano a fenomeni di occlusione (più precoci se la neoplasia interessa il colon di sinistra) e perché il sanguinamento è di grado diffi cil-mente rilevabile, comunque modesto. Nelle donne in età fertile, oltre alle situazioni sopra menzio-nate, la causa più frequente va ricercata in eventuali emor-ragie dell’apparato genitale (alterazioni del ciclo mestruale, metrorragie da cause organiche, meccaniche o ormonali).
1 Che gli spinaci ne siano particolarmente ricchi è però una leggenda nata da un errore nella compilazione delle prime tabelle del contenuto di ferro di vari alimenti.
C0240.indd 1054C0240.indd 1054 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Capitolo 48 - ANEMIE
81055
Giova prendere in considerazione anche eziologie meno comuni, quali epistassi frequenti, emorragie in varia sede da disordini emocoagulativi, gravi ematurie, varie forme di emoglobinuria, stillicidio intestinale da anchilostoma o da Necator americanus . Infi ne, è importante segnalare anche l’anemia sideropenica, possibile nei donatori di sangue. Vi sono poi numerosi casi che non rientrano necessa-riamente in una delle quattro cause principali esposte sopra, ma in un loro reciproco combinarsi. Per esempio, la gravidanza, in cui all’instaurarsi dell’anemia ferro-priva concorrono sia un diminuito apporto sia un aumentato fabbisogno ( Tab. 48.5 ).
Fisiopatologia Il termine sideropenia è impiegato per defi nire una con-dizione nella quale tutto il contenuto in ferro dell’orga-nismo è ridotto, senza menzione alcuna delle cause. Dal momento che le riserve di ferro possono esaurirsi prima che si manifesti una riduzione dell’eritropoiesi, l’anemia è uno stadio tardivo della sideropenia.
In altre parole, si può parlare di una sideropenia prelaten-te quando, prima ancora di avere una diminuzione del ferro circolante, la presenza di emosiderina a livello del sistema reticolo-endoteliale midollare è notevolmente ridotta. Non sembra esserci evidenza biochimica di questo stato, fatta eccezione, forse, per una modifi-cazione dei livelli di ferritina sierica. In questa fase si assiste a un aumento compensatorio dell’assorbimento del ferro. C’è poi uno stato di sideropenia latente , con cui si intende la sideropenia che si accompagna a una diminuzione del ferro plasmatico, ma non ad alterazioni ematologiche. Da ultimo, si verifi ca uno stadio di eritropoiesi sideropeni-ca , nel quale la produzione di emoglobina è via via sem-pre più limitata dalla ridotta quantità di ferro plasmatico ( Tab. 48.6 ). Questa limitazione avviene regolarmente ogni qualvolta la saturazione della transferrina scende sotto il 16%. Non sempre, però, ridotti livelli ematici signifi cano eritropoiesi sideropenica, dal momento che talora questi dipendono da anormalità del metabolismo del ferro anche in assenza di sideropenia, come nel caso dell’anemia delle malattie croniche. Una volta sviluppatasi la sideropenia, con il passare dei mesi compare una microcitosi, perché nella maturazione della serie eritrocitaria le duplicazioni avvengono fi no a quando il contenuto in Hb non abbia raggiunto una certa quantità. Se vi è carenza di ferro la duplicazione procede ugualmente, ma con perdita di massa cellulare e immissio-ne nel torrente circolatorio di eritrociti più piccoli. Si tratta, però, di una microcitemia che, inizialmente, interessa solo una minoranza delle cellule, almeno in questa fase, non si accompagna a una diminuzione signifi cativa dell’MCV, la quale, viceversa, compare quando la sideropenia perdura ormai da lungo tempo. Perché vengano prodotti eritrociti maturi occorre dunque che negli eritroblasti venga formata una congrua quantità di Hb. Se le scorte di ferro (che è costituente essenziale di Hb) sono esaurite, il singolo eritrocita avrà quantità in-suffi cienti di Hb al suo interno. È opportuno sottolineare l’espressione “se le scorte di ferro sono esaurite”; infatti, l’organismo intacca le proprie risorse prima di palesare un difetto nella produzione di Hb. Al microscopio ottico, con i comuni metodi di colorazione, si osserva così ipocromia dapprima ristretta a pochi elementi e, solamente più tardi, una diffusa riduzione della concentrazione emoglobinica. In condizioni ordinarie si tratta dunque di una situazione caratterizzata da ipocromia, microcitosi e minore traspor-to di ossigeno ai tessuti.
Apporto inadeguato � Dieta Aumentato fabbisogno � Infanzia � Gravidanza � Lattazione Assorbimento inadeguato � Acloridria � Chirurgia gastrointestinale � Malattia celiaca � Altre malattie che provocano malassorbimento Perdita protratta di ferro � Sanguinamento a partenza gastrointestinale (causa più
frequente nel sesso maschile) � Menorragia (causa più frequente nel sesso femminile in età
fertile) � Donatori di sangue � Emoglobinuria � Sindrome di Goodpasture � Teleangectasia emorragica ereditaria � Disturbi dell’emostasi
*Possibile sovrapposizione di più fattori eziologici contemporaneamente nello stesso paziente.
Tabella 48.5 Fattori eziologici della sideropenia*
Tabella 48.6 Diversi livelli di sideropenia
Normale Sideropenia prelatente
Sideropenia latente
Anemia iniziale
Anemia severa
Ferro del sistema reticoloendoteliale
Normale Ridotto Assente Assente Assente
Sideremia Normale Normale Ridotta Ridotta RidottaAnemia No No No Sì (modesta) Sì (grave)Ipocromia/microcitosi No No No Sì (alcune cellule) Sì (diffusa)Aumentato assorbimento di Fe No Sì Sì Sì SìAlterazioni epiteliali No No No No SìMCV/MCHC Normali Normali Normali Normali RidottiMCV (Mean Corpuscular Volume) = volume corpuscolare medio; MCHC (Mean Corpuscolar Hemoglobin Concentration) = concentrazione corpuscolare media di emoglobina.
C0240.indd 1055C0240.indd 1055 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1056
Il rene, però, risponde a questa ipossigenazione secernen-do EPO , che a sua volta stimola il midollo. Si tratta però di un compenso apparente; in realtà le emazie in via di maturazione che hanno diffi coltà a fabbricare e incorpo-rare l’Hb nel proprio citoplasma sono anche più fragili e più difficilmente arrivano a completa maturazione. Infatti, una quota di GR può morire nel midollo stesso (eritropoiesi ineffi cace) , rendendo vano il compenso do-vuto a un’aumentata produzione numerica degli elementi della serie rossa. In linea teorica non ci dovrebbero essere modifi cazioni a carico degli altri stipiti cellulari (globuli bianchi e piastrine); in realtà non è raro il riscontro di granulocitopenia e trombocitosi, di oscura interpreta-zione. Tuttavia, il metabolismo del ferro non riguarda esclusivamente l’Hb e, infatti, entra nella mioglobina e in molti coenzimi, principalmente nei citocromi, nelle catalasi, perossidasi e nelle metallofl avoproteine. Un suo defi cit, quindi, provoca conseguenze soprattutto in quelle cellule caratterizzate da rapido turnover. Si notano inoltre alterazioni a livello di epiteli di rivestimento, annessi cutanei, cavo orale e mucosa gastrica.
Manifestazioni cliniche La maggior parte dei pazienti presenta una progressione estremamente lenta e graduale della sintomatologia. Il paziente affetto da ulcera peptica , per esempio, o da neoplasia dell’apparato digerente, o ancora la donna che lamenta mestruazioni abbondanti o frequenti, quando si rivolgono al medico curante, presentano già dei valori di Hb inferiori a 8 g/dL. Clinicamente il paziente lamenta disturbi generali comuni a tutte le forme di anemia quali cefalea, astenia ingrave-scente, tachicardia, dispnea da sforzo, facile irritabilità, insonnia, labilità emotiva, pallore. Caratteristica invece dell’anemia sideropenica sembra essere la “voglia” di particolari sostanze (pica) , di argilla, di amido da bucato o di ghiaccio (pagofagia) . In una parte dei pazienti, soprattutto in quelli in cui l’ane-mia dura da tempo, possono comparire lesioni a carico della cute, degli annessi cutanei e delle mucose. La fre-quenza di questi segni (peculiari anch’essi dell’anemia sideropenica) è diversa a seconda della popolazione presa in esame. In Europa e negli Stati Uniti sono piuttosto rari, a differenza del Sud-Est asiatico, ma le ragioni di questa curiosa distribuzione non sono ben chiare. La cute diven-ta secca, anelastica, i capelli sono sottili, fragili, radi e le unghie opache, fragili, rigate longitudinalmente, talvolta appiattite o addirittura concave (coilonichia) . I disturbi a carico delle mucose riguardano soprattutto il cavo orale e, più raramente, il faringe. Le labbra presentano piccole ragadi alle commissure (chei-lite angolare); la mucosa orale è arrossata, la lingua liscia, levigata, pallida. Al confi ne tra ipofaringe ed esofago si possono notare creste mucose che sporgono all’interno del lume, riducendone il calibro. Ciò causa disturbi della deglutizione (disfagia sideropenica) . La triade sintomato-logica comprendente anemia, glossite ed esofagite viene defi nita sindrome di Plummer-Vinson o Paterson-Kelly . Si possono registrare anche disturbi gastrici. A tutt’oggi i rapporti sideropenia-atrofi a della mucosa gastrica con acloridria non sono ancora del tutto chiariti. I dati esistenti, infatti, divergono soprattutto in ragione
del sistema impiegato per stimolare la secrezione gastri-ca. Oggi si può affermare che la presenza di una gastrite riduce l’assorbimento del ferro, la cui carenza peggiora ulteriormente la sintomatologia gastrica. Nelle donne è frequente l’atrofi a della mucosa vaginale.
Esami di laboratorio Quando il quadro clinico è conclamato, l’anemia è tipica-mente microcitica. Il patrimonio emoglobinico e l’emato-crito sono sempre inferiori alla norma. Nel caso dell’Hb si possono toccare valori molto bassi, ma più comunemente ci si mantiene attorno agli 8 g/dL. Quanto ai GR, raramente scendono sotto i 3 × 10 12 /L e nelle forme lievi si mantengono ai limiti inferiori della norma. Tutti diminuiti, invece, sono i parametri corpu-scolari. Al microscopio si osserva una particolare diafania della parte centrale dei GR (ipocromia), che sono anche più pic-coli (microcitemia). Inoltre, proprio perché l’eritropoiesi è anomala, si troveranno emazie di grandezza differente (anisocitosi) e di forme bizzarre (poichilocitosi). L’entità delle alterazioni morfologiche è proporzionale alla gravità dell’anemia. Il numero dei reticolociti può essere aumentato, se l’attività del midollo stimolato su-pera la quota di eritropoiesi ineffi cace; in caso contrario sono diminuiti, anche se più spesso si mantengono nella norma. Il parametro ematochimico fondamentale ai fi ni della diagnosi è la concentrazione di ferro nel siero, cioè la sideremia. Nelle anemie sideropeniche si possono avere valori molto bassi (5-10 � g/dL). Il ferro nel siero è legato alla transferrina, la quale viene dosata solitamente come TIBC, che, confrontata con la concentrazione di ferro sierico, permette di valutare la per-centuale di transferrina saturata dal ferro, normalmente il 20-35%. La concentrazione di transferrina nel siero è inversamente proporzionale ai depositi di ferro, per cui in caso di sideropenia l’aumentata sintesi di transferrina, che risulta scarsamente saturata, porta ad aumento del valore della TIBC e della LIBC . Un altro dato determinante ai fi ni diagnostici è rap-presentato dalla ferritina . Può circolare nel plasma in piccole quantità (12-300 � g/L) e bene si correla con la quantità di ferro presente nell’organismo. Solitamente la diagnosi di anemia sideropenica non richiede il puntato sternale a fi ni diagnostici. Comunque, nel caso venga eseguito, si potrebbero osservare una perdita dei depositi di ferro (negativa la colorazione con il blu di Prussia per la presenza di emosiderina) e una quota ridotta di sideroblasti. Il numero dei leucociti è nella norma, ma talvolta può ridursi; le piastrine possono arrivare a 600-1000 × 10 9 /L e il meccanismo responsabile dell’aumento non è noto.
Diagnosi La diagnosi di anemia sideropenica è basata essenzial-mente sui dati di laboratorio. Esistono peraltro anemie microcitiche ipocromiche che possono essere scambiate per una carenza di ferro. Nel caso di anemie sideroblasti-che e talassemie, la distinzione è suffi cientemente facile, dal momento che si tratta di patologie associate a una concentrazione di ferro normale o elevata. Nelle anemie
C0240.indd 1056C0240.indd 1056 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Capitolo 48 - ANEMIE
81057
delle malattie croniche, invece, in cui è presente una ri-duzione della sideremia, la diagnosi differenziale è posta grazie alla transferrinemia e alla ferritinemia. Nelle anemie sideropeniche, infatti, si avranno sideremia e ferritinemia diminuite, ma transferrina aumentata; se si dosa poi la TIBC, risulterà un aumento assoluto della transferrinemia. Invece, nel caso dell’anemia delle malattie croniche, side-remia e transferrinemia sono ridotte, mentre la ferritina è normale o aumentata. Va sottolineato che, per quanto riguarda la ferritina, un suo tasso sierico inferiore a 12 � g/L è correlato si-curamente a una carenza di ferro, mentre il riscontro di un valore normale o anche aumentato non è da ri-tenere signifi cativo per escludere un’eventuale carenza marziale, perché il livello di ferritina si modifica nel corso di infezioni o infi ammazioni (come sopra citato, per esempio, nel caso delle malattie croniche), per cui sul valore fi nale possono interferire due meccanismi di segno opposto. Si può ricorrere, infi ne, anche al puntato sternale e alla valutazione dei depositi di ferro con metodo citochimico; nell’anemia sideropenica, come si è detto, si osserva una loro deplezione.
Prognosi Si può parlare di prognosi solo facendo riferimento alla causa che ha provocato l’anemia, diversa nel caso di un carcinoma gastrico sanguinante o di emorroidi. Raramen-te il paziente giunge a morte per l’anemia in quanto tale,
ma piuttosto per la causa di essa. Frequenti sono le ricadu-te (33% dei pazienti donne e il 25% degli uomini).
Terapia Il trattamento di elezione è rappresentato dal ferro (vi sono più di cento preparati in commercio). Quasi tutti i pazienti con anemia sideropenica rispondono ai preparati per os. Nelle gastriti atrofiche si som-ministra solfato ferroso, che non necessita di acido cloridrico per essere assorbito. Conviene somministrare ferro per via endovenosa (o intramuscolare) solo nei casi documentati di ma-lassorbimento. A eccezione di quei pazienti con perdita di ferro co-spicua e continua, la correzione dello stato anemico può essere raggiunta in circa 2 mesi. Una volta corretta l’anemia, si deve continuare la terapia per altri 6 mesi, allo scopo di ripristinare le riserve del ferro. Quanto alle lesioni epiteliali, quelle riguardanti le un-ghie e la lingua si risolvono entro 3-6 mesi, ma già do-po 2 settimane si osservano segni di miglioramento. Non responsiva del tutto (o meno responsiva) sem-bra essere la disfagia sideropenica, come anche la gastrite e i difetti di secrezione gastrica, soprattutto negli adulti.
L’anemia delle malattie infi ammatorie croniche è di ri-scontro abbastanza frequente, isolata o associata ad altre forme di anemia; essa si riscontra in pazienti portatori di neoplasie, infezioni croniche, malattie infiammatorie croniche, tipica tra queste l’artrite reumatoide . Si tratta di un’anemia ipoproliferativa da lieve a mo-derata con iposideremia, transferrinemia ridotta, basso indice reticolocitario, aumento della protoporfi rina delle emazie, depositi reticoloendoteliali di ferro normali o aumentati, con assenza o notevole riduzione dei side-roblasti.
Eziopatogenesi Non è defi nitivamente chiarita, ammettendosi la respon-sabilità di tre meccanismi principali tra loro interagenti in misura diversa: ridotta sopravvivenza delle emazie, ridotta attività proliferativa del midollo e alterata libera-zione del ferro nel plasma, per blocco a livello del sistema reticolo-endoteliale. Il punto chiave sembra l’incapacità del midollo di incre-mentare la produzione di emazie per sopperire alla perdita
dovuta alla modesta emolisi, per cui una situazione emo-litica di grado lieve è suffi ciente a provocare anemia. In ricerche sperimentali con colture di midollo si è dimo-strato che i macrofagi attivati, e non i macrofagi in stato di quiete, sopprimono la formazione di colonie eritroidi; ciò si verifi ca anche in presenza di un piccolo numero di macrofagi, indicando così la responsabilità di un fattore solubile. Questo è da identifi care con il Tumor Necrosis Factor (TNF) o cachettina, che inibisce lo sviluppo delle cellule ematopoietiche a livello di CFU-GEMM, CFU-GM, BFU-E e CFU-E. Per l’inibizione delle BFU-E e CFU-E si sono dimostrate effi caci concentrazioni picomolari, simili a quelle prodotte spontaneamente da monociti isolati di pazienti anemici portatori di neoplasia. Infi ne, per la patogenesi di questa forma di anemia, viene tra-dizionalmente riconosciuta l’importanza del cosiddetto “blocco reticolo-endoteliale del ferro”. Con metodiche di ferrocinetica si è effettivamente dimostrata una difettosa capacità del sistema reticolo-endoteliale di reimmettere il ferro in circolo e di renderlo in tal modo disponibile al midollo, ma il defi cit è di entità modesta e non tale da rendere completamente conto dell’anemia. Tale blocco, tuttavia, può spiegare l’iposideremia di questa particolare forma di anemia. La transferrinemia è bassa non solo per la prevalenza del catabolismo proteico, ma anche perché, di fatto, le scorte di ferro nelle cellule del sistema reticolo-endoteliale sono aumentate ( Fig. 48.5 ).
Anemia delle malattie infi ammatorie croniche 2
2 Questa anemia era tradizionalmente chiamata “anemia delle malattie croniche”. Il nome è stato abbandonato perché potrebbe generare confusione; l’ipertensione arteriosa e il diabete infatti sono malattie croniche nelle quali non si verifi ca questa anemia. In realtà, anche la denominazione attuale non è perfetta perché, oltre che nelle infi amma-zioni croniche, questa anemia si può osservare anche nelle neoplasie.
C0240.indd 1057C0240.indd 1057 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1058
Manifestazioni cliniche Le manifestazioni cliniche sono soprattutto quelle riferibili alla malattia di base; affaticabilità, perdita di peso, astenia, anoressia, pallore possono instaurarsi insidiosamente nella gran parte delle affezioni croniche. È perciò diffi cile stabi-lire il reale apporto dello stato anemico a questi sintomi. Raramente una ridotta quantità di Hb rappresenta il pri-mo riscontro di una malattia di base fi no a quel momento ignorata. Esiste comunque una correlazione grossolana tra entità dello stato anemico e attività della malattia di base
qual è indicata dalla febbre, infi ammazione, suppurazione o calo ponderale. L’anemia può essere normocitica e normocromica quando prevalgono la diminuita sopravvivenza degli eritrociti e la ridotta attività midollare, oppure microcitica se viceversa prevale la tendenza a trattenere ferro nei depositi. L’Hb è solitamente compresa tra 8-12 g/dL, i reticoloci-ti nella norma o di poco ridotti. Caratteristicamente vi sono iposideremia, marcata diminuzione della TIBC e normale o moderata riduzione della saturazione di tran-sferrina, RDW normale (si veda in precedenza, Diagnosi differenziale). La ferritina sierica e il ferro midollare sono normali o aumentati, mentre i sideroblasti nel midollo sono ridotti o assenti. Da segnalare, inoltre, è un discreto aumento della cu-premia, della protoporfi rina libera eritrocitaria e della escrezione urinaria di coproporfi rine. È importante sottolineare come lo studio della crasi ema-tica sia di scarso aiuto nel precisare l’esatta natura della malattia cronica presente. Si tratta infatti di un segno aspecifi co comparabile all’aumento del numero dei granu-lociti o alla velocità di eritrosedimentazione elevata. L’unica terapia effi cace è quella diretta nei confronti della malattia cronica esistente.
L’anemia presente nei pazienti con insuffi cienza renale cronica è di grado variabile ed è correlata con i valori di creatininemia. È un’anemia iporigenerativa ed è conse-guente alla ridotta attività endocrino-secretrice di secre-zione di eritropoietina a livello renale proporzionale all’entità del defi cit di attività escretrice, condizionando una riduzione dell’emivita degli eritrociti. Le caratteristi-che ematologiche dell’anemia consistono in un patterrn normocromico, con eritrociti dismorfi ci (“echinociti”) per irregolarità del profi lo citoplasmatico; la conta reticoloci-taria è diminuita e l’assetto marziale può essere sovrappo-nibile al quadro di sideropenia, in caso di concomitanti
perdite ematiche croniche prevalentemente dal tratto gastroenterico per ridotta funzionalità piastrinica nell’am-bito dello stato uremico; in alternativa, questo assetto può essere simile a quello dell’anemia degli stati infi ammato-ri cronici, se concomitano quadri di flogosi croniche, come pielonefriti croniche. La terapia di questa forma di anemia è costituita dall’eri-tropoietina umana ricombinante (rhEPO); il dosaggio del recettore solubile della transferrina (TfR) costituisce un indicatore precoce sensibile di risposta favorevole alla terapia, in quanto è direttamente proporzionale alla massa eritroblastica midollare.
Anemia dell’insuffi cienza renale cronica M.D. Cappellini
Costituiscono un raro gruppo di anemie iporigenerative familiari, trasmesse come carattere autosomico recessivo (tipo I e II) o dominante (tipo III), caratterizzate da un’ane-mia di grado da lieve a moderato, con ittero ed epato-splenomegalia di entità moderate. La patogenesi si basa sulla precoce distruzione intramidollare degli eritroblasti con conseguente eritropoiesi ineffi cace; raramente con-comita emolisi cronica. • CDA tipo I : eredità autosomicà recessiva;
caratterizzata da macrocitosi con eritroblasti con nucleo frammentato o con ponti cromatinici, anisopoichilocitosi, sovraccarico marziale
e negatività del test anti-I (antigene marcatore dell’eritropoiesi fetale) e del test del siero acidifi cato, da cui l’acronimo HEMPAS (Hereditary Erythroblastic Multinuclearity with a Positive Acidifi ed Serum test).
• CDA tipo II : eredità autosomica recessiva; caratterizzata da normocrocitosi con eritroblasti bi- o multinucleati sottoposti a fenomeni di carioressi, anisopoichilocitosi, assenza di sovraccarico marziale e positività del test anti-I e del test del siero acidifi cato.
• CDA tipo III : eredità autosomica dominante; caratterizzata da macrocitosi con eritroblasti
Anemie diseritropoietiche congenite M.D. Cappellini
N AS AMC
T
T
TS
S S
Figura 48.5 Relazioni tra
sideremia (S) e concentrazione
di transferrina (T) nel siero, nel normale
(N), nell’anemia sideropenica (AS)
e nell’anemia delle malattie infi ammatorie
croniche (AMC).
C0240.indd 1058C0240.indd 1058 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Capitolo 48 - ANEMIE
81059
multinucleati giganti sottoposti a fenomeni di carioressi, anisopoichilocitosi, sovraccarico marziale e negatività del test anti-I e del test del siero acidifi cato.
Nell’ambito delle anemie diseritropoietiche congenite è inoltre compresa l’anemia sideroblastica congenita , trasmessa come carattere X-linked, caratterizzata da
sideroblasti e siderociti, microcitosi, ipocromia e ipersi-deremia, con alterato metabolismo porfi rinico. Questo tipo di anemia risponde alla terapia con piridossina a dosi elevate; nei casi resistenti viene posta indicazione alla terapia trasfusionale e, di conseguenza, ferro-chelante. Nella CDA II la splenectomia in alcuni casi può limitare o eliminare il supporto trasfusionale.
Le anemie sideroblastiche sono un gruppo eterogeneo di disordini caratterizzati dalla presenza di eccessive quan-tità di ferro nei mitocondri perinucleari dei normoblasti. Questa anomalia si rende evidente negli strisci midollari colorati per il ferro (blu di Prussia), dove compaiono eri-troblasti con un anello più o meno completo attorno al nucleo (sideroblasti ad anello) .
Eziologia Molti sono i disordini associati all’anemia siderobla-stica e altrettanto numerose le classifi cazioni proposte ( Tab. 48.7 ). Una prima suddivisione riguarda la presenza di una forma acquisita e di una ereditaria. Quest’ultima è trasmessa come un’anomalia legata al cromosoma X, con penetran-za variabile (ci sono segnalazioni anche in favore di una trasmissione autosomica-recessiva). La forma acquisita (molto più frequente) può osservarsi in associazione con una grande varietà di farmaci, di infi ammazioni croniche, di neoplasie. Spesso può capi-tare di non riuscire a identifi care una possibile causa e allora si parla di anemia sideroblastica acquisita idiopa-tica refrattaria; essa appartiene alle mielodisplasie e sarà descritta più avanti.
Patogenesi L’abnorme accumulo di ferro nei mitocondri degli eritro-blasti ha fatto sì che l’interesse si concentrasse sugli aspetti mitocondriali della sintesi dell’eme (per la regolazione della sintesi dell’eme e il metabolismo delle porfi rine si veda Fig. 69.3 nel Capitolo 69 ). La prima tappa comporta la formazione di acido Δ -ami-nolevulinico, a partire da glicina e succinil CoA. Questa reazione è catalizzata da un enzima mitocondriale, l’acido Δ -aminolevulinico-sintetasi (ALA-sintetasi) , che è l’unico enzima della catena metabolica limitante la velocità di reazione e richiede l’intervento del piridossal-5-fosfato, che è la forma coenzimatica attiva della vitamina B 6 . Esiste quindi una relazione tra vitamina B 6 e sintesi di Hb. La trasformazione dell’acido Δ -aminolevulinico in porfobili-nogeno, uroporfi rinogeno III, coproporfi rinogeno e proto-porfi rina IX si verifi ca a livello delle creste mitocondriali o nel citoplasma, mentre la coniugazione tra le molecole di ferro e le protoporfi rine è mediata dall’eme-sintetasi, un enzima mitocondriale. Esiste infi ne un meccanismo a feedback negativo sull’at-tività dell’ALA-sintetasi; se c’è molto eme si accentua l’inibizione, se l’eme manca si riduce l’inibizione. La difettosa sintesi dell’eme, ritenuta il meccanismo patogenetico principale, può conseguire dunque ad al-terazioni del metabolismo della vitamina B 6 e a modifi -cazioni di attività dell’ALA-sintetasi, dell’eme-sintetasi o di qualcuno degli altri enzimi implicati nella sintesi porfi rinica. Tra i casi ereditari , complessivamente rari, la forma più fre-quente, trasmessa come carattere legato al cromosoma X a penetranza variabile, sembra potersi ascrivere a un defi cit di ALA-sintetasi oppure di coproporfi rinogeno-ossidasi, l’enzima che catalizza il passaggio da coproporfi rinogeno III a protoporfi rinogeno IX. Come si è accennato, l’ anemia acquisita idiopatica refrat-taria appartiene al gruppo delle mielodisplasie e, quindi, delle malattie delle cellule staminali ematopoietiche a evoluzione clonale, analogamente alle leucemie. Nel caso delle forme acquisite secondarie a numerosi farmaci e ad alcolismo, l’alterazione principale è a livello del me-tabolismo della vitamina B 6 , per il frequente verifi carsi di un defi cit condizionato da un aumento del fabbisogno e non da un diminuito apporto. Infi ne, nell’ intossicazione da piombo il quadro ematologico può essere correlato a diversi effetti biochimici. In primo luogo si ha inibizione dell’acido Δ -aminolevulinico-deidra-tasi, della coproporfi rinogeno-ossidasi e dell’eme-sintetasi, enzimi, come si è visto, interessati alla sintesi dell’eme. Ma
Anemie sideroblastiche G. Gromo
Ereditarie � Legata al cromosoma X (defi cit di Δ-aminolevulinico-sintetasi) � Autosomica recessiva
Acquisite � Idiopatica refrattaria (una delle forme di mielodisplasia) � Secondaria a:
– farmaci antagonisti della vitamina B6 (isoniazide, L-dopa, ecc.); tossine mitocondriali (cloramfenicolo ecc.); meccanismo d’azione ignoto (azatioprina ecc.)
– alcolismo – intossicazione da piombo – malattie mieloproliferative (per esempio, mielofi brosi,
leucemia) – malattie linfoproliferative (per esempio, linfoma, mieloma)
� Cause varie (per esempio, tubercolosi, artrite reumatoide, malattie della tiroide, porfi rie, neoplasie ecc.)
Tabella 48.7 Classifi cazione delle anemie sideroblastiche
C0240.indd 1059C0240.indd 1059 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1060
il piombo interferisce anche sulla pompa del sodio della membrana dei GR inibendo l’ATPasi (ridotta sopravviven-za delle emazie; si veda a proposito della sferocitosi), nel metabolismo del DNA e dell’RNA (inibizione della pirimi-din-nucleotidasi) e infi ne nei processi calcio-dipendenti, compresa la respirazione mitocondriale; quest’ultima alterazione, unita alla compromissione della sintesi dell’eme, può contribuire alla formazione di sideroblasti ad anello.
Fisiopatologia Le anemie sideroblastiche sono caratterizzate da una difficoltosa formazione di emoglobina, in quanto il ferro che viene normalmente assunto dagli eritroblasti, a causa delle alterazioni enzimatiche citate, non viene regolarmente incorporato nelle molecole di emoglo-bina e si deposita all’interno degli eritroblasti stessi, in particolare nei mitocondri perinucleari. L’anemia si presenta, in una parte di casi, con gli stessi caratteri dell’anemia sideropenica, con ipocromia e microcitosi a distribuzione non uniforme; in altri casi il volume corpuscolare medio è normale e talvolta addirittura aumentato. Questa variabilità di reperti indica, oltre all’esistenza di un disturbo nella sintesi dell’emoglo-bina, anche un’alterata maturazione degli eritroblasti, che si accompagna a una certa quota di eritropoiesi anomala e ineffi cace.
Manifestazioni cliniche Nella forma ereditaria l’anemia può essere presente nei primi mesi di vita, ma di solito viene diagnosticata per la prima volta nei giovani adulti di sesso maschi-le, anche se non è chiara la ragione di questo ritardo diagnostico. L’anemia acquisita idiopatica refrattaria si verifi ca soprat-tutto nei soggetti di oltre 50 anni (età media 66 anni); è comunque possibile che sia sottodiagnosticata e come tale molto più frequente di quanto non si pensi. Le forme ereditarie e quelle acquisite (a eccezione di quella idiopatica) si comportano in maniera abbastanza simile e non si discostano, per quanto riguarda la sintomatologia, dalle altre anemie. Il grado di anemizzazione (mediamente 6,5/g/dL di Hb), come pure l’ipocromia e la microcitosi, è notevole. Al microscopio si osservano anisopoichilocitosi, cellule a bersaglio , granulazioni basofi le e un relativo dimorfi smo eritrocitario. Per confermare la diagnosi è necessario l’esa-me di un preparato midollare colorato con blu di Prussia, per la dimostrazione del ferro; mentre il midollo normale presenta circa il 30-50% di eritroblasti contenenti alcuni granuli di ferro disposti nel citoplasma in modo del tutto casuale (sideroblasti), in condizioni patologiche sono stati descritti due tipi di sideroblasti. Un primo tipo è caratterizzato da un aumento generalizzato del numero e del diametro dei granuli citoplasmatici e si riscontra in numerose affezioni quali emosiderosi, emocromatosi, ta-lassemie, anemie ipoplastiche ed emolitiche; un secondo tipo, proprio delle anemie sideroblastiche, mostra il ferro localizzato elettivamente nei mitocondri perinucleari (sideroblasti ad anello).
Il quadro midollare presenta anche, con discreta fre-quenza, un’iperplasia eritroblastica con prevalenza degli elementi basofili e talvolta una piccola quota di megaloblasti. La sideremia è elevata, la transferrinemia bassa. Al quadro sopra descritto fa eccezione la forma idiopa-tica refrattaria, che presenta alcune peculiarità. L’esame obiettivo è generalmente nella norma, a eccezione di un’epatosplenomegalia modesta (40% dei casi). Quanto alle costanti ematologiche si rileva, più di frequente, una macrocitosi (MCV 100 fL), talvolta associata a piastrino-penia e granulocitopenia. Da segnalare, inoltre, a livello midollare un’iperplasia eritroide (M:E = 1:1), modifi ca-zioni in senso megaloblasticoo e il 43-95% di sideroblasti ad anello. La percentuale di saturazione della transferrina è notevol-mente aumentata, come pure la ferritina. Nel 50% dei casi ci sono anomalie cromosomiche. La sopravvivenza mediana di questi pazienti è di 10 anni, destinata comunque ad aumentare nei casi in cui non si richiede terapia trasfusionale. Nel 7% l’evento terminale è una leucemia mielomonoblastica acuta.
Diagnosi Le anemie sideroblastiche devono essere distinte da altre cause di anemia microcitica ipocromica. Nel corso di una carenza di ferro, sideremia e ferritina sono ridotte, TIBC aumentata e, negli strisci midollari, macrofagi ed eritroblasti sono privi di ferro colorabile con il blu di Prussia. L’anemia delle malattie croniche è caratterizzata da sideremia bassa, ridotta TIBC, ferritina normale e depositi di ferro midollare normali o modestamente aumentati. Quanto ai sideroblasti patologici, pur essendo presenti, non hanno mai la caratte-ristica struttura “ad anello”. Nell’anemia sideroblastica idiopatica refrattaria la ma-crocitosi e la presenza di megaloblasti a livello midollare potrebbero ricordare una carenza primaria di B 12 o acido folico. Anche se i sideroblasti ad anello possono essere presenti in queste forme, tuttavia il loro numero è note-volmente inferiore. Resta pur sempre la possibilità di una sovrapposizione, cioè di una carenza di folati secondaria ad anemia sideroblastica.
Terapia La terapia è possibile nei casi in cui esista un deficit condizionato di vitamina B 6 , quindi nelle forme ac-quisite secondarie ad altre malattie, a farmaci o ad agenti tossici. Assai di frequente, però, le dosi cura-tive sono molto elevate, nell’ordine delle centinaia di milligrammi. Le forme ereditarie non sempre rispondono alla pi-ridossina e non di rado si osservano comportamen-ti diversi tra i membri affetti di uno stesso nucleo familiare. Nella forma idiopatica, viceversa, non vi è pratica-mente nulla da fare, anche se si registrano alcuni casi sensibili alla somministrazione di piridossal-5-fosfato (supposto deficit di piridossal-chinasi).
C0240.indd 1060C0240.indd 1060 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Capitolo 48 - ANEMIE
81061
Le talassemie sono un gruppo di anemie ereditarie carat-terizzate dalla ridotta o abolita produzione di una o più catene peptidiche che costituiscono la porzione globinica dell’emoglobina. Questa situazione comporta un alterato rapporto tra ca-tene peptidiche globiniche di diverso tipo, precipitazione intracellulare delle catene peptidiche, che risultano in eccesso, e alterazioni sia dell’eritropoiesi sia della soprav-vivenza degli eritrociti che in queste condizioni vengono prodotti. Le talassemie sono molto comuni in una fascia geografi ca che va dalle coste del Mediterraneo ai Balcani, al Medio Oriente, all’India, all’Asia sudorientale, fino alla Nuo-va Guinea. In Italia una forma particolare di talassemia ( � -talassemia) è particolarmente comune nelle regioni in-sulari, nel meridione della penisola e nel delta padano (con particolare concentrazione nella provincia di Ferrara).
Regolazione genetica della sintesi di emoglobina La porzione proteica della molecola emoglobinica, o glo-bina, è formata da quattro catene peptidiche a due a due uguali ( Fig. 48.6 ). Nell’adulto la maggior parte dell’emo-globina comprende due catene di 141 aminoacidi, dette � , e due catene di 146 aminoacidi, dette � . La costituzione dell’emoglobina dell’adulto (HbA) viene perciò indicata come � 2 � 2 . In realtà, i geni strutturali che codificano la sintesi dell’emoglobina sono ben più di due (uno per le catene � e uno per le catene � ) e possono essere distinti in due gruppi. Il primo, che per comodità sarà chiamato di tipo � , include due geni strutturali presenti sul cromosoma 16, indicati (dal nome della catena peptidica che codifi cano) con le lettere greche zeta ( � ) e alfa ( � ). Questi ultimi sono presenti in due copie su ciascun cro-mosoma 16. Il secondo gruppo, che per comodità sarà chiamato di tipo non � , include ben quattro geni strutturali, presenti sul cromosoma 11, indicati con le lettere greche epsilon ( ε ), gamma ( � ), delta ( � ) e beta ( � ). I geni � e � sono con-tigui e sono presenti ciascuno in una singola copia. Per le catene � esiste, in realtà, un blocco di geni strutturali defi niti rispettivamente G � 1 , G � 2 e A � , così che le catene � sintetizzate sono in realtà una miscela eterogenea. Le differenze sono però così trascurabili che, ai fi ni pratici, si possono considerare le catene � come se fossero omo-genee e codifi cate da un singolo gene. L’organismo utilizza tutti questi geni nelle varie tappe del suo sviluppo, alcuni di essi in successione in una specie di staffetta, dando origine a vari tipi di Hb in base alla regola che si ha una combinazione due a due tra le ca-tene di tipo � e le catene di tipo non � prodotte in un particolare momento. Nella vita embrionale vengono utilizzati, sul cromosoma 16, prima i geni che codifi cano le catene � e poi i geni che codifi cano le catene � , mentre, sul cromosoma 11, vengo-no utilizzati prima i geni che codifi cano le catene ε e poi i geni che codifi cano le catene � . Il risultato è che vengono prodotte in successione prima un’emoglobina defi nita Hb Gower 1 ( � 2 ε 2 ), poi una miscela tra un’emoglobina defi nita
Hb Gower 2 ( � 2 ε 2 ) e un’emoglobina defi nita Hb Portland ( � 2 � 2 ) , e infi ne un’emoglobina defi nita Hb F ( � 2 � 2 ) o emo-globina fetale; questa denominazione dipende dal fatto che questa emoglobina predomina nettamente durante tutta la vita fetale. Nella vita fetale , tuttavia, inizia l’utilizzo di altri due geni strutturali del cromosoma 11, prima quelli che codifi -cano le catene � e poi quelli che codifi cano le catene � . Può perciò iniziare la formazione dell’emoglobina di tipo adulto o HbA ( � 2 � 2 ) e di HbA 2 ( � 2 � 2 ). L’utilizzo del gene che codifi ca le catene � cresce con il tempo e va di pari passo con una minore espressione del gene che codifi ca le catene � . Ne consegue che all’aumentare della forma-zione di HbA corrisponde una riduzione nel tempo della formazione di HbF. Alla nascita l’HbF è circa il 60% di tutta l’Hb, mentre al termine del primo anno di età è ridotta a quantità tra-scurabili e tale resterà per il resto della vita (circa 0,5%). La formazione di catene � prosegue nella vita adulta, ma sempre in misura modesta, così che l’HbA 2 si aggira normalmente sul 2,5% del totale. Perciò, nella vita adul-ta l’HbA costituisce normalmente circa il 97% di tutta l’emoglobina. Uno schema con la successione temporale della sintesi delle varie Hb è presentato nella fi gura 48.7 .
Eziopatogenesi Le talassemie dipendono da un’anomalia di uno o più geni codifi canti le catene peptidiche della globina, la cui
Talassemie F. Pellò
Cromosoma 16 ζ αε γ
Cromosoma 11
Embrione Feto 1° anno dopo il 1° anno
100%0,5%60%
97,0%40%
2,5%
βδ Figura 48.7
Successione temporale della sintesi delle varie emoglobine.
Cromosoma 16Due copie di una tra le seguenti catene di tipo α:ζ, α
Cromosoma 11Due copie di una tra le seguenti catene di tipo non α:ε, γ, δ, β
Figura 48.6 Rappresentazione schematica della struttura delle emoglobine.
Gli spicchi rappresentano le catene peptidiche della globina. I cerchi con il segno + i gruppi eme, uno per ciascuna catena peptidica. A fi anco sono indicate le possibili catene peptidiche dei due tipi che, nelle emoglobine normali, sono sempre a due a due uguali.
C0240.indd 1061C0240.indd 1061 6/9/10 3:08:59 PM6/9/10 3:08:59 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1062
produzione viene abolita del tutto o in parte. In teoria, il difetto può interessare uno qualsiasi di questi geni, ma in realtà i difetti che contano sono a carico dei geni co-difi canti le catene � ( � -talassemia ), dei geni codifi canti le catene � ( � -talassemia ), o, in forma combinata, dei geni codifi canti le catene � e quelle � ( � - � -talassemia) .
Natura dell’alterazione genetica La normale espres-sione dei geni delle catene peptidiche dell’Hb comporta la loro trascrizione primaria in lunghe molecole di RNA (detto RNA eterogeneo nucleare o HnRNA). Nel nucleo degli eritroblasti, queste molecole sono tra-sformate nella forma più piccola e defi nitiva dell’RNA messaggero (mRNA) grazie a un’operazione defi nita pro-cessing , che consiste nell’eliminazione di questi segmenti dell’HnRNA, i quali corrispondono agli introni (porzioni di DNA non destinate a essere tradotte) e nella congiun-zione ( splicing ) dei segmenti che corrispondono agli esoni (porzioni di DNA destinate a essere tradotte). Successiva-mente, l’mRNA è tradotto nella globina sui poliribosomi del citoplasma con coinvolgimento di RNA transfer, che trasporta gli aminoacidi specifi ci sui polipeptidi globinici in crescita. Le catene di tipo � e di tipo non � complete, che vengono costruite su ribosomi separati, vengono liberate, si inserisce il gruppo eme e si forma il tetramero di emoglobina. Molte forme di � -talassemia e alcuni tipi di � - o � - � -talassemia dipendono dalla delezione totale o parziale dei geni cor-rispondenti. Nella maggior parte dei casi di � -talassemia, l’alterazione genetica è di tipo differente ed è per lo più legata a mutazioni puntiformi che interferiscono con una qualsiasi delle tappe che conducono alla trascrizione e alla traduzione dei geni così mutati, o che condizionano la formazione di catene globiniche altamente instabili, distrutte subito dopo essere formate. Le possibilità sono varie, per cui va ricordato che, sul piano della genetica molecolare, esistono diverse forme di � -talassemia e per molte di queste il meccanismo molecolare non è ancora stato accertato. Un caso particolare è rappresentato dal gene Lepore, che deriva da un crossing-over asimmetrico tra il gene che codi-fi ca le catene � e quello che codifi ca le catene � ( Fig. 48.8 ); l’Hb Lepore ha normali catene � combinate con catene derivanti dalla fusione tra i residui NH 2 terminali della catena e i residui COOH terminali della catena � .
Il punto di fusione è variabile, per cui si hanno tipi diversi di Hb Lepore, per diversa lunghezza dei residui � e � nella catena non � . Il gene Lepore sintetizza la corrispondente catena pep-tidica in misura quantitativamente ridotta rispetto alle normali catene � , da qui deriva la classifi cazione di questa anomalia tra le talassemie. Le emoglobine anti-Lepore sono i prodotti reciproci, in cui persistono un gene � e un gene � completo più il prodotto di fusione consistente delle porzioni di geni � e � persi nell’Hb Lepore; le principali sono l’Hb Miyada e l’Hb P-Nilotic. Una situazione correlata alle talassemie è la persistenza ereditaria di Hb fetale (HPFH, Hereditary Persistence of Fetal Haemoglobin) , in cui la produzione di quantità re-lativamente grandi di Hb fetale continua nella vita adulta, in assenza di anomalie ematologiche importanti. L’HbF è distribuita uniformemente nelle emazie, al con-trario di quanto avviene nella � -talassemia; la condizione è dovuta al mancato switch perinatale F → A.
Varianti genetiche • Difetto di catene � : come si è visto, l’ � -talassemia
è dovuta, nella maggior parte dei casi, a delezione genetica. Essendo presenti in due copie sui cromosomi 16 i geni che codifi cano le catene � , ne consegue che esistono due alterazioni possibili a livello del singolo cromosoma: 1) delezione di entrambe le copie del gene della catena � (situazione indicata con la notazione � 0 ) e 2) delezione di una sola copia del gene della catena � (situazione indicata con la notazione � + ). Le combinazioni genetiche possibili sono indicate nella tabella 48.8 . Come si vede, l’assenza di una sola delle quattro copie normalmente disponibili del gene per le catene � (due per ciascuno dei due cromosomi) è priva di equivalente clinico e comporta solo lo stato di portatore silente. L’assenza di due copie del gene determina un’alterazione ematologica lieve compatibile con un buono stato di salute, defi nita trait � -talassemico (si veda oltre). L’assenza di tre copie del gene è invece accompagnata da un quadro clinico molto grave con presenza nelle emazie di un’emoglobina anomala formata da tetrameri di catene � (HbH = � 4 ). L’assenza di tutte e quattro le copie del gene è incompatibile con la vita, entrando le catene � nella costituzione di tutte le emoglobine a partire dalla vita fetale. Ne
Geni Genotipi
� � = � (normale)
� � / � – , ( � � + ) portatore silente
� – = � + � – / � – , ( � + / � + ) trait talassemico
– – = � ° � � /– –, ( � / � °) trait talassemico
– –/ � – , ( � °/ � + ) malattia da HbH ( � 4 )– –/– –, ( � °/ � °) idrope fetale con Hb Barts ( � 4 )
Geni Genotipi
Tabella 48.8 Combinazioni genetiche possibili nella � -talassemia e corrispettivi clinici
Figura 48.8 Crossing-over asimmetrico
che conduce a formazione del
gene Hb Lepore e anti-Lepore.
C0240.indd 1062C0240.indd 1062 6/9/10 3:09:00 PM6/9/10 3:09:00 PM

Capitolo 48 - ANEMIE
81063
risulta la morte intrauterina con il quadro dell’idrope fetale. La scarsa quantità di emoglobina che si forma in queste condizioni è anomala e formata da tetrameri di catene � (Hb Barts = � 4 ) . Una variante di � -talassemia importante per la conferma del modello genetico è quella associata alla presenza nel sangue della cosiddetta emoglobina Constant-Spring (HbCS) , che contiene catene � normali e una catena � abnormemente lunga, con 172 (CSI) o 169 (CSII) aminoacidi (in luogo dei normali 141) situati all’estremità C terminale della catena � CS . L’HbCS è sintetizzata in quantità ridotta; allo stato omozigote vi è il 5-6% di HbCS, HbA 2 normale, tracce di Hb Barts e il resto è costituito da HbA, a indicare che esistono � loci normali nell’omozigosi CS e quindi che il locus � è duplice ( � CS � / � CS � ). L’emoglobina Constant-Spring è diffusa nel Sud-Est asiatico, dove è presente nel 3% della popolazione.
• Difetto di catene � (semplice o associato) : si è visto che i meccanismi molecolari alla base della � -talassemia sono molteplici e diversi da un caso all’altro. Tuttavia, fortunatamente per il clinico, i risultati possono essere solo due: 1) assenza completa di sintesi di catene � , e i geni corrispondenti vengono indicati con la notazione � 0 ; 2) ridotta sintesi di catene � , e i geni corrispondenti vengono indicati con la notazione � + . Lo stesso può dirsi per la � - � -talassemia, nella quale il condizionamento genetico può comportare la mancata produzione di entrambe le catene peptidiche ( � - � ) 0 o la loro produzione in quantità ridotta ( � - � ) + (è questa una forma che infl uenza la sintesi delle catene globiniche in forma molto lieve e che ha importanza soprattutto per la discendenza). È da segnalare che in alcune forme di � -talassemia con il gene � + e in tutte le forme di � - � -talassemia con il gene ( � - � ) 0 si ha una persistenza a livelli superiori alla norma di HbF; ciò si traduce in una minore gravità del quadro clinico in caso di omozigosi. Le combinazioni genetiche possibili in queste forme morbose sono indicate nella tabella 48.9 . Il signifi cato dei termini ivi impiegati verrà spiegato in seguito (si veda oltre, Manifestazioni
cliniche delle � -talassemie ). Risulta perciò chiaro che i pazienti con le forme più lievi di talassemia sono eterozigoti e quelli con le forme più gravi omozigoti. Nel caso di matrimonio di un eterozigote con un normale, esiste una probabilità del 50% che nascano fi gli talassemici, essi pure con la forma eterozigote della malattia. Nel caso del matrimonio di due eterozigoti esiste una probabilità del 25% che nascano fi gli con la forma omozigote della � -talassemia e del 50% che nascano fi gli con la forma eterozigote. Perciò, i pazienti affetti da talassemia major dovrebbero avere entrambi i genitori affetti dalla forma più lieve della malattia. In casi nei quali solo uno dei genitori è apparso in realtà affetto, lo studio della sintesi delle catene globiniche negli eritroblasti del genitore apparentemente sano ha dimostrato che anche questi presenta un difetto che può essere ricondotto a una forma eterozigote di ( � - � ) + talassemia. Deve essere aggiunto che, derivando per lo più il difetto genetico della � -talassemia da mutazione del gene per la catena peptidica corrispondente (e non da delezione genetica), è facile che questa forma sia diffusa in aree geografi che dove coesistono altre anomalie ereditarie della sintesi dell’emoglobina, sempre prodotte da mutazione del gene delle catene � , ma che conducono alla sua sintesi in forma qualitativamente alterata (emoglobinopatie). Si possono perciò avere casi di doppia eterozigosi tra il gene della � -talassemia e quello dell’emoglobina anomala.
Fisiopatologia L’emopoiesi talassemica è dominata dallo sbilancio sin-tetico � /non � . Nelle forme più gravi di � -talassemia (un solo gene � funzionante) si ha un eccesso di catene � che formano tetrameri di HbH ( � 4 ); questa Hb è altamente instabile e può precipitare nelle cellule. Nella � -talassemia si ha diminuita produzione di ca-tene globiniche � ed eccesso relativo di catene � che, quando non vengono incorporate in HbA o in HbF, sono relativamente instabili in soluzione e tendono ad aggregarsi e a precipitare; in quantità relativamente pic-cole si trovano anche catene � libere. Nei pazienti non talassemici il rapporto � : � è all’incirca 1; nei reticolociti della � -talassemia eterozigote esso è intorno a 2, mentre negli omozigoti, quando le catene � sono dimostrabili, varia tra 5 e 25. Il materiale � globinico aggregato può essere visto alla mi-croscopia elettronica sia nei nuclei sia nel citoplasma degli eritroblasti e nei reticolociti. La precipitazione dell’eccesso di catene � ha luogo quando procede l’emoglobinizzazione degli eritroblasti e compare già negli eritroblasti policro-matofi li. La quota di cellule che contengono inclusi au-menta progressivamente con la maturazione eritroblastica; in alcuni casi di grave defi cit delle catene � le inclusioni possono assumere le dimensioni del nucleo picnotico dei normoblasti maturi. Gli effetti della precipitazione intracellulare dell’eccesso di catene � possono essere così riassunti: • eccessiva liberazione di radicali liberi che provocano
la perossidazione della membrana, formazione di metaemoglobina e danno a diversi processi
Gene Omozigote Eterozigote � + Talassemia major
o intermediaTalassemia minor o minima
� ° Talassemia major Talassemia minor o intermedia
( � - � )° Talassemia intermedia Talassemia minor
( � - � ) + Non descritta Portatore * silente
Lepore Talassemia major con Hb Lepore
Talassemia minor con Hb Lepore
* Menomazione della sintesi delle catene � e � molto lieve e che ha importanza solo nella discendenza qualora il coniuge sia portatore di � -talassemia (si veda il testo).
Tabella 48.9 Combinazioni genetiche possibili nella � -talassemia e corrispettivi clinici
C0240.indd 1063C0240.indd 1063 6/9/10 3:09:00 PM6/9/10 3:09:00 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1064
metabolici che hanno come effetto un aumento della rigidità dell’emazia;
• inibizione della proliferazione delle cellule eritroidi, con blocco in fase G 1 e morte in situ (eritropoiesi ineffi cace);
• alterata fosforilazione delle proteine della membrana che causa una spiccata distorsione dell’emazia; queste emazie con membrana difettosa, unitamente agli eritrociti provvisti di corpi inclusi, che vengono ulteriormente danneggiati dal passaggio attraverso la milza (con asportazione dei corpi inclusi), causano un rallentamento a livello del microcircolo.
La mancanza di catene � e l’eccesso dannoso di catene � sono parzialmente compensati dalla riattivazione della sintesi delle catene � e � , le quali sono in grado di aumen-tare l’emoglobinizzazione del GR con la formazione di Hb F e A 2 e la “neutralizzazione” di alcune delle catene � libere. Ugualmente, nella � - � -talassemia e nella HPFH la mancanza della sintesi � –globinica ha un minore effetto sul bilancio � /non � , perché la sintesi di catene � com-pensa in parte quella di catene � . La fi siopatologia dei sintomi della talassemia ha come punto centrale l’anemia ( Fig. 48.9 ), dovuta a prematura mortalità intramidollare degli eritroblasti (eritropoiesi ineffi cace), al minor volume delle emazie (microcitosi), al minor conte-nuto corpuscolare medio di emoglobina (ipocromia), alla diminuita sopravvivenza eritrocitaria per esaltata emocate-resi splenica secondaria al danno di membrana. L’anemia cronica stimola l’eritropoiesi che si espande a livello midollare in aree dalle quali essa è normalmente assente, come le ossa lunghe, e anche a livello extra-midollare, nel fegato, nella milza, nei linfonodi. Nella
talassemia major questo si verifica in una fase molto precoce dello sviluppo, con conseguenti alterazioni della struttura e della forma delle ossa, aventi come risultato la facies simil-mongoloide. Successivamente il midollo può uscire attraverso i fori va-scolari, provocando formazioni pseudotumorali vicino al luogo della sua origine, di solito in sede paravertebrale. I precursori eritroidi � -talassemici contenenti le inclusioni di aggregati di catene � vanno incontro in buona parte a morte intramidollare (nel midollo si è osservata la fagocitosi di interi eritroblasti); quando essi giungono a maturazione, i corpi inclusi nelle emazie vengono elettivamente “snocciolati” dalla milza e si originano in questo modo eritrociti dalla forma bizzarra (schistociti, eritrociti “a lacrima” ecc.). In sostanza, il destino di ogni cellula dipende dal grado del danno subito durante i vari stadi di sviluppo; quelle più gravemente lese sono incapaci di abbandonare il midollo e muoiono in situ , mentre quelle con minori precipitati nel passaggio attraverso la milza possono essere private degli inclusi e ritornare in circolo oppure venire distrutte. La milza va incontro ad aumento di volume per iperplasia della componente reticolare macrofagica; di conseguenza aumenta il sequestro splenico delle emazie, da cui emolisi più intensa, instaurando così un circolo vizioso. Nell’organismo talassemico si viene a creare un accumulo marziale dovuto al processo emolitico, all’aumentato assorbimento di ferro che si ha quando è stimolata l’eri-tropoiesi (si veda in precedenza, Metabolismo del ferro) e all’apporto di ferro coincidente con la terapia emotrasfu-sionale, necessaria nei casi più gravi. La conseguenza è una siderosi dei vari parenchimi, in particolare del miocardio, del fegato, del pancreas.
Alterato sviluppo e accrescimento
MiocardiosiderosiEpatosiderosi
ACCUMULO DI FERRO
Terapiaemotrasfusionale
Aumentato assorbimentodi ferro
Processo emolitico
+
+
Modificazioni ossee
Altre
Espansioneeritropoietica
Stimolo all’eritropoiesi
Prematura mortalità intramidollaredegli eritroblasti
(eritropoiesi inefficace)
Poche cellule in circolo, microciticheipocromiche, talora con corpi inclusi
Ridotta sopravvivenza delle emazie
ANEMIA CRONICA SPLENOMEGALIA
+
Figura 48.9 Schema
sintetico della fi siopatologia
dei sintomi delle talassemie.
C0240.indd 1064C0240.indd 1064 6/9/10 3:09:00 PM6/9/10 3:09:00 PM

Capitolo 48 - ANEMIE
81065
Comuni sono anche le alterazioni endocrine per il dan-neggiamento di pancreas, tiroide, paratiroidi e surreni, che sono le complicanze più comuni per accumulo di ferro nei pazienti adulti. Negli adolescenti l’accumulo di ferro nell’ipofi si anteriore disturba la maturazione sessuale in circa il 50% dei casi.
Manifestazioni cliniche delle � -talassemie Qui di seguito verranno affrontate prima la � -talassemia e poi la � -talassemia; la � -talassemia è infatti diffusa nell’area mediterranea e in Italia, mentre l’ � -talassemia lo è soprattutto nel Sud-Est asiatico. Nella descrizione clinica delle singole forme talassemiche occorre una precisazione terminologica. La più ampia va-rietà di forme cliniche è stata descritta per la � -talassemia, per la quale si sono considerate tre possibilità: • anemia grave con sopravvivenza impossibile
oltre l’età infantile in assenza di emotrasfusioni, talassemia major;
• anemia grave, ma con sopravvivenza possibile fi no all’età adulta, anche in assenza di emotrasfusioni, talassemia intermedia;
• anemia modesta spesso compatibile con uno stato di salute normale, trait talassemico, tradizionalmente indicato nella letteratura italiana come talassemia minor o malattia di Greppi-Rietti-Micheli.
Tuttavia, possono esserci forme di talassemia minor to-talmente irrilevanti e altre di qualche signifi cato; queste sono defi nite nella letteratura anglosassone come stati di silent carrier (portatore silente) ed erano indicate tra-dizionalmente nella letteratura italiana con il termine di talassemia minima . Si ritiene perciò giustifi cato adottare per le prime la dizione, da alcuni abbandonata, di talas-semia minima (o trait talassemico) .
Considerazioni generali Le manifestazioni cliniche della � -talassemia hanno intensità diversa in rapporto a: • riduzione della HbA normale; • età di comparsa di ridotta sintesi di emoglobina; • entità dello sbilanciamento sintetico � /non � , con
presenza più o meno rilevante di catene � libere con i caratteristici precipitati che danno luogo ai corpi inclusi;
• presenza di altre Hb che possono esercitare una funzione di relativo compenso, in particolare HbF.
Com’è indicato nella tabella 48.9 , i tre principali quadri clinici di talassemia major (o malattia di Cooley), talas-semia intermedia e talassemia minor (o malattia di Rietti-Greppi-Micheli) riconoscono una pluralità di genotipi, anche se le forme più gravi sono preferenzialmente asso-ciate all’omozigosi e la talassemia minor all’eterozigosi. A questi tre fenotipi principali vanno ancora aggiunti la talassemia minima (o trait talassemico) e lo stato di portatore silente. Va però sottolineato che l’eccessivo schematismo non inquadra tutti i genotipi possibili, tenendo conto anche dei casi di doppia eterozigosi tra due geni talassemici o tra un gene talassemico e un gene responsabile di emoglobi-nopatia. D’altra parte, poiché la fi siopatologia del processo
talassemico è unitaria, il quadro clinico ed ematologico è caratterizzato da alcuni sintomi fondamentali presenti, con diversa intensità e con passaggio sfumato e graduale, nelle forme di talassemia di diversa gravità.
� -talassemia major (malattia di Cooley) È la forma più grave di anemia emolitica congenita. Le manifestazioni cliniche compaiono a 4-6 mesi di vita, quando nel sog-getto normale ha luogo il passaggio dalla prevalenza della produzione di catene � alla prevalenza della produzione di catene � e � . I pazienti hanno i sintomi di una grave anemia. All’esame obiettivo si nota ipoevolutismo somatico, con peso e sta-tura inferiori alla norma. La cute è pallida con un colore caratteristico dovuto alla combinazione di ittero e aumen-tata deposizione di melanina. La facies è mongoloide, con ossa zigomatiche prominenti, ossa del cranio ispessite, radice del naso infossata, palpebre tumide, allargamento delle mandibole con malocclusione. È costante una cardiomegalia, per il sommarsi dell’ipossia cronica e della siderosi miocardica, con segni di scom-penso di circolo; costante è anche l’epatosplenomegalia. L’epatomegalia di solito è meno spiccata della spleno-megalia, ma raggiunge dimensioni notevoli dopo sple-nectomia, esercitando una specie di funzione vicariante della milza per quanto riguarda le funzioni fi ltranti e di emopoiesi extramidollare. Si possono anche avere epatiti da trasfusione, epatopatia cronica da sovraccarico mar-ziale, da scompenso di circolo congestizio, da defi cienze nutritive. La splenomegalia, per l’ingombro addominale, può de-terminare eventuale compressione dello stomaco e del rene sinistro; inoltre, la milza ingrandita può andare in-contro, anche se raramente, a infarto splenico e, infi ne, può instaurarsi un ipersplenismo con pancitopenia (si veda il Capitolo 55 ). La maggior parte di questi segni può essere notevolmente ridotta, se il paziente è regolarmente e adeguatamente trasfuso; in questo caso, tuttavia, risultano particolar-mente gravi i segni derivanti dalla deposizione di ferro nel miocardio e in altri parenchimi.
Esami di laboratorio L’esame emocromocitometri-co rivela un’anemia molto spiccata, con diminuzione dell’emoglobina e del numero dei GR, microcitosi con MCV spiccatamente diminuito e ipocromia con MCH ridotto e MCHC normale. È di fondamentale importanza l’esame morfologico dello striscio del sangue periferico; i GR hanno un aspetto estremamente polimorfo e questo è il reperto più caratteristico della malattia di Cooley, cioè l’estrema anisopoichilocitosi. Si osservano infatti normociti, macrociti, leptociti ipocromici quasi traspa-renti, sferociti, cellule a bersaglio, eritrociti “a lacrima” (specialmente dopo che la milza ha “snocciolato” i corpi inclusi costituiti dai precipitati di catene � ), eritrociti con punteggiatura basofi la o policromatofi la, eritrociti “a biscotto”, schistociti ecc. È di frequente riscontro la presenza in circolo di eritroblasti, per lo più ortocromatici, ma talvolta anche policromatofi li e perfi no basofi li. L’elettroforesi dell’emoglobina dà reperti diversi a seconda del genotipo responsabile della talassemia major, com’è indicato dalla tabella 48.10 .
C0240.indd 1065C0240.indd 1065 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1066
L’esame del midollo dimostra una ricchezza cellulare con iperplasia della serie eritroide costituita da eritroblasti, per lo più di piccole dimensioni (microeritroblasti), e curva maturativa eritroblastica stimolata. Lo studio delle resistenze globulari all’ipotonia salina ri-vela un’aumentata resistenza osmotica, con una notevole quota di cellule intatte sino a concentrazioni di NaCl più basse della soglia emolitica media normale di 4,0-4,45 g/L. La diminuita fragilità osmotica è dovuta alla presenza di emazie appiattite (leptociti) in cui il rapporto volume/superfi cie è molto diminuito. Gli altri dati di laboratorio dimostrano: 1) aumento del-la bilirubina non coniugata e modesto aumento dei re-ticolociti, secondario all’iperemolisi; 2) aumento della sideremia e diminuzione della transferrinemia totale; 3)parametri caratteristici dell’eritropoiesi ineffi cace, cioè un aumentato turnover del ferro plasmatico e una diminuita incorporazione del 59 Fe nelle emazie. La radiografi a delle ossa, specie di quelle corte e piatte, di-mostra un assottigliamento della trabecola più esterna e la presenza di una striatura perpendicolare tra i tavolati; nella porzione alta dell’osso frontale e nell’osso parietale del cranio le trabecole si dispongono perpendicolarmente alla superfi cie, dando luogo al cosiddetto “cranio a spazzola”. Nelle vertebre ci sono aspetti particolari detti a ragnatela, per il reticolo a maglie larghe formato dalle trabecole.
Decorso e prognosi La malattia di Cooley in senso stretto è una malattia grave, caratterizzata da una cronica per cui spiccata anemia, per cui il paziente arriva con diffi coltà all’adolescenza. Questi pazienti, dipendenti dalla terapia emotrasfusionale, vanno incontro a un sovraccarico marziale iatrogeno che si aggiunge, come si è visto, al ferro derivante dall’aumentato assorbimento e dal processo emolitico. Ne consegue, in
assenza di altre complicanze intercorrenti come infezioni o emorragie, una situazione di emocromatosi secondaria con siderosi miocardica e scompenso di circolo irreversibile.
Talassemia intermedia Questo termine è utilizzato per indicare un gruppo di pazienti che presenta un quadro clinico ed ematologico di gravità intermedia tra la forma major e la forma minor di talassemia. I genotipi responsabili della talassemia sono indicati nella tabella 48.10 ; a questi vanno aggiunte alcune situazioni di doppia eterozigosi e, precisamente, la doppia eterozi-gosi per la � - e � - � -talassemia e la doppia eterozigosi per la � -talassemia e la persistenza ereditaria di emoglobina fetale. Si suppone che alcuni pazienti omozigoti per la � -talassemia possano avere una capacità aumentata di degradare le catene � globiniche libere prevenendone le conseguenze dannose, mentre il contrario sembra avve-nire in pazienti eterozigoti che presentano importanti precipitati � -globulinici a livello degli eritroblasti. Lo sviluppo psichico e fi sico di questi pazienti è normale; le deformazioni ossee sono scarse o assenti, per cui la facies microcitemica è molto sfumata e l’intensità del pallore è variabile a seconda della gravità dell’anemia. Si ha costan-te epatosplenomegalia con siderosi epatica; frequente è la colelitiasi, mentre rare sono le ulcere trofi che malleolari. Il quadro ematologico è qualitativamente analogo a quel-lo descritto per la forma di talassemia major, però l’anemia ipocromica microcitica è meno spiccata ed è quasi sempre associata a elevata reticolocitosi. La sopravvivenza è variabile, data la variabilità del quadro clinico ed ematologico; in alcuni pazienti essa è abbastan-za lunga, anche in assenza di terapia emotrasfusionale.
Talassemia minor (malattia di Rietti-Greppi-Micheli) È la forma eterozigote della � -talassemia, i cui possibili genotipi sono elencati nella tabella 48.10 . Il paziente è un soggetto cronicamente anemico sin dall’infanzia, con anemia non grave; spesso essa è rico-nosciuta solo occasionalmente in coincidenza di un esame di laboratorio eseguito per altri motivi. Talvolta la sintomatologia è limitata ai sintomi e segni generali dell’anemia; in altri casi si può osservare colorito giallognolo della cute per l’ittero secondario alla compo-nente emolitica, la quale può anche portare alla colelitiasi da calcoli di bilirubina. In questi pazienti la colelitiasi, a cui paiono predisposti, può anche essere costituita da calcoli di colesterolo. Altri dati obiettivi sono la splenomegalia, non costante e comunque non cospicua, e la presenza, rara, di ulcere ai malleoli, fenomeno caratteristico di tutte le anemie emolitiche croniche.
Esami di laboratorio L’esame emocromocitometrico dimostra un’anemia da lieve a media (Hb dai 9 ai 12 g/dL) con numero di emazie spesso aumentato (poliglobulia), con volume corpuscolare medio ridotto (microcitosi) e contenuto corpuscolare medio di emoglobina ridotto (ipocromia). I reticolociti sono notevolmente aumentati, a valori oltre il doppio del normale. L’esame dello striscio del sangue periferico mette in evi-denza l’anisopoichilocitosi tipica di queste malattie, ma
Tipo di talassemia
Omozigote Eterozigote
� + Talassemia Talassemia majorHba + F + A 2
Talassemia minorAumento HbA2
� + Talassemia con alta F
Talassemia intermedia
Talassemia minor
HbA + F + A 2 5-12% HbF
� ° Talassemia Talassemia major
Talassemia minor o intermedia
HbF + A 2 Aumento HbA 2
( � - � )° Talassemia o F talas-semia
Talassemia intermediaSolo HbF
Talassemia minor
HbF 5-15%HbA2 normale
( � - � )+ Talassemia Non de-scritta
Portatore silente
HbF e A 2 normali
Lepore Talassemia major
Talassemia minor
HbF + Lepo-re HbA
+ Lepore + A 2
Tabella 48.10 Emoglobine presenti nei vari tipi di � -talassemia
C0240.indd 1066C0240.indd 1066 6/9/10 3:09:01 PM6/9/10 3:09:01 PM
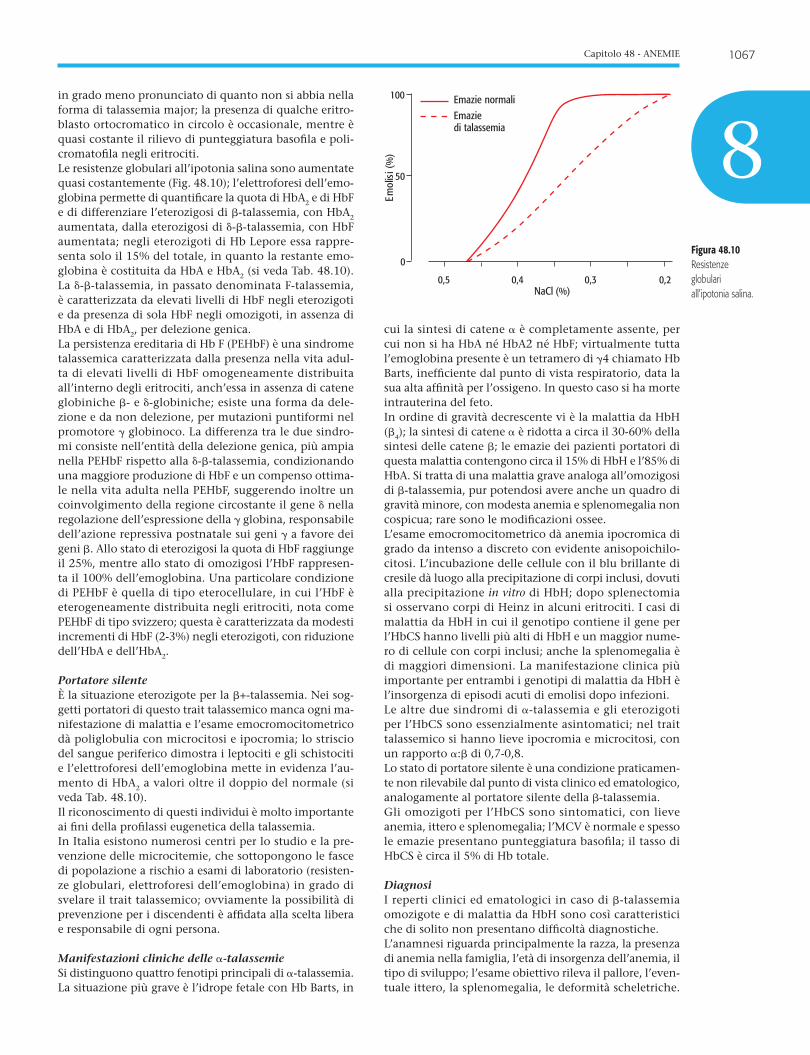
Capitolo 48 - ANEMIE
81067
in grado meno pronunciato di quanto non si abbia nella forma di talassemia major; la presenza di qualche eritro-blasto ortocromatico in circolo è occasionale, mentre è quasi costante il rilievo di punteggiatura basofi la e poli-cromatofi la negli eritrociti. Le resistenze globulari all’ipotonia salina sono aumentate quasi costantemente ( Fig. 48.10 ); l’elettroforesi dell’emo-globina permette di quantifi care la quota di HbA 2 e di HbF e di differenziare l’eterozigosi di � -talassemia, con HbA 2 aumentata, dalla eterozigosi di � - � -talassemia, con HbF aumentata; negli eterozigoti di Hb Lepore essa rappre-senta solo il 15% del totale, in quanto la restante emo-globina è costituita da HbA e HbA 2 (si veda Tab. 48.10 ). La � - � -talassemia, in passato denominata F-talassemia, è caratterizzata da elevati livelli di HbF negli eterozigoti e da presenza di sola HbF negli omozigoti, in assenza di HbA e di HbA 2 , per delezione genica. La persistenza ereditaria di Hb F (PEHbF) è una sindrome talassemica caratterizzata dalla presenza nella vita adul-ta di elevati livelli di HbF omogeneamente distribuita all’interno degli eritrociti, anch’essa in assenza di catene globiniche � - e � -globiniche; esiste una forma da dele-zione e da non delezione, per mutazioni puntiformi nel promotore � globinoco. La differenza tra le due sindro-mi consiste nell’entità della delezione genica, più ampia nella PEHbF rispetto alla � - � -talassemia, condizionando una maggiore produzione di HbF e un compenso ottima-le nella vita adulta nella PEHbF, suggerendo inoltre un coinvolgimento della regione circostante il gene � nella regolazione dell’espressione della � globina, responsabile dell’azione repressiva postnatale sui geni � a favore dei geni � . Allo stato di eterozigosi la quota di HbF raggiunge il 25%, mentre allo stato di omozigosi l’HbF rappresen-ta il 100% dell’emoglobina. Una particolare condizione di PEHbF è quella di tipo eterocellulare, in cui l’HbF è eterogeneamente distribuita negli eritrociti, nota come PEHbF di tipo svizzero; questa è caratterizzata da modesti incrementi di HbF (2-3%) negli eterozigoti, con riduzione dell’HbA e dell’HbA 2 .
Portatore silente È la situazione eterozigote per la � + -talassemia. Nei sog-getti portatori di questo trait talassemico manca ogni ma-nifestazione di malattia e l’esame emocromocitometrico dà poliglobulia con microcitosi e ipocromia; lo striscio del sangue periferico dimostra i leptociti e gli schistociti e l’elettroforesi dell’emoglobina mette in evidenza l’au-mento di HbA 2 a valori oltre il doppio del normale (si veda Tab. 48.10 ). Il riconoscimento di questi individui è molto importante ai fi ni della profi lassi eugenetica della talassemia. In Italia esistono numerosi centri per lo studio e la pre-venzione delle microcitemie, che sottopongono le fasce di popolazione a rischio a esami di laboratorio (resisten-ze globulari, elettroforesi dell’emoglobina) in grado di svelare il trait talassemico; ovviamente la possibilità di prevenzione per i discendenti è affi data alla scelta libera e responsabile di ogni persona.
Manifestazioni cliniche delle � -talassemie Si distinguono quattro fenotipi principali di � -talassemia. La situazione più grave è l’idrope fetale con Hb Barts, in
cui la sintesi di catene � è completamente assente, per cui non si ha HbA né HbA 2 né HbF; virtualmente tutta l’emoglobina presente è un tetramero di � 4 chiamato Hb Barts , ineffi ciente dal punto di vista respiratorio, data la sua alta affi nità per l’ossigeno. In questo caso si ha morte intrauterina del feto. In ordine di gravità decrescente vi è la malattia da HbH ( � 4 ); la sintesi di catene � è ridotta a circa il 30-60% della sintesi delle catene � ; le emazie dei pazienti portatori di questa malattia contengono circa il 15% di HbH e l’85% di HbA. Si tratta di una malattia grave analoga all’omozigosi di � -talassemia, pur potendosi avere anche un quadro di gravità minore, con modesta anemia e splenomegalia non cospicua; rare sono le modifi cazioni ossee. L’esame emocromocitometrico dà anemia ipocromica di grado da intenso a discreto con evidente anisopoichilo-citosi. L’incubazione delle cellule con il blu brillante di cresile dà luogo alla precipitazione di corpi inclusi, dovuti alla precipitazione in vitro di HbH; dopo splenectomia si osservano corpi di Heinz in alcuni eritrociti. I casi di malattia da HbH in cui il genotipo contiene il gene per l’HbCS hanno livelli più alti di HbH e un maggior nume-ro di cellule con corpi inclusi; anche la splenomegalia è di maggiori dimensioni. La manifestazione clinica più importante per entrambi i genotipi di malattia da HbH è l’insorgenza di episodi acuti di emolisi dopo infezioni. Le altre due sindromi di � -talassemia e gli eterozigoti per l’HbCS sono essenzialmente asintomatici; nel trait talassemico si hanno lieve ipocromia e microcitosi, con un rapporto � : � di 0,7-0,8. Lo stato di portatore silente è una condizione praticamen-te non rilevabile dal punto di vista clinico ed ematologico, analogamente al portatore silente della � -talassemia. Gli omozigoti per l’HbCS sono sintomatici, con lieve anemia, ittero e splenomegalia; l’MCV è normale e spesso le emazie presentano punteggiatura basofi la; il tasso di HbCS è circa il 5% di Hb totale.
Diagnosi I reperti clinici ed ematologici in caso di � -talassemia omozigote e di malattia da HbH sono così caratteristici che di solito non presentano diffi coltà diagnostiche. L’anamnesi riguarda principalmente la razza, la presenza di anemia nella famiglia, l’età di insorgenza dell’anemia, il tipo di sviluppo; l’esame obiettivo rileva il pallore, l’even-tuale ittero, la splenomegalia, le deformità scheletriche.
100
50
0
(%)
isilomE
Emazie normali
Emaziedi talassemia
0,5 0,4 0,3 0,2NaCl (%)
Figura 48.10 Resistenze globulari all’ipotonia salina.
C0240.indd 1067C0240.indd 1067 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1068
L’esame emocromocitometrico, l’esame dello striscio del sangue periferico e l’elettroforesi dell’emoglobina danno i reperti già descritti. Gli stati di eterozigosi presentano maggiori diffi coltà dia-gnostiche, però le alterazioni caratteristiche nelle emoglo-bine F e A 2 di solito chiarifi cano la situazione. Molto importante è la diagnosi differenziale con l’anemia sideropenica , ugualmente ipocromica e microcitica; i criteri differenziali più importanti in quest’ultima forma sono la bassa sideremia con bassa saturazione della tran-sferrina, il basso valore di reticolociti (prima della terapia marziale) e la normalità del quadro emoglobinico.
Diagnosi prenatale Le cellule o i tessuti per la diagnosi prenatale possono essere ottenuti mediante biopsia dei villi coriali (per via transcervicale o mediante aspirazione per via transaddominale), oppure mediante amniocentesi o fetoscopia. La biopsia dei villi coriali permette di ottenere il DNA fetale nel primo trimestre di gravidanza (9-12 settimane di gesta-zione), l’amniocentesi in periodo più tardivo (17-18 setti-mane di gestazione); per la fetoscopia l’epoca del prelievo è a 19-22 settimane. Tutte queste metodiche comportano un rischio di perdita del feto, con percentuali che variano tra le diverse casistiche, comunque intorno al 4% per la biopsia dei villi coriali e intorno al 2% con la tecnica dell’amnio-centesi. La fetoscopia presenta il rischio maggiore. L’analisi del DNA può essere eseguita con metodo indiret-to o diretto; il primo si basa sullo studio dei polimorfi smi di siti per enzimi di restrizione (enzimi batterici che ta-gliano il DNA a livello di sequenze molto specifi che) del gruppo dei geni globinici, per mezzo dei quali è possibile marcare un carattere genetico e seguirne l’ereditarietà. Essendo basato su studi di linkage 3 è necessario studiare preventivamente non soltanto la coppia di genitori, ma anche un altro fi glio affetto o normale. Il metodo diretto si basa sull’identifi cazione del difetto molecolare (preventivamente noto); le delezioni e le mu-tazioni puntiformi possono essere riconosciute diretta-mente attraverso analisi con enzimi di restrizione, o con oligonucleotidi sintetici marcati con 32 P e usati in test di ibridizzazione per il DNA normale e talassemico. In epoca tardiva di gestazione si ricorre al prelievo di sangue fetale, su cui viene eseguita l’analisi quantitativa delle catene globiniche fetali sintetizzate in vitro . Nelle forme omozigoti per � 0 talassemia non si evidenzia alcuna sintesi di catene globiniche, mentre nelle forme omozigoti per � + talassemia esse vengono prodotte in quantità molto ridotta rispetto al soggetto normale, con possibilità di sovrapposizione tra gli omozigoti � + con produzione di catene globiniche relativamente alta ed eterozigoti con bassa produzione; il rischio di errore dia-gnostico è intorno all’1%.
Terapia I pazienti gravemente anemici, come nella � -talassemia omozigote o in alcune forme di talassemia intermedia, devono essere sottoposti a trattamento trasfusionale. In
passato era stato raccomandato il cosiddetto trattamen-to “ipertrasfusionale”, allo scopo non solo di correggere l’anemia, ma anche di sopprimere l’eritropoiesi (il che è utile per evitare i danni dell’eritropoiesi inefficace) e di inibire l’assorbimento gastrointestinale di ferro. Come si vedrà, infatti, l’accumulo di ferro nell’organismo è il principale problema di questi pazienti, indipendente-mente dal fatto che siano trattati con trasfusioni. Oggi si è visto che l’eccesso di trasfusioni, di per sé, è in gra-do di generare un sovraccarico di ferro nell’organismo e perciò si preferisce aderire allo schema proposto da autori italiani (Cazzola et al., 1997), che hanno racco-mandato un approccio più prudente, suggerendo di trasfondere sangue solo se, prima di ogni trasfusione, i livelli di Hb non superano i 9,5 g/dL. Si è visto che questo regime è associato a un’adeguata soppressione midollare e a un inferiore accumulo di ferro. Comunque, il problema dell’accumulo di ferro resta e deve essere affrontato con la regolare somministrazione di un chelante. A questo scopo il farmaco ordinaria-mente impiegato è la desferoxamina, che deve essere somministrata per via parenterale, endovena, sottocute o intramuscolo. Il farmaco è sicuramente efficace e, in pazienti già affetti da fibrosi epatica per eccessivo accumulo di ferro, si è dimostrato in grado di impedire la progressione in cirrosi. La somministrazione di de-sferoxamina deve essere regolata in base all’accumulo di ferro nell’organismo e ciò viene comunemente fatto basandosi sui livelli ematici di ferritina. Questo è un parametro impreciso, perché si stima che indichi in maniera affidabile le scorte organiche di ferro sola-mente in circa il 50% dei casi. Un metodo molto più preciso, che non può però essere eseguito con troppa frequenza, è la valutazione del ferro in biopsie epatiche, considerando che un carico di questo elemento non superiore a 15 mg/g di peso secco diminuisce notevol-mente il rischio di manifestazioni cliniche da iperaccu-mulo di ferro. L’impiego di immagini ottenute con la risonanza magnetica è invece inaffidabile. Esiste, per la verità, una tecnica incruenta per misurare la quantità di ferro depositata nel fegato, chiamata “suscettometria magnetica”. Tuttavia, le apparecchiature in grado di praticarla sono poche in tutto il mondo. Un trattamento più radicale della malattia di Cooley è il trapianto di midollo da donatore HLA-identico. Il successo di questa misura è tanto più probabile quanto minori sono i danni già instauratisi per ac-cumulo di ferro. In bambini indenni da questi danni la sopravvivenza senza segni di malattia supera il 90% a 3 anni dal trapianto, mentre in quelli che già li presentano e nella maggior parte degli adulti la percentuale scende al 60%. Sono allo studio terapie sperimentali, come l’impiego di un chelante attivo per somministrazione orale, il deferiprone, che però è risultato deludente sia dal punto di vista dell’efficacia sia da quello degli effetti collaterali, oppure come i tentativi di aumentare la sintesi di emoglobina fetale con l’uso di vari farmaci, per lo più chemioterapici con discreta tossicità. Anche questi ultimi tentativi sono stati finora deludenti. 3 Associazione su uno stesso cromosoma di geni che segregano insieme
nella meiosi.
C0240.indd 1068C0240.indd 1068 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Capitolo 48 - ANEMIE
81069
Un nuovo approccio terapeutico, di tipo eziopatoge-netico, si attende dalle ricerche di biologia moleco-lare, che hanno portato un notevole contributo alla comprensione delle fini alterazioni genetiche delle talassemie e dei conseguenti meccanismi di alterata sintesi delle catene globiniche. Poiché, come si è visto, lo sbilanciamento sintetico � /non � è di importanza preminente per la fisiopatologia del processo talasse-mico, mediante metodiche di manipolazione genetica potrebbe essere indotta la riattivazione della sintesi delle catene globiniche � e � o la soppressione dell’ec-cesso di sintesi di catene � ; inoltre, il gene patologico potrebbe essere sostituito con un gene purificato.
Esiste una condizione naturale a riprova dell’importanza che lo sbilanciamento sintetico tra le catene globiniche riveste per la patogenesi del quadro clinico della � -talas-semia. Questa condizione si verifica nei soggetti portatori del doppio trait � -talassemico e � -talassemico; il rapporto � : � , che è 2,5 nel trait � -talassemico, in questi casi è 1. La sintesi globinica bilanciata porta a un’alterazione minore delle emazie e il volume corpuscolare medio è normale. Infine, la recente scoperta di sottopopolazioni di eri-troblasti capaci di sintetizzare preferenzialmente HbF potrebbe portare all’identificazione di farmaci in grado di incentivare la proliferazione di cellule sintetizzanti HbF, che possiede una buona attività funzionale.
Sono condizioni ereditarie caratterizzate dalla sintesi di Hb strutturalmente anomale. Sono state descritte almeno 1000 varianti di emoglobine anomale, ma solo in un terzo dei casi l’alterazione strutturale comporta “malat-tia”. Più del 90% delle emoglobinopatie è attribuibile alla sostituzione di un solo aminoacido nella struttura prima-ria di Hb per mutazione puntiforme nella corrispondente tripletta del gene strutturale. Altre varianti si caratterizza-no per la sostituzione di uno o più aminoacidi, per la produzione di Hb a catena allungata o per la fusione di geni differenti dopo crossing-over (per esempio, Hb Le-pore e anti-Lepore). Sono malattie trasmesse con carattere autosomico a do-minanza parziale; è cioè possibile la presenza nello stesso individuo di due varianti emoglobiniche (ereditate l’una dal padre e l’altra dalla madre). Le emoglobine patologiche, per convenzione, vengono indicate con lettere dell’alfabeto o nome del luogo dove sono state scoperte, seguite dalla lettera dell’alfabeto gre-co che dà il nome alla catena polipeptidica della globina interessata dall’anomalia strutturale, dal numero che pre-cisa la posizione nella catena polipeptidica occupata dal residuo mutato e dal nome del nuovo aminoacido (per esempio, Alabama � 39 Gln → Lys). Le emoglobine patolo-giche sono caratterizzate da quattro alterazioni funzionali principali, responsabili di altrettanti quadri clinici: • alterata affi nità per l’ossigeno, che provoca cianosi o
poliglobulia a seconda di una diminuita o aumentata affi nità per l’ossigeno;
• formazione di HbM con metaemoglobinemia e cianosi;
• presenza di emoglobine instabili accompagnate da corpi di Heinz ed emolisi;
• HbS e HbC (meno frequenti HbD, E, O) con formazione di tattoidi.
Emoglobinopatie dovute a emoglobine con alterata affi nità per O 2
Alcune varianti strutturali sono caratterizzate da uno spostamento dell’equilibrio tra ossi- e desossi-emoglobina verso la forma ossigenata o non ossigenata.
Aumentata affi nità In questo caso l’emoglobina ha una struttura più stabile nella forma ossigenata, il che signifi ca una minore cessio-ne di ossigeno ai tessuti. La produzione di eritropoietina è aumentata e ne consegue eritrocitosi di compenso. Quando la massa di GR è cresciuta in modo tale da com-pensare l’ipossia tissutale, anche l’eritropoietina torna a valori normali. Si tratta comunque di una patologia rara (45 varianti in poco più di 200 casi segnalati) e limitata a gruppi familiari ben distinti. I pazienti sono pletorici, asintomatici, con alti valori di Hb ( > 16 g/dL) e di Hct (45-65%), e raramente necessitano di salassi. Piastrine e leucociti sono nella norma (si veda Poliglobulie o eritrocitosi secondarie , Capitolo 49 ). La diagnosi non è facile e non sempre è possibile grazie a una comune elettroforesi di Hb, dal momento che più del 50% delle varianti ad alta affi nità migra in modo del tutto regolare. Viceversa, la curva di dissociazione dell’ossiemo-globina, completamente deviata a sinistra, è diagnostica ( Fig. 48.11 ).
Diminuita affi nità La struttura di Hb è più stabile nella forma deossigenata, per questo motivo l’ossigeno viene rilasciato ai tessuti assai rapidamente. Si tratta comunque di un’anomalia ancora più rara delle precedenti. I pazienti sono ciano-tici, talvolta c’è modesta anemia, riferibile a diminuita eritropoietina e a una ridotta sopravvivenza delle emazie dovuta alla contemporanea presenza di Hb instabile.
Emoglobinopatie da HbM
Prima di entrare nel dettaglio della patologia conviene fa-re una breve digressione sul signifi cato della cianosi . Per cianosi si intende una colorazione azzurrognola o bluastra della cute o delle mucose, prodotta da un incremento della concentrazione assoluta di Hb ridotta nei capillari o dalla presenza di composti anomali di Hb (metaemoglobina o sol-foemoglobina ). La cianosi diventa manifesta quando sono presenti 5 g o più di Hb ridotta per decilitro di sangue ovvero 1,5-2 g/dL di metaemoglobina o 0,5 g/dL di solfoemoglobi-na, composta da emoglobina e solfuri solubili instabili.
Emoglobinopatie G. Gromo, M. Storti
C0240.indd 1069C0240.indd 1069 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1070
Le emoglobinopatie da HbM sono caratterizzate da una so-stituzione aminoacidica (nella catena � o � ) responsabile dell’irreversibile ossidazione del ferro emico a ferro ferri-co, in uno stato cioè non più suscettibile di riduzione a opera della NADH metaemoglobin-reduttasi o di qualsiasi altro sistema di metaemoglobin-reduttasi del GR. Si tratta dunque di una metaemoglobinemia congenita, in cui solo lo stato di eterozigosi permette la sopravvivenza. Rara in tutto il mondo, è comunque l’emoglobinopatia più importante in Giappone. Le forme acquisite di HbM, più frequenti rispetto alle congenite, derivano dall’esposi-zione a farmaci o a sostanze tossiche in grado di ossidare direttamente l’eme. Tra i farmaci in grado di indurre la formazione di metaemoglobina ci sono i sulfamidici, la fenazopirina e alcuni anestetici locali, mentre tra le sostanze tossiche sono inclusi l’acqua ricca di nitrati e alcuni pesticidi. I pazienti hanno un quadro clinico variabile, a seconda della sostituzione specifi ca. Negli eterozigoti, solo il 25-30% dell’Hb totale è HbM ; ciò è dovuto verosimilmente alla diminuita velocità di sintesi di Hb anomala. Vi può essere metaemoglobinemia anche in presenza di emo-globina instabile; tuttavia questa è transitoria e precede il momento della precipitazione (si veda oltre, Emoglobi-nopatie da emoglobine instabili ). La cianosi compare dopo la nascita se il difetto è sulla ca-tena � (interessa infatti anche l’HbF), ma solo 3-6 mesi più tardi se è la catena � a essere interessata. Si tratta di una condizione destinata a durare tutta la vita, asintomatica e per la quale il trattamento non è né indicato né possibile. Utili a fi ni diagnostici sono l’esame spettrofotometrico di un emolisato acido e l’elettroforesi delle emoglobine (a pH 7,1 migrano più rapidamente dell’HbA). Si deve inoltre porre la diagnosi differenziale con la meta-emoglobinemia da defi cit di NADH-reduttasi e da agenti tossici (per esempio, nitrati, chinoni, clorati).
Emoglobinopatie da Hb instabili
Sono caratterizzate dalla precipitazione intracellulare dell’Hb, che causa la formazione di inclusi (i corpi di Heinz) ed emolisi. L’affezione è pienamente espressa nel-lo stato eterozigote; l’omozigosi è incompatibile con la vita. I GR alterati dalla precipitazione dell’emoglobina han-no ridotta deformabilità, incontrano diffi coltà a supe-rare il microcircolo e vengono trattenuti nel passaggio dai cordoni splenici ai sinusoidi. In molti casi i corpi di Heinz sono rilasciati nella milza (fenomeno del pitting); la parte restante del GR richiude la sua membrana e ritorna in circolo, ma solitamente è più suscettibile ai processi emolitici. Da qui il riscontro, dopo splenecto-mia, di un numero superiore di GR contenenti corpi di Heinz. Il quadro clinico è molto variabile, soprattutto a causa delle notevoli differenze strutturali fra le varianti di Hb instabili. Ne sono state identifi cate più di 70, comune-mente classificate in quattro gruppi in relazione alla gravità dei segni e dei sintomi: da una modesta emolisi senza anemia ad anemia emolitica intensa, con retico-lociti aumentati, ittero, splenomegalia ed epatomegalia. Per dimostrare la presenza dei corpi di Heinz si incubano le emazie con coloranti ossidanti come il cristalvioletto e il blu di metilene. Talvolta è necessaria l’aggiunta di un altro agente ossidante come l’acetilfenilidrazina. La diagnosi di anemia emolitica secondaria a Hb instabile può essere fatta con il riscontro dei corpi di Heinz e con alcuni test specifi ci (che possono essere reperiti su manuali specialistici). L’elettroforesi di Hb è indicativa solo nella metà dei casi, dal momento che l’anomalia strutturale implica una modificazione di carica della molecola non sempre evidenziabile con una “normale elettroforesi”.
Sa02 (%)
100
80
60
40
20
0
0 20 40 60 80 100
Pa02 (mmHg)
Figura 48.11 Curva di
dissociazione ossiemoglobinica.
C0240.indd 1070C0240.indd 1070 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Capitolo 48 - ANEMIE
81071
Emoglobinopatie da HbS, C, D, E
Emoglobinopatia da HbS È una malattia ereditaria dovuta alla presenza nei GR di HbS, che polimerizza in particolari condizioni di ipos-sigenazione e deforma i GR impartendo loro una con-formazione falciforme. In inglese si chiama sickling cell disease e da sickle (falce) viene la lettera S con cui è indicata l’emoglobinopatia. L’HbS è presente con un polimorfi smo bilanciato 4 e una frequenza del 10-40% in alcune popolazioni esposte alla malaria endemica, particolarmente tra i neri dell’Africa centrale e occidentale, tra i loro discendenti emigrati e in alcune popolazioni caucasiche del Mediterraneo del Nord, Medio Oriente e India. Attualmente l’8% dei neri nordame-ricani, latinoamericani e dei Caraibi ha un trait falcemico, lo 0,16% ha anemia drepanocitica e lo 0,18% una doppia eterozigosi (HbSC 0,06%; HbS � -talassemia 0,12%). Fra le popolazioni di origine mediterranea, l’HbS/ � -talassemia è la più comune forma di malattia drepanocitica. In Italia le regioni più interessate sono la Calabria e la Sicilia.
Eziopatogenesi L’HbS differisce dall’HbA soltanto per la sostituzione dell’acido glutammico in posizione 6 della catena � con la valina. Esiste dunque una catena � alterata codifi cata da un gene anomalo. Questo gene può essere allo stato omozigote per l’HbS e dare anemia a cellule falciformi , eterozigote per l’HbS (trait falciforme) , di doppia eterozigosi HbS e HbC o HbD ovvero HbS/ � -talassemia (microdrepanocitosi) . Si osserva il fenomeno della falcizzazione ogni qualvolta la pressione parziale di O 2 scende sotto i 50-60 mmHg. La desossi-HbS, infatti, è meno solubile della desossi-HbA e tende a polimerizzare perdendo la propria disposizione secondo una geometria casuale e formando, all’interno dell’eritrocita, fasci di fi bre tubulari a decorso parallelo dette “tattoidi” ( Fig. 48.12 ). Ciascuna fi bra è a sua volta composta da 6-8 fi lamenti disposti a spirale lungo un asse centrale. Ogni fi lamento è costituito da numerosi tetra-meri di HbS polimerizzati a formare una struttura rigida e ordinata, longitudinale, di molecole impilate. Questi fasci deformano la membrana del GR, che assume l’aspetto a falce. Il fenomeno è osservabile direttamente con la mi-croscopia elettronica e indirettamente misurando le alte-razioni di viscosità e fi ltrabilità. L’equilibrio di HbS fra la sua fase amorfa (liquida, irregolare, casuale) e quella solida (cristallina, regolare) è condizionato, come si diceva, dalla diminuita pressione parziale di O 2 . Tuttavia, intervengono anche altri fattori: • la disidratazione o l’aumento dell’osmolarità
plasmatica, che favoriscono la fuoriuscita di H 2 O dalle emazie e l’aumento della concentrazione di HbS nelle cellule;
• le alte temperature, che aumentano la velocità di polimerizzazione di HbS; dal momento, però, che i valori cui bisogna arrivare sono al di fuori
di quelli fi siologici, si tratta di un dato a esclusivo uso “di laboratorio”;
• la concentrazione di HbS (se è inferiore al 50% non c’è falcizzazione) e il tipo di Hb che è associata all’HbS nel GR; se l’HbS si trova associata a HbF, il tasso di falcizzazione è notevolmente ridotto perché in questa mescolanza viene inibita la polimerizzazione.
Con l’HbA (l’eterozigosi di riscontro più comune) la si-tuazione si discosta di poco dalla norma. Viceversa, il quadro si presenta molto grave con grado di falcizzazione estremamente elevato nei soggetti portatori di HbS/C (situazione, questa, non rara, dal momento che entrambe le Hb sono diffuse nelle medesime zone geografi che). In caso di doppia eterozigosi HbS/ � -talassemia si registra un aggravamento della sintomatologia anemica, più di quanto non avvenga nei soggetti portatori di talassemia minor.
Fisiopatologia I GR rigidi e deformati dopo che ha avuto luogo il pro-cesso di falcizzazione sono intrappolati nel microcircolo, soprattutto nella milza, dove la fi ltrazione si verifi ca in condizioni di ipossia, acidosi e lento fl usso ematico. La trasformazione a falce dei GR provoca un incremento della viscosità del sangue, ne ostacola il fl usso nei piccoli vasi, aggrava l’anossia locale, favorisce un’ulteriore falciz-zazione e innesca in tal modo un circolo vizioso. Il fenomeno della falcizzazione è inizialmente reversi-bile e in presenza di una normale pressione di ossigeno l’HbS si depolimerizza e le cellule riassumono una forma normale. Ciascun ciclo di falcizzazione provoca però una perdita, da parte del GR, di acqua, potassio e calcio, per cui esso alla fi ne non sarà più in grado di riprendere la sua forma regolare biconcava. Infatti, negli individui
4 Questo signifi ca che gli eterozigoti hanno un vantaggio sugli omo-zigoti sia per il gene normale sia per quello patologico. Nel caso della HbS il vantaggio potrebbe essere derivato da una maggiore resistenza delle loro emazie al parassita malarico.
O2
Figura 48.12 Formazione dei tattoidi in coincidenza di una riduzione della pressione parziale di O 2 .
C0240.indd 1071C0240.indd 1071 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1072
con anemia falciforme (omozigosi per il gene HbS), è sempre presente una quota di cellule (dal 4 al 44%) che non riacquista la forma normale allorché il sangue viene riossigenato, cioè una quota di GR che rimane irrever-sibilmente falciforme. Defi nite Irreversibly Sickled Cell (ISC), sono quelle cellule che si vedono negli strisci di sangue periferico allestiti senza particolari accorgimenti (si veda oltre, Esami di laboratorio ). Le emazie così alte-rate hanno una sopravvivenza nettamente abbreviata e, per questa ragione, nei soggetti nei quali si ripetono nel tempo varie crisi di falcizzazione interviene anche un’anemia emolitica. Inoltre, la presenza nell’emazia di HbS non solo provoca le profonde alterazioni di forma del GR in condizioni di ipossigenazione, ma è anche causa di effetti secondari che ne modifi cano le proprietà della membrana a livello dei fosfolipidi e delle proteine. Si è osservato che l’emazia a falce ha una netta tendenza ad aderire in modo abnorme alle cellule endoteliali, ai mo-nociti e ai macrofagi; l’abnorme adesività è stata attribuita alle modifi cazioni di membrana che, accanto all’aumento di viscosità e alla presenza di emazie deformate e rigide, è responsabile dei fenomeni di occlusione vascolare tipici della malattia.
HbS/S (omozigosi) Manifestazioni cliniche La malattia esordisce solitamente nel corso del primo an-no di vita. Quando il bambino nasce presenta all’incirca il 50% di HbF e il 50% di HbS; con il passare dei mesi la quota di HbF, protettiva ai fi ni delle crisi di polimeriz-zazione dell’emoglobina S, diminuisce; aumenta invece quella di HbS e i primi segni dell’anemia emolitica cronica cominciano a manifestarsi. Due aspetti sono particolarmente interessanti: l’aumenta-ta suscettibilità a contrarre infezioni e le crisi dolorose. Per quanto riguarda il primo punto, principale respon-sabile di un’elevata mortalità infantile (10% dei casi nel primo anno di vita), si riconoscono numerose cause tra le quali viene soprattutto attribuita importanza a un dan-neggiamento della microcircolazione splenica in cui le cellule falcizzate ristagnano. Con il passare del tempo questo organo, sottoposto a una serie di numerosissimi infarti, diviene fi broso e si atrofi zza. La milza, cioè, non è più in grado, fi n dai primi anni di vita, di assolvere alle sue consuete funzioni di fi ltro dei microrganismi, anti-corpopoiesi, emocateresi ecc.; si verifi ca sostanzialmente una condizione di asplenismo. Le crisi dolorose sono invece dovute a fenomeni di oc-clusione vascolare acuta a opera delle ISC e delle cellule falciformi reversibili che più a lungo si trattengono nel microcircolo. Non è viceversa dimostrato che la trombosi in quanto tale svolga un ruolo primario nella produzione di infarti tissutali, anche se l’aumentata distruzione di emazie rilascia in circolo materiali ad attività trombopla-stinica (si veda oltre, Emoglobinuria parossistica notturna ). Le crisi dolorose possono essere avvertite in varie sedi: torace, addome, più spesso agli arti, alle dita delle mani soprattutto nei bambini, o dei piedi. La frequenza delle crisi è variabile da giorni ad anni, così come la durata, da ore a giorni. Sono scatenate da fattori occasionali: ipossia, stress emotivi, malattie respiratorie, episodi infettivi acuti,
abbassamento della temperatura corporea o disidratazio-ne. I ricorrenti episodi ischemici provocano, con l’andare del tempo, danni irreversibili agli organi e agli apparati interessati. • Cuore . Nella circolazione coronarica vi è sempre una
considerevole estrazione di ossigeno dai GR, perciò facilmente si avrà comparsa di falcizzazione, ingorgo dei vasi e arresto del fl usso ematico. Nell’80% dei casi, dopo i 5 anni di età, si osserva cardiomegalia, dopo i 40 anni scompenso cardiaco congestizio e raramente infarto. I reperti auscultatori mimano quelli dell’insuffi cienza mitralica.
• Polmone . È l’organo più frequentemente colpito. Anche in pazienti asintomatici, con le prove di funzionalità respiratoria si dimostrano alterazioni di perfusione e di diffusione dovute verosimilmente al danno endoteliale esercitato cronicamente sulle pareti vascolari da parte dei GR rigidi e deformati. La malattia polmonare cronica, che spesso provoca cuore polmonare cronico, è la più frequente causa di morte negli adulti.
• Rene . La midollare del rene, a causa dell’iperosmolarità, è particolarmente suscettibile all’infarto da falcizzazione. Frequente è la comparsa di ematuria spontanea non dolorosa. Il difetto principale consiste in una ridotta capacità di concentrare le urine; nella maggior parte dei pazienti vi è isostenuria .
• Ossa . Possono verifi carsi infarti ossei, soprattutto alle ossa lunghe in vicinanza delle articolazioni e ai corpi vertebrali. Una complicanza tutt’altro che rara è l’osteomielite . Gli agenti patogeni più frequentemente responsabili sono le salmonelle e lo stafi lococco. Possono verifi carsi anche fratture patologiche e schiacciamenti. Probabilmente la deformità più invalidante è la coxopatia cronica secondaria a necrosi asettica del femore (fabbisogno di O 2 aumentato in relazione all’accrescimento da un lato, insuffi ciente vascolarizzazione dall’altro). Meno frequente è la necrosi asettica dell’omero; entrambe le lesioni sono generalmente bilaterali. Una complicanza possibile durante l’infanzia è la cosiddetta sindrome mano-piede , dovuta a microinfarti della midollare del carpo e del tarso. Le lesioni scompaiono senza terapia, ma lasciano residui radiologici simili all’osteomielite.
• Occhi . Vi sono aree di degenerazione retinica con infarti, emorragie del vitreo, distacco di retina e retinite proliferativa. Nel 90% dei casi si riscontrano microaneurismi nei vasi superfi ciali della congiuntiva bulbare.
• Cute . Ulcere atrofi che degli arti inferiori, soprattutto alla superfi cie mediale della tibia e della caviglia.
• Apparato genitale maschile . Di non raro riscontro è il priapismo .
• Sistema nervoso . Circa il 25% dei pazienti affetti da anemia falciforme presenta lesioni neurologiche, che comprendono emiplegia, amaurosi, paralisi dei nervi cranici. Di solito queste manifestazioni si verifi cano prima dei 15 anni. Più raramente il quadro si complica fi no alla comparsa di episodi convulsivi, stato soporoso, coma.
C0240.indd 1072C0240.indd 1072 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Capitolo 48 - ANEMIE
81073
• Fegato e vie biliari . Un terzo dei pazienti sviluppa colelitiasi a metà adolescenza, solo il 10% di essi presenta ostruzione biliare. Sono descritti anche necrosi e infarti splenici. L’epatomegalia è costante.
L’ anemia ha un andamento cronico, esacerbato da crisi emolitiche e di aplasia. Le crisi aplastiche frequentemente dovute al parvovirus provocano una franca anemizzazio-ne con reticolociti diminuiti e ipoplasia midollare. L’aplasia può durare dai 7 ai 10 giorni; al termine, la ripre-sa dell’emopoiesi è segnalata da una crisi reticolocitaria e dalla comparsa di eritroblasti in circolo. Talvolta si instaura carenza di acido folico con megaloblastosi midollare ed eritropoiesi ineffi cace. Il quadro è attenuato dall’elevata percentuale di HbF che non solo inibisce la polimerizzazio-ne di HbS, ma possiede anche un’elevata affi nità per l’O 2 .
Esami di laboratorio Si tratta di un’anemia emolitica normocitica normocro-mica, con Hb fra i 6-9 g/dL, salvo riduzioni ulteriori in occasione di crisi emolitiche o di aplasia. A partire dai 6 anni di vita, quando iniziano la fi brosi splenica e l’autosplenectomia, si osservano percentuali variabili di ISC. La loro sopravvivenza in circolo è breve, hanno basso MCV e alta MCHC. I reticolociti sono au-mentati (10-20%) e talvolta ci sono eritroblasti. Le piastri-ne sono normali, discretamente aumentati invece sono i granulociti neutrofi li. A dispetto dell’anemia, la velocità di eritrosedimentazione è normale per le anomalie mor-fologiche degli eritrociti.
Diagnosi Il classico test di falcizzazione su vetrino consiste nell’in-cubare del sangue con una sostanza riducente (metabisol-fi to di sodio) al di sotto di un coprioggetto, sigillato per favorire lo stato di ipossia dei GR. Un altro test indicativo, ma non sicuramente specifi co, è quello della precipitazione di Hb (rilevabile attraverso un intorbidamento della soluzione), quando un emolizzato che contiene questa emoglobina viene diluito con una soluzione contenente un’alta concentrazione di fosfati. D’altra parte, l’elettroforesi dell’emoglobina in condizioni standard non consente, in quanto tale, la diagnosi di anemia drepanocitica, dal momento che sono più di 50 le varianti emoglobiniche che presentano la medesima migrazione elettroforetica di HbS. La diagnosi è tuttavia possibile con tec-niche particolari (per le quali si rinvia ai testi specialistici).
Decorso e prognosi Con una buona nutrizione e il rapido controllo dei proces-si infettivi, la lenta crescita prepubere dei soggetti affetti da anemia falciforme è compensata da una pubertà ritar-data, per cui lo sviluppo alla fi ne è normale. Nei pazienti che superano il primo decennio di vita la prognosi è mi-gliore e la mortalità inferiore al 5%. Raramente il paziente sopravvive fi no a 50-60 anni. La gravità del quadro clinico è variabile e la malattia de-corre con peggioramenti improvvisi o crisi, intervallati da periodi di relativo benessere. Le infezioni e gli infarti tissutali rappresentano spesso un problema clinico di notevole entità, molto più dell’ane-mia in quanto tale.
Terapia Non esiste terapia specifica in grado di migliorare le manifestazioni cliniche della malattia. Infatti, l’im-piego di sostanze che inibiscono la gelificazione di HbS e bloccano la falcizzazione si è rivelato finora inefficace, se non addirittura tossico. Le crisi dolorose devono essere trattate tempestivamente sommini-strando sufficienti quantità di liquidi, ossigeno in modo intermittente e analgesici. Benché non esistano studi controllati, un’exsangui-notrasfusione parziale (ogni 3-6 settimane) con GR HbA/A concentrati riduce i GR HbS/S al di sotto del 50%, fa cessare la crisi o previene ulteriori infarti. Si dovrebbe limitare comunque questo tipo di tratta-mento a situazioni di crisi dolorose gravi, persistenti e refrattarie, e alle donne in stato di gravidanza.
Eterozigosi per HbS (trait falcemico) Il trait falcemico consiste in una condizione solitamente asintomatica, senza emolisi o alterazioni clinicamente signifi cative. Si è già detto in precedenza della distribuzione geografi ca, tuttavia è utile ricordare come l’elevata presenza del gene S sia dovuta a un vantaggio selettivo dello stato eterozigo-te (AS) rispetto agli stati omozigoti (AA e SS), cioè a un po-limorfi smo bilanciato. Il vantaggio è probabilmente legato a una protezione contro la malaria. Infatti, il plasmodio sopravvive meno all’interno degli eritrociti HbS. La sopravvivenza eritrocitaria è normale perché la percentua-le di HbS contenuta nelle emazie non è suffi ciente a innescare la falcizzazione. In condizioni normali gli unici effetti notati sono lesioni alla midollare del rene (ematuria e ipostenuria per danno tubulare), oltre a saltuari infarti splenici. Questo può verifi carsi nel corso di esercizi fi sici intensi, voli ad alta quota in aerei non pressurizzati, immersioni marine, anestesia ecc. La durata della vita media dei pazienti portatori di trait non è signifi cativamente diminuita. L’Hb nel trait è costi-tuita dal 25-45% di HbS, dal 55-75% di HbA, da quantità normali di HbF e HbA 2 . Il test di falcizzazione e quello di solubilità di Hb sono positivi.
HbS/ � -talassemia Se il gene della � -talassemia è un � 0 o un � + che comporta un’alterazione grave dell’eritropoiesi, il quadro clinico può essere indistinguibile da quello della forma omozigote HbS/S . Se invece il gene della � -talassemia è un � + che comporta una forma lieve, il quadro clinico che ne risulta è una forma lieve di anemia a cellule falciformi.
HbS/C Il gene C prevale nelle popolazioni dell’Africa occidentale. Il quadro è simile a quello dell’omozigosi HbS/S, anche se di entità più lieve. Le complicanze sono dovute principalmente all’alta viscosità ematica e consistono in crisi dolorose, lesio-ni retiniche, necrosi asettiche della testa del femore, ematu-ria e priapismo. Il grado di anemia è modesto, frequente è la splenomegalia, mentre le ISC sono rare nello striscio. L’emoglobina è costituita dal 50% di HbS, 50% di HbC; A 2 e F sono normali.
C0240.indd 1073C0240.indd 1073 6/9/10 3:09:01 PM6/9/10 3:09:01 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1074
HbS/D L’Hb D (Punjab) proviene dal nord dell’India. L’anemia è assai modesta, poco frequenti sono le complicanze infettive e le crisi dolorose. Al tracciato elettroforetico di routine su acetato di cellulosa non può venire distinta di HbS/S.
Emoglobinopatie da HbC L’HbC è dovuta alla sostituzione dell’acido glutammico con la lisina nella posizione 6 della catena � dell’emo-globina. È frequente soprattutto fra le popolazioni nere dell’Africa occidentale (40% nel nord del Ghana). Nei neri americani l’incidenza del trait si aggira attorno al 2%.
Manifestazioni cliniche dello stato omozigote (HbC/C) La malattia da omozigosi causa di solito un’anemia emoli-tica cronica di lieve entità, il più delle volte asintomatica. La splenomegalia è costante. Vi può essere ittero e la co-lelitiasi è frequente nell’adulto. La fragilità osmotica dei GR è diminuita. Lo striscio di sangue contiene numerose (50-90%) cellule a bersaglio ben emoglobinizzate. Per la diagnosi defi nitiva occorre identifi care la presenza di HbC (la quasi totalità) e l’assenza di HbA o di altre emoglobine anomale all’elettroforesi. La C migra lentamente insieme con la A 2 . Non comporta una mortalità più elevata della norma.
Manifestazioni cliniche dello stato eterozigote I pazienti sono asintomatici e non presentano anemia. Vi sono molte cellule a bersaglio (20-50%) nel periferico e la fragilità osmotica appare diminuita. L’HbC raggiunge il 30-40% del totale.
Emoglobinopatie da HbD La variante più comune, D Punjab, è dovuta alla sosti-tuzione dell’acido glutammico con la glutammina nella posizione 121 della catena � . La frequenza di questo gene in India è del 3%; vi sono solo rarissime segnalazioni di omozigosi per il gene D. In questo caso si osserva solo lieve anisocitosi, ma non anemia, ittero o splenomegalia. L’eterozigosi per l’emoglobina D è asintomatica. La fragi-lità osmotica è ridotta, mentre è aumentata l’affi nità per l’O 2 se confrontata con quella dell’HbA. Da segnalare infi ne che la solubilità di HbD è simile (allo stato deossigenato) a quella di HbA, perciò i test di fal-cizzazione e di solubilità di Hb sono negativi. Quando è ereditata insieme con HbS, attenua in modo signifi cativo le manifestazioni cliniche di quest’ultima.
Emoglobinopatia da HbE È dovuta alla sostituzione dell’acido glutammico con la lisina nella posizione 26 della catena � . Si trova nel Sud-Est asiatico (in Thailandia raggiunge il 30%) e viene ereditata come trait autosomico dominante. L’elevata fre-quenza, poi, nelle stesse regioni della � - o � -talassemia com-porta alte probabilità di associazione tra i due disordini. Lo stato omozigote può avere lieve anemia con microcito-si ed emazie a bersaglio. L’emoglobina E è instabile e può quindi andare incontro a emolisi accelerata, se esposta ad agenti ossidanti. Lo stato eterozigote è asintomatico. Nel caso di un doppio eterozigote tra HbE e � -talassemia, condizione diffusa tanto da essere considerata la più co-mune seria emoglobinopatia in tutto il mondo, si possono avere quadri clinici molto variabili, che vanno da una condizione non distinguibile dalla talassemia major a for-me molto più lievi che non necessitano di trasfusioni.
Negli ultimi anni sono state identifi cate numerose altera-zioni dello scheletro della membrana eritrocitaria; alcune di esse consistono in alterazioni primitive, di tipo strut-turale o funzionale, delle proteine costituenti lo scheletro della membrana, risultanti in anomalie morfologiche dell’emazia, con o senza ridotta sopravvivenza eritrocita-ria. Attualmente sono sicuramente ascrivibili a questo gruppo la sferocitosi ereditaria, l’ellissocitosi ereditaria con le sue varianti e la piropoichilocitosi ereditaria, a esse strettamente collegata. Prima di trattare delle malattie da difetto primitivo della membrana, viene premessa una breve descrizione della struttura della membrana eritrocitaria. Se mediante lisi ipotonica si vuota l’emazia del suo conte-nuto (Hb), l’ombra della membrana che rimane è denomi-nata con la parola inglese ghost per descriverne l’aspetto evanescente, appunto come un fantasma. Essa è costituita per circa il 41% di lipidi, per il 52% di proteine e per il 7% di carboidrati. I lipidi sono costitu-iti in parti uguali da fosfolipidi e da colesterolo libero; i fosfolipidi appartengono a quattro classi principali: sfi ngomielina, fosfatidilcolina, fosfatidiletanolamina e fosfatidilserina.
I fosfolipidi formano intorno all’emazia un doppio strato, con le estremità polari in contatto con la fase acquosa (ci-toplasma o plasma) e le catene degli acidi grassi orientate verso la faccia interna dello strato stesso. Le proteine della membrana sono divise in due gruppi, le proteine periferiche , che formano un reticolo all’interno del-lo strato lipidico, rivolto verso il citoplasma, e le proteine integrali , che attraversano i lipidi sia dal lato intracellulare sia dal lato extracellulare, e che sono perciò anche dette proteine transmembranose. Queste proteine sono indicate con particolari denomina-zioni o con numeri che si riferiscono alla loro mobilità elet-troforetica su gel di poliacrilamide (la quale è dipendente dal peso molecolare delle proteine che vengono fatte mi-grare), contando dall’estremità catodica a quella anodica del gel. Con il termine di scheletro della membrana si indica l’in-sieme di quelle proteine che residuano dopo estrazione dei ghost con il detergente non ionico Triton X-100, che allontana i lipidi. A differenza del citoscheletro delle più complesse cellule eucariotiche, che forma strutture intra-cellulari (microfi lamenti, fi lamenti intermedi, microtu-buli), nei GR lo scheletro è molto più semplice e fa parte
Alterazioni dello scheletro della membrana eritrocitaria F. Pellò
C0240.indd 1074C0240.indd 1074 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Capitolo 48 - ANEMIE
81075
della superfi cie citoplasmatica della membrana cellulare; perciò è chiamato scheletro della membrana. La proteina principale di questo scheletro è chiamata spectrina ed è un dimero (SpD) formato da due proteine fi lamentose, le proteine 1 e 2 (dette anche, più comu-nemente � e � ). Le due proteine decorrono affi ancate con l’estremità amino-terminale della catena � posta accanto alla estremità carbossi-terminale della catena � ( Fig. 48.13 ). Queste due estremità appaiate formano quella che viene chiamata la “testa” della spectrina (e perciò le estremità opposte rappresentano la “coda”). Due unità di spectrina possono associarsi testa contro testa formando dei tetrameri. Le code delle unità di spec-trina non possono associarsi direttamente, ma riescono a farlo legandosi a un’altra proteina dello scheletro della membrana, la F-actina (proteina 5). Questo legame è notevolmente stabilizzato dal coinvolgimento di un’al-tra proteina, che è indicata con il numero 4.1. Perciò, sia per collegamento diretto tra di loro delle unità di spectrina, sia grazie all’intermediazione del complesso F-actina-proteina 4.1, sulla superficie citoplasmatica della membrana eritrocitaria si forma una rete compo-sta da molteplici unità di spectrina. Questa rete deve essere ancorata alle proteine integrali della membrana e, attraverso di esse, al doppio strato lipidico. L’anco-raggio avviene in due modi; la proteina 4.1 (che, come si è visto, costituisce uno dei punti nodali della rete) si lega con la porzione intracitoplasmatica della proteina integrale glicoforina A . Le catene della spectrina sono in grado di legarsi a una proteina detta anchirina o protei-na 2.1 (il legame avviene in prossimità delle giunzioni testa-testa delle unità di spectrina) che, a sua volta, si attacca alla porzione intracitoplasmatica della proteina integrale designata con il numero 3. Tanto la glicoforina A quanto la proteina 3 sono glicoproteine. La glicoforina A determina la specifi cità dei gruppi sanguigni M e N a seconda dei residui aminoacidici situati in posizione 1 e 5. La proteina 3 è implicata nel trasporto del glucosio e degli anioni. Quindi, le proteine principali dello schele-tro interagiscono tra loro secondo un piano orizzontale, parallelo alla membrana, e secondo un piano verticale, perpendicolare alla membrana. Le interazioni secondo
il piano orizzontale, cioè le interazioni tra i dimeri di spectrina, e tra i dimeri di spectrina, la proteina 4.1 e la F-actina servono ad assicurare la continuità del reticolo proteico submembranoso, organizzato in una struttura bidimensionale; le interazioni verticali, tra spectrina, anchirina e proteina 3 e tra spectrina, proteina 4.1 e glicoforina assicurano la connessione del reticolo dello scheletro ai costituenti integrali della membrana. È probabile che alterazioni delle interazioni proteiche secondo i due diversi piani siano responsabili di modifi -cazioni di forma differenti del GR.
Sferocitosi ereditaria
Si tratta di un’anemia causata da un’anomalia intrinseca della membrana eritrocitaria che ne modifi ca la forma da biconcava in sferica, ne riduce la deformabilità e ne favorisce l’intrappolamento a livello splenico. È una ma-lattia congenita con modalità classica di trasmissione ereditaria di tipo autosomico dominante, raramente di tipo autosomico recessivo. Più comune tra le popolazioni del Nord Europa, è tuttavia segnalata in tutte le razze. Colpisce i due sessi con uguale frequenza e la prevalenza è di 20 casi/100.000 persone.
Eziopatogenesi Il difetto primario, trasmesso ereditariamente, è a livello della membrana dei GR; nel 25% dei casi le alterazioni della membrana sono di riscontro apparentemente sporadico. I tipi di defi cit sono due, la mancanza totale o parziale di una proteina dello scheletro della membrana e l’in-terazione anomala delle proteine periferiche tra loro o con una proteina integrale. Le anomalie di riscontro più frequente sono • una diminuzione della spectrina; • una riduzione della proteina 3 associata alla
riduzione di spectrina; • un difetto molecolare all’estremità distale (coda)
della spectrina, risultante in un legame difettoso con la proteina 4.1;
• un assemblaggio alterato dei dimeri della spectrina.
InterazioneSp-actina 4.1
ProfilamentoF-actina
Proteina4.1
Glicoforina-A
InterazioneSp-4.1 Glic.-A
InterazioneSp-2.1-3
Ankirina
InterazioneSpD-D
Doppiostrato lipidico
Proteina 3
Catena Catena Spectrina Proteina
4.1
Figura 48.13 Modello dello scheletro della membrana del GR. (Si veda il testo.)
C0240.indd 1075C0240.indd 1075 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1076
È stata osservata, inoltre, anche una netta riduzione dei fosfolipidi e del colesterolo della membrana. Si ammette che la modifi cazione della forma del GR da discocita a sferocita sia dovuta a una riduzione del rap-porto superfi cie/volume, per riduzione della superfi cie da instabilità e perdita di membrana, dovuta a sua volta ai vari difetti della spectrina. Infatti, il defi cit di spectrina fa sì che vi siano aree in cui il doppio strato lipidico, privo di sostegno, va incontro a perdita di materiale, con conseguente instabilità anche delle proteine integrali. I GR a tipo di sferociti vanno incontro a importanti mo-difi cazioni delle loro proprietà, cioè acquisiscono una diminuita plasticità e deformabilità, un alterato metaboli-smo cellulare, soprattutto a livello del trasporto degli ioni attraverso la membrana, un’aumentata fragilità osmotica, un aumento della viscosità del citoplasma con aumento della concentrazione di emoglobina, attribuita a lieve disidratazione cellulare. L’alterato metabolismo cellulare è rappresentato princi-palmente da un aumento della permeabilità eritrocita-ria agli ioni sodio e potassio. Perché la concentrazione intracellulare di sodio e potassio si mantenga nei limiti fi siologici, è necessario che nei GR di questi soggetti si abbia un incremento di attività del sistema enzimatico noto come pompa sodio/potassio. Esso è legato alla mem-brana dell’emazia ed è stato descritto come ATPasi Na + /K + dipendente, volendo con ciò indicare che si tratta di un sistema che idrolizza l’ATP a una velocità che dipende dalla concentrazione dei cationi; si rende quindi neces-saria un’elevata produzione di ATP, che negli sferociti si realizza grazie a un incremento della glicolisi anaerobia. È per questo aumento di funzione della pompa Na + /K + che le cellule si deprivano di ATP in assenza di glucosio. Non è però perfettamente nota la natura di questa ano-mala permeabilità dei GR al sodio in corso di sferocitosi ereditaria. Nelle emazie di SE si è anche dimostrato un basso livello di 2,3-difosfoglicerato unitamente a una riduzione del pH, che ne è probabilmente la conseguenza. L’aumentata fragilità osmotica si ritiene sia dovuta al fatto che la confi gurazione di sferocita, cioè con un volume dell’emazia aumentato rispetto alla superfi cie, predispone di per sé alla lisi osmotica in presenza di concentrazioni più alte di cloruro di sodio rispetto a quanto si verifi ca nei GR normali a forma di discocita; infatti anche i discociti, prima di andare incontro alla lisi, assumono la forma di sferocita.
Fisiopatologia Il punto chiave nella sferocitosi ereditaria è, come si è visto, un’anomalia intrinseca all’emazia che la rende su-scettibile a una distruzione prematura nella milza. La diminuita sopravvivenza dei GR nella sferocitosi ere-ditaria è causata dal loro intrappolamento nei sinusoidi splenici; dopo splenectomia, infatti, migliora sensibil-mente l’anemia e la sopravvivenza degli eritrociti ritorna a valori pressoché normali. Durante il passaggio nella polpa rossa della milza, i GR devono deformarsi per at-traversare il microcircolo splenico, superare i cordoni, i seni e passare poi nel circolo venoso. Gli sferociti, in ragione della loro ridotta deformabilità, si trattengono
a lungo nella milza, dove non trovano una quantità di glucosio (energia) suffi ciente a mantenere il loro elevato metabolismo, esauriscono progressivamente il contenuto in ATP e perdono i lipidi di membrana. Si forma così una popolazione di microsferociti fagocitati dai macrofagi e destinati a una rapida emolisi. È importante ricordare, infi ne, come gli eritrociti di pazienti affetti da sferocitosi ereditaria abbiano una sopravvivenza ridotta quando vengono trasfusi in soggetti sani, normale quando invece il ricevente è splenectomizzato. Come pure è normale la vita media di emazie isogruppo trasfuse a un soggetto con sferocitosi. Ciò signifi ca che il difetto è intrinseco al GR e che vi è emolisi solo in presenza della milza. Pertanto, la sferocitosi ereditaria è caratterizzata da un’emolisi extravascolare e quindi da iperbilirubinemia indiretta con urobilinuria. Il midollo può incrementare la sua attività eritropoietica in misura tale da mantenere in termini modesti l’anemia, anche se in certi periodi questo compenso può essere difettoso e si possono sviluppare epi-sodi di più marcata anemizzazione (crisi “aplastiche”).
Manifestazioni cliniche La triade sintomatologica della malattia è rappresenta-ta da anemia emolitica con sferocitosi periferica, ittero, splenomegalia. Tuttavia. l’espressività dei sintomi e segni è assai variabile, a seconda della gravità della malattia. Si distinguono infatti tre forme principali: • una forma lieve, che interessa circa un quarto
dei pazienti, senza anemia, perché la produzione midollare di eritrociti è in grado di compensare completamente la distruzione. Gli altri membri della famiglia possono anch’essi presentare questa forma lieve oppure essere affetti dalla forma grave. I segni di emolisi e la splenomegalia possono essere modesti o assenti, ma la situazione può aggravarsi in occasione di malattie virali (per esempio, mononucleosi infettiva), di uno sforzo fi sico importante, della gravidanza;
• una forma moderata che interessa circa i due terzi dei pazienti, con distruzione emolitica degli eritrociti non compensata e periodi intermittenti di ittero, spesso collegati a infezioni virali; la splenomegalia è di solito presente, di entità variabile;
• una forma grave, che interessa una piccola quota di pazienti ( < 10%), con anemia grave, spesso richiedente emotrasfusioni di concentrati di GR; questi pazienti possono presentare un ritardo di crescita e andare incontro a modifi cazioni nella struttura delle ossa del cranio, con la tipica turricefalia (per ispessimento delle ossa frontali e parietali). Nelle forme più gravi possono comparire anche ulcerazioni agli arti inferiori, soprattutto perimalleolari, simili a quelle di facile riscontro nella talassemia minor e nella drepanocitosi.
Frequente è la colelitiasi, complicanza peraltro di molti disturbi emolitici cronici. Tutti i pazienti, ma con probabilità e frequenza maggiori quelli affetti dalla forma grave, possono andare incontro a due tipi di crisi: emolitica e aplastica. La crisi emolitica può essere precipitata da un’infezione o dallo stress ed è caratterizzata da aumento della bilirubi-
C0240.indd 1076C0240.indd 1076 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Capitolo 48 - ANEMIE
81077
nemia indiretta, splenomegalia e anemia; di solito non è molto grave e richiede una terapia trasfusionale. Le crisi aplastiche sono molto più gravi e possono anche avere esito fatale; si presentano con febbre, dolore ad-dominale, vomito, reticolocitopenia e anemia, talvolta anche neutropenia e piastrinopenia. La crisi dura da 10 a 14 giorni e la fi ne è contrassegnata dalla comparsa dei reticolociti nel sangue periferico. L’eziologia della crisi aplastica è molto verosimilmente virale, in particolare da parvovirus. I parvovirus sono comuni agenti di malattia negli animali; il loro nome (da parvus, piccolo) è dovuto al fatto che sono i più piccoli virus (15-28 nm di diametro alla microscopia elettronica) contenenti DNA in grado di infettare cellule animali. Il parvovirus fu descritto nel 1975 e si è rivelato l’agente eziologico, nelle persone normali, della quinta malattia nei bambini e di una sindrome di tipo poliartralgico negli adulti. L’infezione da parvovirus è molto contagiosa e il riscontro, nel 50% degli adulti normali, di anticorpi antiparvovi-rus nel siero presuppone un’immunità acquisita nell’età scolare. La nomenclatura uffi ciale è parvovirus B19 , che è il ter-mine impiegato dallo scopritore del virus. Questo virus è importante in ematologia perché la prima malattia che è stata associata all’infezione da parvovirus B19 è la crisi aplastica transitoria osservata in pazienti con drepanocitosi; successivamente si è dimostrato che il parvovirus B19 può indurre crisi aplastiche transitorie in pazienti affetti da numerosi processi emolitici di base: la già ricordata drepanocitosi, la sferocitosi ereditaria, la talassemia, l’anemia emolitica da defi cit degli enzimi eritrocitari, l’anemia emolitica autoimmune. Talvolta la crisi aplastica rappresenta l’esordio clinico di un’anemia emolitica misconosciuta. Le crisi aplastiche transitorie so-no caratterizzate dalla brusca insorgenza di grave anemia, talvolta accompagnata da granulocitopenia e piastrinope-nia, con assenza di reticolociti e presenza nel midollo di scarsi precursori eritroidi. I pazienti con crisi aplastiche spesso non hanno altri sin-tomi, oppure possono presentare febbre con sintomi di infezione delle vie aeree superiori, gastroenterite o malat-tia simil-infl uenzale. Nelle prove in vitro con colonie in mezzo semisolido, l’effetto inibente del parvovirus B19 si è dimostrato alta-mente specifi co per la linea eritropoietica e privo di effetto sul numero di colonie derivate dalla CFUGM. Vi sono, infi ne, alcune segnalazioni molto recenti che in soggetti immunodepressi, per esempio portatori di sindromi di immunodefi cienza congenita, di neoplasie in terapia chemioterapica, di AIDS, gli episodi di improvviso defi cit midollare che talvolta si verifi cano possono essere dovuti a infezione da parvovirus.
Esami di laboratorio I valori di emoglobina sono general-mente compresi tra 8 e 11 g/dL, anche se, in occasione delle crisi “aplastiche”, l’anemia può aggravarsi notevolmente. Lo striscio di sangue periferico presenta circa il 20-30% della popolazione eritrocitaria con una forma rotondeg-giante, intensamente e omogeneamente colorata (priva cioè della consueta area centrale chiara).
L’anisopoichilocitosi è marcata; il volume corpuscolare medio è di solito normale, mentre la CHCM il più delle volte si presenta elevata. Vi è una reticolocitosi (5-20%) che può anche essere notevolmente superiore, soprat-tutto nel periodo che segue immediatamente una crisi “aplastica”. Nel midollo emopoietico si osserva una spiccata iperpla-sia della serie rossa, talvolta con inversione del rapporto leuco/eritroblastico. La bilirubina indiretta nel siero è aumentata, l’aptoglobina sierica assente o diminuita. L’escrezione urinaria di bilinogeno è aumentata, le feci, poi, hanno elevatissimi quantitativi di bilinogeno. Non esistono esami di per sé specifi ci della sferocitosi ereditaria. La diagnosi poggia quindi su di una costella-zione di dati che, presi nel loro complesso, suggeriscono l’esistenza della malattia. Tra questi, di particolare importanza sono il test di fragilità osmotica e il test di autoemolisi . Gli sferociti, rispetto agli eritrociti normali, presentano minore superfi cie per uni-tà di volume e quando vengono incubati con soluzioni ipotoniche si lisano più facilmente. Il sangue di un pa-ziente con sferocitosi ereditaria comincia a emolizzare alle concentrazioni 0,5-0,7% di NaCl (normalmente l’emolisi inizia con NaCl allo 0,45%). La presenza poi di una “co-da” osmoticamente resistente rifl ette la percentuale di reticolociti presenti. La sferocitosi presenta una netta riduzione delle resistenze globulari osmotiche, quando i GR vengono preincubati a 37 °C per 24 ore e poi sottoposti allo stress iposmotico (infatti c’è un’ulteriore deplezione di ATP). Nei casi lievi quest’ultima avvertenza permette di “sensibilizzare il siste-ma” ed evidenziare ugualmente la presenza dell’anomalia, altrimenti non dimostrabile. Nel test di autoemolisi il sangue è incubato a 37 °C, ste-rilmente, per 48 ore. In condizioni normali il rilascio di Hb arriva al massimo al 4%; nella sferocitosi ereditaria, viceversa, può raggiungere anche il 40%. Questa non è la sola malattia nella quale l’autoemolisi sia incrementata; tuttavia è possibile restringere ulterior-mente il campo delle ipotesi aggiungendo ATP o glucosio nel mezzo di sospensione. Una marcata riduzione del fenomeno dopo aggiunta di queste sostanze è fortemente suggestiva di sferocitosi ereditaria, anche se un risultato analogo è osservabile nell’anemia emolitica da defi cit di triosofosfato-isomerasi. La sopravvivenza eritrocitaria, infi ne, con emazie marcate con 51 Cr è diminuita nei pazienti non splenectomizzati e normale in quelli che sono stati sottoposti all’intervento chirurgico. Ulteriori conferme alla diagnosi si ottengono con uno studio della famiglia del paziente. Lo screening può essere fatto molto semplicemente con emocromo-citometria, conta dei reticolociti, esame dello striscio di sangue periferico.
Diagnosi Nell’anemia emolitica autoimmune è possibile che si ve-rifi chi una certa sferocitosi a carico degli eritrociti. Questa forma è però facilmente differenziabile dalla sferocitosi ereditaria per la positività del test di Coombs e per la normalità del test di autoemolisi. Un possibile problema, in presenza di ittero evidente e lieve anemia, come può capitare nella sferocitosi ereditaria, è la differenziazione da
C0240.indd 1077C0240.indd 1077 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1078
una sindrome di Gilbert (si veda il Capitolo 30 ). Quest’ul-tima, tuttavia, non si accompagna di regola né a urobili-nuria né ad alcuna delle stigmate ematologiche proprie della sferocitosi.
Decorso e prognosi La malattia ha un andamento cronico, con saltuarie esa-cerbazioni dell’anemia, caratterizzate da splenomegalia dolente, ittero franco, reticolocitosi, iperbilirubinemia indiretta in occasione delle crisi emolitiche. Come si è visto, raramente insorgono crisi aplastiche con carattere di maggiore gravità. Complessivamente la prognosi è buona, perché si ha a disposizione una terapia patogenetica in grado di nor-malizzare il quadro ematologico, rappresentata dalla sple-nectomia.
Terapia La splenectomia è l’unica terapia utile. Una volta eseguita, il difetto intrinseco dei GR persiste; tuttavia il quadro ematologico si normalizza a eccezione della comparsa di lieve trombocitosi e leucocitosi. Talvolta la mancanza di guarigione è ascrivibile alla presenza di milze accessorie, più spesso a diagnosi non esatta. L’aumentata frequenza di batteriemie che si osserva nei bambini di età inferiore a 5 anni dopo splenecto-mia impone che l’intervento sia ritardato per quanto possibile (almeno dopo il decimo anno di età). Non di rado l’emolisi cronica e l’eritropoiesi accelerata che ne consegue richiedono l’istituzione di una terapia con acido folico.
Ellissocitosi ereditaria
Il nome della malattia deriva dall’aspetto ellittico dei GR nel sangue periferico. Si tratta di una malattia eteroge-nea, sia in termini di espressione clinica sia di morfologia delle emazie. Sulla base della forma dei GR si possono distinguere tre gruppi principali: 1) la forma comune in
cui l’emazia è biconcava; 2) la forma sferocitica, che rap-presenta un fenotipo ibrido tra la sferocitosi ereditaria e l’ellissocitosi; 3) la forma stomatocitica (emazie a forma di scodella), comune nelle popolazioni della Malesia che, a differenza delle altre due forme di ellissocitosi, conferisce resistenza all’infestazione malarica. L’ellissocitosi comune, la più frequente delle tre forme, si trasmette ereditariamente come carattere autosomico a dominanza incompleta; allo stato eterozigote costituisce o una condizione asintomatica con normale sopravvi-venza delle emazie o si esprime con un’emolisi da lieve a moderata. Gli omozigoti hanno di solito grave anemia emolitica con evidenti micropoichilocitosi, microellisso-citosi e microsferocitosi. La piropoichilocitosi ereditaria , malattia strettamente corre-lata all’ellissocitosi, è trasmessa con modalità autosomica recessiva, caratterizzata da spiccatissima micropoichiloci-tosi e instabilità termica delle emazie. Il difetto molecolare alla base dell’ellissocitosi e della piropoichilocitosi ha luogo a livello delle proteine del citoscheletro della membrana, che possono essere qua-litativamente alterate, con defi cit funzionale, o ridotte quantitativamente. Il difetto più comune riscontrato in circa un terzo dei pa-zienti con ellissocitosi ereditaria e in quasi tutti i pazienti con piropoichilocitosi è costituito da un difetto di assem-blaggio dei dimeri della spectrina in tetrameri e oligomeri, per un difetto strutturale delle catene � della spectrina. La notevole instabilità delle emazie riscontrata nelle forme gravi di ellissocitosi e piropoichilocitosi, con conseguen-te frammentazione dei GR, poichilocitosi ed emolisi, è verosimilmente dovuta a instabilità dello scheletro della membrana. Non è completamente chiarito il meccanismo di formazio-ne degli ellissociti; si suppone che i GR normali assumano transitoriamente una forma ellittica quando sottoposti allo stress dovuto all’attrito da scivolamento sulla parete endoteliale; poiché, come si è già visto, l’alterazione a livello dello scheletro della membrana ne riduce la fl es-sibilità e la plasticità, cioè lo rende più rigido, esso non sarebbe in grado di riprendere la forma iniziale al venir meno dello stress.
Si tratta di anemie emolitiche ereditarie secondarie a ri-dotta attività di uno degli enzimi necessari al metabolismo glucidico eritrocitario, ovvero gli enzimi della via glicoli-tica di Embden-Meyerhof e quelli dello shunt dell’esoso-monofosfato. Le forme di più comune riscontro sono quelle causate da defi cit di glucosio-6-fosfato-deidrogena-si (G-6-PD) e di piruvato-chinasi (PK).
Cenni sul metabolismo eritrocitario Durante il suo sviluppo, il GR perde molte delle “vie biochi-miche” presenti nella cellula nucleata. Il GR maturo, infatti, non può sintetizzare acidi nucleici, lipidi, enzimi, glucidi ed eme. Non possiede gli enzimi del ciclo degli acidi tricar-bossilici, la catena respiratoria e la fosforilazione ossidativa. Gli eritrociti maturi mantengono unicamente integri la glicolisi anaerobia e il ciclo del glucosio-6-fosfato.
Infatti, nonostante un’attività metabolica molto limita-ta, l’eritrocita richiede energia per mantenere fl essibile la membrana cellulare, regolare il fl usso degli ioni sodio e potassio, far funzionare il sistema ossido-riduttivo capace di proteggere la cellula dall’azione di sostanze os-sidanti endogene ed esogene. Questi obiettivi vengono raggiunti grazie alla glicolisi, in particolare mantenen-do appropriati livelli intracellulari di alcune molecole specifi che quali ATP, GSH, NADH, NADPH e 2,3-DPG ( Tab. 48.11 ). Circa il 90% del glucosio che giunge nell’eritrocita viene me-tabolizzato ad acido lattico attraverso la glicolisi anaerobia. È questa la sola via utilizzata dal GR per produrre ATP. Per ogni mole di glucosio vengono prodotte due moli di ATP suffi cienti a mantenere attive le funzioni ATP-dipen-denti, cioè il trasporto attivo attraverso la membrana di
Anemie da defi cit degli enzimi eritrocitari G. Gromo
C0240.indd 1078C0240.indd 1078 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Capitolo 48 - ANEMIE
81079
ioni calcio, sodio e potassio, la fosforilazione delle protei-ne di membrana e la glicolisi stessa. Attraverso la glicolisi anaerobia si produce anche NADH, un cofattore di fonda-mentale importanza nelle reazioni di ossidoriduzione. Lo shunt di Rapaport-Luebering, la via alternativa nel pas-saggio da 1,3-difosfoglicerato a 3-fosfoglicerato, permette poi la sintesi e la degradazione di 2,3-DPG, un importante regolatore del processo di ossidazione dell’emoglobina. Solo il 10% del glucosio che penetra nel GR viene normal-mente catabolizzato attraverso il ciclo dei pentosi.
La principale funzione di questa via è di produrre NADPH e di mantenere così il glutatione in forma ridotta (GSH) ( Fig. 48.14 ). Il GSH protegge i GR dalle sostanze ossidanti (ioni superossido e perossido di idrogeno) liberate da macrofagi e granulociti durante la fagocitosi o prodotte dagli stessi GR dopo assunzione di determinati farmaci. Non viene generato, invece, ATP. La quantità di glucosio che entra nel ciclo dei pentosi non è costante, ma è in funzione della quantità di NADP e può aumentare fi no a 30 volte.
Anemie da difetto degli enzimi della glicolisi anaerobia
Sono anemie rare. Il 90% dei casi è provocato da defi cit di PK, il 4% da defi cit di glucosiofosfato-isomerasi (GPI, GlucosePhosphate Isomerase) e i restanti casi dalla caren-za di altri enzimi della via glicolitica anaerobia.
Anemia emolitica da defi cit di piruvato-chinasi (PK) Si tratta di una malattia diffusa in tutto il mondo, anche se la gran parte dei casi è stata segnalata nella popolazione europea.
Eziopatogenesi Il difetto enzimatico è ereditato come carattere autoso-mico recessivo; maschi e femmine vengono interessati in egual misura. La malattia conclamata si ha solo negli omozigoti ed è associata a un’attività dell’enzima PK pari al 5-25% dell’at-tività normale. Negli eterozigoti l’attività della PK è ridotta di circa il 50% senza che vi sia un’evidenza clinica del difetto; tuttavia
Composto Funzione principaleATP Fornisce energia per il trasporto attraverso
la membrana di ioni sodio, potassio e calcio; trasferisce gruppi fosforici ad alta energia alle proteine di membrana
2,3-DPG Riduce l’affi nità di Hb per l’O 2 e consente una maggiore cessione di O 2 ai tessuti
GSH Fornisce gruppi sulfi drilici utilizzabilil per metabolizzare le sostanze endogene ed esoge-ne ossidanti; contribuisce alla detossifi cazione della H 2 O 2
NADPH Fornisce protoni per rigenerare il glutatione ridotto dal glutatione ossidato
NADH Cofattore nella reazione che previene l’accumulo di metaemoglobina
Tabella 48.11 Composti intermedi metabolici di fondamentale importanza nel globulo rosso
LATTATO
PIRUVATO
Piruvico-chinasi
GLUCOSIO
GLUCOSIO-6-FOSFATO Glucosio-6-fosfato-deidrogenasi
Glutatione-perossidasi
Glutatione-perossidasi
6-FOSFOGLUCONATOShunt dell’esosomonofosfato
PENTOSOFOSFATO
6-fosfogluco-deidrogenasi
NADP NADPH
GSSHGSH
H2O2 H2O2
aiboreana isilocilG
Figura 48.14 Ruolo della glucosio-6-fosfato-deidrogenasi nella produzione di NADPH e nel mantenimento del glutatione in forma ridotta (GSH).
C0240.indd 1079C0240.indd 1079 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1080
una sintomatologia, se pur meno marcata, è presente an-che in alcuni pazienti con eterozigosi per due mutazioni distinte e interagenti (doppia eterozigosi). È importante inoltre ricordare che nei tessuti umani sono presenti tre isoenzimi della PK; il tipo I è contenuto nei GR e nel fe-gato, il tipo II nei reni e il tipo III in leucociti, piastrine, fegato, reni, muscoli e cervello. Non sempre esiste una correlazione fra entità dell’anemia ed entità del defi cit enzimatico. La ridotta attività della PK è dovuta, nella gran parte dei casi, a un difetto quantitati-vo; in altri casi sono state riconosciute varianti strutturali con alterate proprietà cinetiche.
Fisiopatologia La PK eritrocitaria è un tetramero con un peso molecolare di 225.400. Catalizza la conversione da fosfoenolpiruvato in piruvato e interviene così in una delle due reazioni glicolitiche capaci di produrre ATP (l’altra è catalizzata dalla fosfoglicero-chinasi). La carenza di PK, quindi, provoca una riduzione della sintesi di ATP, un accumulo dei metaboliti che precedono questa tappa nella sequenza glicolitica (PEP; 3 PG; 2,3-DPG) e una diminuzione dei substrati a valle (piruvato e lattato). Il danno metabolico principale dei GR carenti di PK è la diminuita produzione di ATP con conseguente riduzione di tutte le funzioni ATP-dipendenti. Compaiono lesioni funzionali della membrana eritro-citaria che provocano riduzione della concentrazione intraeritrocitaria di potassio e accumulo di ioni calcio. La membrana diviene più rigida, meno elastica, e possono comparire, alla superfi cie, protuberanze e spicole (acan-tocitosi) . Le emazie così modifi cate vengono riconosciute dal sistema reticolo-endoteliale del fegato e della milza e fagocitate. Da segnalare che anche i reticolociti (capaci di sfruttare la fosforilazione ossidativa mitocondriale per produrre ATP) sono distrutti nella milza, dal momento che quest’organo costituisce un ambiente sfavorevole (pH basso, ridotta PO 2 , bassa concentrazione di glucosio) per il metabolismo mitocondriale dei reticolociti.
Manifestazioni cliniche L’espressione della malattia è variabile; talvolta si manife-sta alla nascita con grave ittero neonatale, altre volte il de-fi cit di PK viene diagnosticato, del tutto occasionalmente, durante l’età adulta. In genere, comunque, anemia e ittero compaiono nel corso dell’infanzia. La sintomatologia è, con buona approssimazione, simile a quella osservata nel-le anemie emolitiche croniche (anemia, ittero, reticolociti aumentati, splenomegalia). L ’incidenza di litiasi biliare è elevata, mentre rare sono le ulcere agli arti inferiori, come pure le modifi cazioni ossee (soprattutto bozze frontali) associate a iperplasia del midollo emopoietico, quali si osservano in altre forme di anemie emolitiche croniche ereditarie.
Esami di laboratorio L’anemia normocitica normocromica è di gravità variabi-le, il tasso di Hb può variare da 6 a 12 g/dL e l’Hct dal 17 al 37%. Non esistono solitamente fl uttuazioni nell’entità dell’anemia; rare sono le esacerbazioni in occasione di in-fezioni o interventi chirurgici. Tutti i parametri morfologi-ci caratteristici di un’eritropoiesi accelerata sono presenti
nello striscio periferico: policromasia, anisocitosi, poichi-locitosi e GR nucleati. Talvolta sono osservati acantociti ed echinociti. La reticolocitosi è spiccata (5-15%) e aumenta ulteriormente dopo splenectomia (50%). Normali sono i leucociti e le piastrine. La bilirubina indiretta è aumentata, la sideremia normale, le urine presentano un’elevata escre-zione di urobilinogeno, il midollo osseo è ipercellulare con intensa iperplasia della serie eritropoietica.
Diagnosi In passato, il primo esame di screening da eseguire nel sospetto di un defi cit di PK era il test di autoemolisi. Si può infatti osservare una percentuale di emolisi del 20-30% (valore normale 4%), corretta dall’aggiunta di ATP ma non di glucosio (a differenza di quanto avviene nella sferocito-si ereditaria). Oggi si preferisce dosare direttamente l’atti-vità dell’enzima presente nei GR del paziente, calcolando la velocità di ossidazione dell’NADH a NAD. Nei pazienti con defi cit di PK dei GR (isoenzima I) è anco-ra presente la PK leucocitaria (isoenzima III). Si deve porre quindi particolare attenzione nell’allontanare i leucociti, dal momento che l’attività della PK leucocitaria è 300 volte maggiore di quella dei GR e potrebbe infi ciare il risultato del test. Il defi cit di PK si presenta comunemente come un’anemia emolitica normocitica normocromica. La diagnosi differenziale, dopo avere escluso i più co-muni processi emolitici, deve comprendere le anemie emolitiche ereditarie da emoglobina instabile e le altre enzimopatie ereditarie dei GR. La presenza di emoglobina instabile può essere dimostrata ricorrendo al test della stabilità al calore o a quello della precipitazione con isoproterenolo, le altre enzimopatie vengono dimostrate con dosaggi quantitativi dell’attività dei diversi enzimi. Quanto poi alla sferocitosi, il quadro periferico e le resistenze globulari dovrebbero essere suf-fi cienti a dirimere eventuali dubbi diagnostici.
Terapia Non esiste terapia specifica e le cure sono essen-zialmente di supporto: acido folico per compensare l’aumentato consumo dovuto alla marcata eritropo-iesi, terapia trasfusionale nelle forme più gravi. La splenectomia spesso migliora il quadro clinico, ben-ché raramente si assista a una completa risoluzione dell’anemia emolitica.
Anemie emolitiche da defi cienza di altri enzimi della glicolisi anaerobia Sono forme molto rare. Il defi cit è trasmesso solitamente come carattere autosomico recessivo ed è causa di anemia solo se è presente allo stato omozigote. Unica eccezione è la carenza di fosfoglicero-chinasi (PGK) , che è trasmessa come carattere legato al sesso. I maschi sono affetti da grave anemia, mentre le femmine no. In alcuni casi il difetto è localizzato solo nei GR (defi cit di esochinasi) , altre volte anche nei leucociti, che peraltro mantengono le loro normali funzioni (defi cit di fosfogli-cero-chinasi e glucosio-fosfato-isomerasi).
C0240.indd 1080C0240.indd 1080 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Capitolo 48 - ANEMIE
81081
Da ricordare, poi, il caso della triosofosfato-isomerasi , in cui l’enzima è diminuito nei leucociti, nelle cellule muscolari e nel liquido cefalorachidiano. L’anemia sarà allora accompagnata da frequenti infezioni e gravi disturbi cardiaci e neurologici. La diagnosi di questi deficit si ottiene determinando l’attività degli enzimi glicolitici eritrocitari. La terapia è sintomatica.
Anemie da difetto degli enzimi dello shunt dei pentosofosfati
Il defi cit di glucosio-6-fosfato-deidrogenasi (G-6-PD) è l’anomalia più importante dello shunt dei pentosi. Si calcola che oltre 100 milioni di persone nel mondo ne siano portatrici. Gli altri difetti enzimatici sono rarissimi e solo sporadi-camente sono stati documentati casi di carenza di gluta-tione-sintetasi e glutatione-reduttasi.
Anemia emolitica da defi cit di glucosio-6-fosfato-deidrogenasi (G-6-PD) Il defi cit di G-6-PD nei GR maturi è il risultato della sintesi di un enzima instabile e con emivita ridotta. Meno co-munemente il defi cit è dovuto a una ridotta sintesi o alla sintesi di un enzima inattivo o cineticamente anomalo. Questo difetto enzimatico è stato riscontrato in tutte le popolazioni, ma con particolare frequenza in una zona geografi ca che comprende il bacino mediterraneo, l’Africa occidentale e centrale, l’Asia minore e quella sudorientale. In Europa l’incidenza è dello 0,1% e in Italia dello 0,4%; fra le popolazioni nere del Nord America raggiunge il 10%. La distribuzione geografi ca è simile a quella della talas-semia, della malaria e della drepanocitosi. Si ritiene che questo difetto enzimatico costituisca un vantaggio se-lettivo nei confronti della malaria da Plasmodium falci-parum . Infatti, i GR con defi cit di G-6-PD sono meno frequente-mente parassitati rispetto ai GR normali in vitro . Il gene per la G-6-PD è posto sul cromosoma X, la tra-smissione ereditaria è pertanto legata al sesso. Quindi, i maschi che portano il gene per il defi cit di G-6-PD sono emizigoti. In questi soggetti esistono solo geni anomali per la G-6-PD, da cui deriva un interessamento generale della popolazione eritrocitaria. Le donne che presentano il gene anomalo, invece, sono comunemente eterozigoti. Infatti, ogni cellula eritropoietica presenta, a caso, uno solo dei due cromosomi X attivo, mentre l’altro è inatti-vato precocemente durante la vita embrionale (ipotesi di Lyon). Una femmina eterozigote per il defi cit di G-6-PD ha quindi due popolazioni di GR, una normale e l’altra carente. Tuttavia, poiché il processo di inattivazione del cromosoma X è casuale (l’ampiezza di ciascuna popola-zione, infatti, può non essere esattamente del 50%), la gravità dell’anemia dipenderà dal numero complessivo di GR carenti di G-6-PD.
Eziopatogenesi La ridotta effi cienza dell’enzima è provocata, nella quasi totalità dei casi, dalla presenza di varianti strutturali di G-6-PD con caratteristiche biochimiche e cinetiche alte-
rate, quali la stabilità al calore, l’affi nità per i substrati, il grado di inibizione prodotto dal NADPH, il pH ottimale di attività, la mobilità elettroforetica e cromatografi ca. La variante corrispondente a un’attività enzimatica “nor-male”, defi nita G-6-PD B + , è la più comune ed è conside-rata lo standard di riferimento. La variante seconda in ordine di frequenza e ad attività enzimatica solo lievemente ridotta (pari all’84%) è la G-6-PD A + . Rispetto alla B + è elettroforeticamente più veloce e differisce strutturalmente per un singolo aminoacido. Si trova nel 30% della popolazione nera maschile sta-tunitense e, in genere, tra quanti originano dall’Africa occidentale e centrale. Le varianti patologiche più comuni sono la G-6-PD A – e la G-6-PD Mediterranea. La G-6-PD A – si osserva in individui di discendenza afri-cana e ha un’attività pari al 5-15% di quella dell’enzima normale. La migrazione elettroforetica è sovrapponibile a quella della G-6-PD A + , ma la G-6-PD A – è estremamente instabile e con caratteristiche cinetiche anomale. La G-6-PD Mediterranea è maggiormente frequente tra i caucasici. L’attività enzimatica nei GR maturi degli indi-vidui con questa variante è quasi assente per defi ciente sintesi dell’enzima stesso. La mobilità elettroforetica è identica a quella della G-6-PD + . Altre varianti a ridotta attività che si osservano con re-lativa frequenza sono la G-6-PD Canton e la G-6-PD De-brousse . La G-6-PD Oklahoma è caratterizzata da ridotta affi nità dell’enzima per il suo substrato. Sono state descritte fi nora più di 160 varianti, raggrup-pabili, in base alle caratteristiche biochimiche e ai quadri clinici che determinano, in diverse classi. • Classe I: varianti con grave defi cienza enzimatica
associata ad anemia emolitica cronica. • Classe II: varianti con grave defi cienza enzimatica
non associata ad anemia emolitica cronica. Tra queste è compresa la Mediterranea. In questa classe alcuni farmaci, infezioni e alimenti possono scatenare un’emolisi acuta.
• Classe III: varianti con modesto defi cit enzimatico. La più frequente è la A – .
• Classe IV: varianti con attività enzimatica normale o quasi, tra cui la A + e la B + .
• Classe V: varianti con attività enzimatica aumentata.
Fisiopatologia Nei soggetti normali l’attività della G-6-PD decresce len-tamente e progressivamente durante l’invecchiamento fi siologico dei GR; tuttavia, dopo 120 giorni, è ancora suffi ciente a mantenere una quantità di NADPH e di GSH tale da preservare l’integrità dell’eritrocita. Diverso, in-vece, è il comportamento del GR con defi cit di G-6-PD. Infatti, le varianti patologiche comportano un decremen-to di attività enzimatica più rapido, così da raggiungere prima dei 120 giorni il livello critico oltre il quale il GR viene rimosso dal sistema reticolo-endoteliale (emolisi extravascolare). In alcune varianti quali A – e Mediterranea questo processo avviene quando il GR ha 90-100 giorni di età, il che comporta un quadro di emolisi spontanea, che però è del tutto compensato dall’aumento dell’at-tività eritropoietica. Invece altre varianti (per esempio,
C0240.indd 1081C0240.indd 1081 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1082
New York o Oklahoma) presentano una diminuzione critica dell’enzima dopo soli 20 giorni. In questi casi vi è sempre anemia emolitica cronica, più o meno grave a seconda del tipo di variante. La carenza di G-6-PD è anche causa di crisi emolitiche acute. Infatti, quando i GR sono sottoposti a uno stress ossidativo (farmaci, infezioni, alimenti), l’attività di G-6-PD richiesta a difesa dell’integrità e funzionalità della membrana è più elevata. Tuttavia gli eritrociti normali, anche nel corso di queste evenienze, non sono distrutti. Infatti, le sostanze ossidanti sono ridotte nel GR dal sistema del glutatione in cui sia GSH sia NADPH fungono da donatori di H + . In carenza di G-6-PD vengono meno questi substrati e le emazie diven-gono sensibili all’azione tossica delle sostanze ossidanti. Da parte sua, il sistema delle catalasi non è suffi ciente da solo a proteggere le cellule. Alcuni farmaci ( Tab. 48.12 ), direttamente o generando pe-rossido di idrogeno, ossidano i lipidi di membrana, il gruppo emico di Hb e le catene globiniche. La conversione, poi, di Hb in meta-Hb insieme alla produzione di ione superossido comporta un effetto dannoso per la membrana. La molecola dell’emoglobina diventa instabile e precipita nel GR sotto forma di corpo di Heinz. I corpi di Heinz (vi-sibili al microscopio ottico in uno striscio di sangue dopo colorazione con cristalvioletto o altri coloranti sopravitali) sono dei microprecipitati delle dimensioni di 0,2-2 � . In una prima fase si trovano al centro del citoplasma eritrocitario, poi aderiscono alla membrana alterandone il trasporto ionico. Le emazie che contengono i corpi di Heinz vengono trattenute nella microcircolazione spleni-ca. Prima di essere nuovamente rilasciate in circolo vengo-no liberate da questi corpi inclusi ( pitting splenico), il che porta a un ulteriore danno di membrana. Anche durante la fagocitosi, in occasione di processi infettivi, vengono liberate sostanze ossidanti dai granulociti e dai macrofagi. Ovvero l’emolisi potrebbe essere provocata dall’azione ossidante di sostanze endogene prodotte in corso di al-cune malattie. Per quanto riguarda gli alimenti, sono soprattutto da ricordare le fave. Da questi legumi è stata isolata una sostanza capace di interferire nel metabolismo del glutatione; si tratta del dopachinone, un metabolita della L-dopa, che catalizza l’ossidazione del GSH.
Manifestazioni cliniche La carenza di G-6-PD può dare origine a: • anemia emolitica cronica non sferocitica; • crisi emolitica acuta in occasione di assunzione
di farmaci; • crisi emolitica acuta in corso di infezioni;
• crisi emolitica acuta in occasione di assunzione di particolari alimenti;
• ittero neonatale.
Anemia emolitica cronica non sferocitica Le varianti associate a questo quadro sono circa 50. Di recente sono stati segnalati alcuni casi anche tra i portatori della varian-te Mediterranea. Il decorso è sovrapponibile a quello di altre anemie emolitiche croniche. Può esordire alla nascita con ittero emolitico neonatale e talvolta complicarsi con improvvise crisi di emolisi, che compaiono in occasione di infezioni o dopo assunzione di alcuni farmaci.
Crisi emolitica acuta secondaria a farmaci Sono assai numerosi i farmaci in grado di scatenare una crisi emoli-tica in un paziente portatore di defi cit di G-6-PD. Uno stesso farmaco, poi, può scatenare crisi in alcune va-rianti e non in altre. Per esempio, il CAF induce emolisi nei portatori di variante Mediterranea ma non in quelli di variante A – . La crisi di solito si manifesta improvvisamente dopo 1-3 giorni dall’assunzione del composto. L’anemia è molto marcata, l’emoglobinuria intensa, compaiono i corpi di Heinz, aumenta la bilirubinemia e l’ittero si fa manifesto. Spesso tutto questo si accompagna a senso di profondo ma-lessere, brividi, nausea, vomito, febbre, dolori addominali e lombari. Dopo 5 giorni circa compare intensa reticolocitosi, cui fa seguito un sostanziale miglioramento dell’anemia. Nella variante A – l’emolisi è di breve durata e autolimitante, cioè il quadro emolitico non peggiora anche se continua la somministrazione del farmaco alle medesime dosi che hanno scatenato la crisi. Infatti, dopo la brusca emolisi iniziale della metà più vecchia dei GR, il processo emolitico si riduce perché le cellule più suscettibili sono state ormai distrutte. D’altra parte, le emazie con meno di 50 giorni di vita dispongono di una maggiore quantità di enzima G-6-PD e sono meno suscettibili, o del tutto insensibili, ai danni ossidativi. A questo punto l’emolisi è presente, ma solo una piccolissima percentuale di emazie è distrutta giornalmente, per cui può essere compensata dall’eritropoiesi midollare. Nelle varianti a defi cit enzimatico più grave (per esempio, Mediterranea), invece, l’anemia si aggrava se il composto non viene subito sospeso. Infatti, in queste varianti l’at-tività enzimatica è inadeguata a fronteggiare le richieste aumentate anche nei GR giovani e nei reticolociti. Tra una crisi emolitica e l’altra il quadro ematologico è nella norma, senza reticolocitosi o splenomegalia.
Crisi emolitica acuta secondaria a infezioni Sono state documentate con certezza crisi emolitiche in corso di polmonite lobare, insuffi cienza renale acuta , infl uenza , epatite , chetoacidosi diabetica , salmonellosi . Il miglio-ramento è lento a causa di una diminuita produzione di reticolociti e sovente inizia dopo che l’infezione è guarita o l’alterazione metabolica è stata corretta.
Crisi emolitica acuta secondaria ad alimenti (favismo) Che l’esposizione alle fave ( Vicia fava ) fosse tossica e tal-volta mortale è cosa risaputa fi n dai tempi di Pitagora, ma solo di recente si è scoperto che questo rischio riguarda esclusivamente alcuni portatori di variante Mediterranea, mentre non è mai stato riscontrato nei portatori di va-riante A – o di altre varianti.
� Sulfamidici (sulfacetamide, sulfanilamide ecc.) � Antipiretici e analgesici (aminopirina, acetofene ecc.) � Antimalarici (primachina, pamachina) � Nitrofurani (nitrofurantoina, furazolidone ecc.) � Varie (vitamina K, isoniazide, probenecid, acido nalidixico,
CAF ecc.)
Tabella 48.12 Farmaci responsabili di emolisi negli eritrociti con defi cit di G-6-PD
C0240.indd 1082C0240.indd 1082 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Capitolo 48 - ANEMIE
81083
L’emolisi può essere molto grave e inizia solitamente alcu-ne ore dopo l’esposizione ai pollini o 2 giorni circa dopo l’ingestione di fave. Circa il 90% degli attacchi di favismo acuto è causato da ingestione di semi freschi o cotti della pianta, mentre i restanti casi sono dovuti all’esposizione a pollini di fave (soprattutto in primavera), particolarmente in Sardegna, ove l’incidenza della variante G-6-PD Medi-terranea raggiunge il 35%. In alcuni casi la crisi è mortale. Si pensa a una predisposizione genetica, dal momento che solo pochi individui con variante Mediterranea, per lo più appartenenti allo stesso nucleo familiare, presentano sensibilità alle fave.
Ittero neonatale Il defi cit di G-6-PD è la causa più fre-quente di ittero neonatale dopo la malattia emolitica del neonato (incompatibilità materno-fetale per il fattore Rh). L’abnorme suscettibilità all’emolisi (talvolta così grave da rendere necessaria l’exanguinotrasfusione) può essere dovuta a ipoglicemia, a ridotta attività della glutation-perossidasi, all’azione ossidante della vitamina K talora somministrata ai neonati. L’ittero neonatale è raro nella mutazione G-6-PD A – Africana.
Diagnosi L’anemia emolitica da defi cit di G-6-PD non presenta un quadro ematologico caratteristico. Il mezzo più specifi co per porre diagnosi è la valutazione dell’attività dell’enzima mediante dosaggio quantitativo spettrofotometrico della velocità di riduzione del NADP a NADPH. Questo esame può dare falsi negativi in pazienti ammalati se è effettua-to subito dopo la crisi emolitica, quando la popolazione eritrocitaria residua è molto giovane. Le femmine eterozigoti rappresentano un problema dia-gnostico, dal momento che coesistono cellule normali
e cellule carenti di G-6-PD e questo può mascherare la presenza del difetto. Le due principali classi di anoma-lie che possono simulare un deficit di G-6-PD sono la riduzione della concentrazione intracellulare di GSH e le emoglobine instabili. Le cause, oltre al defi cit di G-6-PD, che provocano la riduzio-ne di GSH intracellulare sono molto rare. È suffi ciente ricor-dare il defi cit di glutation-sintetasi o di � -glutamil-sintetasi. Le emoglobine instabili, da parte loro, possono essere facil-mente differenziate mediante la dimostrazione di un’alterata mobilità elettroforetica o con test di precipitazione.
Terapia Il più importante provvedimento consiste nell’evitare la somministrazione di farmaci che causano emolisi per stress ossidativo nei GR carenti di G-6-PD. La splenectomia raramente si dimostra efficace; talvolta è necessaria la terapia trasfusionale nelle crisi emo-litiche di pazienti portatori della variante Mediter-ranea.
Anemie emolitiche da defi cienza di altri enzimi dello shunt dei pentosi Sono stati segnalati, benché assai rari, defi cit congeniti della � -glutamil-cisteina-sintetasi e della glutation-sintetasi. Poiché questi due enzimi partecipano alla sintesi del gluta-tione, queste affezioni presentano quadri clinici analoghi a quelli del defi cit di G-6-PD. Alcuni episodi di ittero emolitico neonatale vengono poi attribuiti a defi cit di glutation-perossidasi.
L’emoglobinuria parossistica notturna (EPN), o sindrome di Marchiafava-Micheli, è un’anemia emolitica causata da un difetto acquisito della membrana eritrocitaria che rende una popolazione di GR notevolmente sensibile all’azione litica del complemento , comunque esso venga attivato. Clinicamente è caratterizzata, nella forma clas-sica, da attacchi di emolisi intravascolare ed emoglobinu-ria, talora notturni. Nella gran parte dei pazienti, tuttavia, si osservano anemia emolitica cronica senza una precisa correlazione con la notte, pancitopenia, sideropenia e ricorrenti episodi trombotici. La EPN colpisce indifferentemente individui di ambo i sessi, per lo più tra i 20 e 40 anni; raramente la si incon-tra nell’infanzia e tra gli anziani. Non vi è familiarità né predisposizione razziale.
Eziopatogenesi La EPN è caratterizzata da un’anomala sensibilità di GR, piastrine e granulociti alla azione litica del complemento. Questo è facilmente dimostrabile, in vitro , con l’impiego di un antisiero contenente anticorpi antieritrociti fi ssan-ti il complemento. I GR EPN fissano più C1 per unità
di anticorpo presente alla loro superfi cie di quanto non facciano le cellule normali. A sua volta, il C1 promuove una maggiore fi ssazione del C3 (per molecola di C1) (per l’attivazione della cascata del complemento, si veda il Capitolo 73 ). Tuttavia, la presenza dell’anticorpo non è necessaria per la lisi dei GR di pazienti con EPN; infatti, il C3 si fi ssa di preferenza attraverso la via alternativa (properdinica). Non tutte le emazie in corso di EPN sembrano risentire della lisi complemento-mediata. Dopo attente ricerche si sono potute identifi care tre sottopopolazioni: EPN I normale, EPN II, sensibile 6-7 volte più del normale al complemento, e infi ne EPN III, 20-25 volte più sensibile. Le tre popolazioni possono coesistere nel sangue dei pa-zienti in tutte le combinazioni possibili, dando così vita a differenti quadri clinici, riferibili, comunque, soprattut-to alla percentuale di EPN III presente. Tuttavia, in una buona parte di pazienti affetti da EPN viene riscontrata nel sangue una semplice miscela di cellule EPN I e EPN II. L’aumentata sensibilità delle cellule EPN II e EPN III al complemento non ha però ancora trovato una spiegazio-ne davvero soddisfacente.
Emoglobinuria parossistica notturna G. Gromo, M. Storti
C0240.indd 1083C0240.indd 1083 6/9/10 3:09:02 PM6/9/10 3:09:02 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1084
Le ricerche intraprese al fi ne di accertare eventuali alte-razioni a livello di membrana non hanno fornito ancora dati certi, a eccezione di una ridotta o assente attività acetilcolinesterasica di membrana nelle popolazioni EPN II e III. Studi più recenti sembrano aver identifi cato come base patogenetica il defi cit alla superfi cie delle emazie di due proteine importanti per tenere a freno l’attivazio-ne del complemento; una è indicata con la sigla MIRL (Membrane Inhibitor of Reactive Lysis) o CD59, e l’altra è contrassegnata dalla sigla DAF (Decay Accelerating Factor) o CD55. In realtà è suffi ciente la carenza della prima proteina, che antagonizza le tappe avanzate della cascata del complemento, per rendere le emazie suscet-tibili alla lisi. Queste due proteine (e altre) sono vincolate alla superfi cie cellulare in una maniera particolare. Infatti, non attra-versano la membrana cellulare a tutto spessore grazie a un segmento idrofobico della loro catena peptidica, ma sono tenute ancorate grazie al legame con un oligosac-caride contenente GPI e perciò detto GPI-àncora. Questa molecola è immersa nello strato esterno del doppio fo-glietto lipidico e la sua assenza fa sì che tutte le molecole proteiche che le sono ordinariamente collegate non siano più trattenute alla superfi cie cellulare. Perciò, nella EPN il difetto fondamentale riguarda la sintesi della GPI-àncora, che avviene in maniera difettosa in conseguenza di mu-tazioni somatiche. Sono stati individuati vari geni importanti per la sintesi della GPI-àncora. Il più importante è chiamato PIG-A, è situato sul cromosoma X ed è necessario per il primo passo della sintesi della GPI-àncora, ossia l’aggiunta di N-acetil glucosamina a fosfatidil inositolo. La EPN può essere assimilata al gruppo di forme morbose classifi cate come “sindromi mielodisplastiche”. Si tratta di malattie che presentano alterazioni in tutte e tre le linee staminali, ma – a differenza delle forme mie-loproliferative – sono caratterizzate da una situazione “ipoproliferativa” (si veda il Capitolo 51 ). Interconversioni tra le diverse patologie sopra elencate non sono rare, e il rapporto EPN/anemia aplastica ne è un esempio. Tutte queste forme, a loro volta, possono poi esi-tare in leucemia mieloide acuta (si veda il Capitolo 51 ).
Fisiopatologia L’alterazione più caratteristica della EPN è l’insorgenza di crisi notturne intravascolari con emoglobinuria . La frequenza di queste crisi è variabile e in larga misura dipende dalla percentuale di GR EPN III patologicamente sensibili al C. Se sono inferiori al 20%, l’emolisi, seppure rilevabile, sarà di modestissima entità; tra il 20-50% si presenta emoglobinuria saltuaria, che diventa costante quando superano il 50%. In pazienti con eritrociti appar-tenenti alla popolazione EPN II, raramente si ha emoglo-binuria. Tali crisi sembrano essere legate più propriamente al sonno. Infatti, avvengono di giorno in quei soggetti che presentano un ritmo sonno/veglia invertito, dal momento che svolgono la loro attività nel corso della notte. Anni fa si ipotizzava che la ridotta attività dei centri respiratori durante il sonno, con relativo aumento della pressione parziale di CO 2 ematica e lieve riduzione del pH, favo-risse l’attivazione della via alternativa del C e l’emolisi delle popolazioni EPN II e III. Oggi questa ipotesi sembra
raccogliere però meno consensi. Analoghe spiegazioni sono state prese in considerazione in relazione al ritmo circadiano di secrezione del cortisolo. Anche lo sforzo fisico continuato e l’acidosi che ne deriva potrebbero scatenare la crisi di EPN. Un’altra caratteristica dei pazienti con EPN è un’aumen-tata propensione alle trombosi venose . Anche per questa alterazione è stato supposto un ruolo della carenza di una proteina, questa volta alla superfi cie dei leucociti e delle piastrine. Si tratterebbe del recettore per l’attivatore del plasminogeno a opera dell’urochinasi (uPAR, urokinase Plasminogen Activator Receptor), che è importante per la generazione di plasmina e perciò per la fi brinolisi. Un punto importante è la riconosciuta associazione della EPN con l’anemia aplastica. A questo proposito si è supposto che in tale condizione, immunologicamente mediata, le cellule staminali emopoietiche, alterate per la mutazione somatica che conduce alla EPN, godrebbe-ro di un vantaggio selettivo, in quanto più resistenti ai meccanismi di distribuzione immunologica e, in modo particolare, all’azione delle cellule natural killer (NK). Infi ne, i pazienti con EPN hanno una notevole propen-sione alle infezioni batteriche , attribuibile in parte alla leucopenia (per la frequente associazione con anemia aplastica) e in parte a menomazione delle funzioni dei granulociti, probabilmente in conseguenza del fatto che, per la maggior parte, sono derivati da un clone abnor-me di cellule ematopoietiche (lo stesso che dà origine agli eritrociti particolarmente suscettibili all’azione del complemento).
Manifestazioni cliniche L’emoglobinuria è presente all’esordio solo in un quarto dei casi, ma nel decorso della malattia compare quasi in tutti i pazienti, talora con esacerbazioni emolitiche notturne. In questi pazienti, oltre alla ritmicità del sonno/veglia degli episodi emolitici, si registrano crisi di emolisi acuta di varia durata (pochi giorni/qualche settimana) con anemizzazione intensa. Solitamente la causa di queste crisi rimane sconosciuta, anche se talvolta esse si accom-pagnano a infezioni virali, trasfusioni, mestruazioni, interventi chirurgici, vaccinazioni o assunzione di sali di ferro. L’emoglobinuria non è mai correlata all’espo-sizione al freddo. I sintomi di queste crisi sono dolori retrosternali, lombari (forse dovuti all’iperattività delle cellule dei tubuli prossimali nel riassorbire l’Hb, con iperemia renale), addominali (a tipo colica, della durata di 1-2 giorni). Compaiono inoltre astenia, pallore, febbre, cefalea e subit-tero. Talvolta nausea e vomito (si suppone per via rifl essa, dopo distensione della capsula renale). Il quadro clinico delle crisi è in parte sovrapponibile a quello di una rea-zione da incompatibilità trasfusionale. Le complicanze più frequenti sono aplasia midollare, trombosi venose e infezioni. Quanto all’anemia apla-stica, la sua associazione con la EPN è stata documen-tata almeno in tre circostanze: nell’anemia di Fanco-ni, nell’anemia aplastica farmacoindotta e nell’anemia anemia aplastica idiopatica. Di solito la diagnosi di EPN segue quella di pancitopenia e solo raramente si verifi ca l’opposto.
C0240.indd 1084C0240.indd 1084 6/9/10 3:09:03 PM6/9/10 3:09:03 PM

Capitolo 48 - ANEMIE
81085
Le trombosi venose rappresentano la principale causa di morte dei pazienti con EPN (50%). Vengono interessati i distretti degli arti inferiori, il sistema portale, il circolo cerebrale e quello mesenterico. Importante, dunque, è porre la massima attenzione ai dolori addominali talvolta segnalati dal paziente, i quali, in assenza di emolisi acuta, trovano una possibile eziologia nella trombosi. In particolare va presa in considerazione l’ipotesi di una trombosi delle vene epatiche (nausea, coliche addominali, epatomegalia improvvisa, ascite), un fatto questo che, destinato a essere rapidamente fatale, si verifi ca in circa il 30% dei pazienti con EPN. Anche la cefalea potrebbe essere dovuta a piccole trombosi o rappresentare il segno premonitore di una complicanza cerebrovascolare. Va segnalata, inoltre, la presenza di gravi disfagie e odi-nofagie in correlazione con le crisi emolitiche. Studi sulla peristalsi hanno infatti evidenziato contrazioni esofagee 10 volte superiori a quelle normali. La patogenesi di que-sto fenomeno è sconosciuta. Le infezioni sono assai fre-quenti; il 10% dei decessi può essere ascritto alle infezioni, perché, anche se di scarsa entità, favoriscono comunque l’insorgenza di un processo emolitico ovvero di una crisi di aplasia midollare.
Esami di laboratorio Il quadro ematologico è spesso grave, con Hb inferiore a 6 g/dL. I GR solitamente sono macrocitici, ma l’anemia può anche essere normocitica o microcitica per le note-voli quantità di ferro perse nel corso delle crisi di emoglo-binuria. I reticolociti sono percentualmente aumentati, anche se in valore assoluto si mantengono su valori bassi a causa della gravità dell’anemia e per il sovrapporsi di un’anemia aplastica. Nel 50% dei casi si ha leucopenia con possibile neutropenia e relativa linfocitosi. La vita media dei GR è ridotta, mentre è normale quella dei neu-trofi li, con riduzione della fosfatasi alcalina leucocitaria, chemiotassi, fagocitosi e attività acetilcolinesterasica. Anche la trombocitopenia è assai frequente, ma vita me-dia e funzioni piastriniche appaiono del tutto normali, il che sembra in contraddizione con le segnalazioni di alterazioni di membrana che ne modifi cherebbero la sen-sibilità al C. Sembra dunque esserci, per quanto riguarda piastrine e leucociti, un problema di produzione ridotta piuttosto che di aumentata distruzione. Durante la crisi il plasma si presenta color bruno-oro e rifl ette la presenza di elevati livelli di bilirubina non co-niugata, emoglobina e metemalbumina. La sideremia è bassa, il test di Coombs negativo, le LDH molto elevate. Nelle urine l’urobilinogeno aumenta e talvolta c’è emo-globinuria; le cellule dei tubuli prossimali metabolizzano l’Hb dopo averla riassorbita ed eliminano il ferro sotto forma di emosiderina . Questo spiega perché l’emoglo-binuria non sia un segno sempre costante al pari della emosiderinuria. Utile si rivela strisciare su di un vetrino parte del sedimen-to urinario, colorarlo con il blu di Prussia e confermare così la presenza o meno di granuli di emosiderina. Alta, infi ne, è l’incidenza di alterazioni renali, sia funzionali sia anatomiche (ematuria, ipostenuria, alterazioni tubulari, ridotta clearance della creatinina). Il midollo osseo mostra ampia variabilità di reperti, dall’iperplasia normoblastica con lieve riduzione dei
megacariociti all’aplasia parziale o totale. Studi cito-genetici non hanno evidenziato alcuna alterazione cromosomica diagnostica di EPN, anche se le segnala-zioni sono numerosissime (delezione della Y, trisomia 9 ecc.).
Diagnosi La diagnosi di EPN deve essere considerata in ogni pa-ziente che presenta i segni di emolisi intravascolare, in presenza di: emoglobinuria ed emosiderinuria; pancito-penia in associazione con emolisi, con midollo sia ipo- sia normocellulare; persistente e grave iposideremia soprat-tutto se in concomitanza di crisi emolitiche; ricorrenti trombosi venose, specialmente addominali; inspiegabili ma ricorrenti coliche addominali; cefalee accompagnate da un quadro di emolisi cronica. Deve comunque essere differenziata dalle anemie emolitiche immuni e dall’emo-globinuria parossistica a frigore .
Esami diagnostici La diagnosi di EPN si fonda sulla possibilità di stabilire la presenza di “emazie troppo sensibili al C”. Il classico test in vitro per la diagnosi di EPN consiste nell’incubare i GR del paziente in presenza di siero umano acidifi cato (test di Ham) . In ambiente acido, infatti, il C viene attivato per la via secondaria e provoca emolisi. È importante risospendere le emazie nel siero di un altro soggetto compatibile, perché quello del paziente può avere i fattori del C consumati, data la ripetuta attivazione sui GR sensibili. Tra gli altri test in vitro che permettono di svelare la ma-lattia, il più comunemente usato è la lisi al saccarosio . Quando il siero e i GR sono incubati in una soluzione di carboidrati a debole forza ionica (per esempio, il sac-carosio), si attiva il C sia per la via classica sia per quella alternativa. Dal momento che l’attivazione è pur sempre di modesta entità, soltanto le emazie EPN subiranno una certa emolisi, non quelle normali. C’è poi il test della trombina o test di Crosby . Una sospen-sione acidifi cata di emazie viene messa a contatto con trombina bovina. L’emolisi che si verifi ca è attribuibile alla presenza di anticorpi emolitici eterofi li contaminanti la preparazione di trombina bovina. Il test è di più facile realizzazione dei precedenti, ma meno specifi co. Da ul-timo, il test al calore: se il sangue di un paziente affetto da EPN è lasciato a 37 °C per 1-3 ore si verifi ca emolisi; si tratta di una prova aspecifi ca, dal momento che risulta positiva anche con gli sferociti e con le emazie di pazienti affetti da alcuni tipi di anemie emolitiche autoimmuni. D’altra parte, la sua negatività costituisce una prova a sfavore della diagnosi di EPN.
Decorso e prognosi Si tratta di una malattia cronica con sopravvivenza media-na intorno ai 10 anni. La gravità del quadro e la prognosi dipendono soprattutto da tre fattori: • ampiezza della popolazione patologica (EPN II/III); • grado di ipoplasia midollare; • frequenza delle complicanze trombotiche e infettive.
In una piccolissima percentuale di pazienti la malattia re-gredisce completamente, mentre in un 50% si registra un
C0240.indd 1085C0240.indd 1085 6/9/10 3:09:03 PM6/9/10 3:09:03 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1086
discreto miglioramento della popolazione complemento-sensibile, anche se persiste la positività ai test sierologici specifi ci. Nella maggioranza dei casi, però, l’evento termi-nale è un’aplasia midollare globale; occasionalmente la EPN esita in leucemia mieloide acuta o in mielofi brosi.
Terapia A eccezione del trapianto di midollo osseo, non vi è terapia specifica, ma solo sostitutiva, dell’anemia. Sono stati tentati, fino a oggi, pochi trapianti. In caso
di attecchimento, comunque, scompaiono i cloni sensibili all’azione del complemento. Gli androgeni si sono rivelati solo saltuariamente utili (si veda in precedenza, Anemia aplastica ), mentre i corticosteroidi a dosi elevate possono arrestare una crisi emoglobinurica. Le trasfusioni di GR lavati in soluzione fisiologica correggono momentaneamente l’anemia, perché le emazie trasfuse vivono regolarmente nel paziente e inoltre sopprimono transitoriamente la produzione di cloni EPN II/III.
Con il termine immunoemolisi si indica un processo per il quale le emazie sono prematuramente distrutte a causa dell’interazione con anticorpi diretti contro antigeni pro-pri della loro membrana o contro antigeni estranei all’or-ganismo (farmaci), in grado di stabilire con la membrana eritrocitaria vari tipi di associazione. La produzione degli anticorpi responsabili dell’immuno-emolisi può far parte della normale risposta immunitaria verso gli antigeni riconosciuti come non facenti parte del self. Dato che molti antigeni sulla membrana delle emazie presentano un polimorfi smo (ossia sono, per condizio-namento genetico, diversamente ripartiti tra gli indivi-dui), è possibile che questo avvenga quando le emazie veicolanti un particolare antigene vengono a contatto con il sistema immunitario di un soggetto che è sprov-visto di quell’antigene e che perciò lo riconosce come estraneo a se stesso. Le reazioni immunoemolitiche di questo tipo vengono dette da alloimmunizzazione o, più comunemente, da isoimmunizzazione e sono provocate da trasfusione di sangue incompatibile o da incompatibilità materno-fetale. Un processo immunoemolitico può anche essere provo-cato da anticorpi prodotti dallo stesso soggetto cui appar-tengono le emazie, per un errore del sistema immunitario nella discriminazione tra self e non self. Le reazioni di questo tipo vengono dette da autoimmunizzazione . I farmaci possono indurre reazioni emolitiche principal-mente in quanto riconosciuti come sostanze estranee all’organismo. Si vedrà, tuttavia, che non è impossibile che provochino genuine reazioni autoimmunitarie verso costituenti della membrana eritrocitaria. Tenendo conto di tutto questo, le anemie immunoemolitiche sono perciò distinte come segue ( Tab. 48.13 ): • anemie emolitiche da isoimmunizzazione; • anemie emolitiche autoimmuni (tra le quali possono
essere incluse anemie immunoemolitiche indotte da farmaci).
Le anemie immunoemolitiche sono normocitiche nor-mocromiche, presentano aumento del numero dei reti-colociti, sferocitosi, iperbilirubinemia indiretta, iperplasia eritroide del midollo osseo e splenomegalia. La sopravvi-venza eritrocitaria è diminuita con emazie sia autologhe
sia di donatore compatibile. È invece normale quando gli eritrociti di un paziente affetto da anemia emolitica autoimmune vengono trasfusi in un soggetto sano. Si tratta di malattie che nella forma pura (non coesistendo altre forme morbose) sono piuttosto rare. Più frequenti, viceversa, sono le forme associate ad altre malattie.
Considerazioni generali Le variabili importanti per la comprensione delle caratte-ristiche peculiari delle anemie immunoemolitiche sono: • natura degli antigeni implicati; • proprietà degli anticorpi; • meccanismo dell’emolisi.
Antigeni Come si è detto, gli antigeni che sono ber-saglio delle reazioni immunoemolitiche possono essere costituenti propri della membrana eritrocitaria o sostanze estranee all’organismo. La natura chimica degli antigeni propri della membrana eritrocitaria è nota molto im-perfettamente. Una distinzione utile è però tra antigeni polisaccaridici e antigeni proteici. Gli antigeni polisaccaridici sono più resistenti ai proces-si di estrazione e purifi cazione e possono essere meglio
Anemie immunoemolitiche G. Gromo
Anemie immunoemolitiche da isoanticorpi � Reazioni emolitiche post-trasfusionali � Malattia emolitica del neonato da isoimmunizzazione
materno-fetale
Anemie immunoemolitiche da autoanticorpi � Anemie da anticorpi incompleti caldi
– Idiopatiche – Sintomatiche di malattie linfoproliferative, malattie
autoimmuni e processi infettivi – Secondarie all’assunzione di farmaci
� Anemie da anticorpi completi freddi – Idiopatiche – Sintomatiche di malattie linfoproliferative e processi infettivi
� Anemie da emolisine bifasiche
Tabella 48.13 Classifi cazione delle anemie immunoemolitiche
C0240.indd 1086C0240.indd 1086 6/9/10 3:09:03 PM6/9/10 3:09:03 PM

Capitolo 48 - ANEMIE
81087
caratterizzati chimicamente. In realtà si tratta di sostanze largamente diffuse in natura e presenti anche in strutture come la parete cellulare dei batteri, le cellule vegetali ecc. Dato che questi antigeni sono resistenti ai processi di digestione, essi, quando sono liberati da microrganismi intestinali o sono ingeriti con alimenti vegetali, possono essere assorbiti come tali nell’intestino e stimolare il si-stema immunitario. Questo processo avviene naturalmente in maniera di-versa da un individuo all’altro, nel senso che ciascun individuo reagisce solo agli antigeni che non possiede alla superfi cie delle proprie cellule. A causa dell’ampia distribuzione in natura di queste sostanze e del loro polimorfi smo, sono moltissimi coloro che posseggono nel sangue anticorpi di questo tipo che, non essendo derivati da un’immunizzazione manifesta con cellule incompatibili, sono chiamati anticorpi naturali . Sono antigeni polisaccaridici quelli del sistema ABH-Lewis e del gruppo P (glicolipidi), e quelli indicati come I e i (glicoproteine). Gli antigeni proteici sono sostanze chimicamente molto meno note, a causa della facilità con la quale vengono alterate nel processo di purifi cazione. Non esistono anti-corpi naturali contro questi antigeni; d’altro canto, anche se fossero largamente diffusi in natura, questi antigeni non potrebbero indurne la formazione perché sono de-gradati dai processi digestivi. Sono antigeni proteici quelli dei sistemi Rh, Kell e Duffy.
Anticorpi Gli anticorpi implicati nelle reazioni immu-noemolitiche possono essere distinti in isoanticorpi se reagiscono con antigeni eritrocitari propri della specie, ma assenti nell’individuo che li ha prodotti, e autoanticorpi se reagiscono con antigeni eritrocitari posseduti dall’indi-viduo che li ha prodotti. Alcune caratteristiche di questi anticorpi meritano di essere considerate. • Classe immunoglobulinica : molto spesso una risposta
anticorpale è inizialmente e transitoriamente con anticorpi della classe IgM, e successivamente e persistentemente con anticorpi della classe IgG (o IgA). Il passaggio dall’una all’altra classe immunoglobulinica, ferma restando la specifi cità anticorpale, viene indicato con il termine inglese switch . Nel caso degli anticorpi impegnati nelle reazioni immunoemolitiche, lo switch avviene limitatamente quando sono diretti contro gli antigeni polisaccaridici. Perciò gli anticorpi di questo tipo, siano essi naturali o immuni, sono generalmente, ma non esclusivamente, della classe IgM. Dato che gli anticorpi della classe IgM sono incapaci di attraversare la placenta, ne consegue che le incompatibilità materno-fetali per antigeni polisaccaridici provocano raramente anemia emolitica del neonato. Lo switch avviene regolarmente quando gli anticorpi sono diretti contro antigeni proteici della membrana eritrocitaria e contro farmaci a essa associati. Gli anticorpi di questo tipo (che, come si è visto, sono sempre immuni) sono perciò in larga parte della classe immunoglobulinica IgG e sono capaci di attraversare la placenta e di provocare anemia emolitica del neonato nel caso di incompatibilità materno-fetale.
• Termicità : una caratteristica importante degli anticorpi impegnati nelle reazioni immunoemolitiche è la temperatura ottimale alla quale essi più facilmente reagiscono. Gli anticorpi diretti contro gli antigeni polisaccaridici (che, come si è visto, sono più spesso della classe IgM) reagiscono in genere meglio a temperature più basse di 37 °C e sono detti anticorpi freddi . Si pensa che questo fatto sia dipendente da alterazioni di conformazione, o degli antigeni o dei siti ricognitivi anticorpali, indotte dalla bassa temperatura. L’ambito di termicità è molto variabile da un anticorpo all’altro. Gli anticorpi diretti contro gli antigeni del sistema ABH, pur reagendo meglio a temperature più basse, lo fanno comunque in maniera suffi ciente anche a 37 °C. Gli altri anticorpi di questo tipo, tuttavia, reagiscono signifi cativamente solo al di sotto di 32 °C e questo limite viene assunto per defi nire “freddo” un anticorpo. Gli anticorpi diretti contro gli antigeni proteici e contro i farmaci reagiscono meglio a 37 °C e sono perciò detti anticorpi caldi .
• Capacità di agglutinare le emazie : fi siologicamente gli eritrociti sono in sospensione nel plasma e non agglutinano fi no a che non subentra “qualcosa” a far da ponte tra l’uno e l’altro. Gli anticorpi sono detti completi o incompleti a seconda che siano o meno capaci di agglutinare direttamente le emazie. La maggior parte dei GR, infatti, è dotata alla superfi cie di carica elettrica negativa, presenta cioè un potenziale detto z di circa 15 mV che determina un reciproco allontanamento. Le IgM, che sono pentameri, riescono a superare la repulsione tra GR e a provocare agglutinazione e sono perciò dette anticorpi completi. Le IgG, invece, pur essendo bivalenti, non riescono a vincere il potenziale z e ad avvicinare due GR e sono perciò dette anticorpi incompleti ( Fig. 48.15 ).
Figura 48.15 Rappresentazione schematica di anticorpi incompleti IgG (a) che non riescono a vincere la repulsione delle cariche elettriche negative sulle emazie. La repulsione può essere invece vinta dagli anticorpi IgM (b), detti perciò completi.
15 mV
b
a
C0240.indd 1087C0240.indd 1087 6/9/10 3:09:03 PM6/9/10 3:09:03 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1088
Tuttavia, si è potuto ugualmente dimostrare la presenza di anticorpi incompleti tipo IgG alla superfi cie dei GR o nel siero nonostante non siano in grado di agglutinare spontaneamente. Ciò è dovuto a un test di emoagglutinazione in vitro chiamato test di Coombs . Con il test di Coombs diretto si rileva la presenza di anticorpi (o di frazioni del complemento) adesi alle emazie, con il test di Coombs indiretto , la presenza di anticorpi liberi nel siero ( Fig. 48.16 ). Come schematizzato nella fi gura 48.16 a, nel test di Coombs diretto i GR del paziente sono già ricoperti di anticorpi incompleti caldi (IgG antieritrociti) ( 1 ); dopo essere stati opportunamente lavati, sono incubati con siero di coniglio anti-immunoglobuline (Ig) umane (siero di Coombs) o con sieri specifi ci anti-IgG, anti-IgA, anti-IgM, anti-C 3 o anti-C 4 ( 2 ). Se sulle membrane
dei GR sono adesi gli anticorpi o le frazioni complementari corrispondenti, si avrà reazione con gli anticorpi presenti nel siero di Coombs, e questi ultimi riusciranno a formare ponti tra le singole cellule e a indurre agglutinazione ( 3 ). Come schematizzato nella fi gura 48.16 b, nel test di Coombs indiretto, gli anticorpi sono liberi nel siero del paziente ( 1 ); il siero in esame viene incubato con emazie di altro soggetto dotate dell’antigene nei riguardi del quale sono diretti gli anticorpi di cui si va alla ricerca ( 2 ). Se gli anticorpi sono presenti nel siero in esame, essi si legheranno alla superfi cie delle emazie, per cui la successiva aggiunta del siero di Coombs provocherà agglutinazione con lo stesso meccanismo con cui questo avviene nel test diretto ( 3 ).
• Capacità di fi ssare il complemento : la reazione tra antigeni e anticorpi attiva la sequenza del complemento per la via classica, cioè a partire dai componenti della frazione C1. Perché la fi ssazione del complemento possa avvenire, occorre che due unità anticorpali giustapposte reagiscano con l’antigene. Per gli anticorpi della classe IgG, che sono monomeri, questo ha poca probabilità di accadere se la concentrazione anticorpale nel siero non è molto elevata. Infatti, gli anticorpi si distribuiscono casualmente alla superfi cie delle emazie e la probabilità che ve ne sia un numero signifi cativo di così strettamente adiacenti da realizzare la giustapposizione necessaria dipende dalla loro distanza media. La probabilità che questo accada è, ovviamente, molto più elevata se la concentrazione di anticorpi nel siero è alta ( Fig. 48.17 ). Diverso è il caso degli anticorpi della classe IgM, che sono dei pentameri e perciò possono
a b
Figura 48.16 Test di Coombs
diretto (a) e indiretto (b).
a
b c Figura 48.17 Capacità di
anticorpi IgG di fi ssare il
complemento sulle emazie.
La probabilità che questo avvenga è bassa se il titolo anticorpale è modesto (a), mentre con titolo più elevato (b) è più probabile che due molecole anticorpali siano giustapposte e venga attivata la sequenza complementare (c).
C0240.indd 1088C0240.indd 1088 6/9/10 3:09:03 PM6/9/10 3:09:03 PM

Capitolo 48 - ANEMIE
81089
facilmente fi ssare il complemento grazie alla reazione con l’antigene di due subunità della loro molecola. Tuttavia, la termicità degli anticorpi ha molta importanza ai fi ni dello sviluppo della cascata del complemento, una volta che si sono realizzate le condizioni per il suo inizio. Infatti, l’attivazione del complemento procede bene a temperature intorno ai 37 °C e si verifi ca in misura molto minore a temperature inferiori. Ne consegue che gli anticorpi della classe IgG, che sono caldi e quindi dotati della termicità adatta per l’attivazione del complemento, possono fi ssare il complemento raramente, e cioè solo quando sono molto concentrati nel siero. Al contrario, gli anticorpi della classe IgM, che facilmente fi ssano il complemento, sono di regola freddi e perciò agiscono a temperature poco adatte per l’attivazione complementare.
Meccanismo dell’emolisi L’immunoemolisi può verifi carsi tanto con un meccani-smo intravascolare quanto con un meccanismo extrava-scolare. L’ emolisi intravascolare è possibile solo quando la sequenza complementare alla superfi cie delle emazie si svolge fi no all’attivazione delle frazioni C8 e C9. Come si è visto, questo può avvenire solo se la fi ssazione del complemento è adeguata e se il processo della sua atti-vazione avviene a 37 °C. In pratica questi due requisiti, per le ragioni precedentemente esposte, sono soddisfatti solo per gli anticorpi della classe IgG a concentrazione molto elevata nel plasma e per gli anticorpi della classe IgM diretti contro gli antigeni del sistema ABH, che, pur essendo defi nibili in senso lato anticorpi freddi, si legano signifi cativamente alle emazie anche a 37 °C. Nella maggior parte dei casi si verifi ca perciò un’ emolisi extravascolare , che dipende da un’accelerata distruzione negli organi emocateretici delle emazie ricoperte di im-munoglobuline e/o frazioni del complemento (si veda Fig. 48.17 ). Gli anticorpi della classe IgG sono incomple-ti, ossia incapaci di agglutinare le emazie. Essi possono tuttavia essere adesi con i loro siti leganti l’antigene alle emazie normalmente circolanti, esponendo perciò alla superfi cie di queste cellule la regione della loro molecola che corrisponde al frammento Fc. Quando le emazie così ricoperte attraversano gli organi emocateretici tendono a essere trattenute e distrutte dai macrofagi, i quali sono dotati di una struttura di superfi cie complementare alla regione Fc delle IgG (cosiddetto recettore per Fc). Questo processo avviene prevalentemente nella milza, dove i GR sono normalmente concentrati e, se ricoperti con IgG, benefi ciano meno dell’azione competitiva che le IgG cir-colanti nel plasma possono esercitare per il recettore per il Fc dei macrofagi (azione che interferisce con l’emolisi e risulta protettiva). L’attività distruttiva dei macrofagi, o eritrofagocitosi, può essere completa o solo parziale; in questo caso viene rimossa una porzione più o meno estesa della membrana delle emazie e ne risultano cellule frammentate, sferociti, poichilociti che vengono seque-strati quando passano attraverso la milza o il fegato. Può anche capitare che i GR circolanti siano ricoperti da fra-zioni complementari. Questo può dipendere dall’effetto di anticorpi della classe IgG suffi cientemente concentrati da iniziare la fi ssazione del complemento, ma non tanto
da provocare l’emolisi intravascolare (e in questo caso le emazie saranno ricoperte tanto da IgG che da frazioni complementari). Può dipendere anche dall’azione di an-ticorpi freddi della classe IgM, che fi ssano il complemento nel microcircolo periferico di quelle regioni corporee (viso, mani) dove, per esposizioni ambientali, può essere raggiunta la temperatura adatta per la loro azione. In questo caso, mobilizzandosi verso la circolazione ge-nerale e riportandosi a 37 °C, le emazie si liberano dal legame con questi anticorpi, però rimangono ricoperte di frazioni del complemento che era stato fissato, ma non suffi cientemente attivato (e in questo caso le emazie saranno ricoperte solo da frazioni complementari, e non da anticorpi). La frazione complementare importante a questo riguar-do è il C3b (il C3 attivato si scinde in C3b, che resta attaccato alle emazie, e C3a, che passa in soluzione). I macrofagi posseggono, infatti, un recettore per il C3b e le emazie ricoperte da questa frazione complementare sono preferenzialmente trattenute e distrutte negli organi emocateretici. Questo processo si differenzia da quello analogo che ha luogo per le emazie ricoperte da IgG per due caratteristi-che. In primo luogo, non esistendo C3b circolante nel plasma, manca l’azione competitiva per il recettore dei macrofagi e la distruzione delle emazie ha luogo anche al di fuori della milza; in realtà, essa avviene largamente nel fegato a opera delle cellule di Kupffer. In secondo luogo, quando le cellule sono ricoperte solo da C3b, l’emolisi è molto meno efficiente di quando sono ricoperte da IgG. Ciò dipende anche dal fatto che, nella gran parte dei casi, prima che l’emazia venga fago-citata, il C3b si scinde in C3c e C3d grazie a un enzima fi siologicamente presente nel plasma (C3b inattivato-re). La frazione C3c rimane adesa al macrofago, quella C3d (riconosciuta dal siero anti-C3 nel test di Coombs diretto) al GR. Perciò, l’emolisi dipende sia dal numero di frazioni C3b adese, sia dal grado di effi cacia del C3b inattivatore. Per un’emolisi massiva la fi ssazione del C deve essere rapida e “superare la protezione” dell’inattivatore. Quando i GR sono ricoperti tanto da IgG che da C3b, l’emolisi è invece di intensità maggiore rispetto a quando sono ricoperti solo da IgG, dato che si addizionano i due meccanismi che i macrofagi mettono in opera per trattenerli e distruggerli negli organi emocateretici.
Anemie immunoemolitiche da isoanticorpi
Considerazioni generali sui gruppi sanguigni I gruppi sanguigni sono caratterizzati da antigeni presenti alla superfi cie dei GR. Ne esistono più di 300, raggruppati in sistemi, e vengono trasmessi come caratteri autoso-mici codominanti. Il solo che presenta una trasmissio-ne legata al sesso (cromosoma X) è quello denominato Xg a . È possibile suddividere i principali sistemi antigenici ( Tab. 48.14 ) in relazione alla loro associazione con isoan-ticorpi immuni o naturali. Gli isoanticorpi naturali sono immunoglobuline la cui ori-gine è ancora oggetto di discussione, ma che compaiono al di fuori di ogni stimolo antigenico dimostrabile. Alcuni anticorpi (Ab) sono presenti sistematicamente nel plasma
C0240.indd 1089C0240.indd 1089 6/9/10 3:09:04 PM6/9/10 3:09:04 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1090
di individui le cui emazie sono portatrici di determinati antigeni. È questo il caso degli anti-A, anti-B, e anti-AB, sempre dimostrati negli individui di gruppo B, A, 0, e detti Ab naturali regolari. Anche se generalmente appartengono alla classe IgM, possono essere anche IgG o IgA. In genere i soggetti di gruppo A e di gruppo B posseggono pressoché esclusi-vamente Ab naturali della classe IgM, mentre quelli di gruppo 0 hanno anche una signifi cativa quantità di Ab naturali della classe IgG. Altri Ab sono più rari, come gli anti-P, riscontrati nei soggetti P 1 anti-Le a,b,x nei soggetti Le, e sono defi niti Ab naturali irregolari. Gli isoanticorpi immuni compaiono in risposta a stimo-li antigenici noti. Moltissimi Ab di gruppi sanguigni sono di questo tipo. Gravidanze e trasfusioni sono re-sponsabili dell’isoimmunizzazione anti-Rh, Kell, Duffy, Kidd. Gli Ab formati in questo modo risultano di regola prevalentemente della classe IgG. Una prima immuniz-zazione induce la produzione transitoria di IgM, men-tre il persistere del medesimo stimolo antigenico o più stimolazioni ripetute determinano la sintesi di IgG. Nel discorso che segue si parlerà esclusivamente dei sistemi AB0 e Rhesus .
Sistema AB0 L’espressione fenotipica degli antigeni (Ag) A e B è sotto la dipendenza di due geni interdipendenti, il gene H e il gene A o B . Il gene H (il cui allele inattivo è il gene h ), presente nella gran parte della popolazione umana, produce un enzima che permette, grazie alla fi ssazione di un L-fucoso su di un mucopolisaccaride “di base”, la formazione dell’Ag o sostanza H. I soggetti che hanno sul secondo gene l’allele A producono a loro volta un enzima capace di fi ssare la N-acetilgalattosamina al complesso polisaccaride-L-fucoso (o sostanza H) originando la sostanza A. Tutti i soggetti di gruppo A hanno in comune una stessa sostanza di gruppo A, ma l’80% di essi ha inoltre una sostanza sup-plementare A 1 . Costoro vengono chiamati di gruppo A 1 , mentre il restante 20% che possiede A ma non A 1 viene chiamato A 2 . I soggetti A 2 o A 2 B presentano in circolo un Ab naturale anti-A 1 .
I soggetti che hanno sul secondo gene l’allele B presen-tano una galattosio-transferasi in grado di annettere al complesso polisaccaride-L-fucoso il galattosio, originando così la sostanza B. Coloro che non hanno né l’allele A né l’allele B sono incapa-ci di modifi care la sostanza H e sono perciò di gruppo 0. Ci sono poi alcuni soggetti che non possiedono il gene H (sono cioè omozigoti per h ). Costoro, anche se ereditano il gene A o B, non posseggono sui loro GR gli antigeni (Ag) A o B corrispondenti, poiché sono privi del precursore H. Peraltro, possono trasmettere l’antigenicità A o B alla pro-le, nel caso che questa acquisisca la capacità di produrre la sostanza H dall’altro genitore. Sono detti di fenotipo Bombay e presentano nel siero Ab anti-A, anti-A 1 , anti-B e anti-H. Per quanto riguarda gli anticorpi, gli anti-A e anti-B sono anche detti agglutinine, rispettivamente, � e � ; il titolo anticorpale può variare, per l’anti-A tra 1:64 e 1:512, per l’anti-B tra 1:16 e 1:64. Nei soggetti di gruppo 0, oltre agli Ab anti-A e anti-B, esistono anticorpi la cui molecola è in grado di reagire sia con l’antigene A sia con quello B (attività cross-reattiva anti-AB), Ab anti-A, A 1 , B e H.
Sistema Rhesus Il sistema Rhesus (Rh) riveste un’importanza capitale in biologia umana e comprende determinanti antigenici principali CDE-ce . Un sistema genetico complesso control-la la presenza di tali sostanze sui GR. I geni DCE sembrano essere situati in tale ordine sul medesimo cromosoma C, c (come pure E, e), sono alleli e si esprimono separatamente, cioè il soggetto eterozigote C/c avrà le due sostanze sui GR, mentre gli omozigoti ne avranno una sola. Quanto a d , invece, designa semplicemente l’assenza di D. Non esistono quindi Ab anti-d. D è l’antigene più immunogeno di questo e di tutti i siste-mi di gruppi caratterizzati dalla produzione di anticorpi immaturi (si veda Tab. 48.14 ). L’85% dei soggetti di razza bianca è Rh-positivo, cioè è omozigote o eterozigote per l’AgD, mentre il 15% è Rh-negativo, cioè non presenta l’antigene D. L’immunogenicità dell’antigene D è molto importante, perché, dopo una sola trasfusione di GR Rh-positivi (o D+) AB0 compatibile, il 30-50% dei riceventi Rh-negativi (o D–) sviluppa un anti-D nei 6 mesi successivi. Questa frequenza dipende dalla quantità di emazie trasfuse. Il rischio è minore per gli antigeni c, E (1,5%) e minimo per C, e (inferiore allo 0,05%). Le tecniche di ricerca degli anticorpi del sistema Rh de-vono tenere conto della classe di questi anticorpi: IgM, dopo la prima stimolazione, ma più spesso IgG e solo eccezionalmente IgA.
Altri sistemi di gruppi sanguigni Oltre al sistema AB0 e a Rh, esistono numerosi altri sistemi di gruppi sanguigni che costituiscono allotipi o marcatori genetici, vale a dire Ag di cui nella specie umana esistono molte varianti; è questo un esempio di polimorfismo genetico (si veda Tab. 48.14 ). Alcuni di questi sistemi come Kell , Duffy , Kidd assumono una grande importanza nella pratica trasfusionale; per esempio, il rischio di immunizzazione è particolarmente elevato per gli antigeni del sistema Kell (5%). Altri siste-mi, come il Lutheran , rappresentano eccellenti marcatori
Gruppi Antigeni principali Caratterizzati da anticorpi naturali AB0 A 1 A 2 BHLewis Le a LeP 1 P 1 P k Ii IiMNSs MNSs Caratterizzati da anticorpi immuni Rh CDEecKell KkJs a Kidd Jk a Jk b Duffy Fy a Fy b Lutheran Lu a Lu b MNSs MNSs
Tabella 48.14 Principali sistemi di gruppi sanguigni
C0240.indd 1090C0240.indd 1090 6/9/10 3:09:04 PM6/9/10 3:09:04 PM

Capitolo 48 - ANEMIE
81091
genetici negli studi familiari e delle popolazioni. Infi ne, come già detto, il sistema Xg è, fi no a oggi, l’unico tra i sistemi di gruppi sanguigni conosciuti che sia controllato dal cromosoma sessuale X.
Reazioni trasfusionali Nonostante i suoi benefi ci, la trasfusione di emocom-ponenti può dare luogo a complicanze importanti, che possono essere suddivise in due gruppi principali: • reazioni di tipo non immune; • reazioni di tipo immune.
Reazioni di tipo non immune Queste reazioni, che esulano dalla trattazione del presente capitolo, si possono manifestare durante la terapia infusionale come shock settico da contaminazione batterica massiva del sangue trasfuso (evenienza divenuta rara da quando si im-piegano materiali di plastica monouso), scompenso cardiaco congestizio, oppure a distanza come infezioni batteriche o virali (per esempio, epatite virale , AIDS , sifi lide , malaria , to-xoplasmosi , brucellosi , infezioni da cytomegalovirus ), danni da sovraccarico di ferro (emosiderosi) o di citrato (ipocalce-mia) quando vengono infuse grandi quantità di sangue.
Reazioni di tipo immune Comprendono invece la sindrome brivido-ipertermia (secondaria alla presenza di leucoagglutinine acquisite per immunizzazione precedente del paziente; analoga situa-zione si ha nel caso di isoimmunizzazione antipiastrine), la porpora trombocitopenica post-trasfusionale, le reazio-ni allergiche (orticaria, prurito, edema di Quincke, crisi di asma), ma soprattutto le reazioni emolitiche trasfusionali . Benché serie e spesso fatali, esse sono oggi abbastanza rare. Sono provocate dalla distruzione delle emazie del donatore o del ricevente in seguito all’infusione di sangue immunologicamente incompatibile.
Distruzione dei GR del donatore Nella maggior parte dei casi è provocata da isoanticorpi immuni e si verifi ca soprattutto in pazienti precedentemente immunizzati (politrasfusi, pluripare). Va sottolineata, a questo propo-sito, l’importanza degli Ab anti-Rh, Kell, Duffy e Kidd. Trattandosi di IgG e data la dispersione degli Ag Rh sulla membrana eritrocitaria, l’emolisi è extravascolare. La gravità della reazione dipende dalla quantità di emazie trasfuse, come pure dal titolo anticorpale (in questo caso Ab immuni). Questo principio è valido per ogni tipo di reazione trasfusionale emolitica. La sintomatologia è caratterizzata da nausea, brivido e febbre. Shock e insuffi cienza renale sono assai rari. Talvol-ta l’emolisi può essere provocata da isoanticorpi naturali (errore di tipizzazione del gruppo, scambio delle sacche di sangue) nell’ambito del sistema AB0. Trattandosi di IgM, sono capaci di fi ssare il C e provocare quindi emolisi intra-vascolare. Immediatamente dopo l’inizio della trasfusione il malato riferisce cefalea, dolori lombari, brividi, palpitazioni, tachipnea, nausea e vomito. L’ipotensione è la regola, il rial-zo termico invece è lievemente ritardato. Talvolta compare il quadro della coagulazione intravascolare disseminata. Sono presenti tutti i segni di emolisi intravascolare: emo-globinemia elevata, emoglobinuria senza ematuria, metal-buminemia, diminuzione del tasso di aptoglobina.
In un secondo tempo, entro 12 ore, compare ittero franco. Quando l’emolisi è massiva, il rischio più grave è quello di shock irreversibile con insuffi cienza renale acuta da necrosi tubulare.
Distruzione dei GR del ricevente Si tratta di un’evenien-za molto più rara della precedente, che tuttavia può avere conseguenze assai gravi quando, per errore, in caso di estrema necessità, sangue di gruppo 0 contenente Ab im-muni anti-A o anti-B ad alto titolo (superiore a 1:200/300) viene trasfuso a un paziente di gruppo A o B. Quando, invece, gli anticorpi anti-A e anti-B sono presenti a basso titolo nel sangue trasfuso, essi possono venire assorbiti dalle sostanze A e B solubili eventualmente presenti in circolo nel ricevente. È importante ricordare, infi ne, come talora la reazione emolitica sia ritardata (da 3 a 15 giorni dopo la trasfusio-ne) e si manifesti con riduzione dell’emoglobina e ittero. Si verifi ca in persone già immunizzate (gravidanze, trasfu-sioni) con isoanticorpi a titolo molto basso non accertabili con le comuni tecniche di gruppaggio.
Terapia Il sospetto di incidente trasfusionale impone la so-spensione immediata della trasfusione di sangue. Le reazioni allergiche in genere regrediscono dopo antistaminici e antipiretici, talvolta è necessario ricor-rere all’uso di corticosteroidi. Per i provvedimenti da adottare in caso di shock e insufficienza renale acuta si vedano i Capitoli 3 e 39. L’emorragia secondaria a coagulazione intravascolare disseminata può essere controllata con eparina e in-fusione di fattori plasmatici della coagulazione, oltre che con antitrombina III, piastrine e fibrinogeno.
Isoimmunizzazione materno-fetale Si defi nisce isoimmunizzazione materno-fetale l’insieme delle manifestazioni patologiche che hanno come causa l‘immunizzazione della madre determinata da un alloan-tigene cellulare presente nel feto e, come conseguenza, la malattia più o meno grave di quest’ultimo. È la causa più frequente di ittero neonatale non fi siolo-gico. Il passaggio attraverso la placenta di IgG materne provoca nel feto la distruzione delle sue emazie qualora presen-tino un antigene di gruppo ematico ereditato dal padre ed estraneo alla madre. La quasi totalità dei casi osservati riguarda incompatibilità nell’ambito dei sistemi Rhesus e AB0. Quest’ultimo tipo di isoimmunizzazione, due volte più frequente, presenta però un quadro clinico assai sfu-mato e non sempre rilevabile.
Malattia emolitica del neonato da incompatibilità Rh Eziologia e patogenesi Si osserva una malattia emolitica del neonato (MEN) ogni qualvolta la madre è Rh-negativa e il feto Rh-positivo. Più
C0240.indd 1091C0240.indd 1091 6/9/10 3:09:04 PM6/9/10 3:09:04 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1092
spesso è interessato l’antigene D (90% dei casi), ma vengo-no segnalati casi di incompatibilità per gli antigeni c ed E. Questa osservazione avviene soprattutto da quando viene eseguita di routine, al momento del parto, la profi lassi materna con Ig anti-D. Non trattandosi di anticorpi naturali, la presenza di im-munoglobuline anti-D (o Rh) nella madre è necessaria-mente secondaria a precedente immunizzazione. Il passaggio dei GR attraverso la placenta può verifi carsi a partire dalle 5 settimane di gravidanza, ma è soprattutto al momento del parto che queste “trasfusioni” sono di entità notevole. Sarebbero favorite da manovre ostetriche: versione ester-na, prelievi di liquido amniotico, scollamento manuale della placenta. Oltre alla via placentare, il passaggio di emazie dal feto alla madre sarebbe possibile attraverso i capillari mesenterici e attraverso quelli delle tube del Falloppio (in caso di parto cesareo). Infi ne, questo passaggio avviene in donne Rh-negative che sono state trasfuse inavvertitamente con sangue Rh-positivo. Il rischio di immunizzazione della madre è in funzione del numero dei GR che attraversano la barriera placentare. Il test di Kleihauer (eluizione di Hb adulta mediante acido citrico a pH 3,3) ha consentito di mettere in evidenza (in percentuale) la presenza di GR fetali sullo striscio di sangue materno. Un limite di questa prova è la presenza fi siologica di HbF (0,5-1,0%) nell’adulto. Approssimativamente 1 GR fetale per 100.000 GR materni corrisponde a 0,02 mL. Recenti studi eseguiti in donne primipare con fi glio Rh-incompatibile segnalano alcuni dati: • passaggio non rilevabile di GR del feto: rischio
di immunizzazione pari a 1,3%; • passaggio di circa 0,25 mL di GR del feto: rischio dell’8%; • passaggio di circa 0,25-3 mL di GR del feto: rischio
del 20%; • passaggio superiore a 3 mL di GR nel feto: rischio
del 50%.
Vi è tuttavia notevole variabilità; infatti circa il 25-30% delle donne Rh-negative, nonostante ripetute stimolazio-ni, non è in grado di sintetizzare anticorpi anti-Rh. L’incompatibilità AB0 possiede un certo effetto protettivo e previene l’isoimmunizzazione, se pur in modo non del tutto completo. Verosimilmente in questo caso le emazie fetali vengono rapidamente distrutte dagli “anticorpi na-turali” della partoriente, prima che si possa sviluppare una risposta anticorpale per il sistema Rh; la protezione sembra più completa in presenza di anticorpi anti-A (90%) che non anti-B (55%). Il rischio di MEN nel corso della prima gravidanza è mi-nimo, sia perché il passaggio dei GR del feto alla madre avviene prevalentemente al momento del parto, sia per-ché la risposta immune primaria provoca la sintesi di IgM che non attraversano la placenta. Nel caso di una seconda gravidanza incompatibile, le prime emazie fe-tali che entrano nel circolo materno determinano una nuova stimolazione, provocando una rapida produzione di anticorpi, questa volta di tipo IgG (tutte le sottoclassi sono interessate). Le IgG anti-D materne si fi ssano alla superfi cie delle emazie fetali e ne facilitano la distruzione (per lo più extravascolare).
Fisiopatologia L’anemia che consegue alla distruzione delle emazie fe-tali comporta iperplasia della serie eritroide midollare ed eritropoiesi extramidollare compensatoria. I reticolociti saranno aumentati di numero e si avrà pre-senza in circolo di eritroblasti. Qualora l’emolisi sia di entità tale da superare le capacità di compenso del feto, compare marcata ipossia tissutale che si traduce in gravi lesioni epatiche, cardiache e turbe della funzione di scam-bio a livello placentare. L’ittero alla nascita generalmente è assente perché la biliru-bina viene eliminata attraverso la placenta. Compare però nelle prime 24 ore di vita. La bilirubina indiretta aumenta poi rapidamente sia perché il processo emolitico continua, sia perché l’attività della glucuronil-transferasi epatica nel neonato è molto bassa. Quando il pigmento biliare rag-giunge valori superiori ai 20 mg/dL soverchia la capacità legante dell’albumina e si deposita nei tessuti ricchi di lipidi (per esempio, nell’adipe sottocutaneo, nella corti-cale del surrene), ma soprattutto nei nuclei grigi centrali del cervello, in quanto la barriera ematoencefalica non è ancora capace di impedire il passaggio del pigmento. Questo porta a gravi complicazioni neurologiche di tipo extrapiramidale (ittero nucleare), capaci di determinare la morte del neonato oppure complicanze psicomotorie drammatiche e irreversibili.
Manifestazioni cliniche ed esami di laboratorio Le manifestazioni cliniche più importanti della MEN sono rappresentate da anemia, ittero, epatosplenomegalia e, nei pazienti non trattati, ittero nucleare (o kernittero) . Le conseguenze della malattia vanno da un processo emo-litico non sempre evidenziabile, nonostante la positività per il test di Coombs (30-40% dei casi), a malattia emo-litica grave più o meno tardiva (40-50%), a idrope fetale subito dopo il parto (5%), a morte intrauterina del feto a 25-35 settimane (15%). • Idrope fetale : qualora l’emolisi fetale sia estremamente
grave, dapprima si registra un incremento compensatorio dell’eritropoiesi epatica, destinata a sovvertire, entro breve, la struttura parenchimale del fegato. Compaiono poi ipertensione della vena porta e di quella ombelicale, edema placentare, ipertrofi a trofoblastica. Come risultato fi nale si hanno gravi turbe della circolazione feto-placentare con riduzione dell’apporto nutritivo al feto. Si registrano edemi generalizzati, versamenti ascitici, pleurici e pericardici con abnorme distensione dell’addome e del torace (anasarca). L’epatosplenomegalia è imponente. I piccoli pazienti presentano inoltre estese soffusioni emorragiche cutanee secondarie a defi cit di sintesi dei fattori epatici della coagulazione. I feti, in genere, vengono partoriti prevalentemente a 7-8 mesi e le possibilità di sopravvivenza non superano il 10-20%.
• Anemia emolitica grave : qualora non la si affronti tempestivamente, si tratta di una situazione spesso mortale. La maggior parte dei pazienti affetti da MEN alla nascita non presenta particolare pallore e la concentrazione emoglobinica nel cordone ombelicale spesso è solo ai limiti inferiori della norma. Tuttavia, entro le prime 24 ore di vita i valori
C0240.indd 1092C0240.indd 1092 6/9/10 3:09:04 PM6/9/10 3:09:04 PM

Capitolo 48 - ANEMIE
81093
scendono a livelli estremamente bassi. Compare ittero che raggiunge il massimo di intensità nella quarta giornata. La bilirubina supera i 20 mg/dLl e, tra il secondo e il quinto giorno, possono comparire segni di ittero nucleare (ipotonia, modifi cazione della tonalità del pianto, sonnolenza, perdita del rifl esso della suzione e della prensione, irregolarità nella respirazione). Subentrano poi, qualora la sintomatologia si aggravi, opistotono e contrazioni. Nella maggior parte dei casi, però, i neonati non muoiono, ma sviluppano nel tempo paralisi cerebrale con disturbi motori, paresi con movimenti involontari distonici o atetosici, ipoacusia e disturbi dell’intelligenza. I parametri ematologici evidenziano la presenza di anemia, reticolocitosi marcata e GR nucleati in circolo. L’anemia è macrocitica con intensa poichilocitosi; la sferocitosi è assente, caratteristica della MEN da incompatibilità AB0. I reticolociti possono raggiungere valori del 60%. Uno degli aspetti più caratteristici dei neonati affetti da MEN è però sicuramente il riscontro in circolo di precursori dei GR nucleati (eritroblastosi fetale). A fronte dei normali 0,2-2 × 10 9 /L nei bambini nati a termine o prematuri, nei casi di MEN il numero degli elementi nucleati della serie rossa raggiunge i 10-100 × 10 9 /L. Sono aumentati di volume ma non si tratta di megaloblasti, come in passato alcuni autori hanno sostenuto. Compare anche leucocitosi (30 × 10 9 /L) e bene si correla alla gravità dell’anemia. Si deve tuttavia ricordare che alla nascita i valori normali sono compresi tra 15 e 20 × 10 9 /L. Aumentano soprattutto i granulociti neutrofi li. Il numero delle piastrine invece è normale. Occasionalmente il quadro di anemia emolitica compare dopo alcune settimane dal parto. Non è ancora del tutto chiarito il meccanismo patogenetico responsabile di questa anemia “tardiva”, ma sembra dovuto alla persistenza in circolo di anticorpi anti-D o a momentaneo “esaurimento” midollare. Quest’ultima ipotesi è in parte negata dalla reticolocitosi quasi sempre rilevabile. L’anemia è di grado modesto, l’ittero assente, come pure assenti sono gli eritroblasti in circolo. Il quadro solitamente è transitorio e regredisce spontaneamente a 8 settimane.
Diagnosi perinatale e postnatale La diagnosi in corso di gravidanza si basa sulla presenza di isoanticorpi anti-D nel siero della madre e sull’esame del liquido amniotico. La sensibilizzazione materna e la MEN sono rare nel corso della prima gravidanza, salvo che la donna non sia stata immunizzata in precedenza con trasfusioni Rh-positive. Perciò solo l’1,5-2% delle primipare presenta anticorpi anti-D al termine della gravidanza. L’immu-nizzazione avviene soprattutto al momento del parto e gli anticorpi IgM divengono titolabili solo circa dopo 8 settimane. La concentrazione anticorpale rimane comunque bassa fi no alla seconda gravidanza, allorquando un nuovo pas-saggio di sangue fetale (di solito nel secondo trimestre) provoca l’aumento del titolo anticorpale.
Questa volta gli anticorpi sono IgG. Si effettua una prima determinazione a 16 settimane e una seconda a 28-32 setti-mane. Qualora si registri un incremento di anticorpi anti-Rh si eseguono ulteriori controlli ogni 1-2 settimane. Nella gran parte dei casi l’entità è correlabile alla gravità del-la MEN. Ulteriori informazioni si possono ricavare a 28-32 settimane eseguendo un prelievo di liquido amniotico e va-lutandone il contenuto di bilirubina e l’eventuale presenza di anticorpi anti-Rh. In genere lo studio spettrofotometrico del liquido amniotico permette di valutare con precisione la concentrazione di pigmento presente (in gran parte bili-rubina) e quindi l’aumentata emolisi fetale. Da questo si può dedurre quale condotta adottare: asten-sione terapeutica, parto provocato o trasfusione intrau-terina. La diagnosi alla nascita è soprattutto immunologica e si basa sull’incompatibilità materno-fetale (AB0, Rh) e sul test di Coombs diretto, la cui positività dimostra l’avve-nuta sensibilizzazione delle emazie del neonato. Il test di Coombs va eseguito sul sangue del cordone ombelicale. Il test di Coombs indiretto effettuato sul sangue materno invece è positivo. La diagnosi è anche biologica, dal momento che si fonda sull’anemia ingravescente, sul dosaggio della bilirubina indiretta e sulla comparsa di ittero.
Terapia In corso di gravidanza , quando i dati di laboratorio indicano la presenza di una grave forma di MEN, è utile anticipare il travaglio anche a 34 settimane. Qualora invece si manifesti rischio di morte intrau-terina prima di 34 settimane, si deve provvedere alla plasmaferesi del sangue materno (per eliminare gli anticorpi circolanti) e alla trasfusione intrauterina del feto. Così facendo si ottengono risultati soddisfacenti nel 60-70% dei casi. Alla nascita , in tutti quei casi che presentano Hb in-feriore ai 12 g/dL e bilirubina maggiore di 5 mg/dL nel sangue del cordone ombelicale, si deve eseguire exsanguinotrasfusione. In questo modo si rimuovono dal circolo del neonato i GR ricoperti di anticorpi, si corregge l’anemia e si riduce l’iperbilirubinemia. La selezione del sangue da trasfondere ovviamente è in rapporto ai gruppi sanguigni della madre e del neonato. La somministrazione di albumina (capace di fissare una certa quantità di bilirubina) e la fototerapia (espo-sizione del bambino ai raggi ultravioletti) costituisco-no importanti supporti alla terapia trasfusionale.
Profi lassi La profilassi della MEN si realizza principalmente nel prevenire, al momento del parto, l’immunizzazione del-la madre nei confronti degli Ag fetali del sistema RH. Si inoculano 300 g di � -globuline anti-D entro le prime 72 ore dal parto alle madri Rh-negative, non immunizzate nei confronti dell’antigene D, con fi glio Rh-positivo (ivi compresi i fi gli con D debole).
C0240.indd 1093C0240.indd 1093 6/9/10 3:09:04 PM6/9/10 3:09:04 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1094
Si ritiene che le gammaglobuline anti-D rimuovano con rapidità i GR Rh-positivi dal circolo materno, ovvero bloc-chino direttamente i siti antigenici dei GR fetali. La prevenzione è oggi concepita come un fenomeno tempo-raneo e quindi va ripetuta a ogni gravidanza incompatibile. La profi lassi è indicata anche nei casi di aborto, di am-niocentesi o quando sussiste il fondato sospetto di una emorragia transplacentare.
Malattia emolitica del neonato (MEN) da incompatibilità AB0 Eziologia e patogenesi Si tratta di un evento non eccezionale (due volte più frequente della MEN da incompatibilità Rh) dovuto alla distruzione intravascolare delle emazie di un feto di gruppo per lo più A (talvolta B) fi glio di madre di gruppo 0. Infatti, solo le donne di gruppo 0 (curiosamente) pos-sono sintetizzare anticorpi anti-A di tipo IgG che pas-sano la placenta, entrano nel circolo fetale e lisano le emazie. Statisticamente circa il 20% di tutte le gravidanze presenta incompatibilità AB0 del tipo che potrebbe portare a MEN, ma l’incidenza di fatti emolitici significativi è di 1:150 nascite, e circa un quinto dei bambini a rischio presenta ittero.
Manifestazioni cliniche L’ittero di media gravità compare entro il primo giorno di vita e non causa quasi mai sintomatologia neurologica tipo kernittero. Anche l’anemia è modesta. Di particolare interesse è la sferocitosi assai spiccata. Compaiono inoltre iperplasia eritroide a livello midollare e reticolocitosi. Il test di Coombs diretto, espletato sul sangue ombelicale, spesso è negativo, perché gli anticorpi, dato il loro esiguo numero, occupano siti distanti sulla membrana delle emazie. Nella gran parte dei casi il decorso è benigno e la prognosi favorevole. Si differenzia dalla MEN da incompatibilità Rh per i se-guenti motivi: • pari frequenza di interessamento del primo nato
e dei seguenti; • né il rischio né la gravità della malattia aumentano
nelle maternità successive se il primogenito è affetto da MEN;
• media gravità della sintomatologia clinica.
Principi di terapia Raramente trova giustifi cazione l’exsanguinotrasfusione; di solito è suffi ciente la fototerapia.
Anemie emolitiche autoimmuni (o anemie immunoemolitiche da autoanticorpi)
Sulla base delle caratteristiche chimicofi siche degli au-toanticorpi è possibile stabilire una classifi cazione delle anemie emolitiche autoimmuni (AEA): • AEA da anticorpi incompleti caldi; • AEA da anticorpi completi freddi; • AEA da emolisine bifasiche.
AEA da anticorpi incompleti caldi Sono anemie emolitiche caratterizzate da un’emolisi se-condaria alla formazione di anticorpi diretti contro eritro-citi normali (autoanticorpi antieritrociti). Gli anticorpi caldi appartengono solitamente alla classe di IgG, più raramente a quella IgA. Di solito sono diretti specifi camente contro determinanti antigenici del sistema Rh (soprattutto il D). In genere si distinguono forme idiopatiche e seconda-rie. Queste ultime compaiono in associazione a malattie immunoproliferative, autoimmuni, infezioni batteriche, virali o vengono provocate da farmaci ( Tab. 48.15 ).
Epidemiologia Non sembra esserci una particolare predisposizione razzia-le, anche se la maggior parte degli studi riguarda la razza caucasica. L’incidenza annuale si aggira su 1 caso/80.000 individui. Vengono colpite tutte le età (picco di incidenza sotto i 40 anni), con una leggera prevalenza per il sesso femminile, soprattutto nelle forme cosiddette idiopatiche. Queste ultime costituiscono circa il 40% delle AEA, anche se l’incidenza reale (20-81%) risulta assai diffi cile da valutare con precisione a causa della diversa importanza attribuita a malattie concomitanti e soprattutto al periodo di osser-vazione. Le forme sintomatiche, che interessano di norma una popolazione di età superiore ai 45 anni, rappresentano circa il 60%, di cui un 20% è secondario all’assunzione di farmaci.
Patogenesi Il meccanismo fondamentale consiste nell’autoimmunizza-zione verso qualche antigene dei GR. In alcuni casi si tratta di autoantigeni, in altri di antigeni estranei che si trovano occasionalmente sui GR e che evocano una risposta anti-corpale. Le emazie sono per lo più coperte solo da IgG e non c’è complemento, perché la disposizione casuale degli anticorpi rende poco probabile che ve ne sia una signifi ca-tiva quantità di contigui l’uno all’altro (per attivare il com-plemento, infatti, sono necessari almeno due anticorpi, molto vicini tra loro, fi ssati al bersaglio). Viceversa, quando
Farmaci che agiscono provocando la formazione di autoanticorpi
� a-metildopa � Levodopa � Acido mefenamico
Farmaci che agiscono come apteni � Penicilline � Cefalosporine
Farmaci che agiscono mediante la formazione di immunocomplessi (tipo “astante innocente”)
� Chinidina � Fenacetina � Acido para-aminosalicilico � Sulfonamidi � Isoniazide � Altri
Tabella 48.15 Farmaci che provocano anemie immunoemolitiche
C0240.indd 1094C0240.indd 1094 6/9/10 3:09:04 PM6/9/10 3:09:04 PM

Capitolo 48 - ANEMIE
81095
la concentrazione anticorpale è molto elevata si può fi ssare il complemento con le IgG. Le cause e i meccanismi che determinano la formazione di autoanticorpi antieritrocitari sono in gran parte sconosciuti, anche se numerose ipotesi sono state formulate a questo proposito. Nelle anemie emolitiche da anticorpi caldi l’emolisi è intrasplenica, come è stato spiegato nella trattazione gene-rale di questo argomento. Raramente l’emolisi può essere di tipo intravascolare. Questo si verifi ca in presenza di un titolo particolarmente alto di anticorpi, quando è comple-tata l’intera sequenza di attivazione del complemento.
Manifestazioni cliniche Esami di laboratorio L’esordio e il decorso variano da caso a caso. Il quadro clinico non è particolare, ma ri-chiama quello già descritto per altre anemie. Più spesso la malattia si sviluppa in modo insidioso con un’anemia emolitica di modesta gravità che si accompagna a subit-tero e lieve splenomegalia. Si tratta di un’anemia normo-citica normocromica; la lieve macrocitosi saltuariamente presente è riferibile alla reticolocitosi (i reticolociti sono infatti del 10% più grandi delle normali emazie). Nei casi più gravi, a esordio fulminante e spesso letale (emolisi improvvisa, febbre, quadro di shock), si possono trovare nel sangue periferico segni di un’intensa rigenerazione midollare dei GR quali, a carico delle emazie, la presenza di punteggiature basofi le, una colorazione grigiastra anzi-ché rosa (al May-Grünwald Giemsa). Inoltre, sullo striscio periferico ci sono numerosi sferociti. Infatti, il GR con adese le IgG può non essere completamente distrutto dal macrofago, fagocitato in parte, alterato nella sua normale confi gurazione e tendere alla sferocitosi. La sopravviven-za eritrocitaria è sempre ridotta e le emazie da donatore sano hanno anch’esse emivita ridotta. Nella gran parte dei casi sono presenti iperbilirubinemia indiretta, elevata escrezione di bilinogeni urinari e fecali, e una diminu-zione dell’aptoglobina sierica. La sideremia può essere elevata. Una volta accertata la presenza di un’anemia di tipo emolitico, se ne dimostra con maggior precisione la natura grazie al test di Coombs diretto, capace di rilevare la presenza o meno di anticorpi e/o di frazioni comple-mentari sulle emazie del paziente in esame. Nei casi più lievi può anche mancare l’anemia e l’unico sintomo è la positività per il test di Coombs diretto per le IgG. Solita-mente, invece, è presente anemia di media gravità con test di Coombs diretto positivo per le IgG e negativo per la frazione C3 del complemento. Il test di Coombs indiretto, invece, è per lo più negativo. Quando invece l’anemia è grave, il test di Coombs diretto è positivo sia per le IgG sia per il C3; infatti, il numero di Ig fi ssate al GR è alto e le IgG, occupando siti vicini, permettono la fi ssazione del complemento. In alcuni casi si può positivizzare anche il test di Coombs indiretto a testimonianza della presenza tanto di anticorpi adesi alle emazie quanto di anticorpi circolanti. Da segnalare, infi ne, come un ristretto numero di pazienti affetti da AEA presenti test di Coombs diretto negativo. Ciò si verifi ca in circa il 3% dei casi e sembra dovuto al basso numero di anticorpi adesi all’eritrocita. Con gli antisieri in commercio, infatti, il test di Coombs risulta positivo quando il numero di anticorpi adesi a cia-scuna emazia è compreso tra 500 e 100. I “casi negativi” ne presentano solitamente meno di 10.
Il più delle volte, comunque, si usano sieri di Coombs di-retti contro globuline umane totali, scarsamente sensibili, che reagendo sia con le frazioni complementari (soprat-tutto C3b) sia con le IgG non permettono di differenziare ciò che è fi ssato in realtà alle emazie.
Decorso e prognosi L’andamento della malattia è cronico ed è contraddistinto da un alternarsi di remissioni e riacutizzazioni del processo emolitico. Nel 20-30% dei casi l’evoluzione è fatale (emo-lisi particolarmente acuta, embolie polmonari, processi trombotici); la sopravvivenza a 5 anni è del 70% circa. Gli episodi infettivi riguardano in particolare i pazienti splenectomizzati o sottoposti a terapia con citostatici.
Terapia I pazienti con test di Coombs positivo ma senza segni clinici di emolisi solitamente non richiedono alcuna terapia. Viceversa, nei casi caratterizzati da importan-te emolisi il trattamento di elezione è rappresentato dai corticosteroidi. 5 Nel 70% (alcune statistiche parlano del 90%) dei casi il quadro ematologico migliora nettamente, con il 20-30% di remissioni definitive. Il più delle volte, tuttavia, è necessario proseguire la terapia di man-tenimento a dosi scalari per lunghi periodi (mesi o anni), perché un’eventuale sospensione è seguita immediatamente da ricadute. Nei pazienti che non hanno tratto alcun beneficio dalla terapia steroidea (20%) e in quelli che hanno bisogno di elevate terapie di mantenimento, si ricorre anche all’impiego di farmaci immunosoppressori. La splenectomia è destinata a migliorare l’emolisi nel 50% dei casi, percentuale che aumenta qualora la sede di distruzione dei GR sia prevalentemente splenica (e non epatica, per esempio). Le trasfusioni devono essere praticate solo nei casi di estrema ne-cessità e comunque presentano un’utilità limitata, dal momento che i GR trasfusi vengono agglutinati e lisati. Recentemente si è dimostrato che la sommi-nistrazione endovenosa di � -globuline umane ad alte dosi può essere utile.
Forme sintomatiche di malattie linfoproliferative autoimmuni e di processi infettivi Si possono sviluppare in corso di linfomi maligni , leucemie (soprattutto linfatica cronica), mielomi, più raramente altre neoplasie (carcinomi renali, della prostata, del colon). Talvolta si associano ad artrite reumatoide , epatite cronica attiva, colite ulcerosa , ma soprattutto lupus eritematoso sistemico .
5 La AEA idiopatica trae benefi cio dai corticosteroidi non tanto per la loro generica attività immunosoppressiva, quanto perché interferisco-no con le capacità fagocitarie dei macrofagi, riducendo il numero dei loro recettori per Fc e C3.
C0240.indd 1095C0240.indd 1095 6/9/10 3:09:05 PM6/9/10 3:09:05 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1096
Da ultimo, sono presenti processi infettivi, batterici, virali e fungini. I quadri ematologico e sierologico sono del tutto sovrapponibili a quelli dell’AEA idiopatica, anche se talvolta la sintomatologia è largamente infl uenzata dal tipo di patologia associata. Da ricordare, infi ne, come l’anemia emolitica possa anticipare di mesi o anni la ma-lattia di base, costituendone l’unico segno rilevabile.
Forme secondarie all’assunzione di farmaci (si veda Tab. 48.15 ) Diversi sono i farmaci che possono scatenare un’emolisi di tipo autoimmune. � -metildopa È un agente antipertensivo di largo uso. Si ritiene che il farmaco stabilisca un legame con un auto-Ag, determinando per questa via la cooperazione dei linfociti T e quindi l’autoimmunizzazione. L’auto-Ag sarebbe un antigene di superfi cie delle emazie di solito appartenente al sistema Rh. Il farmaco e i suoi metaboliti non partecipano direttamente alla reazione emolitica. Una volta che si sono formati, gli anticorpi, infatti, si legano alle emazie anche in assenza del composto. Oltre agli anticorpi anti-GR è possi-bile dimostrare, nei pazienti che fanno uso di � -metildopa, la presenza di altri autoanticorpi quali ANA, fattore reuma-toide, anticorpi antimucosa gastrica. Si tratterebbe dunque dell’induzione di un fenomeno autoimmunitario da parte di un fattore estrinseco che non sembra prendere poi parte direttamente al processo immunitario generato. Non tutto è stato ancora chiarito circa il meccanismo re-sponsabile dell’induzione di questi autoanticorpi. Dati recenti dimostrerebbero che l’ � -metildopa possiede attività inibente sui linfociti soppressori sia in vivo sia in vitro , come pure sulla risposta dei linfociti ai mitogeni policlonali. Questi effetti, che perdurano a lungo anche dopo sospensio-ne del farmaco, sarebbero mediati da un persistente aumento dell’AMP ciclico intralinfocitario. I fattori genetici, poi, sem-brano rivestire un qualche ruolo nel determinare o meno una suscettibilità a questo o quel farmaco. Infatti, una larga parte di coloro che sviluppano una positività per il test di Coombs presenta l’antigene di istocompatibilità HLA B27 . L’uso della � -metildopa rende positivo il test di Coombs diretto con siero anti-Ig totali o con siero anti-IgG nel 10-30% dei casi, ma solo l’1% (o meno) accusa anemia emolitica. Questo dipende dagli autoanticorpi e dall’af-fi nità tra autoanticorpi e auto-Ag (fattori entrambi varia-bili). Di solito il test di Coombs diviene positivo dopo 3-6 mesi dall’inizio del trattamento. Questo intervallo di tempo rimane invariato anche quando il farmaco viene somministrato a un paziente che abbia già avuto in una precedente occasione una positività del test di Coombs indotta da � -metildopa. Sospendendo la terapia si assiste a una negativizzazione del test di Coombs in un tempo che varia da poche setti-mane a 2 anni. Altri farmaci si comportano con meccani-smo analogo alla � -metildopa e sono la L-dopa e l’acido mefenamico. La L-dopa non determina comparsa di anemia emolitica, anche se viene riscontrata positività per il test di Coombs nel 6-8% dei soggetti che ne fanno uso. Il quadro clinico dell’anemia emolitica autoimmune pro-vocata dalla � -metildopa (e farmaci a comportamento simile) presenta un esordio insidioso e un decorso lento mai ingravescente.
Non sono stati descritti quadri clinici particolari e la sinto-matologia è del tutto sovrapponibile a quella già descritta per la AEA idiopatica da anticorpi incompleti caldi. La reticolocitosi è modesta, come pure l’iperbilirubine-mia. Di solito la sospensione del farmaco è suffi ciente ad arrestare l’emolisi.
Penicillina È un preziosissimo antibiotico usato per un numero elevato di infezioni. Si lega alla membrana del GR e causa la produzione di anticorpi (IgG) diretti contro il com-plesso farmaco-emazia. La penicillina si comporta da aptene, per cui non sarebbe in grado di determinare la produzione di anticorpi se non si legasse alle proteine di membrana dei GR dando luogo a un Ag completo; ovvero gli anticorpi emolizzano solo gli eritrociti che hanno il farmaco legato alla superfi cie. Tra gli effetti collaterali della penicillina, l’AEA è di gran lunga meno frequente della comparsa di fenomeni allergici (talvolta fi no allo shock anafi lattico) o della malattia da siero, fenomeni dose-indipendenti. L’AEA può invece venire indotta soltanto in caso di trattamento prolungato e a dosi estremamente elevate (20.000.000 U o più nelle 24 ore), il che si verifi ca nella sola endocardite infettiva. Circa il 3% dei pazienti che ricevono dosi analoghe per via endovenosa presenta un test di Coombs diretto positivo per le IgG, ma solo una percentuale minima sviluppa AEA. Alla sospensione del trattamento il test di Coombs si negativizza prontamente. L’emolisi non è mediata dall’attivazione del complemento (test di Coombs diretto per il complemento negativo), ma è secondaria a sequestro splenico ed eritrofagocitosi. Il quadro clinico dell’AEA da penicillina è improvviso e si manifesta pressoché esclusivamente nei pazienti sottopo-sti a terapia con dosi molto alte di penicillina. L’emolisi è prevalentemente extravascolare. All’interruzione della terapia segue una rapida guarigione e il test di Coombs diretto diventa negativo nel giro di qualche giorno o set-timana.
Cefalosporine Anche gli antibiotici della classe delle cefalosporine provocano raramente AEA con meccanismo analogo a quello della penicillina. Tuttavia, le cefalospori-ne, quando sono impiegate ad alte dosi e/o sono sommi-nistrate negli uremici, possono rendere positivo il test di Coombs (in assenza di anemia emolitica) anche con un altro meccanismo. Questi farmaci, infatti, modifi cano (se somministrati ad alte dosi), con meccanismi non immu-nologici, le membrane eritrocitarie in modo tale che a esse si legano per assorbimento passivo varie proteine sieriche (albumina, fi brina, immunoglobuline, talvolta frazioni del complemento). Questa sensibilizzazione aspecifi ca non sembra però indurre emolisi.
Chinidina Si tratta di un composto abbastanza frequen-temente impiegato in cardiologia. Il farmaco, dopo essersi legato a una proteina plasmatica, induce la produzione di anticorpi del tipo IgM, IgG o entrambe. L’immunocomplesso, per ragioni non del tutto chiarite, tende a fi ssarsi alla membrana dei GR. Responsabile di questo legame sarebbe l’attivazione del complemento a opera degli immunocomplessi, dato che le emazie hanno affi nità per il C3b, allo stesso modo dei macrofagi. Il più delle volte l’immunocomplesso, dopo attivazione del
C0240.indd 1096C0240.indd 1096 6/9/10 3:09:05 PM6/9/10 3:09:05 PM

Capitolo 48 - ANEMIE
81097
complemento, si stacca dalla membrana dell’eritrocita ed è libero di agire con altre cellule. Le emazie dunque sono “astanti innocenti”, dal momento che vengono emoliz-zate senza neppure partecipare alla reazione immunitaria. Questo fatto rende ragione di due fenomeni: • positività del test di Coombs diretto per il
complemento e non per IgM e IgG; • assenza di correlazioni tra quantità di farmaco
ed entità dell’emolisi.
Reazioni analoghe sono state documentate per numerosi altri farmaci (si veda Tab. 48.15 ). Il quadro di anemia si manifesta all’improvviso in modo particolarmente grave e, come detto, non è correlato alla dose di farmaco assunta. L’emolisi è prevalentemente intravascolare con emoglobi-nemia, emoglobinuria e intensa reticolocitosi. Da segnalare la possibile comparsa di trombocitopenia e/o neutropenia, insufficienza renale (50% dei casi) e coagulazione intravascolare disseminata. Si deve sospendere immediatamente il farmaco responsa-bile ed eventualmente ricorrere a terapia trasfusionale. Gli steroidi in questo caso non presentano alcuna utilità, dal momento che l’emolisi è prevalentemente intravascolare.
AEA da anticorpi completi freddi (crioagglutinine) Sono anemie emolitiche caratterizzate da un’emolisi se-condaria alla formazione di anticorpi diretti contro eritro-citi normali (autoanticorpi antieritrocitari). Gli anticorpi freddi appartengono alla classe di IgM. Si legano ai GR tra 0 e 4 °C e tendono a staccarsi a temperature superiori ai 32 °C. Fissano il C, la cui sequenza di attivazione viene però raramente completata. La maggior parte presenta una specifi cità per i determinanti antigeni del sistema I/i e, più raramente, per Ag H, M, N. Ogni essere alla nascita presenta alla superfi cie eritrocitaria una sostanza polisaccaridica, le cui qualità antigeniche vengono identifi cate dalla lettera i. Con il passare del tem-po la massima parte degli individui cambia questo antige-ne in una sostanza diversa chiamata I, che può essere con-siderata, per la sua diffusione, un “antigene pubblico”. Come si è detto, nella maggior parte dei casi gli autoanti-corpi in questa forma morbosa reagiscono o con l’antige-ne I presente nelle emazie della maggior parte degli adulti o con l’antigene i del sangue del cordone ombelicale. Esistono pazienti che hanno anticorpi che reagiscono con le emazie di ambedue i tipi e che perdono la capacità di reagire, se le emazie sono trattate con enzimi proteolitici. Gli anticorpi di questo tipo sono chiamati anti-Pr. Di questa forma di AEA esistono forme idiopatiche e for-me sintomatiche e l’andamento clinico può essere acuto o cronico.
Forme idiopatiche L’autoanticorpo è monoclonale (a differenza della gran parte delle altre forme); presenta infatti, per cause im-precisate, un solo tipo di catena leggera, più spesso k . La malattia sembra dovuta al fatto che una cellula capace di produrre questo particolare autoanticorpo (linfocita B) sfugge ai meccanismi di regolazione e si mette a proliferare autonomamente. Si realizza così, in breve tempo, un clone
cellulare di dimensioni tali da produrre quantità elevate, in proporzione, di questo particolare autoanticorpo. Questa AEA da anticorpi freddi è di più facile riscontro nei soggetti anziani (maggiori di 50 anni) e presenta un andamento per lo più cronico.
Forme sintomatiche di malattie linfoproliferative Si hanno in corso di macroglobulinemia di Waldenström , linfomi , leucemia linfatica cronica e mieloma . In questi casi le agglutinine possono essere specifi che sia per l’an-tigene I sia per l’Ag i. Spesso sono monoclonali e possono formare crioprecipitati in vitro .
Forme sintomatiche di infezioni In queste circostanze si verifi ca la produzione di crioagglu-tinine di tipo policlonale. Nelle infezioni da Mycoplasma pneumoniae (PPLO), la produzione di crioagglutinine (spe-cifi camente dirette contro l’Ag eritrocitario I) è comune, ma raramente compare emolisi. Il Mycoplasma provoca più di una reazione autoimmune e si ritiene lo faccia attraverso reazioni crociate. Nella mononucleosi infettiva , invece, le crio-agglutinine sono dirette contro un Ag eritrocitario. Tuttavia, poiché questo Ag si trova di solito nei GR del feto, mentre ne sono sprovvisti gli eritrociti dell’adulto, l’emolisi non dovrebbe verifi carsi. In realtà, anche se rara, può capitare.
Patogenesi Le crioagglutinine e il complemento si fi ssano ai GR nelle aree periferiche (punta del naso, mani, piedi, lobi delle orec-chie) della circolazione superfi ciale, dove la temperatura è più bassa e facilmente si ha vasocostrizione e quindi stasi. Quando gli eritrociti fanno ritorno nei tessuti a tempe-ratura più elevata, l’anticorpo si stacca dai GR, mentre il complemento vi rimane legato. La capacità delle crioag-glutinine è massima a 4 °C e decresce progressivamente fi no ai 31 °C. Al contrario, il complemento comincia a fi ssarsi a 12 °C e raggiunge il massimo della sua attivazio-ne a 37 °C. Quindi, a 37 °C il test di Coombs diretto per le IgM sarà negativo, mentre sarà positivo quello per il complemento (si veda Fig. 48.17 ). Nella gran parte dei casi la sequenza di attivazione non è completa, ma si arresta allo stadio C3b ( Fig. 48.18 ).
C3aC3b
C3cC3d
Figura 48.18 Scissione della frazione C3 del complemento durante l’attivazione della sequenza complementare. Alla superfi cie delle emazie è fi ssata prima la frazione C3b, poi residua solo la frazione C3d.
C0240.indd 1097C0240.indd 1097 6/9/10 3:09:05 PM6/9/10 3:09:05 PM

Parte 8 - MALATTIE DEL SANGUE E DEGLI ORGANI EMOLINFOPOIETICI 1098
I GR ricoperti di C3b sono riconosciuti soprattutto dalle cellule del Kupffer e distrutti (in misura minore, però, rispetto a quando sono ricoperti da Ig). La presenza, poi, di C3d sulla superfi cie dei GR, che residua dopo la scissione del C3b ad opera del C3 inattivatore, li protegge da una nuova attivazione della sequenza del complemento indotta dagli autoanticorpi e li preserva dalla lisi.
Manifestazioni cliniche In corso di polmonite da M. pneumoniae , di mononucleosi infettiva, di infl uenza spesso vengono prodotte crioagglu-tinine; tuttavia, raramente la loro concentrazione è tale da provocare emolisi. Quando ciò avviene, il quadro sintoma-tologico è quello di anemia emolitica acuta: pallore, febbre, ittero, vomito, diarrea, dolori lombari e addominali, retico-locitosi, leucocitosi neutrofi la, lieve epatosplenomegalia. Nelle forme idiopatiche e sintomatiche di malattia lin-foproliferativa, l’anemia insorge gradualmente e la sin-tomatologia tende ad aggravarsi con la stagione fredda. Spesso l’unico sintomo è rappresentato dall’acrocianosi , secondaria all’agglutinazione di emazie nei capillari cutanei delle estremità. Il fenomeno raramente porta alla gangrena ischemica e regredisce di solito con il riscaldamento. Il test di Coombs diretto è positivo per il complemento e negativo per le IgM. È possibile comunque dimostrare direttamente la presenza di anticorpi completi freddi indu-cendo l’agglutinazione delle emazie in presenza del siero del paziente a freddo (a 4 °C). Si usano GR presi dal cordo-ne ombelicale per ricercare anticorpi anti-i ed eritrociti di adulto per gli anticorpi anti-I. L’agglutinazione è reversibile, cioè scompare, riportando le emazie incubate con il siero del paziente a 37 °C. Solitamente il titolo non è molto alto (1:32; 1:128), tranne che nelle forme idiopatiche (1:1000; 1:256.000 e oltre). L’agglutinazione spontanea delle emazie in vitro rende diffi cile o impossibile il conteggio eritrocitario e confe-risce un aspetto tipico agli strisci di sangue in cui tutti i GR sono impilati. Il numero dei reticolociti è nor-male, talvolta è presente iperbilirubinemia. Il midollo osseo presenta un aumento del numero assoluto degli eritroblasti e spesso dei linfociti. Da segnalare, a que-sto proposito, come un quadro di linfocitosi midollare associato a iper- � -globulinemia di tipo M monoclona-le avvicini la forma cronica idiopatica alla malattia di Waldenström.
Terapia Il paziente deve evitare esposizioni al freddo, così da precludere l’insorgenza di crisi emolitiche. Le forme sintomatiche di malattie infettive guari-scono spontaneamente e pertanto non richiedono alcun trattamento. Le forme idiopatiche e sintomatiche di processi linfoproliferativi talvolta rendono necessario un trattamento volto a limitare il processo emolitico. Splenectomia e corticosteroidi raramente si sono rivelati utili.
Infatti, i cortisonici intervengono nel rapporto regione Fc delle IgG/macrofagi, che, come si è detto, ha luogo essenzialmente nella milza, mentre nelle anemie di questo tipo gli anticorpi della classe IgG non sono implicati. Qualche risultato in più sembrano invece ottenere gli schemi terapeutici che ricorrono ai farma-ci immunosoppressivi. In alcuni casi è stata utilizzata, con successo, la plasmaferesi, che riduce il tasso di anticorpi circolanti.
AEA da emolisine bifasiche (emoglobinuria parossistica a frigore ) È una rara forma di AEA caratterizzata dalla presenza di anticorpi bifasici (emolisina di Donath-Landsteiner) che appartengono alle IgG. È da rilevare che si tratta di anticorpi freddi, che cioè fanno eccezione alla regola che vede prevalenti gli anticorpi della classe IgM tra quelli con queste caratteristiche termiche. Sono specifi ci per gli antigeni eritrocitari del sistema Pp (ma soprattutto P). La malattia può essere idiopatica, sintomatica di sifi lide (acquisita o congenita), di infezioni virali quali rosolia , parotite , varicella , infl uenza e morbillo . Il mec-canismo di formazione di questi particolari anticorpi è sconosciuto.
Patogenesi A 37 °C l’emolisina bifasica è priva di qualsiasi attivi-tà sulle emazie. Solo in presenza di basse temperature, nel microcircolo delle aree corporee periferiche in cui il raffreddamento può raggiungere il sangue circolante, ha luogo il legame dell’anticorpo con le emazie, e di solito il titolo anticorpale è suffi cientemente elevato da fi ssare il C. Trattandosi di un anticorpo incompleto, non si ha nes-suna agglutinazione delle emazie e nessuna stasi di queste cellule nelle aree fredde. Il rapido passaggio dei GR che hanno interagito con l’anticorpo nella circo-lazione generale, a 37 °C, consente il veloce comple-tamento della attivazione complementare e l’emolisi intravascolare.
Manifestazioni cliniche La malattia non presenta preferenza né di sesso né di età. La sintomatologia è caratterizzata da crisi emolitiche accessionali cui seguono periodi di remissione completa. In corrispondenza dell’esposizione di tutto il corpo o di parti di esso alle basse temperature o anche alcune ore dopo (fi no a un massimo di 8), si ha comparsa di brivi-di, febbre, dolori lombari, crampi addominali, vomito e diarrea. Dopo l’attacco compare emoglobinuria (urine color lava-tura di carne), che dura per circa 24 ore. Il grado di anemizzazione, reticolocitosi e iperbilirubine-mia indiretta dipende dall’entità della crisi, che solita-mente non si prolunga oltre le 2 ore. Ben presto compare leucopenia, cui segue granulocitosi.
Talora è presente splenomegalia e possono presentarsi disturbi tipo fenomeno di Raynaud.
C0240.indd 1098C0240.indd 1098 6/9/10 3:09:05 PM6/9/10 3:09:05 PM

Capitolo 48 - ANEMIE
81099
Il test di Coombs diretto per le IgG non è positivo, dato l’allontanamento delle Ig dagli eritrociti alla temperatura di 37 °C. Positivo invece è il test di Coombs diretto per il complemento. La diagnosi si basa sul riscontro diretto dell’anticorpo di D-L e su alcune prove quali il test omonimo, che consiste nella dimostrazione di emolisi in vitro riscaldando a 37 °C per 30 min un campione di sangue in precedenza tenuto a 0 °C per 10 min. La diagnosi differenziale è posta soprattutto con l’emoglo-binuria da marcia, l’emoglobinuria parossistica notturna, la mioglobinuria.
Terapia La prognosi è molto buona nel caso di una forma sintomatica di morbillo e di parotite; al cessare della malattia si avrà infatti completa reversibilità del qua-dro ematologico senza alcuna terapia specifica. La forma idiopatica (molto rara) tende ad avere anda-mento cronico, mentre quella sintomatica di sifilide si attenua curando l’infezione. La splenectomia e l’uso di corticosteroidi sono inutili; spesso l’unica terapia possibile è di evitare le esposi-zioni del paziente al freddo.
Un meccanismo emolitico contribuisce alla patogenesi di molte delle anemie di cui si è parlato in precedenza e per alcune è l’esclusivo fattore patogenetico. Nella maggior parte dei casi dipende da fattori intraglobulari, ossia da anomalie insite nelle emazie che ne accorciano la soprav-vivenza. Nel caso delle anemie immunoemolitiche le emazie sono normali, ma è qualcosa che agisce dall’ester-no che le conduce a una distruzione precoce. Questo è un esempio di anemia emolitica da fattori extraglobulari. Ne esistono vari altri elencati nella tabella 48.16 . Dell’anemia emolitica in corso di ipersplenismo si è par-lato nel Capitolo 32 . Tra le altre anemie emolitiche ci si sofferma in modo particolare sull’anemia emolitica micro-angiopatica. Questa è dovuta a rottura dei GR circolanti per l’impatto traumatico contro trombi che si vengono a formare nella microcircolazione. Esistono tre cause di anemia emolitica microangiopatica: • coagulazione intravascolare disseminata (si veda
il Capitolo 56 ); • anomalie della parete vasale nel corso di situazioni
come l’ipertensione maligna e la sclerodermia (microangiopatia trombotica secondaria), l’eclampsia , il rigetto di un trapianto renale e le vasculiti;
• microangiopatia trombotica primitiva (porpora trombotica trombocitopenica , sindrome emolitico-uremica), di cui si parlerà nel Capitolo 56 .
Dal punto di vista clinico, l’anemia emolitica microan-giopatica è normocromica normocitica, con reticolocitosi e aumento della LDH nel siero e diminuzione dell’ap-toglobina. La sua caratteristica distintiva è la presenza sullo striscio del sangue periferico di eritrociti deformati o frammentati (schistociti).
Altre anemie emolitiche
Brodsky R A . Advances in the diagnosis and therapy of PNH . Blood Rev 2008 ; 22 ( 2 ) : 65 – 74 .
Gallagher P G . Update in the clinical spectrum and genetics of red cell membrane disorders . Curr Hematol Rep 2004 ; 3 : 85 – 91 .
Hoffman P C . Immune haemolytic anemia-selected topics . Am Soc Hematol Educ Program 2006 : 13 – 8 .
Iolascon A , De Falco L , Beaumont C . Molecular basis of inherited microcytic anemia due to defects in iron acquisition or heme
synthesis . Haematologica 2009 Mar ; 94 ( 3 ) : 395 – 408 . Epub 2009 Jan 30. Review.
Nemeth E . Iron regulation and erythropoiesis . Curr Opin Hematol 2008 ; 15 ( 3 ) : 169 – 75 .
Weatherall DJ, Clegg JB The thalassaemia syndromes. 3rd ed. Oxford: Blackwell Scientifi c; 1981.
Bibliografi a
Anemie immunoemolitiche
Anemia in corso di splenomegalia (ipersplenismo)
Anemia da effetti tossici diretti sulle emazie � Da veleni chimici � Da veleni di serpenti � Da agenti biologici (infezione da clostridi, malaria)
Anemie da fattori fi sici � Ustioni � Radiazioni � Trauma meccanico
— Emoglobinuria da marcia — Turbolenza nelle cavità cardiache (protesi valvolari, stenosi
aortica calcifi ca) — Anemia emolitica microangiopatica
Tabella 48.16 Anemie emolitiche da fattori extraglobulari
C0240.indd 1099C0240.indd 1099 6/9/10 3:09:05 PM6/9/10 3:09:05 PM

C0240.indd 1100C0240.indd 1100 6/9/10 3:09:05 PM6/9/10 3:09:05 PM