chimerascan mi-kyoung seo 2014.11.25. outline fusion gene in cancer workflow for fusion gene...
TRANSCRIPT

ChimeraScan
Mi-kyoung Seo2014.11.25

Outline
• Fusion gene in cancer• Workflow for Fusion gene discovery using RNA-
seq• Fusion gene discovery (fusion finder tools)– Alignment strategy– Filtering strategy
• ChimeraScan Method• ChimeraScan Running & Result• Discussion

Fusion genes in Cancer
• Fusion genes are hybrid genes that combine parts of two or more original genes
• Somatic fusion genes are regarded as one of the major drivers behind cancer initiation and progression
• The biological significance of fusion genes to-gether with their specificity to cancer cells has made them into excellent targets for molecu-lar therapy
• Fusion genes are used as diagnostic, prognos-tic and subtype marker
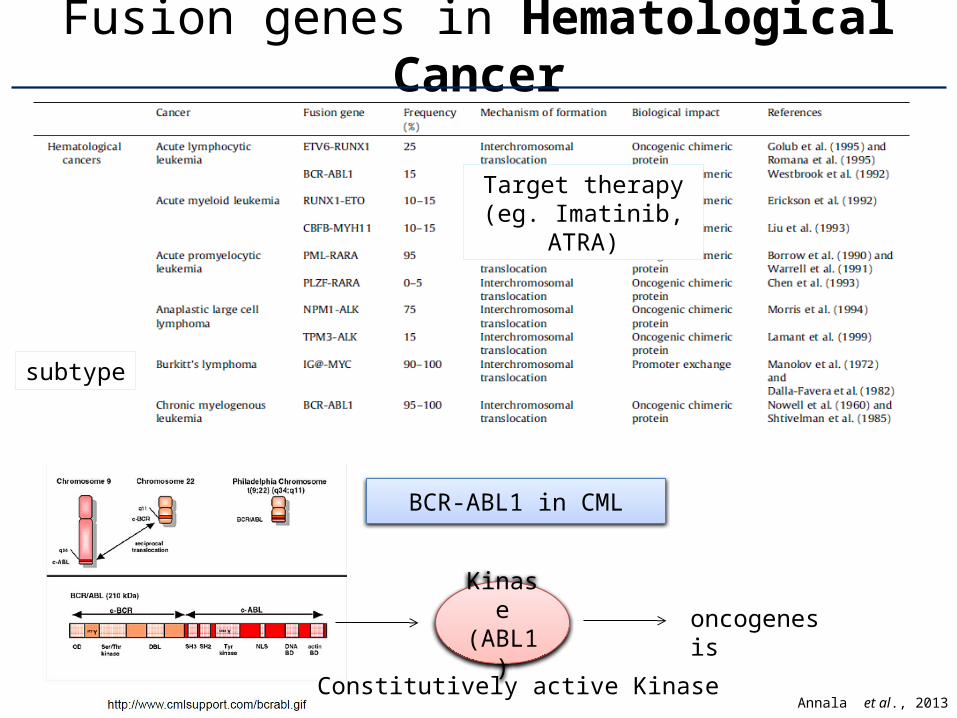
Fusion genes in Hematological Cancer
BCR-ABL1 in CML
Ki-nase(ABL1
)
subtype
Target therapy(eg. Imatinib,
ATRA)
Annala et al., 2013Constitutively active Kinase
oncogene-sis

Fusion genes in Solid Cancer
Parker BC and Zhang W, 2013
Annala et al., 2013

Hiroyuki Mano, 2013
Biological impact of fusion genes
Chimera transcript
Overexpression of Oncogene Oncogenic chimeric protein
Promoter exchangeRegulatory element
Escape from microRNA regu-lation
Parker et al. 2013Arul Chinnaiyan, 2014
Constitutively active Kinase
5’ 3’5’ 3’
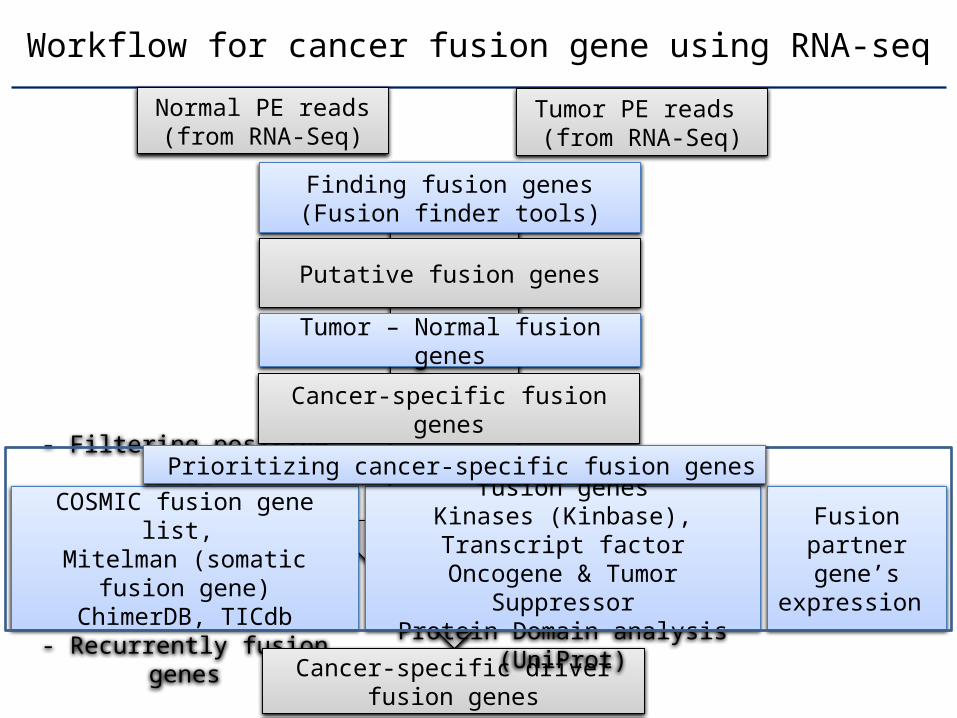
Workflow for cancer fusion gene using RNA-seq
Normal PE reads (from RNA-Seq)
Finding fusion genes(Fusion finder tools)
Putative fusion genes
- Filtering positive listCOSMIC fusion gene list, Mitelman (somatic fusion
gene)ChimerDB, TICdb
- Recurrently fusion genes
Tumor PE reads (from RNA-Seq)
Cancer-specific fusion genes
Cancer-specific driver fusion genes
Fusion part-ner gene’s expression
Tumor – Normal fusion genes
- Functional profiling of fusion genes
Kinases (Kinbase), Transcript factor
Oncogene & Tumor SuppressorProtein Domain analysis (U-
niProt)
Prioritizing cancer-specific fusion genes

Fusion finder tools
Alignment strategy- Whole paired-end based tool- Paired-end & fragmentation based tool- Direct fragmentation based tool
Beccuti et al., 2013
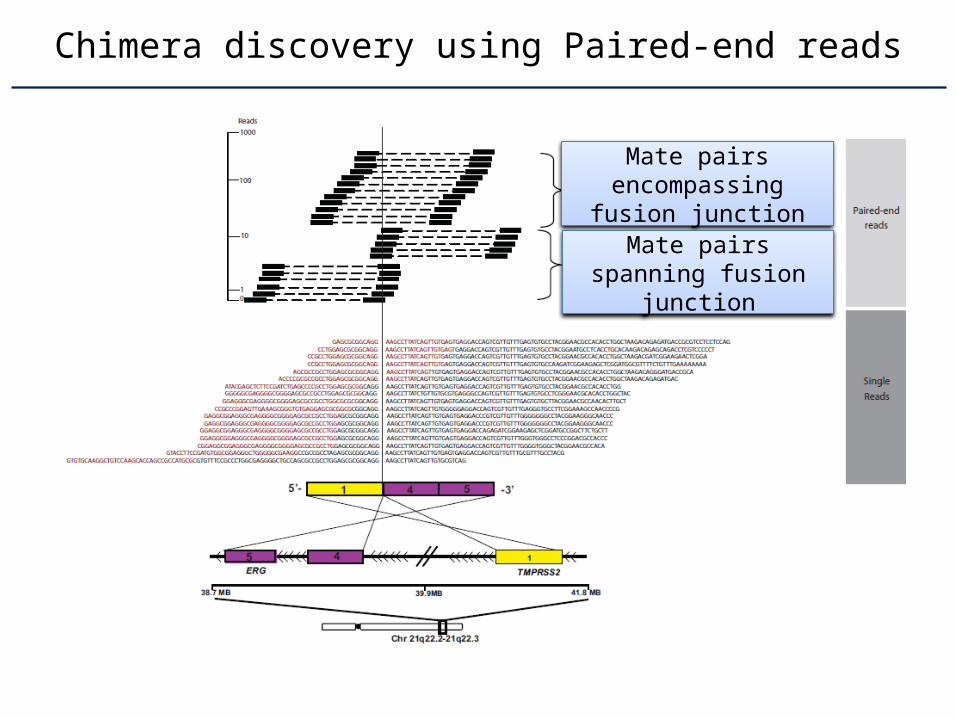
Chimera discovery using Paired-end reads
Mate pairs encom-passing fusion junc-
tion
Mate pairs spanning fusion junction
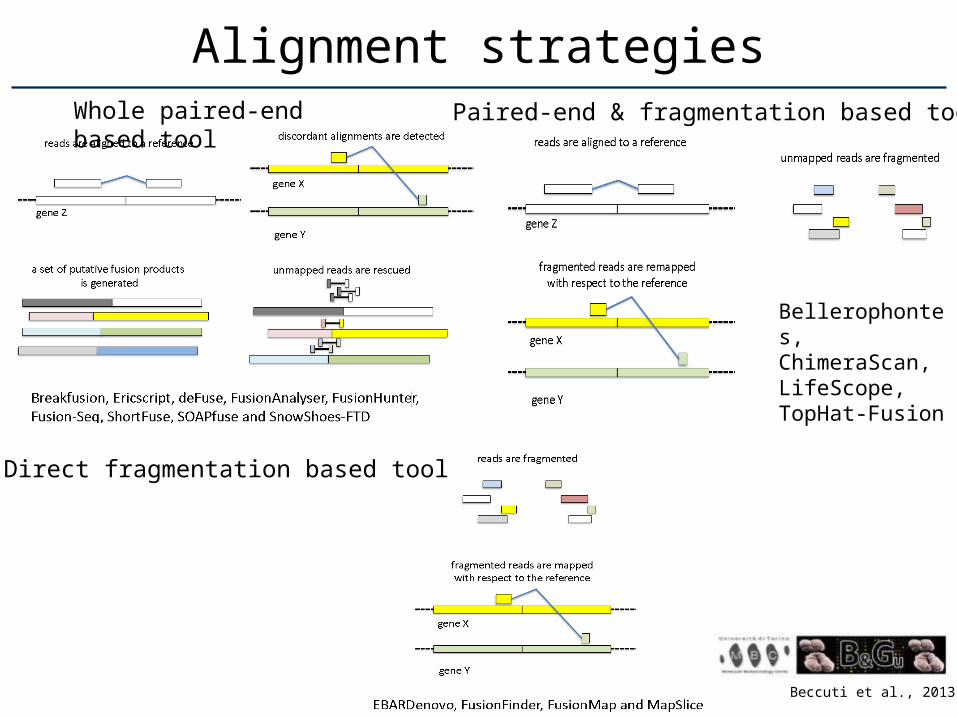
Alignment strategies
Beccuti et al., 2013
Whole paired-end based tool
Paired-end & fragmentation based tool
Bellerophontes, ChimeraScan, LifeScope, TopHat-Fusion
Direct fragmentation based tool

Filtering strategies
Homology filtersPaired-End information filters
Anchor Length filters
Encompassing/junction Spanning Reads filters
PCR Artifact filters
Beccuti et al., 2013
Quality based filters
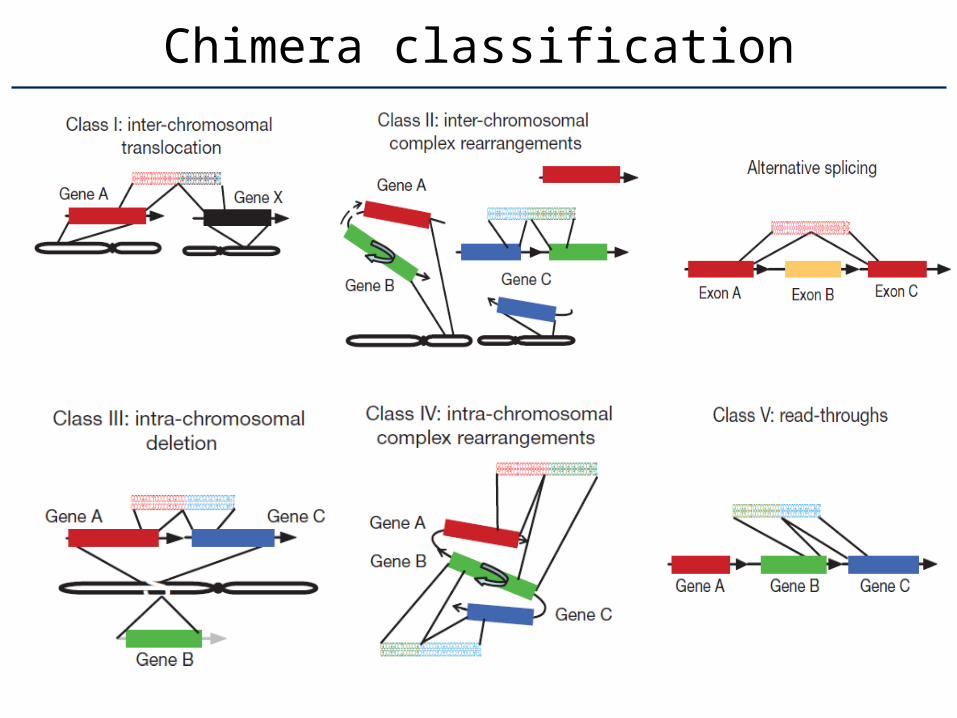
Chimera classification

Software information
• Purpose– To search high-throughput RNA sequencing (RNA-
seq) data for transcription of chimeric genes.
• Software URL– https://code.google.com/p/chimerascan/
• Category– Fusion Gene Detection
• License– The GNU General Public License is a free (GNU GPL
v3)

ChimeraScan

Workflow
Initial paired-end alignment
Second alignment

ChimeraScan Pipeline Workflow
• Step 0: Create alignment index– chimerascan_index.py
• Step 1: Prepare reads for alignment– ChimeraScan parses FASTQ• 1) converts all quality scores to Sanger format (Phred + 33)• 2) converts the qname for the reads from an ar-
bitrarily long string to a number (1/1, 1/2 for PE reads)

ChimeraScan
• Step 2: Align paired-end reads – Paired-end reads are aligned to a combined
genome and transcriptome reference. (--mul-timaps, default: 40)
Concordant reads align as a pair to the genome or a sin-gle gene
* Initial paired-end alignment (using Bowtie)- both reads in a pair must align within a distance range (default: 0-1000bp)- specified by the user with the --min-fragment-length and --max-fragment-length flags.1. Read trimming (--trim5 and --trim3) trims a specified number of bases from the 5' or 3‘ end of all reads.2. Number of mismatches (--mismatches) tolerated in alignments3. Additional arguments (--bowtie-args) can be directly passed to Bowtie. NOTE It is not recommended to use this unless the user is absolutely sure about it!
ChimeraScan
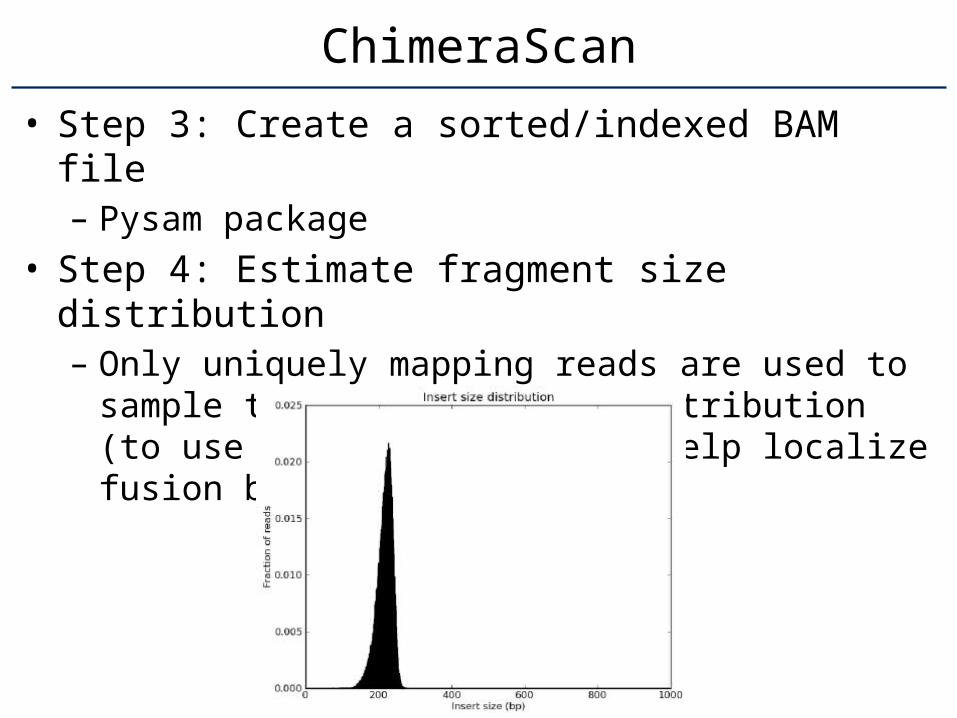
• Step 3: Create a sorted/indexed BAM file– Pysam package
• Step 4: Estimate fragment size distribution– Only uniquely mapping reads are used to sam-
ple the fragment size distribution (to use in fu-ture steps to help localize fusion breakpoints).

ChimeraScan
• Step 5: Realign initially unmapped reads
• Step 6: Discover discordant reads
All of the initially unmapped reads are treated as single reads and re-aligned.Additionally, the reads are trimmed such that only the sequences at the ends of the fragment are aligned (default=25bp).* command-line options of BowtieMismatches (default=3)
* Second alignment
ChimeraScan
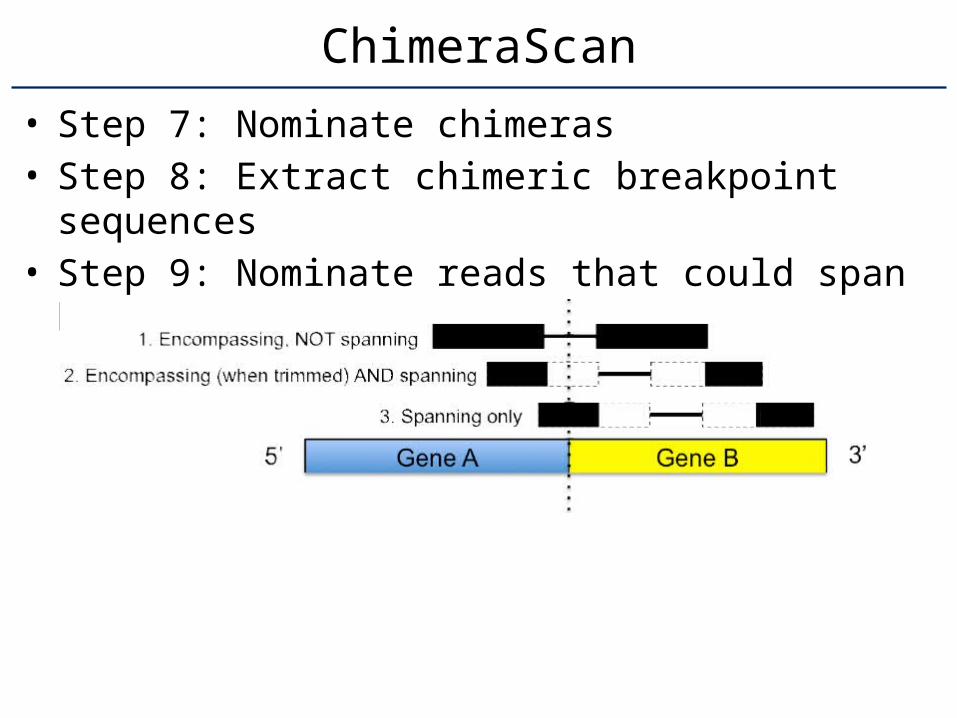
• Step 7: Nominate chimeras• Step 8: Extract chimeric breakpoint sequences• Step 9: Nominate reads that could span
breakpoints

ChimeraScan
• Step 10: Align against breakpoint sequence database (Step 8)
• Step 11: Assess breakpoint spanning align-ment results
Reads that align to the breakpoint sequence index are discarded if the overlap is small (less than anchor_min bases) or have larger overlap but contain mismatches (red reads). Reads overlapping the breakpoint by more than anchor_length bases are retained (green read).

ChimeraScan
• Step 12: Filter chimeras
• Step 13: Produce a text output file (BEDPE file)• Step 14: Produce an HTML web site (optional) – chimerascan_html_table.py
The chimeras are passed through a number of filters in order to remove erroneous artifacts. These include:1. Chimeras with very low coverage (specified with --filter-unique-frags, default 2) may have arisen from ligation artifacts during library prepara-tion2. Chimeric transcript expression may be much lower than the expres-sion of one or both of the wild-type transcripts in the sample (specified with --filter-isoform-fraction, default 0.10).3. Reads that support chimeras may not agree with the fragment size distribution of the library (specified with --filter-isize-percentile, default 99%).4. Users may have a list of known false positives available (specified with --filter-false-pos as a path to a file containing false positives)

ChimeraScan’s ability to identify high-quality chimeras
VCaP (prostate can-cer) 2x53bp
LNCaP (prostate can-cer) 2x34bp
MCF7 (breast can-cer) 2x35bp
Experimentally vali-dated candidates 9/10 4/4 12/13
Novel candidates
78 105 152
NDUFAF2-MAST4(2 encompassing
& 1 spanning reads)
TBL1XR1-RGS17(Ruan et al., 2007)
Tools deFuse (McPher-son et al., 2011)
ChimeraScan (Iyer et al.,
2011)
MapSplice (Wang et al.,
2010)
shortFuse (Kin-sella et al., 2011)
# call 가장 적음 78 400 245
True positive 60% 90% 60% 70%
Comparison of fusion finder tools

Running & Result
0. Running the chimerascan indexerpython chimerascan_index.py <reference_genome.fa> <gene_models.txt> <index_output_dir>
1. Finding fusion genespython chimerascan_run.py -v --quals illumina < index_output_dir > Read_1.fq Read_2.fq Output_dir
2. Creating an HTML page for output visualization (optional)python chimerascan_run.py –o chimeras.html /Output_dir/chimeras.bedpe
Result: chimeras.html (chimeras.bedpe)

Optimizing Removal of False Positive Fusions
Filtering- lack of spanning read13346681(1917)-Presence of intronic sequences in the fusion681249 (1717)
FF: FusionFinder; THF: TopHat-fusion; MS:MapSplice; FM: FusionMap; FH:FusionHunter;DF: defuse; BF: Bellerophontes; CS: ChimeraS-can.

Discussion
• Considering of biological function of fusion gene in cancer
• Filtering & annotation strategies for custom purpose
• Minimizing false positive