chapter ii anion templated synthesis o anion templated...
TRANSCRIPT

Chapter II
Anion Templated Synthesis Anion Templated Synthesis Anion Templated Synthesis Anion Templated Synthesis oooof f f f Calix[4]Pyrroles Calix[4]Pyrroles Calix[4]Pyrroles Calix[4]Pyrroles aaaand nd nd nd NNNN----Confused Confused Confused Confused
Calix[4]PyrrolesCalix[4]PyrrolesCalix[4]PyrrolesCalix[4]Pyrroles

56
CHAPTER II
ANION TEMPLATED SYNTHESIS OF CALIX[4]PYRROLES
AND N-CONFUSED CALIX[4]PYRROLES
2.1. Introduction
Anions play essential roles in biological systems and are important environmental
pollution such as nuclear waste produced during the reprocessing of nuclear fuel.
Anions participate 70% of all enzymatic binding events, serving as either enzyme
substrates or cofactors. Further, excess phosphate and nitrate as the result of agricultural
run-off and urban use are responsible for eutrophication in water-ways.
Simmons and Park reported the first synthetic polyammonium-based anion receptors in
1968.1 Since then especially over the course of the last two decades numerous anion
receptors have been synthesized and studied. Over the past several decades a large
number of acyclic and macrocyclic compounds have been synthesized as cation
receptors and evaluated for their abilities to recognize cations.2 In recent years as the
importance of anions in biological and environmental systems has become increasingly
recognized and attention has been directed towards the design and construction of anion
receptors. As a consequence both cation and anion recognition are now well-established
branches of supramolecular chemistry.3,4 To effect anion recognition most ion pair
receptors take advantage of hydrogen bonding donors (urea, amide, imidazolium,
pyrrole, and hydroxyl group), Lewis acidic sites (boron, aluminum and uranyl) and
positively charged polyammonium groups.
Figure 2.1: Ion-pair interaction to receptor mediated ion-recognition

57
2.2. Calix[4]pyrrole as a anion receptor based on pyrroles
The tetrapyrrolic macrocycle calix[4]pyrrole was found in 1996 by Sessler et al. to be
able to bind certain anions in organic solvents.5 In 2005 Moyer, Sessler and Gale et al
reported that calix[4]pyrroles can act as an ion pair receptor for various cesium salts and
certain organic halide salts in the solid state.6 X-ray crystal structures of several cesium
and organic cation-containing anion complexes of calix[4]pyrroles were solved, Taken
in concert they revealed that the anions are bound to the pyrrolic NH protons via
hydrogen bonding. These interactions which were expected on the basis of prior studies
serve to fix the calix[4]pyrrole in the cone conformation. This conformational locking
in turn, provides an electron-rich bowl-shaped cavity into which, e.g., cesium cation is
bound via a combination of π-metal and dipole interactions. Further evidence that
calix[4]pyrrole is effective for ion pair recognition came from liquid-liquid extraction
studies carried out by Wintergerst et al.7 This study demonstrated that compound can
extract CsCl and CsBr, but not CsNO3, from an aqueous phase into nitrobenzene, a
relatively polar organic phase. The solvent extraction process was modeled in terms of
three thermochemical steps. The first of these steps involves a partitioning of the
cesium cations and the halide anions into the nitrobenzene phase from the water phase.
The second step involves a conformational change of the calix[4]pyrrole such that it
adopts the cone conformation, a geometry it maintain as the results of halide anions
binding. The third step involves the cesium cation binding within the bowl-shaped calix
cavity created as the result of the conformational change takes place in 2nd step. While
thermodynamically equivalent in terms of the final state it was appreciated that these
steps could be taking place concurrently. The key point is that under conditions of this
extraction calix[4]pyrrole binds both the cesium cation and a halide anions (Cl¯or Br¯).
2.3. History and structure of calix[4]pyrroles
The white crystalline material first synthesized by Baeyer in 1886, by the
cyclocondensation of pyrrole with acetone in the presence of hydrochloric acid.8 Later
Chelintzev and Tronov repeated the reaction and proposed the structure of the
compound which was named as α,β,γ,δ-octamethylporphinogen by Fischer.9 In 1955,

58
Rothemund and Gage improved this synthesis by using methanesulfonic acid as the
catalyst.10 Other than these important findings this class of compounds (e.g.,
octamethylcalix[4]pyrrole) were only studied sporadically in the 100 years following
their discovery, most of which merely focused on the refined synthesis of these
macrocycles and their meso-substituted derivatives but not their applications.11 This
situation changed in the early 1990s when Floriani and coworkers began exploring their
metal coordination chemistry extensively, thus reviving interest in these venerable
macrocycles after a long dormant period.12
Calix[4]pyrroles, originally named “pyrrole-acetone” and formally known as meso-
octaalkylporphyrinogens are macrocycles composed of four pyrrole units linked via
four di-substituted meso-carbons at the α positions. Figure 2.2 describe the basic
skeleton of calix[4]pyrrole where bracket number indicates the repeating pyrrolic units
linked to each other by sp3 hybridized carbon atoms. calix[4]pyrroles are not readily
oxidized to form porphyrin-like structures. As a consequence the four pyrrole units are
all in their neutral NH-bearing forms in calix[4]pyrroles and no electron delocalization
exists within the macrocyclic system as a whole.
´
NH
NH HN
HN
H3C
H3C
H3C CH3
CH3
CH3
H3C CH3
1
2
3
4
5
6
7
8910
1112
14
13
15
20
22
2124
23
1819
17
16
n
Figure 2.2: Structure of calix[4]pyrrole
Calix[4]pyrrole can adopt four different confirmations: 1,3-alternate, 1,2-alternate,
partial cone and cone, similar to calix[4]arene which has been observed experimentally
and noted theoretically.13

59
OROR
OROR
RO
OR OR
OR OR
OR
OR OROROR ROOR
1,3-alternate 1,2-alternate partial cone cone
HN H
NHN N
HN
NH
NH
HN
H
N
H
N
H
N
H
N
H
HN H
NHN
N
H
0.0 (0.0) 6.7 (4.4) 4.8(3.7) 16.0 (11.4)
Figure 2.3: Four possible conformations of calix[4]arenes (upper) and calix[4]
pyrrole (lower)
2.4. Synthesis of calix[4]pyrroles and related macrocycles
Calix[4]pyrroles or meso-substituted porphyrinogens are an important class of neutral
macrocycles receptor capable of binding anions, transition metals14 and neutral
molecules15 in both solution and solid states.16 The synthesis of calix[4]pyrroles bears
analogy to that of porphyrin in that they obtained from the electrophilic α-substitution
of pyrrole with ketones in the presence of acid catalyst.
There are three general method for the synthesis of calix[4]pyrroles:
(a) one-pot [1+1+1+1+1] condensation;
(b) [2+2] condensation and
(c) [3+1] condensation.
HN
R1
R2
O
+
HN
R1
R2
OH
HN
OH
R1
R2
HN
OH2
R1
R2
HN
R1
R2
HN
+3
R1
R2
O
+3
NH
NHHN
NH
R1
R2
R1
R2
R1 R2
R1 R2
NH
NH HN
HN
R1 R
2
R1
R2
R1
R2
R2
R1
[H+]
3
1 2
Scheme 2.1: Mechanistic presentation of the synthesis of calix[4]pyrrole

60
Although calix[4]pyrroles have been synthesized 100 years ago by the acid catalyzed
condensation of ketones and pyrrole, after that a number of strategies have been
employed to synthesized and characterized various functionalized calix[4]pyrroles by
using a wide varieties of catalyst including HCl,17,18 Lewis acids,19,20 chlorzinc,21 p-
toluenesulfonic acid or zeolites22 to improve the yield of specific calix[4]pyrroles.
Among which an alternate synthetic protocol using acidic zeolites catalyst gave a facile
and efficient method for the synthesis of variety of calix[4]pyrroles.23 The graphite
oxide and reduced graphene oxide has been used as solid acid catalysts in organic and
aqueous solutions for the preparation of 5,5-dimethyldipyrromethane and
octamethylcalix[4]pyrrole at room temperature.24
NH
R2R1
O
NH HN
R1 R2
NH
NH HN
HN
R2
R1
R1 R2
R1
R2
R2 R1
HNNH
NH HN
R1
R2
R2
R1
R1 R2
R2R1
GO
+
AI-MCM-41
+
1 2
3 4
3
Scheme 2.2: Selective synthesis of calix[4]pyrrole and dipyrromethane using
different catalyst
Interestingly, meso-octamethylcalix[4]pyrrole (OMCP) or acetonpyrrole was first
reported by Baeyer, and its structure is shown in Figure 2.2 and N-confused
calix[4]pyrrole (Figure 2.3) both macrocycles are position isomers. While in the regular
calix[4]pyrrole the meso-carbons are always connected to C2 and C5 position of the
pyrrole moieties, the N-confused isomer comprises at least one pyrrole moiety
connected to the rest of the macrocycle via C2, C4-connection.

61
NH
NH
HN
HN
H3C
H3C
H3C CH3
CH3
CH3
H3C CH3
5
Figure 2.4: Structure of N-confused calix[4]pyrrole
In the early 1990s, first reports emerged describing the identification of porphyrin
isomers, in which one or more of the four pyrrole moieties are inverted.25,26 The
inverted pyrrole in these N-confused porphyrins is connected to the rest of the
macrocycle via one α-carbon (C2-position) and one β-carbon (C4-position) as opposed
to α,α’-carbons (C2, C5-positions) in regular porphyrins.
Similarly to the formation of N-confused porphyrins, the condensation of ketones and
pyrrole also produces a certain number of C2, C4-pyrrole connections giving rise to N-
confused calix[4]pyrroles. The first N-confused calixpyrrole synthesis was reported in
1975 although the structural assignment may not have been correct, as the NMR spectra
were not recorded at the time. 27 Inspired by the earlier report and by further reports on
N-confused porphyrins, Dehaen and co-workers mounted the first systematic effort
aimed at the preparation of N-confused calix[4]pyrroles which provided the first
rational synthesis of this calix[4]pyrrole isomer.28
2.5. Conformations of calix[4]pyrrole and their relative energies
Calix[4]pyrrole shows geometrical resemblance to calix[4]arene and that it would be
expected to adopt four major conformations like calix[4]arene. Energies optimization
by BLYP/6-31G, and BLYP/6-31+G** methods reveals that the predicted orders of
stabilities for the gas phase and CH2Cl2 solution are namely 1,3-alternate > partial cone
> 1,2-alternate> cone. Molecular mechanics calculations indicate that the 1,3-alternate
conformation is most stable conformation in case of calix[4]pyrrole having torsional
interaction of about 1-2 kcal/mol smaller than that of the other conformations. Each set
of adjacent pyrrole rings in calix[4]pyrrole has an inherent propensity to exist in an anti
rather than syn orientation. In the 1,3-alternate conformation each adjacent pyrrole pair

62
is anti as a result this particular conformation benefits the most from this electrostatic
effect. On the other hand, the pyrroles in the cone conformation are point to the same
direction and as a result this conformation suffers the most from electrostatic repulsion.
2.6. Analysis of anion-binding modes of calix[4]pyrrole
Experimental results indicate that meso-alkyl substituted calix[4]pyrrole form 1:1
complexes with fluoride and chloride anions. In these anion complexes the
calix[4]pyrrole macrocycles are observed to adopt the cone conformation with the anion
sitting above the cone and forming four hydrogen bonds with the four pyrrole N-Hs.29
On the other hand neutral molecules such as alcohols and amides form 2:1 complexes
with calix[4]pyrroles. Optimization of all the putative anion-binding structures for all
the four limiting conformations of calix[4]pyrrole showed that 1,3-alternate structure is
unstable and is converted into the cone structure upon geometry optimization.30
HN H
NHN N
H
N
NHNH
HN
HX
NNN N
HH H H
X
Organic Solvent
Figure 2.5: Conversion of 1,3-alternate conformation into cone in the presence of
anion
In the case of cone structure it has four equivalent NH---F bonds of about 1.68 Å. Due
to the short NH---F hydrogen bonds, the calix[4]pyrrole become flattened. This is
indicated by the decrease of N1-C2-C3-C4 dihedral angles from -76° in F-free cone
structure 6 to -64.8° in cone-F- complex. In contrast to the cone complex, the partial
cone complex forms three NH- - -F- hydrogen bonds.
NNN N
HN
H
N
HN
H
N
H
NNN
N
H
X1.385
X
H H H
1.4621.534
X
HH H
1.627
1,2-alternate (Cs)22.0 (20.4)
partial cone (Cs)10.6 (9.4)
cone (C4v)0.0 (0.0)
Figure 2.6: Calculated structure of calix[4]pyrrole-F and their relative energies at
BLYP/6-31-G**level

63
Table 2.1: Binding energy (kcal/mol) of 1:1 fluoride anion binding modes of
calix[4]pyrrole calculated at the blyp/6-31+g** level
Entry 1,2-alternate
∆Ebinding
Partial cone
∆Ebinding
Cone
∆Ebinding
Gas phase -44.6 -56.0 -66.6
Solution -7.0 -17.5 -27.6
There is a considerable change in geometry observed upon fluoride anion binding, the
flat pyrrole in partial cone tilts upward while the other three pyrroles become relatively
flattened. The cone complex is calculated to be most stable whereas the partial cone
complex is predicted to be the second most stable. In the gas phase partial cone anion
complex is higher in energy by 10.6 kcal/mol than that of cone complex. When the
solvent effect by dichloromethane is accounted for the destabilization of partial cone as
compared to cone is decreased to9.3 kcal/mol. The 1,2-alternate complex is predicted
to be less stable than the cone structure by 25.4 and 20.4 kcal/mol in the gas phase and
in CH2Cl2 solution respectively. The relative stabilities reflect the number of hydrogen
bonds in these complexes indicating that the in ionic hydrogen bonds in these
complexes are very strong.
2.7. β-Substituent effects on anion-binding abilities of calix[4]pyrrole
One simple way to strengthen the anion-binding is to increase the binding force i.e.
hydrogen bond strength between the host and anion. Modification of the hydrogen bond
acceptor (host part) is apparently a possible approach to tune the binding ability of
calix[4]pyrroles. In the anion-free pyrroles the bond length of N-H is about 1.01-1.02 Å
at the BLYP/6-31+G** level. When fluoro anion is involved, the N-H bond is
elongated to1.24-1.46 Å varying in different substituted structures. The stronger
electron-withdrawing group normally elongates the N-H bond more while the H-F bond
is significantly shortened. This implies that the basicity of F- is stronger than those of
the pyrrole anions in the gas phase.

64
N
R1
R2
H N
R1
R2
H
R1=R2= CH3O, CH3, H, F, Cl, Br, CN
7 8 3 9 10 11 12= X
6
Scheme 2.3: β-Substitution effect on calix[4]pyrrole--anion binding
It appears that both σ and π effects of the substituents affect the anion binding energy.
Methoxy group is a strong π donor and a weak σ acceptor. While the π-donating factor
decreases the binding energy, the σ-accepting factor increases the binding energy. As a
result, the methoxy group has little effect on the binding energy. Fluoro, chloro, and
bromo groups are stronger σ-acceptors and behave as electron-acceptors. Therefore they
all increase the binding energy to similar extents. Cyano group is a strong electron-
acceptor and it can significantly increase the binding energy.31
Table 2.2: Calculated anion-binding energies in kcal/mol of different β-
disubstituted calix[4]pyrrole with fluoride ion
β-substitutedCP-X- BLYP 6-31+G** BLYP6-31+G**(sol.)
R1=R2= OCH3 -40.4 -13.3
R1=R2= CH3 -38.9 -13.1
R1=R2= H -38.6 -13.4
R1=R2= F -48.4 -18.7
R1=R2= Cl -51.6 -19.0
R1=R2= Br -52.9 -19.2
R1=R2= CN -68.3 -27.5
2.8. Synthesis of conformational isomer of calix[4]pyrrole
Introduction of aryl or other rigid groups into the meso-like positions of calix[4]pyrroles
could serve not only to change the intrinsic anion selectivity of the calix[4]pyrrole
skeleton but also in appropriate cases to introduce secondary binding sites that might

65
allow for selective recognition of cationic, anionic or neutral guests. The aryl ketone,
pyrrole and acid were dissolved in methanol and stirred at room temperature in inert
atmosphere to give four conformational isomers namely αααα, αααβ, ααββ and αβαβ.32
In the specific case of the αααα isomer of a deep cavity structure was observed in the
solid state, wherein the calixpyrrole core is in a so-called cone conformation.5
Structures of lower symmetry are seen in the case of the other isomers with
conformations other than pure cone (e.g., 1,3-alternate, partial cone) being observed in
certain instances (e.g., the αααβ isomer). Proton NMR spectroscopy was used to
confirm the configurational assignments and to analyze conformational effects in
solution. As a general rule it was found that free rotation around the pyrrole-bridging
meso-like carbons and ipso-aryl bonds pertains at room temperature in acetonitrile-d3-
D2O (99.5:0.5; v/v) in the absence of an added anionic substrate. Thus the symmetries
of the various isomers were reflected directly in their 1H NMR spectra particularly in
the number of pyrrole CH resonances. The αααα isomers have one type of CH group
represented by an “a” in Scheme 2.4 , while the ααββ isomer has two types of pyrrolic
CH group (represented by “a” and “b”), and the αααβ isomer possesses four different
CH groups (“a”,“b”, “c” and “d”) and in all cases the requisite number of signals were
observed.
NH
NH HN
HN
NH
COCH3
H3C
CH3
CH3
H3C
+
NH
NH HN
HN
H3C
CH3
CH3
H3C
NH
NH HN
HN
H3C
CH3
CH3
H3C
NH
NH HN
HN
H3C
CH3
CH3
H3C
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
αααααααααααααααα
a
a aa
a
aaa
a
a
aa
bb
bb
a
a
a
aa
a
aa
ααααββββααααββββ
ααααααααββββββββ
ααααααααααααββββ
ab
a
b
c
d
cd
CH3SO3H
Dry methanol
1 13
14 15
16 17
Scheme 2.4: Synthesis of conformational isomer of calix[4]pyrrole

66
2.9. Results and discussion
The discovery of octamethyl calix[4]pyrrole as an anion receptor has prompted the
synthesis of a variety of calix[4]pyrrole derivatives,33 expanded calixpyrroles,34
strapped calixpyrroles35 and N-confused calixpyrroles36 to understand the binding of
different anions with specific calix[4]pyrrole. From chemical viewpoint calix[4]pyrroles
are being explored as catalyst in organic synthesis37 as well as precursor of important
organic molecules such as calixpyridines and calixpyridinopyrrole.38 Beside these, they
have been found applications as fluorescent,39 colorimetric,40 electrochemical signaling
device41 and new solid supports capable of separating anion mixtures.42 This
macrocycles are also used as anion transporter across lipid bilayer membrane.43
Moreover, in contrast to the well-studied templating properties of cationic44 and neutral
species45 anion templates have been very little exploited in synthetic chemistry. This
relative lack of anion-templated processes is partially attributed to some of the intrinsic
properties of anions such as their diffuse nature, pH sensitivity, and their relative high
solvation free energies.46 However, as the increasing number of anion-directed
assemblies reported in the last few years, these limitations are not as critical as initially
thought. They act as template in the coordination, supramolecular and inorganic hybrid
structures.47,48 The use of anions templates to prepare cyclic structures is not exclusive
confined to metal-containing assemblies,49 but it is also found in organic systems. For
example Alcalde et al., have reported the halide-directed synthesis of a series of
[14]imidazoliophanes,50 trihalogenated-templates in the synthesis of calix[6]pyrroles51
and solution phase anion directed self-assembly of meso-functionalized
calix[4]pyrroles.52 Owing to their tremendous applications in the area of supramolecular
chemistry they have gained significant importance. The synthesis of calix[4]pyrroles
and N-confused calix[4]pyrroles in higher yields have been discussed in the following
subsection:
2.9.1. Synthesis of calix[4]pyrroles and N-confused calix[4]pyrroles
The reaction of equimolar quantity of pyrrole (1) with acetone (2a) in the presence of
tetrabutylammonium fluoride in dichloromethane at 25˚C afforded a isomeric mixture

67
of octamethylcalix[4]pyrrole (OMCP) (3a) and N-confused octamethylcalix[4]pyrrole
(NC-OMCP) (4a) in 81% and 18% yield respectively. (Scheme 2.5)
NH
NH HN
HN
R1
R2
R1 R2
R1
R2
R1 R2
NH
+ R1 R2
O
1 2 3 5
Bu4N+X-
NH
NH
HN
HN
R1
R2
R1 R2
R1
R2
R1 R2
2a. R1=R2 = CH3 3a. R1=R2 = CH3 5a. R1=R2 = CH3
2b. R1=R2 = C2H5 3b. R1=R2 = C2H5
2c. R1= CH3, R2 = C2H5 3c. R1= CH3, R2 = C2H5 5c. R1= CH3, R2 = C2H5
2d. R1-R2 = (CH2)4 3d. R1-R2 = (CH2)4 5d. R1-R2 = (CH5)4
2e. R1-R2 = (CH2)5 3e. R1-R2 = (CH2)5 5e. R1-R2 = (CH2)5
2f. R1-R2 = (CH2)6 3f. R1-R2 = (CH2)6 5f. R1-R2 = (CH2)6
X- = F, Cl, I, AcO
Scheme 2.5: Anion-templated synthesis of calix[4]pyrroles and N-confused calix[4]
pyrroles
Using the catalytic amount of anion along with .01 equivalent of methanesulfonic acid
fasten the reaction as well as improved the yield of the products. To analyze the role of
anions in the reaction the cyclocondensation reaction of the pyrrole and acetone was
performed using different solvent and acid catalyst with and without the use of anion
and the results obtained were summarized in table 2.2. It was observed that when the
reaction of equimolar quantity of pyrrole (1) and acetone (2) was performed using
ethanol as a solvent and methanesulfonic acid catalyst the reaction was completed in
four hours at refluxing temperature giving 65% of OMCP (3a) and 15% of NC-OMCP
(5a) along with some polymeric product. Whereas using ytterbium triflate in ionic
liquid for the cyclocondensation of pyrrole and acetone gave octamethylcalix[4]pyrrole
as the only product in excellent yield.53 Cyclocondensation reaction of pyrrole with
acetone without using anions gave acceptable results taking Amberlyst™-15 as a
catalyst in 8 hours at ambient temperature. 54

68
Table 2.2: Synthesis of meso-octamethylcalix[4]pyrrole (3a) and N-confused
octamethylcalix[4]pyrrole (5a) using different catalyst and solvent
S.No. Solvent Catalyst Anions Reaction
time
(mins)
Conversion
of pyrrole
%
yield
of 3a
%
yield
of 5a
1 Ethanol Conc. HCl - 300 62a 56 -
2 Ethanol CH3SO3H - 120 90a 68 15
3 Ethanol BF3OEt2 - 200 85a 65 12
4 Ethanol CF3COOH - 140 87a 60 10
5 CH2Cl2 CH3SO3H - 140 85a 80 3
6 CH2Cl2 BF3OEt2 - 260 80a 63 4
7 CH2Cl2 CF3COOH - 150 94a 56 8
8 [bmim][BF4] Yb(OTf)3 - 90 95b 92 -
9 CH2Cl2 Amberlyst™-15 - 480 99 b 83 14
10 CH2Cl2 CH3SO3H F - 5 100 b 81 17
11 Toluene CH3SO3H F - 10 87 b 75 8
12 CHCl3 CH3SO3H F - 7 98 b 80 15
(a) at refluxing temperature; (b) at 25˚C
The structure of 3a and 5a were confirmed by IR, 1H-NMR, 13C-NMR and ESI-MS
spectroscopic data. In fact 1H NMR-spectroscopy is particularly useful for the
differentiation of the OMCP (3a) and NC-OMCP (5a). The 1NMR-spectrum of
compound 3a, showed a sharp singlet at δ 1.52 ppm were assigned for the twenty four
methyl group protons at meso position, a doublet at δ 5.90 ppm for the eight β-pyrrolic
protons and a broad resonance at δ 7.08 ppm for the four pyrrolic N-H protons (Figure
2.7). The integration ratio of the three peaks areas were 6:2:1, in agreement with the
empirical formula and D4 symmetry of the compound 3a.
Figure 2.7: 1H NMR spectrum of 5,5,10,10,15,15,20,20-octamethylcalix[4]pyrrole

69
On the contrary, in the 1NMR-spectrum of 5a, multiplet appeared at δ 1.53-1.56 ppm
for the 24 methyl protons. Signals for the α-pyrrole hydrogen (1H) and β-pyrrole
hydrogen (1H) of the 2,4-disubstituted pyrrole appear at δ 6.51(doublet) and 5.09,
respectively, which was well apart from those of the β-hydrogens atoms (6H) of the 2,5-
disubstituted pyrrole (δ. 5.82-5.97). The coupling constant between the band α-
hydrogen atoms of the confused pyrrole in 5a has a typical value of 2.0 Hz. Four
distinct broad resonances at δ 7.00 (1H), 7.15 (1H), 7.54 (1H) and 7.78 (1H) ppm is due
to four pyrrolic N-H protons indicates the reduction of symmetry in case of N-
confused-octamethylcalix[4]pyrrole (5a) as compared to octamethylcalix[4]pyrrole.
The higher polarity and non-zero dipole moment of the compound 4a have been
attributed to their lower symmetry. The highly unsymmetrical nature of the compound
5a is also seen in its carbon spectrum which gives total of twenty four peaks. The four
peaks at δ 28.94, 29.34, 29.67 and 30.30 ppm is due to CH3 carbons, at δ 31.40, 31.90,
34.59 and 35.32 ppm for four meso carbons, at δ 101.55, 101.77, 102.13, 102.75,
103.33, 103.89 and 104.21 ppm for β-pyrrolic carbon, at δ 111.61 ppm for the α-carbon
of inverted pyrrole ring and at δ 133.19, 137.59, 137.82, 138.28, 138.45, 138.77,
138.89, 139.48 and 141.13 ppm for α-pyrrolic carbon atoms.
Figure 2.8: 1H NMR spectrum of N-confused 5,5,10,10,15,15,20,20-octamethyl
calix[4]pyrrole in CDCl3
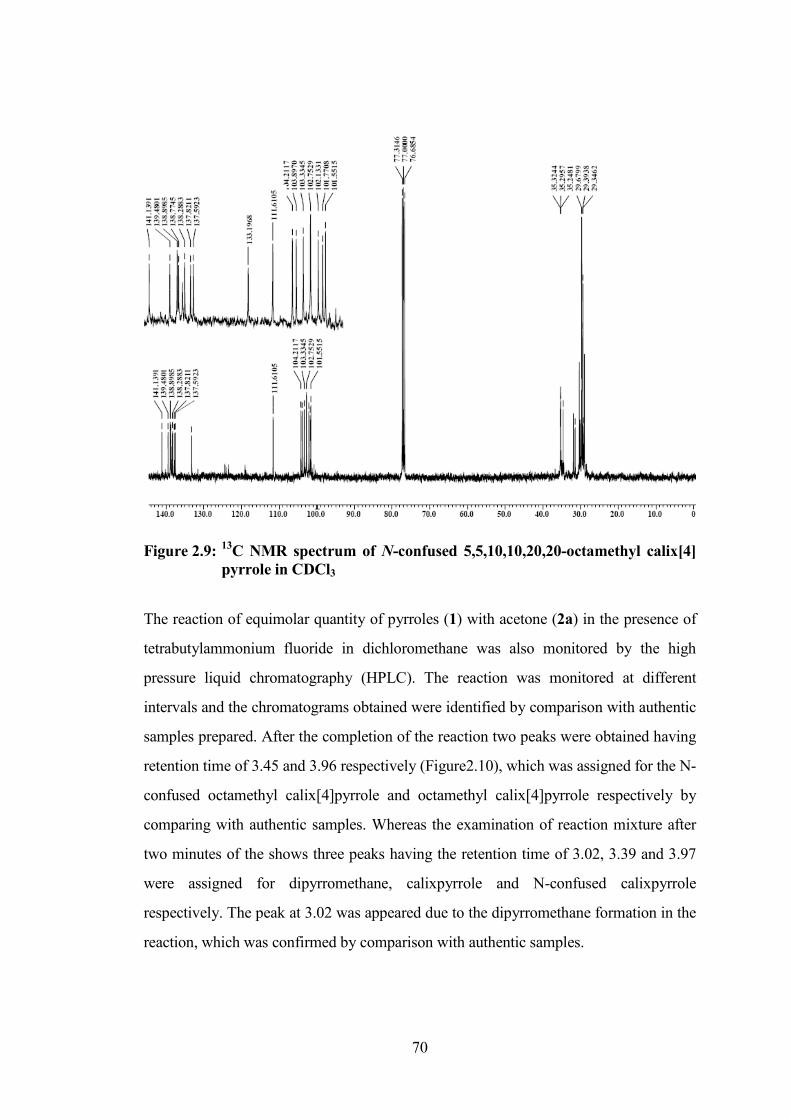
70
Figure 2.9: 13
C NMR spectrum of N-confused 5,5,10,10,20,20-octamethyl calix[4]
pyrrole in CDCl3
The reaction of equimolar quantity of pyrroles (1) with acetone (2a) in the presence of
tetrabutylammonium fluoride in dichloromethane was also monitored by the high
pressure liquid chromatography (HPLC). The reaction was monitored at different
intervals and the chromatograms obtained were identified by comparison with authentic
samples prepared. After the completion of the reaction two peaks were obtained having
retention time of 3.45 and 3.96 respectively (Figure2.10), which was assigned for the N-
confused octamethyl calix[4]pyrrole and octamethyl calix[4]pyrrole respectively by
comparing with authentic samples. Whereas the examination of reaction mixture after
two minutes of the shows three peaks having the retention time of 3.02, 3.39 and 3.97
were assigned for dipyrromethane, calixpyrrole and N-confused calixpyrrole
respectively. The peak at 3.02 was appeared due to the dipyrromethane formation in the
reaction, which was confirmed by comparison with authentic samples.

71
Figure 2.10: High pressure liquid chromatogram of (A) dipyrromethane, (B)
5,5,10,10,15,15,20,20-octamethyl calix[4]pyrrole
Figure 2.11: High pressure liquid chromatogram of reaction mixture
Figure 2.12: High pressure liquid chromatogram of N-confused 5,5,10,10,15,
15,20,20-octamethyl calix[4]pyrrole

72
Owing to our curiosity and high yields of compound 3a and 5a in the presence of
anions prompted us to examine the role of anions in the cyclization of other ketones 2b-
2h with pyrrole (Table 2.5). The reaction of cyclohexanone with pyrrole in the
presence of tetrabutylammonium fluoride in dichloromethane gave solid precipitate
within minutes indicates formation of two products on TLC with Rf values 0.85 and
0.42 respectively. Crystal of the compound was grown by the slow evaporation of the
CHCl3 solution of the crude mixture without any further purification. Examination of
the crystalline product shows the association of the fluoride ion with the NH proton of
the calix[4]pyrrole (3e). The crystal structure of the complex between calix[4]pyrrole
and tetrabutylammonium fluoride reveals the role of tetrabutylammonium fluoride as
the template. The fluoride atom of the guest is anchored to the cavity of the
calix[4]pyrrole through the hydrogen bonds between the fluoride atom of the guest and
hydrogen atom of the NH of four pyrrole rings. The crystal of N-confused-cyclohexyl
calix[4]pyrrole with anion cloud not be obtained from the mixture this could be
attributed to the lower percentage of N-confused-cyclohexyl calix[4]pyrrole in the
reaction mixture as compared to the percentage of cyclohexyl calix[4]pyrrole. In 1H
NMR spectrum of 5,5,10,10,15,15,20,20-tetraspiro-cyclohexylcalix [4]pyrrole (3e) two
multiplets at 1.46 and 1.89 ppm were assigned for the cyclohexyl methylene group
protons, and a doublet at 5.88 ppm with coupling constant J = 2.7 Hz was assigned for
the eight β-pyrrolic protons and a broad singlet at 7.04 ppm was assigned for four
pyrrolic NH protons. 13C NMR spectrum confirmed the symmetry of the molecule in
13C NMR spectrum of compound 3e showed three peaks at 22.69, 25.96, and 37.13
ppm for cyclohexyl carbon, peak at 39.55 ppm for the meso-carbon, and two peaks
appeared at 103.41 and 136.39 ppm for β-pyrrolic carbon and α-pyrrolic carbon
respectively, confirmed the formation of compound 3e. A band appeared at 3445 cm-1
in IR spectrum of compound 3e which indicates the presence of NH group in the
molecule.
In a given templated reaction the size and shape of the directing anions are the main
factors that determine the reaction. It was seen that when the reaction was performed in
the presence of anions other than fluoride the yield of the reaction decreases as

73
compared to that in the presence of fluoride ion as well as reaction time also increases
table 2.3. This could be attributed to the stronger binding affinity of fluoride ion
towards the compound 3a and 5a. Apart from its strong affinity towards calix[4]pyrrole
and N-confused calix[4]pyrrole, fluoride ion also favors the reaction as a template due
to its smaller size.
Figure 2.13: ESI-MS spectrum of crude reaction mixture of 1 and 2 after completion
of reaction
Table 2.3: Anion catalyzed condensation of pyrroles and acetone in dichloromethane
Entry Anions % Yield of 3a % Yield of 5a Reaction time
(mins)
1 F- 81 17 5
2 Cl- 80 15 7
3 I- 71 10 20
4 AcO- 70 9 30
Further the reaction of pyrrole and acetone was also examined in different solvent. A
subsequent solvent screening revealed that low-polarity solvents led to higher yield,
with CH2Cl2 providing the best results (Table 2.4). Reaction carried out in toluene and
benzene gave comparatively low yield with prolonged reaction time.
74
Table 2.4: Anion templated synthesis of octamethylcalix[4]pyrrole and N-confused
octamethylcalix[4]pyrrole in the presence of fluoride ion in different
solvents
Entry Solvents % Yield of 3a % Yield of 5a Anions
1 MeOH (EtOH) 65 10 -
2 CHCl3 80 15 F-
3 CH2Cl2 81 17 F-
4 CH3CN 78 14 F-
The reactions of different ketones with pyrrole were performed in the presence of
fluoride ion and the results were summarized in table 2.5. The reaction of
cycloheptanone with pyrrole in the presence of fluoride ion gave the corresponding
calix[4]pyrrole and N-confused calix[4]pyrrole in thirty minutes at room temperature
and the isolated yields were 70% and 10% respectively, but the time required for the
conversion was relatively long. This could be attributed to the bulkiness of
corresponding ketone. The 1NMR-spectrum of compound 5f shows two multiplets
appeared at δ 1.46-1.63 ppm and2.00-2.08 ppm for the methylene protons. Signals for
the α-pyrrole hydrogen (1H) and β-pyrrole hydrogen (1H) of the 2,4-disubstituted
pyrrole appear at δ 6.41(doublet, J=2 Hz) and 5.74, respectively. Whereas multiplet
appears for β-hydrogens atoms (6H) of the 2,5-disubstituted pyrroles at δ. 5.82-5.97).
Four distinct broad resonances at δ 6.82 (1H), 6.97 (1H), 7.43 (1H) and 7.65 (1H) ppm
is due to four pyrrolic N-H protons.
Table 2.5: Anion templated synthesis of different calix[4]pyrroles and N-confused
calix[4]pyrroles in the presence of fluoride ion
Entry 3, 5 % Yields Time (min)
1 3a, 5a 81, 17 5
2 3b 61 30
3 3c, 5c 78, 13 15
4 3d, 5d 86, 12 20
5 3e, 5e 79, 20 3
6 3f, 5f 70, 10 30

75
Figure 2.14: 1H NMR spectrum of 5,5,10,10,15,15,20,20-tetraspiro-N-confused
cycloheptylcalix [4]pyrrole
2.9.2. Synthesis of meso-unsubstituted porphyrinogens and N-confused
porphyrinogens in the presence of anions
One equivalent of sodium borohydride was added portion wise to the solution of
pyrrole-2-carboxaldehyde in 20 ml of THF under nitrogen atmosphere. The mixture
was stirred for 2-3 hours under nitrogen atmosphere. After completion of the reaction,
reaction mixture was treated with water for complete dissolution of the suspension of
NaBH4 and the mixture was extracted with dichloromethane. The combined organic
phase was further washed with water and dried over anhydrous sodium sulfate.
Triethylamine (2-3 drops) was added and solvent was evaporated under reduced
pressure to give yellow oil. Further 1-2 drop of triethylamine was added and the pure
compound was distilled off under reduced pressure as colorless liquid. The pure
compound was stored in the refrigerator to avoid polymerization. The structure of
compound was confirmed by IR and 1H-NMR spectroscopic data.

76
NH
CHONH
OH
NaBH4/THF
N
N N
N
Bu4N+X-
CDCl3, [H+]F
H H
HH
N
N
HN
N
FH H
HH
18 19 20 21
Scheme 2.6: Synthesis of porphyrinogen and N-confused porphyrinogen
Figure 2.15: 1H NMR spectrum of 2-Hydroxymethylpyrrole
Further acidic cyclocondensation reaction of the 2-hydroxymethylpyrrole (19a) was
performed in the presence of tetrabutylammonium fluoride (TBAF) in deuterated
chloroform under nitrogen atmosphere and completion of the reaction was
monitored by TLC. After the completion of reaction the mixture was examined by
1H NMR spectroscopy. In 1H NMR spectrum of the reaction mixture showed four
broad resonances at δ 8.60, 8.76, 8.96 and 9.22 which can be assigned for the N-
confused porphyrinogen and porphyrinogen respectively. Downfield shift in the
peak of NH proton can be attributed to the fluoride ion binding to the NH-proton of
the porphyrinogen. Appearance of the three broad peaks in the reaction mixture can
be assigned as the presence of N-confused porphyrinogen in the mixture. The peak
appeared at 4.45 ppm was assigned for the meso-methylene protons of
porphyrinogen.
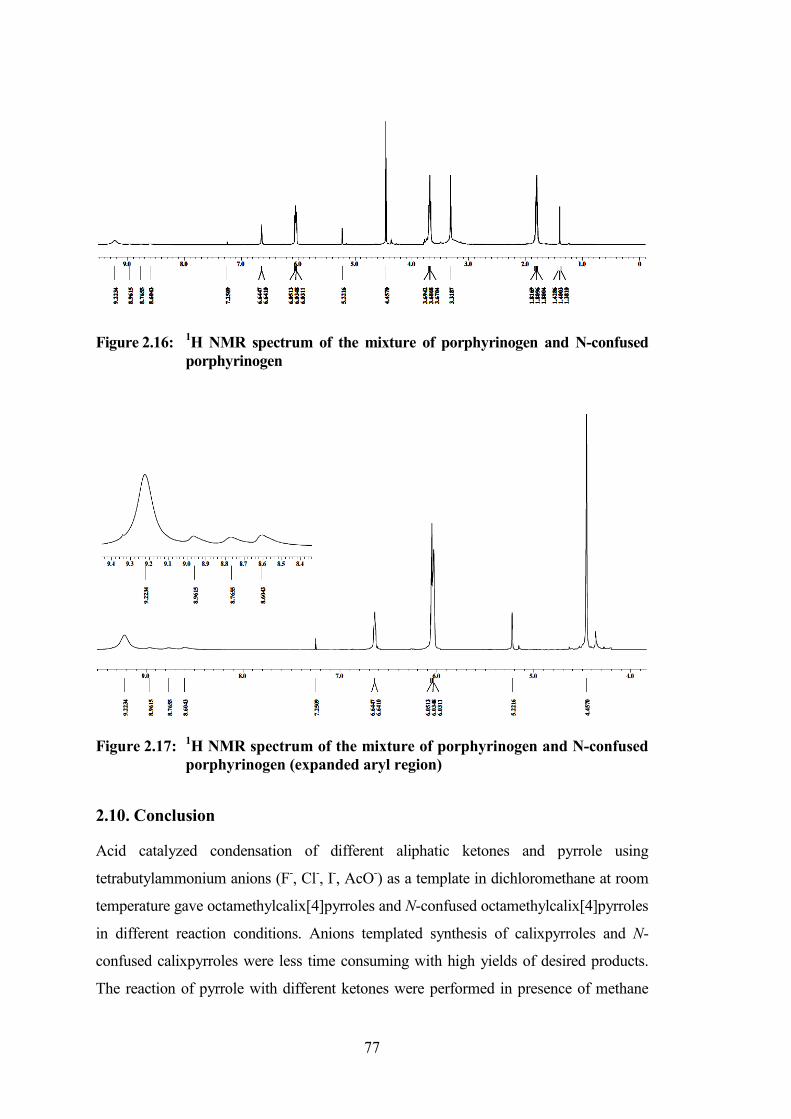
77
Figure 2.16: 1H NMR spectrum of the mixture of porphyrinogen and N-confused
porphyrinogen
Figure 2.17: 1H NMR spectrum of the mixture of porphyrinogen and N-confused
porphyrinogen (expanded aryl region)
2.10. Conclusion
Acid catalyzed condensation of different aliphatic ketones and pyrrole using
tetrabutylammonium anions (F-, Cl-, I-, AcO-) as a template in dichloromethane at room
temperature gave octamethylcalix[4]pyrroles and N-confused octamethylcalix[4]pyrroles
in different reaction conditions. Anions templated synthesis of calixpyrroles and N-
confused calixpyrroles were less time consuming with high yields of desired products.
The reaction of pyrrole with different ketones were performed in presence of methane

78
sulfonic acid in dichloromethane using different anions as template and best results were
obtained when F anion was used as a template due to the smaller size and greater affinity
to bind with calixpyrrole and N-confused calixpyrrole. The reaction of bulky alkyl and
cycloalkyl ketones with pyrrole gave the calix[4]pyrroles in good yields whereas N-
confused calix]4[pyrroles in low yields but in case of cyclohexanone the yield of
calixpyrrole and N-confused calixpyrrole is equal to like acetone this is due to the greater
reactivity of cyclohexanone than other cyclic ketones.
The meso-unsubstituted porphyrinogen or calixpyrrole and N-confused calixpyrroles
are highly unstable and readily oxidized to corresponding stable porphyrins due to this
effect
The meso-unsubstituted porphyrinogen or calixpyrrole and N-confused calixpyrrole
were also formed in situ by the acid catalyzed tetramerization of 2-hydroxymethyl
pyrrole which was synthesized by the reaction of pyrrole-2-carboxyldehyde with
sodium borohydride, in presence of F anion in deuterated chloroform at room
temperature and formation of the products were confirmed by 1H NMR spectroscopy.
2.11. Experimental
All melting points are uncorrected and expressed in degree centigrade and were
recorded on Thomas Hoover Unimelt capillary melting point apparatus. The infrared
spectra were recorded on Perkin-Elmer FT-2000 spectrometer and νmax are expressed in
cm-l. 1H NMR was recorded on Jeol-delta-400 spectrometer using tetramethylsilane
(TMS) as an internal standard and chemical shifts (δ) are expressed in ppm. ESI-MS
were recorded by LC-TOF (KC-455) mass spectrometer of Waters. The starting
materials such as pyrrole, acetone, diethyl ketone, ethylmethyl ketone, cyclopentanone,
cyclohexanone and methane sulphonic acid were purchased from Spectrochem
Chemicals India. The quaternary ammonium salts; normal tetrabutylammonium halides
(n-Bu4NF, n-Bu4NCl, n-Bu4NBr, n-Bu4NI), and normal tetrabutylammonium acetate
(n-Bu4NAcO) were purchased from Aldrich. The pyrrole was distilled prior to use and
the solvents used were of analytical reagent grade. The compounds synthesized were
separated by column chromatography using neutral alumina and characterized by
melting points, 1H NMR, 13C NMR, IR and ESI-MS techniques.

79
2.11.1. 5,5,10,10,15,15,20,20-Octamethyl calix[4]pyrrole (3a)
Equimolar amount of pyrrole (1 ml, 14 mmol) and acetone (1.04 ml, 14 mmol) were
taken up in dry dichloromethane (10 mL). Tetrabutylammonium fluoride (0.95 mg,
0.003mmol) was added to the reaction mixture, which was stirred at 25˚C for a minute
and then methanesulfonic acid (0.1 mmol) was added. The progress of reaction was
monitored by thin layer chromatography (TLC). After the completion of reaction, the
reaction mixture was extracted by dichloromethane and washed several time with water.
Organic phase was separated and dried over sodium sulfate. The filtrate was
concentrated at vacuum to gave the crude product, which was subjected to column
chromatography over neutral alumina eluting with petroleum ether-chloroform (9:1,
v/v) to afford pure meso octamethylcalix[4]pyrrole (3a).
5,10,15,20-Octamethyl-calix[4]pyrrole (3a)
Physical state: White solid
Yield: 2.05 g (81%)
mp: 295 ºC (Lit. mp. 296 ºC)
Rf = 0.8 (SiO2, 1:1 petroleum ether/chloroform)
IR (KBr pellet, cm-1
): 3444(br, pyrrole NH), 2973, 2932, 2870, 1578, 1448, 1414,
1279, 1233, 1044, 759
1H NMR (400 MHz, CDCl3): δ = 1.52 (s, 24H, CH3), 5.90 (d, J = 2.92 Hz, 8H, β-
pyrrole CH), 7.08 (brs, 4H, Pyrrole NH)
13C NMR (100 MHz, CDCl3) δ = 29.08 (CH3), 35.15 (meso C), 102.67 (β-pyrrole
CH), 138.55 (α-pyrrole C)
HRMS (ESI-MS) for C28H36N4 [M+H]+ : calcd, 429.2862; found, 429.2860.
2.11.2. 5,5,10,10,15,15,20,20-Octamethyl-N-confused calix[4]pyrrole (5a)
Further elution of the column with petroleum ether-chloroform (3:2, v/v) gave the N-
confused isomer of octamethylcalix[4]pyrrole (5a).
Physical state: white solid
Yield: 250 mg (17%)

80
mp 184 ºC
Rf = 0.45 (SiO2, 1:1 petroleum ether/chloroform);
IR (KBr pellet, cm-1): 3430, 2910, 2845, 1521, 1616, 1179, 749.
1H NMR (400 MHz, CDCl3) δ = 1.48-1.54 (m, 24H, CH3), 5.07 (brs, 1H, β-pyrrole
CH), 5.87 (m, 2H, β-pyrrole CH), 5.89 (brs, 2H, β-pyrrole CH), 5.90 (brs, 2H, β-pyrrole
CH), 6.51 (s, 1H, α-pyrrole CH), 6.97 (brs, 1H, pyrrole NH), 7.13 (brs, 1H, pyrroles,
NH), 7.48 (brs, 1H, pyrrole NH),7.76 (brs, 1H, pyrrole NH);
13C NMR (100 MHz, CDCl3) δ = 29.0, 29.4, 29.6, 30.3 (4x CH3), 34.6, 35.3, 35.8,
35.9 (4x meso C), 101.6, 101.8, 102.1, 102.8, 103.3, 103.9, 104.2 (7x β-pyrrole CH),
111.6 (α-pyrrole CH), 133.2 (β-pyrrole C), 137.5, 137.8, 138.2, 138.7, 138.8, 139.4,
141.1 (7x α-pyrrole C)
ESI-MS for C28H36N4 [M+H]: calcd, 429.2862; found, 429.2860.
2.11.3. Synthesis of 5,5,10,10,15,15,20,20-octaethyl calix[4]pyrrole (3b)
Similar procedure was used for the synthesis of the compound as used above for the 3a
Physical state: white solid
Yield: 1.97 g (61%)
Mp: 224 °C
Rf = 0.76 SiO2, 1:1 petroleum ether/chloroform)
1H NMR (400 MHz, CDCl3) δ = 0.64-0.68 (t, 24 H, CH3-), 1.79-1.57 (q, 16H, -CH2-),
5.89 (d, J =2.80 Hz, 8H, β-pyrrole CH), 7.09 (brs, 4H, NH)
13C NMR (100 MHz, CDCl3) δ = 8.04 (CH3), 29.18 (-CH2-), 42.63 (meso-C), 104.98
(β-pyrrolic C), 135.78 (α-pyrrolic C)
ESI-MS for C36H52N4 [M+H]-: calcd, 539.4113; found, 539.4120.
2.11.4. Synthesis of 5,10,15,20-tetraethyl-5,10,15,20-tetramethyl-calix[4]pyrrole (3c)
Similar procedure was used for the synthesis of the compound as used above for the 3a
Physical state: white solid
Yield: 1.86 g (78%)

81
mp 144 ºC
Rf = 0.76 SiO2, 1:1 petroleum ether/chloroform)
IR (KBr pellet, cm-1): 3435 (br, pyrrole NH), 2968, 2931, 2876, 1413, 1297, 1211,
1040, 761, 706
1H NMR (400 MHz, CDCl3) δ = 0.68-0.71 (t, 12H, CH3-), 1.36-1.40 (q, 8H, -CH2-),
1.85 (brs, 12H, CH3), 5.88 (d, J = 1.44 Hz, 8H, β-pyrrole), 7.08 (brs, 4H, NH)
13
C NMR (100 MHz, CDCl3, TMS): δ = 8.6 (-CH3), 29.9 (CH3-), 33.1(-CH2),
39.1(meso-C), 103.7(β-pyrrole CH), 137.2(α-pyrrole C)
ESI-MS for C32H44N4 [M+H]: calcd, 484.1162; found, 483.2840.
2.11.5. 5,10,15,20-tetraethyl-5,10,15,20-tetramethyl N-confused-calix[4]pyrrole (5c)
Physical state: yellow solid
Yield: 180 mg (13%)
mp 120 ºC
Rf = 0.4 SiO2, 1:1 petroleum ether/chloroform)
IR (KBr pellet, cm-1): 3435 (br, pyrrole NH), 2968, 2931, 2876, 1413, 1297, 1211,
1040, 761, 706
1H NMR (400 MHz, CDCl3) δ= 1.83-1.12 (m, 29H, - CH2CH3), 1.92 (3H, s, CH3),
5.53 (1H, br), 5.78 (m, 2H), 5.88 (brs, 2H,), 6.03 (br, 2H, β-pyrrole), 6.40 (d, J = 2 Hz,
1H, α-pyrrole CH), NH: 7.35 (br, 2H), 7.53 (br, 1H), 7.63 (br, 1H)
ESI-MS for C32H44N4 [M+H]: calcd, 484.1162; found, 483.3482.
2.11.6. 5,5,10,10,15,15,20,20-Tetrakis spirocyclopentyl calix[4]pyrrole (3d)
Physical state:
Yield: 2.26 g (76%)
mp 235 ºC;
Rf = 0.63( SiO2, 1:1 petroleum ether/chloroform);
IR (KBr pellet, cm-1
): 3496 (brs, pyrrole NH), 2959, 2870, 1630, 1580, 1414, 1254,
1042, 765;

82
1H NMR (400 MHz, CDCl3) δ= 1.71-1.69 (m, 16H, CH2), 2.03-2.00 (m, 16H, CH2),
5.86 (d, J = 2.8 Hz, 8H, β-pyrrole), 7.10 (brs, 4H, NH);
13C NMR (100 MHz, CDCl3, TMS): δ = 23.76, 38.89 (cyclopentyl C), 46.79 (meso
C), 102.89 (pyrrole β-CH), 137.14 (pyrroles α-CH);
ESI-MS for C32H44N4 [M+H]: calcd: 531.3488; found: 531.3486.
2.11.7. 5,5,10,10,15,15,20,20-Tetrakis spirocyclopentyl N-confused calix[4]pyrrole (5d)
Physical state: white solid
Yield: 350 mg (12%)
mp 199 ºC
Rf = 0.42 SiO2, 1:1 petroleum ether/chloroform)
IR (KBr pellet, cm-1
): 3496 (br, pyrrole NH), 2959, 2870, 1630, 1580, 1414, 1254,
1042, 765
1H NMR (400 MHz, CDCl3) δ= 1.50-1.20 (m, 16H, CH2). 2.25-1.98 (m, 16H, CH2),
5.58 (br, 1H, β-pyrrole -CH), 5.88 (m, 2H, β-pyrrole -CH), 5.90 (br, 2H, β-pyrrole -
CH), 6.00 (br, 2H, β-pyrrole -CH), 6.42 (d, J =1.97 Hz, 1H, α-pyrrole), 7.00 (brs, 2H,
NH), 7.29 (brs, 1H, NH), 7.48 (brs, 1H, NH).
2.11.8. 5,5,10,10,15,15,20,20-Tetrakis spirocyclohexyl calix[4]pyrrole (3e)
Physical state: white solid
Yield: 2.79 g (80%)
mp 273 °C (lit28-271-272 ºC)
Rf = 0.85 SiO2, 1:1 petroleum ether/chloroform)
IR (KBr pellet, cm-1
): 3445 (br, pyrrole NH), 2924, 2849, 1577, 1413, 1184, 752
1H NMR (400 MHz, CDCl3) δ= 1.40-1.47 (m, 24H, CH2), 1.89-1.92 (m, 16H, CH2),
5.88 (d, J = 2.7 Hz, 8H, β-pyrrole), 7.04 (brs, 4H, NH)
13C NMR (100 MHz, CDCl3, TMS): δ = 22.69, 25.96, 37.13, 39.55 (hexyl C), 103.41
(pyrrole β-CH), 136.39 (pyrroles α-CH)
(ESI-MS) for C40H52N4 [M+H]- : calcd, 587.4114; found, 587.3860.

83
2.11.9. 5,5,10,10,15,15,20,20-Tetrakis spirocyclohexyl N-confused calix[4]pyrrole (5e)
Physical state: white solid
Yield: 600 mg (20%)
mp 223 ºC (lit28-223.2-223.6 ºC)
Rf = 0.42 SiO2, 1:1 petroleum ether/chloroform)
IR (KBr pellet, cm-1
): 3468 (br, pyrrole NH), 1423, 1280
1H NMR (400 MHz, CDCl3) δ=1.20-1.60 (m, 24H, cyclohexyl, CH), 1.70-2.10 (m,
16H, cyclohexyl, CH), 5.50 (br, 1H; pyrrole β-H), 5.82 (m, 2H; pyrrole β-H), 5.97 (br,
2H; pyrrole β-H), 6.03 (br, 2H; pyrrole β-H), 6.42 (d, J =1.97 Hz, 1H; pyrrole α-H),
7.10 (br, 2H; pyrrole NH), 7.44 (brs, 1H; pyrrole NH), 7.63 (brs, 1H, pyrrole NH)
13
C NMR (100 MHz, CDCl3, TMS): δ = 22.77, 25.91, 25.99, 26.36, 36.51, 37.0, 37.4,
37.67, 38.36, 39.31, 39.70, 39.72, (hexyl C), 101.3, 102.2, 103.9 (pyrrole β-CH), 112.8
(pyrrole α-CH), 130.1 (pyrrole β-C), 133.7, 134.6, 136.5, 137.8, 139.1, 140.3 (pyrrole
α-C)
(ESI-MS) for C40H52N4 [M+H]- : calcd, 587.4114; found, 587.3860.
2.11.10. Synthesis of 5,10,15,20-tetrakis spirocycloheptyl calix[4]pyrrole
Physical state: white solid
Yield: 2.5 g (76%)
Mp: 163 ˚C
Rf: .67 (SiO2, 1:1 petroleum ether/chloroform))
1H NMR (400 MHz, CDCl3, TMS): δ 1.52-167 (m, 32H, cyclo-heptyl, CH); 1.92-2.02
(m, 16H, cyclo-heptyl, CH), 5.84 (d, 8H, β-pyrrolic), 6.87 (brs, 4H, NH);
13C NMR (100 MHz, CDCl3, TMS): δ 23.12, 29.77, 39.33 (cycloheptyl); 42.69 (meso
C); 102.88 (β-pyrrolic CH); 138.35 (α-pyrrolic CH)
(ESI-MS) for C44H60N4 [M+H]- = calcd, 644.4818; found, 645.4824.

84
2.11.11. Synthesis of 5,10,15,20-tetrakis spirocycloheptyl N-confused calix[4]pyrrole
Physical state: white solid
Yield: 382 mg 10%
Rf: 0.49 (SiO2, 1:1 petroleum ether/chloroform))
1H NMR (400 MHz, CDCl3, TMS): δ 1.46-1.67 (m, 32H, cyclo-heptyl, CH); 2.00-
2.07 (m, 24H, cyclo-heptyl, CH); 5.18 (brs, 1H; pyrrole β-H), 5.74-5.95 (m, 6H; pyrrole
β-H), 6.41 (d, J =1.97 Hz, 1H; pyrrole α-H), 6.82 (br, 1H; pyrrole NH), 6.96 (brs, 1H;
pyrrole NH), 7.52 (brs, 1H, pyrrole NH); 7.65 (brs, 1H, pyrrole NH)
13C NMR (100 MHz, CDCl3, TMS): δ = 23.04, 23.30, 29.68, 29.87, 30.01, 38.64, 40.78,
42.19, 42.46, (heptyl C), 100.11, 101.69, 102.27, 104.48 (pyrrole β-CH), 114.63 (pyrrole
α-CH), 137.09 (pyrrole β-C), 138.43, 139.29, 139.60, 141.06, 143.05 (pyrrole α-C)
(ESI-MS) for C44H60N4 [M+H]- = calcd, 644.4819; found, 645.4820.
2.11.12. Synthesis of 2-hydroxymethylpyrrole
A three-necked flask equipped with magnetic stirrer pyrrole-2-carbaldehyde (500mg,
5.25mmol) was dissolved in THF (15mL) under nitrogen atmosphere. Sodium
borohydride (208mg, 5.5 mmol) was added to the solution in 2-3 fractions resulting in
warming of the reaction flask and gas evolution. The mixture was stirred for 4h until the
reaction was complete. Completion of the reaction was monitored by TLC (n-
hexane/ethyl acetate 1:1) and after completion of the reaction the mixture was treated
with water (25 mL). After the NaBH4 in suspension was completely dissolved, the
mixture was extracted with dichloromethane (4×50 mL). The combined organic extracts
were washed with water and dried over Na2SO4. Triethylamine (2 drops) was added and
the solution was concentrated with a rotary evaporator to yield light-yellow oil. The
yellow oil was further purified by vacuum distillation (90 ˚C) to give a final product as
a colorless liquid and requires storage in the refrigerator to avoid polymerization.
Yield: 80 (400µl).
Rf = 0.32 (SiO2, 1:1 n-Hexane: ethylacetate);
1H NMR (400 MHz, CDCl3) δ= 7.98 (brs, 1H, NH), 7.68 (m, 2H, α-pyrrole), 7.25 (m,
2H, β-pyrrole), 6.95 (d, 2H, β-pyrrole), 4.93 (s, 2H, -CH2OH), 2.43 (s, 1H, OH).

85
2.11.13. Synthesis of meso-unsubstituted porphyrinogen and N-confused
porphyrinogen
Tetrabutylammonium fluoride (.03mmol) was dissolved in the solution of 2-
hydroxymethylpyrrole (195 mg, 2mmol) in deuterated chloroform 2ml. Reaction
mixture was stirred for 2 minutes under nitrogen atmosphere and then methanesulfonic
acid (10µl, 0.1mmol) was added to the mixture. Reaction was stirred at room
temperature for further 2 minutes and completion of reaction was monitored by TLC
(1:1, n-hexane:chloroform). After completion of the reaction the mixture was directly
given for the 1H NMR analysis.
1H NMR (400 MHz, CDCl3) δ= 9.22 (brs, 4H, NH), 8.96 (brs, 1H, NH), 8.77 (brs, 1H,
NH), 8.60 (brs, 1H, NH), 6.64 (d, 2H, J= 1.64 Hz, β-pyrrole), 6.03 (m, β-pyrrole), 5.22
(s, 1H, α-pyrrolic) 4.45 (s, meso-CH2).

86
2.13. References
1 Park, C. H.; Simmons, H. E. Macrobicyclic amines. 111. encapsulation of
halide ions by in,in-l,(k + 2)-diazabicyclo[k.l.m]alkaneammonium ions J.
Am. Chem. Soc. 1968, 90, 2431-2432.
2 (a) Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives; VCH,
Weinheim, 1995. (b) Steed, J. W.; Atwood, J. L. Supramolecular chemistry:
An introduction, Wiley, Chichester, 2000.
3 (a) Gale, P. A.; García-Garrido, S. E.; Garric, Anion receptor based on organic
frameworks: highlights from 2005 and 2006., J. Chem. Soc. Rev. 2008, 37,
151-190. (b) Gale, P. A.; Quesada, R. Anion coordination and anion-
templated assembly: highlights from 2002 to 2004, Coord. Chem. Rev. 2006,
250, 3219-3244.
4 (a) Beer, P. D.; Gale, P. A. Anion recognition and sensing: the state of the art
and future perspectives, Angew. Chem., Int. Ed. 2001, 40, 486-516. (b)
Caltagirone, C.; Gale, P. A. Anion receptor chemistry: highlights from 2007,
Chem. Soc. Rev. 2009, 38, 520-563. (c) Martinez-Manez, R.; Sancenan, F.
Fluorogenic and chromogenic chemosensors and reagents for anions, Chem.
Rev. 2003, 103, 4419-4476.
5 Gale, P. A.; Sessler, J. L.; Kral, V.; Lynch, V. Calix[4]pyrroles: old yet new
anion-binding agents, J. Am. Chem. Soc. 1996, 118, 5140-5141.
6 Custelcean, R.; Delmau, L. H.; Moyer, B. A.; Sessler, J. L.; Cho, W.-S.; Gross,
D.; Bates, G. W.; Brooks, S. J.; Light, M. E.; Gale, P. A. Calix[4]pyrrole: an
old yet new ion-pair receptor, Angew. Chem., Int. Ed. 2005, 44, 2537-2542.
7 Wintergerst, M. P.; Levitskaia, T. G.; Moyer, B. A.; Sessler, J. L.; Delmau, L.
H. Calix[4]pyrrole: a new ion-pair receptor as demonstrated by
liquid−liquid extraction, J. Am. Chem. Soc. 2008, 130, 4129-4139.
8 Beayer, A. Ueber ein condensation product von pyrrol mit acetone, Ber.
Dtsch. Chem. Ges., 1886, 19, 2184-2185.

87
9 Chelintzev, V. V.; Tronov, B. V. J. Process of condensation of pyrrole and
acetone. Constitution of the resulting products, Russ. Phys. Chem. Soc. 1916,
48, 105-155.
10 Rothemund, P.; Gage, C. L. Concerning the structure of ‘‘acetonepyrrole”, J.
Am. Chem. Soc. 1955, 77, 3340-3342.
11 Brown, W. H.; Hutchinson, B. J.; MacKinnon, M. H. The condensation of
cyclohexanone with furan and pyrrole, Can. J. Chem. 1971, 49, 4017-4022.
12 Sessler J. L. and Gale, P.A. The Porphyrin Handbook, in: K.M. Kadish, K.M.
Smith, R. Guilard (Eds.), vol. 6, Academic Press, San Diego, CA, 2000, pp.
257-278.
13 van Hoorn, W. P.; Jorgensen, W. L. Selective anion complexation by a
calix[4]pyrrole investigated by monte carlo simulations, J. Org. Chem. 1999,
64, 7439-7444.
14 C. Floriani, The porphyrinogen-porphyrin relationship: the discovery of
artificial porphyrins, Chem Commun., 1996, 1257-1263.
15 P. A. Gale, J. L. Sessler and V. Kral, Calixpyrrole, Chem Commun., 1998, 1-8.
16 P. A. Gale, J. L. Sessler, V. Lynch and P. I. Sansom, Synthesis of a new
cylindrical calix[4]arenexalix[4]pyrrole pseudo dimer, Tetrahedron Lett.
1996, 37, 7881-7884.
17 Shao, S. J.; Yu, X. D.; Cao, S. Q. Synthesis of calix[4]pyrroles: A class of new
molecular receptor. Chin. Chem. Lett. 1999, 10, 193-194.
18 Dey, S.; Pal, K.; Sarkar, S. An efficient and eco-friendly protocol to
synthesize calix[4]pyrroles, Tetrahedron Lett., 2006, 47, 5851-5854.
19 Farfan, I. M.; Celedon, C. C.; Verduzco, J. A.; Garcia, L. C. An efficient
synthesis of calix[4]pyrroles under lewis acid conditions. Org. Chem. Lett.,
2008, 5, 237-239.
20 Shriver, J. A.; Westphal, S. G. Calix[4]pyrrole: synthesis and anion-binding
properties. an organic chemistry laboratory experiment. J. Chem. Educ.,
2006, 83, 1330-1332.

88
21 Raghavan, K. V. Kulkarni, S. J. Radhakishan, M. Srinivas, N. U.S. Patent,
6605194, 2003.
22 Kishan, M. R.; Srinivas, N.; Raghavan, K. V.; Kulkarni, S. J.; Sarma, A. R. P.;
Vairamani, M. A novel, shape-selective, zeolite-catalyzed synthesis of
calix(4)pyrroles. Chem. Commun., 2001, 2226-2227.
23 Kishan, M. R.; Rani, V. R.; Kulkarni, S. J.; Raghavan, K. V. A new
environmentally friendly method for the synthesis of calix(4)pyrroles over
molecular sieve catalysts. J. Mol. Cat.s A: Chem. 2005, 237, 155-160.
24 Chauhan, S. M. S.; Mishra, S. Graphite oxide and graphene oxide in the
synthesis of dipyrromethane and calix[4]pyrrole, Molecules, 2011, 16, 7256-
7266.
25 Chmielewski, P. J. Latos-Grazynski, L. Rachlewicz, K. Glowiak, T. Tetra-p-
tolylporphyrin with an inverted pyrrole ring: a novel isomer of porphyrin,
Angew. Chem. Int. Ed. 1994, 33, 779-781.
26 Furuta, H.; Asano, T.; Ogawa, T. Exploring the fate of water molecules
striking concentrated sulfuric acid: scattering versus solvation, J. Am.
Chem. Soc. 1994, 116, 779-780.
27 Tsuge, O. Tashiro, M. Kiryu, Y. A new porphyrinogen type of compound,
Org. Prep. Proc. Int. 1975, 7, 39-42.
28 Depraetere, S.; Smet, M.; Dehaen, W. N-Confused calix[4]pyrroles, Angew.
Chem. Int. Ed. 1999, 38, 3359-3361.
29 (a) Allen, W. E.; Gale, A.; Brown, C. T.; Lynch, V.; Sessler, J. L. Binding of
neutral substrates by calix[4]pyrroles, J. Am. Chem. Soc. 1996, 118, 12471-
12472. (b) Anzenbacher, P. Jr.; Try, A. C.; Miyaji, H.; Jursıkova, K.; Lynch, V.
M.; Marquez, M.; Sessler, J. L. Fluorinated calix[4]pyrrole and
dipyrrolylquinoxaline: neutral anion receptors with augmented affinities
and enhanced selectivities, J. Am. Chem. Soc., 2000, 122, 10268-10272.
30 Wu, Y-D.; Wang, Di-F.; Sessler, J. L. Conformational features and anion-
binding properties of calix[4]pyrrole: a theoretical study, J. Org. Chem.
2001, 66, 3739-3746.

89
31 Wanga, D-F.; Wub, Y. Density functional theory studies of β-substituent
effect on conformational preference and anion binding ability of
calix[4]pyrroles, Arkivoc, 2004, 96-110.
32 Anzenbacher, Jr. P.; Jursikova, K.; Lynch, V. M.; Gale, P. A.; Sessler, J. L.
Calix[4]pyrroles containing deep cavities and fixed walls synthesis,
structural studies, and anion binding properties of the isomeric products
derived from the condensation of p-hydroxyacetophenone and pyrrole, J.
Am. Chem. Soc. 1999, 121, 11020-11021.
33 Chauhan, S. M. S.; Kumari, P.; Sinha, N.; Chauhan, P. Isolation, synthesis and
biomimetic reactions of metalloporphyrinoids in ionic liquids, Curr. Org.
Synth., 2011, 8, 393-437.
34 Sessler, J. L.; Cai, J.; Gong, H-Y.; Yang, X.; Arambula, J. F.; Hay, B. P. A
pyrrolyl-based triazolophane: a macrocyclic receptor with CH and NH
donor groups that exhibits a preference for pyrophosphate anions, J. Am.
Chem. Soc., 2010, 132, 14058-14060.
35 Kim, S. K.; Sessler, J. L.; Gross, D. E.; Lee, C-H.; Kim, J. S.; Lynch, V. M.;
Delmau, L. H.; Hay, B. P. A calix[4]arene strapped calix[4]pyrrole: an ion-
pair receptor displaying three different cesium cation recognition modes, J.
Am. Chem. Soc., 2010, 132, 5827-5836.
36 Dehaen, W. Anion recognition by N-confused calix[4]pyrrole-a-
carbaldehyde and its Knoevenagel reaction derivatives, New. J. Chem.,
2007, 31, 691-696.
37 Buranaprasertsuk, P.; Tangsakol, Y.; Chavasiri, W. Epoxidation of alkenes
catalyzed by cobalt(II) calix[4]pyrrole, Catal. Commun., 2007, 8, 310-314.
38 Kral, V.; Gale, P. A.; Anzenbacher, P., Jr.; Jursikova, K.; Lynch, V.; Sessler, J.
L. Calix[4]pyridine a new arrival in the heterocalixarene family, Chem.
Commun. 1998, 9-10.
39 Miyazu, H.; Anzenbacher, P.; Sessler, J. L.; Bleasdale, E. R.; Gale, P. A.
Anthracene-linked calix[4]pyrroles: fluorescent chemosensors for anions,
Chem. Commun. 1999, 1723-1724.

90
40 Gale, P. A.; Twyman, L. J.; Handlin, C. I.; Sessler, J. L. A colourimetric
calix[4]pyrrole: 4-nitrophenolate based anion sensor, Chem. Commun. 1999,
1851-1852.
41 Gale, P. A.; Hursthouse, M. B.; Light, M. E.; Sessler, J. L.; Warriner, C. N.;
Zimmerman, R. S. Ferrocene-substituted calix[4]pyrrole: a new
electrochemical sensor for anions involving CH···anion hydrogen bonds,
Tetrahedron Lett. 2001, 42, 6759-6762.
42 He, L. J.; Cai, Q. S.; Shao, S. J.; Jiang, S. X. Effect of calix[4]pyrrole as
addition reagent on anion separation in capillary zone electrophoresis
(CZE), Chin. Chem. Lett. 2001, 12, 511-512.
43 Gale, P. A. From anion receptors to transporters. Acc. Chem. Res. 2011, 44,
216-226.
44 Hancock, L. M.; Beer, P. D. Sodium and barium cation-templated synthesis
and cation-induced molecular pirouetting of a pyridine N-oxide containing
[2]rotaxane, Chem. Commun., 2011,47, 6012–6014.
45 (a) Hoss, R.; Vkgtle, F. Template synthesis, Angew. Chem. Int. Ed. Engl. 1994,
33, 375-384. (b) Templated Organic Synthesis (Ed.: F. Diederich, P. J. Stang),
Wiley-VCH, Weinheim, 2000.
46 Moyer, B. A.; Bonnesen, P. V. Bonnesen in supramolecular chemistry of
anions (Eds.: A. Bianchi, K. Bowman-James, E. GarcTa- EspaUa), Wiley-
VCH, Weinheim, 1997, 1-41.
47 Gimeno, N.; Vilar, R. Anion as templates in coordination and
supramolecular chemistry. Coord. Chem. Rev., 2006, 250, 3161-3189.
48 An, H.; Xu, T.; Zheng, H.; Han, Z. A New double-keggin-anion-templated
organic-inorganic hybrid architecture: synthesis crystal structure and
photo luminescence. Inorg. Chem. Commun., 2010, 13, 302-305.
49 Vilar, R. Anion-templated synthesis Angew. Chem. Int. Ed. 2003, 42, 1460-
1477.

91
50 (a) Alcalde, E.; Ramos, S.; Pwrez-Garcta, L. Anion template-directed
synthesis of dicationic [14]imidazoliophanes, Org. Lett., 1999, 1, 1035-1038;
(b) Alcalde, E.; Mesquida, N.; Perez-Garcia, L.; Alvarez-Rua, C.; Garcia-
Granda, S.; Garcia-Rodriguez, E. Hydrogen bonded driven anion binding by
dicationic [14]imidazoliophanes, Chem. Commun. 1999, 295-296.
51 Turner, B.; Shterenberg, A.; Kapon, M.; Suwinska, K.; Eichen, Y. The role of
template in the synthesis of meso-hexamethylmeso-hexaphenyl-
calix[6]pyrrole: trihalogenated compounds as templates for the assembly of
a host with a trigonal cavity. Chem. Commun., 2002, 404-405.
52 Garg, B.; Bisht, T.; Chauhan, S. M. S. meso-Functional calix[4]pyrrole: a
solution phase study of anion directed self-assembly. J Incl Phenom
Macrocycl Chem DOI 10.1007/s10847-010-9849-6.
53 Kumar, A.; Ahmad, I.; Rao, M. S. Ytterbium(III) triflate catalyzed synthesis
of calix[4]pyrroles in ionic liquids. Can. J. Chem., 2008, 86, 899-902.
54 Chauhan, S. M. S.; Garg, B.; Bisht, T. Syntheses of calix[4]pyrroles by
amberlyst-15 catalyzed cyclocondensations of pyrrole with selected ketones.
Molecules, 2007, 12, 2458-2466.