chapter 4 mechanisms of organic reactions -...
TRANSCRIPT

Chapter 4 Mechanisms of Organic Reactions

Table of Contents
108
Introduction & Motivation
1. Carbon as the Basis of Organic Chemistry
2. The Nature of the Covalent Bond 2.1. Atomic Orbitals and Hybridisation 2.2. Fomation of Single Bonds 2.3. Formation of Multiple Bonds 2.4. Electron Delocalization, Aromaticity, and Resonance Structures
3. Molecular Structure 3.1. Basic Rules of Nomenclature 3.2. Isomerism
4. Mechanisms of Organic Reactions 4.1. Reaction Thermodynamics and Kinetics 4.2. Nucleophilic Substitution Reactions 4.3. Electrophilic Additions 4.4. Electrophilic Substitutions 4.5. Eliminations 4.6. Radical Reactions
5. Selected Classes of Organic Compounds Relevant for Materials Science
1 h
1 h
8 h 2 h 1 h 1 h 4 h 4 h 2 h 2 h 8 h 1 h 3 h 1 h 1 h 1 h 1 h 6 h

4.1 Thermodynamics, Kinetics, and Categorization
of Organic Reactions

Net Reaction and Mechanism
110
acid alcohol water
net reaction
reaction mechanism
starting materials products
elementary steps
catalyst
RO
OH+ HO R' R
O
O+
R'H2O
H
ROH
OH
H
HO R'R
OH
OHOH
R'R
O
OHOR'
HH
RO
OH
R'
+ H–
H~+ H2O–
ester
• net reaction describes the starting materials and the products of a reaction • reaction mechanisms describes the individual elementary steps of the reaction • catalyst takes part in the reaction mechanism but is retained unchanged

• reaction thermodyamics are concerned with the overall energy balance of chemical reactions
Thermodynamics of Chemical Reactions
111
• Gibbs’ free reaction energy ΔGR determines whether and in which direction the reaction runs • standard Gibbs’ free reaction energy ΔG°R at standard conditions (1 bar, 25°C, all reactants 1 mol/L)
RO
OH+ HO R' R
O
O+
R'H2O
¢GR =¢G�R +RT ln
[R–COOR’][H2O][R–COOH][R’–OH]
¢GR > 0
¢GR < 0
¢GR = 0
exergonic reaction, runs from left to right
endergonic reaction, runs from right to left
reaction is in equilibrium

• all chemical reactions in a closed system progress until they reach the thermodynamic equilibrium
The Chemical Equilibrium
112
• equilibrium constant KR is the ratio of reactant concentrations in equilibrium • standard free reaction energy ΔG°R determines the position of the equilibrium (at given temperature)
¢GR =¢G�R +RT ln
[R–COOR’]eq[H2O]eq
[R–COOH]eq[R’–OH]eq
= 0
KR =[R–COOR’]eq[H2O]eq
[R–COOH]eq[R’–OH]eq
pKR =� logKR
¢G⇥R =�RT lnKR pKR /
¢G™R
RT
RO
OH+ HO R' R
O
O+
R'H2O

• Gibbs-Helmoltz equation dissects free reaction enthalpy into enthalpic and entropic contribution
Reaction Enthalpy and Entropy
113
• standard reaction enthalpy ΔH°R is negative (advantageous) if bond energies in products are higher • standard reaction entropy ΔS°R is positive (advantageous) if the disorder of the system increases
Gibbs-Helmholtz Equation¢G⇥R =¢H⇥
R �T¢S⇥R
exothermic reactions, sum of all bond energy changes negative
endothermic reactions, sum of all bond energy changes positive
exotropic reactions, disorder, degrees of freedom decrease
endotropic reactions, disorder, degrees of freedom increase¢S�R > 0
¢S�R < 0
¢H�R > 0
¢H�R < 0
RO
OH+ HO R' R
O
O+
R'H2O

• reaction kinetics describe “how fast” reactions proceed from the initial state towards the equilibrium
Kinetics of Chemical Reactions
114
• reaction rates r = dci/dt describe the change of the reactant / product concentrations ci over time • rate laws describe the relation between reaction rates ri and substrate concentrations ci
• rate laws are differential equations, solutions (by integration) are polynomial or exponential functions
E
t
RO
OH+ HO R' R
O
O+
R'H2O
r = = −k ⋅ c(t ⋅ c(t = −k ⋅ c(t dc(t)RCOOH
dt)RCOOH )ROH )2
RCOOH
r = ∫ = −k ∫ dt dc(t)RCOOH
c(t)2RCOOH
c(t = )RCOOH
11 + kc(0 t)RCOOH
cRCOOR’ = cH2O
cRCOOH = cR’OH

• reaction rates r proportional to the product of all reactant concentrations according to their molecularity • proportionality factor is called rate constant k
Reaction Order and Molecularity of Chemical Reactions
115
• molecularity is the number of molecules of each type actually involved in an elementary reaction • reaction order is the sum of all exponents of the concentrations of all reactants in the rate law • for simple, single-step reactions, the molecularity strictly determines the reaction order
A B
A + B C + D
A + B + C D
r1r = k1r · [B ]r1f = k1f · [A]
r2r = k2r · [C ][D]r2f = k2f · [A][B ]
r3f = k3f · [A][B ][C ]
r4f = k4f · [A]2[C ]
r3r = k3r · [D]
first order monomolecular monomolecular first order
second order bimolecular bimolecular second order
third order trimolecular monomolecular first order
third order trimolecular bimolecular second orderr4r = k4r · [C ]22 A + B 2 C

• in thermodyamic equilibrium, concentrations of all reactants / products do not change anymore • hence, the rates of forward and reverse reactions must be equal
Relation of Reaction Theromdynamics and Kinetics
116
• ratio of rate constants of forward and reverse reactions determines equilbrium constant K • the faster the forward (relative to the the reverse) reaction, the larger is K • the faster the forward (relative to the the reverse) reaction, the more the equilibrium is on product side
A + B C + D r2r = k2r · [C ][D]r2f = k2f · [A][B ]
r2f = r2r
k2f · [A][B ] = k2r · [C ][D]
k2fk2r
= [C ][D][A][B ]
= K
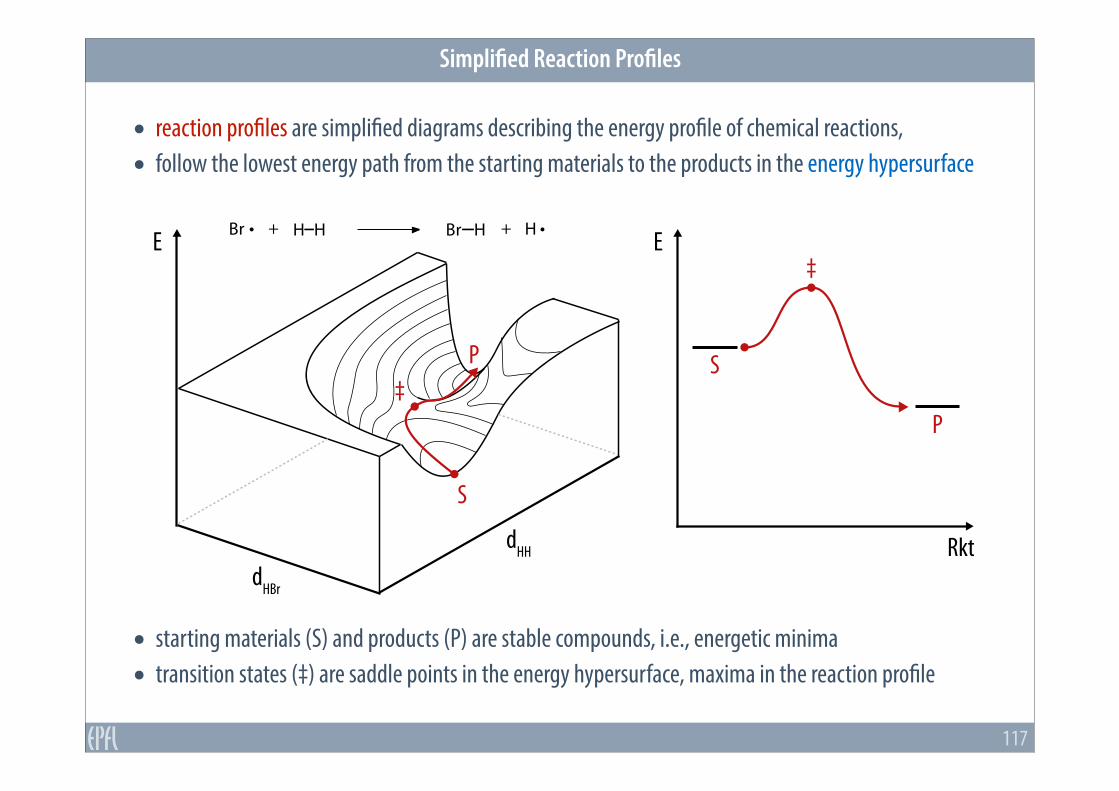
• reaction profiles are simplified diagrams describing the energy profile of chemical reactions, • follow the lowest energy path from the starting materials to the products in the energy hypersurface
Simplified Reaction Profiles
117
• starting materials (S) and products (P) are stable compounds, i.e., energetic minima • transition states (‡) are saddle points in the energy hypersurface, maxima in the reaction profile
P‡
S
E
dHBr
dHH
Br + H H H+Br H
S
‡E
Rkt
P

• reaction profiles are simplified diagrams describing the energy profile of chemical reactions, • follow the lowest energy path from the starting materials to the products in the energy hypersurface
Simplified Reaction Profiles
118
• starting materials (S) and products (P) are stable compounds, i.e., energetic minima • transition states (‡) are saddle points in the energy hypersurface, maxima in the reaction profile
P‡
S
E
dHBr
dHH
Br + H H H+Br H
‡
S
E
Rkt
P

• reaction profiles illustrate both thermodynamics and kinetics of chemical reactions
Relation of Reaction Profiles, Thermodynamics, and Kinetics
119
• standard free reaction energy ΔG° is difference between (S) and (P) energies • ΔG° is also equal to difference between free transition energies ΔG‡ pf forward and reverse reaction • reaction rates k depends on activation energies EA of chemical reactions (approximately equal to ΔG‡)
‡
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
∆Gf‡ < ∆Gr
‡
KR =k f
kr
E A,r ⇥¢G‡r =�RT lnkr
E A, f ⇥¢G‡f =�RT lnk f
exergonic, fast
¢G⇥R =¢G‡
f �¢G‡r
endergonic, slow
¢G⇥R =�RT lnKR

• molecules at a given temperature T have energies according to the Boltzmann probability distribution p
Reaction Kinetics and Thermal Energy
120
• at increasing temperature, an increasingly large fraction of molecules overcomes activation energy EA
• both forward and reverse reaction are accelerated, but more so the forward reaction (if exergonic)
p
E
T1
T2
T3
p(E) = exp( ) ( )8kT
3/2( )E
π
1/2 – E
kT
Rkt
S
E
P
∆Gf° = – ∆Gr° < 0
‡

• a more exergonic reaction will be more shifted towards the product side
Kinetic Interpretation of the Equilibrium
121
• ratio of activation energies changes, forward reaction accelerated, reverse reaction decelerated • for given temeprature, larger/smaller fraction of molecules has energy >EA of forward/reverse reaction
¢G⇥R =�RT lnKR
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
‡
Rkt
S
E
P
∆Gf° = – ∆Gr° < 0
‡
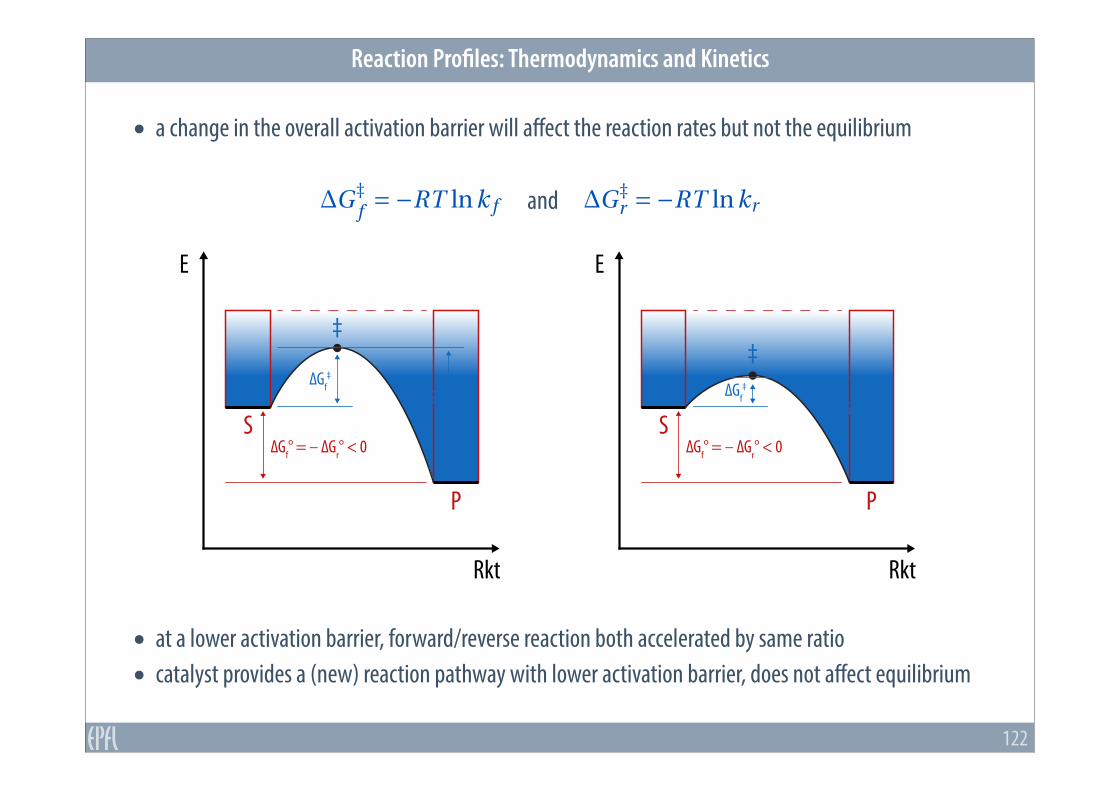
• a change in the overall activation barrier will affect the reaction rates but not the equilibrium
Reaction Profiles: Thermodynamics and Kinetics
122
• at a lower activation barrier, forward/reverse reaction both accelerated by same ratio • catalyst provides a (new) reaction pathway with lower activation barrier, does not affect equilibrium
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
‡∆Gf
‡ ∆Gr
‡
¢G‡f =�RT lnk f ¢G‡
r =�RT lnkrand
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
‡
∆Gf‡
∆Gr‡

• if the activation barrier id far above the thermal energy, the equilibrium cannot be established
Metastable States
123
• for “very high” activation barriers, both forward and reverse reaction become infinitesimally slow • even higher energy reactants are “kinetically stable”, “kinetically trapped”, “metastable”
metastable
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
∆Gf‡
∆Gr‡
‡
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
‡
∆Gf‡
∆Gr‡
¢G‡f =�RT lnk f ¢G‡
r =�RT lnkrand
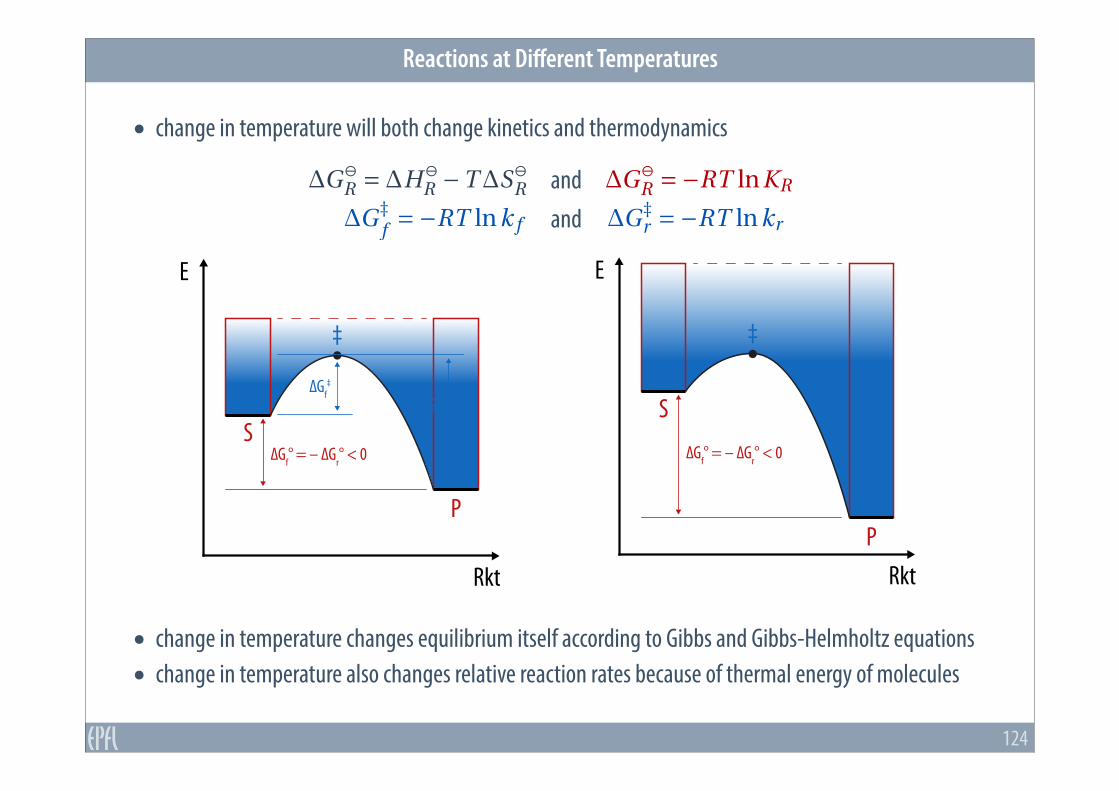
• change in temperature will both change kinetics and thermodynamics
Reactions at Different Temperatures
124
• change in temperature changes equilibrium itself according to Gibbs and Gibbs-Helmholtz equations • change in temperature also changes relative reaction rates because of thermal energy of molecules
S
E
Rkt
P
∆Gf° = – ∆Gr° < 0
‡
∆Gf‡
∆Gr‡
Rkt
S
E
P
∆Gf° = – ∆Gr° < 0
‡
¢G‡f =�RT lnk f ¢G‡
r =�RT lnkrandand ¢G⇥
R =�RT lnKR¢G⇥R =¢H⇥
R �T¢S⇥R

• Polanyi Principle and Hammond Postulate for mechanistically similar, single-step reactions
Hammond Postulate and Polanyi Principle
125
• Polanyi Principle: difference in activation energies proportional to difference in free reaction energies • Hammond Postulate: energetically more similar states are also geometrically more similar
‡1
‡2
‡3
S
E
Rkt
P1
P2
P3
∆G3f° > ∆G2f° = 0 > ∆G3f°
∆G3f‡ > ∆G2f
‡ = ∆G2r‡ > ∆G3f
‡
exergonic
endergonic
“late” transition state higher activation energy
“early” transition state lower activation energy
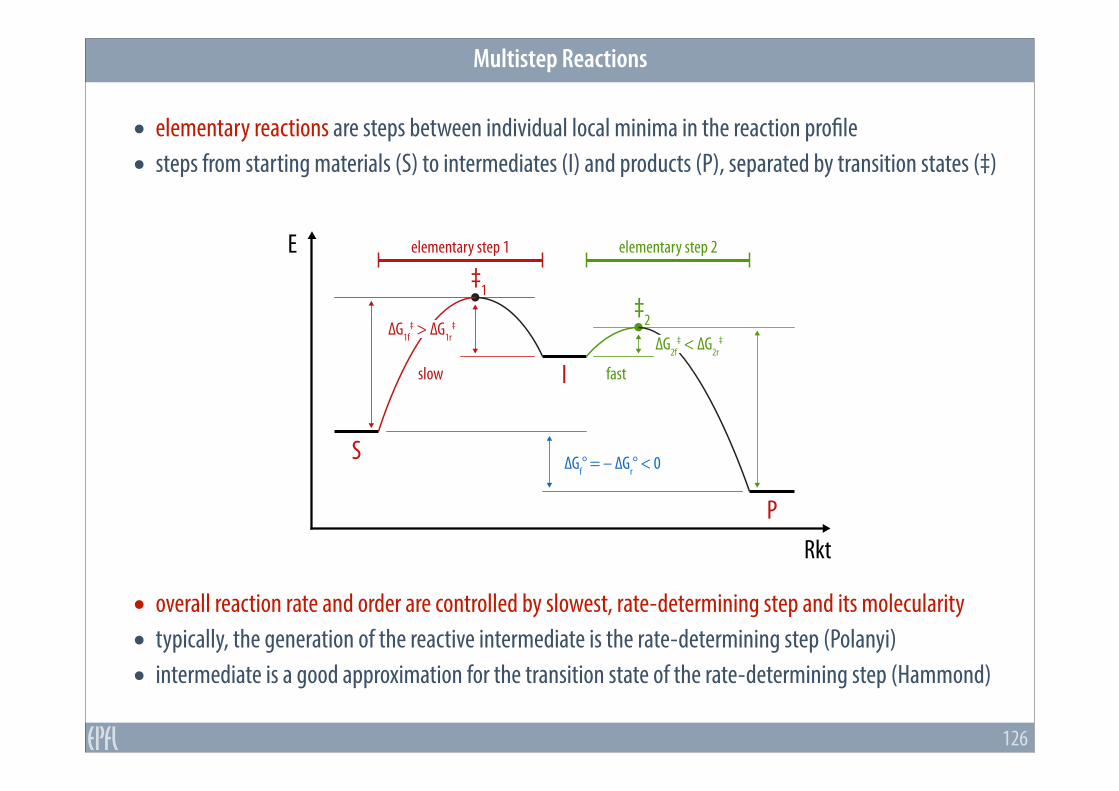
• elementary reactions are steps between individual local minima in the reaction profile • steps from starting materials (S) to intermediates (I) and products (P), separated by transition states (‡)
Multistep Reactions
126
• overall reaction rate and order are controlled by slowest, rate-determining step and its molecularity • typically, the generation of the reactive intermediate is the rate-determining step (Polanyi) • intermediate is a good approximation for the transition state of the rate-determining step (Hammond)
‡1
I
E
Rkt
S
‡2
P
∆Gf° = – ∆Gr° < 0
∆G2f‡ < ∆G2r
‡
elementary step 1 elementary step 2
slow fast
∆G1f‡ > ∆G1r
‡

• classification according to reaction type, i.e., the type of changes to molecular topology
Classification of Organic Reactions
127
R1X R2R3
+ YR1
YR2 R3
+XSubstitution
Substitution
R3R1
R2 R4
Y
X
R2 R3R4
R1+ X + YAddition
Elimination
R3R1
R2 R4+ Y
Y
R2R3R4
R1
Addition
Elimination
R1X Y R3R2
X Y R3R2
R1
Rearrangement
Rearrangement

128

4.2 Nucleophilic Substitutions

Nucleophilic Substitutions (SN Reactions)
130
• reaction of a nucleophile (an electron pair donor) with an electrophilic center (an electron pair acceptor)
R1C LG
R2 R3
+NuR1
CNuR2R3
+ LG
R1
R2R3
R1
R2R3
Nu LG
NuLG– +
nucleophilic substitutionnucleophile leaving group
electrophilic center
SN1 Mechanism: leaving group leaves first (and allows nucleophile to come in subsequently
SN2 Mechanism: nucleophile attacks (and forces leaving group to leave simultaneously)
transition state
intermediate

SN1 Reactions
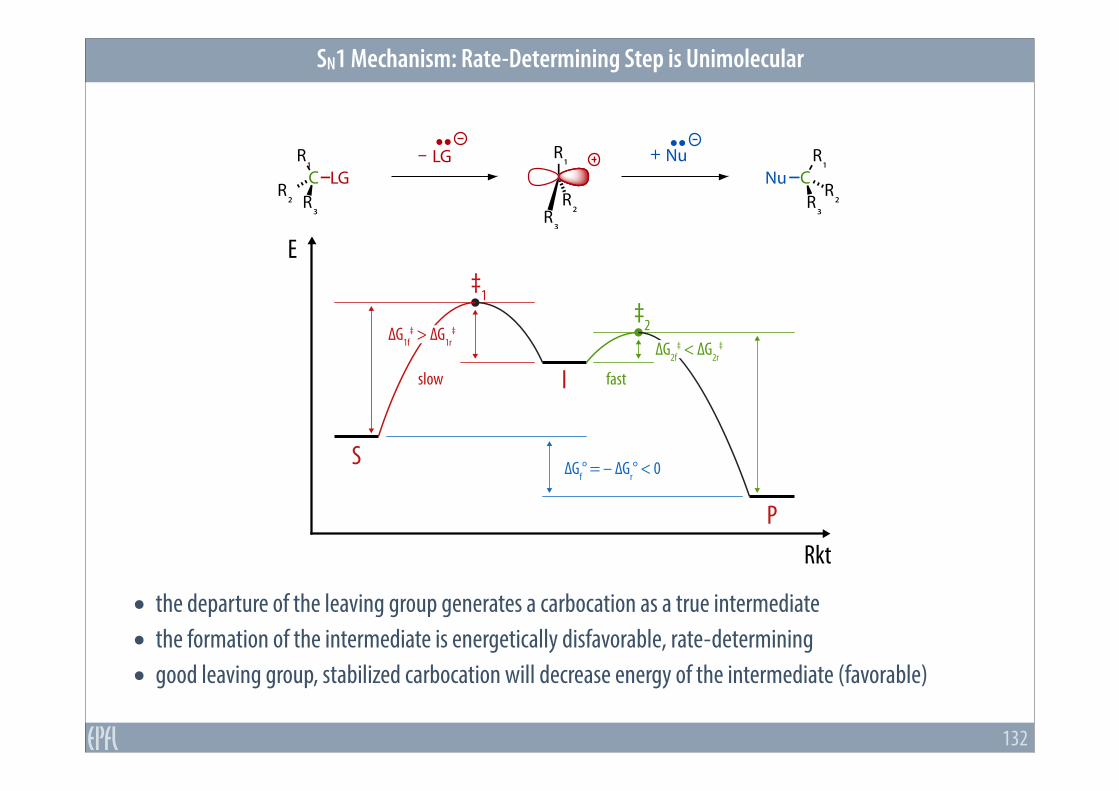
SN1 Mechanism: Rate-Determining Step is Unimolecular
132
• the departure of the leaving group generates a carbocation as a true intermediate • the formation of the intermediate is energetically disfavorable, rate-determining • good leaving group, stabilized carbocation will decrease energy of the intermediate (favorable)
‡1
I
E
Rkt
S
‡2
P
∆Gf° = – ∆Gr° < 0
∆G2f‡ < ∆G2r
‡
slow fast
∆G1f‡ > ∆G1r
‡
R 1C LG
R2 R 3
R 1CNu
R2R 3
R 1
R2R 3
LG– Nu+

• if the electrophilic center is a stereocenter, and the starting material is a pure enantiomer:
SN1 Mechanism: Loss of Stereochemical Information
133
• the departure of the leaving group generates a carbocation that is planar, achiral, sp2-hybridized • attack of the incoming nucleophile can occur from any side with equal probability • product still contains an electrophilic center that is a stereocenter; but is generated as a racemic mixture
R1C LG
R2 R3
R1CNu
R2R3
+R1
R2R3
sp3 sp3sp2
LG–
Nu+
R1C Nu
R2 R3
sp3
or
racemic mixturepure enantiomer planar, achiral

• SN1 reactions are cation-anion dissociation reactions very similar to acid-base reactions
Analogy of SN1 Reactions and Acid-Base Reactions
134
• pKA values are a measure of the strength of a Brønsted acid • the lower the pKA value, the more is the equilibrium on the side of the dissociated ions • pKA values of the corresponding acids are therefore a measure for leaving group quality (lower is better)
pK A = –logK A =� log[H+][LG�]
H–LG
R 1LGR 2 R 3
+ LG
R 1
R 2R 3
H LG + LGH
SN1 reaction
acid-base reaction
Brønsted acid
Lewis acid leaving group
conjugate base

• leaving group quality is approximately inverse to the basicity of the corresponding anion • pKA values of the corresponding acids provide a scale to estimate leaving group quality (lower is better)
Leaving Group Quality
135
• residues that correspond to acids with pKA < 0 are excellent leaving groups • residues that correspond to acids with pKA < 10 are good leaving groups • residues that correspond to acids with pKA < 20 are poor leaving groups • residues that correspond to acids with pKA > 20 are not leaving groups at all
OH
5
O
Me>OH
–1
O
CF3>OH
–10
SO
Me>
–15
O
OH SO
CF3
O
>OH
–7
SO O
FHClHBrHIH >>>
–10 –9 –7 3
FH OHH NH2H CH3H> > >
3 16 38 48

Trivial Names and Abbreviations of Important Leaving Groups
136
OR
O
Me>OR
O
CF3>OR SO
Me>
O
OR SO
CF3
O
>OR SO O
OAcROTFAROMsROTfR OTsR
trifluoromethanesulfonate triflate
methanesulfonate mesylate
4-toluenesulfonate tosylate
trifluoroacetate acetate

• the carbocation intermediate is electron-deficient, must be stabilized by electron-donating groups
Stabilization of the Carbocation Intermediate
137
• SN1 reactions very favorable in benzyl or allyl position (in particular with donor atoms) • SN1 reactions also observed on highly substituted sp3 carbons • SN1 reactions never observed in phenyl position (or other sp2 or sp hybridized carbons)
triphenylmethyl trityl
diphenylmethyl
phenylmethyl benzyl
ethenylmethyl allyl
tertiary carbon
secondary carbon
primary carbon
phenyl (sp2)
H
H
H
R R
R
R R
H
R H
H> > > > >
R Si R
R
>>> CH
H>> >
>>
R Sn R
R
>>
>
H
H
D
H
H
>
A
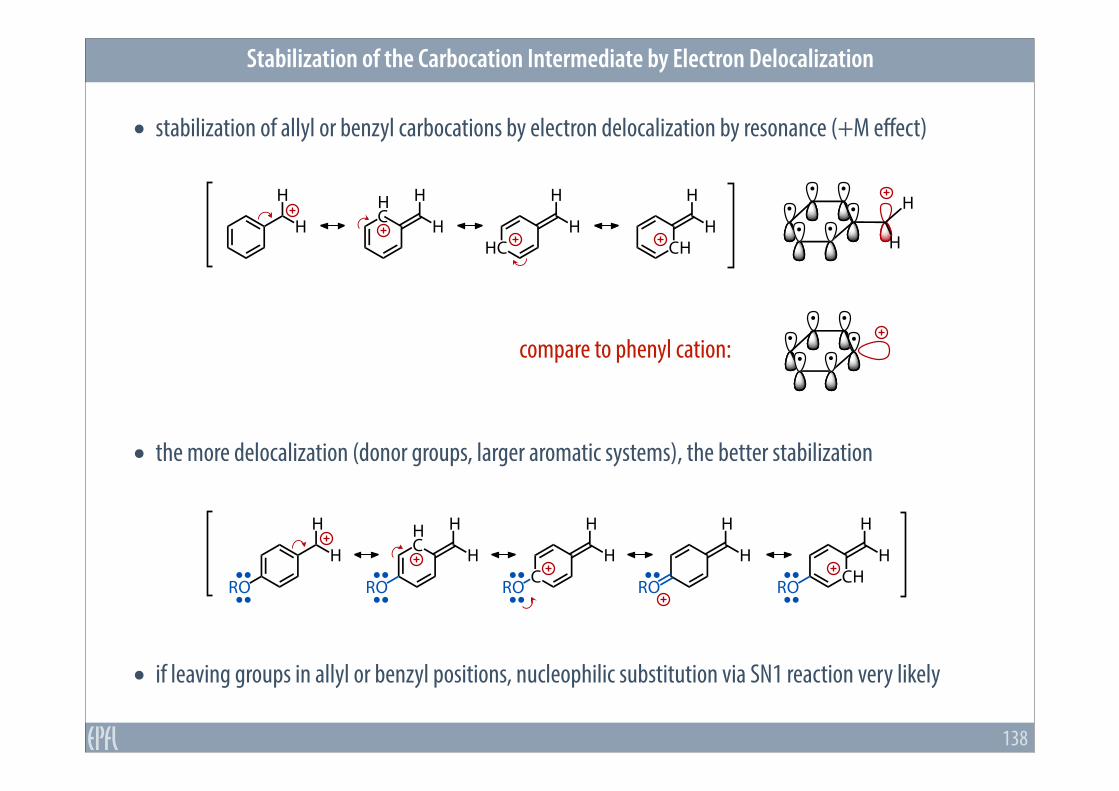
• the more delocalization (donor groups, larger aromatic systems), the better stabilization
• stabilization of allyl or benzyl carbocations by electron delocalization by resonance (+M effect)
Stabilization of the Carbocation Intermediate by Electron Delocalization
138
• if leaving groups in allyl or benzyl positions, nucleophilic substitution via SN1 reaction very likely
H
HHC
H
HHC
H
HCH
H
HH
H
compare to phenyl cation:
H
HHC
H
HC
H
H
H
H
RO RO RO RO CH
H
H
RO

• stabilization by inductive effects (+I effect); conceptual explanation by “hyperconjugation”
Stabilization of the Carbocation Intermediate by Hyperconjugation and on Electropositive Elements
139
• the higher substituted the electrophilic center, the better stabilized is carbocation • in particular, electropositive atoms such as silion are good electrophilic centers for SN1 reactions
R Sn R
R
R Si R
R
R C R
R> >
C CH
HH
δ–
δ+
<
<
<
δ+
R
R< C C
H
HH
R
RC CH
HH
R
RC CH
HH
R
RC CH
HH
R
R
EN 1.9 EN 1.9 EN 2.5
• stabilization by decreasing electronegativity (and size) of cationic center

Examples of SN1 Reactions
140
Cl + MeOH– Cl
+ MeOH OMe
H– H OMe
trityl chloride methanol
good leaving group
excellently stabilized cation
moderate nucleophile
OTf +O
O
CH3NaOAc
–TfO CH2
Br Br
+
Na
OAc
Br
– Na
benzylbromotriflate sodium acetate
excellent leaving group
well-stabilized cation
moderate nucleophile

SN2 Reactions

SN2 Mechanism: Rate-Determining Step is Unimolecular
142
• the attack of the nucleophile cannot result in a stable intermediate (would be pentavalent carbon) • SN2 reactions are single-step reactions that pass through a “pentavalent” transition state • the rate-determining step is hence bimolecular, favored by good nucleophile and electrophilic center
R1C LG
R2 R3
sp3R1
R2R3
Nu LGLG– R1
CNuR2R3
sp3
Nu
‡E
Rkt
S
P
ΔGf° = – ΔGr° < 0
ΔG1f‡ > ΔG1r
‡

• the “pentavalent” transition state is possible because of simultaneous bond formation and cleavage
Molecular Oribtal View of the Reaction and the Transition State
143
• nucleophile electron pair interacts with the empty, antibonding σ* orbital of the C–LG bond • hence back-side attack required, and concerted departure of leaving group inevitable • “early” transition state (more like starting material; Hammond) to avoid “pentavalent” state • good nucleophile (and good leaving group) will favor SN2 reaction
‡σ*
σ
σ*
σ
ENu LG
R1C
R2 R3
LGNuR1
CR2R3
LGNuR1C
R2 R3
Nu LGR1
CR2R3
ΣE/2(S)
(P)

• if the electrophilic center is a stereocenter, and the starting material is a pure enantiomer
Stereochemical Inversion During the SN2 Reaction
144
• due to back-side attack, nucleophile and leaving group on opposite sides of the electrophilic center • transition state has “trigonal-bipyramidal” geometry, R1–R3 in the same plane, then flip to other side • stereoconfiguration is inverted in the process (Walden Umkehr), stereochemical information preserved
pure enantiomer pure enantiomer stereoinversion
R1C LG
R2 R3
sp3R1
R2R3
Nu LGLG– R1
CNuR2R3
sp3
Nu
pure enantiomer

• determination of relative nucleophilicity n according to Pearson:
Nucleophilicity
145
• nucleophiles are also bases; but nucleophilicity is a kinetic parameter, basicity is a thermodynamic one • trends are clear but, different from leaving group quality, there is no simple, logic nucleophilicity scale!
n =� logkNu
kMeOH
kNu
kMeOH
Pearson et al., J. Am. Chem. Soc. 1968, 90, 3319.
Nu + H 3C I CH3Nu + I
MeOH + H 3C I CH3MeO + H I
• nucleophilicity decreases with increasing electronegativity and polarizability (and against basicity)
I > Br Cl > F> R 3C > R2N RO > F>> >>
• anionic nucleophiles always stronger than neutral ones; nucleophilicity decreases with steric hindrance
R2P > R2N R 3P > R 3N>
RS > RO R2S > R2O>
C OR
RR
>C OH
RR
>C OH
RH
>C OH
HH
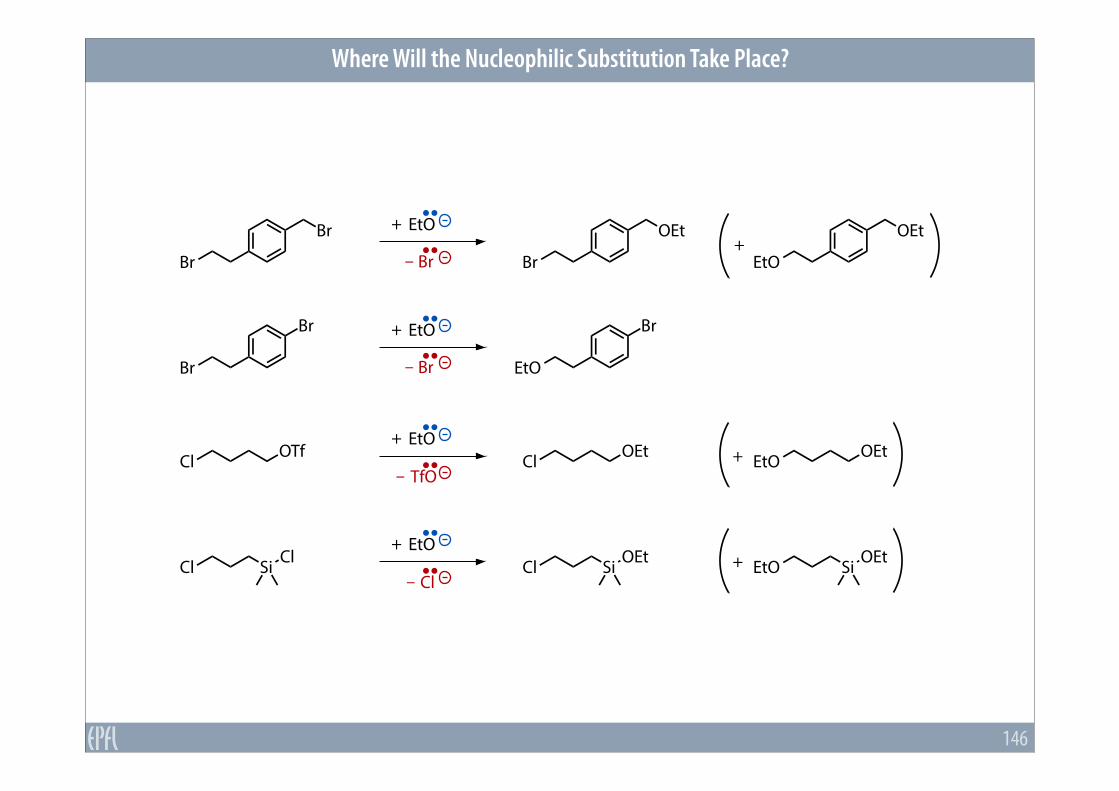
Where Will the Nucleophilic Substitution Take Place?
146
Br
Br
EtO OEt
Br
+
Br
Br
EtO Br
EtO
+
OTfClEtO+
OEtCl
Br–
Br–
TfO–
Si ClClEtO+
Si OEtClCl–
+
+
+
OEt
EtO
OEtEtO
Si OEtEtO

• Williamson synthesis of ethers
Examples for SN2 Reactions
147
• if the reaction proceeds according to the SN2 mechanism, the stereochemistry must be respected
• Gabriel synthesis of primary amines
BrN
O
ON
O
O
NH2
HNHN
O
O
H2N NH2
–
hydrazine
K
–KBr
* *
S-amphetamine
RO OR
Br
HO OH
HO
2 +
pKA 11
pKA 17
+ 2 NEt 3
RO OR
OO
HO
ORRO
base pKA 11
– 2 HNEt 3Br

• consider leaving group quality, stabilization of the carbocation, and nucleophilicity of the nucleophile
Does the Nucleophilic Substitution Follow the SN1 or SN2 Mechanism?
148
• if you decide for a mechanism, give the arguments for your choice • consider explicitly the stereochemical consequences (also in nomenclature of the products if required)
excellent leaving group well-stabilized carbocation
moderate nucleophileSN1 loss of stereochemical information
OTf + NaOAc
– NaOTf
OAc OAc
*
OAc
+ OAc
OAc
*CH3
Cl*
+ NaSAc
– NaCl
OAc
*CH3
SAc*
moderate leaving group non-stabilized carbocation
good nucleophileSN2 inversion of stereoconfiguration
racemic mixture

4.3 Nucleophilic Substitutions on Carbonyl Groups

• carbonyl carbon atoms are inherently very reactive electrophilic centers
Nucleophilic Reactions on Carbonyl Compounds
150
• oxygen (high electronegativity) gives positive partial charge (–I effect) • resonance structures of the C=O π-bond give additional positive formal charge (–M effect) • empty π* molecular orbital (LUMO) has large lobe on carbon that protrudes from molecular plane
R C R
O
R C R
O R
RO
R
RO
π (HOMO) π* (LUMO)
H C H
O
δ+
δ– –
π (HOMO) π* (LUMO)

Nucleophilic Substitution on Carbonyl Compounds: Addition–Elimination Mechanism (SAE)
151
• carbonyl carbons are tetravalent, but sp2 hybridized and therefore coordinatively unsaturated • addition of the nucleophile prior to cleavage of the leaving group is possible, results in real intermediate • attack of the nucleophile results in racemic mixture of intermediates, but no consequences for product
OCNu LGR
+
sp2 sp3
CO
LGR
Nu+
or
OC NuLGR
sp3
LG–
sp2
CO
LGR
Nu+
‡1
I
E
Rkt
S
‡2
P
∆Gf° = – ∆Gr° < 0
∆G2f‡ < ∆G2r
‡
slow fast
∆G1f‡ > ∆G1r
‡

Reactivity of Carbonyl Compounds
152
• amides, acids, and carboxylates have very poor leaving groups, do not easily undergo substitution • aldehydes and ketones have no leaving groups (H, R’) but are in fact reactive for nucleophile addition!
acid halides acid anhydrides ketones aldehydes esters amides acids carboxylates
• reactivity in the first step (for a given nucleophile) depends on electrophilicity of the carbonyl carbon
• both substituents of the carbonyl carbon and further substituents on the potential leaving group matter
R O
OAlkylR O
O
R O
O FF
F
FF
R O
O NO
ONO
O
R
O
O
≈ ≈ > >
Alkyl X
O
X
O
X
O
X
O
D
X
O
A
>>A X
O> > > >>
D X
O>>
active esters regular esters
–M and/or –I effect +M effect
R R'
O
R OR'
O> > >
R H
O
R OH
O
R O
O> >
R' NR'2
O
R O
O> >
R Hal
O O
R>>>

Reactivity of Carbonyl Compounds
153
• amides, acids, and carboxylates have very poor leaving groups, do not easily undergo substitution • since aldehydes and ketones have no leaving groups, they cannot complete nucleophilic substitution
acid halides acid anhydrides (active) esters amides acids carboxylates aldehydes ketones
• reactivity in the second step depends on leaving group quality (see SN1) of the carbonyl substituent
–M and/or –I effect
R OR'
O>
R OH
O
R O
O> >
R' NR'2
O
R O
O>
R Hal
O O
R>>
R R'
O
R H
O>>>> > > >>>
acidic proton deprotonation
pKA (OH–) 23 pKA (H2) 35 pKA (CH4) 48no leaving group
Cl
O
R
O
O
R
O
R O
O
R
FF
F
FF
NO
O
R
O
O
O
FF
F
FF
C
O
FF
F
FF
etc.
NO
O
O
NO
O
O
etc.
acid chlorides (pKA HCl –7)
acid anhydrides (pKA RCOOH 4) pentafluorophenyl esters (pKA PfPOH 6)
N-succinylimidyl esters (pKA SuOH 10)
R
O
ORO
O

Trivial Names and Acronyms of Important Reactants
154
Cl
O
O
O
H3C
O
CH3
SOSO
F3C
O
CF3
O O
ClSOO
H3C
benzoyl chloride (BzCl)
tosyl chloride (TsCl)
acetic anhydride (AcOAc, Ac2O)
mesyl chloride (MsCl)
Triflic anhydride (TfOTf, Tf2O)
ClSO
H3C
O
Cl
O
H3C
acetyl chloride (AcCl)
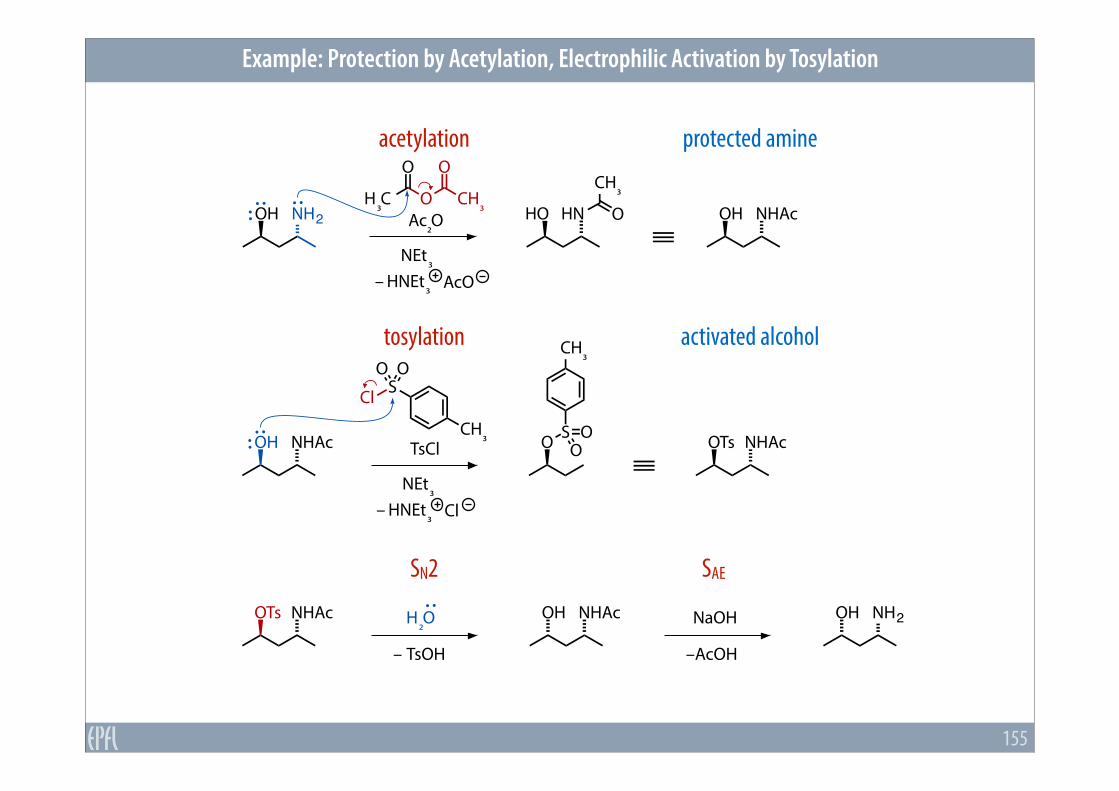
Example: Protection by Acetylation, Electrophilic Activation by Tosylation
155
acetylation protected amine
OH
Cl SO O
CH3
NEt 3– HNEt 3 Cl
OTsClS
OO
CH3
OTsNHAc NHAc
tosylation activated alcohol
SN2 SAE
OTs
– TsOH
OHNHAc NHAcH2O
–AcOH
NaOH OH NH2
NH2
NEt 3– HNEt 3 AcO
HNAc2O NHAcO
O
H 3C
O
CH3CH3
OOH HO OH

• solution: electrophilic activation with peptide coupling reagents
• no amide (peptide) formation between carboxylic acid and amine:
Example: Peptide Coupling Reactions
156
dicyclohexylcarbodiimide (DCC)
R
O
OH+ H2N R'
R
O
O+ H3N R'
R
O
OH
N C NNEt3
– HNEt3R
O
O
NHCN
O
O
R
+ HNEt3NEt3–
H2N R'NH
O
R R' +NH
CNH
O
peptide

Example: Esterification
157
acid alcohol water
reaction mechanism
ester
• Since OH– is a very poor leaving group, acid catalysis is required for electrophilic activation • reaction proceeds under proton migration, any of the two oxygens of the starting material can leave
RO
OH+ HO R' R
O
O+
R'H2O
H
ROH
OH
H
HO R'R
OH
OHOH
R'R
O
OHOR'
HH
RO
OH
R'
+ H–
H~+ H2O–
H
net reaction
electrophilic activation
catalyst regeneration
addition proton migration
elimination

Example for Reactions of Ketones and Aldehydes: Acetalization
158
aldehyde alcohol water
net reaction
reaction mechanism
acetalR
O
H+ HO R'
R
R'O OR'+H2O
H
ROH
H
H
HO R'R
OH
HOH
R'R
O
HOR'
HH
RO
H
R'
+
H~+ H2O–
2H
HO R'+R
OR'
HOH
R'
H–
• Since H– is not a leaving group at all, another reaction pathway is taken after first addition • electrophilic activation and proton migration convert carbonyl oxygen into leaving group • reaction sequence is terminated with a second addition of an alcohol
addition proton migration
elimination addition

4.4 Electrophilic Additions to Multiple Bonds

• olefins are (weak) nucleophiles and react with electrophiles to form carbocations
Reactions of Olefins with Electrophiles
160
R REl El
HH
H
H
HH
Nu+ REl
H
HHNu
R R COOH Cl> > >R
PhRR
RR
RROR >>>>
decreasing electron density (±M or ±I effect)
• reactivity order
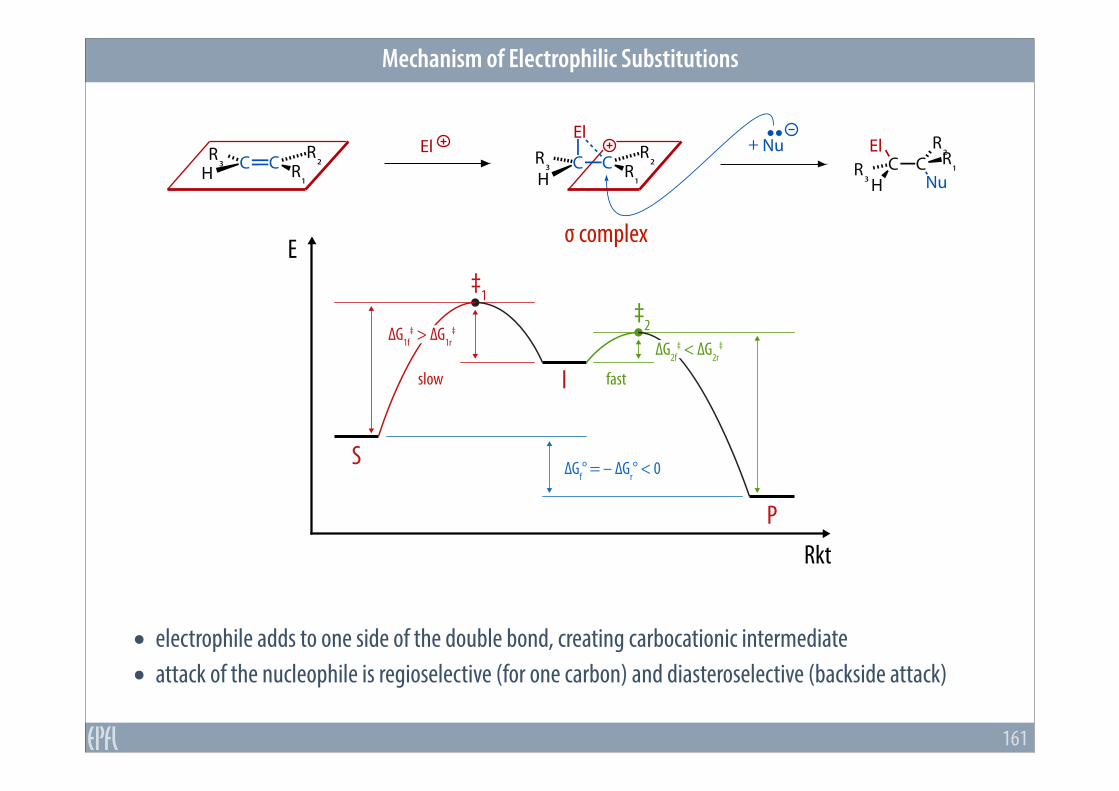
Mechanism of Electrophilic Substitutions
161
• electrophile adds to one side of the double bond, creating carbocationic intermediate • attack of the nucleophile is regioselective (for one carbon) and diasteroselective (backside attack)
‡1
I
E
Rkt
S
‡2
P
∆Gf° = – ∆Gr° < 0
∆G2f‡ < ∆G2r
‡
slow fast
∆G1f‡ > ∆G1r
‡
CR 3H C R 1
R2 CR 3H
C R 1
R2
ElEl Nu+
C C R 1El R2
HR 3 Nu
σ complex

Regioselectivity of Electrophilic Additions
162
• electrophile addition to the double bond preferred so that more stable carbocation is formed • Markovnikov rule: in HX addition, X is added to the “higher substituted” carbon
‡1
I
I’
E
Rkt
S
‡2
‡1’ ‡2’
P
P’ΔG°
ΔG°
slow
ΔG1‡
ΔG1’‡
H
HMeH
H
H
Me
H Br H H Me
H
Br
H Br
Br
MeH
H
H
H H Me
HBr
Br

• backside attack of the nucleophile enforces trans addition
Stereoselectivity of Electrophilic Additions
163
• electrophilic additions are diastereospecific reactions • olefin with given E or Z configuration will be transformed into one diastereomer (a pair of enantiomers)
MeHH Ph
Br
MeH
HPh
Br
+Br Br
(R)(R)(S)(S)
MeHBr
Br
PhH
CHMe C H
PhBr
(S)(S)(R)(R)
HMe
Br
Br
HPh
+
MePhH H
Br
MePh
HH
Br
+Br Br
(R)(R)(R)(R)
MeHBr
Br
HPh
CHMe C Ph
HBr
(S)(S)(S)(S)
HMe
Br
Br
PhH
+
(E)
(Z)

• hydrohalogenation of olefins
Examples
164
HOH
H HSO4
H2O
HBr
H Br
BrBr
Br Br
• hydration
• halogen addition

4.5 Electrophilic Substitutions on Aromatic Compounds

• aromatic π systems are also weak nucelophiles; electrophiles add to one of the double bonds
Multiple Bonds in Aromatic Compounds Do Not React Like Olefins
166
• addition of the nucleophile to complete electrophilic addition would lead to loss of aromaticity • instead, proton elimination to re-establish aromatic π system
ElEl
HH
H
Nu+
H
HEl
El
H HNu
H–
Elelctrophilic Addition
Elelctrophilic Substitution

Mechanism of Electrophilic Aromatic Substitutions
167
• π complex allows electrophile to find most favorable reaction path towards σ complex • electrophilic substitution is regioselective: “most acidic” proton is replaced
σ complex
addition
substitution
‡1
E
Rkt
πS
‡2
P
P’
∆Gf° = – ∆Gr° < 0
∆G2f‡ < ∆G2r
‡
slow fast
∆G1f‡ > ∆G1r
‡
‡0σ
ElH
HElH–El
HH
H
El
π complex

• regioselectivity in electrophilic substitutions is directed by previous substituents
Regioselectivity in Electrophilic Aromatic Substitutions
168
• substituents with +M effect direct the electrophile into para (and ortho) positions • substituents with +M effect increase electron density, and hence reactivity towards electrophile
RO
HC
RO
CH
HCRORO
RO
RHN>
RO>
I>
Br>
Cl>
F>
para major
meta none
ortho minor
BrBr Br
+ FeCl3RO RO+
RO+
ROBrBr
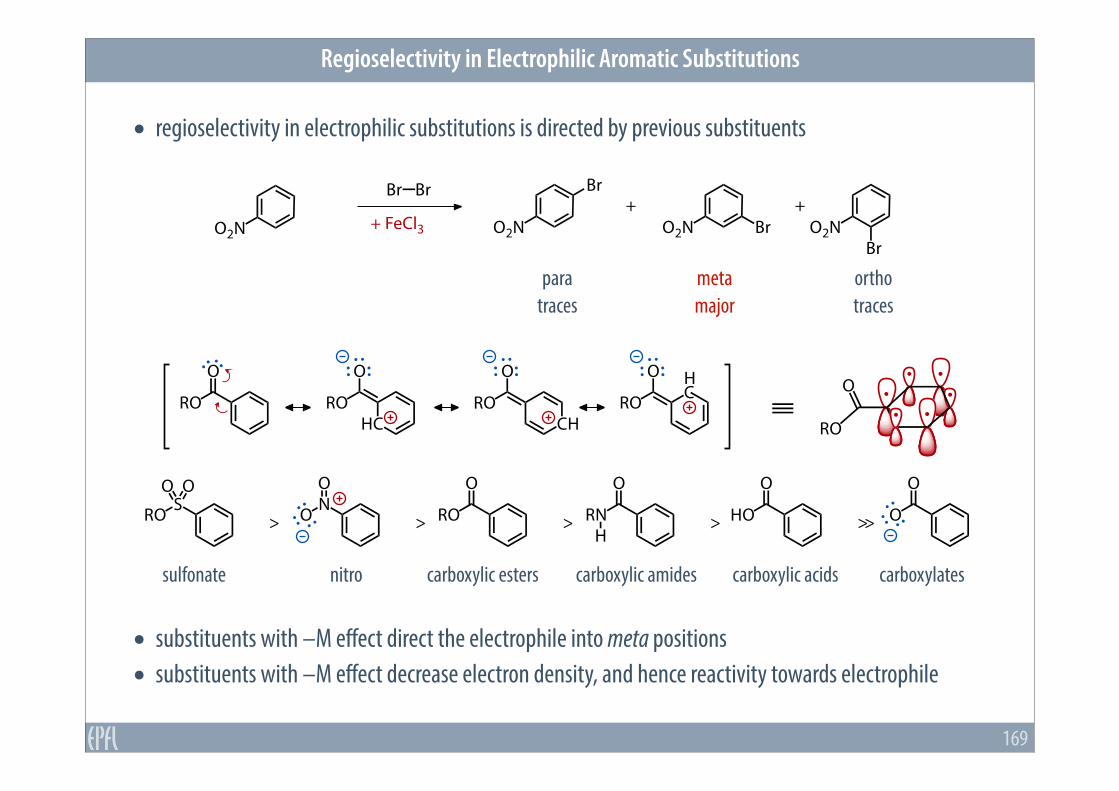
• regioselectivity in electrophilic substitutions is directed by previous substituents
Regioselectivity in Electrophilic Aromatic Substitutions
169
• substituents with –M effect direct the electrophile into meta positions • substituents with –M effect decrease electron density, and hence reactivity towards electrophile
para traces
meta major
ortho traces
BrBr Br
+ FeCl3O2N O2N+
O2N+
O2NBrBr
HC CH
HC
O
RO
O
RO
O
RO
O
ROO
RO
S>
N> > > >>
O
RO
O O
O
O
RO
O
RN
O
HO
O
OH
sulfonate nitro carboxylic esters carboxylic amides carboxylic acids carboxylates

• bromination
Examples
170
H Br+ AlBrCl3
R R
H ClR R
O O
Br AlCl3δ–δ+
R
Cl AlCl3δ–δ+
R
O
+ AlCl3
+ AlCl3+ AlCl4
H BrBr Br
+ FeCl3Br Br FeCl3
δ–δ+
+ FeBrCl3
H SO3HH2SO4
– H2O
H NO2HNO3 / H2SO4
– H2ONO2 H3O 2 HSO4
HSO3 HSO4
• Friedl-Crafts alkylation and acylation
• sulfonation and nitration

4.6 Elimination Reactions
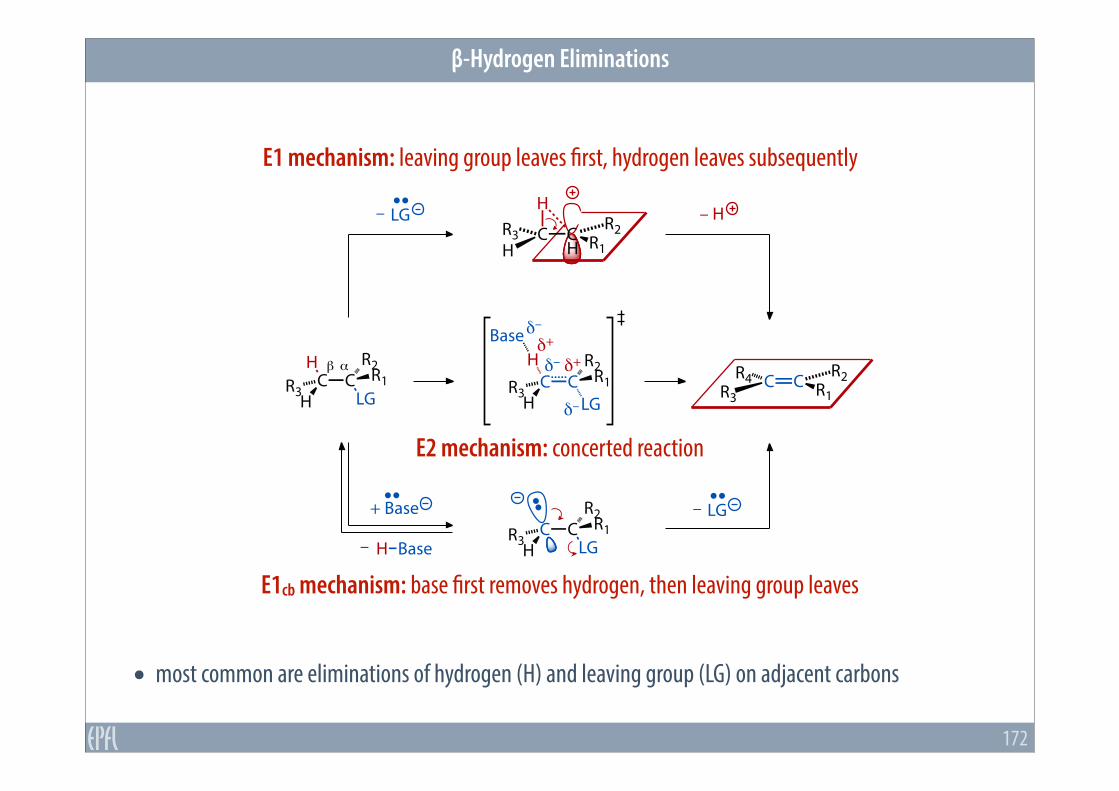
β-Hydrogen Eliminations
172
• most common are eliminations of hydrogen (H) and leaving group (LG) on adjacent carbons
E1 mechanism: leaving group leaves first, hydrogen leaves subsequently
E1cb mechanism: base first removes hydrogen, then leaving group leaves
E2 mechanism: concerted reaction
LG–
CR4R3
C R1
R2
CR3H
CH R1
R2
H – H
C C R1H R2
HR3 LG
+ Base
H Base–C C R1
R2
HR3 LG
C C R1
H R2
HR3
LG
Baseδ–
δ+
δ+δ–
δ–
αβ
LG–

• E1 reactions require good/excellent leaving group and adjacent proton, absence of a base/nucleophile
E1 Reactions and Competition with SN1 Substitutions
173
• first step of E1 reaction is carbocation generation, identical conditions and intermediate as SN1 reaction • E1 eliminations are inevitable side reactions of SN1 reactions • E1: absence of a nucleophile, only weak base (if any), many acidic β-hydrogens, high temperature (!) • SN1: presence of a good (not too basic) nucleophile, few or non-acidic β-hydrogens
LG–CR3
HCH R1
R2
H
C C R1H R2
HR3 LG
– H
Nu+
CR4R3
C R1
R2
C C R2
H
R1HR3
NuC C R1
H R2
HR3 Nu
+
(base)
E1 Elimination
SN1 Substitution
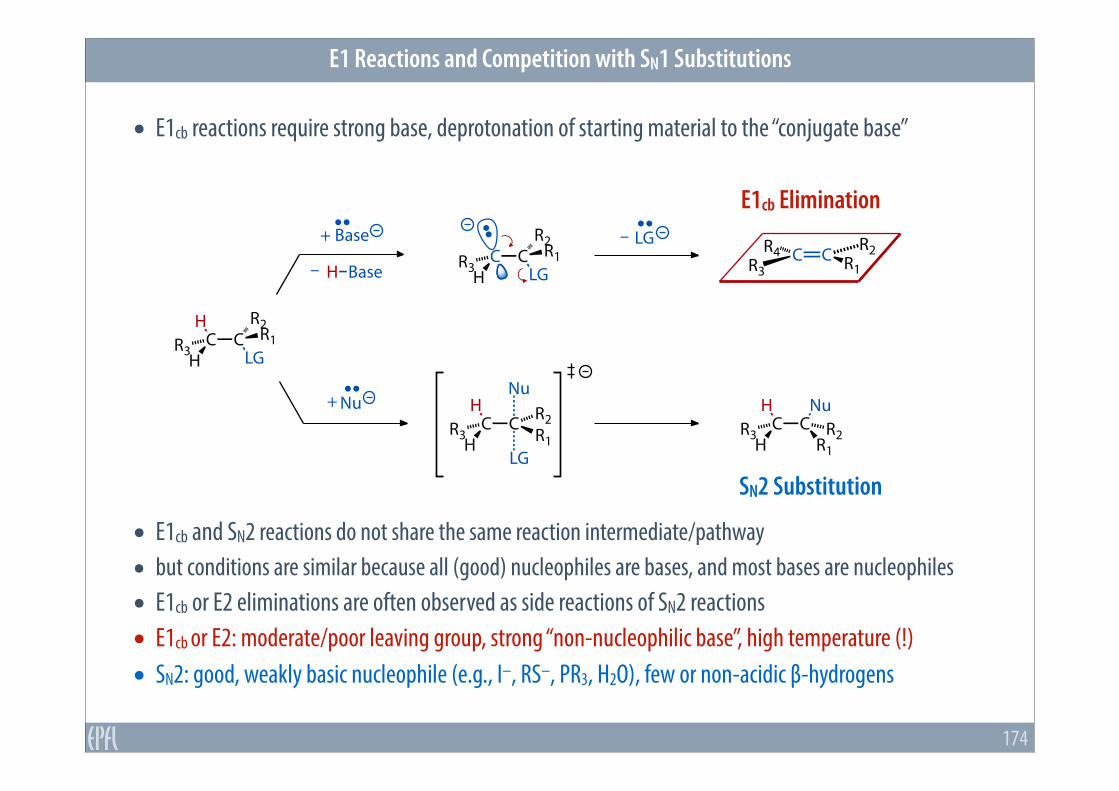
• E1cb reactions require strong base, deprotonation of starting material to the “conjugate base”
E1 Reactions and Competition with SN1 Substitutions
174
• E1cb and SN2 reactions do not share the same reaction intermediate/pathway • but conditions are similar because all (good) nucleophiles are bases, and most bases are nucleophiles • E1cb or E2 eliminations are often observed as side reactions of SN2 reactions • E1cb or E2: moderate/poor leaving group, strong “non-nucleophilic base”, high temperature (!) • SN2: good, weakly basic nucleophile (e.g., I–, RS–, PR3, H2O), few or non-acidic β-hydrogens
E1cb Elimination
SN2 Substitution
C C R1H R2
HR3 LG
+ Base
H Base–C C R1
R2
HR3 LG
CR4R3
C R1
R2
Nu+C C
R1
H R2
HR3
LG
Nu
C C R2
H
R1HR3
Nu
LG–

Non-Nucleophilic Bases
175
• non-nucleophilic bases have a high pKA (of conjugate acid BH) but are strongly sterically hindered!
lithium diisopropylamide LDA, pKA ≈ 40
lithium hexamethyldisilazide LHMDS, pKA ≈ 40
diisopropylethylamine DIEA, pKA ≈ 10
triisopropylamine TIPA, pKA ≈ 10
1,8-bis(dimethylamino)naphthalene proton sponge, pKA ≈ 12
1,4-diazabicyclo[2.2.2]octane DABCO, pKA ≈ 12
1,5-diazabicyclo[4.3.0]non-5-ene DBN, pKA ≈ 12
1,8-Diazabicyclo[5.4.0]undec-7-ene DBU, pKA ≈ 12
NLi
Si N Si
Li
N
N
N
N
N
N
N N
N N

176

4.7 Radical Reactions
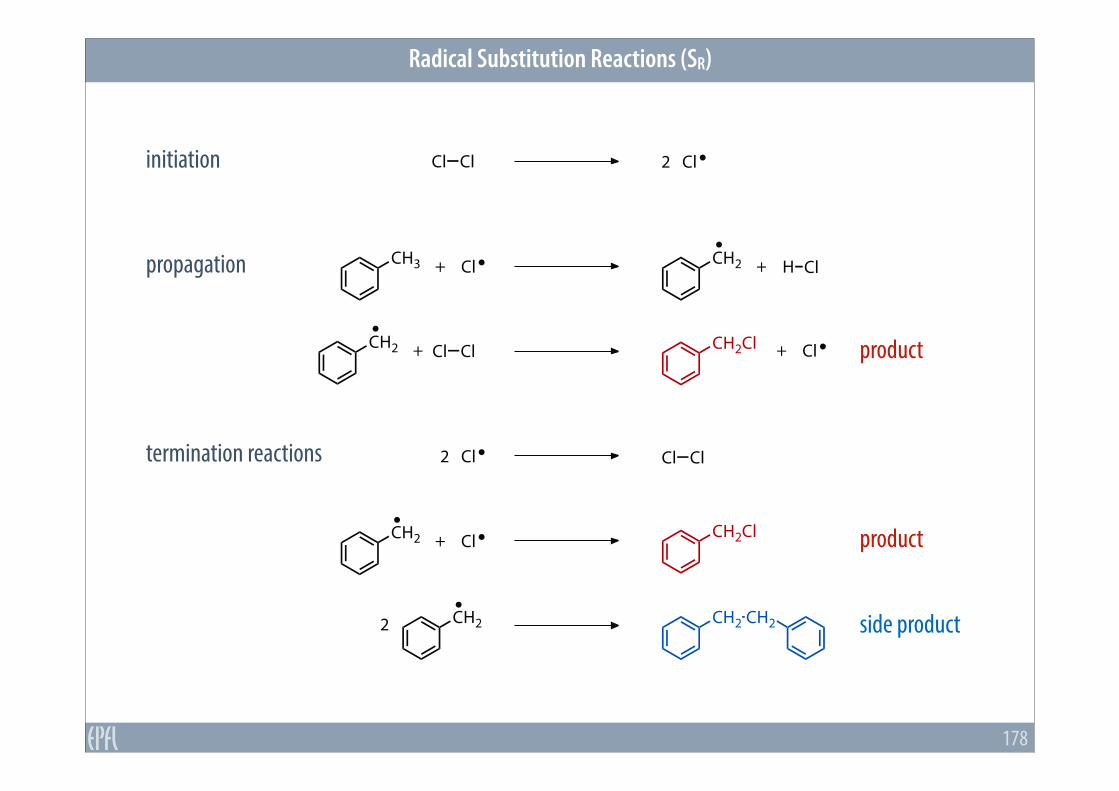
Radical Substitution Reactions (SR)
178
initiation
CH3
Cl Cl 2 Cl
+ Cl CH2 + H Cl
CH2 + Cl Cl CH2Cl + Cl
2 Cl Cl Cl
CH2 + Cl CH2Cl
CH22 CH2 CH2
propagation
termination reactions
product
side product
product

Radical Addition Reactions (AR)
179
initiation
propagation
termination reactions
Br Br 2 Br
+ BrHC
+ Br Br
2 Br Br Br
+ Br
2 CH CH
Br
HC Br + Br
BrBr
BrBr
HC Br
HC Br
BrH2C CH2Br
product
side product
product

180