chapter 313 chapter 18
TRANSCRIPT

333
■ Lipoprotein Metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . .333Lipoprotein Classification and Composition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .333
Transport of Dietary Lipids (Exogenous Pathway) . . . . . . . . . . . . . . . . . . . . . . . . . . . . .334
Transport of Hepatic Lipids (Endogenous Pathway) . . . . . . . . . . . . . . . . . . . . . . . . . . . .335
HDL Metabolism and Reverse Cholesterol Transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .336
■ Disorders of Lipoprotein Metabolism . . . . . . . . . . . . . . . . . . .337Primary Disorders of ApoB-Containing Lipoprotein Biosynthesis Causing Low PlasmaCholesterol Levels (Known Etiology) . . . . . . . . . . . . . . . . . .337
Primary Disorders of ApoB-Containing Lipoprotein Catabolism Causing Elevated Plasma Cholesterol Levels (Known Etiology) . . . . . . . . . . . .338
Primary Disorders of ApoB-Containing Lipoprotein Metabolism (Unknown Etiology) . . . . . . . . . . . .342
Genetic Disorders of HDL Metabolism (Known Etiology) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .344
Primary Disorders of HDL Metabolism (Unknown Etiology) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .345
Secondary Disorders of Lipoprotein Metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .345
Screening . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .347■ Further Readings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .353
Lipoproteins are complexes of lipids and proteins thatare essential for the transport of cholesterol, triglyc-erides, and fat-soluble vitamins. Until recently, lipopro-tein disorders were the purview of lipidologists, but thedemonstration that lipid-lowering therapy significantlyreduces the clinical complications of atheroscleroticcardiovascular disease (ASCVD) has brought the diagno-sis and treatment of these disorders into the domain ofthe general internist. The metabolic consequences asso-ciated with changes in diet and lifestyle have increasedthe number of hyperlipidemic individuals who couldbenefit from lipid-lowering therapy.The development of
safe, effective, and well-tolerated pharmacologic agentshas greatly expanded the therapeutic armamentariumavailable to the physician to treat disorders of lipid me-tabolism.Therefore, the appropriate diagnosis and man-agement of lipid disorders is critically important to thepractice of medicine. This chapter reviews normallipoprotein physiology, the pathophysiology of theknown single-gene disorders of lipoprotein metabolism,the environmental factors that influence lipoprotein me-tabolism, and the practical approaches to their diagnosisand management.
LIPOPROTEIN METABOLISM
LIPOPROTEIN CLASSIFICATION AND COMPOSITION
Lipoproteins are large, mostly spherical complexes thattransport lipids (primarily triglycerides, cholesteryl esters,and fat-soluble vitamins) through body fluids (plasma,interstitial fluid, and lymph) to and from tissues. Lipo-proteins play an essential role in the absorption of dietarycholesterol, long-chain fatty acids, and fat-soluble vitamins;the transport of triglycerides, cholesterol, and fat-solublevitamins from the liver to peripheral tissues; and thetransport of cholesterol from peripheral tissues to the liver.
Lipoproteins contain a core of hydrophobic lipids(triglycerides and cholesteryl esters) surrounded by hy-drophilic lipids (phospholipids, unesterified cholesterol)and proteins that interact with body fluids. The plasmalipoproteins are divided into five major classes based ontheir relative densities (Fig. 18-1 and Table 18-1):chylomicrons, very low density lipoproteins (VLDL),intermediate-density lipoproteins (IDL), low-densitylipoproteins (LDL), and high-density lipoproteins(HDL). Each lipoprotein class comprises a family of
Chapter 313CHAPTER 18CHAPTER 18
DISORDERS OF LIPOPROTEIN METABOLISM
Daniel J. RaderHelen H. Hobbs
46128_18_p333-354 2/1/06 2:42 PM Page 333

particles that vary slightly in density, size, migrationduring electrophoresis, and protein composition. Thedensity of a lipoprotein is determined by the amount oflipid and protein per particle. HDL is the smallest andmost dense lipoprotein, whereas chylomicrons andVLDL are the largest and least dense lipoprotein parti-cles. Most triglyceride is transported in chylomicrons orVLDL, and most cholesterol is carried as cholesterylesters in LDL and HDL.
The apolipoproteins are required for the assemblyand structure of lipoproteins (Table 18-2). Apo-
lipoproteins also serve to activate enzymes impor-tant in lipoprotein metabolism and to mediate thebinding of lipoproteins to cell-surface receptors.ApoA-I, which is synthesized in the liver and intestine,is found on virtually all HDL particles. ApoA-II is thesecond most abundant HDL apolipoprotein and isfound on approximately two-thirds of all HDL parti-cles. ApoB is the major structural protein of chylomi-crons,VLDL, IDL, and LDL; one molecule of apoB, ei-ther apoB-48 (chylomicrons) or apoB-100 (VLDL,IDL, or LDL), is present on each lipoprotein particle.The human liver makes only apoB-100, and the intestinemakes apoB-48, which is derived from the same geneby mRNA editing. ApoE is present in multiple copieson chylomicrons, VLDL, and IDL and plays a criticalrole in the metabolism and clearance of triglyceride-rich particles. Three apolipoproteins of the C-series(apoC-I, -II, and -III) also participate in the metabo-lism of triglyceride-rich lipoproteins. The otherapolipoproteins are listed in Table 18-2.
TRANSPORT OF DIETARY LIPIDS(EXOGENOUS PATHWAY)
The exogenous pathway of lipoprotein metabolismpermits efficient transport of dietary lipids (Fig. 18-2).Dietary triglycerides are hydrolyzed by pancreatic li-pases within the intestinal lumen and are emulsifiedwith bile acids to form micelles. Dietary cholesteroland retinol are esterified (by the addition of a fattyacid) in the enterocyte to form cholesteryl esters and
334 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
Den
sity
, g/m
L
1.20
5 10
Diameter, mm
1.10
1.02
1.06
20 40 60 80 1000
1.006
0.95
HDL
LDL
IDL
VLDL
Chylomicronremnants
Chylomicron
FIGURE 18-1The density and size-distribution of the major classes oflipoprotein particles. Lipoproteins are classified by densityand size, which are inversely related. VLDL, very low densitylipoproteins; IDL, intermediate-density lipoproteins; LDL,low-density lipoproteins; HDL, high-density lipoproteins.
TABLE 18-1
MAJOR LIPOPROTEIN CLASSESa
APOLIPOPROTEINS ELECTROPHORETIC OTHER
LIPOPROTEIN DENSITY, G/MLb SIZE NMc MOBILITYd MAJOR OTHER CONSTITUENTS
Chylomicrons 0.930 75–1200 Origin ApoB-48 A-I, A-IV, C-I, Retinyl estersC-II, C-III
Chylomicron 0.930–1.006 30–80 Slow pre-� ApoB-48 E, A-I, A-IV, C-I, Retinyl estersremnants C-II, C-III
VLDL 0.930–1.006 30–80 Pre-� ApoB-100 E, A-I, A-II, A-V, Vitamin EC-I, C-II, C-III
IDL 1.006–1.019 25–35 Slow pre-� ApoB-100 E, C-I, C-II, C-III Vitamin ELDL 1.019–1.063 18–25 � ApoB-100 Vitamin EHDL 1.063–1.210 5–12 � ApoA-I A-II, A-IV, E, LCAT, CETP
C-III paroxonaseLp(a) 1.050–1.120 25 Pre-� ApoB-100 Apo(a)
aAll of the lipoprotein classes contain phospholipids, esterified andunesterified cholesterol, and triglycerides to varying degrees.bThe density of the particle is determined by ultracentrifugation.cThe size of the particle is measured using gel electrophoresis.dThe electrophoretic mobility of the particle on agarose gel electrophoresis reflects the size and surface charge of the par-
ticle, with � being the position of LDL and � the position of HDL.Note: VLDL, very low density lipoprotein; IDL, intermediate-densitylipoprotein; LDL, low-density lipoprotein: HDL, high-density lipopro-tein; Lp(a), lipoprotein A; LCAT, lecithin-cholesterol acyltransferase;CETP, cholesteryl ester transfer protein.
46128_18_p333-354 1/31/06 10:23 AM Page 334

retinyl esters, respectively. Longer-chain fatty acids(�12 carbons) are incorporated into triglycerides andpackaged with apoB-48, cholesteryl esters, retinyl es-ters, phospholipids, and cholesterol to form chylomi-crons. Nascent chylomicrons are secreted into the in-testinal lymph and delivered directly to the systemiccirculation, where they are extensively processed byperipheral tissues before reaching the liver. The parti-cles encounter lipoprotein lipase (LPL), which is an-chored to proteoglycans that decorate the capillary en-dothelial surfaces of adipose tissue, heart, and skeletalmuscle (Fig. 18-2). The triglycerides of chylomicronsare hydrolyzed by LPL, and free fatty acids are released;apoC-II, which is transferred to circulating chylomi-crons, acts as a cofactor for LPL in this reaction. Thereleased free fatty acids are taken up by adjacent myo-cytes or adipocytes and either oxidized or reesterifiedand stored as triglyceride. Some free fatty acids bindalbumin and are transported to other tissues, especiallythe liver. The chylomicron particle progressivelyshrinks in size as the hydrophobic core is hydrolyzedand the hydrophilic lipids (cholesterol and phospho-lipids) on the particle surface are transferred to HDL.The resultant smaller, more cholesterol ester – rich par-ticles are referred to as chylomicron remnants. The rem-nant particles are rapidly removed from the circulation
by the liver in a process that requires apoE. Conse-quently, few, if any, chylomicrons are present in theblood after a 12-h fast, except in individuals with dis-orders of chylomicron metabolism.
TRANSPORT OF HEPATIC LIPIDS(ENDOGENOUS PATHWAY)
The endogenous pathway of lipoprotein metabolism refers tothe hepatic secretion and metabolism of VLDL to IDLand LDL (Fig. 18-2).VLDL particles resemble chylomi-crons in protein composition but contain apoB-100rather than apoB-48 and have a higher ratio of choles-terol to triglyceride (�1 mg of cholesterol for every 5mg of triglyceride). The triglycerides of VLDL arederived predominantly from the esterification of long-chain fatty acids.The packaging of hepatic triglycerideswith the other major components of the nascent VLDLparticle (apoB-100, cholesteryl esters, phospholipids,and vitamin E) requires the action of the enzyme mi-crosomal transfer protein (MTP). After secretion intothe plasma,VLDL acquires multiple copies of apoE andapolipoproteins of the C series. The triglycerides ofVLDL are hydrolyzed by LPL, especially in muscle andadipose tissue. As VLDL remnants undergo further hy-drolysis, they continue to shrink in size and become
Chapter 18 Disorders of Lipoprotein Metabolism 335
TABLE 18-2
MAJOR APOLIPOPROTEINS
APOLIPOPROTEIN PRIMARY SOURCE LIPOPROTEIN ASSOCIATION FUNCTION
ApoA-I Intestine, liver HDL, chylomicrons Structural protein for HDL, activates LCAT
ApoA-II Liver HDL, chylomicrons Structural protein for HDLApoA-IV Intestine HDL, chylomicrons UnknownApoA-V Liver VLDL UnknownApoB-48 Intestine Chylomicrons Structural protein for
chylomicronsApoB-100 Liver VLDL, IDL, LDL, LP(a) Structural protein for VLDL,
LDL, IDL, LP(a); ligand for binding to LDL, receptor
ApoC-I Liver Chylomicrons VLDL, HDL UnknownApoC-II Liver Chylomicrons VLDL, HDL Cofactor for LPLApoC-III Liver Chylomicrons VLDL, HDL Inhibits lipoprotein binding
to receptorsApoD Spleen, brain, HDL Unknown
testes, adrenalsApoE Liver Chylomicron remnants, IDL, Ligand for binding to LDL
HDL receptorApoH Liver Chylomicrons VLDL, LDL, HDL B2 glycoprotein IApoJ Liver HDL UnknownApoL Unknown HDL UnknownApo(a) Liver Lp(a) Unknown
Note: HDL, high-density lipoprotein; LCAT, lecithin-cholesterol acyl-transferase; VLDL, very low density lipoprotein; IDL, intermediate-
density lipoprotein; LDL, low-density lipoprotein; Lp(a), lipoprotein A;LPL, lipoprotein lipase.
46128_18_p333-354 1/31/06 10:23 AM Page 335

IDL, which contain similar amounts of cholesterol andtriglyceride. The liver removes �40 to 60% of VLDLremnants and IDL by LDL receptor–mediated endocy-tosis via binding to apoE. The remainder of IDL is re-modeled by hepatic lipase (HL) to form LDL; duringthis process, most of the triglyceride in the particle ishydrolyzed and all apolipoproteins except apoB-100 aretransferred to other lipoproteins. The cholesterol inLDL accounts for �70% of the plasma cholesterol inmost individuals. Approximately 70% of circulatingLDLs are cleared by LDL receptor–mediated endocy-tosis in the liver. Lipoprotein(a) [Lp(a)] is a lipoproteinsimilar to LDL in lipid and protein composition, but itcontains an additional protein called apolipoprotein(a)[apo(a)]. Apo(a) is synthesized in the liver and is at-tached to apoB-100 by a disulfide linkage.The mecha-nism by which Lp(a) is removed from the circulation isnot known.
HDL METABOLISM AND REVERSECHOLESTEROL TRANSPORT
All nucleated cells synthesize cholesterol but only he-patocytes can efficiently metabolize and excrete cho-lesterol from the body. The predominant route ofcholesterol elimination is by excretion into the bile,either directly or after conversion to bile acids. Cho-lesterol in peripheral cells is transported from theplasma membranes of peripheral cells to the liver byan HDL-mediated process termed reverse cholesteroltransport (Fig. 18-3).
Nascent HDL particles are synthesized by the intes-tine and the liver. The newly formed discoidal HDLparticles contain apoA-I and phospholipids (mainlylecithin) but rapidly acquire unesterified cholesteroland additional phospholipids from peripheral tissuesvia transport by the membrane protein ATP-binding
336 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
Smallintestines
Exogenous Endogenous
Bile acids+
cholesterol
Peripheraltissues
ApoE
ApoB
LDLR
ApoC's
Dietary lipids
Chylomicron VLDL IDLChylomicron
remnant
LPLFFA
Muscle Adipose
LPLFFA
Muscle Adipose
FIGURE 18-2The exogenous and endogenous lipoprotein metabolic pathways. The exogenouspathway transports dietary lipids to the periphery and the liver. The exogenous pathwaytransports hepatic lipids to the periphery. LPL, lipoprotein lipase; FFA, free fatty acids;VLDL, very low density lipoproteins; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LDLR, low-density lipoprotein receptor.
46128_18_p333-354 1/31/06 10:23 AM Page 336

cassette protein A1 (ABCA1). Once incorporated inthe HDL particle, cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT), a plasma enzymeassociated with HDL. As HDL acquires more choles-teryl ester it becomes spherical, and additionalapolipoproteins and lipids are transferred to the parti-cles from the surfaces of chylomicrons and VLDL dur-ing lipolysis.
HDL cholesterol is transported to hepatoctyes byboth an indirect and a direct pathway. HDL cholesterylesters are transferred to apoB-containing lipoproteins inexchange for triglyceride by the cholesteryl ester trans-fer protein (CETP). The cholesteryl esters are thenremoved from the circulation by LDL receptor–mediated endocytosis. HDL cholesterol can also betaken up directly by hepatocytes via the scavengerreceptor class BI (SR-BI), a cell-surface receptor thatmediates the selective transfer of lipids to cells.
HDL particles undergo extensive remodeling withinthe plasma compartment as they transfer lipids andproteins to lipoproteins and cells. For example, afterCETP-mediated lipid exchange, the triglyceride-enriched HDL becomes a substrate for HL, whichhydrolyzes the triglycerides and phospholipids to gener-ate smaller HDL particles.
DISORDERS OF LIPOPROTEIN METABOLISM
The identification and characterization of genes respon-sible for the genetic forms of hyperlipidemia haveprovided important molecular insight into the criticalroles of apolipoproteins, enzymes, and receptors in lipidmetabolism.
PRIMARY DISORDERS OF ApoB-CONTAINING LIPOPROTEINBIOSYNTHESIS CAUSING LOW PLASMACHOLESTEROL LEVELS (KNOWN ETIOLOGY)
The synthesis and secretion of apoB-containing lipopro-teins in the enterocytes of the proximal small bowel andin the hepatocytes of the liver involve a complex seriesof events that coordinate the coupling of various lipidswith apoB-48 and apoB-100, respectively.
Abetalipoproteinemia
Abetalipoproteinemia is a rare autosomal recessivedisease caused by mutations in the gene encodingMTP, which transfers lipids to nascent chylomicrons
Chapter 18 Disorders of Lipoprotein Metabolism 337
LDLR
Mature LDL
ChylomicronsPeripheral cells
LCAT CETP
NascentHDL
ApoA1
CETP
Freecholesterol
Macrophage
FIGURE 18-3HDL metabolism and reverse cholesterol transport. This pathway transports excesscholesterol from the periphery back to the liver for excretion in the bile. The liver and theintestine produce nascent HDL. Free cholesterol is acquired from macrophages andother peripheral cells and esterfied by LCAT, forming mature HDL. HDL cholesterol canbe selectively taken up by the liver via SR-BI. Alternatively, HDL cholesteryl ester can betransferred by CETP from HDL to VLDL and chylomicrons, which can then be taken upby the liver. LCAT, lecithin-cholesterol acyltransferase; CETP, cholesteryl ester transferprotein; VLDL, very low density lipoproteins; IDL, intermediate-density lipoproteins; LDL,low-density lipoproteins; HDL, high-density lipoproteins; LDLR, low-density lipoproteinreceptor; TG, triglycerides; SR-B1, scavenger receptor class B1.
46128_18_p333-354 1/31/06 10:23 AM Page 337

and VLDL in the intestine and liver, respectively.Plasma cholesterol and triglyceride levels are ex-tremely low in this disorder, and no chylomicrons,VLDL, LDL, or apoB are detectable. The parents ofpatients with abetalipoproteinemia (who are obligateheterozygotes) have normal plasma lipid and apoBlevels. Abetalipoproteinemia usually presents in earlychildhood with diarrhea and failure to thrive and ischaracterized clinically by fat malabsorption, spi-nocerebellar degeneration, pigmented retinopathy, andacanthocytosis. The initial neurologic manifestationsare loss of deep-tendon reflexes, followed by decreaseddistal lower extremity vibratory and proprioceptivesense, dysmetria, ataxia, and the development of aspastic gait, often by the third or fourth decade. Pa-tients with abetalipoproteinemia also develop a pro-gressive pigmented retinopathy presenting withdecreased night and color vision, followed by reduc-tions in daytime visual acuity and ultimately progress-ing to near blindness. The presence of spinocerebellardegeneration and pigmented retinopathy in this dis-ease has resulted in misdiagnosis of Friedreich’s ataxia.Rarely, patients with abetalipoproteinemia develop acardiomyopathy with associated life-threateningarrhythmias.
Most clinical manifestations of abetalipoproteinemiaresult from defects in the absorption and transport offat-soluble vitamins. Vitamin E and retinyl esters arenormally transported from enterocytes to the liver bychylomicrons, and vitamin E is dependent on VLDL fortransport out of the liver and into the circulation. Pa-tients with abetalipoproteinemia are markedly deficientin vitamin E and are also mildly to moderately deficientin vitamin A and vitamin K.Treatment of abetalipopro-teinemia consists of a low-fat, high-caloric, vitamin-enriched diet accompanied by large supplemental dosesof vitamin E. It is imperative for treatment to be initi-ated as soon as possible to obviate the development ofneurologic sequelae.
Familial Hypobetalipoproteinemia
Familial homozygous hypobetalipoproteinemia has aclinical picture similar to abetalipoproteinemia but is au-tosomal codominant in inheritance pattern. The diseasecan be differentiated from abetalipoproteinemia sincethe parents of the probands with this disorder have levelsof plasma LDL-C and apoB that are less than half of thenormal levels. Mutations in the gene encoding apoB-100 that interfere with protein synthesis are commoncauses of this disorder. These patients, like those withabetalipoproteinemia, should be referred to specializedcenters for confirmation of the diagnosis and appropri-ate therapy.
PRIMARY DISORDERS OF ApoB-CONTAININGLIPOPROTEIN CATABOLISM CAUSINGELEVATED PLASMA CHOLESTEROL LEVELS(KNOWN ETIOLOGY)
Single-gene defects can result in the accumulation of spe-cific classes of lipoprotein particles. Mutations in genesencoding key proteins in the metabolism and clearance ofapoB-containing lipoproteins cause type I (chylomicrone-mia), type II (elevations in LDL) and type III (elevationsin IDL) hyperlipoproteinemias (Table 18-3).
Lipoprotein Lipase and ApoC-II Deficiency (Familial Chylomicronemia Syndrome; Type IHyperlipoproteinemia)
LPL is required for the hydrolysis of triglycerides in chy-lomicrons and VLDL.ApoC-II is a cofactor for LPL (Fig.18-2). Genetic deficiency of either LPL or apoC-II re-sults in impaired lipolysis and profound elevations inplasma chylomicrons. These patients also have elevationsin plasma VLDL, but chylomicronemia predominates.Normally chylomicrons are delipidated and removedfrom the circulation within 12 h of the last meal, but inLPL-deficient patients, the triglyceride-rich chylomi-crons persist in the circulation for days. The fastingplasma is turbid, and if left at 40C for a few hours, thechylomicrons float to the top and form a creamy super-natant. In these disorders, called familial chylomicronemiasyndromes, fasting triglyceride levels are almost invariably�11.3 �mol/L (1000 mg/dL). Fasting cholesterol levelsare also usually elevated, but to a much less severe degree.
LPL deficiency is autosomal recessive and has a popu-lation frequency of �1 in 1 million.ApoC-II deficiencyis also recessive in inheritance pattern and is even lesscommon than LPL deficiency. Multiple mutations in theLPL and apoC-II genes cause these diseases. ObligateLPL heterozygotes have normal or mild to moderate el-evations in plasma triglyceride levels, whereas individu-als heterozygous for mutation in apoC-II are not hyper-triglyceridemic.
Both LPL and apoC-II deficiency usually present inchildhood with recurrent episodes of severe abdominalpain caused by acute pancreatitis. On fundoscopic ex-amination the retinal blood vessels are opalescent(lipemia retinalis). Eruptive xanthomas, which are smallyellowish-white papules, often appear in clusters on theback, buttocks, and extensor surfaces of the arms andlegs. These typically painless skin lesions may becomepruritic as they regress. Hepatosplenomegaly resultsfrom the uptake of circulating chylomicrons by reticu-loendothelial cells in the liver and spleen. For reasonsunknown, some patients with persistent and pro-nounced chylomicronemia never develop pancreatitis,eruptive xanthomas, or hepatosplenomegaly. Premature
338 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
46128_18_p333-354 1/31/06 10:23 AM Page 338

ASCVD has not been consistently demonstrated to be afeature of familial chylomicronemia syndromes.
The diagnoses of LPL and apoC-II deficiency are es-tablished enzymatically by assaying triglyceride lipolyticactivity in post-heparin plasma. Blood is sampled afteran intravenous heparin injection to release the endothelial-bound lipases. LPL activity is profoundly reduced inboth LPL and apoC-II deficiency; in patients withapoC-II deficiency, the addition of normal pre-heparinplasma (a source of apoC-II) normalizes LPL activity,but this correction does not occur in patients with LPLdeficiency.
The major therapeutic intervention in familial chy-lomicronemia syndromes is dietary fat restriction (to aslittle as 15 g/d) with fat-soluble vitamin supplementa-tion. Consultation with a registered dietician familiarwith this disorder is essential. Caloric supplementationwith medium-chain triglycerides, which are absorbeddirectly into the portal circulation, can be useful butmay be associated with hepatic fibrosis if used for pro-longed periods. If dietary fat restriction alone is not suc-cessful in resolving the chylomicronemia, fish oils havebeen effective in some patients. In patients with apoC-IIdeficiency, apoC-II can be provided by infusing fresh-frozen plasma to resolve the chylomicronemia. Manage-ment of patients with familial chylomicronemia syn-drome is particularly challenging during pregnancy
when VLDL production is increased. Plasmapheresis maybe required if pancreatitis develops and the chylomi-cronemia is not responsive to diet therapy.
Hepatic Lipase Deficiency
HL is a member of the same gene family as LPL and hy-drolyzes triglycerides and phospholipids in remnantlipoproteins and HDL. HL deficiency is a very rare au-tosomal recessive disorder characterized by elevatedplasma cholesterol and triglycerides (mixed hyperlipi-demia) due to the accumulation of lipoprotein rem-nants. HDL-C is normal or elevated. The diagnosis isconfirmed by measuring HL activity in post-heparinplasma. Due to the small number of patients with HLdeficiency, the association of this genetic defect withASCVD is not known, but lipid-lowering therapy isrecommended.
Familial Dysbetalipoproteinemia (Type IIIHyperlipoproteinemia)
Like HL deficiency, familial dysbetalipoproteinemia(FDBL) (also known as type III hyperlipoproteinemia or fa-milial broad � disease) is characterized by a mixed hyper-lipidemia due to the accumulation of remnant lipopro-tein particles. ApoE is present in multiple copies on
Chapter 18 Disorders of Lipoprotein Metabolism 339
TABLE 18-3
PRIMARY HYPERLIPOPROTEINEMIAS CAUSED BY KNOWN SINGLE-GENE MUTATIONS
LIPOPROTEINS GENETIC ESTIMATED GENETIC DISORDER GENE DEFECT ELEVATED CLINICAL FINDINGS TRANSMISSION INCIDENCE
Lipoprotein lipase LPL(LPL) Chylomicrons Eruptive xanthomas, AR 1/1,000,000deficiency hepatosplenomegaly
pancreatitisFamilial apolipoprotein ApoC-II (APOC2) Chylomicrons Eruptive xanthomas, AR �1/1,000,000C-II deficiency hepatosplenomegaly
pancreatitisFamilial hepatic lipase Hepatic lipase VLDL remnants Premature AR �1/1,000,000deficiency (LIPC) atherosclerosis
Familial ApoE(APOE) Chylomicron and Palmar and AR 1/10,000dysbetalipoproteinemia VLDL remnants tuberoeruptive AD
xanthomas, CHD, PVD
Familial LDL receptor LDL Tendon xanthomas, AD 1/500hypercholesterolemia (LDLR) CHD
Familial defective ApoB-100 (APOB) LDL Tendon xanthomas, AD 1/1000apoB-100 (Arg1500 : GIn) CHD
Autosomal recessive ARH (ARH) LDL Tendon xanthomas, AR �1/1,000,000hypercholesterolemia CHD
Sitosterolemia ABCG5 or LDL Tendon xanthomas, AR �1/1,000,000ABCG8 CHD
Note: AR, autosomal recessive; AD, autosomal dominant; VLDL, very low density lipoprotein; CHD, coronary heart
disease; PVD, peripheral vascular disease; LDL, low-densitylipoprotein.
46128_18_p333-354 1/31/06 10:23 AM Page 339

chylomicron and VLDL remnants and mediates their re-moval via hepatic lipoprotein receptors (Fig. 18-2).FDBL is due to genetic variations in apoE that interferewith its ability to bind lipoprotein receptors.The APOEgene is polymorphic in sequence resulting in the ex-pression of three common isoforms: apoE3, apoE2, andapoE4. Although associated with slightly higher LDL-Clevels and increased coronary heart disease (CHD) risk,the apoE4 allele is not associated with FDBL. Patientswith apoE4 have an increased incidence of late-onsetAlzheimer disease. ApoE2 has a lower affinity for theLDL receptor. Therefore, chylomicron and VLDL rem-nants containing apoE2 are removed from plasma at aslower rate. Individuals who are homozygous for the E2allele (the E2/E2 genotype) comprise the most com-mon subset of patients with FDBL.
Approximately 1% of the general population areapoE2/E2 homozygotes but only a small minority ofthese individuals develop FDBL. In most cases an addi-tional, identifiable factor precipitates the development ofhyperlipoproteinemia. The most common precipitatingfactors are a high-caloric, high-fat diet, diabetes mellitus,obesity, hypothyroidism, renal disease, estrogen defi-ciency, alcohol use, or the presence of another geneticform of hyperlipidemia, most commonly familial com-bined hyperlipidemia (FCHL) or familial hypercholes-terolemia (FH). Rare mutations in apoE cause dominantforms of FDBL; in this case the hyperlipidemia is fullymanifest in the heterozygous state.
Patients with FDBL usually present in adulthoodwith xanthomas and premature coronary and peripheralvascular disease. The disease seldom presents in womenbefore menopause.Two distinctive types of xanthomas areseen in FDBL patients: tuberoeruptive and palmar xan-thomas. Tuberoeruptive xanthomas begin as clusters of smallpapules on the elbows, knees, or buttocks and can growto the size of small grapes. Palmar xanthoma (alternativelycalled xanthomata striata palmaris) are orange-yellowdiscolorations of the creases in the palms. In FDBL, theplasma cholesterol and triglyceride are elevated to a rel-atively similar degree until the triglyceride levels reach�5.6 mol/L (�500 mg/dL), and then the triglyceridestends to be greater than cholesterol.
The traditional approach to diagnose this disorder isto use lipoprotein electrophoresis; in FDBL, the remnantlipoproteins accumulate in a broad � band. The pre-ferred method to confirm the diagnosis of FDBL is tomeasure VLDL-C by ultracentrifugation and determinethe ratio of VLDL-C to total plasma triglyceride; a ratio�0.30 is consistent with the diagnosis of FDBL. Proteinmethods (apoE phenotyping) or DNA-based methods(apoE genotyping) can be performed to confirm ho-mozygosity for apoE2. However, absence of theapoE2/2 genotype does not rule out the diagnosis of
FDBL, since other mutations in apoE can cause thiscondition.
Because FDBL is associated with increased risk ofpremature ASCVD, it should be treated aggressively.Other metabolic conditions that can worsen the hyper-lipidemia (see above) should be actively treated. Patientswith FDBL are typically very diet responsive and canrespond dramatically to weight reduction and to low-cholesterol, low-fat diets. Alcohol intake should be cur-tailed. In postmenopausal women with FDBL, thedyslipidemia responds to estrogen-replacement therapy.HMG-CoA reductase inhibitors, fibrates, and niacin areall generally effective in the treatment of FDBL, andcombination drug therapy is sometimes required.
Familial Hypercholesterolemia
FH is an autosomal codominant disorder characterizedby elevated plasma LDL-C with normal triglycerides,tendon xanthomas, and premature coronary atheroscle-rosis. FH is caused by �750 mutations in the LDL re-ceptor gene and has a higher incidence in certain popu-lations, such as Afrikaners, Christian Lebanese, andFrench Canadians, due to the founder effect. The ele-vated levels of LDL-C in FH are due to delayed catabo-lism of LDL and its precursor particles from the blood,resulting in increased rates of LDL production.There isa major gene dose effect, in that individuals with twomutated LDL receptor alleles (FH homozygotes) aremuch more affected than those with one mutant allele(FH heterozygotes).
Homozygous FH occurs in approximately 1 in 1 mil-lion persons world-wide. Patients with homozygous FHcan be classified into one of two groups based on theamount of LDL receptor activity measured in their skinfibroblasts: those patients with �2% of normal LDL re-ceptor activity (receptor negative) and those patientswith 2 to 25% of normal LDL receptor activity (recep-tor defective). Most patients with homozygous FH pre-sent in childhood with cutaneous xanthomas on thehands, wrists, elbows, knees, heels, or buttocks. Arcuscornea is usually present and some patients have xanthe-lasmas. Total cholesterol levels are usually �12.93mmol/L (500 mg/dL) and can be �25.86 mmol/L(1000 mg/dL). Accelerated atherosclerosis is a devastat-ing complication of homozygous FH and can result indisability and death in childhood. Atherosclerosis oftendevelops first in the aortic root and can cause aorticvalvular or supravalvular stenosis and typically extendsinto the coronary ostia. Children with homozygous FHoften develop symptomatic vascular disease before pu-berty, when symptoms can be atypical and sudden deathis common. Untreated, receptor-negative patients withhomozygous FH rarely survive beyond the second
340 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
46128_18_p333-354 1/31/06 10:23 AM Page 340

decade; patients with receptor-defective LDL receptordefects have a better prognosis but almost invariably de-velop clinically apparent atherosclerotic vascular diseaseby age 30, and often much sooner. Carotid and femoraldisease develop later in life and are usually not clinicallysignificant.
A careful family history should be taken, and plasmalipid levels should be measured in the parents and otherfirst-degree relatives of patients with homozygous FH.The diagnosis can be confirmed by obtaining a skinbiopsy and measuring LDL receptor activity in culturedskin fibroblasts or by quantifying the number of LDLreceptors on the surfaces of lymphocytes using cell-sortingtechnology.
Combination therapy with an HMG-CoA reductaseinhibitor and a bile acid sequestrant sometimes results inmodest reductions in plasma LDL-C in the FH ho-mozygote. Patients with homozygous FH invariably re-quire additional lipid-lowering therapy. Since the liver isquantitatively the most important tissue for removingcirculating LDL via the LDL receptor, liver transplanta-tion is effective in decreasing plasma LDL-C levels inthis disorder. Liver transplantation is, however, associatedwith substantial risks, including the requirement forlong-term immunosuppression. The current treatmentof choice for homozygous FH is LDL apheresis (aprocess where the LDL particles are selectively removedfrom the circulation), which can promote regression ofxanthomas and may slow the progression of atheroscle-rosis. Initiation of LDL apheresis should be delayed until�5 years of age except when evidence of atheroscleroticvascular disease is present.
Heterozygous FH is caused by the inheritance of onemutant LDL receptor allele and occurs in �1 in 500persons worldwide, making it one of the most commonsingle-gene disorders. It is characterized by elevatedplasma LDL-C [usually 5.17 to 10.34 �mol/L (200 to400 mg/dL)] and normal triglyceride levels. Patientswith heterozygous FH have hypercholesterolemia frombirth, although the disease is often not detected untiladulthood, usually due to the detection of hypercholes-terolemia on routine screening, the appearance of ten-don xanthomas, or the premature development of symp-tomatic coronary atherosclerotic disease. Since thedisease is codominant in inheritance and has a high pen-etrance (�90%), one parent and �50% of the patient’ssiblings are usually hypercholesterolemic.The family his-tory is frequently positive for premature ASCVD on oneside of the family, particularly among male relatives.Corneal arcus is common, and tendon xanthomas in-volving the dorsum of the hands, elbows, knees, and es-pecially the Achilles tendons are present in �75% of pa-tients.The age of onset of ASCVD is highly variable anddepends in part on the molecular defect in the LDL
receptor gene and other coexisting cardiac risk factors.FH heterozygotes with elevated plasma Lp(a) appear tobe at greater risk for cardiovascular complications. Un-treated men with heterozygous FH have an �50%chance of having a myocardial infarction before age 60.Although the age of onset of atherosclerotic heart dis-ease is later in women with FH, coronary disease is sig-nificantly more common in women with FH than inthe general female population.
No definitive diagnostic test for heterozygous FH isavailable. Although FH heterozygotes tend to have re-duced levels of LDL receptor function in skin fibrob-lasts, there is significant overlap with the levels in normalfibroblasts.The clinical diagnosis is usually not problem-atic, but it is critical that hypothyroidism, nephrotic syn-drome, and obstructive liver disease be excluded beforeinitiating therapy.
FH patients should be treated aggressively to lowerplasma levels of LDL-C. Initiation of a low-cholesterol,low-fat diet is recommended, but heterozygous FH pa-tients inevitably require lipid-lowering drug therapy.HMG-CoA reductase inhibitors are especially effective inheterozygous FH, inducing upregulation of the normalLDL receptor allele in the liver. Many heterozygous FHpatients can achieve desired LDL-C levels with HMG-CoA reductase inhibitor therapy alone, but combinationdrug therapy with the addition of a bile acid sequestrantor nicotinic acid is frequently required. Heterozygous FHpatients who cannot be adequately controlled on combi-nation drug therapy are candidates for LDL apheresis.
Familial Defective ApoB-100
Familial defective apoB-100 (FDB) is a dominantly in-herited disorder that clinically resembles heterozygousFH. FDB occurs with a frequency of �1 in 1000 inwestern populations.The disease is characterized by ele-vated plasma LDL-C levels with normal triglycerides,tendon xanthomas, and an increased incidence of pre-mature ASCVD. FDB is caused by mutations in the LDLreceptor–binding domain of apoB-100. Almost all pa-tients with FDB have a substitution of glutamine forarginine at position 3500 in apoB-100, although otherrarer mutations have been reported to cause this disease.As a consequence of the mutation in apoB-100, LDLbinds the LDL receptor with reduced affinity and LDLis removed from the circulation at a reduced rate. Pa-tients with FDB cannot be clinically distinguished frompatients with heterozygous FH, although patients withFDB tend to have lower plasma LDL-C than FH het-erozygotes. The apoB-100 gene mutation can be de-tected directly, but currently genetic diagnosis is not en-couraged since the recommended management of FDBand heterozygous FH is identical.
Chapter 18 Disorders of Lipoprotein Metabolism 341
46128_18_p333-354 1/31/06 10:23 AM Page 341

Autosomal Recessive Hypercholesterolemia
Autosomal recessive hypercholesterolemia (ARH) is arare disorder (except in Sardinia) due to mutations in aprotein (ARH) involved in LDL receptor–mediated en-docytosis in the liver. ARH clinically resembles homozy-gous FH and is characterized by hypercholesterolemia,tendon xanthomas, and premature coronary artery dis-ease. The hypercholesterolemia tends to be intermediatebetween the levels seen in FH homozygotes and FH het-erozygotes. LDL receptor function in cultured fibroblastsis normal or only modestly reduced in ARH, whereasLDL receptor function in lymphocytes and the liver isnegligible. Unlike FH homozygotes, the hyperlipidemiaresponds partially to treatment with HMG-CoA reduc-tase inhibitors, but these patients usually require LDLapheresis to lower plasma LDL-C to recommended levels.
Wolman Disease and Cholesteryl Ester Storage Disease
Wolman disease is an autosomal recessive disordercaused by complete deficiency of lysosomal acid lipase.After LDL is taken up from the cell surface by LDLreceptor–mediated endocytosis, it is delivered from en-dosomes to lysosomes. In the acidic environment of theendosome, the particle dissociates from the receptor,which recycles to the cell surface. In the lysosome,apoB-100 is degraded and the cholesteryl esters andtriglycerides of LDL are hydrolyzed by lysosomal acidlipase. Patients with Wolman disease fail to hydrolyze theneutral lipids, resulting in their accumulation withincells. The disease presents within the first weeks of lifewith hepatosplenomegaly, steatorrhea, adrenal calcifica-tion, and failure to thrive. The disease is usually fatalwithin the first year of life and can be diagnosed bymeasuring acid lipase activity in fibroblasts or liver tissuebiopsy specimens. Cholesteryl ester storage disease is aless severe form of the same genetic disorder in whichthere is low, but detectable, acid lipase activity. Patientswith this disorder sometimes present in childhood withhepatomegaly and a mixed hyperlipidemia, due to eleva-tions in the levels of plasma LDL and VLDL. Other pa-tients present later in life with hepatic fibrosis, portal hy-pertension, or with premature atherosclerosis.
Sitosterolemia
Sitosterolemia is a rare autosomal recessive diseasecaused by mutations in one of two members of theadenosine triphosphate (ATP)-binding cassette trans-porter family, ABCG5 and ABCG8.These genes are ex-pressed in the intestine and liver, where they form afunctional complex to limit intestinal absorption andpromote biliary excretion of plant- and animal-derived
neutral sterols. In normal individuals, �5% of dietaryplant sterols, of which sitosterol is the most plentiful, areabsorbed by the proximal small intestine and deliveredto the liver. Plant sterols in the liver are preferentially se-creted into the bile, and plasma plant sterol levels arenormally very low. In sitosterolemia, the intestinal ab-sorption of plant sterols is increased and biliary excre-tion of the sterols is reduced, resulting in increasedplasma levels of sitosterol and other plant sterols. Thetrafficking of cholesterol is also impaired. Patients withsitosterolemia can have either normal or elevated plasmalevels of cholesterol. Irrespective of the plasma choles-terol level, these patients develop cutaneous and tendonxanthomas as well as premature atherosclerosis. Episodesof hemolysis, presumably secondary to the incorporationof plant sterols into the red blood cell membrane, are adistinctive clinical feature of this disease.The hypercho-lesterolemia in patients with sitosterolemia is unusuallyresponsive to reductions in dietary cholesterol content.Sitosterolemia should be suspected when the plasmacholesterol level falls by �40% on a low-cholesterol diet(without associated weight loss).
Sitosterolemia is confirmed by demonstrating an ele-vated plasma sitosterol level. The hypercholesterolemiadoes not respond to HMG-CoA reductase inhibitors,but bile acid sequestrants and cholesterol-absorption in-hibitors, such as ezetimibe, are effective in reducingplasma sterol levels in these patients.
PRIMARY DISORDERS OF ApoB-CONTAINING LIPOPROTEIN METABOLISM(UNKNOWN ETIOLOGY)
A large proportion of patients with elevated levels ofapoB-containing lipoproteins have disorders in whichthe molecular defect has not been defined, largely be-cause multiple other genetic and nongenetic factorscontribute to the hyperlipidemia.
Familial Hypertriglyceridemia
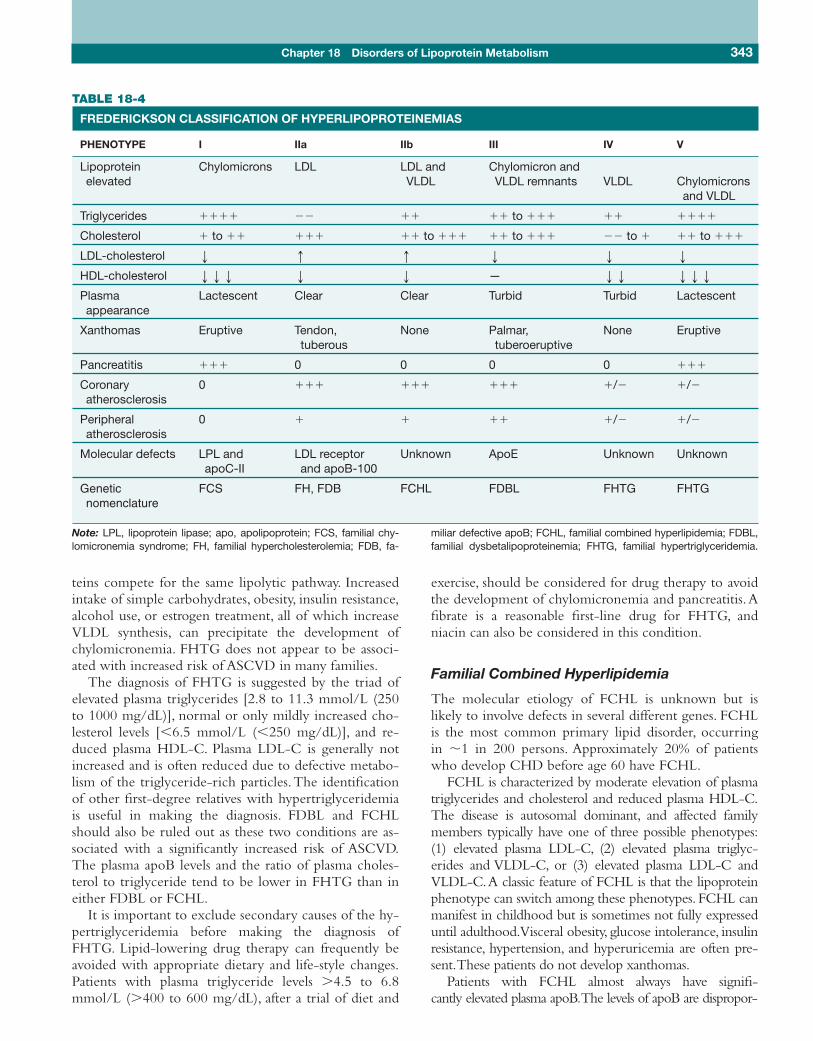
Familial hypertriglyceridemia (FHTG) is a relativelycommon (1 in 500) autosomal dominant disorder of un-known etiology characterized by moderately elevatedplasma triglycerides accompanied by more modest ele-vations in cholesterol. VLDL is the major class oflipoproteins elevated in this disorder, which is often re-ferred to as type IV hyperlipoproteinemia (Fredericksonclassification, Table 18-4).The elevated plasma VLDL isdue to increased VLDL production, impaired VLDL ca-tabolism, or a combination of the two. Some patientswith FHTG have a more severe form of hyperlipidemiain which both VLDL and chylomicrons are elevated(type V hyperlipidemia), as these two classes of lipopro-
342 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
46128_18_p333-354 1/31/06 10:23 AM Page 342
teins compete for the same lipolytic pathway. Increasedintake of simple carbohydrates, obesity, insulin resistance,alcohol use, or estrogen treatment, all of which increaseVLDL synthesis, can precipitate the development ofchylomicronemia. FHTG does not appear to be associ-ated with increased risk of ASCVD in many families.
The diagnosis of FHTG is suggested by the triad ofelevated plasma triglycerides [2.8 to 11.3 mmol/L (250to 1000 mg/dL)], normal or only mildly increased cho-lesterol levels [�6.5 mmol/L (�250 mg/dL)], and re-duced plasma HDL-C. Plasma LDL-C is generally notincreased and is often reduced due to defective metabo-lism of the triglyceride-rich particles.The identificationof other first-degree relatives with hypertriglyceridemiais useful in making the diagnosis. FDBL and FCHLshould also be ruled out as these two conditions are as-sociated with a significantly increased risk of ASCVD.The plasma apoB levels and the ratio of plasma choles-terol to triglyceride tend to be lower in FHTG than ineither FDBL or FCHL.
It is important to exclude secondary causes of the hy-pertriglyceridemia before making the diagnosis ofFHTG. Lipid-lowering drug therapy can frequently beavoided with appropriate dietary and life-style changes.Patients with plasma triglyceride levels �4.5 to 6.8mmol/L (�400 to 600 mg/dL), after a trial of diet and
exercise, should be considered for drug therapy to avoidthe development of chylomicronemia and pancreatitis.Afibrate is a reasonable first-line drug for FHTG, andniacin can also be considered in this condition.
Familial Combined Hyperlipidemia
The molecular etiology of FCHL is unknown but islikely to involve defects in several different genes. FCHLis the most common primary lipid disorder, occurringin �1 in 200 persons. Approximately 20% of patientswho develop CHD before age 60 have FCHL.
FCHL is characterized by moderate elevation of plasmatriglycerides and cholesterol and reduced plasma HDL-C.The disease is autosomal dominant, and affected familymembers typically have one of three possible phenotypes:(1) elevated plasma LDL-C, (2) elevated plasma triglyc-erides and VLDL-C, or (3) elevated plasma LDL-C andVLDL-C.A classic feature of FCHL is that the lipoproteinphenotype can switch among these phenotypes. FCHL canmanifest in childhood but is sometimes not fully expresseduntil adulthood.Visceral obesity, glucose intolerance, insulinresistance, hypertension, and hyperuricemia are often pre-sent.These patients do not develop xanthomas.
Patients with FCHL almost always have signifi-cantly elevated plasma apoB.The levels of apoB are dispropor-
Chapter 18 Disorders of Lipoprotein Metabolism 343
TABLE 18-4
FREDERICKSON CLASSIFICATION OF HYPERLIPOPROTEINEMIAS
PHENOTYPE I IIa IIb III IV V
Lipoprotein Chylomicrons LDL LDL and Chylomicron and elevated VLDL VLDL remnants VLDL Chylomicrons
and VLDL
Triglycerides ���� �� �� �� to ��� �� ����
Cholesterol � to �� ��� �� to ��� �� to ��� �� to � �� to ���
LDL-cholesterol p q q p p p
HDL-cholesterol ppp p p — pp ppp
Plasma Lactescent Clear Clear Turbid Turbid Lactescentappearance
Xanthomas Eruptive Tendon, None Palmar, None Eruptivetuberous tuberoeruptive
Pancreatitis ��� 0 0 0 0 ���
Coronary 0 ��� ��� ��� �/� �/�atherosclerosis
Peripheral 0 � � �� �/� �/�atherosclerosis
Molecular defects LPL and LDL receptor Unknown ApoE Unknown UnknownapoC-II and apoB-100
Genetic FCS FH, FDB FCHL FDBL FHTG FHTGnomenclature
Note: LPL, lipoprotein lipase; apo, apolipoprotein; FCS, familial chy-lomicronemia syndrome; FH, familial hypercholesterolemia; FDB, fa-
miliar defective apoB; FCHL, familial combined hyperlipidemia; FDBL,familial dysbetalipoproteinemia; FHTG, familial hypertriglyceridemia.
46128_18_p333-354 1/31/06 10:23 AM Page 343

tionately high relative to plasma LDL-C due to the pres-ence of small dense LDL particles, which are characteristicof this syndrome and are highly atherogenic. Hyperapobe-talipoproteinemia has been used as a term to de-scribe thecoupling of elevated plasma apoB with normal plasmacholesterol, and is probably a form of FCHL.
A mixed dyslipidemia [plasma triglyceride levels be-tween 2.3 and 9.0 mmol/L (200 and 800 mg/dL), cho-lesterol levels between 5.2 and 10.3 mmol/L (200 and400 mg/dL), and HDL-C levels �10.3 mmol/L (�40mg/dL)] and a family history of hyperlipidemia and/orpremature CHD suggests the diagnosis of FCHL.An el-evated plasma apoB level or an increased number ofsmall dense LDL particles in the plasma supports this di-agnosis. FDBL should be considered and ruled out bybeta-quantification in suspected patients with a mixedhyperlipidemia.
Individuals with FCHL should be treated aggressivelydue to significantly increased risk of premature CHD.Decreased dietary intake of saturated fat and simple car-bohydrates, aerobic exercise, and weight loss have benefi-cial effects on the lipid profile. Patients with diabetesshould be aggressively treated to maintain good glucosecontrol. Most patients with FCHL require lipid-loweringdrug therapy to reduce lipoprotein levels to the recom-mended range. HMG-CoA reductase inhibitors are veryeffective in lowering plasma levels of LDL-C and can alsosignificantly reduce VLDL-C. Nicotinic acid decreasesboth LDL-C and VLDL-C, while raising plasma HDL-C,and is frequently effective for this condition when used incombination with HMG-CoA reductase inhibitors.
Polygenic Hypercholesterolemia
Polygenic hypercholesterolemia is characterized by hy-percholesterolemia with a normal plasma triglyceride inthe absence of secondary causes of hypercholesterolemia.Plasma LDL-C levels are not as elevated as they are in FHand FDB. Family studies are useful to differentiate poly-genic hypercholesterolemia from the single-gene disor-ders described above; half of the first-degree relatives ofpatients with FH and FDB are hypercholesterolemic,whereas �10% of first-degree relatives of patients withpolygenic hypercholesterolemia are hypercholesterolemic.Treatment of polygenic hypercholesterolemia is identicalto that of other forms of hypercholesterolemia.
GENETIC DISORDERS OF HDLMETABOLISM (KNOWN ETIOLOGY)
Mutations in certain genes encoding critical proteins inHDL synthesis and catabolism cause marked variationsin plasma HDL-C levels. Unlike the genetic forms ofhypercholesterolemia, which are invariably associated
with premature coronary atherosclerosis, genetic formsof hypoalphalipoproteinemia (low HDL-C) are not al-ways associated with accelerated atherosclerosis.Whereashigh plasma LDL-C is invariably associated with in-creased atherosclerosis, the risk associated with lowplasma levels of HDL-C depends on the underlyingmechanism. Analysis of the genetic disorders of HDLmetabolism has provided insights into the less well un-derstood etiologic relationship between plasma HDL-Clevels and atherosclerosis.
ApoA-I Deficiency and ApoA-I Mutations
Complete genetic deficiency of apoA-I due to muta-tions in the apoA-I gene results in the virtual absence ofHDL from the plasma. The genes encoding apoA-I,apoC-III, apoA-IV, and apoA-V are clustered togetheron chromosome 11, and some patients with completeabsence of apoA-I have deletions that include more thanone of these genes. Because apoA-I is required forLCAT function, plasma and tissue levels of free choles-terol are increased, resulting in the development ofcorneal opacities and planar xanthomas. Clinicallyapparent coronary atherosclerosis typically appearsbetween the fourth and seventh decade in the apoA-I-deficient patient.
Although missense mutations in the apoA-I genehave been identified in selected patients with lowplasma HDL [usually 0.39 to 0.78 mmol/L (15 to 30mg/dL)], they are very rare causes of low HDL-C levelsin the general population. Patients with apoA-IMilano
have very low plasma levels of HDL due to the rapid ca-tabolism of the apolipoprotein, but these patients do nothave an increased risk of premature CHD. Other thancorneal opacities, most individuals with low plasmaHDL-C levels due to missense mutations in apoA-Ihave no clinical sequelae. A few specific mutations inapoA-I cause systemic amyloidosis, and the mutantapoA-I has been found as a component of the amyloidplaque.
Tangier Disease
Tangier disease is a rare autosomal codominant form oflow plasma HDL-C caused by mutations in the gene en-coding ABCA1, a cellular transporter that facilitates effluxof unesterified cholesterol and phospholipids from cells toapoA-I (Fig. 18-3).ABCA1 plays a critical role in the gen-eration and stabilization of the mature HDL particle. In itsabsence, HDL is rapidly cleared from the circulation. Pa-tients with Tangier disease have plasma HDL-C levels�0.13 mmol/L (�5 mg/dL) and extremely low circulat-ing levels of apoA-I.The disease is associated with choles-terol accumulation in the reticuloendothelial system,
344 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
46128_18_p333-354 1/31/06 10:23 AM Page 344

resulting in hepatosplenomegaly and pathognomonic en-larged, grayish yellow or orange tonsils. An intermittentperipheral neuropathy (mononeuritis multiplex) or asphingomyelia-like neurologic disorder can also be seen inthis disorder. Tangier disease is associated with prematureatherosclerotic disease, but the risk is not as high as mightbe anticipated given the markedly decreased plasma HDL-C and apoA-I. Plasma LDL-C is also low and this may at-tenuate the atherosclerotic risk. Obligate heterozygotes forABCA1 mutations have moderately reduced plasmaHDL-C levels and are also at increased risk of prematureCHD.
LCAT Deficiency
LCAT deficiency is a rare disorder caused by mutationsin lecithin-cholesterol acyltransferase (Fig. 18-3). LCATis synthesized in the liver and secreted into the plasma,where it circulates associated with lipoproteins. Becausethe enzyme mediates the esterification of cholesterol, theproportion of free cholesterol in circulating lipoproteinsis greatly increased (from �25% to �70% of total plasmacholesterol). Lack of normal cholesterol esterificationimpairs the formation of mature HDL particles and leadsto rapid catabolism of circulating apoA-I. Two geneticforms of LCAT deficiency have been described inhumans—complete deficiency (also called classic LCATdeficiency) and partial deficiency (also called fish-eye disease).Progressive corneal opacification due to the depositionof free cholesterol in the lens, very low plasma HDL-C[usually �0.26 mmol/L (�10 mg/dL)], and variable hy-pertriglyceridemia are characteristic of both types. Inpartial LCAT deficiency, there are no other known clini-cal sequelae. In contrast, complete LCAT deficiency ischaracterized by a hemolytic anemia and progressive re-nal insufficiency that eventually leads to end-stage renaldisease (ESRD). Despite the extremely low plasma levelsof HDL-C and apoA-I, premature ASCVD is not a fea-ture of either complete or partial LCAT deficiency, onceagain exemplifying the complex relationship betweenlow plasma levels of HDL-C and the development ofASCVD. The diagnosis can be confirmed by assayingLCAT activity in the plasma.
CETP Deficiency
Mutations in the gene encoding cholesteryl ester trans-fer protein (CETP) cause a high HDL-C conditioncalled CETP deficiency. CETP facilitates the transfer ofcholesteryl esters among lipoproteins, especially fromHDL to apoB-containing lipoproteins in exchange fortriglycerides (Fig. 18-3). Homozygous deficiency ofCETP, which occurs predominantly in Japan, results invery high plasma HDL-C [�3.88 mmol/L (�150
mg/dL)] due to accumulation of large, cholesterol-richHDL particles. Heterozygotes for CETP deficiency haveonly modestly elevated HDL-C. The relationship ofCETP deficiency to risk of ASCVD remains a matter ofdebate.
PRIMARY DISORDERS OF HDLMETABOLISM (UNKNOWN ETIOLOGY)
The gene defect in other individuals with either veryhigh or very low plasma HDL-C is not known.
Primary Hypoalphalipoproteinemia
The most common inherited cause of low plasmaHDL-C is termed primary or familial hypoalphalipopro-teinemia. Hypoalphalipoproteinemia is defined as aplasma HDL-C level below the 10th percentile in thesetting of relatively normal cholesterol and triglyceridelevels, no apparent secondary causes of low plasmaHDL-C, and no clinical signs of LCAT deficiency orTangier disease. This syndrome is often referred to as“isolated low HDL.”A family history of low HDL-C fa-cilitates the diagnosis of an inherited condition, whichusually follows an autosomal dominant pattern. Themetabolic etiology of this disease appears to be primar-ily accelerated catabolism of HDL and its apolipopro-teins. Several kindreds with primary hypoalphalipopro-teinemia have been described in association with anincreased incidence of premature ASCVD.
Familial Hyperalphalipoproteinemia
Familial hyperalphalipoproteinemia has a dominant in-heritance pattern. Plasma HDL-C is usually �2.07mmol/L (80 mg/dL) in affected women and �1.81mmol/L (70 mg/dL) in affected men.The genetic basisof primary hyperalphalipoproteinemia is not known,and the condition may be associated with decreased riskof CHD and increased longevity in some cases.
SECONDARY DISORDERS OF LIPOPROTEINMETABOLISM
Significant changes in plasma levels of lipoproteins areseen in a variety of diseases. It is critical that secondarycauses of hyperlipidemias (Table 18-5) are consideredprior to initiation of lipid-lowering therapy.
Obesity
Obesity is frequently, though not invariably, accompaniedby hyperlipidemia. The increase in adipocyte mass andaccompanying decrease in insulin sensitivity associatedwith obesity have multiple effects on lipid metabolism.
Chapter 18 Disorders of Lipoprotein Metabolism 345
46128_18_p333-354 1/31/06 10:23 AM Page 345

More free fatty acids are delivered from the expandedadipose tissue to the liver where they are re-esterified inhepatocytes to form triglycerides, which are packagedinto VLDL for secretion into the circulation. High di-etary intake of simple carbohydrates also drives hepaticproduction of VLDL, leading to increases in VLDLand/or LDL in some obese individuals. Plasma HDL-Ctends to be low in obesity.Weight loss is often associatedwith a reduction of plasma apoB-containing lipopro-teins and an increase of plasma HDL-C.
Diabetes Mellitus
Patients with type 1 diabetes mellitus are generally nothyperlipidemic if they are under good glycemic control.Diabetic ketoacidosis is frequently accompanied by hy-pertriglyceridemia due to increased hepatic influx offree fatty acids from adipose tissue. The hypertriglyc-eridemia responds dramatically to administration of in-sulin in the insulinopenic diabetic.
Patients with type 2 diabetes mellitus are usually dys-lipidemic, even if under relatively good glycemic con-trol.The high levels of insulin and insulin resistance as-sociated with type 2 diabetes have multiple effects onfat metabolism: (1) a decrease in LPL activity resultingin reduced catabolism of chylomicrons and VLDL,
(2) an increase in the release of free fatty acid from theadipose tissue, (3) an increase in fatty acid synthesis in theliver, and (4) an increase in hepatic VLDL production.Patients with type 2 diabetes mellitus have several lipidabnormalities, including elevated plasma triglycerides(due to increased VLDL and lipoprotein remnants), ele-vated dense LDL, and decreased HDL-C. In some dia-betic patients, especially those with a genetic defect inlipid metabolism, the triglycerides can be extremely ele-vated. Elevated plasma LDL-C levels are usually not afeature of diabetes mellitus and suggest the presence ofan underlying lipoprotein abnormality or may indicatethe development of diabetic nephropathy. Patients withlipodystrophy, who have profound insulin resistance, havemarkedly elevated VLDL and chylomicrons.
Thyroid Disease
Hypothyroidism is associated with elevated plasmaLDL-C due primarily to a reduction in hepatic LDL re-ceptor function and delayed clearance of LDL. Con-versely, plasma LDL-C is often reduced in the hyperthy-roid patient. Hypothyroid patients may have increasedcirculating IDL, and some are mildly hypertriglyceri-demic [�3.34 �mol/L (�300 mg/dL)]. Because hy-pothyroidism is easily overlooked, all patients presenting
346 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
TABLE 18-5
SECONDARY FORMS OF HYPERLIPIDEMIA
LDL HDL
VLDL IDL CHYLOMICRONS LP(A) ELEVATED REDUCED ELEVATED REDUCED ELEVATED ELEVATED ELEVATED ELEVATED
Note: LDL, low-density lipoprotein; HDL, high-density lipoprotein;VLDL, very low density lipoprotein; IDL, intermediate-density
lipoprotein; Lp(a), lipoprotein A; DM, diabetes mellitus.
Renalinsufficiency
Inflammation
Menopause
Orchidectomy
Hypothyroidism
Acromegaly
Nephrosis
Drugs: growthhormone
Hypothyroidism
Nephroticsyndrome
Cholestasis
Acuteintermittentporphyria
Anorexianervosa
Hepatoma
Drugs:thiazides,cyclosporine,tegretol
Severe liverdisease
Malabsorption
Malnutrition
Gaucher disease
Chronic infectiousdisease
Hyperthyroidisim
Drugs: niacintoxicity
Smoking
DM type 2
Obesity
Malnutrition
Gaucherdisease
Drugs:anabolicsteroids,betablockers
Obesity
DM type 2
Glycogen storagedisease
Hepatitis
Alcohol
Renal failure
Sepsis
Stress
Cushing syndrome
Pregnancy
Acromegaly
Lipodystrophy
Drugs: estrogen, betablockers, furosemide,glucocrticoids, bileacid –bindingresins, retinoicacid, HIV proteaseinhibitors
Multiplemyeloma
Monoclonalgammopathy
Autoimmunedisease
Hypothyroidism
Autoimmunedisease
Drug:Isotretinoin
Alcohol
Exercise
Exposure tochlorinatedhydrocarbons
Drugs:estrogen
46128_18_p333-354 1/31/06 10:23 AM Page 346

with elevated plasma LDL-C or IDL should be screenedfor hypothyroidism.Thyroid replacement therapy usuallyameliorates the hypercholesterolemia.
Renal Disorders
Nephrotic syndrome is associated with hyperlipoproteine-mia, which is usually mixed but can manifest as hypercho-lesterolemia or hypertriglyceridemia alone.The hyperlipi-demia of nephrotic syndrome appears to be due to acombination of increased hepatic production and de-creased clearance of VLDL, with increased LDL produc-tion. Effective treatment of the underlying renal diseasenormalizes the lipid profile, but most patients with chronicnephrotic syndrome require lipid-lowering drug therapy.
ESRD is often associated with mild hypertriglyc-eridemia [�3.34 �mol/L (�300 mg/dL)] due to theaccumulation of VLDL and remnant lipoproteins in thecirculation. Triglyceride lipolysis and remnant clearanceare both reduced in patients with renal failure. Becausethe risk of ASCVD is increased in hyperlipidemic pa-tients with ESRD, they should be treated aggressivelywith lipid-lowering agents.
Patients with renal transplants are usually hyperlipi-demic due to immunosuppression drugs (cyclosporineand glucocorticoids); they present a difficult manage-ment problem as HMG-CoA reductase inhibitors mustbe used cautiously in these patients.
Liver Disorders
Because the liver is the principal site of formation andclearance of lipoproteins, it is not surprising that liver dis-eases can profoundly affect plasma lipid levels in a varietyof ways. Hepatitis due to infection, drugs, or alcohol is of-ten associated with increased VLDL synthesis and mild tomoderate hypertriglyceridemia. Severe hepatitis and liverfailure are associated with dramatic reductions in plasmacholesterol and triglycerides due to reduced lipoproteinbiosynthetic capacity. Cholestasis is associated with hyperc-holesterolemia, which sometimes can be very severe.Themajor pathway by which cholesterol is excreted is via se-cretion into bile, either directly or after conversion to bileacids. Cholestasis blocks this critical excretory pathway. Incholestasis, free cholesterol coupled with phospholipids aresecreted into the plasma as constituents of a lamellar parti-cle called Lp(X). These particles can deposit in skin folds,producing lesions resembling those seen in patients withFDBL (xanthomata strata palmaris). Planar and eruptivexanthomas can also be seen in patients with cholestasis.
Alcohol
Regular alcohol consumption has a variable effect onplasma lipid levels.The most common effect of alcohol is
to increase plasma triglyceride levels. Alcohol consump-tion stimulates hepatic secretion of VLDL, possibly by in-hibiting the hepatic oxidation of free fatty acids, whichthen promote hepatic triglyceride synthesis and VLDLsecretion. The usual lipoprotein pattern seen with alco-hol consumption is type IV (increased VLDL), but per-sons with an underlying primary lipid disorder may de-velop severe hypertriglyceridemia (type V) if they drinkalcohol. Regular alcohol use is also associated with amild to moderate increase in plasma levels of HDL-C.
Estrogen
Estrogen administration is associated with increasedVLDL and HDL synthesis resulting in elevated plasmatriglycerides and HDL-C. This lipoprotein pattern isdistinctive since the levels of plasma triglyceride andHDL-C are typically inversely related. Estrogen treat-ment may convert a person with type IV to type V hy-perlipidemia. Plasma triglyceride levels should be moni-tored when birth control pills or estrogen replacementtherapy is initiated. Use of low-dose estrogen prepara-tions or the estrogen patch can minimize the effect ofexogenous estrogen on lipids.
Glycogen Storage Diseases
Other rarer causes of secondary hyperlipidemias includeglycogen storage diseases such as von Gierke’s disease,which is caused by mutations in glucose-6-phosphatase.The inability to mobilize hepatic glucose during fastingresults in hypoinsulinemia and increased release of freefatty acids from adipose tissue. Hepatic fatty acids syn-thesis is also increased, resulting in fat accumulation inthe liver and increased VLDL secretion. The hyperlipi-demia associated with this disease can be very severe butresponds well to treatment of the underlying disorder.
Cushing Syndrome
Glucocorticoid excess is associated with increased VLDLsynthesis and hypertriglyceridemia. Patients with Cushingsyndrome can also have mild elevations in plasma LDL-C.
Drugs
Many drugs have a significant impact on lipid metabo-lism and can result in significant alterations in thelipoprotein profile (Table 18-5).
SCREENING
Guidelines for the screening and management of lipiddisorders have been provided by an expert Adult Treat-ment Panel (ATP) convened by the National Cholesterol
Chapter 18 Disorders of Lipoprotein Metabolism 347
46128_18_p333-354 1/31/06 10:23 AM Page 347

Education Program (NCEP) of the National HeartLung and Blood Institute.The NCEP ATPIII guidelinespublished in 2001 recommend that all adults over age 20have plasma levels of cholesterol, triglyceride, LDL-C,and HDL-C measured after a 12-h overnight fast. Inmost clinical laboratories, the total cholesterol andtriglycerides in the plasma are measured enzymaticallyand then the cholesterol in the supernatant is measuredafter precipitation of apoB-containing lipoproteins todetermine the HDL-C. The LDL-C is estimated usingthe following equation:
LDL-C � total cholesterol � (triglycerides/5) � HDL-C
The VLDL-C is estimated by dividing the plasmatriglyceride by 5, reflecting the ratio of cholesterol totriglyceride in VLDL particles.This formula is reason-ably accurate if test results are obtained on fastingplasma and if the triglyceride level ��4.0 �mol/L(350 mg/dL). The accurate determination of LDL-Clevels in patients with triglyceride levels greater thanthis requires application of ultracentrifugation tech-niques (beta quantification), although direct assays forLDL-C are also available in some laboratories.
348 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
TREATMENT FOR CHDMultiple epidemiologic studies have demonstrateda strong relationship between serum cholesteroland CHD. Randomized controlled clinical trialshave unequivocally documented that loweringplasma cholesterol reduces the risk of clinicalevents due to atherosclerosis.Although the propor-tional benefit accrued from reducing plasma LDL-C is similar over the entire range of LDL-C val-ues, the absolute risk reduction depends on thebaseline LDL-C, the presence of established CHD,and other cardiovascular risk factors.
Elevated plasma triglyceride levels are also asso-ciated with increased risk of CHD, but this rela-tionship weakens considerably when statistical cor-rections are made for the plasma levels of LDL-Cand HDL-C. Plasma levels of HDL-C are stronglyand consistently inversely related to the prevalenceand incidence of CHD, and yet no clinical trialdata are available demonstrating that increasingplasma levels of HDL-C reduces the frequency ofcardiovascular events. No pharmacologic agentsare available that exclusively either lower plasmatriglyceride levels or increase plasma HDL-Clevels, contributing to the dearth of clinical trial
data addressing the role of treatment of these lipidabnormalities in CHD prevention. Since both hy-pertriglyceridemia and low plasma levels of HDL-C confer higher ASCVD risk, the NCEP ATPIIIrecommends more aggressive therapy to lower theplasma LDL-C in patients with these dyslipi-demias.
Nonpharmacologic TreatmentDIET
Dietary modification is an important componentin the management of hyperlipidemia. In the hy-percholesterolemic patient, dietary saturated fatand cholesterol should be restricted. For patientswho are hypertriglyceridemic, the intake of sim-ple sugars should also be curtailed. For severe hy-pertriglyceridemia [�11.3 mmol/L (�1000mg/dL)], restriction of total fat intake is critical.The most widely used diet to lower the LDL-Clevel is the “Step 1 diet” developed by the Ameri-can Heart Association. Most patients have a rela-tively modest (�10%) decrease in plasma levels ofLDL-C on a step I diet in the absence of any as-sociated weight loss. Almost all persons experi-ence a decrease in plasma HDL-C levels with areduction in the amount of total and saturated fatin their diet.
FOODS AND ADDITIVES
Certain foods and dietary additives are associatedwith modest reductions in plasma cholesterol lev-els. Plant stanol and sterol esters are available in avariety of foods such as spreads, salad dressings, andsnack bars.They interfere with cholesterol absorp-tion and reduce plasma LDL-C levels by �10 to15% when taken three times per day.The additionto the diet of psyllium, soy protein, or Chinese redyeast rice (which contains lovastatin) can havemodest cholesterol-lowering effects. Other herbalapproaches such as guggulipid require furtherstudy to assess their effectiveness.
WEIGHT LOSS AND EXERCISE
The treatment of obesity, if present, can have a fa-vorable impact on plasma lipid levels and shouldbe actively encouraged. Plasma triglyceride andLDL-C levels tend to fall and HDL-C levels tendto increase in obese persons who lose weight.Aer-obic exercise has a very modest elevating effect onplasma levels of HDL-C in most individuals buthas cardiovascular benefits that extend beyond theeffects on plasma lipid levels.
46128_18_p333-354 1/31/06 10:23 AM Page 348

Chapter 18 Disorders of Lipoprotein Metabolism 349
Pharmacologic Treatment
The decision to use drug therapy depends on thecardiovascular risk. An effective way to estimateabsolute risk of a cardiovascular event over 10years is to use a scoring system based on theFramingham Heart Study database. Patients with a10-year absolute CHD risk of �20% are consid-ered “CHD risk equivalents.” Current NCEP AT-PIII guidelines call for drug therapy to reduceLDL-C to �2.6 mmol/L (�100 mg/dL) in pa-tients with established CHD, other ASCVD (aortic aneurysm, peripheral vascular disease, orcerebrovascular disease), diabetes mellitus, orCHD risk equivalents. Based on these guidelines,most CHD and CHD risk–equivalent patientsrequire cholesterol-lowering drug therapy. Mod-erate risk patients with two or more risk factorsand a 10-year absolute risk of 10 to 20% shouldbe treated to a goal LDL-C of �3.4 mmol/L(�130 mg/dL).All other individuals have a goal ofLDL-C �4.1 mmol/L (�160 mg/dL), but not allpersons are candidates for drug therapy to achievethis goal.
Persons with markedly elevated plasma LDL-Clevels [�4.9 mmol/L (�190 mg/dL)] should beconsidered for drug therapy even if their 10-yearabsolute CHD risk is not particularly elevated.The decision to initiate drug treatment in indi-viduals with plasma LDL-C levels between 3.4and 4.9 mmol/L (130 and 190 mg/dL) can bedifficult. Although it is desirable to avoid drugtreatment in patients who are unlikely to developCHD, a very high proportion of patients whoeventually develop CHD have plasma LDL-Clevels that are in this range. Other clinical infor-mation can assist in the decision-making process.For example, a low plasma HDL-C [�1.0mmol/L (�40 mg/dL)] supports a decision in fa-vor of more aggressive therapy. The diagnosis ofthe metabolic syndrome also identifies a higherrisk individual who should be targeted for thera-peutic life-style changes and might be a candidatefor more aggressive drug therapy. Other labora-tory tests, such as an elevated plasma Lp(a) orhigh-sensitivity C-reactive protein, may help toidentify additional high-risk individuals. In per-sons at low risk, the emphasis should primarily beon dietary and life-style modification.
Drug treatment is also indicated in patientswith triglycerides �11.3 mmol/L (�1000 mg/dL)who have been screened and treated for secondarycauses of chylomicronemia. The goal is to reduce
plasma triglycerides to �4.5 mmol/L (400 mg/dL)to prevent the risk of acute pancreatitis. Mostmajor clinical end-point trials with statins haveexcluded persons with triglyceride levels �3.9 to5.1 mmol/L (�350 to 450 mg/dL), and thereforethere are few data regarding the effectiveness ofstatins in reducing cardiovascular risk in personswith triglycerides higher than this threshold.Combination therapy is often required for optimalcontrol of mixed dyslipidemia.
HMG-COA REDUCTASE INHIBITORS
3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA reductase) is the rate-limiting step in choles-terol biosynthesis, and inhibition of this enzyme de-creases cholesterol synthesis. By inhibitingcholesterol biosynthesis, HMG-CoA reductase in-hibitors (statins) lead to increased hepatic LDL re-ceptor activity and accelerated clearance of circulat-ing LDL, resulting in a dose-dependent reductionin plasma LDL-C. There is wide interindividualvariation in the initial response to a statin, but oncea patient is on the medication, the doubling of thedose produces a 6% further reduction of plasmaLDL-C. The HMG-CoA reductase inhibitors cur-rently available differ in their LDL-C reducingeffects (Table 18-6). HMG-CoA reductase in-hibitors also reduce plasma triglycerides in a dose-dependent fashion, which is proportional to theirLDL-C lowering effects [if the triglycerides are�3.9 mmol/L (�350 mg/dL)]. HMG-CoA reduc-tase inhibitors have a modest HDL-raising effect (5to 10%), and this effect is not dose-dependent.
HMG-CoA reductase inhibitors are well toler-ated and can be taken in tablet form once a day.Potential side effects include dyspepsia, headaches,fatigue, and muscle or joint pains. Severe myopa-thy and even rhabdomyolysis occurs rarely. Therisk of myopathy is increased by the presence ofrenal insufficiency and by coadministration ofdrugs that interfere with the metabolism ofHMG-CoA reductase inhibitors, such as ery-thromycin and related antibiotics, antifungalagents, immunosuppressive drugs, and fibric acidderivatives. Severe myopathy can usually beavoided by careful patient selection, avoidance ofinteracting drugs, and by instructing the patient tocontact the physician immediately in the event ofunexplained muscle pain. In the event of musclesymptoms, the plasma creatine phosphokinase(CPK) level should be obtained to document themyopathy, but serum CPK levels do not need to
46128_18_p333-354 1/31/06 10:23 AM Page 349

350 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
TABLE 18-6
SUMMARY OF THE MAJOR DRUGS USED FOR THE TREATMENT OF HYPERLIPIDEMIA
DRUG MAJOR INDICATIONS STARTING DOSE MAXIMAL DOSE MECHANISM COMMON SIDE EFFECTS
HMG-CoA reductase Elevated LDL p Cholesterol Myalgias, arthralgias, inhibitors (statins) synthesis, elevated transaminases,Lovastatin 20 mg daily 80 mg daily p hepatic LDL dyspepsiaPravastatin 40 mg qhs 80 mg qhs receptors Simvastatin 20 mg qhs 80 mg qhsFluvastatin 20 mg qhs 80 mg qhs p VLDL productionAtorvastatin 10 mg qhs 80 mg qhsRosuvastatin 10 mg qhs 40 mg qhs
Bile acid sequestrants Elevated LDL q Bile acid excretion Bloating, constipation, Cholestyramine 4 g daily 32 g daily q LDL receptors elevated trigylceridesColestipol 5 g daily 40 g dailyColesevelam 3750 mg daily 4375 mg daily
Nicotinic acid Elevated LDL, low p VLDL hepatic Cutaneous flushing; GI Immediate-release HDL, elevated TG 100 mg tid 2 g tid synthesis upset; elevated glucose, Sustained-release 250 mg bid 1.5 g bid uric acid, and liver Extended-release 500 mg qhs 2 g qhs function tests
Fibric acid derivatives Elevated TG, elevated q LPL Dyspepsia, myalgia, Gemfibrozil remnants 600 mg bid 600 mg bid p VLDL gallstones, elevated Fenofibrate 160 mg qd 160 mg qd synthesis transaminases
Fish oils Severely elevated TG 3 g daily 12 g daily p Chylomicron and Dyspepsia, diarrhea, fishy VLDL production odor to breath
Cholesterol absorption inhibitors p Intenstinal cholesterol Elevated transaminasesEzetimibe Elevated LDL 10 mg daily 10 mg daily absorption
be monitored on a routine basis as an elevatedCPK in the absence of symptoms does not predictthe development of myopathy and does not neces-sarily suggest the need for discontinuing the drug.
Another side effect of HMG-CoA reductase in-hibitor therapy is hepatitis. Liver transaminases(ALT and AST) should be checked before startingtherapy, at 8 weeks, and then every 6 months. Sub-stantial (�3 � upper limit of normal) elevation intransaminases is relatively rare, and mild to moder-ate (1 to 3 � normal) elevation in transaminases inthe absence of symptoms need not mandate dis-continuing the medication. Severe clinical hepatitisassociated with HMG-CoA reductase inhibitors isexceedingly rare, and the trend is toward less fre-quent monitoring of transaminases in patients tak-ing HMG-CoA reductase inhibitors. The HMG-CoA reductase inhibitor–associated elevation inliver enzymes resolves after discontinuation of themedication.
Overall, HMG-CoA reductase inhibitors ap-pear to be remarkably safe. Over 50,000 patientshave been treated with HMG-CoA reductaseinhibitors for over 5 to 6 years as a part of large
randomized controlled clinical trials and no in-crease in any major noncardiac diseases have beenseen in these individuals. HMG-CoA reductaseinhibitors are the drug class of choice for LDL-Creduction and are by far the most widely usedclass of lipid-lowering drugs.
BILE ACID SEQUESTRANTS (RESINS)
Bile acid sequestrants bind bile acids in the intes-tine and promote their excretion in the stool. Inorder to maintain an adequate bile acid pool, theliver diverts cholesterol to bile acid synthesis. Thedecreased hepatic intracellular cholesterol contentupregulates the LDL receptor and enhances LDLclearance from the plasma. Bile acid sequestrants,including cholestyramine, colestipol, and coleseve-lam (Table 18-6), primarily reduce plasma LDL-Clevels but can increase plasma triglycerides.There-fore, patients with hypertriglyceridemia should notbe treated with bile acid–binding resins.
Cholestyramine and colestipol are insolubleresins that must be mixed with liquids. Colestipolis also available in large tablets but multiple tabletsmust be taken to achieve significant lowering of
Note: LDL, low-density lipoprotein; VLDL, very low density lipoprotein; HDL, high-density lipoprotein; TG, triglycerides; LPL, lipoprotein lipase.
46128_18_p333-354 2/3/06 3:18 PM Page 350

Chapter 18 Disorders of Lipoprotein Metabolism 351
plasma LDL-C levels.The newest bile acid seques-trant, colesevelam, has greater bile acid–bindingcapacity than traditional resins. The colesevelamtablets are smaller, and fewer tablets per day are re-quired. Most side effects of resins are limited to thegastrointestinal tract and include bloating and con-stipation. Bile acid sequestrants may bind otherdrugs (e.g., digoxin, warfarin) and interfere withtheir absorption. Therefore, all other medicationsshould be taken either 1 h before or 4 h after thebile acid sequestrants.
Bile acid sequestrants are not systemically ab-sorbed and are very safe.They are the cholesterol-lowering drug of choice in children and inwomen of childbearing age who are lactating,pregnant, or could become pregnant. These drugscan also be useful in young, well-motivated pa-tients with moderate hypercholesterolemia whowish to avoid systemic drug therapy. This class ofdrugs is also useful in combination with HMG-CoA reductase inhibitors in patients who are un-able to reach their LDL-C goal on HMG-CoAreductase inhibitor monotherapy and have rela-tively normal triglyceride levels.
NICOTINIC ACID (NIACIN)
Nicotinic acid, or niacin, is a B-complex vitaminthat reduces plasma triglyceride and LDL-C lev-els and raises the plasma HDL-C (Table 18-6) inhigh doses. Niacin is the only currently availablelipid-lowering drug that significantly reducesplasma levels of Lp(a). If properly prescribed andmonitored, niacin is a safe and effective lipid-lowering agent.
The cheapest form of niacin is immediate-release crystalline niacin. Niacin should be startedat a low dose (100 mg three times a day) and takenwith meals to delay absorption.The dose of niacinshould be increased every 4 to 7 days by 100 mguntil a dose of 500 mg tid is obtained. After 1month on this dose, lipids and pertinentchemistries (glucose, uric acid, liver transaminases)should be measured. The dose can be further in-creased as needed up to a total dose of 6 g/d.Themost frequent side effect is cutaneous flushing, butthis improves with continued administration. Inmany patients, taking an aspirin 30 min prior tothe niacin prevents flushing. Over-the-countersustained-release forms of niacin are generally ad-ministered twice a day and are associated with lessflushing, but some have been associated with rarecases of severe hepatitis.A clue to the development
of niacin-induced hepatitis is a sudden, precipitousdrop in the plasma lipid levels.A prescription formof extended-release niacin that is administeredonce daily at bedtime has not been associated withsevere hepatic toxicity. Mild elevations in transami-nases occur in up to 15% of patients treated withany form of niacin, but these elevations rarely re-quire discontinuation of the medication. Niacinpotentiates the effect of warfarin, and these twodrugs should be prescribed together with caution.Acanthosis nigricans and maculopathy are infre-quent side effects of niacin. Niacin is contraindi-cated in patients with peptic ulcer disease and canexacerbate the symptoms of esophageal reflux.Niacin can raise plasma levels of uric acid and pre-cipitate gouty attacks in susceptible patients.
Niacin can raise fasting plasma glucose levels,but concerns regarding the use of niacin in dia-betic patients have been allayed by the results oftwo studies. In one study, short-acting niacin treat-ment of dyslipidemia was associated with only aslight increase in fasting glucose and no significantchange from baseline in the HbA1c. In the other,low-dose niacin was found to reduce triglycerideseffectively and raise HDL-C in diabetics withoutadversely impacting glycemic control.
Successful therapy with niacin requires carefuleducation and motivation of the patient. Its advan-tages are its low cost and long-term safety. It is themost effective drug currently available for raisingHDL-C levels. It is particularly useful in patientswith combined hyperlipidemia and low plasmalevels of HDL-C and is effective in combinationwith statins.
FIBRIC ACID DERIVATIVES (FIBRATES)
Fibric acid derivatives, or fibrates, are agonists ofPPAR�, a nuclear receptor involved in the regula-tion of carbohydrate and lipid metabolism. Fibratesstimulate LPL activity (enhancing triglyceride hy-drolysis), reduce apoC-III synthesis (enhancinglipoprotein remnant clearance), and may reduceVLDL production. Fibrates are the most effectivedrugs available for reducing triglyceride levels, andthey also raise HDL-C levels (Table 18-6). Fibrateshave variable effects on LDL-C, and in hyper-triglyceridemic patients can sometimes be associ-ated with increases in plasma LDL-C levels.
Fibrates are generally very well tolerated. Themost common side effect is dyspepsia. Myopathyand hepatitis occur rarely in the absence of otherlipid-lowering agents. Fibrates promote cholesterol
46128_18_p333-354 1/31/06 10:23 AM Page 351

352 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
secretion into bile and are associated with an in-creased risk of gallstones. Importantly, fibrates canpotentiate the effect of warfarin and certain oralhypoglycemic agents; the anticoagulation statusand plasma glucose levels should be closely moni-tored in patients on these agents.
Fibrates are the drug class of choice in patientswith severe hypertriglyceridemia [11.3 mmol/L(�1000 mg/dL)] and are a reasonable considera-tion in patients with moderate hypertriglyc-eridemia [4.5 to 11.3 mmol/L (400 to 1000mg/dL)]. The Veterans Affairs High-DensityLipoprotein Intervention Trial study also suggeststhat they may have a role in high-risk patientswith well-controlled LDL-C levels but elevatedplasma triglyceride levels and low plasma levels ofHDL-C. The relative indications of fibrates vs.statins and the role of combined therapy will bedetermined by ongoing and future trials.
OMEGA-3 FATTY ACIDS (FISH OILS)
N-3 polyunsaturated fatty acids (PUFAs) are pre-sent in high concentration in fish and in flax seeds.The most widely used n-3 PUFAs for the treat-ment of hyperlipidemias are the two active mole-cules in fish oil, eicosapentanoic acid (EPA) anddecohexanoic acid (DHA). N-3 PUFAs have beenconcentrated into tablets and in doses of 3 to 6g/d decrease fasting and postprandial triglycerides.At least 6 g/d is usually required for a substantialtriglyceride-lowering effect, and many patients re-quire 9 to 12 g/d. Fish oil treatment of hyper-triglyceridemia can be associated with a significantincrease in plasma LDL-C levels. Fish oil supple-ments can be used in combination with fibrates,niacin, or statins to treat hypertriglyceridemia. Ingeneral, fish oils are well tolerated and appear tobe safe, at least at doses up to 3 g.The large num-ber of capsules required for a therapeutic effect,the associated dyspepsia, and fishy aftertaste havelimited the clinical use of these agents. Althoughfish oil administration is associated with a prolon-gation in the bleeding time, no increase in bleed-ing has been seen in clinical trials.
CHOLESTEROL ABSORPTION INHIBITORS
A new mechanism of cholesterol-lowering is theinhibition of intestinal cholesterol absorption. Eze-timibe (Table 18-6) inhibits the absorption of di-etary and biliary cholesterol from the intestinal lu-men. It reduces LDL-C cholesterol levels by�18% as monotherapy or in combination with a
statin. Cholesterol absorption inhibitors are partic-ularly useful in combination with a statin in pa-tients unable to reach their LDL-C goal on statinmonotherapy.
COMBINATION DRUG THERAPY
Combination drug therapy is frequently used inthe following situations: (1) patients unable toreach their LDL-C goal on a single drug, (2) pa-tients with combined hypertriglyceridemia andhypercholesterolemia that cannot be adequatelycontrolled with a single drug, and (3) patients withelevated LDL-C and low HDL-C levels. Inabilityto achieve LDL-C goal is not uncommon onstatin monotherapy. If the patient has a normalplasma triglyceride level, a bile acid sequestrantcan be added. A cholesterol absorption inhibitorcan also be used in this setting. Combination ofniacin with a statin is an attractive option for high-risk patients who do not attain their target LDL-Clevel on statin monotherapy and who have anHDL-C � 10.3 mmol/L (�40 mg/dL). Oneproduct currently available offers the combinationof lovastatin and extended-release niacin in a sin-gle tablet.
Patients with combined hyperlipidemia fre-quently have persistent hypertriglyceridemia onstatin monotherapy. Addition of niacin or a fibratecan reduce the plasma triglyceride level in thesepatients. Conversely, hypertriglyceridemic patientstreated with a fibrate often fail to reach theirLDL-C goal and are therefore candidates for theaddition of a statin. Coadministration of statins andfibrates has obvious appeal in patients with com-bined hyperlipidemia, but no clinical trials have as-sessed the effectiveness of a statin-fibrate combina-tion compared with either a statin or a fibratealone in reducing cardiovascular events, and thelong-term safety of this combination is notknown. Statin-fibrate combinations are known tobe associated with an increased incidence of severemyopathy (up to 2.5%) and rhabdomyolysis, andpatients treated with these two drugs must becarefully counseled and monitored.This combina-tion of drugs should be used cautiously in patientswith underlying renal or hepatic insufficiency; inthe elderly, frail, and chronically ill; and in those onmultiple medications.
Other Approaches
Occasionally, patients cannot tolerate any of theexisting lipid-lowering drugs at doses required for
46128_18_p333-354 1/31/06 10:23 AM Page 352

FURTHER READINGS
Chapter 18 Disorders of Lipoprotein Metabolism 353
adequate control of their lipid levels. Some pa-tients, mostly those with genetic lipid disorders,remain significantly hypercholesterolemic despitecombination drug therapy. These patients are athigh risk for development or progression of CHDand clinical CHD events.
LDL APHERESIS
The preferred option for management of patientswith refractory or drug-resistant hypercholes-terolemia is LDL apheresis. In this process, the pa-tient’s plasma is passed over a column that selec-tively removes the LDL, and the LDL-depletedplasma is returned to the patient. Patients on max-imally tolerated combination drug therapy whohave CHD and a plasma LDL-C level �5.2mmol/L (�200 mg/dL) or no CHD and a plasmaLDL-C level �7.8 mmol/L (�300 mg/dL), arecandidates for every-other-week LDL apheresis.
PARTIAL ILEAL BYPASS
Partial ileal bypass interrupts the enterohepatic cir-culation of bile acids, resulting in upregulation ofthe hepatic LDL receptor and reduction in plasmaLDL-C levels. Diarrhea is a common side effect,and the incidence of kidney stones, gallstones, andintestinal obstruction is increased after ileal bypasssurgery.The clinical utility of partial ileal bypass atthis time is limited to severely hypercholes-terolemic patients with normal triglycerides whocannot tolerate existing lipid-lowering medica-tions and do not have access to LDL apheresis.Partial ileal bypass has not been proven effective inpatients with severe hypercholesterolemia whohave not responded adequately to statins.
Management of Low HDL-C
Severely reduced plasma HDL-C [�0.5 mmol/L(�20 mg/dL)] accompanied by triglycerides �4.5mmol/L (�400 mg/dL) usually indicates the pres-ence of a genetic disorder, such as a mutation inapoA-I, LCAT deficiency, or Tangier disease. HDL-C levels �0.5 mmol/L (�20 mg/dL) are commonin the setting of severe hypertriglyceridemia, inwhich case the primary focus should be on themanagement of the triglycerides. Secondary causesof more moderate reductions in plasma HDL [0.5to 10.3 mmol/L (20 to 40 mg/dL)] should be con-sidered (Table 18-5). Smoking should be discontin-ued, obese persons should be encouraged to loseweight, sedentary persons should be encouraged toexercise, and diabetes should be optimally con-trolled.When possible, medications associated with
reduced plasma levels of HDL-C should be discon-tinued. The presence of an isolated low plasmaHDL-C level in a patient with a borderline plasmaLDL-C should prompt consideration of LDL-lowering drug therapy in high-risk individuals.Statins increase plasma levels of HDL-C only mod-estly (�5 to 10%). Fibrates also have a modest effecton plasma HDL-C levels (increasing levels �5 to15%) except in patients with coexisting hyper-triglyceridemia, where they can be more effective.Niacin is the most effective therapeutic agent andcan increase plasma HDL-C levels by up to �30%.
The issue of whether pharmacologic inter-vention should be used to specifically raise HDL-C levels has not been adequately addressed inclinical trials. Pending these studies, it may be rea-sonable to initiate therapy (with a fibrate orniacin) directed specifically at reducing plasmatriglyceride levels and raising the plasma HDL-Clevel in persons with established CHD and lowHDL-C levels whose plasma LDL-C levels are ator below the goal.
� BROUSSEAU ME et al: Effects of an inhibitor of cholesteryl es-ter transfer protein on HDL cholesterol. N Engl J Med350:1505, 2004
Inhibition of cholesteryl ester transfer protein (CETP)has been proposed as a strategy to raise HDL choles-terol levels. This study examined the effects of torce-trapib, a potent inhibitor of CETP, on plasma lipopro-tein levels in subjects with low HDL cholesterol.Treatment with 120 mg of torcetrapib daily markedlyincreased HDL cholesterol levels and also decreasedLDL cholesterol levels.
� CANNON CP et al: Intensive versus moderate lipid loweringwith statins after acute coronary syndromes. N Engl J Med350:1495, 2004
This study compared standard versus intensive lipid-lowering therapy with statins in 4162 patients who hadbeen hospitalized for an acute coronary syndrome.The intensive lipid-lowering statin regimen providedgreater protection against death or major cardiovascu-lar events, suggesting that such patients benefit fromearly and continued lowering of LDL cholesterol to lev-els substantially below current target levels.
46128_18_p333-354 1/31/06 10:23 AM Page 353

LAROSA JC et al: Past, present, and future standards for managementof dyslipidemia.Am J Med 116 Suppl 6A:3S, 2004
LIMA JA et al: Statin-induced cholesterol lowering and plaque regres-sion after 6 months of magnetic resonance imaging-monitoredtherapy. Circulation 110:2336, 2004
354 Section III Diabetes Mellitus, Obesity, Lipoprotein Metabolism
� GRUNDY SM et al: Implications of recent clinical trials for theNational Cholesterol Education Program Adult TreatmentPanel III Guidelines. J Am Coll Cardiol 44:720, 2004
Heart Protection Study Collaborative Group:MRC/BHF Heart Protection Study of cholesterol low-ering with simvastatin in 20,536 high-risk individuals: Arandomised placebo-controlled trial. Lancet 360:7,2002
� Muldoon MF et al: Cholesterol reduction and non-illnessmortality: Meta-analysis of randomised clinical trials. BMJ322:11, 2001
Executive summary of the third report of the NationalCholesterol Education Program (NCEP) expert panelon detection, evaluation, and treatment of high bloodcholesterol in adults (Adult Treatment Panel III). JAMA285:2486, 2001
� NISSEN SE et al: Statin therapy, LDL cholesterol, C-reactiveprotein, and coronary artery disease. N Engl J Med 352:29,2005.
Recent trials have demonstrated better outcomes withintensive than with moderate statin treatment. Thisstudy of patients with coronary artery disease con-firmed a reduced rate of progression of atherosclerosisassociated with intensive statin treatment, and docu-mented a correlation with greater reductions in the lev-els of both atherogenic lipoproteins and CRP.
46128_18_p333-354 1/31/06 10:23 AM Page 354