chapter 1 thermodynamics - university of …vvanchur/phys4031/chapter1.pdfsolution: binary...
TRANSCRIPT

Chapter 1
Thermodynamics
1.1 PreliminariesDegree of freedom (d.o.f) is a number of parameters necessary toformulate the initial value problem divided by two.
For a harmonic oscillator the initial value problem is specified with onlytwo parameters: position coordinate (or angle) and velocity coordinate (orangular velocity) which together correspond to only a single d.o.f.
In Hamiltonian mechanics the position q and momentum p coordinatescome in conjugate pairs (q, p). Thus the number of d.o.f. is the number of
3

CHAPTER 1. THERMODYNAMICS 4
such pairs or one half of the total dimensionality of the phase space.
Problem: How many d.o.f. in a problem of binary collision ofprotons in the LHC (Large Hadron Collider)?Solution: Binary collisions involve collisions of only two parti-cles. Each particle is specified by position and velocity vectorcoordinates. This makes the number of d.o.f. (2 · 3 + 2 · 3)/2 = 6.
Our main objective in this course is to understand the behavior of systemswith a very large number of d.o.f. N ∫ 1 (e.g. NA = 6.02214 ◊ 1023
molecules in a box). The task is (in some sense) much more ambitious thanthe problems we are used to in physics courses where the number of d.o.f. isusually small. For N . 10 analytical methods may be useful; for N . 1010
computer may work; for N ≥ 1googol = 10100 statistical physics may be theonly tool.
There are two standard ways to study the large N limit:
• phenomenological (e.g. thermodynamics) and
• fundamental (e.g. statistical mechanics).
We start with phenomenological approach and later will use a kinetic theoryto justify a more fundamental (?) approach.
Thermodynamics is a phenomenological theory of systems withmany d.o.f.
The main idea is that only a small number of measurable parameters (e.g.volume V , pressure P , temperature T , etc.) should be su�cient for describingthe so-called equilibrium states.
Equilibrium state is the state whose thermodynamic parametersdo not change with time.
It is an important experimental fact that in equilibrium all of the parame-ters are either extensive or intensive. For example, volume is extensive, butpressure is intensive.
Extensive parameter is proportional to the amount of substanceand intensive parameter is independent on the amount of sub-stance.Problem: Give an example of neither extensive nor intensivequantity. Explain.
CHAPTER 1. THERMODYNAMICS 5
Solution: When two wires with electrical resistances R1
andR
2
are connected in series than the total resistance is an ex-tensive quantity Rtotal = R
1
+ R2
, but when the wires are con-nected in parallel the total resistance is not extensive 1/Rtotal =1/R
1
+ 1/R2
. Therefore, electrical resistance is not an extensivenor intensive quantity.
Not all of the thermodynamic parameters are independent of each other.Equation of state,
f(P, V, T ) = 0, (1.1)describes the dependence and reduces the number of independent parameters.For example, the equation of state for an ideal gas (su�ciently diluted gas)is given by,
PV = NkT, (1.2)where N is the number of molecules and k is the Boltzmann’s constant.
Because of a universal character of ideal gases (1.2) can be used to set arelative temperature scale (but to set an absolute scale and to give a meaningto T = 0 it is necessary to postulate the Third Law of thermodynamics to bediscussed later in the course). For example, we can measure P V
Nkwhen water
boils and denote this temperature by T = 0, and then we can measure P VNk
when water freezes and denote this temperature by T = 100. Then we canuse linear interpolation and extrapolation to assign temperature to arbitraryvalues of P V
Nk. This is the Celsius scale.

CHAPTER 1. THERMODYNAMICS 6
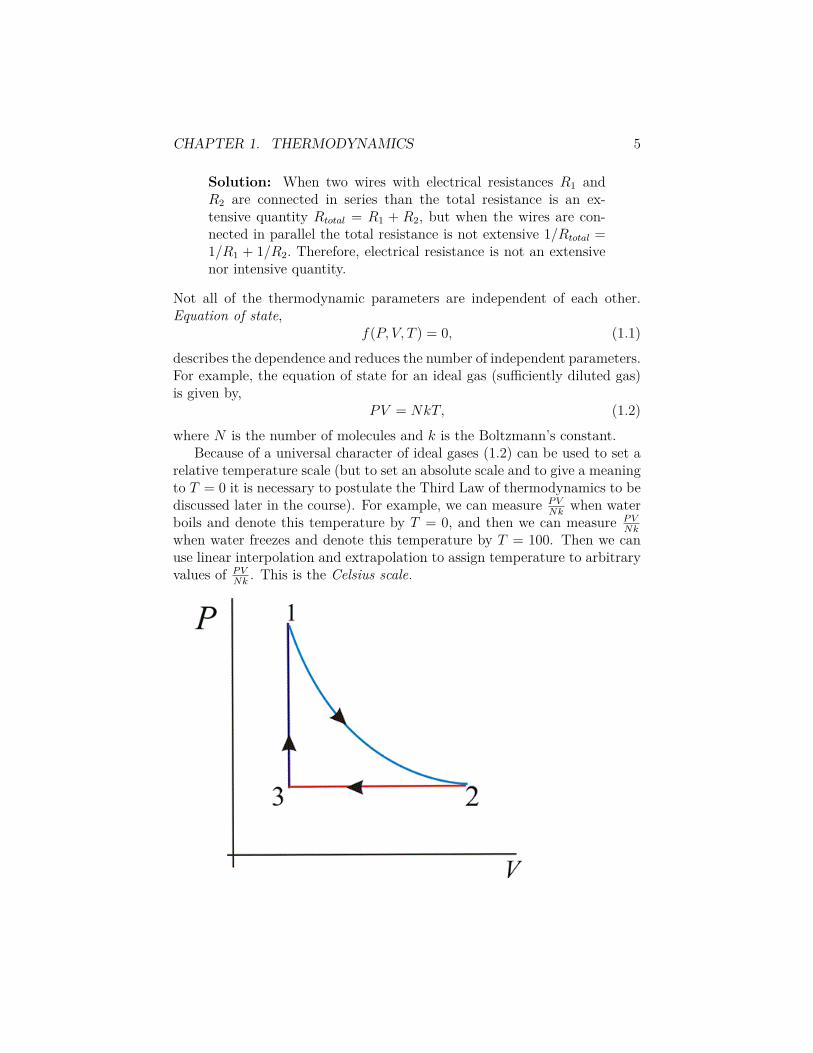
It is convenient to think of f(P, V, T ) as a 3D function which describesa given thermodynamical system and of (P, V, T ) as a point in 3D whichdescribes a given state of the system. The projection of the surface of theequation of state on P ≠ V plane is known as the P ≠ V diagram.
Reversible transformation of di�erent types such as
• 1-2. isothermal (i.e. constant temperature)
• 2-3. isobaric (i.e. constant pressure)
• 3-1. isochoric (i.e. constant volume)
are described by di�erent paths on the P ≠ V diagram.
1.2 Zeroth LawEvery theory is based on a number of statements which cannot be proved(not that we can prove anything in physics). In mathematics such statementsare called axioms, but in physics they go by di�erent names: principles, laws,assumptions, etc. With this respect thermodynamics is not an exception asit is based on four laws of thermodynamics numbered from 0 to 3 (for purelyhistorical reasons).
Zeroth Law in words: If systems A and C are eachin thermal equilibrium with system B, then A is inthermal equilibrium with system C.Zeroth Law in symbols:
A ≥ B and C ≥ B ∆ A ≥ C (1.3)
where ≥ is a relation between systems such that “A ≥ B” readsas “A is in a thermal equilibrium with B”.Problem: Under assumption that every system is in equilibriumwith itself, prove that ≥ is an equivalence relation.Solution: Equivalence relation must satisfy three properties:1) Reflexivity: By assumption, A ≥ A.2) Symmetry: Let A ≥ B. From Reflexivity B ≥ B. UsingZeroth Law, B ≥ B and A ≥ B ∆ B ≥ A.3) Transitivity: Let A ≥ B and B ≥ C. From Symmetry C ≥ B.From Zeroth Law, A ≥ B and C ≥ B ∆ A ≥ C.

CHAPTER 1. THERMODYNAMICS 7
An important consequence of the equivalence relation “~” is that it divides allof the system into equivalence classes which can be labeled by temperature.Of course, this does not tell us why the temperature should be a real numberas opposed to, for example, integers, complex or even more exotic p-adicnumber.
1.3 First LawThe Zeroth Law does not tell us why temperature is a real number. Thusadditional laws must be postulated before this fact can be established notonly experimentally (e.g. using equation of state for ideal gases), but alsomore theoretically. The next assumption is the First Law which should beviewed as a statement about conservation of energy.
First Law in words: The increment in the internal en-ergy U of a system is equal to the difference betweenthe increment of heat Q accumulated by the systemand the increment of work W done by it.First Law in symbols:
dU = dQ ≠ dW, (1.4)
where dU is an exact di�erential.Exact di�erential dX is a di�erential whose integral
sdX depends
only on the limits of integration, but not on the path.The First law is used to define the state function U which isan extensive quantity: doubling the mass doubles the internalenergy. For quasi-static (or su�ciently slowly varying) processesthe work done by the system dW = PdV such that
dU = dQ ≠ PdV. (1.5)
Problem: Are the di�erentials dQ and dW exact or not?Solution: W =
sPdV is not a full derivative and therefore de-
pends not only on the initial and final points but also on the path.This can be shown explicitly by integrating along di�erent pathfrom (P
1
, V1
) to (P2
, V2
) assuming that P1
< P2
and V1
< V2
:
⁄(P1,V2)
(P1,V1)
PdV +⁄
(P2,V2)
(P1,V2)
PdV = P1
(V2
≠V1
) ”= P2
(V2
≠V1
) =⁄
(P2,V1)
(P1,V1)
PdV +⁄
(P2,V2)
(P2,V1)
PdV.

CHAPTER 1. THERMODYNAMICS 8
Therefore, dW is not an exact di�erential and since dU is exactdue to the First Law dQ = dU ≠ dW is also not exact.
It is convenient to think of the conjugate pair P and V as a generalizedforce (intensive quantity) and generalized displacement (extensive quantity)respectively. Other commonly used conjugate pairs include temperature Tand entropy S, chemical potential µi and number of particles Ni of type i,etc. Then the total change in internal energy can be expressed as the sumof the products of generalized forces and generalized displacements:
dU = TdS ≠ PdV +ÿ
i
µidNi. (1.6)
In other words, when a given generalized force is not balanced it causes ageneralized displacement whose product equals to the energy transfer in orout of the system.
1.4 Second LawSecond Law in Kelvin words: No process is possible in which thesole result is the absorption of heat from a reservoir and itscomplete conversion into work.Second Law in Clausius words: No process is possible whose soleresult is the transfer of heat from a body of lower tempera-ture to a body of higher temperature.
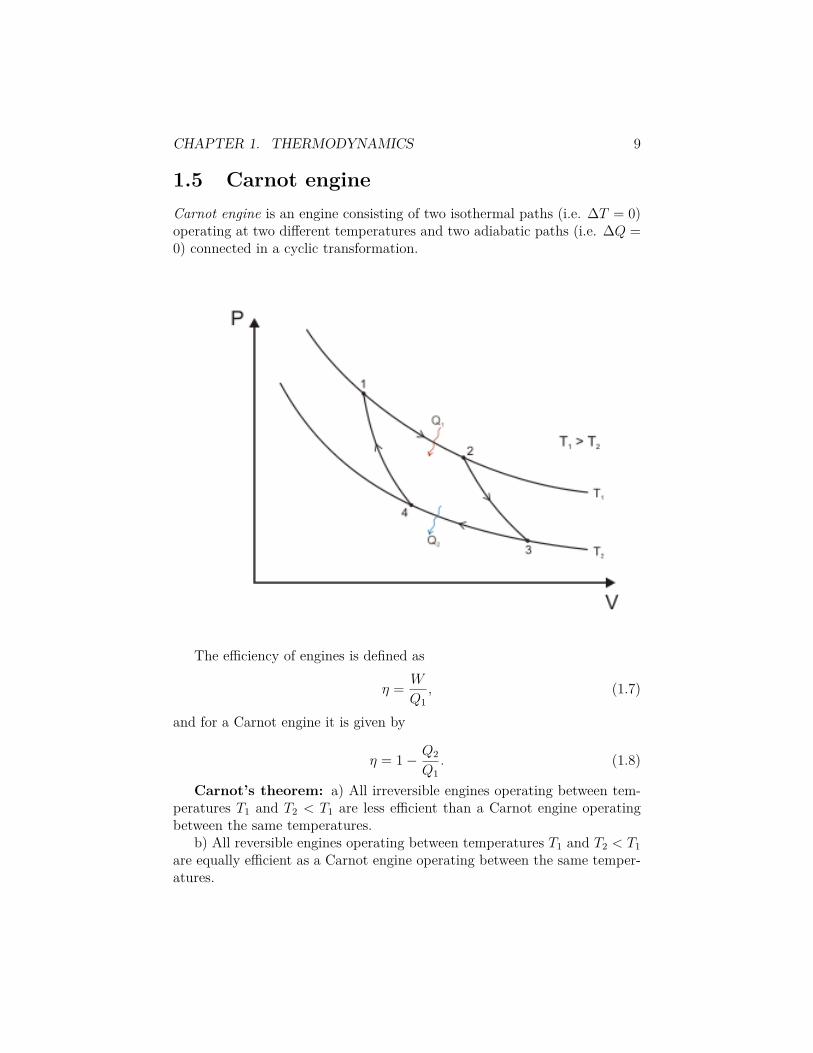
To show the equality of the two statements it will be useful to define a heatengine. Heat engine is a system which undergoes a cyclic transformation thattakes heat Q
1
from a warmer reservoir, converts some of it to work W andrejects the rest Q
2
= Q1
≠ W to a colder reservoir. In contrast, refrigerator,is a heat engine running backwards in time: use work to extract heat from acolder reservoir and to reject it to a warmer reservoir.
Problem: Prove that the Kelvin and the Clausius statements ofthe Second Law are equivalent.Solution: Assume that Kelvin statement is false ∆ Extract Qof heat from a reservoir at temperature T
2
and convert it entirelyto work W = Q. Then convert this work back into heat Q = Wand transfer it to reservoir at temperature T
1
> T2
∆Clausiusstatement is false.Assume that Clausius statement is false ∆ Extract Q of heatfrom a reservoir at temperature T
2
and transfer it to reservoir attemperature T
1
> T2
∆ Operate an engine between temperaturesT
1
and T2
designed such that W = Q ∆Kelvin statement is false.
CHAPTER 1. THERMODYNAMICS 9
1.5 Carnot engineCarnot engine is an engine consisting of two isothermal paths (i.e. �T = 0)operating at two di�erent temperatures and two adiabatic paths (i.e. �Q =0) connected in a cyclic transformation.
The e�ciency of engines is defined as
÷ = W
Q1
, (1.7)
and for a Carnot engine it is given by
÷ = 1 ≠ Q2
Q1
. (1.8)
Carnot’s theorem: a) All irreversible engines operating between tem-peratures T
1
and T2
< T1
are less e�cient than a Carnot engine operatingbetween the same temperatures.
b) All reversible engines operating between temperatures T1
and T2
< T1
are equally e�cient as a Carnot engine operating between the same temper-atures.

CHAPTER 1. THERMODYNAMICS 10
Proof: a) Combine an arbitrary heat engine whose e�ciency is ÷ with areversed Carnot engine whose e�ciency is ÷Õ so that there is no work doneby the combined system. If ÷ = ÷Õ then the process does nothing in conflictwith irreversibility assumption. If ÷ > ÷Õ then the process is in the conflictwith Clausius statement of the Second Law, i.e.
W = Wc ∆ ÷Q = ÷ÕQÕ ∆ �Q = ÷
÷Õ Q ≠ Q > 0.
Thus, ÷ Æ ÷c. Together ÷ Æ ÷c and ÷ ”= ÷cgive us ÷ < ÷c.
b) We have already proved that ÷ Æ ÷Õ regardless of reversibility. Now wecan reverse the process by combining a reversed heat engine with a Carnotengine which leads to conclusion ÷Õ Æ ÷. Together ÷ Æ ÷Õ and ÷Õ Æ ÷ give us÷ = ÷Õ.
According to Carnot’s theorem the e�ciency of reversible process betweenany two temperatures is a universal number, i.e. ÷(T
1
, T2
). This allows usto define not only relative, but also absolute temperature scale. Considerthree Carnot cycles 1-2, 2-3 and 1-3 operating between di�erent temperaturesT
1
and T2
, T2
and T3
, T1
and T3
respectively, where without loss of generalitywe assume that T
1
> T2
> T3
. We can now construct a combine cycle such

CHAPTER 1. THERMODYNAMICS 11
that the heat Q2
rejected by 1-2 is absorbed by 2-3 which is a reversible andthus a cycle 1-3 by Carnot’s theorem. Then the heat absorbed by reservoirat T
3
must satisfy both
Q3
= Q1
≠ W13
= Q1
(1 ≠ ÷(T1
, T3
))and
Q3
= Q2
≠W23
= Q2
(1≠÷(T2
, T3
)) = (Q1
≠W12
)(1≠÷(T2
, T3
)) = Q1
(1≠÷(T1
, T2
))(1≠÷(T2
, T3
)).
Therefore(1 ≠ ÷(T
1
, T3
)) = (1 ≠ ÷(T1
, T2
))(1 ≠ ÷(T2
, T3
))and
1 ≠ ÷(T1
, T2
) = f(T2
)f(T
1
) (1.9)
for an arbitrary function f(T ) which is by convention is set to be a linearfunction, i.e.
÷ = 1 ≠ T2
T1
(1.10)
and from the definition of e�ciency
Q2
Q1
= T2
T1
. (1.11)
1.6 EntropyThe Second Law suggests a new thermodynamical quantity, called entropyand usually denoted by S. It is conveniently introduced using Clausius’stheorem.
Clausius’s Theorem: For any cyclic transformationj dQ
TÆ 0. (1.12)
The equality holds for reversible transformations.Proof: Subdivide the cycle into infinitesimal transformationswhere the temperature T remains roughly constant. During eachtransformation the system receives dQ of heat and does dWS
of work. Arrange a series of Carnot cycles operating between

CHAPTER 1. THERMODYNAMICS 12
temperatures T and TR where is the temperature of an arbitraryreservoir. The whole purpose of each Carnot cycle is to take dQR
of heat from reservoir and deliver to the system dQ of heat andto do dWC of work. Then according to the absolute definition oftemperature
dQR
dQ= dQ + dW
dQ= TR
T. (1.13)
By the Kelvin’s statement of the Second Law the total work mustbe non-positive
idW =
i(dWC + dWS) =
s(dQ + dWC) Æ 0
and thus,j
(dQ + dWC) = TR
j dQ
TÆ 0 ∆
j dQ
TÆ 0 (1.14)
since TR > 0. For a reversible cycle we can run the process inopposite direction to show that
i dQT
Ø 0 which together withi dQ
TÆ 0 implies
i dQT
= 0.
An immediate consequence of the Clausius’s Theorem is that for reversibletransformations between two states A and B the integral
s BA
dQT
does not de-pend on the path. Indeed if we take two distinct reversible paths parametrizesby dQ and dQÕ then together they would form a reversible cycle, i.e.
⁄ B
A
dQ
T+
⁄ A
B
dQÕ
T= 0 (1.15)
and thus, ⁄ B
A
dQ
T=
⁄ B
A
dQÕ
T. (1.16)
This suggests that for a reversible transformation we can define and exactdi�erential
dS © dQ
T. (1.17)
whose integral defines relative entropy up to an arbitrary constant of integra-tion. This also produces a new pair of conjugate thermodynamic variablesgeneralized force T and generalized displacement S, i.e.
dU = TdS ≠ PdV. (1.18)Although the entropy was defined using only reversible processes (as a
reference), the Clausius’s Theorem implies that for all systems

CHAPTER 1. THERMODYNAMICS 13
S(B) ≠ S(A) Ø⁄ B
A
dQ
T. (1.19)
(This can be seen by connecting the irreversible process from A to B withany reversible process from B to A such that
i dQT
=s B
AdQT
+s A
BdQT
=s B
AdQT
+S(B) ≠ S(A) Æ 0.) Moreover, for thermally isolated systems (i.e. dQ = 0)
S(B) ≠ S(A) Ø 0 (1.20)
where the equalities hold for only reversible transformations. This shows thatin a thermal equilibrium (when the state of the system does not change) thesystem must be in a state of maximal entropy.
Second Law in symbols:dS
dtØ 0 (1.21)
Not all of the processes we observe in nature are reversible. Many processesare irreversible, which is a rather surprising fact given that the fundamentallaws of physics (as we know them) are usually time symmetric. Then, whydoes the nature always picks some initial conditions and not other? Whydon’t we see a thermal state? Why is there an asymmetry between past andfuture? Why do we see arrow of time?
1.7 dQ equationsConsider a system specified by three parameters P, V, T any two of whichare independent variables due to equation of state. Then, the di�erential ofinternal energy can be expressed in three possible forms
dU(P, V ) =A
ˆU
ˆP
B
V
dP +A
ˆU
ˆV
B
P
dV (1.22)
dU(T, V ) =A
ˆU
ˆT
B
V
dT +A
ˆU
ˆV
B
T
dV (1.23)
dU(P, T ) =A
ˆU
ˆP
B
T
dP +A
ˆU
ˆT
B
P
dT. (1.24)
Using the First Law given by Eq. (1.5) we obtain can the so-called ”Qequations:
dQ(P, V ) = dU(P, V ) + PdV =A
ˆU
ˆP
B
V
dP +AA
ˆU
ˆV
B
P
+ P
B
dV (1.25)

CHAPTER 1. THERMODYNAMICS 14
dQ(T, V ) = dU(T, V ) + PdV =A
ˆU
ˆT
B
V
.dT +AA
ˆU
ˆV
B
T
+ P
B
dV (1.26)
dQ(P, T ) = dU(P, T )+PdV =AA
ˆU
ˆP
B
T
+ P
AˆV
ˆP
B
T
B
dP+AA
ˆU
ˆT
B
P
+ P
AˆV
ˆT
B
P
B
dT
(1.27)where the heat capacities at constant pressure and volume are defined by
CP ©A
ˆQ
ˆT
B
P
=A
ˆU
ˆT
B
P
+ P
AˆV
ˆT
B
P
(1.28)
CV ©A
ˆQ
ˆT
B
V
=A
ˆU
ˆT
B
V
. (1.29)
Roughly speaking the heat capacities describe how much heat can be storedin a system as its temperature is increased.
Problem: Under assumption that U(T ) is a function of temper-ature alone and CV does not depend on temperature, prove thatfor an ideal gas it is more e�cient (requires less energy) to heatup the system at constant volume than at constant pressure.Solution: Since U is not a function of P nor V ,
AˆU
ˆT
B
P
=A
ˆU
ˆT
B
V
= dU
dT
and the di�erence between specific heat capacities (1.28) and(1.29) is given by
CP ≠ CV = P
AˆV
ˆT
B
P
=A
ˆ(PV )ˆT
B
P
= Nk.
where the last equality is obtained from the equation of statefor an ideal gas (1.2). Since both the number of molecules andthe Boltzmann’s constant are positive number we conclude thatCP ≠ CV > 0 or that it is easier heat up the system at constantpressure rather than at constant volume.
The dQ equation in their present form are not very useful. Using the SecondLaw we can rewrite equations (1.26) and (1.27)
dQ
T= dS = 1
T
AˆU
ˆT
B
V
dT + 1T
AAˆU
ˆV
B
T
+ P
B
dV (1.30)

CHAPTER 1. THERMODYNAMICS 15
dQ
T= dS =
A1T
AˆU
ˆP
B
T
+ P
T
AˆV
ˆP
B
T
B
dP +A
1T
AˆU
ˆT
B
P
+ P
T
AˆV
ˆT
B
P
B
dT
(1.31)and use the exactness of dS, i.e.
dS =A
ˆS
ˆT
B
V
dT +A
ˆS
ˆV
B
T
dV ∆A
ˆ
ˆV
AˆS
ˆT
B
V
B
T
=A
ˆ
ˆT
AˆS
ˆV
B
T
B
V
to derive the following relations
ˆ
ˆV
A1T
AˆU
ˆT
B
V
B
T
= ˆ
ˆT
A1T
AAˆU
ˆV
B
T
+ P
BB
V
(1.32)
ˆ
ˆP
A1T
AˆU
ˆT
B
P
+ P
T
AˆV
ˆT
B
P
B
T
= ˆ
ˆT
A1T
AˆU
ˆP
B
T
+ P
T
AˆV
ˆP
B
T
B
P
(1.33)or
AˆU
ˆV
B
T
+P = T
AˆP
ˆT
B
V
and ≠T
AˆV
ˆT
B
P
=A
ˆU
ˆP
B
T
+P
AˆV
ˆP
B
T
. (1.34)
Then one can use these expressions to simplify the dQ equations
TdS = CV dT + T
AˆP
ˆT
B
V
dV (1.35)
TdS = CP dT ≠ T
AˆV
ˆT
B
P
dP. (1.36)
1.8 Chemical potentialChemical potential, denoted byµ, is defined as work required to increase thenumber of particle by one. The name - chemical potential - is due to thefact that µ, describes the tendency of particles to move from higher densitiesto lower densities. Paired together with a number of particles N it formsanother conjugate pair which appears in the most fundamental equation ofthermodynamics:
dU = TdS ≠ PdV + µdN (1.37)

CHAPTER 1. THERMODYNAMICS 16
which combines in a single equations the First and Second laws.As was already noted T ,P and µ are intensive quantities and S, V and
N are extensive. This can be expressed as a scaling lawU(⁄S, ⁄V, ⁄N) = ⁄U(S, V, N) (1.38)
which can di�erentiated with respect to ⁄ at ⁄ = 1,A
ˆU
ˆS
B
V,N
S ≠A
ˆU
ˆV
B
S,N
V +A
ˆU
ˆN
B
S,V
N = U(S, V, N). (1.39)
By combined with (1.37) we obtain Euler equationU = ST ≠ PV + µN (1.40)
as well as the Gibbs-Duhem equationSdT ≠ V dP + µdN = 0. (1.41)
In a more general form it reads as
SdT ≠ V dP +ÿ
i
µidNi = 0, (1.42)
where µi is a chemical potential and Ni is the number of particle of type i.
1.9 Perpetual motion machinesIf the First Law could be violated then it would open an exciting possibility ofperpetual motion machine of the first kind: do work without input of energy.Such machines would be in a conflict with conservation of energy, but thereis certainly no prove that they cannot exist. Moreover it is well known thatfor gravitational (more precisely general relativistic) systems the mass andenergy is not always well defined.
Although the perpetual motion machines of the first kind are not allowedby the First Law, there are perpetual motion machines of the second kindwhich are not in conflict with conservation of energy, yet they have neverbeen constructed. It is an experimental fact that not all of the processesallowed by the conservation of energy are observed in nature. For example,we see ice cubes melting in warm water to cool it down, but we never see atime-reversed process where cool water splits into warm water with ice cubes.Such processes are called irreversible, in contrast to reversible processes whichcan run backwards in time if its initial and final conditions are interchanged.In thermodynamic such processes are prohibited by postulating the SecondLaw. If the Second Law could be violated it would allow perpetual motionmachines of the second kind: do work using thermal energy.

CHAPTER 1. THERMODYNAMICS 17
1.10 Thermodynamic potentialsAs we have seen the extrema (maximum) of entropy corresponds to an equi-librium state of an isolated system (i.e. dQ = dW = 0)
dS
dt= 0. (1.43)
Can this concept be generalized to open systems? For this purpose inaddition to U we define three new thermodynamic potentials: Enthalpy,Helmholtz free energy, Gibbs free energy and Landau free energy.
• EnthalpyH = U + PV (1.44)
It is convenient to express H in di�erential form as a function of S andP
dH = dU + PdV + V dP = TdS + V dP. (1.45)
When there is no heat exchange (i.e. dQ = 0) and the external force isconstant (i.e. P = const),
dH = dU + PdV + V dP = dW + PdV Æ 0. (1.46)
where the equality corresponds to quasi-static processes (i.e. dW = ≠PdV ).
• Helmholtz free energyA = U ≠ TS (1.47)
It is convenient to express A in di�erential from as a function of V andT
dA = dU ≠ TdS ≠ SdT = ≠PdV ≠ SdT. (1.48)
When there is no external work (i.e. dW = 0) and the temperature remainsconstant (i.e. T = const),
dA = dU ≠ SdT ≠ TdS = dQ ≠ TdS Æ 0. (1.49)where the equality corresponds to reversible processes (i.e. dQ = TdS).
As an example of the variational principle consider gas at a constanttemperature T in a box of volume V divided by a sliding piston into V
1
and

CHAPTER 1. THERMODYNAMICS 18
V2
. In the equilibrium state
0 = dA =A
ˆA
ˆV1
B
T
dV1
+A
ˆA
ˆV2
B
T
dV2
(1.50)
=A
ˆA
ˆV1
B
T
dV1
+A
ˆA
ˆV2
B
T
d(V ≠ V1
)
=AA
ˆA
ˆV1
B
T
≠A
ˆA
ˆV2
B
T
B
dV1
or AˆA
ˆV1
B
T
=A
ˆA
ˆV2
B
T
and using (1.48) we can conclude that in the equilibrium state the pressureson both sides of the piston must be equal.
• Gibbs free energyG = U ≠ TS + PV (1.51)
It is convenient to express G in di�erential form as a function of P andT since
dG = dU ≠ SdT ≠ TdS + PdV + V dP = V dP ≠ SdT. (1.52)
When the external force (i.e. P = const) and temperature remains constant(i.e. T = const) ,
dG = dU≠SdT≠TdS+PdV +V dP = dU+PdV ≠TdS = dQ≠dW+PdV ≠TdS Æ 0.(1.53)
where the equality corresponds to reversible (i.e. dQ = TdS) and quasi-static(i.e. dW = ≠PdV ) processes.
Note that the exactness of di�erential dH, dA, and dG, impliesA
ˆT
ˆP
B
S
=A
ˆV
ˆS
B
P
.
AˆP
ˆT
B
V
=A
ˆS
ˆV
B
T
.
andA
ˆV
ˆT
B
P
= ≠A
ˆS
ˆP
B
T
.

CHAPTER 1. THERMODYNAMICS 19
Other thermodynamic relations are conveniently summarized by the so-called Maxwell or thermodynamic square:
For a thermodynamic potential of interest (in bold) whose arguments areplaced in the neighboring corners (in italic) the derivative of the potentialwith respect to one of its argument with other argument held fixed is deter-mined by going along a diagonal line either with or against the direction ofthe arrow. Going against the arrow yields a minus sign (e.g. P = ≠
1ˆUˆV
2
S,
S = ≠1
ˆGˆT
2
P).
1.11 Third LawEntropy of a system is defined for all states using a reference state by con-necting it with a reversible transformation. However, if the surface definedby the equation of state is disconnected then the reversible transformationsmight not exist. Moreover it is important to have definition of entropy fordistinct systems which may later come into contact with each other. Therole of the Third Law is to defines an absolute scale of entropy which definesuniquely the entropy of an arbitrary equilibrium state of any system.
Third Law in words: The entropy of any system atabsolute zero is zero.Third Law in symbols:
limT æ0
S(T ) = 0 (1.54)
Consequences of the Third Law:
1. Partial derivative of S at T = 0 with respect to every thermodynamic

CHAPTER 1. THERMODYNAMICS 20
parameter X vanishes:
limT æ0
AˆS
ˆX
B
T
= 0. (1.55)
2. Heat capacity CX at T = 0 with fixed thermodynamic parameter Xvanishes:
limT æ0
CX = limT æ0
AˆQ
ˆT
B
X
= limT æ0
T
AˆS
ˆT
B
X
= 0. (1.56)
3. Absolute zero cannot be reached in a finite number of steps. Thisstatement is often used as an alternative definition of the third law.We will come back to it at the end of the course when the quantumstatistical mechanics is introduced.
Physical theories are always based on assumptions which very often turn outto be false (sooner or later). Now, that you have seen all of the theoreti-cal assumption (laws of thermodynamics) which go into a phenomenologicaltheory of thermodynamics it is a good time to ask which of the assumptionis likely to be wrong?
Zeroth Law (Universality of Temperature):
A ≥ B and C ≥ B ∆ A ≥ C (1.57)
First Law (Conservation of Energy):
dU = dQ ≠ dW. (1.58)
Second Law (Arrow of Time):
dS
dtØ 0 (1.59)
Third Law (Quantum Mechanics):
limT æ0
S(T ) = 0 (1.60)