cellulose chemistry v2
TRANSCRIPT

D. Klemm, B. Philipp, T. Heinze, U. Heinze,W. Wagenknecht
Comprehensive Cellulose ChemistryVolume 2
Functionalization of Cellulose
WILEY-VCHComprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

D. Klemm, B. Philipp, T. Heinze,U. Heinze, W. Wagenknecht
ComprehensiveCellulose ChemistryVolume 2Functionalization of Cellulose
® WILEY-VCHWeinheim · New York · Chichester · Brisbane · Singapore · Toronto

Prof. Dr. D. KlemmFriedrich-Schiller-Universität JenaInstitut für Organische undMakromolekulare ChemieHumboldtstraße 1007743 JenaGermany
Prof. Dr. B. PhilippMax-Planck-Institut für Kolloid-und GrenzflächenforschungKantstraße 5514513 Teltow-SeehofGermany
Dr. T. HeinzeFriedrich-Schiller-Universität JenaInstitut für Organische undMakromolekulare ChemieHumboldtstraße 1007743 JenaGermany
Dr. U HeinzeFriedrich-Schiller-Universität JenaInstitut für Organische undMakromolekulare ChemieHumboldtstraße 1007743 JenaGermany
Dr. W. WagenknechtMax-Planck-Institut für Kolloid-und GrenzflächenforschungKantstraße 5514513 Teltow-SeehofGermany
This book was carefully produced. Nevertheless, authors, editors and publisher do not warrant the informationcontained therein to be free of errors. Readers are advised to keep in mind that statements, data, illustrations,procedural details or other items may inadvertently be inaccurate.
Library of Congress Card No. applied for.
A catalogue record for this book is available from the British Library
Die Deutsche Bibliothek - CIP-Einheitsaufnahmeapplied for
© WILEY-VCH Verlag GmbH, D-69469 Weinheim (Federal Republic of Germany), 1998
Printed on acid-free and low chlorine paper
All rights reserved (including those of translation into other languages). No part of this book may be reproduced inany form - by photoprinting, microfilm, or any other means - nor transmitted or translated into machine-radablelanguage without written permission from the publishers. Registered names, trademarks, etc. used in this book, evenwhen not specifically marked as such, are not to be considered unprotected by law.Composition: Graphik & Textstudio, D-93164 Laaber-WaldetzenbergPrinting: betz-druck, D-64291 DarmstadtBookbinding: W Osswald, D-67433 Neustadt

Preface
Cellulose, as the most abundant organic polymer, has served mankind for thou-sands of years as an indispensable material for clothing and housing, and hasformed a large part of human culture since the Egyptian papyri. In contrast withcellulose application as a natural product, the use of this polymer as a chemicalraw material started just 150 years ago with the discovery of the first cellulosederivatives, but subsequently developed to a production volume of more than5 million tons annually during this century. At the same time the classical areasof processing cellulose as a natural product by mechanical technologies, forexample the manufacture of textile goods from cotton, received a strong impetusby combining them with chemical processes to improve product quality. Thisline of progress is closely related to and often originated from the developmentof a systematic chemistry of cellulose comprising predominantly the chemicaltransformation of the macromolecule.
The knowledge acquired in this area was compiled during this century in anumber of monographs and text books still serving as a valuable scientific back-ground in today's cellulose research. But most of these books were publishedseveral decades ago, and thus could not take into account recent developments,for example the relevance of ecological problems in cellulose processing, dis-cussion of the advantages and shortcomings of natural resources in general, ortoday's boom in synthetic organic and supramolecular chemistry. Besides this,some of these books consider only a special field or reflect a rather special pointof view. In the authors' opinion, no text book or monograph on the organicchemistry of cellulose is available now, that presents in a comprehensive andstill conveniently readable manner the theoretical background and the experi-mental state of the art at the end of this century.
It is the intention to fill this gap by the two volumes of this book, centered onthe routes and the mechanisms of cellulose functionalization, but covering alsothe close interrelation between a heterogeneous cellulose reaction and the su-pramolecular structure of this polymer. Special emphasis has been put on distri-bution of functional groups in relation to reaction conditions and on analyticaltechniques for their characterization. Not only recent efforts in cellulose researchand development are presented and cited but also important results on the lastcenturies actual up to now are included in order to give a comprehensive de-scription of the chemistry of cellulose.

VI Preface
The authors are indebted to WILEY-VCH Verlag for agreeing to a two-volume presentation, allowing accuracy and readability of the text to be com-bined, and also leaving enough space for numerous experimental procedures,that are suitable for making a graduate student familiar with the practical labo-ratory work in cellulose chemistry. From a didactic point of view, as well as forthe sake of convenient information retrieval, the authors found it appropriate tosurvey in the first volume some aspects of cellulose of a more general naturerelevant to chemical reactions. Included are e.g. its properties and structure inrelation to reactivity, the processes of swelling and dissolution, with their conse-quences to chemical reactions, and the pathways of cellulose degradation ac-companying chemical transformations of this polymer. Special emphasis isgiven in this part to aspects of physical chemistry and colloid chemistry. Arather detailed presentation of cellulose analytics for characterizing the originalpolymer and its derivatives at the various structural levels is also included inVolume I. Volume II deals with the various classes of cellulose derivatives, withemphasis on the reaction mechanisms and distribution of functional groups, in-cluding, in addition, in each of the chapters also a brief abridgment of relevantindustrial processes and an overview of properties and areas of application of theproducts in question. In both volumes results obtained by the authors' groups areadequately accentuated, especially with regard to Figures and Tables.
It is hoped that the two volumes of this book will be accepted as auseful textbook by graduate students in science, with special interest in cellulo-sics, and that it will serve as a comprehensive source of information for chem-ists, physicists, biologists, and engineers professionally engaged with this poly-mer. The authors' work would find its best appreciation, if the book helps tostimulate young scientists to professional activities in cellulose chemistry, whichoffers a challenge to innovative ideas and new experimental pathways, also intothe next century.

Contents
Volume 1: Chapters 1 to 3
1 Introduction 1
2 General Considerations on Structure and Reactivityof Cellulose 9
2.1 Structure and Properties of Cellulose 92.1.1 The molecular structure 92.1.2 The supramolecular structure 152.1.3 The morphological structure 222.1.4 Pore structure and inner surface 252.1.5 The accessibility of cellulose 292.1.6 Alien substances associated with the cellulose matrix 322.1.7 Macroscopic properties of cellulose 332.1.7M General properties and gross morphology 332.1.7.2 Mechanical properties of cellulose 352.1.7.3 Electrical, optical and thermal properties of cellulose 372.1.7.4 Chemical and environmental properties of cellulose 39
2.2 Swelling and Dissolution of Cellulose 432.2.1 Limited swelling of cellulose 442.2.1.1 Swelling of cellulose in water 452.2.1.2 Limited swelling of cellulose in some organic liquids in
comparison with water 512.2.1.3 Swelling of cellulose in aqueous solutions of sodium hydroxide
and in related systems 562.2.1.4 Interaction of cellulose in media in the transition range between
solvent and swelling agent 582.2.2 Dissolution of cellulose 602.2.2.1 Some general comments on cellulose dissolution 602.2.2.2 Systematic description of important classes of cellulose solvent
systems 622.2.2.3 Structure formation of cellulose and cellulose derivatives 732.2.3 Concluding remarks 79

VIII Contents
2.3 Degradation of Cellulose 832.3.1 Hydrolytic degradation of cellulose 842.3.1.1 Acid hydrolysis of cellulose 852.3.1.2 Enzymatic hydrolysis 932.3.2 Degradation of cellulose by aqueous alkali 992.3.3 Oxidative degradation of cellulose 1012.3.4 Mechanical degradation of cellulose 1042.3.5 Thermal degradation of cellulose and cellulose derivatives 1072.3.6 Radiation degradation of cellulose 1182.3.7 Consequences of degradation of cellulose on its chemical
processing 124
2.4 Principles of Cellulose Reactions 1302.4.1 Some principles of polymer reactions 1302.4.2 Survey of important reaction types of cellulose 1352.4.2.1 Principles and characteristics of cellulose reactions under
homogeneous conditions 1412.4.2.2 Principles and characteristics of cellulose reactions under
heterogeneous conditions 1452.4.2.3 Activation of cellulose 1502.4.3 Advantages and limitations of cellulose reactions in DMA/LiCl
solution 155
3 Analytical Methods in Cellulose Chemistry 1673.1 Determination of the Degree of Polymerization of
Cellulose and its Derivatives 1683.2 Chemical Analysis (Elemental Analysis and Functional Group
Analysis) of Cellulose and Cellulose Derivatives 1733.3 Application of Instrumental Analysis in Cellulose Chemistry 1813.4 Techniques of Polymer Fractionation and Chromatographie
Separation in Cellulose Analysis 1953.5 Summary of Analytical Routes to Total DS and Substituent
Distribution 2023.6 Characterization of the Structure of Cellulosics in the Solid
State 2043.7 Characterization of Cellulose-Liquid Interaction on Swelling
and Dissolution 2133.8 Outlook for the Future Development of Cellulose Analysis 217

Contents IX
Appendix I Experimental Protocols for the Analysis of Cellulose 223Fractionation of cellulose nitrate 227Preparation of:
level-off DP cellulose 232decrystallized cellulose 232cellulose tricarbanilate 233
Determination of:DP of cellulose 234DS of cellulose acetate 235carbonyl group content of cellulose 236carboxyl group content 236water retention value of cellulose 237DS of cellulose xanthogenate 238DS of carboxymethylcellulose 240DS of trity!cellulose 241
Structure analysis of thexyldimethylsily!celluloses by NMR spectroscopyand HPLC 241
Alkali resistance of cellulosic materials 243Alkali solubility of cellulose materials 247
Subject index 253

X Contents
Volume 2; Chapters 4 and 5
4 Systematics of Cellulose Derivatization 14.1 Formation and Modification of the Polymer Skeleton of
Cellulose 14.1.1 Synthesis of the polymer skeleton of cellulose 24.1.2 Covalent crosslinking of cellulose 64.1.2.1 Principles of cellulose crosslinking 64.1.2.2 Chemical routes to crosslinking of cellulose 64.1.2.3 Role of supramolecular and morphological structure in
cellulose crosslinking 144.1.2.4 Material properties of crosslinked cellulose 154.1.2.5 Applications of cellulose crosslinking 164.1.3 Grafting onto cellulose chains 174.1.3.1 Relevance of grafting 174.1.3.2 Chemistry of cellulose graft copolymer formation 174.1.3.3 Some effects of supramolecular and morphological structure 224.1.3.4 Properties and applications of graft copolymers of cellulose 244.1.4 Synthesis of cellulose block copolymers 27
4.2 Interaction of Cellulose with Basic Compounds 314.2.1 Preparation and properties of alkali cellulosates 324.2.2 Interaction of cellulose with aqueous and alcoholic solutions of
alkali hydroxides 334.2.2.1 General comments on the process of interaction and on product
properties 334.2.2.2 Swelling and dissolution of cellulose in alkali hydroxide
solutions 344.2.2.3 Chemical processes of interaction between cellulose and
alkali hydroxide solutions 354.2.2.4 Role of cellulose physical structure in cellulose-alkali
hydroxide interaction 404.2.2.5 Cocepts for understanding cellulose-alkali hydroxide interaction .... 464.2.2.6 Survey of commercisl processes based on cellulose-alkali
hydroxide interaction 494.2.2.7 Properties and application of alkali cellulose 504.2.3 Interaction of cellulose with tetraalkylammonium hydroxides 514.2.3.1 Swelling and dissolution of cellulose in solutions of
tetraalkylammonium hydroxides 524.2.3.2 Chemical interaction between cellulose and tetraalkylammonium
hydroxides 524.2.3.3 Changes in cellulose structure and 54

Contents XI
4.2.4 Interaction of cellulose with guanidinium hydroxide 544.2.5 Interaction of cellulose with ammonia and hydrazine 574.2.6 Interaction of cellulose with aliphatic mono- and diamines 624.2.7 Concluding remarks 66
4.3 Metal Complexes of Cellulose 714.3.1 General routes of cellulose-metal atom interaction 714.3.2 Chemistry of cellulose-metal complex formation 734.3.2.1 Copper complexes of cellulose with N-containing ligands 744.3.2.2 Other aqueous cellulose solvents based on transition metal-
amine complexes 784.3.2.3 Transition metal-alkali-tartaric acid complexes of cellulose 824.3.2.4 Interaction of cellulose with metal hydroxo compounds 854.3.2.5 Interaction of cellulose with some inorganic salts 864.3.3 Supramolecular and morphological aspects of cellulose-metal-
complex formation 904.3.4 Properties of cellulose-metal complexes 924.3.5 Application of cellulose-metal complexes 934.3.5.1 Filament and film formation from cellulose-metal-complex
solutions 934.3.5.2 Covalent functionalization of cellulose dissolved in metal-
complex systems 944.3.5.3 Characterization of cellulose in metal-complex systems 954.3.5.4 Determination of foreign substances in cellulosic products by
means of metal-complex solvents 954.3.6 Future problems of cellulose-metal complex research 96
4.4 Esterification of Cellulose 994.4.1 Esters of cellulose with inorganic acids 1004.4.1.1 Cellulose nitrate 1014.4.1.2 Cellulose nitrite 1124.4.1.3 Cellulose sulfates 1154.4.1.4 Cellulose phosphate and other phosphorus-containing cellulose
derivatives 1334.4.1.5 Cellulose borates 1404.4.1.6 Desoxycelluloses 1424.4.2 Cellulose esters with reagents derived from carbonic acid
(H2CO3) 1454.4.2.1 Cellulose esters of monothiocarbonic acid (H2CSO2) 1454.4.2.2 Cellulose dithiocarbonate esters 1474.4.2.3 Cellulose carbamate 1614.4.3 Esterification with organic acids 1644.4.3.1 General remarks 164

XII Contents
4.4.3.2 Cellulose formate 1664.4.3.3 Cellulose acetate 1684.4.3.4 Cellulose esters of higher aliphatic acids 1824.4.3.5 Esters of cellulose with substituted monocarboxylic
aliphatic acids 1864.4.3.6 Esters of cellulose with di- and tricarboxylic aliphatic acids and
their derivatives 1894.4.3.7 Cellulose esters with aromatic acids 1904.4.3.8 Esters of cellulose with organic acids carrying sulfonic or
phosphonic acid groups 1944.4.3.9 Phenylcarbamates of cellulose 1964.4.4 Concluding remarks on cellulose esterification 197
4.5 Etherification of Cellulose 2074.5.1 General remarks on etherification 2074.5.2 Aliphatic ethers of cellulose 2104.5.2.1 Alkyl ethers of cellulose 2104.5.2.2 Carboxymethylcellulose and related anionic cellulose ethers 2214.5.2.3 Hydroxyalkyl ethers of cellulose 2344.5.3 Various functionalized alkyl ethers of cellulose 2494.5.3.1 Cyanoethylcellulose and related cellulose ethers 2504.5.3.2 Functionalized cellulose ethers with basic N-functions 2554.5.3.3 Sulfoalkyl ethers of cellulose 2604.5.3.4 Miscellaneous functionalized alkyl ethers of cellulose 2614.5.4 Aralkylethers and arylethers 2624.5.4.1 Arylmethyl ethers 2624.5.4.2 TriphenylmethylCtrityl') and related ethers 2634.5.4.3 Arylethers 2734.5.5 Silyl ethers of cellulose 2744.5.5.1 Heterogeneous silylation of cellulose 2784.5.5.2 Homogeneous silylation of cellulose 2794.5.5.3 Properties and structure characterization 2804.5.5.4 Subsequent reactions of silylcelluloses 2854.5.5.5 Formation of supramolecular structures using silylcelluloses 2904.5.6 Summary and outlook 294
4.6 Oxidation of Cellulose 3024.6.1 Oxidation of primary hydroxy groups 3044.6.2 Oxidation of secondary hydroxy groups 308
5 Outlook onto Future Developments in Cellulose Chemistry 3155.1 Cellulose as a Raw Material for Chemical Conversion 3165.2 The Relevance of Intermolecular Interactions 318

Contents XIII
5.3 New Cellulosic Compounds 3195.4 Commercial Processes of Chemical Conversion
of Cellulose 3215.5 Supramolecular Architectures 322
Appendix II Experimental Procedures for the Functionalizationof Cellulose 327
Preparation of FeTNa solvent for cellulose 331Dissolution of cellulose in TV^-dimethylacetamde (DMA)TLiCl 331Preparation of a cellulose trinitrate without significant chain degradation 332Sulfation of cellulose with SO3-DMF 332Cellulose sulfate, synthesis via cellulose trifluoroacetate in DMF 334Cellulose sulfate, synthesis via trimethylsilylcellulose in THF 335Preferentially C-6-substituted cellulose sulfate via an acetate sulfate
mixed ester 336Predominantly C-2/C-3-substituted cellulose sulfates 337Cellulose phosphate from a partially substituted cellulose acetate 338Preparation of a cellulose fiber xanthogenate and a cellulose
xanthogenate solution 339Cellulose tricarbanilate 340Cellulose phenylcarbamate, synthesis via cellulose trifluoroacetate
inpyridine 341Cellulose formate, synthesis in HCOOHTPOCl3 342Laboratory procedure for the preparation of cellulose triacetate
by fiber acetylation 343Acetylation of bacterial cellulose 344Site-selective deacetylation of cellulose triacetate 344Cellulose dichloroacetate, synthesis with dichloroacetic acid/POC!3 345Cellulose trifluoroacetate (DS = 1.5), synthesis with TFA/TFAA ........Ϊ.'.'.'.'.'.'.'! 346Cellulose methoxyacetates, synthesis in DMA/LiCl 347Cellulose-4-nitrobenzoate, synthesis via cellulose trifluoroacetate
catalyzed with p-tosyl chloride 348Cellulose-4-nitrobenzoate, synthesis via cellulose trifluoroacetate with
4-nitro-benzoic acid imidazolide 349Cellulose tosylate, homogeneous synthesis in DMA/LiCl 3502,3-Di-O-methylcellulose 352Carboxymethylcellulose, heterogeneous synthesis in isopropanol/water 353Carboxymethylcellulose, synthesis in DMA/LiCl 355Carboxymethylcellulose, synthesis via cellulose trifluoroacetate
in DMSO 3576-O-Tripheny!methyl (trityl) cellulose, homogeneous synthesis
in DMA/LiCl 3592,3-O-Carboxymethyl-6-O-triphenylmethylcellulose, synthesis via
6-O-tritylcellulose in DMSO 361

XIV Contents
Detritylation of 2,3-O-carboxymethyl-6-O-triphenylmethyl cellulose 362Crosslinking of cellulose powder with epichlorohydrin 363Organosoluble cyanoethylcellulose 364Trimethylsilylcellulose, synthesis in pyridine/THF 365Trimethylsilylcellulose, synthesis in DMA/LiCl 367Celluloses esters, synthesis via trimethylsilylcellulose, general
procedure without solvents 3686-O-Thexyldimethylsilylcellulose 3702,6-Di-O-thexyldimethylsilylcellulose 3716-O-Thexyldimethylsilyl-2,3-di-O-methylcellulose 372Trimethylsilylcellulose methoxyacetate. synthesis via cellulose
methoxyacetate in DMA 3736-Carboxycellulose, homogeneous synthesis with phosphoric acid 374
Subject index 377

List of Abbreviations for Volumes 1 and 2
AGUBnCadoxenCMCCOSYCP-MASCTACTFAGuamCuenDMADMAPDMFDMSODPDPn
DPV
DPW
DSD5Ac
D5N
DSpD5S
DS$iDSx
DTADVSEDAEtFeTNaFTGPCg-tGuOH
acetic acid anhydrideanhydroglucopyranose unit(s)benzylcadmiumethylenediamin chelatecarboxymethylcellulosehomonuclear chemical shift correlation spectroscopycross-polarization magic angle spinningcellulose triacetatecellulose trifluoroacetatecuprammonium hydroxide [Cu(NH3)4]OHcupriethylenediamine chelate7V,Af-dimethylacetamideA^Af-dimethylaminopyridineAf,W-dimethylformamidedimethyl sulfoxidedegree of polymerizationnumber- average degree of polymerizationviscosity-average degree of polymerizationweight average degree of polymerizationdegree of substitutiondegree of substitution of acetyl groupsdegree of substitution of chlorine atomsdegree of substitution determined by means of HPLCdegree of substitution of nitrogen atomsdegree of substitution of phosphorus atomsdegree of substitution of sulfur atomsdegree of substitution of silyl groupsdegree of substitution of xanthogenate groupsdifferential thermal analysisdivinyl sulfoneelectron donor-acceptorethylferric sodium tartrateFourier transformgel-permeation chromatographygauche-transguanidinium hydroxide

XVI Abbreviations
H-CMCHECHMPTHPCLBLODPLRVM.W.mesylateMeMFMSNioxamNioxenNMMNONMPr.h.rts (index)SAXSSECSEMSERSTDMS celluloseTDMSClTEATEMTGt-gTHETMSTMS-ClTPCtriflatWAXSWRV
(index)
the free acid of carboxymethylcellulosehydroxyethylcellulosehexamethylphosphoric acid triamidehydroxypropylcelluloseLangmuir-B lodgettlevel-off degree of polymerizationliquid retention valuemolecular weightmethylsulfonatemethylmole fractionmolar substitutionnickel ammonium hydroxidenickel ethylenediamine chelateAf-methylmorpholine-TV-oxide7V-methylpyrrolidonerelative humidityroom temperaturesubstitutedsmall-angle X-ray scatteringsize-exclusion chromatographyscanning electron microscopySurface enhanced Raman spectroscopythexyldimethylsilyl cellulosethexyldimethylchlorosilanetriethylaminetransmission electron microscopythermogravimetrytrans-gauchetetrahydrofurantrimethylsilyltrimethylsilyl chloridetriphenylcarbinoltrifluoromethanesulfonatewide-angle X-ray scatteringwater retention valueneighbour C-atom

4 Systematics of Cellulose Functionalization
The following systematics of cellulose functionalization will be structured ac-cording to the typical reaction types of hydroxy groups, i.e. esterification,etherification and oxidation. Specific characteristics of the cellulose macromole-cule will also be considered, i.e. the formation of addition compounds with basicsubstances and the formation of metal complexes, as well as changes of thepolymer skeleton by grafting or crosslinking. Each of the chapters will be prod-uct-centered, describing primarily the chemistry of formation of the derivative,its subsequent modification and considering also properties and applications ofthe product formed. Special emphasis will be given to the kinetics and mecha-nism of the derivatization reaction and on the role of cellulose supramolecularstructure. For products of commercial relevance, a brief description of the tech-nical process is included. For selected products of scientific and/or practicalinterest, a laboratory procedure for synthesis, purification and characterizationwill be given in the Appendix of this volume.
4.1 Formation and Modificationof the Polymer Skeleton of Cellulose
Before turning to the functionalization of cellulose at the hydroxy groups, it isappropriate to survey briefly some routes of formation and modification of thepolymer skeleton of cellulose, considering the following topics:(i) synthesis of the ß-l,4-glucan chain,(ii) covalent crosslinking between cellulose chains,(iii) combination of ß-l,4-glucan sequences with synthetic macromolecules bygrafting and by synthesis of block copolymers.(iv) modification of the cellulose skeleton by formation of cyclic ethers acrossthe AGU, and subsequent changes in the configuration of the functional groups,
Most of the work published in the whole subject area is concerned withgrafting and crosslinking of cellulose, the latter topic being of great practicalrelevance in connection with the finishing of cellulosic textiles. Results on thechemical synthesis of the cellulose molecule are still rather scarce and have metwith limited success only in comparison with the perfect achievement of nature.But the first successful regio- and stereoselective synthesis of nature-identical
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

2 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
cellulose without enzymes or microorganisms in 1996 (Nakatsubo et al. 1996),was an intellectually important result and a principally novel way to preparefunctionalized celluloses. Changes in the configuration of the macromolecule viainner ether formation, as well as the block copolymerization of cellulose, arepresently considered as areas of limited interest only, but of scientific relevance.
4.1.1 Synthesis of the polymer skeleton of celluloseThree routes of synthesis of the cellulose chain have to be considered here, i.e.(i) biosynthesis in living organisms,(ii) in vitro enzymatic synthesis,(iii)chemical synthesis by polymerization of suitable monomers.
Cell wall
Pore subunit
Crystallizationsubunit
jeftjzs
\CyCytoplasm
Microfibril Plasma membrane
Figure 4.1.1. Hypothetical model of a cellulose synthase complex in the plasma mem-brane (Delmer and Amor, 1995).
Until the middle of this century, cellulose was taken for granted as a polymerdelivered by nature, and the research activities were centered on its chemical andphysical processing and on the elucidation of its structure. But this situation haschanged in more recent decades, due to the rapid developments in biochemistry.The course and the mechanism of biosynthesis of cellulose has received growinginterest in academic research, as demonstrated by the rapidly growing number ofrelevant publications, which however up to now have remained without techno-logical consequences.
The very complex process of cellulose biosynthesis comprises not only thestepwise formation of the ß-l,4-glucan chain, but also the establishment of a

4.1.1 Synthesis of the polymer skeleton of cellulose 3
well-defined supramolecular order and fibrillar architecture in the solid polymerformed. Furthermore, different mechanisms have to be assumed for the forma-tion of cellulose in higher plants on the one hand, and in bacteria and algae onthe other. According to Brown (1996) and Delmer and Amor (1995) this is ac-complished by a complex of proteins with different enzymic and other func-tional activities (see Fig. 4.1.1). A detailed description of cellulose biosynthesiswas published by Colvin (1985) and Tarchevsky and Marchenko (1991).
In the last few years, the biosynthesis of cellulose using bacteria such as Ace-tobacter xylinum has been extended as the synthesis of partially functionalizedcelluloses. According to Ogawa and Tokura (1992a, b), the copolymerization of ß-D-glucose with 7V-acetylglucosamine by Acetobacter xylinum leads to the incor-poration of the amino sugar into the cellulose skeleton of up to 4 mol %.
The enzymatic in vitro synthesis was investigated in recent years along tworoutes:(i) reacting UDP(uridine-diphosphat)-glucose with purified cellulose synthase;(ii) condensation of glucose or its derivatives by cellulases.
Achievements along the first route are summarized by Lin and Brown Jr.(1989), (see also Amikan and Benziman, 1989; Kudlicka et al., 1996; Blantonand Northcote, 1990). A simplified scheme of this route is shown in Fig. 4.1.2(Kobayashi et al., 1995).
The enzymatic in vitro synthesis of 'short chain' cellulose of DP 22 has beendescribed (Kobayashi et al., 1992; 1995; 1996). ß-Cellobiosyl fluoride was con-densed as the substrate in a mixed solvent of acetonitrile and an aqueous buffer(pH 5) by means of purified cellulase from Trichoderma viride, an enzyme sys-tem well known for its hydrolysis activity on the glycosidic linkages of long-chain cellulose (see chapter 2.2). The reaction system changed from a homoge-neous to a heterogeneous state during the 12 h of treatment, and the reactionproduct obtained was characterized as a linear ß-l,4-glucopyran, identical withcellulose, by 13C NMR- and IR spectroscopy, as well as by conversion to cellu-lose triacetate after previous deactivation of the enzyme system. With a purifiedcellulase, Lee et al. (1994) succeeded in assembling the ß-l,4-glucan chainsduring their synthesis to a defined supramolecular structure resembling celluloseI. It was assumed that a micellar aggregation of the partially purified enzymeoccurs and that in the substrate, in an organic/aqueous solvent system, there isalignment of glucan chains with the same polarity and extended chain confor-mation favored. Included in the enzymatic in vitro synthesis is the preparation offunctionalized celluloses, e.g. of the methyl ether starting from 6-0-methyl-ß-cellobiosyl fluoride.
Since the early attempts by Schlubach (Schlubach and Luhrs, 1941) numerousresearch efforts have been devoted to the chemical synthesis of the cellulosemacromolecule by polycondensation or by ring opening polymerization, but all

4 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
of these studies had limited success, obviously due to the difficulties of obtain-ing a strictly linear stereoregular chain structure. So, for example, the condensa-tion of 2,3,6-glucose tricarbanilate with ?2θ5 in a mixture of CHCl3 and DMSOresulted in a cellulose-like but branched polymer, containing about 1 % phos-phorus. Also, by a cationic polymerization of l,4-anhydro-2,3,6-0-benzyl-oc-D-glucopyranose with various Lewis acids, no stereoregular 1,4-glucopyran couldbe obtained (Micheel et al., 1974; Micheel and Broode, 1974 and 1975; Uryu etal., 1985). Obviously, the choice of suitable protecting groups in the monomer isthe decisive point, as demonstrated by Uryu et al. (1981) by the synthesis of a ß-1,4-D-ribopyran by a cationic ring opening polymerization. For this topic thereader is also referred to a comprehensive review by Kotchetkov (1987) on thesynthesis of polysaccharides with a regular structure.
OH
O OIl Il
,Ps. J\
I 0^1O O
UDP-glucoseHO OH
cellulosesynthase
OH
HOOH
cellulose
Figure 4.1.2. Simplified scheme of enzymatic in vitro synthesis of cellulose startingfrom UDP-glucoe (Kobayashi et al, 1995).
Recently, Nakatsubo et al. (1996) succeeded in synthesizing cellulose mole-cules by cationic ring-opening polymerization of 3,6-di-O-benzyl-a-D-glucose-1,2,4-0-pivalate to 3,6-di-0-benzyl-2-O-pivaloyl-ß-D-glucopyran, and subse-quent removal of the protecting ether and ester groups. The presence of adequateether groups, preferably benzyl groups, in the 3-O-position is considered to beessential for achieving a stereoregular structure, and the presence of estergroups, preferably pivaloyl groups in the 2-O-position, is required for securing aß-glucosidic linking of the monomer units. A simplified scheme of the synthesisis presented in Fig. 4.1.3.

4.1.1 Synthesis of the polymer skeleton of cellulose 5
R1 R
-" n
Figure 4.1.3. Simplified scheme of cellulose synthesis by cationic ring-opening polym-erization (Nakatsubo et al., 1996).
Λ^Λ^-carbonyldiimidazole served as a dehydrating agent in ortho ester synthe-sis. This reagent, frequently employed in glucoside and peptide synthesis, pref-erentially attacks a hydroxy group that is more acidic than 4-OH to give a 1-0-carbonylimidazole derivative. This is further converted to a dioxocarbenium ionintermediate, by removal of the carbonyl imidazole group and then to an ortho-ester by intramolecular attack of 4-OH. Polymerization of the ortho-ester can becatalyzed by BF3 · Et2O, by (phenyl)3
+CSbC!6~ or most efficiently by(phenyl)3+CBF4~ in methylene chloride as the medium. The ß-l,4-glucopyranstructure of the compound obtained with a DPn of about 20 was confirmed by13C NMR spectroscopy. The transformation of this compound to cellulose wasachieved via the triacetate by converting it at first to the 2-O-acetyl derivativewith MeONa in tetrahydrofuran (THF)/MeOH, and subsequently with acetanhy-dride in pyridine, followed by debenzylation with Pd/H2 under pressure andacetylation of the free hydroxy groups with acetic anhydride in pyridine. Nodepolymerization was observed during this procedure. After deacetylation withMeONa in THF, finally a cellulose showing the X-ray pattern of cellulose II wasobtained. This route of synthesis described here in some detail obviously repre-sents the present 'state of the art' and simultaneously gives an impression of thedifficulties and problems to be overcome in regio- and stereoselective cellulosechemosynthesis.
An interesting route to a highly branched cellulose macromolecule was re-cently reported by Franzier et al. (1996). An anhydrous solution of cellulose inDMA/LiCl was treated with hydrogen fluoride in pyridine at a low HF concen-

6 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
tration, resulting in long-chain branching of the polymer, which is obviouslycaused by transglycosidation via glycosyl fluoride groups as intermediates.
4.1.2 Covalent crosslinking of cellulose4.1.2.1 Principles of cellulose crosslinking
From the commercial point of view, the formation of covalent crosslinks be-tween the cellulose chains is the most important route to modify the polymerskeleton of this polysaccharide. It is widely employed on a large, industrial scaleto improve the performance of cellulosic textiles. Although structure and mate-rial properties of cellulose in the solid state are largely determined by a self-crosslinking via intermolecular hydrogen bonds, this intermolecular interactionis partially reversible in the presence of water and is completely overcome byconventional cellulose solvents like aqueous Guam. Thus covalent crosslinkingis required to avoid undesirable changes of cellulosic goods in the wet state.
Since Eschalier's reported crosslinking of cellulose by the action of formalde-hyde (Eschalier, 1906 and 1907) at the beginning of this century, numerouscrosslinking agents and crosslinking reactions have been described, most ofthem being based on the formation of ether bonds by alkylation of hydroxygroups at neighboring cellulose chains. Material properties after crosslinkingwere found to depend on the constitution and length distribution of thecrosslinks on the one hand, and on the crosslink density (average distance be-tween two crosslink points along the cellulose chain) and the distribution of thiscrosslink density within the fiber structure on the other. This implies a stronginfluence of cellulose supramolecular and morphological structure on the effectsof crosslinking with the reagent employed.
In this subchapter, the chemistry of crosslinking will be considered first,turning then to the interplay between crosslinking and physical structure, andfinally surveying the changes in the material properties obtained and the indus-trial application of covalent crosslinking.
4.1.2.2 Chemical routes to crosslinking of cellulose
There are various routes to crosslinking the polymer by covalent or ionic reac-tions.
• Recombination of cellulose macroradicals formed chemically or by irradiation.• Reaction of anionic cellulose derivatives by at least divalent metal cations.• Oxidative crosslinking by formation of disulfide bridges from mercapto groups
attached to cellulose.

4.1.2 Covalent crosslinking of cellulose 1
• Formation via urethane bridges by reaction of cellulosic hydroxy groups withisocyanates.
• Crosslinking via ester groups formed by reaction with polycarboxylic acids.• Formation of ether bonds with an at least difunctional etherifying agent.
Covalent or ionic reactions can take place either intermolecularly, i.e. betweenreactive sites of two or more different macromolecules, or intramolecularly, i.e.between suitable sites along the same polymer chain. Both processes usuallyoccur simultaneously to a varying extent. The analytical characterization of thecrosslinked products still poses serious problems: usually only an average num-ber of crosslinks per unit chain length (crosslink density) can be estimated fromthe amount of heteroatoms like nitrogen or sulfur introduced, or from a determi-nation of the gain in weight of the sample, due to addition of the crosslinkingagent. Information on the distribution of the crosslinks and on details of theirstructure is still rather scarce and mostly obtained by indirect methods, such asfor example characterization of physicochemical bulk properties of thecrosslinked products, such as for example swelling or solubility.
Macroradicals suitable for crosslink formation by recombination can be gen-erated from cellulose chains either by high-energy irradiation leading to homo-lytic bond cleavage, or by transfer reactions from a radical source outside themacromolecules. Kriss et al. (1985) reported the photolytic generation of ligandradicals of Mn3+ complexes with acetyl acetonate, and the subsequent formationof cellulosic macroradicals by a transfer reaction, finally resulting in crosslink-ing, and a predominant crosslinking in comparison with cellulose chain degra-dation is assumed by Philipp et al. (1982) after electron-beam irradiation of cel-lulose at a low dose rate.
Ionic crosslinking requires the presence of anionic groups, like carboxymethylgroups or sulfuric acid half-ester groups. As suitable crosslinking agents FeCl3,Al2(SO4^ or Cr2(SO4)3 are known (Heinze et al. 1990). Further details on ioniccrosslinking and application of the gels obtained will be described in connectionwith carboxymethylcellulose (see chapter 4.5) and with carboxycellulose (seechapter 4.6).
Crosslinking of cellulose by oxidative coupling of mercapto groups to disulfidebridges was studied (Sakamoto et al., 1970), comparing samples with the mercaptogroups directly bound to the cellulose chain with those with the mercapto groupstethered to the polymer backbone via a long spacer. In the latter case a completeand fully reversible oxidative crosslinking could be easily achieved due to themobility of the mercapto groups, while with these groups directly bound to thebackbone only a small fraction could be converted to disulfide bridges.
The reaction of cellulosic hydroxy groups with diisocyanates usually poses noproblems (Sakamoto et al., 1970). This route is not practiced in textile finishing

8 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
due to the toxicological hazards involved. A combination of the activities of theisocyanate group and a vinylic C=C double bond in cellulose crosslinking wasrealized by employing acrylic isocyanate as the crosslinking agent.
Crosslinking by ester bond formation occurs in the reaction of cellulose with asuitable di- and polycarboxylic acid. According to recent infrared studies (Yangand Wang, 1996) five-membered cyclic anhydrides are formed as intermediatesin the thermally activated crosslinking of cotton fabrics with suitable polycar-boxylic acids. Comparing the crosslinking performance of different polycarbox-ylic acids, those carrying their carboxyl groups at adjacent C atoms, and thusbeing capable of forming five-membered anhydrides, were found to be moreeffective in cellulose crosslinking than those carrying their carboxyl groups atalternating C atoms of the polymeric acid chain. The only six-membered cyclicanhydride formed and detected on the treated cotton was that of poly aery lie acid.Self-crosslinking via intermolecular esterification can take place with anioniccellulose derivatives, especially carboxymethylcellulose, at low pH and elevatedtemperature, due to reaction of acid groups with free hydroxy groups of neigh-boring polymer chains.
The numerous routes to crosslinking cellulose via acetal resp. ether bonds willnow be considered in some detail due to their scientific and commercial rele-vance:
• Acetalization of hydroxy groups with formaldehyde• Acetalization with glyoxal or its homologs• Reaction with N-derivatives of formaldehyde like dimethylol urea• Michael addition of divinylic compounds with hydroxy groups• Etherification of hydroxy groups by aliphatic di- or tri-halogenated com-
pounds like dichloroethane• Alkylation by epoxides like 1,2,3,4-diepoxibutane• Etherification by epichlorohydrin
Crosslinking with formaldehyde proceeds as a two-step reaction via a cellu-lose hemiacetal (methylolcellulose) as an intermediate according to the generalscheme
CeII-OH + CH2O · - CeII-O-CH2OH
CeII-O-CH2OH + CeII-OH · - CeII-O-CH2-O-CeII + H2O
In reality, the formation of acetal bridges - usually taking place in an aqueousacid medium - is considerably more complicated by the fact that:

4.1.2 Covalent crosslinking of cellulose 9
(i) both steps proceed as equilibrium reactions, and the acetal bridges exhibit alimited stability only and can split-off formaldehyde under suitable conditions;(ii) crosslinking in the acid medium is inevitably accompanied by some chaindegradation due to acid hydrolysis of glycosidic linkages, which becomes morepronounced with increasing reaction temperature;(iii) the kinetics of the crosslinking reaction is governed by a specific acid ca-talysis, with the rate of formaldehyde add-on increasing with increasing H+ orH3O
+ concentration (Fig. 4.1.4).
_ © fast _ ®CeII-O-CH2-OH + H ^=^ CeII-O-CH2-OH2
_ © slow _ Θ ©CeII-O-CH2-OH2 ^=±: [CeII-O-CH2 -CeII-O = CH2J+ H2O
H- θ fast Ie
CeII-O-CH2+ CeII-OH ^=^ CeII-O-CH2-O-CeII
HΙΘ fast Ä
CeII-O-CH2-O-CeII ^=± CeII-O-CH2-O-CeII + He
Figure 4.1.4. Scheme of acid catalysis in formaldehyde crosslinking of cellulose (takenfrom Meyer et al., 1976).
Meyer et al. (1976) mentions in his detailed studies on the kinetics and themechanism of this process that a specific catalysis by H+ or H3O
+ is responsiblefor more than 98 % of the crosslinks formed by formaldehyde. It was assumedthat added metal salts like MgCl2 co-catalyze the process by increasing theH3O
+ concentration and not by a catalytic action of the metal cation itself. Theoverall course of the reaction was determined by one of the chemical reactionsteps or by swelling and diffusion processes, depending on reaction conditionsand structure of the cellulose sample. In practise, crosslinking with formalde-hyde can be performed as a wet process by treating the specimen with an aque-ous acidic formaldehyde solution at room temperature and subsequent curing at100-130 0C, with the crosslinking taking place within minutes during this dry-ing process. An alternative is the so-called dry process, with the specimensoaked at first with aqueous boric acid followed by drying and subsequently bythe crosslinking action of paraformaldehyde vapor.

10 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
The inconvenient handling of free formaldehyde can be avoided and the struc-ture of the crosslinks can be varied within wide limits by employing as crosslink-ing agents the methylol or alkoxymethyl derivatives of different N-containingcompounds (urea, cyclic ureas, carbamates, acid amides or triazines) forming ace-tal bridges between the cellulose chains as indicated by the scheme (Fig. 4.1.5):
OM H®
2 CeII-OH + HO-CH2-N-C-N-CH2-OHI - ι ' "" -2W 2 O
H HOIl
CeII-O-CH2-N-C-N-CH2-O-CeIII I
H H
OI l H®
2 CeII-OH + H3CO-CH2-N-C-N-CH2-OCH3 —I I -2 CH3OH
H H
Figure 4.1.5. Crosslinking of cellulose with urea derivatives.
Besides the crosslinking reaction proper, self-condensation of the agent as wellas liberation of formaldehyde have to be taken into account in this usually acid-catalyzed process governed by interdependent chemical equilibria. The structuraltype of CH2O binding in these crosslinkers can vary widely, resulting in largedifferences in stability against formaldehyde liberation (Petersen and Petri, 1985):
•^s
Λ N—GH,—Ο—Cell · CeII-O-CH2-O-CeIII
ο
^N-CH,-OH · CeII-O-CH2-OH
J-CH2-OR · CH2O , HO-CH2-OH ,
' R - AlkylO O
AΝ —CH2-N'

4.1.2 Cov alent er o s slinking of cellulose 11
O
HOCH2-N^N-CH2OH
O
AHOCH2-N NH + CH2O
100
χ 10,-5
50
2 4. 6 8 pH 10
Figure 4.1.6. pH-dependent stability of a methylol group in a cyclic urea (Peterson andPetri, 1985).
As illustrated by the example in Fig. 4.1.6, the stability of methylol groupsagainst acid or alkaline hydrolysis is largest near the neutral point, with the rateconstant of hydrolysis increasing steeply to both sides of the pH scale.
The kinetics and the mechanism of these crosslinking processes have beenthoroughly studied over the last 30 years (Peterson and Petri, 1985). The reac-tivity of the agents and the stability of the crosslinks formed against formalde-hyde liberation could be correlated to their constitution. By techniques of mo-lecular modeling and statistical design, high-performance crosslinkers have beendeveloped with only a minimal tendency to liberate CH2O during processing andstorage of the textile goods subjected to this crosslinking treatment.
While formaldehyde must be considered as difunctional in forming acetalbridges between cellulose chains, glyoxal can act as a tetrafunctional crosslinker,connecting two cellulose chains already at the hemiacetal formation stage of thereaction. Model experiments with low molecular alcohols (Sangsari et al., 1990)on competitive hemiacetal and acetal crosslinking, led to the conclusion thatalcohols with two vicinal hydroxy groups are much more effective in hemiacetalformation than those with isolated hydroxy groups, while the subsequent cata-lyzed acetal formation proceeded preferentially with isolated alcoholic hydroxygroups. A predominant formation of dioxan bisacetal structures was reported inthis study, and the reaction mechanism derived was assumed to hold true in

12 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
principle also for crosslinking of cellulose by glyoxal. Glycol aldehyde and gly-col were reported to be effective co-reagents. In agreement herewith also, theproperties of an hydroxyalkyl ether of cellulose like hydroxyethylcellulose in anaqueous medium can be efficiently changed by crosslinking with glyoxal.
Crosslinking by Michael addition of cellulosic hydroxy groups onto vinyliccarbon-carbon double bonds is preferably performed with divinyl sulfone(DVS) in an aqueous alkaline system according to:
2 CeII-OH + CH2 = CH-SO2-CH = CH2
CeII-O-CH2-CH2-SO2-CH2-CH2-O-CeII
The formation of hydrogels from mixtures of CMC and hydroxyethylcellulosein aqueous alkaline solution (0.02 M KOH) by crosslinking with DVS may besighted (Esposito et al., 1996). The crosslinking density, defined the ratio be-tween the maximal number of reacted sites (based on DVS input) and the totalnumber of reactive sites, was varied within wide limits via the molar ratio ofDVS to polymer. Crosslinking densities above 1 indicate a partially monofunc-tional mode of reaction of the difunctional crosslinker.
Formation of ether crosslinks by reaction of cellulose with alkyl halides orepoxides proceeds along the conventional routes of cellulose etherification (seechapter 4.5), as indicated by the examples in Fig. 4.1.7.
OH0
2 CeII-OH + CICH2-CH2CI ^- 2 HCI
CeII-O-CH2-CH2-O-CeII
OH0
2 CeII-OH + CH2-CH-CHp-CH >\ / \ /
O O
CeII-O-CH2-CH-CH-CH2-O-CeIII I
OH OH
Figure 4.1.7. Crosslinking of cellulose by 1,2-dichloroethane and 1,2,3,4-diepoxybutane.
While the reaction between halide functions and the hydroxy groups requiresa strongly alkaline medium, the ring opening and subsequent formation of etherbonds with diepoxides is catalyzed already by a low alkali concentration. Alsoacid catalysis of this reaction has been reported. According to Benerito et al.

4.1.2 Covalent crossünking of cellulose 13
(1961) the change of cotton properties by crosslinking with diepoxides dependslargely on the ratio of Zn(B F4)2 as an acidic catalyst per mol of AGU.
CH2-CH-CH2CI\ /
O
(OH®)
CeII-OH +ΘCHo-CH-CHoCI
ΙΟΙ,θ
CeII-O-CH2-CH-CH2CI
OH
θCH2-CH-CH2CI
1 θΙΟΙ
CeII-O-CH2-CH-CH2CI
OH
> CeII-O-CH2-CH-CH2 +
O
Cell-O-CH2-CH-CH2+Cell-OH\ /
CeII-O-CH2-CH-CH2-O-CeII
OH
Side reactions
CeII-O-CHp-CH-CHpCII
OH
CH2-CH-CH2CI
O
CH-CH-CH2CI
O
H2O / ΟΗΘ
CeII-O-CH2-CH-CH2OH
OH
CeII-O-CH2-CH-CH2CI
-*- CH2-CH-CH2I l I
OH OH OH
Q-CH2-CH-CH2CI
OH
Figure 4.1.8. Scheme of cellulose crosslinking with epichlorohydrin.
A combination of the halide function and the epoxide function is realized inthe frequently employed crosslinking agent epichlorohydrin. According to thereaction scheme presented in Fig. 4.1.8, the epoxide ring is cleaved in the alka-line reaction medium with subsequent formation of a l-chloro-2-hydroxypropylether of cellulose. Then the Cl atom is split-off as a chloride anion in the pres-ence of the strong alkali and a 1,2-epoxide is formed which, after cleavage, re-acts with a second hydroxy group of cellulose to give a 2-hydroxypropyl ethercrosslink. A direct reaction between the chlorine atom and a cellulosic hydroxygroup (ether formation by Williamson reaction) is obviously impeded under thestrongly alkaline conditions employed, favoring epoxy ring formation. As a sidereaction, saponification of the l-chloro-2-hydroxypropyl ether to a 1,2-dihydroxypropyl ether of cellulose can take place. Furthermore, epichlorohydrincan be saponified to glycerine, or further molecules of the crosslinker can be

14 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
added to the hydroxy group of the l-chloro-2-hydroxypropyl ether resulting inlonger crosslinking bridges. The reactivity of cellulosic hydroxy groups in epi-chlorohydrin crosslinking decreases in the order OH-2 > OH-6 > OH-3 (Luby etal., 1979).
The presence of a sufficient amount of water and of a NaOH concentration ofat least 9 % (Dautzenberg et al., 1980) have to be considered as necessary pre-requisites for successful epichlorohydrin crosslinking, which is performed usu-ally by steeping in the epichlorohydrin-containing alkaline liquid, by sprayingthis liquid onto the cellulose sample, or by treatment with epichlorohydrin vaporafter alkaline steeping. A reaction time of 2 h and a reaction temperature of60 0C were found to be adequate in crosslinking of cellulose powder (Fanter,1980). A very large amount of water present and a low temperature of reactionhave been reported to favor 1,2-dihydroxypropyl ether formation and thus todecrease the reagent yield for crosslinking which can be 80-90 % under optimalreaction conditions. The degree of crosslinking can be varied within wide limitsup to about 1.5 via the molar ratio of epichlorohydrin per AGU.
4.1.2.3 Role of supramolecular and morphological structure incellulose crosslinking
The number of crosslinks formed and their distribution within the cellulose sam-ple depends largely on its structure. This holds true for acid-catalyzed formalde-hyde or methylol urea crosslinking, as well as for the action of diepoxides or ofepichlorohydrin in a strongly alkaline medium. An important factor controllingcrosslink density and distribution, and thus also the changes in material proper-ties, is the state of swelling of the sample prior to or during the crosslinking pro-cess.
The distribution of formaldehyde crosslinks was assessed by a special dyingtechnique with rhodamine B (Kokot et al., 1975). A different distribution ofdimethylol urea derivatives in cotton was reported after previous NH3 treatmenton the one hand, and mercerization with NaOH on the other, with this differentdistribution also being reflected in the material properties of the crosslinkedsamples (Zeronian et al., 1990).
On crosslinking with epichlorohydrin, the crystallinity of cellulose I is af-fected only after previous transformation to sodium cellulose. After neutraliza-tion and drying of the crosslinked sample a rather diffuse X-ray pattern in-between the lattice types of sodium cellulose and cellulose II was observed dueto the spacing action of the ether crosslinks impeding the formation of a well-defined cellulose II lattice (Dautzenberg et al., 1980). The mode of alkali treat-ment and the structural changes resulting therefrom were found to influencelargely the course of epichlorohydrin crosslinking. The gross morphology of
4.1.2 Covalent crosslinking of cellulose 15
cellulose powder particles exhibited only minor changes after epichlorohydrincrosslinking, and the altered morphology on the fibrillar level revealed by scan-ning electron microscopy seemed to be caused mainly by subsequent deswellingand shrinking and not by the crosslinking reaction itself.
4.1.2.4 Material properties of crosslinked cellulose
Just as with other linear polymers, cellulose is rendered insoluble in its commonsolvents by crosslinking to a sufficiently high density. The solubility in Guam ofepichlorohydrin-crosslinked !inters powder was found to decrease sharply, wellbelow a degree of crosslinking of 0.1 in the case of a uniform crosslinked distri-bution throughout the cellulose structure. The presence of non-crosslinked re-gions shifted the onset of solubility decrease to a somewhat higher degree ofcrosslinking.
80
x102
60
ιgI2 0
a)§200
"100fe
I0.2 0.6 1.0
Degree of crosslinking
0.01 0.03 0.05Mole crosslink /mole cellulose
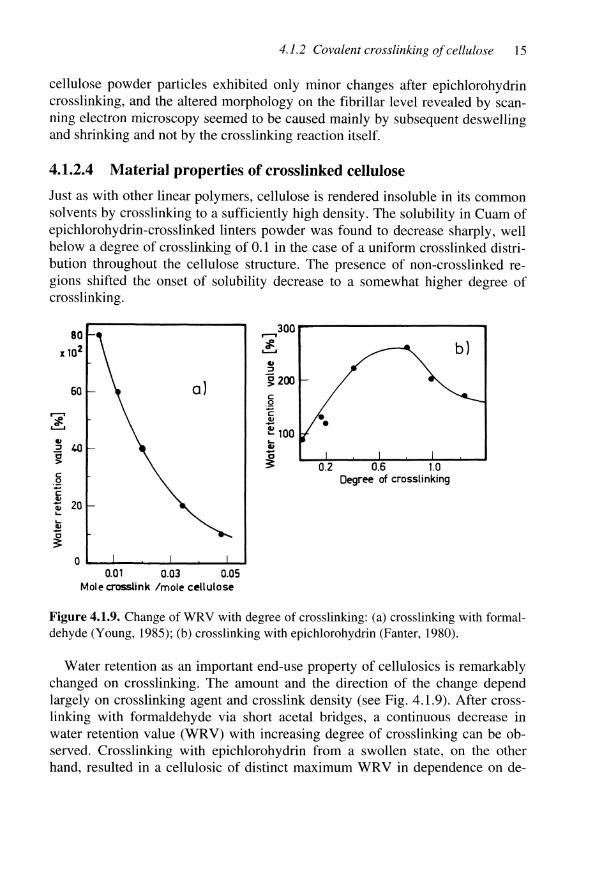
Figure 4.1.9. Change of WRV with degree of crosslinking: (a) crosslinking with formal-dehyde (Young, 1985); (b) crosslinking with epichlorohydrin (Fanter, 1980).
Water retention as an important end-use property of cellulosics is remarkablychanged on crosslinking. The amount and the direction of the change dependlargely on crosslinking agent and crosslink density (see Fig. 4.1.9). After cross-linking with formaldehyde via short acetal bridges, a continuous decrease inwater retention value (WRV) with increasing degree of crosslinking can be ob-served. Crosslinking with epichlorohydrin from a swollen state, on the otherhand, resulted in a cellulosic of distinct maximum WRV in dependence on de-

16 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
gree of crosslinking. Obviously the spacer action and the hydrophilicity of 1,2-dihydroxypropyl ether chains formed dominates at low and medium crosslinkdensity and enhances the WRV, before it decreases again at high crosslink den-sity (see chapters 2.2 and 2.3). It is interesting to note that the susceptibility toenzymatic or acid hydrolysis of glycosidic bonds also passed a distinct maxi-mum with increasing degree of crosslinking.
Crosslinking, especially with formaldehyde or formaldehyde urea compounds,affects decisively the mechanical properties of cellulose fibers and threads.These effects are the basis of commercial application of cellulose crosslinking inthe textile industry. The stiffness and wrinkle resistance of cellulosic threads aresignificantly enhanced by crosslinking, while strength and extensibility are di-minished. According to Cowan and Hurwitz (1982) this strength loss is largelyreversible after de-crosslinking by cleaving the acetal bridges with alkali, andthus is not to be traced back to the inevitable loss of DP connected with the pro-cess, but is caused by the crosslinking itself.
4.1.2.5 Applications of cellulose crosslinking
The crosslinking of cellulose finds its most important commercial application intextile finishing of cellulose-based fabrics for conveying to them some end-useproperties relevant for the consumer, like e.g. wrinkle resistance, permanentpress and easy care properties, or a special handle. These developments startedwith the integration of a formaldehyde treatment into the viscose process andwas later expanded to the treatment of textile goods from cotton. Today pre-dominantly methylolated or alkoxymethylated urea compounds are employed ascrosslinking agents. Usually the fabric is soaked with the aqueous crosslinkingsystem in a continuous process at room temperature and at a speed of 60-100 m/min and then continuously cured at a temperature between 100 and130 0C. The actual development aims to have the crosslinking agents liberating aminimum of formaldehyde in processing as well as in storage and use of thefabrics, a partially methylated dimethylol urea derivative being sighted as anexample (Petersen, 1990).
Crosslinking by epichlorohydrin was employed to modify the pore structureand the swelling behavior of cellulose beads (Loth and Philipp, 1989). Forma-tion of hydrogels by crosslinking water-soluble cellulose ethers with variouscrosslinking agents has been proposed for the preparation of Chromatographiematerials. Especially the crosslinking of carboxymethylcellulose along variousroutes has been widely studied in order to open up new areas of application, forexample as dental glue after partial self-crosslinking between hydroxy and car-boxyl groups, or as a component in sanitary goods making use of the high swel-ling and high water-binding capacity of CMC, rendered insoluble in water bycovalent crosslinking (Klemm et al., 1985; Young, 1985; Heinze et al., 1990).

4.1.3 Grafting onto cellulose chains 17
4.1.3 Grafting onto cellulose chains4.1.3.1 Relevance of grafting
Grafting of synthetic polymers onto the macromolecule cellulose has been am-ply studied in the second half of this century as a scientific challenge based onprinciples of cellulose chemistry as well as on general polymer chemistry, and asa promising route to combine the advantages of the material properties of cellu-lose with those of synthetic polymers. The 'state of the art' about 10 years agohas been comprehensively described by Helbreich and Guthrie (1981). Gener-ally all the routes of polymer synthesis known today can be employed for a co-valent attachment of polymer side chains onto a cellulose backbone, but freeradical polymerization of vinylic compounds initiated by a redox system or byhigh-energy radiation dominates by far. Mostly the grafting is performed ontocellulosic materials in the solid state applying liquid or gaseous monomers, withthe consequence of a strong influence of the supramolecular and morphologicalstructure of the cellulosic substrate on the course of the grafting reaction. De-spite the remarkable and often favorable changes in the material properties ofcellulosics obtainable by grafting, and despite several promising developmentsreaching the pilot plant level, the commercial application of cellulose graftingremained behind the optimistic expectations announced two or three decadesago, obviously mainly for economical reasons.
Within this subchapter, the chemical principles of cellulose grafting will beconsidered first, in connection with the relevant reaction parameters and thestructural parameters employed for cellulose graft copolymer characterization.Subsequently, some effects of supramolecular and morphological structure ofthe substrate on the course of grafting will be surveyed briefly, turning then fi-nally to the material properties and some areas of application of cellulose graftcopolymers.
4.1.3.2 Chemistry of cellulose graft copolymer formation
Ushakov (1943) first attempted to copolymerize allyl and vinyl derivatives ofcellulose with acrylic acid esters, resulting in the formation of insoluble graftedpolymers. Table 4.1.1 summarizes typical routes of cellulose grafting. But quitepredominantly the free radical polymerization of vinylic compounds has beenused in studying cellulose grafting (Berlin and Kislenko, 1992).
As shown in the scheme below, cellulose graft polymerization is inevitablycombined with some homopolymerization of the monomer. The analytical char-acterization of a cellulose graft copolymer therefore requires, besides the deter-mination of the so-called add-on, i.e. the amount of monomer transformed topolymer, a separate assessment of the homopolymer formed via its extraction, inorder to obtain the grafting efficiency. Furthermore, the length of the grafted

18 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
side chains and their number per backbone molecule of average chain length canvary within wide limits and can be estimated after hydrolysis of the cellulosebackbone by performing a macromolecular characterization of the side chains. Itmust be emphasized, however, that graft copolymer analysis poses many prob-lems and uncertainties in its practical realization.
Table 4.1.1. Routes to graft copolymers of cellulose
Route Example
Free radical polymerization
Anionic polymerization
Cationic polymerization
Ring opening polymerizationPolyadditionPolycondensationCoupling of preformed macro-molecules onto cellulose
Styrene after redox initiationAcrylonitrile after high-energyirradiationAcrylonitrile onto alkalicellulosate'Cardanol' after initiation withBF3 etherate (John and Pillai,1989)CaprolactamEthylene oxide + NaOHaq
Amino carbonic acid chloridesPolyamide, polyesters
Initiation (irradiation) Propagation
C — C— C'
M — M*— M*
C + M —CM
C* + M* — CM* or C*+ M
CM*+ nM —CMn+1
M* + nM —- MO+1
Chain transfer
C* + S — C +
S* + M S +
S
M'
TerminationC* + M * — CMCM*,+CM;— c2Mm+n
CM;+CM;— CMn-CMm
Mn* + M* -Mn+1
A broad variety of cellulosic materials has in the meantime been employed assubstrates for grafting. Besides cotton and other natural fibers, wood pulp andviscose filaments and fabrics, also lignocellulosic materials like straw or cellu-

4.1.3 Grafting onto cellulose chains 19
lose derivatives like cellulose acetates have been used. Some monomers, fre-quently reported as suitable for cellulose grafting, are:
• Styrene · Acrylic acid · Vinylpyridine• Acrylonitrile · Na-vinyl sulfonate · Dimethylaminoethyl• Acrylic acid esters methacrylate• Methacrylic acid esters• Acrylamide• Fluorinated methacrylate
Quite predominantly, the grafting is conducted in a heterogeneous systemwith the solid polymer and with the monomer being present in the liquid state,often in the presence of water or organic liquid. But also grafting under homo-geneous conditions has been reported, e.g. in the DMA/LiCl system.
For starting a graft side chain, a radical site at the cellulose backbone is defi-nitely required. These radical sites can originate from the homolytic bond cleav-age within the AGU, for example after high-energy irradiation, from the decom-position of a suitable functional group at the macromolecule, e.g. a peroxidegroup, or from a radical transfer reaction initiated by a radical formed outsidethe macromolecule, for example by a redox reaction. Important radical-genera-ting systems used in cellulose grafting (Young, 1977; Krässig, 1971) are Ce(III)/Ce(IV), Mn(II)Mn(III), and Fe(II)/H2O2/xanthogenate group.
They have the advantage of being applicable in aqueous media. The rather com-plex action of Ce4+ on cellulose can be formulated in a highly simplified manner as
CeII-H +Ce4+ — Cell· + Ce3++ H+
(Stannett and Hopfenberg, 1971). Grafting of vinylacetate onto a sulfite-dissolving pulp by means of the redox system Fe(II)/H2O2 has been recentlyreported (Zara et al., 1995).
Mn3+ leads to the oxidation of the aldehyde groups and the 1,2-glycol moie-ties at the chain ends and of the 2,3-diole units of the AGU within the macro-molecule (Ränby, 1981). A simplified reaction scheme for the xanthogenateredox system is presented in Fig 4.1.10.
According to Krässig (1971) this method leads to grafts with numerous andrather short side chains, and the reaction can be easily controlled via the amountof xanthogenate groups previously introduced and the monomer concentration.The 'xanthogenate method' is also well suited to grafting onto lignocellulosicmaterials like mechanical pulp (Hornof et al., 1977).

O
20 4. l Formation and Modification of the Polymer Skeleton of Cellulose
For attaching various types of cationic side chains onto cellulose, a furtherroute to free radical grafting was investigated (Bojanic, 1996). Cellulosic hy-droxy groups are at first transformed to an acrylic ester by reaction with acryloylchloride, and subsequently a conventional free radical polymerization is startedat the C=C bonds introduced in the first step.
I Il ^Fe I M .FeLCH2-O-C-S + HO· — * hCH-O-C-S + H2O
S , S
Il Il^CH2-O-C-SH +HO· — hCH-O-C-S· + H
- Subsequent grafting with vinyl monomers (CH2 = CHX) e.g.styrene, acrylonitrile-
OH
KC-CH2-CHxJcH2-CHXi-CH2-CH2X and1 H L Jn
I " r ihCH2-O-C-S-CH2-CHX4CH2-CHX + CH2-I " - J n
Figure 4.1.10. Reaction scheme of cellulose grafting by the xanthogenate method(Krässig, 1971).
Grafting of vinyl monomers as e.g. styrene onto cellulose derivatives withstructopendant unsaturated ester moieties, especially onto cellulose cinnamate,has been reported (Zhang and McCormick, 1997), employing AIBN (azobisiso-butyronitrile) as an initiator in this homogeneous free radical graft polymeriza-tion in DMA/LiCl.
After mechanochemical treatment of cellulose, three types of radicals suitablefor a subsequent graft copolymerization could be detected by a combination ofscanning calorimetry and ESR spectrometry. These are alkoxy radicals formedat C-4 by glucosidic bond cleavage, carbon radicals at C-I and carbon radicals atC-2 and C-3 due to carbon bond scission between these two C atoms. The alk-oxy radicals proved to be rather stable at ambient temperature and inert againstoxygen, while the C radicals form peroxyradicals in the presence of oxygen.

4.1.3 Grafting onto cellulose chains 21
Radiation grafting of cellulose is generally performed with high-energy elec-tron-beam or γ-irradiation, although an initiation by corona discharge or by UVradiation is mentioned too in the literature. In spite of its high susceptibility tochain cleavage by high-energy radiation, cellulose is one of the most frequentlyradiation-grafted polymers. The grafting is performed either by a pre-irradiationtechnique, i.e. a two-step process consisting of irradiation of the substrate as thefirst step and the interaction of the pre-irradiated material with the monomer asthe second. Also, the so-called simultane technique, by applying irradiation tothe monomer-soaked cellulose material, was used.
Fig. 4.1.11 gives an example of the increase of mass of the sample due tografting by the two-step technique in dependency on radiation dose in the pre-irradiation step at otherwise constant reaction conditions. A steep increase ofadd-on occurs already at a rather low dose, followed by a levelling-off. Thisindicates the advantage of a rather low irradiation dose for an efficient grafting,while a further increase of the dose mainly promotes chain scission without im-proving the graft yield. In order to secure a high efficiency of grafting, the tran-sition time between pre-irradiation and grafting must be kept short, as the add-onis proportional to the actual radical concentration and decreases steeply withincreasing transition time (Fig. 4.1.12).
The course of radiation grafting is strongly influenced by the moisture contentof the cellulose sample, as well as by its supramolecular structure (see the fol-lowing section).
16
£13
ο 10
2 6 10 UDose [RGy]
Figure 4.1.11. Increase in mass of sample in dependence on radiation dose in two stepradiation grafting (other reaction conditions kept costant) (Rätzsch et al, 1990).
In conclusion, the structure of the grafted polymer and the material propertiesdependent thereon are influenced by a large number of parameters, combiningthe degrees of freedom of the cellulose reaction with those of the free radicalpolymerization. So, for example, the number of side chains and their distribution

22 4. l Formation and Modification of the Polymer Skeleton of Cellulose
depends on the initiation technique and the monomer employed, as well as oncellulose supramolecular and morphological structure. The length of the sidechains is mainly determined by the reaction system employed, but can addition-ally be controlled by the presence of a 'chain regulator' like CCl4. Side chainsrepresenting alternating copolymers can be grafted onto cellulose by a suitablechoice of two monomers forming electron donator-acceptor complexes (Gailord,1976). Monomers with two carbon-carbon double bonds can of course also beapplied to cellulose grafting, but the probability of an irregular course of reac-tion and of crosslink formation is considerably increased here. Besides the pa-rameters given by the reaction components, also the external reaction conditions,such as concentration ratios, reaction temperature and reaction time, are of highrelevance in determining the structure of a cellulose graft copolymer.
22
E 16
ω K(ΛO
ε 12υ
_c
10
810 20
Transition time [min]30
Figure 4.1.12. Decrease of add-on (increase of mass) with transition time in two-stepradiation grafting of cellulose (other reaction conditions kept constant) (Rätzsch et al.,1990).
4.1.3.3 Effects of supramolecular and morphological structure oncellulose grafting
The supramolecular and morphological structure of the cellulose sample stronglyinfluences the course of a grafting reaction, as well as the structure and proper-ties of the graft material, via the spatial distribution, the mobility and the stabil-ity of the radicals formed, as well as via the transport rate of the monomer intothe fiber wall. By an appropriate choice of the grafting system and the reactionconditions, either a rather uniform grafting throughout the cellulose fiber or apreferential surface grafting can be achieved. These general statements hold truefor chemical as well as radiation-initiated grafting. A Mn3+-initiated grafting ofvarious acrylic acid esters onto soft-wood pulp starts at the fiber surface andthen proceeds gradually into the interior of the fiber (Ränby, 1981). With meth-

4.1.3 Grafting onto cellulose chains 23
ylacrylate, the diffusion of the initiator proved to be the limiting factor, whilewith the more voluminous butyl acrylate an impeded monomer diffusion limitedthe grafting to the fiber surface. The high surface selectivity in the Ce(IV) graftcopolymerization of acryl amide and a cationic monomer onto wood pulp fiberswas emphasized (Gruber and Granzow, 1996).
In radiation grafting the course of reaction significantly depends on the mois-ture content of the substrate. Radiation grafting of a completely dry pre-irradiated cellulose did not start until the temperature of thermal polymerizationof the monomer was reached, while the starting temperature was significantlydecreased by stepwise enhancement of the water content up to a level between 5and 20 % (Plotnikov and Lesins, 1981). The mobility of the radicals formedincreases with the moisture content in the less well ordered regions of a pulp orcotton fiber, resulting in an increase in polymer add-on with the moisture con-tent in a grafting experiment employing the 'simultaneous method', and the de-cay rate of the radicals also increases with the content of H2O. A much higherstability of radicals trapped in the crystalline regions of the fiber as comparedwith those located in the amorphous regions was emphasized (Rätzsch et al.,1990).
Stannett and Hopfenberg (1971) demonstrated the influence of swelling of acellulose substrate, in connection with the gel effect of radical polymerization,by the dependency of molar mass of the graft and of polymer add-on by graftingof cellulose 2,5-acetate in styrene/pyridine mixtures of increasing swellingpower (see Fig. 4.1.13).
20 40 60Pyridine in styrene [%]
80 100
Figure 4.1.13. Effect of swelling on the yields and molecular weights of the grafted sidechains for the mutual radiation grafting of styrene to cellulose acetate films · 0.0025mm; O 0.025 mm thickness. Dose of 10 Mrad at 0.35 Mrad/h at 25 0C (Stannett andHopfenberg, 1971).

24 4. l Formation and Modification of the Polymer Skeleton of Cellulose
A maximum in both parameters is found at a medium degree of swelling,permitting a sufficiently fast excess of the monomer entering the substrate butsecuring a sufficiently large gel effect to impede side chain termination.
The mutual interaction between fiber morphology and course of grafting in-volves, however, not only the effect of fiber morphology on the grafting reactionbut also the change of this morphology due to grafting. The morphologicalchanges of a cotton fiber on radiation grafting with various vinyl monomerssignificantly depend on the molar volume of the monomer applied (Arthur,1976). For example, side chains of poly (methyl methacrylate) were uniformlydistributed in a collapsed fiber structure, while in the case of poly(butyl methac-rylate) and higher poly(alkyl acrylates) a fiber opening and layering effect wasobserved. By appropriate timing of irradiation and swelling, either a uniformgrafting throughout the fiber or a skin/core grafting can be achieved. A cationicgraft copolymer can exhibit quite a different morphology depending on graftingtechnique (pre-irradiation or simultaneous method) (Rätzsch et al., 1990). Thepreradiation technique was recommended for surface grafting, especially of beechpulp as the substrate, while the simultane technique resulted in a more uniformgrafting across the fiber.
4.1.3.4 Properties and applications of graft copolymers of cellulose
Graft copolymerization of cellulose with appropriate monomers frequently re-sults in decisive changes of the chemical and physical properties as well as innumerous more or less qualitatively evaluated end-use properties of the polymer.The expectations promoting research in this area, i.e. an advantageous combina-tion of properties of natural and synthetic polymers, could be widely realized ata laboratory or a small-sized technical scale. But in contrast to the large numberof publications dealing with the effects of grafting on macromolecular structure(see for example Table 4.1.2; Krässig, 1971), investigations correlating, in aquantitative manner, end-use properties to grafting systems and grafting condi-tions and the structural changes resulting therefrom, are comparatively scarce.An example is given in Table 4.1.3 (Rogowin, 1972), regarding the glass transi-tion temperature of styrene-grafted cotton.
Most of the information available today on property changes by grafting con-cerns fibers, filaments and fabrics, and more recently also to some extent cellu-lose-based membranes. Properties of cellulose fibers affected by grafting are:
4.1.3 Grafting onto cellulose chains 25
Fiber finenessTensile strengthElongation at breakElastic modulusWater vapor uptakeWater inbibitionThermoplasticityDimensional stabilityAbrasion resistance
DegradabilityPermanent press behaviorWrinkle resistanceWater repellencyOil repellencySoil releaseMicrobial resistanceFlame retardancy
Generally, the property changes observed can be traced back to a varying extent tochanges in the chemical structure of the macromolecules by the covalently attachedsynthetic side chains on the one hand, and to an altered supramolecular and morpho-logical structure on the other. In the case of water inbibition, a parameter relevant tocellulose textiles as well as to membranes, a prevailing effect of supramolecular andmorphological structure has been assumed, with the constitution of the side chainsplaying a minor role only. Cationic side chains, however, were reported to bind lesswater than anionic ones under comparable conditions (Mukherjee et al., 1983).
Table 4.1.2. Examples of the relation between grafting conditions and structure ofcellulose graft polymers(Krassig, 1971)
Backbonepolymer
Cotton(DP-900)
Cotton(DP- 1200)
Methodof initia-tion
Post irra-diationgrafting
Simulta-neousirradia-tiongraftingRedoxreaction
Grafting con-ditions
(Mrad)
0.32styrene3.24styrene0.32 1styrene
3.24styrene0.02 MCe(IV);acrylonitrile
Add-on
(%)22.6
83.7
19.6
47.5
27.5
Homo-poly-mer(%)24
18
39
35
20
M.W. ofsidechains(x 106)
3.02
2.26
1.07
0.31
0.06
Sidechainsper AGU
0.02
0.08
0.04
0.35
1.13
As can be expected from the broad spectrum of cellulose properties that canbe changed by grafting, a host of applications for cellulose graft copolymers has

26 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
been proposed, especially during the 1970s. These include, besides the modifi-cation of textile yarns and fabrics of cellulose, grafting onto cellulose derivativeslike cellulose acetate or onto lignocelluloses like straw, with the polymer add-onbeing much less at the lignin than at the cellulose component (Fanta et al.,1987). Further examples are the use of special monomers like perfluorinatedcompounds or various cationic acrylics, and last but not least the application ofgrafted products outside the textile field, for example in ion-exchange and fil-tering processes (Duntsch et al., 1989), in membrane separation processes foroil/water mixtures, or in soil conditioning and seed planting (Stannett, 1985).
Table 4.1.3. Effect of grafting on the glass transition of cellulose (Rogowin, 1972)
Copolymer
Cellulose-polystyrene
Compositionproducts
Cellulose(%)606074.872.9
of reaction
Grafted polymer(%)404025.227.1
M. W. of thegrafted chain
158.00074.1507.8004.150
Glass transitiontemperature
12610210496
But despite all these achievements of research and development, only a few ofthe grafting procedures and graft product applications proposed arrived at thestage of pilot-scale production or even industrial manufacture. Obviously short-comings in process economy, problems in subsequent processing steps and alack of market acceptance may be the main reasons for this disappointing situa-tion, which led to a significant decline of research activities in this area duringthe last 15 years. Nevertheless, some of these developments will be surveyedbriefly at the end of this subchapter.
Much effort has been spent on preparing cellulose-based super-absorbing materi-als by grafting anionic side chains onto the cellulose backbone, but at least up to nowthese products could not compete efficiently with the crosslinked acryl-based syn-thetic materials dominating the market (Stannett, 1985). An interesting combinationof properties of cellulose and acrylonitrile fibers has been achieved by Rogowin(1974), who investigated the grafting of various monomers onto viscose before,during and after the spinning step and developed a technical process of grafting ac-rylonitrile onto freshly spun viscose fibers in aggregates, producing nearly l t ofgraft material per batch. This process, however, depended on temporary regionaleconomic conditions and therefore was later abandoned.
Another process developed to the pilot scale was an antimicrobial finish ofcellulosic fabrics by grafting with acrylic or methacrylic acid to a grafting de-gree of 2-3 % and subsequent binding of copper ions to the carboxyl groups at

4.1.4 Synthesis of cellulose block copolymers 27
the side chains (Heger, 1990). The product obtained, and primarily intended forhospital laundry, exhibited a satisfactory antimicrobial behavior of good perma-nency, but its poor handling and color impeded acceptance in the market.
Last but not least, the combination of an acid-catalyzed crosslinking of cellu-losics by methylol acrylamides and a subsequent free radical grafting shall bementioned, which was the first industrial application of radiation grafting forconveying permanent press properties, high wrinkle recovery and shrink resis-tance to cellulosic textiles.
4.1.4 Synthesis of cellulose block copolymersIn principle, cellulose block copolymer synthesis starts from a cellulosic prepo-lymer of usually low DP provided with reactive end groups and with protectedhydroxy groups at the C-2, C-3 and C-6 position of the AGU to avoid side chaingrafting. These reactive end groups can then be used either to initiate the forma-tion of a block of a synthetic polymer or to form a covalent linkage to a syn-thetic macromer. Two- and three-block copolymers, as well as star-shaped blockcopolymers synthesized along these routes have been described.
Attempts reviewed by Rogowin and Galbraich (1983) to provide reactiveradical end groups by homolytic chain scission via the input of mechanical en-ergy (ball milling, vibration milling) succeeded in the combination of cellulosicsegments with those of e.g. polyamides, but the copolymers obtained were ofrather ill-defined structure. Examples of synthesis of a polymer sequence ontocellulose end groups by free radical or cationic polymerization, resulting in well-defined structures, have been described (Feger and Cantow, 1980 and 1982). Apolymeric photoinitiator suitable for starting a subsequent free radical polymeri-zation of vinylic monomers has been obtained by coupling a strictly monofunc-tional hydroxy-end-group-terminated sequence of a cellulose triester (acetate,propionate, butyrate) with bis-4-isocyanatophenyl disulfide. Cellulose-derivative-terminated three-block copolymers of defined structure were preparedby a macroinitiator-started free radical polymerization, the latter being consid-ered more suitable for block formation onto cellulosics than a living anionicpolymerization. A route to linear or star-shaped block copolymers containingsequences of trimethylcellulose and of polyoxytetramethylene was realized via acationic polymerization of THF (Mezger and Cantow, 1983). Trimethylcellulosewas partially cleaved by acid hydrolytic scission of the glycosidic bonds to ob-tain chain fragments with a reactive end group, from which a cationic polymeri-zation of THF was started with AgSbF6 as a catalyst and finally terminated byaddition of KCN in methanolic KOH. Solution properties of these copolymerswere governed by an incompatibility of the two kinds of blocks.

28 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
Coupling a synthetic prepolymer with suitable end groups to those of a cellu-losic sequence protected at the C-2, C-3 and C-6 position using the highly reac-tive isocyanate group has been successfully employed. As an example, the com-bination of low DP cellulose triacetate with polypropylene glycol via an endgroup reaction with toluene diisocyanate in the presence of stannous octanoateas a catalyst (Amick et al., 1980) shall be cited. Just as in other routes of blockcopolymer synthesis, starting from cellulose triacetate sequences, the protectinggroups can be subsequently removed by an appropriate saponification procedure.As rather special applications of this principle in cellulosic copolymer synthesis,the reaction of cellulose triacetate oligomers with diisocyanates to cellulosetriacetate chains with urethane links at regular distances and the formation ofsome kind of alternating copolymer with urea and urethane linkages obtained byreacting glucosamine with a suitable diisocyanate may be mentioned.
Examples of recombining the two wood components, cellulose and lignin, bysimultaneous block and graft copolymerization were recently given (de Oliveiraand Glasser, 1994; Demaret and Glasser, 1989). Segments of cellulose triacetateand cellulose tripropionate in the DP range between 5 and 60 after end groupfunctionalization by isocyanate groups were reacted with hydroxypropyl lignin.A strong dependency of the shape of the macromolecules in solution, as well asof the morphology of the copolymers, on the length of the cellulosic segmentswas observed.
As grafting, block copolymerization of cellulosics represents a route to athorough modification of the material properties of the polymer, and severalareas of application have been proposed, for example enhancement of the biode-gradability of synthetic polymers (Kim et al., 1976), but so far none of these hasbeen realized on an industrial scale.
ReferencesAmick, R., Gilbert, R.D., Stannett, V.T., Polymer 1980, 27, 648-650.Amikan, D., Benziman, M., /. Bacteriol 1989, 777, 6649-6655.Arthur, J.C., /. Macromol Sci.-Chem. 1976, AlO, 653-670.Benerito, R.R., Webre, B.C., McKelvey, J.B., Textile Res. J. 1961, 37, 757.Berlin, A.A., Kislenko, V.N., Prog. Polym. ScL 1992, 77, 765-825.Blanton, R.L., Northcote, D.H., Planta 1990, 7SO, 324-332.Bojanic, V., /. AppL Polym. ScL 1996, 60, 1719-1725.Brown Jr., R.M.,/. Macromol. ScL, Pure Appl. Chem. 1996, A33, 1345-1373.Colvin, J.R., in Encyclopedia of Polymer Science and Engineering, Vol. 3,Mark, H.F., Bikales, N.M., Overberger, G.G., Menges, G., Kroschwitz, JJ.(Eds.), New York: John Wiley & Sons, 1985, pp. 60-68.

References 29
Cowan, S.L., Hurwitz, M.D., Ind. Eng. Chem. Prod. Res. Dev. 1982, 27, 629-632.
Dautzenberg, H., Fanter, C., Fink, H.-P., Philipp, B., Cellul Chem. Technol.1980,14, 633-653.
de Oliveira, W., Glasser, W.G., Polymer 1994, 35, 1977-1985.Delmer, D.P., Amor, Y., Plant Cell 1995, 7, 987-1000.Demaret, V., Glasser, W.G., Polymer 1989, 30, 570-575.Duntsch, L., Petzold, G., Rätzsch, M., Heger, A., Jacobasch, H.-J., Petr, A.,
Patent DD 269 561, 1989; Chem. Abstr. 1990, 772, 38829.Eschalier, X., British Patent 1906, 25, 647.Eschalier, X., /. Soc. Chem. Ind. 1907, 26, 821.Esposito, F., DeNobile, M.A., Mensitieri, G., Nicolais, L., /. Appl. Polym. Sei.
1996, 60, 2403-2407.Fanta, G.F., Burr, R.C., Doane, W.M., 7. Appl. Polym. Sei. 1987, 33, 899-906.Fanter, C., Ph.D. Thesis, Academy of Science (GDR) 1980.Feger, C., Cantow, HJ., Polym. Bull. 1980, 3, 407-413.Feger, C., Cantow, HJ., Polym. Bull. 1982, 6, 321-326 and 583-588.Franzier, Ch.E., Wendler, St.L., Glasser, W.G., Carbohydr. Polym. 1996, 3l,
11-18.Gailord, N.G., /. Macromol. Sci-Chem. 1976, A 10, 737-757.Gruber, E., Granzow, C., Papier (Darmstadt) 1996, 50, 293-299.Helbreich, A., Guthrie, J.T., in The Chemistry and Technology of Cellulosic
Copolymer, Berlin: Springer Verlag, 1981.Heger, A., in Technologie der Strahlenchemie von Polymeren, Berlin: Akademie
Verlag, 1990.Heinze, Th., Klemm, D., Loth, F., Philipp, B., Acta Polym. 1990, 41, 259-269.Hornof, V., Danesault, C., Kokta, B.V., Valade, J.L., /. Appl Polym. Sei. 1977,
27, 2991-3002.John, G., Pillai, C.K.S., Polym. Bull. 1989, 22, 89-94.Kim, S., Stannett, V.T., Gilbert, R.D., /. Macromol Sci.-Chem. 1976, AlO,671-679.
Klemm, D., Schnabelrauch, M., Geschwend, G., Wiss. Zeitschr. Friedrich-Schiller-Univ. Jena, Naturwiss. R. 1985, 34, 813-820.
Kobayashi, S., Kashiwa, K., Shimada, J., Kawasaki, T., Shoda, S., Makromol.Chem., Macromol Symp. 1992, 54/55, 509-518.
Kobayashi, S., Shoda, S., Uyama, H., Adv. Polym. Sei. 1995, 727, 1-30.Kobayashi, S., Okamoto, E., Wen, X., Shoda, S., J. Macromol Sei., Pure Appl.
Chem. 1996, A33, 1375-1384.Kokot, S., Komatsu, K., Meyer, U., Zollinger, H., Textile Res. J. 1975, 45, 673-
681.Kotchetkov, Tetrahedron 1987, 43, 2389-2436.

30 4.1 Formation and Modification of the Polymer Skeleton of Cellulose
Krässig, H., Sven. Papperstidn. 1971, 74, 417-428.Kriss, E., Bukhtiyarov, V.K., Kryukov, A.J., Tkachenko, Z.A., Shrets, D.J.,
Teor. Prikl Khim. beta-Diketonatov. Met. 1985, 101-110.Kudlicka, K., Lee, J.H., Brown Jr., R.M., Am. J. Bot. 1996, 83, 274-284.Lee, J.H., Brown Jr., R.M., Kuga, S., Shoda, S.-L, Kobayashi, S., Proc. NatlAcad. ScL U.S.A. 1994, 91, 7425-7429.
Lin, F.C., Brown Jr., R.M., in Cellulose and Wood-Chemistry and Technology,Schnerch, C. (Ed.), New York: John Wiley & Sons, 1989, pp. 473-492.
Loth, F., Philipp, B., Makromol. Chem., Macromol. Symp. 1989, 30, 273-287.Luby, P., Kuniak, L., Fanter, C., Makromol Chem. 1979,180, 2379-2386.Meyer, U., Müller, K., Zollinger, H., Text. Res. J. 1976, 46, 756-762.Mezger, T., Cantow, H.-J., Makromol. Chem. 1983,110, 13-27.Micheel, F., Broode, O.-E., Liebigs Ann. Chem. 1974, 702.Micheel, F., Broode, O.-E., Liebigs Ann. Chem. 1975, 1107.Micheel, F., Broode, O.-E., Reinking, K., Liebigs Ann. Chem. 1974, 124.Mukherjee, A.K., Sayal, S., Siddhartha, S., Cellul Chem. Technol. 1983, 178,
141-153.Nakatsubo, F., Kamitakahara, H., Hori, M., /. Am. Chem. Soc. 1996, 118,
1677-1681.Ogawa, R., Tokura, S., Carbohydr. Polym. 1992a, 19, 171-178.Ogawa, R., Tokura, S., Int. J. Biol. Macromol. 1992b, 14, 343-347.Petersen, H., Petri, N., Melliand Textilber. 1985, 66, 217-222; 285-295; 363-
369.Petersen, H., Colour. Annu. 1990, 61-65.Philipp, B., Dan, D.C., Jacopian, V., Heger, Α., Acta Polym. 1982, 33, 542-545.Plotnikov, O.V., Lesins, A., Khim. Drev. 1981,1, 111-112.Ränby, B., Int. Symp. Wood Pulping Chem., Ekman.Days, 1981, Stockholm:
SPCI, 1981, Vol. 4, 111-117.Rätzsch, M., Dunsch, L., Petzold, G., Petr, A., Heger, Α., Acta Polym. 1990, 41,
620-627.Rogowin, Z.A., /. Polym. ScL, Part C 1972, 37, 221-237.Rogowin, Z.A., Tappi 1974, 57, 65-68.Rogowin, Z. A., Galbraich, L. S., in Die chemische Behandlung und
Modifizierung der Zellulose, Stuttgart: Thieme, 1983.Sakamoto, M., Takeda, J., Yamada, Y., Tonami, H., J. Polym. Sei. Part A-I
1970, 8, 2139-2149.Sangsari, F.H., Chastrette, F., Chastrette, M., Blanc, A., Mattioda, G., Reel.
Trav. Chim. Pays-Bas 1990, 709, 419-424.Schlubach, H.M., Luhrs, L., Liebigs Ann. 1941, 547, 73.

References 31
Stannett, V.T., Hopfenberg, H.B., in Cellulose and Cellulose Derivatives,Bikales, N.N.M., Segal, L. (Eds.), New York: John Wiley & Sons, 1971, PartV, pp. 907-936.
Stannett, V.T., in Cellulose and Its Derivatives, Kennedy, J.F. (Ed.), Chichester,UK: Ellis Horwood, 1985, pp. 387-399.
Tarchevsky, J.A., Marchenko, G.N., Cellulose, Biosynthesis and Structure,Berlin: Springer Verlag, 1991.
Uryu, T., Kitano, K., Ito, K., Yamanouchi, J., Matsuzaki, K., Macromolecules1981,74, 1.
Uryu, T., Yamanouchi, J., Kato, T., Higuchi, S., Matsuzaki, K., /. Am. Chem.Soc. 1983, 6865.
Uryu, T., Yamaguchi, C., Morikawa, K., Terui, K., Kanai, T., Matsuzaki, K.,Macromolecules 1985, 18, 599.
Ushakov, S.N., Fiz.-Mat. Nauk (USSR) 1943, 7, 35.Yang, C.Q., Wang, X.L., /. Polym. ScL, Part A - Polym. Chem. 1996, 34,
1573-1580.Young, R.A., J.Agric. Food Chem. 1977, 25, 138.Young, R.A., in Absorbency, Chaterjce, P.K. (Ed.), Amsterdam: Eisevier Sei.Publ, 1985, pp. 217.
Zara, L., Erdelyi, J., Hell, Z., Borbely, E., Rusznäk, L, Tappi J. 1995, 78, 131-134.
Zeronian, S.H., Bertoniere, N.R., Alger, K.W., Xie, Q., /. Text. lust. 1990, 87,310-318.
Zhang, Z.B., McCormick, C.L., J. Appl. Polym. ScL 1997, 66, 307-317.
4.2 Interaction of Cellulose with Basic Compounds
This chapter will be centered on various classes of 'addition compounds' ofcellulose, i.e. compounds formed without covalent derivatization of the macro-molecule but nevertheless representing chemical entities by themselves, withchemical and physical properties differing often decisively from that of unmodi-fied cellulose. Quite predominantly, processes of interaction of solid celluloseare the topic of this text. Thus the interdependency between the chemical inter-action and the supramolecular and morphological structure of the cellulose sam-ple plays a decisive role. After considering briefly the so-called alkali cellulo-sates this subchapter will be structured according to the reagent employed inpreparing the various addition compounds with cellulose, i.e. aqueous and alco-holic solutions of alkali and tetraalkylammonium hydroxides, guanidinium hy-droxide, hydrazine, ammonia and aliphatic amines.

References 31
Stannett, V.T., Hopfenberg, H.B., in Cellulose and Cellulose Derivatives,Bikales, N.N.M., Segal, L. (Eds.), New York: John Wiley & Sons, 1971, PartV, pp. 907-936.
Stannett, V.T., in Cellulose and Its Derivatives, Kennedy, J.F. (Ed.), Chichester,UK: Ellis Horwood, 1985, pp. 387-399.
Tarchevsky, J.A., Marchenko, G.N., Cellulose, Biosynthesis and Structure,Berlin: Springer Verlag, 1991.
Uryu, T., Kitano, K., Ito, K., Yamanouchi, J., Matsuzaki, K., Macromolecules1981,74, 1.
Uryu, T., Yamanouchi, J., Kato, T., Higuchi, S., Matsuzaki, K., /. Am. Chem.Soc. 1983, 6865.
Uryu, T., Yamaguchi, C., Morikawa, K., Terui, K., Kanai, T., Matsuzaki, K.,Macromolecules 1985, 18, 599.
Ushakov, S.N., Fiz.-Mat. Nauk (USSR) 1943, 7, 35.Yang, C.Q., Wang, X.L., /. Polym. ScL, Part A - Polym. Chem. 1996, 34,
1573-1580.Young, R.A., J.Agric. Food Chem. 1977, 25, 138.Young, R.A., in Absorbency, Chaterjce, P.K. (Ed.), Amsterdam: Eisevier Sei.Publ, 1985, pp. 217.
Zara, L., Erdelyi, J., Hell, Z., Borbely, E., Rusznäk, L, Tappi J. 1995, 78, 131-134.
Zeronian, S.H., Bertoniere, N.R., Alger, K.W., Xie, Q., /. Text. Inst. 1990, 81,310-318.
Zhang, Z.B., McCormick, C.L., J. Appl. Polym. ScL 1997, 66, 307-317.
4.2 Interaction of Cellulose with Basic Compounds
This chapter will be centered on various classes of 'addition compounds' ofcellulose, i.e. compounds formed without covalent derivatization of the macro-molecule but nevertheless representing chemical entities by themselves, withchemical and physical properties differing often decisively from that of unmodi-fied cellulose. Quite predominantly, processes of interaction of solid celluloseare the topic of this text. Thus the interdependency between the chemical inter-action and the supramolecular and morphological structure of the cellulose sam-ple plays a decisive role. After considering briefly the so-called alkali cellulo-sates this subchapter will be structured according to the reagent employed inpreparing the various addition compounds with cellulose, i.e. aqueous and alco-holic solutions of alkali and tetraalkylammonium hydroxides, guanidinium hy-droxide, hydrazine, ammonia and aliphatic amines.
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

32 4.2 Interaction of Cellulose with Basic Compounds
4.2.1 Preparation and properties of alkali cellulosatesAlkali cellulosates as analogues of alkali alcoholates (alkali alkoxides) can beprepared by reacting the polymer with the alkali metals Li, Na or K in liquidammonia, as first shown by Scherer (Scherer and Hussey, 1931) for K-cellulosate. According to Schmid and Becker (1925), Schmid et al. (1928) andMuskat (1934) the reaction proceeds at -35 to -50 0C within some hours, withthe evolution of hydrogen probably via the formation of alkali amide as the re-active intermediate, and can be considerably accelerated by addition of sodiumchloride. With sodium metal, a trisubstituted cellulosate was obtained, whilewith potassium or lithium only a DS below 3 was reached and calcium proved tobe unsatisfactory as a reagent. Bredereck (Bredereck and Vlachopoulos, 198Oa)prepared a lithium cellulosate of DS = 3 by reacting an ammonia cellulose -obtained from cotton - with lithium in liquid NH3. A fast cellulosate formationin the disordered regions of the ammonia cellulose was observed with all threealkali metals, potassium, sodium and lithium, but a subsequent rather fast first-order reaction within the lattice layers of the addition compound was observedwith lithium only. Sodium reacted much more slowly and potassium did notpenetrate the lattice at all.
The reactivity of alkali alkoxides obviously is insufficient to convert celluloseinto cellulosates, while with thallium alkoxide in diethyl ether or benzene a par-tial introduction of cellulosate groups (DS < 3) could be achieved (Harris andPurves, 1940). On the other hand, the route to cellulosates via the correspondingalkoxides proved to be successful with tetraalkylammonium compounds: byreaction of a suspension of native cellulose I with the methoxides of the tet-ramethylammonium and the benzyltrimethylammonium cation in anhydrousMeOH or DMSO, the corresponding cellulosates with a DS of up to 0.7 havebeen prepared. The DS increased with the concentration of the methoxide anddecreased with the molar volume of the tetraalkylammonium cation under givenreaction conditions (Bredereck and Thi Bach Phnong Dau, 198Ob).
As to be expected, all the cellulosates so far prepared exhibit a very high re-activity and can be converted to cellulose esters by reaction with acid anhydridesor acid chlorides or to cellulose ethers with alkyl halides. Xanthation with CS2
(see chapter 4.4) proceeds rapidly in the presence of a small amount of water(Scherer and Gotsch, 1939). According to Bredereck and Thi Bach Phnong Dau(198Ob) the reactivity of various cellulosates with a DS of 0.4 in a subsequentmethylation increases in the order of the cations Li+ < Na+ < Me4N
+ <Me3BnN+.
All cellulosates are highly basic and rapidly decomposed by water or by CO2
from the air, and they can be kept for some time only with strict exclusion ofmoisture.

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 33
4.2.2 Interaction of cellulose with aqueous and alcoholicsolutions of alkali hydroxides
4.2.2.1 General comments
Since John Mercer observed about 150 years ago the swelling of cotton fibers inaqueous sodium hydroxide and the changes in physical fiber properties afterremoval of the lye, the interaction between cellulose and aqueous alkali hy-droxides, especially NaOH, has been one of the principal research topics in thechemistry and physics of cellulose, leading to decisive progress in understandingcellulose structure and reactivity and resulting in large-scale technical processes.
The interaction between cellulose fibers and aqueous alkali hydroxides ischaracterized by an uptake of alkali hydroxide and water onto the fiber resultingin a decrease of lye concentration in the surrounding medium, by a strong lateralswelling of the fiber and by a change in X-ray lattice dimensions in the orderedregions above a specific lye concentration. The binding state of the alkali hy-droxide onto the cellulose can still not be exactly defined: obviously someanionization of the hydroxy groups occurs without a true 'cellulosate' (alcoho-late) being formed. In the interaction of cellulose with aqueous alkali hydroxidesolutions the hydration shell of the alkali hydroxide ion dipoles and its changewith lye concentration plays a dominant part, regarding alkali hydroxide andwater uptake by the fiber. This course of alkali uptake with lye concentrationdepends strongly on the supramolecular structure of the sample, resembling atypical heterogeneous course of reaction with highly ordered cellulose fibers. Onremoval of the alkali hydroxide by washing or by neutralization, cellulose in thelattice modification of cellulose II is regenerated from all the alkali cellulosesformed, with a degree of order usually lower than that of the starting material.
Among the reaction products of cellulose with various aqueous alkali hy-droxides, only the so-called sodium cellulose is of practical relevance as an in-termediate of limited stability: it decomposes rather rapidly by sorption of CO2
from the air, and it is depolymerized by the oxidative action of air oxygen (seechapter 2.3). Two routes of application of this reactive intermediate are realizedtoday in large-scale processes, i.e.(i) the transformation of native cellulose I to mercerized cellulose (cellulose II)with changed textile properties via sodium cellulose.(ii) the transformation of native cellulose I into sodium cellulose as the startingmaterial for subsequent large-scale esterification or etherification of cellulose,especially xanthation and carboxymethylation.

34 4.2 Interaction of Cellulose "with Basic Compounds
4.2.2.2 Swelling and dissolution of cellulose in alkali hydroxidesolutions
The most striking phenomenon in cellulose-alkali hydroxide interaction is thestrong and fast lateral swelling of cellulose fibers in aqueous alkali hydroxidesolutions. If performed without tension this lateral swelling is connected with adecrease in fiber length, and in any case the tensile strength of the fiber signifi-cantly decreases. The swelling takes place on a time scale of seconds to a fewminutes and obviously is diffusion-controlled.
As already discussed in the chapter 2.2, the swelling power of the lye passes amaximum in dependency on lye concentration, which is shifted to higher alkalihydroxide concentration with increasing atomic weight of the alkali cation, butcorresponds in all cases to about the same molar alkali hydroxide concentration(Heuser and Bartunek, 1925). From LiOH to CsOH the steepness and absoluteheight of the maximum decrease in correspondence to a decreasing hydrationshell of the alkali cation.
Comprehensive work on swelling of cellulose in aqueous sodium hydroxide hasshown that the increase in fiber diameter not only depends on lye concentrationbut also on the physical structure of the sample. A lowering of the steeping tem-perature generally results in a higher degree of swelling and favors the dissolutionof low DP cellulose from the accessible parts of the sample (see chapter 2.2).
The increase in solubility by lowering the temperature due to an exothermicheat of cellulose dissolution in aqueous NaOH has been investigated thoroughlyin recent years (e.g. Yamashiki et al., 1990; Lang and Laskowski, 1991). Opti-mal results were obtained within 9-10 % NaOH at a temperature of about -10 0C. Rather clear solutions with a cellulose content up to 5 % could beobtained from degraded cellulose samples with a DP up to 200, while at higherDP a partial solubility only was observed (compare Fig. 4.2.1). The mode ofdegradation is obviously of minor influence here (Fig. 4.2.1).
100
r__,80
^6OI 40
ω 20
200 400 600 800DP
Figure 4.2.1. Solubility of degraded spruce sulfite pulp samples in 10% aqueous NaOHat -10 0C in dependence on DP. Mode of degradation: · thermal treatment; O acid hy-drolysis; · electron beam irradiation; Δ irradiation and thermal treatment (Lang andLaskowski, 1991).

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 35
A problem impeding technical application for film spinning arises from theinstability of these solutions, which form coherent gels on standing. Accordingto Lang et al. (1989) these gels can be redissolved by a suitable transient eleva-tion of temperature. A cyclic cooling and heating procedure was found to bemost suitable to obtain fiber-free solutions. Completeness of dissolution as wellas the stability of these solutions can be enhanced by addition of zinc oxideand/or urea. The phenomena observed are interpreted by Lang and Laskowski(1991) as being due to an interaction of NaOH with the cellulose via an incorpo-ration of cellulosic hydroxy groups into the solvation shell of the NaOH sol-vates, which despite their high hydration number are stabilized at the low tem-perature and thus provide a spacer action to separate the cellulose chains (seealso chapter 4.3). A 1H- and 13C NMR study of degraded cellulose (DP-15)dissolved in NaOD/U2O was centred on hydroxy group dissociation in depend-ence on NaOD concentration (4-30%). The hydroxy group at C-3 proved to bethe most resistant one to dissociation. According to this study, cellulose macro-molecules dissolved in NaOH behave different from those in a highly swollenstate (Isogai, 1997).
4.2.2.3 Chemical processes of interaction between cellulose andalkali hydroxide solutions
As observed already at the beginning of this century (Heuser and Bartunek,1925), all the alkali hydroxides from LiOH to CsOH are strongly chemisorbedfrom their aqueous solution onto cellulose, with a stepwise sorption isothermbeing found with highly ordered cotton cellulose. The plateaus of these iso-therms indicate a constant molar ratio of alkali sorbed per AGU over a ratherwide range of lye concentration. Subsequent studies of alkali sorption were pre-dominantly concerned with aqueous NaOH and in some cases also with KOH(Mori, 1991). Employing a more sophisticated technique (Schwarzkopf, 1932)with an inert salt of negligible sorption tendency added to the lye, the step iso-therm was confirmed for NaOH and KOH. As demonstrated in Fig. 4.2.2 for theNaOH sorption from aqueous lye by spruce sulfite pulp, the alder values of theso-called apparent alkali uptake are misleading in so far as they neglect the si-multaneous uptake of water by the cellulose moiety, which is adequately consid-ered, however, by determining the so-called true alkali uptake.
The plateau of true alkali uptake appearing between 15 and 20 % NaOH byweight corresponds to a NaOH sorption of 1 mol of NaOH/mol of AGU, i.e. aone-to-one addition compound, and shows a further increase above this concen-tration. The water sorption was found to pass a pronounced maximum corre-sponding to a water uptake of 4-5 mol/mol of AGU. The uptake of alkali andwater proceeds very rapidly on about the same time scale as the lateral fiber swel-ling, and is practically «complete after 10-20 min, with the initial rate showing a

36 4.2 Interaction of Cellulose with Basic Compounds
maximum at a lye concentration of 15-20 %. Obviously this process of alkalisorption is also diffusion-controlled.
I01.0
"δ
·—Ό.5χο
5<οε
ι|10 20
NaOH [Wt %]30
Figure 4.2.2. Equilibrium values of NaOH uptake at room temperature (·, true uptakeafter pulp redrying at 20 0C; O, at 105 0C; D, apparent uptake) and water uptake (·)(Philipp, 1955).
In order to understand the course of alkali uptake with lye concentration andthe mechanism of cellulose-alkali hydroxide interaction, the structure of aque-ous alkali hydroxide solutions as well as the physical structure of the polymermust be included in the consideration.
The present structural concept for aqueous alkali hydroxide solutions is basedon the assumption of a hydrogen-bonded water structure with some monomo-lecular H2O besides the water clusters, and a disturbance of this water structureby dissolved ions tightly associated with water molecules in their Α-shell ofhydration and more loosely associated with water molecules of the B-shell. Inthe series of alkali hydroxides, the hydration shell of the cation decreases drasti-cally with increasing atomic weight, i.e. from 120 mol of H2OTLi+ to 13 mol ofH2OTCs+ (Dobbins, 1973); Li+ and Na+ are usually classified as structure-forming ions, while K+, Rb+ and Cs+ are assumed to be structure-breaking ones.For the isolated OH~ ion, a stable hydration shell with three water molecules isdescribed (Hinton and Amis, 1967; Eigen, 1963). At higher lye concentration, aninsertion of the OH~ ion into the hydration shell of the cation is assumed, re-sulting in a hydrated ion dipole. In dependence on lye concentration, a ratherlarge number of defined hydration states has been postulated for sodium hy-droxide, while a much smaller one is assumed for KOH. Experimental evidenceon several defined hydration states for NaOH has been obtained from measure-ments of the line width of the 23Na NMR signal (Kunze et al., 1985; Fig. 4.2.3).The tendency of association to an ion dipole corresponding to a decrease in de-gree of dissociation of the alkali hydroxide in dilute solution, increases in theorder KOH < NaOH < LiOH.

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 37
10 20 30NaOH concentration [wt%]
40
Figure 4.2.3. Viscosity reduced line width of the 23Na NMR signal of aqueous NaOHsolutions at different temperatures (· 268 K; O 303 K; D 323 K) (Kunze et aL, 1985).
From the site of the polymer, the following reasoning is based on a two-phaseconcept for cellulose with ordered and disordered regions (see chapter 2.1), withthis supramolecular order being stabilized by intra- and intermolecular hydrogenbonds. On interaction with water, a first layer of H2O molecules is associatedvery tightly with cellulosic hydroxy groups in the disordered regions while fur-ther sorption occurs more loosely, comparable to the A- and B-shell of hydrationin the case of ions. In connection with water, cellulose is considered to be struc-ture-breaking.
The interaction between cellulose and aqueous alkali hydroxides resulting inswelling and specific uptake of alkali and water must be considered as a verycomplex process comprising destruction of hydrogen bonds within the cellulosemoiety as well as within the aqueous lye phase, as a decrease in supramolecularorder of the polymer, changes in the structure of hydration shells as well as inthe chain conformation of cellulose, and finally as a partial anionization of cel-lulosic hydroxy groups. Despite a host of experimental evidence obtainedmainly with cotton cellulose and represented here by a few examples only, afinal separate evaluation of all these factors with regard to their relevance for thewhole process is not yet possible. Nevertheless, some considerations on themechanism of cellulose-alkali interaction depending on lye concentration andtype of alkali cations shall be subsequently presented, while for further experi-mental data the reader is referred to the comprehensive reviews of Warwicker etal. (1966), Zeronian and Cabradilla (1973), and to other publications (Philipp etal., 1983 and 1985). The following context is centered on interaction with NaOHbut deals also with a comparison of NaOH and KOH and with the effect of sub-stituting an aqueous medium by an alcoholic one.
According to present concepts, free monomolecular water penetrates first intothe cellulose structure, destroying intermolecular hydrogen bonds in the less

38 4.2 Interaction of Cellulose with Basic Compounds
ordered regions. So-called s welling-active NaOH ion dipoles (Heuser and Bar-tunek, 1925) and/or hydroxy anions are assumed to promote the interaction inthe ordered regions above an NaOH concentration of about 9 %, being partiallyor totally depleted of their hydration shell in this process and thus providing afurther amount of monomolecular water (Bartunek, 1956). Usually the hydroxyanions are seen to be responsible for the primary interaction with the cellulosichydroxy groups in the ordered regions of the structure, while the hydrated cationis seen to be responsible for the resulting swelling. Progressively, the originalstabilization of the cellulose structure by inter- and intramolecular hydrogenbonds is thus substituted by a stabilization via addition complexes between cel-lulosic hydroxy groups, NaOH ion dipoles and water molecules, with cellulosichydroxy groups being included in the hydration shell of the ion dipoles, andwater molecules being released from this shell. At a lye concentration between35 and 40 %, a stable tetra-solvate with two H2O molecules and two hydroxygroups for example, has been concluded from the experimental evidence avail-able. On the molecular level no binding of NaOH onto the cellulose chains wasdetected up to a lye concentration of about 9 %, while rather dramatic changestake place in the concentration range between 9 and 15 %, characterized by thespecific uptake of NaOH and water in the disordered as well as in the orderedregions, changes in chain conformation, with a preference for twisted conforma-tions at the glycosidic bond between C-I and C-4, and a change in lattice dimen-sions of the ordered regions (see next section). At about 15 % NaOH, the trans-formation to sodium cellulose I is completed, resulting in a still rather highlyordered structure despite some loss of X-ray crystallinity in this conversion pro-cess. A rather uniform chain conformation and an overall chemical compositionof 1 mol of NaOH and 4 to 5 mol of H2O/mol of AGU remains nearly constantup to a lye concentration of about 22 %. A site-preferential interaction of NaOHwith the hydroxy groups at C-2 and C-3 is assumed (Fink et al., 1995). Still openremains the question of anionization of cellulosic hydroxy groups: obviously astate of binding in between an addition compound with completely intact cellu-losic hydroxy groups and an anionization to an alcoholate anion has to be con-sidered. From 23Na NMR line width measurements, after stepwise depletion ofalkali cellulose samples from adhering lye by pressing, three rather well-definedstates of NaOH binding can be concluded, i.e. a delocalized binding in the disor-dered regions, a localized binding in the disordered regions and a localizedbinding in the crystalline regions, with a rapid exchange obviously taking placebetween the tightly bound and the loosely bound Na+ ions (Kunze, 1983). At anNaOH concentration of about 25 %, with the lye being already depleted of freewater, a further significant change in cellulose-alkali structure becomes visibleby NMR and WAXS measurements: a still tighter interaction between Na+ andO-atoms at C-2 and C-3 takes place, the overall chain conformation beingchanged from a two-fold to a three-fold screw axis, with the lattice spacing re-

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 39
sembling that of a sodium cellulose II, and also a conformational change beingobserved for the primary CH2OH group. These changes are mainly coursed byadditional breakage of hydrogen bonds. At still higher NaOH concentration (upto 50 %) a further decrease in supramolecular order takes place in connectionwith a rather wide spread of conformational states of the primary CH2OH group,which probably now also interacts more intensely with the alkali. In summary,the interaction between cellulose and aqueous NaOH up to a lye concentration of50 % can be considered to be based on spatial and conformational changes of thepolymer chains by destruction of the original hydrogen-bond pattern in connec-tion with concentration-dependent specific interaction between cellulosic hy-droxy groups and hydrated NaOH ion dipoles. NaOH uptake in the first part ofthe sorption isotherm (up to 15 % NaOH) in the lye is obviously governed by anincomplete accessibility of the cellulose structure to alkali cellulose formation,while the further NaOH uptake above a lye concentration of 20 % is probablyconnected with changes in hydration of the NaOH and further insertion of hy-droxy groups into the hydration shell of the ion dipoles.
The effect of temperature on cellulose interaction with aqueous NaOH as adiffusion-controlled reaction is rather small. A lowering of the temperature fromthe range 20-40 0C, employed for sodium cellulose formation in the viscoseprocess, down to about O 0C, results in a somewhat stronger binding of Na+ ontothe polymer, as revealed by the shape of the 23Na NMR signal, besides a higherswelling due to stabilization of the NaOH hydration shell.
Comparing the action of aqueous NaOH, and aqueous KOH on the other, ontocellulose, two points of difference have to be emphasized besides many simi-larities: KOH penetrates into the ordered regions of cellulose at a somewhatlower molar concentration than NaOH, and KOH uptake is higher than that ofNaOH up to a lye concentration of about 4.4 N, while above that concentrationNaOH uptake exceeds that of KOH. Probably the different behavior of KOH iscaused by its somewhat higher basic strength and its lower tendency to ion di-pole formation, resulting in a stronger partial anionization of cellulosic hydroxygroups, in agreement with the stronger ionic character of potassium alcoholate ascompared with sodium alcoholate. It seems worth mentioning that the reactivityof potassium cellulose in a subsequent cyan ethylation exceeds that of sodiumcellulose. The second point of difference between the action of KOH and ofNaOH is connected with the lower hydration of KOH and its lack of swelling-active hydrates, resulting in swelling values only half as high as those obtainedwith NaOH and also resulting in an incomplete conversion of the ordered re-gions of the cellulose moiety into potassium cellulose according to Mori (1991).
The high relevance of solvation in the interaction between cellulose and alkalihydroxides becomes clearly visible also by comparing aqueous and ethanolicNaOH, as in the latter case the interaction proceeds much more slowly and withmuch less swelling of the fibers (Philipp et al., 1987a), and the changes in eel-

40 4.2 Interaction of Cellulose with Basic Compounds
lulose physical structure differ significantly from those observed with aqueouslye (see next section). The alkalization effect obtained with NaOH dissolved in amixture of water and isopropanol resembles that observed with an aqueous lyeof much higher concentration, obviously due to formation of a cellulose/NaOH/water phase with a high alkali concentration at the expense of alkali and watercontent of the surrounding alcoholic phase. Furthermore, some competition be-tween alcohol molecules and NaOH ion dipoles for H2O molecules can be as-sumed, resulting in a decrease of the NaOH hydration shell and in consequencein a mode of cellulose/NaOH interaction observed with aqueous lye of muchhigher concentration.
4.2.2.4 Role of cellulose physical structure in cellulose-alkali hydroxideinteraction
The complex interaction between cellulose and dissolved alkali hydroxides af-fects all the structural levels of the polymer and vice versa is influenced bychanges in any of the structural levels. The subdivision employed here into'chemical interactions' and 'role of physical structure' mainly serves the purposeof clearness without being necessarily the result of scientific reasoning.
Changes in supramolecular structure on alkali treatment of cellulose havebeen predominantly investigated by WAXS, supplemented by solid state CP-MAS 13C NMR spectroscopy and by IR spectroscopy, with the effect of NaOHconcentration on degree of crystallinity, crystallite size and lattice dimensions ofthe ordered regions being the most frequent topic of research.
With !inters as the starting material the lattice transition from cellulose I to thatof Na-cellulose I, and after neutralization to cellulose II, begins at a lye concentra-tion of about 10 % and is completed at about 14 % NaOH (see Fig. 4.2.4).
10 12 14 16NaOH concentration [wt%]
Figure 4.2.4. Content of sodium cellulose and cellulose II, dependent on the aqueousNaOH concentration.

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 41
The difference observed between the percentage of sodium cellulose I formedat a given lye concentration and the amount of cellulose II obtained after neu-tralization indicates a partial reversibility of sodium cellulose formation, i.e. itspartial retransformation to cellulose I on neutralization (Philipp et al., 1985;Hayashi, 1976). This partial reversibility is obviously caused by an incompletetransformational change of a part of the macromolecules and led to the assump-tion of two different sodium cellulose I modifications, i.e. Na-cellulose I1 with abent 4Ci conformation retransformable to cellulose I, and Na-cellulose I2 with abent and twisted 4Ci conformation yielding cellulose II on neutralization. At asufficiently high lye concentration, all the cellulose chains have taken the bentand twisted conformation of Na-cellulose I2, and a 100 % yield of cellulose II isobserved on neutralization. A detailed analysis of the X-ray patterns in depend-ence on lye concentration revealed a preferential conversion of the smallerand/or less well-ordered crystallites within the transition interval and definitelyindicated only a moderate decrease in crystallinity on alkali treatment, with thehighly swollen sodium cellulose still exhibiting a remarkable crystalline order.Substitution of NaOH by KOH proved to be of minor influence only on the lat-tice transition curve based on molar lye concentrations, with the beginning of thetransition obviously starting at a somewhat lower molar concentration in thecase of KOH. According to Zeronian and Cabradilla (1973), fiber swelling aloneis not a sufficient prerequisite for lattice transformation, the start of which de-pends on the alkali cation, the reaction temperature and the medium, besides thelye concentration. At a lye concentration of 5 N, lattice conversion was found tobe completed with LiOH, NaOH and KOH, while differences between thesealkali hydroxides were observed at lower concentration. The WAXS resultsoutlined here are corroborated by recent solid state CP-MAS 13C NMR data,indicating the beginning of conformational changes at a lye concentration ofabout 9 %, with the most significant changes in signal position and shape occur-ring at up to 15 % NaOH and indicating an increasing preference for twistedconformations (Fink et al., 1995).
With NaOH of 15 % by weight, a complete lattice transformation to Na-cellulose I2 is achieved at room temperature in a fast diffusion-controlled latticelayer reaction (so-called permodoid reaction), resulting in the complete accessi-bility of the hydroxy groups in the crystalline regions to consecutive reactions.But the conversion of the cellulose I lattice to sodium cellulose I by no means isthe only one observed by WAXS, and already about 50 years ago Sobue et al.(1939) published a phase diagram of sodium cellulose modifications in depend-ence on steeping lye concentration and steeping temperature, together with theunit cell dimensions of the various phases (see Table 4.2.1 and Fig. 4.2.5).
Although with the dependence on cellulose starting material and conditions ofpreparation of the alkali cellulose somewhat deviating WAXS data may be ob-

42 4.2 Interaction of Cellulose with Basic Compounds
tained, the results of Sobue et al. (1939) can still be considered a valid basis forpractical work.
The lattice transition curve from cellulose I to cellulose II via sodium cellu-lose I (percentage of cellulose II versus lye concentration) depends significantlyon the supramolecular structure of the starting material: cotton !inters require ahigher lye concentration for this lattice conversion than wood pulp (see Fig.4.2.6), and even between different spruce sulfite dissolving pulps, significantdifferences in the course of the curve have been reported by Philipp et al. (1959).
Table 4.2.1. Unit cell dimensions of various Na-cellulose modifications (Sobue et al., 1939).
Modification
Na-cellulose INa-cellulose IINa-cellulose IIINa-cellulose IVNa-cellulose VCellulose I (for comparison)
a(A)
25.610.0022.2010.0313.958.23
b(k)
13.210.009.179.98
13.957.84
C(A)
20.5015.410.2610.315.310.28
Tf40°60°90°52°41°40'84°
c = fiber axis.
Temperature [0C]100
10 20 30
NaOH - Concentration [Weight-%]
Figure 4.2.5. Phase diagram of the sodium cellulose compound depending on the NaOHconcentration and temperature (Sobue et al., 1939, compare also Krässig, 1993).

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 43
100 -
50
9 11 13NoOH [%]
Figure 4.2.6. Lattice transition curve from cellulose I to cellulose II of cotton !inters (·)and a spruce sulfite pulp (O) in dependence on steeping lye concentration at room tem-perature (Philipp et al., 1959).
Activation of !inters cellulose with liquid NH3 prior to the alkali treatment re-sults in a definite shift of the transition curve to lower alkali concentration (seeFig. 4.2.7), and a similar shift to lower alkali concentration is observed for thefirst step in the curve of alkali uptake versus lye concentration (Loth et al.,1984).
According to Käufer (1984) the rate of steeping lye diffusion differs betweenordered and disordered regions and depends on crystallite size, with the appro-priate consequences on the kinetics of sodium cellulose formation.
r-,100 ~
3 6 9 12NaOH concentration [%]
Figure 4.2.7. Lattice transition of cellulose I to cellulose II of a spruce sulfite pulp sam-ple before (*) and after activation (Δ) with NH3 (Schleicher et al., 1973 and 1974).
Corresponding to the changes on the supramolecular level so far considered,remarkable effects are also observed in the fibrillar morphology of cellulosesamples on treatment with alkali hydroxides (see Fig. 4.2.8). Purz et al. (1995)compared in a recent morphological study the action of aqueous and ethanolic

44 4.2 Interaction of Cellulose with Basic Compounds
Spruce sulfite pulp: (a) untreated; (b) 10 % NaOH; (c) 11 % NaOH; (d) 12 % NaOH.
Cotton !inters: (a) untreated; (b) 12 % NaOH; (c) 15 % NaOH; (d) 25 % NaOH.
Bacterial cellulose: (a) untreated; (b) 10 % NaOH; (c) 12 % NaOH; (d) 15 % NaOH.
Figure 4.2.8. Changes of the microfibril structure of cellulose treated with aqueous NaOHfor l h at room temperature revealed by REM (Philipp and Purz, 1983; Purz et al., 1995).

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 45
NaOH on cotton !inters, spruce sulfite pulp and bacterial cellulose from Aceto-bacter xylinum.
The microfibrillar structure of the wood pulp and the bacterial cellulose wasfound to be destroyed by aqueous NaOH above a concentration corresponding toan almost complete lattice transition, and after regeneration to cellulose II nofine fibrillar structure could be resolved in the electron microscopic image. Thelye concentration required proved to be higher with the bacterial cellulose thanwith the sulfite pulp, in agreement with the higher lye concentration necessaryfor lattice transformation, due to the high crystallinity and larger crystallite di-mensions of the bacterial cellulose. With !inters, on the other hand, a fine fibril-lar structure prevailed throughout the whole process and could be definitelyresolved after regeneration to cellulose II, obviously due to a higher fibrillarorganization of !inters cellulose compared with wood cellulose. All these mor-phological changes occurred within 1 h, depending somewhat on the history ofthe sample, and by lowering the temperature of treatment from 20 0C to O 0C thelimiting concentration of NaOH required was shifted to somewhat lower values.
The cellulose II recovered from alkali cellulose by washing and/or neutraliza-tion differs from the original cellulose I sample not only with regard to latticedimensions but also with regard to degree of order, fibrillar morphology, andpore and void structure, as well as with regard to water vapor sorption and liquidwater retention. The degree of crystallinity xc is generally somewhat diminishedafter conversion of high molecular cellulose I samples to cellulose II, but can beenhanced with low DP cellulose due to short-chain extraction from the amor-phous regions by the alkali and due to excessive recrystallization on washingand drying, as observed with LODP !inters by Fink et al. (1992). The changes infibrillar morphology already discussed find their counterpart in an altered poreand void structure: according to Fink et al. (1992) the total pore volume as wellas the total inner pore surface was considerably enhanced by conversion ofLODP !inters to cellulose II via sodium cellulose, while the average pore di-ameter was found in this SAXS study to be significantly diminished. As possiblecauses, a partial collapse of pores on deswelling as well as the formation of newsmall pores, in combination with an enlargement of already existing pores to asize outside the range of the SAXS method, have been discussed.
Transformation to cellulose II via alkali cellulose generally results in a con-siderable reduction of the LODP after hydrolysis down to a limiting value ofabout 70 after thorough mercerization. This drop in LODP was observed byZeronian and Cabradilla (1973) to increase under comparable conditions in theorder of alkali hydroxides of LiOH < NaOH < KOH. Water regain (sorption ofwater at 65 % relative humidity) is increased by conversion to cellulose II tonearly twice the original value for high DP cotton !inters, in agreement with thechanges in degree of order and pore structure, but this increase in regain obvi-

46 4.2 Interaction of Cellulose with Basic Compounds
ously cannot be directly correlated with the previous swelling during cellulose-alkali interaction. The increase in water retention value generally observed afterinteraction of cellulose with aqueous alkali and subsequent neutralization de-pends largely on steeping lye concentration and type of alkali employed (seeFig. 4.2.9), as well as on the physical structure of the original sample and theprocedures of alkalization, neutralization and drying. The mechanical tensionapplied on the sample during alkali treatment also exerts an influence on thecriteria considered here, as well as on the mechanical properties of the regener-ated fibers (Warwicker et al., 1966).
UO
120
|100
I 80
60
4 8 12Να 0 H [vol %]
15
Figure 4.2.9. Change of the WRV of cotton !inters (DP = 890; means slope of thetwo regions of the curve) after treatment with aqueous NaOH and subsequent neutraliza-tion (Jayme and Roffael, 1970).
4.2.2.5 Concepts for understanding cellulose-alkali hydroxide interaction
The complex chemical and physical structural changes of cellulose on interac-tion with alkali hydroxides, and the interdependency of effects occurring at dif-ferent structural levels, justify an overview of previous and present concepts andmodels for understanding these processes.
The viewpoint of the organic chemist was represented by Z. A. Rogowin, whoassumed a preferential anionization of the hydroxy group at C-2 due to its higheracidity and could explain the behavior of alkali celluloses in consecutive de-rivatization reactions, but neglected widely the role of supramolecular structure.
The viewpoint of Neale (1929; 1930; 1931) and of Pennings (Pennings et al.,1961; Pennings and Prins, 1962), on the other hand, was determined by princi-ples of colloid chemistry and membrane theory, assuming a Donnan equilibriumbetween an external phase of aqueous sodium hydroxide solution and an internalphase of the cellulose-alkali hydroxide water moiety and giving a plausible in-terpretation of cellulose fiber swelling on interaction with aqueous alkali. The

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 47
cellulose in the minor phase is considered here as a weak monobasic acid par-tially forming a sodium salt with NaOH according to the mass action law in adynamic equilibrium
CeII-OH +NaOH- CeII-ONa +H2O
To balance the nonequilibrium between inner and outer phases, water pene-trates into the inner phase and swells the cellulose until a swelling pressure dueto cohesive forces in the polymer structure is reached to compensate the osmoticforces.
Sobue et al. (1939) founded their considerations mainly on WAXS results andemphasized the role of supramolecular structure in the alkalization process, ar-riving at the concept of a permotoid lattice layer reaction comprising amorphousas well as crystalline structural regions. This concept proved to be suitable forunderstanding the enhanced reactivity of alkali cellulose and the appearance ofdifferent WAXS phases varying in lattice dimensions and composition, but thereremained some discrepancy between results on alkali uptake by chemical analy-sis and the X-ray data on crystalline phase composition.
This discrepancy can be reconciled by the concept of the 'reactive structuralfractions' (RSF concept) published by Fink et al. (Fink et al., 1986; Fig. 4.2.10),at least in the practically important region of up to 20 % aqueous NaOH.
100
80
^40
20
O
Cell
8 12 16 20NaOH [wt%]
Figure 4.2.10. Reactive structural fractions (RSF) versus concentration of NaOH.
The concept is based on the two-phase model of the cellulose structure withcrystalline and amorphous regions, and is centered on the statements backed byexperimental evidence from sorption and WAXS studies that:(i) the X-ray crystalline fraction of sodium cellulose I has a constant composi-tion of maximal 0.5 mol NaOH, to minimal 3.5 mol H2O/AGU, up to a lye con-centration of about 20 %, while the water and alkali content of the amorphousfraction varies with the lye concentration and can reach a value of about 2 molNaOH/ AGU at a sufficiently high lye concentration;

48 4.2 Interaction of Cellulose with Basic Compounds
(ii) integral sorption values of NaOH and H2O in the lye concentration range upto 15 %, i.e. in the range of lattice transition, should be replaced by a so-calledspecific uptake considering, besides the fully accessible amorphous regions, alsopart of the crystalline regions, which has already been transformed to the cellu-lose I lattice.
Application of this concept permits a plausible interpretation of the swellingmaximum of cellulose in aqueous lye and results in a good compatibility ofsorption and WAXS data.
Despite its qualitative and at that time rather hypothetical character, the so-called 'hydrate shell explosion' theory of Heuser and Bartunek (1925) openedup a new and very promising route to understanding cellulose-alkali hydroxideinteraction, as it focused for the first time on the important role of NaOH hydra-tion and of the so-called free water on swelling, alkali uptake and lattice transi-tion of cellulose interacting with aqueous NaOH.
A more recent concept consistent with ample experimental evidence and rep-resented with slight variations by several groups' (Fink et al., 1995) is centeredon the breaking of defined inter- and intramolecular hydrogen bonds within thesolid state structure of cellulose by hydrated NaOH ion dipoles, resulting inconformational changes of the macromolecules with a preference for twistedconformations at higher lye concentration.
0 (2 )
Figure 4.2.11. Scheme of Na-cellulose I structure according to Fink et al. (1995).
Figure 4.2.11 shows the various possibilities of interaction, including the hy-drogen bonds involved. At lower NaOH concentration, e.g. 18 % (resulting inNa-cellulose I formation), the interaction preferentially takes place at C-2 and C-6, and not until arriving at a higher concentration of > 22 % NaOH does it occurat C-3. Due to cleavage of the C-3---O-5 hydrogen bond, also the two-foldscrew-axis of the polymer backbone gets lost. By using this concept, accentuat-ing the important role of defined alkali hydroxide hydrates on a more modern

4.2.2 Aqueous and alcoholic solutions of alkali hydroxides 49
level, the effects of lye concentration, of type of alkali as well as of the liquidmedium (water or alcohol), can be understood. The concept implies a preferen-tial cellulose NaOH interaction at the C-2 position, although other opinions(Fengel and Wegener, 1989) have been published too.
As an open question remains the state of binding of NaOH at the differentpositions of the AGU, which cannot yet be exactly defined and probably is situ-ated somewhere in between the borderline cases of an alcoholate and an additioncompound stabilized by intermolecular forces only.
4.2.2.6 Survey of commercial processes based on cellulose-alkalihydroxide interaction
Mercerization of cotton originally consists of the transformation of the native cel-lulose I of cotton fabrics to cellulose II ('mercerized' cellulose) via the intermedi-ate formation of sodium cellulose by the action of aqueous NaOH under mechani-cal tension. The process has the purpose of enhancing dyeability and gloss of thecotton fabric and can be conducted as a so-called 'cold mercerization' or as a 'hotmercerization'. In cold mercerization the fabric is drawn through aqueous NaOHof about 30 % concentration at a temperature of about 20 0C at a speed of 30-40 m/min with a residence time of some minutes in the alkaline bath. The fabric isthen washed free of alkali with water in a stepwise counter-current process stillunder tension, eventually with the addition of some acetic acid to neutralize thelast traces of lye. In hot mercerization a temperature of 60 to 70 0C is employed ata significantly lower lye concentration of about 22-24 % NaOH, also under me-chanical tension. Elimination of alkali by washing with water can be helped byaddition of acetic acid in the last step here too, but this may complicate the recy-cling of the washing liquid. The know-how in both mercerization processes mainlyconsists of the optimal adaptation of mechanical tension and in the most economi-cal use of water in the washing steps including recycling. Further development isproposed to increase the velocity of the moving fabric through the alkaline bath upto about 100 m/min in cold mercerization.
A process analogous to mercerization, developed specifically for viscoserayon staple fabric in order to increase dyeability, is the treatment of this fabricwith aqueous NaOH of about 6 % concentration at 70-80 0C also under me-chanical tension with subsequent washing. These milder conditions take intoconsideration the much lower alkali resistance of the rayon staple in comparisonwith cotton. The increase in dyeability achieved by all three modes of the mer-cerization process can be traced back to the altered pore and void structure of thepolymer regenerated after alkaline treatment.
An alkali cellulose in the form of sodium cellulose I suitable for subsequentxanthation in the viscose process is generally obtained by the action of aqueousNaOH of about 18 % concentration at a temperature between 20 and 40 0C onto

50 4.2 Interaction of Cellulose with Basic Compounds
a hard wood or soft wood dissolving pulp in the form of sheets, rolls or flocks.In an older mode of the process now barley practised, pulp sheets fixed betweenperforated iron plates were treated in a chest-like iron 'steeping press' with lyeof appropriate concentration for about 1 h, then pressed to a press weight ratio ofabout 3.2:1 and then shredded to fibrous flakes suitable for subsequent xan-thation after adequate oxidative depolymerization ('preripening'). A standardalkali cellulose from spruce sulfite pulp had a composition of 32-34 % cellulose,15-17 % NaOH and about 50 % water, and contained less than 1 % Na2CO3 inthe freshly prepared state. Today, generally a slurry steeping process is practisedin the viscose plants, mainly to saving on man-power. The continuous slurrysteeping process proceeds by mixing and beating the pulp with the lye usually ata temperature of about 40 0C for a maximum of l h and subsequent automatedpressing to the press weight ratio required, followed by shredding and preripen-ing. The capacity of today's slurry steeping reactors, made of stainless steel, isabout 10m3. The enhancement of steeping temperature to about 40 0C in com-parison with about 20 0C in the classical steeping press process has no signifi-cant bearing on the chemical reactions and structural changes in alkali celluloseformation, but mainly serves as a viscosity reduction of the lye for better han-dling. A so-called hot alkalization at about 100 0C has been proposed, especiallyfor beach pulp, by Pavlov et al. (1983) in order to enhance pulp reactivity inxanthation and viscose quality for spinning, but to the authors knowledge thisprocess is not practised in industry probably due to a high loss of polymer bydegradation to soluble products and an unsatisfactory control of oxidative degra-dation before the scheduled preripening step.
Alkali cellulose production in the viscose process is now often performed inthe presence of a small amount of a nonionic or anionic surfactant, which doesnot significantly interfere with the course of alkali cellulose I formation(Schleicher et al., 1967), but promotes a smooth xanthation and a good filter-ability of the viscose solution.
Alkali celluloses for subsequent manufacture of cellulose alkyl ethers or car-boxymethylcellulose are in principle prepared also by a slurry steeping process,now centered in its further development on a drastic reduction of liquid-to-solidratio in the steeping reactor for ecological reasons. In contrast with alkali cellu-lose production in the viscose process, the steeping is performed here with asignificantly higher NaOH concentration of between 30 and 40 % NaOH de-pending on type of cellulose ether and procedure of etherification, and resultingin an alkali cellulose of considerably higher cellulose and NaOH contents.
4.2.2.7 Properties and applications of alkali cellulose
Alkali celluloses employed as intermediates in cellulose derivatization can becharacterized as a white-to-yellowish slippery fibrous mass of highly alkaline

4.2.3 Interaction of cellulose with tetraalkylammonium hydroxides 51
nature. All alkali celluloses are unstable in so far as on residence in the open airthey undergo a rather fast oxidative depolymerization, and are decomposed bythe CO2 in the air finally to a degraded cellulose II and sodium carbonate. In thepresence of an excess of water cellulose II is formed from alkali cellulose viaintermediate, unstable addition-compound structures (Sobu et al., 1939). Onheating, alkali cellulose is rapidly decomposed by alkaline degradation of thepolymer to low molecular products.
The products of interaction between cellulose and alkali hydroxides are em-ployed as intermediates only, with sodium celluloses being the only products ofindustrial relevance.
The complete solubility of low DP cellulose in aqueous NaOH under specialconditions has become the basis of an alternative process for cellulose fiberspinning, which is now in development but so far has not been practised in in-dustry (see chapters 2.2 and 4.2.2.2). The filaments obtained here without a tran-sient covalent derivatization resemble in their structure and their textile proper-ties more those obtained by the amine oxide process than those manufactured bythe viscose process. The main problems still impeding cellulose fiber spinningfrom aqueous NaOH solutions are the necessity to employ a cellulose of too lowa DP for achieving optimal textile properties and an uncontrollable instability ofthe solutions at a sufficiently high polymer content.
A recent study on the supramolecular structure and the mechanical propertiesof filament spun from aqueous NaOH solution (Yamane et al., 1996) indicated ahigh crystallinity similar to amine-oxide-spun fibers and a low crystal orienta-tion due to a low draft and stretching ratio, and more strongly developed in-tramolecular than intermolecular hydrogen bonds. Tensile strength and elonga-tion were reported to be comparable to those of viscose fibers.
Besides being the basis of intermediate products, cellulose-alkali hydroxideinteraction is employed in cellulose analysis for determining the alkali-solublepart of pulps and for chain-length fractionation by extraction in the low DPrange (see chapter 3).
4.2.3 Interaction of cellulose with tetraalkylammoniumhydroxides
Due to their highly basic character and the ability to form hydrated ion dipolesin aqueous solution, tetraalkylammonium hydroxides with the general formulaR4NOH interact with cellulose in quite a similar manner to alkali hydroxides,with the only significant difference of being not only swelling agents, but alsogood solvents for cellulose on appropriate choice of the substituents R. For theoverview of R4NOH-cellulose interaction it is therefore appropriate to followthe same route of presentation as that pursued with alkali hydroxides.

4.2.3 Interaction of cellulose with tetraalkylammonium hydroxides 51
nature. All alkali celluloses are unstable in so far as on residence in the open airthey undergo a rather fast oxidative depolymerization, and are decomposed bythe CO2 in the air finally to a degraded cellulose II and sodium carbonate. In thepresence of an excess of water cellulose II is formed from alkali cellulose viaintermediate, unstable addition-compound structures (Sobu et al., 1939). Onheating, alkali cellulose is rapidly decomposed by alkaline degradation of thepolymer to low molecular products.
The products of interaction between cellulose and alkali hydroxides are em-ployed as intermediates only, with sodium celluloses being the only products ofindustrial relevance.
The complete solubility of low DP cellulose in aqueous NaOH under specialconditions has become the basis of an alternative process for cellulose fiberspinning, which is now in development but so far has not been practised in in-dustry (see chapters 2.2 and 4.2.2.2). The filaments obtained here without a tran-sient covalent derivatization resemble in their structure and their textile proper-ties more those obtained by the amine oxide process than those manufactured bythe viscose process. The main problems still impeding cellulose fiber spinningfrom aqueous NaOH solutions are the necessity to employ a cellulose of too lowa DP for achieving optimal textile properties and an uncontrollable instability ofthe solutions at a sufficiently high polymer content.
A recent study on the supramolecular structure and the mechanical propertiesof filament spun from aqueous NaOH solution (Yamane et al., 1996) indicated ahigh crystallinity similar to amine-oxide-spun fibers and a low crystal orienta-tion due to a low draft and stretching ratio, and more strongly developed in-tramolecular than intermolecular hydrogen bonds. Tensile strength and elonga-tion were reported to be comparable to those of viscose fibers.
Besides being the basis of intermediate products, cellulose-alkali hydroxideinteraction is employed in cellulose analysis for determining the alkali-solublepart of pulps and for chain-length fractionation by extraction in the low DPrange (see chapter 3).
4.2.3 Interaction of cellulose with tetraalkylammoniumhydroxides
Due to their highly basic character and the ability to form hydrated ion dipolesin aqueous solution, tetraalkylammonium hydroxides with the general formulaR4NOH interact with cellulose in quite a similar manner to alkali hydroxides,with the only significant difference of being not only swelling agents, but alsogood solvents for cellulose on appropriate choice of the substituents R. For theoverview of R4NOH-cellulose interaction it is therefore appropriate to followthe same route of presentation as that pursued with alkali hydroxides.
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

52 4.2 Interaction of Cellulose with Basic Compounds
4.2.3.1 Swelling and dissolution of cellulose in solutions oftetraalkylammonium hydroxides
All the compounds considered here are, in aqueous solution, strong swellingagents for cellulose, with the effect of swelling generally increasing with in-creasing concentration of the base, and also with the total molar volume of thesubstituents R at a given base concentration, due to an increasing spacer effect ofthe substituent groups.
Swelling in aqueous tetraethylammonium hydroxide, measured by the in-crease in thickness of pulp sheets, reached its final value after a few minutesdepending somewhat on the drying history of the sample, and followed a ratelaw dQ/dt = k (£L - Q)2 (Schwabe and Philipp, 1955).
Aqueous solutions of tetraalkylammonium hydroxides with sufficiently largesubstituents act as solvents for cellulose, if the base concentration exceeds alimiting value decreasing with increasing molar mass of the substituents (Lieser,1937). Lieser and Leckzyck (1936) mention a minimal molar mass of thetetraalkylammonium hydroxide of 150 as a prerequisite for solvent action, whileStrepicheev et al. (1957) assume a somewhat higher value of molar mass, andtetraethylammonium hydroxide as well as trimethylbenzylammonium hydroxideare not classified as solvents. Triethylbenzylammonium hydroxide, as well asdimethyldibenzylammonium hydroxide, however, are explicitly recommendedas good solvents for cellulose.
4.2.3.2 Chemical interaction between cellulose andtetraalkylammonium hydroxides
The quite similar uptake of base from aqueous solution of tetraethylammoniumhydroxide on the one hand, and NaOH on the other, is demonstrated by Fig.4.2.12, indicating a 'step isotherm' in both cases with a steep increase at aboutthe same molar fraction of base and a subsequent plateau corresponding to theaddition compound of 1 mol of base/mol of AGU, if the so-called 'true uptake ofbase' according to Schwarzkopf (1932) (see also chapter 2.1.2) is taken as thecriterion.
The maximal water uptake was found here (2-3 mol of H2O/mol of AGU) to besomewhat lower than with NaOH (about 4 mol of H2O/mol of AGU). Decrystalli-zation and depolymerization of the pulp sample employed by ball milling resultedin significant changes in the curve of base uptake versus base concentration, whichthen resembles more a distribution curve for a solute between two liquid phasesthan the step isotherm that is typical for a heterogeneous reaction.
The uptake of base from aqueous solution proceeds very rapidly on about thesame time scale as the swelling of the sample, and is obviously diffusion-controlled, arriving at its final value within 10 min.

4.2.3 Interaction of cellulose with tetraalkylammonium hydroxides 53
NaOH
1.5
1.0
0.5
TEOH
0.05 0.10 0.05 0.10MoI fraction
Figure 4.2.12. True base uptake' of NaOH and tetraethylammonium hydroxide (TEOH)by spruce sulfite pulp in dependence on mole fraction of base in aqueous solution (Schwabeand Philipp, 1955).
The interaction between cellulose and tetraalkylammonium hydroxides inaqueous solution is assumed by Strepicheev et al. (1957) to proceed via a hy-drate complex between cellulosic hydroxy groups, water molecules and the polarend of the R4NW OH^~) ion dipole, with the nonpolar substituents R acting asspacers to promote the separation of polymer chains. Pasteka (1984) proposed amodel for dissolution of cellulose in triethylbenzylammonium hydroxide whichis based on a sheet lattice structure for the crystalline regions of native cellulose,with the hydroxy groups caring for intersheet cohesion via hydrogen bonds andnonpolar forces for cohesion of the macromolecules within the sheets, and whichis centered on the idea that the polar part of the tetraalkylammonium base dis-rupts the intersheet hydrogen bonds, while the nonpolar parts penetrate betweenthe cellulose chains within each sheet and separates them.
As demonstrated by the results obtained by Schwabe and Philipp (1955) withsolutions of tetraethylammonium hydroxide, the substitution of water by metha-nol or rc-pentanol as a solvent for the base decisively diminishes the equilibriumvalues of swelling and of base uptake and decreases the rate of both these proc-esses by about two orders of magnitude (see Fig. 4.2.13).
100
Figure 4.2.13. Kinetics of the tetraethylammonium hydroxide uptake from methanol(TEOH = tetraethylammonium hydroxide).

54 4.2 Interaction of Cellulose with Basic Compounds
4.2.3.3 Changes in cellulose structure and applications
As shown by Sisson and Saner (1939) for several of these compounds with dif-ferent substituents R, tetraalkylammonium hydroxides in aqueous solutionspenetrate at sufficiently high base concentration into the crystalline regions ofcellulose to form addition compounds exhibiting crystal lattice dimensions dif-ferent from those of the starting material. Simultaneously the crystalline order ofthe sample is significantly decreased. Depending on the route of decompositionof these compounds by washing and/or neutralization, cellulose II as well ascellulose I can be recovered: with cotton cellulose swollen in trimethylben-zylammonium hydroxide a rather well decrystallized cellulose II was recoveredin the presence of organic liquids, while on recrystallization with water celluloseI was obtained (Vigo et al, 1969, 1970 and 1972).
Trimethy!benzyl (Triton B) and dimethyl dibenzyl (Triton F) ammonium hy-droxide were recommended in the past as solvents for viscosity determination ofcellulose, but obviously are not widely used now.
Cellulose solutions of higher concentration in aqueous tetraalkylammoniumhydroxides have been transformed into cellulose filaments with acceptable tex-tile properties by spinning in an acid bath, but this route cannot compete eco-nomically with other ones as an alternative for the viscose process.
Of some interest in the organic chemistry of cellulose functionalization is the useof tetraalkylammonium hydroxides like dimethyldibenzylammonium hydroxide ortriethylbenzylammonium hydroxide as solvents for cellulose for performing etherifi-cation reactions, especially alkylations under homogeneous conditions, taking ad-vantage of a more uniform substituent distribution along and between the polymerchains. This route, however, is suitable on the laboratory scale only, without thechance of industrial realization due to the high price of the solvent and the problemsof recycling and/or disposal. A complete derivatization of all the hydroxy groups to atrixanthogenate of DS = 3 has been reported by Lieser and Leckzyck (1936) by re-acting cellulose dissolved in Et4NOH with an excess of CS2.
A decrystallization of cellulose via the intermediate formation of an adductwith tetraalkylammonium hydroxide for enhancing cellulose reactivity by thisactivation process has also been considered and practised on a laboratory scale,but probably the effects obtainable are inferior to those of ammonia treatment.
4.2.4 Interaction of cellulose with guanidinium hydroxide
Guanidinium hydroxide (GuOH) formed from guanidine in aqueous solutionaccording to the scheme
,NH2
NH-C + H2OΊ2
Cl-NH9
"NH2 _|OH'

4.2.4 Interaction of cellulose with guanidinium hydroxide 55
is a strong base, comparable to alkali hydroxides, and is additionally capable offorming hydrogen bonds or interacting with them via the amino groups of itsmesomeric stabilized cation. Just as with alkali hydroxides, a formation of iondipoles can be observed in concentrated aqueous solution. Already about 70years ago Dehnert and König (1925) reported a mercerization-like action ofaqueous solutions of guanidine on cellulose in their study of interactions be-tween this polymer and various onium compounds. A more detailed investiga-tion has been published (Koura et al., 1975; Philipp et al., 1987b), which coversswelling, base uptake and X-ray patterns of cellulose on interaction with aque-ous GuOH solutions, as well as the LODP and the water retention value of thecellulose samples regenerated by washing and neutralization. Subsequently, abrief survey of these results will be given.
Aqueous solutions of GuOH cause a strong swelling of cellulose, increasingwith the base concentration up to at least 50 %, but not resulting in dissolution ofthe polymer even at the highest concentration investigated of 53 % GuOH. Thelatter fact can probably be traced back to the formation of hydrogen bonds be-tween cellulosic hydroxy groups and the N functions of the guanidinium cationacting as a 'trifunctional hydrogen-bond crosslinker'.
The curve of base uptake versus base concentration indicates a strong sorptionof GuOH from the surrounding solution and confirms the adduct formation al-ready mentioned by Dehnert and König (1925), arriving at a molar ratio of about1.5 GuOH/AGU at the highest base concentration investigated of 53 % (ca.8.5 mol/1). But in contrast with aqueous sodium hydroxide, this 'true base up-take' increases rather continuously with base concentration without showing adefinite step in this sorption isotherm. Water sorption passes a maximum of ca. 2H2O/mol of AGU at a base concentration of about 50 %.
Interaction between cellulose and GuOH probably proceeds similarly to thatof alkali hydroxides via a complex formation between cellulosic hydroxy groupsand the hydrated GuOH ion dipoles, but is supplemented here by the formationof hydrogen bonds between the amino groups of the base and the hydroxygroups of the polymer. This type of hydrogen bond is known to be stronger thanthose formed between hydroxy groups.
The peculiar course of GuOH sorption onto a sample of highly ordered !interscellulose resembles more the distribution equilibrium of a solute between twoliquid phases than the step isotherm of a heterogeneous reaction, and is similarto that encountered in NaOH sorption onto a well decrystallized cellulose. Thisapparent contradiction is reconciled by evaluating and comparing the appropri-ate WAXS patterns: while in the case of NaOH a still rather highly ordered andwell-defined WAXS phase of sodium cellulose is formed, a complete loss ofsupramolecular order must be concluded from the pattern of an originally highlycrystalline cellulose after loading with aqueous GuOH in the range of base con-

56 4.2 Interaction of Cellulose with Basic Compounds
centration between 25 and 30 %. Already at lower base concentration a signifi-cant lowering of supramolecular structure can be observed (Fig. 4.2.14).
Figure 4.2.14. WAXS diagram of !inters cellulose treated with guanidinium hydroxide(GuOH): (a) 10 % GuOH, (b) 10 % GuOH plus regeneration (Philipp et al, 1987b)
At very high base concentrations this amorphization is not so complete, pos-sibly due to the very high viscosity of the GuOH solution impeding a completepenetration of the crystalline regions of the cellulose moiety during the reactiontime employed. In the low range of base concentration of up to about 20 %GuOH, the WAXS pattern of cellulose I is fairly well retained indicating, how-ever, a continuous loss of supramolecular order with increasing base concentra-tion.
The disordered structure of cellulose is widely retained after decomposition ofthe cellulose GuOH adduct by washing with water, neutralization with aceticacid, solvent exchange and drying, and is not significantly changed even afterboiling the sample with water. The X-ray patterns of the regenerated samplesindicated in some cases a poorly ordered cellulose I, and in others a poorly or-dered cellulose II, but so far no clear cut correlation could be derived betweenthe conditions of regeneration and the cellulose modification obtained. Solidstate CP-MAS 13C NMR spectra of the regenerated samples confirmed the lowdegree of order and indicated the coexistence of various chain conformations(Philipp et al., 1987b).
The regenerated samples showed a decisive increase in accessibility, with thelargest effects being obtained by treatment with a GuOH solution of about 25-30 %, i.e. in the same range where the most pronounced amorphization of theGuOH-loaded cellulose has been observed. These regenerated samples had awater retention value of about twice the original one, and their LODP haddropped from about 160 to about 100. Remarkable is the extremely good acces-sibility of these samples to enzymatic degradation (Dan, 1981), which may be

4.2.5 Interaction of cellulose with ammonia and hydrazine 57
traced back to the low degree of order and the conformational nonuniformity, aswell as to the morphological changes at the fibrillar level observed in electronmicroscopic studies: in the scanning electron microscopy (SEM) micrographs ofa cotton !inters sample treated with 40 % aqueous GuOH the original regular,fine fibrillar structure of the fiber surface was widely destroyed and replaced byrather nonstructured lumps of cellulosic matter and deep holes, and only somedisarranged and twisted fibril bundles remained of the original structure.
The interaction between cellulose and guanidinium hydroxide so far finds nopractical application. However, GuOH treatment of cellulose with subsequentregeneration of the sample might be of interest in the laboratory as an effectiveactivation technique for enhancing the reactivity and accessibility of cellulose.
4.2.5 Interaction of cellulose with ammonia andhydrazine
Ammonia and hydrazine are much less basic than the agents considered so far inthis chapter on adduct formation between cellulose and basic compounds, as
their basicity constants in aqueous solutions amount to only 2 x 10~5 and 2 χ 10~6, respectively. Thus the interaction between the polymer and NH3 or N2H4 canbe supposed to occur predominantly by breaking O ··· H ··· O bonds and replac-ing them by N ··· H ··· O hydrogen bonds, and not by interaction between cellu-losic hydroxy groups and hydrated ion dipoles of the base. Nevertheless, NH3 aswell as N2H4 are able to penetrate even into the highly ordered regions of cellu-lose and to form well-defined addition compounds resulting in significantchanges of cellulose structure after decomposition the addition compound andregeneration of the polymer. Subsequently, the interaction of cellulose with am-monia and its consequences on cellulose structure will be described in somedetail with respect to its relevance for organic reactions and the textile process-ing of this polymer, followed by a brief survey of results relatetd to hydrazine.
Interaction of ammonia with cellulose, resulting in structural changes of thepolymer, can take place with NH3 in the liquid state or in the gaseous state atsufficiently high pressure, and also with solutions of the base in water or in polarliquids above a minimum level of NH3 concentration. Systematic studies onadduct formation and lattice transitions have predominantly been performed withliquid NH3.
Liquid NH3 (boiling point -33 0C) is a moderate swelling agent for cellulose,with a swelling power in between that of water and that of aqueous NaOH ofoptimal swelling concentration. It can be turned into a cellulose solvent by add-ing suitable inorganic salts like isothiocyanates or iodides as a second solventcomponent.

58 4.2 Interaction of Cellulose with Basic Compounds
The formation of at least two defined addition compounds between celluloseand liquid NH3 has been reported already about 60 years ago by Hess and co-workers (Hess and Trogus, 1935), with the results of the studies still represent-ing the actual state of knowledge, i.e.(i) a 1 : 1 complex obtained after the evaporation of all free liquid NH3 at a tem-perature above -33 0C;(ii) a complex consisting of 2 mol of NH3/mol of AGU formed below -33 0C.
Furthermore, a 6 : 1 complex is claimed to be formed according to Hess andGundermann (1937). As indicated by the corresponding crystal lattice dimen-sions obtained by WAXS at -20 to -33 0C for ammonia cellulose I and at a tem-perature below -33 0C for ammonia cellulose II, the interaction and adduct for-mation takes place in the highly ordered regions of the cellulose structure too.The 1-0-1 lattice spacing is considerably enhanced compared with the startingmaterial, cellulose I.
On standing under anhydrous conditions, ammonia cellulose I slowly decom-poses with evolution of NH3 to cellulose III, a modification resembling celluloseII, but being transformed, however, on treatment with water to cellulose I. In thisway, cellulose I can be reversibly converted to cellulose III according to
NH3CeIII · CeIIIII
H2O
Starting from a cellulose II sample, cellulose II is regenerated via the interme-diate transitions to ammonia cellulose and cellulose III, indicating some memoryeffect of the intermediates for the structure of the original sample.
All the cellulose samples regenerated after treatment with liquid NH3 have alower degree of order than the original one, the decrystallization effect dependingwidely on the procedure of ammonia treatment as well as that of regeneration.Under suitable conditions, samples exhibiting no crystalline X-ray pattern at allcan be prepared from cellulose I as well as from cellulose II, which, however, aresusceptible to recrystallization after a longer time of residence, especially in thepresence of moisture. The fibrillar architecture is significantly damaged by anammonia treatment, too, as can be seen by a loosening and distortion of the con-centric rings of fibrils in the TEM micrograph of the fiber cross section and by theappearance of deep clefts and fissures partially covered by fibril strings in theSEM micrograph of the fiber surface. This decrease in supramolecular order andfibrillar regularity is reflected also by a significant increase in water retentionvalue and water regain at 65 % relative humidity, as well as by a decrease of theLODP from an original value of about 160 to about 90 for cotton !inters celluloseand in a considerable increase in the initial rate of acetylation with acetanhydride.An even higher enhancement of accessibility has been reported for the synergisticaction of liquid ammonia and aqueous NaOH in consecutive treatment steps (Vigo

4.2.5 Interaction of cellulose with ammonia and hydrazine 59
et al., 1972). An activating pretreatment with liquid ammonia has also beenclaimed to promote the conversion of cellulose to soluble cellulose ethers (HoechstAG, 1984). A shift of the concentration required for the Cell I -> Na-CeIl I -^ CellII transition to lower values by pretreating the sample with liquid NH3 (Schleicheret al., 1973 and 1974) has already been mentioned. Also, dissolution of celluloseby emulsion xanthation was found to take place at lower concentration of NaOHafter activation with liquid NH3. In a subsequent silylation of cellulose with tri-methylsilyl chloride, a significant difference in the effect of activation with liquidammonia has been observed by Wagenknecht et al. (1992) in dependence on acti-vation temperature, an activation at -60 0C results in a smoother derivatizationthan a pretreatment at about -30 0C. Obviously it can make a difference in a sub-sequent derivatization reaction whether ammonia cellulose II or ammonia celluloseI is formed in the pretreatment step.
Ammonia at a pressure of 0.5-0.7 MPa at room temperature shows similareffects to liquid ammonia in the low temperature range and converts cellulose Ito cellulose III of a significantly lower degree of order, with the effect beingenhanced by the action of CO2, SO2 or acetic anhydride in a subsequent treat-ment step at the same level of pressure (Prusakov, 1982).
Much more convenient and less hazardous than an activation with liquid ammo-nia, but of comparable efficiency is the pretreatment of cellulose I with highly con-centrated solutions of NH3 in suitable solvents, as shown in a comprehensive study(Koura et al., 1973; Koura and Schleicher, 1973) with !inters cellulose and byWagenknecht et al. (1992) in connection with a subsequent silylation of cellulose.While solutions of NH3 in alcohols like ethanol or glycol proved to be ineffectiveover the whole range of concentrations, mixtures of NH3 with water, DMSO orformamide brought about significant structural changes and considerable activationeffects at a molar ratio of NH3-to-solvent > 1, and with solvents containing aminofunctions, like ethanolamine or morpholine, an even smaller molar ratio of about 0.7was required for this purpose. Within the structural criteria employed, the NH3 con-centration required was lowest for an increase in WRV and successively higher foran increase in water regain and a drop in LODP or an accelerated acetylation. No-ticeable is the distinct maximum in WRV of more than twice the original value ob-tained by treatment of !inters cellulose with a mixture of 1 mol of NH3 and 2 mol ofethanolamine (Koura et al., 1973; Koura and Schleicher, 1973). Especially for asubsequent silylation, an activation procedure employing a saturated solution of NH3
in DMF or THF at -10 to -15 0C has been reported by Wagenknecht et al. (1992).Addition of an NH3TDMF mixture instead of the two single components to the pre-dried and precooled cellulose was found to be essential for this route of activation.An activation time of about 2 h proved to be sufficient for achieving the maximaleffect, before accelerating the etherification by raising the temperature slowly toabout 60 0C (see chapter 4.5). Too early an increase in temperature was observed tobe detrimental to the activation intended.

60 4.2 Interaction of Cellulose with Basic Compounds
The examples cited here demonstrate the relevance of activation by NH3 inthe organic chemistry of cellulose. The structural changes resulting in the su-permolecular and the fibrillar level (see Fig. 4.2.15) from interaction of NH3
with cellulose are of consequence also in the material properties of the polymer.Therefore, this interaction has also become the basis of textile processes for
improving properties of cotton and viscose fabrics, especially with regard to dye-ability and handling. These processes show some similarity to mercerization asboth consist of a lowering of supramolecular order and a loosening of morphologi-cal structure, although differences do exist with regard to the details of these ef-fects. In comparison with mercerization with NaOH, these treatments with liquidammonia have the advantage of an easy elimination of the reagent, especially inthe so-called dry process, but include the hazards of handling liquid ammonia. In arecent review (Brederik and Blüher, 1991) a so-called 'dry' and a so-called 'wet'process of liquid NH3/cellulose interaction for the pretreatment of cotton fabricprior to easy care treatment have been compared with regard to structural changesof the polymer: in the 'dry' process with elimination of ammonia by evaporation,cellulose III, besides cellulose I, was found in the final product, while in the 'wet'process with the NH3 being washed out by water the final product consisted exclu-sively of cellulose I. In both cases a decrease in degree of order and in crystallitesize was observed after the NH3 treatment, and the pore and void structure provedto be more uniform than in the untreated fabric.
Figure 4.2.15. Changes in the microfibrillar structure of bacterial cellulose by treatmentwith liquid NH3 (-65 0C, 30 min) revealed by TEM: (a) untreated; (b) solvent exchangeand treatment with liquid NH3; (c) mechanically disintegrated, solvent exchange andNH3 treatment (micrographs by HJ. Purz, Teltow-Seehof).

4.2.5 Interaction of cellulose with ammonia and hydrazine 61
Interaction of cellulose with hydrazine exhibits similarities, but also somedifferences from that with ammonia. In contrast with NH3, anhydrous N2H4 wasfound to be a solvent for cellulose at elevated temperature, despite its still lower
basicity constant of about 2 χ 10~6, acting without covalent derivatization bybreaking down H ··· O ··· H bonds in the cellulose structure and replacing themby N — H — O bonds in the cellulose-solvent complex (Litt and Kumar, 1977).From this solution, cellulose II with a lamellar morphology could be regenerated(Kolpak et al., 1977), and on extruding the hot solution, cellulose threads withquite a special texture were obtained after elimination of the N2H4 (Lee andBlackwell, 1981).
In a comprehensive WAXS study of ramie (cellulose I), mercerized ramie(cellulose II) and fortisan fiber (cellulose II), after soaking with nearly anhy-drous N2H4 of 97 % concentration, after evaporation of excess N2H4 and afterdecomposition of the cellulose hydrazine complexes by water vapor, Lee andBlackwell (1981) confirmed the intracrystalline swelling on interaction of N2H4
with cellulose and arrived at different WAXS patterns for the hydrazine com-plexes formed with cellulose I on the one hand, and cellulose II on the other. Amolar ratio of 0.5 N2H4/mol of AGU and of 1.5 mol of N2H4/mol of AGU werereported by the authors for the complexes formed with mercerized ramie andwith fortisan, respectively. For native ramie, no change in X-ray crystallinitywas observed along the transition route from cellulose I via the hydrazine-cel-lulose complex to again cellulose I.
Similar to aqueous solutions of NH3, interaction of solutions of N2H4 in waterof sufficiently high base concentration results in changes in supramolecularstructure and a significant enhancement of accessibility: according to Trogus andHess (1931), the action of a 60 % aqueous N2H4 solution (molar ratio,1 N2H4 : 1.3 H2O) leads to a strong intracrystalline swelling and a change inunit cell dimensions in the crystalline regions, which again are different for cel-lulose I and cellulose II as the starting material in agreement with the recentobservations by Blackwell employing nearly anhydrous N2H4. In a study onactivation of cotton !inters and LODP !inters by aqueous solutions of N2H4
(Koura et al., 1975), a significant increase in WRV was already observed at aconcentration as low as 5.9 % N2H4, corresponding to a molar ratio of 1 N2H4 :28 H2O. At a molar ratio of about 1 N2H4 : 2 H2O, i.e. 1 H2N group to 1 H2Omolecule, about the same activation effect with regard to WRV and LODP wasobtained at 20 0C, as with an aqueous NH3 solution of a molar ratio of 1 : 1 at-20 0C. Lowering the temperature of treatment from 20 to -20 0C, or increasingthe N2H4 concentration to a molar ratio of about 1 N2H4 : 1 H2O (hydrazinehydrate), did not significantly change the effects obtained.
From a practical point of view, aqueous solutions of N2H4 present no advan-tages as an activating agent for cellulose in comparison with NH3 in water. Thepossibility of dissolving cellulose in anhydrous N2H4 and of forming threads

62 4.2 Interaction of Cellulose with Basic Compounds
from these solutions is of scientific interest regarding correlations between sol-vent action and filament structure obtained from the solution, but will probablynot find any practical application due to the hazards of handling anhydrousN2H4.
4.2.6 Interaction of cellulose with aliphatic mono- anddiamines
Just as ammonia or hydrazine, aliphatic mono- and diamines can penetrate evenin the highly ordered regions of cellulose and form addition compounds, result-ing in a change in crystalline lattice dimensions determined by WAXS and in anincrease in accessibility and reactivity after decomposition of the complex andregeneration of the cellulose. The driving force here also consists of the re-placement of O ··· H ··· O bonds between cellulosic hydroxy groups with thestronger N — H — O bonds between cellulose and amine. Research activities inthis area have been centered on addition-compound structure and lattice dimen-sions in relation to amine structure on the one hand, and on the activation effectsobtained via a transient amine adduct formation on the other.
Primary aliphatic amines act as rather strong swelling agents on cellulosewithout being solvents by themselves or being turned into solvents by additionof salts (Wagenknecht, 1976; Davis et al., 1943; Lokhande, 1966; Howsman andSisson, 1954). A unique case is the solvent action of binary systems of methyl-amine and DMSO (see also section 2.2). As demonstrated by the examples inTable 4.2.2, swelling of cellulose in primary aliphatic monoamines is favored bya low temperature and proceeds rather slowly already with ft-propylamine, whilewith ethylene diamine the final value is reached within 1 h.
Table 4.2.2. Liquid retention value (LRV) of cotton !inters inaliphatic amines (Wagenknecht, 1976).
Amine T(0C) LRV (%) after
C3H7NH2
C4H9NH2
H2N(CH2)2NH2
H2N(CH2)2OHfor comparison:HO(CH2)2OHDMF
20O
20O
2020
2020
I h
641115896
12996
5945
1 day
8313366
13111795
--
4 days
9413062
155--
6045

4.2.6 Interaction of cellulose with aliphatic mono- and diamines 63
Regarding amine chemical structure, at least one primary amino group is re-quired for addition-compound formation in the highly ordered regions, whilesecondary and tertiary amines are ineffective and also ethanolamine obviouslydoes not penetrate into the crystallites. Polyamines containing primary as well assecondary amino groups, on the other hand, can penetrate after suitable preswel-ling, with the secondary amino groups obviously also being active in hydrogen-bond interaction (Creely and Wade, 1978). Steric hindrance of the primaryamino group in hydrogen-bond formation can impede penetration into the cel-lulose structure, as demonstrated by the action of isopropylamine or secondarybutylamine in comparison with the corresponding η-compounds (Creely, 1971),but this obstacle can be overcome by pretreatment with a suitable swellingagent. The addition complexes with methyl-, ethyl- and rc-propylamine can beobtained by direct interaction between dry cellulose I or cellulose II and theappropriate amine, while the adducts with the higher amines from C4 to C7 re-quire a two-step procedure, i.e. a preswelling with e.g. ethylamine and subse-quent substitution of this primary swelling agent by the higher amine. As illus-trated by Fig. 4.2.16, the 1-0-1 lattice spacing, indicating the distance betweenadjacent lattice layers in the crystalline regions, increases steadily with the num-ber of C atoms of the linear primary aliphatic amine.
3,0-
2,5-
2,0-
0,5-
0,03 4
No. of C-atoms
Figure 4.2.16. 1-0-1 lattice layer spacing of cellulose amine complexes dependent onthe number of C-atoms of the amine (see Creely, 1971).
Aliphatic diamines with terminal H2N groups at both ends of the carbon chaincan accomplish intracrystalline swelling and addition-compound formation

64 4.2 Interaction of Cellulose with Basic Compounds
throughout the cellulose structure, as experimentally studied with these com-pounds up to octamethylene diamine, and the action of ethylene diamine has beencomprehensively investigated (Creely et al., 1959; Creely, 1977). Concerningaddition complex stoichiometry, the idea of crosslinking action of one ethylenediamine molecule between two hydroxy groups in adjacent lattice layers resultingin a ratio of 1 amine/2 AGU seems logical, but most experimental evidence avail-able today is in favor of a rather stable 1 : 1 complex, which, however, does notexclude some interlayer crosslinking. According to Howsman and Sisson (1954)some freedom of rotation of monomer chain units around the glycosidic bond,resulting in a twisted conformation, is assumed for monoamines, while in the caseof diamines this rotation is restricted by the hydrogen-bond crosslinks.
All the cellulose amine complexes can be decomposed by water yielding cel-lulose I in the case of native cellulose as the starting material, and cellulose II ifa cellulose II sample had been used. On decomposition of a complex obtainedfrom native cellulose by evaporation of the amine, cellulose III was found in thecase of a monoamine, while cellulose I was obtained from the complex with adiamine (Trogus and Hess, 1931). These results comply well with the modeloutlined above for the mode of swelling of these two classes of compounds.Decomposition of the addition compounds by nonaqueous media may lead to analternative lattice modification too, as shown by Lokhande et al. (1976) for eth-ylene diamine treated cotton, where on regeneration of the cellulose with water amixture of cellulose I and cellulose II was obtained, while decomposition bymethanol yielded cellulose III. Besides the lattice modification obtained on cel-lulose regeneration, the activation effect can also be influenced by the mode ofdecomposition of the adduct.
After passing intermediate adduct formation with a mono- or diamine, thedegree of order of a cellulose sample is decreased, and its accessibility and reac-tivity is enhanced, as demonstrated by some data on X-ray crystallinity, watervapor regain and reactivity in esterification in Table 4.2.3 after treatment withethylamine and ethylene diamine.
Table 4.2.3. Ratio of disorder after-to-before amine treatment of cottoncellulose (Venkataraman et al., 1979; Warwicker et al., 1966)
Criterion Disorder ratio after treatment with:ethylamine ethylene diamine
l-;cc 2.33 1.94H2O regain 1.4 1.3Reactivity in esterification ca. 2a 1.3
a bacetylation; formylation.
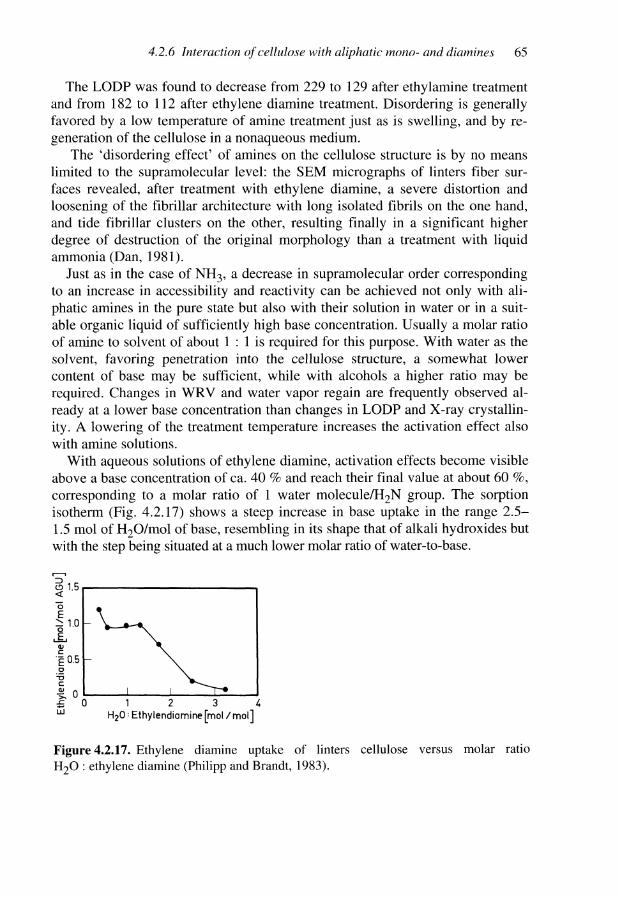
4.2.6 Interaction of cellulose with aliphatic mono- and diamines 65
The LODP was found to decrease from 229 to 129 after ethylamine treatmentand from 182 to 112 after ethylene diamine treatment. Disordering is generallyfavored by a low temperature of amine treatment just as is swelling, and by re-generation of the cellulose in a nonaqueous medium.
The 'disordering effect' of amines on the cellulose structure is by no meanslimited to the supramolecular level: the SEM micrographs of !inters fiber sur-faces revealed, after treatment with ethylene diamine, a severe distortion andloosening of the fibrillar architecture with long isolated fibrils on the one hand,and tide fibrillar clusters on the other, resulting finally in a significant higherdegree of destruction of the original morphology than a treatment with liquidammonia (Dan, 1981).
Just as in the case of NH3, a decrease in supramolecular order correspondingto an increase in accessibility and reactivity can be achieved not only with ali-phatic amines in the pure state but also with their solution in water or in a suit-able organic liquid of sufficiently high base concentration. Usually a molar ratioof amine to solvent of about 1 : 1 is required for this purpose. With water as thesolvent, favoring penetration into the cellulose structure, a somewhat lowercontent of base may be sufficient, while with alcohols a higher ratio may berequired. Changes in WRV and water vapor regain are frequently observed al-ready at a lower base concentration than changes in LODP and X-ray crystallin-ity. A lowering of the treatment temperature increases the activation effect alsowith amine solutions.
With aqueous solutions of ethylene diamine, activation effects become visibleabove a base concentration of ca. 40 % and reach their final value at about 60 %,corresponding to a molar ratio of 1 water molecule/H2N group. The sorptionisotherm (Fig. 4.2.17) shows a steep increase in base uptake in the range 2.5-1.5 mol of H2O/mol of base, resembling in its shape that of alkali hydroxides butwith the step being situated at a much lower molar ratio of water-to-base.
1 2 3 iH2O: Ethylendiomine [mol / mol]
Figure 4.2.17. Ethylene diamine uptake of !inters cellulose versus molar ratioH2O : ethylene diamine (Philipp and Brandt, 1983).

66 4.2 Interaction of Cellulose with Basic Compounds
Also in contrast with alkali hydroxides or guanidinium hydroxide, the uptake ofethylene diamine was not accompanied by a specific water sorption at the begin-ning or within the plateau of base uptake corresponding to a molar ratio of 0.9-1.0 mol of ethylene diamine/mol of AGU. Ethanolamine on the other hand, obvi-ously cannot penetrate the ordered regions of the cellulose structure, as con-firmed by a sorption of only about 0.1 mol/mol of AGU over the whole range ofconcentration of the ethanolamine/water mixture. But, ethanolamine can eventu-ally increase the activation effect of an aliphatic amine above that obtained withthe pure amine, as demonstrated by the course of WRV observed after treatingcotton !inters with methylamine/ethanolamine mixtures of increasing methyl-amine concentration (see Table 4.2.4).
In a comprehensive study on the activating action of amine/solvent mixturesonto cotton !inters, from which some selected data are presented in Table 4.2.4,Koura et al. (1973) arrived at the conclusion that the intermolecular interactionsin the ternary systems of cellulose aliphatic amine and solvent are governed by:(i) the competition between O ··· H ··· O and N ··· H ··· O bond formation withthe latter being significantly stronger especially at low temperature;(ii) the potential hydrogen-bond density of the amine (or the amine solvent asso-ciate) besides its molecular volume and geometrical shape;(iii) the interaction of the amine with the solvent or with solvent associates, an exam-ple being the destruction of self-associates of ethanolamine molecules by hydrogen-bond interaction between the hydroxy group and an amino group with the effect ofenhanced swelling of the cellulose by the newly formed ethanolamine associate.
Especially this last point accentuates again the relevance of activeagent/solvent interaction for understanding swelling and activation of cellulosein these binary mixtures.
Cellulose-amine complexes find no application as products, but are of scien-tific interest as intermediates in special routes of cellulose activation.
4.2.7 Concluding remarksThis chapter deals with the cellulose chemistry of intermolecular interaction,resulting in addition compounds of limited stability and sometimes ill-definedcomposition. These compounds originate either from complex formation be-tween a cellulosic hydroxy group and a hydrated ion dipole of an alkali ortetraalkylammonium hydroxide, or from hydrogen-bond interaction by replacingO ··· H ··· O bonds between cellulosic hydroxy groups with the strongerO ··· H ··· N bonds between cellulosic hydroxy groups and a suitable basic com-pound like NH3, N2H4 or an aliphatic amine. In the first case the uptake of baseis combined with a specific water sorption, while in the second case obviouslyno bound water is included in the complex.

Tab
le 4
.2.4
. In
crea
se in
the
acce
ssib
ilit
y of
cot
ton
!int
ers
(DP
130
0) b
y tr
eatm
ent w
ith a
min
e co
ntai
ning
sol
vent
mix
ture
s (K
oura
et a
l., 1
973)
.
Am
ine
wit
hout
CH
3NH
2
CH
3NH
2
CH
3NH
2
CH
3NH
2
CH
3NH
2
CH
3NH
2
CH
3NH
2
H2N
(CH
2)2N
H2
H2N
(CH
2)2N
H2
H2N
(CH
2)2N
H2
H2N
(CH
2)2N
H2
H2N
(CH
2)2N
H2
H2N
(CH
2)2N
H2
Solv
ent
with
out
H2N
(CH
2)2O
HH
2N(C
H2)
2OH
H2N
(CH
2)2O
HH
2N(C
H2)
2OH
H2N
(CH
2)2O
H(C
H3)
2SO
(CH
3)2S
OH
2N(C
H2)
2OH
H2N
(CH
2)2O
H(C
H3)
2SO
(CH
3)2S
OH
2OH
2O
Mol
arra
tio
Am
ine:
Solv
ent
— 1:3
1:2
1:2
2:3
1:1
1:3
1:1
1:3
1:1
1:3
1:1
1:2
1:1
Tem
pera
ture
/tim
e°C
/h
- 0/1
0/1
0/24
0/1
0/1
0/1
0/1
20/1
20/1
20/1
20/1
20/1
20/1
WR
V(%
)
52 78 100
118
112
107 70 96 78 106 73 105 89 94
H2O
vapo
rre
spir
.(%
)6.
8- - - 9.
29.
3- 9.
27.
79.
3- 9.
0- -
LO
OP
(Gua
m)
160
160
132
100
127
100
160 88 140 99 150
104
115 88
Ace
tyl3
cont
ent
(%)
22 24 - - 45 (47)
28 46 32 40 34 42 - -
a ace
tyla
tion:
3 m
in a
t 25 0
C w
ith
acet
anhy
drid
e/H
C!O
4.
K)
X) S'
OQ 1 a S-

68 4.2 Interaction of Cellulose with Basic Compounds
On interaction of cellulose with guanidinium hydroxide, obviously both theseprinciples are realized. Addition-compound formation in water as the reactionmedium proceeds very rapidly and is usually diffusion-controlled, but can bedelayed and slowed down in nonaqueous media, e.g. alcohols. Aqueous solu-tions of the compounds considered here lead to complex formation even in thehighly ordered regions of the cellulose structure, above a limiting base concen-tration in the solution, corresponding to a change in WAXS pattern and fre-quently a steep increase in base uptake within a step-like sorption isotherm typi-cal for a heterogeneous type of reaction.
After decomposition of the complexes by the action of e.g. water, or byevaporation of the base in the case of volatile agents, cellulose I, II or III oflower degree of order and an enhanced accessibility and reactivity, with an al-tered fibrillar architecture, is obtained. The lattice modification and the magni-tude of the decrystallization effect depend on the complex-forming system in-volved and on the procedure of regeneration of the cellulose from the complex.
The complexes formed between cellulose and basic compounds find no appli-cation as products, but are of high scientific and practical relevance as interme-diates in activating pretreatments of the polymer for subsequent covalent de-rivatization.
ReferencesBartunek, R., Kolloid-Z. 1956,146, 35.Bredereck, K., Vlachopoulos, G.,Angew. Makromol Chem. 198Oa, 84, 81-96.Bredereck, K., Thi Bach Phnong Dau, Angew. Makromol Chem. 198Ob, 89,
167-181.Bredereck, K., Blüher, Α., Melliand Textilber. 1991, 72, 46-54.Creely, J.J., Segal, L., Loeb, L., /. Polym. ScL 1959, 36, 205-214.Creely, J.J., Text. Res. J. 1971, 41, 274-275.Creely, J.J., J. Polym. ScL9 Polym. Chem. Ed. Al 1977, 75, 521-522.Creely, JJ., Wade, R.H., J. Polym. ScI, Polym. Lett. Ed. 1978, 76, 291-295.Dan, D.C., Ph.D. Thesis, Academy of Science (GDR) 1981.Davis, W.E.A., Barry, A.J., Peterson, F.C., King, A.J., /. Am. Chem. Soc. 1943,
63, 1294-1299.Dehnert, F., König, W., Cellul-Chem. 1925, 6, 1.Dobbins, R.J., Tappi 1973, 53, 2284-2290.Eigen, M., Angew. Chem. 1963, 75, 489.Fengel, D., Wegener, G., Wood, Chemistry, Ultrastructure, Reactions, Berlin: de
Gruyter & Co., 1989.Fink, H.-P., Dautzenberg, H., Kunze, J., Philipp, B., Polymer 1986, 27, 944-
948.

References 69
Fink, H.-P., Philipp, B., Zschunke, C., Hayn, M., Acta Polym. 1992, 43, 266-269.
Fink, H.-P., Walenta, E., Kunze, J., Mann, G., in Cellulose and CelluloseDerivatives, Physico-chemical Aspects and Industrial Applications, Kennedy,J.F., Phillips, G.O., Williams, P.A., Piculell, L. (Eds.), Cambridge: WoodheadPubl. Ltd., 1995, pp. 523-528.
Harris, C.A., Purves, C.B., Pap. Trade J. 1940, 770, 29.Hayashi, J., Sen'i Gakkaishi, 1976, 32, P37.Hess, K., Trogus, C., Ber. Dtsch. Chem. Ges. 1935, 68, 1986.Hess, K., Gundermann, J., Ber. Dtsch. Chem. Ges. 1937, 70, 1788.Heuser, E., Bartunek, R., Cellul.-Chem. 1925, 6, 19-26.Hinton, J.F., Amis, E.S., Chem. Rev. 1967, 67, 367.Hoechst AG, Patent DE 3241720, 1984; Chem. Abstr. 1984, 707, 74631.Howsman, J.A., Sisson, W.A., in Cellulose and Cellulose Derivatives, Ott, E.,
Spurlin, H.M., Graffein, M.W. (Eds.), New York: Interscience Publ. Inc., 1954,pp. 328.
Isogai, A., Cellulose, 1997, 4, 99-107.Jayme, G., Roffael, E., Papier (Darmstadt) 1970, 24, 181-186.Käufer, M., Papier (Darmstadt) 1984, 38, 583-589.Kolpak, F.J., Blackwell, J., Litt, M., /. Polym. ScL, Polym. Lett. Ed. 1977, 75,
655.Koura, A., Schleicher, H., Faserforsch. Textiltech. 1973, 24, 82-86.Koura, Α., Schleicher, H., Philipp, B., Faserforsch. Textiltech. 1973, 24(5),
187-194.Koura, A., Philipp, B., Schleicher, H., Wagenknecht, W., Faserforsch.
Textiltech. 1975, 26, 514-515.Krässig, H.A., in Cellulose - Structure, Accessibility and Reactivity, Krässig,
W.A. (Ed.), Yverdon: Gordon and Breach Sei. Publ. S.A., 1993.Kunze, J., Ph.D. Thesis, Academy of Science (GDR) 1983.Kunze, J., Ebert, A., Lang, H., Philipp, B., Z. Phys. Chem. (Leipzig) 1985, 266,49-58.
Lang, H., Bertram, D., Loth, F., Patent DD 260190, 1988; Chem. Abstr. 1989,770, 156358.
Lang, H., Laskowski, I., Cellul Chem. Technol 1991, 25, 143-153.Lee, D.M., Blackwell, J., /. Polym. ScL, Polym. Phys. Ed. 1981, 79, 459-465.Lieser, Th., Leckzyck, E., Ann. 1936, 522, 56.Lieser, Th., Liebigs Ann. Chem. 1937, 528, 276.Litt, M., Kumar, N.G., Patent US 4 028 132, 1976; Chem. Abstr. 1977, 87,
186315.Lokhande, H.T., Bombay Technol. 1966, 76, 22-26.

70 4.2 Interaction of Cellulose with Basic Compounds
Lokhande, H.T., Shukla, S.R., Chidambareswaran, P.K., Paul, RB., /. Polym.ScL, Polym. Lett. Ed. 1976, 14, 747-749.
Loth, F., Philipp, B., Dautzenberg, H., Acta Polym. 1984, 35, 483-486.Mori, U., Ph.D. Thesis, University of Jena 1991.Muskat, I.E., /. Am. Chem. Soc. 1934, 56, 693; 2449.Neale, S.M., /. Text. Inst. 1929, 20, T373.Neale, S.M., /. Text. Inst. 1930, 27, T255.Neale, S.M., /. Text. Inst. 1931, 22, T320; T349.Pasteka, M., Cellul. Chem. Technol 1984,18, 379-387.Pavlov, P., Makazchieva, V., Simeonov, N., Dimov, K., Cellul. Chem. Technol.1983, 77, 575-583; 585-592.
Pennings, A.J., Prins, W., Hale, R.D., Ränby, B.C., /. Appl. Polym. ScL 1961, 5,676.
Pennings, A.J., Prins, W., J. Polym. Sei. 1962, 58, 229-248.Philipp, B., Faserforsch. Textiltech. 1955, 6, 180-181.Philipp, B., Lehmann, R., Ruscher, Ch., Faserforsch. Textiltech. 1959, 70, 22-35.Philipp, B., Brandt, A., Cellul Chem. Technol 1983, 77, 323-332.Philipp, B., Purz, HJ., Papier (Darmstadt) 1983, 37, V1-V13.Philipp, B., Fink, H.-P., Kunze, J., Purz, HJ., Tappi Proc., Int. Dissolv.
Specialty Pulps 1983, 177-183.Philipp, B., Fink, H.-P., Kunze, J., Frigge, K., Ann. Phys. (Leipzig) 1985, 42,507-523.
Philipp, B., Kunze, J., Fink, H.-P., Structures of Cellulose, ACS Symp. Ser.1987a, 340, 178.
Philipp, B., Kunze, J., Loth, F., Fink, H.-P., Acta Polym. 1987b, 38, 31-36.Prusakov, V.V., Chim. Drew. 1982, 4, 112-113.Purz, HJ., Graf, H., Fink, H.-P., Papier (Darmstadt) 1995, 49, 714-730.Scherer, P.C., Hussey, R.E., J. Am. Chem. Soc. 1931, 53, 2344.Scherer, P.C., Gotsch, L.P., Bull Va. Polytech. Inst. 1939, 32.Schleicher, H., Philipp, B., Ruscher, Ch., Faserforsch. Textiltech. 1967, 78, 1-4.Schleicher, H., Daniels, C., Philipp, B., Faserforsch. Textiltech. 1973, 24, 371-376.
Schleicher, H., Daniels, C., Philipp, B., J. Polym. ScL, Symp. 1974, 47, 251-260.Schmid, L., Becker, B., Berichte 1925, 585, 1966.Schmid, L., Waschkaw, A., Ludwig, E., Monatsh. 1928, 49, 107.Schwabe, K., Philipp, B., Holzforschung 1955, 9, 104-109.Schwarzkopf, O., Z Elektrochem. 1932, 38, 353-458.Sisson, W.A., Saner, W.R., /. Phys. Chem. 1939, 43, 687.Sobue, H., Kiessig, H., Hess, K., Z Phys. Chem. 1939, B43, 309-328.Strepicheev, A.A., Klunjanc, J.L., Nikolaeva, N.S., Mogilevskij, E.M., 7zv.Akad. Nauk SSSR, Otd. Chim. Nauk 1957, 6, 750-754.

4.3. l General routes of cellulose-metal atom interaction 71
Trogus, C., Hess, K., Z. Phys. Chem. (Leipzig) 1931, B14, 387.Venkataraman, A., Subramanian, D.R., Maniunath, B.R., Padkye, M.R., Indian
J. Text. Res. 1979, 4, 106-110.Vigo, T.L., Wade, R.H., Mitcham, R., Welch, C.M., Text. Res. J. 1969, 39, 305-
316.Vigo, T.L., Mitcham, R., Welch, C.M., /. Polym. Sd. 1970, 8, 385-393.Vigo, T.L., Mitcham, R., Welch, C.M., Text. Res. J. 1972, 42, 96.Wagenknecht, W., Ph.D. Thesis, Academy of Science (GDR) 1976.Wagenknecht, W., Nehls, L, Stein, A., Klemm, D., Philipp, B., Acta Polym.
1992, 43, 266-269.Warwicker, J.O., Jeffries, R., Colbrain, R.L., Robinson, R.N., in The Cotton Silk
and Man-Made Fibers Research Association, Didsburg: Shirley InstitutePamphlet, 1966, No. 93.
Yamane, J., Mori, M., Saito, M., Okajima, K., Polym. J. 1996, 20, 1039-1047.Yamashiki, T., Kamide, K., Okajima, K., in Cellul. Sources Exploit., Kennedy,J.F., Phillips, G.O. (Eds.), London: Ellis Horwood, 1990, pp. 197-202.
Zeronian, S.H., Cabradilla, K.E., /. Appl. Polym. ScL 1973, 77, 539-552.
4.3 Metal Complexes of Cellulose
4.3.1 General routes of cellulose-metal ion interactionCellulose-metal ion interaction has many features, ranging from the sorption anddesorption of calcium ions during wood pulp manufacture and artificial fiberspinning using cellulose cuprammonium hydroxide solutions, to the design andpreparation of sophisticated cellulosic materials with e.g. catalytic properties.Two main routes to metal-ion-containing cellulose products have to be consid-ered, i.e.(i) the use of the cellulose backbone as a polymeric carrier of functional groupsdeliberately introduced by covalent reaction and subsequently employed for theinteraction with metal ions;(ii) the engagement of hydroxy groups of the polyhydroxy compound 'cellulose'as a polymer ligand coordinated to a metal cation acting as the center of com-plexation.
A detailed discussion of the first one of these routes would by far surpass thescope of this book, and only two examples will therefore be mentioned brieflyfor illustration: the binding of Ca2+ or Fe2+ to cellulose powders containingvarying numbers of carboxyl groups. The interaction was found to be governedby the concentration ratio of [M2+] to [H+] on the one hand, and the level ofcarboxyl content on the other, without a full saturation of all carboxylic sites bythe metal cation being obtained under the conditions investigated (Jacopian et

4.3. l General routes of cellulose-metal atom interaction 71
Trogus, C., Hess, K., Z. Phys. Chem. (Leipzig) 1931, B14, 387.Venkataraman, A., Subramanian, D.R., Maniunath, B.R., Padkye, M.R., Indian
J. Text. Res. 1979, 4, 106-110.Vigo, T.L., Wade, R.H., Mitcham, R., Welch, C.M., Text. Res. J. 1969, 39, 305-
316.Vigo, T.L., Mitcham, R., Welch, C.M., /. Polym. Sd. 1970, 8, 385-393.Vigo, T.L., Mitcham, R., Welch, C.M., Text. Res. J. 1972, 42, 96.Wagenknecht, W., Ph.D. Thesis, Academy of Science (GDR) 1976.Wagenknecht, W., Nehls, L, Stein, A., Klemm, D., Philipp, B., Acta Polym.
1992, 43, 266-269.Warwicker, J.O., Jeffries, R., Colbrain, R.L., Robinson, R.N., in The Cotton Silk
and Man-Made Fibers Research Association, Didsburg: Shirley InstitutePamphlet, 1966, No. 93.
Yamane, J., Mori, M., Saito, M., Okajima, K., Polym. J. 1996, 20, 1039-1047.Yamashiki, T., Kamide, K., Okajima, K., in Cellul. Sources Exploit., Kennedy,J.F., Phillips, G.O. (Eds.), London: Ellis Horwood, 1990, pp. 197-202.
Zeronian, S.H., Cabradilla, K.E., /. Appl. Polym. ScL 1973, 77, 539-552.
4.3 Metal Complexes of Cellulose
4.3.1 General routes of cellulose-metal ion interactionCellulose-metal ion interaction has many features, ranging from the sorption anddesorption of calcium ions during wood pulp manufacture and artificial fiberspinning using cellulose cuprammonium hydroxide solutions, to the design andpreparation of sophisticated cellulosic materials with e.g. catalytic properties.Two main routes to metal-ion-containing cellulose products have to be consid-ered, i.e.(i) the use of the cellulose backbone as a polymeric carrier of functional groupsdeliberately introduced by covalent reaction and subsequently employed for theinteraction with metal ions;(ii) the engagement of hydroxy groups of the polyhydroxy compound 'cellulose'as a polymer ligand coordinated to a metal cation acting as the center of com-plexation.
A detailed discussion of the first one of these routes would by far surpass thescope of this book, and only two examples will therefore be mentioned brieflyfor illustration: the binding of Ca2+ or Fe2+ to cellulose powders containingvarying numbers of carboxyl groups. The interaction was found to be governedby the concentration ratio of [M2+] to [H+] on the one hand, and the level ofcarboxyl content on the other, without a full saturation of all carboxylic sites bythe metal cation being obtained under the conditions investigated (Jacopian et
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

72 4.3 Metal Complexes of Cellulose
al., 1975). Maekawa and Koshijima (1990) describe the synthesis of a cellulose-based polymer hydroxamic acid via 2,3-dicarboxycellulose, in connection with asubsequent complexation of various transition metal cations to the hydroxamicacid functions.
Table 4.3.1. Survey of cellulose-metal complexes.
Type of complex Medium
Transition metal complexes with WaterNH3 or amine as ligands
Transition metal complexes with Watertartaric acid as ligand
Metal hydroxo complexes WaterNeutral salts of special structure Water,(with or without NH3) dipolar aprotic liquid
The second route with cellulose acting as a polymeric polyhydroxy ligand inmetal complex formation will be the topic of this chapter, with the somewhatarbitrary subdivisions employed here being given in Table 4.3.1.
The systems considered here are predominantly aqueous ones containing thepolymer in the dissolved or highly swollen state. The first three classes of com-plexes and their routes of formation can be understood by the principles of com-plex chemistry with cellulosic hydroxy groups in the deprotonated or non-deprotonated state acting as ligands to a central atom, and with ligand-exchangeprocesses playing a dominant role. In the last mentioned case, polymer metalcation interaction is considerably weaker and can be interpreted either as electrondonor-acceptor complex formation between the salt and the cellulose, or as partici-pation of cellulosic hydroxy groups in the solvation of the ion dipole of the salt.
Subsequently, the chemistry of cellulose-metal complex formation will bepresented in some detail, accentuating adequately the scientifically and practi-cally important copper complexes and putting some emphasis on the many stillopen questions. Effects of supramolecular structure of the polymer on complexformation will be considered in connection with processes of cellulose swellingand dissolution, as many cellulose-metal complexes are well suited to get thepolymer to form an aqueous solution. The subchapter on cellulose-metal com-plex application (4.3.5) is therefore also centered on their action as cellulosesolvents, considering especially the commercial cuprammonium process for themanufacture of filaments, fibers and films of regenerated cellulose. This chapteris closed by an outlook on the questions still open and promising routes of futureresearch.

4.3.2 Chemistry of cellulose-metal complex formation 73
4.3.2 Chemistry of cellulose-metal complex formationCellulose-metal cation interaction represents a broad variety of phenomena re-garding type and strength of binding between cellulose hydroxy groups and themetal moiety, type of complex-forming metal atom and its position in the peri-odic table, as well as the structure of ligands coordinated to the central atom.Most of the knowledge acquired up to now results from studies of cellulose-copper complexes with ammonia or amines as further ligands, which willtherefore be treated in a separate section, before than comparing them with othertransition metal-amine complexes.
Although the complex chemistry of cellulose is primarily concerned with theAGU along the macromolecule and the three hydroxy groups in each of the re-peating units, it can be understood and efficiently promoted only in connectionwith the intra- and intermolecular hydrogen bond system of cellulose and itschanges during cellulose-metal ion interaction. Fig. 2.1.6 in chapter 2.1.2 em-phasizes this point and is presented here again for the readers convenience (Fig.4.3.1).
cellulose I cellulose
Figure 4.3.1. Most probable hydrogen bond patterns of cellulose allomorphs(Kroon-Batenburg et al., 1990).

74 4.3 Metal Complexes of Cellulose
4.3.2.1 Copper complexes of cellulose with N-containing ligands
As early as 1857, Schweizer described the dissolution of cellulosic materials in asolution of cupric hydroxide and aqueous ammonia, and this 'Guam-cellulosesystem' is still the number one among cellulose-metal complexes with regard topractical use and scientific challenge. Due to its technical relevance for artificialcellulose filament production, this system was studied in the first half of thiscentury by numerous groups world wide. It was recognized rather early on that[Cu(NH3)4](OH)2 is the active species, which strongly interacts with the cellulo-sic hydroxy groups at the C-2 and C-3 positions, and that a small amount ofalkali hydroxide promoted dissolution and complexation of the polymer. Anexcess of alkali resulted in precipitation of a swollen compound, usually de-
scribed as the Normann compound, with the formula NaJC[Cu(C6H8O5)2] (χ ~ 2).Increasing the copper content in a given system, the cellulose showed at firstlimited swelling, then partial dissolution, and finally complete dissolution to ahomogeneous medium, with the different states of dispersion depending also onNH3 concentration, temperature, polymer-to-liquid ratio, and DP of the cellu-lose. At a copper concentration of between 15 and 30 g/1 and an ammonia con-centration not below 15 %, even high DP cellulose was found to dissolvequickly and completely. The copper atom in the dissolved complex was assumedto be coordinated to the hydroxy groups at C-2 and C-3 on the one hand and totwo NH3 molecules on the other. But these early investigations performed bye.g. optical rotation measurements, dialysis, electrolysis or ion exchange, led to acontroversial discussion on the real structure of the cellulose-copper complexformed, centering frequently on the question of a cationic or an anionic nature ofthe polymer-copper containing species.
More recent work on the chemical structure of the cellulose-copper complexshall be surveyed by mentioning the ESR study of Baugh et al. (1968) regardingthe spatial position of the copper atoms in relation to the polymer chain, and thecircular dichroism (CD) measurements of Miyamoto et al. (1996), who relatedthe two cotton effects observed to the state of copper binding, and assumed anequilibrium between copper-substituted monomer units, unsubstituted AGU andcopper tetramine cations in the aqueous Cuam-cellulose system, with an averagecopper-to-AGU ratio of 0.6-0.8.
Decisive progress in the elucidation of the complex structure was achievedintroducing the techniques and the reasoning of modern inorganic complexchemistry into this area of cellulose chemistry (Kettenbach et al., 1997).Burchardt succeeded for the first time in performing static and dynamic lightscattering measurements in the deeply colored cellulose-Cuam system (Bur-chardt et al., 1994). From the results obtained with low molecular polyols asmodels, as well as with cellulose itself, it can be concluded that:

4.3.2 Chemistry of cellulose-metal complex formation 75
(i) a very stable polyolato complex is formed by interaction of [Cu(NH3)4](OH)2
with cellulose, with two coordination sites of the copper atom being occupied bythe deprotonated O atoms at C-2 and C-3 of the AGU, and the other two sitesbinding NH3 molecules (see Fig. 4.3.2), approaching a degree of complexationof nearly 100 % of the AGU with decreasing diol concentration in the system;
--HO
m Cu(NH3Jn(OH)2
-2m H2O, -m (n-2)NH3
H3N NH3
Figure 4.3.2. Scheme of Cuam-cellulose complex structure (Burchardt et al., 1994).
(ii) even at high copper and NH3 concentration, a small amount of a copper bis-diolato complex (see Fig. 4.3.3) can be formed, resulting in intra- or intermo-lecular crosslinking, which is favored by a high OH~ ion concentration and canfinally lead to precipitation of compounds of the Normann type;(iii) despite the breaking of the major part of the hydrogen bonds existing in thestarting polymer, during complexation and dissolution, strong hydrogen bondsof the type OH--·Ο~ can be formed between the primary hydroxy group at C-6and the deprotonated O-2 atom of an adjacent AGU without interfering with thechain conformation, thus leading to an increased chain stiffness.

76 4.3 Metal Complexes of Cellulose
The complexation of one copper atom and two NH3 ligands to each diol unitleads to an increase in molar mass of these AGUs from 162 to 258. The remark-able chain stiffness of the complex is reflected by a Kuhn segment length of25.6 nm corresponding to about 50 monomer units (compared with about 2 nmand 10 monomer units in the case of a polystyrene chain). It is interesting to notethat in a dilute Cuam-cellulose solution, the increase in radius of gyration and inhydrodynamic radius of the polymer coils with DP is slowed down, obviouslydue to some 'back-coiling', via formation of intramolecular bisdiolate (cuprate)crosslinks (Burchardt et al., 1994). The gel formation observed with cellulose-Cuam solutions of higher concentration after a long residence time may possiblybe traced back to the same causes on the intermolecular level. Cellulose com-plexed and dissolved by Guam is rather susceptible to oxidative chain degrada-tion, with nitrite ions formed from the ammonia present acting as the active in-termediate in oxygen transfer.
a)
π 2+
b)CH
-NH2 O-\ ^y
CuCH O2^NH
\
-CHI
-CHcellulose chain
c)*HC-°\ /°-CH'
I CuHC- \
ι 2-
-CH
Figure 4.3.3. Structures a) ethlene diamine copper complex, Cuen, b) Cuen cellulosecomplex, c) cellulose cuprate (Burchardt et al., 1994).
Other copper complex-based aqueous cellulose solvents can be prepared withethylene diamine (en) or 1,3-diaminopropane (pren) as ligands to the centralcopper atom. These systems show similarities to the Guam system in so far asdiolato copper complexes with the hydroxy groups at C-2 and C-3 are formedhere too, but also some differences exist regarding complex structure and bind-

4.3.2 Chemistry of cellulose-metal complex formation 77
ing strength: the Cuen system is prepared by dissolving cupric hydroxide in justthe sufficient amount of aqueous ethylene diamine to form the complex[Cu(Cn)2](OH)2 as the active species, while the Guam solution always contains alarge excess of ammonia. This solvent, first described by Traube (1911), issomewhat less efficient at complexing cellulose than Guam, as a higher amountof copper per AGU is required, obviously due to the fact that the bidentate Ii-gand ethylene diamine forms a stable five-membered ring with the central atomand therefore exhibits some resistance to ligand exchange, with the C-2/C-3 diolstructure of the AGU necessary for complexation and dissolution of the cellu-lose. But also here the formation of a heteroleptic copper complex with a biden-tate diolato ligand on the one hand, and a bidentate ethylene diamine ligand onthe other, can be assumed to be the driving force for cellulose dissolution. Alsohere, the formation of bisdiolato crosslinks (cuprate structures) with the elimina-tion of ethylene diamine as a ligand must be taken into account (see Fig. 4.3.3).Cellulose degradation in Cuen was observed to be much less than in Guam.
An efficient copper complex-based cellulose solvent described by Gadd(1982) is obtained by dissolving freshly precipitated Cu(OH)2 in a slight excessof 1,3-diaminopropane, resulting in a copper cation complexed by two bidentatediaminopropane units as the prevailing active solvent species. The six-membered rings formed in this ligand coordination are less stable than the five-membered ones in the case of ethylene diamine, and a ligand exchange with apartially deprotonated diol structure of the AGU can take place rather easily, asindicated by the scheme in Fig. 4.3.4.
CH2OH
HO OH
6CH2OH
5>-0
H2N ^
H2N NH2
2+
+20ΗΘ
Ο--- + H2N NH2 + H2Oθ
O OH\ /
Cu/ N
H2N' YlH2
Figure 4.3.4. Scheme of cellulose 1,3-diaminopropane copper complex structure (Gadd,1982).

78 4.3 Metal Complexes of Cellulose
A copper-to-AGU ratio of 1 : 1 was observed above a limiting copper con-centration, and at a high pH formation of a bisdiolato complex without residualdiamine ligands is reported here also. Gadd (1982) emphasizes the importance ofan adequate stability of the primary homoleptic cationic complex, permitting apartial but not a complete ligand exchange with the diol units of cellulose, andhe emphasizes further the necessity of an at least partial deprotonation of thesediol units. With ethanolamine as a ligand to copper, obviously no cellulose sol-vent system can be realized due to the very strong binding of the deprotonatedligand to the central Cu atom, which impedes subsequent ligand exchange withthe cellulosic diol moiety.
Finally, the Cu(OH)2-biuret-alkali complexes shall be mentioned briefly,which have been described as solvents for cellulose already by Schiff (1898) andlater were thoroughly investigated by Jayme and Lang (1957). Employing amolar ratio of 1 Cu to 2 biuret the best results on cellulose dissolution were ob-tained with KOH as the alkali added, and a highly viscous, clear violet solutionwith a concentration of up to 8 % cellulose of a DP of about 800 could be pre-pared. A probable formula for the active species is presented in Fig. 4.3.5.
O H H Οθ
\\ \ / /C-N N = C
/ \ / \HN Cu NH 2 K®· 4 H9O
\ / \ /C = N N — C
4 / \ \\. Οθ H H O _
Figure 4.3.5. Proposed structure of the cellulose-biuret-copper complex (Jayme andLang, 1957).
Structural details of the cellulose complex formed in the solvent have not beenpublished so far, but a ligand exchange with formation of a diolato complexseems to be probable also here.
4.3.2.2 Other aqueous cellulose solvents based on transitionmetal-amine complexes
The chemistry of cellulose-metal complexes received an important impetus inthe middle of this century, when Jayme and his group (Jayme, 1971) discoverednumerous new cellulose solvents based on cationic complexes of zinc, cadmium,cobalt and nickel, with ethylene diamine or ammonia as ligands. An overview ofthese solvents and their active species is presented in Table 4.3.2.

4.3.2 Chemistry of cellulose-metal complex formation 79
The efficiency of these solvents is mostly lower than that of the copper-basedones, and a higher metal concentration is required to get e.g. a dissolving pulpinto solution. So, for example, a cobalt concentration of about 70 g/1 or a zincconcentration of about 80 g/1 are mentioned as optimal in the publications ofJayme's group, compared with about 15 g copper in the case of a Guam solution.The solvent power of Nioxam was found to increase with the nickel as well aswith the NH3 content of the system, ca. 1.5 % Ni (at high ammonia concentra-tion) and 15 % NH3 (at high nickel concentration) representing the minimumvalues for dissolution (Jayme and Neuschäffer, 1955). From a cellulose solutionin Nioxen, a precipitate with a Ni-to-AGU ratio of 0.87 and an ethylene dia-mine-to-Ni ratio of up to 2.7 could be isolated by precipitation with /i-propanol(Jayme and Neuschäffer, 1955), indicating a binding of about one Ni cation fromthe solution to each monomer unit of cellulose. According to Hoelkeskamp(1964) no real complex formation occurs with Nioxen.
More detailed studies on the relation between solvent preparation and compo-sition, and the solvent power, as well as on the mode of cellulose-solvent inter-action have been performed with Cadoxen, which, as a colorless solvent ofrather high solvent power, found wide application in the analytical characteriza-tion of cellulosic products. Figure 4.3.6 illustrates the dependency of solventpower on solvent composition and Cd starting compound.
Table 4.3.2. Transition metal complex solvents for cellulose.
Solvent Active species
Guam [Cu (NH3)4](OH)2
Cuen [Cu (H2N-(CH2)2-NH2)2](OH)2Cupren [Cu (H2N-(CH2)3-NH2)2](OH)2Pd-en [Pd (H2N-(CH2)2-NH2)](OH)2
Cooxen [Co (H2N-(CH2)2-NH2)2](OH)2
Zincoxen [Zn (H2N-(CH2)2-NH2)2](OH)2
Cadoxen [Cd (H2N-(CH2)2-NH2)3](OH)2
Nioxam [Ni (NH3)6](OH)2
Nioxen [Ni (H2N-(CH2)2-NH2)3](OH)2
Nitren [Ni (NH2CH2CH2)3N](OH)2
Regarding the binding of Cd to cellulose, Jayme originally presumed by anal-ogy to Cuam or Cuen, a complex interaction with the C-2 and C-3 hydroxygroup of the AGU. From conductivity measurements in dependence on cadmiumand cellulose concentration, a rather strong binding of Cd to cellulose was con-cluded with a sterically feasible sorption of 2 Cd/3 AGU (Hugglins, 1987). Di-alysis experiments, on the other hand, as well as 13C and 113Cd measurements,(Bain et al., 1980) and 13C NMR studies (Nehls et al., 1995), led to the conclu-

80 4.3 Metal Complexes of Cellulose
sion that Cadoxen is a noncoordinating cellulose solvent forming no chelatecomplex with the diol moiety of the AGU, but interacting with cellulose ac-cording to an acid-base principle similar to aqueous alkali. This view is corrobo-rated by the increase in solvent power observed on enhancing the Cd concentra-tion and on adding some NaOH to the system. This is confirmed in a recentpublication (Burger et al, 1995), who assumes for all the transition metal-aminecomplex solvents listed in Table 4.3.2, not a diolato complex formation withcellulose, but rather an acid-base interaction similar to alkali-cellulose forma-tion, with an additional chain separating effect of the voluminous amine-complex cation persisting as a homoleptic cationic complex in the system.
80
•"60
.2
l202-
Cellulosesolubility
Completelysoluble
Partiallysoluble
Insoluble
Initial cadmium compoundCadmium Cadmium hydroxide Basic cadmium
oxide low inchloride chloride
D
D
Area in which lie thecompositions that dissolv«cellulose at 2O0C
6 8 10 12Cadmium content [wt%]
U 16
Figure 4.3.6. Solubility of cellulose in Cadoxen solutions of various compositions(Jayme, 1971).
The same publication describes two new transition metal complex solventswhich really dissolve the cellulose by formation of a diolato complex: a solventsystem composed of nickel nitrate with a slight molar excess of tris-2-aminoethylamine ftren') and the double molar quantity of NaOH-dissolvedcellulose to a blue viscous solution under formation of a diolato complex withthe structure presented in Fig. 4.3.7.
This heteroleptic complex with a tetradentate amine ligand, exhibited strikingdifferences to the cellulose Nioxen system, for example the Ni-cellulose-trencomplex was decomposed by addition of ethylene diamine with precipitation ofcellulose due to the preference of nickel for amine ligands. The Ni-tren systemproved to be an effective solvent also for other polysaccharides, e.g. amylose orchitosan.

4.3.2 Chemistry of cellulose-metal complex formation 81
NH5
<^yFigure 4.3.7. Structure of cellulose units in Ni-tren (Burger et al., 1995).
An interesting new metal complex solvent suitable for NMR and light scat-tering work on cellulose has been presented on the basis of [Pd(II)(en)]2+(OH")2
as the active species (Airichs et al., 1998). In the absence of excess ethylenediamine cellulose is dissolved slowly but completely with formation of a het-eroleptic diolato complex (Fig. 4.3.8).
Figure 4.3.8. Structure of cellulose units in Pd-en (Burger et al., 1995).
Due to the high binding strength of Pd(II) for amine ligands and its rather low af-finity for the CT ligand, this cellulose complex persists even in a strongly alkalinesolution without crosslinking to a Normann compound like bisdiolato complex.
In conclusion, some more general reasoning on transition metal complex for-mation and cellulose dissolution shall be summarized: the systems in questionare governed by pH-dependent coordination equilibria on the one hand, and asplitting and reformation of cellulosic hydroxy bonds on the other. Systemsleading to deprotonation of cellulosic hydroxy groups can act as solvents only ifcrosslinking between the cellulose chains via Normann compounds like bisdio-

82 4.3 Metal Complexes of Cellulose
lato structures is avoided. In the case of transition metal-amine complex sol-vents this can be achieved along two routes, i.e.(i) persistence of a voluminous homoleptic cationic complex, with amine ligandsacting as a spacer between cellulose chains like the cationic part of a tetraalkylammonium hydroxide ion dipole, as realized e.g. in the solvent Cadoxen;(ii) formation of a heteroleptic complex with a diolato ligand, i.e. the deproto-nated C-2 and C-3 hydroxy groups of cellulose on the one hand, and one ormore amine ligands on the other, requiring a balanced affinity of diol and amineligands to the central atom and realized e.g. in the Cuen solvent.
In the latter case, precipitation can occur at high alkalinity by interchaincrosslinking via bisdiolato complex formation due to substitution of the residualamino ligands by a second deprotonated diol moiety. Despite the general breakdown of the inter- and intramolecular hydrogen bond system of cellulose duringswelling and dissolution, new, strong isolated hydrogen bonds of the OH— O" type,bridging adjacent AGU, can be formed with complexed deprotonated hydroxygroups, resulting in a remarkable stiffening of the cellulose chains in solution.
4.3.2.3 Transition metal-alkali-tartaric acid complexes of cellulose
An effective, strongly alkaline solvent for cellulose, composed of Fe(OH)3, so-dium hydrogen tartrate and sodium hydroxide in aqueous solution, was de-scribed (Verbürg, 1951), assuming a complex with the structure depicted in Fig.4.3.9, which easily hydrolyzes in water, but is stabilized by an excess of NaOH.
By a systematic variation of the component ratio, the rather small area of sol-vent composition, dissolving even high DP cellulose rapidly and completely,could be defined (Fig. 4.3.10).
o-c
YHC
HO/ \O ~°
O5O
I
\.CH
O = (T\c
6 Na®
Figure 4.3.9. Probable structure of the FeTNa complex with a ratio of Fe(OH)3/tartaricacid/NaOH of 1 : 3 : 6 (Jayme, 1971).

4.3.2 Chemistry of cellulose-metal complex formation 83
20 40 60 80tortaric acid [%]
Figure 4.3.10. Location within the system Fe(OH)3/tartaric acid/NaOH of the FeTNacomplex capable of dissolving cellulose, shaded area indicates the effective compositionfor cellulose dissolution (Jayme, 1971).
A total solids content of about 350 g/1 including an excess of NaOH of 1-3mol/1 was recommended as an effective cellulose solvent.
Besides the original procedure (Jayme and Bergmann, 1954) starting fromfreshly precipitated, purified Fe(OH)3, several more convenient routes to preparethe FeTNa solvent have been recommended. They either employ the isolatedcomplex [(C4H3O^)3Fe]Na6 or an isolated ferric tartaric acid with the formula[(C4H2O6)Fe]H as a storable intermediate, or proceed via a direct mixing offerric salt, sodium tartrate, NaOH and water to give a solvent ready for use. Asuitable system is composed e.g. of 190 g/1 FeTNa complex, 5 g/1 excess sodiumtartrate and 1.5 mol/1 excess NaOH for stabilization (Valtasaari, 1957). A proce-dure successfully practiced by us in preparing the FeTNa solvent is presented inthe Appendix of Vol. 2.
FeTNa solutions with a complex concentration up to 480 g/1 and an adequateamount of excess NaOH can be easily prepared via tartaric ferric acid. The mo-lar ratio of tartrate to ferric ion in the complex can in principle be varied be-tween 1 : 1 and 4.5 : 1, with an optimum for cellulose dissolution at about 3 :1 ,and its significant decrease in solvent power at the highest molar ratio (Bayer etal., 1965). The presence of nitrate ions (from ferric nitrate as the starting mate-rial) was found to decrease the solvent power, to prolong the time required fordissolution of cellulose, and to enhance the sensitivity to cellulose degradationby air oxygen. The intrinsic viscosity of cellulose dissolved in FeTNa depends,according to Moiseev and Ivanov (1984), on the tartaric acid concentration, themolar ratio of tartaric acid to ferric ion, and on the total ionic strength of thesystem. Cellulose degradation by air oxygen is negligibly small in a suitablyprepared FeTNa solution, with the rate constant of chain cleavage amounting to

84 4.3 Metal Complexes of Cellulose
only about 1/10 of that observed in Cuen. A considerable faster chain cleavagetakes place, however, in FeTNa, with oxidized celluloses containing carbonylgroups due to the high alkalinity.
Cellulose-FeTNa interaction was at first considered as an enhanced alkaliswelling of the polymer without a chemical reaction (Jayme and Verbürg, 1954),but in the meantime experimental evidence has been acquired in favor of a com-plex binding of the AGU to the central Fe atom. A strong chemical interactionbetween cellulose and FeTNa is indicated also by an enthalpy of -11 and -13 kJ/hydroxy group reported in a thermochemical study of Ivanov et al. (1984).A comparison of the 13C NMR spectra of aqueous solutions of sodium tartrate,FeTNa, and cellulose dissolved in FeTNa (Nehls et al., 1995) revealed a down-field shift of about 15 ppm for the 13C signals of the tartrate due to complexationto ferric ions, and a downfield shift of the same magnitude was observed for 13Csignals of cellulose after dissolution without further changes in shape and posi-tion of the tartrate signals. This downfield shift of the cellulose signals in com-parison with those obtained in e.g. aqueous NaOH or tetraalkyl ammonium hy-droxides indicates a strong chemical interaction. According to an assumption(Bayer et al., 1965) based on the interpretation of its own results, the bindingtendency of the central iron atom for glycol units is 'still unsatisfied' in the pres-ence of three tartrate molecules, and a maximal uptake of 4.5 glycol units ispostulated. This, however, seems rather improbable from the viewpoint of to-days' inorganic coordination chemistry. In the above authors' opinion, this re-sidual glycol binding tendency of the FeTNa solution, with a ratio of threemolecules of tartrate per Fe(III), leads to an insertion of C-2/C-3 hydroxy groupsof AGU into the complex, and the cellulose is dissolved by formation of a newcomplex with the FeTNa complex already existing. A different viewpoint ispresented by Dale (1980) assuming a competition between tartrate moleculesand the glycol moiety of the AGU for the maximally six coordination sites of theiron central atom.
Replacement of Na+ by K+ in the FeTNa complex considerably diminished,but in principle maintained the solvent power of the system for cellulose,whereas a replacement of the tartrate ligands by chemically related compoundssuch as oxalic acid, lactic acid, citric acid, salicylic acid, or glycol, did not resultin cellulose-dissolving systems. A FeTNa-analogous copper complex with tar-trate and NaOH as further components in aqueous solutions did not dissolvecellulose despite a broad variation of the component ratio.
The FeTNa solvent is supposed to dissolve cellulose to a molecularly dis-persed system up to a level of ca. 0.3 % of polymer, to give a solution of re-markably high viscosity, while at a polymer concentration above 2 % FeTNacellulose gels are formed. Besides a widespread application of the FeTNa sol-vent in the analytical characterization of cellulosic materials, it has been used byPlisko and Danilov (1962) as a medium for etherification of dissolved cellulose

4.3.2 Chemistry of cellulose-metal complex formation 85
with phenyl sulfonic acid methyl or ethyl ester at 30-70 0C and 2.5 % polymer con-centration, arriving at methyl- or ethylcelluloses soluble in 1 % aqueous NaOH.
According to Seger et al. (1996), a dilute solution of cellulose in FeTNashows the behavior of a molecularly dissolved semirigid polymer. Above theoverlap concentration c*, fibrillar chain aggregates were observed in contrastwith amylose, forming a loosely entangled chain molecule (Fig. 4.3.11). Thisdifference is assumed to be caused by different hydrogen bond formation be-tween the macromolecules.
Figure 4.3.11. Structure of a highly concentrated solution of cellulose (left) and amylose(right) in FeTNa (Seger et al., 1996).
4.3.2.4 Interaction of cellulose with metal hydroxo compounds
Swelling and dissolution of cellulose in aqueous NaOH can be impeded as wellas enhanced in the presence of metal hydroxo compounds, in dependence onsubstance added. These opposite effects already indicate two different modes ofaction: addition of cuprate anions diminishes swelling and solubility and finallyresults in the formation of the Normann compound already mentioned, whichalso can be obtained via an elimination of ammonia ligands by addition of so-dium hydroxide to a solution of cellulose in Guam. A crosslinking of cellulosechains by formation of a bisdiolato complex between copper as the central atomand deprotonated glycol moieties of the AGU results in a compound that isswellable but not soluble in aqueous alkali, and resembles somewhat a sodiumcellulose (see chapter 4.2). The Normann compound shows a WAXS pattern ofits own described by Normann (1906) and Trogus and Hess (1929), and accord-ing to (Burchardt et al., 1994; Burger et al., 1995) is represented by the formulaNa2[Cu(C6H8O5)2] (H2O)^. Similar crosslinking diolato complexes are probablyformed between cellulose and Fe(III), but not with Fe(II): Jacopian et al. (1975)reported Fe(III) sorption exceeding the carboxyl content by about one order ofmagnitude for a slightly oxidized cellulose sample, while the Fe(II) sorptionalways remained significantly below the level of the carboxyl content. In con-trast with Fe(II), the Fe(III) could not be desorbed even at a pH value of 2, butthe iron content decreased significantly at this pH after partial reduction of thecellulose-bound Fe(III) to Fe(II). Furthermore, it is well known that aqueous

86 4.3 Metal Complexes of Cellulose
sodium hydroxide can be made free of traces of Fe(III) by filtration through apad of cotton fibers.
On the other hand, the solubility of wood pulp in aqueous alkali of maximumswelling power (8-11 % of NaOH) can be markedly increased up to complete-ness by addition of ZnO to the lye at a level of 1-5 %, resulting in the formationof zincate anions. This increase in cellulose solubility studied by various groups(e.g. Sharkov and Amosov, 1975; Garves, 1974; Ramalingam, 1979) can befurther enhanced by addition of urea, due to the hydrotropic action of this com-pound, and is observed not only with cellulose itself but also with low substi-tuted cellulose ethers and esters, including cellulose xanthogenate and cellulosecarbamate (Lang and Laskowski, 1990). Similar but usually smaller effects oncellulose solubility have been reported for various other hydroxo compounds,e.g. aluminate, plumbate and berrylate, added to the sodium hydroxide lye.
Regarding cellulose zincate interaction, no preferential binding or transport of zincat the cellulose moiety was observed in tracer studies (Borgin, 1949; Borgin andStamm, 1950), contradicting formation of a stable complex. Much more likely is aninteraction via strong hydrogen bonds between the cellulose chains and the zin-cate, with the anionic species, i.e. Zn(OH)4
2" or ZnO22- being employed as vo-
luminous additional spacers between the macromolecules. This viewpoint iscorroborated by the fact that a low temperature favors an increase in solubility,and that this increase occurred also with KOH and LiOH as alkali hydroxides.
4.3.2.5 Interaction of cellulose with inorganic salts
As observed by von Weimarn (1912) and by Katz and Derksen (1931), variousneutral salts in concentrated aqueous solution are able to swell and even to dis-solve cellulose. The solvent power of these systems, however, was found to belimited. A rather low DP and degree of lateral order, and a temperature above50 0C favored chain separation. As suitable cations Li+, Ca2+ and Zn2+, as ani-ons SCN~, I", [HgI4]
2" and [ZnCl4]2" were explicitly mentioned. Lithium iso-
thiocyanate obviously represents an especially suitable cation-anion combina-tion, for which even the formation of a crystalline addition compound with awell-defined WAXS pattern of its own has been reported.
A common feature of all these solvent-active salts is the combination of astrongly hydrated and strongly polarizing cation with a rather voluminous weaklyhydrated and easily polarizable anion, and several of the solvent-active combina-tions are known to form oxonium compounds with alcoholic hydroxy groups. Forexample, LiCl in a water/n-butanol system exhibits a distribution of the salt be-tween the two liquid phases caused by the competition of water and alcohol mole-cules for a site in the solvation shell of the Li+ ion. Cellulose dissolution by the iondipoles of e.g. LiSCN in a concentrated aqueous solution can be understood as a

4.3.2 Chemistry of cellulose-metal complex formation 87
participation of cellulosic hydroxy groups in the solvation of the Li+ cation, withthe voluminous anionic part of the ion dipole acting as a chain-separating spacer.
The dissolution of cellulose to give a macroscopic homogeneous system in themelt of various isothiocyanates at a temperature between 100 and 200 0C hasbeen described (Lukanoff et al., 1983). A melt composed of NaSCN and KSCNwas found to dissolve even high DP cellulose samples in the presence of a smallamount of Ca(SCN)2-SH2O or of DMSO. The best effects were obtained alsohere with LiSCN. The polymer samples regenerated from these melt solutionsexhibited the WAXS pattern of cellulose II. The LODP of the regenerated sam-ple was found between 40 and 70 depending on the melt system, and indicatingespecially in the NaSCN/KSCN melts a state of dispersion above the macromo-lecular level. The spinning of threads from all these isothiocyanate salt systems,including aqueous ones, met with little success due to the strong osmotic forcesinhibiting the formation of a well-ordered and oriented thread structure.
Lithium-salt-based cellulose solvents can also be obtained by dissolving LiCl orLiBr in a suitable dipolar aprotic liquid, especially in DMA, but usually require anadequate preactivation of the cellulose sample. Table 4.3.3 presents an overviewtaken from Morgenstern and Kammer (1996) on the solvent systems in question,which, according to 13C NMR studies (Nehls et al., 1994), have to be classified asnonderivatizing systems dissolving the polymer via hydrogen-bond complexation.
The decisive point is an interaction between the intra- and intermolecularhydrogen-bond system of the cellulose, and the hydrogen-bond complex be-tween e.g. LiCl and DMA, in connection with the fact that Li+ and Cl~ are fairlytightly connected, forming an ion dipole with a strongly solvated cationic and aweakly solvated anionic site. Three possible structures of this primaryDMA/LiCl complex are depicted in Figs. 4.3.12-14. A model implies a strongercomplex formation with DMA than with DMF (Fig. 4.3.14).
A-C7
\ /N
/ \HgC
Figure 4.3.12. Structure of the DMA/LiCl complex (Morgenstern and Kammer, 1996).

88 4.3 Metal Complexes of Cellulose
Table 4.3.3. Cellulose solvents of a Li-salt and a dipolar aprotic liquid.
Solvent Cellulose activation Typical composition Reference(wt%)a
DMA/LiCl
NMP/LiCl
DMF/LiCl
DMEU/LiCl
DMPU/LiCl
DMA/LiBr
NMP/LiBr
HMPT/LiCl
DMSO/LiCl
All known methods
All known methods
Swelling in liquidammonia, followedby solvent exchangeHeating in the sol-vent;Swelling in water,followed by solventexchangeSwelling in water,followed by solventexchangeHeating in the solvent
Heating in the solvent
Heating in the solvent
Solvent exchange
Large variability
6/8.5/85.5
3/10/87
2/5.5/92.55/5/90
3.5/5/91.5
3/20/77
1/18/81
5/11/84
3/8/89
Turbak et al.,1980McCormickand Callais,1987El-Kafrawy,1983Morgensternand Kammer,1996Herlinger andHengstberger,1985
Herlinger andHengstberger,1985Turbak andSakthivel, 1990Turbak andSakthivel, 1990Turbak andSakthivel, 1990Petrus et al.,1995
a The composition is given in the sequence: cellulose/salt/solvent.Abbreviations: DMEU, dimethylethylene urea; DMPU, dimethylpropylene urea;HMPT, hexamethylphosphoric acid triamide.
A binding of up to 4 DMA molecules per Li+ is assumed by Morgenstern andKammer (1996) and Herlinger et al. (1990). In these publications the authorsresumed the various structural models proposed in the literature for theDMA/LiCl/cellulose complex and favor the model shown in Fig. 4.3.13, empha-sizing the tight ion pairing between Li+ and Cl~ in this aprotic system.

4.3.2 Chemistry of cellulose-metal complex formation 89
HSyT N
CeII-O SCI θ
Figure 4.3.13. Proposed structure of the cellulose/DMA/LiCl complex.
From the viewpoint of coordination chemistry, both these structures are con-sidered more or less hypothetically, and the structure depicted in Fig. 4.3.14 isconsidered more probable.
H3C\ Φ /~
/N=\H3C R
= H, CH3
Figure 4.3.14. Most probable structure for the DMA/LiCl and DMF/LiCl complex.
Li+ obviously interacts strongly with cellulosic hydroxy groups, as indicatedby the continuous shift of the 7Li NMR signal with increasing cellulose concen-tration. A hydrogen-bond-complex ligand exchange according to the equilibrium
CI-Li-(DMA)x+ Cell - OH = Cl -Li -(CH -Cell) (DMA)x-1+DMA
has been proposed as the driving force for cellulose dissolution. This ligand-exchange mechanism in the coordination sphere of Li+ requires an adjusted me-dium strength of the primary complex between the lithium salt and the dipolaraprotic liquid and an adjuvating action of the Cl~ site of the dipole for weaken-ing the hydrogen-bond system of the cellulose by its basicity. Furthermore, anadequate spatial structure of the primary solvent complex is necessary for fittingcellulose hydroxy groups into the coordination sphere. The better solvent actionof DMA/LiCl in comparison with DMF/LiCl could be qualitatively explained bythis reasoning. More detailed and more quantitative information on the dissolu-tion process of cellulose in the systems considered are expected from compre-hensive thermodynamic studies.

90 4.3 Metal Complexes of Cellulose
4.3.3 Supramolecular and morphological aspects ofcellulose-metal complex formation
Cellulose-metal complex formation has quite predominantly been studied inconnection with cellulose dissolution and cellulose solution properties, implyinga thorough interaction with all polymer chains irrespective of supramolecularorder and fibrillar architecture. In agreement herewith, a quite well-ordered cel-lulose II is generally obtained on regeneration of the polymer from a metal com-plex solution of cellulose I. In the very few cases of solid complex compoundsbeing described, e.g. the copper-based Normann compound or a cellu-lose/LiSCN adduct, these compounds exhibit a WAXS pattern of their own. Inan recent publication Miyamoto (1996) emphasizes the effect of the OH~ con-centration in a Guam solution on the structure of the regenerated cellulose: whileat low OH~ concentration only the amorphous regions are affected, a high OH~concentration leads to a crosslinking of cellulose chains via copper atoms evenin the crystallites.
While the copper-based complex solvents and FeTNa dissolve even high DPcellulose samples completely, the solvent power appears to be limited in someother more weakly complexing solvents like Zincoxen. In the case of Cadoxen, apreswelling of the cellulose sample in an aliphatic amine or with urea in DMSOwas reported to be beneficial for the subsequent dissolution (van ZyI, 1983).DMA/LiCl and related systems frequently require an adequate preactivation ofthe cellulose sample by, e.g., preswelling in water and subsequent solvent ex-change for achieving a homogeneous cellulose solution free of gel particles.
The supramolecular and morphological structure of cellulose regeneratedfrom metal-amine complex solutions is obviously significantly influenced bythe type of metal complex, as demonstrated by the change in cellulose mem-brane structure and performance due to addition of a small amount of Zincoxencellulose solution to a spinning dope of cellulose in Guam (Zhang et al, 1991).By Ramalingam (1979) the effect of addition of zincate to a viscose spinningsolution on filament structure and properties is reported, which, however, maybe primarily caused by an effect of the Zn2+ ions on cellulose xanthogenate de-composition in the acid spinning bath.
At a metal-complex concentration too low for cellulose dissolution, a limitedswelling of the polymer takes place, often revealing details of the fiber morphol-ogy, and resulting in an increase in accessibility for e.g. a subsequent enzymaticdegradation (Hamilton et al., 1984). These swelling processes in various com-plex solvents, especially in Cuen, Cadoxen and FeTNa, have been amply em-ployed to study the morphological structure of native cellulose fibers or the in-fluence of various production parameters on the morphology of rayon filamentand staple, but little information only is available on the course of swelling ofthe same sample in different metal-complex systems. The rather slow swelling in

4.3.3 Cellulose-metal complex formation 91
FeTNa compared with Cuen or Cadoxen, favoring qualitative and quantitativeinvestigation on swelling kinetics, is mentioned in various publications (e.g.Evans and Jeffries, 1970). By Casperson et al. (1969) the swelling of normal-wood and tension-wood cellulose in phosphoric acid on the one hand, and Cuen,Cadoxen and FeTNa on the other, has been compared. The solvent power of thesystems investigated, with the dissolved part of the cellulose fibers at the end ofthe swelling process used as the criterion, increased in the order phosphoric acid< Cadoxen < Cuen < FeTNa. The morphological swelling patterns differedlargely between H3PO4 and the metal-complex systems, but to a small extentonly within the latter group. Some difference was found with regard to the num-ber of fibers exhibiting ballooning, which was higher with FeTNa than withCuen. Worth mentioning is the pronounced heterogeneity of morphologicalswelling pattern within the same sample in the same medium, especially in thefast-swelling Cuen, which may possibly be traced back to a different level ofprimary wall destruction during pulping and bleaching and/or to the presence ofearly wood and late wood within one sample.
The morphological changes connected with the swelling of cellulose fibers(cotton, wood pulp) in FeTNa on the one hand, and DMA/LiCl on the other,have been investigated by Unger et al. (1995) using light microscopy, and byPiontek et al. (1996) using electron microscopy. By Unger et al. (1995) thecourse of swelling was assessed by a combination of phase-contrast microscopy,videographic image storage and computer-aided image processing, employing asquantitative criteria the average rate of increase in fiber thickness and the timeinterval to rupture of the fibers fixed at both ends. In FeTNa, swelling proceededmuch faster than in DMA/LiCl, the rate of swelling surpassing that of dissolu-tion, and an especially strong swelling of the S2 layer was observed in connec-tion with a considerable resistance of the primary wall and the Sl layer to dis-solution (see chapter 2.1). With DMA/LiCl on the other hand, swelling and dis-solution proceeded simultaneously across the whole fiber, accompanied by aloosening and widening of the fibrillar network.
Furthermore, a differentiation between various pulp samples, as well as cor-relations between swelling data and those of supramolecular structure and poredimensions, became much more clearly visible in FeTNa than in DMA/LiCl.The differences in mode of swelling in both systems are probably caused pre-dominantly by the different routes of polymer-solvent interaction on the mac-romolecular level, but differences in osmotic pressure within the fiber may alsoplay some role. SEM and TEM investigations by Piontek (Piontek et al., 1996)on the initial stage of spruce pulp and !inters dissolution in FeTNa and inDMA/LiCl are in agreement with the above statements. Interaction withDMA/LiCl is characterized by a lamellar separation in the case of cotton Untersand by the appearance of isolated fibrils of the spruce pulp.

92 43 Metal Complexes of Cellulose
4.3.4 Properties of cellulose-metal complexes
In most of the systems considered here, cellulose-metal complex formation isconnected with dissolution of the polymer, and thus the properties of these cel-lulosic compounds have been quite predominantly investigated in the state ofsolution, which is also the basis for their industrial and scientific application.Isolation of cellulose-metal complexes in the solid state by precipitation withlower aliphatic alcohols has been described in some cases, but the question re-mains open of whether or not the composition of the compound remains thesame as in solution.
Cellulose solutions in systems based on copper, iron, cobalt or nickel as thecentral atom are deeply colored, limiting their applicability to physicochemicalinvestigations of the polymer in solution. Cellulose in metal-complex solventsusually exhibits a high solution viscosity due to chain stiffening effects of poly-mer-solvent interaction. The outstanding high solution viscosity of cellulose inFeTNa and in some nonaqueous salt-containing systems is additionally caused bythe high viscosity of the solvent itself, besides a rather extreme chain stiffening.This is demonstrated in Table 4.3.4 by the intrinsic viscosity ratios of some metal-complex solvents in comparison with cellulose carbanilate in acetone. Worth men-tioning in connection with these results (Linow et al., 1972) is the different be-havior of samples of cellulose I in comparison with samples of regenerated cellu-lose (cellulose II). This indicates an influence of supramolecular structure on thestructure of the cellulose-metal-complex solution. The relationship between intrin-
sic viscosity and molar mass or intrinsic viscosity and DP according to [^=
has been presented already in Table 3.1 in chapter 3 (Volume 1).
Table 4.3.4. Averaged ratio of intrinsic viscosity ofcellulose cellulose I samples, DP 670-11800 in metalcomplex solvents: [^!complex to intrinsic viscosity ofcellulose tricarbanilates [η]carbanilate (Linow et al., 1972).
Metal complex Wcomplex/Wcarbanilatesolvent
Guam 1.19Cadoxen 1.19Cuen 1.33FeTNa 1.65
All the cellulose-metal complexes considered here exhibit high stability in anexcess of the solvent, but are decomposed by addition of a large amount of wa-ter. Acidification of the aqueous alkaline systems like cellulose/Cuen or cellu-lose/FeTNa results in a rapid destruction of the complex and a precipitation of

4.3.5 Application of cellulose-metal complexes 93
the polymer in the modification of cellulose II. Oxidative chain degradation ofcellulose dissolved in metal complex solvents plays a significant role only in thecopper-based systems, where it is triggered by the copper-catalyzed oxidation ofNH3 or amine ligands to e.g. nitrite or oxime groups.
The toxicity of the metal-complex solvents is mainly determined by the tran-sition metal employed and is rather high in the case of Cadoxen, which has to betaken into account on handling this solvent in the analysis and characterizationof cellulosics.
4.3.5 Application of cellulose-metal complexesMetal complex solvents for cellulose find wide application in the processing andin the characterization of cellulose and cellulosic products along the routes of(i) formation of threads or films of regenerated cellulose by decomposition ofthe complex and precipitation of the polymer;(ii) covalent functionalization of cellulose under homogeneous conditions insome of the solvent systems in question;(iii) physicochemical characterization of cellulose on the macromolecular levelespecially with regard to average molar mass, molar mass distribution and chainstiffness;(iv) assessment and characterization of foreign substances in cellulosic products.
4.3.5.1 Filament and film formation from cellulose-metal complexsolutions
After the spinning of cellulose filaments from Guam solution had been estab-lished as an industrial process at the end of the previous century, other cellulose-metal complex systems have been studied for this purpose too, for example cel-lulose dissolved in FeTNa or in DMA/LiCl. But so far none of these investiga-tions have reached the pilot scale due to technological, economical or ecologicalproblems. The Guam process, on the other hand, still keeps its mark in the pro-duction of cellulose filament, staple fiber and membranes, despite a significantreduction in production capacity during the last decades. Filament spinning bythe Guam process shall now be discussed briefly (Krässig et al., 1986). Bleachedcotton !inters (DP 1000-1200) or refined wood pulp with low hemicellulosecontent (DP 800-1000) are fluffed and reacted with cupric hydroxide or a basiccopper salt and concentrated aqueous ammonia to give a highly viscous solutionwith a viscosity of about 200 Pa s, employing adequately strong mixing blades.The solution is filtered through stainless steel sieves with a mesh size of 40-70μιη and de-aerated, loosing here a considerable amount of ammonia. Dependingon spinning technology, the solution contains 4-11 % of cellulose, 4-6% ofcopper and 6-10 % of ammonia. Copper input amounts to about 0.4 kg/kg of

94 4.3 Metal Complexes of Cellulose
cellulose, that of ammonia to 0.65-0.80 kg of anhydrous NH3/kg of cellulose.The process of cellulose dissolution can be controlled by the amount of ammo-nia. Reductants like Na2SO3 or glucose may be added to minimize oxidativecellulose chain degradation.
Filament spinning can be performed in a one-bath process, but usually a two-bath process is used. In the first bath the cellulose-copper complex is precipi-tated by desalted water as a soft precipitant, and in the second bath the celluloseis regenerated by the action of 1.5-3 % aqueous H2SO4 at 20-25 0C. Due to thestability and the very high stretchability of the filaments formed in the first bath,a funnel spinning process can be employed with the stretch applied to the fila-ments by the increasing streaming velocity of the bath (water at a temperature of35-45 0C) in the glass or plastic funnels (Krässig et al., 1986).
Total stretch applied amounts to 10000-15000 %, and rather large spinneretbores can therefore be used. The filaments emerging from the funnel are thenguided into the second bath. One-bath spinning is employed for cord filaments,sometimes with aqueous NaOH as the precipitant. The spinning speed amountsat present to about 150 m/min but an increase to more than 500 m/min appears tobe feasible with special conveying equipment for the freshly spun filaments. Forstaple fiber spinning through spinnerets with 2000-3000 bores, the ammoniacontent in the spinning dope is reduced by 1-2 % to avoid adhesive gluing offilaments. The after treatment, i.e. the depletion of the cellulose from salts andlast traces of copper and the sizing and drying, is performed here with a filamenttow or after cutting to staple. More than 99 % of the copper input and up to 50 %of the ammonia are recovered from the process.
Unique features of cellulose filaments spun from Guam are the higher finenessobtainable in comparison with viscose filaments and the silk-like gloss and silk-like handling due to the circular cross section and the smooth surface of thethreads. Hollow fibers spun from Guam solution are widely used as membranesfor hemodialyses. By Zhang et al. (1995) the spinning of cellulose/casein blendfilaments with cellulose peptide bonds from a Guam solution containing bothpolymers has been reported.
4.3.5.2 Covalent functionalization of cellulose dissolved in metal-complex systems
Cellulose etherification in the alkaline medium of copper-complex solvents hasbeen studied widely in the first half of this century (Henkel AG, 1959). Also anetherification of cellulose in FeTNa has been reported more recently (Plisko andDanilov, 1962). A new and very successful route to homogeneous etherificationas well as esterification of cellulose under aprotic conditions has been opened upby the discovery of the system DMA/LiCl as a solvent for cellulose. Relevantresults so far obtained will be presented in chapter 2.4 (Volume 1).

4.3.5 Application of cellulose-metal complexes 95
4.3.5.3 Characterization of cellulose in metal-complex systems
For a macromolecular characterization of cellulose chains in solution, the intrin-sic viscosity of cellulose samples at different DP levels has been determined invarious metal-complex solvents such as Guam, Cuen, Cadoxen or FeTNa and
compared with [77] of the corresponding cellulose carbanilates (Linow et al.,
1972). Relations between molar mass or DP and the intrinsic viscosity [η] were
established directly or indirectly by [77] comparison (see Table 3.1). Still widely
used is the Guam system with a copper content of about 13 g/1 and a NH3 con-tent of about 200 g/1, being a suitable solvent according to our experience. Forassessing the molecular weight distribution of cellulose samples, GPC tech-niques were adapted to FeTNa and to Cadoxen (Jayme, 1978), employing frac-togel as the stationary phase in the latter case and covering a DP range between400 and 2000. Information on molar mass distribution of cellulose samples wasalso acquired by fractional dissolution with FeTNa and with zincate in aqueousNaOH (Bergner and Philipp, 1986), in the latter case for the short-chain part ofpulp samples and regenerated alkali celluloses up to DP 200. Cadoxen wasfound to be very suitable for the macromolecular characterization of low-substituted alkali-stable cellulose derivatives like low DS carboxymethylcellu-lose not soluble in water or aqueous alkali. A technique for assessing the degreeof crosslinking in cellulose products has been developed by extracting a soluble'sol phase' with FeTNa as the solvent and determining the percentage of poly-mer in the sol phase and in the gel phase.
Numerous metal-complex systems, preferably Cuen, Cadoxen, FeTNa andrecently DMA/LiCl, served to revealed details of cellulose fiber morphology bystudying the course of swelling with optical microscopy or electron microscopy(see chapter 3, Volume 1). The course of fiber elongation underload in FeTNahas been employed for assessing the influence of various parameters of spinningand after-treatment of crimped viscose staple fibers on fiber structure and prop-erties (Hampe and Philipp, 1972). Philipp et al. (1984) developed a FeTNa sol-vent-based so-called reaction morphometry by assessing the change of number,size and shape of fiber particles with reaction time (time of dissolution) in astreaming fiber suspension by means of a light scattering technique, and em-ployed the data obtained for quantifying dissolution kinetics, revealing signifi-cant differences in the kinetic parameters between various pulp samples.
4.3.5.4 Determination of foreign substances in cellulosic productsby means of metal-complex solvents
Two different analytical routes have been successfully pursued here in recentyears: one consists of an optical or conductometric assessment of the particlenumber and the particle size of undissolved residues after dissolution of the eel-

96 4.3 Metal Complexes of Cellulose
lulose in Cadoxen or in FeTNa. Insoluble gels as well as inorganic impuritiesconsisting of e.g. SiO2 have been determined in this way in various celluloseproducts (Arnold et al., 1970 and 1971).
The second route consists of a spectrophotometric investigation of the origi-nally colorless Cadoxen solvent after dissolving a cellulose product containingalien substances soluble or colloidally dispersible in the solvent (SjOstrom andEnström, 1966). This technique has been adapted to assess the dye content incellulose threads and fabrics or to characterize wood pulps for paper manufac-ture with regard to their content of impurities, especially lignin (Jayme, 1971).
For further details concerning the applications surveyed briefly in sections4.2.5.1 to 4.2.5.4 the reader is referred to the comprehensive review given byJayme on applications of Cadoxen and FeTNa (Jayme, 1978).
4.3.6 Future problems of cellulose-metal complexresearch
Cellulose-metal complexes, as one of the oldest areas of cellulose chemistry,which was neglected for many decades, enjoyed a revival in recent years by theimpact of modern inorganic complex chemistry and by the discovery of numer-ous nonaqueous solvent systems composed of a dipolar aprotic liquid and e.g.lithium chloride. One important route of future research doubtless is the em-ployment of the cellulose chain as a macromolecular carrier for metals in variousbinding states, arriving at special functional polymers. This route of research canprovide new cellulose-based high-tech materials with interesting catalytic, opti-cal and magnetic properties, and also precursors for specially shaped inorganicmaterials obtained after thermal decomposition. Further progress can be ex-pected in understanding the principles and driving forces of complex formationand in preparing new classes of stable cellulose transition metal complexes. Re-garding industrial application, the Guam spinning process will keep its place dueto the unique textile properties of the filaments obtained and very probably willnot be substituted in the foreseeable future, neither by metal ion free alternativeprocesses nor by other metal-complex solvents. As an open question remains thefuture progress of research on neutral salt-based aqueous and nonaqueous sys-tems complexing cellulose via hydrogen bonds or by insertion of hydroxygroups into the solvation shell. The development of new processes for filamentspinning or film casting on the basis of future knowledge acquired here is ratherimprobable due to osmotic problems in regenerating structure formation and torecovery problems of the chemicals. But possibly solvents of this kind will re-ceive increasing interest as reaction media for special covalent cellulose de-rivatizations under homogeneous conditions of reaction.

References 97
ReferencesAirichs, R., Ballauf, M., Eichkorn, K., Hanemann, O., Kettenbach, G., Klüfers, P.,
Chem. Eur. J. 1998, 4, in press.Arnold, A., Philipp, B., Schleicher, H., Faserforsch. Textiltech. 1970, 27(9),
361-366.Arnold, A., Philipp, B., Schleicher, H., Faserforsch. Textiltech. 1971, 22(1), 41-
42.Bain, A.D., Eaton, D.R., Hux, R.A., Tong, J.P., Carbohydr. Rev. 1980, 84, 1.Baugh, PJ., Hinojosa, O., Arthur, Jr., J.C., Mares, T., /. Appl. Polym. Sei. 1968,
72(2), 249-265.Bayer, F., Green, J.W., Johnson, D.C., Tappi 1965, 48, 557.Bergner, Ch., Philipp, B., Cellul Chem. Technol. 1986, 20, 591-600.Borgin, K., Nor. Skogind. 1949, 3, 96.Borgin, K., Stamm, A.J., Z. Phys. Colloid Chem. 1950, 54, 772.Burchardt, W., Habermann, N., Klüfers, P., Seger, B., Wilhelm, U., Angew.
Chem. 1994, 706, 936-939.Burger, J., Kettenbach, G., Klüfers, P., Macromol Symp. 1995, 99, 113-126.Casperson, G., Philipp, B., Jacopian, V., Hoyme, E., Faserforsch. Textiltech.
1969, 20(2), 61-70.Dale, J., J. Polym. ScL, Polym. Chem. Ed. 1980,18, 3163-75.El-Kafrawy, A., Lenzinger Ber. 1983, 55, 44-47.Evans, G.M., Jeffries, R., /. Appl. Polym. Sei. 1970,14(3), 633-653.Gadd, K.F., Polymer 1982, 23, 1867-1869.Garves, K., Holzforschung 1974, 28, 168-171.Hamilton, TJ., Dale, B.E., Ladisch, M.R., Tsao, G.T., Biotechnol. Bioeng.
1984, 26(7), 781-787.Hampe, H., Philipp, B., Cellul Chem. Technol. 1972, 6, 447-471.Henkel AG, Patent, DT AS 1068685, 1959.Herlinger, H., Hengstberger, M., Lenzinger Ber. 1985, 59, 96.Herlinger, H., Grynaeus, P., Hirt, P., Koch, W., Hengstberger, M., Rembold, S.,
Günzel, K.H., Lenzinger Ber. 1990, 65-72.Hoelkeskamp, F., Papier (Darmstadt) 1964,18, 201-204.Hugglins, M.B., Wood Cellul. 1987, 119-126.Ivanov, A.V., Soholov, V.V., Tsvetkov, V.G., Poltoratskii, G.M., in Probl
Kalorim. Khim. Thermodin., Dokl. Vses. Konf., 10th, Emanuel (Ed.),Chernogolovka, USSR: Akad. Nauk SSSR, 1984, Vol. l, pp. 316-318.
Jacopian, V., Philipp, B., Mehnert, H., Schulze, H., Dautzenberg, H.,Faserforsch. Textiltech./Z. Polymerforsch. 1975, 26, 153-158.
Jayme, G., Verbürg, W., Reyon, Zellwolle, Andere Chem.-Fasern 1954, 32,193-275.

98 4.3 Metal Complexes of Cellulose
Jayme, G., Neuschäffer, K., Papier (Darmstadt) 1955, 9, 563.Jayme, G., Lang, F., Kolloid Z. 1957, 750, 5.Jayme, G., in Cellulose and Cellulose Derivatives, Bikales, N.M., Segal., L.
(Eds.), New York: John Wiley & Sons, 1971, pp. 381-410.Jayme, G., Papier (Darmstadt) 1978, 32(4), 145-149.Katz, J.R., Derksen, J.C., Red. Trav. Chim. Pays-Bas 1931, 50, 149; 736.Kettenbach, G., Klüfers, P., Mayer, P., Macromol Symp. 1997, 720, 291-302.Krässig, H., Steadman, R.G., Schliefer, K., Albrecht, W., in Ullmann's
Encyclopedia of Industrial Chemistry, Gerhartz, W., Yamamoto, Y.S.,Campbell, F.T., Pfefferkorn, R., Rounsaville, J.F. (Eds.), Weinheim: VCHVerlagsgesellschaft mbH, 1986, Vol. A5, pp. 375-418.
Kroon-Batenburg, L.M.J., Kroon, J., Nordholt, M.G., Polym. Commun. 1986,27, 290-292.
Kroon-Batenburg, L.M.J., Kroon, J., Nordholt, M.G., Papier (Darmstadt) 1990,44, 640-645.
Lang, H., Laskowski, L, CeIIuL Chem. Technol 1990, 25, 143-153.Linow, K.-J., Koura, A., Philipp, B., Schleicher, H., Faserforsch. Textiltech.
1972,23(7), 286-291.Lukanoff, B., Schleicher, H., Philipp, B., Cellul Chem. Technol 1983, 77, 593-
600.Maekawa, E., Koshijima, T., /. Appl Polym. Sei. 1990, 40, 1601-1630.McCormick, C.L., Callais, P.A., Polymer 1987, 28, 2317-2323.Miyamoto, T., Polym. J. 1996, 28(3), 273-81.Miyamoto, I., Matsuoka, Y., Matsui, T., Saito, M., Okajima, K., Polym. J.
1996,28,276-281.Moiseev, B.A., Ivanov, M.A., Khim. Drev. 1984, 2, 72-77.Morgenstern, B., Kammer, H.-W., Trip 1996, 4(3), 87-92.Nehls, L, Wagenknecht, W., Philipp, B., Stscherbina, D., Prog. Polym. Sei.
1994, 79, 29-78.Nehls, L, Wagenknecht, W., Philipp, B., Cellul. Chem. Technol. 1995, 29, 243-
251.Normann, W., Chem. Z. 1906, 20, 584.Petrus, L., Gray, D.G., BeMiller, J.N., Carbohydr. Res. 1995, 268, 319-323.Philipp, B., Linow, K.-J., Unger, E.W., Fischer, K., Anders, W., Zellst. Pap.
1984, 6, 203-207.Piontek, H., Berger, W., Morgenstern, B., Fengel, D., Cellulose 1996, 3, 127-
139.Plisko, E.A., Danilov, S.N., Zh. Prikl. Khim. 1962, 35, 2112.Ramalingam, K.V., Man-Made Text. India 1979, 22, 410-412.Schiff, H., Ann. 1898, 299, 238.Schweizer, E., /. Prakt. Chem. 1857, 72, 109-111.

References 99
Seger, B., Aberle, T., Burchard, W., Carbohydr. Polym. 1996, 31, 105-112.Sharkov, V.J., Amosov, V.Α., Tr. Vses. Nauchno-Issled. Inst. Tsellyul-Bum.
PromstL 1975, 65, 119-124.Sjöström, E., Enström, B., Sven. Papperstidn. 1966, 69, 469.Traube, W., Ber. Dtsch. Chem. Ges. 1911, 44, 3319.Trogus, C., Hess, K., Z Physikal Chem. 1929, B6, 1.Turbak, H.F., Hammer, R.B., Davies, R.E., Hergert, H.L., Chem. Tech. 1980,
70,51.Turbak, H.F., Sakthivel, A., Chem. Tech. 1990, 20, 444.Unger, E.W., Fink, H.-P., Philipp, B., Papier (Darmstadt) 1995, 49(6), 297-
307.Valtasaari, L., Pap. PUU 1957, 39, 243.van ZyI, J.D., Pap. PUU 1983, 65(4), 293-294; 296-298.Verbürg, W., Ph.D. Thesis, TH Darmstadt 1951.Weimarn, P.P.V., Kolloid-Z. 1912,11, 41.Zhang, L., Yang, G., Fang, W., /. Membr. Sei. 1991, 56(2), 207-215.Zhang, L., Yang, G., Xiao, L., /. Membr. Sei. 1995,103(1-2), 65-71.
4.4 Esterification of Cellulose
Esters of cellulose with inorganic and organic acids were the first covalentlymodified cellulose derivatives to be synthesized in the laboratory. Cellulosenitrate, cellulose acetate and cellulose xanthogenate had been produced on anindustrial scale already in the second half of the previous century and comprisetoday more than 90 % of the production capacity in the chemical processing ofcellulose (Table 4.4.1).
Table 4.4.1. Production capacity of commercial cellulose esters(average values of world production, t/a).
Ester Production capacity(t/a)
Cellulose xanthogenate 3200,000 (as intermediate)Cellulose acetate 850,000Cellulose nitrate 200,000
This chapter presents an overview of the general course of reaction, the routes ofsynthesis, the product properties, and the areas of application of cellulose estersof scientific and/or practical interest, without the claim of completeness. In-cluded is an abridgment of the technical processes in the case of cellulose ni-

References 99
Seger, B., Aberle, T., Burchard, W., Carbohydr. Polym. 1996, 31, 105-112.Sharkov, V.J., Amosov, V.Α., Tr. Vses. Nauchno-Issled. Inst. Tsellyul-Bum.
PromstL 1975, 65, 119-124.Sjöström, E., Enström, B., Sven. Papperstidn. 1966, 69, 469.Traube, W., Ber. Dtsch. Chem. Ges. 1911, 44, 3319.Trogus, C., Hess, K., Z. Physikal. Chem. 1929, B6, 1.Turbak, H.F., Hammer, R.B., Davies, R.E., Hergert, H.L., Chem. Tech. 1980,
70,51.Turbak, H.F., Sakthivel, A., Chem. Tech. 1990, 20, 444.Unger, E.W., Fink, H.-P., Philipp, B., Papier (Darmstadt) 1995, 49(6), 297-
307.Valtasaari, L., Pap. PUU 1957, 39, 243.van ZyI, J.D., Pap. PUU 1983, 65(4), 293-294; 296-298.Verbürg, W., Ph.D. Thesis, TH Darmstadt 1951.Weimarn, P.P.V., Kolloid-Z. 1912, 11, 41.Zhang, L., Yang, G., Fang, W., /. Membr. Sei. 1991, 56(2), 207-215.Zhang, L., Yang, G., Xiao, L., /. Membr. Sei. 1995,103(1-2), 65-71.
4.4 Esterification of Cellulose
Esters of cellulose with inorganic and organic acids were the first covalentlymodified cellulose derivatives to be synthesized in the laboratory. Cellulosenitrate, cellulose acetate and cellulose xanthogenate had been produced on anindustrial scale already in the second half of the previous century and comprisetoday more than 90 % of the production capacity in the chemical processing ofcellulose (Table 4.4.1).
Table 4.4.1. Production capacity of commercial cellulose esters(average values of world production, t/a).
Ester Production capacity(t/a)
Cellulose xanthogenate 3200,000 (as intermediate)Cellulose acetate 850,000Cellulose nitrate 200,000
This chapter presents an overview of the general course of reaction, the routes ofsynthesis, the product properties, and the areas of application of cellulose estersof scientific and/or practical interest, without the claim of completeness. In-cluded is an abridgment of the technical processes in the case of cellulose ni-
Comprehemive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

100 4.4 Esterification of Cellulose
träte, cellulose xanthogenate and cellulose acetate. The chapter starts with cel-lulose esters of inorganic acids, turning then to those of organic acids preparedby conventional esterification, as well as by special reactions. The esters withanalogues or derivatives of carbonic acid F^CC^, especially the cellulose xan-thogenates, are treated in a separate section with regard to their technical im-portance and their special position between inorganic and organic esters of cel-lulose.
4.4.1 Esters of cellulose with inorganic acidsAmong the numerous inorganic acids known today, only a few have been em-ployed as final products or as a reactive derivative in systematic studies of cel-lulose esterification, and a still smaller number of inorganic cellulose esters isproduced on a commercial scale. Especially of interest are some oxygen-containing acids of the elements nitrogen, phosphorus, sulfur and boron. Cellu-lose carbonates have not be isolated up to now despite many experimental stud-ies, obviously due to their instability (see section 4.4.2). Cellulose esters of oxy-gen-containing halogen acids have not been prepared until now, probably inconsequence of the explosion hazards expected, although a feasible route to acellulose perchlorate can be imagined via reacting the polymer with nitrosylperchlorate in DMF (see section 4.4.1.2). Cellulose esters with hydrogen acidsof halogens and of sulfur are known as halodesoxycelluloses and thiodesoxy-celluloses, but play a rather marginal role in cellulose chemistry. They will besurveyed in section 4.4.1.6 together with other types of desoxycelluloses.
Besides the formation of covalent esters, the existence of more or less well-defined addition compounds between cellulose and several strong inorganicacids has been described. Especially to be mentioned here is the so-calledKnecht compound formed by the action of 65 % aqueous HNC^ and an additioncompound with ca. 9 M aqueous perchloric acid. The isotherm of acid uptake independence on acid concentration resembles somewhat the step isotherm ofNaOH sorption (see chapter 4.2). Both these cellulose-acid addition compoundshave been studied earlier as examples of heterogeneous cellulose reactions(Knecht, 1904), especially by a joint evaluation of WAXS data and chemicalanalysis. These products are presently considered as oxonium compoundsformed in competition between alcoholic hydroxy groups and water moleculesin binding H+ according to
ROH + HoO+^=^ HpO + ROHo+
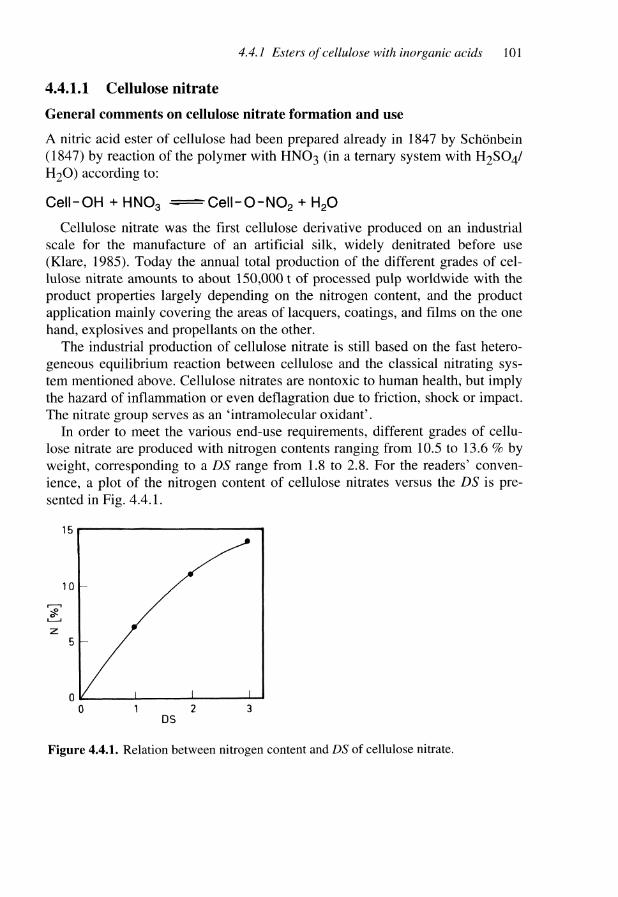
4.4.1 Esters of cellulose with inorganic acids 101
4.4.1.1 Cellulose nitrate
General comments on cellulose nitrate formation and use
A nitric acid ester of cellulose had been prepared already in 1847 by Schönbein(1847) by reaction of the polymer with HNÜ3 (in a ternary system with K^SC^/H^O) according to:
CeII-OH + HNO3 =^Cell-O-NO2 + H2O
Cellulose nitrate was the first cellulose derivative produced on an industrialscale for the manufacture of an artificial silk, widely denitrated before use(Klare, 1985). Today the annual total production of the different grades of cel-lulose nitrate amounts to about 150,000 t of processed pulp worldwide with theproduct properties largely depending on the nitrogen content, and the productapplication mainly covering the areas of lacquers, coatings, and films on the onehand, explosives and propellants on the other.
The industrial production of cellulose nitrate is still based on the fast hetero-geneous equilibrium reaction between cellulose and the classical nitrating sys-tem mentioned above. Cellulose nitrates are nontoxic to human health, but implythe hazard of inflammation or even deflagration due to friction, shock or impact.The nitrate group serves as an 'intramolecular oxidant'.
In order to meet the various end-use requirements, different grades of cellu-lose nitrate are produced with nitrogen contents ranging from 10.5 to 13.6 % byweight, corresponding to a DS range from 1.8 to 2.8. For the readers' conven-ience, a plot of the nitrogen content of cellulose nitrates versus the DS is pre-sented in Fig. 4.4.1.
15
10
1 2DS
Figure 4.4.1. Relation between nitrogen content and DS of cellulose nitrate.

102 4.4 Esterification of Cellulose
Subsequently, the chemistry of cellulose nitrate formation and decompositionwill be described in some detail, considering adequately also the role of cellulosestructure in this heterogeneous reaction preserving the fibrous state of the polymer,and then briefly surveying the industrial process of cellulose nitration as well asthe end-use properties and some main areas of application of the products.
The chemistry of cellulose nitrate formation and decomposition
Besides the classical 'nitrating acid mixture' of HNO3, H2SO4 and H2O, whichis still quite predominantly employed in the industrial manufacture of cellulosenitrate, numerous other systems have been studied (see Table 4.4.2) in order todevelop more favorable alternative production processes, or to prepare specialsamples of cellulose nitrates, and to elucidate the mechanism of the nitrationreaction. As an alternative process, avoiding sulfate group formation and facili-tating recovery of chemicals and waste disposal, the nitration with a ternarymixture of 45-94 % by weight HNO3, 3.7-34 % by weight Mg(NO3)2 and 3-21 % by weight H2O has been proposed, with the magnesium nitrate taking therole of a water binding agent (Hercules Powder, 1957 and 1962).
Table 4.4.2. Nitrating of cellulose under different conditions (Baiser et al., 1986)
Nitrating agent
HNO3/H2O < 75 % HNO3
78-85 % HNO3
86-89 % HNO3
90-100 % HNO3
HNO3/H2SO4/H2OHNO3/inorg. nitrateHNO3 vaporHNO3 vapor + NOx
HNO3/H3PO4/P2O5
N2O5
N2O5XCCl4
HNO3XCH2Cl2
HNO3XCH3-NO2
HNO3XCH3COOHXAc2OHNO3Xpropionic acidXbutyric acid
Cellulose nitrate(N % by weight)
81013.313.713.913.7513.8
14.04; 14.12a
14.1214.1414.014.0
14.08; 14.14a
14.0
Comment
Addition compound('Knecht'), unstabledissolutiongelatinizationno swellingIndustrial nitration
Slow reaction
Rapid reaction
Trinitrate
Dissolution
After extraction with methanol.

4.4.1 Esters of cellulose with inorganic acids 103
Very high nitrogen contents, up to or near to the theoretical value of 14.14 %,can be realized without significant chain degradation by the systemsHN(VCH2Cl2 at O to -30 0C, HNO3/H3PO4/P2O5 and HNO3/acetic acid/ ace-tic anhydride, and these mixtures are preferentially employed for a nearly poly-mer-analogous nitration of cellulose samples and their subsequent macromo-lecular characterization.
Relevant information on the mechanism of cellulose nitration was obtained bystudying the action of HNO3 vapor as well as of N2C^. Also, nitronium saltslike [NO2J
+[BF4]- and KNO3 in 98 % H2SO4 (Miles, 1955) were found to in-troduce a limited amount of nitrate groups to the cellulose chain. Formation ofcellulose nitrate was also claimed to occur on heating a solution of the polymerin N2O4/DMF (Clermont and Bender, 1972; Schweiger, 1974), but this resultcould not be confirmed in studies of our own, probably due to differences inwater content of the system. According to Torgashov et al. (1988) the action ofN2O4 on cellulose results in cellulose nitrite formation on the one hand, andformation of an addition compound between cellulose and HNO3 (Knecht com-pound) on the other. This addition compound is also obtained by the action ofaqueous HNO3 at a concentration below 75 %, whereas at higher HNO3 con-centration covalent esterification begins, connected with dissolution or gelatini-zation of the polymer at an acid concentration up to 90 %.
For the classical nitrating system to be considered now in detail, the nitrogencontent of the cellulose nitrate can be varied within wide limits via the compo-nent ratio of the HNO3/H2SO4/H2O mixture (Fig. 4.4.2).
80
60 0Figure 4.4.2. Nitrogen content dependent on composition (mol %) of the HNO3/H2SO4/H2O mixture (Miles, 1955). Numbers within the figure denote the N-content of cellulosenitrates. Hathed area denotes the region of strong swelling or dissolution. — Chedin lineseparating the region of NO2
+ content at low H2O concentration from the region withoutformation of NO2
+; xxx indicates the variation of the N-content of cellulose nitrate viathe ratio of H2SO4 : H2O at a constant concentration of HNO3 of 25% by weight.

104 4.4 Esterification of Cellulose
The highest nitrogen content, of about 13.4 % (DS^ = 2.7), is obtained at amolar ratio of HNO3/H2SO4/H2O = 1 : 2 : 2 , corresponding to an acid composi-tion of 21.36 % HNO3, 66.44 % H2SO4 and 12.20 % H2O. In cellulose nitratemanufacture, the nitrogen content is usually controlled by varying the ratio ofH2O to H2SO4 at a constant HNO3 concentration of about 25 %, as shown forvarious grades of cellulose nitrate by the data in Table 4.4.3. Figure 4.4.3 illus-trates the decrease in DS^ with increasing water content of the system at a con-stant HNO3 concentration of 21.36 % by weight.
Table 4.4.3. Typical nitrating acid compositions for various grades of cellulose nitrate.
Nitrating acid
% HNO3
2525
25
25
% H2SO4
55.856.6
59.5
66.5
%H2O
19.218.4
15.5
8.5
Cellulose nitrate
Type %
Celluloid gradeLacquer grade,EtOH solubleLacquer grade,ester solubleGun cotton
N by weight
10.911.3
12.3
13.4
DSN
1.952.05
2.35
2.70
= Degree of substitution of nitrate groups.
3.0
2.6
I 2.2
1.8
1.410 14 18 22
Water content [%]
Figure 4.4.3. Dependence of the degree of esterification (DS) on the water content of theoptimal nitrating mixture (HNOß : H2SC^ = 1 : 2 ) (Baiser et al., 1986).
Despite the heterogeneous system, cellulose nitrate formation proceeds as a fastequilibrium reaction. This equilibrium can be shifted to higher or lower nitrogencontent of the product by appropriately changing the nitration acid composition,taking into account some hysteresis on partial denitration via an increase of thewater content. In consequence, the theoretical nitrogen content of 14.14 % cannotbe obtained with this system for thermodynamic reasons, in full agreement with amaximal nitrogen content of about 13.5 % realized experimentally.

4.4.1 Esters of cellulose with inorganic acids 105
From the experimental evidence available and by analogy to the nitration ofother organic compounds like benzene and further aromatic compounds underSE reaction conditions, the NO2
+ cation is generally assumed today to be thekey intermediate in cellulose nitrate formation by the HNO3/H2SO4/H2O mix-ture. The nitronium cation can be formed via several routes, and this cation or itshydrated form, H2NO3+, can react with cellulosic hydroxy groups in the free orprotonized form in several ways, e.g.:
2 HNO3 =^ H2NO3
+ + NO3"
H2NO3
+ ^=^ NO2
++ H2O
HNO3 + H2SO4 ^=^ NO2
+ + HSO4" + H2O
NO2
+ + NO3" ~ N2O5
CeII-OH + NO2
+ ^=* CeII-O-NO2 + H+
CeII-OH + H+ ^=^ CeII-OH2
+
CeII-OH2
+ + N2O5 ^=^ CeII-O-NO2 + NO2
+ + H2O
CeII-OH2
+ + NO3" ^=^ CeII-OH - HNO3 =^ CeII-O-NO2 + H2O
Cell-ΟΝΟ + HNO3 ^=* CeII-O-NO2 + HNO2
CeII-OH + [NO2J+[BF4]" =^ CeII-O-NO2 + HBF4
As a further nitrating agent N2U5 has to be considered which forms in situfrom HNO3 within the cellulose fiber, and which probably is at least partiallyresponsible for the very fast rate of the nitration reaction. According to Ramanspectroscopic studies, NO2
+ is present in anhydrous HNO3 at an amount ofabout 3 %, but nearly all the HNO3 *s converted to NO2
+ by an excess ofH2SO4. Thus an increase in £!2804 concentration in the nitrating acid mixturefavors NO2
+ formation, while an increase in water content decreases its molarratio with respect to the other components (see Fig. 4.4.4).
Cellulose nitrate formation observed with 0.5 M [NO2]+[BF4]- in sulfolan
(tetrahydrothiophene dioxide), as well as with KNO3 in 98 % H2SO4, forming ameasurable amount of NO2
+, is quite in line with the decisive role of NO2
+ incellulose nitration (Miles, 1955). As a further possible reaction route, cellulosenitrate formation via the labile nitrite has been proposed in order to explain thepositive catalytic effect of NOx on cellulose nitration with HNO3/H2SO4/H2O.
Finalizing this abridgment of reaction mechanisms, two facts have to be men-tioned which complicate the course of cellulose nitration with the HNO3/H2SO4/H2O system.

106 4A Esterification of Cellulose
HNO3
Limit of nitrationof nitrobenzene
NO2OH not detectablespectroscopically
75 50H2O [mol %]
25^ H2SOA
Figure 4.4.4. NO2+ concentration (mol/1000g) in dependence on nitrating acid compo-
sition (Albright, 1981).
(i) The presence of a large amount of H2S 04 in the nitrating mixture favors theformation of sulfate groups besides the nitrate groups. These sulfate groups canbe present at an amount of up to 3 % of the polymer at low to medium nitrogencontent (DS^ < 2), up to 80 % of this amount existing in the form of -OSC^Hhalf-ester groups, whereas at a high degree of nitration only 0.2 to 0.5 % of sul-fate groups has been observed. The saponification and elimination of these sul-fate groups by treatment with dilute aqueous acid at elevated temperature is thepurpose of the so-called stabilization process (see the next section),(ii) The equilibrium constant K of the nitration reaction differs largely betweenthe three possible positions within the AGU and depends significantly also onthe nitrating system (see Table 4.4.4).
Table 4.4.4. Equilibrium constant K of the hydroxy groups of the AGU in nitration withHN03/H2S04/H20.
System K value of: Reference
HNCVH2SO4TH2OHNO3TH2O
OH-21.80.26
OH-31.00.12
OH-65.812.6
Wu (1980)Cicirovetal. (1990)
With both mixtures, the O-6 position is preferentially nitrated, resulting in re-gioselectively O-6 cellulose nitrates up to a rather high nitrogen content. Anincrease in the water content of the system was observed to favor O-2 nitrationin comparison with O-3. The system of nitration also affects the uniformity ofsubstituent distribution along and between the polymer chains, as revealed bythe data in Table 4.4.5, comparing the systems HNO3/H2SO4/H2O and

4 A. 1 Esters of cellulose with inorganic acids 107
HNO3/CH2C12 at nearly equal levels of DS^, and showing a considerableamount of nonmodified AGUs even at a DS of 2.10 with the usual ternary mix-ture, probably caused by some nonuniformity in H2SC^ distribution within thefiber moiety.
Table 4.4.5. Pattern of substitution of various cellulose nitrates prepared in HNO3/H2SO4/H2O (A) and HNO3Oi2Cl2 (B) (Short and Munro, 1989; Short et al., 1989).
System of nitration NMR % of AGU nitrated in position:
ABAB
1.1.2,2,
.80
.95
.10
.19
2,3,6
36234932
2,6
22.38.1839
I
.5
.5
3,6
15.5191016
6
9.15.6
13
unmodified
,5.5
16.4
17O
.5
As already emphasized, nitration of cellulose in the HNO3/H2SO4/H2O sys-tem proceeds very fast, and the equilibrium nitrogen content is usually obtainedwithin 10 min at room temperature with a thoroughly dispersed fiber sample.The course of reaction is obviously diffusion controlled, depending decisivelyon the degree and uniformity of swelling and thus on the macro and micro mor-phology of the sample. After surface structure modification due to drying at105 0C, the initial rate of nitration of a sulfite pulp was found to decrease toabout 1/2 of that of the sample dried at 20 0C in vacuo, in good agreement withthe significantly decreased swelling rate in water or aqueous alkali (Philipp,1958). Decreasing the reaction temperature of nitration from 30 to -10 0C sig-nificantly changes the course of nitrate formation (Fig. 4.4.5), indicating a dis-tinct difference in nitration rate between regions of low and high accessibility ata low temperature of reaction.
Time [min]10 60 1000
Figure 4.4.5. Nitrogen content versus reaction time on nitration of spruce sulfite pulp(predried at 20 0C) in dependence on reaction temperature.

108 4.4 Esterification of Cellulose
Obviously a fast penetration of the nitrating acid into the ordered regions isimpeded by its high-viscosity at low temperature, and only the surface near re-gions of the fibrils are nitrated quickly, while after a sufficiently long reactiontime the same DS^ is reached as at room temperature. A very fast course ofnitration at room temperature is obviously not a peculiarity of theHNO3/H2SO4/H2O system, but can also be realized with HNO3/CH2C12 at20 0C with a sufficiently high molar ratio of HNO3 to solvent.
The chemical conversion of cellulose to cellulose nitrate is accompanied bysignificant changes in supramolecular structure. The reaction is generally as-sumed to proceed 'intramicellar' or 'permutoid' by nitration of the lattice layersin a quasi-homogeneous way, implying a consideration of not only the nitratingpower but also the swelling power in optimizing a nitration acid system. The7-0-7 lattice distance increases, on nitration to DS^ = 2.8, from 0.66 to 0.73 nm,without reversibility by denitration (Miles, 1955), and the OH-6· · -OH-3 inter-molecular hydrogen bond is assumed to be preferentially broken in this process,in agreement with the observed preferential O-6 nitration. Up to a nitrogen con-tent of 7.5 % (DS = 1.14) only cellulose II but not the pattern of cellulose nitrateis found in the WAXS diagram, followed by a nearly amorphous pattern up to10.5 % N (DS = 1.8), from which then gradually the cellulose nitrate patternemerges which is fully developed above a nitrogen content of 12.8 % (DS = 2.5)(Miles, 1955). An increase in water content of the nitrating system obviouslyfavors cellulose II formation, as well as decrystallization and structure homog-enization, while a fast conversion to a high nitrogen content leaves no time foran intermediate formation of a cellulose II lattice. Together with the crystallinelattice chain conformation also the orientation and conformation of the sidegroups are changed on nitration. On the morphological level, the cell wall layerS2 is much more easily nitrated than the layers P and Sl, and the fibrillar surfacemorphology and the surface porosity are significantly changed, especially athigh nitrogen contents (see chapter 2.1).
Degradation of cellulose nitrate can take place by acid hydrolysis of glyco-sidic linkages, by saponification of the ester groups and by decomposition of thenitrate substituents. Some decrease in chain length due to acid hydrolysis is in-evitable in the strongly acidic medium of nitrate formation, and is used deliber-ately in the later production steps for product viscosity adjustment. The nitrategroups are rather stable against saponification in a moderately acidic medium,but more susceptible to cleavage under alkaline conditions. Decomposition ofthe nitrate groups can be started by the action of aliphatic amines, H2S or alkalisulfides, an aqueous solution of Na2S being well known as an effective deni-trating agent. A thorough denitration under rather mild conditions can be per-formed with Na2S in EtOH.
Decomposition via radical formation of the nitrate groups by UV light orhigh-energy radiation has been mentioned already (see chapter 2.3.6). Of great

4.4 J Esters of cellulose with inorganic acids 109
practical interest is the thermal decomposition of cellulose nitrate, which starts ata temperature above 130 0C (in the case of well-stabilized lacquer nitrates,above 180 0C) corresponding to deflagration by formation of NC^+ radicals,which initiate a strongly exothermic radical chain reaction, resulting finally inCOx, NOx, N2, H2U and CH^O as end products. The fast autocatalytic chainreaction of thermal decomposition can lead to deflagration, and is the basis forthe use of cellulose nitrates as explosives. As stabilizers against thermal decom-position, radical scavengers like diphenylamine, and also phosphoric acid ortartaric acid are used.
Industrial production of cellulose nitrate
The industrial production process of cellulose nitrate can be reviewed onlybriefly within the framework of this book; for further details the reader is re-ferred to Baiser et al. (1986). A general scheme of the process is presented inFig. 4.4.6.
Cellulose HNO., I I HpSCyOleum
ShreddingPretreatme
II
3ntNitratingacid
I
I Nitration |
ιI Separation |—"
Acidrecovery
I Prestabilization |
Digestionunder pressure
Poststabilization |
I Alcohols I I SeparationI
Ή Dehydration
CNalcohol-wet
I Plasticizer |
|Gelatinization|— ι
CNwater-wet
CNchips
Figure 4.4.6. Diagram of cellulose nitrate (CN) production (Baiser et al., 1986).

110 4.4 Esterification of Cellulose
As a starting material, bleached and scoured cotton !inters or a refined soft-wood or hardwood pulp, with an α-cellulose content of at least 92 %, up toabout 96 %, is used. Linters quality with regard to nitrogen content under givennitration conditions is obtained with highly refined prehydrolysis sulfate pulps
of about 96 % α-content. Important criteria are a low hemicellulose and lignincontent and a low ash content, especially with regard to Ca2+ ions possiblyforming calcium sulfate precipitates in the production process. The DP of thestarting material largely determines the viscosity level of the product. Besidesthese chemical criteria, the macro and micro morphology of the pulp, especiallythe surface porosity, are important with regard to the swelling behavior of thepulp as a decisive process parameter determining nitration rate and uniformity aswell as acid retention. Before nitration, the cellulose is disintegrated to fluff,shreds or chips or used in the form of crepe paper with about 20 g/m2. The cel-lulose is used for nitration without drying, often with a moisture content of up to50 %. The packing of the material in the reactor played a significant role in de-termining reaction rate and acid retention.
Nitration itself is still generally performed with the classical ternary mixtureof HNO3/H2SO4/H2O, either in a batch process or in a continuous reactor. Inthe batch process the cellulose is reacted in a stainless steel tube reactor with thenitrating acid for about 30 min at a solid-to-liquid ratio of about 1 : 20 to 1 : 50at a temperature between 10 and 35 0C, a low temperature being employed forhighly nitrated products. The product yield remains about 15 % below the theo-retical one due to formation of side products like oxalic acid. After centrifuga-tion the reaction mass contains about 100-130 % nitrating acid in the case ofcotton !inters, and up to 300 % in the case of wood pulp. For rapid displacementof the strong acid, the crude cellulose nitrate is dispersed in a large excess ofcold water at a solid-to-liquid ratio of about 1 : 100. The continuous processpractised since about 1960, and resulting in higher product uniformity andhigher safety, employs a series of straight-run vats or tubes with conveyers or apressurized reaction loop, cutting down the reaction time to 6-12 min, followedby continuous centrifugation of the reaction mass.
Stabilization and viscosity adjustment of the cellulose nitrate is performed bya series of washes and cooking, the cooking steps being at first with water con-taining 0.1-1 % acid and finally with water adjusted to pH 7 for elimination ofthe last traces of acid. Highly nitrated products for explosives require an espe-cially careful stabilization in order to avoid uncontrolled decomposition. Forreducing the time consumed in stabilization, pressure cooking at 130-150 0C isemployed with low- and medium-nitrated products. Cellulose nitrates for cellu-loid or lacquer production are shipped as fibers or flakes, products for use asexplosives after mechanical disintegration by wet beating, and in both cases witha residual water content of about 25-35 %. All these steps of cellulose nitrate

4.4.1 Esters of cellulose with inorganic acids 111
processing require a continuous elimination of NO^ vapors in order to avoid anuncontrolled autocatalyzed decomposition.
For further processing, the water is displaced by alcohol in the case of lacqueror celluloid nitrates. The products can also be gelatinized by incorporation ofsofteners like phthalic acid esters, or aqueous dispersions of softened cellulosenitrate are prepared for further use in coating.
Properties of cellulose nitrates
Cellulose nitrates in the DS range of commercial interest, between 1.8 and 2.8,are white, transparent, odorless, nontoxic and rather hydrophobic solids, thephysical and chemical properties of which depend significantly on the nitrogencontent. This holds true for the density, which increases in this DS range fromabout 1.5 to above 1.7 g/cm3, and especially for the course of thermal degrada-tion (with the deflagration tendency increasing with the nitrogen content), aswell as for the solubility in organic liquids. Products with a nitrogen contentbetween 10.9 and 11.3 % dissolve readily in ethanol. They are soluble in otheralcohols, ketones and esters in a transition range between 11.4 and 11.1 % N,while at a nitrogen content above 11.8-13.7 %, organic esters are the most fa-vorable solvents.
With a dielectric constant of about 7 and a specific resistance of10n-1012 Ω/cm, commercial cellulose nitrates are considered as good insula-tors. They show excellent film-forming properties from solution after evapora-tion of the solvent, the films exhibiting breaking elongations between 3 and30 % and a breaking strength between 50 and 100 N/mm2.
Commercial cellulose nitrates can be plasticized with a variety of conven-tional softeners such as adipates, phthalates, organic phosphates and vegetableoils, and they are compatible with a large number of synthetic polymers likealkyd and ketone resins, formaldehyde/urea condensates, or poly aery lates.
Application of cellulose nitrates
The first application of cellulose nitrate was the Old-timer' celluloid. It ismanufactured by kneading cellulose nitrate with a nitrogen content of10.5-11.0 % with ethanol and camphor (softener) to a mass containing 70-75 %cellulose nitrate, which can be shaped very precisely by pressing at elevatedtemperature. Celluloid still holds a marked share in cellulose nitrate applicationfor special products like combs, hair ornaments, drawing equipment and pingpong balls. An important and actual application of cellulose nitrate are explo-sives, i.e. blasting, detonating, propellant, shooting, igniting, and pyrotechnicalagents with a high nitrogen content, usually of or above 12.6 %.

112 4.4 Esterification of Cellulose
The excellent mechanical and adhesive properties of cellulose nitrate filmsand coatings still promote the widespread application of cellulose nitrate lac-quers containing 10-13 % polymer. They can be processed by spraying withcompressed air, casting, rowling, doctor knives coating or dipping. They areused e.g. as wood lacquers for furniture, as metal and paper lacquers and also assealing lacquers for cellophane plastic and metal foils, as well as for the prepa-ration of printing inks, e.g. for flexo printing. Cellulose nitrate membranes stillplay a role as filter and separation media and recently found an interesting newfield of application as nuclear track detectors in high-energy physics and geol-ogy, with the tracks obtained by radiation degradation of the nitrate groups beingdeveloped and made visible microscopically by treatment with aqueous NaOH(Watjen et al., 1993).
Finally, the very promising and rapidly expanding area of cellulose nitratedispersion lacquers shall be mentioned with the aim of replacing organic sol-vents with the ecologically safe water. Products of this kind are available todayas cellulose nitrate/softener dispersions for absorbing media like leather, as wellas for continuous film formation on non-absorbing media containing in this casean adequate amount of coalescing agent (Baiser et al., 1986).
4.4.1.2 Cellulose nitrite
In contrast with cellulose nitrate, the nitrite of cellulose cannot be prepared byesterification with the appropriate acid due to the low acidity and the low stabil-ity of nitrous acid, HNO2. However, a highly substituted nitrite of cellulose canbe obtained by reacting the polymer with N2O4, NOCl or several salts like nitro-sylic compounds under anhydrous conditions in a suitable dipolar aprotic solventlike DMF. With N2O4 as the reagent, the reaction proceeds according to theoverall process: CeIl-OH + N2O4 -> CeIl-O-NO + HNO3, requiring at least3 mol OfN2O4 for a complete esterification of the hydroxy groups.
Routes of synthesis of cellulose nitrite and course of the reaction
In combination with a suitable dipolar aprotic solvent like DMF or DMSO,N2O4 has long been known to dissolve even high molecular cellulose quickly,i.e. within 10-30 min, and completely at room temperature, if at least 3 mol ofN2O4/mol of AGU are present in the system. Since the early publication ofFowler et al. (1947), the question of whether or not this dissolution takes placetogether with the formation of a cellulose nitrite, has been the subject of contro-versial discussions for several decades and has decisively stimulated studies oncellulose nitrite formation and stability. Most of the investigators favored a co-valent derivatization by ester formation, assuming a heterolytic cleavage of theN2O4 molecule according to

4ΛΛ Esters of cellulose with inorganic acids 113
N2O4 -> NO+ + NO3-
as the primary step followed by the esterification reaction
CeIl-OH + NO+ + NO3~ -> CeIl-O-NO + HNO3
But well-founded arguments were published in favor of an addition compoundof cellulose and N2O4, solvated by complex formation with the dipolar aproticliquid (Golova et al., 1986).
This controversy was settled quite recently by in situ 13CX1H two-dimensionalNMR studies (Wagenknecht et al., 1992a), showing that under strictly anhy-drous conditions (water content < 0.01 %), a complete derivatization to cellulosetrinitrite takes place in the systems cellulose/DMF/N2O4 and cellu-lose/DMSO/N2O4 with an excess of N2O4. In the presence of a small amount(0.1-1 %) of water, only a partial derivatization occurs, the O-6 position beingpreferentially esterified in this case. On addition of about 1 mol/mol AGU ofmethanol or water to a cellulose trinitrite solution in DMF or DMSO, a consid-erable cleavage of nitrite groups was observed already at or below room tem-perature. The stability of the nitrite groups at the different positions increases inthe order C-2 < C-3 « C-6. In any case, the O-6 position is preferentiallymodified by nitrite formation. On the other hand, the reaction of cellulose previ-ously dissolved in DMA/LiCl with an excess of N2O4 resulted in a DS of estergroups below 1.5 (partial DS at C-2 0.25, at C-3 0.35, at C-6 0.75), obviouslydue to a partial inactivation of cellulosic hydroxy groups by formation of hydro-gen-bond complexes with the solvent system (Wagenknecht et al., 1992a).
Various authors reported the isolation of cellulose nitrite with DS values up to3.0 from the cellulose/N2O4/DMF system by precipitation with a liquid of lowpolarity. A beneficial action of tertiary amines on preserving a high DS was no-ticed in some of these studies (Schweiger, 1974). Our own experiments resultedin a complete or nearly complete denitrosation on addition of water, ethanol oracetone to the above-mentioned system, while on addition of diethyl ether, acellulose nitrite with a DS of only 0.3 was obtained. Cellulose nitrites with DSvalues between 2.5 and 2.95 could be isolated after addition of triethylamine byprecipitation with a mixture of diethyl ether and methylene chloride at O 0C.From the system cellulose/NOCl/DMF (Wagenknecht et al., 1976) a cellulosenitrite with a DS of 2.6 and only a very small content of Cl CC)S1Q ~ 0.02) wasprepared by the same procedure (see Table 4.4.6).
By using an excess of nitrosylic compounds, i.e. nitrosyl sulfuric acid, nitro-syl tetrafluoroborate and nitrosyl hexachloroantimonate, in DMF, cellulose isdissolved nearly as quickly and completely as by using N2O4 itself(Wagenknecht et al., 1976). From the analytical data of the precipitates obtainedwith TEA/diethyl ether/methylene chloride on the one hand, and with H2O onthe other, it can be concluded that both the cationic and the anionic part of the
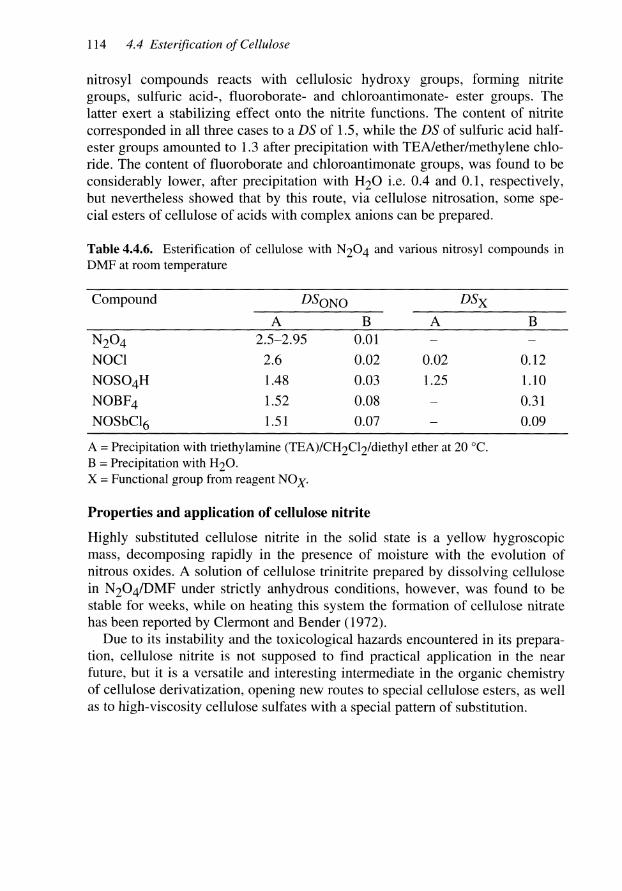
114 4.4 Esterification of Cellulose
nitrosyl compounds reacts with cellulosic hydroxy groups, forming nitritegroups, sulfuric acid-, fluoroborate- and chloroantimonate- ester groups. Thelatter exert a stabilizing effect onto the nitrite functions. The content of nitritecorresponded in all three cases to a DS of 1.5, while the DS of sulfuric acid half-ester groups amounted to 1.3 after precipitation with TEA/ether/methylene chlo-ride. The content of fluoroborate and chloroantimonate groups, was found to beconsiderably lower, after precipitation with I O i.e. 0.4 and 0.1, respectively,but nevertheless showed that by this route, via cellulose nitrosation, some spe-cial esters of cellulose of acids with complex anions can be prepared.
Table 4.4.6. Esterification of cellulose with N2C^ and various nitrosyl compounds inDMF at room temperature
Compound £^ONO £>5χ
N2O4
NOCl
NOSO4H
NOBF4
NOSbCl6
A2.5-2.95
2.6
1.48
1.52
1.51
B0.01
0.02
0.03
0.08
0.07
A-
0.02
1.25
-
-
B-
0.12
1.10
0.31
0.09
A = Precipitation with triethylamine (TEA)/CH2Cl2/diethyl ether at 20 0C.B = Precipitation with H2O.X = Functional group from reagent
Properties and application of cellulose nitrite
Highly substituted cellulose nitrite in the solid state is a yellow hygroscopicmass, decomposing rapidly in the presence of moisture with the evolution ofnitrous oxides. A solution of cellulose trinitrite prepared by dissolving cellulosein N2O^DMF under strictly anhydrous conditions, however, was found to bestable for weeks, while on heating this system the formation of cellulose nitratehas been reported by Clermont and Bender (1972).
Due to its instability and the toxicological hazards encountered in its prepara-tion, cellulose nitrite is not supposed to find practical application in the nearfuture, but it is a versatile and interesting intermediate in the organic chemistryof cellulose derivatization, opening new routes to special cellulose esters, as wellas to high- viscosity cellulose sulfates with a special pattern of substitution.

4.4.1 Esters of cellulose with inorganic acids 115
4.4.1.3 Cellulose sulfates
General comments on synthesis and product
The esterification of hydroxy groups of cellulose according to
CeIl-OH + SO3 -> CeIl-OSO3HCeIl-OH + XSO3H -» CeIl-OSO3H + XH (X = H2N, HO, Cl)
generally proceeds to the acid half-ester, which can be converted to a neutralsodium salt soluble in water above a DS$ of 0.2-0.3. Generally, the term 'cellu-lose sulfate' will be used to denote the acid half-ester or its sodium salt. Forma-tion of the full ester is obviously negligible with nearly all procedures so farreported. The synthesis of medium to high DS cellulose sulfates with theSO3TDMSO or the SO3/DMF complex has been comprehensively investigatedby Whistler et al. (1968) and Schweiger (1966 and 1972). Most frequently sulfu-ric acid, sulfur trioxide or chlorosulfonic acid have been employed as sulfatingagents, either as the only reaction component besides cellulose, or in combina-tion with alcohols, amines or inert media like chlorinated hydrocarbons. Via thechoice of the reaction system and the adaptation of reaction conditions like timeand temperature of reaction and molar ratio of agent to AGU, the whole range ofDS between O and 3 can be realized in cellulose sulfate formation. A generalproblem is the rather excessive chain degradation due to hydrolytic cleavage ofglycosidic bonds accompanying cellulose sulfate formation in many of thestrongly acidic media.
Principle routes of synthesis of cellulose sulfates to be considered subse-quently in more detail are:(i) sulfation of hydroxy groups of unmodified cellulose, usually starting in aheterogeneous system;(ii) sulfation of free hydroxy groups in partially functionalized cellulose esters orethers with the primary substituent acting as a protecting group;(iii) sulfation by displacement of an ester or ether group already present in themacromolecule.
Along the routes (ii) and (iii) regioselectively functionalized cellulose sulfatescan be obtained. Along all the three routes a conversion of the acid half-ester toa neutral salt, usually the sodium salt, is necessary for arriving at a stable prod-uct not susceptible to a hydrolysis catalyzed by the strongly acidic OSO3Hgroup.
In the following sections the three routes of synthesis will be presented insome detail, considering advantages and short-comings of the procedures withregard to product properties. These properties will then be surveyed in the solidstate as well as in aqueous solution, and finally an overview will be given onpresent and future areas of application of these products.

116 4.4 Esterification of Cellulose
Routes of cellulose sulfate synthesis
Sulfation of unmodified cellulose
Since the early publication of Bracannot (1819) on the reaction between cellu-lose and sulfuric acid, resulting in a severely degraded product with a DS be-tween 1 and 2, a large variety of sulfating reagents for unmodified cellulose havebeen studied. Besides H2SO4, SO3 and ClSO3H, also SO2Cl2, FSO3H,ClSO2-OC2H5, CH3-CO-SO4H and NO-SO4H were employed in these one-,two- or multicomponent systems. Some of these are listed in Table 4.4.7 to-gether with the state of dispersity and the DS range obtained.
Table 4.4.7. Heterogeneous sulfation of cellulose in various systems(Philipp and Wagenknecht, 1983)
System
H2SO4
H2SO4XSO2
H2SO4/chlorinated hydrocarbonsH2SO4/diethyl etherH2SO4/low aliphatic alcoholClSO3HXSO2
ClSO3HXpyridineClSO3HXpyridineXtolueneClSO3HXformamideSO3XSO2
SO3XCS2
SO3Xdiethyl etherSO3XDMSOSO3XDMFSO3XpyridineSO3Xtriethyl phosphateSO2Cl2XDMF, formamide
Range of DS8
1-2-0.9-0.3
0.2-0.40.1-1.0-1.8
1.9-2.8-2.8
up to 3.0-2.2
up to 3.01.3-2.11.3-2.01.5-2.6
up to 2.2up to 3.00.2-0.5
State ofdispersity
BAAAAABABAAABBBBA
, degree of substitution of sulfate groups.A Heterogeneous during the whole course of sulfation.B Transition from a heterogeneous to a homogeneous system during the
sulfation reaction.
As can be seen from these data, a broad range of DS§ values can be coveredby systems remaining strictly heterogeneous during the whole course of reactionand also by those showing a transition from the heterogeneous to the homogene-ous state during the course of sulfation. As to be expected, SO3 and ClSO3H

4.4.1 Esters of cellulose with inorganic acids 117
generally exhibit a higher reactivity than H2SO4. With many reagents, sulfationalong and between the polymer chains obviously proceeds rather nonuniformly,resulting in a poor or no solubility of the sodium cellulose sulfate in water evenat a DS value above 0.30. If water-soluble products are obtained at all, the spe-cific solution viscosity is generally low, even with !inters as starting material,due to an excessive hydrolytic chain degradation during sulfation.
As illustrated by Fig. 4.4.7, with results of the system H2SO4/diethyl ether atroom temperature, the final DS can be reached within a few minutes, if the reac-tion in this nonswelling medium is limited to easily accessible regions of thestructure, and comes to a stop at a DS of about 0.3, arriving at a water-insolublesodium cellulose sulfate (Philipp and Wagenknecht, 1983).
0.3
0.2
ωtoQ
0.1
O 20 40 60 80 1OCTime of reaction [min]
Figure 4.4.7. Increase in DS with time of reaction in the heterogeneous sulfation ofcellulose powder in the H2SO4/diethyl ether system (22 0C, molar ratio H2SO4 : diethylether = 2.8 : 1) (Philipp and Wagenknecht, 1983).
In the stronger swelling medium H2SO4/isopropanol/toluene, which was recentlyinvestigated (Lukanoff and Dautzenberg, 1994) with respect to process development,a considerably higher DS of about 0.7 can be obtained (Petropavlovski, 1973), re-sulting in a partial or even total solubility of the sodium cellulose sulfate in water(see Fig. 4.4.8). The course of reaction here is largely determined by the equilibriumof propylsulfuric acid formation. With increasing DS§, resulting from a higher reac-tion time and/or reaction temperature and/or sulfuric acid input, a general decrease ofthe insoluble part of the sulfated sample is observed.
This increase in solubility, however, due to an enhancement of DS has to be repaidby a significant decrease in specific solution viscosity of the soluble part due to amore extensive chain degradation. Starting from unmodified cellulose as the solidphase, a sodium cellulose sulfate completely soluble already at low DS obviouslycannot be realized without excessive chain degradation, i.e. a low solution viscosity.
After dissolution of the polymer in a nonaqueous, nonderivatizing solventmedium, a fast sulfation of free hydroxy groups up to a high DS should be ex-

118 4.4 Esterification of Cellulose
pected. But the results obtained by us with sulfuric acid, its anhydride and itsacid chlorides were disappointing, in so far as a smooth course of reaction wasimpeded by an early coagulation of the system and/or by sometimes violent in-teractions between the sulfating agent and components of the solvent. In theO-basic binary mixture A^methylmorpholine 7V-oxide/DMF, a DS§ exceeding0.1 was obtained only with SC^C^, accompanied by severe chain degradation(Wagenknecht et al., 1985). Some results of sulfation experiments withSO3/DMF in several solvent media, with and without addition of TEA, aresummarized in Table 4.4.8, indicating a positive effect of the presence of TEA.
0.6
0.5
0.4
0.3
0.2
0.1
O60 120 180
Time [min]24-0
Figure 4.4.8. DS dependence of cellulose sulfation on reaction time (O 0C, molar ratioH2SC>4/isopropanol/toluene) (Lukanoff and Dautzenberg, 1994).
Table 4.4.8. Sulfation of cellulose dissolved in nonaqueous solventswith SO3-DMF (2 h, room temperature, excess of reagent)
Solvent and state of dispersity
NMMNO/DMFHMPT/LiClDMA/LiCl
without TEA0.06 H0.64 C->H0.40 C
with TEA0.14 C0.70 C^H0.56 C
H Homogeneously during the course of reaction.C Coagulation after reagent addition.C—>H Redissolution after primary coagulation.HMPT hexamethy!phosphoric acid triamide.
In spite of rather high DS§ values obtained in HMPT/LiCl and DMA/LiCl, theproducts exhibited only a partial solubility in water. Among the sulfating agentstested, ClSC^H proved to be the most suitable in systems of this kind, as shownby the rather high DS values achieved with this reagent at elevated temperature

4.4. l Esters of cellulose with inorganic acids 119
with !inters cellulose dissolved in TEA/SO2/formamide. Also, under these con-ditions a continues gel on reagent addition is formed, and it shows a smoothcourse of reaction, with the dependency of DS§ on molar ratio of ClSC^H-to-AGU and on temperature as illustrated in Fig. 4.4.9.
1.2
0.8ω
ωQ
0.4
O 2 4 6 8 10Molar ratio of HSO3Cl: AGU
Figure 4.4.9. Dependence of DS on molar ratio of HSC^Cl: AGU in sulfation of !interscellulose in TEA/SC^/formamide/HSOßCl (time of reaction 2 h) (Philipp andWagenknecht, 1983).
But even here sodiumcellulose sulfates of only partial water solubility wereobtained. In summary, the sulfation of cellulose dissolved in dipolar aproticnonderivatizing media cannot be recommended as a route to soluble cellulosesulfates, as these conditions don't show any advantage in comparison with e.g. asuspension of cellulose in DMF or DMSO reacting with SOß rather smoothlywith gradual transition to a homogeneous medium.
Sulfation of free hydroxy groups in partially derivatized cellulose
The free hydroxy groups of various partially modified cellulose esters and etherscan be sulfated by conventional sulfating agents in a suitable dipolar aproticmedium often rather rapidly and completely, while the primary substituent actsas a protecting group in the anhydrous acid reaction system and is not attackeditself by the reagent. A regioselective pattern of substitution of the primaryfunctional group offers in this case a route to cellulose sulfates with a definedsite-selective distribution of the sulfate groups within the AGU. The acetylgroup of partially esterified cellulose acetates proved to be a very suitable pro-tecting group in a subsequent sulfation, as it is definitely stable in the anhydrousacid reaction medium, in contrast with the more mobile formyl group(Wagenknecht et al., 199Ia). Moreover, it can be easily and completely split offafter the reaction in an alkaline protic medium without impeding the DS of sulfu-ric acid half-ester groups as the only substituent in the final product. The fol-lowing considerations will therefore be centered on the role of the acetyl groupas an intermediate protecting group in sulfation, especially in regioselectivesulfation of cellulose. But of course the route of synthesis outlined here can also

120 4.4 Esterification of Cellulose
be employed to arrive at products containing sulfuric acid half-ester groups aswell as the primary protecting group.
Sulfation of cellulose acetates with a DS^c in the range 0.8-2.5 is preferen-tially performed under homogeneous conditions with DMF acting as the solventfor the polymer and as the reaction medium employing one of the conventionalsulfating agents listed in Table 4.4.9.
Table 4.4.9. Sulfating agents for cellulose acetate dissolved in DMF.
Sulfur trioxide 803Oleum with 33 % SO3
66 % SO3
Chlorosulfonic acid ClSO3HSulfuryl chloride SO2Cl2
Amidosulfonic acid H2NSO3HAcetylsulfuric acid CH3-CO-SO4H
The reactivity of these agents in cellulose acetate sulfation decreased in theorderSO3 > oleum > ClSO3H > SO2Cl2 > CH3-CO-SO4H > H2NSO3H.The SO3-DMF complex, oleum with 33 % SO3 or 66 % SO3, and especially ahigh-quality chlorosulfonic acid, act very fast in these esterifications and thereaction is nearly completed within half an hour even at O 0C. This high reactionrate can cause problems with regard to product uniformity, due to an unevenreagent distribution, if larger charges of the viscous cellulose acetate solutionwith a polymer content between 10 and 20 % are to be processed (Wagenknechtand Schwarz, 1996) to a low degree of sulfation. Acetylsulfuric acid exhibits amore moderate reactivity but still higher than that of amidosulfonic acid. Thelatter proved to be a very convenient sulfating agent at a reaction temperaturebetween 50 and 80 0C (see the DSs-time plot in Fig. 4.4.10).
A conceivable amination at the cellulose chain by this reagent has never beenobserved in employing amidosulfonic acid for sulfation of cellulose acetates. Asan essential point of cellulose sulfate synthesis with all these reagents, the re-quirement of a strictly anhydrous medium with a residual water content lowerthan 0.05 % must be emphasized.
As demonstrated by Fig. 4.4.11, summarizing the results of sulfation experi-ments with cellulose-2,5-acetate in DMF and SO3 or ClSO3H, the acetyl groupreally acts as a reliable protecting group, as the DS$, with values between 0.4and 0.5, does not exceed the amount of free hydroxy groups even at a large ex-cess of sulfating agent.
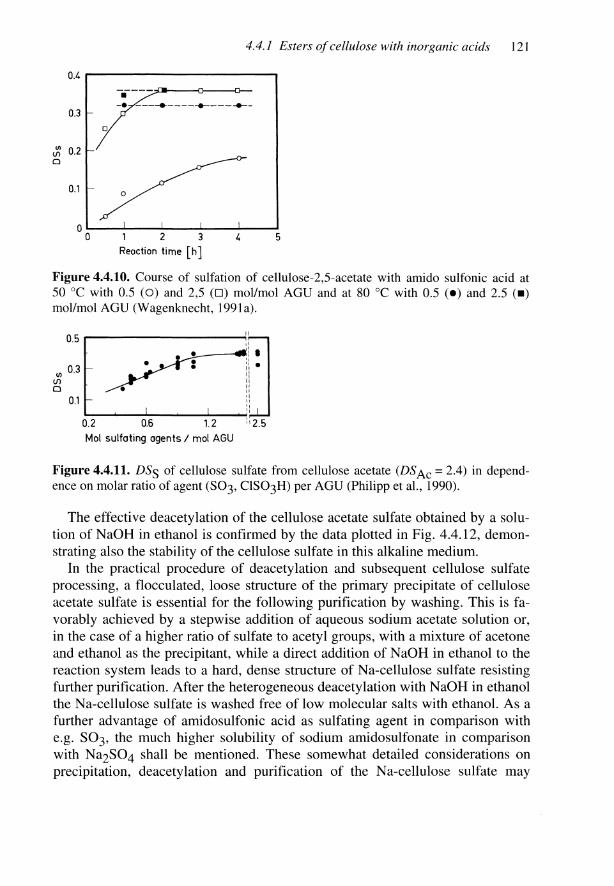
4AJ Esters of cellulose with inorganic acids 121
(U
0.3
ω 0.2Q
0.1
0 1 2 3 4 5
Reaction time [h]
Figure 4.4.10. Course of sulfation of cellulose-2,5-acetate with amido sulfonic acid at50 0C with 0.5 (O) and 2,5 (D) mol/mol AGU and at 80 0C with 0.5 (·) and 2.5 (·)mol/mol AGU (Wagenknecht, 1991a).
0.5
0.3
0.1
0.2 0.6 1.2Mol sulfoting agents / mol AGU
Figure 4.4.11. DS§ of cellulose sulfate from cellulose acetate (DSp^c = 2.4) in depend-ence on molar ratio of agent (SO3, ClSO3H) per AGU (Philipp et al., 1990).
The effective deacetylation of the cellulose acetate sulfate obtained by a solu-tion of NaOH in ethanol is confirmed by the data plotted in Fig. 4.4.12, demon-strating also the stability of the cellulose sulfate in this alkaline medium.
In the practical procedure of deacetylation and subsequent cellulose sulfateprocessing, a flocculated, loose structure of the primary precipitate of celluloseacetate sulfate is essential for the following purification by washing. This is fa-vorably achieved by a stepwise addition of aqueous sodium acetate solution or,in the case of a higher ratio of sulfate to acetyl groups, with a mixture of acetoneand ethanol as the precipitant, while a direct addition of NaOH in ethanol to thereaction system leads to a hard, dense structure of Na-cellulose sulfate resistingfurther purification. After the heterogeneous deacetylation with NaOH in ethanolthe Na-cellulose sulfate is washed free of low molecular salts with ethanol. As afurther advantage of amidosulfonic acid as sulfating agent in comparison withe.g. SO3, the much higher solubility of sodium amidosulfonate in comparisonwith Na2SU4 shall be mentioned. These somewhat detailed considerations onprecipitation, deacetylation and purification of the Na-cellulose sulfate may
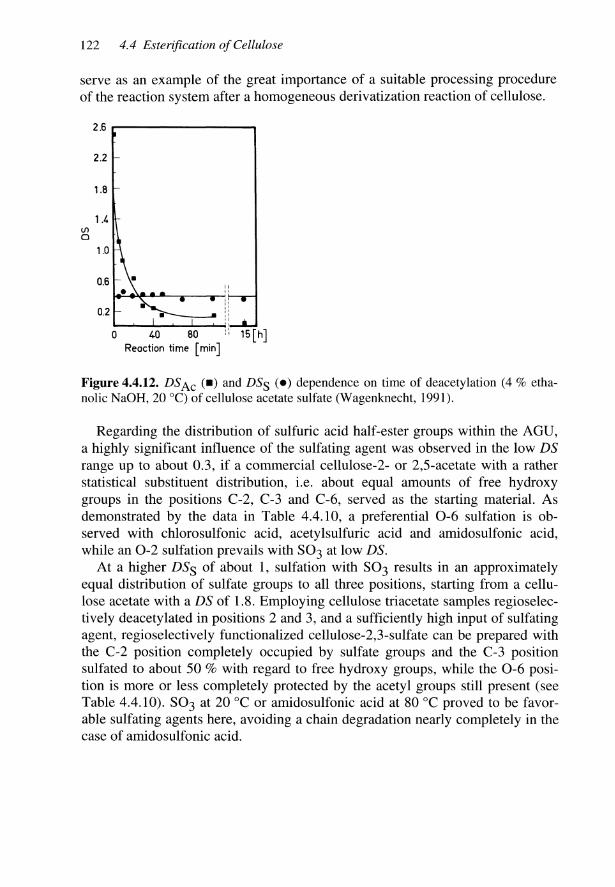
122 4.4 Esterification of Cellulose
serve as an example of the great importance of a suitable processing procedureof the reaction system after a homogeneous derivatization reaction of cellulose.
O 40 80Reaction time [min]
Figure 4.4.12. DS^C (·) and DS$ (·) dependence on time of deacetylation (4 % etha-nolic NaOH, 20 0C) of cellulose acetate sulfate (Wagenknecht, 1991).
Regarding the distribution of sulfuric acid half-ester groups within the AGU,a highly significant influence of the sulfating agent was observed in the low DSrange up to about 0.3, if a commercial cellulose-2- or 2,5-acetate with a ratherstatistical substituent distribution, i.e. about equal amounts of free hydroxygroups in the positions C-2, C-3 and C-6, served as the starting material. Asdemonstrated by the data in Table 4.4.10, a preferential O-6 sulfation is ob-served with chlorosulfonic acid, acetylsulfuric acid and amidosulfonic acid,while an O-2 sulfation prevails with 803 at low DS.
At a higher DS§ of about 1, sulfation with 803 results in an approximatelyequal distribution of sulfate groups to all three positions, starting from a cellu-lose acetate with a DS of 1.8. Employing cellulose triacetate samples regioselec-tively deacetylated in positions 2 and 3, and a sufficiently high input of sulfatingagent, regioselectively functionalized cellulose-2,3-sulfate can be prepared withthe C-2 position completely occupied by sulfate groups and the C-3 positionsulfated to about 50 % with regard to free hydroxy groups, while the O-6 posi-tion is more or less completely protected by the acetyl groups still present (seeTable 4.4.10). SO3 at 20 0C or amidosulfonic acid at 80 0C proved to be favor-able sulfating agents here, avoiding a chain degradation nearly completely in thecase of amidosulfonic acid.

4.4.1 Esters of cellulose with inorganic acids 123
Table 4.4.10. Sulfation of statistically (S) and regioselectively (R) deacetylated celluloseacetate samples (Philipp et al., 1995).
Cellulose acetate Sulfating agentType
S
RRR
DS8
DS AC Agent mol/mol
2.38
2.641.861.48
AGUSO3
ClSO3HH2NSO3HH2NSO3HH2NSO3HH2NSO3HH2NSO3H
0.40.50.51.0123
0.350.220.350.520.250.951.15
Partial DSS in positionC-26
0.200.040.110.170.170.550.74
C-3
0.00.00.040.150.080.200.15
C-6
0.150.180.200.200.00.200.26
% in C-
43825738
O2123
A convenient route to regioselectively or preferentially C-6-substituted cellu-lose sulfates, employing also the cellulose acetate sulfate as intermediate, con-sists of the competitive esterification of cellulose suspended in DMF with amixture of acetic anhydride and SOß or CISOßH, and a subsequent deacetylationwith NaOH in ethanol. After preparation at room temperature, the system isheated to about 50 0C, and esterification takes place during 30 min to 4 h withgradual and finally complete dissolution of the polymer. The DS$ obtained afterdeacetylation depends primarily on the molar ratio of sulfating agent to acidanhydride and can reach an upper value of about 1.5. An exclusive sulfation ofthe C-6 position was indicated by the 13C NMR spectrum up to a DS§ of about0.8, CISOßH showing a somewhat higher regioselectivity than 803. For a reli-able control of DS§, an input of about 8 mol of acid anhydride/mol of AGU andan appropriate adjustment of the amount of sulfation agent added was found tobe favorable. The results of some of these experiments are summarized in Table4.4.11.
The sodium cellulose sulfates, prepared via acetosulfation, were completelywater-soluble at and above a DS of 0.3. This somewhat higher minimal DS$, ascompared with samples obtained in a strictly homogeneous course of reactionfrom partially substituted cellulose acetates, is obviously caused by a less uni-form sulfate group distribution along and between the polymer chains due to theinitially heterogeneous reaction system. On the other hand, this route of aceto-sulfation permits the synthesis of Na-cellulose sulfates of higher solution vis-cosity (about 200 mPa s in 1 % aqueous solution) with a high DP cotton !inters(1400) as the starting material. The solution viscosity of samples prepared frompartially substituted cellulose acetate is limited to less than 15 mPa s due to therather low DP (about 250) of the starting material.

124 4.4 Esterification of Cellulose
Table 4.4.11. Acetosulfation of !inters cellulose (Philipp et al, 1995).
mol of AC2Ü/mol of AGU
16888
mol of ClSO3H/ DS5 O-6 esterification (%)mol of AGU
1.40.723
0.200.500.751.30
100959558
The exact mechanism of this acetosulfation with a transition from the hetero-geneous to the homogeneous system is not yet clear. The mechanistic concept ofdissolution acetylation with £[2804 as the catalyst and the intermediate intro-duction of some sulfate groups obviously cannot be transferred to all routes ofacetosulfation due to the other ratio of acetyl to sulfate groups (see chapter4.4.3). We assume in our system of acetosulfation a rather fast reaction of easilyaccessible C-6 hydroxy groups, combined with a gradual esterification of hy-droxy groups in all the three positions by acetanhydride.
As already indicated above, the protecting action of ester or ether groups al-ready present, together with a sufficiently large number of free hydroxy groups,can be used to synthesize various sulfated ethers and esters of cellulose evenwith a regioselective pattern of substitution, and can thus provide routes to newdoubly functionalized cellulose derivatives with interesting applicational prop-erties. The cellulose acetosulfates frequently cited above showed a high waterbinding capacity and were successfully tested as sanitary supersorbers. Doublymodified derivatives with interesting surfactant properties were obtained by usby sulfating the 6-position of a 2,3-0-laurylcellulose or preferentially the 2,3-position of predominantly 6-O-tosyl or -tritylcelluloses, employing 803 as thesulfating agent and DMF or pyridine as the reaction medium.
The sulfation of CMC in the DS range of 0.5-2.0 in the usual manner, i.e.with SO3 in a dipolar aprotic solvent under homogeneous conditions, posedproblems due to an only minimal solubility of the polymer in these solvents.These difficulties could be overcome, however, by presenting the CMC in a veryfine dispersed state to the reagent, either by previous dissolution in water, subse-quent precipitation with an excess of DMF and elimination of the water byazeotropic distillation, or still better by preparing a highly swollen slurry ofCMC in a mixture of dimethylacetamide and /?-toluenesulfonic acid prior tosulfation with SO3 in this system (Vogt et al., 1995 and 1996).
Sulfation of cellulose via displacement of labile ester or ether groups
In contrast with the acetyl group with its well-established protecting actionagainst sulfating agents in a dipolar aprotic medium, the very labile nitrite group

4.4.1 Esters of cellulose with inorganic acids 125
is displaced rather easily by various sulfating agents from its position in theAGU. In this way, a reaction system of cellulose dissolved with an excess ofN2O4 in DMF (> 3 mol of N2O4/mol of AGU) to a cellulose trinitrite and con-taining an excess of N2O4 and HNO3 as further components, can be directlysulfated without isolation of the cellulose trinitrite (Schweiger, 1974;Wagenknecht et al., 1993) according to the scheme in Fig. 4.4.13 by the sulfat-ing agents indicated.
Cellulose/N2O4/DMF
(excess of N2O4, HNO3)
SO3
SO2
NOSO4H
CISO3H
SO2CI2
H2NSO3H
CeII-OSO3H + N2O4
CeII-OSO3NO
CeII-OSO3H + N2O3
CeII-OSO3NO+ HNO2
ΓΏΙΙ OCO U -ι- ΜΟΓΜUeil WoVJ3M + iNvJUl
CeII-OSO2CI + NOCI
r*aii_r>cr> u j. M t α. H2O
Figure 4.4.13. Scheme of possible reactions in the system cellulose/N2O4/DMF onaddition of different sulfating agents (Wagenknecht et al., 1993).
SO2 here reacts via an intermediate formation of NOSO4H with the HNO3
present in the system. The DS$ obtained depends, under comparable externalconditions, significantly on the sulfating agent employed and covers a rangebetween 0.3 with NOSO4H and 1.6 with SO2Cl2, with this large difference ob-viously been caused by a different position of the transesterification equilibrium.With all the sulfating agents studied except H2SO4, water-soluble Na-cellulosesulfates were obtained above a DS$ of 0.2-0.25 after elimination of residualnitrite groups by hydrolysis in a protic medium and subsequent neutralizationand purification of the cellulose sulfate half-ester (Wagenknecht et al., 1993). Byminimizing hydrolytic chain degradation during this product processing, cellu-lose sulfates with very high solution viscosity, up to 2500 mPa s (1 % aqueoussolution), could be synthesized from cotton !inters, DP 1400.
Due to a site-selective transesterification reactivity of the nitrite groups independence on sulfating agents and reaction temperature, a wide variety of sub-stitution patterns of Na-cellulose sulfates can be realized, covering the rangefrom 100 % C-6 substitution with NOSO4H, down to < 20 % in the case of SO3
at low reaction temperature (see Table 4.4.12).
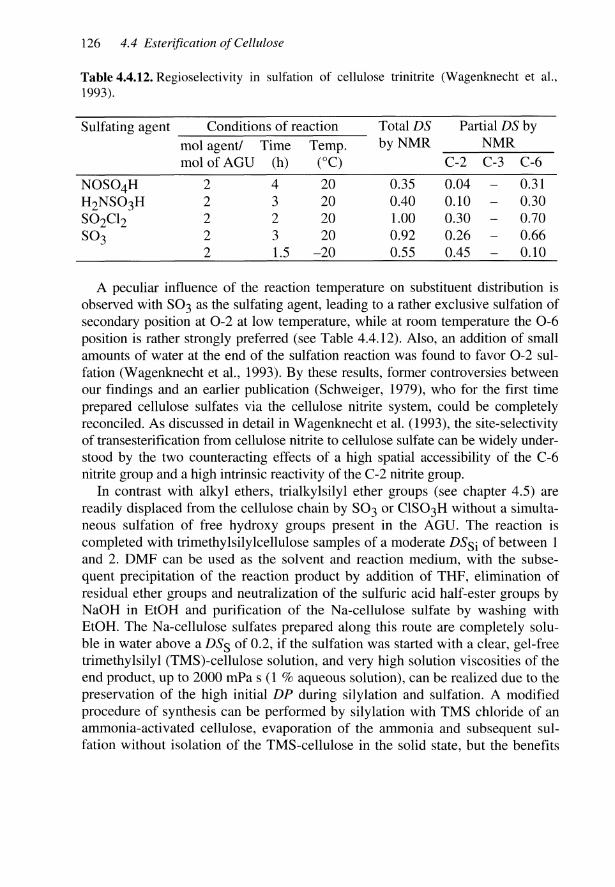
126 4Λ Esterification of Cellulose
Table 4.4.12. Regioselectivity in sulfation of cellulose trinitrite (Wagenknecht et aL1993).
Sulfating agent Conditions of reaction
NOSO4HH2NSO3HSO2Cl2
SO3
mol agent/mol of AGU
22222
Time(h)
43231.5
Temp.(0Q20202020
-20
Total DSby NMR
0.350.401.000.920.55
Partial DS byNMR
C-2
0.040.100.300.260.45
C-3 C-6
- 0.31- 0.30- 0.70- 0.66- 0.10
A peculiar influence of the reaction temperature on substituent distribution isobserved with 803 as the sulfating agent, leading to a rather exclusive sulfation ofsecondary position at O-2 at low temperature, while at room temperature the O-6position is rather strongly preferred (see Table 4.4.12). Also, an addition of smallamounts of water at the end of the sulfation reaction was found to favor O-2 sul-fation (Wagenknecht et al., 1993). By these results, former controversies betweenour findings and an earlier publication (Schweiger, 1979), who for the first timeprepared cellulose sulfates via the cellulose nitrite system, could be completelyreconciled. As discussed in detail in Wagenknecht et al. (1993), the site-selectivityof transesterification from cellulose nitrite to cellulose sulfate can be widely under-stood by the two counteracting effects of a high spatial accessibility of the C-6nitrite group and a high intrinsic reactivity of the C-2 nitrite group.
In contrast with alkyl ethers, trialkylsilyl ether groups (see chapter 4.5) arereadily displaced from the cellulose chain by 803 or ClSC^H without a simulta-neous sulfation of free hydroxy groups present in the AGU. The reaction iscompleted with trimethylsily!cellulose samples of a moderate DS^ of between 1and 2. DMF can be used as the solvent and reaction medium, with the subse-quent precipitation of the reaction product by addition of THF, elimination ofresidual ether groups and neutralization of the sulfuric acid half-ester groups byNaOH in EtOH and purification of the Na-cellulose sulfate by washing withEtOH. The Na-cellulose sulfates prepared along this route are completely solu-ble in water above a DS§ of 0.2, if the sulfation was started with a clear, gel-freetrimethylsilyl (TMS)-cellulose solution, and very high solution viscosities of theend product, up to 2000 mPa s (1 % aqueous solution), can be realized due to thepreservation of the high initial DP during silylation and sulfation. A modifiedprocedure of synthesis can be performed by silylation with TMS chloride of anammonia-activated cellulose, evaporation of the ammonia and subsequent sul-fation without isolation of the TMS-cellulose in the solid state, but the benefits

4.4.1 Esters of cellulose with inorganic acids 127
of this simplified synthesis have to be repaid by additional efforts in processingand purifying the reaction products (Wagenknecht et al., 1992b).
The DS§ of the final product depends of course on the molar ratio of sulfatingagent to AGU, but is limited by the level of the previous silylation, and was notfound to exceed the DS^. Figure 4.4.14 illustrates this by the results of sulfationof two TMS-cellulose samples of different
2.5
«n 1.5ωQ
0.5
O 2 A 6 8 10MoI sulfcrting agents / mol AGU
Figure 4.4.14. DS$ dependence on the amount of sulfating agent (SOs ·; ClSOsH ·)during homogeneous sulfation of TMS-cellulose with DS 1.5 and 2.4 at 20 0C, 3 h(Wagenknecht et al., 1992b, reprinted with permission from Elsevier Science).
With highly substituted silylcelluloses, DS$ values of 2.5 and higher could berealized, but a definitely trisubstituted cellulose sulfate has not yet been preparedalong this route.
In the sulfation of TMS-cellulose, a remarkable preference of the C-6 positionis generally observed, resulting in an exclusively C-6-substituted cellulose sul-fate up to a DS of 0.95, with chlorosulfonic acid as sulfating agent. At higher DSvalues also the C-2 position, and to a smaller extent additionally the C-3 posi-tion, is occupied by sulfate groups. Addition of pyridine as a weak base to thereaction mixture on sulfation results in a decrease of DS§ and a somewhat morepronounced esterification of the C-2 position.
Regarding the mechanism of silylcellulose sulfation, the trialkylsilyl ether groupdefinitely acts as a leaving group. Even under rather mild conditions, i.e. on sul-fation with amidosulfonic acid in the presence of an excess of TEA, no protectingaction of the silyl groups could be detected, as observed in the esterification ofTMS-cellulose with carbonic acid chlorides (see chapter 4.4.3 and 4.5), and againthe O-6 position was preferentially esterified in 2,6-O-TMS-cellulose of DS^ of1.5. Still an open question remains, as to why the DS$ was never found to exceedthe DS$i due to sulfation of residual free hydroxy groups in moderately high sub-stituted silylcelluloses. Some kind of 'steric shielding' of the hydroxy groups bythe large voluminous reaction complex between the trialkylsilyl groups and thesulfating agent could be discussed as a possible cause. Concerning the interactionbetween the trialkylsilyl ether group and 803, a primary cellulose derivative con-

128 4.4 Esterification of Cellulose
taining approximately equal molar amounts of sulfur and silicium has been iso-lated from the reaction mixture under anhydrous aprotic conditions. From theanalytical data obtained (Wagenknecht et al, 1992b; Nehls, 1994) we assumed aninsertion reaction of 803 between the cellulose chain and the trialkylsilyl groupwith the primary formation of the cellulose silylsulfate with a subsequent splittingoff of the silyl moiety as a trialkylsilanol, rapidly forming hexaalkyldisiloxane.This route of reaction is known from low molecular analogues (Bott et al., 1965).The sulfuric acid haifester groups are rather stable in an aqueous alkaline medium,but are saponified much faster in an aqueous or alcoholic acid milieu. A preferen-tial loss of sulfate groups at C-2 has been observed in the "methylation analysis" ofcellulose sulfates (Gohdes et al., 1997).
Just as with other cellulose-related polysaccharides, xylans can be convertedto sulfate half-esters too. With a beech wood xylan (DP ~ 140, 80-83 % pento-san content) dissolved after appropriate activation in the ^04/DMF system, aDS§ of 0.2 was obtained with SÜ2 (via NOSC^H formed in situ) and a DS$ of0.55 reached with 803 as the sulfating agent (Philipp et al., 1987).
Summary of routes to cellulose sulfates with defined patterns of substitution
The following Table 4.4.13 gives an overview of the various procedures forsynthesizing cellulose sulfates with a defined pattern of functionalization withinthe AGU including the range of DS§ realized.
Table 4.4.13. Overview of routes to regioselectively functionalized Na-cellulose sul-fates.
Site of sulfation
C-6
C-2C-6/C-2
C-2/C-3
Intermediate
NitriteSilyl etherCelluloseNitriteNitriteSilyl etherAcetate
Sulfating agent
NOSO4H; SO2
ClSO3HAc2O + ClSO3HSO3
SO3; SO2Cl2SO3;C1SO3HSO3; H2NSO3H
Range of DS$realized0.3-0.60.3-1.00.3-0.80.3-1.01.0-2.00.5-2.00.3-1.4
Properties of cellulose sulfates
Cellulose sulfuric acid half-esters in the acid form (H+ form) can be isolatedfrom the appropriate reaction mixture as a white hygroscopic mass soluble inwater and in rather polar organic liquids at a DS above 0.2-0.3. Due to thestrongly acidic character of the SOßH groups the products are unstable in the

4.4.1 Esters of cellulose with inorganic acids 129
solid state as well as in solution, as they are susceptible to a fast 'autohydrolytic'chain degradation and a splitting off of the half-ester groups.
The stable form of cellulose sulfuric acid half-esters generally employed inapplication is the sodium salt, a white, odorless and tasteless powder that can betransformed to clear films via an aqueous solution, and which exhibits a goodthermal stability up to 150 0C for a short time, and up to 100 0C for a longertime, in the purified, acid-free state.
In dependence on DP, Na-cellulose sulfates are completely water-soluble abovea DS of 0.2-0.3, the limiting value depending on the uniformity of substituentdistribution along and between the polymer chains, in consequence of the proce-dure of synthesis. Furthermore, a predominant C-6 substitution obviously favorssolubility as compared with the positions C-2 and C-3. For water-soluble Na-cellulose sulfates in the low DS range, between 0.2 and 0.45, an [i]]-Mw relation-
ship of [77] = 1.365 x 10'2 M^Y0-94, with [77] given in ml/g, has been reported byAnger et al. (1987). Very high solution viscosities of up to about 5000 mPa s forthe 1 % aqueous solution have been reported by Schweiger (1979) for Na-cellulose sulfate samples in the low DS range, between 0.3 and 0.5, prepared viacellulose nitrite from a high molecular cellulose (cotton) avoiding significant deg-radation, while in the DS range above 1, only viscosities of about 1000 mPa s wererealized by the same author. In our work, solution viscosities up to 2000 mPa swere measured with samples prepared from cotton !inters via cellulose trinitrite orTMS-cellulose, while with wood dissolving pulps as the starting material, the so-lution viscosity of comparable samples did not exceed a value of about 500 mPa s.Na-cellulose sulfate solutions of higher concentration exhibit pseudoplastic(thixotropic) behavior, with the thixotropic effect increasing with decreasing DS.Aqueous solutions of Na-cellulose sulfate show a remarkably good resistanceagainst thermodegradation and shear degradation (Schweiger, 1979). A viscosityreduction of only 25 % was supported after 25 h of thermal treatment at 100 0C;and chain degradation on continuous shearing proved to be much less than withother polysaccharides, including conventional cellulose ethers. A special rheologi-cal phenomenon observed with aqueous solutions of Na-cellulose sulfates is theformation of thermoreversible gels (Holzapfel et al., 1986; Dautzenberg et al.,1994), first reported (Schweiger, 1972) with high DS samples in the presence ofpotassium ions. According to our results (Dawydoff et al., 1984) aqueous Na-cellulose sulfate solutions with a polymer content of 1 % and a DS between 0.25and 0.40, form stiff thermoreversible gels with a melting point of about 65 0C inthe presence of 2 % KCl after isolating the Η-form of the ester by water-freemethanol from the homogeneous N2O4/SO2/DMF medium. For the samples ofvery low DS, between 0.15 and 0.20, thermoreversible gels can be obtained with-out addition of K+ by dissolving the polymer in hot water and subsequent coolingof the 1 % solution (Philipp et al., 1985).

Tab
le 4
.4.1
4. C
ellu
lose
sul
fate
: re
latio
nshi
p be
twee
n ce
rtai
n pr
oper
ties
and
appl
icat
ions
(Sc
hwei
ger,
197
9).
Vis
cosi
ty
Pseu
do-
Solu
bil-
E
nzym
e Sh
ear
Tem
p-
Susp
en-
Film
C
ross
- C
ross
- Pr
otei
n So
lven
tpl
astic
- ity
re
sist
ance
re
sist
ance
er
atur
e si
on
form
a-
linki
ng
linki
ng
reac
- to
lera
nce
ity a
nd
stab
ility
st
abili
ty
tion
film
so
lutio
n tiv
ityyi
eld
poin
t
Ter
tiary
oil
+ +
+re
cove
ryO
il-w
ell
+ +
+ +
+ +
drill
ing
Pain
ts
+ +
+ +
Pape
r +
+ +
+ +
Tex
tiles
+
+ +
+E
xplo
sive
s +
+Ph
otog
raph
y +
+ +
+C
osm
etic
s +
+ +
+ +
Too
thpa
ste
+ +
+Fo
od
+ +
+ +
+ +

4.4.1 Esters of cellulose with inorganic acids 131
Na-cellulose sulfate behaves as a strong polyelectrolyte. The aqueous solutionshows a considerable compatibility with some organic liquids, e.g. lower ali-phatic alcohols, which increases somewhat with the DS in the range between 0.3and 1.0. Na-cellulose sulfate samples with a DS between 0.3 and 1.5 are notprecipitated from their aqueous solution by mono-, di- or trivalent metal cations.As an anionic polyelectrolyte, Na-cellulose sulfate forms polyelectrolyte com-plexes, including insoluble polysalts with cationic polyelectrolytes (Philipp etal., 1989), and also with the cationic sites of proteins. In the presence of a suffi-ciently large amount of hydrophobic acetyl groups, i.e. in the case of Na-cellulose acetate sulfates, the water solubility of the product gets loss, but a re-markably high water-binding power of up to 1000 % and more still remains,unfortunately, however, combined with a low salt tolerance due to the ioniccharacter of the hydrophilic sulfate groups.
Na-cellulose sulfate can be degraded by cellulolytic enzymes or cellulase-producing microorganisms up to a DS of about 1, the limiting value dependingsomewhat on the uniformity of substituent distribution along the polymer chains(Schweiger, 1972). High-purity Na-cellulose sulfates, free of acid residues andtoxic heavy-metal ions, definitely exhibit no cytotoxicity (Dautzenberg et al.,1985a; 1996a and 1996b). In line with other sulfate-group-bearing polyelectro-lytes, Na-cellulose sulfate can show biological activity on interaction with hu-man blood (heparinoid effects) (Okajima et al., 1982).
For further details on the properties of cellulose sulfates the reader is referredto the comprehensive overview given by Schweiger (1972).
Application of cellulose sulfate
Up to now, Na-cellulose sulfate has not been a commodity derivative of cellu-lose but is still a specialty despite numerous promising areas of application. Thismay be caused mainly by the fact that technologically, economically and ecol-ogically feasible routes of synthesis, avoiding excessive chain degradation, havenot been published before about 1980, while a broad variety of water-solublecellulose ethers had already established its market.
An overview of possible areas of application in relation to product propertieshas been published (Schweiger, 1972) and is presented in Table 4.4.14. Thenumerous areas of application already tested or proposed can be systematizedaccording to(i) film-forming properties of Na-cellulose sulfates;(ii) special rheological effects of Na-cellulose sulfates in aqueous solution;(iii) behavior of Na-cellulose sulfates as anionic polyelectrolytes;(iv) biological activity of Na-cellulose sulfate.
The film-forming properties of Na-cellulose sulfates have been proposed for ap-plication in coatings, especially in the paper industry, taking into account the possi-

132 4Λ Esterification of Cellulose
ble modification by subsequent crosslinking of the highly accessible, free hydroxygroups by conventional crosslinking agents for cellulose, e.g. formaldehyde.
The high solution viscosity of adequately synthesized Na-cellulose sulfates inwater means these products are recommended as thickeners and viscosity en-hancers in many industrial and domestic areas, and the high efficiency of thesesolutions in stabilizing suspensions of e.g. TiU2 has been emphasized. Their gel-forming properties in connection with nontoxicity and good compatibility withother polysaccharides make cellulose sulfates of an appropriate DS well suitedfor preparing thermoreversible gels as required e.g. in microbiology, either as asingle component or in a gel blend with other polysaccharides.
The anionic component in polyelectrolyte complexes of Na-cellulose sulfatefinds promising applications in the membrane area: pervaporation membranescomposed of a low DS cellulose sulfate with a special substitution pattern andpolydimethyldialylammonium chloride have combined mechanical stability, highflux rate and good selectivity in the separation of lower aliphatic alcohols fromtheir mixture with water (Richau et al., 1996). The interface reaction between Na-cellulose sulfate in aqueous solution and a solution of a suitable cationic polyelec-trolyte can be used to encapsulate biological materials under quasi-physiologicalconditions without impeding their biological activity, as shown in comprehensiveinvestigations (Dautzenberg et al., 1985a) with enzymes, living cells, microorgan-isms or cell organelles. Interaction between Na-cellulose sulfates and proteins canbe employed to enhance the viscosity of these system and/or to separate specialproteins from aqueous solution (Schwenke et al., 1988).
Last but not least, the heparinoid action (i.e. anticlotting activity of humanblood) of special Na-cellulose sulfates must be mentioned here. Heparinoid ac-tivity was observed (Okajima et al., 1982) with highly substituted products andshown to depend especially on a high degree of substitution in the C-2/C-3 posi-tion also at moderate total DS$ (Klemm et al., 1997). Table 4.4.15 demonstratesthis with some results.
Table 4.4.15. Anticlotting activity of Na-cellulose sulfates (NaCS) with different pat-terns of substitution (25 μg of NaCS/ml of blood) (Klemm et al., 1997).
Total DSS
0.950.951.14Blank experiment
Partial D5§ at position:
C-2 C-3 C-60.00 0.00 0.950.30 0.30 0.350.74 0.09 0.31
TT
(S)
18.929.0
> 600.017.5
PTT
(s)
80.80136.5
> 600.035.0
TT Thrombin time.PTT Partial thromboplastin time.

4.4.1 Esters of cellulose with inorganic acids 133
Similar anticlotting effects are known for xylan sulfates with a DS$ of 1.5-2(Kindness et al., 1979, 1980; Philipp et al., 1987; Stscherbina and Philipp,1991). Also, some other polysaccharide sulfates, cellulose and xylan sulfateswere reported to stimulate immunological defense, to inhibit growth of cancerand to show beneficial effects against HIV infections (Hatanaka et al., 1991).
4.4.1.4 Cellulose phosphate and other phosphorus-containingcellulose derivatives
General comments on phosphorylation reactions and products obtained
The element phosphorus can be covalently attached to the cellulose chain via areaction of hydroxy groups to give:
phosphate groups CeIl-O-P(O)(OH)2
phosphite groups CeIl-O-P(OH)2
phosphonic acid groups CeIl-P(O)(OH)2
Many of the reactions involved are not quite clear yet regarding their course andmechanism, as well as the pattern of substitution. The products obtained arefrequently insoluble due to crosslinking and rather ill-defined, and are oftencharacterized by their phosphorus content only.
Most frequently employed are derivatives of pentavalent phosphorus, i.e.^ΡΟφ Ρ2θ5, and POC^. Compared with the corresponding compounds ofhexavalent sulfur, these phosphorylating agents, usually leading to anionic cel-lulose phosphates, show a lower reactivity in esterification and lead to much lesschain degradation during this process. As a peculiarity of cellulose phosphoryla-tion by the above-mentioned reagents, a tendency to form oligophosphate sidechains has to be mentioned, frequently resulting in crosslinking between cellu-lose chains, and thus impeding product solubility.
Phosphorylation of cellulose is performed either by reaction at the hydroxygroups of the original polymer, or by a second-hand derivatization of a celluloseether or ester already formed. In the former case the reaction usually starts in aheterogeneous system or employs a cellulose solution in a nonderivatizing sol-vent system; in the latter case a homogeneous system is generally preferred inorder to arrive at soluble products. Regioselective patterns of substitution can inprinciple be realized along both of these routes.
As reaction products, usually anionic cellulose derivatives are obtained. Theircomplete solubility in water or aqueous alkali, however, is, in contrast with cel-lulose sulfate synthesis, rather more the exception than the rule, due to theabove-mentioned crosslinking reaction, and requires special procedures for thereaction itself and for the subsequent product isolation and purification. Appli-cations of cellulose phosphorylation already practised are the preparation ofcellulose-based cation exchangers and the flame proofing of cellulosic textiles.

134 4.4 Esterification of Cellulose
Reaction routes and systems for cellulose phosphorylation
Highly concentrated or water-free orthophosphoric acid has been widely used asan effective phosphating agent, and various procedures have been reported forpreparing soluble as well as insoluble cellulose phosphates with phosphoruscontents of about 10 % (Nuessle et al., 1956). According to Touey (1956), wa-ter-soluble cellulose phosphates of rather high DP can be prepared with water-free ί ΡΟφ For enhancing phosphorylation reactivity, mixtures of HßPC^ with^2^5 have been employed. As to be expected, the degree of substitution ofphosphorus atoms (DSp) increases with the molar ratio of reagent per AGU andthe time of reaction, but chain degradation is enhanced too. Water-soluble cel-lulose phosphates have been synthesized in ternary systems of Η^ΡΟφ ?2θ5 andDMSO, connected with severe chain degradation down to a DP of about 200,with cotton cellulose as the starting material, and also with ternary systems ofΙ^ΡΟφ ^2^5 and aliphatic alcohols with 4 to 8 C-atoms, arriving at productswith up to 6 % phosphorus, corresponding to a DSp of < 0.2 (Nuessle, 1956;Touey, 1956). The reaction of cellulose with a melt solution of t^PC^ and urearesulted in the formation of a soluble, but strongly degraded, cellulose mono-phosphate monoammonium salt. The same system was employed by Nehls andLoth (1991) at a lower temperature of 120 0C for the phosphorylation of beadcellulose and cellulose powders to highly swellable but still water-insolubleproducts with DSp values between 0.3 and 0.6. The nitrogen content of thesecellulose phosphates was very low (0.1-0.2 %). A very preferential C-6 substi-tution could be concluded from the 13C NMR spectra. A significantly higherphosphorus content than that corresponding to the DSp calculated from the13C NMR spectrum indicates the formation of cellulose oligophosphates, whichobviously form crosslinks impeding solubility. A hydrogen bond stabilizedcomplex between Η^ΡΟφ urea and cellulose, according to the scheme in Fig.4.4.15, is assumed as the transition state in this cellulose phosphorylation.
HI
.Ox
CeII-CH2^ XH
HOx !x A/P-OJ IxNH-C-NH2
HO Il ^HX MO O
Figure 4.4.15. Scheme of reaction complex in cellulose phosphorylation withand urea (Nehls and Loth, 1991).

4.4.1 Esters of cellulose with inorganic acids 135
Phosphorus oxychloride (POCl3) is known as an effective phosphating agent forcellulose from numerous studies, starting from a cellulose suspension in DMF orpyridine, or from a cellulose solution in a nonderivatizing solvent system. Usuallyonly partially soluble products are obtained by the procedures described, andphosphorylation is frequently accompanied by an excessive chlorination, i.e. for-mation of desoxycellulose entities. According to Vigo and Welch (1973) im-midinium compounds can be formed in systems containing POCl3 or PCl3 andDMF (see scheme in Fig. 4.4.16) which promote cellulose chlorination.
T ?H
PCI3 + 20 = C-N(CH3)2 Ce||_o-P-OHCellulosephosphite
+ H2O
ClI
CeII-O-P-OH + CeII-CIChlorodesoxycellulose
+ CeII-OH I - DMFH
CI-P
I θX0-C=N(CH3)2
'O-C=N(CH3)2
H
Cl+ CeII-OH ι θ
2 Cle > Ce|,_o_p_o__Cz:N(CH3)2Cle
- HC/, - DMF
- HCI, - DMF + H2O
OHI
CeII-O-P-OHCellulosephosphite
Figure 4.4.16. Scheme of reaction of PCl3 in DMF with cellulose (Vigo and Welch,1973; Wagenknecht et al., 1979).
As shown by Wagenknecht et al. (1979) for the action of PCl5, POCl3 andPCl3 on cellulose in formamide and dimethylformamide as the medium, thephosphorylation to partially soluble, considerably degraded products with DSpvalues of about 0.3 is accompanied by an excessive chlorination, up to a degreeof substitution of chlorine atoms (DS(^) of 0.7 in DMF, whereas the productsobtained in formamide contained only very small amounts of chlorine (DS^i< 0.05). The problem of simultaneous phosphorylation and chlorination of cel-lulose by POCl3 was comprehensively studied by Zeronian et al. (1980) in de-pendence on various reaction parameters.
The reaction of cellulose dissolved in nonderivatizing systems like NMMNO,LiCl/HMPT or DMA/LiCl results in a spontaneous coagulation and rather inho-

136 4A Esterification of Cellulose
mogeneous reaction products containing phosphorus as well as chlorine that areonly partially soluble in water and rather heavily degraded. As compared withcellulose suspensions as the starting system, these initially homogeneous sys-tems exhibit no advantages, if the preparation of soluble high molecular cellu-lose phosphates is intended.
Trivalent phosphorus can be introduced into the cellulose molecule by reac-tion with PC13 (Vigo and Welch, 1973) or by transesterification with dimethylphosphite, arriving at hydrolysis-susceptible phosphite esters of cellulose(Yuldashev et al., 1965). Synthesis of cellulose phosphites has also been re-ported, employing mixed anhydrides of hydrophosphorus and acetic acid andarriving at phosphorus contents of up to 8 % (Predvoditelev et al., 1966). Ex-perimental routes to cellulose phosphonates with the phosphorus directly boundto a C-atom of the polymer are either an esterification with methyl or phenyl-phosphonic anhydride (Yuldashev and Muratova, 1966; Petrov et al., 1965), or atwo-step reaction consisting of chlorination of the polymer with SOC12 to givechlorodesoxycellulose with a high Cl content (up to 16 %) and the subsequentreaction of this compound with triethylphosphite to the cellulose phosphonatevia an Apruzov rearrangement. Also the preparation of cellulose phosphoniteshas been reported (Kiselev and Danilov, 1962).
Completely or partially substituted cellulose derivatives have been phos-phated by various acids or acid chlorides of pentavalent phosphorus, usuallystarting from a homogeneous system and rather frequently arriving at solubleproducts. Stable ether groups like the carboxymethyl groups and also the acetylgroup of cellulose acetates act as efficient protecting groups in the nonaqueoussystems involved, and only free hydroxy groups are converted to phosphategroups. CMC with a DS of 0.8 was converted to an ether ester with a DSp of 0.3in the system F^PC^/urea with the phosphate groups again preferentially locatedat C-6 (Nehls and Loth, 1991). With a sample of hydroxyethy!cellulose (MS ~ 2)a considerable higher DSp of 0.6 was obtained with the same system under com-parable conditions of reaction, obviously due to the participation of the hydroxyend groups of the side chains in esterification.
A somewhat more detailed consideration is deserved by the phosphorylationof cellulose acetates, as different patterns of substitution of cellulose phophatescan be realized here after splitting off the acetate groups in aqueous alkalinemedium without significantly affecting the phosphate groups. The preparation ofsoluble cellulose acetate phosphates by reacting the cellulose acetate after dis-solution in acetone with POC^ in the presence of an aliphatic amine has beenreported. Whistler and To wie (1969) used polytetraphosphoric acid in combina-tion with tri-ft-butylamine in DMF to esterify free hydroxy groups of a low-DScellulose acetate at 120 0C to a DSp of about 1, arriving at a water-soluble prod-uct after elimination of the acetyl groups. From a comparison of different phos-phating agents, i.e. diphosphoryl tetrachloride, phosphorus oxychloride, dichlo-

4Λ.1 Esters of cellulose with inorganic acids 137
rophosphoric acid, and polytetraphosphoric acid, in phosphating partially sub-stituted cellulose acetates in DMF in the presence of an aliphatic amine, it can beconcluded that all the chlorine-containing agents, especially diphosphoryl tetra-chloride, lead to an early coagulation of the initially homogeneous reaction sys-tem, resulting in cellulose phosphates of poor solubility in spite of a rather highDSp of between 0.5 and 1.0 (see Table 4.4.16).
Table 4.4.16. Comparison of different phosphating agents in the presence of tn-n-butylamine, in the phosphorylation of commercial cellulose 2-acetate (reaction time 6 h;deacetylation in NaOH/EtOH) (Philipp et al., 1995).
Phosphating agent(mol/mol AGU)
HPO2Cl2
P2O3Cl4
(2.0)(1.5)(1.5)
Amine(mol/mol
AGU)15153
T(0C)
2020
120
DSp
0.590.450.78
% Cl Solubility
0.070.17
2 N NaOH
GelInsolubleSoluble
H2O
GelInsolubleSoluble
With polytetraphosphoric acid, on the other hand, the system remained homo-geneous during the whole reaction, and water- or alkali-soluble cellulose phos-phates could be isolated under suitable conditions after deacetylation(Wagenknecht, 1996). The advantages of the combination polytetraphosphoricacid/tri-ft-butylamine were fully confirmed in this study, and this combinationhas been employed for phosphating partially substituted cellulose acetates over awide range of DS and with different patterns of substitution. Some results ob-tained with statistically and with regioselectively in C-6-substituted acetates aresummarized in Table 4.4.17.
The pattern of substitution of the resulting cellulose phosphates resembles aninverse image of that of the original acetate, with the DSp increasing generallywith decreasing DS^C. But it must be emphasized that in contrast with sulfation,not all of the free hydroxy groups could be converted to phosphate groups, thedifference increasing with the increasing amount of free hydroxy groups. As canbe seen also from the data in this Table, phosphate groups in the C-6 positionobviously promote product solubility in aqueous media much more than anequal amount of ester groups in the C-2/C-3 position.
Unstable primary substituents (ether or ester groups) can act as the leavinggroup in a subsequent phosphorylation with ?2θ5 or POClß in the absence of anamine, as shown by our results in the cellulose nitrite system or in case of TMS-cellulose. TMS-cellulose of DS 1.5 with the silyl groups predominantly in the O-6 position could be reacted with an excess of phosphating agent in DMF/TEA togive an insoluble cellulose phosphate with a DSp of 0.3-0.6 (Klemm et al.,
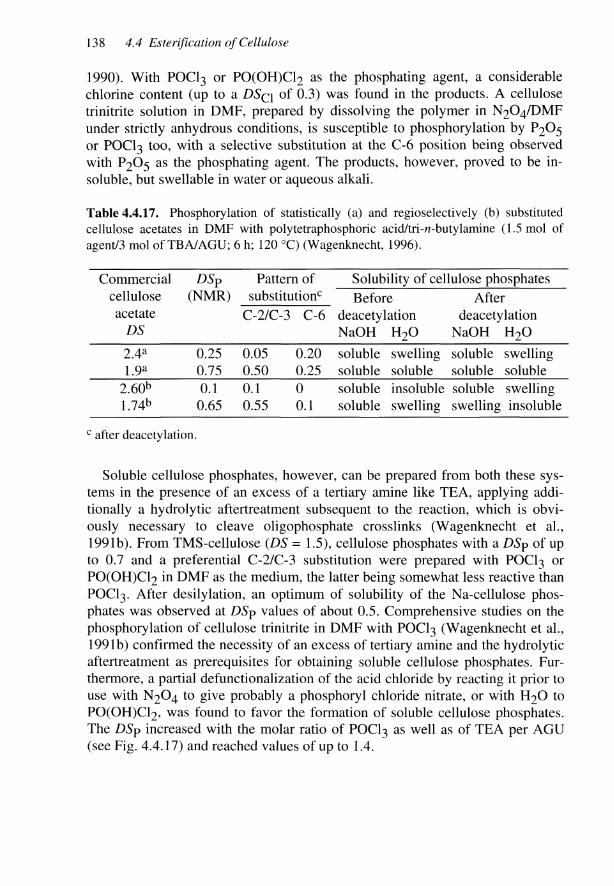
138 4.4 Esterification of Cellulose
1990). With POCl3 or PO(OH)Cl2 as the phosphating agent, a considerablechlorine content (up to a DS^\ of 0.3) was found in the products. A cellulosetrinitrite solution in DMF, prepared by dissolving the polymer in N2U4/DMFunder strictly anhydrous conditions, is susceptible to phosphorylation by P2U5or POCl3 too, with a selective substitution at the C-6 position being observedwith P205 as the phosphating agent. The products, however, proved to be in-soluble, but swellable in water or aqueous alkali.
Table 4.4.17. Phosphorylation of statistically (a) and regioselectively (b) substitutedcellulose acetates in DMF with polytetraphosphoric acid/tri-n-butylamine (1.5mol ofagent/3 mol of TBA/AGU; 6 h; 120 0C) (Wagenknecht, 1996).
Commercialcelluloseacetate
DS
2.4a
1.9a
2.60b
1.74b
DSp(NMR)
Pattern ofsubstitution0
C-2/C-3 C-6
O.O.O
O.
2575.165
0.050.500.10.55
O.O.OO.
2025
1
Solubility of cellulose phosphatesBefore
deacetylationNaOH H2O
solublesolublesolublesoluble
swellingsolubleinsolubleswelling
Afterdeacetylation
NaOH H2O
solublesolublesolubleswelling
swellingsolubleswellinginsoluble
; after deacetylation.
Soluble cellulose phosphates, however, can be prepared from both these sys-tems in the presence of an excess of a tertiary amine like TEA, applying addi-tionally a hydrolytic aftertreatment subsequent to the reaction, which is obvi-ously necessary to cleave oligophosphate crosslinks (Wagenknecht et al.,199Ib). From TMS-cellulose (DS = 1.5), cellulose phosphates with a DSp of upto 0.7 and a preferential C-2/C-3 substitution were prepared with POCl3 orPO(OH)Cl2 in DMF as the medium, the latter being somewhat less reactive thanPOCl3. After desilylation, an optimum of solubility of the Na-cellulose phos-phates was observed at DSp values of about 0.5. Comprehensive studies on thephosphorylation of cellulose trinitrite in DMF with POCl3 (Wagenknecht et al.,199Ib) confirmed the necessity of an excess of tertiary amine and the hydrolyticaftertreatment as prerequisites for obtaining soluble cellulose phosphates. Fur-thermore, a partial defunctionalization of the acid chloride by reacting it prior touse with N2U4 to give probably a phosphoryl chloride nitrate, or with H2O toPO(OH)Cl2, was found to favor the formation of soluble cellulose phosphates.The DSp increased with the molar ratio of POCl3 as well as of TEA per AGU(see Fig. 4.4.17) and reached values of up to 1.4.

4.4.1 Esters of cellulose with inorganic acids 139
0.8
0.6
0.2
0.6
0.2
0 2 4 - 6MoI POCl3XmOlAGU
0 4 . 8 12 16 20Mol TEA/molAGU
Figure 4.4.17. Effect of POC13 (a) and TEA (b) input on cellulose phosphorylation inN2O4/DMF at 20 0C (Wagenknecht et al., 199Ib).
The Cl content of the product depended significantly on the order of additionof POC13 and amine, and was much higher (DSQ up to 0.2) with the POC^added before the amine. According to our experience, a long residence time of astrongly acidic phosphating system with an acid chloride as the agent generallyfavors chlorination, while the presence of the amine exerts some buffering ac-tion, besides its effect as an adjuvant base for enhancing the reactivity of theagent. From the NMR spectra of the phosphates, a preferential location of theester groups in the C-2/C-3 position could be concluded. Optimal solubility wasobserved also here at a DSp level of about 0.5, this range being broadenedsomewhat by employing a difunctionalized POQ^. Obviously, the solubility ofcellulose phosphates, prepared by this as well as by other procedures in water oraqueous alkali, is determined by two counteracting effects, increasing with DSp,i.e. an increasing hydrophilicity due to the anionic substituents, and an increas-ing tendency to crosslinking. Probably some kind of optimal balance is obtainedin the DSp region of about 0.5.
Finalizing this presentation of experimental routes to cellulose phosphates, theheterogeneous reaction of alkali cellulose with POC^ in the presence of benzeneshall be mentioned as a modification of the Schotten-Baumann reaction for es-terification, leading here, according to Reid and Mazzeno (1949), to a consid-erably degraded cellulose phosphate.
Properties of cellulose phosphates
The attachment of phosphorus atoms to the cellulose chain significantly de-creases the inflammability of cellulose threads due to less formation of inflam-mable volatiles on thermal degradation. This flame retardation is still increasedby the presence of chlorine atoms frequently introduced in side reactions ofphosphorylation such as chlorodesoxycellulose units.

140 4.4 Esterification of Cellulose
By introducing the anionic phosphate groups into the cellulose molecule, ca-tion-exchange properties are conveyed to the polymer and its hydrophilicity isenhanced. The H+ form of the phosphate group shows a moderate acidity onlyand can be stored for some time without significant hydrolytic chain cleavage, incontrast with cellulose sulfate. At a DSp above 0.2, sodium cellulose phosphatescan be, but do not necessarily have to be, water- or alkali-soluble. As demon-strated, especially by the regioselectively substituted cellulose phosphates pre-pared via cellulose acetates, the site of substitution is also relevant to solubility,C-6-substituted products showing a much better solubility. With soluble sodiumcellulose phosphates, very high solution viscosities can be obtained if excessivechain degradation during esterification is avoided. Probably also strong inter-molecular interactions via phosphate groups and/or oligophosphate side chainscontribute to this high viscosity.
Application of cellulose phosphates
Phosphorylation of cellulose threads is employed to convey flame retardancy tocellulosic textiles for special, mostly technical, use, taking into account somedeterioration of textile mechanical properties and textile handling. Celluloseparticles of different sizes and shapes bearing phosphate groups, find wide ap-plication as weak cation exchangers, especially in biochemical separation proc-esses. Soluble cellulose phosphates have been recommended as viscosity en-hancers and thickeners in aqueous systems, with the nontoxicity of these prod-ucts being an advantage. Regioselectively (in the C-2/C-3 position) substitutedcellulose phosphates were recently observed to inhibit the activation of detri-mental blood proteins in hemodialysis after incorporation in hemodialysis mem-branes (Wagenknecht, 1996).
4.4.1.5 Cellulose borates
Boron-containing cellulose derivatives have been studied predominantly in orderto improve special applicational properties of cellulosic materials, for exampleflame retardancy or heat stability. Systematic chemical investigations on thecourse and mechanism of cellulose borylation are rather scarce and are obvi-ously impeded by ill-defined products due to crosslinking and formation ofoligo- and polyborate moieties. These tendencies being more pronounced than inthe case of phosphorylation.
Two main routes of synthesis have being employed rather frequently to pre-pare boronic acid esters of cellulose, i.e.(i) the direct esterification of cellulosic hydroxy groups with orthoboric or me-taboric acid according to
CeIl(OH)3 + H3BO3 -»(CeIlO)3B

4.4.1 Esters of cellulose with inorganic acids 141
(ii) a transesterification of cellulose with boronic acid esters of lower aliphaticalcohols (boron alkoxides)
CeIl(OH)3 + B(OR)3 -> (CeIlO)3B
Due to the strong crosslinking tendency of the borylation agents indicated in theborderline schemes of reaction, a meaningful assessment of the DS requires ad-ditional assumptions on reagent functionality realized in the reaction, and theproducts are therefore usually characterized just by their boron content.
A direct borylation of cellulosic hydroxy groups has usually been performedwith ortho- or metaboric acid in a melt of urea at 150-200 0C. Ermolenko (Er-molenko et al, 197Ia) reports a boron content of 1.8 % after reacting cellulosewith HBO2/urea at 220 0C for 1 h. This boron content corresponds to a formalDS of about 0.7, assuming a trifunctional mode of reaction. A parallelism be-tween this borylation reaction and a phosphorylation with HPO3/urea at 150 0Cto a DSp of about 1 is emphasized in the above-mentioned publication. Thepreparation of a mixed borate/phosphate of cellulose by subsequently reactingthe polymer with H3PO3/urea and with Η4Ρ2θ7 or HPO3/urea in the tempera-ture range 100-200 0C has been described in Ermolenko et al. (197Ib). Ac-cording to Ermolenko et al. (197Ia) cellulose acetate can been converted to anacetate borate mixed ester by treatment with H3BO3 at 260 0C, obviously via theintermediate formation of poly boric acids.
The transesterification of cellulose with boron trialkoxides [B(OR)3, with R =Me, Et, Pr] can be performed at considerably lower temperature, for example inbenzene as the medium (Gertsev et al., 1990). According to Arthur and Bains(1974) a boron content of 6.8 % could be obtained by this procedure, corre-sponding to trisubstitution of the cellulosic hydroxy groups assuming again atrifunctional reaction. Also, graft copolymers of cellulose can be borylated withboron trialkoxides, as demonstrated by Tyuganova and Butylkina (1992) or graftcopolymers of cellulose with 2-methyl-5-vinylpyridine.
As a rather special route to cellulose borates the reaction of cellulose as ahydroxy group containing polymer with trialkylboranes has to be mentioned,which, according to
BR3 + R'-OH -» ROBR2 + RH
is applied to the analytical determination of active hydrogen atoms (Koester etal., 1971). As described by Dahlhoff et al. (1988), a per-O-diethyl-borylatedamylose or cellulose can be regioselectively reduced by an ethyl diborane to aboron-substituted polyanhydroglycitol.

142 4.4 Esterification of Cellulose
The formation of five-membered ring complexes between vicinal hydroxygroups of polysaccharides including cellulose with boric acid in aqueous sys-tems has already been reported many years ago. More recently, a reversible gelformation of a well-degraded 2,3-dihydroxypropylcellulose of DP 20 with boraxin aqueous solution has been studied, and the formation constants of the prob-able five-membered ring complexes have been determined (Sato et al., 1992).
Regarding now special product properties of cellulose borates, the attachmentof borate groups conveys to the cellulose chain a cation-exchange capacity andan enhanced thermal stability due to a decreased rate of thermal oxidation (Ar-thur and Bains, 1975). According to Ermolenko and Luneva (1977) the nontoxicborate group exhibits antibacterial and antifungal as well as hemostatic activi-ties. Important for several areas of application is the strong crosslinking ten-dency during borylation of cellulose. The stability of the borate ester group tohydrolysis or alcoholysis is discussed with some degree of controversy in theliterature, probably due to different amounts of crosslinking in the products in-vestigated.
Based on the above-mentioned properties, various areas of application of bo-rylated cellulose have been proposed: the crosslinking tendency on borylationwas claimed to be advantageous in packaging and micro-encapsulation. Theenhanced thermal stability of borylated cellulose has been considered advanta-geous in the preparation of e.g. insulating paper. The broad antibacterial activityof cellulose borates was emphasized as a basis for medical use.
4.4.1.6 Desoxycelluloses
The term 'desoxycellulose' denotes cellulose derivatives resulting from the sub-stitution of a hydroxy group by halogen, sulfur or nitrogen or even carbon, withthe hetero- or carbon atom directly bound to a carbon atom of the AGU. Halo-,pseudohalo- and thiodesoxycelluloses can be formally considered as celluloseesters of the appropriate hydrogen halides, hydrogen pseudohalides or of hydro-gen sulfide. Of special relevance to the organic chemistry of cellulose up to noware the chloro- and the iododesoxycelluloses. But a systematic investigation ofthe synthesis of desoxycelluloses is an open field of cellulose chemistry.
A route to desoxycelluloses starts from the cellulose esters with p-toluenesulfonic acid (tosylcellulose) or with methanesulfonic acid (mesylcellu-lose), usually reacting these esters with inorganic salts containing the group tobe introduced as the nucleophilic reagent in this displacement reaction. Someexamples are presented in Table 4.4.18.
According to Titcombe et al. (1989) the use of tetraalkylammonium fluoridesproved to be successful for reaching a high degree of substitution. Chlorodes-oxycellulose is most conveniently prepared by reacting cellulose with SOC^ inpyridine (Carre and Manclere, 1931), DMF (Polyakov and Rogowin, 1963),

4.4.1 Esters of cellulose with inorganic acids 143
CC14 (Fumasoni and Schippa, 1963) or CHCl3, arriving at DS^ values of up to1.0. But frequently also some sulfur (DSg up to 0.1) is introduced into the mac-romolecule, probably via cyclic sulfides (Carre and Manclere, 1931). Also,SO2C12 can be employed to prepare chlorodesoxycelluloses with DS values of0.4-0.8 (Wagenknecht et al., 1979). A homogeneous route to chlorodesoxycel-lulose was described by Furuhata et al. (1992), starting from a solution of thepolymer in DMA/LiCl and reacting with W-chlorosuccinimide and triphenyl-phosphine. A homogeneous chlorination can also be performed with methylsul-furyl chloride after dissolving the polymer in the system chloral/DMF. For pre-paring fluorodesoxycellulose, a treatment of mesylcellulose with an aqueousNaF solution has been described earlier by Pascu and Schwenker (1957) andKrylova (1987). But this route leads to a very low DS only, due to dissolutionproblems.
Table 4.4.18. Preparation of desoxycelluloses via (A) tosyl- or (B) mesylcellulose.
Desoxy group Reagents and conditions
Fluoro- B NaF in H2O ~ 0.05Chloro- A Tosyl chloride and pyridine at 0.4-0.9
high temperatureA LiCl in acetylacetone ~ 1.00
(2 h at 130 0C)Bromo- B NaBr in H2O ~ 0.1
A NaBr in acetylacetone ~ 1.0(2 h at 130 0C)
Iodo- A/B NaI in acetylacetone -1.0(2 h at 130 0C)
Mercapto- A H2S in pyridine ~ 0.28(8 h at 40 0C, then 70 h at
room temperature)A Na2S2O3 in DMSO
Cyano- A KCN in DMF or methanol ~ 0.41(100-150 0C)
Thiocyanato- A NaSCN in acetonylacetone ~ 1.03(11 h at 110 0C)
Azido- A NaN3 in DMSO 0.19(110-13O0C) 0.43
^Desoxy = degree of substitution of desoxy groups.

144 4.4 Esterification of Cellulose
According to Ishii et al. (1977) a fast reaction takes place at the C-6 position,followed by C-3, whereas no chlorination was observed at C-2. A complete ex-change of the tosylate groups at C-6 with chlorodesoxy groups was recentlyreported by Rahn (1997), who reacted a cellulose tosylate (prepared under ho-mogeneous conditions; see chapter 4.3) with LiCl in acetylacetone for 2 h at130 0C. Bromodesoxycellulose can be obtained by analogy to the chloro com-pound with TV-bromosuccinimide and triphenylphosphine (Tseng et al., 1995).Also, the nucleophilic exchange of tosylate groups with bromodesoxy groups byreacting tosylcellulose with NaBr in acetylacetone can be recommended (Rahn,1997).
A tosylation and subsequent iodination to iododesoxycellulose was often for-mally employed to assess the amount of free hydroxy groups at the C-6 in par-tially substituted cellulose derivatives, because only the tosylate groups in thisposition were selectively replaced by iodine (Malm et al., 1948; Heuser et al.,1950). According to Rahn (1997) this procedure is somewhat questionable as aquantitative method, as deviations in the DS balance have been observed. Simi-lar displacement reactions can be performed with cellulose nitrate, as only thenitrate groups in the C-6 position are substituted by iodine. Sulfur bound directlyto this C-atom can be introduced by reacting tosylcellulose with ^28203 inDMSO to a 'Bunte-salt' of tosylcellulose, which is subsequently oxidized to adisulfide bridge with e.g. F^C^ in an alkaline medium (Camacho Gomez, 1997).Just as described for the halodesoxycelluloses, pseudohalodesoxy derivativescan be obtained, and the same holds true for nitrodesoxycellulose (CeIl-NC^)prepared by reacting tosylcellulose with NaNC^.
Tosylcellulose is employed as the starting material also for preparing amino-desoxycellulose by reacting it with Nt^, aliphatic amines, or hydrazine (Teshi-rogi et al., 1979; Engelskirchen, 1987). An alternative route starts from a highlysubstituted cellulose nitrate, which is reacted with NaNH2 in liquid NF^. Prod-ucts with a DS of nitrogen of up to 1 were obtained, which were soluble in F^Oand dilute aqueous acids, but not in organic liquids (Scherer and Feild, 1941).
Desoxycelluloses can be considered as promising starting materials for subse-quent steps of cellulose functionalization: an acidodesoxycellulose obtained byreaction of tosylcellulose with sodium, can be cleared by UV irradiation, open-ing a route to a selectively oxidized 6-aldehydecellulose (Clode and Horton,1971). The binding of a rather complex functional group directly to the skeletonof cellulose was demonstrated recently by Rahn (1997) by reacting tosylcellu-lose for 8 h at 100 0C in a DMF/water medium with the sodium salt of iminodi-acetic acid. About 50 % of the toslyate groups at C-6 were substituted by theiminodiacetic acid group attached to the polymer skeleton via a C-N bond. Atransformation of 6-chlorodesoxycellulose to hydrazlnodesoxycellulose and asubstituted hydrazlnodesoxycellulose was recently employed by Nakamura andAmano (1997).

4.4.2 Cellulose esters with reagents derived from carbonic acid (H2 COj) 145
Halodesoxycelluloses, especially chlorodesoxycellulose, exhibit a rather highthermal stability, with the temperature of beginning thermal decomposition de-creasing in the order chloro- > bromo- > iododesoxycellulose. Thermal decom-position takes place with a liberation of the appropriate hydrogen halide. Quitesimilar to phosphorylation, chlorination of cellulose results in increased charformation and decreased evolution of inflammable volatiles in thermal decom-position (Jain et al., 1987a), and therefore has found some attention in the flameproofing of cellulosic textiles.
A route to the attachment of long alkyl side chain on the cellulose moleculevia C-N-C bonds is the reaction of 6-chlorodesoxycellulose with n-alkylamines(CH3(CH2)nNH2; N = 5, 11, 17) yielding alkylaminodesoxycelluloses (Naka-muraetal., 1997).
4.4.2 Cellulose esters with reagents derived fromcarbonic acid (H2CO3)
Despite much experimental effort, cellulose esters of carbonic acid (cellulosecarbonates) have not been isolated up to now, obviously due to the instability ofthese compounds. But cellulose esters of the thio analogue of t^CC^, i.e. ofmonothiocarbonic acid and dithiocarbonic acid are well known, the cellulosehalf-ester of dithiocarbonic acid ('cellulose xanthogenate'), as its Na salt, repre-senting the key intermediate in artificial fiber spinning by the commercial vis-cose process. Furthermore, esters of cellulose with carbamic acid in recent yearshave been amply studied in connection with an alternative process of artificialfiber manufacture. These three classes of compound only are of interest as proc-ess intermediates and not as final products, and therefore will be subsequentlyconsidered with regard to their chemistry of formation as well as that of decom-position and subsequent reactions.
4.4.2.1 Cellulose esters of monothiocarbonic acid (H2CSO2)
Carbonyl sulfide (COS), the moderately stable anhydride of the presumablyextremely unstable and not yet isolated monothiocarbonic acid P^CSC^, reactswith anionized alcoholic hydroxy groups to give alkyl monothiocarbonic acidhalf-ester anions
COS + RO- -> ROCOS-
This bimolecular reaction proceeds about three orders of magnitude faster thanthe corresponding one between COS and hydroxy ions leading to monothiocar-bonate anions. In contrast with the esterification reactions with inorganic acidanhydrides considered so far, the esterification with COS requires an activation

4.4.2 Cellulose esters with reagents derived from carbonic acid (H2 COj) 145
Halodesoxycelluloses, especially chlorodesoxycellulose, exhibit a rather highthermal stability, with the temperature of beginning thermal decomposition de-creasing in the order chloro- > bromo- > iododesoxycellulose. Thermal decom-position takes place with a liberation of the appropriate hydrogen halide. Quitesimilar to phosphorylation, chlorination of cellulose results in increased charformation and decreased evolution of inflammable volatiles in thermal decom-position (Jain et al., 1987a), and therefore has found some attention in the flameproofing of cellulosic textiles.
A route to the attachment of long alkyl side chain on the cellulose moleculevia C-N-C bonds is the reaction of 6-chlorodesoxycellulose with n-alkylamines(CH3(CH2)nNH2; N = 5, 11, 17) yielding alkylaminodesoxycelluloses (Naka-muraetal., 1997).
4.4.2 Cellulose esters with reagents derived fromcarbonic acid (H2CO3)
Despite much experimental effort, cellulose esters of carbonic acid (cellulosecarbonates) have not been isolated up to now, obviously due to the instability ofthese compounds. But cellulose esters of the thio analogue of t^CC^, i.e. ofmonothiocarbonic acid and dithiocarbonic acid are well known, the cellulosehalf-ester of dithiocarbonic acid ('cellulose xanthogenate'), as its Na salt, repre-senting the key intermediate in artificial fiber spinning by the commercial vis-cose process. Furthermore, esters of cellulose with carbamic acid in recent yearshave been amply studied in connection with an alternative process of artificialfiber manufacture. These three classes of compound only are of interest as proc-ess intermediates and not as final products, and therefore will be subsequentlyconsidered with regard to their chemistry of formation as well as that of decom-position and subsequent reactions.
4.4.2.1 Cellulose esters of monothiocarbonic acid (H2CSC^)
Carbonyl sulfide (COS), the moderately stable anhydride of the presumablyextremely unstable and not yet isolated monothiocarbonic acid P^CSC^, reactswith anionized alcoholic hydroxy groups to give alkyl monothiocarbonic acidhalf-ester anions
COS + RO- -> ROCOS-
This bimolecular reaction proceeds about three orders of magnitude faster thanthe corresponding one between COS and hydroxy ions leading to monothiocar-bonate anions. In contrast with the esterification reactions with inorganic acidanhydrides considered so far, the esterification with COS requires an activation
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

146 4.4 Esterification of Cellulose
of the alcoholic component by anionization of the hydroxy groups in the mannerof a Schotten-Baumann reaction. In an aqueous alkaline medium, alkyl mono-thiocarbonates are considerably more stable than monothiocarbonate itself,yielding SH~, CO^2~ and ROH as end products of decomposition.
0.5
I0 5 10 15 20 25 30
NaOH [wt.%]
Figure 4.4.18. Maximal DS in the reaction of alkali cellulose with carbonyl sulfide independence on steeping lye concentration (Philipp, 1957a).
As first reported by Hess and Grotjahn (1952), alkali cellulose (sodium saltcellulose I) can be converted by reaction with COS at about O 0C to a solid-fibersalt of cellulose monothiocarbonic acid half-ester with a limiting DS$ of about 1,which can be rather completely dissolved in dilute aqueous alkali without, how-ever, yielding a fiber-free solution. Studies of our own (Philipp, 1957a) con-firmed these findings of Hess and Grotjahn and revealed a close correlation be-tween the so-called true alkali uptake of the alkali cellulose employed as startingmaterial and the maximal DS$ of the greenish-gray cellulose monothiocarbonatehalf-ester salt, with a rather constant level of DS$ between 0.8 and 0.9 being ob-served in the range of alkali-cellulose steeping-lye concentrations of 14-20 % (seeFig. 4.4.18).
Throughout this range of lye concentration, sodium cellulose I is formed witha nearly constant true alkali uptake of 1 mol of NaOH/mol of AGU, which inthis heterogeneous reaction obviously sets an upper limit for substitution of hy-droxy groups by monothiocarbonate residues. Furthermore, it could be con-cluded from these experiments that in the fibrous cellulose monothiocarbonate,as well as in its aqueous alkaline solution, a rather fast transesterification be-tween cellulosic hydroxy groups via free COS has to be assumed, as expressedby the equilibrium
CeII-O' + COS CeII-O-COS'
Na cellulose monothiocarbonate and its aqueous alkaline solutions decom-posed rather rapidly to sulfide, carbonate and cellulose and can be handled onlyat low temperatures of about O 0C. Due to its high rate of formation and decom-position, and the fast transesterification mentioned above, Na cellulose mono-

4 Λ.2 Cellulose esters with reagents derived from carbonic acid (H2CO 3) 147
thiocarbonate ('COS xanthogenate') plays some role as an intermediate in cel-lulose xanthation and transxanthation during the viscose process (see section2.3.2.2).
4.4.2.2 Cellulose dithiocarbonate esters
General comments on reaction and product properties
Just as with the alkoxy anions of low molecular alcohols, carbon disulfide (CS2)reacts with anionized cellulosic hydroxy groups to give a moderately stable cel-lulose dithiocarbonic acid half-ester anion according to
CeIl-O- Na+ + CS2 -> CeIl-O-CSS- Na+
which in principle can be subsequently reacted to a full ester with an alkyl hal-ide. Of practical relevance, however, is the sodium salt of the half-ester only, asthe introduction of a sufficient amount of anionic dithiocarbonate groups to thecellulose chain makes the polymer water- or alkali-soluble by transforming it toa poly electrolyte. Thus, therewith, is the purpose of converting the cellulosefibers to the homogeneous dissolved polymer component of an aqueous spinningsolution in the manufacture of artificial cellulose fibers via the viscose process.The chemistry of this process is, however, not so simple, as indicated by theabove equation, as only about 70 % of the CS2 input is converted to cellulosexanthogenate. The rest is consumed by formation of inorganic sulfidic products,and as xanthogenate formation and decomposition, taking place simultaneouslyin an aqueous alkaline medium, and as finally the conversion of the alkalinecellulose xanthogenate solution to a filament of cellulose II by spinning in anacid bath representing a complex chemical process too, which is largely affectedby the previous steps of xanthation and xanthogenate dissolution. Subsequently,the chemistry of cellulose xanthogenate formation and decomposition will bedescribed in some detail, together with the results of model experiments, turningthen to the role of alkali-cellulose structure, and finally giving an overview ofthe present state of the viscose process for manufacturing artificial cellulosefibers and filaments via cellulose xanthogenate.
The chemistry of xanthogenate formation and decomposition inaqueous media
As industrial cellulose xanthogenate formation and decomposition takes place insystems containing between 50 and 85 % of water, a brief survey of the generalchemistry of xanthogenate and by-product formation in aqueous media, as wellas on decomposition of xanthogenates in dependence on pH, with reference tohomogeneous model systems, seems appropriate to make the reader familiar

148 4.4 Esterification of Cellulose
with the complex reaction mechanism before turning to the characteristics ofcellulose xanthation.
As can be seen from the reaction rate constants in Table 4.4.19, the conver-sion of alkoxy anions to xanthogenate anions is generally highly favored incomparison with dithiocarbonate formation with hydroxy ions.
Table 4.4.19. Parameters of the reactions of CS2 with various anions at10 0C in 0.1-1.5 N aqueous NaOH.
Reaction
CS2 + OH-CS2 + SH-CS2 + RO-CS2 + CS2O
2-CS2 + CSO2
2-
Rate constant(mnrM-moH)
0.0090.0854.75.92.6
(kcal/mol)
20-21211615.6-
(cal/mol-0C)5.1
10.50.3
-0.5-
= Entropy of activation.
The dithiocarbonate, as a reactive intermediate, gives rise to consecutive re-actions, finally leaving to sulfide, carbonate and trithiocarbonate (CS3) as stableend products, but also involving the formation and subsequent decomposition ofcarbonyl sulfide. The latter reacts by analogy to €82 independently with RO~anions, as well as with hydroxy anions, to give alkyl monothiocarbonate andthiocarbonate, respectively, with rate constants about three orders of magnitudehigher than those of the corresponding reactions with CS2- The nucleophilicityof the anions in question increases in the order OH~ < SH~ < RO~ = CS2O2~.The energy of activation was found to be significantly lower for RO~ andCS2O2~ compared with the other anions, possibly due to an asymmetry of thehydration shell. The entropy of activation decreases in the order S2~ > OH~> RO~ (R = C2H5) > CS2O2~, indicating an increasing demand for specialorientation of the anion in order to form the reaction complex with CS2(Dautzenberg and Philipp, 1969). Due to the limited solubility of CS2 inaqueous systems (1.4 g = 18 mmol/1 in pure water), the course of reaction canbecome diffusion controlled with an excess of CS2 present as a separate liquidphase, as shown by Philipp (1955) for the 'limiting system' H2O/NaOH/CS2,where the transport of €82 to the aqueous phase was found to be ratedetermining above 30 0C. In this system, sulfide was formed as the only stableend product up to about pH 10, while at higher alkalinity an increasing amountof CS32~ was observed, passing a maximum at about 5 N NaOH and thendecreasing rapidly above 7.5 N NaOH due to changes in the hydration shell ofthe NaOH dipoles (see chapter 4.2). In the presence of air or oxidants, sulfide isoxidized to disulfide, which reacts very rapidly with CS2 to perthiocarbonate,

4.4.2 Cellulose esters with reagents derived from carbonic acid (H2CO 3) 149
rapidly with CS2 to perthiocarbonate, and also some thiosulfate is formed, allthese compounds playing a role as minor by-products in the viscose process.
The rate of xanthogenate formation between an alkoxy group and C$2 largelydepends on the chemical constitution of the alcohol in question and can differbetween low molecular aliphatic alcohols by about two orders of magnitude,taking as examples the slow reaction of isopropanol and the fast reaction ofglycerine. The rate and the mechanism of xanthogenate decomposition are gov-erned by the chemical constitution of the alcohol, but also depend decisively onthe pH of the medium.
In the acid region of the pH scale rapid decomposition to ROH and CS2 viafree xanthogenic acid takes place with any xanthogenate, with only a small ratedifference between so-called stable xanthogenates like ethylxanthogenate orunstable xanthogenates like glycerine xanthogenate. At the other end of the pHscale, i.e. at and above pH 14, a steep increase in xanthogenate decompositionrate takes place too, with dithiocarbonate being the predominant primary de-composition product, and a considerably larger rate difference between variousxanthogenates being observed than in acid decomposition. Intermediate forma-tion of an orthoxanthogenate by addition of a hydroxy ion to the CS double bondhas been proposed to explain the course of this reaction. Remarkable differencesin stability between ethyl or 1,4-butandiol xanthogenate on the one hand, andglycol and glycerine xanthogenate on the other, have been observed in the pHrange 7-13 (Philipp and Fichte, 1960): the so-called stable xanthogenates men-tioned first are slowly decomposed at a nearly constant rate over a wide range ofpH with evolution of free CS2 as the predominant product of decomposition dueto reaction with water molecules probably forming primarily a hydration com-plex with the xanthogenate anions. With glycol or glycerine xanthogenate, how-ever, a rather large amount of carbonyl sulfide, bound at least partially as alkylmonothiocarbonate, was already observed at a pH of about 9, besides formationof a large amount of sulfide; a stepwise desulfuration of the xanthogenate wasproposed as a possible mechanism for this route of dexanthation. The decisivepoint of difference between the two groups of xanthogenates is not the numberbut the mutual position of the hydroxy groups. Obviously, the above-mentionedstepwise desulfuration is favored by a vicinal hydroxy group, at least in the caseof aliphatic alcohols.
The course of xanthation and dexanthation of mono- and polysaccharidestakes an intermediate position between the so-called 'stable' and the 'unstable'xanthogenates, but resembles more that of the stable ones despite the existenceof vicinal hydroxy groups in the saccharide molecule. The location of these hy-droxy groups within an anhydropyranose ring obviously exerts a stabilizingaction on the xanthogenates formed. According to Philipp (1957b) primary aswell as secondary hydroxy groups of monosaccharides can be xanthogenated,the maximal level of xanthogenate formation being of course lower with xylose

150 4.4 Esterification of Cellulose
than with glucose. Studies on emulsion xanthation of various polysaccharides,i.e. a short-chain cellulose (ß-cellulose), a beech xylan, an ivory nut mannaneand an alginate at 4 % polymer concentration in 5 N NaOH at 28 0C with anexcess of CS2 (see Fig. 4.4.19), revealed a rather similar course of reaction for ß-cellulose and xylan, except for the very plausible fact that the maximal DS of 0.95with xylan amounted to two sorts only of that of ß-cellulose with a DS of 1.45.
With the mannan on the other hand, a significantly faster formation and de-composition of the xanthogenate with a maximal DS of 1.6 can be concludedfrom the data shown in Fig. 4.4.23 (see later). Obviously, the cis position of thehydroxy groups at C-2 and C-3, in contrast with the trans position in cellulose,leads to a faster decomposition, similar to that observed with glycol xanthogen-ate, quite in agreement with the observed shift of the ratio of trithiocarbonate tosulfide formed as by-products in favor of sulfide. On xanthation of alginate un-der the conditions used here, a definitely lower maximal DS (ca. 0.7) than withxylan was found, possibly due to a shielding action of the anionic group alreadypresent in the C-6 position. From the viewpoint of the industrial viscose process,a moderate amount of xylan units in the dissolving pulp obviously does not dis-turb the xanthation reaction, but mannose units consume more than the adequateamount of CS2 and transform it rather quickly to undesired by-products.
Figure 4.4.19. Course of xanthation of various polysaccharides: (a) low DP cellulose;(b) beech xylan; (c) ivory nut mannan (Philipp, 1957b), γ-value = 100 - DS.
Characteristics of cellulose xanthogenate formation and decomposition
Generally, xanthation of cellulose complies with the principles of this reactionoutlined above: xanthation and CS2 hydrolysis proceed independently in a reac-tion-controlled process. The formation of by-products, especially trithiocarbon-ate, increasing with the temperature of reaction due to the difference in activa-tion energies (EA =13 kcal/mol for xanthation, Ξ 21 kcal/mol for CS2 hydroly-

4.4.2 Cellulose esters with reagents derived from carbonic acid (7/2COj) 151
sis). All three hydroxy groups of the AGU can participate in the reaction. Afterthe pioneering work of Hess et al. (1951) on the heterogeneous course of cellu-lose xanthation, and of Matthes (1952) on transxanthation via free CS2> decisiveprogress in understanding the mechanism and the kinetics of cellulose xan-thation and dexanthation was achieved in the late 1950s and 1960s. Especially tobe mentioned are the comprehensive studies of the groups of Samuelson (e.g.Samuelson, 1948; Dunbrant and Samuelson, 1965) on xanthogenate group sta-bility and its spectrophotometric assessment, of the group of Treiber (e.g. Trei-ber et al., 1955 and 1956; Treiber and Fex, 1956) on the colloid chemistry ofcellulose xanthation and xanthogenate solution, of Hovenkamp (e.g. Hovenk-amp, 1963 and 1965) on the role of sodium dithiocarbonite in the xanthationprocess, and of Dautzenberg (e.g. Dautzenberg et al., 1972) on the formation oflow molecular sulfidic products during xanthation and dexanthation. Two im-portant characteristics, have to be considered in connection with cellulose xan-thogenate formation and decomposition, i.e.(i) the influence of polymer supramolecular structure on maximal DS obtainable,and on substituent distribution;(ii) the existence of a quasi-equilibrium of dexanthation and rexanthation viafree CS2 as the active agent in aqueous alkaline solutions of this 'moderatelyunstable' xanthogenate.
With regard to supramolecular order or 'state of dispersity' of the polymer,two borderline cases can be realized:(i) a xanthation of low DP cellulose homogeneously dissolved in aqueous alkaliwith liquid CS2;(ii) a xanthation of rather well-ordered fibrous sodium cellulose with gaseous orliquid CS2 (so-called fiber xanthation).
A so-called 'emulsion xanthation', i.e. the reaction of a cellulose suspensionin aqueous NaOH with liquid CS2, leading to gradual dissolution of the polymerduring reaction, takes an intermediate position between these borderline cases.The industrial xanthation process usually corresponds quite closely to a fiberxanthation. In homogeneous xanthation, a strongly preferential C-6 substitutiontakes place. With sufficiently high CS2/NaOH input all three hydroxy groups ofthe AGU can be xanthogenated up to a DS of nearly 3 in a homogeneous oremulsion xanthation (Geiger and Weiss, 1953). With increasing substitution ofhydroxy groups by xanthogenate groups, the rate constant of homogeneousxanthation decreases, while that of dexanthation remains nearly constant. Anenhanced hydroxy concentration in homogeneous xanthation leads to an in-creased xanthation rate constant especially for the C-6 position, obviously due toa further breakdown of intra- and/or intermolecular cellulosic hydrogen bonds.
Fiber xanthation of an alkali cellulose, on the other hand, is characterized by alimited maximal DS of about 0.9-1.0 even with a large excess of €82 and by apreferential substitution at the C-2 position. CS2 physically dissolved in the

152 4 Λ Ε st erification of Cellulose
adhering lye is the active agent also in the xanthation of fibrous alkali cellulose,and the reaction rate increases with increasing €82 pressure according to Grot-jahn (1953).
Figure 4.4.20 presents the course of DS with time of reaction for differenttemperatures in the range of practical interest, employing a large excess of CS2-From a quantitative evaluation of the kinetic data can be concluded that the re-action proceeds according to the scheme
Na-CeIIk-1
Cell-xanthogenatek-2
Cell Il
with cellulose xanthogenate as a moderately stable intermediate and the ratio ofthe rate constants k\lk^ being about 10 (Philipp, 1956). The experimentally ob-served maximal DSx values of about 0.9-1.0 in fiber xanthation in connectionwith this ratio of rate constants, indicate that obviously the so-called true alkaliuptake of 1 mol of NaOH/mol of AGU of the alkali cellulose employed is thelimiting factor for the DSx value, which comes rather close to a value of DS = 1due to slow decomposition of the xanthogenate during its process of formation.A quantitative calculation presented in Philipp (1956) confirms this assumption.Furthermore, the maximal DSx remains constant within the total range of sodiumcellulose I formation, i.e. at steeping lye concentrations between 14 and 22 %(see Fig. 4.4.21), and the same holds true for the rate constant of xanthogenateformation and decomposition.
2 3 4Time[h]
Figure 4.4.20. Course of γ-values on alkali cellulose fiber xanthation at different tem-peratures (O 20 0C, Δ 28 0C, D 35 0C) (Philipp, 1957c).
At a steeping lye concentration above 22 %, the maximal DSx decreases dueto a strongly diminished xanthation rate caused by lack of free water as solventfor the CS2 (Bartunek, 1953). At a steeping lye concentration below 14 %, themaximal DSx, as well as the true alkali uptake, decrease sharply indicating an

4.4.2 Cellulose esters with reagents derived from carbonic acid (H2 CO^) 153
alkali-cellulose formation in the less well-ordered regions only (see chapter 4.2).Simultaneously, the rate constant of xanthation increases significantly and theenergy of activation decreases: at a concentration of steeping lye of 6 % NaOH,the maximal DS amounts to 0.24, the energy of activation to only about 7kcal/mol, and the rate constant of xanthation is twice the value observed aftersteeping with 18 % NaOH. The low-ordered regions of alkali cellulose are obvi-ously much more rapidly xanthogenated in possibly a diffusion-controlled proc-ess than the crystalline regions. Xanthation of the crystalline regions of sodiumcellulose I can be classified as a so-called lattice layer reaction, with the 1-0-1lattice distance gradually increasing during the reaction, but with some delay atthe beginning in comparison with the course of DSx (Hess et al., 1951), indicat-ing again a faster xanthation of the less well-ordered regions.
1Q ^NoOH-uptake
0.5
O 5 10 15 20 25NaOH [wt.%]
Figure 4.4.21. Maximal DS of fiber xanthogenate and 'true' NaOH uptake in molNaOH/mol AGU(see chapter 4.2) of alkali cellulose in dependence on steeping lye con-centration.
A comparison of alkali-cellulose samples prepared by steeping with 18 %NaOH of different pulps resulted in significant differences in the rate constant offiber xanthation of up to 25 %, with alkali cellulose from !inters exhibiting thelowest value (Philipp, 1956).
Table 4.4.20. Xanthogenate group distribution in fiber xanthogenate and viscose.
Sample
Fiber xanthogenate (DS 0.61)Viscose, non-ripened (DS 0.58)Viscose, moderately ripened (DS 0.49)Viscose, extensively ripened (DS 0.28)
DS at C-2/C-3
0.380.340.16O
DS at C-6
0.170.240.320.32
Technique: Preparation of DA-xanthogenate, tosylation, iodination.Total DS via N-content of DS-xanthogenate.Distribution via analysis of iodinated sample.

154 4.4 Esterification of Cellulose
The preferential substitution at the C-2 position in xanthation with a limitedamount of CS2 (see Table 4.4.20) has been correlated in early work with thehigher acidity of this hydroxy group, but is obviously mainly caused by a lowavailability of the C-6 hydroxy group in the ordered structure of the alkali cel-lulose during fiber xanthation, while on homogeneous xanthation with freelyavailable hydroxy groups in all three positions the C-6 position is obviouslyfavored.
Cellulose fiber xanthogenates at a DSx level of about 0.5 can be easily andcompletely dissolved in 1-2 molar aqueous NaOH to give a viscous polymersolution containing, besides the cellulose xanthogenate, trithiocarbonate, car-bonate and sulfide, as well as small amounts of di- and monothiocarbonate, per-thiocarbonate and thiosulfate as by-products. Also, some monothiocarbonatesubstituents at a DS level below 0.04, have been detected in the cellulose xan-thogenate moiety (Bernhardt, 1926). This cellulose xanthogenate solution un-dergoes rather complex chemical and colloidal changes on standing ('ripening'),which are of high relevance to viscose preparation and spinning. The overallDSx decreases continuously during this ripening process. A fast decrease is ob-served in the number of xanthogenate groups at C-2, while the level of partialDSx at C-6 remains rather constant over a long period or was even found to betemporarily enhanced (Fig. 4.4.22).
60
COQ
20
O 5 10 15 20 25Time[h]
Figure 4.4.22. Course of partial DSx during viscose ripening (· C-6, · C-2, A C-3)DS [%] = % of total DS (König et al., 1993).
Furthermore, a rather constant level of free CS2 of about 1 % of the totalamount bound to cellulose could be detected in these cellulose xanthogenatesolutions and was found to appear again even after precipitation throughoutwashing and redissolution of the cellulose xanthogenate (Philipp and Dautzen-berg, 1967). From a quantitative evaluation of these facts and other observations,most researchers including the authors group assume a quasi-equilibrium ofdexanthation and rexanthation in these aqueous alkaline cellulose xanthogenatesolutions resulting in a redistribution of xanthogenate groups by transxanthation,

4.4.2 Cellulose esters with reagents derived from carbonic acid (H2CO 3) 155
although other opinions have been published (König et al., 1993). This quasi-equilibrium of de- and rexanthation can be formulated according to
CeII-OH + OH'+ CS2 Cell-O-CSS' + H2O
[CeIl-O-CSS-J[H2Q]K ~ [CeIl-OH][CS2][OH"]
with the quasi-equilibrium constant K being about 105 for the comparativelystable C-6 xanthogenate, and about 103 for the more labile C-2/C-3 xanthogen-ate in 1-2 N NaOH at room temperature. From model experiments can be con-cluded that the rate constant of dexanthation at the C-2/C-3 position is about 16times higher than the corresponding one at C-6, while the rate constant of rex-anthation at C-6 is about 4 times higher than that at C-2 in 1-2 N aqueous NaOHat 20 0C. From this quasi-equilibrium, some free CS2 is continuously drained byirreversible reactions with OH or SH ions. While liberation of CS2 by reactionof xanthogenate with water molecules is the dominating route of dexanthation upto an NaOH concentration of about 2 N, xanthogenate decomposition to dithio-carbonate prevails at higher alkali concentrations, and no transxanthation canoccur e.g. with 10 N NaOH as the medium (Table 4.4.21).
Table 4.4.21. Decomposition of cellulose xanthogenate in aqueous NaOH.
MoI ofNaOH/1
0.11.02.06.5
10.0
% CS2a
999870-9014-18
O
%CS202-a
12
10-3082-86
100a Percentage relative to xanthogenate.
In consequence of the transxanthation process (Matthes, 1952) outlined herebriefly, xanthogenate substituents are not only transferred from the C-2 to the C-6 position but also are more evenly distributed along and between the cellulosechains, resulting in a drop in solution viscosity. During ripening, the concentra-tion of low molecular by-products, especially of Na2CS3, steadily increases atthe expense of hydroxy ions, and the tendency of the system to coagulate onaddition of electrolytes (NaCl, M^Cl) is enhanced. After extensive overall dex-anthation, the viscosity increases rather steeply due to loss of hydrophilic ani-onic groups until syneresis of the system takes place (Götze, 1967).

156 4.4 Esterification of Cellulose
The course of cellulose xanthogenate decomposition in solution can be re-tarded or accelerated by adding compounds interfering with the transxanthationvia free CS2*. the irreversible drain of free CS2 can be enhanced by addition ofH2Ü2 or retarded by addition of Na2SU3 via the level of disulfide formed in thesystem, which reacts very rapidly with CS2 to give perthiocarbonate. Additionof small amounts of polyhydric alcohols with vicinal hydroxy groups, like gly-col or glycerine, results in fast, irreversible C$2 consumption and the formationof rather large amounts of COS, leading to an increased overall decompositionrate of the xanthogenate. Besides this, a fast drop in viscosity is observed proba-bly due to the participation of COS in the exchange of substituents, resulting in amore uniform substituent distribution. A similar effect, i.e. a significantly re-duced viscosity of the system, has been observed according to Philipp (1957a)on addition of a few percent COS to the liquid CS2 employed in xanthation.
Like all other xanthogenates, cellulose xanthogenate is rapidly decomposed ina strongly acidic medium, e.g. in 1 N Η^βΟφ via free cellulose xanthogenicacid, to give CS2 and cellulose II, the physical structure of which largely de-pends on the chemical and physical state of the xanthogenate solution and on theconditions of acid treatment, and which can be influenced, via the ratio of co-agulation rate to decomposition rate, by the presence of zinc ions and specialadditives (see the next section). Cellulose xanthogenate is rather easily decom-posed already at a temperature of 90-100 0C with evolution of CS2- Cellulosexanthogenate is precipitated from its aqueous alkaline solution by numeroustransition metal cations, especially those forming widely insoluble sulfides likeZn2+, Hg2+ or Ag+. Some of these xanthogenates show a spontaneous decompo-sition to the corresponding metal sulfide as observed for example with silver ormercury salts of cellulose xanthogenate.
Cellulose xanthogenate is a rather reactive cellulose ester well suited for sub-sequent steps of derivatization. Some typical routes are indicated in Fig. 4.4.23.Reaction of cellulose xanthogenate with nonsubstituted or substituted alkyl hal-ides leads to full esters of cellulose xanthogenic acid and permits the attachmentof various functional groups onto the cellulose chain. These full esters are muchmore stable than the cellulose xanthogenate itself, and they are soluble in vari-ous organic liquids. Of special analytical interest in determining the pattern ofsubstitution is the stable and organosoluble ester formed with Λ^,Λ^-diethyl chlo-roacetamide (Matthes, 1952). Some reactions proceeding with the elimination ofone sulfur atom or of CS2 open up a route to special cellulose derivatives likearyl-substituted cellulose thiocarbamates or cyanoethylcellulose. Furthermore,the xanthogenate group can be employed for covalent crosslinking between thecellulose chains or for preparing radical sites on the cellulose chains for subse-quent grafting (see chapter 4.1). Of special analytical interest for a convenienttitration of xanthogenate groups is the oxidation of two SH functions to a disul-fide bridge by iodine.

4.4.2 Cellulose esters with reagents derived from carbonic acid f//2C(9jj 157
R = Alkyl, CH2-COOH, CI-R
2 — CON(C2H5)2, l * Cell-O-C;
\O
,S
CeII-O-C'^SeNa®
H2C-CH-CN, H2O
- CS2, - NaOH
H9N-R1
* CeH-O-CH2-CH2-CN
- /VaSHCeII-O-C'
SNHR1
H2CN2, H2OCeII-O-CH33- CS2, - NaOH, - N2
I2
CeII-O-C
V-sx
Figure 4.4.23. Consecutive reactions of cellulose xanthogenate.
Survey of the commercial viscose process
After its invention by Cross, Bevan and Beadle in 1893 (Cross et al., 1893) theviscose process of manufacturing cellulose rayon filament and staple fiber hasbeen practised for many decades as the only one and later as the dominating onein the commercial production of chemical fibers. Important aspects of the proc-ess are its versatility and adaptability to end-use requirements and one century ofprocess engineering experience. As severe shortcomings, from the ecologicalhazards connected with the handling and disposal of CS2 and S, to the lowspeed of spinning in comparison with melt-spun synthetic fibers have to bementioned. These disadvantages, however, could be at least partially compen-sated by recent developments, which will be adequately emphasized in the fol-lowing context. The scheme in Fig. 4.4.24 gives an overview of the numeroussteps of chemical and physical treatment of cellulose during the viscose process.
Hard wood as well as soft wood sulfite or prehydrolysis sulfate pulp with an α-cellulose content of between 91 and 96 %, an ash content of < 0.1 %, a verylow content of calcium and heavy metal ions and a high uniformity at all threestructural levels is used today as the starting materials. The conversion to alkalicellulose (sodium cellulose I) is usually performed by continuous slurry steepingwith aqueous NaOH of about 18 % concentration and subsequent continuouspressing to a cellulose content of 32-35 % and an alkali content of between 15and 16%. After shredding and oxidative depolymerization ('preripening', seechapter 2.3) to the appropriate level of DP, xanthation of the alkali cellulosetakes place in a dry or wet churn process (or frequently in a hybrid processstarting with dry alkali cellulose followed by subsequent addition of aqueous

158 4.4 Esterification of Cellulose
NaOH) with a total amount of 28-30 % CS2, at a temperature of about 30 0C forseveral hours. The cellulose xanthogenate, with a DS of about 0.5, is dissolvedin dilute aqueous NaOH, usually under high-intensity mechanical agitation, togive a viscous solution containing about 8 % cellulose and about 6-7 % totalNaOH. Besides Na cellulose xanthogenate and free NaOH, this viscose solutioncontains trithiocarbonate and carbonate at the 1 % level, sulfide and perthiocar-bonate at the 0.1 % level, and small amounts of thiosulfate, dlthiocarbonate andmonothiocarbonate. The subsequent viscose ripening for 1-3 days at constanttemperature, at the level of or below room temperature, serves the purpose ofadjusting the degree of substitution and the viscosity of the solution to the level
Steeping with 18% NaOH
Alkali cellulose shredding
CeII-OH + NaOH—>CeII-O0Na0 + H2O
Preripening of alkali cellulose
Xanthogenation with CS2
(sulfidation)
Oxy 'dative depot y-merization
CeII-O0Na0+ CS2
CeII-O-C-S0Na0
IlS
Dissolution of xanthogenatein dilute aqueous NaOH
Viscose ripeningCeII-O-C-S0Na0+ H2O 5
CeII-OH + Na0OH0 + CS2
Filtration of the spinningsolution
Spinning into acidbath (H2SO4, Na2SO4)
Filament aftertreatment
CeII-O-C-S0Na0+ H0
SIl Ä m
CeII-O-C-S0H0
CeII-OH + CS2
Figure 4.4.24. Scheme of the viscose process.

4.4.2 Cellulose esters with reagents derived from carbonic acid (H2CO^) 159
desired for the spinning process, and to redistribute the xanthogenate substitu-ents for achieving a higher uniformity of substitution along and between thechains. This step is often combined with several procedures of filtration in orderto eliminate persistent fiber fragments and to reduce the gel particle content ofthe system to a sufficiently low level.
Much research and development work has been put into reducing the CS2input from about 35 % 20 years ago, to less than 30 % today, and about 25 %being the goal of further development. This reduction in CS2 input necessary forecological reasons has to be performed without impeding the quality of the spin-ning solution by a higher content of fiber fragments and gel particles due to alower DS of the xanthogenate. To solve this problem, a better yield of the CS2input for xanthation (less by-product formation) and a higher uniformity of thexanthogenate had to be striven for. Two routes have been successfully pursuedfor this purpose in recent years. The first one consists of the supply of a highlyreactive pulp obtained by special pulping procedures, by loading the pulp withsurfactants, facilitating xanthation and by providing a pulp with a higher chain-length uniformity, for example by radiation depolymerization. As recentlyshown by Fischer et al., (1996) the high-DP part of a dissolving pulp usuallycarries less than the adequate amount of xanthogenate groups and thus can leadto difficulties in viscose filtration and spinning due to the presence of low sub-stituted fiber fragments and gels. The second route is characterized by a stillbetter mutual adaptation of the various steps of viscose preparation.
The principle of the conventional viscose-spinning process consists of press-ing the viscose solution through corrosion-resistant spinnerets with about 100holes in the case of rayon filaments spinning, several 1000 holes in the case ofrayon staple spinning, with a hole diameter of between 50 and 100 μηι, into anacid bath of aqueous t SC^ and Na2SC>4 at a temperature of about 40 0C, andconveying the thread of cellulose II successively formed via godets onto a bob-bin. Structure formation of the thread is governed by the rate ratio of cellulosexanthogenate coagulation and decomposition on the one hand, and the mechani-cal forces exerted on the forming thread at various stages of the spinning processon the other. But also the state of ripening of the spinning solution has a strongbearing on the fiber structure formed due to its close interconnection with bothof the factors mentioned. Via the process parameters of spinning and aftertreat-ment, the mechanical properties of the threads can be varied within wide limits.For further details the reader is referred to Götze (1967). But only one pointshall be mentioned briefly: by the presence of zinc ions in the spinning bath,often in combination with special additives based on amines and/or polyethyleneoxides, so-called skin-core-filaments with a different fibrillar architecture in theouter skin and the inner core can be produced resulting in textile properties thatare outstanding for special applications. The effects obtained are based on a hin-dered diffusion of the H^O ions into the fiber structure due to the clogging of

160 4.4 Esterification of Cellulose
micropores by sulfidic zinc compounds (Klare and Grobe, 1964). Recent devel-opments are aiming to reduce this zinc content for ecological reasons withoutcompromising filament properties, and to produce a significant increase of thespinning velocity above its present level of about 170 m/min. This poses, ofcourse, new physicochemical problems with regard to xanthogenate decomposi-tion and filament structure formation, as well as engineering problems concern-ing for example a computerized and automated starting of the spinning process,or the hydrodynamics of conveying the forming thread through the spinningbath.
From the present state of development, the forecast seems justified that de-spite the existence of alternative processes and despite ecological problems notyet fully solved, the viscose process will keep its place in the foreseeable futuredue to its versatility and due to the fact that viscose rayon filament and staplefiber are still indispensable in many areas of the textile industry.
Properties of cellulose xanthogenate
Cellulose fiber xanthogenate at the conventional DS level of about 0.5 is a yel-lowish fibrous mass, easily soluble in water or dilute aqueous alkali, exhibitingthe properties of a poly electrolyte in these solutions. It can be precipitated fromthese aqueous systems by lower aliphatic alcohols or other water-miscible or-ganic liquids, by salting out with low molecular electrolytes or by adding cationsof heavy metals. Cellulose xanthogenate is unstable in aqueous media over thewhole range of pH and is rapidly decomposed by acids with the evolution ofCS2. Its thermal stability is rather low, decomposition starting already below100 0C with the liberation of CS2.
Applications of cellulose xanthogenate
Concerning annual production capacity, cellulose xanthogenate is the numberone among cellulose derivatives, but it is used as an intermediate only and not asa final product of chemical cellulose processing. Its quite predominant applica-tion is a transient solubilization of cellulose for converting the short wood-pulpfibers into endless filaments or staple fibers of cellulose II. But also films ofcellulose II, especially for food packaging purposes, are still manufactured inmany countries from aqueous alkaline cellulose xanthogenate solutions (vis-cose). Besides this, cellulose xanthogenate solutions can be employed to convertcellulose into specially shaped products, by putting the viscose into the appro-priate form before decomposition. An example of commercial relevance is theproduction of cellulose sponges (macroporous sponges) by thermal decomposi-tion of viscose in chest-like forms after previous addition of crystalline Na2SC^.Finally, the preparation of macroporous cellulose beads from viscose shall bementioned as a recent development in the area of carrier and separation materi-

4 A.2 Cellulose esters with reagents derived from carbonic acid (7/2^(9 3) 161
als. In this process, drops of viscose are coagulated and decomposed to cellulose IIbeads in an organic liquid of suitable density and boiling point, which is inmis-cible with water, at a temperature of about 90 0C (Dautzenberg et al., 1985a andb). Chlorobenzene was found to be especially suitable for this process. After thedecomposition, the low molecular by-products are washed out with water. Byvariation of composition and state of ripening of the viscose, as well as of theconditions of decomposition, the pore structure of the beads obtained can bevaried within wide limits and adapted to special end-use requirements.
4.4.2.3 Cellulose carbamate
General comments on formation and decomposition of cellulose carbamateand its possible applications
Cellulose carbamates with a low DS of about 0.3 have received considerableattention in recent years as alkali-soluble intermediates in an alternative processof artificial cellulose-fiber spinning, the so-called carbamate process (Segal andEggerton, 1961; Ekman, 1984; Lang et al., 1986). Cellulose carbamates areformed in a high-temperature reaction between cellulose and urea via isocyanicacid as active intermediate. From their aqueous alkaline solution these carba-mates can be spun in an acid bath to filaments, subsequently decarbaminated byalkali to threads of cellulose II. The chemistry of this process looks very simple,but in reality is probably still more complicated than that of the viscose process,as numerous condensation equilibria of C-N bond formation and cleavage haveto be considered. Furthermore, only a small DS range, between 0.2 and 0.3, isavailable for preparing alkali-soluble cellulose carbamates, because with in-creasing DS a growing tendency of crosslink formation counteracts the solubi-lizing action of the hydrophilic substituents. Due to these facts and still unsolvedproblems of decarbamation, the process is now practised on a pilot scale only,despite looking very promising at first.
Chemistry of cellulose carbamate formation and decomposition
On heating cellulose with urea above its melting point of 133 0C, carbamateester groups can be introduced into the cellulose chain by reaction of hydroxygroups with isocyanic acid formed as an active intermediate on decomposition ofurea (Fig. 4.4.25, A). This reaction is catalyzed by metal salts, especially zincsulfate. Suitable external conditions have to been chosen in order to eliminatethe ammonia formed as by-product of urea decomposition and to minimize iso-merization of isocyanic acid to cyanic acid, as the latter favors crosslinking bycondensation reactions. But also the isocyanic acid can give rise to condensationstructures, for example by biuret formation (Fig. 4.4.25, B). Generally,crosslinking between polymer chains impeding solubility is enhanced by in-

162 4.4 Esterification of Cellulose
creasing the temperature and the time of reaction, but, on the other hand, a suffi-ciently large number of hydrophilic substituents must be introduced to cleave theinterchain hydrogen bonds in the subsequent process of dissolution. The extentof crosslinking can be estimated by comparing the DS obtained by mass increaseof the purified product and the DS obtained via its nitrogen content, with thelatter usually having the lower value. According to recent 13C NMR studies(Nehls et al., 1994), reaction of hydroxy groups by carbamate ester groups takesplace exclusively at the 2 position. Due to the low DS level set by the crosslink-ing tendency, a intimate contact between cellulose and urea and an equal tem-perature throughout the whole mass are necessary to ensure a sufficiently uni-form substituent distribution along and between the polymer chains. For cleav-ing crosslinks formed via CN bonds, also a treatment of the reaction mass withliquid ammonia has been considered besides for the main purpose of extractingexcess urea.
H2N\ 14O0C —
A) C=O ^ HN = C = O + NH3
H |sj lsocyanic acid
~~ "~ CeII-O-C-NH2
- IlO
CeII-O-C-NH2 _ im^ CeII-OH- NH3, Na2CO3
H2N
C = O— / + CeII-OH Cross/inked
B) HN=C = O — ΗΝχ %e//u/ose
H2N +
7
C = 0
C = O H2N Biuret/
H2N
Figure 4.4.25. Scheme of the carbamate process.
Cellulose carbamate with a DS of between 0.2 and 0.3 can be dissolved inaqueous NaOH of optimal solvent power, i.e. a concentration between 10 and11%, eventually containing additionally some zincate or berylate (see chapter4.3). In this alkaline medium the carbamate groups are irreversibly decomposed
CeII-OH + HN = C = O

4.4.2 Cellulose esters with reagents derived from carbonic acid (T^COjj 163
to carbonate and ammonia at a rate depending on NaOH concentration and tem-perature, and unsubstituted cellulose is formed which can eventually coagulateto a low ordered cellulose II after sufficient decarbamation. A redistribution ofsubstituents cannot take place in this system, as the decomposition is irreversi-ble, in contrast with transxanthation via free CS2 in the viscose process. Also, incontrast with cellulose xanthogenate, cellulose carbamate is rather stable in anacid medium and can be coagulated as a cellulose carbamate thread by spinningin an acid bath.
Brief description of the cellulose carbamate process of fiber spinning
As the starting material, a highly reactive dissolving pulp at a DP level of about300 is employed, the latter being obtained either by the pulping process itself orby irradiation depolymerization, or by an intermediate alkalization and oxidativedepolymerization of the alkali cellulose. For an intimate contact between cellu-lose and reagent, the pulp is swollen in an aqueous solution of urea of about40 % concentration at a solid-to-liquid ratio of about 1 : 3 for several hours atambient temperature, then pressed off, milled and dried. The mass containing alarge excess of urea and eventually some zinc sulfate as catalyst is than reactedfor 1 to 2 h at 140-150 0C, either in a rotating kiln in the presence of air, or in astirred reactor with an inert medium (hydrocarbon) caring for the uniformtransmission of heat and impeding isomerization of isocyanic to cyanic acid.
The yellowish-to-brown colored reaction product, with a DS between 0.25and 0.30, is extracted with water or with liquid ammonia to recover excess ureaand eliminate colored by-products. A hydrolysis step at high pressure and tem-perature may be included for partial cleavage of crosslinks before the product isdissolved in aqueous sodium hydroxide of 10-11 % concentration and kept forsome time at a temperature of about 5 0C for gradually reducing the DS withoutan early coagulation of the system. The viscosity of the solution decreases ini-tially during this process but increases again with further lowering of the DS.After excessive filtration, the solution with a polymer content of 6-7 % is spunin an acid bath with about the same speed as in the viscose process to obtain acellulose carbamate thread. In order to produce artificial fibers of sufficientlyhigh wet tenacity, the carbamate groups have to be eliminated by a subsequentalkaline treatment as far as possible, followed by an acid treatment step for de-swelling. The problem of eliminating the last residual carbamate groups from thecellulose chains within the fiber structure limits at present the textile quality ofthreads spun by the carbamate process, but probably the quality level of a con-ventional rayon staple fiber can be obtained.
In comparison with the viscose process, the carbamate process has the advan-tage of a better ecological compatibility, and much of the conventional equip-ment of a viscose plant can be used, but on the other hand it still lacks the versa-

164 4.4 Esterification of Cellulose
tility and the ultimate quality level of rayon filament and staple spun from vis-cose. The present technology of the carbamate process resembles some kind of'via rope walk', as in several points it has to find an acceptable balance betweencounteracting effects.
Purified cellulose carbamate with a DS of about 0.3 is a white mass, contain-ing some covalent crosslinks, and is soluble in aqueous alkali under slow de-composition to cellulose II, carbonate and ammonia, whereas it is insoluble andrather stable in dilute aqueous acid. The only application of cellulose carbamateknown so far is its use as an intermediate for solubilizing cellulose in the carba-mate process for artificial fiber spinning, practised now on a pilot scale.
4.4.3 Esters of cellulose with organic acids
4.4.3.1 General remarksThe formation of cellulose esters of organic acids proceeds along the routes ofesterification of alcoholic hydroxy groups, well known from low molecular or-ganic chemistry: usually the anhydride or the chloride of the acid in question isemployed as the agent, while the reactivity of the acid itself suffices only insome cases to obtain an appreciable degree of esterification, even at large ex-cess. An acyl cation RCO+ can be generally assumed as the active intermediate,the formation of which is favored either by an acid catalysis in the case of thefree acid as the reagent, according to
RCOOH + H+ -> RCO+ + H2O
or by the adjuvant action of a tertiary base like TEA or pyridine, with the acidderivatives serving as the agent, according to
RCOCl + NR'3 -^ RCO-NR3+ CI-
As a very effective adjuvant base, 4-dimethylaminopyridine was recommendedfor various esterifications of cellulosic hydroxy groups in homogeneous systems(Philipp et al., 1983). Esterification of alcoholic hydroxy groups generally takesplace as an equilibrium reaction, with the ester bonds formed being susceptibleto hydrolytic cleavage in an aqueous acid medium, and at sufficiently high alka-linity an irreversible saponification of the ester groups can occur.
While practically any aliphatic or aromatic acid chloride can be reacted withcellulosic hydroxy groups at 100 0C in the presence of pyridine as a free base,application of pyridinium hydrochloride is frequently more favorable in the caseof an acid anhydride as the esterifying agent, as it promotes formation of theacid chloride as an active intermediate. Especially in synthesizing long-chainaliphatic or aromatic esters of cellulose, the use of a chlorinated acid anhydridelike chloroacetanhydride, in combination with the free acid to be esterified, can

164 4.4 Esterification of Cellulose
tility and the ultimate quality level of rayon filament and staple spun from vis-cose. The present technology of the carbamate process resembles some kind of'via rope walk', as in several points it has to find an acceptable balance betweencounteracting effects.
Purified cellulose carbamate with a DS of about 0.3 is a white mass, contain-ing some covalent crosslinks, and is soluble in aqueous alkali under slow de-composition to cellulose II, carbonate and ammonia, whereas it is insoluble andrather stable in dilute aqueous acid. The only application of cellulose carbamateknown so far is its use as an intermediate for solubilizing cellulose in the carba-mate process for artificial fiber spinning, practised now on a pilot scale.
4.4.3 Esters of cellulose with organic acids
4.4.3.1 General remarksThe formation of cellulose esters of organic acids proceeds along the routes ofesterification of alcoholic hydroxy groups, well known from low molecular or-ganic chemistry: usually the anhydride or the chloride of the acid in question isemployed as the agent, while the reactivity of the acid itself suffices only insome cases to obtain an appreciable degree of esterification, even at large ex-cess. An acyl cation RCO+ can be generally assumed as the active intermediate,the formation of which is favored either by an acid catalysis in the case of thefree acid as the reagent, according to
RCOOH + H+ -> RCO+ + H2O
or by the adjuvant action of a tertiary base like TEA or pyridine, with the acidderivatives serving as the agent, according to
RCOCl + NR'3 -^ RCO-NR3+ CI-
As a very effective adjuvant base, 4-dimethylaminopyridine was recommendedfor various esterifications of cellulosic hydroxy groups in homogeneous systems(Philipp et al., 1983). Esterification of alcoholic hydroxy groups generally takesplace as an equilibrium reaction, with the ester bonds formed being susceptibleto hydrolytic cleavage in an aqueous acid medium, and at sufficiently high alka-linity an irreversible saponification of the ester groups can occur.
While practically any aliphatic or aromatic acid chloride can be reacted withcellulosic hydroxy groups at 100 0C in the presence of pyridine as a free base,application of pyridinium hydrochloride is frequently more favorable in the caseof an acid anhydride as the esterifying agent, as it promotes formation of theacid chloride as an active intermediate. Especially in synthesizing long-chainaliphatic or aromatic esters of cellulose, the use of a chlorinated acid anhydridelike chloroacetanhydride, in combination with the free acid to be esterified, can
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

4.4.3 Esters of cellulose "with organic acids 165
be of technical advantage: the chlorinated acid anhydride acts as an 'impellingagent' according to
(ClAc)2O + 2 RCOOH -» R(CO)2O + 2 ClCH2COOH
and the preparation and excess application of an expensive acid anhydride canthus be avoided. With acid chlorides of higher aliphatic or of aromatic acids,esterification of cellulose can be performed also according to the Schotten-Baumann reaction with alkali cellulose as the starting material, but a largeexcess of reagent is required here of course.
Besides the direct action of free carbonic acids, their anhydrides or their acidchlorides on cellulosic hydroxy groups, transesterification reactions frequentlyprovide a suitable route to cellulose esters. These reactions proceed either byinteraction of hydroxy groups of the polymer with a labile ester or a salt of theacid in question, or by reaction of acid anhydrides or chlorides with labile esteror ether groups attached to the polymer and serving as a leaving group in esteri-fication.
As a technique of the future, enzymatic esterification of cellulose may beenvisaged, although it has not been realized up to now with the polymer itself,but only with pyranosidic and furanosidic low molecular saccharides: a pancrea-tin-catalyzed transesterification reaction between saccharidic hydroxy groupsand vinyl acetate in a THF/TEA mixture was reported (Lay et al., 1996) result-ing in a site-specific acetylation of the C-6 position of the saccharide.
Besides the well-known principles of esterification, some characteristics haveto be kept in mind in connection with cellulose ester formation. Also, esters car-rying two different substituents ('mixed esters') or ether-esters of cellulose canbe prepared, the former playing a role also on the commercial scale. The degreeof substitution obtained and the distribution of the substituents along and be-tween the macromolecules is largely governed by the accessibility of the hy-droxy groups, just as in other reactions of the polymer. In a thoroughly hetero-geneous system with a low degree of cellulose swelling, the reaction can be lim-ited to the surface of the fiber material resulting in a very low average DS. Insystems exhibiting a high degree of swelling or turning from the heterogeneousto the homogeneous state in the course of reaction, a DSof 3, or of nearly 3, canbe reached under suitable conditions frequently, and the same holds true, ofcourse, after previous dissolution of the polymer in a nonderivatizing solventlike DMA/LiCl. The preference of substitution within the AGU at low DS de-pends largely on the reaction system considered, but often a substitution reactionat C-6 is found to be favored.
Although in principle any aliphatic or aromatic acid residue can be attached tothe cellulose backbone by esterification, only a limited number of these deriva-tives has been thoroughly studied and only rather few, especially the acetate andsome mixed esters with acetate groups, have gained commercial relevance. So

166 4.4 Esterification of Cellulose
far, esterification of cellulose has mainly served the purpose of modifying thematerial properties of the polymer, and rather high DS values were required withthe question of arriving at a DS of exactly 3 often being of scientific and practi-cal interest. In connection with the recent trend of cellulose chemistry to tailoredderivatives as building blocks for defined complex supramolecular structures,the preparation of regioselectively substituted organic esters receives increasingattention and will be adequately considered in this subchapter.
Subsequently, a brief systematic survey shall be presented on the characteris-tics of formation and decomposition of the various classes of organic celluloseesters, on the role of supramolecular structure in these processes as well as onproperties and areas of application of the products. This survey will be struc-tured according to the classes of organic acids in question, i.e. unsubstituted andsubstituted aliphatic carbonic acids, aromatic carbonic acids and sulfonic acids,and it will also include the urethanes of cellulose, especially the carbanilate, as aclass of derivatives closely related to the esters.
4.4.3.2 Cellulose formate
The formylation of cellulose belongs to the rather few esterification reactions ofthis polymer with organic acids proceeding to high DS values with the free aciditself. As illustrated by Fig. 4.4.26, DS values of about 2.5 are obtained with 98-100 % formic acid at room temperature after a reaction time of about 2 weeks,with the formic acid simultaneously acting as a 'derivatizing solvent', leadingfinally to a macroscopically homogeneous medium (Takahashi et al., 1986;Philipp et al., 1990).
fe2ΛQ
1
O Λ 8 12Reaction time [d]
Figure 4.4.26. Course of cellulose formylation in HCOOH (Philipp et al., 1990).
Addition of F^SC , HCl or ZnC^ was found by these authors to increase deci-sively the rate of esterification (see Fig. 4.4.26). A lowering of the formic acidconcentration and/or an increase in water content result in a decrease in formylgroup content, as to be concluded from the preparation of DMS O- soluble cellu-lose formates of a DS of about 0.6 in a reaction system consisting of formic acid,phosphoric acid, water, and cellulose, and becoming homogeneous after about

4.43 Esters of cellulose with organic acids 167
24 h (Schnabelrauch et al., 1992). In several studies, a preferential reaction at the6 position has been reported, followed by one at C-2. Esterification of cellulosewith concentrated formic acid combined with dissolution of the polymer is gen-erally accompanied by a severe hydrolytic chain cleavage by scission of theglycosidic bonds.
A special route to cellulose formate, interesting from the viewpoint of syn-thetic organic chemistry, has been described by Vigo et al. (1972), by reactingcellulose in DMF with thionyl chloride, and employing a formimminium com-pound of cellulose as an intermediate, according to the scheme in Fig. 4.4.27.
HI CeII-CI
SOCI2 + 0 = C-N(CH3)2 Chlordesoxycellulose
- SO2
Il ι θ +CeII-OH ι +CeII-OH °CI-S-O-C=N(CH3)2Cle ^CeII-O-S = O *· CeII-CI+ CeII-O-S —OH
- HCI, - DMF
-HCI \ + H2O
OIl
Cell—O —S-OH __Cellulose sulfite CeII-CI
- DMF
H HI θ + CeII-OH l Φ
^ CI-C=N(CH3)2Cle * Cell-0-C=N(CH3)2Cle
- SO2 - HC/
- (CH3J2NH2CI I + H2O
OIl
CeII-O-C-H
Cellulose formate
Figure 4.4.27. Scheme of reaction route to cellulose formate via formimminium com-pounds (Vigo et al., 1972).
As compared with cellulose acetate and the higher fatty acid esters, celluloseformate has to be classified as an unstable derivative: already the moisture con-tent of the air leads to a slow liberation of formic acid, and according to Fuji-moto et al. (1986) a cellulose formate with a DS of 2-2.5 is completely decom-posed by 10 h boiling with water. From studies on sulfation of cellulose for-mates with a DS of 2-2.5 in DMF it could be concluded that sulfation takesplace not only at the free hydroxy groups but probably also by a transesterifica-tion of formyl groups, in contrast with the behavior of cellulose acetate (Philipp

168 4.4 Esterification of Cellulose
et al., 1990). Also, a comparatively low thermal stability had been assessed byDTA measurements in the same study.
The course of cellulose formate formation is strongly affected by the su-pramolecular order of the polymer: the rate of esterification/dissolution in concen-trated formic acid was found to increase in the order cotton !inters < woodpulp < viscose rayon, and could be considerably enhanced by a suitable preactivationof the cellulose sample. The course of a strictly heterogeneous formylation with90 % aqueous formic acid was observed to depend strongly on cellulose physicalstructure on the one hand, and on the reaction temperature on the other, and has beenemployed by several authors to obtain a so-called lateral order spectrum of the sam-ple in question (Marchessault and Howsmon, 1957; Philipp and Baudisch, 1965).According to these studies, structural regions of the sample of successively loweraccessibility are made available for formylation by a stepwise increase of reactiontemperature in the range between -5 0C and +40 0C, resulting, under otherwise con-stant conditions, in characteristic lateral order patterns for different samples. It has tobe emphasized that these patterns can by no means be considered as absolute lateralorder spectra but only as a kind of 'finger print' for classifying various cellulosematerials, especially different types of viscose filaments.
From the viewpoint of material properties, cellulose formates are character-ized by their high susceptibility to hydrolytic ester cleavage, as already men-tioned, and by a good solubility over a wide range of DS in various polar sol-vents like DMSO, DMF, concentrated formic and acetic acid, and dichloropro-pionic acid (Philipp et al., 1990). According to Schnabelrauch et al. (1992) solu-bility in DMSO was already observed at a DS of 0.6.
Up to now, cellulose formates have not been produced on an industrial scaleor applied commercially, and also the attempts to employ this unstable deriva-tive as an intermediate in artificial cellulose fiber spinning so far have not metwith practical success.
4.4.3.3 Cellulose acetate
General comments on reactions and products
Cellulose acetate was described as the first organic ester of cellulose already bySchutzenberger (1865 and 1869), who reacted cotton cellulose with acetanhy-dride in a sealed tube at 180 0C and arrived at an ethanol-soluble product. Four-teen years later, Franchimont (1879) recognized the catalytic efficiency ofH2SÜ4 and also of HC1U4 in this process. Both these observations provided thebasis for commercial cellulose acetate manufacture starting already at the begin-ning of this century and performed up to now with acetanhydride as the esteri-fying agent and sulfuric acid (or in special cases also perchloric acid) as thecatalyst. The raw materials are cotton !inters or refined wood pulp. Acetylsulfuricacid, formed by reaction between the agent and a catalyst, acts as an important

4.4.3 Esters of cellulose with organic acids 169
intermediate in this process providing the necessary level of acetyl cations foresterification. A considerable decrease in chain length due to hydrolytic cleav-age of glycosidic bonds is an inevitable consequence of the strongly acidic sys-tem employed, resulting in a DP level of about 300 for the cellulose acetate,with a starting material of DP of 800-1600.
Acetylation of cellulose is industrially performed either retaining the grossmorphology of the fibers ('fiber acetylation'), or with a transition from an ini-tially heterogeneous to a homogeneous reaction system ('solution acetylation').In both cases a fully substituted cellulose triacetate (CTA) is obtained, as thereaction product in the fibrous state or dissolved in the reaction system, respec-tively. This derivatization to a DS of 3 is necessary in order to secure completeorganosolubility of the product, as a lower average DS leads to an inhomogene-ous distribution of acetyl groups along and between the polymer chains.
While a fiber CTA is directly used for film casting or filament spinning afterdissolution in e.g. CH^C^, the CTA obtained by solution acetylation is usuallyconverted without isolation to a product with a DS of about 2.5 by partialdeacetylation in a homogeneous acid system containing some water. In this waya homogeneous distribution of acetyl groups is obtained, and the so-called 'sec-ondary cellulose acetate' or 'cellulose 2,5-acetate' is easily and completely solu-ble in the convenient solvent acetone, and can be converted to filaments or filmsby a so-called dry spinning process.
Regarding material properties, cellulose acetate resembles more a syntheticplastic than a cellulosic, showing some similarities to cellulose trinitrate, butwithout the inflammability hazards of the latter. CTA and secondary acetateexhibit good mechanical properties and good stability under atmospheric condi-tions, including rot and water resistance. It can be processed, however, only viathe solution state or in the presence of a large amount of plasticizer. Melting isaccompanied by decomposition due to the high melting point of 225-250 0C forcellulose 2,5-acetate and above 300 0C for cellulose triacetate. Most of the ap-prox. 0.9 million tonnes produced annually is employed for the production offilaments, fibers, films, membranes and cigarette filters.
Besides its industrial relevance, acetylation of cellulose plays an importantscientific role as a model reaction in elaborating new routes of synthesis for re-gioselectively substituted cellulose esters, and new analytical techniques fortheir comprehensive characterization.
Chemistry of cellulose acetylation and deacetylation, including effects ofcellulose accessibility on the course of reaction
Cellulose, i.e. !inters or wood pulp, can be acetylated either by direct esterifica-tion of hydroxy groups or by a transesterification, employing a labile primarysubstituent, e.g. a nitrite group (Mansson and Westfeld, 1980), as the leaving

170 4.4 Esterification of Cellulose
group. The reaction can be performed in a strictly heterogeneous way, retainingthe gross morphology of the original fiber, with transition from a heterogeneousto a homogeneous state in a system capable of dissolving the CTA form, or in astrictly homogeneous way after previous dissolution of the unsubstituted poly-mer in a derivatizing or nonderivatizing solvent system. The first two routes areof industrial relevance in manufacturing CTA as a large-scale product, while thehomogeneous route has been amply studied in recent years to prepare well-defined, partially acetylated products.
In contrast with formic acid, acetic acid is not capable to esterify cellulose to asignificant extent, and the more reactive acetanhydride is quite predominantlyemployed, mostly as a liquid, in special cases, also in the vapor phase. Ketene(CH2=C=O) can in principle also be used, if the course of reaction permits theintermediate formation of acetanhydride. Acetyl chloride represents a still morereactive esterifying agent, which is frequently employed in scientific studies,especially in homogeneous acetylation, in combination with a tertiary amine asan adjuvant base. As an interesting variation of the general procedure, acetyla-tion of cellulose in DMF/pyridine with an alkali or alkaline earth salt of aceticacid in the presence of p-toluenesulfonyl chloride, has been reported (Shimizuand Hayashi, 1988). Introduction of acetyl groups by transesterification has alsobeen achieved with ethylene diacetate, with the cellulose dissolved in the system/?-formaldehyde/DMSO at elevated temperature (Johnson, 1969; Johnson andNicholson, 1976).
Just like any esterification, acetylation of cellulose is an equilibrium reaction,which can be shifted to the ester side by applying an adequate excess of reagentand by minimizing the water content in the system, and which can be decisivelyaccelerated by the presence of a suitable catalyst, promoting formation of theacetyl cation CH^CO+ as the reactive intermediate. Minimization of the watercontent is performed here by the interaction between water and acetanhydride(or acetyl chloride). Formation of acetyl cations is promoted in the case ofacetanhydride as the reagent by adding t^SC^ (5-10 % of the weight of thecellulose) or HC1O4 (1-2 % of the cellulose weight) as an acid catalyst, formingacetylsulfuric acid or acetylperchloric acid, respectively. Methanesulfonic acidcan be used, too, but is less effective, just as afe some Lewis acids like ZnC^.With acetyl chloride as the agent, tertiary amines like TEA or pyridine are wellsuited as the adjuvant base, forming an acylium complex according to:
R-C-Cl + N-R2
6 R,
R-C-N-RIl \O R
/nD'2
3
cr

4.4.3 Esters of cellulose with organic acids 111
Still more effective, especially in a homogeneous system with a nonpolar re-action medium, is the stronger, basic 4-dimethylaminopyridine (Philipp et al,1983). The acid-catalyzed acetylation with acetic anhydride results in a dramaticdrop in DP due to hydrolytic chain cleavage, for example from a DP of 1500-2000 of the bleached and scoured cotton !inters employed, down to DP values of350-500 for a CTA prepared by the dissolution process. The preparation ofhigh-DP cellulose acetates with DP values > 1200 was reported by Kulakova etal. (1971) in a system consisting of Ac2O/acetyl chloride and acetic acid.
Turning now more closely to acetylation of cellulose in the acetic anhy-dride/acetic acid system, i.e. a heterogeneous system at least at the beginning ofesterification, the decisive effect of cellulose accessibility, determined by thesupramolecular and morphological structure of the polymer, must be emphasizedfirst: the course of esterification is not only determined by the chemical reactionitself, but also depends largely on sorption, swelling and diffusion phenomena,which affect reaction rate and product quality. Similar to formylation, highlyaccessible regions are esterified first and/or under milder conditions, but in con-trast with formic acid, swelling in acetanhydride is rather small and the activeintermediate, i.e. acetylsulfuric acid or acetylperchloric acid, possesses a largermolar volume than formic acid, thus making penetration of the fiber moietymore difficult. Generally, the disordered regions of the fiber structure are con-sidered to be more rapidly acetylated than the crystalline regions.
Regarding the chemical interaction between cellulose, acetanhydride, aceticacid and catalyst (£[2804 or HClO4) the following statements may be condensedfrom the large number of experimental studies published already in the first halfof this century (Malm and Hiatt, 1954).(i) A prerequisite of any thorough and uniform acetylation is the adequate acti-vation of the cellulose fibers, predominantly performed by using acetic acid,(ii) The catalyst is strongly chemisorbed onto the fibers, as to be concluded fromthe large heat of sorption, which increases in the order of catalytic activity, i.e.ZnCl2 < H2SO4 < HClO4.(iii) Sulfuric acid as a catalyst not only promotes the formation of acetyl cationsas the reactive species in esterification, but also leads to introduction of sulfatehalf-ester groups at a level of some percent of the acetyl groups, which must beremoved in a subsequent step of stabilization (see chapter 4.1). In contrast withH2SO4, perchloric acid as a catalyst does not lead to an analogous esterification,but is known to cause a more severe chain-length degradation than H2SO4.(iv) Acetylation as well as sulfation obviously occur at a higher rate at the C-6position compared with C-2/C-3.(v) The state of interaction between acetanhydride and catalyst influences theoverall course of reaction, as shown by a comparison between a fresh and anaged acetylation mixture.

172 4.4 Esterification of Cellulose
During acetylation of cellulose with Ac2O/HAc/catalyst, either a two-phasesystem can be maintained during the whole course of reaction, resulting in a so-called fiber CTA, or the two-phase system can be gradually transformed to ahomogeneous one, yielding a so-called solution CTA. In fiber acetylation thepreactivated cellulose is reacted with a large excess of Ac2U in the presence ofHAc, usually with HC1Ü4 as the catalyst at slightly elevated temperature for 1 toseveral hours. Formation of the triacetate is indicated by a change in the bire-fringence in the fibers from a positive to a negative value. Also, Ac2U vapor canbe employed for fiber acetylation, with the crystal modification of the fiber CTAdepending on reaction temperature (CTA I below 80 0C, CTA II above 80 0C).This vapor process can be modified by adding some propionic or butyric acidanhydride to the vapor phase, obtaining the appropriate mixed ester, i.e. cellu-lose acetopropionate or acetobutyrate. Besides the preparation of the triester,fiber acetylation can be employed to give a morphologically limited partial ace-tylation, e.g. of only the surface of paper sheets, by suitable adaptation of reac-tion conditions. Also, an acetylation of whole wood fibers from Southern Pinewith AC2Ü has been reported by Rowell (1982) and Shiraishi and Yoshioka(1986), resulting in a 20 % add-on, with a preferential introduction of acetylgroups into the lignin component compared with the holocellulose. The gradualtransition from a heterogeneous to a homogeneous system in solution acetylationis achieved by a large excess of glacial acetic acid acting as a solvent for CTA,applying a moderate excess of Ac2U and !!2804 as the catalyst at a reactiontemperature of about 50 0C and a reaction time of several hours. Complete dis-solution does not occur until a DS of almost 3 is reached. Swelling and dissolu-tion of the polymer is facilitated by the presence of methylene chloride, substi-tuting some of the glacial acetic acid with this good solvent for CTA. Both theseprocedures again require an adequate preactivation of the polymer by acetic acidtreatment with or without part of the catalyst. Both procedures are practisedindustrially to obtain a CTA solution that can be converted to an acetone-solubleproduct by homogeneous deacetylation to a DS of about 2.5 without intermedi-ate isolation of the CTA. Some further details of the technical process will bepresented in the subsequent section.
Fiber CTA and solution CTA differ with regard to solubility in various media,colloid chemical behavior and rate of deacetylation, even at the same averageDP and DS. This difference is obviously caused by a stronger interchain cohe-sion in the case of fiber CTA, resulting in larger supramolecular aggregates,even in dilute solutions of fiber acetates, compared with solution acetates(Bischoff, 1963).
The commercial relevance of the chemical process of acetylation has pro-moted a mathematical modeling of the course of reaction on a predominantlyphenomenological level, providing useful interpolation data on the change of DSwith time of reaction in dependence on various reaction parameters.

4.4.3 Esters of cellulose with organic acids 173
Homogeneous acetylation of cellulose and cellulose derivatives in varioussystems has been amply studied for the last 30 years in connection with the ap-plication of new organic solvent systems, new routes of synthesis for regioselec-tively substituted cellulose derivatives, and new approaches for their compre-hensive analytical characterization. From the results of this work it can be con-cluded that the reactivity of cellulosic hydroxy groups in homogeneous acetyla-tion can vary widely in dependence on the system considered, and that a prefer-ential or even a regioselective acetylation of one or two of the sites within theAGU can be achieved by an appropriate procedure of synthesis: homogeneousacetylation of free hydroxy groups in a partially acetylated sample of DS\c = 1-2 indicated a strong influence of the esterifying agent, as with acetanhydride apreferential substitution at C-6, with acetyl chloride a preferential substitution atC-2, and with the C-3 position showing the lowest reactivity in both cases(Nehls et al., 1994).
Among the numerous nonderivatizing solvent systems for cellulose, so faronly a solution in DMA/LiCl and a melt solution in jV-ethylpyridinium chloridehave been successfully employed for acetylation of this polymer (McCormickand Chen, 1982; Miyamoto et al., 1984 and 1985; Kamide et al., 1987; Huse-mann and Siefert, 1969 and 1970), indicating a preferential substitution at the C-6 position, With many other systems a smooth acetylation with the conventionalreagents Ac2U and acetyl chloride is inhibited by a violent interaction betweenone of the solvent components and the agent. These detrimental side reactionscan be widely avoided by employing derivatizing solvent systems. So, for ex-ample, the cellulose trinitrite formed on dissolving the polymer in theN2O4/DMF system could be transesterified with Ac2U to a cellulose acetate ofDS = 2 with the C-2 position reacting the fastest (Mansson and Westfelt, 1980).In the paraformaldehyde/DMSO solvent system, obviously all the hydroxy endgroups of the methylol side chains are preferentially acetylated withAc2O/pyridine. A high DS of the acetyl groups could also be obtained in thissystem by transesterification of ethylene diacetate in the presence of Na acetateat 90 0C (Seymor and Johnson, 1978). In the system chloral/DMF/pyridine, cel-lulose was found to dissolve with complete substitution of the hydroxy groupsby the appropriate half-acetal groups, which could be acetylated to a DS of 2.5by Ac2U or acetyl chloride (Clermont and Manery, 1974).
The free hydroxy groups within the AGU of rather stable partially substitutedcellulose derivatives can be acetylated to an extent depending on the systemconsidered. A complete substitution of all residual free hydroxy groups has beenreported for tosyl cellulose (DS 0.9-2.3) by reaction with 3 mol of Ac2U permol of hydroxy groups in the presence of sodium acetate (10 % Ac2U) in pyri-dine at 60 0C for 6 h (Heinze et al., 1996a), and for TMS-cellulose (DS = 2) withan excess of Ac2U (Stein and Klemm, 1988). Acetylation of the free hydroxygroups in a benzyl ether of cellulose with DS = 2, in benzene with Ac2U in the

174 4.4 Esterification of Cellulose
presence of TEA has been studied by Philipp et al. (1983); an addition of DMAPto the system was found to increase the DS of acetyl groups from 0.1 to 0.35,while a further increase of Ac2O input did not lead to any significant effect.Starting from 6-O-trity!cellulose with a DS of 0.98, acetylation withAc2O/pyridine resulted in a regioselectively substituted cellulose acetate withpartial DS values of 0.15 at C-2, 0.10 at C-3 and 0.0 at C-6 after detritylationwith gaseous HCl in CF^C^ (Yasuda and Yoneda, 1995). Some results of ourown on the preparation of regioselectively (in the C-2 and C-3 positions) sub-stituted cellulose acetates via 6-0-silylcellulose lead to a DS value of 1.1 startingfrom TMS-cellulose with DS = 1.9. The complete desilylation without deacety-lation takes place with 1 N HCl in THF within 15 min.
Ac2Ü proved to be superior to acetyl chloride in avoiding an early loss ofprimary substituent groups, which could be selectively removed after acetylationby HCl in an aprotic medium like THF (Stein and Klemm, 1988).
As already emphasized, acetylation of cellulose is an equilibrium reaction,deacetylation occurring with an excess of water in the presence of an acid cata-lyst providing a sufficiently high accessibility of the acetyl groups. A homoge-neous partial deacetylation of CTA in aqueous acetic acid, with ^804 as thecatalyst, is practised on an industrial scale in order to reduce the DS^C to about2.5. Energies of activation of 16.6 kcal/mol or 18.3 kcal/mol (Eicher, 1986) havebeen reported for this process in the case of solution CTA, while a much highervalue of 25.2 kcal/mol was observed by Bischoff (1963) for fiber triacetates,which was assumed to be caused by a dissociation of supramolecular clusterswith increasing temperature enhancing the availability of the acetyl groups forhydrolysis. In an aqueous acid system, deacetylation at the C-6 position obvi-ously proceeds faster than at C-2/C-3. A preferential deacetylation at these sec-ondary C atoms, on the other hand, can be performed in amine-containing sys-tems of special composition: Miyamoto reported a preferential deacetylation atC-2 and C-3 in the presence of hydrazine (Miyamoto et al., 1985).
The data summarized in Table 4.4.22 illustrate that preferentially C-6-substituted cellulose acetates can be obtained from CTA by the action of a ter-nary mixture of DMSO/water and an aliphatic amine like e.g. dimethylamine, orhexamethylenediamine, which rather selectively deacetylates the two secondarypositions. Hydrazine, on the other hand, proved to be less effective under theconditions employed. During a homogeneous aminolysis of CTA by ethylenediamine after dissolution in dimethylacetamide in a water free system, Deus etal. (1991) observed a very uniform deacetylation at all three positions of theAGU in comparison to other routes of deacetylation. The joint relevance of thetwo prerequisites for deacetylation, i.e. the presence of an aqueous medium andthe accessibility of the acetyl groups to hydrolysis, are illustrated by the behav-ior of powdered cellulose acetate (DS > 2.5) against water and acetone: the DSof an aqueous suspension of the powder remains unchanged for a long period of

4.4.3 Esters of cellulose with organic acids 175
time due to the hydrophobicity of the particles. The acetate powder dissolved indry acetone also exhibits no detectable change in DS over a long period. Suspen-sion of the particles in a water/acetone mixture, accompanied by considerableswelling, however, results in a significant decrease in the acetyl content within afew hours at room temperature. This is obviously promoted by a small numberof acid groups present in the polymer (Ludwig and Philipp, 1990). The irre-versible alkaline saponification of acetyl groups in solid samples depends ontheir accessibility, i.e. the state of swelling too, as demonstrated in Fig. 4.4.28for the decrease in acetyl DS on treatment with 0.1 N NaOH in water/acetonemixtures of increasing acetone content. In our laboratory-scale studies, an effi-cient hetero-saponification of acetyl groups was achieved by treatment of theswollen sample with 1 N KOH in EtOH.
Table 4.4.22. Homogeneous deacetylation of CTA in amine-containing media at 80 0C(Wagenknecht, 1996; Deus and Fribolin, 1991).
AmineExample mol/mol
ofAGUHMDA 2.3
DMA 4.5
t(h)
2.54.59
1424
511152024
DSAca
2.602.411.871.330.75
2.552.061.841.591.45
DSAc
(NMR)
2.652.41.951.50.75
2.552.01.81.61.2
Pattern of substitutionC-2
0.80.650.450.20.05
0.750.50.350.30.2
C-3
0.850.750.550.450.1
0.80.50.50.40.3
C-6
1.01.00.90.850.6
1.01.00.950.90.7
a Functional group analysis.HMDA NH2-(CH2)6-NH2.DMA HN(CH3)2.
In the absence of water, the acetyl groups of cellulose acetates dissolved in anaprotic liquid are rather stable even in the presence of acid anhydrides or acidchlorides at room or slightly elevated temperature. Within these limits, the acetylgroup serves as an effective protecting group in a subsequent homogeneous es-terification of residual hydroxy groups, as demonstrated e.g. in our studies onsulfation of cellulose 2,5-acetate with SO3 or ClSO3H, resulting in a completesulfation of residual hydroxy groups without loss of acetyl groups. A trans-esterification with elimination of the acetyl group obviously takes place only

176 4.4 Esterification of Cellulose
with high boiling acid chlorides at high temperatures, as indicated by Frautschiet al. (1983) for the reaction of cellulose acetate with palmitoyl chloride at120 0C under nitrogen, resulting in a 50 % conversion. On heating of celluloseacetate under DTA conditions, deacetylation was observed besides dehydrationand glycosidic bond cleavage already at an early stage of thermal decomposition(Jain et al., 1986 and 1987b).
20 4.0 60Acetone [mol%]
Figure 4.4.28. Effect of acetone content and temperature (· 30 0C, · 40 0C, A 50 0C)on the course of saponification of cellulose acetate (DS = 2.9) in 0.1 N NaOH (3 h, liq-uid-to-solid ratio 200 : 1) (Lukanoff et al., 1969).
Finalizing this section on the chemistry of acetylation and deacetylation ofcellulose, the important role of modern techniques of instrumental analysis for acomprehensive characterization of the samples involved must be accentuated.Examples mentioned here explicitly are the 1H and 13C NMR spectroscopicstudies by the group of Kamide, including a complete signal assignment (Ka-mide and Saito, 1994), and the combined solid state NMR and Raman spectro-scopic investigations (VanderHart et al, 1996) on the polymorphs of CTA withnew aspects of correlating the molecular and supramolecular structure of cellu-lose acetates.
Survey of the industrial process of cellulose acetylation
All the industrial processes practised today are aimed at the manufacture of afully substituted cellulose triacetate (DS > 2.9) as the primary product. This iseither isolated and processed as it is, or converted to a so-called secondary ace-tate with a DS of between 1.8 and 2.5 (predominantly near 2.5), by a partial

4.4.3 Esters of cellulose with organic acids 111
deacetylation under homogeneous conditions. A heterogeneous procedureyielding a uniform secondary acetate is not yet available. Most of the 832,000tons of cellulose acetate produced worldwide in 1988, mainly in the USA(50 %), in Western Europe (16 %) and Japan (13 %), is manufactured by theprocess of solution acetylation and subsequently converted to secondary acetate,while only a minor amount is obtained by fiber acetylation.
For both these processes, high-ΖλΡ, scoured and bleached cotton !inters arepredominantly employed as the raw material, but also an adequately refined soft-wood sulfite pulp or even a special great prehydrolysis sulfate pulp can be used.Formation of cellulose II on alkali refining of the pulp should be avoided as itmay impede a smooth course of acetylation. A low ash content, a very low con-tent of alkaline-earth and heavy-metal cations, a low content of organosolubleextractives, as well as a low content of pentosans and mannans, are further re-quirements to be met by an acetate great wood pulp. Studies on acetylation of anonrefined pulp, with 87 % α-cellulose content (Matsumura and Saka, 1992),indicated the formation of a considerable amount of glucomannan acetate, in-soluble in glacial acetic acid, employed as the solvent for CTA. The raw mate-rial is dried to a residual water content of 4-7 % and then preactivated by treat-ment with glacial acetic acid (30-100 % of the cellulose weight), eventually inthe presence of some F^SC . In solution acetylation with either glacial aceticacid alone or in combination with methylene chloride is employed as a solventfor the CTA formed, arriving finally at a polymer concentration of between 10and 20 % in the CTA solution.
Acetylation is generally performed with an excess of acetic anhydride (10-40 % above the amount needed for CTA formation) in the presence of l-^SC^ asthe catalyst. In the 'acetic acid process' the exothermic reaction is performed ina kneader equipped with effective cooling and mixing facilities, adding the ace-tic anhydride stepwise and employing 2-5 % H^SC^ (calculated on celluloseweight) as the catalyst. The mixture, passing gradually from a fiber suspensionto a viscous solution, is kept for several hours at a temperature of 50 0C, reactiontemperature and reaction time determining the decrease in DP. The CTA formedis subsequently converted to secondary acetate without isolation of the CTA byadding water or dilute aqueous acetic acid to the system at an excess of 5-10 %Ü2O above that needed to decompose excess acetanhydride. This excess of wa-ter is sufficient for an effective decomposition of most of the sulfate half-estergroups in the cellulose chain and to decrease the DS of acetyl groups to the levelrequired. But it is still low enough to keep the cellulose acetate in solution. Aftersome hours of treatment at 40-80 0C the reaction mass is buffered with magne-sium acetate. Then the cellulose acetate is precipitated with water under stirring,subsequently cooked under pressure with aqueous 1 % mineral acid for furtherstabilization, washed and then dried under vacuum to a moisture content of 1-3 %. The yield amounts to about 95 % of the theoretical one.

178 4.4 Esterification of Cellulose
In the methylene chloride process, a mixture of about 2 parts of CI^C^ and 1part of glacial acetic acid is employed as the medium for swelling and dissolvingthe activated polymer. The level of the t^SC^ concentration can be kept here atabout 1 %, calculated on cellulose, due to a faster and higher swelling of thereaction mass. The acetylation itself is performed under similar conditions as inthe acetic acid process, with the low-boiling CH2C12 serving as an internalthermostat to keep the reaction temperature at a level of 50 0C in the mixingvessel equipped with stirring facilities. Desulfation and partial deacetylation arein principle performed as already mentioned, with the exception that the CI^C^is distilled off and recovered before precipitation of the cellulose acetate. Anadvantage of the methylene chloride process is the much lower amount of diluteaqueous acetic acid to be disposed of as a waste product of the process.
In fiber acetylation, the activated cellulose is reacted with an excess ofacetanhydride in the presence of a large amount of a nonsolvent for CTA (CC^,benzene, toluene) and about 1 % HC1O4 (calculated on cellulose) in a rotatingsieve-drum, mounted in a stainless steel vessel at a temperature of up to 50 0Cfor one to several hours. After the reaction the CTA is separated from the liquidphase, buffered, washed and freed from residual nonsolvent by steaming, anddried to a low residual moisture content of about 1 %. A stabilization step isusually not necessary here as no perchloric acid ester groups are bound to thecellulose.
Continuous processes for cellulose acetylation have been described too, butobviously are scarcely practised due to the inferior uniformity and general qual-ity of the products obtained.
Properties of cellulose acetate
Cellulose triacetate is a semicrystalline polymer, crystallizing in the two allo-morphs of CTA I and CTA II. Ample research efforts have been made to eluci-date the detailed structure of these modifications and their conditions of forma-tion (Buchanan et al., 1987). By VanderHart et al. (1996) the structural differ-ence is traced back to a different backbone conformation, and a different chainpolarity, i.e. parallel in CTA I and antiparallel in CTA II is considered as prob-able. In dependence on polymer concentration, DP, temperature and solvent,CTA can form various liquid crystalline phases. For details the reader is referredto the comprehensive work of Zugenmaier (1994) and Guo and Gray (1994).
Commercial cellulose acetates, i.e. CTA and cellulose 2,5-acetate, are high-melting, high-strength and tough polymer materials, exhibiting a high UV sta-bility and film transparency, combined with low inflammability. CTA melts at306 0C, and cellulose 2,5-acetate at 225-250 0C with decomposition. The pres-ence of butyrate groups besides the acetate groups decreases the melting point of

4.4.3 Esters of cellulose with organic acids 179
cellulose acetates considerably, and the solubility and the compatibility withother polymers are enhanced.
Concerning the scientifically interesting and technically important point ofcellulose acetate solubility, the hydrophobicity and the high resistance to hydro-carbons are to be mentioned as characteristic material properties of high-DScellulose acetates. Table 4.4.23 presents an overview of solvents for the variouscellulose acetates in dependence on DS.
Table 4.4.23. Solubility of cellulose acetate with different patterns of substitution invarious liquids.
Liquid DSAC range of solubility for partially deacetylatedcellulose acetate
WaterDMFAcetone (< 0.01 % H2O)Acetone (1 % H2O)PyridinePyridine/H2O (1 :1 v/v)Ethyl lactate
in C-2/-3/-6 position3
0.8-1.01.8-2.7insoluble2.3-2.60.8-2.70.6-2.01.6-2.7
in C-2/-3 position13
insoluble1.3-2.8insoluble2.5-2.61.2-2.81.2-1.62.6-2.8
Deacetylation of cellulose triacetate:a with CH3COOHTH2SO4 (Deus and Fribolin, 1991).b with amine/DMSO/H2O (Philipp et al., 1995).
This classification of course only holds true on the prerequisite of a suffi-ciently uniform acetyl-group distribution along and between the polymer chains.Otherwise no complete solubility at all can be expected. Besides this decisivefactor, the substituent distribution within the AGU plays an important role too:from the results published in Miyamoto et al. (1985), Deus and Fribolin (1991)and Philipp et al. (1995) it can be concluded that the hydrophile/hydrophoberatio, i.e. the ratio of hydroxy to acetyl groups at the C-6 position, predomi-nantly determines the solubility of the sample in various solvents. So, for exam-ple, solubility in water has been observed for low-DS cellulose acetates in theDS region of around 0.8 only for samples carrying a large amount of hydroxygroups at C-6 in the case of statistical acetyl group distribution, whereas afterregioselective deacetylation of a CTA at the C-2 and C-3 positions the reactionproducts remained insoluble in water in the same DS region. Worth mentioningin this connection is our observation that commercial cellulose 2,5-acetate, withits free hydroxy groups rather equally distributed on the three positions at C-2,C-3 and C-6, proved to be insoluble in an absolutely dry acetone, with a water

180 4A Esterification of Cellulose
content below 0.02 %, whereas it readily dissolved in standard-grade acetonewith a water content of about 1 %. Another point of interest to be traced back,however, to a different course of reaction, is the difference in solubility betweensolution CTA and fiber CTA described in Bischoff (1963): while the solubilityof a solution CTA in acetone at -40 0C reached the 20 % level, a fiber CTAexhibited a solubility of less than 5 %, obviously due to a stronger chain aggre-gation persisting from the supramolecular structure of the native cellulose.
The rheology of cellulose acetate solutions at various levels of polymer con-centration, DS and solvent has been widely studied, including that of liquidcrystalline systems and of thermoreversible gels formed in e.g. water/dioxane asthe solvent (Altena et al., 1986). From the numerous investigations published bymany groups, promoted by the industrial relevance of the dissolved state forcellulose acetate processing, only two rather arbitrarily chosen examples shall bepresented here in order to illustrate the broad spectrum of topics studied:Klenkova and Khlebosolova (1977) compared the rheological behavior of CTAwith that of cellulose tripropanoate and cellulose tributyrate, concluding fromtheir results a high chain flexibility of CTA and a predominance of the DP abovethe DS in determining the rheological properties of CTA in solution. Burchardand Schulz (1989) studied the intermolecular interaction between cellulose ace-tate macromolecules by employing globular proteins as the probe and concludedfrom their results the presence of reversible as well as irreversible supramolecu-lar aggregates, assuming the existence of some kind of fringed micelle for CTAin solution. Obviously, the state of solution of cellulose acetates still presentsnumerous open problems to polymer and colloid science due to the large numberof variables involved, but also offers further approaches to give defined su-pramolecular structures by employing cellulose acetates with a tailored substitu-ent distribution.
Application of cellulose acetate
Cellulose acetate is commercially available either as the triacetate (DS 2.9-3.0;acetyl content ca. 45 %) or as cellulose 2,5-acetate (DS 2.4-2.5; acetyl contentca. 40 %), and as a specialty also in the DS range 1.85-2.0 (acetyl content ca.35 %). Besides products carrying the acetyl group as the only ester group, mixedesters with a varying amount of propanoic or butanoic ester groups are manu-factured in order to improve melt processibility.
The classical areas of cellulose acetate application are the manufacture offilaments for textile use and of films via a solution of the secondary acetate(DS ~ 2.5) in acetone. The filaments are formed in a so-called 'dry spinningprocess' by evaporation of the low-boiling solvent during thread formation be-tween spinneret and godet. A spinning dope containing between 20 and 30 %polymer (preferentially 25 %) is pressed through a spinneret with 20-100 holes

4.4.3 Esters of cellulose with organic acids 181
in a 4-6 m long spinning column and exposed to a stream of hot air at 80-100 0C resulting in formation of the solid filament by solvent evaporation. Thefilaments are stretched in a still plastic state to enhance their mechanicalproperties. A spinning speed between 300 and 800 m/min is generally employed.In spinning cellulose triacetate filaments from a solution in methylene chlo-ride/methanol, the stretched filaments showing a core shell structure due to par-tial crystallization are heat-set at 180-220 0C for some minutes or seconds inorder to reduce water retention and water absorption and to improve the washand wear properties of the finished goods.
Cellulose triacetate (fiber triacetate) finds its predominant application in theproduction of high-quality cine film as it exhibits an excellent dimensional sta-bility combined with very low flammability, in contrast with films from cellu-lose nitrate. Besides films, textiles from CTA filaments are on the market, pro-duced by dry spinning of a CTA solution in e.g. a methylene chloride/methanolmixture. With regard to textile properties, cellulose acetate filaments take anintermediate position between rayon and synthetics, resembling much more thelatter. Due to this competition with synthetics, no growth in production and mar-ket share can be expected in the future, but textiles from cellulose acetate willkeep their place despite their rather high production cost due to some specialassets regarding e.g. handle and dyeability. About 130,000 tonnes per year ofcellulose acetate filaments are still produced, especially for linings and women'sapparel wear.
As a third, also a classical area of cellulose acetate application, its use as aplastic material, must be mentioned. Especially mixed esters containing butyrate,besides the acetate groups (cellulose acetobutyrate) can be melt processed, espe-cially by injection molding to produce consumers goods with attractive me-chanical properties and attractive appearance; but also in this field celluloseacetate stands in hard competition with synthetic plastics.
Thermoplastic processing of cellulose acetate to high-quality consumer goodsis realized today along the two routes of:(i) thermoplastic shaping of cellulose acetate proper in combination with about30 % softener (mostly phthalates);(ii) melt processing of cellulose acetobutyrates of varying ester-group ratio.
A growing market for cellulose acetate can be seen, however, in two more re-cent areas of application, i.e. as a material for cigarette filters and for separationmembranes. As a material for cigarette filters, cellulose acetate obviously meets inan unique manner the requirements of filtering efficiency and taste quality.
During the recent decades, cellulose acetate (DS 2.5-3) has found a new, in-teresting and prosperous area of application in the manufacture of separationmembranes for ultrafiltration, reverse osmosis and hemodialysis. These mem-branes are prepared from solutions of the polymer in a suitable liquid or a mixedsolvent, combining solvent evaporation, polymer precipitation by a nonsolvent

182 4 A Esterification of Cellulose
and eventually subsequent annealing of the solid product. By these numerousdegrees of freedom, the pore size of the membrane can be varied within ratherwide limits and adapted to the special end-use intended. In any case the porestructure is asymmetric, exhibiting a pore-size gradient across the membranewith a fine porous 'separation active' layer at one side (pore size in the nm
range) and a coarse, porous supporting layer (pore size in the μιη range) at theother. In reverse osmosis predominantly employed for desalting of sea water,these cellulose acetate membranes have the advantage of good stability againstchlorine chemicals in the necessary disinfection cycles, but show a lower fluxrate as compared with the competing synthetic products from aromatic polyam-ides. Also, in hemodialysis in the so-called 'artificial kidney', cellulose acetatemembranes are still widely employed due to their good blood compatibility.
4.4.3.4 Cellulose esters of higher aliphatic acids
In principle, cellulose esters of higher aliphatic acids are synthesized along thesame routes as described for cellulose acetate, i.e. employing the acid anhydridewith a suitable catalyst or the acid chloride in the presence of a tertiary base asthe predominant acylation systems. It must be taken into account that the higheracid chlorides and acid anhydrides are less reactive than e.g. acetyl chloride andacetic anhydride, and that these higher anhydrides and chlorides are rather spe-cial and therefore expensive chemicals. The 'impeller technique' employing theappropriate carbonic acid in combination with chloroacetic anhydride is of spe-cial interest in connection with the higher cellulose esters, and effort has beenmade to find catalysts of very high efficiency.
The propionylation of cellulose of course resembles most closely acetylation,and can be performed as a solution propionylation with the anhydride and anacid catalyst. Also, a cellulose suspension in dioxane/pyridine can be employedfor propionylation to high DS, in this case with propionic acid chloride as theagent. Farvardin and Howard (1985) studied the heterogeneous propionylationof cellulose in systems consisting of propionic acid, propionic anhydride and anappropriate metal chloride as the catalyst in an aprotic solvent, comparing vari-ous metal chlorides and solvents with regard to their effect on reaction rate. Thekinetics was described by two consecutive first-order reactions with the secondone proceeding faster than the first one. For a homogeneous acylation of cellu-lose to esters, with an aliphatic chain length of between three and eight carbonatoms, a 2 % polymer solution in DMA/LiCl with 9 % LiCl, and a mixture ofthe appropriate acid with its anhydride in the presence of dimethylcyclohexyl-carbodiimide or pyrrolidinopyridine as the catalyst was employed (Sama-ranayake and Glasser, 1993). A very low excess of reagent was reported to benecessary for reaching high DS values, with the sites at C-6 and C-2 being morereactive than that at C-3. Regioselectively substituted propionylcelluloses with

4.4.3 Esters of cellulose with organic acids 183
the ester groups in the C-2/C-3 positions have been prepared from 6-0-trimethylsilyl and 6-0-tritylcelluloses by reacting this compounds with an ex-cess of propionic anhydride in the presence of pyridine and subsequent desilyla-tion or detritylation with HCl (Iwata et al., 1992). The 2,3-propionates proved tobe more stable in the acid medium than the 2,3-acetates and could be isolatedwithout loss of ester groups. The propionylation of partially substituted methyl-and ethylcelluloses to give stable ether-esters has been reported by Guo andGray (1994), with free hydroxy groups in the C-6 position being preferentiallyesterified during the homogeneous reaction.
Mixed esters containing aliphatic residues from C-3 to C-5, besides acetylgroups, can be prepared in the conventional way with the appropriate acid anhy-drides in the presence of Ρ^Οφ
For the preparation of higher aliphatic cellulose esters care must be taken indrying the solvent employed. A preactivation of the polymer with an aliphaticamine was found to be advantageous in synthesizing higher esters from the bu-tyrate up to the stearate, with the acid chloride or the anhydride as the agent. Thehydrophobicity of the product increased with DS and with the molar volume ofthe substituent. On esterification of a hydrolyzed cellulose ('microcrystallinecellulose') with pelargonic acid chloride, up to a DS of 3 has been reported byBattista et al. (1978). In a medium of DMF and pyridine, a mixture of p-toluenesulfonyl chloride and the Na-salt of the appropriate aliphatic or aromaticacid was found to be effective in preparing higher cellulose esters (Shimizu etal., 1993a). A homogeneous transesterification was reported in Shimizu et al.(1993b) for a cellulose trinitrite by lauroyl chloride in a N2Ü4/DMF solution ofholocellulose (delignified wood consisting of cellulose and hemicelluloses).
A special route to higher aliphatic esters of cellulose has been proposed(Kwatra et al., 1992): in this 'vacuum acid chloride technique' the cellulose isreacted directly with the appropriate acid chloride without the presence of a sol-vent at a sufficiently high temperature. The HCl formed is eliminated from thesystem continuously by vacuum. A palmitoyl ester of cellulose was obtainedwith a yield of 90 %. The reaction was found to be chemically and not diffusioncontrolled; adequate kinetic models were reported.
A full signal assignment of the 1H and 13CNMR spectra of cellulosetriacetate, tripropionate and tributyrate has been published (Buchanan et al.,1987) with the conclusion that only small changes in chemical shift (usuallywithin 1 ppm) take place depending on the size of the ester group. An NMRspectroscopic study of two regioselectively substituted, mixed cellulose triesters,i.e. 6-0-acetyl-2,3-0-propionylcellulose and 6-0-propionyl-2,3-0-acetylcellu-lose has been published by Iwata et al. (1996).
Some physical properties of higher aliphatic cellulose esters in the solid stateare presented in Table 4.4.24 in comparison with cellulose acetate. Obviouslythe intermolecular interaction between the polymer chains decreases with in-

184 4.4 Esterification of Cellulose
creasing length of the ester side chain, as indicated for example by the change inmelting point and in the elastic modulus of the crystalline regions (Nishino et al.,1995) of these semicrystalline solids. According to Buchanan et al. (1989) noprinciple change in polymer backbone conformation is induced by increasing thelength of the ester side chains. Already the cellulose butyrate melts without de-composition at 192 0C and thus can be melt processed.
The higher esters of cellulose, as investigated in the range from C-3 to C-18of side chain length, are increasingly hydrophobic, but soluble in many organicliquids of medium to low polarity. Methylene chloride is a good solvent formany of these cellulose derivatives. An especially broad spectrum of solvents isknown for esters of medium chain length, e.g. the valerate and the caproate.Many of these higher aliphatic esters of cellulose form liquid crystalline systemswith suitable solvents, and especially the higher members of the homologousseries, e.g. the octadecanoate, were found to be suitable for the preparation ofLangmuir-Blodgett monolayers and multilayers (Kawaguchi et al., 1985).
Rheological studies (Klenkova and Khlebosolova, 1977) on semiconcentratedsolutions of cellulose triacetate, tributyrate and acetobutyrate, in dependence onDP, substituent group and DS, indicated a predominant effect of DP, with cel-lulose triacetate showing the highest value of T]Q under comparable conditions.Correlations between the rheological properties of these solutions and the physi-cal properties of threads and films prepared therefrom were concluded from thisstudy. Rheological investigations of dilute solutions of cellulose tripropionate(Casay et al., 1995) lead to the assumption of worm-like chains in these systems,in between the limiting models of a random coil and a rigid rod.
100
80
60
40
20
Acetate
O 0.5 1.0 1.5 2.0 2.5 3.0DS
Figure 4.4.29. Complement (C 5a) activation (y-axis in %) by cellulose based-membranes with various ester substituents (Vienken et al., 1995). Complement activationas a criterion of membrane hemocompatibility is given in relation to a nonmodified cel-lulose standard (= 100%).

Tab
le 4
.4.2
4. S
ome
prop
ertie
s of
hig
her r
c-al
ipha
tic tr
iest
ers
of c
ellu
lose
in c
ompa
riso
n w
ith
cellu
lose
ace
tate
Tri
este
r
(2,5
-Ace
tate
)A
ceta
tePr
opio
nate
But
yrat
eV
aler
ate
Cap
roat
eH
epta
noat
eL
aura
teM
yris
tate
Palm
itate
Mel
ting
poin
t(0C
)22
5-25
030
623
418
312
2 94 88 91 106
105
Cha
rpo
int
(0C)
ca. 2
3031
5>
315
> 31
5>
315
> 31
529
0>
315
315
315
Den
sity
(g/m
l)
1.30
1.28
1.23
1.17
1.13
1.10
1.07
1.00
0.99
0.99
%
Moi
s-tu
re r
egai
n(7
5 %
r.h.
)6-
6.5c
3.8
1.5
0.7
0.3
0.2
0.2
0.1
0.1
0.1
Ten
sile
So
lubi
litie
s in
b
stre
ngth
3 M
ethy
lene
A
ceto
ne(k
g/m
m2)
chlo
ride
+ +
7.3
+4.
9 +
+3.
1 +
+1.
9 +
+1.
4 +
+1.
1 +
+0.
6 +
0.6
+0.
5 +
Eth
yl
Tol
uene
acet
ate
— —
— —
+ + + +
++
+
+
+ + +a M
easu
rem
ents
on
film
s; b
sol
uble
(+)
, ins
olub
le (-
); c
65
% r.
h. (
Mal
m a
nd H
iatt,
195
4).

186 4.4 Esterification of Cellulose
Higher aliphatic esters of cellulose find application as specialty plastics, pre-dominantly as mixed esters, especially as acetobutyrates of cellulose, which canbe melt processed. Furthermore, cellulose acetobutyrates are used as componentsin melt coatings for paper. Cellulose propionate has been proposed for the prepa-ration of microspheres for the encapsulation of antibiotics. Higher members ofthe series find current interest in the preparation of Langmuir-Blodgett layers.Introduction of palmitoyl groups into a cellulose acetate was found to increasealbumin binding and hemocompatibility of films formed therefrom. Systematicstudies of the effect of aliphatic ester group and DS on the hemocompatibility ofcellulose-based hemodialysis membranes (Vienken et al., 1995) indicated a DSoptimum for each system investigated, which was shifted to lower DS valueswith increasing side chain length, with an optimal compatibility being obtainedwith a low-substituted cellulose stearate (Fig. 4.4.29).
4.4.3.5 Esters of cellulose with substituted monocarboxylic aliphaticacids
Most of the work published in this area has been performed with chlorinated orfluorinated acetic acids or their anhydrides or chlorides, respectively. Halogena-tion at the methyl group generally increases the reactivity in esterification. Ap-plication of chloroacetic anhydride as a catalyst to esterification of cellulosewith other less reactive agents has already been mentioned. Bludova et al. (1984)compared the heterogeneous course of reaction of cellulose with formic acid,acetic acid, trichloroacetic acid and trifluoroacetic acid, and reported forCFßCOOH a thorough reaction of amorphous as well as of crystalline regions,whereas with CC^COOH only the amorphous regions were found to be acylatedand dissolved. A preferential substitution at the C-6 position was observed inboth cases. According to Pikler et al. (1980) a monochloroacetate of cellulosecan be prepared by reaction of alkali cellulose with an excess of chloroacetylchloride in DMF. The chlorine content of the product was reported to increasestrongly with the reaction temperature between 70 and 100 0C under otherwisefixed reaction conditions, and an energy of activation of 80 kJ/mol was calcu-lated from this dependency. In contrast with propionic acid itself, 1,2-dichloropropionic acid can be directly reacted with cellulose in the presence ofHC1O4 as a catalyst (Jain et al., 1980). 2,2-Dichloropropionic acid esters of par-tially substituted carboxymethylcellulose were obtained by reacting the un-modified cellulosic hydroxy groups with the appropriate acid chloride in pyri-dine at 20 0C for 4 h, employing a fine suspension of CMC in the reaction sys-tem (Schnabelrauch et al., 1990). A more convenient and effective procedure forsubsequent modifications of CMC has been described (Vogt et al., 1996). CMCwas treated in a dipolar-aprotic solvent like DMA or DMSO with p-toluenesulfonic acid, yielding a highly reactive gel-suspension of the polymer.

4.4,3 Esters of cellulose wiih organic acids 187
This mixture allows the direct esterification of unmodified hydroxy groups ofCMC, as exemplified by acylation with carbonic acid chlorides or anhydridesand with isocyanates as well as by sulfation, phosphatation and silylation.
Of some relevance to cellulose derivatization as well as to cellulose dissolu-tion is the interaction between the polymer and CF3COOH of 98-100 % con-centration. This acid dissolves cellulose already at room temperature withoutconsiderable degradation, and regenerated cellulose without any ester groups canbe recovered from these solutions by precipitation in an aqueous medium. Therewas some discussion on whether or not the cellulose is esterified on dissolutionin CFßCOOH, which could be settled by a 13C NMR spectroscopic study (Nehlset al., 1995): On dissolving cellulose in concentrated trifluoroacetic acid, most ofthe derivatization does not occur before a clear solution is obtained. As shownby the 13C NMR spectra in Fig. 4.4.30, at first only the C-6 position is affected,followed later on by the C-2 and to a smaller extent also the C-3 position, arriv-ing after about 28 days at a total DS of 1.6.
C-2.3,5
100 90 80 70 60ό [ppm]
Figure 4.4.30. 13C NMR spectra of cellulose after different times of reaction in trifluo-roacetic acid (Nehls et al., 1995): (a) 10 h; (b) 2 days; (c) 28 days; index ' means esteri-fied position, index " means influenced by C'.
Several routes of synthesis to cellulose trifluoroacetates were recently devel-oped and compared by Liebert et al. (1994), i.e.(i) esterification with a mixture of CF3COOH and trifluoroacetic acid anhydride;(ii) esterification with CF3COOH and partially hydrolyzed POC^, arriving at aDS of TFA groups of up to 1.6 for the reaction product soluble in DMF, DMSO,or pyridine;

188 4.4 Esterification of Cellulose
(iii) reaction of cellulose with phenyltrifluoroacetate resulting in insoluble prod-ucts with a DS of TFA groups of 0.3 only;(iv) reaction of TMS-cellulose of DSsi = 2.8 with CF3COOH and partially hy-drolyzed POCl3 in Ct^C^, with the TMS groups obviously acting as the leav-ing groups and arriving at a DS of trifluoroacetate groups of up to 2.4, withcomplete elimination of the silyl substituents, the products being soluble inDMF, THF and acetone.
Cellulose trifluoroacetates of high purity in the DS range 1.5-2.1 could beprepared along route (i) and a subsequent 'thermal purification' at 150 °C/80 Pafor elimination of excess reagent, solvents and by-products. The esterificationwas accompanied by a moderate chain degradation from e.g. DP 1400 to DP800. The trifluoroacetates exhibited good solubility in DMF, DMSO, THF andpyridine and thermal stability up to 250 0C. The 13C NMR data revealed again apreferential substitution at the C-6 position. Contact with water at room tem-perature led to a quick and complete decomposition to regenerated cellulose. Inthe authors opinion, cellulose trifluoroacetates can be considered as versatileintermediates for subsequent steps of derivatization.
Methacrylate esters of cellulose with a DS of up to 2.0 have been preparedwith methacryloyl chloride as the agent in the presence of pyridine in DMF asthe medium (Svistunova et al., 1964). The free hydroxy group could be subse-quently acetylated, and an analogous route of synthesis was described for mixedoleate/acetate esters of cellulose (Iodannidis et al., 1966). Another route tomixed cellulose esters containing acetyl and methacryloyl groups was describedin Pohjola and Aarmikoivu (1976) and Pohjola et al. (1976), starting from a meltsolution of cellulose in A^-ethylpyridinium chloride, which was reacted with0-10 mol of acetanhydride and 3-7.5 mol of methacryloyl chloride per mol ofAGU in the presence of pyridine, arriving at products with a total DS between0.5 and 2.5 and a DS of vinyl groups between 0.1 and 0.9. As shown quite re-cently by Zhang and McCormick (1997), the DMA/LiCl system is well suitedfor a homogeneous esterification of dissolved cellulose with various unsaturatedcarbonic acids or their anhydrides, e.g. crotonic, methacrylic, vinylacetic or cin-namic acid. A^TV'-Dicyclohexylcarbodiimide was employed as a condensationagent and 4-dimethylaminopyridine (or 4-pyrrolidinopyridine) as an acylationcatalyst. Reaction products obtained with crotonic or methacrylic acid (or theiranhydrides) exhibited poor solubility, due to side reactions favoring the highreaction temperature required here. The reaction with vinylacetic or cinnamicacid, however, proceeded facile to products readily soluble in DMSO.
A direct route to acetoacetates of cellulose was recently published in Edgar etal. (1995) by reacting a cellulose solution in DMA/LiCl with bis-/-butylacetoacetate or acetoacetic acid chloride arriving at esters with a DS of upto 3. Solubility in various media was determined by the level of DS, low-DS

4.4.3 Esters of cellulose with organic acids 189
products being dissolved in t^O. Also, the preparation of levolinic acid esters ofcellulose with DS values up to 1 has been reported (Vladimirova et al., 1965).
So far, cellulose esters with substituted monocarboxylic aliphatic acids havenot been manufactured on a commercial scale and have found, with the excep-tion of the trifluoroacetates, only limited scientific interest in the organic chem-istry of cellulose.
4.4.3.6 Esters of cellulose with di- and tricarboxylic aliphatic acidsand their derivatives
Publications in this area dealing mostly with compounds carrying oxalic, malo-nic, maleic or succinic acid residues. A comprehensive review has been pub-lished (Allen and Cuculo, 1973). The routes of synthesis are analogous to thosepresented for monocarboxylic acid esters. They start from cellulose or a partiallysubstituted cellulose ester or ether in a heterogeneous or a homogeneous system.They employ the acid anhydride or acid chloride as the esterifying agent, withthe peculiarity that these agents can react bifunctionally with the result ofcrosslinked and therefore insoluble products. Crosslink formation may be re-duced by masking one of the acid functions with a less reactive group like anester or amide moiety.
Cellulose oxalates with up to 2 acid equivalents bound per mol of AGU andprobably considerable crosslinking were obtained in a heterogeneous reaction ofspruce sulfite pulp with oxalyl chloride in glacial acetic acid or DMF. The pres-ence of 4-dimethylaminopyridine enhanced the add-on considerably in the caseof native pulp, but reduced it significantly in the case of mercerized pulp, proba-bly due to the changed pore structure of the sample impeding penetration of thevoluminous acid chloride-DMAP complex (Philipp et al., 1983). Organosolublecellulose oxalates could be obtained by reacting the polymer with an oxalic half-ester acid chloride ROOC-COCl in the presence of pyridine in nitrobenzene(Frank and Caro, 1930). The synthesis of a cellulose trimethoxalate has beendescribed by Rebek and Jurkowisch (1977), who reacted cotton cellulose withmethoxalic acid anhydride in the presence of pyridine at 60 0C, arriving after 4 hat a DS of 2.9.
A rapidly proceeding succinylation of cellulose with succinic anhydride inmethanesulfonic acid at 25 0C has been described by Hirabayashi (1984), lead-ing to only a small amount of crosslinking, which however rendered the prod-ucts incompletely soluble. Esterification of cellulose with e.g. succinamic, ma-leamic (and phthalamic) acid by a pad bake technique in the presence of ammo-nium sulfamate to DS values between 0.5 and 1, has been described (Cuculo,1971; Allen and Cuculo, 1976):

190 4 A Esterification of Cellulose
O OIl Il
CeII-OH + H2N-C-(CH2)2 —C-OH
75O0C Aqueous medium
Cell-O-C — (CH2)2-C-OH + NH3
O O
The products proved to be soluble in 5-12 % aqueous NaOH with the amidegroup being saponified, and a carboxylated crosslinkable cellulosic compoundbeing formed. As curiosities, the preparation of a cellulose furoate by esterifica-tion with furoic acid anhydride and pyridine in a dipolar aprotic solvent, and of acellulose citrate with a rather large amount of free carboxyl groups shall bementioned (Shaposhnikova et al., 1965; Touey and Kiefer, 1956).
Esterification of free hydroxy groups in partially substituted cellulose acetateswith a DS^c between 2 and 3 has been accomplished with various dicarboxylicacid chlorides or anhydrides in the presence of a tertiary amine like pyridine anda metal acetate as catalyst, leading to soluble as well as insoluble products de-pending on reaction conditions (Malm and Fordyce, 1940).
A promising route to new cellulose derivatives consists in the attachment ofunsaturated ester groups with C-C double or triple bonds onto the polymerskeleton, as shown recently (Klemm and Vogt, 1995) by the esterification of freehydroxy groups in carboxymethylcellulose with maleic acid anhydride in a di-polar aprotic solvent, or by introduction of C-C triple bonds via esterificationwith acetylene dicarboxylic acid methyl ester after dissolving the cellulose inDMA/LiCl.
4.4.3.7 Cellulose esters with aromatic acids
In contrast with the broad variety of aliphatic esters experimentally studied, thespectrum of aromatic esters of cellulose investigated so far in some detail israther small and quite predominantly limited to the synthesis and characteriza-tion of cellulose benzoates (including ring-substituted products) and phthalates.Besides the esters of aromatic carboxylic acids, that of /?-toluenesulfonic acid,known as tosylcellulose, will be considered here in some detail as an interestingintermediate in cellulose derivatization chemistry. With regard to the high boil-ing point of the acid anhydrides and acid chlorides in question, which are em-ployed also as the esterifying agents, aromatic ester synthesis sometimes can beperformed at rather high temperature with an excess of agent serving as the re-action medium.

4.4.3 Esters of cellulose with organic acids 191
According to Braun and Bahlig (1994) a cellulose tribenzoate with a DS be-tween 2.8 and 2.9 is obtained in a one-step reaction with benzoyl chloride in thepresence of pyridine. By Mannschreck and Wernicke (1990) nitrobenzene isrecommended as a medium for preparing a tribenzoate of cellulose with benzoylchloride in the presence of pyridine at 130-140 0C, while a monobenzoate couldbe conveniently prepared by reacting alkali cellulose with an appropriate amountof benzoyl chloride. Also, higher substituted products were obtained in a Schot-ten-Baumann-type reaction with NaOH and benzoyl chloride, but pyridine as abase proved to be more effective for this purpose. An unconventional route ofsynthesis has been described by Isogai et al. (1988), who obtained a cellulosebenzoate of DS 2.5 and a DP of about 800 by ozonization of a cellulose triben-zyl ether of DP 1200.
Cellulose tribenzoate is a hard and brittle solid with a glass transition tem-perature of 155 0C and a melting temperature of 274 0C, and soluble in DMF,CHCl3 and CH2Cl2 (Braun and Bahlig, 1994). For a smooth film formation, atleast 20 % softener is required. The tribenzoate was found to be thermally stableup to 250 0C. Differential scanning calorimetry and TG data between 20 and450 0C were published by Jain et al. (1986), indicating debenzoylation and radi-cal formation at high temperature. According to Mannschreck (1990) cellulosetribenzoate is a versatile sorbent for separating various enantiomers.
Derivatization to benzene-ring-substituted cellulose benzoates of high DS hasbeen accomplished with the appropriate free acids containing -NO2, -Cl, or-OCH3 in the presence of pyridine and p-toluenesulfonyl chloride. The positionof the substituent within the benzene ring proved to be of minor importance forthe course of reaction (Shimizu et al., 1993a, b). A remarkable catalytic effect of4-dimethylaminopyridine was observed in the esterification of the free hydroxygroups of a cellulose benzyl ether of DS = 2 with 1 mol of 4-nitrobenzoyl chlo-ride in the presence of 1 mol of TEA per mol of AGU at room temperature inbenzene as the reaction medium. By addition of 0.2 mol of DMAP/mol of AGUto the system, the DS of benzoate ester groups increased from less than 0.01 to0.3-0.4 in this homogeneous reaction (Philipp et al., 1983). As illustrated by thedata in Table 4.4.25, see also Fig. 4.4.31, the TMS group acts as a leaving groupat or above 100 0C, with an excess of benzoyl chloride serving as the reactionmedium, and the benzoate ester groups are prelimanary introduced at the C-6position with elimination of the volatile trimethylchlorosilane. At low tempera-ture in the presence of a tertiary amine, on the other hand, the TMS group is aneffective protecting group, and free hydroxy groups in the C-2/C-3 position arebenzoylated (Stein and Klemm, 1988; Klemm et al., 1990).
Cinnamates of cellulose with a DS of up to 3 have been prepared in a homo-geneous reaction of the polymer dissolved in DMA/LiCl with cinnamoyl chlo-ride in the presence of pyridine at 30-60 0C, with a preferential substitutionbeing observed at the C-6 position (Ishizu et al., 1991). This homogeneous reac-

192 4.4 Esterification of Cellulose
tion was compared in Ishizu et al. (1991) with the heterogeneous one of a cellu-lose suspension in cinnamoyl chloride and pyridine, and with a Schotten-Bau-mann-type reaction with aqueous NaOH.
Table 4.4.25. Conditions and results of the benzoylation of TMS-cellulose with acidchlorides (Stein and Klemm, 1988; Klemm et al., 1990).
TMS- Acid chloride Amine Reactioncellulose R-COCl tempera-
(DS) (mol/molof AGU) ture(0C)
,Z. 4-Ό ο / \— MO — AoU
(2.5)
(5.0)2·62 R=^jV-CH2-CH2-Br ~& 16°
(3.5)
i-55 R=-/=VNO TEAb 25
1-99 R = -( )-NO2
TEAC 25
1.99 R=^Q-CH2-CH2-Br TEAC ^
a Standard reaction conditions: without solvent, 30 min, N2.b Solvent DMF, 4 h.c Solvent benzene, catalyst DMAP.
OH ^O 0 OH/ ^ 80 -16O0C /
f o i l ι Γϊ Γ* ~* /^nIIL/elL + n U >' L/eiL
OSi(CH3)3 Cl 3 3 O — C — R1 1
/° °(Et3N)" 'Cl
O OI l I l
,°-C-R HC,/«,, S'*-"
Celluloseester(DS)
1.57
2.30
2.53
0.56
0.43
0.39
XOSi(CH3)3
25°C>2°mi" OH
Figure 4.4.31. Conversion of TMS-cellulose to cellulose esters by different routes.

4.4.3 Esters of cellulose with organic acids 193
Ample research work has been invested in the phthaloylation of cellulose andsome of its derivatives like partially substituted ethylcellulose and especiallypartially substituted cellulose acetates (Fig. 4.4.32).
CeII-OH CeII-OHO
(H3C- C=0)>
(H3C-C = O)
(OH)
Figure 4.4.32. Scheme of phthaloylation of cellulose and of cellulose acetate.
Cellulose phthalate half-esters show a pH-dependent solubility in aqueousmedia that is useful for the manufacture of process auxiliaries. Phthalic anhy-dride is generally employed as the agent in the presence of a basic catalyst. ByLevesque et al. (1987) the esterification of cellulose and chitosan with the anhy-drides of phthalic acid, nitrophthalic acid and trimellitic acid in the presence ofTEA and 4-dimethylaminopyridine in DMSO is described, soluble productsbeing obtained at a molar ratio of anhydride per AGU of > 1.
Numerous publications are dealing with the synthesis and application of thecommercially relevant cellulose acetophthalates. These products are generallyobtained by reacting cellulose acetate in the DS range between 1.7 and 2.5 withphthalic acid anhydride in the presence of a basic catalyst like pyridine or TEA,in a dipolar aprotic or rather nonpolar medium (DMSO, DMF, dioxane, acetone,benzene). Also, tetrahydro- or hexahydrophthalic acid anhydride have been re-ported as esterifying agents, and besides the catalysts mentioned above also pi-coline, lutidine, 4-dimethylaminopyridine and 1,4-diazadicyclo-2,2,2-octanehave been employed. Furthermore, the phthaloylation of cellulose acetate in amelt of the anhydride has been performed. Malm et al. (1957) phthaloylated acellulose acetate of DS 1.8 in glacial acetic acid in the presence of sodium ace-tate. The authors emphasized the significant effect of the water content in thereaction system on reagent yield. Wagenknecht et al. (1987) investigated thephthaloylation of several cellulose acetates with phthalic acid anhydride in diox-ane at 100 0C, and in acetone at 56 0C, varying the DS of the starting material,the catalyst, the reagent input ratio and the time of reaction. With acetates of a

194 4.4 Esterification of Cellulose
DS of 2 and 2.5 (strictly homogeneous course of reaction) a complete substitu-tion of all the free hydroxy groups was accomplished, whereas at a lower DS ofacetyl groups and an at least partially heterogeneous course of reaction, the DSof phthaloyl groups remained below the amount of free hydroxy groups, result-ing in a total DS of less than 3. DMAP and l,4-diazadicyclo-2,2,2-octane provedto be superior to pyridine and TEA as catalysts. The decisive effect of the watercontent in the reaction system on the DS of phthaloyl groups obtained undergiven conditions is confirmed by the data in Table 4.4.26.
Table 4.4.26. Effect of water content on the phthaloylation of cellulose acetate (DS = 2)in acetone (Wagenknecht et al., 1987) (2.4 mol of phthalic acid anhydride and 1 mol ofTEA/AGU; 56 0C, 4 h).
% H2Ü in acetone
1.80.30.030.03
State of drying of CA
oven-dryoven-dryair-dryoven-dry
DS
0.610.710.800.98
Cellulose acetate phthalates are produced commercially as a specialty prod-uct, and are mainly employed in coatings for e.g. tablets, and as a process auxil-iary in the photographic industry.
4.4.3.8 Esters of cellulose with organic acids carrying sulfonic orphosphonic acid groups
Besides the cellulose esters with carboxylic acids considered so far, esters of thepolymer can be formed also with sulfonic or phosphonic acid groups or the ap-propriate acid chlorides. Phosphonic acid esters like the methylphosphonate ofcellulose have been discussed already in section 4.4.1 of this chapter, and alsothe esters with aliphatic sulfonic acids like methylsulfonic acids, leading to so-called 'mesy!cellulose', shall only be mentioned here, as they are without scien-tific or commercial relevance. The esters of cellulose with /7-toluenesulfonicacid, the so-called tosylcelluloses, on the other hand, deserve some more atten-tion as they form versatile intermediates in the organic chemistry of cellulosederivatization.
Cellulose tosylates can be prepared in a heterogeneous as well as a homoge-neous system of reaction. In both cases a preferential substitution at the C-6position is observed, without a pronounced regioselectivity being detected at lowand medium DS values (Takahashi et al., 1986).

4.4. 3 Esters of cellulose with organic acids 195
A heterogeneous procedure has been employed in former studies, reacting acellulose suspension in pyridine at room temperature up to 80 0C with a largeexcess of p-toluenesulfonyl chloride (Hess and Ljubitch, 1932; Honeyman,1947) according to
CeII-OH + H 3 C - ^ S O 2 C I CeIhO-SO-
Besides requiring a long reaction time of even days, and a high reagent-to-cellulose ratio of up to 40 : 1, this 'heterogeneous procedure' has the disadvan-tage of excessive side reactions, i.e. chlorination and eventually also formationof aminodesoxy groups. The chlorination to chlorodesoxycellulose additionallyis favored by a high reaction temperature. Furthermore, the products obtainedusually exhibit a poor solubility. These shortcomings can be avoided by per-forming the esterification in a thoroughly homogeneous system after previousdissolution of the polymer in a nonderivatizing solvent system, e.g. DMA/LiCl.A suitable homogeneous procedure in this solvent system has been recently de-scribed by Rahn et al. (1996). In this study 0.6-9 mol of tosyl chloride/mol ofAGU were employed, starting from cellulose with a DP between 280 and 5100,arriving (in the presence of TEA as the base) at tosylcelluloses of DS valuesbetween 0.4 and 2.3. Reaction time was 24 h at 8 0C. The higher reaction rate ofthe C-6 position as compared with those at C-2 and C-3 has been confirmed inthis study. The reaction products were soluble in DMSO irrespective of the DSobtained, while the solubility in other solvents like DMF, acetone, THF, or chlo-roform was found to depend on DS. The range of solubility of these homogene-ously prepared cellulose tosylates is significantly broader than that of conven-tionally synthesized ones, and the former show good film-forming propertiese.g. for the preparation of membranes, and can be processed by means of a ther-mally induced phase separation process. According to this, tosylcelluloses ofgood and uniform solubility can only be prepared by this homogeneous process,which takes place with only minimal side reactions (DS^\ = 0.01-0.02) (Rahn etal., 1996; Heinze, 1997).
In a broad variety of subsequent reaction steps, the tosylate function can beemployed as a protective group as well as a leaving group. Employing the tosy-late group as a protecting group, aliphatic and aromatic, and also unsaturatedmixed esters of cellulose could be obtained by esterification of free hydroxygroups. The appropriate anhydride instead of the acid chlorides is recommendedfor this purpose in order to avoid chlorination of the polymer chains. A completeacylation of all the free hydroxy groups of cellulose tosylates was observed withacetic anhydride or propionic anhydride without a decrease in the DS of tosylategroups, and even introduction of stearyl groups was accomplished of up to 84 %of the total amount of free hydroxy groups. By acylation of free hydroxy groups,

196 4.4 Esterification of Cellulose
soluble cellulose derivatives with a controlled hydrophile/hydrophobe ratio canbe synthesized. Besides this, amphiphilic esters (phthalates, trimellitates andsulfates) of cellulose tosylates with unusual solubility properties can be ob-tained. Sulfation of the tosylates results in water-soluble cellulose sulfate half-esters with reactive tosylate groups, which are well suited for the design andexperimental realization of new supramolecular cellulosic structures (Heinze andRahn, 1996a). A protective action of tosylate groups has been observed, too, inthe reaction with isocyanates.
The function of the tosylate group as a leaving group has long been known inthe synthesis of desoxycelluloses, e.g. by reaction with NaI to a 6-O-iododesoxycellulose according to
Cell-O-SO2-^)-CH3 + NaI - CeII-I + Na-O-SO2
Acetylacetone has been proposed as reaction medium (Rahn et al., 1996),which permits rather short times of reaction due to its high boiling point. Theavailability of cellulosic compounds with reduced functionality and controlledreactivity along this route has been emphasized recently (Heinze et al., 1996b;see also chapter 4.4.1.6).
Cellulose tosylates generally show a satisfactory chemical and thermal stabil-ity. A comprehensive thermoanalytic characterization of homogeneously pre-pared tosylcelluloses from 20 0C up to 500 0C has been published by Heinze etal. (1996a), indicating a lower temperature of decomposition as compared withcellulose itself due to detosylation and simultaneous backbone degradation. Anintegration of tosylate groups into a partially substituted cellulose acetate re-sulted in a lowering of the decomposition temperature and an increased charyield (Takahashi et al., 1986; Heinze et al., 1996b).
4.4.3.9 Phenyl carbamates of cellulose
Cellulose can be smoothly reacted with phenyl isocyanate according to
—CeIl-OH+ ~
in a dipolar aprotic medium in the presence of pyridine, yielding a trisubstitutedproduct ('cellulose tricarbanilate') with a sufficiently high excess of reagentafter about 10 h reaction time at 70-100 0C. The reaction system changes gradu-ally from a heterogeneous one to a homogeneous one, and this carbanilation isaccompanied by only a negligible chain degradation. It thus represents a suitableroute to convert cellulose 'polymer-analogues' to a soluble derivative for subse-

4.4.4 Concluding remarks on cellulose esterification 197
quent characterization in solution. After the reaction the excess isocyanate canbe decomposed by addition of dry methanol. After precipitation with a wa-ter/methanol mixture the reaction product is recovered as a white solid, which issoluble in various dipolar aprotic solvents like DMF, DMSO, THF or acetone(Burchard and Husemann, 1961; Schroeder and Haigh, 1979; Rantanen et al.,1986). A laboratory procedure published by Burchard and Husemann (Burchardand Husemann, 1961) for preparing cellulose tricarbanilate is given in the Ap-pendix. A strictly homogeneous route to cellulose tricarbanilate is available by dis-solving the sample in DMA/LiCl and reacting it with an adequate amount of phenylisocyanate in the presence of some pyridine as catalyst (Terbojevich et al., 1995).
Besides the well-established position of cellulose tricarbanilates in generalsolution characterization of cellulosics (Burchard and Schulz, 1989), and espe-cially in the determination of the molar mass distribution of cellulose samples byGPC (Saake et al., 1991), some ring-substituted phenyl carbamates of cellulosehave recently gained interest as sorbents for the Chromatographie separation ofenantiomers. A photocontrolled chiral recognition was reported by Yashima etal. (1995) with 4-phenyl-azo-phenyl carbamates of cellulose and amylose inconnection with a photoresponsive cisltrans isomerization, the trans isomershowing a higher selectivity. The optical resolving ability of two regioselec-tively carbanilated cellulose and amylose samples has been compared by Kaidaand Okamoto (1993), one of the samples carrying a 3,5-dimethylphenyl carba-mate residue in the C-2/C-3 position and a 3,5-dichlorophenyl carbamate residuein the C-6 position, while the other sample had attached the chloro-substitutedresidue in the 2,3-position and the methyl-substituted one in the C-6 position. Acomprehensive macromolecular characterization of samples of bis-3,5-dimethylphenyl carbamates of cellulose with molar masses between 2 χ ΙΟ4 and
4 χ 106 in dilute solution in l-methyl-2-pyrrolidone has recently been presentedby Tsuboi et al. (1995). The authors emphasized the remarkable optical anisot-ropy of this polymer and concluded from their light scattering, sedimentationand viscosity data a worm-like chain behavior in solution.
Concentrated solutions of regioselectively functionalized cellulose phenylcar-bamates ('cellulose carbanilates') can form lyotropic liquid crystallinemesophases. Their optical properties depend on the pattern of substitution withinthe AGU as well as on the specific substitution within the phenylring by CHs-,F- or Cl- (Derleth and Zugenmaier, 1997)
4.4.4 Concluding remarks on cellulose esterification
The esterification of cellulose plays a central role in chemical conversion of thispolymer. From the scientific point of view it represents a very broad spectrum ofchemical compounds and material properties, and it is the most important point

4.4.4 Concluding remarks on cellulose esterification 197
quent characterization in solution. After the reaction the excess isocyanate canbe decomposed by addition of dry methanol. After precipitation with a wa-ter/methanol mixture the reaction product is recovered as a white solid, which issoluble in various dipolar aprotic solvents like DMF, DMSO, THF or acetone(Burchard and Husemann, 1961; Schroeder and Haigh, 1979; Rantanen et al.,1986). A laboratory procedure published by Burchard and Husemann (Burchardand Husemann, 1961) for preparing cellulose tricarbanilate is given in the Ap-pendix. A strictly homogeneous route to cellulose tricarbanilate is available by dis-solving the sample in DMA/LiCl and reacting it with an adequate amount of phenylisocyanate in the presence of some pyridine as catalyst (Terbojevich et al., 1995).
Besides the well-established position of cellulose tricarbanilates in generalsolution characterization of cellulosics (Burchard and Schulz, 1989), and espe-cially in the determination of the molar mass distribution of cellulose samples byGPC (Saake et al., 1991), some ring-substituted phenyl carbamates of cellulosehave recently gained interest as sorbents for the Chromatographie separation ofenantiomers. A photocontrolled chiral recognition was reported by Yashima etal. (1995) with 4-phenyl-azo-phenyl carbamates of cellulose and amylose inconnection with a photoresponsive cisltrans isomerization, the trans isomershowing a higher selectivity. The optical resolving ability of two regioselec-tively carbanilated cellulose and amylose samples has been compared by Kaidaand Okamoto (1993), one of the samples carrying a 3,5-dimethylphenyl carba-mate residue in the C-2/C-3 position and a 3,5-dichlorophenyl carbamate residuein the C-6 position, while the other sample had attached the chloro-substitutedresidue in the 2,3-position and the methyl-substituted one in the C-6 position. Acomprehensive macromolecular characterization of samples of bis-3,5-dimethylphenyl carbamates of cellulose with molar masses between 2 χ ΙΟ4 and
4 χ 106 in dilute solution in l-methyl-2-pyrrolidone has recently been presentedby Tsuboi et al. (1995). The authors emphasized the remarkable optical anisot-ropy of this polymer and concluded from their light scattering, sedimentationand viscosity data a worm-like chain behavior in solution.
Concentrated solutions of regioselectively functionalized cellulose phenylcar-bamates ('cellulose carbanilates') can form lyotropic liquid crystallinemesophases. Their optical properties depend on the pattern of substitution withinthe AGU as well as on the specific substitution within the phenylring by CHs-,F- or Cl- (Derleth and Zugenmaier, 1997)
4.4.4 Concluding remarks on cellulose esterification
The esterification of cellulose plays a central role in chemical conversion of thispolymer. From the scientific point of view it represents a very broad spectrum ofchemical compounds and material properties, and it is the most important point
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

198 4.4 Esterification of Cellulose
of intersection between general organic chemistry and the special chemistry ofcellulose derivatization, promoting the introduction of modern reaction theoryinto cellulose chemistry. Furthermore, esters like the nitrate or the carbanilateare indispensable for the macromolecular characterization of cellulose in solu-tion and for assessing molar mass distribution. From the commercial point ofview, esterification of this polymer is by far the most widely employed route.Cellulose xanthogenate in about 90 % of the total production of cellulose de-rivatives.
The area of organic cellulose esters has been investigated rather thoroughlyover many decades, and discoveries of really new types of compounds havebeen rather scarce in recent years. The inorganic esters, on the other hand, havebeen studied more, emphasizing definitely the nitrate and to some extent thesulfate, and leaving ample space for further exploration.
At present, future developments in cellulose esterification are envisaged bythe authors as the synthesis of compounds with well-defined and pre-set patternsof substitution along the polymer chain, as well as within the single AGU, in-cluding double and triple substitution with different groups, in order to providemacromolecular entities for the design of well-defined supramolecular cellulose-based architectures. For achieving this goal in an adequate extension, homoge-neous and heterogeneous reactions, as well as combinations of both, will have tobe pursued, implying a deeper insight into the relations between chemical reac-tivity and physical structure of cellulose, besides the application of the full rep-ertoire of theoretical principles and experimental techniques of modern organicchemistry in the field of cellulose esterification.
References
Albright, L.F., in Encyclopedia of Chemical Technology, New York: John Wiley& Sons, 1981, 3rd. Edn., Vol. 15, pp. 841-853.
Allen, T.C., Cuculo, J.A., /. Polym. ScL, Macromol Rev. 1973, 7, 189-262.Allen, T.C., Cuculo, J.A.,Appl. Polym. Symp. 1976, 28, 811-829.Altena, F.W., Schroder, J.S., Van de Hüls, R., Smolders, C.A., /. Polym. ScL,
Part B, Polym. Phys. 1986, 24, 1725-1734.Anger, H., Berth, G., Wagenknecht, W., Linow, K.J., Acta Polym. 1987, 38,
201-202.Arthur, J.C., Bains, M.S., Patent US 3790562, 1974; Chem. Abstr. 1974, 8I9
39285.Arthur, J.C., Bains, M.S., Patent US 3891621, 1975; Chem. Abstr. 1975, 83,
114817.

References 199
Baiser, K., Hoppe, L., Eichler, T., Wandel, M., Astheimer, H.-J., in Ullmann'sEncyclopedia of Industrial Chemistry, Gerhartz, W., Yamamoto, Y.S.,Campbell, F.T., Pfefferkorn, R., Rounsaville, J.F. (Eds.), Weinheim: VCH,1986, Vol. A5, pp. 419-459.
Bartunek, R., Papier (Darmstadt) 1953, 7, 153-158.Battista, O.A., Armstrong, A.T., Radchenko, S.S., Polym. Prepr., Am. Chem.
Soc., Div. Polym. Chem. 1978, 79, 567-571.Bernhardt, R., Kunstseide 1926, 8, 173-75; 211-213; 257-260; 313-319.Bischoff, K.H., Ph.D. Thesis, University of Leipzig 1963.Bludova, O.S., Klenkova, N.I., Matveeva, N.A., Kutsenko, L.I., Volkova, L.A.,
Borisova, T.I., Zh. Prikl. Khim. 1984, 57, 603-610.Bott, R.W., Eaborn, C., Hashimoto, T., /. Organomet. Chem. 1965, 3, 442-447.Bracannot, H., Ann. Chim. Phys. 1819, 72, 185.Braun, D., Bahlig, K.H., Angew. Makromol. Chem. 1994, 220, 199-207.Buchanan, C.M., Hyatt, J.H., Lowman, D.W., Macromolecules 1987, 20, 2750-
2754.Buchanan, C.M., Hyatt, J.A., Lowman, D.W., 7. Am. Chem. Soc. 1989, 777,
7312-7319.Burchard, W., Husemann, E., Makromol. Chem. 1961, 44, 358-387.Burchard, W., Schulz, L., Papier (Darmstadt) 1989, 43, 665-674.Camacho Gomez, J.A., Ph.D. Thesis, University of Jena 1997.Carre, P., Manclere, P., Compt. Rend 1931, 792, 1567.Casay, G.A., George, A., Hadjichristidas, N., Lindner, J.S., Mays, J.W., Peiffer,
D.G., Wilson, W.W., 7. Polym. ScL, Part B, Polym. Phys. 1995,33, 1537-1544.Cicirov, A.A., Kuznecov, A.V., Kargin, Ju.M., Klockov, V.Vm., Marcenko,
G.N., Garifzjanov, G.G., Vysokomol. Soedin., Ser. A 1990, 32, 502-506.Clermont, L.P., Bender, F., J. Polym. ScL A-I 1972, 70, 1669-1677.Clermont, L.P., Manery, N., /. Appl. Polym. ScL 1974, 78, 2773-2384.Clode, D.M., Horton, D., Carbohydr. Res. 1971, 79, 329.Cross, C.F., Bevan, B.T., Beadle, C., Ber. Dtsch. Chem. Ges. 1893, 26, 1096
and 2520.Cuculo, J.A., Text. Res. J. 1971, 41, 321-326; 375-378.Dahlhoff, W.V., Imre, J., Koester, R., Macromolecules 1988, 27, 3342-3343.Dautzenberg, H., Philipp, B., Faserforsch. Textiltech. 1969, 20, 213-218.Dautzenberg, H., Philipp, B., Schumann, J., Faserforsch. Textiltech. 1972, 23,
192-198.Dautzenberg, H., Loth, F., Wagenknecht, W., Philipp, B., Papier (Darmstadt)
1985a, 39, 601-607.Dautzenberg, H., Loth, F., Borrmeister, B., Bertram, D., Lettau, H., Mende, M.,
Stamberg, J., Peska, J., Makromol. Chem. Suppl. 1985b, 9, 211.

200 4A Esterification of Cellulose
Dautzenberg, H., Jaeger, W., Kotz, J., Philipp, B., Seidel, Ch., Stscherbina, D.,in Poly electrolytes - Formation, Characterization and Application, München:Hanser Publishers, 1994.
Dautzenberg, H., Arnold, G., Tiersch, B., Likanov, B., Eckert, U., Prog.Colloid Polym. ScL 1996a, 707, 149-156.
Dautzenberg, H., Lukanoff, B., Eckert, U., Tiersch, B., Schuldt, U., Ber.Bunsenges. Phys. Chem. 1996b, 700, 1045-1053.
Dawydoff, W., Linow, K.-J., Philipp, B., Nahrung 1984, 28, 241-260.Deus, C., Fribolin, H. Siefert, E., Makromol Chem. 1991, 792, 75-83.Derleth, C., Zugenmaier, P., Macromol Chem. Phys. 1997,198, 3799-3814.Dunbrant, St., Samuelson, O., 7. Appl. Polym. Sei. 1965, 9, 2489-2499.Edgar, K.J., Arnold, K.M., Blount, W.W., Lawniczak, J.E., Lowman, D.W.,
Macromolecules 1995, 28, 4122-4128.Eicher, T., in Ullmann's Encyclopedia of Industrial Chemistry, Gerhartz, W.,
Yamamoto, Y.S., Campbell, F.T., Pfefferkorn, R., Rounsaville, J.F. (Eds.),Weinheim: VCH, 1986, Vol. A5.
Ekman, K., Chemiefasern 1984, 34/86, 399-400.Engelskirchen, K., in Methoden der Organischen Chemie, Stuttgart: Georg
Thieme, Houben-Weyl, 1987, E20, pp. 2126.Ermolenko, J.M., Vorob'eva, N.K., Kofman, A.E., Zonov, Yu.G., hv. Akad.
NaukB. SSR, Ser. Khim. Nauk 1971a, 6, 59-63.Ermolenko, J.N.,Skorynina, J.S., Vorob'eva, N.K., Dokl. Akad. Nauk B. SSR
1971b, 75, 244-246.Ermolenko, J.N., Luneva, N.R., Cellul. Chem. Technol. 1977, 77, 647-653.Farvardin, G.R., Howard, P., in Cellulose and its Derivatives, Kennedy, J.F.
(Ed.), Chichester: Ellis Horwood, 1985, pp. 227-236.Fischer, K., Hintze, H., Schmidt, L, Papier (Darmstadt) 1996, 50, 682-690.Fowler, W.F., Unruh, C.C., McGee, P.A., Kenyon, W.O., J. Am. Chem. Soc.1947, 69, 1636-1640.
Franchimont, A., Compt. Rend. 1879, 89,111.Frank, G.V., Caro, W., Ber. Dtsch. Chem. Ges. 1930, 63, 1532-1543.Frautschi, J.R., Munro, M.S., Lloyd, D.R., Eberhart, R.C., Trans.-Am. Soc.
Artif. Intern. Organs 1983, 29, 242-244.Fujimoto, T., Takahashi, S., Tsuji, M., Miyamoto, T., Inagaki, H., /. Polym.
Sei., Polym Lett. 1986, 24, 495.Fumasoni, S., Schippa, G., Ann. Chim. (Rome) 1963, 53, 894.Furubeppu, S., Kondo, T., Ishizu, A., Sen9i Gakkaishi 1991, 47, 592-597.Furuhata, K., Chang, H.-S., Aoki, N., Sakamoto, M., Carbohydr. Res. 1992,
230, 151-164.Geiger, E., Weiss, B.J., HeIv. Chim. Acta 1953, 36, 2009-2014.

References 201
Gertsev, V.V., Nesterenko, L.Yu., Romanov, Yu., Khim. Farm. Zh. 1990, 24,36-38.
Götze, K., in Chemiefasern nach dem Viskoseverfahren, Heidelberg: Springer,1967.
Golova, L.K., Kulicichin, V.S., Papkov, S.P., Vysokomol. Soedin., Ser. A 1986,28, 1795-1809.
Grotjahn, H., Z. Elektrochim. 1953, 57, 305-317.Guo, J.-X., Gray, D.G., /. Polym. Sei., Part A, Polym. Chem. 1994, 32, 889-
896.Hatanaka, K., Nakajima, L, Yoshida, T., Uryu, T., Yoshida, O., Yamamoto, N.,
Mimura, T., Kaneko, Y., J. Carbohydr. Chem. 1991, 70, 681-690.Heinze, Th., Habilitation Thesis, Friedrich Schiller University of Jena 1997.Heinze, Th., Rahn, K., Jaspers, M., Berghmans, H., J. Appl. Polym. Sei. 1996a,
60(11), 1891-1900.Heinze, Th., Rahn, K., Jaspers, M., Berghmans, H., Macromol. Chem.-Phys.
1996b, 797, 4207-4227.Heinze, Th., Rahn, K., Macromol. Rapid Commun. 1996a, 77, 675-681.Heinze, Th., Rahn, K., Papier (Darmstadt) 1996b, 50, 721-729.Hercules Powder, Patent US 2776965, 1957.Hercules Powder, Patent US 3063981,1962.Hess, K., Ljubitch, N., Liebigs Ann. Chem. 1932, 507, 62.Hess, K., Kiessig, H., Koblitz, W., Z. Elektrochim. 1951, 55, 697-708.Hess, K., Grotjahn, H., Z. Elektrochem. 1952, 56, 58-61.Heuser, E., Heath, M., Shockley, W.H., J. Am. Chem. Soc. 1950, 72, 670-674.Hirabayashi, Y., Macromol. Chem. 1984 785, 2371-2376.Holzapfel, G., Linow, K.-J., Philipp, B.: Wulf, K., Wagenknecht, W., Acta
Polym. 1986, 37, 553-557.Honeyman, J., /. Chem. Soc. (London) 1947, 168.Hovenkamp, S.G., /. Polym. Sei. 1963, C2, 341-355.Hovenkamp, S.G., PH.D. Thesis, Delft 1965.Husemann, E., Siefert, E., Makromol. Chem. 1969,128, 288-291.Husemann, E., Siefert, E., Bull. Inst. Politeh. lasi 1970,16, 47.lodannidis, O.K., Pogasov, Y.L., Aikhodzhaev, B.I., Rozyankhunov, R.,
Kryazhev, V.N., Gurkovskaya, L.V., Khim. Volokna 1966, 58.Ishii, T., Ishizu, A., Nakamo, J., Carbohydr. Res. 1977, 59, 115.Ishizu, A., Isogai, A., Tomikawa, M., Nakamo, J., Mokuzai Gakkaishi 1991, 37,
829-833.Isogai, A., Ishizu, A., Nakano, J., Sen'i Gakkaishi 1988, 44, 312-315.Iwata, T., Azuma, J.I., Okamura, K., Muramoto, M., Chun, B., Carbohydr. Res.
1992, 224, 277-283.Iwata, T., Okamura, K., Azuma, J., Tanaka, F., Cellulose 1996, 3(2), 91-106.

202 4.4 Esterification of Cellulose
Jain, R.K., LaI, K., Bhatnagar, H.L., J. Indian Chem. Soc. 1980, 57(6), 620-623.
Jain, R.K., LaI, K., Bhatnagar, H.L., Eur. Polym. J. 1986, 22, 993-1000.Jain, R.K., LaI, K., Bhatnagar, A.L., J. Appl. Polym. Sd. 1987a, 33, 247-282.Jain, R.K., LaI, K., Bhatnagar, H.L., Thermochim. Acta 1987b, 777, 187-199.Johnson, D.C., Patent US 344 7939, 1969.Johnson, D.C., Nicholson, M.D., Appl. Polym. Symp. 1976, 28, 931.Kaida, Y., Okamoto, Y., Bull. Chem. Soc. Jpn. 1993, 66, 2225-2232.Kamide, K., Okajima, K., Kowsaka, K., Matsui, M., Polym. J. 1987, 79, 1405-
1412.Kamide, K., Saito, M., Macromol. Symp. 1994, 83, 233-271.Kawaguchi, T., Nakahara, H., Fukuda, K., Thin Solid Films 1985, 733, 29-38.Kindness, G., Williamson, F.B., Long, W.F., Biochem. Biophys. Res. Commun.
1979, 88, 1062-1068.Kindness, G., Williamson, F.B., Long, W.F., Biochem. Soc. Transactions
1980, 8, 85-86.Kiselev, A.D., Danilov, S.N., Patent SU 159524,1962.Klare, H., Grobe, Α., Oesterr. Chem.-Ztg. 1964, 65, 218.Klare, H., in Geschichte der Chemiefaserforschung, Berlin: Akademieverlag,
1985.Klemm, D., Schnabelrauch, M., Stein, A., Philipp, B., Wagenknecht, W., Nehls,
L, Papier (Darmstadt) 1990, 44, 624-632.Klemm, D., Vogt, S., in Physico-Chemical Aspects and Industrial Applications,
Kennedy, J.F., Phillips, G.O., Williams, P.A., Piculell, L. (Eds.), Cambridge:Woodhead Publ. Ltd., 1995, pp. 169-176.
Klemm, D., Heinze, Th., Philipp, B., Wagenknecht, W., Acta Polym. 1997, 48,277-297.
Klenkova, N.I., Khlebosolova, E.N., CeIM. Chem. Technol. 1977, 77, 191-208.Knecht, E., Ber. Dtsch. Chem. Ges. 1904, 37, 549.König, L., Döring, R., Postel, F., Papier (Darmstadt) 1993, 47, 641.Koester, R., Amen, K.L., Bellut, H., Fenzl, W., Angew. Chem., Int. Ed. Engl.
1971,10, 748-750.Kulakova, O.M., Klenkova, N.I., Tsimara, N.D., Khim. Tekhnol. Proizvod.
Tsellyl. 1971, 83-88.Krylova, R.G., RUSS. Chem. Rev. 1987, 56, 97.Kwatra, H.S., Caruthers, J.M., Tao, B.Y., Ind. Eng. Chem. Res. 1992, 37(72),
2647-2651.Lang, H., Laskowski, J., Lukanoff, B., IV Int. Symposium on Μάη-Made Fibers,
Kalinin, 1986, Preprints, Vol. 2, pp. 212-223.Lay, L., Panza, L., Riva, S., Khitri, M., Tirendi, S., Carbohydr. Res. 1996, 297,
197-204.

References 203
Levesque, G., Chiron, G., Roux, O., Makromol Chem. 1987,188, 1659-1664.Liebert, T., Schnabelrauch, M., Klemm, D., Erler, U., Cellulose 1994, 7, 249.Loth, F., Philipp, B., Macromol Symp. 1989, 30, 273-287.Ludwig, J., Philipp, B., Acta Polym. 1990, 4I9 230-233.Lukanoff, T., Linow, K.-J., Philipp, B., Faserforsch. Textiltech. 1969, 20, 383-
387.Lukanoff, B., Dautzenberg, H., Papier (Darmstadt) 1994, 48, 287-298.Malm, C.J., Fordyce, C.R., Ind. Eng. Chem. 1940, 32, 405-408.Malm, C.J., Tanghe, L.J., Laird, B.C., /. Am. Chem. Soc. 1948, 70, 2740-2747.Malm, C.J., Hiatt, G.D., in Cellulose, Ott, E., Spurlin, H.M., Graffin, M.W.
(Eds.), New York: Interscience, 1954, pp. 763-824.Malm, C.J., Mench, J.W., Hiatt, G.D., Ind. Eng. Chem. 1957, 49, 84-88.Mannschreck, A., Wernicke, R., Labor. Praxis 1990,14, 730-738.Mansson, P., Westfeld, L., Cellul Chem. Technol 1980,14, 13-17.Marchessault, R.H., Howsmon, J.A., Text. Res. J. 1957, 27, 30.Matsumara, H., Saka, S., Mokuzai Gakkaishi 1992, 38, 270-276; 862-868.Matthes, A., Faserforsch. Textiltech. 1952, 3, 127-141.McCormick, C.L., Chen, T.S., in Macromolecular Solutions, Solvent-Property
Relationships in Polymers, Seymor, R.B., Stahl, G.A. (Eds.), New York:Pergamon Press, 1982, pp. 101-107.
Miles, F.D., in Cellulose Nitrate, The Physical Chemistry of Nitrocellulose, itsFormation and Use, London: Oliver and Boyd, 1955.
Miyamoto, T., Sato, Y., Shibata, T., Inagaki, H., /. Polym. Sd., Polym. Chem.Ed. 1984, 22, 2362-2370.
Miyamoto, T., Sato, Y., Shibata, T., Tanahashi, M., Inagaki, H., /. Polym. ScL,Polym. Chem. Ed. 1985, 23, 1373-1381.
Nakamura, S., Amano, M., J. Polym. ScL Part A: Polym. Chem. 1997, 35, 3359-3363.
Nakamura, S., Sanada, N., Sen-I Gokkoishi 1997, 53, 467-470.Nehls, L, Loth, F., Acta Polym. 1991, 42, 233-235.Nehls, L, Habil. Thesis, University of Potsdam 1994.Nehls, I., Wagenknecht, W., Philipp, B., Stscherbina, D., Prog. Polym. ScL
1994,19, 29-78.Nehls, I. Wagenknecht, W., Philipp, B., Cellul Chem. Technol. 1995, 29, 243-
251.Nishino, T., Takano, K., Nakamae, K., Saitaka, K., Hakura, S., Azuma, J.,
Nuessle, C., Ford, P.M., Hall, W.P., Lippert, A.L., Text. Res. J. 1956, 26, 32.Okajima, K., Kamide, K., Matsui, T., Patent EP 53473, 1982; Chem. Abstr.
1982, 97, 133577.Okamura, K., /. Polym. ScI9 Part B, Polym. Phys. 1995, 33, 611-618.Pascu, E., Schwenker, R.F., Text. Res. J. 1957, 27, 173.

204 4.4 Esterification of Cellulose
Petropavlovski, G.A., Faserforsch. Textiltech. 1973, 24, 49-57.Petrov, K.A., Sopikova, JJ., Nifant'ev, E.E., Vysokomol. Soedin. 1965, 7, 667.Philipp, B., Faserforsch. Textiltech. 1955, 6, 509-520.Philipp, B., Ph.D. Thesis, Technical University of Dresden 1956.Philipp, B., Faserforsch. Textiltech. 1957a, 8, 91-98.Philipp, B., Faserforsch. Textiltech. 1957b, 8, 21-27.Philipp, B., Faserforsch. Textiltech. 1957c, 8, 45-53.Philipp, B., Faserforsch. Textiltech. 1958, 9, 520-526.Philipp, B., Fichte, Ch., Faserforsch. Textiltech. 1960, 77, 118-124; 172-179.Philipp, B., Baudisch, J., Papier (Darmstadt) 1965, 79, 749-757.Philipp, B., Dautzenberg, H., Papier (Darmstadt) 1967, 27, 118-124.Philipp, B., Wagenknecht, W., Cellul. Chem. Technol. 1983, 77, 443-459.Philipp, B., Fanter, C., Wagenknecht, W., Hartmann, M., Klemm, D.,
Geschwend, G., Schumann, P., Cellul. Chem. Technol. 1983, 77, 341-353.Philipp, B., Wagenknecht, W., Holzapfel, G., Cellul. Chem. Technol. 1985, 79,
331-339.Philipp, B., Nehls, L, Wagenknecht, W., Schnabelrauch, M., Carbohydr. Res.
1987,764, 107-116.Philipp, B., Dautzenberg, H., Linow, K.-J., Kotz, J., Dawydoff, W., Prog.
Polym. Sei. 1989,14, 91-172.Philipp, B., Wagenknecht, W., Nehls, L, Ludwig, J., Schnabelrauch, M., Kim Ho
Rim, Klemm, D., Cellul. Chem. Technol. 1990, 24, 667-678.Philipp, B., Klemm, D., in Abschlußbericht zum Förderprojekt BEO 22-
0310375A (BMFT) 1994.Philipp, B., Klemm, D., Wagenknecht, W., Wagenknecht, M., Nehls, L, Stein,
A., Heinze, Th. Heinze, U., Heibig, K., Camacho, J., Papier (Darmstadt)1995, 49, 3-7; 58-64.
Pikler, A., Jurasek, A., Jasova, V., Piklerova, A., Cellul. Chem. Technol. 1980,14(5), 697-701.
Pohjola, L., Aarnikoivu, P.L., Pap. PUU 1976, 58, 331.Pohjola, L., Riala, R., Tammela, V., Pap. PUU 1976, 58, 198.Polyakov, A.I., Rogowin, Z.A., Vysokomol. Soedin. 1963, 5, 11.Predvoditelev, D.A., Nifant'ev, E.E., Rogowin, Z.A., Vysokomol. Soedin. 1966,
S, 76.Rahn, K., Diamatoglou, M., Klemm, D., Berghmans, H., Heinze, Th., Angew.
Makromol. Chem. 1996, 238, 143-163.Rahn, K., Ph.D. Thesis, University of Jena 1997.Rantanen, T., Farm, P., Sundquist, J., papper o. Trä. 1986, 68, 634.Rebek, M., Jurkowisch, B., Papier (Darmstadt) 1977, 30, 372-374.Reid, J.D., Mazzeno, L.W., Ind. Eng. Chem. 1949, 41, 2828-2831.

References 205
Richau, K., Schwarz, H.H., Apostel., R., Paul, D., /. Membr. ScL 1996, 113,31-41.
Rowell, R.M., Wood Sei. 1982, 75, 172-182.Saake, B., Patt, R., Puls, J., Philipp, B., Papier (Darmstadt) 1991, 45, 727-735.Samaranayake, G., Glasser, W.G., Carbohydr. Polym. 1993, 22, 1-7; 79-86.Samuelson, O., Cellulosa och Papper 1948, 295-325.Sato, T., Tsujii, Y., Fukuda, T., Miyamoto, T., Macromolecules 1992, 25,
3890-3895.Scherer, P.S., Feild, J.M., Rayon Melliand Text. Mon. 1941, 22, 607.Schnabelrauch, M., Geschwend, G., Klemm, D., /. Appl. Polym. Sei. 1990, 39,
621-628.Schnabelrauch, M., Vogt, S., Klemm, D., Nehls, L, Philipp, B., Angew.
Macromol. Chem. 1992,198, 155-164.Schönbein, C.F., Ber. Natuforsch. Ges. Basel 1847, 7, 27.Schroeder, L.R., Haigh, F.C., Tappi 1979, 62, 103.Schützenberger, P., Compt. Rend. 1865, 61, 485.Schützenberger, P., Ber. Dtsch. Chem. Ges. 1869, 2, 163.Schweiger, R.G., Chem. Ind. (London) 1966, 22, 900.Schweiger, R.G., Carbohydr. Res. 1972 27, 219-228.Schweiger, R.G., Tappi J. 1974 57, 86-90.Schweiger, R.G., Carbohydr. Res. 1979, 70, 185-198.Schwenke, K.D., Augustat, B., Wagenknecht, W., Nahrung 1988, 32, 393-407.Segal, L., Eggerton, P.V., Text. Res. J. 1961, 37, 460-471; 991-992.Seymor, R.B., Johnson, E.L., /. Polym. Sei., Polym. Chem. Ed. 1978, 76, 1-11.Shaposhnikova, S.T., Pogosov, Y.L., Aikhodzhaev, B.I., Vysokomol. Soedin.
1965, 7, 1314.Shimizu, Y., Hayashi, J., Sen'i Gakkaishi 1988, 44, 451-456.Shimizu,Y., Nakayama, A., Hayashi, J., in Cellulosics, Chemical, Biochemical
Material Aspects, Kennedy, J.F., Phillips, G.O., Williams, D.A. (Eds.),Chichester: Ellis Horwood, 1993a, pp. 369-374.
Shimizu, Y., Nakayama, A., Hayashi, J., Sen'i Gakkaishi 1993b, 49(7), 352-356.
Shiraishi, N., Yoshioka, M., Sen'i Gakkaishi 1986, 42, T346-T355.Short, R.D., Munro, H.S., Polym. Commun. 1989, 30, 366-368.Short, R.D., Munro, H.S., Matthews, R., Pritchard, T., Polym. Commun. 1989,
30, 217-220.Stein, A., Klemm, D., Makromol. Chem., Rapid Commun. 1988, 9, 569-573.Stscherbina, D., Philipp, B., Acta Polym. 1991, 42, 345-351.Svistunova, R.P., Aikhodzhaev, B.I., Pogosov, Y.L., Pakhimova, I.V., Patent SU
173739,1964. '

206 4Λ Esterification of Cellulose
Takahashi, S.-L, Fujimoto, T., Barua, B.M., Miyamoto, T., Inagaki, H., /.Polym. ScL, Part A, Polym. Chem. 1986, 24(11), 2981-2993.
Terbojevich, M., Cosani, A., Camilot, M., Focher, B., /. Appl. Polym. ScL 1995,55, 1663-1671.
Teshirogi, T., Yamamoto, H., Sakamoto, M., Tonami, H., Sen'i Gakkaishi 1979,35, T525.
Titkombe, L.A., Bremner, J.B., Burgar, M.I., Ridd, MJ., French, J., Maddern,K.N., Appita 1989, 42, 282-286.
Toney, G.P., Kiefer, J.E., Patent US 2759787,1956.Torgashov, V.J., Gert, E.V., Bildyukevich, A.V., Kapuckij, F.N., Chem. Drev.
1988, 7, 14-19.Touey, G.P., Patent US 2759924,1956.Treiber, E., Fex, O.F., Rehnström, J., Piova, M., Svensk Papperstidn 1955, 58,287-295.Treiber, E., Fex, O.F., Sven. Papperstidn. 1956, 59, 51-57.Treiber, E., Gierer, J., Rehnström, J., Schurz, J., Holzforschung 1956, 70, 36-
42.Tseng, H., Furuhata, K., Sakamoto, M., Carbohydr. Res. 1995, 270, 149-161.Tsuboi, A., Yamazaki, M., Norisuye, T., Teramoto, A., Polym. J. 1995, 27,
1219-1229.Tyuganova, M.A., Butylkina, N.G., Khim. Drev. 1992, 4/5, 25-30.VanderHart, D.L., Hyatt, J.A., Atalla, R.H., Tirumalai, V.C., Macromolecules
1996, 29, 730-739.Vienken, J., Diamatoglou, M., Hahn, C., Kamusewitz, H., Paul, D., Artif.
Organs 1995, 79, 398-406.Vigo, T.L., Daighly, B.J., Welch, C.M., /. Polym. ScL, Part B, Polym. Phys.
1972, 70, 397^06.Vigo, T.L., Welch, C.M. Textilveredelung 1973, 8, 93-97.Vladimirova, T.V., Galbraich, L.S., Peker, K.S., Rogowin, Z.A., Vysokomol.
Soedin. 1965, 7, 786.Vogt, S., Heinze, Th., Röttig, K., Klemm, D., Carbohydr. Res. 1995, 266, 315-
320.Vogt, S., Klemm, D., Heinze, Th., Polym. Bull. 1996, 36, 549-555.Wagenknecht, W., Philipp, B., Schleicher, H., Beierlein, L, Faserforsch.
Textiltech. 1976,27, 111-117.Wagenknecht, W., Philipp, B., Schleicher, H., Acta Polym. 1979, 30(2), 108-
112.Wagenknecht, W., Philipp, B., Keck, M., Acta Polym. 1985, 36, 697-698.Wagenknecht, W., Paul, D., Philipp, B., Ludwig, T., Acta Polym. 1987, 38, 551-
554.

References 207
Wagenknecht, W., Nehls, L, Kotz, J., Ludwig, J., Cellul Chem. Technol 1991a,25, 343-354.
Wagenknecht, W., Philipp, B., Nehls, L, Schnabelrauch, M., Klemm, D.,Hartmann, M., Acta Polym. 1991b, 42, 213-216; 554-560.
Wagenknecht, W., Nehls, L, Philipp, B., Carbohydr. Res. 1992a, 237, 211-222.Wagenknecht, W., Nehls, L, Stein, A., Klemm, D., Philipp, B., Acta Polym.
1992b,43, 266-268.Wagenknecht, W., Nehls, L, Philipp, B., Carbohydr. Res. 1993, 240, 245-252.Wagenknecht, W., Papier (Darmstadt) 1996, 50, 712-720.Wagenknecht, W., Schwarz, H.H., Patent DE 4435180, 1996; Chem. Abstr.
1996, 725, 61285.Watjen, U., Kriews, M., Dannecker, W., Nucl. Instrum. Methods Phys. Res.,
Part B, 1993,75, 257-261.Whistler, R.L., Unruh, P.O., Ruffini, G., Arch Biochem. Biophys. 1968, 726,
647.Whistler, R.L., Towle, P.A., Arch. Biochem. Biophys. 1969, 735, 396.Wu, T.K., Macromolecules 1980,13, 74-79.Yashima, E., Naguchi, J., Okamoto, Y., Macromolecules 1995, 28, 8368-8374.Yasuda, M., Yoneda, H., Patent JP 07070202, 1995; Chem. Abstr. 1995, 723,
35587.Yuldashev, A., Muratova, U.M., Askarov, M.A., Vysokomol. Soedin. 1965, 7,
1923.Yuldashev, A., Muratova, U.M., Dokl. Akad. Nauk Uzb. SSR 1966, 23, 42.Zhang, Z.B., McCormick, C.L., /. Appl. Polym. Sei. 1997, 66, 293-305.Zeronian, S.H., Adams, S.A., Alger, K., Lipsha, A.E., /. Appl Polym. Sei. 1980,
25,519-528.Zugenmaier, P., in Cellulosic Polymers, Blends and Composites, Gilbert, R.D.
(Ed.), Munich: Hanser Publ., 1994, pp. 71-94.
4.5 Etherification of Cellulose
4.5.1 General remarks on etherificationCellulose etherification is a very important branch of commercial cellulose de-rivatization that started considerably later than the conversion of the polymer toesters. Preparation of a cellulose ether was reported for the first time in 1905 bySuida, who reacted the polymer with dimethyl sulfate to give a methylcellulose.The first patent claiming the preparation of soluble nonionic alkyl ethers of cel-lulose was issued in 1912 to Lilienfeld, and by 1920 the synthesis of someother important classes of cellulose ethers like carboxymethylcellulose, benzyl-

References 207
Wagenknecht, W., Nehls, L, Kotz, J., Ludwig, J., Cellul Chem. Technol 1991a,25, 343-354.
Wagenknecht, W., Philipp, B., Nehls, L, Schnabelrauch, M., Klemm, D.,Hartmann, M., Acta Polym. 1991b, 42, 213-216; 554-560.
Wagenknecht, W., Nehls, L, Philipp, B., Carbohydr. Res. 1992a, 237, 211-222.Wagenknecht, W., Nehls, L, Stein, A., Klemm, D., Philipp, B., Acta Polym.
1992b,43, 266-268.Wagenknecht, W., Nehls, L, Philipp, B., Carbohydr. Res. 1993, 240, 245-252.Wagenknecht, W., Papier (Darmstadt) 1996, 50, 712-720.Wagenknecht, W., Schwarz, H.H., Patent DE 4435180, 1996; Chem. Abstr.
1996, 725, 61285.Watjen, U., Kriews, M., Dannecker, W., Nucl. Instrum. Methods Phys. Res.,
Part B, 1993,75, 257-261.Whistler, R.L., Unruh, P.O., Ruffini, G., Arch Biochem. Biophys. 1968, 726,
647.Whistler, R.L., Towle, P.A., Arch. Biochem. Biophys. 1969, 735, 396.Wu, T.K., Macromolecules 1980,13, 74-79.Yashima, E., Naguchi, J., Okamoto, Y., Macromolecules 1995, 28, 8368-8374.Yasuda, M., Yoneda, H., Patent JP 07070202, 1995; Chem. Abstr. 1995, 723,
35587.Yuldashev, A., Muratova, U.M., Askarov, M.A., Vysokomol. Soedin. 1965, 7,
1923.Yuldashev, A., Muratova, U.M., Dokl. Akad. Nauk Uzb. SSR 1966, 23, 42.Zhang, Z.B., McCormick, C.L., /. Appl. Polym. Sei. 1997, 66, 293-305.Zeronian, S.H., Adams, S.A., Alger, K., Lipsha, A.E., /. Appl Polym. Sei. 1980,
25,519-528.Zugenmaier, P., in Cellulosic Polymers, Blends and Composites, Gilbert, R.D.
(Ed.), Munich: Hanser Publ., 1994, pp. 71-94.
4.5 Etherification of Cellulose
4.5.1 General remarks on etherificationCellulose etherification is a very important branch of commercial cellulose de-rivatization that started considerably later than the conversion of the polymer toesters. Preparation of a cellulose ether was reported for the first time in 1905 bySuida, who reacted the polymer with dimethyl sulfate to give a methylcellulose.The first patent claiming the preparation of soluble nonionic alkyl ethers of cel-lulose was issued in 1912 to Lilienfeld, and by 1920 the synthesis of someother important classes of cellulose ethers like carboxymethylcellulose, benzyl-
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

208 4.5 Etherification of Cellulose
cellulose or hydroxyethylcellulose had been described. Industrial productionstarted in the two decades between 1920 and 1940, beginning with carboxy-methylcellulose (CMC) in the early 1920s in Germany. The worldwide indus-trial manufacture of cellulose ethers has presently arrived at a level of about halfa million tons annually, with CMC dominating by far, followed by methy!cellu-lose and hydroxyethylcellulose (Table 4.5.1).
Table 4.5.1. Production capacity (t/a) of economically importantcellulose ethers (Brandt, 1986).
Ether Production capacity
Carboxymethylcellulose 300,000 t/aMethylcellulose 70,000 t/aHydroxyethylcellulose 54,000 t/a
Among the various routes to synthesis of cellulose ethers, which will be de-scribed in detail in the following sections, only two are of commercial relevance,i.e.(i) the reaction of hydroxy groups with an alkyl chloride in the presence ofstrong alkali-metal hydroxides, according to the Williamson ether synthesis,consuming 1 mol of alkali/mol of alkyl chloride reacted;(ii) the ring-opening reaction of an alkylene oxide with the hydroxy groups,which is catalyzed by alkali-metal hydroxides without significant alkali con-sumption, and which often results in longer side chains due to further add-on ofalkylene oxide onto the newly formed hydroxy groups.
Industrial etherification of cellulose is exclusively performed in a heterogene-ous system, starting from alkali cellulose. Due to side reactions with the waterpresent in the aqueous system in large excess (calculated on a molar basis) andcompeting with the cellulosic hydroxy groups for the etherifying agent, reagentyield remains considerably below the 100 % margin, and a further processing toremove by-products from the crude cellulose ether is usually required for high-purity products. The etherification of cellulose in the dissolved state can be re-alized too and is of scientific interest today in connection with the control of thefunctionalization patterns of the polymers and with the synthesis of new types ofcellulose ethers.
The abundant variability of cellulose ether structures described up to now andthe remarkable broad spectrum of cellulose ethers commercially available can betraced back to two characteristics besides the well-known possibility of varyingthe DS and the distribution of the substituents: firstly, the chemical constitutionof the alkyl halide and to some extent also of the alkylene oxide can be changed,making anionic and cationic cellulose ethers available besides the neutral ones.

4.5.1 General remarks on etherification 209
Moreover, not only carbon-based cellulose ethers can be prepared, but also vari-ous silyl ethers have been synthesized by reaction of the polymer especially withtrialkylchlorosilanes (see section 4.5.5). Secondly (and this point is still morerelevant in connection with commercial cellulose ethers), the two routes of ethersynthesis outlined above can be combined by adding simultaneously or con-secutively an alkyl chloride and an alkylene oxide to the aqueous alkaline reac-tion system, arriving at so-called mixed ethers of cellulose with two or eventhree different ether functions. Furthermore, numerous routes of a subsequentfunctionalization of cellulose ethers considerably increases the number of struc-tures and products available.
Research and development activities in recent decades have been centered onthe full exploitation of this 'mixed ether principle' for tailoring properties to thebroad variety of end-use requirements. Besides this, the minimization of chaindegradation during the process in order to obtain a high solution viscosity of theproduct and the enhancement of reagent yield with the option to decrease theinput of chemicals for ecological reasons, played a major role.
Cellulose ethers on a commercial scale are generally used as end-products,but serve as interesting intermediates too. In laboratory-scale research they areused either for further chemical modification of the ether group primarily intro-duced, or for subsequent reaction of remaining hydroxy groups present in a par-tially substituted cellulose ether.
The most important properties of cellulose ethers are their solubility com-bined with chemical stability and non-toxicity. Water solubility and/or organo-solubility can be controlled within wide limits via the constitution and the com-bination of ether groups at the cellulose chain, as well as via the DS, and to someextent via the pattern of substitution. Accordingly, cellulose ethers are generallyapplied, in the dissolved or highly swollen state, to many areas of industry anddomestic life, with the spectrum of applications ranging from auxiliaries inlarge-scale emulsion or suspension polymerization, through to additives forpaints and wall paper adhesives, to viscosity enhancers in cosmetics and food-stuffs.
For the sake of clearness and conciseness, the following chapter is structuredaccording to the constitution of the ether group: the first and most voluminoussection deals with aliphatic cellulose ethers, comprising alkyl ethers, substitutedalkyl ethers, hydroxyalkyl ethers and mixed aliphatic ethers of cellulose. Thefollowing section on aryl and aralkyl ethers of cellulose is centered on tri-phenylmethylcellulose and related substances as interesting intermediates intoday's cellulose chemistry. As a special feature of this book, the third sectiondescribes in a rather detailed manner the preparation, properties and subsequentreaction routes of silyl ethers of cellulose, emphasizing adequately the authors'work in this area. Each of the sections begins with a comprehensive discussionof the chemical aspects, followed by a brief consideration of the role of cellulose

210 4.5 Etherification of Cellulose
supramolecular structure by etherification, turning then to the properties and themain areas of application of the various classes of cellulose ethers. A brief de-scription of the industrial process is included for some ethers.
4.5.2 Aliphatic ethers of celluloseAliphatic ethers of cellulose have been extensively investigated since the begin-ning of this century, and comprise also large-scale industrial products of thisclass of cellulose derivatives like methylcellulose, carboxymethylcellulose(CMC), and hydroxyethylcellulose (HEC). Aliphatic cellulose ethers can beclassified in various ways, i.e.(i) from an applicational point of view into nonionic (methylcellulose, HEC) andionic (CMC) ones;(ii) on the basis of the main routes of synthesis, into those obtained by the Wil-liamson synthesis with consuming one mol of base per mol of ether groups in-troduced and those obtained by ring-opening reactions of epoxides as reagentswith a catalytic amount of alkali;(iii) from the viewpoint of a systematic description according to the type offunctional groups attached to the backbone.
In this context the last-mentioned route will be followed, structuring the sec-tion according to alkyl ethers, carboxyalkyl ethers, hydroxyalkyl ethers andethers with special functional groups.
4.5.2.1 Alkyl ethers of cellulose
Chemistry of cellulose alkylation
By far the most important representative of this class of cellulose ethers carryingan unsubstituted alkyl group is methylcellulose, which is available over thewhole DS range 0-3 along various routes of synthesis.
The commercial products with a DS between 1.5 and 2.0 are obtained by aWilliamson reaction of alkali cellulose with gaseous or liquid CH3Cl. The lyeemployed for cellulose alkalization contains at least 40 % NaOH (in contrastwith about 18 % in the viscose process). The methylation of cellulose, which isusually classified as an SN2 reaction, is the result of the nucleophilic attack ofthe cellulosic alkoxido group on the acceptor C atom of the methyl chloride.
CeII-OH + Na+ OH' —- CeII-OI Na+ + H2O
CeII-OI Na+ + C+H3-CI - CeII-Q-CH3 + Na+ Cl'
The etherification of cellulose in the presence of alkali hydroxide is, however,accompanied by the hydrolysis of methyl chloride, with the water present in the

210 4.5 Etherification of Cellulose
supramolecular structure by etherification, turning then to the properties and themain areas of application of the various classes of cellulose ethers. A brief de-scription of the industrial process is included for some ethers.
4.5.2 Aliphatic ethers of celluloseAliphatic ethers of cellulose have been extensively investigated since the begin-ning of this century, and comprise also large-scale industrial products of thisclass of cellulose derivatives like methylcellulose, carboxymethylcellulose(CMC), and hydroxyethylcellulose (HEC). Aliphatic cellulose ethers can beclassified in various ways, i.e.(i) from an applicational point of view into nonionic (methylcellulose, HEC) andionic (CMC) ones;(ii) on the basis of the main routes of synthesis, into those obtained by the Wil-liamson synthesis with consuming one mol of base per mol of ether groups in-troduced and those obtained by ring-opening reactions of epoxides as reagentswith a catalytic amount of alkali;(iii) from the viewpoint of a systematic description according to the type offunctional groups attached to the backbone.
In this context the last-mentioned route will be followed, structuring the sec-tion according to alkyl ethers, carboxyalkyl ethers, hydroxyalkyl ethers andethers with special functional groups.
4.5.2.1 Alkyl ethers of cellulose
Chemistry of cellulose alkylation
By far the most important representative of this class of cellulose ethers carryingan unsubstituted alkyl group is methylcellulose, which is available over thewhole DS range 0-3 along various routes of synthesis.
The commercial products with a DS between 1.5 and 2.0 are obtained by aWilliamson reaction of alkali cellulose with gaseous or liquid CH3Cl. The lyeemployed for cellulose alkalization contains at least 40 % NaOH (in contrastwith about 18 % in the viscose process). The methylation of cellulose, which isusually classified as an SN2 reaction, is the result of the nucleophilic attack ofthe cellulosic alkoxido group on the acceptor C atom of the methyl chloride.
CeII-OH + Na+ OH' —- CeII-OI Na+ + H2O
CeII-OI Na+ + C+H3-CI - CeII-Q-CH3 + Na+ Cl'
The etherification of cellulose in the presence of alkali hydroxide is, however,accompanied by the hydrolysis of methyl chloride, with the water present in the
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

4.5.2 Aliphatic ethers of cellulose 211
system at large molar excess leading to methanol, which can react further withmethyl chloride to form dimethyl ether. This by-product formation accounts for20-30 % of the CH3Cl consumption, resulting in a reagent yield for etherifica-tion of maximally 80 %. For etherification, as well as for the by-product forma-tion, 1 mol of NaOH is consumed per mol of CH3Cl converted, and besides theorganic by-products, a large amount of NaCl is inevitably produced in this process.
Methylation of cellulose by the Williamson reaction is generally performed atelevated temperature with cellulose in the solid state (see section 4RoIe of cellu-lose supramolecular structure in alkylation'). As demonstrated by the results oflaboratory-scale methylation (Philipp et al., 1979) in Fig. 4.5.1, the course ofreaction is characterized by a fast initial state, followed by a slow leveling off ofthe DS just below 2 even at a large excess of methyl chloride.
2.0
1.5
1.0
0.5
100 200 300 400Reaction time [min]
100 200 300Reaction time [min]
400
Figure 4.5.1. Course of total NaOH consumption (left) and degree of substitution ofmethyl groups (right) with time of methylation of alkali cellulose with an excess ofCH3Cl at different temperatures: · 70 0C, O 80 0C, · 90 0C (Philipp et al., 1979).
While an increase in reaction temperature from 70 to 80 0C considerably en-hances the initial reaction rate, a further increase to 90 0C has a minor effectonly. This is consistent with the assumption that the overall rate is determined bythe chemical reaction only at lower temperature, while above 80 0C the reagenttransport rate across the phase boundary and within the alkali cellulose moiety isthe dominating factor. At a reaction temperature above 80 0C, as employed inthe technical process, methyl chloride is known to be a more efficient etherifica-tion agent than the corresponding bromide or iodide due to a higher molar vol-ume. This agrees well with a diffusion-controlled reaction.
Regarding the substituent distribution within the AGU of partially methylatedproducts, obtained by the Williamson reaction, a slight preference for the C-2position compared with C-6 is generally reported, while the C-3 position con-tains ether groups to a definitely lower extent (Dönges, 1990). According to

212 4.5 Etherification of Cellulose
Rosell (1988) a partially methylated sample exhibited a methylation of 70.0 % atO-2, 61.5 % at O-6 and only 35.4 % at O-3 position. The substitution patternalong the chain obviously depends on the procedure of synthesis employed andmay deviate from a statistical distribution (Arisz et al., 1996).
Table 4.5.2. Systems employed in laboratory methylation of cellulose.
Methylation system References
DMSO/NaOH/CH3I Ciucanu and Kerek, 1984;Needs and Selvendran, 1993
DMSO/LiH/CH3I Hakamori, 1964;D'AmbraetaL, 1988
DMF, THF/NaH/CH3I Klemm and Stein, 1995CH2Cl2/2,6-di-i-buty lpyridine/ Mischnick, 1991;(CH3)3O+[BF4] Kulshin et al., 1991(CH3)3PO4/2,6-di-i-butylpyridine/ Prehm, 1980;CF3SO3CH3 (methyltriflate) Mischnick, 1991
The course of cellulose methylation can be widely modified by varying themethylation agent, the base which is required in any case, the reaction medium,and the state of dispersion of the system. Instead of methyl chloride, also methyliodide, dimethyl sulfate, diazomethane (prepared in situ from nitrosomethylurea)or special agents like trimethyloxonium tetrafluoroborate, or methyltriflate (seeTable 4.5.2) can be employed. Methyl iodide proved to be very suitable for themethylation of cellulosic hydroxy groups in a homogeneous medium of thepolymer (e.g. after dissolution in DMA/LiCl or in tetraalkylammonium bro-mide), the reaction medium and the alkyl halide. As a base, NaH or LiH, metal-lic Na dispersed in an ammonia cellulose NH3 system, or di-i-butylpyridine havebeen proposed (see Table 4.5.2). Methylation of cellulose has also been per-formed in aqueous solutions of tetraalkylammonium hydroxides, with the poly-mer in the highly swollen or dissolved state, arriving here at water-soluble prod-ucts already at a DS of about 0.6, due to a more equal ether-group distributionalong the polymer chain (Bock, 1937). Besides water, also dipolar aprotic liq-uids like DMSO, DMF or THF served as reaction media in cellulose methyla-tion. Some of these systems, suitable for laboratory-scale etherification, arelisted in Table 4.5.2.
According to the authors' experience, systems of CH3I and NaH in THF or ofCH3I and finally powdered NaOH in DMSO proved to be very suitable for thepermethylation of free hydroxy groups of partially substituted tritylcelluloses(Camacho Gomez et al., 1996) or trialkylsilylcelluloses (Erler et al., 1992a),

4.5.2 Aliphatic ethers of cellulose 213
without loss of the substituent already present. CD3I can be employed as well, ifit is advantageous for the subsequent instrumental analysis of the product.
Methylation can also be performed with the polymer dissolved in an aproticsystem like DMSO/paraformaldehyde (Nickelson and Johnson, 1977) orDMA/LiCl. In the latter system, copolymers with a nonstatistical distribution ofthe different repeating units were prepared using finely powdered NaOH as thebase and CH3I as the agent (Liebert and Heinze, 1997). The etherification couldtake place only at the points of contact between the NaOH particles and thepolymer.
The alkyl halide/NaOH/DMSO system has been successfully employed inrecent years for preparing regioselectively or completely substituted methylcel-lulose with a homogeneous solution of the polymer: cellulose acetates could beconverted to highly substituted methylcelluloses by a simultaneous deacetylationand etherification (Kondo and Gray, 1990). A regioselectively substituted 2,3-dimethylcellulose was prepared by alkylation of 6-O-tritylcellulose and subse-quent detritylation with HCl (Kondo and Gray, 1991). A small amount of waterin the system proved to be essential for obtaining full methylation of the O-2 andO-3 positions. The detritylated product could be further alkylated to various 6-O-derivatives of 2,3-0-methylcellulose (Kondo, 1993).
Turning now to ethylcellulose and higher alkyl ethers it must be stated firstthat the Williamson ether synthesis under heterogeneous starting conditionsbecomes more and more inefficient with increasing molar volume of the alkylhalide. In the appropriate range of reaction temperature the process is diffusion-controlled and by-product formation prevails with increasing alkyl chain length.Ethylation can still be performed by analogy to methylation by reacting alkalicellulose with ethyl chloride, arriving at a substitution pattern with about equalpartial DS at C-2 and C-6, and again a low degree of etherification at C-3(Dönges, 1990). An activation energy of 10.3 kcal/mol at a reaction temperaturebelow 30 0C, and of 4.4 kcal/mol at higher temperature were reported (Chak-rabarti et al., 1986), indicating again the dominant role of diffusion in the lattercase. An efficient propylation required either a previous partial methylation for'widening' the polymer structure, or employing a tetraalkylammonium hydrox-ide of high swelling power in aqueous solution as the base and reaction medium(Schenck, 1936; Timell, 1950).
The synthesis of alkyl ethers of cellulose with longer side chains usually re-quires nonaqueous systems, more severe basic reaction conditions, and ratherlong reaction times often at elevated temperature. The preparation of long-chainalkyl ethers of cellulose by reaction of cellulose acetate with the appropriatealkyl bromide in the presence of NaOH in DMSO as the reaction medium isdescribed by Basque et al. (1996). Table 4.5.3 presents a survey of some long-chain alkyl ethers and their preparation, starting from a suspension of dry cellu-

214 4.5 Etherification of Cellulose
lose in isopropanol or in DMSO and reacting it with the appropriate alkyl bro-mide in the presence of NaOH or NaH (Blasutto, 1995).
Table 4.5.3. Reaction conditions for long-chain cellulose etherpreparation in DMSO, taken from Blasutto et al. (1995).
Reagent1 -Bromooctadecane1 -Bromohexadecane1 -Bromotetradecane1 -Bromooctadecane1 -Bromododecane1 -Bromotetradecane1 -Bromohexadecane1-Bromooctane1-Bromooctane1 -Bromooctadecane1 -Bromotetradecane
Base Reaction time (h)NaOHNaOHNaOHNaHNaHNaHNaHNaHNaHNaHNaH
9248
133721525a
193
252626
a The suspension of NaH in DMSO was heated to 40 0C to accele-rate the formation of the anion (CH3-SO-CH2)". The cellulose wasadded after cooling at room temperature.
Due to the chemical stability of the ether group, and a DS-dependent solubilityin various media, partially substituted methyl- and ethylcelluloses are well suitedto serving as the starting material for a subsequent functionalization of residualhydroxy groups under homogeneous conditions of reaction. Examples are thepreparation of various organic ester ethers of cellulose from an ethylcellulose ofDS = 2 with various acylanhydrides and acyl chlorides in benzene in the pres-ence of 4-dimethylaminopyridine as the catalyst (Philipp et al., 1983), and thepreparation of mixed methyl/allyl ethers of cellulose from a methylcellulose ofDS = 1.6 by reacting it with allyl chloride or methallyl chloride in the presenceof NaOH in DMSO (Kondo et al., 1987). The same authors also succeeded inthe synthesis of a triallylcellulose by deacetylation/etherification of a celluloseacetate of DS = 1.8 with allyl chloride and NaOH in DMSO.
The laboratory procedure for the methylation of cellulose is presented in theAppendix.
Role of cellulose supramolecular structure in alkylation
Methylation of alkali cellulose with CH3Cl represents a typical 'heterogeneousderivatization reaction', with the accessibility of the cellulose chains to the rea-gent determining the course of conversion in this diffusion-controlled process. A

4.5.2 Aliphatic ethers of cellulose 215
still larger influence of accessibility on the course of reaction was observed withmore voluminous alkylating agents such as dimethyl sulfate or ethyl chloride.The high steeping lye concentration required in alkali-cellulose formation for aneffective etherification, not only supplies the necessary alkalinity for the chemi-cal reaction, but also enhances the availability of the cellulose molecules to thereagent by a further decrease in overall supramolecular order (Fink et al., 1995).
2.0
1.5
co 1.0Q
0.5
20 40 60 80NaOH consumption
100
Figure 4.5.2. Relation between total NaOH consumption and degree of substitution ofmethyl groups on methylation of alkali cellulose (31.8 % cellulose, 30 % NaOH) at dif-ferent temperatures: · 70 0C, O 80 0C, · 90 0C (Philipp et al., 1979).
The strongly heterogeneous character of alkali-cellulose methylation had beenemphasized already by Hess and co-workers (Hess et al., 1933), who observedthe WAXS reflexes of trimethylcellulose already at a low overall DS, and as-sumed a high DS in near-surface areas of the fiber and a negligibly low DS intheir center at an early stage of reaction. In the presence of a sufficient amountof methyl chloride and NaOH, the still existing hydrogen bond system betweenthe cellulose chains is further disturbed on methylation, and the hydrophiliccharacter of the still free hydroxy groups is irreversibly liberated by partialetherification (Dönges, 1990). The overall reaction, however, can come to aquasi-standstill before complete consumption of the NaOH due to diffusion hin-drance, as demonstrated in Fig. 4.5.2, by the limiting DS of about 1.7 reachedafter an alkali consumption of about 80 % (Philipp et al., 1979).
On the other hand, rather small differences in the course of alkali-cellulosemethylation were found between a spruce sulfite pulp and bleached cotton !in-ters. Also, preactivation of the cellulose was found to be of minor influenceonly. This is understandable in so far as the process of alkali-cellulose forma-tion, especially at high steeping lye concentrations, results in a leveling ofstructural differences between the different cellulose materials. This holds trueespecially also for the industrial process of methylation, as there, alkalization is

216 4.5 Etherification of Cellulose
usually preceded by a dry grinding, which by itself already decreases differencesin e.g. X-ray crystallinity (Fink and Walenta, 1994).
Completely substituted trimethylcellulose exhibits a well-defined WAXSfiber diagram, with a period of 10.3 A in the fiber-axis direction, and it can bebrought to crystallization from solution or from its melt (Hess et al., 1928). Thestate of supramolecular order and the side chain conformation of liquid crystal-line systems of ethy!cellulose in CHCl3 have been studied by NMR (Yim et al.,1992).
Survey of the technical process of cellulose methylation (Dönges, 1990;Brandt, 1986)
After dry grinding or chopping, normal-grade wood dissolving pulp is trans-formed into alkali cellulose by treatment with 35-70 % aqueous NaOH (3-4 mol/mol of AGU), and after an eventual preripening for viscosity reduction(see preripening process) the alkali cellulose is methylated with an excess ofCH3Cl employed either in the gaseous or in the liquid state.
In the 'gaseous process' the alkali cellulose is warmed with part of the CH3Clto about 50 0C in a corrosion-resistant pressure vessel by an effective stirringdevice. A reaction temperature of between 60 and 100 0C is maintained for somehours. Reagent, evaporating together with the by-products, is removed, con-densed and recycled into the reactor, together with fresh reagent, in order tokeep a constant concentration of methyl chloride in the reaction system.
The 'liquid methyl chloride process' can be performed as a continuous proc-ess requiring a reaction time of less than 1 h. In this process the alkali celluloseis slurried in excess reagent, and this slurry then is pumped through a partiallyheated reaction tube. By-products and excess reagent are evaporated. The 'liquidprocess' can also be operated in the presence of an inert organic liquid, e.g. di-methyl ether, dimethylglycol or toluene, in order to reduce the reaction pressurein the case of higher boiling liquids and/or to reduce by-product formation.
Both processes can also be employed for the production of mixed ethers, withthe second reagent added before or after methylation, and the course of the reac-tion temperature being program-controlled.
In many of the technical procedures the alkali is completely consumed, oth-erwise a neutralization step is necessary before washing the product with hotwater of 80-90 0C, i.e. well above the gelation temperature, for removal of so-dium chloride and other by-products. In this way, the NaCl content is decreasedto about 1 % for normal grade and about 0.1 % for high-quality methylcellulose.Eventually, the product crosslinks to a low degree with glyoxal for a retardeddissolution in water by slow hydrolysis of the crosslinks. Drying of the productis performed in conventional equipment.

4.5.2 Aliphatic ethers of cellulose 217
Ethylcellulose is manufactured analogously to methylcellulose, with ethylchloride as the reagent, but at a higher temperature, usually above 110 0C. Areaction time of 8-16 h is required, and about half of the reagent input is con-sumed for side reactions, i.e. of ethanol and diethyl ether. Reagent yield is re-ported to increase with the steeping lye concentration (55-76 % NaOH), and astepwise addition of the lye in the process was found to be advantageous. Fur-ther product processing is performed as described for methylcellulose.
Properties of alkylcelluloses, especially methylcellulose
Alkylcelluloses are white-to-yellowish nontoxic solids, exhibiting a gradedsolubility in various media, in dependence on substituent and DS. Hydropho-bicity increases with the length of the alkyl chains and with the DS. Commercialmethylcelluloses in the DS range 1.5-2.0 are to be classified as amphiphilic,while commercial ethy!celluloses with a DS above 2 are definitely hydrophobic.Methylcellulose is chemically very stable and the viscosity of an aqueous solu-tion is independent of pH in the range from pH 2 to 12. Ethylcellulose can formperoxides in the presence of oxygen and light, and eventually needs stabilizationby an antioxidant.
Methylcellulose in the DS range up to 2 proved to be biodegradable in thepresence of water: According to Seneker and Glass (1996) sequences of at leastsix AGU with an unsubstituted C-2 position are required for an enzymatic at-tack, demonstrating once more the relevance of regioselectivity of substitution tointeraction with biological systems. Ethylcellulose and the higher alkyl ethersare hardly degraded by cellulolytic enzymes even at much lower DS.
For a fully substituted methylcellulose, a melting range between 227 and 240 0Cunder decomposition has been reported (Hess et al., 1935). Commercial ethylcel-luloses with a DS above 2 are thermoplastic and can be extruded to give films at asoftening temperature of 130 0C and a flow temperature of 140-160 0C.
The most relevant applicational properties of methyl- and ethylcelluloses arethe solubility and solution properties. As can be seen from Table 4.5.4, methyl-celluloses of increasing DS, i.e. of increasing hydrophobicity, exhibit solubilityin liquids of decreasing polarity, and the same holds true for ethylcellulose, tak-ing into consideration the more hydrophobic nature of the substituent. Thesestatements, however, are valid only for products manufactured by the conven-tional etherification of alkali cellulose in a heterogeneous system, and thereforeexhibiting a nonuniformity of substituent distribution along the polymer chains.A more even distribution, as realized by methylation of cellulose dissolved in atetraalkylammonium hydroxide, results in complete water solubility already at aDS of about 0.6. The effect of regioselectivity on physical product properties, ase.g. solubility and crystallinity, was recently studied by Kondo (1997). He com-pared 6-O-methy!cellulose with non-regioselectively methylated samples and

218 4.5 Etherification of Cellulose
correlated differences in product properties to differences in their hydrogen bondsystems. The excellent solubility and poor crystallinity of 6-0-methylcellulosewere traced back to a lack of interchain hydrogen bonds, while intramolecularhydrogen bonds were assumed to persist even after dissolution of the sample.The optical transparency of aqueous methylcellulose solutions can be enhancedby using a mixed ether with a small amount of hydroxyalkyl groups.
Table 4.5.4. Solubility of methyl- and ethylcelluloses in dependence
Cellulose ether Solvent DS range of solubility
MethylcelluloseMethylcelluloseMethylcelluloseMethylcelluloseMethylcelluloseEthylcelluloseEthylcellulose
Aqueous NaOHWaterEthanolAcetoneTolueneWaterOrganic liquids
0.25-1.01.4-2.0>2.1>2.4>2.70.7-1.7> 1.5; preferably >2
A phenomenon of high scientific and practical relevance is the gelation ofaqueous solutions of methylcellulose with a DS in the range between 1.7 and 2.3at elevated temperatures. Commercial products of DS 1.8 form gels at 54-56 0C.This gelation is reversible along a hysteresis loop of gelation and redissolution,and it plays an important role in methylcellulose purification and processing (seescheme in Fig. 4.5.3).
100
σQ_<Λ
σ
"- Cteer point λ
Dissolving
- kRedissolution
point
-Cloud point
Flocculation
Coagulationpoint
IA
Temperature
Figure 4.5.3. Flocculation of hydrophobic substituents bearing cellulose ethers, indi-cated by the temperature-dependent transparency of an aqueous solution.

4.5.2 Aliphatic ethers of cellulose 219
The gel formation is generally considered to be due to a depletion of the mac-romolecules from their hydration sheets, facilitating hydrophobic crosslinkingvia heavily substituted sequences. This agrees with the observation of a de-creasing gelling temperature with increasing DS of methyl groups and also withthe increasing electrolyte content of the system disturbing the hydration sheetsof the macromolecules. A high solution viscosity, on the other hand, obviouslyimpedes gel formation and thus increases the gelling temperature (Dönges,1990). The degree of nonuniformity of substituent distribution along the chainsdefinitely influences the gelling temperature too at a practically constant DS of1.75 (Arisz et al., 1996), concluding therefore that a balanced hydropho-bic/hydrophilic (via hydrogen bonds) interaction is essential for the gel formation.
An analogous reversible gel formation can also take place with ethylcellulosein aqueous solution, but starts already at a considerably lower temperature ofabout 30 0C.
The [T]J-M relationship of aqueous solutions has been reported (Dönges,1990) to be, formethylcellulose: η = 2.92 X l O - 2 X DpO-905
η = 2.8 χ 10-3χ Mn
0-63
Applications of methyl- and ethylcellulose
The DS ranges of methyl- and ethylcelluloses relevant for commercial productsare listed in Table 4.5.5, and for the readers' convenience the relationship be-tween DS and the methoxyl content of methy!celluloses is depicted in Fig. 4.5.4,as commercial methylcelluloses are often characterized by their methoxyl content.
Table 4.5.5. Main types of commercial cellulose alkyl ethers.
Type
MethylcelluloseMethylcelluloseEthylcelluloseEthylcellulose
DS range
0.25-1.01.5-2.00.7-1.72.2-2.6
Soluble in
AlkaliWaterWaterOrganic solvents
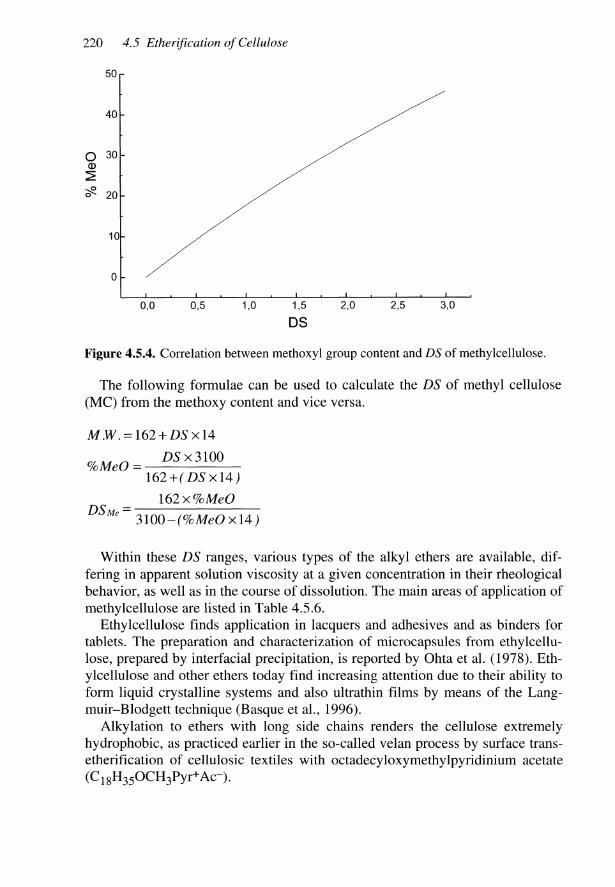
220 4.5 Ethenfication of Cellulose
50Γ
40
O 30
CD
^ 20
10
0,0 0,5 1,0 1,5 2,0 2,5 3,0
DS
Figure 4.5.4. Correlation between methoxyl group content and DS of methylcellulose.
The following formulae can be used to calculate the DS of methyl cellulose(MC) from the methoxy content and vice versa.
%M,0- DSX31°°162 + (DSxHj
l62x%MeO
3lOO-(%MeOxl4)
Within these DS ranges, various types of the alkyl ethers are available, dif-fering in apparent solution viscosity at a given concentration in their rheologicalbehavior, as well as in the course of dissolution. The main areas of application ofmethylcellulose are listed in Table 4.5.6.
Ethylcellulose finds application in lacquers and adhesives and as binders fortablets. The preparation and characterization of microcapsules from ethy!cellu-lose, prepared by interfacial precipitation, is reported by Ohta et al. (1978). Eth-ylcellulose and other ethers today find increasing attention due to their ability toform liquid crystalline systems and also ultrathin films by means of the Lang-muir-Blodgett technique (Basque et al., 1996).
Alkylation to ethers with long side chains renders the cellulose extremelyhydrophobic, as practiced earlier in the so-called velan process by surface trans-etherification of cellulosic textiles with octadecyloxymethylpyridinium acetate(C18H35OCH3Pyr+Ac-).

4.5.2 Aliphatic ethers of cellulose 221
Table 4.5.6. Application of methy!cellulose (Dönges, 1990)Total production worldwide ca. 70 kt (including mixed ethers).
Application area Proportion ( %)
Building industry 47Dispersion paints 21Wall paper paints 14Cosmetics 5Polymerization 5Detergents 4Other 4
4.5.2.2 Carboxymethylcellulose and related anionic cellulose ethers
Carboxymethylcellulose represents, with an annual production of about300,000 t worldwide, the commercially most important cellulose ether, and hasfound ample scientific attention, especially due to its character as a polyelectro-lyte.
The basic chemistry of carboxymethylation is rather simple and has long beenwell known; recent effort has been directed mainly toward process optimizationand rationalization. Scientific progress has been achieved predominantly in thechemical modification of CMC, its analytical characterization as a partially sub-stituted cellulose derivative, and in the understanding of its nature as an anionicpolyelectrolyte, especially in aqueous solution.
Chemistry of carboxymethylation
In principle, carboxymethylation of cellulose proceeds along the same route asmethylation, i.e. by Williamson etherification of alkali cellulose with sodiumchloroacetate in an aqueous or aqueous-alcoholic system according to:CeIl-OH-NaOH + ClCH2COONa -> CeIl-O-CH2COONa + NaCl + H2OJust as in cellulose methylation, a considerable amount of the etherifying agent,i.e. up to 30 %, is consumed in side reactions with the aqueous NaOH, formingpredominantly sodium glycolate by hydrolysis of the chloroacetate (Feddersenand Thorp, 1993).
But in contrast with methylation, the etherifying agent (monochloroacetic acidor sodium monochloroacetate) is water-soluble and nonvolatile, thus avoidingproblems of reagent transport across a phase boundary and permitting the reactionto proceed at atmospheric pressure. Furthermore, only a DS of between 0.4 and 0.8is necessary in the technical process of CMC manufacture in order to meet therequirements of application, which usually takes place in an aqueous solution.

222 4.5 Etherification of Cellulose
The classical process of CMC preparation starts from an alkali cellulose ob-tained by steeping with aqueous NaOH of 20-30 % (by weight) concentrationand subsequent pressing. Etherification is then performed by reaction of thisalkali cellulose with sodium monochloroacetate or monochloroacetic acid:
1 mol or 2 mol respectively of NaOH are consumed in this process per mol ofetherifying agent converted to carboxymethyl groups at the polymer and to so-dium glycolate. At 50-70 0C the reaction takes place over several hours as anexothermic process.
An activation energy of 21 kcal/mol and 22.5 kcal/mol has been reported inKishida and Okimasu (1976) for the main and the side reaction respectively,with the conclusion that a rather low temperature of reaction increases the rea-gent yield for carboxymethylation. Also, a strong pressing of the alkali celluloseprior to etherification is favorable, as it reduces the content of the free aqueousNaOH responsible for the side reaction. Results of a statistical optimization ofalkali cellulose composition for preparing CMC in the DS range between 0.5 and0.7 have been published by Olaru et al. (1978).
The original two-step 'dry' process of alkalization and subsequent etherifica-tion has now been widely substituted by a one-step slurry process, taking placewith the cellulose suspended in a mixture of NaOH, sodium monochloroacetate,water and an excess of isopropanol or ί-butanol. Also, inert liquids like benzeneor acetone have been mentioned as components, while methanol or ethanol haveproved to be unfavorable. By decreasing the DS of the CMC at a given mono-chloroacetate input, the presence of the alcohol promotes an even distribution ofthe monochloroacetate in the reaction mass, resulting in an additional enrich-ment of NaOH in the cellulose phase, favoring a further decrease in su-pramolecular order and a more uniform etherification. On the other hand, ofcourse, the alcoholic hydroxy groups compete with the cellulosic ones foretherification and form low molecular ethers of the structure ROCH2COO" Na+.Monochloroacetate consumption by this type of side reaction is however muchsmaller with isopropanol than with e.g. methanol, due to the lower hydroxygroup reactivity of the former. A less extensive cellulose degradation by alkalineoxidative chain degradation can be considered as a further advantage of theslurry process, especially if high-viscosity types of CMC are required. The labo-ratory procedure for synthesizing CMC by the slurry process is given in the Ap-pendix.
A comprehensive kinetic study of the alkaline hydrolysis of monochloroace-tate and dichloroacetate in the presence of various organic liquids, e.g. alcohols,has been published (Dautzenberg and Philipp, 1979a), employing NaOH con-centrations between 0.1 and 5 mol/1. The results were discussed on the basis ofHammetts H-function, emphasizing the role of free water in the system.
While the preparation of CMC with a DS of up to 1, or slightly higher, by theroutes described above causes no problems, highly substituted samples with a DS

4.5.2 Aliphatic ethers of cellulose 223
above 2 are more difficult to obtain than in the case of methylation. The synthesisof a fully substituted CMC with a DS of 3 is still a matter of discussion. Only byrepeated alkalization-etherification steps DS values above 1.5 can be obtained,with a quite severe chain degradation being inevitable. This was recently empha-sized by Kulicke (Kulicke et al, 1996; Ghannam and Nabil Esmail, 1997) whoprepared CMC samples with a DS of between 0.7 and nearly 3.0 by a slurry pro-cedure with isopropanol, but mentioned that possibly somewhat too high total DSvalues were indicated by the 13C NMR technique employed. In Iwata et al. (1985)the preparation of CMC in the DS range between 2.2 and 2.6 in DMSO in thepresence of tetramethylurea was reported. By Perrier and Benerito (1973) a non-aqueous procedure of carboxymethylation of mercerized cotton cloth to low DS(up to 0.3), by formation of Na-cellulosate in a methanol solution of MeONa andits subsequent conversion to a carboxymethylated product, with sodium mono-chloroacetate in DMSO at room temperature, was described.
The degree of substitution of CMC is usually assessed via the counterionbound to the carboxyl group (precipitation with uranyl acetate), determination asNa2SO4 after wet combustion in the case of salt-free products, acidimetric titra-tion after ashing to Na2O, NaOH, Na2CO3 (Hoeye, 1977), or by a direct titrationof the carboxyl group (alkalimetric titration of H+CMC or titration of Na-CMCwith a cationic poly electrolyte; Hong et al., 1978), or by summation of the par-tial DS values usually obtained by 13C NMR spectroscopy (Baar et al., 1994).Results obtained by different techniques have already been compared in chapter3. The pattern of substitution within the AGU, as determined by 13C NMRspectroscopy, or after hydrolytic chain degradation and 1H NMR measurements(Reuben and Conner, 1983; Granski and Hellmann, 1987), usually shows aslight preference for the C-2 position compared with that of C-6 and a compara-ble low substitution of the C-3 position for samples obtained by the dry processor by the slurry process. A reactivity ratio of O-2/O-3/O-6 of 3.0 : 1.0 : 2.1 wasreported in (Baar et al., 1994) for samples of DS > 1 obtained by a slurry processwith isopropanol (Table 4.5.7).
Table 4.5.7. Substituent distribution (partial DSvalues at positions 2, 3 and 6) in CMC samples ofvarying DS (Baar et al., 1994).
DS2
0.370.430.640.870.971.00
DS3
0.120.220.300.630.750.92
DS6
0.220.340.470.790.941.05
DS
0.710.991.412.292.662.97

224 4.5 Etherification of Cellulose
But also a preferential 6-substitution can be realized in a slurry process, asshown by Cheng (Cheng et al., 1996) (for samples of DS < 1 obtained by a two-phase process with benzene and ethanol as components of the system), who con-cluded a reactivity ratio of O-2/O-3/O-6 = 1.45 : 1.0 : 2.5. Considerable efforthas been spent in recent years on assessing the distribution of carboxymethylgroups along the polymer chains by combining various techniques of degrada-tion, separation and fragment characterization (see chapter 3).
A regioselective carboxymethylation of the C-2 and the C-3 position up to atotal DS of 1.9 could be realized by reacting a 6-0-tritylcellulose or 6-0-(4-methoxy)tritylcellulose in DMSO with monochloroacetate and NaOH at 70 0Cfor 15-29 h and subsequent detritylation with HCl. The NaOH was present in afinely powdered solid state. 6-0-(4-Methoxy)tritylcellulose proved to be moresuitable than tritylcellulose itself with respect to a faster detritylation undermilder conditions. The Na-CMC samples finally obtained were completely solu-ble in water and contained predominantly 2,3-disubstituted AGU (Heinze et al.,1994a).
CMC samples of medium DS, prepared in the DMA/LiCl system in the pres-ence of solid NaOH, exhibited a higher nonuniformity of substitution along thepolymer chains than samples obtained by the conventional slurry technique, asrevealed by a definitely higher fraction of nonsubstituted and trisubstituted AGUafter hydrolysis and Chromatographie separation (Heinze et al., 1994b). Thisprinciple of obtaining block-like CMC structures, by limiting the reaction topoints of contact between the dissolved polymer and the solid NaOH particles,has recently been generalized to a promising concept of synthesis of block-likecellulose ethers, especially cellulose ethers like carboxymethyl-, methyl- andethylcellulose. Also various organosoluble cellulose derivatives of limited sta-bility like acetates, trifluoroacetates or formates and trimethylsilylcellulose wereconverted to CMC with a block-like structure and a DS up to 2.2 by reactionwith monochloroacetic acid and solid NaOH in a suitable dipolar aprotic me-dium like DMSO (Liebert et al., 1996). These samples exhibited a peculiar sub-stituent distribution also at the level of the single AGU, as the C-6 positiondominated, followed by C-3 and then by C-2. The macromolecular productstructure could be varied via the stability of the primary ester substituent, i.e. theease of its saponification in the alkaline system, and the water content present inthe dipolar aprotic liquid. Some of the results obtained are summarized in Table4.5.8 and Fig. 4.5.5.
It may be expected that this new synthesis concept, using a phase-separationprocess in order to gain a reactive microstructure, will become a general methodfor new cellulosics with unconventional patterns of functionalization (Heinze,1997).

4.5.2 Aliphatic ethers of cellulose 225
Table 4.5.8. Conditions and results of carboxymethylation of cellulose dissolved inDMATLiCl, as well as cellulose trifluoroacetate (CTFA), cellulose formate (CF), cellu-lose acetate (CA) and trimethylsilylcellulose (TMSC) via induced phase separation withNaOH particles (size < 0.25 mm) (Liebert and Heinze, 1997).
Starting cellulosicmaterial
Molar ratioa Reactiontime (h)b
CarboxymethylcelluloseNo DS,HPLC Solubility
in waterCellulose in 1
DMA/LiCl 1
CTFA 1
CF 1111
CA 11
TMSC 111
2 : 44 : 85: 105: 1010:2010:2010 : 20d
10 : 20e
10 : 20f
10:2010:2015:3020:4010:2010:2010:2010:2010:20
484848
24
164222442240.512
7a7b7c8a8b8c8d8e8f9a9b9c9d
1Oa1ObUalibUc
1.131.88 +2.07 +0.111.86 +1.54 +1.360.620.971.46 +1.91 +1.362.21 +0.360.452.04 +1.91 +1.97 +
a Molar ratio: Modified AGU : ClCH2COO(H)Na : NaOH.b Reaction temperature 70 0C.c AS1HPLO degree of substitution determined by means of HPLC (see Heinze et al., 1994b).d First addition of ClCH2COONa and subsequent phase separation with solid NaOH
particles.e NaOH particle size: 0.63-1.00 mm.f NaOH particle size: 0.25-0.63 mm.

226 4.5 Etherification of Cellulose
1HPLC
Figure 4.5.5. Mole fractions of repeating units (D glucose, O mono- O-carboxy methyl-,Δ di-O-carboxymethyl-, and V 2,3,6-tri-O-carboxymethylated glucose) in hydrolyzedCMC samples (No. see Table 4.5.8) plotted as a function of the degree of substitutiondetermined by means of HPLC (^HPLC) (Liebert and Heinze, 1997).
Cellulose ethers with a chemical structure related to CMC, carboxyethyl-cellulose and dicarboxymethylcellulose shall be mentioned here briefly. Car-boxyethylcellulose can in principle be obtained along the same route as CMC,but is usually prepared by an alkaline hydrolysis of the nitrile group of a cyano-ethylcellulose (see subsequent section). A preferential substitution at the C-6position was found with samples prepared in this way, up to a DS of about 0.5,by 13C NMR spectroscopy (Nehls et al., 1994). The acid strength of the car-boxyl group is assumed to be somewhat lower than in CMC due to its furtherdistance from the activating ether linkage. Dicarboxymethylcellulose has beenprepared from cellulose and bromomalonic acid according to
CeII-OH + BrCH(COOH)2 Cell-0-CH(COO-)2 + Br'
with DS values up to 1.5 (Kotz et al., 1991). The procedure of etherification isquite analogous to that for CMC: cellulose is reacted with aqueous alkali andchloro- or preferably bromomalonic acid or its sodium salt in the presence ofisopropanol. A substitution at the C-3 and C-6 position was concluded from13C NMR spectroscopic studies (Kotz et al., 1991). In comparison with CMC, asignificantly lower overall pA^a value, indicating a higher acid strength was ob-served with dicarboxymethylcellulose.

4.5.2 Aliphatic ethers of cellulose 227
Carboxymethylcellulose has been modified along various routes by subse-quent steps of derivatization either at the carboxyl group itself or at the free hy-droxy groups of partially carboxymethylated samples. The water or alkali solu-bility of CMC can be of advantage in aqueous reaction systems, but the insolu-bility in aprotic organic liquids up to a high DS often requires the use of a CMCsuspension after adequate physical activation (Vogt et al., 1995; Vogt et al.,1996). Some of these routes of chemical modification of CMC are of consider-able commercial relevance, others of analytical or scientific interest: the car-boxyl group can be esterified by a direct methylation with dimethyl sulfate ine.g. DMSO or after intermediate conversion to the acid chloride of CMC. Thelatter can be obtained by reaction of CMC with a 5-fold excess of SOCl2 at9O0C for I h with a chlorine content of 22 mol % (Nishiuchi et al., 1981).Probably some chlorodesoxycellulose formation must be taken into accounthere. This acid chloride of CMC can then be subjected to a conventional esterifi-cation with a low molecular alcohol or to formation of an acid amide by reactionwith a primary or secondary amine. An alternative route to esterification of theCMC carboxyl groups was reported by Klemm and Geschwend (1989) by O-alkylation and crosslinking in the case of bifunctional bromo derivatives withbromoacetic acid ester. This enzymatically removable crosslinking is importantfor both the controlled change of solubility and the gradual release of incorpo-rated bioactive agents in dependence on the esterase activity.
The hydrophobic modification of CMC by amidation of the carboxy lie groupswith hexadecylamine was studied by Charpentier et al. (1997) along severalroutes. Hydrolytic chain degradation was found to be smallest on coupling theamine with the acid form of CMC activated by W, W'-dicyclohexylcarbodiimidein dry DMSO. As a route to moderate and reversible crosslinking of CMC theintermolecular lactone formation between carboxyl groups and free hydroxygroups according to the scheme in Fig. 4.5.6 has found practical interest formodifying the material properties of CMC with regard to dissolution. Theselactone crosslinks are formed in a neutral to slightly acid medium at elevatedtemperature and are reversibly cleaved again in an alkaline aqueous system.
Q \
CH2-C-O-/ + H2O
Figure 4.5.6. Crosslinking of CMC by intermolecular formation of lactone groups.
The free hydroxy groups of partially substituted CMC can be etherified oresterified along conventional routes with the carboxymethyl groups remainingintact due to their stability. Mixed ethers have been prepared with e.g. methyl

228 4.5 Etherification of Cellulose
halide, ethylene oxide or the sodium salt of vinylsulfonic acid. Cellulose etheresters with carboxymethyl and acetyl groups can be used in tablet coating. Wa-ter-soluble crotonates and methacrylates from partially substituted CMC havebeen obtained with the appropriate acyl chloride in benzene at 40 0C in the pres-ence of pyridine (Plisko et al., 1982). Sulfation of CMC with the SO3/DMFcomplex in dipolar aprotic liquids like DMF has been studied (Vogt et al., 1995;Vogt et al., 1996), arriving at a complete substitution of all hydroxy groups. Arepresentative sample showed a DS of 1.9 of carboxymethyl groups and of 1.1 ofsulfuric acid half-ester groups. Also, regioselectively substituted carboxymethylsulfates could be prepared, starting from a C-2/C-3-substituted CMC. An ade-quate physical activation of the solid CMC, remaining as a finely dispersed,separate phase in the reaction system, proved to be necessary to obtain a high DSof sulfate groups. A sophisticated route to the 13C NMR spectrographic assess-ment of CMC substitution patterns was recently published in Tezuka et al.(1996): in a first step the carboxymethyl groups were converted to methyl estergroups with dimethyl sulfate in DMSO at 40 0C, and subsequently all the freehydroxy groups were propanoylated with the acid chloride in DMA/LiCl at100 0C in the presence of dimethylaminopyridine. The product was soluble inthe complete DS range in DMSO, and its substitution pattern could be deter-mined by 13C NMR spectroscopy in DMSO-J6 employing the well-separatedsignals of the C=O groups of the propanoyl residues in the positions C-2/C-3and C-6.
Covalent crosslinking of CMC has been achieved via ether bonds with e.g.formaldehyde, formaldehyde urea resin precursors, epichlorohydrin or divinylsulfone, or via polyurethane linkages with diisocyanates. Crosslinking domi-nated by Coulombic interactions can take place with polyvalent cations likeLa3+, Al3+ and Fe3+(Heinze et al., 1989; Heinze et al., 1990; Prasad and Kalya-nasundaram, 1993), by polyelectrolyte complex formation with a cationic poly-electrolyte like polydimethyldiallylammonium chloride (Philipp et al., 1989), orby reacting CMC with H3PO4 and aliphatic diamines at pH 3-5 at 160 0C (Pet-ropavslovskii et al., 1984). As another route to modify the polymer skeleton,grafting onto CMC has been widely studied, which can be performed in anaqueous solution of the polymer, e.g. with methacrylates or acrylamide asmonomers and Ce4+ ions as the initiator at 35 0C.
Role of cellulose supramolecular structure in carboxymethylcelluloseformation
Just as with any derivatization reaction with cellulose in the solid state, car-boxymethylation is affected by the accessibility of the polymer chains too. Butthis influence is of minor relevance here for two reasons:

4.5.2 Aliphatic ethers of cellulose 229
(i) the etherification proceeds at a state of high intracrystalline swelling with thesupramolecular order being diminished and differences in supramolecular orderbeing leveled by the high alkali concentration at the site of reaction and, addi-tionally, by the preceding milling frequently employed in the technical process(Fink and Walenta, 1994);(ii) effects of accessibility on the course of reaction are superseded by evensmall differences in reagent distribution, especially in the so-called dry process.
Thus, cotton !inters, as well as a large variety of wood pulps and even wastecellulose (Buytenhuys and Bonn, 1977) can be converted to CMC without spe-cial pretreatment. Conventional carboxymethylation of mechanical wood pulpresulted in about 50 % conversion of the polysaccharides, and the lignin wasfound to inhibit only dissolution, but not carboxymethylation itself (Thi BachTuyetetal., 1981).
In agreement with point (i) cited above, only a four times faster rate of car-boxymethylation with a low DP cellulose dissolved in the aqueous reaction sys-tem was observed compared with the conventional etherification of an alkalicellulose in the solid state, and also a preactivation with liquid ammonia showeda small effect only (Dautzenberg and Philipp, 1979b; Dautzenberg et al., 198Oa).Nevertheless, the influence of accessibility can be observed regarding the courseof enzymatic degradation of CMC (Kasulke et al., 1983) and the limiting DS ofwater solubility, both criteria obviously depending largely on the uniformity ofsubstituent distribution along the chains. A detailed 'supramolecular chemistryof carboxymethylation', i.e. of the changes in hydrogen bond structure in thecourse of reaction, is still a wide open problem to research. From studies oncarboxyethylcellulose with a DS of 0.4 in the solid state, Kamide et al. (1988)concluded a preferential breaking of intramolecular hydrogen bonds on car-boxyethylation, with the intermolecular ones remaining widely intact.
Survey of the technical process of carboxymethylation
The raw material employed in carboxymethylcellulose manufacture largely de-pends on the product quality required: for high-viscosity types, cotton !interswith a DP up to 4000 are used, and oxygen must be excluded to avoid chaindegradation in the strongly alkaline reaction system. Mostly dissolving pulpsfrom hard or soft woods or even from annual plants with a rather low degree ofrefinement are used.
Two process routes are practised commercially, i.e. the so-called dry processand the slurry process. In any case at least 0.8 mol of NaOH/mol of AGU arerequired for commercial CMC types if etherification takes place with sodiummonochloroacetate. If free monochloroacetic acid is employed, one extra moleof NaOH must be added for neutralization. An alkali excess of at least 5 % overthat required for conversion of the monochloroacetate input is considered neces-

230 4.5 Etherification of Cellulose
sary to secure a sufficiently fast reaction. The reagent yield for carboxymethyla-tion generally amounts to 60-80 % of the monochloroacetate input. The exo-thermic reaction requires no initial heating but frequently some cooling tomaintain a reaction temperature in the range 25-70 0C.
The dry process is usually performed in a shredder equipped with toothed,sigma-shaped plates. Mostly an alkali cellulose is prepared first, and then themonochloroacetate is added, often in the solid state. But also the reverse proce-dure, i.e. soaking the cellulose with monochloroacetate solution at the first step,has been practised. Organic liquids like isopropanol, ί-butanol or acetone can beadded in smaller quantities also in the dry process in order to increase reagent dif-fusion and to enhance product uniformity and solubility. In the slurry process, thecellulose, aqueous alkali, monochloroacetate and an excess of e.g. isopropanol aremixed in a conventional reaction vessel equipped with an efficient stirrer, andreacted for one to several hours at a temperature in the range cited above. Also,continuous processes employing a double screw drive have been developed.
The reaction product remains in the solid state throughout the process and isfinally neutralized by e.g. HCl. The crude product contains up to 40 % of lowmolecular salts (on a dry basis), which is washed out by methanol or metha-nol/water mixtures. The level of salt content is about 1 % in technical grades ofCMC and less than 0.1 % in high-quality products for e.g. nutritional use. Thework-up procedure can be combined with a viscosity reduction by H2C^ or withsome crosslinking for modifying the course of dissolution and the rheologicalproperties of aqueous CMC solutions.
Properties of CMC, especially in aqueous solution
All commercial grades of CMC are white, odorless and nontoxic powders, pre-dominantly consisting of the Na salt of CMC and not of the free acid (H-CMC).H-CMC is thermally decomposed without melting or softening. In solution itbehaves as an anionic polyelectrolyte with a weakly acidic group of pKa = 3-4(Kotz et al., 1990). CMC is easily biodegradable up to a DS of about 1, and it isadmitted as a food additive.
The water solubility of CMC as the most relevant applicational property de-pends primarily on the DS, but is largely influenced also by the procedure ofpreparation and the DP, as indicated by the wide range of lower limiting DSvalues for solubility in various media (see Table 4.5.9). Recent results are pub-lished by Liu et al. (1997) and Heinze (1998).
Carboxyethylcellulose with its longer alkyl chain acting as a more efficientspacer than the carboxymethyl group was found to become water-soluble al-ready at a DS of 0.15-0.20, if prepared under homogeneous conditions of reac-tion, securing a rather uniform substituent distribution along the polymer chains(Schleicher et al., 1980).

4.5.2 Aliphatic ethers of cellulose 231
Table 4.5.9. Region of lower DS limit of solubility of CMC.
Solvent Region of limiting DS
4-8 % aq. NaOH >0.15Cold water 0.3-0.6
The state of a dilute aqueous CMC solution by no means resembles a com-plete dispersion of the polymer down to the level of the single macromolecule(Dautzenberg et al., 1978a and 1978b). A macroscopic, clear solution at the 1 %level usually contains, besides single macromolecules, temporary chain aggre-gates held together by hydrogen bonds and larger gel particles persisting fromthe raw material. A considerable part of the macroscopically dissolved polymercould be separated from the solution by centrifugation. As demonstrated in Ta-ble 4.5.10, this part decreased with increasing DS but also depended on the rawmaterial and on the procedure of CMC preparation via differences in substitutionpattern along the chains.
Table 4.5.10. Amount and molar mass of CMC separated fromaqueous solutions by centrifugation.
DS
0.61.11.51.5
Raw material
AAAB
% separated
335
2113
Mw χ ΙΟ-6
5030.420.80
A LODP cellulose powder (DP =160).B Linters cellulose (DP = 1400).
This ill-defined structural state of aqueous CMC solutions makes the [η]-Μ
relationships so far reported somewhat questionable. According to Lavrenko etal. (1986) the DP of CMC can be assessed from viscosity measurements in Ca-
doxen as the solvent by the equation: [η] - 1.93 x 10~3 x MW
LO which is rather
insensitive to DS in the range 0.86-1.1. The above [η]-Μ relationship was
elaborated in the DS range 0.86-1.1 and can be considered to be widely inde-pendent of DS in Cadoxen solvent in the range of commercial interest. Higherconcentrated aqueous solutions of CMC exhibit pronounced thixotropy and vis-coelastic behavior. The apparent viscosity increases with c4·3 and M3·9 at a DSof 1.0 at 25 0C (Kulicke et al., 1996).Besides the facts so far considered, the character of CMC as an anionic poly-electrolyte has to be taken into account when dealing with solutions of thispolymer. The apparent viscosity of aqueous solutions decreases significantly on

232 4.5 Etherification of Cellulose
increasing the content of low molecular electrolytes. In dependence on pH theapparent viscosity passes a maximum (see Fig. 4.5.7): the viscosity increase inthe lower pH range is caused by an uncoiling of the macromolecules due toanionization, while an increasing ionic strength at high pH results in a loweringof viscosity. For further details on the poly electrolyte behavior of CMC solu-tions the reader is referred to the comprehensive studies of Rinaudo (1995) andBerthold et al. (1994). A detailed study on the state of solution of Na-CMC inwater in dependence on molar mass, DS and ionic strength was recently pub-lished by Kästner et al. (1997) based on rheological and on electrical birefrin-gence experiments. The authors discern between 4 ranges of polymer concentra-tion, differing in state of solution with regard to chain coiling and chain entan-glement, with these ranges being strongly influenced by DP, DS of the sampleand by the ionic strength of the system.
7 pH
Figure 4.5.7. Dependence of the viscosity of an aqueous CMC solution on its pH.
By lowering the pH to < 4, or by adding polyvalent metal cations, CMC isprecipitated from aqueous solutions, in the former case as nondissociated H-CMC, in the latter due to formation of salt crosslinks. Gels of H-CMC werefound to show rheological aging due to structural changes (Hakert et al., 1989).On the macroscopic scale, H-CMC films are of rather low strength, but flexibleat higher relative humidity. Compared with films directly cast from aqueousCMC solution, an intermediate xanthation before acid precipitation resulted inlarger and more uniform pores in the H-CMC film (Dautzenberg et al., 198Oband 198Oc), demonstrating again the role of an intermediate derivatization ofcellulosics in solid state structures precipitated from solution.
Slight salt crosslinking of CMC in aqueous solution by e.g. Ca2+ or Al3+ re-sulted in significant changes in the apparent viscosity, which were found to de-pend strongly on the mode of CMC preparation via differences in the pattern ofsubstitution (Heinze et al., 1994c).

4.5.2 Aliphatic ethers of cellulose 233
Areas of application of CMC
Carboxymethylcellulose is presently produced worldwide at a level of 300,000 tannually and this holds by far the first place among cellulose ethers. About two-thirds of the amount manufactured is of standard quality, about one-third ismade up of special high-quality types. Commercial CMC production comprisesthe DS range from 0.3 to 0.9 and a large number of types differing in solutionviscosity and other rheological properties.
CMC is generally applied in aqueous solutions as a thickening and dispersionstabilizing agent in many areas of industry and domestic life. Due to its anioniccharge, CMC acts as a soil redeposition inhibitor against the predominantlynegatively charged soil particles on the surfaces of e.g. textile fibers. The non-toxicity and biocompatibility of CMC permits its use in food products andPharmaceuticals. Due to its character as a poly electrolyte, CMC exhibits a lim-ited 'salt stability' only, i.e. the polyelectrolyte shows a decrease in viscosityand dispersion-stabilizing power in the presence of low molecular electrolytes,which can be a disadvantage in e.g. oil drilling. Mixed ethers, containing hy-droxyethyl groups besides carboxymethyl groups, can combine the excellent saltstability of hydroxyethylcellulose with the excellent dispersion stability effect ofCMC, and are therefore commercially produced too.
An overview on the main areas of commercial application of CMC is pre-sented in the following Table 4.5.11 showing that the use in detergents domi-nates by far.
Table 4.5.11. Areas of application of CMC (Bikales and Segal, 1971).
Area of application
DetergentsFood productsOil drilling mudsTextiles (e.g. warp size)Paper and paper bound sizePharmaceuticalsPaintsOther
Percentage of totalamount applied
38-471413118835
Special uses of CMC in combination with other materials have been proposed,for example the application of CMC containing polyelectrolyte complexes forsoil stabilization, or the use of a CMC-Pt complex in a catalyst for hydrogena-tion of aromatics (Tang et al., 1996).

234 4.5 Etherification of Cellulose
Besides its numerous areas of commercial application, CMC plays an impor-tant role in the organic and physical chemistry of cellulose as a precursor forsubsequent steps of derivatization and as a model of an anionic polyelectrolyte.The DS is often expressed in 'mmol of COOH/g of sample', the relation betweenthese two criteria is given in Fig. 4.5.8.
10r
IO 6OO
Γ
0,0 0,5 1,0 1,5 2,0 2,5 3,0
DS
Figure 4.5.8. Correlation between mmol/g carboxyl groups and the DS of carboxy-methy !cellulose.
The following formulae can be employed for calculation of DS of CMC fromcarbonyl group content and vice versa.
MW. =162+ DS χ 58
/ / / DSxWOO(mmol / g
l62x(mmol/g)COOHDSCMC - 100() _ mmol / g )COOH χ 5gJ
4.5.2.3 Hydroxyalkyl ethers of cellulose
A representative of the hydroxyalkylation of cellulose is the formation of hy-droxyethylcellulose (HEC) and hydroxypropylcellulose (HPC) as commerciallyrelevant derivatives, by reaction of the polymer with ethylene oxide and propyl-ene oxide respectively. Furthermore, the crosslinking of cellulose by using epi-

4.5.2 Aliphatic ethers of cellulose 235
chlorohydrin and the acetalization of cellulosic hydroxy groups by using alde-hydes have to be considered here.
Chemistry of hydroxyalkylation by epoxides
Two important points of difference to the alkylation and carboxymethylation ofcellulose described above have to be mentioned, i.e.(i) hydroxyalkylation with epoxides does not require a stoichiometric, but inprinciple only a catalytic amount of OH~ ions for the cleavage of the epoxy ringand the formation of the C-O bond between the reagent and the alcohol (seescheme in Fig 4.5.9);
(NaOH)HOH - H+ + HO-
HO' + CH2-CH2 SlOW HO-CH2-CH2-O'
\ /O
footHO-CH2-CH2-Q- + H+ 12SL—HO-CH2-CH2-OH
(NaOH)R-OH · · RQ- + H+
RO- + CH2-CH2 SlOW RO-CH2-CH2-Q-
\ /O
foot
RO-CH2-CH2-Q- + H+ asi RO-CH2-CH2-OH
Figure 4.5.9. Scheme of 'hydroxyethylation' of water and of an alcohol.
(ii) hydroxyalkylation is not limited to the hydroxy groups originally present inthe system, but can proceed further at the newly formed hydroxy groups result-ing in hydroxyalkyl chains of varying length (see Fig. 4.5.10);
As indicated in the schemes above, the alkali-catalyzed hydroxyethyl etherformation is accompanied by the reaction of water molecules with ethylene ox-ide to glycol and to polyglycols, with the reagent yield for cellulose etherifica-tion amounting to 50-70 % of the ethylene oxide input. An acid-catalyzedcleavage of the epoxy ring is possible too, but promotes homopolymerizationinstead of the intended etherification and leads to a detrimental hydrolytic cleav-age of the cellulose chains. These are therefore generally hydroxy alky lated in anaqueous alkaline system.

236 4.5 Etherification of Cellulose
OH"CeII-OH + CH2-CH2
O
CeII-O-CH2-CH2-OH + CH2-CH2 -^- CeII-O-CH2-CH2-O-CH2-CH2-OH
O
H2O+CH2-CH2 -^-HO-CH2-CH2-OH
O
HO-CH2-CH2-OH+ CH2-CH2 - L· HO-CH2-CH2-O-CH2-CH2-OH
O
Figure 4.5.10. Scheme of reactions occurring in hydroxyethylation of cellulose in anaqueous alkaline medium.
, CH2OCH2CH2OCH2CH2OCH2CH2OH
,O
^^HOCH2CH2OCH2OCH2CH2OCH2CH2OH
DS = 3MS = 6
Figure 4.5.11. Illustration of the meaning of 'degree of substitution' (DS) and 'molecu-lar substitution' (MS) at one AGU of hydroxyethy!cellulose.
Due to the possible growth of hydroxyalkyl side chains by further add-on ofalkylene oxide, two criteria are necessary for a macromolecular characterizationof HEC and HPC, i.e. the degree of substitution (DS) denoting the average num-ber of cellulosic hydroxy groups per AGU involved in the reaction, and the mo-lecular substitution (MS) denoting the average number of alkylene oxide mole-cules added per AGU, as illustrated by Fig. 4.5.11.
The MS always exceeds the DS, at sufficient high reagent input, the MS growsfaster than the DS in the early stages of reaction, and the ratio MSIDS increasesuntil reaching a level usually between 1.5 and 2.5 denoting the average length ofthe side chains (Fig. 4.5.12).
The MS value of most commercial HEC types covers the range between 1.5and 3.0, corresponding to a DS range between 0.8 and 1.2. A lower MS limit ofabout 1.0 is frequently assumed to be necessary to obtain water solubility of theHEC.

4.5.2 Aliphatic ethers of cellulose 237
1.6
CO
Q U
il.2
1.00.5 1.0 1.5 2.0 2.5
MS
Figure 4.5.12. Course of the ratio MSIDS with the MS of HEC (Arisz et al., 1996).
Hydroxyalkylation of cellulose is generally performed in a thoroughly hetero-geneous course of reaction with the weight ratio of NaOH/cellulose varyingwithin the wide limits of between 0.3 : 1 to 1 : 1, and that of H2O/cellulose be-tween 1.2 : 1 to 3.5 : 1. Frequently a slurry process with an excess of a fairlyinert diluent like /-propanol, ί-butanol or acetone is employed in hydroxyethyla-tion. The reaction proceeds at 30-80 0C within some hours, and the MS is de-termined by the ethylene oxide input. The reaction rate increases with the NaOHand the ethylene oxide concentrations for the main, as well as for the side reac-tion, and a low concentration seems desirable with regard to reagent yield for themain reaction to obtain a uniform product. At higher alkali concentration thereaction can become diffusion-controlled and the uniformity of reagent distribu-tion plays a major part. This agrees with the observation of a significantly higherenergy of activation at a low alkali concentration of 5 % in comparison with thatof 14 % (Mansour et al., 1993). On the other hand, a higher alkali concentrationensures a better and more uniform accessibility of the cellulose chains within thefiber, and usually the industrial reaction conditions are adapted to either a ratherlow or a rather high NaOH concentration (Dönges, 1990). A suspension hy-droxyethylation at rather high temperature and pressure and a large amount ofdiluent was compared with the hydroxyethylation of alkali cellulose with gase-ous ethylene oxide at low temperature and pressure without diluent (Asandei etal., 1995).
Considerations on the pattern of substitution of hydroxyalkylcelluloses haveto taken into account, along with the DS, as well as the MS. The MS can be ob-tained by a modified Zeisel procedure, and the partial DS values, as well as thetotal DS, are usually assessed by NMR spectroscopic or Chromatographie tech-niques. Earlier studies revealed a high reactivity of the hydroxy groups, at the C-6 position and at the side chains ('C-X position'), and a very low reactivity atthe C-3 position. A reactivity ratio of C-2/C-3/C-6/C-X = 3 : 1 : 10 : 10 had beenreported (Wirick, 1968). More recently, a significant effect of the MS alreadypresent on the one hand, and of the alkali concentration on the other, on thisreactivity ratio has been observed: with increasing MS, the relative rate constantof the C-X position decreases markedly and thus slows down the further growth

238 4.5 Etherification of Cellulose
of the side chains, whereas the reactivity ratio of C-2/C-3/C-6 remains nearlyconstant (see Fig. 4.5.13).
-2 3ω J
cO
S i
Χ. I
£ O 0.5 1.0 1.5 2.0 2.5* MS
Figure 4.5.13. Effect of MS on the normalized reactivity at the positions at 2, 3, 6 and Xin hydroxyethylation of cellulose, relative reaction constants at positions 2 ·, 3 O, 6 ·,and X D; X = OH groups at side chains (Arisz et al., 1996).
A higher alkali content results in an increase of reactivity at all the positions,but is much more pronounced at C-6 and C-X than at C-2 and C-3. This resultsin a much higher reactivity at C-6 and of course of C-X at high alkali contentcompared with C-2, while at low alkali concentration the C-2 position is thepreferred site of substitution compared with C-6 (see Fig. 4.5.14).
According to Arisz et al. (1996) intermolecular interactions of the side chainsbetween themselves or with NaOH and with the organic diluent can influencethe reactivity by hydrophobic interaction, as well as by activation due to prefer-ential NaOH binding.
Hydroxypropylation of cellulose proceeds in a similar way to hydroxyethyla-tion according to
OHOH'
CeII-OH + CH2-CH-CH3\2 / 3
O
CeII-O-CHp-CH-CHo
resulting in a more hydrophobic product with secondary hydroxy groups. Anorder of reactivity of C-6 > C-2 > C-3 has been observed. A MS value of about 4is considered necessary here to obtain water solubility.
Starting from a conventional alkali cellulose prepared by steeping with 18 %aqueous NaOH and subsequent pressing, two procedures of hydroxypropylationwere compared by Asandei et al. (1995):(i) a slurry procedure with an organic diluent, employing rc-hexane, i-butanoland alkali cellulose at a ratio of 1.54 : 0.5 : 1 and a propylene oxide/AGU ratioof 7-11 at 60 0C for 2-6 h under pressure;

4.5.2 Aliphatic ethers of cellulose 239
U
12
10>s
I 8
I6CL
4
2
O0.5 1.0
mol NoOHmol AGU
1.5
Figure 4.5.14. Effect of alkali concentration on the reactivity of the positions X, 6, 2,and 3 (top to bottom) in the hydroxyethylation of cellulose (Dönges, 1990).
(ii) a procedure without diluent, employing gaseous propylene oxide at a ratioof 2-6/AGU at 40-50 0C and low pressure for 2-6 h.
With the first procedure MS values up to 4.2, and with the second procedureup to 4.0, were obtained, despite the much lower reagent input in the latter case.
The hydroxy groups in hydroxyalkylcellulose can be employed for subse-quent esterification or etherification. Acetylation has been used for analyticalpurposes in assessing the pattern of functionalization. Etherification of HEC bymethylation, ethylation or hydroxypropylation is performed on a technical scalein the manufacture of mixed cellulose ethers. The introduction of tertiary aminogroups and quaternary ammonium groups into HEC is described by Katsura etal. (1992). The efficiency of phase-transfer catalysis in the hydrophobic modifi-cation of HEC by introduction of dodecylphenylglycetyl ether groups has beendiscussed by Emett (1996).
By Lee and Kwei (1996) the reaction of HPC with hexyl-, octyl-, dodecyl-and octadecyl isocyanate is described, together with the supramolecular andmorphological properties of the products. The simultaneous reaction of alkalicellulose with CS2 and ethylene oxide has been investigated (Lukanoff andPhilipp, 1967): a presumed spacing effect of hydroxy ethyl groups was obviouslyovercompensated by an enhanced side product formation via the ethylene glycolformed in the system and by a reduction of CS2 activity in the presence of ethyl-ene oxide, resulting in a significant decrease in the degree of substitution ofxanthogenate groups and indicating a detrimental effect for the viscose processinstead of the expected beneficial one. Covalent crosslinking of hydroxyalkyl-cellulose can be achieved by conventional bifunctional agents like glyoxal, re-

240 4.5 Etherification of Cellulose
suiting in simultaneous crosslinking and chain scission. Besides these subse-quent covalent reactions presented here in some detail, the intermolecular inter-action between hydroxyalkylcelluloses and surfactants, e.g. sodium dodecylsulfate, has been studied (e.g. Zugenmaier and Aust, 1990).
Role of the supramolecular structure of cellulose on hydroxyalkylation
As a heterogeneous process, the hydroxyalkylation of cellulose is affected by thesupramolecular structure of the polymer too. This influence is diminished bystrong swelling in the alkaline system, by the spacing action of the hydroxyalkylside chains, and by the high reactivity of the C-6 position promoting a moreuniform substituent distribution along the polymer chains, and it is additionallycovered by the strong effect of reagent distribution within the reaction system(Dönges, 1990). Yokota (1986) compared cellulose hydroxypropylation in aslurry process with organic diluent at a low alkali concentration of 0.4 mol/molof AGU on the one hand, and with more concentrated aqueous alkali at a lowliquor ratio on the other. A very heterogeneous progress of reaction from fiber tofiber in the first case was observed, and a more uniform higher state of order inthe second. The introduction of the substituents resulted in an increased 1-0-1lattice spacing, which appeared to be more uniform across and along the fibrilsin the case of the more concentrated aqueous alkali.
Survey of the technical process of cellulose hydroxyalkylation
In commercial hydroxyalkylation the shredded or milled material is reacted withethylene oxide and NaOH, usually in a slurry process with /-propanol, i-butanolor acetone as the diluent, employing 0.5-1.5 mol of NaOH/mol of AGU. This isadded either before the diluent or by pouring directly into the suspension. Withisopropanol as the diluent usually 10-12 mol of H2O/mol of AGU are present inthe reaction system. The reaction proceeds at 30-80 0C for 1-4 h, with the MScontrolled by the amount of reagent applied. The reagent yield for the main re-action is reported to be about 70 % but decreases down to 50 % at high MS. Inorder to decrease the ethylene oxide consumption for the side reactions and toenhance product uniformity, a two-stage process can be practised, arriving at alow-substituted water-insoluble product in the first stage, and then performingthe second stage with only a catalytic amount of alkali and the main part of theethylene oxide, making use of the hydroxyethylene groups introduced in the firststage as a spacer. After neutralization with e.g. HCl, the low molecular by-products are washed out by water/alcohol mixtures.
Hydroxypropylcellulose is manufactured in a slurry process similar to HEC,but requires a higher reaction temperature of up to or above 100 0C, and a longerreaction time due to its lower reaction rate. It is manufactured under pressurewith liquid propylene oxide or hexane as the reaction medium. Purification can

4.5.2 Aliphatic ethers of cellulose 241
be accomplished by washing with hot water, as HPC exhibits gelling in hot wa-ter like methylcellulose.
In the manufacture of HEC-based mixed ethers, usually the hydroxyethylationis accomplished first, followed by etherification with the second reagent.
Properties of hydroxyalkylcelluloses
HEC and HPC are white, odorless, physiologically inert powders, the solubilityof which depends largely on the kind of substituent and the pattern of substitu-tion. The biodegradability of HEC decreases with increasing DS, while thelength of the side chains is of minor relevance to enzymatic attack. HPC is muchmore hydrophobic than HEC and can be extruded without a softener at 160 0C,whereas HEC is not thermoplastic and is decomposed in aqueous solution al-ready above 100 0C.
HEC exhibits solubility in cold, as well as in hot water at an MS above 1.0,and becomes soluble at higher MS also in mixtures of water with some polarorganic liquids like lower alcohols. HPC requires an MS of about 4 for solubilityin cold water and shows gelling at about 40 0C and precipitation at about 45 0C.
The apparent viscosity of aqueous solutions of HEC and HPC depends on theDP, the polymer concentration and the shear rate of the solution.
The [7]]-M relationship has been reported (Dönges, 1990) to be forHEC: 77 =1.1 x 10-2DP0-87
η = l χ ίο-3 Mw°·7
An impression of the concentration and shear rate dependency of the apparentviscosity of nonionic cellulose ethers in general is presented for 2 % aqueoussolutions in Fig. 4.5.15. The figure indicates the broad spread of the viscosityrange for commercial ether types with the appropriate viscosity level usuallybeing included in the designation of the type in question. Besides the strongdecrease of apparent viscosity with increasing shear rate most of these productsexhibit a pronounced viscoelastic behavior in aqueous solution.
As a peculiar feature, the tendency of HEC, and especially of HPC, to formliquid crystalline aqueous systems has to be mentioned. These systems and theiroptical behavior have been comprehensively studied in recent years (e.g. Gias-son et al., 1991). Crosslinked films of HPC from aqueous solutions by 7-irradiation and drying were prepared, and the TEM micrographs of these filmsrevealed a persistence of chiral nematic structures from the solution to the solidfilm. Promising routes to preserve the liquid crystalline order of hydroxyalkyl-cellulose solution in the solid state consist of either preparing the liquid crystal-line system in a polymerizable liquid, e.g. acrylamide, with subsequent forma-tion of a solid matrix by radiation polymerization, or using a mixed ether with asubstituent susceptible to polymerization crosslinking (Hohn and Tieke, 1997).

242 4.5 Etherification of Cellulose
100.000
40.000
10.000
1 .000
'g 400U(Λ
> 100
40
10
10.000 4.0001.000
10.000
4.000
?1.000
^ 400>>'S 1008~ 40
104 10 10*
Rotational speed [s"1]
2 4 6Concentration [wt %]
Figure 4.5.15. General scheme of the concentration (left) and shear dependency (right)of the apparent viscosity of nonionic cellulose ethers (Dönges, 1990).
Areas of application of hydroxyalkylcelluloses
The first place is kept by hydroxyethylcellulose with an annual production of54,000 t worldwide (Dönges, 1990). Some other hydroxyalkylated products ofcommercial relevance are listed in Table 4.5.12, together with the appropriateDS values.
Table 4.5.12. Commercial nonionic mixed ethers of cellulose (Dönges, 1990)
Sample Formula DS
Hydroxybutylmethylcellulose
Ethylhydroxyethylcellulose
Hydroxyethylhydroxypropylcellulose
-OCH3 2-Q-CH2-CHOH-CH2-CH3 0.05-Q-C2H5 0.7-1.2-OCH2-CH2OH 0.8-2.7-OCH2CH2OH 0.8-1.2-OCH2CHOH-CH3 0.65-0.9
HEC is predominantly used in aqueous systems as a thickener or binder, or as aprotective colloid and suspension stabilizer. It exhibits an excellent salt compati-bility and can be converted to transparent films from aqueous solution. Numeroustypes covering a wide range of apparent viscosity are commercially available.

4.5.2 Aliphatic ethers of cellulose 243
The main areas of application of HEC are:
• Dispersion (Latex) paints · Pigment carrier• Ceramic binder · Textile size• Adhesives · Emulsion polymerization• Oil exploitation• Paper sheet formation (wet strength additive together with glyoxal as
crosslinker)
Besides this widespread use of HEC as a product, hydroxyethylation to low DShas been considered for hydrophilizing cellulose and for loosening its physicalstructure by the spacer action of the hydroxyethyl side chains. An interestingdevelopment of an ecocompatible artificial fiber from low DS (DS ca. 0.2) al-kali-soluble HEC (Diacik et al., 1977), finally failed, as obviously completealkali solubility to a gel-free spinning dope and sufficiently high wet strength ofthe fibers obtained proved to be incompatible.
Areas of application of the less hydrophilic, thermoplastic and organosolublehydroxypropylcellulose are known in the food industry and pharmaceuticals dueto the high biocompatibility of this product, which is also of potential interest asa speciality product in the electronics industry. This spacer effect of hydroxy-propylation to give a low DS has been successfully employed to convert macro-porous bead cellulose to a more uniform, rather continuous, network structure bycombining hydroxypropylation with subsequent crosslinking with epichlorohy-drin(Loth, 1991).
Ether bond crosslinking of cellulose with epichlorohydrin
l-Chloro-2,3-epoxypropane (epichlorohydrin) combines the reactivity of analkyl halide with that of an alkylene epoxide towards cellulosic hydroxy groups.Due to this bifunctionality, it acts as an efficient crosslinking agent in an aque-ous alkaline medium according to the reaction scheme depicted in Fig. 4.5.16.
Besides the catalytic amount of NaOH required for epoxy ring cleavage, thestoichiometric amount of 1 mol/mol of epichlorohydrin is necessary here for theepoxide formation. A considerable part of the epichlorohydrin is consumed inthe formation of low molecular by-products, especially of glycerol (see Fig.4.5.17), and the part of the reagent reacting with cellulose is used for bifunc-tional crosslinking, as well as for the monofunctional formation of 1,2-dihydroxypropylcellulose.
Thus, the crosslinking efficiency of epichlorohydrin lags far behind the totalconsumption, and the number and distribution of crosslinks formed dependslargely on the detailed procedure of epichlorohydrin application. Three differentprocedures were compared with regard to the resulting structural changes of a

244 4.5 Etherification of Cellulose
cellulose powder (Dautzenberg et al., 198Od). The decrease of Guam solubilitywith increasing degree of crosslinking (defined as the average number of hy-droxy groups/AGU involved in ether crosslinks) depended significantly on theprocedure employed (see Fig. 4.5.18).
CeII-OH + CH2-CH2-CH2-CI -CeII-CH2-CH-CH2\ / ι ιO OH Cl
OH' CeII-O-CH2-CH-CH2
O
CeII-O-CH2-CH-CH2 + CeII-OH -CeII-O-CH2-CH-CH2-O-CeII
V OH
CeII-O-CH2-CH-CH2+ H2O —-CeII-CH2-CH-CH2\ I ' '
OOH OH
CH2-CH-CH2-CI + 2H2O-QTq-CH2OH-CHOH-CH2OH\ / ' HCI
Figure 4.5.16. Scheme of cellulose crosslinking by epichlorohydrin
100
α.
2 Wcου
c 40
•ο
ο 20υ
•α.LU
O
2 4 6Time [h]
Figure 4.5.17. Course of epichlorohydrin consumption for cellulose etherification (·)and for total consumption (O) (Dautzenberg et al., 198Od).

4.5.2 Aliphatic ethers of cellulose 245
100
.75
50
25
0.2 0.6 1.0Degree of crosslinking
1.2
Figure 4.5.18. Decrease of Guam solubility of a cellulose powder on crosslinking withepichlorohydrin by different procedures of alkalization: (O) high liquid ratio, acetone asdiluent, two liquid phases; (·) high liquid ratio, aqueous NaOH (25 % w/w), no diluent;(·) low liquid ratio, aqueous NaOH (25 % w/w), spray procedure (Dautzenberg et al.,198Od).
Alkali solubility and WRV, on the other hand, did not decrease uniformlywith progressive crosslinking, but passed a maximum with all the proceduresemployed due to the counteracting effects of 'structure widening' by introduc-tion of covalent spacer at a high state of swelling and 'structure tightening' byformation of covalent crosslinks (see Fig. 4.5.19 and 4.5.20).
300
200
100
0.2 0.6 1.0
Degree of crosslinking
Figure 4.5.19. WRV of a cellulose powder versus degree of crosslinking with epichlorohydrin(Dautzenberg et al., 198Od).
On the supramolecular level the reaction with epichlorohydrin resulted in anincrease in the 1-0-1 lattice distance after neutralization and in a fairly severedestruction of the fibrillar architecture of the particles. The interplay between thehydrophilic spacing and the structure tightening crosslinking by etherification

246 4.5 Etherification of Cellulose
with epichlorohydrin has also been emphasized in a study on modification ofbead cellulose (Loth and Philipp, 1989).
60
OCO
20
0.1 0.3 0.5 0.7Degree of crosslinking
Figure 4.5.20. Solubility of cellulose powder in 5 % NaOH versus degree of cross-linking with epichlorohydrin (Dautzenberg et al., 198Od).
Etherification by epoxidation has also been performed with cellulose dis-solved in DMA/LiCl. Diamantoglou reported DS values of about 0.5 with pow-dered NaOH or with LiOH as the base and propylene oxide or epichlorohydrinas the reagent, whereas carboxymethylation with monochloroacetic acid arrivedat a DS < 0.1 only, in the same system (Diamatoglou and Kühne, 1988). Thesefindings demonstrate again the difference in reaction mechanism between a car-boxymethylation requiring a stoichiometric amount, and an epoxidation needingonly a catalytic amount. On the other hand, using an excess of NaOH andClCH2COONa, CMC of high DS of up to 2.3 can be synthesized (Heinze et al.,1994b).
Etherification of cellulose by epoxidation has also been employed to intro-duce functionalized side chains via ether linkages into the macromolecule. Thiswas studied especially as a route to synthesize cellulose derivatives with cationicnitrogen functions attached (see section 4.5.3.2).
Formation and reactions of hydroxymethyl(4methyloP)cellulose andrelated derivatives
Formally, hydroxymethylcellulose can be considered as the first member in aseries of hydroxyalkyl ethers of cellulose, but regarding the mode of formationand the instability of methylolcellulose with the simplified formula CeIl-O-CH2OH, it represents a half-acetal of the polymer. Methylolcellulose was iden-tified and characterized in connection with the discovery of the solvent systemDMSO/paraformaldehyde or DMSO/formaldehyde (Johnson et al., 1976; Bakeret al., 1981) about 20 years ago. At elevated temperature usually of about

4.5.2 Aliphatic ethers of cellulose 247
140 0C, the DMSO/paraformaldehyde system dissolves even high molecularcellulose quickly and completely without significant chain degradation. It hastherefore been studied as a possible route to artificial fiber spinning, as a solventfor cellulose characterization in solution (Gruber and Gruber, 1978), and as asystem for subsequent cellulose derivatization in solution.
The present state of knowledge of cellulose methylolation can be summarizedas follows: due to several chemical equilibria existing in systems of cellu-lose/formaldehyde/polar aprotic liquid, and interacting with each other (see Fig.4.5.21), methylolcellulose is not a well-defined cellulose derivative. Its compo-sition depends largely on parameters like component ratio, mode of componentaddition, rate of heating, and final reaction temperature and reaction time.
(CH20)n^^ n CH2O
CeII-OH + CH2O =^ Cell-O-CH2OH
CeII-OH +(CH2O)x =^ Cell-O-(CH2O)x-H
2 CeII-OH + CH2O =* Cell-O-CH2-O-CeII + H2O
Figure 4.5.21. Scheme of reaction involved in the methylolation of cellulose.
Methylolation can take place with a large excess of formaldehyde above80 0C, or, more comfortably, with paraformaldehyde in DMSO at 135-140 0C.A methylolated cellulose obtained in various polar liquids at elevated tempera-ture with gaseous CH2O, as well as with paraformaldehyde, was reported (Bakeret al., 1981). The authors characterized the product by 1HNMR spectroscopyafter complete acetylation and isolation of the stable acetates. A high MS of be-tween 15 and 25 was required for cellulose dissolution in the various solventsemployed, which, however, could be subsequently lowered to a MS of between0.5 and 3.0 without precipitation of the polymer (see Table 4.5.13).
Table 4.5.13. Initial and final MS of methylolcelluloseprepared in different solvents (Baker et al., 1981).
Solvent
DMSODMFNMPDMAPyridine
Initial MS
18.823.621.920.915.1
Final MS
0.52.01.51.53.0
NMP, W-methylpyrrolidone.

248 4.5 Etherification of Cellulose
Obviously, an initially nonuniform distribution of long methyl side chainschanges gradually to a more uniform one of shorter side chains by cleavage ofCH2O entities. The C-6 position was shown to be the preferred site of reaction,followed by the C-2 position (Nehls et al., 1994). Even a DS of 3 can be realizedaccording to Kinstle and Irving (1983). A molecular substitution of 1.5-2.4 wasfound to be necessary for dissolving cellulose at 85 0C in DMSO with an excessof formaldehyde, but the level of formaldehyde concentration subsequentlycould be lowered considerably before precipitation occurred (Baker et al., 1981).The MS level required obviously depends also on the polar liquid employed.Besides DMSO, DMA/LiCl and DMF/LiCl represent good solvents for methy-lolcellulose.
The methylol groups are easily split-off by water or methanol. Already asmall amount of water is reported to increase significantly the gel content of amethylolcellulose solution in DMSO/paraformaldehyde (Gruber and Gruber,1978). Methylolcellulose in the dissolved state with its strongly solvated butchemically unstable hydroxymethyl groups, has been employed in several stud-ies for subsequent steps of derivatization. By Kinstle and Irving (1983) acetyla-tion with acetyl chloride, ionic grafting of acrylonitrile with sodium hydride, andthe synthesis of a methylolcellulose octadecylcarbamate by reaction with oc-tadecyl isocyanate in the presence of stannic octoate at 50 0C in DMF/LiCl asthe solvent are reported. Sulfuric acid half-ester formation can be accomplishedwith the SO3/DMF complex, but predominantly takes place at the methylol hy-droxy end groups, and most of the sulfur introduced is split-off with the sidegroups in an aqueous medium. Periodate oxidation of methylolcellulose in anaqueous medium was reported to proceed to a high degree of 2,3-dialdehydecellulose formation with gradual decomposition of the methylol groups(Morooka et al., 1989).
Methylolcellulose in DMSO/paraformaldehyde exhibits a very high intrinsicviscosity, exceeding that of cellulose in FeTNa (Gruber and Gruber, 1978). Therelation
[η] (cm3/g) = 3.38 χ K)-2 DP °·84W
has been reported (Baker et al., 1981). The existence of chain aggregates, evenin very dilute solutions, cannot be excluded (Gruber and Gruber, 1978). At apolymer concentration above 18 %, solutions of methylolcellulose in DMSOexhibit interesting liquid crystalline properties (Gilbert and Fornes, 1989), withthe optical data confirming the heterogeneity of the chemical structure along thepolymer chains. A methylolcellulose-based route to artificial fibers has beendeveloped with the cellulose/DMSO/paraformaldehyde system on a semitechni-cal scale, but did not reach the level of industrial production, and obviously can-not compete today with the development of the amine oxide spun fibers.

4.5.3 Various functionalized alkyl ethers of cellulose 249
Methylolcelluloses functionalized at the acetal group have been synthesizedwith various aldehydes or aldehyde derivatives: trichloroacetaldehyde (chloral)is known to dissolve cellulose in the presence of a dipolar aprotic liquid withformation of a substituted methylol derivative.
QCeII-OH + CCI3-C* CeIhO-CH-OH
π ιCCI3
The formation of various cellulose hemiacetals by reaction of the polymer dis-solved in DMA/LiCl with the dimethylacetals of benzaldehyde, cyclohexanoneand acetaldehyde has been studied (Ikeda et al., 1990), e.g.
CeII-OH + CH CeII-O-CH-OMe +MeOH
)
On increasing the temperature to 70 0C and removal of the methanol formed invacuo, a DS of acetal groups of up to 2 could be obtained. A reversible bimol-ecular reaction between cellulosic hydroxy groups and the dimethylacetal isassumed.
Finally, the formaldehyde viscose spinning process shall be mentioned brieflywith the 5-methylol derivative of cellulose xanthogenate:
CeII-O-C-S-CH2OHS
affecting filament formation and filament structure. This compound, as well assome transient -Q-CH2-O- ether crosslinks are formed on adding formaldehydeat the 0.1-1 % level to the viscose spinning dope or to the spin bath. In this way,cellulose xanthogenate decomposition is retarded, and the stretchability of thenascent filament is enhanced, resulting finally in a high tear strength and achanged morphology of the threads (Bartsch et al., 1974).
4.5.3 Various functionalized alkyl ethers of cellulose
Besides the hydroxyalkylcelluloses and CMC, several other functionalized alkylethers of cellulose, e.g.
cyanoethylcellulose: CeIl-O-CH2-CH2-C=Naminoethylcellulose: CeIl-O-CH2-CH2-NH2
sulfoethylcellulose: CeIl-O-CH2-CH2-SO3Hphosphoromethylcellulose: CeIl-O-CH2-PO3H2

4.5.3 Various functionalized alkyl ethers of cellulose 249
Methylolcelluloses functionalized at the acetal group have been synthesizedwith various aldehydes or aldehyde derivatives: trichloroacetaldehyde (chloral)is known to dissolve cellulose in the presence of a dipolar aprotic liquid withformation of a substituted methylol derivative.
QCeII-OH + CCI3-C* CeIhO-CH-OH
π ιCCI3
The formation of various cellulose hemiacetals by reaction of the polymer dis-solved in DMA/LiCl with the dimethylacetals of benzaldehyde, cyclohexanoneand acetaldehyde has been studied (Ikeda et al., 1990), e.g.
CeII-OH + CH CeII-O-CH-OMe +MeOH
)
On increasing the temperature to 70 0C and removal of the methanol formed invacuo, a DS of acetal groups of up to 2 could be obtained. A reversible bimol-ecular reaction between cellulosic hydroxy groups and the dimethylacetal isassumed.
Finally, the formaldehyde viscose spinning process shall be mentioned brieflywith the 5-methylol derivative of cellulose xanthogenate:
CeN-O-C-S-CH2OHS
affecting filament formation and filament structure. This compound, as well assome transient -Q-CH2-O- ether crosslinks are formed on adding formaldehydeat the 0.1-1 % level to the viscose spinning dope or to the spin bath. In this way,cellulose xanthogenate decomposition is retarded, and the stretchability of thenascent filament is enhanced, resulting finally in a high tear strength and achanged morphology of the threads (Bartsch et al., 1974).
4.5.3 Various functionalized alkyl ethers of cellulose
Besides the hydroxyalkylcelluloses and CMC, several other functionalized alkylethers of cellulose, e.g.
cyanoethylcellulose: CeIl-O-CH2-CH2-C=Naminoethylcellulose: CeIl-O-CH2-CH2-NH2
sulfoethylcellulose: CeIl-O-CH2-CH2-SO3Hphosphoromethylcellulose: CeIl-O-CH2-PO3H2
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

250 4.5 Etherification of Cellulose
and related compounds have been studied. Some of them have found a limitedpractical application. The synthesis of these functionalized ethers takes placealong the routes already discussed, i.e. by reaction with an alkyl halide, or by aMichael addition of a compound with an activated C=C bond onto a cellulosichydroxy group. Ordered according to the functional group in the end-product,this section presents an overview on the mode of preparation, the consecutive reac-tions, the properties, and the potential areas of application of these cellulose ethers.
4.5.3.1 Cyanoethylcellulose and related cellulose ethers
Among the functionalized alkyl ethers of cellulose to be considered in this sec-tion, cyanoethylcellulose and some related derivatives have been most widelystudied due to their scientific relevance for cellulose ether formation and cellu-lose ether consecutive reactions and due to their practical importance in the fur-nishing of cellulosic textiles.
Cyanoethylation is the classical example of cellulose etherification by Mi-chael addition of an activated C=C bond to a partially anionized cellulosic hy-droxy group in an aqueous alkaline medium. A simple scheme of this reaction ispresented in Fig. 4.5.22.
The mesomeric structure of the anionic ether primarily formed should in prin-ciple permit a subsequent anionic grafting of acrylonitrile onto the polymerbackbone, which, however, is inhibited by the very fast addition of a proton toform the neutral cyanoethyl ether.
— OH® ^CeII-OH ^=^ CeII-OI +H2O
H® ~
CeII-OI + CH2-CH-C = N ^ CeII-O-CH2-CH-C = N
+ H®
CeII-O-CH2-CH2-C = N
Figure 4.5.22. Scheme of cyanoethylcellulose formation.
Besides the C=N group, several other substituents are able to activate the C=Cbond to an extent sufficient for the addition reaction. Typical examples of rea-gents are:
• Acrylonitrile · Acrylamide• Methacrylonitrile · oc-Methyleneglutaronitrile• oc-Chloroacrylonitrile · irarcs-Crotonitrile• Allyl cyanide · Vinyl sulfonate, acrylic acid esters

4.5.3 Various functionalized alky I ethers of cellulose 251
A decreasing order of reactivity has been reported according to acrylonitrile > a-methylene glutaronitrile = croton nitrile = allyl cyanide > methacrylonitrile,corresponding to a decreasing polarizability of the C=C bond (Lukanoff et al.,1967). α-Chloroacrylonitrile was also found to be less reactive than acrylonitrileitself (Lukanoff et al., 1969).
Numerous systematic investigations have been performed on laboratory-scalecyanoethylation of cellulose in order to assess the relevance of the various reac-tion parameters and to elucidate the interaction between cyanoethylcelluloseformation and its various routes of decomposition. The pioneering work of Bi-kales shall be mentioned explicitly. He reported (Bikales, 1974) the preparationof a fibrous cyanoethylcellulose of DS = 2.75 (12.6 % N) from regenerated cel-lulose with acrylonitrile and aqueous NaOH at 50 0C. The results of numeroussubsequent studies can be summarized as follows: cyanoethylation of celluloseproceeds as a equilibrium reaction usually with the polymer remaining in thesolid state and can be performed either by simultaneous (one-step process) or bysubsequent (two-step process) addition of the components aqueous NaOH andacrylonitrile, usually at a temperature between 30 and 50 0C, within some hours.At low alkali concentration and a temperature not higher than 30 0C a hydrolytic
cleavage of the C=N bond can be widely avoided, which is favored at higherOH~ concentration and higher temperature. As a low molecular by-product, 2,2 '-dicyanodiethylether is formed by cyanoethylation of water to an extent largelydepending on the reaction conditions. In the low DS range, a preferential substitutionat the C-6 position has been reported (Nehls et al., 1994).
Cyanoethylation of cellulose in the fibrous state at a low alkali concentrationof e.g. 2-4 % was found to depend considerably on cellulose physical structurewith regard to rate and final DS of etherification (Lukanoff et al., 1979). Thereaction rate increased significantly after a preactivation of the cellulose sampleby mercerization with 18 % NaOH or by pretreatment with liquid NH3 (see Fig.4.5.23), and could also be enhanced by addition of DMSO to the system forincreasing the solubility of acrylonitrile (Schleicher et al., 1974).
The efficiency of the activating pretreatment increased with its effect on su-pramolecular order and with decreasing NaOH concentration in the system. Arate difference of about one order of magnitude has been observed between astrictly homogeneous and a strictly heterogeneous course of reaction, while theorder of reactivity of various vinylic compounds remained the same in bothcases. An energy of activation of 15.5 kcal/mol for the homogeneous and of11.7 kcal/mol for the heterogeneous course of reaction was reported (Lukanoffet al., 1979).

252 4.5 Etherification of Cellulose
3,0
2,0
COQ
1,0
10 30 50 70 90Time [min]
110
Figure 4.5.23. Course of cyanoethylation of beech sulfite pulp in 2 % aqueous NaOH:(·) pulp without preactivation, (O) pulp activated with liquid NH3 (Lukanoff et al, 1979).
The heterogeneous course of cyanoethylation of a preactivated cellulose sam-ple can turn to a homogeneous one after a brief initial reaction phase due to for-mation of an alkali-soluble cyanoethyl ether above a DSN of 0.3-0.4, which canthen be further etherified under homogeneous conditions, until above a DS ofabout 1.2 precipitation occurs in the aqueous system and further cyanoethylationtakes place at the solid polymer (Koura et al., 1977). A completely homogene-ous course of reaction up to a DS^ of 0.8 could be realized after dissolving thecellulose in 7V-methylmorpholine TV-oxide with the small amount of N-methylmorpholine present in the system already sufficing for the basic catalysisof cyanoethylation (Philipp et al., 1986). It is worth mentioning that alkali-soluble products in the DS region of about 0.4 and water-soluble cyanoethylcel-lulose in the DS range between 0.7 and 1.0 required for their preparation a suit-able preactivation of the starting material for securing a sufficiently uniformsubstituent distribution along the polymer chains. Cyanoethylcellulose obtainedin a thoroughly homogeneous procedure exhibited organosolubility already at aDS above 0.8, while a DS above 2.0 was necessary along a heterogeneous routeof synthesis. It can be concluded from these results that the solubility of cyano-ethylcellulose in various liquids is primarily governed by the DS, as to be ex-pected, but also that the substituent distribution plays an important role.
A combined cyanoethylation and xanthation by simultaneous action of acry-lonitrile and CS2 onto a conventional alkali cellulose resulted in an enhancedxanthation rate and a higher final xanthogenate DS, possibly due to a combinedspacing and CS2 solubilizing action of cyanoethyl side groups, as demonstratedby a DSx of > 0.9 and a D5N of 0.3, with a reagent input of 4 mol of CS2 and1 mol of acrylonitrile/mol of AGU (Lukanoff et al., 1969).
Acrylamide can be added to cellulose in a similar manner as acrylonitrile withformation of the carbamoylethyl ether of cellulose:

4.5.3 Various functionalized alkyl ethers of cellulose 253
ο ο'' OH" "
CeII-OH + CH2 = CH-C^ -^- CeII-O-CH2-CH2-C^NH2 NH2
The reactivity, however, of acrylamide in a Michael addition is significantlylower than that of acrylonitrile, and the amide group is more easily saponified toa carboxyl group than the nitrile group.
The functional groups of cyanoethyl and carbamoylethylcellulose are suscep-tible to various consecutive reactions. Most important is the decomposition in anaqueous medium of higher alkalinity at elevated temperature to give car-boxyethylcellulose as the stable end-product, with free acrylonitrile and car-bamoylethylcellulose acting as intermediates (see scheme in Fig. 4.5.24).
Main reaction(NaOH)
CeII-OH + CH2 = CH-C=N · CeII-O-CH2-CH2-C=N
Side reaction(NaOH) *°
/^U -OU-^ = M - »- OU -/"^U- Γ*L/n2-On W — IN u ^ ΟΠ2-ΌΠ O^
-^MU + NaOH' CH2=CH-COONa + NH3INlM2
xx° (NaOH) *°CeII-OH + CH2 = CH-C. — CeII-O-CH2-CH2-C.
NH2 NH2
CeII-O-CH2-CH2-Ct. + NaOH CeII-O-CH2-CH2-COONa + NH3NH2
CH2 = CH-C = N +H2O (NaQH) HO-CH2-CH2-C = N
(NaOH)2 CH2 = CH-C = N +H2O ^ }— (NCCH2CH2J2O
Figure 4.5.24. Main and side reactions in the formation of cyanoethylcellulose.
The rate constant of cyanoethylcellulose cleavage was observed to increaselinearly with the sodium hydroxide concentration (see Fig. 4.5.25).
As another route from cyanoethylcellulose to carbamoylethylcellulose, oxida-tion with H2O2 in an aqueous alkaline system has been reported.

254 4.5 Etherification of Cellulose
E x103
CJOω§
Ä 0,5 1,0 1,875 2,5oz NaOH concentration [mol/l]
Figure 4.5.25. Rate constant of cyanoethylcellulose cleavage versus NaOH concentra-tion (Lukanoff et al., 1977).
According to Englebretsen and Harding (1992) the nitrile group of cyanoeth-ylcellulose can be reduced to an aminopropyl substituent with diborane. Anamidoxime has been prepared from carbamoylethylcellulose with hydroxyla-mine in a neutral aqueous system at 70 0C (Kubota and Shigehisa, 1995).
The technical process of cyanoethylation usually aims to produce not a com-mercial product 'cyanoethylcellulose', but a furnishing of cellulosic textilegoods or of paper. In the so-called 'two-step process' the cellulose is firstly im-pregnated with aqueous NaOH of e.g. 2 % concentration for l h at 50 0C, andthen reacted with an excess of acrylonitrile at the same temperature. The reac-tion is stopped by addition of acid and the fibrillar material, usually containing3-4 % nitrogen corresponding to a DS of about 0.5, is washed free of by-products and dried. In the One-step process' often employed for obtaininghigher degrees of cyanoethylation, the cellulose is soaked with NaOH and acry-lonitrile at low temperature, which is then raised to a level of about 50 0C for theetherification. The One-step process' was found suitable to obtain DS valuesabove 2 resulting in organosoluble products. Modifications of these two princi-ple process routes are a continuous procedure for cotton cloth with an impregna-tion with aqueous NaOH as the first step and an etherification with acrylonitrilein the gaseous state in a reaction chamber as the second one, and also a so-calledhigh solids process with small amounts of acrylonitrile and aqueous NaOH beingreacted with the cellulose.
Regarding material properties, cyanoethylation results in an improved rotresistance already at low DS, in an enhanced thermoresistance, and more favor-able dielectric properties compared with cellulose. Cyanoethyl ethers of cellu-lose exhibit a graded solubility in aqueous NaOH, water and polar organic liq-uids, including acrylonitrile, depending on the DS and on the procedure ofetherification. Cyanoethylcellulose of high DS becomes thermoplastic at a tem-perature of about 160 0C and is very hydrophobic. The latter property can stillbe enhanced by introduction of additional fluorinated substituents.
The main area of application of cyanoethylation is the rot proof furnishing ofcellulosic textiles, especially of cotton cloth. Besides this, cyanoethylated prod-

4.5.3 Various functionalized alkyl ethers of cellulose 255
ucts are used for speciality papers. Films cast from cyanoethy!cellulose solutionshave been recommended as separation membranes due to their enhanced rottingand chemical stability compared with e.g. cellulose acetate (Chen et al, 1991).The blood clotting efficiency of water-soluble carboxyethyl-carbamoylethylcellulose has been studied (Kamide et al., 1987).
4.5.3.2 Functionalized cellulose ethers with basic N-functions
The chemically most simple route to cellulose ethers with an amino group is thereaction with ethylene imine in an aqueous alkaline medium arriving at amino-ethylcellulose according to
CeII-OH + CH2-CH2 ^ CeII-O-CH2-CH2-NH2\ /NH
quite by analogy to hydroxyethylation with ethylene oxide. This route was prac-tised in the first half of this century for an amination of viscose rayon to low DSin order to improve dyeability, but is now abundant due to the toxicologicalhazards involved.
Subsequently, the aminoethylation of cellulose found limited attention in thepreparation of weak anion exchangers to be used in various Chromatographietechniques, e.g. affinity chromatography, or in connection with enzyme immo-bilization. The aminoethyl ether group was introduced by reaction with eitherethylene imine or 2-aminoethyl sulfate. A procedure for the first route is de-scribed by Podgornyi and Gur'ev (1981) employing cellulose suspension intoluene and reacting it with ethylene imine in the presence of benzyl chloride inan autoclave at 70 0C for 1Oh. Aminoethylation with 2-aminoethyl sulfate in thepresence of aqueous NaOH at a temperature between 70 and 120 0C was claimedfor obtaining ion-exchange materials from cellulose powders (Bischoff andDautzenberg, 1977). The reaction proceeds according toCeIl-OH + NaO3SO-CH2-CH2-NH2 + NaOH -»CeIl-O-CH2-CH2-NH2 + Na2SO4
The primary amino group of aminoethy!cellulose can serve as a reactive site insubsequent transformations for the reaction with N-acetylhomocysteinethio-lactone in order to obtain tailored Chromatographie sorbents (Podgornyi andGur'ev, 1981).
More recent developments were centered not so much on compounds withprimary amino groups, but predominantly on cationic alkyl ethers with tertiaryamino functions or quaternary ammonium groups. A large number of synthesisroutes and a broad variety of products has been described. Amino functionaliza-tion of cellulose became a very attractive area of organic cellulose chemistry,

256 4.5 Etherification of Cellulose
although only a very limited number of products is produced commercially as aspeciality in rather small amounts.
The most important route to cationic cellulose ethers is still the coupling of anN-functionalized compound onto the polymer via displacement of a labile halo-gen atom (Fig. 4.5.26). But also a coupling of cationic groups onto the polymervia an epoxidation is widely used (see Fig. 4.5.27).
Representatives of the first mentioned route are diethylamino-ß-chloroethaneemployed in the preparation of diethylaminoethylcellulose and 3-chloro-2-hydroxypropyltrimethylammonium chloride for introducing propyltrimethyl-ammonium chloride side chains into the polymer.
C2H5
CeII-OH + NaOH + CI-CH2-CH2-Nx
r μ C2H5^2M5
CeII-O-CH2-CH2-Nx + NaCI + H2OC2H5
CeII-OH + CI-CH2-CH-CH2-N(CHg)3 Cl' Na°H
OHCeII-O-CH2-CH-CH2-N(CHg)3
+ Cl' + NaCI + H2O
OH
CeII-O-CH2-C^1+ H2N-(CH2Jn-NH2L/l
o n>2
Cell-O-CH2-C-NH-(CH2)n- NH2+ HCI
CeII-CI + H2N-CH2-CH2-NH2
NaOH
CeII-NH-CH2-CH2-NH2 + NaCI + H2O
CeII-OH + Br-(CH2Jn-Br + NaOH
Cell-O-(CH2)n-Br + NaBr + H2O
CeII-O-(CH2X1-Br + H2N-(CH2Xn-NH2
Cell-O-(CH2)n-NH-(CH2)m-NH2 + HBr
Figure 4.5.26. Introduction of amino groups into cellulose via halogen functions (sim-plified scheme).

4.5.3 Various functionalized alkyl ethers of cellulose 257
The other routes shown above are primarily of scientific interest. They permithowever, the introduction of N-functionalized side chains with one or two aminofunctions and a controlled spacer length for potential application in Chroma-tographie techniques.
CeII-OH + CH2-CH-CH2 + NR3 — CeII-O-CH2-CH-CH2-NR3+ Cl'
Cl OH
CeII-OH + CH2-CH cat.+ oligomer —- CeII-O-CH2-CH cat.+ oligomer\/ OH
CeII-O-CH2-CH-CH2 + H2N-(CH2Jn-NH2
Cell-O-CH2-CH-CH2-NH-(CH2)n-NH2
OH
Figure 4.5.27. Routes of formation of cationic cellulose ether by linkage via epoxygroups (Gruber et al., 1996).
N-functionalization via an 4epoxy coupling' is often performed with gly-cidyltrimethylammonium chloride employing NaOH as a catalyst. A one-stepprocedure for modifying cellulose by substitution with quaternary ammoniumfunctions was recently published by Gruber et al. (1996), who reacted the poly-mer with epichlorohydrin and a tertiary amine in the presence of a stericallyhindered amine as the catalyst (Fig. 4.5.2.7). Also, cationic oligomers with ep-oxy coupling groups were employed by this author for pulp cationization (Fig.4.5.28).
Figure 4.5.28. Cationic epoxide oligomer for cationic cellulose ether formation (Gruberet al., 1996).
The introduction of tertiary and quaternary N-functions can also be realizedby the Michael addition of cationic acryl and methacryl esters or the corre-sponding substituted amides:

258 4.5 Etherification of Cellulose
CH2=CH /Me
O=C-O-CH2-CH2-N'x
CH2=C-CH3 Me
JH2-CH2-CH2-N
Me
O=C-NH-CH2-CH2-CH2-Nx
CH2—C CHg
O=C-NH-CH2-CH2-CH2-NMe3+ Cl"
Employing a low-DS Na-cellulose sulfate in aqueous alkaline solution(0.02 M NaOH) after a reaction time of 3 days at 35-60 0C, maximal D5N val-ues of 0.36 for the tertiary N-function and 0.27 for the quaternary ones wereobtained. Most probably substitution occurred preferentially at the C-2/C-3 po-sition. It must be emphasized that the reaction proceeded smoothly in an aque-ous system only, while in aprotic liquids like DMSO or DMF the D5N remainedbelow 0.1 (Wagenknecht, 1996).
Finally, two possible routes to aminoalkylation starting from cyanoethylcel-lulose shall be mentioned (see Fig. 4.5.29), which are of scientific interest butdemonstrate the feasibility of two well-known pathways of low molecular or-ganic chemistry at the cellulose macromolecule.
Cell-O-CH2-CH2-ctMUNn2
H2O " ^NaOB^
CeII-O-CH2-CH2-C^Nv THF CeII-O-CH2-CH2-NH2
BH3-SMeX
CeII-O-CH2-CH2-CH2-NH2
Figure 4.5.29. Reaction routes from cyanoethylcellulose to aminoalky!cellulose.
The products synthesized by the various routes of N-functionalization havealso been subjected to subsequent reaction steps, e.g. a quaternization of tertiaryamino groups by alkyl halide or dimethyl sulfate, a crosslinking by epichloro-hydrin or an additional substitution with anionic groups by carboxymethylation.
Besides cellulose itself, partially substituted cellulose esters and ethers, espe-cially HEC, hemicelluloses (predominantly xylans), and other polysaccharideslike amylose, have been N-functionalized, preferentially with diethylamino-ß-

4.5.3 Various functionalized alkyl ethers of cellulose 259
chloroethane or 3-chloro-2-hydroxypropyltrimethylammonium chloride. Ac-cording to Ebringerovä and Hromädkovä (1996), 2-hydroxypropyltrimethyl-ammonium groups were introduced into a beech wood hemicellulose dissolvedin NaOH and a DS of up to 1.0 has been arrived with the 2 position being thepreferred site at low DS. Katsura et al. (1992) prepared cationic ethers with terti-ary and quaternary N-functions from HEC, amylose and amylopectin dissolvedin aqueous NaOH, arriving at DS^ values up to 0.5 and concluding a preferentialsubstitution at C-2 from NMR results. A fairly regioselective cationization wasperformed with regioselectively substituted Na-cellulose sulfates dissolved inaqueous NaOH by reaction with 3-chloro-2-hydroxypropyltrimethy!ammoniumchloride (Wagenknecht, 1996). The results obtained with a preferentially C-2/C-3 substituted and a preferentially C-6-substituted cellulose sulfate in comparisonwith samples with a rather statistical distribution of the ester groups within theAGU are summarized in Table 4.5.14.
Table 4.5.14. Etherification of cellulose sulfuric acidhalf-ester with Cl-CH2-CHOH-CH2-NMe3Cl in excess.
Cellulose sulfateDS$ Preferential site of reaction
0.25 C-60.72 C-60.22 C-2/C-30.25 C-2/C-3/C-60.45 C-2/C-3/C-60.70 C-2/C-3/C-6
DSN
0.760.310.390.800.400.26
For preparing these cellulosic ampholytes, 4-8 mol of NaOH and 3-6 mol ofetherifying agent were employed in a reaction time of 4 h at 60 0C. Thus thereaction proceeds much faster than the Michael addition of cationic acryl estersonto the same cellulose sulfates. Also, from this study a preferential etherifica-tion at the C-2/C-3 position can be assumed.
Application-oriented research and development in the N-functionalization ofcellulose has so far been pursued by three routes:(i) preparation of anion-exchanging sorbents for Chromatographie purposes oflow DS (usually 0.1-0.2), starting from alkali cellulose or a slurry with e.g. iso-propanol added, retaining the solid structure of the polymer throughout the proc-ess and arriving at a water-insoluble product which eventually is additionallycrosslinked for reduced swelling;(ii) cationization of cotton or wood pulp for changing the surface properties in e.g.sorption processes, and maintaining, of course, the solid state of the polymer;

260 4.5 Etherification of Cellulose
(iii) synthesis of water-soluble cationic cellulose ethers as process auxiliaries ine.g. the paper industry or in water processing, usually proceeding in a homoge-neous system for securing a uniform substituent distribution along the chains,and either arriving at a rather high DS or employing already a water-solublestarting material like HEC.
A comprehensive contribution to (ii) was recently published by Gruber et al.(1996) who compared three routes of pulp cationization, i.e. the above-mentioned one-step quaternization with epichlorohydrin and a tertiary amine, theattachment of cationic oligomers with epoxy end-groups via ether linkages, andthe competing route of cellulose radical grafting with e.g. a combination of ac-rylamide and diallyldimethylammonium chloride, initiated with Ce4+. Theycompared these routes with regard to filler retention effect, beatability and sheetstrength in paper making.
Today's commercial manufacture of water-soluble cationized cellulose ethersas speciality products usually starts from a water-soluble HEC, which is chemi-cally modified either with glycidyltrimethylammonium chloride in an aqueousalkaline medium, or by radical grafting, employing preferentially diallyldimeth-ylammonium chloride as a cationic monomer. These products can be consideredas special types of hydroxyalkylcelluloses, which due to their cationic chargescan form polyelectrolyte complexes with anionic polymers or surfactants. So forexample a 200-fold increase in solution viscosity of an aqueous solution of so-dium dodecyl sulfate at the critical micelle concentration, by addition of a 1 %aqueous solution of a cationic cellulose ether, was reported (Gruber and Kree-ger, 1996). Hair cosmetics is considered today as a particular field of applicationof these cationic cellulose products.
4.5.3.3 Sulfoalkyl ethers of cellulose
Sulfoalkyl ethers of cellulose are prepared with cellulose in the presence of al-kali or with alkali cellulose at elevated temperature with(i) chloroalkane sulfonate according to
CeII-OH + CI-CH2-CH2-SO3 Na NaOH
CeII-O-CH2-CH2-SO3Na + H2O + NaCI
(ii) alkylation with, e.g. propane sultone, according to
CeII-OH + CH2-CH2^CH2 NaOH CeII-O-CH2-CH2-CH2-SO3Na +H2O
Xo-so2

4.5.3 Various functionalized alky I ethers of cellulose 261
(iii) ethylene sulfonate (vinyl sulfonate) by Michael addition according to
CeIhOH + CH2 = CH-SO3 H NaOH
CeIhO-CH2-CH2-SO3Na +H2O
Ebringerovä and Pastyr (1980) compared these three routes with delignifiedwood as the starting material and arrived at sulfur contents of 3.3-5.3 %, corre-sponding to a range of DS§ from 0.1 to 0.3 and an order of reactivity of theagents of propane sulfone < chloroalkyl sulfonate < vinyl sulfonate. An increasein the NaOH concentration and/or the temperature of reaction (> 65 0C) resultedin a higher DSS. The procedure of alkalization was found to be essential for thecourse of sulfoalkylation.
Sulfomethylcellulose has been prepared with Cl-CH2SO3Na in the presenceof aqueous NaOH of higher concentration at 60-90 0C, and was proposed as acation-exchanger.
Sulfoethylcellulose is synthesized by reaction of cellulose with Cl-CH2-CH2-SO3Na in the presence of strong alkali or by Michael addition of CH2=CH-SO3Na in an aqueous alkali medium. Also, a thionic acid HSO3-O-CH2-CH2SO3H has been proposed as an etherifying agent. The Na salt of Sulfoethyl-cellulose is soluble in water above a DS of 0.3, and less sensitive to precipitationby low molecular electrolytes than carboxymethylcellulose. Thermal degrada-tion of Sulfoethylcellulose and some related compounds has been studied up to60O0C (Oppermann, 1995; Sazanov et al., 1981). Etherification of cellulosewith divinyl sulfone CH2=CH-SO2-CH=CH2 in an aqueous alkali medium re-sults in an efficient crosslinking via CH2-CH2-SO2-CH2-CH2- ether linkages(Anbergen and Oppermann, 1990). For the preparation of sulfopropylcellulose,usually the route of propane sultone ring cleavage and addition in an aqueousalkaline medium is employed. Sulfoalkylcelluloses find limited application ascation-exchanger materials in Chromatographie techniques.
4.5.3.4 Miscellaneous functionalized alkyl ethers of cellulose
Phosphonomethylcellulose CeIl-O-CH2PO3H2 can be prepared by reaction ofalkali cellulose with Cl-CH2PO3H2 at 100-120 0C with a DS of up to 0.5. Theanionic ether becomes water-soluble above a DS of 0.15, but the solution is re-ported to be sensitive to low molecular electrolytes, e.g. H+ and OH" ions ofhigher concentration. Above pH = 10 the disodium salt is formed (Brandt, 1986).Also, HEC has been transformed to a mixed ether containing phosphoromethylgroups.
The formation of halogen-containing cellulose ethers of the structureCeIl-OH-(CH2)^-X, where n = 3, 4; X = Cl, Br, was reported by Klavins and

262 4.5 Etherification of Cellulose
Prikulis (1982) by using the dihalogenoalkane in dioxane or DMF at 80-100 0C.In a subsequent step, the ether was allowed to react with 1,6-diaminohexane inorder to obtain Chromatographie materials for affinity chromatography.
4.5.4 Aralkyl ethers and aryl ethers of cellulose
4.5.4.1 Arylmethyl ethers
As the most important ether of this type benzylcellulose was first reported byLeuchs in 1917. Further ary!methyl ethers contain different types of alkyl resi-dues (e.g. Koenig and Roberts, 1974), halogen substituents (e.g. Frazier andGlasser, 1995), and functional groups like methoxy, nitro, and amino groupsmainly in para-position of the benzyl units or in some cases aryl groups otherthan phenyl (Harkness and Gray, 1991; Isogai et al., 1985). The typical synthesispathway consists in the reaction of cellulose with the corresponding arylmethylhalogenides in presence of a base (e.g. Isogai et al., 1984, 1985):
CeII-OH+ HaI-CH2-Arbase
CeII-O-CHp-Ar
Ar =
CH(CH3)2
CH = CHn
OCHo
NH,
The reaction proceeds under heterogeneous conditions (Braun and Meuret, 1989;Obayashi et al., 1992; Zhdanov et al., 1993) using sodium hydroxide in water or,on the other hand, in an homogeneous medium, e.g. with sodium hydroxide inDMA/LiCl (McCormick and Shen, 1981; McCormick, 1978; Isogai et al., 1984;Kim, 1987). In some cases phase transfer catalyse techniques have been used(Daly et al., 1979; 1982; 1984).

262 4.5 Etherification of Cellulose
Prikulis (1982) by using the dihalogenoalkane in dioxane or DMF at 80-100 0C.In a subsequent step, the ether was allowed to react with 1,6-diaminohexane inorder to obtain Chromatographie materials for affinity chromatography.
4.5.4 Aralkyl ethers and aryl ethers of cellulose
4.5.4.1 Arylmethyl ethers
As the most important ether of this type benzylcellulose was first reported byLeuchs in 1917. Further ary!methyl ethers contain different types of alkyl resi-dues (e.g. Koenig and Roberts, 1974), halogen substituents (e.g. Frazier andGlasser, 1995), and functional groups like methoxy, nitro, and amino groupsmainly in para-position of the benzyl units or in some cases aryl groups otherthan phenyl (Harkness and Gray, 1991; Isogai et al., 1985). The typical synthesispathway consists in the reaction of cellulose with the corresponding arylmethylhalogenides in presence of a base (e.g. Isogai et al., 1984, 1985):
CeII-OH+ HaI-CH2-Arbase
CeII-O-CHp-Ar
Ar =
CH(CH3)2
CH = CHn
OCHo
NH,
The reaction proceeds under heterogeneous conditions (Braun and Meuret, 1989;Obayashi et al., 1992; Zhdanov et al., 1993) using sodium hydroxide in water or,on the other hand, in an homogeneous medium, e.g. with sodium hydroxide inDMA/LiCl (McCormick and Shen, 1981; McCormick, 1978; Isogai et al., 1984;Kim, 1987). In some cases phase transfer catalyse techniques have been used(Daly et al., 1979; 1982; 1984).
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

4.5.4 Aralkyl ethers and aryl ethers of cellulose 263
Moreover, benzylcellulose has been prepared by etherification of celluloseacetate (DS > 2) under simultaneous deacetylation (Shibata et al., 1983; Naka-mura, 1984) and primarily introduced substituents at the aryl units were subse-quently modified, e.g. nitro groups reduced to amino groups.
In the field of arylmethyl ethers benzylcellulose was of commercial impor-tance in the 1940th, particularly in Europe. It is a thermoplastic cellulose de-rivative with a melting range of 90 - 155 0C, insoluble in water, stable againstwater and strong bases and acids, and is soluble in organic solvents such as es-ters, hydrocarbons, and chlorinated carbons. Synthesis and properties of benzyl-cellulose have been described in detail in previous reviews (Braun and Meuret,1989; Engelskirchen, 1987). The typical heterogeneous synthesis leads to DSvalues of about 2.0, the homogeneous reactions to products with DS values up to3.0. Similar results have been obtained in case of benzylcelluloses containingfurther functional groups in the aryl moieties.
In the last years benzylcelluloses with a regioselective distribution of the ethergroups have been prepared using protective group methods (Isogai et al., 1984;Kondo, 1993; 1994). Based on such types of cellulose ethers with a well-definedmolecular structure, investigation on hydrogen bond systems were carried out byFTIR- and solid-state CP/MAS 13C NMR spectroscopy. Together with com-pletely etherified benzylcellulose and p-methylbenzylcellulose the 2,3-di-O-ether has been used in research on thermotropic mesophase properties(Kageyama et al., 1985).
To determined the degree of substitution in benzylcellulose and other cellu-lose derivatives with low DS (e.g. < 0.1) spectroscopic methods (UV/vis, Ra-man, near-IR) using multivariant data analysis have been described (Sollingerand Diamantoglou, 1996). Surface imaging of benzylcellulose containing in acellulosic dialysis membrane was carried out with confocal Raman microscopy.
4.5.4.2 Triphenylmethyl (6trityP) and related ethers
Tripheny!methyl ('trityl') cellulose is the most important ether of a class of or-ganosoluble cellulose derivatives that contain aromatic groups in the substitu-ents. Trity!cellulose, as well as benzhydryl(diphenylmethyl)cellulose, ben-zyl(phenylmethyl)cellulose and phenylcellulose, generally are formed by thereaction of phenylmethyl halides with cellulose in the presence of an organicbase. In order to gain considerable degrees of substitution, a sufficient activationof the starting cellulose material is absolutely necessary.
Only benzylcellulose was commercially available in the mid-1930s in largequantities and was applied as a basic substance to lacquers. At present thesecellulose ethers are mainly of scientific interest. Since the first preparation of O-tritylcellulose by Helfrich and Koester (1924), triphenylmethylation (tritylation)has become an intensively studied and useful method for a preferred substitution

264 4.5 Etherification of Cellulose
of primary hydroxy groups in cellulose chemistry. The products have been usedas intermediates in the synthesis of a number of selectively substituted cellulosederivatives as shown in detail below.
It was found already in the 1940s (Hearon et al., 1943) that heterogeneous trityla-tion in pyridine, using decrystallized cellulose regenerated from viscose, from cu-prammonium solutions, or especially from cellulose acetate by treatment with 15 %aqueous ammonia, yields colorless products with DS values from 0.81 to 1.21.
To evaluate the extent to which the trityl group is specific for the O-6 posi-tion, a chemical analysis was carried out. Following tritylation, the remainingfree hydroxy groups are covered by carbanilation with phenyl isocyanate. Sub-sequent detritylation and /?-toluenesulfonylation of the free hydroxy groupsformed leads to a completely substituted tosylcellulose carbanilate, which wastreated with sodium iodide for displacing the tosyl groups. Provided that thenucleophilic displacement takes place only at the tosyl esters of primary hydroxygroups (see also chapter 4.4.3.8), the content of iodine indicates the amount ofO-6 tritylation in the starting product. Starting from a sample of DS of tritylgroups of 1.03, shows that at least 90 % of the primary hydroxy groups weretritylated. It is noteworthy in this context that the selectivity of the reaction canbe influenced by the reaction time and the molar ratio of anhydroglucoseunits/trityl reagent, as illustrated by Honeyman (1947). The rate of reaction isinitially 58 times faster at the primary than at the secondary hydroxy groups.However, with increasing conversion of the primary hydroxy group this ratiodecreases and become equal at small amounts of remaining primary hydroxygroups. A summary of the early results was given by Green (1963) (see Table4.5.15).
Table 4.5.15. Selectivity of tritylation of regenerated cellulose.a
Reaction conditions Degree of substitution at:
Mol/molb
1.5
9.0
Timec
148
9614
2448
0-60.160.480.760.700.510.990.990.99
0-2/0-30.000.030.070.260.020.100.580.90
a Adopted from Green (1963, with permission), from cellulose acetate.b MoI of trityl chloride per mol of AGU.c Reaction temperature 100 0C.

4.5.4 Aralkyl ethers and aryl ethers of cellulose 265
Based on the mentioned investigations, Gray and Harkness (199Oa and 199Ob)synthesized 6- O-trity!cellulose heterogeneously in pyridine, starting from regen-erated, commercial cellulose acetate, which was deacetylated for 19 days with a15 % aqueous solution of ammonium hydroxide. It was verified by the absenceof a carbonyl stretching band in the infrared spectrum that no acetyl group re-mains on the polymer, as assumed by other authors (Hall and Home, 1973; Ha-giwara et al., 1981). In the Appendix an example of the preparation of 6-0-trity!cellulose with DS 0,97 is given (Gray and Harkness, 199Ob).
As described before, typical tritylation procedures for regioselective 6-O pro-tection of the AGU of cellulose use activated or regenerated cellulose as thestarting material and heterogeneous starting conditions (Helfrich and Koester,1924; Green, 1963; Yalpani, 1985). In this case, the tritylated celluloses becomesoluble during the reaction. In order to compare the reactivities of unsubstitutedand increasingly methoxy-functionalized trityl chlorides with each other, and toexclude some of the problems of a heterogeneous start of the reaction (solubilityof the polymers and accessibility of the hydroxy groups, for instance), CamachoGomez et al. (1996) used a homogeneous derivatization procedure. For this rea-son, the well-investigated DMA/LiCl cellulose solvent system (Dawsey andMcCormick, 1990) was selected (Fig. 4.5.30).
DMA / LiCI, pyridine
250C or 7O0C
R1
H H H
H H OCH3
H OCH3 OCH3
OCH, OCH, OCHo
R4 = H or C(C6H4R1) (C6H4R
2) (C6H4R3)
Figure 4.5.30. Tritylation of cellulose with trityl chloride and methoxy-substituted tritylchlorides.
As shown for the corresponding 4-methoxy-substituted diphenylmethyl chlo-rides (Erler et al., 1992b), the insertion of electron-donating substituents into thearyl moieties of triphenylmethyl chloride increases the rate of the reaction dras-tically. So, the reaction of unsubstituted trityl chloride with cellulose at 25 0C

266 4.5 Etherification of Cellulose
and at comparable reaction times leads to products with very low DS values,being insoluble in organic solvents. The insertion of one methoxy substituentinto the aryl moiety of the triphenylmethyl chloride, results in soluble cellulosederivatives (DS 0.7) after 24 h at 25 0C. The di- and trisubstituted chlorides givesoluble derivatives with DS values of about 1 after 24 h at 25 0C. In this way, thetritylation of cellulose at room temperature is possible for the first time. Table4.5.16 summarizes results of the homogeneous tritylation at 70 0C and of acidicdetritylation at room temperature.
Figure 4.5.31 shows the 13C NMR spectra of cellulose ethers with DS valuesof about 1, synthesized at 70 0C. The peak assignments were carried out basedon the assignments of Takahashi et al. (1986) for trityl- and methylcelluloses, aswell as on the peak assignments of the corresponding cellulose diphenyl ethers.
As no significant splitting of the peaks for the C-6 and C-I carbon atomscould be observed, it may be concluded that the tritylation in all cases proceedswith high 6-O-regioselectivity. Figure 4.5.32 shows the complete spectrum ofthe monomethoxytrityl ether.
Table 4.5.16. Tritylation of cellulose with methoxy-substituted trityl chlorides (3 mol ofreagent/mol of AGU, DMA/LiCl, 70 0C) and detritylation (37 % HCl aq. in THF, 1 : 25v/v at 25 0C) after subsequent permethylation (Camacho Gomez et al., 1996).
Substituent
Trityl
4-Monomethoxytrityl
4,4 '-Dimethoxy trityl
4,4',4"-Trimethoxytrityl
TritylationTime(h)
424484
2448
424484
2448
DS*
0.410.921.05
0.960.920.89
0.971.050.90
0.960.920.93
DSb
0.430.831.12
1.03—
1.03-1.17
—
—
—
-
Relativerate
1
2
2 XlO 5
6 XlO 6
Relativedetritylation
rate
1
18
100
590
a DS calculated from elemental analysis.b DS determined by gravimetry.
The described results demonstrated that soluble 6-0-triphenylmethy!cellulosewith DS values of about 1 were obtained at 25 0C using methoxy-substituted

4.5.4 Aralkyl ethers and aryl ethers of cellulose 267
triphenylmethyl chlorides in DMA/LiCl/pyridine. The tritylation of these de-rivatives is higher than 96 % at the 6-O position. Even after relatively long reac-tion times, using an excess of the reactant and/or at a higher temperature, thesubstitution at the 2-O and 3-O positions was less than 11 %, and comparablewith that of unsubstituted triphenylmethyl ethers of cellulose. The 4-methoxy,4,4'-dimethoxy- and 4,4',4"-trimethoxytriphenylmethyl groups can be removedfaster and under milder conditions than the unsubstituted trityl group. The intro-duction of just one 4-methoxy substituent into the trityl group caused a 10 timesfaster tritylation, as well as a 20 times faster detritylation. The 4-methoxy-substituted trityl groups are, therefore, useful tools for subsequent reactions inbasic media and for the synthesis of regioselectively 2-O- and 3-O-substitutedderivatives of cellulose. Typical examples are given in the Appendix for regio-selective carboxymethylation of cellulose.
C-2,3,5
100 80 606 [ppm]
Figure 4.5.31. 13CNMR spectra (DS ~ 1) of: (a) trityl-, (b) 4-monomethoxytrityl-, (c)4,4'-dimethoxytrityl-, and (d) 4,4',4"-trimethoxytrity!cellulose (Camacho Gomez et al.,1996).

268 4.5 Etherification of Cellulose
C-9,10,11 C-10'
lC-91
C-12
C-11'
C-7
C-1
LJL ~>
C-2,3,5
C-6
I . I . I . I , I , I
160 UO 120 100 80 60
6 [ppm]
Figure 4.5.32. 13C NMR spectrum of 4-(monomethoxtrityl)cellulose (Camacho Gomezet al., 1996).
A triphenylcarbinol (TPC)-moiety-containing cellulose derivative (TPC cellu-lose) was prepared by a two-step reaction (Arai and Kawabata, 1995). First,microcrystalline cellulose was dissolved in SO2/diethylamine/DMSO and ho-mogeneously reacted with /?-bromobenzyl bromide to obtain tn-O-(p-bromobenzyl)cellulose. Secondly, the tri-(9-(p-bromobenzyl)cellulose was re-acted with buty!lithium and then with Michlers ketone. The DS of the obtainedethanol-soluble TPC cellulose was up to 0.56. The leuco form of the TPC moi-ety in this cellulose showed a small extent of ionic dissociation in ethanol under
irradiation with UV light of λ > 290 nm, accompanied by a large degree of de-
composition of the structure. With additions of acid and then of alkali, the TPCstructure was reversibly isomerized from the leuco form to the colored form andthen from the colored form to the leuco form. However, repeated cycles of theadditions of acid and alkali resulted in considerable fatigue with the number ofcycles (Arai and Kawabata, 1995).
The preparation, as well as certain fine structural and thermal properties ofpartially benzhydrylated cotton cellulose (DS 0.31-1.22), have been described(Stanonis and King, 1960; Stanonis and Conrad, 1966; Stanonis et al., 1967;Cannizzaro et al., 1973). Their method employs mercerization and solvent ex-change to 2,3-lutidine/DMF before treatment with benzhydryl bromide at tem-peratures of 120 0C for various intervals of time.
Whereas the tritylation of cellulose with triphenylmethyl chloride inDMA/LiCl solution at 70 0C and with pyridine as a base proceeds over 48 h tosoluble products with DS values of up to 1 (see above), the reactivity of di-phenylmethyl chloride is insufficient under these conditions (see Fig. 4.5.33 andTable 4.5.17).

4.5.4 Aralkyl ethers and aryl ethers of cellulose 269
OCH3
10 30Time [h]
Figure 4.5.33. Conversion plots of some diphenylmethyl chlorides and triphenylmethylchlorides for cellulose etherification in DMA/LiCl at 70 0C using pyridine as a base.Broken lines denote tangents to curves at the origin (Erler et al., 1992b).
Provided that diphenylmethyl etherification of cellulose proceeds via a carbe-nium ion at the reaction center, the insertion of electron-donating substituentswill increase the rate of the reaction. The results obtained with mono- and di-methoxy-substituted diphenylmethyl chlorides, shown in Fig. 4.5.32 and Table4.5.20 (see later), give clear evidence for the influence of the methoxy substitu-ents.
The correlation of the reaction rate at the beginning of the conversion (tangent
to the conversion plot, -log A(DS)/AO versus the sum of the substituent con-
stants according to the Hammett equation (Σσΐ) results in a straight line. In
addition, p-chloro-substituted benzhydryl chloride does not react because of the
positive σΐ-value of the chloro substituent.
Furthermore, use of the most reactive dimethoxy compound enables the syn-thesis of diphenylmethyl ethers of cellulose in the DMA/LiCl solvent system at70 0C within an acceptable time. As shown with this reagent, THF- and 1,4-dioxane-soluble products were obtained after 8 h at 70 0C (DS = 0.83).

270 4.5 Etherification of Cellulose
Table 4.5.17. Comparison of the reactivity of some chlorides in the homogeneousetherification of cellulose (Erler et al., 1992b).
CeIl-OH + CI-C(R1R2R3) PMA/LiC' CeII-Q-C(R1R2R3)v ' (pyridme) 70 C v '
R1 R2 R3 DSa A(DS) h Ιοα A(DS
At
T=V /=V- H 0.02 3.00 χ 10"4 - 3.52
CH3OnQ- Q- H 0.33 2.25x10-2 -1.65
CH30V=V CH30V=V. H 1.01 0.66 -0.18
Q1V=V /°\_ H <0.01
/""V /"V. T=V. 0.83 0.13 -0.88
QP O- °·02 -
:) Σσ+ c
P
O
-0.78
-1.56
0.11
O
O
a DS values of the cellulose ethers formed after 24 h.b Calculated as demonstrated in Fig. 4.5.33.c Sum of substituted constants of the Hammett equation.
Figure 4.5.34 shows the ring carbon spectra of tritylcellulose and the meth-
oxy-substituteddiphenylmethylcellulose.
C-7C-U C-2.3.5
C-1
no 100 90 806 [ppm]
70 60
Figure 4.5.34. Ring carbon NMR spectra of bis(4-methoxyphenyl)methyl ethers (DS =1.06) of cellulose: for the peak data of C-atoms 8-12 of the aralkyl groups see Table4.5.18 (Erler et al., 1992b).
The peak assignments were carried out based on the assignment by Takahashi
et al. (1986) for trityl- and methy!celluloses. It may be concluded that the trity-

4.5 A Aralkyl ethers and aryl ethers of cellulose 271
lated polymer is close to 6-O-trity!cellulose, which confirms that trityl chloridemainly reacts with primary hydroxy groups. Since the splitting of the peaks ofC-I and C-4 carbon atoms (C- V and C-4") indicates substituents at C-2 and C-3, the spectrum of the dipheny!methylated ether demonstrates a less regioselec-tive substitution in the case of bis(4-methoxyphenyl)methyl chloride.
The comparison of the signal intensities of each spectrum with one anotherenables the estimation of DS values to be made. The results are in agreementwith those obtained by means of elemental analysis. Furthermore, this estimationindicates a more effective substitution at C-6 than at C-2 and C-3 of the AGU ofthe diphenylmethyl ether. This limited regioselectivity of substitution may beattributed to the less steric hindrance of the modified diphenylmethyl chloride incomparison with trityl chloride during the reaction, as well as to the higher reac-tivity of the diphenylmethyl derivative caused by the electron-donating methoxysubstituents. The data for the 13C NMR peaks of the substituents in the cellulosederivatives are given in Table 4.5.18.
Soluble 4,4'-bis(dimethylamino)diphenylmethyl ethers of cellulose with a DSfrom 0.5 to 1.0 were prepared by etherification of cellulose dissolved inDMA/LiCl.
The preparation was performed by reacting cellulose in DMA/LiCl with 4,4'-bis(dimethylamino)diphenylmethyl chloride at 50 0C within 24 h, using trieth-ylamine as a base. Depending on the molar ratio of the etherification agent perAGU, soluble products of DS = 0.54 (2 mol/mol of AGU; soluble in DMF, andDMSO) and DS = 1.05 (4 mol/mol of AGU; soluble in DMF, DMSO, THF,acetonitrile and 1,4-dioxane) were obtained. As already shown, the unsubstituteddiphenylmethyl chloride does not react with cellulose under the same reactionconditions (Erler et al, 1992b). Thus the electron-donating dimethylamino sub-stituents at the aryl moieties increase the rate of reaction.
In comparison with pure cellulose two further new peaks at δ = 80.5 and δ =82.9 ppm indicate a C-2 and C-3 substitution. Furthermore, the splitting of thepeaks of C-I and C-4 carbon atoms (C-I" and C-4") demonstrates the substitu-tion at C-2 and C-3 position, too. The comparison of the intensities of each sig-nal with one another enables the estimation of the substituent distribution withinthe AGU. This estimation indicates a more effective etherification at C-6 than atC-2 or C-3. The lower regioselectivity (in comparison with the well-known C-6regioselectively reacting tripheny!methyl chloride; Camacho Gomez et al., 1996)is due to less steric hindrance and higher reactivity caused by the electron-donating dimethylamino substituents.
These ethers show typical absorption spectra with an absorption maximum at265 nm. They are stable in neutral and basic media: under acidic conditions asignificant cleavage of the ether bonds occurs and thereby the formation ofMichlers Hydrol Blue (absorption maximum at 604 nm). Moreover, the dimeth-ylamino-substituted diphenylmethyl ether of cellulose was found to be photo-

K)
<l
K)
Tab
le 4
.5.1
8. R
esu
lts
of 1
3C
NM
R s
pec
tro
sco
py
of
solu
ble
cell
ulo
se e
ther
s (D
S =
0.5
8; 1
.06;
1.1
0);
δ in
ppm
,
in D
MF
-J7 (
Erl
er e
t a
l, 1
992b
).
7 1
Ce
ll -O
-7C
OC
H3
Cel
lulo
se e
ther
Dip
heny
lmet
hyl
4-M
etho
xydi
phen
yl
Num
beri
ng o
f C
ato
ms
of t
he a
ralk
yl g
roup
s7
8
9
10
1
1
12
8'
84 8414
4.7
129.
5
129.
0
128.
5a 55
.4-
55
.413
513
5.5 .6
912
912
8
' .6a
.9
10'
114.
211
4.2
11
'15
9.5
159.
4m
ethy
lB
is(4
-met
hoxy
- 87
14
4.7
12
9.4
12
8.5
12
7.5a
-ph
enyl
)met
hyl
a G
roup
of p
eaks

4.5.4 Aralkyl ethers and aryl ethers of cellulose 273
conducting, as film cast from a DMF solution for instance. Preliminary studieson the behavior of solutions under irradiation by means of flash photolysis andUV spectroscopy were carried out as well.
4.5.4.3 Arylethers
The introduction of an aromatic substituent via an ether bond leads to cellulosearyl ethers, especially to phenyl- and substituted phenylcelluloses. Two synthetispathways are suitable: the etherification of cellulose with activated aryl halo-genides (A) and the displacement reaction of cellulose tosylate with corre-sponding phenolates (B). Typical examples are presented in the followingscheme (Gavlik and Tokar, 1989; Harper, 1982; Mair et al, 1986; Farah andAwdeh, 1972; Avny et al., 1972; Strauss et al., 1987a, b):
K ο op
CeIl-OH+Ar-HaI CeII-O-Ar (A)
TosCI
Cell-O-Tos + Ar-O0
Ar =
-TosO®(B)
NH,
COOH

274 4.5 Etherification of Cellulose
4.5.5 Silyl ethers of celluloseSilicon compounds are well established in organic chemistry and especially inorganic synthesis. In general, the silylation of an organic compound leads to aremarkable increase in its lipophilic behavior, as well as to a drastic increase inthermal stability. With regard to these properties, silylated compounds are verysuitable for gas Chromatographie analyses. The total silylation of, e.g., D-glucose, leads to a pentasilyl ether that is suitable for distillation.
Because of the simple and selective removal of the silicon-containing struc-tural units from the original organic compounds, different types of silyl groupsare well known as regio- and stereoselective protecting groups in organic syn-thesis.
The silylation of cellulose and cellulose derivatives is a suitable way to pre-pare triorganosilyl ethers of different DS values and of different regioselectivity.The most important silylation agent is chlorotrimethylsilane (trimethylsilyl chlo-ride TMS-Cl), well-known and accessible from Müller Rochow synthesis. Butup to now, a wide range of further types of silylation reagents have been used incellulose silylation. Table 4.5.19 summarizes typical examples including reac-tion conditions and DS values.
The reaction of cellulose with TMS-Cl in the presence of pyridine has beendescribed for the first time by Schuyten et al. (1948) preparing O-trimethylsilylcellulose as an insoluble but swellable polymer. It is assumed thatthe insolubility was the result of crosslinking with higher chlorinated silanes asimpurities in the reagent used. With purified TMS-Cl completely soluble tri-methylsilylcelluloses (TMS celluloses) are formed.
Using the chlorosilane method and pyridine as the HCl acceptor TMS cellu-lose with DS values of 2.4 to 3.0 are accessible (see Table 4.5.19). In addition,Klebe and Finkbeiner (1969) have prepared cyanopropyldimethyl-, pheny!di-methyl- and diphenylmethylsilylcelluloses (see Table 4.5.19). The silylation ofpartially modified cellulose acetate with TMS-Cl/pyridine proceeded only at thefree hydroxy groups. In liquid ammonia as the reaction media, Greber andPaschinger (1981a and 198Ib) prepared partially etherified TMS cellulose withDS values of around 1.5. Because of the precipitation of the increasingly lipo-philic silylated polymer from the highly polar medium no further silylation takesplace even with a large excess of the reagent (see Table 4.5.19). As described byGreen (1983) and Schempp et al. (1984), hexamethyldisilazane is a convenientreagent for the trimethylsilylation of cellulose. The reaction requires polar sol-vents and the catalysis of NH4Cl or TMS-Cl/pyridine. To avoid a spontaneousdesilylation under the described silylation conditions, residual pyridinium chlo-ride, as well as ammonium chloride have to be removed, e.g., by dried sodiumcarbonate.
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

4.5.5 Silyl ethers of cellulose 275
With regard to a regioselective modification of cellulose via trialkylsilyl in-termediates, the synthesis of soluble trialkylsilylcelluloses with a wide and ad-justable DS and regiocontrol of the silyl group distribution was of importance.Therefore, the influence of the dispersity of cellulose in the reaction media, ofsolvents, of temperature, of the molar ratio of reagent, and of different types oftrialkylchlorosilanes were under investigation (Stein, 1991). From this point ofview, especially the silylation of cellulose decrystallized with ammonia andsuspended in an aprotic dipolar solvent, has been compared with the silylation ofcellulose dissolved in DMA/LiCl solution.

Tab
le 4
.5.1
9. T
ypic
al e
xam
ples
of
synt
hesi
s pa
ths
for
trio
rgan
osily
!cel
lulo
ses
and
subs
eque
nt d
eriv
ativ
es.
Sily
latio
n ag
ent
Am
ount
3 R
eact
ion
cond
ition
sD
SR
efer
ence
sSo
lven
t T
empe
ratu
re T
ime
(0C)
(h)
(CH
3)3S
iCl/p
yrid
ine
(CH
3)3S
iCl/p
yrid
ine
(CH
3)3S
iCl/p
yrid
ine
(CH
3)3S
iCl/p
yrid
ine
(CH
3)3S
iCl/p
yrid
ine
(CH
3)3S
iCl/N
H3(
liqui
d)
(CH
3)3S
iCl/N
H3(
liqui
d)
NC
(CH
2)3S
i(C
H3)
2Cl/p
yr
2.7
2.7
1.5
3C 2 1 3
idin
e 2.
4
pyri
dine
pyri
dine
xyle
neto
luen
epe
trol
eum
ethe
rN
H3(
liqui
d)
NH
3(liq
uid)
pyri
dine
110
110
110
105-
110
15
-70
-70 13
0-14
0
3 3 4 1 3 4 4 5
2.0
0.65
/0. 1
6b
2.46
2.82
2.9-
3.0
1.46
1.50
2.64
Schu
yten
et a
l., 1
948
Schu
yten
et a
l., 1
948
Kle
be,
1968
aK
lebe
, 19
68b
Kei
lich
et a
l., 1
968
Gre
ber
and
Pasc
h-in
ger,
198
IaG
rebe
r an
d Pa
sch-
inge
r, 19
8 Ib
Kle
be a
nd F
inkb
eine
r,
(C6H
5)2S
i(C
H3)
Cl/p
yrid
ine
1.3
(C6H
5)2S
i(C
H3)
Cl/p
yrid
ine
1.3
[(C
H3)
3Si]
2NH
/(C
H3)
3SiC
l/ 1.
3py
ridi
ne[(
CH
3)3S
i]2N
H/N
H4C
l 1.
1
pyri
dine
13
0-14
0 2
2.5
pyri
dine
13
0—14
0 15
2.
8
DM
F
DM
F
100
1-3
1.1-
1.6
110
8 2.
19
1969
; Fin
kbei
ner a
ndK
lebe
, 196
9K
lebe
and
Fin
kbei
ner,
1969
; Fin
kbei
ner
and
Kle
be, 1
969
Kle
be a
nd F
inkb
eine
r,19
69; F
inkb
eine
r an
dK
lebe
, 196
9C
oope
r et
al.,
198
1
Gre
en 1
983
Tab
le 4
.5.1
9. (c
ont.)
Sily
latio
n ag
ent
Am
ount
a R
eact
ion
cond
itio
nsD
SR
efer
ence
sSo
lven
t T
empe
ratu
re
Tim
e(0C
) (h
)
[(C
H3)
3Si]
2NH
/LiC
l/DM
F(C
H3)
3Si-
NH
-CO
-CH
3
NC
(CH
2)3S
i(C
H3)
2-N
(CH
3)-
CO
-CH
3
(C6H
5)2S
i(C
H3)
-NH
-CO
-CH
3
(CH
3)3S
i-N
=C
-CH
3
(CH
^Si-
O
1.1
3.3
1.25
1.3
1 1.05
LiC
l/DM
Fm
elt
./V-m
ethy
l-py
rrol
idon
e
W-m
ethy
l-py
rrol
idon
e
xyle
ne
benz
ene
80 170-
180
150
160-
170
150
85-9
6
1 1.5-
62 2 1 1
2.7-
2.9
2.65
-2.9
52.
64
2.5
2.73
0.65
d
Sche
mpp
et a
l., 1
984
Bre
dere
ck e
t al.,
196
9K
lebe
and
Fin
kbei
ner,
1969
; Fin
kbei
ner
and
Kle
be,
1969
Kle
be a
nd F
inkb
eine
r,19
69; F
inkb
eine
r an
dK
lebe
, 19
69K
lebe
and
Fin
kbei
ner,
1969
; Fin
kbei
ner
and
Kle
be,
1969
a
MoI
of
sily
latio
n ag
ent/m
ol o
f O
H g
roup
s;d
star
ting
mat
eria
l et
hylc
ellu
lose
DS -
2.25
.st
artin
g m
ater
ial
cellu
lose
ace
tate
DS -
2.30
/2.9
0; c
pre
trea
tmen
t w
ith
sacc
haro
se;

278 4.5 Etherification of Cellulose
4.5.5.1 Heterogeneous silylation of cellulose
Cellulose suspended and preswollen in W-methylpyrrolidone (NMP) at 80 0Chas been activated at -25 0C with ammonia dissolved in the mixture. After addi-tion of the bulky silylating agent thexyldimethylchlorosilane (TDMSCl), thecorresponding thexyldimethylsilylamine was formed. This intermediate shows asufficient silylation activity demonstrated by the reaction of cellulose dissolvedin DMA/LiCl after isolation and characterization of the silylamine. During thesubsequent silylation in the NH3TNMP medium the polymer dissolved. Afterisolation it is soluble in NMP and pyridine and swellable in organic solvents likeDMF and THF. The structure characterization demonstrated high 6-O regiose-lectivity, without detectable silylation at secondary hydroxy groups. As de-scribed earlier (Klemm and Stein, 1995; Klemm et al., 1995), DS values up to1.0 have been observed under similar conditions. In this case of high regioselec-tive heterogeneous reaction of cellulose, we assume a controlled activation andswelling suitable for preferred reactivity of the 6-OH groups with the bulky rea-gent. In the case of trimethylsilylation under the described conditions, and usingDMF, NMP or THF as solvent, the DS can be adjusted by the mole ratio ofTMS-C1/AGU with an exactness of 0.1 DS units (Fig. 4.5.35).
1 mol CI-Si(CH3)3/AGUCeII-OH - Cell-O-Si(CH3)3
(DMF/NH3) DS = Q 8
3,0 Ί
2,5-
2,0-
COQ
1,0-
0,0-1 2 3 4 5 6
MoI TMS-CI/mol AGU
Figure 4.5.35. Control of DS during silylation of cellulose in ammonia containing DMF(Stein, 1991).

4.5.5 Silyl ethers of cellulose 279
During the reaction, the increasingly silylated polymers dissolve. Using DMF,for instance, further silylation leads to precipitation in a highly swollen state.The effects are caused by the growing hydrophobicity of the polymer in relationto the polarity of the reaction medium. The DS range of the solubility of TMScelluloses changes from 0.4-1.6 (DMF) to 0.8-1.9 C/V-methylpyrrolidone) and1.8-2.9 (THF). These trimethylsilylations show a remarkable O-6 selectivity in alower DS range but the reaction proceeds to up to DS values of about 3.
4.5.5.2 Homogeneous silylation of cellulose
Cellulose was dissolved in DMA/LiCl (Dawsey and McCormick, 1990) andsilylated in the presence of pyridine with 1.2-6.0 mol of TDMSCl/mol of AGUat temperatures of 25 0C and 50 0C (see Table 4.5.20).
OH
' HO
Figure 4.5.36. Reaction scheme of regioselective silylation of cellulose with thexyl-dimethylchlorosilane.

280 4.5 Etherification of Cellulose
Up to DS values of 1.5 the products (Fig. 4.5.36) are soluble in the reactionmixture.
The structure characterization resulted in low regioselectivity of the homoge-neous silylation in comparison with the heterogeneous reaction as describedabove. All polymers prepared in DMA/LiCl solution contain increasing amountsof 2,6-di-O-thexyldimethylsilylated units (see Figs. 4.5.40 and 4.5.41).
This means, in the dissolved state even the bulky TDMSCl reacts with thesecondary groups at position 2. The DS values (D5EA, ^^HPLC) giyen in Table4.5.20 show the differences, especially at a higher level. The reason is the cal-culation of £>SEA by using the silicon content determined by elemental analysis.An excess of TDMSCl leads to thexyldimethyldisiloxane during the work-upprocedure, which forms a host-guest compound with the cellulose derivative,and a complete removal of thexyldimethyldisiloxane is impossible.
Table 4.5.20. Silylation of cellulose in DMA/LiCl solution (reaction time 24 h).
Reaction conditionsTDMSCl Temperature Yield Reprecipitation EAa
(mol/mol (0C) ( %) fromAGU)1.22.53.53.56.0
2525255050
82.082.581.076.296.0
DMFTHFTHFTHFTHF
1.031.531.942.112.11
DSHPLCb
1.071.511.641.771.90
a Based on the Si content determined by elemental analysis; based on thepermethylation analysis and calculation according to DS = 2 MFdi + MFmono
(MF = mole fraction).
4.5.5.3 Properties and structure characterization
The trimethylsily!celluloses are soluble in common organic solvents. Underconditions of spin coating and Langmuir-Blodgett techniques (see below) theyform films and ultrathin layers. Using a common spinneret, fibers are formed(Greber and Paschinger, 198Ic). In relation to DS^ they show solubility instrong polar solvents like DMSO and in nonpolar solvents like n-hexane (Fig.4.5.37).
The thexyldimethylsilyl (TDMS) celluloses were soluble in DMF (DS < 1),THF (DS > 1), and chloroform (DS > 1.5).
For characterization of regioselectivity the knowledge of the distribution offunctional groups within the AGU and along the polymer chains is essential.Therefore, suitable methods of structure analysis by 1H and 13C NMR spectros-

4.5.5 Silyl ethers of cellulose 281
copy, as well as by HPLC, after complete methylation and complete chain deg-radation have been developed (Erler et al., 1992a).
n-Hexan\
CH2Cl2-
B- Acetone,ethylacetate
O 0.5 1.0 15 2.0 2.5DS
Figure 4.5.37. Solubility of TMS celluloses in relation to D% (Klemm et al., 199Oa).
In the case of TMS cellulose, e.g., with DSsi 1.55 (Fig. 4.5.38), a complete6-O silylation and an additional O-2 and O-3 silylation have been observed by13C NMR spectroscopy.
C-2.2',3,5
C-1"
C-1
100 80 60 40 20 O6 [ppm]
Figure 4.5.38. 13CNMR spectrum of trimethylsilylcellulose, DS = 1.55, in DMF-J7
(Klemmetal., 199Oa).
In the case of silylcelluloses with bulky alkyl groups, an O-6 silylation andfree hydroxy groups in positions 2 and 3 could be demonstrated. As a typicalexample Fig. 4.5.39 shows the 13C NMR spectrum of teri.-butyldimethylsilyl-cellulose.
An important result of NMR spectroscopy in structural analysis of function-alized celluloses consists of two-dimensional 1H/1!! NMR techniques after sub-sequent derivatization of the original polymers. This method is suitable even forproducts with high molecular weight. In the case of the previously describedTDMS celluloses, the polymers were treated with sodium hydride and methyl

282 4.5 Etherification of Cellulose
iodide in THF solution, which leads to the completely methylated products (seeFig. 4.5.36). In this case it is additionally necessary to substitute all silyl groupsby acetate residues to get a better resolution of the spectra so that the1Hy1H COSY (homonuclear chemical shift correlation spectroscopy) techniqueis useful for peak identification. From this point of view, the obtained methyl-ated polymers were treated with tetrabutylammonium fluoride to remove theTDMS groups completely. The obtained methylcelluloses could be completelyacetylated with acetic anhydride in pyridine.
C-61
100 90 80 70 60ό [ppm]
Figure 4.5.39. 13CNMR spectrum of teri.-butyldimethylsilylcellulose, DS = 0.96, inDMF-J7 (Klemm et al., 199Oa).
In the case of HPLC analysis, the methylated silylcelluloses lead, with trifluo-roacetic acid and water, to the corresponding methylglucoses (mixture of ano-mers) by desilylation and chain degradation. The determination of the OSHPLCwas carried out after separating the methylglucoses by reversed-phase HPLC ona LiChrospher-aminephase column, which separates them into groups of un-modified, mono- and dlfunctionalized AGU. The mole fractions (MF) were cal-culated after integrating the corresponding peaks, £>SHpLc according to2 MFdi + MFmono. For instance, the peaks of 2,3-di-O-methylglucose indicatesilylation at position 6, that of 3-O-methylglucose indicating 2,6-di-O-silylatedAGU.
Figure 4.5.40 shows the structural characterization of two typical TDMS cel-luloses after the described subsequent modification. The NMR results demon-strate that the heterogeneously synthesized polymer 6-O-thexydimethylsilyl-cellulose (DS = 0.69, e.g.) leads to 6-O-acetyl-2,3-di-O-methylcellulose afterstepwise derivatization.
Only one peak is detectable of all the AGU protons. The double peak of theH-6 proton is caused by the diastereotopic effect of the neighboring asymmetriccarbon atom. The homogeneously prepared polymer 2,6-di-O-TDMS cellulose(DS, e.g. 1.02) leads to a nonuniform 6-O-acetyl-2,3-di-O-methyl-co-[2,6-di-<9-

4.5.5 Silyl ethers of cellulose 283
acetyl-3-0-methyl]cellulose after modification. In the 1H NMR spectrum par-tially acetylated hydroxy groups in position 2 lead to a second signal of thisproton which is shifted to higher field.
-2.8
-3.0
-3.2
-3.4
^3.6
-3.8
-4.0
-4.2
-4.4
ppm
10 ι 20 [min] 25 PPm 4.0 3.5
[mV]
80
60
40
20
O Ul__J
_^ttMkiH-21 H-1 H-34 H-3 H-2
8
·/.
$
y·&§8
ίβο
20 [min] 25 PPm
Figure 4.5.40. Structure analysis of heterogeneously (top) and homogeneously (bottom)synthesized TDMS celluloses by (right) 1H COSY NMR (CDCl3, 4O0C) after subsequentmethylation, desilylation and acetylation, as well as by (left) HPLC after complete meth-ylation and additional chain degradation: (1) silicon-containing noncellulosic by-products, (2) 3-<9-methylglucose, (3) 2,3-di-<9-methylglucose, (4) 2,3,6-tri-O-methyl-glucose (Koschella and Klemm, 1997).
Typical proton shifts result from methylated O-2 (2.89 ppm), acetylated O-2(4.79 ppm), methylated O-3 (3.20 ppm) and acetylated O-3 (3.34 ppm) posi-tions. In the corresponding chromatogram of the 6-O-silyl ether (Fig. 4.5.40,top) the peaks of 2,3-di-O-methylglucose show the exclusive silylation of theprimary hydroxy groups. Other positions are not affected by the TDMSCl. 2,3-Di-O-methylglucose indicates unmodified AGU in the polymer (with respect to
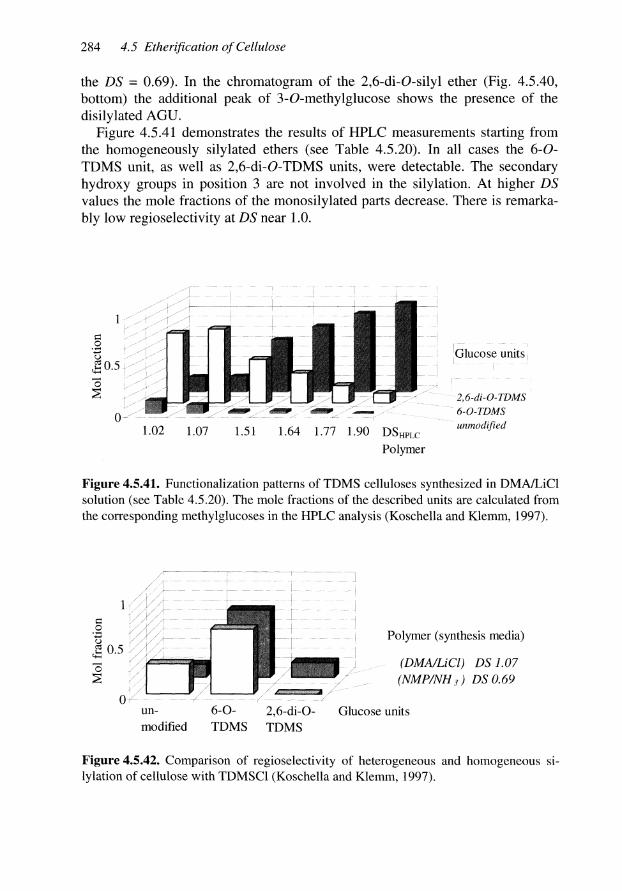
284 4.5 Etherification of Cellulose
the DS = 0.69). In the chromatogram of the 2,6-di-O-silyl ether (Fig. 4.5.40,bottom) the additional peak of 3-O-methylglucose shows the presence of thedisilylated AGU.
Figure 4.5.41 demonstrates the results of HPLC measurements starting fromthe homogeneously silylated ethers (see Table 4.5.20). In all cases the 6-0-TDMS unit, as well as 2,6-di-O-TDMS units, were detectable. The secondaryhydroxy groups in position 3 are not involved in the silylation. At higher DSvalues the mole fractions of the monosilylated parts decrease. There is remarka-bly low regioselectivity at DS near 1.0.
0.5Glucose units.
2,6-di-O-TDMS6-O-TDMSunmodified
Figure 4.5.41. Functionalization patterns of TDMS celluloses synthesized in DMA/LiClsolution (see Table 4.5.20). The mole fractions of the described units are calculated fromthe corresponding methylglucoses in the HPLC analysis (Koschella and Klemm, 1997).
0.5Polymer (synthesis media)
(DMA/LiCl) DS 1.07(NMP/NH3) DS 0.69
un- 6-0- 2,6-di-O-modified TDMS TDMS
Glucose units
Figure 4.5.42. Comparison of regioselectivity of heterogeneous and homogeneous si-lylation of cellulose with TDMSCl (Koschella and Klemm, 1997).

4.5.5 Süyl ethers of cellulose 285
The influence of the dispersity in the reaction media is shown in Fig. 4.5.42. Itcompares the silylation under heterogeneous and homogeneous conditions. Themost important result of this comparison is the absence of any 2,6-di-O-TDMSunits in the heterogeneously prepared polymer indicating the high regioselectiv-ity of silylation under these conditions.
In recent years the silylation of cellulose in liquid ammonia at higher tem-perature has been investigated (Mormann and Wagner, 1995). Using hex-amethyldisilazane as silylating agent, DS values are controlled by the hex-amethyldisilazane/OH equivalent. The reaction is completely heterogeneous andleads to pure trimethylsily!celluloses with DS values of up to 3. As by-productsonly ammonia is formed.
4.5.5.4 Subsequent reactions of silylcelluloses
As described before for analytical investigations, free hydroxy groups in thesilyl ethers of cellulose can be modified to ethers and ester functions withoutdesilylation. From this point of view the silyl residues can be used as protectinggroups.
Therefore, 2,3-substituted ethers of cellulose with DS values between 1.5 and2.0 can be advantageously prepared from 6-0-i-butyl and 6-O-TDMS celluloseby reaction with an appropriate alkyl halide in an aprotic solvent like DMSO orTHF, in the presence of a strong base like sodium hydride. A subsequent effi-cient desilylation of the bulky i-butyl- and TDMS ethers was achieved by tetra-butylammonium fluoride in THF.
Using acyl chlorides in the presence of a tertiary amine, acyl groups are intro-duced to the free hydroxy groups of 6-0-silylated celluloses. A typical exampleis given in Fig. 4.5.43.
The ether ester intermediates could by isolated as soluble polymers in anaprotic work-up procedure under weakly basic conditions, e.g. by precipitationwith 2 % aqueous NaHCO3 solution. Using 1 N aqueous hydrochloric acid atroom temperature a complete desilylation of the trimethylsilyl group proceeds,forming the corresponding cellulose esters (see Fig. 4.5.43).
A high content of ester groups can be obtained in the presence of acylationcatalysts like 4-dimethylaminopyridine. Table 4.5.21 shows typical examples forthis procedure. The results demonstrate the possibility of synthesizing TMScellulose esters with a high DS (up to 2.7), starting from TMS cellulose in theDS region of 2.0.

286 4.5 Etherification of Cellulose
OJV
Cell/\
OH Ο,Λ/
OSiMe3
DMF> 25°c' 4h
Polymer
TMS - Cell
Etherester
Ester
DegreeSi
1.55
1.43
<0.02
of substitutionEster
-
0.59*
0.56
precipitated in 2% aqu. NaHCO3
Cell
Cell
1 N HCI/DMF2 : 3 (v/v)
Figure 4.5.43. Acylation of TMS cellulose with 3,5-dinitrobenzoyl chloride in the pres-ence of triethylamine (Klemm et al., 199Oa).
Table 4.5.21. Influence of catalytic amounts of 4-dimethylaminopyridine (DMAP) onesterification of TMS cellulose with 3,5-dinitrobenzoyl chloride (Klemm et al., 199Oa).
TMS celluloseDSSi
1.681.681.991.992.432.43
DMAP TMS cellulose esterDSSi1.63
+ 1.621.90
+ 1.952.39
+ 2.45
D5ester
0.560.950.170.430.120.27
Additional reaction conditions: 3 mol of acyl chloride and 3 mol of triethylamine/AGU;THF, 25 0C, 4 h; precipitation in 2 % NaHCO3 solution in water.
The described homogeneous acylation of TMS celluloses has been used forsynthesis of different types of aliphatic and aromatic cellulose esters as summa-rized in Table 4.5.22.

4.5.5 Silyl ethers of cellulose 287
Table 4.5.22. Acylation of TMS cellulose with carbonic acid chlorides RCOCl, in thepresence of triethylamine or pyridine (Klemm et al., 199Oa)
TMS celluloseDSSi R
1.551.551.551.99
1.99
1.99
-CH=CH-C6H5
-(CH2)J4-CH3
-CH2Cl-(4)-C6H4-N02
-(4)-C6H4-N02
-(4)-C6H4-N02
Medium Precipi- After- Cellulose estertation in treatment DSc-
DMFTNEt3
DMFTNEt3
DMF/PyBenzene/NEt3
DMAPBenzene/NEt3
DMAPBenzene/NEt3
DMAP
ABAC
C
C
1.1.1.1.
1 N HCl > O
1 N HCl > O.
31.27.2895
.02
.02
^-*ester0.981.051.550.46
0.43
0.39
(A) 2 % NaHCO3 in H2O, (B) C2H5OH, (C) acetone. Additional reaction conditions: 3mol of acyl chloride and tertiary amine/AGU. Py, pyridine; DMAP, 4-dimethylamino-pyridine.
Table 4.5.22 contains examples with and without subsequent desilylation, aswell as the influence of the type of the tertiary amine and the work-up procedureon the structure of products. Using triethylamine as the base, TMS celluloseesters without desilylation can be isolated as described above. In the case ofpyridine as the base, a complete desilylation takes place during precipitation ofthe polymer with water, caused by the acidic pyridinium hydrochloride (seeTable 4.5.22). By acylation with acid chlorides, which are more stable under thework-up procedure, an additional esterification of hydroxy groups resulting fromthe desilylation reaction could be observed.
In contrast with these results, the reaction of TMS celluloses with acid chlo-rides, without addition of tertiary amines, catalysts or solvents, leads to differentresults. Depending on the reactivity of the acid chlorides in the range of 80-160 0C, an acylation takes place at a high rate within 0.5-1 h and simultaneouslychlorotrimethylsilane is liberated. The yield of chlorotrimethylsilane obtained bydistillation is equivalent to the amount of ester groups introduced.
OH
Cell\
R-C\
OSi(CH3)3Cl
OH80-160°C
-(CH3J3SiCICell\
0-C-RIl
O

288 4.5 Etherificatwn of Cellulose
Even when a higher excess of acid chloride was applied, the DS of acylation didnot ever exceed the DS of trimethylsilylation of the starting material.
These results demonstrate that acylation takes place at the oxygen atoms ofthe trimethylsilyloxy groups of the silylated celluloses. The hydroxy groups donot react under these conditions. The TMS celluloses themselves proved to bestable in air up to 280 0C. In the absence of oxygen, TMS celluloses withDS > 2.0 melt at 320-340 0C without decomposition (Cooper et al., 1981).
This new method has been applied to various acid chlorides, including sub-stituted and higher aliphatic and aromatic acid chlorides (Table 4.5.22). In allcases, very high rates and corresponding degrees of substitution were obtained.The use of solvents, for instance nitrobenzene, is possible. The degree of substi-tution of the resulting esters is controlled by the number of trimethylsilyl groupsin the starting polymer, as well as the amount of acid chlorides. The subsequentaddition of various acid chlorides leads to the corresponding mixed celluloseesters.
The isolation of the products is very simple. After complete distillation of thechlorotrimethylsilane (b.p. 57 0C) and the excess of acid chlorides at highertemperature under vacuum, cellulose esters result with high-purity. Chlorotri-methylsilane and acid chlorides, recovered in the manner described, are suitableto be applied to the synthesis without further purification. Remaining trimethyl-silyl groups can be split off completely in aqueous methanol under acidic condi-tions.
The composition and structure of the cellulose esters were confirmed by ele-mental analyses (Table 4.5.23) and IR spectroscopy. The IR spectra show car-bonyl absorptions (VC_Q) at 1765, 1735 and 1730cm"1, as well as the typicalabsorptions of the cellulose residue and the additional functional groups of theester substituents.
The described acylation of TMS celluloses with acid chlorides represents anew route to cellulose esters of higher aliphatic and aromatic acids with high andcontrolled degrees of substitution. The reaction proceeds without solvents andcatalysts by simply heating the TMS celluloses with the acid chlorides underrecycling of the distilled chlorotrimethylsilane and excess of acid chlorides.
Furthermore, the subsequent reactions of silylcelluloses form a suitable wayto prepare water-soluble cellulose sulfuric acid half-esters varying widely in DP,DS and pattern of substitution (see chapter 4.4.1.3).

Tab
le 4
.5.2
3. C
on
dit
ion
s an
d re
sult
s o
f th
e ac
ylat
ion
of
TM
S c
ellu
lose
wit
h a
cid
chlo
rid
esa
(Ste
in a
nd
Kle
mm
, 19
88).
TM
Sce
llu
lose
(D5b)
1.99
1.99
2.46
2.46
2.46
2.62
Ac
id c
hlo
rid
e M
oI
acid
mol
TM
S
R =
CC
l 2C
H3
R =
CC
l 3
R=
-(""
VN
O,
V^
ι/
<i
R =
(C
H2)
14C
H3
R =
—4
^-C
H2C
H2B
r
2. 5. 2.
chlo
rid
e/
Rea
ctio
nC
ellu
lose
est
er
cel
lulo
se
tem
per
atu
re
Yie
ld
(0C
) (
%)
.5 .0 .55.
0
3. 3.
.0 .5
80 90 160
160
160
160
94 95 96 92 96 95
DS
b
1.17
1.87
1.57
2.30
2.50
2.53
Ele
men
tal
an
aly
sis0
calc
.
fou
nd
calc
.
fou
nd
calc
.
fou
nd
calc
.
fou
nd
fou
nd
calc
.
fou
nd
C C C C C C C C C C C
37
.05
36.6
12
6.9
6
26
.54
51.5
051
.07
52
.55
52
.32
72
.85
49
.64
50.1
2
H H H H H H H H H H H
4.03
3.78
1.89
1.69
3.74
3.82
3.37
3.56
11.3
0
4.01
4.33
a
Sta
nda
rd r
eact
ion
co
nd
itio
ns:
wit
ho
ut
solv
ent,
30
min
, n
itro
gen
atm
osph
ere;
fo
r R
see
fo
rmu
la s
chem
e.b
C
alcu
late
d fr
om t
he
con
ten
t o
f S
i o
r C
l, N
, C,
Br,
det
erm
ined
by
elem
enta
l an
alys
is.
c
Cal
cula
ted
from
DS'
, si
lico
n c
onte
nt
of
3 (±
0.0
1) %
.

290 4.5 Etherification of Cellulose
4.5.5.5 Formation of supramolecular structures using silylcelluloses
By Klemm et al. (1990) the synthesis of photoreactive silicon-containing cellu-loses with comb-like structures and asymmetric membranes prepared thereof hasbeen described. For this purpose TMS and i-butyldimethylsilylcellulose wereesterified with cinnamic acid, 4-nitrocinnamic acid, 4-(hexyloxy)cinnamic acid,4-(hexadecyloxy)cinnamic acid and W-cinnamoyl-ll-aminoundecanoic acid. Indetail, the modification of trialkylsilylcelluloses with photosensitive side chainscan be performed in three different ways. The photosensitive group may be at-tached directly to the polysaccharide chain (A) via a spacer group (B), or incombination with a long side chain (C). These three types of comb-like cellulosederivatives are summarized in Fig. 4.5.44.
IA) (B) (C)
"7ΪΓ1 1 1TFPPP
Figure 4.5.44. Representation of modified cellulose chains with photosensitive groups(P) in the side chains (Klemm et al., 199Ob).
Synthesis of the cinnamic-acid-containing side chains was performed by re-action of the acid chloride with 1 1-aminoundecanoic acid yielding TV-cinnamoyl-11-aminoundecanoic acid. After the treatment with SOCl2 the corresponding N-cinnamoyl-11-aminoundecanoyl chloride was obtained:
O
V V-CH = CH-C-CI + H2N- (CH2)10-COOH
O OIl n
CH = CH-C- NH-(CH2)10 — C-OH
O OIl Il
CH = CH-C- NH-(CH2J10- C-CI
In addition, 4-hydroxycinnamic acid was alkylated with hexyl and hexadecylbromide. The products 4-(hexyloxy)- and 4-(hexadecyloxy)cinnamic acid weretransformed to the acid chlorides with SOCl2 yielding 4-hexyloxycinnamoylchloride and 4-hexadecyloxycinnamoyl chloride, respectively:
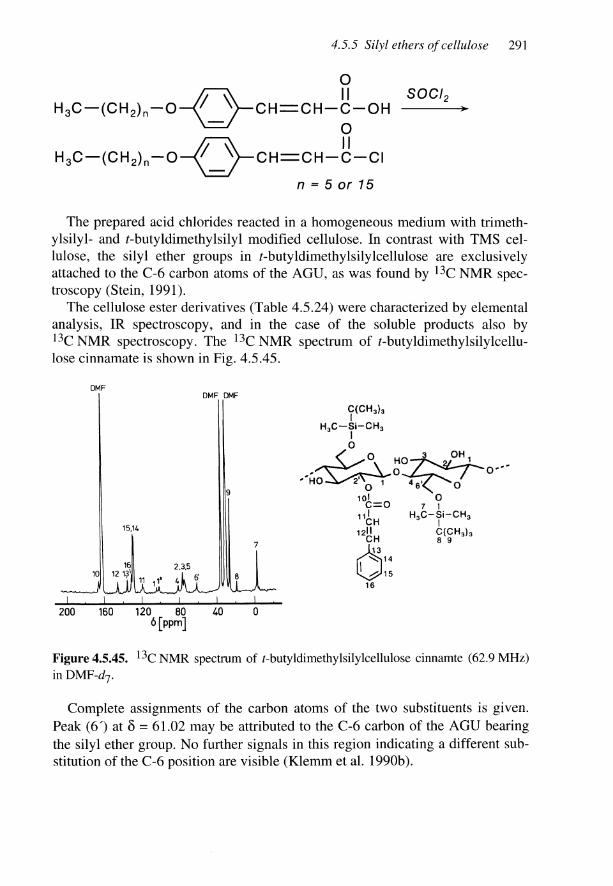
4.5.5 Silyl ethers of cellulose 291
H3C-(CH2)n-<
H3C-(CH2)n-<
V ^-CH = CH-C-OH
O
V ^-CH = CH-C-CI
n = 5 or 15
The prepared acid chlorides reacted in a homogeneous medium with trimeth-ylsilyl- and i-butyldimethylsilyl modified cellulose. In contrast with TMS cel-lulose, the silyl ether groups in ί-butyldimethylsilylcellulose are exclusivelyattached to the C-6 carbon atoms of the AGU, as was found by 13C NMR spec-troscopy (Stein, 1991).
The cellulose ester derivatives (Table 4.5.24) were characterized by elementalanalysis, IR spectroscopy, and in the case of the soluble products also by13C NMR spectroscopy. The 13C NMR spectrum of i-butyldimethylsilylcellu-lose cinnamate is shown in Fig. 4.5.45.
DMFDMF DMF
15,1/»
12 1312,3.5
C(CH3)3
200 160 120 80 40 O6[ppm]
Figure 4.5.45. 13CNMR spectrum of ί-butyldimethylsilylcellulose cinnamte (62.9 MHz)in DMF-J7.
Complete assignments of the carbon atoms of the two substituents is given.Peak (60 at δ = 61.02 may be attributed to the C-6 carbon of the AGU bearingthe silyl ether group. No further signals in this region indicating a different sub-stitution of the C-6 position are visible (Klemm et al. 199Ob).
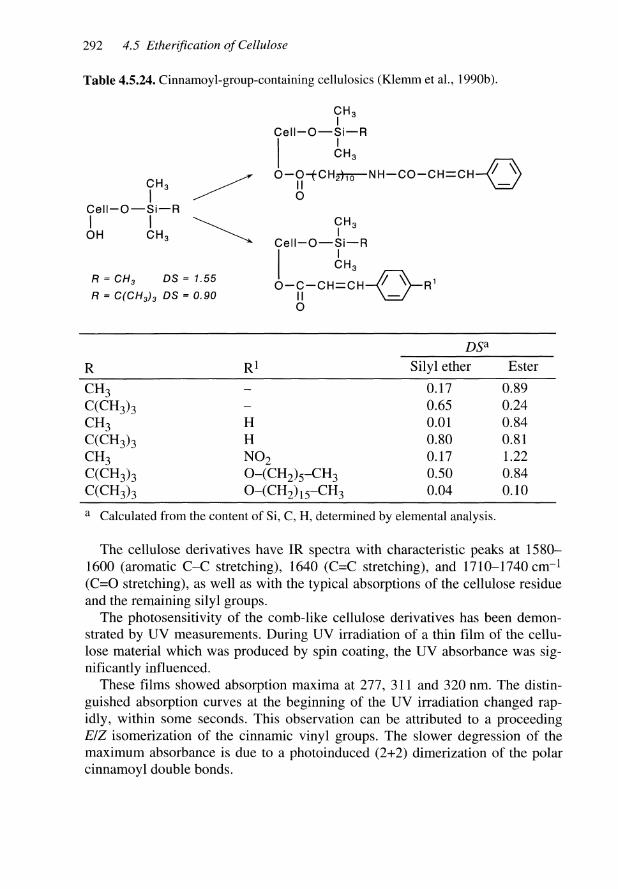
292 4.5 Etherification of Cellulose
Table 4.5.24. Cinnamoyl-group-containing cellulosics (Klemm et al., 199Ob).
CH3
CeII-O-Si—R
CH3
CeII-O—Si-RI I
OH CH3
R = CH3 DS = 1.55
R = C(CH3)3 DS = 0.90
CH3
O-0-eCHöfrö— NH-CO-CH = CIlO
CH3
CeII-O-Si—R
RCH3
C(CH3)3
CH3
C(CH3)3
CH3
C(CH3)3
C(CH3)3
Rl
HHNO2
O-(CH2)5-CH3
0-(CH2)15-CH3
Silyl ether
0.170.650.010.800.170.500.04
Ester
0.890.240.840.811.220.840.10
a Calculated from the content of Si, C, H, determined by elemental analysis.
The cellulose derivatives have IR spectra with characteristic peaks at 1580-1600 (aromatic C-C stretching), 1640 (C=C stretching), and 1710-1740 cm-1
(C=O stretching), as well as with the typical absorptions of the cellulose residueand the remaining silyl groups.
The photosensitivity of the comb-like cellulose derivatives has been demon-strated by UV measurements. During UV irradiation of a thin film of the cellu-lose material which was produced by spin coating, the UV absorbance was sig-nificantly influenced.
These films showed absorption maxima at 277, 311 and 320 nm. The distin-guished absorption curves at the beginning of the UV irradiation changed rap-idly, within some seconds. This observation can be attributed to a proceedingEIZ isomerization of the cinnamic vinyl groups. The slower degression of themaximum absorbance is due to a photoinduced (2+2) dimerization of the polarcinnamoyl double bonds.

4.5.5 Silyl ethers of cellulose 293
Scanning electron micrographs showed the possibility of preparing asymmet-ric membranes from the soluble cinnamic-acid-ester-containing silylcelluloses.
On the other hand, TMS celluloses (DS 2.6-2.9) have been used as solubleintermediates for building up well-defined mono- and multilayered ultrathinfilms of regenerated cellulose. Spreading of the silylcelluloses from chloroformor n-hexane solutions on a water surface and compressing the polymer mole-cules on the water/air interface by the Langmuir-Blodgett (LB) technique(Schaub et al., 1993) forms monolayers up to a surface pressure of 24 N/m. Athigher surface pressures, a plateau region is reached and the monofilm collapses.After transfer of the layer onto hydrophobized glass slides, silicon wafers, orgold surfaces, mono- and multifilms of the silylcelluloses resulted. A subsequentdesilylation can be carried out in a simple way with gaseous HCl within 30 s.
In the case of n-octyldimethylsilylcellulose, a comparable formation ofmonolayers on the air/water interface can be observed, but the low surface pres-sure of 10 mN/m does not allow the transfer of these layers onto a substrate.Further investigation reported the derivatization of the cellulose in ultrathinfilms, e.g., with succinic anhydride, and the utilization of the regenerated andmodified cellulose films for adsorption studies. Information on the film fluidnesscan be obtained by X-ray reflectometry and by using the evaluation of the peri-odical intensity modulations as described by Buchholz et al. (1996).
An investigation of a series of TMS cellulose LB films (DS = 2.7) depositedon silicon wafers reveals that the film thickness increases proportionally with thenumber of layers transferred to the substrate, and the slope of the linear fit indi-cates a spacing of 9.9 A per layer (Fig. 4.5.46).
1200
1000
800
60°4°°200
O20 40 60 80
Number of layers100 120
Figure 4.5.46. Thickness dependence for: TMS cellulose LB films (O), and the corres-ponding regenerated cellulose films (·) (values obtained from X-ray reflectometrymeasurements) (Buchholz et al., 1996).
The corresponding regenerated cellulose films obtained after exposure to HClshow similar behavior, and the spacing per layer was determined to be 4.2 A.This value is consistent with the spacings of different cellulose chains in the

294 4.5 Etherification of Cellulose
corresponding crystalline modifications of cellulose (Walton and Blackwell,1973). Therefore the film thickness decreases by 58 % during the regenerationprocess, but, as can be seen from the appearance of the Kiessig fringes in the X-ray reflection curve and the film roughness (Rieutord et al., 1987), the regener-ated cellulose film is still regular and covers the substrate homogeneously(Schaub et al., 1993).
In contrast with the necessarily hydrophobic LB films of hairy-rod polymers,the films of regenerated cellulose obtained by this method have hydrophilicsurface properties, as is evident from the static contact angle with water (78° forthe hydrophobic TMS cellulose and 23° for the hydrophilic regenerated cellu-lose). In addition, the cellulose multilayer systems are insoluble in most com-mon organic solvents and water, although such films are somewhat swellable inthe latter, and are stable against oxidation and thermal degradation (Schaub etal., 1993). Thus the regeneration of TMS cellulose LB films leads to well-defined thin-film architectures of cellulose, and these may be used as substratesfor many different studies such as investigations of adsorption processes.
4.5.6 Summary and outlookThe etherification of cellulose is usually performed in an aqueous alkaline me-dium with the polymer remaining in a highly swollen but solid state throughoutthe reaction, along the routes of the Williamson ether synthesis, the addition ofepoxy compounds via ring cleavage and the Michael addition of compoundswith activated double bonds onto the cellulose chains. Most cellulose ethersexhibit a high chemical stability, and the broad variability of product structure ispredominantly achieved by choice of the substituent attached, or the combina-tion of substituents in the case of mixed ethers, and by the DS which is deter-mined by reagent-to-cellulose ratio and external reaction conditions. Thesestatements holds true for all commercial cellulose ethers, for which alternativeroutes of synthesis, e.g. via cellulose in the dissolved state, can be widely ruledout today. Present research and development work is primarily aimed at processoptimization, with respect to economy and ecology, to give a tailored adaptationof product properties to end-use requirements.
As interesting and promising characteristics within the cellulose ethers, thetrityl and trialkylsilyl ethers shall be explicitly mentioned: alternative routes ofsynthesis and a wide spectrum of consecutive reactions can be realized here witha good chance of feasibility on a larger scale. Especially the silyl ethers of cel-lulose can be considered today as a kind of turntable in the chemistry of cellu-lose derivatization and as a challenge to further research.

294 4.5 Etherification of Cellulose
corresponding crystalline modifications of cellulose (Walton and Blackwell,1973). Therefore the film thickness decreases by 58 % during the regenerationprocess, but, as can be seen from the appearance of the Kiessig fringes in the X-ray reflection curve and the film roughness (Rieutord et al., 1987), the regener-ated cellulose film is still regular and covers the substrate homogeneously(Schaub et al., 1993).
In contrast with the necessarily hydrophobic LB films of hairy-rod polymers,the films of regenerated cellulose obtained by this method have hydrophilicsurface properties, as is evident from the static contact angle with water (78° forthe hydrophobic TMS cellulose and 23° for the hydrophilic regenerated cellu-lose). In addition, the cellulose multilayer systems are insoluble in most com-mon organic solvents and water, although such films are somewhat swellable inthe latter, and are stable against oxidation and thermal degradation (Schaub etal., 1993). Thus the regeneration of TMS cellulose LB films leads to well-defined thin-film architectures of cellulose, and these may be used as substratesfor many different studies such as investigations of adsorption processes.
4.5.6 Summary and outlookThe etherification of cellulose is usually performed in an aqueous alkaline me-dium with the polymer remaining in a highly swollen but solid state throughoutthe reaction, along the routes of the Williamson ether synthesis, the addition ofepoxy compounds via ring cleavage and the Michael addition of compoundswith activated double bonds onto the cellulose chains. Most cellulose ethersexhibit a high chemical stability, and the broad variability of product structure ispredominantly achieved by choice of the substituent attached, or the combina-tion of substituents in the case of mixed ethers, and by the DS which is deter-mined by reagent-to-cellulose ratio and external reaction conditions. Thesestatements holds true for all commercial cellulose ethers, for which alternativeroutes of synthesis, e.g. via cellulose in the dissolved state, can be widely ruledout today. Present research and development work is primarily aimed at processoptimization, with respect to economy and ecology, to give a tailored adaptationof product properties to end-use requirements.
As interesting and promising characteristics within the cellulose ethers, thetrityl and trialkylsilyl ethers shall be explicitly mentioned: alternative routes ofsynthesis and a wide spectrum of consecutive reactions can be realized here witha good chance of feasibility on a larger scale. Especially the silyl ethers of cel-lulose can be considered today as a kind of turntable in the chemistry of cellu-lose derivatization and as a challenge to further research.
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

References 295
ReferencesAnbergen, U., Oppermann, W., Polymer 1990, 31, 1854-1858.Arai, K., Kawabata, Y., Macromol Chem. Phys. 1995, 796, 2139-2147.Arisz, P.W., Thai, H.T.T., Boon, J.J., Salomons, W.G., Cellulose 1996, 3, 45-
61.Asandei, N., Perju, N., Nicolescu, R., Ciovica, S., Cellul. Chem. Technol 1995,
29,261-271.Avny, J., Rahman, R., Zilkha, A., J. Macromol. ScL, Macromol. Chem. 1972, 6,
177-189.Baar, A., Kulicke, W.-M., Szablikowski, K., Kiesewetter, R., Macromol. Chem.
Phys. 1994, 795, 1483-1492.Baker, TJ., Schroeder, L.R., Johnson, D.C., Cellul Chem. Technol. 1981, 75,
311-320.Bartsch, D., Purz, H.-J., Paul, D., Gensrich, H.-J., Hicke, H.-G., Faserforsch.
Textiltech. 1974,25, 184-189.Basque, P., de Gunzbourg, A., Rondeau, P., Ritcey, A.M., Langmuir 1996, 72,
5614-5619.Berthold, J., Desbrieres, J., Rinaudo, M., Salman, L., Polymer 1994, 35, 5729-
5736.Bikales, N.M., Segal, L. (Eds.), in Cellulose and Cellulose Derivatives, New
York: Wiley-Interscience, 1971, Part V, p. 790.Bikales, N.M., Macromol Synth. 1974, 5, 35-38.Bischoff, K.-H., Dautzenberg, H., Patent DD 124419, GER. (East) 1977; Chem.
Abstr. 1978, 88, 24439.Blasutto, M., Delben, F., Milost, R., Painter, TJ., Carbohydr. Polym. 1995, 27,
53-62.Bock, L.H., Ind. Eng. Chem. 1937, 29, 985-987.Brandt, L., in Ullmanris Encyclopedia of Industrial Chemistry, Gerhartz, W.,
Yamamoto, Y.S., Campbell, F.T., Pfefferkorn, R., Rounsaville, J.F. (Eds.),Weinheim: VCH , 1986, Vol. A5, pp. 461-488.
Braun, D., Meuret, B., Papier (Darmstadt) 1989, 43, 688-694.Bredereck, K., Strunk, K., Menrad, H., Makromol Chem. 1969, 726, 139.Buchholz, V., Wegner, G., Stemme, S., Ödberg, L., Adv. Mater. 1996, 8, 399-
402.Buytenhuys, P.A., Bonn, R., Papier (Darmstadt) 1977, 31, 525-527.Camacho Gomez, J.A., Erler, U.W., Klemm, D.O., Macromol. Chem. Phys.
1996, 797, 953-964.Cannizzaro, A.M., Rollins, M.L., Stanonis, DJ., Text. Res. J. 1973, 43, 397.Chakrabarti, A., Navle, P.B., Chanhari, R.V., Indian J. Technol. 1986, 24, 256-
259.

296 4.5 Etherification of Cellulose
Charpentier, D., Mocano, G., Carpov, A., Chapelle, S., Merle, L., Müller, G.,Carbohydr. Polym. 1997, 33, 177-186.
Chen, L., Yuan, X., Guo, Q., Water Treat. 1991, 6, 283-292.Cheng, F., Li, G., Feng, J., Zhang, J., /. Appl. Polym. ScL 1996, 61, 1831-1838.Ciucanu, I., Kerek, F., Carbohydr. Res. 1984,131, 209-217.Cooper, G.K., Sandberg, K.R., Hink, J.F., J. Appl. Polym. Sei. 1981, 26, 3827.Daly, W.H., Caldwell, /. Polym. ScL, Polym. Lett. Ed. 1979,17, 55-63.Daly, W.H., Caldwell, J.D., van Phung, K., Tang, R., Polym. Prep. (Am. Chem.
Soc., Div. Polym. Chem.) 1982, 23, 145-146.Daly, W.H., Caldwell, J.D., van Phung, K., Tang, R., Polym. ScL Technol. 1984,
24, 45^7.D'Ambra, A.J., Rice, MJ., Zeller, S.G., Gruber, P.R., Gray, G.R., Carbohydr.
Res. 1988,777, 111-116.Dautzenberg, H., Dautzenberg, H., Linow, K.-J., Faserforsch. Textiltech. 1978a,
29, 538-543.Dautzenberg, H., Dautzenberg, H., Linow, K.-J., Faserforsch. Textiltech. 1978b,
29, 593-598.Dautzenberg, H., Philipp, B., Z. Phys. Chem. 1979a, 260, 289-297; 298-308.Dautzenberg, H., Philipp, B., Acta Polym. 1979b, 30, 231-235.Dautzenberg, H. Fink, H.-P., Laskowski, L, Philipp, B., Acta Polym. 198Oa, 31,
662-667.Dautzenberg, H., Philipp, B., Purz, H.-J., Hesse, W., Acta Polym. 198Ob, 31,
11-16.Dautzenberg, H., Philipp, B., Purz, H.-J., Hesse, W., Acta Polym. 198Oc, 31,
137-141.Dautzenberg, H., Fanter, C., Fink, H.-P., Philipp, B., CeIM. Chem. Technol
198Od, 14, 633-653.Dawsey, T.R., McCormick, C.L., J. Macromol. ScL, Rev. Macromol. Chem.
Phys. 1990, C30, 405.Diacik, I., Jambrich, M., Jancarik, V., Kollär, L, Pechärova, L, Lenzinger Her.
1977,42, 118-126.Diamantoglou, M., Kühne, H., Papier (Darmstadt) 1988, 42, 690-696.Dönges, R., Br. Polym. J. 1990, 23, 315-326.Ebringerovä, A., Pastyr, J., CeIM. Chem. Technol 1980,14, 885-892.Ebringerovä, A., Hromädkovä, Z., Angew. Makromol. Chem. 1996, 242, 97-
104.Emett, P.M., Polym. Mater. Sei. Eng. 1996, 75, 381-382.Engelskirchen, K., 1987, in Houben-Weyl, Mehtoden der organischen Chemie,
Teil 3, Bd. E20, p. 2051-2092.Englebretsen, D.R., Harding, D.R.K., Int. J. Pept. Protein Res. 1992, 40, 487-
496.

References 297
Erler, U., Mischnick, P., Stein., Α., Klemm, D., Polym. Bull. 1992a, 29, 349.Erler, U., Klemm, D., Nehls, L, Makromol. Chem., Rapid Commun. 1992b, 13,
195.Farah, F.S., Awdeh, Z.L., /. Immunol Mehtods 1972, l, 353-361.Feddersen, R.L., Thorp, S.N., in Industrial Gums, Polysaccharides and Their
Derivatives, Whistler, R.L., BeMiller, J.N. (Eds.), San Diego: AcademicPress, Harcourt Javanovich, 1993, 3rd Edn., pp. 537-573.
Fink, H.P., Walenta, E., Papier (Darmstadt) 1994, 48, 739-748.Fink, H.P., Walenta, E., Kunze, J., Mann, G., in Cellulose and Cellulose
Derivatives: Physico-chemical Aspects and Industrial Applications, Kennedy,J.F., Phillips, G.O., Williams, P.A., Piculell, L. (Eds.), Cambridge: WoodheadPublishing, 1995, pp. 523-528.
Finkbeiner, H.L., Klebe, J.F., Patent US Pat. 3432488, 1969; Chem. Abstr.1969,70, 116405.
Frazier, C., Glasser, W. G.., J. Appl. Polym. ScL 1995, 58, 1063-1075.Gavlik, J., Tokar, O., 1989, CS patent 259795, 10.05.1989, CA 113, 8291.Ghannam, M.T., Nabil Esmail, M., J. Appl. Polym. ScL 1997, 64, 289.Giasson, J.I., Revol, J.F., Gray, D.G., St.-Pierre, J., Macromolecules 1991, 24,
1694-1696.Gilbert, R.D., Fornes, R.E., /. Polym. ScL, Part B: Polym. Phys. 1989, 27, 1949.Granski, W., Hellmann, G., Papier (Darmstadt) 1987, 41, 668.Gray, D.G., Harkness, B.R., Can. J. Chem. 199Oa, 68, 1135-1139.Gray, D.G., Harkness, B.R., Macromolecules 199Ob, 23, 1452-1457.Greber, G., Paschinger, O., Patent French Pat. 2477157 Al, 1981a; Chem.
Abstr. 1982, 96, 8425.Greber, G., Paschinger, O., Patent DE-OS 3104531, 1981b.Greber, G., Paschinger, O., Papier (Darmstadt) 1981c, 35, 547.Green, J.W., Methods Carbohydr. Chem. 1963, 3, 327-331.Green, J.G., Patent US Pat. 4390692 A, 1983; Chem. Abstr. 1983, 99, 72434.Gruber, E., Gruber, R., Cellul Chem. Technol. 1978, 72, 345-353.Gruber, J.V., Kreeger, R.L., in Polymeric Materials, Encyclopedia, Salamone,
J.C. (Ed.), Boca Raton: CRC Press, 1996, pp. 1113-1118.Gruber, E., Granzov, C., Ott, Th., Papier (Darmstadt) 1996 50, 729-734.Hagiwara, J., Shiraishi, N., Yokota, T., Norimoto, M., Hayashi, Y., /. Wood
Chem. Technol. 1981,1, 93-109.Hakamori, S., /. Biochem. (Tokyo) 1964, 55, 205-208.Hakert, H., Eckert, Th., Müller, T., Colloid Polym. ScL 1989, 267, 226-229.Hall, D.M., Hörne, J.R., J. Appl. Polym. Sd. 1973,17, 2891-2896.Harkness, B.R., Gray, D.G., Macromolecules 1991, 24, 1800-1805.Harper, R.J., Text. Res. J. 1982, 52, 714-720.

298 4.5 Etherification of Cellulose
Hearon, W.M., Hiatt, G.D., Fordyce, C.R., /. Am. Chem. Soc. 1943, 64, 2449-2452.
Heinze, Th., Habilitation Thesis, Friedrich Schiller University of Jena, 1997.Heinze, Th., Klemm, D., Winkelmann, H., Linß, W., Angew. Makromol. Chem.
1989, 769, 69-82.Heinze, Th., Klemm, D., Loth, F., Philipp, B., Acta Polym. 1990, 41, 259-269.Heinze, Th., Röttig, K., Nehls, L, Macromol Rapid Commun. 1994a, 75, 311-
317.Heinze, Th., Erler, U., Nehls, L, Klemm, D., Angew. Makromol. Chem. 1994b,
275, 93-106.Heinze, Th. Heinze, U., Klemm, D., Angew. Makromol. Chem. 1994c, 220, 123-
132.Heinze, U., Ph.D. Thesis, Friedrich Schiller University of Jena, 1998.Helfrich, B., Koester, H., Ber. Dtsch. Chem. Ges. 1924, 57, 587-591.Hess, K., Trogus, C., Friese, H., Liebigs Ann. Chem. 1928, 466, 80.Hess, K., Trogus, C., Eveking, W., Garthe, E., Liebigs Ann. Chem. 1933, 506,
260.Hess, K., Trogus, C., Abel, G., Cellul.-Chem. 1935, 76, 79.Hoeye, J., Nor. Skogind. 1977, 37, 69-70.Hohn, W., Tieke, B., Macromol. Chem. Phys. 1997, 188, 703-715.Hong, L.T., Borrmeister, B., Dautzenberg, H., Philipp, B., Zeiht. Pap. 1978, 27,
207-210.Honeyman, J., /. Chem. Soc. 1947, 168-173.Ikeda, L, Kurata, S., Suzuki, K., in 33rd IUPAC Int. Symp. on Macromolecules,
Montreal, July 7990, AbstractsIsogai, Α., Ishizu, Α., Nakano, J., Sen-I Gakkaishi 1984 a, 40, T504-T511.Isogai, A., Ishizu, J., Nakano, J., /. Appl. Polym. Sei. 1984 b, 29, 3873-3882.Isogai, A., Ishizu,A., Nakano, J., /. Appl. Polym. Sei. 1985, 30,345-353.Iwata, S., Narui, T., Takahashi, K., Shibata, S., Carbohydr. Res. 1985, 145,
160-162.Johnson, D.L., Nicholson, M.D., Haigh, F.C., /. Appl. Polym. Sei,. Polym. Symp.
1976, 2S, 931-943.Kamide, K., Okajima, K., Matsui, T., Ohnishi, M., Polym. J. 1987, 79, 347-356.Kamide, K., Yasuda, K., Okajima, K., Polym. J. 1988, 20, 259-268.Kasulke, K., Dautzenberg, H., Folter, E., Philipp, B., Cellul. Chem. Technol.
1983,77,423-432.Katsura, S., Isogai, A., Onabe, F., Usuda, M., Carbohydr. Polym. 1992, 18,
283-288.Kayeyama, K., Isogai, A., Hyama, K., Nakano, J., Mokuzai Gakkaishi 1985, 37,
274-279.Keilich, G., Thilarik, K., Husemann, E., Makromol. Chem. 1968, 720, 87.

References 299
Kim, Y.H., Han'Guk Somyu Konghakkoechi 1987, 24, 279-285, CA 107,219327.
Kinstle, J.F., Irving, N.M., Polym. ScL Technol 1983, 27, 221-227.Kishida, N, Okimasu, S., Hiroshima Joshi Daigaku Kascigakubu Kiyo 1976, 77,
37-43.Klavins, M., Prikulis, A., Latv. PSR Zinat. Akad. Vestis., Kim. Ser. 1982, 3,
341-342.Klebe, J.F., Patent US Pat. 3418313,1968a; Chem. Abstr. 1969, 70, 59081.Klebe, J.F., Patent US Pat. 3418312,1968b; Chem. Abstr. 1969, 70, 59084.Klebe, J.F., Finkbeiner, H.L., /. Polym. ScL, Part A-I 1969, 7, 1947.Klemm, D., Geschwend, G., Angew. Makromol. Chem. 1989, 769, 175-184.Klemm, D., Schnabelrauch, M., Stein, Α., Niemann, M., Ritter, H., Makromol.
Chem. 1990, 797, 2985-2991.Klemm, D., Schnabelrauch, M., Stein, A., Philipp, B., Wagenknecht, W., Nehls, L,
Papier (Darmstadt) 199Oa, 44, 624-632.Klemm, D., Stein, A., /. Macromol. Sei., Pure Appl. Chem. 1995, A32, 899-
904.Klemm, D., Heinze, Th., Stein, A., Liebert, T., Macromol. Symp. 1995, 99, 129.Koenig, H.S., Roberts, C.W., J. Appl Polym. Sei. 1974, IS9 651-666.Kondo, T., J. Polym. ScL, Part B: Polym. Phys. 1997, 35, 717-723.Kondo, T., J. Polym. ScL, Part B: Polym. Phys. 1994, 32, 1229-1236.Kondo, T., Gray, D.G., in 33rd IUPAC Int. Symposium on Macromolecules,
Montreal, Canada, July 1990.Kondo, T., Gray, D.G., Carbohydr. Res. 1991, 220, 173-183.Kondo, T., Carbohydr. Res. 1993, 238, 231-240.Kondo, T., Isogai, A., Ishizu, A., Nakano, J., /. Appl. Polym. Sei. 1987, 34, 55-
63.Kotz, J., Philipp, B., Nehls, L, Heinze, Th., Klemm, D., Acta Polym. 1990, 41,
333-338.Kotz, J., Nehls, L, Philipp, B., Diamantoglou, M., Papier (Darmstadt) 1991, 45,
226-231.Koschella, A., Klemm, D., Macromol Symp. 1997, 720, 115-125.Koura, A., Lukanoff, B., Philipp, B., Schleicher, H., Faserforsch. Textiltech.
1977, 28, 63-65.Kubota, H., Shigehisa, Y., /. Appl. Polym. Sei. 1995, 56, 147-151.Kulicke, W.-M., KuIl, A.H., KuIl, W., Thielking, H., Engelhardt, J., Pannek,
J.-B., Polymer 1996, 37, 2723-2731.Kulshin, V.A., Zähringer, U., Lindner, B., Jäger, K.-E., Dimitriev, B.A.,
Rietschel, E.Th., Eur. J. Biochem. 1991, 79S, 697-704.Lavrenko, P.N., Okatova, O.V., Filippova, T.V., Shtennikova, I.N., Tsvetkov,
V.N., Dautzenberg, H., Philipp, B., Acta Polym. 1986, 37, 663-670.

300 4.5 Etherification of Cellulose
Lee, J.L., Kwei, T.K., Polym. Prepr., Am. Chem. Soc., Div. Polym. Chem. 1996,37, 787-788.
Liebert, T., Klemm, D., Heinze, Th., /. Macromol. ScL, Pure Appl. Chem. 1996,A31J, 613-626.
Liebert, T., Heinze, Th., in Cellulose Derivatives: Synthesis, Characterization,and Nanostructures, Heinze, Th., Glasser, W.G. (Eds.), ACS Symp. Ser.,1997, in press.
Loth, F., Philipp, B., Macromol. Chem., Macromol. Symp. 1989, 30, 273-287.Loth, F., Patent WP 9109878 Al; Chem. Abstr. 1991, 775, 235055.Lukanoff, B., Schleicher, H., Fischer, K., Wilke, M., WP C 08 BI 290 569 1.Lukanoff, T., Philipp, B., Faserforsch. Textiltech. 1967, 18, 407^13.Lukanoff, T., Zielske, L., Philipp, B., Cellul Chem. Technol. 1967, 7, 287-299.Lukanoff, T., Philipp, B., Loth, F., Faserforsch. Textiltech. 1969, 20, 481-490.Lukanoff, B., Dautzenberg, H., Philipp, B., Schleicher, H., Faserforsch.
Textiltech. 1977, 28, 449-453.Lukanoff, B., Dautzenberg, H., Philipp, B., Acta Polym. 1979, 30, 569-572.Mair, P., Bahadir, M., Körte, F., /. Appl. Polym. Sei. 1986, 32, 5273-5277.Mansour, O.Y., Basta, A.H., Atwa, A.I., Polym. Plast. Technol. Eng. 1993, 32,
415-430.McCormick, C.L. 1978 U.S. patent 78-929749, 31.07.1978, CA 95, 171345.McCormick, C.L., Shen, F.C., Org. Coat. Plast. Chem. 1981, 45, 335-338.Mischnick, P., 7. Carbohydr. Chem. 1991, 70, 711-722.Mormann, W., Wagner, T., 7. Polym. ScL, Part A: Polym. Chem. 1995, 33,
1119.Morooka, T., Norimoto, M., Yamada, T., J. Appl. Polym. Sd. 1989, 38, 849-
858.Nakamura, H., 1984, Jpn. patent 84-68903, 06.04.84, CA: 104, 11651.Needs, P.W., Selvendran, R.R., Carbohydr. Res. 1993, 245, 1-10.Nehls, L, Wagenknecht, W., Philipp, B., Stscherbina, D., Prog. Polym. Sei.
1994, 79, 29-78.Nicholson, M.D., Johnson, D.C., Cellul Chem. Technol. 1978, 77, 349-359.Nickolson, E.D., Johnson, D.C., Cellul Chem. Technol. 1977, 77, 349-59.Nishiuchi, T., Kyoikugakubu, K.D., Hokoku, K., Koichi Daigaku Kyoikugakubu
Kenkyu Hokoku, Dai-3-bu 1981, 33, 1-7.Obayashi, T., Shigetami, S., Hayashi, T., Yamadad, N., Kimura, Y., 1992 Jpn.
patent 92-129009, 21.05.1992, CA 120, 219906.Ohta, Y., Arakawa, M., Kondo, T., Maku 1978, 3, 283-284.Olaru, N., Steinberg, S., Schneider, E., Asandei, N., Cellul. Chem. Technol.
1978, 72, 373-379.Oppermann, W., Papier (Darmstadt) 1995, 49, 765-769.Perrier, D.M., Benerito, R.R., /. Appl. Polym. ScL 1973, 77, 3375-3389.

References 301
Petropavlovskii, G.A., Karina, I.E., Borisova, T.L, Cellul Chem. Technol. 1984,IS9 283-292.
Philipp, B., Bischoff, K.-H., Loth, F., Cellul. Chem. Technol. 1979,13, 23-33.Philipp, B., Fanter, C., Wagenknecht, W., Hartmann, M., Klemm, D.,
Geschwend, G., Schumann, P., Cellul Chem. Technol. 1983, 77, 341-353.Philipp, B., Lukanoff, B., Schleicher, H., Wagenknecht, W., Z. Chem. 1986, 26,
50-58.Philipp, B., Dautzenberg, H., Linow, K.-J., Kotz, J., Dawydoff, W., Prog.
Polym. Sei. 1989,14, 91-172.Plisko, E.A., Nud'ga, L.A., Petropavlovskii, G.A., Zh. Prikl Khim. 1982, 55,
2133-2136.Podgornyi, V.F., Gur'ev, V.P., in Immobilizovannye Proteoliticheskie Fermenty
Lech. Gnoino-Nekroticheskikh Protessov, Novosibirsk: Akad. Nauk SSSR,Sib. Otd., Inst. Tsitol. Genet., 1981, pp. 124-131.
Prasad, M.P., Kalyanasundaram, M., /. Appl. Polym. Sei. 1993, 49, 2075-2079.Prehm, P., Carbohydr. Res. 1980, 78, 372-374.Reuben, J., Conner, H.T., Carbohydr. Res. 1983, 775, 1.Rieutord, F., Benattar, J.J., Bosio, L., Blot, C., de Kouchkovsky, R., /. Phys.
1987, 48, 679.Rinaudo, M., in Cellulose and Cellulose Derivatives: Physico-chemical aspects
and industrial aaplications, Kennedy, J.F., Phillips, G.O., Williams, P.A.,Piculell, L. (Eds.), Cambridge: Woodhead Publishing, 1995, chapter 35, pp.257-264.
Resell, K.G., /. Carbohydr. Chem. 1988, 7, 525-36.Sazanov, Yu.N., Plisko, E.A., Nud'ga, L.A., Petropavlovskii, G.A., Petrova,
V.A., Fedorova, G.N., Zh. Prikl Khim. (Leningrad) 1981, 54, 691-697.Schaub, M., Wenz, G., Wegner, G., Stein, A., Klemm, D., Adv. Mater. 1993 5,
919.Schempp, W., Krause, Th., Seifried, U., Koura, Α., Papier (Darmstadt) 1984,
38, 607.Schenck, H., Thesis 1936.Schleicher, H., Lukanoff, B., Philipp, B., Faserforsch. Textiltech. 1974, 25, 179-
183.Schleicher, H., Lukanoff, B., Philipp, B., Tapi ÖCEPA-Meeting, Wien, 1980, pp.
147-152.Schuyten, H.A., Weaver, J.W., Reid, J.D., Jürgens, J.F., J. Am. Chem. Soc.
1948, 70, 1919.Shibata, T., Nakamura, H., Okamoto, L, 1983, Jpn. patent 83-226527, 30.11.83,
CA: 104, 50643.Seneker, S.D., Glass, I.E., Adv. Chem. Ser. 1996, 248, 125-137.Sollinger, S., Diamantoglou, M.,. Papier (Darmstadt) 1996, 50, 691-700.

302 4.6 Oxidation of Cellulose
Stanonis, DJ., King, W.D., Text. Res. J. 1960, 30, 802.Stanonis, DJ., Conrad, C.M., Appl. Polym. Symp. 1966, 2, 121.Stanonis, DJ., King, W.D., Harbrink, P., /. Appl. Polym. ScL 1967, 77, 817.Stein, A., Klemm. D., Macromol Chem., Rapid Commun. 1988, 9, 569-573.Stein, A., Ph.D. Thesis, University of Jena 1991.Strauss, MJ., Torres, R., Carignan, Y., Tetrahedron Lett. 1987, 28, 159-162.Strauss, MJ., Torres, R., Phelan, J., Can. J. Chem. 1987, 65, 1891-1900.Takahashi, S.I., Fujimoto, T., Barua, B.M., Miyamoto, T., Inagaki, H., /. Polym.
ScL, Polym. Chem. Ed. 1986, 24, 2981.Tang, L., Huang, M., Jiang, Y., Chin. J. Polym. Sd. 1996,14, 199-205.Tezuka, Y., Tsuchiya, Y., Shiomi, T., Carbohydr. Res. 1996, 297, 99-108.Thi Bach Tuyet, L., Ishizu, A., Nakano, J., Jpn. Tappi 1981, 35, 798-804.Timell, T., Studies on Cellulose Reactions, Stockholm: Esselte Akt, 1950.Vogt, S., Heinze, Th., Röttig, K., Klemm, D., Carbohydr. Res. 1995, 266, 315-320.Vogt, S., Klemm, D., Heinze, Th., Polym. Bull. 1996, 36, 549-555.Wagenknecht, W., Papier (Darmstadt) 1996, 50, 712-720.Walton, A.G., Blackwell, J., in Biopolymers, London: Academic Press, 1973.Wirick, M.G., J. Polym. Sd. 1968, 6, 1705-.Yalpani, M., Tetrahedron 1985, 41, 2957.Yim, C.T., Gilson, D.F.R., Kondo, T. Gray. D.G., Macromolecules 1992, 25,
3377-3380.Yokota, H., Cellul. Chem. Technol. 1986, 20, 315-325.Zhadanov, Y. A., Aleksoeev, Y.E., Alekseeva, V.G., Vysokomol. Soedin. A
1993,35, 1436-1441.Zugenmaier, P., Aust, N., Makromol. Chem., Rapid Commun. 1990, 77, 95-100.
4.6 Oxidation of Cellulose
The complete oxidation of cellulose converts it into carbon dioxide and water.This chapter, however, is concerned with much less drastic oxidation, in whichproducts with new functional groups, namely carboxy, aldehyde and ketogroups, are formed while the glycosidic linkages remain intact. The productsobtained are often mentioned as oxycelluloses. This term is inconsistent withmodern chemical nomenclature and should be replaced by more appropriateterms, as proposed here. The use of systematic names, on the other hand, seemsto be too complicated and cannot be recommended. These partial oxidation pro-cesses of cellulose are a long-standing goal in cellulose chemistry, since theyprovide access to various novel products and intermediates with valuable prop-erties. Oxidation processes are also of considerable industrial relevance in isola-tion and purification of cellulose from wood, for example, and in the manufac-

302 4.6 Oxidation of Cellulose
Stanonis, DJ., King, W.D., Text. Res. J. 1960, 30, 802.Stanonis, DJ., Conrad, C.M., Appl. Polym. Symp. 1966, 2, 121.Stanonis, DJ., King, W.D., Harbrink, P., /. Appl. Polym. ScL 1967, 77, 817.Stein, A., Klemm. D., Macromol Chem., Rapid Commun. 1988, 9, 569-573.Stein, A., Ph.D. Thesis, University of Jena 1991.Strauss, MJ., Torres, R., Carignan, Y., Tetrahedron Lett. 1987, 28, 159-162.Strauss, MJ., Torres, R., Phelan, J., Can. J. Chem. 1987, 65, 1891-1900.Takahashi, S.I., Fujimoto, T., Barua, B.M., Miyamoto, T., Inagaki, H., /. Polym.
ScL, Polym. Chem. Ed. 1986, 24, 2981.Tang, L., Huang, M., Jiang, Y., Chin. J. Polym. Sd. 1996,14, 199-205.Tezuka, Y., Tsuchiya, Y., Shiomi, T., Carbohydr. Res. 1996, 297, 99-108.Thi Bach Tuyet, L., Ishizu, A., Nakano, J., Jpn. Tappi 1981, 35, 798-804.Timell, T., Studies on Cellulose Reactions, Stockholm: Esselte Akt, 1950.Vogt, S., Heinze, Th., Röttig, K., Klemm, D., Carbohydr. Res. 1995, 266, 315-320.Vogt, S., Klemm, D., Heinze, Th., Polym. Bull. 1996, 36, 549-555.Wagenknecht, W., Papier (Darmstadt) 1996, 50, 712-720.Walton, A.G., Blackwell, J., in Biopolymers, London: Academic Press, 1973.Wirick, M.G., J. Polym. Sd. 1968, 6, 1705-.Yalpani, M., Tetrahedron 1985, 41, 2957.Yim, C.T., Gilson, D.F.R., Kondo, T. Gray. D.G., Macromolecules 1992, 25,
3377-3380.Yokota, H., Cellul. Chem. Technol. 1986, 20, 315-325.Zhadanov, Y. A., Aleksoeev, Y.E., Alekseeva, V.G., Vysokomol. Soedin. A
1993,35, 1436-1441.Zugenmaier, P., Aust, N., Makromol. Chem., Rapid Commun. 1990, 77, 95-100.
4.6 Oxidation of Cellulose
The complete oxidation of cellulose converts it into carbon dioxide and water.This chapter, however, is concerned with much less drastic oxidation, in whichproducts with new functional groups, namely carboxy, aldehyde and ketogroups, are formed while the glycosidic linkages remain intact. The productsobtained are often mentioned as oxycelluloses. This term is inconsistent withmodern chemical nomenclature and should be replaced by more appropriateterms, as proposed here. The use of systematic names, on the other hand, seemsto be too complicated and cannot be recommended. These partial oxidation pro-cesses of cellulose are a long-standing goal in cellulose chemistry, since theyprovide access to various novel products and intermediates with valuable prop-erties. Oxidation processes are also of considerable industrial relevance in isola-tion and purification of cellulose from wood, for example, and in the manufac-
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

4.6 Oxidation of Cellulose 303
ture and application of cellulose derivatives. In many cases the oxidation repre-sents an undesired but unavoidable side reaction in alkaline oxygen bleachingand pulping. The most important reactions in this context, induced by oxygenradicals, are the formation of carbonyl groups at the C-2 position or at C-3, or atboth positions of the AGU (Sjöström, 1981a; 198Ib). Glycosidic bond cleavagesmay occur by ß-alkoxy elimination. In the case of the 2,3-diketo structure, afurther conversion yield of carboxy furanoside groups may occur, which mayeasily be degraded in the alkaline medium (Malinen and Sjöström, 1975a and1975b). By these processes of oxidative depolymerization from the reducingend-groups a typical peeling of the polymer occurs.
Oxidation reactions in an acidic medium are also important. The extent ofoxidation reactions depends on various conditions, such as alkali concentration,type and amount of active oxygen, and temperature. Also during acidic pulpingand bleaching, oxidation occurs simultaneously with hydrolytic chain degrada-tion. The formation of glucuronic acid is usually observed in acidic pulping(Pfister and Sjöström, 1977). Many review articles are available on this subject(Fengel and Wegener, 1989).
For the determination of carbonyl and carboxyl groups, several methods wereproposed (see chapter 3). The determination of carbonyl groups is generallybased on oxidation (e.g. copper; Zellcheming Merkblatt IV/8/70) or reductione.g. with NaBH4. The carboxyl groups in cellulose are determined e.g. by titra-tion of the acidic groups in the presence of strong acidic salts or by the determi-nation of the amount of bound cations to the material, as the well-known meth-ylene blue method (Wilson and Mandel, 1961). A direct titration of the carboxylgroups is possible in the non-aqueous solvent system DMSO/methylamine/ethanolamine (Dautzenberg and Philipp, 1974).
It should be mentioned that the oxidation of the cellulose not only leads todegradation but may have also some stabilization effects. The effect is especiallydue to the conversion of the reducing end-groups to aldonic acid end-groups(Sjöström, 198 Ia, b). Moreover, the reduction of chain length during aging (so-called ripening) of alkali cellulose includes various oxidation reactions as welland being used in today's large-scale productions for improving the workabilityof the resulting products.
However, for a controlled chemical modification of the polymer, these oxida-tion processes mentioned are, of course, unsuitable. Therefore, in what followsthe principal idea of partial oxidation and properties of the available productswill be discussed. Some information about the qualitative and quantitative de-termination of the functional groups as well as experimental procedures are in-cluded, which the authors can recommend according to the present state ofknowledge.

304 4.6 Oxidation of Cellulose
O
x , x _HO·—AX^ \ HO-
OH OH
CH2OH CH2OH
O
δ ,c*o O OHO HO
w v\\
Figure 4.6.1. Different repeating units of oxidized cellulose.
On principle, cellulose as a polyhydroxy compound bearing primary and sec-ondary hydroxy groups, can be oxidized to 6-aldehyde- and 6-carboxycellulose(II), as well as to 2-keto- (III), 3-keto- (IV) or 2,3-diketocellulose (V), as illus-trated in Fig. 4.6.1, neglecting the transformation of end groups. Moreover, 2,3-dialdehyde cellulose (VI) may be obtained by the well-known glycol cleavage
oxidation of α,β-diol units with periodate, which can be easily further oxidized
to 2,3-dicarboxycellulose (VII). The tendency of these oxidations depends sub-stantially on the nature of the oxidants. It is rather complicated to gain both aselective and complete oxidation of a desired position, and therefore, as in thecase of cellulose functionalization in general, copolymers are formed.
Most of the known oxidants from organic chemistry produce both carbonyland carboxylate functions in varying proportions. Even the so-called selectiveoxidants may form different functions, depending on experimental parameters,such as e.g. pH, temperature, time, and state of activation of the starting cellu-lose. Moreover, many oxidation reactions result in more or less depolymer-ization of the macromolecules.
4.6.1 Oxidation of primary hydroxy groups
There is no direct and selective oxidation method available at this time for thetransformation of the primary hydroxy groups to the aldehyde functions. In an

4.6.1 Oxidation of primary hydroxy groups 305
elaborate stepwise procedure, 6-aldehyde cellulose, by photolysis of the 6-azido-6-deoxy derivative, was prepared (Clode and Horton, 1971).
Cellulose can be oxidized directly at C-6 to yield 6-carboxycellulose by oxi-dation with nitrogen dioxide in a nonpolar solvent such as tetrachloromethane(Yackel and Kenyon, 1942). The nitrogen dioxide oxidation of cellulose undervarious conditions has been extensively studied (Neveil, 1963). Possibly becauseof the heterogeneous reaction conditions, which lead to a lower accessibility ofthe reaction sites, depolymerization is severe and the products contain nitrogen.An improved procedure of the well-known nitrogen dioxide method has beendeveloped using phosphoric acid and sodium nitrite as the oxidizing agents (Fig.4.6.2; Painter, 1977).
OH
HO
O\
•
OH
(i) 85% H3POVNaNO2
(ii) HCOOH
(iii) NaBH4
COOH
HOOH
Figure 4.6.2. Reaction scheme of C-6 oxidation (Painter, 1977).
The content of the formed carboxy groups depends not only on the reactiontime but also drastically on the degree of polymerization of the starting cellulosematerial (Fig. 4.6.3). Surprisingly, the extent of oxidation increases with risingmolecular weight of the starting material (Heinze et al., 1993).
Con
tent
of C
OO
H g
roup
s (%
)
80-
70-
60-
50-
40-
30-
20-
10-
n_
: -. ; · :
• Cellulose powder, DP 1 60τ · Viscose staple fibre, DP 300
± Spruce sulfite pulp, DP 600τ Cotton !inters, DP 1400
•
6 8
Reaction time (h)
10
Figure 4.6.3. Content of formed carboxy groups of cellulose with different DP afteroxidation with NaNO2^3PO4, depending on the reaction time (Heinze et al., 1993).

306 4.6 Oxidation of Cellulose
Because of the high viscosity of the solutions, the liberated oxidizing agentN2O3 generates a foam which guarantees the contact between the cellulose andthe oxidizing agent. The foam also prevents loss of the gaseous oxidizing agent.It was found that the stability of the foam increases on increasing the molecularweight of the cellulose and that is why the degree of oxidation increases in thesame direction provided comparable reaction times of at least 5 h are considered.
The 13C NMR spectra of the sodium carboxycelluloses in D2O solution showconsiderable changes in the chemical shift values of the C-4, C-5 and C-6 atomsignals in comparison with the spectra of the starting celluloses. Fig. 4.6.4 showsa spectrum of a nearly completely oxidized sample (Fig. 4.6.4b, degree of oxi-dation = 0.82), as well as a spectrum of 6-carboxycellulose with remaining pri-mary hydroxy groups (Fig. 4.6.4a, degree of oxidation = 0.62). In addition tospectra of cellulose, a new signal occurs at 175.5 ppm, which can be assigned toa carboxyl group in the C-6 position of the anhydroglucose unit. In the case ofpreparing the sodium carboxycellulose with sodium hydroxide, a further signalwas found at 165.1 ppm without changes of the chemical shift values of theother signals.
C-6 1
C-6 1
180 UO 100ό [ppm]
60
Figure 4.6.4. 13C NMR spectrum of sodium carboxycellulose (conversion to the sodiumsalt by NaBH4) degree of oxidation: a = 0.62; b = 0.82 (Nehls et al., 1991).

4.6.1 Oxidation of primary hydroxy groups 307
This signal is caused by the presence of formic acid ester groups which areformed during the destruction of the excessive oxidizing agent N2O3, achievedpreferentially by addition of the formic acid. If one uses sodium borohydride toprepare the sodium salt, the carboxycellulose is free of formic acid ester groups(which means the cellulose formed is selectively oxidized only in the C-6 posi-tion). The existence of keto groups, as assumed by Painter et al. (1985), couldnot be confirmed because of the absence of NMR signals typical for CO groups(NehlsetaL, 1991).
Sodium carboxy celluloses show a high tendency to form ionotropic gels,even in a spherical shape, with calcium ions (Heinze et al., 1990). Physico-chemical properties in aqueous solutions were studied by potentiometry andmicrocalorimetry (Cesaro et al., 1987). The pK§ of the free acid form of 6-carboxycellulose was determined to be 2.8 ±0.1, indicating that the cellulosederivative is rather a strong acid (Kotz et al., 1990).
Cellulose, dissolved in formic acid, was treated with several oxidizing agentsincluding those important in pulp bleaching. Kinetic and viscometric measure-ments show that chlorine, bromine, nitric acid and hydrogen peroxide moder-ately accelerate the depolymerization of the polymer. Periodic acid, chromicacid and hydrogen peroxide/ferrous sulfate initiate a fast degradation, followedby a slower reaction. Considerable formation of carbonyl and carboxyl groupsoccurs, but no regioselective oxidation, except for periodic acid, was found(Graves, 1993).
A new method for the selective oxidation of primary hydroxy groups waspublished by using hypobromite as the oxidizing agent, mediated by 2,2,6,6-tetramethyl-1-piperidinyoxy (de Nooy et al. 1994). At the optimum pH value,between 10 and 11, water-soluble polyglucans like potato starch and pullulancould be converted into the 6-carboxy derivatives with a selectivity of more than95 % (de Nooy et al. 1995). The primary alcohol groups of various poly-saccharides with widely differing structures and water solubility, including cel-lulose, were oxidized by using the new 2,2,6,6-tetramethyl-l-piperidinyoxymethod (Chang and Robyt, 1996). Some open questions of this oxidationmethod concerning the water-insoluble cellulose were discussed recently (Be-semeretal. 1997).
Carboxycelluloses, of various contents of carboxy groups, have been suc-cessfully used in wound healing to prevent post-surgical adhesions (Dimitri-jevich et al., 1990). The subsequent periodate oxidation of 6-carboxycellulosewas used to evaluate the conformational interpretation of hemiacetal stability(Painter, 1977).
After a suitable pretreatment, consisting of the precipitation of an aqueous 6-carboxycellulose solution in Af,A^dimethylformamide and removing the waterfrom the highly swollen gel, a sulfation of the polymer with SO3 or HSO3Cl yields

308 4.6 Oxidation of Cellulose
the unstable acidic sulfate half-esters. Subsequent neutralization leads to the water-soluble sodium salts of the corresponding esters (Schnabelrauch et al., 1991). Suchpolyelectrolytes are interesting materials due to their gel- and symplex-formingtendency and their potential biological activity as heparin-like anticoagulationagents. They may used to build up special supramolecular architectures.
The same activation procedure can be used for subsequent modifications ofthe carboxy groups. 6-Carboxycellulose reacts with SOCl2 to give the corre-sponding acid chloride with a nearly complete conversion. The subsequent reac-tion with benzylamine/pyridine, e.g., yields 6-carboxybenzyl amides of cellulosewith additional glucuronic acid and glucose residues in the polymer backbone(Rahnetal., 1995).
The oxidation with ruthenium tetroxide, e.g., has found to form productscontaining both carbonyl and carboxyl functions (Daneault et al., 1983). Obvi-ously, the application of heterogeneous catalysis in combination with undis-solved cellulose leads to serious problems. A challenge for future developmentsis the use of either homogeneous catalyst (van Bekkum, 1991) or solutions ofunmodified cellulose in appropriate new solvents, or combinations of both.
4.6.2 Oxidation of secondary hydroxy groupsThe oxidation of secondary hydroxy groups without a C-C bond cleavage may
yield 2-keto- (III), 3-keto- (IV) or 2,3-diketocellulose (V) (see Fig. 4.6.1). Usingthe mild oxidizing agent acetic anhydride/DMSO, unmodified cellulose dis-solved in DMSO/paraformaldehyde is transformed into 3-ketocellulose (Bossoet al., 1982). It is assumed that this path is attributed to a reversible formation ofhydroxymethyl- and poly(oxymethylene)ol groups at O-2 and O-6. On the otherhand, protected cellulose derivatives like 6-O-triphenylmethyl- and 6-O-acetylcellulose are oxidized mainly at the C-2 position with the same reagentsystem, as well as with a mixture of DMSO/dicyclohexylcarbodiimide/pyri-dine/trifluoroacetic acid (the so-called Pfitzner-Moffatt reagent) at certain con-centrations (Bredereck, 1967). The keto group content reached values of up to0.8 and the degree of polymerization was approximately 150. More detailedinvestigations show that by oxidizing 6-O-triphenylmethylcellulose withDMSO/acetanhydride both 2-keto- (54 %) and 3-ketocellulose (36 %) is formed(Defaye and Gadelle, 1977).
Oxidation of cellulose in HClO4 solutions with Mn(III) as the oxidizing agentwas found to be of first order with respect to the oxidizing agent. The oxidationproducts, using electrogenerated Mn(III), which acts as an electron-transfer me-diator for the reaction, was ketocellulose, as revealed primarily by a new IRband at 1729 cm-1 (Zhang and Park, 1995).
The selectively oxidized ketocelluloses could be further modified e.g. by re-ductive amination using sodium cyanoborohydride (Yalpani et al., 1984).

4.6.2 Oxidation of secondary hydroxy groups 309
The most selective process of cellulose oxidation is the treatment of the polymerwith periodic acid and its salts under aqueous conditions forming 2,3-dialdehydecellulose (Nevell and Zeronian, 1962). Under suitable conditions, the periodateoxidation of cellulose and many other polysaccharides may be controlled simplyby the reaction time, and may be conducted in a quantitative manner (Table 4.6.1;Maekawa and Koshijima, 1984). However, it has been noted that under specialconditions, periodate oxidation of cellulose may lead to products containing highlevels of carboxyl functions or acidic endiol groups (Perlin, 1980). In order toavoid radical-induced depolymerization reactions, especially in laboratory-scalepreparations, it is recommended to carry out the reaction in the dark and to useradical scavengers. The initial periodate oxidation of cellulose is largely limited tothe readily accessible regions, i.e. the amorphous region, and has been used there-fore to determine the accessibility of cellulose starting materials (Lai, 1996). Tominimize the polymer degradation a homogeneous periodate oxidation wasachieved via methylolcellulose. The freshly prepared methylolcellulose, producedby dissolution of cellulose in paraformaldehyde/DMSO and subsequent precipita-tion in methanol (Johnson and Nicholson, 1976), was regenerated in aqueous peri-odate solution under simultaneous oxidation (Morooka et al., 1989). The oxidationlevel reached nearly 100 % within 1Oh, while the DP remained unchanged. In theheterogeneous procedure, the polymer degradation can be reduced by a stepwiseoxidation. After any oxidation step, the hemiacetals formed are destroyed by re-duction with NaBH4, and moreover a radical scavenger like propan-1-ol is added(Painter, 1988).
Table 4.6.1. Preparation of 2,3-dialdehyde cellulose by periodate oxidation of sprucesulfite pulp (DP = 650) with 0.25 M aqueous NaIO4 solution at 60 0C (Rahn and Heinze,1997).
Reaction time(h) Yield
(g)
2 8.94 7.85 8.26 8.78 8.0
2,3-Dialdehyde celluloseRecovery3
(%)
9482879284
Conversion1*mmolofCHO
groups/ 100 g of cellulose713±3775±9808±6929±17
1914±9
(%)
5762657381
a Recovery: Quotient (x 100) of actual yield of polymer isolated to the theoretical weightof 2,3-dialdehyde cellulose from 9.6 g of cellulose.
b Determined according to Pommerening et al. (1992).

310 4.6 Oxidation of Cellulose
The aldehyde groups of 2,3-dialdehyde cellulose may undergo a variety ofsubsequent reactions under aqueous conditions, well known from the low mo-lecular chemistry of aldehydes. Hydration and the formation of different acetalstructures yield a masking of the groups. In the presence of even traces of waterthe carbonyl functions are hydrated and therefore no typical carbonyl absorp-
tions (vco) can be detected. 2,3-Dialdehyde cellulose, as a similar dialdehyde, is
very sensitive to alkaline solutions where, besides degradation, an internal Can-nizzaro rearrangement reaction occurs which may be used for the determinationof the content of oxidized functions (Pommerening et al., 1992).
2,3-Dialdehyde cellulose has found considerable interest for various subse-quent reactions. Reductive amination was used to synthesize ion-exchange mate-rials with chelate groups (Csanady et al., 1989). The oxidized materials, even ofspherical bead cellulose, were used to immobilize enzymes (Valentova et al.,1981; Turkova et al., 1979). An interesting extension of the periodate oxidationis based on the subsequent borohydride reduction of the 2,3-dialdehyde cellu-lose, yielding a new type of acyclic stereoregular polymer of 2,4,5-tris(hydroxy-methyl)-l,3-dioxopentamethylene simply called 2,3-dialcohol cellulose (Fig.4.6.5). These water-soluble polymers are useful for the determination of theparent insoluble 2,3-dialdehyde cellulose by means of 1H and 13C NMR spec-troscopy (Fig. 4.6.6; Maekawa, 1991).
OH OHr 3l β>
/^LJ r^LJΟΠο ΟΠο
-4C 5C O-U U UI ιH H
H
ι "\s
|2CH2
IOH
ο _*
OH
"3COOH 6CH2
4 Ι 5 >O Γ* OO U Uι ι
. H H
H1P OL/ U
2COOH
(a) (b)
Figure 4.6.5. Structural formula of (a) poly[(2A,45,5/?)-2,4,5-tris(hydroxymethyl)-l,3-dioxopentamethylene and (b) the corresponding oxidized material.
The treatment of 2,3-dialdehyde cellulose with aqueous sodium bisulfite af-forded new water-soluble cellulose-based poly electrolytes with a maximal con-version of one of the two aldehyde groups per oxidized repeating unit (Rahn andHeinze, 1997). The formation of the aldehyde bisulfite adduct may be an alter-native method for assessing the aldehyde group content by subsequent elementalanalysis. A common method used to determine the content of dialdehyde groupsconsists of an internal Cannizzaro reaction induced by 0.5 M sodium hydroxide

4.6.2 Oxidation of secondary hydroxy groups 311
solution, and a subsequent backtitration of the remaining lye (Pommerening etal., 1992).
2,3-Dialdehyde cellulose with a low degree of oxidation (oxygen consump-tion 2.45 per 100 AGU) shows a drop in tensile strength of 34 % compared withthe starting cotton cellulose (Buschle-Diller and Zeronian, 1993). The sameholds true for the further oxidized 2,3-dicarboxycellulose and for the reducedproduct (2,3-dialcohol cellulose).
WVV*Jw*+*sri+^^
180 UO 100
ό [ppm]
60
Figure 4.6.6. 13C-NMR spectra of (a) 2,3-carboxycellulose and (b) 2,3-dialcohol cellu-lose (Rahn and Heinze, 1997; Maekova, 1991).
The corresponding triacetates were obtained by conventional acetylation ofthe polyalcohol, i.e. by treatment with pyridine/acetic anhydride for 6 h at 70 0C(Casu et al., 1985). The molecular structures of 2,3-dialcohol cellulose and ofthe corresponding polytriacetates of different content of acyclic repeating unitswas proven by means of 1H and 13C NMR spectroscopy (Maekawa, 1991).
Starting from 2,3-dialdehyde cellulose, the corresponding dicarboxy deriva-tives may be obtained by mild oxidation with sodium chlorite (Maekawa andKoshijima, 1984). A typical 13C NMR spectrum of a complete oxidized sampleis shown in Fig. 4.6.6. Products of a nearly complete oxidation are readily solu-

312 4.6 Oxidation of Cellulose
ble in water. The combined use of sodium chlorite and hydrogen peroxide (twomoles of each per mole of dialdehyde moieties) reduces oxidant costs, avoids theevolution of toxic chlorine dioxide and yields more selectively products of highmolecular weight (Floor et al., 1989). Studies on the viscosity and flocculationof multivalent salts confirmed the typical polyelectrolyte properties (Varma andChavan, 1995). 2,3-Dicarboxycellulose possesses interesting complexing prop-erties for metal cations such as copper, cobalt, nickel and calcium. The interac-tion of aqueous solutions yields gel-like products. Due to the calcium-bindingproperties the products are potentially attractive co-builders in phosphate-freedetergents. Dicarboxy polysaccharides, which contain sugar blocks in the poly-mer chain, show a better biodegradability than completely oxidized products.The builder performance in detergent formulations depends on the dicarboxycontent (Matsumura et al., 1993).
ReferencesBesemer, A.C., in Cellulose Derivatives: Synthesis, Characterization, and
Nanostructures, Heinze, Th., Glasser, W.G. (Eds.), ACS Symp. Ser., 1997, inpress.
Bosso, C., Defaye, J., Gadelle, A., Wong, C.C., Pedersen, C., /. Chem. Soc.,Perkin Trans. 11982, 1579-1585.
Bredereck, K., Tetrahedron Lett. 1967, 8, 695-698.Buschle-Diller, G., Zeronian, S.H., /. Appl. Polym. ScL 1993, 47, 1319-1328.Casu, B., Naggi, A., Torri, G., Allegra, G., Meille, S.V., Cosani, A.,
Terbojevich, M., Macromolecules 1985,18, 2762-2767.Cesaro, A., Delben, F., Flaibani, A., Paoletti, S., Carbohydr. Res. 1987, 760,
355-368.Chang, P.S., Robyt, J.F., /. Carbohydr. Chem. 1996, 75, 819-830.Clode, D.M., Horton, D., Carbohydr. Res. 1971, 79, 329-337.Csanady, G., Narayanan, P., Müller, K., Wegscheider, W., Knapp, G., Angew.
Makromol. Chem. 1989, 770, 159-172.Daneault, C., Kokta, B.V., Cheradame, H., J. Wood Chem. Technol 1983, 3,
459-472.Dautzenberg, H., Philipp, B., Faserforsch. Textiltech. 1974, 25, 422-425.Defaye, J., Gadelle, A., Carbohydr. Res. 1977, 56, 411.de Nooy, A.E.J., Besemer, A.C., van Bekkum, H., Red. Trav. Chim. Pays-Bas
1994, 773, 165-166.de Nooy, A.E.J., Besemer, A.C., van Bekkum, H., Carbohydr. Res. 1995, 269,
89-98.Dimitrijevich, S.D., Tatarko, M., Gracy, R.W., Linsky, C.B., Olsen, C.,
Carbohydr. Res. 1990, 795, 247-256.

References 313
Fengel, D., Wegener, G., Wood: Chemistry, Ultrastructure, Reactions, Berlin:Walter de Gruyter, 1989.
Floor, M., Hofsteede, L.P.M., Groenland, W.P.T., Verhaar, L.A.Th., Kieboom,A.P.G., van Bekkum, H., Red. Trav. Chim. Pays-Bas 1989, 108, 384-392.
Graves, K., Holzforschung 1993 47, 149-154.Heinze, Th., Klemm, D., Loth, F., Nehls, L, Angew. Makromol Chem. 1990,
178, 95-107.Heinze, Th., Klemm, D., Schnabelrauch, M., Nehls, L, in Cellulosics: Chemical,
Biochemical and Material Aspects, Kennedy, J.F., Phillips, G.O., Williams,P.A. (Eds.), New York: Ellis Horwood, 1993, pp. 349-355.
Johnson, D.C., Nicholson, Μ.Ό.,ΑρρΙ. Polym. Symp. 1976, 28, 931.Kotz, J., Philipp, B., Nehls, L, Heinze, Th., Klemm, D., Acta Polym. 1990, 41,
333.Lai, Y.-Z., in Chemical Modification of Lignocellulosic Materials, Hon, N.-S.
(Ed.), New York: Marcel Dekker, 1996, pp. 35-96.Maekawa, E., /. Appl. Polym. ScL 1991, 43, 417-422.Maekawa, E., Koshijima, T., J. Appl Polym. ScL 1984, 29, 2289-2297.Mahnen, R., Sjöström, E., Cellul Chem. Technol 1975a, 9, 651-655.Malinen, R., Sjöström, E., Pap. Puu 1975b, 57, 5-13.Matsumura, S., Nishioka, M., Shigeno, H., Tanaka, T., Yoshikawa, S., Angew.
Makromol. Chem. 1993, 205, 117-129.Morooka, T., Norimoto, M., Yamada, T., /. Appl. Polym. ScL 1989, 38, 849-
858.Nehls, L, Heinze, Th., Philipp, B., Klemm, D., Ebringerova, Α., Acta Polym.
1991, 42, 339.Nevell, T.P., Zeronian, H., /. Text. Inst. 1962, 53, T90.Nevell, T.P., in Methods in Carbohydrate Chemistry, Whistler, R.L., BeMiller,
J.N. (Eds.), New York: Academic Press, 1963, Vol. 3, pp. 164.Painter, TJ., Carbohydr. Res. 1977 55, 95-103.Painter, T.J., Cesaro, Α., Delben, F., Paoletti, S., Carbohydr. Res. 1985, 140,
61-68.Painter, TJ., Carbohydr. Res. 1988, 779, 259.Perlin, S.A., in The Carbohydrates, Chemistry and Biochemistry, Bigman, W.,
Horton, D. (Eds.), 1980, Vol. 16, pp. 1167-1215.Pfister, K., Sjöström, E., Pap. Puu 1977, 59, 711-720.Pommerening, K., Rein, H., Bertram, D., Müller, R., Carbohydr. Res. 1992, 233,
219.Rahn, K., Heinze, Th., Klemm, D., in Cellulose and Cellulose Derivatives,
Physico-Chemical Aspects and Industrial Applications, Kennedy, J.F.,Phillips, G.O., Williams, P.A., Piculell, L. (Eds.), New York: Ellis Horwood,1995, pp. 213-218.

314 4.6 Oxidation of Cellulose
Rahn, K., Heinze, Th., Cellul. Chem. Technol. 1997, in press.Schnabelrauch, M., Heinze, Th., Klemm, D., Nehls, L, Kotz, J., Polym. Bull
1991, 27, 147-153.Sjöström, E., in Wood Chemistry. Fundamentals and Applications, New York:
Academic Press, 1981a.Sjöström, E., Pap. Puu 1981b, 63, 438-442.Turkova, J., Vajcnev, J., Vancurova, D., Stamberg, J., Collect. Czech. Chem.
Commun. 1979,44,3411.Valentova, O., Marek, M., Svec, F., Stamberg, J., Vodrazka, Z., Biotechnol
Bioeng. 1981, 23, 2093.van Bekkum, H., in Carbohydrates as Organic Raw Materials, Lichtenthaler,
F.W. (Ed.), Weinheim: VCH, 1991, p. 289.Varma, AJ., Chavan, V.B., Carbohydr. Polym. 1995, 27, 63-67.Wilson, W.K., Mandel, J., Tappi 1961, 44, 131-137.Yackel, B.C., Kenyon, W., /. Am. Chem. Soc. 1942, 64, 121.Yalpani, M., Hall, L.D., Defaye, J., Gadelle, A., Can. J. Chem. 1984, 62, 260-
262.Zhang, H., Park, S.-M., Carbohydr. Res. 1995, 266, 129-142.

5 Future Developments in CelluloseChemistry - An Outlook
It can be stated that future research on chemical transformation of cellulose willhave to consider the macromolecular, the supramolecular, and the morphologicalstructure level of this polymer. This holds true for the starting material, thecourse of its chemical conversion and the processing of the reaction productsand their application. Besides the synthesis of tailored cellulose-based macro-molecules, the design and experimental realization and the appropriate applica-tion of artificially ordered supramolecular architectures must be considered as anintegral part of future cellulose chemistry. An efficient realization of this inno-vative concept definitely requires a still closer cooperation of cellulose chemis-try with adjacent areas of science, e.g. general polymer and colloid science, bi-ology, and engineering sciences, in order to explore and use adequately the re-markable innovative potential of this polymer. It requires also a closer coopera-tion between basic research and application in the industry active in the field ofcellulose chemistry and technology.
Future innovation in the chemical conversion of cellulose will be primarilybased on the synthesis of functionalized cellulosic compounds with a well-defined and preset primary structure at the macromolecular level. The furtherprocessing of these entities will be used to give defined supramolecular archi-tectures adapted to end-uses, especially in the conventional, the high-tech andthe biomedical areas. Regioselectively modified derivatives with a defined pat-tern of functionalization within the AGU and along the macromolecule, andnonconventional functional groups, e.g. chromophores or fluorophores, redoxand photoactive groups, as well as functional groups with special magnetic oroptical properties and biologically activity, are going to play a dominant rolehere. Moreover, nucleophilic displacement reactions with suitable derivativeslike cellulose sulfonates or halogendeoxy derivatives are interesting routes tonew functionalized polysaccharides, including the use of different paths of syn-thesis (Heinze and Rahn, 1996; Klemm et al., 1996; Heinze and Glasser, 1997;Rahn, 1997; Nakamura and Amana, 1997; Nakamura and Sanada, 1997; Heinze,1997).
Besides this exciting vision of progress, justified by actual experimental re-sults, the conventional esterification and etherification of cellulose for manu-facturing artificial fibers and process auxiliaries will require a lot of experimen-
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

316 5 Outlook onto Future Developments in Cellulose Chemistry
tal and theoretical research and new technologies in order to arrive at processesof better eco-compatibility at lower cost and improved product quality. In orderto make both these routes really prosperous in practice, a thorough investigationand a deeper understanding of non-covalent intermolecular interactions of cel-lulosic macromolecules in the original and in the derivatized state will be neces-sary. This holds true for process of cellulose dissolution, as well as for the 'stateof solution' of cellulosic compounds, including the formation of liquid crystal-line systems.
For a more detailed discussion of this future development in cellulose chem-istry, it is necessary to consider the whole route from the starting material to thefinal product, and its applicational properties in dependence on its molecular,supramolecular and morphological structure. This implies an integrative consid-eration of all these three structural levels, even in organic-chemistry-centeredresearch work on cellulosics, and an adequate development of analytical tech-niques, balanced and adapted to progress in the synthesis and application of newcompounds and supramolecular entities.
5.1 Cellulose as a Raw Material for ChemicalConversion
In the foreseeable future, cellulose delivered by nature and obtained by me-chanical and chemical processing of wood, and to some extent also that fromannual plant stalks and cotton seed hairs, will remain the starting material forchemical conversion. The chemical synthesis of pure, high-ΖλΡ cellulose surelyrepresents a scientific challenge (Nakatsubo et al., 1996), but from the presentpoint of view does not offers advantage as compared with the natural polymer.The biosynthesis of cellulose, on the other hand, presents numerous open prob-lems regarding e.g. the formation of an ordered supramolecular structure simul-taneously with the growth of the polymer chains. Its further elucidation mayhave practical consequences for the breeding of natural cellulose sources, andmay give impulse to the artificial preparation of supramolecular structures ofcellulosics.
Regarding the processing of natural cellulose, especially from wood, to a suit-able starting material for dissolution and/or derivatization, three major openproblems require future research.(i) In contrast with synthetic polymers with their more or less tailored molar-mass distribution, a wide and often bi- or polymodal molar-mass distribution hasso far been taken for granted in the case of dissolving pulp, leading to the openquestion of the effect of a narrow distribution on processing and product proper-ties. The relevance of this problem has been emphasized in recent years by Al-

5.1 Cellulose as a Raw Material for Chemical Conversion 317
brecht (1987, 1997). The beneficial effect of a narrow molar-mass distributionon viscose preparation and the homogeneity of the spinning solution has beenshown (Fischer et al., 1996). A lot of further research work will be required onthis topic, not only in connection with the viscose process, but also regarding theeffect of a narrow molar-mass distribution on manufacture and product proper-ties of other commercial esters and ethers of celluloses.(ii) The impact of new nonaqueous pulping and new eco-compatible bleachingprocesses on wood pulp production in general will affect also dissolving pulp asthe starting material for chemical conversion. It may open up new routes tohigher purity and better-suited macromolecular properties of this starting mate-rial (Franzreb et al., 1989).(iii) Especially for the manufacture of partially functionalized esters and ethersof cellulose the search for still better and site-selective techniques of celluloseactivation will remain a profitable goal in order to reduce reagent input and ef-fluent output in commercial derivatization processes by enhancing the accessi-bility of the cellulosic hydroxy groups.
Furthermore, the old question of cellulose purity, i.e. the tolerable content ofalien polysaccharides (hemicelluloses like xylans), other functional groups likecarboxyl and carbonyl groups, and accessory compounds like various metal ionsand organosoluble waxy substances, has to be raised again in connection withnew pulping and bleaching processes on the one hand, and more sophisticatedend-use requirements on cellulosic products on the other. Closely related to thisproblem of cellulose purity is the challenge of a more complex utilization ofwood as a chemical raw material. The relevance of this point repeatedly empha-sized by Albrecht (1997) is illustrated by the numerous routes to xylan function-alization experimentally realized on the laboratory scale but not yet commer-cially used (Stscherbina and Philipp, 1991).
Special cellulose morphologies supplied by nature or prepared artificially, forexample the special fibrillar structure of bacterial cellulose from, e.g., Acetobacterxylinum, or uniplanar structures of regenerated cellulose, can be of future interesttoo, in preparing cellulose derivatives for defined supramolecular architectures.Especially bacterial cellulose presents exciting problems to the cellulose chemistsand physicists, regarding not only a deeper understanding of cellulose biosynthe-sis, but also a deliberate manipulation of its structure at the various levels; specialmorphologies, which are of interest for hollow fiber preparation, have already beenrealized experimentally (Yamanaka et al., 1990; Geyer et al., 1994). On the su-pramolecular level the ratio of the two submodifications Ia : Iß can obviously beinfluenced by suitable polymer additives, like sodium carboxymethylcellulose, tothe medium of bacterial growth (Yamamoto et al., 1996). The idea of breeding analready functionalized bacterial cellulose is doubtless still rather visionary, butseems worthwhile for further consideration by cellulose chemists and biologists(Ogawa and Tokura, 1992a and 1992b, Lee et al., 1997).

318 5 Outlook onto Future Developments in Cellulose Chemistry
5.2 The Relevance of Intermolecular Interactions
Functionalization of cellulose to give new products with a preset and well-defined primary structure, as well as the advanced manufacture of conventionalesters and ethers of cellulose, require a much deeper insight into the relationbetween intra- and interchain hydrogen bonds in the course of derivatization,especially in a heterogeneous system. This deeper insight is also necessary tounderstand and to forecast the dissolving action of liquid systems on celluloseand to arrive finally at a comprehensive theory of cellulose dissolution (Bergeret al., 1985; Spange et al., 1997). Furthermore, the elucidation of open problemsof cellulose structure, like that of chain direction and its change on derivatiza-tion, as well as a better understanding of supramolecular structures of cellulosicsin solution and of solid state structure formation from solution, depend on amore comprehensive knowledge of noncovalent interaction along and betweenthe polymer chains. This concerns primarily the various types of hydrogenbonds differing in bonding strength, but also includes other categories of inter-action like hydrophobic forces (e.g. Itagaki et. al., 1997) and nonpolar interac-tions, so far occasionally mentioned but not systematically explored.
Interesting results on the interplay between the hydrogen bond system of cel-lulose and the course of cellulose derivatization have been published in connec-tion with cellulose acetylation (Kamide and Saito, 1994). Hydrogen bond for-mation in regioselectively substituted cellulose ether, especially 2,3- and 6-0-methy!cellulose has been quite recently investigated (Kondo, 1994). Consideringthe broad spectrum of possible chemical transformations at the polymer chain,these results are still rather punctual and represent much more of a challengethan a solved problem. The same statement holds true also for a full under-standing of the effects of the medium in homogeneous and heterogeneous reac-tions, which is closely related to the action of intermolecular forces.
In solving the problems outlined here, progress in the analytical techniquesavailable for assessing rapidly, unambiguously and quantitatively these inter-molecular interactions will play a decisive role. In contrast with recent progressachieved in describing the structure of cellulosics on the macromolecular levelby applying e.g. NMR spectroscopy and Chromatographie techniques, the char-acterization of intermolecular interactions still lags behind and often makes theseinteractions a topic of speculation.

5.3 New Cellulosic Compounds 319
5.3 New Cellulosic Compounds
For the synthesis of new cellulose derivatives with a preset, well-defined pri-mary structure, four routes are presently considered and already experimentallystudied:(i) polymer-analogous reactions at the cellulose chain, centered on regioselec-tively functionalized products and boosted by employing the full repertoire ofexperimental techniques of modern organic chemistry (some more details of thisroute are discussed below);(ii) the enzymatically catalyzed regioselective functionalization, e.g. ester-ification or oxidation of the cellulose macromolecule, as already experimentallyrealized by the lipase-catalyzed acylation of low molecular saccharides (Theri-sod and Klibanov, 1986; Riva et al., 1988; Geyer et al., 1995);(iii) the enzyme-catalyzed polymerization of glucose derivatives to give macro-molecules of preset structure, a route of still rather visionary character (Kobaya-shi et al., 1995, 1997);(iv) an automated stepwise construction of sequential cellulose-based macro-molecules from glucose derivatives by analogy to protein synthesis, whichdoubtless is a very tedious route to produce polymer chains of a defined func-tionalization pattern along the macromolecule, as well as within the single AGU.
Concerning the chemical modification of cellulose by polymer-analogousreactions, the regioselective introduction of two or three different substituents,the use of a controlled substituent migration, the feasibility of a thermoreversiblesubstitution, as well as the search for new protecting or activating substituents,are considered as promising goals of experimental studies at present and for thenear future. A controlled balance of energetic and geometric factors in cellulosereactions can be expected to result in new compounds suitable for the binding oftoxic or valuable substances from liquid systems. Stereoregular reactions oncellulose molecules with chiral reagents may possibly provide auxiliary com-pounds for the synthesis of optically pure low-molecular enantiomers (seeOhinishi and Shibata, 1997).
Regarding site specificity of reaction within the AGU, frequently no 'abso-lute' regioselectivity, e.g. an explicit functionalization at C-6 or C-2, is requiredto arrive at unexpected properties of the compounds in question. Often a moder-ate preference of a site of substitution rather suffices to modify physicochemicalproperties, depending on intermolecular interaction with a solvent or with livingmatter, as demonstrated recently by Klemm et al. (1997).
Besides regioselective derivatives with quite a large amount of free OHgroups, fully substituted compounds (DS = 3) are of interest with regard to theirmaterial properties. They still pose principle and experimental problems withregard to synthesis. For instance, the molar volume of the substituent is defi-

320 5 Outlook onto Future Developments in Cellulose Chemistry
nitely of high relevance in determining whether or not compounds with a DS of3 can be obtained.
Last but not least, the relevance of cellulose-metal complexes in the cellu-lose chemistry of the future shall be emphasized. Cellulose-metal-complexchemistry began about 150 years ago with the discovery of Schweizers reagentas a good solvent for cellulose, resulting in a large-scale process of artificialcellulose fiber spinning and a broad application of metal-complex solvents incellulose analysis. Now, we are on the verge of a full understanding of thereactions involved and the binding states realized in cellulose-metal complexformation. Recently results on the complexation of copper by low-molecularsaccharides were published (Burchard et al., 1994; Burger et al., 1995), andthis promising concept, of considering cellulose as a polyolato ligand from theviewpoint of inorganic metal-complex chemistry, indicates the beginning of arenaissance in this area, which has been rather neglected during the previoushalf century. Cellulose as a poly functional alkoxido ligand can be used to pre-pare compounds with exceptional optical, electrical, magnetic and catalyticproperties along the two routes of:(i) attaching metal atoms to the chiral polymer backbone after suitable function-alization, leading to compounds of controlled stability and controlled chain stiff-ness, which are interesting for transition metal catalysis,(ii) taking cellulose as a macromolecular polyol, forming stable complexes in thedeprotonated state with various metal ions, which can subsequently be trans-formed to coordination polymers with a defined supramolecular architecture orcan even be combined with low-molecular polyol complexes.
Two points may be emphasized again in connection with the synthesis ofcellulose derivatives with a defined macromolecular structure: The relevance ofwork-up steps of isolation and purification of the reaction products before char-acterization and application is going to increase with the level of chemical uni-formity and purity of the primary structure required to arrive at tailored end-useproperties for refined applications. This implies the necessity of a fast and thor-ough analytical characterization of the chemical structure of the compoundsformed in synthesis, and requires further development and adaptation, especiallyof NMR, Fourier transform IR and HPLC techniques to the specific problems ofcellulose derivatives. Besides the thorough chemical characterization of isolatedsamples, the analytical monitoring of a course of derivatization of cellulosegains in relevance at the laboratory scale as well as in industrial processes. As anexample, the continuous monitoring of the phthaloylation of dissolved celluloseacetate by means of a spectroscopic sensor inserted into the reaction vessel maybe cited here (Sollinger and Diamantoglou, 1996).

5.4 Commercial Processes of Chemical Conversion of Cellulose 321
5.4 Commercial Processes of Chemical Conversionof Cellulose
In the comments on the future development of commercial processes of chemi-cal conversion of cellulose, have to be considered the manufacture of artificialfibers, films and specially shaped solid state products on the one hand, and theconversion of the polymer to soluble process auxiliaries on the other. Table5.4.1, presenting the actual worldwide production capacity of cellulose chemicalconversion along various routes, conveys an impression of the economical im-pact of this branch of the chemical industry, justifying adequate future effort inresearch and development.
Table 5.4.1. Production and use of dissolving pulp (Engelhardt, 1995).
Product Production capacity (106 t)
Dissolving pulp + processed cotton !inters 6.0Regenerated cellulose + cellulose powder 3.9Cellulose esters 1.4-organic esters 1.1-inorganic esters 0.3Cellulose ether 0.5
The open question of highest priority doubtless is that of a future partial ortotal replacement of the viscose process by alternative routes of artificial cellu-lose fiber spinning via dissolved cellulosics. Among the various routes pursuedduring the last two decades, the amine oxide process has achieved a highly fa-vored position, while the other choices, e.g. the carbamate method with an alka-line solution of cellulose carbamate as the spinning dope, or the thiocyanateroute employing a combination of ammonia and thiocyanate, take a more mar-ginal position today. But it is still a matter of discussion as to whether the amineoxide process will substitute or just supplement the viscose process, due to theprincipally changed supramolecular and morphological structure and thechanged textile properties of the filaments obtained. Besides its growing impacton the chemical fiber market, the amine oxide process, employing an N-methylmorpholine TV-oxide melt solution of cellulose as the spinning dope, actstoday as a stimulus to research in cellulose structure formation from solution andas a catalyst in getting people from fundamental and applied research togetherfor joint effort (Couly and Smith, 1996). Also, the viscose process itself requiresfurther research and development, as the decision on its future in the fiber mar-ket is still open. The main efforts are directed here now and will be in the nearfuture to a still better ecocompatibility, by cutting down the CS2 input and the

322 5 Outlook onto Future Developments in Cellulose Chemistry
output of toxic emissions, but also on the optimization of the total process econ-omy with an accentuation of fine titres from the product side.
In large-scale cellulose esterification and etherification, the reduction of by-product formation by increasing reagent yield for the main reaction and thesearch for new pathways syntheses with minimal by-product formation is gener-ally considered as promising goals. The same holds true for new technologies toprepare final products with a well-defined polymer morphology. The amount ofsolvent and/or the reaction medium employed will probably be further cut downfor economical and ecological reasons, with a solvent-free process being alreadydiscussed as the ultimate goal of the future.
With cellulose-based polymer materials, a 'cradle-to-grave' philosophy, im-plying ecocompatible manufacture, easy processing of high productivity, highperformance in use and subsequent safe disposal, is supposed to gain in rele-vance (Engelhardt, 1995). A combination of biodegradability and thermoplastic-ity is emphasized as a prosperous approach for the future (Engelhardt, 1996),centering on so-called mixed derivatives, e.g. ether esters of cellulose. Hy-droxyethyl or hydroxypropyl ether groups, in combination with phthalic acidester groups, with a possible posed esterification of free carboxyl groups, areexplicitly mentioned as interesting substituents. Cellulose-based surface coatingsof high compatibility are considered as an expanding area. Of high importanceare innovations in new application fields in medicine, biology and pharmacy.Cellulose grafting had met very limited success in the fiber field so far, but maypossibly be of future interest in connection with soluble cellulosics or with thesurface modification of paper pulp.
It can generally be assumed that large-scale cellulose esterification andetherification to conventional products will be performed also in the futuremainly in a heterogeneous system of reaction, at least at the beginning of theprocess. This again accentuates the necessity of still more effective and site-selective procedures of activation for the cellulose raw material. But in the nearfuture successful investigations of new cellulose solvents and the effective func-tionalization in these media to products with a highly uniform distribution offunctional groups along the polymer chains will result in the industrial applica-tion of this part of cellulose chemistry too.
5.5 Supramolecular Architectures
Today's successful engagement of numerous research groups in nano-structuresand defined colloids (Wegner, 1991 and 1992; Wegner et al., 1993; Schaub etal., 1995), in host-guest interaction and other principles of self-organization ofpolymers, with geometrical aspects gaining in relevance, must be envisaged by

5.5 New Supramolecular Architectures 323
cellulose chemists for tackling the preparation and characterization of morecomplex colloid systems and solid state structures. Some possible starting pointsand promising routes specific to cellulose shall now be considered briefly.
The study of the kinetics and the thermodynamics of dissolution of cellulosederivatives with a well-defined primary structure can help to elucidate the courseand the mechanism of dissolution of this polymer in large-scale processes and togain a deeper insight into the state of solution of these systems. Rheological, X-ray and light-scattering studies of cellulosic compounds of well-defined primarystructure in various solvents can provide data on persistence length and cluster-ing of the chains in dependence on polymer primary structure and concentration,and on solvent composition (Seger and Burchard, 1994). These data may beuseful for comparing experimental data with results obtained by molecular mod-eling.
A problem of high actuality with polymer solutions in general is the formationof liquid crystalline systems with lyotropic or thermotropic mesophases in de-pendence on polymer structure and concentration, solvent composition and tem-perature, and the characterization of these liquid crystalline systems predomi-nantly by optical and rheological techniques. For cellulosics, the relevance ofliquid crystalline phases with regard to film formation and fiber spinning is stillan open question. It is widely discussed today with respect to cellulose dissolvedin amine oxides or with respect to cellulose acetate solutions in various solvents.The study of mesophase formation and mesophase transition of cellulose deriva-tives with a well-defined primary structure and narrow chain length distributioncan provide a deeper insight into the structure-property relations of cellulose-based liquid crystalline systems, and can supply knowledge for answering someopen questions on commercial cellulose processing. Furthermore, cellulose-based liquid crystalline systems may constitute an interesting starting materialfor subsequent covalent or complex-forming reactions, especially crosslinkingprocesses (Guo and Gray, 1994; Zugenmaier, 1994; Müller et al., 1997).
Design and experimental realization of artificially ordered supramolecularstructures with one or more components based on tailored cellulose compoundsare liable to provide a deeper insight into intermolecular interactions betweencellulosic chains, and they can be employed to prepare cellulose-based Lang-muir-Blodgett layers or charged layers of defined architecture. Supramolecularstructures of this kind can find possible applications for example as sensors,light-wave conductors or selective membranes. On the other hand, they providereally exciting starting materials for subsequent chemical transformations withthe aim of attaching e.g. fluorophores and antibodies. Also, the immobilizationof enzymes, the preparation of sophisticated microcapsules and tailored medicaldevices like drug delivery systems can be mentioned here as possible goals ofapplication-oriented research in cellulosics.

324 5 Outlook onto Future Developments in Cellulose Chemistry
ReferencesAlbrecht, W., Forstarchiv 1987, 58, 254-255.Albrecht, W., Papier (Darmstadt) 1997, 57, 627-629.Berger, W., Keck, M., Philipp, B., Schleicher, H., Lenzinger Ber. 1985, 1-8.Burchard, W., Habermann, N., Klüfers, P., Seger, B., Wilhelm, U., Angew.
Chem. 1994, 706, 936-939.Burger, J., Kettenbach, G., Klüfers, P., Macromol Symp. 1995, 99, 113-126.Couley H., Smith, S., Lenzinger Ber. 1996, 75, 51-61.Engelhardt, J., Carbohydr. Eur. 1995, 72, 5-14.Engelhardt, J., Papier (Darmstadt) 1996, 50, 701-711.Fischer, K., Hinze, H., Schmitt, L, Papier (Darmstadt) 1996, 50, 682-688.Franzreb, J.P., Papier (Darmstadt) 1989, 43, V94-V97, V123.Geyer, U., Heinze, Th. Stein, A., Klemm, D., Marsch, S., Schumann, D.,
Schmauder, H.-P., Int. J. Biol Macromol 1994, 76, 343-347.Geyer, U., Klemm, D., Pavel, K., Ritter, H., Macromol. Rapid Commun. 1995,
76,337-341.Guo, J.-X., Gray, D.G., in Cellulosic Polymers, Blends and Composites, Gilbert,
R.D. (Ed.), Munich: Hanser, 1994, pp. 25-41.Heinze, Th., Habilitation Thesis, Friedrich Schiller University of Jena, 1997.Heinze, Th., Rahn, K., Macromol. Rapid Commun. 1996, 77, 675-681.Heinze, Th., Glasser, W.G., in Recent Advances in Cellulose Derivatives:
Heinze, Th., Glasser, W.G. (Eds.), ACS Symp. Ser., 1997, in press.Itagaki, H., Tokai, M., Kondo, T., Polymer 1997, 38, 4201-4205.Kamide, K., Saito, M., Macromol. Symp. 1994, 83, 233-271.Klemm, D., Stein, A., Heinze, Th., Philipp, B., Wagenknecht, W., in Polymeric
Materials Encyclopedia: Synthesis, Properties and Application, Salamone,J.C. (Ed.), Boca Raton, FL: CRC Press, 1996, pp. 1043-1054.
Klemm, D., Heinze, Th., Philipp, B., Wagenknecht, W., Acta Polym. 1997, 48,277-297.
Kobayashi, S., Shoda, S., Uyama, H., Adv. Polym. Sd. 1995, 727, 1-30.Kondo, T., /. Polym. ScL Part B: Polym. Phys. 1994, 32, 1229-1236.Lee, J.W., Yeomans, W.G., Allen, A.L., Kaplan, D.L., Deng, F., Gross, R.A.,
Con. J. Microbiol. 1997, 43, 149-156.Müller, M., Zentel, R., Keller, H., Adv. Mater. 1997, 9, 159-162.Nakamura, S., Amano, M., J. Polym. ScL Part A: Polym. Chem. 1997, 35, 3359-
3363.Nakamura S., Sanada, N., Sen-I Gakkaishi 1997, 53, 467-470Nakatsubo, F., Kamitakahara, H., Hori, M., J. Am. Chem. Soc. 1996, 77S,
1677-1681.Ogawa, R., Tokura, S., Carbohydr. Polym. 1992a, 79, 171-178.

References 325
Ogawa, R., Tokura, S., Int. J. Biol Macromol 1992b, 14, 343-347.Ohnishi, A., Shibata, T., Cell Commun. 1997, 4, 2-6.Okamoto, E., Kiyosado, T., Shoda, S., Kobayashi, S., Cellulose 1997, 4, 161-172.Rahn, K., Ph.D. Thesis, University of Jena 1997.Riva, S., Chopineau, J., Kieboom, A.P.G., Klibanov, A.M., /. Am. Chem. Soc.
1988,770,584.Schaub, M., Fakirov, C., Schmidt, A., Lieser, G., Wenz, G., Wegner, G.,
Albouy, P.-A., Wu, H., Foster, M.D., Majrkzak, M., Satijy, S.,Macromolecules 1995, 28, 1221.
Seger, S., Burchard, W., Macromol Symp. 1994, 83, 291-310.Sollinger, S., Diamantoglou, M., Papier (Darmstadt) 1996, 50, 691-700.Spange, S., Reuter, Α., Vilmeier, E., Keutel, D., Heinze, Th., Linert, W.,
Polymer 1997, in press.Stscherbina, D., Philipp, B., Acta Polym. 1991, 42, 345-351.Therisod, M., Klibanov, A.M., J. Am. Chem. Soc. 1986, 708, 5683.Wegner, G., Ber. Bunsenges. Phys. Chem. 1991, 95, 1326.Wegner, G., Mol Cryst. Liq. Cryst. 1992, 276, 7.Wegner, G., Schaub, M., Wenz, G., Stein, A., Klemm, D., Adv. Mater. 1993, 5,
919.Yamamoto, H., Horii, F., Hirai, A., Cellulose (London) 1996, 3, 225-242.Yamanaka, S., Ono, E., Katanabe, K., Kusakabe, M., Suzuki, Y., Patent EU
0396344,1990; Chem. Abstr. 1992,114, 235093.
Zugenmaier, P., in Cellulosic Polymers, Blends and Composites, Gilbert, R.D.(Ed.), Munich: Hanser, 1994, pp. 71-93.

Appendix to Volume 2:
Experimental Proceduresfor the Functionalization of Cellulose
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, T. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

Preparation of FeTNa solvent for cellulose 331Dissolution of cellulose in A^W-dimethylacetamde (DMA)/LiCl 331Preparation of a cellulose trinitrate without significant chain degradation 332Sulfation of cellulose with SO3-DMF 332Cellulose sulfate, synthesis via cellulose trifluoroacetate in DMF 334Cellulose sulfate, synthesis via trimethylsilylcellulose in THF 335Preferentially C-6-substituted cellulose sulfate via an acetate sulfate
mixed ester 336Predominantly C-2/C-3-substituted cellulose sulfates 337Cellulose phosphate from a partially substituted cellulose acetate 338Preparation of a cellulose fiber xanthogenate and a cellulose
xanthogenate solution 339Cellulose tricarbanilate 340Cellulose phenylcarbamate, synthesis via cellulose trifluoroacetate
inpyridine 341Cellulose formate, synthesis in HCOOHTPOCl3 342Laboratory procedure for the preparation of cellulose triacetate
by fiber acetylation 343Acetylation of bacterial cellulose 344Site-selective deacetylation of cellulose triacetate 344Cellulose dichloroacetate, synthesis with dichloroacetic acid/POC!3 345Cellulose trifluoroacetate (DS = 1.5), synthesis with TFA/TFAA 346Cellulose methoxyacetates, synthesis inDMA/LiCl 347Cellulose-4-nitrobenzoate, synthesis via cellulose trifluoroacetate
catalyzed withp-tosyl chloride 348Cellulose-4-nitrobenzoate, synthesis via cellulose trifluoroacetate with
4-nitro-benzoic acid imidazolide 349Cellulose tosylate, homogeneous synthesis in DMA/LiCl 3502,3-Di-O-methylcellulose 352Carboxymethy!cellulose, heterogeneous synthesis in isopropanol/water 353Carboxymethylcellulose, synthesis in DMA/LiCl 355Carboxymethylcellulose, synthesis via cellulose trifluoroacetate
in DMSO 3576-O-Triphenylmethyl (trityl) cellulose, homogeneous synthesis
in DMA/LiCl 3592,3-0-Carboxymethyl-6-O-triphenylmethylcellulose, synthesis via
6-O-tritylcellulose in DMSO 361Detritylation of 2,3-0-carboxymethyl-6-0-tripheny!methyl cellulose 362Crosslinking of cellulose powder with epichlorohydrin 363Organosoluble cyanoethylcellulose 364Trimethylsilylcellulose, synthesis in pyridine/THF 365Trimethylsilylcellulose, synthesis in DMA/LiCl 367Celluloses esters, synthesis via trimethylsilylcellulose, general
procedure without solvents 368

6-O-Thexyldimethylsilylcellulose 3702,6-Di-O-thexyldimethylsilylcellulose 3716-O-Thexyldimethylsilyl-2,3-di-O-methylcellulose 372Trimethylsilylcellulose methoxyacetate. synthesis via cellulose
methoxyacetate in DMA 3736-Carboxycellulose, homogeneous synthesis with phosphoric acid 374

Appendix (Volume 2) 331
Preparation of the FeTNa solvent for cellulose
For the preparation of 1 1 of FeTNa solvent, 217.1 g of sodium tartrate dihydrate[Na2(C4O4H6)^H2O] are dissolved in 550 ml of distilled water with vigorousstirring under exclusion of light. FeCl3-OH2O (81.1 g) is added and completelydissolved in the mixture with continuous stirring. Subsequently, a cooled solu-tion of 96 g of solid NaOH in 180 ml of distilled water is added dropwise from adropping funnel, cooling the mixture with iced water and keeping its tempera-ture below 20 0C. The solution is then transferred to a 1 1 calibrated flask, filledup to 1 1 with distilled water and is then immediately shaken for complete mix-ing in order to avoid hydrolysis of the complex at the water/lye interface. Thesolvent is then kept in a brown flask in a refrigerator.
ReferenceNagler, H., Ph.D. Thesis, Technical University of Dresden 1994.
Dissolution of cellulose in Λ^ΛΤ-dimethylacetamide (DMA)TLiCl
Method A [2.5 % (w/w) cellulose to DMA/LiCl]Dried cellulose (5 g, 30.9 mmol; at 100 0C under vacuum) was suspended in200 ml of DMA and kept at 130 0C for 2 h under stirring. After the slurry hadbeen allowed to cool to 100 0C, 10 g of anhydrous LiCl (dried at 130 0C for 2 hunder vacuum) were added. By cooling to room temperature under stirring thecellulose dissolved completely.
Method B [4.3 % (w/w) cellulose to DMA/LiCl]Air-dry cellulose [20 g, 5 % (w/w) water, 117 mmol of AGU] was suspended in470 ml of DMA and kept at 160 0C for l h under stirring. In order to replace thecellulose-bound water, about 40 ml of DMA were removed by distillation undera nitrogen atmosphere. After the slurry had been allowed to cool to 100 0C, 40 gof anhydrous LiCl were added. By cooling down to room temperature understirring, the cellulose dissolved completely within some hours.
ReferenceRahn, K., Diamantoglou, M., Klemm, D., Berghmans, H., Heinze, Th., Angew.
Makromol. Chem. 1996, 238, 143-163.

332 Appendix (Volume 2)
Preparation of a cellulose trinitrate without significant chain degra-dation
The preparation of a cellulose trinitrate for subsequent physicochemical investi-gations is performed at O 0C in a mixture of concentrated nitric acid and dichlo-romethane according to the following procedure.
Concentrated nitric acid (white fuming nitric acid, 20 ml) and dichlo-romethane (20 ml) are mixed in a beaker and cooled to O 0C. A 0.75 g sample ofdry cellulose is added and the mixture is kept at O 0C with occasional stirring for30-90 min. A reaction time of 30 min is adequate for e.g. a hydrolytically de-graded cellulose powder, and 90 min for e.g. high-DP cotton !inters. The fibrouscellulose trinitrate is filtered off on a coarse sintered-glass filtering crucible,washed three times with CH2Cl2, three times with methanol, once with water of40-50 0C, and finally again with methanol. The cellulose trinitrate is dried at20 0C under vacuum.
ReferenceLaboratory procedure of Fraunhofer Institute of Applied Polymer Research.
Sulfation of cellulose with SO3-DMF
Preparation of SO3-DMF complex (also commercially available)MA^Dimethylformamide (DMF, 1500 ml) was cooled by stirring in a 3000 ml,three-necked round-bottomed flask immersed in an ice bath. The flask wasequipped with a mechanical stirrer, a CaCl2 tube, and a dropping funnel. Sulfurtrioxide (900 g) was then added dropwise during 2-3 h. The reaction was highlyexothermic, and care had to be taken to maintain the temperature below about40 0C. The DMF-SO3 complex was obtained as a yellowish, crystalline mass,wet with excess DMF. This mixture of complex and DMF was stored underrefrigeration and used in the following sulfations without filtration or furtherpurification. The amounts of complex given below refer to this mixture and notto the actual amount of complex.
Sulfation of celluloseCellulose (100 g), dried for 3 h at 100 0C, was mixed with 300-700 ml of DMFand kept for several h at -25 0C. The mixture was then cooled in a refrigerator,placed in a jacketed Day Mixer, and 450-500 g of the DMF-SO3 complex,cooled to 5 0C, was added to three equal portions. The mixer was cooled bycirculating iced water through the jacket. The temperature was maintained below15 0C throughout the reaction. The total reaction time was about 3 h. The reac-

Appendix (Volume 2) 333
tion mixture was dissolved in iced water, neutralized with dilute sodium hy-droxide, and filtered through a Büchner funnel. The product was precipitated bypouring the solution slowly into 1 volume of methanol, and was pressed out anddried. For further purification, the product was redissolved in water and repre-cipitated with methanol.
Determination of the DSThe product was dissolved and dialyzed against water for 48 h. The dialyzatewas concentrated to low volume and the product was precipitated by methanol,and dried in vacuo at 80 0C. An aliquot was dissolved in 10 % hydrochloric acidand the solution was refluxed overnight. After filtration, the sulfuric acid wasprecipitated with barium chloride and weighed as barium sulfate, whose weightindicated the DS. Amounts of barium sulfate indicating DS values of 1, 2 and 3were calculated, and the values were plotted on a curve versus the DS. The DSvalues of the products were taken from this curve. The procedure is suitable toobtain DS§ values of up to 2.8.
ReferenceSchweiger, R.G., Carbohydr. Res. 1972, 27, 219-228.

334 Appendix (Volume 2)
Cellulose sulfate, synthesis via cellulose trifluoroacetate in DMF
^ pyridine / SO3x 'RO-~_
OR
R = H, COCF3
according to DS
(DMF), 10°- 150C, 3h NaO3SO- _OR
R ' = H , SO3Na
according to DS
CTFA (1 g, DS 1.5) was dissolved in 17 ml of DMF under cooling to O 0C andan inert gas atmosphere. Pyridine-SO3 complex (2.1 g, 4 mol/mol of AGU) wasadded and the reaction mixture was stirred for 3 h at 10- 15 0C: 30 ml of waterwere added. The mixture was neutralized with 5 % (w/w) NaOH and precipi-tated in 150 ml of methanol.
Yield: 1.14 g of the product with DS 0.98.FTIR (KBr): 809 cm-1 (S-O), 1242 cm-1 (S-O)13C NMR (D2O): δ = 60-61 ppm unsubstituted C-6 atom
δ = 70-101 ppm C-I, -2, -3 and -4 atom signals are all split
The sample was soluble in water. Cellulose sulfate with DS 0.41 via cellulosedichloroacetate is obtained by the same procedure. The sample is soluble in wa-ter. Cellulose sulfate with DS 0.56 via cellulose formate are obtained by thesame procedure. This sample is also soluble in water.
ReferenceKlemm, D., Heinze, Th., Stein, A., Liebert, T., Macromol Symp. 1995, 99, 129-
140.

Appendix (Volume 2) 335
Cellulose sulfate, synthesis via trimethylsilylcellulose in THF
OSi(CH3)3
O
OSi(CH3)3
(i) THF, rt
(H) NaOH/methanol ROOR
= H, SO3
0 A/a®
To a solution of 15.57 g (0.042 mol) of trimethylsilylcellulose (Buckeye !intersDP 1470) in 360 ml of dry THF, a solution of 17.36 g (0.113 mol) of sulfur tri-oxide/dimethylformamide complex in 100 ml of dry DMF was added. Afterstirring for 2.5 h at room temperature, the reaction mixture, with the precipitatedproduct, was poured into a solution of 10.7 g (0.267 mol) of sodium hydroxidein 21 of methanol. The precipitate was filtered off, carefully washed withmethanol, dissolved in 500 ml of water, and reprecipitated into ethanol. Afterfiltration and washing with ethanol, the sample was dried at 50 0C under vac-uum.
Yield: 8.04 g (69 %) of the pure product with DS 1.13.FTIR (KBr): 806 (δ S-O), 1240 (ν SO2) cnr1
13CNMR: 66.9 (C-6, completely sulfated), 73.3-79.0 (C-2, C-3, C-4, C-5),100.9 (C-I, sulfatedat C-2), 102.8 (C-I, unsulfated at C-2) ppm.
The degree of substitution based on the elemental analysis was calculated ac-cording to the equation:
MAGU-m%S
The sample was soluble in water.

336 Appendix (Volume 2)
TMS-CelluloseSample no.
12
Molar ratioSO3-DMF/AGU
2.510
Time(h)
2.56
DSS*
1.132.04
S
(%)
13.0817.52
a DS calculated on the basis of sulfur analysis.
ReferenceKlemm, D., Schnabelrauch, M., Stein, Α., Philipp, B., Wagenknecht, W., Nehls,
L, Das Papier 1990, 44 (72), 624-632.
Preferentially C-6-substituted cellulose sulfate via an acetate sulfatemixed ester
A 5 g sample of scoured and bleached cotton !inters is suspended in 250 ml ofdry DMF (water content 0.01-0.02 %). Then 9 g of acetic acid anhydride and7 g of chlorosulfonic acid are added and the mixture is reacted for 8 h at 50 0Cunder stirring. The polymer is dissolved during this procedure and a clear solu-tion is obtained. For precipitation of the polymer, the reaction mixture is pouredslowly into a large excess of ethanol containing 4 % (by weight) sodium acetate.The precipitate is filtered off on a glass filter disk and washed free of sulfate ionswith ethanol containing 20-30 % water. The resulting cellulose acetate sulfate isfurther purified by washing with ethanol and then suspended in 4 % (by weight)NaOH in ethanol, employing a molar ratio of 3 NaOH/mol of AGU, corres-ponding to 90-100 ml of the above-mentioned solution. This suspension isstirred for l h at room temperature and then left for about 15 h before filteringoff the deacetylated product and washing it with ethanol. Finally, the suspensionof the product in ethanol is neutralized to pH = 8 with acetic acid in order toobtain a completely neutral sodium salt of the cellulose sulfuric acid half-ester.Finally the product is again washed with ethanol and dried at 50 0C.
ReferenceWagenknecht, W., Procedure of the Fraunhofer Institute of Applied Polymer
Research, Teltow-Seehof.

Appendix (Volume 2) 337
Predominantly C-2/C-3-substituted cellulose sulfates
Site-specific sulfation is achieved by reacting a predominantly C-6-substitutedcellulose acetate with amidosulfonic acid in a polar aprotic medium, employingthe acetyl groups as protecting groups.
A predominantly C-6-substituted cellulose acetate (15 g) of DS 1.5-2.0 isdried at 105 0C and then dissolved in 100 ml of dry DMF at 80 0C. Subsequently17 g of NH2SO3H dissolved in 70 ml of DMF are added within 5 min, and themixture is stirred for 90 min at 80 0C. The resulting polymer is then precipitatedwith ethanol containing 3 % (by weight) sodium acetate, washed with ethanolfree from low molecular salts and then dried at 60 0C. The cellulose mixed esterobtained exhibited about the same DS level of acetyl groups as the starting mate-rial, e.g. DSAc = 1.55, and a DS of sulfate half-ester groups of up to 1.
For a quantitative deacetylation of the mixed ester a solution containing 4 %(by weight) NaOH and 8 % (by weight) water in ethanol was employed. Thecellulose ester was suspended in 300 ml of this mixture at 20 0C for 24 h to se-cure a complete deacetylation without affecting the sulfate half-ester groups.Subsequently the system was neutralized to pH 7-8, and the sodium-cellulosesulfate was filtered off and washed with ethanol, and finally dried at 50 0C.
An exemplary product obtained by this route from a cellulose acetate with aDS of 1.55 had a DS of sulfate half-ester groups of 1.1, with partial DS values of0.75 at C-2, 0.15 at C-3 and 0.2 at C-6.
ReferenceWagenknecht, W., Papier (Darmstadt) 1996, 50, 712-720.

338 Appendix (Volume 2)
Cellulose phosphate from a partially substituted cellulose acetate
Cellulose acetate (15 g, DS 1.5-2) is dried at 105 0C and dissolved in 150 ml offreshly distilled dry DMF (water content 0.01-0.02 %) at 80 0C with vigorousstirring. For a phosphation of the free hydroxy groups, a solution of 30 g ofpolytetraphosphoric acid (P4O13H6) in 200 ml of DMF and subsequently 67 g oftri-W-butylamine are added, and the system is kept for 6 h at 120 0C under stir-ring. The mixed cellulose, containing acetate and phosphate ester groups, is pre-cipitated by pouring the reaction system into 3 times the volume of a 2 % (byweight) solution of sodium acetate in ethanol. The precipitate is filtered off andwashed with ethanol acidified to pH 2 by addition of HCl until the filtrate is freeof phosphate ions. Then the product is reneutralized to pH 7-8 with NaOH inethanol, washed with ethanol again, and then dried at 50 0C. This mixed ester isthen deacetylated for the preparation of a cellulose phosphate via a mixed ester.The resulting anionic phosphate ester of cellulose exhibited a DSp between 0.5and 1, the DS value being obtained by elemental analysis.
ReferenceWagenknecht, W., Procedure of the Fraunhofer Institute of Applied Polymer
Research, Teltow-Seehof.

Appendix (Volume 2) 339
Preparation of a cellulose fiber xanthogenate and a cellulosexanthogenate solution
Preparation of alkali celluloseAir-dry cotton !inters or wood pulp (100 g) is dispersed under stirring at roomtemperature (ca. 20 0C) in 2 1 of aqueous NaOH of 18 % concentration (byweight), and the slurry is kept for l h at room temperature for complete trans-formation to sodium cellulose. Then the fibrous polymer is filtered off andpressed in a suitable processing device to a press weight ratio of 1 : 2.8 (± 0.1).The press cake of alkali cellulose is disintegrated in a shredder and then kept in aclosed bottle for 2 days at room temperature in order to reduce the DP to a levelof about 400 by alkaline oxidative degradation (see chapter 2.3). An alkali-cellulose composition of 32-34 % cellulose, 15-16 % total NaOH and less than1 % Na2CO3 is adequate for the subsequent xanthogenation. The alkali contentis determined by acidimetric titration after suspending a weighed sample inCO2-free water and heating to boiling, the cellulose content being obtained bydecomposing a weighed sample with acetic acid and assessing the dry weight ofcellulose after thorough washing.
XanthogenationXanthogenation is performed in a round-bottomed flask equipped with a closedfunnel with stop cock and a stop-cocked outlet for evacuation. A 100 ml flask isadequate for the xanthogenation of about 5 g of alkali cellulose. The amount ofCS2 to be added depends on the DS level of the xanthogenate intended: 40 % CS2
by weight (on the basis of dry cellulose input) is adequate for a DS level of 0.5,while 150-200 % is necessary for reaching the maximal DS level of 1. The flaskwith the alkali cellulose is at first evacuated, and then the required amount of CS2
is sucked into the flask by the vacuum. Xanthogenation is found to be almost com-plete after 2 h at 30 0C, with the reaction mass being slowly rotated or occasionallyshaken by hand. If necessary for subsequent reactions, the fibrous cellulose xan-thogenate can be purified from low-molecular by-products like Na2CS3 and Na2Sby kneading with ice-cold saturated, aqueous, ammonium chloride solution.
The cellulose xanthogenate can be dissolved in 4 % (by weight) aqueousNaOH during 1-2 h at a temperature below 20 0C, employing a stainless steel orglass vessel equipped with a sufficiently powerful stirrer. After the above-mentioned partial chain degradation of the alkali cellulose, cellulose xanthogen-ate solutions with a cellulose content of 6-8 % can be obtained without diffi-culty with a CS2 input of 40 % based on dry cellulose.

340 Appendix (Volume 2)
ReferencesTreiber, E., Fex, O.F., Rehnström, J., Piova, M., Sven. Papperstidn. 1955, 58,
287-295.Treiber, E., Bergstedt, S., Rehnström, J., Stephan, A., Papier (Darmstadt) 1957,
77, 133-139 and 194-203.Treiber, E., Rehnström, J., Ameen, Ch., Kolos, F., Papier (Darmstadt) 1962, 76,
85-94.
Cellulose tricarbanilate
1 Heterogeneous synthesisThe cellulose sample is suspended in an excess of pyridine, with the amount ofpyridine depending on the DP of the sample. In the case of the high-DP !inters,about 11 of dry pyridine per g of cellulose is recommended. Then twice the stoi-chiometric amount of phenyl isocyanate required is added, and the mixture isstirred for 10-12 h at 100 0C. In the case of high-DP cellulose samples, addition ofabout the same volume of dry DMF and an increase of the reaction temperature to120 0C is advantageous. During the reaction the cellulose sample is completelydissolved. For isolation of the reaction product the mixture is poured into metha-nol, the precipitate is filtered off, washed with methanol and dried, and for furtherpurification reprecipitated from a solution in acetone. The DS of the tricarbanilateis determined by elemental analysis (nitrogen content) and is above 2.8.
2 Homogeneous synthesisA weighed amount of DMA (15-20 ml) was added to a weighed amount of cellu-lose (-200 mg). The mixture was heated to the reflux temperature for 20-30 min;after cooling to 100 0C, a weighed amount of LiCl was added under stirring (5 %).For samples at a degree of polymerization, stirring was continued for 2 more hoursat 70 0C. A catalytic amount of pyridine (0.4-1 ml) and phenyl isocyanate (2 ml)were added and the reaction was carried at 60-70 0C for 2-3 h. After cooling, drymethanol (2 ml) was added to eliminate excess phenyl isocyanate and the mixturewas precipitated in methanol or a water/methanol mixture (30 : 70). After washingwith water, the cellulose carbanilates were dried under vacuum.
ReferencesBurchardt, W., Husemann, E., Macromol Chem. 1961, 44, 358-387.Terbojevich, M., Cosani, A., Camilot, M., Focher, B., /. Appl. Polym. ScL 1995,
55, 1663-1671.

Appendix (Volume 2) 341
Cellulose phenylcarbamate, synthesis via cellulose trifluoroacetate inpyridine
Cellulose trifluoroacetate (Ig, DS= 1.5) was dissolved in 17 ml of pyridineunder cooling and an inert gas atmosphere. Then 1.42 ml (4 mol/mol of AGU) ofphenyl isocyanate and about 0.01 g of dibutyltin dilaurate were added. Afterstirring the reaction mixture for 16 h at room temperature under an inert gasatmosphere, the product was precipitated in 150 ml of water. The filtrate wasextracted for 72 h with ethanol.
OCOCF3
O
0,, +
''RO^V1^—^OR
R = H, COCF3
according to DS
dibutyltin dUautrate
(pyridine) , rt, 16h '
R'= H ,
according to DS
Yield: 0.84 g (80.78 %) of the pure product with DS 1.3.FTIR (KBr): 1724 crrr1 v (C=O)13C NMR (DMSO-i/6 at 70 0C): δ = 118.8 ppm, 122.3 ppm, 128.3 ppm and
138.7 ppm (C-Harom);6 = 153.1 ppm (O-CO-NH-C6H5)
The sample was soluble in dimethyl sulfoxide.
ReferenceLiebert, T., Ph.D. Thesis, University of Jena 1995.

OCOHO
342 Appendix (Volume 2)
Cellulose formate, synthesis in HCOOHTPOCl3
OHO
+ HCOOH / POCI3 ^
R = H ,COH
according to DS
Spruce sulfite pulp (Ig, 6.2 mmol), dried 48 h over ?2θ5 at room temperature,was suspended in 25 ml of formic acid and stirred for 20 min at room tempera-ture. Then 2.3 ml of POCl3 (4 mol/mol of AGU) was added within about 10 minat O 0C. The reaction mixture was stirred at 100 rpm for 5 h at room temperature.After this time the solution was homogeneous. The reaction product was pre-cipitated in 70 ml of diethyl ether, washed twice with 100 ml of acetone, driedand washed again with 70 ml of acetone. In the case of spruce sulfite pulp, driedat higher temperature, the POCl3 used (3 ml) was partially hydrolyzed with 0.89ml of water within 30 min at O 0C, and within 24 h at room temperature understirring.
Yield: 1.09 g (79 %) of the pure product with AS 2.2.FTIR (KBr): 1730 cm-1 v (C=O)13C NMR (DMF-J7 at 70 0C): δ = 163.3 ppm (C=O), δ = 61.9 ppm substituted
C-6 atom
The sample was soluble in Λ^,Λ^-dimethylformamide, dimethyl sulfoxide andpyridine.
ReferenceLiebert, T., Klemm, D., Heinze, Th., /. Macromol ScL9 Pure AppL Chem. 1996,
A3 3(5), 613-626.

Appendix (Volume 2) 343
Laboratory procedure for the preparation of cellulose triacetate byfiber acetylation
Air-dry cotton !inters (25 g) are activated by swelling in ca. 750 ml of glacial ace-tic acid in a wide-necked bottle and rotated for 5 h at room temperature. Aftersucking and pressing off the acetic acid, the swollen cellulose is returned to thebottle, and an acetylating mixture (prepared as follows) is added immediately:235 ml of acetic anhydride and 225 ml of benzene (free of thiophene) are mixedand cooled to -20 0C, and 0.15 ml of perchloric acid (70 % by weight) and 0.15 mlof concentrated sulfuric acid are added. The complete reaction system in the bottleis rotated again at room temperature (ca. 20 0C) with the reaction temperaturegradually increasing to 26-28 0C. At a higher external temperature some cooling isrequired to avoid a reaction temperature above 29 0C, resulting in severe yieldlosses of product. The progress of acetylation is followed by polarization micros-copy: close to the formation of the cellulose triacetate the positive birefringence ofthe cellulose fibers changes to the negative birefringence of the triacetate fibers. Assoon as the sample exhibits rather uniform and negative birefringence (after aboutl h of reaction), samples of the fibrous material of about 2 g are withdrawn every30 min, washed free of acid with benzene, and after sucking and pressing off thebenzene boiled out with water, solvent exchanged with methanol and diethyl etherand dried. A I g sample of the product is dissolved in 25 ml of a 9 : 1 mixture ofmethylene chloride and methanol, and the solution viscosity is measured. A nearlylinear plot of log η versus reaction time is obtained, permitting an extrapolation tothe product viscosity intended. After reaching this point with the whole reactionsystem, the acidity of the catalyst is buffered by a saturated solution of potassiumacetate in acetic acid (about 150 % of the amount theoretically required). Then150 ml of benzene are added, the liquid phase is drawn off, and the fibrous productis washed free of acid with benzene, and subsequently the benzene is removed bya water-vapor distillation. After a final washing with distilled water to eliminatethe last traces of acid, the product is dried at 110 0C to a residual water content ofabout 3 %. The DS of acetyl groups is controlled by saponification with an excessof alkali and back-titration of the excess with acid. According to the followingprocedure: a cellulose acetate sample of 50 mg is swollen for 24 h at room tem-perature in an acetone/water mixture (1 : 1 by volume). Then 12.5 ml of 1 N KOHin ethanol are added, and after a residence time of 24 h at room temperature, theexcess of alkali is titrated with 0.5 N HCl, using phenolphthalein as indicator. Toassess the last traces of alkali still adhering to the sample, again 2 ml of 0.5 N HClare added and after 2 h titrated with 0.5 N NaOH. A blank titration without cel-lulose acetate is recommended.
ReferenceBischoff, K.H., Ph.D. Thesis, University of Leipzig 1963.

344 Appendix (Volume 2)
Acetylation of bacterial cellulose
OHCH3COOH/ Ac2OfH2SO4
Dry bacterial cellulose (1.5 g, 9.3 mmol) was immersed in glacial acetic acid for15 min. After filtration, 100 ml of glacial acetic acid containing 1.2 ml of sulfu-ric acid was added. The flask was intensively agitated for l min, 6 ml(105 mmol) of acetic anhydride were added, and the suspension was stirred for 6h, to reached a DS of between 2.0 and 2.5 (for determination of acetyl contentsee acyl group analysis). Other DS values were possible by variation of reactiontime. Under stirring, a solution of 4 ml of water and 9 ml of acetic acid wasadded to the mixture. After 30 min, the mixture was dispersed in water and fil-tered. The solids were washed with aqueous sodium bicarbonate and water. Theproduct was dried at 60 0C in vacuo.FTIR (KBr): 1750 (v C=O) cm"1, typical absorptions of cellulose backbone
ReferenceDicke, R., Diploma Thesis, Friedrich-Schiller-University of Jena, 1996.
Site-selective deacetylation of cellulose triacetate
Commercial triacetate (200 g, 0.7 mol) with a DS of 2.9 are dissolved in 3.6 1 ofDMSO at 80 0C under vigorous stirring in vacuum. For partial deacetylation, amixture of 188g of hexamethylene diamine (1.6 mol) and 280ml of water(15.5 mol) is added within 5 min. The reaction system is then kept for 14 h at80 0C with continuous stirring. After precipitation in an excess of ethanol,washing of the precipitate with ethanol and drying at 50 0C, a partially deacety-lated product with a total DS^C of 1.5 is obtained. The partial DS values assessedby 13C NMR spectroscopy in the different positions were 0.2 at C-2, 0.45 at C-3and 0.85 at C-6.
ReferenceWagenknecht, W., Procedure of the Fraunhofer Institute of Applied Polymer
Research, Teltow-Seehof.

Appendix (Volume 2) 345
Cellulose dichloroacetate, synthesis with dichloroacetic acid/POC!3
OHO
+ CHCI2COOH / POCI3
rt, 14h
= H , COCHCI2
according to DS
Spruce sulfite pulp (1 g, 6.2 mmol), dried for 48 h over P2O5 at room temp-erature, was suspended in 25 ml of dichloroacetic acid and stirred for 20 min atroom temperature. Then 5.75 ml of POCl3 (10 mol/mol of AGU) were addedwithin about 10 min at O 0C. The reaction mixture was stirred at 100 rpm for14 h at room temperature until the solution was homogeneous. The reactionproduct was precipitated in ether and reprecipitated twice with acetone and hex-ane. The product was dried for 40 min at 105 0C under vacuum.
Yield: 1.42 g (67.94 %) of the pure product with DS 1.6.FTIR (KBr): 1762 cnr1 v (C=O)13C NMR (DMSO-J6): δ 64.8 ppm (Q-CO-CHCl2); δ 164.1 ppm (O-CO-
CHCl2)
ReferenceLiebert, T., Klemm, D., Acta Polym. 1997, in press.

346 Appendix (Volume 2)
Cellulose trifluoroacetate (DS = 1.5), synthesis with TFA/TFAA
. CF3COOH / (CF3CO)2O
Trifluoracetic acid (TFA 20 ml) was added to l g (6.2 mmol) of cellulose andthe mixture was kept for 20 min at room temperature. Then 10 ml of TFAA (tri-fluoroacetic acid anhydide) were added and the mixture was stirred at roomtemperature for 4 h. Within 2-3 h the solution was homogeneous. In the case ofcotton !inters, 40 ml of TFA and 20 ml of TFAA are used. Diethyl ether (200ml) is passed through the solution to precipitate the reaction product. The whiteprecipitate is filtered off, washed with diethyl ether, and dried at room tempera-ture for at least 20 h under vacuum. The crude product still contains traces ofTFA and diethyl ether. These impurities can be removed by heating the productto 150 0C for 40 min under vacuum.
Yield: 1.78 g (94.2 %) of the pure product with DS 1.5.
FTIR (KBr): 1790 cm-1 v (C=O)13C NMR (DMF-J7 at 70 0C): δ = 67.9 ppm substituted C-6 atom
δ = 116.6 ppm (G-CO-CF3)
The sample was soluble in Λ^,,/V-dimethylformamide, dimethyl sulfoxide, tri-methyl phosphate and pyridine.
ReferenceLiebert, T., Schnabelrauch, M., Klemm, D., Erler, U., Cellulose 1994, 7, 249-
258.

Appendix (Volume 2) 347
Cellulose methoxyacetates; synthesis in DMA/LiCl
To a solution of 1.1 g (6.67 mmol) of Avicel in 50 ml of DMA and 3.3 g of LiCl(dissolution procedure A) in a three-necked flask equipped with a stirrer, a mixtureof 10 ml (0.12 mol) of dried pyridine and 20 ml of DMA was added under inertatmosphere. Methoxyacetyl chloride (6.5 g, 0.06 mol) was added within 30 min.The stirring was continued at room temperature. Then the solution was left tostand overnight and stirred for a further 6 h at 30 0C. The homogeneous reactionmixture was precipitated into 250 ml of methanol, filtered off, suspended in 96 %(w/w) ethanol and carefully dispersed. After filtration and washing with ethanol(four times with approx. 25 ml of ethanol), the sample was dried at 50 0C undervacuum.
OH
HO
O\
** ,
OH
pyridine
+ COCICH2OCH3
OCOCH2OCH3
O
(DMA/LiCl)24h, r t / 6h, 3O0C
OR
R = H, COCH2OCH3
according to DS
Yield: 2.93g (94.7 %) of the pure product with DS 3.0.FTIR (KBr): 2950 (v O-H), 1770 (v C=O) cm-1
The value of DS, 3, was determined by saponification with 0.5 N NaOH andfollowing titration.
Solubility of cellulose methoxyacetates with several DS values
DSa Molar ratipb
acyl chloride/AGUSolubility
3.02.21.70.8
9.04.03.01.5
DMSO, DMA, CH2Cl2, CHCl3DMSO, DMF, DMADMSO, DMF, DMADMF, DMA
a Determined by saponification.b 6.67 mol of Avicel, 2 mol pyridine/mol of methoxyacetyl chloride.

348 Appendix (Volume 2)
ReferencesSiegmund, G., Diploma thesis, Friedrich-Schiller-University of Jena, 1993.Tanghe, L.J., Genung, L.B., Mench, J.W., Methods in Carbohydrate Chemistry
1963, Vol. 3, New York: Academic Press.
Cellulose-4-nitrobenzoate, synthesis via cellulose trifluoroacetatecatalyzed with/?-tosyl chloride
' ROOR
R = H, COCF3
according to DS
(pyridine) , 7O0C, 7 h
ORO
O^
CTFA (Ig) was dissolved in 40 ml of pyridine under an inert gas atmosphereand 0.54 g of 4-nitrobenzoic acid (4 mol/mol of AGU) was added. After 30 min,2.49 g of p-tosyl chloride was added and the reaction mixture was heated to70 0C for 7 h. Then the reaction product was precipitated into 150 ml of water,washed with acetone and extracted with diethyl ether.
Yield: 0.75 g (86.21 %) of the pure product with DS 0.71.FTIR (KBr): 1727 cm-1 v (C=O)13C NMR (DMSO-J6): δ = 164.1 ppm (-CO-); δ - 123.8 ppm, 130.8 ppm,
134.8 ppm, 150 ppm (C-Harom)
ReferenceLiebert, T., Ph.D. Thesis, University of Jena 1995.

Appendix (Volume 2) 349
Cellulose-4-nitrobenzoate, synthesis via cellulose trifluoroacetatewith 4-nitrobenzoic acid imidazolide
OCOCF,
OR
R = H, COCF3
according to DS
(DMF) , 6O0C, 9h
+ NO2^/ \> CO-N
4-Nitrobenzoic acid (2.18g, 4 mol/mol of AGU) was dissolved in 20 ml ofDMF, and 2.11 g of TV, ./V-carbonyldiimidazole was added at room temperatureand stirred until the evolution of CO2 stopped. The solution was mixed with asolution of l g of CTFA in 17 ml of DMF, and was stirred at 60 0C for 9 h andthen 16 h at room temperature. The reaction product was precipitated into 150ml of water and the product was washed with acetone and extracted for 48 hwith diethyl ether.
Yield: 0.89 g (91.75 %) of the pure product with DS 0.91.
FTIR (KBr): 1727 cm-1 v (C=O); 1540 cm-1 vas (NO2); 1350 cnr1 vs (NO2)13C NMR (DMSO-J6): δ = 164.1 ppm (-CO-); δ = 123.8 ppm, 130.8 ppm,
134.8 ppm, 150.3 ppm (C-Harom)
The sample was soluble in W, 7V-dimethylformamide and dimethyl sulfoxide.
ReferenceLiebert, T., Ph.D. Thesis, University of Jena 1995.

350 Appendix (Volume 2)
Cellulose tosylate, homogeneous synthesis in DMA/LiCl
OHO
+ Cl-Tosx HO- _
OH
TEA
(DMA/LiCI) , 80C, 24h
R = H , Tosaccording to DS
Tos =
To a solution of 20 g (1 18.7 mmol, calculated for the water- free product/air-dry)of cellulose (see Table below) in 430 ml of DMA and 40 g of LiCl, a mixture of99.2 ml (712 mmol) of triethylamine and 68 ml of DMA was added in a cylin-drical glass reactor equipped with a stirrer, at room temperature. After cooling to8 0C a solution of 67.9 g (356 mmol) of /?-toluenesulfonyl chloride (3 mol/molof AGU) in 100 ml of DMA was added within 30 min. The homogeneous reac-tion mixture was stirred for a further 24 h at 8 0C and then slowly passed into 5 1of iced water. The white precipitate was filtered off, carefully washed with about15 1 of distilled water and 2 1 of ethanol, redissolved in 1 1 of acetone and repre-cipitated in 3 1 of distilled water. After filtration and washing with ethanol, thesample was dried at 50 0C under vacuum.
Yield: 45.2 g (87 %) of the pure product with DS 1. 79.FTIR (KBr): 3523 (v OH), 3072 (v C-Harom), 2891 (v CH), 1598, 1500, 1453
(v C-C8101n), 1364 (vas SO2), 1177 (vs SO2), 814 (δ C-Harom)cm"1
13CNMR: 20.7 (CH3); 59.9-105.0 (cellulose backbone); 125.3-144.7(C-Harom) ppm
The degree of substitution DS$ = 1. 79 was determined by ultimate analysis onthe basis of sulfur content (S = 13.05 %) and calculated according to the equa-tion:

DS=
Appendix (Volume 2) 351
ΜΑΠΙΓMS · 100% - Mtosyl group'S(%)
The chlorine content (0.45 %) of the sample 1 was determined by ultimateanalysis (see Table below). The intrinsic viscosity [17] = 1.18 dl/g, was deter-mined in dimethyl sulfoxide solution with an Ostwald viscometer (Schott AG,Mainz) at 32 0C.
Sample 1 was soluble, for example in acetone, acetylacetone, dimethyl sul-foxide, ^V,7V-dimethylacetamide, Λ^,Λ^-dimethylformamide, THF, dichloro-methane and dioxane.
Cellulosesample no.
1234
Molar ratio3
Tos-Cl/AGU
3.01.50.90.6
DSs
b
1.790.930.590.38
S(%)
13.059.756.235.69
Cl(%)
0.450.450.400.35
a 0.12 mol of AGU, 4.3 % (w/w) solution, 2 mol triethylamine/mol /?-toluenesulfonylchloride (Tos-Cl).b DS: calculated on the basis of sulfur analysis.
ReferenceRahn, K., Diamantoglou, M., Klemm, D., Berghmans, H., Heinze, Th., Angew.
Makromol Chem. 1996, 238, 143-163.

352 Appendix (Volume 2)
2,3-Di-0-methylcellulose
OTDMS
O^ + TBAF · 3 H2O
OCH3 TBAF = tetrabutylammoniumfluoride
OHO
THF, 7Gf, 5O0C H3CO- UUM
2,3-Di-O-methyl-6-0-thexyldimethylsilylcellulose (45.Og, 0.138mol, DSSi0.9,DSMe2.1) was dissolved in 500ml of THF. Tetrabutylammonium fluoride(78.5 g, 0.25 mol, 2 mol/mol of silyl groups) was added. The mixture was stirredfor 1 day at 50 0C. After cooling down to 25 0C, nearly 250 ml of THF was dis-tilled off by using a rotary evaporator. The solution was poured into 500 ml ofdiethyl ether and the precipitation was completed by addition of 250 ml of hex-ane. The polymer was collected and dried under vacuum. For purification it wassuspended in 0.57 1 of water at least 4 times, stirred and centrifuged. The solidpolymer was dried over potassium hydroxide at 50 0C and p < O. l Torr. Furtherpurification was carried out by dissolving the polymer in chloroform/methanol(4:1, v/v) and precipitation in acetone. The polymer was collected, washed withacetone and dried as described.
Yield: 6.67 gFTIR (KBr): 3450 cm-1 v(OH); 2938-2837 cm-1 V(CH2, CH3); 1461cm-1
5(CH2, CH3); 1072 cm-1 v(C-O-C)
ReferenceA. Koschella, Klemm, D., Macromol Symp. 1997, 720, 115-125.

Appendix (Volume 2) 353
Carboxymethylcellulose, heterogeneous synthesis inisopropanol/water
Air-dry cellulose [15 g, 92.6 mmol, calculated for the water-free product; vis-cose staple fiber (DP - 320), spruce sulfite pulp (DP = 600), cotton !inters(DP = 140O)] was vigorously stirred with 400 ml of isopropanol in a cylindricalglass reactor equipped with a rigid stirrer, while 40 ml of 30 % (w/w) aqueoussodium hydroxide were added dropwise during 30 min at room temperature.After stirring for another hour 18 g (190 mmol) of monochloroacetic acid wereadded during a 30-min period. The mixture was stirred for a further 3 h at 55 0C,and then filtered, suspended in 1 1 of 80 % (w/w) aqueous methanol, and neu-tralized with acetic acid. After filtration, the product was washed three timeswith 80 % (w/w) aqueous methanol, twice with absolute methanol, and dried at55 0C under vacuum. A second and a third carboxymethylation step was runwith a similar procedure.
' HO,^ + CICH2COOH
NaOH
(isopropanol/H2O), SS0C, 3h ' R O ^ OCH2COONa
R = H ,CH2COONaaccording to DS
FTIR spectra (KBr) 1630 cm-1 vas (-COO )1410 cm-1 vs (-COO )
The products are soluble in water already at a DS of 0.4.

354 Appendix (Volume 2)
Startingcellulose3
SSPSSPSSPSSPSSPSSPVSFCL
Molar ratioC1CH2COOH/AGU
1.42.03.05.0b
6.0b
9.0C
3.03.0
Degree of substitution
Uranylmethod0.851.071.311.701.942.42Θ.681.13
HPLC
0.931.291.442.392.322.630.691.35
13C NMR
1.09—
1.352.051.74
a SSP spruce sulfite pulp, VSF viscose staple fiber, CL cotton !inters.b Carboxymethylation twice.c Carboxymethylation three times.
ReferenceHeinze, T., Erler, U., Nehls, L, Klemm, D., Angew. Makromol. Chem. 1994,
275, 93-106.

Appendix (Volume 2) 355
Carboxymethylcellulose, synthesis in DMA/LiCl
OHO
_ _ _ + CICH9COOH' HO ~_
OH
OCH2COONaS
NaOH
(DMA/ LiCI) , 7O0C, 48h ' RO ^V-/H
R = H , CH2COONaaccording to DS
Cellulose [1 g, 6.2 mmol, calculated for the water-free spruce sulfite pulp(DP = 600), dried at 100 0C for 1 h] was dissolved in DMA/LiCl as. Afterstanding overnight, a suspension of dried (45 0C, 30 min under vacuum) mono-chloroacetic acid in 20 ml of DMA was added within 10 min, followed by asuspension of dried (45 0C, 30 min in vacuum) and pulverized NaOH in 20 ml ofDMA within 10 min, under vigorous stirring at room temperature. The reactionmixture was heated to 70 0C for 48 h and then cooled down to room temperatureand precipitated into 300 ml of ethanol. The precipitate was filtered off, dis-solved in 75 ml of distilled water, neutralized with acetic acid and reprecipitatedinto 300 ml of ethanol. After filtration, the product was washed with ethanol anddried under vacuum at 50 0C.
FTIR (KBr): 1630 cm'1 vas (COQ-), 1410 cm"1 vs (COQ-)
The products are insoluble in organic solvents but soluble in water already at DSof 1.4.

356 Appendix (Volume 2)
Molar ratio3
AGU/ClCH2COONa/NaOH
1 21 21 41 21 41 31 41 41 5
4624868810
Reactiontime (h)
102767482448724848
Degree of substitutionUranyl method HPLC
0.240.600.650.900.991.441.601.471.62
0.330.680.921.131.291.671.841.882.07
a Reaction temperature 70 0C.
ReferenceHeinze, T., Erler, U., Nehls, L, Klemm, D., Angew. Makromol. Chem. 1994,
275, 93-106.

Appendix (Volume 2) 357
Carboxymethylcellulose, synthesis via cellulose trifluoroacetate inDMSO
OCOCF3
.0+ CICH2COONa
''RO- _OR
R = H, COCF3
according to DS
NaOH
(DMSO) , 7O0C, 2h ' R Ό ^^\^^OR
R' = H , CH2COONa
according to DS
Cellulose trifluoroacetate (CTFA; 1 g) was dissolved in 18 ml of DMSO undernitrogen. To the solution a suspension of dried, pulverized NaOH (2.47 g, driedunder vacuum at 45 0C, 20 mol/mol of AGU) in 10 ml of DMSO was addedwithin 10 min, followed by 3.6 g of dried monochloroacetate (dried under vac-uum at 45 0C) under vigorous stirring. The temperature was raised to 70 0C.After various reaction times the reaction mixture was cooled down to room tem-perature and precipitated in 75 ml of methanol. The precipitate was filtered off,dissolved in water, neutralized with acetic acid and precipitated into 112.5 ml of80 % (v/v) aqueous ethanol.
Yield: 2.55 g (68 %) of the pure product with DS 1.38.FTIR (KBr): 1618 cm-1 vas (COQ-)
The sample was soluble in water.
Startingcellulose
CTFACTFACTFACTFA
Molar ratioa
1111
: 10:: 10:: 10:: 10:
20202020
Reactiontimeb (h)
0.51.02.04.0
Degree
Uranyl111
.17
.48
.341.69
of substitution
HPLC 1H-NMR1111
.60
.62
.92
.86
11
1
.32
.62—.66
a Modified AGU/monochloroacetate/NaOH.bReaction temperature 70 0C.

358 Appendix (Volume 2)
Carboxymethylcellulose via cellulose formiate is obtained by the same proce-dure.
ReferenceLiebert, T., Klemm, D., Heinze, Th., /. Macromol. ScL, Pure Appl. Chem.
1996, A33(5), 613-626.

Appendix (Volume 2) 359
6-0-Triphenylmethyl (trityl) cellulose, homogeneous synthesis inDMA/LiCl
AVICEL PH-101R (5 g, 30.9 mmol, dried at 100 0C for 1 h, Fluka) is dissolvedin 100 ml of DMA and 7.5 g of LiCl (method A). After standing overnight, 11 g(139.1 mmol) of pyridine (4.5 mol per mol of AGU) were added within 30 min,followed by a mixture of 25.8 g (92.7 mmol) of triphenylchloromethane (3 molper mol of AGU), and 50 ml of DMA within 30 min under stirring at room tem-perature. The homogeneous reaction mixtures were stirred for 48 h additionallyat 70 0C (see Table below), then cooled to room temperature, precipitated into750 ml of methanol, filtered off, dissolved in 100 ml of DMF, and reprecipitatedinto 500 ml of methanol. After filtration and washing with 300 ml of methanol,the sample was air-dried and then dried at 40 0C under vacuum.
R'
pyridine
(DMA / LiCI) , 7O0C, 4Sh
or 250C, 72h ORR = H, Tritylaccording to DS
Trityl = CabC
d
R'
H
H
H
OCH3
R"
H
H
OCH3
OCH3
R'"
H
OCH3
OCH3
OCH3

360 Appendix (Volume 2)
Yield: 90 % pure product with DS 1.05.FTIR (KBr): 3085 and 3055 v(C-Harom), 1500 v(C-Carom) cm-1
13CNMR(DMF-J7): 5=63.8 (C-6); 8=14.9 (C-2,3,5); 6=78.9 (C-4); 6=104.8(C-I); 6=87.0 (C-7); 6=127.5-144.7 (C-Harom)
The sample is soluble in DMA, DMF, DMSO, 1,4-dioxane and THF. Methoxy-substituted tritylcellulose samples can be prepared by an analogous procedure(see Table b-d).
Poly-mer
a
b
bC
C
d
d
T(0C)
70
7025
70
25
70
t(h)
48
244872964
244848
2424484
DegreeEA*
0.410.610.921.050.981.000.960.930.980.970.991.051.090.990.96
of substitutionUV HPLC
1.100.800.900.94 1.031.091.000.981.031.10 1.171.201.18 1.121.13
(DS) SolubilityGravi- DMF 1,4- THFmetry Dioxane0.43 + - -0.67 + (+)** -
0.83 + (+) (+)1.12 + + +
+ + ++ + ++ + ++ + ++ + ++ + ++ + ++ + ++ + ++ + ++ + +
*C-, H- elemental analysis.**Swollen but not soluble.
ReferenceCamacho Gomez, J.A., Erler, U.W., Klemm, D.O., Macromol Chem. Phys.
1996, 797, 953.

Appendix (Volume 2) 361
293-O-Carboxymethyl-6-O-triphenylmethylcellulose, synthesis via6-0-tritylcellulose in DMSO
Tritylcellulose (10 g, 24 mmol, DS = 1.05) was dissolved in 300 ml of DMSOwith stirring at room temperature. After standing overnight, 24.4 g (610 mmol)of powdered sodium hydroxide was dispersed in the solution. After 3 h stirringat room temperature, a mixture of 35.9 g (310 mmol) of monochloroacetate(dried under vacuum at 45 0C) in 30 ml of DMSO was added. The temperaturewas increased to 70 0C. After 4 h, a further mixture of 22.8 g (190 mmol) ofmonochloroacetate in DMSO was added. The addition of monochloroacetate(12.8 g, 110 mmol) was repeated after 16 h. After a total reaction time of 29 h,the mixture was cooled to room temperature and then poured into 2 1 of acetone.The precipitate was filtered and dispersed in 200 ml of water, neutralized withaqueous hydrogen chloride, washed three times with water and ethanol, anddried under vacuum at 50 0C.
+ CICH2COOH
NaOH
(DMSO) , 7O0C, 29hOR
R=H, CH2COONa
according to DS
Yield: 94 % with D5CMC = 1.89FTIR (KBr): 3085 and 3055 v (C-Harom), 1500 v (C-Carom)
1610 vas (-COO-), 1410 vs (-COO) cnr1
The sample is insoluble in water and common organic solvents.
ReferenceHeinze, Th., Röttig, K., Nehls, L, Makromol. Rapid Commun. 1994, 35, 311-
317.

(CH2CI2) , O0C, 45min
362 Appendix (Volume 2)
Detritylation of 2,3-O-carboxymethyl-6-O-triphenylmethyl cellulose
2,3-O-Carboxymethyl-6-O-tritylcellulose (2 g, DS (CMC) 1.89, DS (trityl) 1.05)was treated with HCl gas in 75 ml of dichloromethane for 45 min at O 0C. Themixture was filtered, washed with acetone and dried at 65 0C under vacuum.
HCIgaseous
+ Trityl—OH
R=H, CH2COOH
according to DS Trityl = C
Yield: 89 % with DS (CMC) 1.75.FTIR (KBr): aromatic bands disappear, 1720 crrr1 v (COOH)13C NMR (D2O): 5=71.2 and 71.9 (CH2); δ=60.5 (C-6); 6=80.8 and 81.6
(C'-2,3)
The sample is soluble in dilute aqueous sodium hydroxide, forming the sodiumsalt of CMC. The sodium salt can be isolated by precipitation and is soluble inwater.
ReferenceHeinze, Th., Röttig, K., Nehls, I., Makromol. Rapid Commun. 1994, 35, 311-
317.

Appendix (Volume 2) 363
Crosslinking of cellulose powder with epichlorohydrin
Dry cellulose powder (50 g) is suspended in a three-necked flask with 75 ml of amixture of 85 % (by volume) isopropanol or acetone and 15 % water, and 30 mlof aqueous NaOH (45 % by weight) are added under gentle stirring during 5-10 min. After alkalization for 30 min, epichlorohydrin is added and reactedduring 1-8 h at 60 0C under reflux. The epichlorohydrin input can be variedwithin wide limits between 0.06 mol/mol of AGU and 2 mol/mol of AGU,depending on the degree of crosslinking to be achieved. The etherification isstopped by neutralization of the reaction system with 10 % (by weight) HCl inthe above-mentioned acetone/water or ispropanol/water mixture. The polymer isfiltered off on a sintered-glass disk and washed with the above-mentionedacetone/water or isopropanol/water mixture, respectively. After a final washingwith pure acetone or isopropanol, the reaction product is air-dried at roomtemperature.
Depending on the amount of epichlorohydrin employed, the degree of cross-linking can be varied between 0.1 and 1.5. It is defined here as the number ofhydroxy groups per AGU involved in crosslink formation, and can be deter-mined via the total add-on of epichlorohydrin, taking into account the 1,2-dihydroxypropyl side chains to be assessed separately. The total add-on is de-termined via the difference in sample weight after and before the reaction withepichlorohydrin. The amount of 1,2-dihydroxypropyl side chains is assessed bya gravimetric determination of the formaldehyde formed after periodate oxida-tion, by means of the so-called Dimedon method (Dimedon = 5,5-dimethylcyclohexanedione-1,3). The following procedure was found to be suit-able by the authors groups:
The oxidative glycol cleavage with periodate proceeds formally according to
CeII-O-CH2-CH-CH2 + 1O4" - CeII-O-CH2-CH + CH2I l I l Il
OH OH + H2O + 1O3' O O
and is performed by treating 0.1-2 g of the dry crosslinked sample in a 200 mlErlenmeyer flask with 25 ml of 0.05 M aqueous sodium periodate solution forl h at 50 0C. The polymer is filtered off and washed with 20 ml of water. To thetotal filtrate 10 ml of Dimedon reagent solution is added. This solution is pre-pared by dissolving 25 g of Dimedon, 71.6 g of disodium hydrogen phosphateand 0.6 g of citric acid in aqueous ethanol (50 : 50 by volume) and making upthe solution to 1 1 with 50 : 50 aqueous ethanol. 10 ml of this solution and 0.1 mlof concentrated aqueous HCl are added to the filtrate, and the condensation be-tween Dimedon and formaldehyde takes place then at room temperature over-

364 Appendix (Volume 2)
night. The insoluble crystalline reaction product is filtered off, washed with wa-ter and weighed after 4 h drying at 95 0C.
The amount of formaldehyde formed is obtained by dividing the condensationproduct by 0.73. From this amount of formaldehyde the amount of 1,2-dihydroxypropyl side chains per g of crosslinked sample can be calculated in theconventional manner.
ReferencesMitchell, J., in Organic Analysis, New York: Interscience, Vol. 1, pp. 280,1953Roberts, J., in Starch Chemistry and Technology, Whistler, R., Parschall, P.P.
(Eds.), New York: Academic Press, 1965, Vol. 1, pp. 482.
Organosolublecyanoethylcellulose
A two-step process is used for this route of synthesis, with the first step consist-ing of alkalization of the cellulose and the second one consisting of etherifica-tion of the alkali cellulose in a large excess of acrylonitrile.
Air-dry !inters or wood pulp (20 g) is disintegrated by shredding or millingand subsequently soaked in 100 ml of aqueous NaOH (12 % by weight) for l hat 20 0C. The alkali cellulose is pressed onto a sintered glass filter disk to apress/weight ratio of 3.0.
Then the alkali cellulose is dispersed in a three-necked 1 1 flask in 570 ml ofacrylonitrile and reacted at 60 0C for 30 min. During this procedure the cyano-ethylcellulose formed is dissolved in an excess of reagent to a homogeneoussolution. The reaction is stopped by adding an excess of 10 % aqueous aceticacid, and the polymer is subsequently precipitated from the still homogeneousreaction mass by an excess of an ethanol/water mixture (1 : 1 by volume). Thereaction product is filtered off, subsequently washed at first with hot then withcold water, and dried in the open air. DS values of cyanoethyl groups of 2.5-2.9were obtained by this procedure with the cyanoethylcellulose being soluble inacetone, DMF and DMSO. The DS is determined by the nitrogen content, as-sessed by elemental analysis. The excess of acrylonitrile not consumed in thereaction can be retrieved by distillation.
ReferenceLukanoff, B., Ph.D. Thesis, Academy of Science, GDR 1977.

Appendix (Volume 2) 365
Trimethylsilylcellulose, synthesis in pyridine/THF
OHO CH3
CK, + CI-Si-CH3'
.CKpyridine/THF ''RO - ^" \ 1^ri.8/, °R
R = H, Si(CH3)3
according to DS 1.99 - 2.62
Dried cellulose (16.2 g, 0.1 mol) in 100 ml of dry pyridine was refluxed for 1 h.After cooling to room temperature, 300 ml of THF and a solution of 32.6 g (0.3mol) of chlorotrimethylsilane in 100 ml of THF was added. The addition pro-ceeded dropwise with stirring over a period of 30 min. Stirring was continued atroom temperature for 8 h and a viscous solution, containing suspended pyridinehydrochloride, was obtained. After separation of pyridine hydrochloride by cen-trifugation, the reaction mixture was poured into methanol, collected, washedwith methanol and dried at 50 0C under vacuum.
Yield: 28.5 g (93 %) of the pure product with DS 1.99.FTIR (KBr): 1250 (6S Si-CH3); 840 (v Si-C) cm-1.
The DS was determined gravimetrically on the basis of SiO2 content and calcu-lated according to the equation:
DS= MAGU
100%(MAGU - MH)AGU Hm%Si02
The sample 1 was soluble in chloroform, THF and benzene, samples 2 and 3 aresoluble in hexane.

366 Appendix (Volume 2)
CelluloseSample no.
123
Molar ratioTMS-C1/AGU
356
DSs*
1.992.462.62
Si
18.2820.3420.95
a DS calculated on the basis of SiC^ content.
ReferencesStein, A., Klemm, D., Makromol Chem., Rapid Commun. 1988, 9, 569-573.Stein, A., Thesis, Friedrich-Schiller-University Jena, 1991, p. 82.

Appendix (Volume 2) 367
Trimethylsilylcellulose, synthesis in DMA/LiCl
OHCH3 CH3
^ + CH3-Si-NH-Si-CH3
CH3 CH3
i - Cl
(DMA/LiCl) , WO0C, 24h -'
according to DS 2.91
Dried cellulose (24.0 g, 0.148 mol, Avicel) was dissolved in 600 ml of DMAand 36.0 g of LiCl (dissolution procedure B). After addition of 0.5 ml of chloro-trimethylsilane, 139.6ml (0.666 mol, 4.5 mol/mol of AGU) of hexamethyl-disilazane was added dropwise. The mixture was stirred for 24 h at 100 0C.Within this time the trimethylsilylcellulose precipitated. After cooling down toroom temperature the mixture was poured into methanol and dispersed. Thepolymer was separated and washed with water and ethanol. It was dried undervacuum at room temperature and up to 100 0C over potassium hydroxide.
Yield: 50.8 g (92 %) of the pure product with DS 2.91.
FTIR (KBr): 1251 (δ Si-C), 842 (ν Si-C) cm-1
The degree of substitution was determined gravimetrically on the basis of SiO2
content and calculated by the equation:
DS = , MAGU
• 100%-(MAGU-MH)
m%SiO2
The sample is soluble in hexane.
ReferencesSchempp, W., Krause, Th., Seifried, U., Koura, Α., Das Papier 1984, 38 (12),
607-610.Stein, A., Thesis, Friedrich-Schiller-University Jena, 1991, p. 82.

368 Appendix (Volume 2)
Cellulose esters, synthesis via trimethylsilylcellulose; general procedurewithout solvents
.OSi(CH3)3
' RO
80 - 76O0C
-(CH3)3SiCi 'R" o
Trimethylsilylcellulose (5.0 g; DS see Table below) was added under nitrogenatmosphere to 2.5-5.0 equivalents of acid chloride, liquid at room temperatureor molten at 80 0C. The mixture was heated for 30 min at 80-160 0C (see Tablebelow) and the resulting chlorotrimethylsilane was completely distilled off. Af-ter washing the residue with aqueous methanol (polymer 1, 2; see Table) or pre-cipitating with methanol/water from an organic solution (polymers 3 and 4 innitrobenzene, 5 in THF and 6 in acetone), the resulting cellulose esters weredried at 50 0C under vacuum.
For yield, DS and elemental analyses, see Table.FTIR (KBr): v C-O at 1765 (1, 2), 1735 (3, 4), 1750 (5) and 1730 (6) cnr1
ReferenceStein, A., Klemm, D., Makromol Chem., Rapid Commun. 1988, 9, 596-573.

Tri
met
hyls
ilyl-
A
cid
chlo
ride
cellu
lose
(M
olar
ratio
)(D
S)
1.99
1.99
2.46
2.46
2.46
2.62
R =
CC
l 2C
H3
(2.5
)C
Cl 3
(5.0
)C
6H4N
O2
(2.5
)C
6H4N
O2
(5.0
)(C
H2)
14C
H3
(3.0
)C
6H4(
CH
2)2B
r(3
.5)
Rea
ctio
nte
mpe
ratu
re(0C
)
80 90 160
160
160
160
No. 1 2 3 4 5 6
Yie
ld
94 95 96 92 96 95
Cel
lulo
se e
ster
DSa
Ele
men
tal
anal
ysis
'5
1.17
1.87
1.57
2.30
2.50
2.53
calc
.fo
und
calc
.fo
und
calc
.fo
und
calc
.fo
und
foun
d
calc
.fo
und
C 3
7.05
C 3
6.61
C 2
6.96
C 2
6.54
C 5
1.50
C 5
1.07
C 5
2.55
C 5
2.32
C 7
2.85
C 4
9.64
C 5
0.12
H 4
.03
H 3
.78
H 1
.89
H 1
.69
H 3
.74
H 3
.82
H 3
.37
H 3
.56
H 1
1.3
H 4
.01
H 4
.33
a C
alcu
late
d fr
om t
he c
onte
nt o
f C
l, N
, C o
r B
r de
term
ined
by
elem
enta
l an
alys
es.
b C
alcu
late
d fr
om A
S; s
ilico
n co
nten
t <
0.0
1 %
.

370 Appendix (Volume 2)
6-O-Thexyldimethylsilylcellulose
TDMS-CI
(i) -250C
(H) +8O0C
NH3/ NMP ' HO
S' )-chlorosilane according to DS
TDMS-CI = S' - = thexyldimethyl- R = H or TDMS
Cellulose (16.2 g, 100 mmol, AVICEL PH-IOl, dried for 1 day over potassiumhydroxide under vacuum at 105 0C) was suspended in 65 ml of NMP and stirredat 80 0C for 1 h. After cooling down to -25 0C, 80 ml of NMP, saturated withammonia, was added. The mixture was stirred for 1 h, and a solution of 39.25 ml(200 mmol, 2 mol/mol of AGU) of TDMSCl in 40 ml of NMP was added drop-wise. The formation of a precipitate (ammonium chloride) occurred. After stir-ring for 45 min at -25 0C, the mixture was slowly warmed up to 40 0C. It wasallowed to stand overnight and was stirred for 6.5 h at 80 0C. The highly viscoussolution was poured into 4 1 of buffer solution (pH 7). The polymer was filteredoff, washed with water and dried carefully at p < O. l Torr over potassium hy-droxide, with successively increasing temperature, from 25 0C to 80 0C. Forpurification it was dissolved in NMP, precipitated in buffer solution, washed anddried as described.
Yield: 21.77 g of the pure product with DSS[ 0.78
FTIR (KBr): 3456 cm-1 v(OH); 2958cm-1 V(CH2, CH3); 1465cm-1 8(CH2,CH3); 1252cm-1 6(Si-C); 1110-1037 cm-1 v(C-O-C); 833cm-1
V(Si-C)
The sample was soluble in NMP.The degree of substitution was determined gravimetrically on the basis of SiO2
content (£>%).
ReferenceKoschella, A., Klemm, D., Macromol Symp. 1997, 720, 115-125.

Appendix (Volume 2) 371
296-Di-O-thexyldimethylsilylcellulose
OH
' HOΟ,. + TDMS-CI
OH
TDMS-CI =
OΛ ,
N
H
O^OTDMS
thexyldimethyl-chlorosilane
Cellulose (15.0 g, 92.6 mmol, AVICEL PH-IOl, dried for 1 day over potassiumhydroxide under vacuum at 105 0C) was suspended in 300 ml of N,N-dimethylacetamide (DMA) and stirred for 2 h at 120 0C. After cooling down to100 0C, 22.5 g of LiCl (dried for 1 day over potassium hydroxide under vacuumat 150 0C) was added. The mixture was stirred at 25 0C until a clear solution wasobtained. Imidazole (30.29 g, 445 mmol, 4.8 mol/mol of AGU) was dissolved inDMA and added to the cellulose solution. Thexyldimethylchlorosilane (66.2 g,371 mmol, 4 mol/mol of AGU) was added dropwise. The mixture was stirred for24 h at 25 0C. After some hours precipitation of the silylated cellulose occurs.The mixture was poured into 3 1 of pH 7 buffer solution. The separated polymerwas carefully washed with water and dried under vacuum, first at 25 0C then at100 0C. Further purification was carried out by reprecipitation from THF solu-tion in pH 7 buffer.
Yield: 42 g
FTIR(KBr)^SOSCm-1 v(OH); 2958, 2871cm-1 V(CH2, CH3); 1466cm-1
6(CH2, CH3); 1252cm-1 8(Si-C); 1119-1037 cm-1 v(C-O-C);
833 cm-1 v (Si-C)
The polymer is soluble in THF, hexane and chloroform.

372 Appendix (Volume 2)
6-O-Thexyldimethylsilyl-293-di-O-methylcellulose
NaHO^ + CH3I
" THF, Id1 250C "H3CO3d, 5O0C
6-O-Thexyldimethylsilylcellulose (50.Og, 0.172mol, DSSi 0.9) was suspendedin 1.5 1 of THF. After addition of 41.3 g (1.72 mol, lOmol/mol of modifiedAGU) of sodium hydride, 107 ml (1.72 mol, 10 mol/mol of modified AGU) ofmethyl iodide was added slowly during 1.5 h. Nearly l h after the start of themethyl iodide addition, an exothermic reaction occurs and it was necessary tocool the flask with ice. The mixture was stirred overnight at 25 0C and for 3 daysat 50 0C. After cooling down at room temperature the inorganic salts were sepa-rated by centrifugation. The clear solution was concentrated using a rotaryevaporator and precipitated into 6 1 of pH 7 buffer solution. The polymer wasseparated, washed and dried carefully.
Yield: 45. 17 gFTIR(KBr): 2957cm-1 V(CH2, CH3); 1466cm-1 5(CH2, CH3); 1252cm-1
5(Si-C); 1126-1041 cm-1 v(C-O-C); 831 cm-1 v(Si-C)
ReferenceA. Koschella, Klemm, D., Macromol Symp. 1997, 720, 115-125.

Appendix (Volume 2) 373
Trimethylsilylcellulose methoxyacetate, synthesis via cellulosemethoxyacetate in DMA
OCOCH2OCH3
.0
TMS-CI
(DMA)
15min, rt / 5h, 8O0C
(CH3)3Si — NH"-Si(CH3)3
OCOCH2OCH3
O
OR
R ' = COCH2OCH3 , Si(CH3J3
according to DS
Dried cellulose methoxyacetate (Ig, DS - 1.1, determined by saponificationanalysis) was dissolved in 50 ml of DMA under stirring and an inert atmosphere.Then 1.96 ml (0.019 mol) of hexamethyldisilazane were added at room tem-perature within 15 min, as well as TMS-Cl, at catalytic concentration. The reac-tion mixture was stirred for 5 h at 80 0C. The excess of hexamethyldisilazanewas removed with a water-jet vacuum pump at higher temperature. The reactionproduct was precipitated in buffer solution of pH 7, dispersed, filtered off andwashed with distilled water many times until the filtrate was free of chlorideions. The product was dried at 50 0C under vacuum.
Yield: 1.5 g (94 %) of pure product with D5Si =1.9 and DS ester groups =1.1.FTIR (KBr): 1766 (v OOester), 1252 (δ Si-C), 842 (ν Si-C) cm'1
The DS of ester groups was based on analysis by alkaline saponification. TheDS §i was determined by gravimetric analysis (SiO2 content). The sample wassoluble in THF, dichloromethane, chloroform and toluene.
ReferencesSiegmund, G., Diploma thesis, Friedrich-Schiller-University of Jena, 1993.Stein, A., Thesis, Friedrich-Schiller-University of Jena, 1991, p. 82.

374 Appendix (Volume 2)
6-Carboxycellulose, homogeneous synthesis with phosphoric acid
1. NaNO2/ H3PO4 a 2 . .O
2.NaBH4
(1) 5.0 g of cellulose were dissolved (using a special flask, content 1.5 1, height45 cm) in 200 ml of 85 % phosphoric acid. After 2 h at room temperature, 5.0 gof powdered sodium nitrite were added under vigorous stirring during 15 min.Within 5 h without stirring, a stable foam was formed. It was destroyed by vig-orous stirring and another 5.0 g of sodium nitrite were added. The addition wasrepeated after 3 h. After a total reaction time of 10 h, 50 ml of 85 % formic acidwere added in order to destroy the excess sodium nitrite. All escaping gaseswere absorbed with ethanol. If the reaction time is 8 h, three portions (5.0 g ofsodium nitrite were added) after 4 and 6.4 h. The polymer was precipitated with800 ml of ice-cold acetone and transferred into a beaker. Precipitation was com-pleted by addition of 2 1 of ice-cold ether (caution, very exothermic reaction).After filtration the material was washed with distilled water until the liquid be-comes neutral and then first with 0.5 1 of 50 % ethanol and with 0.5 1 of absoluteethanol. The product was dried under vacuum at 5O0C.
Fourier transform (FT)IR ( KBr ) ACOOH 174° cm"1
(2) 4.0 g of carboxycellulose was added during 3 h to a 10 % NaBH4 aq. solution(50 ml) under stirring at room temperature. After 16 h, without stirring, thissolution was neutralized with acetic acid. The precipitated product was separatedby centrifugation (15 min at 2000 g). The sodium salt was precipitated in 600 mlof acetone and dried under vacuum at 50 0C.
FTIR (KBr) vcoo_ 1580-1630 cm 1A

Appendix (Volume 2) 375
Cellulose starting material DP
Cellulose powder 160
Viscose staple fiber 300
Spruce sulfite pulp 600
Cotton !inters 1400
Reactiontime(h)358103581035810345810
Content of carboxy groups(%)
7.757.962.063.060.160.465.067.962.068.973.575.035.752.373.478.081.0
ReferencesHeinze, Th., Klemm, D., Loth, F., Nehls, L, Angew. Makromol. Chem. 1990,
178, 95-107.Heinze, Th., Klemm, D., Schnabelrauch, M., Nehls, L, in Cellulosics: Chemical,
Biochemical and Material Aspects, Kennedy, J.F., Phillips, G.O., Williams,P.A. (Eds.), New York: Ellis Horwood, 1993, pp. 349-355.

Subject index
accessibility 171, 214acetal structures 310acetate borate ester 141acetophthalates 193acetosulfation 123facetylation 170ff, 176ff- acid catalyst 170- cellulose trinitrite 173-DMA/LiCl 173- W-ethylpyridinium chloride 173- fiber acetylation 178- industrial process 176- mathematical modeling 172- methylene chloride process 178- partial acetylation 172- preactivation 177- preferential substitution 173-rate 171- raw material 177- reactivity of cellulosic hydroxy
groups 173-reagent 170- solution acetylation 172- technical process 172- two-phase system 172- vapor process 1726-0-acetyl-2,3-di-O-methyl-co-[2,6-di-
O-acetyl-3-O-methyl]cellulose 282f- 1H NMR spectrum 283-HPLC 283acrylamide 252activation 43,61, 171,317,322- activating agent 61- with liquid NH3 43acylation 182, 286ff- heterogeneous 182-homogeneous 1826-aldehydecellulose 304aldonic acid end-groups 303aliphatic ethers 210, 213f, 221, 234- carboxymethy!cellulose 221- hydroxyalkyl ethers 234
- long-chain alkyl ethers 213- methy!cellulose 210-preparation 214- subsequent functionalization 214alkali cellulosates 32- preparation 32- properties 32alkali cellulose 49ff, 216, 339- applications 50- preparation 339- properties 50alkali uptake 146ß-alkoxy elimination 303alkyl ethers 207alkylation 21Of, 214- product formation 211- role of cellulose supramolecular
structure 214-SN2 reaction 210alky !cellulose 217ff-applications 219-commercial 219-ASrange 217-Relation 218- hydrophobicity 217- liquid crystalline systems 220- microcapsules 220-properties 217-solubility 217- solution properties 217- ultrathin films 220-viscosity 217amidoxime 254amine oxide process 321amino groups 256- introduction into cellulose 256aminodesoxycellulose 144aminoethylation 255aminoethylcellulose 249ammonia cellulose 58amphiphilic esters 196amylose 258anion-exchanging sorbents 259
Comprehensive Cellulose Chemistry; Volume 2: Functionalization of Cellulose
D. Klemm, B. Philipp, Ύ. Heinze, U. Heinze, W. Wagenknecht
Copyright © 1998 WILEY-VCH Verlag GmbH, Weinheim
ISBN: 3-527-29489-9

378 Subject index
anticlotting activity 132aromatic esters of cellulose 190ff- acetate 193-benzoates 190- cinnamates 191- homogeneous reaction 191-phthalates 190,193artificial fibers 163asymmetric membranes 293
bacterial cellulose 317, 344- acetylation 344base uptake 55bead cellulose 243benzhydryl(diphenylmethyl)cellulose 263benzoylation 192benzylcellulose 263benzyl(phenylmethyl)cellulose 263bleaching 317block copolymers 27f- biodegradability 28- cellulose and lignin 28- cellulose triacetate 28- principle 27- synthesis 27- synthetic prepolymer 28bromodesoxy cellulose 144f-butyldimethylsilylcellulose 281i-butyldimethylsilylcellulosecinnamate
291
C-6 oxidation 305Cadoxen 90, 95, 231, 79ffCannizzaro reaction 310carbamate method 321carbamate process 161, 163- fiber spinning 163carbamoylethy!cellulose 253carbanilation 196f, 264- regioselective 197carbonyl sulfide 145- esterification with 145carboxy groups 3056-carboxycellulose 304, 374
- synthesis 374carboxycellulose 306f-13CNMR 306- ionotropic gels 307-p^s 307- sulfation 307carboxyethylcarbamoylethylcellulose
255carboxyethylcellulose 230, 253carboxyl groups 303- determination 303carboxymethylation 22Iff, 229f, 267- continuous process 230-DMA/LiCl 225- dry process 229- kinetic study 222- laboratory procedure 222- phase-separation process 224- regioselective 224- slurry process 222,229- technical process 229carboxymethy!cellulose 186, 22Iff,
227ff, 353, 355, 357- acid chloride 227-activation 228- anionic polyelectrolyte 234- application 233- block-like structures 224-13CNMR 228- crosslinking 227,228- degree of substitution 223-DP 231-ASrange 233- gel particles 231- heterogeneous synthesis 353- highly substituted 222- intermediate derivatization 232- lactone formation 227- molar mass 231- pattern of substitution 223- preparation 222- properties 230-quality 233- reactivity ratio 223- subsequent derivatization 227- substituent distribution 229

Subject index 379
-sulfation 228- synthesis in DMA/Lid 355- synthesis via cellulose trifluoroacetate
357- water solubility 2302,3-0-carboxymethyl-
6- O- triphenylme thy !cellulose 36 If- detritylation 362- synthesis via 6-O-trity!cellulose 361cation exchangers 140cationic cellulose 257cationization 257, 259- regioselective 259celluloid 111cellulose 331- dissolution in DMA/LiCl 331cellulose 2,5-acetate 169cellulose acetate 121,138,176, 178ff, 274-application 180- deacetylation 121-films 180- liquid crystalline phases 178-NMR 176- phosphory lation 138- plastic material 181-properties 178- Raman spectroscopy 176-rheology 180- separation membranes 181-solubility 179- sulfation 121- supramolecular aggregates 180- textile properties 181cellulose acetate phosphate 136- polytetraphosphoric acid 136cellulose acetate sulfate 121, 123- competitive esterification 123- heterogeneous deacetylation 121- regioselectivity 123cellulose acetobutyrates 186cellulose borate 140, 142- application 142- properties 142- synthesis 140- thermal stability 142cellulose carbamate 16If
- crosslinking 161- decomposition 161-formation 161- substituent distribution 162cellulose citrate 190cellulose dichloroacetate 345- synthesis 345cellulose dithiocarbonic acid 147cellulose ester 182ff, 186, 192- application 186- liquid crystalline systems 184-NMR 183- palmitoyl ester 183- physical properties 183cellulose ethers 208, 256, 318- cationic 256- production capacity 208-regioselective 318cellulose formate 166ff, 225, 342-preparation 166-properties 168- synthesis 342- thermal stability 168- transesterification 167cellulose furoate 190cellulose gels 84cellulose grafting 322cellulose hemiacetals 249cellulose I 41ff, 61cellulose II 41ff, 61cellulose III 58cellulose methoxyacetates 347- synthesis 347cellulose monothiocarbonate 146- maximal DS 146cellulose nitrate lOlff, 108ff, 144- acid hydrolysis 108- application 111- decomposition 102, 108- degradation 108- film-forming properties 111-formation 102,105- industrial production 101, 109- nitrating acid 104- nitrating agent 102- nitrogen content 101, 103

380 Subject index
- properties 111- softeners 111- solubility in organic liquids 111- stabilization 110- viscosity adjustment 110cellulose nitrite 112ff- addition compound 113-application 114-isolation 113- nitrosyl compounds 114- NMR studies 113-properties 114- stability of the nitrite groups 113-synthesis 112cellulose oligophosphate 134cellulose oxalates 189cellulose phenylcarbamate 341- synthesis 341cellulose phosphate 133ff, 137, 139f,
338- application 140-NMR 139- pattern of substitution 137- preparation 338-properties 139- reaction routes 134- Schotten-Baumann reaction 139- solubility 139- solution viscosities 140cellulose phosphite 136cellulose phosphonate 136cellulose phosphonite 136cellulose purity 317cellulose solvents 88cellulose sulfate 116f, 12If9 125, 128f,
131f,332, 334ff- acidic character 128-application 131- biological activity 131- C-2/C-3-substituted 337- C-6-substituted 336- chain degradation 125- defined patterns of substitution 128- distribution of sulfuric acid half-ester
122-DS 111
- DS from cellulose acetate 121- film-forming properties 131- gelforming properties 132- [η]-Mw relationship 129- preparation 336-properties 128-purification 121-SO3-DMF 332-solubility 117- solution viscosities 129- substitution patterns 125-synthesis 116- synthesis via cellulose trifluoroacetate
334- synthesis via trimethylsily!cellulose 335- thermal stability 129- thermoreversible 129cellulose tosylate 350- homogeneous synthesis 350cellulose triacetate 169, 176, 343f- preparation 343- selective deacetylation 344cellulose tribenzoate 191-benzene-ring-substituted 191cellulose tricarbanilate 196f, 340- homogeneous route 197- laboratory procedure 197- molar mass distribution 197cellulose trifluoroacetate 188, 225, 346- synthesis 346cellulose trimethoxalate 189cellulose triniträte 332- preparation 332cellulose trinitrite 138- phosphory lation 138cellulose xanthogenate 339- preparation 339cellulose xanthogenic acid 156cellulose-4-nitrobenzoate 348f- synthesis via cellulose trifluoroacetate
348fcellulose-metal complexes 320cellulosic ampholytes 259cellulosics 292- cinnamoyl-group-containing 292chain degradation 93

Subject index 381
- oxidative 93chain stiffening 82,92chemical synthesis 3f, 6- cationic polymerization 4- polycondensation 3- protecting groups 4- ring opening polymerization 3- stereoregular chain structure 4- transglycosidation 6chlorination 135, 143- homogeneous route 143chlorodesoxycellulose 143churn process 157CMC see carboxymethylcellulose13CNMR 266,28013C NMR spectroscopy 272colloid chemistry 151comb-like cellulose derivatives 290commercial processes 321- artificial fibers, films 321commercial viscose process 157, 159- alkali cellulose 157- ecological hazards 157- skin-core-filaments 159-spinning 159- structure formation 159controlled activation 278copolymerization of ß-D-glucose 3- Acetobacter xylinum 3- 7V-acetylglucosamine 3copper complexes 72ff, 76fCOS see carbonyl sulfidecrosslinking 6ff, 14ff, 243f, 261, 323,
363-applications 16- covalent 6- crosslink density 6- diisocyanates 7- divinyl sulfone 261- epichlorohydrin 244, 363- ester bond 8- ether bonds 6- formaldehyde 6- ionic 7- macroradicals 7- material properties 15
- mechanical properties 16- morphological structure 14- oxidative coupling 7- polycarboxylic acids 8- principles 6- self-crosslinking 6- solubility in Cuam 15- supramolecular structure 14- water retention 15crosslinking by Michael addition 12, 16- di vinyl sulfone 12-hydrogels 12,16crosslinking with alkyl halides and
epoxides 12f-epichlorohydrin 13crosslinking with formaldehyde 8ff, 16- acetal bridges 8- dry process 9- formaldehyde liberation 10- high-performance crosslinker 11-kinetics 9, 11- mechanism 9, 11- methylol derivatives !Off- methylolated 16- tetrafunctional crosslinker 11- urea compounds 16- urea derivatives 10- wet process 9crystalline order 54crystallinity 40, 45- crystallite size 40- lattice dimensions 40Cuam 74ff, 93ffCuen 77,84cyanoethylation 250ff- heterogeneous 252- homogeneous 252- Michael addition 250- reaction rate 251- reactivity 251cyanoethylcellulose 226, 249ff, 258, 364- application 254- consecutive reactions 253- decomposition 251 ff- formation 250- material properties 254

382 Subject index
- oxidation 253- preparation 364cyanopropyldimethylsilylcelluloses 274
deacetylation 174f- alkaline saponification 175-homogeneous 175decrystallization 54, 58desilylation 287desoxy cellulose 142ff, 196- iminodiacetic acid group 144- preparation 143- subsequent functionalization 144- thermal decomposition 145detritylation 264, 2662,3-dialcohol cellulose 31Of-13CNMR 311dialdehyde cellulose 3042,3-dialdehyde cellulose 309- preparation 3092,3-dicarboxycellulose 304, 31 If-13CNMR 311- complexing properties 312dicarboxylic acid methyl ester 190dicarboxymethylcellulose 226diethylaminoethy!cellulose 2562,3-diketocellulose 3044,4' -dimethoxytripheny!methyl groups
2674,4' -dimethoxytrity!cellulose 2674-dimethylaminopyridine 164, 171, 2852,3-dimethy!cellulose 2132,3-Di-O-methylcellulose 352diphenylmethyl ethers 269diphenylmethylsilylcelluloses 274direct esterification 169f- homogeneous 170dissolution 34, 52, 72, 90- dimethyldibenzylammonium hydroxide
52- triethylbenzy!ammonium hydroxide 52dissolving pulp 3212,6-Di-O-thexyldimethylsilylcellulose 371DMA/LiCl 87,93f- structures 87
donor-acceptor complex 72DP see degree of polymerization
emulsion xanthation 150enzymatic esterification 165epoxidation 246equilibrium reaction 164esterification 99f, 186ff- 1,2-dichloropropionic acid 186- inorganic acids 100- TMS-cellulose 188- trifluoroacetic acid 187esters of cellulose 99ff, 112, 115, 133,
140ff, 161, 164, 166, 168ff, 182, 186,189ff
- aromatic acids 190- cellulose acetate 168- cellulose borates 140- cellulose carbamate 161- cellulose formate 166- cellulose nitrate 101- cellulose nitrite 112- cellulose phosphate 133- cellulose sulfates 115- desoxycellulose 142- di- and tricarboxylic aliphatic acids
189- dithiocarbonate esters 147- higher aliphatic acids 182- inorganic acids 100- mesylcellulose 194- monothiocarbonic acid 145- organic acids 164- pheny 1 carbamates 196- phosphonic acid esters 194- production capacity 99- substituted monocarboxylic aliphatic
acids 186- tosy !cellulose 194etherification 207, 246, 270- reactivity 270ethers 285- 2,3-substituted 285ethers of cellulose 210- aliphatic ethers 210

Subject index 383
ethylation 213- activation energy 213ethylcellulose 213,216- liquid crystalline systems 216
FeTNa 82ff,93f, 331- cellulose interaction 84- characterization of cellulosic materials
84- complex binding 84- degradation 83- intrinsic viscosity 83- medium for etherification 84- molecularly dispersed system 84-preparation 331- replacement of Na+ by K+ 84fiber acetylation 172fiber xanthation 151 ff- lattice layer reaction 153- lye concentration 152- maximal DS 151- rate constant 153filament spinning 93film formation 93flame retardation 139flash photolysis 273flocculation 218fluorodesoxy cellulose 143formylation 166ff- hydrolytic cleavage 167- preferential reaction 167-rate of 168functionalized alkyl ethers 249, 255- with quaternary ammonium groups
255- with tertiary amino functions 255
ggraft copolymers 17f, 24, 26, 141- acrylonitrile fibers 26-analysis 18- antimicrobial finish 26- applications 24- cellulose fibers 24- filtering processes 26
-homopolymer 17- ion-exchange 26- properties 24- routes 18- side chains 18- super-absorbing materials 26grafting 17, 19ff- cationic acrylics 26- conditions 25- effect of swelling 23- mechanochemical treatment 20- monomers 19- morphological structure 17, 22- perfluorinated compounds 26- radiation grafting 21 f- radical polymerization 17- radical site 19- redox reaction 19- supramolecular structure 17, 22- surface grafting 22
Hammett equation 269hemodialysis 1401HNMR 2801W1HCOSY 282hollow fibers 94HPLC 226,281HPLC see high performance liquidchromatographyhydrogen bond system 73hydrogen bonds 318hydrophile/hydrophobe ratio 179hydroxyalkylation 234f, 237, 240- by epoxides 235- heterogeneous process 240- heterogeneous reaction 237- hydroxyalkyl chains 235- reaction rate 237- reagent yield 235,237- spacing action 240- technical process 240- two-stage process 240hydroxyalkylcellulose 237, 239, 24If- application 242- liquid crystalline systems 241

384 Subject index
- pattern of substitution 237-properties 241- reaction with isocyanate 239- reactivity ratio 237- subsequent esterification 239- subsequent etherification 239-viscosity 241hydroxyethylation 235hydroxyethylcellulose 234, 236, 242f- application 242- ecocompatible artificial fiber 243- length of the side chains 236-MS 236hydroxymethylcellulose 246- formation 246hydroxypropylation 237f- relative rate 237- slurry procedure 238hydroxypropylcellulose 234, 236-MS 236
iimpeller technique 182impelling agent 165in vitro synthesis 3- cellulase 3- functionalized cellulose 3- micellar aggregation 3induced phase separation 225interaction with aliphatic amines 62ff- accessibility 64- addition complexes 63- addition compounds 62- degree of order 64- diamines 63- ethanolamine 63- ethylene diamine uptake 65- higher amines 63- increase in accessibility 62- methylamine and DMSO 62- polyamines 63- primary amines 62- steric hindrance 63- water sorption 66interaction with alkali hydroxides 33,
35ff, 43, 47
- alkali uptake 35- chain conformation 38- chemical processes 35- comparison of NaOH and KOH 37- conformational changes 39- diffusion-controlled reaction 39- effect of temperature 39- fibrillar morphology 43- general comments 33- hydration states 36-23NaNMR 36,38- NaOH ion dipoles 38- reactive structural fractions 47- sorption isotherm 35- water uptake 36interaction with ammonia 57ff- addition compounds 57- degree of order 58- dry process 60- dyeability 60- fibrillar architecture 58- handling 60- lattice transitions 57- NH3/DMF mixture 59- recrystallization 58- structure 57- swelling power 57- textile processes 60- textile processing 57- water regain 58- wet process 60interaction with guanidinium hydroxide
54ff- accessibility 56- activation technique 57- adduct formation 55- base uptake 55- fibrillar structure 57- regenerated samples 56- solutions of GuOH 55- swelling 55- water sorption 55- X-ray patterns 55interaction with hydrazine 61interaction with inorganic salts 86f- spinning of threads 87

Subject index 385
- suitable cations 86- thiocyanate 86interaction with tetraalkylammonium
hydroxides 5 Iff- applications 54- hydrate complex 53- model of dissolution 53- structure 54- uptake of base 52intermolecular interaction 240intracrystalline swelling 61iododesoxycellulose 142IR spectroscopy 288isomerization 292
ketocellulose 308- selectively oxidized 3082-ketocellulose 3043-ketocellulose 304
1Langmuir-Blodgett layers 184,323Langmuir-Blodgett technique 293lateral order spectrum 168LB see Langmuir-Blodgettlevel-off degree of polymerization 45,
58, 65, 87liquid crystalline systems 323LODP see level-off degree of
polymerization
mmercerization 49- cold mercerization 49- hot mercerization 49mesophase 323mesy !cellulose 142metal complexes 7Iff, 76ff, 85f, 90, 92ff- acid-base interaction 80- application 93- bisdiolato complex 85- bisdiolato crosslinks 77- cellulose zincate interaction 86- characterization of cellulose 95- colored 92
- coordination equilibria 81- covalent functionalization 94- cuprate anions 85- determination of foreign substances 95- formation 72f- heteroleptic complex 82- heteroleptic copper complex 77- homoleptic cationic complex 78- hydroxamic acid functions 72- hydroxy bonds 81- interchain crosslinking 82- ligand exchange 77- ligand-exchange processes 72-main routes 71- precipitation 92- properties 92-reformation 81- solution viscosity 92- spectrophotometric investigation 96- state of solution 92- supramolecular aspects 90- toxicity 93-type 90-with 1,3-diaminopropane 76- with ethylene diamine 76- zincate 90methacrylate ester 188methoxyl group content 2204-methoxytripheny!methyl groups 267methylation 21 Iff, 216f-agent 212- diffusion-controlled reaction 211- gaseous process 216-laboratory 212- liquid methyl chloride process 216- reaction temperature 211- reagent yield 217- regioselective 213- technical process 216methylcellulose 207, 21Of- degree of substitution 211- substituent distribution 211Λ^-methylmorpholine TV-oxide 321methylolcellulose see hydroxymethyl-
cellulose- artificial fibers 248

386 Subject index
- distribution of side chains 248- liquid crystalline properties 248-MS 247- subsequent derivatization 248- viscosity 248Michael addition 257microfibril structure 44mixed cellulose esters 288mixed esters 183mixed ethers 227, 233molar-mass distribution 316mole fractions 226molecular modeling 323molecular weight distribution 954-(methoxytrityl)cellulose 268MS see molecular substitution
Na-cellulose 40ff, 48- kinetics 43-modifications 41- permodoid reaction 41- phase diagram 42Na-cellulose see sodium cellulosenano-structure s 322Nioxam 79Nioxen 79fnitrating system 103nitration 103, 105ff, 110-action of N2O4 103- batch process 110- changes in supramolecular structure
108- continous process 110- course of reaction 107- equilibrium constant 106-mechanism 103- nitronium cation 105- nitronium salts 103- reaction temperature 107- stabilization process 106- substitution pattern 107- sulfate groups 106Ni-tren 8Ofnonionic mixed ethers 242Normann compound 74, 85, 90
ft-octyldimethylsilylcellulose 293oligophosphate crosslinks 138organic ester ethers 214oxidation 302ff, 308ff- content 305- formation of carbonyl groups 303- formation of carboxy, aldehyde and
keto groups 302- heterogeneous 309- homogeneous 309-partial 302- primary hydroxy groups 304- ruthenium tetroxide 308- secondary hydroxy groups 308- selective 304-with Mn(III) 308- with nitrogen dioxide 305- with periodate 304,309- with phosphoric acid 305- with sodium chlorite 311- with sodium nitrite 305oxidized cellulose 304oxycelluloses 302,312- poly electrolyte properties 312-viscosity 312
paper making 260Pd-en 81permethylation 212pervaporation membranes 132phenylcellulose 263phenyldimethylsilylcelluloses 274phosphating agents 137phosphonomethy!cellulose 261phosphoromethylcellulose 249phosphorylating agents 133phosphorylation 133ff- cellulose acetate 136- crosslinking 133- H3PO4 and urea 134- hydroxyethy !cellulose 136- with ternary systems 134photoconducting 273photosensitive side chains 290

Subject index 387
photosensitivity 292phthaloylation 193physical structure 40polyelectrolytes 131, 160, 221, 232, 308- anionic 131polymer degradation 309polymer skeleton 2ff- biosynthesis 2ff- chemical synthesis 2ff- enzymatic synthesis 2ffpolymer-analogous reactions 319polymerization 319- enzyme-catalyzed 319polyolato complex 75pore and void structure 45, 60preactivation 183, 251preferential substitution at the C-6 194process auxiliaries 260propionylcellulose 182- regioselectively substituted 182propylation 213protecting group 120, 175, 285pulping 317
quaternization 258
radical grafting 260raw material 316reductive amination 310regioselective functionalization 319- enzymatically catalyzed 319regioselectivity 271ripening 154
salt stability 233Schotten-Baumann reaction 146, 165Schotten-Baumann-type reaction 192selective membranes 323separation membranes 255silyl ethers 274silylamine 278silylation 59, 274f, 278f-control of DS 278
-DMA/LiCl 275- heterogeneous 278- homogeneous 279- regioselective 275- with chlorotrimethylsilane 274- with hexamethyldisilazane 275- with thexyldimethylchlorosilane 279silylation reagents 2746-O-sily !cellulose 174silylcellulose 127- sulfation 127silylcellulose sulfation 127f-DS 127- insertion reaction 128- mechanism 127silylethers 282, 284f- functionalization patterns 284- structural characterization 282- subsequent reactions 285sodium cellulose 4Of-formation 41sodium glycolate 221solvent 246- DMSO/paraformaldehyde 246stereoregular reactions 319structure formation 318submodifications 317substituent migration 319sulfating agents 120-reactivity 120sulfation 11 off, 124, 126, 332- chain degradation 117- chlorosulfonic acid 119- heterogeneous 116- labile ether groups 124-of CMC 124- protecting group 119- reaction rate 120- reagent distribution 120- regioselective 118-SO3/DMF 118- solvents 118- sulfating reagents 116- via nitrite groups 124- via trialkylsilyl ether groups 126sulfoalkyl ethers 260

388 Subject index
sulfoethylcellulose 249, 261sulfomethy!cellulose 261sulfopropy!cellulose 261supramolecular architectures 322fsupramolecular structures 290, 318swelling 34ff, 52, 72, 90, 165, 171- alkali uptake 35-aqueous KOH 39- chain conformation 38-13C NMR spectrum 291- ethanolic NaOH 39- hydration shell 34, 36- increase in fiber diameter 34- NaOH ion dipoles 38- steeping temperature 34- swelling agents 52- water uptake 36
tthermoreversible substitution 319thexyldimethylchlorosilane 2786-O-Thexyldimethylsilylcellulose 3706-O-Thexyldimethylsilyl-
2,3-di-O-methylcellulose 372thiocyanate route 321TMS cellulose 137, 173, 192, 225, 274,
279ff, 286, 293f, 365, 367- acylation 286- DS range of the solubility 279-LB films 293f- phosphorylation of 137- properties 280-solubility 281-structure 280- synthesis in DMA/LiCl 367- synthesis in pyridine/THF 365tosylate group 195- leaving group 195- protecting group 195tosylcellulose 142, 173, 190, 195f- acylation 195- heterogeneous procedure 195- homogeneous procedure 195- thermoanalytic characterization 196transesterification 125, 141, 146, 165,
169, 183
transxanthation 154trially!cellulose 214tri-0-(/?-bromobenzyl)cellulose 2684,4' ,4'' -trimethoxytriphenylmethyl
groups 2674,4' ,4 " -trimethoxy tritylcellulose
267trimethy!cellulose 215-WAXS 215trimethylsilylcellulose methoxyacetate
373- synthesis 373triorganosilylcelluloses 276- subsequent derivatives 276triphenylcarbinol 268triphenylmethylcellulose 263ff2,4,5-tris(hydroxymethyl)-
1,3-dioxopentamethylene 310tritylation 264ff, 269-DMA/LiCl 269- heterogeneous 264- homogeneous 265- regioselectivity 266- selectivity 264- with methoxy-substituted trityl
chlorides 265- with trityl chloride 2656-O-tritylcellulose 265, 359- preparation 265- synthesis 359tritylcellulose 263ff, 267- subsequent reactions 267two-dimensional 1W1H NMR 281
ultrathin films 293
viscose 51, 154-ripening 154viscose process 49f, 147, 321- preripening 50- slurry steeping process 50- standard alkali cellulose 50viscose ripening 158viscosity 231

Subject index 389
wwater retention value 46water uptake 52WAXS 40ff, 55, 58,61, 108WAXS see wide-angle X-ray scatteringwide-angle X-ray scattering 4OffWilliamson ether synthesis 208Williamson etherification 221WRV 61,65,245WRV see water retention value
xanthation 149ff- heterogeneous 151-kinetics 151-mechanism 151- mono- and polysaccharides 149- various polysaccharides 150xanthogenate 147, 150, 156f, 160- applications 160
- consecutive reactions 157- decomposition 147- formation 147- maximal DS 150- model experiments 147- pattern of substitution 156-properties 160- subsequent derivatization 156xanthogenate decomposition 149- rate 149xanthogenate formation 147f- energy of activation 148- industrial 147- rate constant 148xanthogenate group distribution 153xanthogenation 339xylans 258
Zincoxen 90